diplomado en validación
TRANSCRIPT

1
Validación
Marzo 2008

2
Plan
• Validación– Propósito – Origen– Objetivos– Definiciones– Marco regulatorio

3
El Principal El Principal ObjetivoObjetivo de la de la ValidaciónValidación eses protegerproteger al al
PACIENTEPACIENTE

4
Pero hay otros beneficios
Un proceso validado es un proceso robusto y capaz, que incrementa la calidad, reduce desperdicios y en consecuencia reduce costos.

5
Origenes• Establecimiento de las cGMP´s (Actuales Practicas
de Fabricación) por parte de la FDA (1963)
• La aplicación de la validación en la industría farmacéutica inició a mediados de los 70´s
• No adquirió carácter obligatorio hasta principios de los 80´s
• Inició enfocada en los procesos más críticos (Parenterales) y fue extendiendose a todos los aspectos relativos a la calidad del producto (Software y certificación de proveedores)

6
Origenes
• En 1987 emite la “Guía de Principios Generales para la Validación de Procesos
• En 1998 la Secretaría de Salud emite la “NOM-059-SSA1-1993” Buenas prácticas de fabricación para establecimientos de la industria químico farmacéutica dedicados a la fabricación de medicamentos.
• En Octubre de 2005 se emite el Proyecto de Norma NOM-059-SSA1-2004, la cual establece (Sección 1.1) los requerimientos mínimos necesarios para el proceso de los medicamentos

7
Validación ¿vs? Calificación

8
Calificación• Definiciones:
– Calificación, a la evaluación de las características de los elementos del proceso.
– Calificación del diseño, la evidencia documentada de que el diseño propuesto de las instalaciones, sistemas y equipo es conveniente para el propósito proyectado
– Calificación de la instalación, la evidencia documentada de que las instalaciones, sistemas y equipo se han instalado de acuerdo a las especificaciones de diseñopreviamente establecidas
– Calificación operacional, la evidencia documentada de que el equipo, las instalaciones y los sistemas operan consistentemente de acuerdo a las especificaciones de diseño establecidas.
– Calificación de la ejecución o desempeño, la evidencia documentada de que las instalaciones, sistemas y equipo, se desempeñan cumpliendo los criterios de aceptación previamente establecidos

9
Validación (Definiciones):– VALIDACIÓN: Es la evidencia documentada que demuestra
que a través de un proceso específico se obtiene un producto que cumple consistentemente con las especificaciones de calidad establecidos. (NOM 059)
– Plan Maestro de Validación, documento que especifica la información para la validación de la compañía, donde se definen detalles y escalas de tiempo para cada trabajo de validación a realizar. Las responsabilidades relacionadas con dicho plan deben ser establecidas.
– Validación Retrospectiva: Aplica a sistemas que han sido utilizados por muchos años y que cuentan con gran cantidad de evidencia de que funcionan adecuadamente. Solo aplicable cuando el proceso ha sido robusto estadisticamente. DESCARTADA

10
Calificación vs Validación• Calificación
– Orientada a verificar y recabar evidencia documental de que un equipo funciona conforme a lo establecido
• Validación– Verifica y recaba evidencia de que un SISTEMA o
PROCESO funciona conforme a lo establecido
Calificación Validación
Pero no a la inversa

11
¿Que debe
validarse?

12
• Métodos Analíticos¿?
¿Que debe validarse?
–Sístemas de información¿?

13
–Sistemas Críticos¿?
¿Que debe validarse?
• Limpiezas¿?

14
• Procesos¿?
¿Que debe validarse?
–Instalaciones
¿?

15
–Equipo
¿?
¿Que debe validarse?
–¿Personal?

16
¿Como validar /calificar todo?
• Plan• Unidades• Pruebas• Resultados• Revisión• Reporte.

17
Costos de Validación
• Por tiempo invertido en:– Documentación– Elaboración de protocolos– Elaboración de PNO´s– Trabajo en campo– Colección de datos– Análisis de la información– Etc.

18
Marco Regulatorio• Normas Oficales Mexicanas
– NOM-059-SSA1-1993, Buenas prácticas de fabricación para establecimientos de la industria químico farmacéutica dedicada a la fabricación de medicamentos.
– PROY-NOM-059-SSA1-2004. Buenas prácticas de fabricación para establecimientos de la industria químico farmacéutica dedicados a la fabricación de medicamentos
– NOM-164-SSA1-1998, Buenas prácticas de fabricación para fármacos
• Internacionales– Normas ISO– Agencias regulatorias nacionales (FDA, INVIMA,
ANVISA, etc.)– OMS

19
Validación de Procesos NO Asépticos
Marzo 2008

20
NOM 059-SSA1-2006
Validación3.82 Validación, a la evidencia documentada quedemuestra que a través de un proceso específico se obtiene un producto que cumple consistentementecon las especificaciones de calidad establecidas(NOM 059)

21
Procesos• Proceso: Método, sistema adoptado para llegar a
un determinado fin (Pequeño Larrouse Ilustrado)• Proceso NO Aséptico
– Aquel que no requiere de condiciones que involucren esterilidad, ejemplos:
• Sintesis de activos• Fabricación Tabletas• Fabricación Cápsulas• Fabricación Suspensiones y Jarabes• Fabricación Cremas y Tópicos• Acondicionamiento Primario• Acondicionamiento Secundario

22
Validación de procesos• Objetivo
– Cumplir expectativas del cliente• Cumplir marco regulatorio• Asegurar calidad de producto• Reducir costo
• Método– Controlar las fases del proceso que son
significativas para la calidad del producto o servicio
– Parte de un programa de calidad

23
• ¿Evidencia documentada?– Da un alto grado de aseguramiento
– La validación NO es absoluta, un proceso validado tiene cierta posibilidad de falla
– Hay que definir cual es el grado aceptable de certidumbre
NOM 059-SSA1-2006

24
NOM 059-SSA1-2006• Protocolos
– Calificación de Diseño (DQ)– Calificación de Instalación (IQ)– Calificación de Operación (OQ)– Calificación de Funcionamiento (PQ)– Validación de Procesos
• Aspectos a considerar al elaborar protocolo:– Personal– Áreas– Materias primas– Equipo– Sistemas generales
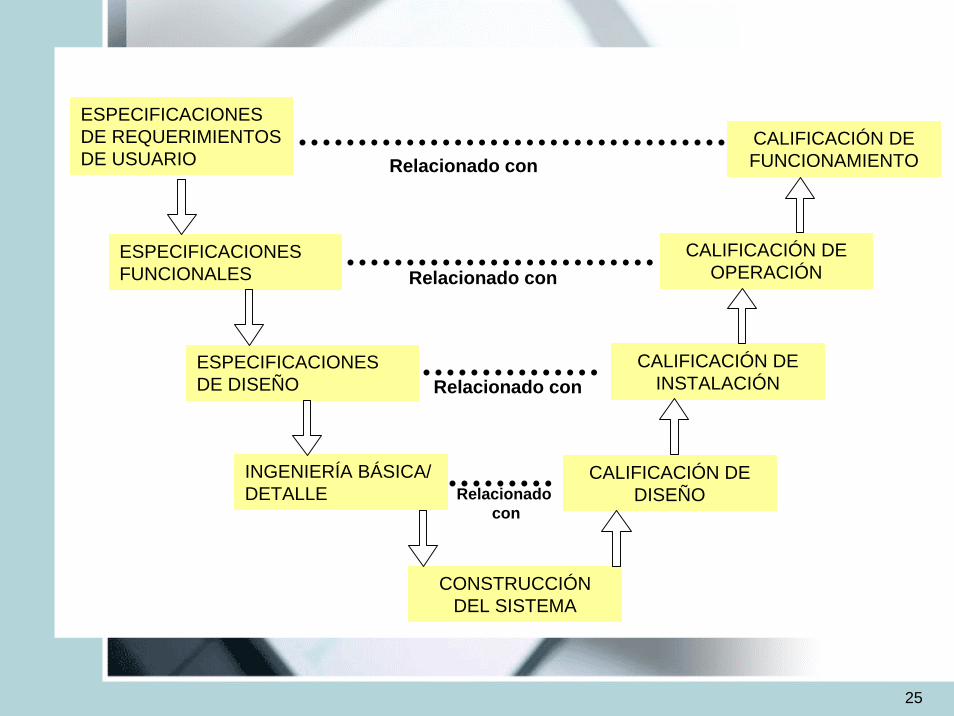
25
CALIFICACIÓN DE FUNCIONAMIENTO
CALIFICACIÓN DE OPERACIÓN
CALIFICACIÓN DE INSTALACIÓN
CALIFICACIÓN DE DISEÑO
CONSTRUCCIÓN DEL SISTEMA
ESPECIFICACIONES DE REQUERIMIENTOS DE USUARIO
ESPECIFICACIONES FUNCIONALES
ESPECIFICACIONES DE DISEÑO
INGENIERÍA BÁSICA/ DETALLE
Relacionado con
Relacionado con
Relacionado con
Relacionadocon

26
Especificaciones de Requerimientos de Usuario
• Tips para elaborarlos– Cada requerimiento enunciado, deberá ser únicamente
referenciado y no más largo de 250 palabras. – Los requerimientos no deberán repetirse o contradecirse– Las ERU deberán expresar requerimientos y no soluciones de
diseño– Cada requerimiento deberá ser susceptible de probarse. – Deberá distinguirse entre requerimientos
mandatorios/regulatorios y características deseadas. – Revisión conjunta entre proveedor y usuario para verificar la
correcta interpretación de los requerimientos

27
Epecificaciones de Requerimientos de Usuario• Tips para elaborarlos
– Evite ambigüedades o lenguaje coloquial. – No deben repetirse o contradecirse– Las ERU deberán expresar requerimientos y
no soluciones de diseño– Cada requerimiento deberá ser susceptible de
probarse.

28
Protocolos
• Objetivo• Alcance• Antecedentes• Documentos de referencia• Procedimiento• Criterio de aceptación.• Responsabilidades• Planeación de la validación• Anexos

29
Protocolo de Validación• Se basa en un conocimiento claro y
profundo del proceso a validar
• Debe indicar un número suficiente de lotes o corridas para demostrar la reproducibilidad
• Se debe cubrir un análisis de riesgo que permita afrontar los retos reales al sistema o proceso

30
Planeación y análisis del proceso de la validación
• Entrada - Proceso - Salida (EPS)• Plan de validación.

31
Calificación de diseño
Objetivo• Asegurar que existe evidencia documental que se
cubre con los criterios de calidad en el diseño del equipo o sistema
• Establecer que sea apropiado para lograr losresultados requeridos de forma segura, confiabley robusta
• Adicionalmente es una forma de registrar losconocimientos adquiridos por el equipo de proyecto y el usuario final
• Debe contar con la revisión del equipo definidoen el PMV

32
Calificación de diseño
• Esta calificación se aplicará en equipos/ sistemas/ procesos nuevos o modificados y consistirá en contrastar el documento de ingeniería básica o las especificaciones técnicas del equipo/ sistema /proceso contra dos aspectos:– Requerimientos de usuarios– Métodos de cálculo– BPX´s y códigos de observancia para cada
empresa

33
Calificación de diseño• Requerimientos • Cualquier necesidad o expectación de un sistema
debe estar detallados y reflejar como se usará el sistema y donde estará localizado
• Punto de partida tanto para la validación, como para la verificación.
• Los requerimientos reflejan las necesidades establecidas o implícitas del cliente.
• Tipos de requerimientos: de diseño, funcionales, de implementación, de desempeño o físicos

34
Calificación de diseño• El desarrollo de los requerimientos incluye la
identificación, análisis, y documentación de la información acerca del dispositivo y su uso propuesto.
• Considerar: distribución de las funciones del sistema, condiciones de operación, características del usuario, riesgos potenciales, y tareas anticipadas.
• Especificar lo siguiente: • Todas el rango de entradas al proceso• Todas los requerimientos de salidas del sistema• Todas las funciones que el sistema desarrollará.

35
Calificación de diseño• Ingeniería básica• Documentación proveniente del proveedor
/diseñador en la fase de ingeniería de detalle paradescribir los detalles de cosntrucción y funcionalidadde la maquina
• Aspectos que deben presentarse en el documento:– Descripción Funcional – Requerimientos de Servicio – Diagramas de Proceso e Instrumentación – Plano general de ubicación – Clave de identificación del equipo– Especificación de componentes Mecánicos– Lista de los lubricantes con posibilidad de contacto
incidental

36
Calificación de diseño• Ingeniería básica• Aspectos que deben presentarse en el documento
(cont.):– Planos eléctricos y neumáticos – Planos de localización de instrumentos – Sistemas de cómputo validados – Especificaciones funcionales de diseño– Especificaciones de diseño de sistemas de cómputo
(Hardware /Software) – Lista preliminar de partes de repuesto

37
Calificación de diseño• Aspectos que deben revisarse en la CD:
– Sistemas de seguridad• Protección de energía elcéctrica almacenada que
provoque accidentes• Protección de superficies calientes• Dispositivos de Bloqueo• Alarmas• Válvulas de alivio
– Aspectos de BPM• De fácil limpieza• Retención de residuos con filtros• Acabados sanitarios sin zonas donde el producto
pueda retenerse

38
Calificación de diseño• Aspectos que deben revisarse en la CD:
– Estándares de construcción• Soldadura• Arreglos• Servicios• Accesos• Materiales de construcción• Acabados
– Requerimientos de usuario• Capacidad• Temperatura• Presión• Volumen • Visibilidad• Cualquier requerimiento adicional

39
Calificación de diseño
• Aspectos que deben revisarse en la CD:– Instrumentos críticos
• Listado de instrumentos críticos con su incertidumbre, tolerancia y rango de operación consistente
– Sistema eléctrico• Diagrama de cableado• Distribución de potencia• Motores• Relevadores

40
Calificación de diseño• Aspectos que deben revisarse en la CD:
– Sistema de control• Cotrolador• Sensores de Instrumentación
– Interfase Hombre-Maquina• Pantallas de programación (Displays)• Niveles de acceso• Alarmas
– Aspectos de mantenimiento• Partes de repuesto• Diagnóstico de fallas• Programa sugerido de mantenimiento
– Documentación de soporte• Manuales

41
Calificación de Instalación• Verificará que todos los criterios de diseño e
instalación hallan sido concluidossatisfactoriamente
• El procedimiento consiste por regla general por un chequeo inicial de la maquina porparte del fabricante (en presencia del validador) bajo sus propios esquemas de revisión y posteriormente se ejecutan laspruebas requeridas en el protocolo del site

42
Calificación de Instalación
• Cada prueba a realizar debe cumplir con un criterio de aceptación
• En caso de sistemas que se construyan en la planta deberá realizarse auditorías aplicando los PNO´s del constructor para verificar la correcta construcción

43
Calificación de Instalación• Deberán asimismo verificarse:
– Características del material de construcción con certificados de análisis
– Calibración vigente y en rango de operación– Sistemas instalados conforme a planos y diagramas– Pasivación
• Toda prueba que no obtenga calificación aprobatoria (No Conformidad) debe documentarse y la calificación no se considerará concluida hasta que se corrija o se justifique que no afectará la calidad del proceso

44
EjemploPruebaNo.
Nombre de la Prueba
Método de Prueba Criterio deAceptación
Aceptado No Aceptado
Iniciales Fecha
1.1
ConexionesEléctricas
Verificar la tensión en conexión, la frequencia, el númerode fase presencia de neutral y tierra en la maquina
Los datos de conexióneléctrica correspondencon los definidos en las especificaciones, en la placa eléctrica de la máquina y con el manual del sistemaeléctrico suministradoManual del SistemaTension 415 V; Frequencia 50 Hz; Fases: 3; Neutral (Si/No) Si; Tierra(Si/No) Si.
RMP 22/Oct//06

45
Calificación de Operación• Debe describir el propósito, método y
criterios de aceptación en detalle de cadaprueba a realizar durante la fase de Calificación de operación
• Incluye:– Pruebas de funcionamiento de operación– Pruebas de paros de emergencia– Sistemas de seguridad– Panel de control– Parámetros vs especificaciones– Pruebas de falta de energía (paro y
rearranque)

46
Calificación de Operación• Estos sistemas se retaran conforme al criterio de
peor de los casos (what if?)• Adicionalmente deberán obtenerse las
condiciones de trabajo normal* (parámetros de operación) sobre las que se redactarán los PNO´s, incluyendo mantenimiento preventivo y limpiezas.
• Otro aspecto a retar son las capacidades máximas y mínimas del equipo
• La capacitación de los futuros operadores del equipo se realiza en esta etapa
* De ser posible se deberían realizar las pruebas con el De ser posible se deberían realizar las pruebas con el productoproducto

47
Calificación de Funcionamiento / Validación de Proceso• Se realiza con operación normal• Tres lotes (o ciclos de operación) como mínimo• Requiere una definición de las variables de
proceso críticas • Establecer intervalos de operación de variables
de control• Realizar con muestreo de nivel alto del producto
que permita definir muestreo requerido para contar con Control estadístico de procesos (CEP) apropiado

48
¿ Qué causa variación ?
Manufac. Tabletas
Entradas –Variables de Operación
Entradas –Variables de Operación
Salidas - medidasde desempeño
Salidas - medidasde desempeño
ProcesoProceso
• Todo puede ser visto como el producto de un proceso.•Los factores que afectan el proceso causan variaciones en el producto de dicho proceso.
Ruido (Causascomunes de Variación)
Ruido (Causascomunes de Variación)
Variables no controlables(Causas especialesde Variación)
Variables no controlables(Causas especialesde Variación)

49
Si no medimos,nunca podremos
mejorar

50
Entendiendo las VariacionesInterpretación de Histogramas
Cambio en el Proceso2 fuentes afectan el proceso

51
• CAUSAS COMUNES– Variación natural, inherente (causada por el diseño del sistema)– Histograma - Muestra una distribución Normal– Run Chart - No hay tendencias - No hay patrones– Un proceso que solo tiene causas comunes de variación se lo
denomina ESTABLE o en control estadístico, es PREDECIBLE
Existen dos causas de variaciones distintas
Histogram
0
2
4
6
8
10
7.3 to <=7.625
7.625 to<= 7.95
7.95 to <=8.275
8.275 to<= 8.6
8.6 to <=8.925
8.925 to<= 9.25
9.25 to <=9.575
9.575 to<= 9.9
Class
# O
bser
vatio
ns
Normal Distribution Mean = 8.5539Std Dev = 0.6469KS Test p-value = .6950
Run Chart
6
6.5
7
7.5
8
8.5
9
9.5
10
10.5
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40
X Axis
Y A
xis

52
• CAUSAS ESPECIALES DE VARIACION– Variación Anormal –Histograma - Muestra una distribución No-normal–Run Chart - Patrones, espinas , tendencias, variaciones / cambiosabruptos–Un proceso bajo la influencia de Causas Especiales de variación se denomina INESTABLE, fuera de control estadístico, IMPREDECIBLE
Existen dos causas de variaciones distintas
Histogram
0
2
4
6
8
10
12
14
16
7.3 to <=8.025
8.025 to<= 8.75
8.75 to <=9.475
9.475 to<= 10.2
10.2 to <=10.925
10.925 to<= 11.65
11.65 to<= 12.375
12.375 to<= 13.1
Class
# O
bser
vatio
ns
Normal Distribution Mean = 9.7779Std Dev = 1.5198KS Test p-value = .0839
Run Chart
6
7
8
9
10
11
12
13
14
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46
X Axis
Y A
xis

53
Centro del Proceso:
2σ=σ
130 140 150 160 170
Distribución normal
n)yy(n
1i
2
i2∑=
−=σ
ny
y i
n
1i=Σ
=
Varianza del proceso:
Nro de veces que se presentó
Desviación standard del proceso:

54
Distribución NormalAncho natural del proceso otolerancias naturales: + 3 sigma
1 sigma 68 %2 sigmas 95 %3 sigmas 99.73 %
¿Por qué medir la calidad en sigmas?

55
Capacidad de un proceso
• La habilidad del proceso de generar productos que se encuentran dentro de los límites de especificación
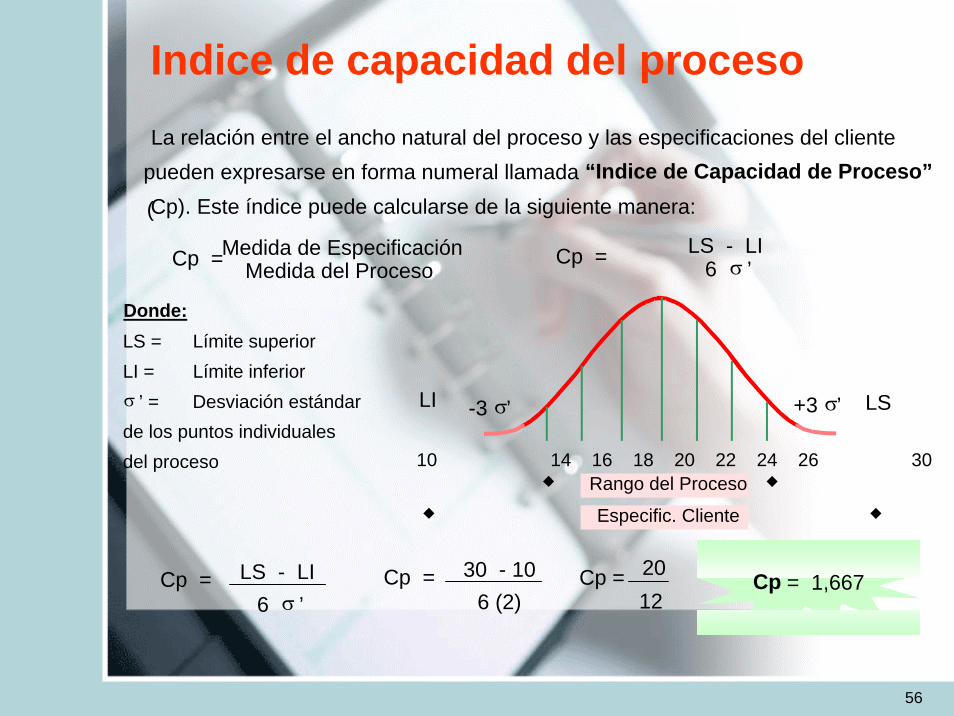
56
Indice de capacidad del procesoLa relación entre el ancho natural del proceso y las especificaciones del cliente
pueden expresarse en forma numeral llamada “Indice de Capacidad de Proceso”
(Cp). Este índice puede calcularse de la siguiente manera:
Cp =Medida de EspecificaciónMedida del Proceso
Cp = LS - LI6 σ ’
Donde:LS = Límite superiorLI = Límite inferiorσ ’ = Desviación estándarde los puntos individualesdel proceso 10 14 16 18 20 22 24 26 30
Rango del Proceso
Especific. Cliente
LI LS-3 σ’ +3 σ’
Cp = LS - LI6 σ ’
Cp = 30 - 106 (2)
Cp = 2012
Cp = 1,667

57
Indice de capacidad del proceso20 40
Cp = 2025
= 0,8
20 40
Cp = 2010
= 2,00
¿Qué es seis sigma?¿Qué es seis sigma?

58
Histogram
0
2
4
6
8
10
7.3 to <=7.625
7.625 to<= 7.95
7.95 to <=8.275
8.275 to<= 8.6
8.6 to <=8.925
8.925 to<= 9.25
9.25 to <=9.575
9.575 to<= 9.9
Class
# O
bser
vatio
ns
Normal Distribution Mean = 8.5539Std Dev = 0.6469KS Test p-value = .6950
Histogram
0
2
4
6
8
10
7.3 to <=7.625
7.625 to<= 7.95
7.95 to <=8.275
8.275 to<= 8.6
8.6 to <=8.925
8.925 to<= 9.25
9.25 to <=9.575
9.575 to<= 9.9
Class
# O
bserv
ati
on
s
Normal Distribution Mean = 8.5539Std Dev = 0.6469KS Test p-value = .6950
Histogram
0
2
4
6
8
10
12
14
16
7.3 to <=8.025
8.025 to<= 8.75
8.75 to <=9.475
9.475 to<= 10.2
10.2 to <=10.925
10.925 to<= 11.65
11.65 to<= 12.375
12.375 to<= 13.1
Class
# O
bser
vatio
ns
Normal Distribution Mean = 9.7779Std Dev = 1.5198KS Test p-value = .0839
Histogram
0
2
4
6
8
10
12
14
16
7.3 to <=8.025
8.025 to<= 8.75
8.75 to <=9.475
9.475 to<= 10.2
10.2 to <=10.925
10.925 to<= 11.65
11.65 to<= 12.375
12.375 to<= 13.1
Class
# O
bserv
ati
on
s
Normal Distribution Mean = 9.7779Std Dev = 1.5198KS Test p-value = .0839
ESTA
BLE
INES
TAB
LE
Buena capacidadMala capacidad
Capacidad vs. Estabilidad (En control estadístico)

59
Validación de Formas Sólidas
Marzo 2008

60
Prerrequisitos• Primero que nada las instalaciones y equipos deben estar
calificados , de modo que aseguremos que todo el equipo y los sistemas de control estan propiamente instalados y funcionan conforme al diseño para soportar cualquier proceso a validar
• Una vez cubierto este aspecto, cada proceso a desarrollar en esa instalación deberá validarse para asegurar que manufactura entregará producto consistentemente
• Finalmente es necesario realizar , especialmente en este caso donde se presentan dificultades adicionales, una validación de la limpieza y sanitización que permita asegurar la eliminación de trazas de activo

61
Aspectos a cubrir en instalacionesServicios Requerimiento
Aire comprimido Sistema validado con al menos la misma calidad y con los mismos requerimientos que el aire del cuarto. Pruebas de integridad de filtros terminales
Agua Purificada Sistema validado con instrumentos calibrados, programa de monitoreo microbiológico /Fisicoqímico. Pruebas de integridad en flitros de venteo
Agua potable Con sistema de cloración y monitoreo microbiológico
Aire Acondicionado /Cuarto
Sistema validado y conforme a la clase E del proyecto NOM 059, programa de monitoreo microbiológico /Fisicoqímico. Pruebas de integridad de filtros terminales

62
Aspectos a cubrir en proceso
Aspecto RequerimientoDispensado Biombos instalados, balanzas calibradas/ verificadas, monitoreos
del área confrome al programa. Procedimientos de limpieza de áreas, accesorios y recipientes de almacenamiento. Condiciones y materiales de recipientes verificados.
Granulación líquida
Tamaño del lote, tipo de mezclador, incluyendo posición de cuchillas, mamparas y desmunuzador, velocidad de impulsor y granulador. Temperatura, cantidad forma y velocidad de adición de la solución aglutinante. Secuencia de carga de materias primas. Método de determinación de fin de granulación: Método de descarga, incluyendo el raspado. Verficación de homogeneidad
Secado Tamaño de lote, volumen, temperatura y humedad del aire a la entrada y en la salida. Temperatura final del producto, tiempo de secado, frecuencia de agitación y contenido de humedad del gránulo

63
Aspectos a cubrir en procesoAspecto RequerimientoCribado y molienda del gránulo
Tamaño de malla, tipo de molino, velocidad y dirección de la rotación, velocidad de alimentación. Sistema de transferencia almezclador. Determinación de la distribución del tamaño de partícula
Mezclador Tamaño del mezclador, forma, velocidad de rotación. Tamaño del lote, métodos de carga y descarga. Procedimiento de premezclado, cuando aplique. Verificación de uniformidad de contenido en varios puntos. Rendimiento
Compresión / Tableteadora
Verificar uniformidad de contenido, de peso, dureza, espesor, friabilidad y desintegración. Muestrear estratificadamente para detectar demezclado. Verificación de fase final de contenido de activos
Velocidad de la maquina, forma de alimentación, parámetros de precompresión y compresión. Punzones y matrices
Sistemas de rechazo con base en monitoreo de fuerzas compactación.
Detector de metales funcional
Rendimiento

64
Aspectos a cubrir en proceso
Aspecto RequerimientoRecubrimiento Tamaño de lote, volumen, temperatura y humedad del aire a la
entrada y en la salida. Temperatura del producto en las diferentes etapas, velocidad de alimentación de las suspensión, presión de aire de atomización, tipo y números de las pistolas, verificación de estado de boquillas. Velocidad de rotación del bombo. Duración de las etapas. Verificación de apariencia, peso, desintegración y disolución
Blister Método y velocidad de alimentaciónVelocidad de la máquina, temperatura y presión de sellado.Integridad del empaque (hermeticidad).Detectores de código de barras, detección de fallas en llenado.Sensores de paro.Equipo para detección de pinholes

65
Validación de Formas Semisólidas
Marzo 2008

66
Aspectos a cubrir en procesoAspecto RequerimientoDispensado Biombos instalados, balanzas calibradas/ verificadas, monitoreos
del área confrome al programa. Procedimientos de limpieza de áreas, accesorios y recipientes de almacenamiento. Condiciones y materiales de recipientes verificados. Manejo de parafina caliente (chaqueta de calentamiento verificada)
Recipientes Auxiliares de grasas
Materiales de manufactura, sellos, perfiles de calentamiento/ enfriamiento (con placebo), velocidad de agitador, tiempos definidos de fundido de grasas pesada y de las de bajo punto de fusión. Desempeño del motor. Procedimiento de Limpieza x
Tanque de proceso
Revisar, material de manufactura de tanque, agitador, sellos y raspadores. Perfiles de enfriamiento/ calentamiento, incluyendo temp. máximas y mínimas de operación. Revisión características de mezclador, parámetros de operación y rangos. Aforo del tanque (o calibrado de celdas de carga). Calibración de instrumentos: termómetros, manómetros, velocidad, consumo eléctrico. Materiales y operación deválvulas. Consumo eléctrico durante el proceso. Revisión de homogeneidad de la mezcla. Limpieza

67
Aspectos a cubrir en proceso
Aspecto RequerimientoMolino coloidal / Mezclador de alta potencia
Material del equipo, revisión de homogeneidad de contenido, control de temperatura. Procedimiento de limpieza
Bahías de limpieza Pruebas de espreado, ciclos/ procesos de limpieza, verificación de trazas, monitoreo microbiológico, drenaje sanitario
Máquina llenadora Limpieza de tubos, consistencia de llenado, detectores de tubo vacio, lectores de código de barras, verificación de sellado, alineación de tubos. Calidad del aire en punto de llenado
Tiempo de almacenamiento
Condiciones de temperatura, almacenamiento y tiempos máximos en los cuales pueda estar el granel sin empacarse

68
Validación de Sistemas Críticos
Marzo 2008

69
¿Cuáles sistemas son críticos?
• Agua purificada• Agua para inyección• Vapor “puro”• Aire comprimido• Aire acondicionado

•• ProcedimientoProcedimiento–– Calificación de Diseño :Calificación de Diseño :
•• Bases de cálculo consistentes a lo requeridoBases de cálculo consistentes a lo requerido•• Especificaciones conforme a las guías de diseño Especificaciones conforme a las guías de diseño
que se establezcan como estandar (FDA, ISO, que se establezcan como estandar (FDA, ISO, ISPE)ISPE)
•• Manejo de Materiales, Códigos, estándares, Manejo de Materiales, Códigos, estándares, Procedimientos de operación y formatos para las Procedimientos de operación y formatos para las verificaciones del sistemaverificaciones del sistema
•• Fijar los instrumentos críticos y verificar sus Fijar los instrumentos críticos y verificar sus característicascaracterísticas
VALIDACIÓN DE SISTEMAS CRÍTICOS

••ProcedimientoProcedimiento–– Calificación de Instalación :Calificación de Instalación :
•• Planos finales del sistema y Manuales de instalaciónPlanos finales del sistema y Manuales de instalación•• Auditorias en el área con base en manuales y Auditorias en el área con base en manuales y
procedimientos con aval deprocedimientos con aval del área de Calidadl área de Calidad•• Verificación de estado final del sistema contra los planosVerificación de estado final del sistema contra los planos•• Realización y reporte de la prueba hidrostáticaRealización y reporte de la prueba hidrostática
VALIDACIÓN DE SISTEMAS CRÍTICOS

•• ProcedimientoProcedimiento–– Calificación de Operación :Calificación de Operación :
•• Verificación funcional del sistemaVerificación funcional del sistema•• Establecimiento de intervalos de operación y AlarmasEstablecimiento de intervalos de operación y Alarmas•• Procedimientos de operación del sistemaProcedimientos de operación del sistema•• Pruebas de Alarmas y ParosPruebas de Alarmas y Paros•• Filtros de venteoFiltros de venteo
VALIDACIÓN DE SISTEMAS CRÍTICOSVALIDACIÓN DE SISTEMAS CRÍTICOS

••ProcedimientoProcedimiento–– Calificación de Funcionamiento :Calificación de Funcionamiento :
•• Establecimiento niveles de controlEstablecimiento niveles de control•• Procedimientos de operación del sistemaProcedimientos de operación del sistema•• Parámetros a seguir por operadores (Bitácoras) y Aseguramiento Parámetros a seguir por operadores (Bitácoras) y Aseguramiento
(Programa de monitoreos)(Programa de monitoreos)
VALIDACIÓN DE SISTEMAS CRÍTICOSVALIDACIÓN DE SISTEMAS CRÍTICOS
Sistema de Agua Destilada

••Puntos a considerarPuntos a considerar–– Verificación de agua potable surtida a la plantaVerificación de agua potable surtida a la planta–– Pretratamiento de agua potable (clorado, suavizado)Pretratamiento de agua potable (clorado, suavizado)–– Filtros de venteo Filtros de venteo (tipo, procedimiento para instalación y (tipo, procedimiento para instalación y
esterilización)esterilización)–– Parámetros para la regeneración de sistemas de Parámetros para la regeneración de sistemas de
tratamiento (Columnas de intercambio iónico)tratamiento (Columnas de intercambio iónico)–– Contar con datos diferenciados del sistema de generación y Contar con datos diferenciados del sistema de generación y
del de distribucióndel de distribución–– Instrumentos calibradosInstrumentos calibrados (TOC y conductímetro (TOC y conductímetro
especialmente)especialmente)–– PNO´s consistentes con el reporte de ValidaciónPNO´s consistentes con el reporte de Validación–– Verificar la drenabilidad del sistemaVerificar la drenabilidad del sistema
VALIDACIÓN DE SISTEMAS DE AGUAVALIDACIÓN DE SISTEMAS DE AGUA

••Puntos a considerarPuntos a considerar–– Capacidad del compresorCapacidad del compresor–– Cuidar que el compresor sea libre de aceiteCuidar que el compresor sea libre de aceite–– Servicios requeridos (agua de enfriamiento)Servicios requeridos (agua de enfriamiento)–– Pruebas y periodos de cambio de filtrosPruebas y periodos de cambio de filtros–– Proceso de regeneración de los secadores de aireProceso de regeneración de los secadores de aire
VALIDACIÓN DE AIRE COMPRIMIDOVALIDACIÓN DE AIRE COMPRIMIDO
76
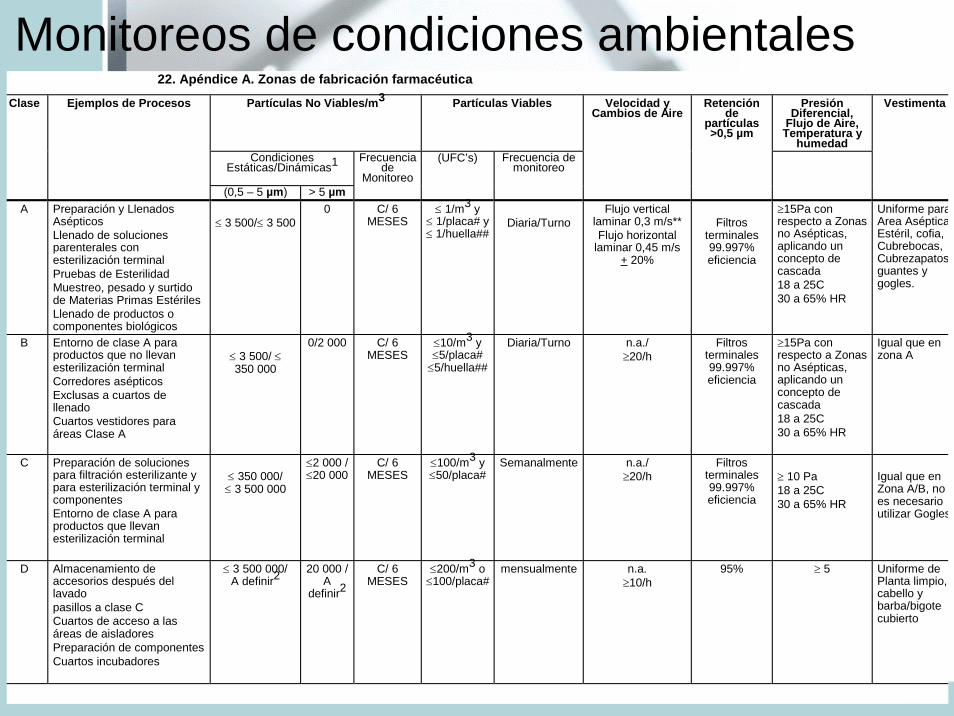
Monitoreos de condiciones ambientales22. Apéndice A. Zonas de fabricación farmacéutica
Clase
Ejemplos de Procesos
Partículas No Viables/m3 Partículas Viables Velocidad y Cambios de Aire
Retención de
partículas >0,5 µm
Presión Diferencial,
Flujo de Aire, Temperatura y
humedad
Vestimenta
Condiciones Estáticas/Dinámicas1
Frecuencia de
Monitoreo
(UFC’s) Frecuencia de monitoreo
(0,5 – 5 µm) > 5 µm A
Preparación y Llenados Asépticos Llenado de soluciones parenterales con esterilización terminal Pruebas de Esterilidad Muestreo, pesado y surtido de Materias Primas Estériles Llenado de productos o componentes biológicos
≤ 3 500/≤ 3 500
0 C/ 6 MESES
≤ 1/m3 y ≤ 1/placa# y≤ 1/huella##
Diaria/Turno
Flujo vertical laminar 0,3 m/s** Flujo horizontal
laminar 0,45 m/s + 20%
Filtros
terminales 99.997% eficiencia
≥15Pa con respecto a Zonas no Asépticas, aplicando un concepto de cascada 18 a 25C 30 a 65% HR
Uniforme paraArea AsépticaEstéril, cofia, Cubrebocas, Cubrezapatosguantes y gogles.
B
Entorno de clase A para productos que no llevan esterilización terminal Corredores asépticos Exclusas a cuartos de llenado Cuartos vestidores para áreas Clase A
≤ 3 500/ ≤350 000
0/2 000 C/ 6 MESES
≤10/m3 y≤5/placa#
≤5/huella##
Diaria/Turno n.a./ ≥20/h
Filtros terminales 99.997% eficiencia
≥15Pa con respecto a Zonas no Asépticas, aplicando un concepto de cascada 18 a 25C 30 a 65% HR
Igual que en zona A
C
Preparación de soluciones para filtración esterilizante y para esterilización terminal y componentes Entorno de clase A para productos que llevan esterilización terminal
≤ 350 000/
≤ 3 500 000
≤2 000 / ≤20 000
C/ 6 MESES
≤100/m3 y≤50/placa#
Semanalmente
n.a./ ≥20/h
Filtros terminales 99.997% eficiencia
≥ 10 Pa 18 a 25C 30 a 65% HR
Igual que en Zona A/B, no es necesario utilizar Gogles
D Almacenamiento de accesorios después del lavado pasillos a clase C Cuartos de acceso a las áreas de aisladores Preparación de componentes Cuartos incubadores
≤ 3 500 000/A definir2
20 000 / A
definir2
C/ 6 MESES
≤200/m3 o≤100/placa#
mensualmente
n.a. ≥10/h
95% ≥ 5 Uniforme de Planta limpio, cabello y barba/bigote cubierto
77
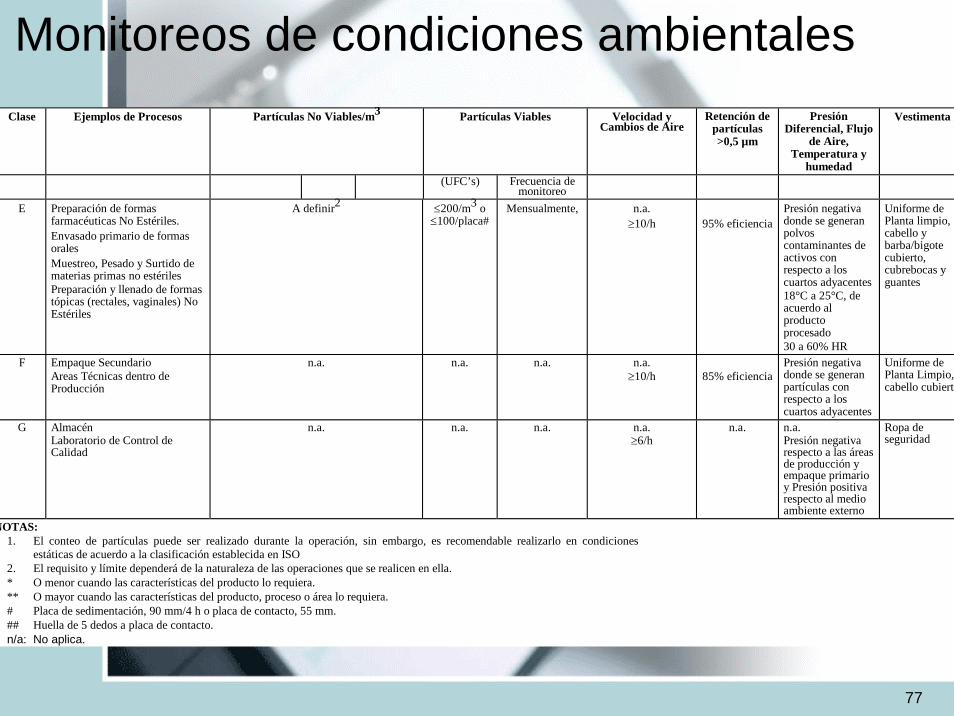
Monitoreos de condiciones ambientalesClase
Ejemplos de Procesos
Partículas No Viables/m3 Partículas Viables Velocidad y
Cambios de Aire
Retención de partículas >0,5 µm
Presión Diferencial, Flujo
de Aire, Temperatura y
humedad
Vestimenta
(UFC’s) Frecuencia de monitoreo
E Preparación de formas farmacéuticas No Estériles. Envasado primario de formas orales Muestreo, Pesado y Surtido de materias primas no estériles Preparación y llenado de formas tópicas (rectales, vaginales) No Estériles
A definir2 ≤200/m3 o≤100/placa#
Mensualmente,
n.a. ≥10/h
95% eficiencia
Presión negativa donde se generan polvos contaminantes de activos con respecto a los cuartos adyacentes 18°C a 25°C, de acuerdo al producto procesado 30 a 60% HR
Uniforme de Planta limpio, cabello y barba/bigote cubierto, cubrebocas y guantes
F Empaque Secundario Areas Técnicas dentro de Producción
n.a. n.a. n.a. n.a. ≥10/h
85% eficiencia
Presión negativa donde se generan partículas con respecto a los cuartos adyacentes
Uniforme de Planta Limpio, cabello cubierto
G Almacén Laboratorio de Control de Calidad
n.a. n.a. n.a. n.a. ≥6/h
n.a.
n.a. Presión negativa respecto a las áreas de producción y empaque primario y Presión positiva respecto al medio ambiente externo
Ropa de seguridad
NOTAS: 1. El conteo de partículas puede ser realizado durante la operación, sin embargo, es recomendable realizarlo en condiciones
estáticas de acuerdo a la clasificación establecida en ISO 2. El requisito y límite dependerá de la naturaleza de las operaciones que se realicen en ella. * O menor cuando las características del producto lo requiera. ** O mayor cuando las características del producto, proceso o área lo requiera. # Placa de sedimentación, 90 mm/4 h o placa de contacto, 55 mm. ## Huella de 5 dedos a placa de contacto. n/a: No aplica.

78
• Mantenimiento de la Validación• Programa de Acabados Sanitarios • Programa de Monitoreo Completo
P. DiferencialesConteo de Partículas viables y no viablesCambios por hora
• Pruebas especialesPruebas de integridad Pruebas de Unidireccionalidad C. de partículas en CNO en áreas estériles
Condiciones Ambientales y Calificación de áreas

79
Monitoreos de condiciones ambientales
• Acabados Sanitarios– Piso
Sin grietas
– TechoSuperficie no descarapelada, sin grietas ni escamasSi separación con marcos de los gabinetes, filtros y rejillas
– LuminariasCon su respectiva micaMicas sin roturas y bien colocadasFuncionando correctamente

80
• Conteo de partículas no viables– Condiciones de muestreo
Condiciones de descanso: sin personal pero con servicios funcionandoCondiciones de operación: con personal, equipos y servicios funcionando
Monitoreos de condiciones ambientales

81
PLAN MAESTRO DE VALIDACIÓNMARZO 2008

82
PLAN MAESTRO DE VALIDACIÓN• Documentos que incluye (14.2.2):
– Política de validación– Estuctura organizacional de las actividades de
Validación (Recursos humanos necesarios)– Resumen de instalaciones, sistemas, equipos y
procesos a validar– Formato a usarse para protocolos y reportes– Planeación y Programación – Control de cambios– Referencia a documentos existentes (políticas /
Normas / códigos a aplicar)

83
Plan Maestro de Validación
• Debe indicar:
– Vigencia– Objetivo– Alcance– Mantenimiento del estado validado

84
Plan Maestro de Validación
• Debe llevar:– Criterios de aceptación– Responsabilidades– Documentación– Programas de validación (crítico)– Autorizaciónes– Revisiones.

85
PLAN MAESTRO DE VALIDACIÓN• Prioridad de validación *
– Parenterales de alto volumen– Parenterales de bajo volumen– Oftálmicos y biológicos– Sólidos Estériles– Sólidos orales de baja
dosificación y alta potencia– Restantes tabletas y cápsulas– Líquidos orales y tópicos
* * Validation of Aseptic Pharmaceutical Processes. F.J. Carleton & Validation of Aseptic Pharmaceutical Processes. F.J. Carleton & J.P. AgallocoJ.P. Agalloco
Procesos Asépticos
Procesos NO Asépticos

86
Ciclo de Validación
• Planeación y análisis del proceso de validación
• Validación Inicial• Control de Cambios• Revisión de la Validación (Frecuencia de
la validación).

87
Comité de Validación
• Multifuncional– Calidad– Ingeniería– Producción– Informática– Logística
• Revisa y aprueba PMV, protocolos y reportes

88
Control de Cambios

89
Control de Cambios
• Es un proceso para realizar cambios quetengan un potencial impacto en la calidadde los materiales o productos, con el fin de asegurar que:
– La calidad del producto, no se vea afectadaadversamente por el cambio
– Cumplimiento de los requerimientos legales y regulatorios
– Involucramiento de todas las áreas– Seguimiento de los cambios.

90
Control de Cambios
–Con el objetivo de lograr losresultados y beneficiosesperados.

91
Control de Cambios
– No deben realizarse cambiosque impacten la calidad sin teneruna revisión formal y aprobaciónpor medio del procedimiento de control de cambios.

92
Control de Cambios:
• Cambios en:– Procedimientos– Registros maestros– Formulaciones– Métodos de prueba– Especificaciones– Materiales (materia prima, materiales de
empaque)– Frecuencias de calibración.

93
Control de Cambios:
• Cambios en:– Sistemas de cómputo– Métodos de limpieza– Métodos analíticos– Areas de manufactura– Servicios críticos– O cualquier otra cosa que pueda afectar la calidad.

94
Control de Cambios
• 1) Necesidad de un cambio• 2) Llenar formato de solicitud• 3) Envío al coordinador de control de
cambios• 4) Registra y envía al comité de control de
cambios• 5) Revisa y autoriza con acciones.

95
Control de Cambios
• 6) Envío al líder del proyecto e involucrados
• 7) Realizar acciones, líder con grupo de trabajo (protocolo, pruebas, etc.).
• 8) Elabora reporte• 9) Aprobación de Aseguramiento y otras
áreas• 10) Realizar el cambio.

96
Formato Control de Cambios
• Debe llevar:– Número de identificación– Tipo de cambio– Título– En que consiste– Situación actual– Justificación– Solicitado por.

97
Buenas y Malas Prácticas de Validación

98
Buenas prácticas de validación
1. Un proceso se considerará adulterado sino se siguen las Buenas Prácticas de Fabricación durante su proceso, sin importar la calidad final del mismo
2. La documentación completa, veraz, clara oportuna y rastreable es la única evidencia histórica que demuestra que el producto se fabricó conforme a las Buenas Prácticas de Fabricación

99
Buenas prácticas de validación
• “Di lo que haces y haz lo que dices”
• “El trabajo no termina hasta que termina la documentación”
• “Si no esta documentado es solo un rumor”

100
Buenas prácticas de validación
3. Cada empresa establece sus propios procedimientos y controles de acuerdo a la naturaleza de sus productos
4. La calidad de los productos debe estar basada en programas que apoyen la operación de manufactura

101
Malas prácticas de validación
• No se da capacitación correcta y documentada al personal
• Investigaciones superficiales y no bien documentadas
• Servicios que no cumplen con los requerimientos operacionales del equipo
• Cableado sin identificar

102
Malas prácticas de validación• Equipos con capacidad insuficiente
• Documentación incompleta al momento de compra de equipos o servicios
• Falta de evaluación de las bases de diseño
• Establecer políticas de validación que no podemos cumplir
• Mala planeación de actividades. Solo realizar una actividad a la vez

103
Malas prácticas de validación• Dar poca importancia a las desviaciones
• No documentar cambios
• Parámetros funcionales fuera de límites
• Alarmas que no funcionan
• Mala selección de tecnología
• Fallas en muestreo:– No hay toma de muestra
– Muestras insuficientes / dañadas