enfermedades del sistema inmunitario
TRANSCRIPT

Universidad Autónoma del Estado de Baja
California
Escuela de Ciencias de la Salud
Unidad Valle Dorado
Enfermedades Del sistema
inmunitario.
Patología Básica, Dra. Wendolyn Flores.BELTRÁN RAMÍREZ DANIELA F.
CÓRDOVA ULBRICH RAÚL
LEM QUIROZ ROGELIO
ORTIZ FLORES HÉCTOR M.
ROBLES RIDAURA KAREN GPE.
Respuesta inmunitaria
normal Respuesta inmunitaria
normal
Inmunidad innata
Inmunidad adaptativa
Primera línea de defensa Por mecanismos
de estimulación
Lem Quiroz Rogelio
Inmunidad innata
Componentes :
Barreras epiteliales
Células fagociticas
Células dendríticas,
natural killers , sistema de
complemento
Lem Quiroz Rogelio
Inmunidad innata Dos reacciones celulares: inflamación y
defensa antimicrobiana (virus, parásitos etc.)
Patrones moleculares asociados al peligro
Patrones moleculares asociados a patógenos
Receptores celulares. Receptores para el reconocimientos de patrones
TLR
Lem Quiroz Rogelio
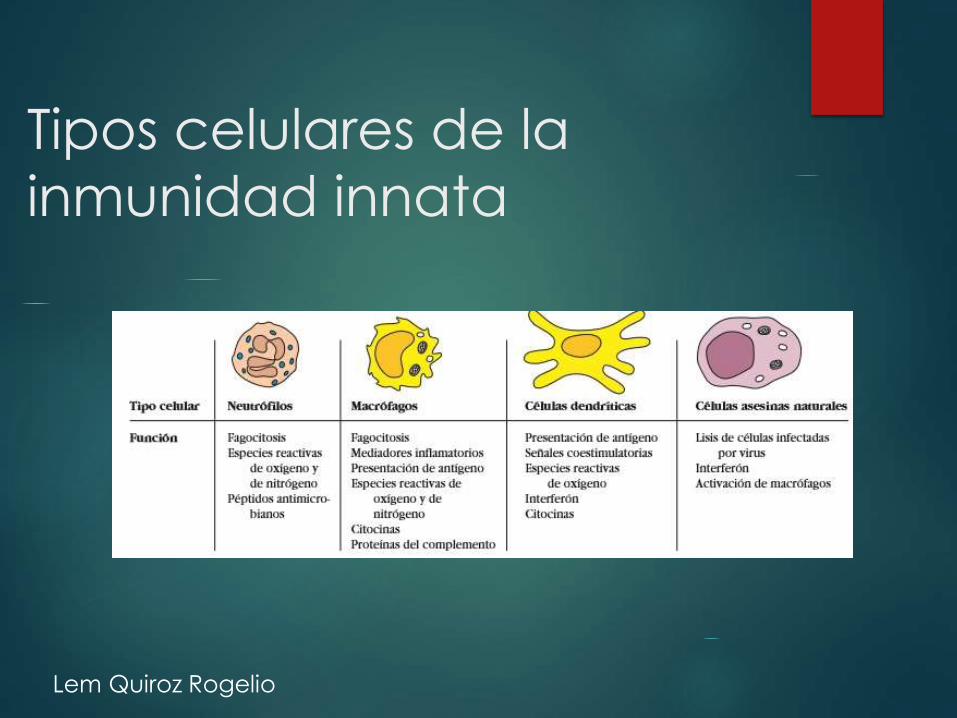
Tipos celulares de la
inmunidad innata
Lem Quiroz Rogelio

Inmunidad adaptativa Formado por linfocitos y sus productos como
los anticuerpos.
Dos tipos:
Humoral• Protege contra microorganismos,
mediada por linfocitos B
Celular • Responsable de la defensa de microorganismos intracelulares
Lem Quiroz Rogelio
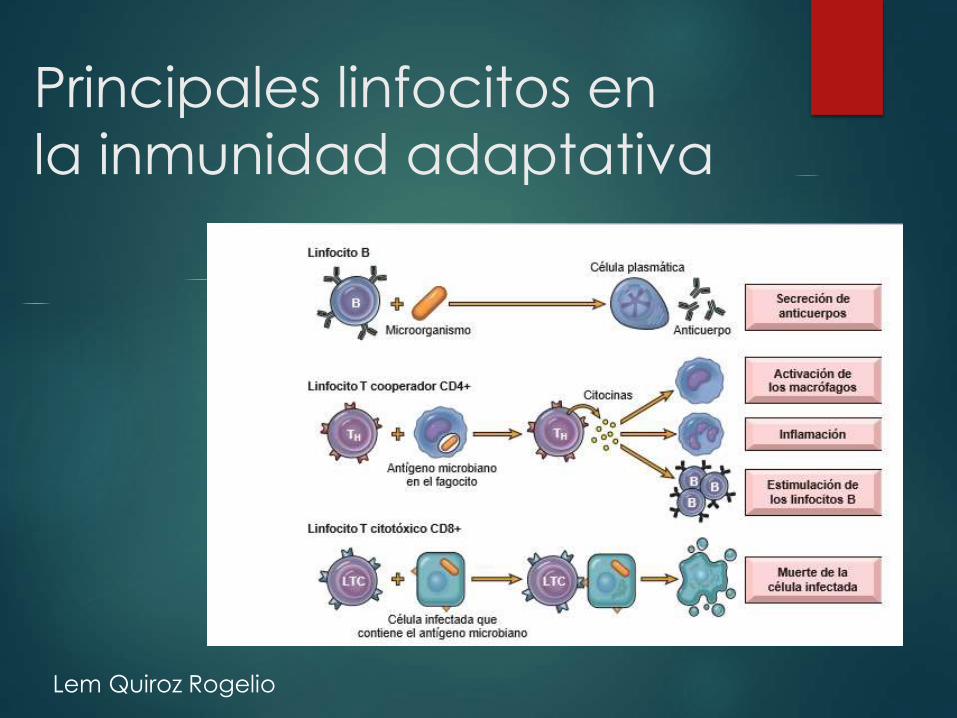
Principales linfocitos en
la inmunidad adaptativa
Lem Quiroz Rogelio

Componentes
del sistema
inmunitario:
células,
tejidos y
moléculas
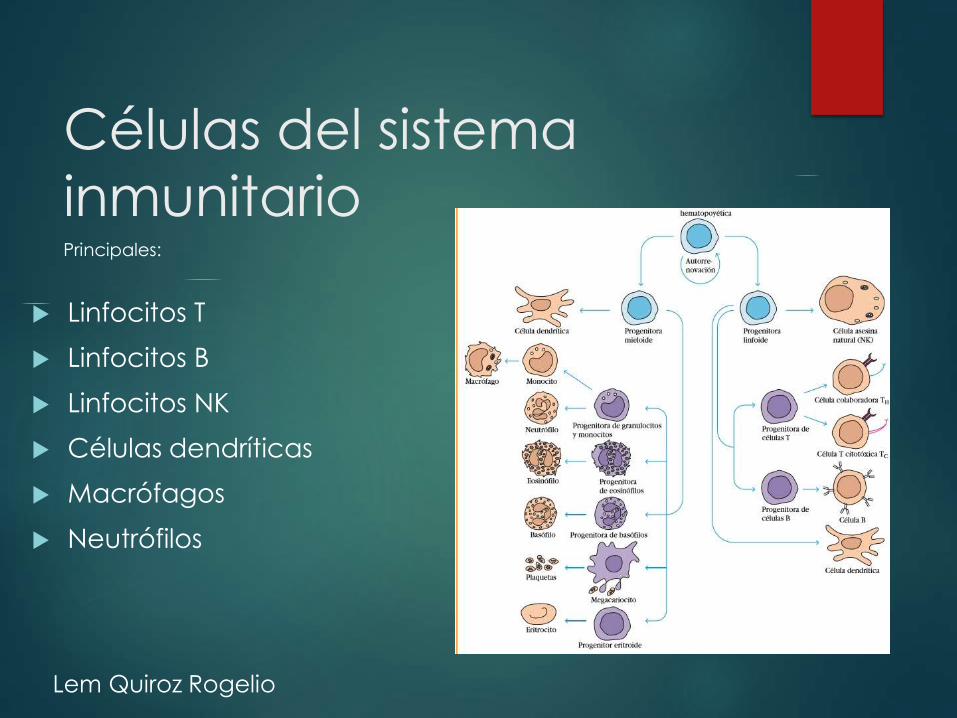
Células del sistema
inmunitario
Linfocitos T
Linfocitos B
Linfocitos NK
Células dendríticas
Macrófagos
Neutrófilos
Principales:
Lem Quiroz Rogelio
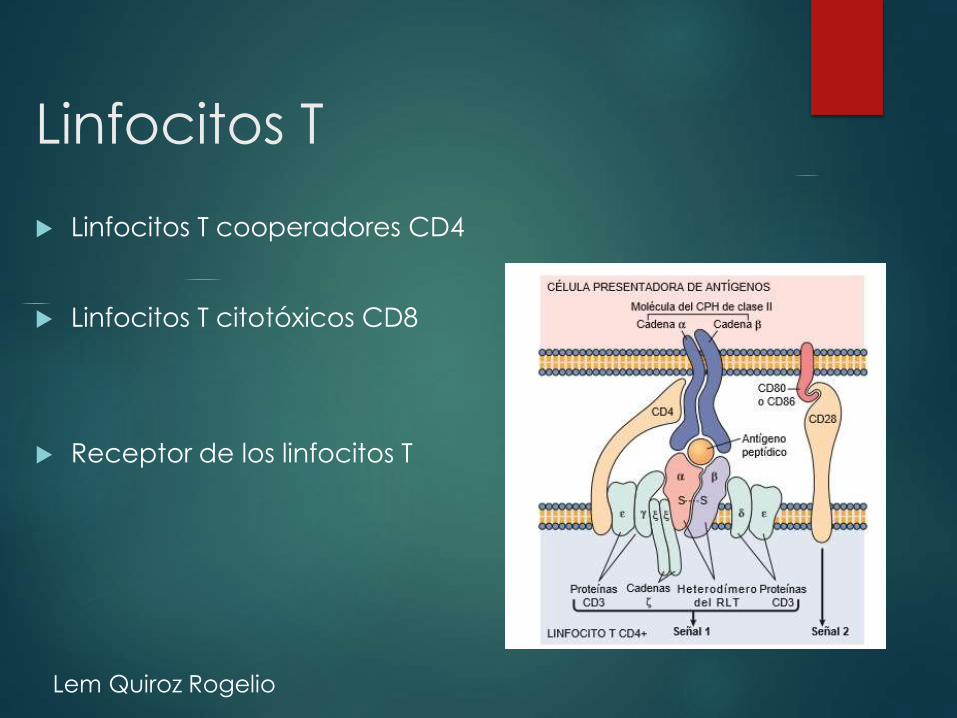
Linfocitos T
Linfocitos T cooperadores CD4
Linfocitos T citotóxicos CD8
Receptor de los linfocitos T
Lem Quiroz Rogelio
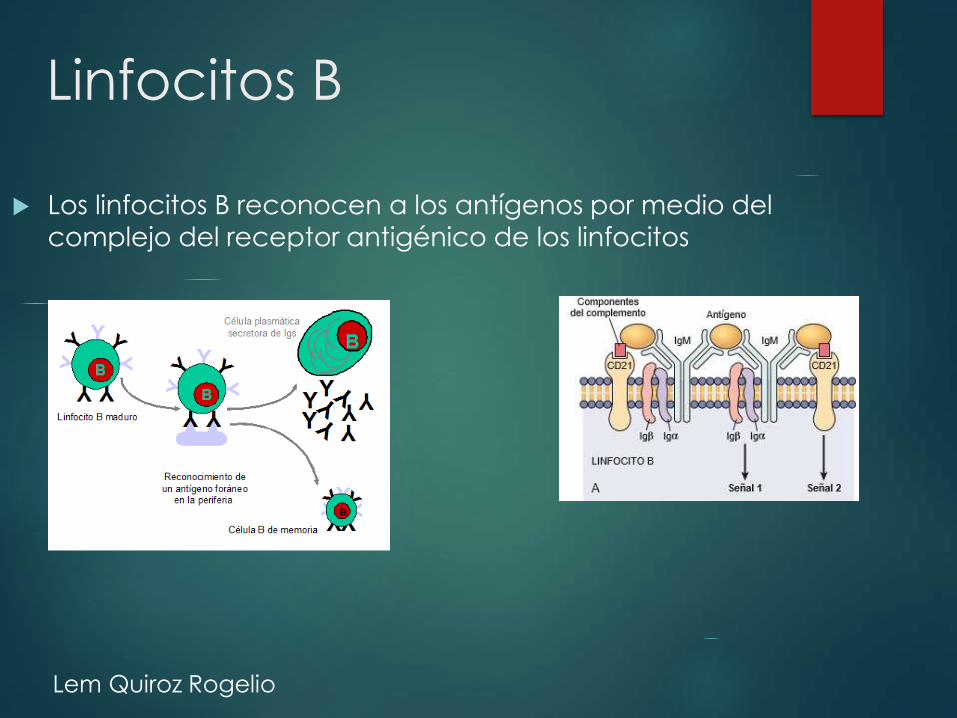
Linfocitos B
Los linfocitos B reconocen a los antígenos por medio del
complejo del receptor antigénico de los linfocitos
Lem Quiroz Rogelio

Células dendríticas Células dendríticas interdigitadas presentadoras
de antígenos
Características y funciones:
Se encuentran debajo de los epitelios, expresan receptores y son atraídas por linfocitos T.
Células dendríticas foliculares participan en la presentación a linfocitos B
Lem Quiroz Rogelio

macrófagos Forma parte del sistema fagocitico
mononuclear
Funciones
Participan en la activación de
linfocitos
Destruir microorganismos
intracelulares
Fase efectora de la inmunidad
celular
Lem Quiroz Rogelio

Linfocitos citolíticos
naturales De 10 a 15 % de los linfocitos en sangre
Son la primera línea de defensa frente a las infecciones
víricas
Tienen 2 moléculas que los identifican: CD16 CD56
Receptor Fc para IgG
Lem Quiroz Rogelio
Tejidos del
sistema inmune
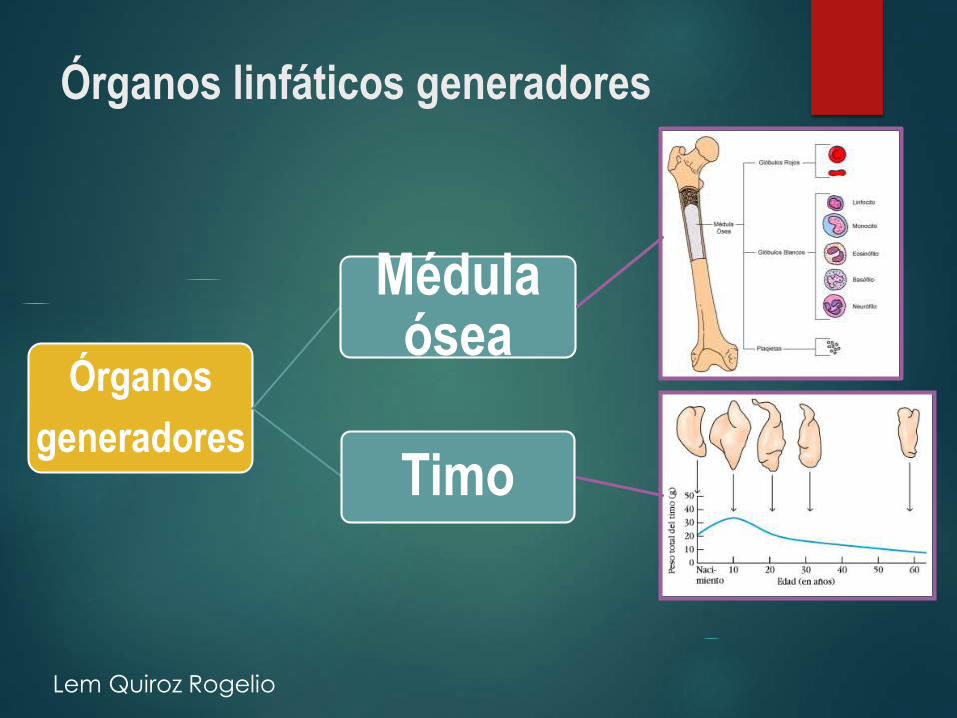
Órganos linfáticos generadores
Órganos
generadores
Médula ósea
Timo
Lem Quiroz Rogelio
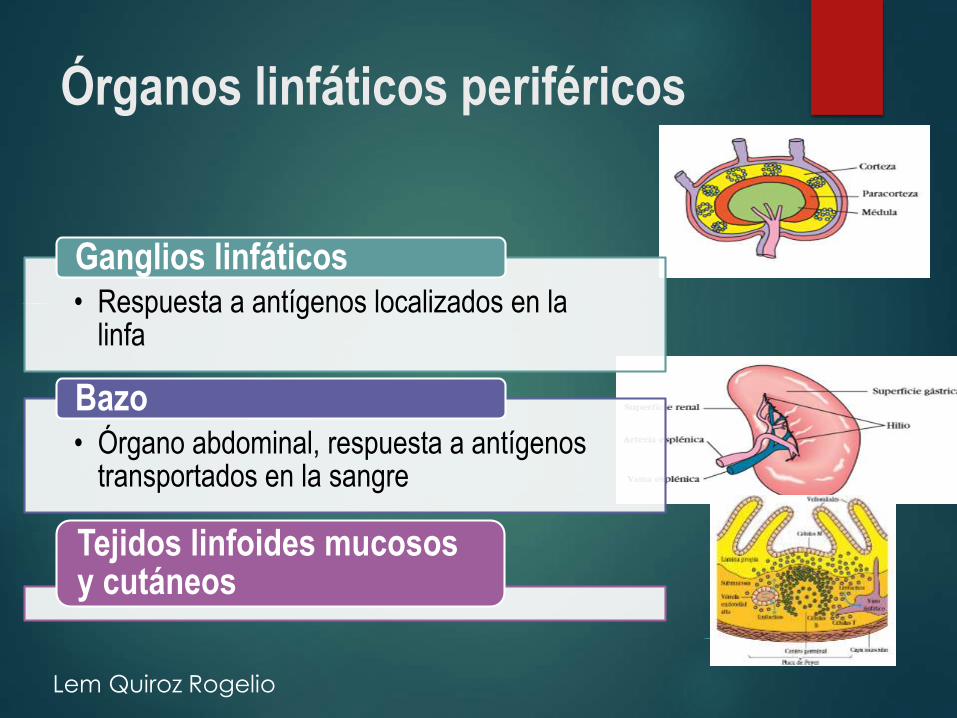
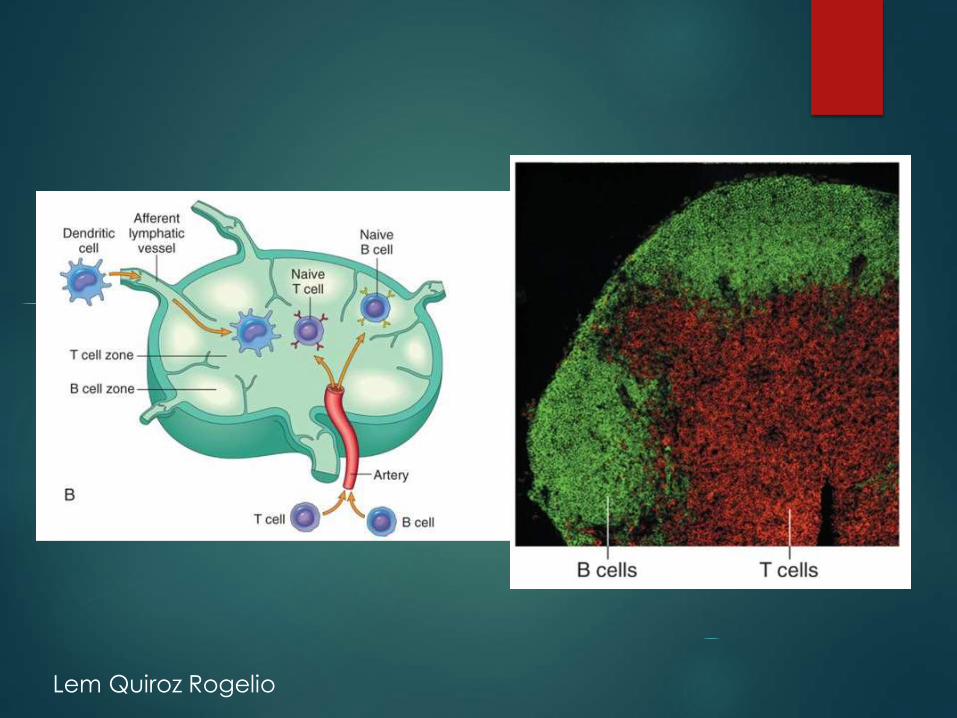
Órganos linfáticos periféricos
• Respuesta a antígenos localizados en la linfa
Ganglios linfáticos
• Órgano abdominal, respuesta a antígenos transportados en la sangre
Bazo
Tejidos linfoides mucosos y cutáneos
Lem Quiroz Rogelio
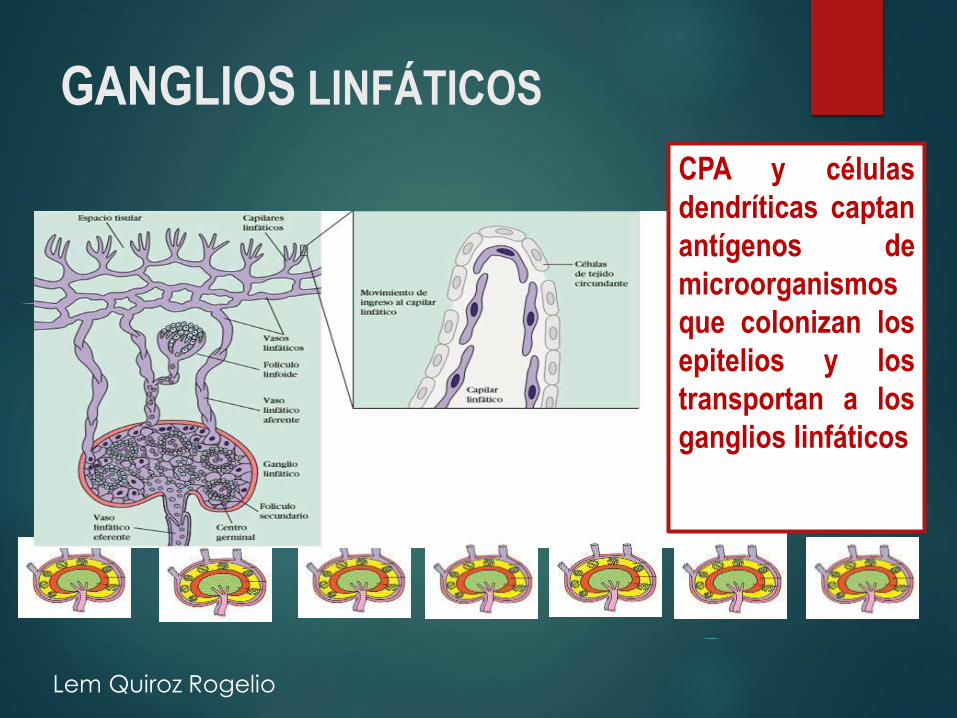
GANGLIOS LINFÁTICOS
CPA y células
dendríticas captan
antígenos de
microorganismos
que colonizan los
epitelios y los
transportan a los
ganglios linfáticos
Lem Quiroz Rogelio
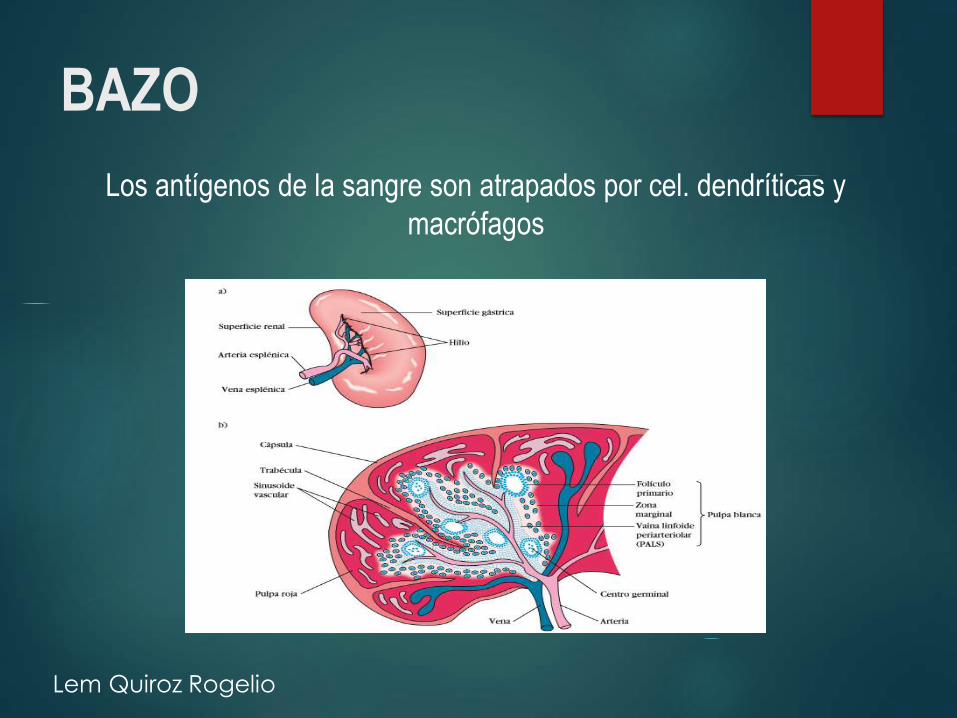
BAZO
Los antígenos de la sangre son atrapados por cel. dendríticas y
macrófagos
Lem Quiroz Rogelio

TEJIDOS LINFOIDES MUCOSOS Y
CUTANEOS MALT BALT Y GALT
Responden a antígenos
que entran a través de
aberturas de los epitelios.
Lem Quiroz Rogelio
Lem Quiroz Rogelio
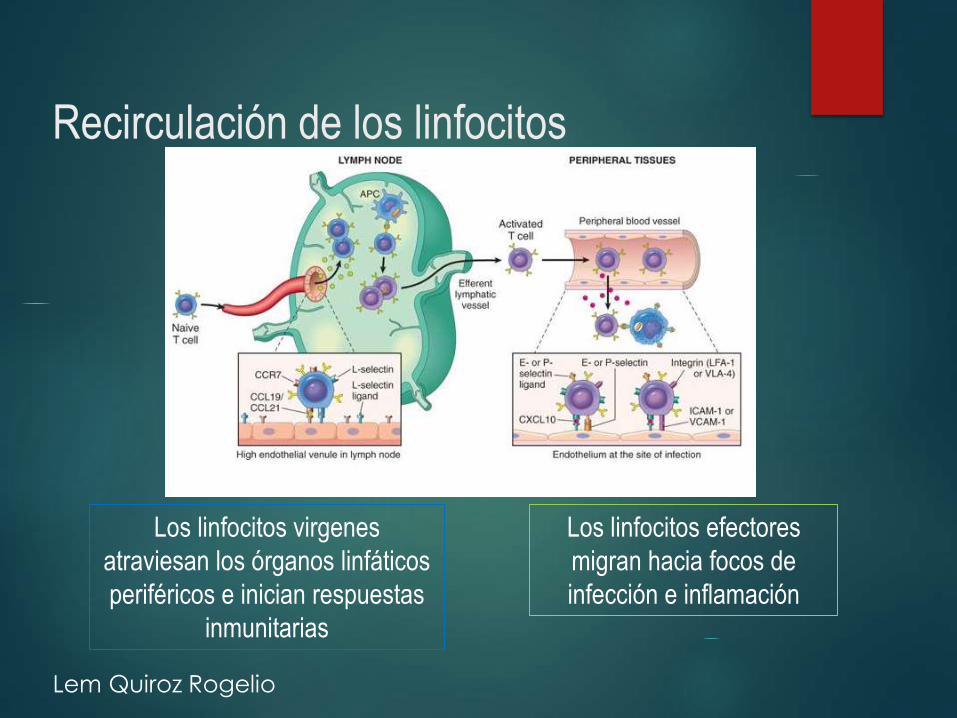
Recirculación de los linfocitos
Los linfocitos virgenes
atraviesan los órganos linfáticos
periféricos e inician respuestas
inmunitarias
Los linfocitos efectores
migran hacia focos de
infección e inflamación
Lem Quiroz Rogelio

Complejo Mayor de
HistocompatibilidadPresentación de péptidos
de la inmunidad adaptativa
Lem Quiroz Rogelio
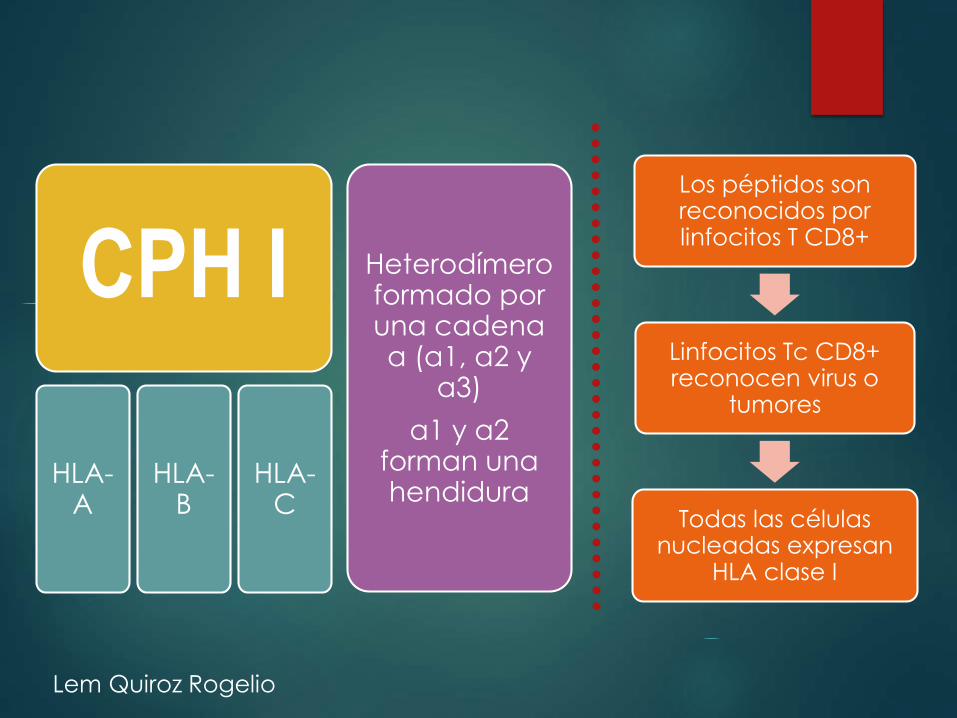
CPH I
HLA-A
HLA-B
HLA-C
Heterodímeroformado por una cadena α (α1, α2 y
α3)
α1 y α2 forman una hendidura
Los péptidos son reconocidos por linfocitos T CD8+
Linfocitos Tc CD8+ reconocen virus o
tumores
Todas las células nucleadas expresan
HLA clase I
Lem Quiroz Rogelio
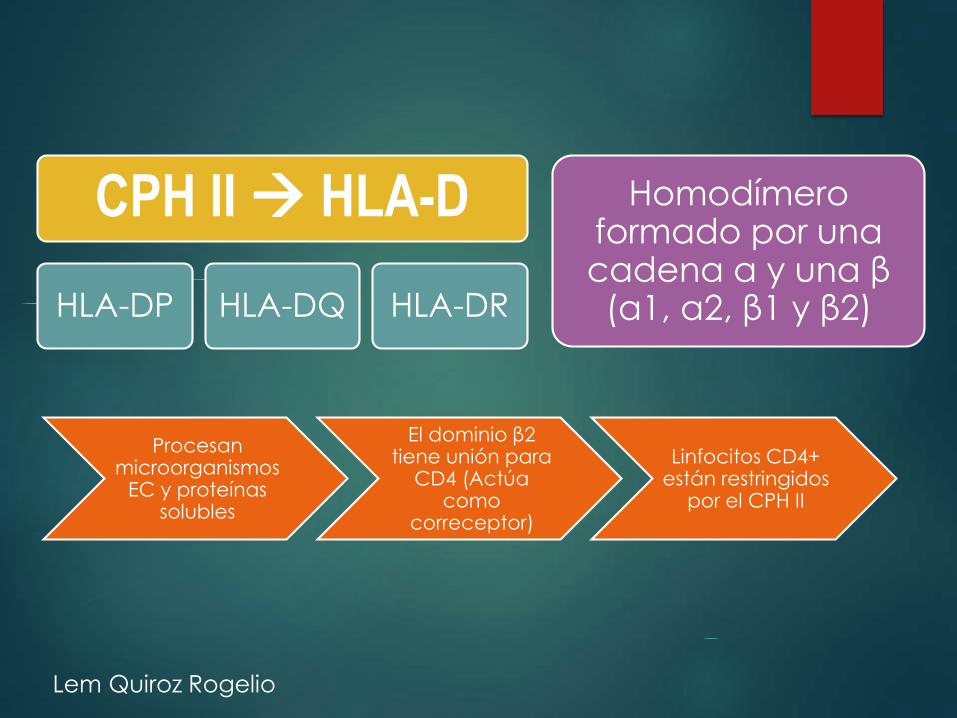
CPH II HLA-D
HLA-DP HLA-DQ HLA-DR
Homodímeroformado por una cadena α y una β(α1, α2, β1 y β2)
Procesan microorganismos
EC y proteínas solubles
El dominio β2 tiene unión para
CD4 (Actúa como
correceptor)
Linfocitos CD4+ están restringidos
por el CPH II
Lem Quiroz Rogelio
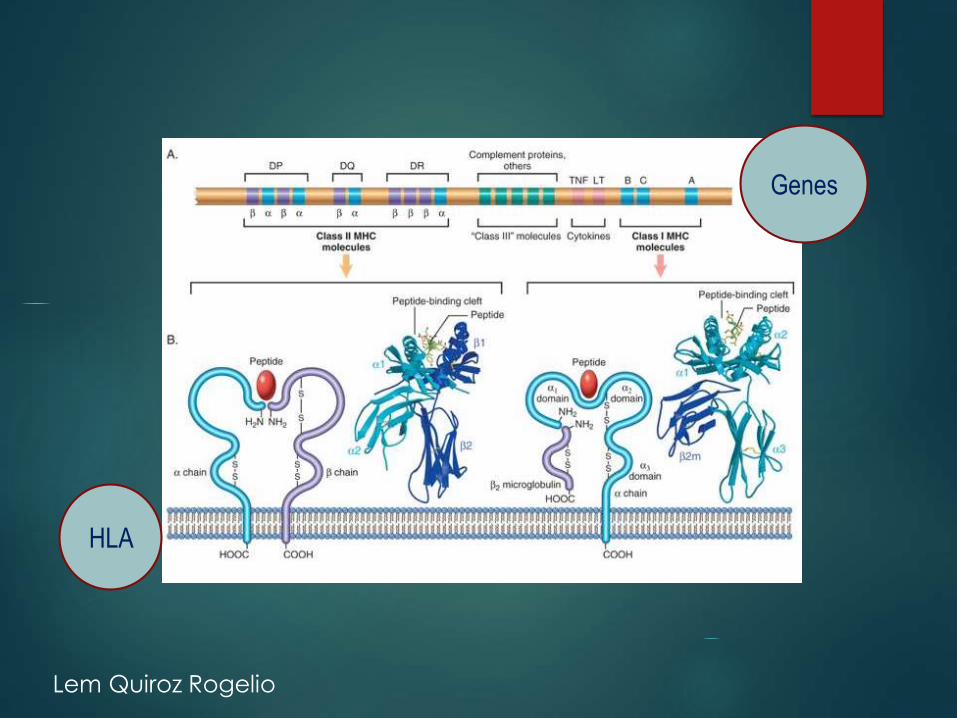
HLA
Genes
Lem Quiroz Rogelio
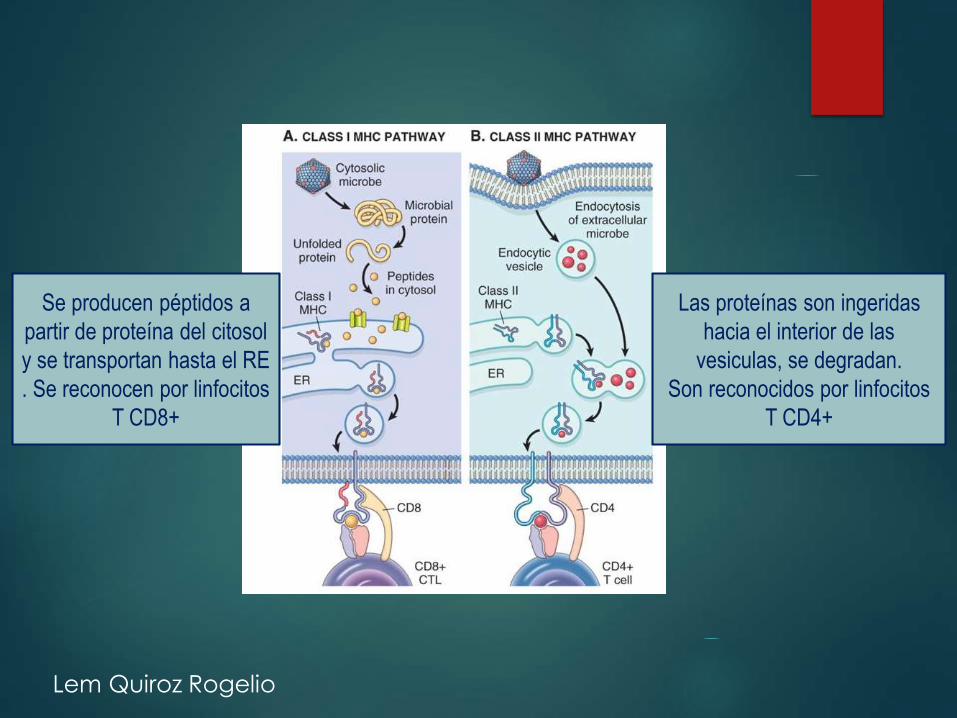
Se producen péptidos a
partir de proteína del citosol
y se transportan hasta el RE
. Se reconocen por linfocitos
T CD8+
Las proteínas son ingeridas
hacia el interior de las
vesiculas, se degradan.
Son reconocidos por linfocitos
T CD4+
Lem Quiroz Rogelio
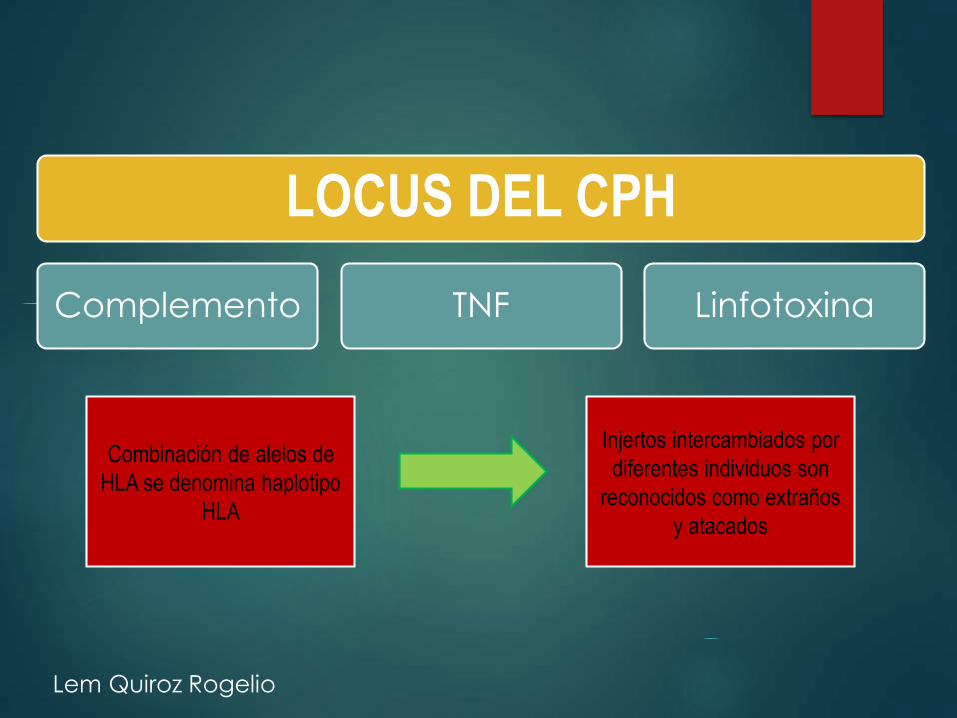
LOCUS DEL CPH
Complemento TNF Linfotoxina
Combinación de alelos de
HLA se denomina haplotipo
HLA
Injertos intercambiados por
diferentes individuos son
reconocidos como extraños
y atacados
Lem Quiroz Rogelio

HLA Asociación con enfermedades
Asociación de los alelos HLA con enfermedades inflamatorias
Enfermedad Alelo HLA Riesgo relativo (%)
Espondilitis anquilosante B27 90 - 100
Artritis posgonocócica B27 14
Uveítis anterior aguda B27 14
Artritis reumatoide DR4 4
Hepatitis crónica activa DR3 13
Sx de Sjorgren primario DR3 9
DM tipo 1 DR3
DR4
DR3/DR4
5
6
20
Lem Quiroz Rogelio

CITOCINAS
Moléculas mensajeras
del sistema inmunitario
Lem Quiroz Rogelio
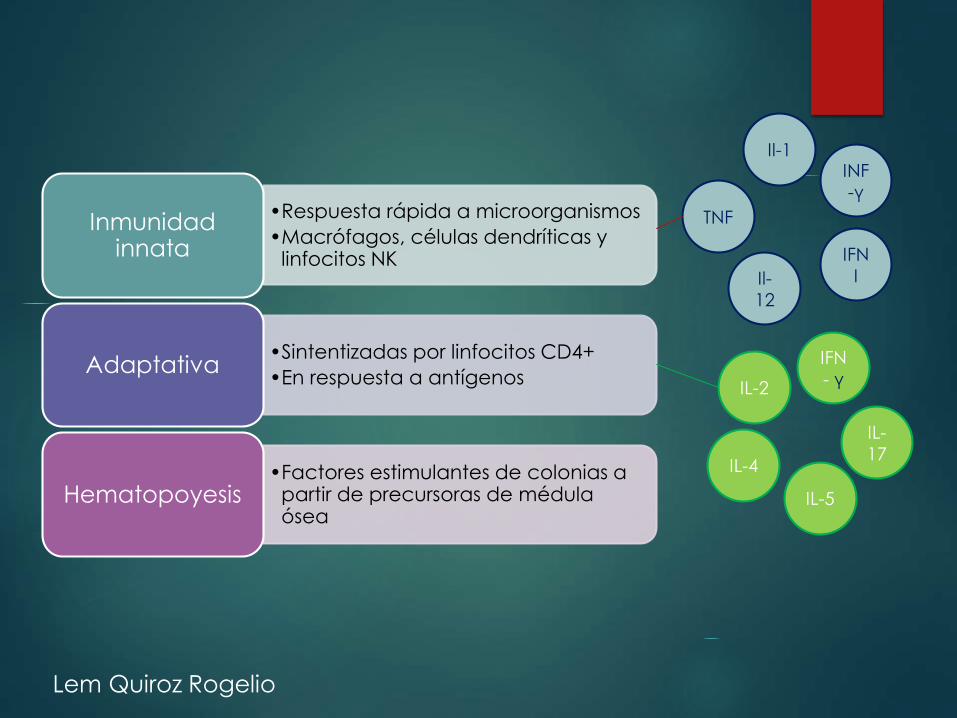
•Respuesta rápida a microorganismos
•Macrófagos, células dendríticas y linfocitos NK
Inmunidadinnata
•Sintentizadas por linfocitos CD4+
•En respuesta a antígenosAdaptativa
•Factores estimulantes de colonias a partir de precursoras de médulaósea
Hematopoyesis
TNF
Il-12
Il-1INF-γ
IFN I
IL-2
IL-4
IL-5
IL-17
IFN- γ
Lem Quiroz Rogelio
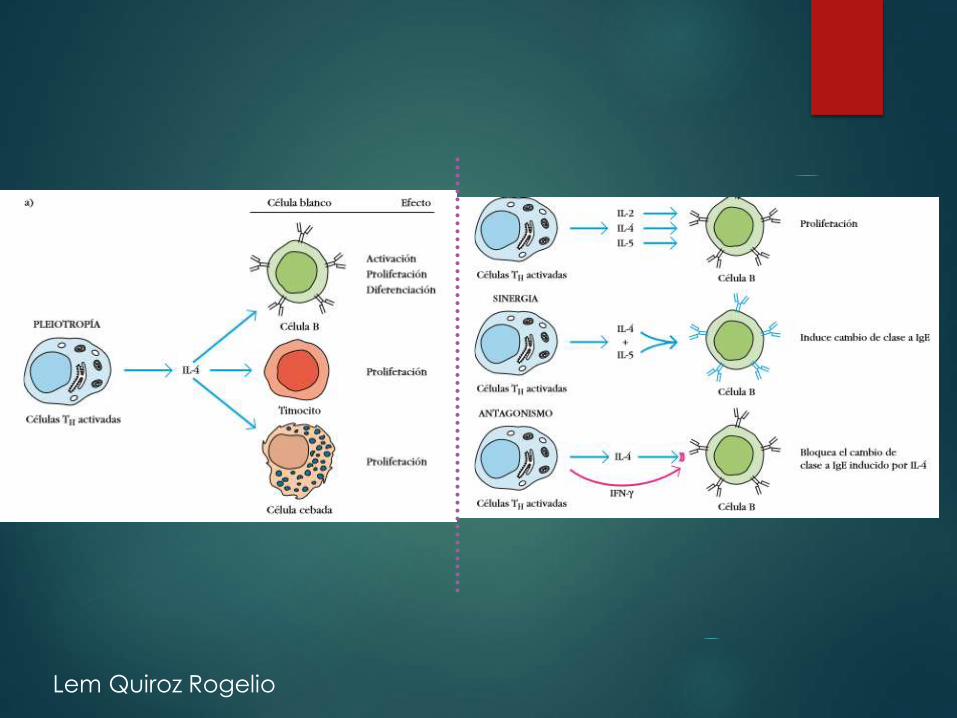
Lem Quiroz Rogelio

Activación de
linfocitos y
respuestas
inmunitarias
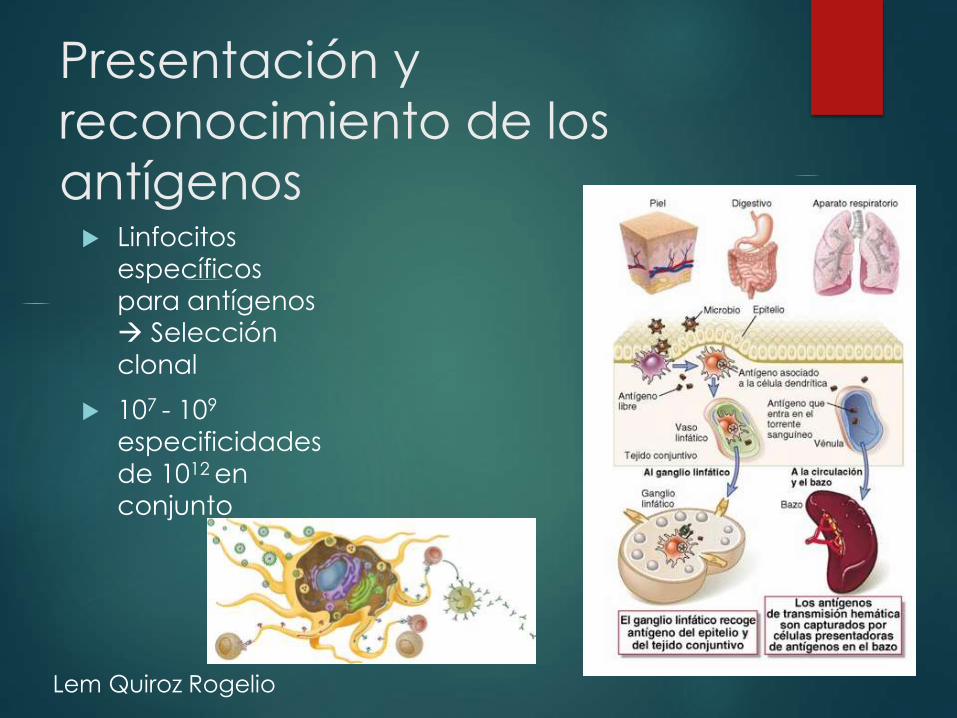
Presentación y
reconocimiento de los
antígenos Linfocitos
específicos
para antígenos Selección
clonal
107 - 109
especificidades
de 1012 en
conjunto
Lem Quiroz Rogelio
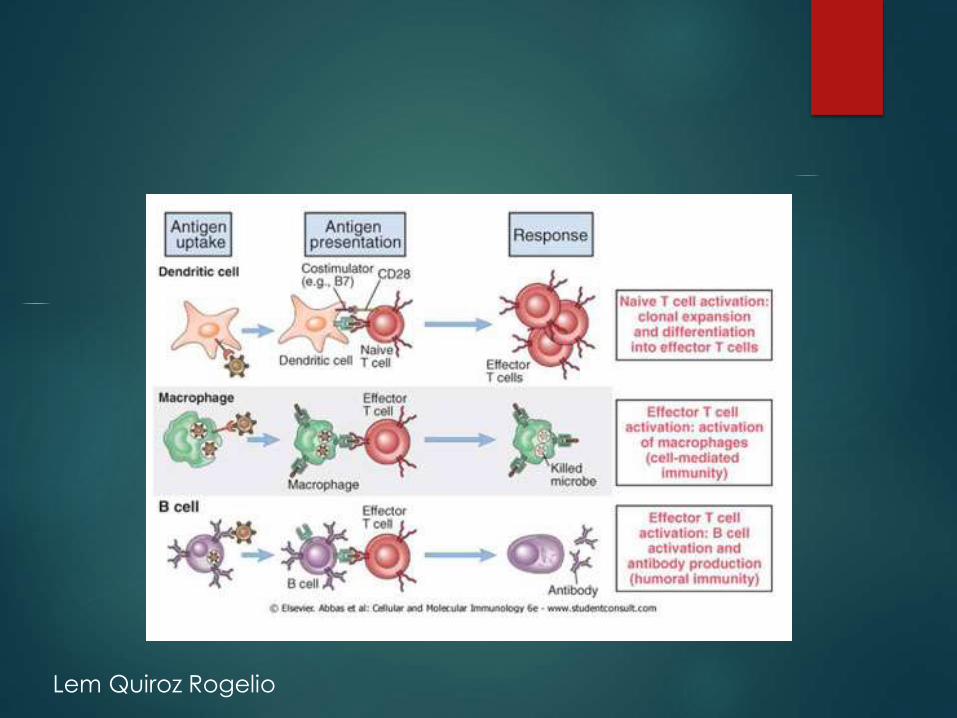
Lem Quiroz Rogelio
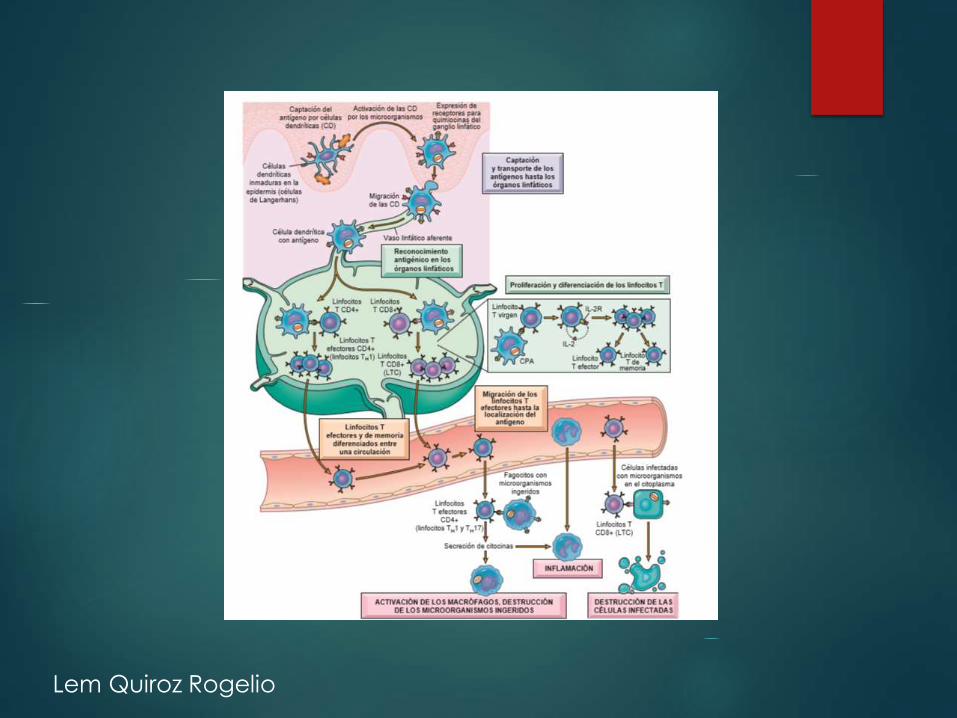
Lem Quiroz Rogelio
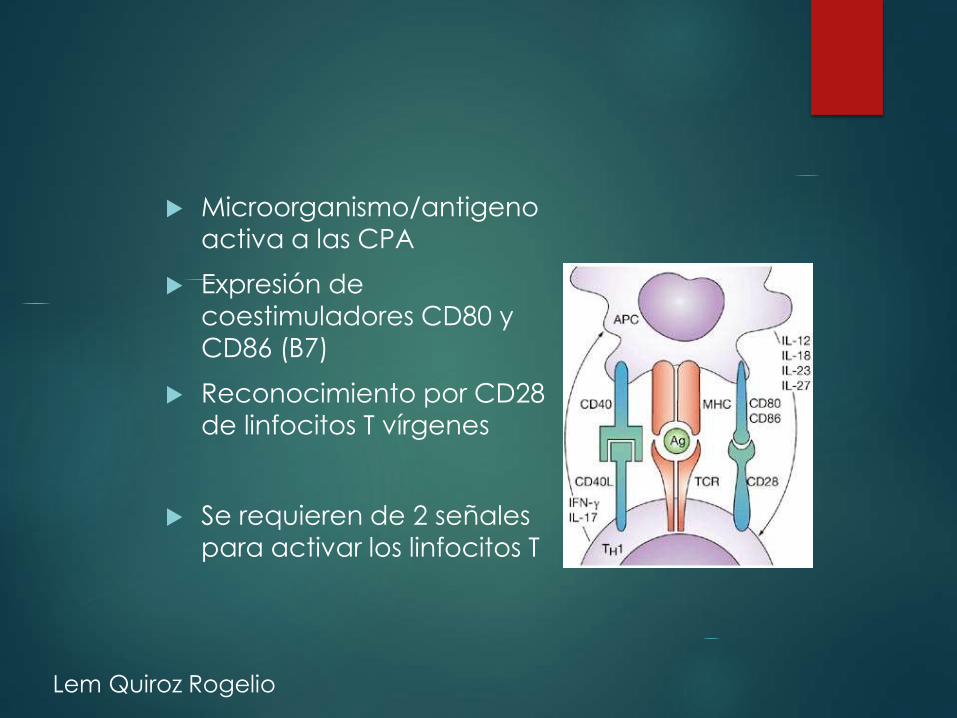
Microorganismo/antigeno
activa a las CPA
Expresión de coestimuladores CD80 y
CD86 (B7)
Reconocimiento por CD28
de linfocitos T vírgenes
Se requieren de 2 señales
para activar los linfocitos T
Lem Quiroz Rogelio

Inmunidad celular
Son activados y diferenciados a celulas
efectoras
Migración a tejidos
Linfocitos CD4+ IL-2 proliferación
CD40 + CD40L + Citocinas
CD4+(CD40L) + Macrófago/Linf.B(CD40)
Activación
Lem Quiroz Rogelio
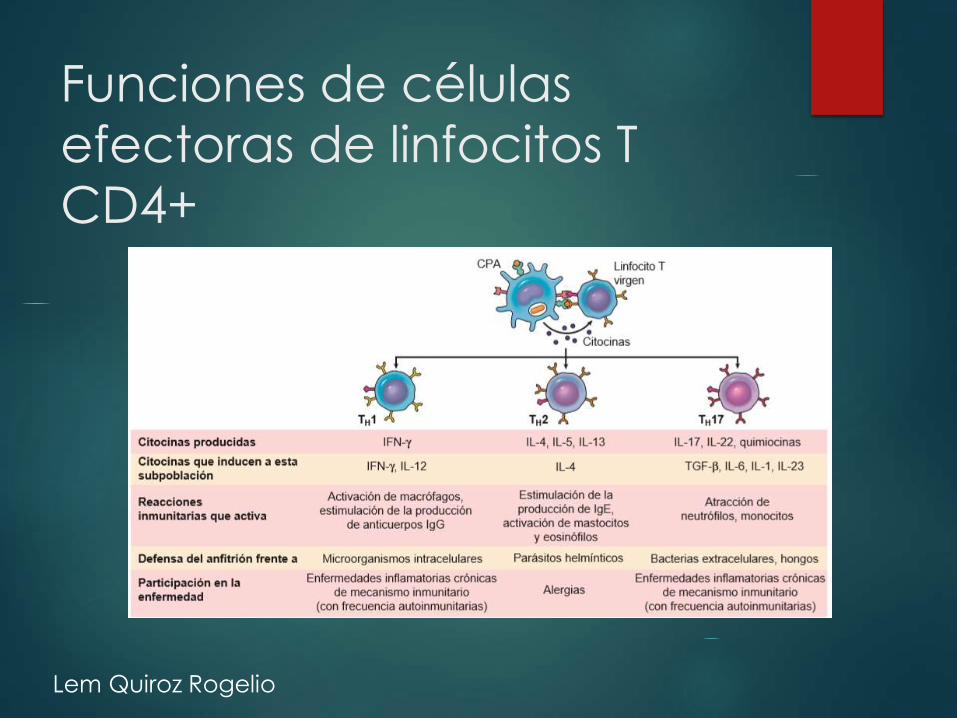
Funciones de células
efectoras de linfocitos T
CD4+
Lem Quiroz Rogelio

Inmunidad humoral
Antígenos polisacáridos y lipídicos Epítopos
Antígenos proteicos MHC
Activación por CD4+ con CD40L y citocinas
Cambio de isotipo IFN-γ e IL-4
Maduración de la afinidadCentros
germinativos
IgG OpsonizacionIgG e IgM ComplementoIgA Tejidos mucososIgE Parasitos
Lem Quiroz Rogelio
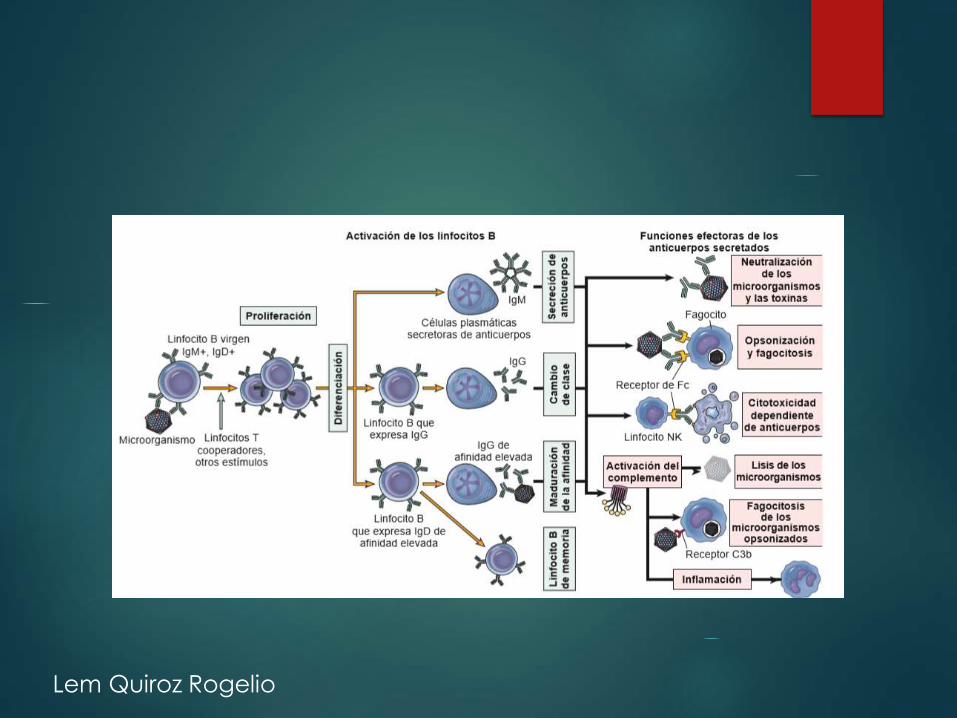
Lem Quiroz Rogelio
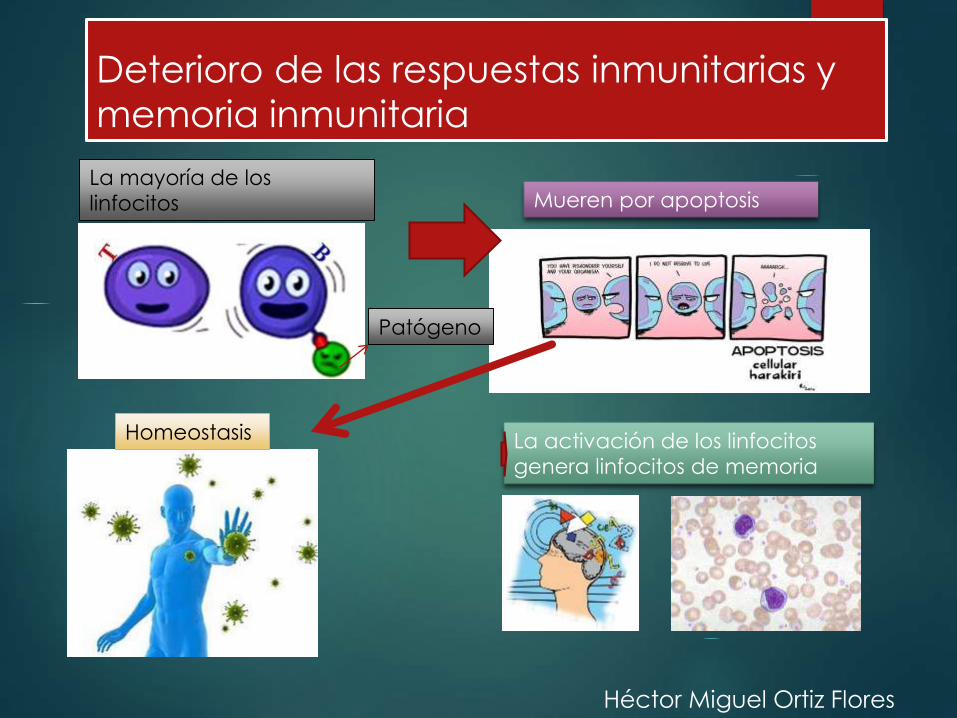
Deterioro de las respuestas inmunitarias y
memoria inmunitaria
La mayoría de los
linfocitos
Patógeno
Mueren por apoptosis
Homeostasis La activación de los linfocitos
genera linfocitos de memoria
Héctor Miguel Ortiz Flores
336902

Hipersensibilidad y trastornos
inmunitarios
Héctor Miguel Ortiz Flores
336902

Hipersensibilidad: Cuando individuo con
exposición repetida a un antígeno
desencadena una reacción patológica
Los trastornos de hipersensibilidad tienen varias
características generales
Héctor Miguel Ortiz Flores
336902

Antígenos tanto exógenos como endógenos pueden
desencadenar reacciones de hipersensibilidad
Vivimos en un entorno Abundan
sustancias Respuesta
inmune
Exógenos
Polvo, polenes
alimentos,
fármacos
Endógenos
Por el sistema
inmune,
generan
enfermedades
autoinmunitarias

La aparición de enfermedades por hipersensibilidad con
frecuencia se asocia a la herencia de determinados
genes de susceptibilidad
Se a implicado a genes de
los antígenos HLA y otros
genes distintos a los del
sistema HLA
Las enfermedades por hipersensibilidad se pueden clasificar
según el mecanismo inmunitario que media la enfermedad

Los principales tipos de hipersensibilidad son los siguientes
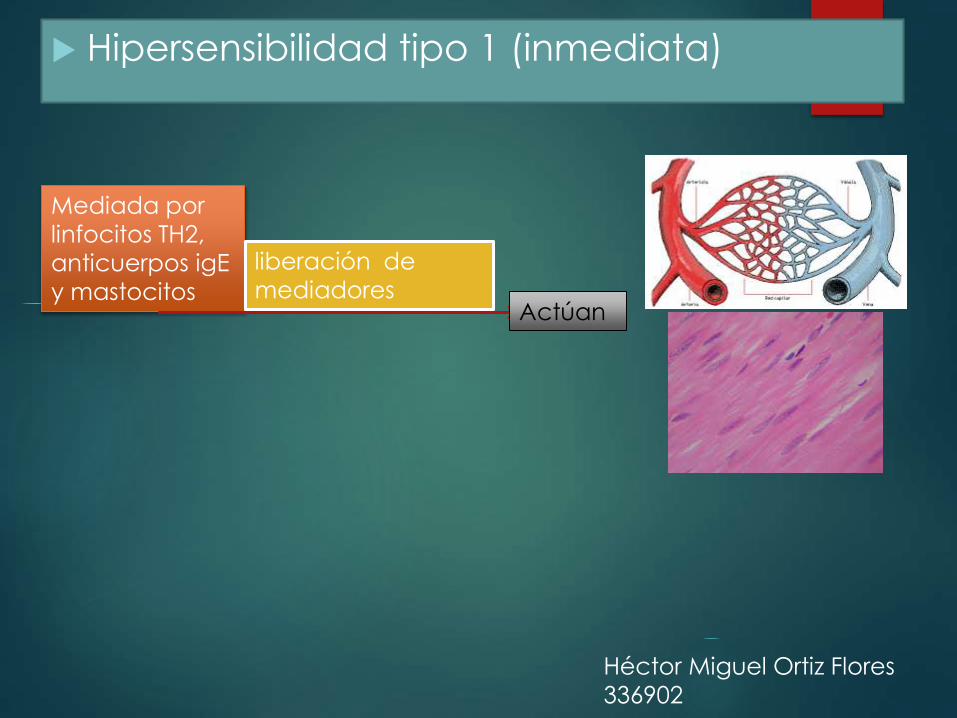
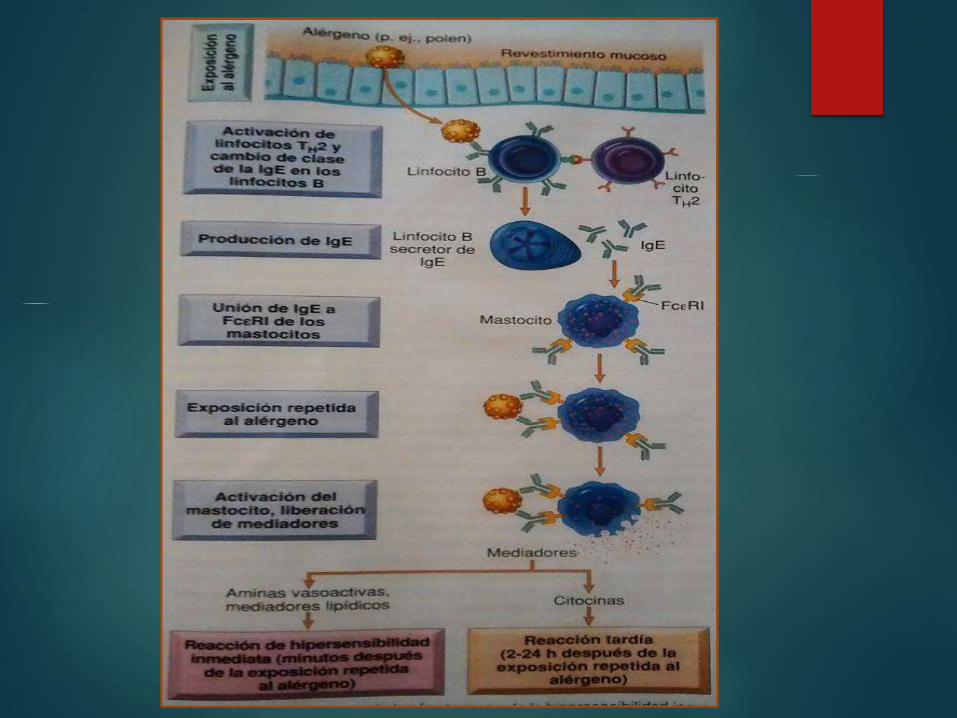
Hipersensibilidad tipo 1 (inmediata)
Mediada por
linfocitos TH2,
anticuerpos igE
y mastocitos
liberación de
mediadores Actúan
Héctor Miguel Ortiz Flores
336902

Hipersensibilidad tipo 2 (trastornos mediados por anticuerpos)
igG e IgM
Participan
Favoreciendo su
fagocitosis Induciend
o
inflamació
n en los
tejidos
lesionados
Héctor Miguel Ortiz Flores
336902
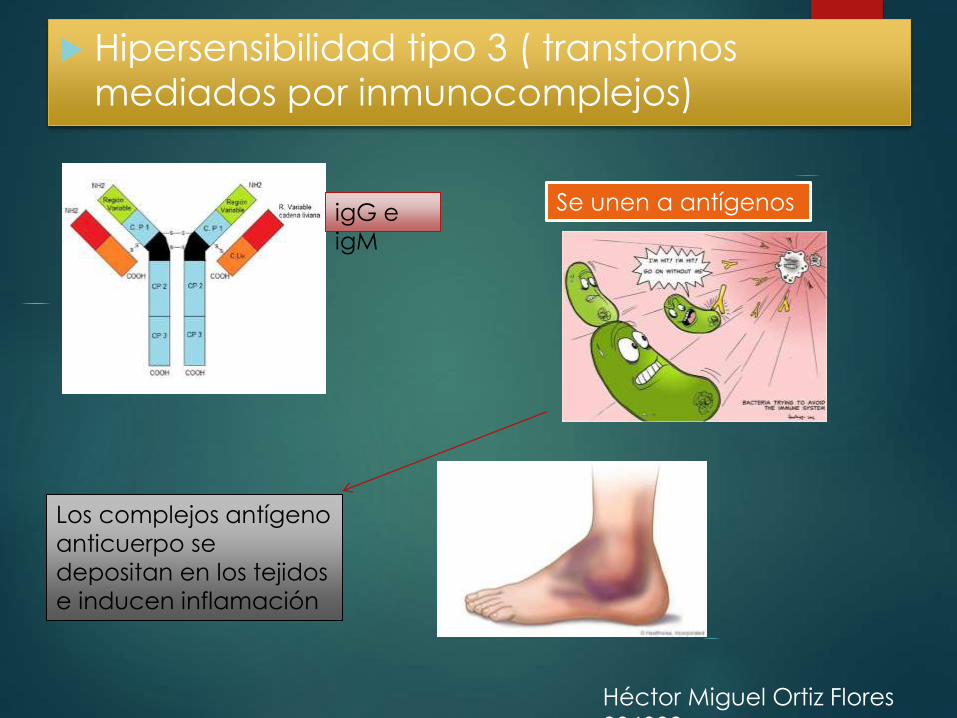
Hipersensibilidad tipo 3 ( transtornos
mediados por inmunocomplejos)
igG e
igM
Se unen a antígenos
Los complejos antígeno
anticuerpo se
depositan en los tejidos
e inducen inflamación
Héctor Miguel Ortiz Flores
336902
Hipersensibilidad tipo 4 ( trastornos inmunitarios mediados por
células)
Linfocitos t
sensibilizados
(linfocito th1 y el
th17 y LTC)
Son la causa de
lesión celular tisular
Héctor Miguel Ortiz Flores
336902
Hipersensibilidad tipo 1
Se caracteriza por
vasodilatación aumento
de la permeabilidad
vascular y dependiendo
de la localización espasmo del musculo liso
o secreción glandular
5 A 30 minutos
Alérgeno
Las Reacciones de
hipersensibilidad
inmediata
Activadas por
• Células derivadas de la medula ósea
• Hay abundancia cerca de los vasos sanguíneos, en los nervios y
en tejidos subepiteliales
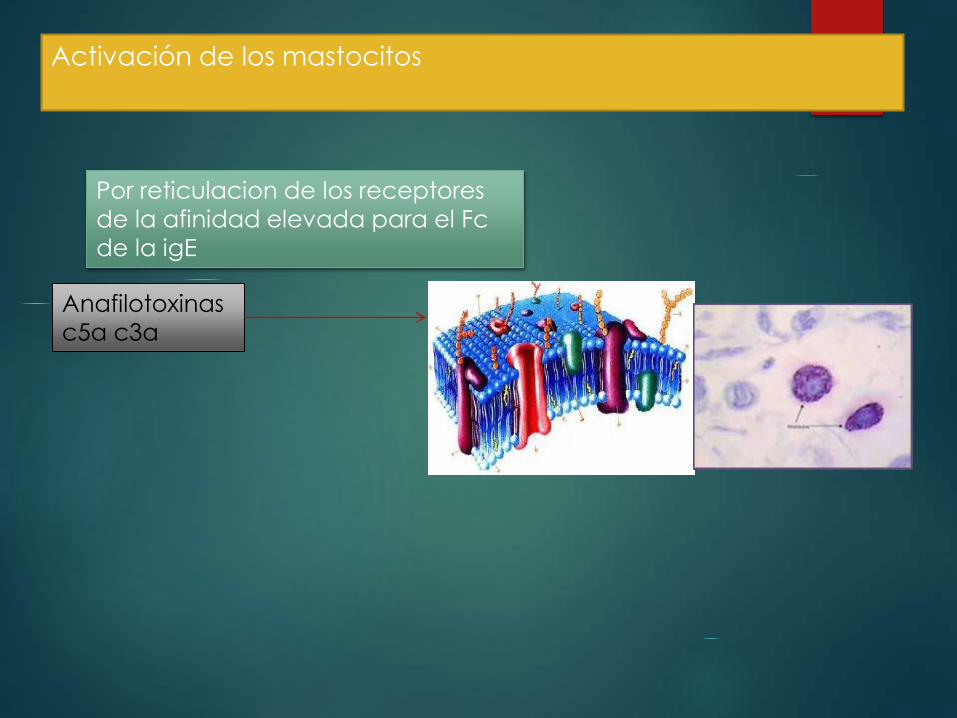
Activación de los mastocitos
Por reticulacion de los receptores
de la afinidad elevada para el Fc
de la igE
Anafilotoxinas
c5a c3a
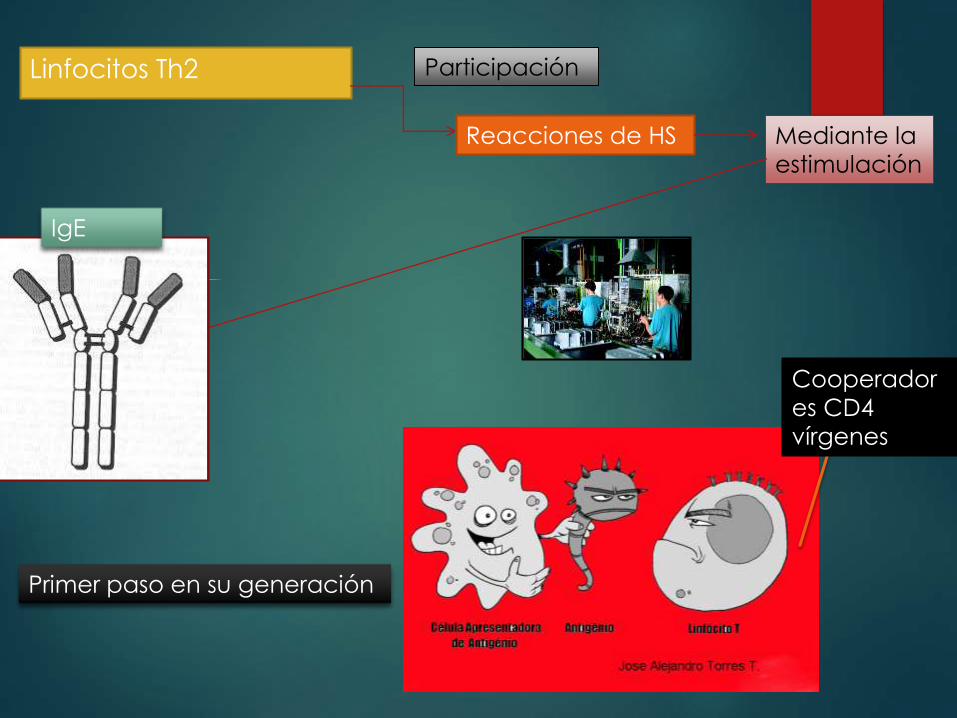
Linfocitos Th2 Participación
Reacciones de HS Mediante la
estimulación
IgE
Primer paso en su generación
Cooperador
es CD4
vírgenes
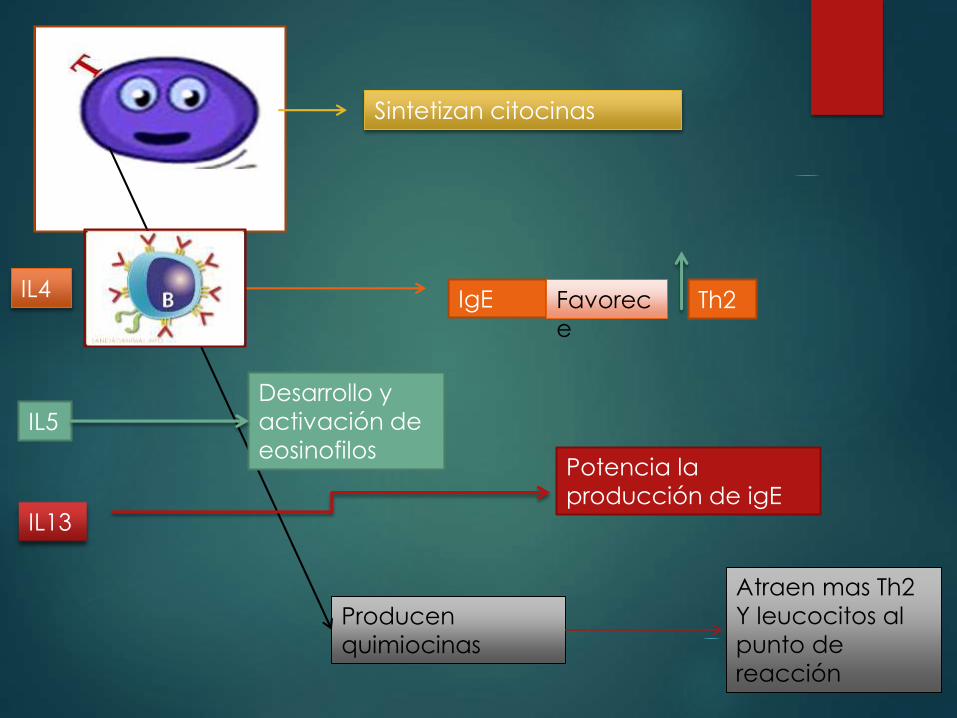
Sintetizan citocinas
Producen
quimiocinas
Atraen mas Th2
Y leucocitos al
punto de
reacción
IgE Favorec
e
Th2 IL4
Desarrollo y
activación de
eosinofilosIL5
IL13
Potencia la
producción de igE
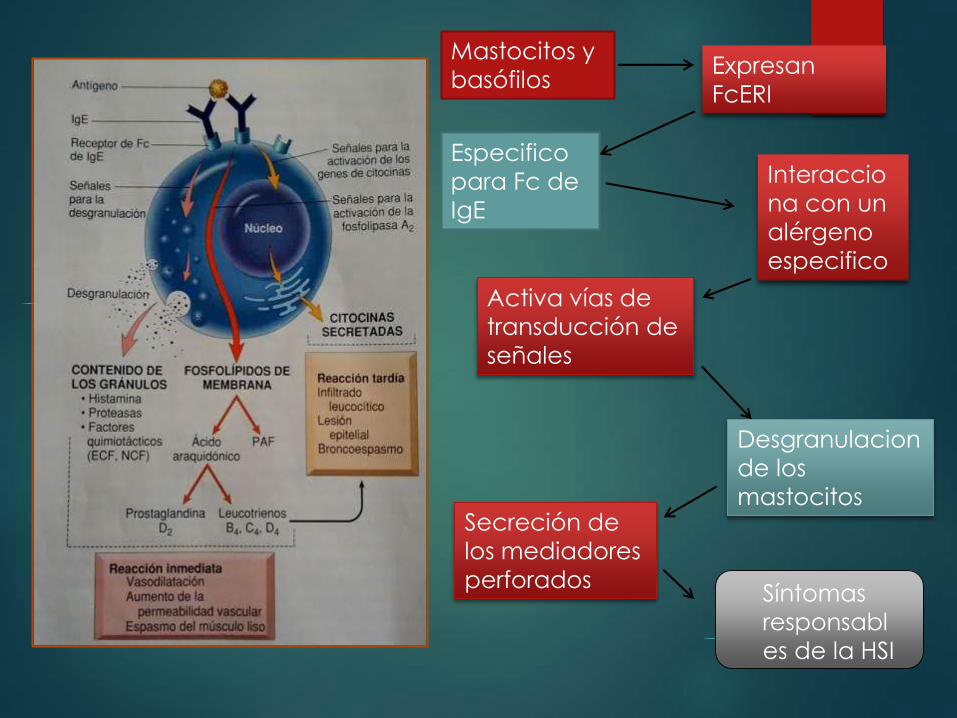
Mastocitos y
basófilos Expresan
FcERI
Especifico
para Fc de
IgE
Interaccio
na con un
alérgeno
especifico
Activa vías de
transducción de
señales
Desgranulacion
de los
mastocitos Secreción de
los mediadores
perforados Síntomas
responsabl
es de la HSI

Mediadores preformados :
Contenidos en los gránulos del mastocito se dividen en 3 categorías
Aminas vasoactivas
Enzimas
Proteoglucanos
Histamina
Proteasas neutras e
hidrolasas acidas
Heparina
Mediadores lipídicos:
Responsables de la activación de la fosfolipasa A2
esta actúa sobre los fosfolípidos de la membrana
para generar acido araquidónico
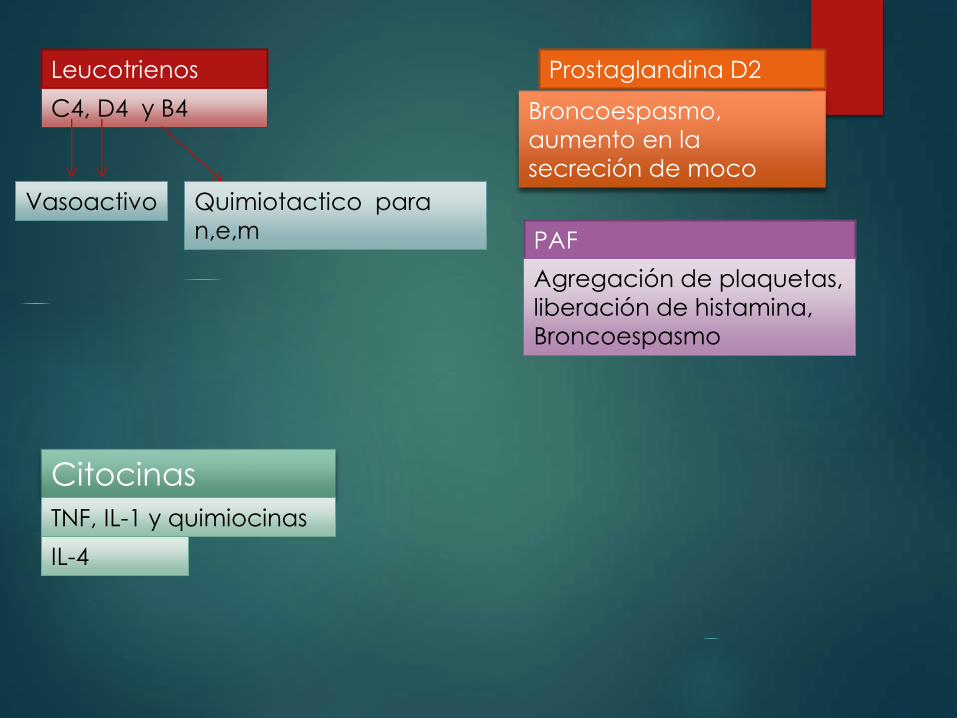
Leucotrienos
C4, D4 y B4
Vasoactivo Quimiotactico para
n,e,m
Prostaglandina D2
Broncoespasmo,
aumento en la
secreción de moco
PAF
Agregación de plaquetas,
liberación de histamina,
Broncoespasmo
Citocinas
TNF, IL-1 y quimiocinas
IL-4

Las reacciones de hipersensibilidad inmediata depende
de las acciones coordinadas de diversos compuestos
quimiotacticos vasoactivos y espasmogenos
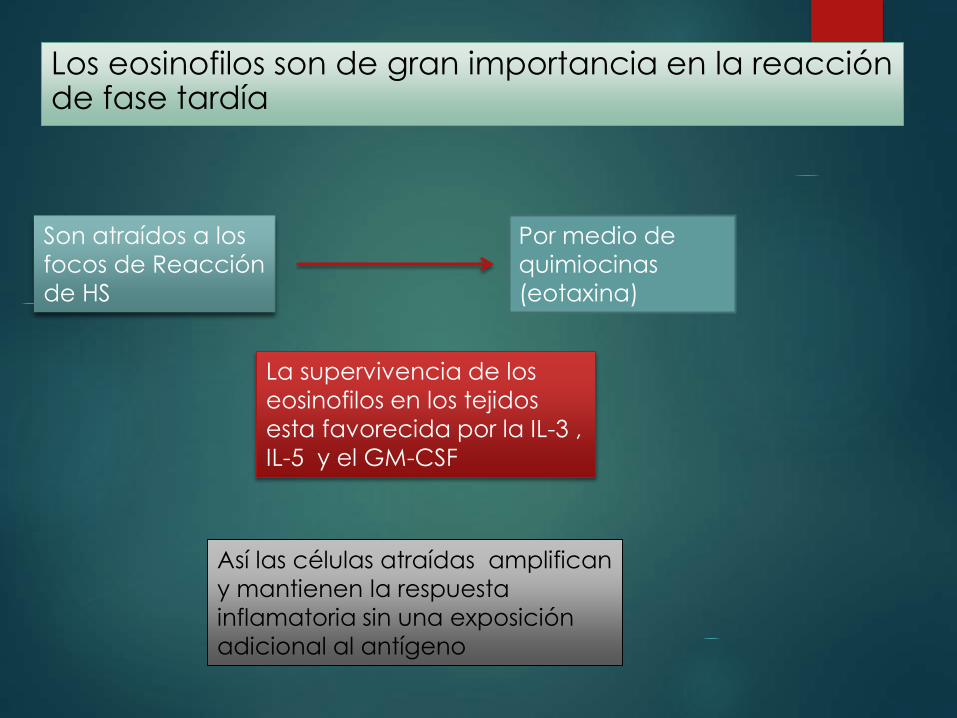
Los eosinofilos son de gran importancia en la reacción de fase tardía
Son atraídos a los
focos de Reacción
de HS
Por medio de
quimiocinas
(eotaxina)
La supervivencia de los
eosinofilos en los tejidos
esta favorecida por la IL-3 ,
IL-5 y el GM-CSF
Así las células atraídas amplifican
y mantienen la respuesta
inflamatoria sin una exposición
adicional al antígeno
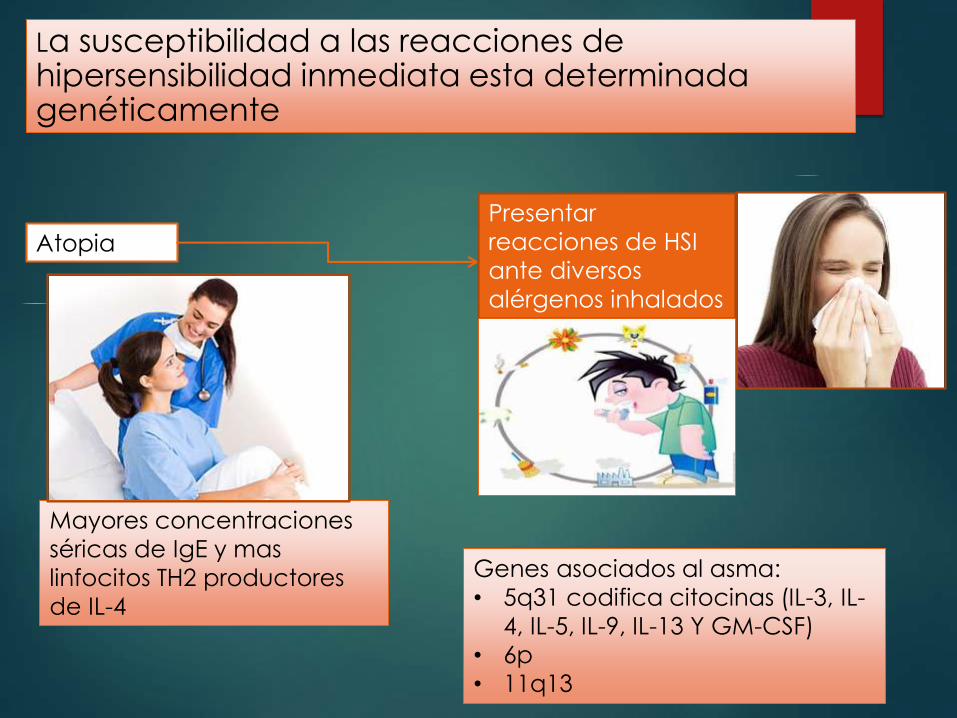
La susceptibilidad a las reacciones de hipersensibilidad inmediata esta determinada genéticamente
Atopia
Presentar
reacciones de HSI
ante diversos
alérgenos inhalados
e ingeridos
Mayores concentraciones
séricas de IgE y mas
linfocitos TH2 productores
de IL-4
Genes asociados al asma:
• 5q31 codifica citocinas (IL-3, IL-
4, IL-5, IL-9, IL-13 Y GM-CSF)
• 6p
• 11q13

Resumiendo la HSI es un
complejo trastorno debido a la:
• Activación de los mastocitos
mediada por la IgE
• Acumulación de células
inflamatorias en los focos de
deposito del antígeno
Regulados por la inducción de
linfocitos T cooperadores TH2 que
estimulan la producción de IgE
(favorece la activación de
mastocitos), producen
acumulación de células
inflamatorias ( Eosinofilos) y
desencadena la secreción de
moco
Las características clínicas se
deben a la liberación de los
mediadores de los mastocitos
así como la inflamación rica en
eosinofilos

Anafilaxia sistémica
Es caracterizada por
Shock vascular
Edema
Dificultad respiratoria
Puede ocurrir tras la
administración de
proteínas ajenas
• Hormonas
• Enzimas
• Polisacáridos
• Fármacos (
penicilina)
Alérgenos
alimentarios como
cacahuates,
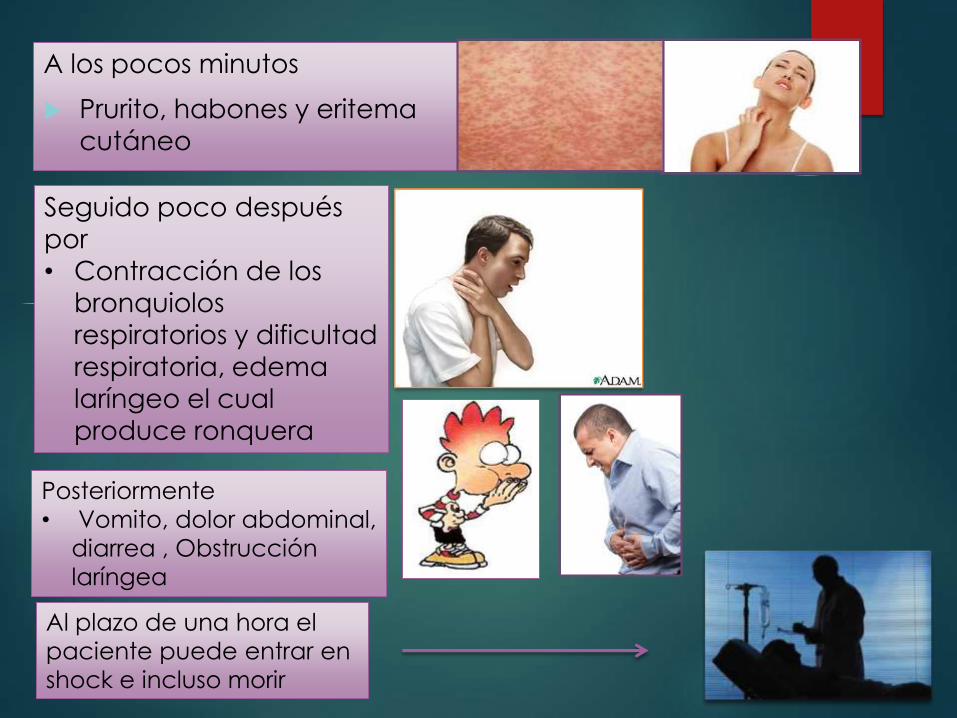
A los pocos minutos
Prurito, habones y eritema
cutáneo
Seguido poco después
por
• Contracción de los
bronquiolos
respiratorios y dificultad
respiratoria, edema
laríngeo el cual
produce ronquera
Posteriormente
• Vomito, dolor abdominal,
diarrea , Obstrucción
laríngea
Al plazo de una hora el
paciente puede entrar en
shock e incluso morir
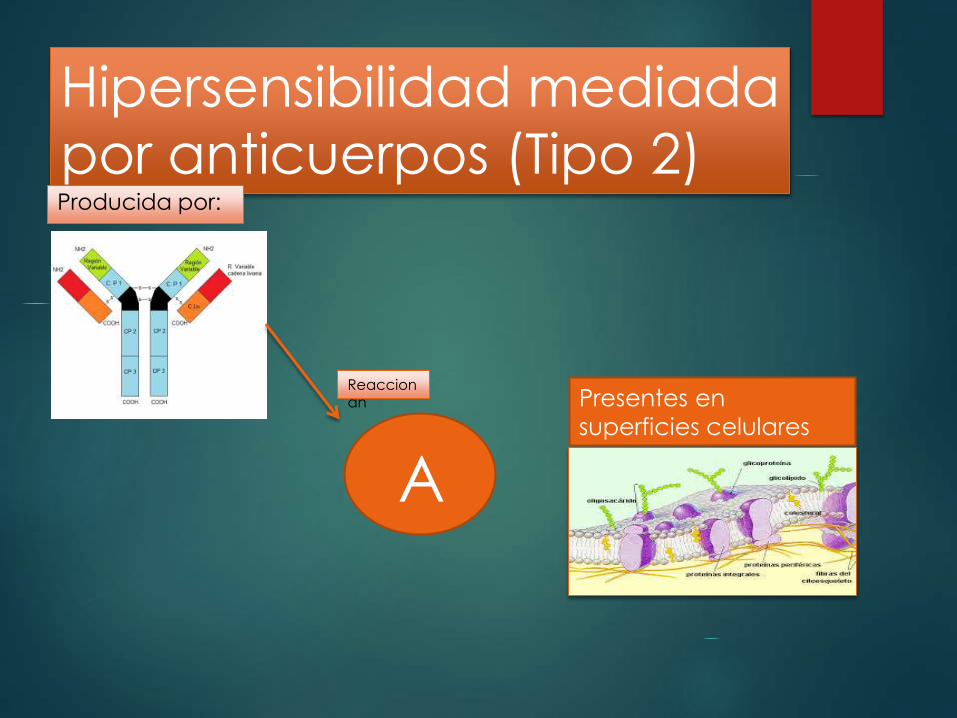
Hipersensibilidad mediada
por anticuerpos (Tipo 2) Producida por:
A
Reaccionan Presentes en
superficies celulares
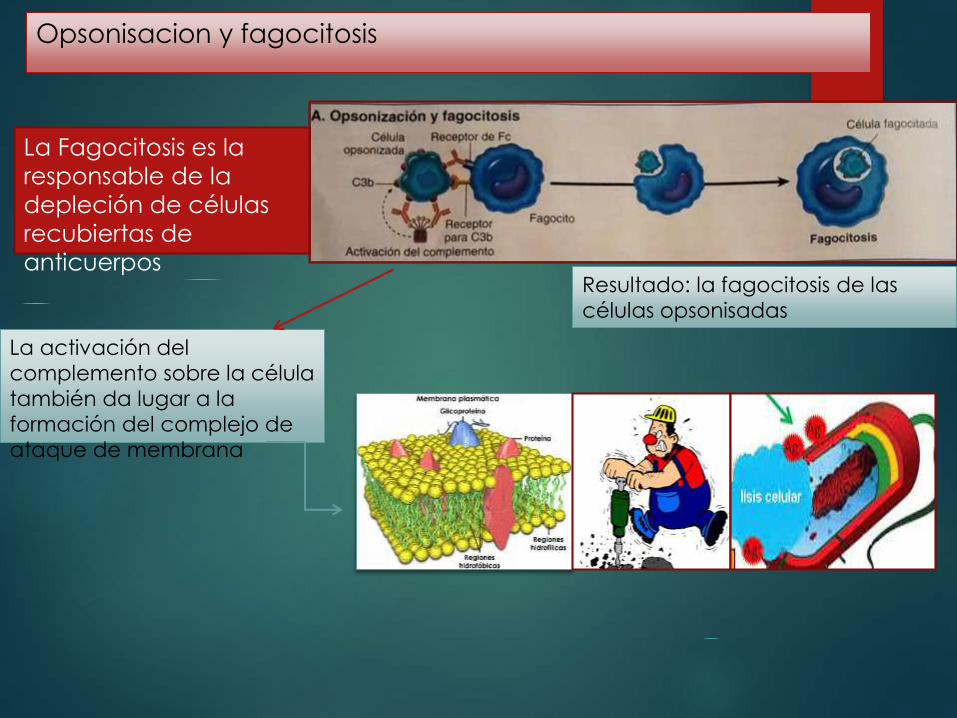
Opsonisacion y fagocitosis
La Fagocitosis es la
responsable de la
depleción de células
recubiertas de
anticuerpos Resultado: la fagocitosis de las
células opsonisadas
La activación del
complemento sobre la célula
también da lugar a la
formación del complejo de
ataque de membrana

¡La destrucción mediada por anticuerpos
puede ocurrir mediante otro proceso!
Citotoxicidad
celular
dependiente
de
anticuerpo
(CCDA)
Sistema de
complement
o
Cooperación de
leucocitos
CCDA esta mediada por:
• Monocitos
• Neutrofilos
• Eosinofilos
• Linfocitos NK
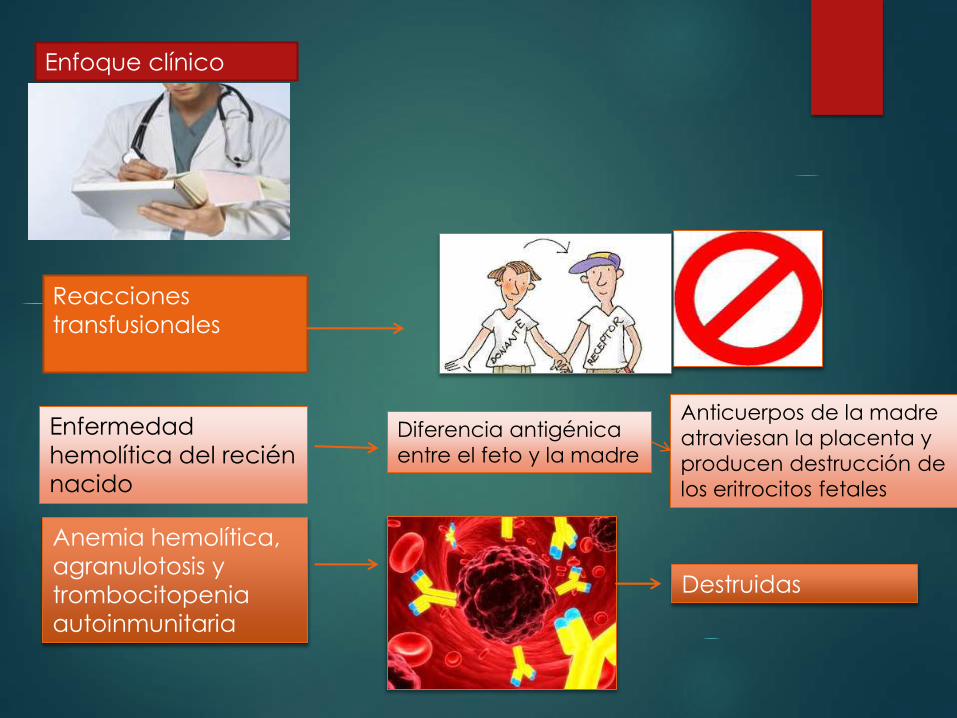
Enfoque clínico
Reacciones
transfusionales
Enfermedad
hemolítica del recién
nacido
Anemia hemolítica,
agranulotosis y
trombocitopenia
autoinmunitaria
Diferencia antigénica
entre el feto y la madre
Anticuerpos de la madre
atraviesan la placenta y
producen destrucción de
los eritrocitos fetales
Destruidas
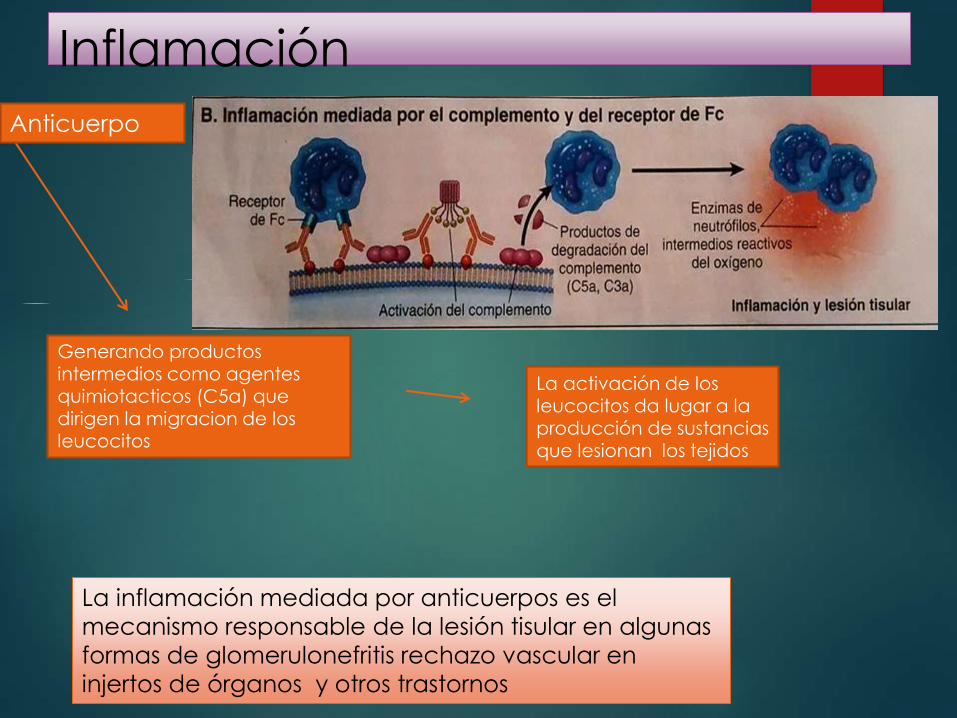
Inflamación
Anticuerpo
Generando productos intermedios como agentes quimiotacticos (C5a) que dirigen la migracion de los leucocitos
La activación de los leucocitos da lugar a la producción de sustancias que lesionan los tejidos
La inflamación mediada por anticuerpos es el
mecanismo responsable de la lesión tisular en algunas
formas de glomerulonefritis rechazo vascular en
injertos de órganos y otros trastornos
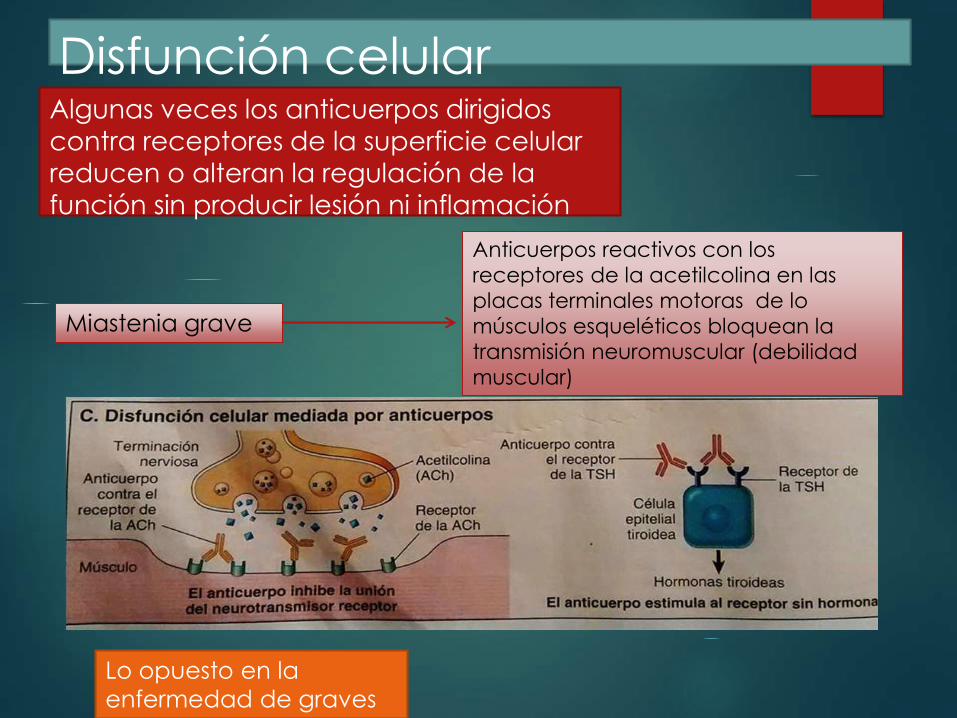
Disfunción celular Algunas veces los anticuerpos dirigidos
contra receptores de la superficie celular
reducen o alteran la regulación de la
función sin producir lesión ni inflamación
Miastenia grave
Anticuerpos reactivos con los
receptores de la acetilcolina en las
placas terminales motoras de lo
músculos esqueléticos bloquean la
transmisión neuromuscular (debilidad
muscular)
Lo opuesto en la
enfermedad de graves

Hipersensibilidad mediada por
inmunocomplejos (tipo 3)
Los complejos
antígeno anticuerpo
producen
Lesión tisular Generando
inflamación
La reacción
patológica inicia:
Antígeno
Los inmunocomplejos se
depositan típicamente en
las paredes vasculares
Localizaciones
extravasculares:
inmunocomplejo
s in situ
Los antígenos que
forman
inmunocomplejos
pueden ser Endógen
os
Las enfermedades mediadas por inmunocomplejos pueden ser
Sistémicas Localizadas
Inmunocomplejo
s que se forman
en la
circulación
Y se
depositan en
muchos
órganos
Órganos
particulares
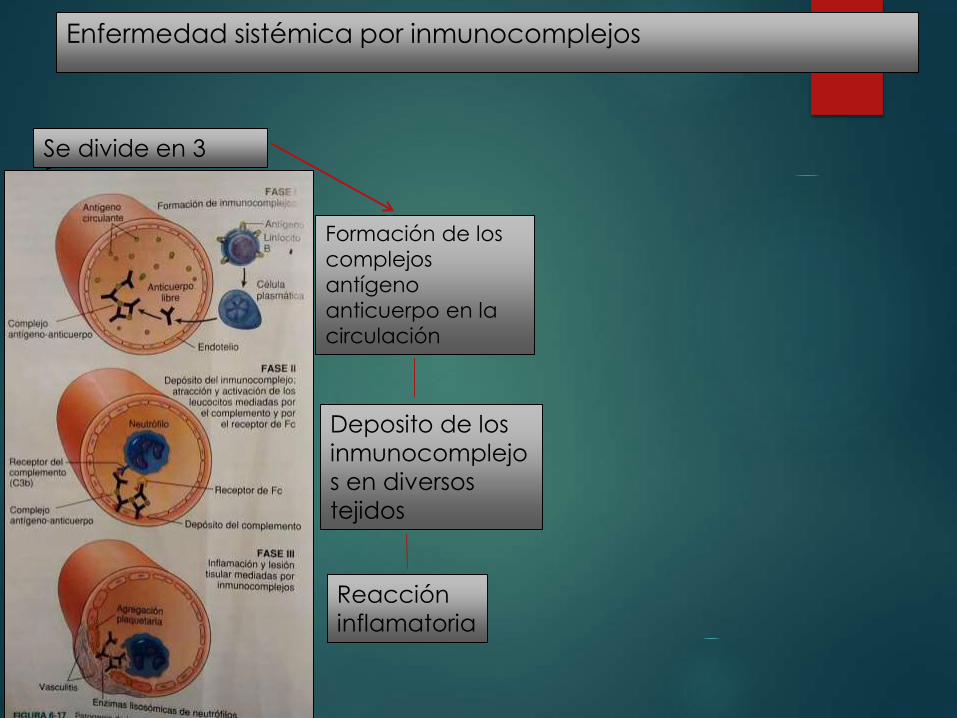
Enfermedad sistémica por inmunocomplejos
Se divide en 3
fases
Formación de los
complejos
antígeno
anticuerpo en la
circulación
Deposito de los
inmunocomplejo
s en diversos
tejidos
Reacción
inflamatoria
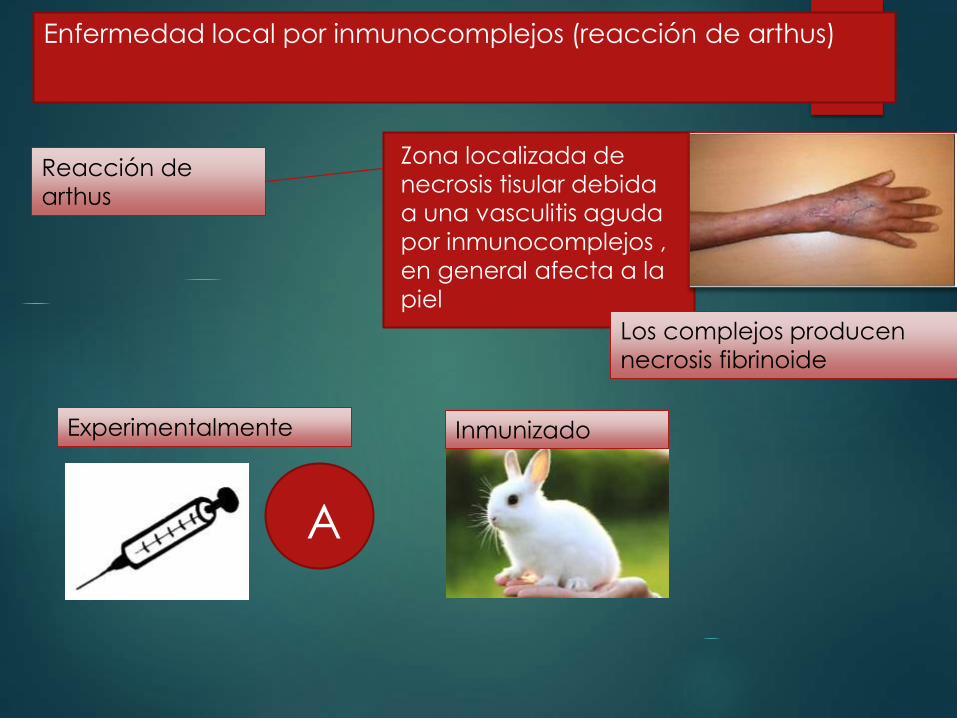
Enfermedad local por inmunocomplejos (reacción de arthus)
Reacción de
arthus
Zona localizada de
necrosis tisular debida
a una vasculitis aguda
por inmunocomplejos ,
en general afecta a la
piel
A
InmunizadoExperimentalmente
Los complejos producen
necrosis fibrinoide
Hipersensibilidad mediada por
linfocitos T (tipo IV)
Se inicia por los linfocitos t activados por el antígeno
Como
linfocitos T,
CD4 Y CD8
HS mediada
por linfocitos
t CD4
Inducid
a por
A
Pueden ser causa
de una
enfermedad
inflamatoria
crónica
En infecciones
víricas los
linfocitos T CD8
pueden ser las
células
efectoras
dominantes
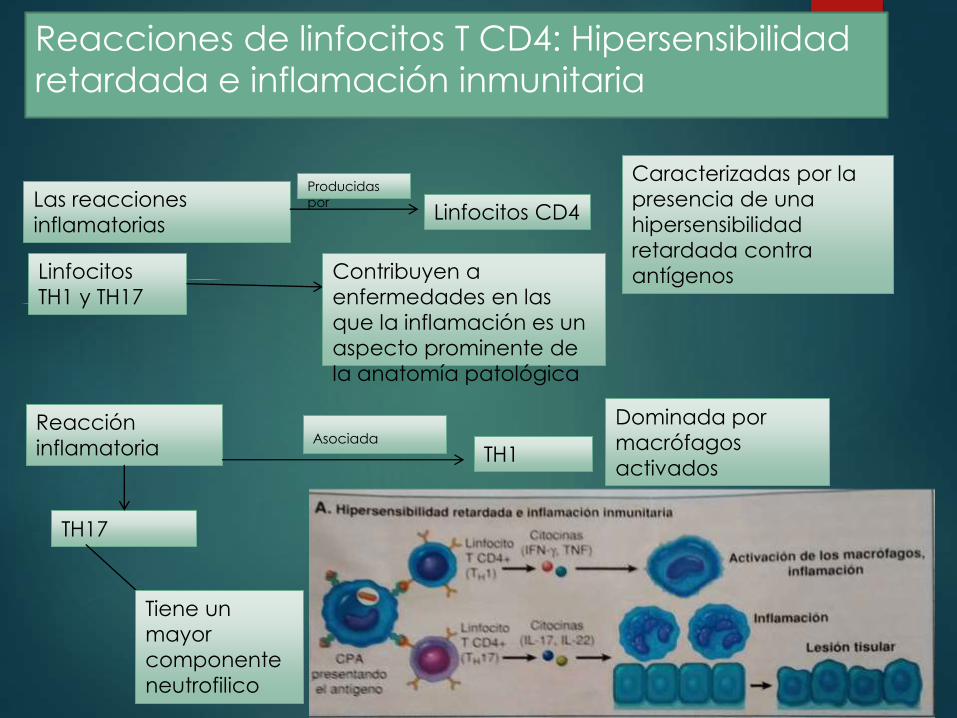
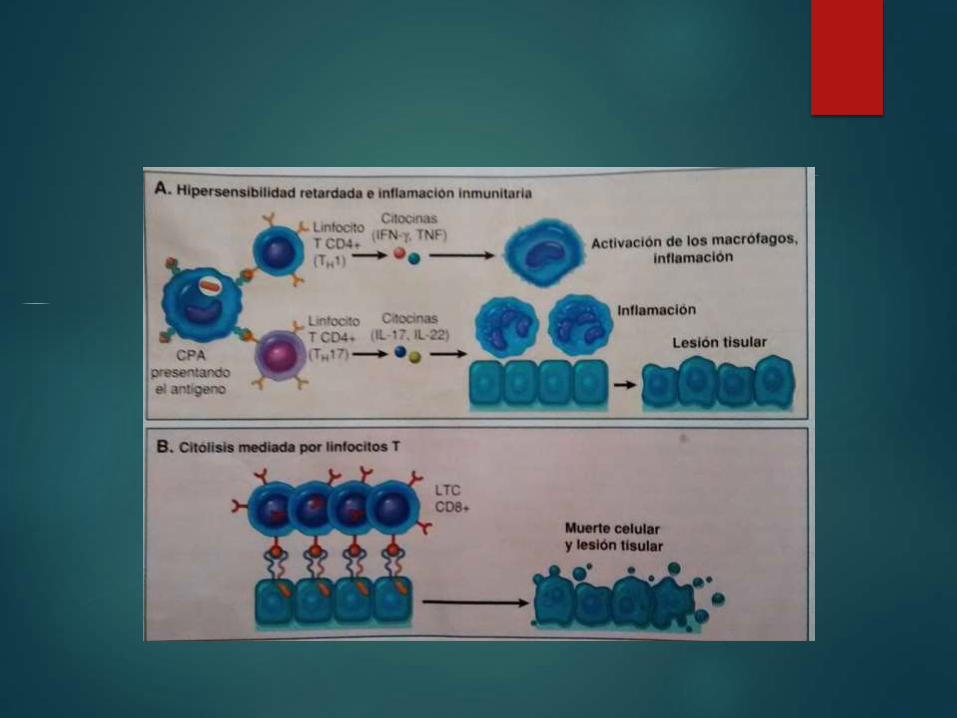
Reacciones de linfocitos T CD4: Hipersensibilidad
retardada e inflamación inmunitaria
Las reacciones
inflamatorias Linfocitos CD4
Producidas
por
Caracterizadas por la
presencia de una
hipersensibilidad
retardada contra
antígenosLinfocitos
TH1 y TH17
Contribuyen a
enfermedades en las
que la inflamación es un
aspecto prominente de
la anatomía patológica
Reacción
inflamatoria TH1Asociada
Dominada por
macrófagos
activados
TH17
Tiene un
mayor
componente
neutrofilico
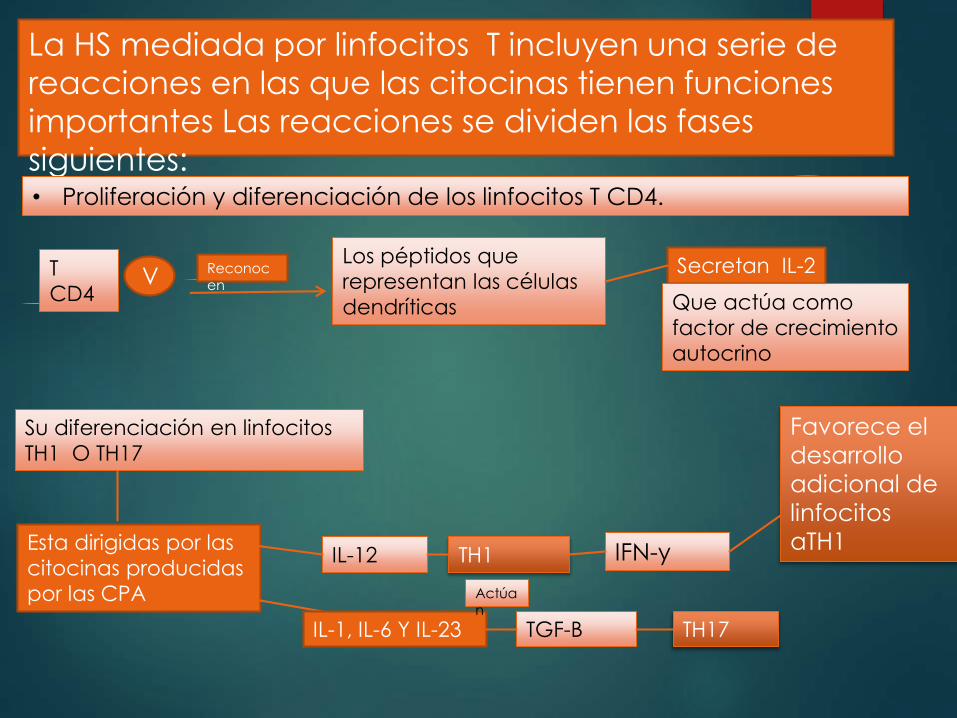
La HS mediada por linfocitos T incluyen una serie de
reacciones en las que las citocinas tienen funciones
importantes Las reacciones se dividen las fases
siguientes: • Proliferación y diferenciación de los linfocitos T CD4.
T
CD4 V
Reconocen
Los péptidos que
representan las células
dendríticas
Secretan IL-2
Que actúa como
factor de crecimiento
autocrino
Su diferenciación en linfocitos
TH1 O TH17
Esta dirigidas por las
citocinas producidas
por las CPA
IL-12 TH1 IFN-y
Favorece el
desarrollo
adicional de
linfocitos
aTH1
IL-1, IL-6 Y IL-23 TGF-B
Actúan
TH17
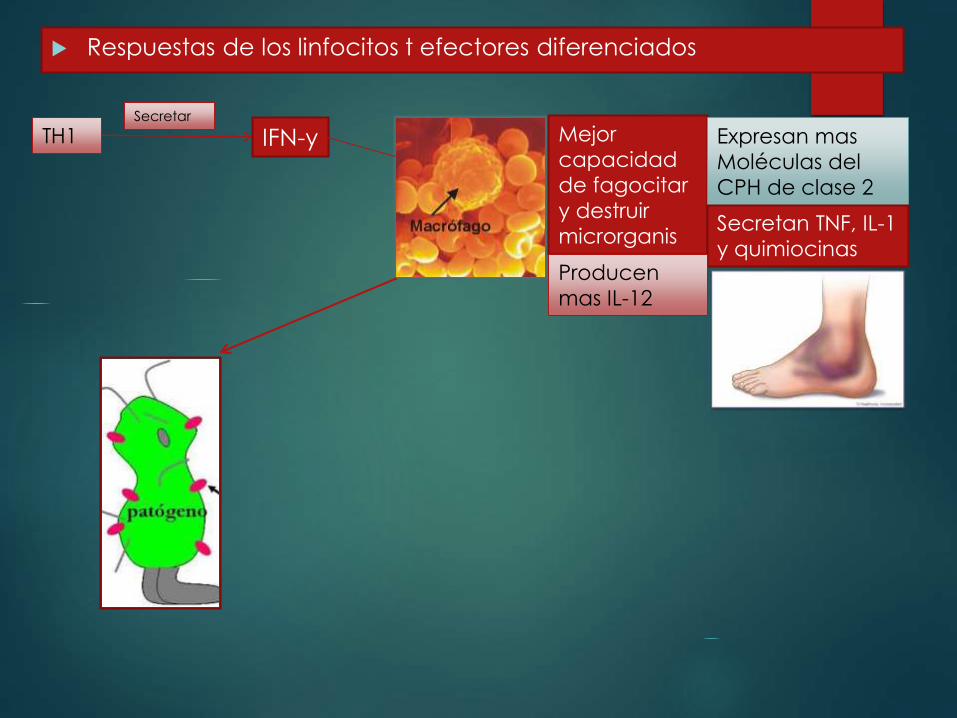
Respuestas de los linfocitos t efectores diferenciados
TH1 IFN-ySecretar
Mejor
capacidad
de fagocitar
y destruir
microrganis
mos
Expresan mas
Moléculas del
CPH de clase 2
Secretan TNF, IL-1
y quimiocinasProducen
mas IL-12
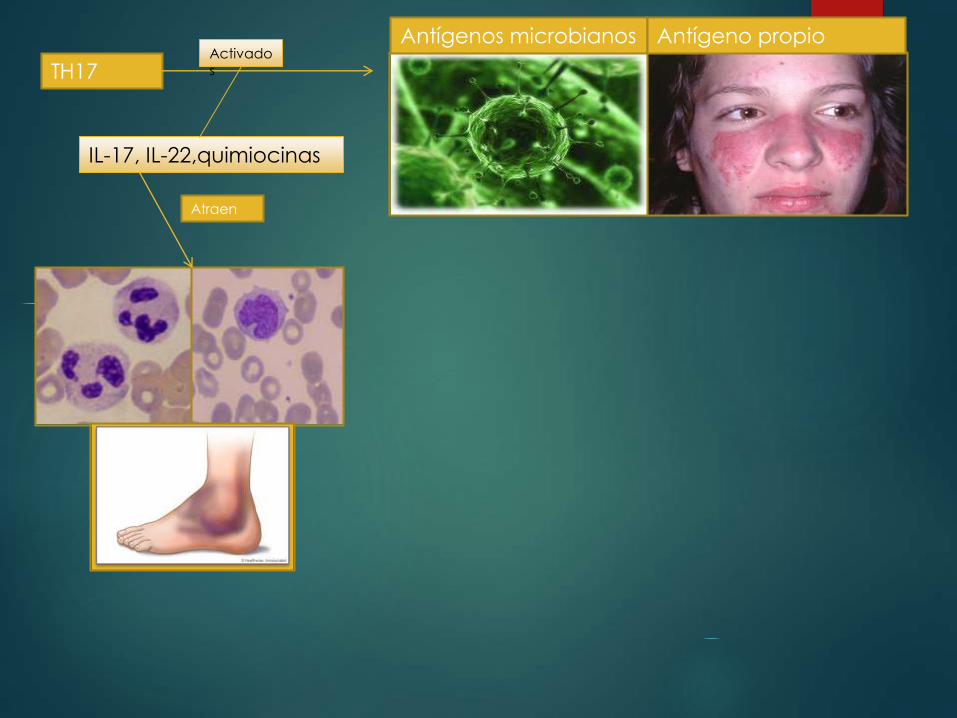
TH17Activados
Antígenos microbianos Antígeno propio
IL-17, IL-22,quimiocinas
Atraen
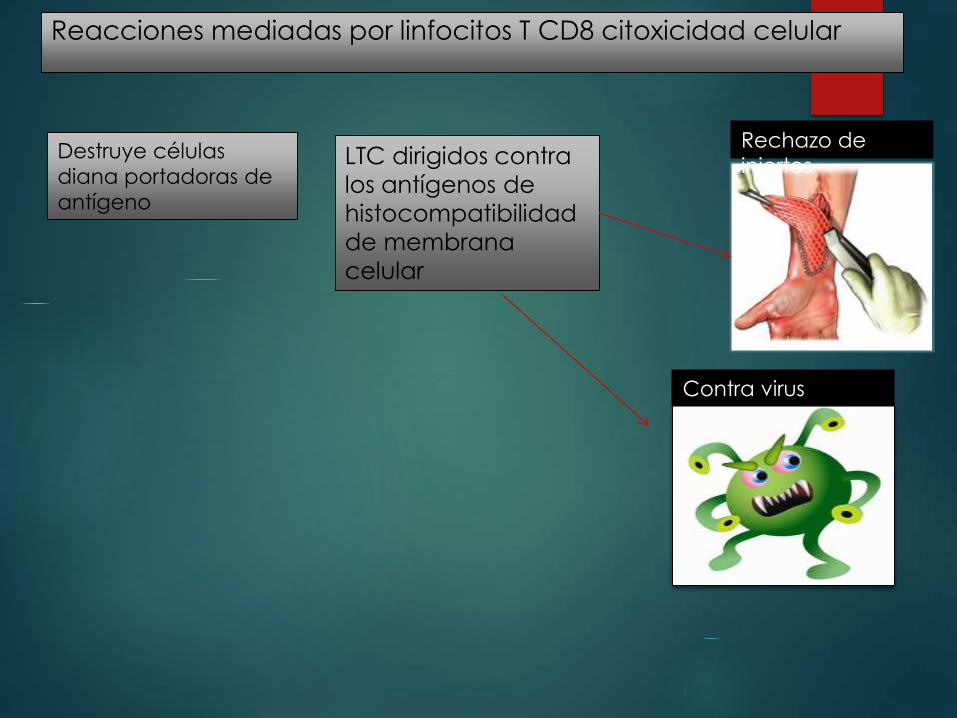
Reacciones mediadas por linfocitos T CD8 citoxicidad celular
Destruye células
diana portadoras de
antígeno
LTC dirigidos contra
los antígenos de
histocompatibilidad
de membrana
celular
Rechazo de
injertos
Contra virus
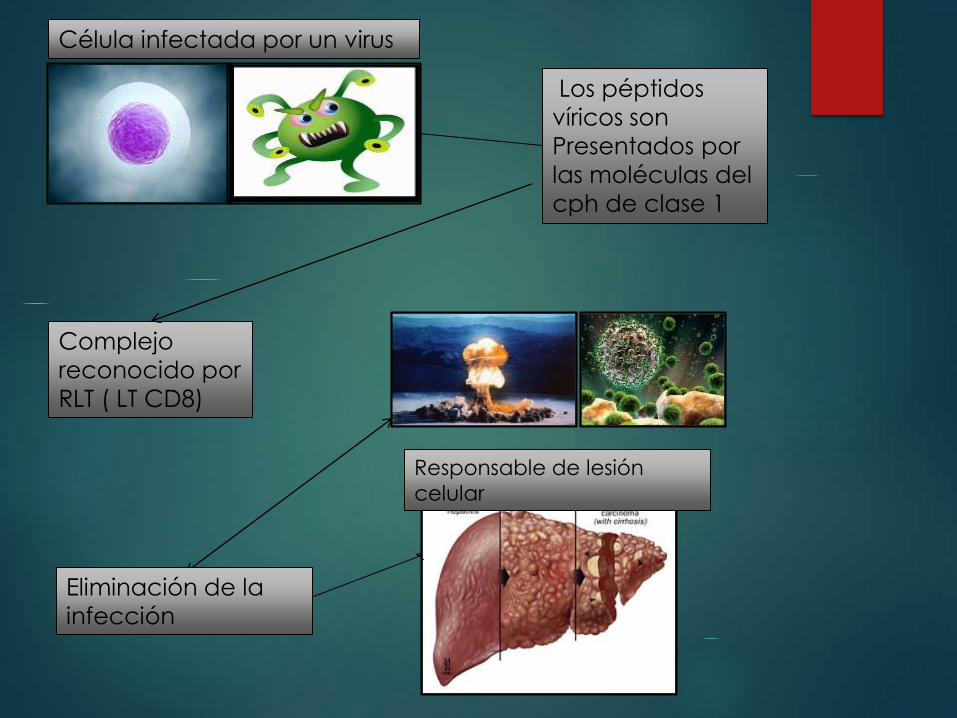
Los péptidos
víricos son
Presentados por
las moléculas del
cph de clase 1
Complejo
reconocido por
RLT ( LT CD8)
Célula infectada por un virus
Eliminación de la
infección
Responsable de lesión
celular
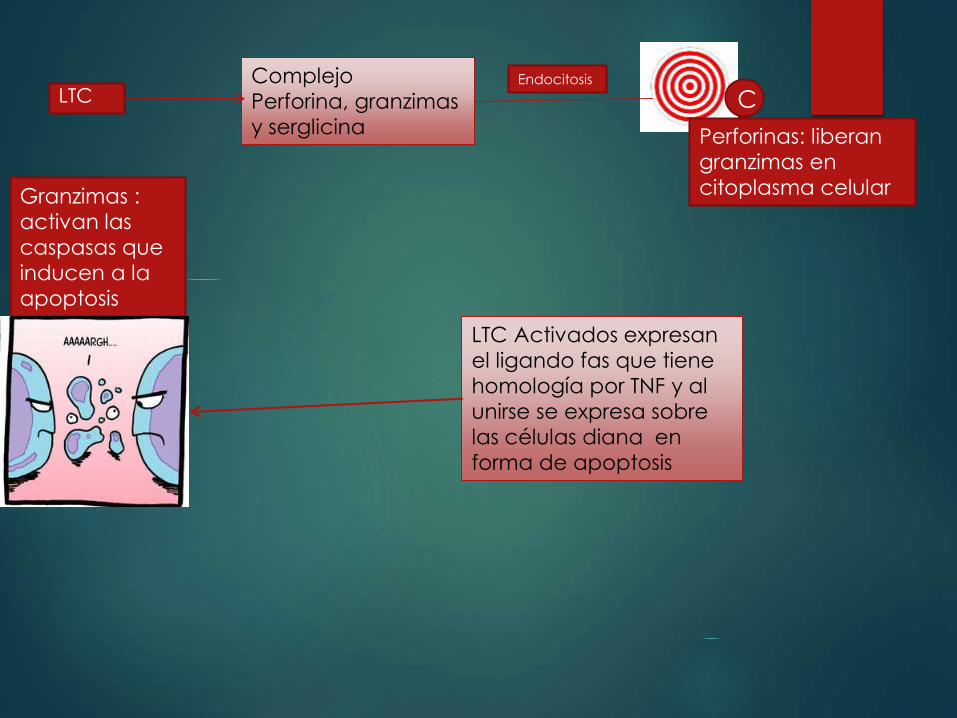
LTC CComplejo
Perforina, granzimas
y serglicina
Endocitosis
Perforinas: liberan
granzimas en
citoplasma celular Granzimas :
activan las
caspasas que
inducen a la
apoptosis
LTC Activados expresan
el ligando fas que tiene
homología por TNF y al
unirse se expresa sobre
las células diana en
forma de apoptosis
Enfermedades autoinmunitarias
Autoinmunidad patológica
• Presencia de una reacción
inmunitaria especifica para
algún antígeno o tejido propio
• Datos de que dicha reacción
no es secundaria a una lesión
tisular
• Ausencia de otra causa bien
definida de la enfermedad
Enfermedades especificas de
órgano
Enfermedades sistémicas o
generalizadas Ejemplo diabetes
mellitus tipo 1 Ejemplo LES
Tolerancia inmunitaria
Es el fenómeno de ausencia de respuesta a un
antígeno como consecuencia de la exposición de los
linfocitos al mismo
Autotolerancia
Los mecanismos
se clasifican en 2
Tolerancia central
Tolerancia periférica
Antígeno
s propios
Tolerancia central
Clones de linfocitos T y B
autorreactivos inmaduros
Que reconocen
antígenos propios
durante su maduración
Linfocitos T Linfocitos B
Son destruidos o se
vuelven inofensivos
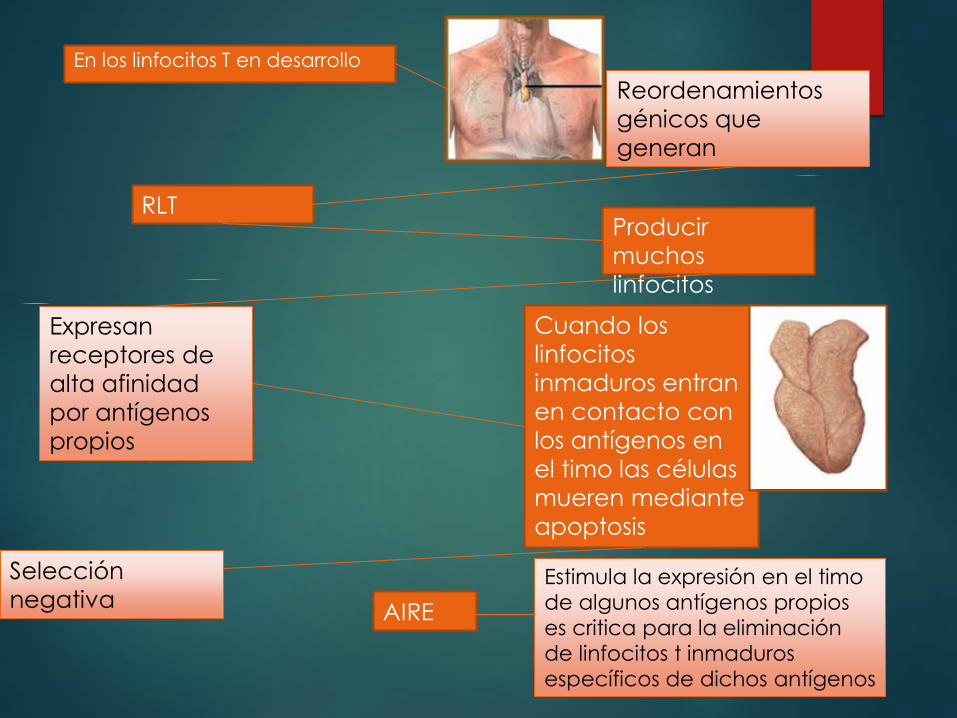
En los linfocitos T en desarrollo
Reordenamientos
génicos que
generan
RLTProducir
muchos
linfocitos
Expresan
receptores de
alta afinidad
por antígenos
propios
Cuando los
linfocitos
inmaduros entran
en contacto con
los antígenos en
el timo las células
mueren mediante
apoptosis
Selección
negativa AIRE
Estimula la expresión en el timo
de algunos antígenos propios
es critica para la eliminación
de linfocitos t inmaduros
específicos de dichos antígenos
cuando los Linfocitos B en
desarrollo
Reconocen de
manera intensa
los antígenos
propios Muchos de ellos reactivan la
maquinaria de
reordenamiento y
comienzan a expresar
nuevos receptores
antigénicos no específicos
de los antígenos propios Proceso
llamado
edición del
receptor Si no se produce
edición del receptor los
linfocitos autoreactivos
sufren apoptosis
Tolerancia periférica
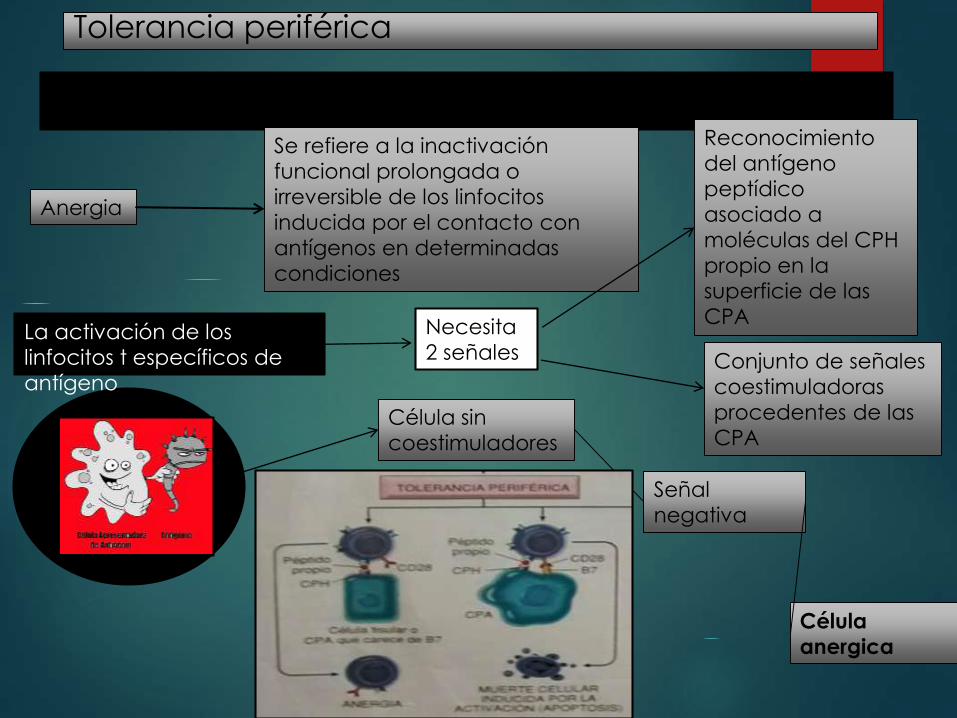
VARIOS MECANISMOS SILENCIAN A LOS LINFOCITOS T Y B POTENCIALMENTE AUTORREACTIVOS EN LOS TEJIDOS PERIFÉRICOS
Anergia
Se refiere a la inactivación
funcional prolongada o
irreversible de los linfocitos
inducida por el contacto con
antígenos en determinadas
condiciones
La activación de los
linfocitos t específicos de
antígeno
Necesita
2 señales
Reconocimiento
del antígeno
peptídico
asociado a
moléculas del CPH
propio en la
superficie de las
CPA
Conjunto de señales
coestimuladoras
procedentes de las
CPA Célula sin
coestimuladores
Señal
negativa
Célula
anergica
Se han demostrado dos mecanismos de anergia de los linfocitos t en
diversos sistemas experimentales
Perdida de la capacidad
de desencadenar
señales bioquímicas
Células
Que reconocen antígenos
propios reciben una señal
inbhidora procedente de
receptores que son
estructuralmente
homólogos a CD28 pero
que tienen funciones
contrarias
Células TCTLA-4
Los linfocitos t necesitan de
CD28 para reconocer las
moléculas de B7 y activarse o
CTLA-4 para reconocer las
mismas moléculas y hacerse
anergicos
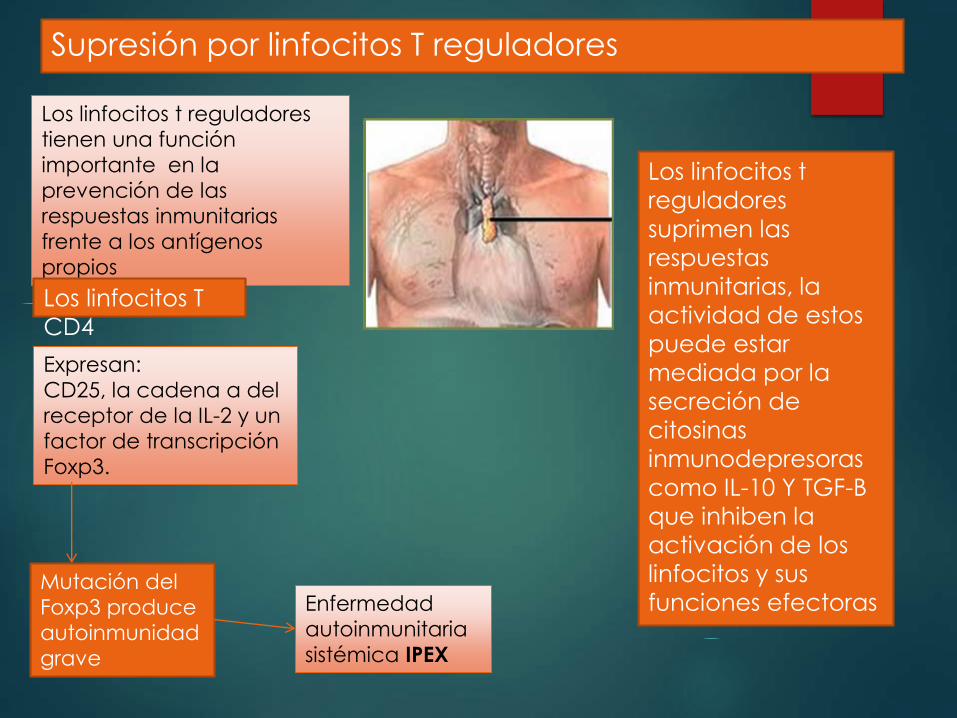
Supresión por linfocitos T reguladores
Los linfocitos t reguladores
tienen una función
importante en la
prevención de las
respuestas inmunitarias
frente a los antígenos
propios
Los linfocitos T
CD4
Expresan:
CD25, la cadena a del
receptor de la IL-2 y un
factor de transcripción
Foxp3.
Mutación del
Foxp3 produce
autoinmunidad
grave
Enfermedad
autoinmunitaria
sistémica IPEX
Los linfocitos t
reguladores
suprimen las
respuestas
inmunitarias, la
actividad de estos
puede estar
mediada por la
secreción de
citosinas
inmunodepresoras
como IL-10 Y TGF-B
que inhiben la
activación de los
linfocitos y sus
funciones efectoras
Eliminación mediante muerte celular inducida por la activación
Los linfocitos T CD4 que
reconocen antígenos
propios pueden recibir
señales que favorecen
su muerte mediante
apoptosis
Muerte celular inducida
por la activación
Se han propuesto dos
mecanismos de muerte celular
inducida por la activación
Si los linfocitos t
reconocen
antígenos propios
Pueden expresar un
miembro proapoptosico
de la familia Bcl (BIM)
Sin miembros antiapoptosicos
de la familia como Bcl-2 y Bcl-x
La acción de BIM no
contrarrestada desencadena la
apoptosis por vía mitocondrial
Los linfocitos
expresan fas, un
miembro de la
familia del
receptor del TNF
Fasl una proteína de membrana
que es estructuralmente
homologa a la citocina TNF se
expresa principalmente en los
linfocitos t activados
La ocupación
de Fas por FasL
Induce a la
apoptosis de los
linfocitos t
activados Por la vía del
receptor de
muerte celular
Se a propuesto que si los antígenos propios ocupan los receptores
antigénicos de los linfocitos t específicos de antígenos propios, se
coexpresan Fas y FasL lo que lleva a la eliminación de los linfocitos
mediante apoptosis mediada por fas

Lupus Eritematoso Sistémico
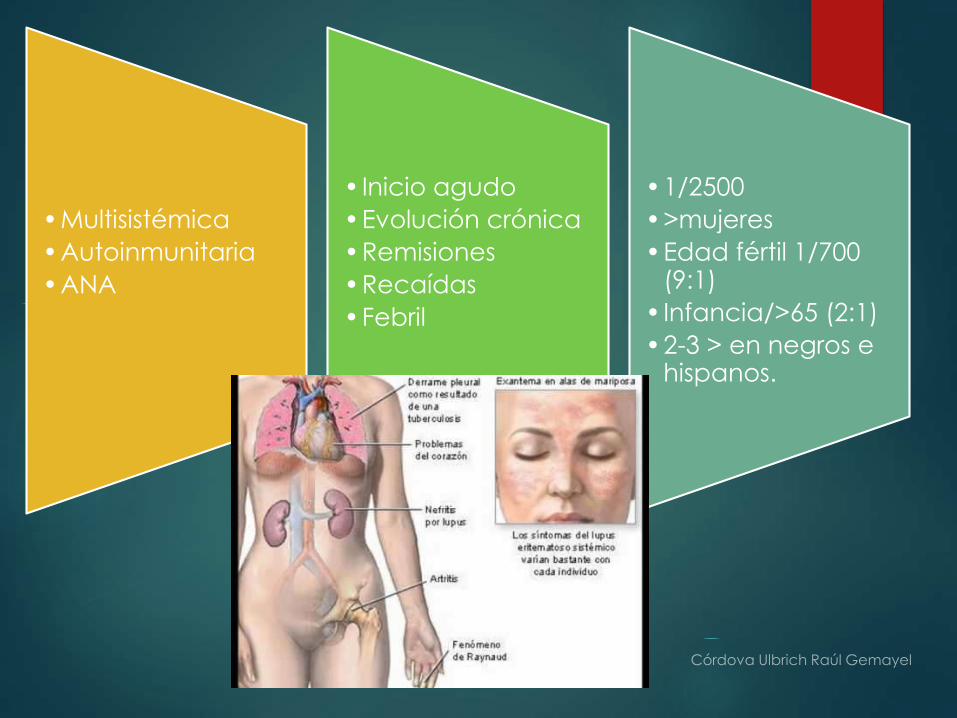
•Multisistémica
•Autoinmunitaria
•ANA
• Inicio agudo
•Evolución crónica
•Remisiones
•Recaídas
•Febril
•1/2500
•>mujeres
•Edad fértil 1/700 (9:1)
• Infancia/>65 (2:1)
•2-3 > en negros e hispanos.

Espectro de autoAc. en LES
Rasgo definitorio
Algunos autoAc reconocen componentes
nucleares y citoplasmáticos. Otros se
dirigen a Ag de sup. de cél. sanguineas.
Útil en el DX y Tx. Importancia patogénica.
• ANA contra Ag nucleares:– Ac contra DNA
– Ac contra histonas
– Ac contra proteinas ≠ de hisonas unidas a RNA
– Ac contra Ag nucleolares

Detección con inmunofluorescencia indirecta.
(sensible pero NO específico)
Patrón fluorescente → tipo de Ac en suero:
Tinción nuclear homogénea o difusa
Tinción anular o periférica
Patrón moteado
Patrón nucleolar
Ac contra AND bicantenario y
Ag Sm → Dx

Además de ANA, otros contra Ag cel. sanguineas. Otros reaccionan con proteinas que forman complejos con fosfolipidos. (40-50% en LE)
Interfieren con pruebas de coagulación (Ac. Anticoagulante lúpico)
Trombosis asociados a abortos espontaneos (Sx. AFL sec)
Sx AFL primario.

Factores Genéticos
• Familia (20% con Ac y alteraciones inmunorreguladoras)
• >en gemelos monocigóticos (>20%)
• Herencia de deficiencia del complemento
Factores Inmunitarios
• Eliminación defectuosa de Linf. B a.reactivos → fallo autotolerancia
• CD4+ escapan de autotolerancia → autoAc patogenicos
• ADN y ARN nucleares activan Linf.B por TLR→ ↑prod. De ANA
• En cel.dendriticas estimulan pruducción de INF , que las activan y favorece resp. De linf Th1→prod. aAc .
• Otras citocinas como TNF y BAFF participan en activacion de linf B y favorece su supervivencia.
Factores ambientales
• Luz UV empeora enfermedad. Induce apoptosis y altera ADN. Modula resp. Inmune.
• Hormonas sexuales. Empeora en menstruación y gestación
• Fármacos como hidralacina, procainamida y D-pricainamida, inducen respuesta similar a LES
Etiología y patogenia• Se desconoce. (fallo de mecanismos que mantiene autolerancia)

Modelo de patogenia del LES
Fact. Ext.
→apoptosis
Alteración de linf
B y T →
autotolerancia
defectuosa. Linf
estimulados por
Ag nucleares
propios (prod. Ac)
Inmunocolmplejo
s se unen a
recptores de Fc.
Ocupación de
TLRs

AutoAc: mediadores. > de lesiones por inmunocomplejos (glomérulo y pequeños vasos)
ANA no pueden penetrar células intactas.
En tejidos, núcleos de cel. Lesionadas reaccionan con ANA
Mecanismo de lesión tisular
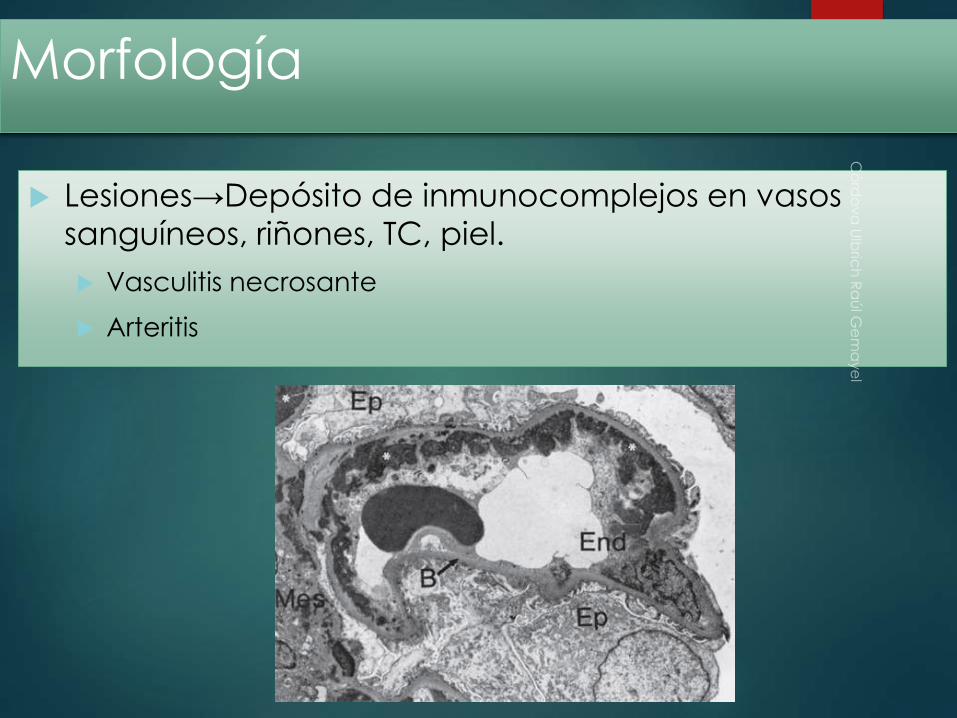
Morfología
Lesiones→Depósito de inmunocomplejos en vasos
sanguíneos, riñones, TC, piel.
Vasculitis necrosante
Arteritis
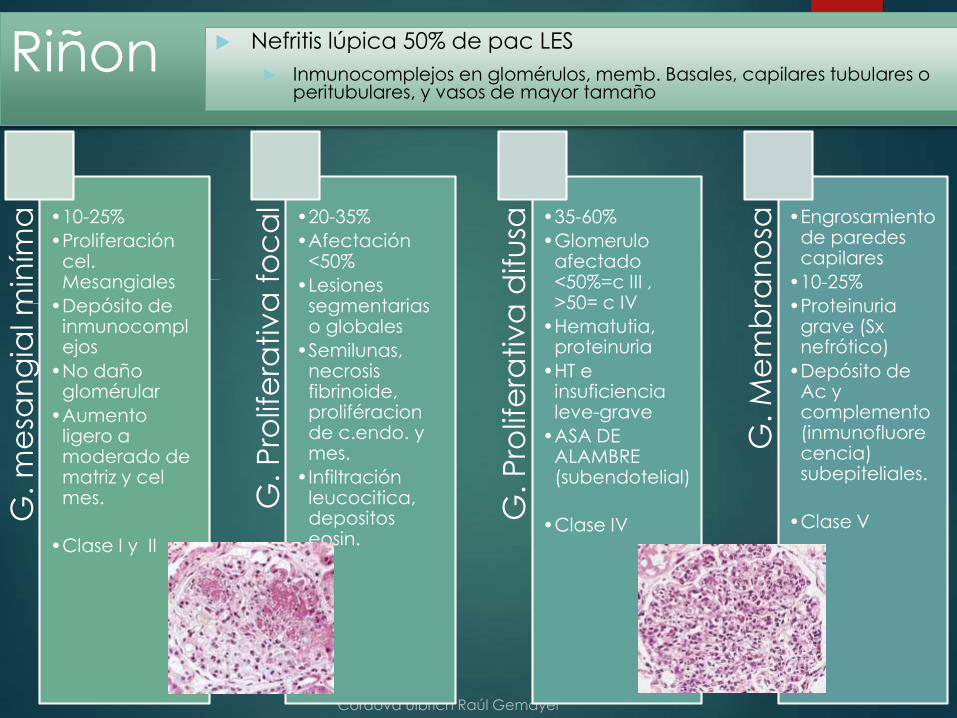
Riñon Nefritis lúpica 50% de pac LES
Inmunocomplejos en glomérulos, memb. Basales, capilares tubulares o peritubulares, y vasos de mayor tamaño
G. m
esa
ng
ialm
iním
a •10-25%
•Proliferación cel. Mesangiales
•Depósito de inmunocomplejos
•No daño glomérular
•Aumento ligero a moderado de matriz y celmes.
•Clase I y II
G. P
rolif
era
tiv
a f
oc
al
•20-35%
•Afectación <50%
•Lesiones segmentarias o globales
•Semilunas, necrosis fibrinoide, proliféracionde c.endo. y mes.
•Infiltración leucocitica, depositoseosin.
G. P
rolif
era
tiv
a d
ifu
sa
•35-60%
•Glomeruloafectado <50%=c III , >50= c IV
•Hematutia, proteinuria
•HT e insuficiencia leve-grave
•ASA DE ALAMBRE (subendotelial)
•Clase IV
G. M
em
bra
no
sa
•Engrosamiento de paredes capilares
•10-25%
•Proteinuria grave (Sxnefrótico)
•Depósito de Ac y complemento (inmunofluorecencia) subepiteliales.
•Clase V
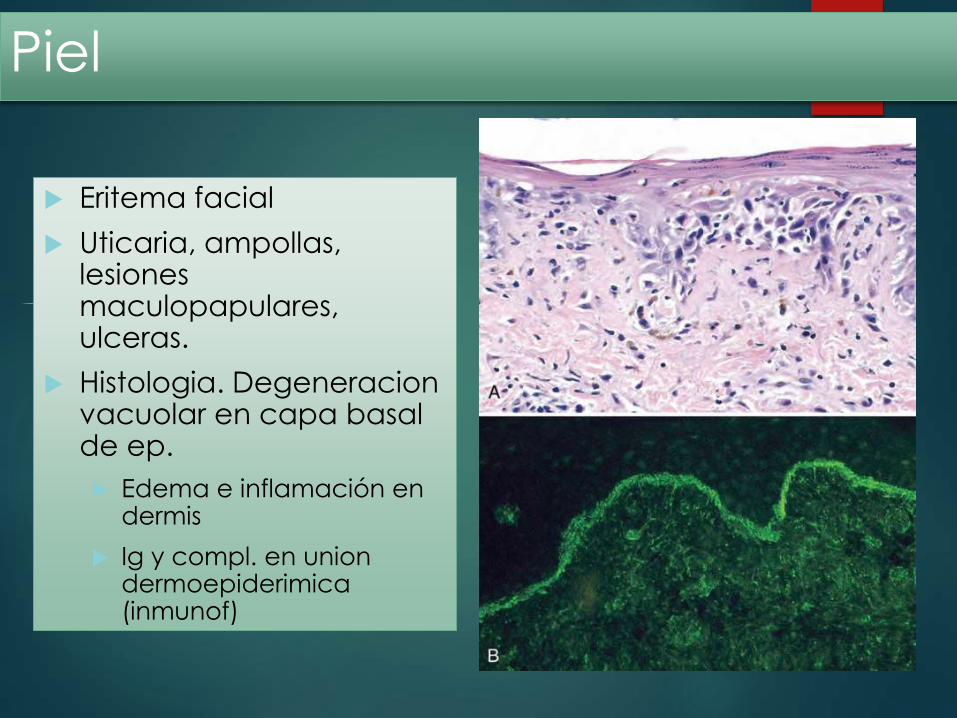
Eritema facial
Uticaria, ampollas, lesiones maculopapulares, ulceras.
Histologia. Degeneracionvacuolar en capa basal de ep.
Edema e inflamación en dermis
Ig y compl. en uniondermoepiderimica(inmunof)
Piel
Sinovitis no erosiva con Poca deformidad
Articulaciones
SNC • Oclusión no inflamatoria de vasos pequeños por prolif.
de la intima por lesión el endotelio por Ac.antiF.
Bazo
Pulmones
• Esplenomegalia, engrosamiento capsular e
hiperplasia folicular
• Pleuritis y derrames pleurales 50%
Otros • Cuerpos de LE en MO y otros org. Ganglios aumentados de
tamaño,
linfadenitis necrosante

Agudas, subagudas, crónicas
Sist. Cardiovascuar (capas) 50% pac.
Miocarditis (<frecuente) → taquicardia
Alteraciones valvulares→engrosamiento
de los velos (disfunción)
Endocarditis de Libman-Sacks
(berrugosa no bacteriana)
Arteriopatia coronaria por
ateroesclerosis
Pericarditis y afección a cavidades serosas

Multisistémica
Mujer joven
Exantema malar, dolor torácico, fotosensible
ANA (100%/no especificos)
Trastorno hematológico, inmunitario, neurológico, renal.
Evolución variable
Casos agudos → muerte en semanas
Con tx se presentan exacerbaciones y remisiones (5-10años)
Insuficiencia renal e infecciones recurrentes, arteriopatias
Características clínicas
• LE discóide crónico (manif. Sistémicas en <frec)– Placas cutaneas con edema,
eritema, descamación, tapones foliculares y atrofia cutanea (borde erit. elevado)
– Ig y c3 en union dermoep.
• LE cutaneosubagudo– Exantema
generaliza-do
– Intermedio LES y LEDC

Hidralacina, procaidemina, isoniacida, D-
penicilamina…
ANA y > nosintomas de LE
>frec Ac contra histonas. Ac contra ADN bic.
<frevuencia
Pac. Con alelo HLA-DR4 >riesgo de LE con admn.
De hidelicina.
LE inducido por fármacos

Artritis reumatoide

Enfermedad multisistémica que afecta principalmente las
articulaciones con sinovitis inflamatoria y proliferativa no
supurativa que progresa a destrucción de cartílago articular y
anquilosis articular.
Causa desconocida. Predisposición genética, ambiente,
autoinmunidad.
Artritis reumatoide

Sx de SJÖGREN
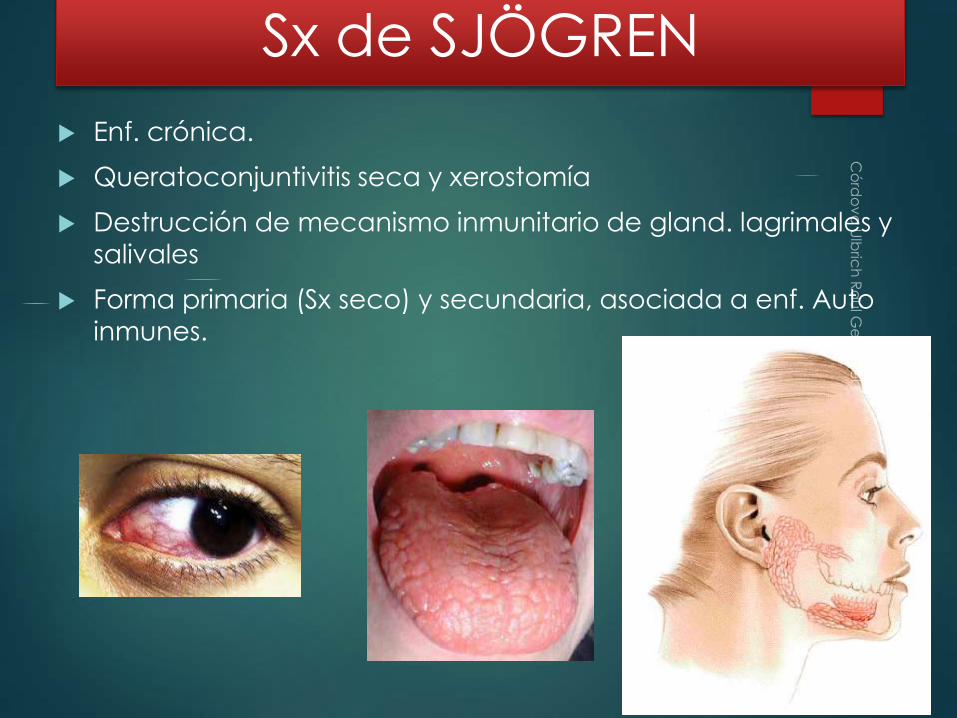
Enf. crónica.
Queratoconjuntivitis seca y xerostomía
Destrucción de mecanismo inmunitario de gland. lagrimales y salivales
Forma primaria (Sx seco) y secundaria, asociada a enf. Auto
inmunes.
Sx de SJÖGREN

↓ de lagrimas y saliva por infiltración linfocítica (CD40, B, c.plas) y fibrosis de las glándulas.
75% pac con factor reumatoide.
ANA en 85%
Ac específicos y no específicos de órgano
Ac contra Ag ribonucleoprotéicos, SS-A(RO) y SS-B(LA) (90% pac)
Asociado a loci HLA-B8, HLA-DR3 y DRW52, HLA-DQA1, HLA-DB1.
Etiología y patogénia
• Patogenia no definida. Implicada activación de linf B y T.
• Desencadenación por inf vírica de G. salivales→muertecel→autoAg
• CD4 y linf B autorreact.→inflamación, lesión tisular→fibrosis

G. salivales, lagrimales; exocrinas en aparato respiratorio,
digestivo y vagina.
Infiltración linfocítica periductal y perivascular.
Infiltrado extenso (folículos linfáticos con centro germinativo)
Hiperplasia de cel epit. de conductos (obstrucción) → atrofia,
fibrosis, hialinización de acinos, sist.de parenquima por grasa
Morfología
• Ausencia de lágrimas →
desecación de epitelio corneal,
mucosa oral puede atrofiar con
fisuras inflamatorias y ulceración
• Sequedad y costras en nariz →
úlceras (perforación de tabique)
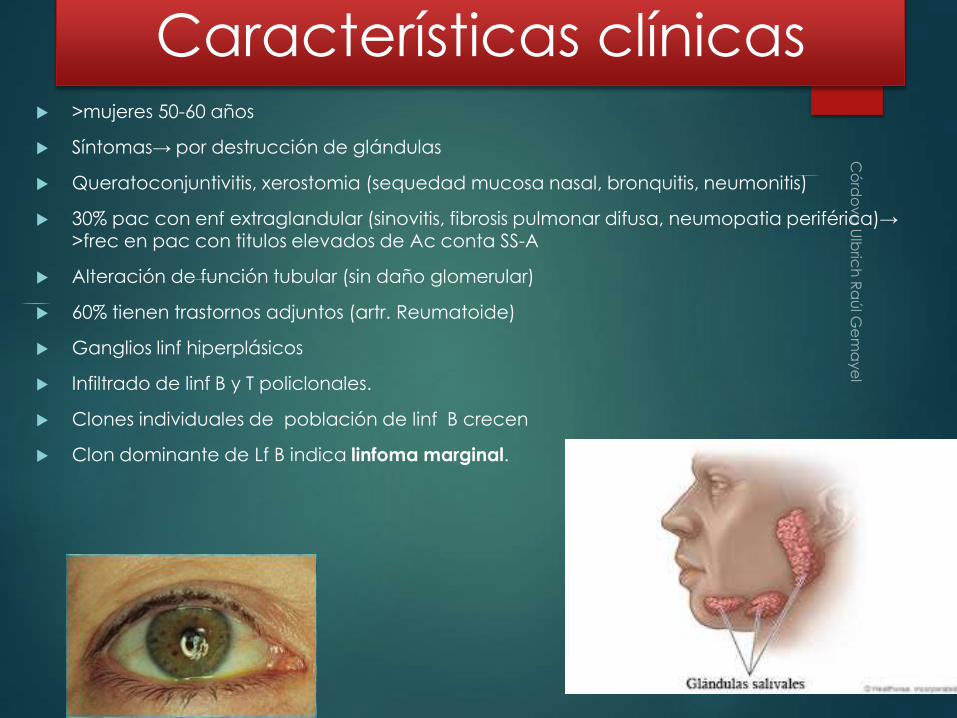
>mujeres 50-60 años
Síntomas→ por destrucción de glándulas
Queratoconjuntivitis, xerostomia (sequedad mucosa nasal, bronquitis, neumonitis)
30% pac con enf extraglandular (sinovitis, fibrosis pulmonar difusa, neumopatia periférica)→ >frec en pac con titulos elevados de Ac conta SS-A
Alteración de función tubular (sin daño glomerular)
60% tienen trastornos adjuntos (artr. Reumatoide)
Ganglios linf hiperplásicos
Infiltrado de linf B y T policlonales.
Clones individuales de población de linf B crecen
Clon dominante de Lf B indica linfoma marginal.
Características clínicas

Esclerosis sistémica (Esclerodermia)
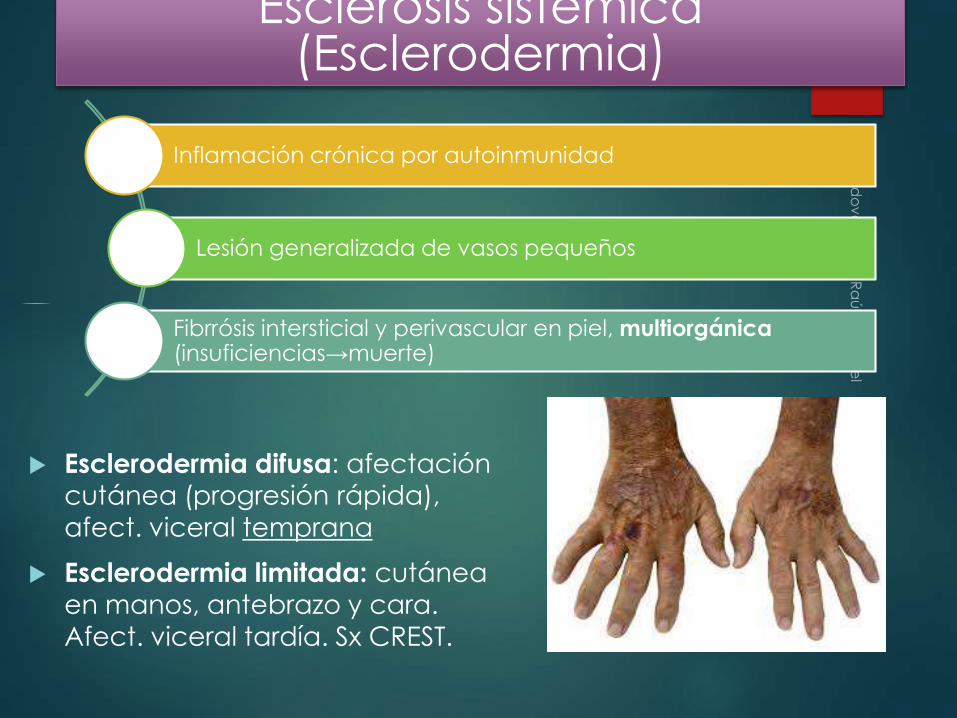
Esclerodermia difusa: afectación cutánea (progresión rápida),
afect. viceral temprana
Esclerodermia limitada: cutánea
en manos, antebrazo y cara.
Afect. viceral tardía. Sx CREST.
Esclerosis sistémica (Esclerodermia)
Inflamación crónica por autoinmunidad
Lesión generalizada de vasos pequeños
Fibrrósis intersticial y perivascular en piel, multiorgánica(insuficiencias→muerte)
Causas desconocidas
Resp. Autoinm., tensión vascular y depósito de colágeno → lesión tisular final
Etiología y patogenia
Respuestas autoinm. anormales
• CD4+ reaccionan y acumulan en piel. Liberan citocinas que activan cel. Linf y fibroblástos.
• Activ. Inadecuada de inm. Humoral. AutoAc.
• ANA contra ADN topoisomerasa I, en 20% con esclerósis dif.
• Ac anticentroméricos(20-30%) → Sx CREST
Lesión vascular
• Lesión microvascularconstante→lesióninicial.
• Proliferación de la intima en arterias digitales.
• Dilatación capilar, >permeabilidad →destrucción capilar
• Lesión por inflamación
• Lesión endo. y agregplaq. → liberación de fact. Plaq. Y endotelial
Fibrósis
• Culminación de alteraciónes
• Alteración en producción de colágeno

Morfología
Piel • Atrófia esclerótica difusa (proximal/distal) >pac
• Edema e infiltración de CD4+, tumefacción y degeneración de
fibras de colágeno.– Engrosamiento de lámina basal en art. pequeñas. (lesión y oclusión)
• Fibrosis en dermis
• > colágeno en dermis. (adelgazamiento de epidermis, perdida de
crestas, atrofia de anexos, engrosamiento de paredes de art y capilares.
• Calcificaciones subcutáneas
• Dedos afilados, limitación de movimiento
articular. Cara de mascara.
Multiorgánico
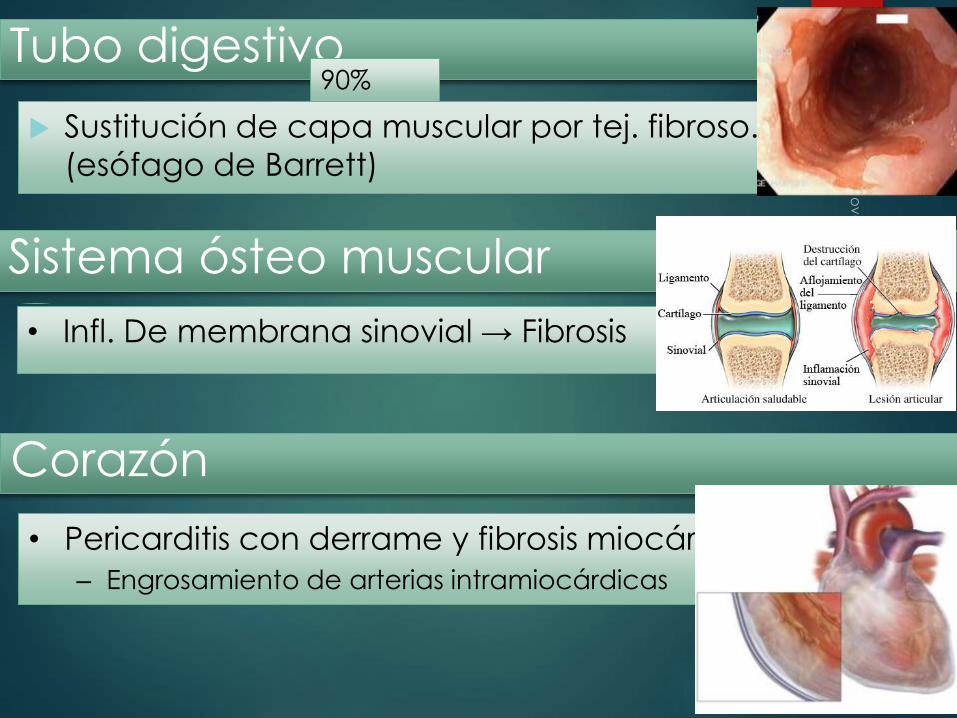
Sustitución de capa muscular por tej. fibroso.
(esófago de Barrett)
Tubo digestivo 90%
Sistema ósteo muscular
• Infl. De membrana sinovial → Fibrosis
Corazón
• Pericarditis con derrame y fibrosis miocárdica
– Engrosamiento de arterias intramiocárdicas
Riñones
• Lesiónes vasculares. Engrosamiento de la intima de arteriasinterlobulillares
• Proliferación de cél. de la intima
• HT en 30%.
• Alteraciones vasculares pronunciadas asociadas a necrosis fibrinoide, trombósis, infarto
• Muerte por insuficiencia renal en 50%
Pulmones
• 50% de afectados manifiestan HT pulmonar y fibrosis intersticial

Mujeres-hombres 3:1 (50-
60años,afroamericanos)
Cambios cutáneos (engrosamiento)
Fenómeno de Raynaud
Disfágia por fibrosis esofágica
Afectación de intestino delgado
Dificultad respiratoria por fibrosis
pulmonar
Características clínicas
• Fibrosis miocardica(arritmias, insuficiencia)
• Proteinuria (30% pac)
• Hipertensión maligna con posterior insuficiencia renal mortal
• Sx CREST (limitado a la piel. Dedos de las manos, antebrazos y cara)
Miopatías Inflamatorias
Son un grupo infrecuente y heterogéneo de trastornos que se
caracterizan por lesión e inflamación de los músculos,
principalmente esqueléticos, probablemente de
mecanismo inmunitario.
En esta categoría se incluyen tres trastornos diferentes:
- Dermatomiositis.
- Polimiositis.
- Miositis por cuerpos de inclusión.
Pueden aparecer de forma aislada o con otras enfermedades
de mecanismo inmunitario, particularmente esclerosis
sistémica.
Karen Robles
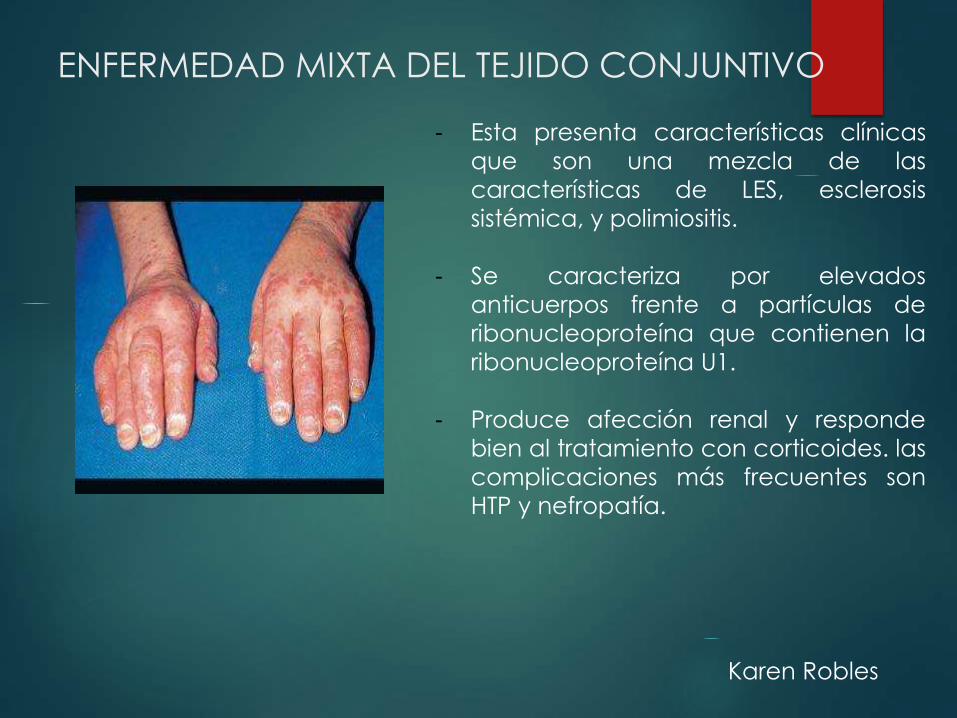
ENFERMEDAD MIXTA DEL TEJIDO CONJUNTIVO
- Esta presenta características clínicas
que son una mezcla de las
características de LES, esclerosis
sistémica, y polimiositis.
- Se caracteriza por elevados
anticuerpos frente a partículas de
ribonucleoproteína que contienen la
ribonucleoproteína U1.
- Produce afección renal y responde
bien al tratamiento con corticoides. las
complicaciones más frecuentes son
HTP y nefropatía.
Karen Robles

Rechazo de los
trasplantes de
órganos
Karen
Robles

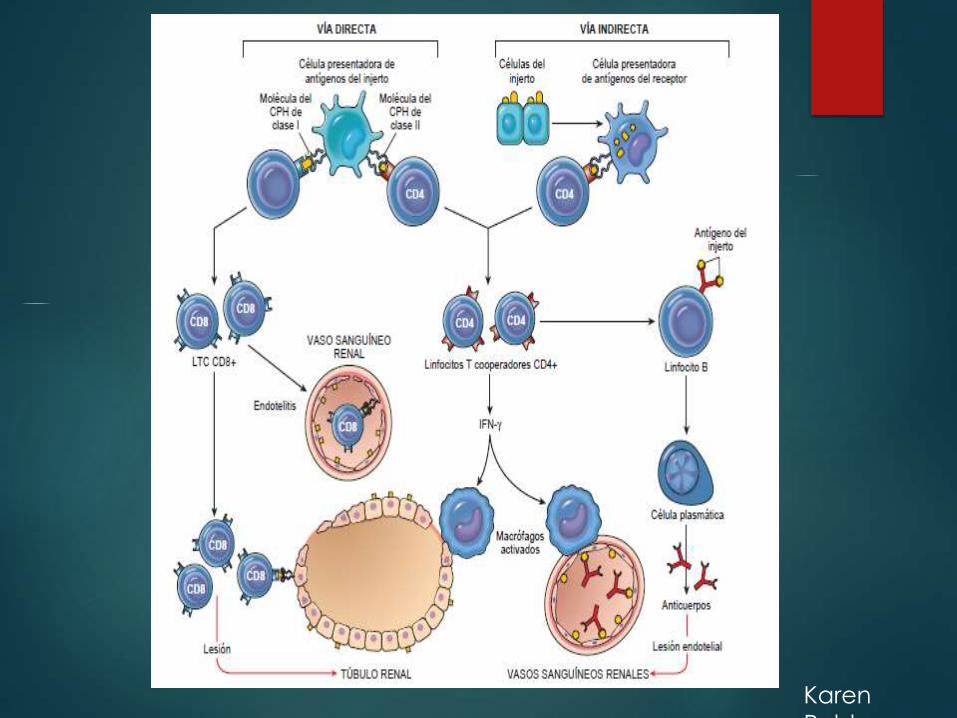
Rechazo de trasplante de
órganosReacciones mediadas por los linfocitos T, se denomina rechazo celular,
donde la destrucción de la célula está dada por los LTC CD8 y por
reacciones de hipersensibilidad retardada (por Lc CD4).
Hay dos vías por las cuales los LT del receptor van a reconocer los
antígenos del donante (alogénicos o aloantígenos).
- Directa.
- Indirecta.
Karen
Robles
Karen
Robles

Reacciones Mediadas por anticuerposRechazo humoral:
- Se produce rechazo hiperagudo
cuando en la circulación del
receptor hay anticuerpos
preformados contra el donante.
- La diana inicial de estos anticuerpos
en el rechazo parece ser la
vasculatura del injerto.
Karen
Robles

Karen
Robles
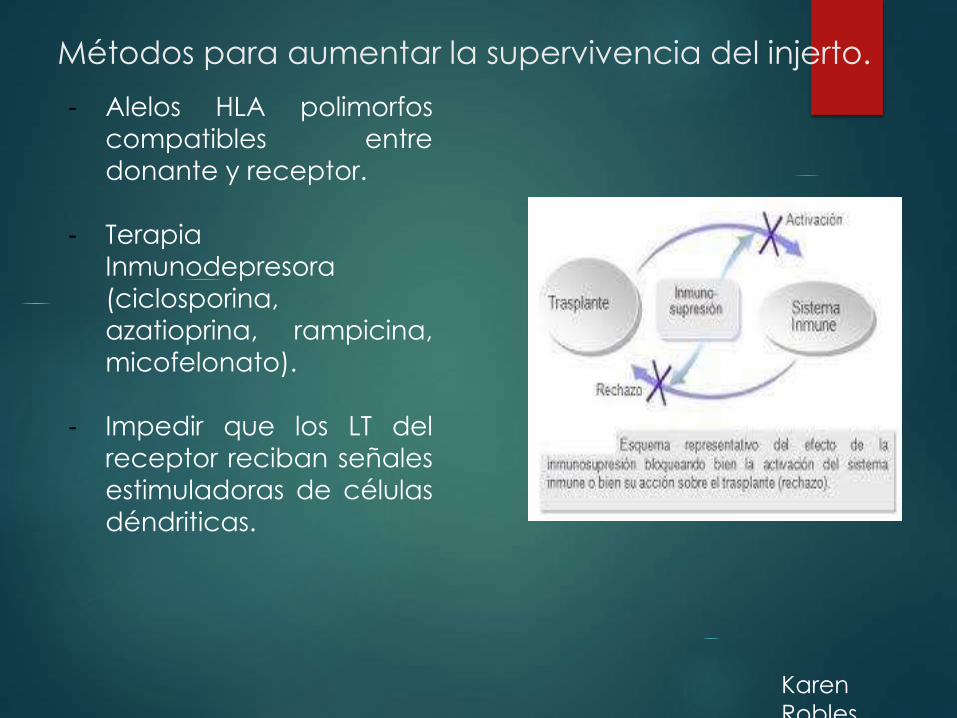
Métodos para aumentar la supervivencia del injerto.
- Alelos HLA polimorfos
compatibles entre
donante y receptor.
- Terapia
Inmunodepresora
(ciclosporina,
azatioprina, rampicina,
micofelonato).
- Impedir que los LT del
receptor reciban señales
estimuladoras de células
déndriticas.
Karen
Robles

Trasplante de órganos sólidos.
En el trasplante
de hígado no
es tan intensa
la reacción de
rechazo.
Se desconoce la
base molecular
de este
privilegio.
Karen
Robles

Trasplante de Células Hematopoyéticas- Para el tratamiento de neoplasias malignas hematológicas, algunos cánceres no hematológicos,
anemias aplásicas, talasemias y algunos estados de inmunodeficiencia.- Para enfermedades que requieren trasplante de médula ósea, se irradia al receptor para destruir
el sistema inmunitario (y, en ocasiones, las células cancerosas) y crear un lecho para el injerto.
- Dos problemas que son exclusivos del trasplante de médula ósea son:- La enfermedad del injerto contra el anfitrión (ICH o rechazo inverso, aguda o crónica).- La inmunodeficiencia.
Karen
Robles

La enfermedad del ICH aguda.
Se produce en un plazo de días a semanas después de un trasplante
de médula ósea alogénica. Aunque puede estar afectado
cualquier órgano, las principales manifestaciones clínicas se deben
a la afectación del sistema inmunitario y de los epitelios de la piel, el
hígado y el intestino.
La enfermedad del ICH crónica.
Después de síndrome agudo o puede aparecer de forma insidiosa.
Estos pacientes tienen una lesión cutánea extensa con destrucción
de los anexos
cutáneos y fibrosis de la dermis. Los cambios pueden recordar a la
esclerosis sistémica.
Karen
Robles
Karen
Robles
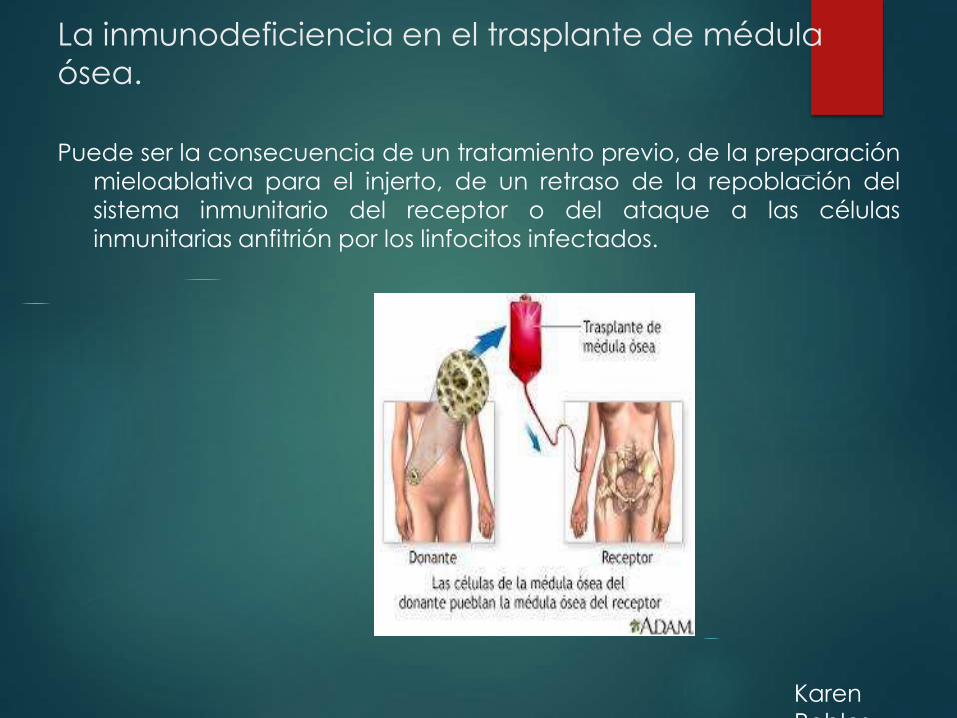
La inmunodeficiencia en el trasplante de médula
ósea.
Puede ser la consecuencia de un tratamiento previo, de la preparación
mieloablativa para el injerto, de un retraso de la repoblación del
sistema inmunitario del receptor o del ataque a las células
inmunitarias anfitrión por los linfocitos infectados.

Síndromes de inmunodeficiencia
Karen
Robles

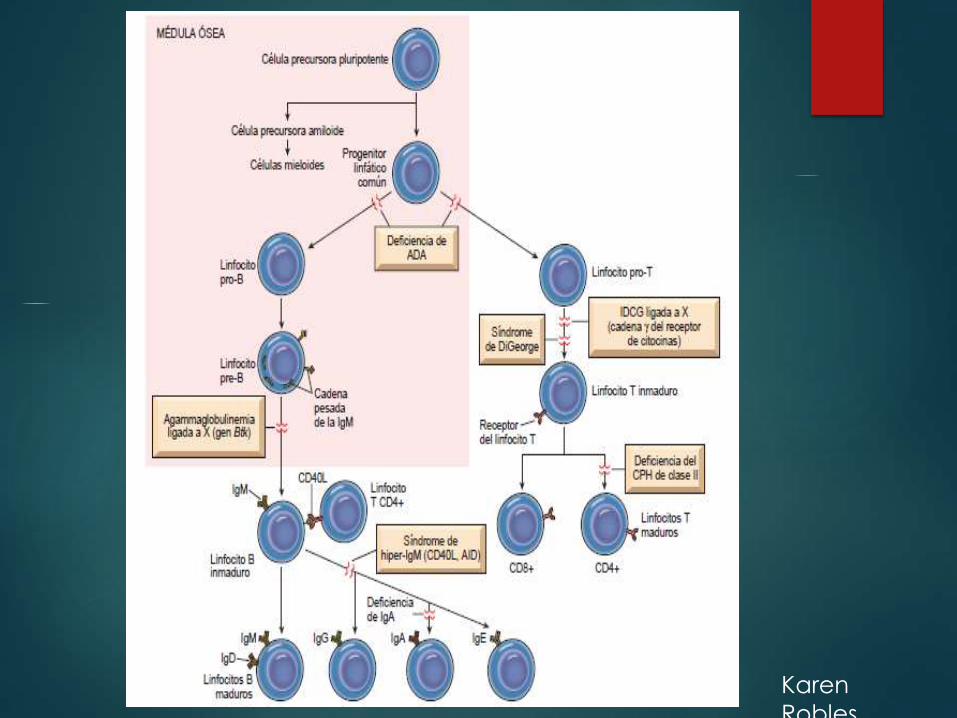
Inmunodeficiencias primarias.
- Están determinadas genéticamente y afectan a las ramas humoral y/o celular de lainmunidad adaptativa o a los mecanismos de defensa de la inmunidad innata.
- Los defectos de la inmunidad adaptativa con frecuencia se clasifican según elprincipal componente afectado.
- La mayoría de las inmunodeficiencias primarias se manifiestan en la lactancia, entre los6 meses y los 2 años de vida, y se detectan porque los lactantes afectados sonsusceptibles a infecciones de repetición.
- Con los avances en los análisis génicos, actualmente se han identificado lasmutaciones responsables de muchas de las inmunodeficiencias primarias frecuentes
Karen
Robles
Karen
Robles

Agammaglobulinemia ligada a X
(agammaglobulinemia de Bruton)
Es una de las formas más frecuentes de inmunodefi ciencia primaria. Se
caracteriza por la ausencia de maduración de los precursores de los
linfocitos B (linfocitos pro-B y linfocitos pre-B) en linfocitos B maduros.
Como enfermedad ligada a X, este trastorno se ve casi totalmente en
varones, aunque se han descrito casos esporádicos en mujeres,
posiblemente producidos por mutaciones en algún otro gen que
actúa por la misma vía. La enfermedad habitualmente no se hace
evidente hasta aproximadamente los 6 meses de edad, cuando se
produce depleción de las inmunoglobulinas maternas.
Karen
Robles

La forma clásica de esta enfermedad tiene las
características siguientes:- Hay ausencia o marcada disminución de linfocitos B en la circulación, y disminución de
la concentración sérica de todas las clases de inmunoglobulinas. Los linfocitos pre-B,que expresan el marcador de la línea B CD19, pero no Ig de membrana, se encuentranen cantidades normales en la médula ósea.
- Los centros germinativos de los ganglios linfáticos, las placas de Peyer, el apéndice y lasamígdalas están desarrollados de forma insuficiente.
- Hay ausencia de células plasmáticas en todo el cuerpo.
- Las reacciones mediadas por linfocitos T son normales.
Karen
Robles

Inmunodeficiencia variable
comúnLa característica común de todos los pacientes es la
hipogammaglobulinemia, que generalmente
afecta a todas las clases de anticuerpos, aunque
en ocasiones sólo a la IgG. El diagnóstico de
inmunodeficiencia variable común se basa en la
exclusión de otras causas bien definidas de
disminución de la producción de anticuerpos
Karen
Robles

Deficiencia aislada de IgAEs una inmunodeficiencia frecuente. En los EE. UU.
aparece aproximadamente en 1 de cada 600
personas de origen europeo. Es mucho menos
frecuente en negros y asiáticos. Las personas
afectadas tienen concentraciones muy bajas de
IgA sérica y secretora. Puede ser familiar o puede
ser adquirida, asociada a toxoplasmosis, parotiditis
o alguna otra infección vírica.
Karen
Robles

Síndrome de DiGeorge (hipoplasia tímica)
Es una deficiencia de linfocitos T que se debe a la
ausencia de desarrollo de la tercera y la cuarta
bolsas faríngeas. Esta última da lugar al timo, las
paratiroides, algunas de las células claras del
tiroides y el cuerpo ultimobranquial.
Karen
Robles

Inmunodeficiencia
combinada graveRepresenta una constelación de síndromes diferentes desde el punto
de vista genético que tienen todos ellos en común defectos de las
respuestas inmunitarias humorales y celulares.
Algunos pacientes presentan exantema morbiliforme poco después del
nacimiento, porque se transfieren linfocitos T maternos a través de la
placenta y afectan al feto, produciendo enfermedad del ICH.
Los pacientes con IDCG son muy susceptibles a infecciones recurrentes
y graves por una amplia gama de patógenos
Karen
Robles

Se han descubierto otras formas menos frecuentes de
IDCG recesiva autosómica:
- Las mutaciones de los genes activadores de la recombinasa
impiden as reordenamientos génicos somáticos esenciales para el
ensamblaje de los receptores de los linfocitos T y de los genes de las
Ig.
- Una cinasa intracelular llamada Jak3 es esencial para la
transducción de señales en la cadena común del receptor de las
citocinas.
- Se han descrito varias mutaciones de las moléculas de
transducción de señales, como las cinasas asociadas al receptor
antigénico de los linfocitos T.
- Las mutaciones que reducen la expresión de las moléculas del CPH
de clase II impiden el desarrollo de los linfocitos T CD4+.
Karen
Robles

Inmunodeficiencia con trombocitopenia y eccema (síndrome de Wiskott-
Aldrich)
Es una enfermedad recesiva ligada a X que se
caracteriza por trombocitopenia, edema y una
marcada vulnerabilidad a infecciones de
repetición, que lleva a la muerte precoz.
Karen
Robles

Deficiencias genéticas del sistema del
complemento
Se asocia a infecciones piógenas recurrentes. El
componente C3 del complemento es necesario
para las vías clásica y alternativa, por lo que una
deficiencia de esta proteína se asocia a
susceptibilidad a infecciones piógenas graves y
recurrentes. También hay aumento de la incidencia
de glomerulonefritis mediada por
inmunocomplejos.
Karen
Robles

INMUNODEFICIENCIAS SECUNDARIAS
- Se pueden encontrar inmunodeficiencias
secundarias en pacientes con cáncer, diabetes y
otras enfermedades metabólicas, malnutrición,
infección crónica y nefropatía.
- En conjunto, las inmunodeficiencias secundarias
son más frecuentes que los trastornos de origen
genético primario. La inmunodeficiencia
secundaria más importante es el sida
Karen
Robles

SÍNDROME DE
INMUNODEFICIENCIA
ADQUIRIDA (SIDA)
Karen
Robles

Es una enfermedad producida por el retrovirus
llamado virus de inmunodeficiencia humana (VIH) y
se caracteriza por una profunda inmunodepresión
que da lugar a infecciones oportunistas, neoplasias
secundarias y manifestaciones neurológicas.
Karen
Robles

Epidemiología
- Los hombres homosexuales o bisexuales.
- Los pacientes que consumen drogas por vía intravenosa.
- Los hemofílicos.
- Los receptores de sangre y hemoderivados que no son
hemofílicos.
- Los contactos heterosexuales de los miembros de otros grupos de riesgo elevado.
- La transmisión sexual.
- Transmisión de madre a hijo.
Karen
Robles
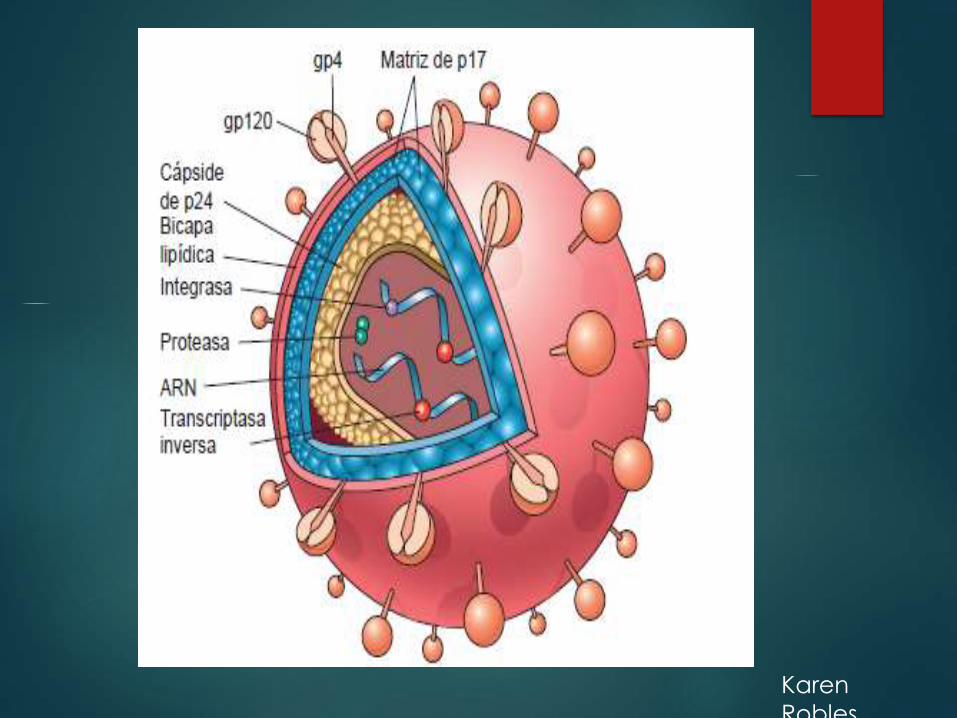
Karen
Robles
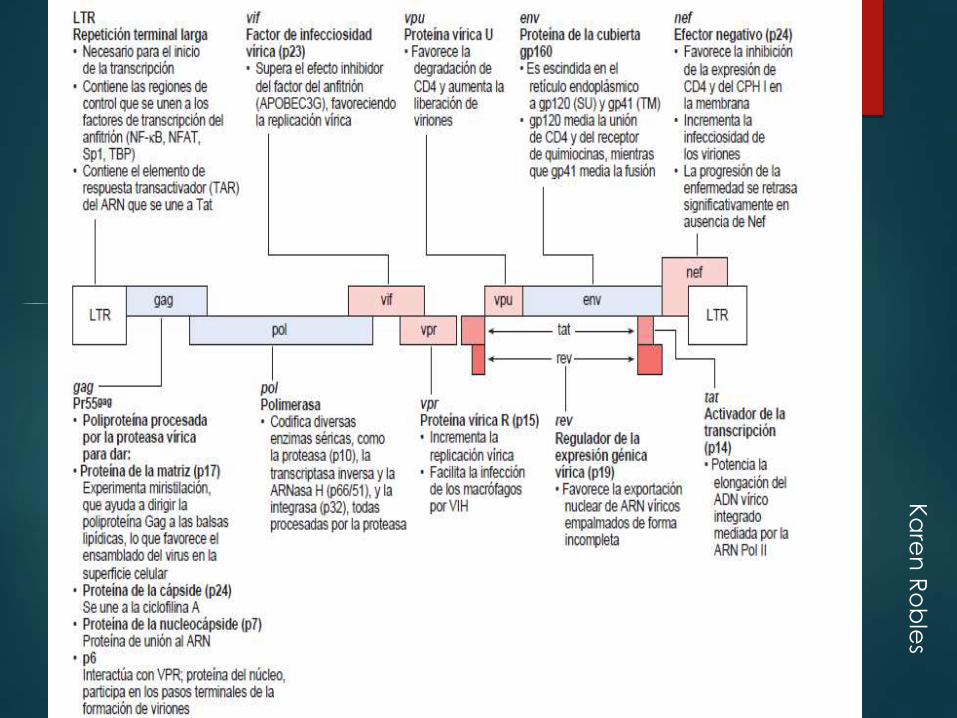
Ka
ren
Ro
ble
s
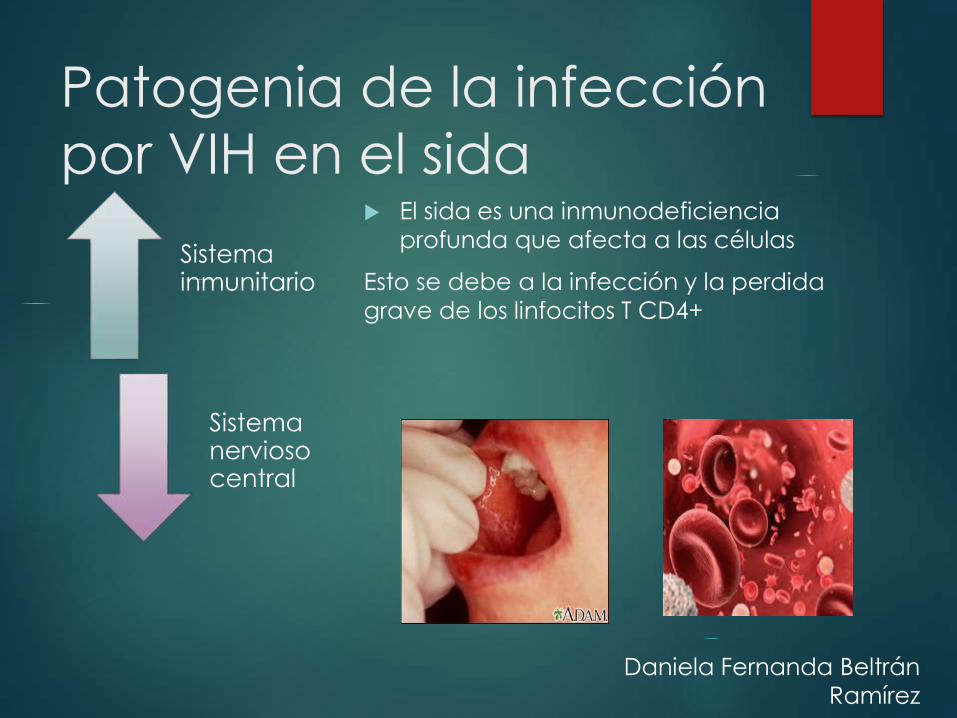
Patogenia de la infección
por VIH en el sida
Sistema inmunitario
Sistema nervioso central
El sida es una inmunodeficiencia
profunda que afecta a las células
Esto se debe a la infección y la perdida
grave de los linfocitos T CD4+
Daniela Fernanda Beltrán
Ramírez

Ciclo vital del VIH
Infección de las células
Integración del provirus en el genoma de la célula anfitriona
Activación de la replicación vírica
Producción y liberación de virus infeccioso
Daniela Fernanda Beltrán
Ramírez
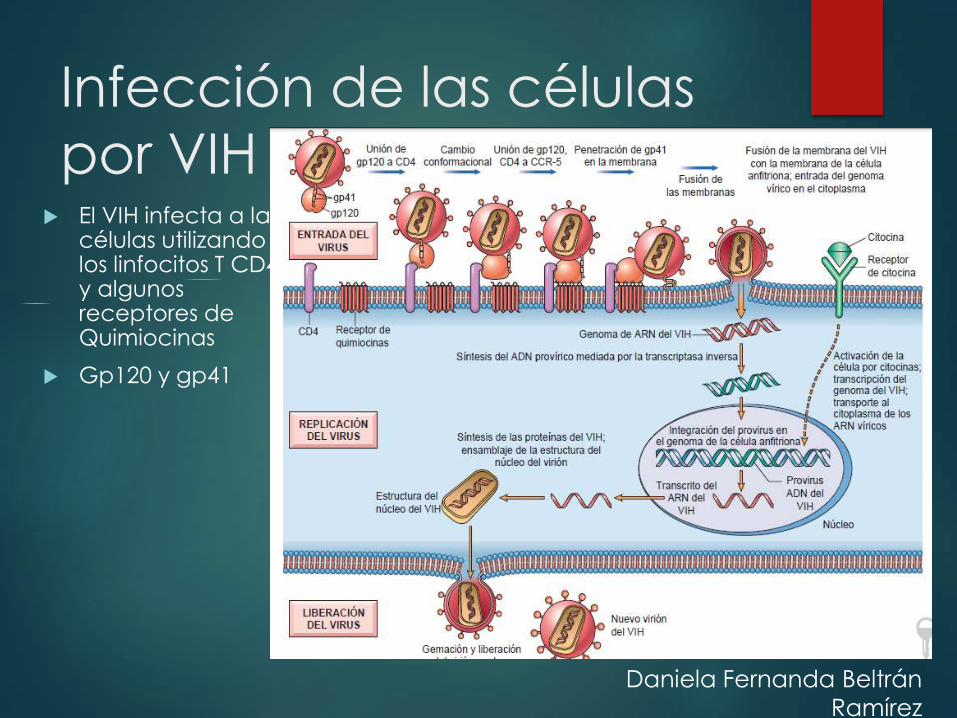
Infección de las células
por VIH El VIH infecta a las
células utilizando a los linfocitos T CD4+ y algunos receptores de Quimiocinas
Gp120 y gp41
Daniela Fernanda Beltrán
Ramírez
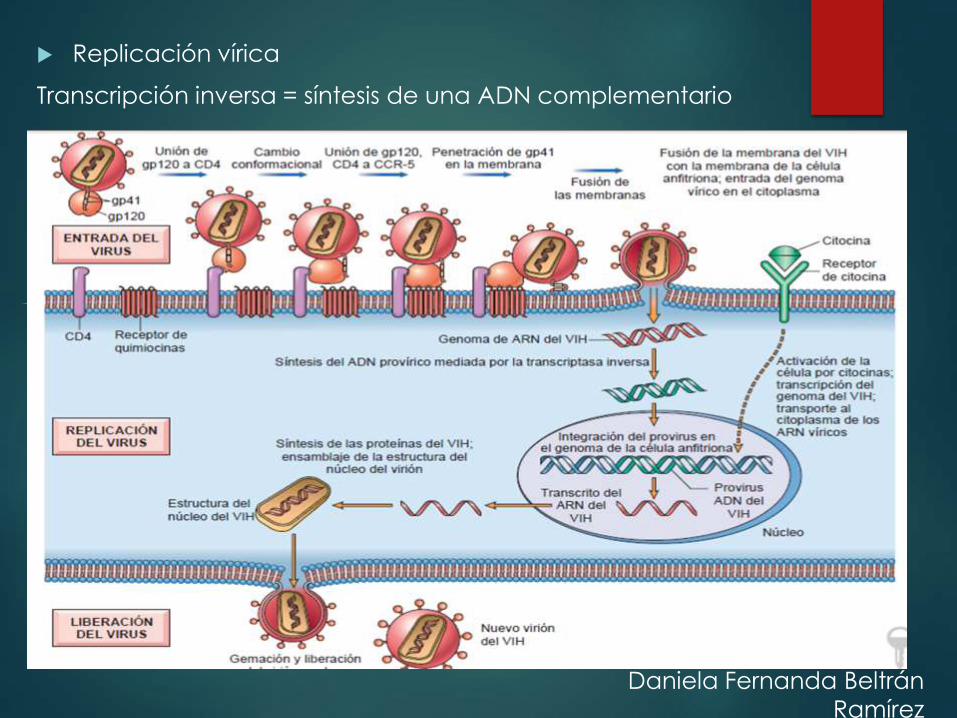
Replicación vírica
Transcripción inversa = síntesis de una ADN complementario
Daniela Fernanda Beltrán
Ramírez
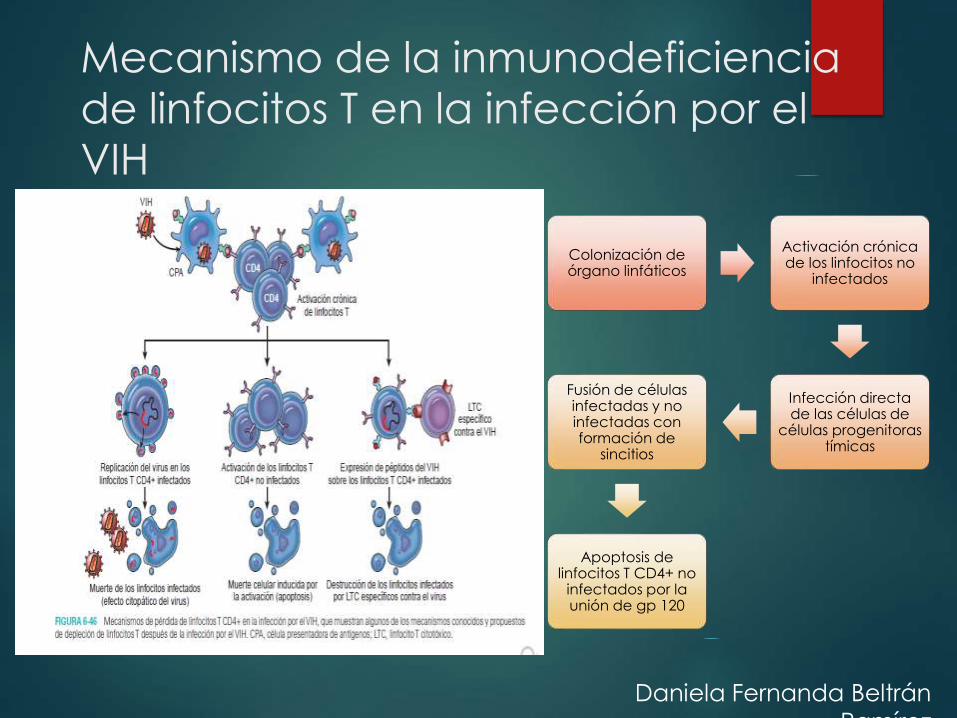
Mecanismo de la inmunodeficiencia
de linfocitos T en la infección por el
VIH
Colonización de órgano linfáticos
Activación crónica de los linfocitos no
infectados
Infección directa de las células de
células progenitoras tímicas
Fusión de células infectadas y no infectadas con formación de
sincitios
Apoptosis de linfocitos T CD4+ no
infectados por la unión de gp 120
Daniela Fernanda Beltrán
Ramírez

Defectos cualitativos de los linfocitos T
Daniela Fernanda Beltrán
Ramírez

Infección por VIH de células distintas a los
linfocitos T
La infección de macrófagos y células dendríticas
también son importantes en la patogenia de la
infección del VIH.
Daniela Fernanda Beltrán
Ramírez
El VIH-1 puede infectar a los macrófagos con diferenciación terminal y puede multiplicarse en ellos.
Macrófagos infectados liberan por gemación cantidades pequeñas de virus, aunque también son resistentes a los efectos citopaticos del VIH
Los macrófagos actúan como porteros de la infección, el 90% de los casos de infección aguda.

• Son infectadas por el virus y lo transportan hasta los ganglios linfáticos regionales donde el virus se transmite a los linfocitos T CD4+
Células dendríticas mucosas
• Son posibles reservorios del virus al igual que los macrófagos.
Células dendríticas foliculares

Patogenia de la afección
del SNC Diana importante de la infección (SN)
Algunos factores solubles: IL-1, TNF e IL-6, oxido nítrico
inducido por gp120.
Daniela Fernanda Beltrán
Ramírez
Monocitos infectados
Macrófagos y microglía

Evolución natural de la
infección por VIH
Infección primaria, diseminación del virus y
síndrome retrovírico agudo
Daniela Fernanda Beltrán
Ramírez
Infección aguda caracterizada por la
infección de los linfocitos T CD4+ de memoria en los
tejidos mucosos
Diseminacióm del virus y aparición de respuestas
inmunitarias
Deteccion de los linfocitos T CD8
Manifestación de la propagación del virus (síndrome retrovírico
agudo)
Niveles de ARN de VIH,marcador de la progresión del virus
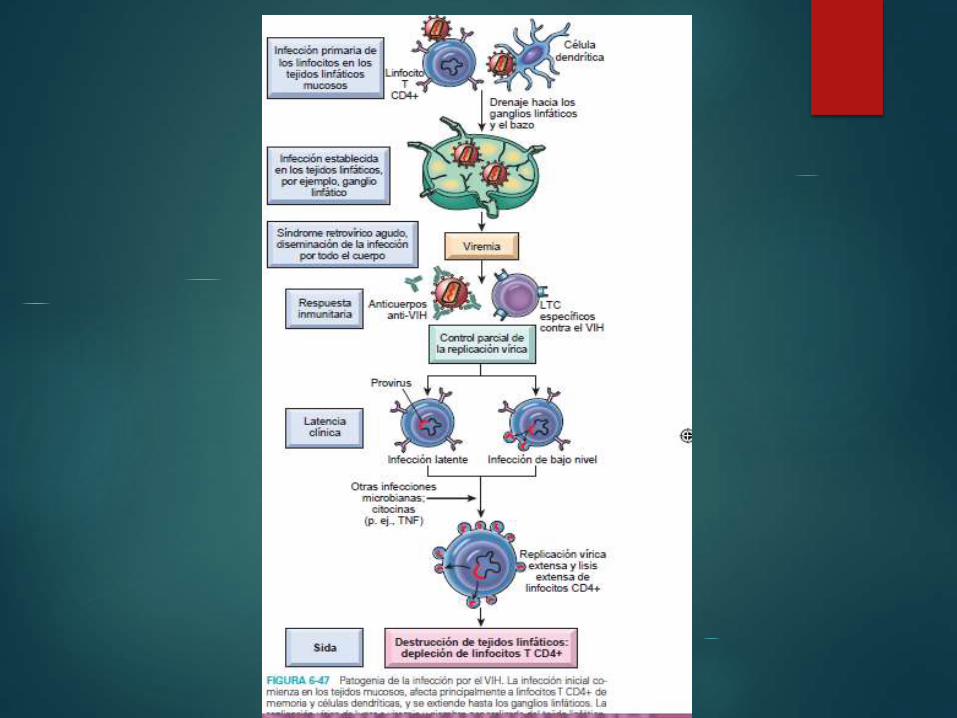
Infección crónica: fase de latencia clínica
- Replicación continua de VIH
- Destrucción celular
Daniela Fernanda Beltrán
Ramírez
• Pocas manifestaciones clínicas o
ninguna
• El 10% de los linfocitos T CD 4
infectados
• Las defensas del anfitrión disminuyes

Sida
Presenta
Daniela Fernanda Beltrán
Ramírez
Desorganización de las defensas del anfitrión,
aumento importante
de la viremia
plasmática y
enfermedad grave o
potencialmente mortal

Características clínicas
del SIDA
Infecciones oportunistas
Son responsables de la
mayoría de las muertes de
pacientes con SIDA no
tratados.
Daniela Fernanda Beltrán
Ramírez
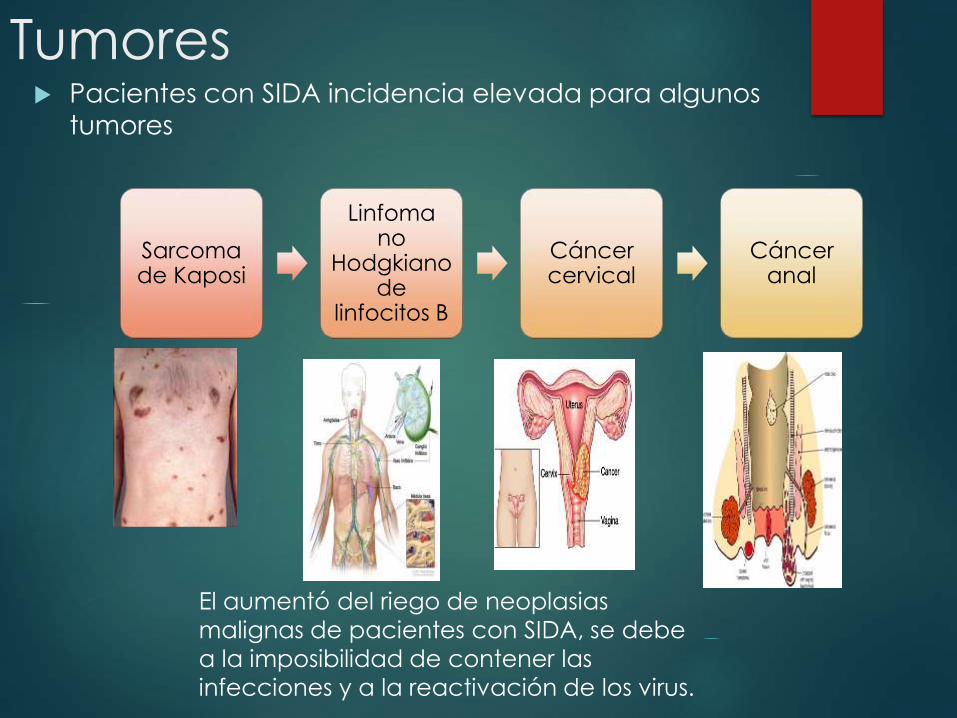
Tumores Pacientes con SIDA incidencia elevada para algunos
tumores
Sarcoma de Kaposi
Linfoma no
Hodgkiano de
linfocitos B
Cáncer cervical
Cáncer anal
El aumentó del riego de neoplasias
malignas de pacientes con SIDA, se debe
a la imposibilidad de contener las
infecciones y a la reactivación de los virus.
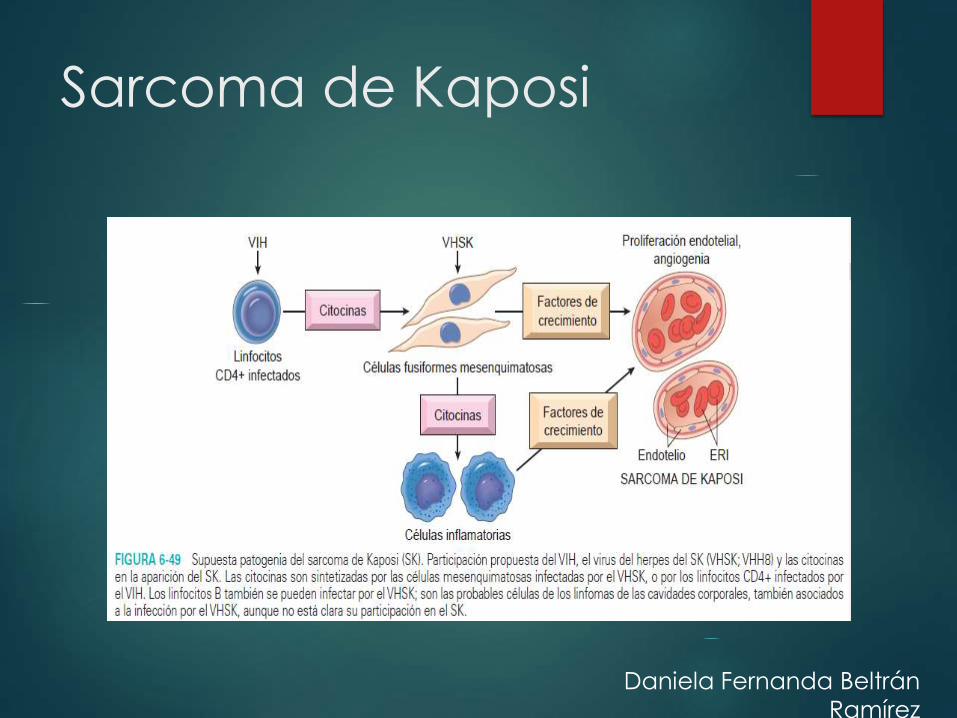
Sarcoma de Kaposi
Daniela Fernanda Beltrán
Ramírez
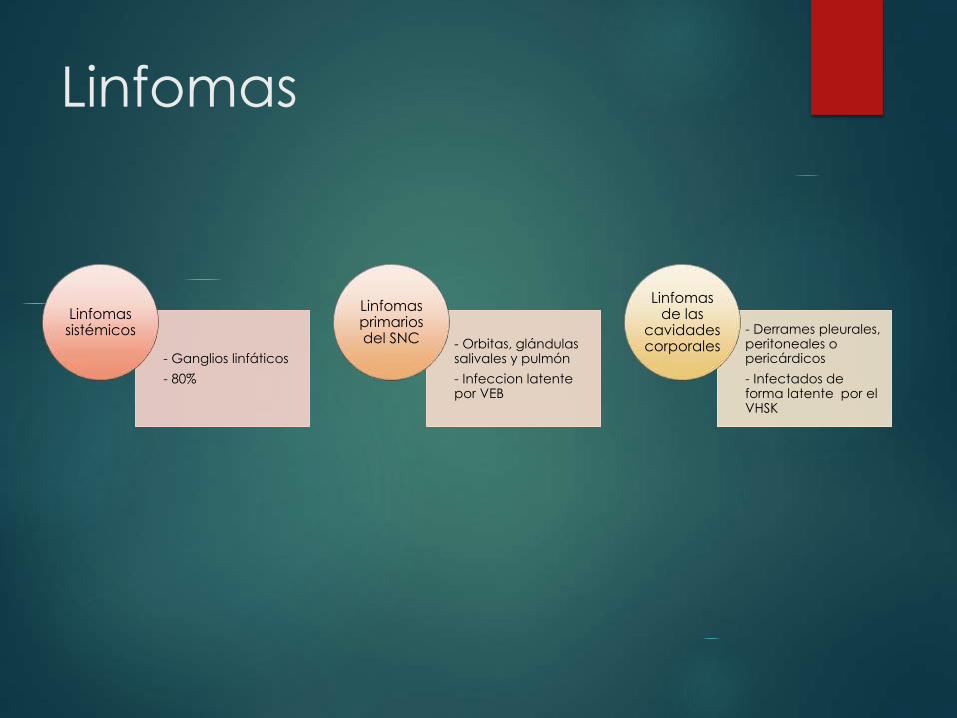
Linfomas
- Ganglios linfáticos
- 80%
Linfomas sistémicos
- Orbitas, glándulas salivales y pulmón
- Infeccion latente por VEB
Linfomas primarios del SNC
- Derrames pleurales, peritoneales o pericárdicos
- Infectados de forma latente por el VHSK
Linfomas de las
cavidades corporales

Efecto del tratamiento
antirretrovírico
Actúan sobre: transcriptasa inversa, proteasa e integrasa del virus
Se administran en combinados para reducir la aparición de mutaciones que presenten resistencia a cualquiera de ellos en forma aislada
Tratamiento antirretrovirico de gran actividad (TARGA) o tratamiento antirretrovirico combinado
Aun cuando aparezca un virus resistente a un fármaco, hay opciones de segunda y tercera línea para suprimir al nuevo virus

Amiloidosis
DANIELA FDA. BELTRÁN RAMÍREZ
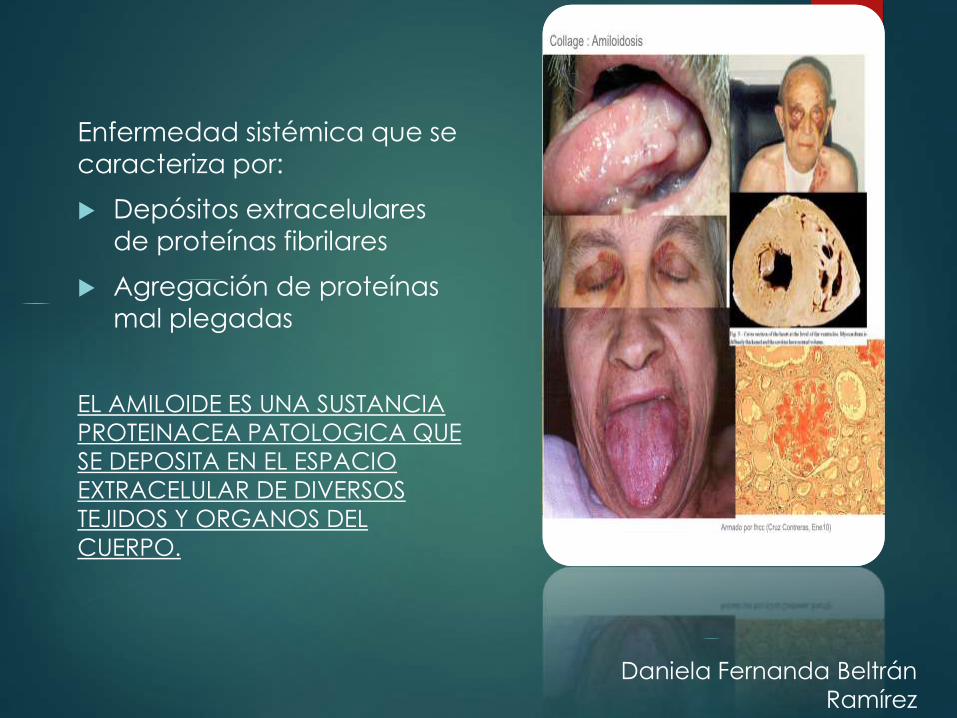
Enfermedad sistémica que se
caracteriza por:
Depósitos extracelulares
de proteínas fibrilares
Agregación de proteínas
mal plegadas
EL AMILOIDE ES UNA SUSTANCIA
PROTEINACEA PATOLOGICA QUE
SE DEPOSITA EN EL ESPACIO
EXTRACELULAR DE DIVERSOS
TEJIDOS Y ORGANOS DEL
CUERPO.
Daniela Fernanda Beltrán
Ramírez
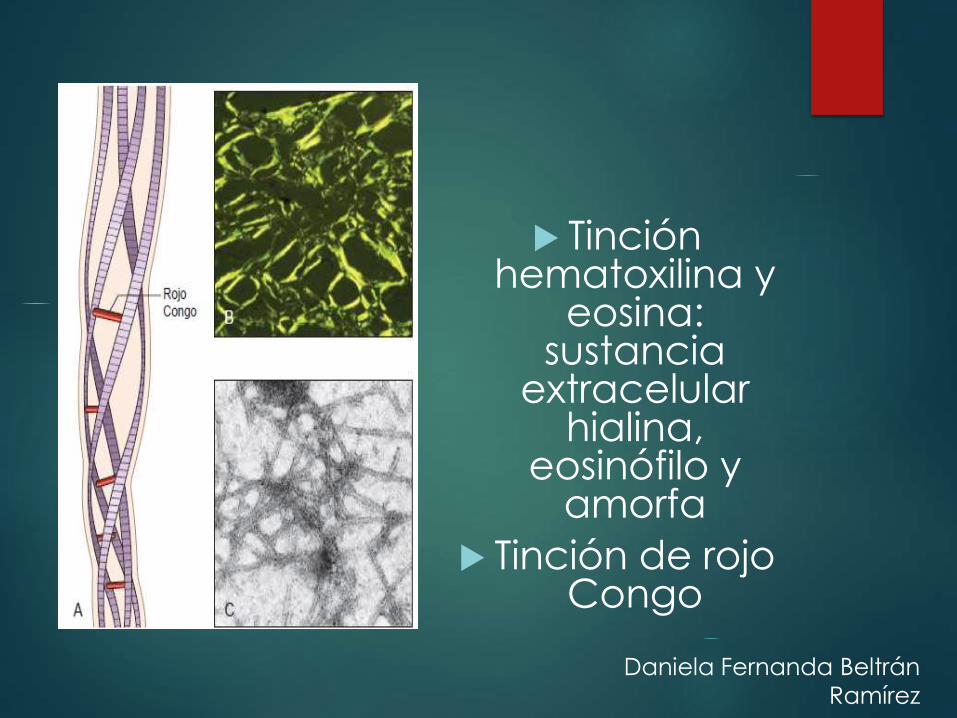
Tinción hematoxilina y
eosina:sustancia
extracelular hialina,
eosinófilo y amorfa
Tinción de rojo Congo
Daniela Fernanda Beltrán
Ramírez

Propiedades de las
proteínas del amiloide
Naturaleza física
Fibras continuas y no ramificadas
Diámetro de 7.5 a 10 nm
Conformación en lamina plegada beta con
reticulacion
Daniela Fernanda Beltrán
Ramírez
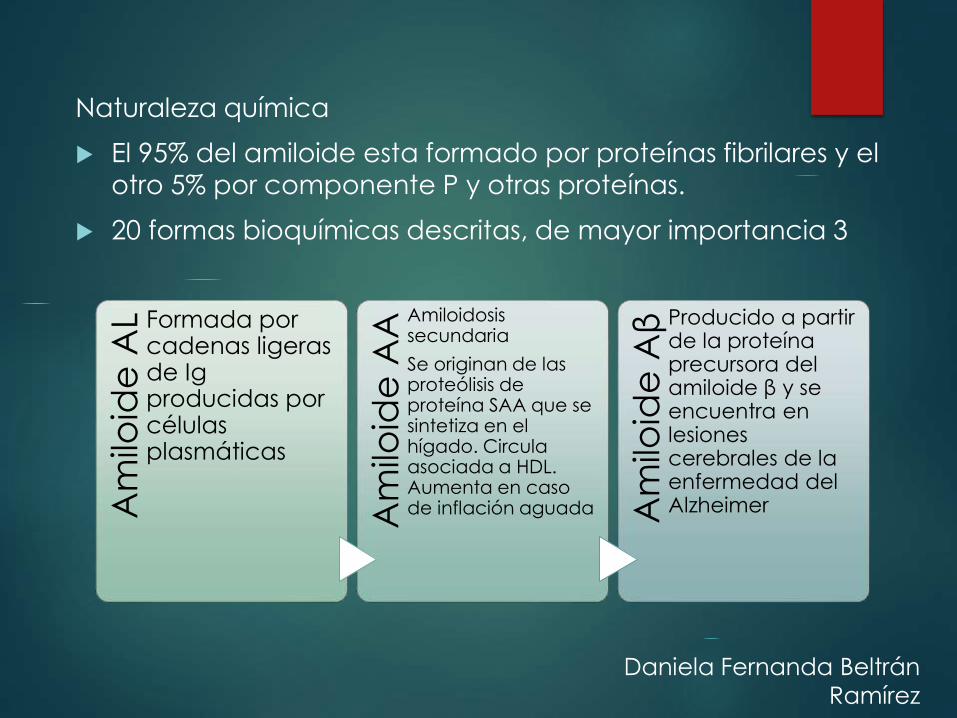
Naturaleza química
El 95% del amiloide esta formado por proteínas fibrilares y el otro 5% por componente P y otras proteínas.
20 formas bioquímicas descritas, de mayor importancia 3
Daniela Fernanda Beltrán
Ramírez
Am
iloid
e A
L Formada por cadenas ligeras de Igproducidas por células plasmáticas
Am
iloid
e A
A
Amiloidosis secundaria
Se originan de las proteólisis de proteína SAA que se sintetiza en el hígado. Circula asociada a HDL. Aumenta en caso de inflación aguada A
milo
ide
Aβ Producido a partir
de la proteína precursora del amiloide β y se encuentra en lesiones cerebrales de la enfermedad del Alzheimer

Otras proteínas Tr
an
stire
tin
a Mutada en polineuropatías amiloidoticas familiares. Normal en Amiloidosis sistémica senil
Β2
-mic
rog
lob
ulin
a
Subunidad de la fibrilla del amiloide de la Amiloidosis en pacientes sometidos a hemodiálisis a largo plazo.
Pro
teín
as
de
l p
rio
n Se agregan en el espacio extracelular y adquieren características de la proteína amiloide .
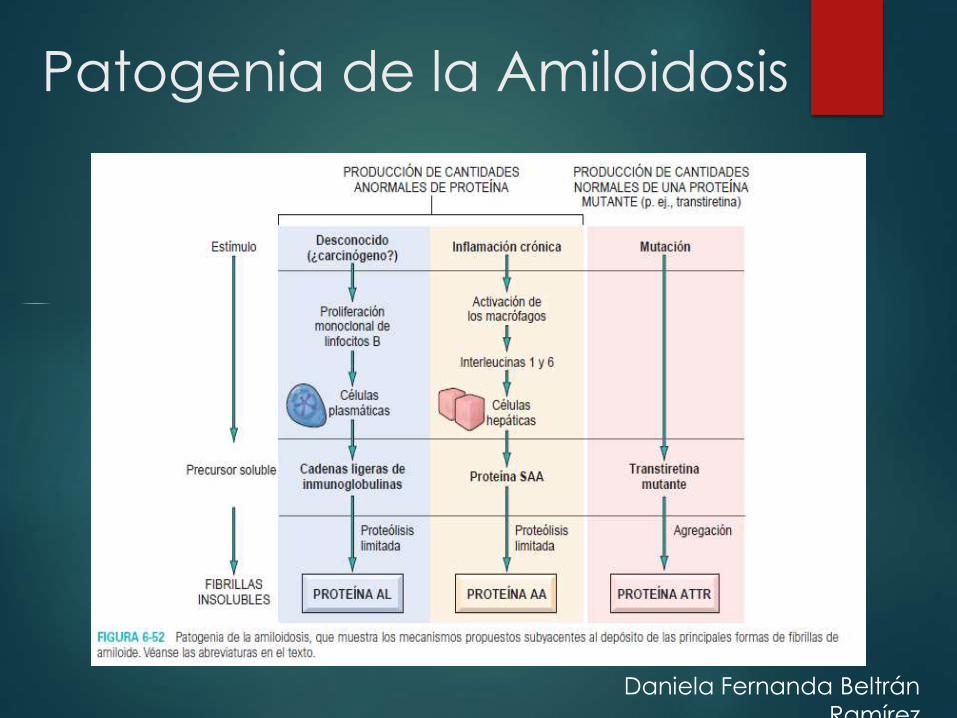
Patogenia de la Amiloidosis
Daniela Fernanda Beltrán
Ramírez

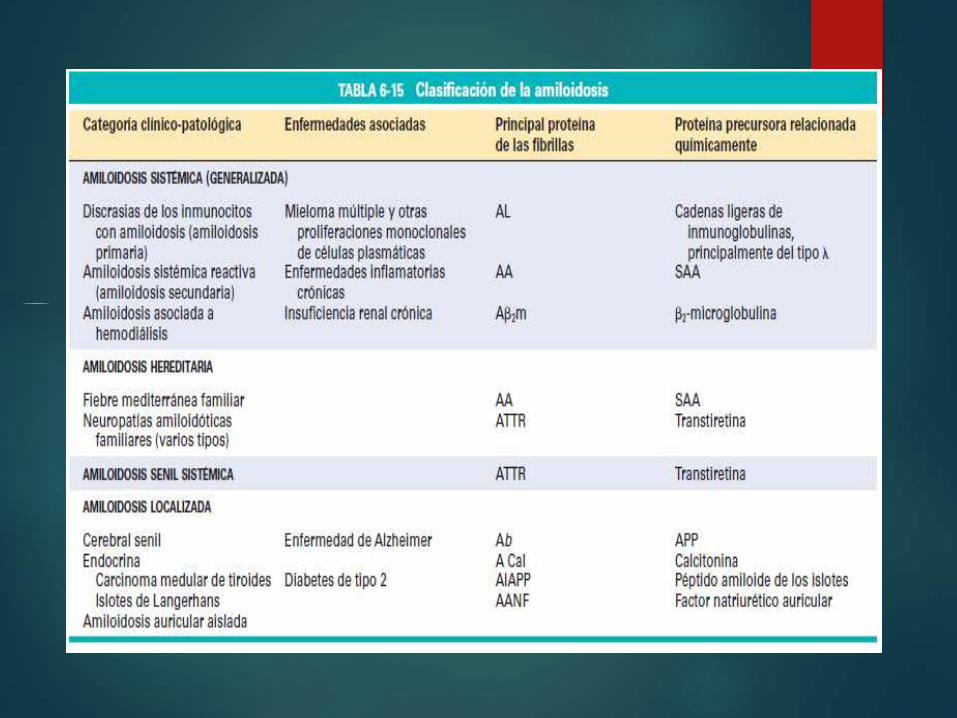
Clasificación de la
AmiloidosisAmiloidosis primaria: discrasia de inmunocitos con amiliodosis
Amiloidosis sistémica reactiva
Amiloidosis asociada a la hemodiálisis
Amiloidosis heredofamiliar
Amiloidosis localizada
Amiloide endocrino
Amiloide senil
Daniela Fernanda Beltrán
Ramírez
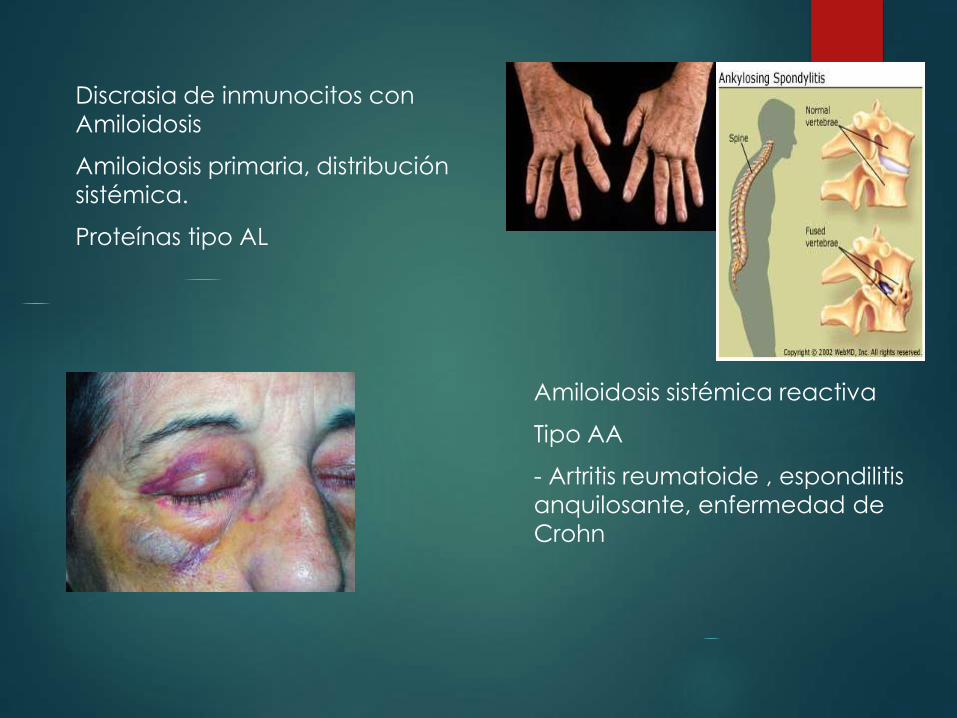
Discrasia de inmunocitos con
Amiloidosis
Amiloidosis primaria, distribución
sistémica.
Proteínas tipo AL
Amiloidosis sistémica reactiva
Tipo AA
- Artritis reumatoide , espondilitis
anquilosante, enfermedad de
Crohn
Amiloidosis asociada a
hemodiálisis
Pacientes con insuficiencia
renal
Consecuencia del depósitos
de β2- microglobulina
síndrome del túnel carpianoAmiloidosis heredofamiliar
Proteinas AA
Fiebre mediterránea
familiar
Gen codifica proteína
pirina
Amilioidosis localizada
Proteima AL
Masas nodulares detectables
macroscópicamente
Se encuentran en : pulmón,
laringe, piel, vejiga urinaria,
etc.
Amiloide senil
Formado TTR
Deposito sistémico amiloide
Pacientes sintomáticos
presentan miocardiopatías
restrictivas y arritmias