control de calidad en el laboratorio de...
TRANSCRIPT

Revista de Hematología Vol. 7, No. 1, 2006
1
Solicitud de reimpresos: Dra. María de los A. Ochoa-Rico, Banco Central de Sangre, Centro Médico Nacional Siglo XXI, Instituto Mexicanodel Seguro Social, Av. Cuauhtémoc No. 330, Col. Doctores, C.P. 06720, México, D.F., México. Tel. 55 19 20 63
Control de Calidad en el Laboratorio de Hemostasia.
María de los Angeles Ochoa-Rico
Banco Central de Sangre, Centro Médico Nacional Siglo XXI, Instituto Mexicano delSeguro Social, México, D.F., México.
a) Solicitud de laboratorio.b) Indicaciones al paciente.c) Identificación del paciente.d) Sitio de punción.e) Manejo de la muestra.f) Transporte y almacenamiento.
a) Solicitud de Laboratorio.Las muestras deben ir acompañadas de una
solicitud debidamente formulada con la siguien-te información: nombre completo, número de re-gistro, edad, sexo, origen étnico, diagnóstico,medicamentos que recibe, última dosis de me-dicamentos anticoagulantes, inhibidores de fibri-nolisis o fibrinolíticos (cuadro 1) (2).b) Indicaciones al paciente.
El paciente debe presentarse a la toma demuestra con un ayuno de cuando menos 4 horas.La última ingesta de alimentos debe de ser bajaen grasas, ya que la lipemia produce turbidezque interfiere con los métodos coagulómetricosy nefelométricos (3).c ) Identificación correcta del paciente.
Es de primordial importancia etiquetar cadamuest ra en presencia de l pac iente coninformación suficiente para evitar confusión conotras muestras.
Obtener una muestra de sangre de unpaciente es mucho más que insertar una agujaen la vena o extraer una gota de sangre de un
INTRODUCCIÓN.Los primeros avances sobre la explicación
de la coagulación sanguínea se iniciaron amediados de 1800, encontrando una relación dela coagulac ión sanguínea a problemashemorrágicos, de flebitis, trombosis arterial yembolismo. El sistema hemostático se conformapor el endotelio vascular, plaqueta, factores dela coagulación y el sistema fibrinoliticos (1).
Los procedimientos de control de calidaden el laboratorio son importantes en su organi-zación y funcionamiento. Su objetivo es propor-cionar exámenes confiables, reproducibles,exactos y ser por sí mismos relevantes para eldiagnóstico y vigilancia clínica de los pacientes.
Para lograr estos objetivos se requiere deuna administración experta que supervise eltrabajo del laboratorio, que garantice que selogre el nivel necesario de las buenas prácticasde labora tor io y que és te se mantengaconstantemente, para lo cual es necesario llevara cabo un programa de aseguramiento decalidad. Este programa contempla 3 aspectos(2):
1. Fase preanalítica.2. Fase analítica.3. Fase postanalítica.
FASE PREANALÍTICA.Es la etapa del estudio que incluye:

2
Revista de Hematología Vol. 7, No. 1, 2006
M de los A Ochoa-Rico.
dedo. Es el primer eslabón de una cadena deeventos que se completan cuando el medicorecibe los resultados de las pruebas de supaciente y es el principio del control de calidaden el laboratorio clínico (4).
La muestra debe tomarse correctamente ybajo condiciones favorables. El paciente debeestar tranquilo, relajado, ya que el estrés y elejercicio afecta los factores de coagulación, asícomo la liberación de plaquetas a la circulación
Cuadro 1Fármacos que afectan las pruebas de laboratorio.
Prueba Medicamento EfectoFactores II, V, Anticonceptivos orales Aumentan la actividadVII, VIII IX,X, XIIAdhesión y Anticonceptivos orales Aumentan la actividadagregaciónPlaquetariaTiempo de Anticoagulantes orales Interfieren con las enzimas queprotrombina Cumarinicos reducen la vitamina K y limitan el
proceso de caboxilación (TP alargado)TTPa o TT Heparina no fraccionada Inhibe al Factor X y II
(TTPa alargado)Plaquetas Aspirina y antiinflamatorios Antiagregantes.Inhiben a la
no esteroides ciclooxigenasa para la síntesis detromboxano A2.
Cuadro 2Alteraciones causadas por el estrés y
ejercicio.Prueba EfectoFactor VIII Aumenta su nivel en
plasmaFunción plaquetaria Afecta la 2a. fase de la
agregación por ADP y Epinefrina
Cuenta de plaquetas AumentaFibrinolisis Aumenta
(cuadro 2)d) Seleccionar el sitio de punción.
Elegir una vena de fácil acceso, como es lavena central, cefálica o radial del antebrazo,evitar áreas de hematomas o de cicatrizaciónextensa.
Se le pide al paciente que cierre el puño yse le frota el brazo de abajo hacia arriba, conobjeto de hacer más visibles las venas. Debeevitarse el ejercicio excesivo de la mano por-que aumentan los factores de coagulación.
La punción debe ser limpia y única y sinexceder del tiempo de ligadura de un minuto;quitar la ligadura tan pronto como la sangrecomience a fluir para evitar la estasis local(cuadro 3).e) Recolección de la muestra.
El anticoagulante de elección es el citratode sódio al 3.8% (0.129 M), en una proporción9 partes de sangre y 1 de anticoagulante,recomendada por el Comité de Trombosis yHemostasia (5).
En tubos de p lás t ico o de v idr iosiliconizado, una vez obtenida la muestra, debe

Revista de Hematología Vol. 7, No. 1, 2006
3Control de calidad en hemostasia.
ser mezclada con el anticoagulante mediantemovimientos de inversión. Se debe evitar lahemólisis de la muestra, debido a que loseritrocitos liberan factores tromboplásticos almedio, que afectan acortando los tiempos decoagulación.
Si el paciente tiene hematocrito menor de30%, no es necesario hacer la corrección de larelación sangre anticoagulante, puesto que elsistema tiene suficiente calcio para evitar lacoagulación. Pero cuando el hematocrito esmayor de 60%, los resultados son afectados porel exceso de anticoagulante, ya que puede quelarel calcio que se emplea durante el procedimientode las pruebas de coagulación, causandoprolongación de los tiempos de coagulación (6).
Por ello, el volumen de anticoagulante debeser ajustado para tener en cuenta la disminucióndel volumen plasmático siguiendo las pautas delcuadro 4, para obtener el volumen necesario deanticoagulante para una muestra de sangre de 5mL.
Otra forma de efectuar la corrección enpacientes que tengan valores de hematocritomayor de 60% es empleando la siguiente formula:
mL anticoagulantes(100 � Hto del paciente )Volumen plasmático normal= mL de anticoagulante para mL de sangre
Ejemplo:0.3 x (100 -67) =0.18 mL anticoagulante + 2.7mL sangre
e) Manejo de la muestra.La estabilidad de las pruebas de coagulación
es crí t ica para el diagnóstico y para elmantenimiento de la terapia anticoagulante.También lo es la temperatura de conservaciónmantenida durante el transporte y almacenamientode las muestras. Los intervalos de tiempo quese recomiendan entre la obtención de lasmuestras y la realización de las pruebas son: 2horas cuando la muestra es mantenida a 22ºC-24°C, 4 horas cuando es almacenada a 4°C, 2semanas a -20°C y 6 meses a - 70°C. Siemprese conserva el tubo tapado hasta su valoraciónanalítica (incluido el tiempo de la centrifugación),para evitar la pérdida de CO2 y la elevación delpH (4).
Para la obtención de plasma pobre enplaquetas (PPP), que se utiliza en la mayoría delas pruebas de coagulación, la muestra de sangrese debe centrifugar a 1500 g durante 15 minutosen centrifuga refrigerada 4°C y para obtener elplasma rico en plaquetas (PRP) la muestra secentrifuga a 150�200 g a temperatura ambiente,durante 10 minutos. Se extrae y realizan laspruebas antes de las 4 horas. Los tubosdestinados a la investigación del anticoagulantelúpico y la mezcla de plasmas normales, se
Cuadro 3Alteraciones causadas por estasis venosa
prolongada.Causa EfectoLiberación de Activación de proteínasFactor Tisular procoagulantes y
anticoagulantesHemólisis Liberación de fosfolipido
activación de factoresSalida de Plaquetas Activación
Cuadro 4Relación sangre anticoagulante con
respecto al hematocrito.
Hematocrito Volumen de Volumen de % anticoagulante sangre
mL mL
60 0.4 4.5 70 0.25 4.75 80 0.2 4.8

4
Revista de Hematología Vol. 7, No. 1, 2006
aconseja someterlos a doble centrifugación, a3 000 g, durante 30 minutos (7).
FASE ANALÍTICA.Es la etapa que considera a las variaciones
relacionadas con el procedimiento técnico en símismo, que afectan los resultados finales del es-tudio del paciente.
El control de calidad interno se utiliza paradeterminar si una serie de técnicas y procedimientosse están realizando correctamente, durante undeterminado periodo de tiempo. Se emplea paraasegurar el buen funcionamiento diario dellaboratorio (8).
Para asegurar que un método está bajocontrol, a diferentes niveles de un dato concreto,es importante incluir muestras de control decalidad con valores normales y anormales. Losmateriales de control de calidad de origenhumano tiene las máximas probabilidades deparecerse a las muestras de pruebas humanas.
La evaluación interna de la calidad estabasada en el uso de una hoja de control deregistró diario (grafica de Levey Jenning) paraobtener el control gráfico de la desviaciónestándar (DE) y calcular el coeficiente devariación (CV), indicadores de precisión de cadatipo de estudio que se realiza a través de uncalibrador o control.
La gráfica de control de calidad se basa enrealizar 20 determinaciones de la prueba adeterminar, utilizando un plasma comercial o defabricación casera de preferencia (mezcla deplasmas). Se representa gráficamente elresultado obtenido del control de cada día deacuerdo a la marca y número de lote del reactivo(figura 1) (7).Preparación del "pool" de plasma.
Se recomienda como mínimo 20 personassanas, que no tomen medicamentos queinterferían con los factores y reacciones de lacoagulación. Se recolecta la sangre de un númeroaproximadamente igual de hombres y mujeres.El rango de edad debe estar entre 20 y 50 años,
una vez obtenida la sangre colocarla en hielo.Durante la preparación del plasma normal(�pool�), centrifugar a 4°C durante 15 minutos,a 2500 g; mezclar en un contenedor de plásticoy poner alícuotas en viales de plástico y congelarinmediatamente en un congelador a -70°C. Hasta6 meses conservan su estabilidad las muestras(9).
Figura 1.- Curva de Levey � Jenning.
El Control de Calidad Externo.Se utiliza para detectar el grado de acuerdo
que hay entre los resultados de un laboratorioy los resultados de otros centros. Permite nosólo conocer el funcionamiento de un laboratorioconcreto, sino también aquellos reactivos ymétodos que producen resultados poco fiableso equívocos.
El Comité Internacional de Estandarizaciónen Hematología, ha definido una preparación dereferencia como aquella sustancia o instrumentocon una o más propiedades suficientementeestablecidas como para ser usado para lacalibración de un instrumento, para el chequeode un método de medición o para asignar valoresde un material.
Existen 3 categorías: estándar primario(internacional), estándar secundario (nacional oregional) y terciario (comercial o local) (7).
En las ciencias biomédicas la autoridad enestándares internacionales es la OrganizaciónMundial de la Salud y más recientemente se hanincorporado a esta actividad el Comité Internacional
ÿ ÿ
15.5
13.5
11.5
Días 1 2 3 4 5 6 7 8 9 1 0 11 12 13 14 15 1 6 17 18 19 20
��ÿ
��ÿ
Mediaÿx
x
x xx
x
xx
x
x
x
x
x
x
xx
x
x
x
M de los A Ochoa-Rico.

Revista de Hematología Vol. 7, No. 1, 2006
5
de Estandarización en Hematología, la FederaciónInternacional de Química Clínica, también elInstituto Nacional para Estándares Biológicos yControl del Reino Unido y el Instituto Nacionalpara Estándar y Tecnologías del Reino Unido(10) . Estas organizaciones son tambiénresponsables de la producción de estándaressecundarios.
Fase postanalítica. Es la confrontación detodas las fases de análisis y tiene la finalidad decorrelacionar los resultados obtenidos con losdiagnósticos de los pacientes (11).
REFERENCIAS.1.- Owen CA Jr. Historical account of tests of hemos-tasis. Am J Clin Pathol 1990; 93:S3-8.
2.- Borzotta AP, Keeling MM. Value of the preoperati-ve history as an indicator of hemostatic disorders. AnnSurg 1984; 200:648-52.
3.- Quintana GS, Martínez-Murillo C, Ambriz FR. Fisio-logía de la Coagulación, En: Hemofilia, Martínez-Mu-rillo C, Quintana GS, Ambriz FR, Kasper C, editores.México: Editorial Prado; 2001. p. 19-42.
4.- NCCLS. Clinical laboratory procedure manual.Approved guideline. NCCLS Document 10 CP2-A. Vi-llanova, PA:NCCLS, 1984; 4 (2):vii + 27�53.
5.- ICSH. Standardization of blood specimen collec-tion procedure for reference value. Clin Lab Haematol1982; 4:83-6.
6.- Martínez-Murillo C, Quintana-González S. Fisiolo-gía de la Hemostasia Primaria En: Manual de Hemosta-sia y Trombosis. Martínez-Murillo C, Quintana-Gon-zález S, editores. México: Editorial Prado; 1996; p. 5-22.
7.- Lewis SM. Standards, reference materials and refe-rence methods. En: Lewis SM, Koepke JA (eds). He-matology: Laboratory Management and Practice.Oxford: Butterworth-Heinemann;1995. p. 129-35.
8.- Koepke JA, Rodgers H, Ollivier MJ. Preanalyticalinstrumental variables in coagulation testing. Am J ClinPathol 1975; 64:591-6.
9.- Kitchen S, Mc Craw A. Diagnóstico de la hemofiliay otros transtornos de la coagulación. FederanciónMundial de Hhemofilia; 2002.
10.- Koepke JA, Bull BS. The intralaboratory controlquality. En: Lewis SM, Koepke JA (eds). Hematology:Laboratory Management and Practice. Oxford: But-terworth-Heinemann; 1995. p. 183-98.
11.- Schman AL, Griner PP. Diagnostic uses of the par-tial thromboplastin time and prothrombin time. AnnIntern Med 1986; 104:810-6.
Control de calidad en hemostasia.

Revista de Hematología Vol. 7, No. 1, 2006
7
Actualización en el diagnóstico y tratamiento de lapúrpura trombocitopénica autoinmune.
Carlos Martinez-Murillo1,2.
1Servicio de Hematología (Unidad 103). Hospital General de México O.D.2Servicio de Hematología y Unidad de Investigación Médica. Hospital General Regional
N° 1, Gabriel Mancera. Ciudad de México, D.F., México.
INTRODUCCIÓN. La púrpura tromboticopénica autoinmune,también denominada púrpura trombocitopénicainmune o idiopática (PTI) es una enfermedadhemorrágica caracterizada por la destrucciónprematura de plaquetas debido a la unión de unautoanticuerpo, habitualmente de la clase IgG, alas glucoproteínas plaquetarias (GPIIb/IIIa) y laposterior depuración del sistema fagocíticomononuclear (1-4).
EPIDEMIOLOGÍA.Incidencia.
La incidencia general se calcula entre 1 a12.5 casos (2.25-2.68) por 100,000 personas(5) o bien otras estadísticas informan 100 casospor 1 millón de individuos por año y en niños seinforma una incidencia de 4 a 5.3 por 100,000personas.
Estas cifras pueden ser mayores, sinembargo, no existen estudios epidemiológicosque estimen la incidencia real de la enfermedad,incluso muchos casos de PTI aguda en niños noreciben atención médica especializada y no sedocumentan los casos estadísticos.
En niños la prevalencia es la misma entrehombres y mujeres, sin embargo, en adultos larelación mujer-hombre es de 2.6-3:1.
En relación a la edad, en los niños laenfermedad no tiene predominio, sin embargo,el pico de prevalencia es de 3 a 5 años.
En los adultos la mayor prevalencia sepresenta entre los 15 y 40 años. Sin embargo,un estudio realizado en Dinamarca encontró queel promedio de edad fue de 56 años, con unincremento progresivo después de los 60 años(5).
Mortalidad y Morbilidad.La primera causa de morbimortalidad de la
PTI es la hemorragia. De hecho la hemorragiaintracraneal, espontánea o postraumática,constituye la principal causa de muerte cuandola cuenta de plaquetas es menor de 10,000/µL.La mortalidad al momento del diagnóstico es del1.5%, sin embargo, la mortalidad en pacientescon menos de 30 x 109/L plaquetas en losprimeros dos años es 4.2 veces mayor (6).
La morbilidad asociada al tratamientopuede ser debido a las complicaciones amediano y largo plazo que provoca el tratamientode la PTI crónica que es recurrente o resistenteal tratamiento, esto ocasionado por el empleode esteroides, esplenectomía, andrógenos oinmunosupresores (7).
Solicitud de reimpresos: Dr. Carlos Martínez-Murillo, Michoacán 18 Casa 1. Col. Miguel Hidalgo, C.P. 14260, México, D.F., México. Tel. 56 06 63 68 E-mail: [email protected]

8
Revista de Hematología Vol. 7, No. 1, 2006
C Martínez-Murillo.
Clasificación.La PTI se clasifica en función del tiempo
de evolución en aguda, cuando la duración esmenor de 6 meses y crónica cuando esta tienemás de 6 meses de evolución después del diag-nóstico. La importancia de determinar si es agudao crónica, fundamentalmente se asocia con laevolución de la enfermedad. Por ejemplo, en losniños el 70% de los casos son agudos y habi-tualmente ocurren después de una evento infec-cioso y tienen un curso autolimitado (8). En con-traste en la población adulta la mayor parte delos casos tienen una evolución a la cronicidad(70 � 80%) (7,9).
Se denomina PTI crónica refractariacuando el paciente no responde a la esplenec-tomía y mantiene cuenta de plaquetas por de-bajo de 20-30 x 109/L plaquetas y requiere deotras modalidades de tratamiento (10,11).
FISIOPATOLOGÍA.Actualmente se conoce que la PTI es me-
diada por autoanticuerpos. Esto se dedujo delas observaciones en neonatos nacidos de mu-jeres afectadas con la enfermedad desarrolla-ban trombocitopenia transitoria. Además estasobservaciones fueron confirmadas sobre el fun-damento de la presencia de trombocitopeniatransitoria en voluntarios sanos, en quienes seles transfundía plasma de individuos afectadoscon la enfermedad. Así, las plaquetas con losautoanticuerpos IgG tienen una depuración ace-lerada por el sistema fagocítico mononuclear, através de los receptores Fcg, que son expresa-dos sobre la superficie de macrófagos tisulares,principalmente de bazo e hígado (1-4).
Los autoanticuerpos que reaccionan a lasplaquetas, principalmente se unen a las gluco-proteínas GPIIb/IIIa, pero también se unen aotros antígenos, como Ib/IX, Ia/IIa, IV y V, asícomo a otros determinantes antigénicos. De he-cho es típica la presencia de anticuerpos contramúltiples antígenos.
La destrucción de plaquetas dentro de las
células presentadoras de antígenos, puede ge-nerar una sucesión de neoantígenos, que resultaen una producción suficiente de autoanticuerposque ocasiona la trombocitopenia.
Los pacientes adultos con PTI tienen linfo-citos T HLA DR (+), incremento en el númerode receptores para interleucina 2 y un perfil decitocinas que sugieren la activación de precur-sores de linfocitos T cooperadores y linfocitosT cooperadores tipo 1. En la PTI, las células Testimulan la síntesis de anticuerpos después dela exposición a fragmentos de glucoproteínas IIb/IIIa.
Se desconoce la razón de la expresión deantígenos criptogénicos y de la activación sos-tenida de linfocitos T.
DATOS CLINICOS.Es importante considerar para el diagnósti-
co de la enfermedad la sintomatología, la evolu-ción de la misma y datos clínicos asociados.
La forma aguda de la enfermedad es la pre-sentación característica en los niños en compa-ración con la presentación crónica de los adul-tos. La PTI en los adultos tiene un inicio insi-dioso y habitualmente no le precede una infec-ción viral u otra enfermedad infecciosa. Los sig-nos y síntomas son muy variables y puede ir des-de presentaciones asintomaticas hasta pacien-tes con hemorragias mucocutáneas.
Los pacientes con cuenta de plaquetas porarriba de 50 x 109/L, el diagnóstico de la PTI esincidental debido a que no presentan sintoma-tología hemorrágica. Por otro lado, los enfer-mos con cuenta de plaquetas entre 30 y 50 x109/L tienen petequias y equimosis al mínimotrauma; en contrate los enfermos con cifras deplaquetas de 10 a 30 x 109/L, tienen petequias,equimosis, epistaxis, gingivorragias y/o metro-rragias espontáneas. Los pacientes con cifra deplaquetas menor a 10 x 109/L tienen un alto ries-go de hemorragias internas, incluyendo hemo-rragia en órganos vitales (v. gr. sistema nervio-so central) (cuadro 1).

Revista de Hematología Vol. 7, No. 1, 2006
9
Cuadro 1DATOS CLÍNICOS
PetequiasEquimosisGingivorragiasHemorragia transvaginal.Hemorragia conjuntivalHemorragia retinianaHematuriaHemorragia de tubo digestivoEsplenomegaliaHemorragia en Sístema Nervioso Central(SNC)
Datos clínicos presentes en los pacientes con PTI. Lapresencia de esplenomegalia se puede llegar a obser-var en el 10% de los pacientes y la hemorragia en SNCse presenta en el 1% (4, 11).
DIAGNÓSTICO.El diagnóstico de la PTI todavía sigue efec-
tuándose por exclusión de otros trastornos queocasionan trombocitopenia. Las presentacionessecundarias pueden asociarse con otros trastor-nos, como lupus eritematoso generalizado, etc.(cuadro 2).
La duración de la hemorragia puede ayudara distinguir entre la forma aguda y la formacrónica de la PTI. Además, la ausencia desíntomas sistémicos apoya la ruta diagnósticahacia una PTI primaria.
La historia familiar es importante porquedistingue entre formas hereditarias como la Púr-pura trombocitopénica cíclica familiar de la ver-dadera PTI (4, 11).
Citometría hemática. La citometría hemáticase marca la presencia de la cuenta baja de pla-quetas y habitualmente el resto de los paráme-tros hematológicos se encuentran dentro de lanormalidad. Sin embargo, en algunos pacientespueden evidenciarse otros trastornos, como lapresencia normocítica normocrómica, que pue-
de ser secundario a la misma hemorragia o mi-crocítica hipocrómica, asociado a deficiencia dehierro o a padecimiento inflamatorio crónico. Enalgunos enfermos puede observarse anemia ma-crocítica que puede estar asociado a datos dedeficiencia de hematínicos o la asociación conanemia hemolítica con reticulocitosis.
Aspirado de Médula Ósea (AMO). Larealización del AMO en los pacientes con PTIha sido controversial. Sin embargo, las guíaspublicadas por la Sociedad Americana deHematología (American Society of HematologyASH) (7), propone que en adultos de menos de
Cuadro 2Enfermedades asociadas a Trombocitopeniasecundaria.
Enfermedades Inmunológicas: Lupus eritematoso generalizado. Artritis reumatoide. Síndrome de anticuerpos antifosfolípidos (SAAF). Anemia hemolítica autoinmune (síndrome de Evans). Enfermedades tiroideas autoinmunes
Estados de InmunodeficienciaSíndromes Linfoproliferativos: Leucemia linfocítica crónica LinfomasInfecciones: Virus inmunodeficiencia humana Virus de la hepatitis C CitomegalovirusAsociada a Medicamentos: Heparina Quinidina AntibióticosTrombocitopenia Congénita Asociado a malformaciones esqueléticas. Púrpura trombocitopénica
amegacariocítica.
Púrpura trombocitopénica autoinmune.

10
Revista de Hematología Vol. 7, No. 1, 2006
60 años, con una presentación típica de laenfermedad, puede evitarse el AMO. Sinembargo, otros autores sugieren que el AMOse realice en individuos de más de 40 años (11).Por otro lado, en los niños se sugiere no realizarAMO si se encuentra bajo vigi lancia ytratamiento con altas dosis de inmunoglobulinaendovenosa (11), excepto en caso de otrasalteraciones hematológicas y presentaciónatípica.
Detección de anticuerpos antiplaquetas. Laprueba de inmunoflorescencia es empleada parainvestigar la presencia de anticuerpos unidos alas plaquetas (PAIgG). El análisis directo de lamedición de los anticuerpos unidos a las pla-quetas tiene una sensibilidad estimada del 49 al66%, con una especificidad del 78 al 92% y unvalor predictivo positivo del 80 al 83% (4, 11).Por otro lado, la detección de anticuerpos li-bres en plasma es de menor utilidad, debido asu mayor variabilidad interlaboratorio y su me-nor sensibilidad y especificidad.
La determinación de anticuerpos específi-cos contra las glucoproteínas plaquetarias GPIIb/IIIa y GPIb/IX son menos sensibles (50 �65%), pero más específicas (90%). El empleode estas pruebas pueden ser de utilidad en ca-sos complejos donde existe duda en el diagnós-tico y pueden auxiliar a determinar si se trata deuna trombocitopenia inmune o no inmune. Nodeben ser empleadas de rutina.
Determinación de trombopoyetina (TPO).Lla determinación de la trombopoyetina puedeser de utilidad en casos complejos, específica-mente para distinguir entre trombocitopenia conpobre producción medular (niveles elevados deTPO) o incremento en du destrucción (nivelesnormales de TPO). Sin embargo, la determina-ción no debe realizarse como rutina, sólo paracasos especiales o para investigación.Helicobacter pylori. Un número de estudios hainformado la presencia de H. pylori en pacientes
con PTI y en algunas series la terapia conantibióticos para erradicar el H. pylori hamejorado casos refractarios de la PTI. A pesarde otros informes contradictorios, puede serrecomendable su determinación en pacientescon PTI crónica refractaria.
TRATAMIENTO.Es importante distinguir el criterio para el
tratamiento de la PTI, que depende fundamen-talmente de la presentación clínica, la cuenta deplaquetas y la evolución de la enfermedad.
Los adultos habitualmente requieren trata-miento al inicio de la enfermedad, debido a quela mayoría de ellos se presentan con cuenta deplaquetas por debajo de 50 x 109/L. El cuadro3 muestra las cifras de plaquetas requeridas parala realización de procedimientos invasivos (12).
Cuadro 3Recomendación británica de cifra de plaque-tas necesarias para realizar procedimientos.
Situación clínica Cifra de plaquetas
Tratamiento dental > 10 x 109/LExtracción dental > 30 x 109/LBloqueo dental regional > 30 x 109/LCirugía Menor > 50 x 109/LCirugía Mayor > 80 x 109/L
Tratamiento inicial (primera línea).Corticosteroides.- La primera línea de trata-miento comprende el empleo de corticosteroi-des, habitualmente prednisona, de 1 a 1.5 mg xkg/día, por 4 a 6 semanas. El porcentaje de res-puestas varía del 50 al 75%, con incremento enla cuenta de plaquetas en las primeras dos a tressemanas de tratamiento. Sin embargo, despuésde la respuesta las recaídas son comunes cuan-do se reduce la dosis del medicamento y hastauna tercera parte de los pacientes puede tener
C Martínez-Murillo.

Revista de Hematología Vol. 7, No. 1, 2006
11
respuestas prolongadas (10-20%).Pacientes quienes presentan falla a este tra-
tamiento o que requieren de altas dosis para man-tener la cuenta de plaquetas segura, deben serconsiderados para esplenectomía (12, 13).
Inmunoglobulina endovenosa.- La inmunoglobulinaendovenosa a altas dosis (IgG AD) es efectiva enelevar la cuenta de plaquetas en el 75% de lospacientes, de los cuales el 50% puede alcanzarcifras normales de plaquetas, sin embargo,produce respuestas transitorias con duración de3 a 4 semanas y posterior descenso en la cuentade plaquetas a niveles pre-tratamiento.
Un estudio prospectivo no ha demostradodiferencias en la respuesta y necesidad de es-plenectomía entre los siguientes tres grupos detratamiento: 1.- IgG AD; 2.- prednisona o 3.- lacombinación. Por otra parte, se ha demostradoque no existe diferencia en los porcentajes derespuesta entre los siguientes esquemas de tra-tamiento: 0.4 g / kg / día x 5 días y/o 1 g / kg /día (dosis única).
El mecanismo de acción involucra variosefectos, como bloqueo de receptores Fc del sis-tema fagocítico mononuclear, bloqueo en la uniónde autoanticuerpos, regulación de la red idioti-po-antiidiotipo y disminución en la producciónde autoanticuerpos (13).
Falla al tratamiento inicial.Se considera falla al tratamiento de prime-
ra línea (corticosteroids y/o IgGAD), a todosaquellos pacientes que requieren altas dosis decorticosteroides para mantener una cifra segurade plaquetas. El porcentaje de estos fluctúanentre 11% y 35% (14). Otro aspecto a consi-derar, es la necesidad de mantener por tiempoprolongado el empleo de corticosteroides a al-tas dosis, por los efectos adversos que ocasio-na a los enfermos. De tal suerte que en caso defalla se debe considerar realizar la siguiente op-ción terapéutica de elección, como la esplenec-tomía. Sin embargo, en ese lapso que el pacien-
te se esplenectomiza, debe mantenerse un nivelseguro de plaquetas mediante una dosis de es-teroides y la combinación de otro fármaco.
Un gran número de medicamentos han sidoempleados para mantener una cifra estable deplaquetas. Esta terapia depende de la edad delenfermo, la gravedad de la presentación, el nivelde plaquetas, tiempo de evolución, estadogeneral del enfermos, etc. Entre las alternativasde tratamiento existen las siguientes opciones;IgG AD, IgG anti-D, danazol, alcaloides de lavinca (cada vez en menor empleo por la pobrerespuesta), etc.
Tratamiento de segunda línea.Esplenectomía. La esplenectomía permaneceaún como la segunda línea de tratamiento cuandohan fallado medidas terapéuticas previas. Elprocedimiento no es estrictamente �curativo�,debido a que el mecanismo inmunológico persistey únicamente se remueve uno de los principalessitios de destrucción.
Hasta el 70% de los pacientes puede tenerrespuesta y alcanzar cifras normales de plaque-tas. Algunos pacientes pueden tener respuestastardías después del procedimiento.
A. Recomendaciones pre-operatorias. Serecomienda que a los pacientes se les incrementalos niveles de plaquetas a base de IgG AD,prednisona o bolos de dexametasona, con objetode disminuir los r iesgos de hemorragiaperioperatoria. Stasi y col. (15) recomiendantratamiento para pacientes con cuenta deplaquetas por debajo de 30 x 109/L.
Es importante la prevención de la infecciónpost-esplenectomía principalmente contrapneumococo, aunque también existen vacunaspara prevenir la infección por Haemophilusinfluenzae y meningococo (vacunas conjugadas),para lo cual es necesaria la administración dossemanas previas a la cirugía de la vacuna (ejem.Pneumovax) y administrarla cada cinco años.
En pacientes esplenectomizados se
Púrpura trombocitopénica autoinmune.

12
Revista de Hematología Vol. 7, No. 1, 2006
recomienda la vacunación anual de la vacunacontra influenza.
Algunos estudios recomiendan el uso de pe-nicilina o eritromicina en los primeros 3 años des-pués de la esplenectomía, sin embargo, no hayestudios que hayan documentado su eficacia enla prevención de la infección por neumococo.Sin embargo, es importante que el paciente siem-pre lo mencione en caso de alguna urgencia uhospitalización.
B. Soporte Operatorio. Aunque el procedimientoes sencillo requiere que el grupo quirúrgico tengaexperiencia en este tipo de enfermos. Se hasugerido que el paciente requiere únicamentesoporte transfusional con plaquetas únicamentemientras se liga la arteria esplénica. Sin embargo,no existen estudios sólidos a este respecto.
C. Cuidados post-operatorios. Las medidaspostoperatorias implican vigilancia de complica-ciones asociadas al procedimiento (22%), entrelas que incluyen embolismo pulmonar, abscesoabdominal, hematoma de la pared abdominal,sepsis y otras. Otros estudios han informado 0%de mortalidad y 7 % de morbilidad.
D. Búsqueda de bazo accesorio. La presenciade un bazo accesorio debe sospecharse enaquellos pacientes que t ienen falla a laesplenectomía o que recaen después de unarespuesta inicial. La mejoría en las técnicasradiológicas ha permitido detectar estos bazoshasta en el 12% de estos pacientes.
E.- Predic tores de la respues ta a laesplenectomía. El mejor predictor de larespuesta a la esplenectomía es el estudio deplaquetas autólogas marcadas con indium, deacuerdo con el estudio de Najean y cols(16) en528 pacientes. Los pacientes donde existedestrucción esplénica, > 90% pueden obtenerremisión. En contraste los pacientes condestrucción plaquetaria hepática o mixta
(hepática y esplénica), el 92% tuvieron falla a laesplenectomía.
Falla a la Esplenectomía.Se considera falla a la esplenectomía a
todos aquellos pacientes que posterior alprocedimiento presentan cuenta de plaquetas<50 x 109/L.
En este caso se denomina PTI Crónica Re-fractaria y requiere de múltiples opciones detratamiento.
A. Corticosteroides.- En caso de falla a la es-plenectomía se recomienda el empleo de corti-costeroides a altas dosis para intentar una remi-sión completa y entre las formas de empleo seencuentra la convencional, con prednisona de 1a 1.5 mg x kg/día, por 2 a 4 semanas, o bien elesquema de altas dosis de dexametasona (17,18), que consiste en 40 mg/día, por 4 días, cada28 días hasta completar 6 ciclos.
También puede ser empleado el esquema debolos de metil prednisolona con el siguiente es-quema: 30 mg/kg/ día por 3 días, seguido de 20mg /kg/ día por 4 días y después 5, 2 y 1 mg/kg/día por 1 semana. La respuesta habitualmentese observa dentro de los primeros 3 a 5 días(19).
B. Altas dosis de Ig.- El empleo de Ig AD a ladosis de 1 g/kg/día, por 2 días consecutivos, enasociación con corticosteroides, incrementa rá-pidamente la cuenta de plaquetas (20, 21) y re-sulta mejor cuando se repite cada tres semanas(22), donde se informa de remisiones comple-tas. Sin embargo, en general, las respuestas conIg AD son transitorias y rara vez produce res-puestas prolongadas (22-24), por lo tanto estetipo de tratamiento habitualmente esta reserva-do para pacientes sintomáticos.C. Inmunoglobulina anti-D.- Desde hace va-rios años se ha empleado la IgG anti-D para eltratamiento de la PTI. Salama y col. (25, 26)identificaron que la infusión de Ig AD en pacien-
C Martínez-Murillo.

Revista de Hematología Vol. 7, No. 1, 2006
13
tes con PTI fue asociado con evidencia de la-boratorio de hemolisis. Entonces hipotetizó queen las preparaciones de inmunoglobulinas exis-tían pequeñas cantidades de anticuerpos anti eri-trocitos que podrían ser los responsables del blo-queo al receptor Fc (FcR) y por lo tanto delincremento en la cifra de plaquetas. Con el ob-jetivo de probar esta hipótesis, administraronIgG anti-D en pacientes con PTI Rh (D+) y ob-tuvieron incremento en la cuenta de plaquetasen la mayoría de los enfermos. Posteriormenteotros autores han documentado el mismo efecto(27-35). Scaradavou y col. (36) informaron queentre 79 y 90% de los adultos que fueron trata-dos con IgG anti-D tuvieron respuesta.
Eritrocitos Opsonizados con IgG anti-D.Desde 1984 (38) se ha publicado la experienciacon el empleo de IgG anti-D en forma deeritrocitos autólogos opsonizados para eltratamiento de la PTI crónica refractaria, dondese han obtenido respuestas de más del 60% (38-42). Estas respuestas han s ido tambiénobtenidas por Ruíz-Arguelles y col. (43) con lamisma forma de tratamiento.
D. Danazol.- El danazol es un andrógenosintético, con pocos efectos virilizantes, que hasido empleado de manera sinérgica con loscorticosteroides. Ahn y col. (44) informó quede 22 pacientes tratados con danazol a las dosisde 200 mg 2 a 4 veces al día, por más de 2meses, el 60% presentó incremento en la cuentade plaquetas por más de 2 meses. El mecanismode acción es desconocido, pero parece ser quedisminuye la expresión de receptores Fc sobreel sistema fagocítico mononuclear del bazo.
E. Inmunosupresores.- La inmunosupresiónpuede ser requerida si fallan los tratamientosprevios. El tratamiento con azatioprina 2 mg/kg/día (máximo 150 mgs) o ciclofosfamida producerespuestas mayores al 25% y la mayoría de ellassostenidas (45). Por su parte, Quiquandon y col.
(46) informaron que de 53 pacientes tratadoscon azatioprina por una media de 18 meses, 64%tuvieron incremento en la cuenta de plaquetas yen 45% tuvo remisiones completas . Laazatioprina tiene un efecto lento y debe seradministrado por un mínimo de 6 meses, antesde considerar que existe falla al tratamiento.
F. Dapsona.- La dapsona es un fármacotradicionalmente empleado para la lepra. Sinembargo, en algunos adultos con PTI crónicatratados con este medicamento, a la dosis de75 a 10 mgs/día, por 21 días, se obtuvo 50%de respuestas (47). El mecanismo de acción dela dapsona es desconocido sin embargo, puedeser debido al bloqueo del sistema fagocíticomononuclear a través del incremento en ladestrucción de glóbulos rojos (48). Parece serque las mejores respuestas a este tratamientoes en casos no graves y que no han sidoesplenectomizados.
G. Anticuerpos monoclonales.- El anti CD20,denominado rituximab ha sido evaluado enalgunas series de pacientes con PTI crónicaref rac tar ia a la dos is de 375mg/m 2 sc ,semanalmente por 4 semanas, en los cuales seobservó algún tipo de respuesta en el 50% (lamitad de ellos con remisión completa), conrespuestas de más de 6 meses. Existe lasugerencia que los pacientes jóvenes respondenmejor al tratamiento (49).
El otro anticuerpo monoclonal que se haempleado es el anti-CD52, denominadoalemtuximab (CAMPATH), el cual ha sidoaprobado para el tratamiento de la leucemialinfocítica crónica y que se ha empleado enpacientes con pancitopenias inmunológicas,incluyendo algunos pacientes con PTI crónicarefractaria, en los cuales se han informadorespuestas después de 3 a 4 semanas detratamiento, con respuestas sostenidas de másde 4 a 9 meses. Sin embargo, el efecto adversomás importante es la inmunodepresión con
Púrpura trombocitopénica autoinmune.

14
Revista de Hematología Vol. 7, No. 1, 2006
linfopenia severa (< 0.1109 / L) y en algunospacientes se ha informado el agravamiento de latrombocitopenia (50).
H. Micofenolato de Mofetil.- Este inmunosu-presor antiproliferativo ha sido aprobado parala prevención del rechazo agudo en pacientestrasplantado. Ha demostrado eficacia en el tra-tamiento de la PTI crónica refractaria, pero re-quiere de mayor comprobación en un mayor nú-mero de enfermos (51).
I . Otras opciones de tratamiento . -Considerando el riesgo beneficio de otrasmodalidades de tratamiento, tales comointerferón alfa, inmunoadsorción con columnasde proteína A, plasmaféresis, doxorrubicinaliposomal y alcaloides de la vinca, estas opcionesterapéuticas ahora no son recomendadas (11).
EVOLUCIÓN.La edad media de la presentación fue a los
39 años y la mayoría de los pacientes presentantrombocitopenia severa. Durante los primeros 2años alunos pacientes pueden manifestar lassiguientes enfermedades; lupus eritematosogeneralizado, artritis reumatoide, síndrome deanticuerpos antifosfolípidos, colitis crónica,linfomas u otros tipos de cáncer, etc.
REFERENCIAS.1.- Karpatkin S. Autoimmune thrombocytopenicpurpura. Semin Hematol 1985; 22: 260-88.
2.- Waters AH. Autoimmune thrombocytopenia:Clinical Aspects. Semin Hematol 1992; 29: 18-25.
3.- George JN, Raskob GE. Idiopathic thrombocytopeniapurpura: A concise summary of the pathophysiologyand diagnosis in children and adults. Semin Hematol1998; 36: 5-8.
4.- Cines DB, Blanchette V. Immune thrombocytopenicpurpura. N Engl J Med 2002; 346:995-1008.
5.- Henrik-Frederiksen H, Schmidt K. The incidence ofidiopathic thrombocytopenic purpura in adul tsincreases with age. Blood 1999; 94: 909-13.
6.- Johanna E, Portielje A, Rudi G, Westendorp J,Hanneke C. Kluin-Nelemans, et al. Morbidity andmortality in adults with idiopathic thrombocytopenicpurpura. Blood 2001; 9:2549-54.
7.- George JN, Woolf SH, Raskob GE, et al. Idiopathicthrombocytopenic purpura: a practice guidelinedeveloped by explicit methods for the AmericanSociety of Hematology. Blood 1996; 88: 3-40.
8.- Calpin C, Dick P, Foon A, Feldman W. Is bonemarrow aspiration needed in acute childhood idiopathicthrombocytopenic purpura to rule out leukemia? ArchPediatr Adolesc Med 1998; 152:345-7.
9.- Chong BH, Keng TB. Advances in the diagnosis ofidiopathic thrombocytopenic purpura. Semin Hematol2000; 37:249-60.
10 . - Taran t ino M. Acute Immune ( id iopa th ic )thrombocytopenic purpura in childhood. Blood Rev2002; 16:19-21.
11.- British Committee for Standards in HaematologyGeneral Haematology Task Force. Guidelines for theinves t iga t ion and management o f id iopa th icthrombocytopenic purpura in adults, children and inpregnancy. Br J Haematol, 2003; 120:574�96.
12.- Pizzuto J, Ambriz R. Therapeutic experience on934 adults with idiopathic thrombocytopenic purpura:Multicentric Trial of the Cooperative Latin Americangroup on Hemostasis and Thrombosis. Blood 1984;64:1179�83.
13.- Warkentin TE, Kelton JG. Current concepts in thetreatment of immune thrombocytopenia. Drugs 1990;40:531�42.
14.- George JN, el-Harake MA, Raskob GE. Chronicidiopathic thrombocytopenic purpura. N Engl J Med1994; 331:1207�11.
15.- Stasi R, Stipa E, Masi M, Cecconi M, Scimo MT,Oliva F, et al. Long-term observation of 208 adults withchronic idiopathic thrombocytopenic purpura. Am JMed 1995; 98:436�42.
C Martínez-Murillo.

Revista de Hematología Vol. 7, No. 1, 2006
15
16.- Najean Y, Rain JD, Billotey C. The site ofdestruction of autologous 111In-labelled platelets andthe efficiency of splenectomy in children and adultswith idiopathic thrombocytopenic purpura: a study of578 patients with 268 splenectomies. Br J Haematol1997; 97: 547�50.
17.- Andersen JC. Response of resistant idiopathicthrombocytopenic purpura to pulsed high-dosedexamethasone therapy. N Engl J Med 1994; 330:1560�64.
18.- Arruda VR, Annichino-Bizzacchi JM. High-dosedexamethasone therapy in chron ic id iopa th icthrombocytopenic purpura. Ann Hematol 1996; 73:175�7.
19.- Akoglu T, Paydas S, Bayik M, Lawrence R, FiratliT. Megadose methylprednisolone pulse therapy inadult idiopathic thrombocytopenic purpura. Lancet1991; 337: 56.
20.- Bussel JB, Hilgartner MW. The use and mechanismof action of intravenous immunoglobulin in thetreatment of immune haematologic disease. Br JHaematol 1984; 56: 1�7.
21.- Imbach P, Wagner HP, Berchtold W, Gaedicke G,Hirt A, Joller P, et al. Intravenous immunoglobulinversus o ra l cor t i cos te ro ids in acu te immunethrombocytopenic purpura in childhood. Lancet 1985;2: 464�8.
22.- Schiavotto C, Ruggeri M, Rodeghiero F. Adversereactions after high-dose intravenous immunoglobulin:incidence in 83 pat ients t reated for id iopathicthrombocytopenic purpura (ITP) and review of theliterature. Haematologica 1993; 78: 35�40.
23.- Schiavotto C, Ruggeri M, Rodeghiero F. Failure ofrepea ted courses o f h igh-dose in t ravenousimmunoglobulin to induce stable remission in patientswith chronic idiopathic thrombocytopenic purpura.Ann Hematol 1995; 70:89�90.
24.- Bussel J. Novel approaches to refractory immunethrombocytopenic purpura. Blood Rev 2002; 16:31-6.
25.- Salama A, Kiefel V, Amberg R, Müeller-Eckhardt C.Treatment of autoimmune thrombocytopenic purpurawith rhesus antibodies (anti-RhO [D]). Blut 1984; 49:29-35.
26.- Salama A, Kiefel V, Mueller--Eckhardt C. Effect ofIgG anti-Rho(D) in adult pat ients with chronicautoimmune thrombocytopenia. Am J Hematol 1986;22: 241-50.
27.- Baglin TP,Smith MP, Boughton BJ. Rapid andcomplete response of immune thrombocytopenicpurpura to a s ingle in jec t ion of rhesus ant i -Dimmunoglobulin. Lancet 1986; 1: 1329-30.
28.- Panzer S, Grumayer ER, Haas OA, Niesner H,Graninger W. Efficacy of rhesus antibodies (Anti-Rho(D)) in autoimmune thrombocytopenia: Correlationwith response to high dose IgG and the degree ofhaemolysis. Blut 1986; 52: 117-21.
29.- Gabra GS, Mitchell R. Anti-D immunoglobulin andimmune thrombocytopenia- a problem of ethics in bloodtransfusion practice. Vox Sang 1988; 54: 246.
30.- Bussel JB, Graziano JN, Kimberly RP, Pahwa S,Aledort LM. Intravenous anti-D treatment of immunethrombocytopenic purpura: Analysis of efficacy,toxicity and mechanism of effect. Blood 1991; 77: 1884-93.
31.- Ware RE, Zimmerman S. Anti-D: Mechanisms ofaction. Semin Hematol 1998; 36: 14-22.
32 . - George JN, E l -Harake MA, As te r RH.Thrombocytopenia due to enhance p la te le tdestruction by immunologic mechanisms. WilliamsHematology 5th ed. New York: Mc Graw-Hill, Inc. 1995.p. 1315-55.
33.- Bussel JB. Recent advances in the treatment ofidiopathic thrombocytopenic purpura: the anti-Dclinical experience. Semin Hematol 1998; 36: 1-4.
34.- Borgna-Pignatti C, Battisti L, Zecca M, LocatelliF. Trea tment o f chron ic ch i ldhood immunethrombocytopenic purpura with intramuscular anti-Dimmunoglobulins. Br J Haematol 1994; 88: 618-20.
35.- Bierling P, Karianakis G, Duedari N, Desaint C,Oksenhendler E, Habibi H. Anti-Rhesus antibodiesimmune thrombocytopenia and human immunodeficiencyvirus infection. Ann Intern Med 1987; 106: 773-4.
36.- Scaradavou A, Woo B, Woloski, BM, Cunningham-Rundles S, Ettinger LJ, Aledort LM, Bussel JB.In t ravenous an t i -D t rea tment o f immunethrombocytopenic purpura: experience in 272 patients.
Púrpura trombocitopénica autoinmune.

16
Revista de Hematología Vol. 7, No. 1, 2006
Blood 1997; 89: 2689�700.
37.- Ambriz R, Pizzuto J, Morales M, Muñoz R,Quintanar G, García N. Treatment of chronic ITP withopsonized erythrocytes with rhesus antibody andlabeled with Tc-99m (Radioimmune method). Blood1984; 64:(suppl):233.
38.- Ambriz R, Quintanar E, Domínguez JL, García EN,Estrada C, Alvarez E, et al. Tratamiento con IgG anti D(Rho) en PTA refractaria, mediante transfusión directa(TD) vs eritrocitos opsonizados "in vitro" (TEOP),Trabajo Multicéntrico AMEH, A.C. Sangre 1987;32:259.
39.- Ambriz R, Muñoz R, Quintanar E, Sigler L, AvilésA, Pizzuto J. Accessory spleen compromising responseto splenectomy for idiopathic thrombocytopenicpurpura. Radiology 1985; 155: 793-796.
40.- Ambriz R, Muñoz R, Quintanar E, Sigler L, AvilésA, Pizzuto J. Accessory spleen compromising responseto splenectomy for idiopathic thrombocytopenicpurpura. The Year Book of Nuclear Medicine. Chicago:Year Book Medical Publishers Inc; 1987. p. 326-7.
41.- Ambriz R, Muñoz R, Pizzuto J, Quintanar E, MoralesM, Avilés A. Low-dose autologous in vitro opsonizederythrocytes. Radioimmune method and autologousopsonized erythrocytes for refractory autoimmunethrombocytopenic purpura in adults. Arch Intern Med1987; 147: 105-8.
42.- Ambriz FR, Martínez-Murillo C, Quintana GS,Collazo-Jaloma J, Bautista JJ. Fc Receptor blockade inpa t ien t s wi th re f rac tory chron ic immunethrombocytopenic purpura. Arch Med Research 2002;33: 536-40.
43. Ruíz-Argüelles GJ, Apreza MMG, Perez-Romano B,Ruiz-Argüelles A. The infusion of Anti-Rho (D)opsonized erythrocytes may be useful in the treatmentof patients, splenectomized or not, with chronic,refractory autoimmune thrombocytopenic purpura. Aprospective study. Am J Hematol 1993; 43: 72-3.
44.- Ahn YS. Rocha R. Mylvaganam R. Garcia R, DuncanR, Harrington WJ. Long-term danazol therapy inau to immune th rombocytopenia : unmain ta inedremission and agedependent response in women. AnnIntern Med 1989; 111:723�9.
45.- Bouroncle BA, Doan CA. Treatment of refractoryidiopathic thrombocytopenic purpura. JAMA 1969;207: 2049�2052.
46.- Quiquandon I, Fenaux P, Caulier MT, Pagniez D,Huart JJ, Bauters F. Re-evaluation of the role ofazath iopr ine in the t rea tment of adul t chronicidiopathic thrombocytopenic purpura. a report on 53cases. Br J Haematol 1990; 74: 223�8.
47.- Godeau B, Durand JM, Roudot-Thoraval F, TennezeA, Oksenhendler E, Kaplanski G, et al. Dapsone forchronic autoimmune thrombocytopenic purpura: areport of 66 cases. Br J Haematol 1997; 97:336-9.
48.- Radaelli F, Calori R, Goldaniga M, Guggiari E,Luciano A. Adult refractory chronic idiopathicthrombocytopenic purpura: can dapsone be proposedas second-line therapy? [Letter]. Br J Haematology1999; 104:641�2.
49.- Stasi R, Pagano A, Stipa E, Amadori S. Rituximabchimaeric anti-CD20 monoclonal antibody treatmentfor adults with chronic idiopathic thrombocytopenicpurpura. Blood 2001; 98:952�7.
50.- Willis F, Marsh JC, Bevan DH, Killick SB, Lucas G,Griffiths R, Ouwehand W, Hale G, Waldmann H, Gordon-Smith EC. The effect of treatment with Campath-1H inpatients with autoimmune cytopenias. Br J Haematol2001; 114:891�8.
51.- Howard J, Hoffbrand AV, Prentice HG, Mehta A.Mycophenolate mofetil for the treatment of refractoryautoimmune haemolytic anaemia and auto-immunethrombocytopenia purpura. Br J Haematology 2002;117: 712�5.
C Martínez-Murillo.

Revista de Hematología Vol. 7, No. 1, 2006
17
INTRODUCCIÓN.La enfermedad de von Willebrand (EvW)
es una enfermedad hemorrágica autosómicahereditaria causada por la deficiencia odisfunción del factor de von Wilebrand (FvW),que se carac ter iza por hemorragiasmucocutáneas de intensidad variable y queafecta primordialmente la hemostasia primaria enla interacción plaqueta, FvW y endotelio.
El FvW es una proteína multimérica quetiene dos funciones en la hemostasia. Es esencialpara la formación del coágulo plaquetario porsus funciones en la adhesión y agregaciónplaquetaria, a través de los grandes multímerosdel factor, y la formación de un complejo con elfactor VIII por medio de una unión no covalente,protegiendo a este factor de la degradaciónenzimát ica . Por lo tan to , cont r ibuyeindirectamente al proceso de coagulación ohemostas ia secundar ia . Es te defec tohemorrágico de origen genético se codifica enel cromosoma 12, cromosoma que se encargade codificar la información para una moléculamadura, con una gran heterogeneidad, queproduce var iac iones b io lógicas en laenfermedad.
En 1926, Erik von Willebrand describió una
enfermedad hemorrágica en una familianumerosa en las Islas Aland, en el golfo deBotnia, en las costas de Finlandia (1). Adiferencia de la hemofilia, en esta enfermedaduno y otro sexo eran afectados y la hemorragiamucocutánea predominaba. Erik von Willebrandasignó el término de �pseudohemofi l iahereditaria� para designar al padecimiento quetenía como característica común, hemorragiasmucocutáneas de intensidad variable, conherencia autosómica y tiempo de hemorragia(TH) prolongado. Posteriormente Jürgenscolabora con Erik von Willebrand y al estudiara los enfermos de las islas Aland consideran eltérmino de �Trombopatía constitucional vonWillebrand-Jürgens� por considerar que setrataba de un defecto plaquetario. En 1957, seinformó que el defecto podía ser corregido porun factor plasmático diferente al factor VIII(FVIII ) , denominándose fac tor de vonWillebrand (FvW) (2). La reducción del FvWcausa reducción del Factor VIII, observando laestrecha relación que tienen ambas proteínas. Lapurificación del FvW y el subsecuente desarrollode reac t ivos sero lógicos y técnicaselectroforéticas especializadas, han permitidoconocer la heterogenicidad del FvW (3-6).
Tratamiento de la enfermedad de von Willebrand.
Sandra Quintana-González1, Carlos Martínez-Murillo2, 3.
1Banco Central de Sangre, Centro Médico Nacional Siglo XXI, Instituto Mexicano delSeguro Social, 2Servicio de Hematología (Unidad 103). Hospital General de México
O.D., 3Servicio de Hematología y Unidad de Investigación Médica. Hospital GeneralRegional N° 1, Gabriel Mancera. Ciudad de México, D.F., México.
Solicitud de reimpresos: Dra. Sandra Quintana-González, Aureliano Rivera No. 1 casa 8, Tizapan San Ángel, C.P. 01090, México, D.F., México. Tel. 56 45 86 13 E-Mail: [email protected]

18
Revista de Hematología Vol. 7, No. 1, 2006
INCIDENCIA.La EvW es la enfermedad hemorrágica
hereditaria más frecuente, con una distribuciónmundial y sin predominio de sexo. Se hainformado una prevalencia de l 0 .9%,aproximadamente 8.2 casos por 1000 habitantesy se ha determinado una prevalencia del 1.3%en población multiétnica (7). De los pacientescon EvW el 70 al 80% son tipo 1, 5 al 15%tienen alguna variedad del tipo 2 y la prevalenciadel tipo 3 (EvW severa) es de 1 a 5 por millónde habitantes en Europa y de 3 por millón enSuecia e Israel. En Alemania el tipo 3 representael 12% de los casos, en Italia el 17% y en Israelhasta el 29% (8-10). En Latinoamérica se hainformado una incidencia de 1.1% en Costa Rica(11).
FACTOR DE VON WILLEBRAND (FvW).El FvW es una glucoproteína de alto peso
molecular s intet izado y almacenado enmegacariocitos y células endoteliales. El geneque codifica el FvW ha sido clonado ylocalizado en el cromosoma 12p13.2. El geneestá compuesto de 178 kilobases con 52 exones.La estructura del FvW está compuesta de unpolipéptido de 270 kD, con una subunidad quecomprende 2,050 residuos de aminoácidos;cada subunidad contiene sitios de unión para lacolágena y para las glicoproteínas (Gp) Ib y
GpIIb/IIIa (fig.1). En vasos sanguíneos intactosel FvW no interactúa con los receptores deplaquetas. Cuando el vaso se daña expone elsubendotelio y se une el FvW. Esta interaccióninduce un cambio conformacional en el FvW,que expone los sitios de unión para que la GpIbde las plaquetas se una al FvW y se lleve a caboel mecanismo de adhesión plaquetaria por mediodel dominio A1 . El FvW se adhiere a la fibrasde colágena de la pared vascular, pero tambiéna otros componentes del subendotelio (12). Porotro lado, en superficies con �high shear stress�se ha demostrado la activación del sitio de uniónde la GpIIb/IIIa (IIb3) sobre la membranaplaquetaria. Esta activación es capaz de unirplaquetas (agregación) por medio del FvW,fibrinógeno, vitronectina y otras proteínas quecontengan la secuencia Arg-Gly-Asp.
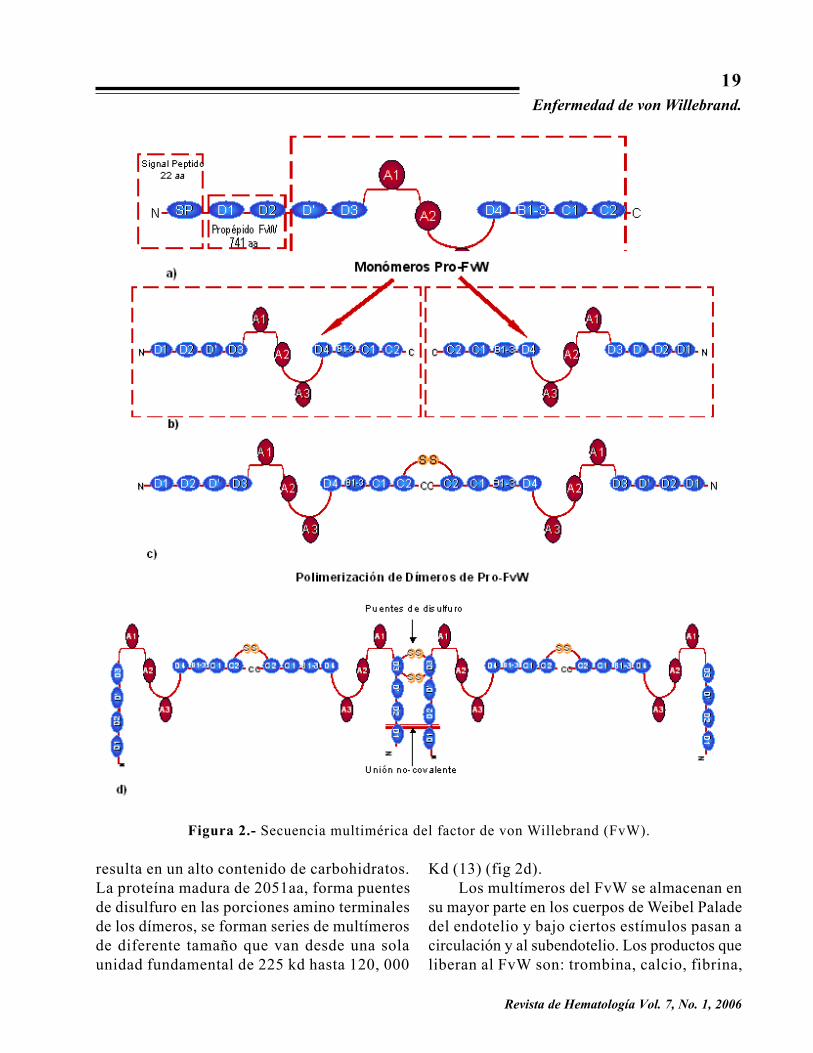
El ARNm codifica para una proteína dealrededor de alrededor de 2,813 aminoácidos(aa) llamada pre-pro-FvW. Este producto inicialde 300 a 350 Kd pierde una fracción llamada�péptido de señal� (SP), que consta de 22 aa,que inicia el proceso de formación de la proteínadel FvW (fig 2a). Después de esta pérdida elpro-polipéptido de 2791 aa (fig 2b), formadímeros a través de la formación de puentesdisulfuro en las porciones carboxi-terminales (fig2c) . Pos ter iormente se l leva a cabo laglucosilación en el aparato de Golgi, lo que
D1 D2 D� D3 A1 A2 A3 D4 B1 B2 B3 C1 C2
ss ss ss ss
Propéptido741 aa
Subunidad Madura2,050 aa
FVIII
Heparina
Colágena
Gp1b αIIb β3
Multímero Dímero
NSP
D1 D2 D� D3 A1 A2 A3 D4 B1 B2 B3 C1 C2
ss ss ss ss
D1 D2 D� D3 A1 A2 A3 D4 B1 B2 B3 C1 C2
ss ssss ssss ss ss ssss ssss ss
Propéptido741 aa
Subunidad Madura2,050 aa
FVIII
Heparina
Colágena
Gp1b αIIb β3
Multímero Dímero
NSP
Fig.1 Estructura del Pro-factor de von Willebrand (FvW). En la figura se señala la organización de los dominiosdel FvW. Estos dominios son definidos y agrupados de acuerdo a su homología interna. Las barras negrasindican la localización de los sitios de unión. La secuencia en el dominio C1 interviene en la unión de la GpIIb/IIIa (IIb/3), pero el estado funcional del dominio D2 permanece desconocida. La unión S-S indica la localizaciónde los puentes disulfuro involucradas en la dimerización y multimerización.
S Quintana-González, C Martínez-Murillo.
Revista de Hematología Vol. 7, No. 1, 2006
19
resulta en un alto contenido de carbohidratos.La proteína madura de 2051aa, forma puentesde disulfuro en las porciones amino terminalesde los dímeros, se forman series de multímerosde diferente tamaño que van desde una solaunidad fundamental de 225 kd hasta 120, 000
Kd (13) (fig 2d).Los multímeros del FvW se almacenan en
su mayor parte en los cuerpos de Weibel Paladedel endotelio y bajo ciertos estímulos pasan acirculación y al subendotelio. Los productos queliberan al FvW son: trombina, calcio, fibrina,
Figura 2.- Secuencia multimérica del factor de von Willebrand (FvW).
Enfermedad de von Willebrand.

20
Revista de Hematología Vol. 7, No. 1, 2006
activador tisular del plasminógeno (t-PA),plasmina, adrenalina bradicinina, interleucina-1,vasopresina y su análogo sintético, la desamino-D-arginina-vasopresina (DDAVP), o del FvW yfavorecen su actividad biológica.
El FvW funciona como el acarreadoresencial del FVIII permitiendo la estabilidad deeste factor en la circulación. El FVIII circula enplasma con el factor de von Willebrand (FvW)para evitar que el factor VIII, el cual es lábil sedestruya. Por lo tanto, el FvW es la moléculaque protege al FVIII de la destrucción de algunasenzimas en plasma y es el factor que le daestabilidad al factor VIII. La unión del factorVIII con el FvW es no covalente y recibe elnombre de Complejo FVIII:C/FvW, el cual esun complejo estable (fig.3).
El FvW se une a la GPIb-IX y establece elcontacto inicial entre las plaquetas y la superficiesubendotelial (colágena), es decir favorece losmecanismos de adhesión plaquetaria. Estoocasiona la activación primaria de la plaqueta.La activación plaquetaria ocasiona la liberaciónde productos almacenados en los gránulos alfay cuerpos densos, incluyendo FvW plaquetarioy el cambio conformacional de la GPIIb-IIIa. ElFvW se une a la GPIIb-IIIa y participa en losmecanismos de interacción plaqueta-plaqueta,
mediante el mecanismo de agregación plaquetaria,donde participa el fibrinógeno y iones de calcio.En la f igura 4 se representa de maneraesquemática los mecanismos de adhesiónplaquetaria. El primer contacto se estableceentre las plaquetas y el FvW por medio delreceptor glucoprotéico Ib. Esta unión se realizaa través del dominio A1 del FvW. Las plaquetasrápidamente pueden unirse a las superficiescubiertas con el FvW, siempre que existancondiciones de flujo y deben tener también altaresistencia a la fuerza de tracción. Después deque las plaquetas se adhieren pueden resistir ala fuerza creada por el flujo que resulta en lasparedes con cizallamiento. La interacción, tieneuna elevada velocidad de disociación intrínseca,resultando en una rápida separación por elmovimiento de rotación impuesto por el flujosanguíneo. Se forman nuevas uniones endiferentes regiones de la membrana de lasplaquetas en rotación, en estrecho contacto conla superficie. La translocación continúa hasta queel receptor GpIIb/IIIa, que inicialmente no sepuede unir con el FvW, posteriormente se activay se une a la secuencia RGDS del dominio C1del FvW. Finalmente, se unirán otras plaquetasa la superficie produciendo el fenómeno deagregación plaquetaria.
Figura 3.- Complejo factor VIII/factor de von Willebrand. El factor VIII se une a la proteínamultimérica del factor de von Willebrand por medio de una unión no covalente.
S Quintana-González, C Martínez-Murillo.

Revista de Hematología Vol. 7, No. 1, 2006
21
En la pared vascular intacta, el flujosanguíneo provoca que los eritrocitos y losleucocitos se encuentren en el centro del vasosanguíneo y las plaquetas se encuentran máscercanas a la pared vascular. Sin embargo, lascélulas endoteliales impiden la interacción deestas plaquetas a la pared intacta del vaso, yaque las fibras de colágena se encuentran en lamatriz subendotelial. Cuando la pared vascularesta intacta y el flujo sanguíneo es normal, elFvW que circula en el plasma y las plaquetaspueden tener mínimas interacciones. En la paredvascular dañada, las fibras de colágena y el FvWse exponen al flujo sanguíneo y a las fuerzas decizallamiento. El FvW plasmático eficientementese une a la colágena expuesta por medio de laGpIa y su estructura se desenrolla, apoyando laadhesión de las plaquetas circulantes en sinergiacon la colágena. La unión del FvW interactúaprimero solamente con el receptor GpIb e iniciala rotación de las plaquetas (fig. 4 y 5b). Estainteracción se disocia rápidamente y la rotaciónde las plaquetas se realiza de acuerdo al flujosanguíneo. Una vez que las plaquetas estánactivadas se forman pseudópodos incrementandola afinidad del factor de von Willebrand y elreceptor GpIIb/IIIa se activa y presenta uncambio conformacional en la superficie de las
plaquetas, lo que ayuda a la interacción deplaqueta-plaqueta (agregación), formando unacoágulo plaquetario a través del FvW y a lascondiciones bajas del flujo sanguíneo y alfibrinógeno (fig. 5c).
CLASIFICACIÓN.La identificación de varios subtipos de la
EvW ha contribuido a su complejidad, ademásde las variaciones en la herencia, manifestacio-nes clínicas y resultados de las pruebas de he-mostasia. El tratamiento de la EvW depende engran medida del subtipo de la enfermedad.
Los progresos recientes en la caracteriza-ción de las mutaciones que causan la EvW, hanproporcionado datos suficientes para reorgani-zar la forma como había sido históricamente cla-sificada la enfermedad. En 1994 se publicó unnuevo sistema de clasificación para la EvW. Estábasada principalmente en el fenotipo de la pro-teína del FvW, que está presente en el plasma yplaquetas del paciente (6). La clasificación iden-tifica dos categorías por alteraciones cuantitati-vas del FvW (Tipos 1 y 3) o por alteracionescualitativas del FvW (Tipo 2).
La deficiencia cuantitativa del FvW enplasma y/o plaquetas identifica a la EvW tipo 1,mientras que la EvW tipo 3 se encuentra ausente
Figura 4.- Adhesión plaquetaria al factor de von Willebrand.
Enfermedad de von Willebrand.

22
Revista de Hematología Vol. 7, No. 1, 2006
Figura 5.- Función del factor de von Willebrand en hemostasia primaria.
o solamente pequeñas cantidades de FvW enplasma y plaquetas se encuentran presentes. Eltipo 1 se diferencia del tipo 3 por la deficiencialeve del FvW (usualmente de 30-40 UI/dL), laherencia autosómica dominante y la presenciade hemorragias leves. Se identifican cuatrosubtipos de la EvW tipo 2. Estos reflejan losmecanismos fisiopatológicos distintos entre cadauno de ellos. El tipo 2A y 2B se caracterizanpor la ausencia de los multímeros de gran tamañoen el plasma; en el tipo 2B, existe un aumentode la afinidad del FvW a la GpIbα.
La ident i f icac ión de las var iantescualitativamente anormales del FvW condisminución de la función dependiente de
plaquetas y la presencia de multímeros normales,ha caracterizado al subtipo 2M, causado pormutaciones que afectan la función del FvW, perono afectan la estructura multimérica. En el tipo2N (Normandy), la estructura multimérica delFvW no está alterada, sin embargo la región N-terminal sobre el FvW no se une al Factor VIII,por lo que solamente se puede identificar por laprueba de unión del FvW/FVIII.
CUADRO CLÍNICO.Clínicamente la enfermedad se caracteriza
por la presencia de hemorragias mucocutáneasde intensidad variable y que tiende a serf luc tuante , es dec i r a l te rnan per íodos
S Quintana-González, C Martínez-Murillo.

Revista de Hematología Vol. 7, No. 1, 2006
23
hemorrágicos con períodos asintomáticos, lo quedificulta el diagnóstico de la enfermedad. Lossíntomas son más intensos en los niños yadolescentes. Además, gran variación en lafrecuencia y severidad de la enfermedad existendentro de las familias afectadas. La expresiónclínica de la EvW usualmente es leve en el tipoI y la severidad aumenta en los tipos 2 y 3. Engeneral , la sever idad de la hemorragiacorrelaciona con el grado de reducción delFVIII:C pero no con el TH.
La epistaxis el principal síntoma en estospacientes, con una frecuencia del 60%; lasmetrorragias constituyen el principal síntoma enlas mujeres adolescentes, cuya frecuencia puedealcanzar cifras hasta del 75% (cuadro 1). Losniños frecuentemente presentan equimosis deaparición espontánea, que sugiere la posibilidadde EvW. Por otra parte, la EvW puede serdiagnosticada después de un procedimientoquirúrgico con hemorragia transoperatoria ypostoperatoria, particularmente después deextracciones dentales o amigdalectomía.Usualmente, el factor VIII se encuentradiscretamente disminuido, por lo tanto, lasmanifestaciones hemorrágicas por alteraciones enla hemostasia secundaria son poco frecuentes en laEvW, excepto en el tipo 3, en donde el factor VIIIse encuentra muy reducido y los pacientes puedentener hematomas y hemartrosis, semejante a lospacientes con hemofilia.
La hemorragia después del parto es rara enlos pacientes con EvW tipo 1, en el cual losniveles del FVIII/FvW son casi normales ygeneralmente se encuentran normales al final delembarazo. En pocos casos, los niveles del FVIII/FvW no son normales durante el embarazo yestas mujeres requieren tratamiento profilácticocon desmopresina o concentrados de FVIII/FvWantes del parto. Las pacientes con EvW tipo 2A,2B y 3 usualmente requieren tratamiento conterapia de reemplazo post-parto.
Cuadro 1Datos clínicos más frecuentes en los enfer-
mos con EvW.Datos Clínicos Frecuencia de
Presentación
Epistaxis 60%Hemorragia transvaginal 50%Hemorragia Post-extrac- 50% ción dentalEquimosis 40%Gingivorragias 35%Hemorragia Post-parto 20%Hemorragia 10% GastrointestinalHematuria 5%Hematomas* 5%Hemartrosis* 3%�/40%�
*Estos defectos ocurren más frecuentemente en eltipo 3. o en la EvW tipo Normandy. �se refiere al tipo 1 y algunos subtipos 2. �se presentan en tres estudios de pacientes con EvWtipo 3.
DIAGNÓSTICO.Los pacientes con EvW manifiestan
síntomas hemorrágicos que son típicos dedefectos de hemostasia primaria. La enfermedaddebe sospecharse en cualquier paciente conhistoria de hemorragia mucocutánea (epistaxis,metrorragias, gingivorragias, etc.) y postoperatoria,especialmente sí la historia familiar sugiere unpatrón de herencia autosómica. Los pacientescon EvW tipo 3 presentan hemorragias quesemejan la hemofilia: hemartrosis, hemorragiasmusculares, etc. (defectos de hemostasiasecundaria).
La interpretación de los valores delaboratorio del FvW es frecuentemente difícil,dado que el diagnóstico se establece con laimagen global de todas las pruebas dehemostasia. Por regla general, no hay un valorde corte aceptado en donde el paciente pueda
Enfermedad de von Willebrand.

24
Revista de Hematología Vol. 7, No. 1, 2006
ser clasificado como EvW en forma definitiva.Existen además variaciones importantes de losniveles del FvW plasmático en el mismopaciente, variables como el ejercicio, eltabaquismo, enfermedad subyacente, fármacos(ejemplo los anticonceptivos orales) y elembarazo pueden modificar los niveles del FvW;el grupo sanguíneo ABO y otros antígenos fueradel sistema ABO como el Lewis. Debido a lavariabilidad biológica de la EvW, el diagnósticoresulta difícil y únicamente logra establecersedespués de varias determinaciones de laspruebas de hemostasia. Por lo tanto, con lavariabilidad del FvW un solo valor normal noexcluye la EvW en el paciente sintomático. Aligual, valores anormales deben confirmarse yrepetir las pruebas posteriormente (14).
Pruebas de Escrutinio. En el cuadro 2 seCuadro 2
Pruebas de laboratorio de la EvW.Pruebas de Escrutinio Pruebas para establecer el tipo de EvWTiempo de hemorragia Agregación plaquetaria inducida por ristocetina (RIPA)TTPa Pruebas de unión al FvW (colágena y FVIII)FvW:RiCof Multímeros del FvWFvW:Ag Pruebas de FvW plaquetarioFVIII:C Análisis de DNAAnalizador de la función plaquetaria (PFA)Grupo sanguíneo ABO
Cuadro 3Nomenclatura del Complejo del Factor VIII/FvW propuesta por la International Society
of Trombosis and Hemostasis.Factor VIII
Proteína VIIIAntígeno VIII:AgFunción VIII:C
Factor de von WillebrandProteína Madura FvWAntígeno FvW:AgActividad Cofactor de Ristocetina FvW:RCoCapacidad de Unión a la Colágena FvW:CBCapacidad de Unión al Factor VIII FvW:FVIIIB
describen las pruebas de laboratorio empleadaspara los pacientes con sospecha de EvW. Elcuadro 3 señala la nomenclatura del complejoFVIII/FvW de acuerdo a las recomendacionesde la Sociedad Internacional de Hemostasia yTrombosis ( In ternat ional Socie ty o fThrombosis and Hemostasis). En las pruebasde escrutinio la cuenta de plaquetas (CP) esusualmente normal, la trombocitopenia levepuede ocurrir en pacientes con tipo 2B. El (TH)usualmente esta prolongado, pero puede estarnormal en pacientes con formas leves de laenfermedad como ocurre en el tipo 1. El tiempode protrombina (TP) es normal y el tiempo detromboplastina parcial activado (TTPa) puedeestar prolongado de acuerdo a la concentracióndel FVIII.
El FvW: Antigénico (FvW:Ag) y el cofactorde ristocetina (FvW:RiCof) son las pruebas
S Quintana-González, C Martínez-Murillo.

Revista de Hematología Vol. 7, No. 1, 2006
25
básicas para la EvW. Estudios adicionales comola agregación plaquetar ia inducida porRistocetina (RIPA) y el estudio de los multímerospermiten caracterizar a la EvW para untratamiento apropiado (cuadro 4).
De acuerdo a la caracterización del la EvW,tenemos las siguientes variedades:
Tipo 1: Es la forma más común (70% de loscasos) que se caracteriza por una disminucióncuantitativa del FvW el cual es funcionalmentenormal y representa un grupo muy heterogéneode enfermedades. La mayoría de los tipos I nose logra explicar su defecto molecular.Clásicamente, el tipo I se hereda en formaautosómica dominante, pero existen algunasexcepciones (18). La EvW tipo 1 se caracterizapor hemorragias leves a moderadas, TH normalo discretamente prolongado y niveles bajos deFvW:Ag, FvW:RiCof y FVIII, con multímerospresentes. Se tienen muchas dificultades paraestablecer los criterios diagnósticos estrictos enesta enfermedad. Un diagnóstico definitivorequiere niveles bajos del FvW en más de unaocasión (usando grupos sanguíneos ajustados alrango normal), historia de hemorragia e historiafamiliar positiva. Sin uno de los dos últimoscriterios el diagnóstico debe considerarse como�probable� (19). Los valores bajos del FvW:Agy FvW:RiCof son difíciles de evaluar, porqueentre otros factores los niveles dependen delgrupo ABO y el nivel de FvW:Ag esta disminuídoaproximadamente en un 25% en personas con
Cuadro 4Hallazgos de laboratorio en los tipos de la EvW.
Tipo FVIII FvW:Ag FvW:Rcof RIPA Patrón Multimérico (plasma)
1 ↓ ↓ ↓ ↓ o normal Todos los tamaños presentes2A ↓ ↓ ↓↓ ↓ Ausencia de multímeros de tamaño
intermedio y grandes2B ↓ o normal Normal o ↓ ↓↓ ↑ Ausencia de grandes multímeros2M ↓ o normal ↓ ↓↓ ↓ o normal Todos los tamaños presentes2N ↓↓ Normal Normal Normal Todos los tamaños presentes3 ↓↓ No detectado No detectado ↓↓ Ausencia total del FvW
grupo sanguíneo �0� comparado con los otrosgrupos . En es tos casos los pac ientescompatibles con el tipo 1 son consideradoscuando los niveles de FvW:Ag y FvW:RiCof seencuentran 2DS más abajo y ajustarlo deacuerdo al grupo sanguíneo.
Tipo 2: Se refiere a deficiencias cualitativas delfactor de von Willebrand. No existen datos so-bre la incidencia correcta de esta enfermedad,sin embargo, se estima que de todos los tiposde EvW del 20-30% pertenecen al tipo 2. Eltipo 2 es muy heterogéneo e incluye a 4 subti-pos; 2A, 2B, 2M y 2N.
2A.- Se hereda en forma autosómica dominante.Las mutaciones se presentan en el dominio A2que interfiere con el ensamblaje y el transporteintracelular de los grandes multímeros. Estospacientes son identificados por niveles bajos onormales del FvW:Ag y marcadamentedisminuídos los niveles de FvW:RiCof, con unpatrón multimérico anormal, caracterizado porpérdida de los multímeros de alto peso moleculary un aumento en la intensidad de los multímerosde ba jo peso molecular. El s i t io demultimerización se localiza actualmente en losdominios D3-A1. El mecanismo detallado de lasmutaciones A2 permanece sin explicación. Otrasmutaciones localizadas en el dominio A2 seasocian con una elevada sensibilización de losmultímeros a la proteólisis en la circulación.Otros pacientes presentan un tipo recesivo de
Enfermedad de von Willebrand.

26
Revista de Hematología Vol. 7, No. 1, 2006
la enfermedad, con mutaciones en el dominio D2,que son compatibles con el papel propuesto delpropéptido en la unión del puente de disulfuro,el cual es necesario para el proceso demultimerización (20).2B. � Se caracteriza por un aumento de laafinidad del FvW por la GPIb de las plaquetas.Se detecta por la agregación plaquetaria a bajasconcentraciones de ristocetina. Al igual que otrossubtipos de la EvW, también es muy heterogéneaen los niveles de FvW:Ag. El patrón multiméricose reporta con deficiencia de los multímeros de altopeso molecular y algunas veces trombocitopenia.Las mutaciones están localizadas en el dominioA1, la mayoría en la región N-terminal del asade unión del puente disulfuro. Se hereda enforma autosómica dominante.
2M (Multímero).- La unión a plaquetas se en-cuentra afectada, pero el patrón multimérico esnormal. Las mutaciones que se observan en estesubtipo están localizadas en la región del exón28 igual que en el tipo 2B, las mutaciones eneste subtipo inactivan el sitio de unión para launión a plaquetas o colágena. Los resultados delaboratorio son similares al subtipo 2A, pero elpatrón multimérico las diferencia
2N (Normandy).- En este subtipo existe unadisminución de la afinidad por el factor VIII,todas las mutaciones se localizan en la regiónN-terminal de la subunidad madura la cual con-tiene el sitio de unión del FVIII, en el dominioD�, aunque algunos casos son encontrados en eldominio D3. La enfermedad se hereda en formarecesiva. La función plaquetaria se encuentranormal, los niveles de FvW:Ag y FvW:RiCof sonnormales, la estructura multimérica es normal,pero los niveles de FVIII se encuentran dismi-nuídos. La hemorragia en estos pacientes es cau-sada principalmente por la disminución delFVIII:C y debe de diferenciarse de la hemofiliaclásica leve.
Tipo 3: La EvW tipo 3 es la variedad que ori-ginalmente informó en 1926 Erick von Wille-brand y se define como la ausencia de FvW:Agcirculante, niveles disminuidos de FVIII:C (1-5%), se hereda en forma autosómica recesiva yes la forma más severa de la EvW. La prevalen-cia se estima en 1:1,000 000 de sujetos. Lashemorragias son caracterizadas no sólo por he-morragia mucocutánea sino también por hemar-trosis y hematomas, como las que se observanen pacientes con hemofilia. Las mutaciones sehan encontrado en el exón 18. Algunos casosdel Tipo 3 resultan de deleciones completas oparciales del gene del FvW. Estos pacientes tie-nen predisposición para desarrollar aloanticuer-pos (5-8%) por la presencia de deleciones, porlo tanto, es importante evaluar el riesgo del de-sarrollo de inhibidores (21).
En México, se llevo a cabo un estudio conla finalidad de confirmar el diagnóstico yclasificar a pacientes con sospecha de EvWmediante el análisis del patrón multimérico.Estudiaron un total de 30 pacientes de los cuales19 tuvieron tipo 1, 8 del tipo 2 y 3 la variedadtipo 3 (22).
Muchas de las mutaciones, las cuales causandiferentes formas de EvW, se han identificado ycorrelacionan sus efectos sobre la estructura yfunción de l FvW. Por o t ro lado , o t rasenfermedades pueden estar relacionadas adefectos cuantitativos o cualitativos en el FvW,como la EvW adquirida (23) y la púrpuratrombocitopénica trombótica recurrente (24). ElFvW se ha asociado también con la trombosisarterial. Además es un marcador plasmático dela activación endotelial en algunas enfermedadesvasculares crónicas, como las angiopatías en lospacientes con diabetes mellitus (25).
TRATAMIENTO.El objetivo del tratamiento en la EvW es
corregir los defectos de la hemostasia. Corregirlas anormalidades en la hemostasia primaria
S Quintana-González, C Martínez-Murillo.

Revista de Hematología Vol. 7, No. 1, 2006
27
(adhesión y agregación plaquetaria) y losdefectos de la hemostasia secundaria. Eltratamiento debe cohibir la hemorragia oprevenirla en caso de un procedimientoquirúrgico. A diferencia de la hemofilia, laprofilaxis regularmente no se utiliza en lospacientes con EvW, porque usualmente lashemorragias son menos severas. Sin embargo,en los pacientes con EvW tipo 3 tienenhemorragias más graves y presentan hemartrosisrecurrentes, lo que ocasiona, al igual que lahemofilia artropatías. Por lo tanto, puede estarindicado en estos pacientes la profilaxis.
La elección del tratamiento depende delsubtipo de la EvW y la naturaleza de la diátesishemorrágica (cuadro 5). A pesar de la altaprevalencia de la EvW, existen pocos estudiosbien controlados sobre la duración e intensidaddel tratamiento. Los niveles de FVIII deben tenerun nivel hemostático adecuado de 30 UI/dL y elobjetivo principal es corregir los defectos de lahemostasia primaria. La corrección del tiempode sangrado y el incremento de los niveles deFvW:RiCof a 50 UI/dL son los parámetros másimportantes. En el caso de la EvW tipo 3, en lacual el comportamiento es semejante a lahemofilia y tienen hemorragia por defectos dehemostasia secundaria, los niveles del FVIIIdebe estar entre 30-50 UI/dL, dependiendo delsitio de la hemorragia. Hay dos tratamientos deelección en la EvW: la desmopresina (DDAVP)
y la terapia transfusional con productossanguíneos. Entre los tratamientos adyuvantesestán los inhibidores de la fibrinolisis, laspreparaciones de estrógenos-progestágenosorales y las fibrinas adhesivas.
Desmopresina (1-Deamino -8-D-Argininavasopresina).
La desmopresina es un derivado sintéticode la hormona antidiurética, originalmentedescubierto para el tratamiento de diabetesinsípida. La desmopresina (DDAVP) es unagonista selectivo para el receptor V2 (V2R).Es probable que la desmopresina actúe sobreuna célula intermedia que libera una hormonaliberadora del FvW, la cual más tarde actúasobre la célula endotelial. En pacientes conhemofilia leve y en algunos pacientes conenfermedad de von Willebrand, la desmopresinaincrementa de manera transitoria los nivelesplasmáticos del Factor VIII y FvW de loscuerpos de Weibel-Palade en las célulasendoteliales. También libera el Factor deplasminógeno tisular (t-PA) e interleucina-8 (IL-8) (cuadro 6).
Las ventajas de utilizar desmopresina es sucosto relat ivamente bajo, con i l imitadadisponibilidad y al ser un medicamento sintéticono transmite enfermedades infecciosas. Ladesmopresina es administrada en niños y adultosa dosis de 0.3 microgramos/Kg de peso, en 20-
Cuadro 5Medidas terapéuticas en la EvW.
Tipo de EvW Tratamiento de Elección Tratamiento Secundario
1 Desmopresina (DDAVP) Concentrado de FVIII-FvWCrioprecipitados
2A, 2M Concentrado de FVIII-FvW CrioprecipitadosDesmopresina (DDAVP)?????
2B Concentrado de FVIII-FvW Crioprecipitados2N Concentrado de FVIII-FvW Desmopresina (DDAVP)3 Concentrados de FVIII-FvW Crioprecipitados
Transfusión de plaquetas
Enfermedad de von Willebrand.

28
Revista de Hematología Vol. 7, No. 1, 2006
30 ml de solución fisiológica, en infusióncontinua, durante 30 minutos por vía intravenosa,en promedio aumentará el Factor VIII y el Factorde von Willebrand de 3-5 veces de lasconcentraciones basales de estos factores, en unlapso de 30-60 minutos. La desmopresina tambiénpuede administrarse por via subcutánea a la mismadosis que la intravenosa y por inhalación nasal(cuadro 7). La administración subcutánea ointranasal son convenientes para tratamientoprofiláctico y tratamiento en casa. La administraciónoral no ha sido evaluada para uso en pacientes conEvW.
Para emplear la desmopresina es muyimportante realizar previamente la prueba a ladesmopresina antes de administrarla para usoterapéutico debido a que hay pacientes que noresponden al tratamiento. Una vez que el pacientese ha comprado la respuesta al tratamiento, estarespuesta generalmente será consistente. La pruebaterapéutica a la desmopresina se debe administrara la misma dosis anteriormente señalada. Se obtieneun valor basal de los niveles de Factor VIII yFvW:RiCof o la prueba de unión a la c. Una vezque el paciente se ha comprado la respuesta altratamiento, esta respuesta generalmente seráconsistente. Se administra la desmopresina y 30-60 minutos después se determinan los factores, paraconocer los niveles máximos de los factores y a las4 horas para obtener la vida media. En caso deque los niveles de Factor VIII y FvW se encuentren
Cuadro 6Efecto farmacológico de la desmopresina (DDAVP).
Incremento del Factor VIIIIncremento del Factor de von Willebrand (FvW)Acorta el Tiempo de HemorragiaMejora la adhesión y agregación plaquetariaIncrementa la GpIb-IX (¿?)Incrementa t-PAIncrementa la IL-8Efectos no conocidos
entre el 10-20% de la actividad son suficientes paracubrir hemorragias leves a moderadas. Sinembargo, se requieren niveles de 30-50% deactividad, para extracciones dentales, pero no paracirugía mayor. Es muy importante evaluar los nivelesde estos factores después de la administración dela desmopresina, para identificar en que tipo dehemorragia puede ser utilizada. La desmopresinapuede emplearse cada 12-24 horas, si es necesario.
S Quintana-González, C Martínez-Murillo.
Cuadro 7Dosis y vías de administración de la des-
mopresina.
Dosis: 0.3 microgramos/Kg peso/dosis en infusión continua durante 20-30 minutos IV o sc
300 microgramos vía Intranasal en adultos 150 microgramos vía intranasal en niños
Niveles de Factor:1.5 veces incrementa los niveles de factorVIII3-5 veces incrementa los niveles de factorde von Willebrand
Niveles máximos:30-60 minutos vía IV90-120 minutos vía intranasal o subcutánea
Vida Media:5-8 horas para factor VIII8-10 horas para FvW

Revista de Hematología Vol. 7, No. 1, 2006
29
No se recomienda administrarla por más de tresdías, por la liberación del t-PA.
La respuesta a la desmopresina varía depen-diendo del tipo de EvW y no en todos los pacien-tes con EvW esta indicado (cuadro 6). Los pa-cientes con EvW tipo 1, en la cual el factor de vonWillebrand es funcionalmente normal, tienen mejorrespuesta que los pacientes con EvW tipo 2. Nose recomienda en el tipo 2A porque habrá un in-cremento disfuncional del FvW y no es efectivo parahemostasia primaria, aunque existen excepciones.La desmopresina está contraindicada en el tipo 2B,porque produce trombocitopenia después de la ad-ministración del medicamento. Los pacientes conel tipo 2M tienen poca respuesta a la desmopresi-na, pero en la práctica debe de hacerse la pruebaterapéutica para decidir el tratamiento. En la EvWtipo 2N, existe aumento de los niveles del FactorVIII, pero la respuesta es a corto tiempo por eldefecto en la unión con el FvW, por lo que no serecomienda como primera opción terapéutica. Lospacientes con EvW tipo 3 no tienen respuesta a ladesmopresina, porque carecen de sitios de alma-cenamiento del FvW.
Los efectos adversos de la desmopresina sonsecundarios a la vasodilatación cutánea, como lacefalea, enrojecimiento facial y tinitus; por lo generalson leves. La presencia de hiponatremia porretención hídrica, causada por el efecto antidiuréticode la desmopresina, es raro pero puede presentarsecon la ingesta abundante de líquidos, por lo que serecomienda, evitar la ingesta excesiva de líquidos.El mayor problema de retención hídrica ehiponatremia con presencia de crisis convulsivas seobserva en niños pequeños por lo que se debe deevitar el medicamento en niños menores de dos añosde edad. No debe administrarse en pacientes conenfermedad arterial coronaria, porque al liberarselos grandes multímeros del FvW puede aumentarla agregación plaquetaria y puede causar infartoagudo del miocardio.
Concentrados de factor VIII.Los pacientes con EvW que no pueden ser
tratados con DDAVP requieren la sustitución deambos factores: factor VIII y FvW. No todos losconcentrados de Factor VIII contienen FvW. Entrelos concentrados de Factor VIII que contienen FvWse encuentra el Humate-P, el cual se ha evaluadoampliamente en estudios clínicos. Contiene grandescantidades de FvW, el concentrado está inactivadoviralmente por medio de pasteurización. Otroconcentrado de factor VIII que contiene grandescantidades de FvW se encuentra el Alfanate, el cuales inactivado viralmente con solvente/detergente yaltas temperaturas. El Inmunate también contienefactor VIII/FvW, derivado del plasma humano, esun concentrado de alta pureza con dobleinactivación viral (tratamiento inicial con polisorbato80 seguido de calentamiento con vapor durante 10horas a 60ºC), el cual se ha demostrado su eficaciaen el tratamiento de los pacientes con EvW26.Existen otros factores de factor VIII con FvW queaunque no existen grandes estudios clínicos, sonefectivos clínicamente, entre ellos se encuentran elOctanate y Fandhi.
Los concentrados monoclonales no contienenFvW al igual que el Factor VIII recombinante, loscuales no deben de emplearse en los pacientes conEvW, porque carecen del FvW.
La dosis de los concentrados de factor VIII/FvW depende del sitio de hemorragia. Serecomiendan en caso de hemorragias leves de 15 a20 UI/Kg. Dependiendo de la severidad de lahemorragia se puede administrar cada 12 horas.En caso de extracción dental se recomienda unadosis de 20-30 UI/Kg. En cirugía de 40-50 UI/Kgdiariamente; en cirugía mayor de 5-10 días detratamiento y en cirugía menor cada tercer díausualmente, 2-4 días.
Crioprecipitados.Los crioprecipitados contienen factor VIII,
factor de von Willebrand, fibrinógeno, factor XIIIy fibronectina. Los crioprecipitados son útiles enlos pacientes con EvW porque contienen laglucoproteína multimérica del FvW. Sin embargo,en la actualidad no se recomiendan como primera
Enfermedad de von Willebrand.

30
Revista de Hematología Vol. 7, No. 1, 2006
elección de tratamiento, porque no se inactivanviralmente y pueden transmitir infecciones. Lascomplicaciones del uso del crioprecipitado son losmismos riesgos de transmisión de enfermedadesque el plasma fresco congelado. Pacientes quereciben grandes cantidades de crioprecipitadospara hemofilia A y por largos periodos de tiempo(ejemplo procedimientos quirúrgicos) han tenidoparadójicamente hemorragia a pesar de adecuadosniveles de FVIII:C y niveles adecuados de FvW.Esta hemorragia está relacionada a altos niveles defibrinógeno, que producen altas cantidades deproductos de degradación de fibrina y alargamientodel TM.
Antifibrinolíticos.Estos productos inhiben la activación del
plasminógeno y la actividad de la plasmina, por lotanto, previenen la lisis del coágulo. Los pacientescon enfermedad de von Willebrand frecuentementepresentan sangrado transvaginal y epistaxis, estose debe a la gran actividad fibrinolítica que sepresenta en estas mucosas. Los pacientes que sesometen a extracciones dentales tienen granactividad fibrinolítica local, que puede incrementarla presencia de hemorragia en estos pacientes.
Los antifibrinolíticos se pueden administrar en for-ma sistémica o local, entre ellos se encuentran al áci-do aminocaproico (Amicar) el cual se indica a dosisde 50-60 mg/Kg c/6 h, el ácido tranexámico (10-15mg/Kg/8 h), puede administrarse VO, IV o tópica.En caso de hemorragias leves pueden usarse solos ocoadyuvantes al tratamiento con concentrados de fac-tor VIII/FvW o desmopresina. Están contraindica-dos en pacientes con hematuria ya que al no lisar elcoágulo éste puede obstruir el tracto urinario.
Estrógenos/progestágenos.La presencia de menorragias es frecuente en
los pacientes con EvW, lo que causa anemia pordeficiencia en hierro. En las pacientes con EvW tipo3, la administración de estrógenos/progestágenosreduce hasta en el 88% la pérdida de sangre. Elmecanismo de acción de estos medicamentos es
hacer menos susceptible al endometrio de sangrar.
Tratamiento en el embarazo.En mujeres con FvW tipo I, los niveles de
factor VIII y FvW aumentan espontáneamentedurante el embarazo y los niveles de estos factoresson normales al termino del embarazo (27). El niveldel Factor VIII es el mejor predictor de hemorragiadurante y después del embarazo. Por lo tanto, serecomienda medirlo al término del embarazo y dossemanas después, cuando los niveles del factor VIIIdisminuyen rápidamente y puede ocurrir hemorragia(28). El riesgo de hemorragia después del partovaginal o cesárea es mínimo cuando los niveles defactor VIII se encuentran entre 30-40% de losniveles normales (28). En las pacientes con EvWtipo 3, no es necesario monitorear los niveles defactor VIII, porque estos no cambian durante elembarazo y la administración diaria de concentradosde Factor VIII/FvW deben ser administrados paraevitar hemorragia (29). La dosis que se recomiendaes 40 UI/Kg/día antes y de 3-4 días post-parto ytener niveles de factor VIII >50% del nivel normal(29).
Tratamiento de pacientes con aloanticuerpos anti-factor de von Willebrand.
Pacientes con EvW tipo 3 pueden presentar alo-anticuerpos contra el FvW después de múltiplestransfusiones que contienen FvW (30). Estosanticuerpos se presentan con una frecuencia del 10-15% de estos enfermos (31). La infusión de losconcentrados de FvW son inefectivos y pueden causaranafilaxia post-transfusión debido a la formación decomplejos inmunes circulantes. Los concentrados quecontienen FvW están contraindicados después de estacomplicación porque la presencia del FvW pone enriesgo la vida del paciente por ocasionar reaccionesanafilácticas, por la activación del complemento y porla formación de los complejos inmunes (32). Paracontrol de las hemorragias en los pacientes conaloanticuerpos, puede usarse concentrados de factorVIII recombinante, que no tienen el FvW. Sinembargo, la vida media de este factor transfundido es
S Quintana-González, C Martínez-Murillo.

Revista de Hematología Vol. 7, No. 1, 2006
31
muy corta (1-2 h) porque carece del FvW y, por lotanto, se degrada rapadamente el FVIII transfundido.La recomendación es administrar el tratamiento eninfusión continua, a grandes dosis, con el objetivode mantener niveles hemostáticos del Factor VIII ycontrolar los fenómenos hemorrágicos (33). Existenestudios del uso del factor VIIa recombinante en estospacientes y la dosis recomendada es de 90microgramos/Kg cada 2 h o 20 microgramos/Kg cadahora, en infusión continua (34).
Enfermedad de von Willebrand adquirida (EvWa).La primera descripción de la forma adquirida
de la EvW fue en 1968 (35). La EvWa es unaenfermedad hemorrágica poco común, conhallazgos clínicos y de laboratorio similares a laforma hereditaria de la enfermedad. Las primerasdescripciones de pacientes con EvWa erancausadas por padecimientos inmunológicos (36). Enlos siguientes años, diferentes enfermedades clínicasprodujeron disminución en los niveles del FvW queprovocaban enfermedad hemorrágica. En lospacientes con EvWa el FvW es sintetizado encantidades normales o sintetizarse en mayor cantidady liberado normalmente a la circulación sanguínea. Losmecanismos propuestos en el desarrollo de la EvWason la presencia de autoanticuerpos que formancomplejos inmunes circulantes, lo que provoca ladestrucción del FvW circulante. Otro mecanismo esla adsorción del FvW sobre las células neoplásicas uotras superficies celulares, aumento en la degradaciónproteolítica del FvW (cuadro 8). Las enfermedadesfrecuentemente asociadas con la EvWa son lasenfermedades linfoproliferativas, autoinmunes oneoplasias.
El tratamiento principal de los pacientes conEvWa es erradicar la causa subyacente. Otras opcio-nes de tratamiento son la administración de DDAVP,concentrados de factor, globulina inmune intravenosa(37).
CONCLUSIONES.Han existido avances importantes en el
diagnostico y tratamiento de los pacientes con EvW.En México es necesario contar con laboratorios dereferencia para completar y diagnosticar los subtiposde la EvW, ya que las opciones terapéuticasanteriormente descritas están disponibles.Actualmente, se encuentra en estudios la preparaciónrecombinante del FvW. La interleucina-11, unacitosina que se ha observado en animales y humanos,incrementar el factor VIII y el FvW, la cual puede seruna nueva forma de tratamiento en el futuro para lospacientes con EvW.
Cuadro 8Mecanismos patogenéticos en la EvWa.
Autoanticuerpos específicos o no específicos que for-man complejos inmunes circulantes e incrementan ladestrucción del FvW:
Enfermedades linfoproliferativasEnfermedades neoplásicasEnfermedades inmunológicas
Adsorción del FvW sobre las clonas de las célulasneoplásicas u otras superficies celulares:
Enfermedades linfoproliferativasEnfermedades neoplásicasEnfermedades mieloproliferativasIncremento del shear stress
Incremento de la degradación proteolítica del FvW:Específico:Enfermedades mieloproliferativasIncremento del shear stressUremiaCiprofloxacinaNo-específico (plasmina):Hiperfibrinolisis primariaHiperfibrinolisis secundariaLisis terapéuticaIncremento Shear Stress:Alteraciones cardiacas congénitasEstenosis aórticaEndocarditisMalformación vascular:
- Telangiectasia Hemorrágica Hereditaria
- Síndrome de Kasabach-MerritAteroesclerosis severaβ-talasemiaSíntesis disminuida:HipotiroidismoDesconocido:Acido valproicoEnfermedad viralTrasplante de hígado
Enfermedad de von Willebrand.

32
Revista de Hematología Vol. 7, No. 1, 2006
REFERENCIAS.1.- Von Willebrand EA. Hereditär pseudohemofili.Finska Läkarsällskapetes Handl 1926; 68:87-112.
2.- Nilsson IM, Blombäck M, Jorpes E, Blombäck B,Johansson SA. Von Willebrand�s disease and itscorrection with human plasma fraction 1-0. Acta MedScand 1957; 159:179-88.
3.- Holmberg L, Nilsson IM. Genetic variants of vonWillebrand�s disease. Br Med J 1972; 3:317-20.
4.- Hoyer LW, Shainoff JR,. Factor VIII-related proteincirculates in normal human plasma as high molecularweight multimers. Blood 1980, 55;1056-59.
5.- Ruggeri ZM, Pareti FI, Manucci PM. Heightenedinteraction between platelets and factor VIII/vonWillebrand factor in a new subtype of von Willebrand�sdisease. N Engl J Med 1980, 302;1047-51.
6.- Sadler JE. A revised classification of von Willebranddisease. Thromb Haemost 1994; 71:520-25.
7.- Rodeghiero F., Castaman C., Dini E. Epidemiologicalinvestigation of the prevalence of von Willebrand�sdisease. Blood; 69:454-459. 1987.
8 . - Ni l s son IM. , Borge L . , Gunnarsson M. ,Kristoffersson AC. Factor VIII related activities inconcentrates. Scand J Haematol; 33, Suppl 41: 157-172.1984.
9.- Mannucci PM, Bloom AL, Larrieu MJ, Nilsson IM,West RR. Artherosclerosis and von Willebrand factor.Prevalence of severe von Willebrand�s disease inwestern Europe and Israel. Br J Haematol 1984; 57:163-9.
10.- Berliner SA, Seligson U., Zivelin A., Zwang E.,Sofferman G. A relatively high frecuency of severe type3 von Willebrand´s disease in Israel. Br J Haematol;1:20-34. 1986.
11. - J iménez C. Es tudio epidemiológico de laenfermedad de von Willebrand en escolares en CostaRica. Memorias de las XVI Jornadas Anuales de laAgrupac ión Mexicana para e l Es tud io de laHematología. Puebla, México, 1995.
12.- De Groot PG, Ottenhof-Rovers M, van Mourik JA,Sixma JJ. Evidence that primary binding site of von
Willebrand factor that mediates platelet adhesion onsubendothelium is not collagen. J Clin Invest 1988;82: 65-73.
13.- Ruggeri ZM , Ware J. The structure and functionof von Willebrand factor. Thromb Haemost 1992; 67:594-599.
14.- Blömback M, Erenoth P, Andersson O, Anvret M.On laboratory problems in diagnosing mild vonWillebrand disease. Am J Hematol 1992; 40:117-20.
15.- Cattaneo M, Federici AB, Lecchi A, Agati B,Lombardi R, Stabile F, et al. Evaluation of the PFA-100system in the diagnosis and therapeutic monitoring ofpatients with Willebrand disease. Thromb Haemost1999; 82: 35-39.
16.- Manucci PM, Lombardi R, Bader R. Heterogeneityof type 1 von Willebrand disease: evidence for asubgroup with abnormal von Willebrand factor. Blood1985; 66:796-802.
17.- Gralnick HR, Rick ME, McKeown LP. Platelet vonWillebrand factor. An important determinant of thebleeding time in type I von Willebrand disease. Blood1986; 68:58-61.
18.- Sadler JE, Matsushita T, Dong Z, Tuley EA,Westfield LA. Molecular mechanism and classificationof von Willebrand disease. Thromb Haemost 1995; 74:161-6.
19.- Battle J, Torea J, Rendal E, Fernández MFL. Theproblem of diagnosing von Willebrand disease. J InternMed 1997; 242: 121-8.
20.- Eikenboom JCJ, Reitsma PH, Peerlinck KMJ, BrietE. Reccesive inheritance of von Willebrand�s diseasetype 1. Lancet 1993; 341: 982-6.
21.- Federici AB and Mannucci PM. Diagnosis andmanagement of von Willebrand disease. Haemophilia1999; 5: 28-37
22.- Martínez-Murillo C, Viveros ME. Enfermedad devon Wi l lebrand En: Manua l de Hemostas ia yTrombosis. Martínez Murillo C, Quintana G, editores.México: Editorial Prado; 1996.
23.- Tefferi A, Nichols WL. Acquired von Willebranddisease: concise review of occurrence, diagnosis,
S Quintana-González, C Martínez-Murillo.

Revista de Hematología Vol. 7, No. 1, 2006
33
pathogenesis, and treatment. Am J Med 1997; 103:536-40.
24.- Furlan M, Robles R, Solenthaler M, Wassmer M,Sandoz P, Laemmie B. Deficient activity of vonWillebrand factor-cleaved protease in chronic relapsingthrombotic thrombocytopenic purpura. Blood 1997;89:3097-103.
25.- Stehouwer CD, Fischer HR, van Kuijk AW, PolakBC, Donker AJ. Endothelial dysfunction precedesdevelopment of microalbuminuria in IDDM. Diabetes1995; 44: 561-4.
26.- Auerswald G, Eberspâcher B, Engl W, Güthner C,Koksch M, Kreuz W, et al. Successful treatment ofpatients with von Wllebrand disease using a high-puri ty double-virus inact ivated factor VIII /vonWillebrand concentrate (Inmunate®). Sem ThrombHemost 2002; 28: 203-213.
27.- Noller KL, Bowie EJ, Kempers RD, Owen CA Jr.Von Willebrand's disease in pregnancy. Obstet Gynecol1973; 41:865-72.
28.- Conti M, Mari D, Conti E, Muggiasca ML,Mannucci PM. Pregnancy in women with differenttypes of von Willebrand disease. Obstet Gynecol 1986;68:282-5.
29.- Mannucci PM. Drug Therapy: Treatment of vonWillebrand�s Disease. N Engl J Med 2004; 351: 683-694
30.- Castaman G, Federici AB, Rodeghiero F, MannucciPM. Von Willebrand�s disease in the year 2003: towardsthe complete identification of gene defects for correctdiagnosis and treatment. Haematologica 2003; 88:94-108.
31.- Mannucci PM, Meyer D, Ruggeri ZM, Koutts J,Ciavarella N, Lavergne JM. Precipitating antibodies invon Willebrand's disease. Nature 1976; 262:141-2.
32.- Bergamaschini L, Mannucci PM, Federici AB,Coppola R, Guzzoni S, Agostoni A. Posttransfusionanaphylactic reaction in a patient with severe vonWil lebrand d i sease : ro le o f complement andalloantibodies to von Willebrand factor. J Lab Clin Med1995; 125:348-55.
33.- Ciavarella N, Schiavoni M, Valenzano E, ManginiF, Inchingolo F. Use of recombinant factor VIIa(NovoSeven) in the treatment of two patients with type
III von Willebrand's disease and an inhibitor againstvon Willebrand factor. Haemostasis 1996;26:Suppl1:150-4.
34.- Lak M, Peyvandi F, Mannucci PM. Clinicalmanifestations and complications of childbirth andreplacement therapy in 385 Iranian patients with type3 von Willebrand disease. Br J Haematol 2000;111:1236-9.
35.- Simone JV, Cornet JA, Abildgaard CF. Acquiredvon Wil lebrand�s Syndrome in sys temic lupuserythematosus. Blood 1968; 31: 806-811.
36.- Ingram GIC, Kingston PJ, Leslie J, Bowie EJW.Four cases of acquired von Willebrand�s syndrome.Br J Haematol 1971; 21: 189-199.
37.- Federici AB, Stabile F, Castaman G, Canciani MT,Mannucci PM. Treatment of acquired von Willebrandsyndrome in patients with monoclonal gammopathyof uncertain significance: comparison of three differenttherapeutic approaches. Blood 1998; 92:2707-11.
Enfermedad de von Willebrand.

Revista de Hematología Vol. 7, No. 1, 2006
35
Solicitud de reimpresos: Dr. Jaime García-Chávez, Pestalozzi No. 635 int. 7, Col. Narvarte, C.P. 03020, México, D.F., México. Tel: 55 23 83 48 E-mail: [email protected]
Inhibidores adquiridos de los factores de la coagulación.
Jaime García-Chávez, Lilia A. García-Stivalet.
Unidad Médica de Alta Especialidad, Hospital de Especialidades del Centro MédicoNacional �La Raza�, Instituto Mexicano del Seguro Social, México, D.F., México.
INTRODUCCIÓN.Los inhibidores adquiridos contra los factores
de la coagulación son una manifestación más deel fenómeno de autoinmunidad, proceso causal deuna morbilidad y mortalidad considerable.Generalmente se trata de anticuerpos dirigidoscontra ciertos determinantes antigénicos de losfactores de la coagulación específicos. Se han descritotres tipos de anticuerpos: autoanticuerpos,aloanticuerpos y xenoanticuerpos. Los aloanticuerposmás comunes son aquellos que surgen enpacientes con deficiencias hereditarias defactores de la coagulación que son tratados conproteínas recombinantes o nativas. Son distintosa los autoanticuerpos que surgen de formaespontánea en personas sin coagulopatíashereditarias. Los xenoanticuerpos se desarrollanen pacientes que se exponen a factores decoagulación de origen animal (1).
Los autoanticuerpos aparecen de formafrecuente en pacientes mayores y son atribuidosa una pérdida de la vigilancia inmunológica poranticuerpos anti-idiotípicos. Se pueden detectartítulos bajos de autoanticuerpos dirigidos contrael factor VIII en pacientes sanos. Generalmenteestán dirigidos contra el sitio activo del factos,
aunque también se han descrito los inhibidorescontra otros sitios, lo que condiciona unacortamiento notable de la vida media (2). Porotro lado, se pueden observar autoanticuerpos,llamados inhibidores espontáneos, responsablesde producir manifestaciones hemorrágicas yalteraciones en las pruebas de coagulación. Lospacientes afectados pueden sufr ir otrasenfermedades de naturaleza autoinmune comoel Lupus Eritematoso Sistémico (LES), aunquefrecuentemente se trata de pacientes previamentesanos. También se puede asociar a otrasenfermedades como cáncer, exposición aalgunas drogas como penicilina (inhibidorescontra FVIII y XII) estreptomicina o gentamicina(inhibidor contra factor V), isoniazida (inhibidorcontra factor XIII). Algunos anticuerpos seencuentran en el posparto inmediato (p.e.inhibidores contra FVIII y FIX). Las pruebasde laboratorio muestran niveles bajos de uno omás factores de la coagulación. El diagnósticose sospecha cuando al adicionar plasma delpaciente a plasma normal, éste sufre unalargamiento de los tiempos de coagulación (1).
Los autoant icuerpos que complicanenfermedades como LES, enfermedades

36
Revista de Hematología Vol. 7, No. 1, 2006
J García-Chávez, LA García-Stivalet.
malignas o secundarias a fármacos, remiten conel tratamiento efectivo de la enfermedad de base.En cambio los que se catalogan como idiopáticospueden desaparecer en forma espontáneamientras que otros persisten por años (1). Detodos los inhibidores contra factores de lacoagulación, aquellos que inactivan al factor VIIIson los más frecuentes.
INHIBIDORES CONTRA FACTOR VIII.
Epidemiología.Los anticuerpos adquiridos contra el factor
VIII son poco frecuentes, mientras la mayoríaaparecen en pacientes mayores sin antecedentesde enfermedades concomitantes. También seasocian a LES, asma, embarazo y enfermedadesneoplásicas. Se han descrito 40 casos deinhibidores contra factor VIII en asociación acáncer. Los más comunes son: leucemialinfocítica crónica, aunque se han descritoasociación con tumores sól idos y otrasenfermedades oncohematológicas (3).
Inmunología.Los inhibidores contra el factor VIII son
inmunoglobulinas del tipos IgG1 e IgG4 (4). Lamayoría son de cadena l igera kappa, ygeneralmente no se unen a complemento.Generalmente se dirigen contra el dominio A2 (enla cadena pesada), contra el dominio C2 (en lacadena ligera), o ambos en la molécula delFVIII . Ensayos de inmunoprecipi taciónmuestran que el 60% de los pacientes presentanmúltiples anticuerpos contra FVIII. Sin embargo,los estudios de neutralización mostraron que serequiere generalmente de un solo fragmento paraque se neutralice la actividad del inhibidor (1).
Los anticuerpos que reaccionan contra eldominio A2 (más específicamente la región entrelos aminoácidos 484 a 509), pueden interferircon la interacción entre esta subunidad del FVIIIy el factor IXa. Se mostró una correlacióninversa entre los niveles de Bethesda y la
concentración de anticuerpo requerida parainhibir la estimulación dependiente de A2 delfactor IXa (1). Los anticuerpos contra eldominio C2 pueden interferir con la unión delFVIII a la fosfatidilserina, contra el factor deVon Willebrand o ambos (5). La actividadanticoagulante de este inhibidor puede resultarde la interferencia con la membrana de unión delFVIIIa, que se requiere para su integración alcomplejo Xasa en formación (6).
Manifestaciones Clínicas.El síntoma de presentación más común es
hemorragias a nivel de piel y músculos. Sinembargo, durante el curso de la enfermedadpueden ocurrir hemorragias en cualquier sitio dela economía. Muchos pacientes presentanhemorragias a nivel de mucosas o membranas;puede existir epistaxis o gingivorragiasrecurrentes. Otros pueden presentar melena ohematoquezia desarrollando síndrome anémicosecundario. También se pueden manifestar comohematuria persistente, en especial asociado ainfección de vías urinarias o enfermedadprostática. Se pueden desarrollar hematomasmusculares en brazos y piernas secundarios atraumatismos leves con complicaciones comocompresión de nervios o compromiso de lairrigación arterial a las extremidades. Lashemartrosis son menos comunes, aunque puedenocurrir. En pacientes en el posparto inmediatola manifestación más importante es la hemorrgiatransvaginal persistente (cuadro 1).
La hemorragia retroperitoneal puede sermasiva y producir la muerte por choquehipovolémico. La hemorragia intracraneal espoco frecuente, pero con consecuenciasdevastadores. Las hemorragias mayores ocurrengeneralmente en más del 80% de los casos, conuna mortalidad aproximada de 20% (1). Enresumen se debe sospechar en un inhibidor deFVIII en pacientes mayores con hemorragiasespontáneas o hemorragia después de un traumamenor.

Revista de Hematología Vol. 7, No. 1, 2006
37Inhibidores adquiridos de la coagulación.
Cuadro 1Sitios frecuentes de hemorragia en
pacientes con inhibidores contrafactor VIII.
Sitio: %
Músculo 44Piel 13Vías urinarias 9Tubo digestivo 9Articulaciones 6Heridas quirúrgicas 6Orofaringe e hipofaringe 4Otors 9
Diagnóstico.El dato pivote en el diagnóstico de
inhibidores contra FVIII es un tiempo detromboplast ina parcial act ivada (TTPa)prolongado, sin alteraciones sobre el tiempode protrombina (TP). Este patrón también se
o b s e r v a e n p a c i e n t e s c o n h e m o f i l i a ,enfermedad de Von Willebrand o deficienciade factores de contacto. Sin embargo, sedistingue de estos realizando correcciones conp l a s m a n o r m a l . E n l o s p a c i e n t e s c o ndeficiencias, la adición de plasma normalcorrige el TTPa, pero cuando se adicionaplasma normal a un plasma que contieneinhibidor, el TTPa se prolonga. Esto se deberealizar inmediatamente después de la mezclay después de la incubación a 37°C por unahora. Este procedimiento permite que losinhibidores débiles que requieren de mástiempo para la incativación del FVIII seanreconocidos (1). Las no corrección con plasma normalproporcionan evidencia de que exis teninhibidores contra el FVIII, pero se disponede pruebas específicas para cada factor ytambién se puede cuantifivcar la potencia demismo por diferentes técnicas como la deBethseda y sucedáneas (figura 1) (1).
TTPa prolongado + TP normal + Cuadro clínico compatible
Figura 1.- Algoritmo para el diagnóstico de inhibidores contra FVIII.
ÿ ��� �ÿ�� �ÿ���� �� ����ÿ
��� ����� ��ÿ�� � �� ��ÿ��ÿ�� ���ÿÿ
�� ���ÿ��� �ÿ
�� ����ÿ��� ���� �ÿÿ
ÿ
���� ���� �� ��ÿ���ÿ�� �� � �ÿ�� ����ÿ

38
Revista de Hematología Vol. 7, No. 1, 2006
Tratamiento (figura 2).Tratamiento durante el evento hemorrágico.
Se cuenta con d i ferentes recursosterapéuticos, cada vez más efectivos, si bienhace apenas unos 5 años el pronóstico era muydistinto. El Factor VIII porcino, fue por muchotiempo el tratamiento de elección, pero ahoraya ha salido del (7). Los efectos adversos eranpoco frecuentes, los más comunes eran mialgiasy dolor lumbar, así como menos frecuentementeanaf i lax ia . La inc idencia repor tada dereacciones adversas es de 10 por 1000infusiones en pacientes que reciben menos de100UI/kg y 82 por 1000 infusiones en pacientesque reciben dosis más altas (1). Otro efectosecundario reportado con el FVIII porcino estrombocitopenia, la cual puede deberse aagregación plaquetaria ex vivo (1).
Cuando el tiempo lo permite se debenesperar los resultados de la prueba de Bethesda,basando el tratamiento en los niveles deinhibidor. Si encontramos un nivel menor a 5U.B., se debe administrar FVIII humanoiniciando con bolos de 100 UI/kg seguido deuna infusión continua de 10 UI/Kg/h hasta elcontrol de la hemorragia o bien que sea claroque el tratamiento no está siendo efectivo. Sedeben determinar concentraciones plasmáticasdel factor a las 4 a 6 h de iniciado el tratamientopara asegurar niveles plasmáticos mayores a 0.25UI/ml. En caso de persistir con hemorragia o conniveles bajos de FVIII, esto sugiere que lapotencia del inhibidor es mayor a la estimadapor la prueba de Bethesda o que el FVIII en elconcentrado es muy susceptible a la inactivaciónpor el inhibidor. El FVIII que se encuentra enconcentrados que tienen niveles altos de factorde von Willebrand (p.e. concentrados de purezaintermedia) pueden encontrarse protegidos dela inactivación por algunos inhibidores, quetienen una mayor especificidad por el dominioC2 del factor VIII (1). Por lo que en estos casosel tratamiento debe ser cambiado a este tipo deconcentrados.
En los casos en los que la hemorragia no secontrola con los concentrados de FVIII existendos alternativas: el factor VII activado humanoy recombinante (rFVIIa) y el concentrado decomplejos activados, que contienen protrombi-na, factor VII, IX y X. El rFVIIa ofrece variasventajas potenciales. Es efectivo iniciando laformación del coagulo cuando se une al factortisular, este complejo subsecuentemente activaal FIX y X. Sin embargo, a dosis terapéuticastambién se puede unir directamente con la fos-fatidilserina plaquetaria de forma independientedel factor tisular (8). Bajo estas circunstanciasse puede generar trombina en la superficie delas plaquetas activadas. Su efectividad hemos-tática es independiente del FVIII y por lo tantono se afecta por los inhibidores del mismo. Otraventaja es que no es un producto de la sangrehumana, por lo que el riesgo de transmisión deagentes infecciosos es bajo.
La experiencia clínica con el rFVIIa se ana-lizó por Glazer y col. (8) en el cual se incluye-ron 1270 episodios de hemorragia en 240 pa-cientes, 18 de estos pacientes con inhibidores.Se observaron respuestas efectivas en 74-100%de los pacientes dependiendo del tipo de hemo-rragia, cuando ésta era considerada como críti-ca la efectividad fue de 91%. Los efectos ad-versos fueron raros e incluyeron hipertensión,rash cutáneo, fiebre, cefalea y epistaxis. La do-sis recomendada fue de 90 µg/kg, tomando encuenta que la vida media del rFVIIa es de sola-mente 2.9 hrs, se debe repetir la dosis cada 2 a3 h por 1 a 2 días o bien hasta la mejoría clíni-ca. También se puede utilizar infusión continuadel rFVIIa a una dosis de 16.5-20 µg/kg/h sien-do segura y efectiva (9).
Los concent rados de l comple joprotrombinasa estan aprobados por la FDA parael tratamiento de pacientes con inhibidores delFVIII (1). Se pueden encontrar 2 productos:Autoplex y FEIBA. Los procoagulantesactivados presentes en Autoplex incluyen losfactores VIIa, IXa, Xa, XIa, y trombina.
J García-Chávez, LA García-Stivalet.

Revista de Hematología Vol. 7, No. 1, 2006
39
Mientras que FEIBA también contiene factorVIIa. La dosis utilizada de Autoplex es de 50U/Kg y de FEIBA 50-75 U/Kg, las dosis se repitencada 8 a 12 h. Con Autoplex se logranrespuestas buenas o excelentes en 87% enpacientes con hemofilia, siendo su uso enpacientes con autoanticuerpos anecdótico peroen general posi t ivo (1 ) . En un es tudioretrospectivo con FEIBA en el que se tratan 60pacientes, 6 de ellos con autoanticuerpos contrael FVIII, se encontraron respuestas favorablesen 81%, presentándose efectos adversos en 5pacientes (1). No se deben administrar agentesantifibrinolíticos en pacientes que recibenconcentrado de complejo protrombinasa. Estosconcentrados son preparados utilizando sangrehumana, por lo que potencialmente puedentransmitir agentes infecciosos.
Inmunomodulación.La administración de globulina hiperinmune
(IVIg) ha inducido disminución de los niveles deinhibidor en algunos pacientes. En un ensayomulticéntrico, 2 de 19 pacientes presentaron unadisminución de los niveles de inhibidor de formarápida, y 4 más tuvieron una respuesta gradualen el transcurso de algunos meses (10). La dosisutilizada fue de 1mg/kg/día por 2 días o 0.4g/Kg día por 5 días. Adicionalmente los pacientesrecibieron prednisolona 1mg/Kg/día por 2semanas con posterior disminución de la dosis.Cuatro pacientes presentaron respuestacompleta , con niveles indetectables deinhibidores al día 21 de tratamiento. Los efectosadversos son poco frecuentes e incluyen mareosy prurito más frecuentemente, aunque ha habidoreportes de insuficiencia renal aguda.
El efecto de la IVIg se atribuye a lapresencia de anticuerpos anti-idiotipo en el
Figura 2.- Algoritmo para el tratamiento de pacientes con inhibidores contra el FVIII.
ÿ������� �� �ÿÿ ÿ����ÿ �������� ���ÿ� � � µ �� ��ÿ��ÿ
���ÿ�� ��� �� ��ÿ
���ÿ���� ��ÿ�� ��ÿ��ÿ����ÿ � ��ÿ�� �� ��ÿ� ���ÿ��ÿ��� �ÿ
���� �ÿ�� � �� �ÿ ��� ��ÿ����� ��ÿ
���ÿ�� ��� �� ��ÿ
ÿ �� ���� ��ÿ��ÿ���� �ÿÿ ������� ����ÿ��ÿ������� �ÿ��� ������ ��ÿ���ÿ� ���� �� � �ÿ
ÿ ��� ���ÿÿ � ��� ��� ����� � � ÿ��� � ������ ��� � �ÿ
� ÿ�� �� �ÿ���ÿ���� �� ���ÿ� �� ��ÿ���� � ��ÿ� ����� ����ÿ� ��� ��ÿ� ���� �ÿ� ��ÿ� ÿ���� ���ÿ
Inhibidores adquiridos de la coagulación.

40
Revista de Hematología Vol. 7, No. 1, 2006
"pool" de inmunoglobulinas utilizado paraprepararla. Estos anticuerpos se dirigen contralos autoanticuerpos del paciente.
Otras opciones incluyen plasmaféresis oinmunoadsorción. La plasmaféresis por sí solano es efectiva debido a que se encuentrangrandes reservor ios extravasculares deautoanticuerpos, por lo que se deben cambiarpor lo menos 4 litros de plasma para tener unimpacto sobre los niveles de anticuerpos. Lainmunoabsorción implica el uso de columnasextracorpóreas que contienen proteínasadsorbentes de inmunoglobulinas unidas asefarosa, con reducciones de 76% de los nivelesde inhibidor posterior a sesiones de 4 h.
Tratamiento a largo plazo.En aproximadamente una tercera parte de
los pacientes, generalmente en aquellos sinenfermedades asociadas, los anticuerposdesaparecen de forma espontánea (1). Sinembargo, la pérdida del anticuerpo puede llevarmeses a años, durante los cuales el paciente seencuentra en riesgo de presentar hemorragiasgraves. Cuando la presencia del anticuerpo seasocia a una enfermedad subyacente, eltratamiento de la misma puede llevar a ladesaparición del anticuerpo. Sin embargo en lamayoría de los pacientes las enfermedadessubyacentes no pueden ser erradicadas (p.e.cáncer). Bajo estas circunstancias se debeintentar eliminar el inhibidor.
Cuando se el diagnóstico de inhibidor seconfirma se debe administrar prednisona 1mg/Kg/día. Una respuesta satisfactoria es ladisminución del nivel de inhibidor en 3 semanasa menos del 50% de su valor inicial. Si estoocurre se debe continuar el tratamiento hasta queel inhibidor desaparezca y se encuentren nivelesnormales de FVIII. Esto se observa en unatercera parte de los pacientes (1). Los pacientesque responden tienen un nivel menor de inhibidorque los pacientes que no responden (3 UBcontra 50 UB respectivamente) y niveles más
altos de FVIII (9% contra 1%). En los 2/3 depacientes que tienen persistencia del inhibidorse encuentra varias opciones:
1. Suspender prednisona e iniciar ciclofosfa-mida a 2mg/Kg/día por 3 a 6 semanas.2. Continuar prednisona y adicionar ciclofos-famida.3. Iniciar ciclosporina con o sin otro agente adosis de 5mg/Kg/día para lograr niveles plas-máticos de 150-350ng/ml4. Uso de quimioterapia de combinación conprednisona (p.e. Ciclofosfamida + vincristina).
Los inhibidores que aparecen en elposparto, casi todos los casos se resuelven enlos 30 meses posteriores, asociándose másfrecuentemente con primeros embarazos yprovocando hemorragias transvaginales u deotro t ipo inmediatamente posparto, contendencia a reaparecer en los siguientesembarazos no ha sido efectivo. El tratamientocon esteroides y ciclofosfamida, auque estetratamiento no ha sido del todo efectivo.
Otros tratamientos.Rituximab: El anticuerpo monoclonal anti
CD-20, rituximab, elimina de forma rápida lamayoría de las células B circulantes. El éxito enel tratamiento de los linfomas con rituximab loha vuelto atractivo como candidato para eltratamiento de las enfermedades benignas queinvolucran los linfocitos B. El concepto centralde esto es la remoción de la fuente celular delos anticuerpos patológicos, se ha utilizado estetipo de tratamiento en enfermedades como PTIcon algunos éxitos. Se han realizado algunosensayos clínicos utilizando rituximab comotratamiento para la hemofilia adquirida,mostrando efectividad como tratamiento deprimera línea, así como en pacientes refractariosa otros tratamientos inmunosupresores. Seencontró depleción de los linfocitos B circulantes,observándose mejores respuestas en pacientes
J García-Chávez, LA García-Stivalet.

Revista de Hematología Vol. 7, No. 1, 2006
41
con títulos de Bethesda bajos (<100BU/ml) condesaparición completa de la actividad delinhibidor en 3 a 12 semanas. Las recaídas seasociaron a un nivel de inhibidor más altos,aunque con el reinicio del tratamiento se lograronnuevas remisiones completas (11).
Por lo que se puede concluir con lainformación con la que se cuenta hasta elmomento que el uso de rituximab comotratamiento es efect ivo, requir iendo sucombinación con otros agentes en pacientes conniveles altos de inhibidor (11).
INHIBIDORES CONTRA FACTOR VONWILLEBRAND.
La deficiencia de factor de von Willebrand(FvW) puede ser congénita o adquirida. Lascausas de deficiencia adquirida incluyen defectosen la síntesis o liberación, absorción de laproteína por superficies celulares (neoplasias)degradación mecánica o proteolítica. Los casosinmunológicos incluyen aloanticuerpos yautoant icuerpos cont ra e l FvW. Losautoanticuerpos pueden ser idiomáticos o bienla primera manifestación de una enfermedadautoinmune como LES o linfomas. El anticuerpogeneralmente se une a sitios no funcionales dela prote ína del FvW, predisponiendo ahemorragias cuando el complejo Ac-FvW eseliminado de forma rápida de la circulación.
El diagnóstico debe ser sospechado enpacientes con episodios de hemorragia deaparición súbita, especialmente en sitiosmucocutáneos como tracto gastrointestinal ogenitourinario. Los laboratorios de rutinamuestran un TTPa prolongado. Estudios másespecíficos mostraran una agregometría aristocetina alterada y disminución de loaconcentración de los multímeros de alto pesomolecular del FvW.
El t ra tamiento en es tos casos condesmopresina frecuentemente controla lahemorragia. En los casos refractarios, se debenadministrar concentrados de FvW. En pacientes
con enfermedades asociadas, p.e. MGUS lainmunoglobulina hiperinmune provee una mejoríamas sostenida. Otro tratamiento utilizado es elrecambio plasmático con buenos resultados, sinembargo el tratamiento a largo plazo se lograsolamente con el tratamiento de la enfermedadsubyacente con desaparición de los anticuerpos.
INHIBIDORES CONTRA FACTOR V.Este tipo de inhibidores son raros, ocurren
de forma espontánea en pacientes de la terceraedad, muy frecuentemente en pacientes que hansido sometidos recientemente a procedimientosquirúrgicos (13). Otras asociaciones encontra-das es la exposición a aminoglucósidos, trans-fusiones y enfermedades malignas. Los inhibi-dores contra el factor V frecuentemente son an-ticuerpos IgG policlonales (1).
El diagnóstico se sospecha con base en unTTPa y TP prolongados los cuales no corrigencon la adición de plasma normal. Se puedenobservar niveles muy bajos de factor V enpacientes con anticoagulante lúpico, coninhibidores específicos contra el factor V.
Las manifestaciones hemorrágicas, enpacientes con inhibidores contra factor V,pueden ser desde triviales hasta poner en riesgola vida. En caso de que se requiera tratamientosuele ser necesario el apoyo transfusional conplaquetas, ya que el factor V plaquetario seprotege de los anticuerpos presentes en lac i rcu lac ión (14) . Otras medidas , comoinmunoglobulina intravenosa, plasmaféresis,esteroides y agentes inmunosupresores, puedenresultar benéficos.
INHIBIDORES CONTRA PROTROMBI-NA Y TROMBINA.
Los anticuerpos dirigidos contra protrombinaocurren en pacientes con anticoagulante lúpico yalgunos pueden forma complejos con protrombinaque acelera su aclaración de la circulación. Lasmanifestaciones hemorrágicas son raras y lapresencia de hemorragia ocurre solamente en los
Inhibidores adquiridos de la coagulación.

42
Revista de Hematología Vol. 7, No. 1, 2006
casos en los que se desarrolle hipoprotrombinemiagrave. Por otro lado, los anticuerpos dirigidoscontra la trombina se asocian a hemorragias. Elanticuerpo descrito es un IgG que se une a lat rombina pero no a la protrombina. Lahemorragia generalmente es de tipo mucocutáneay en sitios de punción.
INHIBIDORES CONTRA FACTOR XIII.Posterior a la activación por la trombina y
calcio, el factor XIII estabiliza la molécula defibrina. Los anticuerpos interfieren con laactividad del factor XIII. Las manifestacioneshemorrágicas ocurren debido a que el coagulorecién formado es frágil, sufriendo lisis temprana.El diagnóstico debe sospecharse en aquellospacientes con manifestaciones hemorrágicas ytiempos de coagulación normales. El diagnósticose debe confirmar con la observación de que loscoágulos formados por la recalcificación delplasma del paciente son solubles en 5M de ureao ácido monocloroacético al 1%.
La mayoría de los pacientes son de edadavanzada, siendo afectados tanto hombres comomujeres. Los anticuerpos formados eran IgG loscuales evitan la estabilización de la fibrina pore l fac tor XII I . El t ra tamiento es conconcentrados del factor XIII lo cual neutralizaal anticuerpo y normaliza la solubilidad delcoagulo.
REFERENCIAS.1.- Eckman MH, Erban JK, Singh SK, Kao GSJ.Screening for the risk for bleeding or thrombosis. AnnIntern Med 2003; 138: W 15-24.
2.- Giles J, Arnout J, Vermylen J, et al. Anti-factor VIIIantibodies of hemophiliac individuals are frequentlydirected towards nonfunctional determinants and donot exibit isotypic restricction. Blood 1993; 82:2452-61.
3.- Sallh S, Wan JY. Inhibition against factor VIII inpatients with cancer. Analysis for 41 patients. Cancer2001; 91:1607-74.
4.- Fulcher CA, Mahoney S, Zimmerman TS. FVIIIinh ib i to r IgG subc lass and FVII I po lypept idespecificity determined by inmunobloting. Blood 1987;69:1475-80.
5.- Pratt KP, Shen BW, Takeshima K et al Structure oftha C2 domain of human factor VIII al 1.5 A resolution.Nature 1999; 402: 439-42.
6.- Scandella D, Gilbert GE, Shima M, et al. Some factorVIII inhibitor antibodies recognize common epitopecorresponding to C2 domain amino acids 2248 through23212 which overlap a phosholipid �binding site. Blood1995; 86:1811-9.
7.- Morrison AE, Ludlam CA, Kessler C. Use of porcinefactor VIII in the treatment of individuals with acquiredhemophilia. Blood 1993; 81:1513-20.
8.- Monroe DM, Hoffman M, Oliver JA, et al. Plateletactivity of high dose factor VIIa is independent oftissue factor. Br J Hematol 1997; 99:542-7.
9.- Santagostino E, Gringeri A, Morfini M, et al.Continuous infusion of recombinant activated factorVIIa for high titer inhibitor patients. Blood 2000;96:267ª
10.- Schwartz RS, Gabriel DAAledort LM, et al. Aprospec t ive s tudy of t rea tment o f acqui red(autoimmune) factor VIII inhibitor with high doseintravenous gama globulin. Blood 1995; 86:797-804.
11.- Stasi R, Brunetti M, Stipa E, et al. Blood 2004;103:4424-8.
12.- Wiestner A, Cho H, Asch A, et al. Blood 2002;100:3426-8.
13.- Feinstein DI, Green D, Federici AB, et al. Diagnosisand managment of pat ients with spontaneouslyacquired inhibitors of coagulation. Hematology1999:192-208.
14.- Schleinitz N, Veit V, Chowguet D, Seux V, ArnouxD, Mokat D, et al. Adquired factor V inhibitor:etiology, bleeding risk and therapeutic managementwith regar to three cases. Rev Med Interne 2001;22:1119-23.
J García-Chávez, LA García-Stivalet.

Revista de Hematología Vol. 7, No. 1, 2006
43
Solicitud de reimpresos: Dr. Abraham Majluf-Cruz, Jaina 41, Col. Letrán Valle, C.P. 03650, México, D.F., México. Tel: 55 39 13 58 E-mail: [email protected]
INTRODUCCIÓN.La coagulación intravascular diseminada
(CID) también se denomina coagulopatía porconsumo. Es un síndrome que siempre essecundario a una enfermedad primaria (1). LaCID representa una de las principales causas demorbilidad y mortalidad en el mundo. Es laprimera causa de muerte en las unidades deterapia intensiva en los Estados Unidos deNorteamérica y la undécima causa de muerte(2). Cada año aparecen más de 750,000 casosnuevos de sepsis grave (>500 muertes/día porseps is grave) . La CID es comple ja eimpredecible, asociada con una mortalidad entre28% y 50%. La población afectada es muyheterogénea y su característica fundamental esla imposibilidad de predecir su curso clínico (3).El incremento en la incidencia se origina delaumento de la población y su edad; el incrementoen la frecuencia de infecciones nosocomiales; laresistencia bacteriana a los antibióticos cada vezmás frecuente, a pesar de que éstos son cadavez más potentes; la elevación del número depacientes inmunocomprometidos secundario alos nuevos tratamientos para el cáncer o lasenfermedades autoinmunes, además de lasnuevas causas de inmunosupresión natural; y el
aumento en el número de cirugías cada vez másextensas y agresivas.
La CID se caracteriza por activación dels is tema de coagulación, que resul ta engeneración intravascular de fibrina que puedeocluir los vasos sanguíneos pequeños y medianosy comprometer el suplemento sanguíneo a losórganos y que puede contribuir a la apariciónde falla orgánica múltiple. El consumo deplaquetas y proteínas del sistema de coagulación,inducen hemorragia de intensidad variable.
La def inic ión de CID es: s índromeadquirido, caracterizado por la activaciónintravascular del sistema de coagulación hastala formación intravascular de fibrina proceso quepuede acompañarse de hiperf ibrinol is issecundaria o de hipofibrinolisis (4).
ENTIDADES CLÍNICAS ASOCIADAS A LACID.
La CID es secundaria a otra enfermedadprimaria. La CID aumenta el riesgo de muertemás allá del de la enfermedad primaria. Además,el control de la causa primaria no necesariamentealivia la CID.
Las infecciones bacterianas son lasenfermedades más frecuentemente asociadas
Coagulación intravascular diseminada.
Abraham Majluf-Cruz.
Depto. de Hematología, Hospital Regional No. 222 "Gabriel Mancera",México, D.F., México.

44
Revista de Hematología Vol. 7, No. 1, 2006
A Majluf-Cruz.
además de las infecciones sistémicas porcualquier otro microorganismo. La CID esresultado de la gravedad de la infección con unarespuesta inflamatoria generalizada, caracterizadapor la liberación citocinas (5). Los traumatismosgraves son otra condición asociada a CID, yaque liberan material tisular a la circulación(grasa o fosfolípidos) y generan hemólisis ydaño endotelial, en especial, los traumatismosa nivel de la cabeza.
Las neoplasias sólidas o hematológicascursan con CID por mecan ismos pocoentendidos, aunque la mayoría de los estudiosimplican al factor tisular. En la leucemiapromielocítica aguda se encuentra una formadistinta de CID, la cual se caracteriza por unestado hiperfibrinolítico grave, aunado a unsistema de coagulación ampliamente activado(6) y aunque predomina la hemorragia, en laautopsia se encuentra trombosis diseminada en
Cuadro 1Condiciones clínicas asociadas a la CID.
Condición clínica Causa
Sepsis o infección grave Potencialmente, cualquier micro-organismo
Traumatismos Los que cursan con lesión tisular grave, traumatismos de lacabeza y embolismo graso
Destrucción de órganos Pancreatitis grave
Neoplasias Sólidas, hematológicas (como la leucemia aguda promielocítica)
Complicaciones obstétricas Placenta abrupta y embolia de líquido amniótico
Alteraciones vasculares Hemangiomas gigantes o aneurismas gigantes.
Falla hepática De cualquier causa: viral, tóxicos, medicamentos
Reacciones tóxicas o Reacción transfusional aguda, rechazo de trasplantes, uso deinmunológicas graves drogas ilícitas y mordedura de serpientes
muchos pacientes.La CID aparece en entidades obstétricas
agresivas: placenta abrupta y embolia de líquidoamniótico (7). Éste activa la coagulación invitro. El grado de separación placentariacorrelaciona con la gravedad de la CID por lafuga de material similar a la tromboplastinaplacentario.
Las alteraciones vasculares, como losaneurismas o los hemangiomas gigantes, puedengenerar una activación local de la coagulación.Los factores hemostáticos activados puedecausar CID, pero es más común la depleciónsistémica de los mismos, así como de lasplaquetas, lo cual puede resultar en unacondición clínica que es indistinguible de laCID.FISIOPATOLOGÍA DE LA CID.
Existen múltiples mecanismos causantes dela CID y ahora se tiene mayor claridad acerca

Revista de Hematología Vol. 7, No. 1, 2006
45Coagulación intravascular diseminada.
de su etiología (figura 1).Directamente, existen mecanismos que
permiten no sólo la generación s ino lapersistencia en la generación de la trombina,enzima clave en el proceso de la CID. Debenconsiderarse los siguientes mecanismos:Generación descontrolada de trombina.
In vivo, el control del sistema hemostáticoes crucial al balancear las actividadesprocoagulante y anticoagulante. La formación defibrina es controlada por el sistema de la proteínaC y por la antitrombina. Este balance hemostáticoque coordina la generación de trombina se pierdeen la CID. La generación de trombina se detectaentre tres y cinco horas luego de la infusión demicroorganismos o endotoxina y el complejofactor tisular/factor VIIa tiene un papel central.No existen cambios en la activación del sistema
de contacto (8) y su inhibición no previene laactivación del sistema hemostático. La eliminacióndel complejo factor tisular/factor VIIa conanticuerpos monoclonales para el factor tisular oel FVIIa inhibe la generación de trombina ypreviene la aparición de la CID y la mortalidad(9).Diseminación y mantenimiento de lageneración de trombina.
Aunque el factor tisular juega un papelfundamental en el inicio de la generación det rombina , o t ros procesos d iseminan lacoagulac ión in t ravascular. Los bro tessecundarios de trombina se generan en la víaintrínseca llevando al consumo y depleción deproteínas anticoagulantes (proteína C yantitrombina). La exposición a fosfolípidosnegativos facilita más el ensamblaje y propagación
Citocinas
Remosión inadecuadade la fibrina
Depósito de fibrinaDisminución de los
factores hemostáticos
Hemorragia
Sepsis
Inhibición de la fibrinolisisdependiente del IaTP-1
Disminución de lossistemas
anticoagulantesnaturales
Activación del sistemade coagulación mediada
por el factor tisular
CID
Figura 1.- Pérdida del balance del sistema de coagulación en la sepsis y la CID. Este imbalance sedebe principalmente a la activación de la hemostasia y de la fibrinolisis y a la caída simultánea de losmecanismos anticoagulantes naturales. IaTP-1: inhibidor del activador tisular del plasminógeno tipo1.

46
Revista de Hematología Vol. 7, No. 1, 2006
de factores hemostáticos (10). Estos mecanismosforman una respuesta expansiva en el tiempo yen el espacio, característica de la CID.
Supres ión de los ant icoagulantesnaturales y deterioro de la fibrinolisis.
Contribuyen a la formación y mantenimientode la generación de fibrina. La concentraciónplasmática de la antitrombina disminuye en elpaciente séptico por consumo dependiente dela misma generac ión de t rombina , dedegradación por elastasa de los neutrófilos y pordisminución de su síntesis hepática. La caída dela antitrombina se asocia con un aumento en lamortalidad. También ocurre una disminución delsistema de la proteína C por disminución en lasíntesis de trombomodulina (11). El factor tisulares normalmente inhibido por el inhibidor de lavía del factor tisular, cuya administración inhibela generación de trombina inducida porendotoxina y disminuye potentemente lamortalidad. Por otra parte, en el momento de laactivación máxima de la hemostasia el sistemafibrinolítico está muy deteriorado. La bacteremiay la endotoxemia aumentan la actividadfibrinolítica, probablemente por liberación deactivadores endoteliales del plasminógeno. Aesta respuesta profibrinolítica sigue unasupresión de la actividad fibrinolítica, conaumento del inhibidor del activador tisular delplasminógeno tipo 1 (12).
Activación inflamatoria.Las alteraciones hemostáticas y fibrinolíticas
dependen de citocinas inflamatorias: FNT-α, lainterleucina 1 (IL-1) e interleucina 6 (IL-6). Elprincipal mediador de la activación de lacoagulación en la CID es la IL-6 (13). El FNT-α influencia la activación por su efecto sobre laIL-6 y es el factor más importante en ladisregulación de los anticoagulantes naturales yla fibrinolisis. Las citocinas anti-inflamatoriastales como la IL-10 modulan la activación de lacoagulación. Una vez activados, el sistemainflamatorio y el de la coagulación amplifican másla respuesta. Mientras que las citocinas y otros
mediadores pro-inflamatorios inducen activaciónde la coagulación, la trombina y otras proteasasinteractúan con receptores localizados sobrecélulas y generan más activación celular con elaumento inflamatorio consecuente. Si el procesose generaliza escapa de la vigilancia localdiseñada para controlar el proceso y el balancefisiológico se rompe, lo que mantiene el círculoinflamación:coagulación.
Activación y disfunción endotelial.La respuesta normal del endotelio regula la
coagulación y la inflamación (14). Su disfuncióny falla puede llevar y quedar marcada por eldesarrollo de la CID. La hipercoagulabilidad yel predominio de la hemorragia o trombosisdependen de factores genéticos y otrosrelacionados con el huésped.DIAGNÓSTICO DE LA CID.
No existe una prueba única para hacer eld iagnós t ico cer te ro . S in embargo, lacombinación de algunas pruebas de laboratorioayuda a establecer con mayor certeza estediagnóstico (15). El diagnóstico debe basarseen la cuenta plaquetaria, evaluación global delcoagulograma (tiempos de tromboplastinaparc ia l ac t ivada y de pro t rombina) ,cuant i f icac ión de la concent rac ión deant i t rombina y de uno o dos fac toreshemostáticos y una prueba para detectarproductos de degradación de la fibrina. Serecomienda utilizar los criterios diagnósticos quese muestran en el cuadro 2.
Cuadro 2Puntaje para el diagnóstico de la CID.
PuntajeVariable 0 1 2
Cuenta plaquetaria >100 <100 <50Metabolito de la no moderado gravefibrina elevadoTiempo de pro- < 3 seg 3 a 6 seg > 6 segtrombina prolongadoFibrinógeno > 1 g/L < 1 g/L
A Majluf-Cruz.

Revista de Hematología Vol. 7, No. 1, 2006
47
Debe calcularse en todo paciente en riesgode tener este problema y que tiene alguna de laspatologías mencionadas en el cuadro 1. Si elpuntaje es >5, se hace el diagnóstico de CIDestablecida y e l puntaje debe repet i rsediariamente. Si es <5, es puntaje es sugestivode CID pero no lo afirma. Debe entoncesrepetirse cada 1 a 2 días.
Realizar las pruebas en serie es más útil queaisladamente. La reducción plaquetaria o la caídasignificativa serial son datos sensibles aunque noespecíficos de CID. El alargamiento delcoagulograma puede reflejar consumo ydepleción de factores, lo que se objetiviza mejoral cuantificar algún factor. La exactitud de laspruebas coagulométricas en un tiempo eslimitada. Cuantificar la antitrombina evalúa elconsumo del inhibidor más importante de latrombina, aunque es caro y requiere tecnologíaespecializada. La cuantificación del fibrinógenopuede sugerir, pero no es muy útil para eldiagnóstico, ya que actúa como un reactante defase aguda, por lo que su concentraciónplasmática puede no disminuir. El dímero D esútil para diferenciar la CID de procesos conplaquetas bajas y alargamiento de los tiemposde coagulación.
El diagnóstico de CID puede hacersecombinando pruebas de laboratorio de rutina(cuadro 2) (15). Los resultados normales noexcluyen una CID, sin embargo, pueden terminaren un acor tamiento de los t iempos decoagulación y en un incremento del fibrinógeno.Por esto, se sugiere la identificación tempranade la CID, no sólo con resultados anormales,sino también identificando su tendencia. Serequieren pruebas más especí f icas querelacionen inflamación y CID que deben sersimples y rápidas.TRATAMIENTO DE LA CID.
Existen múltiples controversias y estudiosclínicos apropiados por la complejidad de la CIDy sus consecuencias. A pesar de esto, la piedraangular del tratamiento es el manejo específico
y vigoroso de la causa de la CID. En algunoscasos, la CID resuelve en las horas siguientes ala resolución de la afección primaria. En otroscasos, la CID puede estar presente por algunosdías aún después de que se insti tuyó eltratamiento. Las medidas de soporte son muynecesarias: sustitución plasmática y plaquetaria,anticoagulantes e inhibidores fisiológicos de lahemostasia.
Terapia con plasma y plaquetas.La disminución de las plaquetas como de la
concentración de los factores aumenta el riesgohemorrágico. La sustitución con plaquetas oplasma no se indica sólo por los resultados delaboratorio y deben emplearse sólo en enfermoscon hemorragia activa, en los que requieren deun procedimiento invasivo y en los que existe unalto riesgo de hemorragia.
Anticoagulantes.Al menos, la heparina inhibe parcialmente
la activación del sistema de coagulación en lasepsis y en otras causas de CID aunque la evi-dencia proviene de estudios de series de casosno controlados y su efecto benéfico sobre cri-terios de respuesta duros nunca se ha demos-trado (16). La seguridad de la heparina es cues-tionable en pacientes con riesgo hemorrágicoalto. El anticoagulante ideal para la CID debeestar dirigido contra el factor tisular. Reciente-mente, se desarrolló un inhibidor específico ypotente del complejo factor tisular/factor VIIa/factor Xa (rNAPc2) que se obtiene de nemáto-dos. Actualmente, se realizan estudios fase II yIII para analizar su efecto en la CID.
Concentrados de inhibidores naturales.Ya que la antitrombina es el inhibidor
fisiológico más importante de la trombina, seinició su uso en humanos. En estudios clínicosen pacientes con sepsis y/o choque séptico, sedemostró mejoría del puntaje y acortamiento dela CID y mejoría orgánica. La caída de laproteína C contribuye en la CID la cual se asociacon resul tados fatales . Se ha intentadosuplementar con proteína C o drotrecogina
Coagulación intravascular diseminada.

48
Revista de Hematología Vol. 7, No. 1, 2006
encontrándose un efecto favorable en estudiosanimales y en humanos (17). Finalmente, en vistadel papel central que tiene el factor tisular en elinicio de todo el proceso fisiopatológico deformación de trombina, puede especularse quela administración del inhibidor de la vía delfactor tisular de tipo recombinante pudiera sermuy útil en estos casos.
REFERENCIAS.1. - Marder VJ , Mar t in SE, Francis CW, et a l .Consumptive thrombohemorrhagic disorders. In:Colman RW, Hirsh J, Marder VJ, et al . Editors.Hemostasis and thrombosis: basic principles andclinical practice. Philadelphia: JB Lippincott; 1987. p.975�1015.
2.- Sands KE, Bates DW, Lanken PN, Graman PS,Hibberd PL, Kahn KL, et al. Epidemiology of sepsissyndrome in 8 academic medical centers. JAMA 1997;278:234-40.
3.- Angus D, Wax R. Epidemiology of sepsis: anupdate. 7Crit Care Med 2001; 29(Suppl 1):S109-S116.
4.- Müller-Berghaus G, Madlener K, Blombäck M,(Eds). DIC: pathogenesis, diagnosis and therapy ofd i ssemina ted in t ravascu la r f ib r in fo rmat ion .Amsterdam: Elsevier Science Publishers BV; 1993.
5.- Levi M, van der Poll T, ten Cate H, et al. Thecytokine-mediated imbalance between coagulant andanticoagulant mechanisms in sepsis and endotoxemia.Eur J Clin Invest 1997; 27:3�9.
6.- Falanga A, Consonni R, Marchetti M, et al. Cancerprocoagulan t and t i s sue fac tor a re d i f fe ren t lymodula ted by a l l - t rans - re t ino ic ac id in acu tepromyelocytic leukemia cells. Blood 1998; 92:143�51.
7.- Martin JN, Stedman CM. Imitators of preeclampsiaand HELLP syndrome. Obstet Gynecol Clin North Am1991; 18:181�98.
8.- van der Poll T, Buller HR, ten Cate H, et al.Activation of coagulation after administration of tumornecrosis factor to normal subjects. N Engl J Med 1990;322:1622�7.
A Majluf-Cruz.
9.- De La Cadena RA, Majluf-Cruz A, Stadnicki A,Colman RW, Suffredini AF. Activation of the contactand f ib r ino ly t ic sys tems a f te r in t ravenousadminis t ra t ion of endotox in to normal humanvolunteers: correlation with the cytokine profile.Immunopharmacology 1996; 33:231-7.
10.- Lobato-Mendizabal E, Majluf-Cruz A. Trombofilia,t romboembol ia y e l uso de las hepar inas nofraccionadas y de bajo peso molecular. Rev Invest Clin2000; 56:346-65.
11.- Conway EM, Rosenberg RD. Tumor necrosis factorsuppresses transcription of the thrombomodulin genein endothelial cells. Mol Cell Biol 1988; 8:5588�92.
12.- Biemond BJ, Levi M, ten Cate H, et al. Endotoxin-induced activation and inhibition of the fibrinolyticsystem: Effects of various interventions in thecytokine and coagulation cascades in experimentalendotoxemia in chimpanzees. Clin Sci 1995; 88:587�94.
13.- van der Poll T, Levi M, Hack CE, et al. Eliminationof interleukin 6 attenuates coagulation activation inexperimental endotoxemia in chimpanzees. J Exp Med1994; 179:1253�9.
14.- Aird WC. The role of the endothelium in severesepsis and multiple organ dysfunction syndrome.Blood 2003; 101:3765-77.
15.- Taylor FB, Toh CH, Hoots WK, Wada H, Levi M.Towards definition, clinical and laboratory criteria anda scoring system for disseminated intravascularcoagulation. Thromb Haemost 2001; 86:1327-30.
16.- Corrigan JJ Jr. Heparin therapy in bacterialsepticemia. J Pediatr 1977; 9:695�700.
17.- Majluf-Cruz A, Bergés-García A, García-Chávez J.Hemofilia. En: Góngora-Biachi RA, editor. Hematología.Actualización 2004. Mérida: Agrupación Mexicana parael Estudio de la Hematología, A.C.; 2004;51-54.