a mió padneó - biblioteca digital exactas
TRANSCRIPT

Di r ecci ó n:Di r ecci ó n: Biblioteca Central Dr. Luis F. Leloir, Facultad de Ciencias Exactas y Naturales, Universidad de Buenos Aires. Intendente Güiraldes 2160 - C1428EGA - Tel. (++54 +11) 4789-9293
Co nta cto :Co nta cto : [email protected]
Tesis de Posgrado
Acerca de la actividad de la Ù-Acerca de la actividad de la Ù-Aminolevulínico dehidrasa, enAminolevulínico dehidrasa, en
relación con su estructurarelación con su estructuramolecular y el contenido de ionesmolecular y el contenido de iones
metálicosmetálicos
Fukuda, Haydeé
1988
Tesis presentada para obtener el grado de Doctor en CienciasQuímicas de la Universidad de Buenos Aires
Este documento forma parte de la colección de tesis doctorales y de maestría de la BibliotecaCentral Dr. Luis Federico Leloir, disponible en digital.bl.fcen.uba.ar. Su utilización debe seracompañada por la cita bibliográfica con reconocimiento de la fuente.
This document is part of the doctoral theses collection of the Central Library Dr. Luis FedericoLeloir, available in digital.bl.fcen.uba.ar. It should be used accompanied by the correspondingcitation acknowledging the source.
Cita tipo APA:Fukuda, Haydeé. (1988). Acerca de la actividad de la Ù-Aminolevulínico dehidrasa, en relacióncon su estructura molecular y el contenido de iones metálicos. Facultad de Ciencias Exactas yNaturales. Universidad de Buenos Aires.http://digital.bl.fcen.uba.ar/Download/Tesis/Tesis_2130_Fukuda.pdf
Cita tipo Chicago:Fukuda, Haydeé. "Acerca de la actividad de la Ù-Aminolevulínico dehidrasa, en relación con suestructura molecular y el contenido de iones metálicos". Tesis de Doctor. Facultad de CienciasExactas y Naturales. Universidad de Buenos Aires. 1988.http://digital.bl.fcen.uba.ar/Download/Tesis/Tesis_2130_Fukuda.pdf

BO”;
B'BL'ÜTECAFACULTAD DE ClENClAS EXACiAo
Y NATURALES I UBA
UNIVERSIDAD DE BUENOS AIRES
FACULTAD DE CIENCIAS EXACTAS Y NATURALES
ACERCA DE LA ACTIVIDAD DE LA ó-AMINOLEVULINICO DEHIDRASA,
EN RELACION CON SU ESTRUCTURA MOLECULAR Y
EL CONTENIDO DE IONES METALICOS
por:
HAYDEE FUKUDA
DIRECTOR:Dra. Alcira M. de] C. BatlTe
CONSEJERO: Dra. Alcira M. de] C. BatTTe
LUGARDE TRABAJO:Centro de Investigaciones sobre Porfirinasy Porfirias (CIPYP). Departamento de Quimica Biológica.FacuTtad de Ciencias Exactas y NaturaTes. Universidad deBuenos Aires. Consejo Naciona] de Investigaciones Cientificasy Técnicas (CONICET)
97“T‘ ‘TEsis presentada para optar a1 titulo de
DOCTOR EN CIENCIAS QUIMICAS
T 9 8 8

a mió padneó

INDICE GENERAL
AGRADECIMIENTOS
ABREVIATURAS
OBJETIVOS
INTRODUCCION
- CAPITULO I: TETRAPIRROLES Y COMPUESTOS RELACIONADOS
1.1. Introducción1.2. Estructura. NomencIatura1.3. PorfirinasI.4. Porfirinógenos1.5. Metanporfirinas1.6. Tetrapirroïes de cadena abiertaI.7. Propiedades1.8. Referencias
- CAPITULO II:
II.II.IIII.II.
LPI-bde
III.III.III.III.III.III.
CAPITULO III:
mm-bwmü
METABOLISMO DE LOS TETRAPIRROLES Y SU
REGULACION
Biosíntesis de tetrapirroiesCatabolismo de] hemoReguIaciónPorfiriasReferencias
6-AMINOLEVULICO DEHIDRASA
IntroducciónCaracteristicas generaIesPropiedades moIecuTaresGrupos sulfhidriIosALA-D y metaTesMecanismo de acción
Página
21
21
51
55
60
62
84
84
86
90
94
100
112

III.
III.III.IIIIIIIIIIIIIII
III
- CAPITULO IV:
IV.T
IV.2IV.3
7.
8.9.
.10.
.11
.12.
.13.
.14.
.15.
Otros factores que afectan 1a actividad deALA-D
Ontogénesis y actividad enzimáticaAspectos genéticosVida media
. Camino metabóiico de] ALARo] de] ALA-Den 1a biosintesis de] hemo
ALA-Dy porfiriasAlgunas patoiogias en ias cuaies e] ALA-Dse encuentra afectadaReferencias
ZINC-ENZIMAS
ETementosinorgánicos en sistemas biológicosMetaioenzimasReferencias
MATERIALES Y METODOS
- CAPITULO I:
o-cHHv-cn-c ..... U'lh(A)N—‘ -....
HHHHHHHH o...
_a o
MATERIALES Y METODOS
Reactivos e instrumenta]Obtención y purificación de enzimasOtras etapas aTternativas de purificaciónEstudios de estructura cuaternariaDeterminación de pesos moiecuTares porfiitración por geiesEiectroforesis en geles de poiiacriiamidaPreparación de apoenzima y metaioenzimasDiáiisis de equiiibrioDeterminación de grupos suifhidriiosTratamiento de ALA-D con DEPMedición de actividad enzimáticaDeterminación de proteinasReferencias
RESULTADOS Y DISCUSION
- CAPITULO I:
1.1.PURIFICACION DE ALA-D
Purificación de ALA-Dde higado porcino
Página
122128
T30
T35
T35
T36
T37
T37
T42
154
154
156
169
T72
T72
172
T73
T74
175
T78
178179
180182182183186
187
188
188
188

1.2. Purificación de ALA-Dde higado bovino1.3. Conclusiones acerca de 1a purificación y
caracterización de1 ALA-Dobtenida a partirde higados porcino y bovino
CAPITULO II: ESTRUCTURA CUATERNARIA Y ACTIVIDAD
II.1.II.2.
ALA-Dde higado porcinoALA-Dde higado bovino. Estudios sobre 1are1ación entre 1a estructura cuaternariay 1a actividad enzimática, mediante HPLC
CAPITULO III: ALA-D Y METALES
III.1.III.2.
ALA-Dde higado porcinoALA-D de higado bovino
CAPITULO IV: GRUPOS FUNCIONALES DEL ALA-D DE HIGADO
BOVINO‘
IV.1. Tio1esIV.2. Histidina
CAPITULO V: MODELO PROPUESTO PARA EL MECANISMO DE
ACCION DEL ALA-D
CAPITULO VI: REFERENCIAS
CAPITULO VII: CONCLUSIONES
VII.1. Purificación y caracterización de 1a enzimaVII.2. Estudios de disociación y reasociaciónVII.3. Función de1 zincVII.4. Sustitución por coba1toVII.5. Grupos su1fhidri105VII.6. Grupos histidinaVII.7. Mecanismo
Página
207
220
222
222
249
249263
284
284
291
303
311
314
314
315316316317
317318

AGRADECIMIENTOS
Quiero expresar mi agradecimiento a la Dra, Alcira M.del C. Batlle quien ha dirigido esta Tesis, brindándome en todomomento su estimulo y dedicación.
Agradezco también a las Dras. Ana Maria Stella y EvaNider por el apoyo que tantas veces me brindaron.
Asimismo, a los Dres. Seiyo Sano y Yutaka Orii, de laFacultad de Medicina de la Universidad de Kyoto, quienes generosamente me recibieron, orientaron y apoyaron durante mi estadiaen Japón.
A la Dra. Eva Kesten, por su colaboración en las determinaciones de zinc.
-A la Lic. Ana Maria Buzaleh, por el esmero y dedicaciónen la transcripción de los manuscritos.
Al CONICET,SUBCYT,la Secretaria de Salud Pública delMinisterio de Bienestar Social y el Banco de la Nación Argentina,por los sUbsidios con que apoyaron la realización de este trabajo.
Al CONICETy al Mombusho, Ministerio de Educación delJapón, por las Becas que me otorgaron.
A la Facultad de Ciencias Exactas y Naturales, por haberme posibilitado los medios para realizar la Tesis.
A todos los integrantes del CIPYP, quienes con sualiento y cariño, me acompañaron durante todos estos años.
Y muy especialmente, al Dr. Sergio Paredes, por su valiosa colaboración en el desarrollo de esta Tesis, dedicándomemuchas horas de su tiempo.

ABREVIATURAS
A.E.ALA
ALA-D
ALA-S
BFP
BFS
B-ME
BrCN
BSA
CPG-asaDEP
DTNB
DTT
EDTA
GSH
HPLC
MMTS
PAGE
PAI
PBG
PBG-asaPCMB
PCT
PM
PMSF
PV
SDS
SDS-PAGE
TCA
TP
TPS 53
UE
UG
Actividad específicaAcido G-aminoïevüïicoAcido G-aminoïevúïico dehidrasaAcido ó-aminoïevülico sintetasaBuffer fosfato de potasioBuffer fosfato de sodioB-Mercaptoetano]Bromuro de cianógenoSeroaïbümina bovinaCoprogenasaDietiïpirocarbonatoDitio-bis-nitrobenzoicoDitiotreito]Eti]endiaminotetraacéticoGlutation reducidoCromatografía 1iquida a alta presiónMetiï-metano-tiosuïfonatoE1ectroforesis en ge] de p01iacri1amidaPorfiria aguda intermitentePorfobi1inógenoPorfobi1inogenasap-CïoromercuribenzoatoPorfiria cutánea tardaPeso moïecuiarF1uoruro de feniI-metiI-suïfoniïoPorfiria variegataDodeciïsuifato de sodioEiectroforesis en ge] de poliacriIamida enpresencia de SDSTricïoroacéticoTiopiridonaThiopropyI-Sepharose GBUnidades enzimáticasUïtrogei

OBJETIVOS
La biosintesis de tetrapirroies es un atributo de todas1as céïuïas vivientes que, comotoda secuencia metabóiica, seencuentra finamente controiada por una serie de enzimas de diferente distribución ceiuïar.
Una de dichas enzimas es 1a 6-amin01evü1ico dehidrasa(ALA-D)que cataiiza 1a condensación de dos mo]édu1as de ácidoó-aminolevüiico (ALA)para formar e] primer intermediario pirrg1ico, e] porfobiiinógeno (PBG).
Es una proteina oïigomérica constituida por ocho subunidades iguaies de peso moiecuiar 35 kDa, con un peso moiecuiarpara e] octámero de 280 kDa. Se ha encontrado, sin embargo, quelas condiciones experimentaïes pueden modificar e] estado de agregación de 1a proteina y, correiativamente, su actividad cataIitica.
Aunque e] mecanismo de sintesis de PBGparece ser muy simiiar en cé1u1as ecuariotas y procariotas, ios requerimientosmetáiicos pueden ser bastante diferentes Asi, en Rp. ¿phenoádes se activa con potasio y otros iones re1acionados, y en mamiferos los agentes que1antes y metales pesados inhiben su actividad. Para higado bovino y eritrocitos de mamíferos, se ha encontrado una significativa participación de] zinc en 1a expresiónde 1a actividad enzimática“
En este proyecto se estudia 1a re1ación entre e] estadomoïecuïar de 1a enzima y su actividad. asi como e] pape] mecanistico que juegan e] zinc y otros iones metáïicos en 1a biosigtesis de] PBG. Partiendo de higado bovino y porcino, se desarrg11ará un método de purificación de 1a enzima que permita obtener una preparación homogénea, aitamente purificada. necesariapara iievar a cabo ios estudios de estructura moiecuiar.
Contando con una preparación purificada de 1a enzima seefectuarán estudios de caracterización. En particuiar, se investigará e] ro] de grupos tioies e iones metá1icos, tanto sobre1a apoenzima como sobre 1a hoïoenzima, empieando para elio agentes quelantes y reactivos de grupos especificos.y no especificos.
Unenfoque interesante-consiste en reempiazar e] meta]en e] sitio activo por otros iones metálicos, tanto por

sustitución a partir de la apoenzima comopor desplazamientoen la enzima nativa, correlacionando el contenido de los metales con la actividad de la enzima resultante. Y sobre este proceso de reactivación, analizar el efecto de protectores de grupos tiolesy atmósfera de incubación.
Se considera de interés ensayar la transformación de laenzima en oligómeros analizando dicha conversión mediante latécnica de la cromatografía liquida a alta presión, empleandoagentes caotrópicos (sulfocianuro de sodio) y disociantes (SDS);y correlacionar la descomposición de la estructura octaméricacon la formación de agregados moleculares menores y, correlativamente con la actividad enzimática.
Dado que se postula que: a) el ALA-Des una zinc-enzima;b) un grupo histidina está involucrado en el sitio activo y c)el dietilpirocarbonato es un reactivo especifico para este aminoácido, es de interés estudiar el-efecto de este reactivo sobre la enzima.
Se realizarán experiencias bajo diferentes condicionesexperimentales, variando la temperatura, pHy_concentraciones deimidazol, zinc y enzima y, en base a los datos obtenidos, se establecerá el númeroy reactividad de residuos histidina esenciales para la actividad enzimática, asi comoel efecto de zinc,,gtros iones metálicos y del sustrato ALA,sobre el proceso de inactivación por el dietilpirocarbonato.
Tratándose de una enzima oligomérica, resulta asimismo deinterés determinar qué rol cumple la estructura cuaternaria enla conformación y función del ALA-D.Para ello se procederá asometer a la proteina a reacciones-de desnaturalización y/o disgciación, con el objeto de obtener información experimental quepermita establecer los mecanismospor los cuales la cadena polipeptidica tiende a plegarse adoptando una conformación especifica. En relación a ésto, el empleo de la enzima inmovilizada esuna herramienta útil para dilucidar los tipos de ensamblamientosde Subunidad, interacciones entre las mismasy relaciones entreestructura y función . En cuanto a este último aspecto, es de interés conocer si las subunidades individuales son activas, contal propósito, se unirá el ALA-Dnativa a la matriz de Sepharose,a través por lo menos de una subunidad, eliminando aquellas subunidades que no se hayan unido covalentemente al gel; lo cualpermitirá el estudio de las propiedades de la subunidad

aisïada e inmoviïizada, asï comotambién su reasociacíón consubunidades soïubïes.

INTRODUCCION

CAPITULO I
TETRAPIRROLES Y COMPUESTOS RELACIONADOS
Introducción
Estructura. Nomenclatura
Porfirinas
Porfirinógenos
Metaioporfirinas
1.5.1. Hemoproteinas1.5.1.1. Hemogiobina1.5.1.2. Mioglobina1.5.1.3. Citocromos1.5.1.4. Citocromo P-4501.5.1.5. Triptofano pirroïasa (Triptofano
2,3-dioxigenasa)1.5.1.6. Peroxidasas y cataïasas
1.5.2. Ciorofiias
1.5.3. Corrinas
Tetrapirroles de cadena abierta
Propiedades
1.7.1. Propiedades químicas
1.7.2. Propiedades fisicas1.7.2.1. Soïubiïidad1.7.2.2. Espectros de absorción1.7.”.3. Fotosensibilidad
Referencias
Página
OCDGDNICD-b—I
12
13
14
17
17
17
17
18
18

I. TETRAPIRROLES Y COMPUESTOS RELACIONADOS
1.1. INTRODUCCION
Todos hemos tenido en aigün momento 1a fantasia de unviaje a través de] tiempo, para retrocederen é] y ver como fueron los comienzbs de 1a vida en 1a Tierra. La curiosidad parainvestigar e] origen, es una necesidad compulsiva de 1a especiehumana, que ha buscado respuestas tanto en 1a Religión como en1a Historia y en 1a Ciencia.
Uno de 105 probiemas más interesantes en e] desarroiiode 1a vida en nuestro pianeta, es ilegar a di1ucidar de qué manera los organismos han obtenido, aimacenado y consumido energia.
Una cé1u1a viva, como 1a Reina Roja de Lewis Caroïl,debe correr a alta veiocidad para permanecer en e] mismo iugar;sin un constante aporte de Energia, ya sea de una fuente externa o de su propia reserva aimacenada, 1a céiuia muere.(Dickerson, 1980).
En e] comienzo, probabiemente se deben haber formadopor medios no bio]ógicos,compuestosde aito contenido de energia,Ios que a1 dar Iugar a moiécuias de menor contenido energéticocomo productos de desecho, consumieron 1a energia iiberada poréstos para poder continuar e] proceso. Debió ser entonces 1afermentación 1a fuente de energia de los primeros organismos,hasta que hicieron su aparición 1a fotosíntesis y 1a respiración(Oparin, 1957).
Apartir de aiii, 1a estructura tetrapirróiica se con;tituyó en una de 1as bases primordiaies para 1a supervivencia deios sistemas biológicos,pues es 1a responsabïe de] color rojo de1a hemoglobina y de] verde de la clorofila, ios pigmentos invoiucrados en ios dos procesos fundamentaies mencionados.
Los tetrapirroies participan entonces, de un gran nümero de reacciones bioiógicas de importancia fisiológica, ta1escomoe] transporte de e1ectrones y energia (citocromos, ciorofilas), gases (hemoglobina y mioglobina), oxidaciones bioiógicas(peroxidasas, catalasas).

1.2. ESTRUCTURA. NOMENCLATURA
Los tetrapirr01es derivan de 2 nücïeos básicos: porfina y cïorina, que contienen 4 anillos pirrólicos (A, B, C,y D)unidos entre si por 4 puentes metenos (a, B, y, 6). La diferencia entre 1a estructura de 1a porfina y 1a c10rina es que e] aniïlo D en ésta ü1tima está reducido (Figura 1.1.).
PORFINA CLORINA
FIGURA1.1.: Estructura de 1a porfina y de 1a clorina
Las dobies 1igaduras conjugadas conforman una estructura resonante pianar sobre 1a cua] pueden haber 8 cadenas 1ateraïes en las posiciones 1 a 8.
Los distintos tetrapirroïes ha11ados en 1a naturaïezaderivan de estos 2 esqueïetos básicos, con aïgunas modificaciones:
a) Sustitución parcia] o tota] de los átomos de carbono externos (1 a 8) de 10s aniïlos pirrólicos, por grupos alquilo.
b) Sustitución o reempïazo sobre Ios puentes meténicos. Los sustituyentes pueden ser.átomos de hidrógeno_(como en 10s porfirinógenos),o un aniïïo cicïopentanona (en 1as cïorofiïas), ocomo en e] caso dela corrina (derivado de 1a vitamina 812)en e] que se ha perdido e] puente meténico 6.
c) Inserción de meta] dando Iugar a 1as metaïoporfirinas; estoscompïejos pueden modificarse por cambios de va1encia, porformación de compuestos de coordinación y por combinacióncon proteinas especificas.
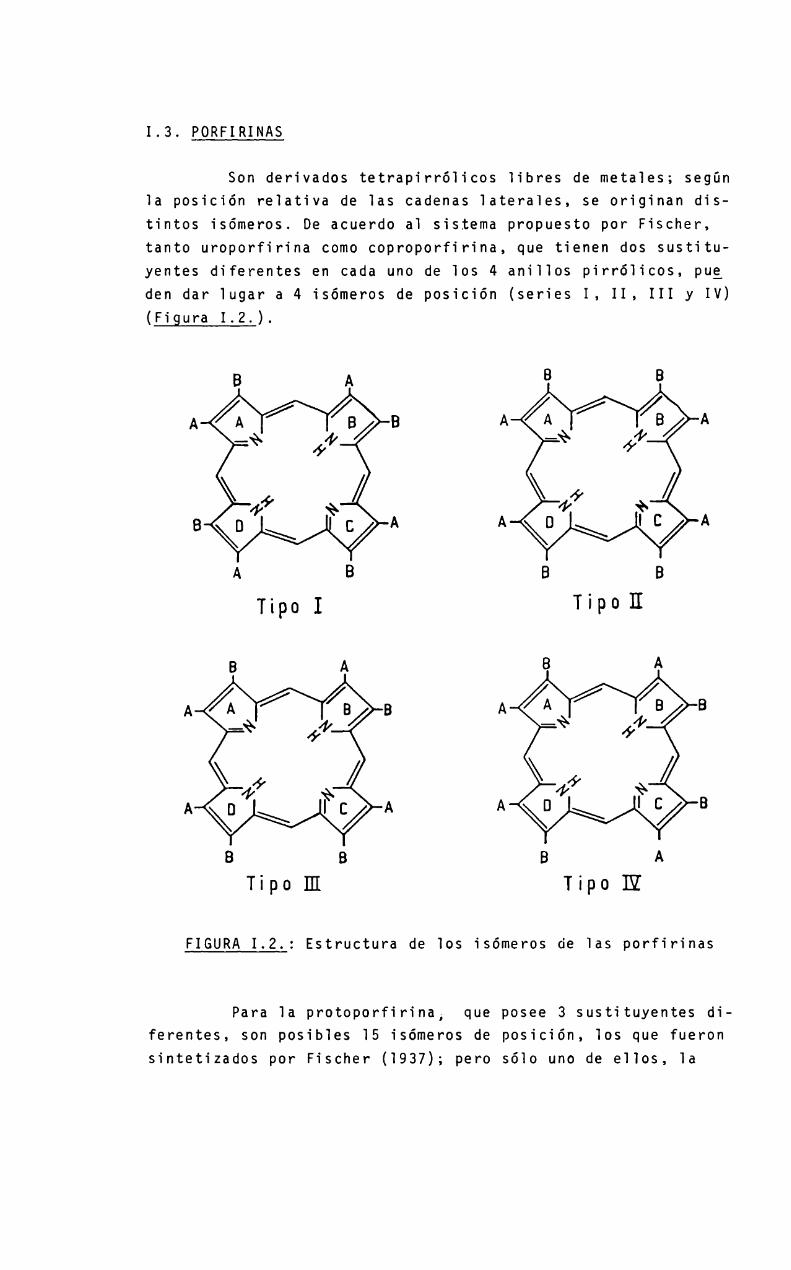
1.3. PORFIRINAS
Son derivados tetrapirróiicos iibres de metales; segün1a posición reiativa de ias cadenas ïaterales, se originan distintos isómeros. De acuerdo a1 sistema propuesto por Fischer,tanto uroporfirina comocoproporfirina, que tienen dos sustituyentes diferentes en cada uno de los 4 aniilos pirróiicos, pueden dar lugar a 4 isómeros de posición (series I, II, III y IV)(Figura 1.2.).
Tipom Tipo m
FIGURA1.2.: Estructura de 105 isómeros de 1as porfirinas
Para 1a protoporfirinai que posee 3 sustituyentes diferentes, son posibies 15 isómeros de posición, los que fueronsintetizados por Fischer (1937); pero sólo uno de ellos, 1a

protoporfirina IX, es e] isómero natura]. Todas 1as porfirinasfisiológicamente activas-pertenecen a 1a serie III (Burnham,1969). Los isómeros de] tipo I, si bien se han detectado enfuentes naturaies, son productos de una sintesis anormai y nopueden ser metabolizados a protoporfirina IX (Figura 1.3.).
V H
V
H
P P
Uroporíirina III Coproporiirina III Proioporfirina IX
A = -CH2-CH3 P = -CH2-CH2-CH3 M = -CH3 V = -CH=CH2
FIGURA1.3.: Porfirinas de interés bioiógico
1.4. PORFIRINOGENOS
E1 tratamiento de ias porfirinas con agentes reductores taies como amaigama de sodio o borohidruro de sodio, conduce a 1a adición de 6 átomos de hidrógeno en e] macrocicio y 1aformación de 105 correspondientes porfirinógenos. Estos son losverdaderos intermediarios en 1a biosintesis de ios tetrapirroles (Bogorad. 1958: Mauzerali y Granick. 1958). Son compuestosincoioros, no pianares, que por 10 tanto no presentan ias caragteristicas de absorción en e] visible que tienen ias porfirinasy se oxidan espontáneamente a 1as correspondientes porfirinas(Figura 1.4.).
1.5. METALOPORFIRINAS
Las porfirinas forman fácilmente queiatos con varios

metaies pero sóio ios compïejos de hierro, magnesio y cobaïtocumpïen roles biológicos importantes. Se han encontrado en 1anaturaieza compiejos de uro y coproporfirina con cobre, cinc ymagnesio, que probabiemente se generan espontáneamente dada 1afaciiidad conque las porfirinas se queian con esos metales. Tanto las porfirinas libres comosus ésteres se combinan con losmetaies formando compiejos soiubies en soiventes orgánicos. Losc0mp1ejos de hierro se denominan genéricamente Hemos. E1 más conocido es e] protohemo o hierro protoporfirina, en e] cua] e]hierro puede estar en forma reducida (hemo) u oxidado (hemina ohematina).
A \ P M P M V
A A M M M M
P P P P p P
Uroporf irinógeno HI Coproporfirinógeno D1 ProtOportirinógeno IX
FIGURA1.4.: Estructura de ios porfirinógenos
E1 hemo, en contacto con e] aire, se oxida fáciimentea hemina o hematina si hay presente iones cioruro. Estos derivados son muy estabies, y es 1a forma usua] bajo 1a cua] seaisla e] grupo prostético de ia hemogiobina.
E1 hierro y ios 4 anilios pirróiicos se ubican en unmismo piano. A] meta] ie quedan 2 posiciones de coordinaciónpor encima y por debajo de] piano, que pueden ser ocupadas pordistintos grupos,dando 1ugar a 10s derivados hemocromoo hemocromógenos. Entre los compuestos que pueden coordinarse podemoscitar: peróxido de hidrógeno, oxigeno, monóxido de carbono, iones cianuro, bases nitrogenadas como la piridina y e] amonio.
En e] caso de 1a hematina, ios complejos similares sedenominan hemicromos o hemicromógenos(filggra I 5;).

3
3
H c CHa3
EHZ ES?¡H2 l 2
COOH COOH
HEMO
/ |‘N
PIRIDINA-HEMOCROMO CLOROHEMINA
FIGURA1.5.: Estructura de] hemo y sus derivados
1.5.1. Hemogroteinas
Constituyen e] grupo de proteinas cuyo grupo prostético es una hierroporfirina. Es una denominación genérica, que noimpiica que e] hierro esté en estado ferroso, ni que sea particu1armente e] protohemo. E1 hierro es uno de ios metaies que seencuentra en mayor abundancia en ios sistemas bioiógicos. Aproximadamente un 70 % de] hierro activo en los mamíferos se encuegtra en forma de complejos de porfirina o hemos en hemogiobina,miogiobina, citocromos, cataiasa, triptofano oxigenasa, peroxidasas; éstas últimas se encuentran particuiarmente en plantas.
En muchas hemoproteinas, e] protohemo se une a 1a

proteina mediante una coordinación entre el átomo de hierro yun átomo de nitrógeno de las cadenas laterales básicas de laproteina, unión que es relativamente fácil de romper, por ejemplo con tratamiento con acetona ácida. En otras hemoproteinas,comoen el caso de los citocromos, existen uniones covalentesadicionales entre las cadenas laterales de la porfirina y losresiduos aminoácidos. En este caso, son necesarios tratamientosmás drásticos para separar la mitad hémica de las proteinas(Marks, 1969).
I.5.l.l. Hemoglobina
Es un componente de los glóbulos rojos, responsabledel transporte de oxigeno desde los pulmones hasta los tejidos;participa también en el transporte de dióxido de carbono desdelos tejidos hasta los pulmones.
Su rol como transportadora de oxigeno es vital, locual se evidencia si consideramos que en su presencia, la capacidad de transporte de oxigeno de l litro de sangre aumenta de5 a 250 ml de oxigeno.
Es una proteina oligomérica de peso molecular 64 KDaconstituida por 4 subunidades, cada una de las cuales posee ungrupo hemo. Dos de las unidades poseen la misma secuencia de gminoácidos y se denominan cadenas a, en tanto que las otras dosse denominan B, formando en conjunto una estructura del tipo
uzgz (Perutz, 1970). Este ordenamiento se ha visto que constitgye un propósito biológico útil.
Cada uno de los 4 átomos de hierro al estado ferrosopuede tomar un mol de oxigeno en forma reversible y se conocecomo oxihemoglobina. Si el átomo de hierro de la hemoglobina seoxida a la forma férrica, el compuesto resultante llamado metahemoglobina, es incapaz de transportar oxigeno.
La unión más importante entre el grupo hemo y la proteina proviene del enlace coordinado entre el hierro del hemoy un átomo de nitrógeno imidazol de un residuo histidina. Existe otro grupo histidina denominado distal que parece jugar uncierto rol en la unión del oxigeno. El grupo hemo, que es muysimilar en su naturaleza gruesa a un hidrocarburo aromático,es también mantenido en su posición por fuerzas de Van der Naalsejercidas por los residuos aminoácidos hidrofóbicos circundantes. Este carácter hidrofóbico del espacio que rodea el hemo

parece estar relacionado con la unión reversible del oxigeno(Perutz y col., l968). A pesar de los numerosos estudios realizados acerca de la estructura espacial y electroquimica de lashemoproteinas oxigenadas, hasta el momentoésta no ha sido determinada univocamente (Jones y col., 1979).
En la linfa de muchos invertebrados también se encuentran hemoproteinas transportadoras de oxigeno. Asi por ejemploalgunas especies de poliquetos del philum Annelida contienenclorocruorina, hemoproteina queen lugar de un vinilo en la posición 2 del anillo porfirinico posee un grupo formilo.
También en las leguminosas se ha identificado la dengminada leghemoglobina,que está presente en las células de losnódulos que contienen la bacteria simbiótica de la especie RhizobLumrelacionada con la fijación de nitrógeno (Lascelles,l964).
1.5.1.2. Mioglobina
Es una hemoproteina semejante a la hemoglobina,que seencuentra en el tejido muscular de vertebrados e invertebrados,actuando como fuente de reserva de oxigeno.
A bajas presiones de oxigeno, la mioglobina tiene mayor afinidad por el oxigeno que la hemoglobina. En esas condiciones (por ejemplo en músculo en ejercicio) la hemoglobina cede el oxigeno a la mioglobina, que lo almacena en las céulasmusculares.
La estructura de la mioglobina, a diferencia de la hemoglobina, consiste en una sola cadena polipeptidica de peso mglecular 17,6 kDay un grupo hemo en el que el átomo de hierroestá unido a un residuo histidina. El protohemo está ubicado enuna región rica en aminoácidos aromáticos, rodeado de cadenaslaterales hidrofóbicas. Los grupos polares se encuentran en laparte externa de la cadena, mientras que los grupos vinilo nopolares están sumergidos en la región interna no polar de la mglécula (Marks, l969).
I.5.l.3. Citocromos
Los Citocromos, término propuesto por Keilin en l925,se definen comohemoproteïnas cuya principal función biológicaes el transporte de electrones en virtud de un cambio reversible

de valencia de su hierro hémico (Keilin, l930).
Se clasifican en grupos a, b, c, y d segün la posicióndel máximo de absorción de sus espectros y el modo de unión delas mitades hémicas a la proteina, identificándose además con
subindices luego de la letra de clase, por ejemplo citocromo b5.- Citocromo a: el grupo prostético es una hierroporfirina llama
da hemo a en la que un grupo formilo reemplaza a un metilo yuna cadena hidrocarbonada reemplaza un grupo vinilo.
- Citocromo b: contiene protohemo que no está unido covalentemente a la proteina.
- Citocromo c: son citocromos con una unión tioéter entre unacadena lateral vinilo y un grupo sulfhidrilo cisteinico de laproteina.
- Citocromo d: contiene hierrodihidroporfirina (clorina) comogrupo prostético.
De todos ellos, el citocromo c por ser pequeño y relativamente fácil de cristalizar,es el que ha sido estudiado endetalle,y Su estructura terciaria ha sido elucidada casi hastaresolución atómica (Dickerson, l972). Se ha encontrado que loscitocromos c de distintas especies (hombre, animales, plantas)son similares unos a otros, y aün más, son idénticos enciertasporciones de la cadena peptidica, y es interesante señalar queesas secuencias de aminoácidos han permanecido invariables durantemillones de años de evolución (Dickerson, l980).
Si bien en un principio se creyó que los citocromos estaban asociados exclusivamente con el transporte de electronesal oxigeno, se encontraron citocromos funcionalmente activos enalgunas bacterias estrictamente anaeróbicas. Por ejemplo enDeáulóovábn40 deóuifiun¿can4, en la que el último aceptor de electrones es el sulfato (Postgate, l959) y en otros organismosque usan nitrato comoaceptor terminal de electrones (Newton yKamen,l96l). También se encontraroncitocromos en cloroplastosde hojas (Lascelles, l964) y en cromatóforos de bacterias fotosintéticas (Vernon y Kamen, l954) , donde se cree están involucrados en el transporte de electrones asociados con la fotosíntesis (Rabinowitch y Govindjee, l965).
Aunquela contribución cuantitativa de los citocromosal contenido total de tetrapirroles en los organismos que contienen hemoglobina o clorofila es pequeño, el rol que cumplen

es vita] (Drabkin, 1951; Lasceiies, 1964). En las céiuias animaies 10s citocromos se iocalizan en ias mitocondrias, donde están invoiucrados en 1a üitima parte de] metaboiismo respiratorio: sistema de transporte e1ectrónico. En e] proceso de cataboiismo, ios sustratos son deshidrogenados y e] átomo de hidrggeno es fijado como NAD(P)H.E1 sistema de transporte de electrones provee 1a ruta por 1a cua] los eiectrones disponibies enforma de NADH(o de otro donor de hidrógeno, como ser ácido sugcinico), son transferidos en forma sucesiva de un componente aotro y finalmente a 1a moiécuia de oxigeno (u otro agente oxidante, ta] comoe] nitrato en organismos anaeróbicos). La secuencia de transferencia de eiectrones en mitocondria de mamiferos es 1a siguiente:
NAD->FAD—>CoQ¡0—>C'it.b—>Cit.c]——>Cit.c —>Cit.a1a3—>02
1.5.1.4. Citocromo P-450
En 1958, en 1a fracción microsoma] de higado de ratas,se encontró una hemoproteina que en su estado reducido formabaun complejo con monóxido de carbonogciuepresentaba una banda deSoret a una iongitud de onda inusuaimente iarga de 450 nm,de 1acua] derivó su nombre.
E1 Citocromo P-450 forma parte de algunas monooxigenasas, enzimas que catalizan reacciones de] tipo:
SH + DH2 + 02-evSOH + D + H20
donde SH es e] sustrato aceptor y DH2es un donor de hidrógeno,que en muchos casos es e] NADHo e] NADPH.
Esta ciase de enzimas se iocaiizan principalmente ene] reticuio endopiásmico de tejidos animaies y vegetales (Coony c01., 1973; Orrenius y Ernster, 1974); también se han encontrado en mitocrondrias de varios órganos de mamíferos tales como higado, riñón y corteza adrena] (Schieyer y c01., 1973), yaün en bacterias (Gunsaïus y c01., 1972).
Son sus sustratos una variedad de compuestos taies comohidrocarburos aïifáticos y aromáticos, ácidos grasos, esteroides y porfirinas. Todos eiios son sustancias dificiies de metaboiizar debido a que soncompuestos reiativamente inertes, singrupos funcionaies e insoiubies o poco soiubies en agua, que es

e] medio por exceiencia en ios organismos. Las monooxigenasasmediante e] oxigeno molecuiar, producen 1a hidroxiiación deaouéiioSpermitiendo asi su metaboiización. Sustancias foráneas ta]es comohidrocarburos haiogenados y diversos fármacos son eiiminados de igua] modo. En consecuencia, estas enzimas son muyimportantes para 105 mamíferos, por cuanto intervienen en iosmecanismos de detoxificación de drogas. También son importantesen los microorganismos,pues por ejempïo hay aigunas bacteriasy ievaduras que pueden crecer con fuentes de carbono tales comohidrocarburos aiifáticos y aromáticos, debido a que las monooxigenasas hacen posible 1a utiiización de estos compuestos.
La diversidad de ias reacciones cataiizadas por e] citocromo P450 es sorprendente, y responden a mecanismos muy diversos. Sin embargo, puede esquematizarse de manera genera] cgmo sigue: 1a enzima forma un compiejo con e] sustrato, y e] hig
rro hémico de] citocromo P450 pasa de su forma oxidada a su estado reducido; e] oxigeno se combina con e] Fe2+ y 10 reoxida
a Fe3+ a 1a vez que se reduce a 02'. Luego e] oxigeno coordinado y reducido, ataca a1 sustrato y e] citocromo P450 se liberaen su estado de Fe3+ (Torres y co]., 1983).
1.5.1.5. Triptofano pirroiasa (Triptofano 2,3-dioxigenasa)
Es 1a ünica dioxigenasa que contiene protohematina IXcomo grupo prostético (Tanaka y Knox, 1959),_pues todas ias otras dioxigenasas caracterizadas contienen hierro no hémico cgmo único cofactor.
Esta enzima fue detectada por primera vez en 1936(Kotake y Masayama, 1936); ha sido aisiada de intestino e higado de mamíferos asi como también de 1a bacteria póeudomonaá¿(uoneócen¿(Feige1son y Brady, 1974). Cataiiza 1a ruptura oxidativa de] aniiio pirróiico de] triptofano a formi]kinurenina,ymediante e] empieo de 1802 se demostró que ambos átomos de oxigeno se incorporan en e] producto (Hayaishi y co]., 1957).
18o NH
PH? u I 2
Ü CHZCHCOOH c CH2CHcoouI 18
N + 0 —> N-C-H
H H 818
Triptofano Formiikinurenina

Para que esta oxidación tenga lugar, es necesaria laformación de un complejo ternario sustrato-enzima-oxigeno, y seha identificado al hemo como el sitio de unión del oxigeno a laenzima (Ishimura y col., l967). Se postula que el hierro probablemente ayude a la deslocalización de los electrones no apareados del oxigeno, y debe pasar del estado férrico (inactivo) alestado ferroso (activo) (Tanaka y Knox, l959).
En higado de rata, se localiza en la fracción soluble,y presenta la caracteristica de que las 2/3 partes de la mismase encuentra como una apoenzima inactiva que puede activarsepor el agregado de hematina (Feigelson y Greengard, l961).
Es una de las pocas enzimas cuya actividad puede aumentarse significativamente por administración ¿n vivo de su sustrato (Knox, l95l). Esto se debe a que el triptofano influye enla partición de la hematina entre la apo-triptofano pirrolasay otras proteinas hémicas, en favor de la primera (Feigelson yGreengard, 1962). Para otros en cambio (Welch y Badaway, 1980;Badawayy col., 1981), el triptofano seria un agente capaz deaumentar la concentración de hemo en higado- y habria una relación inversa entre la saturación por hemode la triptofano pirrolasa y la actividad de la enzima ácido ó-aminolevülico sintgtasa (ALA-S).
1.5.1.6. Peroxidasasycatalasas
Son enzimas que se encuentran ampliamente distribuidasen la naturaleza; las peroxidasas se localizan particularmenteen las plantas.
Catalizan la oxidación de distintos compuestos orgánicos por el peróxido de hidrógeno, en reacciones del tipo
ROOH + AH2—> A + H20 + ROH
donde AH2, el donor de hidrógeno, puede ser un alcohol (fenol,pirogalol, p-cresol, etanol, metanol), una amina primaria, secundaria o terciaria (anilina, p-toluidina), ácido ascórbico oindol. También algunos iones inorgánicos como por ejemplo el igduro, son oxidados por las peroxidasas. En el caso particular
de las catalasas, ROOH= AH2 = H202 y A = 02 y lo que se produce entonces es la descomposición del peróxido de hidrógeno.

El mecanismo de acción general de las catalasas es similar al de las peroxidasas, por lo cual podrian considerarsecomoperoxidasas de diferente especificidad (Dixon y Webb,l964). Asi por ejemplo, cuando se agrega catalasa y etanol a unsistema que produce peróxido de hidrógeno, el alcohol se oxidaa acetaldehido (Keilin y Hartree, l945).
Ambasenzimas interactúan con sus sustratos formandocomplejos intermedios de diferente estabilidad y color (compuegto I, verde; compuesto II, rojo), de manera tal que la reacciónse puede seguir espectrofotométricamente.
Las peroxidasas pueden clasificarse de acuerdo a lanaturaleza de sus grupos prostéticos en peroxidasas flavoproteicas y las que contienen hierro. Estas últimas pueden subdividirse a su vez en dos grupos: las que poseen protohematina ylas que tienen un quelato de hierro de un tetrapirrol aün no identificado (Marks, 1969).
La catalasa proveniente de Rp. ¿pheno¿de4 contiene 4grupos protohematina por mol (Nichols y Schonbaum, l963) y lomismoocurre con las catalasas de higado bovino, eritrocitos decaballo y de humanos (Yamazaki, 1974).
En otros casos, se hanencontrado menos de 4 grupos porfirina por mol, probablemente debido a la degradación de la hematina a pigmentos biliares (Marks, 1969).
Respecto a la función fisiológica de la catalasa, algunos autores consideran que posee un rol protector, ya que destruye el peróxido de hidrógeno, sustancia tóxica para los organismos vivos (Lemberg y Legge, l949) en tanto que otros autoresle asignan una función peroxidativa (Keilin y Hartree,1945).
1.5.2. Clorofilas
Son derivados tetrapirrólicos de la clorina y se diferencian de la estructura del hemo, en los siguientes aspectos:
DJ V Son complejos de magnesio;
b) el anillo D en plantas, y los anillos B y D en bacterias, seencuentran reducidos;
c) tienen un anillo adicional de ciclopentanona (E) formado porciclación de la cadena lateral de ácido propiónico sobre elcarbono 6 con el carbono meténico Y;
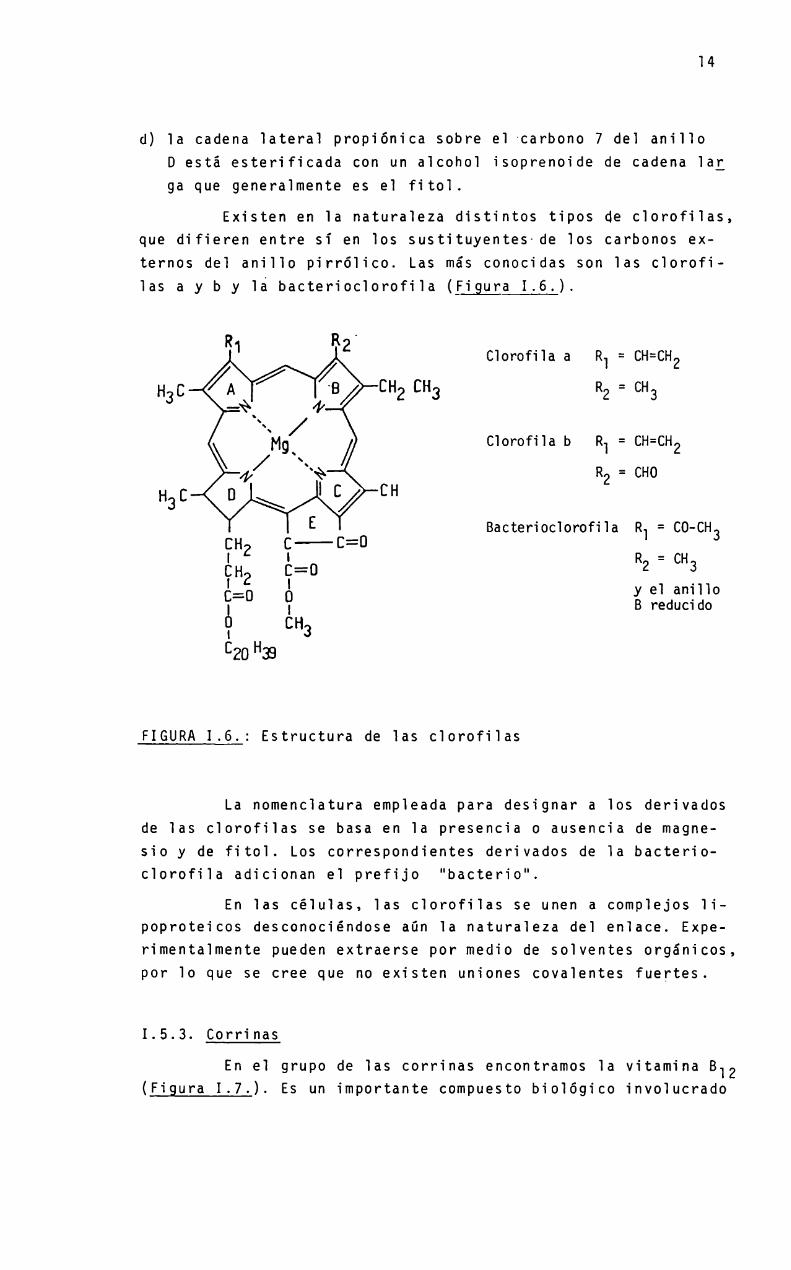
d) la cadena lateral propiónica sobre el carbono 7 del anilloD está esterificada con un alcohol isoprenoide de cadena larga que generalmente es el fitol.
Existen en la naturaleza distintos tipos de clorofilas,que difieren entre si en los sustituyentes-de los carbonos externos del anillo pirrólico. Las más conocidas son las clorofilas a y b y la bacterioclorofila (figura 1.6.).
Clorofila a R = CH=CH2
R = CH
Üomfilab R =CMGQR = CHO
Bacterioclorofila R1 = CO-CH
(IÏHZ [|Z=Ü 2 _ 3= y el anillo
E 0 9 B reducidol CH3
C20 “39
FIGURA1.6.: Estructura de las clorofilas
La nomenclatura empleada para designar a los derivadosde las clorofilas se basa en la presencia o ausencia de magnesio y de fitol. Los correspondientes derivados de la bacterioclorofila adicionan el prefijo "bacterio".
En las células, las clorofilas se unen a complejos lipoproteicos desconociéndose aün la naturaleza del enlace. Experimentalmente pueden extraerse por medio de solventes orgánicos,por lo que se cree que no existen uniones covalentes fuertes.
1.5.3. Corrinas
En el grupo de las corrinas encontramos la vitamina 812(Figura 1.7.). Es un importante compuesto biológico involucrado
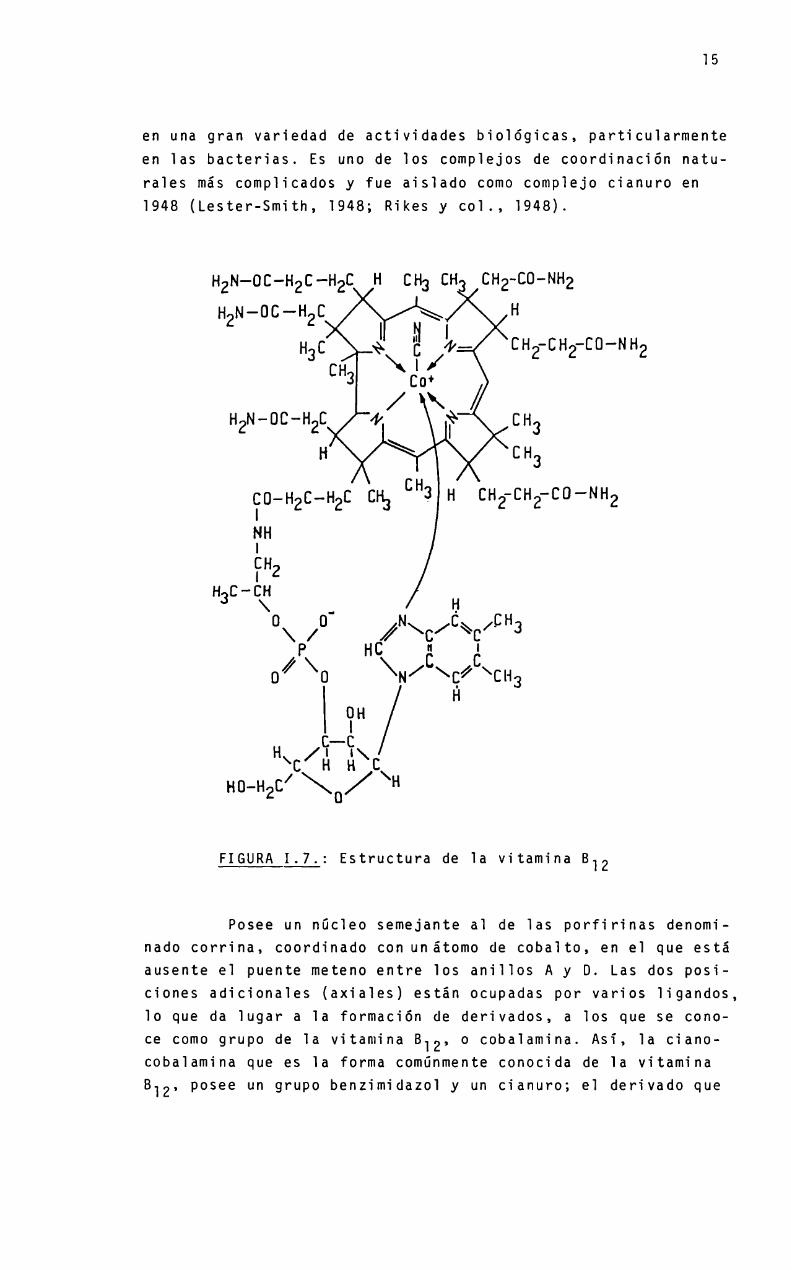
en una gran variedad de actividades bioiógicas, particularmenteen las bacterias. Es uno de ios compiejos de coordinación natura1es más complicados y fue aisiado como complejo cianuro en1948 (Lester-Smith, 1948; Rikes y c01., 1948).
HZN-0C—H2C—H2C H CH3 CH3 GHz-C0—NH2
H
C0-H2C—H2C CH3 .l
HH
i“2H C-CH3 \ _ H
0\ /Ü /N\c/C‘C’CH3P c I
C C
0/ \0 \N/ Mg?\CH3H
pHc-c“xa/i H\c
H0-H2C/\0/ ‘H
FIGURAI.7.: Estructura de 1a vitamina 812
Posee un nücïeo semejante a1 de las porfirinas denominado corrina, coordinado con unátomo de cobaito, en e] que estáausente e] puente meteno entre ios aniiios A y D. Las dos posiciones adicionaies (axiales) están ocupadas por varios ligandos,10 que da Iugar a 1a formación de derivados, a los que se cono
ce como grupo de 1a vitamina 812, o cobalamina. Asi, 1a cianocobalamina que es 1a forma comúnmente conocida de 1a vitamina
B12, posee un grupo benzimidazo] y un cianuro; e] derivado que

posee una molécula de agua en lugar del cianuro se denominaacuocobalamina.
En las formas coenzimas, que son las especies activasen los sistemas biológicos, el grupo cianuro es reemplazado porresiduos alquilo; en la adenosilcoenzima, por un grupo adenosilo.
1.6. TETRAPIRROLES DE CADENA ABIERTA
Son compuestos fuertemente coloreados producidos porla degradación de la hemoglobina; en este caso, se conocen cgmo pigmentos biliares. Tambiénconstituyen el grupo prostéticode las ficobilinas en las algas rojas y azul verdes.
La gran variedad de productos biliares difieren entresi en el grado de hidrogenación de los átomos de carbono queunen los anillos pirrólicos. Comohan perdido la estructura rgsonante del núcleo porfirinico, son inestables yihcïhmnteoxidables. El tipo y la distribución de las cadenas laterales alifíticas son similares a los de las porfirinas, pero poseen algunos restos etilo en lugar de vinilos.
Las ficobilinas de las algas rojas y azul verdes, soncromoproteinas solubles en agua, que poseen pigmentos biliarescomogrupos prostéticos. Se conocen dos tipos de ficobilinas:la ficocianina (azul) y la ficoeritrina (roja) (Figura LLQL.
M E M P P M M E
Il H I l\\ h u ¡fl I (a)HO C N c N CH OHH H2 H H H N
M E M p P M M E
¡I I II II l ,I /I L (b)
0 al CH2 n CH / N CH N 0
FIGURA1.8.: Estructuras propuestas para la ficocianobilina (a)yficoeritrobilina (b).

1.7. EBQPIEDADES
1.7.1. Propiedades químicas
E1 número variabie de grupos carboxilos de ias porfirinas es de gran importancia anaiitica, ya que son principaimenteresponsabies deias diferencias en soiubiiiidad y comportamientocromatográfico.
Los grupos carboxiios pueden ser esterificados, y iosderivados más comúnmentepreparados son Ios ésteres metiiicos.
En ios tejidos y-en los liquidos bioiógicos las porfirinas se encuentran comoporfirinas iibres. Una parte considerabie parece estar en forma de porfirinógenos, ei estado reducido incoioro y no fiuorescente. Los porfirinógenos son oxidadosa porfirinas por medio de] aire y 1a 1uz, o por tratamiento coniodo y es en 1a forma oxidada que se ios trabaja.
Comoya se señaiara, casi todos ios metales pueden combinarse con ios átomos de nitrógeno de ias porfirinas, formandocomplejos metálicos de los cuaies, reiteramos,e1 más conocidoes e] de hierro (hemo). E1 agregado de un meta] en una porfirina provoca cambios en 1a absorción, fiuorescencia y soiubiiidad,que también permiten su identificación.
1.7.2. Propiedades fisicas
1.7.2.1. Soïubilidad
E1 núcleo pirróiico de estructura aromática, le configre propiedades predominantemente hidrofóbicas, ias que se modifican por 1a presencia de ios sutituyentes iateraies aiifáticos,portadores de grupos hidrofiiicos a1coh61icos y carboxiiicos.Sus puntos isoeiéctricos están situados entre pH 3,0 y 4,5. Debido a su carácter anfotéro, son soiubies en medios acuosos tanto básicos comoácidos. También lo son en soiventes orgánicos;ios soiventes empieados comúnmentepara 1a extracción son étery dioxano. Las ciorofiias, debido a1 resto fito], son soiubiestambién en éter de petróieo.
Las porfirinas y sus compiejos metáiicos son fáciimente soiubies en soiventes orgánicos acidificados: acetona-ciorhidrico, éter-ciorhidrico, acetato de etiio-ciorhidrico.

1.7.2.2. Espectros de absorción
E] sistema de dobies iigaduras conjugadas presentes ene] macrocicio, constituye e] grupo cromóforo responsabie de 1aexistencia de fúertes bandas de absorción caracteristicas, enias regiones visible y UVcercano.
E1 espectro de absorción de 10s tetrapirroies es defundamenta] importancia para su identificación y estimacióncuantitativa. Todas las porfirinas exhiben unmáximode absorción en 1a región comprendida entre los 350 y 450 nm, conocidacomobanda de Soret, 1a que es utilizada para 1a cuantificación de estos pigmentos.
Las porfirinas no fiuorescen cuando están dispersadascoioidaimente en medio acuoso y usuaimente tampoco en e] estado sóiido, aunque pueden observarse fiuorescencia cuando estánadsorbidas sobre talco, aiümina o ceiuiosa, en huesos y méduiasde sujetos porfiricos.
La intensidad de fluorescencia también depende de] solvente y de] pH. Es minima en e] punto isoeiéctrico y máxima enácidos mineraies como ácido cïorhidrico a pH 1,0. A] complejarse con metaies ias porfirinas pierden en gran parte,sino tgtaimente, su fiuorescencia. Asi, ios compiejos de Zinc tienenuna fiuorescencia amariiio-naranja, en tanto que ios de hierroy cobre no tienen fiuorescencia visible.
Los porfirinógenos, comoya se comentara, son incoloros y no fluorescen.
E1 método fiuorimétrico es muy sensibie pues permitedetectar porfirinas presentes en cantidades de] orden de 10'8
-1010 M.
M
1.7.2.3. Fotosensibiïidad
Las porfirinas manifiestan una apreciabie sensibiiidadfrente a 1a 1uz, sobretodo a ias radiaciones UV, que son las quemás absorben. Esta fotosensibiiidad disminuye con e1 aumento de]número de grupos carboxiiicos. Asi, 1a uroporfirina es 1a másestabie respecto de las aiteraciones provocadas por 1a iuz. E1pHde] medio tiene particuiarinfiuencia sobre 1a fotosensibiiidad de ias porfirinas, las que en medio fuertemente ácido se alteran con gran faciiidad.

.8. REFERENCIAS
Badaway, A.A.B.; Heïch, A.N. (1981) Biochem. J.12g, 309.
& Morgan, C.J.
Bogorad, L. (1958) J. Bio]. Chem. 233, 516.
B.F.Academic Press,
(1969) en Metabolic Pathways (Ed.NewYork,voi. g, 18.
Burnham, D.M. Greenberg)cap.
Coon, M.J.;w. (1973)
A.P.; P.E.;en Oxidases and Reïated
& Strobe], H.529.
Autor, Lode, E.T.Compounds, p.
Boyer,
R.E. (1972) Sci. Amer. 26, (4), 58.
242, 3.
Dickerson,
Dickerson, R.E. (1980) Sci. Amer.
Dixon, M. & Webb, E.102.
(1964) en Enzymes, 2da. ed. , LongmansLondon, p.
Drabkin, DLL: (1951)'Physi01. Rev. gl, 345.
(1974) en Moiecuïar Mechanisms ofHayaishi), Academic Press, p.87.
Feigeison, P. & Brady, F.A.Oxygen Activation (Ed. 0.
Feigeison, P. & Greengard,0. (1961) J. Bio]. Chem. 236, 153.
Feigeison, P. & Greengard,0.(1962) J. Bio]. Chem. 2 7, 3714.
Fischer, H.(1937) en Die Chemie des Pyrrois, Band II, Leipzig,p. 480.
Gunsaius, I.C. & Lipscomb, J.D. (1972) en Moiecuiar Basis ofeiectron transport (Eds. J. Schultz y B. F. Cameron), AcademicPress, p. 179.
Hayaishi, 0.; Rothberg, S.; Mehier, A.H. & Saito, Y. (1957)J. Bio]. Chem. 229, 889.
Ishimura, Y.; Nozaki, M.; Hayaishi, 0.; Tamura, M. & Yamazaki,I. (1967) J. Bio]. Chem. 242, 2574.
R.D.;Reviews lg,
Summerviiie, D.A. & Basoio, F. (1979) Chem.140.
Jones,
Keiiin, D. (1930) Proc. R. Soc. B log, 418.
Keiiin, D. & Hartree, E.F. (1945) Biochem. J, gg, 293.
Knox, H.E. (1951) Br. J. Exp. Path. gg, 462.
Kotake, Y, & Masayama,T. (1936) Z. physio]. Chem. gig, 237.
Lasceiies, J. (1964) en Tetrapyrroie Biosynthesis and itsRegulation (Ed. Benjamin) New York,

20
Lemberg, R. & Legger,J.N. (1949) en Haematin Compounds andBiïe Pigments. Interscience, NewYork, p. 416.
Lester Smith, E. (1948) Nature 1 1, 638.
Marks, G.S. (1969) en Heme and Chïorophyi (Ed. D. Van NostrandCo.) London.
Mauzerali, D. & Granick, S. (1958) J. Bio]. Chem. 232 1141.
Newton, J.M. & Kamen, M.D. (1961) en The Bacteria (Eds.J.C. Gunsaius y R.Y. Stanier),_I_I_, p. 397.
Academic Press, New York, vo].
Nichoiis, P. & Schonbaum, GgR. (1963) en The Enzymes (Eds. P.D. Boyer, H. Lardy y K. Myrbarck)Academic Press, New York,vo]. 5, p.147.
Oparin, I.A. (1957) en The Origin of Life on the earth.Academic Press, New York.
& Ernster, L.(1974) enMoiecuiarnmchanismof Oxygen215.
Orrenius, S.Activation (Ed.0. Hayaishi), AcademicPress, p.
Perutz, M.F. (1970) Nature 228, 726.
Perutz, M.F.; Muirhead, H.;Nature 219, 131.
Cox, J.M. & Goamani, L.C.G. (1968)
Postgate, J.R. (1959) A. Rev. Microbio]. lg, 505.
Rabinowitch, E.I. & Govindjee, R. (1965) Sci.Amer. 213, 74.
Rikes, E.L.; Brink, N.G.; Koniuszy,K. (1948) Science 191, 396.
F.R.; Wood, T.R. a Foikers,
Schieyer, H.; Cooper, D.Y. & Rosentha], 0. (1973) en Oxidasesand Reiated Redox Systems (Eds. T.E. King, H.S. Mason y M.Morrison), University Park Press, p. 469.
Tanaka, T. & Knox, N.E. (1959) J. Bio]. Chem. 234, 1162.
Torres, H.N.; Carminatti, H. & Cardini, C.E. (1983) en Bioquimica Genera] (Ed. Ateneo), p. 501.
Vernon, L.P. & Kamen, M.D. (1954) J. Bio]. Chem. 1], 643.
Neïch, A.N.& Badaway, A.A.B. (1980) Biochem. J. 192, 403.
(1974) en Moiecular Mechanisms of Oxygen Activation(Ed. 0. Hayaishi) Academic Press, p. 535.Yamazaki, I.

CAPITULO II
METABOLISMO DE LOS TETRAPIRROLES Y SU REGULACION
II.1.11.1.1.
II.1.2.
Biosintesis de tetrapirroiesSuccinii-CoA sintetasaII.1.1.1.II.1.1.2.
PropiedadesMecanismo de acción
6-Amin01evü1icosintetasaII.1.2.1.II.1.2.2.
PropiedadesFormas de aita y baja actividad
II.1.2.3 Mecanismode acciónII.1.2.4 Significado metabóiico de]
ALA-S
[1.1.2L5. Via alternativa para 1a sintesis de ALA
ó-Aminoievülico dehidrasa
PorfobilinogenasaII.1.4.1. PropiedadesII.1.4.2. Mecanismode acciónII.1.4.3. PBG-asay porfiriasDecarboxiiasaII.1.5.1. PropiedadesII.1.5.2. Rutas de decarboxiiación y
mecanismo de acciónII.1.5.3. Decarboxiiasa y PCT
CoprogenasaII.1.6.1. PropiedadesII.1.6.2. Mecanismode acciónII.1.6.3. CPG-asay porfirias
Protogen oxidasaII.1.7.1. PropiedadesII.1.7.2. Mecanismode acciónII.1.7.3. Protogen oxidasa y porfiria
variegata
Página
21
21ñJ
24
25
26
28
29
31
32
34
34
35
36
38
38
39
40
41
42
42
43
45
45
46
46
47

II
II
.4.
.5.
11.1.8.II.1.8.1.II.1.8.2.II.1.8.3.
FerroqueiatasaPropiedadesMecanismo de acciónFerroqueiatasa y porfirias
Cataboïismo de] hemo
Reguïación
11.3.1.II.3.1.1.
II.3.1.2.
II.3.1.4.
II.3.
Porfirias
Referencias
Mecanismos de contro]Contro] a través de cambiosen 1a actividad enzimáticaControi genético a través decambios en 1a cantidad de enzima
Control por especializacióntisuiarControl pOr 10ca1izaciónintraceiuiarContro] por otros constituyentes de] medio ambiente oce1u1arContro] por interconexión conotros caminos metabóïicos
Página
48
49
50
51
51
55
55
55
56
57
58
59
59
60
62

II. METABOLISMO DE LOS TETRAPIRROLES Y SU REGULACION
II.1. BIOSINTESIS DE TETRAPIRROLES
La elucidación de] camino biosintético de los tetrapirroies se debe a una serie de briiiantes trabajos reaiizados en1a década de] 50 por varios grupos de investigadores (Shemin yRittenberg, 1945, 1946, 1951; Cookson y Rimington, 1953; Dreseiy Fa1k, 1953; Granick y Bogorad, 1953; Neuberger y Scott, 1953;Shemin y Russel, 1953; Rimington y Kró], 1955 . Comoresuitadode elios se estabieció que la mayoria de los organismos vivientes son capaces de sintetizar sus propios tetrapirroies a partir de molécuias simples constituyentes de 1a dieta, tales comoe] succinato y 1a glicina, y siguiendo una secuencia de reaccignes catalizadas por varias enzimas, locaiizadas algunas en e]citopiasma y otras en 1a mitocondria (figura 11.1.).
La sintesis de porfirinas sigue una via metabólica idéntica en todas ias céiuïas, hasta 1a formación de protoporfirina IX, que es e] intermediario común de hemos y ciorofiias.En e] caso de ias corrinas, iabifurcación se produce a 1a aïtura de] uroporfirinógeno (urogen) III.
11.1.1. Succinii-CoA sintetasa
La succinii-CoA sintetasa (Suc.CoA-S) (succinato:CoAiigasa, adenosin difosfato E.C. 6.2.1.5.; guanosin difosfatoE.C. 6.2.1.4.), también denominada enzima fosforiiante (Kaufman1955) y succinico tioquinasa (Beinert y co]., 1956), es una enzima que se iocaiiza en 1a fracción particuiada de ias céiuias,y a partir de su descubrimiento por Kaufman(1951), ha sido ai;1ada, detectada y purificada de tejidos animaies (Cha y Parks,1964; Bai] y Nishimura, 1980), vegetaies (Kaufmany Aiivisatos,1955; Nandi y Waygood, 1965; Wider de Xifra y co]., 1971;Schwartz y c01., 1983) y bacterianos (Burnham, 1963; Ramaiey yc01., 1967; Heitzman, 1982). Varios reviews han sido dedicadosa esta enzima (Nishimura y Grinne] , 1972; Bridger, 1974;Nishimura, 1986).
Cataiiza reversiblemente 1a reacción:
2+Succinato + CoA + NTP‘Mï; SucciniI-CoA + NDP+ Pi

22
SUCCINIL CoA 6L|C|NA
ALA-Sintetasa
(ALA-S) (H)co; PPV
ALA 4‘- ...... -- ACIDOJ-AHJIOLEvuuco
(ALA)
NUEVAPORÍIMRAGUDI___. AL¿_Dehidrasa
'_"= J (ALA-0(c)2 H1O
,56 4.- ....... -- PORFOBILINOGEHO
(psc)NH
romannauun¡»mamut _> Domina“ 3(c) unocau l --- >UR0 l
O
Pullillll CUNGENITRtRlÏRUI‘OIlTICA —u. B COOH.————_——. 'Iaomel'aua
mguno lll <--------- -- unoczn lll €02_
a coou
502 (/1 mmm cummmom7 c lll <---- 7 coou ¿ 7 coou ---->7 c I
coz /I ngen t\__ coz6 ‘_ 6 coo“ Decarboxilasa 6 coo” _____6 c 'll '"
c ' (uno-DI
mz {z/q (n rx, chs c lll <--- 5 coou 5 C00" ""' 5 C '
co2 «1/4 f\r Chcorno lll e ------- -- covnoctu lll COPnoceun --—-> corno n
h coou h coon
CUPRÜPORÍ"ui —== col Coprogenasa(CPGaóa’
02 (n)
C02
O N
u noo :l:
'_- PROTOGENlx
moro ¡x """ K Protogen-Oxidasa.._ mmm mmm(PñLOM
“ (H)“ PROÏOPORFIRINA lx
rs.
FeH Perroquelatasa <——-rmrwonlmatnnnwo¡ïua_==
(M
ELFIGURA11.1.: Esquema de] camino bíosíntético de] hemo con 1a
locaïización de 1as enzimas en 1a mitocondria(M) o en e] cítoso] (C). Se señaïan también 1asfa11as enzimáticas primarias de Ias porfírias.

23
donde NTPes un nucieósido trifosfato y M2+un catión metáiicodivaTente, generaïmente magnesio o manganeso (Gibson y c01.,1967). Participa asi en e] ciclo de Krebs a través de Ta fosforiTación de TosnucTeósidos difosfatos, y enla reacción inversa,sintetiza succiniT CoA, precursor deT hemoy de 1a cistationina (Bridger, 1974).
11.1.].1. PropiedadesEs aTtamente especifica para succinato y CoA, no asi
para e] nucieósido trifosfato, en e] que 1a especificidad variasegún e] origen de 1a proteina: mientras que 1a enzima de mamiferos puede empTear GTP o ITP (Cha y Parks, 1964), Ta de plantas utiiiza ATP (Nandi y Waygood, 1965; Nider de Xifra y coi.,1971) y Tas bacterias parecen poder usar cuaTquiera de eiios(Burnham, 1963). Sin embargo recientemente, se ha demostrado unrequerimiento absoiuto por ATPpara 1a enzima de mitocondria demüscuïo de palomas (A11en y Ottaway, 1986).
En cuanto a] peso moiecuiar, se han encontrado vaioresque varian entre 70kDa para 1a enzima de corazón de cerdo (Chay c01., 1967 a) y T41kDa para 1a de E. coi¿ (RamaTey y coT.,1967). Mediante e] empleo de agentes disociantes se confirmóque está compuesta por dos tipos de subunidades: a y 6, de pesomoiecuiar 29,5 KDy 38,5kDa respectivamente (Bridger, 1971), a
signándoseie entonces una estructura tetramérica de] tipo a282a 1a enzima de E. coli (Bridger, 1971), mientras que 1a de mamíferos seria un dimero a8 (Brownie y Bridger, 1972). Se ha reportado además, 1a reconstitución de 1a enzima activa a partirde las subunidades aisiadas (Pearson y Bridger, 1975 a; Bridger,1981).
Presenta 1as caracteristicas de una enzima suifhidriïica, siendo inhibidaporp-cToromercuribenzoato(PCMB) iodobenzoato, iodoacetamida, gÏutatión oxidado y N-etiTmaTeimida(NEMI) (Nishimura y co]., 1973). En aigunos tejidos es necesaria1a presencia deprotectores de grupos -SH como e] gTutatión (GSH)para 1a expresión de su actividad; sin embargo, otros tioies cgmocisteina, tiogTicolato y ditiotreito] (DTT)producen_inactivación (Wider de Xifra y Batlle, T980). Se han detectado 16-18grupos suifhidriios tituTabTes por moTécuTa(Nishimura y co].,1973), de Tos cuaïes tres son importantes para su actividad(CoTTier y Nishimura, 1978; O'Connor-Mc Court y Bridger, 1985).

24
Asimismo, se ha informado que existen tres residuostriptofano en 1a subunidad B (Prasad y c01., 1983), y que 1a mgdificación de uno de elios produce una pérdida compieta de 1aactividad enzimática (Ybarra y c01., 1986).
Buck y co]. (1985) han reportado 1a secuencia primariade 1a enzima de E. coZL. Sus genes se iocaiizarian a1 fina] deuna serie de 9 genesque codifican para otras enzimas de] ciclode Krebs. La expresión de 1a Suc;CoA-S estaria reguiada en forma coordinada con 1a 2-oxogiutarato deshidrogenasa (Buck y co].,1986).
Numerosasevidencias experimentales han establecidoque 1a Suc.CoA-S desempeña un papel de importancia en e] caminobiosintético de ias porfirinas y participa ademásen ios mecanismos que 1a controlan (Labbe y coJ., 1965; Batiie y co].,1975).
II.].1.2. Mecanismode acción
E1 mecanismo de acción de 1a Suc.CoA-S ha sido objetode numerosos estudios, ios cuales poStuian diferentes esquemas,pero todos eiios coinciden en que 1a primer etapa invoiucra lafosforiiación de la proteina enzimática.
Empieando ATP32Py un extracto libre de céiuias, se encontró que 1a mayor parte de 1a radioactividad quedaba asociadaa la fracción proteica que contenia Suc.CoA-S (Ramaiey y co].,1967) y utiiizando técnicas de insoiubiiización se confirmó 1aexistencia de] complejo enzima-P (Wider de Xifra y co]., 1972).
Se ha estabiecido además, que en esta etapa ocurre 1afosforiiación de un residuo histidina (Huitquist y co]., 1966)de 1a subunidad a (Bridger, 197]; Teherani y Nishimura, 1975),siendo 1a subunidad aisiada capaz de fosforiïarse (Pearson yBridger, 1975 b).
En extractos de calios de soya, Nider de Xifra y Batiie(1976) demostraron 1a formación de] compiejo enzima-CoA y observaron que 1a Suc. CoA se combina fácilmente con 1a CoA iibre ocon 1a CoAen presencia de ATPy magnesio, sugiriendo como intermediario de 1a reacción enzimática a] compiejo ternarioE-P-CoA'
E1 sitio de unión para 1a CoAse 10ca1iza en 1a subunidad B (Coiiier y Nishimura, 1978), y en 1a misma subunidad se

encontraría e] sitio para e] succinato y 1a succini] CoA(Pearson y Bridger, 1975 b), 10 que recientemente fue confirmado mediante cromatografía por afinidad en una coiumna de agarosa-hexano-CoA (O'Connor—McCourt y Bridger, 1985).
Para 1a reacción tota] son necesarias las 2 subunidades, a y B, proponiéndose que e] sitio activo se Iocaïiza en e]punto de contacto entre ambos monómeros (Pearson y Bridger,1975 b; Bai] y Nishimura, 1980).
Técnicas de intercambio isotópico empieando 32P(Kaufman y Aïivisatos, 1955; Cha y co]., 1967 a; Gibson y co].,1967), 18o (Cohn, 1951) y 14c (Kaufman y Aiivisatos, 1955; Chay co]., 1967 b; Moyery co]., 1967), confirman 1a reversibilidad de 1a reacción para 1a que se han medido además, constantesde equiïibrio (Kaufman y Aiivisatos, 1955; Lynn y Guyn, 1978).
Teniendo en cuenta todas 1as evidencias experimentaïesacumuiadas hasta entonces, Wider de Xifra y Bat11e (1976) prgpusieron un mecanismo que considera todos ios mecanismos postu1ados planteando Ia posibiiidad de caminos aïternativos, aúnIos no enzimáticos.
Más recientemente, dado que 1a enzima tetramérica posee 2 sitios activos equivalentes, se ha desarroïïado e] concepto de un mecanismo de cooperatividad cata1itica de sitios a1ternativos (Bridger, 1984). Esta hipótesis cuenta con evidenciasbasadas en medidas de intercambio de 1802 (Biid y c01., 1980),resonancia magnética nuc16arcon 31P (Vogeï y Bridger, 1982), estudios cinéticos (Wolodkoy co]., 1983; Nishimura y Mitcheïl,1984, 1985) y de hibridización (No1odko y co]., 1980; O'ConnorMc Court y Bridger, 1985).
11.1.2. ó-Aminoievü1icosintetasa
La formación de ácido 6-amin01evüïico (ALA) a partirde giicina y succini] CoAconstituye 1a primer etapa que conduce especificamente a 1a biosintesis de porfirinas y está catalizada por 1a enzima ó-aminolevüïico sintetasa (ALA-S, succini]CoAzglicina C-succini] transferasa, E.C. 2.3.1.37):
giicina + succini] CoA _Ï¿ÏÉ, ALA+ co2 + CoAPPy

26
Una de las fuentes más activas de esta enzima son lasbacterias del género Athiorhodacea, Rp. ¿pheno¿de4 (Kikuchi ycol., l958 a, b; Burnhamy Lascelles, l963). Se la ha detectado también en bacteriasno-fotosintéticas (Tait, 1973), y otrosmicroorganismos tales como Pnop¿on¿bacten¿um óhehman¿¿ (Menon yShemin, l967), Spánáklum¿tenóaniá (Clark-Walker y col., l967),Rp. paiuótn¿¿ (Viale y col., l980). En tejidos animales: eritrocitos de aves (Gibson y col., 1958), higado de cobayo(Granick y Urata, l963) y de ratas (Marver y col., 1966), glándula de Harder (Tomioy Grinstein, 1968), reticulocitos de conejos (Aoki y col., 1971), higado, eritrocitos (Strand y col.,l970) y plasma humanos (Miyagui y Watson, 1972).
Las evidencias de su existencia en sistemas vegetalesson escasas: se ha reportado en espinaca (Miller y Teng, 1967),callos de soya (Nider de Xifra y col., l97l), cáscara de papas(Ramaswamyy Nair, 1973). También se han publicado resultadosnegativos acerca de los intentos por detectarla en plantas supgriores (Porra e Irving, l970; Beale, l97l; Porra y Grimme, 1973,l974).
En los últimos años, se ha medido su actividad en elalga verdeunicelular Eugiena gnac¿l¿ó, tanto encélulas de la cgpa salvaje comoen las de una mutante aplástida (Beale y col.,l98l) y también se la aisló y purificó en levaduras (Volland yFelix, 1984).
II.l.2.l. EropiedadesEn tejidos mamíferos la enzima se localiza en las mito
condrias.especificamente en la matriz mitocondrial (Scotto ycol., 1983),y en estados normales su nivel es muy bajo; sin embargo, bajo ciertas condiciones experimentales, se ha aisladotambién una forma soluble (Marver y col., l966; Hayashi y col.,1969; Ohashi y Kikuchi, 1972; Whiting y Elliot, 1972; Patton yBeattie, 1973).
Hay evidencias que indican que el ALA-Sse sintetiza enlos polirribosomas microsomales y luego migra a la mitocondria(Scholnick y col., 1969; Srivastava y col., 1983 a), organeladonde exhibe su actividad catalitica (Ohashi y Kikuchi, 1977).
Mediante la preparación de anticuerpos contra ambas enzimas, se comprobó que son inmunoquimicamente idénticas (Whitingy Elliot, l972; Nakakuki y col., l980) apoyando la hipótesis de

que 1a enzima citosóïica es un precursor de 1a particuïada(Yamauchi y co]., 1980).
En 1a mayoria de 1as fuentes estudiadas, se caracteriza por su inestabiiidad y corta vida media:fetal de rata (Woods, 1974), 68 minutos en mitocondria de higado de rata (Marver y c01., 1966), 10 que dificuita su estudio ypurificación. Varios autores adjudican 1a corta vida media a1a acción de proteasas (Aoki y c01., 1975; Aoki, 1978; Nakakukiy c01., 1980; Srivastava y c01., 1983).
E1 peso molecular varia según e] tejido,las condiciones de purificación.
1a especie yUn parámetro que parece afec
tar e1 peso moiecuiar es 1a fuerza iónica de] buffer uti1izadodurante e] proceso de purificación. A baja fuerza iónica, seproducen agregados, principamente Cuando se trabaja con 1a enzima citosóïica (Schoïnick y-c01., 1970). Es asi que se han reportado pesos molecuïares que van de 650kDa a 40kDa (Hayashi y co].1969; Tuboi y co]., 1970 a; Ohashi y Kikuchi,c01., 1972; Nhiting y E11iot, 1972; WhitingNandi y Shemin, 1977).
1972; Schoinick yy Granick, 1976;
Varios autores observaron que e] precursor citoplasmatico posee un peso moïecuïar mayor que e] componente activo mitocondria] (Ohashi yKikuchi, 1977; Srivastava y co]., 1983 b).Es probabïe que las proteinas inactivas que acompañan a1 precursor citopiasmático moduïen 1as propiedades cataiiticas de] componente activo, o tengan un ro] en 1a trasiocación a mitocondrias (Ohashi y Shinohara, 1978). Ambas formas son inmunoquimicamente idénticas, se presentan comodimeros y tienen propiedades cinéticas simiïares; 5610 difieren en e] punto isoeïéctrico10 que podria ser importante en e] proceso de trasïocación a 1amitocondria (Watanabe y co]., 1984). Ambassubunidades de pesomoiecuiar 51 kD, son idénticas (Ohashi y Kikuchi, 1974), 10 quefue confirmado mediante experiencias de entrecruzamiento condimetiïsuberimidato (Nakakuki y co]., 1980).
En 10s últimos años, se han aisiado cDNAque codificanpara e] ALA-Sde higado de embrión de poiio (Borthwick y co].,1984, 1985), higado de poiio (Maguire y c01., 1986) y eritrocitos de polïo (Yamamotoy coï., 1985).
Yamamotoy c01., (1980) sugieren 1a existenciade dos isoenzimas en higado de rata, debido a 1a falta de reactividad cruzada entre ALA-Sde eritrocitos y anticuerpos
34 minutos en higado
o

28
anti ALA-Shepática; proponen ios autores que en ratas debe haber a1 menos dos genes diferentes para esta enzima.
{1.1.2.2. Formas de alta y baja actividad
Desde los primeros intentos de purificar ALA-Sde Rp.aphenoLdeó se habian observado variaciones en 10s niveles de agtividad de 1a enzima (Kikuchi y c01., 1958 a, b), que posteriormente se encontró, se debia a 1a existencia de inhibidores, activadores y diferentes formas enzimáticas.
En céiuias Crecidas semianaeróbicamente a 1a 1u2,Marriot y co]. (1969) observaron una activación espontánea deIos extractos sonicados durante 1a primer hora de aimacenamiento a 4 °C; si las céiulas se oxigenaban antes de ser cosechadas,esta activación espontánea no ocurrïa.Los mismosautores detegtaron en céiuias crecidas semianaeróbicamente una fracción debajo peso moiecuiar termoestabie que era capaz de activar lascéiulas oxigenadas, y de manera simiiar, encéiuias oxigenadas,otra fracción también de bajo peso moiecuiar y termoestabie,que inhibia 1a activación espontánea. Postuiaron entonces, quee] oxigeno aiteraria ias concentraciones de activador e inhibidor.
A] mismo tiempo, e] grupo de Kikuchi aisió un inhibidor especifico de acción reversible; se trataba de un compuesto iábi], sensible a 1a luz, que no era ni hemina ni protoporfirina ni otro compuesto relacionado (Tuboi y co]., 1969). Mástarde, encontraron que ios cuitivos de estas bacterias conteniandos formas de ALA-S: ias fracciones I y II, ias cuaies eran inducidas de manera diferente por variaciones en ias condicionesde crecimiento (Tuboi y c01., 1970 b). La fracción I a su vez,podia existir en una forma activa y otra inactiva. Esta últimapodia activarse a través de aiguna modificación enzimática de1a fracción I (Tuboi y Hayasaka, 1971). Posteriormente, se comprobó que esa conversión requería 1a presencia de un compuestotipo disuifuro (tai comohomocistina o L-cistina), y un componente proteico que fue parciaimente purificado (Tuboi yHayasaka, 1972).
En e] mismo tejido, Fanica-Gagnier y Ciement-Metra](1973) aisiaron y purificaron dos especies de ALA-Sque diferianen su contenido de grupos suifhidriios; ambas presentaban unadiferente compartimentaiización: una era citopiasmática y

29
parecía estar reiacionada con e] metaboiismo respiratorio en o;curidad, en tanto 1a otra se encontraba en cromatóforos y erainducida por 1a 1uz.
E1 grupo de Neuberger, a su vez, postuïó que 1a anu1ación de 1a activación espontánea observada por Marriot, se podía atribuir a1 efecto de] oxigeno sobre e] metaboiismo de iossu1focompuestos (Neuberger y c01., 1973 a). Aisiaron un activador de bajo peso moiecuiar (Neuberger y coi., 1973 b) y sugirigron que compuestos tipo polisuifuros orgánicos controiaban laactividad (Neuberger y c01., 1973 c).
Posteriormente, postuiaron un esquema (Wider de Xifray c01., 1976) para explicar 1as variaciones de actividad deALA-S,según e] cua] 1a actividad enzimática estaría controiadapor 1a concentración de un compuesto de azufre: 1a trisuifocistina.
Unos años después. Inoue y co]. (1979) purificaron unaenzima activante a 1a que identificaron comocistationasa, y encontraron que además de esta enzima activante y 1a L-cistina,se requería un activador de alto peso moiecuiar que sería unaproteína reguiadora (Oyamay Tuboi, 1979).
También en Rp. paiuatn¿4, 1a actividad de ALA-Sestaria controlada por 1a presencia de un compuesto de bajo peso mg1ecu1ar, aunque e] mecanismo de acción de este factor reguiadorsería diferente a1 existente en Rp. ¿phenaádeó (Viale y co].,1980).
En 1a enzima de mamíferos, Irwing y Eiiiot (1969) repo:taron 1a existencia de un inhibidor en mitocondrias de hígadode cobayo. En mitocondria de hígado de rata, Simpson y Beattie(1980) purificaron una proteína capaz de activar e] ALA-Sfavoreciendo 1a formación de agregados de mayor peso moiecuiar, por10 que este activador reguiaría 1a actividad de ALA-Smodificando su estructura cuaternaria.
Se ha postuiado que en caiios de soya (Vázquez y co].,1980) y aún en mamíferos (Vázquez y coi., 1987) podría operarun mecanismo de contro] de 1a actividad de] ALA-S, simiJar a1propuesto para 1a enzima de Rp. ¿phenoádeó.
II.1.2.3. Mecanismode acción
La síntesis de ALAa partir de giicina y succini] CoA

30
va acompañada por 1a pérdida de un grupo carboxiio de 1a giicina como dióxido de carbono y iiberación de CoA.
Es un hecho ya bien establecido 1a participación de]fosfato de piridoxai en esta reacción y que 1a primer etapa consiste en una transaminación entre e] compiejo enzima-fosfato depiridoxa] y 1a giicina (Shemin,1970).
Experiencias iievadas a cabo por e] grupo de Snei](Metzier y c01., 1954) sugirieron que e] ro] primario de] fosfgto de piridoxai en ias reacciones enzimáticas con aminoácidoses e] de 1a activación de] carbono a a través de 1a formaciónde un centro nucieofiiico.
Esto iievó a proponer un mecanismo segün e] cua] e]fosfato de piridoxa] se une a 1a superficie de 1a enzima a través de un grupo suifhidriio dei sitio activo, y se forma una base de Schiff con 1a glicina; 1uego se produce 1a condensaciónde] succinato activado como succinii CoApara dar un ácido inestabie: e] a-amino-B-cetoadipico, que luego se decarboxiïa aALA(Laver y coi., 1959; Granick y Kappas, 1977).
Empieando giicina marcada con 14€, se reaiizaron experiencias que demostraron que e] dióxido de carbono sóio se iiberaba cuando 1a succini] CoAestaba presente en e] medio de incubación, aportando una evidencia indirecta de que 1a activaciónde 1a giicina ocurria por 1a pérdida de un protón (Kikuchi yco]., 1958 b). La fuerte inhibición por e] aminomaionato(Matthew y Neuberger, 1963) y e] hecho de-que ciertos L-aminoácidos son inhibidores más eficaces que sus isómeros D (Gibson yco]., 1958), proveen un apoyo adiciona] para 1a formación de]carbanión.
Años después se prepararon giicinas quirales conteniendo tritio estereoespecificamente iocaiizado en orientación R óS, y se demostró que 1a formación de ALAse produce con 1a iibgración de un protón pro R de 1a giicina (Akhtar y Jordan, 1969;Jordan y Akhtar, 1970; Zamany co]., 1972).
Quedaba por diiucidar si e] a-amino-B-cetoadipico seforma y decarboxiia unido a 1a enzima, o si 10 hace iibre en sgiución, pues se habia encontrado que este ácido se decarboxiiaba espontáneamente en soiución neutra (Laver y coi., 1959), ypor otro iado, si era reaimente un intermediario, ya que era pgsibie que 1a condensación y 1a decarboxiiación ocurriera en

31
forma concertada.
Por otra parte, mediante estudios espectraies, Nandi(1978 a) comprobó 1a formación de una base de Schiff entre 1aenzima y e] fosfato de piridoxa], en 1a que estaria invoiucradaun grupo e-amino de lisina. Luego ocurriría una transaminaciónentre e] compiejo enzima-fosfato de piridoxai y 1a giicina, dando otro complejo de estructura quinoide (Nandi, 1978 b) y seformaría e] carbanión por 1a pérdida de un átomo de hidrógeno ode 1a giicina. En base a estos resuitados, 1a decarboxiiaciónes un proceso enzimático que ocurre 1uego de condensada 1a unidad succini], dando iugar a 1a formación de] ácido a-amino-B-cgtoadipico.
Mediante un procedimiento indirecto empieando ALA-Sy6-aminoievü1ico dehidrasa (ALA-D)purificadas de Rp. ¿phenoáde4,incubadas con giicina tritiada y ios cofactores necesarios, seobtuvo porfobiiinógeno radiactivo, y un anáiisis de este produgto confirmó que e] mecanismo de decarboxiiación ocurre con todos ios intermediarios unidos a 1a enzima (Abboud y co]., 1974).
II.1.2.4. Significado metabólico de] ALA-S
Además de su obvio ro] en 1a sintesis de ALA, una grancantidad de información se ha acumuiado impiicando a1 ALA-Scgmo una enzima clave en e] contro] de 1a biosintesis de tetrapirroies.
En 1a bacteria fotosintética Rp. ¿pheno¿de4, que produce hemo, clorofiia y corrina, eimecanismo de contro] es muy complejo. La bacteria tiene una marcadacapacidad de adaptación avariaciones en las condiciones de crecimiento tales comointensidad de luz y tensión de oxigeno. E1 hemo, a su vez, parece jugar un pape] importante sobre la sintesis de ALA a través deuna inhibición por feed-back (Burnhamy Lasceiies, 1963).y seha sugerido que un mecanismo de des-represión expiicaria ios aumentos en e] nivei de 1a enzima bajo condiciones de intensidadde 1uz reducida y baja tensión de oxigeno (Lasceiies, 1960;Tuboi y co]., 1970 b).
Se han descripto además, como hemos visto, inhibidoresy activadores endógenos (Marriot y c01., 1969; Tuboi y c01.,1969) indicando 1a existencia de otros mecanismos de contro]que operarian a nivei moiecuïar.

En especies mamiferas, el ALA-Ses también una enzimalimitante (Granick, l966; Marver y col., 1966). Los cambios ensu actividad son una de las principales causas que provocan losdisturbios bioquímicos observados en las porfirias tanto hereditarias comoinducidas (Strand y col., 1970).
Sobre la base de elegantes estudios, Granick sugirióque la formación de ALA-Sse encuentra controlada por un mecanismo a nivel de DNAde un sistema operador-represor, en el queel hemo actuaria comoco-represor previniendo la inducción dela enzima (Granick, 1966). Experiencias llevadas a cabo en loslaboratorios de Tschudy apoyan esta hipótesis (Naxmanny col.,l966). La inhibición por hemoocurre probablemente a nivel posttranscripcional (Sassa y Granick, l970; Strand y col., l972;Tyrrel y Marks, 1972; Tomita y col., l974), aunque no puede de;cartarse la posibilidad de que interfiera también a nivel transcripcional (Whiting, 1976; Srivastava y col., l980).
Un nuevo mecanismo de regulación feed-back estaria representado por la inhibición del hemoen la traslocación intracelular de la enzima (Yamauchi y col., 1980; Kikuchi y Hayashi,1981; Hayashi y col., l983).
En levaduras, el nivel de actividad de ALA-Sdependedel estado metabólico de las células, pero aparentemente la sintesis de ALAno es limitante para la producción de hemo(Labbe-Bois y Volland, 1977). Se han encontrado mutantes totalmente deficientes para la sintesis de hemoen las que los niveles de ALA-Sno se hallan modificados (Urban-Grimal y Labbe-Bois,1981). Sin embargo, hay mutantes en las que dicha sintesis estáparcialmente bloqueada, y que presentan un incremento de la actividad de ALA-S(Rytka y col., 1984).
II.l.2.5. Via alternativa para la sintesis de ALA
Existen actualmente numerosas evidencias según las cuales el ALA-Sno es la única enzima capaz de catalizar la formación de ALA. En diversos tejidos se observó la presencia de unatransaminasa que podia producir ALAa partir de un aminoácidodonor y del ácido y-ó-dioxovalérico (DOVA) u otro precursor decinco átomos de carbono como aceptores.
14En 1955, mediante la administración de C-ALAen ratas, Sheminy col. (l955) observaron que la radiactividad

33
aparecia en e] dióxido de carbono respiratorio, e] formiato urlnario y e] grupo ureido de] ácido úrico. Se postuió entonces,e] conocido comocicio de] succinato-giicina o cicio deShemin-Russe], en e] que e] ALAformado por condensación de 1agïicina con e] succinato, se desamina dando DOVA;1uego éstepierde e] grupo carboniio termina] regenerando e] Succinato,dando a su vez un fragmento de un átomo de carbono provenientede] carbono-2 de 1a giicina, que posteriormente es capaz de participar en las reacciones de biosintesis de purinas. Resuitadossimilares se obtuvieron con paiomas y patos (Nemeth y co].,1958) y e] cic10 fue confirmado en Rhodo¿p¿n¿zlum nubnum(Shigesada, 1972).
En 10 que respectaa Iadesaminación de] ALA.en homogenato de higado de mamíferos (Kowalsky co]., 1959) y en extractosde Conynebactenium d¿6phten¿ae (Bagdasarian, 1958) fue detectada una enzima capaz de cataïizar 1a transaminación de] ALAconpiruvato o a-cetogiutarato, produciendo respectivamente aianinay gïutamato.
Gibson y co]. (1961) demostraron que e] equilibrio en1a transaminación estaba desplazado en favor de 1a formación deALAa partir de DOVA.Incubando un extracto enzimático de Rp.Aphenoideó con DOVAy alanina se obtuvo ALAy piruvato, y de esta misma bacteria, Neuberger y Turner(1963) aisiaron una enzimaque caracterizaron comoy-ó-dioxovaiérico transaminasa (DOVA-T).Otro sistema en e] que se midió sintesis de ALAa partir deDOVAfue Chkoneiia vuiganáó (Gassman y co]., 1968); se reaiizaron estudios cinéticos de transaminación y de especificidad deaminoácidos.
Beaie y Casteifranco, trabajando con cotiiedonesde pepinos enverdecidos (1973) y hojas etioladas de cebaday porotos (1974).observaron que partiendo de precursores radiactivos taïes comogiutamato, glutamina y a-cetogiutarato, e] esque1eto intacto de esos cinco átomos de carbono se incorporaba a]ALA, 10 que evidenciaba 1a existencia de una via diferente de]ALA-S, pues si 1a sintesis se produjera a través de] ALA-S, unode los cinco átomos de carbono se liberaria como dióxido decarbono, a1 pasar via a-cetoglutarato a succinato. Se vió además que carbono 1 de] glutamato se convertía en e] carbono 5de] ALA(Beaie y co]., 1975) a través de] a-cetogïutarato y e]DOVA.

34
En hojas de Zeamayó se han purificado parcialmente dosenzimas que catalizan la conversión de a-cetoglutarato a DOVA,ydeDOVAa ALA (Lohr y Friedmann, l976).
Este mecanismo alternativo se piensa que opera en plantas superiores (Beale y Castelfranco, 1974; Lohr y Friedmann,1976) y algas (Gassman y col., 1968; Kipe Nolt y Stevens, 1980),pero se ha detectado también en bacterias (Gibson y col., 1961;Bajkowski y Friedmann, l982) y en tejidos de origen animal(Kowalsky y col., 1959; Vartikowsky y col., 1980). En estos ültimos se ha visto además que la capacidad de la DOVA-Tde sintgtizar ALAparece ser superior a la del ALA-Smitocondrial descgnociéndose aün si ello responde a alguna acción regulatoria. Ambas vias de sintesis de ALAse han detectado también en el algaverde Scenedeómuó obláquub (Klein y Senger, 1978), en Rp. ¿pheno¿de¿ (Kikuchi y col., 1958; Gibson y col., l961) y en E. gnac¿Z¿¿.
II.l.3. 6-Aminolevülicodehidrasa
Esta enzima se tratará exhaustivamente en el CapituloIII.
II.l 4. Porfobilinogenasa
Conel término porfobilinogenasa (PBG-asa) E.C.4.3.l.8)propuesto por Lockwoody Rimington (1957), se designa al complgjo enzimático que cataliza la ciclación de cuatro moléCulas deporfobilinógeno (PBG)para dar urogen III, intermediario fisiglógico en la biosintesis de hemos, clorofilas y corrinas.
Este sistema enzimático está formado por dos proteinas:la uroporfirinógeno I sintetasa o deaminasa, y la uroporfirinógeno III cosintetasa o isomerasa. La primera de ellas conviertecuatro moléculas de PBGen una del tetrapirrol cíclico simétrico urogen I, por medio de una condensación cabeza-cola, con eliminación de amoniaco y cierre final del anillo (Bogorad, 1958).La isomerasa, en cambio, por si sola es incapaz de utilizar elPBGcomo sustrato, tampoco actüa sobre el urogen I, pero asociada a la deaminasa, lleva a la formación del urogen III.
La formación de urogen del tipo III estaria aseguradaporque la cantidad relativa de isomerasa es generalmente muchomayor que la de deaminasa (Frydman y Feinstein, 1974).

Estas enzimas han sido detectadas y aisiadas en diversos organismos que abarcan_toda 1a escaia viviente.
II.1.4.1. Propiedades
Su iocaiización es citoplasmática, aunque aigunos autgres encontraron actividad de PBG-asaasociada a fracciones particuiadas (Bogorad, 1955 a, b; Fa1k y Dresei , 1960; Care] yKahn, 1964; Rossetti, 1978; Rossetti y co]., 1986).
La deaminasa es termoestabie, en tanto que 1a Isomerasa es termoiábii (Higuchi y Bogorad, 1975), propiedad que se haempleado para separar ambos componentes (Liambias y Batiie,1971 a, b).
Tanto 1a PBG-asa como sus componentes se comportan comoenzimas suifhidriiicas (Sancovich y c01., 1969; Liambias yBatile, 1971 a, b; Frydman y Feinstein, 1974; Russel y Rockwei],1980), si bien no se ha estabiecido aün e] modo de acción de10s grupos tioles; se inhiben por compuestos básicos taies comoamonio, hidroxiiamina y alquiiaminas (Bogorad, 1962, 1963;Sancovich y co]., 1969; Frydman y Frydman, 1970; Liambias yBatiie, 1971; Frydmany Feinstein, 1974). En cambio en aigunoscasos las sales de sodio y magnesio tienen un efecto estimulante (Liambias y Batiie, 1971 a, b; Sancovich y coi., 1976;Ciement y co]., 1982). Los estudios cinéticos indican que en algunas fuentes este complejo presenta caracteristicas de proteina aiostérica, observándose en aigunos casos cooperatividadpositiva (Sancovich y co]., 1969; Liambias y co]., 1971) y enotros, negativa (Liambias y Batiie, 1970).
Las determinaciones de pesos moiecuiares han reveiadoque para 1a deaminasa parece haber una uniformidad en cuanto aque e] vaior reportado por diversos autores es de unos 40 kDa(Liambias y Batiie, 1971; Jordan y Shemin, 1973; Higuchiy Bogorad, 1975; Sancovich y co]., 1976; Miyagi y c01., 1979;Anderson y Desnick, 1980; Rossetti y co]., 1980; Wiiiiams yco]., 1981; Fumagaiii y co]., 1985) a excepción de Frydamn yFeinstein (1974) quienes encontraron un peso moiecuiar de 25 kDapara 1a enzima de eritrocitos humanos y Kotier y co]. (1987) deaproximadamente 30 kDapara 1a deaminasa de Rp. patuótnLA.-Encambio para 1a Isomerasa y e] compiejo PBG-asa, se han reportado vaiores que van desde 5 kDahasta 280 kDa,10 que ha 11evado apostuiar que existen fenómenos de asociación-disociación en

36
tre ambos componentes de 1a PBG-asa, asi como con 1a isomerasaaisiada (Rossetti y co]., 1980).
II.1.4.2. Mecanismode acción
Hay evidencias de que 1a formación de] aniiio tetrapirróïico ocurre en dos etapas: primero parece formarse un intermediario poiipirróiico de cadena abierta que en una segunda fase se cic1aria produciendo e] urogen.
En ciertos tejidos se han detectado y aisïado, en condiciones normaies y en presencia de inhibidores, intermediariospoiipirróiicos de cadena abierta que varian en Iongitud y estructura, según 1a fuente y e] tipo de reacción, y que se comportan como intermediarios de 1a reacción pero no como sustratos (Lïambias y Bat11e, 1970 a, b, c,; 1971 a, b; Rossetti yBatiie, 1977; Rossetti y c01., 1977).
En forma paraieia, por sintesis quimica se han obtenido 1a mayoria de Ios pirriimetanos postuiados como intermediarios, asi comoanáiogos con estructuras relacionadas, 11egándose a 1a conclusión de que aquéiios que poseen e] tipo de 1a serie III, pueden actuar comointermediarios normaies en 1a sintgsis de urogen III, mientras que los de 1a serie I, sóio puedenincorporarse en e] urogen I, a 1a vez que inhiben la formaciónde urogen III.
Hasta e] presente, habria acuerdo en cuanto a 1a existencia de 105 intermediarios aminopirriimetanos, pero se difiere fundamentalmente en cuanto a] momento en e] cua] ocurre 1a isomerización.
Aigunos grupos sugirieron que 1a isomerización ocurreen 1a etapa inicia] de condensación de 1as dos primeras moïécu1as de PBG(Liambias y Batiie, 1970 a, b, c,; 1971 a, b; Batiiey Rossetti, 1977; Frydman y Frydman, 1975;Frydman y co]., 1976;Rossetti y c01., 1977; Rossetti, 1978), aunque 10s resultadosmás recientes con E. gnacilia también podrian interpretarse considerando que e] poiipirrol de cadena abierta formado normaimente podria ser un tetrapirriimetano (Batiie y Rossetti, 1977;Rossetti y co]., 1977). .
En contraposición, otros autores (Battersby y McDonaid,1976, 1979; Battersby y co]., 1977 a, b, c) proponen que e] prgceso de isomerización tendria iugar después de 1a formación de

37
un bilano iineai no isomerizado. Según este modeio, 1a reacciónprocede a través de una condensación secuencia] cabeza-cola deios cuatro PBG,para dar'un intermediario abierto identificadocomohidroximetilbilano. La isomerasa actuaría sobre é] produciendo 1a inversión en e] ani110 D,seguido de una rápida ciciación (Battersby y co]., 1979, 1980, 1982).
Por otra parte, e] grupo de Scott postuió 1a formaciónde un compuesto anuiar, e] preuroporfirinógeno, que sería e] sustrato natura] de 1a isomerasa (Burton y co]., 1979; Jordan yco]., 1979; Scott y co]., 1980). En ausencia de 1a cosintetasa,e] preuroporfirinógeno se transformaría químicamente en urogenI.
En trabajos más recientes, se volvió a considerar 1aposibiiidad de] hidroximetilbiiano comoe] intermediario másprobabie de 1a reacción y producto de 1a PBG-asa (Anderson yco]., 1986; Evans y co]., 1986 a, b).
Es de destacar que para 1a deaminasa de eritrocitos humanos se han encontrado müitiples formas (Anderson, 1979;Miyagi y co]., 1979; Desnick, 1980). Mediante una cromatografíade intercambio aniónico y una e1ectroforesis en ge] de poiiacri1amida se obtuvieron cinco formas enzimáticas designadas A, B,C,Dy E, las que representarían las modificaciones que sufre 1aespecie nativa cuando se van formando intermediarios en 1a secuencia de condensación de ias cuatro moiécuias de PBG(Andersony Desnick, 1980) o bien podrian ser isoenzimas con distintascargas (Miyagi y c01., 1979).
A su vez, e] grupo de Jordan reaiizó estudios tendientes a determinar e] modo de unión de] PBGa 1a deaminasa, empieando enzima purificada de Rp. ¿pheno¿de¿. Luego de incubacignes con cantidades estequiométricas crecientes de 14C-PBG,detectaron bandas proteicas con movilidades e1ectroforéticas 1iggramente diferentes, que atribuyeron a intermediarios enzima-sustrato con 1, 2 y 3 moiécuias de PBGunidas (Berry y Jordan,1981). Demostraron que e] compiejo formado entre 1a deaminasa ycantidades estequiométricas de PBG,era un intermediario unidocovaientemente, y que posibiemente sóio e] PBGque forma e] ani110 A de] preuroporfirinógeno, es e] que está unido cova]ente—mente a 1a enzima (Jordan y Berry, 198]).
La deaminasa tendría un ünico sitio cataiítico, que seusaría varias veces (Seehra y Jordan, 1980).

Se han reaiizado también estudios sobre 1a estereoquïmica de 1a reacción, demostrando que ias cuatro posiciones metileno de 105 porfirinógenos de las series III y del hidroximetilbiiano son producidas a través de procesos estereoespecificos(Jones y c01., 1984).
II.1.4.3. PBG-asa1 porfirias
En 1970, Strand y co]. demostraron que en pacientescon porfiria aguda intermitente (PAI), 1a deaminasa hepática estaba reducida un 50 % con respecto a Ios vaïores para individuos normaïes.
En esta porfiria, 1a excreción urinaria de ALAy PBGse encuentra marcadamente aumentada, tanto durante 1a fase aguda como en remisión, en tanto que 10s niveies de porfirinas sonnormales. Esto es 10 que iievó a pensar que e] defecto enzimático primario en 1a PAI se Iocaïiza a nive] de 1a PBG-asa, o de1a deaminasa,disminuyendo en consecuencia 1a veïocidad de sintesis de] hemoy por-de-represión tendria 1ugar un incrementosecundario de 1a actividad de ALA-S.
Si este defecto enzimático está determinado genéticamente, es de esperar que se pudiera detectar en todos los tejidos en Ios cuaïes 1a enzima es activa normaimente. En efecto,estudios posteriores, y e] desarroiïo de métodos simpies. permitieron estabiecer que 1a actividad de deaminasa se encuentratambién reducida en eritrocitos, cuitivos de fibrobïastos y otros tejidos (Boncowskyy c01., 1973; Mayer, 1973;Kreimer-Birnbaum,1975; Nordmany Grandchamp,1976;Astrup, 1978;Batiie y co]., 1978).
Actuaïmente, 1a determinación de deaminasa en sangreconstituye un e1emento fundamenta] para e] diagnóstico de unaPAI, no 5610 en 105 casos clínicamente manifiestos, sino también en portadores asintomáticos de esta anormaiidad genética.
11.1.5. Qegarboxiïasa
La uroporfirinógeno decarboxilasa (URO-D),urporfiringgenogeno III carboxiiiasa (E.C. 4.1.1.3.7) o simpiemente decagboxilasa cataiiza 1a conversión de urogen III (I) en coprogenIII (I) por decarboxiïación de ias cuatro cadenas 1atera1es deácido acético de] sustrato, con 1a consiguiente 1iberación de

cuatro molécuias de dióxido de carbono (Lockwood y Rimington,1957).
Esta enzima citoplasmática ha sido estudiada en diversos organismos: Chtoneiia vuiganta (Bogorad, 1955 a,b), Rp.óphenoádea (Hoare y Heath, 1958 a; Chu y Chu, 1970), bazo de ratón (Romeoy Levin, 1971), giánduia de Harder (Tomio y Grinstein,1968), higado de rata (Aragonés y co]., 1972; Smith y Francis,1979, 1981; Kardich y Woods, 1980); higado de cerdo (Kushner yco]., 1975), higado de poïio (Taira y San Martin de Viaie,1984), reticuiocitos de conejo (Granick y Mauzeraii, 1958 a), gritrocitos de ave.(FaÏk y c01., 1956; Neve y c01., 1956; Hoarey Heath, 1958 b; Batiie y Grinstein, 1964; Tomio y c01., 1970;Garcia y c01., 1973) y humanos (Conford, 1964; Rasmussen yKushner, 1979; de Verneui] y co]., 1980; Eider, 1982).
II.].5.1. PropiedadesE1 oxigeno 1a inhibe en forma significativa, hecho que
se atribuye a 1a oxidación de sus sustratos, ios porfirinóge nos (Mauzeraii y Granick, 1958), si bien no se descarta una agción directa sobre 1a enzima, que ha sido ensayada en condiciones anaeróbicas (Bogorad, 1958; Hoare y Heath, 1958 a, b;Mauzerai] y Granick, 1958; Tomio y c01., 1970) y aeróbicas(Batiie y Grinstein, 1964; San Martin de Viaie y Grinstein,1968; Tomio y c01., 1970).
Hasta e] momentose habia establecido que era una enzima termosensibie (Tomio y co]., 1970; Romeoy Levin, 1971) ypresentaba caracteristicas suifhidriiicas. Asi, e] GSH(Mauzeraii y Granick, 1958; Tomio y coi., 1970) y e] B-mercaptgetano] (Hoare y Heath, 1958 b) 1a activan, en tanto que metaiespesados, iodoacetamida, PCMBy ditionitrobenzoato (DTNB)1a inhiben (Lockwood y Rimington, 1957; Hoare y Heath, 1958 b; Tomioy co]., 1970; Romeoy Levin, 1971; Kushner y c01., 1975;Mukerji y Pimstone, 1986). La enzima de eritrocitos humanos yde aves se inhibe por cationes univaientes (Conford, 1964). Sinembargo, recientemente, si bien seha confirmado 1a naturaiezaSuifhidriiica de 1a URO-D(Straka y Kushner, 1983), se ha desmostrado que 1a actividad de 1a enzima de Rp. pazuatnáó no se aitgra por e] tratamiento con GSH,mercaptoetano] ni calor.
Posee una aita especificidad para los grupos acético,pero no para e] tipo isomérico ya que puede decarboxiiar ios

40
cuatro isómeros de urogen, si bien con diferente veiocidad:III > IV > II > I (Granick y Mauzerail, 1958;Conford, 1964;Smith y Francis, 1979; Koopmann y c01., 1986). Además e] Kmpara e] isómero III es mayor que para e] I (de Verneui] y co].,1980).
Los estudios cinéticos demostraron una inhibición recíproca en 1a decarboxiiación de porfirinógenos de 1a serie IIIpor los de 1a serie I con e] mismo número de carboxiios. En cambio, 1a inhibición fue no recíproca para 1a decarboxiiación de]urogen III por firiaporfirinógeno (firiagen) III y pentaporfirinógeno (pentagen) III. Se sugirió entonces que 10s porfirinógenos de ambas series isoméricas con e] mismo número de carboxiIos son decarboxiiados en e] mismo centro activo, en tanto que1a decarboxiiación‘secuenciai de urogen a coprogen ocurre encuatro sitios diferentes.
Una única proteina es 1a responsabie de estas reaccignes (Batiie, 1973) y estaria constituida por un monómerode40kDa (de Verneuii y colg, 1983; Eider y co]., 1983; Koopmannyco]., 1986) o un dimero de 80 kDa(Kawanishi y c01., 1980) con2 sitios cataïiticos por moiécuia (Elder, 1982; de Verneui] yco]., 1983).
Eider y Urquhart (1984), comparando 1as enzimas de eritrocitos e higado, descartaron 1a existencia de isoenzimas enhumanos, atribuyendo sus diferencias a modificaciones post-traduccionaies debidas a proteóiisis u oxidaciones de grupos tioles.
Se Iogró sintetizar URO-D-enun sistema libre decéiu1as de un 1isado de reticuiocitos, bajo 1a dirección de mRNAsaisiados de higado feta] y reticuiocitos humanos (Grandchampy c01., 1984). Romeo y co]. (1984) cionaron un cDNAde mRNAdeURO-Dde rata de aproximadamente 1.500 nucieótidos. También seha establecido 1a secuencia de un cDNAobtenido de mRNAdeURO-Dde puimón humano (Romeo y c01., 1986) y se demostró queesta enzima está codificada por un 5010 gen.
II.1.5.2. Rutas de decarboxilación y mecanismo de acción
Los hechos experimentaïes indican que 1a decarboXiiación de uroporfirinógenos es un proceso en etapas. Así, en materiales biológicos de pacientes con porfiria cutánea tarda(PCT) (Grinstein ycoi., 1945; Canivet y Rimington, 1953) como

4l
en decarboxilaciones llevadas a cabo ¿n vttno (Falk y col., 1956;Mauzerall y Granick, 1958; Hoare y Heath, 1959; Batlle yGrinstein, 1962), se detectó la presencia de intermediarios con7, 6 y 5 carboxilos. La porfirina heptacarboxilica se denominó firia porfirina (firia) y se demostró que es un intermediario normal en la sintesis de coproporfirinógeno (coprogen)(Batlle y Grinstein, 1962). Las mismas conclusiones se verificaron para los porfirinógenos hexa y penta carboxilicos (SanMartin de Viale y Grinstein, 1968). Las concentraciones de estosintermediarios son normalmente muy bajas,y según Straka y Kushner(1983) dependen del cociente (urogen III)/(enzima).
Garcia y col. (1973) encontraron que la decarboxilación procede en dos etapas: una primera, rápida, en la que ocurre la primer decarboxilación, y la segunda.más lenta,en la queel heptaporfirinógeno se convierte paso a paso en coprogen III.
Se ha conseguido establecer cuáles son las rutas de decarboxilación de los uroporfirinógenos III y I, pero no se hadilucidado cuál es el mecanismo de acción de la enzima. Las sucesivas decarboxilaciones de las cuatro cadenas acetato a loscuatro metilos,pueden ocurrir por 24 secuencias posibles a través de l4 intermediarios. Trabajando en forma conjunta, gruposde investigadores de Inglatera y BuenosAires, sintetizaron estos intermediarios, y determinaron que la reacción tiene lugaren forma ordenada, de manera tal que se inicia en el resto acetato del anillo D, procediendo luego sobre los grupos acetatode los anillos A, B y C hasta formar el coprogen III. Es decir,todo sucederia comosi el urogen III girara en la superficiede la enzima, en el sentido de las agujas del reloj (Jackson ycol., 1976).
En contraste con esta secuenciación ordenada, la deca:boxilación del isómero I tendria lugar por un camino no especifico, pudiendo ocurrir por cualquiera de las dos rutas posiblesexistentes, en forma indistinta (Jackson y col., l977).
Smith y Francis (1979) sugirieron que el reconocimiento y remoción de la cadena acética del anillo D podria ocurriren un sitio distinto de las demás decarboxilaciones.
II.l.5.3. Decarboxilasa y PCT
Comoya se mencionó, entre las observaciones

42
experimentales que sugerian que la decarboxilación enzimáticaes un proceso en etapas, se encontraba el hecho de que las orinasde pacientes con PCTse caracterizaban por la presencia de glevadas cantidades de porfirinas con 8, 7, 6, 5 y 4 carboxilos,y particularmente, uroporfirina (uro) y firia (Dowdley col.,l970;Doss y col., l97l). Se postuló que este cuadro anormal sedebia aun bloqueo a nivel de la decarboxilasa (Kushenr y Barbuto,1975; Kushner y col., 1976; Elder y col., 1978; Felsher y col.,l982; Afonso y col., l984).
Por otro lado, la intoxicación crónica con policloradoscomo por ejemplo el hexaclorobenceno (HCB),provoca en humanos yanimales un tipo de porfiria con sintomatologias clinicas y bigquímicas similares a las observadas en la PCT (San Martin deViale y col., l970).
Tanto en la PCT como en los modelos experimentales, sepostula entonces que el cuadro bioquímico anormal se debe a unadeficiencia enzimática primaria a nivel de la decarboxilasa,que lleva a su vez a un aumento secundario por de-represión dela actividad del ALA-S,produciendo en conjunto el patrón caragteristicode esta porfiria (Elder y col., 1975; San Martin deViale y col., l975).
II.l.6. CoprogenasaLa coproporfirinogenasa , coproporfirinógeno decarbo
xilasa (E.C. 1.3.3.3), coproporfirinógeno oxidasa, o simplemente coprogenasa (CPG-asa) es la enzima que cataliza la conversión de coprogen IIIa protoporfirinógeno IX (protogen IX) mediante la decarboxilación oxidativa de los dos grupos propionilo ubicados en los anillos A y B del núcleo tetrapirrólico, ados residuos vinilo.
II.l.6.l. PropiedadesEs altamente especifica para el coprogen III, no pudieg
do utilizar los isómeros I y II comosustratos, pero si el isómero IV obtenido por sintesis,dando comoproducto final protoporfirina (proto) XIII (Al-Hazimi y col., 1976).
Se localiza en la mitocondria celular (Sano y Granick,196l); Elder y Evans (1978) encontraron que en higado de rataesta enzima está situada en el espacio intermembrana de la

43
mitocondria. Presenta un requerimiento absoiuto por e] oxigenomoiecuiar (Sano y Granick, 1961), que actuaria como aceptor dehidrógeno (Faik y co]., 1953). Es por eiio que en organismos estrictamente anaeróbicos capaces de sintetizar porfirinas, debeexistir aigün otro mecanismo que iieva a 1a formación de protoIX (Mori y Sano, 1968). Asi, en Rp. ¿phenaádea se detectó actividad de CPG-asa en condiciones aeróbicas y anaeróbicas, empleando en este üitimo caso un sistema compuesto por ATP, magnesio y L-metionina comoaceptor de hidrógenos (Tait, 1972).
Los reactivos su1fhidri1icos no a1teran su actividaden forma significativa y e] estudio de su estructura primariadeterminó que se trata de una proteina rica en residuos de aminoácidos aromáticos (Yoshinaga ySano, 1980).
Respecto de su peso moïecuiar, varios autores coinciden en un vaior de 70 a 80 kDa(Bat11e y c01., 1965; Pouison yPoigiase, 1974; Yoshinaga y Sano, 1980a) ymediante e] empieo deagentes disociantes se ha estabiecido que es monomérica(Yoshinaga y Sano, 1980 a)
Ha sido estudiada en diversas fuentes: Chtonetla vuiganLA (Granick, 1955), Chnomatium D (Mori y Sano, 1968; Tait,1972), Rp. ¿phenotdea (Tait, 1972), Eugeena gnactiáa (Granick yMauzeraii, 1958 b), S. ceneu¿4¿ae (Pouison y Poigiase, 1974),hojas de tabaco (Hsu y Miiier, 1970), hemoiizados de eritrocitos de aves (Dresei y Falk, 1956); higado (Batiïe y co]., 1965;Eider y co]., 1978; Grandchamp y co]., 1978) y bazo de rata(Kardish y Woods, 1980), higado vacuno (Sano, 1958; Sano yGranick, 1961; Porra y Faik, 1964) y iinfocitos humanos(Grandchamp y Nordmann, 1977).
Recientemente, Camadroy co], (i986) purificaron a homogeneidad CPG-asa de S. ceneutatae; 1a enzima es un dimero depeso moiecuiar 70kDacon subunidades idénticas; presenta requerimientos de grupos tioies y es activada por fosfoiipidos, a1igua] que 1a enzima de higado bovino (Yoshinaga y Sano, 1980 b).
II.1.6.2. Mecanismode acción
Comoen e] caso de 1a enzima precedente,1a reaccióncatalizada por ia CPG-asa tiene 1ugar por etapas. Esta proposición contó con e] apoyo experimenta] de que en fuentes natura]es se haiió una porfirina tricarboxiiica a 1a que se denominó

44
harderoporfirina por haberse detectado en glándulas de Harderde ratas (Tomioy Grinstein, 1968). El análisis de su estructura que fue confirmada por sintesis quimica (Kennedy y col.,l970), demostró que es un 2-vinilporfirinógeno,intermediario derivado de la coproporfirina (copro) en el que el resto propiónico de la posición 2 del anillo A está Sustituido por un vinilo.Incubando hardero o iso-harderoporfirinógenos (que posee un vinilo en la posición 4) con extractos de E. gnacáiió (Cavaleiroy col., 1973, l974) o hemolizados de eritrocitos de aves (Gamesy col., l976), se observó incorporación de ambos compuestos enproto, con una mayor eficiencia para el primero de ellos. Estosresultados confirmaron que la decarboxilación oxidativa de losresiduos propionato ocurre por etapas, comenzandopor el sustituyente del anillo A y siguiendo luego por el del anillo B. Esdecir, en forma análoga a la decarboxilasa, la coprogenasa actuaria en el sentido de las agujas del reloj.
En lo referente a cómo se lleva a cabo esta decarboxilación oxidativa, Rimington y Tooth en 1961 propusieron que unposible intermediario seria una acrilicoporfirina. Se encontrósin embargo, que el trans 2,4-acrilico deuteroporfirina no actuaba comosustrato (Sano y Granick,.l9ól), quedando la posibilidad de que el isómero cis si lo fuera (Batlle y col., l965).Batlle yRimington (1966) propusieron entonces que en una primera etapa el resto propiónico se oxidaba aa-hidroxipropionatmsesintetizó una mezcla de isómeros de este compuesto y empleandomitocondrias de hígado bovino como fuente enzimática, Sano(l966) demostró su incorporación en protoporfirina.
A su vez, French y col. (1970) aislaron de materialesbiológicos un tetrapirrol de estructura tricarboxilica con unsustituyente cis acrilato en la posición 2 del anillo A, al quese denominóporfirina S-4ll, lo que confirmaria la hipótesis deBatlle y Rimingt0n(1966). Sin embargo, estudios ¿n thno empleando precursores marcados especificamente, indicaron que la S-4llno seria un intermediario normal (Battersby y col., 1972; Zamany col., l972). Jackson y col. (1976) sugirieron entonces que laporfirina S-4ll provendria de un metabolismo anormal y que en reglidad ela-hidroxipropionil derivado en un proceso simultáneo eliminarïa el agua y se decarboxilaria. A su vez, Zamany Ákhtar(l976) propusieron un mecanismo a través de la formación de unabase de Schiff. De todos modos, las evidencias no son

45
suficientes comopara descartar la ruta anteriormente mencionada.
Estudios cinéticos posteriores realizados con preparaciones enzimáticas de diversas fuentes, aportaron evidencias acerca de la formación de intermediarios hidroxilados (Jackson ycol., 1980). Empleando CPG-asa de higado vacuno purificada3.200 veces, Yoshinaga y Sano (1980 b) confirmaron una vez más,que la conversión de los grupos propionilo a vinilo ocurre enetapas, proponiendo a su vez, la formación de un intermediarioB-hidroxipropionato.
II.l.6.3. CPGsasay porfirias
Esta enzima se ve afectada en las coproporfirias hepática y eritropoyética,en las cuales se excretan niveles significativamente elevados de copro urinaria (Dobriner, l936; Watsony col.. l949).
Varios grupos de investigadores observaron que la actividad de la proteina se hallaba disminuida en un 50 % con respecto a controles normales. tanto en pacientes con coproporfiria hereditaria clínicamente manifiesta, comoen portadores deldefecto genético (Elder y col., 1976; Brodie y col., l977).
De acuerdo a esto, la enfermedad se expresa en el estado heterocigota, aunque se han reportado casos de variantes homocigotas (Grandchamp y col., 1980; Nordmanny col., l983).
En los casos de intoxicación por plomo, además delALA-Dtambién la CPG-asa parece estar inhibida, dado que se hanencontrado elevados niveles de ALAy copro en orina (KreimerBirmbaumy Grinstein, l965; Batlle y col., l98l).
II.l.7. Protogen oxidasaLa inserción del hierro en el paso final de la biosin
tesis de hemorequiere que previamente el protoporfirinógeno(protogen) IX se oxide a proto IX. Esta reacción implica la eliminación de seis átomos de hidrógeno del anillo tetrapirrólico,y puede ocurrir no enzimáticamente en presencia de oxigeno molecular (Sano y Granick, l96l).
La naturaleza enzimática de esta oxidación fue confirmada principalmente en higado de ratas y levaduras (Jackson y

46
col., 1974; Poulson y Polglase, 1975; Poulson, l976) y se vióque usa oxigeno como ünico aceptor de hidrógenos, no pudiendodemostrarse requerimiento por ningún cofactor. Sin embargo,Jacobs y Jacobs.(l976) midieron actividad de esta enzima en E.coii en condiciones anaeróbicas,empleando fumarato comoaceptoralternativo de electrones.La oxidación anaeróbica está acoplada a la reducción del fumarato a través de intermediarios de lacadena de transporte de electrones que serian especificamentequinonas (Jacobs y Jacobs, l977 a, b, 1978).
II.l.7.l. PropiedadesLa protoporfirinógeno oxidasa o protogen oxidasa es
una enzima mitocondrial altamente especifica para la proto IX.pues tanto el urogen I o III como el coprogen I ó III, no ac tüan comosustratos.
Es termolábil y sulfhidrilica, postulándose que losrestos sulfhidrilos de la proteina cumplirian un rol en la catílisis uniéndose a los grupos vinilo del protogen IX.
El peso molecular para la enzima de S. cenevióiae y dehigado de rata es de 180 y 35kDa respectivamente (Poulson ycol., 1975; Poulson, l976). Camadroy col., (1985) purificaronesta enzima de higado humano; hallaron que tiene un peso molecular de 32kDay es sensible a la_inhibición por algunas metaloporfirinas tales comocobalto y manganeso-protoporfirina, y enmenor grado por cadmio, níquel y hierro-porfirinas.
Deybachy col. (1985) estudiaron la localización submitocondrial de esta enzima determinando que estaria ubicadadentro de la membrana interna de la organela.
A diferencia de las otras enzimas del camino, en levaduras la protogen oxidasa se sintetizaria en mitocondrias(Poulson y Polglase, 1974 b); sin embargo, en higado de ratas,Kolarov y col. (1983) demostraron que no es sintetizada en mitgcondria, dado que no es inducida por triiodotironina, un inductor de la sintesis de proteinas mitocondriales.
II.l.7.2. Mecanismode acción
En l967, Labbe y col. informaron acerca de la presencia de un pigmento en cultivos de S. cenevtóiae cuyo espectrode absorción y reacciones químicas correspondían a una

47
tetrahidroporfirina; ios autores propusieron que provenía de 1areducción de 1a proto IX, o de 1a autooxidación de] protogen IX.
Años después, Pouison y Poigiase (1973) postuiaron quedicho pigmento (denominadoP-503por presentar una banda típicade absorción en e] espectro visible a 503 nm), constituía un intermediario en Ia biosíntesis de hemo. E1 mismo compuesto fueencontrado también en E. c0¿¿ (Oiden y Hempfiung, 1973), y suestructura fue identificada comouna tetrahidroporfirina IX(Kamitakahara y c01., 1973).
La reacción se verificaría en dos etapas: en 1a primera se formaría e] intermediario P-503 y en un segundo paso cata1izado por 1a protogen oxidasa, ocurriría 1a remoción de cuatro hidrógenos. En apoyo a esta teoría, se observó que en cuitivos de S. ceneu¿¿¿ae en ios que las células se haíiaban reprimldas por acción de 1a giucosa, se producía una acumuiación deP-503 y protogen IX, Ïo que sugería una inhibición o una deficiencia a nivei de 1a protogen oxidasa (P0uison y Poigiase,1974 b).
E1 grupo de Jackson, a su vez,demostró.que e] procesode oxidación es estereoespecífico, pues invoïucra 1a pérdida delos átomos de hidrógeno ubicados en sóio uno de ios lados de]plano de] tetrapirro] (Jackson y c01., 1974; Smith y c01., 1976).
Jones y co]. (1984) sugirieron que de ios cuatro átoamos de hidrógeno en posiciones meso que se pierden durante 1atransformación a proto IX, tres son removidos como hidruros ye] restante comoprotón.
II.1.7.3. Protogen oxidasayporfiria variegata
Una de ias características en e] cuadro bioquímico de1a p0rfiria variegata (PV) es 1a excreción de cantidades significativas de proto en heces, y en menor proporción, copro. Severificó también 1a presencia.de un'compiejo porfirina-péptidono extraíble en éter, conocido comofracción X-porfirina(Rimington y coi., 1968). Este complejo se ha detectado tambiénen bilis y piasma.
En 1976, Smith y co]. propusieron que e] defecto.en esta porfiria podría ubicarse en 1a etapa de oxidación a proto IX.Esta hipótesis contó con e] apoyo de ias evidencias experimentaIes de Brenner y Bioomer(1980).yDeybach y co]. (1981), quienes

48
encontraron una deficiencia del 50 %en la actividad de protogenoxidasa de fibroblastos y linfocitos de pacientes con PV.
II.l.8. FerroguelatasaEsta enzima denominada asi porque cataliza la inser
ción del hierro ferroso en la proto IX; se la conoce también c9mohemosintetasa o protohemo ferroliasa (E.C. 4.9.9.l.l).
La quelación de las porfirinas con los metales es unproceso que puede ocurrir en forma no enzimática a una alta velocidad dependiendo de-las condiciones experimentales (Tokunagay Sano, l972; Kassner y Nalchak, 1973). Es por eso que la existencia de esta enzima fue bastante discutida. Sin embargo, enrepetidas oportunidades,se ha demostrado la sintesis de hemocatalizada por extractos celulares en condiciones en las cuales no habria reacción significativa sin la presencia de material celular (Llambias, l976; Porra y Lascelles (1968),La9activ'des de estos extractos presentaban propiedades que indicaban
.1 o. |m
que la actividad catalitica residía en una proteina. Se ha demostrado además, la inhibición ¿n thno de la sintesis de hemopor anticuerpos de ferroquelatasa bovina (Taketani y Tokunaga,l982). Estas evidencias indican el requerimiento de una enzimaen la última etapa de formación del hemo. De todos modos, no sedescarta la posible contribución de la reacción no enzimática.
Se ha detectado actividad de ferroquelatasa en diversos tejidos: eritrocitos de aves (Goldberg y col., 1956;Krueger y col., 1956; Yoneyamay col., l962; Hanson y Dailey,1984); reticulocitos (Clark y Walsh, 1959), higado, riñón y bazode mamíferos (Lockhead y Goldberg, 1961); higado de cerdo (Porray Jones, l963 a), rata (Labbe y Habbard, 1961; Yoneyamay col.,l965; Llambias, l976) y ratón (Cole y col.; l98l); médula óseahumana (Bottomley, l968); levaduras (Porra y Jones, l963 b;Reithmüller y TUppy, l964; Labbe y col., 1968); bacterias(Porra y Lascelles, 1965; Jones y Jones, 1970); vegetales (Jones,1968; Porra y Lascelles, l968; Goldin y Little, l969), y algas(Browny col., l984). También se ha detectado en glóbulos rojosde sangre humana patológica (Grinstein y col., l959; Lockhead yGoldberg, l96l; Vavra y col., l964) y normal (Langelaan y'col.,l969).

49
II.l.8.l. PropiedadesEn la mayoria de los tejidos estudiados, la reacción
enzimática tiene lugar en anaerobiosis en presencia de agentesreductores tales comoácido ascórbico, GSHo cisteina (Nishiday Labbe, 1959; Labbe y Hubbard, 1961; Porra y Jones 1963 a;Llambias 1976); resultados que sugieren la presencia de grupostioles esenciales (Labbe y Hubbard, 1960).
La naturaleza sulfhidrilica de la enzima fue confirmada por Porra y col. (l967) y Taketaniy Tokunaga (1981) utilizando reactivos de grupos sulfhidrilos tales comoPCMB,arsenito,iodoacetamida.
Es altamenteespecifica para el hierro (Labbe y Hubbard,196l),lo que es de gran importancia, ya que ¿n v¿vo, la enzimadebe seleccionar a este metal de entre una gran variedad decationes divalentes. Sin embargo, además del hierro, puedenser incorporados enzimáticamente a la proto IX otros metales divalentes como el zinc (Camadro y col., l984) y el cobalto(Jones y Jones, 1969; Sinclair y col., l979) ; pero no se ha aclarado aún si la inserción de estos metales es catalizada poruna proteina enzimática o por varias. En higado de cerdo, hay gvidencias de que una sola enzima es la responsable de la inserción de hierro y cobalto en la mesoporfirina (Cánepa, l987).
Respecto de las porfirinas, los grupos vinilo ubicadosen las posiciones 2 y_4, no parecen ser esenciales (Porra yJones 1963 b; Taketani y Tokunaga, l982); en cambio si parecenserlo los grupos carboxilo libres del ácido propiónico (Heinkely col., 1958; Honeyborne y col., 1979). De todos modos, en lacélulasólo se encuentra presente la proto IX, y por lo tanto,no se forman otros hemoderivados (Johnson y Jones, l964; Jonesy Jones, l969). Los porfirinógenos y las porfirinas parcialmegte reducidas, tampoco actúan comosustratos (Porra y Jones,l963 a; Labbe y col., 1968).
Tanto el sustrato porfirina comoel producto hemo, lainhiben (Tephly y col., 1973) y ciertos cationes divalentes comomanganesoy plomola inactivan en forma significativa, al igual que el oxigeno (Porra y Jones, 1963 a; Hazanowska y col.,l966).
Si bien Labbe (Labbe, l976; Labbe y Nielsen, 1976) prgpuso un requerimiento por fosfato de piridoxal que estaria

unido fuertemente a 1a proteina, 1a participación de cofactoresde este tipo en 1a reacción no ha sido aún confirmada (Taketaniy Tokunaga, 1981; Daiiey, 1982).
Maiier y co]. (1980) aisiaron 1a enzima de higado derata, obteniendo una soia banda en SDS-PAGE,y un peso moiecuéiar aperente de63 kDa Taketani y Tokunaga purificaron ferroqueïatasa de higado de rata (1981) y de bovino (1982) utiiizandouna cromatografía en Biue Sepharosa. Obtuvieron proteinas de pgso moiecuiar 240 y 42 kDapara 1a enzima de higado de rata ensus formas nativa y desnaturaiizada respectivamente, y simiiarmente, valores de 200 y 40 kDa paraia de bovino. A su vez, Hansony Dailey (1934) obtuvieron 1a enzima de eritrocitos de poiiopurificada 200 veces con un peso moiecuiar aparente de 42 kDaEstos resuitados sugieren que 1a ferroqueiatasa seria un oiiggmero de 5 ó 6 subunidades.
La ferroqueiatasa se haiia asociada a 1a membrana interna de 1a mitocondria (Mc Kay y coi., 1969),más especificamente en 1a cara interior (Jones y-Jones, 1969), con porciones de1a proteina expuestas a ambos 1ados de 1a membrana, y el sitioactivo orientado hacia su matriz mitocondria] (Harbin y Daiiey,1985). Es por Io tanto una iipoproteina en 1a que e] agregadode soiventes orgánicos y fracciones fosfoiipidicas aumenta suactividad (Yoshikawa y Yoneyama, 1964; Mazarowska y co]., 1966;Sawada y c01., 1969; Simpson y Pouison, 1977; Taketani yTokunaga, 1981; Dailey y Fieming, 1983; Hanson y Daiiey, 1984),y se postuia que un fosfoiipido cargado participaria en e] proceso de activación (Takeshita y c01., 1970). Se ha informado que1a presencia de ácidos grasos también incrementa 1a formaciónde hemo no enzimático (Taketani y Tokunaga, 1984).
Se postuió que existen dos sitios cataiïticos: uno para e] meta] y otro para 1a porfirina (Nishida y Labbe, 1959).Daiiey (1984) propuso un modeio en e] cua] e] hierro se une a1sitio activo a través de dos grupos tioies vecinaies, y 1a porfirina interacciona con residuos arginina (Daiiey y Fleming,1986; Dai1ey y c01., 1986). E1 sitio activo seria un boisiiiohidrofóbico que rodea a] macrocicio por todos sus lados, menosuno (Daiiey, 1985).
II.1.8.2. Mecanismode acción
Daiiey y F1eming (1983) postuiaron un modeïo cinético

51
secuencia] BiBi en e] cua] e] hierro se une a] sitio activo de1a enzima antes que 1a porfirina; 1uego se Iibera e] hemo comoprimer producto, precediendo a 1a 1iberación de Ios dos protones.
II.1.8.3. Ferroqueïatasa y porfirias
En 1961, Magnus y co]. propusieron que e] defecto enzimático primario en 1a protoporfiria eritropoyética (PPE) estabasituado a nive] de la ferroque1atasa. A partir de entonces, esta hipótesis fue confirmada por varios autores (Boncowskyy co].1975; Bottomley y coï., 1975; De Goeij y co]., 1975; Becker yco]., 1977; Bïoomer y c01., 1977; Brodie y c01., 1978; Bioomer,1980; Ste11a y co]., 1981).
En dicha patoïogia, 1a actividad de ferroqueïatasa medida en forma indirecta en cu1tivo de cé1u1as (Bïoomer y c01.,1977; Sassa y c01., 1979) y en forma directa en gïóbuïos rojos(Steïïa y c01., 198]), resuïtó ser un 40-50 % de] nive] norma],resultados concordantes con un estado heterocigota. Sin embargo, mediciones directas en cuïtivos de células mostraron unadisminución de] 90 % respecto de los norma1es (Boncowsky y co].,1975; Bloomer y c01., 1977).
Becker y co]. (1977) y Viljoen y co]. (1979) observaron que 1a actividad de esta enzima se halia disminuida tambiénen pacientes con PV, aunque otros autores (Pimstone y co].,1973; Brodie y c01., 1977; Brenner y Bioomer, 1980; Deybach yc01., 1981) encontraron que los niveies de 1a misma se manteníandentro de 105 valores normaïes.
Por otra parte, 1a acumuïación de proto en gióbuïos r9jos en 105 casos de intoxicación por pïomo, se deberia segúnGoïdberg (1968) a una inhibición de 1a hemo sintetasa. En ratasintoxicadas por este meta], Moorey co]. (1975) encontraron que1a actividad de esta enzima en higado y müscuio cardiaco estaba disminuida.
11.2. CATABOLISMO DEL HEMO
Comoocurre con muchos compuestos bioïógicos, 1a hemoglobina tiene un periodo de vida limitado. Cuando se degrada ene] interior de] organismo, 1a porción proteica, es decir 1a

52
giobina, es reutilizada como tai o bien a través de los aminoácidos que 1a constituyen, en tanto que e] hierro pasa a formarparte de 1a reserva de este meta] para su uiterior utiiización.E1 hemo en cambio, es degradado en e] sistema reticuioendoteiiaide ias céiuias de] higado, bazo y méduia ósea.
En mamíferos, e] hemo es ciivado en una reacción oxidativa que requiere oxigeno molecuiar y NADPH,convirtiéndose enbiiiverdina IX a, que a su vez se reduce a biiirrubina IX a por1a acción combinada de tres enzimas: NADPHcitocromo c reductasa (E.C. 1.6.2.4), hemooxigenasa (hemozNADPHzoxigenoóxido reductasa, E.C. 1.14.99.3) y bi1iverdina reductasa (biiirrubinaNADP+óxido reductasa (Tenhunen y c01., 1969). Las dos primerasse iocalizan en 1a membranade] reticuio endopiásmico constituyendo e] sistema hemooxigenasa microsoma] (Hino y c01., 1979),en tanto que 1a tercera es citosóiica (Tenhunen y c01., 1970).A pesar de esta diferente compartimentaiización, ias tres actúan en forma conjunta, y 1a interacción entre eiias ha sido'estudiada en un sistema reconstituido con preparaciones purificadas a partir de higado y bazo bovinos (Yoshinaga y co]., 1982a, b).
La secuencia de reacciones seria 1a siguiente (F'gura11.2.): 1a hemooxigenasa une e] hemo en una orientación especifica y e] compiejo es reducido por 1a NADPHcitocromo c reductgsa. E1 compiejo hemo-hemooxigenasa funciona además activando e]oxigeno moiecuiar, generando un radica] oxigeno reactivo que asu vez ataca e] aniiio tetrapirróiico en e] carbono metiiénicoentre 10s puentes A y B (Ca), para formar un a-hidroxihemo(Yoshida y co]., 1974; Yoshida y Kikuchi, 1978). Este compuestoreacciona con e] oxigeno produciéndose 1a ruptura de] cicio y1a eliminación de] carbono metiiénico a como monóxido de carbono,dando e] compiejo biiiverdina IX a-hierro. Es probable quee] aniiio de porfirina pueda abrirse y que aún contenga hierro,antes que se separe 1a protoporfirina de 1a giobina (Guzeiian ySwisher, 1979). E1 compiejo ütimo es hidroiizado y reducido poracción de 1a biiiverdinareductasa, que requiere NADPHcomo donorde hidrógenos y se iibera entonces e] átomo de hierro. En eiproceso se consumen 3 moies de oxigeno por mo] de biiirrubinaformada.
Aunque ia mayor actividad enzimática se obtiene concompuestos de protohemina IX, heminas de 1a misma serie asi como

53
otros isómeros (coprohemina I por ejemplo), tienen una ciertaactividad comosustrato. Las porfirinas 1ibres, en cambio, nofunciónan comosustrato; de éstas y otras experiencias reaïizadas con varias metaioporfirinas, se dedujo que e] átomo centra]de hierro conjuntamente con 1a porción proteica eran indispensables para 1a actividad cataiitica (Tenhunen, 1976).
v XX " v H
“kg? “ho? 0 H
H v U M v7’r1 M H r1o, \ v o M
P P P P VCX-H'd 'hemo MProtohemo HEMOOXIGENASA 'm'
MICROSOMAL
P P
forma'oxo‘
Bi|¡verdina lx cx Complejo Fe-Biliverdina
MDP" BILIVERDINA+ REDUCTASA
NADP v M
Bilirubina IX 0€
FIGU A 11.2.: Catab01ismo de] hemo

54
La actividad de la hemooxigenasa es mayor en tejidosvinculados con la destrucción de los glóbulos rojos: bazo, higado, médula ósea y macrófagos (Pimstone y col.,l97l a), que enórganos no involucrados con ese proceso como riñón y pulmones(Pimstone y col., 1971 b).
Los cambios en la disponibilidad de sustrato pueden afectar la actividad de esta enzima. Asi, en estados hemoliticosla actividad de la hemooxigenasa hepática se incrementa de3 a 5veces; por el contrario, en la anemia por pérdida de sangre, suactividad no varia (Tenhuneny col., l970). La administraciónde hemo o hemina produce una estimulación en higado, bazo, macrófagos y otros tejidos (Tenhunen, 1976), probablemente por inducción mediada por sustrato.
En higado se demostró que la hemooxigenasa puede serinducida por diversos compuestos.no*hémicos comoepinefrina einsulina (Bakken y col., 1972), endotoxinas (Bissel y Hammaker,1977), bencenos halogenados (Guzelian y Elshourbagy, l979), cie;tos iones metálicos (Drummondy Kappas, 1981) y agentes inductgres de interferones. La inducción por las tres primeras sustancias estaria mediada por hemo, en tanto que en los casos restantes seria causada por un mecanismo desconocido no relacionadocon el hemo (Kikuchi y Yoshida, 1983). En cambio, la estaño-protoporfirina es un fuerte inhibidor competitivo (Breslow y col.,1986) y podria ser un potencial agente farmacológico para eltratamiento de la hiperbilirrubinemia neonatal,
Experiencias de traducción ¿n vitae indicaron que elsitio principal de sintesis de la hemooxigenasason los polisomas libres, sugiriendo además que la inducción de la enzima estaria mediada por un aumento en la sintesis de mRNA(Shibahara ycol., 1980).
Estos mismos autores clonaron el cDNAde la hemooxigenasa de páncreas de rata y obtuvieron la secuencia nucleotidica.La estructura primaria de la proteina es de 289 aminoácidos.conun segmento hidrofóbico en el extremo carboxilo que podria serimportante para la inserción de la enzima en el reticulo endoplásmico (Shibahara y tol., l985).
Se ha comprobado además, que la NADPHcitocromo c redugtasa por si sola es capaz de degradar al hemo (Masters y Schacter,1976). Sin embargo, la hemo oxigenasa es necesaria en la conversión fisiológica del hemo, para dar pigmentos biliares de la

configuración IX a, ya que e] tipo de producto que se forma está determinado por e] modo en que estas dos enzimas unen e] sustrato (Yoshinaga y c01., 1982 c).
Digamos fina]mente.que en 1a mayoria de los invertebrados se excreta 1a biiiverdina, compuesto inofensivo y fáciimente eiiminabie. En cambio,ios mamiferos ia transforman en biiiverdina que es insoiubie y altamente tóxica,y que debe necesariamente conjugarse con ácido giucurónico para soiubiizarse ypoder asi eiiminarse a través de 1a biiis por e] intestino(O'Carra, 1975).
11.3. REGULACION
La naturaieza esenciaimente dinámica de ios seres vivos determina 1a existencia de mecanismos que aseguran un perfecto equiiibrio entre ias miies de reacciones metabóiicas quetienen lugar en ios organismos.
La sintesis de porfirinas no escapa a esta regia genera], y es notabie e] aito-grado de eficiencia con ei cua] seiieva a cabo,pues en condiciones normaies 1a cantidad de precursores, ALAy PBG, e intermediarios-que se acumuian o excretan,es muy pequeña. E110 es consecuencia de 1a existencia de una seriede mecanismos de contro] estrechamenteiigados.de ta] maneraque si alguno o varios 11egaran a faiiar, se producen serios daños en e] organismo, comoocurre en ias porfirias (Batiie y co].1981).
11.3.1. Mecanismos de contro]
Varios son ios mecanismos propuestos para e] contro] de1a biosintesis de] hemo.
II.3.1.1. Control a través de cambios en 1a actividad enzimática
Uno de los mecanismos de reguiación que posee 1a cé]u—1a es a través de cambios en 1a actividad de ias enzimas iimitantes de 1a veiocidad de un camino metabóiico. En 1a biosintesisde] hemo se ha estabiecido quee] ALA-Ses 1a enzima iimitante(Granick, 1966; Marver y co]., 1966).
Hay a su vez, variosmecanismos que pueden controiar 1a

56
actividad de una enzima. Uno de ellos se realiza a través de laasociación o disociación de las subunidades de una enzima oliggmérica. Unsegundomecanismo que implica cambios más drásticos anivel estructural.es el que ocurre con la activación de los zimógenos, por eliminación enzimática de una cadena polipeptidica.Finalmente, en un gran nümero de procesos enzimáticos, el producto final de una secuencia metabólica ejerce un efecto inhibitorio sobre la primer enzima de ese camino. Este mecanismo seconoce comocontrol por retro-inhibición (feed-back) o por producto final, es de acción rápida, y se basa en efectos alostéricos.
Se ha demostrado que este tipo de control opera en labiosintesis del hemo(Lascelles, l964; Marks, l969) y a niveldel ALA-S.
113.1.2.Control genético a través de cambios en la cantidad deenzima
Otra forma que tiene la célula de regular un camino metabólico es a través del control genético de la sintesis de lasenzimas limitantes. Algunas enzimas se encuentran en cantidadesrelativamente constantes; en cambiootras, sólo se sintetizancuando actúan ciertas sustancias inductoras sobre el materialgenético del nücleo celular que contiene la información necesaria para su sintesis (Amesy Martin, 1964; Kornberg, l965).
En el caso de la biosintesis de porfirinas, existe uncontrol genético de la sintesis de ALA-Sdependiente del DNA,que regula el nivel de la enzima en la célula. Este mecanismopropuesto por Granick (l966).sebasa en la conocida hipótesis derepresión de Jacob y Monod (l96l). En este modelo, el hemo, prgducto final de la secuencia metabólica, se une a una proteina(apo-represor) para formar una molécula de represor. Este actüasobre el gen operador y bloquea la transcripción del gen estrugtural, impidiendo la formación del mRNAdel ALA-S, bloqueandoasi la sintesis de novo de la enzima.
La inhibición por hemoocurriría, según algunos autores, a nivel transcripcional (Whiting, l976; Srivastava y col.,l980; Yamamotoy col., l982), o según otros, a nivel de traducción (Strand y col., 1972; Tomita y col., l974; Yamamotoy col.,l983).
Varios investigadores han desarrollado la hipótesis de

un pool regulatorio de hemointracelular, especificamente invo=lucrado en el control del ALA-S(Granick y col., 1975; Boncowskyy col., l980) que presumiblemente se localiza en el citosol(Badawy, l979).
El hemocontrola asi su propia sintesis, actuando rápidamente por un mecanismo feed-back sobre el ALA-S, y más lentamente por un mecanismo de represión, deteniendo la sintesis prgteica de esta enzima.
Asociado al concepto de que el control de la sintesisde porfirinas depende del nivel de ALA-S, existe la idea de quela cantidad de enzima formada debe variar como respuesta a cie:tos estímulos o necesidades. En efecto, se ha demostrado queel ALA-Ses una enzima inducible por la acción de ciertos compuestos fisiológicos,farmacológicos y quimicos. Este tipo derespuesta se explica en términos generales,considerando que loscompuestos inductores interfieren directa o indirectamente enla combinación del hemo con el apo-represor, o bien alterandoel destino metabólico del hemo.disminuyendo asi su concentración celular y, en consecuencia, haciendo que haya menos co-rgpresor disponible para dicha combinación.
II.3.l.3.Control por especialización tisular
De acuerdo a la información que se dispone de la biosintesis de tetrapirroles en distintos tejidos, parece existiruna correlación simple y directa entre los niveles de actividadde las enzimas de este camino y su capacidad para formar las hgmoproteinas (Sassa y Kappas, 1981).
Por otro lado, se sabe. que los niVeles de-nemoproteinas varian considerablemente en los distintos tejidos, y existen evidencias de que las células de cada tejido son capacesde sintetizar sus propias hemoproteinas de acuerdo además consus necesidades especificas.
Es asi que el higado, en condiciones normales, sintetiza aproximadamente el 15% del hemo que se produce en todo elcuerpo, mientras que la mayor parte del hemo restante lo aportan los eritroblastos de la médula ósea en forma de hemoglobina(Sassa y Kappas, 1981).
Todo esto sugiere que la concentración de las enzimasde este camino determina o regula la cantidad de porfirinas

58
formadas por un dado tejido u órgano.
II.3.lA.Control por localización intracelularSi observamosla localización intracelular de las enzl
mas del camino del hemo, veremos que determinadas etapas tienenlugar dentro de la mitocondria, en tanto que otras suceden en elcitoplasma (Figura II.l.).
Esto significa que los metabolitos intermedios formados se ven forzados a difundir de un compartimento a otro, loque podria constituir en si un mecanismo regulatorio (Sano yGranick, 1961). Asi, la concentración de ALAdentro de la mitocondria juega un papel relevante en la regulación de la sintesis del hemo, ya que determina la cantidad de ALAdisponible enel citoplasma para la sintesis de los tetrapirroles. Se ha post!lado inclusive la existencia de permeasas especificas que controlarian el pasaje de ALAy coprogen a través-de la membranamitocondria], pero esta teoria no cuenta aún con suficiente apgyo experimental (Batlle, 1982).
Se sabe además que el ALA-Sse sintetiza en los polirribosomas microsomales, migrando luego hacia la mitocondria quees su sitio de acción natural (Scholnick y col., l969; Ohashi yKikuchi, 1977). Esta traslocación intracelular de la enzima tendria un rol regulatorio (Kikuchi y Hayashi, l981).
Por otra parte, las tresültimas enzimas de la formaciónde hemo (coprogenasa, prototen oxidasa y ferroquelatasa), aunque con diferentes orientaciones, se encuentran todas en la membrana interna de la mitocondria. Grandchampy col. (l978) subrgyaron la importancia de esta localización submitocondrial parala transferencia de los sustratos a la membranainterna. Se sugiere que las tres enzimas no catalizan sus respectivas reaccignes de una manera totalmente independiente una de otra, sinoque actuarian como un complejo estable en la membrana. Debido aque los sustratos para esas enzimas son relativamente grandes,altamente reactivos, y de caracteristicas no polares, la existencia de tal complejo seria una manera eficiente de procesar talescompuestos, permitiendo que el producto de una enzima sea tomadoinmediatamente comosustrato por la siguiente, sin abandonar enningún momento el entorno de la membrana.
Además,tanto la primer enzima del camino, el ALA-S,

59
comola última, la ferroquelatasa, estarían orientadas hacia lamatriz mitocondrial, sugiriendo que la orientación de la ferroquelatasa también tendría un papel regulatorio (Kappas y col.,1983).
IL3.l.5.Control por otros constituyentes del medio ambienteo celular
La tensión de oxigeno y el estado de óxido-reducciónde la célula afectan las distintas etapas de este camino metabólico: por ejemplo la conversión de coprogen III a protogen IXes una reacción que depende del oxigeno, en tanto que la inserción de hierro en la proto IX está favorecida en condiciones anaeróbicas (Batlle y col., l98l).
En las bacterias fotosintéticas, las condiciones de iluminación desempeñan un papel importante en la regulación de lasíntesis de clorofila Comoocurre con el oxigeno, una elevadaintensidad luminosa reprime la sintesis de ALA-S.Se ha sugerido entonces que en estos organismos,los efectos de la luz y eloxígeno podrian responder a un mecanismo de control común vinculado con la concentración de algún carrier en la cadena detransporte de electrones, y eventualmente con los niveles deATP(Batlle y col., l98l).
El estado de óxido-reducción (Cohen-Bazire y col.,l957), los niveles de NADH(Labbe, l967) y de ATP celular(Gajdos y Gajdos-Türók, l969) son otros factores que participanen el control de este camino.
1L3,1_5 Control por interconexión con otros caminos metabólicos
Los mecanismos de regulación del nemo se encuentrancoordinados por ejemplo, con los de formación de proteinas especificas que constituyen las distintas hemoproteinas, comoen elcaso de la globina.
También existe una estrecha relación con el ciclo delos ácidos tricarboxilicos que regula el nivel de succinil CoA,sustrato necesario para la sintesis de ALA.
En Rp. 4phenoLde4 se ha demostrado que la actividad delALA-Sestá controlada por las actividades relativas de las enzimas cistationasa y rodenasa, ambas vinculadas con el metabolismo de compuestos sulfurados y en la formación y degradación de

60
un trisuifuro que actüa como activador de] ALA-Sen determinadas condiciones (Hider de Xifra y c01., 1976). Un mecanismo similar funciona en callos de soya (Vázquez y co]., 1980) y aünen mamíferos (Vázquez y c01., 1987).
II.4. PORFIRIAS
Las porfirias son un grupo de enfermedades metabólicasproducidas como consecuencia de alteraciones en e] camino metabólico de] hemo. Se caracterizan por 1a formación o acumuiacióny excreción aumentada de porfirinas y/o precursores.
Puedenciasificarse de acuerdo a distintos criterios.En su forma tradiciona], según cuá] fuere e] sitio principa] deexpresión de 1a fa11a metabólica, se dividen en hepáticas y eritropoyéticas. 0 bien, de acuerdo a las principales manifestacignes ciinicas, pueden ser agudas, cutáneas o mixtas. En una propuesta reciente (Batiiey Magnin,1987) se sumó a1 primer critgrio 1a división en agudas y'no agudas, como una forma más satisfactoria de subdividir e identificar a este grupo de enfermedades (Tabia II.1.).
TABLA11.1.: Ciasificación de las porfirias
I. Hepát¿ca¿
Aguda intermitente (PAI) W
Variegata o mixta (PV)Coproporfiria hereditaria (CPH) AGUDAS
Nueva porfiria aguda (NPA) 1
Cutánea tarda: a) Hereditaria (PCT) xb) Adqu1r1da
II. Euuopoyé/a'cws
Congénita eritropoyética- (PCE) ‘Protoporfiria eritropoyética (PPE) N0 AGUDASCoproporfiria eritropoyética (CPE)
III. Hepatoenitaopoyética (PHE) '

61
Unode Ios signos más característicos de las porfirias,especificamente en ias no agudas, es ]a fotosensibilización cutánea resultante de 1a acción de 1a luz sobre ias porfirinasacumuladas en 1a pieT. Este síntoma-no se observa en Ias porfirias ciasificadas comoagudas, 1as cuales.se particularizan porataques agudos con sindrome abdominaï, asociados a una masivaproducción y excreción urinaria de los precursores ALAy PBG.En 1a PV y 1a CPH también pueden acumuiarse porfirinas, y enconsecuencia. puede estar presente ei sindrome cutáneo.
En los üitimos años, e] estudio de ias porfirias ha entrado en una fase genético-enzimológica, asociándose cada clasede porfiria a un defecto enzimático especifico (Figura 11.1.)
La mayoria de las porfirias se heredan con un carácterautosómico dominante, sa1vo 1a PCE y 1a NPA que 10 hacen segúnun modeio recesivo,-y 1a PCTque puede existir en dos formas,hereditaria (dominante)o adquirida.Esta úitima forma está asociada generaïmente a 1a exposición a agentes químicos inductores , en especia] hidrocarburos poiihaiogenados, y también puede estar reiacionada a neopiasias hepáticas.

II.
62
S. REFERENCIAS
Abboud, M.M.; Jordan, P.M. & Akhtar, M. (1974) Chem. Commun.,p. 643.
Afonso, S.; Chinarro, F.S.; Ste11a, A.; Batlie, A.; Lenczner,M.; & Magnin, P. (1984) Rev. Arg. Dermato]. gg, 12.
Akhtar, M. & Jordan, P.M. (1969) Tetrahedron Letters ll, 875.
Ai-Hazimi, H.; Jackson, A.H. & Ryder, D. (1976) J. Chem. Soc.Chem. Comm., 188.
Alien, D.A. & Ottaway, J.H. (1986) FEBS Lett. 194, 171.
Ames, B. & Martin, R. (1964) Ann. Rev. Biochem. 33, 235.
Anderson, P.C.; Battersby, A.R.; Broadbent, H.A.;J.R. & Hart, G.J. (1986) Tetrahedron 5g, 3123.
Fookes, C.
Anderson, P.M. (1979) Tesis Doctora], The City University ofNew York.
Anderson, P.M. & Desnick, R. (1980) J. Bio]. Chem. _á2, 1993.
Aoki, Y. (1978) J. Bio]. Chem. 22;, 2026.
Aoki, Y.; Nada, 0.; Urata, G.; Takaku, F. & Nakao, K. (197])Biochem. Biophys. Res. Commun. gg, 568.
Aoki, Y.; Urata, G.; Takaku, I.Biophys. Res. Commun. Q5, 567.
& Katsunuma, N. (1975) Biochem.
Aragonés, A.; Garcia, R.; San Martin de Viaïe, L.; Tomio, J.M.(1972) An. Asoc. Qca. Arg. gg, 239.
gg,
& Grinstein, M.
Astrup, E.G. (1978) Clin. Sci. M01. Med. 251.
Badawy, A.B. (1979) Biochem. Soc. Trans. 1, 575.
(1958) Nature 181, 1399.Bagdasarian, M.
Bajkowski, A. & Friedmann, H.J. 1982) J. Bio]. Chem. 257,2207.
Bakken, A.F.; Tha1er, M.M. & Schmid, R. (1972) J. Clin. Invest.51, 530.
Bai], D.J. & Nishimura, J.S. (1980) J. Bio]. Chem. 225, 10805.
Eatïïe, A.M. de] C. (1973) Bioquímica Cïïnica Z, 309.
Batlïe, A.M. de] C.mo í, 7.
(1982) Endocrinología Ciinica y Metaboiis

63
Batlïe, A.M. de] C.Acta gg, 197.
& Grinstein, M. (1962) Bíochim. Biophys.
Bat11e, A.M. de] C. & Grínsteín, M. (1964) Biochim. Biophys.Acta gg, 1.
Bat11e, A.M. de] C. & Magnin, P.H. (1987) en Porfirias Humanas y Experimentaïes. Aspectos C1ïnicos y Bioquímicos. ActuaIizaciones Médico-Bioquímicas. Acta Bioquímica Cïínica Latínoamericana, Sup. N° 2.
Bat11e, A.M. de] C.
gación gg, 369.& Rimington, C. (1966) Ciencia e Investi
Bat11e, A.M. de] C. & Rossetti, M.V. (1977) Int. J. Biochem.g, 251.
Bat11e, A.M. de] C.; Benson, A. & Rimington, C. (1965) Biochem.J. gl, 731.
Batlle, A.M. de] C.;_L1ambfas, E.B.C.; Híder de Xifra, E.A. &Tigier, H.A. (1975) Int. J. Biochem. g, 591.
Batïïe, A.M. de] C.; Wider de Xifra, E.A. & SteHad A.M. (1978)Int. J. Bíochem. g, 871.
Bat11e, A.M. de] C.; Hider, E. & Magnín, P.(1981)en PorfírínasyPorfirias. Etiopatogenia, Clínica y Tratamiento (Ed. Eudeba).
Battersby, A.R. & Mc Dona1d, E. (1976) Phi]. Trans. R. Soc.Lond. B. 273, 161.
Battersby, A.R. & MC Donaïd, E. (1979) Acc. Chem. Res. lg, 14.
Battersby, A.R.; Baldas, J.; Coïlins, J.; Grayson, D.; James,K. a Mc Donaïd, E. (1972) J. Chem. Soc. Chem. Comm., 1265.
Battersby, A.R.; McDonald, E.; Hiïïiams,,D.C. &Ninsiger, M.K.w. (1977 a) J. Chem. Soc. Chem. Comm., 113.
Battersby, A.R.; Buckïey, D.G.; Mc Dona1d, E. & Wiïïiams, D.C. (1977 b) J. Chem. Soc. Chem. Comm., 115.
Battersby, A.R.; Johnson, D.w.; Mc Donaïd, E. & Nilïiams, D.C. (1977 c) J. Chem. Soc. Chem. Comm., 117.
Battersby, A.R.; Fookes, C.J.R.; Gustafson-Potter, K.E,;Matcham, G.w.J. a Mc Donaïd, E.
1155.(1979) J. Chem. Soc. Chem.
Commun.,
Battersby, A.R.; Fookes, C.(1980) Nature ¿55, 17.
J.R.; Matcham, G. & Mc Donaïd, E.

64
Battersby, A.R.; Fookes, C.J.R.; Gustafson-Potter. K.E.;Matcham. G.W.J. & Mc Donald. E. (1982) J. Chem. Soc. PerkinTrans l, 2413.
Beale, S.I. (1971) Plant Physiol. gg, 316.
Beale, S.I. & Castelfranco, P.A. (l973) Biochem. Biophys. Res:Commun. gg, l43.
Beale, S.I. & Castelfranco, P.Au (l974) Plant Physiol. g_, 297.
Beale, S.I.; Gough, S.P. & Granick, S. (1975) Proc. Natl.Acad. Sci. USA1;, 27l4.
Beale, S.I.; Foley, T. &Dzelzkalns, V. (l981) Proc. Natl.Acad. Sci. USA, lg, 1666.
Becker, D.; Viljoen, J.;.Katz, K. & Kramer, S. (l977) Br. J.Haemat. ;_, l7l.
Beinert, H.; Green, D.E.; Hele, P.; Hoffman-Ostenhof, 0.;Lynen, F.; Ochoa, S.; PopJack, G. & Ruyssen, R. (l956) Sciencelgí, 614.
Berry, A. & Jordan, P. (l98l) Biochem. Soc. Trans. g, 593.
Bild, G.S.; Janson, C.A. & Boyer, P.D. (l980) J. Biol. Chem.255 , 8109.
Bissel, D.M. & Hammaker, L.E. (l977) Biochem. J. l 6, 30l.
Bloomer, J. (l980) J. Clin. Invest. gg, 321.
Bloomer, J.; Brenner, D. & Mahoney, M. (1977) J. Clin. Invest.gg, 1354.
Bogorad, L. (l955 a) Planta Physiol. gg, XLV.
Bogorad, L. (l955 b) Science lZl, 878.
Bogorad, L. (1958) J. Biol. Chem. 233, 516.
Bogorad, L. (1962) en Methods in Enzymology (Eds. S.P.Collowick y N.0. Kaplan). Academic Press, NewYork, vol. á, p.885.
Bogorad, L. (1963) Ann. N.Y. Acad. Sci. 104, 676.
Boncowsky, H.; Collins, A.; Doherty, J. & Tschudy, D. (1973)Biochim. Biophys. Acta 320, 56l.
Boncowsky, H.; Bloomer, J.; Ebert, P. & Mahoney, M. (l975) J.Clin. Invest. gg, ll39.

65
Boncowsky, H.L.; Healey, J.F.; SincTair, P.R.; Mayer, Y.P. &Erny, R. (T980) Bíochem. J. 188, 289.
Borthwick, I.A.; Srívastava, G.; Hobbs, A.; PiroTa, B.;Brooder, J.; May, B.K. & E11iot, w.H. (1984) Eur. J. Biochem.155, 95.
Borthwíck, I.A.;M.; May, B.K.
Srívastava, G.;& Eïliot, w.H.
Day, A.; Pirola, B.;(T985) Eur. J.
SnowsweTT,
Biochem,lég 48].
Bottomley, S. (1963) BTood, gl, 314.
Bottomley, 5.; Tanaka, M.; Everett, M. (1975) J. Lab. CTin.Med. gg, 126.
Brenner, D. & BToomer, J.(]980) N. Eng]. J. Med. 302, 765.
Bresïow, E.; Chandra, R. & Kappas, A. (T986) J. Bío]. Chem.gol, 3135.
Bridger, w.A. (1971) Biochem. Biophys. Res. Commun.1g, 948.
Bridger, N.A. (1974) en The Enzymes (Ed. P.D. Boyer).AcademicPress, NewYork. vo]. lg, p. 581.
Bridger, w.A. (T981) Can. J. Biochem. gg, 1.
Bridger, N.A. (1984) Curr. Top. CeTT ReguT., 345.
Brodie, M.; Thompson, G.; Moore, M.; Beattie, A. & Goldberg,A. (1977) Quart. J. Med. 5g, 229.
Brodíe, M.; Moore, M.; Thompson, G.; Goldberg, A. & HoTti, G.(1978) CTin. Exp. DermatoT. g, 381.
Brown, S.B.; HoTroyd, J.A.; Vernon, D.I. & Jones, 0.T.G.(T984) Biochem. J. 220, 861.
Browníe, E.R. & Bridger, N.A. (1972) Can. J. Bíochem. gg, 719.
Buck, D.;.Spencer, M.E. & Guest, J.R. (T985) Biochemistry gg,6245.
Buck, D.; Spencer, M.E. & Guest, J.R. (1986) J. Gen. Microbio].13g, 1753.
Burnham, B.F. (1963) Acta Chem. Scand. ll, 5123.
Burnham, B.F. & LasceTTes, J. (1963) Biochem. J. gl, 462.
Burton, G.; Fagerness, P.E.; Hosozawa, 5.; Jordan, P. & Scott,A.I. (1979) J. Chem. Soc. Chem. Commun., 202.
Camadro, J.M.; Ibraham, N.G. & Levere, R.D. (1984) J. Bio].Chem. 259, 5678.

66
Camadro, J.M.; Abraham, N.G. & Levere, R.D. (1985) Arch.Biochem. Biophys. 242, 206.
Camadro,J.M.; Chambon, H.; Joiies, J. & Labbe, P. (1986) Eur.J. Biochem. 159, 579.
Cánepa, E. (1987) Tesis Doctora], UBA.
Canivet, C. & Rimington, C. (1953) Biochem. J. Si, 867.
Care], E.F. á Kahn, J.S, (1964) Arch. Biochem. Biophys. 108, 1.
Cavaïeiro, J.; Kenner, G. á Smith, K. (1973) J. Chem. Soc.Chem. Commun., 183.
Cavaleiro, J.; Kenner, G. & Smith, K. (1974) J. Chem. Soc.Chem. Commun., 1188.
Ciark, R. & Walsh, R.J. (1959) Nature, Lond. lgí, 1730.
Ciark-walker, G.D.; Rittenberg, B. & Lasce11es, J. (1967) J.Bacterio]. gg, 1468.
Cohen-Bazire, G.; Sistrom, W.R. 3 Steiner, R.Y. (1957) J.Ceïï Comp. Physio]. gg, 25.
Cohn, M. (1951) en Phosphorus Metaboiism (Eds. H.D. MCElroyy B. Gïass). John Hopkins Press, vo]. L, p. 374.
Coïe, 5.; Massey, T.; Marks, G. & Racz, w. (1981) Can. J.Physio]. Pharmaco]. gg, 1155.
Coiïier, G.E. a Nishimura, J.S. (1978) J. Bio]. Chem.22;,4938.
Conford, P. (1964) Biochem. J. gl, 64.
Cookson, G.H. & Rimington, C. (1953) Nature ll, 875.
Cha, s. a Parks, R.E. Jr. (1964) J. Bio]. Chem. ggg, 1968.
Cha, 5.; Cha, C.J.M. & Parks, R.E. Jr. (1967 a) J. Bio]. Chem.242, 2577.
Cha, 5.; Cha, C.J.M. á Parks, R.E. Jr. (1967 b) J. Bio]. Chem.gig, 2582.
Chu, T. & Chu, E. (1970) Biochim. Biophys. Acta —I5, 377.
Dailey, .A. (1982) J. Bio]. Chem. 257, 14714.
.A. (1984) J. Bio]. Chem. 259, 2711.
H
Dailey, H
Daiïey, H.A. (1985) Biochemistry 24, 1287.
HDaiïey, .A. ü Fleming, J.E. (1983) J. Bio]. Chem. 258, 11453.

67
Dailey, H.A. & Fleming, J.E. (l986) J. Biol. Chem. 26l, 7902.
Dailey, H.A.; Fleming, J.E. & Harbin, B.M. (l986) J. Bacteriol.1:15"de Goeij, A.;J. Clin.
Christianse, K.5, 397.
& van Steveninck, I. (1975) Eur.Invest.
de Verneuíl, H.; Grandchamp, B. & Nordmann, Y. (l980) Biochim.Biophys. Acta 6ll, l74.
de Verneuil, H.; Sassa, S. & Kappas, M. (1983) J. Biol. Chem.EEE, 2454.
Deybach, J.C.; de Verneuil, H. & Nordmann, Y. (l98l) Hum.Genet. É_, 425.
Deybach, J.C.; Da Silva, V.; Grandchamp, B. & Nordmann, Y.(l985) Eur. J. Biochem. 149, 43l.
Dobríner, K. (l936) Proc. Soc. Exp. Biol. Med. gg, 175.
Doss, M.; Meínhof, N.; Look, D.; Henning, H.; Nawrocki, P.;Dolle, N.; Strohmeyer, G. & Fillippiní, I. South Afr. J. Lab.Clin. Med. 11, 50.
Dowdle, E.; Goldwain, P.; Spong, N. & Eales, L. (l970) Clin.Sci. gg, l47.
Dresel, E.l.B. & Falk, J.E. (1953) Nature l72, ll85.
Dresel, E. & Falk, J. (1956) Biochem. J. 63, 80.
Drummond, G.S. & Kappas, A. (l981) J. Exp. Med. (l98l) 153,245.
Elder, G.H. (1982) Abstracts- VIII th. Tetrapyrrole MeetingSouthampton.
Elder, G.H. & Evans, 0. (l978) Biochem. J. l72, 345.
Elder, G.H. & Urquhart, A.J. (1984) Biochem. Soc. Trans. 1g,663.
Elder, G.H.; Evans, J.0. &Matlin, S. (1975) lst. Int.Porphyrin Meeting, Freiburg l. Br. p. 424.
Elder, G.H.; Evans, J.; Thomas, N.; Cox, R.; Brodie, M.; Moore,M. & Goldberg, A; (1976) Lancet LL, l217.
Elder, 6.; Evans, J.; Jackson, J. & Jackson, A. (1978) Blochem.J. 169, 215.
Elder, G.H.; Tovey, J.A. & Sheppard, D.M. (1983) Biochem. J.215, 4l.

68
Evans. J.N.S.; Davies, R.C.; Boyd, A.S.F.; Ichinose, I.; MacKenzie, N.; Scott, A.I. & Baxter, R.L. (1986 a) Biochemistry22, 896.
Evans, J.N.S.; Burton, G.; Fagerness, P.E. & Mac Kenzie, N. &Scott, A.I. (1986 b) Biochemistry gg, 905.
Falk, J.E. á Drese], E.I.B. (1960) Biochim. Biophys. Acta gg,458.
Faïk, J.E.; Drese], E.I.B. & Rimington, C. (1953) Nature 1 2,292.
Faik, J.E.; Drese], E.I.B.; Benson, A. & Knight, B. (1956)Biochem. J. gg, B7.
Fanica-Gaignier,M. & Ciément Metra], J. (1973) Eur. J.Biochem. gg, 19.
Feïsher, B.; Carpio, N.; Engleking, D. & Nunn, A. (1982) N.Eng]. J. Med. 306, 766.
French, J.; Nicholson, D. & Rimington, C. (1970) Biochem. J.120, 393.
Frydman, R. & Feinstein, G. (1974) Biochim. Biophys. Actagig, 358.
Frydman, R. & Frydman, B. (1970) Arch. Biochem. Biophys. 136,193.
Frydman, R. & Frydman, B. (1975) FEBS Letters gg, 317.
; Valasinas, A.;Soc. London Ser. B.
Frydman, B.; Frydman, R.B.G. (1976) Phi].
Levy, S. & Feinstein,Trans. R. 273, 137.
Fumagaiii, S.A.; Kotler, M.L.; Rossetti, M.V. & Batïie, A.M.de] C. (1985) Int. J. Biochem. ll, 485.
Gajdos, A. & Gajdos-Türük, M. (1969) en Porphyrines etporphyries. Biochimie et clinique (Ed. Masson & Co.).
Games, D.; Jackson, A.H. & Jackson, J.R. (1976) J. Chem. Soc.Chem. Commun., 187.
Garcia, R.C.; San Martin de Viaie, L.; Tomio J. & Grinstein,M. (1973) Biochim. Biophys Acta 309, 203.
Gassman, M.; Pluscec, J. & Bogorad, L. (1968) Piant Physio].4_3_.1411.
Gibson, K.D.; Laver, N. & Neuberger, A. (1958) Biochem. J. 19,71.

69
Gibson, K.D.; Matthew, H.; Neuberger, A. & Tait, G.H. (196])Nature 192, 204.
Gibson, J.; Upper, C.D. & Gunsaïus, I.C. (1967) J. Bio]. Chem.gig, 2474.
Goïdberg, A. (1968) Semin. Hematol. á, 424.
Goïdberg, A.; Ashenbrucker, H.;(1956) Blood ll, 821.
Cartwright, G. & Nintrobe, M.
Goïdin, B. & Littie, H. (1969) Biochim. Biophys. Acta 171, 321.
Grandchamps, B. & Nordman, Y. (1977) Biochem. Biophys. Res.Commun. li, 1029.
Grandchamp, B.; & Nordmann, Y. (1978) Biochem. J.m. 97.
Phung, L.
Grandchamp. B.; Deybach, J.; de Verneui], H. & Nordmann, Y.(1980) Biochim. Biophys. Acta Egg, 577.
de Verneui], H.; Dubart, A.;(1984) Biochem. Biophys.
Grandchamp, B.; Romeo, P.;Goossens, M.; Rosa, J. & Nordmann, Y.Res. Commun. 118, 378.
Granick, S. (1955) en CIBA Foundation Symposium on PorphyrinBiosynthesis and Metaboïism (Ed. G. Woïstenhoïme y E.C.P.Miïïer). London, p. 143.
Granick, S. (1966) J. Bio]. Chem. 241, 1359.
Granick, S. & Bogorad, L. (1953) J. Am. Chem. Soc. lá, 3610.
& Kappas, S. (1977) en Metaboiic Reguïation (Ed.H.J. Vogeï) Academic Press, New York, p. 77.Granick, S.
(1958 a) J. Bio]. Chem. 232, 1119.
(1958 b)
Granick, S. & Mauzeraïï, D.
Granick, S. & Mauzeraïï, D. Feder. Proc. ll, 233.
Granick, S. & Sassa, S. (1971) en Metabolic Pathways (Ed. H.G. Voge1). Academic Press, New York á, p. 77.
Granick, S. & Urata, G. (1963) J. Bio]. Chem. 238, 821.
Granick, S.; Sincïair, P.; Sassa, S. &Grieninger, G. (1975)J. Bio]. Chem. 250, 9215.
Grinstein, M.; Schwartz, S. & Watson, C.J. (1945) J. Bio].Chem. 157, 323.
Grinstein, M.; R.M. & Moore, C.V. (1959) Blood 15,476.
Bannerman,

70
Guzeïian, & Eïshourbagy, N.A. (1979) Arch.178.
P.S. Biochem.9Biophys. 1 ,
Guze1ian, P.S. & Swisher, R. (1979) Biochem. J. 184, 481.
Hanson, J. (1984) Biochem. J. 22, 695.& Dayïey, H.
Harbin, B.M. & Daiiey, H. (1985) Biochemistry 24, 366.
Hayashi, N.; Yoda, B. (1969) Arch.Biophys. 31, 83.
& Kikuchi, G. Biochem.
Hayashi, N.; Watanabe, N. & Kikuchi, G. (1983) Biochem.Biophys. Res. Commun. 115, 700.
Heike], T.; Lockwood, w. & Rimington, C. (1958) Nature,London lgg, 313.
Higuchi, M. & Bogorad, L. (1975) Ann. N. Y. Acad. Sci. 244,401.
Hino, Y.; Asagami, H. &-Minakami,S. (1979) Biochem. J. 178,33].
Hoare, D.S. & Heath, H. (1958 a) Nature l_l, 1592.
Hoare, D.S. & Heath, H. (1958 b) Biochem. J. gg, 51.
Hoare, D.S. & Heath, H. (1959) Biochem. J. 12, 679.
Honeyborne, C.L.; Jackson, J.I. & Jones, 0.T.G. (1979) FEBSLett. gg, 207.
Hsu, w. & Mi11er, G. (1970) Biochem. J. 117, 215.
Huïtquist, D.E.; Moyer, R.w. & Boyer, P.D. (1966) Biochemistryg, 322.
Inoue, 1.; Oyama, H. & Tuboi, S. (1979) J. Biochem. (Tokyo)fi, 477.
Irwing, E. & E11iot, N. (1969) J. Bio]. Chem. g_í, 60.
Jackson, A.H.; Games, D.E.; Couch, P.; Jackson, J.R.; Beïcher,R.B. & Smith, S.G. (1974) Enzyme ll, 81.
Jackson, A.H.; Sancovich, H.A.; Ferramola, A.M.; Evans, N.;Games, D.E.; Matïin, S.A.; Elder, G.H. & Smith, S.G. (1976)Phi]. Trans. R. Soc. London 273, 191.
Jackson, A.H.; Nagaraja Rao, K.R.; Supphayen, D.M. & Smith, S.G. (1977) J. Chem. Soc. Chem. Commm., 696.
Jackson, A.H.; Jones, D.M.; Phiïip, G.; Lash, T.D.; Bat11e, A.M. de] C. & Smith, S.G. (1980) Int. J. Biochem. l_, 681.

71
Jacob, F. & Monod, J. (1961) Symp. Quant. Bio]. gg, 193.
Jacobs, N.J. & Jacobs, J.M. (1976) Biochim. Biophys. Acta¿4.9. 1.
Jacobs, N.J. & Jacobs, J.M. (1977 a) Biochim. Biophys. Acta459, 14].
Jacobs, N.J. & Jacobs, J.M. (1977 b) Biochem. Biophys. Res.Commun. Z_, 429.
Jacobs, N.J. & Jacobs, J.M. (1978) Biochim. Biophys. Acta544, 540.
Johnson, A. & Jones, 0. (1964) Biochim. Biophys. Acta gg, 171.
Jones, C.; Jordan, P.M. & Akhtar, M. (1984) J. Chem. Soc.Perkin Trans. I ll, 2625.
Jones, M.S. & Jones, 0.T.G. (1969) Biochem. J. 113, 507.
Jones, M.S. & Jones, 0.T.G. (1970) Biochem. J. 119, 453.
Jones, 0.T.G. (1968) Biochem. J. 107, 113.
Jordan, P.M. & Akhtar, M. (1970) Biochem. J. 116, 277.
Jordan, P.M. & Berry, A. (1981) Biochem. J. 1 5, 177.
Jordan, P.M. & Shemin, D. (1973) J. Bio]. Chem. 248, 1019.
Jordan, P.M.; Burton, G.; Nordiov, H.; Schneider, M.; Pryde,L. & Scott, A.I. (1979) J. Chem. Soc. Chem. Commun., 204.
Pearistone, J.; Pouison, R. & Poigïase, w.Microb. lg, 1239.
Kamitakahara, R.;(1973) Can. J.
Kappas, A.; (1933) en The MetabolicBasis of Inherited DiseaSes (Eds. Stansbury, Nyngaarden,
Sassa, S. & Anderson, K.
Fredrickson, Goïdstein y Brown), Mc Graw-Hi1] Books, NewYork,p. 1301.
Kardich, R.M. & Woods, J.S. (1980) J. App]. Biochem. g, 168.
Kassner, R.J. & Waichak, H. (1973) Biochim. Biophys. Acta 304,294.
Kaufman, S. (1951) en Phosphorus Metaboiism (Eds. w.o. ¡cElroy y B. Glass). John Hopkins Press, Baltimore, USA, vo]. l,p. 370.
Kaufman, S. (1955) J. Bio]. Chem. 216, 153.
Kaufman, S. & A1ivisatos, S.G. (1955) J. Bio]. Chem. 216, 141.

Kawaníshi, 5.; Seki, Y. & Sano, S. (1983) J. Bio]. Chem. 258,4285.
Kennedy, G.; Jackson, A.H.; Kenner, G. & Suckling, G. (1970)FEBSLetters g, 9.
Kikuchí, G. & Hayashi, N. (1981) M011.Ce11 Biochem. 31, 27.
Kikuchi, G. & Yoshída, T. (1983) M011. Cel] Biochem. 53/54,163.
Kikuchi,G.; Shemin, D.Acta gg, 219.
& Bachman, B. (1958 a) Biochim. Biophys.
Kikuchi, G.; Kumar, A.; Talmage, P.Bío]. Chem. gig, 1214.
& Shemin, D. (1958 b) J.
Kipe-Noït, J. & Stevens, S. (1980) P1ant Physio]. gg, 126.
Kïein, 0. & Senger, H. (1978) Pïant Physio]. gg, 10.
Koïarov, J.; Nelson, B.D.Res. Commun. 119, 383.
& Kuzeïa, S. (1983) Biochem. Biophys.
Koopmann, G.E.; Juknat de Geralnik, A.A. & Bat11e, A.M. de]C. (1986) Int. J. Bíochem. lg, 935.
Kornberg, H. (1965) Symp. Soc. Gen. Microbio]. lá, 8.
Kotïer, M.L.; Fumagaïïi, S.A.; Juknat, A.A. & Bat11e, A.M. de]C. (1987) Comp. Biochem. Physio]. 875, 601.
Kowaïski, E.; Dancewicz, A.; Szot, Z.; Lípinski, B. & Rosiek,0. (1959) Acta Biochim. P01. g, 257.
Kreimer-Birnbaum, M. (1975) Anaïes Asoc. Quím. Arg. gg, 287.
Kreimer-Bírnbaum, M.Acta 111, 110.
& Grinsteín, M. (1965) Biochím. Bíophys.
Krueger, R.G.; Meïnick, I. & Kïein, H.R. (1956) Arch. Biochem.Biophys. g_, 302.
Kushner, J. & Barbuto, A. (1975) C1in. Res. gg, 403A.
Kushner, J.; Barbuto, A. & Lee, G. (1976) J. Cïin. Inv. gg,1089.
Kushner, J.P.; Steinmu11er, D.P. & Lee, G.R. (1975) J. Clin.Invest. _Q, 661.
Labbe-Bois, F. (1967) Lancet ii, 1361.
Labbe-Bois, F.Doss).
(1976) en Porphyrins in Human Diseases (Ed. M.Karger, Base].

73
Labbe-Bois, F. & Hubbard, N. (1960) Bíochim. Biophys. Acta¿L1, 185.
Labbe-Bois, F. & Hubbard, N. (1961) Biochím. Bíophys. Acta gg,130.
Labbe-Bois, F. & Nieïsen, L (1976) en Porphyrins in HumanDiseases (Ed. M. Doss). Kager , Base], p. 141.
Labbe-Boís, F. & Voïïand, C. (1977) Arch. Biochem. Biophys.112. 565.
Labbe-Bois, F.; Kurumada, T. & Onísawa, J. (1965) Biochim.Bíophys. Acta g, 403.
Labbe-Bois, F.; Volïand, C. & Chaix, P. (1967) Biochím.Biophys. Acta 143, 70.
Labbe, B.F.; Voïland, C. & Chaix, P. (1968) Biochim. Bíophys.Acta.159, 527.
Langeïaan, D.E.; Losowsky, M.S. & Toothilï, C. (1969) Bíochem.J. 1_ol, 17 P.
Lasce11es, J. (1960) J. Gen. Microbio]. gg, 487.
Lasceïles, J. (1964) en Tetrapyrroïe Biosynthesis and itsReguïation (Ed. N.D. Benjamin Inc.). New York - Amsterdam.
Laver, w.G.; Neuberger, A. & Underfriend, S. (1958) Biochem.J. m, 4.Laver, w.G.; Neuberger, A. & Scott, J. J. (1959) J. Chem. Soc.1483.
Lockhead, A.C. & Goïdberg, A. (1961) Biochem. J. 1g, 146.
Lockwood, N. & Rimington, C. (1957) Biochem. J. Q_, 8.
Lohr, J.B. & Friedmann, H.C. (1976) Biochem. Biophys. ResCommun. gg, 908.
Lynn, R. & Guynn, R.w. (1978) J. Bioï. Chem. ¿53, 2546.
LLambías, E. (1976) Int. J. Biochem. 1, 83.
Llambías, E. & Bat11e, A.M. de] C. (1970 a) Bíochim. Biophys.Acta 220, 552.
Lïambïas, E.180.
& Batïïe, A.M. de] C. (1970 b) FEBS Letters g,
Llambías, E. A.M. de] C.285.
& Batlle, (1970 c) FEBSLetters g,

Llambias, E. & Batlle, A.M. del C. (l97l a) Biochem. J. l l,327.
Llambias, E. & Batlle, A.M. del C. (197l b) Biochim. Biophys.Acta ggz, 180.
Magnus, I.; Jarret, A.; Prankerd, T. & Rimington, C. (1961)Lancet ii, 448.
Maguire, D.J.; Day, A.R.; Borthwick, I.A.; Srivastava, G.;Nigley, P.M.; May, B.K. & Elliot, w. H. (1986) Nucl. Ac. Res.u, 1379.
Mailer, K.; Poulson, R.; Dolphin D. & Hamilton, A. (1980)Biochem. Biophys. Res. Comm.gg, 777.
Marks, G.S. (1969) en Heme and Clorophyl (Ed. D. Van NostrandCo.) London.
Marriot, J.; Neuberger, A. & Tait, G.H. (1969) Biochem. J.1_1_1, 385.
Marver, H.S.; Tschudy, D.P.; Perlroth, D.P. & Rechcigl, M.(1966) J. Biol. Chem. 24l, 4323.
Masters, B.S.S. & Schacter, B.A. (l976) Ann. Clin. Res. g,suppl. l7, l8.
Matthew,M. & Neuberger, A. (l963) Biochem. J._Z, 60l.
Mauzerall, D. & Granick, S. (1958) J. Biol. Chem. 2 2, ll4l.
Mayer, U.A. (l973) Enzyme lg, 334.
Mazanowska, A.M.; Neuberger, A. & Tait, G.H. (l966) Biochem.J. gg, ll7.Mc Ray, R.; Druyan, R.; Getz, G.5. & Rabinowitz, M. (1969)Biochem. J. ll4, 455.
Menon, I.A.& Shemin, D. (l967) Arch. Biochem. Biophys. l l,304..
Metzler, D.E.; Ikawa, M. & Snell, E.E. (l954) J. Am. Chem.Soc. 1g, 648.
Miller, J.w. & Teng, D. (l967) Proc. 7th. Intern. Congr.Biochemistry, Tokyo, 1059.
Miyagui, K. & Watson, C. (l972) Blood gg, l3.
Miyagi, K.; Petryka, M.; Kawakami, J.; Nakada, F.; Petrika,Z. J. & Watson, C.J. (1979) Proc. Natl. Acad. Sci. USA1g,6l72.

75
Moore, M. ; Meredith, P.; Goïdberg, A.; Carr, K.; Toner, P. &Lawrie, T.D.U. (1975) Cïín. Sci. M01. Med. gg, 337.
Mori, M. & Sano, S. (1968) Biochem. Bíophys. Res. Commun. gg,610.
Moyer, R.w.; Ramaïey, R.F.; But1er, L.C. & Boyer, P.D. (1967)J. Bio]. Chem. 242, 4299.
Mukerji, S,K. & Pimstone, N.R. (1986) Arch. Biochem. Bíophys.255, 619.
Nakakukí, M.; Yamauchí, K.; Hayashi, N. & Kikuchi, G. (1980)J. Bio]. Chem. 255, 1738.
Nandi, D. (1978 a) Arch. Biochem. Biophys. 188, 266.
Nandi, D. (1978 b) J. Bío]. Chem. 253, 8872.
Nandi, D.L. & Shemin, D. (1977) J. Bio]. Chem. 252, 2278.
Nandi, D.L. & Naygood, E.R. (1965) Can. J. Biochem. Physio].93,1605.Nemeth M.; Russel], C.S. & Shemin, D.J. (1958) J. Bio]., A.
Chem. 229, 415.
Neuberger, A. & Scott, J.J. (1953) Nature 172, 1093.
Neuberger, A. & Turner, J.M. (1963) Biochím. Biophys. Acta gl,342.
Neuberger, A.; Sandy, J.D. & Tait, G.H. (1973 a) Biochem. J.12.6., 477
Neuberger, A.; Sandy, J.D. & Tait, G.H. (1973 b) Biochem. J.136, 491.
Neuberger, A.; Sandy, J.D. & Tait, G.H. (1973 c) Enzyme lg,79.
Neve, R.; Labbe, R.; Aldrich, R. (1956) J. Am. Chem. Soc. lg,691.
Nishida, G. & Labbe, R. (1959) Biochim. Biophys. Acta gl,511.
Nishimura, J.S. (1986) en Adv. Enzymo]. Re1at. Areas M01. Bio].(Ed. A. Meister). John Níïey & Son, New York, vo]. gg, p. 141.
Nishimura, J.S. & Grinnelï, F. (1972) en Adv. Enzymo]. (Ed,
A.E. Meister). John Wiley & Son, New York, vo] gg, p. 183.
Nishimura, J.S. & Mitcheïï, T. (1984) J. Bio]. Chem. ¿22,9642.

76
Nishimura, J.S. & Mitchell, T. (1985) J. Bio]. Chem.260,
(1973) J. Bio].
2077.
Nishimura, J.S.;Chem. 248, 743.
Mitcheiï, T. & Grinneli, F.
Nordmann, Y. & Grandchamp, B. (1976) Biochem. Med. lá, 119.
Nordmann, Y.; Grandchmap, B.; de Verneui], H.;(1983) J. Clin.
Phung, L.;Cartigny, B. & Fontaine, G. Invest. 1g, 1139.
O'Carra, P. (1975) en Porphyrins and Metaiïoporphyrins (Ed. K.M. Smith) Amsterdam, Eisevier, p. 123.
0'Connor-Mc Court, M.D.Cel] Bio]. g; 57.
& Bridger, w.A. (1985) Can. J. Biochem.
0e1den, &Hempfïing, (1973) J. Bacterioi. ll}, 914.
0nashi,A. & Kikuchi, G. (1972) Arch. Biochem. Biophys. 15;, 34.
0hashi,A. & Kikuchi, G. (1977) Arch. Biochem. Biophys. 115, 607.
0haShi,A. & Kíkuchi, G. (1979) J. Biochem. (Tokyo) gg, 39.
0hashi,A. & Shinohara, H. (1978) Biochem. Biophys. Res. Commun.8_4, 76.
Oyama, H. & Tuboi, S} (1979) J. Biochem. (Tokyo) gg, 483.
Patton, G.M. & Beattie, D.S. (1973) J. Bio]. Chem. gig, 4467.
Pearson, P.H; & Bridger, N.A. (1975 a) J. Bio]. Chem. 250,4451.
Pearson, P.H.& Bridger, W.A. (1975 b) J. Bio]. Chem. 250,8524.
Pimstone, N.R.; Tenhunen, R.; Seitz, P.T.; Marver, H.S. &Schmid, R. (1971 a) J. Exp. Med. 133, 1264.
Pimstone,N.R.; Enge], P.; Tenhunen, R.; Seitz, P.T.; Marver,H.S. & Schmid, R. (197] b) J. Cïin. Invest. EQ, 2042.
Pimstone, N.R.; Biekkenhorst, G. & Eaies, L. (1973) Enzyme1g,354.
Porra, R.J. & Faik, J.G. (1964) Biochem. J. gg, 69.
Porra, R.J. & Grimme, R.H. (1973) Enzyme lg, 115.
Porra, R.J. & Grimme, R.H. (1974) Arch. Biochem.Biophys. lfií, 312.
Porra, R.J. & Irving, E.A. (1970) Biochem. J. l_g, 42P.
Porra, R.J. & Jones, 0.T.G. (1963 a) Biochem. J. 7, 181.

77
Porra, R.J. & Jones, 0.T.G. (1963 b) Biochem. J. gl, 186.
Porra, R.J. & Lasce11es, J. (1965) Bíochem. J. gg, 120.
Porra, R.J. & Lasce11es, J. (1968) Biochem. J. 19g, 343.
Porra, R.J.; Vitoïs, K.S.; Labbe-Bpís, F. & Newton, N.A.(1967) Biochem. J. 195, 221.
Pouïson, R. (1976) J. Bio]. Chem.gél, 3730.
Poulson, R. & Polgïase, w. (1973) Biochim. Biophys. Acta 329,256.
Pouïson, R. & Polgïase, W.J. (1974 a) J. Bio]. Chem. gig, 6367.
Pouïson, R. & PoïgÏase, J.(1974 b) FEBSLetters gg, 258.
Pouïson, R. & Polgïase, J.(1975) J. Bio]. Chem. ¿59, 2169.
Prasad, A.R.S.; Horowitz, P.M. & Nishimura, J. S. (1983)Biochemistry gg, 4272.
Ramaïey, R.F.; Bridger, N.A.; Moyer, R.w. & Boyer, P.D.(1967)J. Bio]. Chem. gig, 4287.
Ramaswamy, N.K. & Nair, P.M. (1973) Biochim. Biophys. Acta¿23, 269._
Rasmussen, G. & Kushner, J. (1979) J. Lab. CIÍn. Med. gg, 54.
Reithmüïler, G. & Tuppy, H (1964) Bíochem. Z. 519,413.
Rímington, C. & Kro], C. (1955) Nature 115, 629.
Rímíngton, C. & Tooth, B. (1961) J. Biochem. Tokyo 52, 456.
Rimington, C.; Lockwood, w. & Be1cher, R. (1968) Cïin. Sci.gg, 211.
Romeo, G. & Levin, E. (1971) Biochim. Biophys. Acta 230, 330.
Romeo, P.; Dubart, A.; Grandchamp,B.; de Verneui], H.; Rosa,J.; Nordmann, Y. & Goossens, M.(1984) Proc. Nat]. Acad. Sci.USAgl, 3346.
Romeo, P.; Raich, N.; Dubart, A.; Beaupain, D.; Pryor, M.;Kushner, J.; Cohen-SoïaI, M. & Goossens, M. (1986) J. Bio].Chem. 261, 9825.
Rossetti, M.V. (1978) Tesis Doctora], UBA.
Rossetti, M.V. & Batïïe, A.M. de] C. (1977) Int. J. Biochem.g, 277.

78
Rossetti, M.V; Juknat de Gerainik, A.A. & Batiie, A.M. de] C.(1977) Int.J. Biochem. g, 781.
Rossetti, M.V.; Juknat de Gerainik, A.A.; Kotier, M.L.;Fumagaiïi, S.A. & Batiïe, A.M. de] C. (1980) Int. J. Biochem.lg, 761.
Rossetti, M.V.; Lombardo, M.E.; Araujo, L S.; Batiie, A.M. de]C. (1986) Comp. Biochem. Physio]. 858, 451.
Russeïi, C.S. & Rockswelï, F. (1980) FEBSLetters 116, 99.
Rytka, J.; Bilinski, T. & Labbe-Bois, F. (1984) Biochem. J.gig, 405.
Sancovich, H.A.; Batiie, A.M. de] C. & Grinstein, M. (1969 a)Biochim. Biophys. Acta 191, 130.
H.A.;3,
Batiie, A.M.223.
Sancovich, de] C. & Grinstein, M. (1969 b)FEBS Letters
._../.Sancovich, H.A.; Ferramoia, A.M.; Batiie, A.M. de] C.; fKiviievich, A. & Grinstein, M. (1976) Acta Physio]. Latinoam¡/2g, 379.
San Martin de Viaïe, L.C.Biophys. Acta 158, 79.
& Grinstein, M. (1968) Biochim.
San Martin de Viaie, L.; Viaïe, A.A.;M. (1970) Ciin. Chim. Acta gg, 13.
Nacht, S. & Grinstein,
San Martín de Viale, L.; Rios de Moiina, M. de] C.; Hainstok,R. & Tomio, M.J. (1975) lst. Int. Porphyrin Meeting, Freiburgi. Br., p. 445.
Sano, S. (1958) Acta Haemato]. Japan gl, 237.
Sano, S. (1966) J. Bio]. Chem. gil, 5276.
Sano, S. & Granick, S. (1961) J. Bio]. Chem. ¿39, 1173.
Sassa, S. & Granick, S. (1970) Proc. Nat]. Acad. Sci. USA91,517.
Sassa, S.& Kappas, S. (198]) en Advances in HumanGenetics(Ed. H. Hirschom) Pienum Press, NewYork, ll, 12].
Sassa, S.; Za1ar, G.; Poh-Fitzpatrick, M. & Kappas, A. (1979)Trans. ASSOC.Am. Physicians gg, 268.
Sawada, H.; Takeshita, M.; Sugita, Y. & Yoneyama, Y. (1969)Biochim. Biophys. Acta 178, 145.

79
Scott, A.; Burton, 6.; Jordan, P.; Matsumoto, H.; Fagerness,P. & Pryde, L. (1980) J. Chem. Soc. Chem. Comm., 384.
Schoinick, P.L.; Hammaker, L.E. & Marver, H.S. (1969) Proc.Nat]. Acad. Sci. USAgg, 65.
Scho]nick,P.L.; Hammaker, L.E. & Marver, H.S. (1970) Fed. Proc.gg, 542.
Schoinick, P.L.; Hammaker, L.E. & Marver, H.S. (1972) J. Bio].Chem. gil, 4132.
Seehra, J.S. & Jordan, P.M. (1980) J. Am. Chem. Soc. 1_;, 6841.
Shemin, D. (1970) Naturwissenschaften ¿1, 185.
Shemin, D. & Rittenberg, D. (1945) J. Bio]. Chem.lág, 567.
Shemin, D & Rittenberg, D. (1946) J. Bio]. Chem. l_g, 621.
Shemin, D. & Rittenberg, D. (1951) J. Bio]. Chem. 122, 315.
Shemin, D. & Russe11,C.S. (1953) J. Am. Chem. Soc. lá, 4873.
Shemin, D.; Russeiï, C.S. & Abramsky, T. (1955) J. Bio]. Chem.613.Shibahara, 5.; Yoshida, T. & Kikuchi, G. (1980) J. Biochem,88,45.
Shibahara, S.; Mulier, R.; Taguchi, H. &voshida, T. (1985)Proc. Nat]. Acad. Sci. USAgg, 7865.
Simpson, D.M. & Beattie, D.S. (1980) J. Bio]. Chem. 25 , 1630.
& Poulson, R.Simpson, D.M. (1977) Biochim. Biophys. Acta 482,461.
Sinc]air,P.; Gibbs, A.H.; Sinclair, J.F. & De Matteis, F.(1979) Biochem. J. 178, 529.
Smith, A. & Francis, J. (1979) Biochem. J. 1 3, 455.
Smith, A. & Francis, J. (1981) Biochem. J. 195, 241.
Smith, S.G.; Jackson, A.H. & Jackson, J.R. 1976) Ann. C1in.Res. g, Supp]. 7, 53.
Srivastava, G.; Brooker, J.D.; May, B.K. & E11iot, w.H. (1980)Biochem. J. 1 8, 781.
Srivastava, G.; Borthwick, I.A.; Brooker, J.D.; May, B.K. &E11i0t, w.H. (1983 a) Biochem. Biophys. Res. Commun. 110, 23.
Srivastava, G.; Borthwick, I.A.; Brooker, J.D,; May, B.K. &Eiiiot, w.H. (1983 b) Biochem. Biophys. Res. Commun. 117, 344.

80
Stella, A.; Nider, E.;(1981) Rev. Arg.
Lenczner, M.;Derm. gg, 7.
Magnin, P. & Batïïe, A.
Straka, J.G. & Kushner, J.P. (1983) Biochemistry gg, 4664.
Strand, L.J.; Feïsher, B.F.; Redeker, A.G. & Marver, H.S.(1970) Proc. Nat]. Acad. Sci. USAgl, 1315.
Strand, L.J.; Manning, J.& Marver, H.S. (1972) J. Bio]. Chem.¿41, 2820.
Taira, M.C. & San Martín de Viaïe, L. (1984) Enzyme gl, 79.
Tait, G.H. (1972) Biochem. J. 128, 1159.
Tait, G.H. (1973) Biochem. J. 1 1, 389.
Takeshita, M.; Sugita, Y.Biophys. Acta 202, 544.
& Yoneyama, Y. (1970) Biochim.
Taketani, S. & Tokunaga, R. (1981) J. Bio]. Chem.256, 12748.
Taketani, S. & Tokunaga, R. (1982) Eur. J. Biochem. 127, 443.
Taketani, S. 8 Tokunaga, R. (1984) Biochim. Biophys. Acta 798,226.
Teherani, J.A. & Nishímura, J.S. (1975) J. Bio]. Chem. 25 ,3883.
Tenhunen, R. (1976) Annals Cïin. Res. g, 2.
Tenhunen, R.; Marver, H.S. & Schmid, R. (1969) J. Bio]. Chem.233. 6388.
Tenhunen, R.; Marver, H.S. & Schmid, R. (1970) J. Lab. Cïin.Med. Zé, 410.
Tephïy , T.; Webb,_c.; Trussler, P.; Kníffer, F. & Marker(1973) Drug Metabolism l, 259.
Tokunaga, R. & Sano, S. (1972) Biochim. Biophys. Acta ggí, 263.
Tomio, J.M. & Grinstein, M. (1968) Eur. J. Biochem. g, 80.
Tomio, J.M.; García, R.C.; San Martín de Víaïe, LLC. &Grinstein, M. (1970) Biochim. Biophys. Acta 198, 353.
Tomita, Y.; Ohashi, A. &Kikuchi, G. (1974) J. Biochem. (Tokyo)15,1007.Tuboi, S. & Hayasaka, S.(1972) Arch. Biochem. Biophys. 150,690.
Tuboi, 5.; Kim, H.J. & Kikuchi, G. (1969) Arch. Biochem.Biophys. O, 92.

8]
Tuboi, 5.; Kim, H.J.Bíophys. 138, 147.
& Kíkuchi, G. (1970 a) Arch. Biochem.
Tuboi, S.; Kim, H.J. Biochem.Biophys. 138, 155.
& Kikuchi, G. (1970 b) Arch.
Tyrre1,D. & Marks, G. (1972) Biochem. Pharmaco]. gl, 2077.
(1981) M01. 183,Urban-Grima], D. Gen. Genet.85.
& Labbe-Bois, F.
Varticovstki, L.; & Burnham, BQF. (1980) Int. J.Biochem. lg, 739;
Kushner, J.P.
Vavra, J.D.; Mayer, V.K. & Moore, C.V. (1964) J. Lab. Cïin.Med. gg, 736.
Vázquez, E.; Níder de Xífra, E. & Batïïe, A.M. de] C. (1980)Int. J. Biochem._12, 721.
Vázquez, E.; Buzaleh, A.M.; Wider, E. & Batlïe, A.M. de] C.(1987) Int. J. Biochem. lg, 217.
Viale, A.A.; Nider de Xífra, E. & Batlle, A.M. de] C. (1980)Int. J. Biochem. 1g, 729.
Viïjoen, J.; Cayanis, E.; Becker, D.M.; Kramer, S.;Dawson, B. & Bernstein, R. (1979) Am. J. Hemat. g,185.
Vogel, H.J. & Bridger, N.A. (1982) J. Bio]. Chem. 57, 4834.
V011and, C. & Felix, F. (1984) Eur. J. Biochem. 142, 551.
Watanabe, N.; Hayashi, N. & Kikuchi, G. (1984) Arch. Biochen.Biophys. 232, 118.
Watson, C.; Schwartz, 5.; Shuïze, N.; Jacobson, L.& Zagaría,R. (1949) Cïin. Invest. gg, 465.
A.D.;Res.
Naxman, Coïlins, A. & Tschudy, D.P. (1966) Biochem.Commun. 24, 675.Biophys.
P.D. (1982) FEBSLetters 143.Weitzman.237.
& Jaskowska-Hodges, H.
Whiting, H.J; (1976) Biochem. J. 158, 391.
Hhiting, M.J. & Elliot, N.H. (1972) J. Bio]. Chem. 2 7, 6818.
Whiting, M.J. & Granick, S. (1976) J.Bio]. Chem. ¿51, 1340.
Nider de Xifra,128.
E.A. & Batïïe, A.M. de] C. (1973) Enzyme lg,

82
Nider de Xifra, E.A. & Bat11e, A.M. de] C. (1974) Int. J.Biochem. á, 129.
Nider de Xifra, E.A. & Batlle, A.M. de] C. (1976) en Porphyrínsin Human Diseases (Ed. M. Doss y M. Lahn).
Nider de Xifra, E.A. &.Bat1]e, A.M. de] C. (1980) Int. J.Biochem. lg, 717.
Nider de Xifra, E.A. & Tigier, H. (1970) FEBSLetters 2, 30.
Nider de Xifra, E.A.; Bat11e, A.M. de] C. & Tigíer,H.(1971)Bíochim. Biophys. Acta 235, 511.
Níder de Xifra, E.A.; Mendiara, S.(1972) FEBSLetters gl, 275.
& Batïle, A.M. de] C.
Wider de Xifra,(1976) Phi].
E.A.; Sandy, J.; Davies, R.Soc. Lond. B 273, 79.
& Neuberger, A.Trans. R.
Wiïïiams, D.C.; Morgan, G.S.; Mc Donaïd, E.R. (1981) Biochem. J. 193, 301.
& Battersby, A.
Noïodko, w.T.; Brownie, E.R.Bacteriol. 143, 231.
& Bridger, N.A. (1980) J.
Woïodko, w.T.; Brownie, E.R.; O'Connor, M,D.(1983) J.Bio]. Chem. 258, 14116.
a Bridger, w.A.
Woods, J. (1974) J. Mo]. Pharmaco]. lg, 389.
Yamamoto, M.; Hayashi, N. & Kikuchi, G. (1982) Biochem.Bíophys. Res. Commun. 105, 985.
Yamamoto, M.; Hayashi, N. & Kíkuchi, G. (1983) Biochem.Biophys. Res. Common. 11 , 225.
Yamamoto, M.; Yew, N.S.; Federspie], M.; Dodgson, J.B.;Hayashi, N. & Euge], J. . (1985) Proc. Nat]. Acad. Sci.,USAgg, 3702.
Yamamoto, M.; Fujíta, H.; Watanabe, H. ; Hayashi, N.& Kikuchi,G. (1986) Arch. Biochem. Biophys. 2 5, 76.
Yamauchí, K.; Hayashi. N. & Kikuchí, G. (1980)J. Bio]. Chem.ggá, 1746.
Ybarra, J.; Prasad, A.R.S. & Nishimura, J.S. (1986) Biochemistry gg, 7174.
Yoneyama, Y.; Ohyama, H¡; Sugita, Y. & Yoshikawa, H.(1962)Biochim. Biophys. Acta gg, 261.

83
Yoneyama, T.; Tamai, A.; Yasuda, T. & Yoshikawa, H. (1965)Biochim. Biophys. Acta 105, 100.
Yoshida, T. & Kíkuchi, G. (1978) J. Bio]. Chem. 253, 4224.
Yoshída, T.; Takahashi, S. & Kikuchi, G. (1974) J. Biochem.(Tokyo) Zé, 1187.
Yoshikawa, H. & Yoneyama, Y. (1964) en Iron Metaboïism (Ed.F. Gross).Springer-Ver1ag, Berïfn, p. 24
Yoshinaga, T. & Sano, S. (1980 a) J. Bío]. Chem. 2 5, 4722.
Yoshinaga, T. & Sano, S. (1980 b) J. Bío]. Chem. 2 5, 4727.
Yoshinaga, T.; Sassa, S. & Kappas, A. (1982 a) J. Bío]. Chem.221, 7778.
Yoshinaga, T.; Sassa, S. & Kappas, A. (1982 b) J. Bio]. Chem.¿51, 7786.
Yoshinaga, T.; Sassa, S. & Kappas, A. (1982 c) J. Bio]. Chem.¿51, 7794.
Yubísui, T. & Yoneyama, Y. (1972) Arch. Biochem. Biophys. 150,77.
Zaman, Z. & Akhtar, M. (1976)_Eur. J. Biochem. gl, 215.
Zaman, Z.; Abboud, M. & Akhtar, M, (1972) J. Chem. Soc. Chem.Commun, 1263.

CAPITULO III
ó-AMINOLEVULICO DEHIDRASA
III.
III.
III.
III.
III.
III.
III.
Introducción
1.1.1. Antecedentes históricos
Caracteristicas generaiesIII.2.1. Locaiización subceiuiarIII.2.2. Especificidad de sustrato
Propiedades moïecuiares
III.3.1. Peso molecular y estructura cuaternaria
III.3.2. Estructura primaria. Composicióndeaminoácidos
Grupossuïfhidriios
ALA-Dy metaies
[11.5.]. Inhibición por reactivos queiantesde metales
III.5.2. Requerimiento de cationes
Mecanismo de acción
III.6.1. Naturaieza de] sitio activoIII.6.2. Acerca de] númerode sitios activos
Otros factores que afectan 1a actividad deALA-D
III.7.1. BuffersIII.7.2. Inhibidores endógenosIII.7.3. Acción de compuestos de] camino de]
hemo
III.7.3.]. ALAIII.7.3.2. PBGIII.7.3.3. Porfirinas y hemo
Página
84
84
86
87
89
90
90
92
94
100
100
102
112
118120
122
122123
124124
124124

III.
III.
III
III
III
III.
III.
111.15.
8.
9.
.10.
.11.
.12.
14.
III.7.4.III.7.5.
III.7.6.
Sustancias inductoras de porfiriasFosfato de piridoxa] y compuestos deaita energiaUrea y derivados
Ontogénesis y actividad enzimática
Aspectos genéticos
Vida media
Camino metabóiico de] ALA
Ro] de] ALA-Den 1a biosintesis de] hemo
ALA-Dy porfirias
Aigunas patoïogias en ias cuales e] ALA-Dseencuentra afectada
IIIinIII.IIIIII
.14.
.14.14.
.14.
.14.WDWNd
Intoxicación por plomoAlcohoïismoTirosinemia hereditariaNueva porfiria agudaOtras
Referencias
Página
126
127
127
128
130
135
135
136
137
137
137
138139
140
141
142.

84
III. 5-AHINOLEVULICO DEHIDRASA
Este capitulo lo dedicaremos en particular a la enzimaque es objeto de nuestros estudios: la ó-aminolevülico dehidrasa (ALA-D).
Tambiénconocida comoporfobilinógeno sintetasa, cataliza una reacción de caracteristicas interesantes, comoes lade condensar en forma asimétrica dos moléculas de sustrato idégticas: el ALA, llevando a la formación del PBG, primer compuesto cíclico y único monopirrol natural del camino biosintéticodel hemo. En este proceso se pierden dos moléculas de agua, yde ahi su nombre.
En 1964 fue denominada por la Nomenclatura de Enzimasde la International Union of Biochemistry, comoó-aminolevulinato hidroliasa (agregando 5-aminolevulinato y ciclando), y clasificada bajo el número de código E.C. 4.2.1.24. El primer númeroindica que pertenece a la cuarta clase principal de enzimas,que comprende a las liasas, enzimas capaces de eliminar pequeños grupos del sustrato por una ruta no hidrolitica, catalizando la formación de un doble enlace. A su vez, dentro de estegrupo se incluye en la segunda subclase de las carbono oxigenoliasas, queen su mayoria son hidroliasas, indicando el prefijo"hidro" el tipo de reacción.
III.l.l. Antecedenteshistóricos
Casi simultáneamente con el hallazgo de que el ALA, prgducto de la condensación de succinil CoAy glicina, es el intermediario clave en la sintesis de porfirinas y fuente de todoslos átomos de carbono de la molécula tetrapirrólica (Shemin yRussel, 1953), fue elucidada la estructura del PBG(Cookson yRimington, l953; Kennard, 1953), aislado previamente por Westall(l952) de la orina de pacientes con PAI. Comola estructura delprecursor pirrol postulado comoresultado de la condensación dedos moléculas de ALA(Shemin y Russel, l953) y la estructura delPBGaislado de materiales biológicos eran las mismas, quedó clgro que este pirrol es un intermediario en la síntesis de porfirinas, lo que fue confirmado por su conversión directa a

85
porfirina, en numerosos estudios ¿n v¿tno (Bogorad y Granick,i953; DreSe] yFaik, 1953; Faik y c01., 1953; Schmid y Shemin,19:5 a).
Se demostró además que e] PBGsintetizado a partir deC-ALAcontenía 1a acitividad de 14€ teórica, caiculada con 1a
formuiación de que dos moiéculas de ALAse condensan en 1a manera que se muestra en 1a Figura III.1. (Schmid y Sehmin, 1955 ),La estequiometria de esta condensación fue establecida tambiénpor Gibson y co]. (1955) y Granick y Mauzeraiï (1958).
14
co H- co H1 COZH 2 10 c02H 7 8 2
2
3 0% -2 H204 + —————+>
5 ‘\oNH2
NH2
FIGURAIII.1.: Formación de PBGa partir de dosmoiécuias de ALA
Fueron Drese] y Faik (1953) y Granick (1954) quienespostuiaron 1a existencia de una enzima que cataliza 1a conversión de ALAa PBG, y varios 1aboratorios en forma independientedescribieron preparaciones enzimáticas crudas obtenidas a partir de higado de buey, eritrocitos de pollo y eritrocitos de pgto (Dresei y Faik, 1953; Granick, 1954; Gibson y co]., 1955;Schmid y Shemin, 1955 a).
A partir de entonces, se haiió que 1a enzima estabaampiiamente distribuida en 1a naturaieza, y ha sido aisiada yestudiada en diversos organismos.
Asi por ejempio, en bacterias taïes como Conynebacten¿umdiphten¿ae y Bactenáum cadavenió (Gibson y coï., 1955), bacteriapropiónica (Haierych, 1963); Rp. sphcno¿deó (Gajdos y GajdosTorok, 1955, 1966; Lasceiles, 1959, 1960; Burnhamy Lasceiies,1963; Nandi y co]., 1968), Rp. capóuiata (Nandi y Shemin, 1973),S. ¿teiaon¿¿ (Ho y Lasceiies, 1971), Mgcobactencumphlei(Yamasaki y Moriyama, 1971); hongos como U. ¿phaenogena (Komai

86
y Neilands, 1969, 1969) y N. cnaóóa (Muthukrishnan y co]., 19691972); levaduras (Barreiro, 1967, 1969) y otros microorganismoscomo E. gnac¿i¿¿ (Hovenkamp-Obbemay co]., 1974) y Cheonetta(Bogorad y Granick, 1953). También en plantas (Care11 y Kahn,1964; Stobart y Thomas, 1968),hojas de haba (Steer y Gibbs,1969), hojas de trigo (Nandi y Naygood, 1967), tabaco (Shetty yHiïler, 1969 a), espinaca (Bogorad, 1955; Schneider, 1970), cotiiedones de rábano (Shibata yOchiai, 1977) y caiios de soya(Tigier y co]., 1968, iylu); y animaies: higado de buey (Gibsony co]., 1955; Iodice y co]., 1958), higado de vaca (Bat11e ycoi., 1967; Gurba y c01., 1972; Wiïson y co]., 1972), higado deratón (Coïemau, 1966; Doyle, 1971), gïánduia de Harder de ratas(Tomioy c01., 1968), méduia.de ratas (Fujita y co]., 1981),higado de païoma (Schuiman, 1955). eritrocitos de ave (Granick,1954; Shemin y co]., 1954, 1955; Schmid y Shemin, 1955 a;Drese] y Faik, 1956 a, b.; Fa1k y co]., 1956; Granick y Mauzeraïl,1958), eritrocitos de rata y conejo (Fujita y c01., 1981), reticulocitos de conejo (Granick y Mauzera1], 1958), eritrocitos h!manos (Caiissano y c01., 1966; Nakao y c01., 1968; C011ier,197]).
III.2. CARACTERISTICAS GENERALES
En 1a Tabla 111.1. se ha incluido una 1ista de preparaciones de ALA-Daisladas de diversos organismos y tejidos, y algunas de sus propiedades.
E1 ALA-Des una enzima citopïasmática soiubïe, es decir, no unida a organeias (Gibson y co]., 1955; Mauzeraïï yGranick, 1958), cuyo único sustrato es e] ALA.
Se acepta que su peso m01ecu1ar es de 280 kDay es octamérica: presenta caracteristicas de enzimasulfhidriiica, requiriendo reductores tióiicos para una máximaactividad; 105 meta]es pesados y otros inhibidores tióïicos 1a inactivan (Gibsony co]., 1955; Batiie y co]., 1967).
E1 pH óptimo varia entre 6,3 para reticuïocitos de conejo (Granick y Mauzerai], 1958) y 9,5 para 1evaduras (Barreiro,¡967). En genera], e] pH óptimo de p1antas- 7,2-7,8 en hojas detrigo (Nandi y Haygood, 1967); 7,4 en hojas de tabaco (Shetty yMilier, 1968)-y microorganismos- aïrededor de 8,0 en

87
Rp. ¿phenoideó (Burnham y Lascelles, 1963; Nandi y col., 1968)y 9-9,2 en M. phteá (Yamasaki y Moriyama, l97l)-es mayor que elde la enzima de fuentes animales: 6,3-6,7 en ratón (Coleman,1966); 6,8 en higado de vaca (Batlle y col., l967; Wilson ycol., l972).
Es una enzima termoestable, presentando una actividadmáxima¿n vttno a temperaturas de 55 °C (Tigier y col., l968,l970) a 65 ’C (Batlle y col., l967).
Los valores de Kmreportados se encuentran en el rangode 7,7 x lO'5 M (Calissano y col., l966) a 1,0 x lO'3 M (Nandiy Waygood, l967).
Aunque las enzimas de diferentes fuentes no han sidopurificadas en un mismogrado, un análisis de los datos publicados parece indicar que la enzima aislada de bacterias fotosintgticas es esencialmente más activa que la de los organismos eucgriotas 97 UE/mg para Rp. capauiata (Nandi y Shemin, 1973) y120 UE/mgen Rp. ¿phenoideó (Nandi y col., 1968). En estos ültimos, aunque en un rango de variación amplio, se ha llegado aobtener una actividad especifica comparable tanto en higado bovino; 24,3 UE/mg (Tsukamoto, l980); 23 UE/mg (Bevan y col.,1980); 38 UE/mg(Seehra, 1980) como eritrocitos de rata y conejo: 26 UE/mg(Fujita y col., 198l) y eritrocitos humanos:13 UE/mgsin adicionar zinc (Anderson y Desnick, l979; Gibbs yJordan, 198l) y 24 UE/mgen presencia de zinc (Anderson yDesnick, 1979; Gibbs y col., 1985 a).
III.2.l. Localización subcelular
si bien el ALA-Des conocida como una enzima citoplasmática, en plantas superiores se ha demostrado actividad deALA-Den proplástidos y etioplastos (Schneider, 1970; Smith,l970; Sluiters-Scholten y col., 1973; Shibata y Ochiai, 1976).
Granick (l961) reportó la transformación de ALAexógeno a compuestos porfirinicos en proplástidos aislados de hojasde tabaco, habas y cebada. También se demostró que los etioplastos son capaces de sintetizar protoclorofilida marcadaa partirde 14C-ALA(Rebeiz y Castelfranco, l97l). Evidencias éstas queindican que el ALA-Dpara la sintesis de clorofila se localizaen los proplástidos y cloroplastos, y encontrándose presenteademás en el citoplasma (Shibata y Ochiai, l976).

88
TABLAIII.1.: Aigunas propiedades de] ALA-Dde diferentesfuentes
ÜÁCÏEMÁS MW LEVW PLMÏÑS
. Chunta . . . . Hojas d- ullos de Hojas de Hojas de Coflledones Hosz de“de‘mw'm ¡“wm uqulmu LW s'cmvu‘“ tr|uo son tabaco“¿hace de ban uReferencia ly 2 3 4 5 5 7 8 9 ¡0 y H ¡2 IJ
LE. dni“ (') ¡20 97 2.255 0.74 5 0.055 0.28 0.56 522 ".7 6.0
PH (ica) 250 250 316 2m 230 324 232
n’ Subull)dá@5 6 6(PH nba} (50 km)"
Recueriniento -SH - Q o o - v - o
¡INóptlm 3.5 3.0 9.5 7.2-7.B 8.44.8 7.4 3.2 3.0 7.04.0
u (a; 5110" ¡.mo" ¡.sno" mo" 3mm" 6.2110" 2.4110" 3.3.10"lnmblción por
(Mama;
Inhibición porEOL: - O 9 O
Muarimenlon x’ L1’, ‘. ng‘nz' 2' , ng’ N92. ng’uics ¡Aun-db) un}. ngp) 22'Fansub" oaseo. mn" o . .
ERIÏMCITOS GLAM "¡GAM
Avey conejo Himnos DEMR Ratón BovinoEufemia N ¡5 ¡6 ¡7 y IB 19 20 21 y ZZ 23 24 25 26
Li. lili“ (') 0.06-0.07 2.25 13.5 2. 13.3 z. 0.006 ¡2 9 3.5 5.6 15(23.3 con ln ) (2‘.0 con In )
PH (k5!) 230 252 285 270 250 250 ¡Lo 260
h" swmluues a a 6 6 N[PP IDA; (39.5) (41,7) (IE)
¡much-lento -SH o o
DHócum 6.0 6.34.7 6.5 5.7 6.3 6.3
un (n: 3.6110" mu" me“ 4.6.10" 1.5.10" 1.5.10"
lnhinlclbn por
lnnihlclñn por ¡DIA (ICHVI)
¡mari-¡onto un hrion (a; ,nulos MM. Ipurific.)
Fomflón basede Scniff
Referencias: (1) Nandi y c01., 1968; (2) Van Heyningen yShemin, 1971; (3) Nandi y Shemin, 1973; (4) Tamai y col.,1979; (5) Ste11a, 1977; (6) Barreiro, 1967; (7) Nandi yWaygood, 1967; (8) Tigier y c01., 1968; (9) Shetty y Mi11er,
69; (10) Schneider, 1970; (11) Liedgens y c01., 1983;2) Shibata y Ochiai, 1977; (13) Maraïihalii y Bhagwat, 1985;4) Granick y Mauzerall, 1958; (15) Bustos, 1983;6) Anderson y Desnick, 1979; (17) Gibbs y c01., 1985 a;8) Jordan y Gibbs, 1985; (19) Tomio y c01., 1968;0) Coïeman, 1966; (21) Doyïe y Schimke, 1969; (22) Doyïe,71; (23) Gurba y c01., 1972; (24) Gibson y c01., 1955;5) Batïïe y co]., 1967; (26) Wiïson y c01., 1972.) umo1es PBG/h.mg

89
Dado que e] incremento norma] de actividad de.ALAAD_encloropiastos y en citopiasma fue inhibido por cicioheximida,oero no por c10ramfenico]_ni kamamicina durante-e] verdeo de cotiiedones de rábano, se conciuyó que 1a enzima se sintetiza enios ribosomas citopiasmáticos y se incorpora en ios cioropiastosdurante sudesarroïio-(Shibata y Ochiai, 1976). Resuitados simi1ares se obtuvieron en hojas de haba (Siuiter-Schoiten y c01.,1973) y en E. gnac¿¿¿ó (Hovenkamp-Obbema, 1974) y fueron configmados midiendo 1a incorporación de deuterio en ALA-Dde cotiiedones de rábano (Huauit y coi., 1984).
Comoe] ALA-Den ios cioropiastos se libera rápidamente por shock osmótico, 1a enzima estaria en forma soiubie en elestroma, o bien, unida débiimente a 1a mebrana 1ame1ar (Shibatay Ochiai, 1976).
111.2.2. Especificidad de sustrato
Para 1a enzima bovina, no son sustratos ni 1a aminocetona, e-amino-ó-cetohexanoico, ni a-ó-diamino-y-cetopentanoico(Gibson y coi., 1955). Cuando se incubaron mezclas de aminocetona y ALAcon dehidratasa de conejo, no se detectaron productos de condensación (Granick y Mauzeraii, 1958).
Los aminocetoácidos reiacionados con e] ALA, taies comoe] ácido 6-amino-6-cetohexanoico, a,6—amino-s-metiiievuiinico, e] 6-amino-a-metilievuiinico y e] ó-amino-ó-meti11evu1inico,inhibian 1a dehidratasa de Rp. ¿pheno¿de¿ en forma competitiva,y no actuaban comosustratos de 1a enzima (Lartiiiot y Baron,1967).
Aunque e] ácido 1evu1inico y su etiiéster forman una base de Schiff con 1a enzima, sóio'ei ácido 1ibre actúa como uninhibidor competitivo de 1a enzima bacteriana (Nandi y Shemin,1968 b; Ho y Lasceiies, 1971; Yamasaki y Moriyama, 1971).
E1 ácido 1evu1inico y e] Succinato semiaidehido se comportan también como inhibidores competitivos de] ALA-Dde mamigeros (Granick y Mauzeraii, 1958).
La dehidratasa de M. phieá es inhibida de manera compgtitiva por e] ácido 6-hidroxi1evu1inico y e] 6-hidroxi-y-oxo-1norvaiina, aminoácido antitubercuiosis (Yamasaki y Moriyama,1971).

90
Al incubar el metiléster del ALAcon la enzima de Rp.¿pheno¿de4 no se detectó formación del pirrol hasta que ocurríala hidrólisis del éster, indicando que es esencial para el sustrato tener su grupo carboxilo libre (Nandi y col., 1968).
III.3. PROPIEDADES MOLECULARES
III.3.l. Peso molecular y estructura cuaternaria
La enzima aislada de la bacteria fotosíntética Rp.ApheñoLdeA tiene un peso molecular de 220 - 250 kDasegün se determinó por su análisis de sedimentación en gradiente de sacargsa, filtración por geles y equilibrio de sedimentación (Nandiy col., van Heyningen y Sehmin, 1971 a). Se vió que en presencia de iones potasio, la enzima formaba rápidamente agregadosmoleculares, detectándose especies de peso molecular l x 250,2 x 250, 3-4 x 250kDa (Nandi y Shemin, 1968 a). Todas ellas poseían actividad enzimática, tal vez comoresultado de disociaciones bajo las condiciones de ensayo. A su vez se vió que pordilución en ausencia de potasio o en solución de urea lM, la enzima se disociaba en una proteina de peso molecular 120 kDa,yen presencia de SDS en subunidades cuyo peso molecular determinado por PAGEera de unos 40 kDa.Se concluyó entonces que la enzima existe como un hexámero con sus seis subunidades dispuestas con una simetría dihedral en trímeros, que puede disociarse en subunidades monoméricas o agregarse en material de pesomolecular mayor (van Heyningen y Sehmin, 197l b).
Para la enzima de higado de ratón, se obtuvo un pesomolecular de 200 a 270 kDa,obtenidos por sedimentación por difusión, centrifugación en gradiente de sacarosa y filtración porgeles (Coleman, 1966; Doyle y Schimke, l969). El tratamiento conSBS en presencia de B-mercaptoetanol disoció a la enzima nativaen cadenas polipeptidicas de peso molecular 39,5 kDa,medido porPAGEen SDS. Estos resultados sugirieron que la enzima nativa estaba compuesta por seis cadenas polipeptidicas indistinguiblessobre la base de sus pesos moleculares (Doyle, l97l).
En higado bovino, diferentes grupos de investigadores usando diversos métodos reportaron valores entre 250 y 280 kDa(Gurba y col., 1972; Wilson y col., 1972), salvo un reporte de

9l
Batlle y col. (l967) de l40 kDa,el cual probablemente se debe aque fue hallado bajo condiciones que favorecieron la disociaciónde la enzima (Shemin, l974).ya que esos mismos autores (Batlle ycol., l967) en contraron evidencias de una disociación en subunidades de 70 kDa.
Wilson y col. (1972) observaron que en guanidina 7,3 Mconteniendo ditiotreitol (DTT), la enzima se disociaba en subunidades de l7,8 - 18,3 kDa,pero en PAGE-SDSse revelaron subunidades proteicas de 35,5 kDa,valor que es aproximadamente el dobleque el obtenido por el_método de ultracentrifugación en presencia de guanidina. Los autores interpretaron que la enzima sepuede disociar con SDSen siete subunidades diméricas , las quea su vez pueden seguir disociando a formas monoméricas por tratamiento con guanidina.
Cheh y Neilands (1973) hallaron un peso molecular paralas subunidades de 36 kDa,el cual es significativamente menorque el reportado por Gurba y col. (1972) de 40kDa por lo que sugirieron la existencia de siete a ocho subunidades por mol.
Hasta que en l974, Wuy col. (1974) reinvestigaron laestructura cuaternaria del ALA-Dde higado bovino.Mediante unacombinación de microscopía electrónica y determinación de pesosmoleculares, hallaron un peso molecular de 282 - 289 kDa parala enzima nativa, y para las subunidades, valores de 34,9 kDaenguanidina 6 My 35kDa por PAGE-SDS(Wu y col., l974). Por microscopía electronica de la enzima cristalizada, se observabancuatro lóbulos discretos dispuestos en las cuatro esquinas deun cuadrado. Concluyeron entonces que la enzima es un octámerocuyas ocho subunidades se disponen en los vértices de un cubo,
en una simetría dihedral D4.La estructura octamérica fue confirmada por Kreutzer y
col. (l977) en higado bovino y Anderson y Desnick (1979) en eritrocitos humanos;para esta última fuente, Calissano y col.(1966) habian reportado un peso molecular de 283 kDa.
Comovemos, los pesos moleculares de bacterias y tejidos animales están en el rango de 240 - 289 kDa.En plantas sehan reportado valores que están dentro de los mismos rangoszencallos de soya 280kDa (Tigier y col., 1970), en hojas de soya(Scheneider, 1975) y en cotiledones de rábano (Shibata y Ochiai,1977) 282 kDa.En cambio en hojas de espinaca es algo mayorz324 kDa(Schneider, 1970).

92
En este üitimo tejido, una centrifugación en gradientede densidad reveió una masa moiecuiar de 320 kDa para 1a enzimanativa, y de 50 kDa mediante PAG-SBS(Liedgens y co]., 1983).Por micrografia electrónica se observaron particuias de una tamaño ta] que indicaban una masa moïecuiar de 50 kDa (320 kDa),y que corresponde a una moiécuia cúbica de 3;7 nm (6, 7 nm) delado o a una esférica de 4,5 (9,1) nm de diámetro; algunas imágenes daban 1a impresión de un arregio reguiar de tres unidadesde donde los autores interpretaron que 1a moiécuia está compuesta por capas de tres subunidades cada una en una estructura hexamérica (Liedgens y c01., 1983).
Podemosdecir entonces que 1a discrepancia en los vaigres de pesos moïecuiares no se deben tanto a 1a fuente enzimática, sino a ias condiciones experimentaies bajo ias cuaies Serealizan, 1as que influyen en e] estado de agregación de 1a enzima. Asi por ejempio, en higado porcino Stafforini y co].(1980) mediante cromatografía en Sephadex G-200 observaron especies moïecuïares de 140 a 500 kDa, existentes en equiïibrio, dependiendo de las condiciones experimentaïes tales comodimensignes de 1a coiumna, cantidad de proteina sembrada, etc.
Mediante 1a técnica de inmoviiización de enzimas sobresoportes sóïidos, se halïó que e] ALA-Dnativo puede disociarseen subunidades, las que bajo determinadas condiciones pueden rgasociarse y reconstituir 1a enzima nativa (Grune y co]., 1977)y que, aún más, era posible 1a hibridización de 1a enzima de diferentes especies (Batiie y c01., 1978). En esas experienciasquedó c1aro que estructuras menores a 1a de] octámero nativoson enzimáticamente activas (Gurne y c01., 1977) y probabiemente e] dimero sea 1a minima unidad funciona] (Batlle y c01.,1973).
III.3.2. Estructura primaria. Composiciónde aminoácidos
En 1a Tabla III.2. se presentan los datos de 1a composición primaria de] ALA-Dde diversas fuentes.
Podemos observar que 1a composición de aminoácidos delas enzima provenientes de diferentes tejidos de una misma especie, son esenciaïmente idénticas (Fujita y co]., 1986), confirmando 1a identidad inmunoquimica de] ALA-Den diversos tejidos(Chang y co1., 1984; Fujita y co]., 1985; Yamamotoy c01.,1986),10 que sugiere que 1a enzima presente en diversos tejidos, esproducto de un mismo gen.

TABLAIII.2.:ComposicióndeaminoácidosdeALA-Ddedistintostejidosy Aspártico Treonina Serina Gïutámico Proïina Glicina Aïanina Valina Cisteina MetioninaIsoleucina Leucina Tirosina Feniialanina Lisina Histidina Arginina Triptofano
especies
HOJASDE ESPINACI
Res./50kDa
(1)
HIGADO
ERITROCITOS
Ratón
__.__—-—
Res./39.SkDa
(2) 17 14 14 31 IZ 22 296
287 7
298
12 126
14
Rata
Res./35,5kDaRes./260kDa
(fl 24 ll 16 5| 16h¡
(M 14056
126 188 13| 132 204 136 116ND
Bovino
Res./36kDaRes./35kDaRes./3IkDa
(5)
(6) 218
21 28 |9 Z] 33 23 ND
9 832 10 11 10
822 ND
Humanos
(7)
Rata
Res./35,5kDa
(M 24 11 16 54 16
ND:nodeterminado.Referencias:(1)Liedgensyc01.,1983;(2)Doyie,1971; (3)Fujitaycol.,1986;(4)Wilsonyc01.,1972;(5)Kreutzerycol.,1977;(6)Tsukamotoyc01.,1980;(7)AndersonyDesnick,1979;(8)Fujitayc01., 1986.
93

94
Es de destacar e] bajo contenido de residuos cisteinaen e] tejido de espinaca, hecho a] que 10s autores (Liedgens yc01., 1983) atribuyen 1a faita de inactivación por iodoacetamidasobre 1a enzima de este origen, y e] alto contenido de aminoácidos ácidos, aproximadamente e] dobie que en 1a enzima anima];10s autores proponen que estos aminoácidos cumpie un ro] en 1acatáïisis, en reempiazo de Ios residuos cisteina
Un análisis de grupos amino termina] rea1izado en higadode ratón (Doyie, 1971), iuego de disociación de 1a enzima conurea 8 M o con SDS 1 Z reveló que e] residuo amino termina] estaba enmascarado o bien, que estaba de aiguna manera inaccesibie para 1a reacción de dansilación.
E1 bioqueo de los grupos amino fue confirmado en 1a enzima de higado bovino por Kreutzer y co].(1977) y Jaffe y Hanes(1986).
III.4. GRUPOSSULFHIDRILOS
Ya desde ios trabajos de Gibson y co]. (1954) y Granicky Mauzeraii (1958) se demostró que e] ALA-Drequiere de reductgres de grupos tioies para manifestar su máximaactividad ¿nu¿tno.Tanto 1a enzima de mamíferos (Gibson y co]., 1955; Co]eman,1966;Batlie y co]., 1967; Moorey coi., 1971) como de bacterias(Burnham y Lascelles, 1963; Nandi y c01., 1968; Ho y Lascelies,1971; Muthukrishnan, 1972) y piantas (Nandi y Naygood, 1967;Shetty y Milier, 1969 a) requieren una activación preiiminar conreductores de grupos su1fhidriios; siendo en genera] iguaimenteefectivos e] GSH, la cisteina, e] B-mercaptoetano] y e] DTTo ditioeritrito] (DTE). Una excepción 1a constituye 1a enzima purificada de caïios de soya que no depende de 1a presencia de GSHo cisteina para su máximaactividad (Tigier y c01., 1968, 1970),sino que por e] contrario DTTy tiogiicoïato de sodio 1a inhiben parciaimente.
En forma correspondiente, reactivos especificos queblgquean 10s grupos tioles por aiquiiación irreversibie (iodoacetato, iodoacetamida, NEMI),unión con un compuesto órgano-metáiico(PCMÉ,p-hidroximercuribenzoato) o por oxidación a disuifuro(DTNB), o metaies pesados como e] mercurio o e] piomo capaces deformar uniones mercáptidas inhiben a 1a enzima de casi todos losorganismos, reafirmando 1a importancia de ios grupos sulfhidriios

95
para la actividad del ALA-D,y acerca de los cuales hay numerosos trabajos que han investigado su número y función.
Asi por ejemplo, en higado bovino Batlle y col. (l967)reportaron un total de ll grupos tioles por mol de enzima, peroestos autores habian asignado a la enzima un peso molecular del4O kDa. Unos años después, wilson y col. (1972)_por combinacióncon PCMBdetectaron 20 a 26 grupos SH libres por mol de enzima,y mediante carboximetilación e hidrólisis, 56 residuos cisteinapor mol de enzima de peso molecular 280 kDa. Resultados similares fueron obtenidos por un grupo de alemanes quienes titular0nlos grupos sulfhidrilos libres con 4,4'-bis(dimetil-aminodifenilmetanol) y determinaron el número total de grupos tioles apartir de un análisis de aminoácidosy posterior hidrólisis, obteniendo l6 grupos tioles libres por mol,y 56 residuos cisteinapor mol (Kreutzer y col., 1977).
Porotra parte, experiencias de alquilación con iodoacgtato radioactivo revelaron la incorporación de la marca con unaestequiometria de 8 moles de reactivo por octámero, y pérdida deactividad catalitica; el grupo modificado se identificó comocisteina (Jordan y col., l976).
Un análisis cinético mediante la técnica de detención deflujo indicó que por reacción con DTNBla enzima nativa de higgdo bovino presenta l6 grupos sulfhidrilos por mol altamente reagtivos, y 8 adicionales de menor reactividad (Gore y col., 1976).En la enzima carboximetilada con iodoacetato, 8 de los grupos altamente reactivos aün reaccionan, lo que unido a las experiencias de Jordan y col. (l976) sugiere que el iodoacetato reaccionacon un grupo sulfhidrilo muyreactivo esencial para la actividad.
TambiénBarnard y col. (1977) investigaron la inactivación del ALA-Dpor iodoacetato y iodoacetamida y hallaron quesólo si se trataba la enzima previamente con reductores de grupos sulfhidrilos, ocurria la inactivación por estos agentes, locual se debia a la modificación de un residuo cisteina. Observaron además, que: a) el ALAtenia distintos efectos sobre la alquilación segün que se usara iodoacetato o iodoacetamida; b) lascaracteristicas de la unión de las enzimas modificadas por cadareactivo eran diferentes; c) cada reactivo marcaba péptidos diferentes; d) el PBGprotegía en ambas alquilaciones. Estos resultados, sumados a la conocida sensibilidad de ALA-Dfrente a laianctivación por oxidación, llevaron a estos autores a concluir

96
que ios reactivos iodoacetato y iodoacetamida, reaccionan conun residuo cisteina diferente, y que ambostioies estarian ubicados en e] sitio activo, muypróximos entre si, a una distancia que caicuïaron de aproximadamente 5 Á.
Se reaiizaron experiencias simiiares con agentes aiquilantes estructuraimente reiacionados con e] sustrato, tales como los ácidos 5-ha101evu1inico y 3-ha101evu1inico (Seehra yJordan, 1981) (figura III.2.). Tambiénen este caso tuvo iugaruna inactivación de 1a enzima por modificación de una cisteinareactiva situada en e] sitio activo o muy cerca de é]. En cambio1a enzima oxidada no se afectó, resultado que fue consistentecon 1a faita de incorporación de radioactividad de los reactivosmarcados. Las diferencias observadas en 1as propiedades alquiiagtes de las dos series de halocetoácidos sugieren que ambas series reaccionan con diferentes sitios de 1a enzima.
EOOH
GHZ X = C1
GHZ Br
E=0 I
ÉHZ X = NH2 (ALA)X H (ácido ievulinico)
FIGURAIII.2.: Estructura de haiocetoácidos
Tsukamoto y co]. (1979) purificaron 1a enzima de higado bovino y obtuvieron una hoïoenzima que contenía 8 átomos dezinc por mo], en 1a que tituiaron 8 grupos suifhidriios por subunidad de 35 kDa; esta determinación se 11ev6 a cabo por variosmétodos: reacción con DTNB,carboximetilación y oxidación conperfórmico y no se observaron uniones disuifuro.
Sobre 1a base de 1a reactividad hacie e] DTNB,ciasificaron Ios grupos tioies en 3 tipos: dos altamente reactivos, mgdificados en e] término de un minuto (tipo I); tres grupos cuyamodificación se completa en una hora (tipo II) y 3 grupos adicignaies que 5610 se 1iberan por tratamiento con SDS (tipo III).La tituiación de 1a apoenzima reducida, 1a apoenzima oxidada, y

97
1a hoioenzima oxidada con oxigeno, indicó una conversión de unsuifhidriio de] tipo II en tipo I en 1a apoenzima reducida, y Iadesaparición de dos suifhidriios en 1a apoenziam y hoïoenzima oxidadas, io que fue interpretado como 1a formación de una unióndisulfuro intrasubunidad.
Observaron, además, que 1a adición de un equivaientede DTNBpor subunidad iiberaba dos equivaientes de tionitrobenzoico, sugiriendo que 1a pérdida de dos grupos suifhidriios sedebe a 1a formación de una unión disuifuro y que ambos gruposson vecinaïes (Eigura_LLL¿g¿l.
[lggBA III.3.: Reacción de] DTNBcon tioies vecinaies
También haiïaron que Ia enzima conservaba lu actividadmientras ios dos grupos suïfhidriios de] tipo I permanecieran rgducidos, aün en ausencia de] zinc. En cambio 1a actividad enzimítica se pierde compietamente, asi como e] zinc unido, iuego de1a oxidación de los grupos suifhidriios a disuifuros, por modificación de ios tioies por agentes quimicos o por formación demercapturos con metaies pesados. Por 10 que para estos autores,

98
el zinc no seria absolutamente esencial para la actividad, perose requeriria para prevenir la autooxidación de los grupos sulfhidrilos esenciales.
Seehra y col. (1981) a su vez, detectaron la presenciade cuatro grupos sulfhidrilos que exhibian diferente reactividad frente al DTNB:dos grupos altamente reactivos (I y II), quereaccionaban casi instantáneamente (en el orden de 50 y 2000 msggundos respectivamente); uno de mediana reactividad (grupo III),titulado en un periodo de dos minutos, y un grupo de baja reactividad (grupo IV) luego de los 20 minutos.
Cuando la enzima fue titulada con un mol de DTNBporsubunidad, se liberaron dos moles de tionitrobenzoico; la titulación de un mol de subunidad enzimática con 0,5 moles de DTNBpromovió la rápida liberación de un mol de tionitrobenzoico,yla adición de otros 0,5 moles liberaron un mol de tionitrobenzoico. Estos datos sugirieron una reacción inicial de un tiolreactivo con el DTNBpara dar un disulfuro mixto enzima-S-S-tignitrobenzoico, con la liberación de un tionitrobenzoico, seguido de la subsecuente eliminación del tionitrobenzoico unido ala enzima, en una reacción intramolecular con el tiol adyacente, resultando asi en la formación de una unión disulfuro.
Un PAGEde la enzima tratada con DTNBen presencia deSDSpero en ausencia de B-mercaptoetanol, reveló una ünica banda proteica de 35kDa similar a la encontrada para la enzima ngtiva reducida, sugiriendo asi que la unión disulfuro formadapor la acción del DTNBfue intramolecular (intrasubunidad) y nointermolecular (intersubunidad).
Los grupos I y II no estaban presentes en la enzimaoxidada, y su reactividad frente al DTNBera protegida porel PBG.
El grupo III parece estar involucrado en el mantenimiento dela conformación de la enzima nativa, ya que por modificación de este grupo hubo una pérdida de la actividad.
Conestos datos, Seehra y col. (l98l) propusieron elmecanismo de acción del ALA-Dcon el DTNB,que se ilustra en laïüill I-..4.o
Más recientemente, Jaffe y col. (1984) prepararon unaapoenzima estable modificada en sus grupos tioles con

99
metiimetanotiosuifonato (MMTS),un reactivo suïfhidrilico pequeño, que modifica a 1a enzima sin interferir con 1a estructurasecundario o terciaria de 1a proteina (fiigura IILLQL). La modiciación de 1a apoenzima con MMTS-reve16que reaccionaban dos grupos Suifhidriïos por Subunidad; en cambio 1a reacción con 1a hgioenzima reveió 1a modificación de tres grupossuifhidriïos porsuounidad; indicando que en 1a preparación de 1a apoenzima seperdió un grupo suifhidriio. probablemente por oxidación. La apoenzimamodificada es cataïiticamente inactiva, reactivándosepor adición de equivaïentes reductores y ZÍnCu Estos autores señalan que suponiendo que 1as subunidades son idénticas, 1a pérdida observada de un grupo suïfhidriïo por subunidad se deberiaa 1a formación de un-disuïfuro donde cada azufre proviene de unaSubunidad diferente. Si ios átomos de azufre son ligandos de]zinc. esto sugeriria entonces que e] ión metáïico puentea entredos subunidades. En cambio si se acepta que 1a unión disuïfuroes intrasubunidad, elio indicaria que 1as subunidades no son idénticas.
¡ g su (r. su 3.3 su — 5-, su
un, “hs
_ a su _ a su 3.: su És su
g' us I
,4 s-mu 3-5 s- un. ,_s s un. — s s 5"HS M
Mi, MDLus u
5" s-m. 3" s-mn "5 s-nm s 3 su
‘ Hs
[IGURA IILLQL: Mecanismo de acción de] ALA-D conDTNB, según Seehra y co]. (1981).
NBSZ= DTNB
Comovemos, 1as evidencias acumuiadas hasta e] presente puntuaiizan 1a presencia de dos grupos tioies ubicados probablemente en e] sitio activo, y en estrecha proximidad uno con gtro.que forman una unión disulfuro con e] DTNBo en presenciade oxígeno y que esta unión disulfuro puede ser reducida por unaumento de 1a concentración de tioles exógenos, reactivando asi

a 1a enzima.
o
CH3 -- s —- s -— CH3
o
FIGURA_III.5.: Estructura de] MMTS
Sin embargo, una excepción a e11os se ha encontrado en1a enzima de espinaca, pues Liedgens y co]. (1983) observaronque no se producía inactivación por iodoacetamida; tampoco observaron inactivación por diáïisis contra un buffer libre de B-mercaptoetano],y, además haïïaron un bajo número de residuos cisteina (Tab1a_llk¿g¿). Estos haïïazgos Ios 11evó a concluir queios residuos cisteina no participan en 1a catáïisis de] ALA-Dde 1a enzima de espinaca, y que probabiemente, 10 mismo ocurracon 1a enzima de piantas en genera].
III.5. ALA-D Y METALES
III.5.1. Inhibición por reactivos quelantes de metaies
Gibson y co]. (1955) ha11aron que agentes quelantes cgmo 1a y-hidroxiquinoiina-S-ácido suifónico y 8-hidroxi-7-iodoquinolina-S-acido sulfónico, a una concentración de 10'5 Mcausaban una inhibiciónde] 80%-en 1a enzima de higado de buey, sugiriendo que e110 se debia a un efecto de] grupo su1fónico másque a una propiedad queïante de esos compuestos; en apoyo a estasuposición, encontraron que 8-hidroxi-quin01ina, 8-hidroxiquinaldina, ácido quináïdico y ácido picrolónico, no eran inhibitorios.
En cambio, concentraciones de EDTAde] orden de 10'4 Minhibian marcadamente 1a enzima de esa misma fuente (Gibson yc01., 1955) y 1a de reticuïocitos de conejo (Granick, y Mauzeraïï,1958); en este último caso, 1a curva de inhibición era no

101
competitiva, y revertida por diáiisis y por presencia de magnesio. En eritrocitos de poiio, 1a inhibición dependía de la preparación enzimatica (Granick y Mauzeraii, 1958}, sugiriendo iosautores que 1a acción de} EDTApodia deberse a un cambio en 1aconfiguración de 1a enzima o a 1a captura de un meta] esencia]para su actividad.
Preparaciones de ALA-Dde piantas (Nandi y Naygood,1967; Shetty y Miiier, 1969 a; Schneider, 1970; Tigier y co].,1970; Shibata y Ochiai, 1977), levaduras (Barreiro, 1967), hongos (Komai y Neiiands, 1969; Muthukrishnan y c01., 1969) y bacterias (Yamasaki y Moriyama, 1971) son inhibidas por EDTA,conexcepción de Rp. ¿phenoideó y Rp. capóuiata (Burnham y Lasceides,1963; Nandi y Shemin, 1973).
Komaiy Neilands (1969) reportaron 1a inhibición porligados de iones divaientes en U. Aphaenogena. Además de] EDTA,la 1,10-fenantr01ina; 2,2'-bipiridina y 8-hidroxiquinoiina inhibian e] ALA-Dde dicha fuente, 1a que pudo ser parciaimente revertida por zinc (Komaiy Neiiands, 1969). Investigando 1a acción de agentes queiantes de cobre, haiiaron una-potente accióninhibitoria por disulfonato sódico de baticupronia (BCS)y VeOCUpronia(2,9-dimeti1#1,10-fenantroiina) pero sin embargo,e] Cuprizone (bis-cic1ohexanona-oxaidihidrazona) que es especifico para iones cobre (II) y e] dietilditiocarbamato no tuvieron efecto. Por otra parte, e] acetonitriiperciorato de cobre,sustancia usada para 1a regeneración de proteinas cüpricas, mostró una acción inhibitoria; estos resultados junto con ios anáiisis de absorción atómica 11evaron a Komaiy Neiiands (1969)a proponer que e] meta] invoiucrado en 1a actividad de] ALA-Dera e] cobre (I).
En 1a enzima de higado de ratón, e] EDTAa una concentración 10'2 Mpresentó un efecto estimuiante; no asi otros agentes queiantes comoa,u'-dipiridilo odietiiditiocarbamato(Coïeman, 1966). Esta activación también se observó con ionesmercurio, pero e] efecto fue menos pronunciado en 1a medida enque aumentaba e] grado de purificación de ia.enzima; 10 que su?giere que 1a acción de esos dos compuestos podrian deberse a 1ainactivación de una proteina inhibitoria acompañante, que seiria eiiminando en e] curso de 1a purificación. Estudios de sedimentación reveiaron que EDTAy mercurio no actúan disociando unaespecie de a1to peso.m01ecu1ar en subunidades activas menores.

102
Aunque el ALA-Dpurificada de glánuulade Harder de ratas, aparentemente no requiere iones metálicos para su activiuad, la formación de PBGfue inhibida completamente por accióndel EDTAl mH; a esa misma concentración, a,a'-dipiridilo, 8-hidroxiquinolina y B- fenantrolina, no tuvieron efecto (Tomioycol., l968).
El ALA-Daislada de higado bovino y de rata es inhiblda por EDTAy otros agentes quelantes como 8-hidroxiquinolina,o-fenantrolina y 2,2'-bipiridina, sugiriendo que iones metálicos bivalentes están involucrados en el sitio activo de la enzima (Gurba y col., l972). Wilson y col. (1972) hallaron que lainhibición por EDTAlO uMen higado bovino, se revertia por laadición de zinc. Resultados similares fueron reportados porAnderson y Desnick (1979) y Gibbs y col. (1985 a) en eritrocitos humanos.
III.5.2. Requerimiento de cationes
El hecho de que el EDTAinhibe a casi todas las preparaciones de ALA-Dobtenidas de distintas fuentes como se vió en
la sección anterior, llevó a considerar la posibilidad de un requerimiento metálico para la actividad catalitica de la enzima.La bibliografia concerniente a este punto es muyextensa, losresultados son diversos y en muchos casos contradictorios. Asipor ejemplo, Burnhamy Lascelles (l963) y más tarde Nandi y col.(1968) observaron que el ALA-Dparcialmente purificada de Rp.apheno¿dea requiere iones potasio para una máxima actividad abajas concentraciones de sustratoe independientemente de que elanión acompañantefuerafosfato o cloruro. Se determinó que laconcentración óptima de potasio estaba entre l y 3 x lO'2 M. Esta activación alostérica por iones potasio fue confirmada luegopor van Heyningen y Shemin (1970) y el efecto se expresa en unadisminuc1ón del Kmen presencia de dicho 1ón.
Otros cationes monovalentes como litio, rubidio y amonio también ejercen un potente efecto activador (cooperatividadheterotrópica sobre la enzima de Rp. aphenoádea (Nandi y col.,l968). El sodio y el amonio tuvieron en cambio una acción levesobre la enzima vegetal (Schneider, l970).
En cambio, las enzimas aisladas de hojas de trigo(Nandi y Naygood. 1967), hojasde espinaca y tabaco (Shetty y

103
Hiller, 1969), Spánilkum ¿tenaon¿¿ (Ho y Lascelles, l97l), callos de soya (Tigier y col., l970) y M. phtei (Yamasaki yHoriyama, l97l), no requieren potasio para su actividad y aünmás,en la enzima de callos de soya y de M. phleá se observó unainhibición a altas concentraciones de potasio (lO'Ï y l,7 x 10-2Mrespectivamente).
En Rp. ¿phenoidea se observó que además de los metalesmonovalentes, cationes bivalentes como el magnesio y el manganeso restauraban la actividad perdida por diálisis. El magnesioparece activar modificando aparentemente la estructura molecular, en tanto que el manganeso activa a bajas concentraciones ya altas inhibe. Se encontró además, que el ALA-Dde una mutante de Rp. aphenotdea. que a diferencia de la de la cepa salvajeera inhibida por EDTA,podia recuperar la actividad perdida pordiálisis, por adición de iones magnesio. La conformación de laenzima de la cepa mutante seria diferente a la Gt .a cepa salvaje posiblemente debido a un cambio en la estructura primaria,ya que se observó que une el metal menos fuertementeque.en estaúltima (Shemin, l974).
En contraste con la enzima de Rp. ¿phenoLde¿, la de Rp.capóufata es activa aün sin el agregado de potasio, magnesio omanganeso, siendo inhibida por EDTAy además por hierro (II yIII) (Nandi y Shemin, 1973).
En hojas de trigo,Nandi y-Waygood (1967) hallaron queel calcio, magnesio y manganeso activan, mientras que plomo,niquel, cobalto, hierro (II) y sodio no tienen efecto, y cobre(II) y mercurio (II) son inhibitorios: el zinc a una concentración de 10'2 M inhibió un 60 z.
El magnesio parece ser un requerimiento absoluto en algunos tejidos, como por ejemplo M. phleg (Yamasaki y Moriyama,l97l) y hojas de tabaco (Shetty y Miller, 1969 a). En éste ültimo, el manganeso presenta un comportamiento similar que en Rp.¿phenoideó activando a una concentración de hasta 2,5 mM,e inhibiendo a concentraciones Superiores. La actividad enzimáticaperdida por diálisis contra EDTA,se recupera por adición de magnesio o manganeso. siendo mayor el efecto del magnesio; en tantoque el hierro (II), cobre (II) y mercurio (II) resultaron inhibitorios.
La inhibición por reactivos quelantes especificos de cgbre en el hongo u. Aphaenogena (Komai y Neilands, l969) llevó a

104
los autores a proponer al cobre (II) comometal requerido porel ALA-D;sin embargo la actividad perdida no se restauró porla adición de iones cobre, pero si con iones zinc. Ademáselzinc, agregado a cultivos crecidos en presencia de bajas concentraciones de este metal, aumentaba la actividad de ALA-D(Komaiy Neilands, 1968). La adición simultánea-de p-fluorfenilhidrazina o actinomicina D durante el desarrollo impide este aumentode actividad, por lo que la sintesis de ALA-Dparece dependerde iones zinc en la etapa de traslación.
Se encontró que en S. ¿tenóon¿¿ (Ho y Lascelles, l97l),el manganesoactivaba mientras que el zinc, calcio y hierro (II)no tuvieron efecto. En tanto que en callos de soya (Tigier ycol., 1970) el hierro (II) y hierro (III) no modificaron significativamente la actividad de ALA-D,pero el zinc a una concentración de lO'6 Men el medio de cultivo estimulaba su actividad; concentraciones mayores o la ausencia de zinc en el mediode crecimiento dieron comoresultado una menor actividad.
A su vez, Shibata y Ochiai (1977) hallaron que en cotiledones de rábano, el magnesio, manganeso y potasio activabanla enzima, en tanto que el hierro (II) y zinc la inhibian; paraestos autores, el magnesio es un metal esencia] para el ALA-Dde esta fuente. .
En el alga verde Chionclta neguian¿¿ se vió que la actividad de la enzima purificada más de 700 veces, era muy bajaluego de una diálisis contra buffer Tris-HCl conteniendo B-mercaptoetanol, recuperándose por el agregado de magnesio o manganeso; el hierro (II) y el niquel fueron menos efectivos, y elcobalto, zinc, cobre (II) y potasio no tuvieron efecto. Se sugirió que el magnesio era necesario para una máxima actividad enzimática, requiriéndose también para la estabilidad térmica dela enzima (Tamai y col., l979).
Con respecto a la enzima de mamíferos, Coleman (1966)de acuerdo a sus efectos sobre el ALA-Dde higado de ratón, clasificó a los iones metálicos en tres grupos:
a) aquéllos que no tuvieron efecto a concentraciones de hastalO'2 M: magnesio, zinc, cobalto (II), bario, manganeso, hierro (III);
bV aquéllos que inhiben a bajas concentraciones, comocobre (II)
y plomo;

c) los que eran activadores o inhibidores dependiendo de 1a concentración empleada: mercurio (II), hierro (II), piata, queinhibian compietamente si estaban presentes en concentraciones mayores que ias de] GSH, pero que en concentraciones mengres, o bien no tenian efecto (piata) o activaban 1a reacciónmarcadamente (mercurio (II) y hierro (II) ).
Esto último contradecia e] reporte de Gibson y co].(1955) según e] cua] a concentraciones mucho menores que las requeridas para inhibir 1a enzima de higado de ratón, esos tresmetales inhibian 1a enzima de higado de buey y también eran inagtivantes e] zinc y e] magnesio. Para ambas enzimas, aniones taIes como arsenito, fluoruro, cianuro (hasta 10 mM),no tuvieronningún efecto.
En giánduia de Harder de ratas se hailó que piata, me:curio (II), pïomoy cobre (II) a concentraciones entre 0,1 y1 mM,son potentes inhibidores, mientras que el magnesio tieneun efecto más ieve y proporciona] a 1a concentración empleada ya] anión acompañante (Tomio y co]., 1968). E1 ALA-Deritrocitaria de esa misma especie es activada en forma aditiva por e]aïuminio y e] zinc, iogrando e] primero de eiios revertir 1a inhibición por piomo (Meredith y co]., 1974). Según Caiissano yco]. (1965) e] ALA-Dde glóbuios rojos humanos se activa tantocon hierro (II) comocon zinc, en tanto que Coiiier (1971) repo:tó activación por manganeso y Mitchell y co]. (1977) encontraronque e] efecto estimuiante podia ser producido por e] mercurio ,e] cadmio y ei zinc.
Con 1a enzima de eritrocitos humanos purificada 38 mi]veces (Anderson y Desnick, 1979), e] zinc activa a concentracignes entre 20 y 100 pH, e inhibecuando 1a concentración es de]orden miiimoiar; e] piomo es un potente inhibidor que afecta e]Kmy 1a Vmax, y e] hierro (II) inhibe disminuyendo 1a veiocidad.
Por otra parte, Fineiii y co]. (1974) informaron que 1aactividad de ALA-Dde eritrocitos e higado de rata, dependia deios niveies dietarios de zinc y que tanto e] ALA-Dde eritrocitos de ratones controies comointoxicados por piomo, es activada
4¿n v¿tno e ¿n u¿uo por e] zinc, siendo de 10' M 1a concentración que produce 1a máximareactivación (Fineiii y co]., 1975).
Este efecto estimuiante y además antagonistico de] zincsobre 1a inhibición de ALA-Dpor e] piomo, asi como por e] EDTA,

106
fue confirmado iuego por varios autores tanto en eritrocitos deconejo como humanos (Abduila y Haeger-Aronsen, 1971, 1973;Border y co}., 1976 a, b; Haeger-Aronsen y co]., 1976; Cantreiiy co]., 1977; Mitcheii y c01., 1977; Davis y Avram, 1978, 1980).
Empieando hemoiizados de sangre entera de aduitos normaies, Davis y Avram (1978) encontraron que e] cadmio activa auna concentración 100 veces menor que e] zinc, revirtiendo ¿nvátno 1a inhibición por piomo, también a concentraciones 100 vgces menores que las de] zinc,por 10 que, respecto a 1a concentración, es un activador más potente que e] zinc. En cambio, a a1tas concentraciones, e] cadmio inhibe,ocurriendo e] 50 % de inhibición a una concentración apróximadamente 20 veces mayor que1a de] piomo, pero esa inhibición no es revertida por 1a adición¿n uátno de zinc, Sugirieron que ambos metaies tenian un ro] estructura] reiacionado con su unión a grupos suifhidriios perifgricos.
Estos mismos autores, posteriormente (Davis y Avram,1980) extendieron su investigación a 14 iones metáiicos, examinado su efecto sobre 1a enzima norma] e inhibida en un 50 % pore] piomo, asi como también 1a capacidad de una concentración míximamenteestimuiante de] zinc para prevenir ios efectos de estos iones metálicos sobre e] ALA-D.Los resuitados se ciasificgron, entonces, en términos de sus estados de oxidación, númeroy geometria de coordinación y sus caracteristicas de ácidos deLewis biandos o duros, de manera ta] de determinar ias propiedgdes fisicas y químicas asociadas con 1a capacidad de un ión metáiico de activar o inhibir e] ALA-D.
De todos eiios,só]o e] zinc y e] cadmio (que es isoeiégtrico con e] zinc) activan e] ALA-D y previenen 1a inhibición¿n vitae producida por e] piomo. E1 zinc, ácido de Lewis intermedio, revierte 1a inhibición por piomo, cobre (II) y estaño,todos eiios también ácidos de Lewis intermedios, y germanio eindio (ácidos de Lewis duros), pero no previene 1a inhibiciónpor mercurio (II) y piata, y 1a de altas concentraciones de cadmio,siendo los tres ácidos de Lewis biandos. E1 sodio, magnesio,manganeso y aiuminio (ácidos de Lewis duros) no tienen efecto sgbre 1a enzima norma], inhibida por piomo o activada por zinc.
De aCuerdo a estos resuitados, ios autores propusieronque 1a activación o inhibición de] ALA-Dpor iones metálicos sedebe a una interacción con una base de Lewis bianda de 1a enzima.

107
comolo es e] grupo suïfhidriio.
En cambio, en médula ósea de ratas, Dresner y co].(1982) haiiaron que e] cadmio,aün a concentraciones tan bajascomo 5 x 10'6 M, produce una inhibición de] 47 %, y esa mismaconcentración no revierte 1a inhibición por piomo, sino que pore] contrario 1a sinergiza. E1 cobre, manganeso, oro y hierrotambién inhiben; e] zinc revierte 1a inhibiciónpor cadmio y plgmo. Comoe] DTTreactiva la inhibición por cadmio y cobre perono 1a de oro y manganeso, postuiaron que 1a reactividad de iosmismosocurre en sitios diferentes; para cadmio y cobre seriaa nive] de los grupos sulfhidrilos.
En 1a enzima de higado bovino, se habia reportado inicialmente un contenido significativo de cobre (Iodice y co].,1956), pero años más tarde, Gurba y co]. (1972) haiïaron que 1aenzima purificada contenía cantidades estequiométricas de zincen una re1ación de un zinc por mo] de enzima, y pequeñas cantidades noestequiométricas de hierro y cobaito. Dadoque encontraron 6 - 7 sitios de bases de Schiff para e] sustrato por mo] deenzima, pero un solo zinc, ios autores sugirieron 1a posibiiidad de reacciones parciales catalizadas por e] ALA-D,una delas cuaïes requiere ios 6 - 7 sitios de unión de bases de Schiff,y otras , como podria ser 1a eliminación de agua, que requiereun zinc por mo] de enzima; o que alternativamente, e] zinc podia tener un ro] estructura] 1ejos de] sitio activo.
Muypoco tiempo después, Cheh y Neiiands (1973) informaron que e] contenido de zinc era de 0,5 a 2,0 átomogramos dezinc por mo] de enzima, y prepararon una apoenzima que conteníamenos de] 10%de] zinc iniciaïmente presente que presentaba unaactividad casi nu1a. La enzima nativa con 1,2 zinc por mo], tenia un 85% de 1a máxima actividad observada Iuego de 1a saturación por zinc, que se Iograba con 5 a 6 átomogramosde zinc pormo]. La actividad también se recuperó con e] cadmio, en cambiocobaito y manganeso restauraron menos de] 20%mientras que nique], hierro (II y III), cobre (I y II), manganeso, caicio ycromo no fueron efectivos. Los autores consideraron que e] ALA-Dtiene un requerimiento absoluto por e] zinc, describiendola comouna metaioenzima atipica que pierde zinc durante e] curso de 1apurificación.
Paraieiamente, Hiison y co]. (1972) hailaron que e] pigmo es un inhibidor no competitivo y que e] zinc es e] ünico

108
metal que revierte la inhibición por EDTA,pero que a concentraciones superiores a l mHactúa como un inhibidor competitivo,habiendo probado además del zinc. hierro (II), cobre (II) magnesio y calcio, los cuales fueron inefectivos. El cadmio presentóun comportamiento particular ya que a concentraciones de sustrato por debajo de la saturación era inhibitorio, en cambio a concentraciones de sustrato saturantes,era activante.
A fines de la década del 70, Tsukamoto y col. (1979,l930) informaron que el ALA-Dde higado bovino purificado en presencia de zinc y DTTcontenía un átomo de zinc por subunidad de35kDa, teniendo en ese caso un máximo de actividad, pero sugiriendo que el zinc no es absolutamente esencia] para la catálisis, aunque seria necesario para prevenir la autooxidación delos sulfhidrilos. La modificaéión de los grupos Sulfhidrilospor oxidación con aire., reacción con DTNBo l,3-dibromoacetonao formación de mercapturos con metales pesados como el plomo,producia una disminución de la actividad enzimática con la libgración simultánea de zinc. A su vez, por fotooxidación, se destruian residuos histidina esenciales para la actividad(Tsukamoto y col., l975), con una pérdida concomitante de zinc(T5ukamoto y col., 1979), y la apoenzima oxidada necesitaba deun activador tiólico para incorporar zinc. Estos resultados Sugieren que el zinc interacciona con los grupos sulfhidrilos esenciales de mayor reactividad frente al oxigeno o al DTNB,coo:dinándose también con los residuos histidina.
Casi al mismo tiempo, Bevan y col. (1980) hallaron quecon 4 átomos de zinc por octámero, la enzima era completamenteactiva, y para estos autores, el zinc era absolutamente necesario, sugiriendo que la actividad presente en la apoenzima reduciua se debia a una contaminación de zinc en los reactivos. Sinembargo, si estaban de acuerdo con los resultados de Tsukamotoy col. (1979) en que es necesario que los grupos sulfhidrilos dela enzima se encuentren reducidos, para que el zinc interaccione.Analizando el contenido de zinc de varias preparaciones enzimáticas, encontraron generalemnte 2 átomogramo de zinc por octámgro. Los intentos por reactivar la apo-ALA-Dcon otros iones metálicos comoplata, calcio, cobalto, cobre (I), magnesio, manganeso, plomo, aluminio fueron infructuosos; sólo el cadmioactiva la enzima logrando una máxima estimulación con 4 átomogramos de cadmio por mol. Los estudios de unión indicaron

109
que el zinc no se requiere para la unión del sustrato a la enzima, aunque es probable que esté localizado en el sitio activo,y cumpla un rol en la catálisis.
A su vez, Gibbs y Jordan (1981) investigaron la unión65Zn-apoenzima, encontrando una incorporación de 8 átomos dezinc por octámero. La incubación de la 65Zn-holoenzima a temperatura ambiente en anaerobiosis no liberaba la marca, pero habiaun intercambio si se agregaba zinc frio. El cadmio (que se habiareportado como un metal alternativo del zinc) y el plomo desplgzaban la marcación, en cambio metales que no activan ni inhibena la enzima como el magnesio, no. El tratamiento con DTNBo laexposición al oxigeno liberaba 652n a la solución. En consecuencia, el ALA-Dde glóbulos rojos humanos une 8 zinc por octámero e interacciona con los sulfhidrilos reactivos, comoocurrecon la enzima de higado bovino (Tsukamoto y col., 1979).
Sommery Beyersman (l984) reportaron que al aislar laenzima de higado bovino sin el agregado de metal, el contenidode zinc por masa proteica aumentaba durante el curso de purificación, pero que no hay una relación estequiométrica. El zincprodujo una maxima activación a una concentración de 10'5 M;concentraciones superiores son inhibitorias. Mediante una diálisis de equilibrio determinaron una relación de 7,4 átomos dezinc por octámero, con una constante de asociación de5,3 x 10'5 M'Ï y l7 sitios adicionales de unión inespecificaque intervendrian en la inhibición producida por altas concentraciones de zinc. La enzima privada de zinc, logró reactivarseconestemetal y con cadmio; por espectroscopia NMRde la enzima113Cdsustituida, excluyeron una interacción directa del metalcon el sustrato, descartando entonces una participación inmediata del metal en la reacción catalitica, aunque posteriormente(Hasnain y col., 1985) reportaron que ese espectro correspondíaen realidad al complejo 113Cd-EDTAresultante del EDTAcontaminante. Ensayaron además la acción de 23 aniones, entre ellos elcianuro, los cuales no tuvieron efecto, hecho que los autoresinterpretaron comoun apoyo a su anterior propuesta.
De todas formas, una investigación con el reactivo tiólico de zinc, el ácido 2-bromo-3-(5-imidazoil)propiónico, llevóa concluir que el sitio del sustrato, el del zinc y los grupossulfhidrilos esenciales, se encuentran en estrecha proximidad enel sitio activo (Beyersmann y Cox, 1984).

110
Jaffe y co]. (1984) haiiaron que e] tratamiento con e]reactivo MMTSlibera e] zinc unido, dando una apoenzima modificada en sus grupos tioies que es catalïticamente inactiva. Contiene menos de 0,1 átomogramo dezinc por mo] de octámero y reconstituye compietamente su actividad con B-mercaptoetano] yzinc. Ensayaron reconstituir y/o reactivar 1a apoenzima modificada con zinc, cadmio, vanadio, nique], cobaito, piomo y manganeso, y sóio los dos primeros 10 iograron. Coincidieron conBevan y co]. (1980) en que 4 zinc por octámero son necesariospara una actividad cataiitica compiéta, siendo e] zinc absolutamente esencia], y con Tsukamoto y co]. (1979, 1980) en que 1ahoioenzimaaisiada en presencia de zinc contiene 8 átomos por ogtámero, por lo que sugirieron que 10s cuatro sitios de unión adicionaies no serian importantes en 1a catálisis.
Un estudio de] efecto de 105 iones metáiicos sobre 1areactividad de Ios grupos suifhidriios de] ALA-Dde eritrocitoshuamnos, empleando 1a técnica de medición de cinética rápida pordetención de fiujo, reveió que 1a remoción de] zinc aumentaba 1areactividad de ios grupos suifhidridos aitamente reactivos, 10sque en cambio no son afectados por 1a presencia de piomo, porlo que ambosmetaies interactuarian en sitios diferentes (Gibbsy c01., 1985 b). Esto estaria apoyado además por e] hecho deque a aitas concentraciones,e1 zinc inhibe,sugiriendo entoncesque primero ocuparía su propio sitio y iuego a] aumentar 1a concentración un segundo sitio que probabiemnte es e] mismo a1 quese une e] piomo.
E1 sustrato o su anáiogo, e] ácido ievuiinico, o e]producto PBG, no afectan 1a reactividad de 1a enzima frente a1DTNB,por 10 que consideraron que e] zinc participa en forma estructura] y no cataiitica.
También observaron que 1a protección de] zinc frente a1DTNBes instantánea, 10 que sugiere una interacción directa de]meta] con 10s grupos suifhidriios de tipo I. Comoe] zinc también protege de 1a inactivación por oxigeno, en ausencia de tigles exógenos, debe tener un ro] critico en mantener e] residuosuifhidriio esencia] reducido. Resuitados que confirman 10 haiiado por T5ukamoto y c01., 1979, y Bevan y c01., 1980.
En un trabajo reciente se investigó 1a coordinación de]zinc en e] ALA-Dmediante e] estudio de 1a estructura fina porRayos X (exafs), en una muestra que contenia un átomo de zinc

111
por subunidad de 35 kDa (Hasnain y c01., 1985).
Se halló que en la enzima nativa, e] zinc se coordinacon cuatro ligandos: tres azufres y un átomo de bajo Z, que podria ser oxigeno o nitrógeno. A1 oxidarse, la enzima sigue coo:dinada a tres azufres y a un nitrógeno, además de un átomo debajo Z, probabiemente nitrógeno.
E1 ácido 2,bromo-3-(5-imidazoi1)propiónico produce cambios significativos en e] exafs, consistentes con una coordinación con tres átomos de azufre, un nitrógeno y otro nitrógenoque seria de] inhibidor.
La unión de] sustrato a 1a enzima oxidada inactiva, induce un ligero cambio en e] entorno de] zinc, donde ademas deios cinco 1igandos.coordinados en 1a enzima oxidada, estaria presente un sexto átomo de bajo Z.
Si bien e] método no puede detectar si e] zinc está encontacto directo con un oxigeno de] ALA, los autores se inclinanpor considerar que no hay una unión directa de] meta] a1 sustrato de acuerdo con ios ha11azgos de Bevan y co]. (1980) de que e]análogo de] sustrato, e] ácido 1evu1inico, puede unirse a 1a apoenzima iibre de zinc, y a los de Sommer y Beyersmann (1984)de que ios iones zinc tienen una infïuencia iimitada sobre 1asacaración de 1a enzima (1a diferencia de Kmentre 1a apo y 1ahoioenzima es de 60 veces a favor de esta üitima, en tanto que1a Vmaxsóiose dupïica) y de que aniones tales como e] cianurono 1a inhiben.
Los resuïtados son compatibies con 1a idea de que e]sitio de] zincy e] de] azufre están en e] mismo dominio, y en apoyo a eïio, 1a inhibición por e] ácido 2-bromo-3-(5-imidazoi1)propiónico es función de] contenido de zinc de 1a enzima, y esainhibición es prevenida por 1a unión de] ALA(Beyersmann y Cox,1984).
Hasnain y co]. (1985) proponen que e] zinc tiene unro] estructrua1,permitiendo a 1a enzima adoptar 1a conformaciónrequerida para 1a actividad, sin unirse o activar a1 sustratodirectamente.

112
III.6. MECANISMO DE ACCION
La formación de PBG a partir de dos molécuïas de ALA(Granick, 1954; Gibson y co]., 1955; Schmid y Shemin, 1955) esesenciaimente una sintesis de Knorr, en 1a que están invo]ucra—das tres reacciones diferentes: a) una condensación aïdóïica,b) 1a eïiminación de una moïécuia de agua entre e] átomo de carbono que 11eva e] nuevo grupo hidroxiïo formado y un átomo decarbono adyacente y c) 1a formación de una base de Schiff poreliminación de otra moïécuia de agua entre e] carboniio de unamolécuïa de ALAy e] amino de Ia otra.
La reacción tota] requiere 1a ruptura y/o formación depor 10 menos ocho uniones, y como se iïustra en 1a figura III.1.una moiécuia de ALAaporta 1a cadena propiónico de] PBGcon sunitrógeno amino incorporado en e] aniiïo pirróiico, en tanto que1a otra moiécuïa de ALAforma e] lado acetiio de] PBG, reteniendo su grupo amino libre.
Dado que 1a sintesis es cataïizada por una soïa enzima(Gibson y co]., 1955; Neuberger y c01., 1956; Granick y Mauzeraii,1958; Nandi y Shemin, 1968 a),e1 mecanismo de acción debe darcuentano sóïo de 1a catálisis de estas reacciones, sino tambiénde 1a secuencia en que e11as OCUrren,y surgen entonces una seriede consideraciones como ser:
a) Si ambas reacciones, condensación a1dólica y formación de base de Schiff son catalizadas enzimáticamente, ¿cuál de eliasocurre iniciaimente?
b) Si 1a enzima sólo cataïiza 1a condensación aldóïica y 1a formación de la base de Schiff ocurre espontáneamente, ¿éstatiene lugar antes o después de 1a condensación?
c) Si 1a condensación a1dó1ica ocurre iniciaimente, ¿1a pérdidade agua ocurre antes o después de 1a formación de 1a base deSchiff?
d Las tres reacciones ocurren enzimáticamente?\_—
Este análisis fue hecho por Granick y Mauzeralï (1958),pero los datos cinéticos con que contaban entonces eran insuficientes como para predecir un mecanismo, y no fue sino una décgda después que Nandi y Shemin (1968 b) trabajando con 1a enzimaaislada de Rp. ¿phenoide4, encontraron que e] sustrato y anáiogos comoN-acetii-ALA, ácido 1evu1inico y etiiïevuïinato, forman

ll3
una base de Schiff con la enzima. Estos hallazgos sumados al hgcho de que el etil levulinato no inhibe a la enzima en tanto queel levulinico si, y que ambos pueden formar pirroles mixtos conel ALA,permitieron a los autores sugerir un mecanismo razonablepara la formación de PBG, consistente con los datos obtenidos ylas preguntas planteadas, que es el siguiente: se formaría unabase de Schiff entre el grupo carbonilo de la primer molécula deALAy un residuo amino de la enzima. Esto es seguido por un ataque nucleofílico del complejo enzima-sustrato sobre el átomo decarbono carbonilico de la segunda molécula de ALA. El aldol resultante pierde una molécula de agua y el grupo amino libre dela segunda molécula de ALAdesplaza al grupo amino de la enzimapor transiminación o transaldiminación, formando asi el PBG.
El hecho de que la formación de la base de Schiff sedescubriera inicialmente entre los organismos procariotas, y queen ello no se observara el efecto inhibitorio del EDTA,que siocurría en fuentes animales, llevó a la especulación de que elmecanimosoperante en las células procariotas era a través dela formación de una base de Schiff, en tanto que en los organismos eucariotas estaria involucrado un metal. Esta suposición sedebió además a que una situación similar, aunque inversa, ocurría entre las aldólasas de organismos procariotas y eucariotas.
Sin embargo, la situación para el ALA-Dse tornó máscomplicada, pues se encontró que forman también una base deSchiff con el sustrato de la enzima de hojas de espinaca(Liedgens y col., 1983), de maiz (Maralihalli y Bhagwat, 1985),de higado de ratón (Doyle, l97l), de rata (Gorba y col., l972)y de higado bovino (Chaudry y col., 1976; Jordan y Seehra, l980),y de eritrocitos humanos (Jordan y Gibbs, l985).
El aminoácido involucrado en la formación de la base deSchiff con el ALAfue identificado como lisina en Rp. ¿phenoideó(Shemin, 1976) y en eritrocitos humanos (Gibbs y Jordan, 1986),en tanto que se propuso que en la enzima de espinaca era una a:ginina (Liedgens y col., 1983).
Estudios estereoquimicos sobre la formación de PBGllevados a cabo por Chaudry y Jordan (l976) en higado de vaca empleando ALAradioactivo, indicaron que la deprotonación del precursor inmediato del PBGlibre ocurre estereoespecificamentecon la retención del átomo de hidrógeno pro-S de la posición 5del ALAen el producto, lo que implica que esta secuencia también

ll4
está catalizada por la enzima (Akhtar y col., l976).
Dadoque se demostró la formación de pirroles heterólggos entre el levulinico o su éster, y el ALA(Nandi y Shemin,1968 b), y por el hecho de que estos pirroles sólo pueden formarse sólo si el levulinico o su éster forman una-unión base deSchiff con la enzima (figura III.6.), estos autores propusieronque la molécula de ALAque va a dar lugar a la cadena acéticadelPBG, es la que se une a la enzima a través de una unión base deSchiff en lo que llamaron sitio A, en tanto que la segunda molgcula de sustrato va a formar la cadena propiónica y se ubica enel sitio P.
EOOH ÉOOH
COOH CH2 COOH CHI I I l 2
EHZ GHZ CH2 GHZ
ïH2-----ï=0 ÏH2-----ï=0OïF\\ ///CH2 OÏF CH3
CH3 H2 ?“2NH2
(A) (B)
[LQQBA_III.6.: Formación de pirroles heterólogos entre ALAyácido levulfnico.(A) si el ácido levulínico forma una base deSchiff con la enzima; (B) si el ALAes quienforma la base de Schiff con la enzima.
Sin embargo, años más tarde, Jordan y Seehra (l980) empleando técnicas de espectroscopïa NMRcon 13€ y medición de c1nética rápida por el método de detención de flujo, hallaron resultados según los cuales el orden de adición de las moléculasde ALAen la enzima de higado bovino era inverso. Incubaron cantidades equimoleculares de la enzima y 5-13C-ALA,enl a suposición de que uno de los dos sitios de unión del sustrato fuera gcupado preferencialmente por el sustrato marcado. La adición subsecuente de un exceso de ALAfrio fue usada para desplazar lareacción hacia la formación del producto, completando asi un ünico ciclo con respecto al sustrato marcado. De esta manera erade esperar que el PBGformado estuviera marcado en el carbono 2ó en el carbono ll, ya que ambos provienen del carbono 5 del ALA

115
(Neuberger y Scott, 1953; Shemin y Russe], 1933), según que e]sustrato se uniera a un sitio o a otro. E1 resuitado de esta e¿periencia fue que e] PBGobtenido presentó una sola seña] correspondiente a] carbono aromático C-2 y una compieta ausenciade 1a seña] para e] carbono 11. De este modo establecieron que1a primer moiécuia de sustrato que se une a 1a enzima es 1a queva a dar a 1a cadena P de] PBG. Estos datos también puntualizane] hecho de que esa molécuia tiene una afinidad a1 menos un orden de magnintud mayor que 1a de 1a moiécuia que dará iugar a 1acadena acética.
Estos haiiazgos iievaron a ios autores a formuiar un mgcanismo para 1a formación enzimática de PBG(Figura Ill¿l.) enla cua] 1a primer moiécuia de ALAse une a] sitio P formando unabase de Schiff (3) seguido de 1a reacción de 1a segunda moiécuiade sustrato para dar un nuevo intermediario base de Schiff (4)e] cua] es convertido a1 producto a través de ias secuencias4 a 5 + 6.
l I 0. oc ¡nu-om
FIGUBA_LII.7.: Mecanismo de acción de] ALA-Dpropuesto porJordan y Seehra (1980)
Se obtuvieron resultados simiiares con 1a enzima de Rp.¿phen04deó (Jordan y Seehra, 1980) y de eritrocitos humanos(Jordan y Gibbs, 1985 a); y para expiicar estos hechos Jordan yGibbs (1985) propusieron que e] análogo de] sustrato, e]

lló
levulinato,puede-interactuar con ambossitios de unión, el A yel P, actuando como sustrato en el sitio A, pero dado que notiene un grupo amino como el sustrato verdadero, en el sitio Psólo actúa como inhibidor, de manera tal que la inactivación 0currida cuando la enzima se hace reaccionar con borohidruro desodio en presencia de levulinato ¿Nandi y Shemin, 1968 a), es elresultado de la unión del levulinato al sitio P.
Por otra parte, también se estableció que l ó 2 residuos histidina (Tsukamotoy col., l975) y 2 residuos cisteina(Gore y col.; l976; Shemin, 1976; Barnard y col., 1977) son esenciales para la actividad. Barnard y col. (l977) sugirieronque esos grupos podian participar en una catálisis ácido/base,proponiendo entonces un mecanismo que tuvo en cuentas esos hechos, en el Cual ocurren una serie de protonaciones y deprotonaciones a través de grupos tioles y regeneración del estado inicial mediado por el sistema imidazólico de residuos histitina.
Hasta ese momento, sin embargo, no se habia tenido encuenta el rol de los metales, ni la estructura cuaternaria de laenzima. Experiencias realizadas en los laboratorios de Shemin,mostraron que sólo_4 de las 8 subunidades octaméricas, unen lasmoléculas de sustrato, sugiriendo que la enzima exhibe una reagtividad de mitad de sitios.
Estudios de disociación y reasociación de ALA-Dde higado bovino inmovilizada sobre Sepharose (Batlle y col., 1978) parecieron indicar que si bien cada subunidad octamérica contieneun sitio catalitico, la minimaunidad funcional seria la conformación dimérica, en la cual, ambas subunidades si bien de idéntica composición, tendrian sien embargo un rol diferente en lasintesis de PBG.Teniendo en cuenta ésto, Batlle y Stella (l978),en un esquema que respeta los lineamientos básicos propuestospor Shemin en l968), pero que introduce las caracteristicas mencionadas, propusieron un mecanismo en el cual una subunidad forma una base de Schiff con una molécula de ALA, mientras una segunda subunidad está involucrada en una unión no covalente conla otra molécula de Sustrato. Las autoras sugirieron que entreambas subunidades podrian existir sitios de contacto en los queestán involucrados distintos tipos de interacciones, entre losque se incluyen la posibilidad de grupos tioles y no descartanla acción de iones metálicos tales comoel zinc. En este esquema también se tuvo muy en cuenta la participación de un residuohistidina en lugar de los grupos tioles en cada subunidad que

ll7
jugaria un rol importante en el proceso de transporte de protones desde y hacia una región hidrofóbica a una hidrofilica, durante la secuencia de reacciones que llevan a la formación delPBG,y que implican una serie de protonaciones y deprotonaciones.
A su vez, Tsukamoto y.col. (1979) obtuvieron una apoenzima libre de zinc que presentaba la misma actividad que la holoenzima mientras tuviera dos grupos tioles esenciales reducidos; laapoenzima oxidada y la holoenzima con el zin custituido por plgmo, eran inactivas. Sugirieron entonces, una estrecha correlación entre un átomo de zinc, dos residuos cisteina adyacentes ydos residuos histidina, asignando al zinc el rol protector delos grupos tioles esenciales de la autooxidación, presumiblemegte coordinándose a uno de los grupossulfhidrilos vecinales, evltando asi la formación de una unión disulfuro. Esta hipótesis esá apoyada por la observación de que cuando se elimina el zinctiene lugar una rápida formación de'una unión disulfuro. En elmecanismo de acción propuesto (Tsukamoto y col., l980) un grupoimidazol de un residuo histidina en el sitio activo participa enla catálisis ácido-base favoreciendo la formación de un carbanión sobre una primer molécula de ALA. En un segundo paso, elgrupo sulfhidrilo esencial que se encuentra protonado a pH neutro, forma una unión puente de hidrógeno con el carboxilo de lasegunda molécula de agua facilitando su polarización; teniendolugar luego el ataque nucleofilico del carbanión sobre el carbgnilo del segundo sustrato (Figura'III.8.). Y
OXIDIZED NON-OXIDIZED HOLOENZYME Pb-ENZYMEAPOENZYME APOENZYME
(ocliva 0) (ocilvlly 95-IOOl (ccflvivyIOO) (ociiviíyp)
- Zn” =o
cnoerobic
FIGUgfi-LLI.8.: Mecanismo de acción del ALA-Dpropuesto porTsukamoto y col. (1980).

Otros trabajos que apoyan 1a esenciaiidad de] zinc enia reacción cataiitica de] ALA-D,son ios de Bevan y co]. (1980),Beyersmann y Cox (1984), Jaffe y co]. (1984),todos eiios en higado bovino; y Gibbs y co]. (1985 a) en eritrocitos humanos. Sibien Bevan y co]. (1980) indican que e] zinc no parece participar enia unión de] sustrato ni tampoco en e] mantenimiento deia estructura cuaternaria de 1a enzima, estudios de marcaciónpor afinidad con ácido 2-bromo,3w(5-imidazoi1)propiónico, que esun reactivo tióiico y de zinc, indicaron que e] sitio de] zinc,e] de] sustrato y ios grupos suifhidriios esenciaies se encuentran en estrecha proximidad en e] sitio activo (Beyersmann yCox, 1984)._
Más recientemente, Jaffe y Hanes (1986) empiearon unaapoenzima modificada en sus grupos suifhidriios con MMTS,paraestudiar ias primeras etapas de 1a sintesis dei PBG. Observaronque e] borohidruro de sodio lievaba a 1a formación de una basede Schiff entre 1a enzima y e] 14C-ALA, aün bajo condiciones enque no podria formarse ei producto por faita de disponibilidadde zinc (ya que este meta] no podia unirse a 1a enzima) y quee] intermediario también se formaba con 1a apoenzima modificadapor MMTS.Mediante una espectroscopia NMRcon 13€, observarona1 agregar 13C-ALAa 1a hoioenzima una señai que atribuyeron a1PBGunido a ia enzima (Jaffe y Markham, 1987). Para ei conjuntoformado por interacción de] 13C-ALAy ia apoenzima modificadacon MMTS,que no puede formar producto, detectaron una seña] queadjudicaron a1 sustrato unido por una base de Schiff. Si a esecompiejo agregaban cadmio y B-mercaptoetano], obtenían un espectro que coincidia con e] de 1a hoioenzima, y que tenia además1a seña] correspondiente a1 PBGlibre, lo que interpretaron como que se habia iogrado reconstituir a 1a hoioenzima. Con estosresuitados, Jaffe y sus colaboradores consideran queni e] zinc ni ios grupos suifhidriios esenciaies están involucrados en 1a etapa de formación de 1a base se Schiff. Paraestos autores, 1a enzima tiene un absoluto requerimiento porzinc, siendo necesarios los grupos suifhidriios reducidos para1a formación de] sitio de unión de] metal.
III.6.1. Naturaieza de] sitio activo
De io expuesto en los párrafos anteriores, en donde sereseñan todos ios hechos concernientes a1 mecanismo de acción de]

119
ALA-D, que se conocen hasta e] momento, podemos señaïar algunascaracteristicas de] sitio activo de esta enzima:
a)
bV
D.V
Ve
9)
Dado que e] ALA-Dcataliza 1a dimerizaCÏónde dos moïécuïasde ALA,
pendientes para Ios sustratos;e] sitio activo debe tener dos sitios de unión inde
un grupo amino (de un residuo ïisina o arginina) para 1a-fo;mación de una base de Schiff;
un grupo básico sobre e] grupo amino de 1a proteina para ay!dar a 1a extracción de un protón en 1a formación de] carbanión (figura III.9.);un grupo positivo, que forma una unión iónica con e] carboxiIato de] sustrato aumentando 1a afinidad de] y-cetoácido por1a enzima, io que seria consistente con e] hecho de que, aunque el ácido 1evu1inito y su éster pueden reaccionar con e]sitio activo, sólo e] ácido libre puede actuardor competitivo.
como inhibiE1 grupo positivo también orientaria a 1a
molécuia de sustrato de manera ta] de no permitir 1a formae] PBG,
1a cadena propionica en posición de otro pirro] que no sea pues en su ausencia,se podria formar un pirro] conción a y e] grupo amino y otro(Figura III.10.);
propiónico en posición B
un segundo grupo negativo, sugerencia que proviene de 1a observación de que e] a-cetogïutarato, que contiene un grupocarboxiïo en 1ugar de un aminometiïo, no es reducido por e]borohidruro de sodio sobre 1a enzima;
dos grupos sulfhidrilos vecinaïes a1tamente reactivos;
uno o dos residuos histidina que median en 1a transferenciade protones desde y hacia e] sitio activo, sirviendo ademásde ligandos para e] zinc;
un átomo de zinc-cuya función no está aün di1ucidada.
Los puntos c) y d) sóïo tienen aplicación en caso de aceptar e] orden de unión de 1as moiécuïas de ALApropuesto porShemin, y no se descarta 1a posibilidad de que tanto 105 grupossulfhidrilos comolos residuos histidina tengan un ro] en 1a catálisis ácido-base,y Ios átomos de zinc actúen comocentros positivos orientadores.

120
FIGURAIII.9.: Caracteristicas po;tuiadas para e] sitio activo de] ALA-D(Shemin, 1974)
oOOHCH
2NH '
¡ 2 on-CH---—c=0
E'N=/C‘\\sCH N
l 2 H2
‘PH2COOH
FIGURAIII.10.: Formación de] isómero de] PBG.
III.6.2. Acerca de] númerode sitios activos
En un review, Shemin refiriéndose a un trabajo no publicado de Shearer y Wu (Shemin, 1976) indicó que parecia ser que deias 8 subunidades, 5610 4 reaccionan_con e] sustrato para formar1a base de Sehiff (Shemin, 1975). Teniendo en cuenta ésto y 1aevidencia de que 1a enzima se disocia en dimeros funcionaies(Batiie y co]., 1978), Batlie y Steiia (1978) propusieron un

121
mecanismodonde ios sitios activos se encuentran en 1a interfgse de dos subunidades, Ias que, si bien de composición idénticas, tendrian roles diferentes en 1a catáiisis. Unaprueba deesta hipótesis requeriría establecer 1a disimiiitud de ias subunidades.
Wuy co]. (1974) cristalizaron 1a enzima de higado bovino y demostraron que está compuesta por octámeros de subunidades indistinguibies por equiiibrio de sedimentación en guanidina 6 M o por PAGE en presencia de SDS.
Investigando 1a composición de 1as subunidades por unanáiisis de secuencias primarias de 1a enzima de higado bovino,Hohberger (1979) halió que ias secuencias amino termina] estaban bloqueadas y que e] número de péptidos por digestión triptica era igua] a1 que se esperaba de ia composición de aminoácidos. Jaffe y Hanes (1986) confirmaron que las secuencias aminotermina] estaban bioqueadas, aunque existe un reporte de que sólo 1a mitad de los residuos amino terminaies están aciïados,io que implicaría una iigera diferencia en 1a estructura de lasSubunidades (Lingner y Kieinschmidt, i983).
Tsukamoto y co]. (1979) propusieron que 1a enzima dehigado bovino posee 8 sitios activos por octámero y Tschudy y.co]. (1981) en un eiegante estudio de 1a inactivación de la enzima de higado de rata por succiniiacetona conciuyeron, porcomparación con 1a enzima purificada de otras fuentes, que e]número de recambio_(turnover) de 1a enzima de higado de rata esconsistente con 8 sitios activos por octámero.
Investigando 1a inactivación de] ALA-Dpor ios ácidos3 y 5 C1 (3,5 3H )1evu1ïnico, Seehra y Jordan (1981) estabiecigron que 1a reiación entre 1a pérdida de actividad y 1a incorporación de radioactividad a bajos niveies de inactivación Sugeriaque 1a incorporación de 0,5 moies de agente aiquiïante por mo]de subunidad enzimática, conducirían a una compieta inactivación.Sin embargo. en 1a práctica, 1a enzima se inactivaba totaïmentesóio cuando se inc0rporaba un mo] de reactivo por subunidad, sugiriendo que Ia'mitad de los sitios susceptibïes a 1a aiquiiación, son sustanciaimente más reactivos que los otros. Estaaparente reactividad de mitad de sitios hacia ios haiocetoácidospodria deberse a un cambio conformacionai inducido, o ser una manifestación de 1a naturaieza asimétrica de 1a enzima octamérica.

122
El mismo grupo hizo un análisis detallado de la reacción del ALA-Dbovina con DTNBdetectando la presencia de cuatrogrupos tioles por subunidad,de diferente reactividad, mostrandocada uno de ellos diversos grados de reactividad de mitad de sitios (Seehra y col., l98l).
Jaffe y col. (1986) investigaron la inactivación porborohidruro de sodio y marcación con 4-14C-ALAempleando la holgenzima de higado bovino y la apoenzima modificada con MMTSobtgniendo que, bajo condiciones en que era posible el turnover dela enzima, la estequiometria de marcación era de 2,3 14€ por ogtámero, con una inactivación del 50%; en tanto que bajo condiciones en que no es posible el turnover enzimático, la formación de la base de Schiff resultó en una pérdida casi completade actividad (96 %) y 4 moléculas de 14C-ALAatrapadas por octímero. Propusieron que la escasa actividad remanente podria resultar de una pequeña proporción de los cuatro sitios activosque permanecían sin modificar, o que podian ser un reflejo dela reactividad de la mitad de sitios remanentes. Indicaron quesosresultados eran consistentes con 8 sitios activos funcionales, en los que la mitad de los sitios soncapaces de catalizarlareacción a una velocidad, mientras que los otros 4 sitios sonsólo capaces de catalizar la reacción a una velocidad 30 vecesmenor, o que, alternativamente, los 4 sitios secundarios exhiben una constante de unión para el sustrato extremadamente alta(en ese casoseria un fenómenode cooperatividad negativa).
IfD
Asi, existen diferentes opiniones en lo concernientea la naturalezadel sitio activo (la que podria requerir la uniónde dos mitades ubicadas en subunidades adyacentes o ser un sitio completo dentro de una subunidad), y el nümero de sitios agtivos por molécula que para algunos autores es 4 y para otros8.
III.7. OTROS FACTORES QUE AFECTAN LA ACTIVIDAD DE ALA-D
III.7.l. Buffers
En un principio se habia reportado que el ALA-Dde higado bovino era totalmente inactiva en buffer Tris (Gibbs y col.,1955). Experiencias preliminares con la enzima dE'Rp. apheno¿de¿(Burnhamy Lascelles, 1963) mostraron que no tenia actividad en

123
Tris, pero que era activa en mezcïas de Tris y fosfato sugiriendo un requerimiento por fosfato; pero investigaciones posteriores demostraron que en reaiidad e] ión requerido era e] potasio.
E1 ALA-Dde higado de ratón era tan activa en bufferTris como en fosfato (Coieman, 1966) y 1a enzima aisïada deglándula de Harder era un 30 % menos activa en Tris que enbuffer fosfato (Tomioy co]., 1968).
En cambio, e] Tris aumentaba 1a actividad de 1a enzimade caiios de soya (Tigier y c01., 1968) mostrando un efecto opuesto que en mamíferos, en tanto que en hojas de trigo, e] fosfato parecia ser ligeramente inhibitorio (Nandi y Wayggod,1967).
Shetty y Mi]1er_(1969 b) informaron que las dehidratasas de hojas de trigo e higado de buey eran activadas por fosfato, observando un 1eve aumento de actividad en Tris hasta unpH 9,0; atribuyendo entonces 1a baja actividad en Tris a queen ese buffer e] pH óptimo es más alto
A1 respecto cabe aciarar que en muchos casos los pH'sreportados son ios de ios buffers, y no los de 1a mezcia de incubación, y no se puede descartar 1a posibiiidad de que 1a difgrencia de actividades observadas en ios varios buffers podriadeberse a ciertos componentes de] buffer con propiedades queiantes de iones metálicos (Border, 1976 a).
III.7.2. Inhibidores endógenos
Durante e] proceso de purificación de] ALA-Dde caiïosde soya, se detectó un componente de bajo peso moïecuiar que era dificil de e1uir completamente luego de una fiitración porge] (Tigier y c01., 1968). Unextracto iiofiiizado de] mismoinhibió parciaimente a Ta enzima.
También en extractos de N. cnaasa se ha descripto 1apresencia de un inhibidor termoïábii de naturaieza proteica(Muthukrishnan y c01., 1969).
En méduïa ósea de ratas, Kondoy coi. (1981) reportaron1a existencia de un inhibidor termoiábii, sensibie a tripsina ydesnaturaiizado por urea, con un pH óptimo entre 7,5 y 8,0, y unpeso molecuiar de 28 kDa, que produce una inhibición de tipo nocompetitivo. Ese componentese haiió también en céiuias eritroideas de méduia ósea de monos, conejos, vacas y humanos, pero no

124
se encontró en méduia ósea y bazo de ratón. La actividad del ighibidor aumentaba rápidamente 1uego de] nacimiento coincidiendocon un marcado descenso de actividad observado en e] ALA-Dde mgdu1a ósea; este descenso fue menor en e] higado, tejido en e]que no se detectó 1a presencia de] inhibidor.
III.7.3. Acción de compuestos de] camino de] hemo
III.7.3.]. ALA
Con e] objeto de determinar 1a influencia de] mismo sustrato sobre 1a actividad de] ALA-D,se inyectó ALAen ratones,observándose un aumento de 2 a 3 veces de 1a actividad de la enzima hepática comparada con 1a de ios controïes (Russeii yCoieman, 1963). E1 efecto probabTemente se deba a una activacióndirecta de 1a enzima por su sustrato, más que a una sintesis denovo de ia proteina enzimatica (Onisawa y Labbe, 1962).
E1 agregado de ALAa] medio de cuitivo de caïios de sgya, crecidos en oscuridad, produjo una rápida acumuiación de porfirinas y un aumento de actividad de ALA-D(Bat11e y c01.,1975).
III.7.3.2. P_B_G__
En cuitivos de callos de soya, se Ilevaron a cabo expgriencias agregando PBGa] medio de cu1tivo pero se encontróque los caïlos eran impermeables a1 pirro], no pudiendo por 10tando ser utiiizado por e] tejido (Bat11e y c01., 1975).
E1 PBG, que es e] producto de 1a reacción enzimáticade] ALA-D,1a inhibe competitivamente (Seehra y c01., 1981), yen una concentración de 5 mM no previene 1a reacción de] DTNBcon ios grupos tioies de 1a enzima de gióbuios rojos (Gibbs yc01., 1985 a), pero en cambio si 10 hace con 1a enzima de higado bovino (Seehra y c01., 1981).
III.7.3.3. Porfirinas y hemo
La observación de Burnham y Lasceiies (1963) de que e]ALA-Dde Rp. ¿phenoLdeó se inhibia 1evemente por bajas concentraciones de hemina y proto IX, fue refutada por Nandi y co].(1968 a), quienes ha11aron una marcada inhibición.
Otra bacteria fotosintética, 1a Rp. capóulata, es en

l25
cambio relativamente insensible a la acción de estos compuestos(Nandi y Shemin, 1973).
También es débil o no existe el efecto sobre las enzimas de S. ¿tenóon¿¿, M. phieLWy hojas de tabaco (Shetty y Miller,l969 a; Ho y Lascelles, 197l; Yamasaki y Moriyama, 1971).
El ALA-Dde glóbulos rojos puede ser inhibida ¿n uLtnopor proto IX, hemo libre y hemoglobina (Steiner y col., l964;Moore, 1980), pero no lo hacen otras hemoproteinas como citocrgmo c y catalasa (Calissano y col., 1966 a).
La enzima de higado de ratón es inhibida marcadamentepor proto IX, hemina (Coleman, 1966) además de hematina (Doyley Schimke, 1969). Weissberg y Voitek (1974) hallaron que estainhibición en higado y glóbulos rojos de cobayo es de tipo nocompetitiva.
Copro III y coprogen III resultaron serinhibidores másefectivos del ALA-Dde N. cnaóóa que hemina, proto IX o protogen, cuya acción era muy leve (Muthukrishnan y col., l972).
Chandrika y col. (1980) encontraron que haciendo crecer Neuno¿ponacnaaóa en condiciones de deficiencia de hierro,la actividad del ALA-Dera casi indetectable. El agregado dehierro revertia esta situación, pero este efecto podia bloquearse por adición de proto. Se estudió entonces si el hierro actuaba directamente a nivel genético o simplemente eliminado la prgtoporfirina del medio mediante su conversión en hemo. Se encontró que inhibiendo la sintesis de hemoconácido levulinico ocianuro, se inducia la sintesis de ALA-D,mientras que si se inhibia un paso posterior a la formación de proto, por ejemplopor deficiencia de hierro o tratamiento con cobalto, se reprimiala enzima. Cuando los niveles de protoporfirina disminuian pordebajo de su valor óptimo, se inducia el ALA-Dy cuando aumentaban, se reprimia esta enzima. El ácido levulinico y el cianuro pueden inducir a la enzima aün en ausencia de hierro, indicando que este metal sólo actúa convirtiendo la proto en hemo. Enbase a estos datos, los autores sugirieron que la protoporfirina es el regulador fisiológico del ALA-Den N. cnasaa.
También se determinó el efecto de copro, proto y heminasobre la actividad del ALA-Dde Euglena gnacilió ¿n vao e ¿nv¿tno aunque ¿n thno el efecto es mucho mayor, produciendo copro y proto una inhibición del 50 %. Los porfirinógenos

126
correspondientes se comportan en forma anáioga, respondiendo suefecto a un tipo de inhibición por feed-back, comose encontróen otras fuentes (Steiia, 1977).
III.7.4. Sustancias inductoras de porfirias
Las sustancias porfirinogénicas comoe] Sedormid (B-aiiiisopopiiurea), a]iïisopropiiacetamida (AIA) y dicarbetoxidihidrocolidina (DDC), poseen grupos ceto que, a] menos en e] caso de] Sedormid, pueden formar enoiatos y hemiacetaies con iosgrupos suifhidriios libres (Barreiro, 1969). Sin embargo, no tuvieron efecto sobre 1a actividad de ALA-Dde higado y bazo deratón (Coleman, 1966), en contraste con e] marcado aumento deia actividad de estaenzima observado en higado y riñones de conejos y ratas tratadas con AIAy Sedormid (Gibson y co]., 1955;Tancioni y c01., 1964; Marver y c01., 1966; Narisawa y Kikuchi,1966).
Es probabie que estas respuestas sean especificas decada especie, ya que tampoco se observaron efectos en coneji1ios de india tratados con DDC(Granick y Urata, 1963).
A1 agregar AIA a una concentración de 0,2 - 2 % en eimedio de cultivo de tejido de soya, 1a actividad de ALA-Daumegta 1uego de un proiongado periodo de crecimiento; ta] aumentono ocurre si e] AIA se agrega ¿n uitno a1 extracto enzimático(Tigier y c01., 1970).
Repetidas inyecciones de fenilhidrazina producen unasevera condición anémica en conejos y ratones, y un aumento sustancia] de 1a actividad de ALA-Dde bazo, pero poco o ningün efecto sobre 1a enzima hepática (Gibson y coit, 1955; Margoiis yRussei, 1965; Coieman, 1966). E1 efecto en bazo probabiementese deba a un incremento en 1a hematopoiesis extrameduiar(Gibson y co]., 1955).
La administración endovenosa de 4 pg de estradio] a ratas producia cambios irreguiares de ias actividades de ALA-SyALA-Dhepáticas a:io iargo de] tiempo. Cuando se administrabaAIAa ios animales 5 horas antes de matarlos, ocurrian variaciones simiiares que ampliaban ias curvas de actividad eniimática. Mientras que las modificaciones en 1a sintetasa en respuesta a los estrógenos se deben a cambios en 1a cantidad de enzima,no se ha podido demostrar'que ocurra.]o mismo con e] ALA-D(Tschudy y co]., 1967).

127
III.7.5. Fosfato de piridoxa] y compuestos de alta energia
E] fosfato de piridoxa] (PPy), un cofactor de] ALA-S,inhibe alrededor de] 30% e] ALA-Dde Rp. ¿phenoádeó a una concentración de 10'3 M, respondiendo a un tipo de inhibición nocompetitivo. E1 tiempo de incubación no influye sdbre e] gradode inhibición (van Heyningen y Shemin, 1971 b). E1 PPy y 1a piridoxamina tienen e] mismoefecto, pero 1a piridina, e] benzaldehido y 1a piridina-4-carboxa1dehido no modifican 1a actividadde] ALA-D. De manera que 1a acción inhibitoria parece que espropia de 1a moïécula de piridoxa].
Compuestos de aïta energia de origen ce1u1ar tales como pirofosfatos, ATP, UTP, GTP o ITP agregados ¿n v¿tno a 1amezcla de reacción a una concentración de] orden de 3-6 mM, inhiben casi compietamente e] ALA-Dde fuentes vegetaies (Nandi yHaygood, 1967; Tigier y c01., 1970), excepto 1a enzima de tabacoque sóio se afecta 1evemente aün a concentraciones reiativamente aitas (Shetty y Miiier, 1969 a). En caiios de soya, a concegtraciones equivaientes, e] AMPes menos efectivo que e] ADP, e]que a su vez es menor que e] ATP (Tigier y co1., 1970).
III.7.6. Urea y derivados
Nandi (1971) halió que una preparación purificada deALA-Dde Rp. óthüoLdeó era inhibida en forma competitiva porbajas concentraciones de urea, guanidina y meti] derivados de urea.
Estos compuestos podrian interferir con 1a formaciónde] compiejo enzima-sustrato, actuando sobre puentes hidrógenode] sitio activo; se halló que e] sustrato previene 1a inhibiciónpor dichos compuestos, probabiementecompitiendo con 1a urea pore] sitio activo de] ALA-D.
Sin embargo, 1a inhibición es reversibïe, 10 que estaria indicando que 1a inactivación resulta de 1a disociación de1a enzima en subunidades por acción de] agente desnaturaiizante,no descartándose 1a pósibilidad de una acción combinada de ambos efectos. Resuïtados simiïares fueron reportados por Bat11ey co]. (1978) para 1a enzima de higado bovino.

128
III.8. ONTOGENESIS Y ACTIVIDAD ENZIMATICA
La actividad del ALA-Destá estrechamente relacionada
con la madurez general de un organismo, asi como con el tipo detejido y la maduración de sus células. Su actividad es máximaen eritroblastos, eélulas que presentan una elevada sintesis dehemo (Battistini y col., 197l). Dado que no posee mitocondriasel eritrocito humanomaduro no contiene las enzimas particuladas del camino del hemo (Schmid y Shemin, 1955 b; Rimington yGooij, l957, Granick y Levere, l964), pero si contiene las enzimas citoplasmáticas, entre ellas el ALA-D.Si bien la concentración de ALA-Des la misma en los eritroblastos nucleados y enlos eritrocitos anucleados, es probable que en estos últimos laenzima no tenga importancia funcional, ya que en esas célulasno hay sintesis de hemo. Entonces, para ser más precisos, laactividad deberia medirse en las células de la médula ósea, loque, obviamente, presentaria muchas dificultades (Iwanow, l968).
El ALA-Dde glóbulos rojos de diferentes especies muestra una variación considerable (Sassa yBernstein, 1977). Entrelos animales, los conejos presentan la mayor actividad, y losperros la más baja (Abdulla y col., l973). fl
Además la actividad de ALA-Dexhibe cambios marcadosdurante el desarrollo animal: está significativamente elevadaen el higado fetal de ratones (Doyle y Schimke, l969; Weissbergy Voytek, 1974), pero decrece inmediatamente luego del nacimiento para luego aumentar hasta llegar al nivel adulto unas semanas después (Russel y Coleman, l963; Doyle y Schim ke, l969).Puesto que estos cambios son paralelos a las variaciones en elnümero de células eritropoyéticas, es probable que el aumentode actividad en el estado embrionario esté relacionado con lagran proporción de células eritroides en el higado fetal(Doyle y Schimke, 1969; Freshney y Pane, l97l).
Los análisis inmunológicos indican que la enzima fetaly adulta son antigénicamente indistinguibles,pero la primera escon respecto a su efectividad comoantígeno, cataliticamente 2veces más activa que la segunda (Doyle y Schenike, l969). Esadiferencia en actividad específica, expresada comoactividadcatalitica por molécula proteica inmunoquimicamentereactiva,podria deberse a la presencia de un activador en el tejido, auna estructura terciaria más efectiva, o a una modificación en

129
1a estructura primaria que afecte e] aspecto cataiitico pero noe] antigénico. La primera de eiias ha quedado descartada (Doyiey Schimke, 1969) y respecto de ias otras dos , ios estudios deparámetros fisicoquimicos muestran que algunas propiedades sonsimiiares (coeficientes de sedimentación, movilidad eiectroforética, Km)en tanto que otras no (termoestabiiidad, inactivación proteoiitica), por 10 que quedan aún por diiucidarse.
También en ratas aduitas, 1a actividad de ALA-Destáreducida respecto de ia de ios animaies jóvenes (Fineiii y coi.,1974, 1975). Luego de] parto presentan 1a mayor actividad registrada en especies animaies, para decrecer drásticamente durantee] primer periodo de crecimiento, en e] que sin embargo es aüncuatro veces mayor que en ias ratas aduitas (Abduiia y c01.,1978).
En ios humanos, se ha encontrado que 1a actividad ene] recién nacido es e] dobie que en ios aduitos (Abduiia y coi.,1978). En ios gióbuios rojos 1a variación de actividad es tam—.bién un refiejo de los cambios en 1a población eritrocitaria.Hay una actividad de ÁLA-Deritrocitariaeievada en pacientes condesórdenes hemoiiticos que producen reticulocitosis (Anderson ycoi., 1977) y se ha estabiecido una correiación linea] entre e]nivei de actividad de ¡LA-b en gióbuios rojos y 1a cantidad dereticuiocitos. Hay además una gran variación en ios niveies deactividad de ALA-Deritrocitaria en 1a pobiación humana (Sassay c01., 1973) y anima] (Sassa y Bernstein, 1977) que indica uncontro] genético de 1a sensibiiidad de ALA-Dcompatible con unaherencia autosómica dominante (Sassa y co]., 1973).
Los estudios de propiedades taies como peso moiecuiar,tamaño de subunidades, composición de aminoácidos, propiedadescinéticas e inmunorreactividad,reaiizado con ias enzimas purificadas de higado y gióbuios rojos de ratas (Fujita y coi., i986)confirman reportes previos sobre 1a identidad inmunoquimica de]ALA-Dde varios tejidos (Chan y coi., 1984; Fujita y c01., 1985;Yamamotoy c01., 1986), sugiriendo que 1a enzima presente en varios tejidos es producto de un mismo gen.
En piantas y otros organismos fotosintéticos,ia formación de PBGestá controiada por 1a iuz (Steer y Gibbs, 1969;Siuiters-Schotten y coi., 1973; Hovenkampp-Obbemay co]., 1974).
Se ha reportado_que 1a actividad de ALA-Daumenta durante e] desarrolio de ios cioropiastos cuando se produce una

130
rápida acumulación de clorofila causada por la iluminación deplantas etioladas. Asi por ejemplo, en callos de soya se observó un aumento de la actividad de ALA-Dparalelo a un incrementode clorofila cuando callos crecidos en oscuridad se hicieron crgcer en la luz (datlle y col., l97o). Efectos similares se obse:varon en otros tejidos (Stobart y Thomas, 1966; Ebbon y Tait,l969; Steer y Gibbs, 1959; Schneider, 1970, 1971).
En cotiledones de rábano,.el verdeo se completa por iluminación continua durante dos dias; luego de ese periodo elcontenido total de clorofila permanececonstante, pero la actividad de ALA-D-comienzaa decrecer indicando un sistema regulatorio para la actividad de la enzima, que puede participar enla regulación de la sintesis de clorofila (Shibata y Ochiai,l976).
El aumento de la actiVidad de ALA-Dbajo la influenciade la luz se deberia probablemente a una sintesis de la enzimaregulada por la luz más que a una activación de la enzima pre-gxistente (Stobart y Thomas, l968; Schneider, l970). De hecho,en cotiledones de.rábanos se enconró que la luz roja lejana estimula la sintesis de novo del ALA-D(Huault y col., 1984).
III.9. ASPECTOS GENETICOS
Comolo describieron Russell y Coleman (l963), la actividad de ALA-Den tejidos de ratón se encuentra bajo el controlgenético de dos alelos en lo que ha sido llamado "locus levulinato" (Lv). Estos autores observaron que ratones adultos normales pertenecientes a una misma cepa poseian actividades enzimáticas diferentes. Se dividieron asi en dos grupos; la mayor parte de los animales eran homocigotas para el alelo Lv, al que denominaron Lva, en tanto que en ciertas sublineas observaron unamutación denotada va, en los que la actividad enzimática estabadisminuida a un medio o a un tercio de la de las cepas Lva. Lasenzimas de bazo e higado de cada genotipo (Lva/Lva y va/va) pgseian idénticas propiedades fisicas, químicas y enzimáticas, dedonde concluyeron que la estructura primaria de la enzima es lamisma en ambos genotipos y que el gen Lv controlaria la cantidadde enzima (Coleman, 1966).
Combinandotécnicas inmunoquimicas e isotópicas

l3l
demostraron que, efectivamente, las diferentes actividades enzimáticas se debian a distintas cantidades de proteina enzimática,y que el locus Lv regula la cantidad de ALA-Dhepática actuandoa nivel de sintesis de la enzima. La velocidad de degradaciónera la misma en ambas cepas, siendo la vida media de 506 dias(Doyle y Shimke, l969). Se demostró también que el ALA-Dde cada genotipo estaba compuesto por seis subunidades idénticas, cada una con un sitio activo, además los péptidos obtenidos pordegradación con tripsina y la composición de aminoácidos de lasSubunidades en cada genotipo eran idénticas, y si bien no se determinó la secuencia de aminoácidos de la molécula en su totalidad, parecia poco probable que ambas enzimas difirieran en algomás que la velocidad de sintesis (Doyle, l97l).
Hutton y Coleman (1969) clasificaronunas 30 cepas endgcriadas sobre la base de las actividades de ALA-D,encontrandoque en sumayor parte se podian agrupar en tres clases: de altaactividad, Lva/Lva; baja actividad va/Lvb y una intermedia,LvC/LVC.Estos últimos podrian representar una tercer alelo enel locus Lv o una modificación estructural de la enzima. Lascepas con niveles de actividad enzimático atipico fueron consideradas comofuentes potenciales de mutaciones estructurales ycada una fue estudiada cuidadosamente con respecto a varias prgpiedades fisicoquimicas. Se identificó un factor genético queinfluiria sobre la estructura del ALA-D,modificando su labilidad al calor y dado que ambas propiedades, estructura y cantidad, se segregan juntas, se estableció que los factores genéticos que las controlan debian estar relacionados,siendo muyposiblemente alélicos (Coleman, 1971).
La velocidad de sintesis disminuida en los ratones homocigotas va podria resultar de un efecto de dosaje genético.demanera tal que estos ratones tienen menos copias del gen estrugtural de ALA-D.Para este mecanismo se requeriría que la velocidad de sintesis de enzima esté limitada por el número de copiasdel gen estructural. Esta hipótesis explicaría que los nivelesde actividad encontrados para los genotipos Lva, ch y va esténen la relación 3:2:l. Otra alternativa seria que el locus levulinato fuera un elemento controlante localizado muy próximo al locus genético estructural que regule algunos aspectos de la trascripción o traducción del gen estructural, comoocurre con latalasemia humana (Coleman, l97l).

132
Se habia observado, además, que 1a actividad de ALA-Den higado de ratones es 5 veces más aita que en bazo aunque amrbas tienen idénticas propiedades. Sin embargo, e] tratamientode 10s ratones con feniihidrazina aumenta 1a actividad enzimática en bazo, pero no en higado, 1o que sugiere diferentes mecanismos de contro] para 1a sintesis de ALA-Den tejidos hematopoyéticos y no hematopoyéticos (Coïeman, 1966). Se postuió que un gendenominadof/f que causa anemia siderocitica transitoria en ratones recién nacidos, controia 1a actividad de ALA-Den tejidoshematopoyéticos (Margoiis y Russei, 1965),puesto que en estos gnimaies 1a actividad de ALA-Dde bazo no es inducida por feniihidrazinas. Sin embargo, resuitados posteriores demostraron que1a inducciónaocurre pero retrasada en tres o cuatro dias respegto de los ratones normaies (Coieman, 1966). Finaimente se demostró que, contrariamente a 10 que se habia sugerido en un principio, Ios genes Lv y f no están ligados uno a otro, aceptánddseque ios niveies de ALA-Dsóio están controiados por e] gen Lv ye] gen f actúa probabiementecontroiando 1a veiocidad de proiifgración de ias céiulas hematopoyéticas (Coieman y c01., 1969).
Los avances en e] campo de 1a genética en estos.ü1timos años permitieron aisiar varios ciones de] cDNAde] ALA-Dusando un anticuerpo poiicionaï de conejo desarroiiado contra 1asubunidad de 35 kDa purificada a partir de higado de rata. Lahibridización de] cDNAde ALAïDa1 DNAgenómico de ratón indicaque ias diferencias encontradas en 1a actividad enzimática de]ALA-Den cepas de ratones endocriados, proviene de aiteracionesen 1a dosis génica de] ALA-D(Bishop y co]., 1986 a).
Estos autores suponen que ias regiones genéticas de]ALA-Dhan permanecido invariabies, saivo para 1a dosis, o bienque 1a homogeneización genética de] ALA-Dy 1a generación delas diferencias,han ocurrido comparativamente en forma reciente.La inferencia en cuaiquiera de estos dos casos, es que ios genes de ALA-Dde ratón han estado sujetos a una reciente seiección intensiva.
Una comparación de 1a región codificante de] ALA-Ddehigado de rata (Bishop y c01., 1986 b) con 1a de] ALA-Dhumana(Wetmury co1., 1986), muestra un aito porcentaje de homoïogïa(88 %).
Por otra parte, en humanos, Sassa y co]. (1973) hanreportado que 1a actividad en eritrocitos maduros normaies entre

133
un rango de variación bastante amp1io (de 1 a 4 veces).
Battistuzzi y co}. (1981) hicieron un estudio en unapooiación itaiiana,y mediante técnicas eiectroforéticas encontraron tres.fenotipos: una, mayoritaria, a la que denominaronALA-D], otra con una moviiidad menors que e] ALA-D], a 1a quedenominaron ALA-Dz,y una tercera caracterizada por una bandacon una moviiidad intermedia entre 1as dos citadas, a 1a que
denominaron ALA-Dz_]. Estos tres fenotipos se deben a 1a existencia de dos aleios codominantes en un Iocus autosómico, Iosque representarian los dos homocigotas y 1a combinación hetero
cigota de ios aïeios ALA-D1y ALA-D2respectivamente.
Aunque estos autores demostraron 1a presencia de un pgiimorfismo estructura] en e] iocus ALA-D],e110 no contribuiriaa 1a variación cuantitativa tOta], ya que ios tres fenotipos pgseen 1a misma actividad enzimática, 10 que sin embargo, no excluye 1a posibiïidad de 1a existencia de un polimorfismo de unlocus reguiatorio ta] comoe] descripto por Coieman en ratón(Coleman, 1971).
Doss y co]. (1979) presentaron e] caso de dos pacientes no reiacionados cón PAI, que expresaban una deficiencia genética en 1a actividad de] ALA-Dpor debajo de] 1 % de los vaigres normaies, sugiriendo que.este ma] era heredado con rasgosautosómicos recesivos, aunque hubo un caso heterozigota reportgdo por Bird y co]. (1979) y, más recientemente, otro descriptopor Thuneii y co]. (1987).
Para tener mayor información acerca de 1a genética de]ALA-D, Beaumont y co]. (1984) estudiaron 1a 10ca1ización cromosómica de 1a enzima_mediante un método inmunoenzimáico no compgtitivo especifico para ALA-Dhumana, investigando 1a presenciade 1a enzima humanaen iineas ce1u1ares hibridas rodentario-humano, conteniendo varias combinaciones de cromosomas humanos.Mediante esta técnica asignaron e] gen de] ALA-Da1 cromosoma 9.
Estos resuïtados están de acuerdo con los anáïisis deentrecruzamiento reaiizado por Eiberg y co]. (1983), quienes usgron 1a variabiiidad aiéiica de] ALA-Dpara 11evar a cabo un aníIisis de entrecruzamiento en 846 familias danesas, conciuyendoque 1a codificación genética de 1a enzima debe asignarse- a1 crgmosoma 9.
Esta asignación fue confirmada también en ios

134
iaboratorios de Desnick (Hang y coi., 1985), quienes usaron tégnicas de hibridización ceiuiar con híbridos de céiuias de eritroieucemia murina y fibrobiastos humano, en ios que indujeronia diferenciación eritroidea mediante dimetiisuifóxido (DMSO)ydetectaron ALA-Dhumana mediante ensayos de inmunodiscriminaciónusando anticuerpos poiicionaies de ratón contra ALA-Dhumana.
pMás recientemente fueron identificados c ciones de cDNAque codifican para ALA-Dhumana, ios que fueron recionados en
ei bacteriofago M13y subsecuentemente secuenciados (Wetmurycoi., i986).
Uno de ios ciones denominados pEX de 827 pb contienesecuencias de DNAcoiineaiescon cuatro secuencias peptidicas deALA-Dbovina. Ei segundo cion con un inserto de 1.200 pb contigne todos ios aminoácidos que codifican ia información para eiALA-Dhumana, inciuyendo ei N-terminai. Las secuencias nucieotidicas de ios dos ciones de cDNAdifieren en una posición quecodifica dos aminoácidos diferentemente cargados. Esta diferencia de nucieótidos podria ser ia base de ias isoenzimas poiimórficas en carga. Hay también una secuencia rica en cisteinaehistidina, que podria ser ei sitio de unión para ei zinc, y unaregión poco usuai de cargas compiementarias que rodearia ei residuo activo de iisina en ei sitio cataiitico.
Además dei ALA-D,otros tres genes que codifican iasenzimas dei camino dei hemo han sido asignadas a cromosomas especificos o regiones cromosomaies. Asi, ei gen que codifica degminasa se ha iocaiizado en ia región cromosómica iiq23 iiqter(Hangy coi., 1981), y ios genes estructuraies para decarboxiigsa y coprogenasa han sido provisionaimente asignados ai cromosgma i (de Verneuii y coi., 1984) y ai cromosoma 9 (Grandchamp ycoi., i983) respectivamente. Es interesante notar que estos 4genes se iocaiizan en tres diferentes cromosomas: i, 9 y ii. Esdecir, que ios genes biosintéticos de hemo en humanos no estánorganizados en un soio cromosomapara una reguiación coordinada;por ei contrario, ia reguiación de ia biosfntesis de hemohumano requiere uno o varios factores que pueden efectuar ia sintesis de mRNA,ios cuaies se transcriben de por io menos 3 cromosomas diferentes.

135
111.10. _¿g¿_figg¿¿
Berïin y Schimke (1965) propusieron que 1as enzimas queiimitan 1a velocidad de una secuencia bioquímica especifica deben tener un turnover rápido para adaptarse rápidamente a ios requerimientos ce1u1ares. E1 ALA-S, que es 1a enzima que controia1a veiocidad de] camino de] hemo, tiene una vida media corta yestá presente en muy bajas concentraciones (Granick, 1967) cumpliendo asi con Ios requerimientos citados. Pero esto aparentemente no se cump1e con e] ALA-Dhepática de ratón, que segúnboyie y Schimke (1969) tendria una vida media de 5-6 dias. Sinembargo, Granick y Mauzeraïï (1958) reportaron que 1a vida media de] ALA-Dde eritrocitos de poiio y reticuiociots de conejos es de 15 minutos y una hora respectivamente.
111.11. CAMINO METABOLICO DEL ALA
Las etapas iniciaïes o aquélias que constituyen puntosde ramificación de las cadenas metabóïicas son,en genera], centros de reguiación de] camino. Shemin (1954) demostró que 1asporfirinas no son 105 únicos productos metabóïicos de] ALA,puesa partir de é] se pueden formar también e] DOVA,e] cua] viaa-cetogiutarato y succinato puede entrar a1 ciclo de] ácido citrico y a través de Ios carbonos uno contribuir a 1a sintesisde purinas.
E1 ALA-Dse encontraría asi en un punto de ramificación,y podria regular e] destino de] ALAen e] metabolismo intermediopara su uso en 1a sintesis de] hemo o de ias purinas.
Básicamente, e] cicïo de Shemin es una via de degradación de 1a giicina que propone caminos metabóïicos aïternativospara disponer de] exceso de ALAque no se necesita para 1a sintesis de tetrapirroïes, o que no puede consumirse para esos fines cuando e] ALA-Destá bioqueada.
Un 30 % de] ALA formado entra en e] ciclo de Shemin, yun 70 % se empïea para 1a sintesis de porfirinas (Berïin y c01.,1956). La dirección de] ALAhacia uno u otro de ios caminos metabólicos posibïes está controïada por 1a reïación de actividadesde 1as dos enzimas que gobiernan ambas reacciones.
De todos modos, ias evidencias parecen indicar que e]

ALA-Dno tiene una función regulatoria en e] proceso de sintesis de hemo.
111.12. ROL DEL ALA-D EN LA BIOSINTESIS DEL HEMO
E1 ALA-Dno es en genera] una enzima 1imitante de 1asintesis de hemo. La actividad de esta enzima en 1a mayoria de105 tejidos es normaimente mucho más aïta que 1a de ALA-S (unas80 veces en e] higado normaï) (Meyer y Schmid, 1978). De hecho,es muy probabie que ALA-Dsea 1a enzima cuya actividad y cantidad se encuentran en gran exceso comparadas con cuaiquiera de1as otras enzimas de este camino biosintético, por 10 que esnecesaria una profunda inhibición para que afecte significativamente 1a producción de hemo (Baron y Tephiy, 1969; Ebert y c01.,1979).
Que 1a relación ALA-D/ALA-Ses normaïmente a1ta podriadeberse a] hecho de que e] ALAformado en 1a mitocondria debedifundir a1 citoplasma, di1uyéndose en un voiumen ceiuiar mayor,siendo por eiio necesario e1evados niveïes de ALA-Dpara asegurar una máxima conversión de] ALA formado. Esta reïación estáinvertida, sin embargo, en cé1u1as de S. ¿tenóoniá (Ho yLascelies, 1971), pero aún en ese caso 1a dehidrasa estaria prgsente en cantidades no Iimitantes y no tendria un ro] reguiatorio.
En cambio 1evaduras y N. cnaaóa presentan 1a menor actividad de ALA-Dcomparada con 1a de otros organismos, sugiriégdose que en estos organismos 1a enzima tiene un ro] fundamenta]en 1a reguiación de 1a biosintesis de tetrapirroies (Barreiro,1967; Nuthukrishnan y c01., 1972).
También en PaopLoanactenLum Ahenmanáá y E. gnac¿t¿4(Menon y Shemin, 1967; Ebon y Tait, 1969; Ste11a, i977) se hapropuesto que ALA-Dposee un ro] reguïatorio. En E. gnacilió,tanto copro III como proto IX y hemina y los correspondientesporfirinógenos 1a inhiben,siendo su efecto mayor ¿n thno(Steiïa, 1977).
En Rp. ¿phenoLde4, por sus caracteristicas alostéricasy su inhibición por proto y hemina, Nandi y co]. (1968) 1a propusieron como un segundo punto de regulación en 1a sintesis deporfirinas.

137
III.]3. ALA-D Y PORFIRIAS
En genera], se ha visto que en 1as diferentes formas deporfirias humanas hereditarias, e] ALA-Dno tiene un ro] importante. En 1a mayoria de ios casos, e] defecto enzimático primario, se locaiiza a nive1 de otras enzimas de] camino de] hemo,y ios cambios en 1a actividad de ALA-Dson secundarios.
Asi por ejemp10,en pacientes con coproporfiria hereditaria, se encontró que durante ios ataques agudos e] ALA-Den gritrocitos dupiicaba su actividad (Kaufmany Marver, 1970). Enesas condiciones, también hay un aumento de 1a excreción urinaria de ALAy PBG, por 10 que es probabie quee] incremento en 1aactividad de ALA-Dse deba a una mayor concentración de sustrato, y no tuviera una reiación directa con 1a patogenia de 1a enfermedad (Mc Intyre y.c01.,11971).
Se ha encontrado, sin embargo, una significativa a1teración de 1a actividad de esta enzima en aigunas intoxicacionesy condiciones genéticas.
111.14. ALGUNAS PATOLOGIAS EN LAS CUALES EL ALA-D SE ENCUENTRA
AFECTAgA
III.14.1. Intoxicación por piomo
Las primeras evidencias acerca de un efecto inhibitorio directo de] piomo sobre e] ALA-D¿n vátno se debieron aGibson y co]. (1955) y Dresse] y Faik (1956). Unos años más tagde, Lichtman y Feidman (1963) demostraron que 1a actividad enzimática en eritrocitos de pacientes intoxicados por piomo, estaba disminuida.
Se ha encontrado que para un ampiio rango de concentración de piomo en sangre existe una correiación negativa entre 1aactividad de ALA-Dy ios niveïes de plomo (Nakao y co]., 1968;Hernberg y Nikkanen, 1970), de manera ta] que se utiiiza e]ALA-Deritrocitaria como un indice sensible para medir 1a exposición a pïomo ambienta] e industria].
Elpiomo ejerce primariamente una inhibición no competitiva sobre e] ALA-D(Wiison y co]., 1972; Granick y co]., 1973;Anderson y Desnick, 1979; Gibbs y co]., 1985 a) aunque según o

138
tros autores, seria de tipo competitivo (Fujita y col., l936).
En general, se ha intentado explicar el mecanismo de inhibición por plomo en base a interacciones del metal con la enzima, ya sea por un cambio estructura] a nivel de sintesis enzimática (bonsignore y col., l968), o por inactivación de grupos tioles esenciales (Gibson y col., l955; Granick y Mauzerall, l958)mediante la formación de una unión mercáptida (Tsukamoto y col.,l979). Baxter y col. (1979) en base a evidencias halladas utilizando 13C-NMR,se inclinaron por considerar que hay wnainteraccióndel plomo con el sustrato, formando posiblemente un complejo te:nario con la enzima. Hellmutt vBeyersmann (l983) encontraronque el sustrato protege al ALA-Ude la inhibición por plomo,concluyendo que el plomo ataca directamente el sitio activo dela enzima.
Mediante técnicas de radioinmunoensayo, Fujita y col.(l931, l982) determinaron que en las intoxicaciones por plomohay un aumento en la cantidad de ALA-Deritrocitaria, probablemente como respuesta a un aumento en la sintesis de la enzimaen las células de la médula ósea durante la exposición al metal.
La actividad disminuida de ALA-Dpuede recuperarse ¿nv¿tio por tratamiento con GSH(Lichtman y Feldman, 1963), cisteina (Heilmeyer, l966), DTT(Granick y col., 1973); zinc(Finelli y col., l975), zinc y GSH(Mitchell y col., l977),zinc y DTT(Sakai y col., l980, l98l), calentamiento de los hemolizados (Bonsignore y col., 1968; Ushio y col., 1975; Chiba,1976). Resultados todos que sugieren que el plomo ejerce su efecto inhibitorio combinándose con la enzima, y que la eliminación del metal mediante ciertos tratamientos restaura la actividad original.
III.l4.2. AlcoholismoEl etanol produce varios efectos sobre el camino meta
bólico del hemo; entre ellos, aumenta la actividad de ALA-S(Shanley y col., l968) y disminuye la de ALA-D(Moore y col.,l97l). Tambiénes sensible al abuso de alcoholla decarboxilasa(Elder y col., l978) y en la cirrosis alcohólica se encontró disminuida la actividad de la ferroquelatasa (Moore, 1973) asociada a un aumento de protoporfirina eritrocitaria (Kautzsh, 1978).
La actividad de ALA-Des afectada por los niveles de

tioies no proteicos, y se ha propuesto que e] etanol produciriacambios en 1a reiación NADH/NAD+que a su vez incrementan 1asconcentraciones de tioies no proteicos inhibiendo asi a 1a enzima (Moore y c01., 1971).
En ratones, el etanoï produce una disminución de] GSHhepático (Mc Donaid y co]., 1977), hecho que se atribuyó a laformación de acetaidehido, e] cua] es detoxificado por e] GSH(Viña y co]., 198 ). Otros autores sugieren que 1a depieciónde GSh se deberia a 1a oxidación de éste por peróxidos iipidicos producidos por e] etano] (Videia y Vaienzueïa, 1982) o quela acción de] etanol produce un escape de GSHy una inhibiciónde 1a sintesis de] tio] (Speisky y co]., 1985).
Recientemente, se ha demostrado que en ias intoxicacignes agudas por etano], e] contenido de GSHy 1a actividad deALA-Destán reducidas significativamente, y hay evidencias deque e] efecto tóxico de] alcohoi se debe a Su conversión a acetaidehido, que seria e] verdadero inhibidor de] ALA-D(Paredesy c01., 1987).
III.14.3. Tirosinemia hereditaria
Es una enfermedad metabóiica que se produce a raiz deuna faïla enzimática en e] metabolismo de 1a tirosina, debido auna deficiencia en 1a 4-fumari1-acetoacetato-hidr01asa (Lindbiady c01., 1977) y que se trasmite en forma autosómica recesiva(La Du y Gjessing, 1978).
Se caracteriza por tener sintomas neuroiógicos agudossimiiares a ios de una PAI (Gentz y coi., 1969; Kang y Geraid,1970; Strife y c01., 1977). Por esta razón se examinaron ios niveïes de precursores de porfirinas ysehalió que 1a excreción dePBGy porfirinas es norma], pero Ios niveïes de ALAson e1evados (Gentz y co]., 1969; Gauii y c01., 1970; Kang y Gerald, 1970).Se encontró que tanto durante los periodos de remisión como durante Ios ataques agudos, 1a actividad de ALA-Dde estos paciegtes estaba marcadamente disminuida tanto en higado como en g]ó—buios rojos (Lindblad y c01., 1977; Strife y co]., 1977). Sedemostró que e110 se debia a 1a acumuïación en piasma de un derivado de 1a tirosina, e] ácido 4,6-dioxoheptanoico o Succiniiacetona, e] cua] produce un bioqueo a nive] del ALA-D,y con eilo, acumuiación de ALA(Lindbiad y co]., 1977; Christensen yco]., 1981).

140
Se ha demostrado la inhibición ¿n vátno de la succinilacetona sobre el ALA-Sen células de eritroleucemia murina(Ebert y col., 1979), higado de rata (Tschudy y col., l981), higado bovino y de ratón, eritrocitos humanosy cultivo de hepatgcitos de ave (Sassa y Kappas, l983), asi c0mo su acción ¿n v¿vcen ratas (Tschudy y col., 198l).
La succinil acetona produce una inhibición irreversible (Tschudy y col., l981) y de naturaleza competitiva (GrummyFriedmann, l981; Sassa y Kappas, l983), y debido a su estructura similar a la de ALA,se sugiere que actúa en el sitio cataljtico de la enzima (Tschudy y col., 198l; Sassa y Kappas, l983),habiéndose reportado que forma un pirrol con el ALAa través deuna condensación de Knorr (Brummy Friedmann, l98l).
III.l4.4. Nuevaporfiria aguda
En l979, Doss y col. reportaron dos casos de una nuevaforma hereditaria de porfiria que se caracteriza por una disminución del ALA-D, aumento de la excreción urinaria de ALAy sintomas clinicos neurológicos tipicos de una PAI, por lo que sela denominó nueva porfiria aguda (NPA) (Doss y col., l979, l980).
En esta porfiria, la actividad del ALA-Deritrocitariay de médula ósea se encuentra severamente afectada, ya que esde sólo un l3 % respecto de los valores normales de individuosno expuestos a plomo (Doss y col., 1983). Se vió que en los padres de estos pacientes la actividad de ALA-Destaba disminuidaun 50 % respecto de la de sujetos normales, por lo que se infirió que los enfermos son homocigotas y que por lo tanto el gendefectuoso se hereda en forma autosómica recesiva (Doss y col.,1982 a).
Bird y col. (l979) estudiaron los individuos de una familia norteamericana, y observaron que, a pesar de tener disminuida la actividad de ALA-Dentre un 22 y 45 %, no presentabanlos signos característicos de una porfiria aguda, e indicaronque la deficiencia se hereda en forma autosómica dominante.Pocodespués, Doss y col. (l982 b) estudiaron una familia alemana quetambién presentaba una deficiencia enzimática heredada en formaautosómica dominante.Sin embargo, esto no excluye la posibilidadde que la enfermedad metabólica sólo se pueda expresar en condiciones genéticas recesivas.

141
A través de estudios cromatográficos y eiectroforéticos,Brandt y Doss (1981) sugirieron que 1a actividad deficiente de]ALA-Den 1a NPApodria deberse a pequeñas aiteraciones en 1a estructura primaria proteica.
Y más recientemente, Thune11 y co]., (1987) describieron e1 caso de una famiiia con deficiencia en ALA-D,heredadaa 10 iargo de tres generaciones en una forma autosómica dominante, que en un estado heterocigota pareciera no tener significado patoiógico, pero que en 1a forma homocigota conduce a severos sintomas porfiricos y a1 iguai que en e] caso reportado porDoss (Brand y Doss, 1981), 1a manifestación clinica sigue e] esquema genera] de un ma] autosómico recesivo.
III.14.5. Otras
Ademásde 1a intoxicación por plomo, aicoholismo, tirosinemia y NPA, e] ALA-Dse encuentra disminuida en otras condicionestóxicas comoser intoxicación por nicotina (Saiie yZieihuis, 1977), por derivados de petróieo (Rao y Pandya, 1980),tricioroetiieno (Fujita y c01., 1984), y estireno (Fujita y c01.,1986).

142
III.15. REFERENCIAS
- Abduïïa, M. & Haeger-Aronsen, B. (1971) Enzyme 1g, 703.
- Abdulla, M. & haeger-Aronsen, B. (1973) IRCS Int. Res. Commun.Sys. (73-8) 8-]4-1.
- Abdulïa, M.; Svensson, S.; Haeger-Aronsen, B.; Mathur, A. &Haïlenius, K. (1978) Enzyme gg, 170.
- Akhtar, M.; Abboud, M.M.; Barnard, G.; Jordan, P.M. & Zaman,Z. (1976) Philos Trans. R. Soc. London Ser. B. EZ}, 117.
- A1vares, A.P.; Leigh, S.; Cohn, J. & Kappas, A. (1972)J. Exp. Med. 135, 1406.
- Anderson, P.M. & Desnick, R.J. (1979) J. Bío]. Chem. 254,6924.
- Anderson, K.E.; Sassa, S.; Peterson, C.N. & Kappas, A.(1977) Am. J. Med. gg, 359.
- Barnard, G.F.; Itoh, R.; Hohberger, L.H. & Shemin,D. (1977)J. Bio]. Chem. 252, 8965.
- Baron, J. & Tephïy, T.R. (1969) Molecuï. Pharmaco]. g, 10.
Barreiro, 0.L.C. de (1967) Biochím. Biophys. Acta 1 9, 479.
Barreiro, 0.L.C. de (1969) Biochim. Biophys. Acta 178, 412.
Batlïe,0i, 861.Bat11e,Biochem.
Batlïe,Xifra,
Bat11e,Hider de
g, 401.BattistíMoore,
BattistuCaioïa,
Baxter,Pharmaco
E.
M.R.
A.M. de] C. & Ste11a, A.M. (1978)
A.M.J.
de] C.;gi, 80.
Ferramoïa, A M.& Grínste
A.M.
A.
de] 6.; Tigíer, H.A.; L1ambías,
A.M.
Xifra,de] C.;
E.A.Ste11a, A.M.;8 Sancovich,
Ferramoïa,E.A. (1978)
J.J.A.
ni, U.; Morrow,& Goïdberg,
; Ginsburg, D.;(197]) Brit. J.
zi,G. ;S.
Pettrucci, R.; Silvagni, L.;(1981) Ann. Hum. Genet. gg, 233.
Hey, H.E. A.D.477.
C.S.; & Cardin,ï. 47,
Int. J.
in,
E.B.
A.“
M.
.; Sopena,Int.
Thompson,Haemato].
Biochem.
(1967)
& Nider de
(1975) Anaïes Asoc. Quim. Arg. gg, 305.
J. Biochem.
G.;20_, 177.
Urbani,F.R.&
(1979) Toxico]. App].
Y.‘i

143
Beaumont, C.;V.C.;153.
Foubert, C.;M.S.
Grandchamp, B.; Wei], D.; Nguyen,Gross, & Nordmann, Y. (1984) Ann. Hum. Genet. gg,
Berïin, C.M. & Schímke, R.T. (1965) Mo]. Pharmaco]. l, 149.
Berïin, N.I.; Neuberger, A. & Scott, J.J. (1956) Biochem. J.gg, so.
Bevan, D.R.; Bodlaender, P. & Shemín, D. (1980) J. Bio]. Chem.255, ¿030.
Beyersmann, D. & Cox, M. (1984) Biochim. Biophys. Acta 155,162.
Bird, T.; Hamernyík, P.; Nutter, J. & Labbe, R. (1979) Am. J.Hum. Genet. 31, 662.
Bishop, T.R.; Coehn,P.J¿ Boyer, S.H.; Noyes,A.N, & Fre]ín,L.P.(1986 a) Prcc. Nat]. Acad. Sci. USAgg, 5368.
Bishop, T.R.; Frelin, L.P. & Boyer, S.H. (1986 b) Nuc]. Ac.Res. lg, 24.
Bogorad, L. & Granick, S. (1953) Proc. Nat1.Acad. Sci. U.S.A.gg, 1176.
Bonsignore, D.; Cartasegna, C.; & Ardoino, V.(1968)Med. Lavoron, ¿2, 419.
Vergnano, C.
Border, E.A.; Cantreïl, A.C. & Kiïroe-Smith, T.A. (1976 a)Environm. Res. ll, 319.
Border, E.A.; Cantrell, A.C. & Kíïroeásnith,T.A. (1976 b)Br. J. Ind. Med. gg, 35.
Brandt, A. & Doss, M. (1981) Hum. Genet. gg, 194.Brumm, P.J. & Friedmann, H.C. (1981) Biochem. Biophys. Res.Commun. 102, 854.
Burnham, B.F. & Lasceïïes, J. (1963) Biochem. J. gl, 462.
Calissanc,P.; Cartasegna, C.; Bonsígnore, D. (1965) Lav. Um.11, 493.
Calissano, P.; Bonsígnore, D. & Cartasegna, C. (1966 a)Biochem. J. 101, 550.
Caïissano, P.; Cartasegna, C. & Mattein, M. (1966 b) Ita]. J.Biochem. lá, 18.
Cantreï], A.C.; Kíïroe-Smith, T.A.; Simoes, w.M.; Border, E.A. (1977) Br. J. Ind. Med. gg. 110.

Careii, E.F. & Kahn, J.S. (1964) Arch. Biochem. Biophys. 108,i.
Coieman, D.L. (1966) J. Bio]. Chem. gil, 5311.
Coieman, D.L. (1971) Science 11;, 1245.
Coleman, D.L.; Russe11, E.S. & Levin, E.Y. (1969) GeneticsEl. 631.
Coiiier, H.B. (1971) Ciin. Biochem. í, 222.
Cookson, G.H. (1953) Nature 1 1, 875.& Rimington, C.
Chang, C.S.; Sassa, S. & Doyie, D.A.Acta 1g, 297.
(184) Biochim. Biophys.
Chandrika, S.R.; Chandra Kumar, C. & Padmanaban, G. (1980)
Biochim. Biophys. Acta 607, 331. NEhazdry, A.G. & Jordan, P.M. (1976) Biochem. Soc. Trans._í,‘760. Í
Chaudry, A.G.; Gore, M.G. & Jordan, P.M. (1976) Biochem. Soc.Trans. í, 301.
Cheh, A. & Neilands, J.B. (1973) Biochem. Biophys. Res. Commun.52,1050.Chiba, M. (1976) Br. J. Ind. Med. gg, 36.
Christensen, E.; Jacobsen. B.B.; Gregersen, N.; Hjeds, H.;Pedersen, J.B.; Brandt, N.V. & Baekmark, V.B. (1981) Ciin.Chim. Acta 116, 331.
Davis, J.R. & Avram, M.J. (1978) Toxico]. App]. Pharmacoi.55,181.Davis, J.R. & Avram, M.J. (1980) Toxico]. App]. Pharmaco]. gg,281.
Doss, M.; von Tiepermann, R.; Schneider, J. & Schmid, H.(1979)Kiin. Nochenschr. El, 1123.
Doss, H.; von Tiepermann, R. a Schneider, J. (1980) Int. J.Bicohem. 1g, 823.
Doss, M.; Schneider, J., von Tiepermann, R. & Brandt, A.(1982 a) Ciin. Biochem. lá, 52.
Doss, M.; Becker, U.; Sixe], F.; Geisse, S.; Soicher, H. &Schneider, J.; Kufner, G.; Schiege], H. & Stoeppier, M. (1982 b)Klin. Nochenschr. gg, 599.

145
Doss, M.; von Tiepermann, R. & Schneider, J. (1983) Kiin.Nochenschr. gl, 699.
Doyïe, D. (1971) J. Bio]. Chem. g_g, 5449.
Doyïe, D. & Schimke, R.T. (1969) J. Bio]. Chem. ¿_fi, 5449.
Drese], E.I.B. & Falk, J.E. (1953) Nature 11¿, 1185.
Drese], E.I.B. & Falk, J.E. (1956) Biochem. J. 9;, 80.
Dresner, D.L.; Ibrahin, N.G.; Mascarenhas, B.R. & Levere, R.D.(1982) Env. Res. gg, 55.
Ebbon, J.G. & Tait, G.H. (1969) Biochem. J. lll, 573.
Ebert, P.S.; Hess, R.A.; Frykhoim, B.C. & Tschudy, D.P. (1979)Biochem. Biophys. Res. Commun. gg, 1382.
Eiberg, H.; Mohr, J.; Nielson, L.S. (1983) Ciin. Genet. gg,150.
Eïder, G.H.; Lee, G.H. & Tovey, J.A. (1978) New Eng. J. Med.299, 274.
Fa1k, J.E.; Drese], E.I.B. & Rimington, C. (1953) Nature 1 2,292.
Falk, J.E.; Drese], E.I.B.; Benson, A. & Knight, B.C. (1956)Biochem. J. gg, 87.
Finelli, V.N. ; Murthy, L.; Peirano, H.G. & Petering, H.G.(1974) Biochem. Biophys. Res. Comm.gg, 1418,
Finelii, V.N.; Klauder, D.S.; Karaffa, M.A.(1975) Biochem. Biophys. Res. Comm.Q5, 303.
& Petering, H.G.
Freshney, R.I. & Pau], J. (1971) J. Embryo]. Exp. Morph. gg,313.
Fujita, H.; Orii, Y. & Sano, S. (1981) Biochim. Biophys. Actam, 39.Fujita, H.; Sato, K. & Sano, S. (1982) Int. Arch. Occup.Environ. Heaïth gg, 287.
Fujita, H.; KOÍZUmÍ, A.; Yamamoto, M.; Kumai, M.; Sadamoto, T.& Ikeda, M. (1984) Biochim. Biophys. Acta QQQ, 1.
Fujita, H.; Yamamoto, R.; Sato, K. & Ikeda, M.(1985) Toxico].App]. Pharmaco]. ZZ, 66.
Fujita, H.; KOÍzumi, A.; Hayashi, N.Biochim. Biophys. Acta 867, 89.
& Ikeda, M. (1986)

146
Gajdos, A. & Gajdos-Torok, M. (1955) C.R. Soc. Bio]., Paris152, 2138.
Gajdos, A. a Gajdos-Torok, M. (1956) Rev. franc. Etud. Clin.Bio]. l, 966.
Gauii, G.E.; Rassin, D.K.; Soiomon, G.E.; Harris, R.C. &Sturman, J.A. (1970) Pediatr. Res. í, 337.
Gentz, J.; Johansson, S.;Lindb1ad, B.; Lindstedt, S. &Zetterstrom, R. (i969) Clin. Chem. Acta gg, ¿57.
Gibbs, P.N.B.& Jordan,P.M. (1981) Biochem. Soc. Trans. g, 232.
Gibbs, P.N.B. & Jordan, P.M. (1986) Biochem. J. ¿19, 447.I
Gibbs, P.N.B.; Chaudry, A.G. &Jordan, P.M. (1985 a) Biochem.J. ¿39, 25.
Gibbs, P.N.B.; Gore, M.G. & Jordan, P.M. (1985 b) Biochem. J.¿25, 573.
Gibson, K.D.; Neuberger, A. & Scott, S. (1954) J. Biochem.Soc. (Biochem. J.) gg, XLI.
Gibson, K.D.; Neuberger, A. & Scott, J.J. (1955) Biochem. J.gl, 618.
Gore, M.G.; Jordan, P.M. & Chaudry, A.G. (1976) Biochem. Soc.Trans. í, 762.
Granick, S (1954) Science 13g, 1105.
Granick, S (196]) Piant Physio]. Supp. gg, XIVIII.
Granick, S (1966) J. Bio]. Chem. 241, 1359.
Granick, S (1967)en Biochemistry of Chioropiasts (Ed. T.w.Goodwin). Academic Press, New York, p. 373.
Granick, S & Levere, R.D. (1964) Progress in Hemato]. fi, 1.
Granick, S. & Mauzeraii, D. (1958) J. Bio]. Chem. ggg, 1119.
Granick, S & Urata, G. (1963) J. Bio]. Chem. 235, 821.
Granick, J.L.; Sassa, S.; Granick, S.; Levere, R. & Kappas,A. (1973) Biochemica] Medicine g, 149.
Gurba, P.E.; Sennett, R.E. & Kobes, R.D. (1972) Arch. Biochem.Biophys. 150, 130.
Gurne, D. & Shemin, D. (1973) Science 180 , 1188.
Gurne, D.; Chen, J. & Shemin, D. (1977) Proc. Nat]. Acad. Sci.USA ¿3, 1383.

147
Haeger-Arünsen, B.; Abdu11a, M. & Fristedt, B.I. (1971) Arch.Environ. Health g_, 440.
Haeger-Aronsen, B.; Abdu11a, M.; Schütz, A. (1976) Arch.Environ. Heaïth 31, 215.
Hasnaín, S.S.; Hardeïl, E.M.; Garner, D.; Schlosser, M. &Beyersmann, D. (1985) Biochem. J. ggg, 625..
(1966) en Disturbances in HemeSynthesis. Thomas,111inois, p. 151.
Heiïmeyer, L.Springfieïd,
Heïlmutt, M. & Beyersmann, D. (1983) Enzyme gg, 260.
Hernberg, S. & Nikkanen, J. (1970) Lancet,1:63.
Ho,734.
Y.K. & Lasce11es, J. (1971) Arch. Bíochem. Biophys. 1 4,
Honberger, L.H. (1979) Tesis Doctora], Northwestern Univesity.
Hovenkamp-Obbema, R.; Moorman, A. & Stegwee, D. (1974) Z.Pfïanzenphysioï. 1g, 277.
Huault, C.; Bruyant, P. & Baïangé, A.P. (1984) Physio]. P1antg, 469.
HÜcke], D. & Beyersman, D. (1979) Ana]. Bíochem.. gl, 277.
Hutton, J.J. & Coïeman, D.L. (1969) Biochem. Genetics g, 517.
Iodice, A.A.; Richert, D.A. & Schuïman, M.P. (1958) Fed. Proc.ll, 248.
Iwanow, E.D. (1968) Folia Haem. gg, 233.
Jaffe, E.K. & Hanes, D. (1986) J. Bio]. Chem. g_l, 9348.
Jaffe, E.K. & Markham, G.D. (1937) Biochemistry gg, 4258.
Jaffe, E.K.; Saïowes, S.P.; Chen, N.T. & De Haven, P.A. (1984)J. Bio]. Chem. 259, 5032.
Jordan, P.M. & Gibbs, P.N.B. (1985) Biochem. J. 227, 1015.
Jordan, P. & Seehra, J. (1980) FEBSLetters 115, 283.
Jordan, P.M.; Gore, M.G. & Chaudry, A.G. (1976) Biochem. Soc.Trans. A, 762.
Kang, E.S. & Geraïd, P.S. (1970) J. Pediatr. ZZ, 397.
Kaufman, L. & Harver, H.S. (1970) New Eng]. J. Med. ggg, 954.
Kautzch, E. (1978) Acta Hepato-Gastroentero]. gg, 124.
Kennard, 0. (1953) Nature lll, 878.

Komai, H. & Neiïands, J.B. (1968) Arch. Biochem. Biophys. lg_,456.
Komai, H. & Neiïands, J.B. (1969) Biochim. Biophys. Acta 1 1,311.
Kondo, M.; Kajimoto, M.; Kimura, H.; Suzuki, T.; Sasaki,
A.; Niwa; M. & Urata, G. (1981) AVch. Biochem. Biophys. _gg,189.
Kreutzer, M.; Schmidt, M.; Stadïer, E. & Zeitïer, H.J. (1977)Hoppe Seyler's Z. Physio]. Chem. 358, 1081.
La Du, B.N. & Gjessing, L.R. (1978) en TheInherited Disease (Eds. J.B. Stanbury, J.B.Frederickson). Mc Graw-Hi1], NewYork, 4ta.
Metaboïic Basis ofNyngaarden y D.S.edición , p. 256.
Lartiïïot, S. &'Baron, C. (1967) Buïl. Soc. Chim. Bio]. gg, 81.
Lasce11es, J. (1959) Biochem. J. 1;, 50d.
Lasceiles, J. (1960) J. gen. Microbiol. ¿3, 487.
Lichtman, H.C. & Feïdman, F. (1963) J. Clin. Invest. 1g, 830.
Liedgens, H.; Lütz, C. & Schneider, H.A.N. (1983) Eur. J.Biochem. 135, 75.
Lindblad, B.S.; Lindstedt, S.; Steen, G. (1977) Proc. Nat].Acad. Sci. USA11, 4641.
Lingner, B. & Kleinschmidt, T. (1983) Z. Naturforsch. Sect.C. Bio. Sci. gg, 1059.
Maraiihaiïi, G.8. & Bhagwat, A.S. (1986) J. Biosci Z, 359.
Margoïis, F.L. & Russe11, E.S. (1965) Science 139, 496.
Marver, H.S.; Colïins, A.; Tschudy, D.P. & Rechcig], M. (1966)J. Bio]. Chem. gil, 4323.
Mauzeraïi, D. & Granick, S. (1958) J. Bio]. Chem. ggg, 1141.
Mc Donald, C.M.; Dow, J.& Moore, M.R. (1977) Biochem.Pharmaco]. _g, 1529.
Mc Intyre, N.; Pearson, A.J.G.; A11an, D.J.; Craske, S.; West,G.M.L.; Moore, M.R.; Beattie, A D.; Poxton, J. & Goïdberg, A.(1971) Lancet l, 560.
Henon, I.A. & Shemin, D. (1967) Arch. Biochem. Biophys. lgl,304.
Meredith, P.A.; Moore, M.R. & Goïdberg, A. (1974) Biochem.Soc. Trans. g, 1243.

149
Meyer, U.A. & Schmidt, R.I. (1978) en The Metaboïic Basis ofInherited Disease (Eds. J.B. Stanbury, J.Fredrickson). Mc Graw H111 Inc.,
Nyngaarden y D.NewYork, 4ta. edición, p. 1166.
Mitcheïï, R.A.; Drake, J.E.; Nittïin,1977) Clin. Chem. Acta ¿3, 105.
L.A. & Regent, T.A.
Moore, M.R. (1973) Proc. First Int. Symposium on A1coho] andAldehidcfietabolizíng Systems. Stockhoïm, 21.
Moore, M.R. (1980) Cïín. in Haemato]. g, 227.
Moore, M.R.; Beattie, A.D.;(1971) Cïin. Sci. gg, 81.
Thompson, G.G. & Goïdberg, A.
Muthukrishnan, S.; Padmanaban, G.Bio]. Chem. 244, 4241.
& Sarma, P.S. (1969) J.
Muthukríshnan, S.; Na1athi, K.& Padmanaban, G. (1972) Biochem.J. 132. 31.
Nakao, K.; Nada, 0. & Kitamura, T. (1966) Nature ¿13, 838.
Nakao, K.; Hada, 0. & Yano, Y. (1968) Cïin. Chim. Acta lg,319.
Nandi, D.L. (1971) Arch. Biochem. Biophys. 15g, 157.
Nandi, D.L. & Shemin, D. (1968 a) J. Bío]. Chem. ¿53, 1231.
Nandi, D.L. & Shemin, D. (1968 b) J. Bio]. Chem. gig, 1236.
Nandi, D.L. & Shemín, D. (1973) Arch. Bíochem. Biophys. 158,303.
Nandi, D.L. & Waygood, E.R. (1967) Can. J. Biochem. 32, 327.
Nandi, D.L.; Baker-Cohen, K.F. & Shemin, D. (1968) J. Bío].Chem. 24 , 1224.
Narisawa, K. & Kikuchi, G. (1966) Biochim. Bíophys. Acta 123,396.
Neuberger, A. & Scott, J.J. (1953) Nature Lond. 170, 1093.
Neuberger, A.; Scott, J.J. á Shuster, L. (1956) Biochem. J.Q3. 137.
Onisawa, J. a Labbe, B. (1963) Biochím. Biophys. Acta gg, 618.
Paredes, s.R.; Kozicki, P.A.; Fukuda, H.; Rossetti, N.V. &Bat11e, A.M. de] C. (1987) A1c0h01 í, 81.
Rao, G.S. & Pandya, K.P. (1980) Arch. Toxico]. 6, 313.

150
Rebeiz, C.A. & Casteïfranco, P.A. (1971) P1ant Physio]. 31, 24.
Rimington, C. & Booij, H.L. (1957) J. Biochem. gg, 111.
Russeïl, R.L. & Coieman, D.L. (1963) Genetics gg, 1033.
Sakai, T.; Yanagihara, S. & Ushio, K. (1980) C1in. Chem. gg,625.
Sakai, T.; Yanagihara, S. & Ushio, K. (1981) Brit. J. Ind ïed.3_8, 268.
Sa11e, H.J.A. & Zielhuis, R.L. (1977) Int. Arch. Occup.Environ.Health gg, 111.
Sassa, S. & Bernstein, S.E. (1977) Proc. Nat]. Acad. Sci. USA15, 1181.
Sassa, S. ü Kappas, A. (1983) J. Ciin. Invest. ll, 625.
Sassa, S.; Granick, S.; Bickers, D.R.; Levere, R.S. & Kappas,A. (1973) Enzyme lg, 326.
Schmid, R. & Shemin, D. (1955 a) J. Am. Chem. Soc. ZZ, 506.
Schmid, R. & Shemin, D. (1955 b) J. Ciin. Invest. 21, 911.
Schneider, Hj. A.w. (1970) Zschr. Pfianzenphysio]. gg, 328.
Schneider, Hj. A.w. (1971) Phytochem. 1g, 319.
Schneider, Hj.A.w.(1975) Ber. Deutsch. Bot. Ges gg, 83.
Schuiman, M.P. (1955) Fed. Proc. li, 277.
Seehra, J.S. (1980) Tesis Doctora], University of Southampton
Seehra, J.S. & Jordan, P.M. (1981) Eur. J. Biochem. 11;, 435.
Seehra, J.; Gore, M.; Chaudry, A. & Jordan, P. (1981) Eur.J. Biochem. 111, 263.
Shaniey, B.C.; Zoi], S.S. & Joubert, S.M. (1968) Lancet l, 70.
Shemin, D. (1954) Harvey Lectures gg, 258.
Shemin, D. (1974) en The Enzymes, 3ra. edición (Ed. P.0. Boyer)Academic Press,New York, vo] lll p. 323.
Shemin, D. (197o) Ann. N.Y. Acad. Sci. gig, 348.
D. (1976) Phi]. Trans. R. Soc. London B. 109.Shemin, 273,
Shemin, D. ü Russe11, C.S. (1953) J. Am. Chem. 5,Soc. 4873.
Shemin, D.; Abramsky, T. C.S. Chem.1g, 1204.
a Russe11, (1954) J. Am.Soc.

Shemin, D.; RusseTT, C.S. & Abramsky, T. (1955) J. Bio]. Chem.¿12, 613.
Shetty, A.S. & MiTTer, G.w. (1969 a) Biochem. J. 115. 331.
Shetty, A.S. & HiTTer, G.N. (1969 b) Biochim. Biophys. Actalgfi, 458.
Shibata, H. & Ochiai, H. (1976) Plant ¿CeH PhysioTlÏl» 281.
Shibata, H. & Ochiai, H.(1977) PTant &CeTTPhysio]. lg, 421.
STuiters-SchoTten, C.M.T.; van der Berg, F.M. & Stegwee, D.(1973) Z. PfTanzenphysioT. 92, 217.
Smith, H.(1970) Phytochemistry g, 965.
Sommer,R. & Beyersmann,D. (1984) J. Inorg. Biochem. gg, 131.
Speisky, H.; Mac DonaTd, A.; GiTes, G.; Orrego, H.& Israe],Y. (1985) Biochem. J. 225, 565.
Stafforini, D.M.; Polo, C.F.; SteTTa, A.M.; Wider de Xifra,E.A. & BatTTe, A.M. de] C. (1980) Int. J. Biochem. 1g, 757.
Steer, B.T. & Gibbs, M. (T969) PTant Physio]. gg, 781.
Steiner, M.; BaTdini, M.Sci. 119, 548.
& Dameshek, w. (1964) Ann. N Y.Acad.
SteTTa, A.H. (1977) Tesis Doctora], UBA.
Stobart, A.K. & Thomas, D.R. (1968) Phytochemistry Z, 1313.
Strife, C.; Zuroweste, E.L.;Petering, H.G. & Berry, H.K.
Emmett, E.A.;(T977) J.
FineTTi, U.N.;Pediat. gg, 400.
Tamai, H.;gg, 435.
Shioï, Y. & Sasa, T. (1979) Plant &CeTT.Physio].
Tancioni, A.; Tigier, H.A.& Grinstein, M. (1964) Biochem.Pharm. lg, 1095.
Thompson, J.; Jones, D.D. & Beasïey, w.H. (1977) Br.J. Ind.Med. 25, 32.
ThuneTT, 5.; Holmberg, L. & Lundgren, J.(T987) J. Chem. CTin.Biochem. g_, 5.
Tigier, H.A.; BatTTe, A.M. de] C. & Locascio, G. (1968)Biochim. Biophys. Acta 151, 300.
Tigier, H.A.; BatTTe, A.M. de] C. & Locascio, G. (1970)EnzymoTogia gg, 43.

152
Tomio, J.M.; Tuzman, V. & Grinstein, M.(1968) Europ. J.Biochem. g, 84.
Tschudy, D.P.; Waxman, A. & Coïïin, A.(1967) Proc. Nat]. Acad.Sci. USAgg, 1944.
Tschudy, D.P.; F1ess, R.A. & Frykhoïm, C. (1981) J. Bio].Chem. 256, 991.5
Tsukamoto, I. (1980) Tesis Doctora], University of Kyoto.
Tsukamoto, 1.; Yoshinaga, T. & Sano, S. (1976) Biochem.Biophys. Res. Commun.gl, 294.
Tsukamoto, 1.; Yoshinaga, T.& Sano, S. (1979) Biochim.Biophys. Acta 519, 167.
Tsukamoto, 1.; Yoshinaga, T. & Sano, S. (1980)Int. J. Biochem.1g, 751.
Ushio, K.; Sakai; T.; Yanagihara, 5.; Watanabe, H. (1975).Jap.J. Ind. Heaith 11, 475.
Va11ee,B.L. & Ulmer, D.0. (1972) Ann. Rev. Biochem. gl, 91.
van Heyningen, S. & Shemin, D. (1970) Fed. Proc. gg, 937.
van Heyningen, S. & Shemin, D. (1971 a) Biochem. J. lgí, 68 P.
van Heyningen, S. & Shemin, D. (1971 b) Biochemistry 10, 4676.
Videia, L.A. & Va1enzue1a, A.(1982) Life Sci. gl, 2395.
Viña, J.; Estreïa, J.M.; Gerri, C. & Remero, F.J.(1980)Biochem. J. 188, 549.
Waïerych, N. (1963) Acta biochim. P01. 10,
Wang, A.L.; Astrin, K.H.; Anderson, w.F. & Desnick, R.J.(1985) Hum. genet. 19, 6.
Neissberg, B. á Voytek, A. (1974) Biochim. Biophys. Acta 364,304.
Westaïï, R.G. (1952) Nature 170, 614.
Netmur, J.G.(1986)
; Bishop, D.P.;Nat]. Acad.
Canteimo, C. & Desnick, R.J.Proc. Sci. USAgg, 7703.
Nigfield, D.C. & Farant, J.P. (1982) J. App]. Toxico]. g, 139.
Wiïson, M.L.; Iodice, A.A.;(1959) Fed. Proc. 1g, 352.
Schuïman, M.P. & Richert, D.A.
Wiïson, Burger, P.E. & Dowdïe, E.B. (1972) Eur. J.E.L.;Biochem. 29, 563.

153
- Wu, H.H.; Shemin, D.; Richards, K.E. & Níïïiams, R.C. (1974)Proc. Nat]. Acad. Sci. USAll, 1767.
- Yamamoto, M.; Fujita, H.; Watanabe, N.; Hayashi, N. & Kikuchí,G. (1986) Arch. Biochem. Biophys. gig, 76.
- Yamasaki, H. & Moriyama, T. (1971) Biochim. Biophys. Actaggz, 698.

CAPITULO IV
ZINC-ENZIMAS
IV. 3.
Elementos inorgánicos en sistemas biológicosIV.1.].IV.1.2.
Distribución y roies biológicosPropiedades de los iones metáiicos relevantes para 1a catáiisis
Metaioenzimas
IV.2.1. IntroducciónIV.2.2. Nomenciaturay clasificaciónIV.2.3. Zinc-enzimas
IV.2.3.]. Roles biológicos de] zincIV.2.3.2. Aislamiento de zinc-proteinasIV.2.3 3. Efecto de agentes queiantesIV.2 3.4. Propiedades de 1a unión de]
meta]IV.2.3.5 Propiedades espectroscópicasIV.2.3.6 E1 estado entáticoIV.2.3.7 Ligandos que unen zincIV.2.3.8 R01 de] zinc en ias metaloen
zimasIV.2.3.9. Mecanismo de 1as zinc-enzimas
Referencias
Página
154
154
155
156
156
158
159
159
160
160
161
161
162
162
163
165
169

154
IV. ZINC-ENZIMAS
IV.l. ELEMENTOS INORGANICOS EN SISTEMAS BIOLOGICOS
IV l.l. Distribución y roles biológicos
Los elementos presentes en mayor abundancia en los compuestos celulares sont carbono, oxigeno, hidrógeno y nitrógeno.El fósforo es otro elemento inorgánico que constituye un componente estructural mayoritario en los compuestos biológicamenteactivos (Ochiai, 1977).
El segundo grupo importante por su abundancia, comprende: sodio, potasio, magnesio, cloruro, calcio y azufre. Los cuatro primeros son los componentes principales de los fluidos biglógicos y del citoplasma. El sodio y el potasio. si bien similares en cuanto a su comportamiento quimico, son sin embargo difgrentes en cuanto a sus actividades biológicas,siendo hasta antagónicos en algunos aspectos. Ambosactúan en los mecanismos detrasmisión nerviosa, pero el potasio aumenta la respiración entejido muscular y la velocidad de la sintesis proteica, en tanto que el sodio tiene un efecto inhibitorio sobre ambos procgsos. El calcio es uncomponente principal de huesos y caparazones; además participa de la acción hormonal y en la contracciónmuscular, y en algunas proteinas cumple un rol estructural. Elcalcio y el magnesio intervienen en reacciones que involucranATPu otros compuestos con grupos fosfatos.
La tercera categoria de elementos presentes en lascélulas incluye; iodo, selenio y elementos de transición ypost-transición, tales como: hierro, manganeso, cobalto, cobre,zinc y molibdeno. El selenio es esencial en ciertos animales cgmo vacas y gallinas. La única función conocida del iodo en losanimales, es comoconstituyente de las hormonas tiroideas tiroxina y sus derivados. El iodo y el bromo se acumulan en ciertas algas, celenterados y otros organismos marinos. El silicio,comodióxido de silicio, es un componente importante de diatomeas. El boro es esencial para el crecimiento de las plantasmientras que el vanadio lo es para ciertas algas, además de serun componente importante de una proteina contenida en los tunicados.
Unade las caracteristicas de los metales de transición

155
es su capacidad para asumir diferentes estados de oxidación,por lo cual participan en reacciones rédox. El hierro y el cobre, por ejemplo, constituyen el centro activo de varias metalgenzimas, catalizando la transferencia de electrones, reaccionesde oxidación y de oxigenación, y también de proteinas transportadoras de oxigeno. Los iones metálicos, salvo los metales alcalinos, actúan por lo general como ácidos deLewis. Algunos metales de transición y post-transición, comomanganeso, cobalto yzinc, forman parte de los sitios activos de enzimas que catalizan reacciones tales comohidrólisis, hidratación, y decarboxilación de varios compuestos. El molibdeno es componente de enzimas que catalizan reacciones rédox y de fijación de nitrógeno,yel cobalto es componente principal en la vitamina 812. En cambio, a otros elementos pesados tales comoaluminio, estroncio,bario, plomo, cadmio, arsénico, estaño, no se les conoce ninguna función esencial para el hombre u otrosorganismos y,másbien, tienen efectos adversos (inhibidores de enzimas).
IV.l.2. Propiedades de los iones metálicos relevantes para lacatálisisLos iones metálicos, como los protones, son ácidos de
Lewis o electrófilos, pudiendo entonces compartir un par electrónico para formar una unión o. Pueden ser considerados además como-"superácidos", dado que existen en solución neutra,frgcuentemente poseen carga mayor que +l, y en algunos casos pueden aceptar electrones en sus orbitales vacantes de baja energia para formar uniones n. Por lo tanto los metales pueden servir comocatalizadores ácidos generales para reacciones catalizadas por protones. En contraste con éstos, los iones metálicos pueden funcionar como templados tridimensionales para la unión y orientación de bases ya sea independientemente o comoquelatos. Debido a la existencia de orbitales llenos, los ionesmetálicos son más grandes y más polarizables que los protones ypueden por lo tanto donar electrones para formar tanto unionesn como uniones o.
La polarizabilidad de los iones metálicos que se correlaciona con su capacidad de donar electrones para las uniones n,se la ha denominado "blandura". Asi, los iones metálicos “blandos" o polarizables, tienden a ser grandes, tienen varios electrones de valencia desapareados y fácilmente excitables y

156
tienen una carga positiva baja. En cambio,ios iones metálicos"duros" son pequeños, pierden fácilmente eiectrones de valenciadesapareados y pueden tener un estado de oxidación positivo e19vado.
La misma terminoiogia puede ser apiicada a 10s ïigandos, y se correlaciona con su capacidad para aceptar unionesn .Los Iigandos "biandos" o poiarizabies tienden a tener baja elegtronegatividad,y son fáciïmente oxidabies o tienen orbitaies debaja energia vacantes como en ias uniones dobles. Y en forma opuesta, ios ligandos duros tienen baja poiarizabiiidad, alta eiectronegatividad y orbitaies vacantes superiores inaccesibles.
En 1a Tabïa IV.1. (Miidvan, 1970) se detaiian ios iones metáiicos y iigandos "duros", "biandos" e intermedios. Deun examen de ias afinidades de Ios metaies por ios iigandos,Pearson (1966) ha establecido e] principio genera] de que ios"ácidos duros prefieren asociarse con bases duras, y los ácidos b1andos prefieren asociarse con bases biandas". Los casos"borderline" se comportan de un modo intermedio. En 1a Tabla¿[LlL se ve que 1a mayoria de ios metaies y ligandos de interésbioquímico son duros, con algunas pocas excepciones importantes:e] cuproso, Ios tioies y ios hidruros son "biandos"; e] zinc,e] ferroso, cüprico, cobaito e imidazo] son intermedios.
IV.2. METALOENZIMAS
IV.2.]. Introducción
Si bien 1a participación de los iones metáiicos en Iosprocesos bioïógicos se conoce desde tiempos remotos, 1a maneraen 1a cua] eilos ejercen su función no ha sido eiucidada hastadécadas recientes. Se sabe actuaimente que ios metaies jueganroies importantes en funciones cataiiticas hormonaies y otrasreguiatorias; en su contro], en 1a sintesis y estabiiización demacromoiécuias , en 1a contracción muscuiar, conducción nerviosa y transporte.
En ias moïécuias proteicas, ias cadenas 1atera1es polares interaccionan con otros eiectroiitos en soiución, siendoentonces natura] que los iones metálicos tengan frecuentementeun efecto pronunciado sobre 1a actividad cataiítica de las

enzimas.
157
Los varios roies que un ión metáiico puede tener en una reacción enzimática van desde débiles efectos iónicos hastaasociaciones a1tamente especificas,como es e] caso de ias hemoenzimas.
TABLAIV.1.: Ciasificación de ácidos y bases de Lewis deinterés bioquímico
res unión hidrógg
DUROS INTERMEDIOS BLANDOS
Acidos de Lewis H+, Li+, Na+, Be2+ Fe2+, C02+, Ni2+ Cu+, Ag+, Cs+, Hg+
'MgZ+, Ca2+, Sr2+, Cu2+, Zn2+, Pb2+ Pd2+, Cd2+, ng+,n
Mn‘+, Cr3+, Co3+, R3C+ (iones car- RS+, Br+, HO+, 12,
Fe3+, As3+, dono- bonio) Brz, Mo (átomos me
táiicos), CH2(car
2- 2504 , C03 ,
C104 , N03 , NH3
RNH2
N3 ,N2,C1 ,- 2
N02 , 503
no benos)
Bases de Lewis H20, OH', F‘, C6H5NH2,piridi- R25, RSH, Rs‘, Br“,
CH3c02', P043' na, imidazo], I', scn', 52032“,
CN-, RNC, C0,
HsR(carbaniones)
Los iones metáiicos, en genera], ejercen su efecto enios sistemas bioiógicos a muybajas concentraciones, de] ordende trazas. La presencia o ausencia de un eiemento traza particuïar en una matriz bioïógica constituyó un aspecto experimenta] crucia] que preOCUpóa muchos investigadores, convirtiendo1a respuesta a esta Cuestión en un fin en si mismo. Los avances
en disciplinas taies como1a quimica bioiógica, inorgánica, orgánica y fisica, que se apoyan unas en otrasr permitieron resolver gran parte de Ios probiemas anaïiticos experimentaïes queconstituyeronïosprincipaies impedimentospara e] progreso.

158
Asi, ei reconocimiento, purificación y caracterizaciónde 1as metaioenzimas dependió mucho de] progreso de 1a quimicafisica de ias proteinas y 1a metodoiogia para su aisiamiento ycaracterización, asi comode los avances en métodos espectroscópicos, electroquimicos, isotópicos y otros para e] reconocimiento de Ios metaies y sus sitios de unión.
IV.2.2. Nomenciaturay ciasificación
La importancia de ios metaies en Jos sistemas bioïógicos se pone ciaramente de manifiesto si tenemos en cuenta quepor lo menos un tercio de ias enzimas conocidas, contienen metaies en su estructura, requieren metaies para su actividad o sonactivadas por iones metálicos (Dixon y Webb, 1964).
Se considera que una enzima es una verdadera metaioenzima sóio en e] caso en que e] ión metáiico es un participanteesencia] en e] mecanismocataiitico; esto üitimo es extremadamente difici] de demostrar y para eiïo se requieren varios tipos de evidencias experimentaies.
Las metaioenzimas sueien ciasificarse en dos ciases(Vailee, 1955):
a) aquéiias en las que e] meta] está fuertemente unido a 1a enzima;
b) enzimas en ias que ios iones metáiicos son disociabies
Hasta e] momento, se ha dedicado más atención a1 primertipo de enzimas, en tanto que respecto de 1as segundas, a iasque también podemos denominar enzimas activadas por metaies, 1ainformación es más escasa.
Comoya hemos dicho, 1a distinción entre ambas categorias está relacionada con 1a diferente fuerza de unión de] meta] (Maimstrom y Rosenberg, 1959). Las enzimas de] primer grupopueden aislarse y purificarse con e] meta] unido a 1a proteina,y un anáiisis de] contenido metáiico y 1a pureza proteica enlas diferentes etapas de purificación permite establecer una reiación definida entre 1a función enzimática y e] meta]. En e]segundo grupo de enzimas en cambio, e] meta] se disocia durante1a purificación; esto conduce a una pérdida de actividad. y 1ainteracción metaI-enzima surge de] hecho de que 1a actividad serecupera por 1a adición de] ión metáiico.

159
Esto implica que mientras las enzimas del grupo a) nonecesitan el agregado de iones metálicos al medio de ensayo, laactividad de las enzimas del grupo b) depende explícitamente dela concentración de iones metálicos en solución. Sin embargo este solo hecho no basta para clasificar una enzima comometal-agtivada. Es necesario contar con información suficiente, ya queel metal puede influir en la velocidad de reacción sin tener unrol intrínseco en el mecanismo de reacción.
IV.2.3. _inc-en2imas
Comola mayoria de los elementos metálicos el zinc seencuentra en trazas en los materiales biológicos. La quimica analítica del zinc y la imposibilidad para realizar medicionescuantitativas precisas de este metal,fueron sin duda las principales razones que dilataron la asignación del rol biológico quehoy se le reconoce. En la medida que estos problemas se fueronresolviendo, se vió que este elemento se encontraba presente enmayores cantidades que las que se habian presumido; al mismotiempo el conocimiento de su rol critico en procesos y sistemasbiológicos complejos, ha crecido exponencialmente.
Hoyse sostiene que el zinc ejerce su actividad biológica primaria a través de su asociación con proteinas. Hasta ladécada del 50, sin embargo, sólo se conocia una sola zinc-proteina: la anhidrasa_carbónica (Keilin y Mann, 1940), y existiandudas acerca de otros posibles roles biológicos del zinc. La identificación de un gran número de zinc-enzimas durante las ültimas tres décadas ha sido elresultadode losavances conjuntosen la purificación de proteinas y en el desarrollo de técnicasfisicoquimicas y analíticas precisas,empleadas para aislar y cgracterizar las zinc y otras metaloproteinas.
IV.2.3.l. Roles biológicos del zinc
El estudio del rol del zinc en los sistemas biológicosha pasado por cuatro fases.
La primera fue nutricional; y se ha demostrado que elzinc es esencia] para el desarrollo, crecimiento y diferenciación de todas las especies.
La segunda,bioquimica,llevó a demostrar que el zinces un componente integral de varias enzimas, indispensable parasu función catalitica y estabilidad estructural.

160
El reconocimiento de manifestaciones clinicas asociadas con anormalidades en el metabolismo del zinc en todos losvertebrados, incluyendo humanos (Li y Vallee, l980), constituyela tercera fase .en la evolución de la biologia del zinc.
Finalmente, la cuarta fase, que corresponde al rol delzinc en la expresión genética, constituye un campode investigación más reciente (Vallee y Falchuk, l981).
IV.2.3.2. Aislamiento de zinc-proteinas
La interacción de los metales con las proteinas resulta en una metaloproteina o en un complejo metal-proteina, nomegclatura basada, comoya se dijo, en su constante de estabilidad(Vallee, 1955). En las metaloproteinas, el metal se encuentrafirmemente unido y no puede separase durante su aislamiento. Enlos Complejos metal-proteina, el metal se une más débilmente, yla asociación entre un metal particular y una proteina es quimica y biológicamente más lábil.
Ello significa que a lo largo del curso de la purificación, la relación zinc-proteina y zinc-actividad enzimática aumenta para las metaloenzimas, aproximándose a una constante,mientras que para los iones metálicos adventicios no relacionados con la función enzimática, disminuye hasta valores despreciables.
Las proteinas del zinc pueden ser estudiadas por lastécnicas usuales de la quimica de las proteinas y por métodosque responden especificamente a la presencia de ese metal(Vallee y Hacker, 1970), incluyendo su interacción con agentesquelantesde metales y su reemplazo por otros metales.
IV.2 3.3. Efecto de agentes quelantes
Históricamente, la inhibición de metales por agentesquelantes ha jugado un rol principal en el reconocimiento delzinc como constituyente de metaloenzimas, dado que el zinc esun elemento diamagnético con una capa d completa y no tiene otras propiedades fisicas que puedan revelar rápidamente su presencia.
La l-lO fenantrolina y el aa'dipiridilo forman un complejo firme y estable con metales de la primera serie de transición, pero no con los alcalino térreos. En contraste. el EDTAforma complejos estables con metales de ambas series.

161
Generalmente, estos agentes quelantes inhiben las zinc y otrasmetaloenzimas, ya sea eliminandoel metal o por formación de unconjunto con él ¿n Aitu.
Sin embargo, la inhibición de una enzima por un agentequelante no constituye una evidencia concluyente de que estamosfrente a una metaloenzima; y del mismo modo, la falta de inhibición por un agente quelante tampoco es un indice absoluto de laausencia de zinc.
Los agentes quelantes se usan también para separar elmetal de las zinc-proteinas produciendo las correspondientes apoproteinas. Para los distintos agentes quelantes, la efectividad de este proceso varia con la metaloenzima en cuestión.
IV.2.3.4. Propiedades de la unión del metal
La adición de zinc a una apoenzima inactiva establepuede restaurar completamentesu actividad. Esta caracteristicapérdida de actividad enzimática por su eliminación, y la recupgración de la misma por agregado de zinc, constituyen fuertes evidencias de la importancia funcional de este metal en la catálisis. Varios otros cationes divalentes tales comocobalto, manganeso, niquel,hierro, cobre, cadmio y mercurio pueden tambiénreconstituir quimica y funcionalmente varias apoenzimas (Valleey Holmquist, l980). Sin embargo, no puede predecirse cuál de todos estos iones metálicos puede llegar a restaurar la actividadde una zinc-enzima en particular, si bien la sustitución por cgbalto resulta con frecuencia efectiva en la reconstitución deespecies enzimáticamente activas.
IV.2.3.5. Propiedades espectroscópicas
Los métodos de sustitución por otros metales han resultado muy útiles en los estudios de los mecanismos de acción de laszinc-enzimas, ya que se puede reemplazar al zinc espectroscópicamente silente por metales paramagnéticos comoser cobalto, magganeso, niquel, cobre o cadmio.Los metaloderivados asi formadosse investigan entonces por técnicas espectroscópicas tales comoresonancia de spin electrónico (EPR), resonancia magnética nuclear (NMR),dicroismo circular (CD), dicroismo magnetocircular(MCD)y correlación angular perturbada de rayos y. En particular, los espectros de absorción, CDy MCDde las enzimas cobalto sustituidas proveen de información acerca de los sitios de

162
unión de los metales en las zinc-enzimas (Vallee y Holmquist,1980).
IV.2.3.6. El estado entático
Varias de las técnicas espectroscópicas mencionadas enel item anterior indican que las caracteristicas de coordinación del zinc en las metaloenzimas difieren de aquellas encontradas en los complejos simples. En las metaloenzimas, el sitiode unión del metal es siempre altamente asimétrico; comparadacon la de los complejos de iones metálicos, la geometria estádistorsionada ya sea por la longitud inusual de las uniones, opor un nümero impar de ligandos. Se piensa que estas circunstancias significan un estado de tensión o stress, y comoconsecuencia, los espectros de metaloenzimas cataliticamente activosgeneralmente no se asemejan a los de los complejos de iones metálicos, y son entonces atipicos comparados con aquellos modelosconvencionales. Se ha sugerido que el sitio activo de las metaloenzimas es prohibido para la función biológica correspondiente, que es la catálisis, y esta situación se conoce comoestadoentático (Vallee y Williams, 1968).
En este contexto, el término entasis se emplea para indicar una condición de tensión o de stress en las metaloenzimasque existe previa a la interacción con el sustrato. Se cree queel estado entático refleja un área de la enzima con una energiacercana más a la de un estado de transición unimolecular que-alde una molécula estable. Constituye un dominio energéticamente"prohibido", y permite predecir estructuras de coordinación delmetal en el centro activo de la enzima, significativamente difgrentes de los sistemas más simples corrientemente conocidos. N!merosos estudios sobre metaloenzimas por el método de difracción de rayos X confirman la existencia de tales geometrias decoordinación irregulares.
IV.2.3.7. Ligandos que unen zinc
Aunque una amplia variedad de cadenas laterales de aminoácidos pueden actuar como ligandos potenciales del zinc enlas proteinas, se ha encontrado que sólo cuatro de tales aminogcidos son los que efectivamente participan en esta unión. En orden decreciente de frecuencia, ellos son: los nitrógenos del anillo imidazol de la histidina, los azufres de cisteina y las

163
cadenas carboxiiadas de giutamato y aspartato (Dunn, 1975;Chiebowski y Coleman, 1976).
La identificación de los 1igandos que unen un dado meta] por métodos quimicos es dificuitosa. La técnica más efectiva para identificarios es e] análisis cristaïográfico por rayos X,que puede establecerio con un a1to grado de exactitud, sibien es un método que Consume mucho tiempo. Hasta e] presente,han sido determinadas las estructuras de sietezinc-enzimas: a1coho] deshidrogenasa (Eklund y co]., 1976), superóxido dismutasa (Beemy co]., 1977), aspartato transcarbamiïasa (Monacoycoï., 1978), fosfatasa aicaïina (Sowadski y co1., 1981), carboxipeptidasa A (Rees y Lipscomb, 1981), termoiisina (Matthews yHeaver, 1974) y anhidrasa carbónica (Kannany co]., 1975).
Los 1igandos histidina pueden determinarse mediante espectroscopia NMR,y más recientemente, se ha descripto un método reiativamente simpie para determinar e] número de iigandoshistidina, comparando 1a veiocidad de intercambio de ios protones de] carbono 2 de histidina, entre 1a apoenzima y 1a hoioenzima (Gaides y co]., 1980). Aunqueesta técnica ha sido aplicada 5610 a un número limitado de metaioproteinas, entre e11asB-ïactamasa II, superóxido dismutasa y p1astocianina (Va11ee yGaldes, 1984), parece ser bastante promisoria.
Uno de los métodos más simpïes y confiables para asignar 1igandos tioles es a través de sus caracteristicas bandasde transferencia de carga azufre-meta]. Las meta10proteinas quecontienen zinc (II), cadmio (II), cobaito (II), piomo (II) ynique] (II) con Iigandos tioies, exhiben una intensa banda deabsorción a 220, 330, 340 y 360 nm respectivamente..Estas bandas fueron observadas por primera vez en las metaïotioneinas(Kági y Va11ee, 196]),y subsecuentemente fueron demostradas para virtuaïmente todas 1as proteinas que contienen iigandos tio1es y forman mercapturos metálicos.
IV.2.3.8. Ro] de] zinc en las metaioenzimas
En 1as metaloenzimas e] zinc puede cumpiir alguno deios siguientes roïes: cataïitico, estructura], reguïatorio o nocataïitico (Gaides y Vallee, 1982).
Se dice que e] zinc tiene un ro] cataiitico cuando esesencia] por estar directamente invoïucrado en 1a catáiisis, ya

]64
sea formandoparte de] sitio cata]itico o interactuando directamente con e] sustrato, como por ejemp]o en ]a anhidrasa carbónica, carboxipeptidasa A, termo]isina y a]do]asa (Dunn, ]975;Ch]ebowski y Co]eman, ]976; Argos y co]., ]978). La e]iminaciónde] zinc produce una apoenzima inactiva, que retiene su estructura terciaria nativa (Va]]ee y Ga]des, ]984).
Cump]e un ro] estructura] cuando se requiere ünnmmentepara ]a estabi]idad estructura] de ]a proteina, siendo necesario para ]a actividad so]amente en ]a medida en que ]a c0nformación tota] de ]a enzima afecta su acción. Frecuentemente, e]zinc estructura] estabi1iza ]a estructura cuaternaria de enzimas o]igoméricas. Comoocurre por ejemp]o en ]a a-ami]asa de B.óubt¿€¿ó, en ]a cua] e] zinc interviene en ]a dimerización, sinafectar a ]a actividad enzimática (Va]]ee y co]., ]959).
Cuando e] zinc no es esencia] para ]a actividad enzimatica que se mantiene aún en ausencia de] meta], o para ]a estabi]idad de ]a proteina, e] zinc cump]e un pape] regu]atorio, pudiendo actuar comoactivador (aminopeptidasa bovina) o inhibidor (aminopeptidasa porcina) (Va]]e y Ga]des, ]984).
En ciertas meta]oenzimas, como ]a a]coho] deshidrogengsa equina y humana, y ]a fosfatasa a]ca]ina de E. c084, e] zincno está invo]ucrado directamente en ]a catá]isis, ni es esencia] para e] mantenimiento de ]a estructura terciaria de ]a enzima, aunque puede estabi]izar]a,por ]o cua] su función no esc]ara; En estos casos, cuando no se puede definir cómo actüa especificamente e] meta], se dice que cump]e un ro] no cata]itico.
Una dada meta]oenzima puede contener varios números ytipos de átomos de zinc. Asi por ejemp]o, ]a a]coho] deshidroggnasa equina y ]a ]eucina aminopeptidasa contienen un átomo dezinc cata]itico y otro no cata]itico por subunidad.
Los estudios de intercambio de meta] han demostradoque e] reemp]azo de] zinc cata]itico o regu]atorio por otros ignes metá]icos pueden afectar profundamente a ]a enzima, mientras que si e] zinc es estructura] o no cata]itico. ]as consecuencias son menores. Además, ]a geometria de coordinación y/osimetría de] zinc.cata]itico parecendiferir Significativamentede] zinc estructura] y no cata]itico, en tanto que ]as propiedades de coordinación de] zinc reguiatorio aún se desconocen.Los aná]isis de difracción por rayos X han demostrado que e]zinc cata]itico se une por tres ]igandos proteicos y una

molécula de agua, en tanto que el zinc no catalitico y estructural, se coordina con cuatro ligandos proteicos. En el caso delzinc catalitico, la presencia de una molécula de agua significaun sitio de coordinación abierto, que se considera esencia] para la función del zinc en la catálisis Además, para el zinccatalitico, la coordinación es asimétrica y la ge0metria es altamente distorsionada, propiedades que reflejan Su naturalezaentática (Vallee y Williams, l968). Para el zinc no catalitico,en cambio, la geometria de coordinación es más regular y tieneuna mayor simetría, resultando entonces de tipo no entático.
IV.2.3.9. Mecanismo de las zinc-enzimas
El rol más común del zinc en las metaloenzimas es participar directamente en la catálisis; su función estructura] oregulatoria es menos conocida. Ya hemos dicho que la remocióndel zinc catalitico elimina completamente la actividad enzimática, y su re-adición la restaura. Sin embargo, a pesar de los intenSos estudios realizados en un creciente número de zinc-enzimas, el mecanismo detallado es aün desconocido.
El zinc (II) es un ión d10 que electrónicamentees muyestable y, por lo tanto, es poco probable que participeen reacciones de transferencia de electrones. Asi, aunque elzinc se encuentra presente en algunas oxido-reductasascomo porejemplo alcohol deshidrogenasa y superóxido dismutasa, su rolen estas enzimas no es el de actuar como centro rédox (el quees llevado a cabo por el NAD+y el cobre (II) respectivamente),sino más bien el de actuar c0mocentro electrofilico.
Según Dunn (1975), en los sistemas modelos, la catalisis electrofilica del zinc puede ocurrir a través de tres procesos:
a) Comoácido de Lewis, involucrando la activación de unionesquímicas del sustrato.
U' VAumentandola nucleofilicidad del ligando.
Facilitando la catálisis por alineación precisa de los reacÓ V
tantes a través de la coordinación al metal como templado.
Estos tres procesos requieren que-el o los reactantesentren en la esfera de coordinación del metal previo a la catálisis, siendo el zinc (II) un ión adecuado para cumplir ese rol,ya que un ión d no está sujeto a interacciones delO

166
estabiiización por campo ïigando, pudiendo entonces acomodaruna gran variedad de números y geometrias de coordinación, incluyendo aigunas aitamente distorsionadas. Ademáse] zinc secoordina a átomos de oxigeno, nitrógeno y azufre, siendo las veIocidades de sustitución de estos iigandos muy rápidas,y a pHneutro no forma hidroxoderivados (Galdes y Hiii, 1979). Se conocen varios ejempios de catáiisis eiectrofiïica por compiejosde zinc (Dunn, 1975; Goidin y Leigh, 1979) y es razonabie supgner que en las metaloenzimas e] zinc cump1a roies simiiares.Sin embargo, 1as evidencias para este postuïado son escasas, yde ias metaioenzimas conocidas, sólo tres han sido estudiadascon suficiente detaiïe comopara tener aïgunas inferencias mecanisticas, y son: anhidrasa carbónica, carboxipeptidasa Ay a1coho] deshidrogenasa. Todas actúan sobre un grupo C-O; 1a primera como hidratasa, 1a segunda como hidroiasa y 1a tercera comooxido-reductasa. En e11as e] zinc participaria de tres maneras:
a) Poiarizando e] grupo C-O a través de una coordinación directa con e] sustrato, faciiitando asi e] ataque nucieofiiicoen e] átomo de carbono. En este caso e] zinc actúa como ácido de Lewis, faciiitando 1a catálisis ácida a pH neutro y alca1ino. Asi, para 1a carboxipeptidasa, se ha propuesto quee] grupo carbonilo de] sustrato se une a] zinc, y en esteproceso se despïaza una moiécuia de agua unida a] meta] (Reesy Lipscomb, 1981); un mecanismo simiiar se ha demostrado para 1a aicohoi deshidrogenasa (P1app y co]., 1978). En cambioestudios con infrarrojo indican que eiio no ocurre en 1a anhidrasa carbónica (Reipe y Wang, 1967).
b) E1 zinc aumenta 1a nucieofiiicidad de 1a moiécuia de agua,ya sea a través de su coordinación directa a1 meta], o enforma indirecta mediante 1a intervención de un residuo amingácido unido a1 meta]. Esto bajarïa e] pKa de 1a moiécuia deagua de manera ta] de faciiitar 1a catáiisis básica a pH neutro. Este mecanismo es e] que se ha postuiado que opera en1a anhidrasa carbónica,donde ios marcados cambios espectrales observados en 1a cobaito-enzima se cree, refïejan 1a ionización de 1a moiécuia de agua unida a] meta] (Pocker ySarkanen, 1978). Una aïternativa propuesta es que ios cambiosespectraies refïejan 1a ionización de uno de ios ligandoshistidina (Gupta y Pesando, 1975). e] cuaï actúa como una base sustrayendo un protón de una moiécuia de agua en e] sitio activo. Los cáicuïos de orbitaies moiecuiares abinitio

167
favorecen a la primera hipótesis (De Moulin y col., 1977).Los estudios en sistemas modelos muestran que el ión hidróxido unido a metal es un nucleófilo muy reactivo para la hidrólisis de péptidos y ésteres,y un mecanismoque involucra esta especie ha sido propuesto para la carboxipeptidasa A(Vallee y col., 1983).
c) Los dos roles mencionados, no se excluyen mutuamente, y elsustrato puedeunirse al zinc sin desplazar una molécula de agua. En ese caso, se forma un intermediario penta coordinadoen el cual el zinc polariza la unión C-O del sustrato y activa la molécula de agua para un ataque básico. Además, elmetal, a través de su geometria de coordinación flexible,puede actuar como templado para mantener juntos a los reactantes, sugerencia que es consistente con la naturaleza entgtica del metal (Vallee y Williams, 1968). Este modo de acciónse ha postulado que opera en la alcohol desdhidrogenasa(Schmidt y col., 1979). Se propone que los aldehidos se unenal átomo de zinc sin desplazar la molécula de agua. El metalactúa entonces comoácido de Lewis y activa el sustratotransfiriendo un hidruro desde el NADH.El anión resultante,un alcoholato unido al metal, seria protonado por la molécula de agua que actúa como un ácido especifico. Este mecanismo está apoyado por numerosas observaciones cinéticas, perono cuenta aún con evidencias directas.
Mecanismossimilares que involucran intermediarios penta coordinados han sido postulados para la anhidrasa carbónica(Dunn, l975; Kannany col., 1977) y la carboxipeptidasa (Makineny col., 1979; Vallee y col., 1983).
De acuerdo a lo que hemos expuesto, los roles propuestos para el zinc en la catálisistienen un rasgo en común: todos involucran una sustitución de ligandos en el átomo metálico,comoparte integral de la catálisis. Si la geometria del estadode transición asociado con la sustitución de ligando, fuera significativamente diferente de la de la enzima libre, la energiade activación seria muyelevada. Por lo tanto, para una catálisis eficiente, la geometria y la simetría de la enzima libre deben ser muy cercanas a las del estado de transición. Dicho de gtra manera, la catálisis enzimática requiere que en el estadofundamental.el metal tenga una energia más cercana a la del estado de transición que a la de la molécula estable convencional,

168
y está asi "prohibida" para una actividad cataiitica. Este ese] principio fundamenta] de 1a hipótesis entática (Va11e yNiiiiams, 1968) apïicabïe a 1a caracteristica esencia] comúnatodos los mecanismos propuestos.

IV.
169
3. RÉEERENCIAS
Argos, P.; Gravito, R.M.; Eventoff, w.; Rossman, M.G. &Branden, C.I. (1978) J. Mo]. Bio]. 126, Ï4Ïu
Beem, K.M.; Richardson, D.C. & Rajagopaian, K.V. (1977) Biochemistry 1g, 1930.
ChTebowski, J.F. & Coleman, J.E. (1976) en Meta] Ions inBiologicaT Systems (Ed. H. SigeT) Marce] Dekker, NewYork,vo].g, p. 2.
De MouTin, D.; PuTTman, A. & Sarkar, B. (T977) J. Am. Chem.
Soc. gg, 8498.
Dixon, M. & Webb, E.C. (1964) en Enzymes, 2da. edición.Academic Press, New York, p. 672.
Dunn, M.F. (1975) Struct. Bonding. gg, 61.
EkTund, K.; Nordstrom, B.; Zeppezauer, E.; SoderTund, G.;OhTsson, 1.; Boiwe, T.; Soderberg, 8.0.; Tapia, 0.; Branden,C.I. & Akeson, A. (T976) J. MoT. Bio]. 102, 27.
GaTdes, A. & Hi1], H.A. 0. (1979) en SpeciaTist PeridicaTReport (Ed. H.A.O. HiTT). The Chemical Society, London, voT.lp. 317.
GaTdes, A.; Hi1], H.A.0.; Baldwin, G.S.; WaTey, S.G. & Abraham,E.P. (1980) Biochem. J. 187, 789.
GaTdes, A. & VaTTee, B.L. (1982) en Meta] Ions in BioïogicaTSystems (Ed. H. SigeT). Marce] Dekker, NewYork, vo]. lá,p. T.
Goldin, B.T. & Leigh, G.J. (T979) en SpeciaTist PeriodicaTReport (Ed. H.A.O. HiTT) The Chemical Society, London, voT. l,p. 35.
Gupta, R.K. a Pesando, J, M. (1975) J. Bio]. Chem. ¿59, 2630.
Kagi, J.H.R. a VaTTee, B.L. (T961) J. Bio]. Chem. ggg, 2435.
Lovgren, S.; OhTsson,USA lg, 51.
Kannan, K.K.;A. & Petef, M.
Nostrand, B.;(1975) Proc.
Fridborg, K.Nat]. Acad. Sci.
Kannan, K.K.; Petef, M.; Fridborg, K.; Cid-Dresdner, H. &Lovgrer, s. (1977) FEBSLett. 1;, 115.
Keilin, D. & Mann, T. (1940) Biochem. J. gg, 1163.

170
Li, T.K. & Va11ee, B.L. (1980) en Modern Nutrition in Heaithand Disease (Eds. R.S. Goohart y M.E. Shiïs) 6ta. edición.Lea and Febiger, Philadeïphia, p. 408.
L.C.;356.
Makinen, & Shams, J. (1979)J. Bio].
M.w.;Chem.
Kuo, Dymowski, J.J.Q1,
(1959) en Advances inMeister). John Wiïey & Sons, vo]. g_, 131.
Maimstrom, B.G.Enzymoiogy (Ed. A.
& Rosenberg, A.
Matthews, B.w. & Neaver, L.H. (1974) Biochemistry 1}, 1719.
Miïdvan, A.S. (1970) en The Enzymes (Ed. P. Boyer). AcademicPress, vo]. ll, p. 446.
Monaco, H.L.; Crawford, J.L. & Lipscomb, w.N. (1978) Proc.Nat]. Acad. Sci. USAZé, 5276.
Ochiai, E. (1977) en Bioinorganic Chemistry, an Introduction.Aiïyn and Bacon Inc.
Piapp, B.V.; Mo]. Bio].lg¿, 23.
Eklund, H. & Branden, J.C. (1978) J.
Pearson, R.G. (1966) Science 121, 172.
Pocker, Y. & Sarkanen, S.(1978) en Advances in Enzymoïogy(Ed. A. Meister). John Wiley a Sons, vo]. 51, p. 149.
Rees, D.C. Nat]. Acad. Sci.1g, 5455.
& Lipscomb, w.N.(1981) Proc. USA
Reipe, R. & Wang, J.H. (1961) J. Am. Chem. Soc. gg, 4229.
Schmidt, J.;Fariand, J.T.
Chen, J.(1979)
; De_TragÏia, J.; Minke], D. & McJ. Amar. Chem. Soc. 191, 3634.
Sowadski, J.M.;Bio]. líg, 245.
Foster, B.A. & Wyckoff, H.w. (1981) J. M01.
Vaïïee, B.L. (1955) Advances in Protein Chem. lg, 317.
Vaïiee, B.L.B 225, 185.
& Faïchuk, K.H. (198]) Phi]. Trans. R. Soc. Lond.
Valïee, B.L. & Gaides, A. (1984) en Advances in Enzymoïogy(Ed. A. Meister). John Wiley & Sons, vo]. gg, p. 283.
Val]ee,B.L. & H01mquist , B.determining Meta] Ion Environment in ProteinsDarnaïï y R.G. Wiïkins).p. 27.
(1980) en Methods for(Eds.
Eisevier/North Holïand,D.w.
Amsterdam,

171
Valïee, B.L. & Hacker, w.E.C. (1970) en The Proteins (Ed. H.Neurath).Academic Press, NewYork, vo]. y, 103.
Va11ee, B.L. a Hi111ams, R.J.P. (1968) Proc. Natï. Acad. Sci.USA _5_g, 498.
Va11ee, B.L.; Stein, E.A.; Summerweïï, H.N. & Fischer, E.H.(1959) J. Bio]. Chem. 234, 2901.
Va11ee, B.L.; Gaïdes, A.; Auïd, D.S. & Riordan, J.F. (1983)en Meta] Ions in Bioïogy (Ed. T.G. Spiro), Wiley, New York,vo]. 5, p. 26.

MATERIALES Y METODOS

CAPITULO I
MATERIALES Y METODOS
1.2.
1.2.1.
1.2.2
1.3.
1.3.1.
1.3.2
1.4.
1.4.1.
Reactivos e instrumentai
Obtención y purificación de enzimas
Fuentes de enzima1.2.1.1.1.2.1.2.
Higados de cerdo y vacaGióbuios rojos humanos
Purificación de ALA-D1.2.2.1. ALA-Dde higados vacuno y
porcino1.2.2.2. ALA-Dde gióbuios rojos humanos
Otras etapas aiternativas de purificación
Cromatografía por afinidad en ThiopropyiSepharose GB1.3.1.1. Preparación de] ge]1.3.1.2. Inmoviiización dei ALA-D1.3.1.3. Soiubiiización de 1a enzima
inmoviiizadaI 3.1 4 Despiazamiento de grupos tio
piridona (TP)1.3.1 5 Determinación de tioies no
proteicos
Cromatografía hidrofóbica en PhenyiSepharose
Estudios de estructura cuaternaria
Experiencias de disociación y reasociaciónde ALA-D porcina1.4.1.1. Inhibición por urea de] ALA-D
soiubie y su reversibiiidad1.4.1.2. Inmoviiización de ALA-Dsobre
Sepharose 4B1.4.1.3. Disociación de ALA-Dinsoiubili
zada
Página
172
173
173
173173
173
173
173
174
174
174
174
174
174
175
175
175
175
175
176
176

1.4.1.4. Reasociación e hibridización deias subunidades de ALA-Dinsoigbiiizadas
1.4.2. Cromatografía en HPLC
Determinación de pesos moiecuiares por fiitraciónpor geles
E1ectroforesis en geles de poiiacriiamida1.6.1. Eiectroforesis en tubo
1.6.2. Eiectroforesis en p1aca
Preparación de apoenzima y metaioenzimas
1.7.1. Obtención de 1a apoenzima
1.7.2. Obtención de 1a hoioenzima
1.7.3. Obtención de 1a cobaïto-enzima
1.7.4. Determinación de] contenido de zinc ycobaito
Diáiisis de equiiibrio
Determinación de grupos suïfhidriïos
Tratamiento de ALA-D con DEP
1.10.]. Reversión de 1a inactivación por DEP
Medición de actividad enzimática
1.11.1. ALA-Dsoiubïe
1.11.2. ALA-Dinsoïubiiizada sobre Sepharose 4B
1.11.3. ALA-Dinsoiubiiizada sobre TPS 68
1.11.4. Determinación de PBG
1.11.5. Unidad enzimática
Determinación de proteinas
Referencias
Página
176
177
178
178
179
179
179
180
180
180
180
182
182
183
183
183
184
184
185
185
186
187

I. MATERIALES Y METODOS
I.l. REACTIVOS E INSTRUMENTAL
Los siguientes reactivos fueron adquiridos a SigmaChemical Co. (St. Louis, USA): ALA, cisteina, DTT, GSH, fluoruro de fenil-metil-sulfonilo (PMSF),dodecilsulfato de sodio(SDS), dietilpirocarbonato (DEP), seroalbümina bovina (fracciónV).
Se empleó B-mercaptoetanol (BME) y DEAEcelulosa deMerck, bromuro de cianógeno (BrCN) de Fluka A.G. (Suiza) e imidazol de Eastman Kodak.
Los geles de Sephadex G-25, Sephadex G-200, SepharoseCL-4B, Sephacryl 5-300, Phenyl Sepharose, Thiopropyl-SepharoseGB (TPS GB) fueron adquiridos a Pharmacia Fine Chemicals(Uppsala, Suecia), y el Ultrogel AcA34a LKB(Suecia).
Todos los demás reactivos fueron de grado analítico obtenidos de diversas fuentes comerciales.
El material de vidrio utilizado para el análisis de mgtales fue lavado adicionalmente con ácido clorhídrico/ácido nitricoy'enjuagados con agua bidestilada libre de metales. El material de vidrio, las soluciones y reactivos quimicos fueron examinados en forma rutinaria para controlar la contaminación metálica mediante espectrofotometria de absorción atómica (AA).
Los espectros de absorción de las regiones visible yUVse midieron en los siguientes espectrofotómetros: MetrolabRC 325,Union Giken SM 401, Beckman M-35 y Shimadzu UV 2l0.
Para las centrifugaciones se empleó una centrífugaSorvall RCSBrefrigerada, con rotores de ángulo fijo.
Las soluciones stock de DEPse prepararon en etanol-absoluto. La concentración de DEPse determinó antes de cada expgriencia haciendo reaccionar una alícuota de lO ul del reactivocon 3 ml de imidazol lO mM(pH 7,5) a temperatura ambiente, yleyendo la absorbancia del N-carbetoxiimidago] formado a 230 nm(E230 = 3000 M’.I cm“] ; Melchior y Fahrney, l970).

173
1.2. OBTENCION Y PURIFICACION DE ENZIMAS
1.2.1. Fuentes de enzima
1.2.1.1. Higados de cerdo y vaca
Luego de faenados Ios animaies, ios higados fueron mantenidos a 4 °C durante su trasïado. Posteriormente se congeiaron a -20 °C cortados en trozos de aproximadamente 100 g, guardándose a esa temperatura hasta e] momento de su uso.
1.2.1.2. Gióbulos rojos humanos
Se utiiizó sangre venosa de sujetos normaïes, dadoresvoluntarios de] Hospita] RamosMejia. En todos los casos, 1asangre se recogió sobre heparina (0,1 m1 de soiución de heparina de 12,5 mg/m] de soiución fisioiógica, por cada 10 m1 de sangre), se centrifugó a 60O x g , se separó e] piasma de los gigbuios rojos (GR), y éstos se 1avaron con soiución fisioiógica.
1.2.2. Purificación de ALA-D
1.2.2.1. ALA-Dde higados vacuno y porcino
Los procesos de purificación se deta11arán en Resuitados y Discusión.
1.2.2.2. ALA-Dde gïóbuios rojos humanos
Se reaiizó según e] método descripto por Bustos y co].(1980).
Los GR 1avados se 11evana pH 8 con amoniaco concentradoy 1uego se tratan con una mezcla de n-butano] y cioroformo(1:0,4 v/v), en una reïación GRmezcia de 1:0,2]. La mezcïa orgánica se agrega con agitación continua, en baños de temperatura cada vez menores: se pasa de un baño a 0 °C (hieïo), a otroa -5 °C (hieio y sai) y finaïmente a -20 °C. Se continúa agitando durante 15 minutos a 0 °C y se deja en reposo una hora a20 °C (baño de agua). Se centrífuga 20 minutos a 24.000 x g ye] sobrenadante se dia1iza durante 20 horas contra buffer fosfato de sodio (BFS) 0,01 M pH 6,8, usando 1a re1ación de GR abuffer de 1:]00. Finaimente, se reaïiza un fraccionamiento consuïfato de amonio 30-55% de saturación.

174
1.3. OTRAS ETAPAS ALTERNATIVAS DE PURIFICACION
1.3.1. Cromatografía por afinidad en Thiopropy] Sepharose 6B
1.3.1.1. Preparación de] ge]
Se resuspendieron aproximadamente 200 mg de TPS GB (geTseco)en 3,0 m1 de BFS 50 mMpH 6,8. Se dejó hinchar durante 15minutos a temperatura ambiente, Tuego se centrifugó 5 minutos a2.000 rmn y se continuaron Tos Tavados en batch (5 X 2,5 m1)con e] mismo buffer. E1 ge] asi hinchado (0,6 m1) se cargó enuna jeringa de pTástico de 5,0 m1, y se continuaron Tos Tavadosen Ta columnita empTeando una reTación de poTvo seco a voTumende buffer de lavado de T g/200 m1.
1.3.1.2. InmoviTización de ALA-D
Dado que Ta proteina se une a] sitio activo de] ge] através de sus grupos tioTes, se pensó que seria conveniente unapreincubación de Ta fracción enzimática con DTTpara reducir dichos grupos y Tograr asi un buen rendimiento de acopTe. Con este fin, se preincubó Ta soTución proteica con DTT10 mMduranteT5 minutos a 37 °C. Luego se eTiminó eT DTTen exceso por pasajea través de una coTumna de Sephadex G-25 (1,4 x 40 cm), equiTibrada con BFS 50 mM pH 6,8, saturado con nitrógeno y se eTuyócon eT mismo buffer.
La soTución enzimática obtenida de este modo se sembróen una coTumnita de TPS GB (0,4 x 7 cm) y se Tavó con eT buffernitrogenado hasta que en Tos eTuidos no se detectó más proteinas.
1.3.1.3. Soiubiiización de 1a enzima inmoviTizada
Para solubiTizar Ta enzima inmoviTizada se utiTizóBFS 50 mMpH 6,8 nitrogenado, conteniendo DTT 25 mMo un gradiente TineaT de] reactivo tióTico, segün se especifique.
1.3.1.4. DespTazamiento de grupos tiopiridona (TP)
Los umoTes de TP desplazados durante Ta inmoviTizaciónde 1a enzima, se determinaron midiendo Tos valores de absorbancia a 343 nm de cada fracicón e1uida, y utiTizando un coeficiente de extinción moTar de 7,06 x 103 M (Grassetti y Murray, 1967).

T75
1.3.1.5. Determinación de tioies no proteicos
Se iievó a cabo según e] método de Kawata y Suzuki(1983), precipitando las proteinas con TCA5 %y midiendo 1a absorbancia a 412 nm Tuego de 1a adición de DTNBa1 sobrenadante.
1.3.2. Cromatografía hidrofóbica en Phenyi Sepharose
E1 ge] se empaquetó en una coiumna de 2,1 x 15,5 cm gquiiibrada con buffer fosfato de potasio (BFP) 25 mMpH 6,8,conteniendo cioruro de potasio 0,25 My DTT0,25 mM.
Luego de sembrar 1a proteina (aproximadamente 60 mg),se iavó con un voiumen de cama de una soiución de] buffer de equiiibrio sin cloruro de potasio. Se eiuyó con BFP 25 mMpH 7,4 conteniendo DTT 0,25 mMy coiato de sodio 0,5 %. E1 eiuido se recogió en fracciones de 2,5 m] cada una, a una veiocidadde fiujo de 10 m1/h.
Los tubos de mayor actividad especifica se reunieron yprecipitaron con suifato de amonio 55 %de saturación. E1 precipitado se disoïvió en BFP 50 mMpH 7,4 y se diaiizó contra e]mismo buffer conteniendo DTT 1 mM.
1.4. ESTUDIOS DE ESTRUCTURA CUATERNARIA
1.4.1. Experiencias de disociación y reasociación de ALA-Dporcina
1.4.1.1. Inhibición por urea de] ALA-Dsoiubie y sureversibiiidad
La enzima soiubie fue pretratada a 0 - 5 °C con distigtas concentraciones de urea durante 5 minutos; 1uego se determinó 1a actividad enzimática. Cuando 1a actividad enzimática semidió en presencia de urea, se hicieron correcciones debido asu interferencia en 1a determinación de] PBG. Para ellos se realizaron biancos sin ALAy sin enzima, incubando las distintasconcentraciones de urea, en ias condiciones standard. Cuandoseinvestigó 1a reversibilidad de 1a inhibición por urea, 1a enzima fue pretratada según se indicó; iuego se eiiminó e] agente disociante por pasaje a través de una coiumna de Sephadex 6-25 obien, empieando una diáiisis contra BFS 0,01 M pH 6,8.

176
1.4.1.2. Inmoviiización de ALA-Dsobre Sepharose 4 B
Se reaiizó siguiendo 1a técnica descripta por Steiia yco]. (1977), utiiizando 200 mg de BrCN (o 50 mg) por m1 de ge](Sepharose 4 B) decantado, empieándose 1a fracción enzimáticaproveniente de 1a etapa de purificación con suifato de amonio55 % de saturación, o ia proveniente de una cromatografia a través de una columna de Sephadex G-200, según se indique. LaSepnarosa 4B asi activada, se resuspendió en BFS 0,05 M pH 8,0a su voiumen origina], y se ie agregó inmediatamente una soiución de ALA-Dconteniendo 30 mg de proteina por m1 de soiución,en una relación de 1 m1 de enzima por m1 de ge]. La mezcia semantuvo agitando a 4 °C durante 18 horas, a] cabo de ias cuáiesse centrifugó a 2.000 rpm por 5 minutos, y se separó e] sobrenadante. Luego se empaquetó en una coiumna y se 1avó con bufferhasta obtener proteinas negativas en ios eiuidos. La cantidadde proteinas asociadas a1 ge] se determinó por diferencia entre1a cantidad de proteina agregada a1 ge] y 1a cantidad tota] presente en ios eiuidos y en e] sobrenadante de decantación.
1.4.1.3. Disociación de ALA-Dinsoïubiiizada
Se iievó a cabo a 7 °C, haciendo pasar a través de unacoiumna (1,7 x 9 cm) empaquetada con 6 m1 de geI-enzima, o unacoiumna de 0,8 cm de diámetro conteniendo 3 mi de] sistema, unacolución de BFS 0,05 MpH 6,8, con distintas concentraciones deurea, según e] caso; hasta que en ios eiuidos no se detectaronmás proteinas. E1 curso de 1a eiución se 11evó a cabo midiendo1a absorbancia a 280 nm. Tanto 1a actividad enzimática como 1acantidad de proteinas en ios eluidos se determinó previa eiiminación de 1a urea por diáiisis. La detección cuaiitativa de urease reaiizó con ei reactivo de Ehriich, que da un compuesto decoioración amarillenta.
1.4.1.4. Reasociación e hibridización de ias subunidades deALA-Dinsoiubiiizadas '
Los estudios de reasociación e hibridización de 1assubunidades de ALA-Dinsoïubiiizadas obtenidas por desnaturaiización de 1a enzima inmovilizada, se llevaron a cabo haciendopasar una mezcia de buffer renaturaiizante a través de una coiumna (0,8 cm de diámetro) empaquetada con 1 m1 de gei-enzimadisociada. La mezcia de buffer renaturaiizante contenia un gradiente discontinuo de urea en BFS 0,05 M pH 6,8 con 0,025 M de

177
cisteina y una solución proteica de ALA-Dsoluble de higado decerdo (o ALA-Dsoluble de eritrocitos humanos). Los gradientesde urea empleados para las distintas reasociaciones e hibridizaciones, dependieron del tratamiento previo al que fue sometidoel gel. Asi, para el gel tratado con urea 6 M, el gradiente deurea cubrió un rango de concentraciones entre 6 My 0 M; paralos geles disociados con urea 3 M, 2My l M, el rango de concentraciones osciló entre 3 y 0 M, 2 y 0 My l y 0 M, respectivamente. El número total de unidades enzimaticas contenidas en elbuffer reasociante fue de 5 a lO veces mayor que la subunidadesperdidas en la disociación. El volumende buffer renaturalizante empleado guardó una proporción de lO ml de buffer por ml degel disociado. Tanto las reasociaciones comolas hibridizaciones se realizaron a temperatura ambiente, manteniendo un flujode 0,l - 0,2 ml por minuto.
1.4.2. Cromatografía en HPLC
Se empleó un cromatógrafo de HPLCShimadzu LC-3A adicionado de un detector Shimadzu SPD-2A y columnas G4000SwyG30005” de ToyoSoda.(7,5 mmx 60 cm).
Todas las corridas cromatográficas se hicieron a tempgratura ambiente, a una velocidad de flujo de 0,5 ml/minuto y serecogieron fracciones de 0,5 ml.
La enzima purificada proveniente de la fracción UGII,antes de ser sembrada se desaló por una columna de SephadexG-25 (0,9 x l2 cm) equlibrada y eluida con buffer Tris-acetato50 mMpH 7,2 conteniendo DTT l mM.
Usualmente se sembraron lOO a 120 ug de proteina enzimática.
Para las cromatografía de la enzima nativa, la columnase equilibró y eluyó conBFS 50 mMpH 6,8 conteniendo DTT l mMycloruro de sodio O,l M; en cambio para la enzima disociada, seempleó BFS 0,1 M pH 7,4 conteniendo 0,2 % de SDS.
Los radios de Stokes se calcularon de acuerdo a Tanfordy col. (l974).
Las proteinas marcadoras utilizadas fueron: lisozima,inhibidor de tripsina, anhidrasa carbónica, ovoalbümina, seroalbümina, aldolasa y ferritina.

l78
1.5. DETERMINACION DE PESOS MOLECULARES POR FILTRACION POR
GELES
Las columnas de Sephadex 6-200 se prepararon según latécnica de Batlle (l968) y la determinación de pesos moleculares mediante la técnica de filtración por geles se realizó segün el método descripto por Andrews (l964, 1965).
Se usaron las siguientes proteinas marcadoras: citocrgmoc, ovoalbümina, seroalbümina bovina, catalasa, tiroglobulina.
1.6. ELECTRGFORESIS EN GELES DE POLIACRILAMIDA
I.6.l. Electroforesis en tubo
El método empleado se basó en los trabajos descriptospor Ornstein y Davis (1964): para la preparación de los gelesse adicionó lauril sulfato de sodio, cuya concentración finalfue de 0,l % de acuerdo a Weber y Osborn (1969).
Se sembraron lO ml de cada muestra, con una concentración proteica de 50 ug/lO ul, en presencia de sacarosa al lO %y azul de bromofenol (0,1 % de colorante en ácido acético al7 %). El buffer de siembra fue fosfato de sodio 0,l M pH 7,l adicionado con SDS0,l %, al igual que el buffer de corrida. Eltiempo de corrida fue de 5 horas, y se aplicaron 5 mApor tubo.Al cabo de ese tiempo, el gel se coloreó con Coomasie blue (disuelto en una relación de 1,25 mg de colorante en 454 ml de metanol al 50% y 46 ml de ácido acético glacial), dejándolo encontacto l hora. Luego se lavó con agua y finalmente con sucesivos lavados de una mezcla decolorante de acético, metanol y agua (75:50:875). La migración de la proteina se calculó por medio de la fórmula:
d% de migración = —l x lOO
dz
donde d1 es la distancia alcanzada por la proteina al final dela corrida, y d2 es la distancia del frente al final de la cgrrida. La distancia d2 se determinó colocando un pequeño alambre a la altura del colorante azul de bromofenol, antes de lacoloración. Esto se debe a que los tratamientos de coloración y

179
decoioración provocan cambios en 1a longitud de] ge].
Para 1a cuantificación de 1a corrida e1ectroforética,se Ieyó 1a absorbancia de los geïes a 660 nm, empieándose paraello un espectrofotómetro UNICAMSP-BOO, a] que se adicionó unadaptador que permite su registro (Azcurra y c01., 1978).
1.6.2. Electroforesis en placa
Se reaïizó en ge] de poiiacriïamida a1 10 % de 1 mmdeespesor
Las muestras proteicas fueron tratadas con igua] voiumen de soiución disociante (SDS 1 %, DTT 50 mMy gïicerina 20 %en buffer Tris-HC] 10 mMpH 6,8), calentando a baño maria durante 2 minutos; sembrándose aproximadamente 5 ug de proteina.
E1 tiempo de corrida fue de 4 horas a temperatura ambiente, con una intensidad de corriente de 15 mA.
E1 ge] se coloreó con Coomasie Bri11ant Bïue y e] excgso de coïorante se 1avó con 1a mezcia de coiorante de metano]:acéticozagua.
Se corrieron ias siguientes proteinas marcadoras: fosforiïasa b (94 kDa), seroalbümina bovina (67 kDa); ovoaibümina(45 kDa), anhidrasa carbónica (30 kDa), a-iactoaibümina(14,4 kDa).
La densitometria de] ge] teñido se realizó en unChromato Scanner Shimadzu C5900.
1.7. PREPARACION DE APOENZIMA Y METALOENZIMAS
1.7.1. Obtención de 1a apoenzima
La suspensión enzimática (0,5 - 4 mg) en buffer Tris-acetato 50 mM pH 7,2 o BFS 50 mM pH 6,8 se incubó con EDTA
50 mMy DTT 10 mMdurante 30 minutos a 37 °C bajo atmósfera denitrógeno. Luego se diaïizó contra 2 x 200 mi de buffer Tris-acetato 50 mMconteniendo EDTA10 mM por 16 horas, y seguidamente contra 2 x 200 m1 de buffer Tris-acetato 50 mM.Las diálisisse efectuaron a 4 °C bajo atmósfera de argón.
Alternativamente, 1uego de 1a incubación se fiitró inmediatamente por una columna de Sephadex 6-25 (0,9 x 20 cm)

180
equilibrada con BFS 50 mMpH 6,8 nitrogenado. La fracción enzimática recogida se empleó como apoenzima reducida.
1.7.2. Obtención de la holoenzima
Se realizó de manera análoga a la preparación de la apoenzima, sólo que en lugar de EDTAse incubó con acetato dezinc (lO'2 Mconcentración final), y que los buffers de diálisis no contenían EDTA.
1.7.3. Obtención de la cobalto-enzima
La suspensión enzimática (l,8 - 2,7 mg) en bufferTris-acetato 50 mMpH 7,2 se incubó l hora a 37 °C con cobaltolO'2 M, en tubos de Thunberg en los que se evacuó el aire y seincorporó nitrógeno. Luego se dializó con 2 x 200 ml del mismobuffer de incubación, durante 20 horas a 4 °C y bajo atmósferade argón.
1.7.4. Determinación del contenido de zincgy cobalto
Las soluciones enzimáticas se aplicaron directamente alos espectrofotómetros de absorción atómica con llama (ShimadzuAA 650, Varian AA 575) o sin llama (Shimadzu AA 640-13, GFA-Z).Para el análisis cuantitativo se utilizaron soluciones patrónde zinc y cobalto de lOOOppm de Nakarai Chemicals (Kyoto,Japan), o soluciones patrón preparadas a partirde granallas deZlnC.
1.8. DIALISIS DE EQUILIBRIO
Las celdas de diálisis se contruyeron en acrílico siguiendo el diseño de Englund y col. (l969) (figura 141;). Cadacelda consiste en dos compartimentos cilíndricos de 150 ul decapacidad, separados entre si por una membranacircular. Parala preparación de las membranasse emplearon tubos de diálisisVicking 18/32, que se hirvieron durante 5 minutos en una solución de carbonato de sodio 5 %-EDTA50 mMy luego se guardarona 4 °C en etanol 50 %. Previo a su uso, las membranas se hidrataron con agua deionizada. Las celdas se armaron ubicando lamembrana entre las dos cámaras, y el sistema en un soporte

l8l
metálico que se ajusta por compresión. En cada compartimento secolocó una perla de vidrio de l,5 mmde diámetro, para favorecer la agitación. Las soluciones se agregaron con una microjeringa Hamilton, a través de las salidas capilares que se sellaron durante la diálisis con un trozo de cinta Scotch. La suspensión enzimática (2 mg/ml) se puso en contacto con soluciones dezinc de diferentes concentraciones,en un rango entre lO's-lO'3 M.La composición del buffer fue Tris-acetato 50 mHpH 7,2; 5 mMen DTT. El tiempo de diálisis fue de 5-12 horas; se trabajó a25 °C con agitación continua.
“unFIGURAI.l.: Diagrama esquemático
de las celdas de diglisis. El block, quecontiene dos paresde celdas, se montaen un soporte metálicoque se ajusta porcompresión.
Se tomó comoconcentración de zinc libre, la concentración de zinc de la cámara que contenía la proteína enzimática.
Si consideramos que la proteina posee n sitios idénticos independientes, cada uno capaz de unir un ligando particular con una constante de disociación K, de acuerdo a la ley deacción de masas se tiene que:
K = 1L) gn -v )\)
donde L es la concentración de ligando libre (en nuestro caso,el zinc) y v es el número promedio de moléculas de ligando unidas por molécula proteica.
Un reordenamiento de la ecuación conduce a la siguiente

182
expresión (Scatchard, 1949):
v _.—-l(n-v)(L) K
de manera ta] que graficando v/(L) versus v se obtiene una recta de pendiente 1/K, que intercepta 1a abcisa en e] vaior n.
1.9. DETERMINACION DE GRUPOS SULFHIDRILOS
Se efectuó segün e] método de Eliman (1959) con Tigeras modificaciones en cuanto a1 voiumen de incubación (2,0 m1en Tugar de 3,0 m1) y e] buffer empieado (buffer Tris-HCT en lugar de BFP). La reacción se Tievó a cabo a temperatura ambienteempleando 30 - 40 ug de proteina enzimática, midiendo e] aumento de absorbancia a 412 nm.
Para e] cálcuio de] número de grupos suifhidriios, seusó un coeficiente de extinción moiar de 13.600 M'] cm']. Losvaiores de absorbancia fueron corregidos por 1a absorción de1a enzima y e] DTNB; también se tuvo en cuenta e] cambio de v9lumen debido a1 agregado de SDS.
1.10. TRATAMIENTO DE ALA-D CON DEP
La enzima purificada (115 - 230 pg, concentración ensubunidades: 6,6 x 10'6 M a 8,2 x 10'6 M) disuelta en BFP 0,1 MpH 6,3 (o e] que se indique), se activó durante 10 minutos a37 °C con DTT 5 mM.
El resto de] tratamiento se iievó a cabo a 0 °C agregando e] DEPen concentraciones entre 0,1 a 0,35 mM. La reacción se efectuó en las ce1das de un espectrófotómetro ShimadzuUV210 con compartimento refrigerado con una mezcla de hieTo ygliceroi 30 %. A distintos tiempos se sacaron aiicuotas de50 u] en los que se determinó 1a actividad enzimática.
La reacción de etoxiformiTación se detuvo agregando imidazo] 10 mMa pH 7,5; en ensayos previos se habia determinadoque esta concentración de imidazo] no modifica 1a actividad deALA-D.

183
Para medir 1a actividad a tiempo cero, se tomó una a1icuota previo a1 agregado de DEP, que se agregó a1 medio de incubación conteniendo DEPe imidazo1.
Para 10s estudios de protección por sustrato, 1a enzima, una vez activada con DTT, se preincubó con e1 ALAa diferentes concentraciones durante 10 minutos a 0 °C, minimizando asi1a producción de PBG.
La protección por iones zinc se 11evó a cabo pre-tratando 1a enzima activada con DTTpor 10 minutos a 37 °C, previoa 1a modificación por DEP a 0 °C.
E1 nümero de residuos histina modificados se ca1culóen base a 1a absorbancia a 240 nm de acuerdo a Mi1es (1977) con
_ -1 -1un E240 - 3.200 M cm .
1.10.1. Reversión de 1a inactivación por DEP
E1 ALA-D fue modificada con DEP 0,2 mM'a pH 6,8 y 0 °Cdurante 30 minutos. A1 cabo de ese tiempo, se agregó hidroxi1amina (a una concentración fina1 de 0,7 My pH ajustado a 7,0)a 1a enzima modificada y a un contro1 no tratado, y se dejó actuar a1 reactivo a temperatura ambiente durante 30 ó 60 minutos. E1 exceso de hidroxi1amina se e1iminó mediante una fi1tración por geles a través de una co1umna de Sephadex G-25 (0,9 x18 cm) equi1ibrada y e1uida con BFP (Me1chior y Fahrney, 1970).
1.11. MEDICION DE ACTIVIDAD ENZIMATICA
I.11.1. ALA-Dso1ub1e
La actividad enzimática se determinó midiendo 1a cantidad de PBGformado a partir de ALA(Mauzera11 y Granick, 1956).
En condiciones standard1a mezc1a de reacción (1 m1)conteniendo 1a so1ución enzimática (5 - 15 ug). cisteina 12,5 mMo DTT 10 mMy DES 50 mMpH 6,8, se preincubó 15 minutos. 1uego
gfe agregó e1 sustrato ÁLÁW(2,5- 4 mM)y se continuó 1a incubación duranteÏ20 minutos] Tanto 1a preincubación como 1a incubación se 11evaron a cabo a 37 °C en aerobiosis y con agitacióncontinua. La reacción se detuvo confisuïïató_de cobre (so1uciónsaturada); e1 precipitado formadose descartó por centrífugación a 2000 rpm durante 10-15 minutos,y en e1 sobrenadante se

184
determinó el PBGformado.
Alternativamente, la reacción se llevó a cabo en bufferTris-acetato 50 mMpH 7,2 conteniendo DTT lO mMy acetato dezinc z x 10's Men un volumen final de 0,5 ml, y la reacción sedetuvo adicionando igual volumen de TCAlO % conteniendo cloruro mercürico 0,1 M.
Cuandose estudió el efecto de los iones metálicos, seagregó al medio de incubación la solución de la sal del metal,
'y sin efectuar la preincubación se continuó con la reacción enla forma habitual.
Las incubaciones anaeróbicas se realizaron en un bañoDubnoff saturado con atmósfera de nitrógeno.
I.ll.2. ALA-Dinsolubilizada sobre Sepharose 4B
Para la determinación de la actividad enzimática delALA-Dinmovilizada sobre Sepharose 4B, se introdujeron algunasvariantes (Stella, 1977). Se emplearon las mismas concentraciones finales de ALA,cisteina y buffer, excepto que se utilizól ml de la suspensiónd el gel en BFS 0,05 H pH 6,8, manteniendouna relación de gel a volumen final de l:3 (v/v) agitando la me;cla cada 5 minutos durante la incubación; esto último, para asegurar un óptimo contacto de los reactantes con la enzima. Finalizada la incubación, la mezcla se enfrió inmediatamente a0 °C y se centrifugóa 2.000 rpm durante 5 minutos, separando deesta manera el gel. Al sobrenadante se le agregó sulfato de cobre (solución saturada), manteniendo la misma relación que parala enzima soluble; en el nuevo sobrenadante se determinó la cantidad de PBG formado.
I.ll.3. ALA-Dinsolubilizada sobre TPS GB
La determinación de la actividad de la enzima inmovilizada sobre TPS GB se llevó a cabo de manera similar a la de laenzima soluble, salvo que no se le agregó el reactivo tiólico,no se preincubó y se realizó anaeróbicamente. La.reacción en este caso se detuvo por enfriamiento en baño de hielo; mediantecentrifugación a2.000 rpmduranteSnfinutos se separó el gel delsobrenadamente de incubación, y a este último se le hizo el tratamiento ya descripto. El gel se lavó además tres veces con elbuffer de incubación a fin de determinar el PBGque pudiera

185
haber quedado retenido. En este caso, el producto formado se e¿presa por ml de gel decantado.
I.ll.4. Determinación de PBG
La determinación del PBGse basa en su reacción con elreactivo de Ehrlich (2 g de p-dimetilaminobenzaldehido, 25 mlde ácido clorhídrico concentrado y 75 ml de ácido acético glacial) (Moore y Labbe, 1964), con el cual produce un compuestode coloración rojiza. La técnica empleada consiste en mezclarun volumen de muestra con un volumen de reactivo a temperaturaambiente. Luego de la adición del reactivo, el máximodesarrollo del color se alcanza a los 8 minutos, manteniéndose constante hasta los 15 minutos, luego comienza a decaer. El compuesto coloreado tiene un pico de absorción a 555 nm.
La cantidad de PBGformado se calcula mediante la siguiente fórmula:
A555 x dilución x 4 x 103umoles PBG/h.ml =
e x 226 x vE
donde c = 113,6 (Moore y Labbe, 1964)226 = masa molar de PBG
vE = volumen de enzima en el sistema de incubación
El factor 4 se debe a que el tiempo de incubación es de 30 min!tos y a que se empleó una cubeta de 0,5 cm de paso de luz.
Alternativamente, cuando se empleó TCAlO Z para detgner la reacción, se usó el reactivo de Ehrlich modificado (l gde p-dimetilaminobenzaldehido, 16 ml de ácido perclórico al70 %y ácido acético glacial hasta un volumen de 50 ml), y elPBGpresente se calculó aplicando un coeficiente de extinciónmolar de 6,2 x 10'4 M'1 cm’] (Mauzerall y Granick, 1956).
1.11.5. Unidad enzimática
Se expresa en umoles de PBG formados por ml de enzima,en una hora. en las condiciones standard descriptas, y la actividad especifica (A.E.) como el número de unidades por mg deproteina.

1.12. DETERMINACION DE PROTEINAS
E1 contenido proteico se determinó según e] método deLowry y co]. (1951) usando seroalbümina bovina como proteinastandard.
Dado que e] DTTproduce interferencia en e] método deLowry, en ios casos en los que se soiubiiizó 1a enzima con estereactivo, 1a concentración proteica se determinó mediante e] mgtodo de Bradford (1976).
La elución de las proteinas de ias coiumnas se siguiópor ia medición de absorbancia a 280 nm, ó a 260 y 280 nm, deacuerdo a1 método descripto por Narburg y Christian (1957), yempleando 1a siguiente ecuación:
Proteinas (mg/m1) = 1,45 x A280 - 0,74 x A260
Para ios eiuidos de las coiumnas de TPS GB, e] perfilproteico se obtuvo por medición de las densidades ópticas a280 nm, pero como 1a TP interfiere en estas 1ecturas, se aplicó1a siguiente corrección (Grassetti y Murray, 1967);
A280 corregida = A280 - 1,25 x A343

H
187
.l3. REFERENCIAS
Andrews, P. (l964) Biochem. J. gl, 222.
Andrews, P. (l965) Biochem. J. _g, 595.
Azcurra, J.M.; Capasso, J.M.; Carrasco, A.E. & Lacoste, C.L.(l978) Acta Physiol. Latinoam. gg, l63.
(l968) Ciencia e Investigación 24, 242.Batlle, A.M. del C. __
Bradford, N. (1976) Anal. Biochem.lg, 248.
Bustos, N.L.; Stella, A.M.; Nider de Xifra, E.A. & Batlle, A.M. del C. (1980) Int. J. Biochem. lg, 745.
Davis, B.J. (l964) Ann. N.Y. Acad. Sci. l l, 404.
Ellman, G.L. (1959) Arch. Biochem. Biophys. gg, 70.
England, P.T; Huberman, J.A.; Jovin, T.M. & Kornberg, A.(l969) J. Biol. Chem. gig, 3033.
Grassetti, D.R. & Murray, J.F. (1967) Arch. Biochem. Biophys.112, 4l.
Kawata, M. & Su2uki, K. (l983) Toxicol. Lett. lá, l3l.
Lowry, 0.H.; RosebrOugh, N.J.; Farr, A.L. & Randall, R.J.(1951) J. Biol. Chem. Hg, 265.
Mauzerall, D. & Granick, S. (l956) J. Biol. Chem. gig, 435.
(l970) Biochemistry g, 25l.Melchior, w.B. & Fahrney, D.
Miles, E.w. (l977) en Methods in Enzymology (Eds. S.P.Collowick y N.0. Kaplan). Academic Press, NewYork, vol. 51,p. 43l.
Moore, D.J. & Labbe, R.F. (l964) Clin. Chem. lg, llO5.
Ornstein, L. (1964) Ann. N.Y. Acad. Sci. lil, 321.
Stella, A.M. (l977) Tesis Doctoral, UBA.
Stella, A.M.; Wider de Xifra, E.A. & Batlle, A.M. del C.(l977) Moll. Cell Biochem. lg, 97.
Tanford, Ch.; Nozaki, Y.; Reynolds, J.A. & Makino, S. (l974)Biochemistry lg, 2369.
Warburg, 0. & Christian, w. (l942) Biochem. Z. 3l0, 384.
Weber, K. & Osborn, M.(1969) J. Biol. Chem. ¿55, 4406.

RESULTADOS Y DISCUSION

PURIFICACION DE ALA-D
I.1.
I
I
I
I.2.1.I.2.2.
I
.1
.1
.l
.2.
.1.
.2.
.3.
3.
CAPITULO I
Purificación de ALA-Dde higado porcino
Etapas de purificaciónCaracterizaciónI.1.2.1.I.1.2.2.
I.1.2.3.I.1.2.4.I.1.2.5.I.1.2.6.
EstabilidadCantidad de proteina y tiempo deincubaciónCinética de la reacciónEfecto de reactivos tiólicospH óptimoPeso molecular y especiesmoleculares
Métodoalternativo de purificación porcromatografía por afinidadI
I.I.I
.ll.1.
1
3.3.3;
9
hour
.3.1.
Purificación de
IntroducciónExperiencias previasInmovilización de ALA-D en TPS GBPurificación de ALA-D en TPS GB
ALA-D de higado bovino
Etapas de purificaciónCaracterizaciónI.2.2.1.I
HHHH
NNN ...
.2.
2
2
2
23501-50.)
.2.2.2.EstabilidadInfluencia de la cantidad de proteina y el tiempo de incubaciónCinéticaEfecto de reactivos tiólicospH óptimoPeso molecular
Métodoalternativo de purificación porcromatografía hidrofóbica1.2.3.1. Introducción
Página
188
188
191
191
191
191
192
193
193
195
195
198
200
205
207
207209
209
210210210212
213
214214

Página
1.2.3.2. Efecto de] coïato de potasio sobre1a actividad de] ALA-D 215
1.2.3.3. Experiencias previas 216I.2.3.4. Cromatografía en Phenyi-Sepharose 218
1.3. Conclusiones acerca de 1a purificación y caracterización de] ALA-Dobtenida a partir de hígados porcino y bovino 220

188
I. PURIFICACION DE ALA-D
I.l. PURIFICACION DE ALA-D DE HIGADO PORCINO
I.l.l. Etapas de purificaciónLuego de una serie de ensayos preliminares, el esquema
de purificación final resultó el siguiente. Salvo se indiquelo contrario, todas las operaciones se llevaron a cabo a 4 °C.
Extnachón. El higado, usualmente se partió de 200 g,cortado en pequeños trozos, se homogeneizó en una licuadora enporciones de lOO g, con 5 volúmenes de_gfg_20 mMpH 6,8, adicignado de cloruro de sodio 0,15 M; B-ME lO mMy_¿g¿¿_1_mm. El homogenato resultante (fracción H), se centrifugó 3h minutos a13.000 x g y al sobrenadante se le agregó una solución stock dePMSF50 mMen una relación 1:50 v/v (fracción SN).
TnazamLQntoténmtco. El extracto enzimático se llevóa 65 °C mediante agitación constante en baño maría, se mantuvoa esta temperatura durante 2 minutos y luego se enfrió inmediatamente en baño de hielo. Se centrifugó durante l5 minutos al3.000 x g y el precipitado se descartó, guardándose el sobrenadante, al que se le agregó B-MElO mM(fracción HT).
Fnaccionamtcnto con sulfato de amon¿o. Al sistema anterior se le adicionó sulfato de amonio finamente dividido hastallevarlo a 55 % de saturación, manteniendo el PHentre 6,8-7,0mediante el agregado de hidróxido de amonio concentrado. La meícla se dejó agitando durante 45 minutos y luego se cehtrifugó30 minutos a l3.000 x g. El precipitado se disolvió en el minimovolumen de BFS 0,l M pH 6,8; conteniendo B-ME lO mMy PMSF l mM
(fracción 55 %) y se dializó contra 4 litros de BFS 5 mMdepH 6,8; B-ME5 mM. La solución dializada se centrifugó 20 minutos a 27.000 x g, el precipitado se lavó una-vez con el bufferde diálisis, se volvió a centrifugar y se reunieron los sobrenadantes (fracción D).
Caamatogna<ia en DEAE-celuiosa. El dializado, adicionado de PMSF l mM, se aplicó a una columna de DEAE-celulosa(2,5 x 45 cm) equilibrada con BFS 25 mM pH 6,8 y B-ME lO mM. Lacolumna se lavó con 240 ml del buffer de equilibrio al que se leadicionó cloruro de sodio 0,l M; luego se eluyó con un gradiente

189
Iineai de cloruro de sodio de 0,1 a 0,3 Men e] mismo buffer(aproximadamente 400 m] de cada soiución). La veiocidad de f1ujo se mantuvo en 45-55 mi/hora, recogiéndose 10 m1 por fracción.Los e1uidos con actividad especifica no inferior a] 50 %de] miximo vaïor, se reunieron, se supiementaron con B-ME10 mMyPMSF1 mMy se precipitaron con sulfato de amonio 55 %. Luegode 45 minutos de agitación, se coiectó 1a enzima por centrifugación a 13.000 x g por 30 minutos (fracción DEAE).
Caamarogaaáza en Sephacngi 3-300. La enzima proveniente de 1a etapa anterior, disuelta en e] minimo.voiumen de]buffer de equiiibrio, se sembró en una coiumna de Sephacry]S-300 (2,5 x 100 cm) equilibrada con BFS 50 mMpH 6,8 y B-ME10 mM. Se eluyó con e] mismo buffer a una veiocidad de9 4 12 m1/h, recogiéndose 23 m1 por tubo. Las fracciones enzimíticas con actividad especifica no menor de] 90 % de] máximo vaior se reunieron y se llevaron a 55 % de saturación con soluciónsaturada de suifato de amonio neutralizada; se dejó agitando por45 minutos y luego se centrifugó 20 minutos a 27.000 x g. E1 precipitado obtenido (fracción S-300) se guardó a -20 °C hasta e]momento de su uso.
En cada etapa de 1a purificación se determinó e] contenido de zinc (Iagla 1.1.), observándose que este meta] se pierde parciaimente durante e] curso de 1a misma. En consecuencia,1a enzima aisiada-no contiene cantidades estequiométricas dezinc, por 10 cua] 1a enzima nativa aitamente purificada no con;tituye una verdadera hoioenzima, dado que 1a concentración dezinc es de sóio 0,2 átomogramos de zinc_pormsubunidad.
Cuandose midió 1a actividad de las distintas fraccignes obtenidas a 10 1argo de] proceso de purificación, en lascondicionesstandard y agregandozinc (2 x 10'5 Mconcentraciónfina] en 1a mezcia de reacción), se observó en este último casoun aumento reiativo de 1a actividad (Tabïa 1.2.).
Segün podemos ver entonces en 1a Tabia 1.2., medianteei procedimiento descripto se Iogró purificar 1a enzima 654 veces con un rendimiento en unidades de] 14 %y una actividad especifica fina] de 12,4 UE/mgo 14,3 UE/mg, determinado en presencia de zinc.
Por otra parte, si 1a purificación se realiza en presegcia de zinc 0,0] mM,1a actividad especifica fina] es de18,2 UE/mgy 16,1 UE/mg respectivamente según que 1a actividad

190
cata1itica se mida con o sin zinc en e1 medio de incubación.
TABLA1.1.: Contenido de zinc en e1 curso de1a purificación
FRACCION atg Zn atg Zn/mg prot.
H 3.493,8 0,158
SN 2.148,0 0,129
HT 1.516,9 0,419
55 Z 603,5 0,310
D 49,5 0,155
DEAE 8,5 0,282
5-300 7,4 0,532
Las condiciones experimenta1es se describen en e1 texto.
En experiencias pre1iminares, 1uego de1 fraccionamiento con su1fato de amonio, e1 precipitado proteico se pasó poruna co1umna de Sephadex G-200 (2,4 x 54 cm) equi1ibrada y e1uida con BFS 50 mMpH 6,8. Las porciones con actividad enzimáticareunidas, se concentraron con su1fato de amonio 55 %y e1 precipitado se diso1vió en BFS 50 mMpH 6,8 (fracción G-200). En esta etapa se 1ogró una purificación de 2 - 3 veces respecto de1a precedente.
En 1a Figura I.1. podemos observar e1 perfi1 cromatogrífico obtenido en Sephacry1 S-300. Se hace notar que una recromgtografia en 1a misma co1umna no aumentó e1 grado de purificación.
La homogeneidad de esta fracción se contro1ó por e1ectroforesis en PAGE,obteniéndose una banda única (Figura 1.2.).
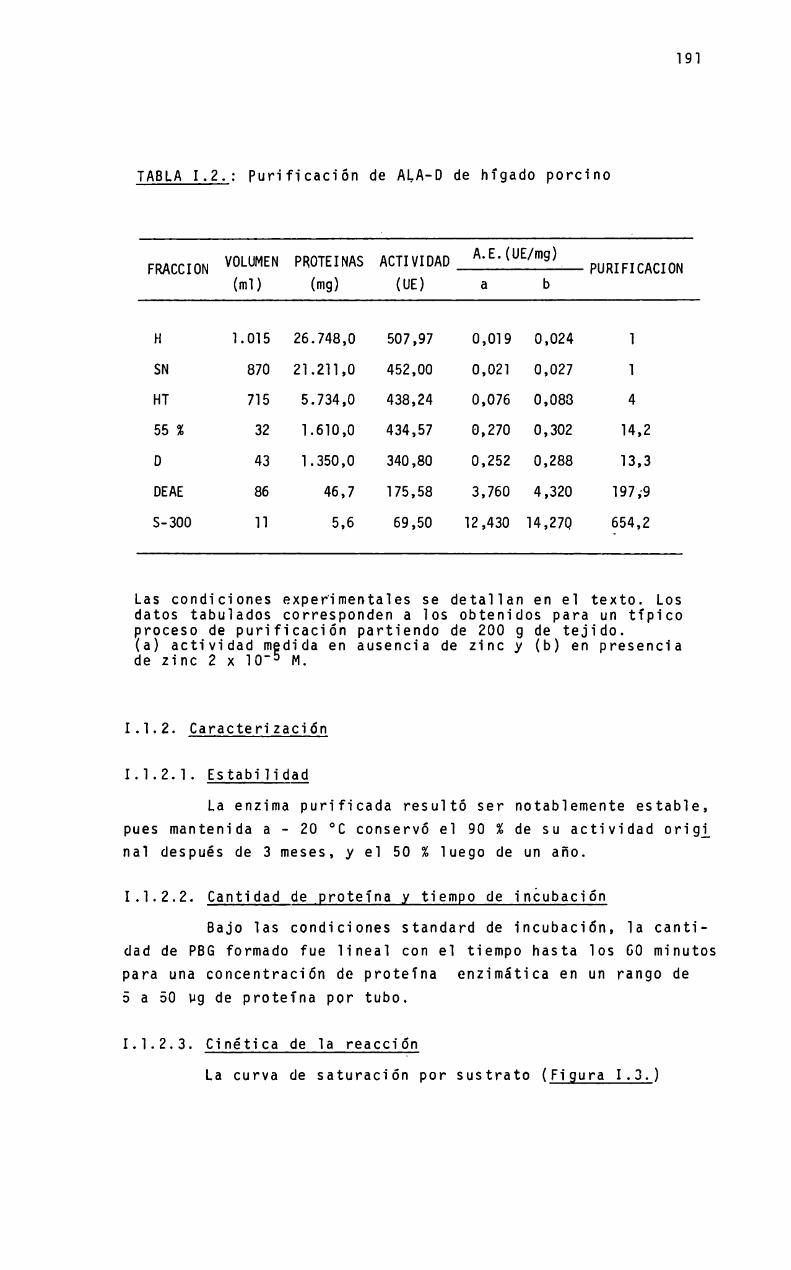
191
TABLA1.2.: Purificación de ALA-Dde higado porcino
FRACCION VOLUMEN PROTEINAS ACTIVIDAD __f¿É¿ÉÉÉfiÏEZ¿__ PURIFICACION(m1) (m9) (UE) a
H 1.015 26.748,0 507,97 0,019 0,024 1
SN 870 21.211,0 452,00 0,021 0,027 1
HT 715 5.734,0 438,24 0,076 0,088 4
55 z 32 1.610,0 434,57 0,270 0,302 14,2
0 43 1.350,0 340,80 0,252 0,288 13,3
DEAE 86 46,7 175,58 3,760 4,320 197,9
s-300 11 5,6 69,50 12,430 14,270 654,2
Las condiciones experimentaies se detaiian en ei texto. Losdatos tabulados corresponden a los obtenidos para un tipicoproceso de purificación partiendo de 200 g de tejido.(a) actividad mgdida en ausencia de zinc y (b) en presenciade zinc 2 x 10' M.
1.1.2. Caracterización
1.1.2.1. EstabilidadLa enzima purificada resultó ser notabiemente estabie,
pues mantenida a - 20 °C conservó e] 90 % de su actividad origina] después de 3 meses, y e] 50 % iuego de un año.
1.1.2.2. Cantidad de proteina y tiempo de incubación
Bajo ias condiciones standard de incubación, 1a cantidad de PBGformado fue iineai con e] tiempo hasta ios 60 minutospara una concentración de proteina enzimática en un rango de5 a 50 ug de proteina por tubo.
1.1.2.3. Cinética de 1a reacción
La curva de saturación por sustrato (Figura 1.3.)

192
muestra que 1a enzima sigue una cinética michaeiiana, iográndode 2,5 mM. E1se 1a saturación con una concentración de ALA
Kmes 0.27 mM(Figura 1.3., inset); este valor es de] mismo orden a] obtenido con preparaciones purificadas de higado bovino(0,15 mM;Wilson y c01., 1972) y de gióbuios rojos humanos(0,27 mM;Anderson y Desnick, 1979).
35 40FRACCION
nm ,
FIGURA1.1.: Cromatografía en Sephacry] 5-300.Las condiciones experimentales se detaïïan enMateriales y Métodos. ( 0 bsorbancia a 28
Absorbancia a 555 nm(o)
1.1.2.4. Efecto de reactivos tióiicos
Comoocurre con 1a enzima proveniente de otras fuentespara 1a máximaexpresión de 1a actividad cataiitica se requiereIa presencia de agentes reductores de grupos suïfhidriios en e]medio de incubación. De los reactivos ensayados, cisteina, B-ME,GSHy DTT, este último resultó ser e] más eficaz,a una concentración de 10 mM(Figura 1.4.).

193
q.) Fosforiiasa b
Seroalbümina‘ bovina
‘v 0voa1bümina
¡.I, Anhidrasacarbonica
Inhibidor de tripsinan a-Lactoalbúmina
FIGURA1.2.: E1ectroforesis SDSPAGE de ALA-D
purificada dehigado porcino.
1.1.2.5. pH óptimoLa curva de actividad enzimática en función de] pH
(Figura 1.5.) muestra un pico único a pH 6,8 tanto con bufferde fosfatos como con buffer Tris, coincidiendo con e] vaior óptimo reportado para 1a dehidrasa de otros tejidos mamíferos(Granick y Mauzeralï, 1958; Coleman, 1966; Bat11e y co]., 1967;Bustos y co].. 1980).
1.1.2.6. Peso moïecuïar y especies moïeculares
En las primeras experiencias, e] peso moiecular (PM),ca1cu1ado por e1 método de fiïtración por geïes, resuitó ser de420 i 20 kDa. Mediante eiectroforesisSDS-PAGE,se determinó quee] PM de cada Subunidad es de 35 t 5 kDa, de manera que 1a enzima de PM420 kDa estaria formada por 12 subunidades, en lugardei octámero que es 1a especie molecuiar identificada en 1a mayoria de Ios casos.
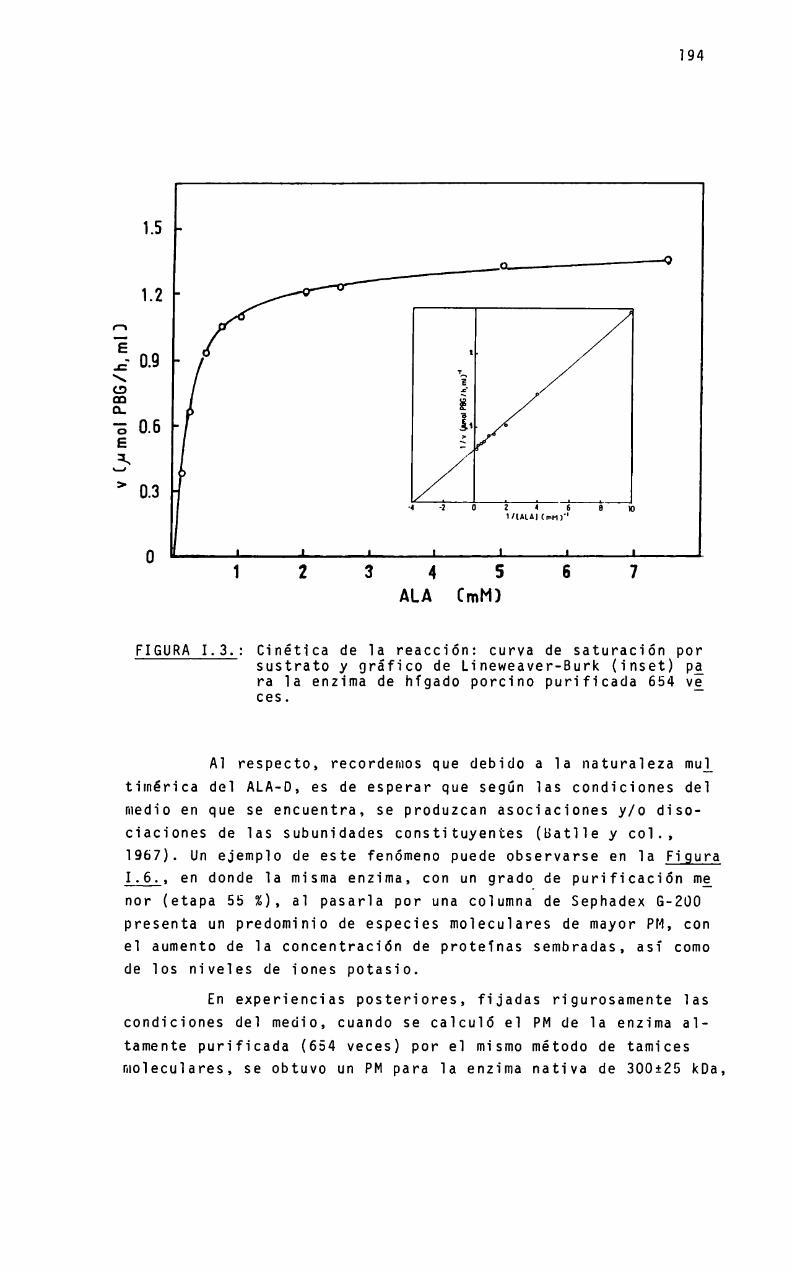
194
1.5
o_______________———-o
1.2 '
Te .
5- 0.9 - TB8-“3 0.6 - {dE :X /
> 0.3 . . e
0 l L 1‘ l J 1 l1 2 3 4 5 6 7
ALA (mM)
FIGURA1.3.: Cinética de 1a reacción: curva de saturación porsustrato y gráfico de Lineweaver-Burk (inset) para 1a enzima de higado porcino purificada 654 vgces.
A] respecto, recordemos que debido a 1a naturaleza multimérica de] ALA-D,es de esperar que según ias condiciones de]medio en que se encuentra, se produzcan asociaciones y/o disociaciones de las subunidades constituyentes (Batïle y co].,1967). Un ejemplo de este fenómeno puede observarse en 1a FiguraLLQL, en donde 1a misma enzima, con un grado de purificación mgnor (etapa 55 %), a1 pasarla por una coiumna de Sephadex G-200presenta un predominio de especies moïecuiares de mayor PM, cone] aumento de 1a concentración de proteinas sembradas, asi comode los niveies de iones potasio.
En experiencias posteriores, fijadas rigurosamente 1ascondiciones de] medio, cuando se calculó e] PMde 1a enzima a1tamente purificada (654 veces) por e] mismo método de tamicesmoïeculares, se obtuvo un PMpara 1a enzima nativa de 300125 kDa,
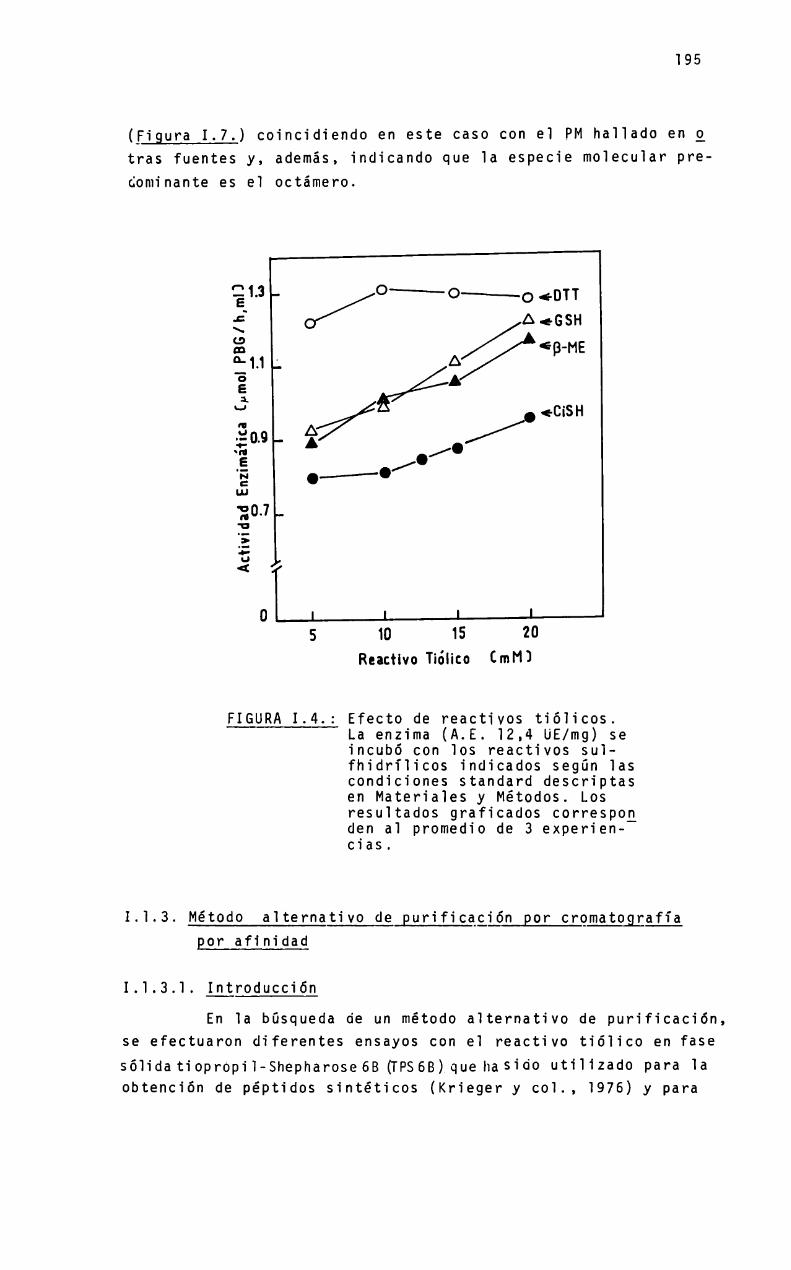
195
(figura I.7.) coincidiendo en este caso con e] PMha11ado en gtras fuentes y, además, indicando que 1a eSpecie molecular predominante es e] octámero.
—h la)l O o
C3/ /A (65HA/ <p-MEf;
/ <-CiSHe /'o <-DTT
-. . -. I
ActividadEnzimáiica(pmolPBG/h,m|)
53 l 0/.0/.___,__.
Q7
0 l l n l
S 10 15 20
Reactlvo Tiólico CmM)
FIGURA1.4.: Efecto de reactivos tióiicos.La enzima (A.E. 12,4 UE/mg) seincubó con los reactivos su]fhidriiicos indicados según iascondiciones standard descriptasen Materiaies y Métodos. Losresuitados graficados corresponden a1 promedio de 3 experienCias.
1.1.3. Método a1ternativo de purificación por cromatografíapor afinidad
1.1.3.1. introducciónEn 1a búsqueda de un método aiternativo de purificación,
se efectuaron diferentes ensayos con ei reactivo tiólico en fasesólidatiopr0pi1-Shepharose68 UPSGB)quehaSÍÓO utÍÏÏZGÓO Pal"a 13obtención de péptidos sintéticos (Krieger y c01., 1976) y para

196
1a purificación de ureasa de semiiias de leguminosas (Carïssony c01., 1976).
A.E.(ua/mg)
FIGURA1.5:: Actividad enzimática de] ALA-Dde higado de cerdo en funciónde] pH.La enzima (A.E. 12,4 UE/mg) seincubó a 105 pH indicados, manteniendo e] resto de 1as condiciones de incubación, según sedetaïïa en Materiales y Métodos.Los pH's indicados corresponden a ios de 1a mezcla de incubación . Los datos graficadoscorresponden a1 promedio de 3experiencias.(C)) Buffer de fosfatos;(la) Buffer Tris; ambos a 50 mM.
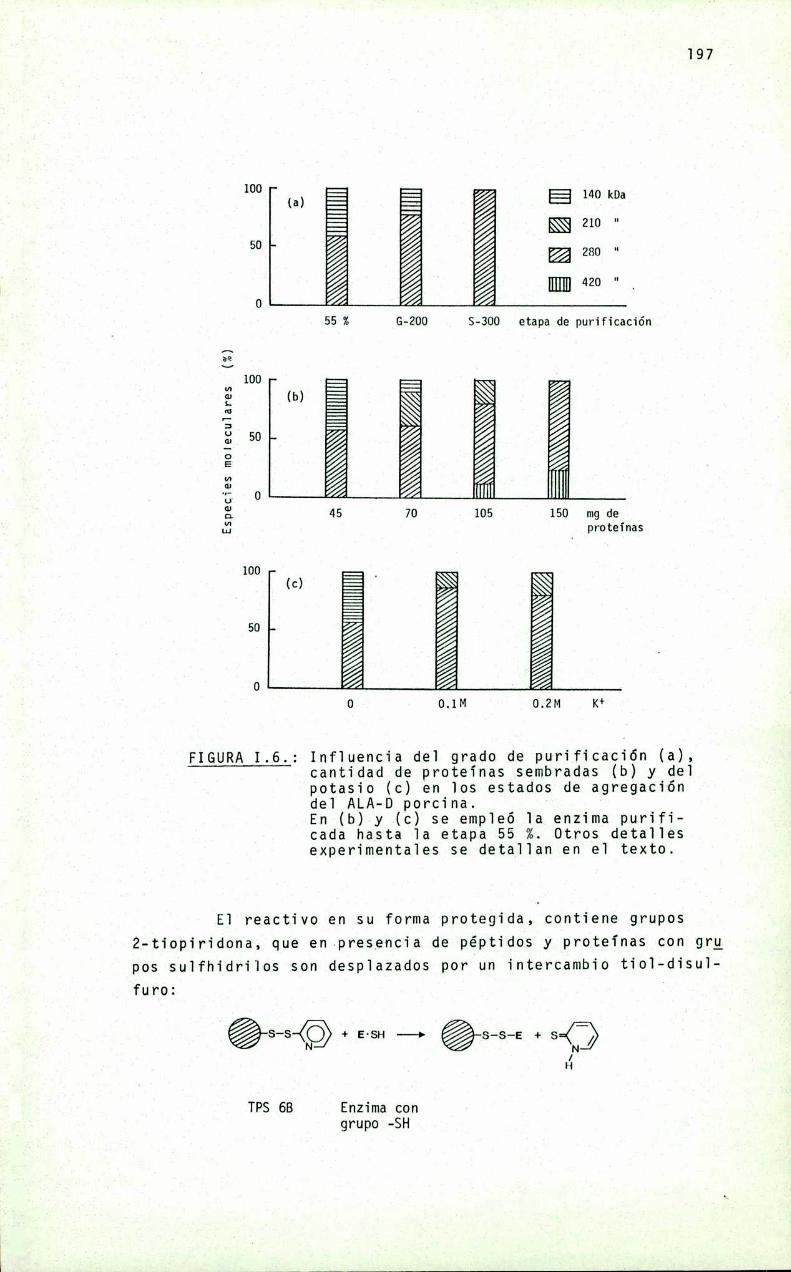
197
100g 140kDa
210 "
7/4 280 "
[[[flfl
(ü
50
420 "
G-200 5-300 etapa de purificación
(%)
mmLtu
3Um
OEm
3U.
g mg deVI au prote] nas
100(d
50
0
O 0.1M K+
FIGURA1.6.: Infiuencia de] grado de purificación (a),cantidad de proteinas sembradas (b) y de]potasio (c) en ios estados de agregaciónde] ALA-Dporcina. 'En (b) y (c) se empieó 1a enzima purificada hasta 1a etapa 55 %. Otros detalïesexperimentaïes se detaiïan en e] texto.
E1 reactivo en su forma protegida, contiene grupos2-tiopiridona, que en presencia de péptidos y proteinas con grgpos suïfhidriïos son desplazados por un intercambio tioï-disulfuro: '
' 8-30 + E-SH——> _ _ O¿225- S) ¿ÉÉ}S S E + S N //
H
TPS GB Enzima congrupo -SH

198
E1 tratamiento con agentes reductores tales como DTT0B-MEpermite separar tanto 1a proteina comoe] grupo protector;obteniéndose asi un tio] 1ibre en fase sóiida:
áS-S-E EH» SH + E-SH+ R-S-S-R
Dadas las caracteristicas suifhidriïicas de] ALA-D,sepensó en utiiizar este reactivo en fase sóiida para una cromatggrafía por afinidad, y estabiecer si era posibïe empïearlo comouna etapa adiciona] en 1a purificación de esta enzima.
&4 0voolbúmína
o 4.Z 0'3 - s lb. . Fibrinógenoeroa urmnag bovina ALA-Dcn (moncmero) Seroolbúmina2 02 _ bovino Catalasa
' ((dfmero)
TiroglobulinoOJ
O l n n n l n n 1 1 n 1 l n l
S 10 20 40 BO4PM x10
FIGURA1.7.: Curva de caïibración de PMen Sephacry] 5-300.
1.1.3.2. Elperiencias previas
Teniendo en cuenta que 1a unión de 1a proteina a] ge]ocurre a través de sus grupos tioies. se consideró convenientepreincubar con DTT1a fracción enzimática a imnoviïizar, con e]objeto de reducir compietamente los grupos sulfhidriios y lograrasi un buen rendimiento de acople. Se 11ev6 a cabo entonces unaexperiencia previa empïeando una coiumna de Sephadex G-25 de
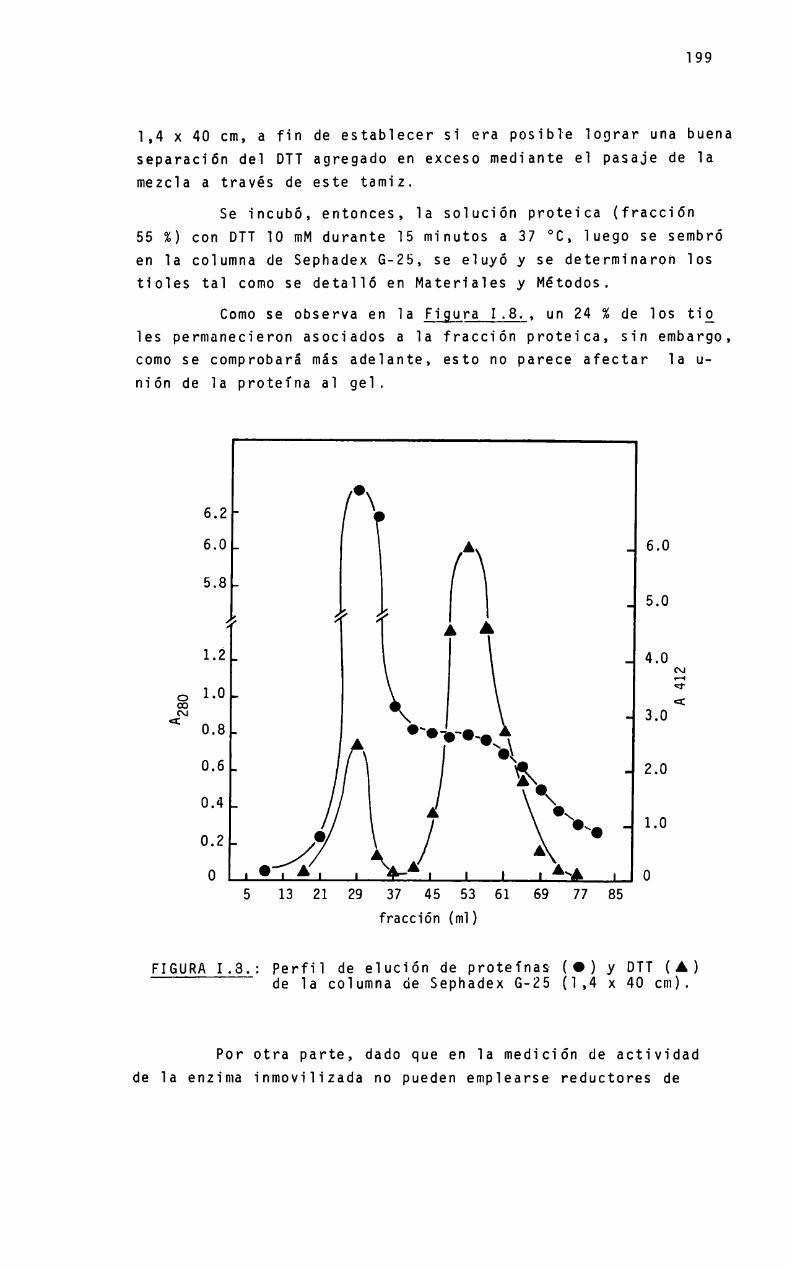
199
1.4 x 40 cm, a fin de estabiecer si era posibie Iograr una buenaseparación de] DTTagregado en exceso mediante e] pasaje de 1amezcia a través de este tamiz.
Se incubó, entonces, 1a solución proteica (fracción55 %) con DTT 10 mMdurante 15 minutos a 37 °C, 1uego se sembróen 1a columna de Sephadex G-25, se eluyó y se determinaron Iostioies ta] comose detaiió en Materiaies y Métodos.
Comose observa en 1a [laura 1.8., un 24 % de 10s tig1es permanecieron asociados a 1a fracción proteica, sin embargo,como se comprobará más adeiante, esto no parece afectar 1a unión de 1a proteina a] ge].
o6.2- \
6 0- A _ 6 0
5.8_- 5.o
¡5 ¿p' A
12 _ _ 4 0S
8 1.0- ZN - 3 o< s
08.. .‘.-...‘.\\ oo 6- ‘ _\2\ 2°o0.4 _ A \. \.\ _ 1.0
0.2- /0 / °A
0 1 . n ‘ 1 1 A\4"'Al l l l“A 05 13 21 29 37 45 53 61 69 77 85
fracción (m1)
FIGURA1.8.: Perfil de e1ución de proteinas (O) y DTT(A)de 1a columna de Sephadex G-25 (1,4 x 40 cm).
Por otra parte, dado que en 1a medición de actividadde 1a enzima inmoviiizada no pueden empiearse reductores de
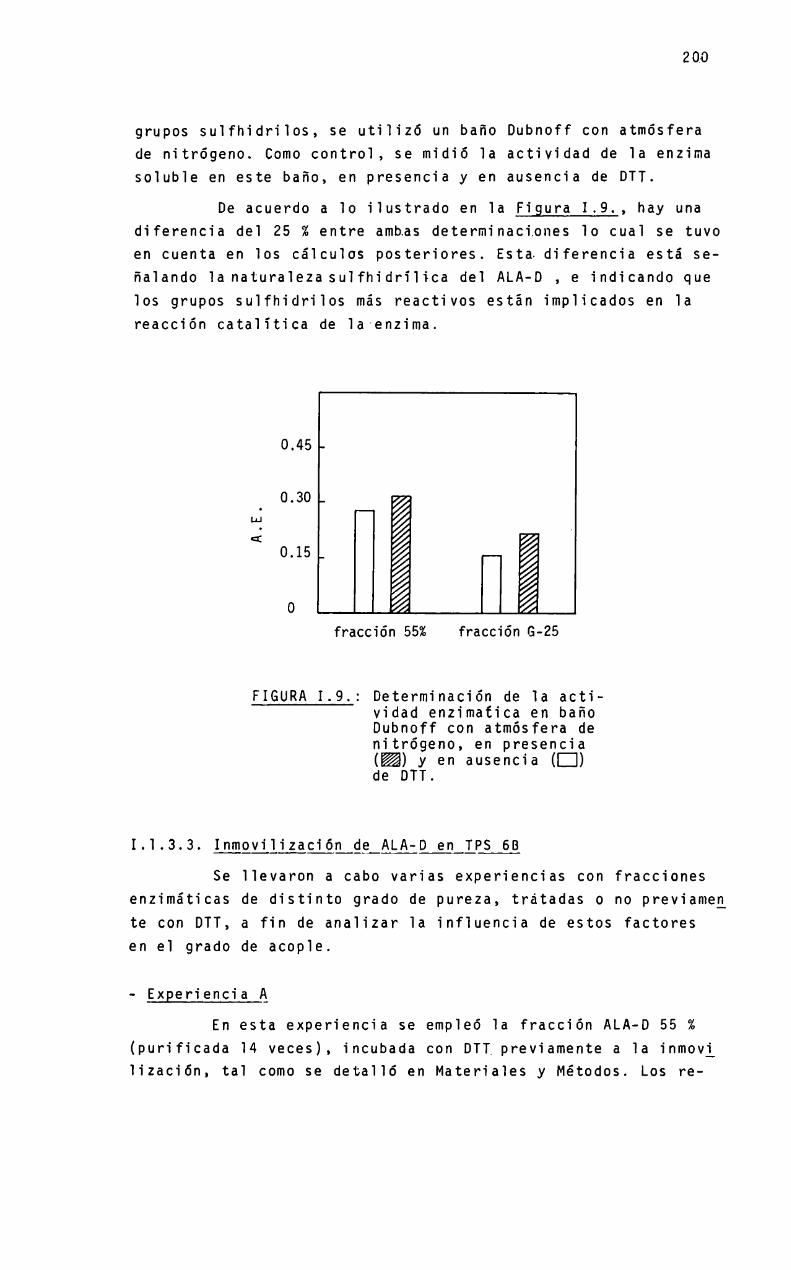
200
grupos suifhidriios, se utiiizó un baño Dubnoff con atmósferade nitrógeno. Comocontro], se midió 1a actividad de 1a enzimasoiubie en este baño, en presencia y en ausencia de DTT.
De acuerdo a io ilustrado en 1a Figura 1.9., hay unadiferencia de] 25 %entre ambas determinaciones io cua] se tuvoen cuenta en los cálcuios posteriores. Esta diferencia está señaiando 1anatura1e2a5u1fhidri1ica de] ALA-D, e indicando queios grupos suifhidriios más reactivos están implicados en 1areacción cataiitica de 1a enzima.
0.45
0.30
i124fracción 55% fracción G-25
FIGURA1.9.: Determinación de 1a actividad enzimafica en bañoDubnoff con atmósfera denitrógeno, en presencia() y en ausencia (El)de DTT.
1.1.3.3. Inmoviiización de ALA-Den TPS 68
Se iievaron a cabo varias experiencias con fraccionesenzimáticas de distinto grado de pureza, tratadas o no previamente con DTT,a fin de analizar 1a infiuencia de estos factoresen e] grado de acopie.
- Experiencia A
En esta experiencia se empleó 1a fracción ALA-D55 %(purificada 14 veces), incubada con DTT_previamente a 1a inmovilización, tai comose detaiió en Materiaies y Métodos. Los re

201
resuitados se resumen en.1a Tabla I.3.
IABLA1.3.: Inmovilización de ALA-Dpurificada 14 veces ypretratada con DTT, en TPS 6 B
CONDICIONES UE/m] ge] mg prot/m] ge] A.E.
% %
Sembradas 118,38' 100: 331,8 100 0,354
Inmovilizadas (a) 71,02 60 153,8 46 0,464
Soïubiïizadas (b) 53,85 45 26,3 17 2,048
Expresadas (c) 1,29 1,1*2,4
Las condiciones.experimentaies se describen en eï texto.(a) Se obtuvieron por diferencia entre las unidades sembradas y las unidades no retenidas por 1a coiumna.(b) Se obtuvieron por tratamiento con DTT25 mM.(c) Representa 1a.actividad de 1a enzima inmoviïizada, medida según se describe en Materiaïes y Métodos.(*) Caïculado respecto a las unidades (b).
Observamos que, casi un 50 % de ias unidades y proteinas sembradas fueron retenidas por e] ge]. Sin embargo, e] tratamiento posterior con DTTparece soïubilizar las unidades deALA-Den forma preferencia] con respecto a otras proteinas contaminantes que también habian sido inmovilizadas, según surgede 1a variación en las acividades especificas.
Vemos también que 5610 un 2,4 % de 1as unidades retenidas se manifestaron cataïiticamente activas; hecho que atribuimos a que 1a enzima queda inmoviiizada a través de sus grupostioies, entre 105 que evidentemente se encuentran los invoïucrados en e] sitio activo.
Cuando 1a enzima inmoviïizada se mantuvo a 4 °C sin tener especia] cuidado en mantener ias condiciones reductoras de]medio, a] cabo de 21 dias perdió toda su actividad.
Observemos además, que por tratamiento con DTT 25 mH

202
se 1iberan unidades soïubïes activas, 10 que pone de manifiestono sóïo 1a presencia de grupos tioles esencíaïes bloqueados porsu unión a] ge], sino que además esta asociación es reversible.
En 1a Figura 1.10. podemos ver que e] exceso de proteinas y unidades no retenidas son eliminados junto con Ios grupostiopiridona despïazados por 1as proteínas inmovilizadas.
10.0 _1.8
-3.08.0 _
_1.26.0
-2.0o l84° Lu
< m 54.o _ fa
-0.6-1.0
2.0
OJO
Voïumen (m1)
s O) y despïazamiegFIGURA1.10.: Perfi] de eïución de proteín (( durante e] pasaje
ato de grupos tiopíridona A.)por 1a coïumna de TPS-68.(I ) Unidades enzimáticas. Las condiciones experimentaïes se detaïïan en Materiales y Métodos.
Un cáïcuïo de Ios umoJes de grupos tíopiridona desplazados durante 1a inmoviïización, arrojó un resultado de 10 umo1es de TP por m1 de ge]. Si tenemos en cuenta que e] contenidotota] de grupos protectores es de 20 umoïes por m1 de geï hinchado, este resultado indica que sólo 1a mitad de] tota] de los si

203
tios iigandos posibies han sido ocupados, a pesar de habersesembrado un exceso de unidades (según vimos en 1a Tabia 1.3.).
- Experiencia B:
Se empleó nuevamente una fracción ALA-D55 %, pero con1a diferencia que se uitracentrifugó durante una hora a 140.000xgcon e] fin de obtener una soiución más limpida, y no se preincubó con DTT. E1 objetivo de esta experiencia era determinar sie] hecho de no reducir los grupos sulfhidriios de ias proteinascontaminantes; podía mejorar 1a unión seiectiva de] ALA-Da]ge], a través de grupos tioies menos reactivos.
Comopuede verse en 1a Tabia 1.4. de] tota] sembradose retuvo un 34 % de unidades y un 23 % en proteinas. 10 que e;taria sugiriendo una mayor selectividad por 1a enzima.
TABLA1.4.: Inmovilización de ALA-Dpurificada 14 veces no tratada con DTT, en TPS-68.
CONDICIONES UE/m] ge] mg prot/m] ge]
% %
Sembradas .79,61 100,0 150,0 100,0
Inmoviiizadas (a) 27,39 34,4 34,3 22,9
Soïubilizadas (b) 12,64 15,9
Expresadas (c) 0 0
Se empleó una fracción 55 Z u1tracentrifugada, sinincubación previa con DTT.(a), (b), (c), como en 1a Tabia 1.3.
Se observa que se puso en contacto con e] ge] una menorcantidad de proteinas que en 1a experiencia A, y que e] porcentaje inmoviiizado es menor, io que podemos atribuirio a] hechode no haber preincubado con DTT.

204
Notamos además que, a pesar de no tener especia] cuidado en mantener ias condiciones anaeróbicas durante 1a experiencia, 1a inmoviiización de 1a enzima tuvo igualmente Iugar. Sinembargo, en estas condiciones, e] ALA-Dinmovilizada resuitó inactiva, pero cuando se produjo 1a disociación por tratamientocon DTT,1a proteina soiubiïizada recuperó su actividad.
- Experiencia C:
Se empleó 1a fracción enzimática DEAE,purificada 137veces,que se preincubó con DTTprevio a su inmovilización.
Para la soiubiiización de 1a enzima inmoyiiizada se aplicó un gradiente de DTTentre 0 y 25 mM(Figura 1.11.), con e]objeto de determinar si con este procedimiento se iograba un grado mayor de purificación.
40- {1 _20I _ 25
¡l/ —3.0/[Í/‘ /
3.0 ._ l/ _20 A _1 5I ll, É
8 I/ h d(N l / E_2.0 :;
I, 152.0 _ / m-l.0
A A ¡z/ I <3l/x/ -10/
z, 10l 1.0 ,_ /// _0.5
/ _ 5’ II"\.! o
’ Ó
/ \ '.\._._.\ Jl .‘.- \.\ lo .SEL, I“Ük n 1 ’yHFA. ‘.14.H.h1’l o o J o
0 6 12 18 24 30 36
Volumen (ml)
[nggfl 1.11.: Perfil de soiubilización de 1a enzima inmoviiizada.Se utiiizó un gradiente de DTTentre O y 25 mM.
(o; absorbancia a 280 nm; (---)DTT (mM);(A. absorbancia a "43 nm; (I) unidades enzimáticas.

205
Comopuede verse en 1a Figura 1.11., inicialmente seproduce un desplazamiento de proteinas no activas y grupos iigandos y posteriormente, a una concentración de DTTde 15 mM,tiene iugar 1a iiberación de 1a proteina enzimática como un único pico.
Se observó debido a 1a baja cantidad de proteinas puestas en contacto, que 1a retención fue tota] (lggla 1.5.). Nuevamente, 1a baja actividad expresada por ias unidades inmoviiizadas nos está indicando 1a participación de Ios grupos tioies esenciales en 1a unión con e] ge]; asimismo e] tratamiento conDTTsoiubiiiza preferenciaimente a] ALA-Dcon notabie actividad.
IABLA_1.5.: Inmovilización de ALA-Dpurificada137 veces y pretratada con DTT, enTPS-68
CONDICIONES UE/m] ge] mg prot/m] ge]
% %
Sembradas 21,25 100 8,7 100
Inmovilizadas (a) 21.25 100 8,7 100
Soiubiiizadas (b) 14,45 68 3,5 40
Expresadas (c) 2,70 13
Se sembró una fracción DEAEpreincubada con DTT.(a) y (c) tienen ios mismos significados que en 1aTabla LLQL(b) se solubiiizó con un gradiente deDTT entre 0 y 25 mM.
1.1.3.4. Purificación de ALA-Den TPS GB
Con ios datos obtenidos en ias experiencias anteriores,se procedió a anaiizar e] grado de purificación logrado mediante esta etapa de inmoviiización de la enzima en TPS GB.
En 1a experiencia A, ias unidades'soiubiiizadas se regnieron en dos fracciones, una con actividad especifica mayor que

206
3,00 UE/mg(FI) y otra con actividad específica menor (FII)(jab1a I.6.).
IAQLA_I.6.: Purificación de ALA-Den TPS-GB. Experiencia A
CONDICIONES UE/m1 ge] mg prot/m1 ge] A.E. PURIFICACION
Sembradas 118,38 331,8 0,35 1
So1ubi1izadas (FI) 17,44 4,5 3,88 11
(FII) 36,12 20,5 1,76 5
Podemos ver entonces que en 1a fracción FI, se a1canzóuna purificación de 11 veces, con una actividad específica fina1de 3,89 UE/mg. Notemos que e1 grado de purificación obtenidopara esta fracción es de1 mismo orden que para 1a etapa de cromatografía en DEAEce1u1osa en e1 esquema convenciona1 (13213I_.2_.>.
En 1a experiencia C, donde se emp1eó una fracción DEAEde actividad especifica 2,44 UE/mg, se obtuvo 1uego de 1a 501!bi1ización una fracción enzimática de actividad especifica4,13 UE/mg, es decir que se 1ogró una purificación de s61o1,7 veces.
Estos resu1tados nos estarian indicando que, aunque 1apurificación por DEAEce1u1osa y por TPS-68 obedecen a fenómenosdiferentes, ambos ge1es serían capaces de e1iminar 1a misma prgporción de proteínas contaminantes. Es decir, 1a TPS-6Bpodriauti1izarse comométodo a1ternativo de purificación, en 1ugar deuna cromatografía en DEAEce1u1osa.

207
1.2. PURIFICACION DE ALA-D DE HIGADO BOVINO
1.2.1. Etapas de purificación
La enzima proveniente de higado bovino se purificó mediante el procedimiento que se detalla a continuación. A menosse indique lo contrario, todas las operaciones se llevaron a cabo a 4 °C.
Extnacc¿ón. El higado fresco, aproximadamente 200 g,cortado en trozos pequeños, se homogeneizó en porciones de 50 gcon l50 ml de BFP 20 mMpH 6,8 adicionado de cloruro de potasio0,15 M, B-ME lO mM, acetato de zinc 0,0l mMy PMSF l mM (bufferI). Se hace notar que, teniendo en cuenta que en el cuadro depurificación de la enzima de higado de cerdo, gradualmente fuedisminuyendo la concentración de zinc, de forma tal que la enzima nativa altamente purificada contenia menos de 0,8 átomogramosdel metal por subunidad, durante la purificación del ALA-Ddehigado bovino se decidió incluir en la composición de los buffersacetato de zinc 0,01 mM.El homogenato (fracción H) se centrifugó 30 minutos a ll.000 x g, y al sobrenadante se le agregó unasolución stock de PMSF 50 mMen una relación de 1:50 v/v (fragción SN).
Tnatam¿ento ténmzco.El extracto enzimático se llevó a65 °C con agitación constante, se mantuvo a esa temperatura durante 2 minutos y se enfrió inmediatamente en baño de hielo.Luego se centrifugó 15 minutos a ll.000 x g, guardándose el sobrenadante (fracción HT).
Fnaccionamáento con bulgata de amon¿o. Al sistema anterior se le agregó sulfato de amonio finamente dividido hastallevarlo a 55 %de saturación. Durante el proceso, se ajustó elpH en 6,8 - 7,0 mediante el agregado de hidróxido de amonio. Lamezcla se dejó agitando 45 minutos y luego se centrifugó 30 minutos a 11.000 x g. El precipitado se disolvió en el minimo volumen de BFP 0,1 M; B-ME lO mM; acetato de zinc 0,01 mMy PMSFl mM(buffer II) (fracción 55 Z), y se dializó hasta el dia siguiente contra 4 litros de BFP 5 mMpH 6,8; B-ME 5 mM, acetatode zinc 0,01 mM(buffer III) La solución dializada se centrifugó 20 minutos a 23.000 x g; el precipitado se lavó una vez conbuffer III, se volvió a centrifugar y se reunieron los sobrenadantes (fracción D).

208
Cnomatoqnaflía en DEAE-cetuloba. El dializado, adicionado de zinc y PMSF, se aplicó a una columna de DEAE-celulosa(4 x 75 cm) equilibrada con BFP 25 mMpH 6,8; B-ME lO mM (buffer
IV). La columna se lavó con 400 ml del buffer de equilibrio conteniendo cloruro de sodio 0,l My luego se eluyó con un gradiente lineal de cloruro de sodio de 0,l a 0,3 Men el buffer IV(aproximadamente 400 ml de cada solución). La velocidad de flujo se mantuvo en 60 - 70 ml /hora, recogiéndose 12 —l5 ml/fragción. Los eluïdos con actividad especifica no inferior al 50 Zdel máximo valor, se combinaron, se suplementaron con zinc yPMSFy se precipitaron con-sulfato de amonio al 55 %. Luego de30 minutos de agitación, se colectó la enzima por centrifugación a 11.000 x g por 30 minutos.
Cnomatagaaáía en Uitaoqei. La columna de Ultrogel AcA34 (2,5 x lOO cm) equilibrada con BFP 50 mMpH 6,8; B-ME l mM(buffer V), se sembró con la enzima disuelta en el buffer de equilibrio adicionado de zinc y PMSF.Se eluyó a una velocidadde 15 - 20 ml/hora, recogiéndose 5 ml /tubo. Se reunieron lasfracciones enzimáticas con actividad especifica no menor al90 % del máximo valor, se llevaron a 55 % de saturación con solgción saturada de sulfato de amonio neutralizada, se dejó agitagdo por 30 minutos y luego se centrifugó 20 minutos a 39.000 x g(fracción UGI). El precipitado se disolvió en el buffer anteriormente citado, y se sometió a una segunda cromatografía enUltrogel. Se empleó el mismo medio eluyante, a un flujo delO - 15 ml/hora y se recogieron 3 ml/fracción. Los eluidos quecumplieron el criterio anteriormente establecido, se colectarony se llevaron a 55 %de saturación consolución saturada de sulfato de amonio. El precipitado, luego de centrifugar a 39.000 x gpor 20 minutos, se guardó a 4 oC hasta su uso (fracción UGII).
La Igglg_L¿l¿ resume los resultados de una purificacióntipica . La enzima se purificó 492 veces, con un rendimiento enunidades del 23 %y una actividad especifica-final de 18,2 umoles de PBGformados por hora, por mg de proteina. En estas condiciones, el contenido de zinc de la enzima purificada en presencia del metal, fue de 0,7 a 0,9 átomogramos de zinc por sub!nidad de 35 kDa.

209
TABLA_}.7.: Purificación de ALA-Dde higado bovino
VOLUMEN PROTEINAS ACTIVIDAD A.E.
FRACCION (m1) (mg) (UE) (UE/mg) PURIFICACION
H 1.028 27.400,0 1.013,8 0,037 1
SN 922 18.917,0 703,7 0,037 1
HT 718 2.975,0 634,3 0,210 5,7
55 % 32 896,5 623,8 0,700 18,9
D 42 817,9 569,7 0,700 18,9
DEAE 121 52,6 529,1 10,000 270,3
UG I 42 14,8 237,3 16,000 432,4
UGII 25 8,8 160,2 18,200 491,9
Los resuitados tabu1ados corresponden a una purificacióntipica a partir de 200 g de higado. La actgvidad enzimatica se midió en presencia de zinc 2 x 10' M.
1.2.2. garacterización
La máxima actividad obtenida (18,2 UE/mg) decayó rápidamente a un va1or de 16 UE/mg, que se mantuvo por 10 dias. A1cabo de dos meses, 1a enzima guardada a 4 °C, retuvo un 70 % de1a actividad origina]. Es interesante ac1arar que cuando 1a enzima conteniendo 0,9 átomogramos de zinc'por subunidad de 35 kDase guardó a -20 °C como precipitado de suifato de amonio, sintener especia1es precauciones por hacer1o en una atmósfera comp1etamente anaeróbica, y luego de un mes se midió e1 contenidode] meta], en 1a fracción obtenida luego de una centrifugacióna 4.000 rpm, se obtuvo un valor de 0.15 átomogramos de zinc porsubunidad. Esto está indicando que durante e] estacionamiento,a1menosen1as condiciones descriptas, también se pierde zinc, y

vimos que disminuye correlativamente su actividad . Es probableque esto se deba a una oxidación de los grupos sulfhidrilos queprovocaría el desprendimiento del metal, de acuerdo con las ideas de Tsukamoto y col. (1979). Recordemos además que la enzimapurificada de higado porcino, con sólo 0,2 átomogramos de zincpor subunidad de 35 kDa, no perdia actividad luego de 3 meses deestacionamiento en condiciones similares, resultados esperadosde acuerdo al comportamiento ahora observado para el ALA-Ddehigado bovino.
1.2.2.2. Influencia de la cantidad de proteina y el tiempo deincubación
Se estudió el efecto del empleo de cantidades variablesde proteina sobre la formación de PBG. Los resultados se ilustran en la Figura_L¿lg¿ Se observa que la formación de productoaumenta linealmente con la cantidad de proteina incubada, hastaaproximadamente 20 ug; con cantidades mayores de proteina ocurreuna desviación de la linealidadw Por otra parte, la actividadespecifica se mantiene constante entre 5 y 20 ug de proteina enla mezcla de reacción. En cuanto al tiempo de incubación, en laFigura I.l3. puede verse que hasta los 60 minutos, la formaciónde PBGaumenta linealmente.
1.2.2.3. i é ic_
Comose observa en la figura I.l4., la cinética de saturación por sustrato es hiperbólica, lográndose la saturacióncon una concentración de ALAde 2,5 a 4 mM, de manera similar alo que ocurría con la enzima oe higado porcino. El Kmobtenidoa partir de la gráfica de las inversas de Lineweaver Burk es0,27 mM,en concordancia con los valores de Kmobtenidos por otros autores para esta mismafuente (Batlle y col., 1967;Wilson y col., 1972).
1.2.2.4. Efecto de reactivos tiólicos
De manera similar a lo observado para el ALA-Dde otrasfuentes y de higado porcino, la presencia de reactivos tiólicostales como: cisteina, B-HE, GSHy DTTestimula la actividad catalitica de la enzima proveniente de higado bovino, obteniéndosesu máxima expresión en presencia de DTT lO mM,

(a) so
A
6'wo- A
unam ICL JO
g oEC
wo
¿Yw
o A l
o ¡o w 30proteína (pg)
'° (b) '
9- sdA
8.
7.
m-
9. A ¿í
3 8’E2' 7
3,0. .3 g_ w' .8'N .
0 .
7x. .
m-
9- o 's‘ '
a.
7.l l L I I I
o s 10 IS 20 zs Jo
proiniu (pg)
FIGURA1.12 Efecto de 1a concentración prgteica sobre (a) 1a formación dePBG y (b) sobre 1a A.E. de 1a egzima, a distintos tiempos de igcubación. Se empïeó 1a fracciónenzimática purificada 300 veces.
211

212
250
20m
rnO. ,'
=100[' 'y'4, , "6,
z ¡9' dso- ‘,n"
¡”.0.‘lo.-/o' A
0 IS 30 45 50
tiempo (min)
FIGURA1.13.: Actividad enzimática en función dedistintos tiemposde incubación.Las mezclas de reagción contenían lassiguientes cantidades de proteina:(0-0) Bug; (H)6,1 ug; (ono) 9,2 ug(r-a) 15,3 ug;(o o)31 ug.Se empleó 1a fracción enzimática purificada 300 veces.
1.2.2.5. pï_gptimo
La ¿iguga_í¿lg¿ muestra e] efecto de] pH sobre la actividad de] ALA-D.Tanto en buffer Tris-acetato como en bufferPIPES se obtuvo un pH óptimo entre 6,3 y 6,8 en concordanciacon e] rango de pH óptimo obtenido por otros autores para e]ALA-Dde esta misma fuente (Batile y co]., 1967; Wilson y co].,1972), y de otros tejidos de mamíferos (Granick y Mauzeralï,

213
1958; Anderson y Desnick, 1979).
1.6
]2-0/e w =.E ï.¿- em 8a CLz; ua ÉE 3j >:1>
0Á1)l l I l l
zs o 25 m zs 10
1HALAJ(mHY1
o l 1‘ l l [1,1 I0 1 2 3 4 10
ALA (mM)
FIGURAI.Lí;: Curva de saturación por sustrato y gráfico de invegsas (inset), para 1a enzima de hígado bovino purificada 490 veces.
1.2.2.6. Beso molgguLag
E1 PMde 1a enzima purificada, medida por 1a técnicade fiitración en geies, por HPLC, fue de 280 t 20 kDa (se detallará en Capituio II de Resuitados y Discusión), y por electroforesis SDS-PAGE,presentó una banda única homogénea de PM3815 kDa (figu¿a_1¿lg¿) Ambosvaiores, en concordancia con losreportados por otros autores para 1a enzima de esta misma fuente (Cheh y Neiiands, 1973; Wuy co]., 1974; Kreutzer y co].,1977).

214
12
o
n 9'- oU‘IE
2C
3 su.i
4
3..
o 1 L L L l l5 6 7 8
pH
FIGURA_I.152:Actividad enzimáticaen función de] pH.Se empleó bufferTris-acetato (O )y buffer PIPES (O),ambos a una concentración 50 mM.E]pH se midió a 25 °Cen 1a mezcia fina]de reacción; Losdatos graficados cgrresponden a1 promgdio de 3 experiencias.
1.2.3. Métodoaiternativo de purificación por cromatografíahidrgfóbtga
1.2.3.1. ¿ngggggcciógDe acuerdo a las caracteristicas de la Phenyi-Sepharose
CL4B, y teniendo en cuenta que, para una interacción hidrofóbica

215
1a eiución de 1a enzima retenida necesitaria de un agente disociante, se investigó primeramente ei efecto de] coiato de potasio sobre 1a actividad de] ALA-Dbovina, con e] objeto de constatar que este detergente podria ser empleado en 1a e1ución deiALA-D.
Fosforiiasa b ,*,.Seroalbümina
abov1n
0voa1bümina -1g,
Ch!
Anhidrasacarbónica "'
a-Lactoaibümina ‘fl‘I
¿LgygA“L¿lg¿zEiectroforesisSDS-PAGE de]ALA-D de higado bovino purificada 492veces.
1.2.3.2. Efecto del colato de potasio sobre 1a actividad de]ALA:H
Se ensayaron entonces concentraciones de colato de potgsio entre Oy.0,5 Z en e] medio de incubación, determinándose laactividad enzimática segün se describió en Materiales y Métodos.
Los resultados se resumen en 1a Tab1a_L¿g¿ De acuerdoa ello, hasta una concentración de 0,1 % de coiato de potasio.no se afecta 1a actividad enzimática de] ALA-D,pudiéndose portanto empiearse para ia eïución de 1a enzima.

1.2.3.3.
216
IABLA1.8.: Efecto de] colato depotasio sobre 1a actividad de] ALA-Dbovina
CONCENTRACION
DE COLATO z DE ACTIVIDADDE POTASIO
(z)
o 100
0,2 ioo
0,3 98
0,4 96
0,5 94
0,6 91
0,8 85
1,0 68
Se empleó 1a enzima purificada250 veces, A.E. = 8,8 UE/mg
EÁR€EÍÉDEÍÉE_EKEXIEE
Se hicieron entonces una serie de ensayos en 105 que sesembró en una columnita de 1,5 x 4 cm unos 4.- 6 mg de proteinaenzimática proveniente de la etapa de purificación DEAE(actividad especifica = 8,8 UE/mg).
En todos los casos, 1as columnas se equiïibraron conBFP 50 mMpH 6,8 conteniendo cioruro de potasio 0,25 My DTT1 mM.
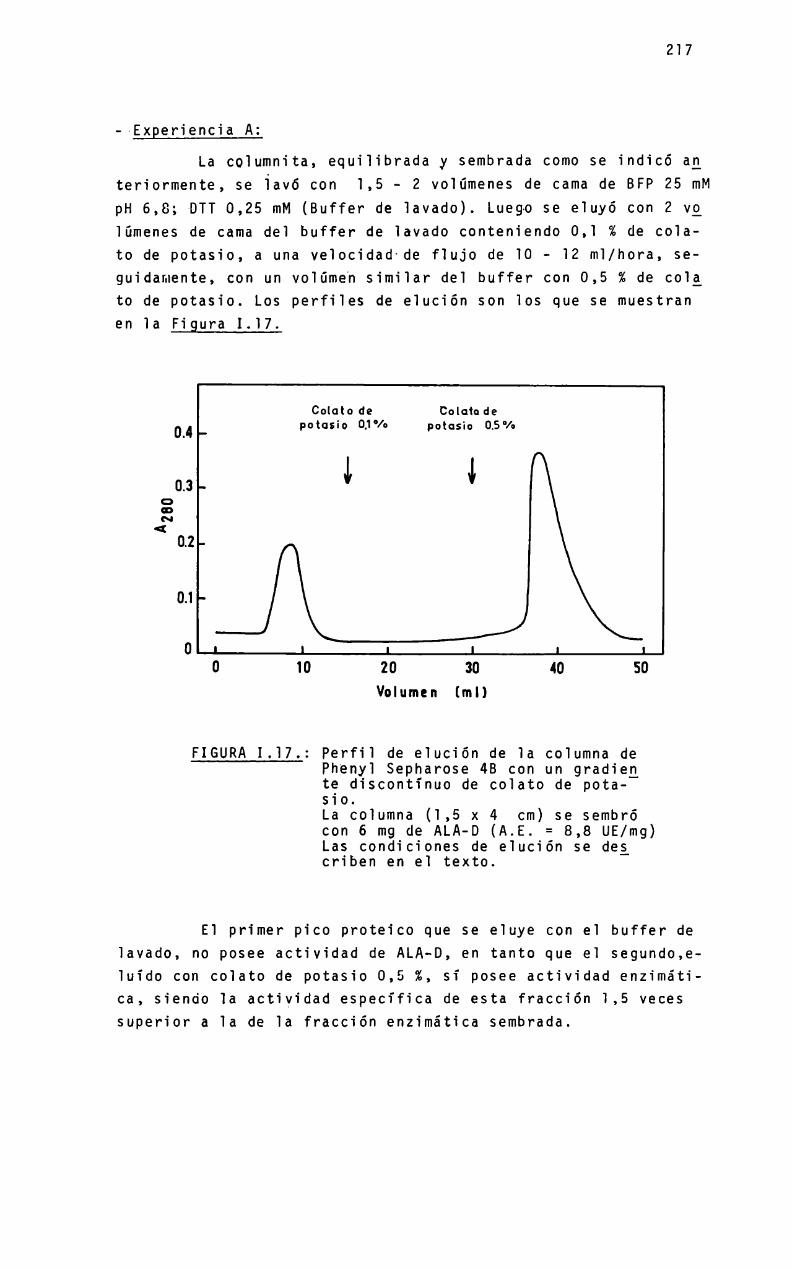
217
--Experiencia A:
La coiumnita, equiiibrada y sembrada como se indicó anteriormente, se Íavó con 1,5 - 2 voiümenes de cama de BFP 25 mMpH 6,8; DTT 0,25 mM(Buffer de lavado). Luego se eiuyó con 2 vgiümenes de cama de] buffer de iavado conteniendo 0,1 % de coiato de potasio, a una veiocidad-de fiujo de 10 - 12 mi/hora, seguidamente, con un volúmen simiiar de] buffer con 0,5 % de c013to de potasio. Los perfiies de eiución son ios que se muestranen 1a Figura 1.17.
Colato de Colqtgde
04,. P°tasí° 0-1°/° potasio 0.5%
0.3- i ioDN<
02
a1
0 l l 4 l | l0 10 20 30 4o so
VMumen (ml)
FIGURA1.17.: Perfii de e1uci6n de 1a coiumna dePheny] Sepharose 4B con un gradiegte discontinuo de coiato de potaSio.La coiumna (1,5 x 4 cm) se sembrócon 6 mg de ALA-D (A.E. = 8,8 UE/mg)Las condiciones de eiución se de;criben en e] texto.
E1 primer pico proteico que se eiuye con e] buffer deiavado, no posee actividad de ALA-D,en tanto que ei segundo,eiuido con coïato de potasio 0,5 Z, si posee actividad enzimática, siendo ia actividad especifica de esta fracción 1,5 vecessuperior a 1a de ia fracción enzimática sembrada.
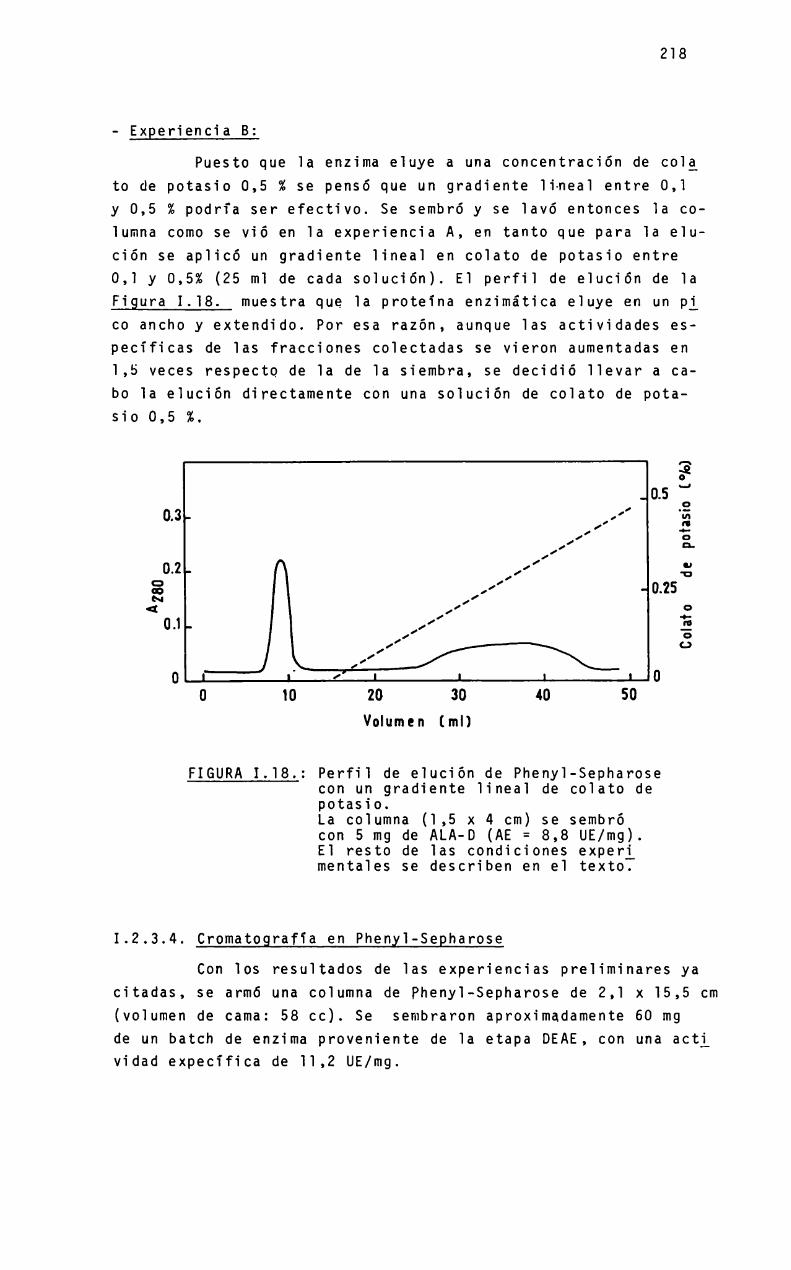
218
- Experiencia B:
Puesto que la enzima eluye a una concentración de colato de potasio 0,5 %se pensó que un gradiente lineal entre 0,ly 0,5 % podria ser efectivo. Se sembró y se lavó entonces la columna como se vió en la experiencia A, en tanto que para la elución se aplicó un gradiente lineal en colato de potasio entre0,] y 0,5% (25 ml de cada solución). El perfil de elución de laFigura 1.18. muestra que la proteina enzimática eluye en un pico ancho y extendido. Por esa razón, aunque las actividades especificas de las fracciones colectadas se vieron aumentadas enl,5 veces respecto de la de la siembra, se decidió llevar a cabo la elución directamente cOn una solución de colato de potasio 0,5 %.
33
0.3- zD.
0.2- g8N
< O0.1 - E
OQ
o
VMumen [mn
FIGURA1.18.: Perfil de elución de Phenyl-Sepharosecon un gradiente lineal de colato depotasio.La columna (l,5 x 4 cm) se sembrócon 5 mg de ALA-D (AE = 8,8 UE/mg).El resto de las condiciones experimentales se describen en el texto.
1.2.3.4. Cromatografía en Phenyl-Sepharose
Con los resultados de las experiencias preliminares yacitadas, se armó una columna de Phenyl-Sepharose de 2,] x l5,5 cm(volumen de cama: 58 cc). Se sembraron aproximadamente 60 mgde un batch de enzima proveniente de la etapa DEAE,con una actividad expecifica de ll,2 UE/mg.

219
La co1umna se equi1ibró y 1avó como se indicó en 1asexperiencias anteriores, y se e1uyó con BFP 25 mM, DTT0,25 mM,co1ato de potasio 0,5%, a una ve1ocidad de 30 m1/h, recogiéndosefracciones de 5 m1.
Se reunieron 1os tubos con mayor actividad especifica,se concentró con su1fato de amonio 55 %, se centrifugó a29.000 x g durante 15 minutos. Luego se disolvió en BFP 50 mMpH 7,4 y se dia1izó 5 horas a 4 °C. E1 dia1izado se sembró enuna coJumna de UGAcA 34 equi1ibrada y e1uida como se deta11óen 1.2.1.
Los resu1tados obtenidos se resumen en 1a Tab1a 1.9.
TABLAI.9.: Purificación de ALA-Da partir de 1a etapaDEAE
FRACCION VOLUMEN PROTEINAS ACTIVIDAD A.E.(m1) (m9) (UE) (UE/mg)
DEAE 10,0 56,0 626,4 11,2
Pheny1-Sepharose 14,6 20,7 342,4 16,5
UG 15,8 10,2 170,3 16,7
Las condiciones experimenta1es se describen en e1 texto
Comopodemos ver, 1a cromatografía hidrofóbica enPheny1-Sepharose conduce a un grado de purificación simi1ar a1obtenido por tamizaje mo1ecu1ar en U1tr0-ge1, pues se 11eva aobtener 1a misma actividad especifica por ambos métodos (Tab1aL¿Z¿. Por otra parte, e1 uso secuencia1 de ambas co1umnas, nomejoró 1a purificación (jab1a 1.9.), por 1o que 1a cromatografíahidrofóbica en Pheny1-Sepharose puede considerarse una a1ternat1va de 1a UGde1 esquema de purificación convenciona1.

220
1.3. CONCLUSIONES ACERCA DE LA PURIFICACION Y CARACTERIZACION
DEL ALA-D OBTENIDA A PARTIR DE HIGADOS PORCINO Y BOVINO
Teniendo en cuenta entonces que durante el curso de lapurificación de la enzima de higado de cerdo ocurrió una pérdidagradual del contenido de zinc, produciendo una fracción purificada con sólo 0,2 átomogramos de zinc por subunidad de 35 kDa, enla secuencia desarrollada para la purificación del ALA-Dde higado bovino, se incluyó siempre a dicho metal en la composición delas diferentes soluciones empleadas. Comose esperaba, el contenido de zinc en la fracción más pura obtenida a partir de higadobovino, estuvo en el orden de l átomogramo de zinc por subunidad de 35 kDa, de acuerdo con los datos obtenidos por Tsukamotoy col. (1979), para la enzima bovina purificada también en presencia de zinc. Estos resultados están además indicando que launión del metal a la enzima es relativamente lábil, y las condiciones experimentales influyen significativamente en la proporción del mismo retenida por la proteina._A su vez, el contenidode zinc y el estado de oxidación de los restos sulfhidrilos guardan una relación directa con la actividad catalitica del ALA-D,que es máxima para una concentración de zinc de l átomogramopor subunidad de 35 kDa, y cuando los grupos sulfhidrilos se hallan totalmente reducidos. La oxidación de estos últimos parecefavorecer el desprendimiento del metal, y lleva a una disminución de la actividad del ALA-D,conforme a la hipótesis deTsukamoto y col. (l979).
Por otra parte, se puede agregar que las propiedadesde las dehidrasas de higado de cerdo y vaca son notablemente similares en cuanto a.su estabilidad, actividad especifica, pHóptimo y Km.
Asi es que se logró un grado de purificación dentro delmismo orden, aün aplicando algunas variantes en la secuencia empleada para cada fuente. Es de notar que la cromatografía porafinidad en TPS GBes-una alternativa interesante quepuede reemplazar con igual eficiencia a la DEAEcelulosa; un idénticocriterio se puede hacer extensiVo al uso de una-cromatografíahidrofóbica con Phenyl-Sepharose. De manera que se pueden sustituir algunas etapas por otras, obteniendo similares resultados.
Las corridas cromatográficas y electroforéticas de lasenzimas nativa y disociada, muestran que,al igual que en otras

221
fuentes, ambas son oligoméricas y 1a especie moïecu1ar predominante es e] octámero, correspondiendo a 1a menor subunidad unPM de 35 kDa.

CAPITULO II
ESTRUCTURA CUATERÑARÍA Y ACTIVIDAD
Página
11.1. ALA-Dde higado porcino 222
11.1.1. ALA-Dsoiubie 222II.1.1.1. Inhibición por urea y su
reversión 22211.1.].2. Identificación de especies
moiecuïares por PAGE 222II.].1.3. Disociación en guanidina y
SDS 224
11.1.2. ALA-Dinmoviïizada 227II.1.2.1. Inmoviïización de ALA-D
sobre Sepharose 4B 227II.1.2.2. Disociación de Seph-ALA-Dcon
urea. Reasociación e hibridización 229
II.2. ALA-Dde higado bovino. Estudios sobre 1a reïación entre 1a estructura cuaternaria y 1a actividad enzimática, mediante HPLC 233
11.2.]. Comportamiento cromatográfico de 1aenzima nativa 234II.2.1.1. Efecto de 1a concentración
proteica 234II,2.I.2. Determinación de] peso
molecular 235II.2.1.3. Radio de Stokes 235
11.2.2. Comportamiento cromatográfico de ALA-Den presencia de SDS' 236II.2.2.1. Patrón cromatográfico en
presencia de distintas concegtraciones de SDS 237
II.2.2.2. Determinación de] peso moïecg1ar de] ALA-Ddisociada 239

Página
II.2.2.3. Análisis de 1os radios deStokes en presencia de SDS 239
11.2.3. Efecto de] SDSsobre_e1 perfíl cromatggráfico en HPLCy-Ia acitvidad enzimática 243
11.2.4. Efecto de] golfocianoro de sodio 24411.2.5. Estudio de Íá actividad específica de Ios
distintos picos obtenidos por HPLC 246

222
II. ESTRUCTURA CUATERNARIA Y ACTIVIDAD
II.I. ALA-D DE HIGADO PORCINO
11.1.1. ALA-Dsoiubie
II.1.1.1. Inhibición por urea y su reversión
Teniendo en cuenta 1a experiencia de nuestro propio iaboratorio (Batiie y co]., 1978), se 11evaron a cabo una seriede experiencias para estudiar e] efecto de 1a urea sobre 1a actibidad de 1a enzima soluble.
Se ensayaron concentraciones crecientes de urea entre0,5 y 6 M. Los resu1tados obtenidos se grafican en 1a Figura11.1. Se usaron preparaciones enzimáticas de diferente contenidoproteico y se observó en todos Ios casos que 1a inhibición esfunción de 1a concentración de urea. Asi, con urea 0,5 M1a inactivación a1canzó a] 75 %, en tanto que concentraciones superiores a 2 M, produjeron 100 % de inhibición.
Por otra parte, se demostró que 1a inhibición por ureaes reversibIe, pues aI eiiminaria se recupera e] 100%de 1a actividad enzimática, para concentraciones de urea 1 y 3 M; cuandose trató con urea 6 M, se restauró e] 90 % de 1a actividad (Eigura 11.2.). Resuïtados que están de acuerdo con 1a hipótesis deNandi (1971) acerca de] dobIe efecto de 1a urea sobre 1a actividad de] ALA-D:como agente disociante e inhibidor competitivo.Debe aciararse que 1a forma más efectiva para eliminar 1a ureafue 1a diálisis; e] desaiado mediante columnas de Sephadex G-25no fue satisfactorio.
[1.1.1.2. Identificación de especies molecuïares por PAGE
Diferentes muestras de ALA-Dde higado de cerdo en suforma soiubie, tanto nativa como en presencia de urea 3 y 6 M,se sometieron a una eiectroforesis PAGE,y se calcuïaron Ios PMde 1as bandas proteicas resuitantes (figgra II.3.).
Se observó que 1a especie octamérica sóio estaba presente en la enzima nativa, no asi en 1as tratadas con urea (Igb1a 11.1.). Aün en e] caso de 1a enzima nativa, se detectó un23 % y un 5% de Ias formas de PM 140 y 70 kDa, respectivamente,

223
pero recordemos la influencia de determinados factores sobre elgrado de asociación del oligómero (Stafforini y col., l980) yla presencia de SDS O,l Men el buffer de siembra y corrida.
100 - g
ÏÏ 75 D<Q
i 50 -l¡C.)<
25 - 4:.\2L '\! oo L . . I ¡í
0 1 2 3
UREA (M)
FIGURAII.l.: Inhibición por urea del ALA-Dsolublede higado de cerdo.Las condiciones de incubación se describieron en Materiales y Métodos.Se grafican los porcentajes de actividad en función de las distintas concentraciones de urea, tomando comolOO Z la actividad de la enzima sinurea. Las fracciones enzimáticas utilizadas contenían: (O) 4,8 mg/ml;(A) 3,7 mg/ml; (I) 2,9 mg/ml y(V) 2,3 mg/ml..
Mediante esta técnica se pudo detectar que la especiemolecular presente en mayor proporción en los sistemas tratadoscon urea, corresponde a la de PM l40 kDa. A medida que aumentala concentración de urea la proporción de la especie de PMl40 kDa decrece, en tanto que aumenta la correspondiente al PM70 kDa. Con urea 6 M aparece una banda correspondiente a la especie molecular 35 kDa, que representa el l6% del total.
Si recordamos que cuando la enzima se somete a la acción de urea 6 M, sólo se restaura el 90% de la actividad
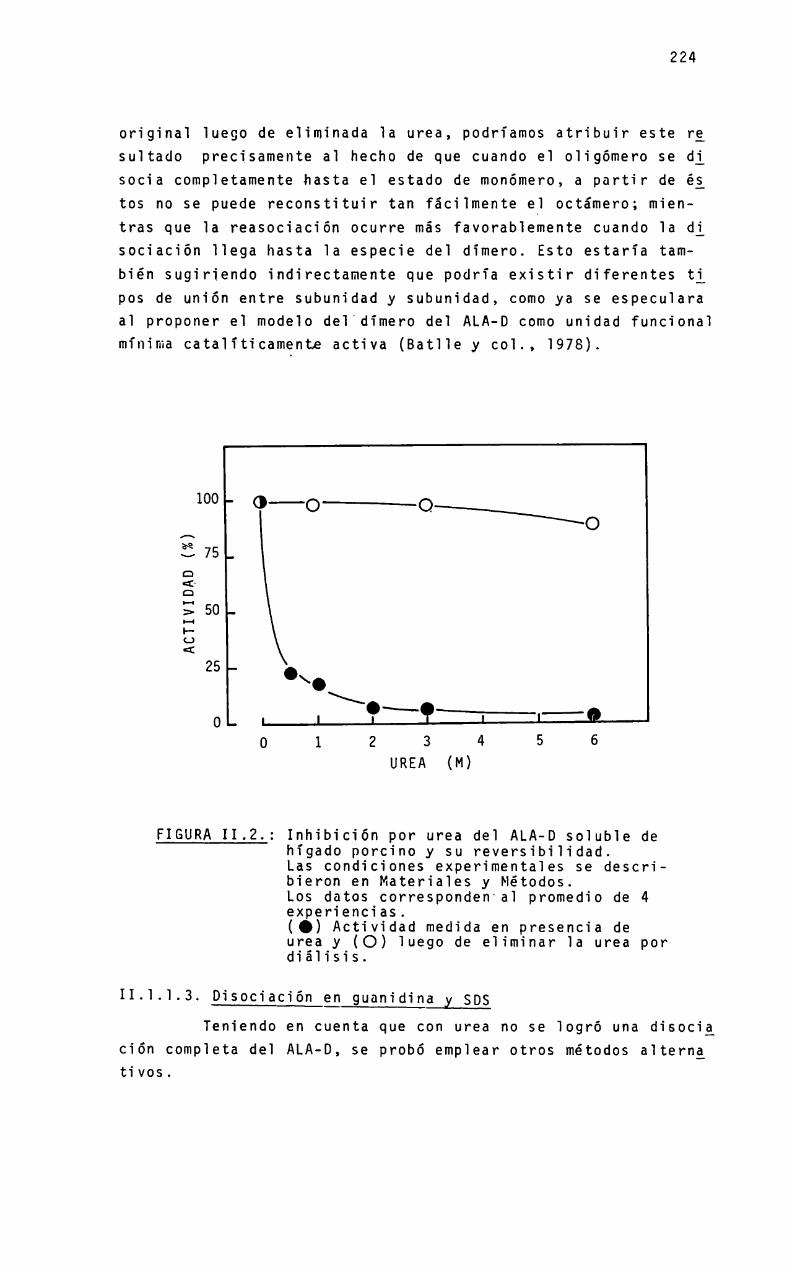
224
original luego de eliminada la urea, podriamos atribuir este resultado precisamente al hecho de que cuando el oligómero se disocia completamente hasta el estado de monómero, a partir de éstos no se puede reconstituir tan fácilmente el octámero; mientras que la reasociación ocurre más favorablemente cuando la disociación llega hasta la especie del dimero. Esto estaria también sugiriendo indirectamente que podria existir diferentes tipos de unión entre subunidad y subunidad, comoya se especularaal proponer el modelo del dimero del ALA-Dcomo unidad funcional
minimacataliticamente activa (Batlle y col., l978).
100- o__——_*._‘““‘*‘s\()
Ei 75 _Q<.D’; 50 _L:L)<
25 _ O\.\0L | l 9 q l l 4L
0 1 2 3 4 5 6
UREA (M)
FIGURAII.2.: Inhibición por urea del ALA-Dsoluble dehigado porcino y su reversibilidad.Las condiciones experimentales se describieron en Materiales y Métodos.Los datos corresponden al promedio de 4experiencias.(O) Actividad medida en presencia deurea y ((3) luego de eliminar la urea pordiálisis.
II.l.l.3. Disociación en guanidina y SDSTeniendo en cuenta que con urea no se logró una disocia
ción completa del ALA-D,se probó emplear otros métodos alternativos.

225
CII300 L <—1 Ez: 2 [::]4 [:216
i -- 3 c:::5 (2:37200 _ 8=
b EN 3MU 6MU
OBSA .<—2,4,6
m2 (dimero)
x 100 -— 0A .
Ï : (dimero) \- <i3,5,7BSA O
+ (monómero)
50 _
0A O_ (monómero)
<—8
30
I l l l
20 40 60 80
migración (%)
FIGURA11.3.: Determinación de los PMde las distintas especies de ALA-DsoiubIe nativa (EN) y tratadacon urea 3 y 6 M (3MU-6MU), obtenidas por eiegtroforesis e identificadas como bandas 1 a1 8(inset).Se representan Ios porcentajes de migración enfunción de] Iogaritmo de PM.BSA: seroaïbümina bovina; 0A: ovoaïbümina
Unade elias consistió en uti1izar guanidina. Para eiio,se trató 1a enzima (entre 0,15 y 0,5 mg de prot/m]; A.E.:3,98 UE/mg) con e] agente disociante y en presencia de DTT5 mM,durante 30 minutos a temperatura ambiente, o durante 10 minutosa 37 °C. Dicha concentración de guanidina produce 1a ruptura to

226
tal de 1a estructura oligomérica, generando 10s monómerosde35 kDa (Hu y co]., 1974). En esas condiciones, tiene iugar unainhibición tota] de 1a actiVidad enzimática, que no se recuperópor diáiisis: Esta se rea1izó a 20 °C durante toda una noche,contra BFS 10 mM pH 6,8; B-ME 1 mM. Debe tenerse en cuenta queparaieiamente se 11evó a cabo un contro] con 1a enzima no tratada con guanidina.
TÁBLA11.1.: Especies moiecuiares presentes en1a enzima soiubie nativa, y en 1aenzima tratada con urea 3 y 6 M
PM (kDa) EN (%) 3MU (%) 6MU (%)
280 72
140 23 65 43
70 5 35 41
35 16
Se tabuian los porcentajes correspondientesa 1as especies moiecuiares presentes en 1aenzima soiuble nativa (EN) y en 1a enzimatratada con urea 3 y 6 M (3MU - 6MU), luegode ser sometidas a una e1ectroforesis PAGE,segün se detalló en Materiaies y Métodos.Los porcentajes de 1as diferentes especies mglecuiares presentes fueron ca1cu1ados empieando e] método de integración de superficies.
Estos resultados confirmarian en principio, 1a idea deque aicanzado e] grado de disociación de] monómero,existen dificuitades para Iograr una correcta reasociación de Ios mismos.
Otro método ensayado, con igua] objetivo, fue e] de utilizar e] SDS1%como agente disociante; a esa concentración gcurre una disociación compieta de 1a moiécuia enzimática, y pérdida tota] de actividad catalitica. Las condiciones experimentaies fueron simiiares a Ias empieadas en los ensayos con guanidina.

227
Dado que por diálisis no fue posible eliminar por completo el agente disociante, se pasó la fracción enzimatica enpresencia de SDS por una columnita de Dowex lX-2 (lOO mg) equilibrada con BFS 50 mMpH 6,8. En los eluidos no se detectó actividad enzimática. El control de la enzima no tratada con SDS,al pasar por la resina intercambiadora, perdió un 40 - 45%deactividad especifica además de habernquedado retenida en la cglumnita un 30 - 40%de proteina enzimática.
Se introdujo entonces una modificación, consistente enequilibrar la columnita de DowexlX-2 con el mismo buffer, perocon el agregado de urea 6 M (Weber y Kuter, l97l). De este modo,el eluido, libre de SDSpero conteniendo urea, se dializó luegotoda una noche contra BFS lO mMpH 6,8; B-ME l mM. En esta formase esperaba conseguir una reasociación gradual de los monómeros.Comoresultado de este tratamiento, se obtuvo una recuperacióndel 50 % de actividad, con respecto al control no tratado conSDS.
Todos estos resultados no hacen más que apoyar nuestrasobservaciones acerca de las dificultades para que ocurra la regsociación de la enzima a su estado nativo, una vez que se disgció totalmente en monómeros, lo que a su vez está de acuerdocon nuestra idea de que podrian existir distintos tipos de unignes entre las subunidades y el dimero seria, necesariamente, launidad funcional minimacataliticamente activa.
Si bien queda por resolver la dificultad de que por este ültimo método, independientemente de la retención de proteinas, se produce una disminución de la actividad enzimática dela enzima, es interesante que por este camino podria llegarse arecomponer en un gran porcentaje la enzima oligomérica a partirde sus especies monoméricas, lo que posibilitaria a su vez, laformación de hibridos enzimáticos a nivel de subunidades funcignales, con monómerosprovenientes de fuentes diferentes.
II.l.2. ALA-Dinmovilizada
II.l.2.l. Inmovilización de ALA-Dsobre Sepharose 4B
En la Tabla II.2. se ilustra una experiencia tipica deinmovilización de ALA-Dde higado porcino, con los valores de gnidades enzimáticas, proteinas y actividad especifica de la enzima antes y después de su insolubilización.

228
TABLA11.2.: Inmoviiización de ALA-Dde higado porcino
FRACCION _ACTIVIDAD UE RETENIDAS PROTEINAS A.E.
'(UE/mi gel) l (Z) (mg/m1 gei) (UE/mg.m1 ge1)
ALA-Dsolubie 7.476 100 30,0-- 249,2
Seph-ALA-D (a) 3.552 48 25,6 138,8
Seph-ALA-D (b) 2.940 39_ 17,0 173,0
Se empieó 1a fracción enzimática proveniente de 1a etapa depurificación 55%de saturación en suifato de amonio.Las UE se expresan en nmoies PBG.h' .La activación de] gei se realizó segün se describe en Materiales y Métodos, con (a) 200 mg y (b) 50 mg de BrCN.
Se observa que cuando se empïearon 200 mg de BrCN, e]porcentaje de unidades enzimáticas y de proteinas retenidas fuesignificantemente mayor que cuando se preparó e] derivado insolubie con 50 mg de BrCN (48 % frente a 39 %, y 85 Z a 50 %, respectivamente). Esto indica que 1a unión de 1a proteina a1 ge]depende de los sitios reactivos presentes en ios componentes aacopiar, los que dependen también de] grado de activación de]ge] (Steiia y coi., 1977; Wider de Xifra y coi., 1978), de mangra ta] que a1 disminuir 1a concentración de BrCN, disminuirianios sitios de unión entre 1a proteína y e] soporte sóiido. Ennuestro caso, esto permitiría mantener a 1a enzimalnásïibremente expuesta a 1a acción de] agente disociante.
Por otra parte, 1a actividad especifica de 1a enzimainmovilizada es menor que 1a de 1a enzima solubie empieada parae] acopie (56 - 69 %), de manera simiiar a 10 encontrado para e]ALA-Dde higadO'bovino (Gurne y co]., 1977; Bat11e y co],, 1978).
Debemos tener en cuenta que cuando se une 1a enzima aun soporte sólido, se 1a está iievando de un estado libre en sgIución, a un micromedio diferente existente sobre 1a superficiey dentro de ios poros de 1a matriz, de manera ta] que se puedenaiterar las propiedadesfisicas y químicas de 1a enzima, además

229
de restringir su movimiento fisico. Puede ocurrir entonces quela unión covalente del ALA-Da la matriz de Sepharose distorsigne Su estructura tridimensional disminuyendo asi su actividadespecifica, o bien que algunas moléculas de enzima se inactivencompletamente, o bien que ocurra una combinación de ambos fenómenos.
II.l.2.2. Disociación de Seph-ALA-Dcon urea, Reasociación ehibridización
Para estudiar el efecto de la urea sobre el ALA-Dinmgvilizada se trató el sistema gel-enzima con diferentes concentraciones del reactivo; luego se lavó con BFSpara eliminar la ureay se midió la actividad del producto resultante en las condicignes ya detalladas en Materiales y Métodos.
Comopodemos ver en la Tabla II.3. en todos los casosy aün con urea 6 Mse detectó actividad catalitica. Esta es menor que la de la enzima inmovilizada nativa y diferente según lamolaridad de urea empleada. A mayor concentración de urea, la agtividad remanente es menor, lo que es un indice de que el gradode disociación depende de la concentración de urea, tal como sevió con la enzima soluble.
Si comparamos las dos primeras columnas, en que se usóla misma fracción enzimática, pero con gel de distinto grado deactivación, vemos que cuando se emplearon 50 mg de BrCN, la actividad remanente en el gel luego del tratamiento con urea, esmenor que cuando se emplearon 200 mg de BrCN. Como ya lo indicamos, esto podria explicarse teniendo en cuenta que cuanto menores el grado de activación del gel, menores son los sitios de unión de la proteina al gel, de manera tal que la enzima se encuentra más expuesta a la activación del agente disociante.
Investigamos también la reversibilidad del proceso dedisociación, pues nos interesaba saber si las subunidades inmovilizadas eran capaces de disociarse con las subunidades solubles agregadas en solución.
Para ello, la Seph-ALA-Ddisociada empaquetada en unacolumnita se trató con una mezcla de buffer conteniendo un gradiente discontfnuo de urea, y ALA-Dsoluble. Luego se lavó conel buffer renaturalizante hasta no detectar proteinas ni urea enlos eluidos, y se midió la actividad del sistema proteina-gelresultante.

230
T BLA11.3.: Disociación, reasociación e hibridizaciónde ALA-Dinmoviiizada
PORCENTAJE DE ACTIVIDAD
FRACCION 30 - 55 % 6-200
50 mg BrCN 200 mg BrCN 50 mg BrCN
Seph-ALA-D nat. 100 100 100
Seph-ALA-D dis-6M 6 12 22Seph-ALA-D reas-GM 14 27 50Seph-ALA-D hibr-6M 22 -
Seph-ALA-D dis-3M 26 33 49Seph-ALA-D reas-3M 29 56 86Seph-ALA-D hibr-3M 33 -
Seph-ALA-D dis-2M 38 49 55Seph-ALA-D reas-ZM 74 65 88Seph-ALA-D hibr-ZM 51 78
Seph-ALA-D dis-1M 67 65SephaALA-D reas-IM 84 81Seph-ALA-D hibr-IM 77 89
Las condiciones experimentales se describen en e} texto;Seph-ALAéD nat: ALA-Dinmoviïizada; Seph-ALA-D dis: ALA-Dinmovilizada y tratada con urea; Seph-ALA-Dreas.: 1uegode] tratamiento con urea, se pasaron subunidades de ALA-Dsoïubïe; Seph-ALA-Dhibr.: 1a reasociación se efectuó consubunidades soïubïes de ALA-Dde GR.
Comoconsecuencia de este tratamiento, vemos en 1a Igbla 1.3. que se produce un aumento de 1a actividad de] compïejogel-enzima. Resultado que sugiere que 1as subunidades unidas a1soporte sólido son capaces de asociarse con 1as subunidades so1ub1es generadas ¿n ¿¿Iu, reconstituyendo una proteina semejante a 1a enzima origina].

El grado de reasociación fue mayor cuanto menor fue elgrado de disociación; lo que es un indice de que la probabilidadde reasociación es mayor Cuando la enzima está menos disociada.
Cuando la enzima está más purificada, vemos que los grados de disociación son menores, en tanto que los de reasociaciónson más elevados, cercanos al 90 % (Tabla 11.3., columna 3).
Por otra parte, y de manera análoga a lo que ocurre conla enzima soluble, para la fracción enzimática 6-200, el tratamiento con urea 6 M conduce a una reasociación menor que paralos otros casos,con tratamientos menos drásticos (urea 2 y 3 M).
Los resultados obtenidos reflejan la importancia de lasinteracciones proteina-proteina, pues sabemosque en la célulalas enzimas no se encuentran aisladas, sino ubicadas en compartimientos o, aún aquellas que son citoplasmáticas, asociadas amembranaso a otras proteinas. En consecuencia, el grado de purificación de una enzima influye significativamente sobre su comportamiento. Asi, cuando se inmoviliza una enzima parcialmentepurificada, puede ocurrir que, debido a la presencia de otrasproteinas contaminantes, éstas competirian por los sitios de unión en el gel con la enzima de manera que no todas las moléculasen este caso del ALA-D,tendrian la posibilidad de establecercontacto y unirse a la matriz del gel, por lo cual podrian llegar a quedar en libertad octámeros completos de la enzima, quefinalmente se perderían en los lavados. Las proteinas contaminantes también podrian ejercer algún tipo de interferencia a nivel de las uniones entre la subunidad ligada al gel y la subunidad asociada a ésta, disociando eventualmente al dimero; sinembargo esta alternativa es la menos posible.
Paralelamente, se llevaron a cabo experiencias similares de reasociación pero empleando subunidades de ALA-Dde eritrocitos humanos, encontrándose también en estos casos, una restauración de la actividad de la Seph-ALA-Ddisociada (Tabla_LL;L,columna l). Esto nos está indicando que es posible la formaciónde hibridos activos entre enzimas de diferentes fuentes Es decir que si bien-pueden existir diferencias entre las secuenciasde aminoácidos de ambas dehidrasas, éstas no parecen ser muy significativas. al menos en lo que respecta a los sitios de contacto entre las subunidades.
Por otra parte, en los eluidos resultantes de la disociación con urea, se detectó actividad enzimática luego de
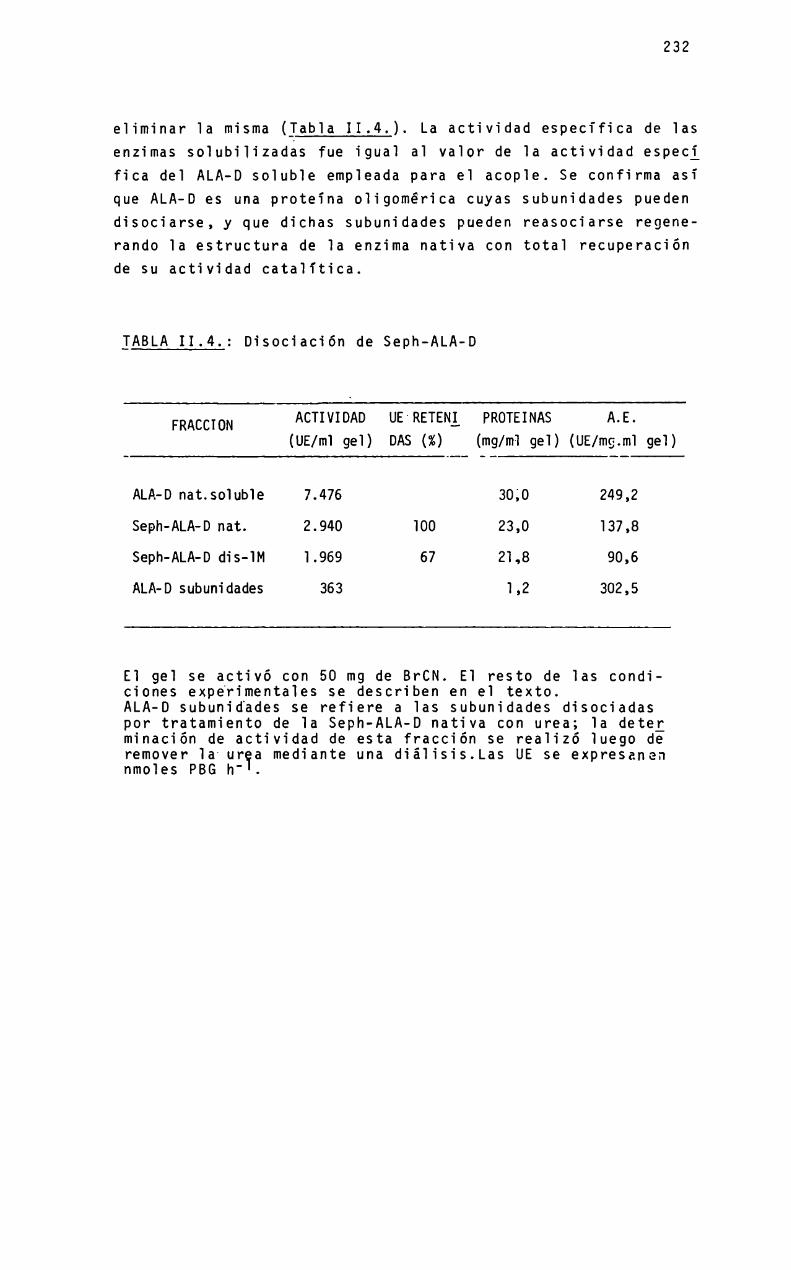
232
eliminar 1a misma (Iabïa 11.4.). La actividad específica de 1asenzimas soiubilizadas fue igua] a1 va10r de 1a actividad especifica de] ALA-Dsoiubie empïeada para e] acople. Se confirma asique ALA-Des una proteina oligomérica cuyas subunidades puedendisociarse, y que dichas subunidades pueden reasociarse regenerando 1a estructura de 1a enzima nativa con tota] recuperaciónde su actividad cataïitica.
IABLA11.4.: Disociación de Seph-ALA-D
FRACCION ACTIVIDAD UE'RETENL PROTEINAS A_E_
(UE/m1 93]) DAS (%) (mg/m1 geï) (UE/mg.m1 ge1)
ALA-Dnat.so]ub1e 7.476 30;0 249,2
Seph-ALA-Dnat. 2.940 100 23,0 137,8
Seph-ALA-Ddis-1M 1.969 67 21,8 90,6
ALA-Dsubunidades 363 1,2 302,5
E1 ge] se activó con 50 mg de BrCN. E1 resto de las condiciones experimentaïes se describen en e] texto.ALA-Dsubunidades se refiere a las subunidades disociadaspor tratamiento de 1a Seph-ALA-Dnativa con urea; 1a determinación de actividad de esta fracción se reaiizó Iuego deremover Ia'urea mediante una diá]isis.Las UEse expresanennmoïes PBG h' .

233
11.2. ALA-D DE HIGADO BOVINO. ESTUDIOS SOBRE LA RELACION ENTRE
LA ESTRUCTURA CUATERNARIA Y LA ACTIVIDAD ENZIMATICA,
MEDIANTE HPLC
La filtración por geles ha sido frecuentemente empleada no sólo para el desalado y fraccionamiento de macromoléculasbiológicas, sino también, comose mencionó anteriormente, parala determinación de pesos moleculares de proteinas desnaturalizadas con reactivos tales comoguanidina yurea (Olson y Liener,l967; Manny Fish, l972). Aunque el agente tensoactivo SDS ha sido empleado con frecuencia para la determinación de pesos moleculares de polipéptidos mediante la técnica de electroforesisPAGE(Heber y Osborn, l969),fueron en realidad muy pocos los intentos realizados para utilizar el SDSen la filtración por ggles (Rosenbusch y Weber, l97l).
La técnica de cromatografía liquida a alta presión(HPLC)ha tenido un avance considerable en esta última década,particularmente para la separación de sustancias biológicas.
Las matrices tales comodextrano, poliacrilamida y agarosa, que son las normalmente usadas en la filtración por geles,han probado ser inadecuadas para HPLC, dado que no resisten lasaltas presiones. La aplicación de la técnica HPLCal campo dela filtración por geles ha sido posible gracias a la apariciónde matrices de sílice o vidrio. Se han desarrollado asi columnas comolasTSK G-4000 y 3000 que han demostrado ser especialmente ütiles para la determinación de pesos moleculares y laseparación de péptidos en presencia de SDS (Imamura y col.,1979; Kato y col., 1980).
Teniendo en cuenta ésto, es que se consideró de interésaplicar esta metodologia a nuestros estudios sobre la relaciónentre la estructura cuaternaria y la actividad enzimática delALA-D,empleando como fuente enzimática la dehidrasa bovina purificada 450 veces.
Usualmente se trabajó con la enzima purificada previamente desalada por pasaje a través de una columna de Sephadex6-25 (0,9 x 12 cm). La muestra proteica (50 - l00 ug) se aplicóen un volumen de inyección de lO a 50 ul, eluyéndose a una velocidad de 0,5 ml/minuto y una presión de 20-30 kg/cm2 a temperatura ambiente; el curso de elución se siguió monitoreando a¿80 nm. Los coeficientes de distribución se calcularon de

234
acuerdo a 1a ecuación:
donde Ve es e] volumen de e1ución de cada proteina.
11.2.1. Comportamiento cromatográfico de 1a enzima nativa
Comopodemos observar en 1a Figura 11.4., 1a proteina giuye en un pico único, que tiene 1a propiedad de ser agudo y simétrico, indice de 1a homogeneidad hidrodinámica de 1a muestraenzimática. Cabe destacar que esta caracteristica se observó en1a enzima recientemente purificada. Luego de un aimacenamientode dos semanas a -20 °C, e] pico proteico eiuyó en forma asimétrica, sugiriendo 1a posibiiidad de ciertas transformaciones en1a estructura oiigomérica tales comoasociaciones o disociaciones de subunidades pero sin ilegar a afectar e] peso moiecuiarde 1a proteina enzimática,ya que e] tiempo de retención no varió,y en ese sentido, todas ias corridas cromatográficas fueron aitamente reproducibïes. Esta expiicación tiene su origen en ios resuitados observados para e] ALA-Dde higado porcino (CapituioI, item I. ).
Cuando e] medio de elución.no contenía cloruro de sodio se observó que 1a proteina quedaba adsorbida a] ge].
La recuperación de proteina fue de aproximadamente un80 - 90 %.
II.2.1.1. Efecto de 1a concentracióngproteica
Se estudió e] efecto de variar 1a concentración proteica de 1a muestra inyectada entre 0,05 y 30 mg/m], 10 que corresponde a una masa de 2 a 1.203 ug. No se observaron variacionesen e] Kd (Iabia I.5.) indicando que en ese rango de concentracignes no se produjeron agregaciones y/o disociaciones de 1a proteina enzimática.
Esto se diferencia de 10 observado en 1a enzima de higado porcino en donde,mediante 1a técnica de fiitración por geles oonvenciona], un parámetro ta] como 1a cantidad de proteinasembrada afectó e] grado de asociación de 1a enzima muitimérica,detectándose asi formas enzimáticas distintas de] octámero nativo.
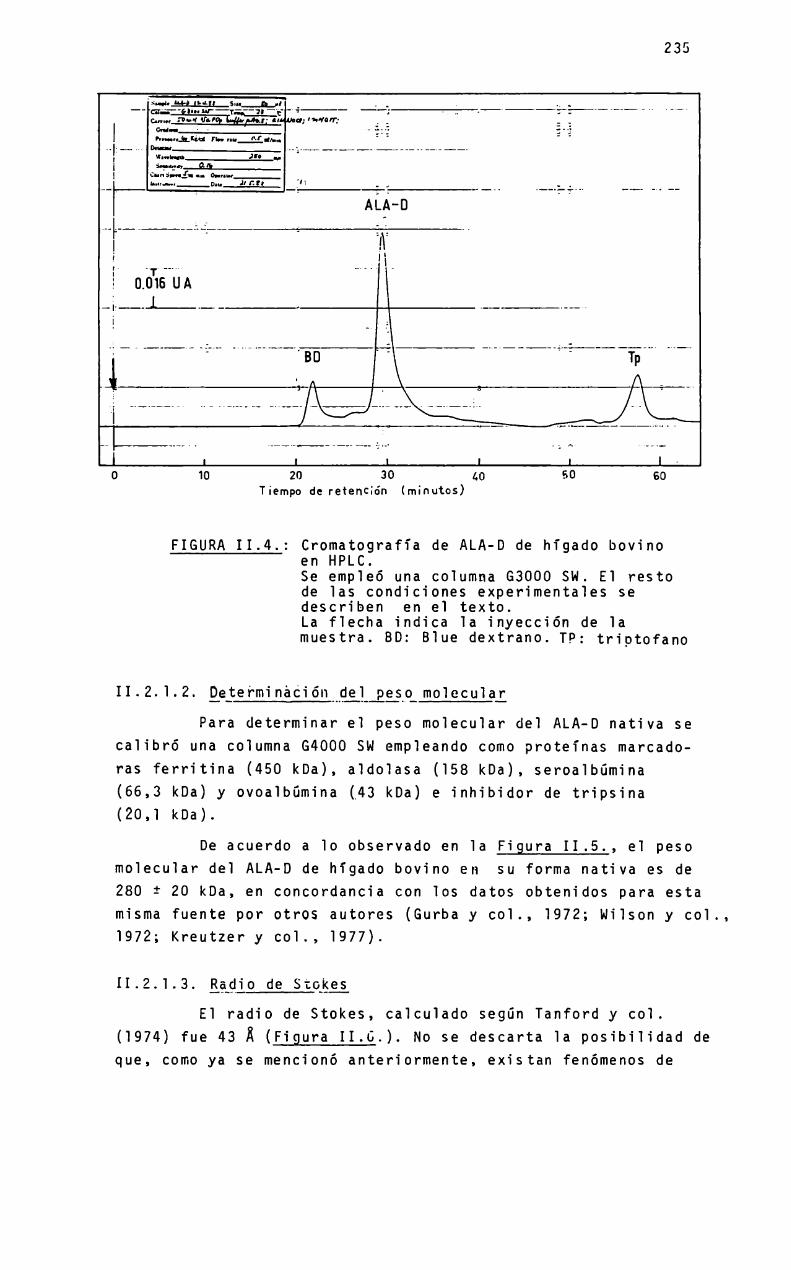
235
.T_wh0.016 UA
Tiempo de retenCIcSn (minutos)
FIGURA11.4.: Cromatografía de ALA-Dde higado bovinoen HPLC.Se empieó una coiumna G3000 SH. E1 restode 1as condiciones experimentaies sedescriben en e] texto.La f1echa indica 1a inyección de 1amuestra. BD: Blue dextrano. TP: triptofano
II.2.1.2. QetermináCión de] peso moïecuiarPara determinar e] peso moiecuiar de] ALA-Dnativa se
caiibró una coïumna 64000 SN empïeando como proteinas marcadoras ferritina (450 kDa), a1d01asa (158 kDa), seroaïbümina(66,3 kDa) y ovoalbümina (43 kDa) e inhibidor de tripsina(20,1 kDa).
De acuerdo a 10 observado en 1a Figura 11.5., e] pesom01ecu1ar de] ALA-Dde higado bovino en su forma nativa es de280 t 20 kDa, en concordancia con 105 datos obtenidos para estamisma fuente por otros autores (Gurba y co]., 1972; Niïson y c01.,1972; Kreutzer y co]., 1977).
{1.2.1.3. 5.3510 de Stojggs
E1 radio de Stokes, caiculado según Tanford y co].(1974) fue 43 Á (Figura II.G.). No se descarta 1a posibiiidad deque, comoya se mencionó anteriormente, existan fenómenos de

236
adsorción, lo que fue notorio cuando a1 buffer de eiución no seagregó cioruro de sodio.
TABLA11.5.: Efecto de 1a concentración proteica sgbre ei perfi] cromïtográfico de] ALA-Dbovina
CANTIDAD DE PROTEINAKd
(ug)
2 0,2514 0,249
24 0,25140 0,247
120 0,2461.200 0,246
Las cantidades_indicadas se inyectaron en un volumen de 40 u]y se eiuyeron con BFS 50 mMpH 6,8; NaC] 0,1 M; DTT 1 mM auna velocidad de 0,5 mi/minuto.
11.2.2. ggmportamiento cromatográfico de ALA-Den presencia de¿g
La exposición a SDScausa desnaturaiización de proteinas y disociación de estructuras oiigoméricas, y se ha demostrado que una variedad de proteinas unen idénticas cantidades deSDS sobre 1a base de peso a peso (Reynoids y Tanford, 1970). Estudios de propiedades hidrodinámicas y ópticas de ios compiejosproteina-SDS resuitantes, indican que se forman particuïas aiargadas en ias cuaies e] tamaño moiecuïar varia únicamente con e]peso moiecuiar de 1a cadena poiipeptídica. Este mecanismo genera]para ias interacciones SBS-proteina, se ha apiicado en 1a interpretación de 105 resultados obtenidos en esta sección.
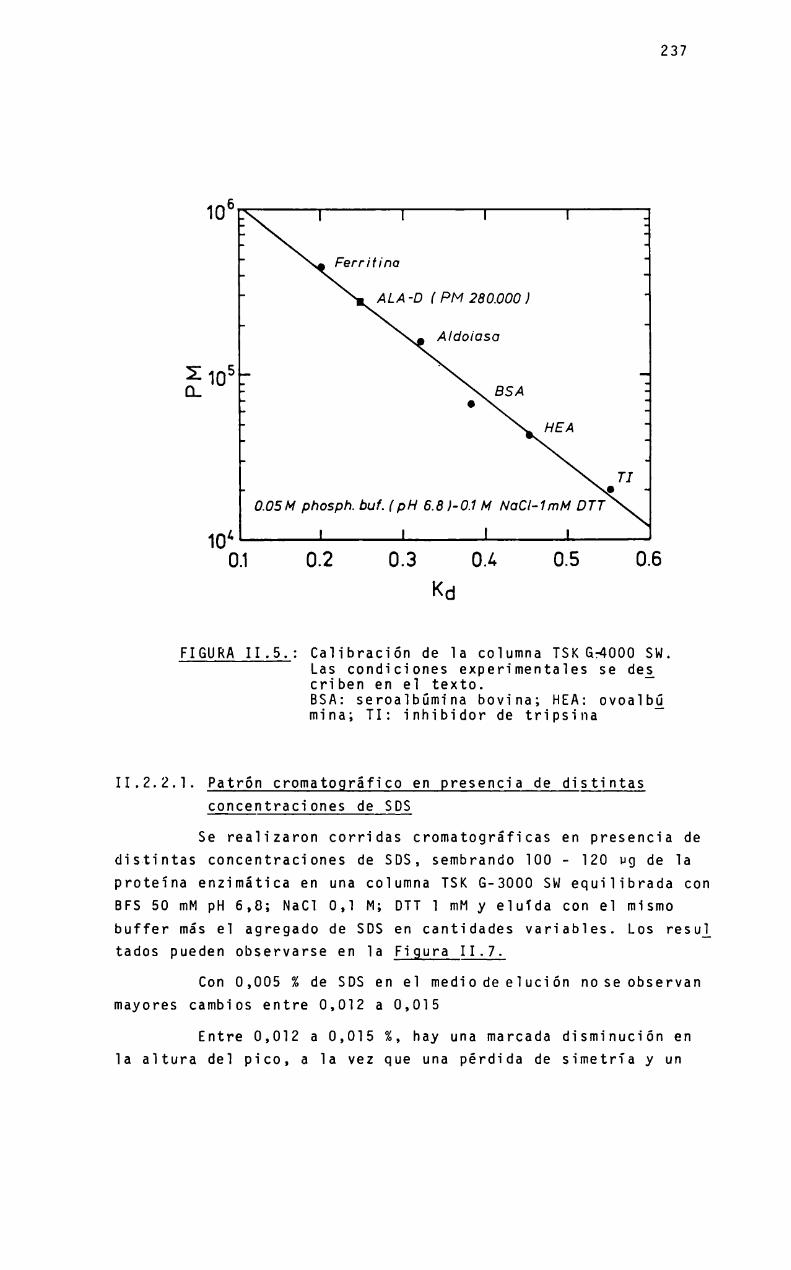
237
610 - l I I l
P Ferritína '
- ALA-D (PM 280.000} '
r A/doiasa '
Z 1 5 — _o í BSA 1
Z ° 2
' HEA
TI.. o ..
0.05M phosph. buf. {pH 6.8 )-0.1 M NaC/—1mMDTT
101. 1 l I L0.1 0.2 0.3 0.1. 0.5 0.6
Kd
FIGURAII.5.: Ca1ibración de 1a co1umna TSK64000 SW.Las condiciones experimenta1es se describen en e1 texto.BSA: seroa1bümina bovina; HEA: ovoa1bgmina; TI: inhibidor de tripsina
II.2.2.1. Patrón cromatográfico en presencia de distintasconcentraciones de SDS
Se rea1izaron corridas cromatográficas en presencia dedistintas concentraciones de SDS, sembrando 100 - 120 ug de 1aproteina enzimática en una co1umna TSK G-3000 SWequi1ibrada conBFS 50 mM pH 6,8; NaC1 0,1 M; DTT 1 mM y e1uida con e1 mismo
buffer más e1 agregado de SDSen cantidades variab1es. Los resultados pueden observarse en 1a Figura L¿¿z¿
Con 0,005 % de SDS en e1 mediode e1ución nose observanmayores cambios entre 0,012 a 0,015
Entre 0,012 a 0,015 %, hay una marcada disminución en1a a1tura de1 pico, a 1a vez que una pérdida de simetría y un

238
iigero corrimiento hacia tiempos de retención menores, indicando 1a disociación de 1a enzima y formación de agregados moieculares menores.
100 1 ¡
LE ¿15AV 75 — 'U1Q)
.x FMÉA3m 50 - —m ALA-D
TJ
o Lyzï5 25 '- (U
CE
l lo0.2 0.1. 0.6 0.8
erfc-1Kd
FIGURA11.6.: Calibración para 1adeterminación dei r9dio de Stokes.Se reaiizó en unacoiumna 6-3000 SH,utiiizando comobufferde e1ución BFS 0,1 MpH 7,4; SDS 0,2%.E1 Kd para ALA-D seobtuvo de 1a Figura11.4.BSA: seroaibümina bgvina, HEA: ovoaibümina; Lyz: lisozima.
Con 0,02 Z de SDS no se observa ningún pico, 10 que se
interpreta comoque 1a enzima se ha disociado totalmente y haquedado adsorbida a1 ge].
A1 aumentar 1a concentración de SDS de 0,025 a 0,2 %, seobserva nuevamente 1a aparición de un pico, cuya aitura va aumentanto a medida que se incrementa 1a concentración de] agente

239
disociante en e] medio de eiución, a 1a vez que hay una adhesión de molécuias de SDS a ias subunidades disociadas de ALA-D,formando agregados molecuiares cada vez mayores, iiegando a saturarse con SDS 0,2 %.
E1 patrón cromatográfico de un soio pico bien definidoindicó 1a existencia de un soio tipo de subunidad, por Io tanto enlas condiciones experimentaies utiiizadas, no se iogró obteneruna coexistencia de especies moiecuiares de diferente peso molecuiar.
Vemos además, que con una concentración de SDS de 0,2 %,se satura 1a proteina produciéndose una disociación tota] deias subunidades, siendo por tanto esta concentración adecuadapara 1a determinación de] peso moiecuiar de 1a enzima disociada.
II.2.2.2. Determinación de] peso moiecuiar de] ALA-Ddisociada
Para 1a determinación de] peso moiecuiar de ias subunidades de ALA-Ddisociadas con SDS, se utiiizó una columna0-3000 SW,y como proteinas standard seroaibümina bovina (66.300),ovoaibümina (43.000), anhidrasa carbónica (30.000), inhibidor detripsina (20.100) y iisozima (14.400). Las muestras se trataronpreviamente con SDS 1% en BFS 0,] M pH 7,4; DTT 1 mM, a 65 °C durante 30 minutos; se enfriaron a temperatura ambiente y se cromatografiaron 5 u] de cada soiución, obteniéndose e] cromatogrgma de 1a Figura II.8.
E1 peso moiecuiar de] ALA-Ddisociada fue de40.000 i 5.000 (40 i 5 kDa), resuitado que concuerda con io obtenido por otros autores (Gurba y c01., 1972; Kreutzer y co].,1977) mediante metodoiogias diferentes aunque basadas en e] mismo principio, y confirmamos que se trata de una enzima homooctamérica (Figura II.9L).
II.2.2.3. Análisis de ios radios de Stokes en presencia de SDS
con ios datos de 1a Figura II.7. y de acuerdo a ios estabiecido por Tanford y co]. (1970), caiculamos ios radios deStokes de] ALA-Dde higado bovino.ensuforma nativa, y disociadacon SDS. Los resultados se grafican en 1a ¿laura II.10.¿observamos que ios radios de Stokes varian con ias concentraciones deSDS. En e] gráfico se ve refiejado e] fenómeno de adsorción que gcurre a concentraciones de SDSde 0,02 %, ya mencionado anteriormente.

240
T0032UA
l ¿ SDS B6)0 027
0005
0012 0]
gg 0.0150.05(m‘—** 002 0025
l l 1 L
20 30 ¿0 20 30 LO
L
Tiempo de retención (minutos)
FIGURAII.7.: Patrón cromatográfico de ALA-Dbovina en presencia de distintas concentraciones de SDS, en unacoiumna G-3000 sw.Los deta11es experimentaïes se describen en e]texto. La fïecha indica 1a e1ución de] Biue Deitrano. UA:unidades arbitrarias de absorbanciaa 280 nm.

r
:'—""'_,'_1_'“.'::'_"‘_P:____J.__ZÏ_—_'
_——-1'...¿,‘-/...-.--v .j... 'II‘“ II'E'I 1'
l
ïr.1')
.-..._‘_........-i_-____.___....__0.-.
...°__ _-__..__..-.._n.1li
n
i|
Tiempoderetencñn(minutos)
FIGURA11.8.:Perfiicromatográficodeproteinasmarcadoras.
Lasproteinasstandardroncomoseindicóen eiuyéndoseconBFS0,1 nuto. (i)Biuedextrano,hidrasacarbónica,(7)triptofano.
(2) (5)
Laflechaindica
(aproximadamente50ugdecadauna)setrata e]textoyseaplicarona1acolumna6-3000SW,MpH7,4SDS1ainyecciónseroanümina inhibidorde
0,2%aunaveiocidadde0,5mï/mlde1amuestra.bovina,(3)ovoaibümina,(4)ag tripsina,(6)iisozima,
241

242
Inicialmente, y hasta una concentración de SDS 0,015 %hay una ligera disminución de los radios de Stokes; y a partirde SDS 0,02 %, un marcado aumento, lo que interpretamos como unfenómeno que refleja la adhesión de las moléculas de SDS a laproteina, y es asi que se obtiene un radio de Stokes aparentepara la enzima disociada de 55 Á, mayor que el correspondienteal octámero nativo original (44 Á).
¿000 I Ï l
3000
ALA-D (PM 40.000}
8 _z 2000OL
1000 —
0.2% SDS- 0.1M phosph.buf. {pH 7.4)
0 l 4L l0.1. 0.5 0.6 0.7 0.8
Kd1/3
FIGURA11.9.: Calibración de la columna G-3000 SW.Las proteinas (2 mg/0,2 ml) se trataron 30 minutos a 60 °C con BFS 0,1 MpH 7,4; DTT l mM; SDS 1% inyectandose luego 5 ul de cada solución.El resto de las condiciones experimentales, como se indicó en Materiales y Métodos. BSA: seroalbüminabovina, HEA: ovoalbümina; CA: anhidrasa carbónica; TI: inhibidor detripsina, Lyz: lisozima.

243
60 l I I
.3 ha.mLO - _Séc) I(ñ
Q)TJ(>20 F ïs(U
CK
0 l l l0 0.05 0.1 0.15 0.2
SDS (%)
FIGURA11.10.: Radios de Stokes aparentes de ALA-DenSDS.Los radios de Stokes se caicuiaron segün Tanford y co]. (1974) a partir deios datos de 1a Figura II.7.
11.2.3. Efecto de] SDSsobre ei perfil cromatográfico en HPLCy 1a actividad enzimática
Se reiacionó e] patrón cromatográfico en HPLCcon SDSy 1a variación de actividad enzimática. Se midió 1a actividadde 1a enzima en un medio de incubación conteniendo ias concentraciones de SDSindicadas (Figura 11.11.)
Se observó una disminución de actividad en 1a medidaenque aumenta 1a concentración de SDS, y cuando ésta 11ega a0,025 % 1a actividad es nula (Figura II.11.b).
Por otra parte, si iuego de incubar 1a enzima con distintas concentraciones de SDS30 minutos a temperatura ambiente,se disminuye 1a concentración de SDS 5 veces por diiución y se

244
mide 1a actividad enzimática, obtuvimos que hasta una concentración de 0,015 %,1a inhibición se revierte. compietamente. Encambio a concentraciones de SDS superiores a] 0,025 % no hay rgcuperación de actividad (Figura II.1].c).
Los cáicuios indican que si se preincuba con SDS 0,02 %y iuego se di1uye 5 veces,1a concentración en e] medio de incubación disminuye a 0,004 %;concentración que segün se ve en 1aFigura II.11.b, no afecta a 1a actividad; sin embargo 1a recupgración rea] es de sóio e] 50 %. Es decir que hasta un cierto iimite, 1a enzima disociada iogra reasociarse y recuperar su actividad.
Vemos además que 1a aitura de pico disminuye a medidaque aumenta 1a concentración de SDS de 0 a 0,02 %; a esta concentración ia enzima quedó totaimente adsorbida a1 ge], probablemente por su disociación tota] (Figura II.11.a). Paraielamente, y en forma coincidente vemos que a esa concentración de SDS,1a actividad de] ALA-Des nu1a (Figura 11.1LLQ).
Entre 0,02 % y 1 % de SDS, aparece nuevamente un picocuya aitura va aumentando, pero 1a actividad de 1a enzima siguesiendo nula.
Es decir, e] SDSdisocia a 1a enzima oiigomérica (octamérica), 10 que está indicado por 1a disminución de] pico proteico, y e110 va acompañado de una pérdida de actividad; y unavez que 1a enzima se ha disociado totaimente, 1as molécuias deSDSse "adhieren" a las subunidades, produciendo agregados moigcuiares cada vez mayores según se vió anteriormente con Ios radios de Stokes aparentes.
11.2.4. Efecto de] suifocianuro de sodio
Se estudió e] efecto de un agente caotrópico como e]suifocianuro de sodio sobre e] perfi] cromatográfico y 1a actividad enzimática de 1a dehidrasa bovina.
Tanto para suifocianuro de sodio 0,5 M como 1 M en e]buffer de elución (BFS 50 mMpH 6,8; Na Ci 0,1 M; DTT 1 mM), seobservó un soio pico proteico simétrico, cuyos pesos molecuiares aparentes fueron 400 y 95 kDa, en tanto que ias actividadesenzimáticas de ias proteinas eiuidas fueron 9 %y cero respectivamente.

245
Altura(°/o)
" l 1 l33 10€) (b) _
g 75 - _E 50 — ...2:3 25 - _<1 0 ' e c é A l
F‘ l I l
..\° 100 m
É, 75 - :2 5° ' “"ó’ 25 - —<1
o é I0 0.025 0.05 0.075 0.1
SDS (%)
FI URA11.11.: Efecto de] SDS sobre e] ALA-D.(a) Aïtura de] pico proteico según distintasconcentraciones de SDS en e] medio de e1ución; (b) ias concentraciones de SDSindicgdas corresponden a 1as presentes en e] medio de incubación, en cambio en (c) corresponden a ias obtenidas en e] medio de incgbación, 1uego de] pretratamiento de 1a enzima con concentraciones de SDS 5 veces superiores.

246
Dadoque e] suifocianuro de sodio debiiita ias unionespuente de hidrógeno, e] aumento de] peso moiecuiar aparente puede deberse a un debiiitamiento de ias interacciones entre iassubunidades, a una variación de 1a estructura secundaria, o aambos efectos simuitáneamente. Comoresuitado de eiio, se produciria una expansión de 1a moiécuia, y formaciónde una estructura funcionalmente desfavorabie, ya que se observó una disminución notoria de 1a actividad cataiitica.
11.2.5. Estudio de 1a actividad especifica de ios distintospicos obtenidos por HPLC
Se sembró 1a enzima purificada en su forma nativa, yen presencia de SDS 0,012 % y 0,025 % en e] medio de eiución,comose detaiió en secciones anteriores, recogiéndose fraccionesde 250 u] en ios que se determinó actividad enzimática y e] contenido proteico. Los resultados se muestran en 1a Figura 11.12.
En ausencia de SDS se obtuvo un pico de actividad especifica homogéneay constante; 1a fracción de actividad especifica constante es un 82 %. Dado que se usó una fracción enzimátirca que iievaba dos semanas aimacenada a -20 °C, es probabie que1a fracción de actividad especifica constante pudiera ser mayorpues como se vió anteriormente, preparaciones que no son fresacas muestran un comportamiento cromatográfico que indica unaprobabie transformación de especies enzimáticas de] octámero nativo origina], a especies moiecuiares agregadas o disociadas, ycorrespondientemente, una 1igera disminución de 1a actividad cataiitica. E1 tiempo de retención indica que e] peso molecuiares 280 kDa (octámero). '
En presencia de SDS 0,012 %, además de una disminuciónde 1a altura de pico se ve un corrimiento hacia tiempos mayores,indicando 1a formación de especies de menor peso moiecuiar. Adiferencia de los que ocurre sin SDS, no se observa un piateauen 1a gráfica de actividad especifica indicando una heterogeneidaden 1a composición de ia muestra.
De acuerdo a 1a recuperación de proteinas, ia actividaden SDS 0,012 % es e] 50 % respecto de 1a actividad en ausenciade] detergente.
Con SDS 0,025 % se obtuvo un pico asimétrico (dato nomostrado) sin actividad.
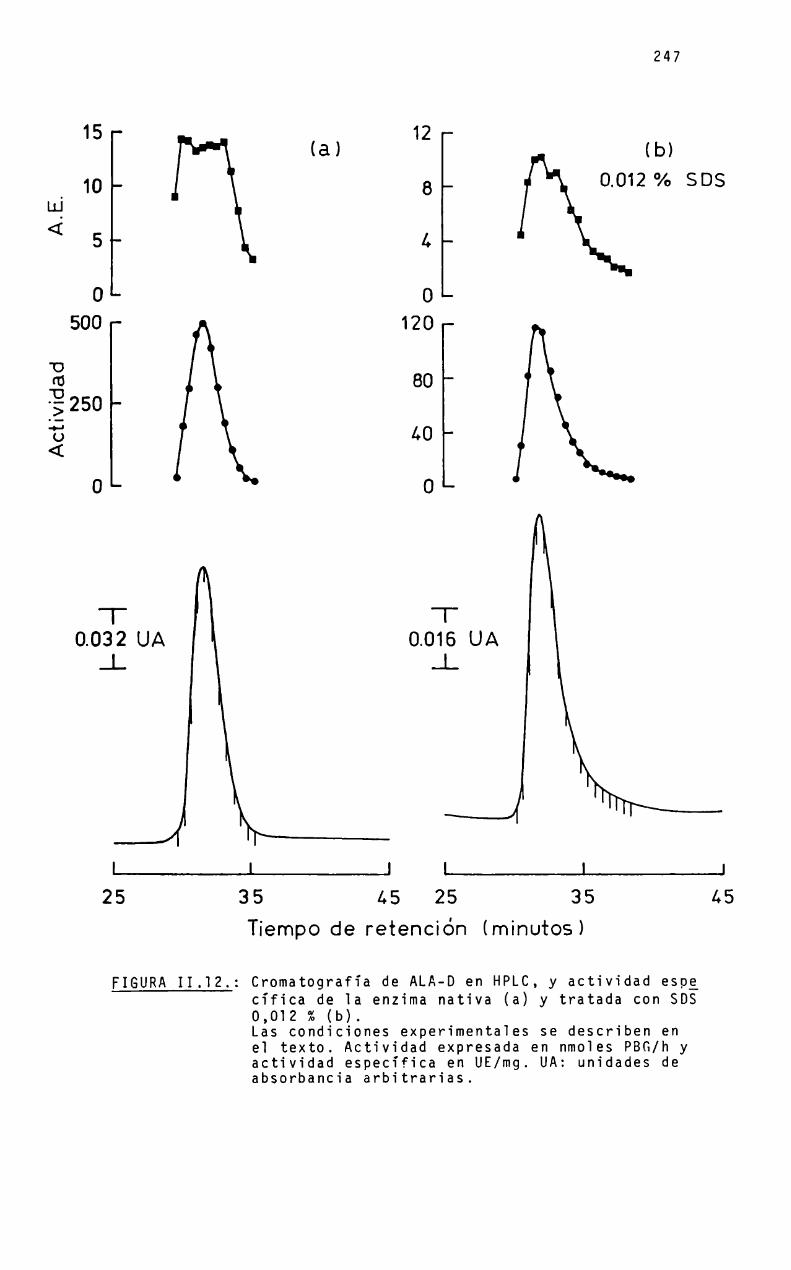
15 - 12 _(a) (b)
10 _ 8 _ 0.012 % sosU4
4 5 - 1. —
0 - o _500 - 120 _
7% eo 13¿5250L'ZS 1.0 <I
0 - o L.
T T0.032 UA 0.016 UA
_L _L_
I 1 J P l J
25 35 ¿.5 25 35 1.5
FIGURA11.12.:
Tiempo de retención (minutos)
Cromatografia de ALA-Den HPLC, y actividad especifica de 1a enzima nativa (a) y tratada con SDS0,012 % (b).Las condiciones experimentaies se describen ene] texto. Actividad expresada en nmoïes PBG/h yactividad especifica en UE/mg. UA: unidades deabsorbancia arbitrarias.

248
Por otra parte, 1a enzima pretratada con SDS 1 %y e]uïda en ausencia de SDS, dió un pico de peso moïecuïar aparenéte de 466 kDa sin actividad en ninguna subfracción. Este picose deberia a 1a formación de especies molecuïares por asociaciónde las moléculas de SDS formando agregados mayores a1 octámeronativo originaï, e indicando una vez más que e] proceso de disgciación de 1a enzima en esas condiciones es irreversibie.

ALA-D Y METALES
III.1. ALA-D
III.1
III.1.
III.1.
III.Ï.
111.1.
III.1.
III.1.
CAPITULO íII
de higado porcino
.1. Efecto dede] ALA-D
PlomoIII.1.2.1.
III.1.2.2.III.1.2.3.
. ZincIII.1.3.1.
III.1.3.2.. Cadmio
III.1.4.1.
III.1.4.2.. Cobalto
III.1.5.1.
III.1.5.2.. Mercurio
III.1.6.1.
III.1.6.2.
. Reversiónde higadoIII.1.7.1.
metaies sobre 1a actividadde higado porcino
Efecto sobre 1a enzimanativaEfecto sobre la apoenzimaCinética de 1a inhibiciónpor piomo
EfectonativaEfecto
sobre la enzima
sobre 1a apoenzima
EfectonativaEfecto
sobre 1a enzima
sobre 1a apoenzima
Efecto sobre 1a enzimanativaEfecto sobre 1a apoenzima
Efecto sobre 1a enzimanativaEfectoen condiciones anaeróbicas
sobre 1a apoenzima
de 1a inactivación de] ALA-Dde cerdo por metaïesAcción de zinc, cadmio ycobalto sobre 1a enzimainhibida por EDTA
Página
249
249
249
249
249
251
252
252252
252
252254
256
256256
256
256
256
258
258

III.2.
III.l.8.
ALA-D de
III.2.l.
III.2.2.
III.l.7.2. Reversiónde la inhibiciónpor plomo
Conclusiones
higado bovino
Acción del zincIII.2.l.l. Efecto del EDTAIII.2.l.2. Reactivación de la apoenzimaIII.2.l.3. Efecto del pHIII.2.l.4. Diálisis de equilibrioAcción del cobaltoIII.2.2.l. Reemplazodel zinc por
cobaltoIII.2.2.2. Modelopropuesto para la
interacción zinc-cobalto enel ALA-D'
Página
260
260
263
263263
263265
266
271
271
278

249
III. ALA-D Y METALES
III.l. ALA-D DE HIGADO PORCINO
III.l.l. Efecto de metales sobre la actividad del ALA-DdeMmmmYa hemos citado extensamente los trabajos realizados a
cerca del efecto de los metales sobre el ALA-Dde diversas fuentes (Capitulo III, Introducción), y vimos que los resultados sonvariados e incluso, en algunos casos, contradictorios. Se investigó, entonces, el efecto de varios metales, bajo condicionesexperimentales diversas, sobre la actividad del ALA-Dde higadoporcino.
Denominaremosenzima nativa, a la enzima purificada enlas condiciones usuales, detalladas en Materiales y Métodos,sin ningún tratamiento ulterior.
En todos los casos, los porcentajes de actividad conel agregado de metales, se expresaron considerando como lOO %la del correspondiente control sin ningún agregado.
III.l.2. Plomo
III.l.2 l. Efecto sobre la enzima nativa
Comovemos en la Figura III.l.a, la enzima nativa seinhibe marcadamente con concentraciones de plomo superiores alO'7 M, llegando a una inhibición casi total a una concentración
del metal de 5 x 10-5 M. El 1050 interpolado del gráfico es de2,5 x 10'6 M, valor similar al hallado para la inhibición delALA-Deritrocitaria de sujetos adultos normales (Davis y Avram,l977).
III.l.2.2. Efecto sobre la apoenzima
Se observa que tanto en anaerobiosis (figura III.l.a)comoaerobiosis (Figura III.l.b) la apoenzima sólo se inhibecon concentraciones de plomo superiores a 5 x 10‘5 M, a diferencia de lo observado para la enzima nativa. También es de notarque en ausencia de DTTen ambas condiciones atmosféricas, el plgmono ejerce ninguna acción sobre la actividad enzimática; estos

Actividad(Z)
Á-A enzimanativa, 02, +DTT (a)Á“-Á apoenzíma, N2, +DTT
¿g ¿>---Z> " , N2, -DTT
'U
3125
'Ü< ¿3
_ ¿ÁIoo- A """""" -—A-_-_ A
‘ " ‘ * * _‘_ ¿SA A 45 ‘*»A
_ A_A--‘\75_ A1” x ¡\\A\\\\
\\\ A\
50h \ \\\\\A‘x A
25- \ \\IL ‘Ák\
o Lgá; 1 l 1 l l
-C -7 -6 '5 'h '3log (Pb++)(M)
(b)
100 A fï”'A'A "" ‘Ét-- A_ : _A—-———_,-_- A ‘—_*s\A 5*‘s\ A \\A
\\¡A‘:\
so - \ AA---A apoenzíma, 02, +DTT \\\A4r--¿S apoenzíma, 02, -DTT ‘
0 n ¡,4L, | 1, l J—m -7 -6 -5 -u —3
FIGURAIII.1.:
log (Pb++) (H)
Efecto de] p1omo sobre 1aenzima nativa y 1a apoenzima, en medio aeróbico yanaeróbico.
250
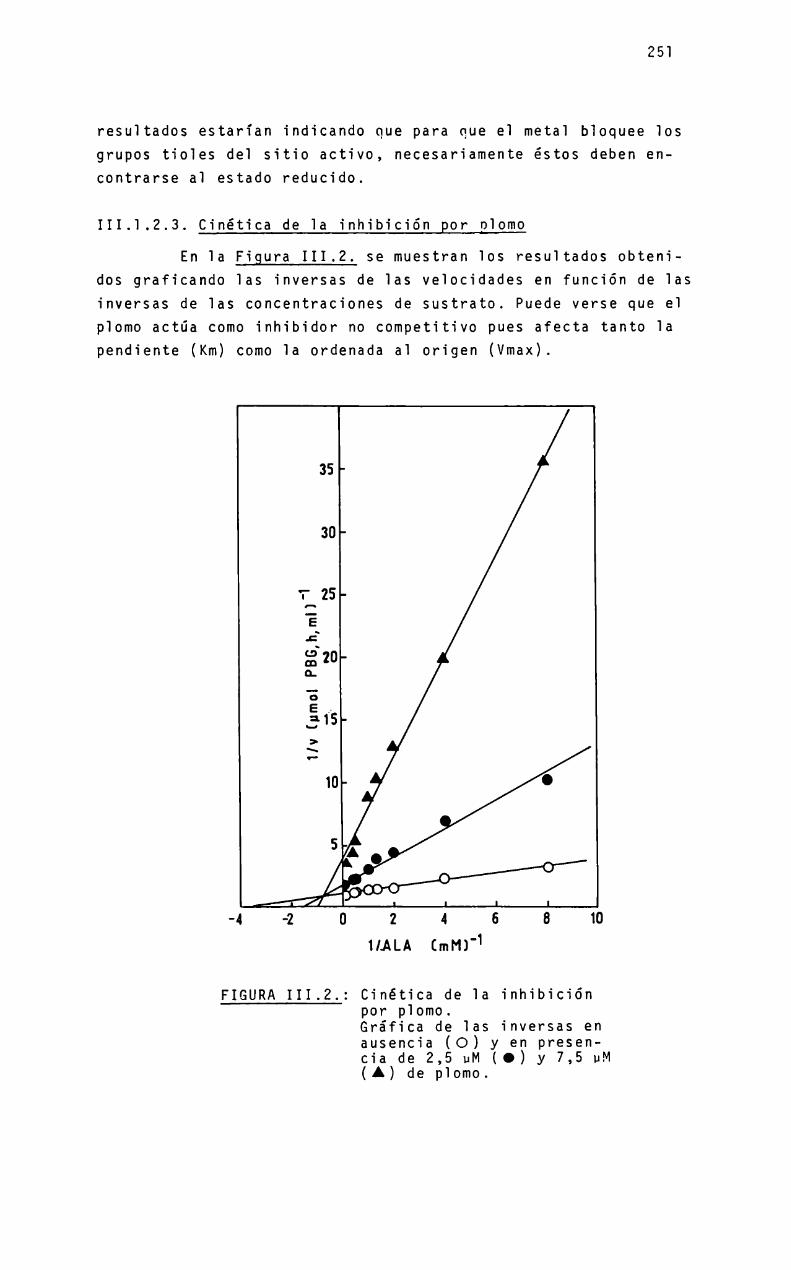
251
resultados estarian indicando que para que e] meta] bioquee 105grupos tioïes de] sitio activo, necesariamente éstos deben encontrarse a1 estado reducido.
III.1.2.3. Cinética de 1a inhibición por 010m0
En 1a Figura III.2. se muestran 105 resuïtados obtenidos graficando 1as inversas de 1as ve1ocidades en función de iasinversas de ias concentraciones de sustrato. Puede verse que e]piomo actúa como inhibidor no competitivo pues afecta tanto 1apendiente (Km) como 1a ordenada a1 origen (Vmax).
35
w o I
N U1l
ilvtumolPBG,h,rnI)q
IIALA num"
FIGURAIII.2.: Cinética de 1a inhibiciónpor piomo.Gráfica de ias inversas enausencia (O) y en presencia de 2,5 uM (O) y 7,5 uM(A) de pïomo.

252
Las constantes de inhibición (Ki) se encuentran entre10-6 y l0'7 M, reafirmando la conocida y significativa afinidadde esta enzima por el plomo.
III.l.3. Zinc
III.l.3.l. Efecto sobre la enzimanativa
Comovemos en la Figura III.};3, entre 10'5 y lO’3 Mse produce una activación, con una estimulación máxima de hastalSO %, a una concentración de 5 x lO'4 M. Concentraciones superiores a lO'3 Mcausan una marcada inhibición.
III.l.3.2. Efecto sobre la apoenzima
En el caso de zinc, a diferencia de lo recién observado con el plomo, el agregado de este metal a la apoenzima, tanto en presencia como en ausencia de DTT, y en aerobiosis comoanaerobiosis provoca una marcada activación (FiguraIII.3.a y b).Sin embargo, se observa que el grado de acción del zinc a concentraciones superiores a las de máximaestimulación para la enzima nativa, es mayor para la apoenzima, aün más en ausencia deDTT, aunque en este caso la disminución del efecto es más brusca que en presencia del agente reductor, señalando que la sensibilidad de la apoenzima a altas concentraciones de zinc es másnotable en ausencia de DTT.
También llama la atención que en todos los casos la mgxima estimulación se produjo con concentraciones de zinc del o:den de 5 x lO'4 M.
III.l.4. Cadmio
III.l.4.l. Efecto sobre la enzimanativa
Con respecto a este metal, los datos existentes en laliteratura son variados y a veces contradictorios. Asi por ejemplo, Kench y Cubb (l970) encontraron que en pollos recién salidos del cascarón, el cadmio produce una estimulación del ALA-Dtanto ¿n vivo como ¿n u¿tno.
En cambio, Nada y col. (l972) reportaron una inhibicióntotal ¿n vitno a una concentración de l mM,en tanto Abdulla yHaeger-Aronsen (l973) hallaron que este metal, a concentracionesdel orden de l mMproduce una inhibición ¿n v¿tno de la enzima.

.—.enz.nativa,02.¡DH(a) .--.mncnllrm,N2,+0" O-'-O" .uz.-nn0‘
¡'00
1I.)
\\ \‘ 300-O'"o
\-_-_
J‘JP 4\\ C
apepmnov
I
OInN
l
8N
(2)mnmv
C. \ b 200
5lJ/"
...\ ¡50..
Ion-o--------'O’
.———.apoenzlma.02'+DTT
apoenzlma,02,-DTT
/ .a__..¿.---.’—
I Ió_,
oll1
..
_.‘ï
-...L_____L____J____l__l__lo
-'I-3-7-c-7-6-5
-u=-7-6-5
Inn(ÏIIH)(H)
FIGURAIII.3.:Efectode]zincsobre1aenzimanativay1aapoenzima,en
-u-3-2
log(ln‘+)(H)
medioaeróbicoyanaeróbico
253

254
En higado bovino, e] cadmio produjo inactivación o estimuiación según que 1a concentración de sustrato fuera baja oaita respectivamente (Wilson y co]., 1972).
En ALA-Dde eritrocitos humanos, una concentración decadmio 4 pMcausó una estimuiación de] 134 % sobre ei contro],en tanto que concentraciones superiores, resultaron progresivamente inhibitorias (Davis y Avram, 1978). Estos autores observaron además que ei cadmio revierte 1a inhibición producida pore] piomo, a una concentración menor que 1a correspondiente dezinc. A1 respecto es interesante señaiar que e] cadmio tiene varias propiedades fisico-químicas simiiares a1 zinc,y sueie acompañar a este üitimo en 1a naturaleza.
En nuestro caso, según se observa en 1a Figura III.4.a,entre 10’7 y 10'4 Me] cadmio no afectó 1a actividad enzimática;concentraciones superibres fueron inhibitorias, con un ID50 de10'3 M.
III.1.4.2. Efecto sobre 1a apoenzima
En medio aeróbico y en presencia de DTT,hasta concentraciones de] orden de 5 x 10'3 Me] cadmio no afecta significativamente 1a actividad enzimática; podriamos considerar que prgduce una iigera activación, que en ningún caso supera e] 125 %(Figura III.4LQ).
En cambio si e] medio es anaeróbico y siempre en presencia de DTT, se produce un aumento notabie de actividad, queiiega hasta un 220 % sobre ei contro], para un rango ampiio deconcentraciones, entre 5 x 10'6 y 5 x 10'3 M (Figura III.4.a).
Tanto en medio aeróbico como anaeróbico, se produce unapronunciada caida de actividad recién a vaiores superiores a5 x 10-3 M de cadmio.
Por otra parte, en ausencia de DTTe] cadmio tambiénproduce una marcada activación, 200 y 350%segün sea en aerobigsis o anaerobiosis respectivamente, a una concentración 10'5 M;una respeuesta simiïar a 1a observada con 1a concentración dezinc que produjo máxima estimuiación.Nuevamente se aprecia quela acción de estos dos metaies, a1 menos sobre e] ALA-Dde higado porcino, es semejante.

I--Iapoenzima.02.#DTT(b) Ü-“Üapoenzima,02.-DTT
350..
I-Ienzimanativa,oz).mnl-"Iapocnlima,N ,+DT‘I'(a)
2
(II-"El" ,N-on
2
(Z) pepwnv300
Í
¡a
‘x
250_
pepunav
l3ON
200—'
I l | l |
I|
150..I'|
| l _l I
I \ \
100_u/
_-- --—-------------—- -—'
—__
1
i.
i
-cc-7-6-5-‘4-3-2
-—7-6—s—4-3
loq(ca‘*)(u)
loq(Cd")(H)
Nl
FIGURAIII.4.:Efectode]cadmiosobre1aenzimanativay1aapoenzima,enmedioaeróbicoyanaeróbíco8
U'l

256
III.1.5. Cobaito
III.1.S.1. Efecto sobre 1a enzima nativa
En presencia de DTT,a concentraciones Superiores a10'5 Me] coba1to produce una significativa y rápida inhibiciónde] ALA-Dque iiega a1 75 z para 10-3 M (Eiggrgnlll¿é¿)- Este gfecto podria deberse a 1a formación de un cobaititioi estabieque inactiva ios grupos suifhidriios esenciaies paraia actividadenzimática.
III.].5.2. Efecto sobre 1a apoenzima
Los resultados obtenidos para ambas atmósferas fueronsimilares. En anaerobiosis y presencia de DTT,hasta una concentración de 5 x 10‘4 M no se producen cambios en 1a actividadenzimática. A vaiores superiores, e] cobalto es inhibitorio,con un IDSOde 5 x 10'3 M (figura III.5.).
Anáiogamente a 10 observado con e] cadmio, en ausenciade DTThay un aumento de actividad en reiación a1 respectivocontro], que 11ega a un máximo de 320.% a una concentración de10-4 M.
Los datos obtenidos en aerobiosis fueron coincidentespor 10 cua] en este caso no inciuimos e] gráfico.
III.1.6. Mercurio
III.1.6.1. Efecto sobre 1a enzima nativa
Se observó que hasta 10'4 Me] mercurio no afectó 1aactividad enzimática; a concentraciones superiores se produjouna inhibición, con un 1050 de 5 x 10'4 M (Figura 111.5.).
III.1.6.2. Efecto sobre 1a apoenzima en condiciones anaeróbicas
A] igua] que con 1a enzima nativa, en presencia de DTTno se observaron variaciones hasta una concentración de 10’4 M,a vaiores mayores, se produjo una rápida y marcada inhibición,que a 10'3 M iiegó a1 100 %, con un 1050 de 2,5 x 10'4 M (Eig_ra III.6.).
En ausencia de DTTse observó una estimulación de]
160 % a 10'5 M de mercurio y iuego una brusca caida, con una inhibición tota] y a 10'4 M. Respecto de este agudo pico de acti
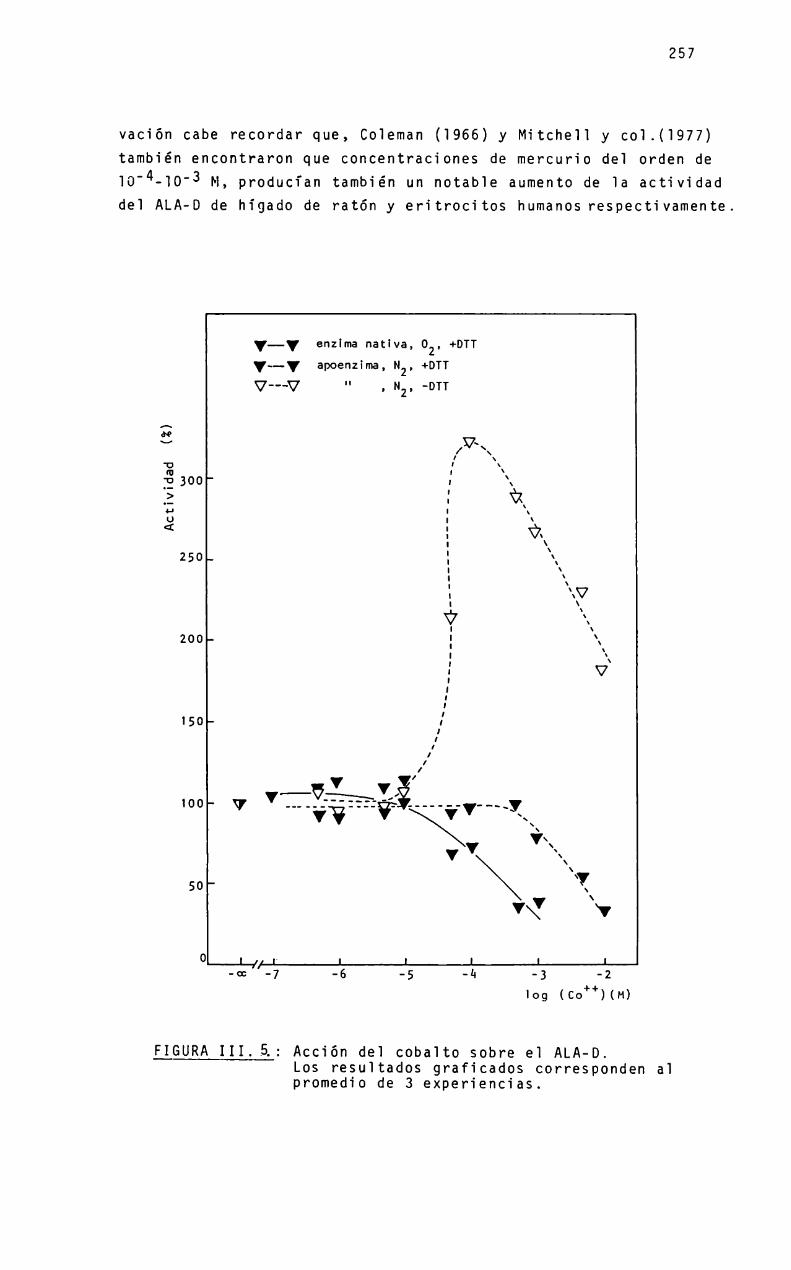
257
vación cabe recordar que, Coleman (1966) y Mitcheïi y co].(1977)también encontraron que concentraciones de mercurio de] orden de10'4-10'3 M, producían también un notabïe aumento de 1a actividadde] ALA-Dde higado de ratón y eritrocitos humanosrespectivamente
'_y enzimanativa, 02. +DTT7-..7 apoenzíma, N2. +DTTV_-_V " , N2, -DTT
){1/ \
I \-o I \ru l ‘\
:9 3 0 0 ’ n \.2 : ix¿J l \\o I \< : v
| \l \250- . X| \
'.| \Vl \\
V xI \
200- g \ \
.' ‘\
'I VIII
150- /
\xs\‘h
V,
100-v “"77”? v "x
o lll l' lI-m '-7 -6 -5 -h -3 -2
log (Co++) (M)
_LGURAIII. í: Acción de] cobaito sobre e] ALA-D.Los resuitados graficados corresponden a]promedio de 3 experiencias.

enzima nativa, 07, +DTT
apoenzima, N2, +DTTí”?¿>69_ , N2, -DTT
Actividad(Z)
100-o :ÏÍQ/Ï.6Jrs\||I
l
\ l\ l
50- x\
\\ \
o l 71,1_L l l o-a -7 -6 -5 -h -3
log (Hg++) (M)
FIGURAIII.4.: Efecto dei mercurio sobre eiALA-D.Los resultados graficados corresponden a] promedio de 3 experiegcias.
III.1.7. Reversión de ia inactivación dei ALA-Dde higado decerdo por metaies
III.1.7.1. Acción de zinc, cadmio y cobaito sobre ia enzimainhibida por EDTA
Para 1a obtención de una apoenzima es clásico ei tratamiento con queiantes, taies como e] EDTA;también se sabe queeste reactivo provoca una notabie disminución de ia actividadde] ALA-D,precisamente por eiiminación de] zinc de] centro de

259
reacción. En base a] efecto observado de] zinc, cadmio y cobalto de reactivar 1a apoenzima, se investigó 1a posibie reversiónde 1a inactivación de] ALA-Dpor EDTApor estos metaies, empïeados a 1as concentraciones de máxima activación. Se encontró(Figura LLL¿1LL que zinc y cadmio fueron capaces de revertircompietamente una inhibición de] 50 % por EDTA,en tanto quepor e] contrario, cobalto diminuyó aún más 1a actividad de 1aenzima.
100
Éï ‘xa O Ifl)
'D
IE
E 75 _
so- C)
C)25_ 'V
o l I l C)
_7 -5 -5 -h
log (EDTA) (M)
FIGURAIII.7.: Reversión de 1a inhibición porEDTA.Los ensayos se reaïizaron sobre1a enzima nativa.Los puntos (O). (I) y (v)correspfinden a1 agregado de zinc5 x 10- M, cadmio-10'5 M y cgbaito 10-4 M respectivamente.Se grafican ios resuitados promedio de 3 experiencias.

260
III.1.7.2. Reversión de 1a inhibición por piomo
Ademásde ios resultados obtenidos acerca de 1a acción
de] zinc sobre 1a enzima nativa y 1a apoenzima, es conocido e]efecto activante de este meta] sobre e] ALA-D,asi como su propiedad de despiazar a] piomo de su unión con Ios restos suïfhidrilos de 1a enzima y concomitantemente revertir 1a inactivaciónde 1a enzima. Empïeando 1a concentración de zinc capaz de provgcar 1a máxima estimuÏación de 1a enzima nativa y de 1a apoenzima, se encontró, según 10 esperado, uan compieta reversión de1a inhibición por e] piomo, a todas 1as concentraciones de] meta] ensayadas (Elgura III.8.).
III.1.8. Concïusiones
De estos resuïtados surge que, a pesar de ias precauciones tomadas, una vez obtenida 1a apoenzima oxida rápidamentesus grupos suifhidriios, pues aún en presencia de DTTya sea anaeróbica como aeróbicamente, 1a actividad residual de 1a misma,sin agregado de ningún meta] osciia alrededor de] 40 % con respecto a Ia de 1a enzima nativa de 1a cua] se ha preparado.
Se acepta en genera] que e] pïomo es capaz de inhibire] ALA-Dpor despïazamiento de] zinc de 1a enzima nativa (¿igura III.8.); sin embargo también hemos observado que en 1a apoenzima cuando este meta] no se encuentra unido a los restos sulfhidrilos protegiéndoios de 1a oxidación, en aerobiosis e] piomo no tiene capacidad para incrementar e] grado de inhibición(Figura III.1.a) y sóïo Io iogra en cierta medida en anaerobiosis (fioura III.].b), cuando e] DTTpuede revertir en parte 1aoxidación de Ios residuos suifhidriios esenciaies. En consecuencia, e] piomo actuaria sobre e] ALA-D,por despiazamiento de]zinc o en ausencia de] zinc sobre ios grupos tioïes de] centroactivo, soiamente cuando éstos están reducidos.
La activación de 1a enzima nativa por zinc 10-6-10'3 M(Figura III.3LQ) puede expiicarse en base a] hecho de que 1apreparación enzimática no posee e] contenido de zinc óptimo, yaque es un hecho conocido y se ha demostrado en este trabajo quedurante 1a purificación se pierde parte de] meta]. La reactivación de 1a apoenzima por agregado de zinc sigue un curso bastante simiiar tanto en aerobiosis (Figura III.3.b) comoen anaerobiosis (Figura III.3.a), pero ya sea en presencia comoen ausencia de DTTes necesario 11egar a una concentración de] orden de

261
10‘4 Mo mayor para apreciar ios cambios, en taies condicionesse iograrïa reconstituir 1a hoioenzima con un óptimo de actividad a una concentración de 5 x 10‘4 M. Un aumento de 1a.cantidadde zinc, parece inhibir 1a enzima, mucho más rápidamente en ausencia de] tio], 10 cual podria deberse a su unión con otros restos suïfhidriios diferentes de Ios de] sitio activo, ya que noocurre 10 mismo en presencia de DTT, provocando ta] vez cambiosestructurales negativos.
1 50 L.
///"‘—_A__‘“‘A*\A\125 - ‘ ‘
100 ..
Actlvndad(Z)
///?75 _ A\\
A
50 _
25 _ A\\
A\o l l J Al
-7 -6 -5 -h
log (Pb++) (n)
FIGURAIII.8.: Reversión de 1a inhibiciónpor pïomo mediante e] zinc.(A)p10mo;ÁA)p10mo mászinc 5 x 10' M.Se grafican ios resuitadospromedio de 3 experiencias.

262
Aparentemente, el cadmio no desplazaria al zinc de susitio de unión, o si lo hace, puede sustituirlo con igual eficacia (figura III.4.a) ya que no se aprecian cambios en la actividad de la enzima nativa por agregado de concentraciones crecientes de ese metal; y, comoel zinc, superado cierto umbral inhibe la enzima.
Quepuede reemplazar al zinc con propiedades activantessimilares lomuestran los datos de las Figuras 111.4. a y b , alos cuales cabria un razonamiento análogo al hecho para el zinc,a excepción de que en aerobiosis y en presencia de DTT, no selogró una reactivación de la apoenzima. Además el cadmio pareceunirse a los grupos sulfhidrilos del sitio activo, aún cuandoéstos se encuentren oxidados, a diferencia del plomo, produciegdo una holoenzima con un máximo de actividad para una concentración de cadmio de 10'5 My que es todavia menor que la del zinc;de manera que si bien el zinc debe ser el metal fisiológico,el cadmio puede sustituirlo ¿n vitae con igual o mayor eficienCia.
El cobalto tampoco desplazaria al zinc de la enzima nativa (figura III.5.), pero podria reactivar la apoenzimacuandose lo agrega en ausencia de DTT. Su incapacidad de reaccionarcon la proteina en presencia de DTTpodria justificarse teniendo en cuenta que este metal forma un complejo cobaltitiol estable con el DTT, que a altas concentraciones aün puede inhibirla enzima.
Finalmente, el comportamiento del mercurio podria asimilarse al del cobalto. De esta forma, a excepción del plomo, bajociertas condiciones los metales cadmio, cobalto y mergurio, serian capaces de reemplaZar al zinc en el ALA-Dde higado porcino, reconstituyendo una metaloenzima tan activa como la zinc-egzima nativa; un resultado no del todo esperado, por cuanto setrata de metales pesados, dos de ellos altamente tóxicos, quereaccionan fácilmente con los restos sulfhidrilos, que sin embaggo no ejercerian su acción a nivel de esta enzima, pero que apgrentemente podrian cumplir una función igualmente protectora delos sulfhidrilos del sitio activo, de acuerdo con la hipótesisde Tsukamoto y col. (l979).

263
III.2. ALA-D DE HIGADO BOVINO
111.2.1. Acción de] zinc
Con 1a hoioenzima a1tamente purificada de] ALA-Dde higado bovino, y su correspondiente apoenzima, se 11evaron a cabouna serie de experiencias tendientes también a obtener más información acerca de] efecto de ciertos metales, en especia] e]zinc, sobrela actividad cataiitica de esta enzima. En particuiarse ha centrado e] interés sobre 1a función que e] zinc juega enesta metaloenzima.
II.2.1.1. Efecto de] EDTA
Como se observa en 1a figura IIILEL, e] EDTA, en concegtraciones entre 10’5 y 10'3 M, produce una marcada inhibición,tanto sobre 1a hoio como sobre 1a apoenzima. En esta üitima, e]efecto inhibitorio es más pronunciado, concordante con 1a notoria activación de] zinc (como veremos más adeiante).
En aerobiosis (figura IILLQLE), en 1a hoïoenzima e]EDTA10'3 M produce una inactivación, en tanto que en 1a apgenzima ese efecto se Iogra ya a una concentración de] agente quelante de 10'5 M.
Resuïtados simiiares se observaron cuando e] ensayo sereaiizó en condiciones anaeróbicas (figura III.9.b), sóio que1as caidas de 1as curvas son algo menos pronunciadas.
III.2.1.2. Reactivación de 1a apoenzima
La apoenzima preparada según se detaiió en Materialesy Métodos, contenía menos de 0,1 átomos de zinc por subunidadde 35 kDa, y resultó compietamente inactiva en aerobiosis, aünen presencia de activadores tióïicos. Sin embargo e] agregadoconjunto de iones zinc y DTTcondujo a 1a recuperación de 1a agtividad origina], es decir, de 1a hoioenzima nativa, ta] comose ooserva en 1a Figura 111.10. Cuando se investigó en deta11ee] efecto de concentraciones variabies de zinc, sobre 1a recongtitución de 1a holoenzima, se observó que ésta ocurria dentrode un rango estrecho que sóio variaba entre 4 y 6 x 10'5 M(Figura III.11.), respondiendo además a una cinética sigmoidea.Esto sugiere que a 1a reactivación de 1a apoenzima por e] zinccorresponde una cinética de tipo cooperativo. Empïeando una

264
serie de ecuaciones teóricas 1a variación en 1a actividad enzimática se reiacionó con las concentraciones de zinc, haiiándoseque a bajas concentraciones, 1a cantidad de zinc que reaccionapor mo] de enzima seria de 8, en tanto que a aitas concentracignes, e] número se eieva a 32. E1 50 % de activación se obtuvocon una concentración de zinc de 5 x T0'° M Cabe aciarar quee] rango de concentraciones de zinc dentro de] cual se producela reactivación, puede sufrir un iigero desplazamiento según e]batch de apoenzima empieado, pero en todos ios casos e] perfilde activación es e] mi smo .
o (u
100 -(D ‘ iooilll
: Ol
n _ g . 75A l AE: g 22
'U '| 'uE | E
.2 5° - E .2 5°3 I O 3< 1 <
l
ó25. i u
\\\\\
‘a>o L Lfi; 1 l N“-C113 o
-: -6 —5 -n .3
log (EDTA) (H) log (EDTA) (H)
FIGURAIII.9.: Efecto de] EDTA.La enzima se incubó con 1as concentraciones deEDTAindicadas,determinandose 1a actividad encondiciones aeróbicas (a) y anaeróbicas (b),como se indico en Materiaies y Métodos.Se grafican ios resuitados promedio de 3 experiencias.(O) Hoioenzima; (O) apoenzima

265
15' -LS
e510- . -1o x.2 3E Sú m¿ d
5' '05 '
l l l
-s -4 -3 -2Log (BTT) M
FIGURAIII.10.: Efecto de distintas concentraciones de DTTsobre 1aactividad de 1a apoenzima,en presencia de zinc4 x 10'J M (o) y en ausencia de dicho meta] (o).
Por otra parte, cuando se investigó e] efecto de] zincsobre ia enzima nativa, observamos que 1a curva de activaciónse despïaza hacia concentraciones de zinc menores, consistentecon e] hecho de que 1a enzima contiene zinc, aún cuando ésta esbaja pues no 11ega a ser una hoioenzima. Además, otro efecto ogservadoes que 1a cinética de reacción deja de ser de tipo cooperativa (figura III.12.).
III.2.1.3. Efecto de] EH
Comoya se observara con 1a enzima de higado de cerdo,a concentraciones de zinc superiores a 10'4 Mtiene 1ugar una inhibición de 1a actividad enzimática (Figura [ll¿lg¿). Tambiénse encontró que esta reacción es pH dependiente (figura IILLlQL).A medida que aumenta e] pH, disminuye 1a actividad tota], y

266
deja de apreciarse e] efecto inhibitorio de] zinc. Se ha encontrado un pKa de 6,5 para este efecto,10 cua] sugiere que en 1ainhibición estarian también invoiucrados residuos histidina de1a proteina enzimática.
2—'l
AE.(uMd/mg)
0 l l l l l l—4.5 —4.4 —4.3 -4.2 -4.1 —4.o
Log Zn2’(M)
FIGUBA*LII.11.: Efecto de] zinc sobre 1a apoenzima.La apoenzima (18 ug) se incubó con distintasconcentraciones de zinc, manteniendo e] restode ias condiciones según se deta11a en Materiaïes y Métodos.Se grafican 10s reSuItados promedio de 4 experiencias.
III.2.1.4. Diálisis de equiiibrioCon e] objeto de lograr mayor información acerca de 1a
interacción zinc-ALA-D, se 11evaron a cabo experiencias de diálisis de equiiibrio, para 10 cua] se puso en contacto 1a suspensión enzimática con distintas concentraciones de zinc, ta] como
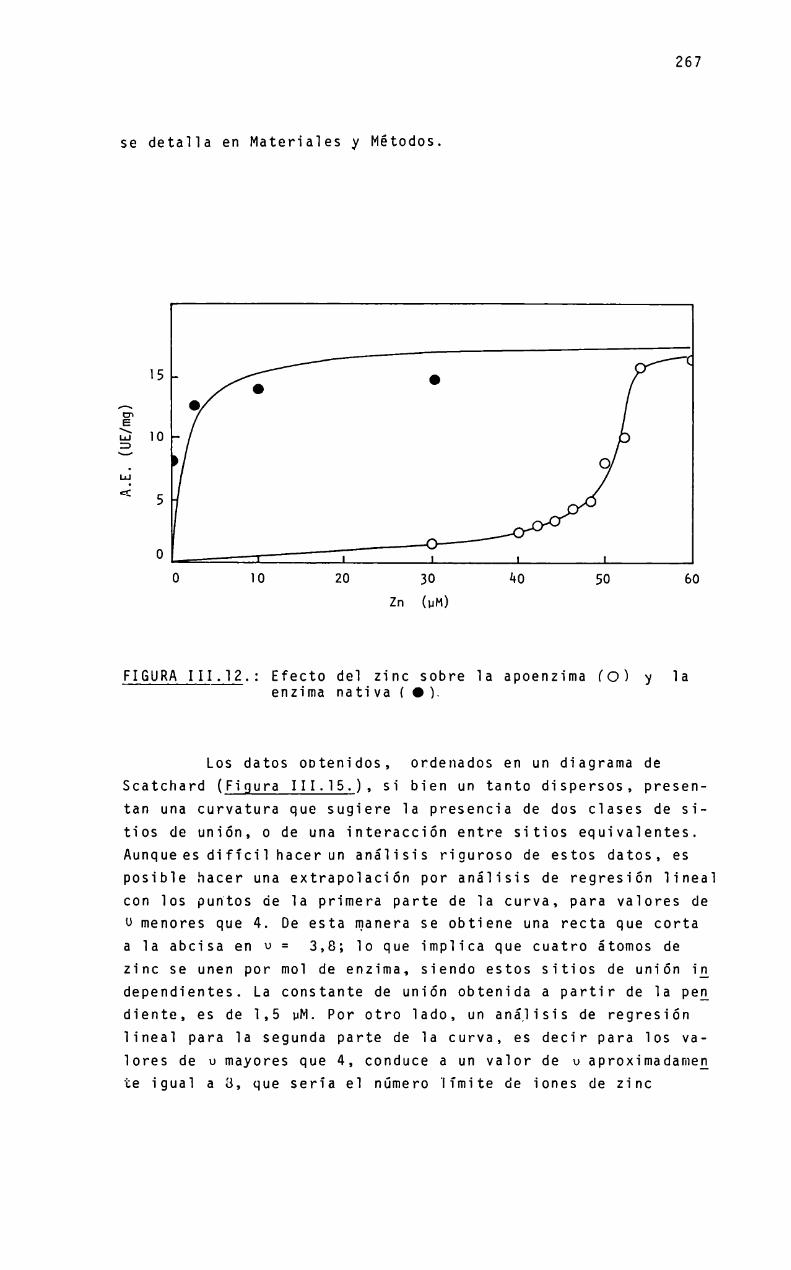
267
se detalla en Materiaies y Métodos.
(UE/mg)
A.E.
Zn (uN)
FIGURAIII.12.: Efecto de] zinc sobre 1a apoenzima (O) y 1aenzima nativa( O).
Los datos ootenidos, ordenados en un diagrama deScatchard (Figura III.15.), si bien un tanto dispersos, presentan una curvatura que sugiere 1a presencia de dos clases de sitios de unión, o de una interacción entre sitios equivalentes.Aunqueesdificiihacer un anáïisis riguroso de estos datos, esposibïe hacer una extrapoïación por análisis de regresión iinea]con los puntos de 1a primera parte de 1a curva, para vaïores deUmenores que 4. De esta manera se obtiene una recta que cortaa 1a abcisa en U = 3,8; 10 que impiica que cuatro átomos dezinc se unen por mo] de enzima, siendo estos sitios de unión independientes. La constante de unión obtenida a partir de 1a pendiente, es de 1,5 uM. Por otro 1ado, un anáJisis de regresiónÏineai para 1a segunda parte de 1a curva, es decir para los va]ores de u mayores que 4, conduce a un vaior de u aproximadamente igua] a 3, que seria e] número ïimite de iones de zinc
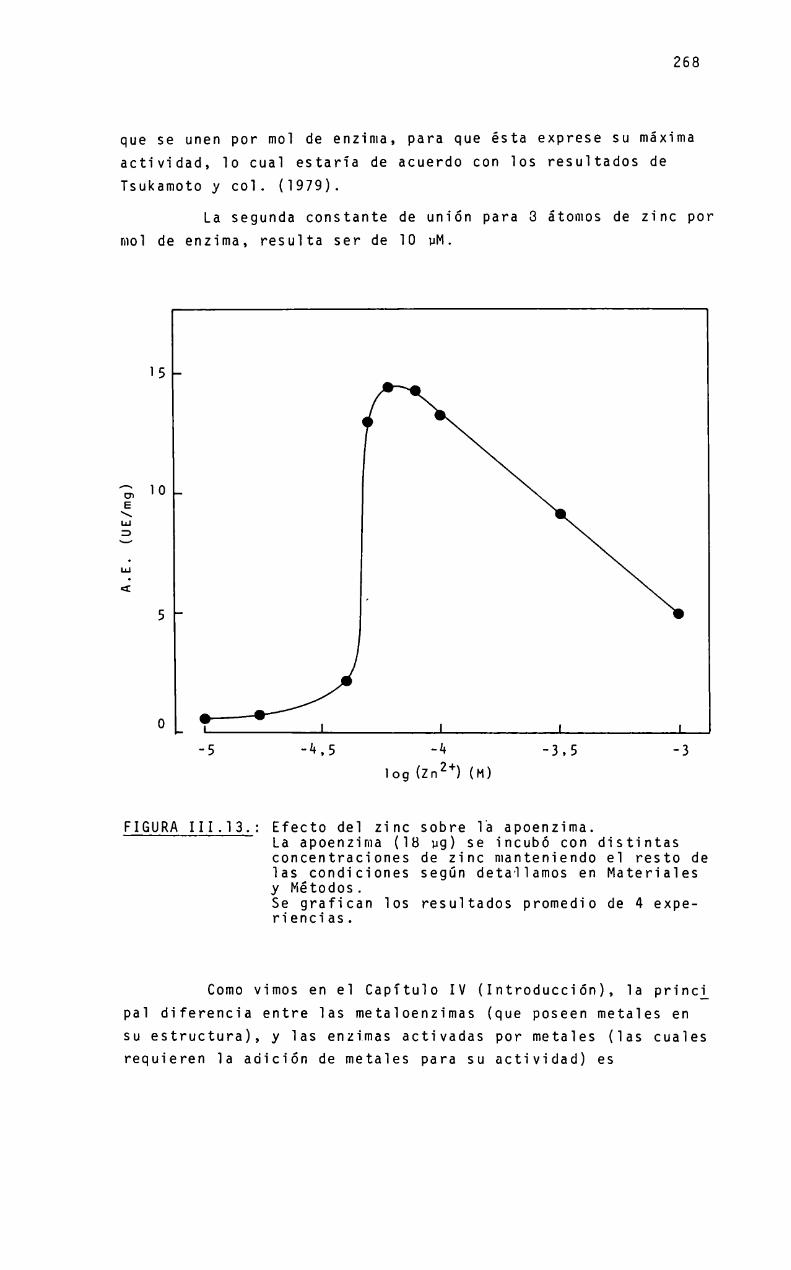
268
que se unen por mo] de enzima, para que ésta exprese su máximaactividad, 10 cua] estaria de acuerdo con 10s re5uitados deTsukamoto y co]. (1979).
La segunda constante de unión para 8 átomos de zinc pormo] de enzima, resuita ser de 10 uM.
¡5
'3,I0_E\
LUD
a]
<1;
S
0 L l 1, l 44L L
-S -hv5 -h -3y5 '3
log (Zn2+) (M)
FIGURAIII.13.: Efecto de] zinc sobre 1a apoenzima.La apoenzima (18 ug) se incubó con distintasconcentraciones de zinc manteniendo e] resto deias condiciones según detaiiamos en Materiaïesy Métodos.Se grafican los resuitados promedio de 4 experiencias.
Comovimos en e] Capituio IV (Introducción), 1a principa] diferencia entre ias metaïoenzimas (que poseen metaïes ensu estructura), y las enzimas activadas por metaïes (las cualesrequieren 1a adición de metaies para su actividad) es
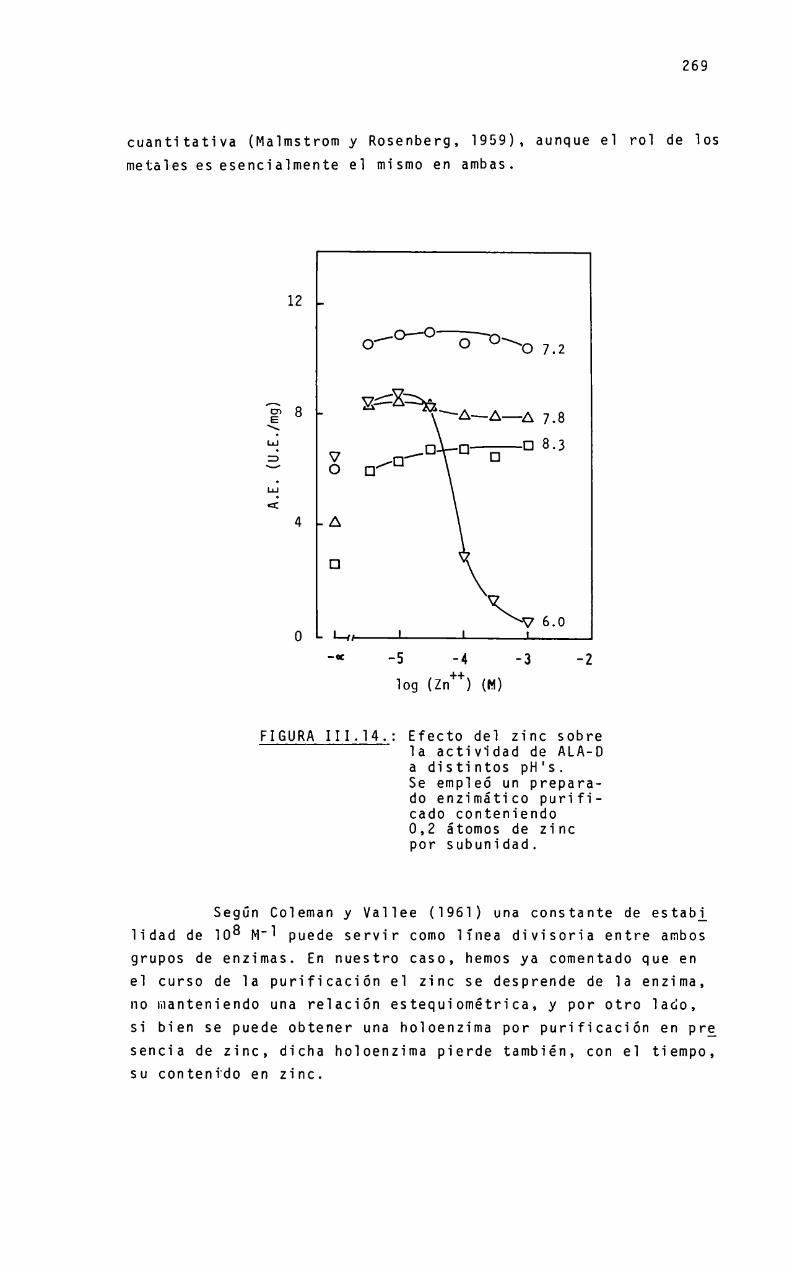
269
cuantitativa (Maimstrom y Rosenberg, 1959), aunque e] ro] de iosmetales esesenciaimente e] mismo en ambas.
12 _
c>—43-——*730/ o \o 7.2
A VíXE” 3 ‘ "‘ Z¿e¿\zl—A—A 7.8\7LAJ. ¡3 E] [18-353 E; D//Ü,,w:d:
4 -¡A
El
L V\v 6 o0 DL I l L
--= -5 -4 -3 -2
iog (Zn++) (M)
FIGURAIII.14.: Efecto de] zinc sobre1a actividad de ALA-Da distintos pH's.Se empieó un preparado enzimático purificado conteniendo0,2 átomos de zincpor subunidad.
Según Coieman y Vaïïee (1961) una constante de estabiiidad de 108 M'1 puede servir como 1inea divisoria entre ambosgrupos de enzimas. En nuestro caso, hemos ya comentado que ene] curso de 1a purificación e] zinc se desprende de 1a enzima,no manteniendo una relación estequiométrica, y por otro iado,si bien se puede obtener una ho1oenzima por purificación en presencia de zinc, dicha hoioenzima pierde también, con e] tiempo,su contenido en zinc.

270
v/(L)(¡m-1)
FIGURAIII.]5.: Gráfico de Scatchard para e]estudio de 1a unión zinc-ALA-D.Las condiciones experimentales se deta11an en Materia1es y Métodos.
Nuestro resuïtado de 1a diáiisis de equiiibrio, de 1acua] obtuvimos una constante de disociación de 1,5 uMo, 1o quees 10 mismo, una constante de asociación de 0,67 x 105 M'Ï indicaria entonces, y.en concordancia con 1o mencionado en e] párrgfo anterior, que ALA-Dpodria ciasificarse dentro de] grupo deIas enzimas meta1—activadas.

271
111.2.2. Acción de] cobaito
III.2.2.1. Reemplazode] zinc por cobaito
Hemosmencionado ya_que 1a sustitución de] zinc, que esespectroscópicamente "silente", por un meta] paramagnético comoe] cobaito, es un método üti] para e] estudio de ios mecanismosde acción de ias zinc-enzimas.
Aunque se habia observado con ei ALA-Dde higado porcino que e] cobaito no seria capaz de reempiazar a1 zinc, inhibiendo 1a actividad cataiitica emkun 75 % a una concentración de5 x 10’4 M(F'gura III.3.), ios resuitados obtenidos para 1a enzima bovina indican que e] mismo efecto se iogra con concentraciones de cobaito diez veces superiores (Figura III.16.),hechoque nos lievó a suponer que, para este caso, 1a sustitución demetaies podria ser posibie.
En consecuencia, se preparó 1a cobaito-enzima como seindiCó en Materiales y Métodos.
a) Agpartir de 1a hoioenzima
Por una parte, se 1a preparó a partir de 1a hoioenzimaobteniéndose Ios resuitados de 1a Tabia III.1.. Se hicieron tresexperiencias, y en todos Ios casos, para ia zinc-enzima (Zn-E)e1contenido de zinc se mantuvo entre 0,8 y 1,0 átomogramos/Subunidad, no detectándose cobaito. En cambio para 1a cobaito-enzima(Co-E), 1a cantidad de cobaito por subunidad estuvo entre 0,6 y1,4, sin que se desprendiera zinc, cuyo contenido permaneció entre 0,6 y 0,8 átomogramos/subunidad. Es de notar que, para 1aCo-E, e] máximo contenido de cobaito coincidió con ei minimo dezinc (Experiencia I).
De acuerdo a 10 que observamos en 1a Tabia III.1., seconfirma que ei cobaito no podria desplazar a1 zinc de su sitio,ubicándose entonces en un segundo sitio para e] meta], ya que entodos 10s casos vemos que 1a Co-E contiene un tota] aproximadode 2 átomos de meta] por subunidad. Podríamos designarla entonescomo una zinc-cobaito-enzima (Co-E-Zn).
Cuando 1a actividad se ensayó en presenciade DTT, 1aCo-E-Zn presentó entre un 59 y un 78 %.de su actividad respectode 1a Zn-E; en cambio en ausencia de DTT, 1a actividad de 1aCo-E-Zn decayó a un 5-12 % respecto de 1a Zn-E a partir de 1acua] se preparó;

A su vez, para 1a Zn-E, 1a ausencia de DTTdisminuyó1a actividad en só1o un 20 % como máximo, si e] ensayo se efectuabainmediatamente después de preparado. Por e] contrario, para 1a Co-E-Zn 1a ausencia de DTTde] medio de incubación, condu
jo a una disminución de] 85 - 90 % de su actividad.
(a)
E'O(U
E>
“.3U
<E
-1
(b)
3'Df0
'D'31:U<
o l l l
-s -u -3 -2 -1
¡09 (Co++ ) (M)
FIGURA111.162: Efecto de] cobaïto sobre e] ALA-Ddehigado bovino.Se ensayó 1a actividad de 1a enzimanativa en presencia de 1as concentraciones de cobalto indicadas, en aergbiosis (a) y en anaerobiosis (b).Los resuitados graficados corresponden a] promedio de 3 experiencias.

273
TABLA111.12: Reemplazo de zinc por cobaito a partir de 1ahoioenzima
EXPERIENCIA I EXPERIENCIA II .EXPERIENCIA III
Zn-E Co-E-Zn Zn-E Co-E-Zn Zn-E Co-E-Zn
A.E.(+ DTT) 13,3 7,9 13,0 10,1 14,1 8,7
A.E.(- 011) 12,9 0,7 10,9 1,3 11,6 1,4
Zn2+/subun. 1,0 0,6 0,8 0,8 0,8 0,7
C02+/subun. 0 1,4 0 1,1 0 0,7
Las actividades enzimáticas se midieron en aerobiosis.Otros deta11es experimentaies se describieron en Materialesy Métodos. A.E. en UE/mg
En 1a Figura III.17. se muestran ias curvas de saturación por sustrato para 1a Zn-E y 1a Co-E-Zn. Vimos ya que 1a hgIoenzima conteniendo aproximadamente 1 átomogramo de zinc porSubunidad se inhibia con EDTA.Un estudio comparativo de] comportamiento de esta Zn-E y 1a Co-E-Zn derivada de e11a , revelóque e] EDTAinhibe a ambas especies enzimáticas, a concentracignes superiores a 10'6 M; con una concentración de 10'5 M, 1a inhibición es prácticamente tota] (figura IIILlQL).
Se investigó además, e] efecto de concentraciones variabies de zinc sobre 1a actividad de ambas metaioenzimas, enpresencia de EDTA10-5 M (13911 III.2.).
Observamos que una concentración de zinc de 10-3 M,que es 1a misma concentración de] EDTAen e] sistema de incubación, revierte totaimente 1a inhibición por e] reactivo queIante, pero concentraciones mayores de zinc, son inhibitorias.
Esta acción inhibitoria de concentraciones aitas de

274
zinc, que ya observáramos también para e] ALA-Dde higado porcino, se confirmó anaiizando en detaiie e] efecto de] zinc sobre1a Zn-E (Tabia III.3.).
v“¿molPBG/h,ml)
ALA (mM)
Elggfifl III.17.: Curvas de saturación por sustrato para 1aZn-E y 1a Co-E-Zn.Las actividades enzimáticas se midieron aergbicamente en ias condiciones detaiiadas enMateriaies y Métodos.(a) Zn-ALA-D: Km = 0,21 mM; Vmax = 13,6 umo
ies PBG/h.ml(b) Co-ALA-D-Zn: Km = 0,49 mM;
Vmax = 9,2 umoies PBG/h. mi
E1 aumento de actividad por e] zinc en una concentración de 1 a 3 x 10'5 M, podria explicarse por e] hecho de queiasZn-E empieadasen estas experiencias no contenían e] máximode zinc, sino 0,8 átomogramos de zinc por Subunidad. Nuevamentevemos que concentraciones de zinc superiores a 10'4 M, son inhibitorias. Estos resuitados se confirmaron empieandodiferentespreparaciones deia hoioenzima, con distintos contenidos de zinc
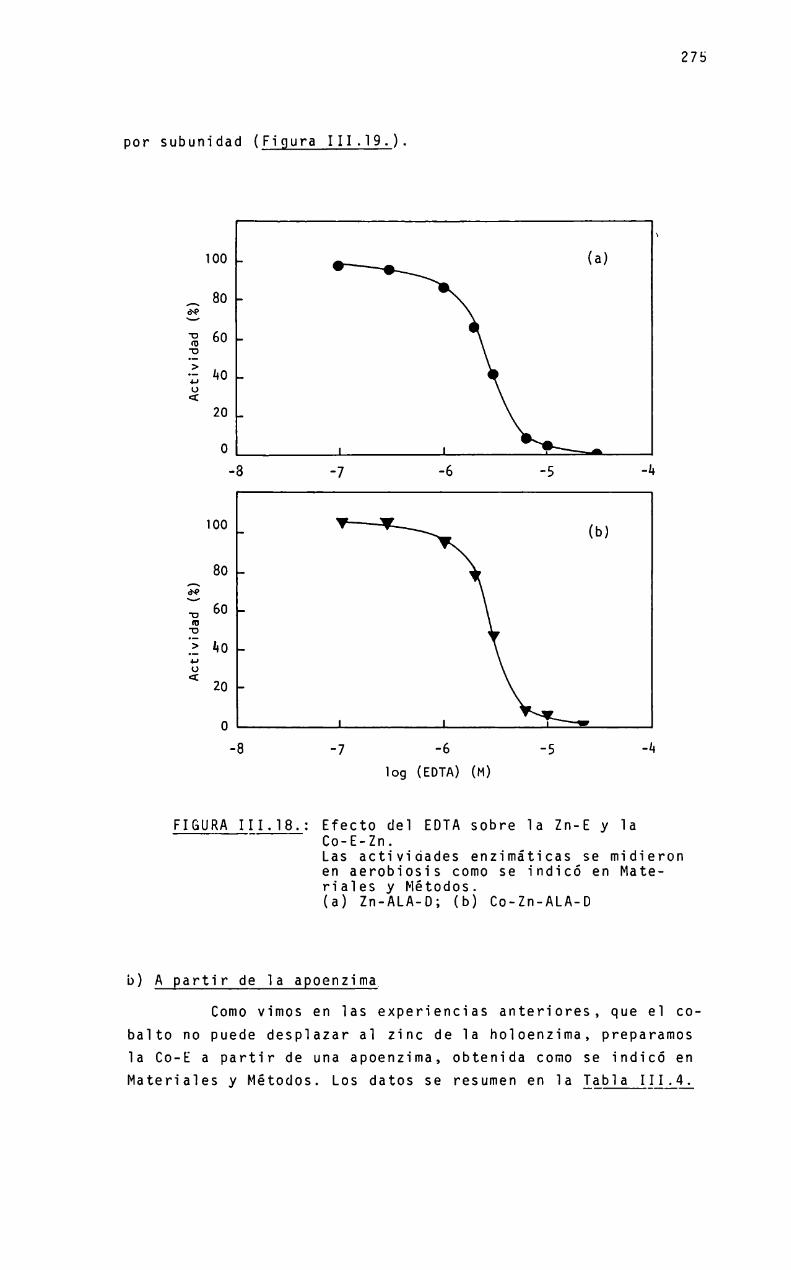
275
por subunidad (Figura III.19.).
80
60
#0
Actividad(2)
20 _
80 _
60 —
Actividad(Z)
0 J L l
-8 -7 -6 -5 -u
log (EDTA) (M)
FIGURAILI.18.: Efecto de] EDTAsobre 1a Zn-E y 1aCo-E-Zn.Las actividades enzimáticas se midieronen aerobiosis como se indicó en Materia1es y Métodos.(a) Zn-ALA-D; (b) Co-Zn-ALA-D
b) A partir de 1a apoenzima
Comovimos en ias experiencias anteriores, que e] cobalto no puede despiazar a1 zinc de 1a hoioenzima, preparamos1a Co-E a partir de una apoenzima, obtenida como se indicó enMateriaies y Métodos. Los datos se resumen en 1a Tabla III.4

276
TABLAIII.2.: Efecto de diferentes concentraciones de zinc sobre iasactividades de 1a Zn-E y iaCo-E-Zn.
¿n-E Co-E-Zn(Zn)
A E z A.E z
o 0,6 0,3
10'5 M 13,7 100 8,1 100
10'4 M 8,6 62 1,4 17
10'3 M 0,4 3 0,1 1
10'2 H 0,3 2 0,1 1
Se midió 1a actividad enzimática como sedetaiió en Materiaies y Métodos, sóio queen presencia de zinc, a ias concentraciones indicadas y de EDTA10-5 M.
Vemosque en este caso, se obtuvouna preparación enzimática con 2 átomogramos de cobaito por subunidad de 35 kDa; esprobable entonces que e] segundo átomo de cobaito haya ocupadoe] sitio de] zinc. Hay un 1igero aumento en e] contenido de zinc
de 1a Co¿-E, que posibiemente se deba a una contaminación, (yasabemos que es muydificil evitarias); de todos modos, eiio noafecta a nuestros resuitados.
La apoenzima iibre de zinc no presentó actividad aün enpresencia de DTT, pero 1a recuperó con concentraciones de zincentre 10'5 y 10'4 M, iiegandoa tener 1a mismaactividad especifica que 1a hoioenzima de 1a cua] se preparó (Igblg_llj¿é¿).

277
TABLAIII.3.: Efecto de] zincsobre 1a Zn-E
(Zn) (M) A.E. %
o 13,30 100
1 x 10‘5 16,00 120
3 x 10'5 15,30 115
1 x 10'4 3,40 25
3 x 10'4 0,80 6
1 x 10'3 0,70 5
3 x 10'3 0,22 2
1 x 10‘2 0,15 1
Se ensayaron ias actividadescon 1as concentraciones de
'zinc indicadas, en las condiciones deta11adas en Materiaies y Métodos.Los resuitados correspondena1 promedio de 3 experiencias.
La Coz-E en presencia de DTT, mostró actividad, si bienésta es aproximadamente 1a mitad de 1a obtenida para ias Co-E deias experiencias anteriores, que de todas maneras recordemos,tenian un contenido de zinc entre 0,6 y 0,8 átomogramos por sugunidad. En ausencia de] reactivo tióiico, 1a Coz-E fue inactiva.Esto indicaria que, cuando e] cobaito ocupa e] sitio de] zinc,no podria reempiazarlo totalmente, dado que 1a actividad cataiitica disminuye, pero pareciera hacerio parciaimente. Además, a1igua] que e] zinc, e] cobaito también ejerceria su acción a través de ios grupos tio]es,ya que 1a Co-E 5610 presenta actividaden presencia de DTT,siendo inactivaen ausencia de] reductor.

278
¡25 —O
¡oo_ o C‘s
75 —ÜfU
'U
Éo; .U<
25 ’
0 __J_#¡41 l-0 -5,o —u,s -h,o -3,5 —3,o —2_5 -2,o
loq (Zn2+) (M)
FIGURAIII.19.: Efecto de] zinc sobre 1a Zn-E.(O) La preparación enzimática contenia 0,8 ¿tomogramos de zinc por subunidad.(O) La preparación enzimática contenía 0,5 átgmogramos de zinc por subunidad.
Efecto de] zinc sobre 1a C02E: como se ve en 1a [lauraIII.20., e] zinc,entre 10'5 y 10'4 M, produce un aumento de 1aactividad de 1a Coz-E. La actividad aumenta hasta a1canzar 1aactividad especifica de 1a Co-E-Zn, que recordemos era aproximadamente 10 UE/mg. Esto indicaria que e] zinc estaria desplazando a] cobaito ubicado en e] sitio dei zinc.
III.2.2.2. Modeiopropuesto para 1a interacción zinc-cobaitogn_e1 ALA¿Q
En base a ios resultados obtenidos de] estudio de 1a interacción zinc-cobaito con 1a apoenzima y las metaioenzimas resuïtantes, se propone ei siguiente modeïo que expiicaria ios

279
cambios en ias actividades y contenido de metaies de 1as especies resuitantes (Figura III.21.).
TABLAIII.4.: Reemplazo de] zincpor cobaito, a pagtir de 1a apoenzima (Apo-E)
A.E.(+ DTT) 0,32 5,20
A.E.(--DTT) 0,20- 0,80
2+Zn /subun. 0,04 0,16
C02+/subun. 2,02
Los detaiies experimentaies sedetailan en e] texto y en Materiaïes y Métodos. ‘
Cuando a 1a hoioenzima nativa identificada como Zn-E(cuyo contenido de zinc es de] orden de 1 átomogramo por subunidad y 1a actividad especifica es de] orden de 14 UE/mg) se trata con EDTA10'5 M, se desprende e] meta] dando como resuitadouna especie de apoenzima con muy bajo contenido de zinc (dei 03den de 0,1 a 0,2 átomogramos/subunidad) y correspondientementeocurre una inactivación cercana a1 90 % (paso I). Cuando esa hgioenzima se 1a trata también con EDTApero en condiciones detegminadas, se puede obtener una verdadera apoenzima con menos de0,04 átomogramos de zinc por subunidad y una actividad especifica de 0,44 UE/mg. Si 1a hoioenzima se trata simultáneamentecon EDTA10'5 My zinc 10'5 M (paso II), o bien 1a apoenzima setrata con zinc 10'4 - 10“5 M(paso III), se reconstituye 1azinc-enzima nativa, recobrando su actividad origina]: 14 UE/mg.

Si a ambas especies, 1a Zn-E o 1a apo-E se ias trata con un exceso de zinc (concentración superior a 10'4 - 10'5 M (pasos IIIy V) se produce una forma menos activa que 1a nativa, posibiemente debido a 1a unión de un átomo de zinc a otro sitio diferentede 1a enzima, en e] cua] podrian encontrarse otros restos suïfhidrilos no funcionaies pero cuya modificación podria 11evar acambios estructuraies negativos con 1a consiguiente caida en 1aactividad cataiïtica ( 14 UE/mg).
TABLAIII.5.: Efecto de]zinc sobre1a apoenzlma
(Zn) (M) A.E.
0 0,44
1 x 10'7 0,34
3 x 10’7 0,38
1 x 10'6 0,27
3 x 10’6 0,39
1 x 10'5 0,61
2 x 10'5 0,92
3 x 10'5 1,43
4 x 10'5 1,68
6 x 10’5 14,60
8 x 10'5 14,40
1 x 10'4 13,50
Se ha demostrado que e] cobaito no puede despïazar a1zinc de 1a hoioenzima de manera que cuando ésta se trata con cgbalto, se obtiene una especie conteniendo como promedio un átomo de zinc y uno de cobalto por subunidad, 1a que podria
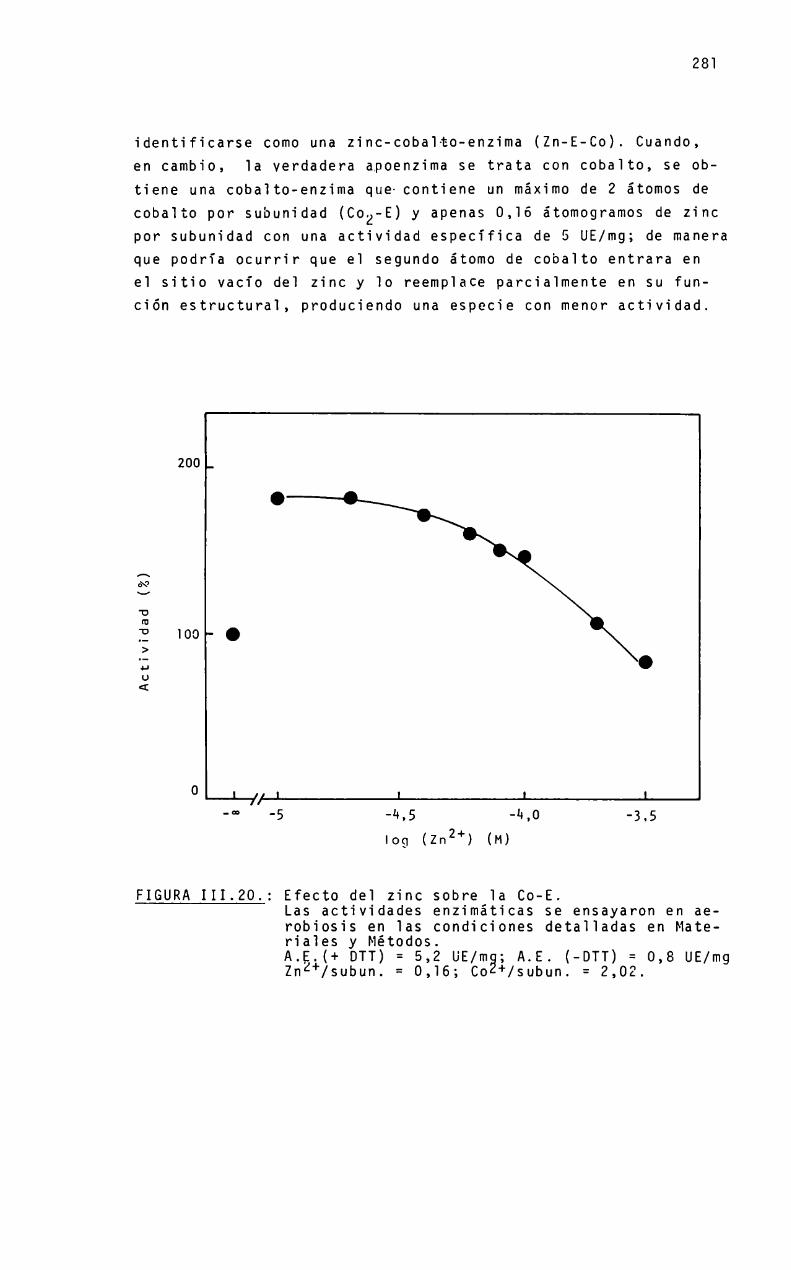
281
identificarse comouna zinc-cobaito-enzima (Zn-E-Co). Cuando,en cambio, 1a verdadera apoenzima se trata con cobaïto, se obtiene una cobaito-enzima que-contiene un máximo de 2 átomos de
cobalto por subunidad (Cog-E) y apenas 0,16 átomogramos de zincpor subunidad con una actividad especifica de S UE/mg; de maneraque podria ocurrir que e] segundo átomo de cobalto entrara ene] sitio vacio de] zinc y 1o reemplace parciaimente en su función estructura], produciendo una especie con menor actividad.
200 L
Cb
33
'Uf0
73 ¡00* o>.5 .U<
o LL_’/ll_l l L l-° -5 -#.5 -ü,0 -3.5
log (Zn2+) (M)
FIGURAIII.20.: Efecto de] zinc sobre 1a Co-E.Las actividades enzimáticas se ensayaron en aerobiosis en 1as condiciones detaïladas en Materiaies y Métodos.A.E.(+ DTT) - 5,2 UE/mgí A.E. (-DTT)/+ _ = 0,8 UE/mgZn¿ /subun. = 0,16; Co subun. = 2,02.

(H HH(HH (VH
(VH) NH“
Zn-Enzhna
*EDTA10'SM #EDTA10'5M‘Zn10'5M
‘EDTA10'5M*Zn>10'SM Co-Enzhna
(Zn-E-Co)
#EDTA10'5M ‘EDTAiO'SM
#Zn106M+EDTA10'5M
*Zn>1O-SM
AEh14AEL<:14 A.E.:9 Alí:OG A.E.:9
——>
Ali:0A ALE:14 AEn<14 A.E.:5 A.E.:7-9
Apo-Enzhna
oZn10“-10‘5M(IV)‘Zn>10"M(V)
Co-Enzhna
(Co-E)
oZn10"-10'5M(IX)
‘Zn>10"M(X)
A.E.<9_> <__A.E.<7
FIGURAIII.21.:Modeïopropuestoparaexplicar1ainteracciónde]zinccon1ahoïoeg
zimatratadaconEDTA,1aapoenzima,y1ascobaito-enzimasobtenidasapartirde1aholoyapoenzima.
282

283
Cuando 1a Zn-E-Co se trata con EDTA(paso VI) se desprende e] zinc y resuita una cobaito-enzima de muybaja actividad (0,3 UE/mg); esto apoya 1a hipótesis antes mencionada de quecuando ei cobaito ocupa e] sitio de] zinc, de aiguna manera puede reempiazario en su función protectora de ios restos suïfhídrlios funcionaies, y comoconsecuencia, se produce una especie enzimática con cierta actividad; io que no puede e] cobaito, reitgramos, es despiazar a1 zinc cuando éste está ubicado en 1a proteina.
Si a 1a Zn-E-Co se 1a somete simuitáneamente a 1a ac
ción de] EDTAy e] zinc (paso VII), este üitimo revierte o impide 1a acción inhibitoria de] queiante y estamos frente a 1a especie de 1a cua] partimos con igual actividad especifica(9 UE/mg) que también puede obtenerse si 1a Coz-E se trata conzinc 10'4 - 10‘5 M; en estas condiciones e] zinc despiaza a1 cgbaito que estaba ocupando su sitio (paso IX).
Si 1a Zn-E-Co, o 1a C02E se tratan ahora con concentraciones de zinc mayores que 10-4 - 10'5 M, Un átomo de zinc podria ubicarse en otro sitio diferente de 1a enzima, comoe] postuiado para ios pasos III y V, con las consecuencias ya discutidas, o también podria suceder que ese exceso de zinc, despiazara a uno o ios dos átomos de cobaito de sus sitios de unión a1a enzima, produciendo una especie menos activa ( 7-9 UE/mg).

CAPITULO IV
3GRUPOS FUNCIONALES DEL ALA-D DE HIGADO BOVINO\ \”(f”° '
J\‘
Página
IV.l. Tioles 284
IV.l.l. Titulación de la enzima reducida con DTT 284
IV.l.2. Titulación de la enzima nativa sinreducir 285
IV.l.3. Conclusiones 285
IV.2. Histidina 29I
IV.2.I. Inactivación del ALA-Dpor DEP 291
IV.2.2. Númerode residuos histidina modificados 294
IV.2.3. Identificación del residuo histidina comoresponsable de la inactivación por DEP 295
IV.2.4. Efecto del pH. Protección por zinc y porsustrato 296
IV.2.5. Efecto de otros metales 299
IV.2.6. Conclusiones 302

284
IV. GRUPOS FUNCIONALES DEL ALA-D DE HIGADO PORCINO
IV.l.log
IV.l.l. litulación de la enzima reducida con DTT
Se empleó la fracción purificada 654 veces (banda única por electroforesis (0,2 átomogramos de zinc por subunidad de35 kDa; actividad: 14,85 UE/ml) totalmente reducida por tratamiento con DTT 20 mMdurante 15 minutos a 37 °C bajo atmósfera
de nitrógeno. Luego se filtró por una columna de Sephadex G-25(0,9 x 12 cm) equilibrada con buffer Tris-HCl 50 mMpH 7,2 saturada con nitrógeno para separar el exceso de DTT. En la solución enzimática resultante se determinó la actividad y el contenido de tioles en función del tiempo, inmediatamente luego deobtenida,a tiempo cero (ER 0h) y luego de mantenida a 4 °C yexpuesta al aire durante 3 y 20 horas (ER 3h; ER 20h).
En una experiencia tipo se trataron aproximadamente60 pg/ml de solución enzimática con DTNB(125 pM) a 25 °C y sesiguió el curso de la reacción por absorbancia a 412 nm (Ellman,1959).
Se observó un rápido aumento de la absorbancia en losprimeros minutos que alcanzó un plateau a los 20 minutos (fig_ra IV.l., Tabla IV.l.).se detectó un grupo sulfhidrilo altamente reactivo, que reaccig
Con la enzima reducida a tiempo cero
nó dentro del primer minuto (tipo I), luego aparecieron dos grupos sulfhidrilos adicionales que reaccionaron más lentamente pgro dentro de los 30 minutos subsiguientes (tipo II) y por último, el tratamiento con SDS0,4%, por desplegamiento de la estructura cuaternaria puso de manifiesto otros tres grupos Sulfhidrilos (tipo III).
La titulación de la enzima expuesta al aire durante 3horas, que ha perdido un 20 % de su actividad original, mostróun comportamiento similar a la anterior aunque parecería ocurrir un ligero aumento de los restos tipo I a expensas del tipoII, en tanto que la variación dees significativa. Esta tendenciapo II a I se define mejor cuandopermanecido 20 horas a 4 °C y al40 % de la actividad inicial. En
los sulfhidrilos tipo III node la conversión de grupos tise titula la enzima que habiaaire, la cual sólo retuvo uneste caso, se detectaron dos

285
restos sulfhidriios tipo I y un tipo II; también apareció unadisminución de los grupos suifhidriios tipo III a un valor cercano a 2, es decir 1a pérdida de uno de estos tioies no expuestos en 1a estructura proteica nativa.
IV.1.2. Titulación de 1a enzima nativa sin reducir
La actividad de 1a enzima nativa purificada 654 vecessin ei agregado de DTTen 1a mezc1a de incubación fue nu1a (Iab1a IV.1.). En presencia de] agente reductor se obtuvo un vaiorde 12,058 UE/mg.
La tituiación de esta fracción enzimática no reducida,reveïó 1a presencia de 2 restos suïfhidriïos tipo I por subunidad de 35 kDa (Figura IV.1., Tabia IV.1.) que reaccionaron dentro de] primer minuto, para 1uego no detectarse cambios en 1aabsorbancia. La adición de SDStituió 2,3 restos suifhidriiostipo III.
IV.1.3. Concïusiones
Los datos obtenidos nos están indicando que en 1a enzima nativa totaimente reducida, pero cuyo contenido de zinc esde sóio 0,2 átomogramos por subunidad, es decir que no se tratade 1a hoioenzima, pero tampoco es una verdadera apoenzima, existe un grupo suïfhidrilo tipo I, dos su1fhidriios tipo II y tressuifhidriïos tipo III. Esta enzima redUCida, a pesar de su bajocontenido de zinc, tiene una actividad a1ta, pues de acuerdo a1a hipótesis de Tsukamoto y c0]. (1979) como hemos dicho, 1afunción de] zinc no seria cataiitica, sino que actuaria protegiendo de 1a oxidación a dos restos suïfhidriios. A medida queesta preparación enzimática se va oxidando, va perdiendo actividad, y uno de sus restos suifhidriïos tipo II se convierte enuno tipo I. Por otro lado, 1a enzima nativa sin reducir.es tota1mente inactiva, sóio se detectan dos restos Sulfhidriïos muyreactivos tipo I, ninguno tipo II y análogamente a 1a preparación anterior envejecida 20 horas a1 aire, también se ha perdidoun resto su1fhidri10 tipo III.

GruposSH/subunidad
_ [a]
_ L l l
_ (b)
F
.. l l l
_ [U
É
L % l l0 30 60
fiempo
FIGURA IV.1.:
(minuios)
Tituiación de grupossuifhidrilos con DTNB.La enzima (60 pg/mï)en buffer Tris-HC]50 mM pH 7,2 se incubócon DTNB (125 pM) durante 30 minutos,y otros30 minutos adicionaïes,1uego dei agregado deSDS 0,4%, a temperaturaambiente. Los vaïoresde absorbancia a 412 nmfueron corregidos por Ioscambios de v01umen por e]agregado de SRS.e = 1,36 x 10 M-1 cm-1(a) Enzima nativa reducida inmediatamente iuegode obtenida y (b) 1uegode estar expuesta a1 airedurante 3 horas,(c) enzimanativa sin reducir.
286

287
Titu1ación de grupos su1fhidri1oscon DTNBy actividad cataliticade ALA-Dde higado de cerdo reducida y 1uego de su exposición a1aire durante diferentes periodos,y de 1a enzima sin reducir
ACTIVIDAD
ENZIMA ENZIMATICA (min) -SH/subunidad(73)
ER 0h 100 0-1 1,001-30 2,00SDS 3,00
ER 3h 80 0-1 1,301-30 1,57SDS 2,83
ER 20h 40 0-1 1,971-30 1,18SDS 2,30
EOX 20h 0 0-1 2,401-30 0,00SDS 2,27
ER: enzima reducida y 1uego expuesta a1 aireEOX: enzima sin reducirLas condiciones experimenta1es se exp1icanen e1 texto.Los resu1tados son promedio de 3 experiencias
Si comparamos nuestros resu1tados con 1a enzima de higado porcino con-105 obtenidos por Tsukamoto y co]. (1979) para1a de higado bovino, veremos que existe una aceptable

288
concordancia. Esos autores habian tituiado en 1a hoioenzima, conteniendo 1 átomogramo de zinc por subunidad, 2 restos tipo I,3 tipo II y 3 tipo III; para ia apoenzima reducida: 4 tipo I;1 tipo II y 3 tipo III y para 1a apoenzima oxidada 2 tipo I, 1tipo II y 3 tipo III. E1 cambio de dos restos suifhidriïos tipoII en dos tipo I a1 pasar de 1a hoioenzima a 1a apoenzima, seatribuyó a que esos dos grupos Suifhidriios estaban asociadosa1 zinc; como se mencionó,e1 ro] asignado a] zinc seria e] demantener esos restos reducidos en 1a hoioenzima, de manera quea1 liberarse e] zinc, en 1a apoenzima oxidada, desaparecen esosdos grupos por formación de unpuente disuifuro.
En nuestro caso, tengamos presente que estamos empïeando una enzima que 5610 contiene 0,2 átomogramos de zinc por sugunidad, de manera que nuestra hipótesis (figura_¿y¿g¿) es quee] átomo de zinc de cada subunidad se encuentra vincuiado con1a subunidad vecina, en e] dimero funciona] (Batiie y Ste11a,1978), a través de dos restos suifhidriios, uno tipo I y uno tipo II provenientes de cada subunidad (figura IV.2LQ). De acuerdo con 1a idea de Tsukamoto y co]. (1979), e] meta] contribuiria a] mantenimiento de] estado reducido de estos tioies, quesi bien estarian ubicados en e] centro activo de 1a enzima, noparticiparian directamente en e] proceso cataiitico; 1a ausencia o muy bajo contenido de zinc iievaria a 1a formación de unpuente entre ambos restos (Figura IV.2.Ql; de esta forma estariamos tituiando un resto tipo I y un resto tipo II menos, enconcordancia con 10 haiiado por Tsukamoto y co]. (1979) y sóiodetectariamos e] único resto suifhidriio aitamente reactivo tipo I ubicado en e] sitio activo y Ios otros dos Sulfhidriios tipo II expuestos y situados en otra región de 1a proteina enzimatica. Estos üitimos grupos suifhidriios, aunque no están reiacionados con 1a actividad, a1 oxidarse 1a enzima e inactivarsepodria modificarse su estructura, y a] exponerse mejor se convertirian en dos restos que reaccionarian rápidamente con e]DTNB,evidenciándose asi comosuifhidriio tipo I (Elgura IV.2.c).
Además, e] uso de preparaciones expuestas a1 aire porperiodos más proiongados aün que ios estudiados por otros autores, nos ha reveiado que los restos suifhidriios ocu1tos (tipoIII) también son susceptibies de oxidación, dado que su contenido es menor en ias muestras estacionadas pormas tiempo a] aireo en 1a enzima nativa oxidada.

289
.oCWULoQonmow;onon<4<Funmusgo»mmgcmx
-omwuWMPcmmmFOWPmeazgmmopmucowocswaoawpFmLmuw—axmaguacpmm3Q0Lamsmzcmm".N.>H<monm
@IÍIÍ@IWNaer:¿32.893®®8
ImmuzmÍImN
22:2:23:3Éazánz325:3¿538m22:23?2:“me..st222.3342:28:;3
6,9;8:9;©/
‘Au ‘\\WSkw\sto
\
@amlemme“mwx®9.a
v5.8N28“AI“2:2 \
\\\\
\.eÑ\Hew\wlm\ zw\m¡lm._
\\&N@_
AU+AUImñYhuzmmu

290
Por oxidación de Ios restos suïfhidriïos tipo I ó III,además de inactivarse 1a enzima se podrian formar puentes disulfuro en e] dimero, inter o intrasubunidades ya sea SI-SI,SIII'SIII ° au" SI'SIII'
Consideramosasimismo, que estos eniaces sulfhidriiicos, deben cumplir un ro] importante en 1a interacción entresubunidades, tanto para formar e] dimero funciona], como paraconstituir e] octámero de máximaactividad cataiitica y es tentador asignar a estos grupos tioïes un pape] orientador y directriz en e] ensambie correcto de 1as dos subunidades de 35 kDa,a si como en 1a unión de ias dos moiécuïas de ALAa1 sitio activo, como ilustraremos más adeiante.

291
IV.2. HISTIDINA
Recordemos qeu se ha propuesto a 1a lisina y, posteriormente, también a 1a histidina y 1a arginina como aminoácidos invoïucrados en e] mecanismo de] ALA-D(Nandi y Shemin, 1968;Tsukamotoy c01., 1975; Batiie y Stelia, 1978; Liedgens y co].,1983).
De elïos, 1a histidina podria ser un iigando de] zinc,ya que según io reportado por Tsukamoto y co]. (1979), por fotooxidación de dichos residuos, disminuye tanto 1a actividad catalitica como 1a cantidad de zinc unido a 1a proteina.
Además, en 1a mayoria de ias zinc-metaioenzimas, sehan detectado residuos histidina esenciales (Ekiund y Branden,1983; Lindskog, 1983). Para acïarar e] ro] de] zinc en unazinc-enzima, es imprescindibie caracterizar Ios grupos funcionaies de] sitio de unión de] meta].
Por otra parte, emp1eandoe] reactivo especifico pararesiduos histidina DEP,Maraïihaïïi y co]. (1985) encontraronque en e] ALA-Dpurificada de maiz se modificaban 7,4 residuoshistidina por mo] de enzima. Esos residuos eran esenciaies para 1a catáïisis enzimática, y el sustrato, ALA,y su anáiogo,e] ácido levuiinico, proteian a 1a enzima de 1a inactivación porDEP, sugiriendo una probabïe modificación en e] sitio activo.Sin embargo, es de hacer notar que estos autores no investigaron una posibie coordinación con iones zinc.
Hay evidencias,además, de que 1a enzima de origen veggta] podria tener aigunas propiedades diferentes a 1a de mamifgros (Liedgens y co]., 1983).
Nosotros hemos estudiado entonces los efectos de 1a m9dificación quimica por DEP,sobre 1a actividad cataïitica de]ALA-Dde higado porcino aïtamente purificada, en soiución acuosa a diferentes pH's,en presencia y en ausencia de iones zinc,obteniendo Ios siguientesresuitados.
IV.2.1. Inactivación de] ALA-Dpor DEP
Se ha observado que e] tratamiento de] ALA-Dcon DEPproduce una inhibición de 1a enzima que es función de 1a tempgratura, tiempo de reacción y concentración de reactivo.
En 1a Figura IV.3.a se grafica e] curso de 1a
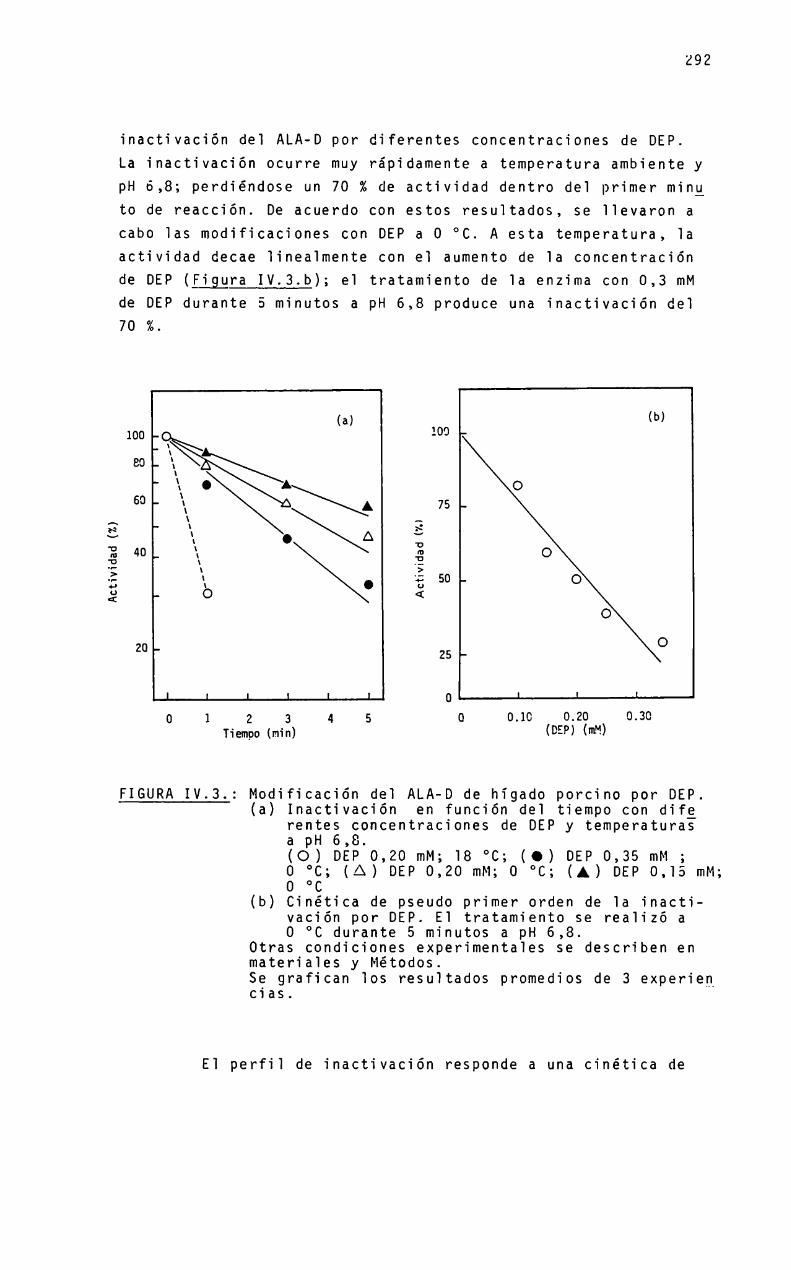
292
inactivación de] ALA-Dpor diferentes concentraciones de DEP.La inactivación ocurre muy rápidamente a temperatura ambiente ypH 6,8; perdiéndose un 70 % de actividad dentro de] primer minuto de reacción. De acuerdo con estos resuitados, se 11evaron acabo las modificaciones con DEP a 0 °C. A esta temperatura, 1aactividad decae Iinealmente con e] aumento de 1a concentraciónde DEP (Figura IV.3.b); e] tratamiento de 1a enzima con 0,3 mMde DEPdurante a minutos a pH 6,8 produce una inactivación de]70 %.
(a)\\\\\\\\
A \ '2N ' eV \\ .\A 'DÉ 40 " \\ Í;
.2 x , s2 - b 2
20
L l L l l 4 o l I l
0 1 2 3 4 5 0 0.10
Tiempo (min)
0.20 0.30(DEP) (¡11"!)
FIGURAIV.3.: Modificación de] ALA-Dde higado porcino por DEP(a) Inactivación en función de] tiempo con dife.
rentes concentraciones de DEPy temperaturasa pH 6 8.(C)) DEP 0,20 mM; 18 °C; (O) DEP 0,35 mM0 °C; (A) DEP 0,20 mM; 0 °C; (A) DEP 0,15 mM;0 °C
(b) Cinética de pseudo primer orden de 1a inactivación por DEP. E1 tratamiento se reaïizó a0 °C durante 5 minutos a pH 6,8.
Otras condiciones experimentaïes se describen enmateriales y Métodos.Se grafican Ios re5u1tados promedios de 3 experiencias.
E] perfi] de inactivación responde a una cinética de

pseudo primer orden. Reordenando ios datos de 1a Figura IV.3.bde acuerdo a1 método de Keech y Farrant (1968) y Roche y McFaldden (1969), se obtienen ios resuitados de 1a Eigura_L1¿fl.E1 tiempo de inactivación media a concentraciones saturantes de
DEP es de 0,5 minutos, con una KDEP = 1,6 mM. De 1a pendientede 1a recta de 1a Figura IV.Q¿b, obtuvimos que e] número de moiécuias de DEPque reaccionan con cada unidad activa, dando uncompiejo inactivo enzima-inhibidor es de 0,87. Este vaior, cercano a 1a unidad, indica que una moiécuia de DEPestá invoiucrada en 1a unión a 1a enzima durante e] proceso de inactivación ypor 10 tanto, habria un soio residuo que reacciona con este mgdificador y cuya integridad es esencia] para 1a actividad enzimática. Además, 1a inactivación por DEPmuestra una cinética desaturación, ta] como 1a que se espera de una unión reversibiede] inhibidor a] sitio activo de 1a enzima.
(a) (b)
6 — o 0 8 _
c I—l
É 4 _ O t 0.6 _ O
S 8'4-3 I
2 _ 0.4 _ y
/V
o L L L 9T 7III l l. lo 2 4 6 o 0.4 0.6 0.8
1/(DEP) (mu-1) iog (DEP)
FI URAIV.4.: (a) Tiempo de inactivación media a concentraciones saturantes de DEP.
(b) Número de moiécuïas de DEP que reaccionan dando e] compiejo inactivo enzima-inhibidor.
Los datos se obtuvieron de 1a Figura IV.g¿, segúnse describe en e] texto.
La incubación dei ALA-D porcina con 0,20 mMde DEP apH 6,8 produce una pérdida de actividad dependiendo de] tiempo,a 1a vez que un concomitante aumento de 1a abosorbancia a 240 nm

correspondiente a 1a formación de 1a unión carbetoxi-histidina(figura IV.5.). En 1a curva de inactivación se observan dos fases: una rápida y otra más 1enta; 1a primera de e11as presenta1a cinética de pseudo primer orden, a concentraciones fijas deDEP.
100
¿ _ .09 _ 3
75 _
_ .06 _ 2
50 _ O
Actividad(1)
AA240nm
dehistidinasmodificadas
a _ o \\\\\.g\\‘\“““*—‘-———- - o3 -1HH0 I I I II I 0 J 0
Tiempo (min)
número
FIGURA V.5.: Curso de inactivación de ALA-Den función de]tiempo y modificación de residuos histidina.La enzima (115 pg) se trató con DEP 0,2 mMa O °Cy pH 6,8. La reacción se siguió espectrofotométricamente a 240 nm (C)). En ios tiempos indicados _se tomaron aiicuotas a ias que, una vez diiuidasen buffer conteniendo imidazo], se ies reaiizaronlas determinciones de actividad (o ). E1 cáicuiode] número de residuos histidina modificados porsubunidad, y otras condiciones experimentaies,son las que se indicaron en Materiaies y Métodos.Se grafican ios resultados promedio de 3 experienCias.
I-l
IV.2.2. Númerode residuos histidina modificados
De acuerdo con ios datos de absorbancia a 240 nm, secalculó que se modifican 3 residuos histidina por subunidad, cada uno de elios con diferente veïocidad de reacción (¿laura IV.5¿1.

295
Es interesante notar que 1a reacción de] primer residuo histidina coincide con 1a primera fase de inactivación rápida. Podemos hacer una correiación entre e] número de residuoshistidina modificados por e] DEP, por subunidad de ALA-D, cone] perfil de inactivación (figura IV.6.) y una extrapoiación de1a primera parte de 1a curva nos indica que un residuo histidina por subunidad es critico para 1a actividad enzimática, e]cua] reacciona más rápidamente que ios otros residuos, no esenciaies.
Actividad(z)
0 ‘I 1 l
0 1 2 3
númerode histidinas modificadas
ELQURAIV.6.: Correiación entre 1a actividad enzimática y e] número de histidinas modifidas por subunidad de ALA-D.Se graficó a partir de iosdatos de 1a Figura IV 3.
IV.Z.3. Identificación de] residuo histidina comoresponsabiede 1a inactivación por DEP
Si bien e] DEPes un reactivo seiectivo para residuos

histidina a pH 6 - 6,8; ocasionalmente pueden reaccionar tambiénotros aminoácidos tales comolisina, tirosina, serina y cisteina (Miles, l977). Por esta razón, resultó importante demostrarque otros grupos que no fueran histidina fueran responsables dela inactivación.
El incremento de absorbancia a 240 nm indica desde yala formación de una unión etoxiformilhistidina durante el cursode la modificación del ALA-Dpor DEP. Además, no se observaroncambios de absorbancia a 280 nm, indicando que no hubo modificación de residuos tirosina. Por otra parte, la modificación deresiduos serina es revertida rápidamente en solución acuosa neutra, efecto no observado en nuestro caso.
Por incubación con hidroxilamina 0,7 Ma pH 6,8 a temperatura ambiente durante l hora, se logró revertir totalmentela inactivacióndel ALA-Dmodificada en un 50 % por DEP 0,2 mM(Tabla I!LÉL¿. La enzima tratada con DEPno recuperó su actividad en ausencia de hidroxilamina. La hidroxilamina reaccionarápidamente con los residuos etoxiformilhistidina, en cambio,la ruptura de las uniones etoxiformillisina con este reactivoes mucho más lenta (Melchior y Fahrney, 1970), y los residuostiosles reaccionan aün 5 veces más lento que la lisina. Por lotanto, nuestros resultados indican que son los residuos histidina los responsables de la inactivación del ALA-Dpor DEP.
IV.2.4. Efecto del pH. Protección por zinc y por sustrato
Teniendoen cuenta que las formas no protonadas de losresiduos histidina se modifican a velocidades mayores que lasformas protonadas, se llevó a cabo un estudio de la inactivación por DEPa diferentes pH's. Comopodemos ver en la FiguraIV.7., esta inactivación es pH dependiente. De los resultadosde esta figura, calculamos las constantes de pseudo primer orden de inactivación (Kapp) para la primera fase (Abdulwajid yHu, l986) los que se graficaron en función del pH, obteniéndoseuna tipica curva de titulación (figura IMLQL)que muestra unpK de 6,8; valor que corresponde muy bien con un residuo histidina. Tsukamotoy col. (l975) obtuvieron un resultado similaren la inactivación de ALA-Dvacuna por fotooxidación.
La presencia de iones zinc en el medio de reacción prgtege la proteina de la reacción deetoxiformilación. Comopodemos observar en la Figura lï¿n¿, el efecto protector del zinc

es mayor a pH menores, efecto que podemos correIacionar con e]hecho de que este meta] ejerce su acción activante sobre e]ALA-D, a pH's menores que 7. Estos haIIazgos también están de acuerdo con 1a hipótesis de HoIbrook e Ingram (1973) quienes prgpusieron que Ias formas no protonadas de histidina son Ias formas reactivas, y con e] trabajo de Cousineau y Meighen (1976)quienes postularon que Ias histidinas no protonadas se modifican a una veiocidad mayor que Ios residuos protonados.
TABLAIV.2.: Reversión de 1a inhibición porDEPmediante hidroxiiamina
ACTIVIDAD DE ALA-D
SISTEMA en BFP 0,] H en NHZOH 0,7 M
30 min 60 min 30 min 60 min
Contro] 100 100
+ NH20H 98 100
+ DEP 50 50 70 100
La enzima se modificó con DEP 0,2 mMa 0 °C,hasta obtener un 50 % de inhibición; Iuego seincubó con hidroxilamina a pH neutro, a tempgratura ambiente por 30 y 60 minutos. La medición de actividad y demas condiciones experimentaïes se detaIIan en Materiaies y Métodos.Se realizó un contro] de Ia enzima sin trataren BFP 0,1 M pH 6,8 con Ios intervaios de tiempo indicados, y un segundo contro] de 1a enzima también sin DEP, pero en presencia de hidroinamina 0,7 Mdurante 30 y 60 minutos.
Es de notar que a pH 7,5 1a inactivación fue mayor quea pH 6,0 y que e] zinc no Iogra prevenir 1a acción de] DEP.
Además 1a protección por zinc es más pronunciada en 1aprimera fase de 1a reacción de inactivación, es decir su aniónse ejerce sobre e] residuo histidina esencia], mientras que noparece tener efecto sobre e] resto de Ios residuos histidina que

298
reaccionan más 1entamente.
100
80 _ \\“
60 - \
4o- \’
’¿’4
(%
y? //??Éí/Actividad
20 _
0 15 30 45 60
Tiempo (min)
FIGURAIV.7.: Inactivación de ALA-Dpor DEP a diferentes vaiores de pH, y protección por zinc.Los detaiies de ias condiciones experimentalesse describieron en Materiaies y Métodos: A Iostiempos indicados se tomaron aiicuotas de 1aenzima tratada con DEP0,2 mM,y se ies determinó 1a actividad. Los resuitados graficadoscorresponden a1 promedio de 3 experiencias.(V) pH 6,0; (v) pH 6,0 y zinc 5 x 10-4 M(A) pH 6,8; (A) pH 6,8 _yzinc 5 x10-4M(o) pH 7,5; (o) pH 7,5 y zinc 5 x10'4 M
E1 sustrato ALAtambién ejerce una protección importante sobre 1a modificación por e] DEP(figura_ll¿g¿). Se hicieronexperiencias preincubando con e] sustrato soiamente, y con e]Sustrato en presencia de zinc. En este üitimo caso, los resuitados indican que ambos efectos son aditivos.
En consecuencia, de todas estas experiencias podemosconcluir que e] residuo histidina, e] sitio de unión de] sustrato y e] de] zinc están en estrecha proximidad en e] centro

299
activo de 1a enzima.
10 rO
A 8-.7 O
C
E
6 _“J O
S /2:4- otuz /
2_ O
J l | I
60 6.5 70 75pH
ELQURA LXLQL: Dependencia de] pH de 1aconstante de veiocidad depseudo primer orden (Kpara 1a inactivación p PpDEP de] ALA-D de higadoporcino.Los vaiores se obtuvieronde 1a Figura IV.7.
)
IV.2.5. Efecto de otros metaies
En 1as metaïoenzimas, 1a Sustitución de] meta] por otros iones metáiicos se utiliza con frecuencia para investigare] ro] de] meta].
Se estudió entonces, 1a posibiiidad de que otros metales , además de] zinc, pudieran interaccionar con ios grupos histidina.
Para elio, se ensayaron ios siguientes metaïes: cadmio,manganeso, cobaito, estaño, seienio y mercurio a cero y 15 minutos, y los resuïtados se grafican en 1a Figugg_gv.10. Para cada
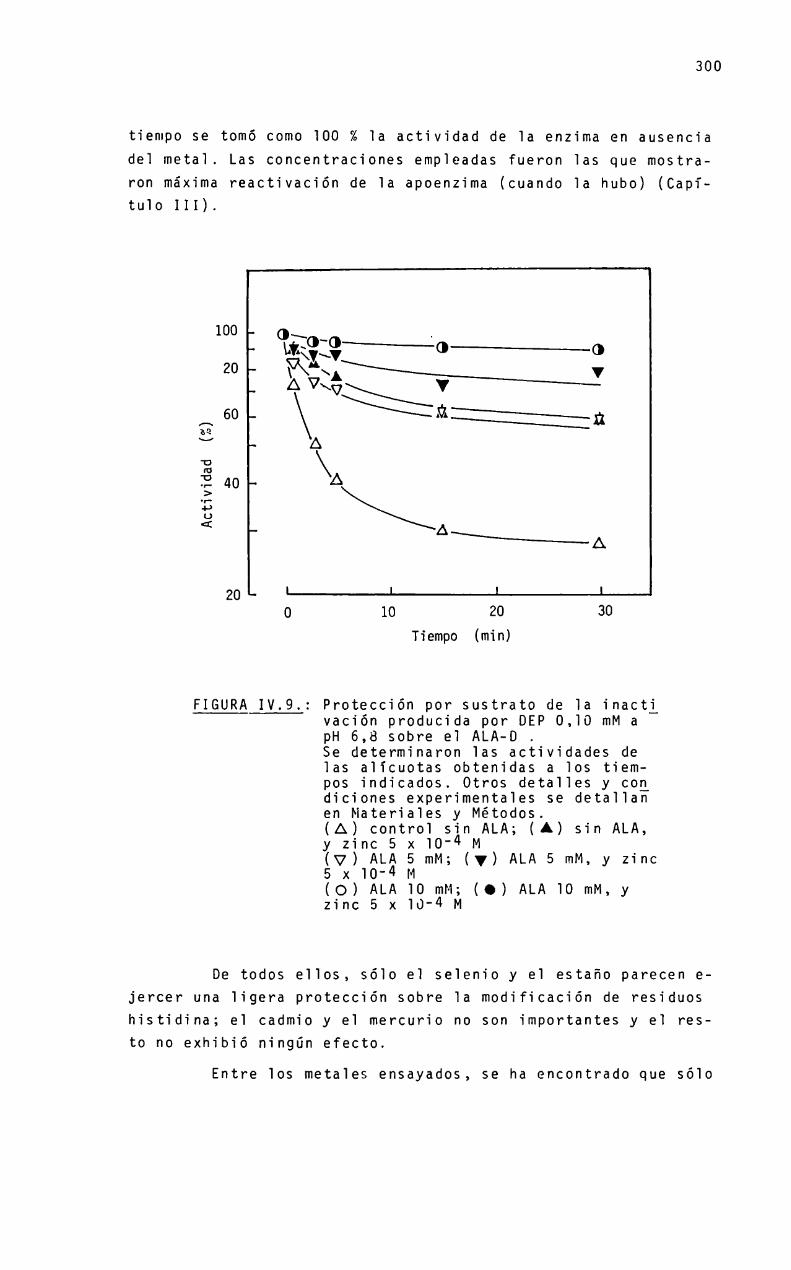
300
tiempo se tomó como 100 % 1a actividad de Ia enzima en ausenciade] meta]. Las concentraciones empieadas fueron ias que mostraron máxima reactivación de 1a apoenzima (cuando 1a hubo) (Capitulo III).
um — o ‘
60_ .v.\n(1 l D'
40
Act1v1dad lD.
\AzoL L I l l
0 10 20 30
Tiempo (min)
FIGURAIV.9.: Protección por sustrato de 1a inactivación producida por DEP 0,10 mMapH 6,8 sobre e] ALA-DSe determinaron ias actividades deias aiicuotas obtenidas a ios tiempos indicados. Otros detaiies y condiciones experimentales se detaiianen Materiales y Métodos.(A) contro] sin ALA; (A) sin ALA,y zinc 5 x 10'4 M(V) ALA 5 mM; (v) ALA 5 mM, _y zinc5 x 10-4 M(o) ALA io mM; (o) ALA 10 mM, yzinc 5 x 10-4 M
De todos eilos, sólo e] selenio y e] estaño parecen ejercer una iigera protección sobre 1a modificación de residuoshistidina; e] cadmio y e] mercurio no son importantes y ei resto no exhibió ningún efecto.
Entre 10s metaies ensayados, se ha encontrado que sóio

301
e] cadmio puede reconstituir una hoioenzima activa; ios resuitados obtenidos en este trabajo indican que 1a interacción de]cadmio con 1a enzima no ocurriría a través de residuos histidina, sino con otros grupos, probabiementerestos tioies (Jaffe yc01., 1984).
200
180
\\WI140 _
120
Actividad(%) 100
90
\\ \\
40 _
20
FIGURA IV.10.:
Hg Cd Mn
Efecto de diferentes metaies sobre 1a inactivación de ALA-D con DEP.La enzima (60 ug) se trató con DEP 0,2 mMa 0 FCy a pH 6,8, en ausencia y presencia de ios meta1es indicados y se midió actividad a tiempo cero(EJ) y 1uego de 15 minutos (EZ). Para cada tiempo se tomó como 100 % 1a actividad de 1a enzimaen ausencia de] meta]. Las concentraciones díios metaies fueron: Cobaito y manganeso: 10' M;zinc: 5 x 10’ M; ca mio, mercurio, estaño,piomo y se1enio: 10’ M.

302
IV.2.6. Conciusiones
- Se ha demostrado que residuos histidina están impiicados ene] mecanismo catalitico de] ALA-Dde higado de cerdo.La enzima se inactiva rápidamente con DEP, y 1a pérdida de agtividad se debe a 1a modificaciónde un residuo histidina porsubunidad situado en,o cerca de] sitio activo.
—Otros residuos nucleofiiicos tales comotirosina, serina, 1isina y grupos su1fhidri10s no están involucrados en 1a inactivación de 1a enzima por DEP.
- La reversión de 1a inactivación de 1a enzima modificada poracción de la hidroxiïamina, y 1a dependencia de 1a inactivación con e] pH apoyan e] hecho de que una histidina es critica para 1a actividad enzimática.
- E1 sustrato ALA,y'el zinc protegen a 1a enzima de 1a inactivación por DEP, indicando que e] residuo histidina y ios sitios de unión de] zinc y de] ALAestán en una región cercanaunos de otros.
- Los resuïtados obtenidos son consistentes con e] mecanismopropuesto por nuestro 1aboratorio (Batiïe y c01., 1978) queasigna un ro] activo a Ios residuos histidina en 1a transferencia de protones entre e] medio acuoso circundante y e] centro activo de carácter hidrofóbico.

CAPITULO V
MODELO PROPUESTO PARA EL MECANISMO DE ACCION DEL ALA-D

303
V.MODELO PROPUESTO PARA EL MECANISMO DE ACCION DEL ALA-D
En este trabajo, empieando preparaciones de ALA-Ddehigado porcino y de vaca, purificadas hasta banda única por eiectroforesis se han determinado y confirmado una serie de propiedades de esta enzima que,sumadas a 1as evidencias experimentaies existentes hasta e] momento, nos permiten proponer un mo
'de10 para expïicar e] mecanismo de reacción de esta enzima sobre ]a base de] esquema postulado por Batlie y Ste11a en 1978.En realidad e] avance introducido ahora está reiacionado con una mayor definición acerca de 1a estructura de] dimero funciona], en cuanto a 1a participación y función de ios restos su]fhidriios y de] zinc, e1 resto de Ios fundamentos mecanisticosson los mismos que ios propuestos en 1978.
E1 ALA-D de mamíferos es un oiigómero de PM 280 kDa,constituida por 8 subunidades de 35 kDa cada una. Se ha demostrado (Batiie y c01., 1978), confirmado ahoray se acepta que1a unidad minima cataïitica es e] dimero. Hemosobservado quecuando 1a disociación de] octámero iiega hasta e] monómero, 1areconstitución de] oiigómero activo no ocurre.
Es indiscutibie que e] ALA-Des una enzima su1fhidri11ca, que se inactiva por oxidación de SUSrestos sulfhidriïos yrecobra su actividad por agregado de un reductor tióiico. Haydatos variabies acerca de] número tota] de grupos suifhidriiospresentes en cada subunidad, pero de acuerdo a determinacionespor anáiisis de aminoácidos y tituiación con DTNBpodriamos yaacordar que existen 8 restos suïfhidriïos por subunidad. De eilos, entre 1 y 3 tioies, son aitamente reactivos; sin embargohay suficientes evidencias como para pensar que soiamente unresto Suïfhidriio está directamente invoiucrado en 1a catáïisisenzimática (Shemin, 1976).Seehra y co].,(]981) han identificadodos grupos muy reactivos, que exhibian reactividad de medio sitio, que estarian situados muy cerca uno de] otro y hay pruebasde 1a existencia de grupos tioïes vecinos en e] ALA-Dde higadobovino (Bat11e y c01., 1967, 1970). Sin embargo, Seehra yJordan (1981),por reacción con distintos reactivos aiquiiantes,estabiecieron que sólo uno de esos Suifhidriios estaria intimamente asociado con e] sitio catalitico. También, Barnard y co].,

304
(1977) habian obtenido datos parecidos, proponiendo que ese resto tiol de cada subunidad participaria en las reacciones de catálisis ácido/base que tienen lugar durante la sintesis del PBG.Sin embargo, aunque no hay dudas de que ese grupo nucleófilo está involucrado en el sitio activo y es esencial, su función catalitica es dudosa. Los grupos Sufhidrilos generalmente participan en la catálisis enzimática a través de la forma de tio-ésteres o tio-hemiacetales y no parecen estar implicados en ningüncaso de catálisis ácido/base (Friedman, 1953).
En consecuencia, el ALA-Dc0ntiene restos Sulfhidrilosde los cuales depende su actividad catalitica. Pero aün no está totalmente aclarado si están o no en el sitio activo, y encaso afirmativo, cuántos de ellos desempeñan un rol primario enla reacción catalitica mismay/o, si la modificación de ciertostioles produce cambios secundarios que llevan a una disminuciónde la actividad.
Nuestros datos acerca de los restos sulfhidrilos en laenzima de higado porcino, están de acuerdo con los de Tsukamotoy col. (l979) para el ALA-Dde higado bovino; pero nuestra propuesta es que solamente uno de esos grupos sulfhidrilos tipo Iparticipa en la catálisis, aunque no por el mecanismopostuladopor Barnard y col. (1977).
El ALA-Des una zinc-enzima, sin embargo es todavia mgtivo de discusión el rol que cumple este metal. De los estudiosrealizados con la holoenzima y la apoenzima de higado bovino(Tsukamotoy col., 1979) y los nuestros, el zinc parecería actuar protegiendo de la oxidación a ciertos restos sulfhidrilosde la enzima, de manera tal que la holoenzima conteniendo un átgmogramo de zinc por subunidad de 35 kDa, exhibiria la máxima agtividad; la pérdida de zinc y oxidación de esos tioles produciría formas menos activas; la apoenzima oxidada es totalmente inactiva, pero puede recuperar su actividad original por tratamiento con un agente reductor como el DTTy zinc. Existe entonces una indiscutible relación entre ciertos restos Sulfhidrilos,que nosotros hemos identificado como una tipo I y tipo II, elzinc, y la actividad de la enzima.
Recordemos también que se ha demostrado que el aminoácido involucrado en la formación de una base deSchiff con unamolécula de ALA, es una lisina (Shemin, l976; Gibbs y Jordan,l986). Pero además de esta lisina del sitio activo, se han

305
identificado otros residuos c0n carga positiva que de algunaforma ejercen su influencia sobre la actividad enzimática (Nandiy col., 1968).
Tsukamoto y col. (l975) habian hallado evidencias deque uno o dos residuos histidina podrian estar involucrados enel sitio activo del ALA-Dde higado bovino, proponiendo luego(Tsukamoto y col., 1978) que esos grupos histidina podrian funcionar comoposibles ligandos del zinc. Es interesante tenerpresente que en la mayoria de las zinc-metaloenzimas se ha encontrado restos histidina (Eklund=y Branden, l983; Lingskog,l983).
Hemos demostrado en este trabajo que en el ALA-Dde higado porcino, existe un residuo histidina que es critico parasu actividad catalitica, y queademás se encuentraubicado en lacercania de los sitios de unión del sustrato y del zinc.
Teniendo en cuenta que en la composición de aminoácidosdel ALA-Dde higado bovino encontramos 9 tirosinas, 4 triptofanos, 6 metioninas por subunidad que no están involucradas enel centro activo, es posible, sin embargo, que estructuralmenteestén situados en la proximidad del mismo, conformando asi uncentro de reacción con un entorno hidrofóbico. Recordando ademas que Hoffe y col. (1967) habian demostrado que los residuoshistidina desempeñaban un rol fundamental en el transporte deprotones desde y hacia el sitio activo de la aldolasa , en elcual también una lisina forma una base de Schiff con su sustrato, ya en l978, sin contar aún con todos los datos obtenidosluego, propusimos (Batlle y Stella, l978) un modelo de reacciónque precisamente postulaba que la transferencia de protones de unmedio acuoso al sitio activo hidrofóbico estaba mediada por residuos histidina ubicados en esa región lipofilica del ALA-D.
Sobre tales bases y estudios de disociación, re-asociación y re-hibridización del ALA-D(Batlle y col., l978) se postuló, por primera vez, la existencia de una unidad funcional minima, constituida por dos subunidades (Eigura_ï¿l¿l, de igualcomposición, pero en la cual cada unidad jugaba un rol diferenteen la sintesis del PBG. Se propuso que la subunidad l estaba involucrada en la formación de una base de Schiff con la primermolécula de sustrato (ALAI), mientras que la subunidad 2 estaba involucrada en la unión no covalente con la segunda moléculade sustrato (ALAII).

306
AxÁ
I QHZ \\\'ÑCH2 cHZl xA 9“ 9:0
NH O:C CH HN2 ¡ ,
CH NH3/NH3 u‘ i
o'AUUn L\/Sb ¿14“
FIGURA V.1.: Modeio de]dimero funcionai minimo de]ALA-D.
Se postuiaba 1a existencia de grupos positivos y negativos en 1a proteina que orientaban 1a ubicación de ias dos molécuias de ALAy se discutia acerca de ios posibies tipos de interacciones existentes entre ios sitios de contacto (A y B) de iasdos subunidades, se pensaba en 1a presencia de uniones iónicas yde hidrógeno entre restos tirosina, triptofano y tioies y hastase iiegó a proponer 1a unión de] zinc entre dos restos suifhidriios aportados por cada subunidad (figura V.2.) pero fundamentaimente se introdujo 1a idea de que un residuo de histidina encada subunidad estaba asociado con 1a actividad cataiitica, en1a trasferencia de protones desde y hacia e] centro activo de 1aenzima.
En e] modeio que ahora proponemos para 1a estructura deese dimero funciona], reafirmamos e] papei asignado a] residuohistidina, y con los datos experimentaies obtenidos, iiegamos a1a esquematización que se muestra en 1a figura_ï¿g¿
Tenemos entonces en cuenta 1a presencia de ios restossuifhidriios tipo I y II impiicados en 1a unión dei zinc

307
(Figuras V.1., V.2. y V.3) 1a de un resto suifhidriio tipo I ubicado en e] sitio activo que actuando comoanión tio1ato, orienta 1a ubicación de ambas moiécuias de ALAen e] centro de reacción de] dimero, a través de 1a formación de] par estabie
tioi-base -S'-—-H-H2N+.Este grupo suifhidriio de] sitio activoseria vecino o estaria en una zona muy cercana a1 resto suifhidrilo (tipo I ó II) que participa en 1a unión con e] zinc o ta]vez a otro resto suifhidriio (tipo III), con e] cua] podria formar un puente disuifuro heterogéneo SI-SIIIintrasubunidad, poroxidación; pues recordemos que en 1a enzima oxidada, totaimenteinactiva, 5610 se tituiaban dos suifhidriios rápido (tipo I, antes tipo II) y dos suifhidriios tipo III, es decir con respectoa 1a reducida se habian perdido dos grupos suifhidriios.
\l I \
:N N:H\
\ Dun\\\ \3333/;sz a 1'71?“1'
FIGURAV.2.: Estructuraposible de]dimero funciona].
E1 residuo histidina también estaria ubicado en e] centro de reacción en una región cercana a los sitios de unión de]sustrato y de] zinc.
Las dos moléculas de ALAterminarian de orientarse correctamente a través de eniaces iónicos entre sus extremos carboxiïos y grupos básicos de 1a enzima. Dado que en ei ALA-Dhay

308
13 residuos iisina por subunidad, estos grupos orientadores cargados positivamente podrian corresponder a grupos iisina, cuyaspropiedades básicas se verian aún más incrementadas, si en suvecindad hubiera también restos tioies, por migración de ios protones de estos últimos hacia 1a iisina. En esta cupia podria intervenir otro de ios tres suifhidriios tipo III no funcionales.
FIGURA V.3.:
na] de] ALA-D.
_ Respecto a ios dos restos suifhidriios tipo II expuestos sobre 1a proteina, pensamos que podrian estar invoiucradosen ia interacción entre dimeros, para formar y/o estabiiizarios tetrámeros y finaimente e] octámero de maiima actividad;también es posibie que estas asociaciones se produzcan a travésde otras uniones como ser de tipo hidrobófico o puentes hidróggno entre residuos tirosina triptofano y otros.
A partir de esta estructura más definida y comprometida que 1a anterior, 1a secuencia y e] mecanismo de ias reacciones que iievarian a 1a sintesis dei PBGseria 1a misma que 1apostuiada hace 10 años (Figura_!¿í.).
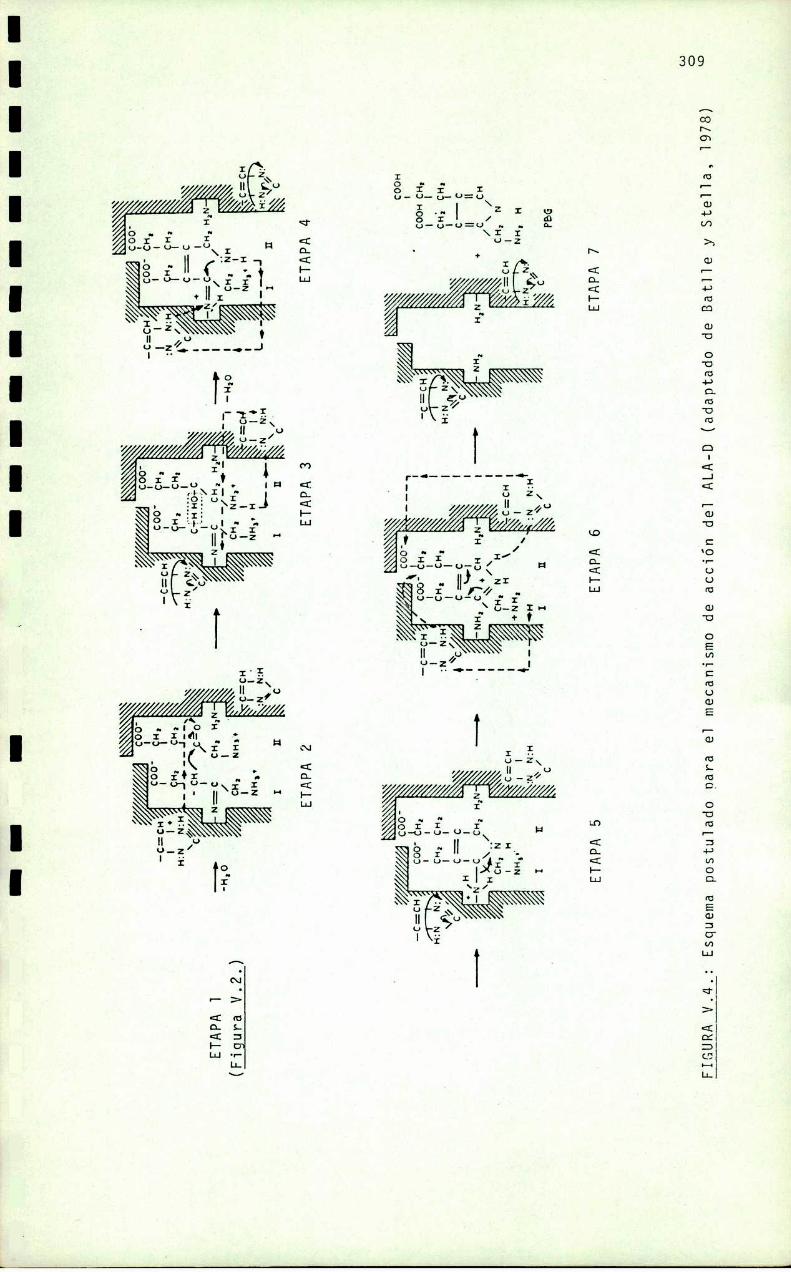
ETAPAT
(FiguraV.2.)
ETAPA3
COOH
l
COOHfu,t
ETAPA5ETAPA6
ETAPA7
FIGURAV.4.:Esquemapostuïadoparae1mecanismo
309
deaccióhde]ALA-D(adaptadodeBatlïeySteïïa,1978)

310
En la primer etapa, se forma una base de Schiff entreel grupo carbonilo del ALAI y el residuo lisina de la Subunidad I, que se ve muy facilitada por la vecindad del resto histidina que transporta un protón desde el carbono 8 del sustratoal centro hidrofóbico, estabilizándose asi tanto el carbaniónintermediario resultante comola base de Schiff.
En la segunda etapa, el carbanión estabilizado efectúaun ataque nucleofilico sobre el grupo y-carbonilo de la molécula II del ALA;en tanto que el residuo histidina de la subunidad l transfiere un protón al oxigeno de ese mismo grupo.
En la etapa siguiente tiene lugar una condensación aldólica, al tiempo que en la histidina de la subunidad l se produce un reordenamiento electrónico, recuperando su estructura inicial y se activa la histidina de la subunidad 2 transportandoun protón del grupo 6-amino del ALAII al N-c amino del grupode la lisina que está formando la base de Schiff con el ALAI.
En la etapa cuarta interviene nuevamente el residuo histidina de la subunidad l en el transporte de otro protón de lamisma molécula del ALAII al nitrógeno de la lisina de su sitioactivo, simultáneamente la histidina de la subunidad recobra suestructura original por reordenamiento de su electrón.
En la etapa quinta, mientras la histidina de la subunidad l también se reordena electrónicamente, ocurre una reacciónde transaminación entre la enzima y el pirrol.
Finalmente, en la etapa sexta el producto formado también experimenta un reordenamiento en el cual participan losresiduos histidina de cada subunidad, dando lugar a la formaciónde una molécula conjugada y no polarizada de PBG, que entoncesse encuentra en condiciones de liberarse fácilmente del sitio agtivo, regenerándose la enzima libre, lista para comenzar un nuevo ciclo de reacciones (etapa VII).

CAPITULO VI
REFERENCIAS

VL
311
BEFERENCIA;
Abduiia, M. & Haeger-Aronsen, B. (1973) Enzyme lg, 708.
Abduiwajid, A.M. & Hu, F.Y.H. (1986) Biochemistry gg, 8167.
Anderson, P.M. & Desnick, R.J. (1979) J. Bio]. Chem. 254,6924.
Barnard, G.F.; Itoh, R.; Hohberger, L.H. & Shemin, D. (1977)J. Bio]. Chem. 252, 8965.
Batile, A.M. de] C. & Steiia, A.M. (1978) Int. J. Biochem. g,861.
Bat11e, A.M. de] C;; Ferramoïa, A.M. & Grinstein, M. (1967)Biochem. J. 104, 244.
Batiie, A.M. de] C.; Ferramoïa, A.M. & Grinstein, N. (1970)en Methods in Enzymoiogy (Ed. Tabor y Tabor), N.Y. and London,Academic Press, vo]. ZZ, p. 216.
Batïie, A.M. de] C.; Ste11a, A.M.; Ferramola, A.M.; Sopena,Y.; Wider de Xifra, E.A. & Sancovich, H.A. (1978) Int. J.Biochem. g, 401.
Bustos, N.L.; Stella, A.M.; Hider de Xifra, E.A. & Bat11e,A.M. (1980) Int. J. Biochem. lg, 745.
Carisson, J.; Oisson, I. & Axén, R. (1976) Acta Chem. Scand.B. gg, 180.
Coieman, D.L. (1966) J. Bio]. Chem. 2 1, 551].
Coïeman, J.E. & Va]1ee,B. (1961) J. Bio]. Chem. 236, 2244.
Cousineau, J. & Meighen, E. (1976) Biochemistry lá, 4992.
Cheh, A. & Neiiands, J.B. (1973) Biochem. Biophys. Res.Commun. gg, 1060.
Davis, J.R. & Avram, M.J. (1978) Toxico]. App]. Pharmaco].gg, 181.
Ekïund, H. & Branden, C. (1983) en Zinc Enzymes (Ed. T.G.Spiro), John Wiïey and Sons, N.Y., p. 123.
Eiiman, G.L. (1959) Arch, Biochem. Biophys. gg, 70.
Friedman, M. (1973) en The Chemistry and Biochemistry of theSuiphydryï group in Aminoacid Peptides and Proteins,
Oxford.Pergamon
Press,

3l2
Gibbs, P.N.B. & Jordan, P.M. (1986) Biochem. J. 229, 447.
Granick, S. & Nauzeraii, D.J. (1958) J. Bio]. Chem. 2 2, 1119.
Gurba, P.E.; Sennet, R.E. & Kobes, R.D. (1972) Arch. Biochem.Biophys. 150, 130.
Gurne, U.; Chen, U1 a Shemin, D. (1977) Proc. Nat]. Acad. Sci.
USA 15, 1383.
Hoffe, P.; Lai, C.; Pugh, E.L. & Horecker, B.L. (1967) PNASBiochemistry Él, 107,
Hoibrook, J.J. & Ingram, V.A. (i973) Biochem. J. 1 1, 729.
Imamura, T.; Konishi, K.; Yokoyama, M. & Konishi, K. (1979)J. Biochem. (Tokyo) gg, 639.
Jaffe, E.K.; Saiowe, S.D.; Chen, N.T. & de Haven, P.A.(1984) J. Bio]. Chem. 259, 5032.
Kato, Y.; Komiya, K,; Sasaki, H. & Hashimoto, T. (1980) J.Chromatog. 193, 29.
Keech, D.B. & Farrant, R.K. (1968) Biochim. Biophys. Acta 151,493.
Kench, J.E. & Gubb, P.J.D. (1970) Biochem. J. 120, 27.
& Zeitier, H.1081.
Kreutzer, M.; Schmidt, M.;Hoppe-Zeyier's Z.
Stadier, E.Chem. l,
(1977)Physioi.
Krieger, D.E.; Erickson, B.w.Nat]. Acad. Sci. USAZ}, 3160.
& Merrifieid, R.B. (1976) Proc.
Liedgens, N.; Lutz, C. & Schneider, H.A.w. (1983) Eur. J.Biochem. 135, 75.
Lindskog, S. (1983) en Zinc Enzymes (Ed. T.G. Spiro) JohnHiiey & Sons, New York. p. 77.
Maimstrom, B.G. & Rosenberg, A. (1959) Advan. Enzymoi. gl,131.
Mann, K,G. & Fish, w. (1972) en Methods in EnZymoiogy (Eds.C.H.w. Hirs y S.N. Timasheff), Academic Press, NewYork, vo].26, 28.
Maraiihaiii, G.B.; Rao, S.R.Chemistry gg, 2533.
& Bhagwat, A.S. (1985) Phyto
Heichior, w.B. & Fahrney, D. (1970) Biochemistry g, 251.
Mitcheii, R.A.; Drake, J.E.;(1977) Ciin. Chem. gg, 105.
Whitiin, L.A. & Rejent, T. A.

3l3
Nandi, D. (1971) Arch. Biochem. Biophys. 13g, 157.
Nandi, D. & Shemin, D. (l968) J. Biol. Chem. 243, l236.
Nandi, D.; Baker-Cohen, K.F. & Shemin, D. (1968) J. Biol.Chem. 24 , l224.
Olson, M.0.J. & Liener, I.E. (1967) Biochemistry g, 3801.
Reynolds, J. & Tandford, C. (l970) Proc. Natl. Acad. Sci.USA gg, 1002.
Roche, T.E. & Mc Faldden, B.A. (1969) Biochem. Biophys. Res.Commun. ¿1, 239.
Rosenbusch, J. & Weber, K. (l97l) J. Biol. Chem. gig, l644.
Seehra, J.S. & Jordan, P.M. (1981) Eur. J. Biochem. ll}, 435.
Seehra, J.S.; Gofe, M.G.; Chaudry, A.G. & Jordan, P.M. (198])Eur. J. Biochem. ll4, 263.
Shemin, D. (l976) Phil. Trans. R. Soc. Lond. B. m_;, l09.
Stafforini, D.M.; Polo, C.F.; Stélla, A.M.; Wider de Xifra,E.A. & Batlle, A.M. del C. (l980) Int. J. Biochem 1g, 757.
Stella, A.M.; Wider de Xifra, E.A. & Batlle, A.M. del C.(1977) Mol. Cell.Biochem. lg, 97.
Tanford, C.; Nozaki, Y.; Reynolds, J. & Makino, S. (1974)Biochemistry lg, 2369.
Tsukamoto, I.; Yoshinaga, T. & Sano, S. (1975) Biochem.Biophys. Res. Commun.gl, 294.
Tsukamoto, 1.; Yoshinaga, T.Acta 570, 167.
& Sano, S. (l979) Biochim.Biophys.
Nada, 0.; Ono, 1.; Maru, R. & Toyokawa, K. (1972) Ind. Healthlg, ll.weber, K. & Kuter, o. (1971) J. Biol. Chem. gig, 4504.
Heber, K. & Osborn, M. (l969) J. Biol. Chem. 244, 4406.
Nider de Xifra, E.A.; Stella, A.M. & Batlle, A.M. del C.(1978) Plant Sci. Lett. ll, 93.
Wilson, E.L.; Burger, P.E. & Dowdle, E.B. (1972) Eur. J.Biochem. gg, 563.
Hu, w.H.; Shemin, D.; Richards, K.E. & Williams, R.C. (l974)Proc. Natl. Acad. Sci. USAll, 1767.

CAPITULO VII
CONCLUSIONES
Página
VII.1. Purificación y caracterización de 1a enzima 314
VII.2. Estudios de disociación y reasociación 315
VII.3. Función de1 zinc 316
VII.4. Sustitución por coba1to 316
VII.5. Grupos su1fhidri1os 317
VII.6. Grupos histidina 317
VII.7. Mecanismo 318

314
VII.CONCLUSIONEE
VII.1.PURIFICACION Y CARACTERIZACION DE LA ENZIMA
En este trabajo se empiearon como fuentes de ALA-D,higados bovino y porcino, ya que si bien ambas provienen de especies fiiogenéticamente cercanas, hay aigunas diferencias interesantes en reiación con Ios parámetros que nos interesaban estudiar.
Se obtuvieron preparaciones aitamente purificadas de ambas enzimas luego de una homogeneización, caientamiento, fraccionamiento salino, diáiisis, intercambio por DEAE-ceiuiosa, ycromatografiaen tamices molecuiares. Mediante estos procedimientos, se logró purificar 1a enzima entre 400 y 600 veces, siendonotable 1a estabiiidad de estos preparados.
E] contenido de zinc por gramo de proteina no guardóuna reiación estequiométrica a 10 1argo de] proceso de purificación, de manera ta] que en ausencia de dicho meta], se obtieneuna fracción purificada con sóio 0,2 átomogramos de zinc por subunidad de 35 kDa. En cambio, a1 inciuir e] zinc en 1a composición de Ias diferentes soiuciones empieadas a 10 iargo de 1apurificación,e1 contenido de zinc en 1a fracción purificada fuede] orden de un átomogramo de zinc/subunidad. Se ha observadosin embargo que con e] tiempo, e] meta] se iibera espontáneamente indicando que 1a unión de] zinc a 1a enzima es reiativamenteiábi]. Además, ias condiciones experimentaies influyen significativamente en 1a proporción de zinc retenida por 1a proteina,asi como también sobre e] estado de oxidación de aïgunos grupossuifhidriios, ya que e] zinc estaria impiicado en mantener red!cidos dichos residuos.
Las propiedades de ambas dehidrasas son simiiares encuanto a su actividad especifica, estabiiidad, pH óptimo y Km.
Las corridas cromatográficas y eiectroforéticas de iasenzimas nativa y disociada, muestran que a1 igual que en otrasfuentes, ambas son oiigoméricas siendo 1a especie moiecuiar predominante e] octámero, y a 1a menor subunidad corresponde unpeso molecuiar de 35 kDa.

315
VII.2.ESTUDIOS DE DISOCIACION Y REASOCIACION
Las experiencias reaiizadas con agentes disociantes taies c0mo urea, guanidina y SDS con 1a enzima soiubie de higadoporcino, indican que una vez.disociada en monómeros, existen dificuitades para iograr una correcta reasociación de ios mismos.
En cambio, mediante 1a técnica de insolubiiización deenzimas, y e] empieo de 1a urea como agente disociante, se confirmó que e] ALA-Des una proteina oiigomérica, cuyas subunidadespueden disociarse, y que dichas subunidades poseen actividad cataiitica. Además, ias subunidades inmoviiizadas son capaces dereasociarse con ias subunidades soiubies generadas ¿n ¿ttu, para reconstituir ia proteina nativa. Empieandosubunidades so]!bies provenientes de] ALA-Dde eritrocitos humanos, se logró obtener un oiigómero activo por reasociación de taies subunidadescon ias subunidades insoiubiizadas de] ALA-Dde higado, 10 cua]demostró 1a posibie formación de híbridos activos entre las enzimas de ambas fuentes. En consecuencia, si bien pueden existirdiferencias entre las secuencias de aminoácidos de ambas dehidrasas, éstas no deben ser significativas, por 10 menos en 10que respecta a] sitio de contacto entre las subunidades.
Los datos obtenidos están de acuerdo con nuestra ideade que podrian existir distintos tipos de uniones entre las subunidades, y que ei dimero seria 1a unidad funciona] minima cataiiticamente actiVa.
En ei caso de] ALA-Dbovina, mediante e] empieo de unacromatografía iiquida a aita presión se haiió que, en condiciones no disociantes, e] peso moiecular era de 280 kDa,de acuerdocon ios vaiores reportados hasta e] momento; no se detectaron asociaciones y/o disociaciones por variación en 1a concentraciónde 1a proteina. E1 tratamiento con SDSprodujo una disminuciónde las formas octaméricas, y, correiativamente, de 1a actividadenzimática. Dichas transformaciones fueron analizadas a travésde los cromatogramas obtenidos, encontrándose una perfecta correlación entre estos üitimos, y ios parámetros anteriormentemencionados.
Para ia dehidrasa_porcina, bajo determinadas condiciones experimentaies, se obtuvieron evidencias de 1a existenciade especies moiecuiares mayores y menores a 1a de] octámero nativo origina], de manera simiiar a io reportado ya para 1a

316
enzima de Rp. apheno¿de4.
VII.3.EgflCION DEL ¿Lflg
A pesar de que se han asignado a] zinc diferentes roiesen e] mecanismo de acción de] ALA-D, ias caracteristicas de 1aunión metal-enzima aün son poco conocidas.
Se reaiizó entonces un estudio de 1a unión metai-enzimamediante una diáiisis de equiiibrio, encontrándose que se unen 4 átomos de zinc por mo] de enzima, con una constante de unión de 1,5 uM, y otros 4 adicionales con una constante de 10 UMen sitios de unión independientes. Se ha estabiecido además que1a unión de] zinc está estrechamente iigada con 1a expresión de1a actividad enzimática.
Se vió que ia apoenzima no presenta actividad en condiciones aeróbicas, ni aún en presencia de activadores tióiicos,pero recupera su actividad origina] si además se adicionan a1medio iones zinc. La reactivación tiene iugar en un rango deconcentraciones de zinc muyestrecho, respondiendo a una cinética sigmoidea, io que indica 1a existencia de una reacción de tipo cooperativo entre 1a apoenzimay'ei meta]. Los re5u1tados sugieren que e] zinc interacciona con ios residuos Suifhidriiosde 1a proteina en dos formas diferentes, según se trate de laactivación de 1a enzima en su forma nativa, o de una renaturaiización de 1a apoenzima.
A concentraciones de zinc superiores a 10'4 M, tieneiugar una inhibición pH dependiente, cuyo pKa está indicando unainteracción de] zinc con residuos histidina.
VIÏ.4.SUSTITUCION POR COBALTO
Los intentos por reemplazar e] zinc por cobaito. nos indicaron que, si bien ei cobalto no es capaz de despiazar a1 zincde su sitio en 1a hoioenzima, puede en cambio ubicarse en su lugar en ia apoenzima, reempiazándoio parciaimente, y ocupar además un segundo sitio para meta]. En base a los datos obtenidosse ha propuesto un modelo que expiicaria 1a acción de] cobaitoen reiación a] zinc y a 1a actividad de ias metaioenzimas resu]tantes.

VII.5.GRUPOS SULFHIDRILOS
La obtención de 1a enzima en un a1to grado de pureza,nos permitió además efectuar un estudio sobre e] ro] de ios grupos suïfhidriios en reiación a 1a actividad catalitica y a 1aintegridad funciona] de 1a enzima.
Empleando DTNB,en presencia y ausencia de agentes disociantes, se determinó entonces e] número y veiocidad de reacción de Ios grupos su1fhidriïos y 1a importancia de su mantenimiento a] estado reducido para 1a actividad catalitica de 1a enzima, asi como su posibie reiación con e] zinc.
Teniendo en cuentaias hipótesis previas de] grupo deSano y 105 resuitados de este trabajo, se ha postuiado un esquema que tiene en cuenta 1a presencia de grupos suïfhidriïos dediferente reactividad y su re1ación con 1a estructura y actividad de] dimero funciona].
VII.6.GRUPOS HISTIDINA
Se compietó e] estudio de ios grupos especificos invglucrados con 1a catáiisis enzimática de] ALA-D,en reiacióncon 1a participación de iones metáiicos, en particuiar e] zinc,utiiizando e] reactivo grupo especifico dietiïpirocarbonato,bajo diversas condicioens experimenta1es, para expiorar 1a posibi1idad de que residuos histidina estén iocaïizados en e] sitioactivo, y que estos grupos estén impiicados en 1a unión de]zinc a 1a proteina. Se llevó a cabo entonces 1a modificación quimica de 1aenzima purificada a partir de higado porcino, conteniendo 0,3 átomogramos de zinc por subunidad.
Se observó que 1a inhibición por DEPes proporciona] asu concentración y a] tiempo de reacción; con respecto a esteüitimo parámetro, se distinguen ciaramente dos etapas: una inicia] de rápido decaimiento de 1a actividad y una segunda más legta. En zinc ejerció un efecto protector sobre 1a enzima, frentea 1a inactivación por e] DEP, este efecto fue sólo importante en1a fase rápida de 1a reacción.
Se determinó e] número de grupos histidina reactivossiguiendo 1a variación de absorbancia a 240 nm. Se tituïaron asi 3 histidinas por subunidad enzimática, cada una de las cuaïes

318
reacciona a diferente velocidad (lO, 20 y 30 minutos). La modificación del primer grupo ocurrió en un tiempo coincidente conla pérdida de actividad enzimática (fase inicial rápida). Por otra parte relacionando el porcentaje de actividad residual conel número de histidinas modificadas, se confirmó que un residuode aminoácido por subunidad está directamente implicado en laactividad catalftica.
Se realizaron además experiencias a distintos pH's, yparacada uno de ellos se investigó el efecto protector del zinc.Se obtuvieron resultados que avalan la hipótesis de otros invegtigadores, según los cuales,sólo las formas no protonadas del imidazol e histidina son reactivas y se modifican a una mayor vglocidad. A su vez, con respecto al zinc, se encontró que estemetal ejerce su acción protectora sobre la histidina, en su forma protonada. I
La reversibilidad de la inhibición por DEPpor acciónde la hidroxilamina nos permitió descartar la posibilidad de unamodificación de residuos lisina.
Se encontró también protección por el sustrato ALA,indicandoque el DEPestaba modificando un residuo ubicado en elsitio activo.
Los resultados obtenidos sugieren entonces que existeun residuo histidina por subunidad, esencial para la actividadenzimática, implicado además en la unión del zinc, e indicanque el sitio de unión del sustrato, el del zinc y de los residuos histidina esenciales se encuentran en estrecha proximidaden el sitio activo. Asimismo,de acuerdo a los resultados de losestudios a diferentes pH's, éstos están de acuerdo con el mecanismo de protonación-deprotonación propuesto por nuestro laboratorio.en 1978.
VII.7.MECANISMO
Finalmente, se ha adoptado y propuesto un mecanismo pgra la reacción de sintesis enzimática‘del PBG, fundamentalmentebasado en esquemas anteriores de nuestro laboratorio, pero adaptado y actualizado en base a los resultados obtenidos en estetrabajo y las evidencias experimentales acumuladas hasta el momento.
fi"
,/ ¿717 ' .