5. análisis de la cinética de transferencia electrónica
TRANSCRIPT

Modificación redox de superficies para electrónicamolecular y electrocatálisis
Ricci, Alejandra Marcela2010
Tesis Doctoral
Facultad de Ciencias Exactas y NaturalesUniversidad de Buenos Aires
www.digital.bl.fcen.uba.ar
Contacto: [email protected]
Este documento forma parte de la colección de tesis doctorales de la Biblioteca Central Dr. LuisFederico Leloir. Su utilización debe ser acompañada por la cita bibliográfica con reconocimiento de lafuente.
This document is part of the doctoral theses collection of the Central Library Dr. Luis Federico Leloir.It should be used accompanied by the corresponding citation acknowledging the source.
Fuente / source: Biblioteca Digital de la Facultad de Ciencias Exactas y Naturales - Universidad de Buenos Aires

UNIVERSIDAD DE BUENOS AIRES
Facultad de Ciencias Exactas y Naturales
Departamento de Química Inorgánica, Analítica y Química Física
MODIFICACIÓN REDOX DE SUPERFICIES PARA ELECTRÓNICA MOLECULAR
Y ELECTROCATÁLISIS
Tesis presentada para optar al título de Doctor de la Universidad de Buenos Aires
en el área Química Inorgánica, Analítica y Química Física
Alejandra Marcela Ricci
Director de tesis: Dr. Ernesto J. Calvo
Consejero de Estudios: Dr. Galo Soller Illia
Lugar de Trabajo: Departamento de Química Inorgánica, Analítica y Química Física ‐
INQUIMAE
Buenos Aires, 2010


A mis padres
A Marcos
A Mariela


“El auténtico conocimiento es conocer la extensión de la propia ignorancia”
Confucio


i
AGRADECIMIENTOS
A mis padres, Alicia y Eduardo, por haberme inculcado la cultura del trabajo y el esfuerzo, las ganas de aprender y por siempre darme los consejos más valiosos en TODOS los órdenes de la vida. A mi hermana, Mariela por ser mi amiga de toda la vida. A los tres, por la familia que tenemos. A mi esposo, Marcos, por ser mi compañero incondicional en TODO, por su bondad, por su paciencia y calma, y por el proyecto de vida que compartimos desde hace ya más de seis años. A Ernesto, por haberme dado un lugar en su grupo para realizar este trabajo de tesis y por haber sido mi guía a lo largo de todo el proceso. A Mario, por ser un gran compañero de trabajo, un guía y consejero excepcional. Por haber leído GRAN parte de esta tesis y estar siempre dispuesto a darme una mano con mis dudas y contratiempos. MUCHOS de los resultados de esta tesis no se hubieran logrado sin tu ayuda y voy a estar eternamente agradecida por eso y por tu amistad. A Edgar, por ser mi AMIGO casi desde el principio, por todos los mates compartidos, por aguantar mi música y mis cantos. Te extraño pelado, volvé! A Lucila, por ayudarme con los espectros PM‐IRRAS, por leer GRAN parte de esta tesis, por las muy buenas correcciones, por la buena onda y amistad. A Ceci, por ayudarme a dar los primeros pasos en esta tesis (y todos los que siguieron), por siempre tener respuesta a mis preguntas y por ser mi “mamá” en el trabajo. A los chicos del labo en general, por aguantar mis malos humores y también los buenos (ustedes sabrán qué es peor!) y por saber apreciar mi talento musical. A Pablo, por su tranquilidad y buena onda, a Néstor, por siempre tener algo descabellado que contar y alegrarnos los días de trabajo y por siempre estar dispuesto a ayudarme con las síntesis. A los chicos que llegaron hace poco: Matías, Walter, Santi, Matías (LN), Jennifer y Verónica por la buena onda de todos los días. A Doris, por su bienvenida cuando llegué al labo. A Mariano y José, por los consejos útiles de cada día y a Julia por ser como una compañera más del grupo. A mis abuelos, Elvira y Tito, y a mí tía Graciela. A mis suegros, Cecilia y Guillermo y mis cuñadas, Pili y Sil, por compartir los domingos en familia que tanto me gustan! A mis amigas, Sol, Guada, Lía, Viqui, Vane, Juli, Flo, Babi y Ceci. Al Dr. Federico Williams, por ayudarme con los espectros de XPS y siempre estar dispuesto a contestar mis preguntas al respecto (y otros respectos). Al Dr. Luis Baraldo y su grupo, por ayudarnos con la síntesis del complejo de osmio. Al Dr. Alex Fainstein y al Dr. Nicolás Tognalli, por medir los espectros Raman resonante. A la Dra. Carolina Vericat, por analizar mis muestras con Microscopía de Efecto Túnel. Al Dr. Damián Scherlis y al Lic. Ezequiel de la Llave por hacer los cálculos teóricos relacionados con los sistemas analizados en esta tesis. Al Dr. Richard Nichols y al Dr. Santiago Martin, por haberme recibido tan bien en Liverpool y por permitirme hacer las medidas de STS in situ en su laboratorio. Al Departamento de Química Inorgánica, Analítica y Química Física y al INQUIMAE por brindar el lugar de trabajo. A Wellcome Trust, a la Agencia de Promoción Científica y Tecnológica y al Consejo de Investigaciones Científicas y Técnicas por las becas que me posibilitaron realizar esta tesis.

ii
RESUMEN
Modificación Redox de Superficies para Electrónica Molecular y Electrocatálisis
a comprensión y control del transporte de carga a través de películas orgánicas de un espesor
nanométrico es de fundamental importancia en diferentes áreas de investigación aplicada como ser
la electrónica molecular, el desarrollo de sensores y biosensores, la protección contra la corrosión, la
electrocatálisis y la fotoconversión solar, entre otras. El conocimiento de cómo la composición y la
estructura química pueden afectar la transferencia electrónica entre un sustrato sólido y una molécula
electroactiva es central en el desarrollo de las mismas.
En esta tesis el objetivo principal consistió en el estudio de fenómenos de transferencia electrónica (TE)
y para ello se prepararon una serie de sistemas modelo basados en electrodos modificados con
monocapas electroactivas. Éstos se obtuvieron por unión del complejo [Os(2,2´‐bpy)2Cl(py‐CH2‐NH2)]+ a
monocapas previamente depositadas sobre electrodos de oro, que se formaron por adsorción de tioles
alifáticos de diferentes longitudes de cadena, un tiol aromático y por electro‐reducción de sales de
diazonio. De esta manera se obtuvieron dos formas de unión a la superficie: Au‐S y Au‐C,
respectivamente. Además, en todos los casos, las moléculas depositadas presentaron grupos ácidos que
reaccionaron con el grupo amino del complejo en una reacción de post‐funcionalización.
Así se obtuvieron sistemas en los que el par Os(II)/Os(III) se encuentra unido a la superficie de un
electrodo de oro a través de puentes moleculares de diferentes longitudes y naturaleza. Todos ellos se
caracterizaron superficialmente empleando espectroscopías infrarrojas (PM‐IRRAS y FT‐IRRAS),
espectroscopía Raman resonante, espectroscopía fotoelectrónica de rayos X y microscopía de efecto
túnel y se observó que el complejo se une a la superficie sin perder su integridad química, formando
efectivamente monocapas electroactivas.
La caracterización electroquímica se llevó a cabo por voltametría cíclica y, en particular, en los estudios
de cinética de transferencia electrónica, se empleó principalmente espectroscopía de impedancia
electroquímica. Los sistemas obtenidos con tioles alifáticos de diferentes longitudes mostraron un
mecanismo de TE consistente con tuneleo a través del enlace. Los sistemas obtenidos a partir de la
reducción de sales de diazonio y con el tiol aromático presentaron velocidades de TE comparables.
Finalmente, se analizaron por espectroscopía de efecto túnel con control electroquímico dos de los
sistemas aromáticos que se diferencian únicamente en el tipo de unión al sustrato de oro (Au‐C y Au‐S)
observándose, en ambos casos, un mecanismo de TE en dos pasos, entre la molécula y los contactos,
con relajación vibracional parcial del centro redox.
Palabras clave: Electrodos modificados, monocapas, sales de diazonio, complejos de osmio,
transferencia electrónica, espectroscopía de impedancia electroquímica, modelo de Smith‐White,
espectrosocopía de efecto túnel con control electroquímico.
L

iii
ABSTRACT
Surface Redox Modification for Molecular Electronics and Electrocatalysis
he understanding and control of charge transport through organic films of nanometric thickness is
of fundamental importance in different areas of applied research such as molecular electronics, the
development of sensors and biosensors, protection against corrosion, electrocatalysis and solar
photoconversion, among others. The knowledge of how the composition and chemical structure can
affect the electron transfer between a solid substrate and an electroactive molecule is central to their
development.
In this thesis the main objective consisted in the study of electron transfer (ET) phenomena, so a series
of model systems, based on modified gold electrodes with electroactive monolayers, were prepared.
These were obtained by binding the complex [Os(2,2'‐bpy)2Cl(py‐CH2‐NH2)]+ to previously formed
monolayers by adsorption of aliphatic thiols of different chain lengths, an aromatic thiol and by electro‐
reduction of diazonium salts. As a consequence, two types of tethers to the surface were obtained: Au‐S
and Au‐C, respectively. Furthermore, in all cases, the deposited monolayers ended in acid groups in
order to react with the amino terminal group in the osmium complex in a post‐functionalization
reaction.
As a result, systems were obtained in which the Os(II)/Os(III) couple was bound to the surface of a gold
electrode through molecular bridges of different lengths and natures. All of them were characterized
using surface infrared spectroscopy (PM‐IRRAS and FT‐IRRAS), resonant Raman spectroscopy, X‐ray
photoelectron spectroscopy and scanning tunneling microscopy. It was found that the complex binds to
the surface without losing its chemical integrity and actually forming electroactive monolayers.
The electrochemical characterization was carried out by cyclic voltammetry and the studies of electron
transfer kinetics were carried on with electrochemical impedance spectroscopy. Systems obtained with
aliphatic thiols of different lengths showed an ET mechanism consistent with through‐bond tunneling.
Systems obtained from the diazonium salts reduction and with the aromatic thiol showed comparable
ET rates.
Finally, two aromatic systems that differ only in the type of bond to the gold substrate (Au‐C and Au‐S)
were analyzed by scanning tunneling spectroscopy with electrochemical control and a two‐step ET
mechanism with partial vibrational relaxation at the redox center was observed in both cases.
Keywords: Modified electrodes, monolayers, diazonium salts, osmium complex, electron transfer,
electrochemical impedance spectroscopy, Smith‐White model, tunneling spectroscopy with
electrochemical control.
T


ÍNDICE
v
CAPÍTULO 1
INTRODUCCIÓN Y OBJETIVOS
1.1 ELECTRÓNICA MOLECULAR................................................... ................................................... .............3
1.2 TRANSFERENCIA ELECTRÓNICA................................................... ................................................... .......4
1.3 ACOPLAMIENTO ELECTRÓNICO DE LARGO ALCANCE ................................................... ..........................5
1.4 ELECTRODOS MODIFICADOS................................................... ................................................... ...........8
1.4.1 Derivatización de Superficies ............................................................................................ ..........8
1.4.2 Técnicas de modificación ......................................... ................................................... ................9
1.4.3 Electrodos modificados y Análisis de Transferencia Electrónica........................................... ....11
1.4.3.1 Monocapas Electroactivas ................................................ ................................................... ......... 12
1.4.4 Junturas Moleculares............................................................................................. ...................14
1.5 ESTADO DEL CONOCIMIENTO................................................... ................................................... .......16
1.6 MOTIVACIÓN Y OBJETIVOS ................................................... ................................................... ..........17
CAPÍTULO 2
MATERIALES Y MÉTODOS
2.1 TÉCNICAS EMPLEADAS: FUNDAMENTOS ................................................... ..........................................23
2.1.1 Técnicas Electroquímicas ........................................................................................... ...............23
2.1.1.1 Reacciones de electrodo........................................... ................................................... ...................... 24 2.1.1.2 Celda electroquímica ......................................................................................... ........................ ........ 24 2.1.1.3 Potenciostato................................................. ................................................... ................................. 26 2.1.1.4 Cronoamperometría ....................................................................................... ....................... ............ 27 2.1.1.5 Voltametría cíclica ............................................ ................................................... ......................... .... 27 2.1.1.6 Impedancia Electroquímica ............................................... ................................................... ............ 31
2.1.2 Técnicas Espectroscópicas ............................................. ................................................... ........35
2.1.2.1 Espectroscopía Infrarroja por Absorción‐Reflexión ........................................... ................................ 35 2.1.2.2 Espectroscopía Infrarroja por Absorción‐Reflexión con Modulación de la Polarización ................... 37 2.1.2.3 Espectroscopía de dispersión Raman resonante ............................................ ................................... 40 2.1.2.4 Espectroscopía fotoelectrónica de rayos X ................................................... ..................................... 42
2.1.3 Técnicas de Microscopía ........................................... ................................................... .............44
2.1.3.1 Microscopía de Efecto Túnel .............................................. ................................................... ............ 44 2.1.3.2 Espectroscopía de Efecto Túnel.............................................. ................................................... ........ 47 2.1.3.3 Espectroscopía de Efecto Túnel con control electroquímico............................................................. 47
2.2 EQUIPAMIENTO UTILIZADO ................................................... ................................................... ..........48
2.2.1 Potenciostato............................................................................................. ..................... ..........48
2.2.2 Electrodos y Celdas ............................................. ................................................... ...................48

ÍNDICE
vi
2.2.3 Espectrómetro Infrarrojo con Transformada de Fourier............................................ ...............49
2.2.4 Espectrómetro de Fotoemisión de Rayos X.................................................. .............................50
2.2.5 Espectrómetro de Dispersión Raman................................................ ........................................50
2.2.6 Microscopio de Efecto Túnel .............................................. ................................................... ....51
2.2.6.1 Microscopía de efecto túnel ............................................... ................................................... ............ 51 2.2.6.2 Espectroscopía de efecto túnel ............................................... ................................................... ....... 51
2.3 PROCEDIMIENTOS EXPERIMENTALES................................................... ...............................................52
2.3.1 Reactivos Químicos y Soluciones.......................................... ................................................... ..52
2.3.2 Limpieza de los sustratos de oro................................................ ...............................................53
2.3.3 Síntesis de Os(2,2’‐bpy)2Cl2 ................................................... ................................................... .53
2.3.4 Síntesis de [Os(2,2´‐bpy)2Cl(py‐CH2‐NH2)]PF6 ................................................... .........................53
2.3.5 Síntesis de tetrafluoroborato de 4‐nitrobencendiazonio ..........................................................54
2.3.6 Síntesis de tetrafluoroborato de 4‐carboxibencendiazonio ......................................................55
2.3.7 Síntesis de tetrafluoroborato de 4‐(2,3,5,6‐tetrafluoro)‐carboxibencendiazonio.....................55
2.3.8 Derivatización de sustratos de oro................................................ ............................................55
2.3.8.1 Electro reducción de sales de diazonio............................................. ................................................. 56 2.3.8.2 Autoensamblado de tioles.............................................. ................................................... ................ 56 2.3.8.3 Post‐funcionalización con [Os(2,2´‐bpy)2Cl(py‐CH2‐NH2)]PF6 ................................................... ........ 56
CAPÍTULO 3
DERIVATIZACIÓN Y CARACTERIZACIÓN SUPERFICIAL
3.1 ESTRATEGIAS DE MODIFICACIÓN DE SUPERFICIES................................................... ............................59
3.1.1 Monocapas Autoensambladas: Compuestos Organosulfurados ..............................................60
3.1.1.1 Enlace Au‐S .................................................. ................................................... ......................... .......... 62 3.1.1.2 Preparación y Cinética de Formación ........................................... ................................................... .. 63 3.1.1.3 Organización superficial......................................... ................................................... ........................ . 63 3.1.1.4 Estabilidad ........................................... ................................................... ........................................... 65
3.1.2 Reducción Electroquímica de Sales de Diazonio ............................................ ...........................65
3.1.2.1 Adsorción reductiva de sales de aril diazonio............................................. ....................................... 66 3.1.2.2 Mecanismo de adsorción........................................... ................................................... ..................... 67 3.1.2.3 Enlace entre la superficie y la capa orgánica ............................................ ......................................... 69 3.1.2.4 Organización superficial: monocapas y multicapas ............................................ ............................... 70
3.1.2.4.1 Mecanismo de formación de capas gruesas .............................................................................. 70
3.2 DERIVATIZACIÓN INICIAL DE SUSTRATOS DE ORO ................................................... .......................71
3.2.1 Derivatización de Superficies de Oro por Autoensamblado de Tioles................................71
3.2.1.1 Determinación del Cubrimiento Superficial......................................... ............................................. 72 3.2.1.2 Caracterización Superficial por FT‐IRRAS................................................. .......................................... 75 3.2.1.3 Caracterización Superficial por PM‐IRRAS ................................................. ........................................ 77

ÍNDICE
vii
3.2.2 Derivatización de Superficies de Oro por Electro‐reducción de Sales de Diazonio ...........79
3.2.2.1 Electro‐deposición de 4‐nitrobenceno ......................................................................................... 79 3.2.2.1.1 Caracterización por FT‐IRRAS................................................. ................................................... . 80
3.2.2.2 Electro‐deposición de Ácido Benzoico ........................................... ............................................... 84 3.2.2.2.1 Caracterización por FT‐IRRAS................................................. ................................................... . 85 3.2.2.2.2 Caracterización por PM‐IRRAS ................................................. .................................................. 87 3.2.2.2.3 Comparación entre capas delgadas y capas gruesas ................................................................. 88
3.2.2.2.3.1 Caracterización por PM‐IRRAS ................................................. .......................................... 88 3.2.2.2.3.2 Caracterización por XPS ................................................. ................................................... . 89
3.2.2.3 Electro‐deposición de Ácido 2, 3 ,5 ,6‐tetrafluorobenzoico ....................................................... 90 3.2.2.3.1 Caracterización por PM‐IRRAS ................................................. .................................................. 91 3.2.2.3.2 Caracterización por XPS ................................................ ................................................... .......... 92
3.3 CONCLUSIONES ................................................... ................................................... ......................... ..95
CAPÍTULO 4
POST‐FUNCIONALIZACIÓN Y CARACTERIZACIÓN
4.1 UNIÓN DEL PAR OS(II)/OS(III) A SUPERFICIES DE ORO ................................................... ...................99
4.2 CARACTERIZACIÓN SUPERFICIAL ................................................... ................................................... .101
4.2.1 Análisis Superficial por FT‐IRRAS................................................. ............................................102
4.2.2 Análisis Superficial por PM‐IRRAS................................................. ..........................................103
4.2.3 Análisis Superficial por Espectroscopia Raman Resonante.....................................................106
4.2.4 Análisis Superficial por Espectroscopia Fotoelectrónica de Rayos X.......................................108
4.2.4.1 Análisis de Au/PhCOOH/Os y Au/SPhCOOH/Os............................................................................... 108 4.2.4.2 Análisis de Au/PhF4COOH/Os .................................................. ................................................... ..... 110
4.2.5 Análisis Superficial por Microscopía de Efecto Túnel .............................................. ................111
4.3 CARACTERIZACIÓN ELECTROQUÍMICA ................................................... ...........................................115
4.4 CONCLUSIONES ................................................... ................................................... ........................122
CAPÍTULO 5
ANÁLISIS DE LA CINÉTICA DE TRANSFERENCIA ELECTRÓNICA
5.1 MARCO TEÓRICO ................................................... ................................................... ......................127
5.1.1 Cinética de Transferencia Electrónica........................................... ..........................................127
5.1.1.1 Ecuación de Nernst................................................................................................ ......................... . 127 5.1.1.2 Modelo de Butler‐Volmer............................................. ................................................... ................ 128 5.1.1.3 Modelo de Marcus............................................... ................................................... ....................... .. 131
5.1.1.3.1 Fundamentos ......................................... ................................................... ........................ ....... 131 5.1.1.3.2 Predicciones de la teoría.............................................. ................................................... ......... 137

ÍNDICE
viii
5.1.1.3.3 Reacciones de electrodo........................................... ................................................... ............ 139
5.1.2 Mecanismos de Transferencia Electrónica........................................... ...................................141
5.2 DETERMINACIÓN DE LA CONSTANTE ESTÁNDAR DE TRANSFERENCIA ELECTRÓNICA............................143
5.2.1 Metodologías empleadas .............................................................................................. .........143
5.2.1.1 Resistencia no Compensada y Capacidad de la Doble Capa: Constante de Celda ........................... 143 5.2.1.2 Voltametría cíclica ............................................ ................................................... ......................... ... 146 5.2.1.3 Espectroscopía de Impedancia Electroquímica ............................................................................... 150
5.3 RESULTADOS ................................................... ................................................... .............................153
5.3.1 Estimación de la distancia Au‐Os................................................. ...........................................153
5.3.2 Medición de k0 por Voltametría Cíclica.............................................. .....................................155
5.3.3 Medición de k0 por Espectroscopía de Impedancia Electroquímica........................................160
5.3.3.1 Transferencia Electrónica en los Sistemas Formados a Partir de Tioles alifáticos .......................... 161 5.3.3.2 Transferencia Electrónica en los Sistemas Aromáticos.................................................................... 164
5.4 CONCLUSIONES ................................................... ................................................... ......................... 169
CAPÍTULO 6
INTERFAZ ELECTRODO‐SOLUCIÓN
6.1 MARCO TEÓRICO ................................................... ................................................... ......................173
6.1.1 Estructura de la Interfase Electrodo‐Solución............................................ .............................173
6.1.2 Teoría de Distribución Interfacial del Potencial ............................................ ..........................175
6.1.2.1 Fundamentos.......................................... ................................................... ....................... ............... 175 6.1.2.2 Predicciones de la Teoría ............................................. ................................................... ................. 179
6.2 EFECTO DE LA DILUCIÓN SUPERFICIAL DEL COMPLEJO DE OSMIO SOBRE LA TRANSFERENCIA
ELECTRÓNICA................................................... ................................................... ...................................180
6.2.1 Preparación de monocapas autoensambladas mixtas y caracterización por desorción
reductiva ............................................................................................... ...........................................180
6.2.2 Post‐funcionalización de monocapas mixtas y análisis por voltametría cíclica......................182
6.2.2.1 Variación del ancho de pico a media altura con el cubrimiento superficial del complejo de Osmio
................................................... ................................................... ......................... ...................................... 184 6.2.2.2 Variación de la constante cinética estándar con el cubrimiento superficial del complejo de Os ... 187
6.3 EFECTO DEL PH Y LA FUERZA IÓNICA SOBRE LA TRANSFERENCIA ELECTRÓNICA..................................190
6.3.1 Análisis de Au/MUA a diferentes pH por PM‐IRRAS ................................................. ..............191
6.3.2 Análisis electroquímico en función del pH con fuerza iónica constante .................................192
6.3.3 Análisis electroquímico a diferentes fuerzas iónicas y pH controlado ....................................196
6.3.4 Análisis de la cinética de transferencia electrónica a distintos pH .........................................199
6.4 CONCLUSIONES ................................................... ................................................... ......................... 200
6.5 APÉNDICE I................................................... ................................................... ................................201

ÍNDICE
ix
6.5.1 Adsorción de un ácido débil a una superficie: equilibrio ácido‐base y potencial electrostático
[445]................................................................................................. ......................... .......................201
6.5.2 Adsorción de un par electroactivo a una superficie: equilibrio redox y potencial electrostático
[428]................................................................................................. ......................... .......................202
6.5.3 Combinación de los equilibrios ácido‐base y de electro‐reducción: Efecto sobre el potencial en
el PTE ................................................ ................................................... ............................................206
CAPÍTULO 7
ESPECTROSCOPÍA DE EFECTO TÚNEL CON CONTROL ELECTROQUÍMICO
7.1 FUNDAMENTOS DE LA TÉCNICA................................................... ................................................... ..209
7.2 MARCO TEÓRICO ................................................... ................................................... .......................211
7.2.1 Moléculas No Electroactivas............................................................................................ .......211
7.2.2 Moléculas Electroactivas: Modelo de Kuznetzov‐Ulstrup ............................................. ..........211
7.2.2.1 Ecuaciones .......................................... ................................................... ........................ .................. 215
7.3 ANÁLISIS DE AU/PHCOOH/OS Y AU/SPHCOOH/OS POR STS CON CONTROL ELECTROQUÍMICO ..217
7.4 CONCLUSIONES ................................................... ................................................... ......................... 220
CAPÍTULO 8
DISCUSIÓN FINAL Y CONCLUSIONES
8.1 TIOLES Y SALES DE DIAZONIO................................................... ................................................... .....223
8.2 TRANSFERENCIA ELECTRÓNICA ................................................... ................................................... ...224
8.2.1 Distancia donor‐aceptor y naturaleza del puente molecular............................................ ......225
8.2.2 Interfaz electrodo‐solución ............................................ ................................................... ......226
8.2.2.1 Efecto del cubrimiento superficial de los centros electroactivos ............................................... ..... 226 8.2.2.2 Efecto del electrolito soporte y del pH ................................................. ........................................... 227
8.2.3 Unión a la superficie: Au‐C vs. Au‐S .................................................. ......................................228
BIBLIOGRAFÍA......................................................................................................... 231


CAPÍTULO 1
INTRODUCCIÓN Y OBJETIVOS


1. INTRODUCCIÓN Y OBJETIVOS
3
ste trabajo de tesis se basa en el estudio de los fenómenos de transporte de carga a
través de películas orgánicas de espesor nanométrico. Para ello se modificaron electrodos
con monocapas electroactivas y se analizó la cinética de la transferencia electrónica entre los
centros redox y la superficie del electrodo.
En el grupo de Electroquímica Molecular se han llevado a cabo estudios detallados sobre la
modificación de electrodos por el método de auto‐ensamblado capa por capa de poli‐
electrolitos electroactivos. El objetivo general ha sido estudiar la respuesta electroquímica de
los mismos y de sus propiedades fisicoquímicas así como su aplicación en el desarrollo de
biosensores. Con esta tesis se buscó abrir una nueva línea de investigación que es de
fundamental importancia en diversas áreas de investigación aplicada como ser la electrónica
molecular, la electrocatálisis y el desarrollo de sensores, entre otros
En este capítulo se presenta el marco conceptual que motivó este trabajo de tesis junto con los
objetivos perseguidos y una breve descripción del contenido de cada uno de los capítulos que
lo componen.
1.1 ELECTRÓNICA MOLECULAR
La electrónica molecular es uno de los campos de la nanociencia que más atención ha recibido
en los últimos años. Se basa en el estudio de los niveles electrónicos moleculares para
comprender ciertos comportamientos propios de las moléculas tales como su capacidad de
unión, reconocimiento y ensamblado que resultan en la formación de estructuras más grandes
que pueden ser aprovechadas en el desarrollo de nuevas tecnologías. A ello se suma el
desarrollo de dispositivos electrónicos que dependen de las propiedades de la materia en la
escala molecular. Por lo tanto, se consideran moléculas individuales, arreglos moleculares y
redes moleculares conectados con otros componentes electrónicos. Principalmente, se busca
el desarrollo de sensores, materiales “inteligentes”, motores moleculares, dispositivos lógicos
y de memoria, transistores moleculares y dispositivos de transducción de energía [1, 2].
Esta disciplina tiene aportes de la química, la ingeniería electrónica, la ingeniería óptica y la
ciencia de estado sólido debido a los objetivos que persigue.
Vale destacar que la electrónica molecular no sólo representa el paso tecnológico final en la
miniaturización de los circuitos electrónicos de computación, sino que también provee nuevas
metodologías para la exploración de la arquitectura de sistemas moleculares, abriendo el
camino para el desarrollo de la ingeniería en la nano escala.
Los primeros estudios sobre transferencia de carga y de energía en moléculas fueron llevados
a cabo en la década de 1940 por Robert Mulliken y Albert Szent‐Gyorgi y estuvieron
E

1. INTRODUCCIÓN Y OJETIVOS
4
relacionados con lo que ellos llamaron sistemas donor‐aceptor [3]. Treinta años después, Mark
Ratner y Ari Aviram publicaron un trabajo titulado “Rectificadores moleculares” [4]. En este
trabajo, los autores habían tomado una molécula orgánica consistente en un sistema ヽ‐dador y
en uno ヽ‐aceptor, separados por un puente de metilenos unidos por enlaces σ. Con ella,
habían calculado cuál sería su respuesta al ser sometida a la acción de un campo eléctrico y
habían hallado que ésta se comportaría como un rectificador. Es decir, Ratner y Aviram habían
vislumbrado el primer dispositivo electrónico molecular. Sin embargo, la falta de métodos que
permitieran hacer contactos eléctricos en esa escala, impidieron medir las características
electrónicas de moléculas individuales, postergando por un tiempo la confirmación de la
predicción de Ratner y Aviram.
Durante la década de 1980, el desarrollo de las metodologías de auto‐ensamblado molecular
[5‐7] y de las técnicas de sonda de barrido [8, 9] permitió la preparación de nano‐estructuras
moleculares y la caracterización de sus propiedades físicas y eléctricas. Con todo ello, la
electrónica molecular ha presentado un rápido progreso que continúa en el presente.
1.2 TRANSFERENCIA ELECTRÓNICA
Mientras que la promesa de nuevos avances tecnológicos ha sido la mayor motivación del
desarrollo de la electrónica molecular, la consideración de sistemas moleculares como
componentes de dispositivos electrónicos ha dado lugar a preguntas más fundamentales. A la
hora de diseñar y fabricar un mecanismo en escala nanométrica lo más importante es la
comprensión y conocimiento del comportamiento y de las propiedades moleculares.
En los últimos años, ha existido un gran progreso en el establecimiento de un marco teórico
para los procesos de transferencia electrónica (TE) en sistemas moleculares, supermoleculares
y biofísicos [10‐12]. La TE provee una base conceptual y técnica para la electrónica molecular,
tanto para los dispositivos como para los materiales moleculares. Es por ello que se ha
expandido el interés en comprender los mecanismos de los procesos de transporte electrónico
en un amplio intervalo de escalas (desde moléculas individuales hasta estructuras nano‐ y
meso‐métricas), de tiempo (hasta el orden del picosegundo) y en diversas fases circundantes
(homogénea e interfacial, molecular, supramolecular) [10, 13‐17].
Hay que destacar que comprender los aspectos fundamentales de la transferencia de carga
interfacial no sólo es importante en electrónica molecular, sino que también lo es en
numerosas aplicaciones tecnológicas como biosensores [18], fotodiodos [19], electrocatálisis
[20], bioelectrocatálisis [21] y fotoconversión solar [22].

1. INTRODUCCIÓN Y OBJETIVOS
5
El progreso en las técnicas de síntesis y de caracterización [23‐28], junto con el creciente
aporte de los métodos computacionales y teóricos [10, 13, 15, 16, 29, 30] han permitido el
avance en el estudio mecanístico de los fenómenos de TE, haciendo posible conocer detalles
del comportamiento dinámico y los aspectos estructurales y energéticos básicos que los
dominan. La perspectiva teórica es fundamental para unificar criterios y marcar las diferencias
en los distintos tipos de transporte de carga.
En general, para llevar adelante los estudios de transferencia electrónica se consideran
sistemas modelo que constan de un donor (D) y un aceptor (A) de electrones, generalmente
separados entre sí por un puente (P). La transferencia electrónica entre D y A, a través de P, se
puede estudiar en fase homogénea [31, 32], en interfases (es decir, como procesos
electroquímicos de óxido‐reducción) [33‐37] y con el pasaje de corriente a través de junturas
moleculares [38‐41]. Se busca comprender la forma en que la velocidad de transferencia
electrónica depende de las propiedades del donor y el aceptor, del solvente y del
acoplamiento electrónico entre los estados involucrados. El análisis se centra en los diferentes
roles que juegan estos factores y la forma en que los mismos afectan los aspectos cualitativos
y cuantitativos del proceso de transporte de carga.
1.3 ACOPLAMIENTO ELECTRÓNICO DE LARGO ALCANCE
El término “cable molecular” ha sido empleado durante décadas para describir el
comportamiento de estructuras que promueven el acoplamiento electrónico fuerte y el
transporte electrónico rápido a través de largas distancias. Estas estructuras juegan un papel
central en muchos ensamblados supramoleculares y se ha propuesto su aplicación en el campo
de la electrónica molecular [42, 43]. Algunos ejemplos que han sido estudiados tanto
experimental como teóricamente incluyen poli‐enos simples [44, 45], poli‐p‐fenilenos [46],
anillos aromáticos fusionados [47], oligómeros de heterociclos aromáticos [48] y nanotubos de
carbono [49].
Los cables moleculares se pueden encontrar dentro de una molécula que posee un dador y un
aceptor de electrones entre los que se transfiere la carga (Figura 1 a)), conectando un grupo
electroactivo con la superficie de un electrodo (Figura 1 b)) y conectando los dos contactos
metálicos de una juntura molecular (Figura 1 c)). En la Figura 1 se muestran esquemáticamente
tres tipos diferentes de ensamblados en los que una cadena molecular discreta (DPA) está
acoplada con baños nucleares o electrónicos. El sistema molecular consiste de un puente, P
(que se toma como una secuencia lineal de subunidades Pj, con j = 1‐n) unido por sus
extremos (P1 y Pn) a los sitios donor (D) y aceptor (A) de electrones. Los diferentes sitios tienen

1. INTRODUCCIÓN Y OJETIVOS
6
asociados estados electrónicos localizados. En cada caso, se muestran ejemplos reportados
por: a) Scott et al. [50], b) Creager y Rowe [51] y c) McCreery [41].
Cuando la transferencia electrónica ocurre en solución (Figura 1 a)), el baño consiste de un
cuasi‐continuo de modos nucleares (es decir, modos de polarización de un solvente polar y
modos vibracionales moleculares) acoplados con los estados locales de D y de A, aunque
también puede existir cierto acoplamiento con los estados asociados a las unidades del
puente. Los estados de D y A, junto con el baño nuclear comprenden los estados vibrónicos
inicial y final en el proceso de TE.
En los estudios de cinética de TE interfacial que emplean electrodos modificados, el sitio
electroactivo (que puede estar en su forma reducida u oxidada) está acoplado al mismo tipo
de baño nuclear que en el caso anterior, mientras que D está acoplado a un electrodo (es
decir, a un baño electrónico) (Figura 1 b)).
En ambos casos, la propiedad de interés es la constante cinética de transferencia electrónica
(kTE) [13, 16] que se puede medir a partir de fenómenos transitorios (como el cambio
exponencial de la población electrónica local) por técnicas de relajación [52, 53].
Alternativamente, se pueden emplear técnicas de estado estacionario (por ejemplo,
voltametría cíclica) [54].
Finalmente, en una juntura molecular (Figura 1 c)) los sitios terminales A y D están acoplados a
baños electrónicos (electrodos) y el proceso de TE se analiza en base a la corriente en estado
estacionario que circula en función de la diferencia de potencial aplicada entre ambos
electrodos, obteniéndose valores de conductividad [13, 15, 16, 29]. En particular, la punta de
Figura 1 Representación esquemática de los elementos involucrados en la transferencia electrónica mediada
por un puente molecular (P). a) TE entre sitios discretos donor y aceptor de electrones (D y A). b) TE entre un
electrodo (D) y un sitio aceptor localizado (A). c) Conductividad entre dos electrodos (D y A): juntura molecular.

1. INTRODUCCIÓN Y OBJETIVOS
7
un microscopio de efecto túnel se puede emplear como un electrodo y reemplazar a uno de
los electrodos de la juntura [55, 56]. De esta manera se pueden analizar SAMs electroactivas
depositadas sobre un electrodo, acercando la punta de un microscopio de efecto túnel (STM,
Scanning Tunneling Microscopy) o de fuerza atómica (AFM, Atomic Force Microscopy), con una
punta conductora, y determinar la corriente que circula a través de un pequeño grupo de
moléculas, y hasta en una molécula individual, en función del potencial aplicado entre el
sustrato y la punta del microscopio.
Cuando la transferencia electrónica entre un dador de electrones y un aceptor ocurre por un
mecanismo de tuneleo electrónico, se espera que el acoplamiento electrónico entre ambas
partes decaiga exponencialmente con la distancia (dDA) y, por ende la constante cinética de TE
también lo haga:
( )oTE DAk k exp βd= − (1.1)
donde β se conoce como parámetro de tuneleo y se puede expresar como:
PP
DP
H2β lna らE
⎛ ⎞⎜ ⎟⎜ ⎟⎝ ⎠= − (1.2)
donde HPP es el acoplamiento electrónico entre las unidades que conforman el puente, a es
una unidad de longitud del puente y らEDP es la energía del estado intermedio en el que el
electrón se encuentra deslocalizado en el puente.
Estas expresiones son válidas para dos mecanismos de transferencia electrónica:
a) tuneleo coherente (también conocido como superintercambio o tuneleo a través del enlace)
en el que los sitios del puente favorecen la TE por el acoplamiento electrónico entre ellos y el
electrón se transfiere a través de los mismos sin residir en el puente.
b) tuneleo directo, en el que el electrón se transfiere a través del espacio, y no a través del
puente molecular.
Existe un tercer mecanismo de transporte de carga: el salto electrónico (electron hopping) en
el que existen transiciones electrónicas consecutivas asistidas por modos vibracionales entre
sitios vecinos en el puente.
El tipo de mecanismo predominante estará determinado por la naturaleza, la magnitud y la
energía de la interacción entre los sitios donor y aceptor de electrones, así como del entorno.

1. INTRODUCCIÓN Y OJETIVOS
8
1.4 ELECTRODOS MODIFICADOS
1.4.1 DERIVATIZACIÓN DE SUPERFICIES
Una superficie o interfaz es una delgada región del espacio (que comprende, en general, unos
pocos nanómetros) en la que las propiedades de un material (composición química, índice de
refracción, fuerza mecánica, conductividad, carga, etc.) son significativamente diferentes
respecto de las que exhibe el seno del mismo. Es esta porción del material la que determina
su reactividad con el entorno y los fenómenos de transporte.
La derivatización de una superficie consiste en su transformación física con el objetivo de
modificar una o varias de sus propiedades físicas, químicas y/o biológicas. En general, la
modificación de las propiedades superficiales de un material se realiza con el objetivo de
cambiar su comportamiento en ciertas condiciones o mejorar su rendimiento en un dado
proceso para poder emplearlo en ciertas aplicaciones o, incluso, para desarrollar nuevas
tecnologías.
Existe una gran variedad de metodologías que permiten alterar las propiedades de los
materiales. Éstas abarcan procesos físicos y químicos. Entre estos últimos vale destacar
aquellos que se basan en la unión directa de moléculas orgánicas ya sea por adsorción física o
por la formación de enlaces químicos con la superficie de interés. La presencia de moléculas
químicamente unidas a una superficie permite controlar las propiedades de la interfaz
(mojado, conductividad, adhesión y reactividad química) logrando un comportamiento
totalmente diferente del que presenta el sustrato desnudo.
Cuando se depositan monocapas o películas moleculares sobre sustratos conductores se
obtienen lo que se conoce generalmente como electrodos modificados. Este campo fue
desarrollado en sus comienzos por Hubbard [57, 58], Murray [59] y Millar [60] y se ha
convertido en un área muy activa de la electroquímica en los últimos años [61‐64].
La modificación de un electrodo consiste en la inmovilización de ciertas moléculas sobre su
superficie con el objetivo de conferirle propiedades químicas, electroquímicas, ópticas u otras
características que posean dichas moléculas. Se pueden preparar de diferentes maneras que
incluyen la adsorción irreversible, la unión covalente de una monocapa molecular y el
recubrimiento del electrodo con películas de polímeros u otros materiales.
El gran desarrollo que ha presentado esta área se debe principalmente a sus posibles
aplicaciones. La electrocatálisis [65, 66] ha sido uno de los puntos principales debido a la
necesidad de desarrollo de materiales adecuados en celdas de combustible [67‐70] y baterías

1. INTRODUCCIÓN Y OBJETIVOS
9
recargables [71, 72]. Asimismo, se ha buscado desarrollar electrodos con propiedades
electrocrómicas que podrían ser útiles en dispositivos tales como las ventanas “inteligentes”
[73‐76]. Otras aplicaciones incluyen la protección contra la corrosión o el ataque químico [77‐
79] y el desarrollo de sensores analíticos y biosensores [80‐82]. Finalmente, existe un creciente
interés en los dispositivos electrónicos moleculares, es decir, aquellos sistemas
electroquímicos que pueden imitar el comportamiento de diodos, transistores y redes
eléctricas [83‐88].
Además de las aplicaciones recién mencionadas, los electrodos modificados abrieron la
posibilidad de investigar la transferencia electrónica sobre su superficie, eliminando los
problemas de transporte de masa, al poder unir a la misma a la especie electroactiva de
interés con las estrategias que se describen a continuación.
1.4.2 TÉCNICAS DE MODIFICACIÓN
Existen diferentes métodos para modificar superficies que permiten generar monocapas o
multicapas moleculares sobre sustratos de diferentes materiales [89]. Entre ellas, vale destacar
el autoensamblado capa por capa de poli‐electrolitos [90, 91], la deposición molecular
empleando el método de Langmuir‐Blodgett [92‐94], el autoensamblado de monocapas
moleculares [95, 96] y la reducción de sales de diazonio [97]. En la Figura 2 se presentan
esquemáticamente cada una de estas técnicas de modificación que se describen brevemente a
continuación.
En el autoensamblado de poli‐electrolitos (Figura 2 a)) un sustrato con una carga neta en su
superficie, por ejemplo positiva, se sumerge en una solución de un poli‐electrolito de carga
opuesta (en este caso sería un poli‐anión) durante unos minutos. De esta manera se forma una
capa delgada del polímero sobre el electrodo y, debido a la sobre‐compensación de cargas, la
superficie ahora presenta una densidad de carga negativa. Luego, el sustrato se lava para
eliminar el exceso del polímero y se coloca en una solución de poli‐catión. Nuevamente, el
polímero se une a la superficie por interacción electrostática con las cargas de la superficie.
Este procedimiento se puede repetir tantas veces como sea necesario hasta obtener una capa
orgánica del espesor deseado con una precisión de ≈ 1 nm. Las principales ventajas de este
método son su simplicidad, la ausencia de equipamiento costoso y la posibilidad de cubrir
prácticamente cualquier superficie.
Las películas obtenidas por el método de Langmuir‐Blodgett (Figura 2 b)) contienen moléculas
anfifílicas (compuestas por una cabeza hidrofílica y una cola hidrofóbica) depositadas sobre un
sustrato sólido. Para ello, estas moléculas primero forman una monocapa en la interfase

1. INTRODUCCIÓN Y OJETIVOS
10
líquido‐aire y luego son transferidas al sustrato por la inmersión (o emersión) del mismo en (o
desde) el líquido. Las películas obtenidas contienen una o más monocapas formadas de esta
manera y, como una monocapa se adsorbe homogéneamente con cada paso de inmersión o
emersión, se pueden obtener capas de un espesor muy controlado.
Las monocapas autoensambladas (SAMs) (Figura 2 c))se pueden preparar usando diferentes
tipos de moléculas y diferentes sustratos [94]. Sin embargo, las SAMs más utilizadas son las
que se obtienen con n‐alcanotioles auto‐ensamblados sobre sustratos de oro. En este caso, la
gran variedad de grupos funcionales que se pueden añadir a estas moléculas y la facilidad del
procedimiento han convertido a esta técnica en una de las favoritas a la hora de derivatizar
superficies.
Por último, la reducción de sales de diazonio (Figura 2 d)) ha cobrado cierta relevancia durante
los últimos años, ya que además de ser una técnica fácil de implementar, genera enlaces más
fuertes entre el sustrato y las moléculas que los obtenidos por las otras técnicas. En este caso,
Figura 2 Diferentes estrategias para modificar electrodos: a) Auto‐ensamblado capa por capa de polielectrolitos,
b) Formación de capas delgadas por el método de Langmuir‐Blodgett, c) Autoensamblado de compuestos
organosulfurados y d) Reducción electroquímica de sales de diazonio.

1. INTRODUCCIÓN Y OBJETIVOS
11
la electro‐reducción de una sal de diazonio genera un radical arilo que reacciona con el
electrodo, formando una unión covalente con el mismo.
Todas las técnicas descriptas permiten preparar la superficie del electrodo para estudios de
transferencia electrónica ya sea por la incorporación de especies electroactivas en la
estructura de las capas orgánicas que se depositan sobre el electrodo o por el bloqueo del
mismo con capas orgánicas suficientemente compactas para analizar un par redox libre en
solución.
1.4.3 ELECTRODOS MODIFICADOS Y ANÁLISIS DE TRANSFERENCIA ELECTRÓNICA
En los estudios de transferencia electrónica que emplean electrodos modificados, el objetivo
principal es evaluar la cinética del transporte de carga entre una molécula electroactiva y el
electrodo, controlando la distancia entre ambos.
Existen dos formas de controlar esta distancia. Cuando las especies electroactivas se
encuentran libres en solución, se emplea un electrodo bloqueado con una monocapa
molecular compacta. En este caso, el acercamiento directo de las moléculas redox a la
superficie del electrodo está restringido por la monocapa que actúa como barrera. La segunda
variante consiste en modificar el electrodo con SAMs electroactivas, es decir, aquellas que
poseen moléculas electroactivas incorporadas en su estructura. En la Figura 3 se muestran
esquemáticamente ambos tipos de sistema.
En cualquiera de las dos modalidades, se reemplaza la capa interna de Helmholtz por una
monocapa molecular disminuyendo, casi en un orden magnitud, la capacidad de la interfase
Figura 3 a) Monocapa autoensamblada de un n‐alcanotiol sobre un electrodo de oro con moléculas
electroactivas unidas a la misma. b) Monocapa de un n‐alcanotiol depositada sobre un electrodo de oro que
actúa como una barrera perfecta que no permite el acercamiento al electrodo de las moléculas redox presentes
en solución.

1. INTRODUCCIÓN Y OJETIVOS
12
electrodo/SAM/electrolito respecto de la que presenta el electrodo desnudo en contacto con
la solución. Además, dicha capacidad se independiza casi totalmente del potencial aplicado al
electrodo. En consecuencia, las constantes de tiempo y los efectos de doble capa de las celdas
disminuyen considerablemente.
A esto puede añadirse el orden tridimensional que impone la monocapa autoensamblada en
las cadenas alifáticas haciendo posible que actúen como puentes moleculares y mantengan
una distancia fija entre el donor (electrodo) y aceptor (par electroactivo) de electrones.
En general, las constantes cinéticas que se desean medir son muy altas y es casi imposible
medirlas con el electrodo desnudo. Es por ello que el uso de electrodos modificados ha
cobrado gran popularidad en este tipo de experimentos. En particular, cuando las especies
electroactivas están incorporadas a la estructura de la SAM también se elimina la componente
de difusión de la corriente medida y esto simplifica los métodos experimentales que se deben
emplear y las ecuaciones que describen las relaciones potencial‐corriente del proceso.
Además, el comportamiento del sistema es menos sensible a los defectos que pueda presentar
la monocapa y el cubrimiento superficial de la especie redox se puede conocer directamente
de la carga medida en una voltametría cíclica del sistema.
1.4.3.1 Monocapas Electroactivas
En los estudios cinéticos que emplean monocapas electroactivas autoensambladas sobre la
superficie de un electrodo se desea que las mismas presenten una estructura compacta y bien
orientada con las moléculas redox ubicadas en la interfase y en pleno contacto con el
electrolito soporte. Además, estas últimas deben estar separadas entre sí para evitar que
exista transferencia electrónica entre ellas. Toda la corriente faradaica debe representar la
transferencia entre los sitios electroactivos y el electrodo. Esta configuración minimiza los
efectos de doble‐capa y permite una rápida transferencia de iones hacia y desde el electrolito
durante los cambios en el estado de oxidación de la monocapa. En estas condiciones las
medidas cinéticas deberían reflejar la verdadera velocidad de transferencia electrónica entre el
electrodo y las moléculas redox, y no otro proceso como el trasporte de iones [98].
Las monocapas electroactivas se pueden preparar de dos formas. Una de ellas consiste en
sintetizar moléculas que poseen un grupo electroactivo y un grupo funcional que presenta alta
afinidad por el sustrato que se desea emplear. La monocapa se forma por adsorción de estas
moléculas con o sin moléculas diluyentes (es decir, moléculas que no poseen el grupo redox)
(Figura 4 a)). El cubrimiento superficial de los grupos redox se puede controlar regulando la
proporción de ambos tipos de moléculas cuando se prepara la monocapa. La desventaja de

1. INTRODUCCIÓN Y OBJETIVOS
13
este método reside en la diferencia de solubilidades que puede existir entre las moléculas
redox y las diluyentes en el solvente de preparación y en la posibilidad de que en la monocapa
se formen dominios de cada una de ellas.
La otra estrategia consiste en formar una monocapa de una molécula terminada en un grupo
funcional que pueda reaccionar con la molécula redox. Por lo tanto, una vez formada la SAM,
se realiza una reacción de post‐funcionalización de la misma con el grupo electroactivo,
incorporándolo de esta manera a su estructura (Figura 4 b)).
Algunos ejemplos de moléculas redox incorporadas a monocapas formadas con alcanotioles,
disulfuros y sulfuros incluyen ferrocenos [24, 33, 51, 98‐107], complejos de penta‐amin rutenio
[108‐115], complejos de poli‐piridil rutenio [116, 117] y pilo‐piridil osmio [118, 119],
viológenos [120‐127], quinonas [123, 128‐133] y porfirinas [123, 134‐138]. El citocromo c y la
azurina se pueden adsorber sobre SAMs terminadas en grupos ionizables o coordinantes [139‐
146]. Las monocapas que contienen complejos de poli‐piridil osmio se pueden formar cuando
uno de los ligandos contiene un nitrógeno aromático (por ejemplo, piridina) [147‐151] o,
cuando uno de los ligandos tiene un grupo amino, por post‐funcionalización de monocapas
terminadas en grupos ácido empleando una carbodiimida, como se verá en esta tesis.
Figura 4 a) Monocapa electroactiva formada por autoensamblado de un alcanotiol terminado en ferroceno.
b) Preparación de una monocapa electroactiva por post‐funcionalización con el complejo redox
([Ru(NH3)5(pyCH2NH2)].

1. INTRODUCCIÓN Y OJETIVOS
14
1.4.4 JUNTURAS MOLECULARES
La Figura 1 c) esquematiza lo que se conoce como juntura molecular. En ella una o varias
moléculas se encuentran entre dos sólidos (metálicos o semiconductores) con los que están en
contacto eléctrico. El fenómeno central en este tipo de dispositivos es el transporte de carga a
través de la o las moléculas ubicadas entre los contactos [152, 153].
Las junturas moleculares se pueden clasificar en varias categorías de acuerdo a la química
involucrada en su fabricación (Langmuir‐Blodgett, SAMs, etc.), a su estructura básica (una
molécula individual, varias moléculas, dos o tres terminales, etc.) o al paradigma experimental
(microscopía de sonda de barrido, juntura de quiebre (obtenidas por ruptura de alambres
modificados con monocapas, en inglés, break junction), etc.). En la Figura 5 se esquematizan
algunos ejemplos.
En particular, el término juntura unimolecular se reserva para aquellas en las que se tiene un
número pequeño de moléculas entre los contactos: no mayor a diez. En cambio, una juntura
de monocapa posee muchas moléculas (103‐1012) en una capa molecular orientada entre los
contactos. Estrictamente hablando, se trata de un dispositivo de capa delgada pero la
monocapa orientada imparte ciertas propiedades que no se observan en las junturas
tradicionales.
La naturaleza del contacto entre los electrodos y la/s molécula/s es muy importante en cuanto
a su efecto sobre el comportamiento de la juntura y está definido por la química empleada en
la unión entre las distintas partes. En general, uno de los electrodos se modifica empleando
alguna de las estrategias de la Figura 2. El contacto superior se puede lograr por evaporación
de una fina película de metal [154‐156] (Figura 5 a)), con electrodos de gota de Hg [157‐160]
(Figura 5 b)) o por quiebre de un electrodo modificado para analizar la conductividad de
aquellas moléculas que hayan quedado estadísticamente atrapadas a través de la juntura
(break junctions) [161‐163] (Figura 5 c)). Finalmente vale mencionar las junturas que emplean
la punta de un microscopio de sonda de barrido (STM o AFM en modo conductor) para
establecer el contacto superior [164‐166] (Figura 5 d)). Esta última interactúa con una o pocas
moléculas y, la molécula individual involucrada en la juntura, no debe estar necesariamente
aislada de sus vecinas.
Las junturas moleculares formadas con la punta de un microscopio de efecto túnel son
particularmente interesantes, ya que al sistema de la Figura 5 d) se puede agregar un
potenciostato, un electrodo de referencia, un contra‐electrodo y una solución electrolítica y,
de esa manera, armar una celda electroquímica. En ella el electrodo de trabajo es el sustrato y

1. INTRODUCCIÓN Y OBJETIVOS
15
su potencial, junto con el de la punta del microscopio, se puede controlar en forma
independiente respecto del electrodo de referencia. Esta técnica se conoce como
Espectroscopía de Efecto Túnel con control electroquímico (Electrochemical Scanning
Tunneling Spectroscopy, E‐STS) y permite analizar la conductividad de moléculas que poseen
grupos electroactivos en función del potencial aplicado [167, 168].
Figura 5 Ilustraciones esquemáticas de diferentes tipos de junturas de monocapa a) Juntura formada por un
sustrato de oro, una película de Langmuir‐Blodgett y una película de oro evaporado. b) Juntura formada con
electrodos de gota de mercurio y una SAM. El electrodo inferior también puede ser de Au. c) Break junction
formada luego de estirar un alambre de oro previamente modificado. d) Juntura molecular formada entre un
sustrato conductor, una monocapa autoensamblada y la punta de un microscopio de sonda de barrido (STM o
AFM conductor).

1. INTRODUCCIÓN Y OJETIVOS
16
1.5 ESTADO DEL CONOCIMIENTO
La electroquímica en la escala nano, de alta resolución ha seguido una línea de evolución
desde fines de la década de 1970 a partir de la interacción de la electroquímica con la física del
estado sólido y la física de superficies [169‐171].
La aparición de técnicas de modificación superficial confiables ha permitido ubicar moléculas
electroactivas sobre electrodos en forma controlada y variar sistemáticamente aquellos
parámetros que afectan directamente a la transferencia electrónica, como ser la distancia
dador‐aceptor de electrones, el entorno químico, la naturaleza del electrolito, la temperatura,
el tipo de electrodo. La gran mayoría de estos estudios se han realizado con monocapas
autoensambladas de tioles alifáticos [102, 105‐107, 111, 113, 172‐174] y de tioles de cadenas
insaturadas [26, 110, 175‐178]. Entre ellos se deben destacar los aportes de Chidsey [24, 52] y
de Finklea [111, 112, 172, 179]. Ambos grupos interpretaron la variación de log (kTE) en función
del sobrepotencial aplicado utilizando la relación de Marcus de la energía libre, y obtuvieron
las constantes cinéticas estándar (k0) y las barreras de energía de reorganización (゜) asociadas
a la transferencia electrónica. Ambos encontraron coeficientes de tuneleo (β) similares y
construyeron los gráficos de Tafel que resultaron ser coherentes con un mecanismo de tuneleo
a través del enlace.
Otros estudios incluyen el análisis de reacciones de transferencia electrónica acopladas con
transferencia de protón [180], el efecto de la solvatación de una especie electroactiva en un
entorno hidrofóbico sobre las reacciones redox [181], el efecto del movimiento del contraión
sobre la TE [51, 98, 104, 182], la dinámica de las moléculas en bicapas lipídicas híbridas [183] y
el efecto de la orientación y conformación de las proteínas electroactivas (citocromo c, glucosa
oxidasa) sobre las velocidades de TE a través de SAMs, un factor importante en la preparación
de sensores electroquímicos para detectar actividad enzimática [184‐187].
En los últimos años la modificación de superficies por reducción de sales de diazonio ha
cobrado importancia ya que no sólo forma capas más estables que los tioles [188], sino que
además se puede emplear con una mayor variedad de materiales [97, 119, 189, 190]. Esta
estrategia también ha sido empleada en algunos estudios de transporte de carga y existe un
interés especial ya que, en este caso, la unión al sustrato es a través de un átomo de carbono
[188, 191‐194].
Al diseño controlado de la superficie de los electrodos se ha sumado el desarrollo de una serie
de técnicas superficiales que incluyen espectroscopía infrarroja [195‐197], Raman [198],
microbalanza de cristal de cuarzo [199] y de modelos teóricos de mecánica estadística y de

1. INTRODUCCIÓN Y OBJETIVOS
17
estructura electrónica de la doble capa [200, 201]. Asimismo, las técnicas STM [170, 202‐204]
y AFM [205] llevaron a la ciencia superficial y a la electroquímica interfacial a un nuevo nivel de
resolución sin precedentes, posibilitando la resolución atómica de las superficies de electrodos
metálicos y semiconductores y de los adsorbatos ubicados sobre las mismas.
Actualmente, los aspectos fundamentales de la transferencia electrónica en solución están
ampliamente entendidos y el desafío está enfocado en aumentar el detalle y la sofisticación
teórica. Por otro lado, en cuanto a los procesos de transferencia electrónica interfacial, el
objetivo es buscar nuevos conceptos que racionalicen el transporte de carga en medios
anisotrópicos y en electrolitos sólidos y aún se buscan nuevos fenómenos de transferencia
electrónica.
1.6 MOTIVACIÓN Y OBJETIVOS
Cuando se inició este trabajo de tesis, en el grupo de Electroquímica Molecular se habían
desarrollado estudios detallados sobre la modificación de electrodos por el método de auto‐
ensamblado capa por capa de poli‐electrolitos electroactivos utilizando poli(alilamina)
modificada con un complejo de osmio piridina‐bipiridina (PAH‐Os) como poli‐catión y glucosa
oxidasa (GOx), poli(vinilsulfonato (PVS) y poli(estirensulfonato) (PSS), como poli‐aniones. Los
estudios llevados a cabo con este tipo de sistemas tenían (y tienen) por objetivo el estudio de
la respuesta electroquímica de los mismos y de sus propiedades fisicoquímicas así como el
desarrollo de biosensores.
En particular, se habían realizado estudios de transferencia electrónica con un par redox
ubicado sobre la superficie de un electrodo a nivel de monocapa, que fueron ciertamente
pioneros pero limitados en cuanto a su alcance [206]. De esta manera, surgió la motivación
principal que marcó el rumbo de este trabajo de tesis: la comprensión y control del transporte
de carga a través de películas orgánicas de un espesor nanométrico. Esto es de fundamental
importancia en electrónica molecular, en el desarrollo de sensores, en protección contra la
corrosión, sólo por nombrar algunas de las áreas de interés. El conocimiento de cómo la
composición y la estructura química pueden afectar la transferencia electrónica entre un
sustrato sólido y una molécula electroactiva es central en el desarrollo de estas áreas de
investigación aplicada. Asimismo, es de mucho interés el estudio de transferencia electrónica
en proteínas y enzimas redox inmovilizadas sobre electrodos [207‐209], que constituyen
sistemas muy complejos en comparación con los que se estudian en esta tesis, que pueden
utilizarse como modelos.

1. INTRODUCCIÓN Y OJETIVOS
18
Cuando se estudian los procesos de transporte de carga algunas de las preguntas que surgen
son: ¿Cuáles son los mecanismos fundamentales que intervienen y cómo dependen de la
distancia y del entorno? ¿Cuáles son las barreras de potencial y cuáles son las velocidades de
transferencia que se pueden alcanzar? Además, este tipo de estudio requiere un amplio
conocimiento de las propiedades de los materiales empleados y genera nuevas preguntas
sobre la naturaleza del contacto entre los mismos. Este tipo de interrogantes fueron el punto
de partida para este trabajo de tesis cuyos contenidos se resumen a continuación. Antes vale
mencionar que en el Capítulo 2, se resumen el detalle experimental de todo el trabajo
realizado y los fundamentos de las técnicas utilizadas.
Con el fin de estudiar fenómenos de transferencia electrónica, se prepararon una serie de
sistemas modelo. Para ello, se prepararon monocapas de tioles alifáticos de diferentes
longitudes de cadena terminados en grupos ácido. Asimismo, se empleó un tiol aromático
(ácido 4‐mercapto benzoico) y se utilizó también la electro‐reducción de sales de diazonio para
generar capas delgadas con el mismo grupo funcional (ácido benzoico y ácido 1‐amino‐2,3,5,6‐
tetrafluorobenzoico). Dado que esta última metodología de derivatización de superficies es
relativamente nueva, uno de los objetivos planteados desde un comienzo fue la determinación
de las condiciones de deposición que permitieran generar capas delgadas en forma controlada.
En el Capítulo 3 se presentan en detalle las dos estrategias de modificación empleadas junto
con las diferentes caracterizaciones que se realizaron de los sistemas obtenidos. En una
segunda etapa, todos ellos fueron post‐funcionalizados con el complejo de osmio [Os(2,2´‐
bpy)2Cl(py‐CH2‐NH2)]+ a partir de la formación de una unión amida entre el grupo amino
presente en el mismo y los grupos ácidos ya presentes en la superficie. De esta manera, se
obtuvieron los sistemas modelo con el par Os(II)/Os(III) unido a la superficie de un electrodo
de oro a través de puentes moleculares de diferentes longitudes y naturaleza. La reacción de
post‐funcionalización se detalla en el Capítulo 4 así como los resultados obtenidos en la
caracterización superficial y electroquímica que se realizó de cada uno de estos sistemas.
El análisis de la cinética de transferencia electrónica fue el punto central de este trabajo de
tesis y se llevó a cabo empleando dos técnicas electroquímicas: voltametría cíclica y
espectroscopía de impedancia electrónica que permitieron obtener valores de la constante
cinética estándar (k0) en cada caso. Estos experimentos y los resultados que arrojaron se
presentan en el Capítulo 5. Asimismo, se examinó la influencia del pH, la fuerza iónica y el
grado de cubrimiento superficial sobre la cinética y la termodinámica del transporte de carga,
teniendo en cuenta que en todos los sistemas estudiados existen grupos ácidos libres (que no

1. INTRODUCCIÓN Y OBJETIVOS
19
reaccionaron con el complejo de osmio) que pueden afectar el comportamiento del par redox.
Todo esto se discute en el Capítulo 6.
Finalmente, en el Capítulo 7 se presentan experimentos llevados a cabo con espectroscopía de
efecto túnel con control electroquímico para profundizar el conocimiento sobre la naturaleza
microscópica de estos sistemas y, en especial, comparar la conductividad molecular cuando el
enlace a la superficie está dado por una unión Au‐S y una unión Au‐C.
Por último, el Capítulo 8 reúne las conclusiones que se desprenden de todo lo expuesto a lo
largo de este trabajo de tesis junto con la discusión final.


CAPÍTULO 2
MATERIALES Y MÉTODOS


2. MATERIALES Y MÉTODOS
23
n este capítulo se presenta una breve descripción de los fundamentos de cada una de las
técnicas utilizadas en este trabajo de tesis. Se detallan las características instrumentales
de los equipos empleados y se describen los experimentos llevados a cabo.
2.1 TÉCNICAS EMPLEADAS: FUNDAMENTOS
2.1.1 TÉCNICAS ELECTROQUÍMICAS
La electroquímica estudia los cambios químicos causados por el pasaje de una corriente
eléctrica y la producción de energía eléctrica a partir de reacciones químicas. Este campo
abarca una gran variedad de fenómenos (por ejemplo electroforesis, corrosión), dispositivos
(como ser arreglos electrocrómicos, sensores electroanalíticos, baterías, celdas de
combustible) y tecnologías (por ejemplo electroplateo de metales, producción a gran escala de
aluminio y cloro) teniendo además aplicación en la caracterización y en la síntesis de
materiales.
Los métodos electroquímicos se pueden dividir en dos categorías dependiendo si se realizan
en condiciones de corriente nula o no. Los métodos potenciométricos utilizan básicamente un
voltímetro para medir el potencial de una muestra respecto de un estándar de potencial
conocido cuando no circula corriente por el sistema. Los métodos voltamétricos emplean un
potenciostato para aplicar un potencial y medir la corriente que circula por el sistema. Las
medidas amperométricas, en las que el potencial, aplicado se mantiene constante y se mide la
corriente en función del tiempo, se incluyen en esta última categoría. En particular, los
métodos voltamétricos son los que tienen mayor aplicación en la caracterización de materiales
y los que han sido utilizados en este trabajo de tesis.
Si el comportamiento del sistema electroquímico en el espacio tridimensional compuesto por
corriente, potencial y tiempo está totalmente especificado, el sistema está completamente
caracterizado desde un punto de vista electroquímico. A partir de esos datos fenomenológicos
se pueden determinar todos los parámetros termodinámicos y cinéticos que controlan al
sistema. Éstos incluyen: 1) los potenciales estándar, 2) la estequiometría electrónica, 3) las
concentraciones de los reactivos, 4) los coeficientes de difusión, 5) las constantes de velocidad
involucradas y 6) el mecanismo de transferencia de carga.
E

2. MATERIALES Y MÉTODOS
24
2.1.1.1 REACCIONES DE ELECTRODO
En los sistemas electroquímicos la atención recae en los procesos y los factores que afectan el
transporte de carga entre diferentes fases químicas (interfase), por ejemplo, entre un
conductor electrónico (electrodo) y un conductor iónico (electrolito).
Una reacción de electrodo es un proceso químico heterogéneo que involucra la transferencia
electrónica hacia o desde una superficie, en general, metálica o semiconductora. Se puede
representar como:
O ne R− ⎯⎯→+ ←⎯⎯ (2.1)
La especie reducida es oxidada por cesión de electrones al electrodo o la especie oxidada es
reducida por ganancia de electrones desde el electrodo. Se considerará a la densidad de
corriente (I) para el proceso anódico (oxidación) positiva, mientras que para el proceso inverso
(reducción), negativa.
La especie electroactiva puede ser una molécula, una especie cargada o un complejo, puede
estar presente en solución o formando una película sobre el electrodo, ser el mismo solvente o
el mismo material de electrodo. El producto de la reacción electroquímica puede pasar a la
solución, a fase gaseosa o formar una nueva fase sobre el electrodo [54, 210].
2.1.1.2 CELDA ELECTROQUÍMICA
Este sistema está formado por dos electrodos separados por, al menos, una fase de electrolito.
El electrodo en el que ocurre la oxidación es el ánodo y aquel en el que ocurre la reducción es
el cátodo. La cantidad de carga en el cátodo debe ser igual a la cantidad de carga que circula
por el ánodo.
Además, para mantener la electroneutralidad en todos los puntos del sistema, los iones deben
moverse a través de la solución entre los electrodos así como los electrones deben pasar por
los cables externos que conectan a estos últimos entre sí.
La reacción neta en la celda está dada por la suma de las hemi‐reacciones que ocurren en cada
electrodo. Cada una de ellas (y por ende, la composición química del sistema cerca de los
electrodos) responde a la diferencia de potencial interfacial en el electrodo correspondiente.
En general, sólo una de estas reacciones es de interés y el electrodo asociado a la misma, se
llama electrodo de trabajo (working electrode, WE). Para observar sólo este proceso, se

2. MATERIALES Y MÉTODOS
25
estandariza la otra mitad de la celda usando un electrodo que tiene fases de composición
esencialmente constante y por ende, potencial constante: el electrodo de referencia (reference
electrode, RE). De esta manera, todos los cambios en la celda se pueden adjudicar al electrodo
de trabajo.
Cuando los dos electrodos de una celda se interconectan por un circuito externo, la reacción
electroquímica ocurrirá espontáneamente sólo si la energía libre asociada a la misma (らG) es
negativa. De no ser así, se debe suministrar energía aplicando una diferencia de potencial
entre los electrodos para poder observar un cambio. Se debe recordar que la relación entre la
energía libre involucrada en una reacción electroquímica global y la diferencia de potencial
entre el cátodo y el ánodo es:
( )C Ae eG nF E EΔ = − − (2.2)
donde n es el número de electrones involucrados en la reacción y F es la constante de Faraday
(9,65 x 104 C mol‐1).
Aún cuando la reacción sea termodinámicamente posible, la velocidad de la misma dependerá
de la velocidad de los procesos en cada electrodo que son, a su vez, funciones del potencial
aplicado.
El potencial adicional (más allá del requerimiento termodinámico) necesario para impulsar la
reacción a una dada velocidad se llama sobrepotencial (η). Además, para que los iones se
muevan a través del electrolito en la celda, se debe suministrar energía. Por lo tanto, el
potencial total de la celda, V, necesario para generar un cambio químico por electrólisis se
puede expresar como:
= − − − −C Ae e A CV E E η η iR (2.3)
donde R es la resistencia de la solución contenida entre los electrodos. Esta ecuación es muy
importante en electroquímica aplicada, ya que en todas las celdas los sobrepotenciales y el
término iR (caída óhmica) representan las ineficiencias energéticas que se deben minimizar.
Si se trabaja en condiciones en las que iR es pequeño se puede utilizar esta celda con dos
electrodos, pero cuando iR es grande, es preferible emplear una celda electroquímica de tres
electrodos. En este caso, el potencial del electrodo de trabajo se mide empleando un electrodo
de referencia y la corriente circula entre el primero y un contra‐electrodo (counter electrode,

2. MATERIALES Y MÉTODOS
26
CE, ó electrodo auxiliar). El dispositivo usado para medir la diferencia de potencial entre estos
últimos tiene una impedancia de entrada alta por lo que el potencial de referencia permanece
constante e igual a su valor de circuito abierto. Este arreglo de tres electrodos es el que se
utiliza en la mayoría de los experimentos electroquímicos [54, 211].
2.1.1.3 POTENCIOSTATO
El potenciostato es un dispositivo de retroalimentación negativa que aplica el potencial
requerido al electrodo de trabajo respecto de un electrodo de referencia. Cuando la caída de
potencial entre ambos se desvía de un valor pre‐seleccionado, aplica una diferencia de
potencial entre el electrodo de trabajo y el electrodo auxiliar, aumentando dicha caída hasta
que el potencial del electrodo de trabajo respecto del de referencia vuelve al valor deseado.
En la Figura 1 se muestra un esquema sencillo de un potenciostato. Consiste en un circuito
eléctrico que controla el potencial de la celda y varía la corriente suministrada en
concordancia. La medida de la corriente se realiza a través de los cambios en la caída de
voltaje sobre una resistencia (Rm en la Figura 1): una resistencia más alta resultará en una
disminución de la corriente y viceversa, de manera de mantener el potencial constante.
El amplificador CA es el responsable de mantener el voltaje entre el RE y el WE lo más cerca
posible del potencial de entrada de la fuente, Ei. Ajusta su salida para controlar
automáticamente la corriente de la celda de manera de satisfacer esta condición de igualdad
[54, 211].
Figura 1 Esquema sencillo de un potenciostato.
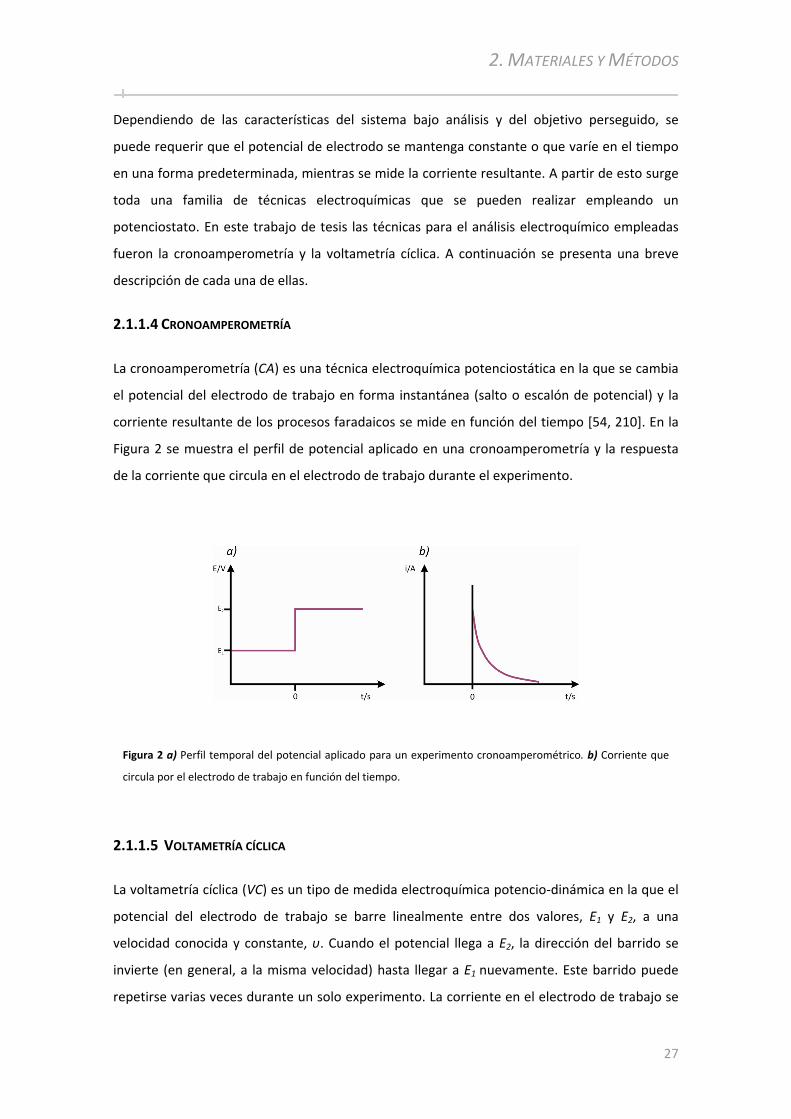
2. MATERIALES Y MÉTODOS
27
Dependiendo de las características del sistema bajo análisis y del objetivo perseguido, se
puede requerir que el potencial de electrodo se mantenga constante o que varíe en el tiempo
en una forma predeterminada, mientras se mide la corriente resultante. A partir de esto surge
toda una familia de técnicas electroquímicas que se pueden realizar empleando un
potenciostato. En este trabajo de tesis las técnicas para el análisis electroquímico empleadas
fueron la cronoamperometría y la voltametría cíclica. A continuación se presenta una breve
descripción de cada una de ellas.
2.1.1.4 CRONOAMPEROMETRÍA
La cronoamperometría (CA) es una técnica electroquímica potenciostática en la que se cambia
el potencial del electrodo de trabajo en forma instantánea (salto o escalón de potencial) y la
corriente resultante de los procesos faradaicos se mide en función del tiempo [54, 210]. En la
Figura 2 se muestra el perfil de potencial aplicado en una cronoamperometría y la respuesta
de la corriente que circula en el electrodo de trabajo durante el experimento.
2.1.1.5 VOLTAMETRÍA CÍCLICA
La voltametría cíclica (VC) es un tipo de medida electroquímica potencio‐dinámica en la que el
potencial del electrodo de trabajo se barre linealmente entre dos valores, E1 y E2, a una
velocidad conocida y constante, 仝. Cuando el potencial llega a E2, la dirección del barrido se
invierte (en general, a la misma velocidad) hasta llegar a E1 nuevamente. Este barrido puede
repetirse varias veces durante un solo experimento. La corriente en el electrodo de trabajo se
Figura 2 a) Perfil temporal del potencial aplicado para un experimento cronoamperométrico. b) Corriente que
circula por el electrodo de trabajo en función del tiempo.
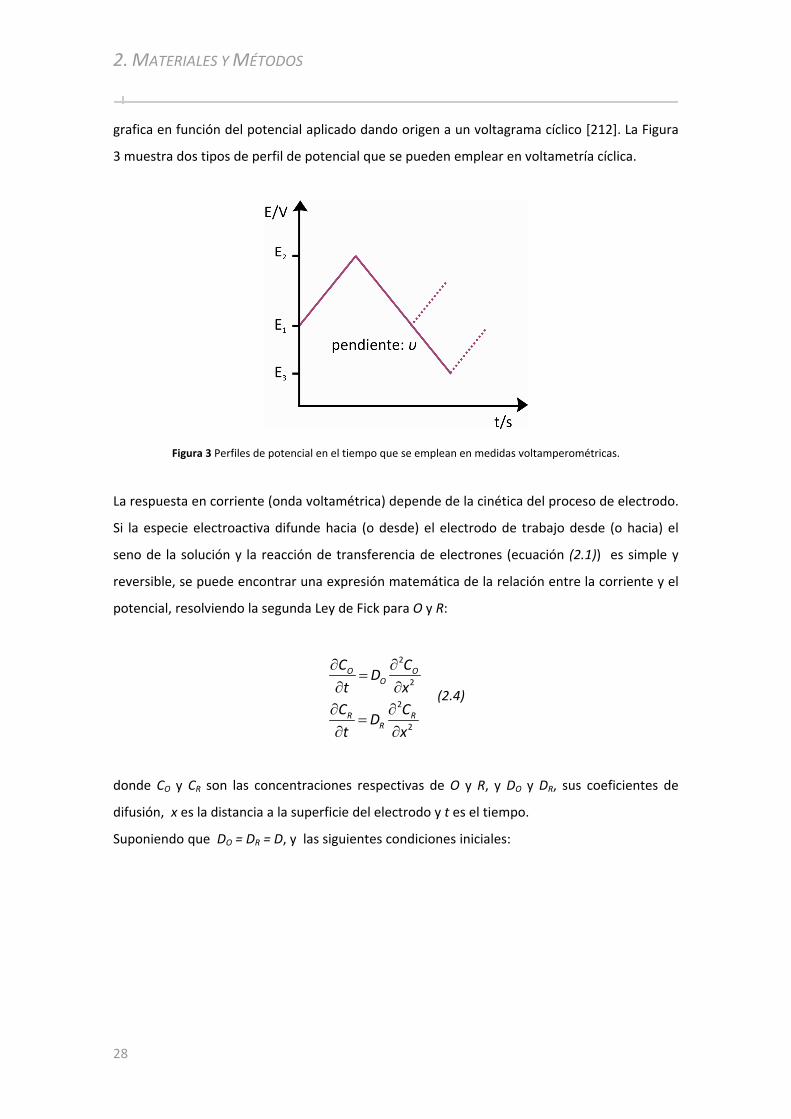
2. MATERIALES Y MÉTODOS
28
grafica en función del potencial aplicado dando origen a un voltagrama cíclico [212]. La Figura
3 muestra dos tipos de perfil de potencial que se pueden emplear en voltametría cíclica.
La respuesta en corriente (onda voltamétrica) depende de la cinética del proceso de electrodo.
Si la especie electroactiva difunde hacia (o desde) el electrodo de trabajo desde (o hacia) el
seno de la solución y la reacción de transferencia de electrones (ecuación (2.1)) es simple y
reversible, se puede encontrar una expresión matemática de la relación entre la corriente y el
potencial, resolviendo la segunda Ley de Fick para O y R:
O OO
R RR
C CD
t x
C CD
t x
∂ ∂=∂ ∂∂ ∂=∂ ∂
2
2
2
2
(2.4)
donde CO y CR son las concentraciones respectivas de O y R, y DO y DR, sus coeficientes de
difusión, x es la distancia a la superficie del electrodo y t es el tiempo.
Suponiendo que DO = DR = D, y las siguientes condiciones iniciales:
Figura 3 Perfiles de potencial en el tiempo que se emplean en medidas voltamperométricas.
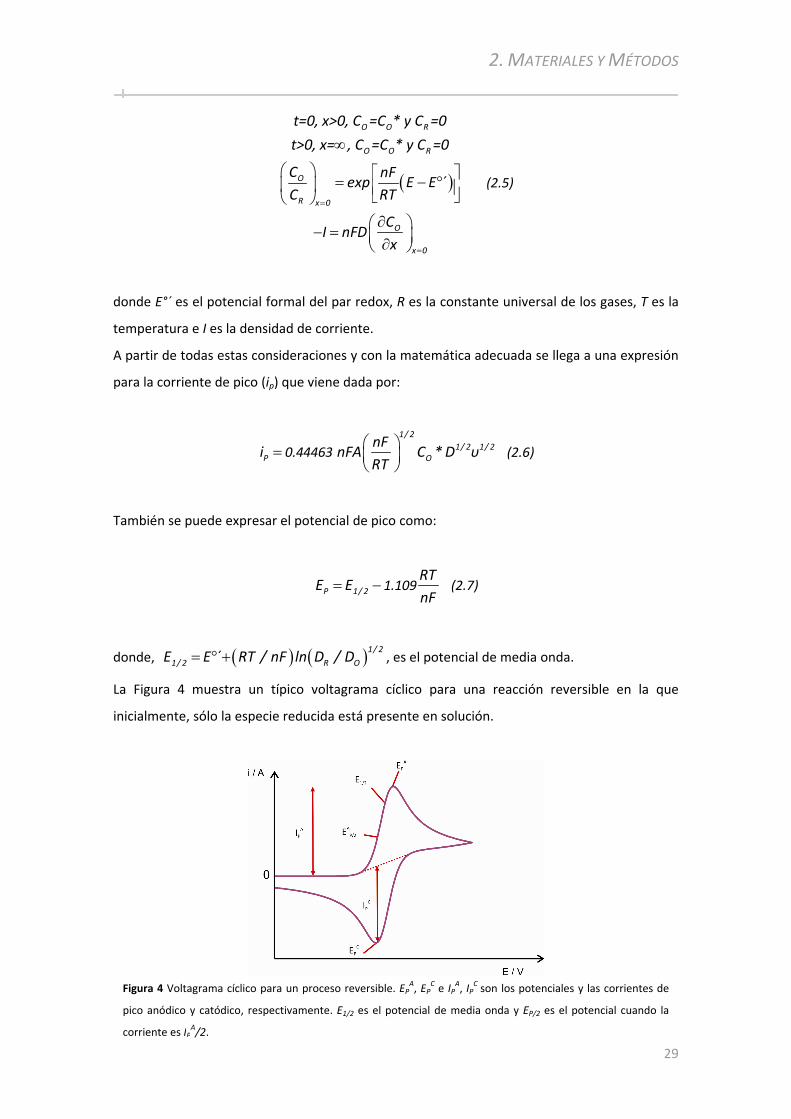
2. MATERIALES Y MÉTODOS
29
( )O O R
O O R
O
R x 0
O
x 0
t=0, x>0, C =C * y C =0
t>0, x= , C =C * y C =0
C nFexp E E ´
C RT
CI nFD
x
=
=
∞⎛ ⎞ ⎡ ⎤= − °⎜ ⎟ ⎢ ⎥⎣ ⎦⎝ ⎠
∂⎛ ⎞− = ⎜ ⎟∂⎝ ⎠
(2.5)
donde E°´ es el potencial formal del par redox, R es la constante universal de los gases, T es la
temperatura e I es la densidad de corriente.
A partir de todas estas consideraciones y con la matemática adecuada se llega a una expresión
para la corriente de pico (ip) que viene dada por:
⎛ ⎞= ⎜ ⎟⎝ ⎠1/ 2
1/ 2 1/ 2P O0.44463
nFi nFA C * D 仝
RT (2.6)
También se puede expresar el potencial de pico como:
= −P 1/ 2 1.109RT
E EnF
(2.7)
donde, ( ) ( )= ° + 1/ 2
1/ 2 R OE E ´ RT / nF ln D / D , es el potencial de media onda.
La Figura 4 muestra un típico voltagrama cíclico para una reacción reversible en la que
inicialmente, sólo la especie reducida está presente en solución.
Figura 4 Voltagrama cíclico para un proceso reversible. EPA, EP
C e IPA, IP
C son los potenciales y las corrientes de
pico anódico y catódico, respectivamente. E1/2 es el potencial de media onda y EP/2 es el potencial cuando la
corriente es IPA/2.

2. MATERIALES Y MÉTODOS
30
Por otro lado, cuando las especies electroactivas están adsorbidas sobre la superficie del
electrodo, la forma del voltagrama se modifica, ya que ahora los reactivos y productos no
difunden hacia y desde el electrodo para transferir la carga [54].
Si se considera que inicialmente, sólo la especie O está presente, la corriente que circula por el
sistema dependerá de la variación de su cubrimiento superficial. Esto se puede expresar de la
siguiente manera:
O R
* *O R
*O R O
よ (t ) よ (t ) i
t t nFA
t=0, よ y よ 0
よ (t ) よ (t ) よ
∂ ∂− = =∂ ∂=
+ = (2.8)
donde よO y よR son los excesos superficiales de O y R, respectivamente y よO* es el exceso
superficial inicial de O. Además, si se suponen isotermas de adsorción de Langmuir para ambas
especies y que la reacción es nernstiana, se puede llegar a expresiones para la corriente en
función del potencial aplicado (2.9), el potencial (2.10) y la corriente de pico (2.11):
( ) ( )( )( ) ( )( ){ }− °⎡ ⎤⎣ ⎦= + − °⎡ ⎤⎣ ⎦
2
2
1
*O O R
O R
仝Aよ b / b exp nF / RT E E ´nFi
RT b / b exp nF / RT E E ´ (2.9)
OP
R
bRTE E ´ ln
nF b
⎛ ⎞⎛ ⎞= ° − ⎜ ⎟⎜ ⎟⎝ ⎠ ⎝ ⎠ (2.10)
= 2 2
4*
P O
n Fi 仝Aよ
RT (2.11)
donde bO = βOよO y bR = βRよR con βi, los parámetros de equilibrio de la isoterma de adsorción
para cada especie.
La Figura 5 muestra una voltametría cíclica para la oxidación y reducción de las especies
adsorbidas. En este caso, la corriente es proporcional a la velocidad de barrido, 仝, a diferencia
del caso anterior en que es proporcional a 仝1/2. Además, el área bajo los picos de oxidación y
reducción representa la carga necesaria para la oxidación y reducción completas de la película
adsorbida sobre el electrodo (nFAよO*). Para una reacción nernstiana ideal bajo las condiciones
de una isoterma de Langmuir, los picos anódico y catódico tienen la misma posición y uno es
imagen especular del otro respecto del eje de potencial, cumpliéndose la siguiente relación:
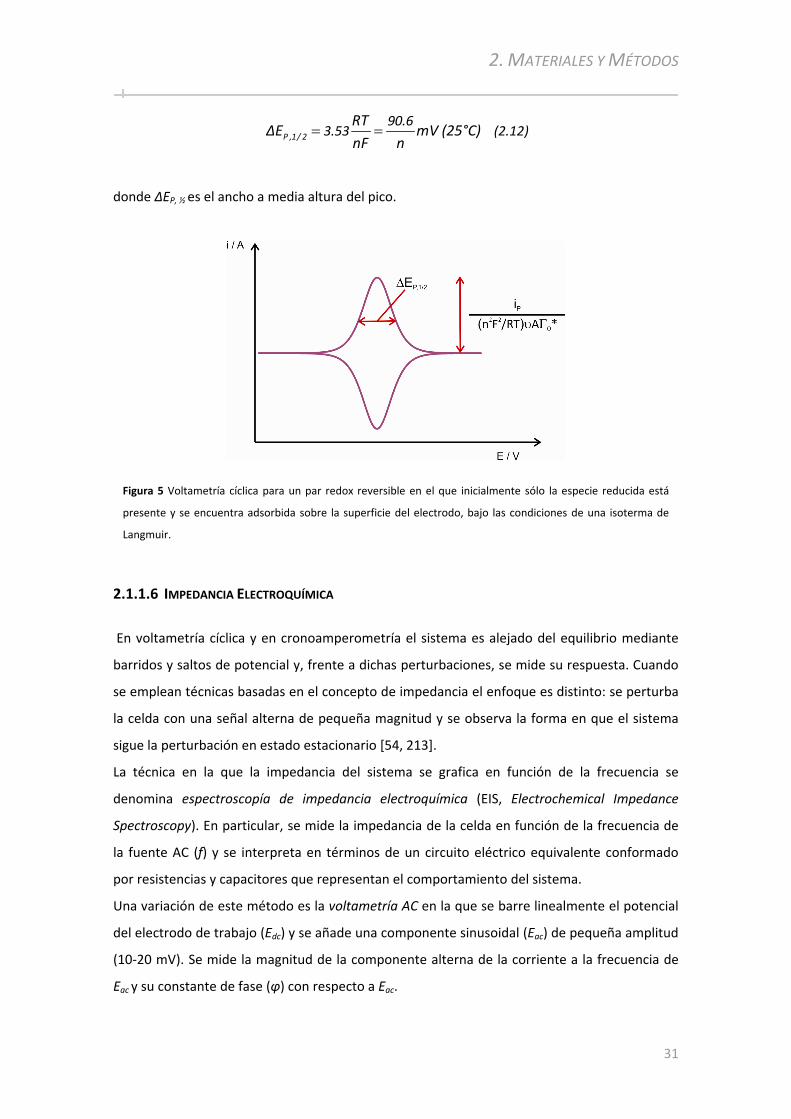
2. MATERIALES Y MÉTODOS
31
= =P ,1/ 2
90.63.53
RTらE mV (25°C)
nF n (2.12)
donde らEP, ½ es el ancho a media altura del pico.
2.1.1.6 IMPEDANCIA ELECTROQUÍMICA
En voltametría cíclica y en cronoamperometría el sistema es alejado del equilibrio mediante
barridos y saltos de potencial y, frente a dichas perturbaciones, se mide su respuesta. Cuando
se emplean técnicas basadas en el concepto de impedancia el enfoque es distinto: se perturba
la celda con una señal alterna de pequeña magnitud y se observa la forma en que el sistema
sigue la perturbación en estado estacionario [54, 213].
La técnica en la que la impedancia del sistema se grafica en función de la frecuencia se
denomina espectroscopía de impedancia electroquímica (EIS, Electrochemical Impedance
Spectroscopy). En particular, se mide la impedancia de la celda en función de la frecuencia de
la fuente AC (f) y se interpreta en términos de un circuito eléctrico equivalente conformado
por resistencias y capacitores que representan el comportamiento del sistema.
Una variación de este método es la voltametría AC en la que se barre linealmente el potencial
del electrodo de trabajo (Edc) y se añade una componente sinusoidal (Eac) de pequeña amplitud
(10‐20 mV). Se mide la magnitud de la componente alterna de la corriente a la frecuencia de
Eac y su constante de fase (φ) con respecto a Eac.
Figura 5 Voltametría cíclica para un par redox reversible en el que inicialmente sólo la especie reducida está
presente y se encuentra adsorbida sobre la superficie del electrodo, bajo las condiciones de una isoterma de
Langmuir.

2. MATERIALES Y MÉTODOS
32
Las dos técnicas emplean una señal de excitación de baja amplitud y se basan en el hecho de
que a bajos sobrepotenciales, la relación corriente‐potencial es prácticamente lineal. En un
sistema que se comporta linealmente, una excitación de frecuencia \ = 2ヽf genera una
corriente también de frecuencia \ (y sólo de frecuencia \). Por otro lado, un sistema que no
presenta una relación i‐E lineal, presenta una respuesta distorsionada que no es realmente
sinusoidal, sino que es una superposición de señales a frecuencias \, 2\, 3\, etc.
Por lo tanto, en un sistema que se comporta linealmente el potencial aplicado y la corriente
resultante se pueden expresar como:
( )( )o
o
E E sen \t
I I sen \t φ
== + (2.13)
donde Eo e Io son las amplitudes del potencial aplicado y la corriente resultante y φ es la
constante de fase entre la corriente y el potencial.
Los resultados que se obtienen en este tipo de análisis se analizan sobre la base de un circuito
electrónico modelo que mejor describe el sistema electroquímico bajo estudio. Por lo tanto, es
necesario conocer cómo se comportan los elementos de un circuito frente a perturbaciones
alternas como las descriptas.
Resistencia: Considerando que la ley de Ohm (E = IR) se cumple siempre, la corriente
resultante es I=(E/R)sen(\t), es decir que no se desfasa respecto del potencial (φ = 0).
Capacitor: Se cumple que q = CdE/dt y por lo tanto:
( ) ヽI \CEcos \t \CEsen \t
2⎛ ⎞⎜ ⎟⎝ ⎠= = + (2.14)
La corriente está desfasada en 90° respecto del potencial. En este caso para simplificar el
tratamiento matemático de estas expresiones se puede utilizar notación compleja. De esta
manera, se puede expresar I como:
jE I
\C−= (2.15)
donde j=√‐1.
Resistencia en serie con un capacitor (circuito RC en serie): Al aplicar un potencial alterno, éste
debe ser igual a la suma de las caídas en cada uno de los elementos:

2. MATERIALES Y MÉTODOS
33
R CE E E
jE I R
\CE IZ
⎛ ⎞⎜ ⎟⎝ ⎠
= += −=
(2.16)
donde Z es el vector impedancia que relaciona el potencial con la corriente. El mismo se puede
representar como:
Re ImZ(\) Z jZ= − (2.17)
donde ZRe y ZIm son las componentes real e imaginaria de la impedancia. En este caso, ZRe = R y
ZIm = 1/\C.
La impedancia es un tipo de resistencia generalizada y la tercera parte de la ecuación (2.16) es
una versión generalizada de la ley de Ohm. El ángulo de fase, φ, expresa el balance entre los
componentes resistivos y capacitivos del circuito en serie. Para una resistencia pura, φ = 0, y
para una capacidad pura, φ = ヽ/2. Para mezclas de ambos, φ toma valores intermedios.
La variación de la impedancia con la frecuencia se puede visualizar de dos formas. En un
gráfico de Bode se presenta la variación de log (|Z|) y de φ en función de log (\). Una forma
alternativa es emplear un gráfico de Nyquist en el que se muestra la variación de ZIm en función
de ZRe para diferentes valores de \. En la Figura 6 se presentan ejemplos de cada uno de estos
gráficos para un circuito RC en serie y uno en paralelo.
Figura 6 Gráficos de Nyquist y de Bode para un circuito RC a) en serie y b) en paralelo.

2. MATERIALES Y MÉTODOS
34
A veces es más útil analizar los circuitos en términos de admitancia, Y, que es la inversa de la
impedancia, 1/Z, y por ende representa un tipo de conductancia.
Una celda electroquímica puede ser considerada como un circuito equivalente de resistencias
y capacitores por los que circula la misma corriente con la misma amplitud y ángulo de fase
que en la celda real bajo una dada excitación. Un circuito frecuentemente empleado es el
circuito de Randles, que se muestra en la Figura 7 a). En él, la capacidad de la doble capa se
representa como un capacitor puro, Cdl. El proceso faradaico es considerado como una
impedancia general, Zf colocada en paralelo con la capacidad de la doble capa. Finalmente, la
resistencia de la solución, RS, está en serie con el conjunto mencionado, ya que toda la
corriente debe pasar la misma.
Cuando se tiene una celda en la que el electrodo de trabajo tiene una película electroactiva
adsorbida, la impedancia faradaica se puede considerar como una resistencia y un capacitor en
serie. La primera representa la transferencia de carga, RCT, y el segundo representa la pseudo‐
capacidad, Cads, de la película adsorbida sobre el electrodo de trabajo. Ambos elementos no
son ideales, a diferencia de RS y Cdl, ya que dependen de la frecuencia. El gráfico de Nyquist
que se obtiene con este tipo de circuito se muestra en la Figura 7 b).
Las expresiones para Cads y RCT son:
2
ads
CT 2ET
F AよC
4RT2RT
RF Aよk
== (2.18)
Figura 7 a) Circuito equivalente de una celda de electroquímica (Circuito de Randles) b) Gráfico de Nyquist para
dicho circuito.

2. MATERIALES Y MÉTODOS
35
donde A es el área del electrodo, よ es el cubrimiento superficial de la especie electroactiva y kET
es la constante cinética del proceso de transferencia electrónica. A partir de estas ecuaciones
se tiene:
ETCT ads
1k
2R C= (2.19)
Por lo tanto, se pueden obtener los valores de よ y kET a partir de estudios de estudios de
espectroscopía de impedancia electroquímica.
2.1.2 TÉCNICAS ESPECTROSCÓPICAS
2.1.2.1 ESPECTROSCOPÍA INFRARROJA POR ABSORCIÓN‐REFLEXIÓN
La espectroscopía infrarroja por absorción‐reflexión (IRRAS, Infrared Reflection‐Absorption
Spectroscopy) es una herramienta poderosa en el estudio de materiales ya que, al involucrar la
absorción de radiación infrarroja (IR) debida a vibraciones de moléculas y átomos vecinos,
brinda información estructural y muy valiosa. En particular, se puede utilizar para caracterizar
las estructuras de monocapas sobre superficies [214‐216].
Un haz de luz IR incidente sobre una superficie pulida a espejo puede separarse en dos
componentes respecto del plano de incidencia: p‐polarizada y s‐polarizada. El campo eléctrico
asociado a la componente p oscila en dirección paralela al plano de incidencia, mientras que
en la componente s, lo hace en la dirección perpendicular. La Figura 8 muestra los vectores
eléctricos, incidentes y reflejados, para cada componente.
Figura 8 Ilustración esquemática de la descomposición de la radiación IR en sus componentes p‐ y s‐
polarizadas. Ep y Es son los vectores eléctricos incidentes y Ep´ y Es´ son los vectores reflejados de las
componente p y s, respectivamente.

2. MATERIALES Y MÉTODOS
36
El cambio de fase de la componente s no exhibe dependencia significativa con el ángulo de
incidencia θ (Figura 9 a)), y es aproximadamente 180° para todos los ángulos de incidencia.
Esto resulta en una amplitud neta nula de la radiación s‐polarizada cerca de la superficie (0 a
10‐2 ゜) (Figura 9 b)). En contraste, el desfasaje de la componente p depende fuertemente del
ángulo de incidencia. La amplitud de dicha componente pasa por un máximo cuando el ángulo
θ es de 88°. En este caso, los campos incidente y reflejado asociados a esta radiación (Ep y Ep´,
respectivamente), se suman dando como resultante un campo de casi el doble de amplitud
que la radiación incidente.
Cuando se quimisorbe una especie a una superficie conductora, existe cierta transferencia de
carga entre ambas. En el caso de sustratos metálicos, la distribución de carga neta inducirá una
distribución de carga imagen (Figura 10).
Si la molécula tiene un momento dipolar, se generará un dipolo imagen en la superficie. Estos
dipolos, a su vez, pueden descomponerse vectorialmente en las direcciones perpendicular y
paralela al plano de la muestra. En este caso, también se observa que, mientras la magnitud de
la componente del dipolo perpendicular a la superficie se duplica por efecto de la carga
imagen, la componente paralela desaparece. Como resultado de las interacciones
constructivas entre el campo del oscilador y las ondas incidentes p‐polarizadas, el aumento de
las intensidades de absorción debería esperarse cuando la orientación de la propagación de la
onda incidente es cercana a la del ángulo rasante.
Figura 9 Dependencia angular a) del cambio de fase (~P y ~S) y b) de los factores de absorción (AP y AS) de las
componentes p y s, respectivamente.

2. MATERIALES Y MÉTODOS
37
Por lo tanto, las vibraciones activas que pueden ser detectadas con esta técnica deben tener
una componente del momento dipolar de transición perpendicular a la superficie de la
muestra. Esto significa que una dada vibración puede no ser observada en un espectro IR de
absorción‐reflexión, aunque la vibración sea activa en el infrarrojo. Estos resultados dan lugar
a la llamada “regla de selección superficial” que es ampliamente utilizada en el estudio de
orientación molecular en películas delgadas o monocapas depositadas sobre metales [217].
Debido a que la cantidad de luz absorbida en esta configuración es pequeña se deben
promediar las señales colectando muchos espectros para aumentar la relación señal‐ruido a
niveles aceptables. El espectro resultante para un sustrato modificado se divide por un
espectro blanco adecuado (tomado sobre un sustrato limpio, por ejemplo) para obtener un
espectro de pseudo‐absorbancia:
= −O
RA(d) 1
R (2.20)
donde R y Ro son las intensidades de reflexión de la muestra y de la referencia,
respectivamente.
2.1.2.2 ESPECTROSCOPÍA INFRARROJA POR ABSORCIÓN‐REFLEXIÓN CON MODULACIÓN DE LA
POLARIZACIÓN
Cuando se analizan capas orgánicas gruesas (espesor > 400 Å) depositadas sobre una
superficie, la técnica IRRAS descripta en la sección anterior funciona muy bien, generando
espectros con bandas intensas. Sin embargo, cuando se tienen capas ultra‐delgadas, en el
orden de una monocapa molecular, esta técnica se acerca a su límite de detección y son
Figura 10 Esquema de la distribución de las cargas real e imagen inducida para a) una molécula con una carga
permanente monopolar y b) una molécula con un momento dipolar dinámico que se puede descomponer
vectorialmente.

2. MATERIALES Y MÉTODOS
38
necesarios tiempos largos de adquisición para mejorar la relación señal/ruido. Durante el
tiempo de medida distintas inestabilidades instrumentales pueden afectar notablemente al
espectro final. Además, la absorción del entorno de la muestra (H2O y CO2 atmosféricos)
también es detectada, y cambios muy pequeños en las condiciones ambientales pueden
dificultar mucho la interpretación de los espectros.
En la espectroscopía infrarroja por absorción‐reflexión con modulación de la polarización
(Polarization Modulation Infrared Reflection Absorption Spectroscopy, PM‐IRRAS), la
polarización del haz incidente se modula entre las dos direcciones ya mencionadas en la
sección anterior: perpendicular (polarización s) y paralela (polarización p) respecto del plano
de incidencia. En la superficie de un metal, el campo eléctrico de la luz s‐polarizada
prácticamente desaparece, mientras que el de la luz p‐polarizada se refuerza. Por lo tanto la
primera componente de la señal modulada es insensible a la presencia de una película de
moléculas sobre la superficie y se puede emplear para obtener el espectro referencia,
mientras que la otra componente se puede utilizar para obtener el espectro de la película. La
modulación entre las polarizaciones s‐ y p‐ se lleva a cabo a una alta frecuencia (50 kHz), y el
espectro de absorción‐reflexión ΔR/ R se obtiene como:
( )S P
S P
R RR
R R R /
−= +Δ
2 (2.21)
donde RS y RP representan las reflectividades de las componentes s‐ y p‐ del haz infrarrojo
modulado en la muestra. Cuando el rendimiento del arreglo experimental es igual para ambos
tipos de polarización, se eliminan aquellos modos vibracionales que son detectados con ambas
componentes. Éste es el caso de las señales correspondientes al H2O y CO2 ambientales.
En la Figura 11 se muestra un esquema del arreglo experimental empleado en esta técnica.

2. MATERIALES Y MÉTODOS
39
Éste consiste de un polarizador estático P y un modulador fotoelástico (PEM). La radiación
incidente puede representarse por dos componentes ortogonales, I0V e I0H, que denotan las
intensidades de las componentes vertical y horizontal de dicha radiación respecto del plano
del banco óptico. Cuando el haz atraviesa el polarizador P, la luz se polariza linealmente en la
dirección paralela al banco óptico, indicada por Il. El eje óptico del modulador está montado
formando un ángulo de 45° con respecto al plano del banco óptico y por ende, el haz
linealmente polarizado a su vez puede descomponerse en dos direcciones perpendiculares
entre sí, Ilx e Ily, definidas según el eje principal del PEM [218].
El PEM consiste de un transductor piezoeléctrico unido a un cristal de ZnSe. El piezo convierte
una onda de potencial en una onda mecánica (acústica) que comprime o expande el cristal.
Este movimiento cambia el índice de refracción en la dirección x e impone un retardo (o una
aceleración) en la componente Ilx del haz incidente linealmente polarizado.
La componente Ily no es afectada y por lo tanto, las componentes se desfasan entre sí en su
paso por el PEM. El retardo impuesto por el PEM depende del potencial aplicado al mismo y de
la longitud de onda del haz incidente. En general, este potencial se elige tal que para una
longitud de onda dada (゜o), la polarización de la radiación cambia y cada 90° de un período, IP o
IS incide en el detector. De esta manera se pueden obtener las señales y calcular el espectro
R / RΔ .
El haz resulta elíptica o circularmente polarizado para longitudes de onda diferentes de ゜o. En
consecuencia, se puede optimizar un experimento para una sola longitud de onda (゜o) y ésta se
elige para que esté en el centro de la región espectral que se desea analizar.
Figura 11 Diagrama simplificado del arreglo experimental empleado en PM‐IRRAS y las orientaciones de los
vectores de campo eléctrico de la radiación electromagnética.

2. MATERIALES Y MÉTODOS
40
La señal en el detector es la resultante de la modulación impuesta por el PEM sobre la señal de
la fuente modulada por el interferómetro de Michelson del espectrógrafo infrarrojo por
transformada de Fourier (FT‐IR). La señal debe demodularse empleando un amplificador Lock‐
in o un demodulador sincrónico para separar ambas contribuciones.
Se emplea la técnica desarrollada por Corn [219] (demodulador sincrónico) y los espectros
obtenidas fueron corregidas por la respuesta del PEM empleando el método descripto por
Frey et al. En la Figura 12 se muestra un espectro PM‐IRRAS de una superficie de oro
modificada con ácido 16‐mercaptohexadecanoico antes (a) y después (b) de la corrección de la
línea de base.
2.1.2.3 ESPECTROSCOPÍA DE DISPERSIÓN RAMAN RESONANTE
La espectroscopía Raman es una técnica espectroscópica basada en la dispersión inelástica de
luz monocromática, en general de una fuente láser de longitud de onda en el visible o en el
infrarrojo cercano. Durante este proceso se intercambia energía entre un fotón y una molécula
que es excitada a un nivel de energía virtual producto de esta interacción. Si la molécula relaja
al estado vibracional inicial, los fotones reemitidos tienen la misma energía que traían
originalmente. Este proceso se denomina dispersión Raylegh. Sin embargo, si la relajación es a
un estado vibracional de mayor o menor energía que la del estado de partida, entonces este
proceso se conoce como dispersión Raman y resulta en una diferencia de energía entre los
fotones incidentes y los reemitidos que es igual a la energía necesaria para excitar a las
Figura 12 a) Espectro PM‐IRRAS de una superficie de oro modificada con ácido 16‐mercaptohexadecanoico
(línea azul) tomado con ゜o = 2900 cm‐1. Se muestra la línea de base obtenida a partir de la respuesta del PEM
(línea roja). b) Espectro resultante cuando se resta la línea de base al espectro mostrado en a).

2. MATERIALES Y MÉTODOS
41
moléculas vibracionalmente [220]. Por lo tanto, este tipo de interacción con la luz permite
observar las vibraciones moleculares.
Desde el comienzo, el trabajo teórico y experimental se centró principalmente en los
fundamentos de la dispersión inelástica y su aplicación en la compresión de la estructura
molecular.
En la Figura 13 se muestran esquemáticamente las transiciones que originan la dispersión
Raman y la probabilidad que tiene cada una de ocurrir. Esta diferencia de probabilidades
influye en la intensidad de cada tipo de línea en un espectro Raman.
En general, la diferencia de energía entre la luz incidente y la luz dispersada se expresa en
unidades de número de onda ( ら` ). Un espectro Raman representa la intensidad de la luz
reemitida en función de ら` . Cuando la energía final es menor a la original, ら` es positiva y
las señales que aparecen se denominan líneas Stokes. Cuando ocurre lo contrario ( ら` < 0), las
líneas del espectro se llaman anti‐Stokes y se deben a la relajación de las moléculas a un nivel
vibracional más bajo que el nivel inicial.
No todas las transiciones generan señal en un espectro Raman. Esto se debe a las reglas de
selección asociadas a este fenómeno, que establecen que sólo aquellas transiciones que
provocan un cambio en la polarizabilidad de la molécula serán observadas. Los espectros
Raman brindan información complementaria a la obtenida por espectroscopía infrarroja.
La espectroscopía Raman se realiza convencionalmente empleando láseres verdes, rojos o del
infrarrojo cercano. Las longitudes de onda están por debajo de las primeras transiciones
electrónicas de la mayoría de las moléculas. Por otro lado, si se ajusta la energía del láser
Figura 13 Esquema que ilustra las transiciones que originan los fenómenos de dispersión Raylegh y dispersión
Stokes y anti‐Stokes. Los espesores de las flechas indican la probabilidad que tiene cada tipo de dispersión de
ocurrir y que determinará la intensidad de cada señal en un espectro Raman.

2. MATERIALES Y MÉTODOS
42
incidente de manera que coincida con la de alguna transición electrónica de la molécula bajo
estudio, los modos vibracionales asociados pueden aumentar la intensidad de sus señales
Raman en un factor de 102‐104 permitiendo trabajar con concentraciones de analito muy bajas
(hasta 10‐8 M). Ésta es la espectroscopía Raman resonante [221].
Este aspecto de la espectroscopía Raman es muy útil para analizar macromoléculas biológicas
con cromóforos en su estructura [222‐225]. En este caso se puede elegir la frecuencia de
excitación de modo que coincida con la banda de absorción de dicho cromóforo y, de esta
manera, observar sólo las vibraciones localizadas dentro del mismo. Esta selectividad es muy
importante a la hora de analizar la relación teórica entre las transiciones electrónicas y las
vibraciones que se realzan por resonancia [226].
La principal desventaja de emplear esta técnica en modo resonante consiste en el riesgo de
observar fluorescencia o fotodegradación de la muestra.
2.1.2.4 ESPECTROSCOPÍA FOTOELECTRÓNICA DE RAYOS X
La espectroscopía fotoelectrónica de rayos X (XPS, X‐Ray Photoelectronic Spectroscopy) es una
técnica de análisis químico superficial que utiliza la foto‐ionización y el análisis de la energía de
los electrones fotoemitidos para estudiar la composición y estado electrónico de la superficie
de una región del sistema bajo estudio [211]. Utiliza fuentes monocromáticas de radiación, en
general, la línea Al Kα (h` =1486.6 eV) o Mg Kα (h` =1253.6 eV). Cuando un fotón es absorbido
por un átomo presente en la muestra, se produce la ionización y la emisión de un electrón de
la capa interna. A partir de este fenómeno se puede medir la distribución de energía cinética
de los electrones fotoemitidos. Para ello, se elige un analizador hemiesférico concéntrico que
usa un campo eléctrico aplicado entre dos superficies hemiesféricas para dispersar los
electrones de acuerdo con su energía cinética. De esta manera se cuentan los fotoelectrones
colectados en función de su energía cinética en un intervalo definido de tiempo. Esto resulta
en un espectro de cuentas electrónicas en función de energía cinética (eV).
Para el proceso global de foto‐ionización:
++ → +A h` A e (2.22)
la energía del fotón incidente está dada por la relación de Einstein:
=E h` (2.23)

2. MATERIALES Y MÉTODOS
43
donde h es la constante de Planck y ` es la frecuencia de la radiación. El principio de
conservación de la energía requiere que se cumpla la siguiente relación:
E(A) h` E(A ) E(e )+ −+ = + (2.24)
donde E(A) y E(A+) denotan las energías correspondientes a un átomo en la muestra antes y
después de ser ionizado por la absorción de un fotón de energía h` y E(e‐) denota la energía
del electrón fotoemitido. Como la energía electrónica viene dada por la energía cinética
únicamente (Ecin), la ecuación (2.24) puede reordenarse para obtener una expresión de dicha
energía:
+⎡ ⎤= − − = −⎣ ⎦cin uniónE h` E(A ) E(A) h` E (2.25)
donde Eunión es la llamada energía de unión que corresponde a la diferencia de energía entre
los átomos ionizados y neutros. Es una medida directa de la energía requerida para remover
un electrón desde su nivel inicial en el átomo y llevarlo al vacío. En este punto debe
considerarse que, cuando se trabaja con sólidos, la energía de unión se mide respecto al nivel
de Fermi correspondiente, por lo que la ecuación (2.25) debe corregirse para considerar la
función trabajo (Φ).
Cada elemento tiene una energía de unión característica asociada a cada orbital de su capa
interna y por ende, cada elemento genera un grupo de picos propios en el espectro
fotoelectrónico, cuya posición está determinada por la energía del fotón incidente y por la
energía de unión. Por lo tanto, la observación de picos a una determinada energía indica la
presencia de un elemento específico cuya concentración, dentro de la zona analizada, está
directamente relacionada con la intensidad de los mismos. Esto convierte a esta
espectroscopía en una técnica de análisis cuantitativo de composición superficial.
Las posiciones y formas de los picos en un espectro XPS pueden también analizarse en mayor
detalle para determinar el estado químico de los elementos constituyentes del material,
incluyendo estado de oxidación, carga parcial y coordinación.
Esta técnica es ampliamente aplicada a todo tipo de sólidos, incluyendo metales [227‐229],
cerámicos [230‐233] y semiconductores [234‐236]. En general, es un método de análisis
superficial no destructivo cuya profundidad nominal de análisis es de 10 nm (10 a 100
monocapas atómicas) con una resolución espacial del orden de 1 a 5 nm.

2. MATERIALES Y MÉTODOS
44
Se pueden analizar películas delgadas con XPS para determinar su espesor. Los métodos típicos
de análisis se basan en medir la composición en función de la profundidad ó del ángulo de
emisión electrónica.
En general se requiere de la aplicación de rutinas de ajuste para distinguir picos de distintos
estados de oxidación o de distintos elementos que pueden solaparse. El uso de patrones con
composición conocida puede ayudar en la asignación de señales.
La principal limitación de XPS es la necesidad de condiciones de ultra alto vacío durante el
análisis. Esto en general limita el tipo de muestras a las que tienen una baja presión de vapor a
temperatura ambiente y un tamaño adecuado para su introducción en la cámara de vacío.
Otra limitación son los efectos de carga en muestras aislantes y semiconductoras. Esto puede
corregirse durante o después de la adquisición de datos, aunque la elección del grado de
corrección no es sencilla.
2.1.3 TÉCNICAS DE MICROSCOPÍA
2.1.3.1 MICROSCOPÍA DE EFECTO TÚNEL
La microscopía de efecto túnel (STM, Scanning Tunneling Microscopy) permite analizar
superficies conductoras a escala atómica y los fenómenos que tienen lugar en ellas. El
principio de esta técnica es muy simple y se basa en acercar a la superficie de una muestra una
punta metálica aguda, denominada “tip”, de manera tal que su distancia a la superficie sea del
orden de unos pocos angstroms. Los electrones pueden moverse dentro del tip y la superficie
respectivamente, pero su propagación a través del espacio está prohibida en la teoría clásica,
ya que su energía total es menor que la energía potencial. Sin embargo, de acuerdo con la
mecánica cuántica, los electrones pueden atravesar la barrera de energía potencial por efecto
túnel dando origen a una corriente túnel medible cuando se aplica una diferencia de potencial
(llamado Ebias, potencial de polarización) entre la punta y la superficie. La probabilidad de
tuneleo es proporcional al solapamiento entre las nubes electrónicas (funciones de onda, /tip
y /S) de la punta y la superficie (ver Figura 14). Este solapamiento disminuye
exponencialmente con la distancia entre ambos. Por lo tanto, la relación entre la corriente
túnel (I) y la distancia punta‐superficie (d) se puede expresar como:
2mφI exp 2 d
ћ
⎛ ⎞∝ −⎜ ⎟⎜ ⎟⎝ ⎠ (2.26)

2. MATERIALES Y MÉTODOS
45
donde m es la masa electrónica, ħ es la constante de Planck y φ es la función trabajo o la altura
de la barrera de tuneleo.
La caída exponencial de la corriente túnel con la distancia resulta en que sólo el átomo de la
punta más cercano a la superficie domina la corriente medida y esto determina la capacidad
de obtener imágenes con resolución atómica por medio de esta técnica. Esto se logra
barriendo la punta sobre la superficie de la muestra y detectando la corriente túnel para
generar un mapa de la misma [237].
La Figura 15 muestra un esquema sencillo del funcionamiento de un microscopio de efecto
túnel. La punta está mecánicamente conectada a un elemento piezoeléctrico que permite
controlar la distancia entre ella y el sustrato ajustando el potencial aplicado al mismo.
Mientras que el sistema de retroalimentación está funcionando, se utilizan otras dos partes del
piezoeléctrico para mover la punta en las direcciones X e Y, paralelas a la superficie. De esta
manera se barre la superficie línea por línea y se obtiene una imagen de la misma.
Figura 14 Efecto túnel entre un sustrato conductor y una punta de STM. EF es el nivel de Fermi, e es la carga del
electrón, Ebias es el potencial de polarización aplicado entre punta y sustrato y /S y /tip son a las funciones de
onda del sustrato y la punta, respectivamente

2. MATERIALES Y MÉTODOS
46
Existen dos formas de operar el microscopio: modo de corriente constante y modo de altura
constante. En el primero, se fija el potencial del piezoeléctrico para establecer un valor dado
de corriente, y la distancia se regula con un circuito electrónico de retroalimentación que mide
continuamente la desviación de la corriente respecto del valor prefijado y aleja la punta de la
superficie cuando la misma es muy alta o lo acerca en caso contrario. En el modo de altura
constante, la posición vertical de la punta no se modifica y la corriente en función del
desplazamiento lateral la misma representa la imagen de la superficie, es decir que en este
caso no se usa la retroalimentación electrónica. Este modo sólo es apropiado para superficies
atómicamente planas, ya que de lo contrario, es inevitable que la punta se estrelle contra la
muestra. Sin embargo, posee la ventaja de permitir la utilización de frecuencias de barrido
mayores que el modo de corriente constante.
Es importante tener en cuenta que las imágenes obtenidas con esta técnica no siempre
pueden interpretarse como mapas topográficos de la superficie ya que la corriente túnel
depende de la densidad de estados de la superficie y de la punta. Esto puede expresarse de la
siguiente manera:
( ) ( )biaseV
t S F bias FI ヾ E eE 0 x ヾT E 0 d0≈ − + +∫0
(2.27)
donde 0 es la energía y ヾS y ヾT son las densidades de estados (DOS, density of states) del
sustrato y de la punta, respectivamente.
La ventaja del STM sobre otras microscopías consiste en que su operación no requiere de
condiciones ambientales especiales. Se puede realizar en el vacío, a altas presiones
Figura 15 Esquema del funcionamiento de un microscopio de efecto túnel (STM).

2. MATERIALES Y MÉTODOS
47
(incluyendo aire) y en líquidos. La elección del ambiente está determinada principalmente por
los requerimientos de la muestra.
La adquisición de imágenes es relativamente simple con los instrumentos modernos. La mayor
parte del tiempo se utiliza en la preparación de la punta y de la muestra.
La microscopía de efecto túnel fue desarrollada a comienzos de la década de 1980 y desde
entonces tanto el instrumental disponible como sus aplicaciones han tenido un gran desarrollo
[211].
2.1.3.2 ESPECTROSCOPÍA DE EFECTO TÚNEL
Los estados electrónicos de una muestra pueden ser caracterizados con un microscopio de
efecto túnel utilizando una técnica que se conoce como Espectroscopía de Efecto Túnel (STS,
Scanning Tunneling Spectroscopy).
De acuerdo a la ecuación (2.27), la corriente túnel resulta de la convolución de las densidades
de estados del sustrato y la punta. Para poder determinar la densidad de estados de la
muestra se debe conocer la correspondiente a la punta, y esto no siempre es posible. Sin
embargo, para un dado intervalo de potenciales, la DOS de la punta puede depender poco de
la energía, 0. Por lo tanto, la ecuación (2.27) se convierte en:
( )tS F bias
dIヾ E eE 0
dV= − + (2.28)
Los experimentos de STS se realizan dejando la punta fija (se apaga el mecanismo de
retroalimentación del microscopio) sobre una región de interés de la muestra y mientras se
mide la corriente túnel, se barre la diferencia de potencial aplicada entre la punta y el sustrato
(Ebias).
Debido a que la corriente túnel es muy sensible a la separación entre la punta y el sustrato, los
datos espectroscópicos medidos, en general, se normalizan por It/V y se expresan como [(V/It)
(dIt/dV)] = [(d ln(It)/d ln(V)].
2.1.3.3 ESPECTROSCOPÍA DE EFECTO TÚNEL CON CONTROL ELECTROQUÍMICO
Esta técnica constituye una aplicación particular de la espectroscopía de efecto túnel. En este
caso se agrega un electrodo de referencia y un contra‐electrodo, y junto con la punta y el
sustrato se sumergen en una solución electrolítica. Por lo tanto se tiene una celda de cuatro
electrodos en la que hay dos electrodos de trabajo: la punta y el sustrato. El potencial de cada

2. MATERIALES Y MÉTODOS
48
uno se aplica con respecto al electrodo de referencia, mientras que el contra‐electrodo mide
los procesos electroquímicos [167].
2.2 EQUIPAMIENTO UTILIZADO
2.2.1 POTENCIOSTATO
Las medidas electroquímicas fueron realizadas con un sistema Autolab PGSTAT V30 (Eco
Chemie, Ultrech, Netherlands) controlado por el programa General Purpose Electrochemical
Software (GPES) y, en algunos casos, el programa NOVA.
Las voltametrías cíclicas obtenidas con altas velocidades de barrido se obtuvieron haciendo
uso de los módulos ADC750 (conversor analógico‐digital de alta velocidad) y Scangen
(generación de rampas de potencial lineales). Todos los experimentos fueron realizados a
temperatura ambiente (20 ± 2 °C).
Las medidas de impedancia electroquímica se realizaron empleando un módulo analizador de
respuesta de frecuencias (FRA 2) aplicando señales de excitación en el intervalo de frecuencias
1‐106 Hz y de una amplitud de 10 mV. En este caso se empleó el programa FRA para controlar
el potenciostato.
2.2.2 ELECTRODOS Y CELDAS
En los experimentos electroquímicos se utilizaron dos tipos de celda, ambas equipadas con un
electrodo de referencia y un electrodo auxiliar: 1) celda de Teflon fabricada a medida que
permite colocar el electrodo de trabajo en posición vertical y aislar el área (0.28 cm2) que se
desea analizar con un ‘o’‐ring, 2) celda de vidrio que permite sumergir el electrodo de trabajo.
En particular, esta última se empleó para la limpieza electroquímica de los electrodos de
trabajo.
El electrodo de referencia empleado fue un electrodo Ag/AgCl, preparado con un alambre de
Ag anodizado en HCl y sumergido en KCl 3 M contenido en un tubo delgado conectado con la
solución a través de una frita de vidrio. Todos los potenciales de electrodo en este trabajo son
referidos al mismo. Por otro lado, se empleó un electrodo de malla de platino de alta área
superficial como electrodo auxiliar.
Se utilizaron sustratos de oro como electrodos de trabajo, que fueron fabricados en el
laboratorio cubriendo sustratos de silicio (100) con una capa de adhesión de titanio (20 nm) y
paladio (20 nm) sobre la que se depositó una capa de oro (200 nm) por evaporación a presión

2. MATERIALES Y MÉTODOS
49
reducida (< 1 x 10‐8 bar). Para ello los metales fueron evaporados térmicamente con una
evaporadora Edwards Auto 306.
Finalmente, en los experimentos de impedancia electroquímica se emplearon electrodos de
gota de oro obtenidos por fusión de alambres de oro de 0.5 mm y 0.15 mm de diámetro. Éstos
fueron limpiados con solución piranha (H2SO4 + H2O2 (3:1)) inmediatamente antes de ser
utilizados.
2.2.3 ESPECTRÓMETRO INFRARROJO CON TRANSFORMADA DE FOURIER
Para medir los espectros infrarrojos por absorción‐reflexión (IRRAS) se utilizó un
espectrómetro FT‐IR Nicolet Magna 560 equipado con un detector criogénico MCT‐A
(refrigerado con N2 líquido), un polarizador de seleniuro de zinc (Nicolet, Spectratech) y un
equipo de purga Whatman 75‐52. Los espectros de las superficies de oro modificadas fueron
obtenidos usando un arreglo experimental diseñado para trabajar en modo absorción‐
reflexión con un ángulo de incidencia de 80°. Una superficie limpia de oro fue usada como
referencia, y se colectaron espectros con radiación p‐ y s‐polarizada, con una resolución de
4 cm‐1 acumulando 200 interferogramas para obtener espectros con una relación señal‐ruido
aceptable.
En los casos en que se empleó modulación de la polarización, se utilizó un espectrómetro
Thermo Nicolet 8700 (Nicolet, Madison, WI) equipado con un banco óptico externo (TOM box)
equipado con un detector MCT‐A (Nicolet), un modulador fotoelástico (PEM, PM‐90 con una
cabeza óptica II/Zs50 ZnSe 50 kHz, Hinds Instruments, Hillsboro, OR), espejos, polarizador y
lente de enfoque sobre el detector de ZnSe. Un demodulador (Syncronous Samplig
Demodulator, SSD, GWC Instruments, Madison, WI) fue usado para separar las señales
provenientes del detector y alimentarlas a la consola de adquisición del espectrógrafo. Los
espectros IR fueron adquiridos con el PEM programado para un retardo de media onda a 2900
cm‐1, para analizar la región del estiramiento del enlace C‐H, a 1450 cm‐1 para analizar los
modos de deformación de los enlaces C‐H y la región aromática y a 1200 cm‐1 para analizar los
modos de estiramiento de los enlaces C‐F.
Se trabajó con un ángulo de incidencia de 80° (espectros en aire) y se adquirieron 1000
interferogramas con una resolución de 4 cm‐1.

2. MATERIALES Y MÉTODOS
50
2.2.4 ESPECTRÓMETRO DE FOTOEMISIÓN DE RAYOS X
Los sistemas Au/PhCOOH, Au/PhF4COOH y Au/PhF4COOH/Os fueron analizados por el Dr.
Federico Williams en el Instituto de Química Física de los Materiales, Medio Ambiente y
Energía. Se empleó un sistema de ultra alto vacío para análisis de superficies equipado con
XPS, espectroscopía fotoelectrónica ultravioleta (UPS) y espectroscopía de dispersión iónica
(ISS) (SPECS, Alemania) operando a una presión de ≈ 3 x 10‐9 mbar. Los espectros fueron
obtenidos con un analizador de energía hemiesférico de 150 mm (PHOIBOS‐150, SPECS) que
permite la detección simultánea de los fotoelectrones en 9 canales y por ende, menores
tiempos de adquisición de datos (0.5 s por dato) con una relación señal‐ruido satisfactoria. A
su vez, se utilizó una fuente de rayos X de ánodo mixto de Al/Mg (XR‐50, SPECS) de una
intensidad estándar. En particular, se empleó la línea Kα del Mg (energía de excitación 1253.6
eV, 250 W)
Por otro lado, el análisis de los sistemas Au/PhCOOH/Os y Au/SPhCOOH/Os fue realizado por la
Dra. Carolina Vericat en el laboratorio del Dr. Roberto Salvarezza en el Instituto de
Investigaciones Fisicoquímicas Teóricas y Aplicadas (INIFTA) de la ciudad de La Plata.
Las medidas de XPS se realizaron en una cámara de ultra alto vacío con una presión de
10‐10 mbar equipada con una fuente de rayos X de ánodo de Mg, línea Kα (1253.6 eV, XR‐50,
SECS GmbH) y un analizador de energía electrónica semiesférico de 100 mm (PHOIBOS 100,
SPECS GmbH). Los espectros se adquirieron con energía de paso de 10 eV y una línea de base
tipo Shirley fue sustraída en cada región de los mismos. Se realizó una calibración con dos
puntos de la escala de energía usando muestras de oro (Au 4 f7/2, energía de unión = 84.00 eV)
y cobre (Cu 2 p3/2, energía de unión = 933.67 eV), cuya limpieza se realizó por bombardeo con
argón. La señal de C 1s a 285 eV fue usada como referencia para eliminar los efectos de carga.
En todos los casos, a la hora de calcular cubrimientos superficiales, las intensidades de las
señales empleadas fueron corregidas por parámetros de calibración adecuados.
2.2.5 ESPECTRÓMETRO DE DISPERSIÓN RAMAN
Estos experimentos fueron realizados por el Dr. Nicolás Tognalli en el laboratorio del Dr.
Alejandro Fainstein en el Instituo Balseiro de la ciudad de San Carlos de Bariloche.
Se utilizó un espectrómetro triple Jobin‐Yvon T64000 operando en modo sustractivo y
equipado con un dispositivo de cargas interconectadas (charge‐coupled device, CCD) enfriado
con N2 líquido. La excitación se realizó con energías entre 1.916 eV (647 nm) y 2.707 eV (458

2. MATERIALES Y MÉTODOS
51
nm) de las 11 líneas de un láser Ar‐Kr. La potencia usada fue 30 mW, concentrada en una
línea, ≈ 2 mm de largo y ≈ 100 ´m de ancho.
Se escogió una línea para reducir la degradación foto inducida del complejo de osmio (foto
blanqueo por intercambio de los ligandos piridina con agua). Los experimentos Raman
resonantes fueron realizados en aire.
2.2.6 MICROSCOPIO DE EFECTO TÚNEL
2.2.6.1 MICROSCOPÍA DE EFECTO TÚNEL
Estos experimentos fueron realizados por la Dra. Carolina Vericat en el laboratorio del Dr.
Roberto Salvarezza en el Instituto de Investigaciones Fisicoquímicas Teóricas y Aplicadas
(INIFTA) en la ciudad de La Plata.
Se utilizaron sustratos de oro manufacturados por la firma Arrandee que fueron recocidos con
llama de hidrógeno a aproximadamente 3000 °C antes de ser modificados.
Las imágenes se tomaron en aire en el modo de corriente constante con un microscopio
Nanoscope IIIa de Veeco Instruments (Santa Barbara, CA) usando puntas de Pt‐Ir. Los
intervalos de corriente túnel, potencial punta‐sustrato y velocidad de barrido empleados
fueron 0.3‐3 nA, 1.0‐1.5 V y 1‐2 Hz, respectivamente.
2.2.6.2 ESPECTROSCOPÍA DE EFECTO TÚNEL
Estos experimentos fueron realizados en el laboratorio del Dr. Richard Nichols en el
Departamento de Química de la Universidad de Liverpool, Inglaterra.
Se emplearon sustratos de oro manufacturados por la firma Arrandee que fueron recocidas a
aproximadamente 800‐1000 °C con un mechero Bunsen inmediatamente antes de ser usados.
Se utilizó un sistema STM Pico2000 (Molecular Imaging) controlado con el programa PicoScan
4.19. Se prepararon puntas de oro por oxidación electroquímica (etching, en inglés) en una
solución HCl : etanol (1:1). Se equipó una celda de tres electrodos fabricada a medida con dos
alambres de oro que actuaron como electrodo auxiliar y de referencia, respectivamente; este
último fue calibrado contra un electrodo de calomel saturado (SCE). El electrolito empleado
fue KClO4 0.1M.

2. MATERIALES Y MÉTODOS
52
2.3 PROCEDIMIENTOS EXPERIMENTALES
2.3.1 REACTIVOS QUÍMICOS Y SOLUCIONES
Todo el material de vidrio y las celdas de teflon utilizados fueron lavados con KMnO4 en medio
ácido y luego con una solución de H2O2 10 % en medio ácido, para eliminar la grasa. Luego se
enjuagaron con abundante agua Milli‐Q® (MilliPore). Todas las soluciones, a su vez, fueron
preparadas con agua Milli‐Q.
A lo largo de todo el trabajo se utilizaron los siguientes reactivos:
Cloruro de sodio (Merck), perclorato de sodio (Merck), fluoruro de sodio (Merck), nitrato de
sodio (Biopack), hidróxido de sodio (Merck), ácido sulfúrico (Merck) y ácido nítrico (Merck)
fueron empleados en la preparación de soluciones.
Para la síntesis y reducción de sales de diazonio se utilizaron los siguientes reactivos: 4‐
nitroanilina (Sigma‐Aldrich), ácido 4‐aminobenzoico (Sigma‐Aldrich), ácido 4‐amino‐2,3,5,6‐
tetrafluoro‐benzoico (Sigma‐Aldrich), nitrito de sodio (Sigma‐Aldrich), nitrito de iso‐amilo
(Sigma‐Aldrich), fluoroborato de sodio (Sigma‐Aldrich), tetrafluoroborato de tetrabutilamonio
(Fluka), acetonitrilo (Merck), etanol (Sintorgan), ácido tetrafluorobórico (solución 50 % , Riedel‐
de Haën), éter etílico (Sintorgan) y ácido clorhídrico (Mallinckrodt).
En la síntesis de los complejos de osmio se usaron: hexacloroosmato de amonio (Anedra), 2,2´‐
bipiridina (Anedra), ditionito de sodio (Merck), hexafluorofosfato de amonio (Fluka) y
etilenglicol (Cicarelli).
Se usaron los siguientes tioles para la modificación de superficies en los casos requeridos:
ácido 4‐mercaptobenzoico, ácido 3‐mercaptopropanoico, ácido 6‐mercaptohexanoico, ácido 8‐
mercaptooctanoico, ácido 11‐mercaptoundecanoico, ácido 16‐mercaptohexadecanoico (todos
Sigma‐Aldrich).
En la post‐funcionalización de las superficies se emplearon: 1‐etil‐3‐(3‐dimetilamino‐propil)‐
carbodiimida (Sigma‐Aldrich), N‐hidroxisuccinimida (Sigma‐Aldrich), ácido 4‐2‐hidroxietil‐1‐
piperazinetansulfónico (HEPES) (Sigma‐Aldrich), nitrato de potasio (Merck).
Las pastillas para la caracterización infrarroja de los productos sintetizados se prepararon con
KBr de la marca Mallinckrodt.
En la caracterización de las muestras por espectroscopía RMN se empleó agua deuterada de la
marca Sigma‐Aldrich.

2. MATERIALES Y MÉTODOS
53
2.3.2 LIMPIEZA DE LOS SUSTRATOS DE ORO
La contaminación superficial de las superficies de oro empleadas como electrodos de trabajo
siempre fue controlada antes de su modificación. Para ello, se realizó una voltametría cíclica
en una solución de H2SO4 2 M, barriendo el potencial entre 0.1 y 1.6 V a 0.1 V s‐1. A partir de
ella, también se estimó el área electroquímicamente activa de los electrodos integrando la
carga del pico de reducción del óxido de oro superficial: se consideró que los sitios de
adsorción de moléculas pequeñas como H2 u O2 son los mismos que participan en las
reacciones electroquímicas de interés. Para calcular el área efectiva del electrodo, se tomó
como referencia, la carga correspondiente a la desorción electroquímica de una monocapa de
oxígeno (pico de reducción del Au) de 400 ´C cm‐2 [238, 239]. En general, el factor de
rugosidad hallado por este método para los electrodos de oro evaporado resultó ser de 1.5‐2.
Los electrodos de gota de oro se limpiaron por inmersión en solución piranha (H2SO4 : H2O2
(3:1)) durante unos minutos y luego se confirmó su limpieza por voltametría cíclica en H2SO4
2M como se describió para los electrodos planos.
2.3.3 SÍNTESIS DE OS(2,2’‐BPY)2CL2
En un balón Schlenk se mezclaron NH4OsCl6 (500 mg, 1.19 mmol) y la 2,2’‐bipiridina (371 mg,
3.38 mmol) en etilenglicol (25 ml). La solución se desgasó tres veces con argón para eliminar el
oxígeno que puede oxidar el Os(II). Se calentó a reflujo durante 50 minutos bajo atmósfera de
argón. Luego, se enfrió la solución a 0°C observándose la formación de una suspensión de
color rojo oscuro.
Se agregaron 10 ml de una solución saturada de Na2S2O4 con agitación y se dejó durante una
noche bajo atmósfera de argón a 4°C.
Finalmente, se filtró el producto con un crisol filtrante con placa sinterizada (galafritte #4), se
lavó con abundante agua y con éter etílico y se secó en desecador.
2.3.4 SÍNTESIS DE [OS(2,2´‐BPY)2CL(PY‐CH2‐NH2)]PF6
En un balón Schlenk se disolvieron 4‐aminometilpiridina (19 mg, 0.178 mmol) y
Os(2,2´‐bpy)2Cl2 (102 mg, 0.178 mmol) en etilenglicol (2 ml). La solución resultante fue
desgasada dos veces con argón y calentada a reflujo durante 3 horas bajo atmósfera de argón.
Luego de enfriar, se agregó 1 ml de solución saturada de NH4PF6 y se agitó durante 30 minutos.
El precipitado resultante se filtró con una membrana Polyamid (Ø 0.45 ´m), se lavó con agua y
se secó al vacío.
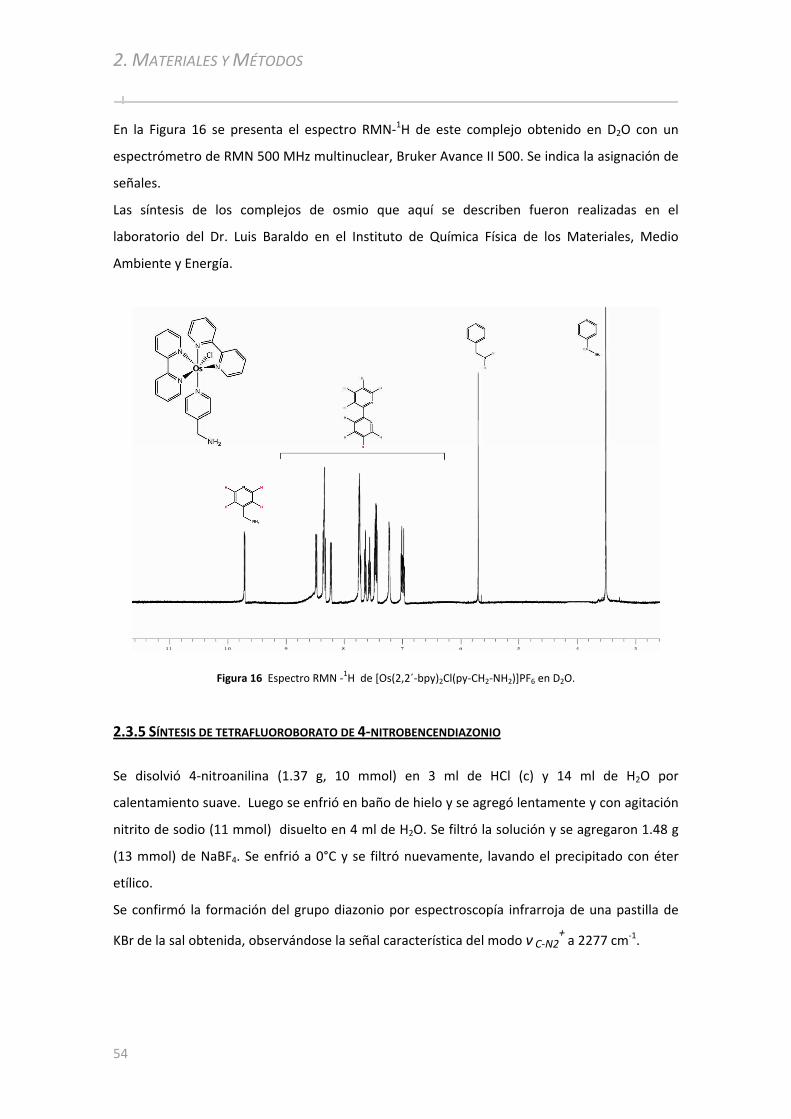
2. MATERIALES Y MÉTODOS
54
En la Figura 16 se presenta el espectro RMN‐1H de este complejo obtenido en D2O con un
espectrómetro de RMN 500 MHz multinuclear, Bruker Avance II 500. Se indica la asignación de
señales.
Las síntesis de los complejos de osmio que aquí se describen fueron realizadas en el
laboratorio del Dr. Luis Baraldo en el Instituto de Química Física de los Materiales, Medio
Ambiente y Energía.
2.3.5 SÍNTESIS DE TETRAFLUOROBORATO DE 4‐NITROBENCENDIAZONIO
Se disolvió 4‐nitroanilina (1.37 g, 10 mmol) en 3 ml de HCl (c) y 14 ml de H2O por
calentamiento suave. Luego se enfrió en baño de hielo y se agregó lentamente y con agitación
nitrito de sodio (11 mmol) disuelto en 4 ml de H2O. Se filtró la solución y se agregaron 1.48 g
(13 mmol) de NaBF4. Se enfrió a 0°C y se filtró nuevamente, lavando el precipitado con éter
etílico.
Se confirmó la formación del grupo diazonio por espectroscopía infrarroja de una pastilla de
KBr de la sal obtenida, observándose la señal característica del modo ` C‐N2+ a 2277 cm‐1.
Figura 16 Espectro RMN ‐1H de [Os(2,2´‐bpy)2Cl(py‐CH2‐NH2)]PF6 en D2O.

2. MATERIALES Y MÉTODOS
55
2.3.6 SÍNTESIS DE TETRAFLUOROBORATO DE 4‐CARBOXIBENCENDIAZONIO
Se disolvió ácido 4‐aminobenzoico (0.4 g, 2.91 mmol) en etanol (1 ml) y se agregaron 0.65 ml
de una solución acuosa de ácido tetrafluorobórico (solución 50% , 3.7 mmol). La mezcla se
enfrió a 0°C en baño de hielo. A continuación, se agregó nitrito de isoamilo (0.6 ml, 4.5 mmol)
por goteo con agitación. La agitación se mantuvo durante 30 minutos. Finalmente, se agregó
éter etílico (30 ml) observándose la precipitación del producto. Se filtró y lavó con abundante
éter etílico [240]. Nuevamente, la observación de una señal a 2287 cm‐1 en el espectro
infrarrojo de una pastilla de la sal obtenida confirmó la formación del producto esperado.
2.3.7 SÍNTESIS DE TETRAFLUOROBORATO DE 4‐(2,3,5,6‐TETRAFLUORO)‐CARBOXIBENCENDIAZONIO
La síntesis de esta sal de diazonio se realizó en forma simultánea con su reducción ya que, una
vez formada, esta sal es muy reactiva. Esto se debe a que los sustituyentes fluoruro ejercen un
efecto atractor de electrones que retira la densidad electrónica del anillo e inestabiliza al grupo
diazonio.
Por lo tanto, se preparó una solución etanólica de ácido tetrafluorobórico (solución 50 % , 0.01
mol) y se agregó nitrito de isoamilo (6 ´l, 0.045 mmol). La mezcla se introdujo en una celda
electroquímica de tres electrodos, con la superficie de oro a modificar conectada como
electrodo de trabajo. Se aplicaron ‐0.7 V, ‐0.2 V y 0 V, según la muestra deseada, y se agregó
una solución etanólica de ácido 1‐amino‐2,3,5,6‐tetrafluorobenzoico (0.02 mmol en 1.5 ml de
etanol). El potencial fue aplicado durante 150 segundos y luego se lavó el sustrato de oro con
abundante acetonitrilo y agua. Finalmente, se secó bajo corriente de N2.
2.3.8 DERIVATIZACIÓN DE SUSTRATOS DE ORO
La preparación de los distintos sistemas bajo estudio en este trabajo se realizó en dos etapas:
1) derivatización de superficies de oro por formación de monocapas orgánicas y
2) post‐funcionalización de dichas monocapas por unión covalente del complejo de osmio a
las mismas.
Las monocapas de moléculas orgánicas se generaron sobre el oro por electro reducción de
sales de diazonio y por autoensamblado de tioles.

2. MATERIALES Y MÉTODOS
56
2.3.8.1 ELECTRO REDUCCIÓN DE SALES DE DIAZONIO
Se derivatizaron superficies de oro con nitrobenceno y con ácido benzoico por reducción
electroquímica de las sales de diazonio derivadas de la 4‐nitroanilina (tetrafluoroborato de
4‐nitrobencendiazonio), y del ácido 4‐aminobenzoico (tetrafluoroborato de 4‐carboxibencen‐
diazonio), respectivamente. Para ello se realizó una cronoamperometría en una celda
equipada con un contra electrodo de Pt, un electrodo de referencia de Ag/AgCl, KCl 3M y con
la superficie de oro a modificar actuando como electrodo de trabajo. Se aplicaron 0.4 V y 0.3 V
vs. Ag/AgCl, respectivamente, durante 5 minutos en una solución de la sal de diazonio
correspondiente en concentración 5 mM en acetonitrilo y tetrafluoroborato de
tetrabutilamonio 0.1 M como electrolito soporte [189, 190, 241‐243].
Una vez finalizado este tratamiento, el electrodo de trabajo fue lavado con abundante
acetonitrilo y con agua y finalmente fue secado bajo corriente de N2.
2.3.8.2 AUTOENSAMBLADO DE TIOLES
Las superficies de oro fueron modificadas con monocapas de los siguientes tioles: ácido 3‐
mercaptopropanoico, ácido 6‐mercaptohexanoico, ácido 8‐mercaptooctanoico, ácido
11‐mercaptoundecanoico, ácido 16‐mercaptohexadecanoico y ácido p‐mercaptobenzoico.
Para ello fueron sumergidas durante 1 hora en soluciones etanólicas 1 mM del tiol
correspondiente. Una vez modificados, los electrodos fueron lavados con abundante etanol y
secados bajo corriente de N2.
2.3.8.3 POST‐FUNCIONALIZACIÓN CON [OS(2,2´‐BPY)2CL(PY‐CH2‐NH2)]PF6
Las superficies previamente derivatizadas con los grupos –COOH se incubaron en una solución
acuosa de 1‐etil‐3‐(3‐dimetilaminopropil)‐carbodiimida (EDC, 40 mM) / N‐hidroxisuccinimida
(NHS, 10 mM) durante 1 hora. Luego, fueron sumergidos durante una noche en una solución
de [Os(2,2´‐bpy)2Cl(py‐CH2‐NH2)]PF6 0.25 mM/buffer de ácido N‐2‐hidroxietilpiperazin‐N´‐2‐
etansulfónico (HEPES, 0.05M), de pH 7.3 y fuerza iónica 0.1 M (KNO3) [191, 244].
De esta manera, se formó una unión amida entre el grupo amino del complejo y los grupos
ácido presentes en la superficie.
En todos los casos, los electrodos fueron lavados con abundante agua y secados con corriente
de N2 una vez finalizada la post‐funcionalización.

CAPÍTULO 3
DERIVATIZACIÓN Y CARACTERIZACIÓN SUPERFICIAL


3. DERIVATIZACIÓN Y CARACTERIZACIÓN SUPERFICIAL
59
n este capítulo se describe la derivatización de superficies de oro empleando dos
estrategias diferentes: 1) adsorción de tioles y 2) reducción electroquímica de sales de
diazonio. Se detallan las características principales de cada una de ellas y se presenta la
caracterización superficial de los sistemas obtenidos con ambas. Las técnicas empleadas
fueron Espectroscopía Infrarroja de Absorción‐Reflexión con Transformada de Fourier (FT‐
IRRAS), Espectroscopía Infrarroja de Absorción‐Reflexión con Modulación de la Polarización
(PM‐IRRAS) y Espectroscopía Fotoelectrónica de Rayos X (XPS). Los fundamentos de todas ellas
han sido presentados en el Capítulo 2.
3.1 ESTRATEGIAS DE MODIFICACIÓN DE SUPERFICIES
Existen relativamente pocos métodos que permiten la derivatización de metales, silicio o
carbono por la formación de un enlace covalente con una capa orgánica, y generalmente éstos
implican condiciones oxidativas. El silicio puede ser modificado por diversos métodos basados
en la activación térmica, química, o fotoquímica de alquenos o de haluros de alquilo, o en la
activación electroquímica de reactivos de Grignard [245] (por ejemplo, Figura 1 a)).
En el caso del carbono, la oxidación electroquímica de aminas [246] y de alcoholes [247]
genera sistemas del tipo de los esquematizados en la Figura 1 b) y c). Otra forma es la
oxidación de ácidos carboxílicos [248] (Figura 1 d)).
E
Figura 1 Diferentes estrategias de derivatización de superficies. a) Modificación de una superficie de silicio
iniciada por una reacción radicalaria. Derivatización de una superficie de carbono por oxidación electroquímica
de b) aminas, c) alcoholes y d) ácidos carboxílicos. e) Autoensamblado de tioles sobre metales.

3. DERIVATIZACIÓN Y CARACTERIZACIÓN SUPERFICIAL
60
Finalmente, los metales pueden derivatizarse por el autoensamblado de monocapas de tioles
(Figura 1 e)). En ese caso se forma una unión covalente relativamente débil, que permite que
las cadenas alquílicas puedan reorganizarse sobre la superficie.
3.1.1 MONOCAPAS AUTOENSAMBLADAS: COMPUESTOS ORGANOSULFURADOS
Las monocapas autoensambladas (self‐assembled monolayers, SAMs) se generan cuando
moléculas surfactantes se adsorben espontáneamente sobre una superficie formando una
capa mono‐molecular, compartiendo todas la misma orientación.
La Figura 2 muestra una imagen idealizada de estas estructuras, en la que las moléculas
poseen un grupo funcional que se une a la superficie (cabeza) y una cola que generalmente
consiste en una cadena alifática extendida. La organización surge de la afinidad por la
superficie y de las interacciones entre las cadenas de la cola.
El campo de trabajo basado en estas estructuras ha presentado un gran crecimiento en cuanto
a los métodos de síntesis y formas de caracterización durante los últimos 20 años, sin
embargo, comenzó a desarrollarse mucho antes de lo que en general se conoce.
En el año 1946 se publicó el primer trabajo que describía la preparación de una monocapa
molecular por adsorción de un surfactante sobre la superficie limpia de un metal [249]. En ese
momento el potencial de esta metodología no fue valorado, y recién en la década de 1980,
comenzó a cobrar la importancia que tiene hoy en día gracias al trabajo de Nuzzo y Allara [6].
Ellos mostraron que se podían preparar SAMs de n‐alcanotioles sobre oro por adsorción de
disulfuros de di‐n‐alquilos desde soluciones diluidas. Poco después, otros grupos
contribuyeron al desarrollo de este nuevo área de conocimiento [250‐256].
Figura 2 Diagrama esquemático de una monocapa ideal de n‐alcanotioles autoensamblada sobre una superficie
de oro.

3. DERIVATIZACIÓN Y CARACTERIZACIÓN SUPERFICIAL
61
La Figura 3 muestra el incremento en el número de publicaciones basadas en este tema con el
correr de los años. El creciente interés se debe a que las SAMs son nanoestructuras con un
gran número de propiedades útiles. Principalmente, su grosor de 1‐3 nm las convierte en el
material en escala nanométrica más elemental que, a su vez, puede emplearse en la
estabilización y funcionalización de objetos nanométricos, como películas delgadas,
nanocables, coloides y otras nanoestructuras. Una de sus características más atractivas es la
facilidad de su preparación, que no necesita equipos sofisticados ni condiciones de trabajo
extremas. Asimismo, pueden utilizarse para acoplar las propiedades electrónicas y ópticas de
estructuras metálicas con su entorno [257, 258], o servir de conexión entre fenómenos que
ocurren a nivel molecular y aquellos de escala macroscópica como el mojado [259, 260], la
adhesión [261] o la fricción [262]. Esto también las convierte en excelentes sistemas modelo
para el análisis de competencia intermolecular, interacciones molécula‐sustrato y molécula‐
solvente y de fenómenos interfaciales.
Además, cabe destacar que pueden incorporar una gran variedad de grupos funcionales, tanto
en la cadena alquílica como en su extremo terminal. Por lo tanto se pueden fabricar diversas
superficies bajo condiciones bien controladas, que pueden tener aplicaciones, por ejemplo, en
la prevención de la corrosión y del desgaste de materiales [263‐265]. Otra característica
notable es la naturaleza biomimética y biocompatible de estas estructuras, lo que las hace
interesantes para aplicaciones en sensores químicos y bioquímicos [222, 266].
Figura 3 Incremento del número de publicaciones sobre monocapas autoensambladas con el correr de los años.
(Fuente: Scopus, www.scopus.com)

3. DERIVATIZACIÓN Y CARACTERIZACIÓN SUPERFICIAL
62
En la literatura se pueden encontrar varios ejemplos de preparación de monocapas
autoensambladas sobre superficies metálicas (Au, Cu, Ag, Pd, Pt, Hg) y semiconductores (Si,
GaAs), utilizando moléculas con grupos funcionales apropiados como –SH, ‐CN, ‐COOH, ‐NH2 y
silanos [95, 267, 268]. En particular, en este trabajo son de interés aquellas obtenidas con
tioles sobre sustratos de oro.
Los compuestos organosulfurados presentan una alta afinidad por diversos metales de
transición como oro, plata, cobre, platino, hierro y mercurio [269‐271]. Esto se debe
probablemente a que pueden formar enlaces con los átomos metálicos superficiales. Existe
una gran variedad de estos compuestos que pueden formar monocapas sobre oro. La Figura 4
muestra algunos ejemplos de los mismos.
Sin embargo, las monocapas más estudiadas, y probablemente más entendidas, son las
formadas por los n‐alcanotioles sobre superficies de Au (111). Ellas se describen a
continuación.
3.1.1.1 ENLACE AU‐S
Las monocapas autoensambladas de los compuestos organosulfurados se forman
espontáneamente por adsorción desde fase líquida o fase gaseosa.
La formación del enlace Au‐S requiere de la activación del enlace S‐H y consiste en la adición
oxidativa del mismo sobre la superficie de oro, seguida de la eliminación del hidrógeno. La
forma en que esto último ocurre no ha sido determinado en forma unánime [272‐274], pero
parece probable que en el vacío se produce la eliminación reductiva del hidrógeno como
H2 (g). En solución existe otra posibilidad: si no ocurre eliminación reductiva, la presencia de
oxígeno en el medio puede llevar a la conversión oxidativa del hidrógeno en agua.
Figura 4 Compuestos organosulfurados que pueden formar monocapas sobre oro.

3. DERIVATIZACIÓN Y CARACTERIZACIÓN SUPERFICIAL
63
En cualquier caso, la interacción Au‐S en el tiolato es suficiente como para evitar su desorción
a temperatura ambiente. La energía de esta unión ha sido estimada en 40‐50 kcal mol‐1 [275,
276].
3.1.1.2 PREPARACIÓN Y CINÉTICA DE FORMACIÓN
La forma más común para la preparación de SAMs sobre oro, plata, paladio, mercurio y otros
materiales consiste en la inmersión de un sustrato limpio en una solución etanólica diluida
(1‐10 mM) del tiol durante 12‐18 horas a temperatura ambiente. Los detalles experimentales
pueden variar según las necesidades particulares del experimento y, es por ello, que hay que
considerar que existen varios factores que pueden repercutir en la estructura de la SAM
resultante y en su velocidad de formación. Ellos son el solvente, la temperatura, la
concentración y pureza del adsorbato, el tiempo de inmersión, la concentración de oxígeno en
solución, la limpieza del sustrato y la longitud de cadena (o en forma más general, la estructura
del adsorbato).
Los estudios cinéticos de la adsorción de alcanotioles sobre superficies de Au han mostrado
que en soluciones relativamente diluidas (1 mM), se observan dos velocidades de adsorción
muy distintas: un paso muy rápido que ocurre en minutos, y uno lento que dura algunas horas.
El paso inicial, descripto por una adsorción de Langmuir controlada por difusión, depende
fuertemente de la concentración del tiol y consiste en la unión del grupo –SH a la superficie. El
segundo paso puede describirse como un proceso de cristalización, en el que las cadenas del
tiol se ordenan formando un cristal líquido bidimensional y depende de las interacciones entre
ellas (Van der Waals, dipolo‐dipolo, etc.) y su movilidad superficial [96, 277].
Se ha observado que existen diferencias significativas entre la cinética de formación de las
monocapas de tioles cortos (menos de 9 metilenos en la cadena) y las de tioles largos (más de
9 metilenos) [278]. Esto se debe a que cuando los tioles son cortos, las interacciones de Van
der Waals entre cadenas son más pequeñas, afectando la velocidad del proceso de
cristalización mencionado, con la formación de monocapas más desordenadas. También se
obtienen monocapas más desordenadas cuando la cola del tiol tiene grupos funcionales
voluminosos.
3.1.1.3 ORGANIZACIÓN SUPERFICIAL
La densidad molecular máxima que puede presentar una monocapa, autoensamblada sobre la
superficie de un dado material, depende de las mínimas distancias entre los átomos que lo

3. DERIVATIZACIÓN Y CARACTERIZACIÓN SUPERFICIAL
64
conforman y del arreglo geométrico que adopten las moléculas que integran dicha monocapa.
Esta densidad molecular bidimensional puede no coincidir con la que correspondería a una
estructura cristalina de las mismas moléculas, es decir, el ordenamiento superficial de las
moléculas no necesariamente maximiza las interacciones entre los componentes orgánicos de
las SAMs. Para minimizar la energía libre de la capa orgánica, las moléculas adoptan
conformaciones que permiten altos grados de interacciones de Van der Waals [279] (y, en
algunos casos, puente de hidrógeno [280‐282]) con moléculas vecinas. Estos arreglos generan
un nivel secundario de organización de la monocapa que es importante en la determinación de
las propiedades macroscópicas de las SAMs.
Existen dos parámetros que se utilizan para describir las variaciones en la orientación de las
moléculas que componen una SAM: el ángulo de inclinación de las cadenas alifáticas de la cola
respecto de la normal a la superficie (α) y el ángulo de rotación alrededor del eje largo de la
molécula (β). El primero puede adoptar valores positivos y negativos y el segundo varía entre
0° y 90°.
Para n‐alcanotioles de cadena larga sobre oro, paladio, plata, cobre, mercurio, platino, las
cadenas alifáticas se extienden en una estructura cuasi cristalina y, casi todas, adoptan la
corformación trans. Las inclinaciones de las mismas varían según el sustrato metálico
empleado: los valores más altos de α se encuentran en oro, donde alcanza un valor absoluto
de 30° [283, 284], mientras que las estructuras más orientadas en la dirección de la normal a la
superficie se encuentran sobre plata (α ≈ 10°) [283, 285, 286] y mercurio (α ≈ 0°) [287]. El valor
promedio de β en oro es aproximadamente 50° [283], mientras que para otros metales, los
valores disponibles indican β ≈ 45° [283, 288]. En el caso del oro, estos datos son consistentes
con un ordenamiento hexagonal de los átomos de azufre con una distancia S‐S de 4.97 Å y un
área ocupada por molécula de 21.4 Å2 [255, 289, 290]. De esta manera, las moléculas forman
una red √3 x √3 R 30° [291, 292]. La literatura también confirma que las cadenas pueden
adoptar un orden secundario de las cadenas correspondiente a una súper red c (4 x 2) [289,
293‐295]. Este tipo de ordenamiento corresponde a un cubrimiento superficial de
≈ 7.5 x 10‐10 mol cm‐2 [95].
En base a este modelo, se ha sugerido que los tiolatos se adsorben tanto en posiciones hollow
(del inglés, hueco) como bridge (del inglés, puente) de la red cristalina del sustrato [296‐298], y
que existe cierto transporte de átomos de oro cuando la adsorción es desde una solución [289,
294]. La migración de los tiolatos entre sitios hollow adyacentes es esencial para la reparación
de defectos en la SAM.

3. DERIVATIZACIÓN Y CARACTERIZACIÓN SUPERFICIAL
65
Sin embargo, es bueno destacar que no todos los tioles adoptan la orientación y organización
superficial de los n‐alcanotioles. Por ejemplo, los compuestos aromáticos como los p‐
bifeniltioles, p‐terfeniltioles y oligo‐(fenilen‐etilen)‐tioles adoptan orientaciones sobre el
Au (111) con un valor de α menor a 30° [299‐302]. De todas maneras, estos sistemas parecen
exhibir un arreglo estructural √3 x √3 R 30° cuando alcanzan el cubrimiento límite de la
superficie. También hay que considerar la orientación de la cara cristalógráfica de la superficie
que se emplea como sustrato. Si bien la mayoría de los estudios han utilizado Au (111), existen
algunos trabajos realizados con Au (100) que demuestran que las SAMs formadas sobre el
mismo generan una sobrecapa c (2x2), cuya densidad de empaquetamiento no podría permitir
una inclinación de las moléculas como la observada en Au (111) [290].
Todo esto sugiere que la química del autoensamblado depende fuertemente de la interacción
metal‐azufre y de las interacciones laterales de los grupos orgánicos.
3.1.1.4 ESTABILIDAD
Las SAMs se forman a partir del ensamblado espontáneo de un adsorbato sobre una
superficie/interfase. Cuando la interacción adsorbato‐sustrato es suficientemente fuerte
(como en el caso de los n‐alcanotioles sobre oro), el sustrato modificado con la monocapa
puede ser removido de la solución usada en la preparación y ser empleado en aplicaciones
posteriores. En este caso, las SAMs son cinéticamente estables en ausencia de un flujo de
adsorbato pero sin embargo, son termodinámicamente inestables. Sólo cuando la velocidad de
desorción es rigurosamente cero se puede esperar que la SAM exista durante un período
indeterminado fuera de la solución en la que fue preparada. Sin embargo, los pares adsorbato‐
sustrato pueden resistir la unión competitiva de las impurezas en la interfase y presentan una
buena estabilidad respecto de la desorción térmica. Aún así, esta estabilidad está limitada por
la fuerza finita del enlace sustrato‐adsorbato y por la susceptibilidad de los tiolatos más
simples a la descomposición (por vía oxidativa u otros caminos disociativos) en ciertos
ambientes en que las SAMs son utilizadas.
Para monocapas simples sobre oro, el límite de estabilidad térmica es ≈ 80 °C [303], moderado
pero suficiente como para poder trabajar con ellas a temperatura ambiente.
3.1.2 REDUCCIÓN ELECTROQUÍMICA DE SALES DE DIAZONIO
La electro‐reducción de sales de diazonio constituye una forma alternativa para modificar
superficies con capas orgánicas y ha captado mucho la atención en los últimos años. Esto se

3. DERIVATIZACIÓN Y CARACTERIZACIÓN SUPERFICIAL
66
debe a dos características significativas: 1) se aplica a una gran variedad de materiales y, 2)
genera uniones muy fuertes entre la superficie y las moléculas orgánicas.
La adsorción reductiva de sales de diazonio fue introducida por Pinson en el año 1992 [304,
305] y posteriormente otros autores contribuyeron a su desarrollo [306‐312]. Esta técnica se
puede aplicar a diferentes materiales: distintas variedades de carbono (carbono vítreo [189,
313], grafito pirolítico altamente orientado (HOPG) [189, 190, 243, 306], fibras de carbono
[313], nanotubos [314]), semiconductores (silicio [315], arseniuro de galio [316]) y metales.
Entre estos últimos no sólo se cuentan metales nobles como oro y platino [317, 318], sino
también metales industriales como hierro, zinc, níquel, cobalto [318], cobre [319] y paladio
[316]. Por lo tanto, esta metodología ofrece una química que puede aplicarse, en principio, a
todo tipo de sustratos. Esto se ve reflejado en la Figura 5 que muestra el aumento en el
número de artículos publicados sobre este tema en los últimos años.
3.1.2.1 ADSORCIÓN REDUCTIVA DE SALES DE ARIL DIAZONIO
La modificación de superficies por reducción electroquímica de sales de diazonio sobre
carbono [189], metales [317] o semiconductores [318] está esquematizada en la Figura 6. Este
procedimiento consiste simplemente en reducir una sal de diazonio ArN2+ (concentración
≈ 1‐10 mM) disuelta en un solvente aprótico con un electrolito soporte (en general,
acetonitrilo + 0.1 M terafluoroborato de tetrabutilamonio, Bu4NBF4) o en medio ácido (H2SO4
2 M, por ejemplo), empleando como cátodo la superficie a ser modificada. El potencial de este
Figura 5 Incremento en el número de publicaciones sobre la adsorción reductiva de sales de diazonio con el
correr de los años. (Fuente: Scopus, www.scopus.com)

3. DERIVATIZACIÓN Y CARACTERIZACIÓN SUPERFICIAL
67
electrodo se fija en el valor del potencial de reducción de la sal correspondiente y se mantiene
durante un tiempo dado (segundos a minutos), o asimismo puede fijarse en un valor más
catódico según el tipo de modificación que se quiera llevar a cabo, como se verá más adelante.
Las sales de aril diazonio (ArN2+BF4
‐) se preparan fácil y rápidamente en un paso a partir de una
gran variedad de aminas aromáticas, la mayoría de ellas, disponible comercialmente. Los
métodos sintéticos están bien establecidos en textos de química orgánica [320]. Por todo ello,
este método de derivatización permite unir una gran variedad de grupos funcionales a la
superficie (alquilo, carboxilo, éster, cianuro, nitro, alcohol, entre otros), ya que éstos pueden
estar presentes como sustituyentes en el anillo aromático de la sal de diazonio.
3.1.2.2 MECANISMO DE ADSORCIÓN
Se puede comenzar el análisis del mecanismo de adsorción reductiva de las sales de diazonio
sobre una superficie observando la Figura 7.
Figura 6 Esquema de la modificación de superficies por reducción electroquímica de sales de diazonio. El
electrodo puede ser metálico, semiconductor o de carbono.
Figura 7 Reducción electroquímica de tetrafluoroborato de 4‐nitrobencendiazonio por voltametría cíclica en una
solución 5 mM O2N‐Ph‐N2BF4/0.1 M Bu4NBF4/ACN. Se realizaron 5 ciclos entre 0.7 y 0.2 V vs. Ag/AgCl a
50 mV s‐1.

3. DERIVATIZACIÓN Y CARACTERIZACIÓN SUPERFICIAL
68
En ella se muestra la reducción por voltametría cíclica de tetrafluoroborato de
4‐nitrobencendiazonio sobre oro, en una solución de Bu4NBF4 0.1 M en acetonitrilo. En el
primer ciclo se observa un pico catódico a 0.4 V (vs. Ag/AgCl) propio de una transferencia
electrónica irreversible que, en este caso, consiste en la eliminación del grupo diazonio como
N2 (g). La desaparición de este pico en los ciclos posteriores puede asociarse al bloqueo del
electrodo por los grupos orgánicos que se han unido a la superficie.
Para hacer una interpretación más detallada de estas observaciones experimentales es
interesante analizar qué ocurre con los haluros de arilo (Ar‐X) cuya estructura química está
relacionada con la de las sales de diazonio y cuya electroquímica está bien establecida. En la
Figura 8 se esquematiza la reacción de estas especies sobre la superficie de un electrodo [97,
321].
El primer paso consiste en la reducción del haluro con la formación de un anión radical
(reacción a) que, a su vez, puede generar un radical arilo.
El anión radical, una vez formado, puede difundir hacia el seno de la solución alejándose del
electrodo. Por lo tanto, dependiendo de la vida media del anión radical, la formación del
radical arilo puede ocurrir cerca o lejos de la superficie del electrodo. Si el radical arilo se
forma lejos del electrodo, no tendrá el tiempo suficiente para difundir hacia él y generar algún
tipo de unión, ya que estas especies tienen tiempos de vida cortos, y pueden desaparecer por
su reducción inmediata (reacción c) o por abstracción de un hidrógeno del solvente (reacción
d).
Por otro lado, si se forma el radical arilo cerca del electrodo o, más aún, si la reducción del
haluro es concertada con la formación del radical (reacción b), entonces este último estará
muy cerca de la superficie o “sobre” la misma, dependiendo del caso. Sin embargo, tampoco
en estos casos se ha observado la unión de los radicales a la superficie. Esto se debe a que
estas especies tienen potenciales de reducción menores que los haluros y por lo tanto, cuando
se generan en estas condiciones, se reducen instantáneamente.
Figura 8 Esquema del mecanismo de reacción de los haluros de arilo (Ar‐X) sobre la superficie de un electrodo.
Svte‐H indica una molécula de solvente, y Svte� y Svte– corresponden a un radical y a un anión formados a partir
de la misma, respectivamente.

3. DERIVATIZACIÓN Y CARACTERIZACIÓN SUPERFICIAL
69
Entonces, ¿por qué en el caso de las sales de diazonio sí se observa la unión de radicales arilo a
la superficie? El éxito de la derivatización de superficies con sales de diazonio reside en el
hecho de que estos compuestos tienen potenciales de reducción muy bajos que no alcanzan
para reducir a los radicales arilo una vez formados. Por lo tanto, el mecanismo propuesto para
la reacción de la Figura 6 consiste en la generación de radicales arilo sobre la superficie del
electrodo por la eliminación de N2 concertada con la reducción de la sal de diazonio [322]
(Figura 9). Debido al bajo potencial al que esto ocurre, los radicales formados pueden
reaccionar con la superficie formando un enlace Sup‐Ar.
El comportamiento particular de las sales de diazonio depende tanto de su fácil reducción
(relacionada con el efecto inductivo atractor del grupo N2+), como del paso concertado de
reducción y eliminación del N2, que asegura que el radical se forme sobre la superficie del
electrodo. La comparación con la electroquímica de los haluros de arilo permite inferir que los
radicales arilo son los responsables de la unión de las moléculas al sustrato.
3.1.2.3 ENLACE ENTRE LA SUPERFICIE Y LA CAPA ORGÁNICA
El enlace formado entre la superficie y la capa orgánica es de naturaleza covalente. Esta
afirmación se deriva de una serie de características propias de estos sistemas. La primera de
ellas es la fuerza de este enlace con superficies metálicas y de carbono, que resiste la
ultrasonicación en diferentes solventes (acetonitrilo, dimetilformamida, dimetilsulfóxido,
benceno, benzonitrilo, acetona, metanol, etanol, diclorometano y cloroformo) y que es estable
durante un mes en condiciones ambiente [189]. El intervalo de potencial en el que estas
películas son estables varía entre 2.6 y 5.6 V dependiendo del sustituyente [323]. Más aún,
sólo pueden ser removidas por pulido mecánico o por raspado con la punta de un microscopio
de fuerza atómica [308]. Todas estas observaciones indican que el enlace formado a partir de
esta derivatización no puede ser un enlace débil por puente hidrógeno o por interacciones de
Van der Waals.
Hasta el momento en que se escribe esta tesis, no existen pruebas espectroscópicas directas
de que el enlace entre la superficie y el anillo aromático sea efectivamente covalente. Sin
Figura 9 Mecanismo esquematizado de electro‐reducción de las sales de diazonio sobre un electrodo. Sup
indica la superficie del electrodo que puede ser metálico, semiconductor o de carbono.

3. DERIVATIZACIÓN Y CARACTERIZACIÓN SUPERFICIAL
70
embargo, existen experimentos que han demostrado en forma indirecta la existencia y
naturaleza de dicho enlace. Estos comprenden, por ejemplo, el análisis de la conductancia de
junturas moleculares formadas entre estos sistemas y un electrodo de mercurio [324] o el
análisis por espectroscopía Raman de estas superficies en función del potencial aplicado a las
mismas [325]. Ambos experimentos prueban la existencia de un alto acoplamiento electrónico
entre el sustrato y la capa orgánica, de manera que es posible inferir la presencia de un enlace
fuerte entre ambas partes. Este tipo de observaciones han sido realizadas en carbono, y
existen algunos ejemplos en la literatura que utilizan sustratos metálicos [188] y
semiconductores [326], aunque todavía es un terreno nuevo que se debe seguir explorando.
3.1.2.4 ORGANIZACIÓN SUPERFICIAL: MONOCAPAS Y MULTICAPAS
Esta estrategia de derivatización permite obtener distintos tipos de deposición según las
condiciones elegidas para la reducción de la sal de diazonio, es decir, el potencial aplicado, el
tiempo y la concentración de la misma en solución.
Una forma de analizar cuánto material orgánico se ha depositado en un sustrato consiste en
medir la concentración superficial de grupos funcionales presentes en las moléculas
aromáticas. Esto se puede hacer mediante diversas técnicas superficiales (espectroscopía
Raman, infrarroja, fotoelectrónica de rayos X, etc.) o por voltametría cíclica, según la
naturaleza del grupo de interés. En general, se considera que una monocapa compacta de de
anillos p‐sustituidos presenta un cubrimiento superficial de ≈ 10‐10 mol cm‐2 [97, 327].
Por otro lado, se ha observado que cuanto más catódico es el potencial aplicado, o mayor es el
tiempo durante el que se lo aplica, mayor es la cantidad de material depositado sobre la
superficie del electrodo, aún cuando se trata de diferentes tipos de sustrato [189, 242, 306,
312]. En muchos casos, en los que el área del pico de reducción de la sal de diazonio indica una
carga correspondiente a la deposición de una monocapa, el análisis por Microscopía de Fuerza
Atómica (AFM) y de Efecto Túnel (STM) muestra la presencia de capas gruesas [243, 308]. Esto
pone de manifiesto que el área del pico de reducción no es un buen indicador directo del tipo
de la capa depositada ya que, como se mencionó, las moléculas pueden depositarse en forma
compacta o no.
3.1.2.4.1 Mecanismo de formación de capas gruesas
La formación de capas gruesas por reducción de sales de diazonio se debe a una reacción de
ramificación que consiste en el ataque de los radicales arilo a moléculas que ya se han unido al
electrodo. Dicha reacción consiste en una sustitución homolítica aromática [97], cuyo

3. DERIVATIZACIÓN Y CARACTERIZACIÓN SUPERFICIAL
71
mecanismo propuesto se presenta en la Figura 10. En un primer paso un radical arilo, formado
por la reducción de una sal de diazonio, se une a la superficie del electrodo como ya fue
descripto. En una segunda etapa, otro radical puede atacarlo generando un radical
ciclohexadienilo. Para recuperar la aromaticidad, este radical debe perder un radical hidrógeno
o, más probablemente, un protón y un electrón. Esta última reacción es posible ya sea por la
transferencia de un electrón al electrodo y pérdida de un protón o por re‐oxidación del radical
por un catión diazonio. La repetición de esta secuencia puede generar capas gruesas de poli‐
fenileno, que son comúnmente llamadas multicapas aunque no deben confundirse con las
multicapas que se obtienen por autoensamblado capa por capa de poli‐electrolitos.
Por otro lado, si dos radicales ciclohexadienilo desproporcionan, la formación de un
ciclohexadieno puede interrumpir la conjugación.
Las velocidades de reacción para la sustitución homolítica en distintas posiciones del anillo son
similares entre sí. Sin embargo, por impedimentos estéricos, es más probable que el ataque
del radical sea en la posición orto al grupo funcional de la molécula ya unida a la superficie.
3.2 DERIVATIZACIÓN INICIAL DE SUSTRATOS DE ORO
3.2.1 DERIVATIZACIÓN DE SUPERFICIES DE ORO POR AUTOENSAMBLADO DE TIOLES
En este trabajo de tesis se emplearon tioles terminados en un grupo ácido carboxílico
(–COOH), necesario para unir el complejo de osmio a la superficie. Se eligieron cinco
Figura 10 Esquema del mecanismo de reacción propuesto para la formación de multicapas orgánicas sobre una
superficie a partir de la reducción electroquímica de una sal de diazonio.

3. DERIVATIZACIÓN Y CARACTERIZACIÓN SUPERFICIAL
72
alcanotioles de diferente largo de cadena y un tiol aromático: ácido 3‐mercaptopropanoico
(MPA), ácido 6‐mercaptohexanoico (MHA), ácido 8‐mercaptooctanoico (MOA),
ácido 11‐mercaptoundecanoico (MUA), ácido 16‐mercaptohexadecanoico (MHDA) y ácido 4‐
mercaptobenzoico (HSPhCOOH). El cubrimiento superficial obtenido en cada caso se
determinó por desorción reductiva y las monocapas obtenidas se caracterizaron por
espectroscopías infrarrojas de superficie, como se detalla a continuación.
3.2.1.1 DETERMINACIÓN DEL CUBRIMIENTO SUPERFICIAL
Las monocapas moleculares formadas por tioles se desorben cuando se aplica un potencial
negativo al electrodo sobre el que están autoensambladas [328‐330]. Este proceso se conoce
como desorción reductiva y la reacción asociada al mismo es:
0M SR + RS + M−− ⎯⎯→ē
Tanto el tiolato como la superficie metálica se solvatan y el tiolato difunde hacia el seno de la
solución. El proceso es reversible: si se detiene la aplicación del potencial negativo puede
ocurrir que los tiolatos se re‐adsorban sobre la superficie, o si el barrido de potencial se
revierte puede existir re‐adsorción oxidativa.
La desorción reductiva de las monocapas de tioles ha sido estudiada en detalle, principalmente
porque la misma depende fuertemente de la estructura y las propiedades de cada SAM. La
forma, magnitud y potencial del pico (Edesorción) asociado a la desorción de un tiol dependen de
un número de factores que incluyen el tipo de molécula, el largo de cadena, el grado de
ordenamiento, el tipo e intensidad de interacciones intermoleculares dentro de la película
orgánica y la cristalinidad del sustrato [331].
A partir del área del pico de desorción de un tiol, se puede estimar cuál era el cubrimiento
superficial del mismo, empleando para ello la carga electroquímica asociada a dicho proceso
[328].
En la Figura 11 se presenta, a modo de ejemplo, la desorción reductiva del ácido
4‐mercaptobenzoico adsorbido sobre Au (111), que se realizó por voltametría cíclica en una
solución de NaOH 0.5 M. En ella se observan dos picos: uno a ‐0.67 V y otro a ‐0.96 V. El
primero de ellos se puede asignar a la desorción reductiva del tiol. El origen del segundo pico
no está totalmente claro y existen dos posibilidades: desorción de átomos de azufre (que se
encuentran como impurezas en los tioles comerciales) [332, 333] o desorción de moléculas de
tiol desde defectos de la superficie de Au (111) [285, 334].

3. DERIVATIZACIÓN Y CARACTERIZACIÓN SUPERFICIAL
73
Figura 11 Curva de desorción reductiva de una monocapa de ácido 4‐mercaptobenzoico depositada sobre una
superficie de Au (111). Se llevó a cabo por voltametría cíclica en una solución de NaOH 0.5 M y la velocidad de
barrido fue de 0.02 V s‐1.
La carga involucrada en la desorción del tiol es de 54 ´C cm‐2 y la del pico observado a
potenciales más negativos es de 13 ´C cm‐2. Ésta última es demasiado alta como para
corresponder a la desorción del ácido 4‐mercaptobenzoico desde defectos presentes en la
superficie. Por lo tanto se considera que corresponde a impurezas presentes en el tiol
comercial.
Tabla 1. Estimación del cubrimiento superficial de sustratos de oro con tioles (Γtiol) por desorción
reductiva.
E desorción / V よtiol / (x 10‐10 mol cm‐2)
HSPhCOOH (Au(111)) ‐0.67 4.5
HSPhCOOH (Au policristalino) ‐0.63 9
MPA ‐0.80 8
MHA ‐0.92 9
MOA ‐0.94 10
MUA ‐0.95 11
MHDA ‐1.28 13
El mismo análisis se realizó con los otros tioles empleados en la derivatización de superficies
de oro. Vale aclarar que en estos casos, la desorción reductiva se llevó a cabo utilizando
sustratos de oro poli‐cristalino, como los empleados en la mayoría de los experimentos
realizados en este trabajo.
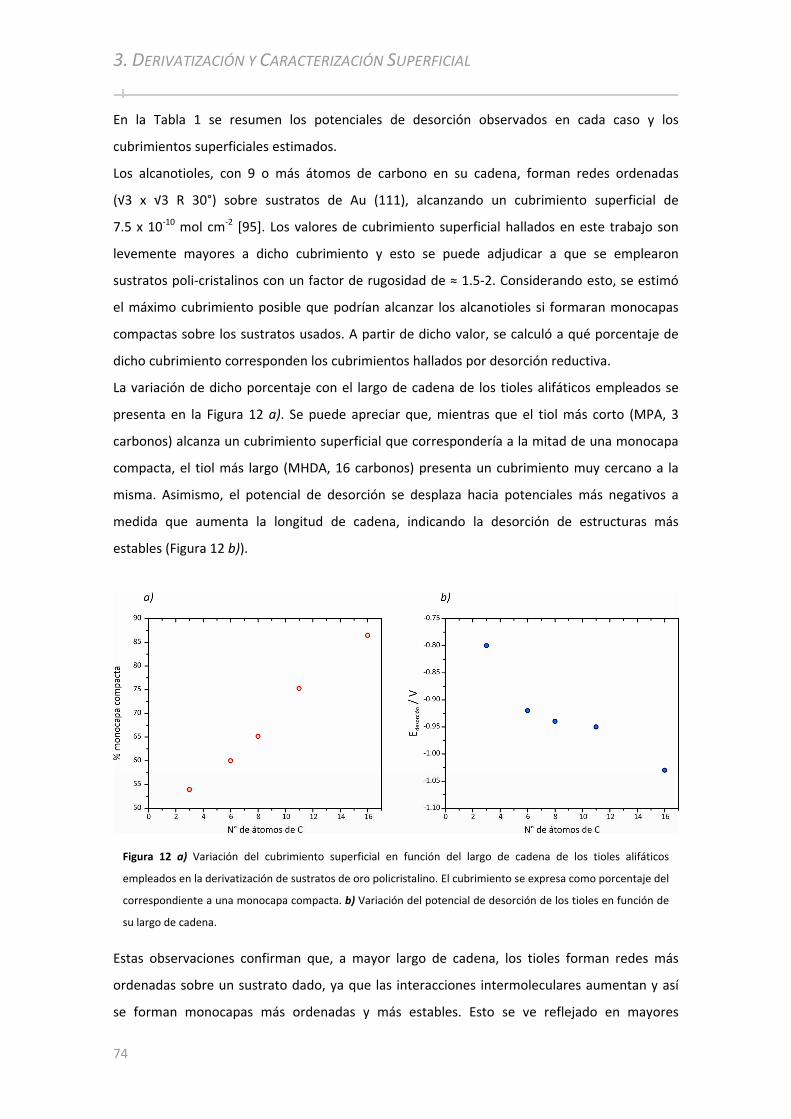
3. DERIVATIZACIÓN Y CARACTERIZACIÓN SUPERFICIAL
74
Figura 12 a) Variación del cubrimiento superficial en función del largo de cadena de los tioles alifáticos
empleados en la derivatización de sustratos de oro policristalino. El cubrimiento se expresa como porcentaje del
correspondiente a una monocapa compacta. b) Variación del potencial de desorción de los tioles en función de
su largo de cadena.
En la Tabla 1 se resumen los potenciales de desorción observados en cada caso y los
cubrimientos superficiales estimados.
Los alcanotioles, con 9 o más átomos de carbono en su cadena, forman redes ordenadas
(√3 x √3 R 30°) sobre sustratos de Au (111), alcanzando un cubrimiento superficial de
7.5 x 10‐10 mol cm‐2 [95]. Los valores de cubrimiento superficial hallados en este trabajo son
levemente mayores a dicho cubrimiento y esto se puede adjudicar a que se emplearon
sustratos poli‐cristalinos con un factor de rugosidad de ≈ 1.5‐2. Considerando esto, se estimó
el máximo cubrimiento posible que podrían alcanzar los alcanotioles si formaran monocapas
compactas sobre los sustratos usados. A partir de dicho valor, se calculó a qué porcentaje de
dicho cubrimiento corresponden los cubrimientos hallados por desorción reductiva.
La variación de dicho porcentaje con el largo de cadena de los tioles alifáticos empleados se
presenta en la Figura 12 a). Se puede apreciar que, mientras que el tiol más corto (MPA, 3
carbonos) alcanza un cubrimiento superficial que correspondería a la mitad de una monocapa
compacta, el tiol más largo (MHDA, 16 carbonos) presenta un cubrimiento muy cercano a la
misma. Asimismo, el potencial de desorción se desplaza hacia potenciales más negativos a
medida que aumenta la longitud de cadena, indicando la desorción de estructuras más
estables (Figura 12 b)).
Estas observaciones confirman que, a mayor largo de cadena, los tioles forman redes más
ordenadas sobre un sustrato dado, ya que las interacciones intermoleculares aumentan y así
se forman monocapas más ordenadas y más estables. Esto se ve reflejado en mayores

3. DERIVATIZACIÓN Y CARACTERIZACIÓN SUPERFICIAL
75
cubrimientos superficiales y en potenciales de desorción reductiva más catódicos, que indican
que la desorción es más difícil debido a la estabilidad creciente.
Finalmente, en la Tabla 1 se han incluido los datos de desorción reductiva del ácido
4‐mercaptobenzoico depositado sobre un sustrato de Au (111) y de Au poli‐cristalino. En el
primer caso, el potencial de desorción y el cubrimiento observados coinciden muy bien con lo
reportado en bibliografía para una monocapa de bencentiol [335, 336].
En cuanto al segundo caso, el cubrimiento hallado es del doble del que presenta el sustrato
plano. Esto es consistente con un factor de rugosidad de 2, como se indicó más arriba.
Por otro lado, se ha reportado en literatura un análisis exhaustivo de este sistema con
sustratos de oro obtenidos por evaporación de oro sobre obleas de Si (111) [337], es decir,
análogos a los utilizados en esta tesis. En dicho trabajo se obtuvo por elipsometría un espesor
de ≈ 9 Å, que coincide muy bien con la altura calculada de una molécula de este compuesto a
partir de consideraciones de longitudes y ángulos de enlace (8.4 Å) [338]. Esta observación
indicaría que las moléculas de ácido 4‐mercaptobenzoico forman una monocapa y se orientan
perpendicularmente a la superficie, aún sobre sustratos que no son atómicamente planos.
3.2.1.2 CARACTERIZACIÓN SUPERFICIAL POR FT‐IRRAS
Algunos de los sistemas obtenidos por autoensamblado de tioles sobre oro se caracterizaron
por FT‐IRRAS para poder asegurar la presencia de los grupos ácido sobre la superficie y
confirmar que, en todos los casos, las capas depositadas estuvieran en el orden de una capa
molecular. Esto es posible si se utiliza radiación polarizada y se aplica la regla de selección
superficial que se presentó en el Capítulo 2.
Los espectros obtenidos se presentan en la Figura 13, dónde sólo se muestran los espectros
obtenidos con radiación p‐polarizada ya que los espectros colectados con luz s‐polarizada no
mostraron señales.
De acuerdo a la regla de selección superficial, sólo los modos vibracionales que tienen una
componente del momento dipolar de transición en la dirección perpendicular a la superficie,
pueden ser observados con luz p‐polarizada. Los modos que no presentan esta característica
no son detectados con esta radiación. Por otro lado, el campo eléctrico de la luz s‐polarizada
se anula sobre la superficie de la muestra y hasta una distancia de unos pocos micrones, por lo
tanto la presencia de monocapas no genera señales. Sólo se observan señales con esta
radiación cuando las moléculas correspondientes se encuentran a distancias mayores y,
siempre y cuando, tengan una componente del momento dipolar de transición paralela a la
superficie.

3. DERIVATIZACIÓN Y CARACTERIZACIÓN SUPERFICIAL
76
Esta regla de selección superficial indica que, a partir de esta técnica, no sólo es posible
obtener información sobre la orientación de las moléculas sobre la superficie sino también,
estimar a qué distancia se hallan de la misma. Cuando se forman multicapas pueden existir
moléculas con orientaciones al azar y a una distancia tal de la superficie que generen señales
en un espectro colectado con radiación s‐polarizada. Teniendo esto en cuenta, esta técnica
permite estimar el grado de cubrimiento de una superficie, haciendo posible diferenciar
cuándo se está en presencia de una monocapa o de multicapas, según las señales observadas
en los espectros obtenidos con ambos tipos de radiación.
En base a esto, la ausencia de señales en los espectros obtenidos con luz s‐polarizada
confirmaría cubrimientos en el orden de la monocapa para todos los sistemas analizados.
Los espectros obtenidos con luz p‐polarizada presentan la señal a ≈ 1740 cm‐1 correspondiente
al estiramiento antisimétrico del carbonilo del grupo –COOH (`a C=O), pero la intensidad de las
mismas es muy débil y, sumado a esto, la presencia de humedad ambiente dificulta la
identificación de más señales que podrían estar presentes. Es por ello que se analizaron todos
estos sistemas por PM‐IRRAS, como se describe a continuación.
Figura 13 Espectros FT‐IRRAS, tomados con radiación p‐polarizada, de sendas superficies de oro modificadas
con MHDA, MUA y HSPhCOOH, respectivamente.

3. DERIVATIZACIÓN Y CARACTERIZACIÓN SUPERFICIAL
77
3.2.1.3 CARACTERIZACIÓN SUPERFICIAL POR PM‐IRRAS
En la Figura 14 se presentan los espectros PM‐IRRAS de las superficies de oro modificadas con
los tioles alifáticos, es decir, con MHDA, MUA, MOA, MHA y MPA, respectivamente. En la parte
a) de dicha figura se muestran los espectros obtenidos con el PEM optimizado en
゜o = 2900 cm‐1 y en la b), en ゜o = 1450 cm‐1 (ver Capítulo 2). Las señales observadas en la Figura
14 a) corresponden a los modos de estiramiento antisimétrico y simétrico de los metilenos
(`a CH2 y `S CH2) de las cadenas alifáticas de los tioles a ≈ 2920 cm‐1 y ≈ 2850 cm‐1,
respectivamente.
Se puede observar que a medida que aumenta el largo de cadena estas bandas se desplazan
hacia menor frecuencia y tienen un menor ancho a media altura. Esta observación fue
reportada por Porter et al. [250] y fue atribuida al hecho de que, a medida que aumenta el
número de metilenos en la cadena alifática de un alcanotiol, aumentan las interacciones entre
las cadenas vecinas en una monocapa autoensamblada del mismo. Esto genera un mayor
orden en la misma que se evidencia en el corrimiento de las bandas de los modos `a CH2 y
`S CH2 hacia menores números de onda. El ancho de las bandas está relacionado con el número
de dominios que existen en la monocapa, cuánto mayor sea este número habrá mayor
dispersión en las frecuencias de los modos mencionados y por ende mayores ancho de banda
[339].
Figura 14 Espectros PM‐IRRAS de Au/MHDA, Au/MUA, Au/MOA, Au/MHA y Au/MPA tomados con
a) ゜o = 2900 cm‐1 y b) ゜o = 1450 cm‐1. El sistema Au/MPA no presentó señales en la región de 2900 cm‐1.
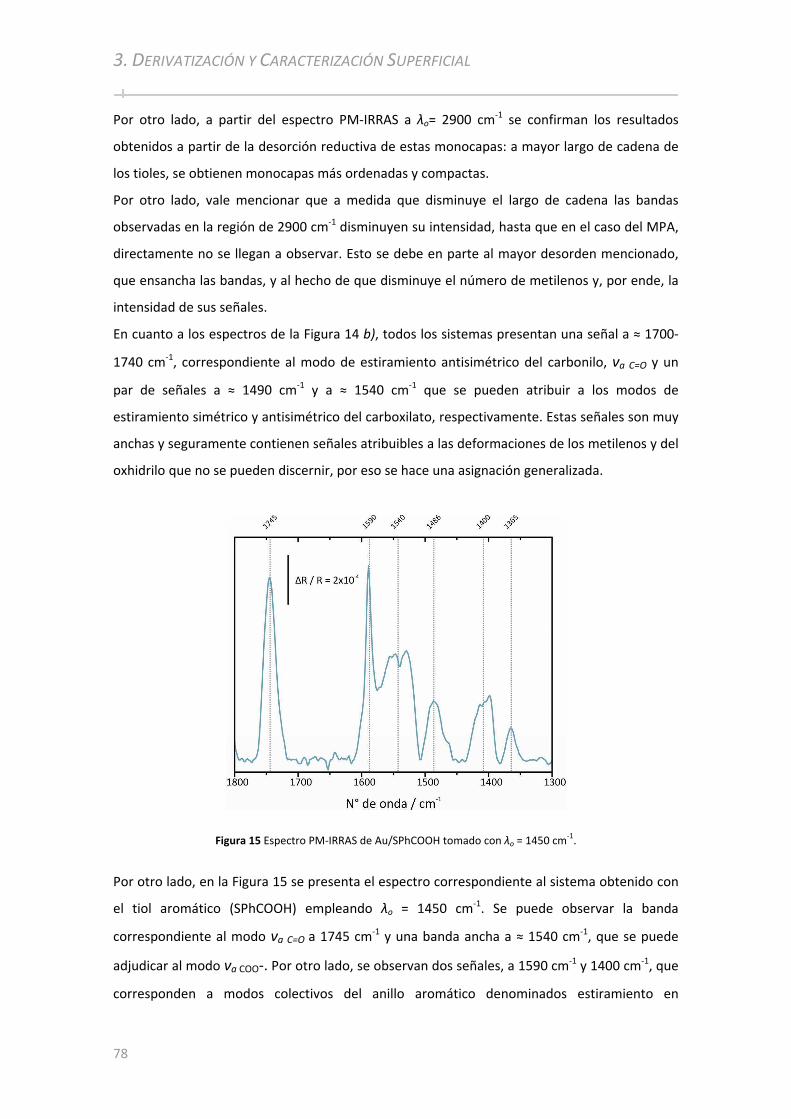
3. DERIVATIZACIÓN Y CARACTERIZACIÓN SUPERFICIAL
78
Por otro lado, a partir del espectro PM‐IRRAS a ゜o= 2900 cm‐1 se confirman los resultados
obtenidos a partir de la desorción reductiva de estas monocapas: a mayor largo de cadena de
los tioles, se obtienen monocapas más ordenadas y compactas.
Por otro lado, vale mencionar que a medida que disminuye el largo de cadena las bandas
observadas en la región de 2900 cm‐1 disminuyen su intensidad, hasta que en el caso del MPA,
directamente no se llegan a observar. Esto se debe en parte al mayor desorden mencionado,
que ensancha las bandas, y al hecho de que disminuye el número de metilenos y, por ende, la
intensidad de sus señales.
En cuanto a los espectros de la Figura 14 b), todos los sistemas presentan una señal a ≈ 1700‐
1740 cm‐1, correspondiente al modo de estiramiento antisimétrico del carbonilo, `a C=O y un
par de señales a ≈ 1490 cm‐1 y a ≈ 1540 cm‐1 que se pueden atribuir a los modos de
estiramiento simétrico y antisimétrico del carboxilato, respectivamente. Estas señales son muy
anchas y seguramente contienen señales atribuibles a las deformaciones de los metilenos y del
oxhidrilo que no se pueden discernir, por eso se hace una asignación generalizada.
Por otro lado, en la Figura 15 se presenta el espectro correspondiente al sistema obtenido con
el tiol aromático (SPhCOOH) empleando ゜o = 1450 cm‐1. Se puede observar la banda
correspondiente al modo `a C=O a 1745 cm‐1 y una banda ancha a ≈ 1540 cm‐1, que se puede
adjudicar al modo `a COO‐. Por otro lado, se observan dos señales, a 1590 cm‐1 y 1400 cm‐1, que
corresponden a modos colectivos del anillo aromático denominados estiramiento en
Figura 15 Espectro PM‐IRRAS de Au/SPhCOOH tomado con ゜o = 1450 cm‐1.

3. DERIVATIZACIÓN Y CARACTERIZACIÓN SUPERFICIAL
79
cuadrante (`q Ph) y estiramiento semicircular (`SC Ph), respectivamente. Las demás señales se
pueden atribuir a las deformaciones del oxhidrilo y del anillo. Estas observaciones concuerdan
muy bien con lo reportado en bibliografía para este sistema [340].
Todas las señales observadas en las Figuras 14 y 15 se resumen en la Tabla 2 junto con sus
asignaciones.
Tabla 2. Asignación de las señales observadas en los espectros PM‐IRRAS tomados luego de la
derivatización inicial de las superficies de oro. Todas las frecuencias están expresadas en cm‐1.
Au/MHDA Au/MUA Au/MOA Au/MHA Au/MPA Au/SPhCOOH Asignación
2920 2922 2926 2929 `a CH2
2850 2854 2854 2858 `s CH2
1741 1741 1714 1726 1722 1745 `a C=O
1590 `q Ph
1531 1529 1545 1545 1530 1540 `a COO‐
1491 1491 1491 1486 `s COO‐
1466 1468 1433 1458 1443 ~CH2
1400 `SC Ph
1365 ~ C‐H
3.2.2 DERIVATIZACIÓN DE SUPERFICIES DE ORO POR ELECTRO‐REDUCCIÓN DE SALES DE DIAZONIO
3.2.2.1 ELECTRO‐DEPOSICIÓN DE 4‐NITROBENCENO
Para derivatizar superficies por electro‐reducción de sales de diazonio, es necesario
determinar las condiciones experimentales particulares para cada sal y cada sustrato. Es por
ello que se empleó la espectroscopía infrarroja de absorción‐reflexión (FT‐IRRAS) para
caracterizar las capas depositadas en diferentes condiciones que se detallarán a continuación.
Para optimizar el procedimiento, se empleó la sal de diazonio derivada de la p‐nitroanilina, ya
que la presencia del grupo nitro genera señales intensas en el infrarrojo que facilitan su
análisis. Luego se realizó la reducción de la sal de diazonio del ácido 4‐aminobenzoico y su
caracterización correspondiente.
La Figura 7 muestra la reducción electroquímica de tetrafluoroborato de 4‐nitrobencen‐
diazonio por voltametría cíclica sobre un electrodo de oro. En ella se observa un pico catódico
a 0.4 V vs. Ag/AgCl que corresponde a la reacción esquematizada en la Figura 6. A partir del

3. DERIVATIZACIÓN Y CARACTERIZACIÓN SUPERFICIAL
80
mismo se eligieron tres condiciones experimentales diferentes para depositar 4‐nitrobenceno
sobre superficies de oro:
1) Voltametría cíclica: tres barridos consecutivos de potencial entre 0.7 y 0.2 V a 5 mV s‐1
(Figura 7).
2) Cronoamperometría: se aplicaron saltos de potencial desde el potencial de circuito abierto
hasta 0.4, 0.1, 0.4 y 0.1 V, respectivamente y en forma consecutiva. En cada caso se mantuvo
el potencial final durante 5 minutos. El objetivo de este tratamiento fue analizar la evolución
de la deposición en condiciones “suaves” de reducción, ya que el potencial aplicado fue
siempre positivo y no se alejó mucho del potencial de reducción de equilibrio.
3) Cronoamperometría: se aplicó un salto de potencial desde el potencial de circuito abierto
hasta ‐0.7 V, manteniendo este potencial durante 5 minutos. En este caso se buscaron
condiciones extremas de deposición con el objetivo de formar multicapas en la superficie.
En todos los casos, la reacción se llevó a cabo en una solución de O2NPhN2BF4 5 mM y Bu4NBF4
0.1 M en acetonitrilo.
3.2.2.1.1 Caracterización por FT‐IRRAS
Los sistemas obtenidos por electro‐reducción de tetrafluoroborato 4‐nitrobencendiazonio en
las distintas condiciones mencionadas fueron caracterizados por espectroscopía infrarroja,
empleando luz s‐ y p‐polarizada.
Figura 16 a) Espectros FT‐IRRAS de una superficie de oro modificada en una solución de tetrafluoroborato de
4‐nitrobencendiazonio por voltametría cíclica. Se muestran los espectros tomados con luz s‐ y p‐polarizada.
b) Espectro FT‐IR de una pastilla de dicha sal en KBr.

3. DERIVATIZACIÓN Y CARACTERIZACIÓN SUPERFICIAL
81
La Figura 16 a) muestra los espectros infrarrojos del sistema obtenido por el tratamiento 1), es
decir, por voltametría cíclica. Mientras que el espectro tomado con luz s‐polarizada no
muestra señales bien definidas, el espectro obtenido con luz p‐polarizada presenta dos señales
intensas a 1528 cm‐1 y 1351 cm‐1, que se pueden adjudicar a los modos de estiramiento
antisimétrico y simétrico del grupo nitro (`a N‐O y `s N‐O), respectivamente. Además en el
mismo, se observa una banda a 1602 cm‐1 que corresponde al modo colectivo en cuadrante
del anillo aromático, así como señales a 1264 cm‐1 y 1111 cm‐1, que son atribuidas a la
deformación ~C=C y, finalmente, una banda a 857 cm‐1 debida a la vibración de los enlaces C‐H
del anillo (w C‐H). Todas ellas están detalladas en la Tabla 3 [341], junto con las señales halladas
en el espectro de transmisión de una pastilla de la sal de diazonio en KBr (Figura 16 b)).
Considerando la regla de selección superficial, se puede estimar que los espectros de la Figura
17 a) indican que las moléculas de 4‐nitrobenceno tienen cierta orientación en la dirección
normal a la superficie y que se encuentran a una corta distancia de la misma (< 1 ´m), ya que
la línea de base del espectro tomado con luz s‐polarizada es ruidosa y, en principio, no se
observan señales en el mismo. Estas observaciones indican que en este caso se ha depositado
una capa delgada de nitrobenceno sobre la superficie de oro.
Por otro lado y a modo comparativo, se muestra el espectro infrarrojo de una pastilla de la sal
de diazonio utilizada en KBr (Figura 16 b)). Se puede apreciar que el espectro tomado luego de
la modificación de la superficie no presenta la señal correspondiente al modo de estiramiento
del enlace C‐N característico del grupo diazonio (`C‐N2+) a 2277 cm‐1, que sí está presente en el
espectro de la pastilla. Esto confirma la pérdida de N2 (g) durante la electro‐reducción y la
formación del radical arilo sobre la superficie de oro.
A su vez, la Figura 17 a) muestra los resultados obtenidos con una superficie de oro que fue
sometida al tratamiento indicado en el punto 2). Se presentan todos los espectros obtenidos
con luz p‐polarizada y el espectro adquirido con radiación s‐polarizada después del primer
salto de potencial, ya que los que se tomaron posteriormente no mostraron diferencias
importantes con el primero.

3. DERIVATIZACIÓN Y CARACTERIZACIÓN SUPERFICIAL
82
Mientras que los espectros obtenidos con luz s‐polarizada resultaron ser silenciosos en la
región 700‐2200 cm‐1, la radiación p‐polarizada muestra las bandas características detalladas
en la Tabla 3.
Luego de la primera cronoamperometría (a 0.4 V), aparecen las señales mencionadas y éstas
muestran un aumento importante en su intensidad luego de la segunda cronoamperometría (a
0.1 V). Esto se observa claramente en la Figura 17 b), en la que sólo se ha incluido la variación
de intensidad de la señal a 1527 cm‐1, que aumenta entre el primer y segundo salto de
potencial y luego, se mantiene prácticamente constante cuando se revierte el potencial a 0.4 V
nuevamente.
Por otro lado, si se comparan los espectros obtenidos con luz s‐polarizada de las Figuras 16 a) y
17 a), se puede observar que el primero presenta indicios de señales 1358 y 857 cm‐1, mientras
que el espectro obtenido luego de una cronoamperometría a 0.4 V no muestra ningún tipo de
señal. Por lo tanto, la espectroscopía infrarroja permite diferenciar a los sistemas obtenidos
por ambos procedimientos, ya que la voltametría cíclica parece generar la deposición de más
capas moleculares que la cronoamperometría realizada en condiciones “suaves” de electro‐
deposición.
Figura 17 a) Espectros FT‐IRRAS de un electrodo de oro modificado por cronoamperometría en una solución de
tetrafluoroborato de 4‐nitrobencendiazonio. Se muestran los espectros tomados con radiación p‐polarizada
luego de cada salto de potencial y el espectro adquirido con luz s‐polarizada luego del primer salto. b) Variación
de la intensidad de la señal observada a 1527 cm‐1 con el potencial aplicado.

3. DERIVATIZACIÓN Y CARACTERIZACIÓN SUPERFICIAL
83
Por último, la Figura 18 muestra el espectro de la superficie modificada en las condiciones más
extremas, es decir, por cronoamperometría a un potencial de ‐0.7 V. En este caso, se observan
señales con ambos tipos de radiación que, además, son mayores que las observadas en los
espectros de la Figura 17 a). Este hecho indica que existen moléculas en la muestra que tienen
una componente del momento de transición dinámico paralela a la superficie y que se
encuentran a una distancia de la misma lo suficientemente grande como para generar señales
con la radiación s‐polarizada. A partir de esto se puede inferir que en estas condiciones ocurre
la ramificación de los anillos aromáticos por el ataque radicalario esquematizado en la Figura
10 con la consiguiente formación de capas gruesas o multicapas sobre la superficie [190, 243].
Figura 18 Espectros FT‐IRRAS de una superficie de oro modificada en una solución de tetrafluoroborato de
4‐nitrobencendiazonio por cronoamperometría. Se aplicó un salto de potencial desde el potencial de circuito
abierto hasta ‐0.7 V. Se muestran los espectros tomados con luz s‐ y p‐polarizada.

3. DERIVATIZACIÓN Y CARACTERIZACIÓN SUPERFICIAL
84
Tabla 3. Asignación de señales observadas en el espectro de transmisión de O2N‐Ph‐N2BF4 (KBr) y en
los espectros FT‐IRRAS de 4‐nitrobenceno depositado sobre oro. Todas las frecuencias están
expresadas en cm‐1.
O2N‐Ph‐N2+BF4
‐ (KBr) Au/PhNO21 Asignación
3038 ` C‐H
3031 ` C‐H
2992 ` C‐H
2277 ` C‐N2+
1610 1602 `q C=C
1531 1537 `a N‐O
1355 1358 `s N‐O
1264 ~ C=C
1111 ~ C=C
1085 ` BF4‐
869 857 w C‐H
744 w C‐H
3.2.2.2 ELECTRO‐DEPOSICIÓN DE ÁCIDO BENZOICO
Las superficies de oro modificadas con ácido benzoico se prepararon por reducción de la sal de
diazonio derivada del ácido 4‐aminobenzoico. Para ello se realizó una voltametría cíclica en
una solución de tetrafluoroborato de 4‐carboxibencendiazonio (Figura 19), a partir de la cual
se identificó el potencial de reducción de esta sal a 0.3 V vs. Ag / AgCl.
A partir de este resultado, se modificó una superficie de oro con ácido benzoico por
cronoamperometría a 0.3 V.
1 4‐nitrobenceno unido a la superficie de oro luego de la reducción de la sal de diazonio
correspondiente.
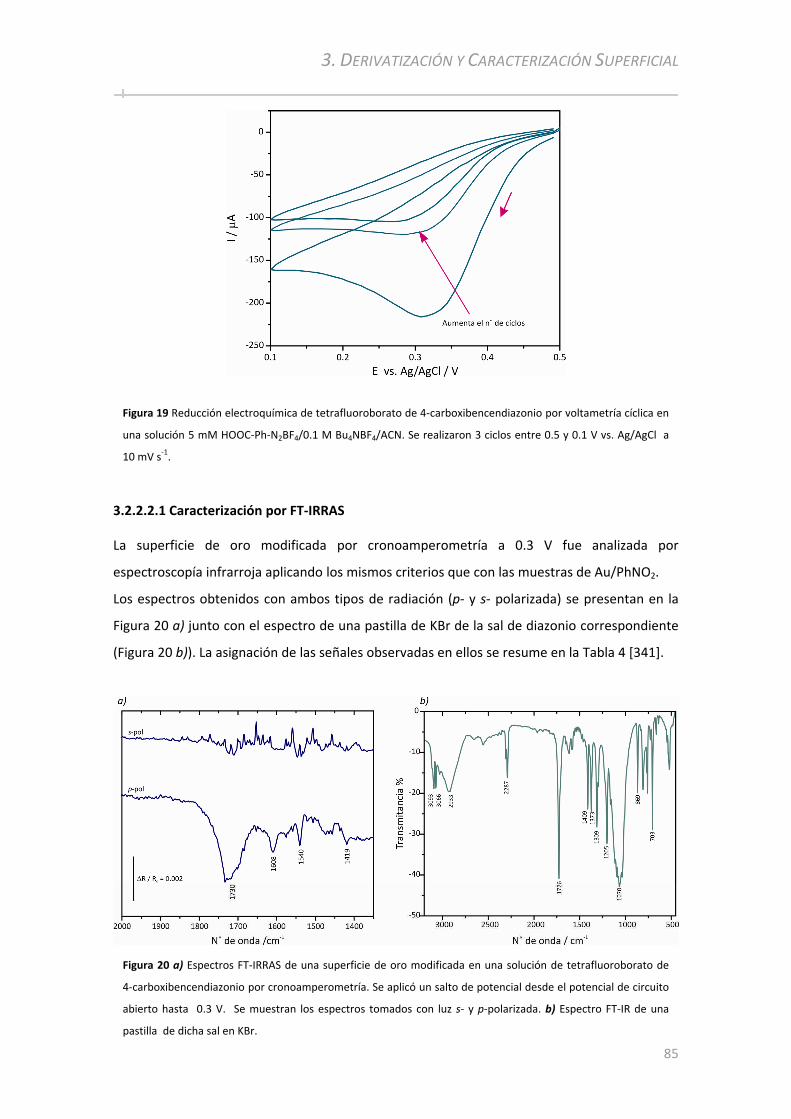
3. DERIVATIZACIÓN Y CARACTERIZACIÓN SUPERFICIAL
85
Figura 20 a) Espectros FT‐IRRAS de una superficie de oro modificada en una solución de tetrafluoroborato de
4‐carboxibencendiazonio por cronoamperometría. Se aplicó un salto de potencial desde el potencial de circuito
abierto hasta 0.3 V. Se muestran los espectros tomados con luz s‐ y p‐polarizada. b) Espectro FT‐IR de una
pastilla de dicha sal en KBr.
3.2.2.2.1 Caracterización por FT‐IRRAS
La superficie de oro modificada por cronoamperometría a 0.3 V fue analizada por
espectroscopía infrarroja aplicando los mismos criterios que con las muestras de Au/PhNO2.
Los espectros obtenidos con ambos tipos de radiación (p‐ y s‐ polarizada) se presentan en la
Figura 20 a) junto con el espectro de una pastilla de KBr de la sal de diazonio correspondiente
(Figura 20 b)). La asignación de las señales observadas en ellos se resume en la Tabla 4 [341].
Figura 19 Reducción electroquímica de tetrafluoroborato de 4‐carboxibencendiazonio por voltametría cíclica en
una solución 5 mM HOOC‐Ph‐N2BF4/0.1 M Bu4NBF4/ACN. Se realizaron 3 ciclos entre 0.5 y 0.1 V vs. Ag/AgCl a
10 mV s‐1.

3. DERIVATIZACIÓN Y CARACTERIZACIÓN SUPERFICIAL
86
Tabla 4. Asignación de señales observadas en el espectro de transmisión de HOOC‐Ph‐N2BF4 (KBr) y en
los espectros FT‐IRRAS de ácido benzoico depositado sobre oro. Todas las frecuencias están
expresadas en cm‐1.
HOOC‐Ph‐N2+BF4
‐ (KBr) Au/PhCOOH 2 Asignación
3093 ` C‐H
3066 ` C‐H
2933 ` O‐H
2287 ` C‐N2+
1726 1730 `a C=O
1608 `q Ph3
1540 `a COO‐
1409 1419 ` sc Ph4
1373 ~ COH en el plano (dímero)
1309 ` C‐O (dímero)
1205 ~ C‐H
1070 ` BF4‐
869 w C‐H5
703 w C‐H4
Como se puede apreciar en la Figura 20 a), el espectro tomado con luz s‐polarizada no tiene
señales definidas, mientras que la luz p‐polarizada revela señales correspondientes al grupo
–COOH y al anillo aromático. Las bandas a 1730 cm‐1 y 1540 cm‐1 corresponden a los modos de
estiramiento antisimétrico del carbonilo y del carboxilato (`a C=O y `a COO‐), respectivamente,
mientras que las señales observadas a 1608 cm‐1 y 1419 cm‐1 se pueden atribuir a modos de
estiramiento colectivos del anillo aromático: en cuadrante (`q Ph) y semicircular (`SC Ph),
respectivamente.
2Ácido benzoico unido a una superficie de oro luego de la reducción de la sal de diazonio correspondiente. 3 Se refiere a un modo de estiramiento colectivo del anillo aromático en cuadrante. 4 Se refiere a un modo de estiramiento semicircular colectivo del anillo aromático. 5 Se refiere a un modo de deformación de los enlaces C‐H del anillo aromático.

3. DERIVATIZACIÓN Y CARACTERIZACIÓN SUPERFICIAL
87
Estos resultados indican que el procedimiento aplicado genera la unión de moléculas de ácido
benzoico a la superficie que tienen una componente del momento de transición en la dirección
normal a la misma. Además, al igual que en el caso del 4‐nitrobenceno, la cantidad de material
depositado corresponde a unas pocas capas moleculares, ya que las moléculas detectadas se
hallan a una corta distancia de la superficie como lo indica la ausencia de señales cuando se
emplea luz s‐polarizada.
3.2.2.2.2 Caracterización por PM‐IRRAS
Si bien en el espectro FT‐IRRAS presentado en la Figura 20 a) muestra las señales esperadas
para una capa delgada de ácido 4‐benzoico sobre oro, dicho sistema también fue analizado por
PM‐IRRAS, de la misma manera que se hizo con los sustratos modificados con tioles. El
espectro obtenido con ゜o = 1450 cm‐1 se muestra en la Figura 21.
Se puede observar la presencia de las bandas correspondientes a los modos `a C=O y `a COO‐
a ≈ 1693 y ≈ 1540 cm‐1, respectivamente. Además, se observan las señales de los modos de
estiramiento en cuadrante y semicircular del anillo aromático a 1610 cm‐1 y a 1406 cm‐1,
respectivamente. Ambas señales están presentes en el espectro obtenido con el sistema
Au/SPhCOOH (Figura 15). En este caso, la primera de ellas está levemente desplazada a menor
frecuencia, 1590 cm‐1. Este desplazamiento se puede explicar teniendo en cuenta que las
frecuencias de vibración de los enlaces C‐X, cuando el carbono pertenece a un anillo aromático
y X es Cl o S, son menores que la correspondiente al caso en que X es un carbono también.
Figura 21 Espectros PM‐IRRAS de Au/PhCOOH tomado con ゜o = 1450 cm‐1.

3. DERIVATIZACIÓN Y CARACTERIZACIÓN SUPERFICIAL
88
Esto se debe a que el enlace C‐X es más débil que el enlace C‐C y por ende vibra a menor
frecuencia [341]. En principio esto indicaría entonces que el enlace entre el C del anillo y un
átomo de Au del sustrato es más fuerte que el enlace C‐S. Finalmente, se observa una banda
correspondiente al modo de deformación del anillo (~C‐H) a 1329 cm‐1.
3.2.2.2.3 Comparación entre capas delgadas y capas gruesas
3.2.2.2.3.1 Caracterización por PM‐IRRAS
En la Figura 22 se presenta el espectro obtenido para un sustrato de oro modificado por
reducción electroquímica de tetrafluoroborato de carboxibencendiazonio (HOOC‐Ph‐N2BF4) a
‐0.7 V, durante 5 minutos. En este caso se espera la formación de capas gruesas sobre el
electrodo debido a que se aplica un potencial muy catódico.
En líneas generales, las señales presentes en el nuevo espectro, coinciden con las ya
observadas para la capa delgada de ácido benzoico. El aspecto más importante de este
espectro es la aparición de una nueva señal a 1377 cm‐1 que no está presente en el espectro
de la capa delgada. La misma puede ser asignada a un modo de estiramiento del grupo azo,
‐N=N‐, de un trans‐azobenceno (`N=N) [341]. La presencia de este tipo de moléculas sobre la
superficie ya ha sido reportada por otros autores y la misma ha sido adjudicada a ya sea 1) la
reacción directa del catión diazonio con grupos funcionales propios de la superficie del
Figura 22 Espectro PM‐IRRAS con ゜o = 1450 cm‐1 de una capa gruesa (multicapas) obtenida por reducción de
tetrafluoroborato de 4‐carboxibencendiazonio a ‐0.7 V.

3. DERIVATIZACIÓN Y CARACTERIZACIÓN SUPERFICIAL
89
electrodo o 2) la formación de un radical azofenilo luego de la reducción de la sal de diazonio y
su unión directa a la superficie del electrodo o a un anillo aromático ya unido a la misma [342].
La primera explicación es más adecuada para un electrodo de carbono [341], mientras que la
segunda es más apropiada para uno de oro [327].
Ambas posibilidades se esquematizan en la Figura 23. En estas condiciones, la capa depositada
consistiría en una mezcla de bifenilos y azobencenos. Por lo tanto, la banda presente en el
espectro PM‐IRRAS a 1377 cm‐1, es consistente con la presencia de azobencenos en las
multicapas de ácido benzoico sobre oro.
3.2.2.2.3.2 Caracterización por XPS
Las superficies de oro modificadas con ácido benzoico por cronoamperometría a 0.3 V y a
‐0.7 V también fueron caracterizadas por XPS. En ambos casos se observaron señales
correspondientes a los niveles C 1s, O 1s y N 1s [327]. En particular, la región centrada en
≈ 400 eV fue la más llamativa debido a que se observó una banda que se puede adjudicar al
nivel N 1s. En la Figura 24 se muestran estas señales y se puede observar que la muestra
obtenida al potencial más catódico (línea roja), presentó una señal más intensa
(aproximadamente, el triple) que la obtenida por reducción a 0.3 V (línea azul).
La presencia de esta señal en el espectro XPS se puede adjudicar a la formación de enlaces
N=N durante la reducción de la sal de diazonio. De esta manera este resultado está en
concordancia con los resultados obtenidos por FT‐IRRAS, que permitió corroborar que cuando
la electro‐reducción de sales de diazonio se lleva a cabo en condiciones muy reductoras, se
Figura 23 Esquema de un mecanismo alternativo de reacción entre una sal de diazonio y la superficie de un
electrodo con la consiguiente formación de grupos azo sobre la superficie.
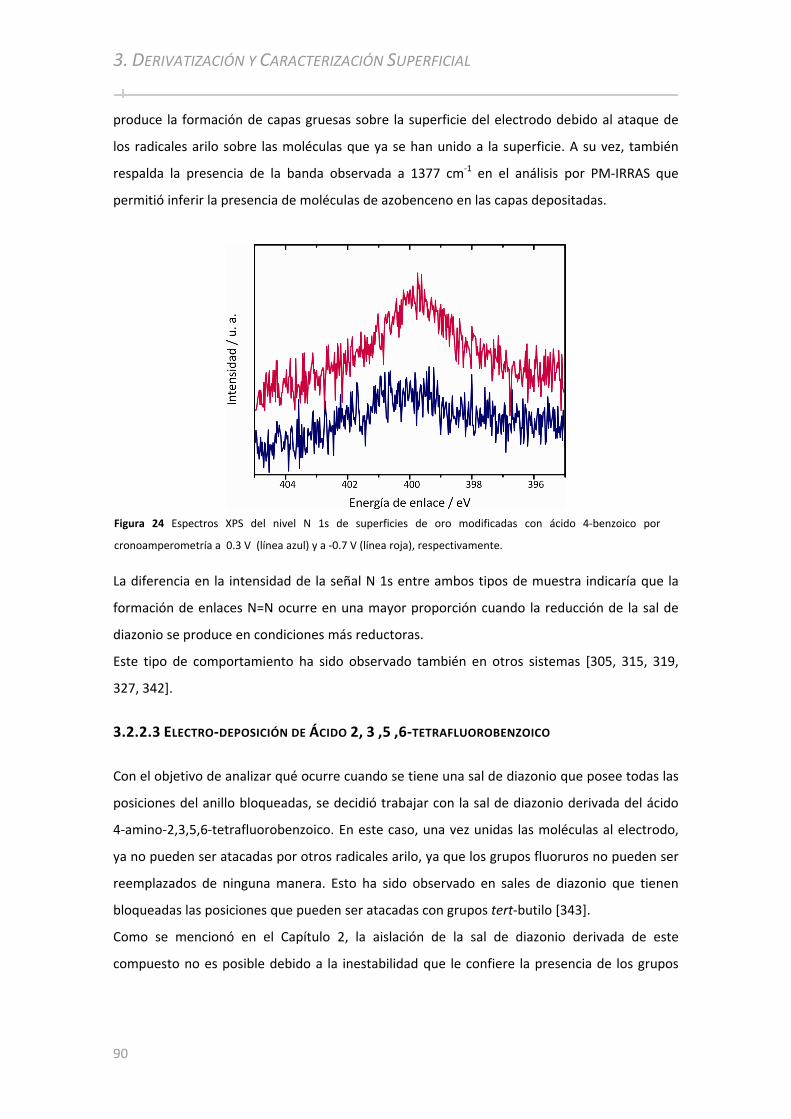
3. DERIVATIZACIÓN Y CARACTERIZACIÓN SUPERFICIAL
90
produce la formación de capas gruesas sobre la superficie del electrodo debido al ataque de
los radicales arilo sobre las moléculas que ya se han unido a la superficie. A su vez, también
respalda la presencia de la banda observada a 1377 cm‐1 en el análisis por PM‐IRRAS que
permitió inferir la presencia de moléculas de azobenceno en las capas depositadas.
La diferencia en la intensidad de la señal N 1s entre ambos tipos de muestra indicaría que la
formación de enlaces N=N ocurre en una mayor proporción cuando la reducción de la sal de
diazonio se produce en condiciones más reductoras.
Este tipo de comportamiento ha sido observado también en otros sistemas [305, 315, 319,
327, 342].
3.2.2.3 ELECTRO‐DEPOSICIÓN DE ÁCIDO 2, 3 ,5 ,6‐TETRAFLUOROBENZOICO
Con el objetivo de analizar qué ocurre cuando se tiene una sal de diazonio que posee todas las
posiciones del anillo bloqueadas, se decidió trabajar con la sal de diazonio derivada del ácido
4‐amino‐2,3,5,6‐tetrafluorobenzoico. En este caso, una vez unidas las moléculas al electrodo,
ya no pueden ser atacadas por otros radicales arilo, ya que los grupos fluoruros no pueden ser
reemplazados de ninguna manera. Esto ha sido observado en sales de diazonio que tienen
bloqueadas las posiciones que pueden ser atacadas con grupos tert‐butilo [343].
Como se mencionó en el Capítulo 2, la aislación de la sal de diazonio derivada de este
compuesto no es posible debido a la inestabilidad que le confiere la presencia de los grupos
Figura 24 Espectros XPS del nivel N 1s de superficies de oro modificadas con ácido 4‐benzoico por
cronoamperometría a 0.3 V (línea azul) y a ‐0.7 V (línea roja), respectivamente.

3. DERIVATIZACIÓN Y CARACTERIZACIÓN SUPERFICIAL
91
fluoruro en el anillo. Por eso se realizó la reacción de diazotación en forma simultánea con la
reducción.
De esta manera, se prepararon superficies modificadas con ácido 2,3,5,6‐tetrafluorobenzoico
por reducción de la sal correspondiente a –0.7 V, ‐0.2 V y 0 V, respectivamente. El objetivo fue
analizar la estructura de las capas depositadas en diferentes condiciones reductoras,
considerando que, aún a ‐0.7 V, en este sistema no se pueden formar multicapas sobre la
superficie. Los sistemas así obtenidos se caracterizaron por PM‐IRRAS y por XPS.
3.2.2.3.1 Caracterización por PM‐IRRAS
En la Figura 25 se presentan los espectros PM‐IRRAS de un sustrato de oro modificado con
ácido 2,3,5,6‐tetrafluorobenzoico a partir de la electro‐reducción de la sal de diazonio
correspondiente por cronoamperometría a ‐0.2 V. Los mismos fueron obtenidos con
゜o = 1200 cm‐1 y ゜o = 1500 cm‐1, respectivamente. Se eligieron estas longitudes de onda para
explorar las regiones del espectro en que aparecen las señales de los modos asociados al
enlace C‐F, al grupo –COOH y al anillo aromático. Ambas regiones se analizaron por separado
para observar mejor las señales de interés. Las muestras obtenidas por reducción de la sal a 0
V y ‐0.7 V presentaron las mismas señales que se observan en la Figura 25.
Las bandas presentes a 1273 cm‐1 y 1203 cm‐1 se pueden adjudicar al modo de estiramiento de
las uniones C‐F (`C‐F). A su vez, la señal presente 1720 cm‐1 corresponde al modo de
estiramiento del carbonilo (`C=O), la señal a 1630 cm‐1 se puede asignar al modo de
estiramiento en cuadrante del anillo aromático (`q Ph) y la señal a 1485 cm‐1, al modo de
estiramiento simétrico del carboxilato (`s COO‐).
Figura 25 Espectros PM‐IRRAS de un sustrato de oro modificado con ácido 2,3,5,6‐tetrafluorobenzoico por
reducción a ‐0.2 V con a) ゜o = 1200 cm‐1 y b) ゜o = 1500 cm‐1

3. DERIVATIZACIÓN Y CARACTERIZACIÓN SUPERFICIAL
92
Finalmente, al igual que en las capas gruesas de ácido benzoico, se observa una señal a
1392 cm‐1 que puede asociarse al modo de estiramiento de un enlace N=N (`N=N). Sin embargo,
en este caso no puede existir sustitución directa en el anillo debido a la presencia de los grupos
fluoruro, y la presencia de grupos azo en la monocapa puede adjudicarse a la unión directa a la
superficie de los anillos a través del grupo N=N como se ejemplifican en la Figura 26.
La señal asignada al modo `N=N fue comparable en las tres muestras, es decir,
independientemente de las condiciones aplicadas en la reducción de la sal de diazonio esta
señal presentó una intensidad similar. Este hecho diferencia a este sistema del sistema
Au/PhCOOH, en el que la unión a la superficie por uniones N=N se minimiza cuando la
reducción de la sal de diazonio se realiza en condiciones “suaves”.
3.2.2.3.2 Caracterización por XPS
Las tres muestras preparadas por reducción de la sal de diazonio derivada del ácido 1‐amino‐
2,3,5,6‐tetrafluorobenzoico a ‐0.7, ‐0.2 y 0 V también fueron analizadas por XPS. En todos los
espectros obtenidos se observaron las bandas correspondientes a los niveles Au 4f, C 1s, O 1s,
N 1s [327] y F 1s [344]. En particular, en la Figura 27, se muestran las señales correspondientes
a estos dos últimos niveles.
Teniendo en cuenta que las señales de C y O pueden presentar una cierta contribución de
contaminación propia de este tipo de muestras, se empleó la señal de F 1s para cuantificar el
cubrimiento superficial de ácido 2,3,5,6‐tetrafluorobenzoico alcanzado en cada caso. Para ello
se empleó la ecuación (3.1):
Figura 26 Formas de unión posibles del ácido 1‐amino‐2,3,5,6‐tetrafluorobenzoico a un sustrato a partir de la
reducción de la sal de diazonio correspondiente.

3. DERIVATIZACIÓN Y CARACTERIZACIÓN SUPERFICIAL
93
a a Au Au Au Aa
Au Au a Au
KE I σ ~ ゜ N cosθよ
KE I σ Ar= (3.1)
donde Гa es el cubrimiento superficial del átomo a adsorbido sobre oro (átomos cm‐2), KE es la
energía cinética de los fotoelectrones, σi son las secciones eficaces de foto‐emisión calculadas
por Scofield [345], Ii es la intensidad de la señal obtenida por integración, ~Au es la densidad del
oro (19.32 g cm‐3), ゜Au es el camino libre medio de los electrones fotoemitidos en el oro
(1.8 x 10‐7 cm), θ es el ángulo de colección de los electrones (30°), ArAu es el peso atómico del
oro (196.97 g mol‐1) y NA es el número de Avogadro. Esta ecuación es válida para estimar
cubrimientos sólo cuando se trata de sustratos atómicamente planos cubiertos con capas lo
suficientemente delgadas como para poder suponer que las señales del sustrato no se ven
atenuadas. Por lo tanto, hay que tener en cuenta que cuando se calculan cubrimientos
superficiales por este método, no se está considerando la rugosidad que puede presentar el
sustrato.
De esta manera, se realizó una estimación del cubrimiento superficial de ácido
2,3,5,6‐tetrafluorobenzoico en las tres muestras que resultó ser en los tres casos
≈ 4.5 x 10‐10 mol cm‐2. Por otro lado, los sustratos empleados en este caso presentaron un
factor de rugosidad de ≈ 1.075 que fue estimado electroquímicamente como se detalló en el
Capítulo 2.
Al comienzo de este capítulo se señaló que el valor esperado para monocapas compactas de
anillos aromáticos p‐sustituidos es de ≈ 10‐10 mol cm‐2 [97, 327]. Sin embargo, en el caso de
Figura 27 Espectros XPS de los niveles a) F 1s y b) N 1s de superficies de oro modificadas con ácido
2,3,5,6‐tetrafluorobenzoico por reducción a ‐0.7 V (línea azul), ‐0.2 V (línea verde) y a 0 V (línea roja).
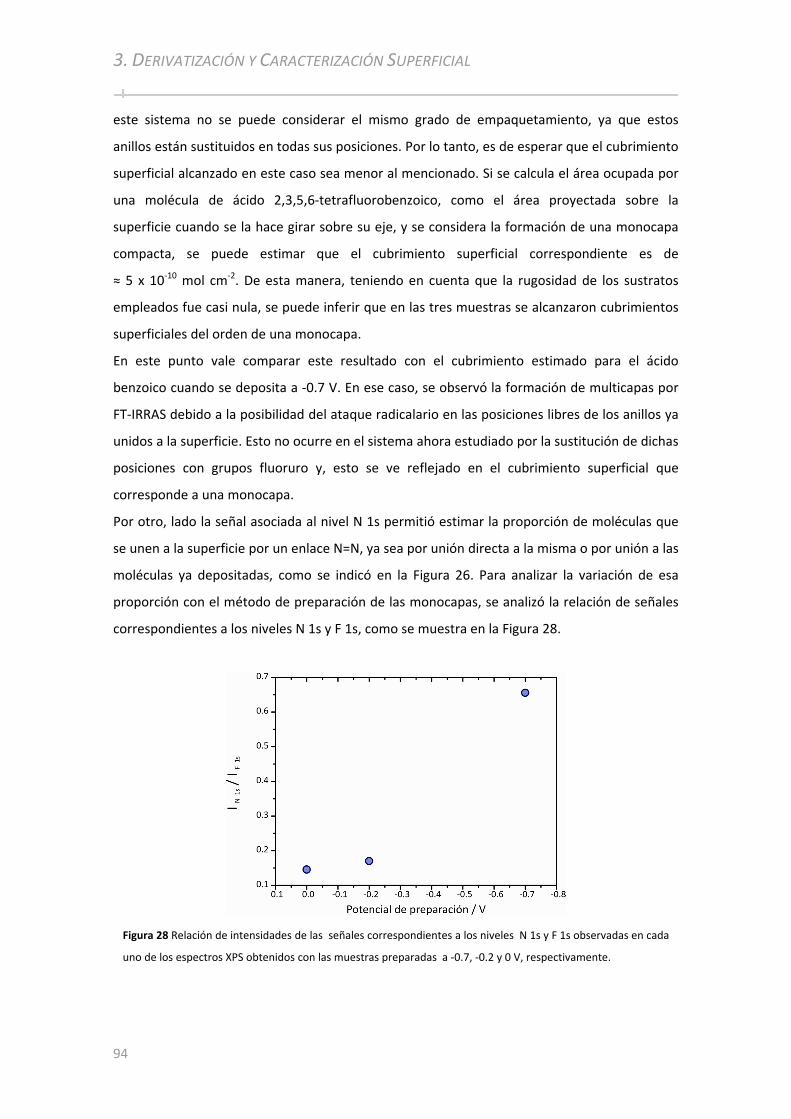
3. DERIVATIZACIÓN Y CARACTERIZACIÓN SUPERFICIAL
94
este sistema no se puede considerar el mismo grado de empaquetamiento, ya que estos
anillos están sustituidos en todas sus posiciones. Por lo tanto, es de esperar que el cubrimiento
superficial alcanzado en este caso sea menor al mencionado. Si se calcula el área ocupada por
una molécula de ácido 2,3,5,6‐tetrafluorobenzoico, como el área proyectada sobre la
superficie cuando se la hace girar sobre su eje, y se considera la formación de una monocapa
compacta, se puede estimar que el cubrimiento superficial correspondiente es de
≈ 5 x 10‐10 mol cm‐2. De esta manera, teniendo en cuenta que la rugosidad de los sustratos
empleados fue casi nula, se puede inferir que en las tres muestras se alcanzaron cubrimientos
superficiales del orden de una monocapa.
En este punto vale comparar este resultado con el cubrimiento estimado para el ácido
benzoico cuando se deposita a ‐0.7 V. En ese caso, se observó la formación de multicapas por
FT‐IRRAS debido a la posibilidad del ataque radicalario en las posiciones libres de los anillos ya
unidos a la superficie. Esto no ocurre en el sistema ahora estudiado por la sustitución de dichas
posiciones con grupos fluoruro y, esto se ve reflejado en el cubrimiento superficial que
corresponde a una monocapa.
Por otro, lado la señal asociada al nivel N 1s permitió estimar la proporción de moléculas que
se unen a la superficie por un enlace N=N, ya sea por unión directa a la misma o por unión a las
moléculas ya depositadas, como se indicó en la Figura 26. Para analizar la variación de esa
proporción con el método de preparación de las monocapas, se analizó la relación de señales
correspondientes a los niveles N 1s y F 1s, como se muestra en la Figura 28.
Figura 28 Relación de intensidades de las señales correspondientes a los niveles N 1s y F 1s observadas en cada
uno de los espectros XPS obtenidos con las muestras preparadas a ‐0.7, ‐0.2 y 0 V, respectivamente.

3. DERIVATIZACIÓN Y CARACTERIZACIÓN SUPERFICIAL
95
Se puede observar claramente que cuando se aplica el potencial más reductor, la proporción
de moléculas con un enlace N=N en su estructura aumenta cuánto más catódico es el potencial
aplicado en la reducción de la sal de diazonio.
Este resultado indica una clara dependencia entre la forma en que las moléculas se unen al
sustrato y el potencial aplicado para que esto ocurra. Este comportamiento también fue
observado en el sistema Au/PhCOOH a partir del aumento en la señal del nivel N 1s al aplicar
‐0.7 V en la reducción de la sal correspondiente.
3.3 CONCLUSIONES
En este capítulo se presentaron dos formas de derivatización de superficies: adsorción de
tioles y electro‐reducción de sales de diazonio. Ambas estrategias fueron empleadas para
funcionalizar sustratos de oro con grupos –COOH con el ulterior objetivo de unir a los mismos
el complejo [Os(2,2´‐bpy)2Cl(py‐CH2‐NH2)]+, como se presentará en el siguiente capítulo.
En el caso de las superficies modificadas con tioles se estimó el cubrimiento superficial por
desorción reductiva y se realizó un análisis espectroscópico con FT‐IRRAS y PM‐IRRAS.
La deposición electroquímica de sales de diazonio también fue caracterizada por estas técnicas
y se agregó el análisis por XPS.
A partir de todo esto se pudo confirmar la formación de capas delgadas (por reducción de
sales de diazonio) y de monocapas (por autoensamblado de tioles). La espectroscopía
infrarroja con modulación de la polarización (PM‐IRRAS) permitió detectar la presencia de los
grupos funcionales sobre la superficie con una alta sensibilidad al eliminar las interferencias
causadas por el agua y el dióxido de carbono atmosféricos. En el caso de los tioles alifáticos,
mediante el análisis por PM‐IRRAS y los datos obtenidos por desorción reductiva, se pudo
observar que cuánto mayor es el largo de cadena, más compactas son las monocapas que
forman debido a que se establecen interacciones de Van der Waals más fuertes entre las
moléculas sobre la superficie.
Finalmente, se debe destacar que se pudo trabajar con una sal de diazonio muy inestable (la
derivada del ácido 1‐amino‐2,3,5,6‐tetrafluorobenzoico) a partir de cuya reducción se
obtuvieron monocapas moleculares. Asimismo, se determinaron las condiciones de electro‐
deposición de capas delgadas y gruesas (o multicapas) para todas las sales de diazonio
utilizadas y se encontró una estrecha relación entre dichas condiciones y la estructura de las
películas obtenidas.


CAPÍTULO 4
POST‐FUNCIONALIZACIÓN Y CARACTERIZACIÓN


4. POST‐FUNCIONALIZACIÓN Y CARACTERIZACIÓN
99
na vez preparados los sustratos de oro derivatizados con moléculas terminadas en
grupos –COOH, se llevó a cabo la post‐funcionalización de los mismos con el complejo
[Os(2,2´‐bpy)2Cl(py‐CH2‐NH2)]PF6. De esta manera se obtuvieron los diferentes sistemas
modelo con el par Os(II)/Os(III) situado a una distancia controlada de la superficie metálica,
uniéndolo a la misma a través de puentes moleculares de distinta longitud y naturaleza.
En este capítulo se presenta la caracterización general de los sistemas preparados. Se
emplearon para ello Espectroscopía Infrarroja de Absorción‐Reflexión con Transformada de
Fourier (FT‐IRRAS), Espectroscopía Infrarroja de Absorción‐Reflexión con Modulación de la
Polarización (PM‐IRRAS), Espectroscopía Raman Resonante, Espectroscopía Fotoelectrónica de
Rayos X (XPS) y Microscopía de Efecto Túnel (STM). También se caracterizaron dichos sistemas
electroquímicamente con voltametría cíclica.
4.1 UNIÓN DEL PAR OS(II)/OS(III) A SUPERFICIES DE ORO
La unión del complejo de osmio a las superficies de oro modificadas con grupos –COOH se
llevó a cabo empleando 1‐etil‐3‐(3‐dimetilaminopropil)‐carbodiimida (EDC) para posibilitar la
reacción de los mismos con el grupo amino del complejo.
Las carbodiimidas son agentes deshidratantes que se utilizan en química orgánica para activar
ácidos carboxílicos en reacciones de sustitución nucleofílica, ya que reemplazan al grupo –OH y
se comportan como mejores grupos salientes. Es por ello que son empleadas en la preparación
de amidas o ésteres [320, 346‐348] y también, en síntesis de péptidos, entrecruzamiento de
proteínas y preparado de inmunoconjugados [349‐352]. En general, se pueden emplear
aditivos como N‐hidroxibenzotriazol o N‐hidroxisuccinimida para aumentar el rendimiento de
estas reacciones y minimizar otros procesos laterales. En este trabajo se utilizó N‐
hidroxisuccinimida (NHS).
La EDC es una carbodiimida soluble en agua que puede reaccionar con un ácido carboxílico
formando un intermediario O‐acilurea. Como se observa en la Figura 1, este intermediario
puede reaccionar con un grupo amino y formar una unión amida entre ambas partes o, debido
a su labilidad, hidrolizarse regenerando el ácido original. Es por ello que se agrega NHS al
medio de reacción, ya que en este caso, se forma un intermediario éster de mayor estabilidad
que aumenta la eficiencia de la reacción de interés.
U

4. POST‐FUNCIONALIZACIÓN Y CARACTERIZACIÓN
100
La post‐funcionalización realizada en este trabajo se puede visualizar en la Figura 1
considerando que la molécula unida al grupo –COOH (representada por 1) corresponde a la
superficie de oro con las cadenas alifáticas o los anillos aromáticos, según sea el caso. A su vez,
la molécula unida al grupo –NH2 (representada por 2) es el complejo de osmio. Por lo tanto,
teniendo en cuenta que la derivatización inicial de los sustratos de oro se realizó con los ácidos
3‐mercaptopropanoico (MPA), 6‐mercaptohexanoico (MHA), 8‐mercaptooctanoico (MOA), 11‐
mercaptoundecanoico (MUA), 16‐mercaptohexadecanoico (MHDA), 4‐mercaptobenzoico
(SPhCOOH), ácido benzoico (PhCOOH) y ácido 2,3,5,6‐tetrafluorobenzoico (PhF4COOH), en la
Figura 2 se presentan los sistemas modelo con los que se trabajó y que serán denominados de
aquí en adelante como: Au/MPA/Os, Au/MHA/Os, Au/MOA/Os, Au/MUA/Os, Au/MHDA/Os,
Au/SPhCOOH/Os, Au/PhCOOH/Os y Au/PhF4COOH/Os. Las siglas MPA, MHA, MOA, MUA,
MHDA, SPhCOOH, PhCOOH y PhF4COOH representan a las moléculas utilizadas en la
derivatización de los sustratos con grupos –COOH, y Os representa al complejo unido a los
mismos a través de la unión amida.
Figura 1 Mecanismo de formación de un enlace amida entre un ácido carboxílico y una amina en presencia de
1‐etil‐3‐(3‐dimetilaminopropil)‐carbodiimida (EDC) y de N‐hidroxisuccinimida (NHS).

4. POST‐FUNCIONALIZACIÓN Y CARACTERIZACIÓN
101
4.2 CARACTERIZACIÓN SUPERFICIAL
Los sistemas que se presentaron en la sección anterior fueron caracterizados por técnicas de
análisis superficial y por voltametría cíclica con el objetivo de confirmar que, efectivamente, la
post‐funcionalización ocurrió según lo representado en la Figura 1. Para ello, se emplearon las
técnicas aplicadas en la caracterización inicial que se presentó en el capítulo anterior (FT‐
IRRAS, PM‐IRRAS y XPS) y, además, Espectroscopia Raman Resonante y Microscopía Túnel.
Figura 2 Representación esquemática de los sistemas modelo empleados en este trabajo para el análisis de los
fenómenos de transferencia de carga entre un sustrato de oro y un centro redox (osmio). Las siglas MHDA,
MUA, MOA, MHA, MPA, SPhCOOH, PhCOOH y PhF4COOH representan a las moléculas utilizadas en la
derivatización de los sustratos con grupos –COOH, y Os representa al complejo unido a los mismos a través de
una unión amida.

4. POST‐FUNCIONALIZACIÓN Y CARACTERIZACIÓN
102
4.2.1 ANÁLISIS SUPERFICIAL POR FT‐IRRAS
En la Figura 3 se presentan los espectros obtenidos con los sistemas Au/MHDA/Os,
Au/MUA/Os, Au/MPA/Os, Au/SPhCOOH/Os y Au/PhCOOH/Os. En la parte a) de la figura se
presenta el espectro infrarrojo en modo transmisión de una pastilla de KBr del complejo de
osmio empleado en la post‐funcionalización ([Os(2,2´‐bpy)2Cl(py‐CH2‐NH2)]PF6) junto con los
espectros obtenidos para cada sistema con luz p‐polarizada (Figura 3 b)). En todos los casos,
los espectros colectados con luz s‐polarizada no exhibieron señales y por eso no se muestran.
Cabe aclarar que el espectro correspondiente al sistema Au/MPA/Os mostró señales muy
débiles en comparación con los otros sistemas y es por eso que el mismo está aumentado 10
veces respecto de los demás espectros.
En la Tabla 1 se presenta la asignación de las señales observadas en cada caso. Sin embargo, al
igual que en la caracterización de las monocapas iniciales, estos espectros son poco claros y la
presencia de bandas correspondientes al H2O atmosférica, dificulta mucho la identificación de
dichas señales. Es por ello que en todos estos sistemas también fueron analizados por PM‐
IRRAS como se describe en la siguiente sección.
Figura 3 a) Espectro infrarrojo en modo transmisión (FT‐IR) de una pastilla de [Os(2,2´‐bpy)2Cl(py‐CH2‐NH2)]PF6
en KBr. b) Espectros infrarrojos en modo absorción‐reflexión (FT‐IRRAS) de Au/MPA/Os, Au/SPhCOOH/Os,
Au/MUA/Os, Au/MHDA/Os y Au/PhCOOH/Os colectados con luz p‐polarizada. Notar que la escala del espectro
correspondiente a Au/MPA/Os está aumentado 10 veces.
4. POST‐FUNCIONALIZACIÓN Y CARACTERIZACIÓN
103
Tabla 1. Asignación de las señales observadas en los espectros FT‐IRRAS de las superficies de oro
modificadas con [Os(2,2´‐bpy)2Cl(py‐CH2‐NH2]+. Todas las frecuencias están en unidades de cm‐1.
Os/KBr6 Au/MPA/Os Au/MUA/Os Au/MHDA/Os Au/SPhCOOH/Os Au/PhCOOH/Os Asignación
1735 1738 1738 1738 `a C=O
1640 1662 1662 ` C=O o amida I
1540 1540 1540 `a COO‐
1604 `q Ph
1475 ` Bpy o `s COO‐
1460 1458 1458 1458 1458 ` Bpy o ~CH2
1419 1419 1419 1419 1419 ` Py
1257 ~C‐H
1016 ~ C‐H
844 w C‐H
764 w C‐H
4.2.2 ANÁLISIS SUPERFICIAL POR PM‐IRRAS
Se procedió de la misma manera descripta en el Capítulo 3 y se optimizó el PEM para
゜o = 2900 cm‐1 y ゜o = 1450 cm‐1. Los espectros obtenidos con los sistemas derivados de los
tioles alifáticos se muestran en la Figura 4 y fueron similares a los obtenidos para las capas
iniciales. A su vez, la Figura 5 presenta las espectros obtenidos con ゜o = 1450 cm‐1 de los
sistemas derivados del ácido 4‐mercaptobenzoico y del ácido benzoico. Todas las señales
presentes en estos espectros se encuentran asignadas en la Tabla 2.
6 Pastilla del complejo [Os(2,2´‐bpy)2Cl(py‐CH2‐NH2)]PF6 en KBr.

4. POST‐FUNCIONALIZACIÓN Y CARACTERIZACIÓN
104
En general, la asignación de las señales en la región de 1450 cm‐1 no es directa ya que, por el
ancho y la forma poco definida de las bandas, es difícil discriminar cuáles de ellas
corresponden a nuevos modos que se incorporaron con el complejo de osmio y cuáles no. En
particular, las señales más importantes son las que aparecen a ≈ 1490 cm‐1 y ≈ 1420 cm‐1. La
primera de ellas puede adjudicarse al modo `s COO‐ o a un modo de estiramiento colectivo de
las bipiridinas, `Bpy. La segunda de ellas se puede asociar a un modo colectivo de la piridina
presente en el complejo.
Figura 4 Espectros PM‐IRRAS de Au/MHDA/Os, Au/MUA/Os, Au/MOA/Os, Au/MHA/Os y Au/MPA/Os obtenidos
con a) ゜o = 2900 cm‐1 y b) ゜o = 1450 cm‐1.
Figura 5 Espectros PM‐IRRAS de Au/PhCOOH/Os y Au/SPhCOOH/Os tomados con ゜o = 1450 cm‐1.

4. POST‐FUNCIONALIZACIÓN Y CARACTERIZACIÓN
105
Todos los sistemas, excepto el sistema Au/SPhCOOH/Os, presentan una banda nueva, a
≈ 1660 cm‐1, luego de la reacción con el complejo. La misma puede adjudicarse al modo de
estiramiento del carbonilo de una amida (`C=O o amida I). Esta señal constituye una prueba
directa de que la post‐funcionalización ocurre según lo planteado en la Figura 1 y que el
complejo está unido a la superficie por una unión amida con los grupos ácido de las capas
delgadas. El hecho de que esta banda no se observe en el caso del tiol aromático puede
deberse a una cuestión de orientación de las moléculas o a que dichas señales están solapadas
con otras. En cualquier caso, no se puede afirmar ni descartar que el complejo esté unido.
Existen varios ejemplos en la literatura que utilizan este tipo de post‐funcionalización con éxito
[110, 191, 353‐355] y no se encuentran razones para pensar que en este caso la reacción no
haya funcionando debidamente.
Por otro lado, las demás señales observadas en todos los sistemas coinciden con las que ya
estaban presentes antes de la post‐funcionalización. En el caso particular de los tioles alifáticos
se debe destacar que las señales de la región de 2900 cm‐1 están en la misma posición que se
observó antes de la post‐funcionalización en el Capítulo 3. Esto confirma que la incorporación
del complejo de osmio a las monocapas es por la unión a los grupos ácidos terminales de los
tioles y que éste no se intercala entre las cadenas alifáticas. Si así lo hiciera, el desorden que
provocaría generaría el desplazamiento de las señales mencionadas hacia mayores números
de onda de acuerdo a lo reportado por Porter et al [250].
Finalmente, vale analizar con más detenimiento lo que sucede con el sistema Au/MPA/Os que
presenta dos señales a 2928 cm‐1 y 2856 cm‐1 mientras que la monocapa de MPA no
presentaba señales apreciables en esta región del espectro. En el Capítulo 3 se atribuyó la
ausencia de estas señales a dos factores: el desorden que puede presentar esta monocapa ya
que se trata de un tiol muy corto (3 C) y el reducido número de metilenos que dificulta la
detección de sus señales. La aparición de señales luego de la post‐funcionalización podría
indicar que la unión del complejo genera un cierto orden en la monocapa de manera que las
bandas disminuyen su ancho a media altura siendo ahora posible diferenciarlas de la línea
base. Por otro lado, el momento dipolar de transición de los modos de estiramiento de los
metilenos de la cadena alifática es perpendicular a la dirección de esta última y un aumento en
la intensidad de las señales sería un indicio del aumento en la interacción del momento dipolar
con la radiación p‐polarizada, es decir, un aumento en el ángulo de inclinación de la cadena (α)
[284].

4. POST‐FUNCIONALIZACIÓN Y CARACTERIZACIÓN
106
Tabla 2. Asignación de las señales observadas en los espectros PM‐IRRAS de las superficies de oro
modificadas con [Os(2,2´‐bpy)2Cl(py‐CH2‐NH2]+. Todas las frecuencias están en unidades de cm‐1.
MHDA/Os MUA/Os MOA/Os MHA/Os MPA/Os SPhCOOH/Os PhCOOH/Os Asignación
2920 2922 2926 2928 2928 `a CH2
2850 2850 2854 2852 2856 `s CH2
1738 1741 1728 1732 1740 1729 1741 `a C=O
1659 1659 1643 1645 1660 1660 ` C=O o amida I
1590 1606 `q Ph
1535 1533 1547 1547 1545 1540 1540 `a COO‐
1487 1489 1485 1487 1479 ` Bpy (o `s COO‐)
1464 1464 1462 1462 1462 1462 ` Bpy (o ~ CH2)
1423 1421 1423 1421 ` Py
1404 ` sc Ph
1369 ~ C‐H
Por lo tanto, estos resultados sugieren que antes de la post‐funcionalización, la monocapa de
MPA presenta un alto grado de desorden con bandas de poca intensidad y anchas de manera
que no se pueden diferenciar de la línea de base. Luego de unir el complejo de osmio, es
posible observarlas claramente y esto se podría atribuir a un aumento en el orden de las
cadenas, con mayor interacción entre ellas y un aumento en su inclinación respecto de la
dirección normal a la superficie.
4.2.3 ANÁLISIS SUPERFICIAL POR ESPECTROSCOPIA RAMAN RESONANTE
Esta técnica espectroscópica fue empleada para profundizar la caracterización de los sistemas
compuestos por ácido benzoico y 4‐mercaptobenzoico, respectivamente, post‐funcionalizados
con el complejo [Os(2,2´‐bpy)2Cl(py‐CH2‐NH2)]PF6.
Las condiciones experimentales aplicadas se eligieron en base a trabajos anteriores realizados
con complejos de osmio similares [356] (ver Capítulo 2). Se utilizaron 11 líneas láser de
longitudes de onda en el intervalo 647‐457 nm para la excitación, observándose un máximo de
intensidad de la dispersión Raman a 524.5 nm. A esta energía la excitación del láser es
resonante con la transición de transferencia de carga desde el osmio a los ligandos (piridina y
2,2´‐bipiridina). La Figura 6 presenta los espectros Raman resonante obtenidos para los

4. POST‐FUNCIONALIZACIÓN Y CARACTERIZACIÓN
107
sistemas mencionados, Au/PhCOOH/Os (línea azul) y Au/SPhCOOH/Os (línea roja). Asimismo,
se muestra el espectro obtenido para un sustrato de oro limpio (línea verde).
Todas las señales observadas se pueden asignar al complejo, excepto aquella presente a
1553 cm‐1 que, como puede apreciarse, también está presente en el espectro del sustrato
limpio. Esta señal no pudo ser identificada pero se utilizó para normalizar las intensidades de
las otras señales.
En cuanto a las señales restantes, la banda presente a 375 cm‐1 está relacionada con el modo
de estiramiento `Os‐N, a 660 cm‐1 se observa un modo de deformación correspondiente al
ligando piridina (~Py) y, finalmente, a 1317, 1477 y 1607 cm‐1, se encuentran diversos modos de
estiramiento asociados a los ligandos bipiridina (`Bpy) [356].
A partir de estos espectros es posible afirmar que en ambos casos el complejo de osmio está
sobre la superficie de oro y que su estructura química está intacta luego de la reacción de post‐
funcionalización. Sin embargo, en el caso del sustrato modificado con ácido 4‐mercapto‐
benzoico, las señales son más débiles en comparación con las observadas en el espectro del
otro sistema. Esto puede atribuirse a que la cantidad de complejo presente en este caso es
menor que para el sistema derivado de la sal de diazonio, como se verá más adelante.
De todas maneras, mediante este análisis se confirma que la post‐funcionalización funcionó en
ambos casos sin dañar la estructura del complejo.
Figura 6 Espectros Raman Resonante de Au/PhCOOH/Os (línea azul) y Au/SPhCOOH/Os (línea roja). La línea
verde corresponde al espectro de un sustrato de oro limpio.

4. POST‐FUNCIONALIZACIÓN Y CARACTERIZACIÓN
108
4.2.4 ANÁLISIS SUPERFICIAL POR ESPECTROSCOPIA FOTOELECTRÓNICA DE RAYOS X
4.2.4.1 ANÁLISIS DE AU/PHCOOH/OS Y AU/SPHCOOH/OS
Los sistemas Au/PhCOOH/Os y Au/SPhCOOH/Os fueron analizados por XPS y en la Figura 7 se
presentan las regiones espectrales de interés. En ambos casos, además de las señales del
sustrato de oro, se observaron señales que indicaron la presencia de carbono, oxígeno,
nitrógeno y osmio. En el caso del tiol también se observaron señales correspondientes al
azufre.
Las señales de Os 4f [357] y N 1s [327] permitieron confirmar la presencia del complejo de
osmio. Si bien la señal de Os 4f se solapa parcialmente con la de Au 5p [344], es evidente que
el complejo se encuentra presente en las dos muestras.
Asimismo, la presencia de la señal de S 2p a 162 eV [358], característica de un tiolato en un
enlace Au‐S, indica que la monocapa de ácido 4‐mercaptobenzoico no se degrada durante la
reacción de post‐funcionalización.
El área ocupada por una molécula del complejo de osmio se estimó mediante cálculos teóricos
basados en la Teoría del Funcional de la Densidad (Density Functional Theory, DFT) [359]. Para
ello, la estructura teórica de la molécula del complejo de osmio unida a la superficie fue
construida a partir de dos fragmentos: el complejo de osmio y el ácido benzoico
(o 4‐mercaptobenzoico) unido a la superficie. La estructura de cada uno de estos fragmentos
se optimizó por separado. Para estimar la geometría del sistema completo, ambos se
combinaron y se dejaron relajar parcialmente para optimizar el enlace entre ellos. Mediante
este cálculo se obtuvo un área ocupada de 120 Å2 por molécula de complejo. Por lo tanto, se
estimó que el cubrimiento superficial de una monocapa compacta del mismo es de
1.38 x 10‐10 mol cm‐2. En este caso, la relación Os:S debería ser ≈ 1:6, si se asume que el ácido
4‐mercaptobenzoico se ordena en una red √3 x √3 R 30° y presenta un cubrimiento de
7.5 x 10‐10 mol cm‐2 [95].

4. POST‐FUNCIONALIZACIÓN Y CARACTERIZACIÓN
109
La relación de señales Os:S fue de ≈ 0.01, indicando un grado de sustitución de los grupos
ácido del 3 % y una eficiencia de la reacción de post‐funcionalización de ≈ 6 % .
A partir de la intensidad de la señal Os 4f se estimó el cubrimiento superficial del complejo de
osmio y se obtuvo un valor de 9 x 10‐12 mol cm‐2.
En cuanto al sistema derivado de la sal de diazonio (Au/PhCOOH/Os) no se realizó esta
estimación ya que en este sistema no se puede emplear la ecuación (3.1) del Capítulo 3. Esto
se debe a que en este caso no se puede suponer que las señales del sustrato no estén
atenuadas por el material depositado sobre él, ya que según lo discutido en el Capítulo 3, no se
puede afirmar que se depositó una monocapa molecular de ácido benzoico en la reducción de
la sal de diazonio.
Finalmente, ambos sistemas presentaron una relación Os:N similar al valor esperado de 1:6
que indica que el complejo no se destruye al ser inmovilizado sobre la superficie.
Figura 7 Espectros XPS de Au/PhCOOH/Os (línea azul) y Au/SPhCOOH/Os (línea roja). En ambos sistemas se
observan a) las señales del Os 4f y b) la señal del N 1s. c) El sistema Au/SPhCOOH/Os presenta además, la señal
correspondiente al S 2p del grupo tiolato unido a la superficie de oro.
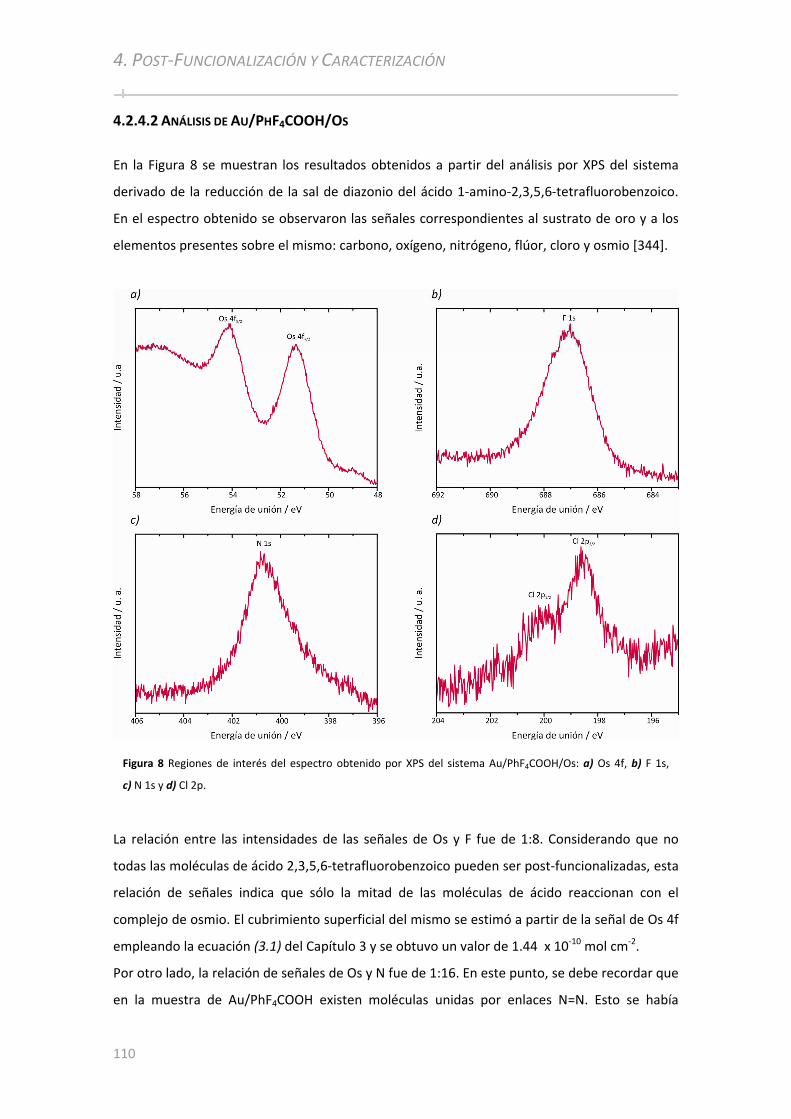
4. POST‐FUNCIONALIZACIÓN Y CARACTERIZACIÓN
110
4.2.4.2 ANÁLISIS DE AU/PHF4COOH/OS
En la Figura 8 se muestran los resultados obtenidos a partir del análisis por XPS del sistema
derivado de la reducción de la sal de diazonio del ácido 1‐amino‐2,3,5,6‐tetrafluorobenzoico.
En el espectro obtenido se observaron las señales correspondientes al sustrato de oro y a los
elementos presentes sobre el mismo: carbono, oxígeno, nitrógeno, flúor, cloro y osmio [344].
La relación entre las intensidades de las señales de Os y F fue de 1:8. Considerando que no
todas las moléculas de ácido 2,3,5,6‐tetrafluorobenzoico pueden ser post‐funcionalizadas, esta
relación de señales indica que sólo la mitad de las moléculas de ácido reaccionan con el
complejo de osmio. El cubrimiento superficial del mismo se estimó a partir de la señal de Os 4f
empleando la ecuación (3.1) del Capítulo 3 y se obtuvo un valor de 1.44 x 10‐10 mol cm‐2.
Por otro lado, la relación de señales de Os y N fue de 1:16. En este punto, se debe recordar que
en la muestra de Au/PhF4COOH existen moléculas unidas por enlaces N=N. Esto se había
Figura 8 Regiones de interés del espectro obtenido por XPS del sistema Au/PhF4COOH/Os: a) Os 4f, b) F 1s,
c) N 1s y d) Cl 2p.

4. POST‐FUNCIONALIZACIÓN Y CARACTERIZACIÓN
111
observado tanto en el análisis por PM‐IRRAS como por el de XPS de este sistema. Al restar la
señal de N 1s observada antes de la post‐funcionalización a la observada luego de la misma, se
halló una relación Os:N de 1:6. Esta relación es consistente con la estructura del complejo y
confirma que el mismo no sufre ningún tipo de degradación cuando se une a la superficie.
Finalmente, este hecho también fue confirmado a partir de la relación de señales Os:Cl que
resultó ser 1:1.
4.2.5 ANÁLISIS SUPERFICIAL POR MICROSCOPÍA DE EFECTO TÚNEL
Las Figuras 10 y 11 muestran el análisis realizado por STM de los sistemas constituidos por
ácido benzoico y 4‐mercaptobenzoico post‐funcionalizados con [Os(2,2´‐bpy)2Cl(Py‐CH2‐NH2)]+,
respectivamente. Inicialmente, en ninguno de los dos se pueden observar las terrazas y
escalones del Au (111) que son características de este tipo de sustrato, sino que un entramado
denso de puntos brillantes (Figura 10 a) y Figura 11 a)) los cubre completamente.
Luego de tomar estas imágenes, en cada caso, se acercó la punta del microscopio a la
superficie de la muestra aplicando un setpoint de 3 nA y un potencial entre la punta y la
muestra (Ebias) de 1 V. Se dejó que el tip barriera un área reducida de la muestra durante cierto
tiempo, luego se abrió el campo de barrido para tomar las imágenes de las Figuras 10 b) y 11
b). En ellas se puede apreciar una ventana abierta por el tip en las capas orgánicas respectivas.
La altura de las mismas se estimó a partir de los perfiles que se muestran en las Figuras 10 c) y
11 c). Ésta resultó ser de 1.35 nm para el sistema obtenido por la reducción de la sal de
diazonio (Au/PhCOOH/Os) y de 1.5 nm para el sistema basado en el tiol aromático
(Au/SPhCOOH/Os).
Por otro lado, se calcularon las alturas de las moléculas resultantes de la reacción del complejo
de osmio con el ácido benzoico y con el ácido 4‐mercaptobenzoico a partir de la optimización
de sus estructuras por cálculos DFT que ya fue mencionada (Figura 9) [359]. Éstas resultaron
ser 1.42 nm y 1.58 nm, respectivamente, y están en concordancia con las alturas medidas a
partir de la litografía realizada con la punta del microscopio. Esto indicaría que, en principio, no
se formaron multicapas del complejo sobre la superficie.
Asimismo, se tomaron imágenes en regiones de las muestras que presentaron un menor
cubrimiento. Las mismas se muestran en las Figuras 10 d) y 11 d) junto con los análisis de
altura de los perfiles correspondientes (Figuras 10 y 11 f) y g), respectivamente). A partir de
los mismos se determinó una altura de los puntos brillantes de (0.80 ± 0.15) nm y
(0.80 ± 0.2) nm para cada sistema, respectivamente. Comparando nuevamente con las
distancias calculadas por DFT, este valor coincide con la longitud teórica de la porción de la

4. POST‐FUNCIONALIZACIÓN Y CARACTERIZACIÓN
112
molécula que comprende desde el grupo C=O hasta el átomo de osmio en ambos casos (Figura
9).
Cabe aclarar que estas imágenes se tomaron aplicando distintos potenciales entre el sustrato y
la punta del microscopio, y no se observaron diferencias entre las mismas.
Asimismo, como se muestra en las Figuras 10 e) y 11 e), se estimó el tamaño promedio de
estos puntos realizando un análisis de las imágenes 10 d) y 11 d), respectivamente. Para ello,
se empleó una aplicación del software del microcopio (power spectral density analysis) luego
de filtrar dichas imágenes. El tamaño promedio resultó ser de ≈ 4.35 nm y ≈ 4.0 nm,
respectivamente para las muestras de Au/PhCOOH/Os y Au/SPhCOOH/Os. Estos valores son
muy grandes para corresponder a moléculas individuales y esta observación puede deberse a
un artefacto de convolución de la punta del microscopio.
Figura 9 Estructura optimizada por DFT del sistema Au/PhCOOH/Os. Se indican las distancias Au‐Os para el
mismo y para el sistema Au/SPhCOOH/Os y la distancia desde el grupo C=O al átomo de osmio.

4. POST‐FUNCIONALIZACIÓN Y CARACTERIZACIÓN
113
Figura 10 Imágenes STM en aire de 500×500 nm2 de Au (111) derivatizado con ácido benzoico y post‐
funcionalizado con [Os(2,2´‐bpy)2Cl(py‐CH2‐NH2)]+, tomadas con un setpoint de 350 nA y Ebias = 1.5 V a) antes y b)
después de abrir una ventana en la capa orgánica por litografía. c) Sección transversal de la imagen b) mostrando
la altura de la capa orgánica. d) Imagen STM en aire de 280×280 nm2 tomada con un setpoint de 500 pA y
Ebias = 1.3 V, en una zona del sustrato de bajo cubrimiento. e) Análisis del tamaño de los puntos observados en d).
f) Región de la imagen d) sobre la que se analizó el corte transversal que se muestra en g).
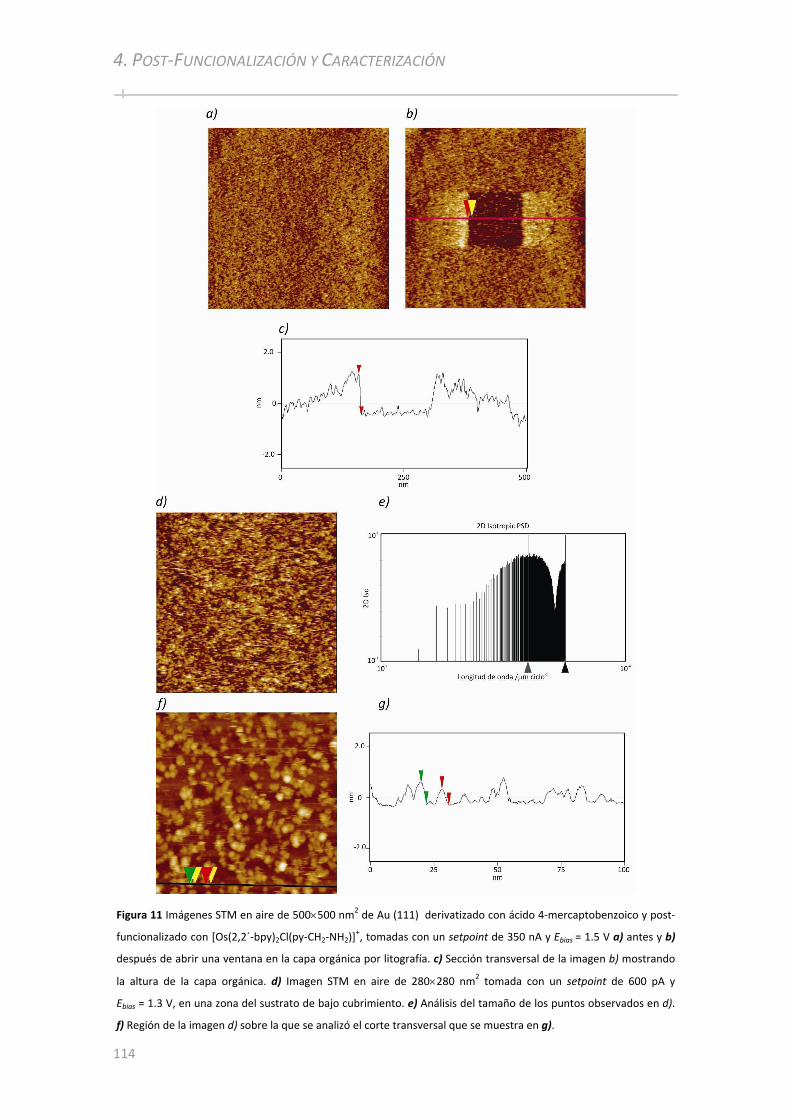
4. POST‐FUNCIONALIZACIÓN Y CARACTERIZACIÓN
114
Figura 11 Imágenes STM en aire de 500×500 nm2 de Au (111) derivatizado con ácido 4‐mercaptobenzoico y post‐
funcionalizado con [Os(2,2´‐bpy)2Cl(py‐CH2‐NH2)]+, tomadas con un setpoint de 350 nA y Ebias = 1.5 V a) antes y b)
después de abrir una ventana en la capa orgánica por litografía. c) Sección transversal de la imagen b) mostrando
la altura de la capa orgánica. d) Imagen STM en aire de 280×280 nm2 tomada con un setpoint de 600 pA y
Ebias = 1.3 V, en una zona del sustrato de bajo cubrimiento. e) Análisis del tamaño de los puntos observados en d).
f) Región de la imagen d) sobre la que se analizó el corte transversal que se muestra en g).

4. POST‐FUNCIONALIZACIÓN Y CARACTERIZACIÓN
115
Finalmente, en la Figura 12 se muestran imágenes obtenidas para sustratos de Au (111)
modificados con a) ácido benzoico y b) 4‐mercaptobenzoico antes de ser post‐funcionalizados
con el complejo de osmio. Se puede observar que estas imágenes tienen baja resolución y
mientras que la muestra de Au/PhCOOH presenta cierta textura, la rugosidad media en ambas
imágenes siempre es menor que en el caso de las capas post‐funcionalizadas.
4.3 CARACTERIZACIÓN ELECTROQUÍMICA
Todos los sistemas preparados se caracterizaron electroquímicamente. Para ello, se realizaron
voltametrías cíclicas con cada uno de ellos y, como se muestra a continuación, se obtuvo el
cubrimiento superficial del complejo de osmio (ГOs) y el potencial formal (E°´) del par
Os(II)/Os(III) en cada caso.
En primer lugar, se confirmó que no existiera desorción de las capas delgadas depositadas en
la derivatización inicial de los sustratos de oro, en las condiciones de trabajo elegidas. En todos
los casos se observó la señal esperada para la carga de una doble capa capacitiva como la que
se muestra en línea roja en la Figura 13, que corresponde a un electrodo de oro modificado
con ácido 11‐mercaptoundecanoico (MUA) en NaCl 0.1 M. En la misma figura también se
incluye la voltametría en H2SO4 2M del mismo electrodo antes de ser derivatizado con el tiol.
Figura 12 Imágenes STM de sustratos de Au (111) modificados con a) ácido benzoico y b) ácido
4‐mercaptobenzoico, respectivamente.
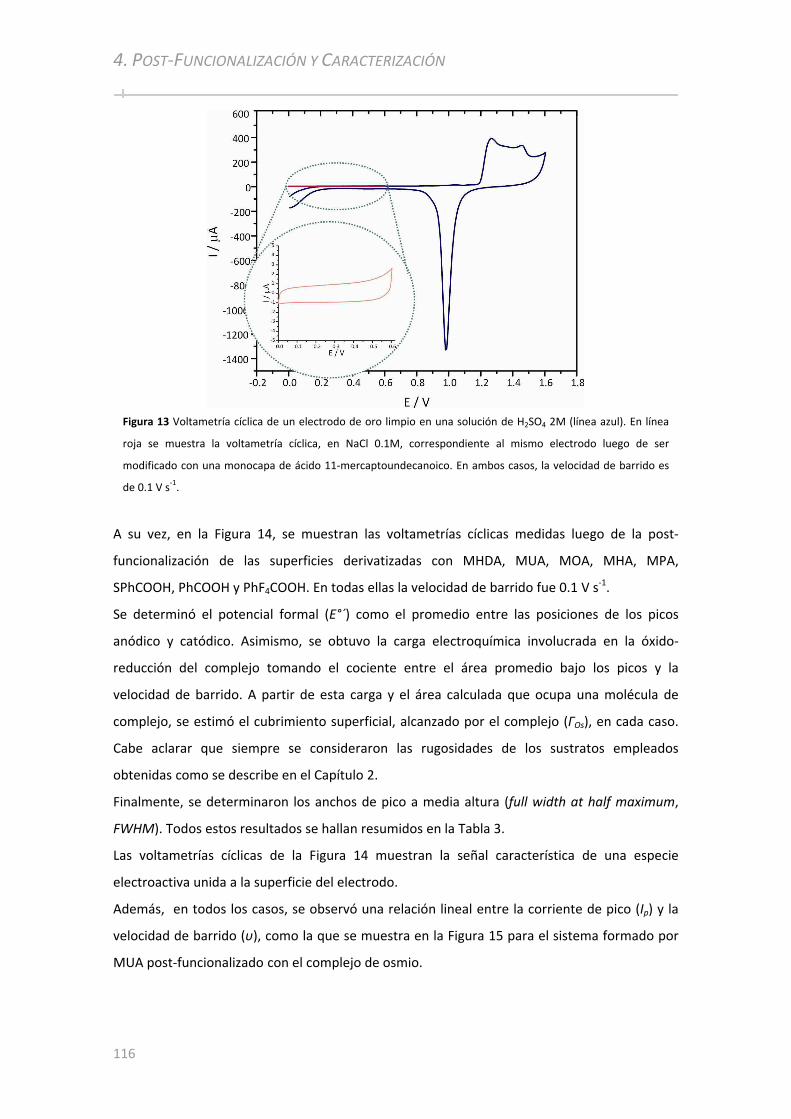
4. POST‐FUNCIONALIZACIÓN Y CARACTERIZACIÓN
116
A su vez, en la Figura 14, se muestran las voltametrías cíclicas medidas luego de la post‐
funcionalización de las superficies derivatizadas con MHDA, MUA, MOA, MHA, MPA,
SPhCOOH, PhCOOH y PhF4COOH. En todas ellas la velocidad de barrido fue 0.1 V s‐1.
Se determinó el potencial formal (E°´) como el promedio entre las posiciones de los picos
anódico y catódico. Asimismo, se obtuvo la carga electroquímica involucrada en la óxido‐
reducción del complejo tomando el cociente entre el área promedio bajo los picos y la
velocidad de barrido. A partir de esta carga y el área calculada que ocupa una molécula de
complejo, se estimó el cubrimiento superficial, alcanzado por el complejo (ГOs), en cada caso.
Cabe aclarar que siempre se consideraron las rugosidades de los sustratos empleados
obtenidas como se describe en el Capítulo 2.
Finalmente, se determinaron los anchos de pico a media altura (full width at half maximum,
FWHM). Todos estos resultados se hallan resumidos en la Tabla 3.
Las voltametrías cíclicas de la Figura 14 muestran la señal característica de una especie
electroactiva unida a la superficie del electrodo.
Además, en todos los casos, se observó una relación lineal entre la corriente de pico (Ip) y la
velocidad de barrido (仝), como la que se muestra en la Figura 15 para el sistema formado por
MUA post‐funcionalizado con el complejo de osmio.
Figura 13 Voltametría cíclica de un electrodo de oro limpio en una solución de H2SO4 2M (línea azul). En línea
roja se muestra la voltametría cíclica, en NaCl 0.1M, correspondiente al mismo electrodo luego de ser
modificado con una monocapa de ácido 11‐mercaptoundecanoico. En ambos casos, la velocidad de barrido es
de 0.1 V s‐1.

4. POST‐FUNCIONALIZACIÓN Y CARACTERIZACIÓN
117
Figura 14 Voltametría cíclica de un electrodo de oro modificado con a) MHDA/Os, b) MUA/Os, c) MOA/Os,
d) MHA/Os, e) MPA/Os, f) SPhCOOH/Os, g) PhCOOH/Os y h) PhF4COOH/Os. Todas las medidas fueron
realizadas en NaNO3 0.1 M, con una de velocidad de barrido de 0.1 V s‐1.
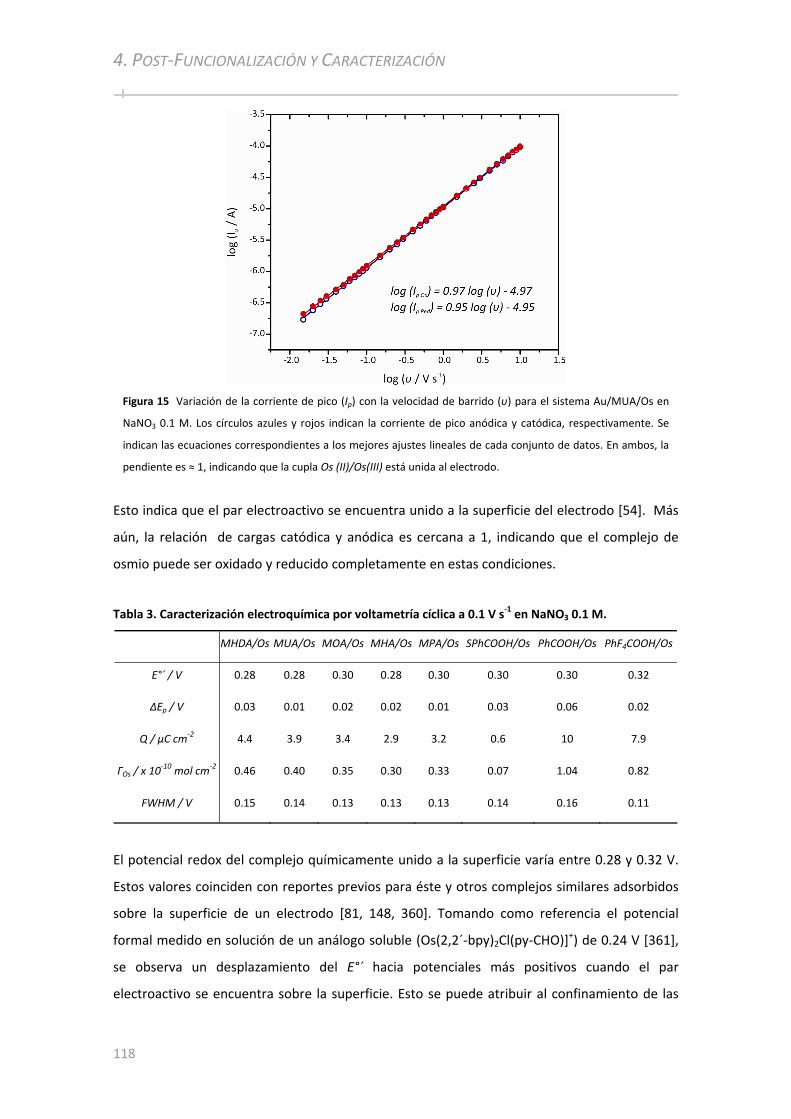
4. POST‐FUNCIONALIZACIÓN Y CARACTERIZACIÓN
118
Esto indica que el par electroactivo se encuentra unido a la superficie del electrodo [54]. Más
aún, la relación de cargas catódica y anódica es cercana a 1, indicando que el complejo de
osmio puede ser oxidado y reducido completamente en estas condiciones.
Tabla 3. Caracterización electroquímica por voltametría cíclica a 0.1 V s‐1 en NaNO3 0.1 M.
MHDA/Os MUA/Os MOA/Os MHA/Os MPA/Os SPhCOOH/Os PhCOOH/Os PhF4COOH/Os
E°´ / V 0.28 0.28 0.30 0.28 0.30 0.30 0.30 0.32
らEp / V 0.03 0.01 0.02 0.02 0.01 0.03 0.06 0.02
Q / ´C cm‐2 4.4 3.9 3.4 2.9 3.2 0.6 10 7.9
ГOs / x 10‐10 mol cm‐2 0.46 0.40 0.35 0.30 0.33 0.07 1.04 0.82
FWHM / V 0.15 0.14 0.13 0.13 0.13 0.14 0.16 0.11
El potencial redox del complejo químicamente unido a la superficie varía entre 0.28 y 0.32 V.
Estos valores coinciden con reportes previos para éste y otros complejos similares adsorbidos
sobre la superficie de un electrodo [81, 148, 360]. Tomando como referencia el potencial
formal medido en solución de un análogo soluble (Os(2,2´‐bpy)2Cl(py‐CHO)]+) de 0.24 V [361],
se observa un desplazamiento del E°´ hacia potenciales más positivos cuando el par
electroactivo se encuentra sobre la superficie. Esto se puede atribuir al confinamiento de las
Figura 15 Variación de la corriente de pico (Ip) con la velocidad de barrido (仝) para el sistema Au/MUA/Os en
NaNO3 0.1 M. Los círculos azules y rojos indican la corriente de pico anódica y catódica, respectivamente. Se
indican las ecuaciones correspondientes a los mejores ajustes lineales de cada conjunto de datos. En ambos, la
pendiente es ≈ 1, indicando que la cupla Os (II)/Os(III) está unida al electrodo.

4. POST‐FUNCIONALIZACIÓN Y CARACTERIZACIÓN
119
moléculas en la superficie que provoca el incremento de las interacciones intermoleculares
debido a la mayor proximidad entre ellas, respecto de lo que ocurre cuando están libres en
solución. Debido a que el complejo posee una carga positiva, tanto en su estado reducido
(Os(II)+) como en su estado oxidado (Os(III)2+), oxidarlo implica aumentar las repulsiones
electrostáticas entre moléculas vecinas y es por ello que el potencial formal en esta
configuración es mayor que el observado cuando el par está en solución.
Sin embargo, vale aclarar que en algunas condiciones se observa lo contrario, es decir, un
corrimiento de E°´ hacia potenciales más negativos respecto del observado en solución. En el
Capítulo 6 se discutirá en detalle este tipo de comportamiento.
Los voltamogramas presentados en la Figura 14 no son ideales como se evidencia en una
separación entre picos no nula (らEp > 0), aún para velocidades de barrido bajas, y en un ancho
de pico a media altura mayor que el esperado para un proceso ideal (90.6 mV), que se puede
asociar con las mencionadas interacciones repulsivas entre los centros electroactivos unidos a
la superficie.
El máximo cubrimiento esperado para una monocapa de moléculas del complejo se puede
calcular a partir del área que ocupa una de estas moléculas. Como ya se mencionó este área
se estimó a partir de la optimización de la geometría de una molécula [Os(2,2´‐bpy)2Cl]py‐CH2‐
NH‐CO‐Ph por DFT, obteniéndose un área ocupada por molécula de 120 Å2. A su vez, en
bibliografía se reporta un valor de ≈ 180 Å2 para este tipo de complejos [360, 361]. Éste se
desprende del tamaño hallado, a partir de datos cristalográficos, para complejos similares
([Os(NH3)N2]Cl2 [148] y [Ru(2,2´‐bpy)3][PF6]2 [362]). Por lo tanto de aquí en adelante, se
trabajará con el tamaño teórico mencionado, ya que éste fue deducido específicamente para
el complejo de interés en este trabajo y no discrepa apreciablemente del reportado por otros
autores.
A partir del área calculada por DFT se desprende que para una monocapa compacta del
complejo utilizado en este trabajo, el cubrimiento superficial es de 1.38 x 10‐10 mol cm‐2, que
corresponde a una densidad de carga de ≈ 1.3 ´C cm‐2.
En literatura existen numerosos ejemplos de modificación de electrodos con complejos de
osmio similares, en los que también se evalúa el cubrimiento superficial de los mismos a partir
de la correspondiente carga electroquímica asociada a su óxido‐reducción. En complejos del
tipo [Os(2,2´‐bpy)2Clpy(CH2)npy]+, en los que la adsorción sobre la superficie de electrodos de
platino y oro ocurre a través del átomo de N de la piridina terminal, el cubrimiento superficial
es de ≈ 0.8 a 2 x 10‐10 mol cm‐2 [360, 361, 363, 364]. A su vez, en complejos en los que el
enlace es a través de un tiol, [Os(2,2´‐bpy)2Cl(py‐CH2)nSH]+, el cubrimiento es levemente

4. POST‐FUNCIONALIZACIÓN Y CARACTERIZACIÓN
120
mayor, 3 x 10‐10 mol cm‐2 [365]. En todos estos casos, los cubrimientos obtenidos están en el
orden de una monocapa.
Por otro lado, existen reportes en los que se ha utilizado el mismo complejo de osmio que en
esta tesis para post‐funcionalizar capas delgadas obtenidas por reducción de las sales de
diazonio derivadas del ácido 3‐(4‐aminofenil)propiónico y del ácido 4‐aminobenzoico. En este
caso se han obtenido cubrimientos en el orden de 2‐3 x 10‐10 mol cm‐2 [81, 119].
Comparando ahora los resultados extraídos de la bibliografía con los correspondientes a los
distintos sistemas analizados aquí y presentados en la Tabla 3, se puede ver que en todos los
casos estos últimos son menores y no alcanzan el valor estimado para una monocapa de
complejo. Para comprender esta diferencia hay que considerar que en todos los casos
reportados en la literatura, excepto el que utiliza sales de diazonio, los complejos de osmio se
unen directamente al electrodo, ya sea a través de un átomo de N o por un grupo tiol, que ya
están presentes en la molécula. Este proceso de autoensamblado resulta en la formación de
monocapas ordenadas sobre el electrodo como fue demostrado por microscopía de efecto
túnel por Hudson y Abruña [365].
En cambio, los sistemas que se estudiaron en esta tesis se prepararon a partir de una reacción
entre el grupo amino del complejo de osmio y los grupos ácido ya unidos a la superficie del
electrodo. El cubrimiento superficial alcanzado por el complejo a partir de esta reacción puede
verse afectado por posibles efectos de impedimento estérico entre moléculas de complejo que
dificulten su unión específica a los grupos ácido presentes en la superficie. Es por ello que es
conveniente definir dos conceptos que serán útiles para el análisis del cubrimiento superficial
del complejo de osmio en cada sistema y algunos de los resultados que se presentan en los
capítulos siguientes. Se define:
1) Grado de sustitución: como el porcentaje de los grupos ácidos que han reaccionado con el
complejo de osmio. Este valor no puede ser del 100 % porque el área que ocupa una molécula
de complejo corresponde a varios sitios ocupados por grupos ácido.
2) Eficiencia de post‐funcionalización: como el rendimiento de la reacción de unión del
complejo a la superficie. Este rendimiento es del 100 % cuando se alcanza el grado máximo de
sustitución de los grupos ácido, es decir, un empaquetamiento compacto de moléculas de
complejo.
Para calcular cada uno de estos valores, se deben tomar los cubrimientos superficiales de los
tioles y del ácido 2,3,5,6‐tetrafluorobenzoico empleados en la derivatización inicial de los
sustratos de oro. Los cubrimientos superficiales de los tioles se estimaron en el Capítulo 3 por
desorción reductiva y en todos ellos, se encontraron valores similares a lo esperado para

4. POST‐FUNCIONALIZACIÓN Y CARACTERIZACIÓN
121
monocapas de cada uno (0.8‐1.3 x 10‐9 mol cm‐2). El cubrimiento del ácido 2,3,5,6‐
tetrafluorobenzoico se estimó a partir de la señal de F 1s observada por XPS y también estuvo
en el orden de lo esperado para una monocapa del mismo (≈ 4.5 x 10‐10 mol cm‐2).
Con estos datos y con el cubrimiento del complejo de osmio obtenido en cada sistema (Tabla
3), se puede estimar el grado de sustitución de los grupos ácido y la eficiencia de post‐
funcionalización.
A partir de los máximos cubrimientos esperados para monocapas compactas de tioles
(7.5 x 10‐10 mol cm‐2 [95]) y de una monocapa de ácido 2,3,5,6‐tetrafluorobenzoico (5 x 10‐10
mol cm‐2) y del máximo cubrimiento del complejo esperado para una monocapa compacta del
mismo (1.38 x 10‐10 mol cm‐2), se puede calcular que el máximo grado de sustitución de los
ácidos carboxílicos esperado es del 19 % , para los tioles y del 28 % para el ácido 2,3,5,6‐
tetrafluorobenzoico.
Comparando el grado de sustitución de los grupos –COOH obtenido en cada caso con estos
valores máximos, se puede estimar la eficiencia del proceso de post‐funcionalización. Esta
información se resume en la Tabla 4.
Tabla 4. Grado de sustitución de los grupos –COOH y eficiencia de la post‐funcionalización con el
complejo [Os(2,2´‐bpy)2Cl(py‐CH2‐NH2)]PF6
% de sustitución
de grupos ‐COOH
% eficiencia
post‐funcionalización
MHDA/Os 3.5 19
MUA/Os 3.6 19
MOA/Os 3.6 19
MHA/Os 3.3 18
MPA/Os 4.1 22
SPhCOOH/Os 0.7 3
PhF4COOH/Os 18 65
Se puede observar que los tioles alifáticos no presentan diferencias significativas en el grado
de sustitución de sus grupos terminales. Sin embargo, el tiol aromático presenta un grado de
sustitución notablemente menor, mientras que el sistema derivado del ácido
2,3,5,6‐ tetrafluorobenzoico es el que presenta el mayor grado de sustitución.

4. POST‐FUNCIONALIZACIÓN Y CARACTERIZACIÓN
122
Una posible explicación para estas observaciones puede surgir si se considera la reacción
química de post‐funcionalización en sí misma. Al tratarse de una reacción de sustitución
nucleofílica de acilo, su rendimiento es función de la reactividad del grupo ácido en la
superficie frente al nucleófilo, que en este caso es el grupo amino del complejo. Dicha
reactividad será mayor, cuánto menor sea la densidad electrónica sobre el ácido.
Por lo tanto, el bajo rendimiento de la reacción observado para el ácido 4‐mercaptobenzoico
se puede explicar considerando el efecto dador de electrones que presenta el grupo tiol que
repercute en la densidad electrónica sobre el grupo ácido, desactivándolo para la reacción de
sustitución nucleofílica de acilo. El efecto exactamente contrario ocurre en el ácido
2,3,5,6‐ tetrafluorobenzoico, en el que la presencia de los sustituyentes fluoruro disminuye la
densidad electrónica en el ácido aumentando su reactividad frente a los grupos amino. Es por
ello que este sistema presenta la mayor eficiencia de reacción.
Finalmente, se pueden comparar los cubrimientos superficiales de los sistemas
Au/SPhCOOH/Os y Au/PhF4COOH/Os, obtenidos por voltametría cíclica, con los estimados a
partir de la señal de Os 4f en los espectros obtenidos con XPS. En el primer caso, el análisis por
XPS se realizó con un sustrato de Au (111) y el cubrimiento obtenido fue de 9 x 10‐12 mol cm‐2.
Para el sistema Au/PhF4COOH/Os, se utilizó un sustrato igual al empleado en el análisis
electroquímico, con un factor de rugosidad de 1.70. El cubrimiento observado por XPS resultó
ser de 1.44 x 10‐10 mol cm‐2, que al ser corregido por el factor mencionado resulta en
8.5 x 10‐11 mol cm‐2. Recordando que el análisis electroquímico arrojó cubrimientos
superficiales de 7 x 10‐12 mol cm‐2 y 8.2 x 10‐11 mol cm‐2 para cada sistema, respectivamente, se
puede decir los valores de cubrimiento superficial del complejo de osmio obtenidos por cada
técnica guardan una muy buena concordancia entre sí.
Por último cabe aclarar que el sistema Au/PhCOOH/Os quedó excluido del análisis precedente
porque en el mismo no se puede afirmar que se forme una monocapa de ácido benzoico
durante la reducción de la sal de diazonio, sino más bien, una capa delgada que está formada
por anillos aromáticos ramificados. Por lo tanto, el hecho de que en este caso los sitios de
unión del complejo estén distribuidos tridimensionalmente, impide que se aplique el mismo
razonamiento que se utilizó para estimar los cubrimientos máximos a partir del simple cálculo
de áreas ocupadas por cada tipo de molécula.
4.4 CONCLUSIONES
En este capítulo presentaron los sistemas que se emplearon como modelos para el análisis de
la cinética de transferencia de carga. Se detalló la forma en la que fueron obtenidos y la

4. POST‐FUNCIONALIZACIÓN Y CARACTERIZACIÓN
123
caracterización tanto superficial como electroquímica. En todos los casos se pudo confirmar la
unión del par redox a la superficie y la conservación de la estructura química del complejo.
En el siguiente capítulo se presentará el análisis de la cinética de transferencia electrónica en
cada uno de estos sistemas y las variaciones observadas según las características de cada uno
de ellos.


CAPÍTULO 5
ANÁLISIS DE LA CINÉTICA DE TRANSFERENCIA
ELECTRÓNICA


5. ANÁLISIS DE LA CINÉTICA DE TRANSFERENCIA ELECTRÓNICA
127
n los capítulos anteriores se detalló la forma en la que se derivatizaron y caracterizaron
sustratos de oro con el complejo [Os(2,2´‐bpy)2Cl(pyCH2NH2)]+ unido a diferentes
moléculas que actúan como puentes entre el par Os(II)/Os(III) y el electrodo. En este capítulo,
a la información obtenida por técnicas de análisis superficial y por voltametría cíclica, se añade
la caracterización de la cinética de transferencia electrónica entre el metal y los sitios
electroactivos a través de los diferentes puentes moleculares.
En la primer parte de este capítulo se describen brevemente el Modelo de Butler‐Volmer, el
Modelo de Marcus y los mecanismos de transferencia electrónica conocidos, que son
conceptos centrales en la interpretación de los resultados obtenidos. Luego se describen las
metodologías empleadas para la determinación de las constantes cinéticas de transferencia
electrónica y, finalmente, se presentan los resultados obtenidos.
5.1 MARCO TEÓRICO
5.1.1 CINÉTICA DE TRANSFERENCIA ELECTRÓNICA
5.1.1.1 ECUACIÓN DE NERNST
Las reacciones de electrodo son procesos químicos heterogéneos en los que la carga puede
fluir entre fases con distinta conductividad eléctrica: electrodo y electrolito. Por lo tanto, su
comportamiento depende fuertemente de las concentraciones superficiales y del potencial
aplicado y puede ser analizado tanto en base a un modelo químico como a uno eléctrico.
Para comenzar se considerará la siguiente reacción genérica:
d
i
k
kO ne R− ⎯⎯→+ ←⎯⎯ (5.1)
en la que las especies O y R están unidas a la superficie del electrodo y están relacionadas por
una reacción elemental unimolecular de óxido‐reducción en la que intercambian n electrones.
Las velocidades de la reacción directa e inversa son, respectivamente:
d d O
i i R
仝 k よ
仝 k よ
== (5.2)
donde よO y よR son los cubrimientos superficiales de O y R, y kd y ki, son las constantes de
velocidad de reducción de O y oxidación de R. Estas constantes se miden en unidades de s‐1 y
E

5. ANÁLISIS DE LA CINÉTICA DE TRANSFERENCIA ELECTRÓNICA
128
las velocidades, en mol cm‐2 s‐1. La velocidad neta de la reacción está dada por la diferencia
entre las velocidades parciales:
d i仝 仝 仝= − (5.3)
Cuando el sistema llega al equilibrio la interconversión neta entre O y R es nula y las
velocidades de la reacción directa e inversa se igualan. La constante de equilibrio, K, es:
dR
O i
kよK
よ k= = (5.4)
En el caso particular de una reacción de electrodo, el equilibrio se puede expresar con la
ecuación de Nernst, que relaciona las concentraciones superficiales de O y R con el potencial
de electrodo:
o´ O
R
よRTE E ln
nF よ
⎛ ⎞= + ⎜ ⎟⎝ ⎠ (5.5)
donde, E°´ es el potencial formal.
5.1.1.2 MODELO DE BUTLER‐VOLMER
La ecuación de Butler‐Volmer [366, 367] es considerada una de las relaciones fundamentales
de la electroquímica y un punto central en el estudio de la cinética de electrodo. Este modelo
considera que ambas reacciones, anódica y catódica, ocurren simultáneamente en el electrodo
y, en base a ello, describe la dependencia de la corriente eléctrica con el potencial aplicado en
el mismo.
La corriente, i, se puede expresar en función de las concentraciones superficiales de O y R en la
superficie del electrodo, よO y よR, y las constantes cinéticas de la reacción directa e inversa, kd y
ki:
[ ]i R d Oi nFA k よ (t ) k よ (t )= − (5.6)

5. ANÁLISIS DE LA CINÉTICA DE TRANSFERENCIA ELECTRÓNICA
129
Es decir, i es directamente proporcional a la diferencia de velocidades de las reacciones que
ocurren en el electrodo y la constante de proporcionalidad viene dada por el número de
electrones involucrados en las mismas y por el área del electrodo, A.
Por otro lado, se considera que las energías de activación para ambas reacciones, らGd‡ y らGi
‡,
varían en forma lineal con el sobrepotencial, η= E – Eo´, aplicado al electrodo:
( )( ) ( )d d
i i
らG らG αF E E ´
らG らG α F E E ´
= + − °= + − − °‡ ‡
0
‡ ‡0 1
(5.7)
Si η es positivo, entonces la nueva energía de activación catódica, らGd‡, es menor que la del
estado estándar, らG0d‡, y la fracción en la que cambió se denota con la letra α (coeficiente de
transferencia), que es una medida de la simetría de la barrera de activación y puede tomar
valores entre 0 y 1. Este modelo considera que α es independiente del potencial del electrodo,
es decir la considera constante. Esto implica que los perfiles de energía libre son lineales en el
intervalo de potencial de interés, como se observa en la Figura 1, donde se muestra la
variación de las barreras de activación con el sobrepotencial aplicado. Sin embargo, en
intervalos de potencial amplios esta aproximación deja de ser válida y α depende del
potencial, como se verá más adelante.
Considerando que las constantes cinéticas tienen la forma de Arrhenius, kd y ki se pueden
escribir como:
( )( ) ( )
d dd d d
iii i i
らG らGk A exp A exp exp αf E E ´
RT RT
らGらGk A exp A exp exp α f E E ´
RT RT
⎛ ⎞ ⎛ ⎞= − = − − − °⎡ ⎤⎜ ⎟ ⎜ ⎟ ⎣ ⎦⎝ ⎠ ⎝ ⎠⎛ ⎞⎛ ⎞= − = − − − °⎡ ⎤⎜ ⎟⎜ ⎟ ⎣ ⎦⎝ ⎠ ⎝ ⎠
‡ ‡0
‡‡0 1
(5.8)
donde f = F/RT.

5. ANÁLISIS DE LA CINÉTICA DE TRANSFERENCIA ELECTRÓNICA
130
Si el electrodo está en equilibrio con una solución que tiene O y R en igual concentración (よO =
よR), entonces el potencial del mismo es el del equilibrio descripto por la ecuación de Nernst (E
= E°´) y la reacción directa e inversa ocurren a la misma velocidad (kdよO = kiよR). En estas
condiciones las constantes de velocidad de las reacciones directa e inversa son iguales y el
valor que tienen recibe el nombre de constante de velocidad estándar, k0. Si se expresan kd y ki
en función de k0, se tiene:
( )( ) ( )d
i
k k exp αf E E ´
k k exp α f E E ´
= − − °⎡ ⎤⎣ ⎦= − − °⎡ ⎤⎣ ⎦
0
0 1 (5.9)
La constante de velocidad estándar es una medida de la facilidad con la que el sistema puede
llegar al equilibrio y, aunque ésta sea pequeña, kd y ki pueden llegar a valores altos si se aplican
potenciales suficientemente extremos respecto de E°´.
Si se reemplazan estas expresiones en la ecuación (5.8) se obtiene una relación completa entre
la corriente y el potencial, que se conoce como la ecuación de Butler‐Volmer:
( α ) f ( E E ´) αf ( E E ´)R Oi nFAk よ (t )e よ (t )e− − ° − − °⎡ ⎤= −⎣ ⎦0 1 (5.10)
Por lo tanto, cuando el sistema está en equilibrio, i = 0, y las corrientes anódica (ia) y catódica
(ic) se compensan entre sí (ia = ‐ic). Se define como corriente de intercambio, io, al valor que
tienen ia e ic en el equilibrio:
Figura 1 Variación de la energía libre de activación de una reacción de electrodo con el sobrepotencial aplicado
sobre el mismo. En este caso, η > 0 y la barrera de activación para la reducción aumenta, mientras que para la
oxidación, disminuye.

5. ANÁLISIS DE LA CINÉTICA DE TRANSFERENCIA ELECTRÓNICA
131
( ) ee α ( E E ´)αf ( E E ´)o O Ri nFAk よ e =nFAk よ − − °− − °= 10 0 (5.11)
Usando la ecuación de Nernst, io se puede reescribir como:
( α ) αo O Ri nFAk よ よ− −= 0 1 (5.12)
Cuando no hay efectos de transferencia de masa, la ecuación (5.10) se reduce a:
αfη ( α ) fηoi i e e− −⎡ ⎤⎣ ⎦= − 1 (5.13)
Como se mencionó anteriormente, este modelo supone que α es constante y por ende, se
puede emplear en aquellos casos en los que el potencial varía en un intervalo pequeño de
valores. En la mayoría de los sistemas en los que las especies redox están en solución, cuando
el potencial está relativamente lejos de E°, la transferencia de masa comienza a limitar el
proceso global (se observa una corriente límite, il), con lo cual no se llega a la situación en la
que α deja de ser constante. Sin embargo, cuando O y R están unidas a la superficie del
electrodo y la transferencia de masa no interviene, se puede trabajar en ventanas de potencial
más amplias y por ende, para analizarlos es necesario considerar la curvatura de los perfiles de
energía libre. Esto fue descripto por el Modelo de Marcus que se presenta a continuación.
5.1.1.3 MODELO DE MARCUS
5.1.1.3.1 Fundamentos
La teoría de Marcus fue desarrollada por Rudolph A. Marcus en 1956 [368, 369] para explicar
las velocidades de las reacciones de transferencia electrónica entre dos especies químicas
(donor y aceptor de electrones). Fue originalmente formulada para reacciones de esfera
externa, en las que las dos especies químicas no están unidas directamente entre sí, pero fue
extendida por Noel S. Hush, a reacciones de esfera interna en las que sí existe una unión
química entre el donor y el aceptor de electrones [370‐372].
Marcus recibió el Premio Nobel de Química del año 1992 por su teoría y ésta es utilizada para
describir una gran variedad de procesos en química y biología, incluyendo fotosíntesis [373‐
375], corrosión [376], ciertos tipos de quimiluminiscencia [377‐380] y separación de cargas en

5. ANÁLISIS DE LA CINÉTICA DE TRANSFERENCIA ELECTRÓNICA
132
algunas celdas solares [381‐383], entre otros. Actualmente, la teoría de Marcus es la teoría
dominante de transferencia electrónica en química.
Marcus se inspiró para desarrollar su modelo en un artículo publicado por William Libby [384]
en el que se empleaba el principio de Franck‐Condon para explicar por qué las reacciones de
auto‐intercambio de cationes pequeños en solución acuosa, (como la que se muestra en la
ecuación (5.14), los asteriscos indican isótopos radioactivos) son relativamente lentas respecto
de las mismas reacciones con cationes grandes como Fe(CN)64‐.
* *Fe Fe Fe Fe+ + + ++ → +2 3 3 2 (5.14)
En su trabajo, Libby estableció que cuando se transfiere un electrón desde una molécula (o
ión) a otra, las dos nuevas moléculas (o iones) formados se encuentran en un entorno de
moléculas de solvente inadecuado ya que las mismas no tuvieron tiempo para reacomodarse
durante el rápido salto electrónico. De esta manera, en la reacción (5.14) el ión Fe2+* se forma
en un entorno apropiado para el ión Fe3+* original y lo mismo ocurre con el nuevo ión Fe3+,
generándose una barrera de energía de solvatación. En cambio, cuando los iones son grandes,
como MnO4‐ o Fe(CN)6
4‐, el cambio de campo eléctrico en la vecindad de cada ión por la
transferencia electrónica debe ser menor y por ende, el entorno de moléculas de solvente no
resulta tan inadecuado para los productos formados. Con este razonamiento Libby predijo una
menor barrera de activación para la reacción de auto‐intercambio con iones voluminosos.
El principio de Franck‐Condon establece que durante una transición electrónica un cambio
desde un nivel vibracional a otro será más probable si las dos funciones de onda vibracionales
tienen un buen solapamiento. Desde un punto de vista clásico, esto quiere decir que una
transición es más probable si durante la misma, no ocurren cambios en la posiciones de los
núcleos de la molécula involucrada y de su entorno [385‐387], es decir, si se trata de una
transición vertical. Este concepto se aplica también a las transferencias electrónicas ya que, al
igual que las transiciones electrónicas, son instantáneas en comparación con el movimiento de
los núcleos de la molécula en cuestión y de la reorganización del solvente. Teniendo en cuenta
todo esto, la idea propuesta por Libby resulta estar incorrecta, o incompleta, ya que para
reacciones que ocurren sin absorción de fotones, la energía no se conserva: los iones deberían
formarse en un entorno de alta energía no adecuado, y esto sólo sería posible por absorción
de luz (transición vertical) y no en la oscuridad.
Por ejemplo, se considera la siguiente reacción de auto‐intercambio:

5. ANÁLISIS DE LA CINÉTICA DE TRANSFERENCIA ELECTRÓNICA
133
[Fe(H O) ] [Fe(H O) ] [Fe(H O) ] [Fe(H O) ]+ + + ++ → +2 3 3 22 6 2 6 2 6 2 6 (5.15)
En este caso, la distancia de equilibrio Fe‐O para Fe (II) es de 2.21 Å, mientras que para Fe (III)
es de 2.05 Å. La Figura 2 muestra las superficies de energía potencial para los reactivos y
productos de esta reacción: si la transferencia electrónica ocurre en sus posiciones de
equilibrio, entonces se tendría un ión Fe (II) comprimido y un ión Fe (III) estirado. Éstos son
estados vibracionales excitados y, si la transferencia electrónica ocurre en la oscuridad,
liberarían energía, con lo que se violaría el principio de conservación. Por lo tanto, estos
reactivos deben igualar su energía antes de la transferencia. Por ende, deben existir
fluctuaciones en todas las coordenadas nucleares y en las coordenadas de orientación de las
moléculas de solvente y en cualquier otra coordenada cuya distribución en el estado final
difiera de la del estado inicial. Marcus denominó a la energía necesaria para que estos cambios
ocurran en el sistema, energía de reorganización (゜).
De esta manera, las coordenadas de reacción pueden tomar valores que satisfagan tanto el
principio de Franck‐Condon como el de conservación de la energía y que lleven al sistema a la
configuración del punto de cruce de las dos parábolas, en el que la transferencia electrónica
puede ocurrir sin absorción ni emisión de energía, es decir en la oscuridad.
En la Figura 3 se muestra el perfil de energía potencial del sistema entero: reactivos y solvente.
Ésta es función de todas las coordenadas del sistema que incluyen la posición y orientación de
las moléculas individuales de solvente, sus momentos dipolares y las coordenadas
Figura 2 Superficies de energía potencial para la reacción de auto‐intercambio de Fe(H2O)62+/3+

5. ANÁLISIS DE LA CINÉTICA DE TRANSFERENCIA ELECTRÓNICA
134
vibracionales de los reactivos, entre otras. La abscisa de esta figura considera todas las
dimensiones mencionadas y representa cualquier movimiento concertado, desde una
configuración que corresponde a la estructura electrónica de los reactivos a una
correspondiente a los productos. La superficie R denota el perfil de energía potencial que tiene
el sistema cuando tiene la configuración electrónica de los reactivos, mientras que P
corresponde a la de los productos.
Si la interacción entre los reactivos es alta, existe un desdoblamiento (en inglés, splitting) de
las dos superficies en su intersección. Si dicha interacción es muy débil, este efecto es
despreciable. En el primer caso, el sistema siempre permanecerá en la superficie de menor
energía a medida que se mueva de izquierda a derecha en la Figura 3, es decir, lo hará
adiabáticamente. Por otro lado, si la partición entre las superficies es despreciable, el sistema
permanecerá en la superficie R a medida que cruza la intersección: movimiento no adiabático.
A partir de esto, la transferencia electrónica puede ocurrir, ya sea porque el sistema remonta
la barrera de energía entre reactivos y productos (comportamiento clásico), o porque la
atraviesa por efecto túnel (comportamiento semi‐clásico). Si este último es despreciable,
entonces la barrera es la energía de activación para la reacción de transferencia de energía.
Figura 3 Perfil de energía potencial de reactivos y productos para una reacción adiabática. HRP es el
acoplamiento electrónico entre el estado inicial y el estado final del sistema. ゜ es la energía de reorganización,
らG0 es cambio total de energía libre para la transferencia electrónica y らG‡ es la barrera de energía asociada a
la misma.

5. ANÁLISIS DE LA CINÉTICA DE TRANSFERENCIA ELECTRÓNICA
135
La energía libre de activación らG‡ en la Figura 3, es la diferencia entre el punto de cruce (para
un proceso no adiabático) y el punto de equilibrio en la superficie de los reactivos. Si se
ignoran los cambios de entropía, la energía libre es igual a la energía potencial. El punto de
cruce entre las superficies corresponde a la cima de la barrera de activación (qC) y es el punto
en que ocurre la transferencia electrónica.
Aproximando las superficies de energía por parábolas se puede llegar a una expresión para la
energía de activación:
( ) ( )P R
P R
k q q らGらG
k q q
⎡ ⎤−= +⎢ ⎥−⎢ ⎥⎣ ⎦2 0
‡
2
21
8 (5.16)
Como ya se mencionó, la energía de reorganización, ゜, consiste en la energía requerida para
todos los ajustes estructurales (en los reactivos y en las moléculas del entorno) para que el
sistema asuma la configuración correcta y ocurra la transferencia electrónica.
Se puede definir como ( )P R
k゜ q q= − 2
2, y con eso se llega a:
゜ らGらG
゜
⎛ ⎞= +⎜ ⎟⎝ ⎠20
‡ 14
(5.17)
Esta ecuación expresa la altura de la barrera, o la energía de activación, para el cruce de dos
curvas adiabáticas en términos de la energía libre total, らG0 (que es nula si se trata de una
reacción de auto‐intercambio) y la energía de reorganización, ゜. A su vez, ゜ tiene una
contribución interna (゜i), que viene dada por la reorganización en los reactivos, y una externa
(゜o), que considera a la del solvente. La energía de reorganización interna esta dada por la
ecuación (5.18):
( )2i j O , j R , jj
1゜ k q q
2= −∑ (5.18)
donde kj son las constantes de fuerza los i modos vibracionales y qi,j son las coordenadas de los
desplazamientos asociados a los mismos. Por otro lado, la energía de reorganización externa
se puede obtener con la siguiente expresión, asumiendo que el solvente es un dieléctrico

5. ANÁLISIS DE LA CINÉTICA DE TRANSFERENCIA ELECTRÓNICA
136
continuo y que los reactivos son esferas de radios a1 y a2, separados entre sí una distancia
d = a1 + a2:
2
o
0 1 2 op s
e 1 1 1 1 1゜
8ヽ0 2a 2a d 0 0
⎛ ⎞⎛ ⎞= + − −⎜ ⎟⎜ ⎟⎜ ⎟⎝ ⎠⎝ ⎠ (5.19)
donde 0op y 0s son las constantes dieléctricas óptica y estática del solvente, respectivamente, 00
es la permitividad del vacío.
A partir de la energía de activación, se puede obtener una expresión para la constante cinética
de transferencia electrónica, kET, usando la relación de Arrhenius:
ET
B
らGk Aexp
k T
⎡ ⎤−= ⎢ ⎥⎣ ⎦‡
(5.20)
donde el prefactor A depende de la naturaleza de la transferencia electrónica (es decir, si es
unimolecular o bimolecular) y らG‡ se expresa según (5.17).
La teoría de Marcus puede extenderse a reacciones de transferencia electrónica heterogéneas
[388]. En ese caso, el cambio de energía libre total, らG0, puede reemplazarse por F(E – E0) en la
ecuación (5.17):
( )F E E ´゜らG
゜
− °⎛ ⎞= +⎜ ⎟⎝ ⎠2
‡ 14
(5.21)
y ゜o puede expresarse como:
2
o
0 O op s
e 1 1 1 1゜
8ヽ0 a d 0 0
⎛ ⎞⎛ ⎞= − −⎜ ⎟⎜ ⎟⎜ ⎟⎝ ⎠⎝ ⎠ (5.22)
donde se considera al reactivo como una esfera de radio ao ubicado a una distancia d = 2ao de
la superficie del electrodo.
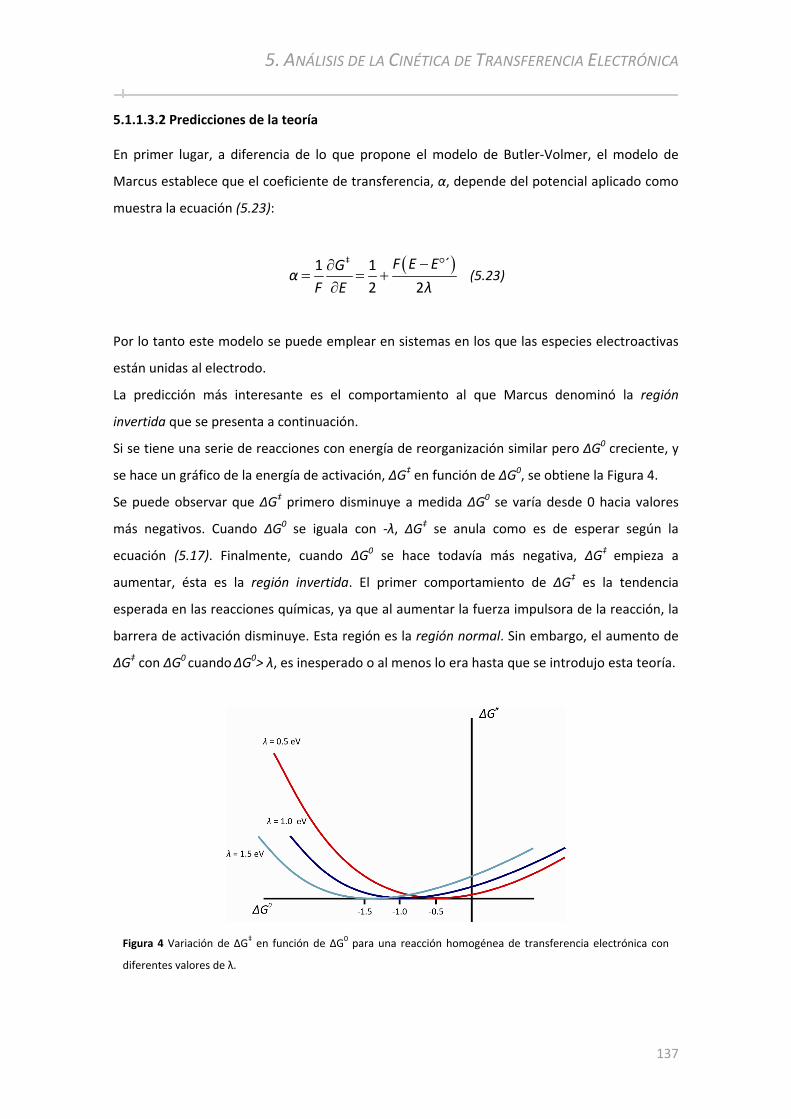
5. ANÁLISIS DE LA CINÉTICA DE TRANSFERENCIA ELECTRÓNICA
137
5.1.1.3.2 Predicciones de la teoría
En primer lugar, a diferencia de lo que propone el modelo de Butler‐Volmer, el modelo de
Marcus establece que el coeficiente de transferencia, α, depende del potencial aplicado como
muestra la ecuación (5.23):
( )F E E ´Gα
F E ゜
− °∂= = +∂‡1 1
2 2 (5.23)
Por lo tanto este modelo se puede emplear en sistemas en los que las especies electroactivas
están unidas al electrodo.
La predicción más interesante es el comportamiento al que Marcus denominó la región
invertida que se presenta a continuación.
Si se tiene una serie de reacciones con energía de reorganización similar pero らG0 creciente, y
se hace un gráfico de la energía de activación, らG‡ en función de らG0, se obtiene la Figura 4.
Se puede observar que らG‡ primero disminuye a medida らG0 se varía desde 0 hacia valores
más negativos. Cuando らG0 se iguala con ‐゜, らG‡ se anula como es de esperar según la
ecuación (5.17). Finalmente, cuando らG0 se hace todavía más negativa, らG‡ empieza a
aumentar, ésta es la región invertida. El primer comportamiento de らG‡ es la tendencia
esperada en las reacciones químicas, ya que al aumentar la fuerza impulsora de la reacción, la
barrera de activación disminuye. Esta región es la región normal. Sin embargo, el aumento de
らG‡ con らG0 cuando らG0> ゜, es inesperado o al menos lo era hasta que se introdujo esta teoría.
Figura 4 Variación de ΔG‡ en función de ΔG0 para una reacción homogénea de transferencia electrónica con
diferentes valores de ゜.

5. ANÁLISIS DE LA CINÉTICA DE TRANSFERENCIA ELECTRÓNICA
138
La región invertida se puede visualizar fácilmente observando la Figura 5. A medida que らG0 se
hace más negativa, bajando la curva de los productos verticalmente respecto de la de los
reactivos, disminuye la barrera de activación, らG‡ (dada por la intersección de las curvas de
reactivos y productos). Esta barrera desaparece cuando らG0 = ‐゜ y luego aumenta nuevamente
[389].
Una explicación posible para este comportamiento consiste en una alta y negativa energía
libre de reacción que implica que los productos deben aceptar la energía liberada muy rápido
en sus modos vibracionales, y la probabilidad de hacerlo disminuye a medida que –らG0 se
hace mayor que ゜.
Finalmente, hay que notar que el efecto de región invertida no debería observarse para una
reacción de electrodo sobre un metal. Esto se debe a que la ecuación (5.21) fue desarrollada
con la idea implícita de que los electrones involucrados en la reaccionan provienen de un
intervalo estrecho de estados correspondientes al nivel de Fermi del electrodo.
Si bien se predice que la velocidad de reacción a estas energías muestra inversión a
sobrepotenciales muy negativos, siempre hay estados ocupados por debajo del nivel de Fermi
y ellos pueden transferir un electrón a la especie oxidada sin inversión. Cualquier vacancia de
baja energía creada en el metal por la transferencia heterogénea será llenada con un electrón
del nivel de Fermi, con disipación de la diferencia de energía en forma de calor.
Figura 5 Prefil de energía libre a lo largo de la coordenada de reacción q para una reacción no adiabática:
a) región normal, donde 0 ≤ ‐らG0; b) la condición para máxima constante de velocidad donde –らG0 = ゜; c) la
“región invertida” donde –らG0> ゜.

5. ANÁLISIS DE LA CINÉTICA DE TRANSFERENCIA ELECTRÓNICA
139
5.1.1.3.3 Reacciones de electrodo
El modelo de Marcus, junto con las contribuciones de Gerischer [390, 391], Memming [392] y
Bard [393], ha permitido comprender mejor los procesos de transferencia electrónica en
electrodos metálicos y semiconductores. La Figura 6 muestra un diagrama que ejemplifica la
aplicación de este modelo a las reacciones de electrodo, en particular, se muestra el caso de
un metal en contacto con una solución de electrolito que contiene las dos especies de un par
electroactivo (O y R).
Lo primero que se debe tener en cuenta al analizar este gráfico es que el eje y representa
energía electrónica referida a la energía necesaria para traer un electrón desde el vacío. No se
muestra el número de estados disponibles para los electrones en las diferentes bandas, sin
embargo, se indica el nivel de Fermi que corresponde al estado más alto ocupado en el metal.
Del lado de la solución, el diagrama describe la cantidad relativa de las especies oxidada y
reducida, respectivamente, con lo cual del lado de la solución el eje x representa el número de
estados cerca del electrodo (N) cuya energía es la que indica el eje y.
Considerando que la óxido‐reducción del par en solución es reversible, entonces en el
equilibrio el intercambio de electrones con el electrodo ocurre en el nivel de Fermi, ya que
éstos son los electrones más energéticos para la reducción y el estado más bajo que puede
ocuparse en la oxidación, se encuentra justo por encima del nivel de Fermi. La solvatación de
las especies O y R para que la transferencia electrónica en estas condiciones sea isoenergética
Figura 6 Densidades de estados de un electrodo metálico en contacto con una solución de R y O en iguales
concentraciones. Los estados llenos del electrodo (coloreados de azul) se solapan con los estados de O (vacíos)
y, por ende, puede ocurrir reducción. Los estados de R (llenos) solapan con los estados llenos del electrodo, por
lo que la oxidación está bloqueada.

5. ANÁLISIS DE LA CINÉTICA DE TRANSFERENCIA ELECTRÓNICA
140
es la energía necesaria para adecuar a la especie oxidada y reducida a la presencia o ausencia
del electrón, es decir es la energía de reorganización. Además, la energía electrónica de la
forma más estable de R es menor que aquella asociada a O y la distribución de estados de cada
una consiste en un pico centrado en estas energías.
En la Figura 6 también se incluye el diagrama de energía libre del sistema en solución, en el
que se observa que, para adoptar la configuración adecuada para la transferencia electrónica,
R debe remontar la parábola hacia mayores energías electrónicas y, luego de la reacción, O
desciende por su parábola hacia energías mayores aún, ya que se comporta como un hueco en
un semiconductor.
A partir de estas consideraciones, se puede llegar a una expresión para la velocidad de
transferencia electrónica entre el electrodo y el par en solución, teniendo en cuenta que ésta
será proporcional a la densidad de estados ocupados en una fase y los desocupados en la otra
fase para una dada energía. Además, hay que tener en cuenta que los estados están
distribuidos sobre un amplio intervalo de energías electrónicas y, por ende, se debe hacer una
integración sobre todos los niveles de energía.
La velocidad para la transferencia electrónica será:
A O red OVelocidad `AN よ (t ) (0)W (゜ ,0) f (0)ヾ(0)d0∞
−∞= ∫ Є (5.24)
donde NA es el número de Avogadro, A es el área expuesta del electrodo, よO(t) es la
concentración de O en la superficie del electrodo, WO(゜,0) es la función de densidad de
probabilidad, Єred(0) es una función de proporcionalidad, f(0) es la fracción de estados
ocupados y ヾ(0) es la densidad de estados del electrodo. En forma análoga se puede obtener
una expresión de la velocidad en función de los parámetros asociados a R. En cualquiera de los
casos, si se divide por la concentración correspondiente se puede obtener una expresión para
la constante cinética de reducción o de oxidación:
[ ]d red O
i red R
k ` (0)W (゜ ,0) f (0)ヾ(0)d0
k ` (0)W ( ゜ ,0) f (0) ヾ(0)d0
∞
−∞∞
−∞
== −
∫∫
Є
Є 1
(5.25)
El desarrollo que lleva a estos resultados se puede encontrar en la bibliografía citada.

5. ANÁLISIS DE LA CINÉTICA DE TRANSFERENCIA ELECTRÓNICA
141
Es importante destacar el papel de la energía de reorganización que afecta a la respuesta
corriente‐potencial esperada. Cuánto más alta es, más se separan las distribuciones de estados
de O y R entre sí y, para un dado sobrepotencial, ocurrirá la reducción u oxidación, según cómo
se alinee el nivel de Fermi con dichas distribuciones.
5.1.2 MECANISMOS DE TRANSFERENCIA ELECTRÓNICA
Se han propuesto tres mecanismos para describir la transferencia electrónica de largo alcance
(> 10 Å) entre un donor y un aceptor de electrones que se hallan unidos entre sí a través de un
puente molecular. El donor y el aceptor pueden formar parte de la misma molécula o el donor
puede ser un electrodo al cual se ha unido una especie capaz de aceptar electrones. Los
electrodos modificados con monocapas que poseen centros electroactivos en su estructura
corresponden a este último caso. En ellos, el puente molecular está formado por las moléculas
que separan dichos centros de la superficie del electrodo.
Estos mecanismos son electron hopping (del inglés, salto electrónico; en adelante se empleará
la terminología inglesa) [13, 394‐396], superintercambio, o tuneleo a través del enlace, [13,
394‐397] y tuneleo directo, o a través del espacio, [398, 399].
El primer mecanismo supone pasos secuenciales de transferencia electrónica entre sitios
vecinos pertenecientes al puente molecular, con relajación de energía total en cada paso
elemental. Los estados electrónicos del puente están cerca del nivel de Fermi del electrodo
donor y la ocupación de los mismos está asistida por fluctuaciones vibracionales. En este caso
la interacción con el entorno es importante.
El término superintercambio (para el segundo mecanismo) implica que en este caso, la
transferencia se da por tuneleo a través de un puente molecular con niveles energéticos
vacantes más altos que la energía del electrón que se transfiere, es decir que están por encima
del nivel de Fermi del donor. Estos niveles favorecen la transferencia por el solapamiento entre
primeros vecinos. En este caso el tuneleo es no resonante y la interacción con el entorno es
más débil que en el caso anterior.
Finalmente, en el tercer mecanismo el puente molecular representa simplemente una barrera
de potencial a través de la cual el electrón se transfiere por tuneleo simple. Esta barrera, sin
embargo, es menor que la que existe en ausencia del puente debido a las interacciones
electrostáticas de largo alcance o interacciones de corto alcance del electrón con el puente, es
decir, con la polarización del mismo. Este mecanismo prevalece cuando los niveles de energía
del puente están muy por encima del nivel de Fermi del donor.

5. ANÁLISIS DE LA CINÉTICA DE TRANSFERENCIA ELECTRÓNICA
142
El mecanismo que gobierna un determinado sistema depende de la estructura electrónica del
puente, del entorno y del electrodo y no siempre se puede determinar unívocamente de cuál
se trata.
En particular, cuando se tiene un electrodo modificado con una película formada por
moléculas saturadas (en general, como ya se ha mencionado, tioles), la misma actúa como una
barrera para el tuneleo desde el electrodo a la especie redox. Se han llevado a cabo numerosos
estudios para investigar la dependencia de la constante cinética de transferencia electrónica
con el espesor de estas películas [27] y se ha hallado que existe una caída exponencial de la
misma con la distancia (d):
0 max βdk k e−∝ (5.26)
donde β es el parámetro de distancia de tuneleo y kmax corresponde al valor de k0 cuando el
centro redox se encuentra en el plano de máximo acercamiento al electrodo. Cuando la
molécula que actúa de puente tiene unidades repetitivas, como un grupo metileno en una
cadena alifática, β se puede expresar respecto del número de unidades.
Una estrategia para determinar este parámetro para moléculas de distintos largos y
estructuras de cadena (especialmente tioles), es formar monocapas autoensambladas de las
mismas entre dos electrodos (de oro y/o mercurio) y medir la variación de la corriente que
circula a través de estas junturas en función del potencial aplicado entre ambos electrodos
[160, 400].
Otra forma, es la determinación de la constante cinética estándar de alcanotioles
funcionalizados con diferentes centros electroactivos y autoensamblados sobre oro, en
función del largo de cadena. Esto se ha realizado con las siguientes especies: ferroceno [52,
102, 105‐107, 110], pyRu(NH3)5 [110, 112, 172], quinonas [401‐403] y Os(2,2´‐bpy)2Clpy [148,
149, 360], entre otras. La pendiente del gráfico semi‐logaritmico de k0 en función de la
distancia es β, cuyo valor está en el orden de ≈ 1 Å‐1 o ≈ 1.0‐1.2 / CH2, independientemente del
par redox involucrado. A su vez, valores de β ≈ 0.3 Å‐1 han sido encontrados para ferroceno
unido a la superficie del electrodo a través de mercapto‐oligo(fenilen‐etilenos) [33, 147, 177].
Según el valor de β se puede inferir si se está en presencia de un mecanismo de tuneleo a
través del enlace o a través del espacio [176]. El criterio aceptado establece que un valor de
β ≈ 0.5‐1 Å‐1 es coherente con un mecanismo de superintercambio [404, 405], mientras que
Finklea y Hanshew han propuesto que valores de β ≈ 1.3‐1.8 Å‐1 corresponden a un mecanismo
de tuneleo simple (a través del espacio) [112]. En el caso de puentes aromáticos entre el par

5. ANÁLISIS DE LA CINÉTICA DE TRANSFERENCIA ELECTRÓNICA
143
redox y el electrodo, el valor de β ≈ 0.3 Å‐1 corresponde a un mecanismo de tuneleo a través
del enlace.
5.2 DETERMINACIÓN DE LA CONSTANTE ESTÁNDAR DE TRANSFERENCIA ELECTRÓNICA
5.2.1 METODOLOGÍAS EMPLEADAS
Las técnicas electroquímicas son muy apropiadas para estudiar el acoplamiento electrónico de
largo alcance y la transferencia electrónica entre electrodos metálicos y moléculas redox
unidas a los mismos [406]. Estos sistemas pueden servir como excelentes modelos para el
análisis del transporte de carga a través de un puente molecular, debido en parte, a la
naturaleza eléctrica de la señal que se genera (una corriente) como resultado de dicho
transporte.
Desde el punto de vista electroquímico estos fenómenos se pueden caracterizar determinando
la constante de velocidad estándar, k0, asociada a la transferencia electrónica. Existen muchos
métodos electroquímicos para caracterizar los procesos de transferencia electrónica
interfacial. Entre ellos se pueden nombrar los siguientes: cronoamperometría [112, 113, 407,
408], barrido lineal del potencial y voltametría cíclica [112, 408‐411], voltametría de corriente
alterna (voltametría AC) [173, 407], espectroscopía de impedancia electroquímica [412], una
variación de voltametría AC/impedancia electroquímica [98, 101, 102, 117, 175, 176, 413‐415]
y por salto de temperatura indirecto inducido por láser (ILIT) [105, 110, 416‐418].
En este trabajo de tesis se emplearon la voltametría cíclica y la espectroscopía de impedancia
electroquímica para determinar k0. Antes de describir el procedimiento particular asociado a
cada una de ellas, es necesario hacer algunas observaciones respecto de cuáles son los
factores que pueden limitar la medición de una constante cinética y de qué forma se puede
superarlos.
5.2.1.1 RESISTENCIA NO COMPENSADA Y CAPACIDAD DE LA DOBLE CAPA: CONSTANTE DE CELDA
Cuando fluye una corriente en una celda electroquímica existe una caída de potencial entre el
electrodo de referencia y el electrodo de trabajo debida a la resistencia no compensada, Ru,
entre ambos. Esta caída de potencial depende de la conductividad del electrolito, la distancia
entre los dos electrodos y la magnitud de la corriente. Usando la ley de Ohm, la caída de
potencial (らEohm ) se puede calcular como el producto entre la corriente (I) y la resistencia de la
solución no compensada (Ru): らEohm = I x Ru. Si se asume que el pasaje de corriente no afecta el

5. ANÁLISIS DE LA CINÉTICA DE TRANSFERENCIA ELECTRÓNICA
144
potencial del electrodo de referencia (idealmente no polarizable), el potencial del electrodo de
trabajo se puede expresar como:
W aplicado ohmE E らE= − (5.27)
donde EW es el potencial efectivo del electrodo de trabajo, Eaplicado es el aplicado y la diferencia
entre ambos es la caída óhmica.
Si bien, en principio, Ru puede ser compensada totalmente para corregir el error que
introduce, esto es muy difícil de realizar en la práctica debido a que la compensación introduce
oscilaciones instrumentales no deseadas en la medida.
A su vez, la capacidad de la doble capa (Cdl) juega un papel importante en la velocidad de
respuesta de una celda ya que, operacionalmente, cambiar el potencial del electrodo de
trabajo consiste en cambiar el estado de carga de la doble capa. Este proceso es un fenómeno
transitorio que decae con una constante de tiempo igual a RuCdl. Por lo tanto, el cambio de
potencial del electrodo de trabajo involucra esta constante de tiempo que recibe el nombre de
constante de tiempo de la celda y determina la menor escala de tiempo en la que se puede
llevar a cabo un experimento. Es decir, se podrán medir procesos faradaicos que ocurran en
una escala de tiempo mayor a RuCdl.
Tanto Cdl como Ru dependen del tamaño del electrodo aunque lo hacen de manera diferente.
La capacidad de la doble capa se puede expresar como:
0dl dlC AC= (5.28)
donde Cdl0, en el modelo más simple, es la capacidad de un condensador de caras paralelas por
unidad del área:
o odl
00C
d= (5.29)
con 0o, la permitividad del vacío, 0, la constante dieléctrica del medio y d el espesor del
dieléctrico. Cuando el electrodo está desnudo y en contacto con electrolito acuoso Cdlo tiene
un valor de 10‐50 ´F cm‐2 [54] y, cuando está cubierto por un alcanotiol, su valor es de 6‐10 ´F
cm‐2 [419, 420]. A partir de esto, se observa que cuánto menor es el tamaño del electrodo de
trabajo, menor es la capacidad de la doble capa.

5. ANÁLISIS DE LA CINÉTICA DE TRANSFERENCIA ELECTRÓNICA
145
Por otro lado, la dependencia de Ru con el tamaño del electrodo es menos clara. El valor
preciso de la misma depende principalmente de la geometría del electrodo de trabajo y de
dónde esté ubicado el electrodo de referencia respecto este último [54, 421]. Por ejemplo,
según la simetría del sistema, Ru se puede expresar como:
u
u0
u0 0
x 1R = para un electrodo plano
A ゛1 2x
R = ln para un electrodo cilíndrico2ヽ ゛ r
1 xR = para un electrodo esférico
4ヽ゛r x + r
⎛ ⎞⎜ ⎟⎜ ⎟⎝ ⎠⎛ ⎞⎜ ⎟⎜ ⎟⎝ ⎠
` (5.30)
donde x es la distancia entre el electrodo de trabajo y el de referencia, r0 es el radio del
electrodo de trabajo, cuando es esférico o cilíndrico, l es la longitud de este último y A es el
área del electrodo plano. En particular, se considera que l >> r0 y x > r0.
Por lo tanto, a partir del producto de la resistencia no compensada y la capacidad de la doble
capa y, teniendo en cuenta las expresiones del área de una esfera y de un cilindro, se puede
obtener la constante de la celda para cada simetría:
odl
u dl
o0u dl dl
0
00u dl dl
0
xCR C para un electrodo plano
゛r 2x
R C ln C para un electrodo cilíndrico゛ r
r xR C C para un electrodo esférico
゛ x r
⎛ ⎞⎜ ⎟⎜ ⎟⎝ ⎠⎛ ⎞⎜ ⎟⎜ ⎟⎝ ⎠
=== +
(5.31)
Observando estas expresiones se puede apreciar que, para un electrodo plano, RuCdl siempre
depende de la distancia entre electrodos, mientras que para el electrodo cilíndrico, esta
dependencia es menos pronunciada cuando x es grande, debido a la función logarítmica. A su
vez, en un electrodo esférico, la dependencia con x de RuCdl disminuye marcadamente a
medida que r0 también lo hace. Además, si r0 es lo suficientemente pequeño se pueden
alcanzar escalas de tiempo lo suficientemente cortas (< 1 ´s) y así estudiar procesos cada vez
más rápidos (k0 > 106 s‐1).
Esto se puede visualizar en la Figura 7 en la que se grafica la variación de la constante RuCdl
para un electrodo de disco (plano) y uno esférico, ambos de radio r0, empleando las
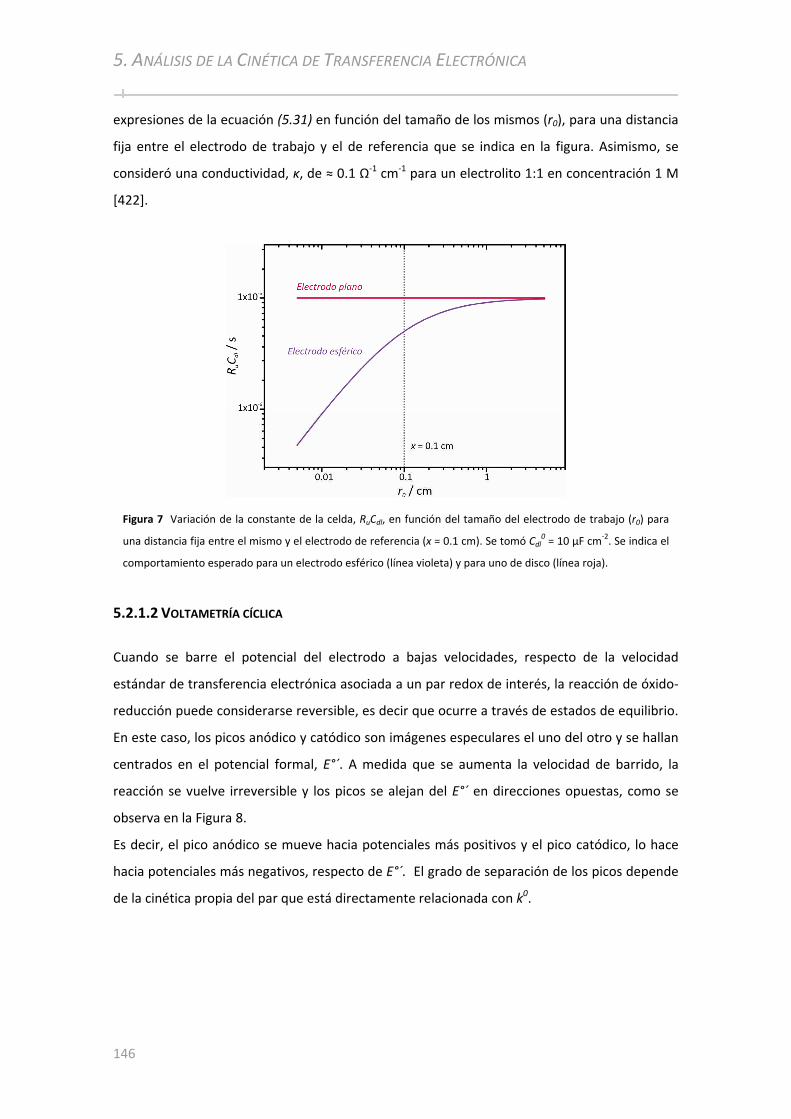
5. ANÁLISIS DE LA CINÉTICA DE TRANSFERENCIA ELECTRÓNICA
146
expresiones de la ecuación (5.31) en función del tamaño de los mismos (r0), para una distancia
fija entre el electrodo de trabajo y el de referencia que se indica en la figura. Asimismo, se
consideró una conductividad, ゛, de ≈ 0.1 Ω‐1 cm‐1 para un electrolito 1:1 en concentración 1 M
[422].
5.2.1.2 VOLTAMETRÍA CÍCLICA
Cuando se barre el potencial del electrodo a bajas velocidades, respecto de la velocidad
estándar de transferencia electrónica asociada a un par redox de interés, la reacción de óxido‐
reducción puede considerarse reversible, es decir que ocurre a través de estados de equilibrio.
En este caso, los picos anódico y catódico son imágenes especulares el uno del otro y se hallan
centrados en el potencial formal, E°´. A medida que se aumenta la velocidad de barrido, la
reacción se vuelve irreversible y los picos se alejan del E°´ en direcciones opuestas, como se
observa en la Figura 8.
Es decir, el pico anódico se mueve hacia potenciales más positivos y el pico catódico, lo hace
hacia potenciales más negativos, respecto de E°´. El grado de separación de los picos depende
de la cinética propia del par que está directamente relacionada con k0.
Figura 7 Variación de la constante de la celda, RuCdl, en función del tamaño del electrodo de trabajo (r0) para
una distancia fija entre el mismo y el electrodo de referencia (x = 0.1 cm). Se tomó Cdl0 = 10 ´F cm‐2. Se indica el
comportamiento esperado para un electrodo esférico (línea violeta) y para uno de disco (línea roja).

5. ANÁLISIS DE LA CINÉTICA DE TRANSFERENCIA ELECTRÓNICA
147
El marco teórico para el análisis de la electroquímica de este tipo de sistemas fue desarrollado
por Laviron en la década de 1970 [423, 424], utilizando el modelo de Butler‐Volmer. Con el
mismo, se puede obtener información cinética empleando k0 y α como parámetros para
simular una voltametría cíclica. Dichos parámetros se varían hasta que el voltamograma
teórico coincide con el experimental. De esta manera se pueden obtener los valores de k0 y α
que mejor caracterizan al sistema de interés.
Figura 9 Trumpet plot simulado en base al modelo de Marcus para un par redox unido a la superficie del
electrodo con E°´= 0.3 V, k0 = 1000 s‐1 y ゜ = 0.5 eV.
Figura 8 Voltametrías cíclicas simuladas para un par electroactivo unido a la superficie de un electrodo con
velocidades de barrido crecientes (0.01, 0.04, 0.09, 0.3, 0.8, 2.5, 7, 20, 60, 150, 500 y 900 V s‐1). Se empleó el
modelo de Marcus con E°´= 0.3 V, k0 = 1000 s‐1 y ゜ = 0.5 eV. En el recuadro se presenta la voltametría simulada a
una velocidad de barrido de 0.01 V s‐1, en la que los picos son imágenes especulares uno de otro.

5. ANÁLISIS DE LA CINÉTICA DE TRANSFERENCIA ELECTRÓNICA
148
Experimentalmente, esto se realiza con un conjunto de voltametrías cíclicas medidas a
velocidades de barrido crecientes y, a partir de las mismas, se grafica respectivamente la
posición de los picos anódico y catódico en función de la velocidad de barrido. De esta manera,
se obtienen curvas como la que se muestra en la Figura 9, que se conoce como trumpet plots
(que en inglés significa gráficos “trompeta”) por la forma característica que presenta cada
rama al evidenciar cómo los picos se separan entre sí a mayores velocidades de barrido
Por lo tanto, se simula cada uno de los voltamogramas según lo propuesto por Laviron y se
varían los valores de k0 y α hasta obtener el mejor ajuste de las curvas experimentales. Cabe
aclarar que la simulación de los voltamogramas se puede hacer en el marco de cualquier
modelo teórico de transferencia electrónica, según el sistema que se analice. Es decir que
también se puede emplear el modelo de Marcus para expresar las constantes de velocidad y,
en este caso, los parámetros son k0 y ゜ [409, 410].
Como se verá a continuación, el análisis de datos obtenidos en esta tesis se realizó empleando
el modelo de Marcus. En la Figura 10 se muestran algunas simulaciones obtenidas con
diferentes valores de k0 (con ゜ = 0.5 eV) y para diferentes valores de ゜ (con k0 = 250 s‐1). Se
puede observar claramente que las curvas teóricas obtenidas con diferentes valores de ゜ en el
intervalo 0.2 a 0.8 eV no varían significativamente. Es por ello que para ajustar las curvas
experimentales se decidió fijar el valor de ゜ en 0.5 eV y variar el valor de k0 hasta obtener la
mejor coincidencia con las curvas teóricas.
Figura 10 Variación de la posición de pico con la velocidad de barrido simulada para a) diferentes constantes
cinéticas estándar de transferencia electrónica: k0 = 20, 50, 100, 250 y 550 s‐1, respectivamente y ゜ = 0.5 eV y
b) para diferentes energías de reorganización: ゜ = 0.3, 0.4, 0.5, 0.6, 0.7 y 0.8 eV, con k0 = 250 s‐1.

5. ANÁLISIS DE LA CINÉTICA DE TRANSFERENCIA ELECTRÓNICA
149
Antes de proseguir, se debe hacer una observación muy importante. La voltametría cíclica es
un método eficiente y conveniente para analizar transferencia electrónica en electrodos
modificados, sin embargo, la caída óhmica es un gran limitante en este tipo de análisis ya que
afecta a la fuerza impulsora de la reacción, es decir, al potencial del electrodo de trabajo. De
hecho, la creciente separación entre los picos anódico y catódico cuando se aumenta la
velocidad de barrido también puede deberse a la resistencia no compensada de la solución.
Ésta es grande cuando las altas velocidades de barrido inducen altas corriente.
Feldberg ha demostrado que la resistencia no compensada y una cinética de electrodo lenta
pueden exhibir un efecto similar sobre la forma característica de los voltagramas y en su
deformación a medida que se aumenta la velocidad de barrido [425]. Por lo tanto, se puede
obtener información engañosa si no se tiene el cuidado suficiente que asegure que el efecto
de la caída óhmica es despreciable.
La resistencia no compensada es importante en solventes orgánicos, como acetonitrilo y
diclorometano, e inclusive en agua cuando se barre el potencial a altas velocidades. Este
efecto se puede compensar experimentalmente, usando por ejemplo una retroalimentación
positiva. De todas maneras, una compensación perfecta es muy difícil de lograr y puede
introducir artefactos no deseados. Alternativamente, se puede medir la caída óhmica
experimentalmente y considerarse en el tratamiento posterior de los datos [426, 427].
Por lo tanto, si bien este tipo de método electroquímico, ha sido ampliamente utilizado en
estudios cinéticos de electrodos modificados por su simplicidad y disponibilidad con
equipamiento electroquímico estándar, la aplicación cuantitativa del mismo se restringe
usualmente a sistemas cuyas constantes de velocidad de transferencia electrónica son
significativamente menores que la inversa de las constantes de tiempo asociadas con los
procesos no faradaicos.
Los métodos en dominio de la frecuencia, como la espectroscopía de impedancia
electroquímica y la voltametría de corriente alterna (voltametría AC), han sido menos
empleados. Estas técnicas permiten detectar la presencia de especies redox aún cuando se
tiene cargas muy pequeñas y esto permite utilizar electrodos más pequeños, disminuyendo la
constante de la celda.
Por lo tanto, en esta tesis, además de utilizar la voltametría cíclica para estimar las constantes
cinéticas, también se utilizó impedancia electroquímica. El procedimiento aplicado se detalla a
continuación.

5. ANÁLISIS DE LA CINÉTICA DE TRANSFERENCIA ELECTRÓNICA
150
5.2.1.3 ESPECTROSCOPÍA DE IMPEDANCIA ELECTROQUÍMICA
En el año 1998 Creager y Wooster presentaron una forma alternativa para medir constantes
cinéticas de transferencia electrónica que se puede aplicar a sistemas con especies
electroactivas inmovilizadas sobre un electrodo [413]. Ésta consiste en medir voltagramas AC a
diferentes frecuencias y observar cómo disminuye el pico de corriente observado en el
potencial formal, a medida que aumenta la frecuencia del potencial alterno aplicado. Esta
disminución es una medida de qué tan rápida es la transferencia electrónica entre el electrodo
y los sitios electroactivos como para responder a una excitación cada vez más rápida y corta en
el tiempo.
En la práctica, se observa cómo varía el módulo del cociente entre la admitancia
correspondiente al pico (Ypico) y la correspondiente a un potencial alejado del mismo (Ybkg) con
el logaritmo de la frecuencia (log (f)). El gráfico obtenido se puede ajustar fácilmente y obtener
la constante cinética de transferencia electrónica para el sistema analizado. En particular, se
obtiene la constante cinética estándar (k0) ya que se evalúa la admitancia en el pico, es decir,
en el potencial formal del par redox.
En esta tesis se utilizó el mismo tipo de análisis pero, en lugar de utilizar voltametría AC, se
empleó espectroscopía de impedancia electroquímica, ya que de esta manera se pueden
obtener más datos y de una forma más automática. El procedimiento empleado consistió en
tomar espectros de impedancia (ZRe + jZIm vs. f) a diferentes potenciales contenidos en una
ventana centrada en E°´ (en general, entre 0 y 0.5 V) y espaciados entre sí por 0.01 V. Para
cada potencial se tomaron espectros a varias frecuencias espaciadas logarítmicamente (en
general, 105‐1 Hz para los sistemas más lentos y 106‐1 Hz para los sistemas más rápidos,
tomando 15 y 25 frecuencias, respectivamente) (Figura 11 a)).
A partir de los espectros se puede obtener la impedancia para cada potencial y cada frecuencia
y por ende construir los gráficos de admitancia en función del potencial aplicado (Figura 11 b)).
Finalmente, con estos últimos se obtienen los valores de Ypico e Ybkg, cuyos cocientes se grafican
en función del log (f) (Figura 11 c)).
Para extraer el valor de k0 de las curvas |Ypico / Ybkg| vs. log (f) se modeló la interfase electrodo‐
solución empleando el circuito equivalente que se muestra en la Figura 12.

5. ANÁLISIS DE LA CINÉTICA DE TRANSFERENCIA ELECTRÓNICA
151
Figura 11 a) Espectros de impedancia tomados a diferentes potenciales aplicados. En particular, la línea roja
corresponde a E = E°´del sistema analizado. b) Variación de la admitancia en función del potencial aplicado. Se
observa un pico para E = E°´. c) Variación de |Ypico/Ybkg| en función de la frecuencia. La línea roja es el mejor
ajuste considerando el circuito equivalente de la Figura 13. En este caso se obtiene una kTE = 15000 s‐1.
Figura 12 Circuito equivalente utilizado para modelar la interfaz electrodo‐solución de los sistemas analizados
por espectroscopía de impedancia electroquímica.

5. ANÁLISIS DE LA CINÉTICA DE TRANSFERENCIA ELECTRÓNICA
152
La impedancia total del sistema se puede expresar como la suma de Z3 y ZP (impedancia del
circuito en paralelo), ya que son equivalentes a dos elementos en serie. Para ello es necesario
hallar las expresiones de cada una de ellas. En particular, Z3 es la resistencia de la solución
(Z3 = RS), mientras que (ZP)‐1 = (Z1)
‐1 + (Z2)‐1. A su vez, Z1 es la impedancia correspondiente a un
circuito RC en serie y Z2 es la impedancia correspondiente a un capacitor puro (ver Capítulo 2)
y se pueden expresar como:
1 CTads
2dl
jZ R
2ヽfCj
Z2ヽfC
= += (5.32)
donde RCT es la resistencia asociada a la transferencia de carga, Cads es la pseudo‐capacidad de
la monocapa redox, Cdl es la capacidad de la doble capa y j 1= − . Por lo tanto, ZP se puede
expresar como:
CTads dl
P
CTads dl
j jR
2ヽfC 2ヽfCZ
j jR
2ヽfC 2ヽfC
⎛ ⎞⎜ ⎟⎜ ⎟⎝ ⎠+= + + (5.33)
Entonces la impedancia total asociada al circuito de la Figura 12 se puede escribir como:
S
CTads dl
total
CTads dl
j jR
2ヽfC 2ヽfCZ R
j jR
2ヽfC 2ヽfC
⎛ ⎞⎜ ⎟⎜ ⎟⎝ ⎠+= + + + (5.34)
Cuando el potencial aplicado corresponde al potencial formal del par (Ypico), la admitancia total
del sistema es la inversa de Ztotal y, cuando el potencial está alejado de E°´ (Ybkg), la admitancia
total es la inversa de (Z2 + Z3).
A partir de las expresiones de las ecuaciones (5.32) a (5.34) y considerando que
RCT = 2RT / F2AよOsk0, según se señaló en el Capítulo 2, es posible ajustar los datos
experimentales como se indica en línea roja en la Figura 11 c). Para ello, los valores de Cdl y RS
se obtienen a partir de los valores de impedancia medidos a alta frecuencia y a un potencial

5. ANÁLISIS DE LA CINÉTICA DE TRANSFERENCIA ELECTRÓNICA
153
alejado de E°´. En particular, RS es el valor de la componente real de la impedancia medida en
esas condiciones (RS = ZRe), mientras que Cdl se obtiene de la componente imaginaria
(Cdl = 1/2ヽfZIm). Con ambos valores, además, se puede estimar la constante de la celda,
verificar que se cumple RuCdl << k0 y confirmar que se está midiendo las constantes en forma
adecuada.
Una vez obtenidos estos valores, el ajuste se lleva a cabo empleando como parámetros a Cads y
k0, que se varían hasta obtener la mejor coincidencia entre la curva experimental y la teórica.
5.3 RESULTADOS
5.3.1 ESTIMACIÓN DE LA DISTANCIA AU‐OS
Al principio de este capítulo se señaló que, en el análisis de los mecanismos de transferencia
de carga mediada por un puente molecular, es muy importante conocer la distancia que media
entre el aceptor y el donor de electrones. En esta tesis, la distancia de interés es la que existe
entre el centro electroactivo de osmio y la superficie del electrodo de oro. Por lo tanto, antes
de detallar los resultados obtenidos por cada técnica, se presentan las distancias estimadas
para cada uno de los sistemas analizados y las consideraciones realizadas para ello.
La distancia considerada entre la superficie del electrodo y el átomo de Os se muestra en la
Figura 13. Para calcular dicha distancia se emplearon consideraciones geométricas y longitudes
y ángulos de enlace conocidos [338].
Figura 13 Estructuras representatitvas de los sistemas bajo estudio en los que se indica la distancia, entre la
superficie del electrodo y los sitios electroactivos, en función de la cual se analizó k0.

5. ANÁLISIS DE LA CINÉTICA DE TRANSFERENCIA ELECTRÓNICA
154
En el Capítulo 3 se señaló que durante la reducción de las sales de diazonio los radicales
formados se pueden unir de diferentes maneras al sustrato. En particular, se observó que en el
caso del ácido 2,3,5,6‐tetrafluorobenzoico algunas moléculas se unen a través de un grupo azo,
como se muestra en la Figura 13. Por lo tanto, al estimar la distancia Au‐Os en este sistema, se
consideró un intervalo de longitudes que contempla ambas posibilidades.
Por otro lado, en el caso del sistema Au/PhCOOH/Os se consideraron todas las moléculas que
pueden existir en la capa molecular que se deposita durante la electro‐reducción de la sal de
diazonio correspondiente. Ellas se muestran en la Figura 14. Considerando que en la superficie
existe una mezcla de estas estructuras, se puede considerar que la distancia promedio entre el
centro osmio y la superficie del electrodo puede adoptar valores en el intervalo
(dPhF4COOH, 1.5 x dmono), donde dPhF4COOH es la distancia calculada para el sistema
Au/PhF4COOH/Os en el que, como se discutió en el Capítulo 3, la mayor parte de las moléculas
se encuentra como se muestra en la segunda estructura idealizada de la Figura 13.
En la Tabla 1 se resumen las distancias estimadas entre el electrodo y el centro redox para
todos los sistemas.
Figura 14 Distancia entre la superficie del electrodo de oro y los sitios electroactivos para las diferentes
estructuras que puede presentar la capa subyacente obtenida por reducción del tetrafluoroborato de
4‐carboxibencendiazonio.

5. ANÁLISIS DE LA CINÉTICA DE TRANSFERENCIA ELECTRÓNICA
155
Tabla 1 Distancia que media entre el par redox Os(III) / Os(II) y la superficie del electrodo de oro.
d / Å
Au/MHDA/Os 29.2
Au/MUA/Os 22.7
Au/MOA/Os 17.8
Au/MHA/Os 16.2
Au/MPA/Os 12.3
Au/SPhCOOH/Os 13.8
Au/PhF4COOH/Os 11.6 – 13.0
Au/PhCOOH/Os 11.6 ‐ 17.4
5.3.2 MEDICIÓN DE K0 POR VOLTAMETRÍA CÍCLICA
Se utilizó este método para estimar la constante estándar de transferencia electrónica de
todos los sistemas en estudio. Para ello, se realizaron voltametrías cíclicas a velocidades de
barrido crecientes y se tomó la separación entre los picos anódico y catódico para cada una de
ellas. Las voltametrías obtenidas con los sistemas Au/MUA/Os y Au/PhCOOH/Os en NaNO3
0.1 M se muestran en la Figura 15, a modo de ejemplo.
Figura 15 Voltametrías cíclicas a velocidades de barrido crecientes en NaNO3 0.1 M de los sistemas
a) Au/MUA/Os y b) Au/PhCOOH/Os. La velocidades de barrido varían entre 0.015 y 100 V s‐1 para el primer
sistema y, entre 0.015 y 50 V s‐1, para el segundo. La corriente está normalizada por el área del electrodo
correspondiente y por la velocidad de barrido.

5. ANÁLISIS DE LA CINÉTICA DE TRANSFERENCIA ELECTRÓNICA
156
Los sistemas restantes mostraron el mismo tipo de comportamiento. A su vez, en cada caso, se
simuló la variación de las posiciones de pico con la velocidad de barrido, empleando el modelo
de Marcus para describir la cinética de electrodo. Es decir, se simularon los voltamogramas a
cada velocidad de barrido, expresando la corriente en función del potencial con las constantes
de oxidación y reducción que se indican en la ecuación (5.25). A partir de esto, se varió el valor
de k0, manteniendo ゜ fija en 0.5 eV, hasta obtener el mejor ajuste de las curvas (Ep vs. log(仝))
experimentales.
En la Figura 16 se presentan los resultados experimentales obtenidos en NaNO3 0.1 M para
cada sistema, junto con el mejor ajuste logrado por simulación. En general, la velocidad de
barrido se varió entre 0.01 y 100 V s‐1. En la Tabla 2 se presentan los valores de k0 obtenidos en
cada caso.
Tabla 2 Constante cinética estándar de transferencia electrónica obtenida por Voltametría Cíclica.
k0 / s‐1
Au/MHDA/Os 10
Au/MUA/Os 140
Au/MOA/Os 140
Au/MHA/Os 150
Au/MPA/Os 190
Au/SPhCOOH/Os 550
Au/PhCOOH/Os 45
Au/PhF4COOH/Os 130
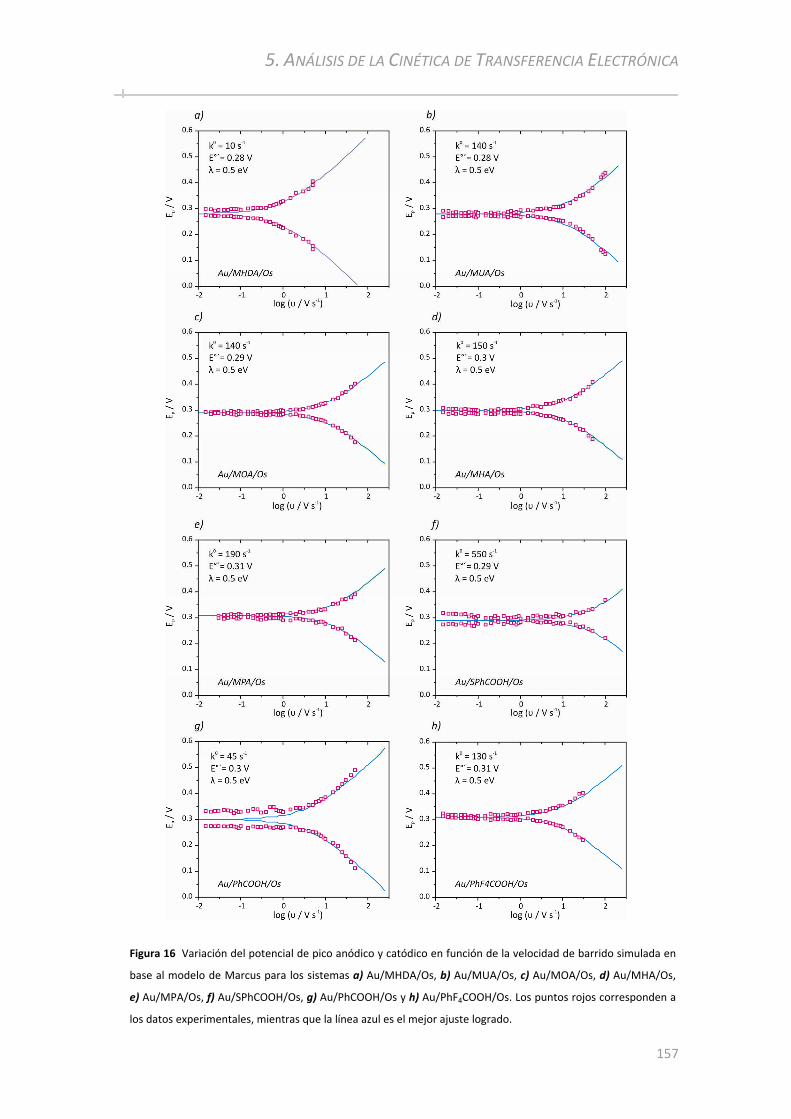
5. ANÁLISIS DE LA CINÉTICA DE TRANSFERENCIA ELECTRÓNICA
157
Figura 16 Variación del potencial de pico anódico y catódico en función de la velocidad de barrido simulada en
base al modelo de Marcus para los sistemas a) Au/MHDA/Os, b) Au/MUA/Os, c) Au/MOA/Os, d) Au/MHA/Os,
e) Au/MPA/Os, f) Au/SPhCOOH/Os, g) Au/PhCOOH/Os y h) Au/PhF4COOH/Os. Los puntos rojos corresponden a
los datos experimentales, mientras que la línea azul es el mejor ajuste logrado.
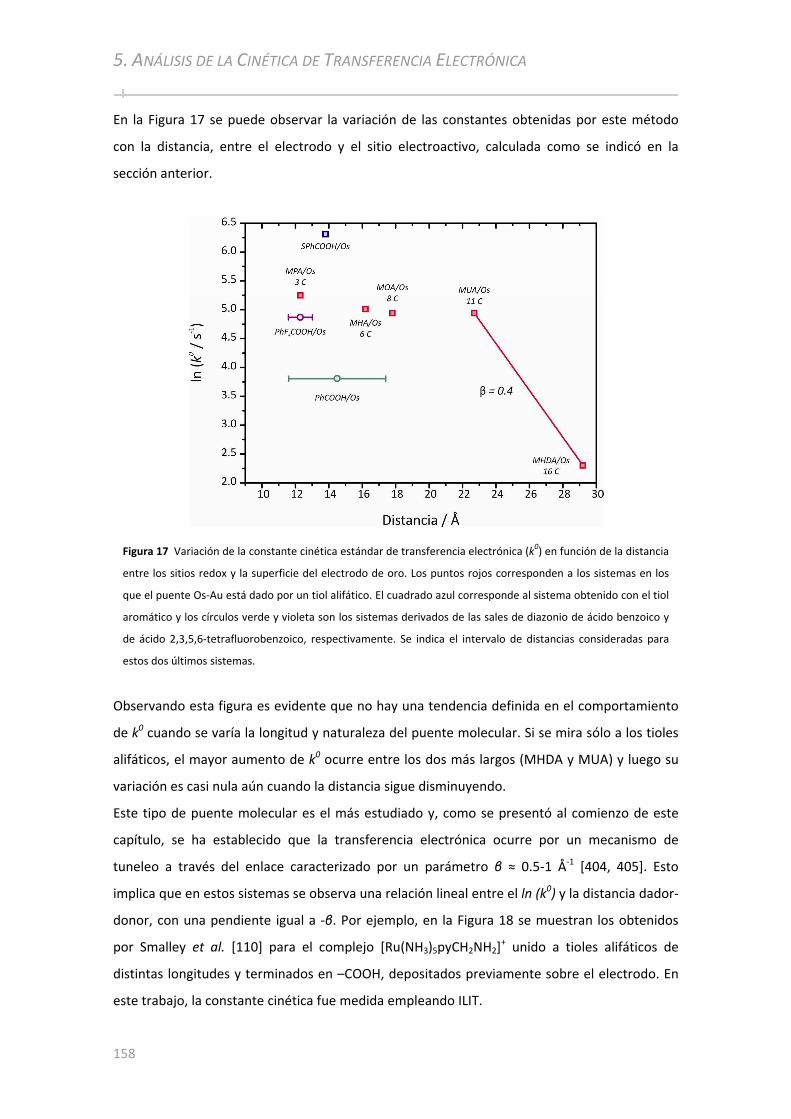
5. ANÁLISIS DE LA CINÉTICA DE TRANSFERENCIA ELECTRÓNICA
158
En la Figura 17 se puede observar la variación de las constantes obtenidas por este método
con la distancia, entre el electrodo y el sitio electroactivo, calculada como se indicó en la
sección anterior.
Observando esta figura es evidente que no hay una tendencia definida en el comportamiento
de k0 cuando se varía la longitud y naturaleza del puente molecular. Si se mira sólo a los tioles
alifáticos, el mayor aumento de k0 ocurre entre los dos más largos (MHDA y MUA) y luego su
variación es casi nula aún cuando la distancia sigue disminuyendo.
Este tipo de puente molecular es el más estudiado y, como se presentó al comienzo de este
capítulo, se ha establecido que la transferencia electrónica ocurre por un mecanismo de
tuneleo a través del enlace caracterizado por un parámetro β ≈ 0.5‐1 Å‐1 [404, 405]. Esto
implica que en estos sistemas se observa una relación lineal entre el ln (k0) y la distancia dador‐
donor, con una pendiente igual a ‐β. Por ejemplo, en la Figura 18 se muestran los obtenidos
por Smalley et al. [110] para el complejo [Ru(NH3)5pyCH2NH2]+ unido a tioles alifáticos de
distintas longitudes y terminados en –COOH, depositados previamente sobre el electrodo. En
este trabajo, la constante cinética fue medida empleando ILIT.
Figura 17 Variación de la constante cinética estándar de transferencia electrónica (k0) en función de la distancia
entre los sitios redox y la superficie del electrodo de oro. Los puntos rojos corresponden a los sistemas en los
que el puente Os‐Au está dado por un tiol alifático. El cuadrado azul corresponde al sistema obtenido con el tiol
aromático y los círculos verde y violeta son los sistemas derivados de las sales de diazonio de ácido benzoico y
de ácido 2,3,5,6‐tetrafluorobenzoico, respectivamente. Se indica el intervalo de distancias consideradas para
estos dos últimos sistemas.

5. ANÁLISIS DE LA CINÉTICA DE TRANSFERENCIA ELECTRÓNICA
159
Asimismo, se muestran nuevamente los datos medidos por voltametría cíclica para facilitar la
comparación.
En ambos sistemas el par electroactivo está unido a la superficie por una unión amida, que se
formó con la misma reacción de post‐funcionalización y las monocapas subyacentes son de la
misma naturaleza. Por lo tanto, ambas familias de sistemas son esencialmente iguales, excepto
por la identidad de la especie redox. Es por ello que no hay razón suficiente para justificar que
en el caso del par Ru(II)+2/Ru(III)+3 se observe un comportamiento coherente con un
mecanismo de tuneleo a través del enlace y que no sea así con los sistemas que aquí se
estudian.
La hipótesis más fuerte, frente a estos datos, es que en las condiciones experimentales
utilizadas, las medidas por voltametría cíclica están limitadas por la constante RuCdl de la celda
empleada que, en todos los casos, fue del orden de 10‐30 ´s. Es decir, la separación de picos
observada se debe a dicha constante y no a que el potencial se barre lo suficientemente rápido
como para volver irreversible la reacción de transferencia electrónica. Es decir que, para los
tioles que tienen menos de 11 C, se midió la constante de la celda en el lugar de k0. Esto
también es cierto para los sistemas obtenidos con el tiol aromático (Au/SPhCOOH/Os) y con la
Figura 18 Comparación con datos de bibliografía (Smalley et al.) de la variación de la constante cinética estándar
con la longitud del puente molecular, que se indica en función del número de átomos de carbono en la cadena
alifática. Se indican los valores del parámetro de tuneleo (β) hallado en cada caso.

5. ANÁLISIS DE LA CINÉTICA DE TRANSFERENCIA ELECTRÓNICA
160
sales de diazonio (Au/PhCOOH/Os y Au/PhF4COOH/Os), que son lo suficientemente cortos
como para inferir que en estos casos la medida también estuvo limitada por RuCdl.
Por todo esto, se procedió a probar un método alternativo para evaluar las constantes
cinéticas, teniendo en cuenta que la prioridad principal era disminuir la constante de tiempo
de la celda. De esta manera, se utilizó la espectroscopía de impedancia electroquímica y se
emplearon electrodos de trabajo más pequeños. Los resultados se presentan a continuación.
5.3.3 MEDICIÓN DE K0 POR ESPECTROSCOPÍA DE IMPEDANCIA ELECTROQUÍMICA
Se emplearon electrodos esféricos de oro, preparados por fusión de un alambre de oro que,
por la tensión superficial del oro líquido, permite formar una pequeña esfera en la punta del
mismo. Se utilizaron alambres de oro de 0.5 mm y 0.15 mm de diámetro con los que se
fabricaron esferas de oro de un área de ≈ 0.02 cm2 y ≈ 0.005 cm2, respectivamente. Los
electrodos más grandes se utilizaron con los sistemas más lentos (los formados con los tioles
de 16, 11 y 8 C) y los más pequeños, con los sistemas restantes. Todas las mediciones se
llevaron a cabo en una solución acuosa de NaNO3 1 M.
En las Figuras 19 a) y b) se muestran, a modo de ejemplo, los gráficos de Nyquist obtenidos
para los sistemas Au/MHDA/Os y Au/MHA/Os por espectroscopía de impedancia
electroquímica. Se tomaron espectros a diferentes potenciales entre 0 y 0.5 V (un espectro
cada 0.01 V) y, a partir de todos ellos, se obtuvo la variación de la admitancia con la frecuencia
que se muestra en las Figuras 19 c) y d), para los sistemas mencionados. Esto se realizó con
todos los sistemas estudiados.
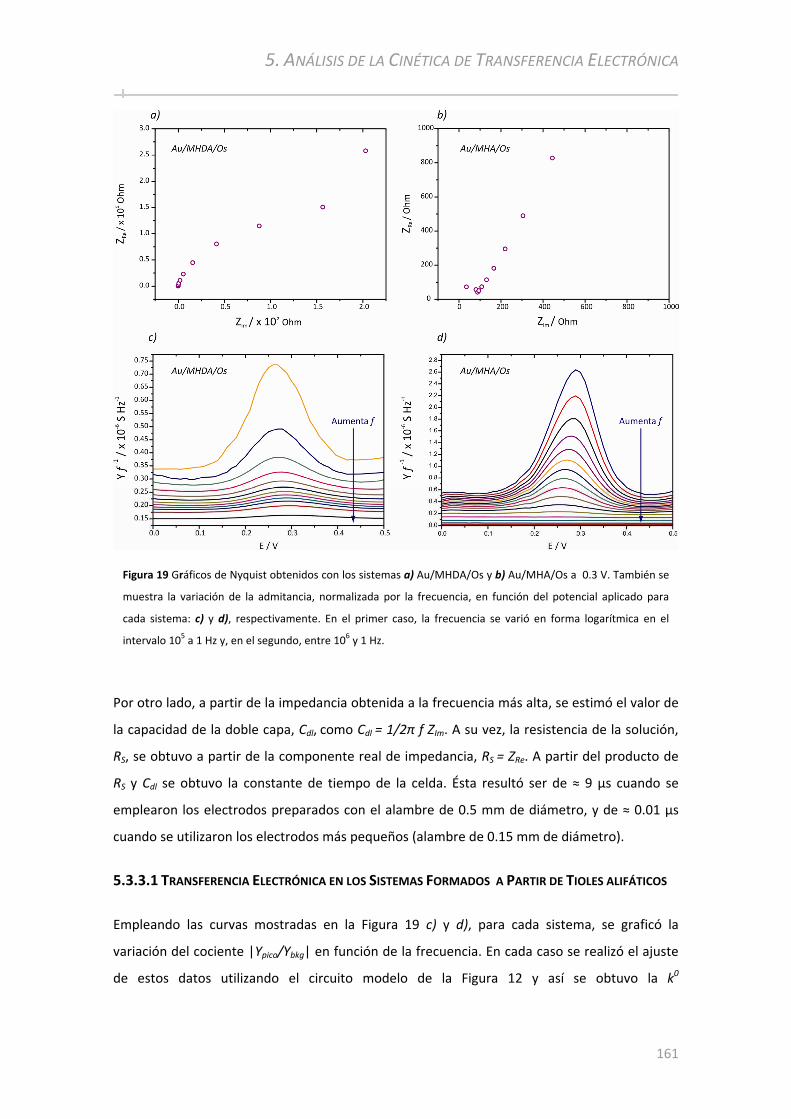
5. ANÁLISIS DE LA CINÉTICA DE TRANSFERENCIA ELECTRÓNICA
161
Por otro lado, a partir de la impedancia obtenida a la frecuencia más alta, se estimó el valor de
la capacidad de la doble capa, Cdl, como Cdl = 1/2ヽ f ZIm. A su vez, la resistencia de la solución,
RS, se obtuvo a partir de la componente real de impedancia, RS = ZRe. A partir del producto de
RS y Cdl se obtuvo la constante de tiempo de la celda. Ésta resultó ser de ≈ 9 ´s cuando se
emplearon los electrodos preparados con el alambre de 0.5 mm de diámetro, y de ≈ 0.01 ´s
cuando se utilizaron los electrodos más pequeños (alambre de 0.15 mm de diámetro).
5.3.3.1 TRANSFERENCIA ELECTRÓNICA EN LOS SISTEMAS FORMADOS A PARTIR DE TIOLES ALIFÁTICOS
Empleando las curvas mostradas en la Figura 19 c) y d), para cada sistema, se graficó la
variación del cociente |Ypico/Ybkg| en función de la frecuencia. En cada caso se realizó el ajuste
de estos datos utilizando el circuito modelo de la Figura 12 y así se obtuvo la k0
Figura 19 Gráficos de Nyquist obtenidos con los sistemas a) Au/MHDA/Os y b) Au/MHA/Os a 0.3 V. También se
muestra la variación de la admitancia, normalizada por la frecuencia, en función del potencial aplicado para
cada sistema: c) y d), respectivamente. En el primer caso, la frecuencia se varió en forma logarítmica en el
intervalo 105 a 1 Hz y, en el segundo, entre 106 y 1 Hz.

5. ANÁLISIS DE LA CINÉTICA DE TRANSFERENCIA ELECTRÓNICA
162
correspondiente. Estos resultados se presentan en la Figura 20 en la que se indica el número
de carbonos en la cadena alifática del tiol correspondiente.
La altura del plateau observado en cada caso depende del valor de la pseudo‐capacidad de la
película electroactiva, Cads (Cads = F2AよOs / 4RT) y, por ende, del cubrimiento superficial de
complejo (よOs) presentado por cada sistema. Es por ello que todas las curvas fueron
normalizadas para facilitar su comparación utilizando la siguiente expresión:
( )pico bkg
pico bkg máx
Y / Y 1¨
Y / Y 1
⎡ ⎤⎣ ⎦⎡ ⎤⎢ ⎥⎣ ⎦
−= − (5.35)
donde (Ypico/Ybkg)máx es el valor más alto de Ypico/Ybkg y ¨ varía entre 0 y 1.
Se puede observar que a medida que disminuye la longitud del tiol, la caída de |Ypico/Ybkg|
ocurre a mayores frecuencias, indicando que la constante cinética de transferencia electrónica
aumenta. En la Tabla 3 se resumen las constantes cinéticas (k0), los valores de cubrimiento
Figura 20 Variación de ¨ con la frecuencia de excitación para todos los sistemas preparados con tioles alifáticos.
Los símbolos corresponden a datos experimentales mientras que las líneas son los mejores ajustes logrados
tomando como modelo el circuito de la Figura 12. Los círculos negros corresponden al sistema Au/MHDA/Os
(16 C), los circulos rojos, a Au/MUA/Os (11 C), los cuadrados verdes se obtuvieron con el sistema Au/MOA/Os
(8 C), los cuadrados azules corresponden a Au/MHA/Os (6 C) y finalmente, los círculos marrones se obtuvieron
con Au/MPA/Os (3 C). Todas las curvas están normalizadas según se indica en la ecuación (5.35).
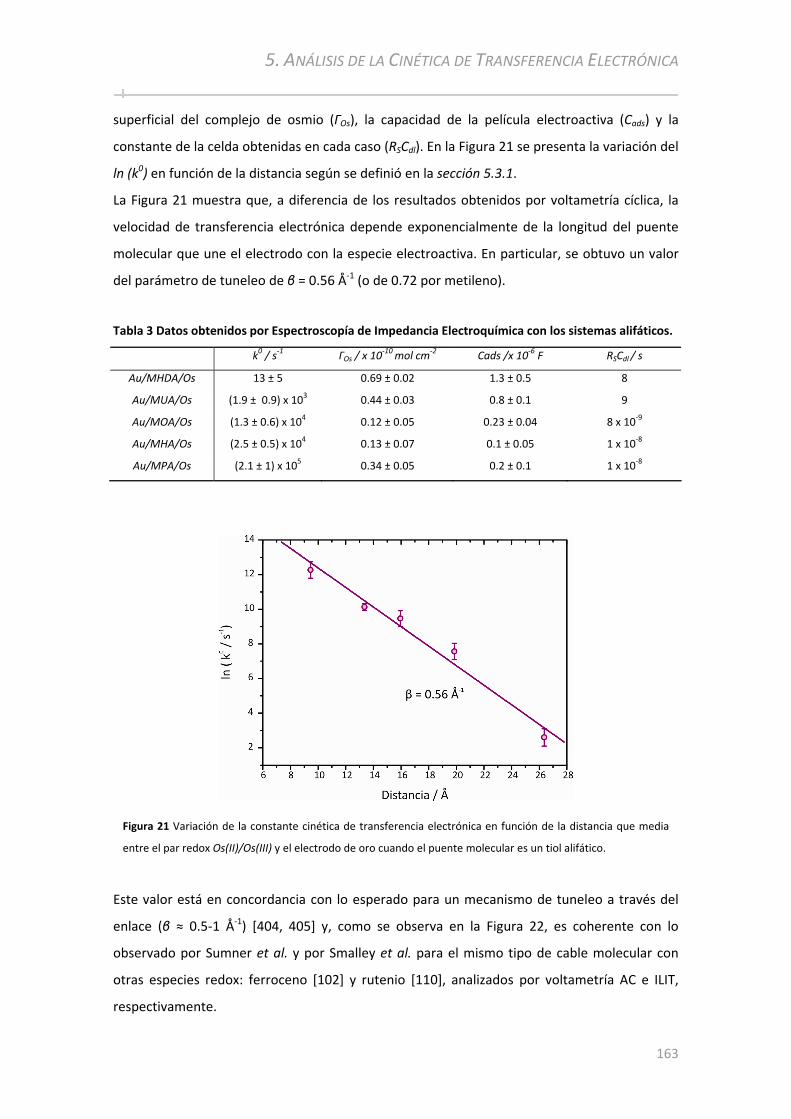
5. ANÁLISIS DE LA CINÉTICA DE TRANSFERENCIA ELECTRÓNICA
163
superficial del complejo de osmio (よOs), la capacidad de la película electroactiva (Cads) y la
constante de la celda obtenidas en cada caso (RSCdl). En la Figura 21 se presenta la variación del
ln (k0) en función de la distancia según se definió en la sección 5.3.1.
La Figura 21 muestra que, a diferencia de los resultados obtenidos por voltametría cíclica, la
velocidad de transferencia electrónica depende exponencialmente de la longitud del puente
molecular que une el electrodo con la especie electroactiva. En particular, se obtuvo un valor
del parámetro de tuneleo de β = 0.56 Å‐1 (o de 0.72 por metileno).
Tabla 3 Datos obtenidos por Espectroscopía de Impedancia Electroquímica con los sistemas alifáticos.
k0 / s‐1 よOs / x 10‐10 mol cm‐2 Cads /x 10‐6 F RSCdl / s
Au/MHDA/Os 13 ± 5 0.69 ± 0.02 1.3 ± 0.5 8
Au/MUA/Os (1.9 ± 0.9) x 103 0.44 ± 0.03 0.8 ± 0.1 9
Au/MOA/Os (1.3 ± 0.6) x 104 0.12 ± 0.05 0.23 ± 0.04 8 x 10‐9
Au/MHA/Os (2.5 ± 0.5) x 104 0.13 ± 0.07 0.1 ± 0.05 1 x 10‐8
Au/MPA/Os (2.1 ± 1) x 105 0.34 ± 0.05 0.2 ± 0.1 1 x 10‐8
Este valor está en concordancia con lo esperado para un mecanismo de tuneleo a través del
enlace (β ≈ 0.5‐1 Å‐1) [404, 405] y, como se observa en la Figura 22, es coherente con lo
observado por Sumner et al. y por Smalley et al. para el mismo tipo de cable molecular con
otras especies redox: ferroceno [102] y rutenio [110], analizados por voltametría AC e ILIT,
respectivamente.
Figura 21 Variación de la constante cinética de transferencia electrónica en función de la distancia que media
entre el par redox Os(II)/Os(III) y el electrodo de oro cuando el puente molecular es un tiol alifático.

5. ANÁLISIS DE LA CINÉTICA DE TRANSFERENCIA ELECTRÓNICA
164
5.3.3.2 TRANSFERENCIA ELECTRÓNICA EN LOS SISTEMAS AROMÁTICOS
El mismo procedimiento descripto para los sistemas formados con tioles alifáticos fue llevado a
cabo con los sistemas obtenidos a partir del tiol aromático y de las dos sales de diazonio.
En la Figura 23 se muestran las curvas de ¨ en función de la frecuencia para los tres:
Au/SPhCOOH/Os, Au/PhCOOH/Os y Au/PhF4COOH/Os y en la Tabla 4 se presentan las
constantes cinéticas (k0) obtenidas con este análisis, junto con los valores de cubrimiento
superficial del complejo de osmio (よOs), la pseudo‐capacidad redox (Cads) y la constante de celda
(RSCdl).
Figura 22 Datos de variación de k0 con la longitud del puente molecular alifático (se indica el número de
carbonos en el mismo) extraídos de bibliografía para dos cuplas distintas: ferroceno/ferrocianuro (cuadrados
violetas) y Ru(II)/Ru(III) (círculos rojos) unidas por el mismo tipo de puente molecular que se empleó en este
trabajo de tesis. Se muestran nuevamente los resultados obtenidos con Os(II)/Os(III) a modo de comparación
(círculos azules).
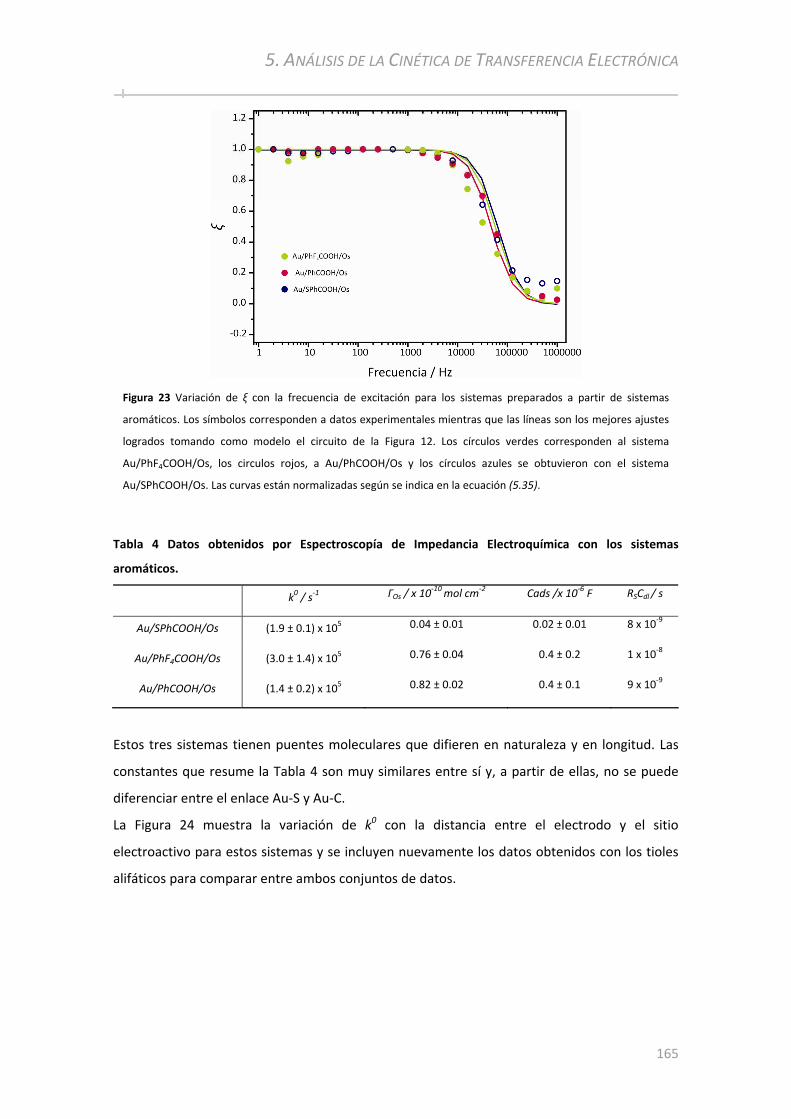
5. ANÁLISIS DE LA CINÉTICA DE TRANSFERENCIA ELECTRÓNICA
165
Tabla 4 Datos obtenidos por Espectroscopía de Impedancia Electroquímica con los sistemas
aromáticos.
k0 / s‐1 よOs / x 10‐10 mol cm‐2 Cads /x 10‐6 F RSCdl / s
Au/SPhCOOH/Os (1.9 ± 0.1) x 105 0.04 ± 0.01 0.02 ± 0.01 8 x 10‐9
Au/PhF4COOH/Os (3.0 ± 1.4) x 105 0.76 ± 0.04 0.4 ± 0.2 1 x 10‐8
Au/PhCOOH/Os (1.4 ± 0.2) x 105 0.82 ± 0.02 0.4 ± 0.1 9 x 10‐9
Estos tres sistemas tienen puentes moleculares que difieren en naturaleza y en longitud. Las
constantes que resume la Tabla 4 son muy similares entre sí y, a partir de ellas, no se puede
diferenciar entre el enlace Au‐S y Au‐C.
La Figura 24 muestra la variación de k0 con la distancia entre el electrodo y el sitio
electroactivo para estos sistemas y se incluyen nuevamente los datos obtenidos con los tioles
alifáticos para comparar entre ambos conjuntos de datos.
Figura 23 Variación de ¨ con la frecuencia de excitación para los sistemas preparados a partir de sistemas
aromáticos. Los símbolos corresponden a datos experimentales mientras que las líneas son los mejores ajustes
logrados tomando como modelo el circuito de la Figura 12. Los círculos verdes corresponden al sistema
Au/PhF4COOH/Os, los circulos rojos, a Au/PhCOOH/Os y los círculos azules se obtuvieron con el sistema
Au/SPhCOOH/Os. Las curvas están normalizadas según se indica en la ecuación (5.35).

5. ANÁLISIS DE LA CINÉTICA DE TRANSFERENCIA ELECTRÓNICA
166
Estos datos no se pueden ajustar linealmente porque el análisis sería incorrecto por el hecho
de que no se trata del mismo puente molecular que varía su longitud, sino que además de la
longitud varía su naturaleza. Sin embargo, se debe destacar que las constantes medidas caen
aproximadamente en la recta de los sistemas alifáticos (β ≈ 0.56 Å‐1) y esto indica que su
orden de magnitud es consistente con la distancia a la que se hallan los centros redox de la
superficie.
Como se mencionó al comienzo de este capítulo se han encontrado valores de β ≈ 0.3 Å‐1 para
ferroceno unido a la superficie del electrodo a través de mercapto‐oligo(fenilen‐etilenos) [33,
147, 177]. Por otro lado, el par Os(II)/Os(III) ha sido analizada cuando está unida a puentes
aromáticos de diferente naturaleza y por distintas técnicas. Forster [147‐149] y Abruña [360] la
han estudiado unida a puentes moleculares del tipo py‐(CH2)n‐py sobre oro por
cronoamperometría y por voltametría cíclica, respectivamente, empleando en ambos casos
microelectrodos de platino. En particular, Forster observó un valor de β = 1.2‐1.6 Å‐1 y lo
atribuyó a un mecanismo de tuneleo a través del espacio.
En la Figura 25 se presentan estos datos extraídos de bibliografía junto con los obtenidos en
este trabajo.
Figura 24 Variación de la constante cinética de transferencia electrónica en función de la distancia que media
entre el par redox Os(II)/Os(III) y el electrodo de oro cuando el puente molecular contiene un anillo aromático
(círculos rojos). Se indican los rangos de distancias considerados en los sistemas Au/PhCOOH/Os
Au/PhF4COOH/Os. En azul se muestran nuevamente los datos obtenidos con los tioles alifáticos.
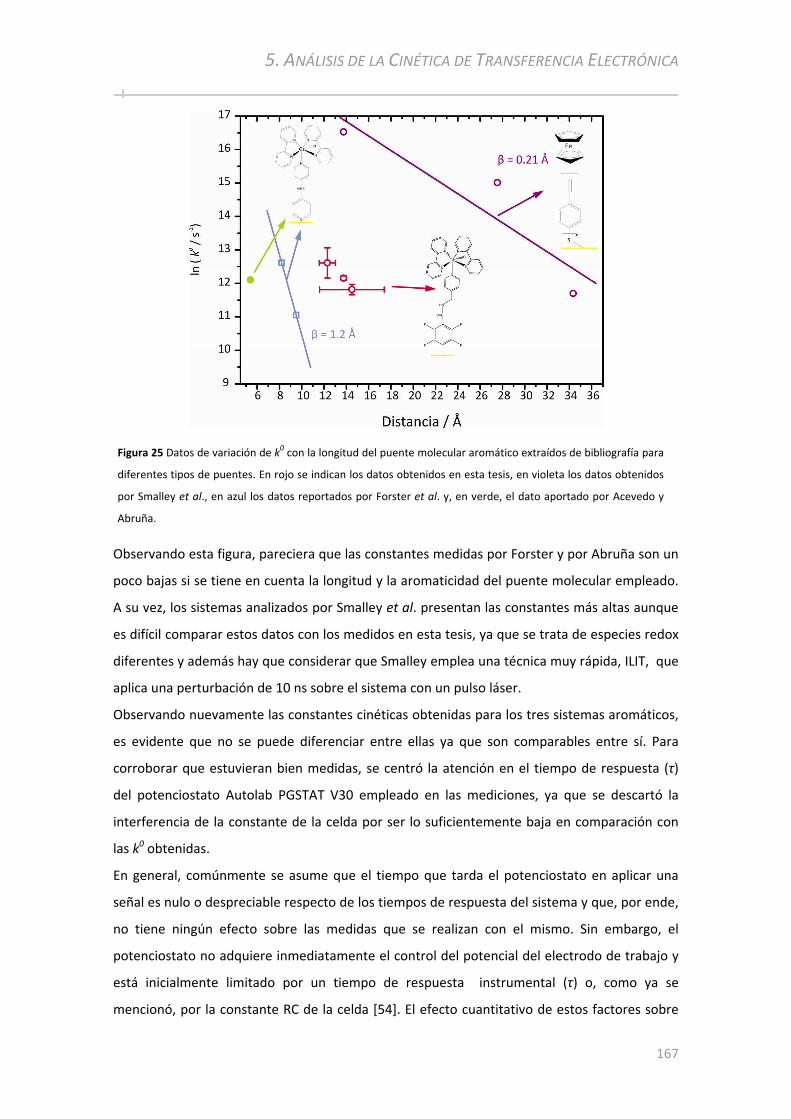
5. ANÁLISIS DE LA CINÉTICA DE TRANSFERENCIA ELECTRÓNICA
167
Observando esta figura, pareciera que las constantes medidas por Forster y por Abruña son un
poco bajas si se tiene en cuenta la longitud y la aromaticidad del puente molecular empleado.
A su vez, los sistemas analizados por Smalley et al. presentan las constantes más altas aunque
es difícil comparar estos datos con los medidos en esta tesis, ya que se trata de especies redox
diferentes y además hay que considerar que Smalley emplea una técnica muy rápida, ILIT, que
aplica una perturbación de 10 ns sobre el sistema con un pulso láser.
Observando nuevamente las constantes cinéticas obtenidas para los tres sistemas aromáticos,
es evidente que no se puede diferenciar entre ellas ya que son comparables entre sí. Para
corroborar que estuvieran bien medidas, se centró la atención en el tiempo de respuesta (〃)
del potenciostato Autolab PGSTAT V30 empleado en las mediciones, ya que se descartó la
interferencia de la constante de la celda por ser lo suficientemente baja en comparación con
las k0 obtenidas.
En general, comúnmente se asume que el tiempo que tarda el potenciostato en aplicar una
señal es nulo o despreciable respecto de los tiempos de respuesta del sistema y que, por ende,
no tiene ningún efecto sobre las medidas que se realizan con el mismo. Sin embargo, el
potenciostato no adquiere inmediatamente el control del potencial del electrodo de trabajo y
está inicialmente limitado por un tiempo de respuesta instrumental (〃) o, como ya se
mencionó, por la constante RC de la celda [54]. El efecto cuantitativo de estos factores sobre
Figura 25 Datos de variación de k0 con la longitud del puente molecular aromático extraídos de bibliografía para
diferentes tipos de puentes. En rojo se indican los datos obtenidos en esta tesis, en violeta los datos obtenidos
por Smalley et al., en azul los datos reportados por Forster et al. y, en verde, el dato aportado por Acevedo y
Abruña.

5. ANÁLISIS DE LA CINÉTICA DE TRANSFERENCIA ELECTRÓNICA
168
las medidas de constantes cinéticas ha sido analizado en profundidad en la literatura y se han
derivado ecuaciones que describen el error cometido cuando las perturbaciones aplicadas a un
sistema electroquímico no son ideales [428‐431].
Para analizar el tiempo de respuesta del potenciostato utilizado en las mediciones de
constantes cinéticas, se aplicó un salto de potencial 1 V sobre la celda electroquímica utilizada
y se registró el perfil de dicha señal con un osciloscopio. El resultado se muestra en la Figura 26
y se puede observar que el tiempo de respuesta del potenciostato es de aproximadamente
5 ´s.
Si se considera que no se pueden estudiar procesos más rápidos que la perturbación que se
aplica sobre un sistema entonces, en este caso, se presentan limitaciones para medir en forma
precisa fenómenos que ocurran en menos de 〃 = 5 ´s después de aplicada la misma. Dicho de
otra forma, la medida de cinéticas más rápidas que 1 / 〃 ≈ 2 x 105 s‐1, tiene cierto error
asociado al tiempo de respuesta del potenciostato.
Los tres sistemas aromáticos y el obtenido con el tiol más corto (MPA) presentaron constantes
cinéticas que estuvieron en el orden de ≈ 1 x 105 s‐1 con lo cual, a partir del análisis precedente,
deberían ser medidas nuevamente con un potenciostato más rápido, con una técnica óptica o
coulostática [432] para corroborar los valores que arrojó este estudio.
Finalmente, vale mencionar el único trabajo reportado en la literatura que analiza monocapas
de ácido 4‐mercaptobenzoico y capas delgadas de ácido benzoico preparadas de la misma
manera que en esta tesis y post‐funcionalizadas con ferrocen‐metilamina (por el mismo
procedimiento aquí empleado) [191]. Este trabajo fue llevado a cabo por Gooding y
Figura 26 Evolución del potencial en el tiempo aplicado por el potenciostato Autolab PGSTAT V30. Se registró
con un osciloscopio y se empleó la misma celda utilizada en las medidas de impedancia electroquímica.

5. ANÁLISIS DE LA CINÉTICA DE TRANSFERENCIA ELECTRÓNICA
169
colaboradores y en él se midieron las constantes cinéticas de transferencia electrónica por
voltametría cíclica. Los valores hallados fueron ≈ 257 s‐1, para la sal de diazonio, y ≈ 103 s‐1, para
el tiol aromático. Estas constantes son muy bajas en comparación con las obtenidas en el
presente trabajo, aún con limitaciones del potenciostato (Tabla 4), y en realidad son similares a
las que se midieron en esta tesis por voltametría cíclica. Esto sugiere que en el trabajo
mencionado se cometió un error apreciable en la medición de k0.
5.4 CONCLUSIONES
A lo largo de este capítulo se realizó un análisis detallado de la transferencia electrónica en los
sistemas modelo que incluyen al complejo de osmio unido a la superficie de un electrodo de
oro a través de un puente alifático, de longitud variable, y de diferentes puentes aromáticos.
Este análisis se realizó por dos técnicas electroquímicas: voltametría cíclica y espectroscopía de
impedancia electroquímica. Se comprobó que en la primera de ellas la limitación de la
constante de celda es importante cuando se quiere analizar sistemas en los que el puente
molecular tiene una longitud menor a 20 Å.
Finalmente, empleando la segunda de las técnicas mencionadas, se pudo observar que en los
sistemas alifáticos el mecanismo de transferencia electrónica es el tuneleo a través del enlace,
como está reportado para este tipo de sistemas.
Por otro lado, en el caso de los sistemas aromáticos, los valores de k0 hallados no permiten
diferenciar entre los distintos tipos de enlace a la superficie del electrodo (Au‐C y Au‐S). Se
deberían validar estos datos empleando un potenciostato más rápido, debido a las
limitaciones presentadas por el que fue utilizado en este análisis.


CAPÍTULO 6
INTERFAZ ELECTRODO‐SOLUCIÓN


6. INTERFAZ ELECTRODO‐SOLUCIÓN
173
n el Capítulo 4 se demostró que los sistemas estudiados en esta tesis poseen grupos
ácidos en la superficie que no reaccionan con el complejo de osmio durante la post‐
funcionalización. Es decir, en los sistemas finales se tiene el par redox rodeado de grupos que
pueden cambiar su estado de carga con el pH y, por ende, la densidad de carga superficial.
En este capítulo se presenta un análisis más detallado de la interfaz electrodo‐solución y de los
factores que la afectan y, que por ende, pueden afectar la cinética de transferencia
electrónica, dado que los grupos redox experimentan un cambio en su estado de carga. Se
estudia el efecto de la concentración superficial de los centros electroactivos, de la fuerza
iónica y del pH de la solución empleada sobre la cinética y termodinámica de la transferencia
electrónica. Los resultados se analizaron en el marco del Modelo de Smith‐White [433] que se
detalla a continuación junto con la descripción de la doble capa.
6.1 MARCO TEÓRICO
6.1.1 ESTRUCTURA DE LA INTERFASE ELECTRODO‐SOLUCIÓN
El primer modelo de la doble capa eléctrica fue desarrollado por Helmholtz [434, 435], quien
modeló la interfase electrodo‐solución como si se tratara de un capacitor simple de caras
paralelas (Figura 1 a)). Este modelo supone que la superficie del electrodo acumula carga ya
que se trata de un conductor, y los iones de la solución se adsorben sobre ella para
compensarla. La limitación de este modelo reside en la suposición de que la capacidad de esta
doble capa es independiente del potencial aplicado al electrodo y de la concentración del
electrolito [54].
Más tarde, Gouy [436, 437] y Chapman [438] plantearon, en forma independiente, un modelo
más completo, considerando que si bien en el electrodo la carga puede confinarse a la
superficie, lo mismo no necesariamente es cierto en la solución. Por lo tanto, propusieron que
los iones forman una capa difusa con una mayor concentración de carga cerca de la superficie
del electrodo, donde las fuerzas electrostáticas pueden superar en alguna medida al desorden
térmico, y disminuye con la distancia a la misma (Figura 1 b)). De esta manera se genera una
separación de cargas en la interfase. El espesor de la capa difusa dependerá de la
concentración del electrolito soporte y del potencial aplicado al electrodo. A mayor potencial
aplicado y/o a mayor concentración, el espesor disminuye y la capacidad de la doble capa
aumenta [54].
La ecuación (6.1) expresa la relación que existe entre la densidad de carga superficial del metal
y la distribución de potencial en la doble capa:
E

6. INTERFAZ ELECTRODO‐SOLUCIÓN
174
( )1/2 0M BS 0 sal
B
zeφσ σ 8k T00 C sinh
2k T
⎛ ⎞⎜ ⎟⎜ ⎟⎝ ⎠= − = (6.1)
donde σM y σS son las densidades de carga en el metal y la solución, respectivamente, 0 es la
constante dieléctrica del medio, 00 es la permitividad del vacío, φ0 es el potencial electrostático
medido entre el electrodo y el seno de la solución, Csal es la concentración del electrolito en el
seno de la solución, kB es la constante de Boltzmann y T es la temperatura. El desarrollo que
lleva a la deducción de esta expresión está bien descripto en la literatura citada [54].
Si bien este modelo es más completo que el propuesto por Helmholtz, todavía presenta ciertas
fallas que se atribuyen a la suposición de que los iones están restringidos a una posición en la
solución. Son considerados como cargas puntuales que pueden acercarse a la superficie del
electrodo hasta una distancia arbitraria y, por eso, cuando se aplican altos potenciales los
iones pueden acercarse tanto como para anular la distancia entre ambas fases (electrodo y
solución). Este comportamiento no es posible ya que los iones tienen un tamaño finito y no
pueden acercarse al electrodo más allá de su radio iónico y, si están solvatados, hay que
considerar también el radio de la esfera de solvatación. Stern [439] sugirió que se debía
considerar este plano de máximo acercamiento y combinó los modelos pensados por
Helmholtz y Gouy‐Chapman en uno solo (Figura 1 c)). Es decir, consideró que la interfase
estaba formada por varias capas. La más cercana al electrodo, la capa interna, contiene
moléculas de solvente (y a veces algunos iones u otras moléculas) que se hallan adsorbidos
específicamente sobre la superficie del electrodo. Esta capa se denomina capa compacta, capa
de Helmholtz (por la similitud con este modelo) o capa de Stern. El plano en el que están
Figura 1 Modelos propuestos para la estructura de la interfaz electrodo‐solución: a) Modelo de Helmholtz,
b) Modelo de Gouy‐Chapman y c) Modelo de Gouy‐Chapman‐Stern.

6. INTERFAZ ELECTRODO‐SOLUCIÓN
175
ubicados los centros de los iones adsorbidos específicamente, se denomina plano interno de
Helmholtz. La segunda capa está compuesta por los iones solvatados que se hallan adsorbidos
no específicamente. Éstos pueden acercarse menos al electrodo e interactúan con él por
medio de fuerzas electrostáticas de largo alcance. El plano en el que se ubican recibe el
nombre de plano externo de Helmholtz. Debido a la agitación térmica en la solución, estos
iones se distribuyen en la capa difusa que se extiende desde el plano externo de Helmholtz
hasta el seno de la solución. A partir de estas consideraciones, la relación entre la densidad de
carga y la distribución de potencial se puede expresar como:
( ) ( )2
B
1/2 1/2 M 2M B BS 0 0 0sal sal
B 0
σ xzeφ zeσ σ 8k T00 C sinh 8k T00 C sinh φk T 2k T 00
⎡ ⎤⎛ ⎞⎛ ⎞ ⎢ ⎥⎜ ⎟⎜ ⎟⎜ ⎟ ⎜ ⎟⎢ ⎥⎝ ⎠ ⎝ ⎠⎣ ⎦= − = = − (6.2)
El desarrollo de esta expresión se encuentra detallado en la bibliografía citada.
6.1.2 TEORÍA DE DISTRIBUCIÓN INTERFACIAL DEL POTENCIAL
6.1.2.1 FUNDAMENTOS
La voltametría cíclica es el método electroquímico más utilizado en la caracterización de
sistemas en los que las especies electroactivas están unidas a la superficie del electrodo de
trabajo. En particular, cuando se trata de películas delgadas, la teoría más simple que describe
estos sistemas [54] permite realizar tres predicciones en cuanto a la forma del voltamograma:
i) la corriente de pico (Ip) es directamente proporcional a la concentración superficial de los
centros redox, よT, y dicho pico aparece al potencial estándar del par redox, E°; ii) la onda
voltamétrica es simétrica respecto del E° y tiene un ancho a media altura de 0.09 V/n (con n, el
número de electrones involucrados en la reacción de electrodo) y iii) la posición de la onda y
su ancho no varían con よT. Sin embargo, frecuentemente en la práctica estas predicciones no
se cumplen. En general, las desviaciones observadas han sido atribuidas a efectos de actividad
superficial, inhomogeneidades en la estructura química de las películas y a efectos de
apareamiento iónico de los centros redox con los contraiones presentes en el electrolito.
Smith y White propusieron una teoría que considera la distribución interfacial del potencial en
la superficie de un electrodo metálico, modificado con una película electroactiva adsorbida
irreversiblemente, y que permite explicar las desviaciones de la idealidad observadas
experimentalmente en este tipo de sistemas [433].

6. INTERFAZ ELECTRODO‐SOLUCIÓN
176
En primer lugar, se debe considerar el esquema del perfil de potencial interfacial que se
muestra en la Figura 2 para un electrodo metálico modificado con una monocapa
electroactiva. En la misma, se asume que los centros redox se encuentran a una distancia finita
de la superficie del electrodo, ubicados en el plano de transferencia electrónica (PTE).
Los potenciales en el metal, el plano de transferencia electrónica y en el seno de la solución
están indicados por φM, φPTE y φS, respectivamente. La caída de potencial desde el metal a la
solución ocurre en dos etapas. La primera caída (φM – φPTE) es lineal, ocurre dentro de la
monocapa molecular y puede modelarse como una caída de potencial dentro de un
dieléctrico. La segunda, (φPTE – φS), ocurre entre el PTE y el seno de la solución, en lo que se
conoce como la capa difusa y que puede ser descripta por el modelo de Gouy‐Chapman
descripto en la sección anterior.
Considerando esta descripción, se evidencia que la respuesta voltamétrica debe depender de
esta distribución de potencial ya que sólo una fracción de la caída de potencial interfacial
(φM – φS) ocurre entre el electrodo y el PTE. A partir de este hecho, se deduce que el potencial
de equilibrio que gobierna las concentraciones superficiales de O y R, será función del
potencial electrostático en el PTE (φPTE). A su vez, este potencial será función de la estructura
molecular de la película y de cualquier factor de la celda que afecte la distribución de potencial
(por ejemplo, la constante dieléctrica del solvente). Además, si las cargas electrostáticas de las
especies electroactivas (zO y zR) no son iguales, φPTE también será función del estado de
Figura 2 Esquema de una película electroactiva adsorbida irreversiblemente sobre un electrodo en contacto con
una solución de electrolito. Los centros O/R se indican con los círculos violeta y están separados de la superficie
por una película dieléctrica de espesor finito. La línea roja indica el perfil de potencial interfacial que cae
linealmente entre el metal y el PTE y no linealmente entre éste y el seno de la solución.

6. INTERFAZ ELECTRODO‐SOLUCIÓN
177
oxidación de la película. Por lo tanto, la fuerza impulsora de la transferencia de carga variará
continuamente durante una voltametría cíclica de una manera difícil de predecir.
La teoría propuesta por Smith y White permite predecir cómo será el comportamiento de este
tipo de sistemas teniendo en cuenta todo lo mencionado. A continuación se presenta la
expresión para la respuesta voltamétrica que surge de la misma. Se remite a la bibliografía
citada y al Apéndice I para mayor detalle.
Para comenzar, se debe tener en cuenta que en estos sistemas la película electroactiva está
adsorbida irreversiblemente y esto implica que: i) el par redox no está presente en solución y ii)
la concentración superficial total de las especies redox (よT = よO + よR) es constante durante el
experimento.
Para poder predecir la relación corriente‐potencial (i‐E) de un dado sistema, primero es
necesario hallar una expresión que relacione el potencial de electrodo con las concentraciones
de equilibrio de O y R (よO y よR), ya que son estas últimas las que lo determinan. A partir de
consideraciones simples de los potenciales electroquímicos ( i ) de todas las especies
presentes en la celda, se llega a:
( ) OPET S
R
よRTE E φ ‐φ ln
nF よ
⎡ ⎤− °− = ⎢ ⎥⎣ ⎦ (6.3)
Para determinar la relación よO / よR es necesario conocer la diferencia de potencial entre el PTE
y el seno de la solución (φPET – φS). Para ello, se considera que i) la única carga en la monocapa
es la asociada a O y R en el PTE, ii) las cargas en el metal (x = 0) y en los centros redox (x = d)
están deslocalizadas, iii) los iones del electrolito sólo se pueden acercar hasta una distancia del
electrodo x = d y iv) la contribución al potencial interfacial por parte de los dipolos moleculares
es despreciable.
A partir de estas consideraciones y empleando la Ley de Gauss para relacionar los potenciales
de cada fase con las cargas correspondientes, se llega a las siguientes expresiones de densidad
de carga:
( )( )
0 1 M PTEM
PTE SS 0 2
M PTE S
0 0 φ ‐φσ
d
ze φ ‐φ2kTσ 0 0 ゛ senh
ze 2kT
σ σ σ 0
=⎛ ⎞⎛ ⎞= − ⎜ ⎟⎜ ⎟⎝ ⎠ ⎝ ⎠
+ + = (6.4)

6. INTERFAZ ELECTRODO‐SOLUCIÓN
178
donde e es la carga del electrón, σM, σPTE y σS son las densidades de carga asociadas al metal, al
plano de transferencia electrónica y a la solución, respectivamente, є0 y є1 y є2 son las
permitividades del vacío, de la película molecular y de la solución, respectivamente y ゛ es la
inversa de la distancia de Debye (゛‐1) que se define como la distancia en que los iones pueden
apantallar un campo eléctrico. En este caso, dicho campo eléctrico viene dado por las cargas
presentes en la capa electroactiva depositada sobre el electrodo. Si se considera un electrolito
acuoso z:z, la distancia de Debye se puede estimar como:
r1 02
o
0 0 RT゛
2F C− = (6.5)
donde 00 es la permitividad del vacío, 0r es la constante dieléctrica, Co es la concentración del
electrolito y R, T y F son las constantes conocidas.
Usando las ecuaciones (6.3) y (6.4) se puede describir la distribución del potencial interfacial
en función del potencial aplicado y de parámetros electroquímicos. Es conveniente describir la
distribución de potencial en función del estado de oxidación de la película, y considerando
E = φM ‐ φAg (φAg corresponde al potencial en el electrodo de referencia, Ag/AgCl):
M PTE PZC
RT 1 fφ ‐φ E ‐E ln
nF f
−⎡ ⎤⎛ ⎞° − ⎜ ⎟ ⎢ ⎥⎝ ⎠ ⎣ ⎦= (6.6)
donde f=よO / よT , es la fracción de moléculas en el estado oxidado y EPZC es el potencial de carga
cero del electrodo desnudo, en ausencia de adsorción de iones de la solución, medido
respecto del electrodo de referencia. Por lo tanto se puede reescribir σM y σPTE reemplazando
(6.6) en (6.4).
Considerando que la corriente voltamétrica total, que se mide en función del potencial
aplicado al electrodo, es la suma de la corriente capacitiva (ic) y de la corriente faradaica (if), se
pueden hallar expresiones para cada una ellas considerando todo lo expuesto hasta ahora.
De esta manera, la corriente capacitiva se puede expresar como:
( )1 1 1c T T 1 S PTE Mi 仝AC con C C C 1 σ / σ− − −= = + + ∂ ∂ (6.7)

6. INTERFAZ ELECTRODO‐SOLUCIÓN
179
donde 仝 es la velocidad de barrido en la voltametría cíclica, A es el área del electrodo, CT es la
capacidad interfacial total y C1 y CS son las capacidades asociadas a la película y a la solución,
respectivamente. A su vez, para la corriente faradaica se tiene:
( )2 2PTE
f T
φn Fi 仝Aよ f 1 f 1
RT E
⎛ ⎞ ∂⎛ ⎞= − −⎜ ⎟ ⎜ ⎟∂⎝ ⎠⎝ ⎠ (6.8)
De esta manera, la respuesta voltamétrica para un par redox adsorbido irreversiblemente
sobre un electrodo se puede expresar como:
PTEt c f T
M
σi i i 仝A 1 C
σ
⎛ ⎞∂= + = +⎜ ⎟∂⎝ ⎠ (6.9)
6.1.2.2 PREDICCIONES DE LA TEORÍA
Cuando se tiene un par del tipo 2 +O + R+ yywxyyē (que es justamente la categoría a la que
pertenece el Os(II)+ / Os(III)+2) adsorbida irreversiblemente sobre la superficie de un electrodo,
el modelo de Smith y White predice que la voltametría cíclica obtenida se desvía del
comportamiento ideal en ciertas condiciones: a) cuando aumenta el cubrimiento superficial
(よT), b) disminuye la concentración de electrolito, c) la película que actúa de dieléctrico entre la
el sitio electroactivo y el metal presenta una mayor relación є1/d1 o d) se emplean solventes
de menor constante dieléctrica. La no idealidad se manifiesta en los picos anódico y catódico
que se ensanchan y se alejan del potencial formal esperado, corriéndose hacia potenciales más
positivos.
La interpretación física de estas observaciones se obtiene de la variación de la distribución del
potencial a través de la película electroactiva a medida que varía el potencial aplicado y, por
ende, el estado de oxidación de esta última. Mientras la mayor parte de la caída de potencial
ocurre dentro de la monocapa, existe una pequeña caída entre el PTE y la solución (φPTE – φS).
Esta caída depende del estado de oxidación de la película y aumenta a medida que ocurre la
oxidación. Este aumento provoca la disminución de la fuerza impulsora de la transferencia
electrónica definida según la ecuación (6.3), afectando la relación i‐E en la forma mencionada.
De esta manera, se pone de manifiesto la importancia de la distribución interfacial de
potencial que debe considerarse a la hora de analizar los voltagramas de este tipo de sistemas,

6. INTERFAZ ELECTRODO‐SOLUCIÓN
180
excepto en aquellos casos en los que se puede asumir en forma razonable que la totalidad de
la caída de potencial ocurre entre el metal y el PTE.
6.2 EFECTO DE LA DILUCIÓN SUPERFICIAL DEL COMPLEJO DE OSMIO SOBRE LA TRANSFERENCIA
ELECTRÓNICA
Con el objetivo de analizar la magnitud de las interacciones entre los sitios electroactivos sobre
la superficie del electrodo y cómo éstas afectan a la cinética de transferencia electrónica, se
prepararon muestras con diferentes cubrimientos superficiales del complejo de osmio. Para
ello, se prepararon monocapas mixtas de tioles terminados en –CH3 y –COOH,
respectivamente, variando la cantidad relativa de cada uno de ellos en la solución etanólica
empleada en su preparación. Luego se realizó la post‐funcionalización con el complejo de
osmio y en cada caso se analizó por voltametría cíclica. Además, se determinaron las
constantes cinéticas de transferencia electrónica por voltametría cíclica y por espectroscopía
de impedancia electroquímica, siguiendo los mismos procedimientos que se detallaron en el
Capítulo 5.
La mezcla de los dos tioles permite la formación de monocapas mixtas permitiendo el control
del cubrimiento del complejo de osmio.
6.2.1 PREPARACIÓN DE MONOCAPAS AUTOENSAMBLADAS MIXTAS Y CARACTERIZACIÓN POR DESORCIÓN
REDUCTIVA
La preparación de monocapas autoensambladas compuestas por dos tioles diferentes se
puede llevar a cabo sumergiendo el sustrato que se quiere derivatizar en una solución que
contiene ambos tioles en la proporción deseada. En general, se emplean soluciones de
concentración total 1 mM.
Sin embargo, se debe considerar que, en general, la composición de la monocapa resultante es
proporcional, pero diferente, a la composición de la solución empleada en la preparación [23],
ya que puede existir cierta segregación entre los tioles de diferente naturaleza [440]. Es por
ello que es necesario determinar cuáles son los cubrimientos superficiales alcanzados por cada
tiol para las diferentes concentraciones relativas en la solución original. Una manera de
hacerlo es por desorción reductiva de las monocapas mixtas obtenidas en cada caso,
estimando los cubrimientos superficiales de cada componente a partir del área del pico de
desorción correspondiente a cada uno.
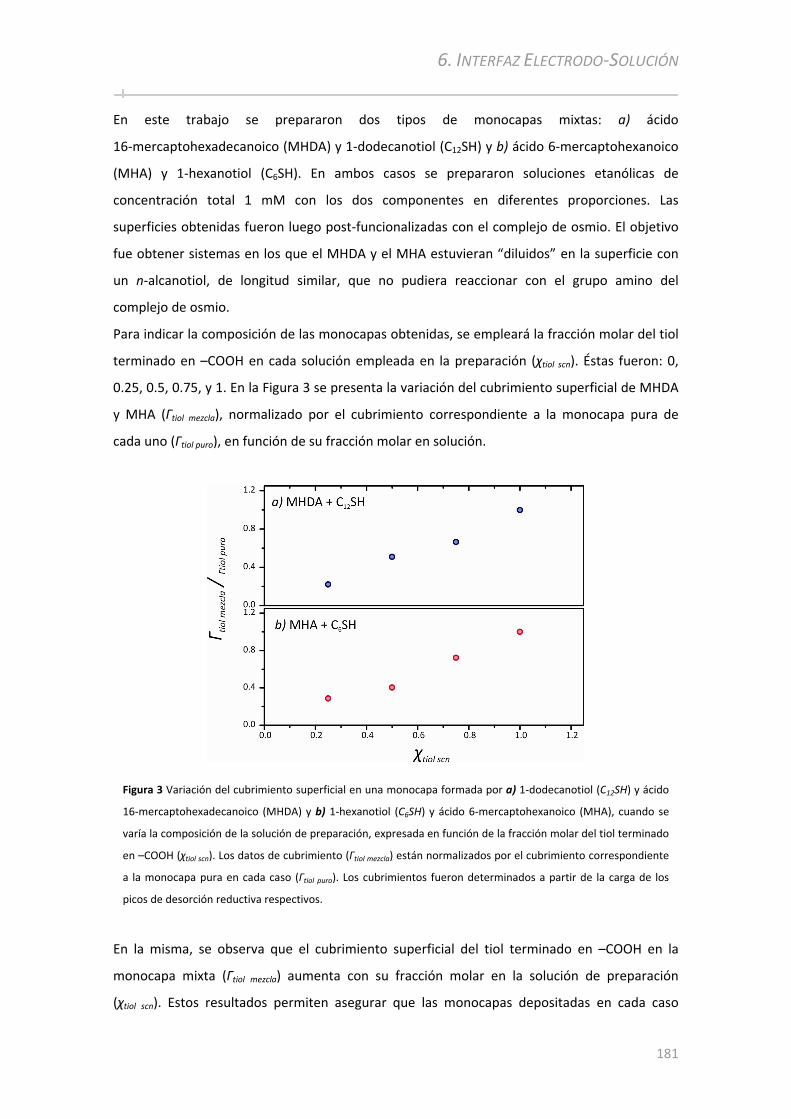
6. INTERFAZ ELECTRODO‐SOLUCIÓN
181
En este trabajo se prepararon dos tipos de monocapas mixtas: a) ácido
16‐mercaptohexadecanoico (MHDA) y 1‐dodecanotiol (C12SH) y b) ácido 6‐mercaptohexanoico
(MHA) y 1‐hexanotiol (C6SH). En ambos casos se prepararon soluciones etanólicas de
concentración total 1 mM con los dos componentes en diferentes proporciones. Las
superficies obtenidas fueron luego post‐funcionalizadas con el complejo de osmio. El objetivo
fue obtener sistemas en los que el MHDA y el MHA estuvieran “diluidos” en la superficie con
un n‐alcanotiol, de longitud similar, que no pudiera reaccionar con el grupo amino del
complejo de osmio.
Para indicar la composición de las monocapas obtenidas, se empleará la fracción molar del tiol
terminado en –COOH en cada solución empleada en la preparación (‐tiol scn). Éstas fueron: 0,
0.25, 0.5, 0.75, y 1. En la Figura 3 se presenta la variación del cubrimiento superficial de MHDA
y MHA (よtiol mezcla), normalizado por el cubrimiento correspondiente a la monocapa pura de
cada uno (よtiol puro), en función de su fracción molar en solución.
En la misma, se observa que el cubrimiento superficial del tiol terminado en –COOH en la
monocapa mixta (よtiol mezcla) aumenta con su fracción molar en la solución de preparación
(‐tiol scn). Estos resultados permiten asegurar que las monocapas depositadas en cada caso
Figura 3 Variación del cubrimiento superficial en una monocapa formada por a) 1‐dodecanotiol (C12SH) y ácido
16‐mercaptohexadecanoico (MHDA) y b) 1‐hexanotiol (C6SH) y ácido 6‐mercaptohexanoico (MHA), cuando se
varía la composición de la solución de preparación, expresada en función de la fracción molar del tiol terminado
en –COOH (‐tiol scn). Los datos de cubrimiento (よtiol mezcla) están normalizados por el cubrimiento correspondiente
a la monocapa pura en cada caso (よtiol puro). Los cubrimientos fueron determinados a partir de la carga de los
picos de desorción reductiva respectivos.
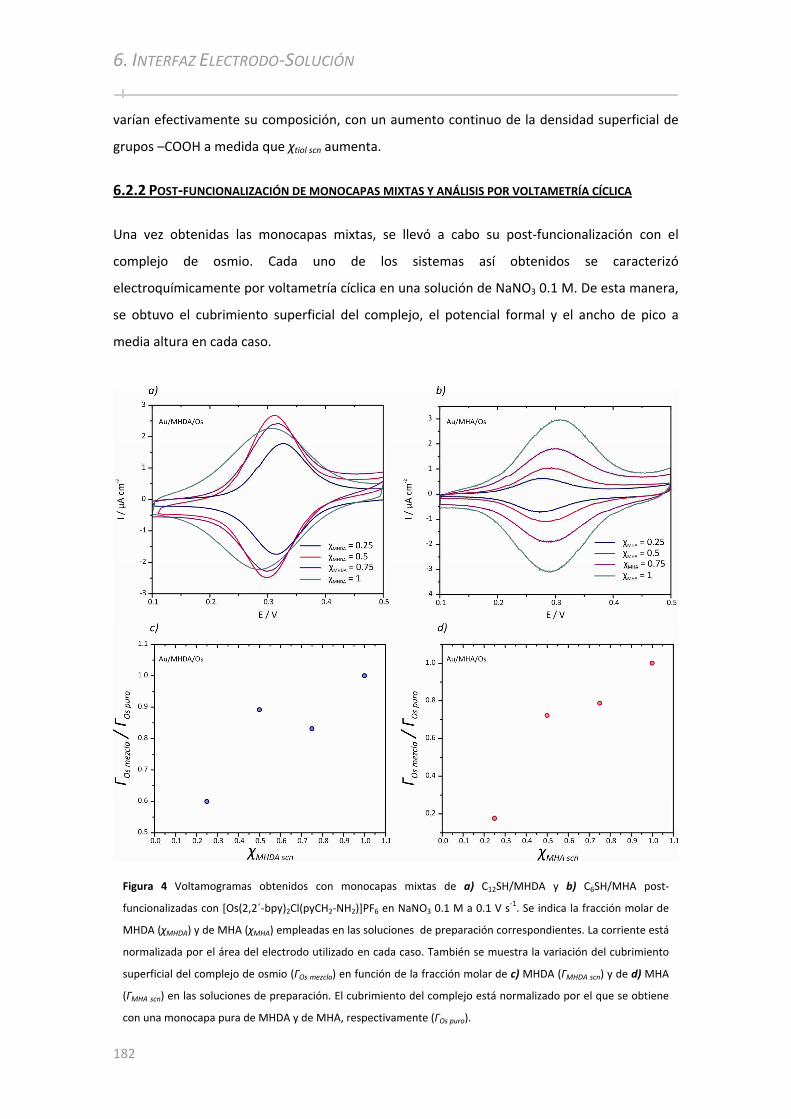
6. INTERFAZ ELECTRODO‐SOLUCIÓN
182
varían efectivamente su composición, con un aumento continuo de la densidad superficial de
grupos –COOH a medida que ‐tiol scn aumenta.
6.2.2 POST‐FUNCIONALIZACIÓN DE MONOCAPAS MIXTAS Y ANÁLISIS POR VOLTAMETRÍA CÍCLICA
Una vez obtenidas las monocapas mixtas, se llevó a cabo su post‐funcionalización con el
complejo de osmio. Cada uno de los sistemas así obtenidos se caracterizó
electroquímicamente por voltametría cíclica en una solución de NaNO3 0.1 M. De esta manera,
se obtuvo el cubrimiento superficial del complejo, el potencial formal y el ancho de pico a
media altura en cada caso.
Figura 4 Voltamogramas obtenidos con monocapas mixtas de a) C12SH/MHDA y b) C6SH/MHA post‐
funcionalizadas con [Os(2,2´‐bpy)2Cl(pyCH2‐NH2)]PF6 en NaNO3 0.1 M a 0.1 V s‐1. Se indica la fracción molar de
MHDA (‐MHDA) y de MHA (‐MHA) empleadas en las soluciones de preparación correspondientes. La corriente está
normalizada por el área del electrodo utilizado en cada caso. También se muestra la variación del cubrimiento
superficial del complejo de osmio (よOs mezcla) en función de la fracción molar de c) MHDA (よMHDA scn) y de d) MHA
(よMHA scn) en las soluciones de preparación. El cubrimiento del complejo está normalizado por el que se obtiene
con una monocapa pura de MHDA y de MHA, respectivamente (よOs puro).
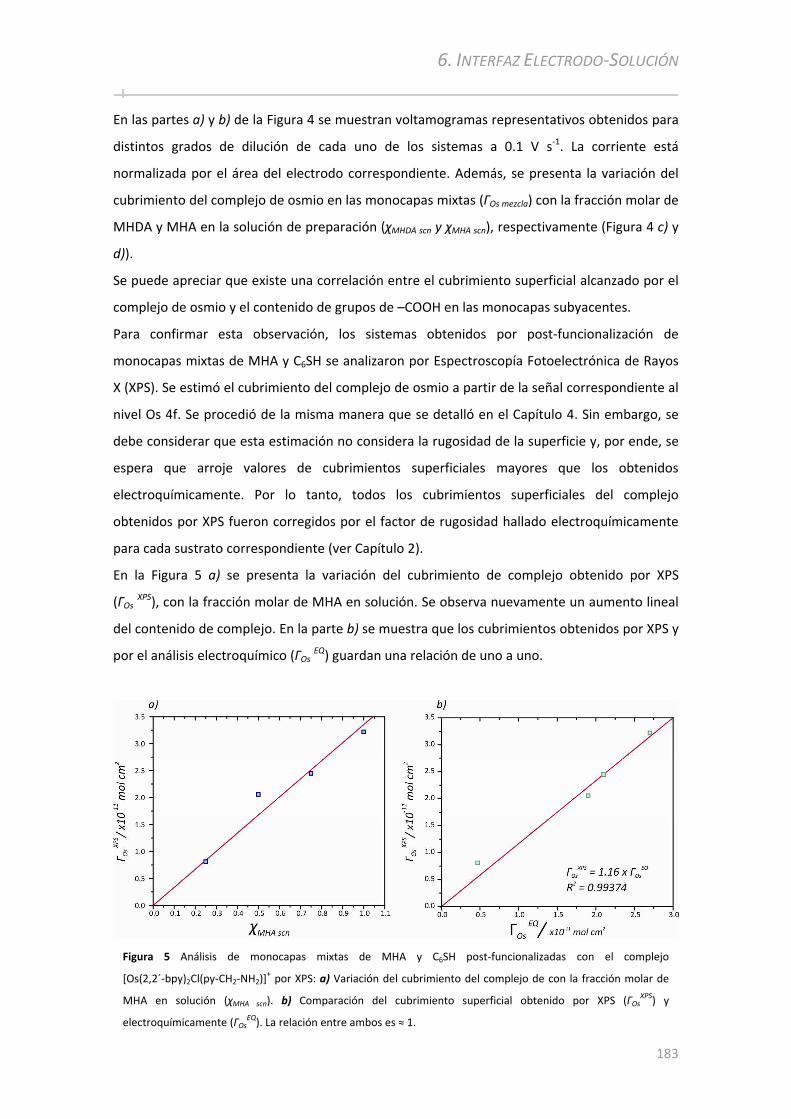
6. INTERFAZ ELECTRODO‐SOLUCIÓN
183
En las partes a) y b) de la Figura 4 se muestran voltamogramas representativos obtenidos para
distintos grados de dilución de cada uno de los sistemas a 0.1 V s‐1. La corriente está
normalizada por el área del electrodo correspondiente. Además, se presenta la variación del
cubrimiento del complejo de osmio en las monocapas mixtas (よOs mezcla) con la fracción molar de
MHDA y MHA en la solución de preparación (‐MHDA scn y ‐MHA scn), respectivamente (Figura 4 c) y
d)).
Se puede apreciar que existe una correlación entre el cubrimiento superficial alcanzado por el
complejo de osmio y el contenido de grupos de –COOH en las monocapas subyacentes.
Para confirmar esta observación, los sistemas obtenidos por post‐funcionalización de
monocapas mixtas de MHA y C6SH se analizaron por Espectroscopía Fotoelectrónica de Rayos
X (XPS). Se estimó el cubrimiento del complejo de osmio a partir de la señal correspondiente al
nivel Os 4f. Se procedió de la misma manera que se detalló en el Capítulo 4. Sin embargo, se
debe considerar que esta estimación no considera la rugosidad de la superficie y, por ende, se
espera que arroje valores de cubrimientos superficiales mayores que los obtenidos
electroquímicamente. Por lo tanto, todos los cubrimientos superficiales del complejo
obtenidos por XPS fueron corregidos por el factor de rugosidad hallado electroquímicamente
para cada sustrato correspondiente (ver Capítulo 2).
En la Figura 5 a) se presenta la variación del cubrimiento de complejo obtenido por XPS
(よOs XPS), con la fracción molar de MHA en solución. Se observa nuevamente un aumento lineal
del contenido de complejo. En la parte b) se muestra que los cubrimientos obtenidos por XPS y
por el análisis electroquímico (よOs EQ) guardan una relación de uno a uno.
Figura 5 Análisis de monocapas mixtas de MHA y C6SH post‐funcionalizadas con el complejo
[Os(2,2´‐bpy)2Cl(py‐CH2‐NH2)]+ por XPS: a) Variación del cubrimiento del complejo de con la fracción molar de
MHA en solución (‐MHA scn). b) Comparación del cubrimiento superficial obtenido por XPS (よOsXPS) y
electroquímicamente (よOsEQ). La relación entre ambos es ≈ 1.

6. INTERFAZ ELECTRODO‐SOLUCIÓN
184
Por lo tanto, a partir de los resultados obtenidos por dos técnicas diferentes, se puede afirmar
que para ambos sistemas se lograron distintos grados de dilución superficial de los centros
electroactivos de osmio.
6.2.2.1 VARIACIÓN DEL ANCHO DE PICO A MEDIA ALTURA CON EL CUBRIMIENTO SUPERFICIAL DEL
COMPLEJO DE OSMIO
La Figura 6 presenta la variación del ancho de pico a media altura en función del cubrimiento
superficial del complejo de osmio para ambos sistemas analizados. En ambos casos, el mismo
aumenta con la concentración superficial del complejo.
Para interpretar este hecho, en primer lugar, se debe tener en cuenta que el ancho de pico a
media altura (FWHM) es una medida de las interacciones intermoleculares entre los sitios
electroactivos. Cuando el sistema se comporta idealmente, el FWHM es de 0.09 V/n (siendo n
el número de electrones asociados a la reacción de electrodo en cuestión) [54]. En el caso de la
óxido‐reducción del complejo de osmio, n = 1 y el FWHM ideal esperado es de 0.09 V. Cuando
se barre el potencial del electrodo en sentido positivo, los centros reducidos (R) se van
oxidando (a O) y, a medida que lo hacen, el entorno de aquellos sitios que aún no se han
oxidado va cambiando, modificándose entonces el potencial formal de los mismos.
Cuando las especies reducida y oxidada tienen carga neta del mismo signo, entonces las
interacciones entre R y O son repulsivas, y a medida que aumenta la proporción de O en la
Figura 6 Variación del ancho de pico a media altura (FWHM) de la cupla Os(II)+/Os(III)+2 en función de la fracción
molar (‐ tiol scn) de MHDA (cuadrados azules) o de MHA (círculos rojos) en la solución de preparación.

6. INTERFAZ ELECTRODO‐SOLUCIÓN
185
superficie, las repulsiones intermoleculares también lo hacen, aumentando el potencial formal
de oxidación de los centros R que aún no se han oxidado.
De esta manera, se genera una distribución de potenciales formales en la superficie que se ve
traducida en un aumento neto del potencial formal y en el ensanchamiento de los picos
observados en una voltametría cíclica. Cualitativamente, se puede decir que la oxidación se
verá desfavorecida cuando el cubrimiento superficial de la especie redox sea mayor, ya que en
ese caso, las repulsiones electrostáticas entre los sitios serán más fuertes.
Otra explicación para la observación de ensanchamientos de pico cuando se analizan
monocapas electroactivas, puede residir en la existencia de efectos de doble capa que describe
el modelo de Smith‐White [433]. Cuando las SAMs son suficientemente gruesas y tienen una
constante dieléctrica suficientemente baja, todos los cambios de potencial aplicado sobre el
electrodo resultan en cambios de potencial dentro del cuerpo de la monocapa, con mínimos
cambios en el plano de transferencia electrónica. Sin embargo, cuando se tienen altos
cubrimientos de los centros redox y uno de los estados de oxidación de los mismos no tiene
carga neta, los efectos de doble capa se hacen particularmente importantes. Éste no es el caso
de los sistemas estudiados aquí.
En el complejo de osmio, R y O corresponden a Os(II)+ y Os(III)+2, es decir que ambas especies
tienen carga neta del mismo signo. Por lo tanto, es de esperar que las interacciones entre ellas
sean repulsivas y que éstas aumenten con el cubrimiento superficial del complejo,
observándose un aumento del FWHM en consecuencia [360].
Por lo tanto, la variación del FWHM indica el aumento de las repulsiones electrostáticas entre
los sitios electroactivos en la superficie. De hecho, para los sistemas en los que el complejo
está más diluido, su valor es de ≈ 0.09 V, tanto para MHDA como para MHA. Esto corresponde
al comportamiento ideal, mientras que para los sistemas con mayor concentración de osmio,
el FWHM es ≈ 0.15 V, señalando una fuerte desviación de la idealidad.
Por las mismas razones que explican el aumento en el ancho de pico con el cubrimiento de
complejo, se esperaría un aumento en el potencial formal. Sin embargo, la variación del mismo
no es importante, como puede observarse en los voltagramas de la Figura 4. Una posible
explicación para este hecho puede ser el apantallamiento de cargas por parte de los iones del
electrolito soporte que se halla en concentración 0.1 M. Otra razón para esta observación
puede encontrarse analizando la eficiencia de la reacción de post‐funcionalización que se
alcanza en cada sistema. Ésta fue definida en el Capítulo 4 y, en la Figura 7, se presenta la
variación de la misma en función de la fracción molar de MHDA o de MHA en la solución de
preparación.

6. INTERFAZ ELECTRODO‐SOLUCIÓN
186
Se puede apreciar que a medida que aumenta el contenido del tiol terminado en –COOH sobre
la superficie, disminuye la eficiencia de la reacción mencionada. Es decir que aumenta,
comparativamente, el número de sitios ácidos que no reaccionan con el complejo. La razón
más probable para este hecho es el impedimento estérico entre las moléculas del complejo de
osmio. Los grupos ácidos no sólo son grupos polares sino que además, al pH del agua, pueden
presentar cierto grado de deprotonación [23]. Por lo tanto, se debe considerar que pueden
ejercer cierto apantallamiento de las cargas sobre la superficie, contrarrestando las
repulsiones electrostáticas entre los centros redox y resultando en la no dependencia de E°´
con el cubrimiento superficial del complejo observada experimentalmente.
La diferencia en la dependencia de E°´ y de FWHM con el cubrimiento superficial de sitios
electroactivos es coherente con el hecho de que el potencial formal de un par redox está
relacionado con las interacciones intermoleculares de las éste participa, mientras que el ancho
de pico en una voltametría cíclica se relaciona con la variación de dichas interacciones con el
potencial aplicado.
El efecto de los grupos –COOH sobre el potencial formal en función del pH se analiza con
mayor detalle en la próxima sección.
En la literatura existen varios ejemplos del análisis de la variación del E°´ y el FWHM con la
dilución superficial de una especie electroactiva de interés. En general, se analizan monocapas
de tioles terminados en ferroceno/ferrocianuro (Fc/Fc+) diluidos con tioles terminados en
metilo [24, 51, 104]. En estos trabajos se observa el aumento de ambas propiedades con el
Figura 7 Variación de la eficiencia de la reacción de post‐funcionalización para cada sistema en función de la
composición de la solución de preparación (‐ tiol scn). Los cuadrados azules corresponden al sistema formado por
MHDA diluido con C12SH y, los círculos rojos, al formado por MHA diluido con C6SH.

6. INTERFAZ ELECTRODO‐SOLUCIÓN
187
cubrimiento superficial de los sitios redox, siguiendo la tendencia esperada para un aumento
de las interacciones intermoleculares sobre la superficie.
Asimismo, Acevedo y Abruña, observan la misma tendencia cuando aumentan el cubrimiento
superficial del complejo [Os(2,2´‐bpy)2Cl(py‐(CH2)2py] sobre un electrodo de platino [360]. Sin
embargo, estos ejemplos se diferencian del sistema analizado en este trabajo en que la
dilución de los centros electroactivos se realiza con grupos no polares o por control de los
tiempos de inmersión del sustrato en la solución de preparación.
Finalmente, existen dos ejemplos en bibliografía de sistemas en los que se realiza una dilución
superficial de los sitios electroactivos con grupos polares. En el trabajo de Eggers et al. se co‐
adsorben tioles terminados en Fc/Fc+ con tioles terminados en –CH3 y en –OH. La cantidad de
ferroceno se mantiene constante mientras que se varían las proporciones relativas de los
grupos diluyentes [441]. El otro ejemplo lo constituye un trabajo del mismo grupo en el que se
modifican sustratos de carbono vítreo y de oro con mezclas de ácido benzoico y benceno por
reducción de las sales de diazonio derivadas de ácido 4‐aminobenzoico y anilina,
respectivamente. Las capas orgánicas así obtenidas se post‐funcionalizan con ferroceno [191].
En ambos trabajos, se observa que a medida que la proporción de grupos polares sobre la
superficie aumenta, también lo hace el potencial formal. Los autores adjudican este
comportamiento a efectos de distribución del potencial interfacial. Sin embargo, en los
mismos el par redox es Fc/Fc+, los grupos polares son neutros y los efectos electrostáticos son
mayores que con el par Os(II)+/Os(III)2+, en el que ambos estados de oxidación tienen carga
neta y los grupos polares pueden tener carga (–COOH y –COO–).
Por lo tanto, en los sistemas aquí analizados, el comportamiento observado para el potencial
formal y el ancho de pico se atribuye a efectos de interacciones laterales entre los sitios
electroactivos teniendo en cuenta el apantallamiento de dichas interacciones por parte de los
grupos –COOH y –COO–.
6.2.2.2 VARIACIÓN DE LA CONSTANTE CINÉTICA ESTÁNDAR CON EL CUBRIMIENTO SUPERFICIAL DEL
COMPLEJO DE OS
Las constantes cinéticas de transferencia electrónica se determinaron por voltametría cíclica y
por espectroscopía de impedancia electroquímica. La primera técnica se empleó con los
sistemas obtenidos a partir de monocapas mixtas de MHDA y C12SH, mientras que los
derivados de mezclas de MHA y C6SH fueron analizados con la segunda. En el Capítulo 5 se
demostró que el sistema Au/MHDA/Os tiene una cinética de TE que es lo suficientemente
lenta como para poder ser medida a partir de la separación de picos en función de la velocidad

6. INTERFAZ ELECTRODO‐SOLUCIÓN
188
de barrido (por voltametría cíclica). En cambio, se observó que la cinética del sistema
Au/MHA/Os es demasiado rápida y por ello fue evaluada por impedancia electroquímica.
Las Figuras 8 a) y b) muestran los resultados obtenidos en cada caso: las curvas de potencial de
pico en función de la velocidad de barrido y las curvas de |Ypico / Ybkg| en función de la
frecuencia para los sistemas derivados de MHDA y MHA, respectivamente. Asimismo, se
presentan los valores de k0 obtenidas con cada uno de ellos para las diferentes composiciones
de las soluciones de preparación de las monocapas mixtas (‐MHDA y ‐MHA, respectivamente).
En ambos casos se observa que la constante cinética estándar de transferencia electrónica
disminuye a medida que la fracción molar del tiol terminado en –COOH es mayor. En las
secciones anteriores se observó que el cubrimiento superficial de complejo de osmio es
Figura 8 a) Curvas de variación del potencial de pico con la velocidad de barrido, obtenidas por voltametría
cíclica, para distintas monocapas mixtas de MHDA y C12SH post‐funcionalizadas con el complejo de osmio.
b) Curvas de ¨ en función de la frecuencia, obtenidas por impedancia electroquímica, para distintas monocapas
mixtas de MHA y C6SH post‐funcionalizadas con el complejo de osmio. En ambos casos se indican las diferentes
fracciones molares de MHDA y de MHA en las correspondientes soluciones de preparación. A su vez, se muestra
la variación de k0 con el cubrimiento superficial del complejo de osmio para cada sistema:
c) Au/MHDA + C12SH/Os y d) Au/MHA + C6SH/Os.

6. INTERFAZ ELECTRODO‐SOLUCIÓN
189
directamente proporcional a dicha fracción molar. Por ende k0 disminuye a medida que el
cubrimiento superficial del complejo aumenta.
Según se observa en las Figuras 8 c) y d), en ambos sistemas, k0 aumenta en un orden de
magnitud cuando se compara la muestra con mayor cubrimiento superficial del complejo con
la de menor cubrimiento. En el sistema Au/MHDA/Os, k0 varía entre 120 s‐1 y 10 s‐1, mientras
que en el sistema Au/MHA/Os, lo hace entre 1.1 x 105 s‐1 y 2.5 x 104 s‐1.
Para interpretar este resultado es necesario considerar la energía de reorganización (゜)
asociada a la transferencia electrónica en cada caso [368, 369]. Ésta viene dada por:
a) los modos vibracionales de la molécula electroactiva: energía de reorganización interna (゜i),
b) el solvente que debe adoptar la configuración adecuada para estabilizar los productos de la
reacción de transferencia electrónica: energía de reorganización externa (゜o).
Cuando un centro electroactivo se encuentra en solución y participa de una reacción de
transferencia de carga de esfera externa (oxidándose o reduciéndose), ゜ está determinada
principalmente por la energía de reorganización externa. Sin embargo, si el centro
electroactivo se encuentra confinado a una superficie, como en este caso, no sólo el solvente
en contacto con el mismo, sino también todas las moléculas unidas a la superficie deberán
sufrir una re‐polarización de sus dipolos para acompañar una posible reacción de transferencia
electrónica. Esto contribuye a la energía de reorganización del sistema y repercute sobre la
magnitud de la constante cinética de transferencia electrónica.
Esta situación se puede comparar de alguna manera con lo que ocurre cuando se analiza la
cinética de transferencia electrónica en diferentes solventes. Las propiedades dinámicas del
solvente pueden jugar un papel importante en cuanto a la velocidad en la que se atraviesa la
barrera de activación en una reacción de transferencia electrónica y, por ende, en la magnitud
de la constante de velocidad asociada a la misma. La activación térmica del reactivo a lo largo
de la coordenada de reacción proviene de transferencias de energía entre el mismo y su
entorno. Uno de los mecanismos principales por el que esto ocurre es a través de las
interacciones dipolo‐dipolo entre el reactivo y el solvente. El tiempo característico para las
mismas está relacionado con el tiempo de relajación longitudinal del solvente, 〃L. Para
reacciones adiabáticas, que involucran la reorganización del solvente en mayor medida que las
vibraciones internas del reactivo, la velocidad de pasaje del mismo a través del estado de
transición está determinada principalmente por este parámetro dinámico del solvente [442].
Es decir, en estos casos, cuánto mayor sea 〃L, menor será la velocidad de transferencia
electrónica, debido a la mayor dificultad del solvente para adoptar la configuración adecuada
para dicha reacción.

6. INTERFAZ ELECTRODO‐SOLUCIÓN
190
Forster y Faulkner observaron que la k0 del par Os(II)/Os(III) depende de la dinámica del
solvente utilizado [149]. Estos autores analizaron monocapas de [Os(2,2´‐bpy)2Clpy(CH2)2 py]+
en acetonitrilo y en cloroformo cuyos tiempos de relajación longitudinal son 0.2 ps y 2.4 ps,
respectivamente. Las constantes cinéticas obtenidas en cada uno fueron k0 = 1.1 x 105 s‐1 y
k0 = 7.4 x 103 s‐1, confirmando que k0 aumenta cuando el tiempo de relajación del solvente
disminuye, es decir, cuando el solvente se re‐orienta más rápidamente para acompañar el
proceso de transferencia electrónica. Los tiempos de relajación longitudinal dependen de la
constante dieléctrica y aumentan cuánto mayor es la misma debido a las mayores
interacciones que existen entre las moléculas de solvente.
Al comienzo de esta sección se mostró que cuánto mayor es el cubrimiento superficial de
osmio, mayor es la cantidad relativa de grupos –COOH que no han reaccionado en la post‐
funcionalización. De esta manera, el entorno de los sitios redox se vuelve cada vez más polar y,
por ende la energía de reorganización aumenta, provocando una disminución en la cinética de
transferencia electrónica.
6.3 EFECTO DEL PH Y LA FUERZA IÓNICA SOBRE LA TRANSFERENCIA ELECTRÓNICA
Como se ha destacado a lo largo de este capítulo, los sistemas analizados presentan una
proporción de grupos –COOH libres, es decir, aquellos que no reaccionaron durante la post‐
funcionalización con el complejo de osmio. Por lo tanto, se debe considerar su equilibrio ácido‐
base que, a diferentes pH, puede modificar la densidad de carga superficial y afectar el proceso
de transferencia de carga entre los centros redox y la superficie. En esta sección se analiza
dicho efecto y cómo el mismo varía cuando se modifica la fuerza iónica de la solución en
contacto con la superficie. En particular, se analizó el comportamiento electroquímico de los
sistemas Au/MUA/Os, Au/SPhCOOH/Os y Au/PhCOOH/Os en función del pH y de la fuerza
iónica de la solución en contacto con los mismos.
Los sistemas estudiados en este trabajo tienen grupos funcionales con carga neta: los sitios
Os(II)+ y Os(III)2+ y los grupos –COO‐ libres. Estos últimos variarán su estado de carga con el pH
y por lo tanto, no se puede analizar la influencia de la fuerza iónica sobre el comportamiento
electroquímico sin tener en cuenta el pH en el que esto se lleva a cabo. Es por ello que se
realizaron cuatro experimentos:
1) análisis de una monocapa de Au/MUA por PM‐IRRAS luego de ser sumergida en soluciones
de pH = 3 y pH = 11, para confirmar espectroscópicamente que, a cada pH, el estado de
ionización de los grupos –COOH sobre la superficie cambia.

6. INTERFAZ ELECTRODO‐SOLUCIÓN
191
2) medida de la variación del potencial formal con el pH a alta y baja fuerza iónica constante
(0.1 M y 4 mM, respectivamente).
3) medida de la variación del potencial formal con la fuerza iónica a pH ácido y básico (3 y 11,
respectivamente).
4) Medida de la constante cinética estándar de transferencia electrónica (k0) a pH 3 y 11 con
fuerza iónica 1 M.
Los experimentos 2) y 3) se realizaron con los sistemas Au/MUA/Os, Au/SPhCOOH/Os y
Au/PhCOOH/Os y el experimento 4) se llevó a cabo con todos los sistemas estudiados en esta
tesis. Los resultados se presentan a continuación.
6.3.1 ANÁLISIS DE AU/MUA A DIFERENTES PH POR PM‐IRRAS
El pKa de un ácido unido a una superficie se define como el pH de la solución en contacto con la
misma, en el que la mitad de los grupos –COOH se hallan ionizados [443]. En particular, el valor
de pKa reportado para monocapas autoensambladas de MUA es de ≈ 6 [23, 444, 445].
Para confirmar que, efectivamente, el estado de ionización del MUA es diferente a los pH en
los que se llevó a cabo la posterior caracterización electroquímica, se realizó un análisis por
Espectroscopía Infrarroja de Absorción‐Reflexión con Modulación de la Polarización (PM‐
IRRAS).
Se tomó un espectro inicial en aire de una superficie de oro modificada con una monocapa
autoensamblada de MUA. Luego, dicha superficie se sumergió en una solución de NaOH 1 mM
y se volvió a tomar un espectro. Finalmente, la misma muestra fue sumergida en una solución
de HNO3 1 mM y se volvió a analizar por PM‐IRRAS. Los espectros obtenidos con ゜o = 1450 cm‐1
se presentan en la Figura 9.
De acuerdo a la asignación de señales realizada en el Capítulo 3, se puede observar que la
inmersión de la muestra en NaOH 1 mM hace desaparecer las señales correspondientes al
grupo –COOH: `a C=O (≈ 1726 cm‐1) y ~OH (≈ 1410 cm‐1), respectivamente. A su vez, existe un leve
aumento de la señal correspondiente al modo `a COO‐ (≈ 1540 cm‐1) y aparece la señal
correspondiente al modo de estiramiento simétrico del carboxilato, `s COO‐ (≈ 1490 cm‐1). Esto
indica que la monocapa presenta un grado de protonación menor al que presentaba
inicialmente. Luego de la inmersión en HNO3 1 mM, la señal a 1726 cm‐1 se recupera
parcialmente, así como la correspondiente al modo ~OH. Este comportamiento indica que
efectivamente el estado de ionización de la monocapa de MUA varía con el pH.
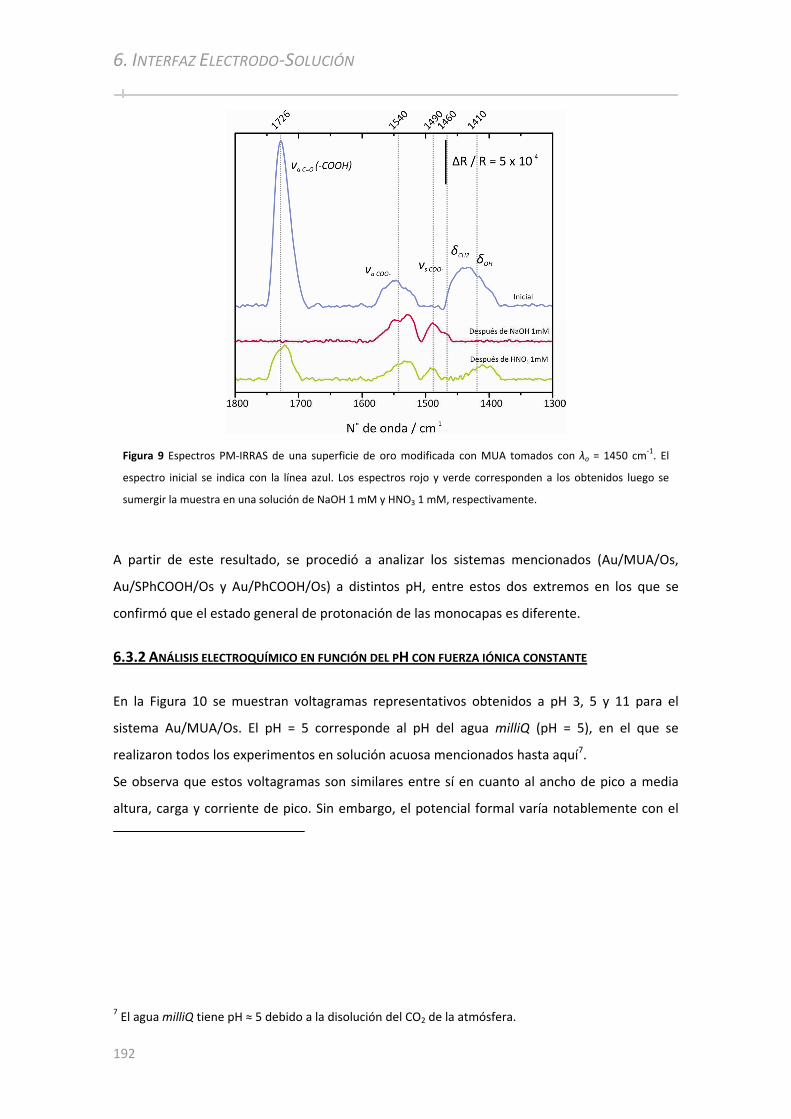
6. INTERFAZ ELECTRODO‐SOLUCIÓN
192
A partir de este resultado, se procedió a analizar los sistemas mencionados (Au/MUA/Os,
Au/SPhCOOH/Os y Au/PhCOOH/Os) a distintos pH, entre estos dos extremos en los que se
confirmó que el estado general de protonación de las monocapas es diferente.
6.3.2 ANÁLISIS ELECTROQUÍMICO EN FUNCIÓN DEL PH CON FUERZA IÓNICA CONSTANTE
En la Figura 10 se muestran voltagramas representativos obtenidos a pH 3, 5 y 11 para el
sistema Au/MUA/Os. El pH = 5 corresponde al pH del agua milliQ (pH = 5), en el que se
realizaron todos los experimentos en solución acuosa mencionados hasta aquí7.
Se observa que estos voltagramas son similares entre sí en cuanto al ancho de pico a media
altura, carga y corriente de pico. Sin embargo, el potencial formal varía notablemente con el
7 El agua milliQ tiene pH ≈ 5 debido a la disolución del CO2 de la atmósfera.
Figura 9 Espectros PM‐IRRAS de una superficie de oro modificada con MUA tomados con ゜o = 1450 cm‐1. El
espectro inicial se indica con la línea azul. Los espectros rojo y verde corresponden a los obtenidos luego se
sumergir la muestra en una solución de NaOH 1 mM y HNO3 1 mM, respectivamente.
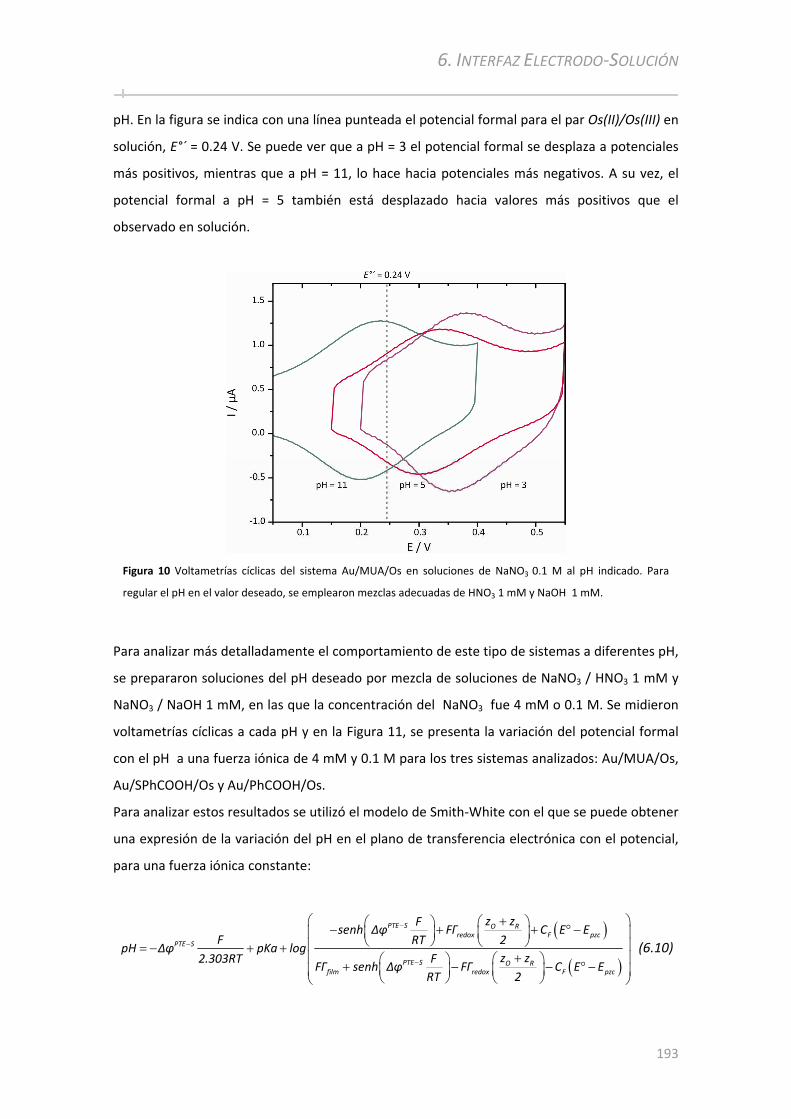
6. INTERFAZ ELECTRODO‐SOLUCIÓN
193
pH. En la figura se indica con una línea punteada el potencial formal para el par Os(II)/Os(III) en
solución, E°´ = 0.24 V. Se puede ver que a pH = 3 el potencial formal se desplaza a potenciales
más positivos, mientras que a pH = 11, lo hace hacia potenciales más negativos. A su vez, el
potencial formal a pH = 5 también está desplazado hacia valores más positivos que el
observado en solución.
Para analizar más detalladamente el comportamiento de este tipo de sistemas a diferentes pH,
se prepararon soluciones del pH deseado por mezcla de soluciones de NaNO3 / HNO3 1 mM y
NaNO3 / NaOH 1 mM, en las que la concentración del NaNO3 fue 4 mM o 0.1 M. Se midieron
voltametrías cíclicas a cada pH y en la Figura 11, se presenta la variación del potencial formal
con el pH a una fuerza iónica de 4 mM y 0.1 M para los tres sistemas analizados: Au/MUA/Os,
Au/SPhCOOH/Os y Au/PhCOOH/Os.
Para analizar estos resultados se utilizó el modelo de Smith‐White con el que se puede obtener
una expresión de la variación del pH en el plano de transferencia electrónica con el potencial,
para una fuerza iónica constante:
( )( )
PTE S O Rredox F pzc
PTE S
PTE S O Rfilm redox F pzc
z zFsenh らφ Fよ C E E
F RT 2pH らφ pKa log
z zF2.303RTFよ senh らφ Fよ C E E
RT 2
−−
−
⎛ + ⎞⎛ ⎞⎛ ⎞− + + °−⎜ ⎟⎜ ⎟ ⎜ ⎟⎝ ⎠ ⎝ ⎠⎜ ⎟= − + + +⎜ ⎟⎛ ⎞⎛ ⎞+ − − °−⎜ ⎟ ⎜ ⎟⎜ ⎟⎝ ⎠ ⎝ ⎠⎝ ⎠ (6.10)
Figura 10 Voltametrías cíclicas del sistema Au/MUA/Os en soluciones de NaNO3 0.1 M al pH indicado. Para
regular el pH en el valor deseado, se emplearon mezclas adecuadas de HNO3 1 mM y NaOH 1 mM.
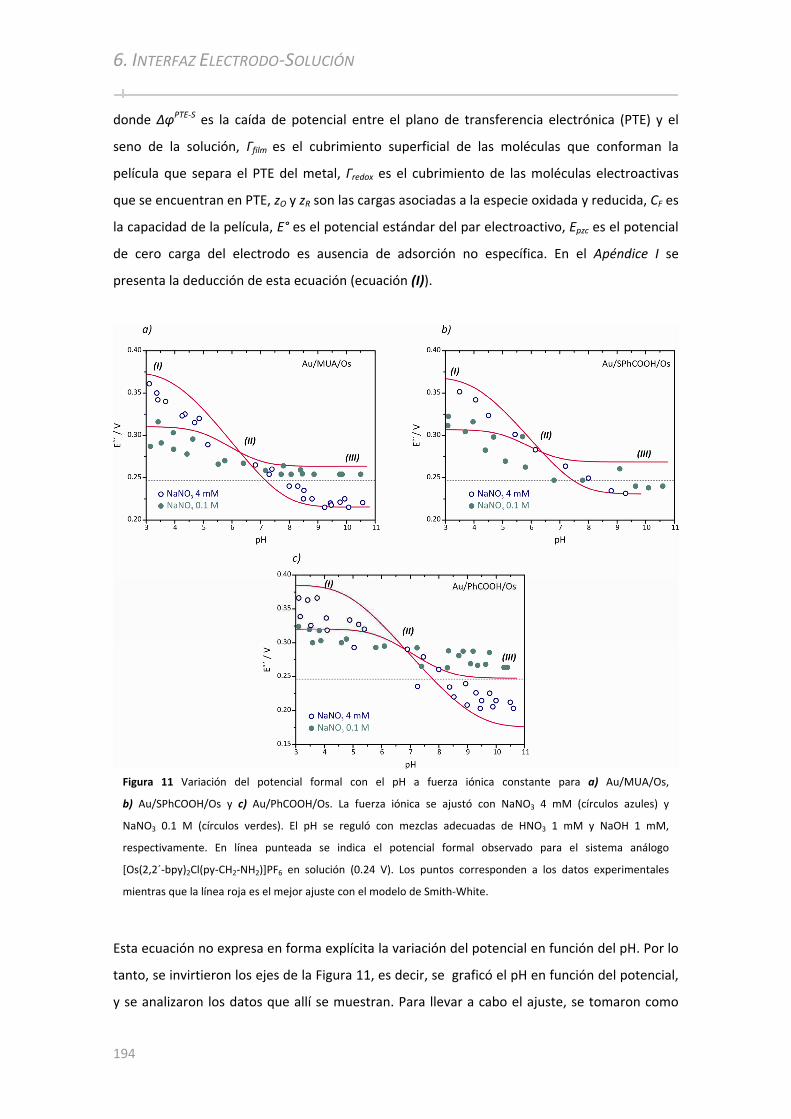
6. INTERFAZ ELECTRODO‐SOLUCIÓN
194
donde らφPTE‐S es la caída de potencial entre el plano de transferencia electrónica (PTE) y el
seno de la solución, よfilm es el cubrimiento superficial de las moléculas que conforman la
película que separa el PTE del metal, よredox es el cubrimiento de las moléculas electroactivas
que se encuentran en PTE, zO y zR son las cargas asociadas a la especie oxidada y reducida, CF es
la capacidad de la película, E° es el potencial estándar del par electroactivo, Epzc es el potencial
de cero carga del electrodo es ausencia de adsorción no específica. En el Apéndice I se
presenta la deducción de esta ecuación (ecuación (I)).
Esta ecuación no expresa en forma explícita la variación del potencial en función del pH. Por lo
tanto, se invirtieron los ejes de la Figura 11, es decir, se graficó el pH en función del potencial,
y se analizaron los datos que allí se muestran. Para llevar a cabo el ajuste, se tomaron como
Figura 11 Variación del potencial formal con el pH a fuerza iónica constante para a) Au/MUA/Os,
b) Au/SPhCOOH/Os y c) Au/PhCOOH/Os. La fuerza iónica se ajustó con NaNO3 4 mM (círculos azules) y
NaNO3 0.1 M (círculos verdes). El pH se reguló con mezclas adecuadas de HNO3 1 mM y NaOH 1 mM,
respectivamente. En línea punteada se indica el potencial formal observado para el sistema análogo
[Os(2,2´‐bpy)2Cl(py‐CH2‐NH2)]PF6 en solución (0.24 V). Los puntos corresponden a los datos experimentales
mientras que la línea roja es el mejor ajuste con el modelo de Smith‐White.

6. INTERFAZ ELECTRODO‐SOLUCIÓN
195
parámetros el potencial estándar (E°), los cubrimientos superficiales de los tioles y del ácido
benzoico (ГOs = Гredox y Гtiol = Гfilm) y la constante de equilibrio ácido‐base (pKa). Se superpusieron
curvas teóricas de らφPTE‐S vs. pH, calculadas con la ecuación (6.10), con las curvas obtenidas
experimentalmente y se variaron los parámetros mencionados hasta obtener la mejor
coincidencia entre experimento y teoría. El mejor ajuste se indica en línea roja en la Figura 11 y
en la Tabla 1 se muestran los valores de los parámetros obtenidos.
Tabla 1 Parámetros obtenidos con el ajuste de curvas E°´ vs. pH con el modelo de Smith‐White.
E° / V pKa ГOs / x 10‐10 mol cm‐2 Гtiol / x 10‐10 mol cm‐2
Au/MUA/Os 0.28 6 0.10 0.37
Au/SPhCOOH/Os 0.28 6 0.08 0.30
Au/PhCOOH/Os 0.29 7 0.10 0.26
Para interpretar los resultados de la Figura 11 vale recordar que los grupos –COOH presentes
en la superficie pueden apantallar las cargas superficiales de los centros electroactivos,
especialmente si están deprotonados. Este apantallamiento disminuye las interacciones
electrostáticas entre los sitios redox positivos que son repulsivas y generan el desplazamiento
del potencial formal observado hacia potenciales más negativos. Además, cuando los grupos
ácido están deprotonados (pH = 11) la densidad de carga en el PTE es negativa y, por ende, el
potencial en el plano de transferencia electrónica (φPTE) también, observándose que el
potencial formal se desplaza hacia potenciales más negativos. Cuando están protonados (pH =
3), la densidad de carga en el PTE y φPTE son positivos, ya que ahora son los centros
electroactivos Os(II)+ y Os(III)+2 los que determinan el estado de carga. En este caso se observa
un desplazamiento positivo del E°´.
En la Figura 11 se puede apreciar que, efectivamente, al variar el pH en el intervalo 3‐11, el
potencial formal se desplaza desde potenciales más positivos que el E°´ observado en solución
(≈ 0.24 V) a potenciales cercanos al mismo.
En particular, se puede observar que las curvas obtenidas para los tres sistemas presentan tres
regiones definidas. La región (I) comprende el intervalo de pH < 3, en el que el número de
cargas no varía en la película ya que a este pH los grupos –COOH se hallan prácticamente todos
protonados. En este caso la carga está dada sólo por los centros redox. La segunda región (II)
comprende el intervalo 3 < pH < 8 y en ella se observa la mayor variación del potencial formal
con el pH. Este comportamiento se puede adjudicar a la deprotonación de los grupos ácidos
con el aumento del pH de la solución.

6. INTERFAZ ELECTRODO‐SOLUCIÓN
196
Finalmente, la región (III) corresponde a pH > 8. En ella nuevamente se observa un potencial
formal prácticamente constante y cercano (o menor) al E°´ observado en solución, ya que a
estos pH, los grupos ácido están casi en su totalidad deprotonados.
En todos los casos se observa una mayor variación de E°´ para la menor concentración de
NaNO3 empleada. Esto es consistente con el hecho de que los iones en solución también
contribuyen al apantallamiento de cargas debido la formación de pares iónicos con los centros
redox. A menor concentración del electrolito soporte, menor es esta contribución y el efecto
del pH sobre el potencial formal es más evidente.
En cuanto al ajuste de estos datos con la ecuación teórica que se desprende del modelo de
Smith‐White, se puede decir que los potenciales estándar y las constantes de equilibrio ácido‐
base obtenidos tienen buena concordancia con los valores esperados. A su vez, el cubrimiento
del complejo de osmio obtenido también es consistente con el medido electroquímicamente,
aunque es un poco menor que lo esperado, especialmente en el caso del sistema
Au/PhCOOH/Os. En el Capítulo 4 se encontraron cubrimientos de 0.4 x 10‐10, 0.07 x 10‐10 y 1.04
x 10‐10 mol cm‐2 para Au/MUA/Os, Au/SPhCOOH/Os y Au/PhCOOH/Os, respectivamente.
Finalmente, el cubrimiento superficial de de los grupos ácidos es muy bajo en comparación con
lo presentado en el Capítulo 3, en el que se observaron cubrimientos superficiales de
1.1 x 10‐10 y 9 x 10‐12 mol cm‐2 para MUA y SPhCOOH, respectivamente. Este hecho ha sido
observado por otros autores [182, 446, 447] que lo atribuyen a la formación de pares iónicos
muy fuertes entre los iones en solución y las cargas en la superficie. Ésta es una posibilidad que
no está considerada en el modelo empleado en el ajuste de datos (Gouy‐Chapman, ver
Apéndice I).
Es interesante destacar que en este experimento el par redox actúa como una sonda que
reporta el pKa de otra especie presente en la superficie.
Cabe mencionar que también se analizó la variación del ancho de pico a media altura con el pH
pero en ningún caso se observó una dependencia clara de su comportamiento, aunque fue
mayor que 0.12 V para todos los sistemas.
6.3.3 ANÁLISIS ELECTROQUÍMICO A DIFERENTES FUERZAS IÓNICAS Y PH CONTROLADO
Para analizar la variación del potencial formal con la fuerza iónica, se prepararon soluciones de
NaOH 1 mM y HNO3 1 mM para fijar el pH en 11 y en 3, respectivamente, y se agregó NaNO3
en la concentración deseada.
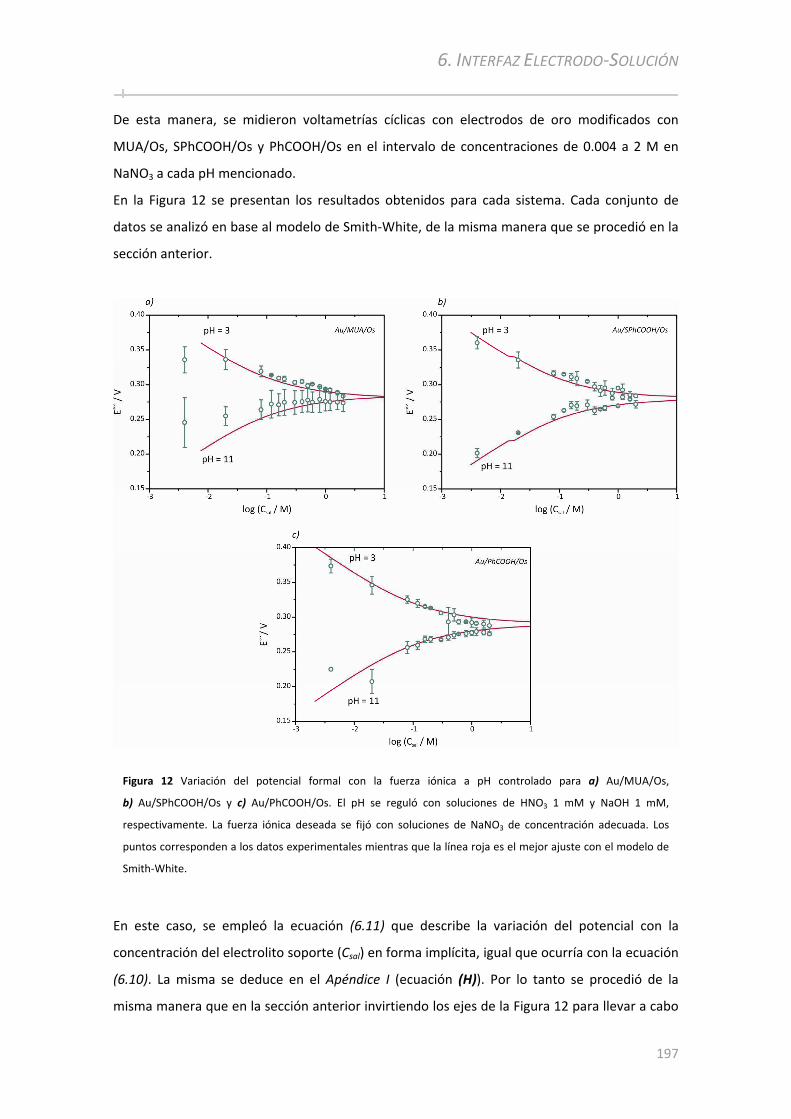
6. INTERFAZ ELECTRODO‐SOLUCIÓN
197
De esta manera, se midieron voltametrías cíclicas con electrodos de oro modificados con
MUA/Os, SPhCOOH/Os y PhCOOH/Os en el intervalo de concentraciones de 0.004 a 2 M en
NaNO3 a cada pH mencionado.
En la Figura 12 se presentan los resultados obtenidos para cada sistema. Cada conjunto de
datos se analizó en base al modelo de Smith‐White, de la misma manera que se procedió en la
sección anterior.
En este caso, se empleó la ecuación (6.11) que describe la variación del potencial con la
concentración del electrolito soporte (Csal) en forma implícita, igual que ocurría con la ecuación
(6.10). La misma se deduce en el Apéndice I (ecuación (H)). Por lo tanto se procedió de la
misma manera que en la sección anterior invirtiendo los ejes de la Figura 12 para llevar a cabo
Figura 12 Variación del potencial formal con la fuerza iónica a pH controlado para a) Au/MUA/Os,
b) Au/SPhCOOH/Os y c) Au/PhCOOH/Os. El pH se reguló con soluciones de HNO3 1 mM y NaOH 1 mM,
respectivamente. La fuerza iónica deseada se fijó con soluciones de NaNO3 de concentración adecuada. Los
puntos corresponden a los datos experimentales mientras que la línea roja es el mejor ajuste con el modelo de
Smith‐White.

6. INTERFAZ ELECTRODO‐SOLUCIÓN
198
el análisis de los datos experimentales. La línea roja en dicha figura es el mejor ajuste de los
datos experimentales a la ecuación (6.11):
( )xO R
tiol redox F pzcx2
salPTE S0 S
z z10よ よ C E E
10 1 2FC
F80 0 RTsenh らφ
RT−
⎡ ⎤⎛ ⎞ +⎛ ⎞ + ° −⎢ ⎥⎜ ⎟ ⎜ ⎟+ ⎝ ⎠⎝ ⎠⎢ ⎥= ⎢ ⎥⎛ ⎞⎜ ⎟⎢ ⎥⎝ ⎠⎣ ⎦ (6.11)
donde PTE Sa
Fx pK pH らφ
2.303RT−= − + + .
En este punto se debe destacar que los ajustes de la sección anterior se realizaron
simultáneamente con los de la Figura 12, buscando los valores de los parámetros que
generaran el mejor ajuste de los dos conjuntos de datos para cada sistema. Esto es muy
importante ya que se pueden ajustar los datos obtenidos en ambos experimentos con el
mismo modelo. Por lo tanto, los valores de los parámetros que se muestran en la Tabla 1 son
los empleados también en esta sección.
En todas las curvas de la Figura 12, se observa el desplazamiento del E°´ a medida que
disminuye la concentración del electrolito soporte. Cuando el pH es 3, dicho desplazamiento es
positivo, mientras que cuando el pH es 11, es negativo. Esto se puede explicar de la misma
manera en que se explicó la dependencia del E°´ con el pH, ya que se trata del mismo
fenómeno. En este caso, para un dado pH, a medida que aumenta la concentración del
electrolito soporte, la formación de pares iónicos disminuye la intensidad de las interacciones
electrostáticas entre las especies confinadas a la superficie. Asimismo, existe un
apantallamiento de la densidad de carga en el plano de transferencia electrónica que atenúa la
diferencia de potencial entre dicho plano (φPTE) y el seno de la solución (φS). Es por todo ello
que, a medida que aumenta la fuerza iónica de la solución, el potencial formal de desplaza
menos respecto del valor que presenta cuando el par redox está libre en solución.
Las pendientes de las curvas indican que la compensación de cargas se satisface con la
adsorción de aniones cuando el exceso superficial es positivo y con la adsorción de cationes,
cuando este exceso es negativo. El fenómeno del potencial de superficie es análogo al
potencial de Donnan o de membrana en películas de un espesor mayor a la distancia de Debye
[448, 449].

6. INTERFAZ ELECTRODO‐SOLUCIÓN
199
6.3.4 ANÁLISIS DE LA CINÉTICA DE TRANSFERENCIA ELECTRÓNICA A DISTINTOS PH
Se midieron las constantes cinéticas estándar de todos los sistemas estudiados en esta tesis a
pH = 3 y a pH = 11 en una solución de NaNO3 1 M. Para ello se utilizó la espectroscopía de
impedancia electroquímica y se procedió de la misma manera que se detalló en el Capítulo 5.
En la Figura 13 se presentan, a modo de ejemplo, las curvas de |Ypico / Ybkg| en función de la
frecuencia para los sistemas Au/MHDA/Os y Au/MPA/Os obtenidas a pH 3 y 11 junto con las
obtenidas sin regular el pH (pH = 5).
En la Figura 13 se observa que no existen diferencias significativas entre los resultados
obtenidos a los diferentes pH. Lo mismo fue observado con los demás sistemas estudiados.
Para interpretar estas observaciones es necesario considerar la distancia de Debye (゛‐1).
Empleando la ecuación (6.5) se puede estimar que la misma es de ≈ 0.3 nm en estas
condiciones. Por otro lado, se puede calcular cuántas moléculas de tiol sin post‐funcionalizar
existen en la superficie. Para ello se deben considerar los cubrimientos superficiales de los
tioles que se estimaron por desorción reductiva en el Capítulo 3 y la eficiencia de post‐
funcionalización estimada en el Capítulo 4. En el caso del MHDA se tiene un cubrimiento de
1.3 x 10‐10 mol cm‐2 y para el MHA, 9 x 10‐10 mol cm‐2. A su vez, el grado de sustitución de los
grupos –COOH fueron 3.5% y 3.3 % , respectivamente. Con estos valores se tiene que en el caso
del tiol más largo, una molécula terminada en –COOH ocupa 13 Å2 y, en el caso del MHA,
ocupa 18 Å2. Esto indica que en ambos casos la distancia promedio entre moléculas cargadas
es de 1.5‐2 nm, distancia comparable con la de Debye. Por lo tanto, debido al apantallamiento
Figura 13 Variación de ¨ en función de la frecuencia para a) Au/MHDA/Os y b) Au/MPA/Os obtenidas a pH 3, 5 y
11, en una solución de NaNO3 1 M. Los puntos son los datos experimentales mientras que las líneas son los
mejores ajustes utilizando el circuito de Randles como modelo (ver Capítulo 5).

6. INTERFAZ ELECTRODO‐SOLUCIÓN
200
de cargas que esto implica, no es de sorprender que no se observen cambios en k0 con el pH
de la solución.
Estos experimentos se llevaron a cabo en NaNO3 1 M a fin de minimizar la resistencia no
compensada (Ru) en la medición de la cinética ya que en soluciones más diluidas la misma y,
por ende, la constante de celda (RuCdl) impediría medir la cinética de transferencia electrónica.
Es por esto que, en principio, no se puede sacar ninguna conclusión respecto de estas
observaciones.
6.4 CONCLUSIONES
A lo largo de este capítulo se realizó un análisis detallado de la influencia de la estructura de la
interfase electrodo‐solución sobre la transferencia electrónica. Para ello se utilizó la
voltametría cíclica y la espectroscopía de impedancia electrónica a fin de estudiar el efecto del
cubrimiento superficial, el pH y la fuerza iónica sobre la termodinámica y la cinética del
transporte de carga entre la superficie del electrodo y los sitios electroactivos.
Se pudo observar el aumento de las interacciones intermoleculares cuando aumenta el
cubrimiento superficial del complejo de osmio, así como también, de la energía de
reorganización que genera una disminución de la constante cinética de transferencia
electrónica.
Asimismo, se observó el acoplamiento de los equilibrios ácido‐base y redox al verificarse la
influencia de los grupos ácidos sobre el potencial formal, que pueden modificar su estado de
carga, y por ende la densidad de carga superficial, afectando de esta manera el potencial en el
plano de transferencia electrónica.
Estos experimentos complementan la caracterización realizada en los capítulos anteriores de
este tipo de sistemas y ponen en relevancia la complejidad que poseen los mismos, al
presentar grupos con propiedades ácido‐base que pueden intervenir en el estado de carga de
la superficie y en las interacciones intermoleculares que existen sobre la misma.

6. INTERFAZ ELECTRODO‐SOLUCIÓN
201
6.5 APÉNDICE I
6.5.1 ADSORCIÓN DE UN ÁCIDO DÉBIL A UNA SUPERFICIE: EQUILIBRIO ÁCIDO‐BASE Y POTENCIAL
ELECTROSTÁTICO [450]
En la Figura 14 se muestra un esquema de un sistema conformado por un ácido débil
adsorbido sobre la superficie de un electrodo que está en contacto con una solución acuosa
electrolítica. En analogía con lo expuesto en el Capítulo 6, se plantea que todos los grupos
ácido están a una distancia finita del electrodo y residen en un plano que se llamará plano de
disociación ácida (PDA).
Para comenzar, se pueden escribir las expresiones de los potenciales electroquímicos
( ( )j 0 j j ji i i i i i´ ´ RT ln a z Fφ , a actividad del ión i en la fase j= + + = ) de cada especie,
considerando todas las fases y los equilibrios entre las mismas.
[ ]
a
0 0
H H
0
A
0
HA
K
S PDAS PDA
H
PDA PDA
A
PDA
HA
HA H A
´ ´ RT ln H Fφ ´ RT ln H Fφ
´ ´ RT ln A Fφ
´ ´ RT ln HA
+ + +
− −
+ −
+ +
−
+
⎡ ⎤ ⎡ ⎤= + + = + +⎣ ⎦ ⎣ ⎦⎡ ⎤= + −⎣ ⎦
= +
yywxyy
Figura 14 Esquema de la distribución de potencial de un sistema formado por un ácido débil adsorbido sobre un
electrodo en contacto con una solución de electrolito. Los grupos HA están separados de la superficie por una
película dieléctrica de espesor finito. La línea roja indica el perfil de potencial interfacial que cae linealmente
entre el metal y el PDA y no linealmente entre éste y el seno de la solución.

6. INTERFAZ ELECTRODO‐SOLUCIÓN
202
donde φS y φPDA son los potenciales electrostáticos en el seno de la solución y en el plano de
disociación ácida, respectivamente. Teniendo en cuenta el equilibrio ácido‐base (con una
constante asociada, Ka), se pueden igualar los potenciales electroquímicos:
[ ] ( )( )
HA H A
S PDAPDA0 0 S 0 PDAHA H A
0 0 0 PDA SHA aH A
PDAS PDA S
a
´ ´ ´
´ RT ln HA ´ RT ln H Fφ ´ RT ln A Fφ
´ ´ ´ 2.303RT logK F φ φ
f2.303RT log H 2.303log F φ φ
1 f
f2.303RTpK 2.303RTpH 2.303RT log
1 f
+ −
+ −
+ −
+ −
+
= +⎡ ⎤ ⎡ ⎤+ = + + + + −⎣ ⎦ ⎣ ⎦
− − = − − =⎡ ⎤⎡ ⎤= + − −⎢ ⎥⎣ ⎦ −⎣ ⎦
⎡ ⎤− = − + ⎢ ⎥−⎣ ⎦PDA
PDA SFらφ −−
donde f=[A‐]PDA/[HA]PDA + [A‐]PDA, es el grado de deprotonación del ácido. Reordenando se llega
a una expresión que relaciona la diferencia de potencial entre el PDA y la solución con el grado
de deprotonación del ácido, teniendo en cuenta su pKa y el pH de la solución en contacto con
el electrodo:
PDA
PDA S 2.303RT fらφ pKa pH log
F 1 f− ⎛ ⎞⎡ ⎤= − +⎜ ⎟⎢ ⎥⎜ ⎟−⎣ ⎦⎝ ⎠ (A)
6.5.2 ADSORCIÓN DE UN PAR ELECTROACTIVO A UNA SUPERFICIE: EQUILIBRIO REDOX Y POTENCIAL
ELECTROSTÁTICO [433]
En la Figura 15 se muestra un esquema de la distribución de potencial interfacial cuando se
tiene la superficie de un electrodo modificada con un par electroactivo, unido
irreversiblemente a una distancia fija de la misma.
En este caso, se considera que las especies redox se hallan en un plano denominado plano de
transferencia electrónica (PTE). Éste es el caso analizado cuando se presentó el modelo de
Smith‐White.
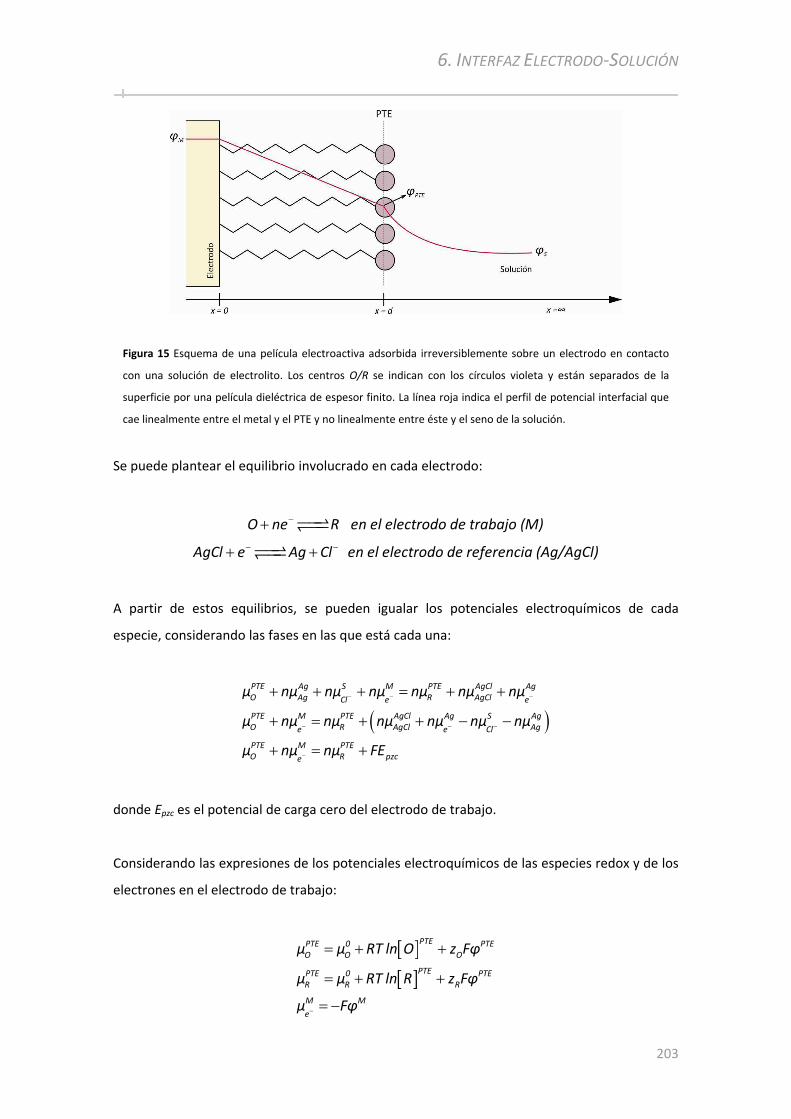
6. INTERFAZ ELECTRODO‐SOLUCIÓN
203
Se puede plantear el equilibrio involucrado en cada electrodo:
O ne R en el electrodo de trabajo (M)
AgCl e Ag Cl en el electrodo de referencia (Ag/AgCl)
−− −+
+ +yywxyy
yywxyy
A partir de estos equilibrios, se pueden igualar los potenciales electroquímicos de cada
especie, considerando las fases en las que está cada una:
( )PTE Ag S M PTE AgCl AgO Ag R AgClCl e e
PTE M PTE AgCl Ag S AgO R AgCl Age e Cl
PTE M PTEO R pzce
´ n´ n´ n´ n´ n´ n´
´ n´ n´ n´ n´ n´ n´
´ n´ n´ FE
− − −
− − −
−
+ + + = + ++ = + + − −+ = +
donde Epzc es el potencial de carga cero del electrodo de trabajo.
Considerando las expresiones de los potenciales electroquímicos de las especies redox y de los
electrones en el electrodo de trabajo:
[ ][ ]
PTEPTE 0 PTEO O O
PTEPTE 0 PTER R R
M M
e
´ ´ RT ln O z Fφ
´ ´ RT ln R z Fφ
´ Fφ−
= + += + += −
Figura 15 Esquema de una película electroactiva adsorbida irreversiblemente sobre un electrodo en contacto
con una solución de electrolito. Los centros O/R se indican con los círculos violeta y están separados de la
superficie por una película dieléctrica de espesor finito. La línea roja indica el perfil de potencial interfacial que
cae linealmente entre el metal y el PTE y no linealmente entre éste y el seno de la solución.

6. INTERFAZ ELECTRODO‐SOLUCIÓN
204
Se puede llegar a:
[ ] [ ]PTE PTE0 PTE M 0 PTEO O R R pzc´ RT ln O z Fφ Fφ ´ RT ln R z Fφ FE+ + − = + + +
Considerando que ´O0 – ´R
0= FE° y que en el caso del par Os(II)+/Os(III)+2, zR ‐ zO=‐1, se tiene
que:
[ ][ ] ( )PTE
0 M PTEpzc R O
RFE RT ln Fφ FE z z Fφ
O
⎛ ⎞= + + + −⎜ ⎟⎜ ⎟⎝ ⎠PTE
0 M PTEOpzc
O
1 fFE RT ln Fらφ FE
f−⎛ ⎞−= + +⎜ ⎟⎝ ⎠ (B)
donde fO = ГO / ГO + ГR es la fracción de especies oxidadas sobre la superficie.
La caída de potencial total entre el electrodo de trabajo y el de referencia (E – Epzc) se puede
expresar como la suma de las caídas parciales entre el primer electrodo y el plano de
transferencia electrónica (らφM‐PTE) y entre éste último y el seno de la solución (らφPTE‐S). A partir
de ello se obtiene una expresión que relaciona el potencial del electrodo de trabajo, el
potencial en el plano de transferencia electrónica y el grado de oxidación de las especies
redox:
M PTE PTE SpzcE E らφ らφ− −− = + (C)
PTE S 0 Opzc pzc
O
0 PTE SO
O
fRTらφ E E E E ln
F 1 f
fRTE E ln らφ
F 1 f
−
−
⎛ ⎞− + + = + + ⎜ ⎟−⎝ ⎠⎛ ⎞= + +⎜ ⎟−⎝ ⎠
Hasta ahora se consideró la dependencia del potencial en el plano de transferencia electrónica
con el equilibrio redox que tiene lugar en el mismo. Para relacionar este equilibrio con la
concentración del electrolito soporte y cómo dicha relación afecta al PTE, se debe tener en
cuenta la estructura de la interfase entre el metal y la solución. Para ello se emplea el modelo
de Gouy‐Chapman.
Considerando que la monocapa que separa el PTE del metal actúa como un dieléctrico, se
puede expresar la densidad de carga del electrodo (σM) como:

6. INTERFAZ ELECTRODO‐SOLUCIÓN
205
F 0 FC 0 0=
M PTE 0 O 0 FM F pzc
O
1 f 0 0RTσ C らφ E E ln
F f d− ⎡ ⎤⎛ ⎞−= = − −⎢ ⎥⎜ ⎟⎝ ⎠⎣ ⎦ (D)
donde CF y 0F es la capacidad y la permitividad de la película, y 00, la permitividad del vacío.
A partir del modelo de Gouy‐Chapman se obtienen las siguientes expresiones de potencial en
el PTE y densidad de carga (σPTE):
( )PTE S PTE M1/ 2
sal S 0
σ σ2RTらφ asenh
F 8C 0 0 RT
− ⎡ ⎤+= ⎢ ⎥⎢ ⎥⎣ ⎦ (E)
( )PTE film redox O O R Rσ Fよ f Fよ f z z z= − + − +⎡ ⎤⎣ ⎦ (F)
donde 0S es la constante dieléctrica de la solución, Csal es la concentración de electrolito
soporte, Гfilm es el cubrimiento superficial de las moléculas que conforman la película que
separa el PTE del metal y Гredox es el cubrimiento de las moléculas electroactivas que se
encuentran en PTE. Considerando que las moléculas separadoras tienen propiedades ácido‐
base, como es el caso de los sistemas estudiados, se considera que f es la fracción de grupos
deprotonados.
Si se considera que el potencial de pico en una voltametría cíclica (tanto de oxidación como de
reducción) corresponde a la situación en que la mitad de los centros electroactivos se han
oxidado (o reducido): fO = 0.5. Teniendo esto en cuenta y reemplazando las ecuaciones (D) y
(F) en la ecuación (E) se obtiene:
( ) ( )( )
O Rfilm redox F pzc
PTE S
1/ 2
sal S 0
z zFよ f Fよ C E E2RT 2らφ asenh
F 8C 0 0 RT
−+⎡ ⎤− + + °−⎢ ⎥= ⎢ ⎥⎢ ⎥⎢ ⎥⎣ ⎦
(G)
Esta expresión relaciona el potencial con la fuerza iónica y tiene en cuenta la dependencia con
el pH de la densidad de carga superficial, ya que considera el equilibrio ácido‐base.

6. INTERFAZ ELECTRODO‐SOLUCIÓN
206
6.5.3 COMBINACIÓN DE LOS EQUILIBRIOS ÁCIDO‐BASE Y DE ELECTRO‐REDUCCIÓN: EFECTO SOBRE EL
POTENCIAL EN EL PTE
Para obtener ecuaciones que expresen el potencial en el PTE (らφPTE‐S) en función del pH y de la
fuerza iónica se deben combinar las ecuaciones que se han ido señalando a lo largo de este
desarrollo como (A) a (F). Además, se debe considerar que el plano de disociación ácida
coincide con el plano de transferencia electrónica.
Las ecuaciones (A) y (G) proveen expresiones del potencial en el PTE en función del pH y de la
fuerza iónica, respectivamente. Si se despeja f (grado de deprotonación de los grupos HA) de
la ecuación (G) y se reemplaza en (A), se obtiene una expresión que contempla la variación del
potencial en función del pH con el efecto del equilibrio redox incluido en f para una dada
fuerza iónica.
A su vez, si se despeja f de la ecuación (A) y se reemplaza en (G) se tiene una expresión del
potencial en función de la fuerza iónica que contempla el estado de protonación de la película
al pH de trabajo.
No existen expresiones analíticas para らφPTE‐S en función de Csal ni para らφPTE‐S en función del
pH, pero sí se puede expresar la fuerza iónica (Csal) y el pH en función de らφPTE‐S como se
presenta a continuación:
( )xO R
tiol redox F pzcx2
salPTE S0 S
z z10よ よ C E E
10 1 2FC
F80 0 RTsenh らφ
RT−
⎡ ⎤⎛ ⎞ +⎛ ⎞ + ° −⎢ ⎥⎜ ⎟ ⎜ ⎟+ ⎝ ⎠⎝ ⎠⎢ ⎥= ⎢ ⎥⎛ ⎞⎜ ⎟⎢ ⎥⎝ ⎠⎣ ⎦ (H)
donde PTE Sa
Fx pK pH らφ
2.303RT−= − + +
( )( )
PTE S O Rredox F pzc
PTE S
PTE S O Rfilm redox F pzc
z zFsenh らφ Fよ C E E
F RT 2pH らφ pKa log
z zF2.303RTFよ senh らφ Fよ C E E
RT 2
−−
−
⎛ + ⎞⎛ ⎞⎛ ⎞− + + °−⎜ ⎟⎜ ⎟ ⎜ ⎟⎝ ⎠ ⎝ ⎠⎜ ⎟= − + + +⎜ ⎟⎛ ⎞⎛ ⎞+ − − °−⎜ ⎟ ⎜ ⎟⎜ ⎟⎝ ⎠ ⎝ ⎠⎝ ⎠ (I)
Las ecuaciones (H) e (I) fueron empleadas para ajustar curvas experimentales de E°´ vs. Csal
(con pH constante) y de E°´ vs. pH (con Csal constante), respectivamente.

CAPÍTULO 7
ESPECTROSCOPÍA DE EFECTO TÚNEL CON CONTROL
ELECTROQUÍMICO


7. ESPECTROSCOPÍA DE EFECTO TÚNEL CON CONTROL ELECTROQUÍMICO
209
n los últimos años, se ha vuelto posible medir el comportamiento eléctrico y
electroquímico de pequeños grupos de moléculas, aún alcanzando el límite de moléculas
individuales [39, 161, 451‐454]. Esto ha permitido el estudio sistemático de la influencia de la
estructura molecular así como de los contactos y el entorno sobre la conductividad de las
junturas moleculares [192, 455]. En general, en las mismas se han utilizado contactos de oro y
las moléculas se han unido a ellos por métodos convencionales de quimisorción, empleando
para ello tioles, aminas, fosfinas, piridinas o nitrilos [39, 403, 456]. Sin embargo, prácticamente
no se han estudiado sistemas formados por moléculas unidas a los contactos por enlaces Au‐C.
Por lo tanto, se buscó analizar el sistema obtenido por reducción de la sal de diazonio derivada
del ácido 4‐aminobenzoico, Au/PhCOOH/Os, ubicando a la molécula en una juntura molecular.
Dicha juntura se formó con la punta de un microscopio de efecto túnel (STM) y la técnica
empleada para analizar la conductividad en este sistema fue la Espectroscopía de Efecto Túnel
con control electroquímico (Electrochemical Scanning Tunneling Spectroscopy, E‐STS). En vista
de que el sistema mencionado se diferencia del Au/SPhCOOH/Os sólo en el átomo de azufre,
también se analizó este último con el objetivo de comparar ambos tipos de enlace con el
contacto metálico.
En este capítulo se presentan los fundamentos de la técnica empleada y una breve descripción
del modelo teórico desarrollado por Ulstrup y Kuznetsov [167, 399] empleado en el análisis de
los resultados. Finalmente, se muestran los resultados obtenidos con los dos sistemas de
interés.
7.1 FUNDAMENTOS DE LA TÉCNICA
La espectroscopía de efecto túnel in situ fue introducida por Kolb [167, 203], Siegenthaler
[202], Nichols [398] e Itaya [457] y sus asociados a fines de la década de 1980. Los conceptos y
formalismos teóricos han sido desarrollados desde principios de la década de 1990 [458] hasta
el presente [167].
Como ya fue mencionado en el Capítulo 2, esta técnica es una aplicación especial de la
microscopía de efecto túnel. En ella se incluyen dos electrodos de trabajo y un electrodo de
referencia. Los primeros dos son el sustrato y la punta del microscopio STM. También se
agrega un cuarto electrodo (el contra‐electrodo) para monitorear los procesos electroquímicos
faradaicos. En la Figura 1 se muestra un esquema del arreglo experimental empleado en este
tipo de experimento que consta del microscopio STM y de un potenciostato y en el que todos
los electrodos están sumergidos en una solución electrolítica.
E

7. ESPECTROSCOPÍA DE EFECTO TÚNEL CON CONTROL ELECTROQUÍMICO
210
De esta manera se puede controlar el potencial del sustrato y de la punta del microscopio, en
forma independiente, respecto de un electrodo de referencia común. En forma simultánea se
puede controlar el proceso faradaico que ocurre en el sustrato (electrodo de trabajo), a partir
de la corriente electroquímica que circula entre este último y el electrodo auxiliar, y la
conductividad de la juntura molecular formada entre el sustrato y el tip, a partir de la corriente
túnel que circula entre ellos.
El experimento consiste en variar el potencial aplicado entre la punta del microscopio y dicho
sustrato, (Ebias = Etip – ES) mientras se registra la corriente túnel (It) que circula entre ambos. El
control electroquímico implica que, al introducir todo este conjunto en una solución
electrolítica y agregar el electrodo de referencia y el contra‐electrodo, se pueden controlar en
forma independiente, los potenciales aplicados al sustrato y al tip respecto de la referencia. La
espectrosocopía de efecto túnel convencional permite analizar la conductividad de un número
reducido de moléculas (o de una molécula individual, single molecule) adsorbidas sobre el
sustrato. Al agregar el control electroquímico, se puede explorar el comportamiento de las
mismas en sus diferentes estados de oxidación, si se trata de especies electroactivas.
En particular, se ha destacado la analogía que existe entre los sistemas nanométricos de tres
electrodos y los transistores de estado sólido [459, 460]. El sustrato y el tip son equivalentes al
emisor y colector de un transistor, mientras que el electrodo de referencia actúa como la base
del mismo. Es por ello que este tipo de junturas recibe el nombre de transistores moleculares
[459, 461].
Las diferencias operacionales, sin embargo, son importantes. Mientras que los transistores de
estado sólido unimoleculares operan en condiciones de ultra alto vacío y a temperaturas
criogénicas [403, 405, 453], las junturas moleculares mencionadas pueden funcionar a
Figura 1 Esquema del arreglo experimental utilizado en STS in situ en la que se tiene control electroquímico
independiente del electrodo de trabajo (sustrato) y del tip con respecto a un electrodo de referencia común.

7. ESPECTROSCOPÍA DE EFECTO TÚNEL CON CONTROL ELECTROQUÍMICO
211
temperatura ambiente y con un entorno de materia condensada [459, 462] que incluye no sólo
soluciones acuosas, sino también, líquidos iónicos [463].
7.2 MARCO TEÓRICO
7.2.1 MOLÉCULAS NO ELECTROACTIVAS
Al confinar moléculas electroquímicamente inactivas en un STM configurado como se describió
en la sección anterior, se espera que las mismas exhiban curvas de It vs. Ebias simples. Los
niveles LUMO y HOMO de este tipo de moléculas (por ejemplo, n‐alcanotioles) están fuera de
resonancia con los niveles de Fermi de los electrodos adyacentes (sustrato y tip) y el
mecanismo que gobierna la transferencia electrónica entre ambos es el tuneleo a través del
enlace (o superintercambio). Es por ello que las relaciones It vs. Ebias e It vs. η (con η = ES – ES°, el
sobrepotencial aplicado al sustrato), son lineales, reflejando el tuneleo a través de una barrera
de potencial.
Esto se debe a que estas moléculas no redox, representan barreras para la conductividad
electrónica y, si bien es probable que no se las considere como componentes principales de un
circuito, comprender sus mecanismos más simples de transferencia electrónica es importante.
De hecho, el estudio tanto experimental como teórico de estos sistemas, ha posibilitado el
avance en diversos aspectos de este área de conocimiento. Éstos incluyen los contactos
molécula‐electrodo, la dinámica del enlace entre ambos y la dinámica de la estructura
molecular en sí misma, que se ve reflejada en la dependencia con la temperatura y la
modulación de la conductividad molecular [398, 464].
7.2.2 MOLÉCULAS ELECTROACTIVAS: MODELO DE KUZNETZOV‐ULSTRUP
Las moléculas electroactivas poseen niveles moleculares más bajos que las moléculas no
electroactivas y, al ser confinadas entre el sustrato y la punta del microscopio, estos niveles
pueden quedar localizados en una ventana de energía entre los niveles de Fermi de los dos
contactos. De esta manera, se abren canales de conducción electrónica más eficientes que el
superintercambio fuera de resonancia mencionado en la sección anterior. Estos mecanismos
generan diferentes respuestas It‐Ebias y convierten a este tipo de moléculas en elementos más
versátiles a la hora de pensar en un circuito electrónico.
La Figura 2 a) muestra un esquema de una molécula ubicada entre un sustrato metálico (por
ejemplo, Au (111)) y la punta del microscopio. En este caso, ambos electrodos metálicos tienen
un continuo de niveles electrónicos, mientras que la molécula tiene niveles de energía

7. ESPECTROSCOPÍA DE EFECTO TÚNEL CON CONTROL ELECTROQUÍMICO
212
discretos. Éstos se ensanchan, por las interacciones del centro redox con el electrolito
circundante y con los electrodos, y fluctúan constantemente alrededor de sus posiciones de
equilibrio promedio debido a variaciones en la energía de solvatación. La magnitud de estas
fluctuaciones viene dada por la energía de reorganización del entorno (゜).
En todo momento existe una corriente túnel directa (no resonante) que fluye entre el tip y el
sustrato, independientemente de las posiciones relativas de los niveles moleculares con
respecto a los niveles de Fermi de los electrodos adyacentes.
Debido a la fluctuación en energía de los niveles moleculares puede ocurrir que el nivel de
Fermi de uno de los electrodos (por ejemplo, el sustrato) se acerque al orbital más bajo
desocupado (LUMO) de la especie redox pero quedando todavía ubicado por debajo del
mismo. El LUMO puede disminuir su energía por fluctuaciones y acercarse al nivel de Fermi del
sustrato causando la primera transferencia electrónica. A partir de aquí hay tres escenarios
posibles:
a) si el electrón se transfiere “inmediatamente”, es decir, sin relajación de la molécula y del
entorno, este proceso corresponde a un mecanismo de tuneleo resonante [442, 465].
Figura 2 a) Esquema de una molécula de PhCONHCH2[py Os(2,2´‐bpy)2Cl] encerrada entre un sustrato de oro y
la punta del microscopio de efecto túnel. b) Diagrama de energía esquemático que muestra la transferencia
electrónica en dos pasos mediada por la molécula redox. El electrón es transferido desde el nivel de Fermi del
sustrato al LUMO del complejo y, luego de una relajación vibracional parcial, se transfiere al nivel de Fermi del
tip.

7. ESPECTROSCOPÍA DE EFECTO TÚNEL CON CONTROL ELECTROQUÍMICO
213
Cuando el centro reducido tiene tiempo suficiente para relajar vibracionalmente, otras dos
situaciones son posibles:
b) En el límite no adiabático (acoplamiento electrónico débil entre el centro redox y ambos
electrodos adyacentes), la velocidad de transferencia electrónica es baja y el centro reducido
puede relajar completamente antes de la segunda transferencia electrónica. El segundo paso
pierde la “coherencia electrónica” respecto del primero, y el camino de reacción prosigue
según una barrera de Franck‐Condon. De esta manera, se produce un pequeño aumento de la
corriente túnel, ya que básicamente sólo un electrón se transfiere por cada fluctuación
molecular inicial [399].
c) En el límite adiabático (acoplamiento electrónico fuerte entre la molécula y los electrodos
adyacentes) la transferencia electrónica es rápida [167, 399]. Esta situación corresponde a un
caso intermedio, es decir, transferencia electrónica en dos pasos con relajación vibracional
parcial y coherencia parcial. Luego del primer paso, la energía del ahora ocupado LUMO
disminuye por relajación vibracional, y el segundo paso de transferencia electrónica ocurre
antes de la completa relajación y antes de que la energía del nivel molecular pase el nivel de
Fermi del segundo electrodo. Por lo tanto, el LUMO vacío empieza a relajar hacia energías
superiores y puede aceptar otro electrón del sustrato antes de pasar su nivel de Fermi,
repitiendo entonces el ciclo. Este proceso puede comprender varios ciclos de pasos de
reducción y re‐oxidación molecular consecutivos por cada fluctuación inicial del nivel
molecular. Como consecuencia de este proceso, la corriente túnel aumenta significativamente.
En la Figura 2 b) se muestra un diagrama de niveles de energía que esquematiza este
mecanismo de transferencia electrónica entre el sustrato y el tip a través de los niveles de
energía de una molécula ubicada entre ellos.
La espectroscopía de efecto túnel con control electroquímico utiliza el potenciostato para
imponer el alineamiento entre los niveles moleculares y los niveles de Fermi de los contactos,
manteniendo el Ebias fijo.
La energía electrónica de los niveles moleculares asociados a la molécula ubicada en la juntura
se modifica cuando cambia el sobrepotencial aplicado al sustrato sobre el cual está adsorbida.
Por lo tanto, si se varía dicho potencial se pueden modular sus estados HOMO y LUMO y
sintonizarlos con los niveles de Fermi de los electrodos adyacentes.
Por ejemplo, si la molécula está en su estado oxidado y se barre el sobrepotencial del sustrato
en sentido negativo, es decir, el potencial aplicado se hace cada vez más catódico, el nivel de
Fermi de dicho electrodo aumentará en energía electrónica hasta alinearse con el LUMO de la
molécula. Una vez alineados se transferirá el primer electrón. Esta alineación es posible

7. ESPECTROSCOPÍA DE EFECTO TÚNEL CON CONTROL ELECTROQUÍMICO
214
cuando el potencial aplicado es cercano al potencial de equilibrio electroquímico, E°, de la
molécula ubicada en la juntura, ya que a ese potencial su nivel redox es térmicamente
accesible.
Por lo tanto, como se mencionó más arriba, cuando el LUMO y el nivel de Fermi del sustrato
igualan su energía, aumenta la corriente túnel que circula a través de la juntura. Por lo
expuesto hasta aquí, dicho aumento se observa cuando el potencial del sustrato es ≈ E°. La
posición exacta del aumento de la corriente depende también de otros aspectos,
particularmente de las caídas de potencial electroquímico y del Ebias en el sitio en el que está el
centro redox dentro de la juntura de tuneleo.
Ahora bien, si el sobrepotencial aplicado sobre el sustrato sigue haciéndose más negativo y
llega a ser menor que E°, el LUMO quedará por debajo del nivel de Fermi perdiéndose la
alineación y, por ende, la corriente túnel (It) caerá nuevamente. Por lo tanto, ésta pasará por
un máximo al barrer el sobrepotencial aplicado sobre el sustrato (η). La relación esperada
entre It y η se muestra esquemáticamente en la Figura 3.
Figura 3 Esquema del comportamiento esperado de la corriente túnel (It) en función del sobrepotencial (η)
aplicado al sustrato para un Ebias fijo.

7. ESPECTROSCOPÍA DE EFECTO TÚNEL CON CONTROL ELECTROQUÍMICO
215
7.2.2.1 ECUACIONES
Las ecuaciones que describen el modelo descripto en la sección anterior están basadas en un
proceso de transferencia electrónica en dos pasos y se pueden derivar de manera similar a las
ecuaciones estándar de cinética electroquímica en la llamada “región normal” [167]. La
corriente túnel total (It) se puede expresar como:
t 1 2
1 1 1I I I= + (7.1)
donde I1 e I2 son las corrientes de transferencia electrónica entre el sustrato y la molécula y
entre esta última y el tip, respectivamente. Ellas se pueden expresar como:
( ) ( )
( ) ( )B
B
2
bias1 1 1 bias
2
bias bias2 2 2 bias
゜ e¨η eáE\I e゛ ヾ eE exp
2ヽ 4゜k T
゜ e¨η eáE eE\I e゛ ヾ eE exp
2ヽ 4゜k T
⎡ ⎤⎢ ⎥⎢ ⎥⎣ ⎦⎡ ⎤⎢ ⎥⎢ ⎥⎣ ⎦
+ += −+ + += −
(7.2)
donde e es la carga elemental, ゛ es el coeficiente de transmisión electrónica, ヾ es la densidad
de estados electrónicos en los electrodos cerca de sus niveles de Fermi, \ es la frecuencia
característica de vibración nuclear, η es el sobrepotencial (η = ES – E°), kB es la constante de
Boltzmann y T es la temperatura. ¨ y á son parámetros que describen el corrimiento efectivo
del potencial del electrodo con la variación de η y Ebias, respectivamente, en la posición en la
que se encuentra el centro redox. Es decir, consideran la distribución no lineal del potencial en
la juntura y varían entre 0 y 1 [399, 466]8.
8 Son equivalentes al factor de simetría, o coeficiente de transferencia de carga electroquímica, y se
refieren a la dependencia del potencial electroquímico y del Ebias, respectivamente.

7. ESPECTROSCOPÍA DE EFECTO TÚNEL CON CONTROL ELECTROQUÍMICO
216
Asumiendo los mismos valores de ゛ y ヾ para ambos pasos de transferencia electrónica
(contactos simétricos), se obtiene la siguiente expresión para It en función de Ebias y de η para
una geometría fija de la juntura de tuneleo:
( ) ( ) ( )B B
1
2 2
t bias bias bias bias
\ e eI e゛ヾ eE exp ゜ ¨η áE exp ゜ E ¨η áE
2ヽ 4゜k T 4゜k T
−⎧ ⎫⎡ ⎤ ⎡ ⎤⎪ ⎪⎨ ⎬⎢ ⎥ ⎢ ⎥⎪ ⎪⎣ ⎦ ⎣ ⎦⎩ ⎭= + + + + − − (7.3)
Cuando el acoplamiento electrónico entre la molécula y los electrodos es fuerte (límite
adiabático) se obtiene la siguiente expresión a partir de la ecuación (7.3) que se convierte en:
( ) ( ) 1
biasbiast bias
B B
1 / 2 á eE e¨η゜ eE1 \i e゛ヾ eE exp cosh
2 2ヽ 4k T 2k T
−⎡ ⎤⎡ ⎤⎛ ⎞⎢ ⎥⎢ ⎥⎜ ⎟⎢ ⎥⎜ ⎟ ⎢ ⎥⎝ ⎠⎢ ⎥⎣ ⎦⎣ ⎦− −−= − (7.4)
Para conocer el detalle de la deducción de estas ecuaciones se remite a la bibliografía citada
[167, 399].
Pobelov et al. [168] halló una expresión numérica de la ecuación (7.3), tomando ゛ = 1 (límite
adibático) y valores típicos de ヾ para oro y de \ para un entorno acuoso. Así se obtiene:
( ) ( ) 12 2
t bias bias bias bias9.73 9.73
I 1820E exp ゜ ¨η áE exp ゜ E ¨η áE゜ ゜
−⎧ ⎫⎡ ⎤ ⎡ ⎤⎪ ⎪⎨ ⎬⎢ ⎥ ⎢ ⎥⎣ ⎦ ⎣ ⎦⎪ ⎪⎩ ⎭= + + + − − (7.5)
Cuando el sobrepotencial aplicado al sustrato y el potencial aplicado entre éste y el tip son
bajos (Ebias, η < < ゜) se puede obtener una ecuación simplificada:
( )( )( )bias bias
t
bias
910E exp 9.73 ゜ EI
cosh 19.4 ¨η á 1 / 2 E
⎡ ⎤⎢ ⎥⎣ ⎦⎡ ⎤⎢ ⎥⎣ ⎦
− += + − (7.6)
donde cosh (x) = (ex+e‐x)/2 es el coseno hiperbólico. Esta ecuación representa la forma
predicha de la corriente túnel en función del sobrepotencial aplicado al sustrato para un Ebias
fijo (Figura 3):

7. ESPECTROSCOPÍA DE EFECTO TÚNEL CON CONTROL ELECTROQUÍMICO
217
( ) ( )( ) ( )
t ,picot S
S pico
biast ,pico picobias bias
II E
cosh 19.4¨ E E
Econ I 910E exp 9.73 ゜ E y E E 1 / 2 á
¨
⎡ ⎤⎢ ⎥⎣ ⎦⎡ ⎤⎢ ⎥⎣ ⎦
= −= − + = °+ −
(7.7)
Epico e It,pico representan la posición y la altura del pico observado en la corriente túnel.
En base la ecuación (7.7) se desprenden dos importantes relaciones. La primera de ellas es que
la dependencia de Epico con Ebias es lineal siendo E° la ordenada al origen. La segunda relación es
la de It,pico con Ebias que se puede expresar como:
( ) bias
t ,pico
bias
Iln 6.81 9.73゜ 9.73E
E
⎛ ⎞⎜ ⎟⎜ ⎟⎝ ⎠ = − − (7.8)
A partir de esta relación es posible estimar la energía de reorganización. Ambas relaciones
permiten contrastar la teoría con los datos experimentales.
7.3 ANÁLISIS DE AU/PHCOOH/OS Y AU/SPHCOOH/OS POR STS CON CONTROL ELECTROQUÍMICO
La espectroscopía túnel con control electroquímico fue empleada para estudiar el transporte
de carga a través del centro redox de osmio y determinar si el potencial electroquímico puede
usarse para regular la transferencia electrónica a través de la molécula unida al sustrato por un
enlace Au‐C o, por el enlace más convencional, Au‐S.
Para realizar este experimento, se prepararon superficies de oro modificadas con los sistemas
PhCOOH/Os y SPhCOOH/Os de la misma manera descripta en el Capítulo 4. Cada una de ellas
fue introducida en el microscopio STM, dentro de una celda equipada con dos electrodos de
oro que actuaron como electrodo de referencia y contra‐electrodo. A éstos se sumó un cuarto
electrodo, el tip del microscopio que se acercó a la superficie, aplicando un setpoint de 1 nA.
Cabe aclarar que al alejar el tip de la superficie se observó una caída de la corriente (0.01 nA)
asegurando que la corriente medida se debía al efecto túnel.
Se aplicó una diferencia de potencial entre el sustrato y el tip (Ebias = ET – ES) que se mantuvo
constante. El potencial inicial del sustrato (ES,i) se fijó lejos del potencial de equilibrio del
complejo (E°). En este punto se apagó el circuito de retroalimentación del microscopio y se
varió el potencial del sustrato (ES) a 50 mV s‐1, en una ventana lo suficientemente amplia como
para pasar por E° tanto con ES como con ET, mientras que se monitoreó la corriente túnel (It).
Este barrido de ES se realizó tanto en sentido positivo como negativo.
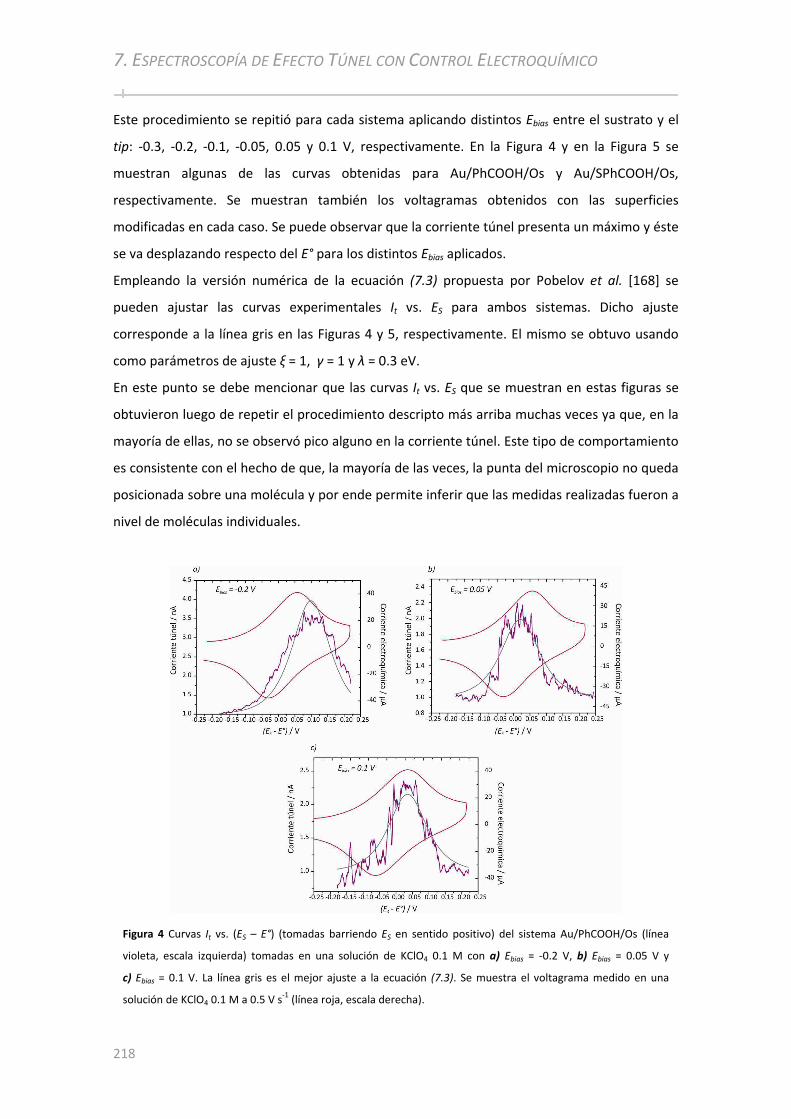
7. ESPECTROSCOPÍA DE EFECTO TÚNEL CON CONTROL ELECTROQUÍMICO
218
Este procedimiento se repitió para cada sistema aplicando distintos Ebias entre el sustrato y el
tip: ‐0.3, ‐0.2, ‐0.1, ‐0.05, 0.05 y 0.1 V, respectivamente. En la Figura 4 y en la Figura 5 se
muestran algunas de las curvas obtenidas para Au/PhCOOH/Os y Au/SPhCOOH/Os,
respectivamente. Se muestran también los voltagramas obtenidos con las superficies
modificadas en cada caso. Se puede observar que la corriente túnel presenta un máximo y éste
se va desplazando respecto del E° para los distintos Ebias aplicados.
Empleando la versión numérica de la ecuación (7.3) propuesta por Pobelov et al. [168] se
pueden ajustar las curvas experimentales It vs. ES para ambos sistemas. Dicho ajuste
corresponde a la línea gris en las Figuras 4 y 5, respectivamente. El mismo se obtuvo usando
como parámetros de ajuste ¨ = 1, á = 1 y ゜ = 0.3 eV.
En este punto se debe mencionar que las curvas It vs. ES que se muestran en estas figuras se
obtuvieron luego de repetir el procedimiento descripto más arriba muchas veces ya que, en la
mayoría de ellas, no se observó pico alguno en la corriente túnel. Este tipo de comportamiento
es consistente con el hecho de que, la mayoría de las veces, la punta del microscopio no queda
posicionada sobre una molécula y por ende permite inferir que las medidas realizadas fueron a
nivel de moléculas individuales.
Figura 4 Curvas It vs. (ES – E°) (tomadas barriendo ES en sentido positivo) del sistema Au/PhCOOH/Os (línea
violeta, escala izquierda) tomadas en una solución de KClO4 0.1 M con a) Ebias = ‐0.2 V, b) Ebias = 0.05 V y
c) Ebias = 0.1 V. La línea gris es el mejor ajuste a la ecuación (7.3). Se muestra el voltagrama medido en una
solución de KClO4 0.1 M a 0.5 V s‐1 (línea roja, escala derecha).
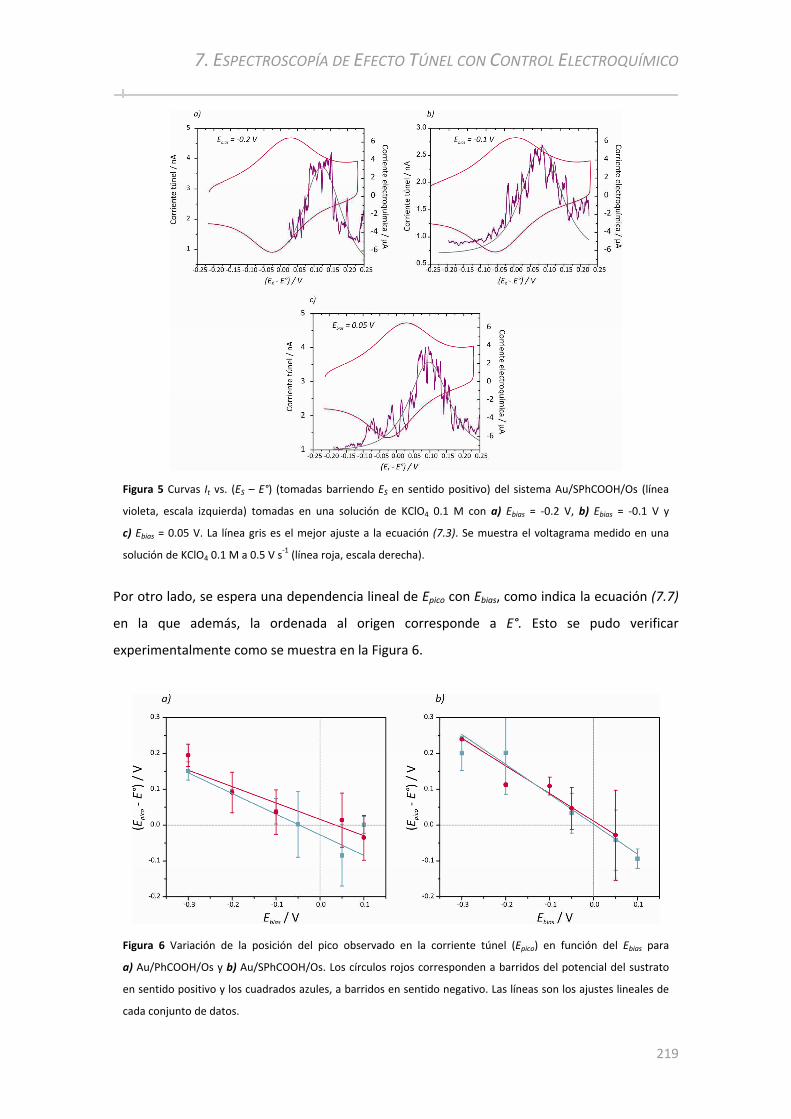
7. ESPECTROSCOPÍA DE EFECTO TÚNEL CON CONTROL ELECTROQUÍMICO
219
Por otro lado, se espera una dependencia lineal de Epico con Ebias, como indica la ecuación (7.7)
en la que además, la ordenada al origen corresponde a E°. Esto se pudo verificar
experimentalmente como se muestra en la Figura 6.
Figura 5 Curvas It vs. (ES – E°) (tomadas barriendo ES en sentido positivo) del sistema Au/SPhCOOH/Os (línea
violeta, escala izquierda) tomadas en una solución de KClO4 0.1 M con a) Ebias = ‐0.2 V, b) Ebias = ‐0.1 V y
c) Ebias = 0.05 V. La línea gris es el mejor ajuste a la ecuación (7.3). Se muestra el voltagrama medido en una
solución de KClO4 0.1 M a 0.5 V s‐1 (línea roja, escala derecha).
Figura 6 Variación de la posición del pico observado en la corriente túnel (Epico) en función del Ebias para
a) Au/PhCOOH/Os y b) Au/SPhCOOH/Os. Los círculos rojos corresponden a barridos del potencial del sustrato
en sentido positivo y los cuadrados azules, a barridos en sentido negativo. Las líneas son los ajustes lineales de
cada conjunto de datos.

7. ESPECTROSCOPÍA DE EFECTO TÚNEL CON CONTROL ELECTROQUÍMICO
220
La ordenada al origen (Epico‐E°) obtenida a partir de los ajustes lineales de los datos que se
muestran en la figura fue cero en todos los casos, indicando que efectivamente se cumple lo
predicho por la ecuación (7.7). Además las pendientes obtenidas fueron ≈ 0.6, un valor cercano
al reportado en literatura por otros autores para otros sistemas [168, 452].
Estas observaciones son consistentes con un mecanismo de transferencia electrónica en dos
pasos entre el sustrato y el tip con relajación vibracional parcial que involucra los niveles
moleculares del complejo de osmio. Esto implica además, que el acoplamiento entre la
molécula y los contactos es fuerte (límite adiabático).
7.4 CONCLUSIONES
Los sistemas Au/PhCOOH/Os y Au/SPhCOOH/Os presentan mecanismos de transferencia
electrónica similares cuando se hallan incluidos en la juntura molecular formada por el
sustrato y el tip del microscopio STM. Es decir, una transferencia electrónica en dos pasos,
entre la molécula y los contactos, con relajación vibracional parcial del centro redox.
Se han reportado resultados similares para complejos de osmio unidos a superficies de oro y
platino a través de una piridina [452], en soluciones acuosas de electrolitos, y a través de un
tiol en un líquido iónico [463]. Sin embargo, ésta es la primera vez que se analiza este tipo de
sistemas unidos por un enlace Au‐C, demostrando además que este enlace mantiene su
funcionalidad electrónica molecular y electroquímica a partir del comportamiento de
transistor molecular observado en este trabajo.

CAPÍTULO 8
DISCUSIÓN FINAL Y CONCLUSIONES


8. DISCUSIÓN FINAL Y CONCLUSIONES
223
l objetivo general de este trabajo de tesis consistió en el análisis de los mecanismos
involucrados en la transferencia electrónica entre un par electroactivo y la superficie de
un electrodo cuando los centros redox se encuentran confinados sobre dicha superficie. Para
ello, se escogió como sistema modelo el par Os(II)/Os(III) (en el complejo [Os(2,2´‐bpy)2Cl(py‐
CH2‐NH2)]+) que se depositó sobre la superficie de electrodos de oro por post‐funcionalización
de capas delgadas o monocapas moleculares previamente formadas sobre los mismos por dos
metodologías diferentes:
a) adsorción de n‐alcanotioles de distintos largos de cadena y de ácido 4‐mercaptobenzoico
(tiol aromático) y,
b) reducción electroquímica de sales de diazonio
Estas dos estrategias permitieron conectar el par Os(II)/Os(III) con la superficie del electrodo a
través de puentes moleculares de distintas longitudes y naturaleza, controlando de esta
manera el acoplamiento electrónico con la misma. Además, ambas formas de derivatización
permitieron formar dos tipos de unión diferentes con el sustrato: a través de un átomo de
azufre, formando un enlace Au‐S, y a través de un átomo de carbono, formando un enlace
Au‐C.
La caracterización de los sistemas obtenidos por electro‐reducción de sales diazonio constituyó
otro de los objetivos principales de esta tesis, ya que esta técnica de modificación de
superficies es relativamente nueva en comparación con la adsorción de tioles [189, 305].
Asimismo, se buscó comparar la cinética de transferencia electrónica de los sistemas
preparados a partir de cada técnica de derivatización inicial. A continuación se enumeran las
observaciones principales que se desprenden de este trabajo.
8.1 TIOLES Y SALES DE DIAZONIO
Todos los sistemas obtenidos por adsorción de tioles o electro‐reducción de sales de diazonio
se caracterizaron superficialmente por técnicas infrarrojas, especialmente PM‐IRRAS. Los
sistemas obtenidos con las sales de diazonio también fueron analizados por XPS.
En todos los casos se pudieron observar señales que indicaron la presencia de los grupos
funcionales esperados sobre la superficie y, con los tioles, se pudieron estimar cubrimientos
coherentes con monocapas moleculares a partir de la carga asociada a su desorción reductiva.
En particular, los resultados obtenidos de este análisis y de los espectros PM‐IRRAS
permitieron confirmar que los tioles alifáticos forman monocapas más ordenadas y compactas
cuánto mayor es el número de metilenos en la cadena [250].
E

8. DISCUSIÓN FINAL Y CONCLUSIONES
224
Por otro lado, se utilizaron las sales de diazonio derivadas del ácido 4‐aminobenzoico y del
ácido 1‐amino‐2,3,5,6‐tetrafluorobenzoico, respectivamente. Esta última es muy inestable
pero aún así se pudo trabajar con ella, llevando a cabo su síntesis en forma simultánea con su
reducción. En particular este sistema es muy interesante por dos motivos. El primero es que, al
tener todas las posiciones del anillo bloqueadas, permite depositar monocapas de ácido
2,3,5,6‐tetrafluorobenzoico sobre el sustrato. Como se mencionó en el Capítulo 3, durante la
electro‐reducción, los radicales arilo pueden unirse a las posiciones orto al grupo –COOH de los
anillos que ya se han unido a la superficie del electrodo. Esto provoca la formación de capas
moleculares ramificadas, que son de un espesor mayor al asociado a una monocapa. Este
ataque radicalario no puede ocurrir cuando el anillo tiene las posiciones bloqueadas [343],
como en este caso.
El segundo motivo de interés en este sistema consiste en la presencia de los fluoruros que
facilitan su análisis por XPS, ya que la señal de F 1s se diferencia perfectamente de las señales
debidas a contaminación superficial, que enmascaran las señales de C 1s y O 1s de la muestra.
Con ambas sales de diazonio se determinaron las condiciones de electro‐deposición de capas
delgadas y gruesas (o multicapas) y se encontró que cuando se aplican potenciales muy
negativos respecto al potencial de equilibrio, las moléculas se pueden unir a través del enlace
Au‐C o a través del enlace Au‐N=N. Cuando el potencial es menos extremo, la señal de N 1s
disminuye indicando que la mayor parte de las moléculas se une por el átomo de carbono y
esto permite pensar en la formación de una monocapa. Este comportamiento también se
confirmó por PM‐IRRAS, a partir de la observación (o no) de una señal a 1300 cm‐1 que se
puede adjudicar al modo ` N=N.
8.2 TRANSFERENCIA ELECTRÓNICA
La velocidad de transferencia electrónica depende del acoplamiento electrónico entre el donor
y el aceptor de electrones (en este caso, la superficie del electrodo y el par electroactivo) y de
diferentes factores que hacen al medio en el que ocurre esta transferencia. El acoplamiento
electrónico puede variar según la naturaleza del donor y del aceptor, la distancia que medie
entre ambos, la naturaleza del puente molecular que los une y del enlace de dicho puente con
cada una de las partes. A su vez cuando los sitios electroactivos se encuentran confinados
sobre una superficie, como en los sistemas aquí analizados, el entorno que rodea a cada uno
de ellos comprende, no sólo el solvente en contacto con el electrodo modificado y el
electrolito soporte, sino también, las especies vecinas que están presentes sobre la superficie.

8. DISCUSIÓN FINAL Y CONCLUSIONES
225
8.2.1 DISTANCIA DONOR‐ACEPTOR Y NATURALEZA DEL PUENTE MOLECULAR
En este trabajo la naturaleza del donor y del aceptor de electrones no se modificó: siempre se
utilizaron superficies de oro y el par Os(II)/Os(III). Sin embargo, sí se analizó la dependencia del
acoplamiento electrónico con la distancia entre ambos y con la naturaleza del puente
molecular. Asimismo, se estudió el efecto del tipo de unión a la superficie (Au‐C vs. Au‐S).
Para llevar a cabo la caracterización de la transferencia electrónica se comenzó utilizando
voltametría cíclica. Sin embargo, se observó que la misma se ve limitada por la constante de
celda cuando se quiere analizar sistemas en los que el puente molecular tiene una longitud
menor a 20 Å. Por lo tanto, se completó el análisis cinético empleando espectroscopía de
impedancia electroquímica y electrodos más pequeños, preparados por fusión de delgados
alambres de oro (en lugar de los obtenidos por electro‐evaporación de películas de oro sobre
obleas de silicio que se venían utilizando).
Figura 1 Comparación entre los datos de cinética de transferencia electrónica obtenidos por Espectroscopía de
Impedancia Electroquímica (círculos verdes) y por Voltametría Cíclica (círculos violetas).

8. DISCUSIÓN FINAL Y CONCLUSIONES
226
En la Figura 1 se compara la variación de k0 con la distancia obtenida con cada una de las
técnicas de caracterización electroquímica. Se observa claramente que la constante cinética
obtenida por voltametría cíclica deja de aumentar a partir del sistema Au/MUA/Os, aún
cuando la distancia entre los sitios electroactivos y la superficie disminuye a medida que se
acorta la longitud del puente. Por el contrario, los datos obtenidos por impedancia
electroquímica muestran un importante aumento de k0 a medida que la distancia electrodo‐
sitio electroactivo disminuye. En particular, se obtuvo un parámetro de tuneleo para los
sistemas alifáticos coherente con un mecanismo de tuneleo a través del enlace.
Por otro lado, las constantes obtenidas con los sistemas aromáticos son coherentes con la
corta longitud de los puentes moleculares pero, si bien los tres sistemas se diferencian entre sí
en la longitud del puente y en el tipo de unión al sustrato, no se evidencian diferencias
significativas entre ellos. Este hecho fue adjudicado a la duración de la perturbación más corta
que puede aplicar el potenciostato empleado en esta tesis. El tiempo de respuesta del mismo
no es lo suficientemente corto como para poder asegurar que las constantes medidas no están
limitadas por el mismo. El desafío a futuro es contar con técnicas más rápidas que permitan
acceder con precisión a procesos que ocurran en una escala de tiempo menor a 10‐5 segundos.
8.2.2 INTERFAZ ELECTRODO‐SOLUCIÓN
En los sistemas estudiados en esta tesis, la transferencia electrónica de cada centro redox se
verá afectada por los sitios redox vecinos y por los grupos –COOH que no fueron post‐
funcionalizados. Para analizar el efecto de cada uno de estos factores, se trabajó con distintos
cubrimientos superficiales del complejo de osmio, diferentes pH y diferentes fuerzas iónicas.
8.2.2.1 EFECTO DEL CUBRIMIENTO SUPERFICIAL DE LOS CENTROS ELECTROACTIVOS
Al post‐funcionalizar monocapas mixtas de ácidos n‐mercapto‐alcanoicos y n‐alcanotioles con
el complejo de osmio, se obtuvieron sistemas con diferentes cubrimientos superficiales del
mismo y con distintas relaciones Os:COOH. Es decir, al aumentar el cubrimiento superficial del
complejo, se observaron eficiencias de post‐funcionalización menores, aumentando la
cantidad relativa de grupos ácidos respecto de sitios electroactivos.
Se trabajó con un tiol largo (16 C, MHDA) y con uno corto (6 C, MHA), diluidos con tioles de
longitudes adecuadas (1‐dodecanotiol y 1‐hexanotiol, respectivamente). Por lo tanto se tuvo
en cuenta que los tioles forman monocapas con distinto grado de empaquetamiento según su

8. DISCUSIÓN FINAL Y CONCLUSIONES
227
largo de cadena y así se consideró el posible efecto que esto pudiera tener sobre el
comportamiento analizado.
De esta manera, se ubicó el complejo de osmio sobre la superficie, a dos distancias diferentes
y rodeado de tioles terminados en –CH3 (por el tiol diluyente) y en –COOH (por los grupos no
post‐funcionalizados). El análisis por voltametría cíclica confirmó que al aumentar el
cubrimiento del complejo en la superficie, aumentan las interacciones laterales entre los sitios
electroactivos, al observarse un aumento del ancho de pico a media altura.
Asimismo, se puso de manifiesto la importancia de la polaridad del entorno que rodea a un
sitio electroactivo a partir de dos hechos que se detallan a continuación. En primer lugar, el
potencial formal del par no varía con el cubrimiento superficial del complejo de osmio ya que,
gracias a la menor eficiencia de la post‐funcionalización, hay una mayor proporción de grupos
ácidos en la superficie que ejercen un mayor apantallamiento de las interacciones entre los
centros de osmio.
El otro hecho significativo es la disminución de la constante cinética de transferencia
electrónica con el cubrimiento superficial de complejo. Esta observación se atribuyó a que una
mayor polaridad de la monocapa tiene una contribución positiva a la energía de
reorganización. Es decir, la energía de reorganización aumenta cuánto mayor es la polaridad
de la película depositada sobre el electrodo, ya que esto implica mayores interacciones
electrostáticas entre las especies que acompañan la electro‐reducción de lo sitios redox. De
esta manera se evidencia la importante contribución de las especies vecinas a la energía de
reorganización necesaria para la transferencia electrónica cuando se tiene sistemas redox
confinados a una superficie.
8.2.2.2 EFECTO DEL ELECTROLITO SOPORTE Y DEL PH
Para completar el análisis del efecto de los grupos ácidos sobre la transferencia electrónica
entre los sitios redox y la superficie del electrodo, se analizó el comportamiento del par a
dieferentes pH y fuerzas iónicas. Esto se consideró necesario dado que estas especies pueden
modificar su estado de ionización con el pH y afectar la densidad de carga de la superficie que,
a su vez, repercute sobre el potencial en el plano de transferencia de carga. Asimismo, la
fuerza iónica sirve para regular este efecto debido al apantallamiento que pueden ejercer los
iones en solución de las cargas confinadas en la superficie.
Este análisis complementa la caracterización electroquímica general realizada de todos los
sistemas bajo estudio y pone de manifiesto la complejidad que poseen los mismos, al
presentar grupos con propiedades ácido‐base que pueden intervenir en el estado de carga de

8. DISCUSIÓN FINAL Y CONCLUSIONES
228
la superficie y en las interacciones intermoleculares que existen sobre la misma. Es decir, en
estos sistemas existe un acoplamiento entre el equilibrio ácido‐base y el equilibrio redox.
8.2.3 UNIÓN A LA SUPERFICIE: AU‐C VS. AU‐S
En esta tesis se empleó Espectroscopía de Efecto Túnel con control electroquímico para
profundizar el análisis de los sistemas Au/PhCOOH/Os y Au/SPhCOOH/Os que se diferencian
entre sí en la unión al sustrato: Au‐C y Au‐S, respectivamente. Esta técnica permitió analizar la
transferencia electrónica a través de un número reducido de moléculas en cada sistema. A
partir de este estudio se pudo establecer que ambos presentan mecanismos similares de TE.
Se demostró que el tuneleo entre la punta del microscopio y el sustrato ocurre a través de las
moléculas que quedan ubicadas entre ambos, observándose una transferencia en dos pasos
con relajación vibracional parcial. Este resultado ha sido reportado previamente para sistemas
que poseen el par Os(II)/Os(III) unida al sustrato por piridinas y tioles [452, 461], pero no
cuando la unión es Au‐C. Por lo tanto, se pudo demostrar que se pueden unir puentes
moleculares directamente a sustratos de oro a través de la unión Au‐C, manteniendo intacto el
comportamiento electrónico y la funcionalidad electroquímica.
Este resultado es consistente con las constantes cinéticas obtenidas por espectroscopía de
impedancia electroquímica, que no tienen diferencias significativas entre sí aunque, como ya
se mencionó, estás constantes deberían ser corroboradas.
En conclusión, en este trabajo de tesis:
1) Se realizó una caracterización superficial a nivel molecular de los sistemas modelo
empleando técnicas infrarrojas de superficie (FT‐IRRAS y PM‐IRRAS), espectroscopía Raman
resonante, espectroscopía XPS y microscopía de efecto túnel.
2) Se verificó la dependencia de la velocidad de transferencia electrónica con la longitud de
cadena en los sistemas formados con tioles alifáticos, observándose un mecanismo de tuneleo
a través del enlace.
3) El análisis cinético de los sistemas derivados de la reducción de sales de diazonio y del tiol
aromático rindió constantes cinéticas cuyo orden de magnitud es consistente con la distancia a
la que se encuentran los sitios redox de la superficie. Sin embargo, dicho análisis posee cierto
error generado por el tiempo de respuesta del potenciostato empleado en el mismo.
Asimismo, se observó que los resultados reportados por Gooding y colaboradores para
sistemas similares a los mencionados, tienen un error apreciable que se puede atribuir a la
limitación de las medidas por la constante de celda (RuCdl).

8. DISCUSIÓN FINAL Y CONCLUSIONES
229
4) Se analizó la regulación de carga en la superficie del electrodo que viene dada por el
acoplamiento entre el equilibrio ácido‐base y el equilibrio redox. La variación del potencial de
electrodo afecta el estado de ionización de los grupos ácidos en la superficie y, a su vez, el pH
de la solución en contacto con la misma modifica el potencial formal de los sitios redox
confinados sobre ella.
5) Se observó que, cuando se tienen especies electroactivas confinadas a una superficie, la
energía de reorganización externa (゜o) tiene contribuciones de las interacciones electrostáticas
no locales (de largo alcance) entre los grupos redox, las especies ácido‐base, los contraiones, y
el solvente en el estado de transición.
6) Se empleó espectroscopía de efecto túnel con control electroquímico para analizar, a nivel
de molécula individual, dos sistemas en los que la única diferencia fue la unión al sustrato
(Au‐S y Au‐C) y se observó un mecanismo de transferencia electrónica en dos etapas con
relajación vibracional parcial del centro redox.
La perspectiva a futuro más importante que surge de esta tesis es superar la limitación
impuesta a las medidas de cinética de transferencia electrónica por el tiempo de respuesta del
potenciostato. Para ello, se está considerando implementar métodos basados en saltos de
temperatura (como el reportado por Smalley et al. [418, 467]) y/o métodos coulostáticos [432,
468, 469].

8. DISCUSIÓN FINAL Y CONCLUSIONES
230
Los resultados de esta tesis han sido parcialmente publicados en los siguientes artículos:
“Electrochemical scanning tunneling spectroscopy of redox active molecules bound by Au‐C
bonds.” A. M. Ricci, E. J. Calvo, S. Martin and R. J. Nichols, J. Am. Chem. Soc., 2010, 132, 2494‐
2495 (artículo elegido como arte de tapa).
“Redox molecule based SERS sensors.” N.G. Tognalli, P. Scodeller, V. Flexer, R. Szamocki, A.
Ricci, M. Tagliazucchi, E. J. Calvo and A. Fainstein, Phys. Chem. Chem. Phys., 2009, 11, 7412–
7423.
“The binding between carbon and the Au(111) surface, and what makes it different from the S‐
Au(111) bond.” E. de la Llave, A.M. Ricci, E.J. Calvo, D. Scherlis, J. Phys. Chem. C, 2008, 112,
17611‐17617.
“Electron transfer at Au surfaces modified by Tethered Osmium bipyridine‐pyridine complexes.”
Ricci A.M., Rothacher S., Rolli C.G., Baraldo L., Bonazzola C., Calvo E.J., Tognalli, N., Fainstein A.,
J. Solid State Electrochem. 2007, 11, 1511‐1520.
“An FT‐IRRAS study of nitrophenyl mono‐ and multilayers electro‐deposited on gold by
reduction of the diazonium salt.” A. Ricci, C. Bonazzola, and E. J. Calvo, Phys. Chem. Chem.
Phys. 2006, 8, 4297‐4299.

BIBLIOGRAFÍA
231
BIBLIOGRAFÍA
[1] Handbook of Nanoscience, Engineering and Technology, W.A. Goddard III, D.W. Brenner,
S.E. Lyshevski, G.J. Iafrate, Taylor & Francis Group, Boca Raton, 2007.
[2] Introducing Molecular Electronics, G. Cuniberti, G. Fagas, K. Richter, Springer, Berlin
Heidelberg, 2005.
[3] Molecular Electronics, J.R. Reimers, C. Picconatto, R. Shashidhar, Academy of Sciences, New
York, 2003.
[4] Molecular rectifiers, A. Aviram, M.A. Ratner, Chem. Phys. Lett., 29, 1974, 277.
[5] Penetration‐controlled reactions in organized monolayer assemblies. 1. Aqueous
permanganate interaction with monolayer and multilayer films of long‐chain surfactants, R.
Maoz, J. Sagiv, Langmuir, 3, 1987, 1034.
[6] Adsorption of bifunctional organic disulfides on gold surfaces, R.G. Nuzzo, D.L. Allara, J. Am.
Chem. Soc., 105, 1983, 4481.
[7] Formation of monolayers by the coadsorption of thiols on gold: variation in the head group,
tail group, and solvent, C.D. Bain, J. Evall, G.M. Whitesides, J. Am. Chem. Soc., 111, 1989, 7155.
[8] Surface Studies by Scanning Tunneling Microscopy, G. Binning, H. Rohrer, C. Gerber, E.
Weibwl, Phys. Rev. Lett., 49, 1982, 57.
[9] Introduction to Scanning Tunneling Microscopy, C.J. Chen, Oxford University Press, Oxford,
UK, 1993.
[10] Advances in Chemical Physics, J. Jortner, M. Bixon, I. Prigogine, S. Rice, Wiley, 106. New
York, 1999.
[11] Charge Transfer in Physics, Chemistry and Biology, A.M. Kuznetsov, Gordon & Breach, New
York, 1995.
[12] Electron Transfer in Chemistry and Biology: An Introduction to the Theory A.M. Kuznetsov,
J. Ulstrup, Wiley, New York, 1998.
[13] Charge Transfer on the Nanoscale: Current Status, D.M. Adams, L. Brus, C.E.D. Chidsey, S.
Creager, C. Creutz, C.R. Kagan, P.V. Kamat, M. Lieberman, S. Lindsay, R.A. Marcus, R.M.
Metzger, M.E. Michel‐Beyerle, J.R. Miller, M.D. Newton, D.R. Rolison, O. Sankey, K.S. Schanze,
J. Yardley, X. Zhu, J. Phys. Chem. B 107, 2003, 6668.
[14] Electron Transfer in Chemistry, V. Balzani, Wiley‐VCH, New York, 2001.

BIBLIOGRAFÍA
232
[15] Electron conduction in molecular wires. II. Application to scanning tunneling microscopy, V.
Mujica, M. Kemp, M.A. Ratner, J. Chem. Phys., 101, 1994, 6856.
[16] Electron Transmission through Molecules and Molecular Interfaces, A. Nitzan, Annu. Rev.
Phys. Chem., 52, 2001, 681.
[17] Activated Conduction in Microscopic Molecular Junctions, D. Segal, A. Nitzan, M.A. Ratner,
W.B. Davis, J. Phys. Chem. B, 104, 2000, 2790.
[18] Functional nanoparticle architectures for sensoric, optoelectronic, and bioelectronic
applications, I. Willner, B. Willner, Pure Appl. Chem., 74, 2002, 1773.
[19] Molecular‐scale interface engineering for polymer light‐emitting diodes, P.K.H. Ho, J.S.
Kim, J.H. Burroughes, H. Becker, S.F.Y. Li, T.M. Brown, F. Cacialli, R.H. Friend, Nature, 404,
2000, 481.
[20] Anion Effect on Mediated Electron Transfer through Ferrocene‐Terminated Self‐Assembled
Monolayers, G. Valincius, G. Niaura, B. Kazakeviciene, Z. Talaikyte, M. Kazemekaite, E. Butkus,
V. Razumas, Langmuir, 20, 2004, 6631.
[21] Bioelectrocatalysis‐based dihydrogen/dioxygen fuel cell operating at physiological pH S.
Tsujimura, M. Fujita, H. Tatsumi, K. Kano, T. Ikeda, Phys. Chem. Chem. Phys., 3, 2001, 1331.
[22] Bridge‐Assisted Ultrafast Interfacial Electron Transfer to Nanocrystalline SnO2 Thin Films,
N.A. Anderson, X. Ai, D.T. Chen, D.L. Mohler, T.Q. Lian, J. Phys. Chem. B, 107, 2003, 14231.
[23] A study by contact angle of the acid‐base behavior of monolayers containing .omega.‐
mercaptocarboxylic acids adsorbed on gold: an example of reactive spreading, C.D. Bain, G.M.
Whitesides, Langmuir 5, 1989, 1370.
[24] Coadsorption of ferrocene‐terminated and unsubstituted alkanethiols on gold:
electroactive self‐assembled monolayers, C.E.D. Chidsey, C.R. Bertozzi, P.T. M., A.M. Mujsce, J.
Am. Chem. Soc., 112, 1990, 4301.
[25] Synthesis, Structure, and Properties of Model Organic Surfaces, D. Dubois, R.G. Nuzzo,
Annu. Rev. Phys. Chem., 43, 1992, 437.
[26] Charge Transport through Self‐Assembled Monolayers of Compounds of Interest in
Molecular Electronics, F.R.F. Fan, J. Yang, L. Cai, D.W. Price, S.M. Dirk, D. Kosynkin, Z. Yao, A.M.
Rawlett, J.M. Tour, A.J. Bard, J. Am. Chem. Soc., 124, 2002, 5550.
[27] Electrochemistry of Organized Monolayers of Thiols and Related Molecules on Electrodes,
Electroanalytical Chemistry, H.O. Finklea, Vol. 19, A.J. Bard, I. Rubinstein, Eds.; Marcel Dekker:
New York, 1996.
[28] Molecular Electronics. Synthesis and Testing of Components, J.M. Tour, Acc. Chem. Res.,
33, 2000, 791.

BIBLIOGRAFÍA
233
[29] Electron conduction in molecular wires. I. A scattering formalism, V. Mujica, M. Kemp,
M.A. Ratner, J. Chem. Phys., 101, 1994, 6849.
[30] First‐principles analysis of molecular conduction using quantum chemistry software, P.
Damle, A.W. Ghosh, S. Datta, Chem. Phys., 281, 2002, 171.
[31] Theory of Electron Transfer Reactions: Insights and Hindsights, N. Sutin, Prog. Inorg.
Chem., 30, 1983, 441.
[32] Electron Transfer Reactions in Condensed Phases, M.D. Newton, N. Sutin, Annu. Rev. Phys.
Chem., 35, 1984, 437.
[33] Interfacial Electron‐Transfer Kinetics of Ferrocene through Oligophenyleneethynylene
Bridges Attached to Gold Electrodes as Constituents of Self‐Assembled Monolayers:
Observation of a Nonmonotonic Distance Dependence, J.F. Smalley, S.B. Sachs, C.E.D. Chidsey,
S.P. Dudek, H.D. Sikes, S.E. Creager, C.J. Yu, S.W. Feldberg, M.D. Newton, J. Am. Chem. Soc.,
126, 2004, 14620.
[34] Investigation of the mass transport process in the voltammetry of cytochrome c at 4,4'‐
bipyridyl disulfide modified stationary and rotated macro‐ and microdisk gold electrodes, A.M.
Bond, A.O. Hill, S. Komorsky‐Lovric, M. Lovric, M.E. McCarthy, I.S.M. Psalti, N.J. Walton, J. Phys.
Chem., 96, 1992, 8100.
[35] Dependence of electron transfer kinetics of the ferrocene/ferrocenium couple on the
viscosity in ambient temperature chloroaluminate ionic liquids, Z.J. Karpinski, S. Song, R.A.
Osteryoung, Inorg. Chim. Acta, 225, 1994, 9.
[36] Modulation of Heterogeneous Electron‐Transfer Dynamics Across the Electrode/Monolayer
Interface, D.A. Walsh, T.E. Keyes, R.J. Forster, J. Phys. Chem. B, 108, 2004, 2631.
[37] Electron Tunneling Across Hexadecanethiolate Monolayers on Mercury Electrodes:
Reorganization Energy, Structure, and Permeability of the Alkane/Water Interface, K. Slowinski,
K.U. Slowinska, M. Majda, J. Phys. Chem. B, 103, 1999, 8544.
[38] Nanowire‐Based Molecular Monolayer Junctions: Synthesis, Assembly, and Electrical
Characterization, L.T. Cai, H. Skulason, J.G. Kushmerick, S.K. Pollack, J. Naciri, R. Shashidhar,
D.L. Allara, T.E. Mallouk, T.S. Mayer, J. Phys. Chem. B, 108, 2004, 2827.
[39] Measurement of Single‐Molecule Resistance by Repeated Formation of Molecular
Junctions, B.Q. Xu, N.J. Tao, Science, 301, 2003, 1221.
[40] Elastic and Inelastic Electron Tunneling in Alkane Self‐Assembled Monolayers, W.Y. Wang,
T. Lee, M.A. Reed, J. Phys. Chem. B, 108, 2004, 18398.
[41] Molecular Electronic Junctions, R. McCreery, Chem. Mater., 16, 2004, 4477.

BIBLIOGRAFÍA
234
[42] Overview of nanoelectronic devices, D. Goldhaber‐Gordon, M.S. Montemerlo, J.C. Love,
G.J. Opiteck, J.C. Ellenbogen, Proceedings of the IEEE, 85, 1997, 521.
[43] Metal‐metal interactions in binuclear complexes exhibiting mixed valency; molecular wires
and switches, M.D. Ward, Chem. Soc. Rev., 24, 1995, 121.
[44] Intervalence Electron Transfer in Mixed Valence Diferrocenylpolyenes. Decay Law of the
Metal‐Metal Coupling with Distance, A.‐C. Ribou, J.‐P. Launay, M.L. Sachtleben, H. Li, C.W.
Spangler, Inorg. Chem., 35, 1996, 3735.
[45] Electron transfer and energy transfer through bridged systems. 4. Intermetallic coupling
and electronic spectra of the bis(penta‐ammineruthenium) complexes of alpha, omega.‐
dipyridyl trans‐polyenes in deuterium oxide, J.R. Reimers, N.S. Hush, Inorg. Chem., 29, 1990,
3686.
[46] Conjugated Macromolecules of Precise Length and Constitution. Organic Synthesis for the
Construction of Nanoarchitectures, J.M. Tour, Chem. Rev., 96, 1996, 537.
[47] Structural and Physical Properties of Delocalized Mixed‐Valent [Cp*M(pentalene)M'Cp*]n+
and [Cp*M(indacene)M'Cp*]n+ (M, M' = Fe, Co, Ni; n = 0, 1, 2) Complexes, J.M. Manriquez,
M.D. Ward, W.M. Reiff, J.C. Calabrese, N.L. Jones, P.J. Carroll, E.E. Bunel, J.S. Miller, J. Am.
Chem. Soc., 117, 1995, 6182.
[48] Intervalence Electron Transfer in Pentaammineruthenium Complexes of Dipyridylpolyenes,
Dipyridylthiophene, and Dipyridylfuran, A.‐C. Ribou, J.‐P. Launay, K. Takahashi, T. Nihira, S.
Tarutani, C.W. Spangler, Inorg. Chem., 33, 1994, 1325.
[49] Chemistry and Physics in One Dimension: Synthesis and Properties of Nanowires and
Nanotubes, J. Hu, T.W. Odom, C.M. Lieber, Acc. Chem. Res., 32, 1999, 435.
[50] Spin‐Selective Charge Transport Pathways through p‐Oligophenylene‐Linked
Donor−Bridge−Acceptor Molecules, A.M. Scott, T. Miura, A.B. Ricks, Z.E.X. Dance, E.M.
Giacobbe, M.T. Colvin, M.R. Wasielewski, J. Am. Chem. Soc., 131, 2009, 17655–17666.
[51] Solvent and double‐layer effects on redox reactions in self‐assembled monolayers of
ferrocenyl—alkanethiolates on gold, S.E. Creager, G.K. Rowe, J. Electroanal. Chem., 420, 1997,
291.
[52] Free Energy and Temperature Dependence of Electron Transfer at the Metal‐Electrolyte
Interface, C.E.D. Chidsey, Science, 251, 1991, 919.
[53] Electroanalytical Chemistry, S.W. Feldberg, M.D. Newton, J.F. Smalley, Vol. A.J. Bard, I.
Rubinstein, Eds.; Marcel Dekker: New York, 2003.
[54] Electrochemical Methods, A.J. Bard, L.R. Faulkner, John Wiley and Sons, New York, 2001.

BIBLIOGRAFÍA
235
[55] Electron Transfer through Organic Molecules, L.A. Bumm, J.J. Arnold, T.D. Dunbar, D.L.
Allara, P.S. Weiss, J. Phys. Chem. B, 103, 1999, 8122.
[56] Insertion, Conductivity, and Structures of Conjugated Organic Oligomers in Self‐Assembled
Alkanethiol Monolayers on Au{111}, M.T. Cygan, T.D. Dunbar, J.J. Arnold, L.A. Bumm, N.F.
Shedlock, T.P. Burgin, L. Jones, D.L. Allara, J.M. Tour, P.S. Weiss, J. Am. Chem. Soc., 120, 1998,
2721.
[57] Electrochemistry of chemisorbed molecules. II. Influence of charged chemisorbed
molecules on the electrode reactions of platinum complexes, R.F. Lane, A.T. Hubbard, J. Phys.
Chem., 77, 1973, 1411.
[58] Electrochemistry of chemisorbed molecules. I. Reactants connected to electrodes through
olefinic substituents, R.F. Lane, A. T. Hubbard, J. Phys. Chem., 77, 1973, 1401.
[59] Chemically modified tin oxide electrode, P.R. Moses, L. Wier, R.W. Murray, Anal.Chem., 47,
1975, 1882.
[60] Chiral electrode, B.F. Watkins, J.R. Behling, E. Kariv, J. Am. Chem. Soc., 97, 1975, 3549.
[61] Chemically modified electrodes, R.W. Murray, Acc. Chem. Res., 13, 1980, 135.
[62] Electroactive Polymers and Macromolecular Electronics, C.E.D. Chidsey, R.W. Murray,
Science, 231, 1986, 25.
[63] Encyclopedia of Electrochemistry, M.Fujihira, I.Rubinstein, J.F.Rusling, Wiley‐VCH Verlag
GmbH & Co., 10. Weinheim, 2007.
[64] Molecular Design of Electrode Surfaces, R.W. Murray, A. Weissberger, Wiley‐Interscience,
22. New York, 1992.
[65] Electrochemical Properties of an Osmium(II) Copolymer Film and Its Electrocatalytic Ability
Towards the Oxidation of Ascorbic Acid in Acidic and Neutral pH, K. Foster, T. McCormac,
Electroanal., 18, 2006, 1097.
[66] Electrocatalytic activity of a cobalt hexacyanoferrate modified glassy carbon electrode
toward ascorbic acid oxidation, C.‐X. Cai, K.‐H. Xue, S.‐M. Xu, J. Electroanal. Chem., 486, 2000,
111.
[67] Biofuel cells‐Recent advances and applications, F. Davis, S.P.J. Higson, Biosens.
Bioelectron., 22, 2007, 1224.
[68] A Miniature Membrane‐less Biofuel Cell Operating at +0.60 V under Physiological
Conditions, N. Mano, F. Mao, A. Heller, Chem. Bio. Chem., 5, 2004, 1703.
[69] A non‐compartmentalized glucose O2 biofuel cell by bioengineered electrode surfaces, E.
Katz, I. Willner, A.B. Kotlya, J. Electroanal. Chem., 479, 1999, 64.

BIBLIOGRAFÍA
236
[70] An enzymatic glucose/O2 biofuel cell: Preparation, characterization and performance in
serum, F. Gao, Y. Yan, L. Su, L. Wang, L. Mao, Electrochem. Commun., 9, 2007, 989.
[71] Self‐assembled monolayers of diphenyl disulphide: a novel cathode material for
rechargeable lithium batteries, T. Maddanimath, Y.B. Khollam, M. Aslam, I.S. Mulla, K.
Vijayamohanan, J. Power Sources, 124, 2003, 133.
[72] Blended polymer electrolytes based on poly(lithium 4‐styrene sulfonate) for the
rechargeable lithium polymer batteries, C.H. Park, Y.‐K. Sun, D.‐W. Kim, Electrochim. Acta, 50,
2004, 375.
[73] Electrochromics for smart windows: Thin films of tungsten oxide and nickel oxide, and
devices based on these, G.A. Niklasson, C.G. Granqvist, J. Mater. Chem., 17, 2007, 127.
[74] Towards the smart window: Progress in electrochromics, C.G. Granqvist, A. Azens, J.
Isidorsson, M. Kharrazi, L. Kullman, T. Lindström, G.A. Niklasson, C.G. Ribbing, D. Rönnow, M.
Stromme Mattsson, M. Veszelei, Journal of Non‐Crystalline Solids, 218, 1997, 273.
[75] Recent advances in electrochromics for smart windows applications, C.G. Granqvist, A.
Azens, A. Hjelm, L. Kullman, G.A. Niklasson, D. Rönnow, M. Stromme Mattsson, M. Veszelei, G.
Vaivars, Solar Energy, 63, 1998, 199.
[76] Electrochromic smart windows: Energy efficiency and device aspects, A. Azens, C.G.
Granqvist, J. Solid State Electrochem., 7, 2003, 64.
[77] Self‐assembled monolayers of p‐toluene and p‐hydroxymethylbenzene moieties adsorbed
on iron by the formation of covalent bonds between carbon and iron atoms for protection of
iron from corrosion, T. Shimura, K. Aramaki, Corros. Sci., 49, 2007, 1378.
[78] Electrochemical properties and corrosion protection of organosilane self‐assembled
monolayers on aluminum 2024‐T3, P.E. Hintze, L.M. Calle, Electrochim. Acta, 51, 2006, 1761.
[79] Corrosion performance of conducting polymer coatings applied on mild steel, P. Ocón, A.B.
Cristobal, P. Herrasti, E. Fatas, Corros. Sci., 47, 2005, 649.
[80] Redox Polymer and Probe DNA Tethered to Gold Electrodes for Enzyme‐Amplified
Amperometric Detection of DNA Hybridization, P. Kavanagh, D. Leech, Anal. Chem., 78, 2006,
2710.
[81] Designing stable redox‐active surfaces: Chemical attachment of an osmium complex to
glassy carbon electrodes prefunctionalized by electrochemical reduction of an in Situ ‐
generated aryldiazonium cation, S. Boland, F. Barriere, D. Leech, Langmuir, 24, 2008, 6351.
[82] Improved stability of redox enzyme layers on glassy carbon electrodes via covalent
grafting, M. Pellissier, F. Barrière, A.J. Downard, D. Leech, Electrochem. Commun., 10, 2008,
835.

BIBLIOGRAFÍA
237
[83] Molecular electronics with single molecules in solid‐state devices, K. Moth‐Poulsen, T.
Björnholm, Nature Nanotech., 4, 2009, 551.
[84] Electrical rectification by a molecule: The advent of unimolecular electronic devices, R.M.
Metzger, Acc. Chem. Res., 32, 1999, 950.
[85] Chemical functionalization of single‐walled carbon nanotube field‐effect transistors as
switches and sensors, S. Liu, Q. Shen, Y. Cao, L. Gan, Z. Wang, M.L. Steigerwald, X. Guo,
Coord.Chem. Rev., 254, 2010, 1101.
[86] Molecular self‐assembled monolayers and multilayers for organic and unconventional
inorganic thin‐film transistor applications, S.A. DiBenedetto, A. Facchetti, M.A. Ratner, T.J.
Marks, Adv. Mater., 21, 2009, 1407.
[87] Light switching of molecules on surfaces, W.R. Browne, B.L. Feringa, Annu. Rev. Phys.
Chem., 60, 2009, 407.
[88] Artificial molecular machines, V. Balzani, A. Credi, F.M. Raymo, J.F. Stoddart, Angew.
Chem. ‐ International Edition, 39, 2000, 3349.
[89] Surface modification in microsystems and nanosystems, S. Prakash, M.B. Karacor, S.
Banerjee, Surf. Sci. Rep., 64, 2009, 233.
[90] Encyclopedia of Electrochemistry, M. Bruening, J. Rusling, Vol. 10, M.Fujihira, I.Rubinstein,
J.F.Rusling, Eds.; Wiley‐VCH Verlag GmbH & Co.: Weinheim, 2007.
[91] Encyclopedia of Electrochemistry, S. Kuwabata, H. Yoneyama, Vol. 10, M.Fujihira,
I.Rubinstein, J.F.Rusling, Eds.; Wiley‐VCH Verlag GmbH & Co. KGaA: Weinheim, 2007.
[92] Encyclopedia of Electrochemistry, M.S. Yokohama, M. Fujihira, Vol. 10, M.Fujihira,
I.Rubinstein, J.F.Rusling, Eds.; Wiley‐VCH Verlag GmbH & Co.: Weinheim, 2007.
[93] Langmuir‐Blodgett Films, G. Roberts, Plenum Press, New York, 1990.
[94] An Introduction to Ultrathin Organic Films from Langmuir‐Blodgett to Self‐Assembly, A.
Ulman, Academic Press, New York, 1991.
[95] Self‐Assembled Monolayers of Thiolates on Metals as a Form of Nanotechnology, J.C. Love,
L.A. Estroff, J.K. Kriebel, R.G. Nuzzo, G.M. Whitesides, Chem. Rev., 105, 2005, 1103.
[96] Self‐assembled monolayers of thiols and dithiols on gold: new challenges for a well‐known
system, C. Vericat, M.E. Vela, G. Benitez, P. Carro, R.C. Salvarezza, Chem. Soc. Rev., 39, 2010,
1805
[97] Attachment of organic layers to conductive or semiconductive surfaces by reduction of
diazonium salts J. Pinson, F. Podvorica, Chem. Soc. Rev., 34, 2005, 429.

BIBLIOGRAFÍA
238
[98] Redox Kinetics in Monolayers on Electrodes: Electron Transfer Is Sluggish for Ferrocene
Groups Buried within the Monolayer Interior, J.J. Sumner, S.E. Creager, J. Phys. Chem. B, 105,
2001, 8739.
[99] “Clicking” Functionality onto Electrode Surfaces, J.P. Collman, N.K. Devaraj, C.E.D. Chidsey,
Langmuir, 20, 2004, 1051.
[100] Effect of Interchain Hydrogen Bonding on Electron Transfer through Alkanethiol
Monolayers Containing Amide Bonds, S. Sek, A. Misicka, R. Bilewicz, J. Phys. Chem. B, 104,
2000, 5399.
[101] Distance Dependence of the Electron Transfer Rate through Oligoglycine Spacers
Introduced into Self‐Assembled Monolayers, S. Sek, A. Sepiol, A. Tolak, A. Misicka, R. Bilewicz, J.
Phys. Chem. B, 108, 2004, 8102.
[102] Long‐Range Heterogeneous Electron Transfer Between Ferrocene and Gold Mediated By
n‐Alkane and N‐Alkyl‐Carboxamide Bridges, J.J. Sumner, K.S. Weber, L.A. Hockett, S.E. Creager,
J. Phys. Chem. B, 104, 2000, 7449.
[103] Influence of Electrolyte Activity on Formal Potentials Measured for Ferrocenylhexanethiol
Monolayers on Gold: Indistinguishable Responses in Aqueous Solutions of HClO4 and NaClO4, J.
Redepenning, J. Flood, Langmuir, 12, 1996, 508.
[104] Interfacial Solvation and Double‐Layer Effects on Redox Reactions in Organized
Assemblies, G.K. Rowe, S.E. Creager, J. Phys. Chem., 98, 1994, 5500.
[105] The kinetics of electron transfer through ferrocene‐terminated alkanethiol monolayers on
gold, J.F. Smalley, S.W. Feldberg, C.E.D. Chidsey, M.R. Linford, M.D. Newton, Y.P. Liu, J. Phys.
Chem., 99, 1995, 13141.
[106] Long‐Range Electronic Coupling between Ferrocene and Gold in Alkanethiolate‐based
Monolayers on Electrodes, K. Weber, L.A. Hockett, S.E. Creager, J. Phys. Chem. B, 101, 1997,
8286.
[107] Submicrosecond Electron Transfer to Monolayer‐Bound Redox Species on Gold Electrodes
at Large Overpotentials, D.B. Robinson, C.E.D. Chidsey, J. Phys. Chem. B, 106, 2002, 10706.
[108] Preparation and reversible behavior of organized thiol monolayers with attached
pentaminepyridineruthenium redox centers, H.O. Finklea, D.D. Hanshew, J. Electroanal. Chem.,
347, 1993, 327.
[109] Electron Transfer in a Hg‐SAM//SAM‐Hg Junction Mediated by Redox Centers, E. Tran,
M.A. Rampi, G.M. Whitesides, Agew. Chem., Int. Ed. Engl., 43, 2004, 3835.
[110] Heterogeneous electron‐transfer kinetics for ruthenium and ferrocene redox moieties
through alkanethiol monolayers on gold, J.F. Smalley, H.O. Finklea, C.E.D. Chidsey, M.R.

BIBLIOGRAFÍA
239
Linford, S.E. Creager, J.P. Ferraris, K. Chalfant, T. Zawodzinsk, S.W. Feldberg, M.D. Newton, J.
Am. Chem. Soc., 125, 2003, 2004.
[111] Electrolyte and temperature effects on long range electron transfer across self‐assembled
monolayers, H.O. Finklea, M.S. Ravenscroft, D.A. Snider, Langmuir, 9, 1993, 223.
[112] Electron‐transfer kinetics in organized thiol monolayers with attached
pentaammine(pyridine)ruthenium redox centers, H.O. Finklea, D.D. Hanshew, J. Am. Chem.
Soc., 114, 1992, 3173.
[113] Kinetics of electron transfer to attached redox centers on gold electrodes in nonaqueous
electrolytes, M.S. Ravenscroft, H.O. Finklea, J. Phys. Chem., 98, 1994, 3843.
[114] Influence of Donnan potentials on apparent formal potentials measured for organized
thiol monolayers with attached pentaamminepyridineruthenium redox centers, J. Redepenning,
H.M. Tunison, H.O. Finklea, Langmuir, 9, 1993, 1404.
[115] Controlling the electron transfer mechanism in metal‐molecules‐metal junctions, E. Tran,
C. Grave, G.M. Whitesides, M.A. Rampi, Electrochim. Acta, 50, 2005, 4850.
[116] Electrochemical and electrogenerated chemiluminescence properties of tris(2,2'‐
bipyridine)ruthenium(II)‐tridecanethiol derivative on ITO and gold electrodes, Y. Sato, K. Uosaki,
J. Electroanal. Chem., 384, 1995, 57.
[117] A new redox site as an alternative to ferrocene to study electron transfer in self‐
assembled monolayers, C. Hortholary, F. Minc, C. Coudret, J. Bonvoisin, J.‐P. Launay, Chem.
Commun., 2002, 1932
[118] Synthesis and Characterization of Redox‐Active Metal Complexes Sequentially Self‐
Assembled onto Gold Electrodes via a New Thiol‐Terpyridine Ligand, M. Maskus, H.D. Abruña,
Langmuir, 12, 1996, 4455.
[119] A stability comparison of redox‐active layers produced by chemical coupling of an
osmium redox complex to pre‐functionalized gold and carbon electrodes, S. Boland, K. Foster,
D. Leech, Electrochim. Acta, 54, 2009, 1986.
[120] Ionic interactions play a major role in determining the electrochemical behavior of self‐
assembling viologen monolayers, H.C. De Long, D.A. Buttry, Langmuir, 6, 1990, 1319.
[121] Environmental effects on redox potentials of viologen groups embedded in electroactive
self‐assembled monolayers, H.C. De Long, D.A. Buttry, Langmuir, 8, 1992, 2491.
[122] Electron transfer in self‐assembled monolayers of N‐methyl‐N'‐carboxyalkyl‐4,4'‐
bipyridinium linked to gold electrodes, E. Katz, N. Itzhak, I. Willner, Langmuir, 9, 1993, 1392.

BIBLIOGRAFÍA
240
[123] Kinetic Separation of Amperometric Responses of Composite Redox‐Active Monolayers
Assembled onto Au Electrodes: Implications to the Monolayers' Structure and Composition, E.
Katz, I. Willner, Langmuir, 13, 1997, 3364.
[124] An electroreflectance and photoelectrochemical study of hypericin at bare and modified
gold electrodes, T. Sagara, H. Kawamura, K. Ezoe, N. Nakashima, J. Electroanal. Chem., 445,
1998, 171.
[125] Estimation of average orientation of surface‐confined chromophores on electrode
surfaces using electroreflectance spectroscopy, T. Sagara, N. Kaba, M. Komatsu, M. Uchida, N.
Nakashima, Electrochim. Acta, 43, 1998, 2183.
[126] A Vibrational Spectroscopic Study of the Structure of Electroactive Self‐Assembled
Monolayers of Viologen Derivatives, X. Tang, T. Schneider, D.A. Buttry, Langmuir, 10, 1994,
2235.
[127] Dimerized‐Complexes in Self‐Assembled Monolayers Containing Viologens: An Origin of
Unusual Wave Shapes in the Voltammetry of Monolayers, X. Tang, T.W. Schneider, J.W.
Walker, D.A. Buttry, Langmuir, 12, 1996, 5921.
[128] Application of quinone thio derivatives as a basis for assembling complex molecular
systems at an electrode surface, E. Katz, V.V. Borovkov, R.P. Evstigneeva, J. Electroanal. Chem.,
326, 1992, 197.
[129] Gold electrode modification with a monolayer of molecular lines consisting of electron
carriers with different redox potentials, E. Katz, H.‐L. Schmidt, J. Electroanal. Chem., 360, 1993,
337.
[130] Gold electrode modification with a monolayer of 1,4,5,8‐naphthalenetetrone as a four‐
electron transfer mediator: electrochemical reduction of dioxygen, E. Katz, H.‐L. Schmidt, J.
Electroanal. Chem., 368, 1994, 87.
[131] Organic Thiosulfates (Bunte Salts):Novel Surface‐Active Sulfur Compounds for the
Preparation of Self‐Assembled Monolayers on Gold, J. Lukkari, M. Meretoja, I. Kartio, K.
Laajalehto, M. Rajamoki, M. Lindstrom, J. Kankare, Langmuir, 15, 1999, 3529.
[132] In Situ Quartz Crystal Microgravimetric Studies of Molecular Adsorbates Containing Thiol
and Hydroquinone Moieties Bound to Au(111) Surfaces in Aqueous Electrolytes, Y. Mo, M.
Sandifer, C. Sukenik, R.J. Barriga, M.P. Soriaga, D. Scherson, Langmuir, 11, 1995, 4626.
[133] Electrochemical Properties of Mixed Self‐Assembled Monolayers on Gold Electrodes
Containing Mercaptooctylhydroquinone and Alkylthiols, A.G. Larsen, K.V. Gothelf, Langmuir,
21, 2004, 1015.

BIBLIOGRAFÍA
241
[134] pH‐dependent photoinduced electron transfer at the gold electrode modified with a self‐
assembled monolayer of a porphyrin‐mercaptoquinone coupling molecule, T. Kondo, M.
Yanagida, S.‐i. Nomura, T. Ito, K. Uosaki, J. Electroanal. Chem., 438, 1997, 121.
[135] Electrocatalytic Activity of an Immobilized Cofacial Diporphyrin Depends on the Electrode
Material, J.E. Hutchison, T.A. Postlethwaite, C.‐h. Chen, K.W. Hathcock, R.S. Ingram, W. Ou,
R.W. Linton, R.W. Murray, D.A. Tyvoll, L.L. Chng, J.P. Collman, Langmuir, 13, 1997, 2143.
[136] Molecular films of thiol‐derivatized tetraphenylporphyrins on gold: film formation and
electrocatalytic dioxygen reduction, J.E. Hutchison, T.A. Postlethwaite, R.W. Murray, Langmuir,
9, 1993, 3277.
[137] Synthesis and Properties of Metalloporphyrin Monolayers and Stacked Multilayers Bound
to an Electrode via Site Specific Axial Ligation to a Self‐Assembled Monolayer, D.A. Offord, S.B.
Sachs, M.S. Ennis, T.A. Eberspacher, J.H. Griffin, C.E.D. Chidsey, J.P. Collman, J. Am. Chem. Soc.,
120, 1998, 4478.
[138] Direct electron transfer between the covalently immobilized enzyme microperoxidase
MP‐11 and a cystamine‐modified gold electrode, T. Lötzbeyer, W. Schuhmann, E. Katz, J. Falter,
H.‐L. Schmidt, J. Electroanal. Chem., 377, 1994, 291.
[139] Investigation of the electrode reaction of cytochrome c through mixed self‐assembled
monolayers of alkanethiols on gold(111) surfaces, S. Arnold, Z.Q. Feng, T. Kakiuchi, W. Knoll, K.
Niki, J. Electroanal.Chem., 438, 1997, 91.
[140] Voltammetric Peak Broadening for Cytochrome c/Alkanethiolate Monolayer Structures:
Dispersion of Formal Potentials, R.A. Clark, E.F. Bowden, Langmuir, 13, 1997, 559.
[141] Mimicking Protein‐ Protein Electron Transfer: Voltammetry of Pseudomonas aeruginosa
Azurin and the Thermus thermophilus CuA Domain at Omega‐Derivatized Self‐Assembled‐
Monolayer Gold Electrodes, K. Fujita, N. Nakamura, H. Ohno, B.S. Leigh, K. Niki, H.B. Gray, J.H.
Richards, J. Am. Chem. Soc., 126, 2004, 13954.
[142] Characterising the formation of a bioelectrochemical interface at a self‐assembled
monolayer using X‐ray photoelectron spectroscopy, L. Jiang, A. Glidle, A. Griffith, C.J. McNeil,
J.M. Cooper, Bioelectroch. Bioener., 42, 1997, 15.
[143] Surface‐Enhanced Resonance Raman Spectroscopic and Electrochemical Study of
Cytochrome c Bound on Electrodes through Coordination with Pyridinyl‐Terminated Self‐
Assembled Monolayers, D.H. Murgida, P. Hildebrandt, J. Wei, Y.F. He, H. Liu, D.H. Waldeck, J.
Phys. Chem. B, 108, 2004, 2261.
[144] Intermolecular biological electron transfer: an electrochemical approach, K. Niki, J.R.
Sprinkle, E. Margoliash, Bioelectrochemistry, 55, 2002, 37.

BIBLIOGRAFÍA
242
[145] Impact of Surface Immobilization and Solution Ionic Strength on the Formal Potential of
Immobilized Cytochrome c, J. Petrovic, R.A. Clark, H. Yue, D.H. Waldeck, E.F. Bowden,
Langmuir, 21, 2005, 6308.
[146] Electron‐Transfer Dynamics of Cytochrome C: A Change in the Reaction Mechanism with
Distance J. Wei, H. Liu, D.E. Khoshtariya, H. Yamamoto, A. Dick, D.H. Waldeck, Angew. Chem.,
Int. Ed. Engl., 41, 2002, 4700.
[147] Heterogeneous Kinetics of Metal‐ and Ligand‐Based Redox Reactions within Adsorbed
Monolayers, R.J. Forster, Inorg. Chem., 35, 1996, 3394.
[148] Electrochemistry of Spontaneously Adsorbed Monolayers. Equilibrium Properties and
Fundamental Electron Transfer Characteristics, R.J. Forster, L.R. Faulkner, J. Am. Chem. Soc.,
116, 1994, 5444.
[149] Electrochemistry of spontaneously adsorbed monolayers. Effects of solvent, potential,
and temperature on electron transfer dynamics, R.J.F. Forster, L.R. Faulkner, J. Am. Chem. Soc.,
116, 1994, 5453.
[150] pH Modulated Heterogeneous Electron Transfer across Metal/Monolayer Interfaces, R.J.
Forster, J.P. O'Kelly, J. Phys. Chem., 100, 1996, 3695.
[151] Hole superexchange across a triazole bridged osmium monolayer/electrode interface R.J.
Forster, J.G. Vos, T.E. Keyes, Analyst, 123, 1998, 1905.
[152] Molecular electronic junctions, R.L. McCreery, Chem. Mater., 16, 2004, 4477.
[153] Electrical Measurements in Molecular Electronics, D.K. James, J.M. Tour, Chem. Mater.,
16, 2004, 4423.
[154] Electronically Configurable Molecular‐Based Logic Gates, C.P. Collier, E.W. Wong, M.
Belohradský, F.M. Raymo, J.F. Stoddart, P.J. Kuekes, R.S. Williams, J.R. Heath, Science, 285,
1999, 391.
[155] Fabrication and Transport Properties of Single‐Molecule‐Thick Electrochemical Junctions,
E.W. Wong, C.P. Collier, M. Behloradský, F.M. Raymo, J.F. Stoddart, J.R. Heath, J. Am. Chem.
Soc., 122, 2000, 5831.
[156] Electrical Rectification by a Monolayer of Hexadecylquinolinium
Tricyanoquinodimethanide Measured between Macroscopic Gold Electrodes, R.M. Metzger, T.
Xu, I.R. Peterson, J. Phys. Chem. B, 105, 2001, 7280.
[157] Electron transfer in a Hg‐SAM//SAM‐Hg junction mediated by redox centers, E. Tran, M.A.
Rampi, G.M. Whitesides, Angew. Chem. ‐ International Edition, 43, 2004, 3835.

BIBLIOGRAFÍA
243
[158] Molecular rectification in a metal‐insulator‐metal junction based on self‐assembled
monolayers, M.L. Chabinyc, X. Chen, R.E. Holmlin, H. Jacobs, H. Skulason, C.D. Frisbie, V.
Mujica, M.A. Ratner, M.A. Rampi, G.M. Whitesides, J. Am. Chem. Soc., 124, 2002, 11730.
[159] Electron transport through thin organic films in metal‐insulator‐metal junctions based on
self‐assembled monolayers, R.E. Holmlin, R. Haag, M.L. Chabinyc, R.F. Ismagilov, A.E. Cohen, A.
Terfort, M.A. Rampi, G.M. Whitesides, J. Am. Chem. Soc., 123, 2001, 5075.
[160] A versatile experimental approach for understanding electron transport through organic
materials, M.A. Rampi, G.M. Whitesides, Chem. Phys., 281, 2002, 373.
[161] Conductance of a Molecular Junction, M.A. Reed, C. Zhou, C.J. Muller, T.P. Burgin, J.M.
Tour, Science, 278, 1997, 252.
[162] Electronic transport through single conjugated molecules, H.B. Weber, J. Reichert, F.
Weigend, R. Ochs, D. Beckmann, M. Mayor, R. Ahlrichs, H.v. Löhneysen, Chem. Phys., 281,
2002, 113.
[163] Driving Current through Single Organic Molecules, J. Reichert, R. Ochs, D. Beckmann, H.B.
Weber, M. Mayor, H.v. Löhneysen, Phys. Rev. Lett., 88, 2002, 176804.
[164] Scanning Probe Studies of Single Nanostructures, G.S. McCarty, P.S. Weiss, Chem. Rev.,
99, 1999, 1983.
[165] "Coulomb Staircase" at Room Temperature in a Self‐Assembled Molecular Nanostructure
R.P. Andres, T. Bein, M. Dorogi, S. Feng, J.I. Henderson, C.P. Kubiak, W. Mahoney, R.G. Osifchin,
R. Reifenberger, Science 272, 1996, 1323.
[166] Conductance Switching in Single Molecules Through Conformational Changes, Z.J.
Donhauser, B.A. Mantooth, K.F. Kelly, L.A. Bumm, J.D. Monnell, J.J. Stapleton, J. D. W. Price,
A.M. Rawlett, D.L. Allara, J.M. Tour, P.S. Weiss, Science, 292, 2001, 2303.
[167] Single‐Molecule Electron Transfer in Electrochemical Environments, J. Zhang, A.M.
Kuznetsov, I.G. Medvedev, Q. Chi, T. Albrecht, P.S. Jensen, J. Ulstrup, Chem. Rev. , 108, 2008,
2737–2791.
[168] Electrolyte Gating in Redox‐Active Tunneling JunctionsAn Electrochemical STM Approach,
I. Pobelov, L. Zhihai, T.H. Wandlowski, J. Am. Chem. Soc., 130, 2009, 16045.
[169] Electronic and Molecular Structure of Electrode‐Electrolyte Interfaces, W.N. Hansen, D.M.
Kolb, D.W. Lynch, Elsevier, Amsterdam, 1983.
[170] Electrochemical Surface Science, D.M. Kolb, Angew. Chem., Int. Ed., 40, 2000, 1162.
[171] Electrochemistry of well‐defined surfaces, A.T. Hubbard, Acc. Chem. Res., 13, 1980, 177.

BIBLIOGRAFÍA
244
[172] Multiple Electron Tunneling Paths across Self‐Assembled Monolayers of Alkanethiols with
Attached Ruthenium(II/III) Redox Centers, H.O. Finklea, L. Liu, S. Ravenscroft, S. Punturi, J. Phys.
Chem., 100, 1996, 18852.
[173] AC voltammetry studies of electron transfer kinetics for a redox couple attached via short
alkanethiols to a gold electrode, D.A. Brevnov, H.O. Finklea, H. Van Ryswyk, J. Electroanal.
Chem., 500, 2001, 100.
[174] Ultrafast Voltammetry for Probing Interfacial Electron Transfer in Molecular Wires C.
Amatore, E. Maisonhaute, B. Schöllhorn, J. Wadhawan, Chem. Phys. Chem, 8, 2007, 1321.
[175] Topological Effects in Bridge‐Mediated Electron Transfer Between Redox Molecules and
Metal Electrodes, J.J. Sumner, S.E. Creager, J. Am. Chem. Soc., 122, 2000, 11914.
[176] Electron Transfer at Electrodes through Conjugated “Molecular Wire” Bridges, S.E.
Creager, C.J. Yu, C. Bamdad, S. O´Connor, T. MacLean, E. Lam, Y. Chong, G.T. Olsen, J. Luo, M.
Gozin, J.F. Kayyem, J. Am. Chem. Soc., 121, 1999, 1059.
[177] Rates of Interfacial Electron Transfer through ヽ‐Conjugated Spacers, S.B. Sachs, S.P.
Dudek, R.P. Hsung, L.R. Sita, J.F. Smalley, M.D. Newton, S.W. Feldberg, C.E.D. Chidsey, J. Am.
Chem. Soc., 119, 1997, 10563.
[178] Rapid Electron Tunneling Through Oligophenylenevinylene Bridges, H.D. Sikes, J.F.
Smalley, S.P. Dudek, A.R. Cook, M.D. Newton, C.E.D. Chidsey, S.W. Feldberg, Science, 291,
2001, 1519.
[179] Effect of the Metal on Electron Transfer across Self‐Assembled Monolayers, H.O. Finklea,
K. Yoon, E. Chamberlain, J. Allen, R. Haddox, J. Phys. Chem. B, 105, 2001, 3088.
[180] Proton‐Coupled Electron Transfer of an Osmium Aquo Complex on a Self‐Assembled
Monolayer on Gold, R.M. Haddox, H.O. Finklea, J. Phys. Chem. B, 108, 2004, 1694.
[181] Cyclic Voltammetric and Scanning Electrochemical Microscopic Study of Menadione
Permeability through a Self‐Assembled Monolayer on a Gold Electrode, C.l. Cannes, F.d.r.
Kanoufi, A.J. Bard, Langmuir, 18, 2002, 8134.
[182] In Situ Monitoring of Diffuse Double Layer Structure Changes of Electrochemically
Addressable Self‐Assembled Monolayers with an Atomic Force Microscope, K. Hu, Z. Chai, J.K.
Whitesell, A.J. Bard, Langmuir, 15, 1999, 3343.
[183] Alternating Current Voltammetry Studies of the Effect of Melittin on Heterogeneous
Electron Transfer across a Hybrid Bilayer Membrane Supported on a Gold Electrode, D.A.
Brevnov, H.O. Finklea, Langmuir, 16, 2000, 5973.
[184] "Plugging into Enzymes": Nanowiring of Redox Enzymes by a Gold Nanoparticle, Y. Xiao,
F. Patolsky, E. Katz, J. F.Hainfeld, I. Willner, Science, 299, 2003, 1877.

BIBLIOGRAFÍA
245
[185] Biomaterial engineered electrodes for bioelectronics V. Pardo‐Yissar, E. Katz, I. Willner,
A.B. Kotlyar, C. Sanders, H. Lill, Faraday Discuss., 116, 2000, 119
[186] Glucose oxidase electrodes via reconstitution of the apo‐enzyme: tailoring of novel
glucose biosensors, E. Katz, A. Riklin, V. Heleg‐Shabtai, I. Willner, A.F. Bückmann, Anal. Chim.
Acta, 385, 1999, 45.
[187] Integration of Layered Redox Proteins and Conductive Supports for Bioelectronic
Applications, I. Willner, E. Katz, Angew. Chem. ‐ International Edition, 39 2000, 1180.
[188] Comparison of Diazonium Salt Derived and Thiol Derived Nitrobenzene Layers on Gold,
D.M. Shewchuk, M.T. McDermott, Langmuir, 25, 2009, 4556.
[189] Covalent Modification of Carbon Surfaces by Aryl Radicals Generated from the
Electrochemical Reduction of Diazonium Salts, P. Allongue, M. Delamar, B. Desbat, O.
Fagebaume, R. Hitmi, J. Pinson, J.‐M. Savéant, J. Am. Chem. Soc., 119, 1997, 201.
[190] Formation of Multilayers on Glassy Carbon Electrodes via the Reduction of Diazonium
Salts, J.K. Kariuki, M.T. McDermott, Langmuir, 17, 2001, 5947.
[191] The modification of glassy carbon and gold electrodes with aryl diazonium salt: The
impact of the electrode materials on the rate of heterogeneous electron transfer, G. Liu, J. Liu,
T. Bocking, P.K. Eggers, J.J. Gooding, Chem. Phys., 319, 2005, 136.
[192] Carbon/molecule/metal molecular electronic junctions: the importance of "contacts", R.L.
McCreery, U. Viswanathan, R.P. Kalakodimi, A.M. Nowak, Faraday Discuss., 131, 2006, 33.
[193] Charge Transfer between electroactive species immobilized on carbon surfaces by aryl
diazonium reduction. SECM investigations, D. Zigah, J.M. Noel, C. Lagrost, P. Hapiot, J. Phys.
Chem. C, 114, 2010, 3075.
[194] EIS study of the redox reaction of Fe (CN)6 3‐ / 4‐ at glassy carbon electrode via diazonium
reduction in aqueous and acetonitrile solutions, M. Khoshroo, A.A. Rostami, J. Electroanal.
Chem., 624, 2008, 205.
[195] In‐situ infrared spectroscopy of the electrode/electrolyte solution interphase, A. Bewick, J.
Electroanal. Chem., 150, 1983, 481.
[196] Internal Reflection Spectroscopic Observation of Electrode‐Solution Interface, W.N.
Hansen, R.A. Osteryoung, T. Kuwana, J. Am. Chem. Soc., 88, 1966, 1062.
[197] Electrochemical and PM‐IRRAS studies of potential controlled transformations of
phospholipid layers on Au(111) electrodes, S.L. Horswell, V. Zamlynny, H.Q. Li, A.R. Merrill, J.
Lipkowski, Faraday Discuss., 121, 2002, 405.
[198] Current understanding of the mechanism of surface enhanced Raman scattering, T.E.
Furtak, J. Electroanal. Chem., 150, 1983, 375.

BIBLIOGRAFÍA
246
[199] The Quartz Microbalance: A Novel Approach to the In‐Situ Investigation of Interfacial
Phenomena at the Solid/Liquid Junction, R. Schumacher, Angew. Chem. ‐ International Edition,
29, 1990, 329.
[200] The Statistical Mechanics of the Electrical Double Layer S.L. Carnie, G.M. Torrie, Adv.
Chem. Phys., 56, 1984, 141.
[201] A simple theory of the electric double layer including solvent effects, D. Henderson, L.
Blum, J. Electroanal. Chem., 132, 1982, 1.
[202] Scanning tunneling microscopy at potential controlled electrode surfaces in electrolytic
environment, P. Lustenberger, H. Rohrer, R. Christoph, H. Siegenthaler, J. Electroanal.Chem.,
243, 1988, 225.
[203] An in‐situ scanning tunneling microscopy study of au (111) with atomic scale resolution, J.
Wiechers, T. Twomey, D.M. Kolb, R.J. Behm, J. Electroanal.Chem., 248, 1988, 451.
[204] An in‐situ STM study of potential‐induced changes in the surface topography of Au(100)
electrodes, R.J. Nichols, O.M. Magnussen, J. Hotlos, T. Twomey, R.J. Behm, D.M. Kolb, J.
Electroanal.Chem., 290, 1990, 21.
[205] Electrochemical Applications of in Situ Scanning Probe Microscopy, A.A. Gewirth, B.K.
Niece, Chem. Rev., 97, 1997, 1129.
[206] Electrostatic interactions at self assembled molecular films of charged thiols on gold, V.
Molinero, E.J. Calvo, J. Electroanal. Chem., 445, 1998, 17.
[207] Thermal fluctuations determine the electron‐transfer rates of cytochrome c in
electrostatic and covalent complexes, H.K. Ly, M.A. Martí, D.F. Martín, D. Alvarez‐Paggi, W.
Meister, A. Kranich, I.M. Weidinger, P. Hildebrandt, D.H. Murgida, Chem. Phys. Chem., 11,
2010, 1225.
[208] Distance‐dependent electron transfer rate of immobilized redox proteins: A statistical
physics approach, S. Georg, J. Kabuss, I.M. Weidinger, D.H. Murgida, P. Hildebrandt, A. Knorr,
M. Richter, Phys. Rev. E, 81, 2010, 046101.
[209] Molecular basis of coupled protein and electron transfer dynamics of cytochrome c in
biomimetic complexes, D. Alvarez‐Paggi, D.F. Martín, P.M. Debiase, P. Hildebrandt, M.A. Martí,
D.H. Murgida, J. Am. Chem. Soc, 132, 2010, 5769.
[210] Instrumental Methods in Electrochemistry, S.E. Group, Horwood Publishing, Chichester,
2001.
[211] Characterization of Materials, E.N. Kaufmann, Wiley‐Interscience, 1 y 2. New Jersey,
2003.

BIBLIOGRAFÍA
247
[212] Electrochemistry, C.H. Hamann, A. Hamnett, W. Vielstich, Wiley‐VCH, Chapter 5.
Weinheim, 1998.
[213] Impedance Spectroscopy Theory, Experiment, and Applications, E. Barsoukov, J.R.
Macdonald, John Wiley & Sons, Inc., Hoboken, 2005.
[214] Monitoring Electron Transfer in an Azobenzene Self‐Assembled Monolayer by in Situ
Infrared Reflection Absorption Spectroscopy, H.‐Z. Yu, H.‐L. Zhang, Z.‐F. Liu, S. Ye, K. Uosaki,
Langmuir, 14, 1998, 619.
[215] Electrodeposition of Au Monolayer on Pt(111) Mediated by Self‐Assembled Monolayers,
Y.‐C. Yang, S.‐L. Yau, Y.‐L. Lee, J. Am. Chem. Soc., 128, 2006, 3677.
[216] Structural Investigation of a Self‐Assembled Monolayer of a p‐Nitroanilino‐Terminated
Thiol, A. Wesch, O. Dannenberger, C. Woll, J.J. Wolff, M. Buck, Langmuir, 12, 1996, 5330.
[217] Vibrational Spectroscopy of Molecules and Macromolecules on Surfaces, M.W. Urban,
J.W.a. Sons, Chapter 1. New York, 1993.
[218] Quantitative SNIFTIRS and PM IRRAS of Organic Molecules at Electrode Surfaces,
Advances in Electrochemical Science and Engineering., V. Zamlynny, J. Lipkowski, Vol. 9, R.C.
Alkire, D.M. Kolb, J. Lipkowski, P.N. Ross, Eds.; Wiley‐VCH: Weinheim, 2006.
[219] Polarization Modulation approaches to Reflection‐Absorption Spectroscopy. In Handbook
of Vibrational Spectroscopy, Handbook of Vibrational Spectroscopy, B.L. Frey, R.M. Corn, S.C.
Weibel, Vol. Eds.; John Wiley & sons: 2001.
[220] Raman Spectra in Inorganic Chemistry, J.H. Hibben, Chem. Rev., 13, 1933, 345.
[221] Resonance Raman Intensities and Charge‐Transfer Reorganization Energies, A.B. Myers,
Chem. Rev., 96, 1996, 911.
[222] Wiring enzymes in nanostructures built with electrostatically self‐assembled thin films,
E.J. Calvo, A. Wolosiuk, Chem. Phys. Chem, 6, 2005, 43.
[223] Vibrational characterization of multiply metal‐metal bondedosmium, molybdenum, and
rhenium porphyrin dimers, C.D. Tait, J.M. Garner, J.P. Collman, A.P. Sattelberger, W.H.
Woodruff, J. Am. Chem. Soc., 111, 1989, 9072.
[224] Functional Evaluation of Heme Vinyl Groups in Myoglobin with Symmetric Protoheme
Isomersa Y. Mie, C. Yamada, G.P.J. Hareau, S. Neya, T. Uno, N. Funasaki, K. Nishiyama, I.
Taniguchi, Biochemistry, 43, 2004, 13149.
[225] Gated Electron Transfer of Yeast Iso‐1 Cytochrome c on Self‐Assembled Monolayer‐
Coated Electrodes, J.‐J. Feng, D.H. Murgida, U. Kuhlmann, T. Utesch, M.A. Mroginski, P.
Hildebrandt, I.M. Weidinger, J. Phys. Chem. B, 112, 2008, 15202.

BIBLIOGRAFÍA
248
[226] Infrared and Raman Spectra of Inorganic and Coordination Compounds Part A, K.
Nakamoto, John Wiley & Sons, New Jersey, 2009.
[227] X‐ray photoelectron spectroscopic examinations of beryllium metal surfaces exposed to
chlorinated solvents, J.C. Birkbeck, N.L. Kuehler, D.L. Williams, W.E. Moddeman, Surf. Interface
Anal., 27, 1999, 273.
[228] An X‐ray photoelectron spectroscopic study of stainless steel electrodes after polarisation
in the regions of transpassivity and secondary passivity, A.F. Povey, R.O. Ansella, T. Dickinson,
P.M.A. Sherwood, J. Electroanal. Chem., 87, 1978, 189.
[229] Electrochemical and X‐ray absorption spectroscopy studies of copper coatings on a
hydrogen storage alloy, R.C. Ambrosio, E.A. Ticianelli, J. Electroanal. Chem., 574, 2005, 251.
[230] XPS studies of PZT films deposited by metallic lead and ceramic PZT dual target co‐
sputtering, W.L. Chang, J.L. He, J. Electroceram., 13, 2004, 47.
[231] The effect of preparation method and Sb content on SnO2‐CuO sintering, I.O. Mazali,
W.C. Las, M. Cilense, J. Mater. Sci., 38, 2003, 3325.
[232] Characterization of ceramic‐metal composite hydrogen separation membranes consisting
of barium oxide, cerium oxide, yttrium oxide and palladium, R.V. Siriwardane, J.A. Poston Jr,
E.P. Fisher, T.H. Lee, S.E. Dorris, U. Balachandran, Appl. Surf. Sci., 217, 2003, 43.
[233] XPS analysis of surface layer of sol‐gel‐derived PZT thin films, O. Sugiyama, K. Murakami,
S. Kaneko, J. Eur. Cer. Soc., 24, 2004, 1157.
[234] Growth and morphology of SnPc films on the S‐GaAs(0 0 1) surface: A combined XPS,
AFM and NEXAFS study, A.R. Vearey‐Roberts, H.J. Steiner, S. Evans, I. Cerrillo, J. Mendez, G.
Cabailh, S. O'Brien, J.W. Wells, I.T. McGovern, D.A. Evans, Appl. Surf. Sci., 234, 2004, 131.
[235] Investigation of the electronic structure of the phosphorus‐doped Si and SiO2: Si quantum
dots by XPS and HREELS methods, A.I. Kovalev, D.L. Wainstein, D.I. Tetelbaum, W. Hornig, Y.N.
Kucherehko, Surf. Interface Anal., 36, 2004, 959.
[236] Enhanced oxidation of Ge(1 1 1) surface precovered with Na: Scanning tunneling
microscopy and X‐ray photoemission spectroscopy study, D. Jeon, T. Sakurai, K.D. Lee, J.W.
Chung, Surf. Sci., 559, 2004, 141.
[237] Electrochemical STM, Instrumentation and Electroanalytical Chemistry, T.P. Moffat, Vol.
3, A. Bard, M. Stratmann, Eds.; Wiley‐VCH GmbH & Co. KGaA: Germany, 2001.
[238] M. Breiter, C.A. Knorr, W. Volkl, Z. Elektrochem., 64, 1955, 681.
[239] A study of the dissolution of platinum, palladium, rhodium and gold electrodes in 1 m
sulphuric acid by cyclic voltammetry, D.A.J. Rand, R. Woods, J. Electroanal. Chem., 35, 1972,
209.

BIBLIOGRAFÍA
249
[240] DNA cleaveage activity, M. Kizil, E.I. Yilmaz, N. Pirincciogluetin, C. Aytekin, Turk. J. Chem.,
27, 2003, 539.
[241] Electrochemical modification of a carbon electrode using aromatic diazonium salts. 2.
Electrochemistry of 4‐nitrophenyl modified glassy carbon electrodes in aqueous media, B. Ortiz,
C. Saby, G.Y. Champagne, D. Bélanger, J. Electroanal. Chem., 455, 1998, 75.
[242] Electrochemical Modification of Glassy Carbon Electrode Using Aromatic Diazonium Salts.
1. Blocking Effect of 4‐Nitrophenyl and 4‐Carboxyphenyl Groups, C. Saby, B. Ortiz, G.Y.
Champagne, D. Bélanger, Langmuir, 13, 1997, 6805.
[243] Nucleation and Growth of Functionalized Aryl Films on Graphite Electrodes, J.K. Kariuki,
M.T. McDermott, Langmuir, 15, 1999, 6534.
[244] A Method for the High Efficiency of Water‐Soluble Carbodiimide‐Mediated Amidation, D.
Sehgal, I. Vijay, Anal. Biochem., 218, 1994, 87.
[245] Organic modification of hydrogen terminated silicon surfaces D.D.M. Wayner, R.A.
Wolkow, J. Chem. Soc., Perkin Trans., 2, 2002, 23
[246] Electrochemical Bonding of Amines to Carbon Fiber Surfaces Toward Improved Carbon‐
Epoxy Composites B. Barbier, J. Pinson, G. Desarmot, M. Sanchez, J. Electrochem. Soc., 137,
1990, 1757.
[247] H. Maeda, Y. Tamauchi, H. Ohmori, Curr. Top. Anal. Chem., 2, 2001, 121.
[248] Derivatization of Carbon Surfaces by Anodic Oxidation of Arylacetates. Electrochemical
Manipulation of the Grafted Films, C.P. Andrieux, F. Gonzalez, J.‐M. Savéant, J. Am. Chem. Soc.,
119, 1997, 4292.
[249] W.C. Bigelow, D.L. Pickett, W.A. Zisman, J. Colloid. Interface Sci., 1, 1946, 513.
[250] Spontaneously organized molecular assemblies. 4. Structural characterization of n‐alkyl
thiol monolayers on gold by optical ellipsometry, infrared spectroscopy, and electrochemistry,
M.D. Porter, T.B. Bright, D.L. Allara, C.E.D. Chidsey, J. Am. Chem. Soc., 109, 1987, 3559.
[251] Blocking oriented monolayers of alkyl mercaptans on gold electrodes, H.O. Finklea, S.
Avery, M. Lynch, T. Fursch, Langmuir, 3, 1987, 409.
[252] Organized self‐assembling monolayers on electrodes. 2. Monolayer‐based
ultramicroelectrodes for the study of very rapid electrode kinetics, E. Sabatini, I. Rubinstein, J.
Phys. Chem., 91, 1987, 6663.
[253] Organized self‐assembling monolayers on electrodes: Part I. Octadecyl derivatives on
gold, E. Sabatini, I. Rubinstein, R. Maoz, J. Sagiv, J. Electroanal. Chem., 219, 1987, 365.
[254] Monolayer films prepared by the spontaneous self‐assembly of symmetrical and
unsymmetrical dialkyl sulfides from solution onto gold substrates: structure, properties, and

BIBLIOGRAFÍA
250
reactivity of constituent functional groups, E.B. Troughton, C.D. Bain, G.M. Whitesides, R.G.
Nuzzo, D.L. Allara, M.D. Porter, Langmuir, 4, 1988, 365.
[255] Structures of self‐assembled monolayer films of organosulfur compounds adsorbed on
gold single crystals: electron diffraction studies, L. Strong, G.M. Whitesides, Langmuir, 4, 1988,
546.
[256] Correlations between wettability and structure in monolayers of alkanethiols adsorbed on
gold, C.D. Bain, G.M. Whitesides, J. Am. Chem. Soc., 110, 1988, 3665.
[257] Recent advances in self‐assembled monolayers based biomolecular electronic devices,
S.K. Arya, P.R. Solanki, M. Datta, B.D. Malhotra, Biosens. Bioelectron., 24, 2009, 2810.
[258] Charge conduction and breakdown mechanisms in self‐assembled nanodielectrics, S.A.
Dibenedetto, A. Facchetti, M.A. Ratner, T.J. Marks, J. Am. Chem. Soc., 131, 2009, 7158.
[259] Microstructure, wettability, and thermal stability of semifluorinated self‐assembled
monolayers (SAMs) on gold, H. Fukushima, S. Seki, T. Nishikawa, H. Takiguchi, K. Tamada, K.
Abe, R. Colorado, M. Graupe, O.E. Shmakova, T.R. Lee, J. Phys. Chem. B, 104, 2000, 7417.
[260] Spiroalkanedithiol‐based SAMs reveal unique insight into the wettabilities and frictional
properties of organic thin films, Y.S. Shon, S. Lee, R. Colorado Jr, S.S. Perry, T.R. Lee, J. Am.
Chem. Soc., 122, 2000, 7556.
[261] Reduction of ice adhesion to metal by using self‐assembling monolayers (SAMs), V.F.
Petrenko, S. Peng, Can. J. Phys., 81, 2003, 387.
[262] Friction properties of OTS SAMs and silicon surface under water lubrication, X. Wang, X.
Zhang, I. Ahmed, Y. Liu, S. Wen, J. Wuhan Univ. Technol., 24, 2009, 273.
[263] Corrosion Inhibition by Thiol‐Derived SAMs for Enhanced Wire Bonding on Cu Surfaces,
C.M. Whelan, M. Kinsella, H.M. Ho, K. Maex, J. Electrochem. Soc., 151, 2004, B33.
[264] Progress in STM study of corrosion inhibitor self‐assembled monolayers (SAMs) on metals
I. Corrosion inhibitor SAMs on noble metals, D.S. Kong, L.J. Wan, S.H. Chen, W. Yang, Corrosion
and Protection, 24, 2003, 415.
[265] Progress in STM study of corrosion inhibitor self‐assembled monolayers (SAMs) on metals
II. Corrosion inhibitor SAMs on engineering metals, D.S. Kong, L.J. Wan, S.H. Chen, W. Yang,
Corrosion and Protection, 24, 2003, 507.
[266] Biomimetics with a self‐assembled monolayer of catalytically active tethered
isoalloxazine on au, E.J. Calvo, M.S. Rothacher, C. Bonazzola, I.R. Wheeldon, R.C. Salvarezza,
M.E. Vela, G. Benitez, Langmuir, 21, 2005, 7907.
[267] Formation and Structure of Self‐Assembled Monolayers, A. Ulman, Chem. Rev., 96, 1996,
1533.

BIBLIOGRAFÍA
251
[268] Self‐assembled monolayers of long‐chain hydroxamic acids on the native oxides of
metals, J.P. Folkers, C.B. Gorman, P.E. Laibinis, S. Buchholz, G.M. Whitesides, R.G. Nuzzo,
Langmuir, 11, 1995, 813.
[269] An electrochemical study of the SAMs of 6‐mercaptopurine (6MP) at Hg and Au(111)
electrodes in alkaline media, R. Madueño, T. Pineda, J.M. Sevilla, M. Blázquez, Langmuir, 18,
2002, 3903.
[270] Adsorption of self‐assembling sulfur compounds through electrochemical reactions:
Effects of potential, acid and oxidizing agents, S. Chon, W.K. Paik, Phys. Chem. Chem. Phys., 3,
2001, 3405.
[271] Self‐assembled monolayer coatings on amorphous iron, O. Rozenfeld, Y. Koltypin, H.
Bamnolker, S. Margel, A. Gedanken, Langmuir, 10, 1994, 3919.
[272] The Fate of Sulfur‐Bound Hydrogen on Formation of Self‐Assembled Thiol Monolayers on
Gold: 1H NMR Spectroscopic Evidence from Solutions of Gold Clusters, M. Hasan, D. Bethell, M.
Brust, J. Am. Chem. Soc., 124, 2002, 1132.
[273] Formation of thiol‐based monolayers on gold: implications from open circuit potential
measurements, C.‐J. Zhong, N.T. Woods, G.B. Dawson, M.D. Porter, Electrochem. Commun., 1,
1999, 17.
[274] Nondissociative Chemisorption of Methanethiol on Ag(110): A Critical Result for Self‐
Assembled Monolayers, J.‐G. Lee, J. Lee, J.T.J. Yates, J. Am. Chem. Soc., 126, 2004, 440.
[275] Fundamental studies of the chemisorption of organosulfur compounds on gold(111).
Implications for molecular self‐assembly on gold surfaces, R.G. Nuzzo, B.R. Zegarski, L.H.
Dubois, J. Am. Chem. Soc., 109, 1987, 733.
[276] Structure and binding of alkanethiolates on gold and silver surfaces: implications for self‐
assembled monolayers, H. Sellers, A. Ulman, Y. Schnidman, J.E. Eilers, J. Am. Chem. Soc., 115,
1993, 9389.
[277] Formation of monolayer films by the spontaneous assembly of organic thiols from
solution onto gold, D. Colin, E. Bain, B.Troughton, Y.T. Tao, J. Evall, G.M. Whitesides, R.G.
Nuzzo, J. Am. Chem. Soc., 111, 1989, 321.
[278] Structure and growth of self‐assembling monolayers, F. Schreiber, Prog. Surf. Sci., 65,
2000, 151.
[279] Collective tilt behavior in dense, substrate‐supported monolayers of long‐chain
molecules: a molecular dynamics study, J.P. Bareman, M.L. Klein, J. Phys. Chem., 94, 1990,
5202.

BIBLIOGRAFÍA
252
[280] Thermal Stability of Self‐Assembled Monolayers: Influence of Lateral Hydrogen Bonding,
R. Valiokas, M. Östblom, S. Svedhem, S.C.T. Svensson, B. Liedberg, J. Phys. Chem. B, 106, 2002,
10401.
[281] Organization of Water Layers at Hydrophilic Interfaces, M.C. Gurau, G. Kim, S.‐M. Lim, F.
Albertorio, H.C. Fleisher, P.S. Cremer, Chem. Phys. Chem., 4, 2003, 1231.
[282] Well‐Ordered Self‐Assembled Monolayers Created via Vapor‐Phase Reactions on a
Monolayer Template, M.K. Ferguson, E.R. Low, J.R. Morris, Langmuir, 20, 2004, 3319.
[283] Comparison of the structures and wetting properties of self‐assembled monolayers of n‐
alkanethiols on the coinage metal surfaces, copper, silver, and gold, P.E. Laibinis, G.M.
Whitesides, D.L. Allara, Y.T. Tao, A.N. Parikh, R.G. Nuzzo, J. Am. Chem. Soc., 113, 1991, 7152.
[284] Methylene Blue Incorporation into Alkanethiol SAMs on Au(111): Effect of Hydrocarbon
Chain Ordering, D. Grumelli, L.P. Méndez De Leo, C. Bonazzola, V. Zamlynny, E.J. Calvo, R.C.
Salvarezza, Langmuir, 26, 2010, 8226.
[285] Structure and interfacial properties of spontaneously adsorbed n‐alkanethiolate
monolayers on evaporated silver surfaces, M.M. Walczak, C. C.hung, S.M. Stole, C.A. Widrig,
M.D. Porter, J. Am. Chem. Soc., 113, 1991, 2370.
[286] Electrochemical and Atomic Force Microscopy Study of Carbon Surface Modification via
Diazonium Reduction in Aqueous and Acetonitrile Solutions, M.A. Bryant, J.E. Pemberton, J.
Am. Chem. Soc., 113, 1991, 8284.
[287] Through‐Bond and Chain‐to‐Chain Coupling. Two Pathways in Electron Tunneling through
Liquid Alkanethiol Monolayers on Mercury Electrodes, K. Slowinski, R.V. Chamberlain, C.J.
Miller, M. Majda, J. Am. Chem. Soc., 119, 1997, 11910.
[288] Formation and Structure of Self‐Assembled Monolayers of Alkanethiolates on Palladium,
J.C. Love, D.B. Wolfe, R. Haasch, M.L. Chabinyc, K.E. Paul, G.M. Whitesides, R.G. Nuzzo, J. Am.
Chem. Soc., 125, 2003, 2597.
[289] Superlattice structure at the surface of a monolayer of octadecanethiol self‐assembled on
Au(111), N. Camillone, C.E.D. Chidsey, G.Y. Liu, G. Scoles, J. Chem. Phys., 98, 1993, 3503.
[290] Molecular ordering of organosulfur compounds on Au(111) and Au(100): Adsorption from
solution and in ultrahigh vacuum, L.H. Dubois, B.R. Zegarski, R.G. Nuzzo, J. Chem. Phys., 98,
1993, 678.
[291] Molecular order at the surface of an organic monolayer studied by low energy helium
diffraction, C.E.D. Chidsey, G.Y. Liu, Y.P. Rowntree, G. Scoles, J. Chem. Phys., 91, 1989, 4421.
[292] Atomic scale imaging of alkanethiolate monolayers at gold surfaces with atomic force
microscopy, C.A. Alves, E.L. Smith, M.D. Porter, J. Am. Chem. Soc., 114, 1994, 1222.

BIBLIOGRAFÍA
253
[293] Mechanism of Formation of Au Vacancy Islands in Alkanethiol Monolayers on Au(111),
G.E. Poirier, Langmuir 13, 1997, 2019.
[294] The c(4X2) Superlattice of n‐Alkanethiol Monolayers Self‐Assembled on Au(111), G.E.
Poirier, M.J. Tarlov, Langmuir, 10, 1994, 2853.
[295] Dynamic characteristics of adsorbed monolayers of 1‐dodecanethiol on gold (111)
terraces from in‐situ scanning tunneling microscopy imaging, F. Terám‐Arce, M.E. Vela, R. C.
Salvarezza, A.J. Arvia, Electrochim. Acta 44, 1998, 1053.
[296] Indirect visualization of defect structures contained within self‐assembled
organomercaptan monolayers: combined use of electrochemistry and scanning tunneling
microscopy, L. Sun, R.M. Crooks, Langmuir, 9, 1993, 1951.
[297] The dynamic behavior of butanethiol and dodecanethiol adsorbates on Au(111) terraces,
F. Terán‐Arce, M.E. Vela, R.C. Salvarezza, A.J. Arvia, J. Chem. Phys., 109, 1998, 5703.
[298] Complex Structural Dynamics at Adsorbed Alkanethiol Layers at Au(111) Single‐Crystal
Domains, F. Terán‐Arce, M.E. Vela, R.C. Salvarezza, A.J. Arvia, Langmuir, 14, 1998, 7203.
[299] Electron‐induced crosslinking of aromatic self‐assembled monolayers: Negative resists for
nanolithography, W. Geyer, V. Stadler, W. Eck, M. Zharnikov, A. Gölzhäuser, M. Grunze, Appl.
Phys. Lett., 75, 1999, 2401.
[300] Structural Characterization of Organothiolate Adlayers on Gold: The Case of Rigid,
Aromatic Backbones, C. Fuxen, W. Azzam, R. Arnold, G. Witte, A. Terfort, C. Wöll, Langmuir, 17,
2001, 3689.
[301] Structure of Thioaromatic Self‐Assembled Monolayers on Gold and Silver, S. Frey, V.
Stadler, K. Heister, W. Eck, M. Zharnikov, M. Grunze, Langmuir, 17 2001, 2408.
[302] Self‐Assembled Oligo(phenylene‐ethynylene) Molecular Electronic Switch Monolayers on
Gold: Structures and Chemical Stability, J.J. Stapleton, P. Harder, T.A. Daniel, M.D. Reinard, Y.
Yao, D.W. Price, J.M. Tour, D.L. Allara, Langmuir, 19, 2003, 8245.
[303] Heat capacity measurements of two‐dimensional self‐assembled hexadecanethiol
monolayers on polycrystalline gold, Z.S. Zhang, O.M. Wilson, M.Y. Efremov, E.A. Olson, P.V.
Braun, W. Senaratne, C.K. Ober, M. Zhang, L.H. Allen, Appl. Phys. Lett. , 84, 2004, 5198.
[304] Immobilization of glucose oxidase on a carbon surface derivatized by electrochemical
reduction of diazonium salts, C. Bourdillon, M. Delamar, C. Demaille, R. Hitmi, J. Miroux, J.
Pinson, J. Electroanal. Chem., 336, 1992, 113.
[305] Covalent Modification of Carbon Surfaces by Grafting of Functionalized Aryl Radicals
Produced from Electrochemical Reduction of Diazonium Salts, M. Delamar, R. Hitmi, J. Pinson,
J.M. Saveant, J. Am. Chem. Soc., 114, 1992, 5883.

BIBLIOGRAFÍA
254
[306] Reactions of organic monolayers on carbon surfaces observed with unenhanced Raman
Spectroscopy, Y.C. Liu, R.L. McCreery, J. Am. Chem. Soc., 117, 1995, 11254.
[307] Raman Spectroscopic Determination of the Structure and Orientation of Organic
Monolayers Chemisorbed on Carbon Electrode Surfaces, Y.C. Liu, R.L. McCreery, Anal. Chem.,
69, 1997, 2091.
[308] Mono‐ and multilayer formation by diazonium reduction on carbon surfaces monitored
with atomic force microscopy "scratching", F. Anariba, S.H. DuVall, R.L. McCreery, Anal. Chem.,
75, 2003, 3837.
[309] Electrochemically Assisted Covalent Modification of Carbon Electrodes, A.J. Downard,
Electroanal., 12, 2000, 1085.
[310] Potential‐Dependence of Self‐Limited Films Formed by Reduction of Aryldiazonium Salts
at Glassy Carbon Electrodes, A.J. Downard, Langmuir, 16, 2000, 9680.
[311] Barrier Properties of Organic Monolayers on Glassy Carbon Electrodes, A.J. Downard, M.J.
Prince, Langmuir, 17, 2001, 5581.
[312] Electrochemical and Atomic Force Microscopy Study of Carbon Surface Modification via
Diazonium Reduction in Aqueous and Acetonitrile Solutions, P.A. Brooksby, A.J. Downard,
Langmuir, 20, 2004, 5038.
[313] Modification of carbon fiber surfaces by electrochemical reduction of aryl diazonium
salts: Application to carbon epoxy composites, M. Delamar, G. Desármot, O. Fagebaume, R.
Hitmi, J. Pinson, J.‐M. Savéant, Carbon, 35, 1997, 801.
[314] Unbundled and Highly Functionalized Carbon Nanotubes from Aqueous Reactions, C.A.
Dyke, J.M. Tour, Nano Lett., 3, 2003, 1215.
[315] Electrochemical Formation of Close‐Packed Phenyl Layers on Si(111), C.H. Villeneuve, J.
Pinson, M.‐C. Bernard, P. Allongue, J. Phys. Chem. B, 101, 1997, 2415.
[316] Direct Covalent Grafting of Conjugated Molecules onto Si, GaAs, and Pd Surfaces from
Aryldiazonium Salts, M.P. Stewart, F. Maya, D.V. Konsynkin, S.M. Dirk, J.J. Stapelton, C.L.
McGuiness, D.L. Allara, J.M. Tour, J. Am. Chem. Soc., 126, 2004, 370.
[317] Covalent Modification of Iron Surfaces by Electrochemical Reduction of Aryldiazonium
Salts, A. Adenier, M.‐C. Bernard, M.M. Chehimi, E. Cabet‐Deliry, B. Desbat, O. Fagebaume, J.
Pinson, F. Podvorica, J. Am. Chem. Soc., 123, 2001, 4541.
[318] Organic Layers Bonded to Industrial, Coinage, and Noble Metals through Electrochemical
Reduction of Aryldiazonium Salts, M.‐C. Bernard, A. Chausse´, E. Cabet‐Deliry, M.M. Chehimi, J.
Pinson, F. Podvorica, C. Vautrin‐Ul, Chem. Mater., 15, 2003, 3450.

BIBLIOGRAFÍA
255
[319] Covalent Bonding of Organic Molecules to Cu and Al Alloy 2024 T3 Surfaces via
Diazonium Ion Reduction B.L. Hurley, R.L. McCreery, J. Electrochem. Soc., 151, 2004, B252.
[320] Organic Chemistry, J. McMurry, Thompson Learning, Belmont, 2008.
[321] Aryl radicals from electrochemical reduction of aryl halides. Addition on olefins, Z. Chami,
l.M. Gareil, J. Pinson, J.‐M. Savéant, A. Thiébault, J. Org. Chem., 56, 1991, 586.
[322] The Standard Redox Potential of the Phenyl Radical/Anion Couple, C.P. Andrieux, J.
Pinson, J. Am. Chem. Soc., 125, 2003, 14801.
[323] Stability of Substituted Phenyl Groups Electrochemically Grafted at Carbon Electrode
Surface, M. D’Amours, D. Bélanger, J. Phys. Chem. B 107, 2003, 4811.
[324] A Mechanism for Conductance Switching in Carbon‐Based Molecular Electronic Junctions,
A.O. Solak, S. Ranganathan, T. Itoh, R.L. McCreery, Electrochem. Solid‐State Lett., 5, 2002, E43.
[325] In Situ Raman Spectroelectrochemistry of Electron Transfer between Glassy Carbon and a
Chemisorbed Nitroazobenzene Monolayer, T. Itoh, R.L. McCreery, J. Am. Chem. Soc., 124, 2002,
10894.
[326] Phenyl layers on H–Si(111) by electrochemical reduction of diazonium salts: monolayer
versus multilayer formation, P. Allongue, C.H.d. Villeneuve, G. Cherouvrier, R. Cortés, M.‐C.
Bernard, J. Electroanal. Chem., 550‐551, 2003, 161
[327] Characterization of the Deposition of Organic Molecules at the Surface of Gold by the
Electrochemical Reduction of Aryldiazonium Cations, A. Laforgue, T. Addou, D. Belanger,
Langmuir, 21, 2005, 6855.
[328] Reductive desorption of alkanethiolate monolayers at gold: a measure of surface
coverage, M.M. Walczak, D.D. Pnpenoe, R.S. Deinhammer, B.D. Lamp, C. Chung, M.D. Porter,
Langmuir, 7, 1991, 2687.
[329] Electrochemical quartz crystal microbalance studies of adsorption and desorption of self‐
assembled monolayers of alkyl thiols on gold, T.W. Schneider, D.A. Buttry, J. Am. Chem. Soc.,
115, 1993, 12391.
[330] The electrochemical desorption of n‐alkanethiol monolayers from polycrystalline Au and
Ag electrodes, C.A. Widrig, C. C.hung, M.D. Porter, J. Electroanal. Chem., 310, 1991, 335.
[331] Electrochemical Quartz Crystal Microbalance Investigation of the Reductive Desorption of
Self‐Assembled Monolayers of Alkanethiols and Mercaptoalkanoic Acids on Au, T. Kawaguchi,
H. Yasuda, K. Shimazu, Langmuir, 16, 2000, 9830.
[332] Electrochemical study on competitive adsorption of pyridinethiol with sulfide onto
Au(111) surfaces, S. Yoshimoto, M. Yoshida, S.‐i. Kobayashi, S. Nozute, T. Miyawaki, Y.
Hashimoto, I. Taniguchi, J. Electroanal. Chem., 473, 1999, 85.

BIBLIOGRAFÍA
256
[333] Electrochemically induced transformations of monolayers formed by self‐assembly of
mercaptoethanol at gold, D.E. Weisshaar, M.M. Walczak, M.D. Porter, Langmuir, 9, 1993, 323.
[334] Voltammetric reductive desorption characteristics of alkanethiolate monolayers at single
crystal Au(111) and (110) electrode surfaces, C.‐J. Zhong, J. Zak, M.D. Porter, J. Electroanal.
Chem., 421, 1997, 9.
[335] Structure Evolution of Aromatic‐Derivatized Thiol Monolayers on Evaporated Gold, Y.‐T.
Tao, C.‐C. Wu, J.‐Y. Eu, W.‐L. Lin, Langmuir, 13, 1997, 4018.
[336] Molecular Orientation and Ordered Structure of Benzenethiol Adsorbed on Gold(111), L.‐
J. Wan, M. Terashima, H. Noda, M. Osawa, J. Phys. Chem. B, 104, 2000, 3563.
[337] 4‐Mercaptophenylboronic Acid SAMs on Gold: Comparison with SAMs Derived from
Thiophenol, 4‐Mercaptophenol, and 4‐Mercaptobenzoic Acid, D. Barriet, C.M. Yam, O.E.
Schmakova, A.C. Jamison, T.R. Lee, Langmuir, 23, 2007, 8866.
[338] Lange´s Handbook of Chemistry, 70th Anniversary Edition, J.G. Speight, McGraw‐Hill
Professional, 2004.
[339] Deposition of Metal Overlayers at End‐Groups‐Functionalized Thiolate Monolayers
Adsorbed at Au. 1. Surface and Interfacial Chemical Characterization of Deposited Cu
Overlayers at Carboxylic Acid‐Terminated Structures, E.L. Smith, C.A. Alves, J.W. Anderegg,
M.D. Porter, Langmuir, 8, 1992, 2707.
[340] Interactions between Organized, Surface‐Confined Monolayers and Vapor‐Phase Probe
Molecules. 9. Structure/Reactivity Relationship between Three Surface‐Confined Isomers of
Mercaptobenzoic Acid and Vapor‐Phase Decylamine, M. Wells, D.L. Dermody, H.C. Yang, T.
Kim, R.M. Crooks, Langmuir, 12, 1996, 1989.
[341] The Handbook of Infrared and Raman Characteristic Frequencies of Organic Molecules, D.
Lin‐Vien, N.B. Colthup, W.G. Fateley, J.G. Grasselli, I. Academic Press, Harcourt, Brace, Janovich
Publishers, San Diego, 1991.
[342] Surface Modification of Conducting Substrates. Existence of Azo Bonds in the Structure of
Organic Layers Obtained from Diazonium Salts, P. Doppelt, G. Hallais, J. Pinson, F. Podvirica, S.
Verneyre, Chem. Mater. , 19, 2007, 4570.
[343] Sterically Hindered Diazonium Salts for the Grafting of a Monolayer on Metals, C.
Combellas, F. Kanoufi, J. Pinson, F. Podvorica, J. Am. Chem. Soc., 130, 2008, 8576.
[344] An Introduction to Surface Analysis by XPS and AES, J.F. Watts, J. Wolstenholme, John
Wiley & Sons, Chichester, 2003.
[345] Theoretical Photoionization Cross Sections from 1 to 1500 keV, J.H. Scofield, Lawrence
Livermore National Laboratory, 1973.

BIBLIOGRAFÍA
257
[346] Synthesis of secorapamycin esters and amides, J.S. Skotnicki, R.M. Kearney, A.L. Smith,
Tetrahedron Lett., 35, 1994, 197.
[347] A single vessel protocol for the efficient formation of amide bonds from esters and
lactones, N.H. Kawahata, J. Brookes, G.M. Makara, Tetrahedron Lett., 43, 2002, 7221.
[348] Kinetics of amide formation through carbodiimide/N‐hydroxybenzotriazole (HOBt)
couplings, L.C. Chan, B.G. Cox, J. Org. Chem., 72, 2007, 8863.
[349] Mechanism of Amide Formation by Carbodiimide for Bioconjugation in Aqueous Media,
N. Nakajima, Y. Ikada, Bioconjugate Chem. , 6, 1995, 123.
[350] Reactions of carbodiimides. III. The reactions of carbodiimides with peptide acids, D.F.
DeTar, R. Silverstein, F.F. Rogers Jr, J. Am. Chem. Soc., 88, 1966, 1024.
[351] Surface‐grafting of phosphates onto a polymer for potential biomimetic functionalization
of biomaterials, Y.G. Ko, P.X. Ma, J. of Colloid Interf. Sci., 330, 2009, 77.
[352] Synthesis and modification of functional poly(lactide) copolymers: Toward biofunctional
materials, D.E. Noga, T.A. Petrie, A. Kumar, M. Weck, A.J. García, D.M. Collard,
Biomacromolecules, 9, 2008, 2056.
[353] Voltammetry of covalently immobilized cytochrome c on self‐assembled monolayer
electrodes, M. Collinson, E.F. Bowden, M.J. Tarlov, Langmuir, 8, 1992, 1247.
[354] Covalent immobilization of glucose oxidase onto graphitic electrodes, H.M. Wu, R. Olier,
N. Jaffrezic‐Renault, P. Clechet, A. Nyamsi, C. Martelet, Electrochim. Acta, 39, 1994, 327.
[355] Tetracarboxylic acid cobalt phthalocyanine SAM on gold: Potential applications as
amperometric sensor for H2O2 and fabrication of glucose biosensor, P.N. Mashazi, K.I.
Ozoemena, T. Nyokong, Electrochim. Acta, 52, 2006, 177.
[356] Resonant Raman spectroscopy of PAH–Os self‐assembled multilayers, N. Tognalli, A.
Fainstein, C. Bonazzola, E.J. Calvo, J. Chem. Phys., 120, 2004, 1905.
[357] Ru, Os and Ru–Os supported on mesoporous silica doped with zirconium as mild thio‐
tolerant catalysts in the hydrogenation and hydrogenolysis/hydrocracking of tetralin, D. Eliche‐
Quesada, J.M. Mérida‐Robles, E. Rodríguez‐Castellón, A. Jiménez‐López, Appl. Catal. A: Gen.,
279, 2005, 209.
[358] Sulfur−Substrate Interactions in Spontaneously Formed Sulfur Adlayers on Au(111), C.
Vericat, M.E. Vela, G. Andreasen, R.C. Salvarezza, Langmuir 17, 2001, 4919.
[359] Este trabajo fue realizado por el Lic. Ezequiel de la Llave y el Dr. Damián Scherlis
[360] Electron‐transfer study and solvent effects on the formal potential of a redox‐active self‐
assembling monolayer, D. Acevedo, H.D. Abruña, J. Phys. Chem., 95, 1991, 9590.

BIBLIOGRAFÍA
258
[361] Osmium complexes bearing functional groups: building blocks for integrated chemical
systems, C. Danilowicz, E. Corton, F. Battaglini, J. Electroanal. Chem., 445, 1998, 89.
[362] Crystal and molecular structure of dinitrogenpentaammineosmium(II) chloride,
[Os(NH3)5N2]Cl2, and related ruthenium complexes, J.E. Fergusson, J.L. Love, W.T. Robinson,
Inorg. Chem., 11, 1972, 1662.
[363] Structure of tris(2,2‐bipyridyl)ruthenium(II) hexafluorophosphate, [Ru(bipy)3][PF6]2; X‐
ray crystallographic determination D.P. Rillema, D.S. Jones, H. Levy, Chem. Commun., 1979,
849.
[364] In Situ Quartz Crystal Microbalance Study of Self‐Assembly and Mass Transfer Processes
of a Redox‐Active Osmium Complex, K. Takada, H.D. Abruña, J. Phys. Chem., 100, 1996, 17909.
[365] STM and ECSTM Study of the Formation and Structure of Self‐Assembling Osmium
Complexes on Pt(111), J.E. Hudson, H.D. Abruña, J. Phys. Chem., 100, 1996, 1036.
[366] Studies in heterogeneous equilibria. Part II. The kinetic interpretation of the nernst theory
of electromotive force J.A.V. Butler, Trans. Faraday Soc., 19, 1924, 729.
[367] T. Erdey‐Grúz, M. Volmer, Z. Phyzik. Chem., 150A, 1930, 45.
[368] On the Theory of Oxidation‐Reduction Reactions Involving Electron Transfer. I, R.A.
Marcus, J. Chem. Phys., 24, 1956, 966.
[369] Electrostatic Free Energy and Other Properties of States Having Nonequilibrium
Polarization. I, R.A. Marcus, J. Chem. Phys., 24, 1956, 979.
[370] N.S. Hush, Z. Elektrochem., 61, 1957, 738.
[371] Adiabatic Rate Processes at Electrodes. I. Energy‐Charge Relationships, N.S. Hush, J.
Chem. Phys., 28, 1958, 962.
[372] Adiabatic theory of outer sphere electron‐transfer reactions in solution N.S. Hush, Trans.
Faraday Soc., 57, 1961, 557.
[373] Coupling of protein modes to electron transfer in bacterial photosynthesis, M. Bixon, J.
Jortner, J. Phys. Chem., 90, 1986, 3795.
[374] Energetics and Kinetics of Primary Charge Separation in Bacterial Photosynthesis, D.N.
LeBard, V. Kapko, D.V. Matyushov, J. Phys. Chem. B, 112, 2008, 10322.
[375] Photoinduced electron transfer in supramolecular systems for artificial photosynthesis,
M.R. Wasielewski, Chem. Rev., 92, 1992, 435.
[376] Investigation of photoelectrochemical corrosion of semiconductors., K.W. Frese, M.J.
Madou, S.R. Morrison, J. Phys. Chem., 84, 1980, 3172.
[377] Electrogenerated Chemiluminescence. 58. Ligand‐Sensitized Electrogenerated
Chemiluminescence in Europium Labels, M.M. Richter, A.J. Bard, Anal. Chem., 68, 1996, 2641.

BIBLIOGRAFÍA
259
[378] Chemiluminescence from electron‐transfer reactions, D.M. Hercules, Acc. Chem. Res. , 2,
1969, 301.
[379] Effect of Long‐Distance Electron Transfer on Chemiluminescence Efficiencies, R.D.
Mussell, D.G. Nocera, J. Am. Chem. Soc., 110, 1988, 2764.
[380] Inverted Region Electron Transfer Demonstrated by Electrogenerated Chemiluminescence
at the Liquid/Liquid Interface, Y. Zu, F.‐R.F. Fan, A.J. Bard, J. Phys. Chem. B, 103, 1999, 6272.
[381] Role of Electrolytes on Charge Recombination in Dye‐Sensitized TiO2 Solar Cell (1): The
Case of Solar Cells Using the I‐/I3‐ Redox Couple, S. Nakade, T. Kanzaki, W. Kubo, T. Kitamura, Y.
Wada, S. Yanagida, J. Phys. Chem. B, 109, 2005, 3480.
[382] Photoinduced Charge Generation and Recombination Dynamics in Model Donor/Acceptor
Pairs for Organic Solar Cell Applications: A Full Quantum‐Chemical Treatment, V. Lemaur, M.
Steel, D. Beljonne, J.‐L. Brdas, J. Cornil, J. Am. Chem. Soc., 127, 2005, 6077.
[383] Dependence of Photocurrent and Conversion Efficiency of Titania‐Based Solar Cell on the
Qy Absorption and One Electron‐Oxidation Potential of Pheophorbide Sensitizer, X.‐F. Wang, Y.
Koyama, H. Nagae, Y. Wada, S.‐i. Sasaki, H. Tamiaki, J. Phys. Chem. C, 112, 2008, 4418.
[384] Theory of Electron Exchange Reactions in Aqueous Solution, W.F. Libby, J. Phys. Chem.,
56, 1952, 863.
[385] A Theory of Intensity Distribution in Band Systems, E. Condon, Phys. Rev. , 28, 1926, 1182.
[386] Elementary processes of photochemical reactions, J. Franck, E.G. Dymond, Trans. Faraday
Soc., 21, 1926, 536.
[387] Nuclear Motions Associated with Electron Transitions in Diatomic Molecules, E. Condon,
Phys. Rev. , 32, 1928, 858.
[388] On the Theory of Eelectrochemical and Chemical Electron Transfer Processes, R.A.
Marcus, Can. J. Chem., 37, 1959, 155.
[389] Photoprocesses in Transition Metal Complexes, Biosystems and Other Molecules:
Experiment and Theory, R.A. Marcus, P. Siddarth, N. KIuwer, Massachusetts, 1992.
[390] H. Gerischer, Z. Phys. Chem. Frankfurt am Main, 26, 1960, 326.
[391] Physical Chemistry: An advance treatise, H. Gerischer, H. Eyring, D. Henderson, W. Jost,
Academic Press, 9A. New York, 1970.
[392] The Role of Energy Levels in Semiconductor‐Electrolyte Solar Cells R. Memming, J.
Electrochem. Soc., 125, 1978, 117.
[393] Photoelectrochemistry and heterogeneous photo‐catalysis at semiconductors, A.J. Bard, J.
Photochem., 10, 1979, 59.

BIBLIOGRAFÍA
260
[394] Charge Transport in DNA‐Based Devices, D. Porath, G. Cuniberti, R. Felice, Top. Curr.
Chem., 237, 2004, 183.
[395] DNA Electron Transfer Processes: Some Theoretical Notions, Y.A. Berlin, I.V. Kurnikov, D.
Beratan, M.A. Ratner, A.L. Burin, Top. Curr. Chem., 237, 2004, 1.
[396] Quantum Chemical Calculation of Donor–Acceptor Coupling for Charge Transfer in DNA,
N. Rösch, A.A. Voityuk, Top. Curr. Chem., 237, 2004, 37.
[397] Tunneling while Pulling: The Dependence of Tunneling Current on End‐to‐End Distance in
a Flexible Molecule, J. Lin, D.N. Beratan, J. Phys. Chem. A, 108, 2004, 5655.
[398] Thermal gating of the single molecule conductance of alkanedithiols W. Haiss, H.v.
Zalinge, D. Bethell, J. Ulstrup, D.J. Schiffrin, R.J. Nichols, Faraday Discuss., 131, 2006, 253
[399] Single molecule tunneling conductance: The temperature and length dependences
controlled by conformational fluctuations, A.A. Kornyshev, A.M. Kuznetsov, Chem. Phys., 324,
2006, 276.
[400] Correlating Electron Transport and Molecular Structure in Organic Thin Films, R.E.
Holmlin, R.F. Ismagilov, R. Haag, V. Mujica, M.A. Ratner, M.A. Rampi, G.M. Whitesides, Angew.
Chem. Int. Ed., 40, 2001, 2316.
[401] Electrochemical Behavior of the Naphtoquinone Anchored onto a Gold Electrode through
the Self‐Assembled Monolayers of Aminoalkanethiol, F. Mukae, H. Takemura, K. Takehara, Bull.
Chem. Soc. Jpn., 69, 1996, 2461.
[402] Mixed monolayers formed by the self‐assembly on gold of thiol‐functionalized
anthraquinones and 1‐alkanethiols, L. Zhang, T. Lu, G.W. Gokel, A.E. Kaifer, Langmuir, 9, 1993,
786.
[403] Electrochemical Characteristics of Hydroquinone‐Terminated Self‐Assembled Monolayers
on Gold, H.‐G. Hong, W. Park, Langmuir, 17, 2001, 2485.
[404] Electron Transfer, Encyclopedia of Electrochemistry, H.O. Finklea, Vol. 10, A.J. Bard, M.
Stratmann, Eds.; Wiley‐VCH Verlag GmbH & Co.: Weinheim, 2007.
[405] Ab initio studies of electron transfer. 2. Pathway analysis for homologous organic
spacers, C. Liang, M.D. Newton, J. Phys. Chem., 97, 1993, 3199.
[406] The Electrode/Electrolyte Interface‐ A Status Report, A.J. Bard, H.D. Abruña, C.E.D.
Chidsey, L.R. Faulkner, S.W. Feldberg, K. Itaya, M. Majda, O. Melroy, R.W. Murray, J. Phys.
Chem., 97, 1993, 7147.
[407] Contribution of Intermolecular Interactions to Electron Transfer through Monolayers of
Alkanethiols Containing Amide Groups, S. Sek, B. Palys, R. Bilewicz, J. Phys. Chem. B, 106, 2002,
5907.

BIBLIOGRAFÍA
261
[408] Effect of Electrode Density of States on the Heterogeneous Electron‐Transfer Dynamics of
Osmium‐Containing Monolayers, R.J. Forster, P. Loughman, T.E. Keyes, J. Am. Chem. Soc., 122,
2000, 11948.
[409] Voltammetry of Redox‐Active Groups Irreversibly Adsorbed onto Electrodes. Treatment
Using the Marcus Relation between Rate and Overpotential, K. Weber, S.E. Creager, Anal.
Chem., 66, 1994, 3164.
[410] Cyclic Voltammetric Analysis of Ferrocene Alkanethiol Monolayer Electrode Kinetics
Based on Marcus Theory, L. Tender, M.T. Carter, R. Murray, Anal. Chem., 66, 1994, 3173.
[411] Linear‐Sweep Voltammetry of Irreversible Electron Transfer in Surface‐Confined Species
Using the Marcus Theory, T.M. Nahir, R.A. Clark, E.F. Bowden, Anal. Chem., 66, 1994, 2595.
[412] Studies of electron transfer through self‐assembled monolayers using impedance
spectroscopy, L.V. Protsailo, W.R. Fawcett, Electrochim. Acta, 45, 2000, 3497.
[413] A New Way of Using ac Voltammetry To Study Redox Kinetics in Electroactive
Monolayers, S.E. Creager, T.T. Wooster, Anal. Chem., 70, 1998, 4257.
[414] Synthesis and Characterization of Bis(S‐acetylthio)‐Derivatized Europium Triple‐Decker
Monomers and Oligomers, K.‐H. Schweikart, V.L. Malinovskii, A.A. Yasseri, J. Li, A.B. Lysenko,
D.F. Bocian, J.S. Lindsey, Inorganic Chemistry, 42, 2003, 7431.
[415] Comparison of Electron‐Transfer and Charge‐Retention Characteristics of Porphyrin‐
Containing Self‐Assembled Monolayers Designed for Molecular Information Storage, K.M. Roth,
D.T. Gryko, C. Clausen, J. Li, J.S. Lindsey, W.G. Kuhr, D.F. Bocian, J. Phys. Chem. B, 106, 2002,
8639.
[416] Interfacial bridge‐mediated electron transfer: Mechanistic analysis based on
electrochemical kinetics and theoretical modelling, M.D. Newton, J.F. Smalley, Phys. Chem.
Chem. Phys., 9, 2007, 555.
[417] Indirect Laser‐Induced Temperature Jump Study of the Chain‐Length Dependence of the
pKa's of ω‐Mercaptoalkanoic Acid Monolayers Self‐Assembled on Gold, J.F. Smalley,
Langmuir, 19, 2003, 9284.
[418] An indirect laser‐induced temperature jump study of the influence of redox couple
adsorption on heterogeneous electron transfer kinetics, J.F. Smalley, L. Geng, A. Chen, S.W.
Feldberg, N.S. Lewis, G. Cali, J. Electroanal. Chem., 549, 2003, 13.
[419] Double‐Layer‐Capacitance Titration of Self‐Assembled Monolayers of ω‐Functionalized
Alkanethiols on Au(111) Surface, T. Kakiuchi, M. Iida, S.‐i. Imabayashi, K. Niki, Langmuir, 16,
2000, 5397.

BIBLIOGRAFÍA
262
[420] Voltammetric Measurement of Interfacial Acid/Base Reactions, H.S. White, J.D. Peterson,
Q. Cui, K.J. Stevenson, J. Phys. Chem. B, 102, 1998, 2930.
[421] Impact of Electrode Geometry, Depth of Immersion and Size on Impedance
Measurements, P. Vanýsek, Can. J. Chem., 75, 1997, 1635.
[422] Fisicoquímica, I.N. Levine, McGraw‐Hill/Interamericana de España, Madrid, 1996.
[423] Adsorption, autoinhibition and autocatalysis in polarography and in linear potential
sweep voltammetry, E. Laviron, J. Electroanal. Chem., 52, 1974, 355.
[424] General expression of the linear potential sweep voltammogram in the case of
diffusionless electrochemical systems, E. Laviron, J. Electroanal. Chem., 101, 1979, 19.
[425] Effect of uncompensated resistance on the cyclic voltammetric response of an
electrochemically reversible surface‐attached redox couple: Uniform current and potential
across the electrode surface, S.W. Feldberg, J. Electroanal. Chem., 624, 2008, 45.
[426] A Simulator for Cyclic Voltammetric Responses, M. Rudolph, D.P. Reddy, S.W. Feldberg,
Anal. Chem., 66, 1994, A589.
[427] Cyclic Voltammetry: Simulation and Analysis of Reaction Mechanisms, D.K. Gosser, Jr. ,
VCH, New York, 1993.
[428] DC Relaxation Techniques for the Characterization of Fast Electrode Reactions, Z. Nagy, R.
Varma, J.R. Selman, The Electrochemical Society, Inc./John Wiley & Sons, Inc., 1987.
[429] On the Theory of the Potential and Voltage Step Relaxation Techniques for the
Investigation of Fast Electrode Reactions, Z. Nagy, Electrochim. Acta, 25, 1979, 575.
[430] Error of the Potential and Voltage Step Relaxation Techniques for the Measurement of
Kinetic of Electrode Reactions, Z. Nagy, J. Electrochem. Soc., 129, 1982, 1943.
[431] On the Effect of Finite Rise Time of Pulse Generators on Some Relazation Techniques for
Electrode Kinetic Investigations, Z. Nagy, J. Electrochem. Soc., 125, 1978, 1809.
[432] An informative subtlety of itemperature‐jump or coulostatic responses for surface‐
attached species, J.F. Smalley, M.D. Newton, S.W. Feldberg, Electrochem. Comm., 2 2000, 832.
[433] Theory of the interfacial potential distribution and reversible voltammetric response of
electrodes coated with electroactive molecular films, C.P. Smith, W.H. S., Anal. Chem., 64,
1992, 2398.
[434] H.L.F. von Helmholtz, Ann. Physik, 89, 1853, 211.
[435] H.L.F. von Helmholtz, Ann. Physik, 7, 1879, 337.
[436] G. Gouy, Compt. Rend., 149, 1910, 654.
[437] G. Gouy, J. Phys. Radium, 9, 1910, 457.
[438] D.L. Chapman, Phil. Mag., 25, 1913, 475.

BIBLIOGRAFÍA
263
[439] O. Stern, Z. Elektrochem., 30, 1924, 508.
[440] Structure of monolayers formed by coadsorption of two n‐alkanethiols of different chain
lengths on gold and its relation to wetting, P.E. Laibinis, R.G. Nuzzo, G.M. Whitesides, J. Phys.
Chem., 96, 1992, 5097.
[441] The Effect of Surface Polarity on the Electrochemical Double Layer and Its Influence on
the Measurement of the Standard Rate Constant of Electron Transfer, P.K. Eggers, D.B. Hibbert,
M.N. Paddon‐Row, J.J. Gooding, J. Phys. Chem. C, 113, 2009, 8964.
[442] Surface Electron Transfer Processes, R.J.D. Miller, G.L. McLendon, A.J. Nozik, W.
Schmickler, F. Willig, VCH Publishers, New York, 1995.
[443] The Wetting of Monolayer Films Exposing Ionizable Acids and Bases, T.R. Lee, R.I. Carey,
H.A. Biebuyck, G.M. Whitesides, Langmuir, 10, 1994, 741.
[444] An Indirect Laser‐Induced Temperature Jump Determination of the Surface pKa of 11‐
Mercaptoundecanoic Acid Monolayers Self‐Assembled on Gold, J.F. Smalley, K. Chalfant, S.W.
Feldberg, T.M.N. Nahir, E.F. Bowden, J. Phys. Chem. B, 103, 1999, 1676.
[445] Contact‐Angle Titrations of Mixed .omega.‐Mercaptoalkanoic Acid/Alkanethiol
Monolayers on Gold. Reactive vs Nonreactive Spreading, and Chain Length Effects on Surface
pKa Values, S.E. Creager, J. Clarke, Langmuir, 10, 1994, 3675.
[446] Direct Atomic Force Microscopic Determination of Surface Charge at the Gold/Electrolyte
InterfaceThe Inadequacy of Classical GCS Theory in Describing the Double‐Layer Charge
Distribution, J. Wang, A.J. Bard, J. Phys. Chem. B, 105, 2001, 5217.
[447] Intermolecular and Surface Forces, J.N. Israelachvili, Academic Press, San Diego, 1992.
[448] Effect of Acid−Base Equilibria on the Donnan Potential of Layer‐by‐Layer Redox
Polyelectrolyte Multilayers, M.E. Tagliazucchi, F.J. Williams, E.J. Calvo, J. Phys. Chem. B, 111,
2007, 8105.
[449] A molecular theory of chemically modified electrodes with self‐assembled redox
polyelectrolye thin films: Reversible cyclic voltammetry, M.E. Tagliazucchi, E.J. Calvo, I. Szleifer,
Electrochim. Acta, 53, 2008, 6740.
[450] Voltammetry of molecular films containing acid/base groups, C.P. Smith, H.S. White,
Langmuir, 9, 1993, 1.
[451] Probing Potential‐Tuned Resonant Tunneling through Redox Molecules with Scanning
Tunneling Microscopy, N. Tao, Phys. Rev. Lett., 76, 1996, 4066.
[452] In situ scanning tunnelling spectroscopy of inorganic transition metal complexes T.
Albrecht, K. Moth‐Poulsen, J.B. Christensen, A. Guckian, T. Bjornholm, J.G. Vos, J. Ulstrup,
Faraday Discuss., 131, 2006, 265.

BIBLIOGRAFÍA
264
[453] Single‐electron transistor of a single organic molecule with access to several redox states
S. Kubatkin, A. Danilov, M. Hjort, J. Cornil, J.‐L. Bredas, N. Stuhrhansen, T. Bjornholm, Nature,
425, 2003, 698.
[454] Reproducible Measurement of Single‐Molecule Conductivity, X.D. Cui, A. Primak, X.
Zarate, J. Tomfohr, O.F. Sankey, A.L. Moore, T.A. Moore, D. Gust, G. Harris, S.M. Lindsay,
Science, 294, 2001, 571.
[455] Covalently Bonded Organic Monolayers on a Carbon Substrate: A New Paradigm for
Molecular Electronics, S. Ranganathan, I. Steidel, F. Anariba, R.L. McCreery, Nano Lett., 1, 2001,
491.
[456] Conductance of single 1,4‐disubstituted benzene molecules anchored to Pt electrodes, M.
Kiguchi, S. Miura, K. Hara, M. Sawamura, K. Murakoshi, Appl. Phys. Lett., 91, 2007, 053110.
[457] In situ scanning tunneling microscopy in electrolyte solutions, K. Itaya, Prog. Surf. Sci., 58,
1998, 121.
[458] The investigation of redox reactions with a scanning tunneling microscope: Experimental
and theoretical aspects, W. Schmickler, C. Widrig, J. Electroanal.Chem., 336, 1992, 213.
[459] Transistor effects and in situ STM of redox molecules at room temperature, T. Albretch, A.
Guckian, J. Ulstrup, J.G. Vos, IEEE Trans. Nanotechnology, 4, 2005, 430.
[460] Electrochemical Gate‐Controlled Conductance of Single Oligo(phenylene ethynylene)s, X.
Xiao, L.A. Nagahara, A.M. Rawlett, N.J. Tao, J. Am. Chem. Soc., 127, 2005, 9235.
[461] Transistor‐like Behavior of Transition Metal Complexes, T. Albrecht, A. Guckian, J.
Ulstrup, J.G. Vos, Nano Lett., 5, 2005, 1451.
[462] Electrochemical Origin of Voltage‐Controlled Molecular Conductance Switching, J. He, S.
Lindsay, J.W. Ciszek, J.M. Tour, J. Am. Chem. Soc., 128, 2006, 14828.
[463] Scanning Tunneling Spectroscopy in an Ionic Liquid, T. Albretch, K. Moth‐Poulsen, J.B.
Christensen, T. Bjornholm, J. Ulstrup, J. Am. Chem. Soc., 128, 2006, 6574.
[464] The development of bioelectrochemistry, H.A.O. Hill, Coord. Chem. Rev., 151, 1996, 115.
[465] Unravelling single metalloprotein electron transfer by scanning probe techniques A.
Alessandrini, S. Corni, P. Facci, Phys. Chem. Chem. Phys., 8, 2006, 4383.
[466] In Situ Scanning Tunneling Microscopy of a Redox Molecule as a Vibrationally Coherent
Electronic Three‐Level Process, E.P. Friis, Y.I. Kharkats, A. Kuznetsov, M., J. Ulstrup, J. Phys.
Chem. A, 102, 1998, 7851.
[467] Indirect Laser‐Induced Temperature Jump Study of the Chain‐Length Dependence of the
pKa's of Mercaptoalkanoic Acid Monolayers Self‐Assembled on Gold, J.F. Smalley, Langmuir,
19, 2003, 9284.

BIBLIOGRAFÍA
265
[468] Coulostatic method for the kinetic study of fast electrode processes. II. Experimental
results, P. Delahay, A. Aramata, J. Phys. Chem., 66, 1962, 2208.
[469] Coulostatic method for the kinetic study of fast electrode processes. I. Theory, P. Delahay,
J. Phys. Chem., 66, 1962, 2204.

