Árboles filogenÉticos - bit.etsia.upm.es · quinto paso: se calcula la distancia del nodo u al...
TRANSCRIPT

ÁRBOLES FILOGENÉTICOS

¿Por qué usar filogenias?
El conocimiento del pasado es importante para poder resolver muchas cuestiones relacionadas con procesos biológicos.
Las filogenias nos permiten obtener relaciones entre ancestros y descendientes mediante la topología del árbol.
Las filogenias nos permiten obtener distancias evolutivas mediante la longitud de las ramas del árbol.

Terminología del árbol filogenético
Nodes = nodos
Branches = ramas
Root = raíz

Topología del árbol: patrón de ramas
Misma topología

Taxón = operational taxonomic unit (OTU) S1-S5
Politomía: árbol no resuelto (se puede intentar añadir más secuencias para resolverlo)

Árboles sin raíz (unrooted) o enraízados (rooted)
Outgroup: secuencia/s relacionadas con las secuencias del árbol (ingroup)
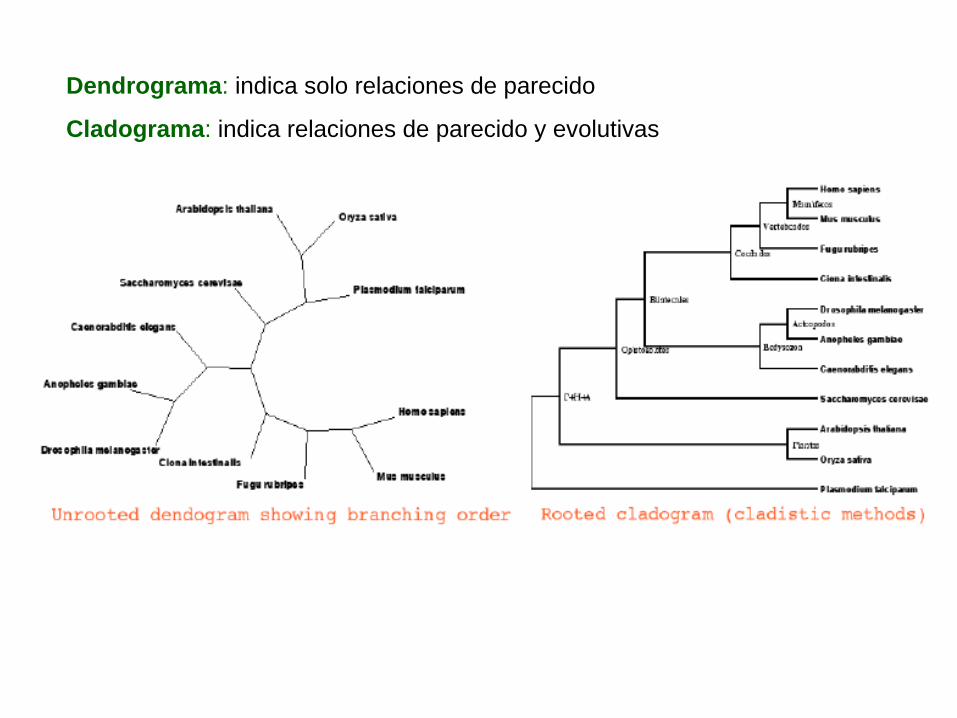
Dendrograma: indica solo relaciones de parecido
Cladograma: indica relaciones de parecido y evolutivas
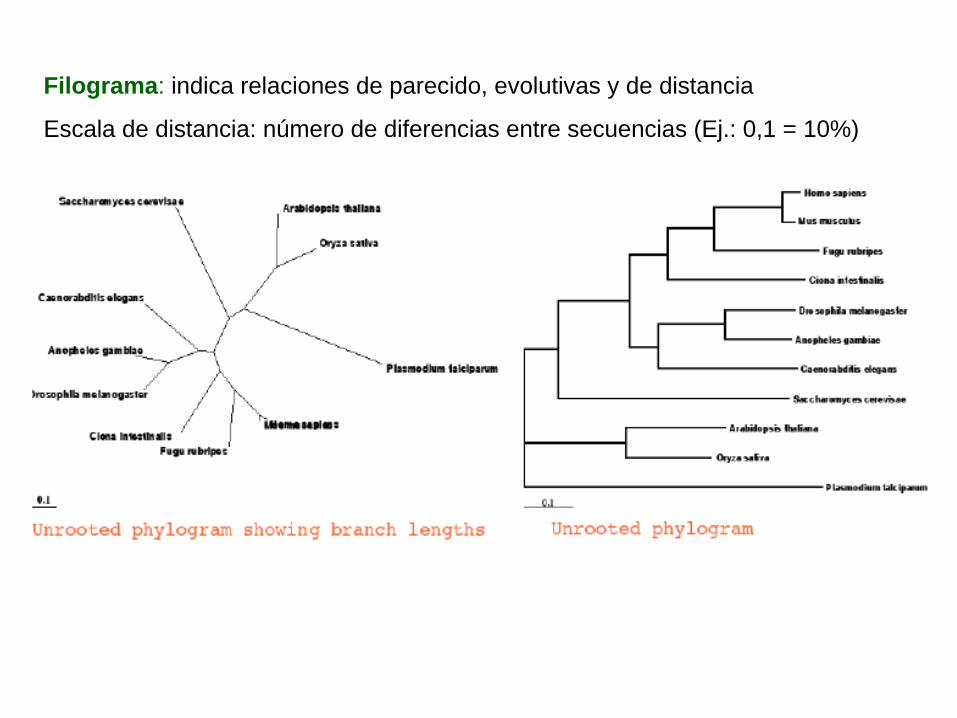
Filograma: indica relaciones de parecido, evolutivas y de distancia
Escala de distancia: número de diferencias entre secuencias (Ej.: 0,1 = 10%)

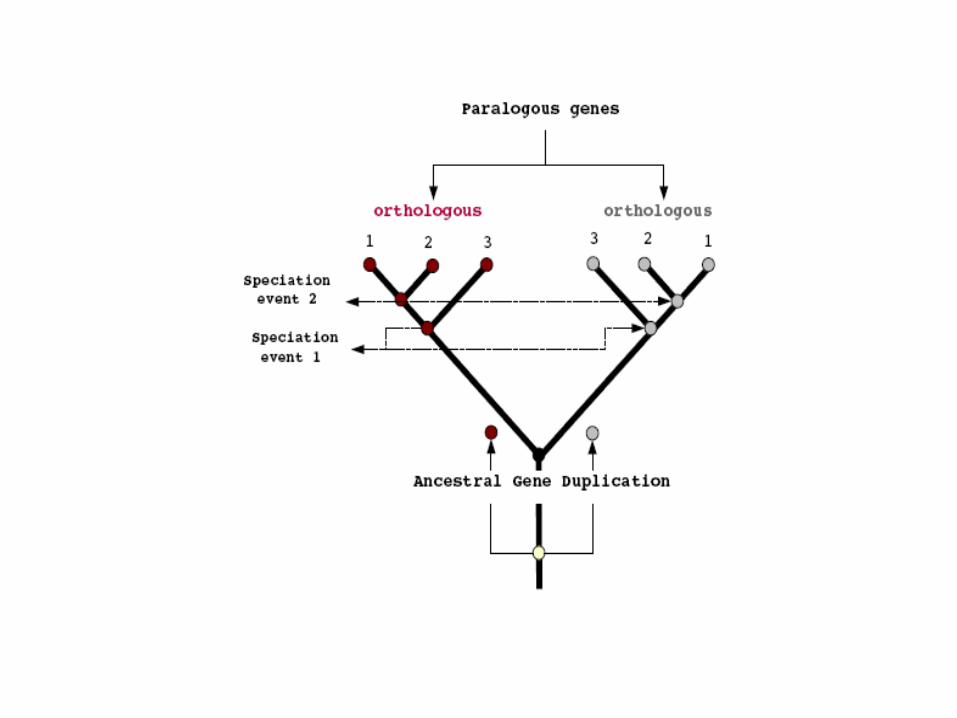

Homoplasia

Homoplasia
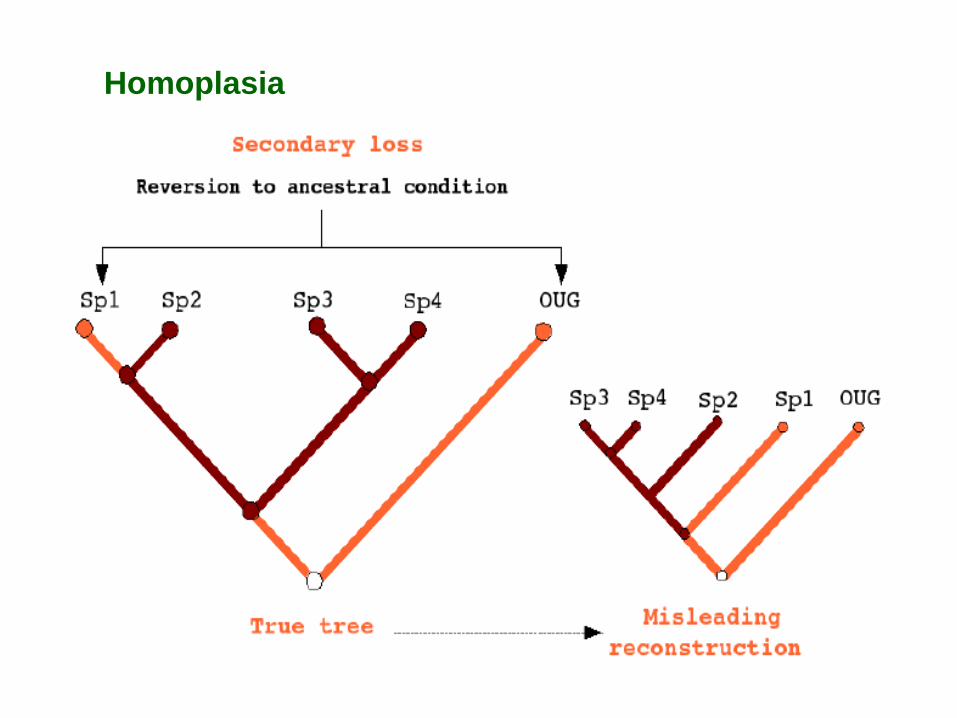
Homoplasia

Modelos de evolución de DNA
La tasa de mutación en una secuencia de DNA depende de:
- La región del genoma
- La posición de la base en el codón



Modelos de sustitución de nucleótidos


Modelos de evolución de proteínas
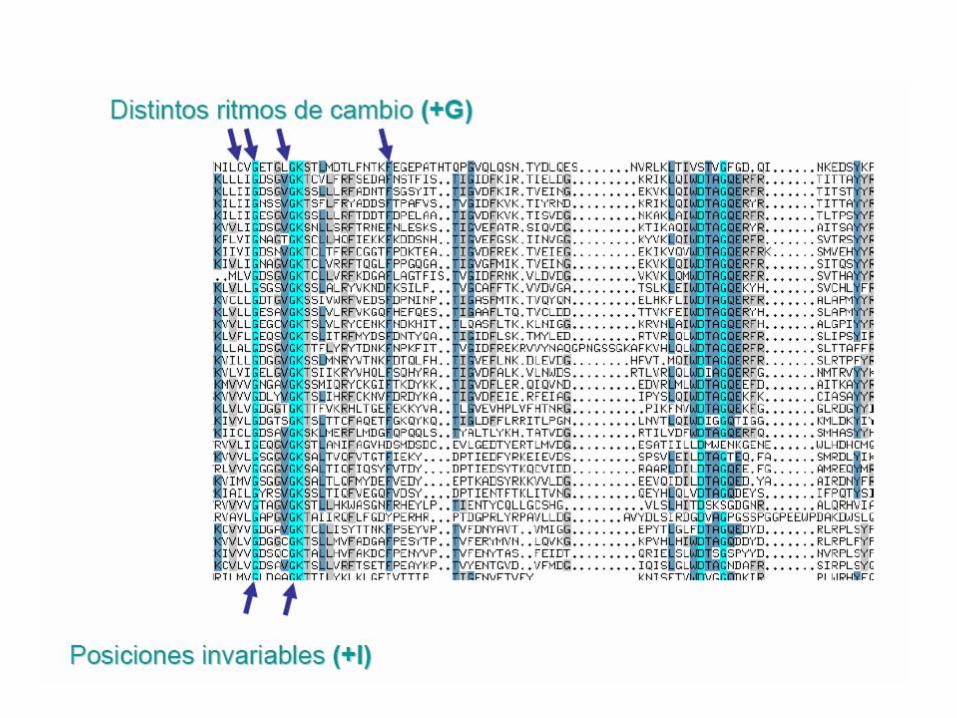

Modelos de evolución
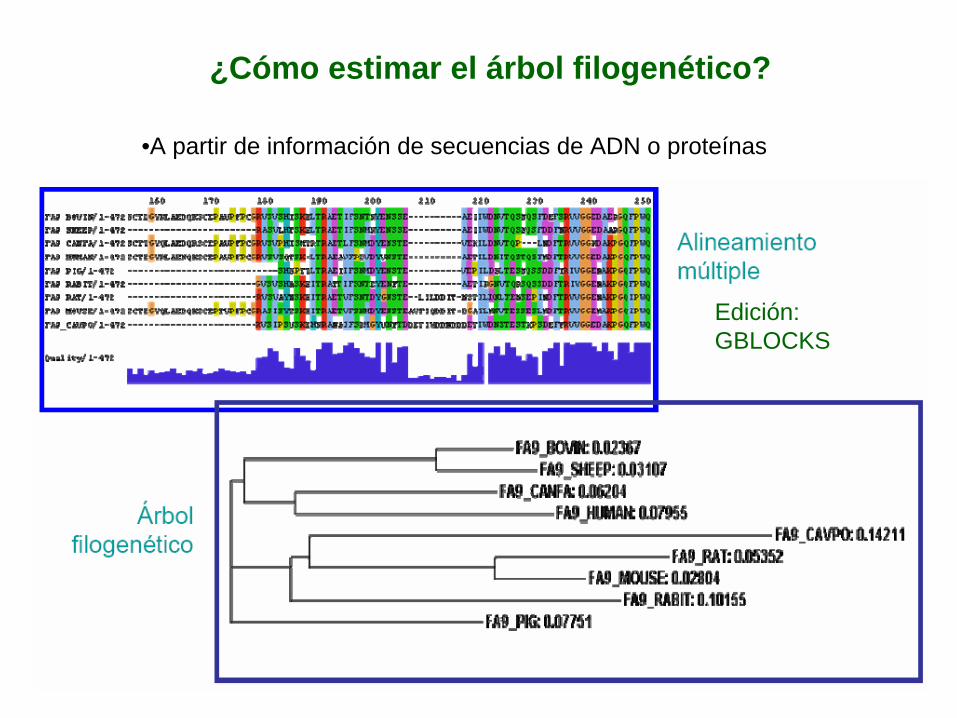
¿Cómo estimar el árbol filogenético?
•A partir de información de secuencias de ADN o proteínas
Edición: GBLOCKS

Evolución molecular: un ejemplo
AAGACTT
AAGACTT
T_GACTT
A_CACTT
AAGGCTT
TAGACCTT_GGGCTT

Evolución molecular: lo que observamos en el presente

Evolución molecular: la hipótesis más probable
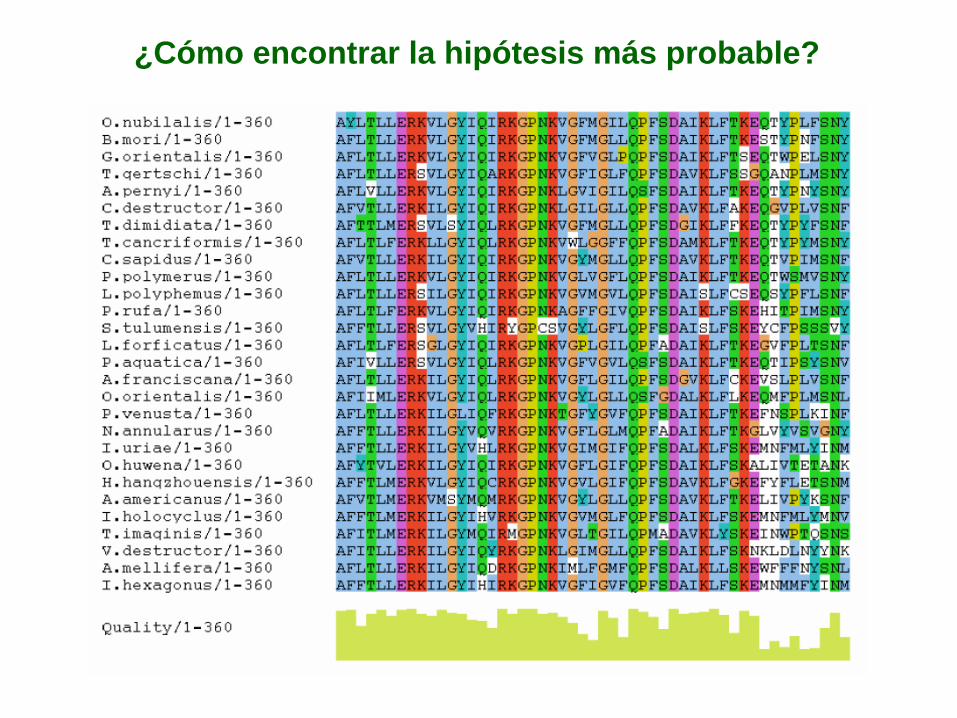
¿Cómo encontrar la hipótesis más probable?

¿Cómo encontrar la hipótesis más probable?

Métodos de reconstrucción filogenética
Basados en distancias:
UPGMA
Neighbor-joining
Basados en caracteres:
Máxima parsimonia
Máxima verosimilitud
Métodos bayesianos

Métodos basados en distancias.
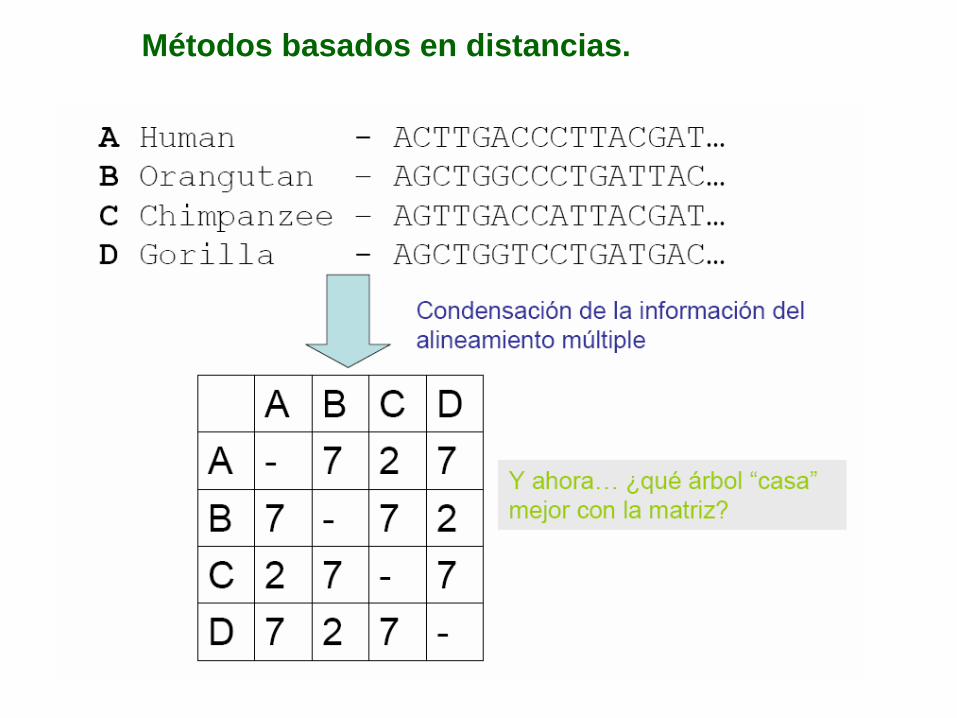
Métodos basados en distancias.

Métodos basados en distancias. UPGMA
• Es el método más simple.
• UPGMA significa Unweighted Pair-Group Method with Arithmetic means(agrupamiento pareado no ponderado utilizando media aritmética).
• Asume la existencia de un reloj molecular evolutivo.
• Ultramétrico, se obtiene un árbol enraizado.

• La hipótesis del reloj molecular supone que para una determinada macromolécula (proteína o gen) el ritmo de cambio es constante y proporcional al tiempo.




UPGMA
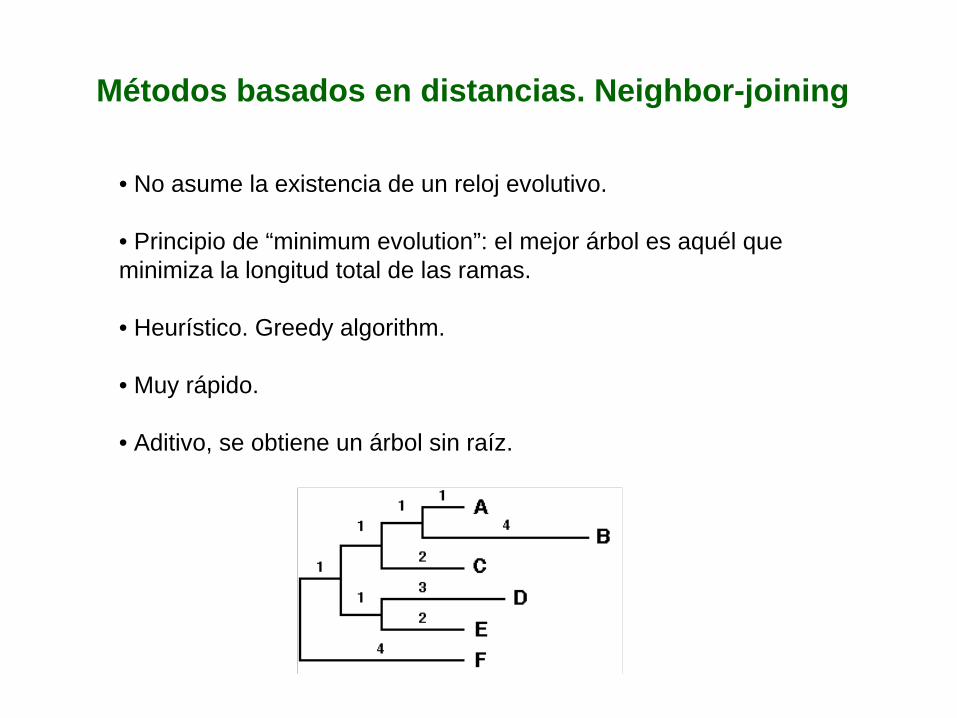
• No asume la existencia de un reloj evolutivo.
• Principio de “minimum evolution”: el mejor árbol es aquél que minimiza la longitud total de las ramas.
• Heurístico. Greedy algorithm.
• Muy rápido.
• Aditivo, se obtiene un árbol sin raíz.
Métodos basados en distancias. Neighbor-joining

Primer paso: se calcula la matriz de distancias
Segundo paso: se calcula la divergencia de cada nodo con el resto de nodos

Tercer paso: se calcula una nueva matriz de distancias usando la fórmula:

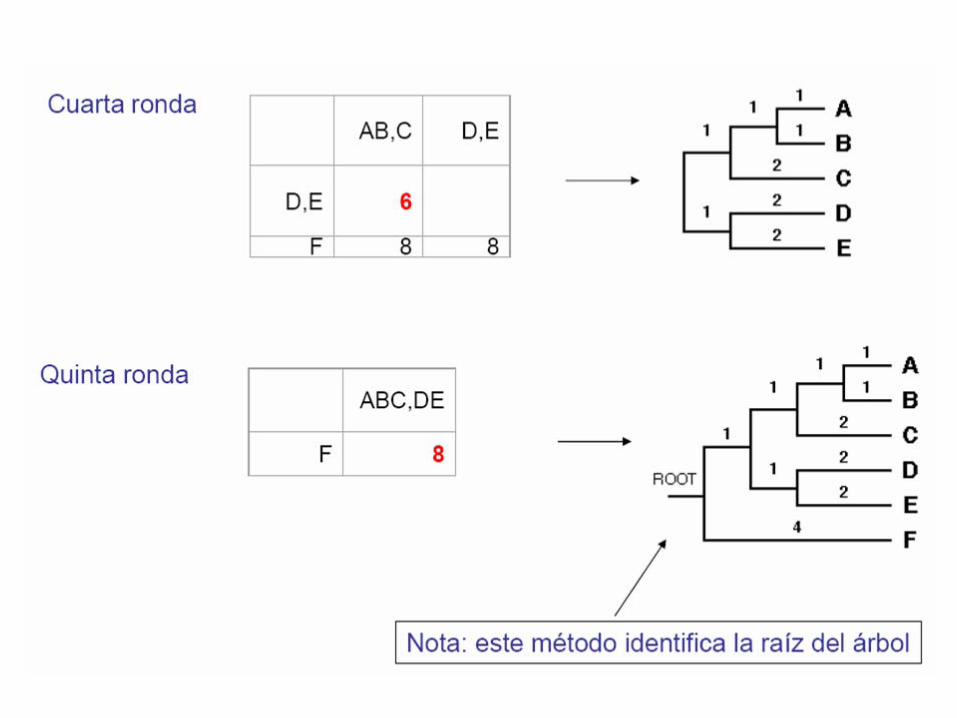
Cuarto paso: se elige la pareja de nodos con menor Mij y se calcula su distancia a un nodo interno U
Quinto paso: se calcula la distancia del nodo U al resto de nodos externos y se construye una nueva matriz de distancias
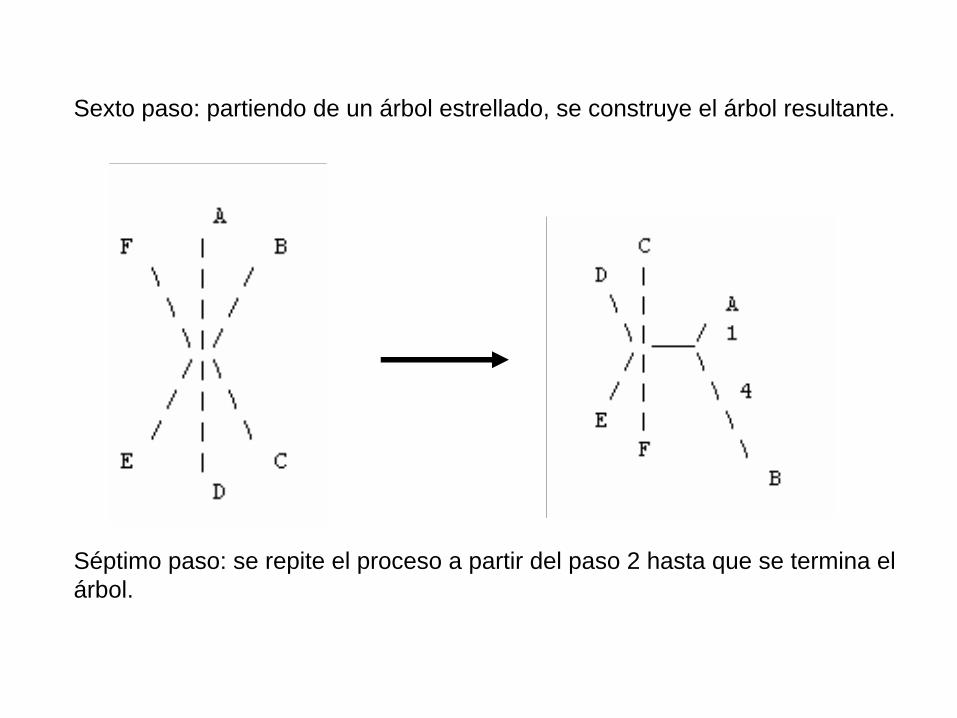
Sexto paso: partiendo de un árbol estrellado, se construye el árbol resultante.
Séptimo paso: se repite el proceso a partir del paso 2 hasta que se termina el árbol.

Neighbour-joining

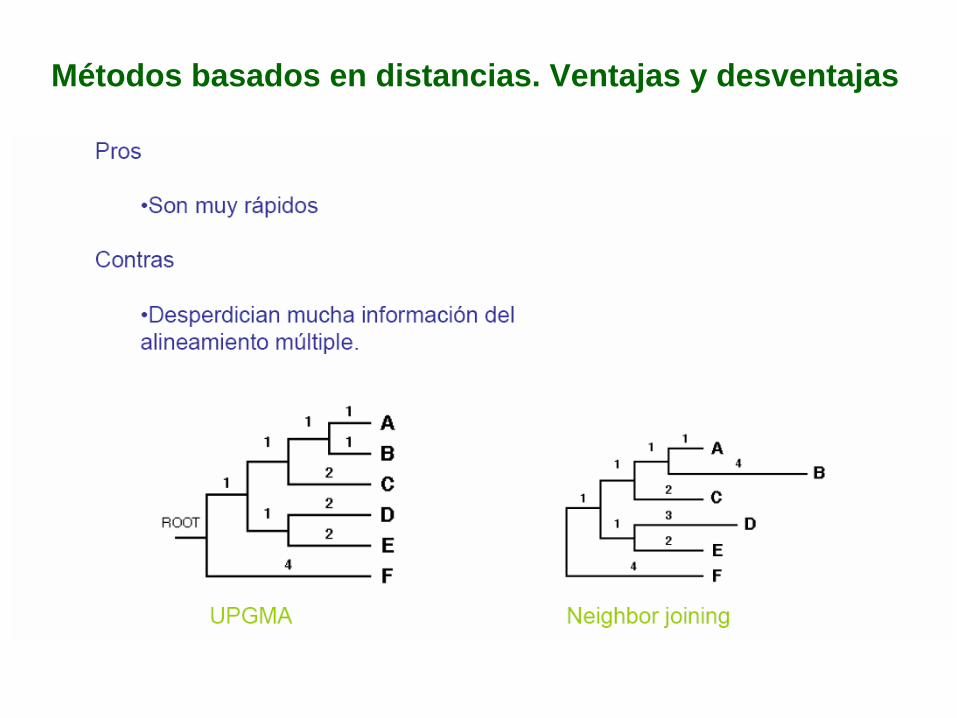
Métodos basados en distancias. Ventajas y desventajas

Métodos basados en caracteres.
Se basan en características del árbol filogenético.
-Máxima parsimonia
-Máxima verosimilitud
- Métodos bayesianos
Búsqueda de árboles

Búsqueda de árboles

Búsqueda de árboles


Métodos heurísticos: stepwise addition

Métodos basados en caracteres. Máxima parsimonia.

Árbol más parsimonioso

Árbol más parsimonioso
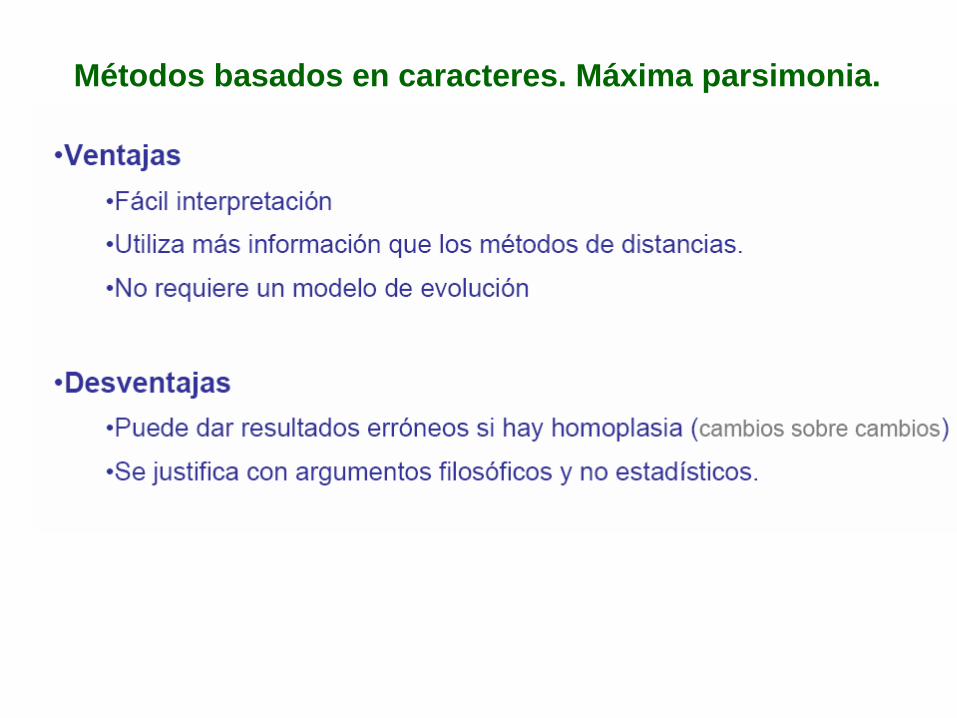
Métodos basados en caracteres. Máxima parsimonia.

Métodos basados en caracteres. Máxima verosimilitud.


Aplicación en filogenia

Métodos basados en caracteres. Máxima verosimilitud.
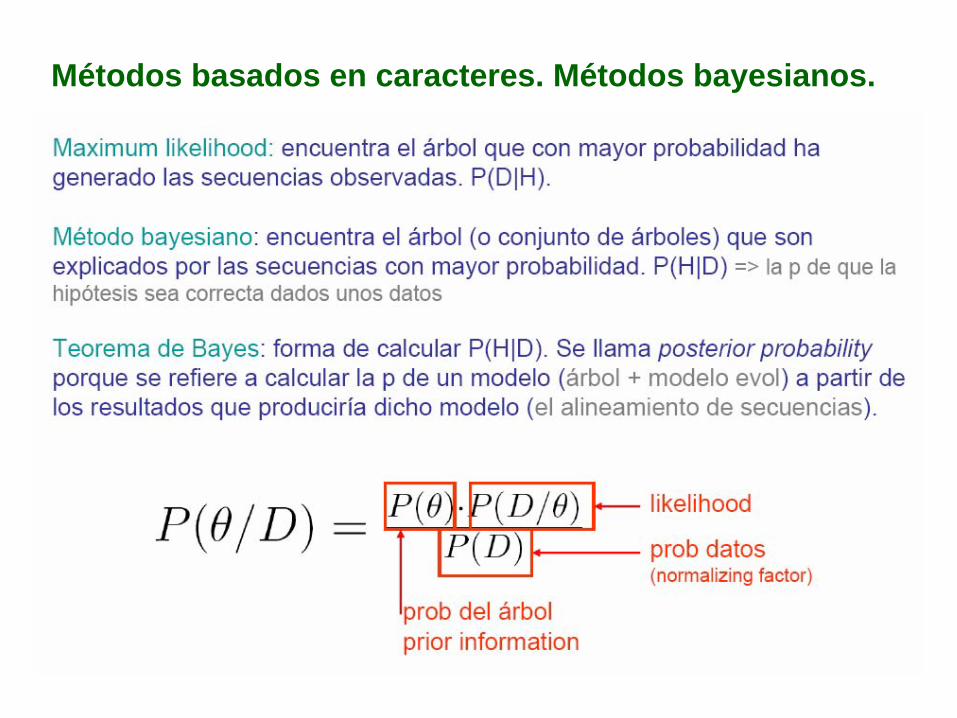
Métodos basados en caracteres. Métodos bayesianos.
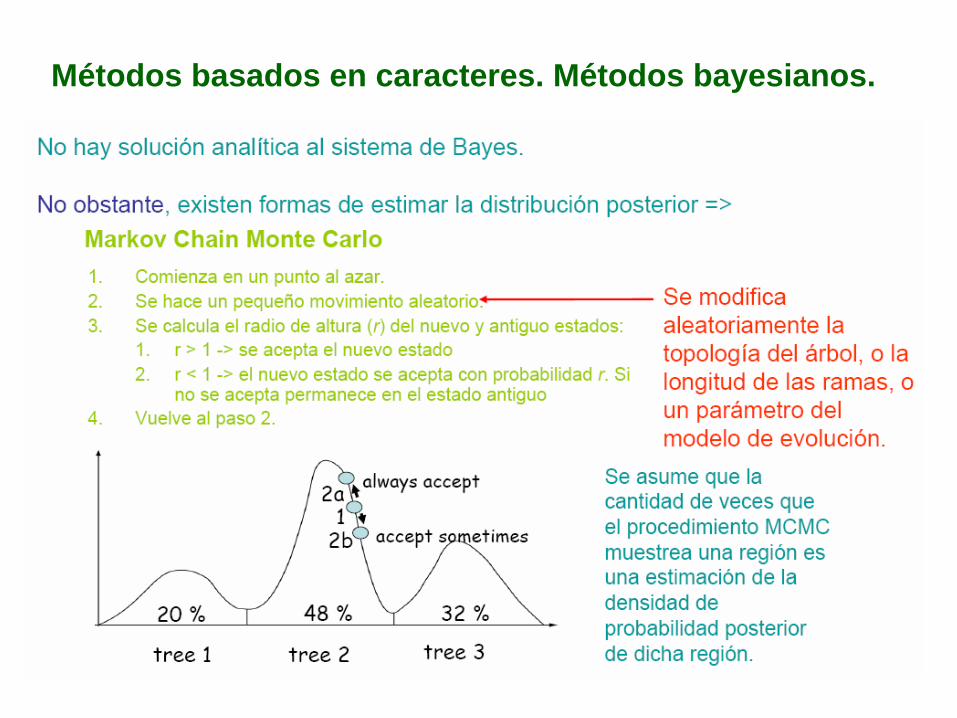
Métodos basados en caracteres. Métodos bayesianos.

Métodos basados en caracteres. Métodos bayesianos.

Contrastes de hipótesis de árboles

Bootstrap: reestimación del árbol mediante la variación en la posición y la repetición de caracteres de la secuencia.
Métodos para comprobar la robustez de un árbol

http://evolution.genetics.washington.edu/phylip/software.html

http://www.megasoftware.net/

http://evolution.genetics.washington.edu/phylip.html

http://paup.csit.fsu.edu/

http://www.phylogeny.fr/

http://www.tree-puzzle.de/
http://atgc.lirmm.fr/phyml/

http://mrbayes.csit.fsu.edu/

http://taxonomy.zoology.gla.ac.uk/rod/treeview.html

http://www.jalview.org/index.html
http://molevol.cmima.csic.es/castresana/Gblocks_server.html

http://www.ebi.ac.uk/cgi-bin/readseq.cgi