abordaje de los errores congÉnitos del …
TRANSCRIPT

CURSO DE FORMACIÓN CONTINUADA A
DISTANCIA 2011-2012
TALLER DEL LABORATORIO CLÍNICO
Nº 4
ABORDAJE DE LOS ERRORES
CONGÉNITOS DEL METABOLISMO
EN EL LABORATORIO CLÍNICO

I.S.S.N.- 1988-7469
Título: Taller del Laboratorio Clínico
Editor: Asociación Española de Biopatología Médica
Maquetación: AEBM
Fecha de Distribución: febrero de 2012

635
Abordaje de los errores congénitos del metabolismo en el laboratorio clínico
Julien S. Crettaz.- BIR 3er año Análisis clínicos. Antonio Rus Martínez.- FEA Análisis clínicos. Área de Diagnóstico Biomédico.
Hospital San Pedro. Logroño. La Rioja.
1. Introducción
Las metabolopatías o errores congénitos del metabolismo (ECM) son enfermedades
genéticas en su gran mayoría autosómicas. Se caracterizan por la alteración de una
proteína (enzima, transportador o cofactor) que produce el bloqueo de un proceso
metabólico con distintas posibles consecuencias: acumulación del sustrato (previo al
paso bloqueado), déficit del producto o activación de rutas alternativas con
acumulación de metabolitos tóxicos.
Las ECM se pueden clasificar en cuanto a su fisiopatología en 3 grupos:
1. Alteración de la síntesis o catabolismo de moléculas complejas. A este grupo
pertenecen las enfermedades lisosomales y peroxisomales así como las
alteraciones intracelulares como la hemocromatosis o el déficit de α1-
antitripsina.
2. Enfermedades por acúmulo de sustancias tóxicas. Comprenden las
aminoacidopatías, las acidurias orgánicas, los defectos del ciclo de la urea y
las intolerancias a azúcares
3. Enfermedades por déficit energético. En este grupo se encuentran los
trastornos de la β-oxidación, de la cadena respiratoria mitocondrial, la
glucogenosis y las acidemias lácticas congénitas.

636
Además existe un conjunto de patologías congénitas que tradicionalmente se han
incluido en el grupo de ECM aunque no sean enfermedades metabólicas
estrictamente hablando. Se suelen definir como enfermedades endocrino-
metabólicas.
Todas los ECM son de baja prevalencia y pertenecen al grupo de “enfermedades
raras”, desde la más común, la fenilcetonuria, hasta otras con sólo unos pocos casos
descritos. Sin embargo debido a su gran número (hay descritas más de 4000) en su
conjunto llegan a presentar una prevalencia notable cercana a 1:300-600 recién
nacidos vivos (RNv) (1).
Se estima que un 50% de ellas se presentan en la edad neonatal pero un 10% lo
hacen entre la pubertad y los 50 años y otro 10% en mayores de 50 años (2). Sus
secuelas más comunes son la desnutrición, las convulsiones y el retraso mental. El
tratamiento es, en muchos casos, de tipo nutricional, basado en dietas con algún tipo
de restricción o con aporte especial de vitaminas y cofactores deficientes.
El abordaje de este tipo particular de patología se puede realizar de 2 maneras
distintas. Por una parte, existen programas de cribado sistemático de estos errores
congénitos para descartar esos cuadros en toda la población. Sin embargo
numerosos ECM no pueden ser detectados incluso con las nuevas técnicas de cribado
ampliado. Por otra, puede existir una sospecha clínica (un síntoma o una alteración
analítica) donde el profesional deberá realizar las pruebas analíticas indicadas y estar
alerta ante posibles alteraciones sugestivas de tales alteraciones.

637
2. Los ECM y el laboratorio
Los datos básicos de laboratorio que se deben estudiar ante la sospecha de un ECM
se presentan en la Tabla 1. En orina podríamos destacar que su olor es característico
de algunos ECM (por ejemplo la enfermedad de la orina con olor a jarabe de arce) y
que la cetonuria en el periodo neonatal siempre es anormal y debe ser investigada.
Los cuerpos reductores están presentes en la galactosemia y la intolerancia a la
fructosa. En sangre el anion gap va a orientar sobre el origen de las acidemias: las
acidemias orgánicas suelen cursar con anion gap aumentado y cetoacidosis mientras
que los defectos del catabolismo lipídico no producen cetoácidos pero sí
hipoglucemia por consumo. Ésta última debe estudiarse en todos los casos cuando
es severa y prolongada en el tiempo ya que es muy común en los ECM al igual que el
aumento de lactato y piruvato. La hipermamonemia puede hacer sospechar de un
defecto del ciclo de la urea o de una acidemia orgánica. Algunas acidurias orgánicas
cursan con granulocitopenia y trombopenia.
Ante cualquier sospecha de ECM, la recogida de muestra es importante. Antes de
iniciar cualquier tratamiento y en fase aguda se debe recoger suero, plasma y orina.
Cabe recordar que el líquido cefalorraquídeo es la muestra de elección para
diagnosticar los ECM de los neurotransmisores y las pterinas. Estas muestras se

638
deben almacenar congeladas para posibles análisis a posteriori. En caso de
fallecimiento se deben recoger biopsias de músculo, hígado y piel para realizar
posibles estudios enzimáticos.
3. El cribado neonatal
Los programas de cribado neonatal (PCN) deben detectar presintomáticamente las
ECM mediante pruebas aplicables a toda la población. Son actuaciones de Salud
Pública cuyo objetivo es la identificación temprana y el tratamiento de los individuos
afectados, de forma que la intervención médica a tiempo reduzca la morbilidad,
mortalidad y las posibles discapacidades asociadas a dichas enfermedades. Tales
programas usan técnicas de alta sensibilidad para seleccionar pacientes con alta
probabilidad de padecer una enfermedad que deberá ser confirmada en muchos
casos mediante uno o varios ensayos diagnósticos.
3.1 El cribado clásico
La selección de las enfermedades susceptibles de ser detectadas mediante tales
programas está en continuo debate. La Organización Mundial de la Salud (OMS)
definió unos criterios clásicos que debe cumplir una enfermedad para ser objeto útil
de un cribado, los criterios Wilson-Junger de 1968 (3).
a) La enfermedad debe tener una alta morbimortalidad si no se diagnostica en el
periodo neonatal.
b) La enfermedad no se detecta por un simple examen físico.
c) El tratamiento precoz existe y mejora significativamente el pronóstico.
d) Su prevalencia es relativamente elevada (>1/10.000-15.000 RNv).
e) Existe un procedimiento analítico de cribado rápido, fiable y de bajo coste.

639
Son sólo 2 las patologías que cumplen estos criterios y por tanto las que se incluyen
en el “cribado clásico”, el hipotiroidismo congénito y la fenilcetonuria. Con el
tiempo estos criterios restrictivos se han ido revisando y otras enfermedades han
sido incluidas en los PCN dependiendo de cada país o sistema sanitario con una
elevada heterogeneidad.
En el cribado clásico se han utilizado diferentes técnicas analíticas como la
fluorimetría, la inmunofluorescencia, los enzimoinmunoensayos (ELISA) y la
cromatografía en capa fina.
Uno de los revulsivos del cribado fue la capacidad de obtener sangre capilar del talón
del recién nacido impregnada en papel absorbente. Este paso ha sido clave para el
desarrollo universal de un cribado neonatal. Esta muestra es estable a temperatura
ambiente y permite su envío por correo tradicional (4). La orina se puede recoger de
la misma manera en papel absorbente.
3.2 El cribado ampliado
En la última década se ha incrementado el uso de la espectrometría de masas en
tándem (TMS) en la identificación de enfermedades metabólicas hereditarias. Esta
técnica se basa en la detección de aminoácidos y acilcarnitinas que, tras su
fragmentación, pueden ser caracterizadas por sus iones resultantes. La capacidad de
detección simultánea de varios analitos con un solo análisis sobre una única muestra
(la misma que el cribado clásico) ha hecho que se considere como una tecnología
muy útil y que se amplíe enormemente las lista de enfermedades detectadas. Con el
tiempo se ha asignado el término “cribado ampliado” a la detección de los ECM
usando esta tecnología. Además la detección de ciertas patologías (fenilcetonuria por
ejemplo) ya contempladas en el cribado clásico presenta claras mejoras si se hace

640
mediante TMS (analizando el índice fenilalanina/tirosina) (1). Uno de los estudios
más amplios realizados a día de hoy es el publicado por Frazier et al. donde el autor
expone los datos de un programa de cribado ampliado con 1 millón de niños
analizados a lo largo de 8 años en Carolina del norte (EE.UU.) diagnosticándose 219
de ellos con una ECM (5). De manera global, este programa de cribado mediante
TMS obtuvo una sensibilidad del 93-100%, una especificidad mayor del 99%, un VPP
del 53% y un VPN del 99,99%.
Aunque el equipamiento de la TMS es caro (500.000 $), puede procesar una muestra
en cuestión de minutos a un coste marginal (10 $ por niño) (6). Sin embargo el
cribado sólo representa una parte del coste total ya que un resultado positivo implica
su seguimiento por especialistas y la realización de otras pruebas diagnósticas.
Sin embargo, la baja prevalencia de las ECM dificulta el consenso sobre valores de
corte lo que unido a la ausencia de guías clínicas y de trabajo hacen que esta
tecnología se encuentre aún en fase de desarrollo pero cada vez más utilizada.
El TMS tiene la capacidad de detectar múltiples alteraciones del metabolismo de los
aminoácidos, del ciclo de la urea, de la oxidación de los ácidos grasos y las acidurias
orgánicas. Sin embargo existen otras ECM de cierta prevalencia que deben ser
cribados con métodos más específicos como las alteraciones de los hidratos de
carbono, del metabolismo de la creatinina o las enfermedades lisosomales entre
otras.
3.3 Situación actual del Cribado Neonatal en España y en el mundo
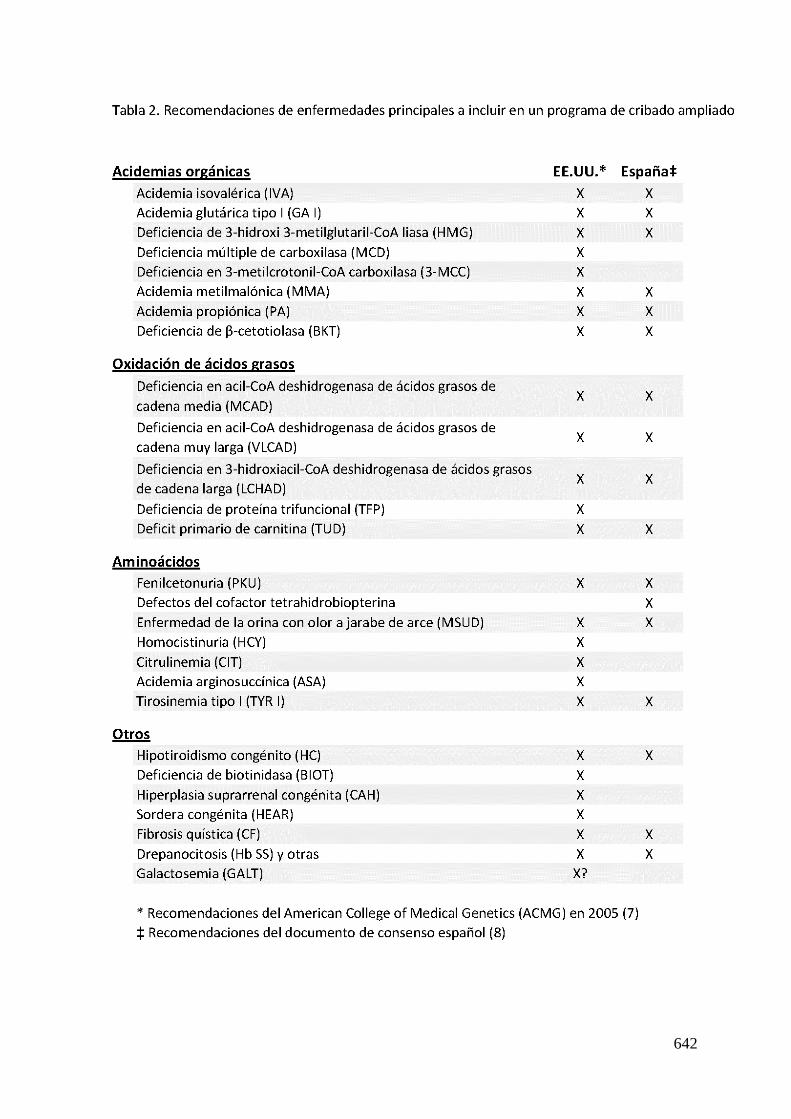
En 2005 el American College of Medical Genetics examinó la evidencia científica y
formuló unas recomendaciones sobre que enfermedades deberían cribarse (7). De
este informe se establecieron 29 alteraciones específicas, muchas de las cuales

641
deben ser analizadas por TMS. A día de hoy este protocolo está implantado en casi
todos los estados. En España se creó un documento de consenso elaborado en el
2009 con la participación de la Asociación Española para el estudio de Errores
Congénitos del Metabolismo (AECOM), de la Asociación Española de Pediatría (AEP) y
de la Sociedad Española de Bioquímica Clínica y Patología Molecular (SEQC) (6). Este
grupo formuló una lista de 19 ECM para su cribado preferente mediante TMS. Las
recomendaciones de los ECM a incluir de manera preferente en un programa de
cribado ampliado de ambos documentos se exponen en la Tabla 2.
Sin embargo, numerosos colectivos han sido críticos con el cribado ampliado
aludiendo a la cantidad de resultados falsos positivos que llevan a la realización de
numerosas pruebas de confirmación a veces invasivas. Además, no es raro obtener
resultados confirmatorios no concluyentes que no pueden establecer un diagnóstico
concreto, dejando a las familias en duda de por vida si su hijo desarrollará algún día
la enfermedad.
A modo de ejemplo, cada año 4 millones de niño en los EE.UU. se someten a un
cribado neonatal ampliado. De todos ellos 13.000 tendrán un cribado positivo, de los
cuales sólo 1000 serán diagnosticados de una enfermedad metabólica mientras los
otros 12.000 habrán tenido un falso positivo (9).
En Europa las diferencias entre países son muy grandes y muchos de ellos
mantienen el cribado clásico aunque la evolución en este campo es muy rápida.
Países como Holanda, Portugal, Austria y Dinamarca ya han implantado un cribado
avanzado parecido al de los EE.UU. (10).
642

643
En España, existen 21 laboratorios para los diferentes programas de cribado neonatal
de las comunidades autónomas. La Asociación Española de Cribado neonatal
(AECNE) publicó un documento informativo a diciembre de 2008 de la situación
actual en España (11).
En este documento se observa que en todas las Comunidades se realiza el cribado
del hipotiroidismo congénito y de las hiperfenilalaninemias; en ocho de la fibrosis
quística; en seis de la hiperplasia suprarrenal congénita; en dos la anemia falciforme
y otras hemoglobinopatías, y en una se realiza la detección precoz de galactosemia y
deficiencia de biotinidasa. Cuatro centros disponen de la tecnología MS/MS para el
cribado de trastornos de aminoácidos, ácidos orgánicos y oxidación de ácidos grasos
pero cada uno criba unas patologías concretas.
4. Métodos de diagnóstico de las ECM
El diagnóstico definitivo de las ECM requiere de la adecuada valoración clínica y de la
cuantificación de metabolitos o enzimas que permitan confirmar o descartar los
resultados iniciales obtenidos en las pruebas de cribado. Los métodos diagnósticos
incluyen diferentes técnicas como la LC-TMS (para aminoácidos y ácidos grasos) o la
CG-MS (para ácidos orgánicos) (12).
El paso final en todo proceso diagnóstico de los ECM debería ser la identificación de
las mutaciones causantes de la enfermedad en genes determinados. La mayor
barrera para su aplicación sistemática al diagnóstico clínico sigue siendo el
desconocimiento de las alteraciones causantes de la enfermedad. Además muchas
ECM son consecuencia de múltiples mutaciones. Otras veces los datos clínicos y de
laboratorio no hacen sospechar de una ECM concreta por lo que buscar sus causas

644
resulta imposible. Los microarrays de ADN pueden ser en un futuro utilizados para la
identificación de múltiples mutaciones de un gran número de genes asociados a los
ECM.
De manera alternativa se puede estudiar la expresión de una proteína o la
funcionalidad de un enzima para diagnosticar los casos dudosos o ciertas patologías
concretas como, por ejemplo, las enfermedades lisosomales o peroxisomales. La
actividad enzimática se puede evaluar monitorizando la producción de algún reactivo
marcado (incluso seguirlo por una ruta metabólica) pero analizar su expresión es
más complicado ya que la mayoría de las ECM no suelen producir una reducción de
la cantidad total de proteína sino modificar su tamaño, estructura o actividad.
5. Defectos del metabolismo de los aminoácidos
Este tipo de ECM se divide en 2 grupos:
1. Las aminoacidopatías donde el aminoácido precursor se acumula en sangre y
filtra a la orina (por ejemplo la fenilcetonuria).
2. Las acidemias orgánicas donde se acumulan los productos catabólicos de las
rutas afectadas (por ejemplo la academia glutárica tipo I).
5.1 Fenilcetonuria
La Fenilcetonuria (PKU del inglés phenylketonuria) o hiperfenilalaninemia, es el más
frecuente de los trastornos metabólicos hereditarios con una frecuencia en España
de unos 1:9200 RNv (8). Su herencia es de tipo autosómica recesiva. En el año 1961
Robert Guthrie desarrolló el primer test para detectar una enfermedad metabólica
hereditaria en niños asintomáticos afectos (13). La enfermedad se caracteriza por la

645
alteración del enzima fenilalanina hidroxilasa encargada de convertir la fenilalanina a
tirosina en el hígado conjuntamente con su cofactor la tetrahidrobiopterina (BH4). El
déficit de esta enzima da lugar a un acumulo patológico de fenilalanina en sangre
que produce alteraciones estructurales del sistema nervioso central.
Estos trastornos pueden prevenirse si se instaura una dieta pobre en fenilalanina
pero este tratamiento dietético ha de iniciarse en los primeros días de vida y antes
de que aparezcan los síntomas clínicos.
La medición de fenilalanina en sangre se lleva a cabo mediante técnicas de
fluorimetría en la mayoría de los centros nacionales y algunos la incluyen en el panel
de analitos de la espectrometría de masas en tándem (5 centros en España, se
estudia el ratio Phe/Tyr).
Los métodos diagnósticos detectan aminoácidos en plasma o ácidos orgánicos en
orina.
5.2 Acidemia glutárica tipo I
Deficiencia autosómica recesiva de glutamil-CoA deshidrogenasa que produce una
alteración en el metabolismo de la lisina, hidroxilisina y triptófano. Su prevalencia en
España es de 1:34.000 RNv (8). Se presenta con una acumulación de algunos
catabolitos de la vía, principalmente ácido glutámico en orina. Se manifiesta con un
cuadro neurológico entre los 6 y 18 meses de edad generalmente desencadenado
por fiebre.
Para el cribado se utiliza la espectrometría de masas en tándem analizando
glutarilcarnitinas en orina (C5-DC). El diagnóstico se realiza cuantificando ácidos
orgánicos en orina y acilcarnitinas en plasma y orina. Mucho menos utilizadas son las

646
pruebas enzimáticas (actividad glutaril-CoA DH en fibroblastos) o genéticas
(mutaciones del gen GCDH).
6. Defectos del metabolismo de los ácidos grasos
Estas ECM suelen manifestarse únicamente cuando existe una gran demanda
energética por parte del cuerpo (fiebre, vómitos e infecciones). Son patologías que
deben ser controladas rápidamente con medidas agresivas para evitar la emergencia
del desarrollo de un cuadro grave. Su presentación clínica más común es la de un
paciente hipoglucémico con alteración de la función hepática.
6.1 Defecto de los ácidos grasos de cadena media (MCAD)
Defecto congénito de acil-CoA deshidrogenasa de cadena media con prevalencia de
1:11.000 RNv en España (8). La sintomatología se suele presentar durante el primer
año de vida desencadenada por el ayuno u otra patología aguda. Se caracteriza por
una hipoglucemia no cetósica, alteraciones hepáticas y neurológicas. Su tratamiento
consiste en evitar periodos de ayuno, una alimentación baja en grasas y
suplementación nutricional con carnitinas.
El cribado se realiza con TMS buscando un perfil característico de acilcarnitinas
(elevación de carnitinas C6, C8 y C10 y aumento del ratio C8/C10). La confirmación
se realiza por pruebas complementarias bioquímicas: confirmación de la
cuantificación de acilcarnitinas en plasma, cuantificación de ácidos orgánicos y
conjugados de glicina en orina, estudios genéticos (principalmente mutación A985G).

647
7. Defectos del metabolismo de los carbohidratos
Estas patología de menor prevalencia dentro de las ECM se presentan con una
elevación de monosacáridos en sangre y en orina. Algunos ejemplos de tales
patologías son los defectos en el almacenamiento de glucógeno o la galactosemia. Se
caracterizan por alteraciones hepáticas e ictericia. Su tratamiento consiste en
eliminar el azúcar en cuestión de la dieta (galactosa, lactosa o fructosa
generalmente).
8. Otros defectos endocrino-metabólicos
Existen otras patologías congénitas que no son defectos metabólicos estrictamente
hablando pero que suelen incluirse ya que se agrupan en los programas de cribado.
Estas enfermedades son el hipotiroidismo congénito, la fibrosis quística, la
hiperplasia suprarrenal congénita, la drepanocitosis, el defecto de biotinidasa y otras.
8.1 Hipotiroidismo congénito
Se caracteriza por un aumento de la TSH y un descenso de la T4 debido a un
funcionamiento anormal del tiroides que tiene efectos en el momento en que el niño
deja de recibir la T4 maternal. Presenta una prevalencia de 1:3000 RNv en nuestro
país. Esta deficiencia no produce sintomatología durante los primeros meses de vida
pero representa una de las causas más importantes de retraso mental.
El cribado suele analizar la TSH al 3er día de vida (nunca antes de las 48h primeras
horas por la elevación fisiológica de la TSH). En España la mayoría de laboratorios
utilizan la inmunofluorescencia a tiempo retardado (DELFIA®) (11).
La confirmación diagnóstica se realiza midiendo tiroxina. La administración de
tiroxina como tratamiento debe realizarse lo antes posible.

648
8.2 Fibrosis quística
La fibrosis quística (FQ) es una enfermedad congénita con herencia autosómica
recesiva ocasionada por la alteración funcional de la proteína CFTR constituyente de
los canales de cloro de la membrana celular. La mutación más frecuente del gen que
codifica esta proteína es la deleción ΔF508 (2/3 de los casos) aunque se hayan
descrito más de 1500 mutaciones distintas. La frecuencia de la FQ en la población
española se encuentra alrededor de 1:3.500 RNv (8).
La clínica más significativa presenta una obstrucción bronquial secundaria al aumento
de viscosidad de las secreciones lo que provoca infecciones de repetición, afectación
de la función pancreática exocrina y otras. La necesidad del cribado neonatal viene
determinada por la inespecificidad de los síntomas en los primeros días de vida. No
existe tratamiento curativo de la enfermedad aunque su diagnóstico precoz permite
mejorar el estado nutricional de los pacientes y prevenir las infecciones respiratorias
recurrentes.
En los últimos años numerosos estudios han planteado la viabilidad y la justificación
del cribado de esta enfermedad (14).
La técnica de cribado consiste en detectar niveles de tripsinógeno inmunoreactivo
(TIR) en sangre capilar recogida sobre papel. En el año 2000 la European Concerted
Action on Cystic Fibrosis (ECACF) estableció un protocolo diagnóstico basado en el
análisis secuencial de TIR en las primeras 48 horas de vida seguido del estudio
genético de CFTR. Este estudio debe detectar al menos un 80% de las mutaciones
descritas (representa unas 30 mutaciones). Este procedimiento presenta un tasa de
falsos negativos del 3,4%. En los casos dudosos (1 mutación) se debe confirmar
mediante el test del sudor.

649
La medición del TIR se realiza mediante inmunoanálisis enzimático (ELISA) o la
inmunofluorescencia a tiempo retardado (DELFIA®, gran mayoría de laboratorios
españoles) (8).
9. Bibliografía
1. Fraga Bermúdez JM, Alonso Fernández JR, Cocho de Juan JA, Bóveda Fontán MD,
Castiñeiras Ramos DE, Colón Mejeras C et al. Memoria de la labor de investigación
galardonada con la dotación para la candidatura española de los Premios Reina Sofía
2008, de Prevención de la Discapacidad. Madrid: Real Patronato sobre Discapacidad;
2009.
2. Saudubray JM, Sedel F. Inborn errors of metabolism in adults. Ann Endocrinol
2009;70(1):14-24.
3. Wilson JMG, Junger G. Principles and practice of screening for disease. Public
Health Papers 34. Geneva: World Health Organisation; 1968.
4. Espada Saenz-Torre M, Dulín Iñiguez E. Procedimiento para la obtención y
recogida de especímenes de sangre sobre papel de filtro en los programas de
detección precoz neonatal de errores congénitos del metabolismo. Química clínica
2001;20:81-88.
5. Frazier DM, Millington DS, McCandless SE, Koeberl DD, Weavil SD, Chaing SH,
Muenzer J. The tandem mass spectrometry newborn screening experience in North
Carolina: 1997-2005. J Inherit Metab Dis 2006;29:76-85.
6. American College of Medical Genetics/American Society of Human Genetics Test
and Technology Transfer Committee Working Group. Tandem mass spectrometry
in newborn screening. Genet Med 2000;2:267-269.

650
7. American College of Medical Genetics. Newborn screening: toward a uniform
screening panel and system. Genet Med. 2006;8:1S-252S.
8. Documento de consenso sobre programas de cribado neonatal en España:
actualizacion y propuestas de futuro. JL Marín Soria, L Aldamiz-Echevarria, DE
Castiñeiras Ramos, J Dalmau Serra, A Fernández Sánchez, D González Lamuño, MªJ
Juan Fita, L M Jiménez Jimenez, C Pérez – Cerdá. 2009.
9. Waisbren SE. Newborn screening for metabolic disorders. JAMA 2006;296:993-
995.
10. Bodamer OA, Hoffmann GF, Lindner M. Expanded newborn screening in Europe
2007. J Inherit Metab Dis 2007;30:439-444.
11. Asociación Española de Cribado Neonatal. II Congreso Nacional de la Asociación
Española de Cribado Neonatal. Valencia 26-28 noviembre 2009.
12. Dietzen DJ, Rinaldo P, Whitley RJ, Rhead WJ, Hannon WH, Garg UC et al.
National Academy of Clinical Biochemistry Laboratory Medicine Practice Guidelines:
Follow-Up Testing for Metabolic Disease Identified by Expanded Newborn Screening
Using Tandem Mass Spectrometry; Executive Summary. Clinical Chemistry 2009;55
1615–1626.
13. Guthrie R. Blood screening for phenylketonuria. JAMA 1961;178:863.
14. Dudding T,Wilcken B, Burgess B,Turner G. Neonatal screening for cystic fibrosis.
Lancet 2000;356:1930.
