coordinación: m.i.q. alejandro torres aldaco. m.i.q ...148.206.53.84/tesiuami/uam3501.pdf ·...
TRANSCRIPT

MANUAL DE ANALISIS DE AGUAS RESIDUALES
Coordinación:
M.I.Q. Alejandro Torres Aldaco. M.I.Q. Judith Cervantes Ruíz.
Elaboró:
Sergio Esteban Vigueras Carmona.
Marzo 1995.
4

A MI MADRE
S

Objetivos
- Desarrollar un manual de análisis de aguas residuales que incluya todas los rubros industriales. - lmplementar en el laboratorio las técnicas incluidas en dicho manual, para facilitar el análisis y la obtención de resultados.
El trabajo tuvo una duración de 6 meses durante el cual se
- Edición de manual - lmplementación en el laboratorio de las técnicas editadas en el manual
Para llegar a la edición del manual se hizo un análisis de las técnicas implementadas para los diferentes ensayos; dependiendo del rubro industrial del que se trataba, de acuerdo a los lineamientos publicados en el Diario Oficial de la Federación, por la Secretaría de Desarrollo Social (Normas oficiales mexicanas en materia de protección ambiental), del 18 de octubre de 1993.
Se realizó un comparativo entre las técnicas y parámetros sugeridos y10 utilizados en otros países u organismos internacionales, de este comparativo se observó que algunas de las técnicas y/o parámetros se ajustan a los primeros y otros sólo se han implementado en México.
Del análisis realizado durante el trabajo de servicio social se obtuvo el siguiente manual, donde se engloban los 33 rubros industriales manejados por la Secretaría de Desarrollo Social (SEDESOL).
realizó: - Revisión bibliográfica
3

Como usar este manual.
Este manual está dirigido a toda la industria que como generadora de efluentes contaminantes está preocupada por la forma de analizar éstos para así poder establecer mecanismos de prevención y tratamiento de sus contaminantes.
En este manual encontrará la información acerca de todas las técnicas de análisis que son requeridas por la SEDESOL para los diferentes rubros industriales.
La forma de uso que recomendamos para dicho manual es la siguiente:
Primeramente el analista debe saber cual es el rubro al que pertenece su empresa, una vez establecido éste, debe de buscar en las tablas de parámetros limites permicibles cuales son los análisis que se piden para sus efluentes y tomar los parámetros máximos y mínimos para el rubro en particular.
Posteriormente debe dirigirse a la parte de muestre0 e identificar como se debe tratar cada una de sus muestras, esta parte es muy importante para que el analista obtenga buenos resultados en sus determinaciones. Sugerimos tenga mucho cuidado y no omita está sección.
Finalmente debe de ubicar cada una de las técnicas particulares establecidas para sus efluentes y leerlas detenidamente, sugiriendo realizar un diagrama de flujo de la técnica para que este convencido de que la ha entendido correctamente. es importante no omitir ninguno de los pies de página ya que contienen información valiosa a cerca de las técnicas - interferencias especiales, recomendaciones y manejo de algunos materiales y sustancias-.
Estamos seguros que este manual servirá, para facilitar la interpretación de las técnicas a si como de los resultados que se obtienen de éstas.
Por Último quiero a gradecer a M.I.Q. Alejandro Torres y a la M.I.Q. Judith Cervantes por su asesoria y apoyo, y muy especialmente a Patricia y mis hermanas Elia y Laura por su colaboración para la Edición de este Manual.

Contenido .................................................................. lntroducclon .............................................................. Muestre0 ................................................................... Normas oficiales NOM-CCA- ...................................... Potencial hidrógeno .................................................. Materia flotante ......................................................... Sólidos suspendidos totales ...................................... Grasas y aceites ........................................................ Determinación de cobre ............................................ Fósforo total .............................................................. Plaguicidas organoclorados ...................................... Determinación de metales ......................................... Demanda bioquímica de oxígeno ............................... Sólidos sedimentales ................................................. Fenoles ..................................................................... Determinación de Sulfuros ........................................ Cromo hexavalente .................................................... Sustancias activas al azul de metileno ....................... Cloruros .................................................................... Cianuros .................................................................... Cadmio ...................................................................... Floruros ..................................................................... Plomo ........................................................................ Nitrógeno total .......................................................... Arsénico .................................................................... Coliformes totales ..................................................... Apendice
..
Demanda química de oxígeno ....................................
Niquel ........................................................................
Requerlmlentos Titulaciones ...........................................................
Objetivos alcanzados y conclución ............................
. . .....................................................
Bibliografía ................................................................
P& 7 8 9 19 40 43 44 46 49 54 62 66 74 83 85 90 94 99 101 106 124 131 136 140 144 151 154 160
163 187 197 199
7

La situación geográfica de los cuerpos de agua y su distribución a lo largo del tiempo, plantean la necesidad de realizar un manejo desde un enfoque regional, más que de división política o sectorial, por lo tanto, es de suma importancia conocer la situación actual de la explotación y sus posibles consecuencias en su calidad y cantidad, con el fin de establecer criterios de carácter científico para su utilización correcta y racional en el corto, mediano y largo plazo.
Para evitar al mínimo posible la contaminación del agua en el territorio mexicano la Dirección General de Prevención y Control de la Contaminación Ambiental, ha establecido dos programas fundamentales que son: - Vigilancia: Permite vigilar y aplicar la legislación vigente en materia de prevención y control de la contaminación ambiental, con el fin de fijar las condiciones particulares de descarga de acuerdo a los usos, capacidad de asimilación y dilución de los cuerpos receptores de las descargas de agua residuales. - Monitoreo de la calidad del agua: Permite conocer en forma sistemática las alteraciones de la calidad de los cuerpos en el tiempo y el espacio, por medio de las determinaciones analíticas de los parametros físicos, químicos, biológicos, microbiológicos y especiales que sirvan para sustentar sobre bases reales las acciones y estratégias en la materia; así como llevar a cabo la evaluación de su aplicación.
Para cumplir con estos programas se han establecido normas para cada tipo de industria donde se establecen los parámetros que deberán analizar y corregir, en caso necesario, con respecto a las aguas residuales que de sus procesos de producción emanen.
El presente manual- está dirigido a las indutrias pertenecientes a los rubros industriales previsto en el diario oficial de 18 de octubre de 1993, para facilitar el análisis de los parámetros que las leyes mexicanas han establecido y fomentar en lo posible la modificación de los procesos para evitar contaminaciones elevadas resultado de la producción de estos materiales.
MUESTREO.
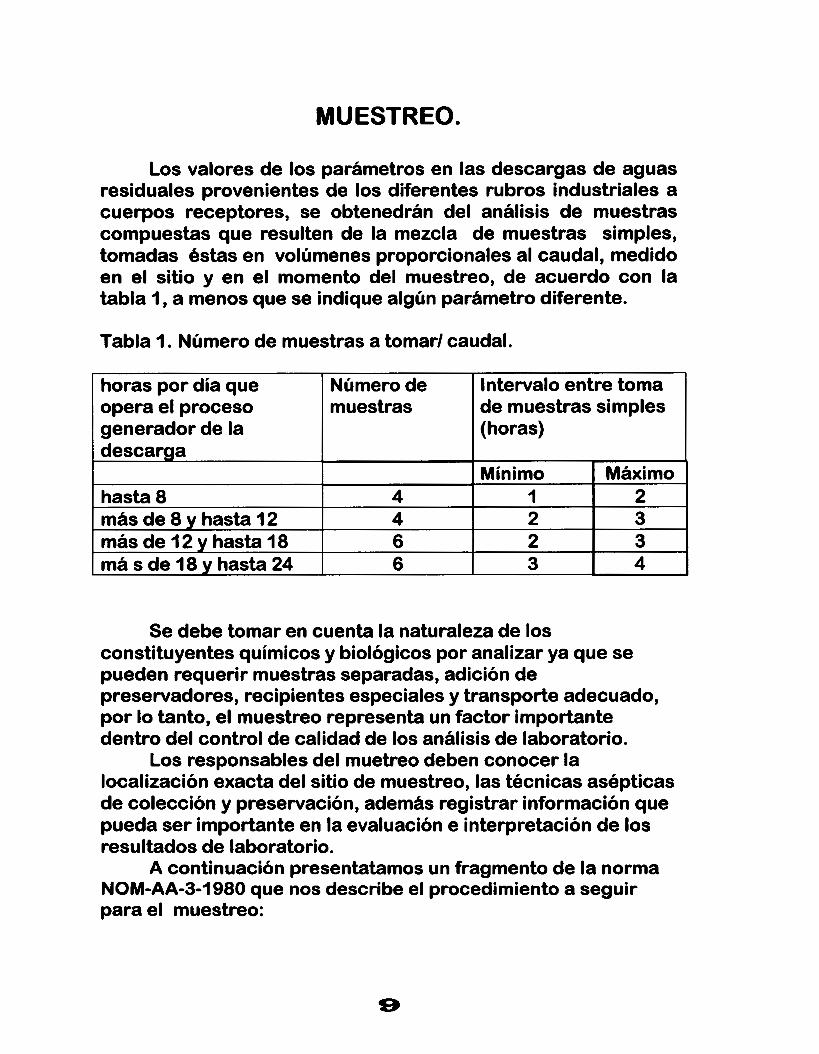
Los valores de los parámetros en las descargas de aguas residuales provenientes de los diferentes rubros industriales a cuerpos receptores, se obtenedrán del análisis de muestras compuestas que resulten de la mezcla de muestras simples, tomadas éstas en volúmenes proporcionales al caudal, medido en el sitio y en el momento del muestreo, de acuerdo con la tabla 1 , a menos que se indique algún parámetro diferente.
Tabla l. Número de muestras a tomarl caudal.
Se debe tomar en cuenta la naturaleza de los constituyentes químicos y biológicos por analizar ya que se pueden requerir muestras separadas, adición de preservadores, recipientes especiales y transporte adecuado, por lo tanto, el muestreo representa un factor importante dentro del control de calidad de los análisis de laboratorio.
Los responsables del muetreo deben conocer la localización exacta del sitio de muestreo, las técnicas asépticas de colección y preservación, además registrar información que pueda ser importante en la evaluación e interpretación de los resultados de laboratorio.
NOM-AA-3-1980 que nos describe el procedimiento a seguir para el muestreo:
A continuación presentatamos un fragmento de la norma

5. PROCEDIMIENTO.
que se aplique a cada caso, debe cumplir los siguientes requisitos.
5.1 .l. Las muestras deben ser representativas de las condiciones que existan en el punto y hora de muestreo y tener el volumen suficiente para efectuar en éI las determinaciones correspondientes.
posible las características del efluente total que se descarga por el conducto que se muestrea.
datos según los incisos 4.1. y 4.2.2. (resumidos en la página 7)
5.1. Cualquiera que sea el método de muestreo específico
5.1.2. Las muestras deben representar lo mejor
5.1.3. AI efectuar el muestreo, deben anotarse los
5.2. Muestreo en tomas. 5.2.1. Se recomienda, se instalen tomas en
conductos a presión o en conductos que permitan el fácil acceso para muestrear al cielo abierto con el objeto de caracterizar debidamente las aguas residuales. las tomas deben tener un diámetro adecuado para muestrear correctamente las aguas residuales en función de los
longitud posible, y procurar situarlas de tal manera que las muestras sean representativas de la descarga. Se recomienda el uso de materiales similares a los del conducto, acero al carbón o de acero inoxidable.
igual a 1 O veces el volumen de la muestra y a continuación se llena el recipiente de muestreo.
materiales que puedan contener, debe ser de la menor
5.2.2. Se deja fluir un volumen aproximadamente
5.3. Muestreo en descargas libres. 5.3.1. Cuando las aguas residuales fluyan libremente
en forma de chorro, debe emplearse el siguiente procedimiento.
5.3.1 .l. El recipiente muestreador debe enjuagarse varias veces antes de efectuar el muestreo. 5.3.1.2. Se introduce el recipiente muestreador en la descarga o de ser posible, se toma
directamente la muestra en su recipiente.
10

5.3.1.3. La muestra se transfiere del recipiente muestreador al recipiente para la muestra cuidando de que éSta siga siendo represen- tativa
5.4. Muestre0 en canales y colectores. 5.4.1. Se recomienda tomar las muestras en el centro
del canal o colector de preferencia en lugares donde el flujo sea turbulento a fin de asegurar un buen mezclado.
5.4.1 .l. Si se va evaluar contenido de grasas y aceites debe de tomar porciones, a diferentes profundidades, cuando no haya mucha turbulencia para asegurar una mayor representatividad.
5.4.2. El recipiente muestreador se debe enjuagar repetidas veces con el agua por muestrear antes de efectuar el muestreo.
5.4.3. El recipiente muestreador, atado con una cuerda y sostenido por la mano de preferencia enguantada, se introduce en el agua residual completamente y se extrae la muestra.
debe cuidar que éSta siga siendo representativa. 5.4.4. Si la muestra se transfiere de recipiente, se
TIPOS DE MUESTRAS
El muestreo debe reflejar un cierto grado de estabilidad en las condiciones durante el período que se recolectan las muestras; de otro modo los resultados serán una muestra heterogénea sin significado alguno. Para el efecto los tipos de muestra se clasifican en:
Muestra simple: Es una porción de agua tomada de un cuerpo receptor en una descarga de aguas residuales. El análisis de una muestra simple, permite conocer la concentración de los componentes del agua a la hora y lugar en que se toma la muestra.
Muestra promedio: Se obtienen a partir del promedio aritmético de los volúmenes de varias muestras simples

colectadas a diferentes intervalos de tiempo, proporcionando un promedio estadístico de la calidad del agua.
Muestra compuesta: Es el resultados de la mezcla de los volúmenes de varias muestras simples. Se pueden tener dos tipos de muestras compuestas, la primera es mezclando varias muestras simples de igual volumen recolecctadas en diferentes sitios sobre la sección transversal y10 distintas profundidades, la segunda, se obtiene mezclando muestras simples según la relación de volumen de flujo a la hora de tomar dichas muestras con el flujo total acumulado, mediante las expresiones :
suma de los flujos de cada muestra Qp = urn_
número de muestras
donde: VR = Volumen de muestra por unidad de gasto (el gasto y
el flujo representan lo mismo, vienen dados en unidad de volumen sobre tiempo)
VT = Volumen total de muestra Qp = Gasto promedio (flujo promedio) N = Número de muestras
donde: VMi = Contribución de volumen total de la muestra i
Ejemplo:
Se desea preparar una muestra compuesta de 4 litros, a partir de cuatro muestras individuales.
El gasto de cada una de ellas en el momento de colectarlas es de 40, 30, 50 y 40 I/s, respectivamente, aplicando

la expresiones anterioriores, se obtiene los militros que deben tomarse para preparar los 4 litros de muestra compuesta.
40 I/s + 30 I/s + 50 Ils + 40 I/s Qp=- = 40 I/s
4
4000 ml
40 I/s 4 vR= = 25 ml S/ I
V M ~ = (25 ml S/ I ) (40 I/s) = 1000 ml V M ~ = (25 ml S/ I ) (30 I/s) = 750 ml V M ~ = (25 ml S/ I ) (50 Ils) = 1250 ml V M ~ = (25 ml S/ I ) (40 Ils) = 1000 ml
Tratamiento de las muestras
A continuación se especifican los tratamientos que se le deben de dar a las diferentes muestra a analizar, dependiendo del tipo de ensayo, si para la técnica a desarrollar no existe una especificación en esta sección sólo se deberá realizar el muestreo indicado y de preferencia analizar las muestras lo más pronto posible o mantenerlas en refrigeración durante ese tiempo
En general el volu'men total para analizar los parámetros fisícuquimícuses de 3 litros, sin embargo, podemos obtener de acuerdo a los análisis fisicoquimicos a realizar una cantidad menor de muestra, éstas deben ser tomadas en frascos de polietileno perfectamente limpios, preservándolos en refrigeración a 4"C, la cantidad muestreada será utilizada para los análisis de pH, sólidos supendidos totales y sólidos sedimentables.
Para realizar el muestreo se puede realizar directamente en la superficie del cuerpo receptor, si las condiciones lo permiten, enjuagando previamente el frasco con el agua a muestrear. Si por las caracteristicas del sitio de muestreo no es

posible hacerlo así, se hace uso de los muestreadores Kemmerer, Winkler, botellas Van D'orn, entre otros; si no se cuenta con ellos, los recipientes pueden ser llenados auxiliándose con cubeta y embudo.
En caso de que las muestras sean tomadas de colectores estos deben tener una válvula de cierre que permita el libre paso de las aguas residuales y de los materiales que pueden contener y proporcionar el cierre hermético de la toma. Esta válvula y los accesorios necesarios para su instalación, deben de ser de materiales similares a los de las tomas ylo los conductos en que éstas se instalen.
Los parámetros que requieren un tratamiento especial de la muestra se enlista a continuación. Es importante que siga cada una de las sugerencias para el tratamiento de las muestras ya que si no se observan éstas existiran interferencias en la técnica que no podrán ser controladas.
DBO. La muestra no debe estar en contacto con el aire, o agitada, puesto que cualquiera de estos factores causaría un cambio en el contenido gaseoso. Las aguas superficiales son muestreadas en botellas para demanda bioquímica de oxígeno, de cuello angosto cónico, con tapón esmerilado de vidrio de 300 ml; se deben de evitar las burbujas en la botella al tomar la muestra, por lo tanto, se sumerge totalmente la botella en otra corriente tapándola inmediatamente después de la toma de muestra.
El uso del muestreador tipo Kemmerer modificado es recomendado para muestreos de más de 1.5 m de profundidad. En este caso la muestra es tomada por un orificio del muestreador que esta comunicado, a la botella de demanda bioquímica de oxígeno, por un tubo que llega a la boca de la misma. Se deben evitar turbulencias.
Conservacíón de la muestra, proceder a la determinación tan pronto como sea posible; de no ser así, la muestra puede conservarse hasta por 8 hr fijando el oxígeno en campo mediante la adición de un mililitro de solución de floruro de potasio, 2 ml de sulfato manganoso, 2 ml de solución alcalina- ioduro-nitruro y 2 ml de ácido sulfúrico de acuerdo a la especificación que se describe en el procedimiento para determinar el oxígeno disuelto.

Determinación de cobre método de la neucoproina. La muestra se deben colectar en frascos de polietileno, preservar añadiendo 5 ml de acido nitric0 concentrado por litro de muestra colectada y refrigerándose a 4 o 5" C.
Coliformes totales y fecales. El procedimiento para la recolección de las muestras de agua para el análisis bacteriológico, depende del tipo de agua que se desee muestrear.
Muestre0 en cuerpos receptores. Las muestras para el análisis bacteriológico, se deben tomar en frascos muestreadores que se hayan lavado con extremo cuidado y esterilizado. En su interior colocar, previo a la esterilización, 0.1 ml de solución triosulfato de sodio al 1% con propósito de inhibir la acción del cloro que pueda contener la muestra, cubriendo el tapón del frasco hasta el cuello con papel aluminio.
Siempre que sea posible, llenar el frasco a 2/3 partes de su capacidad; una cantidad menor sería insuficiente, si fuera mayor, disminuiría el espacio de aire disponible, necesario para hornogenizar la muestra. Las muestras deben de ser representativas del agua en estudio y así mismo no deben contaminarse en forma alguna.
El frasco donde se colecta la muestra no se debe destapar sino hasta el momento donde se efectúe el muestreo. AI muestrear, se debe evitar que el cuello del frasco se ponga en contacto con los dedos o con cualquier otro contaminante.
El examen de la muestra colectada debe realizarse lo más pronto posible, para evitar proliferación o muerte de las bacterias. Cuando el examen se practica dos horas después de tomar la muestra, los resultados empiezan a ser inciertos.
El volumen de muestra suficiente para efectuar el análisis bacteriológico, de preferncia debe de ser aproximadamente 100 ml. Es importante que todas las muestras esten acompañadas de datos completos y exactos de identificación y descripción. N mecanismo de muestreo supeficia/ es e/ siguiente:
Quitar el papel aluminio del cuello del frasco; introducir el frasco aproximadamente 30 ml bajo la superficie del agua.
Despar el frasco dentro del agua. La boca del enbase debe quedar en sentido contrario al flujo de la corriente. Si no existe corriente, en dirección opuesta al movimiento de la mano.
I5

Una vez que la muestra ocupe el volumen correspondiente al frasco (2/3 partes); tapar sin sacarlo del agua teniendo cuidado de que el papel aluminio vuelva a cubrir el cuello de la botella.
Si no es posible la recolección de muestras en las condiciones antes mencionadas, fijar un lastre al frasco, al que se hace decender en el agua.
Para tomar muestrasprofundas en lagos o embalses; usar aparatos especiales que permitan destapar mecánicamente el frasco debajo de la superficie.
Muestre0 en pozos y grifos. Si el pozo está provisto de bomba de mano, bombear durante 5 min. para que el agua fluya libremente, antes la muestra.
Si el pozo está dotado de bomba mecánica, tomar la muestra en una botella previamente flameada de la descarga, dejando que fluya el agua libremente derante 5 minutos antes de timar la muestra.
AI efectuar este muestreo, se debe evitar que el agua escurra fuera del frasco; además se deben de flamear los bordes del frasco y tapón durante el tiempo que dura el muestreo. Esto se hace con objeto de mantener al máximo las condiciones de asepsia.
Si no se cuenta con equipo de bombeo, tomar la muestra directamente del pozo por medio de un frasco estéril con lastre. En este caso se debe evitar la contaminación de la muestra por las natas superficiales.
Si se trata de tomar una muestra de in grifo del sistema de servicio, flamear el grifo y abrirlo completamente, dejando que el agua fluya por 2 ó 3 minutos o el tiempo suficiente para permitir la purga de la línea.
En el momento del muestreo, restringir el flujo de la llave para que se pueda llenar el frasco sin salpicaduras.
Las condiciones de asepsia se debe evitar que el agua escurra fuera del frasco; además, se deben flamear los bordes del frasco y tapón durante el tiempo que dura el muestreo. Esto se hace con objeto de mantener al máximo las condiciones de asepsia.

Niquel. Las muestras se colectan en frascos de plástico. En caso de no efectuar de inmediato el análisis, preservarlas añadiendo 5 ml de ácido nítrico concentrado por litro de muestra. Guardar por un máximo de 6 meses.
Plomo. La muestra se debe colectar en frascos de polietileno y preservarse añadiendo ácido nítrico hasta obtener un pH de 2 a 2.5.
Cadmio. Las muestras se preservan añadiendo ácido nítrico diluido ?:I hasta neutralización, y adicionar un execeso de 5 ml de ácido nítrico concentrado por litro de muestra.
Cianuros. Conservaciún de las muestras. Los agentes oxidantes
tales como cloro, descomponen a la mayoría de los cianuros. Hagase una prueba colocando una muestra en una tira de papel ioduro de potasio (KI) -almidón. Si aparece una decoloración azulada, añadir unos cuantos cristales de tiosulfato de sodio (Na2S203) a la muestra, agitar y repetir la prueba. Si es necesario repetir el procedimiento hasta que no haya decoloración en el papel de prueba; después añadir 0.1 g de Na2S203 al resto de la muestra.
El dióxido de magneso, cloruro de nitrosilo, etc., pueden también decolorar el papel de prueba. De ser posible, llevar a cabo el procedimiento anterior antes de proceder a la conservación de la muestra como se describe posteriormente. Cuando la siguiente prueba indique la presencia de sulfuro, no deben esperarse agentes oxidantes en la muestra.
El sulfuro convertiría rápidamente el CN- en CNS-, especialmente a volres elevados de pH. Probar si hay S2- presente colocando una gota de muestra en papel de prueba de acetato de plomo que haya sido remojado previamente en solución buffer de Acid0 acético, pH4. El obscurecimiento del papel indica la presencia de S2-. Añadir nitrato de cadmio en polvo, Cd (N03)2 4H20, para obtener un precipitado amarillo de sulfuro de cadmio (CdS). Si el contenido de S2- es elevado,

añadir carbonato de cadmio (CdCOS), carbonato de plomo (pbCOS), acetato de plomo o citrato de bismuto. Repetir esta operación hasta que una gota de la muestra previamente tratada no cause obcurimiento en la prueba de papel acidificado de acetato de plomo. Filtrar la muestra para estabilizarla antes de elevar el pH. Cuando se sospeche que existen complejos cianuros-metálicos en forma de partículas, filtrar la solución antes de separar el S2-. Homogeneizar las partículas antes del análisis.
Debido a que la mayoría de cianuros son muy reactivos e inestables, analizar las muestras tan pronto como se pueda. Si la prueba no puede analizarse inmediatamente añadir lentejas de NAOH 6 solución concentrada de NAOH para elevar el valor del pH de la muestra a 12-12.5 y almacenar en un frasco ámbar bien cerrado, en un lugar fresco. Para analizar CNCI, colectar una muestra por separado y omitir la adición de NAOH, debido a que el CNCI se convierte rápidamenten CNO- a valores elevados de pH. Hacer la determinación inmediatamente después del muestreo.
Plaguicidas organoclorados. La muestra se toma únicamente en la suoerficie del cuerpo receptor que se va a analizar. En caso de muestras de aguas profundas se debe utilizar para el muestreo frascos Winkler.
Concervar durante el transporte a 4°C.
Cobre. Las muestras se deben colectar en frasco de polietileno, preservar añadiendo 5 ml de ácido nítrico cocentrado por litro de muestra colectada y refrigerar a 4 o 5OC.

NORMAS OFICIALES MEXICANAS EN MATERIA DE PROTECCION AMBIENTAL (NOM-CCA-
NNN1-ECOUl993)
En esta sección se muestran los parámetros a considerar en el análisis de aguas residuales. Se edita la tabla de la norma correspondiente y se especifican los parámetros mínimos y máximos permisibles por rubro industrial. Además se adicionan las tablas de muestreo para aquellos rubros que no correspondan a los parámetros de muestreos de la tabla 2 (sección de muestreo).
NOM-CCA-001-ECOU1993 Centrales Termoeléctricas Convencionales.
PARAMETROS LIMITES MÁXIMOS PERMISIBLES . I 1 PROMEDIO DIARIO
INSTANTANEO
pH (unidades de I 6 - 9 I 6 - 9 pH)
Sólidos suspendidos
Grasas y aceites totales (mg/l)
80 60
(mg/l)
12 I O Fósforo total I .2 I .o Fierro (mg/l) 1 .o 0.8 Cobre (mgll) 18 15
(mg/l) Zinc (mgll) 2.4 2.0 Bifenilos
policlorados(mg/l) ausente ausente
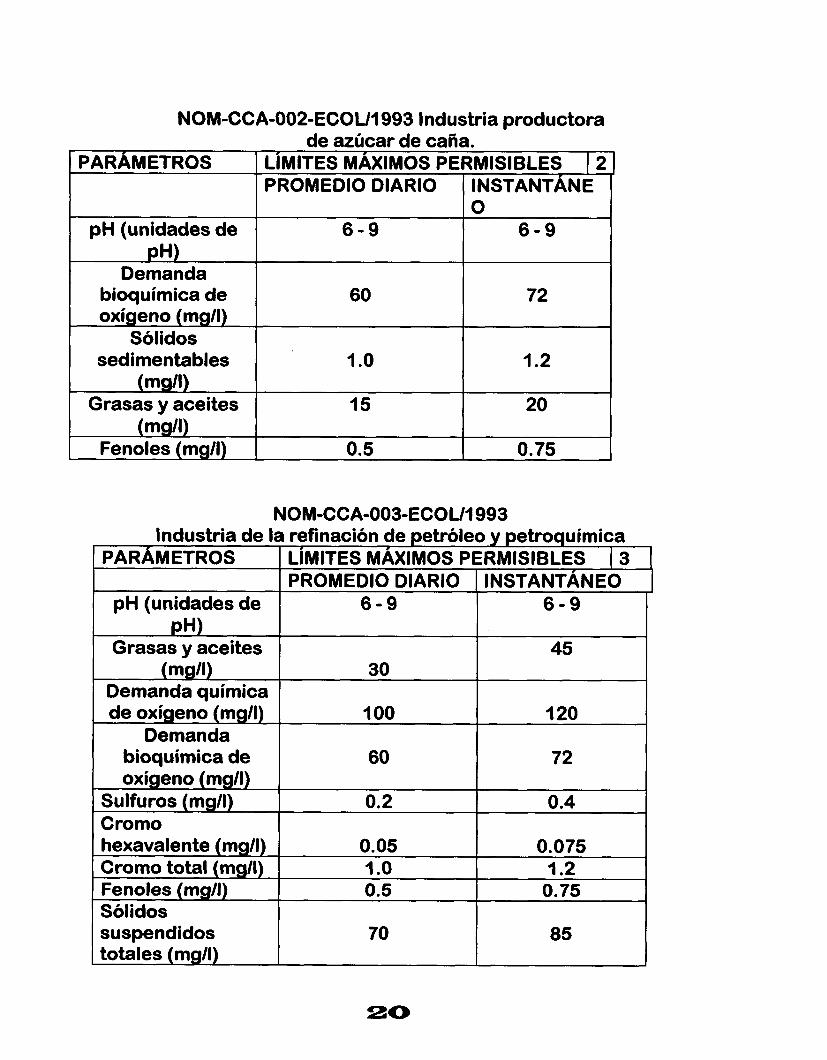
NOM-CCA-002-ECOU1993 Industria productora de azúcar de caña.
PARAMETROS LIMITES MÁXIMOS PERMISIBLES 12 I PROMEDIO DIARIO I INSTANTANE
pH (unidades de I 6 - 9 6 - 9 pH)
Demanda bioquímica de I 60 72 oxígeno (mg/l)
Sólidos sedimentables I I .o 1.2
(mg/l) Grasas y aceites 20 15
( mg/l) Fenoles (mg/l) 0.5 O. 75
NOM-CCA-003-ECOU1993 Industria de la refinación de petró
I PARAMETROS I LIMITES MAXIMOS I PRoMED~oD~ARIO
1 y petr;-;ími;; ,, RMlSlBLES INSTANTANEO
45
120
72
0.4 I
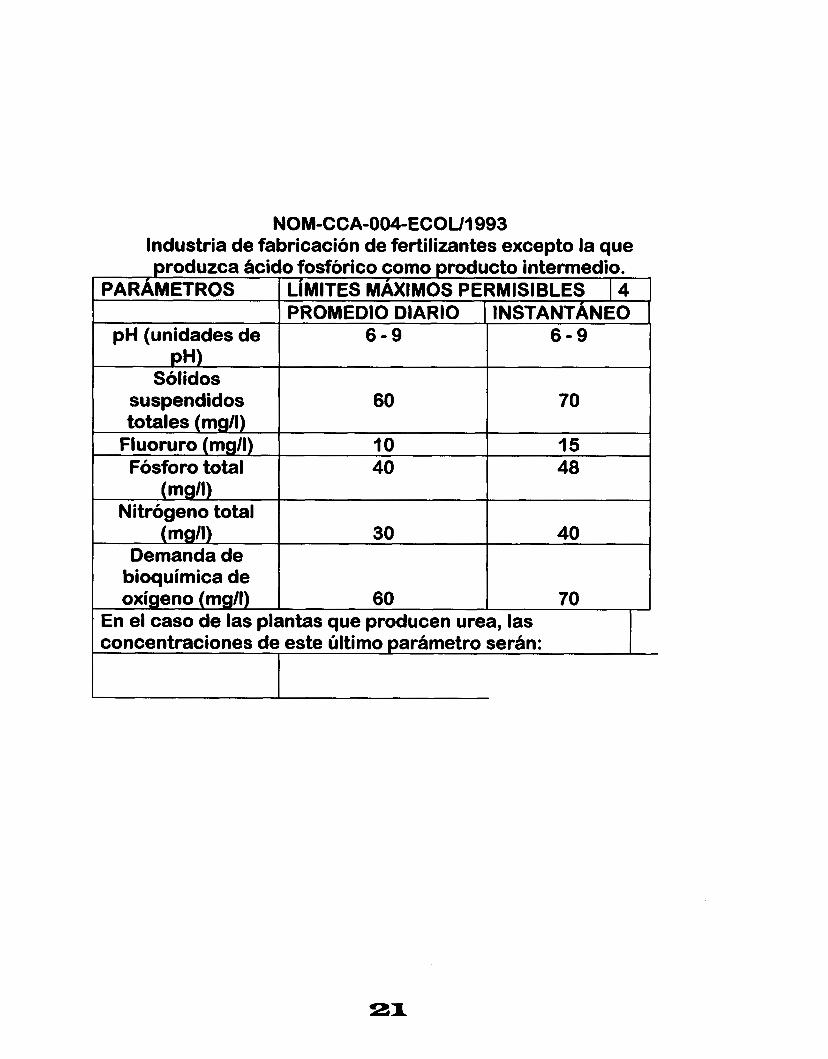
NOM-CCA-OWECOUI 993 Industria de fabricación de fertilizantes excepto la que produzca ácido fosfórico como producto intermedio.
LIMITES MAXIMOS PERMISIBLES 14 PROMEDIO DIARIO I INSTANTÁNEO
PARAMETROS I I
pH (unidades de 6 - 9 6 - 9 pH)
Sólidos suspendidos 70 60 totales (mg/l)
Fluoruro (mgll) 10 15 Fósforo total 40 48
(mgll) Nitrógeno total
(mg/l) 40 30 Demanda de
bioquimica de oxígeno (mg/l) 70 60
En el caso de las plantas que producen urea, las concentraciones de este último oarámetro serán:

NOM-CCA-005-ECOUI 993 Industria de fabricación de productos plásticos
PARAMETROS
pH (unidades de pH)
Sólidos suspendidos totales (mg/l)
Grasas y aceites( mgll)
Sólidos sedimentables(mg
/I) Fluoruros (mg/l)
Demanda química de oxígeno (mgll)
Demanda bioquímica de oxígeno (mg/l) Fenoles (mg/l)
y polimeros sintéticos LIMITES MÁXIMOS PERMISIBLES I 5 PROMEDIO DIARIO
6 - 9 INST~I~NEO ,
70 84
15 20
1 .o 1.2
10 15
200
120 1 O0
240
I
0.5 O. 75 1
NOM-CCA-006-ECOUI 993 Industria de fabricación de harinas.
PARAMETROS . LIMITES MAXIMOS PERMISIBLES I 6 PROMEDIO DIARIO INSTANTANEO
. pH (unidades de
180 Sólidos
6 - 9 6 - 9 pH)
suspendidos 150 totales (mg/l)
Sólidos I .2 sedimentables 1 .o
(mg/l) Demanda
bioquímica de 150 oxígeno (mg/l)
180

NOM-CCA-007-ECOUI 993 Industria de la cerveza y de la malta.
PARAMETROS I LIMITES MAXIMOS PERMISIBLES I 7
pH (unidades de pH)
Sólidos suspendidos
PROMEDIO DIARIO INSTANTÁNEO 6 - 9 6 - 9
I 180 150
totales (mg/l) Sólidos I .2
sedimentables I .o (mgll)
Grasa y aceites 36 30 (rng/l)
Demanda 180 bioquímica de oxígeno (mgll)
150
NOM-CCA-008-ECOUI 993 Industria de fa1
I PARAMETROS ,
t7T pH (unidades de
Sólidos suspendidos totales (mg/l)
Grasas y aceites (mg/l)
Demanda química de oxígeno (mg/l)
Demanda bioquímica de oxígeno (mg/l)
ricación de asbestos de construción. LIMITES MÁXIMOS PERMISIBLES I 8 PROMEDIO DIARIO I INSTANTÁNEO
6 -9 6 - 9
60 70
10
70 60
120 1 O0
15
23
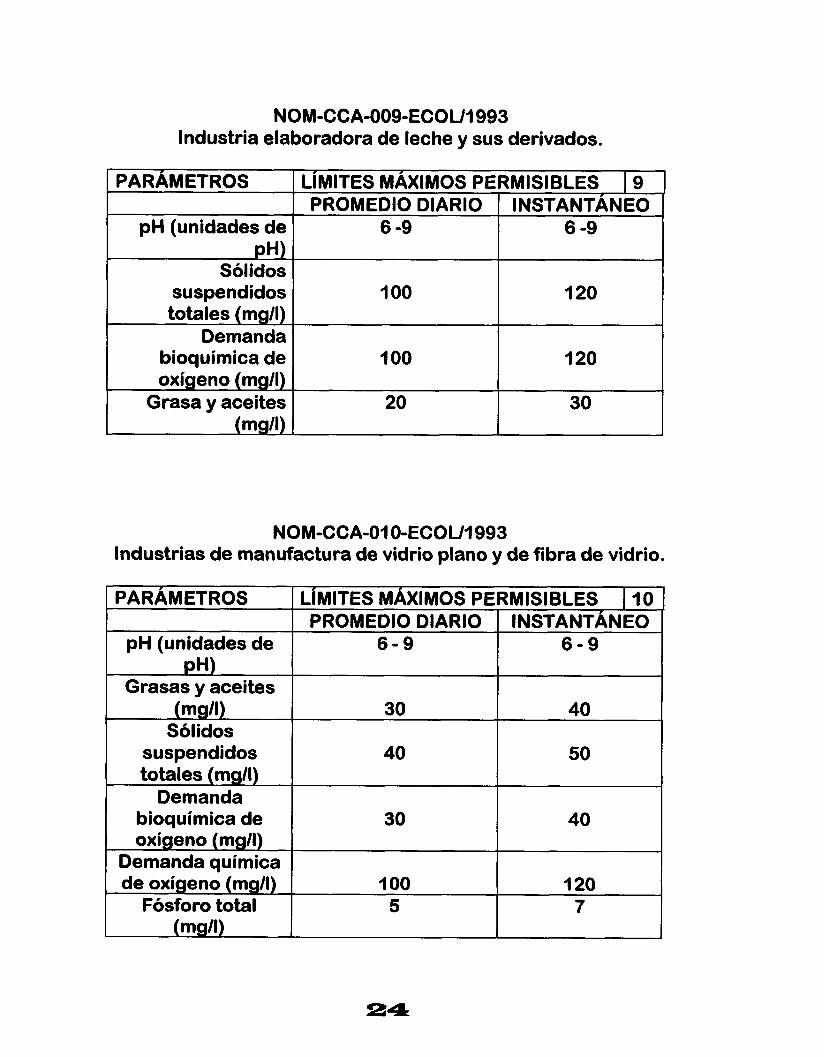
NOM-CCA-009-ECOUI 993 Industria elaboradora de leche y sus derivados.
- PARAMETROS LIMITES MAXIMOS PERMISIBLES I 9
PROMEDIO DIARIO INSTANTÁNEO pH (unidades de 6 -9 6 -9
pH) Sólidos
suspendidos totales (mgll)
120 I O0
Demanda bioquímica de 1 O0 120 oxígeno (mg/l)
Grasa y aceites 20 30 (mg/l)
N Industrias de manu
PARAMETROS
pH (unidades de pH)
Grasas y aceites (mg/l)
Sólidos
3M-CCA-O1 O-ECOUI 993 Factura de vidrio plano y de fibra de vidrio.
LIMITES MÁXIMOS PERMISIBLES
30 40
suspendidos totales (mgll)
50 40
Demanda bioquímica de 30 40 oxígeno (mg/l)
Demanda química de oxígeno (mg/l)
7 5 Fósforo total 120 1 O0
(mg/l) I
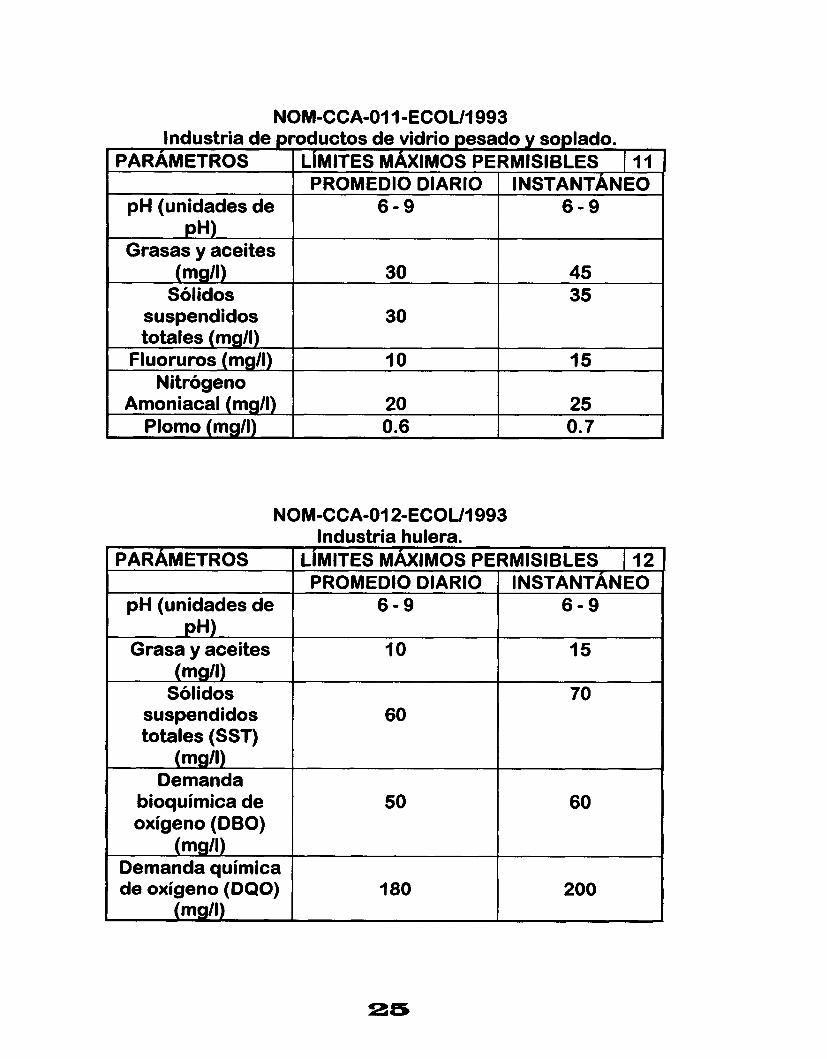
NOM-CCA-O1 I-ECOUI 993 Industria de productos de vidrio pesado y soplado.
PARAMETROS LIMITES MAXIMOS PERMISIBLES I I1 PROMEDIO DIARIO I INSTANTÁNEO
pH (unidades de pH)
6 - 9 6 - 9
Grasas y aceites (mg/l)
Sólidos 45 30 35
suspendidos totales (mg/l)
30
Fluoruros (mg/l) 15 I O Nitrógeno
Amoniacal (mg/l) O. 7 0.6 Plomo (mall) 25 20
N
PARAMETROS
pH (unidades de pH)
Grasa y aceites (mg/l)
Sólidos suspendidos totales (SST)
(mg/l) Demanda
bioquímica de oxígeno (DBO)
(mg/l) Demanda química de oxígeno (DQO)
(mg/l)
)M-CCA-Ol2-ECOUI 993 Industria hulera.
LIMITES MÁXIMOS PERMISIBLES 1 12 ' PROMEDIO DIARIO INSTANTÁNEO
6 - 9
15 10
6 - 9
60 70
50 60
180 200
2s
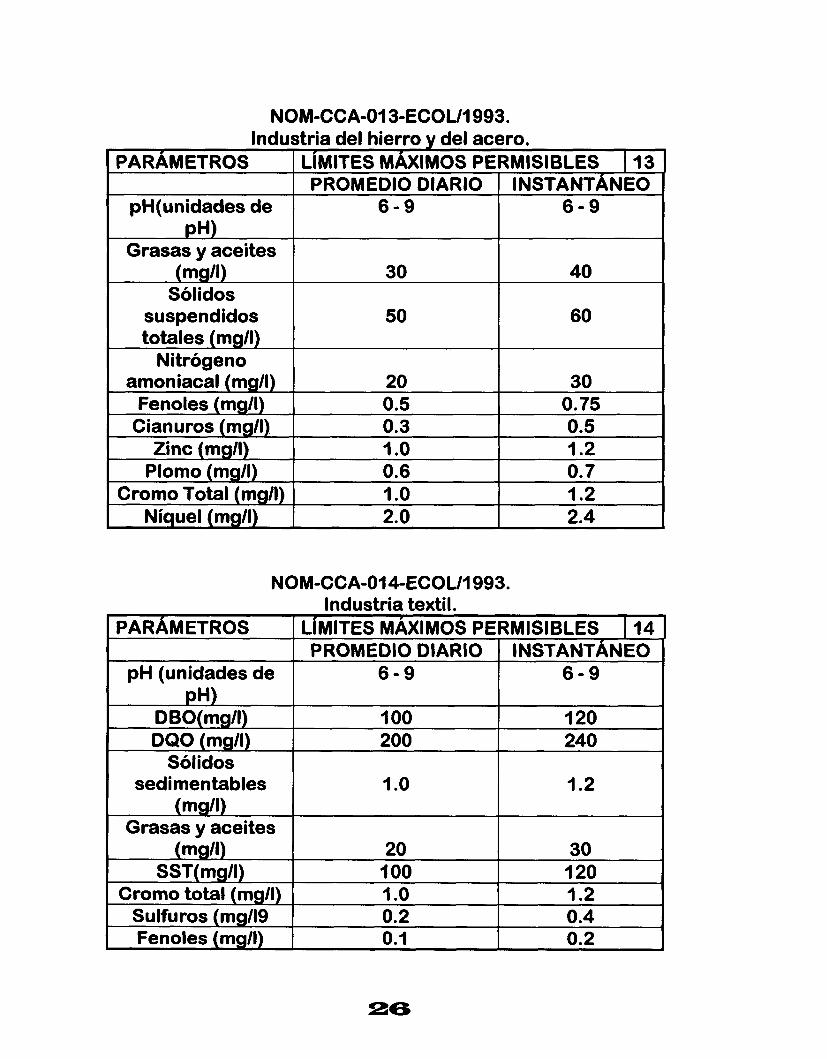
NOM-CCA-013-ECOU1993. Industria del hierro y del acero.
PARAMETROS LíMITES MÁXIMOS PERMISIBLES I 13 PROMEDIO DIARIO INSTANTANEO
pH(unidades de pH)
6 - 9 6 - 9
Grasas y aceites (mg/l) 30 40
Sólidos suspendidos 50 60 totales (mg/l)
Nitrógeno amoniacal (mg/I)
0.5 0.3 Cianuros (mg/l), O. 75 0.5 Fenoles (mgll) 30 20
Zinc (mg/l) I .o 1.2 Plomo (mg/l) 0.6
1.2 1 .o Cromo Total (mgll) O. 7
2.4 2.0 Níquel (mg/l)
NOM-CCA-014-ECOUI 993. Industria textil.
PARAMETROS LíMITES MÁXIMOS PERMISIBLES I 14 PROMEDIO DIARIO I INSTANTANEO
I
pH (unidades de
I .2 I .o sedimentables
240 200 DQO (mg/l) 120 1 O0 DBO(mg/l)
6 - 9 6 - 9 pH)
Sólidos
(mgll) Grasas y aceites
(mg/l) 20 30 SST( mg/l) 1 O0
- 0.2 0.1 Fenoles (mg/l) 0.4 0.2 Sulfuros (mg119 1.2 1 .o Cromo total (mg/l) 120
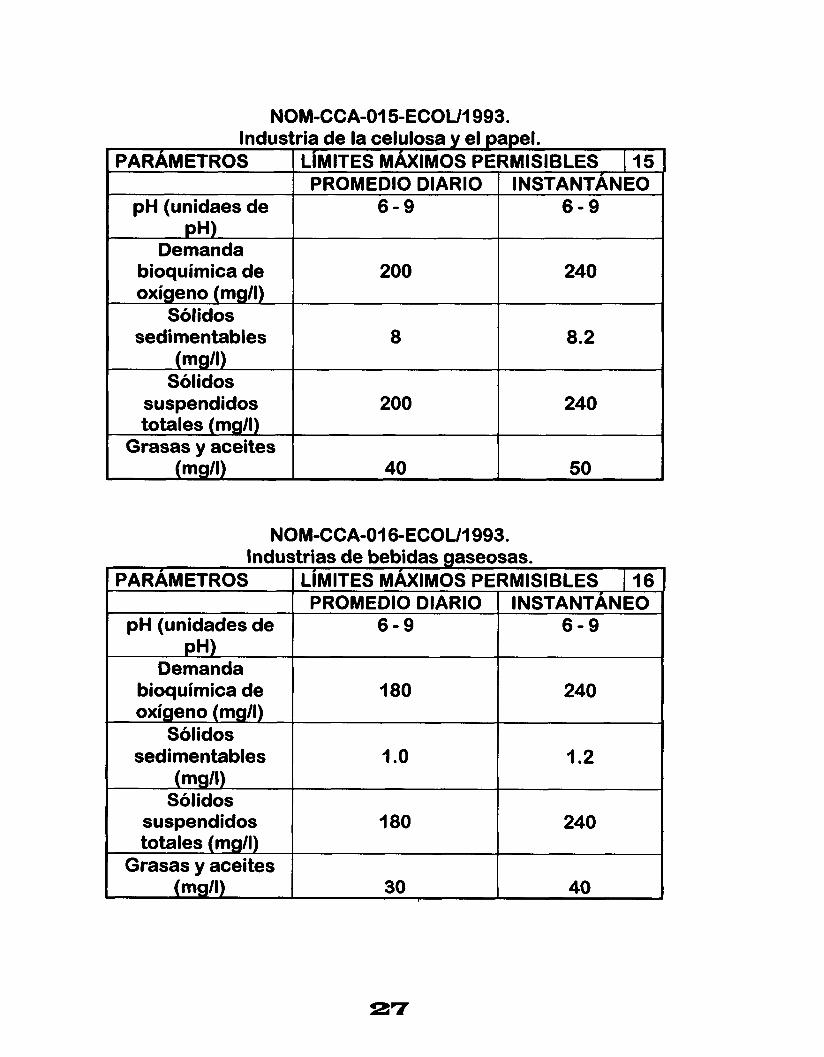
NOM-CCA-015-ECOUI 993. Industria de la celulosa y el papel.
PARAMETROS LIMITES MAXIMOS PERMISIBLES [ 15 PROMEDIO DIARIO I INSTANTANEO
pH (unidaes de I 6-9 I 6 - 9 pH)
Demanda bioquímica de
Sólidos oxígeno (mg/l)
240 200
sedimentables 8 8.2 (mg/l)
Sólidos suspendidos 200 240 totales (mg/l)
Grasas y aceites (mg/l) 50 40
NOM-CCA-03 6-ECOUI 993. Industrias de bebidas gaseosas.
PARAMETROS LIMITES MAXIMOS PERMISIBLES I 16 PROMEDIO DIARIO INSTANTÁNEO
pH (unidades de 6 - 9 6 - 9 pH)
Demanda bioquímica de 180 oxígeno (mg/l)
240
Sólidos sedimentables 1 .o I .2
(mgll) Sólidos
suspendidos 180 240 totales (mg/l)
Grasas y aceites (mg/l) 40 30
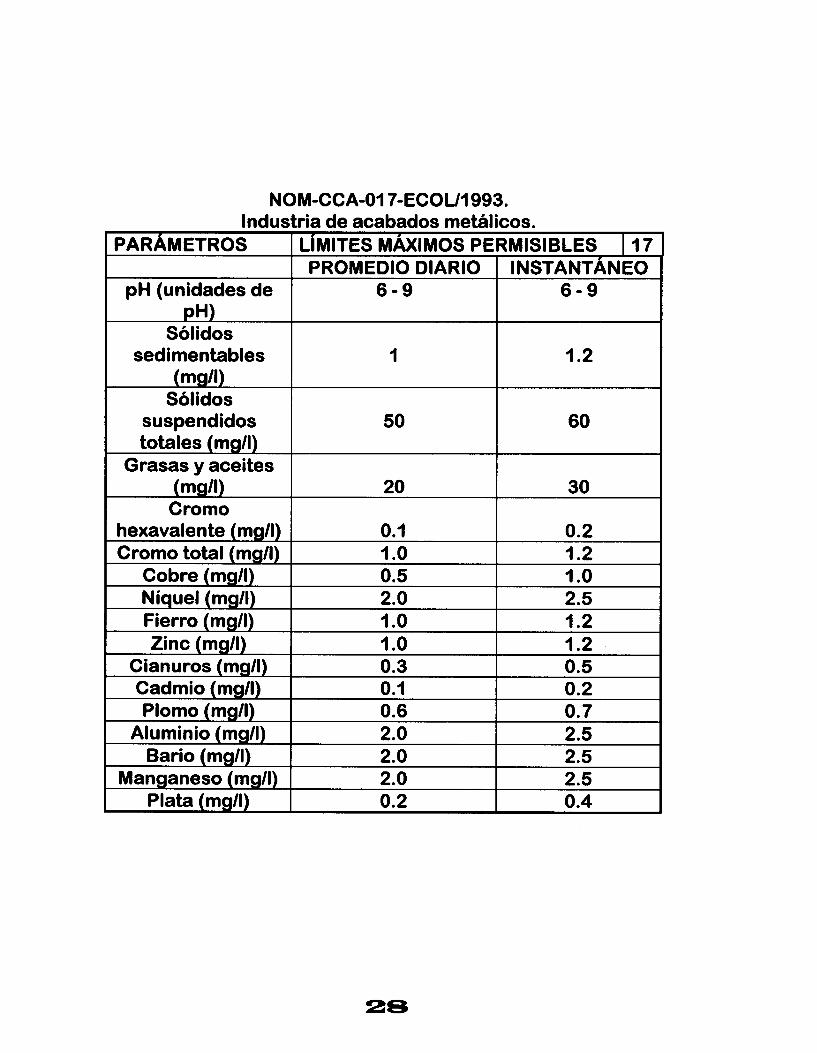
NOM-CCA-017-ECOU1993. Industria de acabados metálicos.
PARAMETROS LIMITES MÁXIMOS PERMISIBLES I 17 PROMEDIO DIARIO I INSTANTÁNEO
pH (unidades de 6 - 9 6 - 9 pH)
Sólidos
(mg/l) Sólidos
suspendidos 50 60 totales (mgll)
Grasas y aceites (mg/l) 20 30 Cromo
hexavalente (mg/l) 0.1 0.2 Cromo total (mg/l) 1 .o 1.2
Cobre (mg/l) 0.5 I .o Níquel (mg/l) 2.0 2.5 Fierro (mg/I) 1 .o
0.5 0.3 Cianuros (mg/l) 1.2 1 .o Zinc (mg/l) 1.2
sedimentables 1.2 1
Cadmio (mg/l)
2.0 Manganeso (mg/l) 2.5 2.0 Bario (mg/l) 2.5 2.0 Aluminio (mgll) 0.7 0.6 Plomo (mg/l) 0.2 0.1
0.4 0.2 Plata (mg/l) 2.5
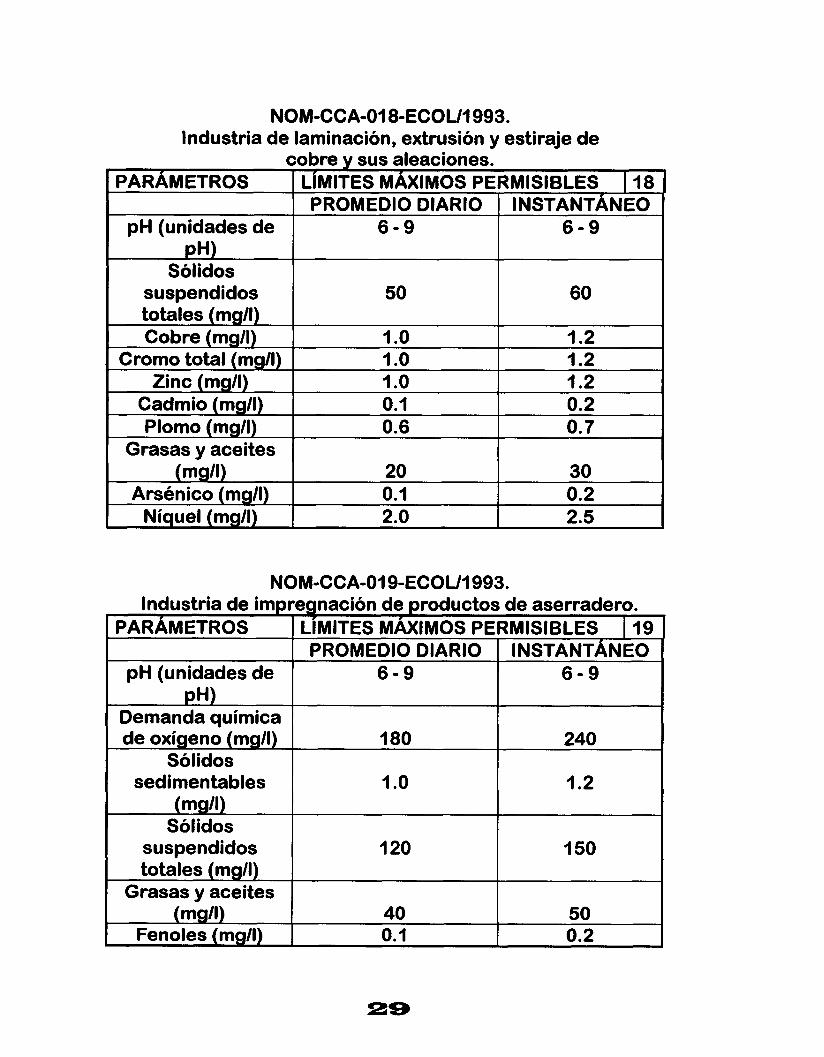
NOM-CCA-018-ECOUI 993. Industria de laminación, extrusión y estiraje de
cobre y sus aleaciones. PARAMETROS LIMITES MAXIMOS PERMISIBLES I 18
PROMEDIO DIARIO INSTANTÁNEO
.
pH (unidades de 6 - 9 6 - 9 pH)
Sólidos suspendidos 50 totales (mg/l)
60
Cobre (mg/l) 1 .o I .2 Cromo total (mg/l) I .o 1.2
Zinc (mg/l)
O. 7 0.6 Plomo (mg/l) 0.2 0.1 Cadmio (mg/l) 1.2 I .o
Grasas y aceites (mg/l) 20 30
Arsénico (mgll) 0.1 0.2 Níquel (mgll) 2. o 2.5
NOM-CCA-019-ECOUl993. Industria de impregnación de productos de aserradero.
PARÁMETROS I LIMITES MAXIMOS PERMISIBLES I 19 PROMEDIO DIARIO INSTANTÁNEO
pH (unidades de 6 - 9 6 - 9 pH)
Demanda química de oxígeno (mg/l) 180 240
Sólidos sedimentables 1 .o I .2
(mg/l) Sólidos
suspendidos 120 150 totales (mgll)
Grasas y aceites (mg/l)
0.2 0.1 Fenoles (mg/l) 50 40

NOM-CCA-020-ECOUI 993. Industria de asbestos textiles,materiales
de fricción y selladores.
PARAMETROS LIMITES MÁXIMOS PERMISIBLES I 20 PROMEDIO DIARIO INSTANTANEO
pH (unidades de 6 - 9 6 - 9 pH)
Sólidos suspendidos 60 totales (mgll)
70
Demanda química de oxígeno (mgll) 1 O0 120
NOM-CCA-021 -ECOUI 993. Industria del curtido y acabado en pieles.
I PARAMETROS I LIMITES MAXIMOS PERMISIBLES I 21 I
I I PROMEDIO DIARIO I INSTANTÁNEO pH (unides de pH) 6 - 9 6 - 9
Demanda bioquímica de 200 oxígeno (mg/l)
240
Sólidos sedimentables 5.0 8.0
(mgll) Sólidos
suspendidos
40 30 Grasas y aceite totales (mgll)
240 200
I .5 I .o Sulfuros (mg/l) 0.2 0.1 hexavalente (mg/l)
Cromo 1.5 1 .o Cromo total (mg/l)
(mg/l)
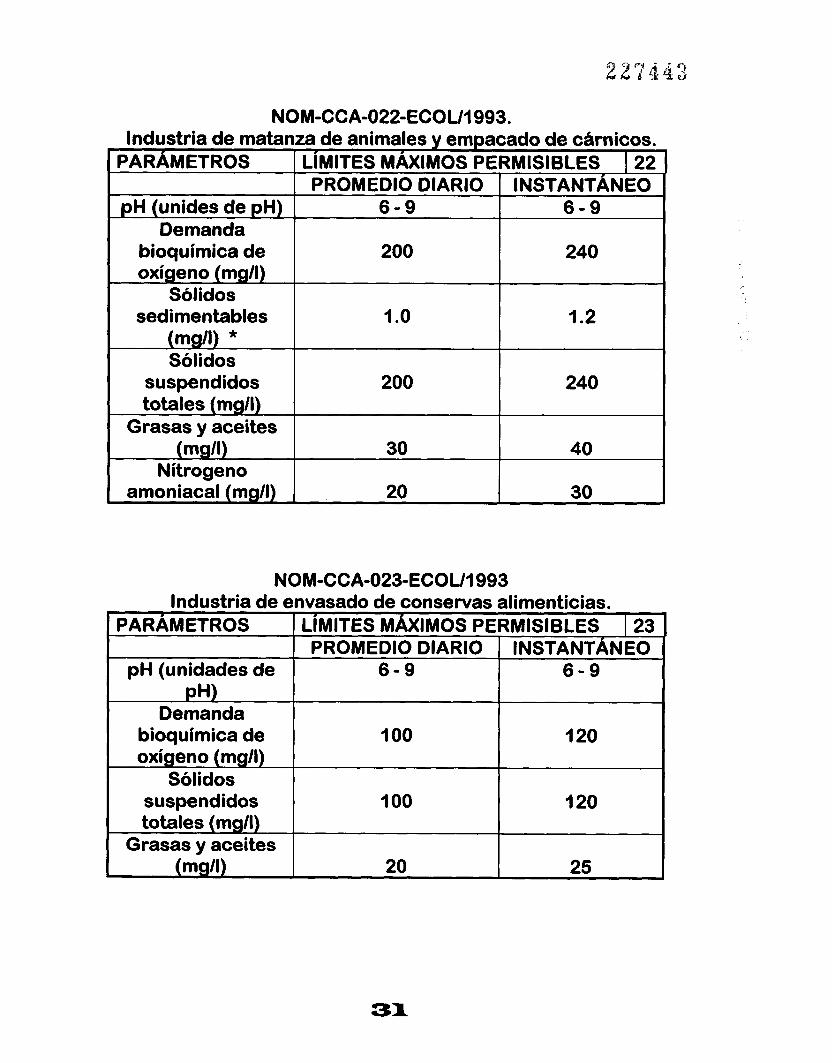
NOM-CCA-022-ECOU1993. Industria de matanza de animales y empacado de cámicos.
PARAMETROS I LIMITES MAXIMOS PERMISIBLES I 22 PROMEDIO DIARIO INSTANTÁNEO
p H (unides de pH) 6 - 9 6 - 9 Demanda
bioquímica de 200 oxígeno (mg/l)
240
Sólidos sedimentables 1 .o I .2
(mg/l) * Sólidos
suspendidos 200 240 totales (mgll)
Grasas y aceites
Nitrogen0 (mgll)
30 20 amoniacal (mg/l)
40 30
NOM-CCA-023-ECOUI 993 Industria de envasado de conservas alimenticias.
PARAMETROS LIMITES MÁXIMOS PERMISIBLES I 23 INSTANTÁNEO PROMEDIO DIARIO
'
pH (unidades de 6 - 9 6 - 9 pH)
Demanda bioquimica de 1 O0 oxígeno (mgll)
120
Sólidos suspendidos 1 O0 120 totales (mgll)
Grasas y aceites (mg/l) 25 20

NOM-CCA-024-ECOUI 993. Industria elabarac
PARAMETROS
pH (unidades de pH)
Demanda bioquímica de oxígeno (mgll)
Sólidos suspendidos totales (mg/l)
Sólidos sedimentables
(mg/l) Grasas y aceites
(mg/l)
llora de papel a partir de celulosa virgen. LIMITES MAXIMOS PERMISIBLES I 24 PROMEDIO DIARIO I INSTANTÁNEO
6 - 9 6” 9
125 150
125 150
4.0 5 0
20 30
NOM-CCA-025-ECOUI 993. Industria elaboradora de papel a partir
de fibra celulósica reciclada. PARAMETROS LIMITES MAXIMOS PERMISIBLES I 25
INSTANTÁNEO PROMEDIO DIARIO pH (unidades de 6 - 9 6 - 9
Demanda bioquímica de 200 240 oxígeno (mg/l)
Sólidos suspendidos I 200 240 totales (mg/l)
Sólidos sedimentables 8.2 8.0
(mgll) Grasas y aceites
(mg/l) 50 40
32

NOM-CCA-026-ECOUI 993. Restaurantes u hoteles.
PARAMETROS LIMITES MAXIMOS PERMISIBLES I 26 INSTANTANEO PROMEDIO DIARIO
.
pH (unidades de 6 - 9 6 - 9 pH)
Demanda bioquimica de 30 oxígeno (mg/l)
45
Grasas y aceites (mg/l) 15 20
Sólidos suspendidos 30 45 totales (mg/l)
Sustancias activas al Azul de Metileno
Coliformes fecales (mg/l)
2,000 1,000 (NMP/lOOml)
6 3
NOM-CCA-027-ECOU1993. Industria del beneficio del café.
PARAMETROS . LIMITES MAXIMOS PERMISIBLES I 27 PROMEDIO DIARIO I INSTANTÁNEO
pH (unidades de
180 150 DBO (mg/l) pH)
6 a 9 6 a 9
Grasas y aceites (mg/l) 10 20
Sólidos sedimentables 1 .o 2.0
(mg/l) SST( mgll) 150 180
Materia flotante I (mgll) ausente ausente
33
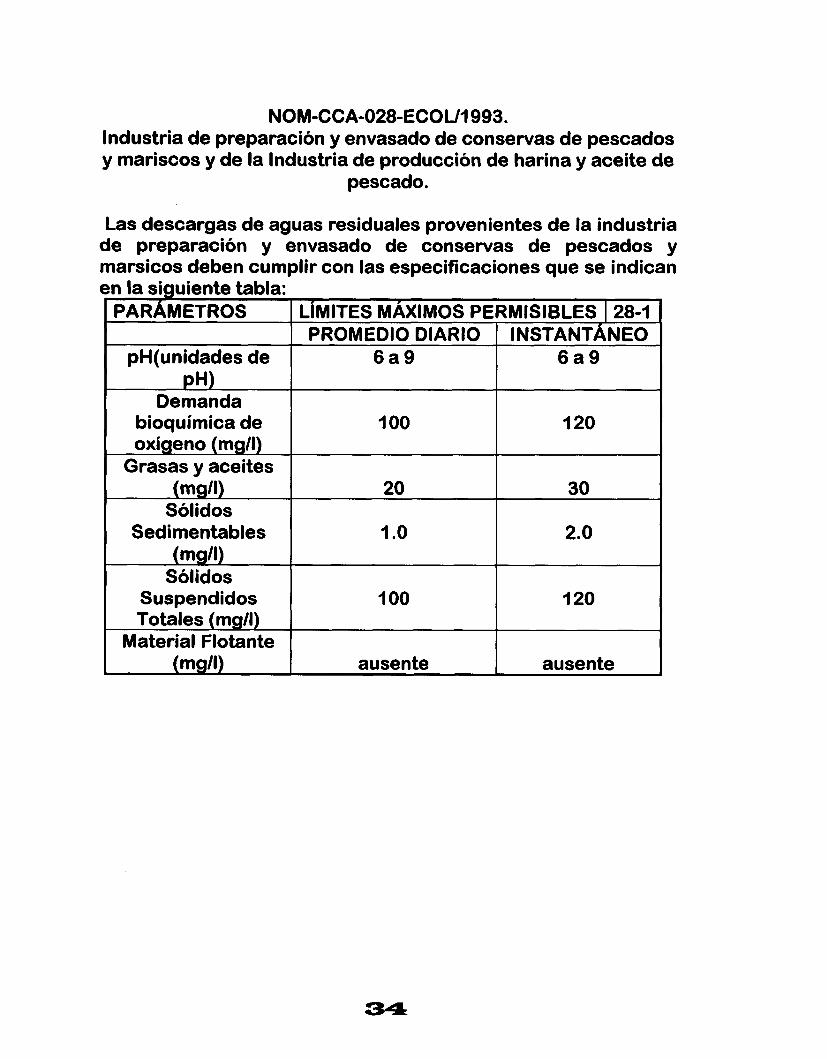
NOM-CCA-028-ECOUI993. Industria de preparación y envasado de conservas de pescados y mariscos y de la Industria de producción de harina y aceite de
pescado.
Las descargas de aguas residuales provenientes de la industria de preparación y envasado de conservas de pescados y marsicos deben cumplir con las especificaciones que se indican en la siguiente tabla:
PARAMETROS LIMITES MAXIMOS PERMISIBLES I 28-1 PROMEDIO DIARIO INSTANTÁNEO
pH(unidades de 6 a 9 6 a 9 pH)
Demanda bioquimica de I O0 oxígeno (mgll)
120
Grasas y aceites (mg/l) 20 30
Sólidos Sedimentables 1 .o 2.0
(mgll) Sólidos
Suspendidos 1 O0 120 Totales (mg/l)
Material Flotante (mg/l) . ausente ausente
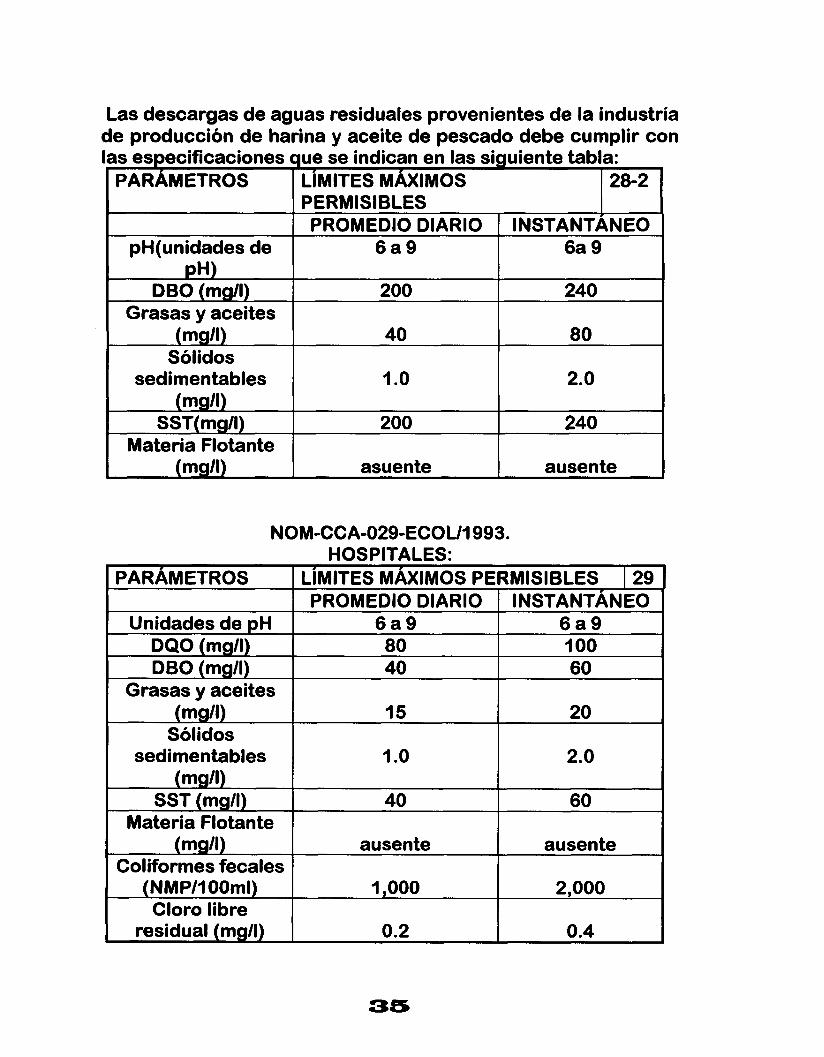
Las descargas de aguas residuales provenientes de la industria de producción de harina y aceite de pescado debe cumplir con as especificaciones que se indican en las siguiente tabla: PARAMETROS LIMITES MAXIMOS 28-2
PERMISIBLES PROMEDIO DIARIO INSTANTANEO
pH(unidades de 6 a 9 6a 9
DBO (mg/l) 200 240
I
pH)
Grasas y aceites (mg/I) 80 40
Sólidos sedimentables
240 200 SST( mg/l)
2. o 1 .o (mgll)
Materia Flotante (msr/l) ausente asuente
NOM-CCA-029-ECOU1993. HOSPITALES:
PARAMETROS LIMITES MAXIMOS PERMISIBLES I 29 PROMEDIO DIARIO INSTANTÁNEO
Unidades de pH
60 40 DBO (mgll) I O0 80 DQO (mg/l) 6 a 9 6 a 9
Grasas y aceites (mg/l) 15 20
Sólidos sedimentables I .o 2.0
(mg/l) SST (mg/l) 40 60
Materia Flotante
Coliformes fecales
Cloro libre
(mg/l) ausente ausente
(NMP/I 00ml)
0.4 0.2 residual (mg/l)
2,000 1,000
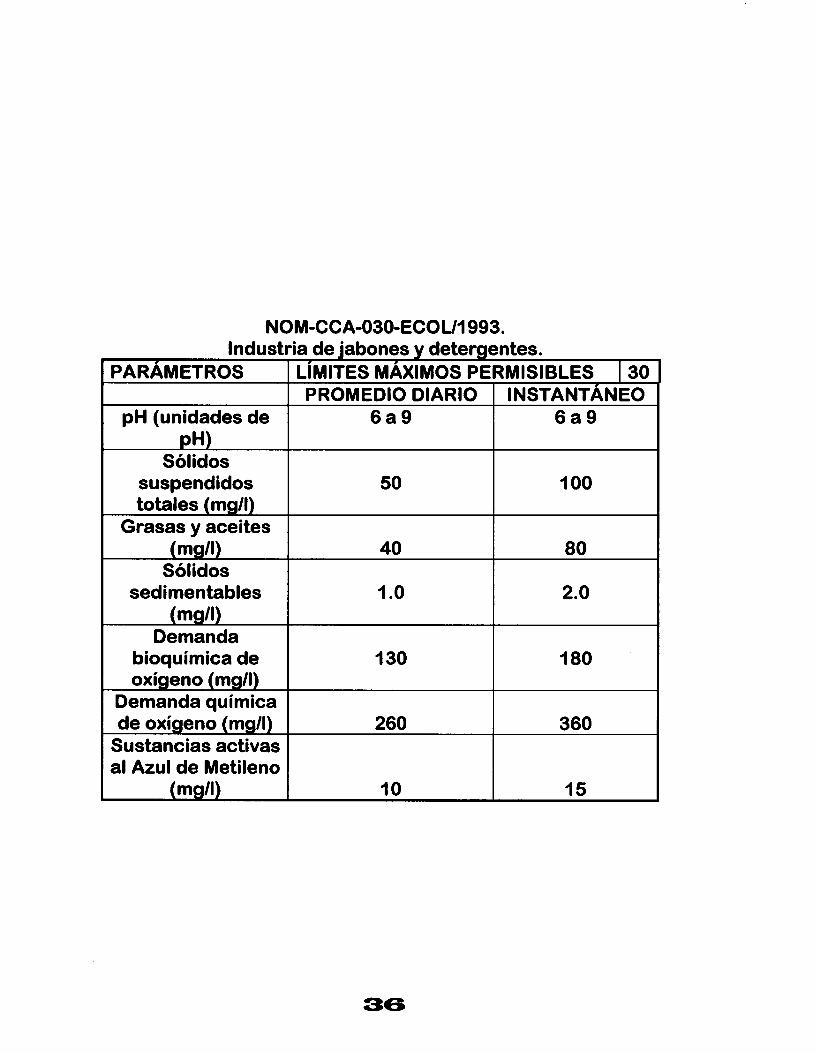
NOM-CCA-030-ECOUI 993. Industria de jabones y detergentes.
PARAMETROS LIMITES MAXIMOS PERMISIBLES I 30 INSTANTANEO PROMEDIO DIARIO
pH (unidades de 6 a 9 6 a 9 pH)
Sólidos suspendidos 50 totales (mg/l)
I O0
Grasas y aceites (mg/l) 40 80
Sólidos sedimentables 1 .o 2.0
(mg/l) Demanda
bioquímica de 130 180 oxígeno (mg/l)
Demanda química de oxígeno (mgll) 260 360
Sustancias activas al Azul de Metileno
(mgll) 15 10

NOM-CCA-031-ECOU1993. Industria, actividades agroindustriales, de servicios y el
tratamiento de drenaje y alcantarillado urbano o municioal. PARAMETROS LIMITES MAXIMOS PERMISIBLES 31
PROMEDIO DIARIO I INSTANTANEO
.
Temperatura ("C) 6 a 9 pH (unidades de
40°C (31 3OK) 0
6 a 9 pH)
Sólidos
(mg/l) Grasas y aceites
(mg/l)
8,000 5,000 eléctri- Conductividad
I O0 60
sedimentables I O 5
ca( micromhos/cm) Aluminio (mgll)
2. o 1 .o Cianuros (mg/l) 1 .o 0.5 Cadmio (mg/l) 1 .o 0.5 Ars6nico (mgll) 20 10
Cobre (mg/l) 5 10
hexavalente (mgll) 0.5 1 .o Cromo total (mgll) 2.5 5.0 Fluoruros (mg/l) 3 6 Mercurio (mg/l) 0.01 0.02 Níquel (mg/l) 4 8 Plata (mg/l) 1 .o 2.0 Plomo (mg/l) 1 .o 2.0 Zinc (mg/l) 12 6
Fenoles (mg/l) 5 I O
Cromo
Sustancias activas al Azul de
Metileno(mg/l) 60 30
37

NOM-CCA-032-ECOU1993. Aguas residuales de origen urbano o municipal para su
disposición mediante riego agrícola. PARAMETROS LíMITES MÁXIMOS PERMISIBLES I 32 pH( unidades de 6.5 a 8.5
pH) Conductividad
ca( micromhoslcm) Demanda
bioquímica de 120 oxígeno (mgll) Aluminio (mgll) 5.0 Arsénico (mgll) 0.1
Boro (mgll) 1.5 Cadmio 0.01
Cianuros (mgll) 0.02 Cobre (mgll) 0.2
Cromo total (mgll) 0.1 Fierro (mgll) 5.0
Fluoruros (mgll) 3.0 Manganeso (mgll) 0.2
Níquel (mgll)
0.02 Selenio (mgll) 5.0 Plomo (mgll) 0.2
elhctri- 2000
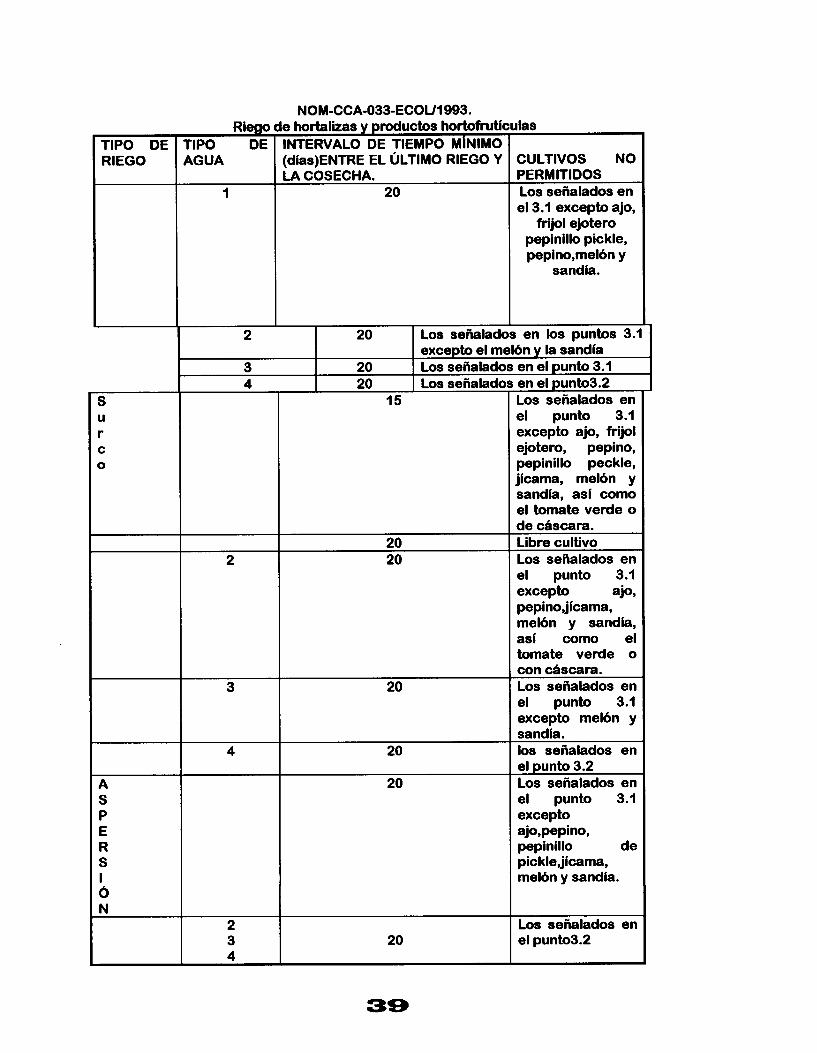
NOM-CCA-033-ECOU1993.
1 TIPO DE RIEGO
A S P E R S I 6 N
Riego rwo DE 4GUA
1
2
4
4
t
e hortalizas y productos h o r t o f r u t i c INTERVALO DE TIEMPO M¡NlMO (dias)ENTRE EL ÚLTIMO RIEGO Y LA COSECHA.
20
20 20
dl :U - v
I
I
I - -
20 Los señalados en los puntos 3.1- excepto el mel6n y la sandia
3 Los señalados en el punto3.2 20 Los señalados en el punto 3.1 20
15
-
-
-
- 2 3 4
20
20
20
llas
CULTIVOS NO PERMITIDOS Los señalados en el 3.1 excepto ajo,
frijol ejotero pepinillo pickle, pepino,mel6n y
sandia.
Los señalados en el punto 3.1 excepto ajo, frijol ejotero, pepino, pepinillo peckle, jicama, mel6n y sandia, asi como el tomate verde o de cascara. Libre cultivo Los señalados en el punto 3.1 excepto ajo, pepinojicama, mel6n y sandia, asi como el tomate verde o con chscara. Los señalados en el punto 3.1 excepto mel6n y sandia. los señalados en el punto 3.2 Los señalados en el punto 3.1 excepto ajo,pepino, pepinillo de picklejicama, mel6n y sandía.
Los señalados en el punto3.2

POTENCIAL HIDRóGENO (pH)
FUNDAMENTO.
El método se basa en que al poner en contacto dos soluciones de diferente concentración de iones hidrógeno, se establece una fuerza electromotriz. Si una de las soluciones tiene una concentración de iones conocida (pH), por medio de la fuerza electromotriz producida, se puede conocer el pH de la otra solución (solución problema), ya que esta fuerza electromotriz es proporcional al pH de la solución problema.
Dentro de la Norma Oficial Mexicana para determinar el pH, se define los siguientes conceptos importantes:
pH: Es el logaritmo negativo de la concentración del ión hidrógeno en una solución acuosa o el logaritmo del recíproco de la concentración de iones hidrógeno.
Acidez: Es la capacidad cuantitativa del agua para reaccionar con los iones hidroxilos.
Alcalinidad: Es la capacidad cuantitativa del agua para reaccionar con los iones hidrógeno.
PREPARACIóN DE SOLUCIONES PATRóN PARA LA CALIBRACI~N.
Los reactivos utilizados deben ser grado analítico, cuando se hable de agua, se entiende agua destilada.
La siguiente tabla muestra las cantidades que se deben utilizar para preparar las soluciones patrón. Las cantidades de productos indicados en la tabla deben prepararse tan pronto se necesiten disolviendo el material en agua a 25°C y diluyendo la solución hastas un litro (1 I). Se usa agua con una conductividad específica de 2 siemens a 25°C y un pH de 5.6 a 6.0.
Para la preparación de soluciones de bórax y fosfatos se hierve y enfría el agua, con el objeto de eliminar el C02 y obtener un pH de 6.7 a 7.3.
El fosfato ácido potásico y el fosfato monosódico se beben secar en una estufa a I 1 0°C - 130°C durante 2 hr.
40
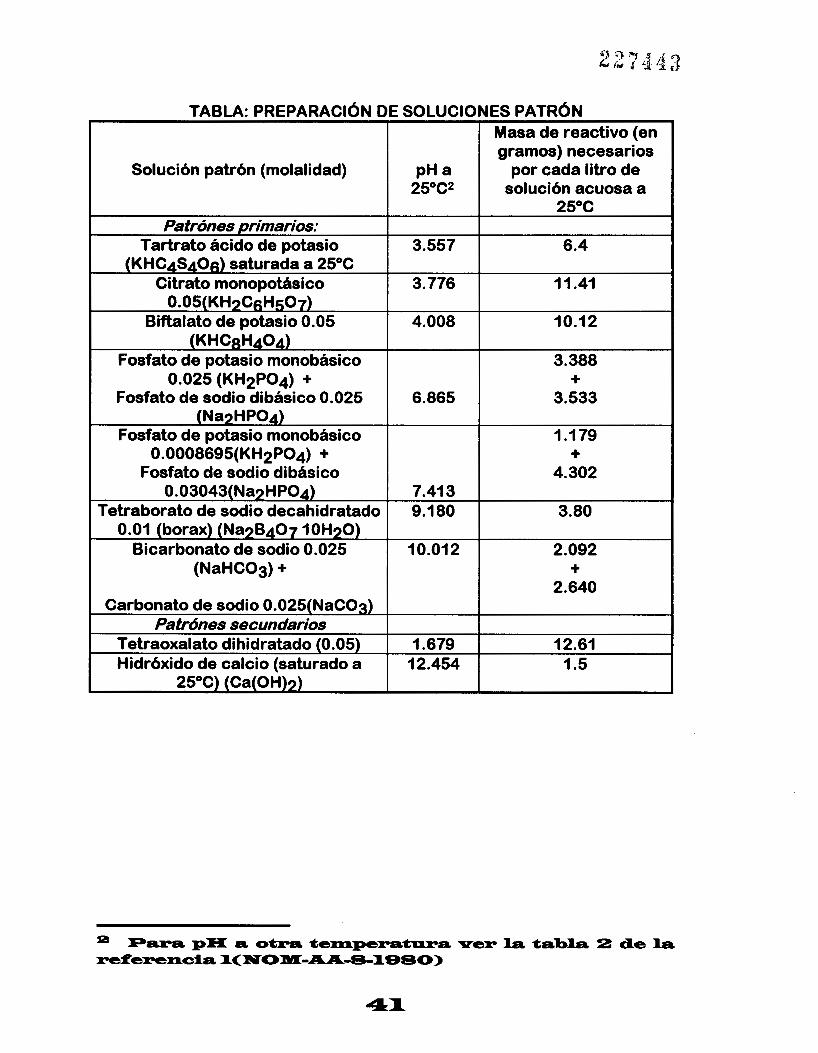
TABLA: PREPARACIdN DE SOLUCIONES PATRdN Masa de reactivo (en gramos) necesarios
Solución patrón (molalidad) solución acuosa a 25"C* por cada litro de PH a
25°C
Tartrato ácido de potasio 6.4 3.557 Patrónes primarios:
(KHCqS406) saturada a 25°C Citrato monopotásico 11.41 3.776
0.05(KHpCgH507) Biftalato de potasio 0.05 10.12 4.008
(KHCaH404) Fosfato de potasio monobásico
3.533 6.865 Fosfato de sodio dibásico 0.025
3.388 0.025 (KH2P04) + +
(NapHP04) Fosfato de potasio monobásico 1 .I 79
0.0008695(KH2P04) + + Fosfato de sodio dibásico 4.302
Tetraborato de sodio decahidratado
2.640
2.092 10.01 2 Bicarbonato de sodio 0.025
3.80 9.1 80 0.03043(NapHP04) 7.41 3
0.01 (borax) (NapB407 lOH7O)
(NaHC03) + +
Carbonato de sodio 0.025(NaC03) Patrónes secundarios
Tetraoxalato dihidratado (0.05) 1.5 12.454 Hidróxido de calcio (saturado a
12.61 1.679
25°C) (Ca(0H)p)
41

PROCEDIMIENTO.
Ajustar y calibrar el potenciómetro siguiendo el procedimiento indicado en el manual del mismo. Para fines de calibración, se permite el empleo de soluciones preparadas o semipreparadas, siendo responsabilidad del usuario de las mismas su correcta concentración.
El potenciómetro debe calibrarse con una solución reguladora patrón cuyo pH se encuentre cerca del que se espera medir, y comprobarse usando cuando menos otras dos soluciones de pH diferentes, uno menor y otro mayor de aquél en que se hizo la calibración. La diferencia entre cualquiera de las tres lecturas y el pH propio de la solución patrón, no debe excederse de 0.1 unidades de pH.3
Para calibrar el potenciómetro solamente se introduce el electrodo en la solución patrón y se ajusta la lectura del aparato al pH de la solución. Una vez hecho esto retirar el recipiente de la solución patrón y lavar los electrodos con agua quitando el exceso con un material adecuado de acuerdo a las instrucciones del fabricante, evite friccionar la superficie del electrodo.
Una vez realizada la calibración se introduce en el recipiente que contiene la muestra los electrodos y se realiza la lectura Ésta se debe realizar a 25°C o a la que fue calibrado el aparato con las soluciones patrón, se puede efectuar determinaciones a otras lecturas siempre y cuando el potenciómetro esté adecuado con los mecanismos compensatorios necesarios.
Se sugiere hacer lecturas por duplicado, la diferencia máxima entre estas dos lecturas no debe ser mayor a 0.1 unidades de pH.

MATERIA FLOTANTE
FUNDAMENTOS
Las definiciones que rigen este análisis son:
Materia flotante : Es el material que flota libremente en la superficie del líquido y que queda retenido en la malla especificada en la norma NOM-06-1973.
PROCEDIMIENTO
Se deja sedimentar la muestra durante 15 minutos. Se vierten aproximadamente las dos terceras partes
superiores de la muestra( ver seccibn de toma de muestras) a través de la malla, teniéndose cuidado de que la materia flotante que sobrenada, quede retenida en dicha mallad.
Se permite el empleo de una cucharilla para arrastrar hacia la malla toda aquella materia flotante que todavía quedara sobre la superficie de la muestra que se está vertiendo o aquella adherida a las paredes del recipiente.
RESULTADOS
Inmediatamente después de filtrada la muestra, se procede al examen de la malla.
La ausencia de material retenido en la malla observado a simple vista, se considera como "ninguna" materia flotante retenida en la malla.

SóLIDOS SUSPENDIDOS TOTALES
FUNDAMENTO.
El método se basa en la evaporación y calcinación de la muestra, en donde los residuos de una y otra operación sirven de base para el cálculo del contenido de sólidos.
Los conceptos importantes que derivan de este análisis según la Norma Oficial Mexicana son:
Sólidos suspendidos totales: Sólidos constituidos por sólidos sedimentables, sólidos en suspensión y sólidos coloidales, cuyo tamaño de partícula no pasa el filtro estándar de fibra de vidrio.
Sólidos sedimentables: Materiales que se depositan en el fondo de un recipiente debido a la operación de sedimentación..
Sedimentación: Operación por medio de la cual, las partículas sólidas suspendidas en un liquido, se asientan debido a la fuerza de la gravedad.
PROCEDIMIENTO.
Para sólidos suspendidos totales, preparar el medio filtrante como sigue:
Colocar un disco de fibra de vidrio en el crisol Gooch con la superficie rugosa hacia arriba, teniendo cuidado de que el disco cubra completamente las perforaciones del Gooch.
Colocar el crisol y el disco en un aparato de filtración aplicando vacío. Lavar el disco con agua, dejando que éSta se drene totalmente.
Suspender el vacío y llevar el crisol a masa constante en la mufla a una temperatura de 550°C 25°C durante un periodo de 15 a 20 minutos. Sacar el crisol, dejar enfriar y determinar su masa (G3).
44

Preparación de la muestra.
Colocar el crisol con el disco en el aparato de filtración y % ~ p Y e;:: 3
Humedecer el disco con agua. *,'. t
Medir con una probeta o pipeta volum&trica según proceda, un volumen adecuado de la cantidad seleccionada de muestra previamente homogenizada, la cual depende de la c L
concentración esperada de sólidos suspendidos. Filtrar la muestra a través del disco y aún aplicando vacío,
lavar el disco tres veces con 10 ml de agua, dejando que el agua drene totalmente en cada lavado.
Suspender el vacío y secar el crisol en la estufa a una temperatura de 103 a 105°C durante una hora. Sacar el crisol, dejar enfriar en un desecador a temperatura ambiente y determinar su masa (G4).
aplicar vacío. 1 '.''$j .*++F.,
j;1;!7:,:'J
5 . Í - , : I
:; _ - r i
_ .
.. , ~ ."' . ' - >
t. - ,
I .
CALCULOS. El contenido de sólidos suspendidos totales, se calculan
con la siguiente fórmula: G4 - G3
V SST=- X 1000
En donde: SST = Sólidos suspendidos totales, en mg/l G4 = Masa del crisol con el residuo, en mg G3 = Masa del crisol con el disco, en mg V = Volumen de muestra en ml
4s

RESUMEN DEL METODO.
El método consiste en acidificar un muestra para extraer las grasas y aceites en solución, la grasa es entonces separada por filtración y extraida con un solvente con ayuda del aparato Soxhlet, posteriormente se evapora el solvente y se cuantifica gravimétricamente el material extraido.
REACTIVOS Y MATERIALES.
Los reactivos que se mencionan, deben ser grado analítico. Cuando se hable de agua debe entenderse agua destilada.
Ácido clorihídrico concentrado. Suspensión de tierra de diatomáceas-sílice ( Hyflo-
supercal o equivalente, 10 gll de agua). Hexano normal con punto de ebullición de 69°C o freón
(1,1,2 tricloro-l,2,3 trifluoretano) de punto de ebullición de 47.5OC .
Cartuchos de extracción (thimbles). Papel filtro de poro medio y de I I cm de diámetro. Discos de tela de muselina de 11 cm de diámetro.
PROCEDIMIENTO.
Es importante que la muestra haya sido tratada como se indica en el procedimiento de muestre0 y preservación de la muestra.
Preparar un filtro con el disco de tela de muselina sobreponiéndole el disco de papel filtro, colocar en el embudo buchner, humedecer la tela y el papel.
Con ayuda del vacío preparar aproximadamente 300 ml de la suspensión de tierra de diatomáceas. (hasta la saturación de los poros); se lava con un litro o menos de agua y aplicar el vacío hasta que toda el agua haya sido filtrada.
46

Pasar la muestra acidificada a través del filtro preparado. Aplicar el vacío hasta que toda el agua haya sido filtrada, recibiéndose en un matraz kitazato de 2000 ml.
Con una pinza transferir a un cartucho de extracción el papel filtro y el material adherido al disco de tela. Limpiar las caras y el fondo del recipiente colector, la tapa y el embudo Buchner con pedazos de papel filtro remojado en el solvente que se va usar, teniendo cuidado de transferir todas las capas de grasas formadas, y de recoger todo el material sólido, agregando los pedazos de papel filtro a dentro del cartucho de extracción, evitando el manejo manual.
El filtrado del kitazato es medido con una probeta para cuantificar el volumen de muestra.
Colocar los cartuchos de extracción en vasos de precipitados llevar a sequedad en una estufa eléctrica a 103°C durante 30 minutos, colocar el cartucho en el aparato de extracción soxhlet, con el matraz al cual previamente se le ha determinado su masa.
Nota: Identificar el número de muestra en el vaso de precipitados nunca en el cartucho.
Adicionar solvente al matraz hasta la mitad de su capacidad. Colocar I cm de altura de algodón en la parte superior del refrigerante. Dejar en reflujo durante 4 h a partir del primer ciclo de recirculación, cambiándole las condiciones de temperatrura hasta que de un ciclo cada 3 minutos aproximadamente. Una vez terminado el tiempo de reflujo vaciar y escurrir el solvente que queda en el extractor al matraz.
Evaporar el solvente en baño maría a 85°C y pasar el matraz a la estufa de vacío a una temperatura de 60°C durante 30 minutos.
Dejar enfriar el matraz en un desecador durante un período de 30 minutos y determinar su masa.
Correr una prueba testigo en las mismas condiciones que se mencionan para una muestra.
47

CALCULOS Y RESULTADOS.
Las cantidad de grasas y aceites se calcula por medio de la siguiente fórmula:
(M2 - MI) X I000 mg
V I Grasas y Aceites = ---- = ----
Donde:
M1 = Masa del matraz vacío a masa constante, en g. M2 = Masa del matraz con muestra, en g.
V = Volumen de muestra, en ml.
Nota: Si la muestra testigo contiene residuos, deberá restarse a la masa de grasa y aceite obtenido.
48

DETERMINACIóN DE COBRE. COLORIMÉTRICO DE LA NEOCUPROíiA
PRINCIPIOS Y FUNDAMENTOS
En soluciones neutras o ligeramente ácidas los iones cuprosos reaccionan con la neocuproína (2,g-dimetil- 1 ,I Ofenantrolina) para dar un complejo de cobre-neocuproína, de color amarillo, el cual se extrae con cloroformo y se cuantifica espectrofotométricamente a una longitud de onda de 457nm.
REACTIVOS
Los reactivos que a continuación se mencionan deben ser de grado analítico a menos que se indique otra cosa y cuando se hable de agua, se debe entender agua bidestilada o desionizada y excenta de cobre. Las soluciones preparadas para este análisis, deben almacenarse en recipientes de polietileno o de vidrio libre de cobre.
hidróxido de amonio concentrado a un litro con agua.
fenantrolina semihidratada en 100 ml de alcohol metílico.
Hidróxido de amonio (NH40H), 5 N. Aforar 330 ml de
Reactivo de neocuproína. Disolver 0.1 O g de 2,9 - dimetill ,I O
NOTA: En las condiciones ordinarias de almacenamiento esta solución es estable sobre un mes.
Solución de clorhidrato de hidroxilamina (NH2 OH HCI). disolver 50 g de clorhidrato de hidroxilamina en 450 ml de agua
Solución de citrato de sodio (Na3CgH507 2H20). Disolver 150 g de citrato sodio en 400 ml de agua, agregar 5 ml de reactivo de clorhidrato de hidroxilamina y 10 ml de reactivo de neocuproína; extraer con 50 ml de cloroformo la impureza de cobre de la solución y desechar la capa de cloroformo.
Solución patrón concentrada de cobre. Pesar 0.200 g de cobre electrolítico pulido y pasarlo a un matraz Erlenmeyer de 250 ml.
4 m

Agregar 10 ml de agua y 5 ml de ácido nítrico concentrado y después de que se estabilice la reacción calentar suavemente para completar la disolución del cobre y hervir hasta el total desprendimiento de los óxidos de nitrógeno.
Enfriar la solución y agregar 50 ml de agua. Transferir el contenido en un matraz volumétrico de un litro y
aforar con agua; 100 ml de esta solución equivale a 0.200 mg de cobre.
Solución patrón diluida de cobre. Transferir 50 ml de la solución patrón concentrada de cobre a un matraz volumétrico de 500 ml y aforar con agua; 1 .O0 ml de esta solución equivale a 0.20 mg de cobre.
INTERFERENCIAS
La determinación del cobre por el procedimiento que se recomienda, se encuentra virtualmente libre de interferencias por otros iones metálicos.
La interferencia del cromo se puede evitar por la adición del ácido sulfúrico para reducir los cromatos y el ión crómico complejo.
En presencia de estaño y de cantidades excesivas de otros iones oxidantes se debe emplear un volumen adicional de clorhidrato de hidroxilamina no mayor de 20 ml.
Las interferencias producidas por el cianuro y el sulfato se eliminan durante el proceso de digestión..
so

PROCEDIMIENTO
Preparación de la curva de calibración.
Preparar un blanco de referencia, colocando 50 ml de agua, 1 ml de ácido sulfúrico concentrado en un embudo de separación de 125 ml.
Agregar 5 ml de solución de clorhidrato de hidroxilamina y 10 ml de solución de citrato de sodio y mezclar bien.
Colocar una serie de embudos de separación de I25 ml y proceder como se indica.
Colocar en una serie de embudos de separación de 125 ml los volúmenes de la solución patrón diluida de cobre indicado en la siguiente tabla:
, VOLúMENES PARA LA CALIBRACION
Solución patrón diluida de Contenido de cobre en (mg)
0.5 0.01 o I .o 0.020 2.0
10.0 0.1 60 8.0 0.1 20 6.0 0.080 4. O 0.040
0.200
cobre (ml)
Diluir a 50 ml con agua, agregando un ml de ácido sulfúrico concentrado y proceder como se indica en la determinación de cobre de la muestra.
Graficar las lecturas de la absorbancia obtenidas con los mg de cobre.
TRATAMIENTO DE LA MUESTRA
Digestión con ácido nítrico- Ácido sulfúrico, (para muestras con materia orgánica fácilmente oxidable).
Tranferir a 100 ml de muestra a un vaso de precipitado de 250 ml, agregar 1 ml de Acid0 sulfúrico concentrado y 5 ml de ácido nítrico concentrado. Evaporar en una parrilla o placa de

calentamiento hasta que aparezcan humos blancos, densos de anhidrido sulfúrico en el vaso de precipitado y no calentar más alla de este punto. Este calentamiento debe hacerse bajo campana de extracción
Si la solución permanece colorida, enfriar y agregar 5 ml de ácido nítrico concentrado y repetir la evaporación hasta la aparición de humos blancos de anhídrido sulfúrico. Se debe tener la seguridad de eliminar completamente el ácido nítrico, según se indique por la claridad de la solución y por la ausencia de humos rojizos en el matraz.
Enfriar la solución a temperatura ambiente y diluir con agua cuidadosamente hasta tener un volumen de 80 ml.
Calentar en placa de calentamiento a ebullición para disolver las sales y enfriar.
Filtrar con crisol de vidrio poroso y enfriar el filtrado a un matraz volumétrico de 1 O0 ml, enfriar y aforar con agua.
Determinación de cobre de la muestra.
Tomar exactamente 50 ml o una porción adecuada de la muestra digerida que contenga 0.004 a 0.2 mg de cobre y transferirla a un embudo de separación de 125 ml. Si ha sido usado un volumen pequeño, diluir a 50 ml con agua.
Agregar 5 m1 de solución de clorhidrato de hidroxilamina y 1 O ml de solución de citrato de sodio y mezclar bien.
Ajustar el pH de la muestra aproximadamente a 4, adicionando incrementos de I ml de hidróxido de amonio 5 N.
Agregar 10 ml de reactivo de neocuproína y 10 ml de cloroformo y agitar vigorosamente durante 30 minutos para extraer el complejo de cobre-neocuproina en el cloroformo.
Dejar que la mezcla se separe en dos capas. Transferir la capa de cloroformo a un matraz volumétrico de
25 ml. Se debe tener cuidado que los extractos no arrastren gotas de agua.
Repetir la extracción de la capa acuosa con una porción adicional de 10 ml de cloroformo y agregar este extracto al anterior.
Diluir los extractos combinados con alcohol metilico y aforar a 25 ml. Tapar y mezclar cuidadosamente.

transferir la porción apropiada de la solución orgánica final a la celda de absorción.
Leer en el espectrofotómetro la absorbancia de esta solución a una longitud de onda de 457 nm usando el blanco como liquido de referencia.
CALCULOS
La concentración de cobre se cálcula por las siguientes fórmulas:
Cálculo directo mg/l de cobre=A
Cálculo por dilución
A X 1000 1 O0
B C mg/l de cobre = ---0 x --

"DETERMINACION DE FOSFORO TOTAL".
DEFINICIONES
Fósforo total.- Es la suma total del fósforo en todas sus diferentes normas .
Aguas Residual.- Líquido de composición variada proveniente de usos municipales, industrial y comercial, agrícola, pública, o privada y que por t a l motivo haya sufrido degradación o alteración en su calidad original.
Agua natural.. Líquido de composición variada, superficial o subterránea, que no haya sufrido degradación o alteración en su calidad original.
FUNDAMENTO
Los métodos se basan en transformar los compuestos fosforados a ortofosfatos, los cuales se hacen relacionar como molibdato de amonio para formar el ácido molibdofosfórico.
En el método de azul de molibdeno o del cloruro estanoso, el ácido molibdofosfórico se reduce para producir el complejo colorido conocido azul de molibdeno. La intencidad de la coloración se determina por espectrofotometría.
Por el método del ácido vanadomoilibdofosfórico, el fósforo en presencia del vanadio da lugar al complejo de fosfovanadomolibdeto produciendo una coloración amarilla cuya intencidad se detertmina por espectrofotometría.
S4

MÉTODO DEL AZUL DEL MOLIBDENO
Todo el material de vidrio empleado en esta determinación debe ser lavado con mezcla crómica, enjuagando dos veces con agua acidulada de HCI (1 :I) caliente y enjuagado dos o tres veces más con agua desionizada. Nunca usar detergentes.
Preparación de soluciones . Solución indicadora de fenolfta1eína.- Disolver 5 g de sal
de fenolftaleína en 500 ml de alcohol etílico al 95% y aforar a llitro con agua. Si es necesario agregar hidróxido de sodio 0.02 N gota a gota hasta que la solución alcance un ligero color rosado.
Solución de ácido fuerte.- Agregar lentamente 300 ml de ácido sulfúrico concentrado a 600 ml de agua, dejar enfriar y agregar 4.0 ml de ácido nítrico 69.71% y diluir a 1 litro .
Solución de molibdato de amonio 1. Disolver 25 g de molibdato de amonio tetrahidratando en 175 ml de agua.
Agregar, con mucho cuidado 280ml de ácido sulfúrico concentrado a 400 ml de agua y dejar enfriar. Añadir la solución de molibdato de amonio.
Solución de molibdato de amonio 11.- Disolver 40.1 g de molibdato de amonio en aproximadamente 500 ml de agua. Lentamente añadir 396 ml de la solución de molibdato de amonio I, enfriar y diluir a 1 litro.
Solución de cloruro estañoso 1.- Disolver 2.5 g de cloruro estañoso dihidratado en 100 ml de glicerol, Asegurando que el reactivo no haya sufrido alteración alguna, usando de preferencia uno nuevo o recientemente abierto.
Solución de cloruro estañoso ILMezclar 8 ml de la solución de cloruro estañoso I con 50 ml de glicerol. Este reactivo es estable durante seis meses.

Solución de ácido sulfúrico (1:2).
Solución de hidróxido de sodio 6N
Solución patrón de fosfatos. Pesar 219.5 mg de fosfato de potacio monobácico anhídrido. Previamente secado en estufa a 378 K (105*C), durante 2 h.
Disolver en agua, transferir la solución a un matraz volumétrico y aforar a 1 litro con agua . De esta solución tomar I O 0 ml, transferir a un matraz aforado de 1000 ml y aforar con agua. Esta solución contiene 5,O pg de P/mI.
Solución alcohólica de ácido sulfúrico.- Añadir cuidadosamente 20 ml de ácido sulfúrico concentrado, a 980 ml de metanol.
Solución de benceno alcohol isopropí1ico.- Agregar 500 ml de alcohol isopropilíco, a 500 ml de benceno.
Procedimiento
Digestión
Efectuar al mismo tiempo e igual procedimiento, una prueba testigo con agua
Tomar una alícuota de muestra de 100 ml o menos, que contenga 200 pg de P como máximo.
Transferir la alícuota de la muestra seleccionada a un vaso de precipitado de 200 ml de forma alta y agregar I ml de ácido sulfúrico concentrado y 5 ml de ácido nítrico concentrado.
Calentar hasta eliminar vapores nitrosos y dejar enfriar. Adicionar apróximadamente 20 ml de agua, una gota de
fenolftaleina. Neutralizar con hidróxido de sodio 6 N hasta ligero color rosa tenue.
Filtrar si es necesario y aforar a 100 ml con agua.
NOTA.- En caso de que la muestra presente interferencias, como color o turbiedad, es necesario llevar a cabo una
S6
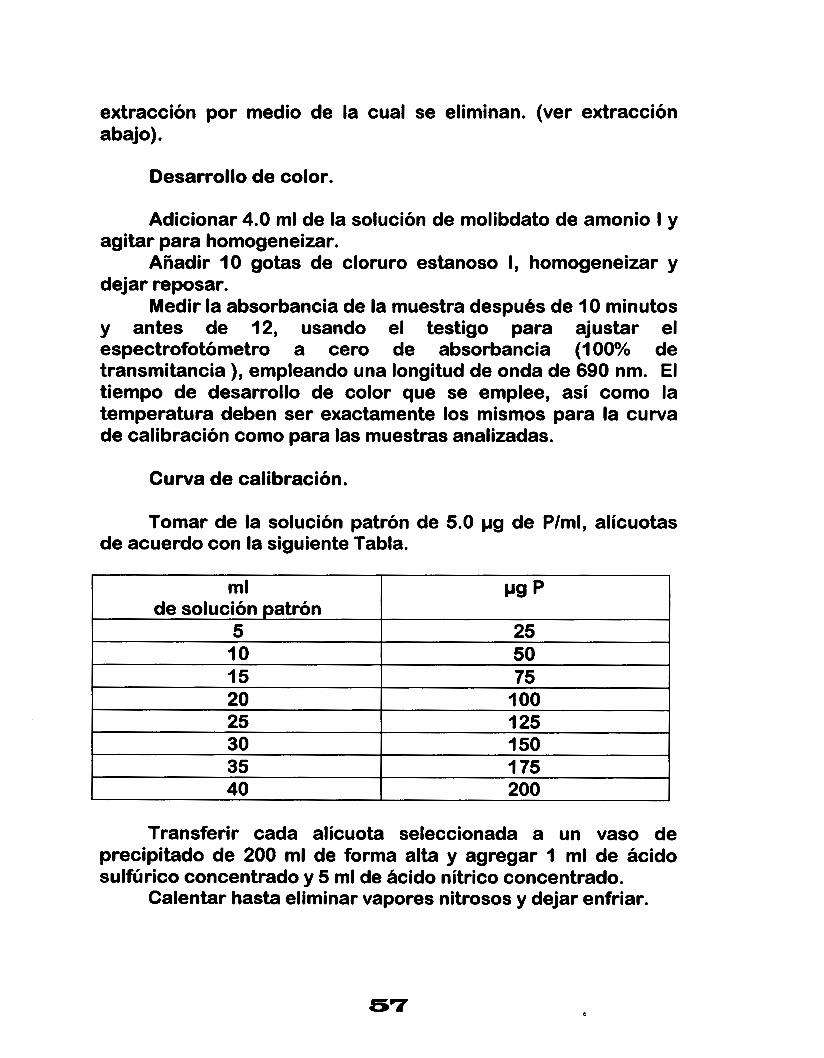
Desarrollo de color.
Adicionar 4.0 ml de la solución de molibdato de amonio I y agitar para homogeneizar.
Añadir 10 gotas de cloruro estanoso I, homogeneizar y dejar reposar.
Medir la absorbancia de la muestra después de 10 minutos y antes de 12, usando el testigo para ajustar el espectrofotómetro a cero de absorbancia (100% de transmitancia ), empleando una longitud de onda de 690 nm. El tiempo de desarrollo de color que se emplee, así como la temperatura deben ser exactamente los mismos para la curva de calibración como para las muestras analizadas.
Curva de calibración.
Tomar de la solución patrón de 5.0 pg de Plml, alícuotas de acuerdo con la siguiente Tabla.
ml Pg p
5 25 I O
200 40 1 75 35 150 30 125 25 1 O0 20 75 15 50
de solución patrón
Transferir cada alícuota seleccionada a un vaso de precipitado de 200 ml de forma alta y agregar 1 ml de ácido sulfúrico concentrado y 5 ml de ácido nítrico concentrado.
Calentar hasta eliminar vapores nitrosos y dejar enfriar.
S7 e

Adicionar apróximadamente 20 ml de agua, una gota de fenolftaleina. Neutralizar con hidróxido de sodio 6 N ligero hasta que tome un color rosa tenue.
Filtrar si es necesario y aforar a 100 ml con agua.
NOTA.- En caso de que la muestra presente interferencias,como color o turbiedad, es necesario llevar a cabo una extracción por medio de la cual se eliminan. (ver extracción ).
Desarrollo de color.
Adicionar 4.0 ml de la solución de molibdato de amonio I y agitar para homogeneizar.
Añadir 10 gotas de cloruro estañoso I, homogeneizar y dejar reposar.
Medir la absorbancia de las alícuotas después de I O minutos y antes de 12, usando el testigo para ajustar el espectrofotómetro a cero de absorbancia (100% de transmitancia ), empleando una longitud de onda de 690 nm. El tiempo de desarrollo de color que se emplee, así como la temperatura deben ser exactamente los mismos para la curva de calibración como para las muestras analizadas.
Elaborar la curva de calibración correspondiente graficando las lecturas de absorción en el eje de las abcisas contra las concentraciones de P en yg, en el eje de las ordenadas.
Extracción (en caso de interferencias).
Una vez que se haya efectuado la digestión tomar una alícuota de 40 ml de muestra, o menor y llevar a 40 ml con agua.
Transferir a un embudo de separación de 500 ml y agregar 50 ml .
Agregar 15 ml de la solución de molibdato de amonio I I, agitar durante 15 segundos y dejar reposar durante 1 O minutos.
Una vez separadas las capas acuosas y orgánica, drenarla capa acuosa.

Tomar 25 ml de la capa orgánica y transferir a un matraz aforado de 50 ml.
Agregar aprbximadamente I 5 ml do solución alcohdica ácida y I O gotas de la solucibn de cloruro de estaño II.
Diluir hast.a el aforo con solución alcohólica ácida y desarrollar el color azul.
Medir en el espectrofotómetro la absorbancia después de 15 minutos, pero antes de 30 minutos a una longitud de onda de 625 nm.
Leer en una curva de calibraci6n hecha con las soluciiines patrón, las cuales se han sometido a una extracción.
Cá1culos.
El contenido del fósforo en la muestra se cálcula mediante la siguiente fórmula:
Donde:
C= Fósforo leído en la gráfica. V= Volumen de la alicuota tomada para ía determinación,
en ml.
La diferiencía entre los resultados obtenidos en pruebas efectuadas por duplicado no debe exceder de f 1 mg/l en caso contrario, se recomienda repetir la combinación.

MÉTODO DE FOSFOVANADOMOLIBDATO.
Reactivos.
Solución de ácido sulfúrico I :2 .Se toma I O ml de ácido sulfúrico concentrado y disolver en 20 ml de agua (47.5 a 48 % en masa ).
Solución vanadato molibdato. Disolver 25 g de molibdato de amonio en 400 ml de agua. Disolver calentando a ebullición 1.25 g de metavanadato de amonio en 300 ml de agua , dejar enfriar hasta temperatura ambiente y adicionar 330 ml de ácido clorhídrico. Agregar la solución de molibdato de amonio a la de metabanadato de amonio y diluir hasta el aforo.
Solución patrón de fosfato. (ver el procedimiento del método anterior
Interferencias
AI calentar la muestra de silicio y el arcénico actuan como interferencias positivas, en tanto que las interferncias negativas provienen de arsenitos, fluoruros, torio , bismuto, sulfuros, tiosulfatos, tiocianatos, o exeso de molibdato.
El ion ferroso provoca un color azul pero este no afecta los resultados si su concentración es menor de 1000 mg /l. La interferencia de sulfuros puede ser eliminada con agua de bromo. Si se usa ácido nítrico en el testigo, los cloruros interfieren de 75 mg/l.
Procedimiento.
Efectuar al mismo tiempo e igual procedimiento una prueba testigo con agua. Tomar 35 ml de muestra la cual contenga de 50 a 1000 pg de fósforo a una alícuota llevada a 35 ml de agua.
Ajustar el pH entre 4 y I O . Si la muestra se tiene a un pH menor que 4 adicionar 50 ml de agua y si es mayor de I O
60

adicionar ácido sulfurico 1:2 empleando solución indicadora de fenoftaleína .
Calentar hasta ebullición durante 60 minutos, adicionando agua con el objeto de mantener el volumen entre 25 y 35 ml .
Restablecer el volumen original y tranferir a un matraz aforado de 50 ml añadir I O ml de reactivo de vanadomolibdato y diluir hasta la marca con agua agitando para homogenizar.
Dejar reposar la muestra durante 10 minutos, después de los cuales se mide la absorbancia de la muestra contra el testigo con el que se ajusta el aparato al00 % de transmitancia a una longitud de onda entre 400 y 490 nm, dependiendo de la sensibilidad deseada. La longuitud de onda a la cual se mide la intencidad de el color depende de la sensibilidad deseada. generalmente se hace a 470 nm y en 1 cm de profundidad óptica.
Para otros casos tenemos:
Límites (mg de fósforo/l) Longitud de onda en nm I .O-5.0 400 2.0-1 0.0 420 4.0-1 8.0 470
Curva de calibracion . De la solución patrón de 50 pg de P/ml tomar alícuotas
deacuerdo a la siguiente tabla:
Solución patrón en ml O 5 10 15 20 25
Pg de p O
250 500 750 I O00 1250
Continuar con el procedimiento de la misma forma que para las muestras. Elaborar la curva de calibracion correspondiente graficando las lecturas de absorbancia en el eje de las abscisas y las concentraciones de fósforo en el eje de las ordenadas.

Cálculos
C
V P= -
Donde:
C = pg de fósforo leidos en la gráfica V = volumen de la alícuota para la determinación en ml.
- DETERMINACION DE PLAGUICIDAS ORGANOCLORADOS - MÉTOOO DE
CROMATOGRAFCA DE GASES.
FUNDAMENTOS
El método consiste en la extracción por partición con un solo disolvente orgánico, de los plaguicidas presentes en el agua y su posterior separación y purificación por cromatografía en columna. Finalmente su cualificación y cuantificación se hace por cromatografiá gas-líquido usando un detector de captura de electrones.
Muestras
Para el control del método de extracción y purificación se efectúa en iguales condiciones y simultaneamente tres muestras: un blanco, un duplicado y una muestra de concentración conocida de plaguicidas organoclorados. Así mismo preparar un estándar de recuperación colocado en un tubo de centrífuga graduado de 15 ml la misma cantidad de

solución de plaguicidas organoclorados que se usa en la preparación de la muestra de concentración conocida. Guardar este tubo en el congelador a -20°C hasta la terminación de la preparación de las muestras y en ese momento llevarlo al mismo volumen de las muestras de concentración conocida de plaguicidas organoclorados.
Extracción
Medir en una probeta de un I de la muestra y transferirla a un embudo de separación de 2 1.
Agregar 100 ml de hexano y agitar vigorosamente al embudo durante 3 minutos, dejar reposar hasta que se separen las fases. Si se forma una emulsión añadir 5 ml de una solución saturada de sulfato de sodio.
Drenar la fase acuosa a la probeta empleada para medir la muestra.
Pasar la fase orgánica a través de una columna que contenga una capa de 5 cm de altura de sulfato de sodio pretratado. Determinar la masa de 300 g de sulfato de sodio, transferirlos a cápsulas de porcelana y calentar a 660°C en una mufla durante 48 horas. Enfriar en una estufa llO°C, transferir a frascos de vidrio ámbar, sellar con parafilm y almacenar a temperatura ambiente; recubrir la face orgánica en un matraz de fondo redondo de 500 ml.
Transferir nuevamente el agua de la probeta al embudo de separación, repetir desde la extracción con porciones de 50 ml de hexano dos veces y colectar los extractaos de hexano en el matraz de 500 ml.
Concentrar lentamente los extractos obtenidos de I a 2 ml con ayuda de vacío en un evaparador rotatorio.
Purificación
En una columna cromatográfica de 19 mm de diámetro interior, 40 cm de longitud y con llave teflón; colocar un tapón de lana de vidrio en el fondo y enjuagar con acetona y hexano, añadir 2 cm de altura de sulfato de sodio pretratado golpeando ligerarmente la columna para tener una superficie uniforme. Siguiendo el mismo procedimiento agregar 15 g de florisil

desactivado al 3 YO y en la parte superior añadir nuevamente 2.5 cm de altura de sulfato de sodio pretratado.
Con una pipeta Pasteur transferir cuantitativamente el concentrado a la columna preparada en el párrafo anterior de la siguiente manera :
Pasar el contenido de un recipiente a otro usando una pipeta Pasteur.
Enjuagar el primer recipiente con 2 ml de hexano y transferir al segundo recipiente. Repetir esta operación por tres veces más.
Esperar a que el disolvente baje a 1 mm de la superficie de la capa superior del sulfato entre cada enjuague.
Nota: nunca permitir que la columna se seque.
Enseguida del Último lavado eluir el extracto que se encuentra en la columna con 200 ml de una mezcla benceno- hexano (1:l). Dejar escurrir el disolvente por la pred de la columna para que no se altere la superficie del sulfato de sodio y recoger el aluato en un matraz balón de 500 ml con boca esmerilada 24/40.
Concentrar lentamente el eluato hasta aproximadamente I ml con ayuda de vacío en un evaporador rotatorio.
Transferir el concentrado anterior cuantitativamente, como se menciona en los incisos marcados con el asterisco (*), a un tubo de centrífuga graduado de 15 ml hasta completar un volumen de 10 ml.
PROCEDIMIENTO
El análisis cromatográfico consiste en identificar los plaguicidas organoclorados presentes en la muestra problema de acuerdo a su tiempo de retención. Se inyectan en el cromatógrafo alternadamente de 2 a 3 pl de las muestras a analizar y los patrones individuales de los plaguicidas bajo las condiciones de operación adecuadas al tipo de aparato con el que se este trabajando.
64

Cualificación
Inyectar primero el blanco, el estándar de recuperación y la muestra de concentración conocida de plaguicidas organoclorados, enseguida, inyectar en forma alternada las muestras problema y los patrones individuales de los plaguicidas cuando menos en dos columnas cromatográficas de empaque con polaridad diferente para identificar con seguridad los compuestos organoclorados que tienen cada una de las muestras.
Cuantificación
La cuantificación se efectúa usando cualquiera de las dos columnas cromatográficas empleadas en la cualificación. Así, simultáneamente se confirma la identidad de cada plaguicida. La cuantificación se hace relancionando las alturas de los picos de las soluciones patrón utilizando la siguiente fórmula:
mg Am. Vie. Ce. X . ATm
I Ae Vim . Vm ATe -=- - "- I o6
En donde:
Am = Altura del pico de la muestra, en cm. Ae = Altrura del pico del patrón, en cm. Vie = Vilumen inyectado del patrón, en TI Vim = Volumen inyectado de la muestra, en TI Ce = Concentración del patrón, en g/ml X = Volumen de dilución de la muestra extraída, en ml Vm = Volumen inicial de la muestra, en ml Tm (*) = Atenuación del amplificador en la muestra ATe (*) = Atenuación del amplificador en el patrón I O 6 = Constante para transformar de g/ml a mg/l
Cuando se trabaja a una misma atenuación no es necesario poner estos factores (*) en la fórmula.

DETERMINACIÓN DE METALES
FUNDAMENTO
Este metodo se basa en la medición de la cantidad de luz monocromática absorbida por el elemento atomizado a determinarse en una flama, por medio de un detector, siendo dicha energía absorbida proporcional a la concentración del elemento.
INTERFERENCIAS
En la determinación de la mayoría de los metales que pueden determinarse por aspiración directa, la interferencia más frecuente es la llamada química que se debe a que el metal por analizar se encuentra en estado molecular y para que exista absorción deberá estar en forma atómica. Otras interferencias se deben a cationes o aniones presentes en la muestra. Para eliminar todo tipo de interferencias en cada elemento será necesario consultar el manual de operación del aparato que se disponga.
Preparación de la muestra
El tratamiento de la muestra dependerá del tipo de metales que se van a determinar. Si se requiere metales totales o extractables no se debe filtrar la muestra; si se desea determinar metales disueltos, la muestra se filtra a través de una membrana de 0.45 micrómetros (previamente lavada con una solución de ácido nítrico I :I).
En cualquier caso acidificar la muestra con ácido nítrico concentrado hasta un pH de 2 o menor.
Si la muestra contiene materiales suspendidos o materia orgánica se requiere otro tratamiento específico que consistirá en una digestión (Continuar calentando cuando sea necesario, añadir ácido nítrico concentrado hasta que la digestión sea completa, esto se indica por un residuo de color claro).
66

Debe ser posible, filtrar y acidificarla muestra en el momento de su extracción en el campo, dependiendo del tipo de análisis que se vaya a efectuar.
Para mercurio, arsénico y selenio se necesita una digestión especial. Algunas aguas contaminadas pueden requerir el uso adicional de ácido sulfúrico ylo perclórico para una digestión completa.
Nota.Se recomienda analizar la muestra lo más rápidamenta posible, de no ser así mantenerla en refrigeración a 278K (5°C).
Tratamiento de la muestra para el análisis de metales totales.
Transferir una porción representativa de la muestra bien mezclada (50 a 100 ml) a un vaso de precipitado, agregar 5 ml de ácido nítrico concentrado y cubrir el vaso con un vidrio de reloj. Evaporar casi a sequedad en una placa de calentamiento asegurándose que la muestra no hierva.
Enfriar el vaso de precipitado y agregar otros 5 ml de ácido nítrico concentrado. Volver a cubrir el vaso y regresarlo a la placa de calentamiento. Aumentar la temperatura hasta reflujo lento.
Continuar calentando cuando sea necesario, añadir ácido nítrico concentrado hasta que la digestión sea completa, esto se indica por un residuo de color claro.
Agregar 1 o 2 ml de ácido nítrico concentrado y calentar el vaso de precipitado lñigeramente para disolver el residuo.
Lavar la paredes del vaso y el vidrio de reloj con agua. Filtrar la muestra para evitar que los silicatos y otros
materiales insolubles obstruyan el atomizador. Aforar el volumen a 50 ml o 100 ml según el volumen de
muestra tomado originalmente. La muestra está lista para ser analizada.
Tratamiento de la muestra para el análisis de metales suspendidos.
Para la determinación de la concentración de metales en material suspendido, se filtra la muestra con un filtro de membrana de 0.45 micrómetros y se produce a la digestión del
6 7

filtro junto con el material que contenga como se indicó en tratamiento anterior.
Corre un blanco de filtro que permita una corrección en la muestra
PROCEDIMIENTO
Operación del Instrumento
Debido a las diferencias que existen entre los distintos espectrofotómetros no es posible formular las instrucciónes aplicables a todos los instrumentos. En general se procede de acuerdo a los siguientes pasos.
Determinación del cadmio, cromo, cobre, plomo, níquel y zinc por aspiración derecta en una flama de aire-acetileno.
Instalar una lámpara de cátodo hueco del metal deseado en el aparato, la cual debe de estar alineada de acuerdo con las instrucciones del fabricante y seleccionar la longitud de onda.
Las longitudes de onda más usuales se dan en la siguiente tabla.
Poner la abertura de la ranura de acuerdo con lo sugerido por el instructivo del aparato para el elemento que se va a medir.
68

Encender el aparato y aplicar a la lámpara de cátodo hueco la corriente sugerida por el fabricante.
Dejar que el aparato se caliente hasta que se estabilice la fuente de energía; normalmente tarda de I O a 20 minutos. Reajustar la corriente como sea necesario después de calentar.
Conectar el aire y ajustar el gasto a lo especificado por el fabricante para obtener la máxima sensibilidad para el metal que se va a medir.
Conectar el acetileno, ajustar el gasto al valor especificado y encender la flama.
Atomizar agua acidificada con 1.5 ml de ácido nítrico concentrado/L; revisar la velocidad de aspiración por in minuto y ajustar a cero el aparoto.
Atomizar un estándar (normalmente de 0.5 mg/l) y ajustar el quemador tanto en sentido vertical, horizontal y ytransversal así como la velocidad de aspiración hasta obtener una respuesta máxima.
El aparato está listo para correr los estándares y las muestras. Cuando se termine de trabajar, extinguir la flama desconectando primero el acetileno y luego el aire.
Determinación de bario por aspiración directa en una flama de óxido nitroso-acetileno.
Instalar una lámpara de cátodo hueco para la determinación de bario, la cual debe de estar alineada de acuerdo con las instrucciones del fabricante y seleccionar la longitud de onda.
Colocar la abertura de la ranura para el bario de cuerdo con lo sugerido por el fabricante.
Encender el aparato y aplicar a la lámpara de cátodo hueco la corriente sugerida por el fabricante.
Dejar que el aparato se caliente hasta que se estabilice la fuente de energía; normalmente tarda de 10 a 20 minutos.
lntalar la cabeza del quemador recomendada para óxido notroso.
Conectar el acetileno (sin encender la flama) y ajustar el gasto al valor especificado por el fabricante para una flama de óxido nitroso-acetileno.
Instalar la cabeza del quemador.
Desconectar el acetileno.
69

Con los suministros de aire y óxido nitroso en funcionamiento, conectar la válvula "t" al óxido nitroso y ajustar el gasto de acuerdo con las especificaciones del fabricante.
Girar la llave de la vávula a la posición del aire y verificar que el gasto sea el mismo.
Conectar el acetileno y encender la flama a un amarillo brillanate.
Con un movimiento rápido, girar la válvula al óxido nitroso. La flama debe ser rosa-rojiza; si no es así, ajustar el paso del combustible para obtener un cono rojo en la flama.
Atomizar agua que contenga 1.5 ml de ácido nítrico concentradolL y revisar la velocidad de aspiración.
Atomizar un estándar del 1 mgll del metal y ajustar el quemador tanto en sentido vertical, horizontal y transversal hasta obtener una respuesta máxima.
El aparato está listo para correr los estándares y las muestras.
Para extinguir la flama, girar la llave de la válvula de óxido nitroso a aire y cerrar la llave del acetileno. Este procedimiento elimina el peligro de un flamazo que podría ocurrir al encender o apagar directamente el óxido nitroso y el acetileno.
Determinación de mercurio por la absorción atómica del vapor frío.
Instalar la lámpara de cátodo hueco para el mercurio en el aparato, la cual debe de estar alineada de acuerdo con las instrucciones del fabricante y seleccionar la longitud de onda.
Poner la abertura de la ranura para mercurio de acuerdo con lo sugerido por el fabricante.
Encender el aparato y aplicar a la lámpara de cátodo hueco la corriente sugerida por el fabricante.
Dejar que el aparato se caliente hasta que se estabilice la fuente de energía ; normalmente tarda de 10 a 20 minutos .
Instalar la celda de adsorción y alinear el paso de la luz para dar una transmisión máxima.
Conectar el equipo asociado a la celda de absorción con tubería de plástico de vinilo como se indica en la figura No. l .
Conectar el aire y ajustar el gasto a 2 L / min y dejar que el aire fluya continuamente.
En matraces Erlenmeyer de 250 ml, preparar soluciónes estándares de 100 ml (ml) cada una que contenga I .0,2.0 y 5.0
7 0

ug/l de mercurio respectivamente y un testigo de 100 ml de agua.
A cada matraz añadir 5 ml de ácido sulfúrico concentrado y 2.5 ml de ácido nítrico concentrado.
A cada matraz añadir 15 ml de solución de permangato de potasio y dejar reposar por 15 minutos.
A cada matraz añadir 8 ml de solución de persulfato de potasio y calentar por 2 horas en un baño maría a temperatura de ebullición. Para mercurio, arsénico y selenio se necesita una digestión especial.Algunas aguas contaminadas pueden requerir el uso adicional de ácido sulfúrico y/o perclórico para una digestión completa.
Enfriar a la temperatura ambiente y añadir 6 ml de solución de sulfato de hidroxilamina-cloruro de sodio para reducir el permanganato de aereación.
A medida que el mercurio se volatiliza y es llevado a la celda de absorción, la absorción espectral aumentará a un máximo en cuestión de segundos. Una vez obtenida la máxima absorción espectral se hace pasar por el filtro con carbón activado para limpiar la línea.
Tan pronto como el registrador regresa aproximadamente a la linea base, quitar el aereador del matraz de reacción y reemplazarlo con un matraz que contenga agua.
Enjuagar el sistema por unos cuantos segundos y correr por el sigiente estándar de la misma manera.
Contruir una curva estándar, gratificando alturas de pico contra microgramos de mercurio.
Correr las muestras tratándolas de la misma manera. Determinación de arsénico y selenio con el atomizador de
grafito de alta temperatura. Principio: La mayoría de los métodos de absorción atómica a
la flama requieren algún tipo de preconcentración de la muetra para obtener la sencibilidad requerida para determinar metales en concentraciones del orden pgll. El atomizador de gráfito permite determinar trazas de metales directamente, y se basa en la técnica de calentar el tubo de gráfito con una corriente eléctrica que es pasada por sus paredes, lográndose así una alta temperatura que permite la atomización de la muestra previamente introducida en el tubo

de gráfito. El tubo de gráfito se pone en dirección longitudinal con respecto al haz de luz del espectrofotómetro. Los extremos de tubo estan conectados a unos electrodos de grafito, y estos a su vez están acoplados a una fuente de energía eléctrica.
El tubo y los electrodos están encerrados en una camisa meMica que permite la circulacvión de agua de enfriamiento.
Para inhibir la oxidación del tubo de grafito, el interior del mismo se purga continuamente con un gas inerte. El tubo de grafito tiene pequeños orificios, unos para que fluya el gas y otro para introducir la muestra. Para la introducción de la muestra se recomienda el uso de pipetas microlíticas con puntas deshechables de plástico. Se recomienda consultar las instrucciones del fabricante para detalles muy específicos en cuanto al uso del equipo, ya que debido a las diferencias que existen entre éstos, no es posible formular instrucciones aplicables a todos ellos.
Introducir la muestra utilizando una geringa o micro pipeta adecuada.
Evaporar el disolvente de la muestra pasando una pequeña corriente eléctrica por el cilindro.
Se necesita convertir la muestra a cenizas, para esto la temperatura en el tubo de grafito debe llevarse a un nivel apropiado hasta que el residuo de la muestra se convierta en cenizas.
Finalmente, se aplica la corriente eléctrica en su totalidad y el elemento de interés se atomiza rápidamente y se registra la señal.
La duración de la señal de absorción es de una fracción de segundos debido a la volatilidad del elemento, por lo cual se debe utilizar un registrador.
En el caso de existir diferencias debido al efecto de absorción de fondo no específica por la dispersión de la luz o absorción molecular, se recomienda consultar la bibliografia existente en cuanto a los métodos disponibles para eliminarla, así como en el caso de diferencias de matriz.
Nota: No se dispone actualmente de suficiente experienciacon este equipo como para hacer generalizaciones.
72

La sencibilidad absoluta de la absorción atómica con el atomizador de grafito es palpablemente mejor que la que se obtiene con el método de nebulizador de flama.
CALCULOS Preparar una curva de calibración obtenida de las lecturas
de los estándares. Determinar el contenido del metal en la muestra, por medio
de la curva, de calibración dependiendo de la absorción espectral leida.
Determinación directa.
Cuando los valores de los metales se determinan en términos de mg/l en la curva de calibración, calcular la concentración de la muestra mediante la siguiente expresión:
mg/l de metal en la muestra = D
mg/lde metal leídos en la curva X --- I O00
Volumen final total en ml
ml de muestra D (Factor de dilución)= --------
73

DEMANDA BlOQUíMlCA DE OXíGENO CJ 3 ‘7 f-
1 -4 4 J
PRINCIPIO.
Este método se basa en la cantidad de oxígeno requerido por los microorganismos para efectuar la oxidación de la materia orgánica presente en el agua y se determina por la diferencia entre el oxígeno disuelto inicial y el oxígeno disuelto al cabo de 5 días de incubación a 20°C.
En la Norma Ofkial Mexicana se dan las siguientes definiciones de importancia:
Oxígeno disuelto: Es la concentración de oxígeno libre que se encuentra presente en el agua, dependiendo dicha concentración de la temperatura, presión, salinidad y otros parámetros.
Demanda bioquímica de oxígeno (DB05): Es una estimación de la cantidad de oxígeno que requiere un población microbiana heterogénea para oxidar la materia orgánica de una muestra de agua.
Sello hidráulico: Capa de agua situada entre el tapón y el borde de la botella de DBO, que sirve para impedir el flujo de gases o cualquier otra sustancia extraña al interior de la botella que pudiera alterar la muestra.
Incubación: Consiste en dejar reposar durante 5 días la muestra a una temperatura constante de 2OoC en un medio seco.
Inóculo: Es un suspención de microorganismos vivos que se han adaptado para reproducirse en un medio específico.
Biota: Es un conjunto de organismos vivos tanto de origen vegetal como animal.
Medio aerobio: Es aquel en el cual se desarrollan microorganismos en presencia de oxígeno molecular.
Medio anaerobio: Es aquel en el cual se desarrollan microorganismos en ausencia de oxígeno molecular.
74

OXíGENO DISUELTO
Para calcular la demanda bioquímica de oxígeno es necesario determinar el oxígeno disuelto de la muestra.
FUNDAMENTO
El método se basa en la oxidación del ión manganoso a ión mangbnico por el oxígeno disuelto en medio fuertemente alcalino. Posteriormente, al acidificar la solución en presencia de un yoduro, el ión mangánico presente oxida el yoduro y libera yodo en una cantidad equivalente al oxígeno disuelto que existía originalmente en la muestra.
El yodo libre se titula con una solución valorada de tiosulfato de sodio.
REACTIVOS
Se deben preparar previamente las siguientes soluciónes: Los reactivos utilizados deben ser grado analítico, cuando
se hable de agua, se entiende agua destilada.
Sulfato manganoso: Disolver 480 g de MnS04.4H20 o bien 400 g de MnS04-2H20 o 364 g de MnS04eH20 en agua, filtrar y aforar a un litro (11). Esta solución se debe utilizar siempre y cuando no dé color con el almidón al adicionarle una solución ácida de ioduro de potasio.
Solución alcali-ioduro-nitruro: Disolver 500 g de NaOH y 136 g de Nal o 700 g de KOH y 150 g de KI en agua, diluir a un litro con agua. A esta solución, agregar 10 g de NaN3 disueltos en 40 ml de agua. Esta solución no debe dar color con solución de almidón cuando se diluya y acidifique.
Solución de floruro de potasio: Disolver 40 g de KF-2H20 en agua y aforar a 1 O0 ml.
7s

Solución indicadora de almidón: Disolver 10 g de almidón en un litro de agua hirviendo, dejar hervir durante 3 o 4 minutos y dejar reposar durante un mínimo de I 2 hr. Usar unicamente el I
líquido sobrante. %, +?(
* '
t??, ~ .;d $?
Solución patrón de tiosulfato de sodio, 1.ON: Disolver 248.2 g de Na2S203-5H20 en agua recién hervida y enfriada, se afora a un litro. Si se desea preservar la solución debe añadirse 5 ml de cloroformo o un gramo de hidróxido de sodio.
Solución valorada de tiosulfato en sodio, 0.025 N: Diluir 25.0 ml de la solución patrón y aforar a un litro. Un mililitro de la solución valorada de tiosulfato 0.025 N es equivalente a 2 mg de oxígeno disuelto.
Solución amortiguadora de fosfato: Disolver 8.5 g de fosfato monobásico de potasio (KHzPO~), 33.4 g de fosfato dibásico de sodio heptahidratado (Na2HP04*7H20), 21.75 g de fosfato dibhsico de potasio (K2HP04) y 1.7 g de cloruro de amonio (NH4CI) en 500 ml de agua y aforar a un litro, el pH de esta solución amortiguadora debe ser 7.2 sin ajuste alguno.
Solución de sulfato de magnesio: Disolver 22.5 g de sulfato de magnesio heptahidratado (MgSO4'7H20) en agua y diluir a un litro.
Solución de cloruro de calcio: Disolver 27.5 g de cloruro de calcio anhidrido (CaC12) en agua y diluir a un litro.
Solución de cloruro ferrico: Disolver 0.23 g de cloruro férrico hexahidratado (FeC13-6H20) en agua y diluir a un litro.
Solución de ácido sulfúrico, 0.1N: Diluir 4.9 g aproximadamente 3 ml de ácido sulfúrico (H2SO4) concentrado de densidad 1.841 g/ml en agua y aforar a un litro.
, .. 1 ' .
Solución de hidróxido de sodio, 0.1 N: Disolver 1.0 g de hidróxido de sodio (NaOH) en agua y aforar a un litro.
7 6

PROCEDIMIENTO PARA CALCULAR EL OXkENO DISUELTO.
(metodo de Winkler, modificado de Alsterberg)
En el mismo frasco para oxígeno disuelto en el cual se recolecto la muestra, se procede de la siguiente manera:
Se agregan 2 ml de sulfato manganoso, 2 ml de solución alcalina-ioduro-nitruro. Ambas adiciones se deben hacer por abajo de la superficie del líquido.
Se coloca el tapón con cuidad para evitar burbujas dentro del frasco, se agita varias veces el frasco por inversión, se deja que se sedimente el precipitado hasta aproximadamente 213 de la altura del frasco.
Se destapa cuidadosamente el frasco y se agrega en seguida 2 ml de ácido sulfúrico concentrado, dejando que este escurra por el cuello del frasco, se tapan nuevamente y se agita hasta que la disolución sea completa.
Se deja reposar 5 minutos y se toma un volumen de 100 ml se titula con tiosulfato de sodio 0.025 N, hasta un color amarillo paja. Se adiciona de 1 a 2 ml de solución de almidón (da un color azul), se continua la titulación hasta la primera desaparición del color azul, se anotan los ml de tiosulfato gastados.
CÁLCULOS Y RESULTADOS
La concentración de oxígeno disuelto debe calcularse en la muestra con la siguiente formula:
donde: a = volumen de tiosulfato gastados (ml). N = normalidad de tiosulfato de sodio. b = Volumen de muestra usados (98.7 ml).
O2 = Oxígeno disuelto.

nota: Los 98.7 ml son por el desplazamiento de volumen con la adición de los reactivos
Observaciones. Se recomienda usar el método electrométrico cuando
haya en la muestra interferencias de color o presencia de sustancias oxidantes y reductoras, ya que se deben considerar los efectos que estos materiales sobre el método de Winkler modificado, debiéndose apegar al instructivo del fabricante del equipo,.
La modificación por floculación con alumbre, debe usarse en presencia de sólidos suspendidos, que interfieren con el desarrollo del método. También se puede llevar a cabo esta floculación por el uso de sulfato de cobre- ácido sulfámico cuando el método se usa en análisis de Iodos activados.
PROC€DIMIENTQ PARA D€T€RMINAR LA DEMANDA 6lOQU.MlCA DE OX¡G€NO.
Método de dilución.
Preparación del agua de dilución. Calcular la cantidad de agua de dilución dependiendo del número de muestras. Efectuar dos diluciones diferentes como mínimo para cada muestra.
En un garrafón previamente lavado con agua, agregar 1 ml de cada una de las siguientes soluciónes: amortiguador de fosfato, sulfato de magnesio, cloruro de calcio y cloruro férrico, para cada litro de agua. Aerear hasta completa saturación.

TABLA DE DILUCIONES RECOMENDADAS
TIPOS DE % DE DILUCIóN DBO5 (ESTIMADA DESECHOS EN mgll)
Desecho industrial 0.1 - 1.0 500-500 concentrado
domésticas
Aguas de río 25.0 - 100.0 5-20 contaminadas -
Aguas residuales
5.0 - 25.0 200 - 100 Efluentes tratados
1 .O - 5.0 1 O0 - 500
Medir directamente en 3 botellas de 300 ml tipo DBO volúmenes apropiados de la muestra por cada dilución, usando un pipeta volumétrica de punta alargada y llenar las botellas con el agua de dilución justamente para que el tapón pueda colocarse sin dejar burbujas de aire.
Determinar el oxígeno disuelto inicial (ODI) en una de las botellas de DBO. A otra de estas botellas determinar el oxígeno disuelto a los I 5 minutos de haber hecho el cálculo a la primera botella
La última botella se mete a la incubadora a 20°C durante 5 dias manteniendo el sello hidráulico en el termino de ese tiempo y determinar la cantidad de oxígeno disuelto en la muestra.
La DBO5 (demanda bioquímica de oxígeno al quinto día) se calcula según la siguiente fórmula:
OD15 - ODF
F. D DBO5 = -
Donde: OD1 5 = oxígeno disuelto a los 15 minutos ODF = oxígeno disuelto final, después de 5 dias de
F.D = factor de dilución, % de dilución expresado en
DBO5 = demanda bioquímica de oxígeno al quinto día
incubación
decimales.

Inoculación.
El objeto del inóculo es introducir en la muestra una población biológica capaz de oxidar la materia orgánica que convenga.
Cuando tales microorganismos ya están presentes, como en las aguas residuales domésticas o efluentes no clorados y en aguas superficiales, no es necesario inocular las muestras.
Cuando haya razón para creer que la muestra contiene muy pocos microorganismos, como resultado de la cloración, temperatura elevada o pH extremo, el agua de dilución debe ser inoculada.
La selección del inóculo adecuado es un factor importante en la determinación de DBO. Muchos desechos industriales contienen compuestos orgánicos que no están sujetos a la oxidación del inóculo comúnmente usado (inóculo de las aguas residuales domésticas ), en estos casos puede usarse un inóculo preparado del suelo, aclimatado y desarrollado en el laboratorio, o el agua receptora colectada abajo del punto de descarga del desecho en particular, (de 3 a 5 Km abajo). Los dos últimos son los de mayores posibilidades. El agua receptora usada como fuente de inóculo indudablemente dará la mejor estimación del efecto de un desecho en tal agua. Debe ser colectada en un punto en donde se haya formado una biota capaz de usar como alimento los compuestos orgánicos particulares presentes. En algunos casos éstos pueden asegurar la selección de un inóculo satisfactorio muchos kilometro abajo del punto de descarga del desecho, lo cual no es práctico con desechos periódicos difícilmente suceptibles a la oxidación biológica, es más práctico formar un inóculo aclimatando el desecho o agua receptora con pequeños incrementos diarios del desecho particular junto con el agua residual doméstica, hasta que se desarrolle un inóculo satisfactorio.
Preparación del agua de dilución con inóculo.
Se prepara el agua de dilución con el inóculo más satisfactorio para el desecho particular en estudio. Solamente experencias pasadas pueden determinar la cantidad efectiva

del inóculo que se agrega por litro, sin embargo, puede servir utilizar 1 - 10 ml de agua residual doméstica por litro de agua de dilución o 10 - 50 ml de agua de río por litro de agua de dilución incubhndose durante 36 hr.
El agua de dilución inoculada debe usarse el mismo día que se prepare.
Incubación con inóculo.
Calcular el porcentaje de inóculo que se requiere para producir por lo menos una DBO (5 días) de 0.5 mgll . Calcular las diluciones del desecho particular del agua como se ilustra en la tabla de diluciones. Se disminuye la concentración del desecho lo suficiente para tomar en cuenta la utilización del oxígeno por el inóculo. Medir la cantidad de desecho que se requiera, al igual que en la tabla de diluciones. Agregar a la muestra aproximadamente la mitad de la cantidad de agua de dilución que se requiere. Esto es necesario para asegurar que el desecho concentrado no es causa de toxicidad a los organismos del inóculo.
Una vez incubado el inóculo determinar ODI, OD15 y ODF, como ya se explico anteriormente, el DBO5 se obtiene:
Corrección por demanda de inóculo.
El valor de la corrección por la demanda del inóculo se obtiene determinando la DBO del mismo. Determinar el abatimiento de oxígeno del inóculo estableciendo una serie separada de diluciones de éste y seleccinando aquella que consuma del 40-70% de oxígeno al quinto día, uno de estos abatimentos se usa luego para calcular la corrección debida a la pequeña cantidad de inóculo al agua de dilución .

81 - B2 DBOC =-
% de dilución
Donde : DBOc = Demanda bioquímica de oxígeno corregida BI = Oxígeno disuelto del agua de dilución inoculada antes
B2 = Oxígeno disuelto del agua de dilución inoculada
El % de dilución se refiere al inóculo.
de la incubación en mg/l . después de la incubación en mgll .
La corrección por inóculo de la demanda bioquímica de oxígeno al quinto día utilizando inóculo queda expresada por :
DB05c = DBO5 - DBOC
Donde:
corregido DB05c = Demanda bioquímica de oxígeno a los cinco días
DBO5 = Demanda bioquímica de oxígeno a los cinco dias DBOc = Demanda bioquímica de oxígeno corregido

DETERMINACIóN DE SÓLIDOS SEDIMENTABLES
FUNDAMENTO.
Los métodos a los que se refiere la Norma Oficial Mexicana, se basan en la propiedad que tienen los materiales sólidos, de acentarse en niveles progresivos de a cuerdo a sus diferentes densidades.
En la norma mexicana se definen los siguientes conceptos de importancia:
Sedimentación.- Es la operación por medio de la cual, las particulas sólidas supendidas en un líquido se asientan debido a la fuerza de la gravedad, dependiendo de su densidad.
Sólidos sedimentab1es.- Son las partículas sólidas que se depositan en el fondo de un recipiente debido a la operación de sedimentación.
PROCEDIMIENTOS.
Determinación volumétrica. - Mezclar cuidadosamente la muestra original a fin de
asegurar una muestra homogénea de sólidos suspendidos a través de todo el cuerpo de líquido.
Una vez mezclada la muestra se llena el cono de sedimentación tipo lmhoff hasta el aforo de un litro y dejar reposar durante 45 minutos para que se acienten las partículas, una vez transcurrido ese tiempo, agitar suavemente los lados del cono con un agitador de vidrio o por rotación para que se sedimenten los sólidos adheridos a las paredes del cono y dejar reposar la muestra otros 15 minutos. Leer directamente en el cono Imhoff.
Determinación gravimétrica.- Se procede de la misma manera que se indica en la
determinación volumétrica, posteriormente se elimina por decantación el agua del cono y se vierte en material sedimentado en un vaso de precipitados, recuperando

totalmente las partículas sedimentadas agregando un poco de agua destilada al cono.
Filtrar el material recuperado a través de un papel filtro previamente tratado, empleando un equipo de vacío.
Pasar el material retenido junto con el papel filtro a una cápsula previamente tarada y secar en una estufa a 100-110°C ( 60 minutos aproximadamente ) inmediatamente introducirla al desecador hasta peso constante; usando la balanza analítica, por diferiencia de peso se conoce el contenido de sólidos sedimentables.
CALCULOS
Para determinar el peso de sólidos sedimentables por litro se utiliza la siguiente fórmula:
A - B S.Se =
V
S.Se = sólidos sedimentales, en mgll . A = peso papel filtro y cápsula con los sólidos
B = peso del papel filtro y cápsula en mg. V = volumen de la muestra original en 1.
Donde:
sedimentables en mg.

FENOLES
PRINCIPIO
Los fenoles purificados reaccionan con la 4 aminoantipirina a un pH de 10 ~f 0.2 en presencia de ferricianuro de potasio para formar una anilina de antipirina, esta anilina es extraida de la solución acuosa con cloroformo.
REACTIVOS
Los reactivos que a continuación se mencionan deben ser grado analítico, cuando se hable de agua se debe entender agua destilada y desionizada, exenta de fenoles y cloro.
Solución de sulfato de cobre (CuS04*5H20).- Disolver
Solución de ácido fosfórico (H3P04).- Diluir I O ml de
Indicador de anaranjado de metilo .- Disolver 0.5 g de
Solución de ácido sulfúrico (H2SO4 ) 1N.- Diluir 26.6 ml
Cloroformo (CHsCI3) o eter etilico (C2H5)2O. Cloruro de Sodio (NaCI). Solución de hidróxido de sodio (NaOH) 2.5N.- Disolver
100 g de hidróxido de sodio en agua y aforar a 1 litro Solución de bromato-bromuro O.IN.- Disolver 2.78 g de
bromato de potacio (KBr03) en agua, agregar 10 g de bromuro de potasio en cristales (KBr), disolver y diluir hasta 1 litro.
Solución de tiosulfato de sodio (Na2S203) 0.25N.- Disolver 6.205 g de tiosulfato de sodio en agua recientemente hervida y enfriada aforar a 1 litro, agregar 5 ml de cloroformo como preservante.
Solución de cloruro de amonio (NHqCI).-Disolver 50 g de cloruro de amonio en agua, diluir a 1 litro
Solución de 4 aminoantipirina .- Disolver I00 ml preparar la solución del día que se va a usar.
IOOg de CuSO4. 5H20 en agua y diluir en I litro.
H3P04 al 85% con 9 ml de agua.
anaranjado de metilo en 1 litro de agua.
de ácido sulfúrico ( densidad = 1.84) con agua y aforar a 1 litro

Solución de ferricianuro de potasio K3Fe (CN)6.- Disolver 8 g de ferricianuro de potasio en agua y diluir a 100 ml. Filtrar si es nesesario. Preparar la solución cada semana.
Sulfato de sodio anhídrido granular (Na2S04). Yoduro de potasio en cristales (KI). Hidróxido de amonio concentrado (NH40H). Solución de almidón .- Preparar una suspencion de 5 g de
almidón en agua fria y verterla en 800 ml de agua en ebullicion, hervir durante 5 minutos con agitación enfriar y diluir a ?litro, dejar reposar de 12 a 24 horas . Como preservante se puede usar 1.25 g de ácido salicílico o gotas de tolueno, esta solución debe preservarse de la luz y en rifregeración.
Solución madre de fenol.0 Disolver 1 g de fenol en agua y diluir a 1 litro.
Normalización de la solución madre de fenol. En un matraz Erlenmeyer, con tapón de vidrio agregar 100
ml de agua, 50 ml de HCI concentrado y agitar el matraz suavemente.
Si el color café del bromo no persiste, agregar porciónes de 10 ml de solución de bromato - bromuro 0.1 N hasta que persista el color.
Dejar reposar 10 minutos y agregar 1 g de KI. Preparar un testigo usando agua y los mismos reactivos
Titular con solución de tiosulfato de sodio (0.025N) usando
Solución intermedia de fenal.- Diluir a ml de la solución
mencionados anteriormente.
solución de almidón como indicador.
madre en 1 litro de agua, un ml de esta solución equivale a IO ug de fenol. Preparar la solución el dia que se va a usar.
Solución patrón de fenol.0 Diluir 50 ml de solución intermedia de fenol en 500 ml de agua; 1ml de esta solución equivale a I ug de fenol. La estabilidad de esta solución es de 2 horas.
INTERFERENCIAS.
Las aguas residuales domésticas e industriales pueden contener interferencias como bacterias en descomposición fenólica sustancias oxidantes, reductoras, y valores alcalinos de pH.

Preparación de la curva de calibración. Tomar diferentes cantidades de la solución patrón de fenol como se muestra en la siguiente tabla:
ml tomados de la solución ug de fenol patrón de fenol
O O 5
30 30 20 20 I O I O 5
40 50
40 50
La solución patrón de cada serie se aforan a 500 ml con agua y se tratan como sigue:
Agregar 10 ml de solución de cloruro de amonio y ajustar el pH a I O 2 0.2 con hidróxido de sodio concentrado. Pasar a un embudo de separación de 1 litro, agregar 3 ml de la solución de 4 aminoantipirina mezclar bien, agregar 3 ml de solución de ferricianuro de potasio. Mezclar nuevamente y dejar que el cloro se desarrolle en 3 minutos.
La solución debe de estar transparente y ligeramente amarilla.
Extraer inmediatamente con 25 ml de cloroformo; agitar el embudo de separación por lo menos 10 veces dejar que el cloroformo se separe agitar de nuevo 10 veces y dejar que el cloroformo se vuelva a separar.
Filtrar el extracto clorofórmico a través del papel filtro que contenga una capa de 5 g de sulfato de sodio anhidrido, con el fin de eliminar la humedad.
Colectar el extracto seco en seldas y medir la absorbancia a 460 nm ajustando el aparato a cero con el primero de la serie y realizar la curva de calibración.
8 7

Destilación de la muestra Tomar 500 ml de la muestra en 1 vaso, llevarla a un pH de
4 aproximadamente si es que no se hizo esto al tomar o preservar la muestra. Agregar 5 ml de la solución de sulfato de cobre pentahidratado y transferirla al aparato de destilación. Correr un testigo usando agua y seguir el mismo procedimiento para la muestra.
Usar una probeta graduada para recivir el destilado. Destilar 450 ml de la muestra, suspender la destilación y
cuando la ebullición cese, esperar de 3 a 5 minutos agregar 50 ml de agua caliente al matraz de destilación , continuar la destilación hasta que se haya conectado colectado un total de 500 ml en la probeta.
Tratamiento para destilados turbios. Una sola destilación casi siempre es suficiente para la
purificación de la muestra. Ocacionalmente el destilador es turbio, en este caso se vuelve a destilar la muestra.
Si el destilado continua turbio aún después de la segunda destilación emplear el siguiente procedimiento.
Tomar 500 ml de la muestra en un vaso agregar 4 gotas de indicador de anaranjado de metilo, acidificar con solución de ácido sulfúrico, y pasarla a un embudo de separación agregar 150 g de cloruro de sodio hasta el vire del indicador.
Agregar 5 porciónes de cloroformo y agitar después de cada adición usando 40 ml en la primera y 3 ml en la segunda; agitar después de cada adición .
Mezclar los extractos alcalinos y calentar en baño maría hasta que el cloroformo sea remobido totalmente, enfriar y diluir a 500 ml con agua .
Destilar nuevamente.
Tratamiento de la muestra después de destilada. Agregar 10 ml de solución de cloruro de amonio y ajustar
el pH a 1 O 0.2 con hidróxido de sodio concentrado. Pasar a un embudo de separación de 1 litro, agregar 3 ml de la solución de 4 aminoantipirina mezclar bien, agregar 3 ml de solución de

ferricianuro de potasio. Mezclar nuevamente y dejar que el cloro se desarrolle en 3 minutos.
La solución debe de estar transparente y ligeramente amarilla.
Extraer inmediatamente con 25 ml de cloroformo; agitar el embudo de separación por lo menos 10 veces dejar que el cloroformo se separe agitar de nuevo 10 veces y dejar que el cloroformo se vuelva a separar.
Filtrar el extracto clorofórmico a través del papel filtro que contenga una capa de 5 g de sulfato de sodio anhídrido, con el fin de eliminar la humedad.
Colectar el extracto seco en seldas y medir la absorbancia a 460 nm ajustando el aparato a cero con el testigo y leer en la curva de calibración.
CALCULOS La concentración de fenoles en aguas se cálcula de la
siguiente manera:
A
B Fenoles = -
Donde: A = ug de fenol leidos en la curva de calibración. B = Volumen de la muestra original, en ml.
Calcular la concentración de fenol como sigue:
F = 7.842(AB-C).
Donde: F = Concentracion de fenol, en mg/l
A = Volumen de la solución de tiosulfato de sodio (0.025N)
B = Volumen de la solución de bromato-bromuro 0.1 N usado
C = Volumen de la solución de tiosulfato de sodio (0.025N)
7.842 = Factor de conversión . para el testigo, en ml
para la muestra y dividido entre I O , en ml.
para la muestra, en ml.

DEMANDA QUíMICA DE OXÍGENO
RESUMEN DEL MÉTODO.
El método se basa en una oxidación enérgica de la materia orgánica y de la inorgánica oxidable que se encuentra en el agua, en un medio fuertemente ácido, con una solución valorada de dicromato de potasio. El execeso del agente oxidante se titula con una solución valorada de sulfato ferroso amónico en presencia de un complejo ferroso de ortofenantrolina como indicador interno.
REACTIVOS.
Los reactivos que se utilicen deben ser de grado analítico. Cuando se hable de agua debe entenderse de agua destilada.
Solución de dicromato de potasio, 0.25 N. Disolver 12.2588 g de dicromato de potasio (previamente secado a 105OC 5 1 "C durante dos horas) aforar con agua a 1 O00 ml en un matraz volumétrico y homogenizar.
Solución de dicromato de potasio 0.025 N. Transferir con pipeta 100 ml de la solución anterior a un matraz volumétrico y aforar a 1 O00 ml y hornogenizar.
Solución de sulfato ferroso amóniaco 0.25 N. Disolver 98.0 g de sulfato ferroso amónico en aproximadamente 800 ml de agua, agregar cuidadosamente 20 ml de ácido sulfúrico concentrado, enfriar, aforar a I000 ml en un matraz volumétrico y hornogenizar.
Normalización de la solución de sulfato ferroso amónico 0.25 N.
Tomar 25 ml de la solución de dicromato de potasio 0.25 N diluir con agua hasta 275 ml, agregar cuidadosamente 50 ml de ácido sulfúrico concentrado hornogenizar, enfriar y titular con la solución de sulfato ferroso amónico 0.25 N utilizando 8 gotas de 1 ,I O fenantrolina (ortofenantrolina) como indicador hasta el cambio de color de azul verdoso a café rojizo.
Solución de sulfato ferroso amónico 0.025 N. Pesar 9.8 g de sulfato ferroso amónico con un exactitud del 0.0001g y Disolverlos en aproximadamente 800 ml de agua, agregar
90

cuidadosamente 20 ml de ácido sulfúrico concentrado, enfriar, aforar a I O00 ml en un matraz volumétrico y hornogenizar.
Normalización de la solución de sulfato ferroso amónico 0.025 N.
Tomar 25 ml de la solución de dicromato de potasio 0.025 N. diluir con con agua hasta 275 ml, agregar cuidadosamente 50 ml de ácido sulfúrico concentrado homogenizar, enfriar y titular con la solución de sulfato ferroso amónico 0.025 N utilizando 8 gotas de 1 , I O fenantrolina (ortofenantrolina) como indicador hasta el cambio de color de azul verdoso a café rojizo.
Solución de ácido sulfúrico-sulfato de plata. Determinar la masa de 9.51 g de sulfato de plata y disolver en 1000 ml de ácido sulfúrico concentrado.
Nota importante: El sulfato de plata requiere un tiempo aproximado de dos días para su completa disolución. La solución formada debe mantenerse en la obscuridad para evitar la descomposición.
Solución indicadora I , I O fenantrolina. Disolver en agua 1.485 g de I , I O fenantrolina y 0.695 g de sulfato ferroso heptahidratado, aforar a 100 ml y hornogenizar.

PROCEDIMIENT0.6
Para niveles mayores de 50 mgll de demanda química de oxígeno. Se transfiere al matraz Erlenmeyer de 500 ml una muestra de 50 ml.
Agregar una cantidad adecuada de sulfato mercúrico. El sulfato mercúrico se agrega para eliminar la interferencia que presentan los cloruros, obteniéndose un complejo de cloruro mercúrico soluble que inhibe la reactividad del ión'cloruro. Si la muestra contiene 40 mg de cloruros o menos, se agregan 0.4 g de sulfato mercúrico, en caso de que la concentración de cloruros sea mayor, se debe agregar sulfato mercúrico en una cantidad tal, que se tenga un relación sulfato mercúrico-cloruro de 1O:l (mg de sulfato mercúricolmg de cloruro).
Por ejemplo si la muestra contiene 60 mg de cloruros se deberá adicionar 600 mg de sulfato mercúrico a la muestra.
Agregar algunas perlas de ebullición, añadir 25.0 ml de la solución de dicromato de potasio 0.25 N y mezclar mediante un movimiento circular (debe enfriarse rapidamente para evitar la perdida de la posible materia volatil que tenga la muestra).
Conectar el matraz Erlenmeyer al condensador tal y como se muestra en la figura 1 y hacer circular el agua de enfriamento.
Por el extremo superior del condensador agregar lentamente 75 ml de la solución de ácido sulfúrico-sulfato de plata7 y agitar con movimiento circular para hornogenizar.
La mezcla debe agitarse perfectamente ya que de no ser así al calentarse ocurren sobrecalentamientos locales que proyectan la mezcla fuera del condensador.

Calentar el matraz que contiene la mezcla y mantener a reflujo durante 2 horas8 a partir del momento en que empieza la ebullición. Dejar enfriar y lavar el condensador con 25 ml de agua.
Añadir agua por el extremo superior del condensador hasta completar un volumen aproximadamente de 300 ml, retirar el matraz del condensador y enfriar a la temperatura ambiente.
Agregar 8 gotas de 1 ,IO fenantrolina como indicador y titular con la solución valorada de sulfato ferroso amónico 0.25 N hasta el cambio de color del azul verdoso a café rojizo.
CALCULOS La demanda química de oxígeno, expresada en mgll, se
calcula con la siguiente ecuación: (VI - V2) X N X 8
DQO = --- x 1000 v3
Donde: DQO = Demanda química de oxígeno, en mgll VI = Volumen de la solución de sulfato ferroso
amónico requerido para la titulación del testigo, en ml V2 = Volumen de la solución de sulfato ferroso
amónico requerido para la titulación de la muestra, en ml V3 = Volumen de la muestra, en ml N = Normalidad de la solución de sulfato ferroso
8 = Equivalente del oxígeno amónico, utilizado en la determinación
Nota: La diferencia entre las determinaciones efectuadas por duplicado no debe exceder del 5% de demanda química de oxígeno. En caso contrario se debe repetir la determinación.

- DETERMINACIóN DE SULFUROS"
FUNDAMENTOS
El método de azul de metileno se basa en la reacción del sulfuro, el cloruro férrico y la dimetil-para- fenilendiamina para producir el azul de metileno. Una vez desarrollado el color, se añade fosfato de amonio para eliminar el color debido al cloruro férrico.
El método iodométrico se efectúa apartir de una titulación basada en la reacción del iodo con el sulfuro en solución ácida, oxidándolo hasta azufre.
INTERFERENCIAS
El método iodométrico adolece de interferncia como las sustancias reductoras que reccionan con el iodo, incluyendo sulfitos, tiosulfatos y varos compuestos orgánicos, sean sólidos y estén disueltos.
En el método de azul de metileno también interfieren los fuertes agentes oxidantes enmascarando la formación del color azul. Cuando la concentración de tiosulfatos es mayor de 10 mg/l, la formación del color se retarda. La misma concentración de sulfuros, si es de varios cientos de miligramos por cada litro, representa una interferencia.
Las interferencias debido a sulfitos, tiosulfatos, ioduros y muchas otras sustancias solubles, exceptuando ferrocianuros, se eliminan adicionando sulfuro de zinc, removiendo el sobre nadante y reemplazándolo por agua. Este mismo procedimiento se usa para concentrar sulfuros, incluso si no hay necesidad de remover las interferencias.
MUESTRAS
Las muestras deben ser tomadas con una aeración mínima. Para determinar sulfuros totales, la muestra debe preservarse
S4

con 4 gotas de solución de acetato de zinc 2 N por cada I 00 ml de muestra, las que se adicionan antes de llenar totalmente el frasco. El almacenamiento debe ser a 4°C.
PROCEDIMIENTO
Método del azul de metileno
Desarrollo del color.
En cada uno de los dos tubos de comparación se colocan 7.5 ml de la muestra, llenándolos hasta sus marcas o usando una pipeta que tenga punta ancha. AI tubo A se le adiciona 0.5 ml de rectivo ácido amino sulfúrico y 0.15 ml de la solución de cloruro férrico; inmediatamente mezclar por inversión una sola vez (si se mezcla en exceso los resultados son bajos por la pérdida de H2S como gas, que no ha tenido tiempo suficiente para reaccionar).
AI tubo B se le adiciona 0.5 ml de solución de H2S04 (I+?) y 0.1 5 ml de solución de FeC13 y mezclar.
Si en el tubo A aparece un color azul, indicará la presencia de sulfuros. Usualmente el desarrollo del color se completa en un minuto; pero por lo regular el tiempo es mayor para devanecer el color rosado que aparece al principio. Después de 5 minutos, a cada tubo, A y B se le añaden 1.6 ml de (NH4)2 HP04 y después de 15 minutos se comparan los colores.
Si se utilizó el acetato de zinc, esperar por lo menos 10 minutos antes de hacer la comparación visual del color.

DETERMlNACldN DEL COLOR.
Estimación visual del color.
AI tubo B se le añade solución de azul de metileno I ó II; dependiendo de la concentración de sulfuros y de la exactitud deseada, gota a gota hasta igualar el color con el tubo A. Si la concentración de sulfur0 excede de 20 mg/l, la prueba debe repetirsed diluyendo la muestra en un décimo. Si la solución I del azul de metileno se justa de manera que 0.05 ml(1 gota) sea igual a 1.0 mg sulfuro/L, y cuando se emplean 7.5 ml de muestra:
mg de suIfuro/L = No. de gotas de la solución I + 0.1 (No. de gotas de la solución 11).
Medida fotomrStrica del color.
Si la concentración de sulfuros está entre 0.1 y 2 mg/l, se debe usar una celda con paso de luz de 1 cm.
Si las concentraciones son mayores o menores, el paso de luz será mayor o menor respectivamente. El límite mayor del método es 20 mgll.
Con una porción de la muestra tratada del tubo B se calibra el instrumento a cero.
Las curvas de calibración se preparan sobre una base de la prueba colimétrica, hecha contra soluciones de Na,S, simultaneamente analizadas por el método iodométrico; se gráfica la concentración sobre la absorbancia. La relación de una línea recta entre la concentración y la absorbancia se puede suponer para una concentración de O a 1.0 mg/l. Leer la concentración de sulfuros de la curva de calibración.
96

MÉTODO IODOMÉTRICO.
En un frasco de 500 ml se mide con bureta una cantidad de iodo en solución, tal que exceda a la concentración de sulfuros presentes. Si es necesario se adiciona agua para completar a 20 ml. Agregar 2 ml de solución de HCI, 6 N: Medir con pipeta 20 ml de muestra y descargar sobre la superficie de la solución que contiene iodo. Si el color de éste desaparece, adicionar más iodo, hasta que permanesca su coloración. Titular con solución valorada de tiosulfato de sodio y almidón como indicador, hasta desaparecer el color azul.
Si el sulfuro se precipitó con zinc y se filtró el ZnS, el filtro con el precipitado debe regresarse a la botella original y adicionar 100 ml de agua. Añadir HCI y la solución de iodo y titular con tiosulfato de sodio.
CALCULOS Si un ml de la solución de iodo 0.0250 N reacciona con 0.4 mg de sulfuro, luego:
mg/l= ( A x B ) - ( C x D ) ~ 1 6 , 0 0 0 ml de la muestra.
A = Solución de iodo, en ml. B = Normalidad de la solución de iodo. C = Tiosulfato de sodio, en ml. D = Normalidaad del tiosulfato de sodio.
Sulfur0 de Hidrógeno no -1onizado.
El sulfuro de hidrógeno y el HS constituyen el sulfuro disuelto y están en equilibrio con los iones Hidrógeno;
H2S ”> H+ + HS’ <
La constante de ionización del H2S se usa para calcular la distribución de sulfuro disuelto entre las dos formas. La constante práctica escrita en forma logaritmica, pK’ es la que
97

se usa. La constante varía con la temperatura y con la fuerza iónica de la solución y 6sta fuerza se puede estimar Fácilmente de la conductividad. El efecto de la temperatura es prácticamente lineal de 15 a 35 OC; se puede usar interpolaciones o extrapolaciones.
Del pH de la muestra y el valor apropiado de pK', se calcula pH - pK' . Leer la proporción de sulfuro disuelto presente como H2S (escala del lado izquierdo de la figura I ). Digamos que esta proporción sea igual a J; luego:
J x sulfuro disuelto = H2S no ionizado, expresado como sulfuro.
La siguiente tabla de algunos valores aproximados de pK para diferentes temperaturas y conductividades.
Valores de pK para el H2S
Valores de pK a la temperatura dada Conductividad a 298K (25OC) 20°C 25OC 30°C pS/cm
- 7.03 0
1 O0 7.08 7.01 6.94 0
700 7.05 6.98 6.91 400 7.06 6.99 6.92 200 7.07 7.00 6.93
7.04 6.97 6.90
50000 6.95 6.88 6.81 22000 6.96 6.89 6.82 14000 6.97 6.90 6.83 I O000 6.98 6.91 6.84 7200 6.99 6.92 6.85 5200 7.00 6.93 6.86 4000 7.01 6.94 6.87 3000 7.02 6.95 6.88 2000 7.03 6.96 6.89 1200
- - _
BS

DETERMINACIóN DE CROMO HEXAVALENTE
FUNDAMENTO
El cromo hexavalente reacciona con la difenilcarbazida en medio ácido para dar un complejo de color violeta, de composición desconocida, el cual se cuantifica a 540 nm.
REACTIVOS
Solución madre de cromo. Disolver 141.4 mg de dicromato de potasio anhídro (KzC1-207) en agua y diluir a 1 litro. Un ml de esta solución equivale a 50pg de cromo hexavalente Cr+6.
Diluir 10 ml de la solución madre de cromo a 100 ml de agua. 1 ml de esta solución equivale a Spg de Cr+6.
Solución de difenilcarbazida. disolver 0.25 g del 1.5 difenilcarbazida en 50 ml de acetona. Almacenar en frascos y eliminar cuando la solución se muestre decolorada.
PROCEDIMIENTO
Preparación y conservación de la muestra.
La alícuota necesaria para hacer el análisis de la muestra debe estar lo más clara posible, por lo que antes de empezar el método debe filtrarse a través de una membrana de 0.45 micras
Se recomienda que el análisis de las muestras se haga lo más pronto posible, en caso contrario, acidificar la muestra con ácido nítrico concnetrado y mantenerla en refrigeración a 4°C.
( M ) *
Preparación de la curva de calibración.
Medir volúmenes de la solución patrón de cromo en el ámbito de 2.0 a 20.0 ml y pasarlos a matraces aforados de 100 ml agregar 2.0 ml de ácido sulfúrico (1 + 1) y 5 gotas de ácido fosforico y diluir a 100 ml. agregar 2.0 ml de la solución de
BB

difenilcarbazida, mezclar y dejar reposar 10 minutos para que se desarrolle totalmente el color.
Medir la absorbancia a 540 nm utilizando una celda de I cm -se recomienda el uso de celdas perfectamente limpias libres de rayaduras y previamente enjuagadas con la muestra antes de efectuar el análisis- correr un testigo usando agua y corregir las lecturas de absorbancia de los patrónes, restando la absorbancia del testigo.
Para construir la curva de calibración se gráfica absorbancia contra pg de Cr+6.
Nota: Se debe construir una nueva curva de calibración cada vez que se usen nuevos reactivos.
Análisis de la muestra.
Filtrar la muestra y tomar la alícuota dependiendo de las características fisicoquímicas de la misma. alícuota para muestras muy claras será de 90 ml, que es la máxima alícuota que se debe tomar.
Agregar 2.0 ml de ácido sulfúrico (1 + 1) y 5 gotas de ácido fosfórico, pasar la solución a un matraz volumétrico de 100 ml y aforar. Añadir 2.0 ml de la solución de difenilcarbazida, mezclar y dejar reposar 10 minutos, para el complejo desarrollo del color.
Medir la absorbancia a 540 nm utilizando una celda de I cm, corregir la lectura restando la absorbancia de un testigo hecho con agua.
De la absorbancia corregida, determinar los pg de Cr+6 presente teniendo como referencia la curva de calibración.
Nota: si la solución es turbia después de diluir a 100 ml tomar la absorbancia antes de la adición de la solución de difenilcarbazida.

CALCULOS
La concentración de cromo hexavalente se cálcula por medio de la siguiente fórmula:
En donde:
A = pg de Cr+6. leídos en la curva V = volumen de la muestra, en ml.
DETERMINACIóN DE SUSTANCIAS AL AZUL DE METILENO.
Los componentes básicos de los detergentes, son compuestos orgánicos con propiedades tensoactivas en solución acuosa.
Los compuestos tensoactivos más empleados en la fabricación de detergentes son los sulfonatos de alquil benceno de sodio (ABS), los sulfonatos de alquil tolueno (ATS) y sus mezclas, cuyas estructuras químicas ramificadas son muy estables y no se degradan, o lo hacen muy lentamente.
Son muchos los efectos causados por un alto contenido de detergente en agua, tales comom: la fromación de espuma, la alta toxicidad de los surfactantes contenidos, que representan un serio peligro a la vida acuática y los fosfatos que propicia el crecimiento desmesurado de la flora acuática.
Este método se basa en la reacción de las sustancias surfactantes con azul de metileno, que dá lugar a la formación de una sal azul, soluble en cloroformo, cuya intensidad de color es directamente proporciónal a su concentración.
La intensidad de color se mide en un espectrofotómetro, a una longitud de onda de 650 a 655 nm.

Surfactante o tensoactivo. Compuesto constituido por una cadena polar alifática que es hidrófila y una parte aromática, no polar que se caracteriza por tener propiedades hidrofóbicas. a esta característica de las moléculas se deben las propiedades humectantes, dispersantes y emulsificantes de los detergentes.
Miscibilidad. es la característica de ciertas sustancias o soluciónes, de poderse mezlcar con otras.
Hidrófilo. Sustancia capaz de absorber y retener agua Hidrófogo. Sustancia que no tiene afinidad por el agua.
INTERFERENCIAS.
En esta determinación interfieren todos los compuestos solubles en cloroformo, capaces de formar una unión con el átomo de nitrógeno del azul de metileno tales como sulfonato, sulfatos orgánicos, carboxilatos, fosfatos y fenoles (sustancias activas al azul de metileno). Estas interferencias se eliminan en los lavados con la solución ácida, la cual las hidroliza retornándolos a la fase acuosa.
REACTIVOS
Los reactivos que sean utilizados deberán de ser grado reactivo, a menos que se indique otra cosa, cuando se hable de agua se debe entender agua destilada.
El material de vidrio utilizado para esta determinación debe laverse con mezcla crómica, enjuagarse dos veces con solución caliente de HCI (1 :I) y enjuagarse 2 o 3 veces más con agua. nunca usar detergentes.
Solución madre de sulfonato de alquilbenceno de sodio (ABS). pesar exactamente I .O000 g de ABS en base al 100% activo, disolver en agua y aforar a un litro con agua. 1 ml de esta solución contiene 1 mg de ABS. Es necesario prepararla cada semana y refrigerarla -se recomienda aforar sólo cuando todo el sulfonato de alquil benceno se haya disuelto y la espuma desaparezca.
Solución patrón de ABS. tomar 10 ml de la solución madre de ABS y aforar a un litro con agua. 1 ml de esta solución contiene 0.010 mg de ABS. esta solución se debe preparar

diariamente.. si no se dispone de cloroformo grado espectrofotométrico se puede utilizar cloroformo grado reactivo-.
Solución alcohólica de fenolftaleína. Disolver 500 mg de fenolftaleína en polvo en 100 ml de alcohol etílico o isopropílico al 50%. neutralizar la solución con hidróxido de sodio aproximadamente 0.02 N, agregando gota a gota hasta la aparición de un ligero color rosado.
Solución de hidróxido de sodio (NaOH) 1 N. disolver 40 g de NaOH en agua y aforar a un litro.
Solución de ácido sulfúrico 1 N. Diluir cuidadosamente 28 ml de ácido sulfúrico concentrado (densidad = 1.84) en agua. Dejar enfriar y aforar a 1 litro.
Reactivo de azul de metileno. Disolver 100 mg de azul de metileno, en 100 ml de agua. De esta solución se transfieren 30 ml a un matraz volumétrico de 1000 ml y agregar 500 ml de agua, 6.8 ml de ácido sulfúrico concentrado y 50 g de fosfato de monosódico monohidratado. Agitar hasta su completa disolución y aforar a 1 litro.
Solución de lavado. En un matraz volumétrico de 1000 ml que contenga 500 ml de agua, agregar 6.8 ml de ácido sulfúrico concentrado y 50 g de fosfato monosódico monohidratado. Agitar hasta su completa disolución y aforar a 1 litro.
Determinación.
El volumen de la muestra de agua para ser analizada, se toma de acuerdo con la concentración probable de ABS, según se indica en la siguiente tabla. Así mismo, efectuar una prueba testigo con agua. Concentración esperada de ABS en Muestra a tomar en
mg/l ml 0.025 - 0.080
1 O0 0.400 - 2.000 250 0.080 - 0.400 400
2.000 - 10.000 2 10.000 - 100.000 20
103

Si el volumen es menor a 100 ml, se debe diluir con agua a este volumen.
Transferir la muestra a un embudo de separación y alcalinizar la solución con hidróxido de sodio 1 N usando solución indicadora de fenolftaleína.
Neutralizar la muestra con solución de ácido sulfúrico 1 N. Agregar 10 ml de cloroformo y 25 ml de azul de metileno.
Agitar vigorosamente durante 30 segundos y dejar en reposo hasta la separación de las fases.
Pasar la fase orgánica a un segundo embudo y lavar el tubo de descarga del primero con un poco de cloroformo - con frecuencia se presenta problemas de emulsificación la cual puede romperse con agitación suave con el extremo plano de una varilla de vidrio. La transferencia de la fase orgánica al segundo embudo de separación se efectúa sólo hasta que las dos fases estén completamente separadas. Si el color azul de la fase acuosa es muy pobre o desaparece después de la primera extracción, agregar 25 ml de solución de azul de metileno-. Repetir la extracción por 3 veces, usando 10 ml de cloroformo en cada ocasión.
Combinar todos los extractos en el segundo embudo de separación, agregar 50 ml de solución de lavado y agitar vigorosamente durante 30 segundos. Dejar reposar y filtrar la capa de cloroformo a través de fibra de vidrio, a un matraz aforado de 100 ml. Repetir el lavado por dos veces empleando 10 ml de solución de lavado en cada ocasión.
Lavar la fibra de vidrio y el embudo con cloroformo, recoger los lavados en el matraz aforado, aforar con cloroformo y mezclar perfectamente.
Determinar la absorción de la solución a 625 nm, contra un testigo -la absorbancia debe medirse después de 15 minutos y antes de 30 minutos de haberse desarrollado el color. Una vez transcurrido este tiempo la solución ya no es estable.
Curva de calibración.
Preparar un serie de tubos con O.O.( testigo) 1.0, 3.0, 5.0, 7.0, 9.0, 11 .O, 13.0, 15.0 y 20 ml de la solución patrón de ABS.
104

Agregar agua hasta un volumen de 100 ml en cada embudo de separación.
Neutralizar la muestra con solución de ácido sulfúrico 1 N. Agregar 10 ml de cloroformo y 25 ml de azul de metileno.
Agitar vigorosamente durante 30 segundos y dejar en reposo hasta la separación de las fases.
Pasar la fase orgánica a un segundo embudo y lavar el tubo de descarga del primero con un poco de cloroformo - con frecuencia se presenta problemas de emulsificación la cual puede romperse con agitación suave con el extremo plano de una varilla de vidrio. La transferencia de la fase orgánica al segundo embudo de separación se efectúa sólo hasta que las dos fases estén completamente separadas. Si el color azul de la fase acuosa es muy pobre o desaparece después de la primera extracción, agregar 25 ml de solución de azul de metileno-. Repetir la extracción por 3 veces, usando 10 ml de cloroformo en cada ocasión.
Combinar todos los extractos en el segundo embudo de separación, agregar 50 ml de solución de lavado y agitar vigorosamente durante 30 segundos. Dejar reposar y filtrar la capa de cloroformo a través de fibra de vidrio, a un m,atraz aforado de 100 ml. Repetir el lavado por dos veces empleando 10 ml de solución de lavado en cada ocasión.
Lavar la fibra de vidrio y el embudo con cloroformo, recoger los lavados en el matraz aforado, aforar con cloroformo y mezclar perfectamente.
Determinar la absorción de la solución a 625 nm, contra un testigo -la absorbancia debe medirse después de 15 minutos y antes de 30 minutos de haberse desarrollado el color. Una vez transcurrido este tiempo la solución ya no es estable.
Trazar la curva de calibración en mg de ABS contra absorbancia.

CALCULOS. El contenido de sulfonato de alquilbenceno de sodio (ABS)
expresado en mg/l, se calcula con la fórmula siguiente:
A
V ABS, en mgll = - X I00
En donde:
A = mg del sulfonato de alquilbenceno de sodio, leidos en
V = Volumen de muestra empleada, en ml la curva de calibración.
Nota: el método tiene una desviación estándar de * 0.2%
CLORUROS.
FUNDAMENTO.
La determinación argentométrica de los cloruros se basa en la formación de cromato de plata de color rojizo, esto ocurre cuando se adicionan al agua iones cromato como indicador, e iones de plata como reactivo precipitante.
Titulando con una solución valorada de nitrato de plata se determina la cantidad necesaria para precipitar todos los cloruros como cloruros de plata, e inmediatamente se observa la formación de cromatos de plata de color rojizo y en ese momento se anota el volumen de solución de nitrato de plata utilizado y se calcula la concentración de cloruros existentes en el agua.

REACTIVOS
Los reactivos que a continuación se mencionan deben ser de grado analítico. Cuando se hable de agua debe entenderse de agua destilada
Solución indicadora de cromato de potasio. Disolver 50 g de cromato de potasio en aproximadamente 500 ml de agua. Agregar solución de nitrato de plata hasta que se forme un precipitado rojo. Dejar reposar 12 horas filtrar y diluir a 1 litro con agua.
Solución valorada de nitrato de plata 0.0141 N. Pesar 824.1 mg de cloruro de sodio previamente secado a 140°C por dos horas, disolver en agua y aforar a 1000 ml, un ml es igual a 0.5 mg de ión cloruro.
Suspención de hidróxido de aluminio. Disolver 125 g de sulfato de aluminio y potasio dodecahidratado (AIK(SO4)2* 12H20) o sulfato de aluminio y amonio dodecahidratado (AINHq(SO4)2*12H20) en un litro de agua.
Calentar a 60°C y agregar 55 ml de hidróxido de amonio (NH4OH) concentrado lentamente con agitación.
Dejar reposar la solución 1 hora aproximadamente, decantar el agua sobre nadante y lavar con agua, dejar reposar y volver a decantar, repetir el procedimiento anterior hasta que se considere libre de cloruros (aproximadamente seis veces).
Solución indicadora de fenoftaleina. (C20H14014). Disolver 0.5 g de fenoftaleina en 50 ml de alcohol etílico y agregar 50 ml de agua. Agregar solución de hidróxido de sodio 0.02 N gota a gota hasta la aparición de una coloración rosa pálido.
Solución de hidróxido de sodio 1 N. Disolver en agua 40 g de hidróxido de sodio y aforar a 1000 ml.
Solución de hidróxido de sodio 0.02 N. Diluir en agua 20 ml de solución de hidróxido de sodio I N y aforar a 1 O00 ml.
Solución de ácido sulfúrico 1 N. Tomar 30 ml de ácido sulfúrico concentrado y diluir a 1 litro.
X 0 7

INTERFERENCIAS.
Ortofosfatos en exceso de 25 mg/l interfieren precipitando
Hierro en exceso de 10 mgll enmascara el punto de vire. lones sulfuro, sulfito y tiosulfato interfieren pero es posible
removerlos por adición de 1 ml de peróxido de hidrógeno (H202) al 30 % y agitar por I minuto.
Bromuros, ioduros y cianuros son determinados como cloruros.
como fosfato de plata.
PROCEDIMIENTO.
Valoración de la solución de nitrato de plata 0.0141 N. Medir un volumen de I O a 20 ml de solución estandar de cloruro de sodio 0.0141 N. Agregar agua hasta completar un volumen de 100 ml. Agregar I ml de solución indicadora de cromato de
Titular con la solución de nitrato de plata hasta vire de amarillo a rojo ladrillo. Calcular la normalidad de la solución de nitrato de plata por medio de la siguiente fórmula:
potasio.
V I x NI NAgNO3 --- -
v2
Donde:
empleada en la titulación.
la titulación.
VI = Volumen de solución estandar de cloruro de sodio
V2 = Volumen de la solución de nitrato de plata utilizada en
N1 = Normalidad de la solución de cloruro de sodio.
Nota: Hacer un mínimo de tres titulaciones y tomar un promedio de las normalidades resultantes.
Tratamiento preliminar de la muestra. Si la muestra presenta coloración o turbiedad tal que interfiera con el punto de vire, trátese como sigue:

Tomar 100 ml de la muestra y agregarle 3 ml de suspención de hidróxido de aluminio. Agitar y dejar reposar hasta sedimentación del precipitado.
Filtrar con papel filtro utilizando un embudo de vidrio. Lavar el precipitado y mezclar esta agua con el filtrado. En caso de que no desaparezca la turbiedad repetir el procedimiento anterior.
Aforar con agua a 100 ml.
Análisis de la muestra. Tomar 100 ml de muestra o una alícuota diluida a 100 ml con agua. Ajustar el pH de la muestra entre 7 y 10 con k i d 0 sulfúrico 1 N ylo hidróxido de sodio 1 N, utilizando un potenciómetro o solución de fenoftaleína.
Agregar 1 ml de solución indicadora de cromato de potasio. Titular con la solución valorada de nitrato de plata hasta el vire de amarillo a rojo ladrillo.
Analizar un testigo con agua en la misma forma que las muestras.
CALCULOS. La concentración de cloruros se determina por medio de
la siguiente fórmula:
- ( A - B ) ~ N x 3 5 4 5 0 CI e”--- “
V
Donde:
empleados en la titulación de la muestra.
empleados en la titulación del testigo.
determinación.
titulación.
A = Volumen en ml de solución de nitrato de plata
B = Volumen en ml de solución de nitrato de plata
V = Volumen de la muestra en ml tomados para la
N = Normalidad del nitrato de palta utilizado en la
109

CIANUROS.
FUNDAMENTOS
El método titulométrico se efectúa a partir del ión cianuro obtenido en la destilación alcalina del tratamiento preliminar de las muestras. Se titula con solución estándar de nitrato de plata (AgN03) para formar el complejo cianurado soluble Ag(CN)2. Una vez que el ión cianuro ha formado el complejo y se ha añadido un ligero exceso del ión plata, p- dimetilaminobenzalrodanina, el que inmediatamente cambia de color amarillo a color salmón. El indicador es sensible a concentraciones de 0.1 mg Agll. Si la titulación muestra que la concentración de cianuros es menor a 1 mg/l, analizar otra porción colorimétricamente.
El método colorimétrico se basa en la reacción del ión cianuro, obtenido en la destilación alcalina del tratamiento preliminar de las muestras, y cloramina - T para producir el cloruro de cianógeno (CNCI) a un pH menor de 8, evitando que se hidrolice a CNO.
Precaución. El cloruro de ciánogeno es un gas tóxico; evítese su inalación.
Una vez que se ha completado la reacción, el CNCI desarrolla una coloración roja azulada al adicionársele un reactivo de piridina - ácido barbitúrico. si el color se conserva en solución acuosa, se lee absorbancia a 578 nm. Para obtener coloraciones de intensidad comparable, mantener la misma concentración de sal en la muestra y en los estándares.
El método titulométrico se aplica para concentraciones de cianuro superiores a 1 mg/l.
El método colorimétrico se aplica para concentraciones de cianuros inferiores de 20 pg/l. Para concentraciones mayores, deberán hacerse las diluciones correspondientes.
Cianuros. Término génerico que incluye a todos los grupos CN correspondientes a los compuestos que los contienen; se clasifican como cianuros simples y cianuros complejos.
Cianuros simples. Compuestos representados por la fórmula A(CN)x , en donde A es un álcali o un metal y x es la Valencia de A, equivalente al número de grupos CN.

Cianuros complejos. Compuestos que se representan por varias fórmulas, sin embargo, los cianuros metálicos alcalinos normalmente se representan por AyM(CN)x donde A es el álcali presente "y veces", M es un metal pesado y "x" es el número de grupos CN; "xvv es igual a la Valencia de A tomada "y veces" más la Valencia del metal pesado.
INTERFERENCIAS.
Los agentes oxidantes pueden destruir la mayoría de vlos cianuros durante el almacenamiento y la manipuación. Añadir tiosulfato de sodio (Na2S203) como se especifica en la sección de muestreo.
El sulfuro destilará junto con el cianuro, y , por consiguiente, afectará los procedimientos colorimétricos y titulométricos. Pruébese la muestra y sepárese el sulfuro conforme se describe anteriormente. tratar 25 ml más de muestra que la requerida, para proveer un volumen suficiente de filtrado.
Loa ácidos grasos que destilan y forman jabones bajo condiciones de titulación alcalina hacen imposible el detectar el punto de vire del indicador. Sepárense los ácidos grasos por extracción. Acidificar la muestra con ácido acético (I + 9) a pH 6- 7.0 (Precaución llevar acabo la operación bajo una campana de extracción y tan rapido como sea posible). Extraer inmediatamente con isooctano, hexano o cloroformo - el orden preferencial es el enlistado-. Usar un volumen de disolvente igual al 20% del volumen de la muestra. Generalmente una extracción es suficiente para reducir la concentración de ácidos grasos abajo de los niveles de interferencia. Evítense las extracciones múltiples o un tiempo de contacto prolongado a valores bajos de pH para minimizar la pérdida de HCN. Cuando la extracción se haya completado, elevar inmediatamente el valor de pH arriba de 12 con solución de NaOH.
Una elevada concentración de carbonato puede afectar la destilación causando gasificación excesiva cuando se añade el ácido. El dióxido de carbono (CO,) liberado puede reducir significativamente el contenido de NaOH en el absorbedor.
Cuando se muestre en efluentes tales como desechos de gasificación de carbón, aguas de lavado de emisiones

atmosféricas, y otrso desechos con alto contenido de carbonatos, utilizar cal hidratada para estabilizar las muestras; añadir lentamente y agitando para elevar el pH de 12 a 12.5. Decantar la muestra a un matraz una vez que el precipitado se haya asentado.
Otras posibles interferencias incluyen sustancias que ocasionen coloración o turbiedad. En la mayoría de los casos, desaparecen con la destilación.
Los aldehídos convierten los cianuros en cianhidrina, que forma nitrilos en las condiciones de destilación. Sólo puede emplearse titulación directa sin destilación, lo que revela solamente los cianurosno complejos. La interferencia ocacionada por formaldehídos es notable en concentraciones que excedan 0.5 mg/l ; eliminarlo añadiendo nitrato de plata (AgN03) a la muestra. utilizar la siguiente prueba preliminar para determinar la presencia o ausencia de aldehídos (límite de detección 0.05 mg/l ).
Conservación de las muestras.
Los agentes oxidantes tales como cloro, descomponen a la mayoría de los cianuros. Hagase una prueba colocando una muestra en una tira de papel ioduro de potasio (KI) -almidón. Si aparece una decoloración azulada, añadir unos cuantos cristales de tiosulfato de sodio (Na2S203) a la muestra, agitar y repetir la prueba. Si es necesario repetir el procedimiento hasta que no haya decoloración en el papel de prueba; después añadir 0.1 g de Na2S203 al resto de la muestra.
El dióxido de magneso, cloruro de nitrosilo, ect., pueden tambien decolorar el papel de prueba. De ser posible, llevar a cabo el procedimiento anterior antes de proceder a la conservación de la muestra como se describe posterior mente. Cuando la siguiente prueba indique la presencia de sulfuro, no deben esperarse agentes oxidantes en la muestra.
El sulfuro convertiría rápidamente el CN- en CNS- especialmente a volres elevados de pH. Probar si hay S' 1 presente colocando una gota de muestra en papel de prueba de acetato de plomo que haya sido remojado previamente en

solución buffer de ácido acético, pH 4. El obscurecimiento del papel indica la presencia de S2" Añadir nitrato de cadmio en polvo, Cd (N03)2-4H20, para obtener un precipitado amarillo de sulfur0 de cadmio (CdS). Si el contenido de S2- es elevado, añadir carbonato de cadmio (CdC03), carbonato de plomo (PbC03), acetato de plomo o citrato de bismuto. Repetir esta operación hasta que una gota de la muestra previamente tratada no cause obcurimiento en la prueba de papel acidificado de acetato de plomo. Filtrar la muestra para estabilizarla antes de elevar el pH. Cuando se sospeche que existen complejos cianuros-metálicos en forma de partículas, filtrar la solución antes de separar el S2" Homogeneizar las partículas antes del análisis.
Debido a que ñla mayoría de cianuros son muy reactivos e inestables, analizar las muestras tan pronto como se pueda. Si la prueba no puede analizarse inmediatamente añadir lentejas de NAOH ó solución concentrada de NAOH para elevar el valor del pH de la muestra a 12-12.5 y almacenar en un frasco ámbar bien cerrado, en un lugar fresco.
Para analizar CNCI, colectar una muestra por separado y omitir la adición de NAOH, debido a que el CNCI se convierte rápidamenten CNO a valores elevados de pH. Hacer la determinación inmediatamente después del muestreo.
Tratamiento preliminar de las muestras.
El tratamiento preliminar varía deacuerdo con la sustancia interferente que esté presente.
Precauci6n.Tener cuidado al manejar las muestras de cianuro, debido a su toxicidad. Llevar a cabo el análisis bajo campana u otra área bien ventilada. Evitese el contacto, inhalación o ingestión. Los sulfuros, ácidos grasos y agentes oxidantes se eliminan por procedimientos especiales. La mayoría de otras sustancias se quitan por destilación.

Reactivos
Los reactivos que se mencionan en esta Norma deben ser grado analítico, a menos quese se indique otra cosa. Cuando se hable de agua debe entenderse agua destilada.
Solución indicadora de MBTH. Disolver 0.05g de hidrocloruro de 3 metil, 2 - benzotiazolona hidrazona en 100 ml de agua.
Solución oxidante del cloruro férrico.- Disolver 1.6 g de ácido sulfámico y I g de FeCI, .6H20 en 100 ml de agua.
Solución de nitrato de plata 0.IN.- Disolver 17.0 g de cristales de AgN0, en agua y diluir a 1 1.
Solución de EDTA 0.1 M.- Disolver 37.2 g de etilendiamintetraacetato disódico en agua diluir a 1 1.
Procedimiento
Colocar una gota de muestra y una gota de agua como blanco en cavidades separadas en un plato blanco de muestreo. Añadir una gota de solución de MBTH y después 1 gota de la solución oxidante de FeCl, a cada una de las cavidades. Dejar que pasen 10 min. para que se desarrolle el color. El cambio de color será de un verde pálido a uno más obscuro, tendiendo a verde azulado si la concentración es alta. Añadir AgNO, 0.1 N gota a gota y repetir la prueba en el plato. Por cada gota de AgNO,, añadir 2 gotas de solución de EDTA. Una concentración de formaldehído de I mg/l en una muestra de 100 ml requerirá aproximadamente 2 gotas de solución de AgNO,,m y 4 gotas de solución de EDTA.
NOTA.- La glucosa y otros azúcares, especialmente al valor de pH empleado para la conservación de la muestra, ocasionan la formación de cianhidrina por la reacción del cianuro con aldosa. La cianhidrina puede reducirse a cianuro con AgNO,, pero el MBTH no es aplicable.

Cianuro total después de la destilación
El ácido cianhídrico (HCN) se libera de la muestra acidificada por destilación y purgado con aire. El HCN.gaseoso se recolecta pasándolo a través de una solución absorbedora de NaOH. La solución de cianuro se determina por procedimientos colorimétricos, titulométricos O
potenciométricos.
Reactivos.
Solución de hidróxido de sodio.Disolver 10 g de NaOH A
Reactivo de cloruro de manganeso. Disolver 510 g de
Solución de ácido sulfúrico. H2S04, 1+1.
1 1.
MgC12*6H20 en agua y diluir a 1 1.
Aparato.
El aparato se muestra en la figura 1 Incluye:
Matraz de destilación. "Claissen" o similar.- Capacidad de 1 litro, con tubo de entrada y adimentado para un condensador enfriado por agua.
Absorbedor de gas.- Con un tubo dispersor de gas equipado con salida provista de un filtro de porosidad media
Elemento de calentamiento ajustable Juntas de vidrio ST.- Recubiertas con teflón o con un
lubricante apropiado para el matraz de destilación y el condensador. Puedan usarse también juntas de hule.
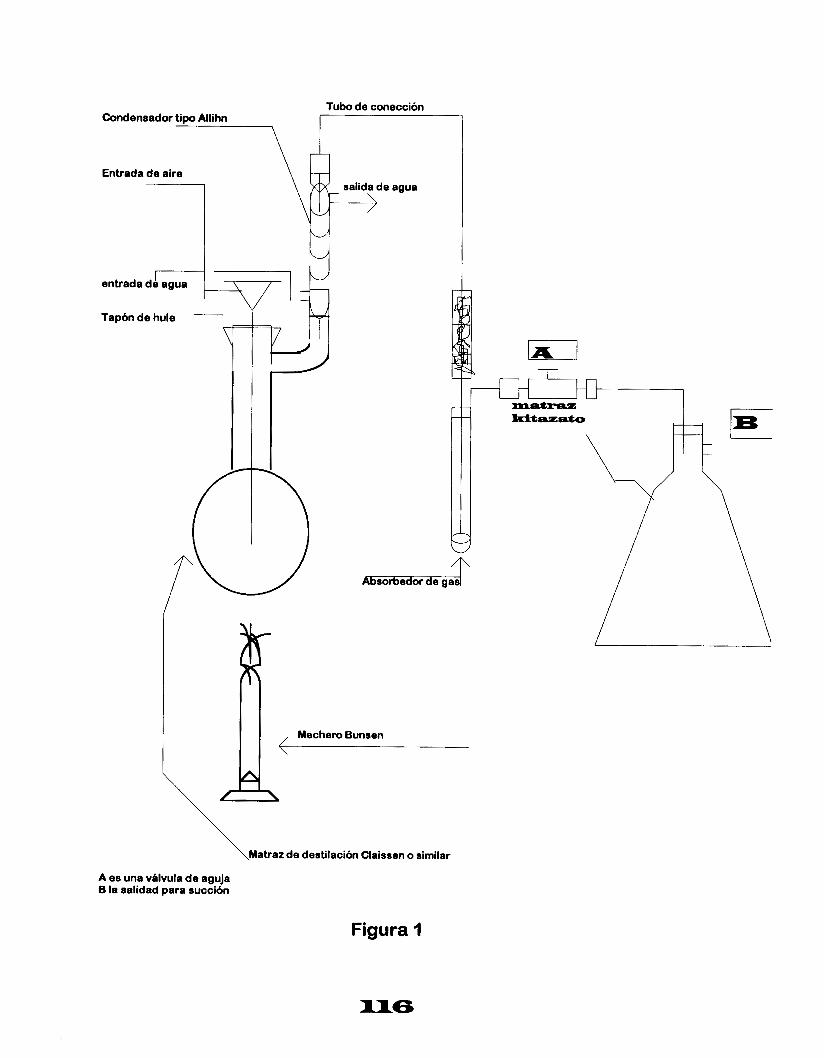
kitazato
I' sor e or e gas
\Matraz de destilación Claissen o similar
A es una vblvula de aguja B la salidad para succión
I
kitazato " jB
Figura 1
U6

Procedimiento.
Añadir 500 ml de la muestra, conteniendo no más de 100 mg CN/litro (diluyase si es necesario con agua) al matraz de destilación. Añadir 50 ml de solución de NaOH al lavador de gases y diluir, si es necesario, con agua para obtener una profundidad adecuada de líquido en el absorbedor. Conectar el tren, consistente en la entrada de aire del matraz de destilación, el matraz, condensador, lavador de gases, trampa de succión y aspirador. Ajustar la succión de modo que entre aproximadamente una burbuja de airels al matraz de ebullición. Este flujo de aire acarreará el HCN del matraz al absorbedor y normalmente evitará la inversión del flujo de HCN a través de la entrada de aire. Si este flujo de aire no evita la inversión del flujo en el tubo de entrega del destilado incrementarlo a 2 burbujas de aire/s. Obsevar la velocidad de la purga de aire en el absorberdor, dado que el nivel del líquido no debe aumentar a más de 6.5 mm - I O mm. Mantener el flujo de aire durante toda la reacción
Añadir 50 ml de solución de ácido sulfúrico (I+?) a través del tubo de entrada de aire. Enjuagar el tubo con agua y permitir que el aire mezcle el contenido del matraz durante 3 min. Añadir 20 ml de reactivo de MgC12 a través de la entrada de aire y lavar con una corriente de agua. Puede formarse un precipitado que se disolverá al calentar.
Calentar a ebullición rápida, pero no se debe permitir que el condensador se inunde o que los vapores se eleven más allá de la parte media del condensador. El nivel de reflujo adecuado es de 40 a 50 gotas /min. Reflujar cuando menos durante 1 h. Quitar el calentamiento pero mantener el flujo de aire. Enfriar durante 15 min y drenar el contenido del lavador de gases en un frasco aparte. Enjuagar el tubo conector entre el condensador y el lavador de gases con agua, añadir el agua al líquido drenado y diluir a 250 ml en un matraz volumétrico.
Determinar el contenido del cianuro por el método titulométrico si la concentración de cianuro excede de 1 mg/l o por el método colorimétrico si la concentración de cianuro es

menor. Utilizar titulación, o bien la prueba preliminar para aproximar la concentración de cianuro.
La destilación da recuperación cuantitativa aún de los cianuros refractarios tales como los complejos de hierro. Para obtener la recuperación completa del cianuro de cabalto emplear un pretratamiento con radiación ultravioleta. Si se sospecha una recuperación incompleta, destilar nuevamente colocando una nueva carga de NaOH en el lavador de gases y reflujar durante una hora más. El cianuro del segundo reflujo, si existe, indicará la totalidad de la recuperación.
Como medida de control de caliudad, probar periódicamente el aparato, reactivos y y otras variables potenciales en el ámbito de concentración de interés. Por ejemplo, debe obtenerse un mínimo de recuperación de 98% para un estándar de I mg CN/litro.
Metodo titulométrico.
Reactivos (ver reactivos en el apéndice de requerimientos)
Solución indicadora. Disilver 20 mg de p-dimetil-amino-
Titulante estándar de nitrato de plata, 0.0192 N. enzalrodanina en 1 O ml de acetona.
Disolver 3.27 g de AgN03 en I litro de agua. Estandarizar contra una solución estándar de NaCI, utilizando el método argentométrico con indicador de K2Cr04, como se indica a continuación:
Medir un volumen de 10 a 20 ml de solución estándar de cloruro de sodio. 0.0192 N.
Agregar agua hasta completar un volumen de 100 ml. Agregar 1.0 ml de solución indicarora de cromato de
Titular con la silución de nitrato de plata hasta un vire de
Calcular la normalidad de la solución de nitrato de plata
patasio.
amarillo a rojo ladrillo.
por medio de la siguiente fórmula:

VI*NI
v2 NAgN03
I
Donde:
VI= Volumen de solución estándar de cloruros de sodio empleado en la titulación. NI= Normalidad de la solución de cloruro de sodio = 0.0192N. V2= Volumen de la solución de AgNO3 empleada en la titulación.
Nota.-hacer un mínimo de 3 titulaciones y tomar un promedio de las normalidades resultantes.
Diluir 500 ml de la solución de AgNO3 de 1 ml sea equivalente a 1 mg de CN.
Procedimiento
De la solución absorbedora, tomar un volumen conocido de muestra de modo que la titulación requiera de aproximadamente 1 a 10 ml de titulante. Diluir a 250 ml o algún otro volumen cuyo uso sea conveniente para todas las titulaciones. Para el caso de que la concentración de cianuros sea baja (>5mg/l) no se diluya. Añadir 0.5 ml de la solución indicadora.
Titular con solución de AgN03 hasta el primer cambio de un color amarillo canario a color salmón. Titular un blanco conteniendo la misma concentración de álcali a agua.
Conforme el analista se familiarice con el punto de vire, los valores de titulante necesarios para el blanco decrecerán hasta alcanzar una gota o menos, con el correspondiente incremento en la precisión.

CALCULOS.
( a - b ) x 1,000 250 mg,,ll = ".. x U" "-
ml de la muestra original ml de la porción utilizada
Donde:
A = Volumen de AgN03 empleado en la titulación de la muestra, en ml. B = Volumen de AgN03 empleado en la titulación del blanco, en ml.
Precisión y exactitud
Para muestras que contienen más de 1 mg CNllitro que han sido destiladas o para muestras que presentan relativamente pocas interferencias, el coeficiente de variación es de 2%. La extracción y la eliminación de S" tienden a aumentar el coeficiente de variación en un grado determinado por la manipulación y el tipo de muestra que trate. El límite de sensibilidad es de aproximadamente 0.1 mg CN/litro, pero a esta concentración el punto de vire es indistinguible. A concentraciones de 0.4 mg/l, el coeficiente de variación es cuatro veces mayor que el obtenido para CN a concentraciones mayores a I .O mgll.
Método de Colorimétrico.
Reactivos (ver reactivos en el apéndice de requerimientos)
Solución de cloramina - T. Disolver 1 .O g en 100 ml de agua. Preparar semanalmente y guardar en refrigeración.
Solución patrón de cianuro. Disolver aproximadamente 2 g de KOH y 2.51 g de KCN en 1 litro de agua.( precaución-El KCN es altamente tóxico; evítese la inhalación o contacto). Estandarizar contra titulante estándar de nitrato de plata (AgN03), como se describe en el procedimiento del método titulométrico, utilizando 25 ml de la solución de KCN. Verificar

la titulación semanalmente, debido a que la solución pierde gradualmente su fuerza; I ml = I mg CN.
Solución Estándar de Cianuro. Basándose en la concentración obtenida de la solución patrón de KCN, calcular el volumen requerido (aproximadamente I O ml) para preparar 1 litro (I) de una solución con una concentración de I O pg CN/ml. Diluir con la solución de 2 g NaOHll (Solución de hidróxido de sodio - Disolver 2 g de NaOH en 1 I de agua. Diluir 10 ml de la solución que contiene 10 pg CN/ml con la solución de 2 g NaOH/I hasta un volumen de 100 ml; 1.0 ml = 1.0 pg CN. Preparar solución fresca diariamente y mantener en un frasco de vidrio bien cerrado. (precaución: Tóxico. Tener cuidado para evitar su ingestión.
Reactivo de piridina - Bcido barbitúrico. Poner 15 g de ácido barbitúrico en un matraz volumétrico de 250 ml y añadir suficiente agua para humedecer las paredes del matraz y el ácido barbitúrico. Añadir 75 ml de piridina y mezclar.Añadir 15 ml de solución concentrada de ácido clorhídrico (HCI), mezclar y dejar enfriar a temperatura ambiente. Aforar con agua, mezclando bien. Este reactivo es estable por un mes; desechar si se obseva la formación de un precipitado.
Solución de bifosfato de sodio, 1 M. Disolver 138 g de NaH2PO4*H2O en 1 I de agua. Guardar en refrigeración.
Solución de hidróxido de sodio. Disolver 2 g de NaOH en I I de agua.
Procedimiento.
Preparación de la curva de calibración.
Prepara una solución blanco de NaOH: A partir de la solución estándar de KCN preparar una serie de diluciones que contengan desde 0.2 hasta 6 pg de CN en 20 ml de solución utilizando la solución de 2 g de NaOH/I para hacer todas las diluciones. Tratar todos los estándares de acuerdo a lo especificado en el DESARROLLO DEL COLOR. Graficar la absorbancia de los estándares contra la concentración de CN (microgramos).

Verificar nuevamente la curva de calibración periódicamente y cada vez que se prepare un nuevo reactivo.
Sobre la base de la primera curva de calibración preparar estándares adicionales que contengan menos de 0.2 pg y más de 6 pg de CN para determinar los límites de medición con el fotómetro utilizado.
Desarrollo del color.
Ajustar el fotómetro a cero de absorbancia cada vez que se use un blanco consistente en la solución de NaOH y todos los reactivos. Tomar una porción del líquido absorbedor obtenido por el método "Cianuro total después de la destilación"; de modo que la concentración de CN esté dentro del ámbito cuantificable, y diluir a 20 ml con solución de NaOH. Colocar esta porción en un matraz volumétrico de 50 ml. Añadir 4 ml de buffer de fosfato y mezclar vigorozamente. Añadir 2.0 ml de solución de cloramina-T y agitar para mezclar. Inmediatamente, añadir 5 ml de la solución de piridina - ácido barbitúrico y agitar levemente. Aforar con agua; mezclar por inversión.
CALCULOS Medir la absorbancia de la solución a 578 nm después de 8 min, pero antes que transcurran 15 min a partir del momento que se añade al reactivo de priridina - ácido barbitúrico. Aún dentro del período de 8 a 15 min, puede haber diferencias en el valor de absorbancia. Para minimizar las variaciones, estandarizar el tiempo para todas las lecturas. Utilizando la curva de calibración y la siguiente ecuación, determinar la concentración de CN en la muestra original.
A x B
C x D CN, ugl ml =

Donde:
A = Peso del CN obtenido a partir de la curva de calibración, en pg (50 ml de volumen final).
B = Volumen total de la solución absorbedora de la
C = Volumen de la muestra original utilizada en la
D = Volumen de la solución absorbedora utilizada en la
destilación, en ml.
destilación, en ml.
determinación colorimétrica, en ml.
PRECISI~N
El análisis de una solución mixta de cianuros conteniendo zinc, sodio, cobre y plata en forma de cianuros en agua, da una precisión dentro del ámbito.
St = 0.11 5 X + 0.031
En donde:
St = Precisión total X = Concentración CN, mgll.

DETERMINACION DE CADMIO-MÉTODO COLORIMÉTRICO DE LA DITIZONA.
FUNDAMENTO
El método se basa en la relación del cadmio presente en el agua con la ditizona para dar un complejo de ditizonato de cadmio de color rojo el cual se extrae con cloroformo y se cuantifica colorimétricamente a una longitud de onda de 515 nm.
INTERFERENCIAS
La mayor interferencia resulta cuando se tienen cantidades elevadas de zinc (relación zinc-cadmio mayor de 500:l) que reducen la posibilidad de extraer las trazas de cadmio y generan resultados bajos.
De otra manera se pueden determinar cantidades de cadmio de 2.5 a 25 pg en presencia de 0.25 mg de cada uno de los siguientes iones cobre, cobalto y zinc y de 2.5 mg de acetato, aluminio, antimonio,arsénico, bismuto, cromo, cianuro, hierro, plomo, manganeso, mercurio, níquel, fosfato, plata, sulfito, tartrato, tiocianato, tiosulfato, estaño y otros iones comunes.
MUESTRE0 Y CONSERVACIóN DE MUESTRAS.
La muestra se debe colectar en frascos de polietileno y preservarse añadiendo ácido nítrico diluido 1 :I hasta neutralización, y adicionar un exceso de 5 ml de ácido nítrico concentrado por I, de muestra.

Preparación de la curva de Calibración.
Colocar en una serie de embudos de separación de 125 ml (color ámbar), los volúmenes de solución patrón diluida de cadmio indicados en la tabla
Volúmenes para la Curva de Calibración Solución patrón diluida de I Contenido de cadmio
cadmio de 2.5pglml (ml) (mg) 0.0 0.0000 1 .o
6.0 0.01 O0 4.0 0.0050 2.0 0.0025
0.0200 8.0 0.01 50
I 10.0 I 0.0250 I
Diluir entre 15 a 20 ml con agua, agregar 1 O ml de solución de tartrato de sodio y potasio y 4.2 ml de hidróxido de sodio 6N. Continuar con los pasos marcados con asterisco (*) de la determinación del cadmio en la muestra.
Transferir una porción adecuada de cada solución final a una celda de absorción de I cm y medir su absorbancia a 515 nm. Usar como referencia tetra cloruro de carbono a un blanco preparado con 20 ml de agua y proseguir siguiendo las indicaciones del parrafo anterior.
Nota= Si el tatracloruro de carbono se usa como referencia, corregir las lecturas de absorbancia de los patrones restando la absorbancia del blanco.
Graficar los valores de absorbancia corregidos contra los pg de cadmio.

Tratamiento de la muestra.
Tomar un volumen de muestra según el contenido de cadmio esperando de acuerdo a la tabla siguiente
Muestra para Análisis Contenido de cadmio Volumen de muestra
esperado (mg/l) recomendable. (ml) < I 1000.0 I 1000.0
1-10 100.0 10-100
1 .o 100-1000 10.0
Nota: Cuando se tienen contenidos bajos de cadmio (< 1 mgll) es recomendable concentrar la muestra evaporando en un mismo baso, porciones de 250 ml hasta obtener un volumen final de 15 a 20 ml.
Digestión
Digestión con ácido nítrico-ácido sulfúrico (para muestras con materia orgánica fácil de oxidar).
Acidificar al anaranjado de metilo a un pH menor de 4.2 con ácido sulfúrico concentrado, agregar 5 ml de ácido nítrico concentrado de 2 ml de peróxido de hidrógeno al 30%.
Evaporar en baño maría o en placa de calentamiento hasta tener un volumen apróximado de 15 a 20 ml, cubrir el recipiente con vidrio de reloj.
Transferir completamente el contenido del recipiente a un matraz Erlenmeyer de 125 ml, enjuagar con 5 ml de ácido nítrico concentrado.
Agregar al matraz unas perlas de vidrio y 10 ml de ácido sulfúrico concentrado.
Evaporar en una parrilla o placa de calentamiento bajo campana de extracción, hasta que aparescan humos densos blancos de anhídrido sulfúrico en el matraz y no calentar más allá de este punto.

Si no se ha clarificado la solución, agregar 10 ml más de ácido nítrico y repetir la evaporación a humos blancos de anhídrido sulfúrico. Se debe tener la seguridad de la eliminación total del ácido nítrico, identificando con la claridad de la solución y por la ausencia de humos rojizos en el matraz.
Enfriar la solución a temperatura ambiente y diluir con agua cuidadosamente hasta tener un volumen cercano a 50 ml.
Calentar en una placa de calentamiento casi a ebullición para disolver las sales solubles y filtrar a través de un crisol Gooch provisto de un disco de fibra de vidrio o en un crisol de vidrio de fondo poroso.
Transferir el filtrado a un matraz volumétrico de 100 ml y enjuagar el filtro con dos porciónes de 5 ml de agua y aforar.
Continuar a partir de la determinación de cadmio en la muestra.
Digestión
Digestión con ácido nítrico-ácido perclórico (para muestras con materia orgánica dificil de oxidar).
Acidificar al anaranjado de metilo a un pH menor de 4.2 con ácido nítrico y agregar un exceso de 5 ml de ácido nítrico concentrado procediendo de la siguiente manera:
Evaporar en baño maría ó en placa de calentamiento hasta tener un volumen aproximado de 15 a 20 ml, cubrir el recipiente con vidrio de reloj.
Transferir completamente el contenido del recipiente a un matraz Erlenmeyer de 125 ml, enjuagar con 5 ml de ácido nítrico concentrado.
Agregar al matraz unas perlas de vidrio y 10 ml de ácido sulfúrico concentrado.
Evaporar en una parrilla ó placa de calentamiento bajo campana de extracción, hasta que aparescan humos densos blancos de anhídrido sulfúrico en el matraz y no calentar más allá de este punto. Si no se ha clarificado la solución, agregar I O ml más de ácido nítrico y repetir la evaporación a humos blancos de anhídrido sulfúrico. Se debe tener la seguridad de la eliminación total del
127

ácido nítrico, identificando con la claridad de la solución y por la ausencia de humos rojizos en el matraz.
Enfriar y adicionar 5 ml de ácido nítrico, I :I, 10 ml de ácido perclórico al 70%, unas perlas de vidrio y evaporar en vaño maría hasta desprendimiento de humos.
Enfriar la solución a temperatura ambiente y diluir con agua cuidadosamente hasta tener un volumen cercano a 50 ml.
Calentar en una placa de calentamiento casi a ebullición para disolver las sales solubles y filtrar a traves de un crisol Gooch provisto de un disco de fibra de vidrio ó en un crisol de vidrio de fondo poroso.
Transferir el filtrado a un matraz volumétrico de 100 ml yenjuagar el filtro con dos porciónes de 5 ml de agua y aforar. Continuar a partir de la determinación de cadmio en la muestra.
NutaEvitar llevar a sequedad debido al peligro de explosión.
Determinación de Cadmio en la Muestra.
Tomar una alícuota de la muestra digerida, que contenga de 25 a 200 pg de cadmio y colocarla en un vaso de precipitados de 150 ml.
Agregar 0.2 ml de ácido clorhídrico concentrado para precipitar la plata presente en la muestra, agitar y dejar reposar por 2 minutos.
Filtrar si es necesario y lavar con agua. AI filtrado y los lavados agregar 5 ml de la solución de tartrato de sodio y potasio, ajustar a un pH de 2 con hcido clorhídrico concentrado o hidróxido de amonio concentrado.
Transferir la solución a un embudo de separación de 125 ml, y efectuar tres extracciones con porciones de 5 ml de solución de ditizona I en cloroformo hasta que la capa de ditizona permanezca verde.
Agitar vigorosamente en cada extracción y desechar los extractos.
Lavar con porciónes de 10 ml de cloroformo hasta que la capa orgánica permanezca incolora, desechar los lavados, finalmente lavar con una porción de 5 ml de tetracloruro de carbono y desechar los lavados.

Transferir la solución acuosa a un vaso de precipitados de 150 ml y añadir 5 ml de solución de tartrato de sodio y potasio.
Ajustar el pH de la solución a 8.5 - 9.0, adicionando hidróxido de amonio concentrado.
Agregar 5 ml de la solución de dimetil-glioxima y agitar vigorasamente durante 30 segundos.
Extraer con, 3 ó más porciónes de I O ml de cloroformo hasta la desaparición de cualquier precipitado blanco de dimetil-glioxima en exceso, desechar los extractos.
Lavar la capa acuosa con 5 ml de tetracloruro de carbono y desechar el lavado.
* Agregar 4.2 ml de hidróxido de sodio 6N.al embudo de separación y mezclar. Enseguida adicionar 5 ml de solución de ditizona II en tetracloruro de carbono y agitar vigorosamente.
Transferir la capa de tetracloruro' de carbono a un embudo de separación limpio de color ámbar.
*Extraer la capa acuosa con una segunda porción de 5 ml de solución de ditizona II en tetracloruro de carbono y combinar las capas orgánicas.
*Continuar extrayendo con porciónes de 3 ml de solución de ditizona II en tetracloruro de carbono hasta que los extractos orgánicos permanezcan incoloros o ligeramente amarillos y agregar estos extractos a los anteriores.
*Lavar dos veces los extractos orgánicos combinados con porciónes de I O ml de hidróxido de sodio 1 N. y 10 ml de agua, y finalmente con 20 ml de agua. Desechar la capa acuosa en cada paso.
*filtrar la solución roja del complejo de ditizonato de cadmio a tavés del papel filtro Whatman núm. 5 o similar y colocar el filtrado en un matraz volumétrico de 25 ml.
*Lavar el papel filtro con una porción de tetracloruro de carbono.
*Aforar con tetracloruro de carbono y mezclar bien.
Nuta.€s recomendable que el material que se maneje sea de color ámbar o de actínica baja, se pueden proteger los embudos de separación y el matraz volumétrico cubriéndolos con papel aluminio, evitando la exposición del extracto a la luz.

"" "
Vransferir una porción adecuada de la solución de tetracloruro de carbono a una celda de absorción de 1 cm.
*Leer la absorbancia de esta solución dentro de los primeros 15 min. siguientes a la extracción, a una longitud de onda de 515 nm usando tetracloruro de carbono puro como liquido de referencia.
CALCULOS
Las concentraciones cadmio se calculan por medio de la siguiente fórmula.
a. Cálculo directo.
mgll de Cd - A/B x 1000
b. Cálculo por dilución
mg/l de Cd = (A/B x 1000) x 1OO/C
Donde: A = Contenido de cadmio leido en la curva de calibración, en mg B = Volumen de muestra, en ml. C = Volumen de la alícuota, en ml. 1 O00 = Factor de conversión. 1 OO/C = factor de dilución.

FLORUROS
RESUMEN DEL METODO.
La concentración de floruros en la muestra se cuantifica mediante la reacción que se lleva a cabo entre estos y los íones círconio del reactivo colorido de SPADNS-ácido circonilo, disociando una porción de éste en un anión complejo incoloro
La decoloración producida por los íones fluor, es proporcional a la concentración de estos y adecuada para mediciones fotométricas.
(ZrF6)-
INTERFERENCIAS
El cloruro residual causa interferencias, por lo que debe ser eliminado totalmente de la muestra mediante una solución de arsenito de sodio (NaAs 02). Los cloruros en cantidades mayores de 700 mg/l causan errores positivos por ello deben ser removidos antes de proceder al desarrollo del color. Causando errores positivos también los hexametafosfatos en concentraciones mayores de 1 .O mgll, de 16 mgll los fosfatos y de 200 mg los sulfatos.
Los errores negativos son debidos a concentraciones mayores de 10 mgll de fierro (Fe3+), 0.1 mg/l de aluminio (AI) y 500 mg/l de alcalinidad como carbonato de calcio (CaC03).
Cuando la alcanidad sea la única interferencia significativa, neutralizar la muestra con ácido clorhídrico o ácido nítrico de concentración adecuada.
La turbiedad y el color presentes en las muestras, sobre todo en agua residuales, impiden el desarrollo y medición del color.
Siempre que una sustancia interferente cause un error de más de 0.1 mg/l la muestra debe ser sometida a una destilación previa al desarollo del color.
La medición de los volúmenes de muestra y de reactivos así como una uniformidad en la temperatura de f2"C en muestras y estándares durante todo el procedimiento, son

factores muy importantes para la obtención de buenos resultados.
REACTIVOS
Los reactivos que a continuación se mencionan deben ser de grado analítico. Cuando se hable de agua debe entenderse de agua destilada
Reactivos para el desarrollo del método.
Solución madre de floruros. Disolver 221.1 mg de fluoruros de sodio anhídrido (NaF) en agua y diluir a I000 ml. 1 .O0 ml de esta solución, equivale a 10.0 pg F.
Solución estándar de floruros de sodio Diluir 1 O0 ml de la solución madre de florururos a 100 ml de agua. 1 .O0 ml de esta solución equivale, a 10.0 ug de F.
Solución de SPADNS. Disolver 958 mg de SPADNS, sal de sodio 2 (para sulfofenilazo) 1.8 dihidroxi-3,6 naftalen disulfonato, también llamado sal trisódica de 4,5 díhidroxi 3- (parasulfafenilazo)-2,7 naftalendisulfónico ácido, en agua y diluir a 500 ml. Esta solución es estable por tiempo indefinido si se le protege de la luz del sol
Reactivos de ácido circonilo- Disolver 133 mg de cloruro de circonilo octohidratado(ZrOC12~8H20) en aproximadamente 25 ml de agua.
Agregar 350 ml de ácido clorhídrico concentrado y diluir a 500 ml de agua.
Reactivo de ácido circonilo-SPADNS.0 Mezclar volúmenes iguales de solución de SPADNS, y reactivo de ácido circonilo
Solución de referencias- Tomar I O ml de solución de SPADNS- y agregar a 100 ml de agua Tomar 7 ml de ácido clorhídrico concentrado y diluir a 10 ml con agua. Agregar a la solución diluida de SPADNS.
Solución de arsenito de sodio (Na As 02)-Disolver 5.0 g de arsenito de sodio en agua y diluir a un litro.

APARATOS
Equipo de destilación.
Matraz de destilación de un litro de capacidad de fondo redondo y cuello largo hecho de vidrio de borocilicato con entrada de 24/40
Tubo de conexion de 12mm de díametro interior Termómetro adaptado al matraz de destilacion con
lecturas hasta 200°C
tuvo de conexión 24/40.
procedimiento.
Adaptador para el matraz de destilacion, termómetro y
Condensador de capacidad especificada en el
Destilación preliminar.
Colocar 400 ml de agua en el matraz de destilación. Agregar con mucho cuidado 200 ml de ácido sulfúrico concentrado y agitar rotando el matraz hasta que el contenido este homógeneo.
Indiciar el calentamiento, primero lentamente y después tan rápidamente como la eficiencia del condesador lo permita. El destilado debe salir siempre frío. Continuar el calentamiento hasta alcanzar una temperatura de 180OC.
Suspender el calentamiento y desechar el destilado, Este proceso elimina contaminaciones por fluoruro en el aparato y ajusta la relación ácido - agua para posteriores destilaciones.
Dejar enfriar la mezcla ácida remanente en el matraz de destilación a 1200C ó menos y agregar 300 ml de la muestra, homogenizar perfectamente y destilar hasta alcanzar una temperatura de 180OC.
Es posible usar la solución de ácido sulfúrico en el matraz repetidas ocaciones, verificando su eficiencia periódicamente

mediante destilación de estándares de floruros. Después de destilar muestra con alto contenido de floruros ó de largos períodos de inactividad, lavar destilando 300 ml de agua.
Desechar cuando el destilado empiece a presentar interferencia ó la recuperación de fluoruros no sea satisfactoria.
Curva de calibración
Preparar una serie de patrones de fluoruros por dilución de los siguientes volúmenes de solución estandar a 50 ml con agua
ml de solución estandar ( O
pF) c
1 I 1 2 I 2 3
5 5 4 4 3
6 I 6 7 I 7
le F mg/l O
~~
1 I 0.2 I 1
0.6 1 0.4
1 1 .o 1 0.8
1 I .4 1 1.2
Tranferir con pipeta de I O ml del reactivo ácido circonilo- SPADNS a cada patrón y mezclar bien.
Colocar el cero de absorbancia del aparato con la solución de referencia y hacer las lecturas inmeditamente.
Trazar la curva de calibración graficando las lecturas de la absorbancia de los patrones contra sus correspondientes concentraciones.
Análisis de las muestras.
Si la muestra contiene cloruro residual agregar una gota (0.5 ml) de solución de arsenito de sodio por cada 0.1 mg de cloruro y mezclar bien.
134

Tomar 50 ml de muestra o una alícuota diluida a 50 ml con agua; puede ser muestra directa o muestra destilada según sea el caso.
Continuar con el procedimiento exactamente en la misma forma que para la curva de calibración:
Tranferir con pipeta de 10 ml del reactivo ácido circonilo-
Colocar el cero de absorbancia del aparato con la solución SPADNS a cada muestra y mezclar bien.
de referencia y hacer las lecturas inmeditamente.
CALCULOS Determinar el contenido de fluoruros en la muestra en la
curva de calibración las concentraciones correspondientes a las absorbancias y aplicando la siguiente fórmula:
mg de F = AIB
Donde:
A = Contenido de F leído en la curva de calibración, en pg. B = Volumen de muestra tomada para el análisis, en ml.

DETERMINACIóN DE PLOMO METODO CLORIMETRICO DE LA DITIZONA.
FUNDAMENTOS
El método se basa en la rección del plomo presente en el agua con la ditizona disuelta en tetracloruro de carbono formando un complejo de ditizonato de plomo de color rosa, cuya intensidad determinada colorimétricamente es proporciónal al contenido de plomo.
INTERFERENCIAS
Los elementos que interfieren en la extracción de plomo a un pH de 8.5 a 9 en presencia de cianuro son: estaño, bismuto y talio.
MUESTRAS
La muetra debe colectar en frascos de polietileno de acuerdo a lo indicado men las normas de muestre0 en vigor, y preservarse añadiendo ácido nítrico hasta obtener un pH de 2 a 2.5.
PROCEDIMIENTO
Digestión de la muestra.
Tomar un volumen de muestra necesaria, acidular al anaranjado de metilo a un pH de 4.2 con ácido sulfúrico y agregar a continuación 5 ml de ácido nítrico y 2 ml de peróxido de hidrógeno.
Evaporar en baño maría hasta tener un volumen aproximado de 15 a 20 ml y cubrir la cápsula con in vidrio de reloj.
Transferir completamente el contenido de la cápsula a un matraz Erlenmyer de 250 ml.

Agregar una perla de vidrio, 5 ml de ácido nítrico y I O ml de ácido sulfúrico y lavar con 2 ó 3 porciónes de agua.
Evaporar en parrilla o placa de calentamiento hasta que aparescan humos densos blancos de anidrido sulfúrico en el matraz.>>Este calentamiento debe realizarse en campana de extracción debido a que los vapores que se producen son tóxicos<<. Si no se ha clarificado la solución, agregar I O ml más de ácido nítrico y repetir la vaporación. Se debe tener la seguridad de la eliminación total del ácido nírtico indicada por la claridad de la solución y por la ausencia de humos rojizos en el matraz.
Enfriar la solución a la temperatura ambiente y diluir cuidadosamente con 50 ml de agua.
Calentar casi a ebullición para disolver las sales y filtrar a través de un crisol Gooch provisto de un disco de fibra de vidrio de fondo poroso, agregar 50 ml de solución de acetato de amonio caliente al crisol Gooch para disolver el posible precipitado de sulfato de plomo formado y recibir los filtrados en un matraz Kitazato.
Transferir el filtrado a un vaso de precipitados de 250 ml y ajustar el volumen a 100 ml con agua y proceder a la determinación de plomo como se indica a continuación.
Determinación de plomo en la muestra:
Agregar de I O a 15 gotas de fenolftaleína y neutralizar con hidróxido de amonio 1 :l.
* Agregar 20 ml de solución de acetato de hidracina o solución de clorhidrato de hidroxilamina, calentar en baño maria a 90 - 95°C durante 1 O minutos y dejar enfriar.
* Agregar 20 ml de solución de tartrato de sodio y ajustar el pH de la solución, aproximadamente a 2.5 con solución de ácido tartárico utilizando potenciómetro.

* Transferir la solución a in embudo de separación, con porciónes de 3 ml de solución, I de ditazona hasta que la capa orgánica tenga un color verde; agitar bien en cada ocasión y separa la capa de cloroformo, la cual se desecha.
* Extraer la solución con 2 porcións de 5 ml de cloroformo paara eliminar la ditazona retenida, y desechar las porciónes de cloroformo.
* Eliminar los residuos de cloroformo extrayendo con una porción de 5 ml de tetracoruro de carbono y deschar esta capa. *Nota.= Si se tiene la seguridad de que los elementos (estaño, bismuto y talio) no están presentes en la muestra, pueden omitirse los puntos marcados con *.
Agregar 10 ml de solución de tartrato de sodio y 5 gotas del indicador de azul de timo1 (Si es necesario, agregar hidróxido de amonio concentrado para hacer que el indicador vire al azul) y I O ml de solución de cianuro de potasio.
Ajustar el pH a 8.5 adicionando hidróxido de amonio 1:l ó solución de ácido tarthrico hasta que el indicador vire al verde.
Agregar 5 ml de solución II de ditizona, agitar bien y pasar cuidadosamente la capa de ditizona a un segundo embudo de separación.
Hacer extracciones sucesivas en la fase acuosa con porciónes de 2 m12 de solución II de ditizona hasta que el color verde persista en la fase acuosa por lo menos en dos extracciónes.
Combinar todos los extractos en el segundo embudo de separación.Al analizar varias muestras, debe tenerse cuidado de hacer el mismo número de extracciónes; debido a que varia la intencidad de color del testigo al aumentar el número de extracciónes.

Agregar 5 ml de tetracloruro de carbono puro al primer embudo de separación y el extracto se recibe en el segundo embudo. %ti@ .?
& Y '
A los extractos combinados de tetracloruro de carbono, agregar 20 ml de la solución alcalina de cianuro de potasio y agitar. y: ,;;
La coloración verde de la ditizona cambia a la coloración rosa <; (7
del ditizonato de plomo, la solución alcalina acuosa se torna *z\ .:: amarillenta, por la formación de sal de ditizona.
,#x! $#I
" x i , ,
c. .T.; "wd r " #
"" * -, :"' ,!5 r- +:..> r . i y{ Cr, ,-
_. C.\
Pasar la capa de tetracloruro de carbono a un matraz ' T
volumétrico de 50 ml. \Y?, .. , , ."
Extraer con porciónes de 2 ml de tetracloruro de carbono y combinar todos los extractos en el matraz volumétrico de 50 ml y aforar hasta la marca con tetracloruro de carbono y agitar. Se debe tener mucho cuidado de que los extractos no arrastren gotas de agua.
Leer la absorbancia de esta solución con tetracloruro de carbono puro como líquido de referencia, en el espectrofotómetro a una longitud de onda de 520 nm.
CALCULOS
La concentración de plomo se cálcula con las siguientes fórmulas:
a) Cálculo directo: A
B mg de plomo / 1 = -- x 100
b) Cálculo de dilución: A 1 O0
B C mg de plomo / 1 = --- x I O00 x ""

Donde:
A: Contenido de plomo leído en la curva de calibración, en mg. B: Volumen de muestra, en ml. C: Volumen de la alícuota, en ml.
- DETERMINACIóN DE NíQUEL
FUNDAMENTO
Para la determinación del níquel, la muestra es sometida a una digestión preliminar con una mezcla ácida para eliminación de inferencias. Posteriormente el cobre y el hierro son separados mediante extracciónes con una solución de cupferrón, el níquel se separa entonces de otros iones por extracción de un complejo de dimetilglioxima con cloroformo, se extrae nuevamente a una face acuosa ácida y se hace reaccionar otra vez con la solución de dimetilglioxima, para el desarrollo del cloro y se mide su absorción espectral fotométricamente.
INTERFERENCIAS
La turbiedad y el color en las muestras causadas principalmente por materia orgánica, sobre todo en aguas residuales, impiden el desarrollo y medición del color. En este caso es necesario hacer una digestión con ácido nítrico-ácido sulfúrico antes del desarrollo del método.
MUESTRAS
Las muestras se colectan en recipientes de plástico. En caso de no efectuar de inmediato el análisis, preservarlas añadiendo 5 ml de ácido nítrico concentrado por litro de muestra. Guardar por un máximo de 6 meses.

Digestión con ácidos nítrico y sulfúrico.
De acuerdo con la concentración esperada del metal tomar un volumen de muestra perfectamente homogenea, de acuerdo a la siguiente tabla:
I Concentración esperada en I Volumen de muestra (ml) I mgll
Mayor que - 1
1 100-1000
1 O00 1 - 1 0 1 O0
Colocar en una cápsula de evaporación y acidificar el anaranjado de metilo con ácido sulfúrico concentrado.
Agregar 5 ml de ácido nítrico concentrado y 2 ml de peróxido de hidrógeno al 30%.
Evaporar a 15 ó 20 ml en un baño de vapor o parrilla de calentamiento, evitando proyecciones.
Transferir la proyección evaporada junto con cualquier sólido nuevamente a un matraz Erlenmeyer de 125 ml.
Lavar la cápsula con 5 ml de ácido nítrico y con 10 ml de ácido sulfúrico concentrados y agregar al matraz.
Agregar unas perlas de vidrio y evaporar hasta desprendimientos de humos densos, blancos de SO3. Suspender el calentamiento.
Si la solución no está clara, agregar 10 ml de ácido nítrico concentrado y repetir la evaporación hasta desprendimientos de humos de SO3.
NOTA: En este punto, todo el ácido nítrico debe haber sido removido, esto se indica por la claridad de la solución y la ausencia de humos cafés en el matraz.
Enfriar a temperatura ambiente y diluir con agua a 50 ml aproximadamente.

Calentar hasta cerca del punto de ebulición para disolver
Filtrar a través de un filtro de fibra de vidrio. Lavar el matraz y el filtro con dos porciónes de 5 ml de
agua. Transferir cuantitativamente el filtrado con los lavados a
un matraz volumétrico de 100 ml. Llevar a la marca con agua y mezclar vigorosamente y de esta de esta solución tomar porciónes para el análisis.
todas las sales solubles.
Preparación de la curva de calibración.
Trasferir con pipetas volumétricas a matraces volumétricos de 100 ml la solución estándar.
ml de solución estándar mg de Ni O
50 5 O
15
25 200 20 150
250
Agregar 25 ml de solución de ácido clorhídrico 1.0 N y 5 ml de agua btomada Enfriar con una corriente de agua fría, agregar 10 ml de hidróxido de amonio concentrado. Agregar 20 ml de la solución de dimetilglioxima sal disódica y 20 ml de alcohol etílico. Aforar con agua y mezclar. Dejar reposar por 10 minutos exactos y medir inmediatamente la absorción espectral de los estándares a 445 nm. Trazar la curva de calibración graficando las absorciones espectrales contra sus respectivas concentraciones.
Tratamiento para separar hierro y cobre.
Tomar una porción de la muestra tratada por digestión con ácido nítrico-hido sulfúrico ,conteniendo entre 50 y 250 mg de Ni . Colocar en un embudo de separación.

Agregar 10 ml de solución de tartrato de sodio, 2 gotas (0.1 ml) del indicador de anaranjado de metilo y gota a gota solución de hidróxido de sodio 6 N hasta el vire del indicador de canela- amarillo. Agregar I ml de ácido acético y enfriar colocando el ambudo en una corriente de agua fría. Añadir 4 ml de cloroformo y mezclar bien. Dejar reposar para separar las fases y continuar agreagando pequeñas contidades de cupferrón hasta que se forme un precipitado blanco (que indica el exceso de cupferrón). Mezclar agitando el embudo, dejar que se separen las fases y desechar la capa de cloroformo. Agregar 1 ml de solución de clorhidrado de hidroxilamina, mezclar y dejar reposar IO minutos y proceder a la separación del níquel.
Separación del níquel
Agregar a la solución del embudo 1Oml de la solución de dimetilglioxima sal disódica y mezclar. Agregar a la solución del embudo 1Oml de la solución de dimetilglioxima sal disódica y mezclar. Pasar la capa de cloroformo a un embudo de separación y repetir esta operación con varias porciones de 10 ml de cloroformo hasta que este sea incoloro, y adicionarla al cloroformo de la primera extracción. Desechar la capa acuosa. Agregar al embudo que contiene el cloroformo, 10 ml de solución de ácido clorhídrico 1.0 N. Mezclar bien y dejar reposar para separar las fases. Regresar la capa de cloroformo al primer ambudo y lavar con 1 O ml de solución de ácido clorhídrico I .O N. Agregar IO ml de ácido clorhídrico concentrado y desechar la capa de cloroformo. Tranferir todas las porciones de ácido clorhídrico a un matraz volumétrico de 100 ml. Continuar con el procedimiento de níquel en las muestras leyendo en la curva de calibración a partir de ; agregar 25 ml de solición de ácido clorhídrico 1 .O N y 5 ml de agua bromada.

CALCULOS
Determinar la concentración de níuel en las muestras leyendo en la curva de calibración la concentración correspondiente a sus absorciones espectrales y aplicando la siguiente fórmula:
A 1 O0 mg/l de Ni = --- x -
B C
Donde: A = Contenido de Ni leído en la curva de calibración, en mg. B = Volumen de muestra tomando para el análisis, en ml. C = Volumen de la porción tomada para el análisis ( después de la digestión con ácido sulfúrico-ácido clorhídrico), en ml.
NITRóGENO TOTAL
FUNDAMENTO
Este método se basa en la determinación de la suma del nitrógeno del amoníaco libre y del nitrógeno orgánico, los cuales son convertidos a sulfato de amonio bajo las condiciones de digestión que aquí se describen.
Mediante digestión en presencia de ácido sulfúrico, sulfato de potasio y sulfato de mercúrico, el nitrógeno de compuestos orgánicos es convertido a sulfato de amonio. El amoníaco es destilado en medio en medio alcalino, absorbido en solución de ácido bórico y determinado por titulación.
DEFINICIONES.
Nitrógeno total. Es la suma de nitrógeno amoniacal y orgánico presente en el agua conocido correctamente como nitrógeno Kjeldehl
Agua residual. Es el líquido de composición variada proveniente de usos municipal, industrial, comercial, agrícola,
144

pecuario o de cualquier otra indole, ya sea pública o privada y que por tal motivo haya sufrido degradación o alteración en su calidad original.
Agua natural. Es el líquido de composición variada dulces y salinas, superficiales y subterraneas que no haya sufrido degradación o alteraciones en su calidad original.
PREPARACI~N DE SOLUCI~NES.
Solución amortiguadora. Agregar 88 ml de una solución de hidróxido de sodio 0.1 N a 500 ml de solución de tetraborato de sodio 0.025 N y aforar a 1 litro.
Solución de sulfato mercúrico. Disolver 8 g de óxido mercúrico rojo en 50 ml de a'cido sulfúrico (15) y aforar a 100 ml con agua.
Solución de ácido sulfúrico-sulfato mercúrico-sulfato de potasio. Disolver I34 g de sulfato de potasio en 650 ml de agua, agregar 200 ml de ácido sulfúrico concentrado.
Añadir 25 ml de solución de sulfato mercúrico y aforar a I litro. Este reactivo debera mantenerse a una temperatura de mayor a 34°C para evitar que cristalice.
Solución de hidróxido de sodio -tiosulfato de sodio. Disolver 500 g de NaOH y 25 g de tiosulfato de sodio en agua y aforar a I litro.
Solución indicadora de fenolftaleína. Disolver 5 g de sal de fenolftaleína disódica en agua y aforar a 1 litro. Si es necesario, añadir hidróxido de sodio 0.02 N gota a gota hasta que la solución alcance un ligero color rosado?
Solución de ácido sulfúrico 0.02 N Solución de hidróxido de sodio 6 N Solución de hidróxido de sodio 0.1 N Solución de hidróxido de sodio 0.02 N

Solución de ácido bórico. Disolver 28 g de ácido bórico en agua agregar 1 O ml de la solución indicadora mixta y aforar a un litro.
Solución indicadora mixta. Se mezclan dos volumenes de rojo de metilo al 0.2% en etanol. Esta solución debe prepararse por lo menos cada 30 días.
Reproducción de la muestra.
La diferencia entre las determinaciones efectuadas por duplicado, no deben exceder de 2 0.03 mg/l, para concentraciones de I a 5 mg/l y de 0.13 mg/l, para concentraciones de 6 a 50 mgll. En caso contrario se recomienda repetir la determinación.
PROCEDIMIENTO
Nitrógeno Amoniacal.
Tomar una muestra dependiendo de las concentraciones esperadas, de acuerdo a la tabla 1 , diluir con agua hasta 50 ml. Preparar un testigo con 500 ml de agua y darle el mismo tratamiento que a la muestra como sigue:
Añadir 25 ml de la solución amortiguadora de boratos y ajustar el pH a 9.5 con solución de hidróxido de sodio 6.ON, utilizando potenciómetro o papel indicador para verificar. Transferir la solución a un matraz Kjeldahl.
Conectar el matraz Kjeldahl al bulbo del aparato de destilación; destilar la muestra cuidando que la temperatura del condensador no pase de 29"C, recolectando el condensado con la punta del tubo del refrigerante sumergido en 50 ml de la solución de ácido bórico del matraz receptor. (ver figura 2).
La destilación se completa cuando se hayan recolectado 300 ml de destilado aproximadamente, incluyendo los 50 ml de la solución de ácido bórico con la solución indicadora mixta.
146

Retirar el matraz colector y titular con solución de ácido sulfúrico 0.02N hasta que la solución vire de un verde esmeralda a un café rojizo.
TABLA 1
Nitrógeno orgánico en
ml/l de nitrógeno. muetra ml de muestra
0-5 500 5-1 O 250 I 0-20 1 O0 20-50 50 50-1 O0 25
Nitrógeno orgánico.
Dejar enfriar el residuo contenido en el matraz Kjeldahl producto de la destilación. Añadir 50 ml de la solución de ácido sulfúrico-sulfato de potasio. Conectar al aparato de digestión. calentar la mezcla en el matraz Kjeldahl a una temperatura que no exceda de 644K (371OC) hasta que los gases de S03 (vapores blancos) sean eliminados y la solución se torne incolora o amarillo pálido, a partir de este momento se mantiene el calentamiento durante 20 minutos más. La digestión se debe efectuar bajo condiciones satisfactorias de ventilación y extracción de gases.
Dejar enfriar la solución, añadir 300 ml de agua y 5 gotas de la solución indicadora de fenolftaleína.
Sostenga el matraz en posición ligeramente inclinada y agregar 50 ml apróximadamente de solución de hidróxido de sodio-sulfato de sodio con lento escurrimiento por las paredes del matraz y sin mezclar, hasta que el matraz de digestión sea conectado al aparato de de destilación, procurando formar dos capas.
147
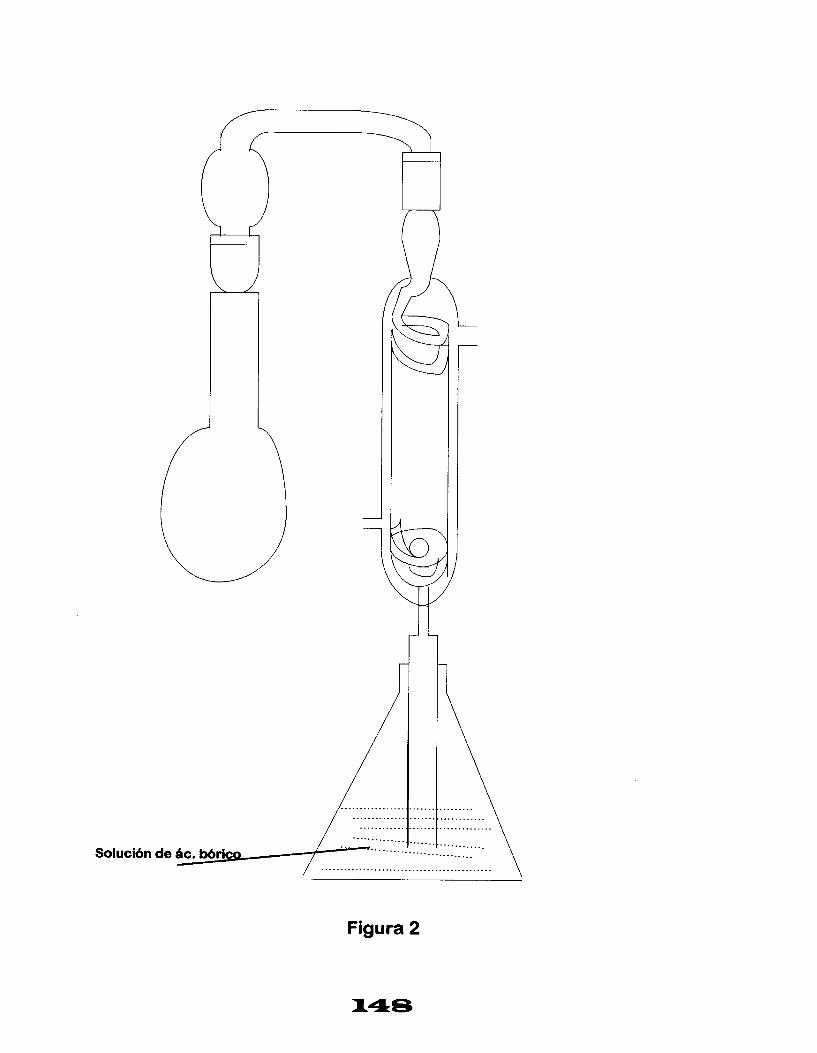
Figura 2

Conectar inmediatamente el matraz al bulbo del aparato de destilación. Agitar y verificar la alcalinidad de la solución de
acuerdo al cambio de la misma (de incoloro a rosa). En caso de que no se haya alcanzado la alcalinidad, agregar un exceso de la solución de hidróxido de sodio-tiosulfato de sodio hasta la obtención de ina coloración rosa. La muestra se destila, cuidando que la temperatura del destilador no se pase de 302K (29"C), recolectando el condensado con la punta del tubo del refrigerante sumergida en 50 ml de la solución de ácido bórico del matraz receptor.
Esta determinación puede hacerse directamente siguiendo el procedimiento descrito anteriormente.
La destilación se completa cuando se hayan recolectado 300 ml de destilado aproximadamente, incluyendo los 50 ml de la solución de hcido bórico con la solución indicadora mixta. Retirar el matraz colector y titular con solución de ácido sulfúrico 0.02 hasta que la solución vire de in color verde esmeralda a un café rojizo.
Nitrógeno total. esta determinación puede hacerse directamente siguiendo el procedimiento descrito anteriormente.

CALCULOS. El nitrógeno amonacal, orgánico y total en mg/l se calcula con la fórmula siguiente:
Nitrógeno total en mg / I I - --X 14 X 1000 (A-B) X N
V
En donde:
A= Volumen de solución de ácido sulfúrico empleado para titular la
muestra, en ml, correspondiente al nitrógeno amoniacal, orgánico o total. B= Volumen de solución de ácido sulfúrico empleado para titular el
N= Normalidad de la solución de ácido sulfúrico. V= Volumen de muetra, en ml. 14= Equivalente del nitrógeno.
testigo, en ml.
En el caso que B sea mayor que A, se repite la prueba. (Se recomienda emplear mayor volumende muestra). O bien, el nitrógeno total en mgll = nitrógeno orgánico en mg/l - nitrógeno amoniacal en mg/l.

DETERMINACIóN DE ARSÉNICO. (MÉTODO ESPECTROFOTOMÉTRICO)~~.
FUNDAMENTO
El arsénico se reduce a arsina por el zinc en solución ácida, la arssina pasada através de un depurador y después a un tubo absorbente que contenga dietil ditio arbamato de plata, para la formación de un complejo rojo soluble cuyo color es proporciónal al contenido de arsénico en la muestra.
Reacciones.
2As + 3Zn + 6HCL ---> 3ZnCI z + 2AsH3
ASH^ + AgSCSN (C2H5)2-- >Complejo rojo
INTERFERENCIAS.
El cromo, cobalto, cobre, mercurio, molibdeno, níquel, platino y plata pueden interferir cuando la concentración presente de alguno de ellos en el agua sea mayor de I O mg/l.
Trazas de sales de antimonio forman la estibina con la arsina que desarrolla un color rojo que interfiere en el análisis.
Las interferencias antes menmcionadas son poco comunes, sin embargo cuando esten presentes deben eliminarse. Para la eliminación de las interferencias, seguir el procedimiento descrito en la Encyclopeda of Industrial Analysis Vol. 6 pag. 237-240 Edit. lnterscience Publishers 1968 N.Y.

Preparación de la curva patrón Tomar volúmenes de la solución patrón (Solución
intermedia de arsénico) como se muestra en la tabla I, diluir con agua a 35 ml tratarlas como se indica en el procedimiento, graficar las lecturas obtenidas contra las concentraciones de arsénico.
PROCEDIMIENTO.
Tomar 35 ml de muestra o una alícuota y pasarla al frasco generador. agregar 5 ml de HCI concentrado y agitar cuidadosamente.
Agregar 2 ml de la solución de KI y 0.4 ml de la solución de SnCIz, agitar cuidadosamente, dejar reposar 15 minutos para que el arsénico se reduzca a estado trivalente.
Impregnar la fibra de vidrio con la solución de acetato de plomo y colocarla en el limpiador.
Montar el aparato generador de arsina (Ver fig. 3). Medir con pipeta 4.0 ml del reactivo de dietil ditio carbonato de plata en el tubo absorbente.
Figura 3
Tubo absorbente
1)

Agregar 3 g de zinc metálico al frasco generador e inmediatamente conectarlo al tubo limpiador asegurándose que estén herméticamente unidos
Dejar durante 30 minutos que se genere la arsina, calentando los primeros 15 minutos de 303 K a 308 K (30 a 35°C). En el tubo absorbedor debe burbujear la arsina, si no hay burbujeo, verificar todas las conexiones; enfriar gradualmente el matraz generador.
Verter la solución del tubo absorbedor a una celda y medir la absorbancia de la solución a 535 nm usando un testigo como referencia. Las celdas deben estar perfectamente limpias y libres de rayaduras, deben enjuagarse con la solución que se va a analizar. N0TA.EI testigo se corre como las muestras, pero únicamente con agua.
TABLA 1
I cm3 de la solución Datrón I T II de arsénico.
I 0.0 0.0 0.1
0.5 0.5 0.1
1 .o 1 .o I 2.0 I 2.0 I 5.0 I 5.0 I 10-0 I 10-0

CALCULOS
Las concentraciones de arsénico se calculan por medio de
A
V
la siguiente fórmula:
C e ""
Donde: C = concentración de arsénico, en mg/l A = cantidad de arsénico leído en la curva, en T g V = volumen de la muestra, en ml
DETERMINACIóN DEL NúMERO MAS PROBABLE (NMP) DE COLIFORMES TOTALES, COLIFORMES
FECALES (TERMOTOLERANTES) Y Escherichia coli PRESUNTIVA.
FUNDAMENTOS
El método se basa en la inoculación de alícuotas de la muestra, diluida o sin diluir, en una serie de tubos de un medio de cultivo líquido conteniendo lactosa.
Los tubos se a las 24 y 48 horas de incubación ya sea a 308 ó 310 K (35 ó 37°C). cada uno de los que muestran turbidez con producción de gas se resiembra en un medio confirmativo más selectivo y, cuando se busca E: coli presuntiva, en un medio en le que se pueda demostrar producción de indol.
Se lleva a cabo la incubación de estos medios confirmativos hasta por 48 horas ya sea 35 o 37OC para la detección de organismos coliformes y a 44°C para organismos coliformes termotolerantes y E. coli.

Mediante tablas estadísticas, se lleva a cabo el cálculo del número más probable (NMP) de organismos coliformes, organosmos coliformes termotolerantes y E. coli que pueda estar presente en 100 ml de muestra, a partir de los números de los tubos que dan resultados confirmativos positivos.
INTERFERENCIAS
Preservación y almacenamiento.
El análisis bacteorológico de la muestra debe practicarse inmediatamente después de su recolección. Es por eso que se recomienda que de no efectuarse así el análisis, se inicie dentro de los dos horas próximas a la recolección de la muestra y en ningún caso, este lapso debe de exceder de 24 horas para agua potable y de seis horas para otro tipo de agua para que sea válido el resultado del análisis. Durante el período que transcurre del muestre0 al análisis, se debe conservar la muestra a 4OC, con objeto de inhibir la actividad bacteriana para no obtener resultados falsos o dudosos.
PROCEDIMIENTO
Pruebas presuntivas.
Preparación de la muestra+O e inoculación del medio.
Antes del examen, mezclar perfectamente la muestra agitándola vigorosamnete para lograr una distribución uniforme de los microorganismos y, dependiendo de la naturaleza del agua y el contenido bacteriano esperado, hacer todas las diluciones esperadas en esta etapa.
Utilizar serie que constan de por lo menos tres diluciones: 10.0 ml, 1 .O ml y 0.1 ml, o bien 1 .O ml, 0.1 ml y 0.01 ml. por cada dilución debe haber 3 ó 5 tubos.

Para diluciones a 10 veces, poner 90 o 9 ml del diluyente en matraces o tubos de dilución esterilizados. Alternativamente, usar volúmenes de diluyente presterilizado en botellas de tapón de rosca. Hacer una o más diluciones a 10 veces transfiriendo un volumen de las muestras de agua a 9 volúmenes de diluyente. Repetir estos pasos cuantas veces sea necesario. Preparar suficiente cantidad de cada dilución para todas las pruebas que se vayan a llevar a cabo con la muestra. Para diluciones diferentes a 10 veces ajustar el volumen de diluyente a la porción de prueba. Inocular los tubos conteniendo medios de aislamiento de doble concentración con porciones de prueba de un volumen mínimo de 5 ml.
Incubación de los tubos.
Incuban los tubos inoculados ya sea a 35 f 1 "C o 37 f 1 "C durante 48 horas.
Examen de los tubos.
Examinar los cultivos de los tubos después de un período de incubación de 18 a 24 horas y considerar como resultados positivos aquellos que muestren turbidez debido al crecimiento bacteriano y formación de gas en los tubos internos invertidos (Durham) junto con producción de ácido si el medio de aislamiento contiene un indicador de pH. Reincubar aquellos tubos que no muestran alguno o todos estos cambios y examinarlos nuevamente para detectar reacciones positivas a las 48 horas.
Pruevas confirmativas.
Inoculación del medio. Resembrar a partir de cada tubo de medio de aislamiento que muestre un resultado positivo en uno o más tubos de medio confirmativo para detectar la producción de gas e indol. Utiluzar uno o más de los siguientes medios confirmativos:

Medios para la producción de gas:
Caldo bilis lactosa verde brillante: Pentena 10.0 g Lactosa 10.0 g Bilis de buey (deshidratada) 20.0 g Verde brillante (0.1% mlm en solución acuosa) 13 ml Agua para llevar a 1000 ml
Disolver la pentena en 500 ml de agua. Añadir los 20 g de bilis de buey deshidratada disueltos en 200 ml de agua; la solución debe tener un pH entre 7.0 y 7.5. Disolver con agua hasta un volumen aproximado de 975 ml. Añadir la lactosa y ajustar el pH a 7.4. Añadir la solución de verde brillante y aforar a 1000 ml con agua. Distribuir volúmenes de 5 ml en tubos de ensayo conteniendo tubos de fermentación invertidos (Durham) y colocar en autoclave a I 1 5°C durante 1 O min.
Nota; Este medio no da resultados reproducibles en todos los casos y se recomineda comprobar sus propiedades inhibitorias antes de usarlo.
Caldo EC: Triptosa 20.0 g Lactosa 5.0 g Mezcla de sales bilianas 1.5 g Fosfato dibásico de potasio 4.0 g (K2H P04) Fosfato monobásico de potasio 1.5 g (NH2P04) Cloruro de sodio (NaCI) 5.0 g Agua para llevar a 1000 ml
Disolver los componentes por separado y agregarlos agitando suavemente. El pH debe ser de 6.9 después de la esterilización. Antes de esterilizar, distribuir en tubos de fermentación con suficiente medio para que el tubo invertido

quede cubierto cuando menos parcialmente después de la esterilización. Como medio confirmativo para coliformes totales, el más generalizado es el caldo bilis lactosa verde brillante (BLVB). Para confirmar la presencia de coliformes fecales se utiliza tanto el BLVB como el caldo EC.
Nota.. Si se usa el medio más inhibitorio de caldo lactosa para aislar, resembrar en alguno de los dos medios confirmativos más selectivos, Caldo Bilis Lactosa Verde Brillante o Caldo EC para efectuar la confirmación.
Incubación y examen.
Para confirmar la presencia de organismos coliformes, incubar un tubo de Caldo Bilis Lactosa Verde Brillante o Caldo EC a 37°C y examinarlo para ver si hay producción de gas dentro de un período de 48 horas.
Para confirmar la presencia de organismos coliformes termotolerantes, incubar otro tubo de Caldo Bilis Lactosa Verde Brillante o Caldo EC a 44°C durante 24 horas para ver si hay producción de gas.
Para confirmar la presencia de E. coli presuntiva, incubar un tubo de agua de triptona para detectar la formación de indol a 317 K (44°C) durante 24 horas. Después añadir de 0.2 a 0.3 ml de reactivo de Kovacs al tubo de agua triptona; el desarrollo de un anillo de color rojo después de agitar suavemente denota la presencia de indol.
Reactivo de Kovacs para indol:
1.4 dimetilaminozadehido 5 g Alcohol amílico (CH3 (CH2),CH) libre de bases orgánicas 75 ml Ácido clorhidrico concentrado (HCI) 25 ml
Disolver el aldehído en el alcohol. Añadir el ácido concentrado con cuidado. Proteger de la luz y almacenar a 4°C.

Nota: El reactivo debe tener una coloración entre amarillo claro y café claro; algunas muestras de alcohol amílico son insatisfactorias y dan una coloración obscura con el aldehído.
Nota 2: La detección de E. coli presuntiva se considera una evidencia satisfactoria de contaminación fecal. Sin embargo, pueden efectuarse mayores pruebas para la confirmación de E. coli si se considera necesario.
Prueba de oxidasa.
Algunas bacterias existentes en el agua pueden conforaerse a la definición de organismos coliformes en muchos aspectos, pero son capaces de producir gas apartir de lactosa solamente a temperaturas inferiores a 37"C, por consiguiente, dan resultados negativos en las pruebas confirmativas estandar para organismos coliformes y su presencia en agua usualmente no se considera significativa. Las especies de Aeromomas, que se encuentran naturalmente en el agua, tienen una temperatura óptima de cresimiento en el rango30 a 35OC, pero apesar de ello son capaces de producir ácido y gas a partir de lactosa a (37OC). Tiene poco significado para efectos sanitarios y se distinguen del grupo de los coliformes por una acción de oxidasa positiva.
Llevar a cabo la prueba con subcultivos puros de los organismos fermentadores de lactosa, cresidos en medio nutriente de agar, como sigue: - Colocar de 2 a 3 gotas de reactivo de oxidasa recientemente preparado en un panel filtro en una caja de Petri.
Reactivo de oxidasa para la prueba de oxidasa: Cloridrato de tetrametil-p-fenilendiamiua 0.1 g Agua 10 ml
Este reactivo no es estable y debe presentarse para usarse en pequeñas cantidades cada vez que sea necesario.
- Con una barra de vidrio o un asa de alambre de platino (no de nicromel), colocar parte del cultivo en el papel filtro preparado.

- Considerar la aparición de un color azul marino purpúreo en un lapso de I O segundos como una reacción positiva. Nota: En cada ocasión que se utitlise el reactivo de oxidasa, llevar a cabo pruebas de control con cultivos de organismos que se sepa dan una reacción positiva (Pseudomomas aeruginosa) y uno que dé una reacción negativa (E. coli) en I O0 ml de la muestra.
Cálculos.
Apartir del número de tubos que dan reacciones positivas en los medios de aislamiento y confirmativo, calcular NMP mediante la siguiente fórmula:
NMP/100 ml= -U"---
No. de tubos positivos x 100
( A X B)"2 """-
A = ml de muestra en tubos negativos B = ml de muestra entodos los tubos
TEMPERATURA
La influencia de la temperatura sobre la calidad del agua es significativa, influye entre otras cosas en el valor de saturación de la concentración de oxígeno disuelto y el metabolismo de la biota acuática, por lo tanto son indispensables mediciones precisas, las que se efectuan por medio de termómetros, termopar o termistor.
Para medir la temperatura se utiliza preferentemente el termómetro de mercurio, que debido a su fragilidad debe ser protegido adecuadamente. El bulbo del termómetro que contiene el mercurio se introduce al agua y se agita suavemente en circulos hasta lograr lecturas estables. Las lecturas deben efectuarse estando el bulbo dentro del agua.
La medición de la temperatura con termopar se basa en la diferencia de potencial que se produce entre dos conductores al aplicarles una temperatura dada; mientras que los
X 6 0

termistores funcionan por cambios en la resistencia eléctrica ocacionada por variaciones de la temperatura.
La norma mexicana para determinar la temperatura en aguas residuales es: NOM-AA-7-1980 Determinación de la la temperatura. Método visual con termometro.
WA TER.-DETERMINA TIÓN OF TEMPERA TURE.
1. OBJETIVO Y CAMPO DE APLICACION. Esta norma oficial establece el método de prueba para determinar la temperatura en aguas.
2. REFERENCIAS. La presente norma se complementa con las Normas Oficiales Mexicanas en vigor siguientes:
2.1. NOM-CH-5 "Termómetros industriales de vidrio". 2.2. NOM-CH-7 "Sistemas termales aplicados como
elementos primarios de medición". 2.3. NOM-AA-3 "Aguas residua1es.-Muestreo". 2.4. NOM-7-1 "Sistema general de unidades de medida.-
sistema (S I) de unidades".
3. FUNDAMENTO. La temperatura se mide con un instrumento apropiado, debidamente calibrado y debe efectuarse en el lugar de muestreo.
4. DEFINICIONES. 4. f. Agua residual. Líquido de composición variada proveniente de uso
municipal, industrial, comercial, agrícola, pecuario o de cualquier
otra indole, ya sea pública o privada, y que por tal motivo haya sufrido degradación o alteración en su calidad original.
4.2. Amp/itud de gama. Diferiencia algebraica entre los limites de medición. 4.3. Descarga.
161

Conjunto de aguas residuales que se vierten o disponen en algún cuerpo receptor.
5. APARATOS Y EQUIPO. 5.1 .Instrumentos medidores de temperatura. Se permite el empleo de instrumentos medidores de
temperatura, siempre y cuando se ajuste a las Normas Oficiales Mexicanas .
NOM-CH-5. " Termómetros industriales de vidrio" y NOM-
primarios de medición " en vigor, y cumplen cuando menos con los requisitos de amplitud de gama Y precisión establecidos para los termómetros de vidrio .
CH-7 "Sistemas termales aplicados como elementos
6. PROCEDIMIENTO. 6.1. Determinar la temperatura de la muestra extraida
según la indicada en la Norma NOM-AA-3 "Aguas residua1es.-Muestreo", y por inmersión directa del instrumento medidor de temperatura, esperar el tiempo suficiente para obtener mediciones constantes.
7.INTERPRETACIÓN DE LOS RESULTADOS. Las lecturas se obtienen directamente de la escala del aparato medidor de temperatura, y se informa en grados Kelvin (K) con aproximación de 0.1 K.
8. INFORME. Deve incluir los siguientes datos:
8.1. Identificación completa de la muestra . 8.2 Referencia a este método. 8.3.Las temperaturas obtenidas, en K. 8.4.Fecha del análisis.
I O . CONCORDANCIA CON NORMAS INTERNACIONALES. No concuerda con ninguna por no existir norma internacional sobre el tema .

APENDICE "A" REQUERIMIENTOS

REQUERIMIENTOS.
Muestre0 Material Botellas para DBO de cuello angosto cónico, tapón esmerilado de vidrio de 300 ml Cubeta y embudo Etiquetas Frascos de polietileno de 1000 ml Frascos de vidrio de I O00 ml Implemento para medir profundidad Libro de registro Muestreadores kemmerer, Winkler, botellas (opcional) Termómetro Equipo Balanza analítica Potenciómetro Refrigerador Reactivos Ácido clorhídrico (HCI) Ácido fosfórico (H3P04) Ácido sulfúrico (H2SO4) Floruro de potasio (KF) Hidróxido de sodio (NaOH) loduro de potasio (KI) loduro de sodio (Nal) o hidróxido de potasio
Nitruro de sodio (NaN3) Sulfato de cobre pentahidratado (CuSO4-
Sulfato manganoso (MnSO4) Tolueno (CgHgCH3)
( K W
5 ~ 2 0 )
Material Agitador de vidrio Espátula Matraz volumétrico de 1000 ml l Pizeta

Termómetro Vaso de precipitados (v)de I000 ml
Balanza analítica Parrilla eléctrica Estufa Potenciómetro Reactivos Bicarbonato de sodio (NaHC03) Biftalato de potasio (KHC8H404) Buffer (pH: 4.0,6.0, 7.0,8.0,10.0. Opcional) Carbonato de sodio (NaC03) Citrato monopotásico (KHzCgH507) Fosfato de potasio monobásico (KH2P04) Fosfato de potasio monobásico (KH2P04) Fosfato de sodio dibásico (Na2HP04) Fosfato de sodio dibásico (Na2HP04) Hidróxido de calcio (Ca(OH)2) Tartrato ácido de potasio (KHCqS406) Tetraborato de sodio decahidratado (bórax) (Na2B407 lOH20) Tetraoxalato dihidratado
Equipo
Sólidos sedimentabl
es Material Agitador de vidrio Cápsula de porcelana Cono de sedimentación tipo lmhoff Desecador Embudo Buchner Matraz Kitazato Papel filtro (Whatman núm 40 de I1 cm de diámetro) Vaso de precipitados (v) de 1 O0 y I O00 ml Equipo Balanza Analítica Equipo de vacío Estufa

Sólidos suspendidos
totales Material Crisol Gooch Desecador Discos de fibra de vidrio Embudo Buchner Matraz Kitazato Pipeta volumétrica de I O y 20 ml Pizeta Probeta de 100 ml Termómetro Equipo Balanza analítica Equipo de vacío Estufa Mufla Reactivos Agua destilada (H20)
Oxígeno disuelto y
DBO Material Agitador de vidrio Botellas DBO de 300 ml Bureta de 50 ml Embudo de tallo largo Espátula Matraz Erlenmeyer de 500 y 100 ml Matraz volumétrico de 1 O00 ml Mosca Papel filtro (Whatman núm 40 de 11 cm de diámetro) Pinzas para bureta Pipeta graduada de 10 ml Pizeta Propipeta Sopote universal Tripie
166

Vaso de precipitados (v) de 1000 y 100 ml Equipo Balanza analítica Estufa Parrilla eléctrica con agitador magnético Potenciómetro Reactivos Ácido sulfúrico (H2SO4 (d=l .U1 g/ml)) Agua destilada (H20) Almidón ((C606ti12)n) Cloruro de amonio (NH4CI) Cloruro de calcio (CaC12) (anhidrido) Cloruro ferroso hexahidratado(FeC13.6H20) Floruro de potasio dihidratado (KFeZHzO) Fosfato dibásico de potasio (K2HP04) Fosfato dibásico de sodio heptahidratado (Na2HP04.7H20) Fosfato monobásico de potasio (KHzP04) Hidróxido de potasio (KOH) Hidróxido de sodio (NaOH) loduro de potasio (KI) loduro de sodio (Nal) Nitruro de sodio (NaN3) Sulfato de magnesio heptahidratado (MgS04.7H20) Sulfato manganoso (MnS04.4H20) Tiosulfato de sodio (Na2S203)
Equipo Autoclave Balanza analítica Equipo de vacío Equipo para filtración con membrana Horno de aire caliente para esterilización con calor seco lncubador o baíío de agua, controlado termostáticamente en un intervalos 35 f 1°C a 44 f 1°C Parrilla eléctrica con agitador magnético Potenciómetro
Coliformes
167

Material Cajas de petri Embudo buchner Embudo de filtración de aproximadamente 50 mm de diámetro Espátula Frascos para toma de muestras, de vidrio o cristal refractario de 250 ml de boca ancha, con tapón de cristal esmerilado, o bolsas de plástico estériles de volumen equivalente Matraz kitazato de 500 ml Matraz volumétrico de 500 y 1 O00 ml Mecheros Membranas filtrantes estériles de aproximadamente 47 mm de diámetro, con características de filtración equivalentes a un tamaño de diámetro nominal de poro de 0.45 tJm* Pinzas de punta redondeada, para manejar membranas Pizeta Tripie Vasos de precipitados de 500 y 3000 ml Reactivos Ácido clorhídrico (HCI) (d=l .I8 g/ml) Agar en forma de polvo o escamas Agua destilada Alcohol amílico ((CH3(CH2)40H (libre de bases orgánicas) azul anilina Azul de bromotimol Azul de bromotimol Bicarbonato de sodio (NaHC03) Cloridrato de tetrametil-p-fenilendiamina Cloruro de 2,3,5-trifenil-tetrazolio (TTC) Cloruro de calcio anhidro (CaC12) Cloruro de magnesio (MgC12) Cloruro de potasio (KCI) Cloruro de sodio (NaCI) Desoxicolato de sodio

Extracto de carne Extracto de levadura Fosfato dibásico de potasio (KzHPO4) Fosfato monobásico de potasio (KH2P03) Fucsina ácida Fucsina básica Hidróxido de sodio L(-)triptofano Lactosa Lauril sulfato de sodio (NaCq 2H25S04) Manitol Paradimetilaminobenzaldehido ((CH312NC6H4CHO) Peptona Peptona proteosa núm. 3 o polipeptona Rojo de fenol Sales biliares núm. 3 o mezcla de sales biliares Sulfito de sodio (NaS03) Tergitol 7 Tiopeptona Tripticasa Triptona Triptosa
Material Frascos de color ambar de 200,500 y 1 O00 ml. Matraz volumétrico de 100,l O00 ml. Espátula Vidrio de reloj Probeta de 100,250 ml. Matraz Erlenmeyer de 500,l O00 ml. Pipetas de 1,5,1 O ml Termómetro Propipeta Fibra de vidrio Frascos de polietileno de 100,200,500 y I000 ml Pizeta Equipo
Arsénico

Espectrofotómetro (535nm) Parrilla eléctrica Refrigerador Generador Gutzeit de arsina y tubo de absorción (ver fig. ) Reactivos Agua destilada Acetato de plomo dihidratado Pb(C2H302)2*3H20 Ácido clorhídrico (HCI) concentrado Yoduro de Potasio (KI) Cloruro estañoso dihidratado(SnC12.2H20) Solución de dietil ditio carbomato de plata AgSCSN(C2&)2 Piridina (CgHgN) Hidróxido de sodio (NaOH) I N Granalla de Zinc de 20 a 30 mallas libre de arsénico Trióxido de arsénico (AszO3)
Material Matraz Kjeldahl de 1000,800 ml. Matraz Erlemmeyer de 100,500 ml. Matraz volumétrico de I O00 ml. Espátula Vidrio de reloj Probeta Pipeta de 1,5 y I O ml Propipeta Agitador de vidrio Termómetro Pizeta Equipo Balnza analítica (0.0001 g) Aparato para la derterminación de NzIKjeldahl Digestor con sistema de extracción de humos Aparato de destilación (ver fig. ) Destilador con sistema de condensación para mantener la temperatura por debajo de 29OC.
Arsénico

Parrilla eléctrica Potenciómetro Refrigerante pinzas para matraz Reactivos Oxido mercúrico rojo (HgO). Ácido sulfúrico concentrado (H2SO4) Sulfato de potasio (K2SO4) Hidróxido de sodio (NaOH) a 6N,0.1 N, 0.02N. Tiosulfato de sodio (Na2S203-5H20) Fenolftaleína disódica. Alcohol etílico ó isopropanol. Ácido bórico (HgBO3). Rojo de metilo Azul de metileno. Tetraborato de sodio (Na26407). Agua destilada Oxido mercúrico rojo (HgO).
Material Termómetro Matraz Aforado de 100ml.y 1000ml. Espátula Pipeta Propipeta Vidrio de reloj Gotero Matraz Elemmeyer 1 O0 y 1 O00 ml. Agitador Probeta Tubos de ensayo Frascos color ambar de 1 O0 y I O00 ml. Equipo Espectofómetro (540nm.) clpaso de luz de 1 cm. Balanza Analítica Refrigerador Celda de 1 cm. Reactivos Agua destilada
Cromo

Azul de Metileno
Dicromato de potasio anhidro (KzCrz07) Ácido Sulfúrico l+l (HzSO4) Ácido Fosfórico al 35% (H3P04) Acetona Solución de difenilcarbazid Ácido Nítrico (H NO3) Menbrana de 0.45 micras (Tm)
Material Matraz Volumétrico 1 O00 ml. Termometro Espatula Embudo de separación (500ml) clllave de teflón Agitador Proveta Gotero Mtraz Elemmeyer 21. Fibra de vidrio Equipo Espectofómetro (640 a 700nm.) Balanza analítica (0.0001g.) Cronómetro Parrilla Eléctrica Reactivos Sulfanato de alkil benceno ( CeH403SNaR) Fenolftaleína (CzoH1404) Hidróxido de sodio (NaOH) Alcohol etilico (CH3-CHz-OH) ó isopropílico
LCH Ácido Sulfúrico (H2SO4) concentrado (d=l.84) Cloroformo (CHC13) grado de espectofotométrico Azul de metileno Fosfato monosódico monohidratado (NaH2P04HzO) Ácido clorhídrico HCI (1:l)
(CHs-CH-CHs)

Mezcla crómica
Material Termómetro Espátula Vidrio de reloj Matraz Erlemmyer 1 O0 1 O00 500ml- Probeta Gotero Plato de muestre0 Matraz volumétrico 250 50 ml. Microbureta Kock, 5 ml. Pipeta Agitador Equipo Colorimétrico Fotómetro de filtro Filtro rojo (570-580nm.) Frasco de vidrio c/tapa
Cianuros
Aparato para destilación de cianuros (ver fig.) Juntas de vidrio ST Matraz de destilación "Claissen" (1 I) Absorbedor de gas Tubo dispersor de gas Filtro de porocidad medía Matraz Kifosato Condesador Válvula de aguja Tira de papel de (KI) Papel de acetato de plomo Filtro Equipo Balanza analítica Parrilla eléctrica Espectofotómetro (575 nm.) Campana de ventilación Reactivos Agua Cristales de tiosulfato de sodio (Na2S203) Solución buffer de ácido acético, pH 4 Nitrato de cadmio, Cd( N03)2=4H20

Carbonato de Plomo (PbC03) Hidróxido de sodio (NaOH) Ácido acético Hidrocloruro de 3metiI 2-benzotiazolonahidrazona Ácido sulfámico Cloruri Férrico exahidratado (FeC13-6H20) Nitrato de Plata (AgN03) Etilendíamintetraacetato disódico (EDTA) Cloruro de magnesio exahidratado (MgC12.6H20) Ácido sulfúrico (H2SO4) ÁIcaIi P-dimetilaminobenzalvodanina Acetina Cloruro de sidio (NaCI) Crimato de potasio (K2CrO4) Solución de cloramina -T Hidróxido de potasio (KOH) Cianuro de potasio (KCN) Piridina Ácido barbitúrico Bifosfato de sodio hidratado (NaH2P04=H20) Buffer de fosfato
Cadmio Material Termómetro Matraz Erlemmeyer de 125,250 ml Espátula Vidrio de reloj Pipeta Propipeta Matraz volumétrico de 1 0 0 , l O00 ml. Tubodeensaye Botella color ámbar de 1000,250 ml. Bureta Embudo de separación de 125,250,500 ml. Agitador de vidrio Frascos de polietileno
174

Embudo de separación color ámbar de 125 ml. Vaso de precipitado de 150,250 ml. Crisol Gooch Disco de fibra de vidrio Equipo Bhscula analítica Refrigerador Espectofómetro (515nm) con celdas de 1 cm. Fotoclorímetro de filtro verde (51 5nm) Celda de absorción de Icm. Placa de calentamiento ó baño maría Campana de extracción Reactivos
Acido clorhídrico (HCI) concentrado Ácido nítrico (H NO3) Ácido perclórico (HC104) Ácido Sulfúrico (H2SO4) concentrado Alcohol etílico (C2H50H) Hidróxido de amonio (NH40H) concentrado Peróxido de hidrógeno (H202) Tetracloruro de carbono (CC14) Solución de hidróxido de amonio (NH40H) Solución de hidróxido de sodio (NaOH) Solución de hidróxido de sodio (NaOH) Solución de dimetil-glioxima (C4H~N202) Difenil ditio carbozona Cloroformo Cristales de ditizona Tartrato de sodio y potasio (KNaCqH406) Cadmio metálico puro Anaranjado de metilo Perlas de vidrio Anidrido sulfúrico
Agua (H20)
Grasas y Aceites
Material

Anillo de hierro, pinzas para refrigerante, pinzas para matraz, espátula, agitador de vidrio Baño María Cartuchos de extracción, algodón Desecador Discos de tela de meselina de 11 cm de diámetro Embudo de Buchner Matraz balón (del equipo Soxhlet) Matraz kitazato de 2000 ml Papel filtro de poro mediano de 11 cm de diámetro Pinzas Pizeta Probeta de 100 ml y 500 ml Refrigerante para tubo de exracción Soxhlet Soporte universal Termómetro Tubo de extracción Soxhlet Vasos de precipitados de 500 ml y 100 ml Equipo Balanza analítica Equipo de vacío Estufa de vacío Estufa Parrilla eléctrica Reactivos Ácido clorhídrico (HCI) Agua destilada Éter de petróleo Hexano (CgH14) o 1 ,I ,2 tricloro-l,2,3 trifluoretano (C2C13F3) Tierra de diatomaceas
Demanda Química de
oxígeno Material Bureta de 50 ml Condensador tipo Friedrichs entranda 24/40
176

Desecador Espátula Manguera latex Matraz Erlenmeyer de 500 ml con boca esmerilada 24/40 Matraz volumétrico de 1 O0 y 1 O00 m1 Perlas de ebullición Pinzas de tres dedos Pinzas para bureta Probeta de 100 y 500 ml Soporte universal Termómetro Vaso de precipitados de 500 ml Equipo Balanza analítica sensibilidad de 0.0001 g Estufa Parrilla eléctrica Reactivos 1 ,I O Fenantrolina (ortofenantrolina) ácido sulfúrico (H2SO4) Agua destilada (H20) Dicromato de potasio (K2Cr207) Sulfato de amonio y fierro hexahidratado (Fe(NH4)2(so4)2'6H20) Sulfato de fierro heptahidratado (FeS04.7H20) Sulfato de plata (Ag2S04)
Cloruros Material Bureta de 50 ml Embudo de tallo largo Espátula Matraz volumétrico de 1 O0 y 1 O00 ml Papel filtro Whatman núm. 40 Pinzas para bureta Pipeta de 5 y I O ml Pizeta Probeta de 50 y 100 ml Soporte universal

Termómetro tripié Vaso de precipitado de 100 y 500 ml Equipo Estufa Parrilla eléctrica Potenciómetro Reactivos Ácido Sulfúrico (H2SO4) Agua destilada (H20) Alcohol etílico (C2H5OH) Cloruro de sodio (NaCI) Cromato de potasio (K2Ct-04) Fenoftaleína Hidróxido de amonio (NH40H ) Hidróxido de sodio (NaOH) Nitrato de plata (AgNO3) Peróxido de hidrógeno (H202) Sulfato de aluminio y potasio dodecahidratado o Sulfato de aluminio y amonio dodecahidratado
Material Adaptador para el matraz de destilación y tubo de conexión 24140 Condensador Espátula Manguera latex Matraz de destilación de 1 O00 ml de fondo redondo y cuello largo hecho de vidrio de borosilicato con entrada 20/40 Matraz volumétrico de 500 y 1 O00 ml pinzas de tres debos Pinzas para matraz Pipeta de I ,5 y1 O ml Probeta de 100 ml Termómetro con lecturas hasta 200°C Tubo de conexión de 12 mm de diámetro interior Tubos de ensaye de 10 mm X 75 mm
Floruros

Vaso de precipitados de 1 O0 y 500 ml Equipo Espectrofotómetro para usarse a 570 nm provisto de un paso de luz de 1 cm como mínimo Fotómetro de filtro provisto de un paso de luz de 1 cm como mínimo y equipado con un filtro amarillo verdoso teniendo una transmitancia máxima entre 550 y 580 nm (opcional) Parrilla eléctrica Reactivos Ácido clorhídrico Ácido sulfúrico (H2SO4) Agua destilada Arsenito de sodio (NaAs02) Arsenito de sodio (NaAs02) Cloruro de circonilo octahidratado (ZrOC12.8H20) Floruro de sodio anhidro (NaF) Sal de sodio 2-para-sulfofenilazo 1,8-dihidroxi 3,6-naftalen-disulfonato (SPADNS) Sulfato de plata (Ag2S04)
Material Baño María Bureta de 50 ml Embudo Buchner Embudo de separación de 1 O00 ml Espátula Matraz de destilación de 500 ml Matraz Erlenmeyer con tapón de vidrio de 250 ml Matraz kitazato Matraz volumétrico de 1 O00 ml Papel filtro Pinzas de tres dedos Pinzas para bureta Pinzas para matraz Pipeta de I O ml Pizeta
Fenoles

Probeta de 50 y 3000 ml Refrigerante Soporte universal Soporte universal Tripié Vaso de presipitados de 100,500 y 1000 ml Equipo Balanza analítica con una presición de
Equipo de vacío Espectrofotómetro para usarse a 460 nm Parrilla eléctrica Potenciómetro Refrigerador Reactivos 4 aminoantipirina Ácido clorhídrico (HCI) Ácido fosfórico (H3P04) Ácido salicilico Ácido sulfúrico (H2SO4) Agua destilada desionizada, exenta de cloruros y fenoles (H20) Almidón ((C606H12)n) Anaranjado de metilo Bromato de potasio (KBrO3) Bromuro de potasio (KBr) Cloroformo (CH3C13) Cloruro de amonio (NH4CI) Cloruro de sodio (NaCI) Fenol (c6H50H) Ferricianuro de potasio (K3Fe(CN)6) Hidróxido de amonio (NH40H) Hidróxido de sodio (NaOH) loduro de potasio (KI) Solución Buffer pH 4 Sulfato de cobre pentahidratado (CuS04.5H20) Sulfato de sodio anhidro granular (Na2S04) Tiosulfato de sodio (Na2S203) Tolueno (CgHgCH3)
0.0001 g

Materia flotante
Material Malla químicamente inerte con abertura de 3 mm Recipiente con boca de 7 cm de ancho aforado entre 2 y 5 litros Embudo de filtración que se adapte al tamaño de la malla Cucharilla de arrastre
Nitrógeno total
Equipo Potenciómetro Digestor con sistema de extracción de humos Parrilla eléctrica Balanza analítica de sensibilidad 0.0001 g Material Frasco de 100,500 y 1 O00 ml Probeta graduada de 50,100 y 500 ml Pipetas de 1 , 5 y 10 ml Matraz Kjeldahl de 800 ml Termómetro Destilador con sistema de condensación para mantener la temperatura abajo de los 29°C Matraz Erlenmeyer de 500 ml Tapón de hule Propipeta Bureta de 50 ml Frascos goteros Soporte universal Pinzas para bureta Agitador magnético (mosca) Reactivos Agua (H20) destilada Oxido mercúrico (HgO) rojo Ácido sulfúrico concentrado (H2SO4) Sulfato de potasio (K2SO4) Hidróxido de sodio (NaOH)

Tiosulfato de sodio pentahidratado (Na2S20395H20) Fenolftaleina disódica Alcohol etílico o isopropanol Ácido bórico (H3B03) Rojo de metilo Azul de metileno (Na2B407)
Equipo Espectrofotómetro con longitud de onda 457 nm, provisto de un paso de luz de 1 cm Potenciómetro Balanza analítica Parrilla eléctrica con agitador magnético Campana de extracción Material Todo el material de vidrio o polietilenoempleado en este método debe enjuagarse con ácido nítrico 1:l y enseguida con agua. Frascos de polietileno de 100,250,500 y I O00 ml Probeta de 50,100,500 y I O00 ml Matraz aforado de 100,500 y 1000 ml Espátula Vidrio de reloj Matraz Erlenmeyer de 250,500 y I000 ml Pipetas de 1,5 y 1 O ml Propipeta Termómetro Agitador magnetic0 (mosca) Embudo de separación de 125 ml Vasos de precipitados de 50,100,250,500 y I000 ml Celdas para espectrofotómetro Reactivos Cloroformo (CHC13) Alcohol metílico (CHsOH) Ácido nítrico (HNO3) Ácido sulfúrico (H2SO4)
Cobre

Hidróxido de amonio (NH40H) 2,9-dimetil-l ,I O-fenantrolina semihidratada Clorhidrato de hidroxilamina (NH20HeHCI) Citrato de sodio (Na3CgH507*2H20) Cobre electrolítico pulido Agua (H20) destilada
Fósforo total
Equipo Espectrofotómetro Balanza analítica Parrilla eléctrica con agitación magnética Estufa Campana de extracción Potenciómetro Material Todo el material de vidrio empleado debe ser lavado con mezcla crómica, enjuagando dos veces con HCI 1:1 caliente y enjuagando dos o tres veces más con agua desionizada. NUNCA USAR DETERGENTE. Matraz volumétrico de 100,500 y 1 O00 ml Espátula Vidrio de reloj Pipetas de 1 , 2 , 5 y 10 ml Probeta de 50,100,250,500 y 1 O00 ml Propipetas Matraz Erlenmeyer de 250,500 y 1000 ml Vasos de precipitados de 100,250,500 y 1 O00 ml Termómetro Gotero Reactivos Agua destilada (H20) Agua desionizada (H20) Ácido silfúrico (H2SO4) Ácido nítrico (HNOS) Molibdato de amonio tetrahidrataso ((NH4)6Mo7024'4H20) Glicerol (C3H803)

Cloruro estañoso dihidratado (SnC12.2H20) Hidróxido de sodio (NaOH) Cloruro mercúrico (HgC12) Fosfato de potasio monobásico (KH2P04) Sal disódica de Fenolftaleína alcohol etílico (C2H60) Metano1 (CH3OH) Benceno (CgH6) Alcohol isopropílico (C3H8O) Cloroformo (CHC13) Metavanadato de sodio
Organo- clorados
Equipo Microbalanza analítica con una precisión de 0.01 mg Parrilla eléctrica con agitación magnética Congelador con temperatura mínima al menos de -1 5°C Mufla que pueda proporciónar un temperatura de al menos 660°C Centrifuga Cromatografo de gases con detector de captura de electrones, columna gas-líquido de vidrio de 180 cm de longitud por 4 mm de diámetro interior o Línea de fase móvil con cartucho de secado con tamiz molecular Refrigerador Bomba de vacío Estufa una temperatura de 130°C Evaporador rotatorio Balanza analítica Material
184

Lavar el material de vidrio con agua y detergente neutro, enjuagar con abundante agua de la llave y sumergirlo en mezcla crómica para eliminar las impurezas orgánicas. Enjuagar sucesivamente con agua de la llave, con agua destilada y acetona grado analítico secar en el horno a 120°C, este material se tapa con papel aluminio y se guarda hasta el momento de su uso. Guantes desechables de hule Mascarilla Pipeta volumétrica de 25 ml Microespátula de acero inoxidable Matraz volumétrico de 1 O, 25,50,100,250,500 y 1000 ml Pipetas de 1 , 2 , 5 y 10 ml Botellas de vidrio de 1000 ml con tapón y contratapón de teflón Embudos de separación de 2000 ml con llave de teflón Columna cromatográfica de 19 mm de diámetro interior, 40 cm de longitud y con llave de teflón Microjeringas de lop1 para cromatografo de gases Parafilm Espátula Vidrio de Reloj Propipeta Pipetas Pasteur Tubos para centrífuga graduadis de 15 ml con tapón esmerilado Frascos Winkler Termómetro Hieleras portatiles Embudo Bückner Matraz kitazato de 250 ml Cápsulas de porcelana Charola Pyrex Frascos de vidrio ambar

Embudo de separación de 2000 ml Probetas de 50,100,250,500 y 1 O00 ml Agitador magnético (mosca) Soporte universal Anillo de hierro Matraz de fondo redondo de 500 ml Tapón de lana de vidrio Matraz balón de 500 ml con boca esmerilada 24/40 Reactivos Hexano (c6H14) Eter de petróleo punto de ebullición 30-6OoC benceno (CgH6) Cloroformo (CH3CI) Acetona (CH3-CO-CH3) Cloruro de metileno (CH2C12) Iso-octano (C8Hq 8) HePbno (C7H.I 6) Tolueno (CgHgCH3 Florisil para croma ! ografía en columna malla 60/1 O0 Sulfato de sodio granular y anhídro (Na2S04) Agua grado plaguicida (ver definiciones para esta técnica) Plaguicidas organoclorados (98% al 100% de pureza) Lindano (O-HCH) Lindano (B-HCH) Heptacloro Dieldrin Endrin Dicloro difenil etano (DDE) DDD o TDE Keltano o dicofol Metoxicloro Toxafeno Agua destilada (H20) Agua desionizada (H20)

APENDICE "6" TITULACIONES

Preparación y valoración del ácido clorhidrico y el hidróxido de sodio
Introducción.
Una solución 1 N de HCI contienen 36.5 g de éste en I000 ml, por lo tanto, una solución 0.1 N contendrb 3.65 g/l. El HCI comercial tiene una pureza aproximada de 37% y una densidad de 1 .I850 glml. Si multiplicamos:
37% X 1 .I850 g/l = 43.847 g/ml
Significa que hay 43.847 g de HCI en 100 ml de solución. Para calcular el volumen de HCI concentrado que equivale a 3.65 g. Tenemos que:
- = mlV.
Por lo tanto:
3.65 g X 100
43.847 glml V=-- = 8.3244ml
Conviene preparar la solución de una concentración ligeramente superior a la deseada, porque se ha evaporado parte del ácido al abrir el recipiente que lo contiene.
La preparación y valoración de la solución de hidróxido de sodio (NaOH) requiere cuidados especiales. La solución debe estar libre de carbonatos pues se trata de una base fuerte y el carbonato presente en el aire como en el agua es absorbido ávidamente por la solución alcalina, por lo que el agua destilada utilizada en su preparación debe haberse hervido recientemente para eliminar este contaminante.
Para preparar un solución aproximadamente 1 N se requiere de 40 g de NaOH.

Preparación de HCI aproximadamente 0.1 N.
Medir de 9 a 10 ml de HCI concentrado con una pipeta y colocarlos en un matraz aforado de 1000 ml que contiene un poco de agua destilada y aforar hasta la marca, agitar y vaciar al recipiente de plástico limpio.
Preparación del NaOH aproximadamente 0.1 N.
Pesar 4 g de NaOH, en un vaso de precipitados (no tocarlo con lasmanos, ni pesarlo directamente en el plato de la balanza) y disolver con un poco de agua destilada previamente hervida, transferirlo a un matraz volumétrico de I000 ml y aforar con el agua destilada y hervida aprovechando para lavar el vaso de precipitados, agitar y vaciar a un recipiente de plástico limpio.
Titulación de la solución preparada de HCI aproximadamente 1 N.
Introducir aproximadamente 2 g de carbonato de sodio anhidro (Na2C03) en un pesa filtro. Secar a 110°C en la estufa durante media hora y enfriar en el desecador.
Pesar 3 porciónes de 0.1 a 0.15 g respectivamente en 3 matraces Erlenmeyer y etiquetar cada uno para su identificación.
Agregar a cada matraz 50 ml de agua destilada previamente hervida y agitar hasta disolver completamente la sal.
Agregar 2 gotas de indicador anaranjado de metilo a cada matraz.
Titular cada uno, añadiendo la solución de HCI, hasta cambio del color o vire del indicador de amarillo a color canela.
Hacer la lectura correspondiente en la bureta anotando el volumen empleado junto con el peso del carbonato de sodio correspondiente.

Realizar la misma operación con los tres matraces. Calcular la normalidad tomando como base el siguiente
ejemplo:
Titulación ml de HCI gastados g de NapC03 1"
31.28 O. 1 725 3' 20.30 0.1 122 2" 23.85 0.1 31 6
Peso equivalente del Nazco3 = 53 Peso meq. del Nazco3 = 0.053
g de Na2C03 NHC~ = --"------
ml de HCI empleados X meq. del Na2C03
1" Titulación :
0.1 31 6 g NHC~ = -- = 0.1079 N
23.85 ml X 0.053
AI realizar el cálculo para las otras dos obtenemos 0.1042 y 0.1040 respectivamente. Obteniendo el promedio de las tres se tiene que la normalidad es de 0.1 053.
Etiquetar el frasco con el nombre de la solución la normalidad y la fecha.
Titulación de la solución preparada de hidróxido de sodio aproximadamente 1 N.
Medir con una pipeta 3 volúmenes de 25 ml de la solución de NaOh que se requiere titular y transferir a 3 matraces Erlenmeyer respectivamente. Etiqueta cada matras para su identificación.
Diluir con agua destilada hasta 50 ml. Agregar dos gotas de indicador fenoftaleína.

Realizar la titulación dejando caer de la bureta la solución de HCI previamente titulada hasta hasta cambio de color o vire del indicador de incoloro a rosa.
Anotar el volumen de HCI gastado junto con el volumen de
Calcular la normalidad mediante la fórmula: NaOH correspondientemente para cada titulación.
v2 Donde:
N1 = Normalidad del HCI. V l = Volumen de HCI. N2 = Normalidad de NaOH. Vp = Volumen de NaOH.

Preparación y valoración de tiosulfato de sodio.
Introducción.
El yodo puede reducirse a ión ioduro en presencia de agentes reductores tales como arsénico (Ill), estaño (11) y tiosulfato de sodio y por consiguiente, pueden ser titulados con una solución de iodo, sin embargo, la solución de iodo presenta dificultades en la detección del punto final, por lo que es preferible agregar un exceso de ioduro de potasio a la solución problema y titular el iodo liberado con un agente reductor, generalmente con un solución valorada de tiosulfato de sodio.
El indicador usado en esta valoración es una solución de almidón soluble que forma un complejo de intenso color azul con el iodo, el cual desaparece al calentarse, pero reaparece al infriarse. Cuando el iodo se titula con tiosulfato el almidón debe agregarse sólamente cuando la mayor parte del iodo ha reaccionado; de otra forma, la coloración en el punto final resulta muy poco definida. La solución de almidón que se usa en esta determinación debe ser preparada en el momento de su empleo porque permite el desarrollo de microorganismos, disminuyendo su poder reductor.
El tiosulfato de sodio es una sal que se puede obtener con un elevado grado de pureza, pero que debido a su tendencia a cristalizarse, el contenido de agua de cristalización es incierto, por cuya caisa n ose puede pesar la cantidad exacta; para hacer una solución de normalidad deseada se pesa la cantidad aproximada que se requiere para obtenerla, valorándose después. El tiosulfato de sodio, como reductor que es, se descompone frente a un oxidante. AI ser oxidado el tiosulfato a tetrationato, la Valencia media del azufre pasa de (-2) a (-2%) y como en la molécula de tiosulfato hay dos átomos de azufre, el cambio total de Valencia es de uno; entonces el peso equivalente de esta sal es igual a su peso molecular, es decir 248.9.
Las soluciónes de tiosulfato de sodio pueden permanecer durante varias semanas sin variar sensiblemente su normalidad, simpre y cuando el agua que se emple para su elaboración este libre de ácido. Es recomendable que el agua que se usa sea hervida previamente para eliminar el gas

carbónico y agregar una pequeña cantidad de carbonatode sodio anhidro (0.lgll) con el fin demantener la solución libre de iones hidrógeno y evitar el desarrollo de de ciertosa microorganismos. Las soluciónes de tiosulfato sufre cierta descomposición cuando son expuestas a la luz, por lo que se recomienda conservarlas en frascos oscuros o cuando no se dispone de ellos, evitar que las soluciónes en frascos claros sean expuestas a la luz.
Valoración de solución de tiosulfato de sodio 0.1 N.
Preparación de la solución de tiosulfato de sodio.
Pesar 25 g de tiosulfato de sodio pentahidratado cristalizado y puro y 0.2 g de carbonato de sodio y disuelvalos en un vaso de precipitado con agua previamente hervida, transfiéralos al matraz de I O00 ml y afore con agua previamente hervida.
Guarde la solución en frasco oscuro tapado perfectamente y etiquetelo.
Preparación de la solución de almidón.
Pese 0.2 g de almidón y agregue unos cuantos ml de agua destilada hasta obtener una pasta no muy viscosa.
Hierva 10 ml de agua destilada y agregue la pasta de almidón con una agitación vigorosa y continúe la ebullición hasta obtener una solución transparente.
Nota: Esta solución debe ser preparada en el momento de su uso.
Valoración de la solución de tiosulfato de sodio 0.1 N.
Mida 20 ml de solución del permanganato de potasio anteriormente valorado y transfiéralos a un matraz erlenmeyer de 300 ml.
Agregue I O ml de ácido clrhídrico I :6

Agregue 10 ml de solución de ioduro de potasio (3 g de ioduro de potasio en 10 ml de agua destilada), agitelo y mezcle perfectamente.
Valore con la solución de tiosulfato de sodio hasta la aparición de un ligero color amarillo.
Agregue 2 ml de la solución de almidón y continúe la titulación hasta la desaparición del color azul.
Repita la operación en 2 muestras adicionales por lo menos.
Calcule la concentración de la solución de tiosulfato de sodio empleando la siguiente fórmula.
PREPARACIóN Y VALORACIóN DE PERMANGANATO DE POTASIO.
Introducción.
Una oxidación se define como el proceso en el que se produce una pérdida de electrones dando como resultado un estado de oxidación mayor (más positivo), y una reducción se define como el proceso en el que se produce una ganancia de electrones dando como resultado un estado de oxidación menor (más negativo).
El permanganato de potasio es un agente oxidante que con frecuencia se emplea como titulante. Actúa como indicador para detectar el punto final y es un agente oxidante de gran fuerza. La solución de permanganato de potasio es estable cuando se toman las precauciones debidas en su preparación. AI preparar la solución las pequeñas cantidades de impurezas reductoras presentes reducen las pequeñas cantidades de Mn04-. En solución neutra el producto de la reducción del

permanganato es Mn02 en vez en vez de Mn+2 que se produce en medio hcido. El Mn02 actúa como catalizador produciendo mayor descomposición del permanganato, que a su vez produce más Mn02, y así sucesivamente. Esto se denomina descomposición catalítica. La solución puede estabilizarse eliminando el Mn02. Por esto antes de valorarla, la solución se hierve para hacer más rapida la oxidación de todas las impurezas, y se deja reposar toda la noche. A continuación se remueve el MnO2 filtrándolo a través de fibra de vidrio. El permanganato de potasio puede valorarse utilizando una sla primaria de oxalato de sodio Na2C204, el cual disuelto en ácido forma Bcido oxálico.
Preparación de permanganato de potasio de 0.02 M.
1. Pesar 2.3 g de permanganato de potasio con exactitud. 2. Disuélvase con agua destilada en un vaso de
3. Vierta la solución en un matraz volumétrico de 1 O00 ml y
4. Caliente la solución final hasta que hierva durante 1
5. Deje reposar 24 horas tapada con u vidrio de reloj. 6. Filtre en un embudo que contenga como medio filtrante
un trozo de lana de vidrio (no use papel filtro). 7. transfiera el filtrado a un frasco ambar y etiquételo.
Valoración de la solución de permanganato de potasio 0.02 M. Pese por triplicado muestras de 0.1 a 0.3 g de oxalato de
sodio. (previamente secado a la estufa a 11 0°C durante una hora).
transfiéralas a tres matraces Erlenmeyer, agregue a cada uno 50 ml de agua destilada.
Agregue 20 ml de ácido sulfúrico 1:4, caliente la muestra a 75°C , valore a esta temperatura dejando caer gota a gota la solución de permanganato de potasio hasta obtener un color rosa pálido que persista 20 segundos.
Obtenga el promedio de las 3 titulaciones y calcule la normalidad del permanganato de potasio usando la fórmula siguiente:
precipitados de 500 ml
afore con agua.
hora en un vaso de precipitados.

g de oxalato de sodio NKm04 =
meq del oxalato de sodio X ml de KMn04
Notas: La bureta debe estar llena y lista para la titulación cuando
la solución llegue a la temperatura deseada. La solución de permanganato de potasio no debe dejarse
en la bureta por períodos largos debido a que podría atacarse la grasa de la llave de paso.
En caso de que la solución adquiera una coloración café significa que falta ácido sulfúrico o que la temperatura no es la adecuada.

OBJETIVOS ALCANZADOS
- Revisión bibliográfica - Edición de manual
RESULTADOS Y CONCLUSIONES
El resultado de este trabajo ha sido la elaboración de un Manual de Análisis de Aguas Residuales en donde se incluye: la descripción de las técnicas de ensayo que recomienda la Secretaría de Desarrollo Social a través de la Dirección General de Normas, recomendaciones para algunas de las técnicas, una sección dedicada exclusivamente a la toma, conservación y tratamiento de muestras, dos apéndices, uno contiene listas detalladas de los requerimientos (material, equipo y reactivos); el otro, las técnicas para la valoración de las soluciones que lo requieren.
Por la forma en que fue estructurado este Manual puede representar una magnífica herramienta , tanto en los laboratorios industrales, como en los que se dedican a la certificación de análisis de aguas residuales.
La parte más valiosa de este trabajo lo es sin duda la experiencia adquirida para estructurar manuales técnicos, que son importantes para el desarrollo de la industria así como del propio profecionista, además en este manual se dan todos los fundamentos en los que están basadas cada una de las técnicas de análisis elemento importante ya que se dan las herramientas necesarias para poder sustituir reactivos o procedimientos en caso de que por alguna causa no se contara con ellos o no se tuviera lo necesario para implementar un procedimiento.
RECOMENDACIONES.
Debido a que la idea original de este Manual era poder anexar en cada técnica una lista de recomendaciones de operación de aquellos puntos que consideramos muestran cierta ambigüedad , es importante implementar éstas para que el objetivo inicial se cumpla.
197

Seria importante que dentro de las licenciaturas de Ingeniería se contara con una UUEEAA dedicada a estudiar los problemas de contaminación de aguas y de efluentes gaseosos vertidos a la atmósfera. De esta forma al llegar a la industria se tendría mhs conciencia de estos problemas y se sabrían las posibles formas de eliminarlo.

BIBLIOGRAFíA
NOM-AA-3-1 980 Aguas residuales muestreo
NOM-AA-4-I 977 Determinación de sólidos sedimentables en aguas residuales. Método del cono imhoff.
NOM-AA-5-1 980 Determinación de grasas y aceites. Método de extracción de soxhlet.
NOM-AA-6-1973 Determinación de materia flotante en aguas residuales. Método visual con malla específica
NOM-AA-7-1 980 Determinación de la la temperatura. Método visual con termómetro.
NOM-AA-8-1 980 Determinación del pH. Método potenciométrico.
NOM-AA-1 2-1 980 Determinación de oxígeno disuelto. Método de Winkler simple o modificado.
NOM-AA-14-1980 Cuerpos receptores muestreo.
NOM-AA-1 7-1 980 Determinación de color. Método espectrofotométrico.
NOM-AA-20-1 980 Determinación de sólidos disueltos totales. Método gravimétrico.
NOM-AA-26-1980 Determinación de nitrógeno total. Método Kieldahl.
NOM-AA-28-1 981 Determinación de demanda bioquímica de oxígeno. Método de incubación por diluciones.
NOM-AA-29-1981 Determinación de fósforo total. Método colorimétrico del azul de molibdeno o cloruro estañoso.

NOM-AA-30-1 981 Análisis de aguas, demanda química de oxígeno. Método de reflujo de dicromato.
NOM-AA-34-3981 Determinación de sólidos en agua. Método gravimétrico.
NOM-AA-36-1 980 Determinación de acidez total y alcalinidad total. Método potenciométrico y volumétrico.
NOM-AA-38-1 981 Determinación de turbiedad del agua. Método turbidimétrico de la bujía patrón.
NOM-AA-39-1 980 Determinación de sustancias activas al azul de metileno (detergentes). Método colorimétrico de azul de metileno.
NOM-AA-42-1981 Determinación del número más probable de coliformes totales y fecales. Método de tubos múltiples de fermentación.
NOM-AA-44-1981 Determinación de cromo hexavalente en agua. Método colorimétrico de la difenilcarbazida.
NOM-AA45-1977 Determinación de color de agua escala platino cobalto. Método de comparación visual.
NOM-AA-46-1971 Determinación del arsénico en aguas. Método espectrofotométrico del dietil ditio (carbamato de plata).
NOM-AA-50-1 978 Determinación de fenoles en agua. Método espectrofotométrico bipirina de la 4-aminoantipirina.
NOM-AA-51-1 981 Determinación de metales. Método espectrofotométrico de absorción atómica.
NOM-AA-53-1 981 Determinación de material extractable con cloroformo. Método gravimétrico.
200

NOM-AA-57-1 981 Determinación del plomo. Método colorimétrico de la ditizona.
NOM-AA-58-1 982 Determinación de cianuros. métodos colorimétrico y titulométrico.
NOM-AA-60-3 981 Determinación de cadmio. Método colorimétrico de la ditizona.
NOM-AA-63-1 981 Determinación del boro. Método potenciométrico con manitol
NOM-AA-64-1 981 Determinación del mercurio. Método colorimétrico de la ditizona.
NOM-AA-65-1 981 Determinación del selenio. Método colorimétrico de la 3,3'-diaminobencidina.
NOM-AA-66-1 981 Determinación de cobre. Método colorimétrico de la neocuproina.
NOM-AA-71-1 981 Determinación de plaguicidas organoclorados. Método cromatográfico de gases.
NOM-AA-72-1983 Determinación de dureza. Método volumétrico con EDTA.
NOM-AA-73-1 981 Determinación de cloruros. Método argentométrico.
NOM-AA-74-1 981 Determinación del ion sulfato. Métodos gravimétrico y turbidimétrico.
NOM-AA-75-1 982 Determinación de sílice. Método colorimétrico y gravimétrico de deshidratación.
NOM-AA-76-1982 Determinación de niquel. Método colorimétrico de dimetilglioxina.

NOM-AA-77-1 982 Determinación de floruros. Método colorimétrico del S.P.A.D.N.S.
NOM-AA-78-1 982 Determinación de zinc. Métodos colorimétricos de ditizana I, la ditizona II y espectrofotometría de absorción atómica.
NOM-AA-79-1 986 Determinación de nitrógeno de nitrato. Método de sulfato de brucina.
NOM-AA-82-1 986 Determinación de nitrógeno. Método espectrofotométrico ultravioleta.
NOM-AA-83-1 982 Determinación de olor. Método empírico de comparación.
NOM-AA-84-1 982 Determinación de sulfuros. Método colorimétrico de azul de metileno e iodométrico.
NOM-AA-8911-3 986 Calidad del agua vocabulario-parte 1- protección al ambiente.
NOM-AA-93-1 984 Determinación de la conductividad eléctrica.
NOM-AA-99-1 987 Determinación de nitrógeno de nitritos en agua.
NOM-AA-1 00-1 987 Determinación de cloro total. Método iodométrico.
NOM-AA-I O1 -1 983 Determinación de estroncio radiactivo. Métodos absorción atómica, gravimétrico y flamometría con espectrofotómetro con aditamento de flama.
NOM-AA-I 02-1 987 Deteccción y enumeración de organismos coliformes termotolerantes y escherichia coli presuntiva. Método de filtración en membrana.
NOM-AA-1 04-1 988 Plaguicidas. Determinación de residuos en agua. Método de toma de muestras

Standar Methods for the Examination of water and wastewater, American Public Health Association. American Water Works, Association and Water Pollution Control Federation. 14Th edition.
Manual de técnicas de muestre0 para agua y aguas residuales; Sebsecretaría de Ecología Centro de Información Documental (SEDUE).
Curso: caracterización y tratamiento de efluentes acuosos industriales, Dr. Julio Landgrave y Dr. Alain Queré. Facultad de Química UNAM Programa universitario de medio ambiente
Reglamento para la prevención y control del medio ambiente
Diario Oficial 18 de octubre de 1993
Química analítica Cuantitativa, Holkova, De. Trillas, México
Manual de aguas para usos industriales, American Society for testing and Materials, Limusa, México 1991.