farmacovigilancia, nom 220 en investigación clínica · seguridad en estudios clínicos y 3) el...
TRANSCRIPT

III Congreso Nacional deInvestigación Clínica
Mayo 17-19, 2017
Farmacovigilancia, NOM 220en Investigación Clínica
QFI. Josué Bautista

1. DECLARACIÓN
Laboró en Pfizer como Líder Regional deFarmacovigilancia.
Soy miembro del comité directivo de la AsociaciónMexicana de Farmacovigilancia.
Las ideas, comentarios y opiniones que aquí expresoson a título personal y no representan la postura de laempresa en la que laborado o las organizaciones en las
que participó.

HISTORIA NATURAL DE LAFARMACOVIGILANCIA EN MEXICO
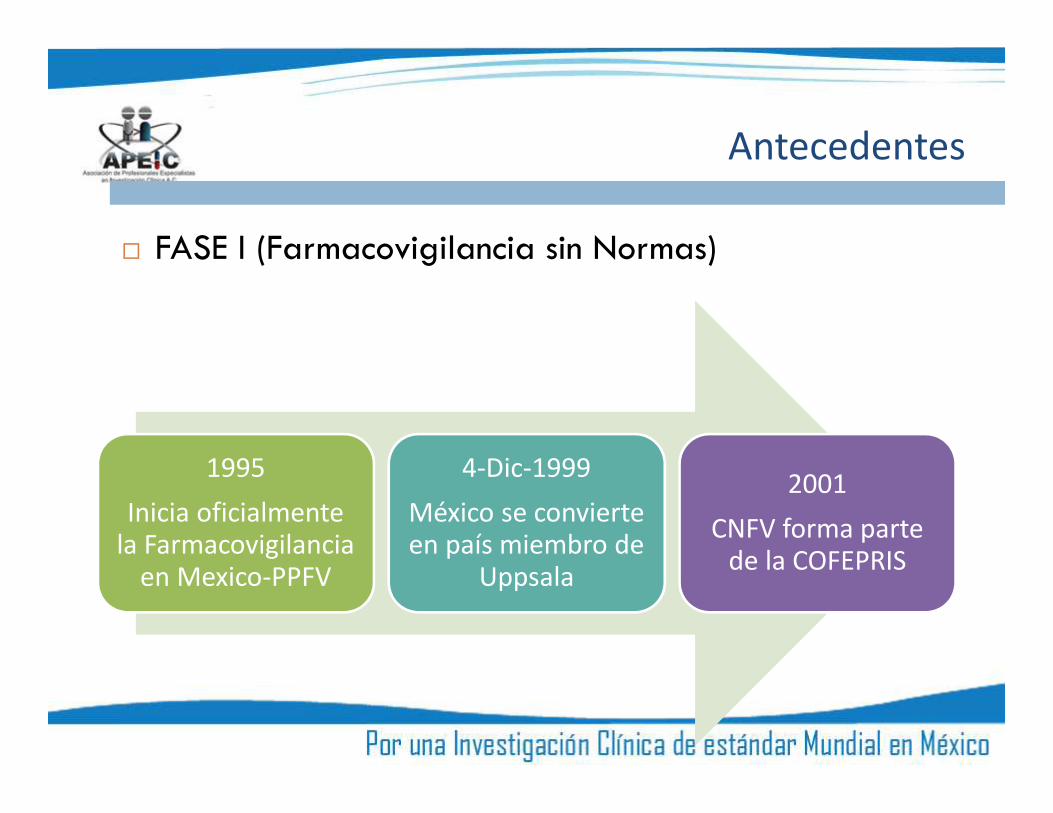
FASE I (Farmacovigilancia sin Normas)
Antecedentes
1995Inicia oficialmente
la Farmacovigilanciaen Mexico-PPFV
4-Dic-1999México se convierteen país miembro de
Uppsala
2001CNFV forma parte
de la COFEPRIS

FASE 2 (Farmacovigilancia con Normas)
Antecedentes
Versión 01 (NOM-220-SSA1-2002)
Publicada el 15 de noviembre de 2004 enel Diario Oficial de la Federación.
En vigor desde el 15 de enero de 2005.
Versión 02 (NOM-220-SSA1-2012)
Publicada el 7 de Enero de 2013 en elDiario Oficial de la Federación.
En vigor desde el 7 de Abril de 2013.

PNN* 2015 y 2016 (DOF:24/04/2015) ModificaciónNorma Oficial MexicanaNOM-220-SSA1-2015,
Instalación y operación dela farmacovigilancia.
Convocatoria de la COFEPRIS agrupo de expertos para la
elaboración o modificación de laNOM
29 de julio 2015
Integración del Subcomité conrepresentantes de COFEPRIS, Cámarasindustriales, Asociaciones Profesionales,
Instituciones Académicas y otrasInstituciones Gubernamentales
1ª reunión en COFEPRIS -14 de agosto 2015
Comités Consultivos Nacionalesen materia de Salud
- De Regulación y FomentoSanitario: SSA1- De Prevención y Control deEnfermedades: SSA2- De Innovación, Desarrollo,Tecnologías e Información enSalud: SSA3*PNN: Programa Nacional deNormalización
AntecedentesFASE 3 (La nueva era)
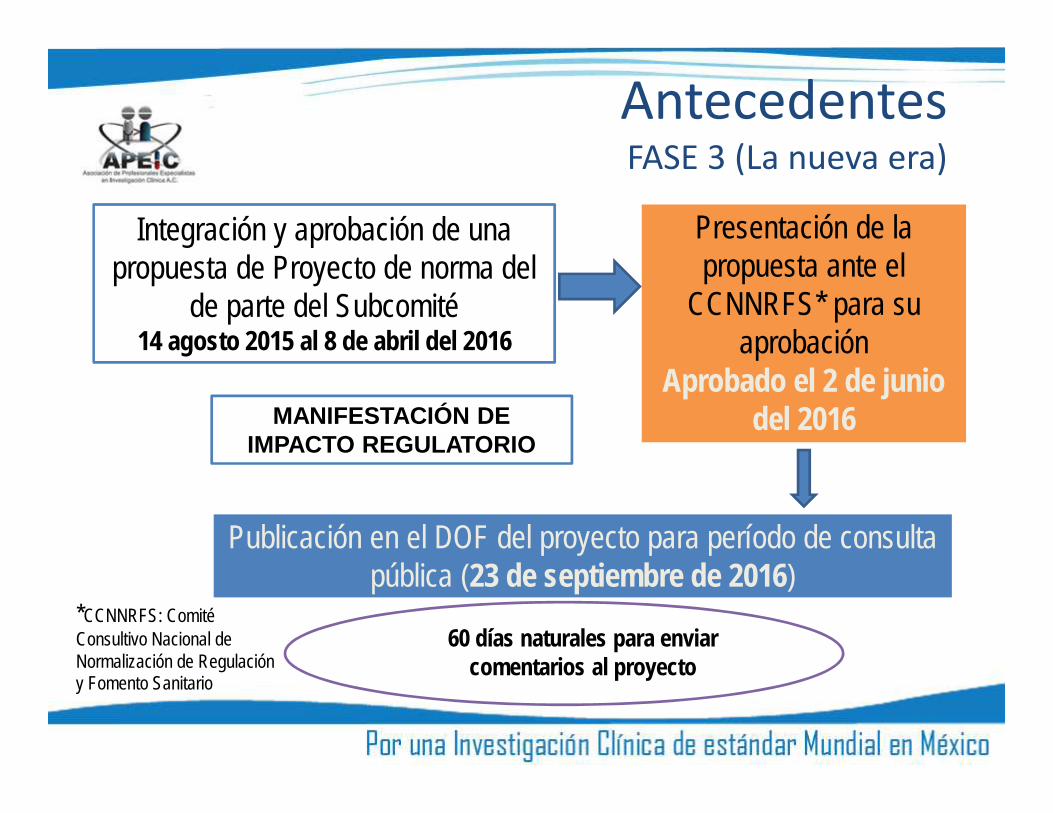
Integración y aprobación de unapropuesta de Proyecto de norma del
de parte del Subcomité14 agosto 2015 al 8 de abril del 2016
Presentación de lapropuesta ante el
CCNNRFS* para suaprobación
Aprobado el 2 de juniodel 2016
Presentación de lapropuesta ante el
CCNNRFS* para suaprobación
Aprobado el 2 de juniodel 2016
Publicación en el DOF del proyecto para período de consultapública (23 de septiembre de 2016)
Publicación en el DOF del proyecto para período de consultapública (23 de septiembre de 2016)
60 días naturales para enviarcomentarios al proyecto
MANIFESTACIÓN DEIMPACTO REGULATORIO
AntecedentesFASE 3 (La nueva era)
*CCNNRFS: ComitéConsultivo Nacional deNormalización de Regulacióny Fomento Sanitario

Revisión de los comentarios por parte del grupo de trabajo deCOFEPRIS
Publicación de la respuesta a comentarios en el DOF
Publicación de la Norma Oficial Mexicana en el DOF(indicará la fecha de entrada en vigor)
Publicación de la Norma Oficial Mexicana en el DOF(indicará la fecha de entrada en vigor)
Presentación de la respuestas a cometarios y la versióndefinitiva de la NOM ante el CCNNRFS para su aprobación
15 de diciembre de 2016
AntecedentesFASE 3 (La nueva era)

LA FARMACOVIGILANCIA APLICADAA LA INVESTIGACIÓN CLÍNICA:LA EVOLUCIÓN

Capitulo 1: Las bases
• El primer modelo de operación de laFarmacovigilanca (FV) en Mexico, NOM-220-SSA1-2002, inició en Noviembre 2004 con laregulación de tres elementos esenciales paraconstruir un sistema de FV:– El reporte individual de casos– El reporte de seguridad en estudios clínicos– El reporte periódico de seguridad

Capitulo 1: Las bases
El reporte de seguridad en estudiosclínicos
Para todos aquellos que cuenten con al menos un centroen México se estableció además del envío expedito de los
casos graves en Mexico, un reporte de frecuenciasemestral así como un reporte final.
Quedando pendiente para una segunda fase la definiciónde los elementos mínimos que debiesen integrarlos y los
periodos límite para su presentación ante el CNFV.

Capitulo 2: El aquí y el ahora
• El segundo modelo de operación de la FV en Mexico,NOM-220-SSA1-2012, se publicó en el DOF el 7 deEnero de 2013– Se precisaron y robustecieron los tres elementos
esenciales para construir un sistema de Farmacovigilancia,1) El reporte individual de casos, 2) El reporte deseguridad en estudios clínicos y 3) El reporte periódico deseguridad
– Se amplió el universo de los participantes y responsablesdel sistema de Farmacovigilancia en México
– Se diferenciaron los elementos y métodos de aplicaciónde la FV entre medicamentos y vacunas
– Se instrumentaron una serie de guías quecomplementarían y favorecerían a la realización de la FVen el país.

Capitulo 2: El aquí y el ahora
• Se incluyó el componente que sin dudas representó yrepresenta ser una de las acciones y retos de mayor impactoen la FV, en principio de cuentas con alcance primario a lasmoléculas nuevas que se pretendan registrar en México:
El Plan de Manejo de Riesgos(RMP por sus siglas en inglés).

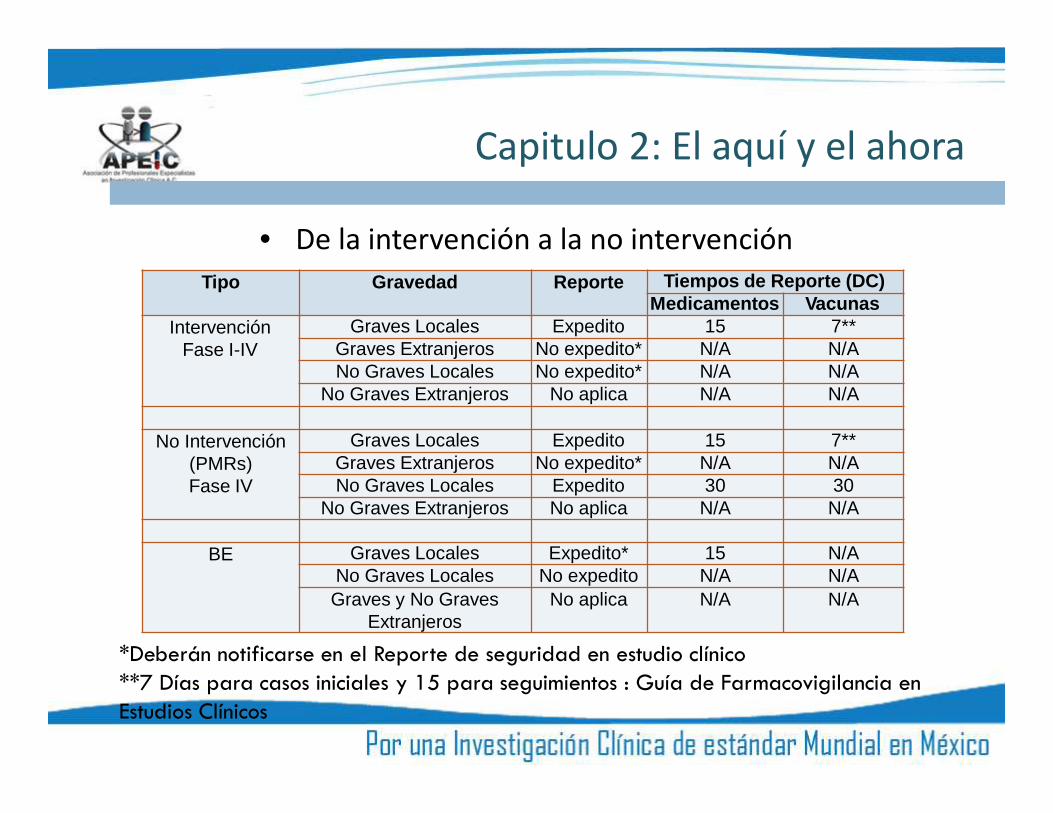
Capitulo 2: El aquí y el ahora
• De la intervención a la no intervenciónTipo Gravedad Reporte Tiempos de Reporte (DC)
Medicamentos VacunasIntervención
Fase I-IVGraves Locales Expedito 15 7**
Graves Extranjeros No expedito* N/A N/ANo Graves Locales No expedito* N/A N/A
No Graves Extranjeros No aplica N/A N/A
No Intervención(PMRs)Fase IV
Graves Locales Expedito 15 7**Graves Extranjeros No expedito* N/A N/ANo Graves Locales Expedito 30 30
No Graves Extranjeros No aplica N/A N/A
BE Graves Locales Expedito* 15 N/ANo Graves Locales No expedito N/A N/AGraves y No Graves
ExtranjerosNo aplica N/A N/A
*Deberán notificarse en el Reporte de seguridad en estudio clínico**7 Días para casos iniciales y 15 para seguimientos : Guía de Farmacovigilancia enEstudios Clínicos

Capitulo 2: El aquí y el ahora
• De la intervención a la no intervención
BE
No
Inte
rven
ción
:Fas
eIV
Inte
rven
ción
:Fas
eI-I
V
Reporte al finaldel estudio
Título del estudio, Númerode protocolo. Objetivos dela Investigación,Descripción de lametodología deinvestigación, Resultadosdel estudio
Tabla de los EAs nograves localesTabla de los EAsgraves localesTabla de los EAs graves(relaciondas) extranjeros
INTERVENCIÓN
Reporte semestraly final
Título del estudio, Fecha deaprobación, Códigoasignado por el CNFV,Periodo del reporte,Número de pacientes a losque se le hayaadministrado eltratamiento, Número dedosis administradas,Eventualidades en eltranscurso, AnálisisEstadístico, Discusión,Conclusiones
Tabla de los EAsgraves/no graveslocalesTabla de los EAsgraves/no gravesextranjeros
NOINTERVENCIÓN
Reporte al finaldel estudio
Título del estudio, númerode protocolo,objetivos de laInvestigación, resultadosdel estudio, análisis yconclusiones.
EAs graves/no graveslocales
BIOEQUIVALENCIA
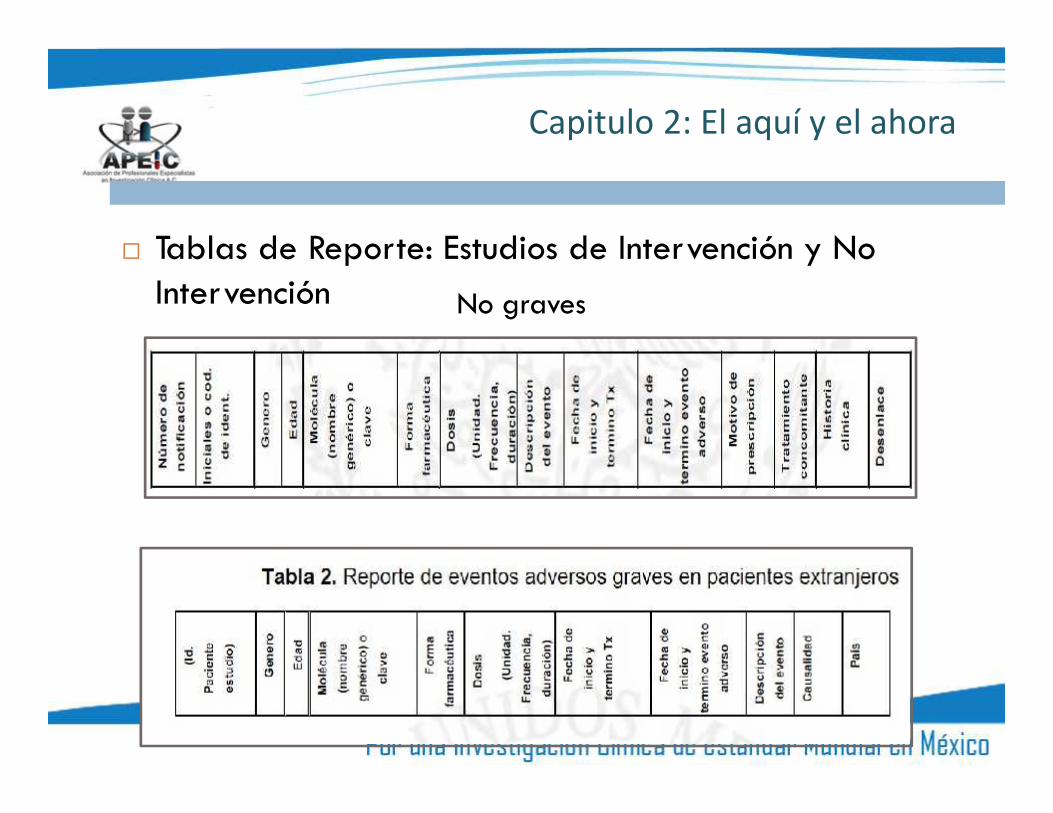
Capitulo 2: El aquí y el ahora
Tablas de Reporte: Estudios de Intervención y NoIntervención No graves

Capitulo 2: El aquí y el ahora
Tablas de Reporte: Estudios de Bioequivalencia

Capitulo 3: Lo que viene
• Notificaciones– Aviso al CNFV de los protocolos de estudios clínicos autorizados por CAS (7.4.2.8)
• Tiempo: No definido en la NOM• Responsable: Titular del Registro Sanitario ó su Representante Legal
– Aviso al CNFV de la cancelación, suspensión, descontinuación y/o reanudación(incluyendo las razones) de todos los estudios clínicos patrocinados y que cuenten con almenos un centro de investigación en México. (7.4.2.10, 7.5.2, 7.5.4)
• Tiempo: 10 15 días hábiles• Responsables: Titular del Registro Sanitario/Representante Legal Y las instituciones
o establecimientos donde ser realiza investigación

Capitulo 3: Lo que viene
• Reporte de seguridad– Alcance– El Reporte de seguridad de Estudios Clínicos, aplica para todos los estudios clínicos,
patrocinados o no, incluyendo tanto a los de bioequivalencia, como a los deFarmacovigilancia que cuenten con al menos un sitio o centro de investigación enMéxico.
• El sometimiento al CNFV de los reportes de seguridad de estudios clínicos, tanto deseguimiento como finales, deberán ser a través del CIS, de conformidad con losiguiente:
– En el caso de estudios patrocinados por titulares de registro o susrepresentantes legales, el sometimiento deberá ser realizado por el titular deregistro sanitario o persona autorizada por éste.
– En el caso de estudios a iniciativa del investigador, el sometimiento deberá serrealizado por el investigador principal responsable del estudio autorizado opersona autorizada.

Capitulo 3: Lo que viene
• Reporte de seguridad– Frecuencia y Contenido
– Responsabilidad de instituciones o establecimientos que realizan investigación oinvestigadores independientes (8.3.2):
• Reportes de seguridad de estudios clínicos de seguimiento• Reportes de seguridad de estudios clínicos finales.
– Mecanismo (8.3.3)• CIS
– Estudios patrocinados por titular de Registro/Representante Legal– Estudios a iniciativa del investigador por Investigador/Persona autorizada

Capitulo 3: Lo que viene
• Reporte de seguridad– Frecuencia y Contenido– Periodicidad (8.3.3.3)
• Anual a partir de la 1ª autorización nacional del estudio• BE solo reporte final• Aviso de finalización de etapa clínica del estudio• Reporte de seguridad final:
– Al finalizar el análisis de la información colectada– En caso de cancelación o descontinuación definitiva del estudio.
– Contenido (8.3.4)
• Referencia: Guía de FV en Investigación Clínica.

