
UNIVERSIDAD DE COSTA RICA SISTEMA DE ESTUDIOS DE POSGRADO
EVALUACIÓN DE LA TALLA FINAL VERSUS PRONÓSTICO DE LA TALLA
FAMILIAR EN PACIENTES PORTADORAS DEL SÍNDROME DE TURNER EN
CONTROL EN EL HOSPITAL NACIONAL DE NIÑOS DURANTE EL PERÍODO
DE 1 ENERO DE 2000 AL 31 DE DICIEMBRE DE 2017
Tesis sometida a la consideración de la Comisión del
Programa de Estudios de Posgrado en Endocrinología
para optar por el grado y título de Especialista en
Endocrinología
JUAN DANIEL SIBAJA JIMÉNEZ
Ciudad Universitaria Rodrigo Facio, Costa Rica
2018

ii
DEDICATORIA
A mi hermano Fernando José, in memoriam, para que su luz me siga acompañando en
este camino, hasta que nos volvamos a ver.
A mis abuelitos Hugo y Efraín, in memoriam, mis modelos a seguir.
A mis padres Liliana y Luis Fernando, cuyas enseñanzas, ejemplo y liderazgo les debo
lo que soy, este logro es de ustedes.
A mi hermana María Gabriela y cuñado Juan Manuel, ejemplo diario de superación, la
cual les admiro.
A mi ahijado José Ignacio, esperando el día que pueda leer estas páginas.
A mis abuelitas Flora y Emilce, motivación diaria para seguir luchando y ejemplo de
vida.
A todos los(a) pacientes con patologías endocrinológicas, cuya atención debo mi
trabajo y esfuerzo.

iii
AGRADECIMIENTOS A la Dra. Laura Ulate y al Dr. Orlando Jaramillo, por toda su dedicación durante la
tutoría de este trabajo final de graduación. Para siempre mi cariño y gratitud.
Al Dr. José Alejandro Cob Sánchez, coordinador nacional del Posgrado en
Endocrinología de la UCR, por toda su paciencia y enseñanzas.
A los(as) coordinadores de la Unidad de Posgrado de Endocrinología de la UCR en los
distintos hospitales de rotación: Dra. Grace Yung Li (Hospital México), Dr. Chih Hao
Chen Ku (Hospital San Juan de Dios), Dra. Maricel Quirós Campos (Hospital Dr. Rafael
Ángel Calderón Guardia) y Dr. Manuel Jiménez Navarrete (Hospital San Vicente de
Paúl). Por su tiempo y dedicación.
A los médicos asistentes endocrinólogos pediatras del Hospital Nacional de Niños: Dr.
Erick Richmond, Dr. Fred Cavallo, Dr. Roberto Bogarín. Por su apoyo y consejo durante
la elaboración de este trabajo final de graduación.

"Esta tesis fue aceptada por la Comisión del Programa de Estudios de Posgrado en Endocrinología de la Universidad de Costa Rica, como requisito parcial para optar por el grado y título de Especialista en Endocrinología"
Dr. Álvaro Morales Ramírez Decano Sistema de Estudios de Posgrado
Dr. José Guillermo Jiménez Montero
Asesor de Tesis
Dr. José Alejandro Cob Sánchez
Director
Programa de Posgrado en Endocrinología
Juan Daniel Sibaja Jiménez Candidato
iv

v
TABLA DE CONTENIDOS
Sección PÁGINA
Portada i
Dedicatoria ii
Agradecimientos iii
Hoja de Aprobación iv
Tabla de Contenidos v
Resumen vi
Lista de Tablas vii
Lista de Figuras ix
Introducción 1
Justificación 2
Antecedentes 3
Planteamiento del problema 3
Marco teórico 4
Materiales y métodos 39
Características del estudio 39
Variables del estudio 41
Fundamento del tipo de estudio 44
Fundamento del tipo de recolección de datos 44
Análisis de datos 44
Reporte de conflicto de intereses 46
Resultados 47
Discusión 68
Conclusiones 82
Recomendaciones 85
Bibliografía 87
Anexos 95

vi
Resumen
El síndrome de Turner es una condición genética que afecta aproximadamente a 1 de cada 2500 nacimientos femeninos, lo que la hace una de las patologías genéticas más comunes encontradas en la práctica pediátrica y siendo una de sus características la presencia de baja talla.
Esta investigación tiene como objetivo evaluar la talla final en comparación con el pronóstico de talla familiar de las pacientes con Síndrome de Turner valoradas en el servicio de Endocrinología del Hospital Nacional de Niños entre enero 2000 y diciembre de 2017. El impacto esperado del estudio es estimular el reconocimiento temprano y la intervención oportuna de la baja talla asociada a esta patología, para mejorar la calidad de vida de estas pacientes.
Se trata de un estudio descriptivo, observacional y retrospectivo de registro médicos, con la totalidad de pacientes con diagnóstico genético de síndrome de Turner, durante el periodo comprendido entre el 1 de enero 2000 y el 31 de diciembre del 2017, y que hayan sido valoradas y documentadas en la base de datos del servicio de Endocrinología del Hospital Nacional de Niños.
Se utilizó estadística descriptiva, distribuciones de frecuencia para las variables cualitativas y medidas de tendencia central y de dispersión para las variables cuantitativas. Se seleccionaron un total de 157 pacientes con síndrome de Turner disponibles en la base de datos del Hospital Nacional de Niños y con atención entre enero 2000 y diciembre de 2017. El análisis principal se realizó con 140 pacientes, correspondiente al 89% de la base de datos. Un total de 17 pacientes, correspondientes a un 11% de la base de datos fueron excluidas del análisis principal debido a la presencia de datos incompletos o bien a que los expedientes clínicos no pudieron ser localizados.
El tratamiento con hormona de crecimiento fue en general bien tolerado y las pacientes tuvieron una buena adherencia al mismo. Esta población de pacientes con síndrome de Turner tuvo una edad promedio al diagnóstico de 8.5 años y una edad promedio de 9.7 años al inicio de tratamiento con hormona de crecimiento, para un retraso en el inicio del mismo de 1.2 años. A su vez la duración promedio del mismo fue de 6.1 años, con una edad promedio al final del tratamiento de 15.8 años.
La talla final promedio fue de 146.2 cm. El pronóstico de talla familiar de estas pacientes fue en promedio de 157.1 cm. Hubo una diferencia estadísticamente significativa de 31 cm en promedio al comparar la talla inicial con la talla final obtenida por estas pacientes, así como en promedio las pacientes tuvieron una talla final estadísticamente significativa de 10.8 cm por debajo del promedio de talla familiar. Al finalizar el tratamiento se encontraron en promedio en el percentilo 3.25 de talla poblacional y alcanzado al final del tratamiento una talla que se encuentra a -2.55 desviaciones estándar de la población de referencia y a -1.60 de la desviación estándar esperable promedio de acuerdo al pronóstico de talla familiar. No se identificó ningún factor como pronóstico de la talla final en esta población, mientras que el pronóstico de talla familiar, la talla inicial y la duración del tratamiento con hormona de crecimiento son factores pronóstico en la ganancia de talla, siendo éste último el de mayor peso estadístico.

vii
LISTA DE TABLAS
TITULO PÁGINA
Tabla 1. Efectos adversos reportados en las pacientes portadoras de
síndrome de Turner en control en el Hospital Nacional de Niños, entre
enero 2000 y diciembre 2017, en tratamiento con hormona del
crecimiento.
51
Tabla 2. Edad de diagnóstico, de inicio y fin de tratamiento con hormona
del crecimiento, duración y dosis al final del este, en las pacientes
portadoras de síndrome de Turner en control en el Hospital Nacional de
Niños, entre enero 2000 y diciembre 2017.
55
Tabla 3. Talla inicial y final, peso final y promedio de talla familiar (PTF)
de las pacientes que completaron tratamiento con hormona de
crecimiento portadoras de síndrome de Turner en control en el Hospital
Nacional de Niños, entre enero2000 y diciembre 2017.
57
Tabla 4. Análisis de la talla al finalizar el tratamiento con hormona del
crecimiento de las pacientes portadoras de síndrome de Turner en
control en el Hospital Nacional de Niños, entre enero 2000 y diciembre
2017, según duración del tratamiento, cariotipo y edad al inicio del
tratamiento.
60
Tabla 5. Análisis de regresión lineal múltiple de la ganancia de talla al
finalizar el tratamiento con hormona del crecimiento con la duración, la
dosis y la edad de inicio del tratamiento, en las pacientes portadoras de
síndrome de Turner en control en el Hospital Nacional de Niños, entre
enero 2000 y diciembre 2017.
61

viii
TITULO PÁGINA
Tabla 6. Comparación de la edad de inicio de tratamiento de las
pacientes portadoras de síndrome de Turner en control en el Hospital
Nacional de Niños, entre enero 2000 y diciembre 2017, según el año de
inicio del tratamiento con hormona del crecimiento.
63
Tabla 7. Comparación de la dosis de hormona de crecimiento al final
del tratamiento de las pacientes portadoras de síndrome de Turner en
control en el Hospital Nacional de Niños, entre enero 2000 y diciembre
2017, según el año de inicio del tratamiento con hormona del
crecimiento.
65
Tabla 8. Comparación de la ganancia de talla en las pacientes
portadoras de síndrome de Turner en control en el Hospital Nacional
de Niños, entre enero 2000 y diciembre 2017, según el año de inicio del
tratamiento con hormona del crecimiento.
67

ix
LISTA DE FIGURAS
TITULO PÁGINA
Figura 1. Distribución de las pacientes portadoras de síndrome de Turner en
control en el Hospital Nacional de Niños, entre enero 2000 y diciembre 2017,
según procedencia geográfica de las pacientes. 48
Figura 2. Distribución de las pacientes portadoras de síndrome de Turner en
control en el Hospital Nacional de Niños, entre enero 2000 y diciembre 2017,
según evolución del tratamiento con hormona del crecimiento. 50
Figura 3. Distribución de las pacientes portadoras de síndrome de Turner en
control en el Hospital Nacional de Niños, entre enero 2000 y diciembre 2017,
según edad al momento del diagnóstico. 52
Figura 4. Distribución de las pacientes portadoras de síndrome de Turner en
control en el Hospital Nacional de Niños, entre enero 2000 y diciembre 2017,
según edad al inicio del tratamiento con hormona del crecimiento. 53
Figura 5. Distribución de las pacientes portadoras de síndrome de Turner en
control en el Hospital Nacional de Niños, entre enero 2000 y diciembre 2017,
según edad al finalizar el tratamiento con hormona del crecimiento. 54
Figura 6. Distribución de la talla inicial y final y de la talla promedio familiar de las
pacientes portadoras de síndrome de Turner en control en el Hospital Nacional
de Niños, entre enero 2000 y diciembre 2017. 58
Figura 7. Distribución de la edad de inicio de tratamiento de las pacientes
portadoras de síndrome de Turner en control en el Hospital Nacional de Niños,
entre enero 2000 y diciembre 2017, según el año de inicio del tratamiento con
hormona del crecimiento.
62
Figura 8. Distribución de la dosis de hormona de crecimiento al final del
tratamiento de las pacientes portadoras de síndrome de Turner en control en el
Hospital Nacional de Niños, entre enero 2000 y diciembre 2017, según el año de
inicio del tratamiento con hormona del crecimiento.
64
Figura 9. Distribución de la ganancia de talla en las pacientes portadoras de
síndrome de Turner en control en el Hospital Nacional de Niños, entre enero 2000
y diciembre 2017, según el año de inicio del tratamiento con hormona del
crecimiento.
66

1
INTRODUCCIÓN
El síndrome de Turner es una patología genética frecuente en la
población femenina, siendo la presencia de baja talla una de sus características
fundamentales.
La sospecha diagnóstica es primordial y esencialmente clínica, siendo
necesaria realizar la confirmación diagnóstica por medio de un análisis de
cariotipo.
Sin tratamiento estas pacientes están destinadas a alcanzar una talla muy
por debajo del pronóstico de talla familiar y en relación a la población general,
surgiendo el tratamiento con hormona de crecimiento como una parte primordial
de la atención integral recomendada para estas pacientes.
El mismo se ha utilizado con buena eficacia y seguridad por
aproximadamente dos décadas, y actualmente se encuentra disponible en la
seguridad social costarricense para el manejo de ésta entidad, entre otras
indicaciones.
Contando el servicio de Endocrinología del Hospital Nacional de Niños
con una base de datos extensa de pacientes con síndrome de Turner, se desea
valorar la talla final comparada al pronóstico de talla familiar de estas pacientes
en el período entre enero 2000 y diciembre 2017, y así determinar la talla final
de las pacientes con esta patología, con la ventaja de tratarse de una población
local.

2
JUSTIFICACIÓN
El Síndrome de Turner es una entidad patológica en la cual las niñas
afectadas presentan una serie de características típicas asociadas a la ausencia
parcial o completa del segundo cromosoma sexual (cromosoma X) con o sin
diversos mosaicismos.
Las pacientes portadoras de esta patología son particularmente
propensas a experimentar comorbilidades las cuales pueden presentarse desde
el momento de su nacimiento (ej. malformaciones cardiacas) o durante el resto
de su vida (patología autoinmune ej tiroiditis o enfermedad celiaca), mismas que
van a determinar en gran medida necesidades de tratamiento y afectar de forma
directa o indirecta su calidad de vida.
Una de las características en las pacientes con Síndrome de Turner es la
presencia de baja talla, por lo que el tratamiento con hormona de crecimiento ha
sido bien estudiado en esta población por más de veinte años.
Existen reportes de metaanálisis con una ganancia de talla en promedio
de 5 a 8 cm con el tratamiento con hormona de crecimiento, mientras que en
pacientes no tratadas alcanzan en promedio 20 cm menos de talla comparado al
pronóstico de talla familiar. Los datos para la población costarricense son
desconocidos.

3
ANTECEDENTES
No hay estudios a la fecha en población costarricense que evalúen
específicamente la talla final en pacientes con síndrome de Turner.
PLANTEAMIENTO DEL PROBLEMA
¿Cuál es la talla final comparada al pronóstico de talla familiar en
pacientes portadoras de síndrome de Turner en control en el Hospital Nacional
de Niños durante el período comprendido entre enero 2000 y diciembre 2017?

4
Marco Teórico
Generalidades
El síndrome de Turner es una condición genética que afecta
aproximadamente a 1 de cada 2500 nacimientos femeninos, lo que la hace una
de las patologías genéticas más comunes encontradas en la práctica
pediátrica.1
Fue descrito originalmente por Henry Turner en 1938, al publicar sobre
siete pacientes con edades entre los 15 y 23 años que fueron referidas por baja
talla y ausencia de desarrollo de características sexuales secundarias.2
Posteriormente se descubrió que estos cambios eran secundarios a variación
cariotípica 45,X en 1959.3
Es causado por una deleción total (monosomía) o parcial (monosomía
parcial) del segundo cromosoma sexual. Múltiples sistemas corporales pueden
verse afectados en grados variables por el síndrome, presentando un reto para
el clínico tanto en su manejo como su diagnóstico.1

5
Diagnóstico
Su diagnóstico es tanto clínico como citogenético. Para que una mujer ó
niña pueda ser considerada como portadora del síndrome debe tenerse la
sospecha clínica inicial, con las características fenotípicas consistentes con el
diagnóstico, como pueden ser por ejemplo la baja talla o la disgenesia o
insuficiencia gonadal.4
Es importante mencionar que todos los individuos con síndrome de
Turner tienen fenotipo femenino; nunca se le debe dar este diagnóstico a un
paciente con fenotipo masculino.4
En aquellas pacientes con sospecha clínica debe realizarse una
confirmación diagnóstica por medio de análisis cromosómico con cariotipo, que
demuestre la ausencia parcial o total de una copia del segundo cromosoma
sexual.4
Variantes Citogenéticas
Algunas pacientes son portadoras de anormalidades estructurales del
cromosoma X, como pueden ser deleciones de una porción del brazo largo o
corto, un cromosoma X en anillo, o isocromosoma X. La mayoría de pacientes
con cromosomas en anillo o isocromosomas tienen características clínicas
similares o indistinguibles de aquellas pacientes con cariotipos clásicos.5

6
Entre los pacientes con deleciones distales del brazo corto del
cromosoma X el diagnóstico es reservado para aquellos portadores de
deleciones que incluyen la banda Xp22.3. Aquellos con deleciones pequeñas del
brazo largo distal a Xq24 pueden tener amenorrea primaria o secundaria sin las
características adicionales del síndrome de Turner. De esta forma, estos
individuos deberían ser diagnosticados más apropiadamente como falla ovárica
prematura.5
Algunos individuos con síndrome de Turner pueden tener cariotipos que
incluyen material del cromosoma Y. Esta circunstancia puede ocurrir como un
mosaicismo para una línea celular 46, XY resultando en un cariotipo 45X/46,XY.
Otros pueden tener material Y en la forma de un fragmento cromosómico
denominado cromosoma marcador.6
Cualquier niña con síndrome de Turner y que tenga un fragmento
cromosómico no identificado debe realizársele hibridación fluorescente in situ o
estudios de ácido desoxirribonucleico para determinar si ese marcador consiste
de material cromosómico Y. La importancia de esta determinación radica a que
en presencia de material cromosómico Y el riesgo de gonadoblastoma es
aproximadamente del 12%, y es recomendada la gonadectomía bilateral como
medida profiláctica.7

7
Diagnóstico Prenatal
Al momento de la concepción, la incidencia del síndrome de Turner es
aproximadamente del 3%, sin embargo la mayoría de estos productos son
abortados espontáneamente, y solamente 1% de estos embriones sobreviven a
término.8 Las anomalías congénitas son detectadas más frecuentemente en
fetos 45,X completos en lugar de mosaicismos. El pronóstico de los fetos con
síndrome de Turner detectado en el período prenatal es relativamente peor que
el pronóstico de aquellos casos observados en la vida postnatal.9
El uso de ultrasonido ha sido reportado como un método confiable en el
diagnóstico prenatal del síndrome de Turner. Algunas de las características
sonográficas comunes del síndrome de Turner son la presencia de higromas
quísticos, incremento del grosos de la translucencia nucal, hidrops fetal no
inmune, así como defectos renales y cardíacos.10
La presencia de estos hallazgos ha llevado al muestreo por
amniocentesis o vellosidades coriónicas. Si se ha realizado un cariotipo
prenatalmente, el estudio debe ser repetido postnatalmente en una muestra
sanguínea del neonato.
El tamizaje neonatal universal para anomalías de los cromosomas
sexuales no ha sido instaurado a la fecha pero es claro que conforme se

8
implemente de acuerdo a las políticas de tamizaje de cada país, el médico a
cargo debe estar preparado en el manejo básico y de dar una información
certera a los padres de familia.
Características Clínicas
Las niñas con síndrome de Turner se pueden presentar al nacimiento con
rasgos dismórficos que incluyen el edema de manos y pies (signo de Bonnevie-
Ullrich), cuello alado, cuarto metacarpianos o metatarsianos cortos, uñas
hiperconvexas, implantación baja de cabello, anomalías craneofaciales como
paladar alto, mandíbula pequeña, y orejas de implantación baja.11
A nivel ocular es posible encontrar pliegue epicántico, ptosis palpebral y
menos comúnmente la presencia de escleras azuladas. En tórax puede
observarse un aspecto en escudo, con distanciamiento inter pezón ampliado.
Otros hallazgos a nivel óseo son la presencia de cúbito valgo, y la deformidad
de Madelung.11
Complicaciones Cardiometabólicas
Muchas niñas se presentan en el período neonatal con diversas
anomalías cardiovasculares y frecuentemente asociados a higromas quísticos,

9
que al resolver dejan esa apariencia de cuello alado, lo que debe hacer
sospechar al clínico de la presencia de una anomalía cardiovascular.12
Estas anomalías cardiovasculares usualmente afectan al tracto de salida
izquierdo. El defecto más común es la válvula aórtica bicúspide, ocurriendo en
aproximadamente 16% de los pacientes y en un 37% de aquellos que se
presentan con cuello alado. Aproximadamente un 11% tienen coartación de
aorta.12
Estudios recientes realizados por medio de resonancia magnética nuclear
han sugerido que el espectro de anormalidades cardiovasculares es mayor. La
comunicación anómala parcial venosa pulmonar está presente en
aproximadamente un 13% de los pacientes. La persistencia de la vena cava
superior izquierda puede ocurrir también. 12
También ha sido demostrada una dilatación generalizada de la aorta y
otros vasos mayores como las arterias carótida y braquial, sugiriendo una
vasculopatía generalizada en pacientes con síndrome de Turner.
Las anomalías de la válvula aórtica, como la válvula aórtica bicúspide, la
coartación de aorta y la hipertensión sistémica, pueden ser factores de riesgo
para una dilatación, disección u ruptura aórtica, complicaciones que aunque

10
raras, son usualmente fatales. Las mismas pueden presentarse inclusive en
ausencia de factores de riesgo.13
Adicional a las anomalías estructurales cardíacas, las pacientes con
síndrome de Turner pueden presentar alteraciones electrocardiográficas, como
por ejemplo la prolongación del intervalo QT. Algunos estudios han demostrado
la presencia de taquicardia en reposo, la cual puede iniciar en la etapa fetal y
afectar hasta un 50% de los mismos. La significancia clínica de estos hallazgos
es incierta.13
Las pacientes con síndrome de Turner tienen un mayor riesgo de
desarrollar hipertensión arterial, una condición que en parte puede ser explicada
por disfunción del sistema autónomo simpático. Estas pacientes deben tener
una evaluación periódica con imágenes que documenten el tamaño de la raíz
aórtica.
La obesidad está presente en la actualidad en muchas pacientes con
síndrome de Turner. Esta condición puede exacerbar otras complicaciones
frecuentes como pueden ser la resistencia a la insulina y la hipertensión.
Estudios recientes han reportado una prevalencia de obesidad en pacientes con
síndrome de Turner mayor a la de población femenina en general, y
particularmente en el rango de entre los 15 y 39 años de edad.15

11
La obesidad no estaba presente como característica en las 7 pacientes
originales descritas por Turner. El aumento de la tendencia a la obesidad en la
población general en los últimos 50 años en todos los grupos etarios tanto de la
población femenina como masculina, como respuestas a los cambios en los
estilos de vida , pareciera explicar mejor la tendencia actual de estas pacientes.
Sistema Genitourinario
Entre el 30-40% de las pacientes con síndrome de Turner tienen
anomalías estructurales renales demostrables por ultrasonido. Su frecuencia
varía por cariotipo. Estas anomalías pueden ser del sistema colector o bien
estructurales o posicionales como riñón en herradura y malrotación renal.16
Las niñas con cariotipo clásico 45, XO son más propensas a desarrollar
anomalías estructurales renales, mientras que aquellas con mosaicismos o con
una estructura anormal del cromosoma X son más propensas a presentar
malformaciones del sistema colector. Estas niñas son más propensas a
desarrollar otras complicaciones a futuro, como pueden ser hipertensión arterial
o infecciones del tracto urinario a repetición.16

12
Trastornos visuales y auditivos
Las pacientes con síndrome de Turner pueden tener una variedad de
anomalías externas oculares y a nivel de oídos así como déficits visuales y
auditivos. Algunas características oculares ya mencionadas en párrafos
anteriores son los pliegues epicanticos, fisuras palpebrales, ptosis e
hipertelorismo. La hiperopía y el estrabismo están presentes entre el 25-35% de
la población.17
Tanto el déficit auditivo neurosensorial como conductivo son
complicaciones severas del síndrome de Turner, ésta última resultante
usualmente de una otitis media crónica, la cual en presencia del síndrome debe
manejarse agresivamente buscando evitar estas posibles complicaciones.17
Dismorfismos
Estas pacientes tienen un patrón característico de desarrollo anormal
craneofacial, de aplanamiento del ángulo de la base del cráneo y una reducción
de la longitud de la base craneal posterior. Aunque la maxila usualmente es
angosta, llevando a un paladar alto, la mandíbula tiende a ser ancha y
micrognática. Esto lleva a diversos grados de maloclusión en estas pacientes.
Así mismo, el desarrollo dental puede ser anormal, presentando una erupción
dental prematura.18

13
Autoinmunidad
Tanto la disfunción tiroidea como la celiaquía ocurren en una proporción
significativa de individuos con síndrome de Turner. El hipotiroidismo es mucho
más común que el hipertiroidismo. Aproximadamente un 4-6% de las pacientes
con Turner desarrollan enfermedad celíaca. Aunque de forma mucho menos
frecuente que las anteriores, la enfermedad de Crohn y la colitis ulcerativa han
sido reportadas con cierta frecuencia en portadoras del síndrome. Es importante
mencionar que todas estas condiciones son importantes de tamizar, dado su
posible impacto en el crecimiento y desarrollo de estas niñas.
Piel
Las niñas con síndrome de Turner tienen una tendencia incrementada a
adquirir nevus melanocíticos. Sin embargo, pareciera que esta condición no les
confiere un riesgo aumentado de melanoma. En el pasado se creía también que
estas pacientes tenían un riesgo aumentado de desarrollar cicatrización
queloide, sin embargo se ha establecido que esta tendencia podría ser producto
de regiones corporales que han sido sometidas a cirugía reconstructiva, como la
cabeza y cuello, más que una diferencia real en la propensión a la curación
normal en pacientes con síndrome de Turner.

14
Pubertad y Embarazo
Algún grado de progresión puberal ocurre espontáneamente en
alrededor de 30% de las pacientes con Síndrome de Turner. Mientras que por
otro lado, el embarazo espontáneo se reporta en un 2-5% de los casos.20
Sin embardo, la falla ovárica ocurre en más del 90% de los casos.
Actualmente se propone iniciar el reemplazo estrogénico (previa medición de
gonadotropinas) alrededor de los 12 años de edad con el fin de coincidir con un
desarrollo puberal normal, y con la evidencia que sugiere que esta práctica no
afecta la talla final de estas niñas.20
Aprendizaje y Conducta psicosocial
La gran mayoría de estas pacientes presentan un desarrollo motor y
cognitivo normal, con inteligencia normal. Sin embargo algunas mujeres con
cromosoma X en anillo pueden presentar algún grado de déficit cognitivo.21
Las niñas con Turner frecuentemente obtienen puntajes más bajos en
medidas de rendimiento no verbal vrs verbales en test estandarizados de
inteligencia, pudiendo estar en riesgo de un menor desempeño en tareas
matemáticas y de percepción visual.21

15
Algunos estudios han demostrado que las niñas con síndrome de Turner
son diagnosticadas más frecuentemente con trastorno de déficit atencional que
la población general. Algunas de las teorías que pudieran explicar esta situación
son el déficit estrogéncio y la haploinsuficiencia de uno o más genes en el
cromosoma X.22
En la adolescencia, estas pacientes tienden a tener dificultades en la
interacción social, mostrando algún grado de inmadurez y ansiedad en
encuentros sociales. El reemplazo puberal podría mitigar algunos de estos
déficits, al percibirse la paciente similar a sus pares.22
Crecimiento y Desarrollo
El crecimiento lineal y la talla adulta son regulados por un complejo
sistema de factores endocrinos, como el eje hormona de crecimiento-factor de
crecimiento similar a insulina, pero también por sistemas intrínsecos no
endocrinos a nivel óseo que garantizan un balance adecuado de proliferación y
diferenciación de los condrocitos para una extensión lineal fisiológica.
La fisiología y los trastornos de esta regulación intrínseca a nivel óseo no
son tan bien comprendidos en la actualidad comparada al sistema endocrino. A
pesar de que se han asociado más de trescientos trastornos monogénicos a
nivel óseo asociados a baja talla y dismorfismos óseos, solo un puñado son lo

16
suficientemente frecuentes como para ser reconocidos de forma relativamente
sencilla por el clínico y el radiólogo, entre ellas la deficiencia del gen SHOX.
La baja talla es la característica clínica más frecuente del síndrome de
Turner, y usualmente el motivo de referencia a una consulta especializada. La
causa de este hallazgo es multifactorial. Se caracteriza por un retardo en el
crecimiento intrauterino, falla para progresar durante la niñez (de inicio inclusive
desde el primer año de vida), ausencia del pico de crecimiento puberal, y
displasias óseas leves.23
A pesar de que las pacientes con síndrome de Turner no son
clásicamente deficientes de hormona de crecimiento, se ha reportado una
disrupción en el eje hormona de crecimiento-factor de crecimiento similar a
insulina como una posible causa de baja talla en estas niñas.24
De manera reciente se ha evidenciado, que uno de las principales
razones para el patrón de crecimiento de estas pacientes es la
haploinsuficiencia del gen SHOX, localizado en la rama distal en Xp22.3 y
Yp11.3 en la región pseudoautosómica.25
La deficiencia de SHOX causa baja talla con un fenotipo altamente
variable el cual es frecuentemente inespecífico en niños y niñas preescolares,
ya que la desproporción en los miembros inferiores y la deformidad de Madelung

17
características se desarrollan con el transcurso del tiempo y aparecen durante la
segunda década de la vida en el caso de que se presenten del todo.26
De esta forma, el espectro clínico observado recuerda distintos fenotipos:
baja talla sin otros hallazgos específicos (llamada baja talla idiopática), baja talla
con hallazgos auxológicos y radiológicos sutiles, y en el otro extremo el
síndrome de Leri-Weill (osteocondrosis). Los signos característicos se presentan
más frecuentemente y de formas más severa en niñas, un hallazgo que podría
ser explicado por la presencia elevada de estrógenos en esta población.27
La deformidad de Madelung es el hallazgo más característico asociado a
la deficiencia de SHOX en las pacientes con síndrome de Turner. Descrita en
1878 por Otto Wilhelm Madelung como una subluxación espontánea hacia
delante de la ulna distal, dándole al brazo y antebrazo una apariencia en
tenedor.28 Existe un efecto similar en miembros inferiores con inclinación de la
tibia y genu valgum.
Adicional a la deformidad de la muñeca, existen otras lesiones menos
específicas de la deficiencia de SHOX en pacientes con síndrome de Turner,
como lo son las ya mencionadas acortamiento del cuarto y quinto
metacarpianos, paladar alto, escoliosis, incremento del ángulo de extensión del
codo y micrognatia. Adicionalmente la hipertrofia muscular de las pantorrillas se
encuentra en aproximadamente un tercio de las pacientes.29

18
Es importante destacar que la ausencia de cualquiera de estos signos,
incluyendo la deformidad de Madelung, no excluye la haploinsuficiencia de
SHOX.
Datos comparativos en talla adulta en individuos de diferentes orígenes
étnicos muestran una diferencia de hasta 20 cm en pacientes con síndrome de
Turner y la media poblacional.30
Tratamiento Baja Talla
Se han utilizada distintas terapias orientadas a impactar la talla final de
niñas con Turner a través del tiempo, dentro de las cuales destaca el uso de
hormona de crecimiento recombinante, el uso de esteroides anabólicos y la
terapia estrogénica.
Esteroides anabólicos
Los esteroides anabólicos se han utilizado durante los últimos treinta y
cinco años con el fin de mejorar la talla final de las pacientes con síndrome de
Turner. En las últimas dos décadas, varios estudios clínicos han sugerido que el
agregar un esteroide débil incapaz de ser aromatizado, la oxandrolona, al
tratamiento convencional con hormona de crecimiento tiene efectos positivos en
la talla adulta.31

19
Una publicación reportó la comparación entre pacientes con síndrome de
Turner tratadas únicamente con hormona de crecimiento más placebo vrs el uso
de hormona de crecimiento junto oxandrolona en dosis bajas (0.03 mgs/kg/día)
o convencionales (0.06 mgs/kg/día) a partir de los 8 años de edad junto con
terapia estrogénica a partir de los 12 años de edad.31
El estudio demostró que el uso de oxandrolona a dosis bajas de 0.03
mgs/kg/día junto con el tratamiento con hormona de crecimiento incrementó la
ganancia de talla (media ± desviación estándar, 9.5 ± 4.7 vrs. 7.2 ± 4.0 cm). Con
el uso de dosis convencionales de oxandrolona, la talla final adulta no tuvo
diferencia estadística significativa compara al tratamiento con hormona de
crecimiento más placebo (media ± desviación estándar, 8.3 ± 4.7 vrs. 7.2 ± 4.0
cm).31
La dosis baja de oxandrolona tuvo un perfil de seguridad aceptable pero
resultó en un ligero retraso en la telarca. Por otro lado el uso de dosis
convencionales de oxandrolona se asoció a virilización en una proporción
grande de pacientes.31
Otros estudios han mostrado resultados variables en cuanto a talla adulta,
por lo que es recomendado balancear el posible beneficio de oxandrolona en
talla comparado a los potenciales efectos adversos.

20
Terapia estrogénica
La falla ovárica es una de las mayores complicaciones asociadas al
síndrome de Turner. El uso de estrógenos para inducir el desarrollo puberal ha
sido estudiado ampliamente en esta población.
Los estudios se han enfocado principalmente en encontrar la edad ideal
de inicio de tratamiento, la vía de administración, formulación y dosis de
reemplazo estrogénico. El efecto de la deficiencia estrogénica en niñas con
síndrome de Turner puede ser observado desde la infancia, pues tienen
elevación de gonadotropinas y retraso en la maduración esquelética.
Datos publicados de un estudio multicéntrico en Estados Unidos,
concluyeron que la administración de estrógenos desde los 8 años de edad y
dosis variables (tan bajas como 25 ng/kg/día) no produjo un beneficio adicional
en la talla final adulta en pacientes con síndrome de Turner.32
Históricamente la práctica habitual ha sido retrasar el reemplazo
estrogénico hasta los 14-15 en pacientes con síndrome de Turner. El UK Turner
Study demostró un efecto positivo en talla adulta con ganancia promedio de 3.8
cm con el uso de etinilestradiol oral en dosis escalonadas (iniciando con 2
mcg/día durante el primer año y llegando hasta 10 mcg/día durante cuatro
meses al final del tercer año) iniciando a los 14 años en lugar de a los 12 años.

21
Esto con el objetivo de evitar asociaciones reportadas previamente entre retraso
de inducción puberal e incremento de talla adulta.33
Algunas publicaciones han sugerido que la práctica de retrasar la terapia
estrogénica debe ser reconsiderada. En una publicación se puso a prueba esta
teoría con ciento cincuenta niñas con síndrome de Turner que fueron divididas
en cuatro grupos de tratamiento: placebo, terapia con hormona de crecimiento,
estrógenos y estrógenos más hormona de crecimiento.34
La dosis utilizada de hormona de crecimiento fue de 0.1 mg/kg tres veces
a la semana. Las dosis de etinilestradiol oral (o placebo) fueron ajustadas de
acuerdo a edad cronológica y status puberal. Las dosis especificadas de
etinilestradiol de 25 ng/kg/día se utilizaron en niñas de entre cinco y ocho años
de edad y 50 ng/kg/día en niñas mayores de ocho años.
Posterior a los doce años, todas las pacientes de los diversos grupos
recibieron dosis escalonadas (100-800 ng/kg/día) de etinilestradiol basados en
la edad.
El estudio concluyó que el efecto en talla adulta fue de 21 cm mayor
(0.32±017 DS) en el grupo tratado tanto con hormona de crecimiento como con
estrógenos comparado a uso de hormona de crecimiento únicamente.34

22
Se sugiere que esta sinergia es posible debido a un incremento local en
la respuesta al factor de crecimiento similar a insulina o un efecto directo de la
hormona de crecimiento sobre la placa esquelética mediado por la dosis ultra
baja de etinilestradiol.
Los resultados también confirmaron que los estrógenos en dosis bajas no
interfieren con los efectos de la hormona de crecimiento, y sugieren que el uso
de estrógenos a dosis bajas puede ser administrado de forma segura a niñas
con síndrome de Turner de forma temprana sin afectar el crecimiento, siendo
actualmente la recomendación, el iniciar la sustitución estrogénica en esas
pacientes entre los 11 y 12 años de edad, aumentando hasta la dosis adulta en
un período de 2-3 años.76
También se sugiere que dicha combinación puede brindar mejorías en la
función neurocognitiva, comportamiento y autoestima, lo cual podría tener un
impacto significativo en la calidad de vida de estas pacientes.34
Uso de Hormona de crecimiento en Síndrome de Turner
La piedra angular en la terapia orientada a la baja talla en el síndrome de
Turner es el uso de hormona de crecimiento humana, aprobada por la agencia
federal de drogas de Estados Unidos en 1996 para uso en niñas con síndrome
de Turner.35

23
Su eficacia es respaldad por más de veinte años de datos de estudios
clínicos, donde ha demostrado mejoría en velocidad de crecimiento y talla adulta
en alrededor de 5-8 cm.35
Inicialmente, la terapia con hormona de crecimiento se iniciaba en la
infancia media-tardía, cuando el paciente estuviera entre dos y tres desviaciones
estándar por debajo del promedio de talla. También los estudios clínicos se
enfocaron en niñas con síndrome de Turner con edades entre los 8-12 años, en
los que la ganancia de talla fue reportada en el rango de 0.8 a 2.1 desviaciones
estándar.36
Un estudio canadiense multicéntrico demostró incrementos en talla en
niñas con síndrome de Turner con edades entre 7 y 13 años, que fueron
aleatorizadas a recibir tratamiento con hormona de crecimiento humana (0.3
mg/kg/semana y máximo 15 mgs/semana) vrs manejo conservador.37
Se siguió a un grupo de 61 pacientes con síndrome de Turner por un
período medio de 5.7 años, en el cual el promedio de talla fue de 7.2 cm por
encima de la talla promedio en el grupo control.37
Como ya se mencionó, la baja talla en las niñas con síndrome de Turner
inicia de forma típica en la etapa prenatal y la desviación estándar de la talla
disminuye progresivamente después del nacimiento.

24
Distintas publicaciones, como las de Stephure et al37 y Davenport et al38,
han demostrado que el tratamiento con hormona de crecimiento durante los
primeros años de la infancia en niñas con síndrome de Turner lleva a
incrementos significativos en la talla final.
El Toddler Turner Study llevado a cabo entre agosto de 1999 y agosto de
2003 reportó que el tratamiento con hormona de crecimiento iniciado entre los 9
meses de edad y los 4 años de vida (edad media de 2 años) logro que las
pacientes alcanzaran una talla en rango normal antes de la edad de 6 años en
93% de las pacientes.38
Es importante mencionar que esta oportunidad de optimizar la talla adulta
en estas pacientes se ve limitada ya que muchas pacientes no son
diagnosticadas hasta la infancia avanzada o adolescencia, con algunas
estimaciones de que hasta un 38% de pacientes con síndrome de Turner son
diagnosticadas en la edad adulta.39
Sin embargo, en la última década, se han desarrollado mejoras en las
guías de diagnóstico y manejo, que han llevado a una detección más temprana,
permitiendo un inicio más expedito del tratamiento.
Datos publicados de un estudio abierto, multicéntrico fase III, con 61
pacientes niñas con síndrome de Turner menores de 4 años de edad recibieron

25
tratamiento con hormona de crecimiento (0.245-0.35 mg/kg/semana) por 4 años
demostró que el 80% de estas niñas fueron capaces de alcanzar una mejoría en
su talla (1.09 desviaciones estándar vrs el grupo control) a una edad media de
6.6 años.40
En la actualidad el consenso es de que el tratamiento con hormona de
crecimiento puede ser iniciado de forma segura a una edad más temprana de la
tradicionalmente considerada para maximizar la talla adulta, sin embargo qué
tan temprano continua siendo algo controversial.
El grupo de consenso Turner Study sugiere empezar la terapia con
hormona de crecimiento tan temprano como se demuestre una falla para
progresar (debajo del percentilo 5).41 Se necesita aún información a largo plazo
en cuanto a eficacia en talla adulta y seguridad en niñas con síndrome de Turner
en las cuales se inicia la terapia en etapas muy tempranas.
Así mismo, la eficacia de la terapia es variable y depende de múltiples
factores, incluyendo el promedio de talla familiar, edad de inicio de la terapia,
duración y dosis de hormona de crecimiento y talla inicial previo inicio del
tratamiento.42

26
Los factores predictores para una mayor talla adulta son tener unos
padres altos, inicio temprano de la terapia y por dosis prolongadas, tallas más
altas al inicio de la terapia y dosis altas de hormonas de crecimiento.42
Dosis y Monitoreo de hormona de crecimiento
El rango de dosis típico de la terapia de hormona de crecimiento en
Estados Unidos es de 50-54 µgs/kg/día (0.35-0.375 mg/kg/semana) y es de 45-
50 µgs/kg/día en Europa. Las últimas guías de manejo recomiendan una dosis
inicial de 45-50 µgs/kg/día (con incrementos hasta una dosis de 68 µgs/kg/día si
la respuesta inicia es subóptima) administrada por vía subcutánea 7 días a la
semana, de preferencia en la noche.43
La talla debe ser monitorizada cada 3-4 meses en el primer año de
tratamiento y cada 4-6 meses posteriormente. El tratamiento puede ser
suspendido posterior a haber alcanzado un crecimiento lineal completo (edad
ósea de aproximadamente 13.5-14 años; velocidad de crecimiento < a 2
cm/año).43
Para superar el retraso en el crecimiento en pacientes con síndrome de
Turner, la mayoría de estudios han demostrado que se requieren dosis
relativamente altas de hormona de crecimiento, indicando la presencia de

27
resistencia a la hormona de crecimiento o al factor de crecimiento similar a
insulina 1.44
La dosis puede ser monitorizada con el patrón de crecimiento del
paciente y los niveles séricos de factor de crecimiento similar a insulina tipo 1
cada 3 a 6 meses.
En un estudio holandés, la ganancia media en cm (desviación estándar)
de talla adulta en respuesta a dosis de hormona de crecimiento de 0.045
mg/kg/día, 0.067 mg/kg/día y 0.089 mg/kg/día fueron de 11.9 (3.6), 15.7 (3.5) y
16.9 (5.2) respectivamente.44
Sin embargo, a mayores dosis de hormona de crecimiento, se alcanzaron
niveles de factor de crecimiento insulínico 1 por encima del límite superior para
la edad en algunos sujetos.45
Beneficios adicionales de la terapia con hormona de crecimiento
Se han descrito efectos beneficiosos en composición corporal, presión
arterial y metabolismo lipídico en niños pequeños nacidos pequeños para edad
gestacional. En un estudio de 6 años de seguimiento con 79 pacientes, las
desviaciones estándar del índice de masa corporal fueron normalizadas en
respuesta a la terapia con hormona de crecimiento.46

28
Se encontró que estos hallazgos se debían a una masa muscular
incrementada más que a cambios en el porcentaje de grasa corporal. El
incremento en masa muscular en asociación a la terapia con hormona de
crecimiento ha sido demostrado también en niños pequeños nacidos pequeños
para edad gestacional en otros estudios.47
A su vez, también se han reportado una tendencia a mejoría en
resistencia a la insulina, incremento en adiponectina de alto peso molecular,
hipertrigliceridemia y una amplificación del déficit de grasa sub cutánea.48
Se han reportado incrementos en masa magra, reducción de adiposidad y
mejoría en la tolerancia a la glucosa durante la terapia con hormona de
crecimiento en niñas con síndrome de Turner.49 De las misma forma se han
reportado también mejorías en el perfil lipídico en niñas con síndrome de Turner
así como en niños pequeños nacidos pequeños para edad gestacional. En
ambos grupos de pacientes estos cambios llevaron a mejorías clínicas
relevantes en el índice aterogénico (relación de colesterol total/lipoproteínas de
alta densidad), así como de niveles de lipoproteína de alta densidad
plasmáticos.50
A nivel óseo, a pesar de que la mejoría en crecimiento lineal está
ampliamente demostrada con la terapia con hormona de crecimiento, varios

29
estudios han descrito mejoría en densidad mineral ósea posterior a la terapia en
niños con síndrome de Prader-Willi y en pacientes con síndrome de Turner.51
Se ha demostrado un incremento en el área de hueso cortical en niños
pequeños nacidos pequeños para edad gestacional tratados con hormona de
crecimiento pero no así en pacientes con síndrome de Turner.
La relevancia clínica de esta posible mejoría en densidad mineral ósea es
desconocida. Sin embargo, la terapia con hormona de crecimiento, puede estar
asociada a mayor resistencia ósea y potencialmente con una reducción en el
riesgo de osteoporosis y fracturas, que ha sido reportada en pacientes adultos
deficientes de hormona de crecimiento.52
El momento de inicio de la terapia podría ser crucial, ya que pacientes
con deficiencia de hormona de crecimiento que recibieron terapia temprana no
presentan un riesgo aumentado de fracturas vertebrales.52 Aún más, el pico de
masa ósea se alcanza de forma tardía en condiciones de retraso puberal o
hipogonadismo, por lo que continuar con la terapia con hormona de crecimiento
por un tiempo posterior a que se haya detenido el crecimiento podría ser una
opción a valorar.53
Adicional a los beneficios en parámetros físicos, la terapia con hormona
de crecimiento se ha asociado en algunos estudios con mejorías en aspectos

30
psicosociales. La baja talla es frecuentemente asociada con estereotipos
negativos y desventajas psicológicas, incluyendo pobre rendimiento escolar,
probemas de conducta y capacidad social disminuida.54
A pesar de que algunos reportes sugieren que estas deficiencias pueden
ser atribuidas exclusivamente a la baja talla del paciente, algunos otros estudios
no muestran un incremento significativo en niños pequeños comparados a sus
pares de estatura normal.55
En cuestionarios sobre calidad de vida en niñas con síndrome de Turner
que recibieron terapia con hormona de crecimiento a largo plazo y esteroides
sexuales se demostró que aquellas que alcanzaron tallas normales para la
población y que además tuvieron un desarrollo puberal acorde tuvieron puntajes
altos en las escalas de calidad de vida, incluyendo en escalas de
funcionamiento social y roles emocionales.56
Seguridad del tratamiento con hormona de crecimiento
La seguridad del tratamiento con hormona de crecimiento ha sido sujeta a
mucha discusión en la literatura. En general, el tratamiento es bien tolerado en
la población pediátrica. La mayoría de efectos adversos reportados son menores
y auto limitados (reacciones locales en el sitio de aplicación, nausea, cefalea y
fiebre). 57

31
Otros efectos adversos como lo son la aparición de edemas y síndrome
del túnel carpal, asociados a retención de líquido mediado por hormona de
crecimiento, son usualmente transitorios y suelen resolver en las primeras
semanas del tratamiento o en respuesta a una reducción de dosis de la
misma.57
Los datos tampoco sugieren que el tratamiento influencie negativamente
los segmentos corporales, acelere la maduración ósea, o altere el inicio o
duración de la pubertad.
Contrario a lo mencionado anteriormente sobre posibles efectos
beneficiosos en el metabolismo de la glucosa, otros estudios reportan
incrementos en los niveles de insulina con respecto al basal, previo inicio de la
terapia tanto en niñas con síndrome de Turner como en niños pequeños para
edad gestacional, aunque se ha observado que los niveles de insulina regresan
a su nivel basal al completar la terapia.58
En un estudio de 37 adultos jóvenes nacidos pequeños para edad
gestacional y tratados con hormona de crecimiento por un período medio de 7.3
años, no hubo un incremento en el riesgo de desarrollar síndrome metabólico o
diabetes mellitus tipo 2 tras un período de seguimiento de 6.5 años posterior a la
suspensión de la terapia comparado a adultos nacidos pequeños para edad
gestacional que no recibieron tratamiento, destacando que en este contexto se

32
utilizan dosis más altas de hormona de crecimiento que para el síndrome de
Turner.58
La mayoría de estos posibles efectos sobre el metabolismo de la glucosa
pueden ser neutralizados por la mejoría en la composición corporal, como la
reducción de la masa grasa visceral.
A pesar de la preocupación de un posible impacto de la terapia con
hormona de crecimiento, vía factor de crecimiento tipo insulina 1, en el
desarrollo de cáncer, algunos datos sugieren una asociación inversa entre los
niveles de este marcador y mortalidad por todas las causas en mujeres.59
No existe evidencia de un riesgo aumentado de recidiva o de una
segunda neoplasia en niños tratados previamente por neoplasias,
particularmente cuando los niveles del factor de crecimiento insulínico se
mantienen dentro del rango normal para la edad.60
Vía de administración y Adherencia
La mayoría de los pacientes prefieren la administración en el muslo, sin
embargo se obtiene efectos farmacocinéticos y farmacodinámicos similares tras
la administración en abdomen, aunque la absorción es ligeramente más rápida
en este sitio.61

33
La administración durante las noches resulta en niveles de hormona de
crecimiento elevados durante este tiempo y niveles bajos durante el día,
simulando el patrón fisiológico de la secreción de hormona de crecimiento.
Adicionalmente, la inyección durante la noche produce incremento en los
efectos lipolíticos y menor stress sobre la sensibilidad insulínica durante el día,
lo cual es preferible desde el punto de vista del perfil metabólico.62
En un estudio de Jens et al, se compararon los efectos sobre el patrón de
secreción circadiano y los niveles circulantes de hormona de crecimiento y sus
metabolitos con la aplicación diurna vrs nocturna de hormona de crecimiento,
además de evaluar algunos efectos metabólicos, encontrándose insulinemias
matutinas significativamente mayores al grupo control, que no recibió hormona
de crecimiento, en aquellos pacientes con aplicación diurna de hormona de
crecimiento.75
La inyección diaria subcutánea, no imita de forma completamente
eficiente, la pulsatilidad nocturna de hormona de crecimiento, ya que las
concentraciones pico posterior a la administración se obtienen en 3-5 horas, y
los niveles circulantes de hormona de crecimiento están constantemente
elevados durante la noche y permanecen en la circulación hasta por 16-20
horas.

34
Estudios comparando la inyección subcutánea diaria durante las noches
con una infusión constante subcutánea de hormona de crecimiento por 4-26
semanas han demostrado efectos metabólicos comparables, evaluados en la
sensibilidad a la insulina, marcadores óseos y lipoproteínas.63
Es así como una preparación de acción prolongada de hormona de
crecimiento humana, que también resulte en niveles elevados de hormona de
crecimiento por un período sostenido podría ser tan efectiva para la promoción
del crecimiento como la inyección diaria, con menos cantidad de aplicaciones.
Se han estudiado en este sentido micro esferas de hormona de
crecimiento, preparaciones cristalinas y conjugadas con hialuronato, resultando
en una concentración sostenida de hormona de crecimiento que puede ser
mantenida por días, permitiendo la administración semanal y hasta bi mensual.64
Las evaluaciones de estas formulaciones en pacientes adultos y
pediátricos con déficit de hormona de crecimiento sugieren que brindan cambios
similares en parámetros antropométricos comparados a la formulación
convencional.65
Podría existir, sin embargo, un incremento más marcado en el factor de
crecimiento similar a insulina con la exposición continua a hormona de

35
crecimiento. Se necesita de mayor experiencia y estudios a largo plazo para
confirmar su seguridad.65
Para alcanzar resultados terapéuticos óptimos, la adherencia continua y a
largo plazo es fundamental. La frecuencia de administración ha sido asociada
con la respuesta de crecimiento. La velocidad de crecimiento en pacientes
pediátricos tratados con hormona de crecimiento que omiten más de 15
inyecciones por mes fue de tan solo un 69% comparado a aquellos que
omitieron entre 4-15 inyecciones al mes en un estudio publicado.66
También en este estudio, se reporta que del 15-24% de 630 niños
omitieron más de 3 inyecciones al mes.66
En otro estudio en 75 pacientes recibiendo tratamiento con hormona de
crecimiento, demostró que un 23% de los pacientes omitió más de 2 inyecciones
por semana y que esto se asoció con una velocidad de crecimiento
significativamente reducida.67
Es por esto que el clínico debe considerar la falta de adherencia como
una causa posible en todos los casos en que se sospeche falla terapéutica. A
pesar de que una falta de adherencia completa es fácilmente detectable con el
enlentecimiento o pausa en el crecimiento, adherencias parciales son más
difíciles de reconocer en ocasiones.

36
Los factores más importantes asociados a la falta de adherencia incluyen
el entendimiento de las consecuencias de omitir las dosis, poca satisfacción ó
falta de percepción sobre el tratamiento, lo cual puede estar influenciado por
expectativas poco realistas, dolor con las inyecciones e inadecuado
entrenamiento en la técnica del dispositivo.68
Debe reconocerse que para alcanzar una adherencia completa es
necesario no solo la cooperación del paciente sino de también del compromiso
total de los padres. En este sentido, la adolescencia se plantea como un
momento retador por la necesidad propia del desarrollo de iniciar un desapego
con los padres.
Factores técnicos y físicos se asocian también con la facilidad de
administración y que pueden tener un impacto en la adherencia. Tiempo de uso
prolongado, medicamentos que son difíciles de preparar y administrar están
asociados a pobre adherencia además de errores potenciales en dosificación.
En un estudio se reporta una preferencia del 98% por una formulación
líquida de hormona de crecimiento vrs una preparación de polvo seco que
requiere de reconstituir previo a su uso. Los pacientes reportaron que la
formulación líquida era menos dolorosa, consume menos tiempo y es más
sencillo de aplicar.69

37
Es esperable que la administración prolongada de un fármaco que está
asociado a dolor y discomfort ejerza un efecto negativo en la aceptación del
tratamiento y sobre la adherencia. El dolor al inyectarse puede estar causado
por poca tolerabilidad al compuesto, al volumen administrado y al sitio de
inyección.70
Los elementos de la formulación que pueden afectar el dolor con la
aplicación incluyen el ph, el tipo de preservante, la tonicidad y el buffer. Las
preparaciones de hormona de crecimiento que contienen fenol (3 mg/ml) como
preservante se asocian con menos reportes de dolor en el sitio de inyección que
aquellas formulaciones que contienen alcohol bencílico.71
El buffer usado para mantener el ph del fármaco en un rango estrecho
también puede afectar la experiencia de aplicación. La histidina y el citrato son
agentes amortiguadores comúnmente utilizados, y se reportan que aquellos que
contienen histidina producen una experiencia menos dolorosa que aquellas que
contienen citrato.71
Se ha documentado que los pacientes en tratamiento con hormona de
crecimiento desean un dispositivo que sea fácil de usar y que requiera pocos
pasos para una administración exitosa.72 Se reporta que la adherencia al
tratamiento con hormona de crecimiento es mayor en aquellos pacientes que se

38
auto inyectan y en aquellos que utilizan un dispositivo automático comparado
con las jeringas convencionales o pre llenadas.73
Reportes de encuestas realizadas tanto por pacientes como por médicos,
destacaron la confianza en el dispositivo como el atributo más importante en un
dispositivo de inyección de hormona de crecimiento.72 La ausencia de dolor fue
considerado por los pacientes como el tercer atributo más importante en un
dispositivo de aplicación.
La flexibilidad para el uso es particularmente importante cuando el
dispositivo está siendo utilizado por un niño o adolescente. Un dispositivo que
requiera pocos pasos en la preparación y con ajuste de dosis flexible puede
contribuir de forma importante en la adherencia.
La ventaja de poder mantener la preparación de hormona de crecimiento
a temperatura ambiente en un sistema de inyección en lapicero de múltiples
dosis debería de tener un efecto beneficioso en la adherencia al tratamiento
debido a la conveniencia de ajustarse a un estilo de vida activo.74

39
Materiales y Métodos
Características del estudio
Tipo de estudio: descriptivo, observacional y retrospectivo.
Propósito del estudio: documentar la talla final comparada al pronóstico
de talla familiar de un grupo amplio de pacientes con síndrome de Turner
valoradas en el servicio de Endocrinología del Hospital Nacional de Niños, en el
período de enero 2000 a diciembre 2017. Una vez que se conozcan estos
valores en esta población portadora de este síndrome en nuestro medio, se
facilitará el manejo y abordaje integral que estas pacientes requieren,
brindándoles información objetiva por ejemplo sobre qué pueden esperar con
respecto a la talla con el uso de hormona de crecimiento.
Objeto de estudio: población con Síndrome de Turner confirmado por
cariotipo, evaluados en el Servicio de Endocrinología del Hospital Nacional de
Niños en el período comprendido entre enero 2000 y diciembre 2017.
Objetivo general: Evaluar la talla final en comparación con el pronóstico
de talla familiar de las pacientes con Síndrome de Turner valoradas en el
servicio de Endocrinología del Hospital Nacional de Niños entre enero 2000 y
diciembre de 2017.

40
Objetivos específicos:
a) Identificar los mosaicismos y anormalidades estructurales presentes
en esta población con síndrome de Turner.
b) Relacionar los mosaicismos y anormalidades estructurales con la talla
final de esta población con Síndrome de Turner.
c) Comparar la talla final alcanzada entre pacientes con síndrome de
Turner tratadas y no tratadas con hormona de crecimiento.
Población: Se incluirá la totalidad de las pacientes valoradas con
síndrome de Turner con confirmación por cariotipo en el Servicio de
Endocrinología del Hospital Nacional de Niños en el período
comprendido entre enero de 2000 y diciembre 2017, número que de
acuerdo a la base de datos disponible en HNN es de
aproximadamente 150 individuos.
Resultados esperables: en función de los resultados se pretende
evaluar la talla final comparada al pronóstico de talla familiar que
caracteriza a las pacientes con síndrome de Turner en la población
costarricense, compararla con los genotipos más comunes, incluyendo
pacientes tratadas y no tratadas con hormona de crecimiento.

41
Variables del estudio:
Variables cualitativas: provincia, tratamiento con hormona de
crecimiento, cariotipo, fecha de diagnóstico, reacciones adversas,
adherencia.
Variables cuantitativas: percentilo poblacional de talla inicial:
percentilo poblacional de talla final, percentilo poblacional de
pronóstico de talla familiar, ganancia de talla en cm, dosis de hormona
de crecimiento diaria final, dosis de hormona de crecimiento
mg/kg/semana, edad cronológica, edad de diagnóstico, edad inicio de
hormona de crecimiento, edad final de tratamiento con hormona de
crecimiento, diferencia pronóstico talla familiar y talla final en cm,
diferencia pronóstico talla familiar y talla final expresado en percentilo,
diferencia pronóstico talla familiar y talla final desviación estándar,
pronóstico de talla familiar, duración del tratamiento con hormona de
crecimiento en años, percentilo de índice de masa corporal.
Criterios de inclusión:
Rango de edad: No existe límite superior o inferior que impida
la inclusión de pacientes a la población por estudiar, recalcando

42
que el síndrome de Turner es una condición genética presente
desde el nacimiento.
Género: Pacientes femeninas con síndrome de Turner
(cariotipo 45X o alguno de sus mosaicismos): se incluyen
únicamente mujeres debido a que el síndrome de Turner es
una patología que por definición se presenta por ausencia
parcial o completa del cromosoma X en mujeres.
Etnia: no hay restricción según etnia.
Inclusión de clases especiales o participantes vulnerables: por
el rango de edad que incluye sujetos desde el nacimiento, se
trabajará con datos de pacientes menores de edad. Debe
quedar claro que se trabajará únicamente con sus registros
médicos, por lo cual no se exponen a algún riesgo físico. Se
guardará tanto para ellos, como para todos los sujetos
incluidos, la confidencialidad de sus registros en el expediente
de salud.
Pruebas de laboratorio y Gabinete: no se efectuarán de forma
prospectiva pruebas de laboratorio o gabinete. Las mismas
serán obtenidas del expediente de salud, e incluyen las
variables suministradas en la sección correspondiente.

43
Criterios de exclusión:
Pacientes con características clínicas de síndrome de Turner,
pero a quienes no se les ha realizado cariotipo pues por
definición no son portadoras de la patología en estudio.
Pacientes con datos incompletos o con expedientes
extraviados.
Pacientes estudiados: Se registraron 157 pacientes con diagnóstico
de Síndrome de Turner confirmado con cariotipo y que recibieron o
reciben hormona de crecimiento en el Hospital Nacional de niños entre
enero 2000 a diciembre 2017. Un total de 17 pacientes fueron
excluidos del análisis principal pues no se pudo ubicar el expediente, o
bien contenían datos incompletos que imposibilitaron ser analizados.
Limitaciones del estudio: Puede existir sesgo de detección al ser un
estudio retrospectivo con análisis de registros médicos donde se
puede perder información no recopilada.

44
Fundamento del tipo de estudio:
Se trata de un estudio observacional, descriptivo, retrospectivo de
registros médicos; por estas características del estudio, no existen
grupos de aleatorización.
Fundamento del tipo de recolección de datos:
La base de datos obtenida de la digitación de las hojas de recolección
permaneció y persistirá bajo la custodia únicamente del investigador
principal, en un documento encriptado y sin que se registre el número
de expediente o nombre del paciente en el mismo; sino que se le
asignó un número único para identificar cada caso.
Análisis de datos
Se utilizó estadística descriptiva, distribuciones de frecuencia para las
variables cualitativas y medidas de tendencia central y de dispersión
para las variables cuantitativas. Para la comparación de promedios de
la talla inicial y final se utilizaron pruebas de T Student para muestras
pareadas, y para la comparación de la talla final con el promedio de la
talla familiar pruebas de T Student para muestras independientes, en
ambos casos se usaron además intervalos de confianza al 95%,
además se aplicó un análisis de varianza para evaluar el promedio de
talla final con respecto a la edad al inicio del tratamiento. Por último,

45
se elaboró un modelo de regresión lineal múltiple para evaluar la
relación entre la talla al finalizar el tratamiento con hormona del
crecimiento y la edad al inicio, dosis y duración del tratamiento. Se
estableció un nivel de significancia estadística del 5%. Para el
desarrollo del análisis se utilizaron los programas informáticos
Microsoft Excel 2016 y SPSS versión 23.

46
Reporte de conflicto de interés:
Tanto el investigador principal como los tutores institucional y
académico no reportan conflictos de interés.
Esta propuesta de investigación fue aprobada por el Comité Ético
Científico del Hospital Nacional de Niños de acuerdo a la sesión CEC-
HNN-011-2018, asignándole el número de protocolo CEC-HNN-006-
2018.

47
Resultados
Distribución y características de los pacientes
Se seleccionaron un total de 157 pacientes con síndrome de Turner
disponibles en la base de datos del Hospital Nacional de Niños y con
atención entre enero 2000 y diciembre de 2017.
Es importante destacar que un total de 17 pacientes, correspondientes
a un 11% de la base de datos fueron excluidas del análisis principal,
debido a una o varias de tres razones: ausencia de información sobre
alguna o algunas de las variables, expedientes que no pudieron ser
localizados o se encontraban depurados o bien datos erróneos que no
correspondieran, como por ejemplo números de cédula u expediente
erróneos. Adicionalmente se decidió excluir a una paciente con
diagnóstico de síndrome de Turner con datos incompletos y que
adicionalmente no resultó candidata a tratamiento con hormona de
crecimiento debido a un síndrome nefrótico. El análisis principal se
realizó por lo tanto con 140 pacientes, correspondiente al 89% de la
base de datos.

48
En la figura 1 se puede observar la distribución de acuerdo a la
procedencia de las pacientes con síndrome de Turner valoradas
durante el período de estudio. La provincia de San José tiene la
representación más alta con un 34% de las pacientes mientras que la
menor representación corresponde a la provincia de Guanacaste con
un 4% de las mismas. También se destaca un 1% de participación de
extranjeros correspondientes a El Salvador y Nicaragua.
Figura 1. Distribución de las pacientes portadoras de síndrome de
Turner en control en el Hospital Nacional de Niños, entre enero 2000 y
diciembre 2017, según procedencia geográfica de las pacientes.
Fuente: Hoja de recolección de datos.
1%
4%
8%
8%
9%
11%
26%
34%
0% 5% 10% 15% 20% 25% 30% 35% 40%
Extranjero
Guanacaste
Heredia
Limón
Puntarenas
Cartago
Alajuela
San José

49
Así mismo es importante mencionar que 77 pacientes con síndrome
de Turner en el período de estudio son portadoras de cariotipo clásico
45,X lo que corresponde a un 55% de las pacientes en la base de
datos. Por otra parte un total de 63 pacientes son portadoras de
mosaicismos, lo que corresponde a un 45% de las pacientes de la
base de datos.
Se puede apreciar la distribución de las pacientes de acuerdo a la
evolución del tratamiento con hormona de crecimiento (figura 2). A
diciembre de 2017, un 68.57 de los pacientes habían completado su
tratamiento, mientras que un 18.57% aún continúan con el mismo.
Adicionalmente, de la totalidad de pacientes que iniciaron tratamiento,
un porcentaje no lo completó por distintas circunstancias que se
incluyen en la figura contemplando al 100% de las pacientes que
iniciaron tratamiento. De acuerdo a las notas del expediente clínico, un
5.0% de las pacientes tuvo un uso irregular del tratamiento y un 5.7%
hicieron abandono del mismo.
También en un 0.72% de las pacientes se determinó que no tuvieron
una respuesta adecuada al tratamiento (definido a criterio del médico
tratante por falta de progresión en la curva de crecimiento). Hubo a su
vez un porcentaje de 1.43% de suspensión por efectos adversos.

50
Figura 2. Distribución de las pacientes portadoras de síndrome de Turner en
control en el Hospital Nacional de Niños, entre enero 2000 y diciembre 2017,
según evolución del tratamiento con hormona del crecimiento.
Fuente: Hoja de recolección de datos.
Un total de 5 pacientes, correspondiente a un 3.57% de las mismas
tuvieron algún tipo de efecto adverso con el tratamiento con hormona
de crecimiento.
La tabla 1 muestra el desglose de los eventos adversos presentados
por las pacientes durante el tratamiento con hormona de crecimiento,
ocurriendo un total de 1 para cada uno de los siguientes efectos
adversos reportados: artralgias, cefaleas, escoliosis, sangrado rectal y
poliposis y cifosis. Las artralgias y cefaleas fueron transitorias y no
ameritaron suspensión del tratamiento, mientras que los casos de
0.72%
1.43%
5,0%
5.71%
18.57%
68.57%
0,0% 10,0% 20,0% 30,0% 40,0% 50,0% 60,0% 70,0% 80,0%
Sin respuesta
S/S Efectos adverso
Uso irregular
Abandono de tx
Aun en Tx
Tx completo

51
escoliosis, sangrado rectal y poliposis y cifosis fueron considerados
graves y ameritaron la suspensión del mismo.
Tabla 1. Total de eventos adversos reportados en las pacientes portadoras de
síndrome de Turner en control en el Hospital Nacional de Niños, entre enero
2000 y diciembre 2017, en tratamiento con hormona del crecimiento.
Eventos adversos al tratamiento Cantidad
Artralgias 1
Cefaleas 1
Escoliosis 1
Sangrado rectal y poliposis 1
Cifosis 1
Fuente: Hoja de recolección de datos.
Se muestra también la distribución de las pacientes con síndrome de
Turner durante el período de estudio de acuerdo al momento del
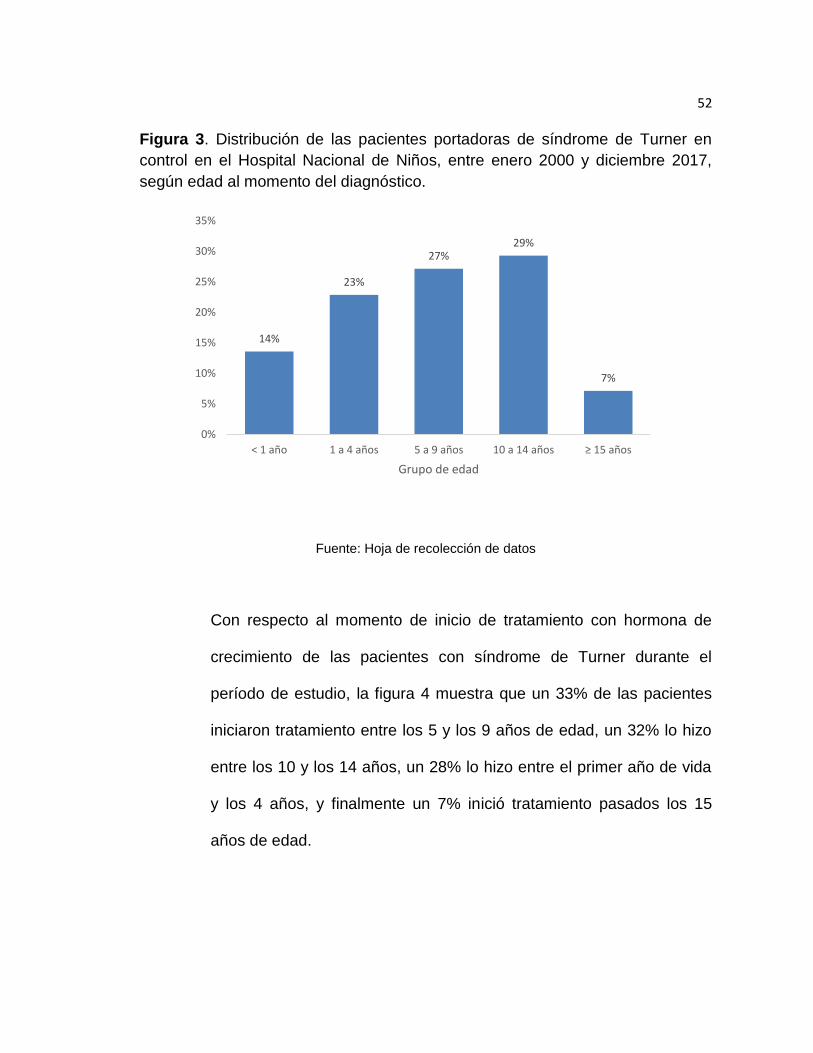
diagnóstico (figura 3). De mayor a menor grupo etario, un 29% de los
casos fueron diagnosticados entre los 10 y los 14 años, un 27% de los
casos entre los 5 y 9 años, un 23% entre el año de edad y los 4 años,
un 14% fueron diagnosticados dentro del primero año de vida, y
finalmente un 7% de los casos fueron diagnosticados a los 15 años o
posteriormente.
52
Figura 3. Distribución de las pacientes portadoras de síndrome de Turner en
control en el Hospital Nacional de Niños, entre enero 2000 y diciembre 2017,
según edad al momento del diagnóstico.
Fuente: Hoja de recolección de datos
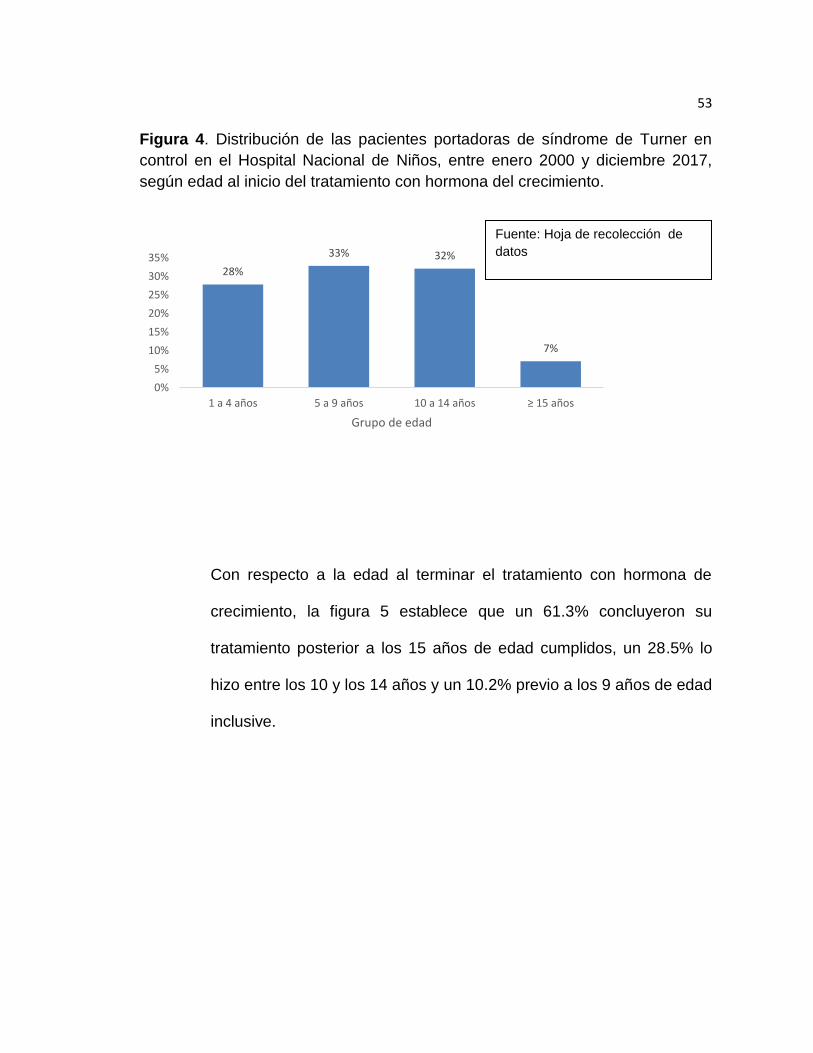
Con respecto al momento de inicio de tratamiento con hormona de
crecimiento de las pacientes con síndrome de Turner durante el
período de estudio, la figura 4 muestra que un 33% de las pacientes
iniciaron tratamiento entre los 5 y los 9 años de edad, un 32% lo hizo
entre los 10 y los 14 años, un 28% lo hizo entre el primer año de vida
y los 4 años, y finalmente un 7% inició tratamiento pasados los 15
años de edad.
14%
23%
27% 29%
7%
0%
5%
10%
15%
20%
25%
30%
35%
< 1 año 1 a 4 años 5 a 9 años 10 a 14 años ≥ 15 años
Grupo de edad
53
Figura 4. Distribución de las pacientes portadoras de síndrome de Turner en
control en el Hospital Nacional de Niños, entre enero 2000 y diciembre 2017,
según edad al inicio del tratamiento con hormona del crecimiento.
Con respecto a la edad al terminar el tratamiento con hormona de
crecimiento, la figura 5 establece que un 61.3% concluyeron su
tratamiento posterior a los 15 años de edad cumplidos, un 28.5% lo
hizo entre los 10 y los 14 años y un 10.2% previo a los 9 años de edad
inclusive.
28%
33% 32%
7%
0%
5%
10%
15%
20%
25%
30%
35%
1 a 4 años 5 a 9 años 10 a 14 años ≥ 15 años
Grupo de edad
Fuente: Hoja de recolección de
datos

54
Figura 5. Distribución de las pacientes portadoras de síndrome de Turner en
control en el Hospital Nacional de Niños, entre enero 2000 y diciembre 2017,
según edad al finalizar el tratamiento con hormona del crecimiento.
Con respecto a las pacientes que completaron el tratamiento con
hormona de crecimiento, la tabla 2 muestra que las mismas tuvieron
una edad promedio de 8.5 años al momento del diagnóstico con una
desviación estándar de -4.7, tuvieron como promedio una edad de 9.7
años de edad de inicio de tratamiento con hormona de crecimiento
con una desviación estándar de -3.9.
Como promedio las pacientes culminaron su tratamiento a los 15.8
años con una desviación estándar de 1.3 y se mantuvieron un total de
6.1 años en tratamiento con hormona de crecimiento con una
desviación estándar de 3.1y utilizaron una dosis final promedio de 1.7
mgs/día de hormona de crecimiento con una desviación estándar de
0.4.
10.2%
28.5%
61.3%
0%
20%
40%
60%
80%
≤ 9 años 10 a 14 años ≥ 15 años
Grupo de edad
Fuente: Hoja de recolección
de datos

55
Tabla 2. Edad de diagnóstico, de inicio y fin de tratamiento con
hormona del crecimiento, duración y dosis al final del este, en las
pacientes portadoras de síndrome de Turner en control en el Hospital
Nacional de Niños, entre enero 2000 y diciembre 2017.
Variable Promedio DS
Edad en años al diagnóstico 8,5 -4,7
Edad en años al inicio del tratamiento
9,7 -3,9
Edad en años al finalizar el tratamiento
15,8 1,3
Duración del tratamiento en años 6,1 3,1
Dosis en mg/dia al final del tratamiento
1,7 0,4
Fuente: Hoja de recolección de datos
De acuerdo a la tabla 3, las pacientes que completaron tratamiento
con hormona de crecimiento tuvieron una talla inicial promedio de
115.4 cm y una talla final promedio de 146.2 cm. El pronóstico de talla
familiar de estas pacientes fue en promedio de 157.1 cm. El peso al
final del tratamiento fue en promedio de 51.1 kg.
Hubo una diferencia estadísticamente significativa de 31 cm en
promedio al comparar la talla inicial con la talla final obtenida por estas
pacientes, así como en promedio las pacientes tuvieron una talla final
estadísticamente significativa de 10.8 cm por debajo del promedio de
talla familiar.

56
Al inicio del tratamiento, las pacientes que completaron tratamiento se
encontraban en promedio en el percentilo 1.79 de talla poblacional y al
finalizar el tratamiento se encontraron en promedio en el percentilo
3.25 de talla poblacional, lo que dio como resultado una diferencia
promedio en percentilo poblacional de talla de 1,47, con una tendencia
hacia la significancia estadística (p igual a 0.054).
El promedio de talla final de esta población se ubicó en el percentilo
18.74, por debajo del promedio esperable de acuerdo al pronóstico de
talla familiar, alcanzando la significancia estadística.
Esta población de pacientes con síndrome de Turner se encontraba al
inicio del tratamiento en promedio -3.24 desviaciones estándar por
debajo de la población de referencia, alcanzado al final del tratamiento
una talla que se encuentra a -2.55 desviaciones estándar de la
población de referencia, dando una diferencia estadísticamente
significativa de 0.95 desviaciones estándar.
Así mismo, esta población de pacientes tiene un promedio de
pronóstico de talla familiar que se encuentra a -0.95 desviaciones
estándar de la población de referencia, obteniendo una talla final que
se encuentra a -1.60 de la desviación estándar esperable promedio de
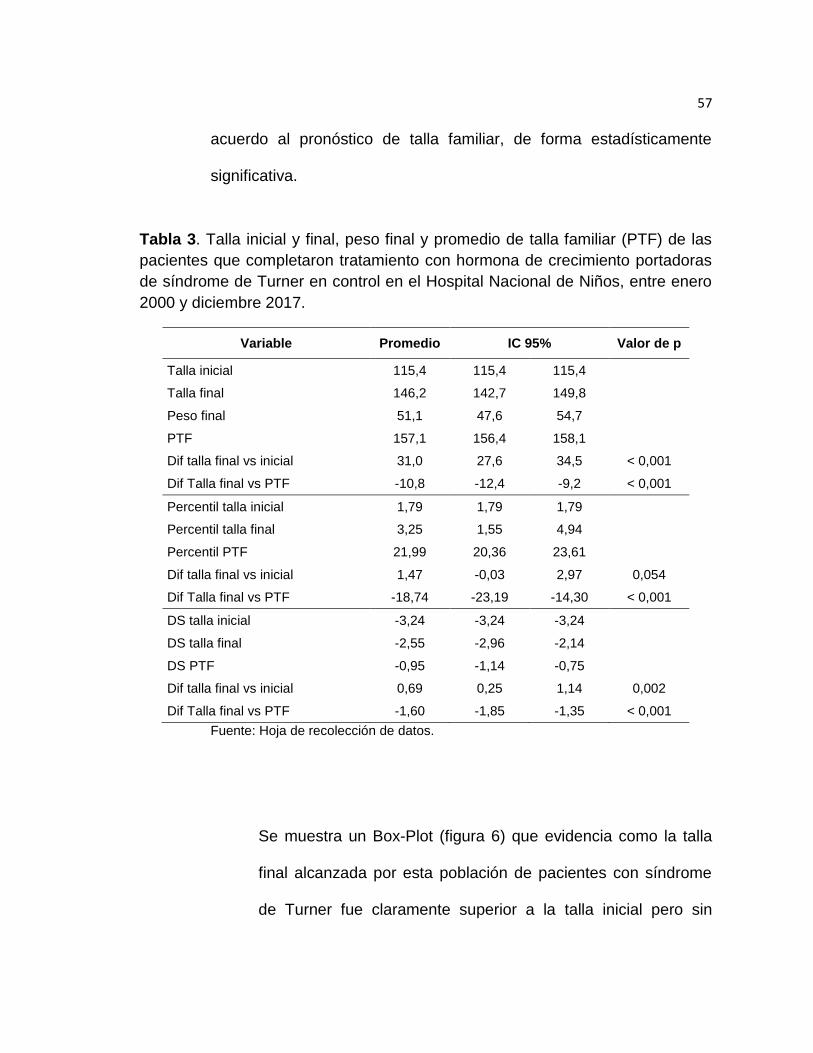
57
acuerdo al pronóstico de talla familiar, de forma estadísticamente
significativa.
Tabla 3. Talla inicial y final, peso final y promedio de talla familiar (PTF) de las
pacientes que completaron tratamiento con hormona de crecimiento portadoras
de síndrome de Turner en control en el Hospital Nacional de Niños, entre enero
2000 y diciembre 2017.
Variable Promedio IC 95% Valor de p
Talla inicial 115,4 115,4 115,4
Talla final 146,2 142,7 149,8
Peso final 51,1 47,6 54,7
PTF 157,1 156,4 158,1
Dif talla final vs inicial 31,0 27,6 34,5 < 0,001
Dif Talla final vs PTF -10,8 -12,4 -9,2 < 0,001
Percentil talla inicial 1,79 1,79 1,79
Percentil talla final 3,25 1,55 4,94
Percentil PTF 21,99 20,36 23,61
Dif talla final vs inicial 1,47 -0,03 2,97 0,054
Dif Talla final vs PTF -18,74 -23,19 -14,30 < 0,001
DS talla inicial -3,24 -3,24 -3,24
DS talla final -2,55 -2,96 -2,14
DS PTF -0,95 -1,14 -0,75
Dif talla final vs inicial 0,69 0,25 1,14 0,002
Dif Talla final vs PTF -1,60 -1,85 -1,35 < 0,001
Fuente: Hoja de recolección de datos.
Se muestra un Box-Plot (figura 6) que evidencia como la talla
final alcanzada por esta población de pacientes con síndrome
de Turner fue claramente superior a la talla inicial pero sin

58
embargo, y a pesar del tratamiento con hormona de
crecimiento, quedó por debajo de la talla esperada de acuerdo
al promedio de talla familiar, con intervalos de confianza
amplios.
Figura 6. Distribución de la talla inicial y final y de la talla promedio familiar de
las pacientes portadoras de síndrome de Turner en control en el Hospital
Nacional de Niños, entre enero 2000 y diciembre 2017.
Fuente: Hoja de recolección de datos.
La tabla 4 muestra un análisis con distintas variables de las
pacientes que completaron el tratamiento con hormona de
crecimiento.
Las pacientes que cumplieron menos de 5 años de tx alcanzaron
en promedio una talla final de 146.1 cm y aquellas que
cumplieron 5 o más años en tratamiento con hormona de
crecimiento alcanzaron una talla promedio de 146.4 cm. Al

59
compararlos se obtuvo una diferencia 0.3 cm a favor de aquellas
que tuvieron 5 o más años de tratamiento, sin alcanzar
significancia estadística.
Aquellas pacientes con cariotipo clásico 45,X tuvieron en
promedio después del tratamiento una talla de 146.2 cm y
aquellas con anormalidades estructurales alcanzaron una talla
promedio de 146.3 cm, dando como resultado una diferencia de
0.1 cm a favor de aquellas pacientes con anormalidad
estructurales sin alcanzar la significancia estadística.
Al finalizar el tratamiento con hormona de crecimiento, aquellas
pacientes que lo iniciaron entre el primer año de vida y los 4 años,
alcanzar una talla final promedio de 145.2 cm, mientras que
aquellas que iniciaron tratamiento entre los 5 y los 9 años
alcanzaron una talla promedio de 146.4 cm.
Las que iniciaron tratamiento entre los 10 y los 14 años,
alcanzaron una talla promedio de 145.7 cm y las que lo
empezaron después de los 15 años inclusive tuvieron una talla
final promedio de 149.5.
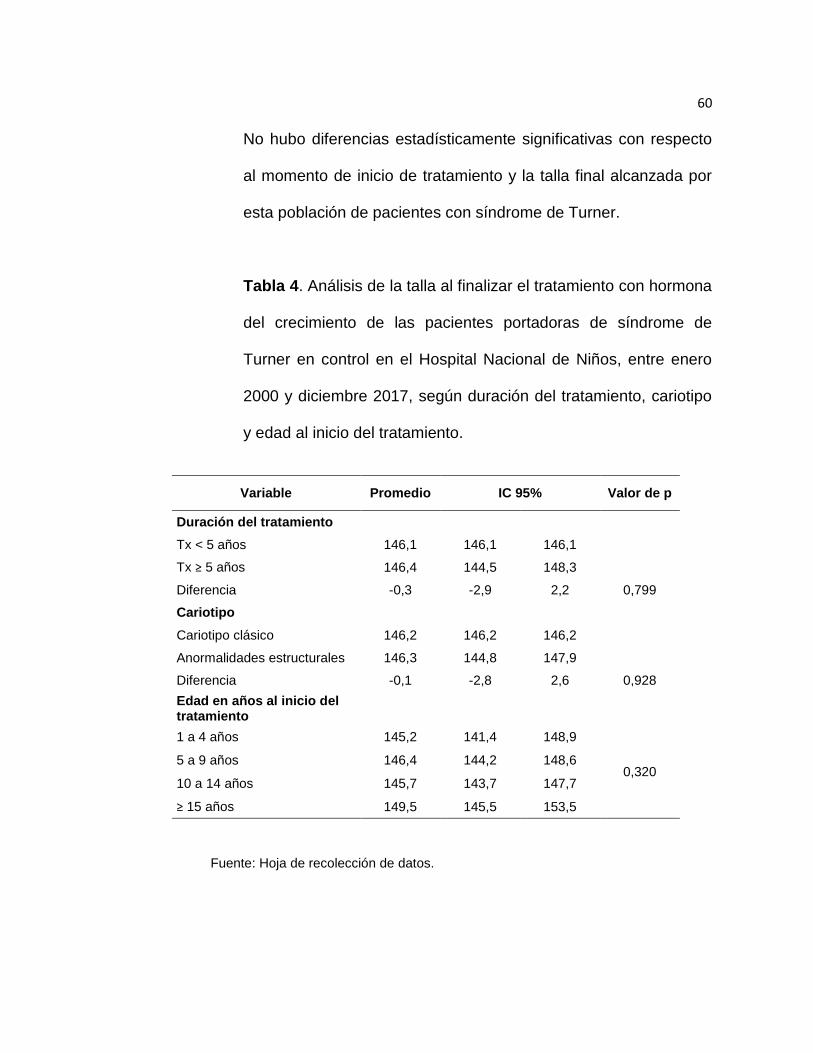
60
No hubo diferencias estadísticamente significativas con respecto
al momento de inicio de tratamiento y la talla final alcanzada por
esta población de pacientes con síndrome de Turner.
Tabla 4. Análisis de la talla al finalizar el tratamiento con hormona
del crecimiento de las pacientes portadoras de síndrome de
Turner en control en el Hospital Nacional de Niños, entre enero
2000 y diciembre 2017, según duración del tratamiento, cariotipo
y edad al inicio del tratamiento.
Variable Promedio IC 95% Valor de p
Duración del tratamiento
Tx < 5 años 146,1 146,1 146,1
Tx ≥ 5 años 146,4 144,5 148,3
Diferencia -0,3 -2,9 2,2 0,799
Cariotipo
Cariotipo clásico 146,2 146,2 146,2
Anormalidades estructurales 146,3 144,8 147,9
Diferencia -0,1 -2,8 2,6 0,928
Edad en años al inicio del tratamiento
1 a 4 años 145,2 141,4 148,9
0,320 5 a 9 años 146,4 144,2 148,6
10 a 14 años 145,7 143,7 147,7
≥ 15 años 149,5 145,5 153,5
Fuente: Hoja de recolección de datos.

61
Se realizó un análisis de regresión lineal múltiple de la ganancia de
talla al finalizar el tratamiento con hormona de crecimiento, mostrado
en la tabla 5, con la duración, la dosis, la edad de inicio del
tratamiento, el cariotipo, el pronóstico de talla familiar y la talla inicial al
tratamiento, obteniendo un coeficiente de 3.16, 0.03, 0.05, 0.75, 0.33 y
-0.40 respectivamente.
Únicamente la talla inicial al tratamiento, el pronóstico de talla familiar
y la duración del tratamiento en años alcanzaron una diferencia
estadísticamente significativa para predecir la ganancia de talla,
siendo la duración del tratamiento en años la variable con mayor peso
en el análisis, de acuerdo al coeficiente de determinación.
Tabla 5. Análisis de regresión lineal múltiple de la ganancia de talla al finalizar el
tratamiento con hormona del crecimiento con la duración, la dosis y la edad de
inicio del tratamiento, en las pacientes portadoras de síndrome de Turner en
control en el Hospital Nacional de Niños, entre enero 2000 y diciembre 2017.
Variable Coeficiente IC 95% Valor de p R^2
Dosis mg/día al final del Tx 0,03 -2,66 2,73 0,980 0,0974
Edad (años) al inicio del Tx 0,05 -0,91 1,02 0,912 0,8732
Talla al inicio del Tx -0,40 -0,59 -0,21 < 0,001 0,8742
PTF 0,33 0,13 0,53 0,002 0,0004
Duración del Tx (años) 3,16 2,15 4,18 < 0,001 0,9131
Cariotipo 0,75 -1,26 2,76 0,462 0,0190
Fuente: Hoja de recolección de datos.

62
Como análisis secundario se exploraron algunas variables en función
del período en años según inicio de tratamiento, siendo así y como
demuestra el Box-Plot en la figura 7, donde se da la distribución según
la edad de inicio de tratamiento de las pacientes con síndrome de
Turner, observando como aquellas pacientes que iniciaron tratamiento
en el quinquenio comprendido entre 2005-2009 fueron mayores que
las pacientes que iniciaron tratamiento en el quinquenio 2000-2004 y
el período 2010-2015. Las pacientes que iniciaron tratamiento entre el
2000-2004 tuvieron una edad similar al inicio de tratamiento que las
pacientes que iniciaron entre el 2010-2015.
Figura 7. Distribución de la edad de inicio de tratamiento de las pacientes portadoras
de síndrome de Turner en control en el Hospital Nacional de Niños, entre enero 2000 y
diciembre 2017, según el año de inicio del tratamiento con hormona del crecimiento.
Fuente: Hoja de recolección de datos.

63
La tabla 6 muestra una comparación de la edad de inicio de
tratamiento en las pacientes con síndrome de Turner, resultando que
aquellas pacientes que iniciaron tratamiento entre los años 2000 y
2004 tuvieron una media de edad de 8,0 años con una desviación
estándar de 3,7 años. Aquellas pacientes que iniciaron tratamiento
entre los años 2005 y 2009 tuvieron una media de edad de 9,7 años
con una desviación estándar de 4,8 años y aquellas que lo iniciaron
entre los años 2010-2015 tuvieron una edad media de 7,4 años con
una desviación estándar de 3,8 años. Así mismo hubo una diferencia
estadísticamente significativa (p=0,023) entre la edad de las pacientes
que iniciaron tratamiento entre los años 2005 y 2009 vrs aquellas que
lo iniciaron ya sea entre los años 2000 y 2004 o entre los años 2010 y
2015.
Tabla 6. Comparación de la edad de inicio de tratamiento de las pacientes
portadoras de síndrome de Turner en control en el Hospital Nacional de Niños,
entre enero 2000 y diciembre 2017, según el año de inicio del tratamiento con
hormona del crecimiento.
Año de inicio de tratamiento
Media (años)
Desviación estándar
Valor de p de la comparación de
las medias
2000 – 2004 8,0 3,7
0,023 2005 – 2009 9,7 4,8
2010 – 2015 7,4 3,8
Fuente: Hoja de recolección de datos.

64
Siguiendo con el análisis secundario, la figura 8 muestra la distribución
de acuerdo a la dosis utilizada de hormona de crecimiento al final del
tratamiento en las pacientes con síndrome de Turner según el período
en años de inicio de tratamiento, resultando que se utilizó una dosis
mayor de hormona de crecimiento al final del tratamiento en el
quinquenio 2000-2004 y una dosis menor en aquellas que finalizaron
el tratamiento en el período 2010-2015,
Figura 8. Distribución de la dosis de hormona de crecimiento al final del
tratamiento de las pacientes portadoras de síndrome de Turner en control en el
Hospital Nacional de Niños, entre enero 2000 y diciembre 2017, según el año de
inicio del tratamiento con hormona del crecimiento.
Fuente: Hoja de recolección de datos.

65
La tabla 7 muestra una comparación de la dosis utilizada de hormona
de crecimiento al final del tratamiento en esta población de pacientes
con síndrome de Turner, siendo que entre el año 2000 y 2004 se
utilizó una dosis media de 1.74 mgs/día con una desviación estándar
de 0,38, durante los años 2005 y 2009 se utilizó una dosis media de
1.62 mgs/día con una desviación estándar de 0,35 y durante los años
2010 y 2015 se utilizó una dosis media de 1,50 mgs/día con una
desviación estándar de 0,29. No hubo una diferencia estadísticamente
significativa entre las dosis medias de hormona de crecimiento
utilizadas al final del tratamiento al realizar la comparación entre
dichos períodos.
Tabla 7. Comparación de la dosis de hormona de crecimiento al final del
tratamiento de las pacientes portadoras de síndrome de Turner en control en el
Hospital Nacional de Niños, entre enero 2000 y diciembre 2017, según el año de
inicio del tratamiento con hormona del crecimiento.
Año de inicio de tratamiento
Media (mgs/día)
Desviación estándar
Valor de p de la comparación de
las medias
2000 - 2004 1,74 0,38
0,098 2005 - 2009 1,62 0,35
2010 - 2015 1,50 0,29
Fuente: Hoja de recolección de datos.

66
Como última variable y como se muestra en la figura 9, se presenta la
distribución de la ganancia de talla por período en años según inicio
de tratamiento con hormona de crecimiento, resultando que en el
quinquenio comprendido entre 2000 y 2004 hubo la mayor ganancia
de talla y siendo el período entre 2010 y 2015 en el que hubo la menor
ganancia de talla.
Figura 9. Distribución de la ganancia de talla en las pacientes portadoras de
síndrome de Turner en control en el Hospital Nacional de Niños, entre enero
2000 y diciembre 2017, según el año de inicio del tratamiento con hormona del
crecimiento.
Fuente: Hoja de recolección de datos.

67
Siguiendo con la variable de ganancia de talla de acuerdo al período
en años según inicio de tratamiento con hormona de crecimiento en
esta población de pacientes con síndrome de Turner, las pacientes
que iniciaron su tratamiento entre el año 2000 y 2004 tuvieron una
media de ganancia de talla de 37.2 centímetros, significativamente
mayor al resto de períodos. Aquellas pacientes que iniciaron su
tratamiento entre 2005 y 2009 tuvieron una ganancia media de talla de
24 centímetros, y aquellas que lo iniciaron entre el 2010 y 2015
tuvieron una media de ganancia de talla de 20,2 centímetros. Estos
últimos dos períodos sin diferencia estadísticamente significativa entre
sí.
Tabla 8. Comparación de la ganancia de talla en las pacientes portadoras de
síndrome de Turner en control en el Hospital Nacional de Niños, entre enero
2000 y diciembre 2017, según el año de inicio del tratamiento con hormona del
crecimiento.
Año de inicio de tratamiento
Media (cms)
Desviación estándar
Valor de p de la comparación de las medias
2000 - 2004 37,2 18,0
<0,001 2005 - 2009 24,0 12,6
2010 - 2015 20,2 8,2
Fuente: Hoja de recolección de datos.

68
Discusión
El síndrome de Turner es una patología frecuente con múltiples
comorbilidades, una de las cuales es la baja talla, que de no ser
tratada, puede llevar a serias repercusiones en el ámbito psicosocial
de las pacientes afectadas.
Entre el período de estudio, se contó con una base de datos que
incluye a 157 pacientes con síndrome de Turner que fueron atendidas
en el servicio de Endocrinología del Hospital Nacional de Niños, centro
de referencia nacional para el manejo de esta patología.
A nuestro entendimiento este trabajo constituye a la fecha, el estudio
con mayor número de pacientes con síndrome de Turner a nivel
nacional y a la vez el más extenso en enfocarse en el parámetro de la
talla final alcanzada con el tratamiento con hormona de crecimiento.
Son pocas las publicaciones sobre síndrome de Turner a nivel
latinoamericano.
Partiendo de una incidencia reportada de 1 en cada 2000-2500
nacimientos femeninos, ésta base de datos debería contar con
aproximadamente 250 pacientes, asumiendo un promedio anual de
nacimientos femeninos de 35000. Por lo que el número de pacientes

69
es menor al esperado y esto podría obedecer a un subdiagnóstico
importante o bien al hecho de que estos pacientes están siendo
manejados en atención primaria o por el pediatra general. Esta
situación da pie a la necesidad de reforzar siempre el diagnóstico de
esta patología, obligación que recae en los médicos de atención
primaria y pediatras generales.
El análisis principal se realizó con el 89% de los pacientes,
destacando la robustez de la base de datos del servicio de
Endocrinología y del soporte de archivo y expediente electrónico con
el que cuenta el Hospital Nacional de Niños. Fueron pocos los
expedientes que no contaban con datos suficientes o que habían sido
depurados.
Como era esperable, más de la mitad de las pacientes con síndrome
de Turner provienen de las dos provincias con mayor cantidad de
población en el país, como lo son San José y Alajuela. Llama la
atención la proporción menor de pacientes de la provincia de Heredia
y de la provincia de Guanacaste.
Se podría considerar que esta tendencia sea normal, aunque también
puede servir para sospechar de un bajo diagnóstico de la patología en

70
estos lugares (siendo que el principal motivo de referencia fue por baja
talla y no por sospecha de Turner) o bien un bajo porcentaje de
referencia por parte del médico de atención primaria o el pediatra.
La provincia de Guanacaste es una región de alta emigración hacia
otras zonas del país, por lo que algunas de estas pacientes podrían
haberse trasladado a provincias de mayor densidad poblacional.
Siguiendo con las características de esta población costarricense de
pacientes con síndrome de Turner, llama la atención la proporción
inversa encontrada de casos de mosaicismos vrs cariotipo clásico,
siendo lo reportado en la literatura la presencia de mosaicismos en
alrededor del 55% de las pacientes. El haber obtenido una proporción
mayor de cariotipos clásicos en nuestra población podría obedecer a
una condición propia de esta población de pacientes.
Sobre la evolución del tratamiento con hormona de crecimiento, es
importante destacar el porcentaje amplio de pacientes que para el
período de estudio habían completado su tratamiento con hormona de
crecimiento, y por lo tanto, se encontraron disponibles para ser
analizados de acuerdo a su talla final.

71
Hubo un poco más de 10% de pacientes los cuales tuvieron falta de
adherencia (establecido por el médico tratante como un uso irregular
del mismo) o bien con abandono del tratamiento. Se conoce que el
apoyo familiar es fundamental durante el tratamiento con hormona de
crecimiento, y este porcentaje de pacientes con esta situación debe
ser siempre tomado en consideración para que en cada visita, el
médico endocrinólogo haga énfasis a los padres sobre la importancia
del tratamiento, que a esta edad la administración del mismo es su
responsabilidad.
Se destaca también que únicamente 2 pacientes ameritaron que se
les suspendiera el tratamiento con hormona de crecimiento, 1 por
presencia de pólipos colónicos con sangrado rectal y otro con
empeoramiento de una cifosis de columna, ambos efectos adversos
posibles con el tratamiento con hormona de crecimiento, y que
representaron un porcentaje mínimo de los pacientes tratados,
destacando la buena respuesta general al tratamiento con hormona de
crecimiento, como está descrito en la literatura.
Solo en el caso de un paciente, se consideró que la respuesta no fue
la adecuada con el tratamiento con hormona de crecimiento y por lo
tanto se le suspendió el mismo, comprobando la alta eficacia de esta

72
terapia en el tratamiento de la baja talla de las pacientes con síndrome
de Turner. Se trató de una niña que inició la terapia a los 6.3 años de
edad y lo mantuvo durante 9 años con buena adherencia, sin embargo
su talla al momento de la suspensión (15.3 años) era de tan solo 135
cm (percentilo poblacional 0.1) y llegando a utilizar una dosis final de
1.63 mgs/dia de hormona de crecimiento
Adicional a estos dos pacientes que ameritaron suspensión del
tratamiento por efectos adversos, únicamente tres más presentaron
reacciones adversas. La cefalea y las artralgias son ampliamente
reconocidas como posibles efectos adversos menores en el
tratamiento con hormona de crecimiento, y que además usualmente
mejoran con la duración del mismo. La escoliosis es per se una
posible comorbilidad del síndrome de Turner por lo que su
documentación no obedece necesariamente a un efecto adverso del
fármaco.
A excepción de un paciente al cual no se le inició hormona de
crecimiento por presentar concomitantemente una enfermedad renal
lo que se consideró iba a perjudicar su talla final indistintamente si
fuera tratada o no, todos los pacientes con síndrome de Turner de los
cuales había datos disponibles fueron tratados en algún momento lo

73
que destaca la amplia disponibilidad del tratamiento en nuestro país
para tratar la baja talla de pacientes con esta condición.
El tratamiento con hormona de crecimiento se encuentra disponible en
la Caja Costarricense del Seguro Social desde 1987 formando parte
de la lista oficial de medicamentos, restringiéndose su uso únicamente
previa aprobación de la Comisión de Hormona de Crecimiento,
formada por los médicos asistentes especialistas endocrinólogos
pediatras del servicio de Endocrinología del Hospital de Niños. La
mayoría del período de estudio, a excepción de 1 año, se utilizó
hormona de crecimiento original con nombre comercial Genotropin®, y
que es la preparación disponible en la actualidad.
Todos estos factores hablan de la solidez de la institución para ofrecer
un tratamiento y seguimiento de calidad a las pacientes con baja talla
no solo con síndrome de Turner, sino también para las distintas
indicaciones con las que cuenta la terapia.
La mayoría de pacientes fueron diagnosticadas en el rango de edad
de entre los 5 y los 14 años, con una edad promedio de 8.5 años.
Existiendo reportes que hablan de un retraso importante en el
diagnóstico del síndrome de Turner, inclusive en la adolescencia y

74
adultez, el promedio obtenido para el diagnóstico resalta la fortaleza
de nuestro sistema de salud con la preparación del personal de
atención primaria y pediatría general, logrando detectar de forma
temprana esta patología para así iniciar su abordaje integral. Mayores
esfuerzos deben realizarse, posiblemente en zonas alejadas de los
centros urbanos para lograr reducir cada vez más la edad de
diagnóstico en busca de un mejor control de las comorbilidades
asociadas.
Este diagnóstico temprano contribuyó a que en promedio el
tratamiento con hormona de crecimiento se iniciara de manera
temprana a los 9.7 años, con la tercera parte iniciándose en el rango
de 5-9 años. Hay que recordar que el inicio temprano de hormona de
crecimiento es un factor pronóstico de la talla final de las pacientes
con síndrome de Turner en terapia con hormona de crecimiento, y que
actualmente las últimas guías de práctica clínica en el manejo del
síndrome de Turner, recomiendan el inicio de hormona de crecimiento
entre los 4-6 años, y de preferencia antes de los 12-13 años.
Hubo un retraso promedio de 1.2 años entre el diagnóstico y el inicio
de la terapia con hormona de crecimiento, un tiempo adecuado para
valorar la velocidad de crecimiento en aquellos pacientes

75
diagnosticados tempranamente, con una edad ósea retrasada y que
aún se mantienen por encima del percentilo 5 de talla poblacional.
El diagnóstico e inicio de tratamiento relativamente temprano, permitió
alcanzar una duración promedio de tratamiento de 6.1 años hasta una
edad promedio de 15.8 años, representando este un tiempo promedio
acorde a lo reportado en la literatura, destacando que la duración del
tratamiento es otro factor pronóstico de la talla final de las pacientes
con síndrome de Turner en tratamiento con hormona de crecimiento,
siendo que se aplican los mismos criterios de otras series publicadas
para conclusión del tratamiento, es decir velocidad de crecimiento
menor a 2 centímetros por año o edad ósea ≥ 14 años.
La edad promedio de suspensión alcanzada es acorde con el tiempo
recomendado para la suspensión del mismo, ya sea por cierre de
epífisis o porque se alcanzó una velocidad de crecimiento menor a 2
cm por año, ambos criterios de crecimiento utilizados en esta serie.
Esta población de pacientes con síndrome de Turner tratadas con
hormona de crecimiento durante el período establecido alcanzaron en
promedio una talla final 146.2 cm con una ganancia promedio de 31
cm, la cual es más alta que lo reportado en la literatura (10-25 cm), sin

76
embargo hay que considerar el diagnóstico temprano de esta
población y por ende la talla inicial menor que permitiría explicar este
fenómeno.
La talla final de estas pacientes se ubicó 10.8 cm por debajo del
esperado para talla familiar, un hallazgo que es esperable a pesar del
tratamiento con hormona de crecimiento, y que por el contrario
demuestra su eficacia, ya que pacientes no tratados se reporta un
promedio de talla de 20 cm por debajo del promedio esperado.
Si bien no hubo una diferencia estadísticamente significativa del
percentil final de talla poblacional alcanzada por las pacientes, existe
una clara tendencia hacia la misma, lo cual puede ser explicado por el
tamaño de la muestra, asumiendo que una muestra mayor permitiera
mostrar esta diferencia.
Las pacientes alcanzaron una desviación estándar de talla final que
fue estadísticamente menor al inicio del tratamiento aunque por
debajo del promedio de talla familiar, resultados que son esperables
de acuerdo a lo reportado sobre la eficacia del tratamiento con
hormona de crecimiento, con desviaciones estándar reportadas al final
de tratamiento entre 1.4 y -2.18.

77
En este estudio ni la duración de tratamiento con hormona de
crecimiento mayor o menor a 5 años ni la edad de inicio de
tratamiento fueron factores que afectaran la talla final de las pacientes
con síndrome de Turner, hallazgo diferente a lo reportado en la
literatura. Fueron relativamente pocos los pacientes que tuvieron
tratamiento tan temprano como antes de los 4 años o por un tiempo
demasiado prolongado lo cual podría explicar la falta de diferencia en
la talla final debido al tamaño de la muestra.
El resultado del cariotipo no influyó en la talla final, algo que es
esperable de acuerdo a lo reportado.
Se estableció un modelo de regresión lineal con el fin de predecir la
ganancia de talla de acuerdo a factores que se han reportado tienen
un efecto sobre la misma, como lo son la dosis de hormona de
crecimiento, la edad al inicio del mismo, la talla inicial, el pronóstico de
talla familiar y la duración del tratamiento.
Únicamente la talla inicial, el pronóstico de talla familiar y la duración
de tratamiento en años tuvieron una diferencia estadísticamente
significativa en esta población, siendo el factor de mayor peso la

78
duración del tratamiento en años como predictor de la ganancia de
talla.
La ausencia como predictores de ganancia de talla, de la dosis al final
del tratamiento y la edad al inicio del mismo en esta población puede
obedecer al uso de una dosis promedio algo menor (0.25
mgs/kg/semana) que la recomendada en la literatura (0.3
mgs/kg/semana) y a que relativamente pocos pacientes iniciaron
tratamiento a edades demasiado tempranas como antes de los 4
años.
El análisis secundario realizado arroja resultados interesantes sobre la
ganancia de talla de esta población con síndrome de Turner en los
años analizados, así como también deja algunas interrogantes.
En primer lugar, no se evidenció un claro patrón de mejoría en
ganancia de talla con el paso de los años y a más experiencia con el
uso de hormona de crecimiento en esta población, pues más bien las
pacientes tratadas inicialmente entre los años 2000 y 2004 tuvieron la
mayor ganancia de talla media.

79
Estos resultados pudieran explicarse al menos parcialmente por el
hecho de que las paciente analizadas durante este período fueron
algo más jóvenes que el grupo tratado en el quinquenio siguiente,
aunque esto difiere del resultado del análisis de regresión lineal,
donde la edad de inicio de tratamiento no fue un predictor
estadísticamente significativo de la ganancia de talla.
Ahora bien, esta mayor ganancia de talla media en estas pacientes
tratadas durante este quinquenio no puede explicarse exclusivamente
por el factor edad de inicio de tratamiento, pues al compararla con la
población que inició tratamiento entre los años 2010-2015 no hubo
una diferencia estadísticamente significativa.
Tampoco puede justificarse esta diferencia en ganancia de talla
encontrada en las pacientes que iniciaron tratamiento entre 2000 y
2004 vrs el resto de períodos por el factor dosis de hormona de
crecimiento al final del tratamiento, pues no hubo una diferencia
estadísticamente significativa entre las dosis utilizadas en ninguno de
los períodos.
Es por esto que esta diferencia en ganancia de talla encontrada en las
pacientes que iniciaron tratamiento en el período 2000-2004 debe

80
obedecer a un conjunto de variables no analizadas y que se escapan
al alcance de esta investigación, entre las cuales podrían estar el
tiempo de seguimiento, factores adicionales en la adherencia, edad de
inicio de estrogenización, o bien dosis de hormona de crecimiento
utilizada al inicio y durante el seguimiento.
Estos factores plantean la posibilidad de una nueva investigación a
futuro o bien una extensión de la presente que explore estas
diferencias en ganancia de talla junto con otras variables de interés.
Este estudio cuenta con algunas limitaciones importantes de
mencionar, siendo la principal de ella el no contar con gráficas de talla
específicos para la población costarricense, teniendo que utilizar
gráficas obtenidas de población de los Estados Unidos, con tallas
evidentemente distintas por cuestiones étnicas y alimentarias, entre
otras.
De esta manera y en el entendido de que la población costarricense
fuera más pequeña, posiblemente se subestime un poco el percentilo
y la desviación estándar de talla en esta población, aunque
representada y comparada adecuadamente con el pronóstico de talla
familiar.

81
Otra limitante es el sesgo de detección establecido por la pérdida de
información no disponible, ya sea por circunstancias administrativas
del manejo del expediente o bien errores de digitación que impidieron
contar con toda la información de la base de datos y que de haber
sido incluidos podrían cambiar el análisis de esta población de
pacientes con síndrome de Turner.

82
Conclusiones
1) El servicio de Endocrinología del Hospital Nacional de Niños
cuenta con una base de datos robusta y completa de las pacientes
con síndrome de Turner que llevaron control entre enero 2000 y
diciembre 2017, lo que permite obtener una información muy
amplia sobre las comorbilidades que acompañan a esta población.
2) La mayoría de las pacientes provienen de grandes áreas de
población nacional, si bien podría tratarse de una distribución
normal, también podría reflejar un menor diagnóstico de estas
pacientes en zonas más marginales.
3) Esta población costarricense de pacientes con síndrome de Turner
incluye una proporción mayor de pacientes con cariotipo clásico
45,X lo cual podría reflejar una característica propia de la población
o bien a un subdiagnóstico de mosaicismos.
4) Existe de manera general una proporción adecuada de pacientes
que cumplen con el tratamiento con hormona de crecimiento,

83
reflejando la calidad en el seguimiento de estas pacientes y
también la buena instrucción y compromiso de los padres.
5) El porcentaje de eventos adversos fue bajo y aún más lo fue la
suspensión del tratamiento por efectos adversos, lo que refuerza la
seguridad de la terapia con hormona de crecimiento.
6) La edad promedio de diagnóstico y por ende también de inicio de
tratamiento se encuentra dentro del rango promedio para países
desarrollados, permitiendo una atención pronta de las
comorbilidades de esta población.
7) La talla final de estas pacientes se ubicó en una desviación
estándar de talla poblacional mejor que la talla inicial pero siempre
por debajo del pronóstico de talla familiar, lo cual es esperable de
acuerdo a su condición.
8) No hubo una diferencia en la talla final al evaluar el uso de
hormona de crecimiento de manera temprana ni con la duración
del mismo mayor o menor a 5 años, hallazgo que podría estar

84
relacionado a la reducida cantidad de pacientes que cumplieron
estas características.
9) La duración del tratamiento con hormona de crecimiento, la talla
inicial y el pronóstico de talla familiar fueron factores pronósticos
en esta población para la ganancia de talla, herramienta que
podría utilizarse como apoyo al abordar las expectativas del
paciente y de sus padres.

85
Recomendaciones
1) Aprovechando la base de datos del servicio de endocrinología del
Hospital Nacional de Niños, se podría ampliar la información de estudio,
en uno de seguimiento y a la vez reportar la frecuencia y seguimiento de
otras comorbilidades de esta población.
2) Se debe reforzar, especialmente en áreas rurales, y por medio de
educación médica continua como pueden ser jornadas de actualización,
el diagnóstico del síndrome de Turner, para garantizar la captación y
atención integral de la mayoría de pacientes con esta patología.
3) La información de la base de datos puede ampliarse con información de
aspectos psicosociales de la población con síndrome de Turner, y
generar estudios que permitan abordar este aspecto de la patología.
4) Reforzar los programas de atención farmacéuticos, instancia que permite
a los padres y los pacientes evacuar dudas y promover de esta forma la
adherencia a terapias como la de uso de hormona de crecimiento.

86
5) La institución debe velar siempre por mantener la calidad de los
medicamentos ofrecidos a los usuarios, y los resultados con el uso de
hormona de crecimiento original de este estudio son un ejemplo de este
esfuerzo.
6) Ampliar el análisis de regresión lineal obtenido, pues permite al médico
estimar la ganancia de talla con los factores de más peso para
pronosticar el éxito de la terapia con hormona de crecimiento.
7) Las autoridades competentes deben establecer gráficos de talla y peso
acorde con las características de nuestra población y llevar a cabo los
estudios epidemiológicos necesarios para este fin.
8) Se debe mantener y reforzar la coordinación con los servicios de
endocrinología de adultos en los hospitales nacionales, regionales y
periféricos, a fin de facilitar la transición adolescente-adulto de las
pacientes con síndrome de Turner.

87
Bibliografía
1. Pinsker JE. Turner syndrome: updating the paradigm of clinical care. J Clin
Endocrinol Metab. 2012; 97: E994–E1003.
2. Turner HH. A syndrome of infantilism, congenital, webbed neck and cubitus
valgus. Endocrinology. 1938; 23:566-574.
3. Chih-Ping CH, Shu-Chin CH. Prenatal Sonographic Features of Turner
Syndrome. J Med Ultrasound. 2007; 15:251-257.
4. Frias JL, Davenport ML. Committee on Genetics and Section on
Endocrinology, Health supervision for children with Turner syndrome.
Pediatrics. 2003; 111:692–702.
5. Hook EB. Exclusion of chromosomal mosaicism: tables of 90%, 95% and
99% confidence limits and comments on use. Am J Hum Genet 1977; 29:
94–97.
6. Freriks K, Timmers HJ, Netea-Maier RT, et al. Buccal cell FISH and blood
PCR-Y detect high rates of X chromosomal mosaicism and Y chromosomal
derivatives in patients with Turner syndrome. Eur J Med Genet 2013; 56:
497–501.
7. Cockwell A, MacKenzie M, Youings S, et al. A cytogenetic and molecular
study of a series of 45,X fetuses and their parents. J Med Genet 1991; 28:
151–5.
8. Sas TC, de Muinck Keizer-Schrama SM. Turner’s syndrome: a paediatric
perspective. Horm Res 2001; 56(Suppl 1):38–43.
9. Bronshtein M, Zimmer EZ, Blazer S. A characteristic cluster of fetal
sonographic markers that are predictive of fetal Turner syndrome in early
pregnancy. Am J Obstet Gynecol 2003; 188:1016–20.
10. Donaldson MD, Gault EJ, Tan KW, Dunger DB. Optimising management in Turner
syndrome: from infancy to adult transfer. Arch Dis Child. 2007; 91:513–520.

88
11. Shankar RK, Backeljauw PF. Current best practice in the management of
Turner syndrome. Therapeutic Advances in Endocrinology and
Metabolism.2017; 9:33 – 40.
12. Dulac Y, Pienkowski C, Abadir S, Tauber M, Acar P. Cardiovascular
abnormalities in Turner's syndrome: what prevention? Arch Cardiovasc
Dis. 2008 Jul-Aug; 101(7-8):485-90.
13. Carlson M, Silberbach M. Dissection of the aorta in Turner syndrome. J Med
Genet. 2007:44:745–749.
14. Loscalzo ML, Van PL, Ho VB. Association between fetal lymphedema and
congenital cardiovascular defects in Turner syndrome. Pediatrics. 2005;
115:732–735.
15. Hanew K, Tanaka T, Horikawa R, Hasegawa T, Fujita K, Yokoya S. Women
with Turner syndrome are at high risk of lifestyle-related disease –From
questionnaire surveys by the Foundation for Growth Science in Japan.
Endocr J. 2016 May 31; 63(5):449-56.
16. Lippe B, Geffner ME, Dietrich RB, Boechat MI, Kangarloo H. Renal
malformations in patients with Turner syndrome: imaging in 141 patients.
Pediatrics. 1988 Dec; 82(6):852-6.
17. Ralli M, Turchetta R, Cianfrone G. Hearing Disorders in Turner’s syndrome.
Otolaryngol Open J. 2016; 2:115-119.
18. Szilágyi A, Keszthelyi G, Nagy G, Madléna M. Oral manifestations of patients
with Turner syndrome. Oral Surg Oral Med Oral Pathol Oral Radiol
Endod. 2000 May; 89(5):577-84.
19. Grossi A, Crinò A, Luciano R, Lombardo A, Cappa M, Fierabracci A.
Endocrine autoimmunity in Turner syndrome. Ital J Pediatr. 2013 Dec 20;
39:79.
20. Hankus M, Soltysik K, Szeliga K, Antosz A, Drosdzol-Cop A, Wilk K, Zachurzok
A, Malecka-Tendera E, Gawlik AM. Prediction of
Spontaneous Puberty in Turner Syndrome Based on Mid-Childhood
Gonadotropin Concentrations, Karyotype, and Ovary Visualization: A

89
Longitudinal Study. Horm Res Paediatr. 2017 Dec 22. doi:
10.1159/000485321. [Epub ahead of print].
21. Freriks K, Verhaak CM, Sas TC, Menke LA, Wit JM, Otten BJ, de Muinck Keizer-
Schrama SM, Smeets DF, Netea-Maier RT, Hermus AR, Kessels RP, Timmers
HJ. Long-term effects of oxandrolone treatment in childhood on
neurocognition, quality of life and social-emotional functioning in young
adults with Turner syndrome. Horm Behav. 2015 Mar; 69:59-67. doi:
10.1016/j.yhbeh.2014.12.008. Epub 2015 Jan 3
22. Lesniak-Karpiak K, Mazzocco MM, Ross JL. Behavioral assessment of social
anxiety in females with Turner or fragile X syndrome. J Autism Dev
Disord. 2003 Feb; 33(1):55-67.
23. Tosson H, Rose SR, Gartner LA. Children with 45, X/46, XY karyotype from
birth to adult height. Horm Res Paediatr 2010; 74:190–200.
24. Spiliotis BE. Recombinant human growth hormone in treatment of Turner
syndrome. Therapeut Clin Risk Manag 2008; 4:1177–1183.
25. Binder G. Short stature due to SHOX deficiency: genotype, phenotype, and
therapy. Horm Res Paediatr 2011; 75:81–89.
26. Binder G, Ranke MB, Martin DD: Auxology is a valuable instrument for the
clinical diagnosis of SHOX haploinsufficiency in schoolage children with
unexplained short stature. J Clin Endocrinol Metab 2003; 88: 4891– 4896.
27. Ogata T, Matsuo N, Nishimura G: SHOX haploinsufficiency and overdosage:
impact of gonadal function status. J Med Genet 2001; 38:1–6.
28. Madelung OW: Die spontane Subluxation der Hand nach vorne. Verh Dtsch
Ges Chir 1878; 7:259–276.
29. Rappold G, Blum WF, Shavrikova EP, Crowe BJ, Roeth R, Quigley CA, Ross JL,
Niesler B: Genotypes and phenotypes in children with short stature: clinical
indicators of SHOX haploinsufficiency. J Med Genet 2007;44: 306–313.

90
30. Ranke MB, Pflu¨ ger H, Rosendahl W, Stubbe P, Enders H, Bierich JR, Majewski
F 1983 Turner syndrome: spontaneous growth in 150 cases and review of
the literature. Eur J Pediatr 141:81–88.
31. Menke LA, Sas TCJ, de Muinck Keizer-Schrama SMPF, et al. Efficacy and
safety of oxandrolone in growth hormone-treated girls with Turner
syndrome. J Clin Endocrinol Metab 2010; 95:1151–1160.
32. Quigley CA, Crowe BJ, Anglin DG, Chipman JJ, and U.S. Turner Syndrome
Study Group. Growth hormone and low dose estrogen in Turner syndrome:
results of a United States Multi-Center Trial to near-final height. J Clin
Endocrinol Metab 2002; 87:2033–2041.
33. Gault EJ, Perry RJ, Cole TJ, et al. Effect of oxandrolone and timing of
pubertal induction on final height in Turner’s syndrome: randomised,
double blind, placebo controlled trial. Br Med J 2011; 342:d1980.
34. Ross JL, Quigley CA, Cao D, et al. Growth hormone plus childhood low-dose
estrogen in Turner’s syndrome. N Engl J Med 2011; 364:1230–1242.
35. Carel JC, Mathivon L, Gendrel C, et al. Near normalization of final height with
adapted doses of growth hormone in Turner’s syndrome. J Clin Endocrinol
Metab 1998; 83:1462–1466.
36. Plotnick L, Attie KM, Blethen SL, Sy JP. Growth hormone treatment of girls
with Turner syndrome: The National Cooperative Growth Study
Experience. Pediatrics 1998; 102:479–481.
37. Stephure DK. The Canadian Growth Hormone Advisory Committee. Impact of
growth hormone supplementation on adult height in Turner syndrome:
results of the Canadian randomized controlled trial. J Clin Endocrinol Metab
2005; 90:3360–3366.
38. Davenport ML, Crowe BJ, Travers SH, et al. Growth hormone treatment of
early growth failure in toddlers with Turner syndrome: a randomized,
controlled, multicenter trial. J Clin Endocrinol Metab 2007; 92:3406–3416.
39. Gravholt CH. Clinical practice in Turner syndrome. Nat Clin Pract Endocrinol
Metab 2005; 1: 41–52.

91
40. Linglart A, Cabrol S, Berlier P, et al. Growth hormone treatment before the
age of 4 years prevents short stature in young girls with Turner syndrome.
Eur J Endocrinol 2011; 164:891–897.
41. Bondy CA. Care of girls and women with Turner syndrome: a guideline of
Turner Syndrome Study Group. J Clin Endocrinol Metab 2007; 92:10–25.
42. Baxter L, Bryant J, Cave CB, et al. Recombinant growth hormone for children
and adolescents with Turner syndrome. Cochrane Database Syst Rev 2007;
CD003887.
43. Gravholt CH, Andersen NH, Conway GS, et al. Clinical practice guidelines for
the care of girls and women with Turner syndrome: proceedings from the
2016 Cincinnati International Turner Syndrome Meeting. Eur J Endocrinol
2017; 177: G1–G70.
44. Y.K. van Pareren, S.M. de Muinck Keizer-Schrama, T. Stijnen, et al. Final
height in girls with Turner syndrome after long-term growth hormone
treatment in three dosages and low dose estrogens. J. Clin. Endocrinol.
Metab. 88 (2003) 1119–1125.
45. P. Park, P. Cohen. The role of insulin-like growth factor I monitoring in
growth hormone-treated children. Horm. Res. 62 (Suppl. 1) (2004) 59–65.
46. T. Sas, W. de Waal, P. Mulder, et al. Growth hormone treatment in children
with short stature born small for gestational age: 5-year results of a
randomized, double-blind, dose–response trial. J. Clin. Endocrinol. Metab. 84
(1999) 3064–3070.
47. V.H. Boonstra, N.J. Arends, T. Stijnen, W.F. Blum, O. Akkerman, A.C.
HokkenKoelega. Food intake of children with short stature born small for
gestational age before and during a randomized GH trial. Horm. Res. 65
(2006) 23–30.
48. L. Ibáñez, A. Lopez-Bermejo, M. Díaz, A. Jaramillo, S. Marín, F. de Zegher,
Growth hormone therapy in short children born small for gestational age:
effects on abdominal fat partitioning and circulating follistatin and high-

92
molecular-weight adiponectin. J. Clin. Endocrinol. Metab. 95 (2010) 2234–
2239.
49. N. Wooten, V.K. Bakalov, S. Hill, C.A. Bondy. Reduced abdominal adiposity
and improved glucose tolerance in growth hormone-treated girls with
Turner syndrome. J. Clin. Endocrinol. Metab. 93 (2008) 2109–2114.
50. A. van Teunenbroek, S.M. de Muinck Keizer-Schrama, H.J. Aanstoot, T. Stijnen,
N. Hoogerbrugge, S.L. Drop. Carbohydrate and lipid metabolism during
various growth hormone dosing regimens in girls with Turner syndrome.
Dutch Working Group on Growth Hormone. Metabolism 48 (1999) 7–14.
51. J. Salles, N. Lounis, G. Diene, F. Conte-Auriol, P. Moulin, M. Tauber. Bone
mineralbassessment in patients with Prader–Willi syndrome. Bone 40
(Suppl 1) (2007) 275.
52. G. Mazziotti, A. Bianchi, S. Bonadonna, et al. Increased prevalence of
radiological spinal deformities in adult patients with GH deficiency:
influence of GH replacement therapy. J. Bone Miner. Res. 21 (2006) 520–528.
53. D. Teegarden, W.R. Proulx, B.R. Martin, et al. Peak bone mass in young
women. J. Bone Miner. Res. 10 (1995) 711–715.
54. D.E. Sandberg, L.D. Voss. The psychosocial consequences of short stature:
a review of the evidence. Best Pract. Res. Clin. Endocrinol. Metab. 16 (2002)
449–463.
55. D.E. Sandberg, W.M. Bukowski, C.M. Fung, R.B. Noll. Height and social
adjustment: are extremes a cause for concern and action? Pediatrics 114
(2004) 744–750.
56. E.M. Bannink, J. van Doorn, T. Stijnen, S.L. Drop, S.M. de Muinck Keizer-
Schrama. Free dissociable insulin-like growth factor I (IGF-I), total IGF-I and
their binding proteins in girls with Turner syndrome during long-term
growth hormone treatment. Clin. Endocrinol. (Oxf.) 65 (2006) 310–319.
57. J. Bryant, C. Cave, R.Milne. Recombinant growth hormone for idiopathic
short staturein children and adolescents. Cochrane Database Syst. Rev. 4
(2003) CD004440; (Update in): Cochrane Database Syst. Rev. 3 (2007)
CD004440.

93
58. W. Jeffcoate. Growth hormone therapy and its relationship to insulin
resistance, glucose intolerance and diabetes mellitus: a review of recent
evidence. Drug Saf. 25 (2002) 199–212.
59. N. Friedrich, R. Haring, M. Nauck, et al. Mortality and serum insulin-like
growth factor (IGF)-I and IGF binding protein 3 concentrations. J. Clin.
Endocrinol. Metab. 94 (2009) 1732–1739.
60. C.A. Sklar, A.C. Mertens, P. Mitby, et al. Risk of disease recurrence and
second neoplasms in survivors of childhood cancer treated with growth
hormone: a report from the Childhood Cancer Survivor Study. J. Clin.
Endocrinol. Metab. 87 (2002) 3136–3141.
61. T. Laursen. Clinical pharmacological aspects of growth hormone
administration. Growth Horm. IGF Res. 14 (2004) 16–44.
62. J.O. Jørgensen. Human growth hormone replacement therapy:
pharmacological and clinical aspects. Endocr. Rev. 12 (1991) 189–207.
63. T. Laursen, J.O. Jørgensen, G. Jakobsen, B.L. Hansen, J.S. Christiansen.
Continuous infusion versus daily injections of growth hormone (GH) for 4
weeks in GHdeficient patients. J. Clin. Endocrinol. Metab. 80 (1995) 2410–
2418.
64. D.R. Clemmons. Long-acting forms of growth hormone-releasing hormone
and growth hormone: effects in normal volunteers and adults with growth
hormone deficiency. Horm. Res. 68 (Suppl. 5) (2007) 178–181.
65. A.R. Hoffman, B.M. Biller, D. Cook, et al. Genentech adult growth hormone
deficiency study group, efficacy of a long-acting growth hormone (GH)
preparation in patients with adult GH deficiency. J. Clin. Endocrinol. Metab.
90 (2005) 6431–6440.
66. P. Desrosiers, F. O'Brien, S. Blethen. Patient outcomes in the GH Monitor: the
effect of delivery device on compliance and growth. Pediatr. Endocrinol.
Rev. 2 (Suppl. 3) (2005) 327–331.
67. R.R. Kapoor, S.A. Burke, S.E. Sparrow, et al. Monitoring of concordance in
growth hormone therapy. Arch. Dis. Child. 93 (2008) 147–148.
68. F. Haverkamp, L. Johansson, H. Dumas, et al. Observations of nonadherence
to recombinant human growth hormone therapy in clinical practice. Clin.
Ther. 30 (2008) 307–316.

94
69. J. Müller, N.E. Skakkebaek, B. Jacobsen, et al. Norditropin® SimpleXx™: a
liquid human growth hormone formulation, a pen system and an auto-
insertion device. Horm. Res. 51 (Suppl. 3) (1999) 109–112.
70. J.T. Jørgensen, J. Romsing, M. Rasmussen, et al. Pain assessment of
subcutaneous injections. Ann. Pharmacother. 30 (1996) 729–732.
71. T. Laursen, B. Hansen, S. Fisker. Pain perception after subcutaneous
injections of media containing different buffers. Basic Clin. Pharmacol.
Toxicol. 98 (2006) 218–221.
72. H. Dumas, P. Panayiotopoulos, D. Parker, V. Pongpairochana. Understanding
and meeting the needs of those using growth hormone injection devices.
BMC Endocr. Disord. 6 (2006) 5.
73. M. Oyarzabal, M. Aliaga, M. Chueca, G. Echarte, A. Ulied. Multicentre survey
on compliance with growth hormone therapy: what can be improved? Acta
Paediatr. 87 (1998) 387–391.
74. B. Hansen, A. Bojesen, T. Laursen. The perception of pain after
subcutaneous injection of five different medias containing different
solutions for use in the dissolution human growth homone (GH) in healthy
volunteers. Horm. Res. 58 (Suppl. 2) (2002) 106.
75. O.L. Jens, Jorgensen, N. Moller, T. Lauritzen, K. G. M. M. Alberti, H. Orskov,
Jens S. Christiansen. Evening versus Morning Injections of Growth
Hormone (GH) in GH-Deficient Patients: Effects on 24-Hour Patterns of
Circulating Hormones and Metabolites Journal of Clinical Endocrinology &
Metabolism. 70(1990):207-14.
76. C H Gravholt and others. Clinical practice guidelines for the care of girls and
women with Turner syndrome: proceedings from the 2016 Cincinnati
International Turner Syndrome Meeting European Journal of
Endocrinology (2017) 177, G1–G70.

95
ANEXOS
Anexo 1. Hoja de recolección de datos
FORMULARIO RECOLECCIÓN DATOS
Protocolo de estudio: “Evaluación de talla final vrs pronóstico de talla familiar
en pacientes portadoras de síndrome de Turner en control en el Hospital
Nacional de Niños durante el período comprendido entre enero 2000 y diciembre
2017
Criterio de inclusión
Cumple: Sí o No
Pacientes femeninas con características clínicas de síndrome de Turner y cariotipo 45X o alguno de sus mosaicismos
Criterio de exclusión
Cumple: Sí o No
Paciente femeninas con características clínicas de síndrome de Turner, sin cariotipo realizado
A) Epidemiológicos
1. Clave #:____ Edad: __________ (años)
2. Edad al diagnóstico: ___________________ (años)
3. Provincia: _______________________
4. Fecha de Diagnóstico: ________________

96
B) Antropométricos
1. Peso al final de tx (Kg):________________
3. Talla más reciente: ____ (cms) DS: _____ Percentilo: ______
4. Pronóstico de talla familiar: ________ (cms)
C) Clínicos
1. Cariotipo Clásico: Sí ( ) No ( )
2. Mosaicismo: Sí ( ) No ( ) Tipo: ________________
D) Tratamiento
1. Uso de GH Sí ( ) No ( ) 2. Duración de tx ________ (años)
3. Edad inicio de tx________ (años) 4. Edad al S/S tx________ (años)
5. Talla inicial: ______ (cms) DS: _____________ Percentilo: __________
6. Talla final: _______ (cms) DS: _____________ Percentilo: __________
7. Ganancia de talla (cms) __________
8. Dosis promedio diaria de HCG __________ (mgs) Dosis promedio diaria
HCG por peso: _____________ (mg/kg).
9. Tratamiento: Completó ( ) Continúa ( ) Suspendió ( )
10. Reacciones adversas ( ) Sí ( ) No Especifique:
11. Adherencia como reportada por el médico:
Nombre y Apellido de la persona que recoge los datos:
Fecha de la recolección:
Firma:








