alt gene en gestacion
TRANSCRIPT

capítulo 13altERacIoNES GENÉtIcaS QuE aFEctaN la GEStacIÓN
Clara Eugenia Arteaga D.
obstetricia integral Siglo XXI

altERacIoNES GENÉtIcaS QuE aFEctaN la GEStacIÓN, Clara Eugenia Arteaga D.
242
obstetricia integral Siglo XXI
En los últimos años la investigación en embriología molecular ha mostrado que una importante proporción del genoma humano
está implicado en procesos de embriogénesis y desarrollo. Se calcula, por ejemplo, que para desarrollar un sistema nervioso son necesarios más del 60% de los genes del genoma humano. Esto supone, con alta probabilidad, que mutaciones genéticas o alteraciones cromosómicas producirán anomalías del desarrollo fetal las cuales dependiendo de su gravedad llevarán a la muerte fetal o a defectos estructurales o fun-cionales evidenciables al nacimiento (anomalías congénitas). Así por ejemplo se sabe que más de la mitad de los abortos espontáneos del primer trimestre son ocasionados por anomalías cromosómicas (1).
El conocimiento de la etiología y la etiopatogenia de las anomalías congénitas (AC) es fundamental, dados sus graves efectos sobre la sa-lud, la expectativa y la calidad de vida de los afectados, sobre su entor-no social y las consecuencias emocionales y económicas. Las AC cons-tituyen la primera causa de mortalidad infantil en los Estados Unidos y las enfermedades genéticas la principal razón de admisión en los hospitales pediátricos; en el año 1999 las AC ocasionaron una tasa de mortalidad infantil (TMI) de 138,2 por cada 100.000 nacimientos, su-perando a la prematurez, cuya TMI fue de 110,9 por cada 100.000 (2).
La mortalidad durante el primer año de vida se puede analizar más de-talladamente observando las muertes ocurridas en el período neonatal y el posneonatal. La mortalidad neonatal (MN) incluye las muertes sucedidas en los primeros 28 días de vida extrauterina. La mortalidad posneonatal (MPN), las muertes acaecidas desde el día 29 hasta los 11
meses. Del análisis separado de cada uno de estos grupos se ha con-cluido que el mayor descenso en las cifras de mortalidad ocurrió a ex-pensas del grupo de MPN, a consecuencia del manejo más adecuado de situaciones como la isoinmunización, el parto complicado, el trau-ma de parto, la inmadurez o la prematurez, etcétera, lo que ha llevado a la disminución de su peso relativo sobre la mortalidad, quedando en los primeros lugares otras causas más difíciles de reducir como las AC. En Colombia la mortalidad infantil ha descendido también acelera-damente durante el presente siglo: de 196 por 100.000 en 1938, a 135 por 100.000 en el periodo de 1950 a 1955 y a 46% en el de 1985 a 1990, mientras que la mortalidad por AC aumentó de 151 a 162 por 100.000 y pasó de ocupar el quinto lugar como causa de mortalidad neonatal en 1979 al cuarto en 1981 y al segundo en 2005 (3-5).
Las AC muestran frecuencias muy estables del 3 al 5% que sólo pa-recen verse modificadas por algunas variaciones geográficas y por el mejoramiento en los métodos diagnósticos y de detección.
La mayoría de los programas de detección de defectos congénitos sur-gen a partir de 1961, cuando es reconocida en Europa la “epidemia de talidomida”, con el propósito de detectar factores ambientales que pudieran tener una influencia adversa sobre la reproducción humana. Estos programas han sido básicamente de tres tipos: seguimiento de una cohorte de gestaciones, registro de defectos congénitos al naci-miento y detección de defectos congénitos a cualquier edad.
En Europa funciona desde 1979 un programa de cooperación interna-cional, logrado a través de registros regionales existentes aún antes de

242
altERacIoNES GENÉtIcaS QuE aFEctaN la GEStacIÓN, Clara Eugenia Arteaga D.
243
obstetricia integral Siglo XXI
1961 denominado Eurocat, que cuenta con el apoyo de la Comunidad Económica Europea (CEE), incluyendo hasta 1981 17 centros en 10 países miembros de la CEE y cubriendo cerca de 200.000 nacimientos al año.
En Estados Unidos el Centro de Control de Enfermedades de Atlanta (CDC) inició en 1967 el MACDP (Programa de Defectos Congénitos del Área Metropolitana de Atlanta), uno de los dos programas inde-pendientes que buscan estudiar la magnitud del problema de las ano-malías congénitas, cubriendo cerca de 24.000 nacimientos anuales.
En Latinoamérica, en 1967, surgió el Estudio Colaborativo Latino-americano de Malformaciones Congénitas (ECLAMC), un programa para la investigación clínica y epidemiológica de los factores de riesgo en la etiología de las anomalías congénitas usando la aproximación metodológica de casos y controles. Hasta 2004 la cobertura incluía to-dos los países de América del Sur, además de Costa Rica y República Dominicana (6,7).
El interés creciente en el estudio del desarrollo embrionario y fetal ha permitido el conocimiento de un gran número de alteraciones de la morfogénesis presentes al nacimiento y producidas o bien por facto-res genéticos o ambientales, o bien por el concurso de ambos. En este proceso la genética clínica ha sido de gran ayuda, puesto que a través del estudio de pacientes con un fenotipo anormal se han podido en muchos casos identificar los genes involucrados y los mecanismos por los cuales la mutación genera el desarrollo anormal, lo cual significará el comienzo de la prevención y del tratamiento de las anomalías con-génitas.
MEcaNISMoS GENÉtIcoS aSocIaDoS a aNoMalíaS coNGÉNItaSVarios son los mecanismos genéticos involucrados en la generación de anomalías del desarrollo. Podemos señalar los siguientes:
Genes que codifican proteínas estructurales: en el caso del colágeno, por ejemplo, las moléculas de procolágeno son enrolladas en estruc-turas tridimensionales para formar la fibra rígida de colágeno. La mu-tación en colágeno 1 causa osteogénesis imperfecta, cuya severidad dependerá de manera predecible de cómo y cuándo ocurra la altera-ción en la formación de las fibrillas; el colágeno tipo IV es necesario en la membrana basal de los riñones y el oído y su mutación ocasiona el síndrome de Alport, caracterizado por sordera y nefritis; el colágeno tipo VII mantiene la dermis y la epidermis juntas y sus alteraciones ocasionan la epidermólisis bullosa.
Genes relacionados con la regulación de la mitosis: Mutaciones en estos genes pueden verse en síndromes asociados con poco crecimien-to o con sobrecrecimiento. En la microcefalia primaria, por ejemplo, se han descrito mutaciones en 5 loci; uno de los más frecuentes es el gen ASPM, el cual determina la orientación del huso mitótico durante la división de las células radiales de la glía de la zona ventricular, y se puede predecir que la mutación del gen lleva en el caso de la microce-falia a una disminución en el número de mitosis y en consecuencia en el número de neuronas de la corteza en desarrollo. Igualmente, exceso de división con apoptosis disminuida ha sido asociado con síndromes de sobrecrecimiento como en el de Proteus o el de Becwith Wiedeman.

altERacIoNES GENÉtIcaS QuE aFEctaN la GEStacIÓN, Clara Eugenia Arteaga D.
244
obstetricia integral Siglo XXI
Genes relacionados con la migración celular: en desórdenes tales como el Hirshprung, donde la migración de las células de la cresta neural no alcanza su localización final para formar los ganglios entéricos. Varias mutaciones génicas han sido asociadas con este mecanismo.
Genes para activación o inactivación de otros genes: esta función es cru-cial en los procesos de diferenciación celular, basados en el control de la transcripción. En algunos síndromes de ambigüedad sexual, verbi-gracia, la mutación en el gen SRY, un factor de transcripción, puede reprimir el desarrollo testicular, impidiendo la activación de otro gen, el SOX9, el cual actúa normalmente diferenciando células de la gónada primitiva en células de Sertoli y en consecuencia desarrollando testí-culo (8).
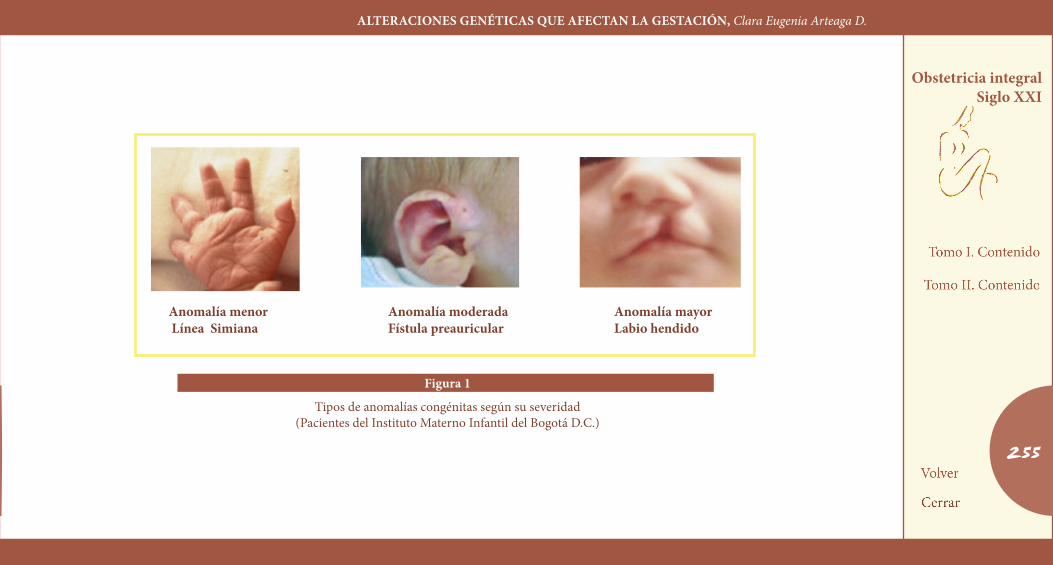
claSIFIcacIÓN DE laS aNoMalíaS coNGÉNItaSLas anomalías del desarrollo pueden presentarse como alteraciones aisladas comprometiendo un órgano o una sola región corporal o for-mando un patrón de múltiples alteraciones. Los defectos congénitos aislados pueden ser subdivididos, a su vez en las categorías de mayores o menores, dependiendo de su severidad (Figura 1).
Las anomalías mayores se definen como aquellas desviaciones del desarrollo que dañan significativamente la función normal corporal o reducen la expectativa normal de vida. Cerca del 3% de los recién nacidos tienen tales anomalías mayores. Alrededor de 2/3 de todos los defectos congénitos mayores son aislados, pero algunos de ellos
pueden ser lo suficientemente severos como para comprometer la vida de los pacientes o producir incapacidad física.
Las anomalías menores son aquellas que no ocasionan daño estético ni funcional grave, ni comprometen la vida de quien las porta. Existe debate con relación al hecho de considerar a las anomalías menores como francas desviaciones del desarrollo, pues son observadas en más del 4% de las personas normales y no ha sido posible establecer su relevancia biológica; algunos las denominan alteraciones de la feno-génesis. Tal consideración ha hecho sugerir que la denominación de anomalía menor se reserve para los casos en los cuales exista un con-texto de anormalidad que en términos prácticos signifique que existen al menos tres de estas anomalías, para explorar activamente la búsque-da de otras anomalías mayores ocultas. Por ejemplo, las hendiduras palpebrales oblicuas, o hendiduras mongoloides, deben considerar-se como anomalía menor en los niños con síndrome de Down, pero como variantes normales en el caso de personas de origen asiático. Por esta razón, aun cuando se presentan en forma aislada, no tienen significado clínico, pero de encontrarse varias en un individuo pueden alertar sobre un cuadro dismórfico (9).
Sin embargo, la definición de anomalía menor ha incluido tradicional-mente a un grupo de alteraciones que, a pesar de no tener importancia estética o funcional, sí constituyen francas desviaciones del desarrollo normal. Un ejemplo de estas alteraciones son las fístulas preauricula-res o las mamilas supernumerarias. Estas alteraciones, a diferencia de las anomalías menores, tienen importancia desde el punto de vista clí-

244
altERacIoNES GENÉtIcaS QuE aFEctaN la GEStacIÓN, Clara Eugenia Arteaga D.
245
obstetricia integral Siglo XXI
nico y deben considerarse para propósitos de asesoramiento genético aun encontrándose en forma aislada. Por esta razón se ha considerado la opción de introducir en esta clasificación una tercera categoría, que se ha denominado anomalías moderadas.
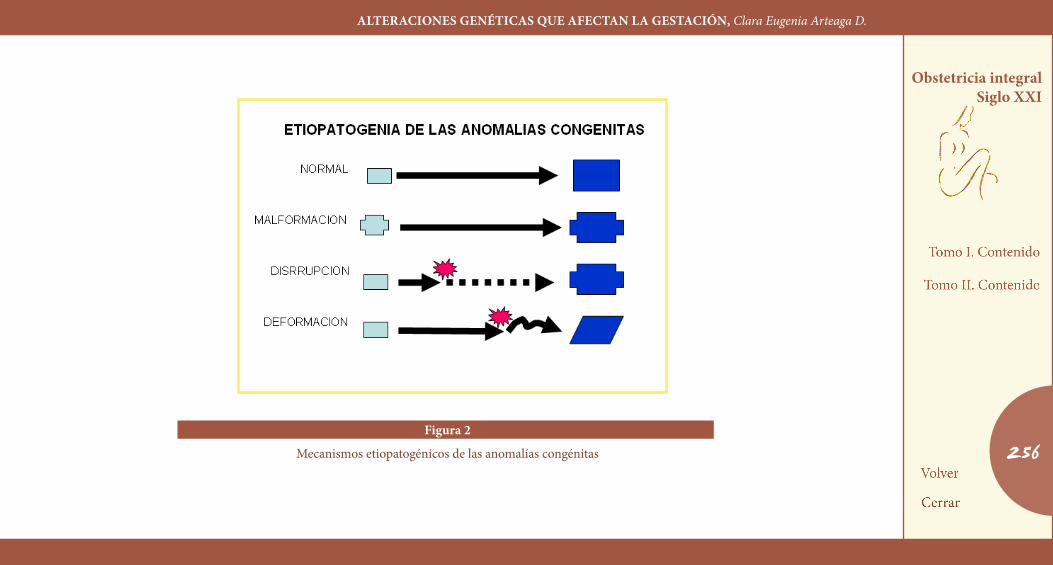
claSIFIcacIÓN EtIopatoGÉNIcaEl conocimiento de la etiopatogenia de las anomalías del desarrollo ha permitido establecer una adecuada denominación y clasificación que ha suplantado a las puramente descriptivas. Utilizamos para este propósito las definiciones recomendadas por un grupo de trabajo in-ternacional (Figura 2) (10).
Esta clasificación parte de considerar el problema de las anomalías congénitas de acuerdo a su origen, es decir, intrínsecas, o producidas por alguna alteración en la información genética del propio embrión, y extrínsecas, esto es, que obedecen a alteraciones externas al embrión, o dependientes del microambiente materno o del macroambiente. De esta consideración surgen dos denominaciones: malformación y dis-rupción.
Malformación es un término asignado a un defecto morfológico de un órgano o de una región más grande del cuerpo, debido a un proceso de desarrollo intrínsecamente anormal. La designación de intrínseco, como lo mencionábamos, hace referencia a los factores hereditarios y determina que la alteración en la morfogénesis se produzca en el pri-mordio de la estructura, como ocurre, digamos, en el labio y paladar
hendidos de origen genético. En contraste con esta definición, aparece la de disrupción, antes llamada malformación secundaria, y que hace referencia a cierto defecto morfológico de un órgano o parte de él, o una región más grande el cuerpo, a consecuencia de una interferencia extrínseca con un proceso de desarrollo originalmente normal. Esta definición excluye la participación etiológica de factores hereditarios, e implica la presencia de factores externos tales como infecciones, dro-gas, agentes físicos, etcétera, como ocurre, por citar un caso, en una hendidura labial secundaria a una brida amniótica. Sin embargo, los factores hereditarios pueden jugar un papel predisponente e influir so-bre el desarrollo de disrupciones.
La denominación de deformación surge como derivada de la defini-ción de disrupción y se refiere a aquellas anomalías causadas por fuer-zas mecánicas inusuales sobre el feto en desarrollo, particularmente en el tercer trimestre. Estas fuerzas mecánicas surgen de varias fuentes, incluyendo la presión ejercida por un gemelo o anomalías uterinas maternas, ologohidramnios, o aun por daños neurológicos del feto que impiden la función muscular normal y el movimiento articular y que podrían llevar, por ejemplo, a una artrogriposis múltiple (10,11).
coMplEJoS DE MÚltIplES aNoMalíaSEn lo que se refiere a los patrones de múltiples anomalías presentes en un individuo, éstas pueden estar etiopatológicamente relacionadas, o ser el producto de un solo proceso patogénico, ocurrir simultánea-

altERacIoNES GENÉtIcaS QuE aFEctaN la GEStacIÓN, Clara Eugenia Arteaga D.
246
obstetricia integral Siglo XXI
mente en un mismo individuo por razones puramente estadísticas o de probabilidad. Con el propósito de expresar cada una de estas posi-bilidades, diversos términos han sido recomendados:
Defecto de campo de desarrollo. Es el patrón de anomalías derivadas de la alteración de un solo campo de desarrollo, definiéndose campo de desarrollo como aquellas partes del embrión en las cuales se con-trolan y coordinan los procesos de desarrollo de estructuras complejas relacionadas e interactuantes entre sí. Esta definición implica un solo proceso malformativo o disruptivo. John Opitz ha postulado que du-rante la blastogénesis temprana el embrión representa una sola unidad morfogenética, es decir, un campo primario de desarrollo. Un ejem-plo de defecto de campo de desarrollo es el complejo de malformacio-nes relacionado con la holoproscencefalia o las anomalías que surgen cuando se altera el campo de desarrollo de la línea media. Martínez Frías y colaboradores consideran que los defectos de lateralidad, tales como las dextrocardias y dextroposiciones, pueden ser consecuencia de defectos en el campo de desarrollo de la línea media (12,13).
Secuencia. Es el patrón de anomalías derivadas de una única, anterior, conocida o presunta anomalía o factor mecánico. La secuencia se con-sidera, entonces, como un concepto patogénico, es decir, hace referen-cia al mecanismo que desencadena la cascada de anomalías, y no como un concepto etiológico. Esto ocurre, por ejemplo, en el complejo de anomalías secundario al oligohidrarmios, conocido como secuencia de Potter, o la secuencia de anomalías derivadas de las valvas uretrales.
Síndrome. Es un patrón de múltiples anomalías relacionadas etioló-gicamente y que no representan ni una secuencia ni un defecto de campo. El síndrome generalmente implica una causa única. En esta definición se incluyen las anomalías cromosómicas y los desórdenes determinados por mutaciones génicas.
Asociación. Es el patrón de anomalías que ocurren juntas en un individuo con una frecuencia mayor que la esperada por la pro-babilidad. De acuerdo con esto, la posibilidad de que un recién nacido tenga dos anomalías congénitas no relacionadas es la pro-babilidad (frecuencia) de que tenga una por la probabilidad (fre-cuencia) de que tenga la otra. Cuando la frecuencia real con la que aparecen estas dos anomalías es mayor que la esperada, hablamos de asociación. La relación entre cada una de las anomalías en una asociación no tiene connotación etiológica o patogénica, sino pu-ramente estadística. El valor diagnóstico de una asociación estri-ba, en primer lugar, en que el reconocimiento de dos o tres de las anomalías estimula a la búsqueda de otras quizá ocultas, y en segundo lugar, en que el riesgo de recurrencia para los otros des-cendientes es muy bajo. Una de las asociaciones más conocidas es la Asociación VATER. Las asociaciones se denominan mediante siglas formadas por la primera letra de los órganos o estructuras comprometidos. Así, VATER deriva de V: anomalías vertebrales, A: ano imperforado, TE: fístula traqueo-esofágica, y R: anomalía renal o radial (14).

246
altERacIoNES GENÉtIcaS QuE aFEctaN la GEStacIÓN, Clara Eugenia Arteaga D.
247
obstetricia integral Siglo XXI
Malformaciones aisladas y herencia multifactorialLas anomalías aisladas representan, sin lugar a dudas, las más frecuen-tes anomalías congénitas, y la mayoría de ellas entran en la categoría de desórdenes de herencia poligénica o multifactorial. Dentro de las más comunes se encuentran los defectos del tubo neural, las anomalías cardiacas, las hendiduras labiopalatinas, la luxación de cadera, el pie equinovaro y otras no detectables al nacimiento que son enfermedades crónicas del adulto, como la diabetes, la enfermedad coronaria, etcé-tera. La herencia multifactorial supone que el componente hereditario de un padecimiento particular depende de varios genes menores que actúan juntos, con la participación de factores ambientales. Los desór-denes multifactoriales tienden a presentarse en grupos familiares en una distribución dispersa, sin un claro patrón mendeliano. Según esto, la heredabilidad dependerá de muchos genes (poligénica) que conjun-tamente producen una susceptibilidad o predisposición a determinada anomalía. La expresión clínica de dicha anomalía se daría cuando un número suficiente de genes en combinación con factores ambientales excede un nivel umbral (15).
Este modelo predice, entonces, que los familiares de un individuo afectado compartirían con él, genes alterados en proporción directa a la cercanía del parentesco. Predice igualmente que el riesgo para los descendientes será mayor si el afectado pertenece al sexo menos fre-cuentemente comprometido.
De acuerdo, también, con este modelo, el riesgo de recurrencia para nuevos descendientes aumenta con el número de parientes afectados y con la gravedad de la anomalía del individuo originalmente afectado.
Una de las más frecuentes anomalías aisladas es el labio hendido con o sin paladar hendido LH (P). El labio hendido ocurre entre la cuarta y octava semana del desarrollo embrionario por una alteración en la formación del paladar primario. Esta alteración puede interferir tam-bién en el desarrollo del paladar secundario dando como resultado el labio y el paladar hendidos. Sin embargo, si la alteración inicial es en el paladar secundario, ésta no interfiere con el desarrollo del paladar primario que ya se ha formado, produciéndose solamente un paladar hendido. Así que existe una diferencia embriológica entre estas dos entidades y, además, una diferencia epidemiológica, por lo cual, para efectos etiológicos, son consideradas como dos entidades indepen-dientes. El LH (P) es uno de los defectos congénitos más comunes y representa la mitad de las anomalías craneofaciales, el 80% de los casos ocurre en el sexo masculino y existe una considerable variación entre grupos étnicos. Así por ejemplo, en caucásicos su frecuencia varía del 1 al 1,27%, en japoneses 1,7% y en negros americanos 0,4%.
En el paladar hendido la frecuencia general es de 0,4%, con muy poca variación étnica y mayor compromiso del sexo femenino. Las hendi-duras labiopalatinas son clasificadas como no sindrómicas, es decir, aisladas y sindrómicas o asociadas a otras anomalías, como ocurre con los más de 400 síndromes monogénicos en los que las hendiduras son parte del cuadro clínico (OMIM). En los casos en los cuales las hendiduras labiopalatinas se encuentran aisladas, la etiología suele ser multifactorial, sin que aún se puedan establecer factores ambientales específicos. Sin embargo, muchas investigaciones indican que los fac-tores genéticos juegan un papel mayor en su etiología, y se han sugeri-

altERacIoNES GENÉtIcaS QuE aFEctaN la GEStacIÓN, Clara Eugenia Arteaga D.
248
obstetricia integral Siglo XXI
do patrones de herencia dominante, recesiva y compleja que suponen la interacción entre varios genes y entre genes y factores ambientales. Dentro de los genes candidatos se han sugerido loci sobre la región 2p13 (TGFA), 4p16 (MSX1), 6p23-25, 14q24 (TGFB3), 17q21 (RARA) y 19q13 (CLPTM1,BCL3), entre otros (8).
Un grupo muy importante —y el más numeroso— de malformaciones aisladas son las cardiopatías congénitas (CC), con una frecuencia de 1% en recién nacidos y del 10% de malformación cardiaca severa en abortos espontáneos. Durante muchos años se atribuyó a las CC una etiología multifactorial, pero hoy se sabe que muchos defectos cardía-cos están relacionados con mutaciones en genes, relacionados directa o indirectamente con la morfogénesis cardiaca. El desarrollo de un órgano tan complejo como el corazón, con sus muchas estructuras asociadas y tipos celulares, supone una serie de eventos moleculares y genéticos que hasta ahora empiezan a ser dilucidados con la utiliza-ción de modelos de desarrollo en vertebrados e invertebrados. Estos modelos han mostrado mecanismos altamente conservados evolutiva-mente, disparados por señales moleculares específicas y mediados por factores igualmente específicos de transcripción de tejidos. Este pro-grama controla la diferenciación a cardiomiocitos de las células stem del mesodermo embrionario y la subsiguiente activación de genes de morfogénesis y de contractilidad. Genes como el de la proteína mor-fogenética del hueso (BMP) y el Nkx2.5, son los más tempranos mar-cadores de morfogénesis cardiaca. Los genes GATA, actuando como factores de transcripción, juegan un papel importante en la formación del tubo cardiaco; y el Sonic hedgehog y Nodal, pertenecientes a la
familia de factores de crecimiento transformante en el establecimiento de la dirección del asa cardiaca y el Ptx-2, en la interpretación de seña-les derecha-izquierda. Así mismo, genes específicos actuando para la formación de las cámaras, los septos, los sistemas de conducción y la vasculatura del corazón, han sido hoy elucidados, y este conocimiento ha permitido empezar a descubrir los eventos que subyacen a la car-diopatía congénita (16,17).
Otro grupo de especial importancia por su frecuencia en RN y en ma-yor proporción en mortinatos, son las anomalías del SNC. Dentro de éstas, los defectos del tubo neural tienen la importancia adicional de contar con la posibilidad de un diagnóstico prenatal preciso, a partir del establecimiento de una población de riesgo, conformada por las parejas con antecedentes de hijos previos afectados y por gestantes con valores elevados (percentil 95) de alfafetoproteína sérica.
Estas alteraciones incluyen la anencefalia (ausencia parcial o completa de bóveda craneana y ausencia de cerebro), meningoceles (protrusión de las meninges a través de defectos de cierre de los arcos vertebrales), encefalocele (protrusión del cerebro y sus membranas a través del crá-neo) y espira bífida (defecto de cierre de los arcos vertebrales).
En estos defectos existe una notable variación geográfica con relación a su frecuencia general; así por ejemplo, la incidencia total señalada para las Islas Británicas es de 4,5 a 5% RN y en EE. UU. la frecuencia señalada es de 1,5 a 2% RN, con menor incidencia en negros y orien-tales. Sin embargo, en sitios considerados como de mayor incidencia, como es el caso de Irlanda oeste, ésta ha descendido desde 1976, cuan-

248
altERacIoNES GENÉtIcaS QuE aFEctaN la GEStacIÓN, Clara Eugenia Arteaga D.
249
obstetricia integral Siglo XXI
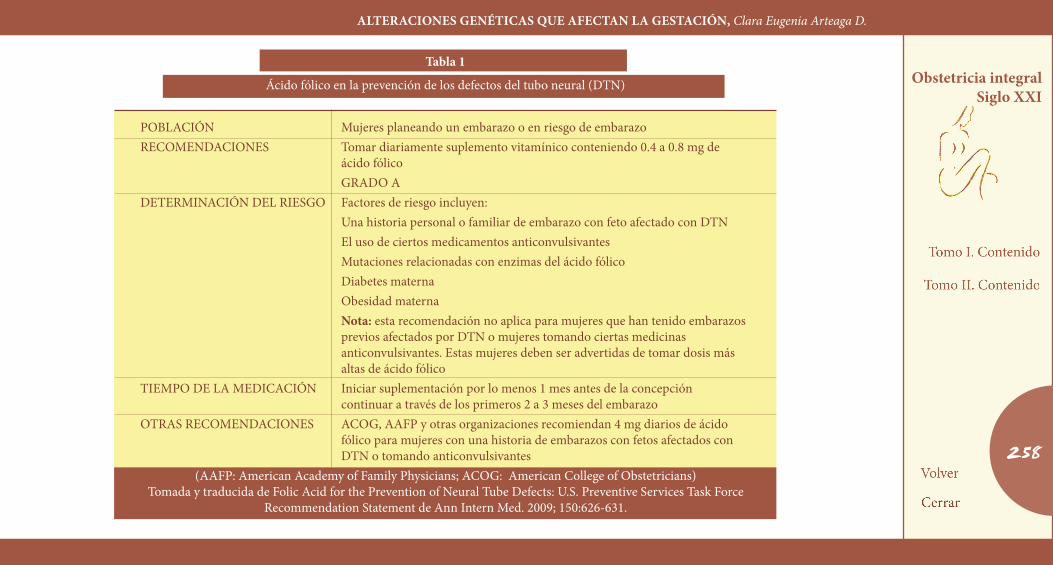
do se implementó el programa de screening masivo para diagnóstico prenatal con la determinación en suero de alfafetoproteína en mujeres gestantes entre las semanas 16 y 20. Adicionalmente había sido es-tablecido que la deficiencia de ácido fólico estaba relacionada con la aparición de defectos del tubo neural. En 1976 la tasa era de 5,1% y en 1980 de 4%, sugiriendo que es posible la prevención primaria de los defectos del tubo neural. En 1996 la U. S. Preventive Services Task For-ce (USPSTF) recomendó, en base a varios estudios que demostraron una reducción estadísticamente significativa de los defectos del tubo neural en mujeres que tomaron ácido fólico en el período periconcep-cional, que quienes estuvieran planeando un embarazo tomaran una dosis diaria de multivitaminas con un contenido de ácido fólico de 0,4 a 0,8 mg, empezando por lo menos 1 mes antes de la concepción y con-tinuando durante el primer trimestre —recomendación grado A— y el consumo de multivitaminas conteniendo una dosis diaria de 0,4 mg de ácido fólico para mujeres en riesgo de embarazo no planeado, con la finalidad de reducir los defectos del tubo neural —recomendación grado B— (Tabla 1) (18).
La hidrocefalia es otra de las anomalías del SNC que puede presen-tarse en forma aislada e implica un riesgo alto de muerte perinatal o de secuelas neurológicas graves. Esta entidad es muy heterogénea en su origen (genética, infecciosa, asociada a prematurez) y se considera que alrededor del 50% de las hidrocefalias infantiles tienen un origen prenatal, muchas con aparición congénita, o sea, con manifestaciones al nacimiento.
La etiología de esta entidad puede ser poligénica, ligada al cromosoma X, autosómica recesiva, o deberse a una alteración cromosómica o a una infección intrauterina (particularmente toxoplasmosis) o a altera-ciones en el desarrollo del SNC como holoproscencefalia, hidranence-falia, encefalomalasia, quistes, papilomas de plexos coroideos, etcéte-ra. La incidencia encontrada en la zona metropolitana de Atlanta entre 1970 y 1983 fue de 0,48 a 0,56% RN.
Otras patologías poligénicas de menor importancia por su más bajo impacto sobre la salud y la expectativa de vida, registran las siguientes incidencias:
•Estenosispilórica 1,52%RN •Pieequinovaro 1,77%,2,56-2,74%RN •Atresiaesofágica 0,31%RN •Luxacióncongénitadecadera 0,79-0,89%RNComplejos de múltiples anomalías: del complejo de malformaciones congénitas consideraremos, como fue señalado, los síndromes, se-cuencias, asociaciones y defectos de campo de desarrollo.
En el grupo de los síndromes, examinaremos los de etiología monogé-nica y las anomalías cromosómicas.
Los desórdenes monogénicos son individualmente entidades muy ra-ras, pero en conjunto pueden afectar al 1% de los RN vivos y al 7% de los mortinatos. La frecuencia total de desórdenes monogénicos es difícil de establecer porque sólo un 25% de ellos se manifiestan al na-cimiento.

altERacIoNES GENÉtIcaS QuE aFEctaN la GEStacIÓN, Clara Eugenia Arteaga D.
250
obstetricia integral Siglo XXI
El primer grupo a considerar son las entidades de herencia autosómica dominante. En este tipo de herencia la enfermedad es expresada en el heterocigoto, es decir, que basta la presencia de un alelo mutante del par para producir una alteración del fenotipo. En términos generales, el producto del gen normal es, la mayoría de las veces, una proteína estructural, no enzimática, tal como el colágeno o proteínas compo-nentes de un receptor de membrana, aun cuando para la gran mayoría de los defectos autosómicos dominantes la naturaleza de la proteína defectuosa o anormal es completamente desconocida.
La probabilidad que tiene un individuo afectado de transmitir el gen a la progenie en cada embarazo es del 50%, siendo igual la proporción de hombres y mujeres afectados. En estos desórdenes la transmisión ocurre verticalmente de padre a hijo; sin embargo, la gravedad de la mayoría de estas enfermedades puede disminuir en los individuos su capacidad reproductiva, haciendo que muchas de estas enfermedades se presenten por primera vez en una familia como el producto de una mutación nueva. Por ejemplo, en la acondroplasia cerca del 80% de los casos corresponden a mutaciones nuevas.
Se señalan las frecuencias en RN de algunas entidades con herencia dominante que pueden manifestarse en el nacimiento:
•Osteogénesisimperfecta 0,04%RN
•Acondroplasia 0,02%RN
•SíndromedeMarfan 0,04%RN
•SíndromedeVanderWoude 0,01½RN
Algunas otras patologías de herencia autosómica dominante pueden ser muy frecuentes aun cuando no dan manifestaciones tempranas, como ocurre en el caso de la hipercolesterolemia familiar, cuya inci-dencia señalada es del 2%.
En las entidades con herencia autosómica recesiva el producto del gen mutante para la mayoría de las entidades descritas hasta la fecha es una enzima defectuosa o en cantidad anormal. Estos desórdenes son clíni-camente aparentes sólo en estado homocigoto, esto es, cuando ambos alelos de un locus particular son anormales.
En dichas patologías los progenitores son sanos, pero portadores de un gen mutado; ambos sexos son igualmente afectados, no ocurre trans-misión vertical y el riesgo para la progenie después de un hijo afectado es del 25%. Son desórdenes que se presentan con más frecuencia en uniones consanguíneas, de tal manera que si el riesgo de base para cualquier anomalía es del 3%, para un hijo de padres consanguíneos, será del 4 al 5% para productos de primos en primer grado.
Una de las patologías recesivas más frecuentemente observadas al na-cimiento es la hiperplasia suprarrenal congénita por deficiencia de la enzima 21 hidroxilasa, la causa más frecuente de ambigüedad sexual; en recién nacidos se señala una frecuencia de 0,2% para esta enferme-dad en general y de 0,03 a 0,06% RN para la forma perdedora de sal.
En otras patologías recesivas manifiestas en el RN, se señalan las si-guientes frecuencias:

250
altERacIoNES GENÉtIcaS QuE aFEctaN la GEStacIÓN, Clara Eugenia Arteaga D.
obstetricia integral Siglo XXI
251
•Fibrosisquística 0,5%RN(caucásicos)
•Fenilcetonuriaclásica 0,1½RN
•Enf.poliquísticarenal 0,1%RN
•SíndromedeMeckel-Gruber 0,3½RN
El último grupo de las patologías monogénicas son los desórdenes li-gados a X. Los genes responsables de ellos están localizados sobre el cromosoma X, de manera que el riesgo clínico y la severidad de la en-fermedad es diferente en cada uno de los dos sexos. Para este grupo el hombre es considerado hemicigótico en relación con los genes ligados a X, puesto que al tener un solo cromosoma X la presencia de un alelo mutante en dicho cromosoma es suficiente para que la enfermedad se exprese; mientras que las mujeres pueden ser homocigotas, es decir, presentar el alelo mutado en ambos locus y padecer la enfermedad, situación muy infrecuente; o ser heterocigotas, esto es, presentar la mutación en un solo locus y tener la condición de portadoras sanas. Cuando es el padre el afectado, todos sus hijos varones serán sanos, pues del padre reciben el cromosoma Y, y todas sus hijas mujeres serán heterocigotas portadoras; para las madres portadoras, los hijos varo-nes tendrán un 50% de riesgo de ser hemicigotos afectados y el 50% hemicigotos sanos, y las hijas mujeres un riesgo del 50% de ser hete-rocigotas portadoras sanas y el 50% de ser homocigotas sanas. Por tal razón las características principales de este tipo de patologías son, en primer lugar, una notoria mayor incidencia en varones, y en segundo lugar, ser transmitida a través de una serie de mujeres portadoras y de hombres afectados (19).
La mayoría de las patologías ligadas a X no se manifiestan en el RN. Dentro del grupo de aquéllas manifiestas al nacimiento, se señalan las siguientes frecuencias:
•HidrocefalialigadaaX 0,1%RN
•IctiosiscongénitaligadaaX 0,1%RN
•RetardomentalligadoaX 0,1%RN
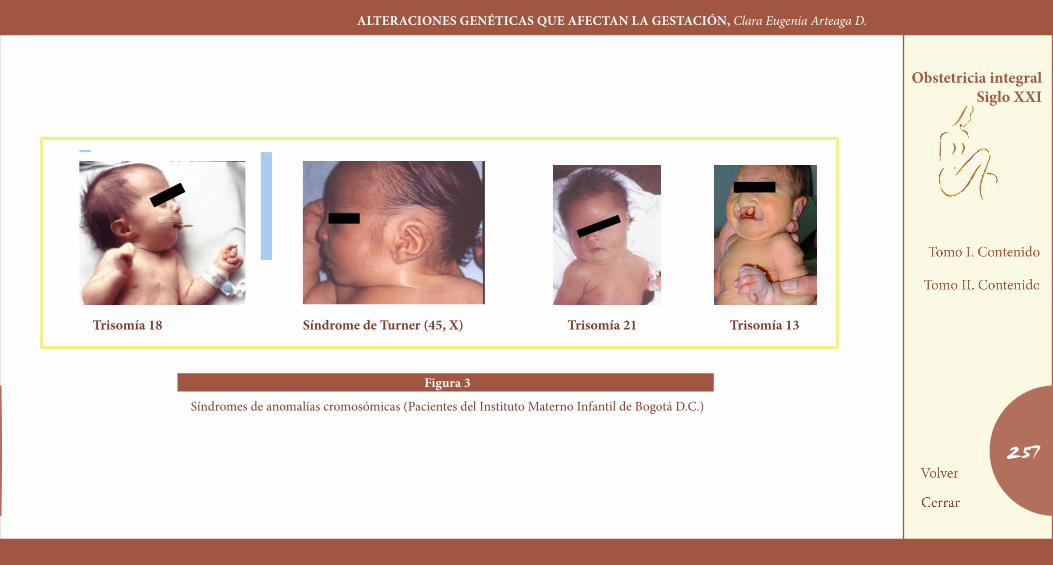
Otro gran grupo de síndromes de múltiples malformaciones lo cons-tituyen las anomalías cromosómicas. Desde los últimos años de la dé-cada de los cincuenta, cuando se creó la técnica que permitió obser-var y contar los cromosomas humanos, hasta el momento actual, en que se dispone de técnicas para identificar con precisión cada uno de los cromosomas e identificar mínimas alteraciones estructurales, ha sido posible acumular suficiente experiencia que resume el impacto de la anomalía cromosómica sobre las malformaciones congénitas, las muertes perinatales y las pérdidas fetales tempranas y tardías.
Dentro de las anomalías cromosómicas se distinguen dos categorías: las anomalías numéricas, casi todas resultado de una mutación nueva que involucra una no disyunción de los cromosomas durante la divi-sión celular, dando como resultado exceso de uno o más cromosomas, o sea, un complemento cromosómico de 47 (trisomía), 48 (tetraso-mía) etcétera, o el déficit de un cromosoma dando un complemento de 45 (monosomía) (Figura 3). Dentro de este grupo cromosómico pueden observarse también duplicaciones completas del set cromosó-mico, produciendo poliploidías.

altERacIoNES GENÉtIcaS QuE aFEctaN la GEStacIÓN, Clara Eugenia Arteaga D.
obstetricia integral Siglo XXI
252
El ejemplo mejor conocido de una trisomía es el de la trisomía 21 o síndrome de Down, la causa más frecuente de retardo mental en niños preescolares. Para este síndrome se señalan cifras de frecuencia del 1,6% RN, 1,45% RN, 1,25% RN, 0,96% RN, etcétera, mostrando un aumento significativo de las frecuencias con el incremento de la edad materna.
La alta frecuencia de síndrome de Down en mujeres sin riesgo previo por edad hizo que metodologías de tamizaje masivo fueran introduci-das en el diagnóstico prenatal. El tamizaje en suero materno para sín-drome de Down ha sido ofrecido en el Reino Unido desde 1980, cuan-do se estableció la asociación de síndrome de Down con niveles bajos de alfafetoproteína y de estriol no conjugado (nE3). Sólo hasta 1992 fue posible determinar que esta metodología era útil y se incorporó a la rutina de diagnóstico prenatal. Las pruebas de tamizaje han evo-lucionado y nuevos métodos ecográficos, como la sonolusencia nucal o bioquímicos como la proteína A del embarazo, han empezado a ser utilizados en la rutina clínica. El tamizaje del primer trimestre solo o en combinación con tamizaje de segundo trimestre mostraba una rata de detección total de casos de síndrome de Down de por lo menos 75% y una rata de falsos positivos del 3% hasta abril de 2007 (20).
Para otras anomalías cromosómicas numéricas se señalan las siguien-tes frecuencias:
•Trisomía18 0,2%RN
•Trisomía13 0,06%RN
•47XXY(Klinefaelter) 1,25-1,58%RNmasculinos
•47XYY 1,4%RN
•45X0(Turner) 0,66%-1,41%RNfemeninos
Las anomalías estructurales surgen como el resultado de rupturas y reuniones anormales dentro de dos cromosomas. Una proporción im-portante de ellas son transmitidas de padres a hijos. Las principales son las translocaciones, las cuales involucran un intercambio de frag-mentos entre dos cromosomas y pueden ser balanceadas cuando el rearreglo no implica ni pérdida ni ganancia de material cromosómico y desbalanceadas cuando el rearreglo conduce a pérdida o ganancia de material cromosómico; las deleciones, que implican pérdida parcial de fragmentos cromosómicos; las duplicaciones, cuando hay ganancia de fragmentos de cromosomas específicos; y otras menos frecuentes, como inversiones y anillos cromosómicos.
Otros complejos de malformaciones congénitas son las asociaciones. Para el caso de la asociación VATER la frecuencia es de de 0,16% RN.
anomalías congénitas y factores ambientalesSe considera que cerca del 10% de todas las anomalías congénitas son el resultado de la exposición a un agente teratogénico in útero. Un teratógeno puede ser definido como una droga, o agente químico, infeccioso o físico, enfermedad materna o estado metabólico mater-no alterado, que actuando sobre el embrión o el feto interrumpen su

252
altERacIoNES GENÉtIcaS QuE aFEctaN la GEStacIÓN, Clara Eugenia Arteaga D.
obstetricia integral Siglo XXI
253
desarrollo normal y causan una anomalía estructural o funcional evi-denciable posnatalmente. La tragedia de la talidomida abrió el campo de la investigación en teratología. Uno de los más grandes estudios sobre la posible asociación entre el uso de drogas durante la gestación y malformaciones congénitas data de 1958, como parte del Proyecto Colaborativo Perinatal, que colectó información de 12 hospitales es-tadounidenses; el reporte terminó en 1965 y fue publicado 12 años después, describiendo el uso de drogas y los efectos sobre el feto en más de 50.000 gestaciones. Muchos estudios han sido publicados des-de entonces.
La exposición a ciertos fármacos y agentes de fuentes ambientales como los químicos de la industria agrícola o petroquímica, son una amenaza creciente. Entre los fármacos con mayor efecto teratogéni-co conocido, además de la talidomida, se encuentran los retinoides. Estas sustancias, incluida la vitamina A, son esenciales en la embrio-génesis normal; mediando procesos como proliferación y diferencia-ción actúan en conjunto con la familia de genes llamada receptores de retinoides, y se considera que su efecto deletéreo sobre el feto puede comprometer cualquier órgano o sistema. El ácido valproico, al igual que casi todos los medicamentos anticonvulsivantes, tiene efecto te-ratogénico, afectando el sistema nervioso y el desarrollo craneofacial, cardiovascular y musculoesquelético. Se calcula que del 1 al 2% de los fetos expuestos a ácido valproico desarrollarán DTN.
A pesar del poco conocimiento sobre las vías moleculares que son alte-radas para generar el efecto teratogénico, nueva tecnología como la de
los microarreglos y el conocimiento creciente sobre la identificación y clasificación de los genes podrán generar información valiosa sobre las alteraciones que ocurren en el genoma global y conocer los genes in-volucrados en estos procesos anormales para posibilitar la prevención en forma racional (21,22).
REFERENcIaS1. Evans Hl. Chromosome anomalies among livebirth. J Med Genet. 1997; 14: 309-
312.2. petrini J, Dannus K, Rusell R. Contribution of birth defects to infant mortality
in the United States. Teratology 2002; 66: 3-6.3. pabón a, Ruiz l. La mortalidad en Colombia, vol 5. Niveles ajustados de
mortalidad por secciones del país 1973-1985 y análisis de causas por sexo y edad 1979-1991. Bogotá: Instituto Nacional de Salud; 1986.
4. Yepez FJ. La salud en Colombia. Documento general. Estudio sectorial de salud, t. 1. Ministerio de Salud y Departamento Nacional de Planeación. Bogotá: Pre-sencia; 1990.
5. Monsalve aM, londoño Ic, ocampo J, cruz DF, Saldarriaga W, Isaza c. Distribución geográfica en Cali, Colombia, de malformaciones congénitas. Hos-pital Universitario del Valle, marzo de 2004-febrero de 2005. Colombia Médica; 2006.
6. castilla EE, orioli lM. ECLAMC: The Latin-American Collaborative Study of Congenital Malformations. Community Genet. 2004; 7: 76-94.
7. Weatherall Jac, DeWals p, lechat MF. Evaluation of information systems for the surveillance of congenital malformations. Int J Epidemiol. 1964; 13: 193-196.
8. lidral ac, Murray Jc. Genetic Approaches to Identify Disease Genes for Birth Defects with Cleft Lip/Palate as a Model. Birth Defects Research 2004; 70: 893-901.

altERacIoNES GENÉtIcaS QuE aFEctaN la GEStacIÓN, Clara Eugenia Arteaga D.
obstetricia integral Siglo XXI
254
9. aase J. Dysmorphologic diagnosis for the pediatric practitioner. Ped Clin North Am. 1992; 39(1): 135-156.
10. Spranger J, Bernischke K, Hall Jo, lenz W, lowry RB, opitz JM, pinsky l et al. Recommendations of an internacional working group. J Ped. 1982; 100(1): 160-165.
11. Merks JHM, Van Karnebeek cDM, caron HN, Hennekam R. Phenotypic Ab-normalities: Terminology and Classification. American Journal of Medical Gene-tics 2003; 123A: 211-230.
12. opitz JM, Zanni G et al. Defects of blastogenesis. Am J Med Genet. 2002; 115(4): 269-286.
13. Martínez Ml, Frías Jl. Primary developmental field III: clinical and epidemio-logical study of blastogenetic anomalies and their relationship to different MCA patterns. Am J Med Genet. 1997; 70(1): 11-15.
14. Shaw-Smith c. Oesophageal atresia, tracheooesophageal fistula, and the VAC-TERL association: review of genetics and epidemiology. J Med Genet. 2006; 43: 545-554.
15. carter co. Genetic of common single malformations. Br Med Bull. 1976; 32.16. Nora JJ, Nora aH. The genetic contribution to congenital heart diseases. In:
Nora JJ, Takao A, eds. Congenital heart diseases: causes and processes. New York: MtKisko;1984.
17. Srivastava D. Genetic regulation of cardiogenesis and congenital heart disease. Ann Rev Pathol Mech Dis. 2006; 1: 199-213.
18. u. S. preventive Services task Force. Clinical Guidelines Folic Acid for the Prevention of Neural Tube Defects: U.S. Preventive Services Task Force Recom-mendation Statement. Ann Intern Med. 2009; 150: 626-631.
19. Motulsky aG. Formal Genetics of Humans: Modes of Inheritance. In: Vogel and Motulsky’s Human Genetics, Problems and Approaches Fourth. Completely Revi-sed Edition; 2010.
20. Gidiri M, McFarlane J, Holding S, lindowa SW. Maternal serum screening for Down syndrome: are women’s perceptions changing? BJOG An International Journal of Obstetrics and Gynaecology; 2007.
21. Finnell RH, Gelineau-van Waes, Eudy JD, Rosenquist tH. Molecular basis of environmentally induced birth defects. Annu Rev Pharmacol Toxicol. 2002; 42: 181-208.
22. Källén Ba. Methodological issues in the epidemiological study of the teratoge-nicity of drugs. Congenital anomalia 1987; 27(1).
254
altERacIoNES GENÉtIcaS QuE aFEctaN la GEStacIÓN, Clara Eugenia Arteaga D.
Volver
obstetricia integral Siglo XXI
255
Figura 1
Tipos de anomalías congénitas según su severidad(Pacientes del Instituto Materno Infantil del Bogotá D.C.)
anomalía menor anomalía moderada anomalía mayor línea Simiana Fístula preauricular labio hendido
altERacIoNES GENÉtIcaS QuE aFEctaN la GEStacIÓN, Clara Eugenia Arteaga D.
Volver
obstetricia integral Siglo XXI
256Figura 2
Mecanismos etiopatogénicos de las anomalías congénitas
256
altERacIoNES GENÉtIcaS QuE aFEctaN la GEStacIÓN, Clara Eugenia Arteaga D.
Volver
obstetricia integral Siglo XXI
257
Figura 3
Síndromes de anomalías cromosómicas (Pacientes del Instituto Materno Infantil de Bogotá D.C.)
trisomía 18 Síndrome de turner (45, X) trisomía 21 trisomía 13
altERacIoNES GENÉtIcaS QuE aFEctaN la GEStacIÓN, Clara Eugenia Arteaga D.
Volver
obstetricia integral Siglo XXI
258
tabla 1
Ácido fólico en la prevención de los defectos del tubo neural (DTN)
POBLACIÓN Mujeres planeando un embarazo o en riesgo de embarazoRECOMENDACIONES Tomar diariamente suplemento vitamínico conteniendo 0.4 a 0.8 mg de ácido fólico GRADO ADETERMINACIÓN DEL RIESGO Factores de riesgo incluyen: Una historia personal o familiar de embarazo con feto afectado con DTN El uso de ciertos medicamentos anticonvulsivantes Mutaciones relacionadas con enzimas del ácido fólico Diabetes materna Obesidad materna Nota: esta recomendación no aplica para mujeres que han tenido embarazos previos afectados por DTN o mujeres tomando ciertas medicinas anticonvulsivantes. Estas mujeres deben ser advertidas de tomar dosis más altas de ácido fólicoTIEMPO DE LA MEDICACIÓN Iniciar suplementación por lo menos 1 mes antes de la concepción continuar a través de los primeros 2 a 3 meses del embarazoOTRAS RECOMENDACIONES ACOG, AAFP y otras organizaciones recomiendan 4 mg diarios de ácido fólico para mujeres con una historia de embarazos con fetos afectados con DTN o tomando anticonvulsivantes
(AAFP: American Academy of Family Physicians; ACOG: American College of Obstetricians)Tomada y traducida de Folic Acid for the Prevention of Neural Tube Defects: U.S. Preventive Services Task Force
Recommendation Statement de Ann Intern Med. 2009; 150:626-631.