bibing.us.esbibing.us.es/proyectos/abreproy/4166/descargar_fichero/pfc+versión... · estudio de...
TRANSCRIPT

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS
DE LAS SUSTANCIAS PRIORITARIAS ORGÁNICAS
DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
Samuel Ordás Naise
Departamento de Ingeniería Química y Ambiental Universidad de Sevilla
Sevilla, a 10 de Julio de 2006

Quisiera agradecer a los profesores Don José Morillo Aguado y Don José Usero García, la paciencia que han tenido en la larga realización de este Proyecto Fin de Carrera, así como a Cristina, que siempre estuvo dispuesta a ayudarme si lo necesitaba.

A mis padres,
que vivieron toda la realización del proyecto

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
1
ÍNDICE DE CONTENIDOS
I. INTRODUCCIÓN Y OBJETIVOS. .................................................................................... 1
II. SUSTANCIAS PRIORITARIAS..................................................................................... 4 1. Introducción. .................................................................................................................. 4 2. Sustancias prioritarias...................................................................................................... 4
III. MÉTODOS CROMATOGRÁFICOS. ............................................................................. 15 1. Introducción. ................................................................................................................ 15 2. Características básicas................................................................................................... 17 3. Magnitudes fundamentales de la separación: selectividad, eficacia y resolución .................. 20
3.1. Selectividad. .......................................................................................................... 24 3.2. Eficacia. ................................................................................................................ 24 3.3. Resolución............................................................................................................. 26
IV. CROMATOGRAFÍA DE LÍQUIDOS DE ALTA EFICACIA............................................... 29 1. Introducción. ................................................................................................................ 29 2. Fase inversa. ................................................................................................................ 29 3. Tipos de relleno ............................................................................................................ 31 4. Modelos de retención .................................................................................................... 32 5. Intercambio iónico. ....................................................................................................... 38 6. Pares iónicos. ............................................................................................................... 39 7. Exclusión molecular....................................................................................................... 41 8. Separaciones quirales.................................................................................................... 43 9. Afinidad. ...................................................................................................................... 44 10. Modos de elusión. ....................................................................................................... 45
V. CROMATOGRAFÍA DE GASES. .................................................................................... 48 1. Introducción. ................................................................................................................ 48 2. Sistemas de inyección de muestras................................................................................. 48
2.1. Modalidades de inyección........................................................................................ 49 2.2. Dispositivos externos para introducción de muestras ................................................. 53
3. La columna.................................................................................................................. 57 3.1. Tipos de columnas y dimensiones............................................................................ 57
3.1.1. Efecto de las dimensiones sobre el comportamiento de las columnas ................... 59 3.1.2. Efecto de las dimensiones sobre la eficacia y resolución. ..................................... 61 3.1.3. Preparación de columnas ................................................................................. 64
3.2. La fase móvil. ........................................................................................................ 65 3.2.1. Naturaleza y propiedades de las fases móviles más usadas. ................................ 66
3.3. Fases estacionarias................................................................................................. 68 3.3.1. Fases estacionarias líquidas. ............................................................................. 69 3.3.2. Fases estacionarias sólidas. .............................................................................. 77
4. Sistemas de detección. .................................................................................................. 79 4.1. Detectores de conductividad térmica........................................................................ 79 4.2. Detectores de ionización ......................................................................................... 80
4.2.1. Detector de ionización de llama (FID)................................................................ 81 4.2.2. Detector termoiónico (TID)............................................................................... 82

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
2
4.2.3. Detector de captura de electrones (DCE). .......................................................... 83 4.2.4. Detector de fotoionización (DFI). ...................................................................... 84
4.3. Detectores electroquímicos. .................................................................................... 85 4.3.1. Detector de conductividad electrolítica (DCEL). .................................................. 85
4.4. Espectógrafo de Masas. .......................................................................................... 85 4.4.1. Sistema de introducción de muestra.................................................................. 86 4.4.2. Cámara de ionización....................................................................................... 89 4.4.3. Analizador....................................................................................................... 91 4.4.4. El Detector...................................................................................................... 93
4.5. Aplicaciones........................................................................................................... 94 5. Control del proceso cromatográfico................................................................................. 95 6. Temperatura programada .............................................................................................. 98
VI. PRECONCENTRACIÓN Y PURIFICACIÓN DE MUESTRAS........................................ 101 1. Introducción. .............................................................................................................. 101 2. Técnicas de extracción con disolventes. ........................................................................ 102 3. Extracción con fluidos supercríticos (McNally, 1996; King, 2002). .................................... 103 4. Técnicas de destilación. ............................................................................................... 103 5. Extracción-destilación simultánea (EDS). ....................................................................... 103 6. Inyección directa. ....................................................................................................... 103 7. Técnicas de fraccionamiento por absorción.................................................................... 104
7.1. Trampas capilares abiertas (TCA). ......................................................................... 105 7.2. Trampas capilares abiertas múltiples (TCAM). ......................................................... 106 7.3. Tubos Rellenos (Packed Beds)............................................................................... 106 7.4. Microextracción en fibra capilar (MEFC).................................................................. 106 7.5. Extracción Dinámica en Fase Sólida (EDFS). ........................................................... 107 7.6. Extracción por Absorción sobre Barra Agitadora (ESBA). .......................................... 108 7.7. Dispersión en Matriz de Fase Sólida (DMFS) ........................................................... 111
VII. MÉTODOS PROPUESTOS. ...................................................................................... 112 1. Análisis. ..................................................................................................................... 112
1.1. Introducción ........................................................................................................ 112 1.2. Pesticidas semivolátiles ......................................................................................... 112 1.3. Hidrocarburos policíclicos. ..................................................................................... 115 1.4. Organoclorados volátiles. ...................................................................................... 118 1.5. Otras sustancias................................................................................................... 120 1.6. Sustancias Prioritarias Orgánicas. .......................................................................... 123
2. Cromatografía de gases con espectrometría de masas (CG-EM). ..................................... 124 2.1. Instrumentación................................................................................................... 125 2.2. Procedimiento...................................................................................................... 126
2.2.1. Material. ....................................................................................................... 127 2.2.2. Factores de influencia. ................................................................................... 128
VIII. CONCLUSIONES................................................................................................... 132
IX. ANEXOS .................................................................................................................. 134
X. BIBLIOGRAFÍA......................................................................................................... 147

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
1
I. INTRODUCCIÓN Y OBJETIVOS.
Una gran cantidad de compuestos orgánicos individuales se ha introducido en las
aguas de ríos, lagos y mares como resultado de las actividades antropogénicas. Las
principales fuentes responsables de la presencia de estos compuestos en las aguas
superficiales son las aguas residuales y emisiones industriales que contienen, entre otros,
diferentes disolventes orgánicos, compuestos orgánicos específicos y sus productos de
degradación; las aguas residuales urbanas (aceites contaminantes, detergentes), la
contaminación asociada a los medios de transporte (aceites contaminantes, compuestos
organometálicos y otros contaminantes específicos) y la actividad agraria (plaguicidas y
sus productos de degradación).
Considerando las vías de transmisión, los contaminantes orgánicos alcanzan los ríos
y embalses a través de fuentes puntuales (descargas de aguas residuales, fugas de aceite
de los barcos y otros vertidos), fuentes difusas (escorrentías en terrenos agrícolas, malas
prácticas de aplicación de plaguicidas, procesos de lixiviado en vertederos) o por
transporte a través de la atmósfera y posterior deposición.
En este contexto es donde surge la Directiva 2000/60/CE, también conocida
como Directiva Marco de Aguas (DMA), y publicada el día 22 de diciembre de 2000 en
el Diario Oficial de las Comunidades Europeas (CE) del Parlamento Europeo y del Consejo
de 23 de octubre de 2000, por la que se establece un marco comunitario para la
protección de las aguas superficiales continentales, de transición, costeras y subterráneas,
para prevenir o reducir su contaminación, promover su uso sostenible, proteger el medio
ambiente, mejorar el estado de los ecosistemas acuáticos y atenuar los efectos de las
inundaciones y las sequías.
La Directiva Marco relativa al Agua (DMA) extiende el campo de la protección
acuática a todas las aguas: establece el objetivo claro de que en el año 2015 debe
conseguirse un “buen estado ecológico” para todas las aguas europeas y el uso sostenible
del agua. La nueva Directiva representa un planteamiento ambicioso e innovador, con
vistas a la gestión del agua. Los elementos principales de la legislación incluyen:

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
2
o La protección de todas las aguas – ríos, lagos, aguas costeras y aguas
freáticas
o El establecimiento de objetivos ambiciosos con el fin de asegurar que todas
las aguas se encuentren en “buen estado ecológico” en el año 2015
o La necesidad de establecer una cooperación transfronteriza entre países, y
también de todas las partes implicadas
o Asegurar la participación activa de todos los interesados, incluidas ONG y
comunidades locales, en todas las actividades de gestión del agua
o Contar con políticas de fijación de precios del agua y garantizar que el que
contamine pague
o Buscar un equilibrio entre los intereses del medio ambiente y los que
dependen de él
Los compuestos que afectan a la calidad del agua poseen propiedades físicas,
químicas y toxicológicas muy diferentes, por lo que la propuesta de medidas efectivas
para proteger las aguas litorales de su impacto adverso requiere disponer de una
información muy completa acerca de su distribución en los diferentes compartimentos
medioambientales. La calidad de la información referente al grado de contaminación de
las aguas depende de la información obtenida a partir de los métodos analíticos aplicados.
El principal problema con el que se encuentran los métodos de análisis de trazas de
compuestos orgánicos en el medio ambiente es la necesidad de cubrir un gran número de
analitos de interés a los bajos niveles de detección requeridos y en presencia de un
número incluso mayor de compuestos que pueden actuar como posibles interferentes
durante el análisis. Es necesario, por tanto, aplicar técnicas instrumentales sofisticadas
como son la cromatografía de gases (CG), la cromatografía líquida de alta resolución
(CLAE) o las llamadas técnicas acopladas en las que se combina una técnica de
separación apropiada con métodos de determinación espectrales como la espectrometría
de masas, la espectroscopia ultravioleta, espectroscopia infrarroja o la espectroscopia de
emisión atómica.

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
3
Por otro lado, si se quieren determinar contaminantes orgánicos a niveles de
concentración tan bajos como los presentes en aguas (ng/L), es inevitable aplicar
procesos de preconcentración previo al análisis de los mismos. El objetivo de estos
procesos de preconcentración es aislar los analitos de interés de la matriz original y
transferirlos a una disolución compatible con el procedimiento de análisis instrumental a
aplicar. El factor de enriquecimiento de este proceso de transferencia puede oscilar entre
uno y cuatro órdenes de magnitud.
El principal objetivo de este proyecto es realizar un estudio crítico de las distintas
técnicas existentes para el análisis de las sustancias prioritarias de la Directiva Marco de
Aguas (2000/69/CE) y, sobre la base de lo anterior, proponer las técnicas más adecuadas
para el análisis de estas sustancias.

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
4
II. SUSTANCIAS PRIORITARIAS.
1. Introducción.
Son objeto de estudio aquellas que aparecen en el Diario Oficial de las Comunidades
Europeas, “Decisión Nº 2455/2001/CE del Parlamento Europeo y del Consejo de 20 de
noviembre de 2001, por la que se aprueba la lista de sustancias prioritarias en el ámbito
de la política de aguas, y por la que se modifica la Directiva 2000/60/CE”.
Estas sustancias presentan métodos normalizados pero que no siempre alcanzan la
sensibilidad que requiere la nueva normativa, por lo que habrá que buscar nuevas
técnicas de análisis con el objeto de llegar a las sensibilidades requeridas.
Características y métodos normalizados para el análisis de los compuestos
prioritarios según la EPA (entre paréntesis aparece un compuesto representativo).
2. Sustancias prioritarias.
1. Alacloro: compuesto organoclorado cuya fórmula química es C14H20ClNO2. Se usa como herbicida en ciertos cultivos, como los de maíz, col y algodón. El alacloro es moderadamente tóxico para los organismos acuáticos. No se bioacumula significativamente. El uso de alacloro está prohibido en muchos Estados Miembros. Actualmente sólo tiene permiso para ser utilizado en cinco de ellos, entre los que se encuentra España, ya que parece ser un producto mejor adaptado a climas cálidos. Se fabrica en nuestro país.
2. Aldrina: compuesto organoclorado cuya fórmula química C12H8Cl6. es un insecticida de amplio espectro que se usa principalmente para combatir plagas del suelo y del algodón y en la lucha contra la langosta. Se genera en el medio ambiente como producto de la descomposición de la dieldrina; en los organismos vivos se transforma en dieldrina por efecto del metabolismo. Presenta una alta toxicidad para los organismos acuáticos y persistencia en el agua.
Estructura química del Alacloro
Estructura química del Aldrina

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
5
3. Antraceno: hidrocarburo aromático
policíclico con fórmula química C6H4(CH)2C6H4. Se usa para síntesis química, creosote y otros preservantes de madera, membranas resistentes al agua y productos relacionados, así como para otros productos industriales y usos científicos específicos. Hace algunos años se empleó para la producción de quinina, en la actualidad se usa en la manufactura de productos del tinte. Además, otros productos que contienen antraceno como aceite de antraceno o alquitrán de carbón se han utilizado en cosmética, Ej.: jabones, lociones, aceites, champú y gel. Se bioacumula en la cadena alimentaria, concretamente en organismos acuáticos y plantas.
4. Atracina: compuesto organonitrogenado cuya fórmula química es C8H14ClN5. Se usa para eliminar malas hierbas en cultivos, y como herbicida no selectivo en tierras no cultivadas. La degradación de esta sustancia en el agua es lenta, pero no se acumula en la cadena alimentaria. Su utilización está permitida en Europa, excepto en Finlandia, Suiza, Dinamarca, Alemania y Austria.
5. Benceno: Hidrocarburo aromático con fórmula química C6H6. La principal utilidad del benceno es la producción de tres de sus derivados: etilbenceno, cumeno y ciclohexano. Y éstos son a su vez utilizados en la manufactura de otros productos. El benceno forma la base de una gran variedad de intermediarios aromáticos y para el grupo de compuestos cicloalipáticos. Es empleado como base en la manufactura de gomas sintéticas, plásticos, resinas, lubricantes, tintes, detergentes, medicamentos y pesticidas. El benceno es poco soluble en agua y no se acumula en plantas o animales.
6. Difeniléteres bromados: compuesto organohalogenado con fórmula química C12HxOBry. De esta familia de compuestos tan sólo se aconseja analizar como representante el Pentabromobifeniléter (penta-BDPE); ya que otros como el octa- y deca- BDPE están aún en estudio bajo la Regulación 793/93. El Penta-BDPE se utiliza en materiales esponjosos de poliuretano y productos de tapicería y
Estructura química del Antraceno
Estructura química del Atracina
Estructura química del Benceno

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
6
aplicaciones automovilísticas; también se usa en la producción de poliuretano no poroso. No se disuelven fácilmente en agua; se adhieren a las partículas y se depositan en el fondo. Pueden acumularse en peces. Otros datos: Penta-BDPE no es producido en la Unión Europea, sino que es importado.
7. Cadmio y sus compuestos: Metal con símbolo químico Cd. Se utiliza en baterías Ni-Cd, en la producción de pigmentos, como estabilizante, como agente de galvanoplastia del metal y en aleaciones. El Cadmio se adhiere fuertemente a partículas en la tierra. Parte del cadmio se disuelve en el agua; no se degrada en el medio ambiente, pero puede cambiar de forma química. Las plantas, peces y otros animales incorporan el cadmio del medio ambiente. Este metal permanece en el organismo por largo tiempo y puede acumularse después de años de exposición a bajos niveles.
8. Parafinas cloradas de cadena corta: compuesto organoclorado con fórmula química: CnH2n+2–zClz (n =10-13; z = 4-10). Son utilizadas como plastificantes de PVC, fluidos para trabajar el metal, retardantes de llama en textiles y caucho, textiles resistentes al agua, pinturas, sellante y adhesivos, cuero, etc. Son muy tóxicas para los organismos acuáticos y pueden provocar efectos nocivos a largo plazo sobre el medio acuático.
9. Clorofenvinfos: compuesto organofosforado con fórmula química: C12H14Cl3O4P. Se usa como aracnicida e insecticida. No parece acumularse en plantas ni animales acuáticos. Los clorofenvinfos están en el mercado y se producen dentro de la Unión Europea.
10. Cloropirifos: compuesto organoclorado con fórmula química C9H11Cl3NO3PS. Se usa Como insecticida, aracnicida y nematicida. Se ha usado ampliamente en viviendas y en agricultura. En el hogar, se usa para controlar cucarachas, pulgas y termitas; también se utiliza en collares de animales domésticos para erradicar pulgas y garrapatas. En agricultura, se emplea para suprimir garrapatas en el ganado y en forma de rocío para el control de plagas en las cosechas. Los clorpirifos se adhiere firmemente a partículas del suelo, además, es poco soluble en agua. Es muy tóxico para los organismos acuáticos.
Estructura química del Clororfenvinfos
Estructura química del Clororpirifos

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
7
11. o, p-DDT: pesticida usado extensamente en el pasado para controlar insectos
en agricultura e insectos que transmiten enfermedades como la malaria. Su fórmula química es C14H9Cl5. Aún se usa en algunos paises, especialmente en zonas tropicales. Sus efectos sobre la salud son graves.
12. p, p-DDT: pesticida usado extensamente en el pasado para controlar insectos en agricultura e insectos que transmiten enfermedades como la malaria. Su fórmula química es C14H9Cl5. Aún se usa en algunos países, especialmente en zonas tropicales. Sus efectos sobre la salud son graves.
13. 1,2-Dicloroetano: compuesto organoclorado cuya fórmula química es ClCH2CH2Cl. El uso más común es la producción de cloruro de vinilo, sustancia que se emplea para manufacturar una gran variedad de productos plásticos y de vinilo, incluyendo cañerías, tapices de muebles y automóviles, cubiertas, artículos para el hogar, piezas de automóviles. Se usa también como solvente y se añade a la gasolina para evitar el uso del plomo como antidetonante. Se degrada muy lentamente en el agua. El 1,2-dicloroetano liberado se evaporará al aire o pasará a través del suelo y entrará al agua subterránea.
14. Diclorometano: Halocarbono, su fórmula química es CH2Cl2. El cloruro de metilo es un solvente industrial. Se utiliza para quitar pintura, para la preparación de plásticos revestidos (adhesivos), en la purificación y aislamiento de intermediarios o en la manufactura de productos químicos finos o farmacéuticos. También puede emplearse como un solvente de procesos para reacciones químicas (producción del ester de la celulosa y formación de cinta fotográfica, producción de policarbonato) y para la extracción de materiales de la fruta y de productos de bebidas (Ej.: descafeinado del café). El cloruro de metilo no se disuelve fácilmente en agua. Es improbable que el cloruro de metilo se acumule en plantas o en animales.
15. Dieldrina: es un insecticida que se utiliza principalmente en los cultivos de algodón. Su fórmula química es C12H8Cl6O. Debido a su excelente solubilidad en el agua, la dieldrina se acumula en los sistemas acuáticos, siendo muy tóxica para sus organismos.
Estructura química del o, p-DDT
Estructura química del p, p-DDT
Estructura química de la Dieldrina

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
8
16. Di(2-etilhexil)ftalato (DEHP): Ftalato con fórmula química C24H38O4. Es
utilizado generalmente como plastificante en polímeros (principalmente en PVC). También es utilizado en aplicaciones no poliméricas como adhesivos y sellantes, pintura y lacas, cartuchos de tinta de impresora y condensadores. También es utilizado en materiales de cerámica avanzados utilizados para aplicaciones electrónicas y estructurales. Generalmente también se añade a los plásticos para hacerlos más flexibles. El DEHP está presente en productos de plástico tales como cubiertas de paredes, manteles, baldosas, tapices de muebles, cocinas de baño, mangueras, forros de piscinas, ropa impermeable, dodotis para bebés, muñecas, juguetes, zapatos, tapices y techos de automóviles, cubierta de alambres y cables, tuberías para uso médico y bolsas para almacenar sangre. No se evapora ni se disuelve fácilmente en agua, especialmente si se encuentran en profundidad. Se disuelve más rápido en agua si ésta contiene gas, aceite o limpiadores de pintura. El DEHP en el suelo o el agua puede ser degradado por microorganismos a compuestos que no son dañinos. Se encuentra en plantas, peces y otros animales, pero los animales que ocupan los lugares más altos en la cadena alimentaria pueden degradar DEHP, de manera que los niveles en sus tejidos son generalmente bajos.
17. Diuron: compuesto organoclorado, su fórmula química es C6H3Cl2NHCON(CH3)2. Se usa como herbicida para el control de la mala hierba en tierras no cosechadas. Muy peligroso para el ambiente acuático. Puede provocar daños a largo plazo. Su utilización está registrada en todos los Estados Miembros, excepto en Finlandia y Suecia.
18. Endosulfán (alpha-endosulfán): compuesto organoclorado con fórmula química C6H6Cl6O3S. Se usa para controlar insectos tanto en cosechas de productos comestibles como no-comestibles, y también como preservativo para madera No se disuelve fácilmente en agua, y además se adhiere a partículas en suspensión o a sedimentos en el fondo. Puede acumularse en los cuerpos de animales que habitan en aguas contaminadas con endosulfán. Es muy tóxico para algunas especies.
19. Endrina: su fórmula química es C12H8Cl6O. Es un pesticida para controlar insectos, roedores y pájaros. La sustancia es muy tóxica para los organismos acuáticos. En la cadena alimentaria referida a los seres humanos tiene lugar bioacumulación.
Estructura química del Endosulfán
Estructura química de la Endrina

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
9
20. Fluoranteno: su fórmula química es C16H10. A lo largo de la cadena
alimentaria de interés para el ser humano, se produce una bioacumulación, especialmente en aceites y grasas.
21. Hexaclorobenceno: compuesto organoclorado, su fórmula química es C6Cl6. Los principales metabolitos son pentaclorofenol, tetraclorohidroquinona y pentaclorotiofenol, pero se han descrito más de 43 metabolitos diferentes. El HCB ya no se produce ni se utiliza intencionadamente en la Unión Europea. El HCB se utilizó anteriormente como funguicida, en la manufactura de barras de aluminio y grafito y como intermediario en la producción de compuestos aromáticos clorados. Se encuentra asimismo presente como impureza en varios pesticidas, como la atracina, y en numerosos procesos químicos, incluyendo la fabricación de percloroetileno, clorobencenos, y otros compuestos orgánicos clorados, como PVC. Es tóxico y se bioacumula.
22. Hexaclorobutadieno: compuesto organoclorado, su fórmula química es Cl2:CClCCl:CCl2. Se utilizaba principalmente como solvente para caucho y otros materiales, como líquido intercambiador de calor, como líquido transformador, fluido hidráulico y como licor para eliminar manchas de hidrocarburos. También ha sido empleado como preparador de semillas y funguicida para varias cosechas. También era utilizado en la manufactura de productos como barras de grafito y aluminio. El cincuenta por ciento del hexaclorobutadieno en el agua puede degradarse a otros productos químicos en aproximadamente 30 días. Los peces y mariscos pueden acumular hexaclorobutadieno. Ya no se produce en la Unión Europea.
23. Hexaclorociclohexano (α-isómero, Lindano): compuesto organoclorado. Hay ocho formas químicas (llamadas isómeros) del HCH. Las más comunes son alfa-, beta-, gama-, y delta-HCH. El más común de los isómeros es el gama-HCH (conocido también como lindano). Este compuesto se usó como insecticida en frutas y cosechas de hortalizas (incluso en tabaco y hortalizas cultivadas en invernaderos) y en plantaciones forestales (incluso árboles de Navidad). Aún se
Estructura química del Fluoranteno
Estructura química del Hexaclorobenceno
Estructura química del Hexaclorobutadieno

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
10
sigue usando en ungüentos para tratar piojos en la cabeza y el cuerpo y contra la sarna. En el suelo, el sedimento y el agua, es degradado a sustancias menos peligrosas por algas, hongos y bacterias. Los isómeros de los HCH se degradan rápidamente en agua; el lindano no permanece en el agua por más de 30 días. Puede acumularse en tejidos grasos de peces. El lindano es tóxico para peces y organismos acuáticos invertebrados. Muchos países prohíben la utilización de lindano.
24. Isodrina: pesticida organoclorado con fórmula química C12H8Cl6.
25. Isoproturón: compuesto organonitrogenado con fórmula química C12H18N2O. Se usa como herbicida sistemático selectivo en el control de malas hierbas. No es relevante la bioacumulación en peces.
26. Plomo y sus compuestos: metal cuyo símbolo químico es Pb. El plomo tiene muchos usos diferentes como fabricación de baterías, productos rodados, compuestos químicos: (estabilizantes, esmaltes de cerámica, cristal para tubos de monitores de ordenador), municiones, pesos, aleaciones (soldaduras, fabricación de cobre amarillo), forros de cable (cables de transmisión) y aditivos en gasolina. El plomo no se degrada, sin embargo los compuestos de plomo son transformados por la luz solar, el aire y el agua. El movimiento del plomo desde el suelo hasta las aguas subterráneas dependerá del tipo de compuesto de plomo y de las características del suelo. El plomo y sus compuestos se acumulan en los seres vivos.
27. Mercurio y sus compuestos: metal cuyo símbolo químico es Hg. El mercurio metálico se utiliza en la producción de gas de cloro y sosa cáustica y en termómetros, implantes dentales y pilas. Las sales de mercurio se emplean en cremas para aclarar la piel y en cremas y ungüentos antisépticos. El
Estructura química del Hexaclorociclohexano
Estructura química del Isoproturón Estructura química de la Isodrina

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
11
metilmercurio puede formarse en el agua y el suelo por la acción de bacterias, y se acumula en los tejidos de peces.
28. Naftaleno: hidrocarburo aromático policíclico, su fórmula química es C10H8. El Naftaleno tiene una gran variedad de aplicaciones: como materia de base del anhídrido ftálico, en la manufactura de ciertos colorantes, creosote (preservantes de madera), repelente de polillas, en la pirotecnia para los efectos especiales de la industria cinematográfica, en la síntesis de algunos productos farmacéuticos y pesticidas, etc. Se disuelve moderadamente en agua; no es bioacumulable. Una de las empresas más relevantes en la producción de Naftaleno se encuentra España.
29. Níquel y sus compuestos: elemento metálico, su símbolo químico es el Ni. Se emplea en unos 300,000 productos aproximadamente para consumo, aplicaciones industriales, militares, de transporte, en la marina y en la arquitectura. Aunque en la mayoría de los casos se utiliza como aleación. El níquel presenta una considerable toxicidad en peces y plantas acuáticas.
30. Nonilfenoles (4-(para)-nonilfenol): hidrocarburo aromático, su fórmula química es C15H24O. Una gran cantidad de nonilfenoles se utilizan para la producción de nonilfenol etoxilato (NPEs). Los nonilfenoles se utilizan como intermediarios químicos para resinas y aditivos, detergentes industriales, procesado de textil y cuero, emulsiones de polimerización y productos agroquímicos. El medio acuático es el más sensible a este tipo de sustancia, los riesgos de envenenamiento son muy elevados. Tienen tendencia a acumularse en los organismos vivos.
31. Octilfenoles (para-tert-octilfenol): hidrocarburo aromático, su fórmula química es C8H17C6H4OH. Usos: Se utiliza en la producción de etoxilatos, como surfactantes, etc. Sustancia potencialmente persistente o muy persistente y tóxica.
Estructura química del Naftaleno
Estructura química del Nonilfenol
Estructura química del Octilfenol

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
12
Estructura química del Pentaclorobenceno
31. Pentaclorobenceno: hidrocarburo aromático, su fórmula química es C6HCl5. Se usa como intermediario químico en la producción del funguicida quintoceno (pentacloronitrobenceno). Es también una impureza en otros productos, como hexaclorobenceno. Es persistente y bioacumulable. Muy tóxico para los organismos acuáticos.
32. Pentaclorofenol: compuesto organoclorado con fórmula química C6Cl5OH. Es usado como preservante de textiles que pueden ser atacados por hongos y bacterias. Los niveles de este compuesto, que se pueden encontrar en organismos acuáticos, son generalmente bajos.
33. Hidrocarburo aromáticos policíclicos (Benzo(a)pireno, Benzo(b)fluoranteno, Benzo(g,h,i)perileno, Benzo(k)fluoranteno, Indenol(1,2,3-cd)pireno): la fórmula química general es CxHyR Los hidrocarburos aromáticos policíclicos son un grupo de más de 100 sustancias químicas Los HHPP se encuentran generalmente como una mezcla de dos o más de estos compuestos, tal como el hollín. Los HHPP se encuentran en el alquitrán, el petróleo crudo, el creosote y alquitrán para techos, algunos se utilizan en medicamentos o para fabricar tintes y pesticidas. La mayoría de los HHPP no se
Estructura química del Pentaclorofenol
Estructura química del Benzo(g,h,i)perileno
Estructura química del Benzo(k)fluoranteno
Estructura química del Indenol(1,2,3-cd)pireno
Estructura química del Benzo(a)pireno
Estructura química del Benzo(b)fluoranteno

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
13
disuelven fácilmente en agua. Los microorganismos pueden degradar los HHPP en el suelo o en el agua después de un período de semanas a meses. En el suelo, los HHPP se adhieran firmemente a partículas; ciertos HHPP se movilizan a través del suelo y contaminan el agua subterránea. La cantidad de HHPP en plantas y en animales puede ser mucho mayor que la cantidad en el suelo o en el agua donde viven estos organismos.
34. Simacina: compuesto organonitrogenado con fórmula química ClC3N3(NHC2H5)2. Es un herbicida que se utiliza para el control de las malas hierbas. Se emplea en cultivos como cereales, frutas (Ej.: uvas y cítricos) y nueces, así como en ciertas aplicaciones no agrícolas como céspedes. La simacina es poco tóxica para organismos acuáticos, sin embargo tiene una toxicidad mayor en plantas.
35. Compuestos de Tributiltín (tributiltin-catión): compuesto organoestánnico con fórmula química CxHyRSn. Se emplea en pinturas para evitar la bioincustración en el fondo de los buques. También se usan como preservantes de la madera; pueden acumularse en peces y otros animales y en plantas; es muy tóxico para éstas. Se adhieren al sedimento y a las partículas en suspensión en el agua.
36. Triclorobencenos (1,2,4-Triclorobenceno): compuesto organoclorado con fórmula química C6H3Cl3. Usados como intermediarios químicos, particularmente para ciertos herbicidas, pigmentos y tintes. También se utilizan como solventes de procesos. En el pasado han sido empleados como portadores de colorantes, lubricantes y aditivos; también en algunos productos farmacéuticos. En aguas, se adsorben en los sedimentos y se bioacumulan en los organismos. Ya no se sigue produciendo en la Unión Europea.
37. Triclorometano (Cloroformo): compuesto trihalometanos, cuya fórmula química es CHCl3. Se utiliza en productos farmacéuticos, prótesis y en los sectores químico y médico, así como sustancia orgánica para productos químicos y petroquímicos. La mayor parte de la producción de cloroformo se emplea para fabricar monoclorodifluorometano (CFC22) que se aplica como refrigerante. Además se utiliza como producto intermedio en la fabricación de tetrafluoretano que luego puede ser polimerizado (PTFE). También encuentra aplicación en la fabricación de colorantes, productos farmacéuticos y pesticidas. Su uso como solvente y como anestésico está disminuyendo rápidamente. En el medio acuático, el cloroformo se descompone con extrema lentitud. La bioacumulación del
Estructura química del Simacina
Estructura química del 1,2,4-Triclorobenceno

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
14
cloroformo es mínima a pesar de su alta liposolubilidad. El agua potable contiene, en ciertas ocasiones, elevadas concentraciones de cloroformo resultante de la cloración del agua. Debido a la distribución universal y a la considerable cantidad que ingresa al medio ambiente todos los años, se debe considerar al cloroformo como una amenaza ecológica. El Cloroformo se produce en 6 de los Estados Miembros, entre ellos España.
38. Trifluralina: compuesto organonitrogenado, su fórmula química es F3C(NO2)2C6H2N(C3H7)2. Es uno de los principales herbicidas y es utilizado en el control de hierbas anuales y para malas hierbas y forrajes, cereales, vegetales, semillas oleaginosas y cosechas de ciertas frutas. La trifluralina es muy tóxica para los peces y otros organismos acuáticos. Se bioacumula moderadamente. Su uso está autorizado en todos los Estados Miembros, excepto en Suecia, Dinamarca y Holanda.
Estructura química del Trifluralina

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
15
III. MÉTODOS CROMATOGRÁFICOS.
1. Introducción.
A principios del siglo XX (Ettre, 1993), el botánico ruso M.S. Tswett había observado
que la clorofila y los pigmentos asociados son solubles en éter de petróleo y ligroína
(fracciones ligeras de la destilación del petróleo, muy usadas como disolventes no polares
en aquella época), pero que estos disolventes no eran capaces de extraerlos de las hojas.
Por el contrario, si se añadía etanol al disolvente, la extracción se producía sin ningún
problema. Concentró la disolución a sequedad, disolvió los pigmentos en ligroína e
impregnó con la disolución un papel de filtro, matriz celulósica de composición afín a los
tejidos de las hojas. Cuando sometió el papel a una extracción con ligroína sólo consiguió
arrastrar los carotenos, permaneciendo la clorofila. La adición de pequeñas cantidades de
etanol a la ligroína producía la inmediata extracción de todos los pigmentos.
Como no existía ninguna afinidad química entre los pigmentos y la celulosa, dedujo
que estos cambios de solubilidad se debían a fenómenos de adsorción, lo que constituye
una notable deducción, teniendo en cuenta que en aquella época muy pocos científicos
poseían conocimientos del tratamiento teórico de la adsorción.
Intuyó las posibilidades que se le presentaban de poner a punto un método de
separación de pigmentos que, quizás, podría extenderse a cualquier otro tipo de
sustancia, y planteó el siguiente experimento: rellenó con un sólido en polvo un tubo de
vidrio de unos 12 cm. de altura y unos 3 cm. de diámetro, acabado en un estrechamiento
taponado por una bola de papel de filtro, pasó ligroína pura para desplazar el aire, e
introdujo un extracto de pigmentos en ligroína. Ayudó el paso de la disolución con algo de
presión o vacío, observando la progresión por los colores de las bandas. Ensayó como
relleno más de un centenar de sólidos, obteniendo buenos resultados con muchos de
ellos, entre los que se encontraban la inulina, el carbonato cálcico y la alúmina. Como
disolventes utilizó ligroína y etanol.
Estos resultados los comunicó al Congreso de la Sección de Biología de la Sociedad
de Ciencias Naturales de Varsovia, en marzo de 1903. Sus dos publicaciones más
importantes sobre el tema aparecieron en 1906 en el Berichte der Deutschen Botanischen
Gesellschaft (Imforme de la sociedad botánica alemana). Puesto que se podía seguir el
proceso visualmente mediante el colorido de las bandas de los pigmentos, dio el nombre

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
16
de cromatografía a la técnica. De esta manera se produjo uno de los descubrimientos más
importantes del siglo XX.
Los trabajos de Tswett fueron prácticamente ignorados durante un periodo de
veinte años. Las razones de este olvido pueden ser las siguientes: en primer lugar, el libro
(Tswett, 1910) en que describe la técnica fue publicado en ruso; además, en 1913 dichos
trabajos sufrieron un feroz ataque por parte de Willstatter y Stoll, que no fueron capaces
de reproducir los experimentos; y, finalmente, en aquella época los químicos orgánicos
estaban interesados fundamentalmente en los productos mayoritarios (procesos de
enriquecimiento y purificación). Fue el fuerte desarrollo posterior de la Bioquímica, más
interesada en componentes menores y en procesos de separación total de mezclas
complejas, el que despertó el interés por la cromatografía.
Éste fue el origen casual de una de las técnicas más importantes de separación que
existen hoy día, de hecho, su uso es muy distinto del de una simple etapa previa de
eliminación de interferentes, ya que es un método cualitativo de análisis al usar como
parámetro los tiempos necesarios para la elutriación, y puede llegar a ser cuantitativo
mediante el uso de detectores apropiados.
Un proceso de separación consiste básicamente en hacer que diferentes sustancias
químicas, que comparten un espacio homogéneo, es decir, cuyas concentraciones en
cualquier parte de dicho espacio son las mismas, pasen a ocupar zonas preferentes en
éste u otros espacios.
Una separación perfecta implicaría la separación total de todos los constituyentes de
forma que en cada una de las zonas preferentes se encuentre solo un compuesto. Lo
normal, sin embargo, es que los resultados de un proceso de separación no sean totales,
sino que se produzcan alteraciones en las concentraciones, ci, de cada componente de la
mezcla. Si consideramos dos zonas del espacio que compartan de forma homogénea las
moléculas de dos compuestos distintos, antes de la separación la concentración de cada
compuesto sería la misma en cada una de las dos zonas consideradas. Una separación
consistiría en conseguir que, después del proceso, exista una diferencia de la
concentración en ambos puntos. Estas expresiones pueden interpretarse como la
existencia de cierta preferencia de los componentes por una de las zonas. A partir de
estos sencillos conceptos puede definirse una magnitud que sirva para describir la calidad

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
17
de una separación: el factor de separación, α, que es la relación de la separación entre
dos componentes, siendo i y j los componentes, y a y b las zonas.
bj
aj
bi
ai
cc
cc
=α
Ecuación del factor de separación
Cuanto más se diferencie α de 1, mejor será la separación. Un ejemplo muy claro
para explicar esta magnitud es una extracción líquido-líquido en un embudo de
decantación: α sería el cociente entre los coeficientes de reparto de los dos solutos. En
una destilación simple las zonas a y b serían el calderín del destilador y el receptáculo del
destilado y, en un proceso cromatográfico, las fases móvil y estacionaria.
2. Características básicas.
Es difícil dar una definición de la cromatografía que sea clara y unívoca. Si se quiere
describir la forma en que se produce, puede decirse que se trata de una técnica de
separación en la que la mezcla a resolver se introduce en un sistema formado por un
fluido (fase móvil), que circula en íntimo contacto con una fase estacionaria (sólida o
líquida), que permanece inmóvil durante el proceso. Cuando una molécula de soluto se
encuentra en la fase móvil, avanza a la misma velocidad media que ésta, pero cuando
entra en la fase estacionaria, su velocidad media es cero. Si los componentes de la mezcla
difieren en su afinidad por alguna de las fases, sus velocidades medias de avance a lo
largo del sistema serán diferentes. Como consecuencia de ello se produce la separación.
Para clasificar los diferentes tipos de cromatografía, el primer criterio que puede
establecerse es la naturaleza de las fases. Como segundo criterio, se utiliza el mecanismo
de separación:
1. Cuando la fase estacionaria es un líquido, el mecanismo de separación predominante es el reparto, por lo que se le denomina cromatografía de reparto.
2. Si la fase estacionaria es un sólido, cabe la posibilidad de varios mecanismos, dependiendo de su naturaleza:

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
18
a. Cromatografía de adsorción. Se basa en la distinta magnitud de las
interacciones de los componentes de la mezcla con la superficie activa de la fase estacionaria.
b. Cromatografía de intercambio iónico. La fase estacionaria es un sólido con capacidad para intercambiar iones con la fase móvil.
c. Cromatografía de exclusión molecular: La fase estacionaria es un sólido reticular con tamaños de retícula del mismo orden que las moléculas de la muestra a separar. Estas moléculas serán capaces de penetrar en más o menos huecos según su tamaño, forma, cargas eléctricas, etc.
d. Cromatografía de afinidad. Se utiliza como mecanismo de separación una interacción específica entre el analito y la fase estacionaria. Si la interacción es de tipo biológico, será cromatografía de bioafinidad.
Otro criterio es la relación de polaridad entre la fase móvil y la estacionaria.
1. Cuando la fase móvil es menos polar que la estacionaria se denomina cromatografía en fase normal (FN).
2. Cuando la fase móvil es la más polar, se denomina cromatografía en fase inversa (FI). Esta última forma, que es la más utilizada actualmente en cromatografía de líquidos, usa como fase estacionaria una molécula no polar (monomérica o polimérica) unida químicamente a un soporte. El comportamiento de este tipo de fase estacionaria es intermedio entre el de un sólido y un líquido, inclinándose más en uno u otro sentido según las condiciones del entorno, fundamentalmente la naturaleza de la fase móvil.
La cromatografía puede clasificarse también de acuerdo con la forma de desarrollar
el proceso cromatográfico:
1. Cromatografía frontal. Se introduce la muestra de forma continua en el sistema cromatográfico, constituyendo ella misma la fase móvil.
2. Cromatografía de desplazamiento. La fase móvil contiene un componente más afín a la fase estacionaria que los componentes de la muestra y es capaz de desplazarlos de ella.
3. Cromatografía de elución. La muestra se introduce en un momento determinado y la fase móvil circula continuamente. Los componentes de la muestra salen de la columna como zonas, que en el caso ideal presentan una distribución gaussiana de concentración y están separadas unas de otras.
Por último, la disposición geométrica de las fases también puede emplearse para
clasificar las técnicas cromatográficas:

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
19
1. Si la fase estacionaria se sitúa en un tubo, se trata de cromatografía en
columna.
2. Si se deposita sobre una superficie abierta (papel, placa de vidrio o metal), se denomina cromatografía en plano.
El contenido de este proyecto se concreta en la cromatografía en columna, aunque
los principios básicos, tanto termodinámicos como de la dinámica del proceso, son
comunes a todas ellas. La forma habitual de desarrollar el proceso en una columna es la
elución, al igual que en el sistema original de Tswett.
La cromatografía en columna se realiza en un equipo instrumental que consta de las
siguientes partes fundamentales:
Figura III.1
Esquema básico de un cromatógrafo.
1. Una fuente de fase móvil, cuya naturaleza depende fundamentalmente del tipo de fluido que se utilice.
2. Un sistema de regulación y medida del caudal o la presión de la fase móvil.
3. Un sistema de inyección de la muestra: se trata de un dispositivo que permite introducir la muestra en la corriente de fase móvil.
4. La columna: es la parte fundamental, ya que en ella se produce la separación. Se trata de un tubo construido en diversos materiales (metal, vidrio, sílice fundida, polímeros, etc.), de una longitud que puede oscilar entre varios centímetros y más de un centenar de metros, y cuyo diámetro interno varía entre varios micrómetros y varios centímetros. La forma de distribución de la fase estacionaria y el valor de la relación de fases, (volumen de fase móvil / volumen de fase estacionaria) dan lugar a los diferentes tipos de columnas.
5. Un recipiente termostatizado para controlar la temperatura de la columna. En cromatografía de líquidos es frecuente trabajar a temperatura ambiente en cuyo

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
20
caso se prescinde de este componente. En cualquier caso, para medidas precisas es imprescindible su uso.
6. Un sistema de detección: es un dispositivo que mide en continuo y transforma en una señal eléctrica una propiedad, generalmente física, de la fase móvil, que se ve ampliamente alterada por la presencia de pequeñas concentraciones de otras sustancias (componentes de la muestra).
7. Un sistema de amplificación y tratamiento de la señal eléctrica generada en el detector. El tratamiento puede incluir la adquisición y almacenamiento de datos y ciertos cálculos mediante un sistema informático
3. Magnitudes fundamentales de la separación: selectividad, eficacia y resolución
La cromatografía es un proceso dinámico, en cuya ejecución es necesario controlar
rigurosamente una serie de variables experimentales si se desea obtener resultados
reproducibles. La magnitud más importante es la velocidad de desplazamiento de los
analitos. Ésta depende, para un sistema cromatográfico dado, de la temperatura y del
gradiente de presión aplicado.
La temperatura influye normalmente en las propiedades de todas las sustancias. Su
control y conocimiento exactos son un importante indicador de la calidad de un
cromatógrafo.
El tiempo necesario para llevar a cabo un análisis está directamente relacionado con
la velocidad con que se mueven los analitos. Esta variable puede controlarse y medirse
fácilmente con bastante precisión. Su valor depende de la naturaleza de las sustancias a
estudiar y de las condiciones experimentales elegidas. Los dispositivos instrumentales que
permiten modificarla son diferentes dependiendo del tipo de fluido utilizado como fase
móvil (líquido, gaseoso o supercrítico). Esta regulación es otro factor primordial para
evaluar la calidad de un cromatógrafo. En cuanto a su medida, puede hacerse a partir del
tiempo de permanencia del soluto en la columna y de la longitud recorrida de la misma.
• Datos experimentales
Cuando se introduce una sustancia en una columna cromatográfica, la zona que
ocupa se va ensanchando durante su progresión a lo largo de la misma y su distribución
de concentraciones se puede asimilar, generalmente, a una función de Gauss.
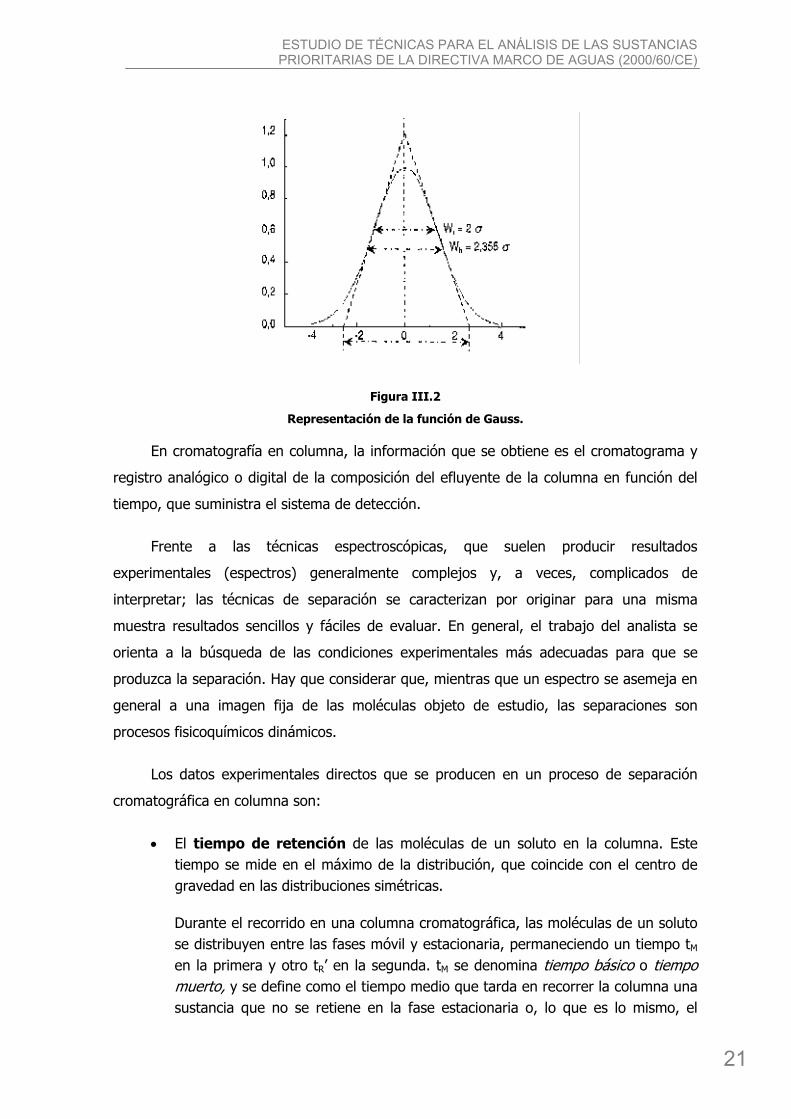
ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
21
Figura III.2
Representación de la función de Gauss.
En cromatografía en columna, la información que se obtiene es el cromatograma y
registro analógico o digital de la composición del efluyente de la columna en función del
tiempo, que suministra el sistema de detección.
Frente a las técnicas espectroscópicas, que suelen producir resultados
experimentales (espectros) generalmente complejos y, a veces, complicados de
interpretar; las técnicas de separación se caracterizan por originar para una misma
muestra resultados sencillos y fáciles de evaluar. En general, el trabajo del analista se
orienta a la búsqueda de las condiciones experimentales más adecuadas para que se
produzca la separación. Hay que considerar que, mientras que un espectro se asemeja en
general a una imagen fija de las moléculas objeto de estudio, las separaciones son
procesos fisicoquímicos dinámicos.
Los datos experimentales directos que se producen en un proceso de separación
cromatográfica en columna son:
• El tiempo de retención de las moléculas de un soluto en la columna. Este tiempo se mide en el máximo de la distribución, que coincide con el centro de gravedad en las distribuciones simétricas.
Durante el recorrido en una columna cromatográfica, las moléculas de un soluto se distribuyen entre las fases móvil y estacionaria, permaneciendo un tiempo tM en la primera y otro tR’ en la segunda. tM se denomina tiempo básico o tiempo muerto, y se define como el tiempo medio que tarda en recorrer la columna una sustancia que no se retiene en la fase estacionaria o, lo que es lo mismo, el
ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
22
tiempo de retención de la fase móvil. A tR’ se le denomina tiempo de retención ajustado y es la magnitud relacionada con el tiempo que las moléculas del soluto permanecen retenidas en la fase estacionaria. Las diferencias de esta magnitud para distintos solutos reflejan la facilidad de su separación en el sistema.
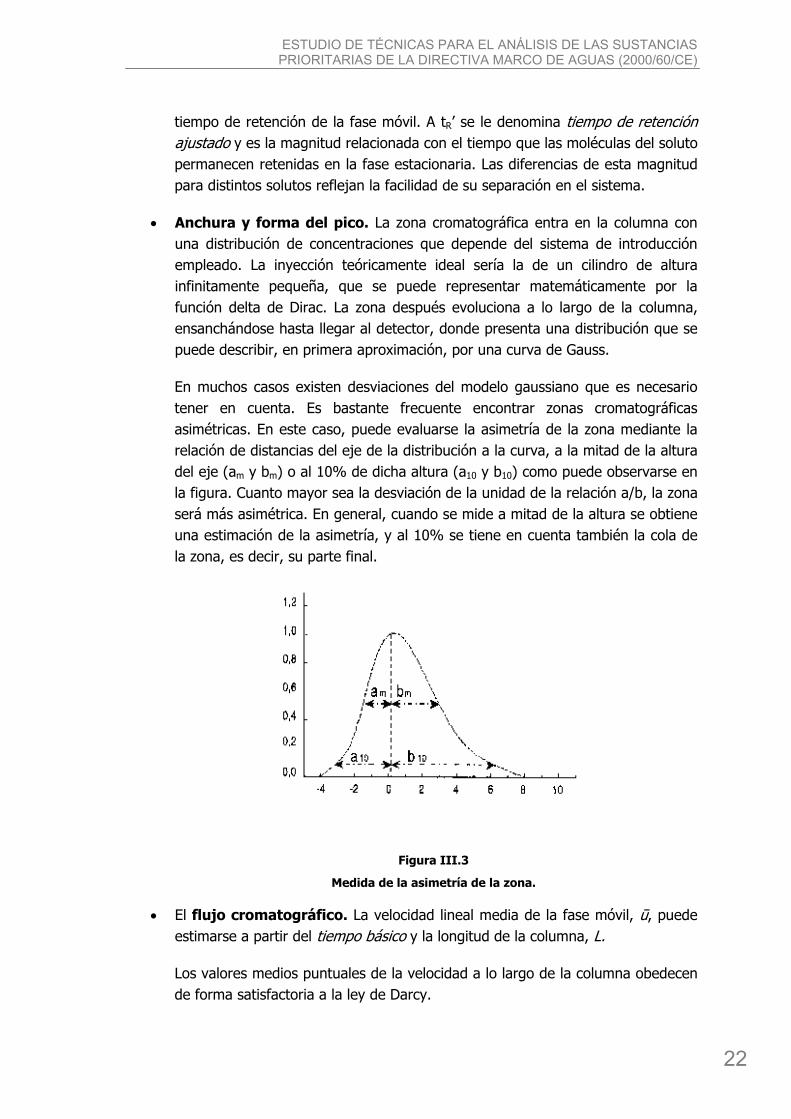
• Anchura y forma del pico. La zona cromatográfica entra en la columna con una distribución de concentraciones que depende del sistema de introducción empleado. La inyección teóricamente ideal sería la de un cilindro de altura infinitamente pequeña, que se puede representar matemáticamente por la función delta de Dirac. La zona después evoluciona a lo largo de la columna, ensanchándose hasta llegar al detector, donde presenta una distribución que se puede describir, en primera aproximación, por una curva de Gauss.
En muchos casos existen desviaciones del modelo gaussiano que es necesario tener en cuenta. Es bastante frecuente encontrar zonas cromatográficas asimétricas. En este caso, puede evaluarse la asimetría de la zona mediante la relación de distancias del eje de la distribución a la curva, a la mitad de la altura del eje (am y bm) o al 10% de dicha altura (a10 y b10) como puede observarse en la figura. Cuanto mayor sea la desviación de la unidad de la relación a/b, la zona será más asimétrica. En general, cuando se mide a mitad de la altura se obtiene una estimación de la asimetría, y al 10% se tiene en cuenta también la cola de la zona, es decir, su parte final.
Figura III.3
Medida de la asimetría de la zona.
• El flujo cromatográfico. La velocidad lineal media de la fase móvil, ū, puede estimarse a partir del tiempo básico y la longitud de la columna, L.
Los valores medios puntuales de la velocidad a lo largo de la columna obedecen de forma satisfactoria a la ley de Darcy.

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
23
Para la integración de la ecuación de Darcy es necesario conocer la ecuación de estado del fluido que constituye la fase móvil. En general, en cromatografía de gases y de fluidos supercríticos suele utilizarse como aproximación válida el modelo de los gases perfectos. Si es necesaria mayor precisión, se utiliza la ecuación del virial para cromatografía de gases, pero ésta no es la práctica normal. En cromatografía de líquidos sirve como aproximación válida en la mayoría de los casos la consideración de la fase móvil como líquido incompresible.
• La retención cromatográfica. Cuando una zona cromatográfica progresa a lo largo de una columna y la fase móvil circula a una velocidad lineal u, la velocidad de la zona, v, será inferior (para las sustancias que se quedan retenidas en la columna) o igual a la de la fase móvil (para las no retenidas). La relación entre ambas velocidades se llama factor de retardo, R, que varía entre cero y 1. Al ser además la longitud fija para una determinada columna, el factor de retardo relaciona también el tiempo muerto y el tiempo de residencia de forma directa.
Este factor es muy usado en la cromatografía plana por su facilidad de medida. En la cromatografía en columna, la retención se suele expresar mediante el factor de retención (V), que se define como la relación entre el tiempo de retención ajustado y el tiempo básico. Desde un punto de vista físico, k representa la relación entre los tiempos medios que una molécula de soluto reside en las fases estacionaria y móvil, es decir, la relación de probabilidades de estar en una u otra fase y, en consecuencia, será también la relación entre el número de moléculas de soluto que, en un instante determinado, están en las fases estacionaria y móvil.
Existe una evidente relación entre k y R, ya que son dos formas de medir el mismo fenómeno.
Se denomina relación de fases, β, al cociente entre los volúmenes de fase móvil y de fase estacionaria de la columna
El volumen de retención, VR, de un compuesto es el volumen de fase móvil que entra en la columna desde el instante de la inyección hasta el momento de la salida del máximo de la distribución de la zona que ocupa. De la misma manera que para el tiempo de retención, puede definirse el volumen de retención ajustado.

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
24
3.1. Selectividad.
La selectividad de un sistema cromatográfico para separar una pareja de sustancias
i y j se puede describir mediante el factor de separación, α, que se definió anteriormente.
El valor de α constituye una estimación de la idoneidad del sistema para separar la pareja
de sustancias. Cuando su valor es la unidad, el sistema es incapaz de producir la
separación y será necesario recurrir a otro sistema distinto. Por el contrario, cuanto más
difiera de la unidad, la separación será más fácil. En cromatografía, si a > 1,14 se
considera que la separación es fácil; si a < 1,01, se estima que la separación es muy
difícil, α posee todas las propiedades termodinámicas de K, magnitud termodinámica que
sólo depende de la naturaleza del soluto y las fases, y de la temperatura, con la ventaja
de que su medida es más fácil y exacta al tratarse de una magnitud que se mide con
relación a otra sustancia.
3.2. Eficacia.
Cada zona cromatográfica no solamente progresa a lo largo de la columna sino que,
además, se ensancha. La magnitud del ensanchamiento se puede medir mediante la
desviación típica (σ) de la zona. Esta magnitud no es función lineal del camino recorrido
en la columna (z), pero sí lo es la varianza (σ 2) a través de la expresión:
σ2 = H·z
donde la constante de proporcionalidad H se denomina altura equivalente a un plato
teórico y constituye una estimación de la eficacia del sistema de separación. Cuanto
menor sea su valor, el sistema será más eficaz ya que el ensanchamiento de la zona por
unidad de longitud de columna recorrida será menor.
H describe fundamentalmente la calidad del sistema, pero no su eficacia total. Para
ello se define otra magnitud, el número de platos teóricos de la columna, N, que es la
relación entre la longitud de la columna y la altura equivalente de un plato teórico.
Como es evidente, dentro de la columna cromatográfica no existen platos, siendo el
origen de estas magnitudes, H y N, la búsqueda de similitud con las descritas para la
destilación. Basándose en este símil, se describe H en una separación cromatográfica
ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
25
como el tramo de columna en el que se produce un equilibrio de distribución entre las
fases
De manera análoga, N sería el número total de equilibrios de distribución que se
producen a lo largo de toda la columna. Aunque estas afirmaciones son difíciles de
comprobar porque la comparación con la destilación es cuestionable, el valor comparativo
de H y N dentro de las técnicas analíticas de separación es innegable
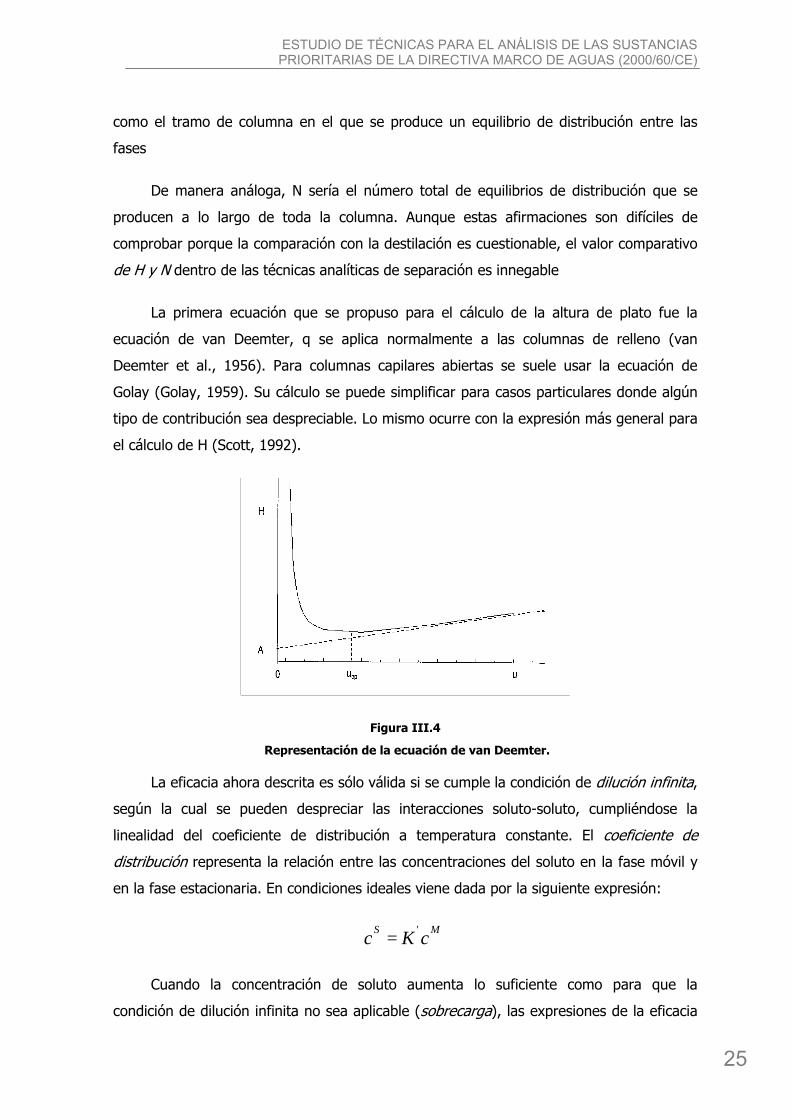
La primera ecuación que se propuso para el cálculo de la altura de plato fue la
ecuación de van Deemter, q se aplica normalmente a las columnas de relleno (van
Deemter et al., 1956). Para columnas capilares abiertas se suele usar la ecuación de
Golay (Golay, 1959). Su cálculo se puede simplificar para casos particulares donde algún
tipo de contribución sea despreciable. Lo mismo ocurre con la expresión más general para
el cálculo de H (Scott, 1992).
Figura III.4
Representación de la ecuación de van Deemter.
La eficacia ahora descrita es sólo válida si se cumple la condición de dilución infinita,
según la cual se pueden despreciar las interacciones soluto-soluto, cumpliéndose la
linealidad del coeficiente de distribución a temperatura constante. El coeficiente de
distribución representa la relación entre las concentraciones del soluto en la fase móvil y
en la fase estacionaria. En condiciones ideales viene dada por la siguiente expresión:
cS = K ' cM
Cuando la concentración de soluto aumenta lo suficiente como para que la
condición de dilución infinita no sea aplicable (sobrecarga), las expresiones de la eficacia

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
26
no son válidas ya que ésta depende de la concentración, variando la velocidad de las
partículas de soluto dependiendo de su posición. Todo esto provoca que la anchura del
pico aumente, y se pueda producir una pérdida de su simetría y una variación de su
posición.
Los factores que más influyen en la aparición de la sobrecarga son el diámetro de la
columna y el mecanismo de separación, sobrecargándose antes el mecanismo de
adsorción que el de reparto. Las columnas de relleno tienen por norma general una
mayor capacidad que las columnas abiertas.
Por último cabe decir, que la eficacia de una columna cromatográfica es
independiente del tipo de cromatografía
3.3. Resolución.
Si se ha inyectado en la columna una pareja de sustancias que avanzan a distintas
velocidades, la distancia entre los máximos de las zonas (Δz) progresará de forma lineal a
lo largo de la columna. Simultáneamente, el ancho de las zonas (4σ) también variará a lo
largo de la columna, de forma que existe un punto de la columna, situado a una distancia
z1 de la entrada, donde Δz = 4σ, es decir, en el que las dos zonas están perfectamente
separadas, aunque unidas por la base. Cuando la distancia recorrida sea menor que z1, la
Figura III.5
Evolución de la distancia entre zonas y de su anchura a lo largo de la columna.
ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
27
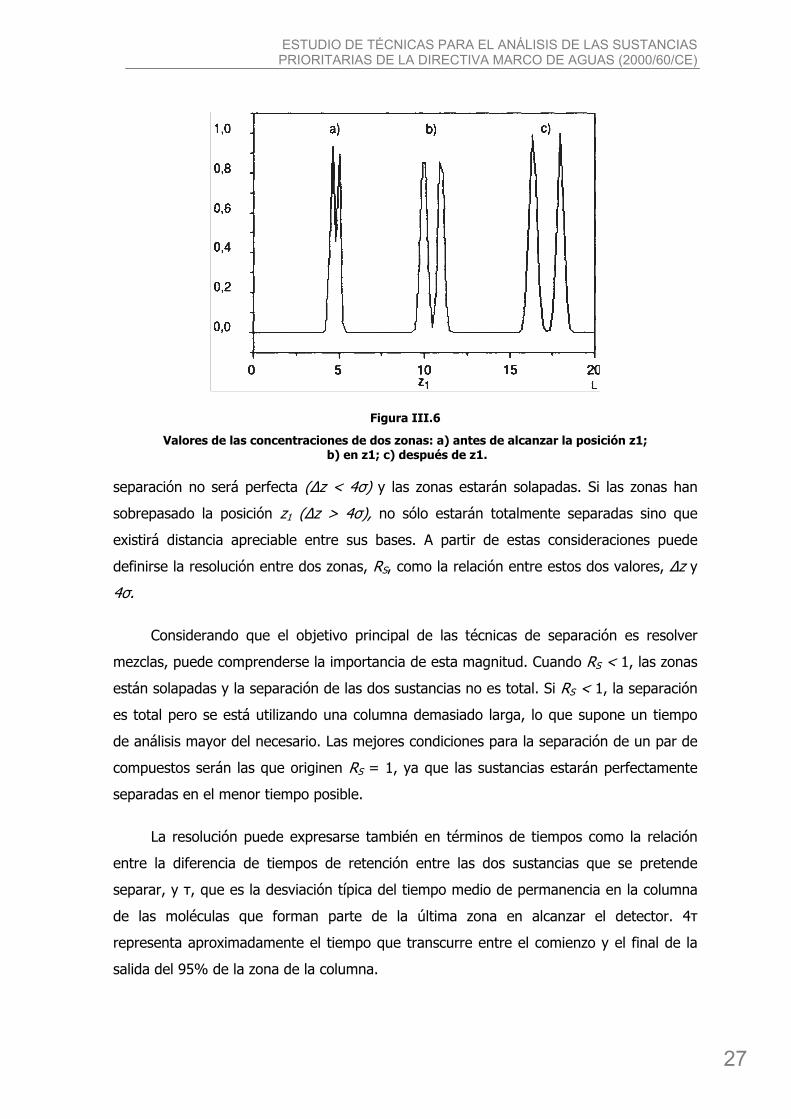
Figura III.6
Valores de las concentraciones de dos zonas: a) antes de alcanzar la posición z1; b) en z1; c) después de z1.
separación no será perfecta (Δz < 4σ) y las zonas estarán solapadas. Si las zonas han
sobrepasado la posición z1 (Δz > 4σ), no sólo estarán totalmente separadas sino que
existirá distancia apreciable entre sus bases. A partir de estas consideraciones puede
definirse la resolución entre dos zonas, RS, como la relación entre estos dos valores, Δz y
4σ.
Considerando que el objetivo principal de las técnicas de separación es resolver
mezclas, puede comprenderse la importancia de esta magnitud. Cuando RS < 1, las zonas
están solapadas y la separación de las dos sustancias no es total. Si RS < 1, la separación
es total pero se está utilizando una columna demasiado larga, lo que supone un tiempo
de análisis mayor del necesario. Las mejores condiciones para la separación de un par de
compuestos serán las que originen RS = 1, ya que las sustancias estarán perfectamente
separadas en el menor tiempo posible.
La resolución puede expresarse también en términos de tiempos como la relación
entre la diferencia de tiempos de retención entre las dos sustancias que se pretende
separar, y τ, que es la desviación típica del tiempo medio de permanencia en la columna
de las moléculas que forman parte de la última zona en alcanzar el detector. 4τ
representa aproximadamente el tiempo que transcurre entre el comienzo y el final de la
salida del 95% de la zona de la columna.

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
28
Puede pensarse con toda lógica que cualquier separación, por muy difícil que sea,
es posible con tal que α≠1 y se disponga de una longitud de columna necesaria. El
importante factor que no tiene en cuenta este razonamiento es el tiempo de análisis que,
en primera aproximación, se define como el tiempo de retención del último componente
en ser eluido.
Puede ocurrir que las condiciones necesarias para obtener RS > 1 sean tales que el
tiempo de análisis sea inviable, o simplemente poco práctico. Por esta razón es
importante definir una nueva magnitud, la rapidez de la columna (Halasz, 1968), que se
puede evaluar mediante la velocidad de generación de platos, N/tR. Esta magnitud, como
la eficacia, para que tenga valor comparativo entre columnas debe medirse para
sustancias que se retengan de forma similar (el mismo valor de k). Existe así una relación
entre la selectividad y eficacia con la resolución.

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
29
IV. CROMATOGRAFÍA DE LÍQUIDOS DE ALTA EFICACIA
1. Introducción.
La cromatografía de líquidos comprende todas las técnicas cromatográficas en las
que la fase móvil es un líquido. Dentro de ellas se encuentran desde las más primitivas de
cromatografía en papel, en capa fina o en columna abierta, hasta los métodos de
cromatografía de líquidos de alta eficacia (CLAE) que vamos a tratar a continuación.
La comparación de los métodos de cromatografía de gases con los de cromatografía
de líquidos pone de manifiesto una clara diferencia en el papel que juega la fase móvil en
ambos tipos. Las separaciones cromatográficas se rigen por la interacción de los solutos
con el sistema cromatográfico, entendiendo como tal el conjunto fase móvil-fase
estacionaria. La fase móvil tiene un papel primordial en el proceso de cromatografía de
líquidos en lo que respecta a las interacciones con el soluto, mientras que en
cromatografía de gases la fase móvil es prácticamente inerte. En CLAE la naturaleza de la
fase móvil se puede cambiar prácticamente de manera casi ilimitada, de forma que su
modificación permite variar ampliamente la selectividad y controlar la separación de los
constituyentes de la muestra, aun con un número limitado de fases estacionarias. De
acuerdo con la naturaleza de estas fases, los modos más utilizados de CLAE se pueden
resumir en fase inversa, intercambio iónico, pares iónicos y exclusión molecular, sin
olvidar la importancia que están adquiriendo otros modos entre los que hay que destacar
la cromatografía de líquidos quiral y la de afinidad.
La naturaleza tan diversa de las fases estacionarias y móviles que se pueden
emplear en CLAE lleva consigo una gran versatilidad en los mecanismos de separación.
Esta versatilidad se ve incrementada por la posibilidad de provocar en el interior de la
columna de separación equilibrios secundarios de tipo ácido-base, de formación de pares
iónicos, de reparto micelar o de formación de complejos. Los modos de separación más
usados en la práctica son los siguientes.
2. Fase inversa.
Esta nomenclatura obedece a razones históricas, como contraposición a la fase
normal, modalidad en desuso en CLAE. La denominación de fase normal o fase inversa
hace referencia a la polaridad relativa de las fases móvil y estacionaria. Mientras que en

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
30
fase normal la fase estacionaria es más polar que la fase móvil, en fase inversa el sistema
cromatográfico está constituido por una fase estacionaria, que generalmente es de
naturaleza apolar, y una fase móvil más polar que la fase estacionaria.
La cromatografía de líquidos en fase inversa es el modo de CLAE más empleado en
la actualidad y será la que tratemos en estos apartados. Se estima que el 50% de las
separaciones que se llevan a cabo en cromatografía de líquidos emplean este modo
(Majors, 1995). Este hecho se debe a la comercialización de rellenos específicos para esta
modalidad, que son estables, reproducibles, de pequeño tamaño de partícula, y que
permiten alcanzar una elevada velocidad de transferencia de materia, lo que da lugar a
columnas con elevada eficacia. Las fases estacionarias más simples desde un punto de
vista conceptual son las basadas en sílice con funcionalidad alquilo, que se obtienen
uniendo mediante enlace covalente un n-alquilsilano a la superficie de una partícula de
sílice. Otros tipos de fases de «arquitectura» más complicada, tales como las oligoméricas
o las poliméricas, también se emplean en este modo de separación. La fase móvil es
generalmente agua con un modificador orgánico (metanol, acetonitrilo o
tetrahidrofurano). Con el fin de optimizar la selectividad de la separación pueden
emplearse mezclas ternarias o cuaternarias de estos disolventes.
El mecanismo por el que tiene lugar la separación en fase inversa está abierto a
debate. Su estudio supone un conocimiento lo más detallado posible de las fuerzas que
intervienen en la interacción del soluto con los componentes de la fase móvil, la
interacción del soluto con las moléculas de la fase estacionaria y la transferencia de la
molécula de soluto de la fase móvil a la fase estacionaria. La dificultad para estimar estas
interacciones reside en los conocimientos limitados que se tienen sobre la termodinámica
de las disoluciones y, sobre todo, en la dificultad para modelizar de manera adecuada las
fases estacionarias en las condiciones de separación. Con este fin se ha estudiado el
efecto que tienen diferentes rellenos de fase inversa sobre la retención y la selectividad
(Antle et al., 1985; Tchapla et al., 1993), concluyendo que sus distintos comportamientos
se deben a las diferencias en la relación de fases entre unas columnas y otras, a la
desigual polaridad de las fases y a la diferente afinidad de los solutos por las fases
empleadas, controlada principalmente por fuerzas de dispersión.
A modo de ejemplo de la dificultad que tiene caracterizar una fase estacionaria, se
ha de saber que la relación de fases en una columna de fase inversa está relacionada con

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
31
la longitud de la cadena hidrocarbonada unida a la sílice, con la densidad superficial de la
fase (μmoles de n-alquilsilano por unidad de superficie de sílice) y con la superficie
específica del material silíceo empleado en su preparación. La fase móvil juega un doble
papel: por una parte, su naturaleza y composición determinan las interacciones con las
moléculas del soluto; por otra, modifica notablemente la organización de la fase
estacionaria, interviniendo indirectamente en las interacciones entre ésta y el soluto. La
interacción entre el soluto y las cadenas hidrocarbonadas de la fase estacionaria no es
sólo función de la naturaleza del soluto, sino también de la densidad superficial y
organización de la fase (Tchapla et al., 1993).
3. Tipos de relleno
Pueden dividirse en tres grandes grupos: monoméricos, oligoméricos y poliméricos.
• Fases monoméricas. Se preparan por reacción de un n-alquildimetil-clorosilano y los grupos silanoles de la superficie de la sílice, dando lugar a una unión covalente a través de un grupo siloxano (Si-O-Si). Desde un punto de vista estructural son las fases más sencillas de las tres citadas, ya que las cadenas n-alquílicas forman una monocapa sobre la superficie de la sílice. El grado de recubrimiento que consiguen los diferentes fabricantes con estas fases varía entre 2 y 4 μmoles/m2, dependiendo de las condiciones de reacción y del tipo de silano usado. Esta característica justifica en parte las diferencias de comportamiento cromatográfico observado entre los diferentes productos comerciales para fase inversa. Se estima que después de la funcionalización todavía quedan aproximadamente un 50% de los grupos silanoles de la superficie de la sílice sin reaccionar, debido a impedimentos estéricos. Parte de los grupos silanoles residuales pueden ser bloqueados (Proceso denominado end-capping) en una reacción posterior del relleno con un silano de menor tamaño, generalmente trimetilclorosilano. A pesar de esta segunda reacción, un buen número de grupos silanoles permanecen sin bloquear y pueden interaccionar con el soluto durante el proceso de separación.
• Fases oligoméricas. Si la superficie de la sílice está saturada con agua y se emplea un n-alquiltriclorosilano como reactivo, éste reacciona simultáneamente con los grupos silanoles de la sílice y con el agua presente en el medio originando una reacción de entrecruzamiento del silano y la formación de un n-alquilpolímero, que quedará unido a la superficie originando un recubrimiento de espesor equivalente a varias monocapas del n-alquil-derivado. Generalmente el espesor de esta multicapa varía de unos puntos a otros de la superficie. La formación del polímero tiene dos consecuencias: la densidad superficial en estas fases es bastante mayor que en las fases monoméricas; y, debido al

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
32
entrecruzamiento del silano, el ordenamiento es también mayor que en las monoméricas.
• Fases poliméricas. Están constituidas por polímeros sintéticos de los que el más empleado es el copolímero estireno-divinilbenceno. Debido a que en este caso toda la partícula está enteramente constituida por polímero, estos rellenos presentan generalmente mayor retención que las fases monoméricas y las oligoméricas y, probablemente, un mayor grado de orden que las anteriormente descritas.
Figura IV.1
Síntesis del copolímero estireno-divinilbenceno (http://www.tecnociencia.es/especiales/intercambio_ionico/introduccion.htm).
De esta descripción se puede deducir que es posible emplear varios tipos de fases
estacionarias, que difieran notablemente en la densidad superficial y la organización de
los restos orgánicos que intervienen en la retención. Por tanto, es lógico pensar que estas
fases pueden interaccionar con el soluto de maneras diferentes, dependiendo de las
condiciones de separación (fase móvil, temperatura, etc.).
4. Modelos de retención
Se han propuesto tres modelos basados respectivamente en el efecto solvófobo, el
reparto del soluto entre la fase móvil y la fase estacionaria, y en la adsorción del soluto
sobre la fase estacionaria.
• Modelo del efecto solvófobo. Este modelo pone de relieve el papel primordial de la fase móvil en cromatografía de líquidos en fase inversa (CLFI) al admitir que el soluto, debido a su hidrofobicidad, es expulsado de la fase móvil hacia la fase estacionaria. Se describe a continuación de forma resumida este efecto desde un punto de vista termodinámico, pudiendo consultarse los trabajos originales (Melander y Horváth., 1980; Horváth y Melander, 1976,1977), para un estudio más detallado.

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
33
El equilibrio que da lugar a la retención de un soluto, i, sobre la fase estacionaria, S, en presencia de la fase móvil, puede escribirse como:
i+S->iS
donde iS es el complejo formado por asociación del soluto adsorbido sobre la fase estacionaria. Como ya se ha indicado, la retención del soluto puede expresarse a través del factor de retención, k, que está relacionado con el coeficiente de distribución.
Para estudiar la variación del potencial químico se puede considerar el proceso
dividido en dos partes: la correspondiente a las interacciones entre i y S en un
gas inerte para originar el complejo iS y debida a la introducción de i, S e iS
dentro del disolvente.
En muchos casos las interacciones entre i y S fuera del disolvente tienen lugar a través de fuerzas de tipo dispersivo, siendo posibles otras interacciones que contribuyan a la retención del soluto si éste es polar (interacciones dipolo-dipolo o dipolo-dipolo inducido) o si la fase estacionaria tiene grupos polares o que permitan transferencia de carga.
Para calcular el valor de la variación del potencial químico se puede suponer que para introducir las especies i, S, o iS en el disolvente es necesario crear un hueco capaz de alojar a cada una de dichas especies. La energía necesaria para crear el hueco es función de su tamaño, que a su vez depende del volumen de la molécula que se pretende introducir y de la tensión superficial del disolvente. Esta energía se denomina término de cavidad, Δμcav. A la energía correspondiente a la interacción de la molécula alojada en el hueco con el disolvente que la rodea, Δμint, contribuyen tres términos: un término que incluye las interacciones de tipo van der Waals entre el soluto y el disolvente, un término electrostático (si la molécula de soluto está cargada eléctricamente) y un término entrópico.
El balance de fuerzas que contribuyen a favor y en contra de la asociación determina que i y S estén más o menos fuertemente asociados y, por tanto, que el soluto esté más o menos retenido por la fase estacionaria. Cuanto mayor sea la tensión superficial, mayor será la retención ya que se requiere más energía para crear la cavidad que aloje a cada especie en la fase móvil. La influencia de ΔA (incremento de área) se justifica al tener en cuenta que el complejo iS presenta al disolvente acuoso una superficie menor que la suma de las que presentan i y S por separado, siendo por tanto el sistema asociado más estable que sin asociar. Si existen cargas no compensadas entre i y S, cuanto mayor sea la constante dieléctrica del medio mayor será la retención.

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
34
Por tanto, según este modelo, en cromatografía en fase inversa el soluto es expulsado hacia la fase estacionaria debido a la tensión superficial de la fase móvil. La asociación entre las moléculas de soluto y de fase estacionaria facilita la estabilidad del complejo iS que controla la retención al disminuir la superficie orgánica en contacto con la fase móvil. Sin embargo, el modelo solvófobo no considera las interacciones del soluto con la fase estacionaria si no es a través del área de contacto ΔA. Esto quiere decir que, de acuerdo con este modelo, la retención del soluto es en buena parte independiente de la fase estacionaria utilizada, lo que está en desacuerdo con un buen grupo de resultados experimentales que demuestran que, para determinadas fases estacionarias, tanto la retención como la selectividad están relacionadas con la naturaleza y la densidad de recubrimiento de la fase estacionaria (Dorsey y Cooper, 1994; Sanders y Wise, 1990; Sentel y Dorsey, 1989).
• Modelo de reparto. Una teoría más reciente sobre la retención en cromatografía de líquidos en fase inversa, que explica en parte el efecto de la fase estacionaria, ha sido desarrollada por Dill et al. (Dill, 1980; Dill et al., 1988; Dorsey y Dill, 1989). Proponen que la retención del soluto se debe fundamentalmente al balance de dos tipos de fuerzas: las originadas por la interacción de las moléculas de soluto con las que la rodean en la fase estacionaria y en la fase móvil, que se pueden expresar a nivel molecular mediante las constantes de interacción binarias; y las que proceden del ordenamiento parcial, con la correspondiente disminución de entropía, que experimentan las cadenas de la fase estacionaria al alojar las moléculas del soluto entre ellas. El modelo se puede describir brevemente de la forma siguiente:
El coeficiente de reparto entre la fase móvil y la fase estacionaria, y por tanto la retención, viene dado por el cociente entre sus concentraciones en las fases estacionaria y móvil, ci
S/ciM, lo cual se puede relacionar con el potencial químico
mientras se considera equilibrio.
a. Interacción de las moléculas de soluto con las que le rodean en la fase estacionaria y en la fase móvil. El modelo supone que cada una de las dos fases es un líquido amorfo y que la retención se puede asemejar al reparto del soluto entre dos fases, una acuosa y otra orgánica. Este reparto se puede cuantificar por el coeficiente de reparto entre una fase acuosa, de la misma composición que la fase móvil cromatográfica, y un hidrocarburo líquido, de la misma naturaleza química que la fase estacionaria cromatográfica pero con mayor número de grados de libertad. En este caso, la fuerza que controla la transferencia del soluto de una fase a otra es la afinidad química relativa de aquél con la fase estacionaria o con la móvil.

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
35
De una forma simple, la fase estacionaria y la fase móvil se representan en el modelo mediante una red tridimensional de celdillas elementales con las que interaccionan las moléculas de soluto. A nivel molecular, el proceso de transferencia de una molécula de soluto de la fase móvil a la fase estacionaria se puede visualizar como la sucesión de tres procesos elementales (Dorsey y Dill, 1989).
Figura IV.2
Representación esquemática de la transferencia de una molécula de soluto: (i) desde la fase móvil; (M) a la fase estacionaria; (S) en el modelo de reparto.
a. Creación de una cavidad del tamaño del soluto en la fase estacionaria.
b. Proceso de transferencia.
c. Llenado de la cavidad dejada por el soluto en la fase móvil.
La fuerza que gobierna la transferencia del soluto procede de las
interacciones necesarias para poner en contacto las moléculas i y j cuando
están separadas una distancia infinita. Diferentes fuerzas contribuyen, por
ejemplo, si las moléculas i y j tienen una carga neta, existe un componente
de tipo electrostático; si i y j tienen dipolos permanentes, el valor es
proporcional al del producto de los momentos dipolares.
Este modelo explica varios hechos importantes observados
experimentalmente en cromatografía en fase inversa:
a. La retención de los solutos es generalmente proporcional al coeficiente de reparto entre la fase móvil y un hidrocarburo líquido de la misma naturaleza química que la fase estacionaria.

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
36
b. El factor de retención es proporcional al tamaño del soluto ya que el
tamaño de cavidad está relacionado con el factor n.
c. lnK debe crecer linealmente con la tensión superficial de la fase móvil, puesto que ésta lo hace con trabajo necesario para poner en contacto dos moléculas de la fase móvil separadas una distancia infinita.
d. Todos los factores que cambian la solubilidad del soluto en la fase estacionaria o en la fase móvil deben modificar su retención; por ejemplo, se observa que aumentando el pH de la fase móvil los solutos ácidos débiles aumentan su carga neta y su afinidad por la fase móvil.
Aunque este modelo elemental se ha desarrollado para un disolvente puro, puede extenderse fácilmente para mezclas de cualquier número de componentes en la fase móvil, como es la práctica habitual en fase inversa.
b. Efecto del ordenamiento de las cadenas de la fase estacionaria al alojar al soluto. El modelo de reparto permite, aplicando cálculos de mecánica estadística, tener en cuenta las variaciones de conformación que se producen en las cadenas hidrocarbonadas de la fase estacionaria al penetrar en ellas las moléculas de soluto.
La creación de la cavidad dentro de la fase estacionaria para alojar al soluto induce una extensión y un mayor orden en las cadenas de la fase estacionaria. Dicho en términos termodinámicos, la introducción del soluto entre las cadenas de la fase estacionaria es entrópicamente desfavorable, lo que obliga a realizar un trabajo para llevar a cabo este proceso. A partir de esta suposición, el modelo permite predecir, en aceptable acuerdo con los resultados experimentales, que la representación del coeficiente de reparto del soluto entre la fase estacionaria y la fase móvil frente a la densidad de recubrimiento del relleno debe pasar por un máximo. A valores bajos de la densidad de recubrimiento, el coeficiente de reparto aumenta linealmente; a valores elevados, el coeficiente de reparto disminuye debido a la dificultad del soluto para penetrar en la fase estacionaria al aproximarse ésta al estado cristalino.
• Modelo de adsorción. Utilizando el mismo modelo de celdillas establecido por Dill para el modelo de reparto, se puede desarrollar un modelo de adsorción si se supone que en las condiciones experimentales no hay penetración del soluto en la fase estacionaria.
Comparando esto con lo dicho antes sobre las interacciones, se llega a la conclusión de que para la misma naturaleza química de soluto y disolvente, como el trabajo de las moléculas con la fase móvil es del mismo tipo en ambos

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
37
mecanismos, la adsorción será más débil que el reparto, ya que la entalpía de transferencia es del orden de z veces menor, según el modelo de la red tridimensional cúbica. Esto se debe a que solamente una fracción 1/z de la molécula de soluto, la que se adsorbe sobre la superficie, intercambia moléculas con la fase móvil que la rodea y el resto permanece invariable.
La evidencia experimental obtenida con fases estacionarias bien definidas y
sistemas de soluto modelo hacen pensar que los tres mecanismos descritos anteriormente
pueden tener lugar en cromatografía en fase inversa. Que la separación esté controlada
por uno u otro mecanismo depende del tipo de fase estacionaria utilizada, de la fase móvil
empleada, de la mayor o menor rigidez de los solutos a separar y de la temperatura
(Tchapla et al., 1993). Las condiciones más probables en las que cada mecanismo tiene
lugar son:
1. La estructura de la fase estacionaria en forma de empalizada sugiere una interacción del soluto con la parte terminal de las cadenas unidas de la fase estacionaria, que puede ser descrita por el modelo de adsorción. Esta situación se ha observado en la retención sobre fases monoméricas con elevada densidad de recubrimiento de solutos de una cierta flexibilidad, como los que contienen largas cadenas hidrocarbonadas, entre los que se encuentran las series homólogas de los alquilbencenos.
Figura IV.3
Modelo de organización de la fase estacionaria: (a) empalizada, (b) cepillo y (c) gavilla de heno.
2. En el caso de la estructura en forma de cepillo puede haber una inserción de las moléculas de soluto dentro de la fase estacionaria, con interacción entre el soluto y la fase estacionaria controlada por fuerzas de tipo solvófobo. Este comportamiento se ha observado en la separación de solutos flexibles sobre fases monoméricas con baja densidad de recubrimiento.
3. En la estructura en forma de gavilla de heno, las moléculas de soluto están inmersas en la fase estacionaria como en un proceso de reparto. Se ha observado que este modelo es más probable en fases oligoméricas, en las que

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
38
los solutos flexibles pueden introducirse fácilmente entre las cadenas hidrocarbonadas.
No hay que olvidar que bajo las cadenas hidrocarbonadas de la fase estacionaria los
grupos silanoles libres pueden intervenir también en la retención, sobre todo de los
solutos básicos, que poseen centros de densidad electrónica alta como nitrógenos con
pares electrónicos no compartidos o cargados positivamente. Por ejemplo, Horváth et al.
(Nahum y Horváth, 1981; Bij et al., 1981) han observado, empleando columnas de alquil-
sílice, variaciones anormales de la retención de solutos polares, como péptidos con grupos
amino no protegidos, en función del contenido en agua de la fase móvil. El
comportamiento vuelve a ser normal si esos grupos amino se bloquean o si se añade una
amina a la fase móvil. Sin embargo, este efecto de los grupos silanoles es en general
pernicioso para la separación, ya que conduce a picos cromatográficos anchos y con
colas. En la medida de lo posible, se trata de evitar esta adsorción parásita causada por
los silanoles libres empleando un aditivo catiónico en la fase móvil que los enmascare, tal
como la trietilamina, o fases especialmente diseñadas para la separación de compuestos
básicos, en las que la interacción entre el soluto y los grupos silanoles de la superficie es
muy pequeña.
5. Intercambio iónico.
La cromatografía de intercambio iónico se usa para el análisis de sustancias
cargadas eléctricamente en las condiciones de trabajo. En este modo de cromatografía se
emplean fases estacionarias con grupos funcionales cargados eléctricamente. El soporte
puede ser sílice o un polímero. La fase móvil contiene un tampón, que controla la carga
de los solutos, y una sal, que compite con los solutos en su interacción con las cargas de
la fase estacionaria. Las cargas permanentemente unidas al soporte cromatográfico están
compensadas por contraiones, es decir, por iones libres de carga opuesta. El paso a
través de la columna de una muestra, o fase móvil, con iones del mismo signo que los
contraiones de la columna puede provocar el desplazamiento de los contraiones y la
retención de los iones de la muestra o fase móvil. La selectividad y la fuerza de elución
dependen de parámetros como el tipo y concentración de iones en la fase móvil y el pH.
Para una misma fase móvil, el aumento de la concentración salina disminuye la retención
de los solutos. Además de la concentración del contraíon, su naturaleza también influye
sobre la retención a través de la constante de equilibrio del intercambio. Su valor refleja
la relación de afinidad del intercambiador iónico por el soluto y el contraión, que

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
39
generalmente es mayor para iones de mayor carga, con menor volumen hidratado y
mayor polarizabilidad. De este modo la retención de los solutos de la muestra aumenta,
en general, con la naturaleza del anión en la fase móvil en el siguiente orden:
citrato < sulfato < oxalato < yoduro < nitrato < cromato < bromuro
< sulfocianuro < cloruro < formiato < acetato < hidroxilo < fluoruro
El aumento en la retención sobre cambiadores catiónicos aumenta con la naturaleza
del ión presente en la fase móvil en el siguiente orden
Ba2+ < Pb2+ < Sr2+ < Ca2+ < Ni2+ < Cd2+ < Cu2+ < Co2+ < Zn2+ < Mg2+ < UO22+
< Tl+ < Ag+ < Cs+ < Rb+ < K+ < NH4+ < Na+ < H+ < Li+
El pH de la fase móvil juega también un papel importante sobre la retención,
controlando fundamentalmente la ionización de la muestra.
Los intercambiadores iónicos pueden clasificarse en catiónicos y aniónicos. Dentro
de cada grupo pueden diferenciarse los intercambiadores fuertes y débiles. Los
intercambiadores catiónicos fuertes contienen generalmente grupos sulfónicos, mientras
que los carboxilatos suelen estar presentes en los intercambiadores catiónicos débiles. Las
sales de amonio cuaternario son los grupos funcionales más usuales en los
intercambiadores aniónicos fuertes, empleándose las aminas primarias en los
intercambiadores aniónicos débiles.
6. Pares iónicos.
Este modo de cromatografía permite la separación de solutos que se encuentran
ionizados en las condiciones de trabajo. En la mayoría de las ocasiones, las separaciones
se llevan a cabo en las mismas columnas que se emplean en fase inversa sin formación
de pares iónicos. La fase móvil consiste en la mezcla de un tampón acuoso y un
modificador, a la que se le añade un contraión de carga opuesta a la del soluto. Para el
análisis de sustancias aniónicas se emplean frecuentemente como contraiones las sales de
amonio cuaternario, mientras que las sales de n-alquilsulfatos se encuentran entre los
contraiones más utilizados para el análisis de sustancias catiónicas.
En el caso más simple de cromatografía de pares iónicos, el soluto i y el contraión c
son solubles en la fase móvil acuosa M, mientras que el par iónico ic es soluble en la fase

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
40
estacionaria orgánica S. El proceso para solutos aniónicos y catiónicos podría
representarse respectivamente de forma esquemática como sigue
i-M+c+M ->icS
i+M+c-M ->icS
Sin embargo, no está probado que éste sea el mecanismo en todos los casos. Hay
algunos autores que describen el proceso como un intercambio iónico, al interpretar que
el contraión está adsorbido sobre la fase estacionaria, mecanismo que parece más
probable en el caso de contraiones de gran tamaño.
La retención está controlada por diversos parámetros, entre los que hay que
destacar la naturaleza y concentración del contraión, la fuerza iónica de la fase móvil y su
pH.
Para un mecanismo de retención simple esquematizado según:
iM+cM->icM->icS
y teniendo en cuenta la relación entre el factor de retención y el coeficiente de
distribución, y la ley de acción de masas, la retención del soluto aumenta linealmente con
la concentración de contraión. En la práctica, esta previsión se cumple en un intervalo
reducido de concentraciones del contraión, debido a que los equilibrios implicados en la
retención son más complejos que los indicados.
La influencia de distintos factores sobre la retención es diferente si la formación del
par iónico tiene lugar en la fase móvil que si ocurre en la fase estacionaria (Horváth et al.,
1977). La relación entre la retención del soluto en presencia del contraión y la que tendría
en su ausencia, definida como el incremento de la retención, es función de la carga del
contraión y del tamaño y la carga del soluto cuando el par iónico se forma en la fase
estacionaria. Cuando la formación del par iónico tiene lugar en la fase móvil, la retención
depende de la superficie de la parte hidrófoba del contraión y de la carga del soluto, pero
no de su tamaño.
La retención del soluto aumenta al hacerlo la constante de equilibrio de formación
del par iónico. De este modo, para el análisis de solutos catiónicos, cuanto más fuerte sea

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
41
el ácido que da lugar al contraión, el equilibrio de formación del par iónico estará más
desplazado hacia la formación del par y mayor será la retención. Otro factor que tiene
influencia sobre la retención es la longitud de la cadena alquílica del contraión que, al
aumentar, produce un incremento de su hidrofobicidad y, como consecuencia, de la
retención.
Junto con la sal correspondiente al contraión puede introducirse otra sal en la fase
móvil con objeto de controlar la fuerza iónica. A bajas fuerzas iónicas puede producirse
una disminución en la retención del soluto respecto a la que tendría sin la adición de la sal
debido a la competencia entre ésta y el soluto por unirse al contraión. Para fuerzas
iónicas elevadas, el aumento de tensión superficial debido al efecto de la concentración
de la sal es responsable del aumento en la retención.
El pH de la fase móvil influye a través de su efecto sobre la ionización del soluto.
Cuanto mayor es el grado de ionización, más se favorece la formación del par iónico, y
mayor es la retención.
7. Exclusión molecular.
En cromatografía de exclusión la separación de los componentes de la muestra está
basada en el tamaño efectivo de sus moléculas.
El soporte cromatográfico actúa como un tamiz molecular. La retención de las
moléculas es función de su tamaño, eluyendo en primer lugar las moléculas mayores, que
no se introducen en los poros del relleno, y correspondiendo el mayor tiempo de
retención a las moléculas más pequeñas, cuyo diámetro les permite entrar y salir de los
poros del relleno cromatográfico.
En cromatografía de exclusión la separación está controlada por el relleno de la
columna, por lo que es aconsejable el empleo de columnas con pequeños tamaños de
partícula para obtener elevadas eficacias. Su naturaleza puede ser polimérica o basada en
sílice, siendo crítica la elección del relleno en función de los tamaños moleculares de los
solutos a separar, y siendo necesario obtener la curva de calibración para los diferentes
materiales de relleno. En la figura puede observarse la representación teórica de una de
estas curvas, en la que a un volumen de retención VR(A), eluyen las moléculas cuyo
elevado tamaño (≥MWA) impide su entrada en los poros del material de relleno, y eluyen

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
42
por tanto en el volumen de exclusión de la columna. A un volumen VR(D), el volumen de
permeación, eluyen todas aquellas moléculas de tamaño suficientemente pequeño
(≤MWD) para poder entrar y salir de todos los poros del material de relleno. En el
intervalo de volumen de elución entre VR(A) y VR(D) pueden separarse las moléculas de
tamaños intermedios. Como puede observarse, cuanto menor sea la pendiente de esta
zona de la curva mayor será la posibilidad de separar compuestos de tamaños
moleculares próximos. Hay que tener en cuenta que estas curvas de calibración se suelen
obtener con compuestos patrón de unas determinadas características químicas y
geométricas. Es evidente que, además del peso molecular, la disposición geométrica de la
molécula tiene gran influencia sobre su tamaño efectivo y sobre la probabilidad de
introducirse en los poros del relleno cromatográfico. Esta consideración es importante
tanto para la elección de la columna apropiada para la separación de los componentes de
una muestra dada, como para calcular el peso molecular de un soluto a partir de una
recta de calibrado y de su tiempo o volumen de retención en una columna de exclusión
molecular.
Figura IV.4
La retención en cromatografía de exclusión.
A diferencia de otros modos de cromatografía de líquidos, en exclusión molecular la
fase móvil no juega un papel específico en la discriminación de los solutos, sino que su
misión es transportarlo a lo largo de la columna. La selección de la fase móvil se basa en
su capacidad para disolver la muestra, su baja viscosidad para evitar altas presiones y su
compatibilidad con el relleno cromatográfico. Además de estas características y como ya
se indicó, la fase móvil debe en ocasiones eliminar las interacciones específicas entre el
soluto y el relleno. La retención no deseable debida a fenómenos como adsorción,

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
43
interacciones iónicas, etc., puede ponerse de manifiesto mediante la presencia de picos
en el cromatograma con tiempos de retención superiores al tiempo de permeación, que
suele corresponder al disolvente.
8. Separaciones quirales.
La importancia que tiene la diferenciación de enantiómeros ha desencadenado un
interés creciente por el desarrollo de procedimientos que permitan su separación. Por ello
se hace aquí una breve referencia a los métodos que permiten la resolución de estas
moléculas.
La separación de enantiómeros mediante cromatografía de líquidos puede abordarse
de diferentes modos. El procedimiento tradicional para la separación de enantiómeros es
su transformación en diastereoisómeros, que pueden separarse en CLAE, generalmente
en fase inversa, sin el empleo de fases quirales. Sin embargo, este procedimiento conlleva
la necesidad de preparar derivados, con el consiguiente riesgo de reacciones secundarias,
y requiere la disponibilidad de reactivos quirales de pureza controlada. Además, el
proceso es largo y laborioso y, en muestras con bajo contenido en enantiómeros, es difícil
lograr un rendimiento cuantitativo. Estos inconvenientes se evitan con la separación
directa de los enantiómeros, que puede realizarse mediante fases estacionarias quirales o
añadiendo aditivos quirales en la fase móvil. La inclusión de estos aditivos es fácil de
llevar a cabo y permite gran número de variaciones.
La preparación de fases estacionarias quirales puede realizarse por recubrimiento
del relleno al hacer pasar la solución a través de la columna, o por unión iónica o
covalente del reactivo quiral al material de relleno de la columna. Dentro de los rellenos
cromatográficos hay que destacar los siguientes grupos:
1. Los que producen interacciones π-π.
2. Los que portan una proteína unida, de modo que los dos enantiómeros experimentan interacciones de distinta magnitud con la proteína durante su paso por la columna.
3. Los que incluyen cavidades quirales, que pueden producirse por polimerización del material de relleno en presencia de un mol de quiral, como se mencionará al hablar de las técnicas de impresión molecular, este grupo comprende también las ciclodextrinas y los éteres corona.
4. Derivados de polisacáridos, como celulosa, amilosa, etc.

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
44
9. Afinidad.
La cromatografía de afinidad se basa en la interacción específica entre una
molécula, o grupo de moléculas, y un ligando, que es una molécula que se fija a la fase
estacionaria para interaccionar con las del soluto. Cuando uno de los componentes del
complejo, generalmente el ligando, se inmoviliza en un soporte cromatográfico, la
formación del complejo específico puede utilizarse como base para la separación. El
mayor auge de la cromatografía de afinidad, desde sus orígenes a finales de los años 60,
ha correspondido a la cromatografía de bioafinidad, en la que las interacciones entre
ligandos y moléculas se basan en reconocimientos de tipo biológico. Este desarrollo se ha
potenciado con la difusión del estudio de biomoléculas en áreas como la biotecnología, la
bioquímica o la farmacología. Diferentes biomoléculas pueden tener características
fisicoquímicas muy similares, por lo que su separación por técnicas basadas en estas
propiedades puede resultar difícil, mientras que sí puede abordarse mediante
cromatografía de bioafinidad al ser ésta una técnica basada en interacciones altamente
específicas entre un ligando y una macromolécula. En este sentido, los antígenos se unen
a los anticuerpos, el RNA mensajero híbrida con el DNA complementario, etc.
La retención en cromatografía de bioafinidad se debe a uniones complejas que
incluyen distintos tipos de interacciones no covalentes, como son fuerzas de van der
Waals, puentes de hidrógeno, fuerzas hidrófobas, dipolo-dipolo, de transferencia de carga
o electrostáticas. La orientación espacial adecuada de los grupos a través de los cuales se
realiza la interacción facilita una elevada combinación cooperativa de fuerzas, lo que
constituye la base de la elevada selectividad y especificidad del enlace en cromatografía
de bioafinidad.
Entre los modos más selectivos de cromatografía de bioafinidad se encuentra la
cromatografía de inmunoafinidad, basada en el reconocimiento entre antígenos y
anticuerpos. Un antígeno es una sustancia que introducida en un organismo origina una
respuesta inmune, es decir, origina la producción de anticuerpos específicos frente a ese
antígeno. Esta respuesta altamente específica de la naturaleza, se ha aprovechado en los
métodos inmunológicos de análisis para emplear los anticuerpos como reactivos
específicos contra los antígenos. Cuando los anticuerpos se inmovilizan en un relleno
cromatográfico, el inmunoadsorbente formado puede emplearse para llevar a cabo
métodos de cromatografía de inmunoafinidad.

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
45
El anticuerpo se encuentra inmovilizado en la columna, a través de la cual se hace
pasar la muestra que contiene el antígeno, es decir el analito de interés. Éste es
capturado y queda retenido en la columna, mientras que el resto de los componentes de
la muestra que no son reconocidos por el anticuerpo eluyen sin retenerse. La detección
de los compuestos retenidos puede realizarse de diferentes formas, siendo la más usual
su elución con un agente de desorción para medir la respuesta en el detector del
cromatógrafo. La principal ventaja de esta técnica es su selectividad, de modo que
eligiendo el anticuerpo adecuado puede analizarse un único compuesto o, si interesa, se
puede analizar conjuntamente toda una familia de compuestos que tengan en común las
zonas de la molécula que son reconocidas por el anticuerpo, es decir que tengan los
mismos epítopos. De manera inversa, pueden analizarse anticuerpos específicos
empleando una columna con el correspondiente antígeno.
Dada la selectividad proporcionada por los distintos reconocimientos específicos que
se dan en la naturaleza, se han desarrollado técnicas para tratar de imitar este
comportamiento para el análisis de otras sustancias, preparando sintéticamente rellenos
cromatográficos para el reconocimiento y análisis de una o varias moléculas dadas. Estas
técnicas se denominan de impresión molecular (Ngo, 1993). La influencia de la
orientación espacial de los grupos funcionales en la selectividad de la interacción impone
un cuidadoso control de las posiciones de unión en la preparación de rellenos
cromatográficos de impresión molecular.
La preparación de polímeros de este tipo comienza mezclándose la molécula que se
quiere reconocer, el molde, con los monómeros adecuados para producir interacciones
específicas. A continuación se polimeriza la mezcla en presencia de concentraciones
elevadas de un agente de entrecruzamiento para formar un polímero rígido e insoluble.
Finalmente se elimina el molde de modo que la red polimérica dispone de microcavidades
con impresiones que son complementarias en forma y en funcionalidad química con el
molde. Si este material polimérico se emplea como relleno en una columna, se retendrán
de forma preferente las moléculas similares al molde.
10. Modos de elusión.
Se denomina fuerza eluyente o poder de elución de una fase móvil a su capacidad
para eluir un soluto dado desde una fase estacionaria determinada (Snyder, 1968). Un
disolvente B tendrá mayor fuerza eluyente que otro disolvente A, si un soluto tiene un

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
46
factor de retención menor, en la misma columna, cuando se emplea B como fase móvil,
que cuando se emplea A. En términos generales, se habla de eluyentes fuertes y
eluyentes débiles, según que su fuerza eluyente sea alta o baja. Así por ejemplo, en
cromatografía de líquidos en fase inversa, cualquier modificador orgánico (metanol,
acetonitrilo o tetrahidrofurano) es un disolvente fuerte en comparación con el agua, que
en esta técnica es siempre el disolvente más débil que se utiliza. En una fase móvil
formada por la mezcla de agua y modificador orgánico, cuanto mayor sea su contenido en
modificador mayor será su fuerza eluyente, ya que los solutos estarán menos retenidos
cuanto mayor sea la concentración de disolvente orgánico en la fase móvil. Por la misma
razón, en cromatografía de intercambio iónico, cuanto mayor es la concentración de sal
en la fase móvil mayor es su poder de elución para solutos iónicos.
Se puede cambiar de forma considerable el factor de retención de las sustancias a
separar, y por tanto el tiempo de duración del análisis, modificando la fuerza eluyente de
la fase móvil. Una parte de las estrategias para optimización de los métodos de
separación en cromatografía de líquidos se basa en los conocimientos que se tienen de
cómo varía la fuerza eluyente de la fase móvil con su composición (Snyder et al., 1968).
En cromatografía de líquidos hay dos estrategias generales para abordar una
separación: la elución isocrática y la elución en gradiente.
• Elución isocrática. La fuerza eluyente de la fase móvil permanece constante durante la separación. Se emplea generalmente cuando los componentes de la muestra tienen factores de retención semejantes. En estos casos casi siempre es posible encontrar una composición de la fase móvil en la que todos los solutos se eluyen en un intervalo de retenciones razonable (1 < k < 10) con una resolución óptima.
• Elución en gradiente. En las separaciones en gradiente se hace variar el poder de elución de la fase móvil a lo largo del análisis cambiando la composición de la fase móvil que entra en la columna, de acuerdo con un programa preestablecido. La elución en gradiente se emplea en la separación de mezclas complejas, formadas por solutos de naturaleza muy distinta y que poseen un amplio intervalo de factores de retención (1 < k < 50). Este modo de elución permite incluir todos los componentes en un solo cromatograma utilizando una opción similar a la que representa la programación de temperatura en cromatografía de gases. Si una muestra compleja se tratase de analizar mediante elución isocrática con una fase móvil de elevado poder de elución con el fin de hacer salir de la columna todos los componentes en un

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
47
tiempo razonable (~30 min), es probable que los componentes de menor retención salieran próximos al tiempo básico y con poca resolución. Por el contrario, si se utilizase una fase móvil de bajo poder de elución para conseguir la resolución de los primeros componentes, los solutos de mayor retención tardarían mucho tiempo en eluirse y originarían picos anchos que, debido a su dilución en la fase móvil, serían difíciles de detectar. En estos casos la elución en gradiente permite incrementar la fuerza eluyente de la fase móvil a lo largo del análisis para provocar una disminución de los factores de retención, tanto más acusada cuanto más retenido esté el compuesto. Los solutos menos retenidos eluyen bien resueltos al comienzo del gradiente, cuando una fase móvil de baja fuerza eluyente entra en la columna; en esta etapa del gradiente los solutos más retenidos avanzan muy lentamente hasta que llega fase móvil de fuerza eluyente creciente en sucesivas etapas. Entonces la velocidad de desplazamiento de los solutos más retenidos se acelera y eluye. En una elución en gradiente, los componentes de la muestra salen de la columna en tiempos razonables y generalmente dan lugar a picos estrechos fácilmente detectables. La diferencia de comportamiento de los solutos en elución isocrática y en gradiente se ilustra en la figura (Snyder, 1980).
Figura IV.5
Fracción de columna recorrida (r) por las bandas de los solutos X, Y, Z en función del tiempo para una separación isocrática (a) y en gradiente de elución (b); c y d
representan los cromatogramas que se originan; tg corresponde al momento en que r = 1; X el soluto menos retenido; Z el soluto más retenido.

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
48
V. CROMATOGRAFÍA DE GASES.
1. Introducción.
Su comienzo data del 1951, año en el que la revista Journal of Biochemistry recibió
un trabajo escrito por James y Martin (1952) sobre cromatografía de reparto gas-líquido,
donde se asentaron las bases teóricas de la técnica, si bien fue van Deemter (1956) quien
realizó el desarrollo teórico más importante. A lo largo de esa década se sucedieron
diversos avances como la programación de temperatura (Griffiths et al., 1952), las
columnas capilares abiertas (Golay, 1958) y los detectores de ionización (McWilliam y
Dejar, 1958; Lovelock, 1958); aunque realmente los avances han continuado desde
entonces haciendo de esta técnica la más utilizada para análisis cuantitativo y cualitativo
para compuestos orgánicos de volatilidad media o alta.
2. Sistemas de inyección de muestras.
La mayor parte de los dispositivos existentes para la introducción de muestras en
cromatografía de gases llevan a cabo su tarea tratando de no alterar las condiciones de
temperatura, presión y flujo, es decir, sin interrumpir el funcionamiento. Al igual que en
otras técnicas, un sistema óptimo debe cumplir una serie de requisitos:
- La composición de la fracción de muestra introducida en la columna cromatográfica debe ser la misma o muy similar a la composición de la muestra original, para poder obtener un correcto análisis cuantitativo; es decir, no deben existir pérdidas de componentes por descomposición, adsorción u otro mecanismo, ni tampoco discriminación entre componentes por otras causas.
- La anchura de zona ocupada por la muestra debe ser mínima para aprovechar al máximo la eficacia de la columna.
- Debe funcionar de manera perfectamente reproducible.

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
49
2.1. Modalidades de inyección.
En los cromatógrafos de gases la muestra debe pasar a la columna directamente o
arrastrada por el gas portador, sin alterar su flujo. Teniendo en cuenta la sensibilidad
deseada y la capacidad de carga de la columna, se introducen cantidades que van desde
los microgramos a los picogramos, empleando volúmenes que pueden oscilar desde los
nanolitros a varios centenares de microlitros; en estos casos, y tratándose de mezclas
muy diluidas, la mayor parte del disolvente se elimina antes de llegar a la columna.
La mayoría de los equipos comerciales van provistos de algún tipo de inyector, que
es básicamente una cámara de pequeño volumen situada a la entrada de la columna,
recorrida por el gas portador y dotada de calefacción independiente. Todos ellos tienen,
además, un pequeño obturador, que consiste generalmente en una membrana de
elastómero llamada septum que se perfora mediante una microjeringa al introducir la
muestra. Las modalidades de inyección más comunes son las siguientes.
Figura V.1
Esquema de inyectores de CG, sin y con divisor de flujo.
• Con evaporación previa
a. Inyección directa:
El esquema se muestra en la figura. La muestra introducida a través del septum se vaporiza bruscamente y es arrastrada por el gas portador hacia la columna. La cámara se mantiene a una temperatura ligeramente superior a la de la columna.

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
50
Este tipo de inyectores tiene un requisito: el gas portador debe circular por la
cámara a una velocidad tal que el ensanchamiento de la zona de muestra sea mínimo, y ello se consigue cuando su velocidad lineal en la cámara es similar o superior a la que lleva en la columna. Por ello este tipo de inyector se utiliza con buenos resultados en columnas de gran diámetro, sean de relleno o abiertas.
En general poseen más ventajas que inconvenientes: el ensanchamiento de banda es inapreciable, no hay pérdidas de ninguna clase, salvo para solutos termolábiles, y la cámara suele ir recubierta de una camisa de vidrio o cuarzo, especialmente desactivada y fácil de limpiar, para minimizar la adsorción. Sin embargo, cuando se utiliza este tipo de dispositivo con columnas de pequeño diámetro, cuyos flujos óptimos son bajos (<1 ml/min), la diferencia de velocidades lineales entre cámara y columna provoca un ensanchamiento incompatible con la eficacia que se espera de tales columnas.
b. Inyección con división de flujo (split) (Grob, 1986):
El esquema básico se muestra en la figura. Se utiliza para columnas de pequeño diámetro, generalmente capilares abiertas.
El gas portador que arrastra la muestra, una vez evaporada, se divide en dos partes: una pasa a la columna y otra se envía al exterior. El objetivo que se persigue es evitar el ensanchamiento de la zona ocasionado por las diferencias de diámetro entre la cámara y la columna. Para conseguirlo, es importante que la velocidad lineal del gas en la cámara sea igual o superior a la velocidad en la columna, lo que se consigue fácilmente si la salida de gas hacia el exterior está situada por detrás de la entrada en la columna, como se observa en la Figura.
Figura V.2
Esquema de control neumático de inyección con división de flujo.
La división de flujo ocasiona una división equivalente de la muestra que, en principio, debe ser proporcional a la relación de división de flujos, también conocida

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
51
por relación de división de flujos y que se define como la relación entre el flujo total y el que pasa por la columna, Ftotal/Fcolumna. Desgraciadamente esta relación no siempre se mantiene estable y representa la principal fuente de problemas de este tipo de inyector.
Cuando se introduce una muestra disuelta, la disolución es lanzada como un surtidor de microgotas: las más pequeñas se evaporan y otras chocan con las paredes calientes de la camisa de vidrio. Dado el poco tiempo que la mezcla de gas y muestra permanece en la cámara (1-5 s), algunas gotas que contienen los componentes menos volátiles pueden ser arrastradas al exterior antes de evaporarse completamente, lo que se pone de manifiesto como una disminución del área en el cromatograma de ciertos componentes discriminados.
Para evitar o minimizar este tipo de problemas se han propuesto diferentes diseños para la camisa de vidrio. Por otra parte, la evaporación brusca del disolvente en un espacio tan reducido provoca una sobrepresión, que altera momentáneamente la relación de división de flujos y, por lo tanto, hace casi imposible calcular con exactitud qué cantidad de muestra se ha enviado realmente a la columna. Esta sobrepresión es también la causa de que el procedimiento no permita introducir grandes volúmenes de muestra, ya que el exceso se enviaría al exterior.
Este tipo de dispositivos suelen llevar una salida de gas portador adicional, llamada purga del septum, destinada a enviar al exterior, sin que contaminen la columna, la mayor parte de los monómeros y demás impurezas que, procedentes del material elastómero del septum, pasan continuamente al gas portador. La inyección con división de flujo es quizá la más utilizada en el trabajo con columnas capilares al ser un procedimiento rápido, cómodo, moderadamente reproducible y fácil de automatizar.
Sus principales inconvenientes son:
La discriminación de ciertos componentes de la muestra cuando se trabaja con mezclas de amplio intervalo de ebullición.
La descomposición de muestras termolábiles.
La dificultad de llevar a cabo un calibrado absoluto del sistema, ya que casi nunca se llega a saber cuánta muestra ha llegado realmente al detector.
c. Inyección sin división/con división interrumpida (splitless):
Se emplea para aumentar la cantidad de muestra que llega al detector, sobre todo cuando se trabaja en el análisis de trazas con columnas capilares.

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
52
Se utilizan los mismos inyectores mencionados anteriormente, pero se opera
del siguiente modo: el divisor de flujo se cierra, se introduce la muestra, y se vuelve a abrir después de un periodo controlado de tiempo del orden de 15 a 60 s. De esta manera la mayor parte de la muestra penetra lentamente en la columna mientras el divisor está cerrado, y los últimos restos son barridos hacia el exterior cuando se abre de nuevo.
Para compensar el ensanchamiento de zona ocasionado se puede operar de dos formas:
• La más frecuente consiste en mantener la columna a temperatura muy baja a fin de que los vapores de la muestra vayan condensando en una zona lo más estrecha posible a medida que van entrando (crioenfoque). Una vez abierto el divisor de flujo se programa la temperatura del horno según requiera el proceso cromatográfico, con lo que se obtiene una eficacia comparable a la de una inyección en modalidad split. Evidentemente, es necesario que exista una diferencia de temperaturas importante entre el punto de ebullición de los solutos y la temperatura inicial de la columna, a fin de que condensen totalmente.
• Otra forma de conseguir la reconcentración es utilizando el llamado efecto del disolvente. En este caso, se trata de lograr que el disolvente condense a la entrada de la columna, formando, junto con la fase estacionaria, una especie de fase mixta de elevado espesor que reduce local y temporalmente la relación de fases, β, lo que supone el aumento del factor de retención de las moléculas de soluto situadas en el frente de la zona cromatográfica y, por tanto, un impedimento para su avance. De esta forma, los solutos quedan atrapados en el primer tramo de la columna, cubierto temporalmente con esta fase mixta; una vez que se evapora el disolvente, los solutos comienzan a eluirse en forma de banda reconcentrada. Es preciso elegir un disolvente que tenga un punto de ebullición lo suficientemente elevado para poder condensar en las condiciones elegidas y que se separe convenientemente de los solutos. Cuando se trabaja con fases no inmovilizadas el disolvente puede acabar arrastrando la fase estacionaria, desnudando la columna interiormente; para evitarlo, se puede intercalar un capilar sin fase antes de la columna, o dejar deliberadamente sin recubrir 1 -2 m en cabeza de columna, lo que se denomina zona sin retención.
Para trabajar en cualquiera de estas dos modalidades se utilizan los mismos dispositivos, llamados inyectores con/sin división de flujo. Los equipos comerciales llevan a cabo el control neumático básicamente de una de estas dos maneras: regulación por la presión de entrada a la columna y un restrictor variable en la salida

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
53
al exterior, o uso de un sistema de retrocontrol de presión a la salida, manteniendo un regulador de flujo a la entrada).
d. Inyector con temperatura programada (PTV) (Schomburg, 1985)
Es básicamente igual que los descritos anteriormente, pero en lugar de trabajar a una temperatura prefijada la muestra se introduce en frío, se deposita sobre un relleno y se calienta rápidamente hasta una temperatura determinada, lo que provoca la evaporación homogénea de la disolución y su arrastre hacia la columna.
La combinación de esta programación de temperatura con la operación de las válvulas de apertura y cierre del divisor de flujo permite eliminar de modo selectivo gran parte del disolvente y aumentar la cantidad de muestra introducida, reduciendo casi por completo la discriminación y disminuyendo la descomposición de muestras lábiles.
• Sin vaporización previa
El procedimiento (Grob, 1987) que cumple esta condición se llama inyección en columna. En él la muestra se deposita directamente en cabeza de la columna, sin vaporización previa ni división de flujo. Para ello se utilizan inyectores con obturador mecánico en vez de septum y jeringas especiales con agujas de pequeño diámetro externo (<0,3 mm), compatible con el interno de las columnas, que suele ser de 0,3-0,5 mm.
Las ventajas son muchas: no hay división de la muestra ni discriminación, no hay precalentamiento excesivo ni contacto con metales a altas temperaturas, se evitan descomposiciones, y desaparecen los problemas típicos del septum tales como la contaminación con siliconas volátiles y con partículas sólidas. También tiene sus inconvenientes: el disolvente puede afectar a la fase estacionaria, las agujas de las jeringas se obstruyen con facilidad y las muestras deben estar lo más exentas posible de compuestos no volátiles, que se depositarían en su totalidad en la columna.
2.2. Dispositivos externos para introducción de muestras
Según sea el estado físico de la muestra, se necesitarán diferentes tipos de dispositivos para tomarla y transferirla al sistema cromatográfico, ya sea directamente a la columna o al inyector, y respetando los requisitos indicados más arriba. Algunos de estos dispositivos son muy simples, como las microjeringas; otros son equipos tan voluminosos y sofisticados como el propio cromatógrafo, y muchos de ellos incluyen toma de muestra e introducción en la columna, reemplazando a los inyectores de tipo básico descritos anteriormente. Se pueden mencionar los que se utilizan para:

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
54
Tabla V.1
Dispositivos externos más usados para la introducción de muestras en CG (Dabrio et al., 2000)
• Introducción de muestras en estado líquido.
Además de las muestras líquidas, una gran parte de las muestras sólidas se introducen disueltas en algún disolvente, lo que hace que el procedimiento más común de inyección de muestras utilice microjeringas de pequeño volumen (1-10 μl). Sin embargo, la búsqueda de mayor sensibilidad y el acoplamiento CL-CG han sido las principales causas del desarrollo de las técnicas de inyección de volúmenes muy superiores.
No es posible la introducción en una columna capilar de volúmenes del orden de 100 μL de líquido o superiores, aunque sería muy ventajoso para el análisis de trazas o cuando se tienen muestras muy diluidas que no es fácil concentrar sin perder parte de los analitos.
Para trabajar en estas condiciones, hay que introducir la muestra en una zona sin retención o en una precolumna, evaporar la mayor parte del disolvente a baja temperatura eliminándolo a través del divisor de flujo o de otro sistema similar, y reconcentrar los solutos mediante efecto de disolvente o simple crioenfoque.
Es recomendable llevar a cabo este tipo de inyecciones mediante algún sistema automático.
Válvulas Muestras gaseosas
Jeringas de gases
Microsondas Muestras sólidas
Pirolizadores
Muestras Líquidas o disueltas en un líquido Microjeringas
Vapor confinado
Arrastre con gas inerte Compuestos volátiles en matrices sólidas
o líquidas
Desorción Térmica

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
55
• Introducción de muestras en estado gaseoso.
En algunos casos este tipo de muestras se puede introducir directamente en la columna, pero muchas veces hay que llevar a cabo una etapa de enriquecimiento o preconcentración. Entre los distintos dispositivos utilizados para la introducción de muestras gaseosas se pueden mencionar:
o Jeringas de gases. Dotadas con un émbolo antifuga, especialmente diseñadas para estos casos.
o Válvulas. Similares a las que se emplean en cromatografía de líquidos.
• Introducción de muestras en estado sólido.
o Microsondas. Muchas son parecidas a las jeringas de líquidos, con la punta preparada para introducir pequeñas cantidades de sólidos que se depositan en ella, bien directamente, bien por evaporación a partir de una disolución. Algunas de ellas pueden utilizarse con los inyectores provistos de septum mencionados para muestras líquidas.
o Pirolizadores. Permiten analizar muestras no vaporizables. Son dispositivos que provocan la descomposición térmica controlada de la muestra y el arrastre de sus fragmentos hacia la columna. Si la pirólisis es reproducible, el análisis de esos fragmentos suministra la información deseada acerca de la composición de la muestra. Este tipo de inyección se aplica a compuestos que no pueden eluirse en cromatografía de gases, como macromoléculas o incluso células.
• Introducción de compuestos volátiles.
o Sistemas de muestreo de vapor confinado (static headspace). Cuando una muestra líquida o sólida que contiene componentes volátiles se mantiene a temperatura constante en un recipiente cerrado, dichos componentes se reparten entre líquido y vapor; cuando se alcanza el equilibrio, la cantidad de cada componente en la fase gaseosa depende de su concentración inicial en la fase líquida o sólida, de la temperatura y del coeficiente de actividad.
La fase gaseosa puede inyectarse directamente en el cromatógrafo de gases. Como suele estar muy diluida se requieren cantidades de muestra relativamente grandes, y es habitual el uso de columnas con

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
56
Figura V.3
Inyección de espacios de cabeza, (a) Vapor confinado, (b) Arrastre con gas inerte.
elevada cantidad de fase estacionaria o columnas de relleno. Existen varios dispositivos comerciales que operan automáticamente, lo que suele ser necesario para conseguir una reproducibilidad satisfactoria.
o Métodos de arrastre con gas inerte. Si se hace pasar una corriente de
gas a través de una muestra líquida o sólida que contiene componentes volátiles, éstos son arrastrados de modo continuo por el gas y pueden llegar a extraerse casi totalmente de la disolución. Para recuperar los analitos es necesario pasar el gas por una trampa que los retenga.
Las trampas se basan en dos modos básicos de captura de solutos: la criocondensación o la adsorción. El posterior calentamiento de las trampas permite introducir los solutos en la columna. Si la desorción de la trampa es lenta, se produce un ensanchamiento de la zona que se debe reconcentrar en cabeza de columna.
o Desorción térmica. Se utiliza para extraer compuestos volátiles de muestras sólidas o de trampas donde aquéllos hayan condensado previamente. La muestra se calienta en una corriente de gas portador que arrastra los componentes volátiles hacia la columna. Habitualmente se requiere una etapa de crioconcentración, pudiendo ser la reconcentración por espacio en cabeza innecesaria si los componentes a determinar tienen factores de retención moderados o altos. Entre sus ventajas pueden citarse la ausencia de disolventes y la sencillez de la etapa de preparación. Su principal inconveniente es la posibilidad de formación de artefactos por descomposición térmica de la matriz o la pérdida de componentes lábiles.

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
57
3. La columna.
Las dimensiones de la columna tienen una trascendencia directa en la eficacia del
sistema, y el material de que está construida condiciona en ciertos casos los parámetros
de operación. Las columnas en Cromatografía Gaseosa se pueden clasificar en dos tipos
principales: abiertas y de relleno. En las primeras, la fase estacionaria se impregna sobre
la pared interna del tubo, dejando en el centro un canal para la fase móvil; en las
segundas, la fase se deposita sobre partículas sólidas que llenan completamente el tubo
en el que van contenidas.
3.1. Tipos de columnas y dimensiones
Los tubos son casi siempre cilíndricos, aunque a veces se han propuesto de sección
rectangular o elíptica. Las columnas analíticas que se utilizan en Cromatografía Gaseosa
tienen una gran variedad de dimensiones, tal como se recoge en la tabla.
Figura V.4
1Columna de relleno de cromatografia gaseosa (http://www.uib.es/depart/dqu/dquo/pau/Cromatograf%92a/columnas.html).
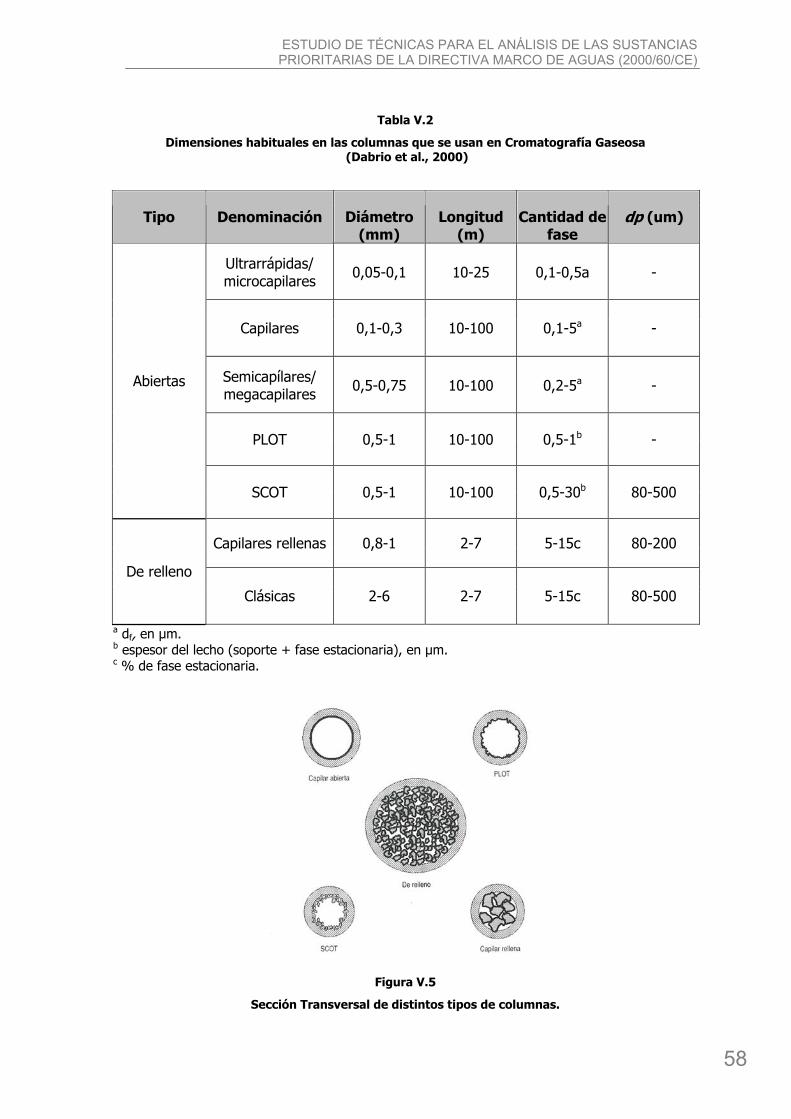
ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
58
Tabla V.2
Dimensiones habituales en las columnas que se usan en Cromatografía Gaseosa (Dabrio et al., 2000)
Tipo Denominación Diámetro (mm)
Longitud (m)
Cantidad de fase
dp (um)
Ultrarrápidas/ microcapilares 0,05-0,1 10-25 0,1-0,5a -
Capilares 0,1-0,3 10-100 0,1-5a -
Semicapílares/ megacapilares 0,5-0,75 10-100 0,2-5a -
PLOT 0,5-1 10-100 0,5-1b -
Abiertas
SCOT 0,5-1 10-100 0,5-30b 80-500
Capilares rellenas 0,8-1 2-7 5-15c 80-200
De relleno
Clásicas 2-6 2-7 5-15c 80-500
a df, en μm. b espesor del lecho (soporte + fase estacionaria), en μm. c % de fase estacionaria.
Figura V.5
Sección Transversal de distintos tipos de columnas.

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
59
En las columnas capilares abiertas de pared recubierta (WCOT, Wall-Coated Open
Tubular) la fase estacionaria se deposita sobre la pared interna del tubo, que es lisa y en
general poco modificada; en las columnas abiertas de pared porosa (PLOT, Porous-Layer Open Tubular) existe una capa porosa sobre la pared interna que se suele conseguir por
corrosión de la pared o por deposición de un producto sólido. La capa porosa puede servir
de soporte para la fase líquida estacionaria o actuar ella misma como fase estacionaria.
Las columnas abiertas recubiertas con soporte (SCOT, Support-Coated Open Tubular) son
similares a las PLOT, sólo que en este caso la capa porosa consta de partículas sólidas
depositadas a partir de una suspensión que también se pueden recubrir de fase
estacionaria.
Figura V.6
Columna Capilar WCOT (http://www.uib.es/depart/dqu/dquo/pau/Cromatograf%92a/columnas.html)
3.1.1. Efecto de las dimensiones sobre el comportamiento de las columnas
Tanto el tipo de columna como sus dimensiones influyen fundamentalmente sobre tres características: la permeabilidad, la relación de fases y la capacidad de carga, que a su vez determinan su distinto comportamiento cromatográfico.
a. Permeabilidad.
La permeabilidad específica, B0, expresa la resistencia de la columna al paso de la fase móvil.
La baja permeabilidad explica la gran caída de presión en las columnas de relleno y limita a menos de 5 m. la longitud que pueden tener cuando se utiliza un intervalo habitual de presión, es decir, menos de 8 kg/cm2, y tamaños de partícula no excesivamente pequeños (hasta 100-120 mesh). En columnas abiertas de diámetro igual o menor que 0,1 mm, la longitud máxima no suele exceder los 15-
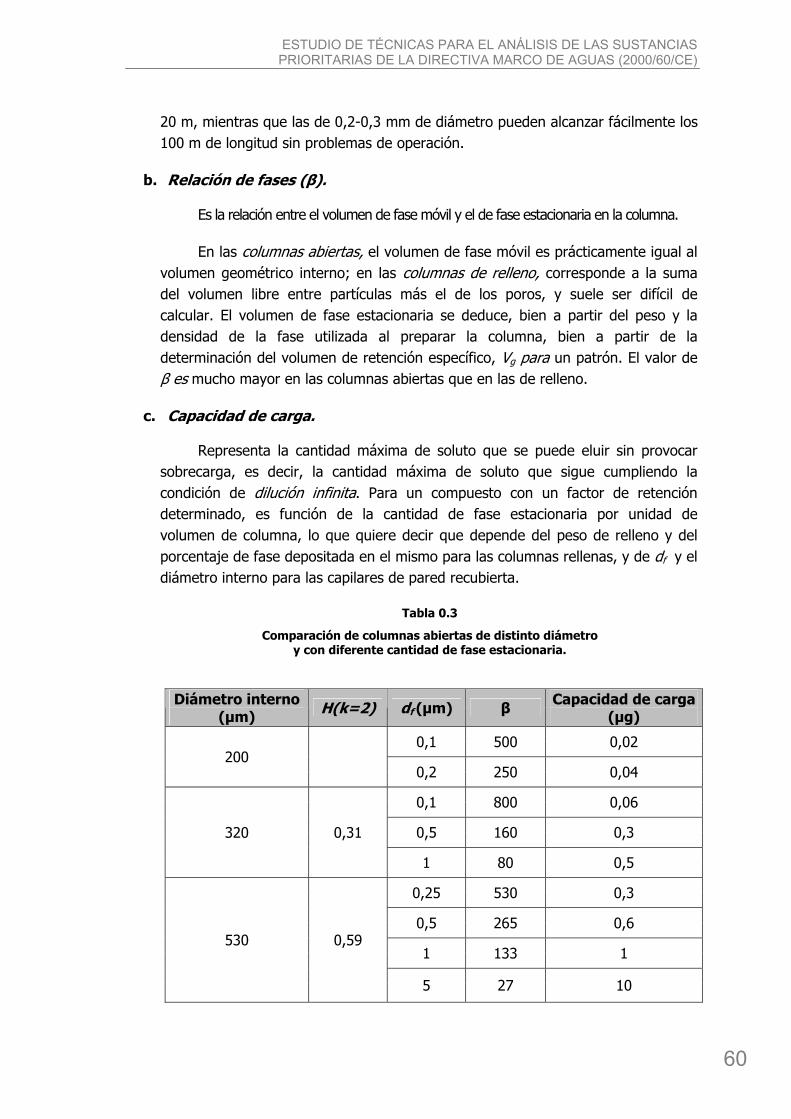
ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
60
20 m, mientras que las de 0,2-0,3 mm de diámetro pueden alcanzar fácilmente los 100 m de longitud sin problemas de operación.
b. Relación de fases (β).
Es la relación entre el volumen de fase móvil y el de fase estacionaria en la columna.
En las columnas abiertas, el volumen de fase móvil es prácticamente igual al volumen geométrico interno; en las columnas de relleno, corresponde a la suma del volumen libre entre partículas más el de los poros, y suele ser difícil de calcular. El volumen de fase estacionaria se deduce, bien a partir del peso y la densidad de la fase utilizada al preparar la columna, bien a partir de la determinación del volumen de retención específico, Vg para un patrón. El valor de β es mucho mayor en las columnas abiertas que en las de relleno.
c. Capacidad de carga.
Representa la cantidad máxima de soluto que se puede eluir sin provocar sobrecarga, es decir, la cantidad máxima de soluto que sigue cumpliendo la condición de dilución infinita. Para un compuesto con un factor de retención determinado, es función de la cantidad de fase estacionaria por unidad de volumen de columna, lo que quiere decir que depende del peso de relleno y del porcentaje de fase depositada en el mismo para las columnas rellenas, y de df y el diámetro interno para las capilares de pared recubierta.
Tabla 0.3
Comparación de columnas abiertas de distinto diámetro y con diferente cantidad de fase estacionaria.
Diámetro interno (μm) H(k=2) df (μm) β Capacidad de carga
(μg)
0,1 500 0,02 200
0,2 250 0,04
0,1 800 0,06
0,5 160 0,3 320 0,31
1 80 0,5
0,25 530 0,3
0,5 265 0,6
1 133 1 530 0,59
5 27 10
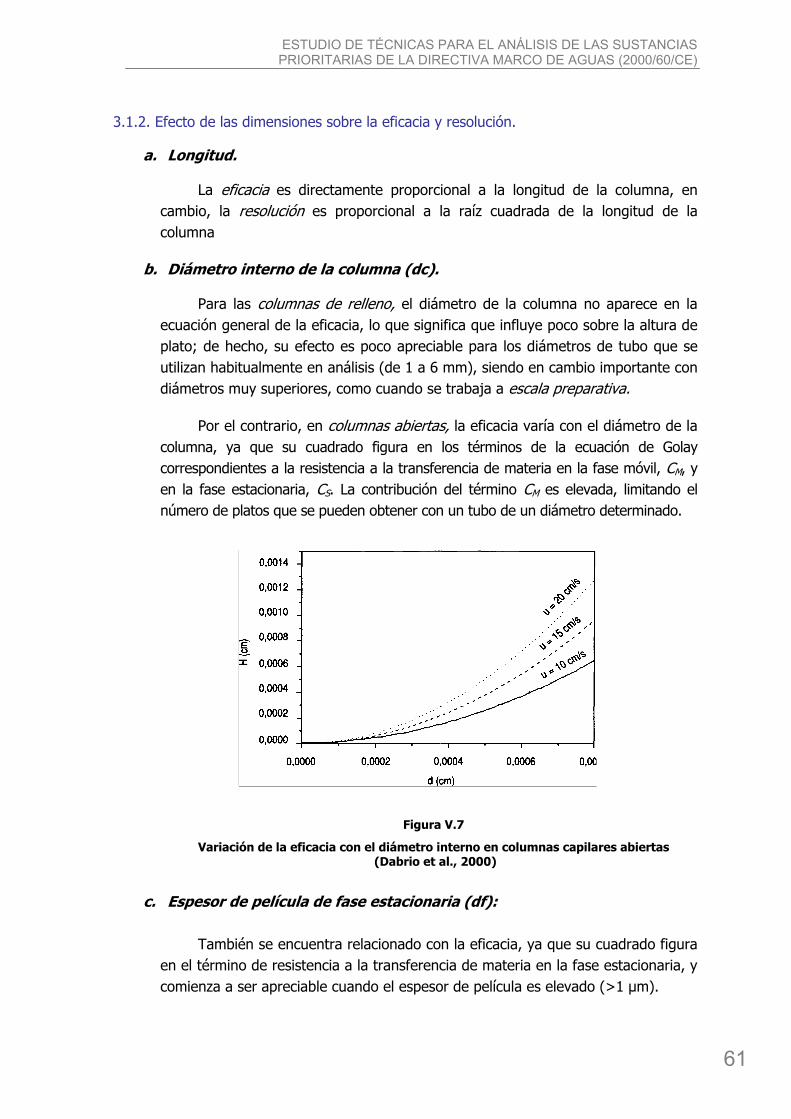
ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
61
3.1.2. Efecto de las dimensiones sobre la eficacia y resolución.
a. Longitud.
La eficacia es directamente proporcional a la longitud de la columna, en cambio, la resolución es proporcional a la raíz cuadrada de la longitud de la columna
b. Diámetro interno de la columna (dc).
Para las columnas de relleno, el diámetro de la columna no aparece en la ecuación general de la eficacia, lo que significa que influye poco sobre la altura de plato; de hecho, su efecto es poco apreciable para los diámetros de tubo que se utilizan habitualmente en análisis (de 1 a 6 mm), siendo en cambio importante con diámetros muy superiores, como cuando se trabaja a escala preparativa.
Por el contrario, en columnas abiertas, la eficacia varía con el diámetro de la columna, ya que su cuadrado figura en los términos de la ecuación de Golay correspondientes a la resistencia a la transferencia de materia en la fase móvil, CM, y en la fase estacionaria, CS. La contribución del término CM es elevada, limitando el número de platos que se pueden obtener con un tubo de un diámetro determinado.
Figura V.7
Variación de la eficacia con el diámetro interno en columnas capilares abiertas (Dabrio et al., 2000)
c. Espesor de película de fase estacionaria (df):
También se encuentra relacionado con la eficacia, ya que su cuadrado figura en el término de resistencia a la transferencia de materia en la fase estacionaria, y comienza a ser apreciable cuando el espesor de película es elevado (>1 μm).

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
62
d. Tamaño de partícula (dp):
Su contribución es importante en las columnas de relleno ya que aparece en dos términos de la ecuación de la eficacia.
A no ser que las partículas sean esféricas y uniformes, su efecto final no es fácil de predecir cuantitativamente (Littiewood, 1962) debido a las dificultades que presenta la estimación de los parámetros relacionados con la estructura geométrica del lecho de partículas (γ, λ, ω, q). La eficacia de las columnas aumenta cuando se utilizan rellenos más finos y uniformes, con el límite práctico para un tamaño de partícula en torno a 100 μm, por debajo del cual la permeabilidad B0 se hace demasiado pequeña y requiere una presión de entrada superior a la que suelen proporcionar los equipos comerciales.
e. Materiales del tubo.
El material ideal debería ser flexible, con buena conductividad térmica, transparente, inerte, irrompible y económico. Como tal material no existe, hay que seleccionar el que posea las especificaciones más adecuadas a cada caso.
Para las columnas de relleno se suele recurrir a materiales como el vidrio y el acero inoxidable, ya que la influencia de la pared en el comportamiento de estas columnas es pequeña: en primer lugar, la superficie interna del tubo es mucho menor que la superficie del relleno; además, la actividad superficial de ambos materiales no es muy elevada, comparable a la del relleno no recubierto por la fase estacionaria. Entre los dos materiales citados más arriba, el vidrio tiene la ventaja de su mayor inercia química frente a posibles acciones catalíticas del acero; su desventaja es la fragilidad.
En el caso de las columnas capilares abiertas, la influencia de la superficie interna del tubo es muy grande, ya que soporta una pequeña cantidad de fase estacionaria y una fracción importante de las moléculas eluidas entran en contacto con dicha pared cuando se disuelven en la fase estacionaria. Por ello la inercia química del tubo, que a veces se refuerza mediante tratamientos químicos de desactivación, es el requisito primordial para la selección, seguido por la resistencia a la rotura. El material más usado actualmente es la sílice vítrea, también llamada sílice fundida, recubierta exteriormente de poliimida, que es un material menos frágil que el vidrio y con una inercia química elevada; otros materiales mucho menos usados son el Ni y el vidrio. La transmisión de calor a través de los tubos de sílice vítrea es especialmente eficiente debido a su reducido espesor de pared (<0,1 mm).

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
63
f. Soportes.
En las columnas de relleno la fase estacionaria se deposita sobre partículas llamadas soporte. La primera columna de cromatografía de gases (James y Martin, 1952) utilizaba un soporte de Celita con aceite de silicona como fase estacionaria. Los materiales propios de la cromatografía en columna, como la sílice o la alúmina, que se utilizaron en los primeros años, fueron pronto reemplazados por otros, como las tierras de diatomeas, carbones, bolas de vidrio, etc., que modernamente tienden a ser a su vez sustituidos por materiales de síntesis tales como sílices, carbones grafitados y polímeros orgánicos.
Un soporte ideal debe ser indeformable a la presión, presentar resistencia a la rotura y al calor, y poseer estabilidad química, inercia química superficial, superficie específica elevada y tamaño y porosidad uniforme. Los soportes vendidos comercialmente se homogeneizan por tamizado y su intervalo de tamaños suele expresarse en unidades de paso de tamiz ASTM.
1. Soportes silíceos naturales. Los soportes silíceos naturales, o tierras de diatomeas, son conglomerados de algas unicelulares y están constituidos por un 90% de SO2, siendo el resto varios óxidos metálicos. Estas tierras dan lugar a dos series de soportes:
• Soportes rosas. Se producen por calcinación a elevadas temperaturas (>1.000 °C), a las cuales se forman óxidos de hierro que le dan el color. Son muy porosos, duros y densos, y soportan hasta un 30% de fase estacionaria líquida. Su superficie es muy activa, por lo que sólo se utilizan para separar compuestos muy inertes.
• Soportes blancos. Se originan por tratamiento de los anteriores con fundentes a unos 900 °C. El material se funde en parte, el hierro forma complejos incoloros y el soporte obtenido posee menor superficie específica, menos densidad, es muy frágil pero bastante más inerte. No es habitual cargarlos con más de un 10-15% de fase líquida.
Ambas series de soportes se lavan con ácidos para eliminar los óxidos más superficiales, indicándose en el nombre comercial con las siglas AW (Acid-washed), y a veces se desactivan por tratamiento con dimetilclorosilano para reducir el número de grupos silanol superficiales, usándose en este caso las siglas DMCS.
2. Soportes silíceos de síntesis. Están constituidos por un 99% de Si02, y se caracterizan por su tamaño y forma regulares y por tener tratamientos de desactivación muy controlados. Muchos de ellos son similares a los que se

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
64
usan en cromatografía de líquidos, con tamaños de partícula entre 50-100 μm, que son ligeramente mayores que en esa modalidad.
3. Otros soportes. Algunos polímeros orgánicos, aunque diseñados primordialmente como fases estacionarias sólidas, se pueden recubrir con fase estacionaria líquida. Existen también soportes mixtos, similares a los de fase inversa en cromatografía de líquidos, constituidos por un núcleo de sílice con una capa de moléculas orgánicas ligadas (C-8, C-18, fenilalquil, diclorofenil, etc.). Esta capa tendría la misión en cromatografía de gases de desactivar la superficie y facilitar la impregnación con la fase estacionaria líquida.
3.1.3. Preparación de columnas
Mientras que la preparación de columnas de relleno es relativamente sencilla y puede llevarse a cabo en cualquier laboratorio, la preparación de columnas abiertas requiere bastante habilidad. A continuación se indican, de forma resumida, los pasos más importantes que hay que seguir para preparar una columna, utilizando tubos, soportes y fases comerciales.
a. Columnas de relleno.
Se pesa una cantidad del soporte elegido algo mayor que la necesaria para llenar la columna, y se le añade una disolución que contenga la cantidad necesaria de fase estacionaria disuelta. Se agita, se desgasifica y se elimina el disolvente por evaporación o filtración. El relleno se seca y se introduce en el tubo limpio mediante presión o vacío, y con vibración suave, para conseguir una distribución lo más compacta posible. Los extremos se obturan con lana de vidrio y rejilla metálica, o fritados. Los tubos se pueden utilizar más de una vez.
b. Columnas abiertas de pared recubierta.
A no ser que el tubo comercial, que debe ser siempre nuevo, esté ya desactivado, hay que llevar a cabo algún tratamiento previo de desactivación, que puede llegar a ser muy laborioso. La fase se disuelve en un disolvente de bajo punto de ebullición (Pentano, didorometano, éter etílico y sus mezclas, son los más usados) y se deposita sobre la pared interna de dos formas (Grob, 1986):
• Método estático. La columna se llena con una disolución diluida de fase estacionaria, se cierra uno de los extremos, y se va haciendo evaporar el disolvente por el otro mediante vacío o temperatura superior a la de ebullición del disolvente, con lo que la fase va quedando depositada en la pared.

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
65
• Método dinámico. Se hace circular por el tubo una pequeña cantidad de
disolución concentrada de fase estacionaria empujada por un gas inerte que va mojando el tubo por el frente y evaporándose por detrás, dejando a su paso una capa muy fina de fase estacionaria.
El método estático es más difícil de llevar a cabo, pero controla perfectamente la cantidad de fase depositada en la columna y el espesor de la capa, pudiendo llegarse a obtener películas muy uniformes de hasta 5 μm. El método dinámico es muy sencillo de realizar, pero la cantidad de fase que se deposita se puede calcular sólo aproximadamente, la uniformidad es difícil de conseguir y el espesor de la película es siempre reducido.
Si se quiere obtener una columna más estable, hay que dar un tratamiento extra de reticulación química de la fase estacionaria, para lo cual se añade a la disolución de llenado el reactivo deseado y la reacción se lleva a cabo, después de la impregnación, mediante calor o irradiación. El exceso de reactivo se elimina por lavado o por arrastre con gas inerte.
3.2. La fase móvil.
En la cromatografía gaseosa, la fase móvil es siempre un gas que suele ser inerte y
que cumple aproximadamente la ecuación de los gases perfectos. Al ser el producto pV
constante a lo largo de la columna, la densidad, presión y la velocidad de la fase móvil
variarán de forma no lineal, siendo sus valores función de la distancia a la entrada de la
columna.
A continuación vamos a comentar las variables más importantes que nos vamos a
encontrar en la cromatografía gaseosa:
• El valor medio de la velocidad lineal se podría obtener tanto de su cálculo a partir de la longitud de la columna y el tiempo básico, como a través de j, factor de corrección de la compresión.
• El flujo de la fase móvil se define como el volumen de fase móvil que pasa por la sección de columna en la unidad de tiempo, y está relacionado con la velocidad lineal a través del área y de la porosidad.
El flujómetro suele estar a temperatura ambiente, por lo que el flujo que obtenemos de él habrá que corregirlo para la temperatura de la columna, lo cual es sencillo al permanecer el resto de parámetros constantes, por lo que para gases ideales se puede aplicar una simple regla de tres.

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
66
• El volumen de retención es el volumen de gas portador necesario para eluir un
compuesto de tiempo de retención tR.
Existen varios tipos de volúmenes de retención:
a. Volumen total de retención (VR): tiene en cuenta la totalidad del gas portador.
b. Volumen de retención ajustado (V’R): tiene en cuenta el volumen básico relativo al tiempo básico.
c. Volumen de retención neto (VN): se obtiene de la corrección del anterior con el factor de la compresibilidad.
• El volumen de retención específico a la temperatura de la columna es el volumen de retención neto por gramo de fase estacionaria.
Pasar al volumen de retención específico a 0ºC resulta sencillo, y como antes, en caso de ser todos los demás parámetros constantes, se puede aplicar proporcionalidad.
Teniendo en cuenta que el volumen de interpartícula, V0, coincide con el volumen
básico corregido, se puede relacionar el coeficiente de distribución con el factor de
retención (k), vinculándose un parámetro puramente cromatográfico con una variable
termodinámica y pudiendo usarse la medida de VN como método para conocer los
coeficientes de reparto de una forma sencilla.
3.2.1. Naturaleza y propiedades de las fases móviles más usadas.
Se suele emplear un gas inerte cuya única función sea arrastrar las moléculas de
soluto a través de la columna, en general se suele emplear nitrógeno, helio o hidrógeno
(este último necesita de medidas especiales en el laboratorio) como gas portador. La
selección de uno u otro suele hacerse atendiendo a criterios económicos y de rapidez.
• Viscosidad (μ).
Influye en la circulación de la fase móvil a través de la ley de Darcy, necesitando un gradiente de presión mayor para viscosidades mayores.
En las condiciones de trabajo que se dan en la cromatografía gaseosa, el valor de la viscosidad no varía con la presión, aunque sí con la temperatura, pudiéndose estimar ésta última mediante una aproximación.
Recientemente se han tabulado valores de μ entre 0 y 400ºC para hidrógeno, helio y nitrógeno (Hinshaw y Ettre. 1997). Lo que se puede observar es que al
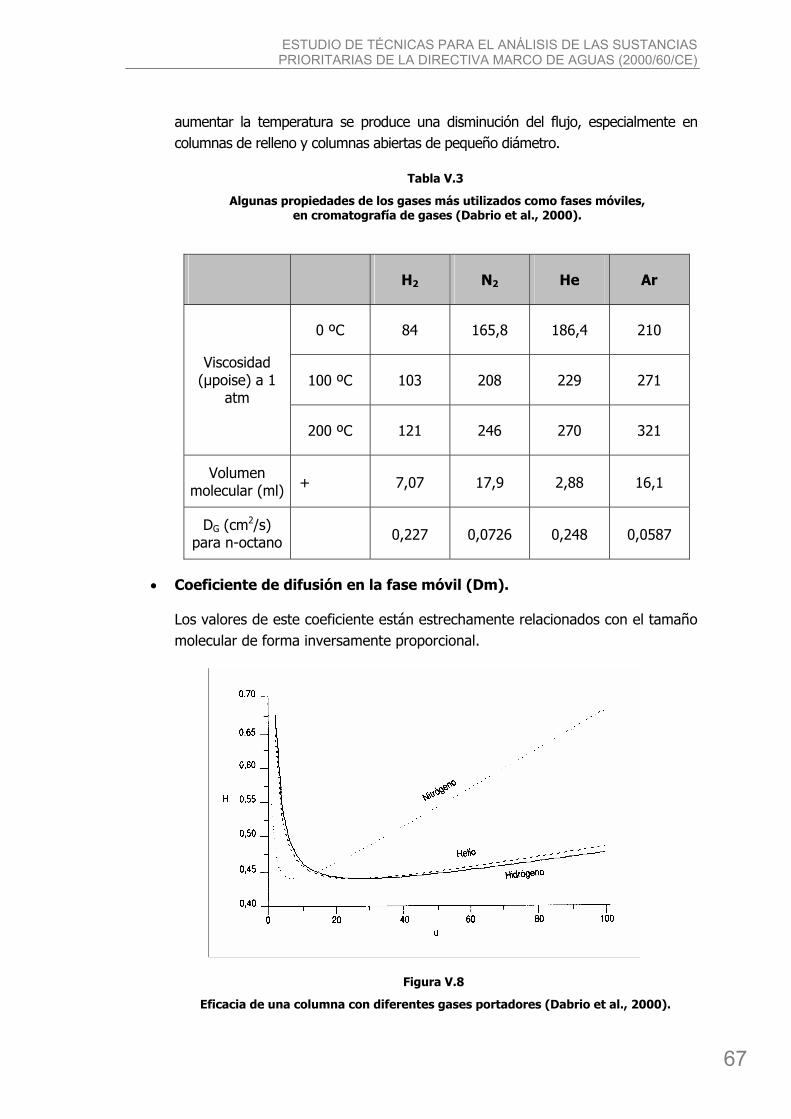
ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
67
aumentar la temperatura se produce una disminución del flujo, especialmente en columnas de relleno y columnas abiertas de pequeño diámetro.
Tabla V.3
Algunas propiedades de los gases más utilizados como fases móviles, en cromatografía de gases (Dabrio et al., 2000).
H2 N2 He Ar
0 ºC 84 165,8 186,4 210
100 ºC 103 208 229 271 Viscosidad
(μpoise) a 1 atm
200 ºC 121 246 270 321
Volumen molecular (ml) + 7,07 17,9 2,88 16,1
DG (cm2/s) para n-octano 0,227 0,0726 0,248 0,0587
• Coeficiente de difusión en la fase móvil (Dm).
Los valores de este coeficiente están estrechamente relacionados con el tamaño molecular de forma inversamente proporcional.
Figura V.8
Eficacia de una columna con diferentes gases portadores (Dabrio et al., 2000).

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
68
El valor de los coeficientes de difusión es un parámetro de gran importancia ya que influye directamente sobre dos términos de la ecuación general de la eficacia.
La naturaleza del gas portador no influye en el valor de Hmin en Cromatografía Gaseosa pudiéndose calcular mediante el uso de expresiones, ocurriendo lo mismo con la velocidad óptima.
La velocidad óptima es mayor para hidrógeno y helio que para el nitrógeno, al ser la difusividad de este último sensiblemente menor. Esto significa que para las mismas condiciones, la eficacia de columnas que usen helio o hidrogeno como gases portadores es mayor que las de nitrógeno a partir de una cierta velocidad, por lo que se pueden realizar los análisis más rápidamente sin pérdidas significativas en la eficacia.
Otra ventaja de la alta difusividad del hidrógeno y del helio es que repercute favorablemente en H, pero también contribuye al mayor ensanchamiento de la zona en los posibles volúmenes extracolumna, sobre todo en el inyector, donde se requiere especial atención a la relación volumen/velocidad de gas portador.
• Pureza.
Si bien la presencia de impurezas inertes no afecta a la separación, si puede resultar perjudicial para la fase estacionaria, principalmente la presencia de agua y oxígeno a altas temperaturas. Otras impurezas pueden dar lugar a la aparición de picos extraños en el cromatograma. Por todo esto se recomienda usar gases portadores con una pureza mínima del 99.998% y el uso de sistemas adicionales de purificación a la entrada del equipo, especialmente si las líneas de conducción del gas son largas.
3.3. Fases estacionarias.
En la cromatografía gaseosa nos encontramos dos tipos de fases estacionarias, las
sólidas, que emplean un mecanismo de adsorción en su interacción con el soluto, y las
líquidas, que usan un mecanismo de reparto.
La selectividad que presenten, se debe sobre todo a la naturaleza de la fase
estacionaria, aunque la temperatura también influye.

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
69
3.3.1. Fases estacionarias líquidas.
• Mecanismos de separación en fases líquidas.
El mecanismo de reparto es el más importante cuando la fase estacionaria es líquida, y consiste en una distribución del soluto entre la fase vapor y la líquida en la cual se disuelven. Si el soluto formara una disolución real con la fase estacionaria, se podría aplicar la ley de Henry.
Teniendo en cuenta otras ecuaciones como la de los gases perfectos y de la densidad, se puede llegar a la conclusión de que las sustancias se eluyen en orden inverso a sus presiones de vapor, o lo que es lo mismo, en orden creciente de puntos de ebullición. Esto es así siempre y cuando los compuestos sean químicamente similares como las series homólogas, ya que si los coeficientes de distribución son muy distintos, la elución no seguirá este orden.
A pesar de lo complicado de prever el comportamiento cromatográfico a partir de valores termodinámicos, se pueden dan algunas normas generales:
a. Los solutos apolares en fases apolares presentan un comportamiento relativamente próximo al ideal, y se eluyen en orden similar al de sus puntos de ebullición.
b. Cuando un soluto con coeficiente de actividad elevado, que tiene sus moléculas fuertemente asociadas, se disuelve en una fase apolar, su interacción con la fase estacionaria será muy débil y su retención menos que la de un soluto apolar con similar presión de vapor.
c. Cuando se eluye un soluto apolar sobre una fase polar, cuyas moléculas están fuertemente asociadas entre sí y rechazan al soluto, la interacción mutua es también débil.
d. Cuando fase y soluto son polares y pueden interaccionar entre sí positivamente, la retención es elevada.
(Dabrio et al., 2000)
En general la elección de la fase estacionaría dependerá de la diferencia entre los puntos de ebullición de las sustancias a separar, de manera que si son muy distintos, una fase estacionaria cualquiera será capaz de llevar a cabo la elución de forma correcta, pero si son similares, necesitaremos usar una fase estacionaria que sea selectiva, interaccionando de forma distinta con las sustancias, lo que da lugar a una diferencia entre los coeficientes de actividad de las mismas. A veces la similitud molecular entre los solutos es tan grande, que esto no es suficiente y necesitamos del uso de propiedades especiales para obtener una elución satisfactoria. Los mecanismos a los que podemos recurrir son:

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
70
a. Interacciones quirales: se usa para la separación de parejas de
isómeros ópticos (enantiómeros) que siempre coeluyen en condiciones normales. Para lograrlo usamos fases que contengan grupos ópticamente activos (quirales, que interaccionan selectivamente con uno de los miembros de la pareja, reteniéndolo en mayor medida que al otro. No obstante, la diferencia de retención no deja de ser pequeña, por lo que requiere del uso de columnas de gran eficacia.
b. Interacciones con mesofases: los cristales líquidos son moléculas alargadas o planas que permanecen en estado líquido dentro de un cierto intervalo de temperatura, pero a diferencia de otras sustancias lo hacen con un determinado orden, en una orientación, de modo que la geometría molecular de los solutos puede usarse como factor discriminatorio, no pudiendo todos alojarse entre las moléculas de la fase estacionaria (mesofases).
c. Formación de complejos: se aprovechan las posibilidades de formación de complejos entre organometálicos que posean iones, como Cu2+, Co2+, o Ni2+, y solutos con grupos donadores de electrones, como olefinas, compuestos aromáticos,…
d. Otros mecanismos mixtos: además de los citados, nos podemos encontrar adsorciones en las interfases gas-líquido, y líquido-sólido. Estas adsorciones suelen tener problemas añadidos como picos sustraídos total o parcialmente, ensanchamientos, colas o pérdidas de simetría.
• Propiedades de las fases líquidas.
La variedad de sustancias que podríamos usar como fase estacionaria es muy grande, pero normalmente se usan compuestos poliméricos. Los criterios para la elección de la fase estacionaria son varios y se encuentran analizados a continuación.
a. Intervalo de temperatura de uso: el límite inferior viene marcado por el punto de fusión o reblandecimiento en su defecto, mientras que el límite superior lo marca o la temperatura a la que la presión de vapor es apreciable (≥0.1 mmHg), o por el aumento de la señal del detector originada por la descomposición térmica de la fase estacionaria. Una pureza insuficiente del gas o la presencia de puntos activos en el soporte o en la pared interna de la columna pueden provocar una disminución de dicho intervalo.
b. Polaridad: está relacionado con el número y tipo de grupos funcionales presentes en la fase estacionaria, pero es difícil de expresar de forma numérica, debiéndose medir en principio a partir de parámetros moleculares. El problema es que muchas de las fases estacionarias usadas no tienen una composición unívoca, si no que son una mezcla compleja de sustancias
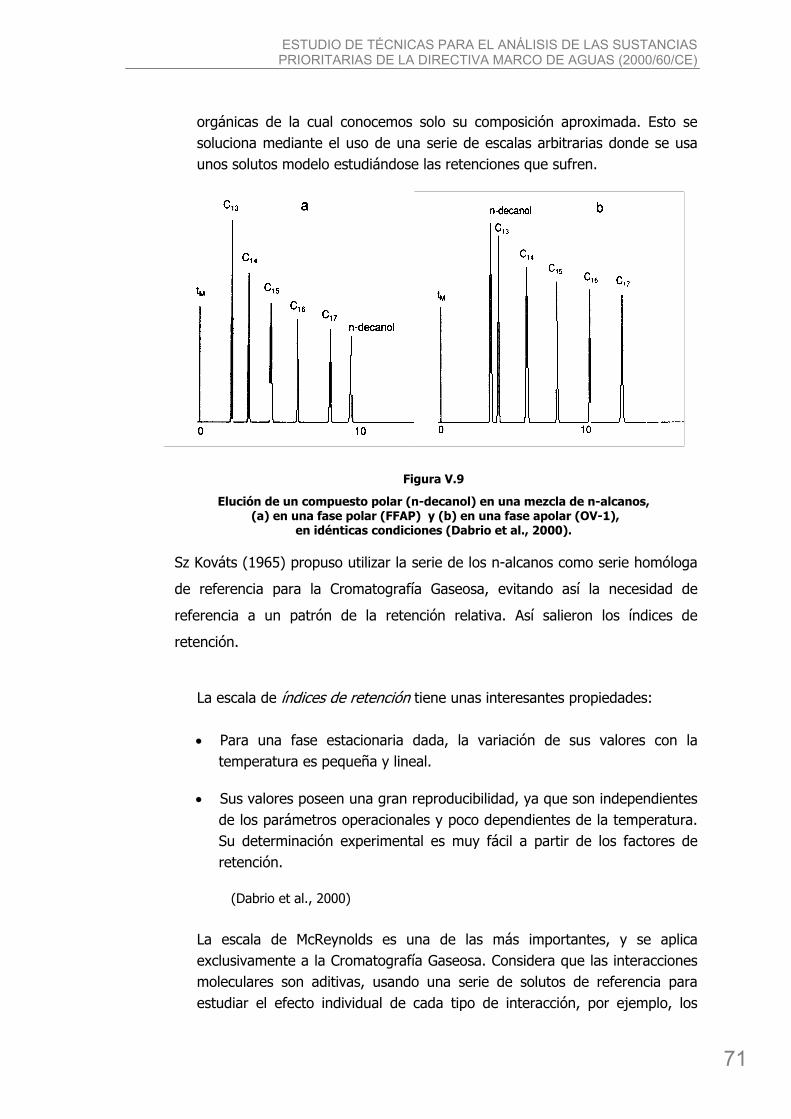
ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
71
orgánicas de la cual conocemos solo su composición aproximada. Esto se soluciona mediante el uso de una serie de escalas arbitrarias donde se usa unos solutos modelo estudiándose las retenciones que sufren.
Figura V.9
Elución de un compuesto polar (n-decanol) en una mezcla de n-alcanos, (a) en una fase polar (FFAP) y (b) en una fase apolar (OV-1),
en idénticas condiciones (Dabrio et al., 2000).
Sz Kováts (1965) propuso utilizar la serie de los n-alcanos como serie homóloga
de referencia para la Cromatografía Gaseosa, evitando así la necesidad de
referencia a un patrón de la retención relativa. Así salieron los índices de
retención.
La escala de índices de retención tiene unas interesantes propiedades:
• Para una fase estacionaria dada, la variación de sus valores con la temperatura es pequeña y lineal.
• Sus valores poseen una gran reproducibilidad, ya que son independientes de los parámetros operacionales y poco dependientes de la temperatura. Su determinación experimental es muy fácil a partir de los factores de retención.
(Dabrio et al., 2000)
La escala de McReynolds es una de las más importantes, y se aplica exclusivamente a la Cromatografía Gaseosa. Considera que las interacciones moleculares son aditivas, usando una serie de solutos de referencia para estudiar el efecto individual de cada tipo de interacción, por ejemplo, los

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
72
alcanos representan las fuerzas de dispersión y el benceno las de inducción. Las contribuciones individuales se representan a través de los índices de retención de cada soluto en la fase de estudio (I), y en escualano (Iesc), que es el soluto que se eligió como referencia (1,6,10,15,19,23-hexametil-docosano).
Tabla V.4
Propiedades de las fases estacionarias utilizadas en cromatografía de gases (Dabrio et al. 2000.
La diferencia entre los índices de retención está relacionada con una serie de parámetros propios de cada compuesto, llamados sondas de McReynolds, y con otra serie de parámetros característicos de la fase que se denominan constantes de McReynolds.
Como el límite superior de uso de las columnas de escualano es muy bajo (120 °C), y es imposible determinar los índices de retención en ciertas fases que no son líquidas a esa temperatura, se ha propuesto utilizar en su lugar un hidrocarburo ramificado, el 24,24-dietil-19,29-dioctadecil-heptatetra-contano, que se conoce como C87, y que se puede calentar hasta 260 °C.
c. Viscosidad: el coeficiente de difusión en un líquido se puede estimar mediante la ecuación de Wilke y Chang, donde se puede observar que el

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
73
coeficiente de difusión de un líquido es inversamente proporcional a la viscosidad del disolvente, por lo que interesaría utilizar líquidos de baja viscosidad como fases estacionarias.
Esta es la razón por la cual se han usado polímeros lineales, ya que poseen una baja viscosidad tanto por ser lineales como por su menor peso molecular. Su inconveniente es que las columnas capilares abiertas se impregnan mal con líquidos de baja viscosidad al formarse gotas en vez de la buscada película, razón por lo que se usan polímeros de peso molecular elevado para estos recubrimientos.
La eficacia que se obtiene con el uso de estos polímeros es, al contrario de lo que se podría pensar, semejante a la obtenida con fases de baja viscosidad, lo cual se puede explicar en parte por el empleo de altas temperaturas que disminuyen la viscosidad, y por la movilidad molecular de estos polímeros, que permiten a las pequeñas moléculas de soluto deslizarse con facilidad entre las largas cadenas de la fase estacionaria.
d. Tensión superficial: buscamos una fase estacionaria capaz de formar una película estable y uniforme sobre un sólido, para lo que su tensión superficial debe ser inferior a la energía superficial del sólido. Este problema suele aparecer sobre todo en las columnas capilares con superficies lisas de baja energía (teflón, vidrio), no teniendo por qué haber problemas en materiales activos (metales) o inertes pero con superficie específica elevada.
e. Pureza: es necesario una elevada pureza para evitar separaciones no reproducibles. Además ciertas impurezas pueden acortar la vida útil de la columna.
• Fases líquidas más usadas.
En los primeros tiempos se utilizaron con éxito toda clase de productos industriales: lubricantes como Apiezon, aceites y gomas de silicona, detergentes como Triton, Alkaterge, etc., anticongelantes como etilenglicoles, y otros muchos.
Actualmente, hay dos tipos fundamentales de fases estacionarias: algunos de los productos mencionados, que se venden parcialmente purificados y sometidos a controles de calidad que garanticen un comportamiento cromatográfico reproducible, y moléculas de síntesis que suelen ir dirigidas a la resolución de problemas específicos y que resultan mucho más fiables.
a. Hidrocarburos: algunas de las fases más antiguas eran de este tipo, por ejemplo, los Apiezones son parafinas que suelen dar buenos resultados en la

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
74
separación de isómeros cis y trans. Actualmente algunos hidrocarburos se utilizan como fases de referencia (como el escualano en la escala de McReynolds).
b. Polisiloxanos: los polisiloxanos o siliconas, debido a sus excelentes propiedades, son el grupo de moléculas más utilizadas.
Figura V.10 Estructura general de los Polisiloxanos.
La longitud de la cadena se relaciona con la viscosidad del producto, que puede variar entre 103 y 106 cpoises (aceites y gomas de silicona, respectivamente). La funcionalidad química de los radicales R determina la polaridad y la estabilidad térmica. Las más usadas tienen sustituyentes metilo, vinilo, trifluoropropilo, fenilo y cianoalquilo (en orden aproximado de menor a mayor polaridad).
Las metil siliconas (con el 100% de R = CH3) son las más estables químicamente; su polaridad es muy baja en la escala de McReynolds, su intervalo de operación es muy amplio, y se emplean como fase estacionaria universal para abordar problemas desconocidos o cualquier tipo de separación que no requiera una selectividad muy elevada. A veces se preparan con un pequeño porcentaje (<5%) de grupos vinilo, lo que eleva su polaridad ligeramente y facilita su reticulación.
La sustitución de parte de los grupos metilo por radicales trifluoropropilo origina las trifluoropropil siliconas, cuya polaridad es relativamente baja, pero poseen una selectividad especial que las hace muy útiles en determinadas aplicaciones (acetatos de alditol, plaguicidas clorados,…).
Los grupos fenilo, que forman parte de las fenil siliconas en porcentajes que alcanzan el 50% como máximo, confieren al polímero una polaridad de tipo medio y buena estabilidad térmica.
Los grupos cianoetilo o cianopropilo originan las fases más polares de que se dispone comercialmente; de hecho, el producto con índice 100 en la escala CP-Index es una cianopropil silicona. La estabilidad térmica de estas fases es

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
75
menor que la de otras siliconas, y además son muy sensibles al oxígeno y a determinados reactivos, por lo que requieren el uso de gas portador de gran pureza y mantener el flujo de gas inerte durante el apagado y encendido del horno, e incluso en el almacenamiento.
c. Polioxiranos: los polietilenglicoles y polipropilenglicoles siguen en popularidad a las siliconas, siendo en este caso cadenas lineales con la siguiente fórmula general:
Se utilizan polímeros de peso molecular muy variable, comprendidos entre 3 x 102 y 4 x 106, pero el más empleado es el conocido como Carbowax 20M, cuyo peso molecular medio está comprendido entre 16.000 y 20.000. Exhiben una polaridad media y su estabilidad térmica es discreta, pero se consideran insustituibles en el análisis de compuestos volátiles, aceites esenciales, alcoholes, etc. Cuando los grupos OH terminales se esterifican con ácido nitrotereftálico se obtienen fases de polaridad y características muy similares, pero capaces de eluir correctamente ácidos libres y de sustraer aldehídos.
d. Fases quirales: en 1966 (Gil-Av et al., 1966) se utilizaron por primera vez derivados de aminoácidos puros que, actuando como fase estacionaria, podían separar enantiómeros de aminoácidos. Posteriormente se han usado dipéptidos y diamidas. Como su estabilidad térmica era muy reducida, se sustituyeron por cianosiliconas poliméricas, donde el grupo ciano se ha hecho reaccionar con amidas.
Otro tipo de fases quirales son los complejos metálicos (Schurig, 1977), que se han utilizado para separar olefinas, alcoholes, aminas y otros compuestos.
En los últimos años han aparecido numerosos derivados de ciclodextrinas (König, 1992), que han permitido separar gran número de pares enantioméricos de estructura química muy diversa. Se puede obtener un amplio intervalo de polaridades según la naturaleza de los sustituyentes que reemplazan los H activos de la ciclodextrina, que pueden ser grupos metilo, pentilo, acetilo, butirilo y algún otro. Según la naturaleza de los sustituyentes varía su estabilidad química y térmica, y proporcionan una gran versatilidad para la separación de compuestos. Muchas de ellas se utilizan disueltas en polisiloxanos, a los que algunas se han unido químicamente. La selectividad de estas fases es también muy distinta para parejas de enantiómeros

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
76
químicamente similares, y no es fácil de predecir. Un ejemplo se muestra en la figura 3.8.
Figura V.11
Separación de enantiómeros de alcoholes y acetatos arílicos bicíclicos utilizando una columna de ciclodextrina sustituida.
e. Fases mixtas: están constituidas por la mezcla de dos o más fases (Molera et al., 1969), y se utilizan por dos motivos: para aprovechar las selectividades de ambas cuando se quieren separar dos parejas o más de compuestos que no se resuelven en una única fase, o para mejorar propiedades físicas, tales como la viscosidad o la tensión superficial, de otra fase muy selectiva. Con este último objetivo, las ciclodextrinas suelen mezclarse con siliconas para su empleo como fases estacionarias, ya que por sí solas no impregnan bien las columnas de pared recubierta.
f. Fases inmovilizadas: inmovilizadas, reticuladas, entrecruzadas, ligadas... todos estos adjetivos se aplican a una fase que no puede ser extraída de la columna por lavado con disolventes. No siempre está claro que esto suceda porque la fase estacionaria se haya unido químicamente a la pared interna de la columna o al soporte, o porque las moléculas de la fase se hayan unido entre sí constituyendo un polímero tridimensional insoluble,; lo que es cierto es que se puede disponer de columnas con fases estacionarias inmovilizadas cuyas propiedades de retención sean prácticamente idénticas a las de las fases solubles originales, y con varias ventajas adicionales: mayor estabilidad frente al tiempo y frente a la inyección de grandes volúmenes de muestra, además de la posibilidad de lavar las impurezas acumuladas con el uso.

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
77
3.3.2. Fases estacionarias sólidas.
Son las que primero se usaron en cromatografía de gases y también las más
sencillas de utilizar en la preparación de una columna (Kiselev y Yashin, 1969).
Actualmente se limita su uso a la resolución de un pequeño porcentaje de problemas,
tales como el análisis de gases permanentes, para los que resultan insustituibles.
El mecanismo básico de separación cuando se emplean fases sólidas es la
adsorción, en condiciones que se pueden considerar de sorción puramente física
(fisisorción): fuerzas de van der Waals con energías de interacción pequeñas, que dan
lugar a procesos de adsorción/desorción muy rápidos. En estas condiciones se forman
monocapas de moléculas que no llegan a cubrir la superficie del adsorbente. Para
conseguir buenas eficacias es muy frecuente utilizar columnas abiertas de pared
recubierta con soporte.
• Ventajas e inconvenientes de las fases sólidas
a. La adsorción es un fenómeno que permite obtener selectividades muy altas capaces de poner de manifiesto diferencias estructurales muy pequeñas. Los isótopos del hidrógeno se separaron ya en 1962 (Mohnke y Saffert, 1962).
b. Los procesos de sorción/desorción suelen ser más rápidos que los de disolución/evaporación y, en consecuencia, se pueden conseguir con facilidad eficacias muy elevadas.
c. Estos procesos tienen lugar en un intervalo de temperaturas muy amplio, sin que se produzcan pérdidas de fase estacionaria por evaporación (sangrado).
d. La falta de linealidad de las isotermas, motivada por la perdida de la condición de dilución infinita antes de formar incluso la primera monocapa, limita la capacidad de carga de estas columnas. Más allá de la zona lineal, habrá que usar modelos para poder describir el proceso. En el caso de adsorción de gases sobre sólidos, la ecuación propuesta por Langmuir resulta adecuada.

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
78
Figura V.12
Isoterma de Langmuir.
e. Los factores de retención son en general altos, lo que es una ventaja para el
análisis de compuestos de bajo peso molecular y un inconveniente para los
de alto peso molecular.
• Tipos de fases sólidas
Se utilizan fases sólidas en forma de partículas pequeñas de superficie específica elevada, y, como en cualquier otro relleno cromatográfico, conviene que sean lo más uniformes posible en cuanto a tamaño, porosidad, actividad superficial y distribución en la columna. Además, deben ser térmicamente estables y químicamente inertes para que no reaccionen con los solutos.
Aunque tradicionalmente se han venido usando productos de origen natural con escaso tratamiento, similares a los soportes que se describen más adelante, actualmente se prefiere utilizar productos de síntesis, que permiten un mejor control de su composición y propiedades:
- Las fases inorgánicas permiten trabajar a temperaturas muy elevadas; entre ellas podemos citar variedades comerciales de sílice (Spherosil, Durasil), carbón grafitado (Carbopack, Graphpack), tamiz de carbón (Carbosieve, Spherocarb), zeolitas y alúmina.
- Las fases orgánicas suelen ser de tipo polimérico; son muy usados los polímeros de estireno/divinilbenceno (Chromosorb serie Century, Porapaks), los de óxido de fenileno (Tenax) y los polifluorocarburos (Kel F).

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
79
4. Sistemas de detección.
Aunque la lista de detectores existentes para cromatografía de gases es muy amplia
(Scott, 1996), los más usados se pueden distribuir, según su fundamento físico, en cuatro
grupos: conductividad térmica, ionización, electroquímicos y espectroscópicos. Cada uno
de estos grupos incluye diferentes dispositivos que pueden funcionar en distintas
modalidades.
Figura V.13 Esquema de detectores de conductividad térmica,
(a) Sistema con dos filamentos, (b) Dispositivo con un solo filamento.
4.1. Detectores de conductividad térmica
Han sido muy utilizados, aunque actualmente su uso ha disminuido. Son detectores
universales de sensibilidad media y que responden a la concentración.
El efluyente de la columna se hace pasar por una cámara que contiene un filamento
caliente; cuando se establece el régimen estacionario, la velocidad de disipación de calor
entre el filamento y la pared de la cámara se hace constante, e igualmente la temperatura
del filamento. Al aparecer un soluto disuelto en el gas portador, varía la conductividad
térmica de la mezcla y, por tanto, la velocidad de disipación de calor en la cámara, lo que
conduce a una modificación momentánea de la temperatura del filamento que, a su vez,
hace variar su resistencia eléctrica. Esta variación de resistencia se mide fácilmente con
un puente de Wheatstone.
El diseño clásico consta de dos dispositivos como los de la figura; por el primero
pasa el efluyente de la columna y por el segundo, de referencia, pasa gas portador puro;
cuando las composiciones son distintas, el puente de Wheatstone se descompensa. El

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
80
diseño moderno lleva un único filamento y un sistema de modulación de la señal que
produce mayor sensibilidad y una compensación más fiable; en este caso el efluyente se
hace pasar alternativamente por las células con o sin filamento.
Los gases portadores más recomendables para este tipo de detector son H2 y He,
que presentan conductividades térmicas del orden de 200-300 veces mayores que la
mayoría de las sustancias orgánicas de peso molecular superior a 50. Así pues, la
respuesta puede ser cuantitativamente diferente de unas sustancias a otras, y su valor
depende también de la geometría de la cámara, de la temperatura seleccionada y del
flujo.
Este detector es muy adecuado para el análisis de sustancias que no den respuesta
satisfactoria en otros detectores, como es, por ejemplo, el caso de los gases
permanentes. Como es un detector no destructivo, es de gran utilidad en operaciones a
escala preparativa y para intercalarlo en serie con otro detector más selectivo.
4.2. Detectores de ionización
Todos ellos tienen en común el hecho de ionizar los solutos, suministrando al
efluyente de la columna una energía que puede ser térmica, química, electromagnética o
radiactiva. La respuesta de este tipo de detectores depende de dos factores: el
rendimiento de producción de iones y la eficacia de captación de los mismos.
Además de la fuente de energía, todos estos detectores tienen en común el empleo
de dos electrodos: el polarizador, que suministra una diferencia de potencial adecuada, y
el colector, que puede ser indistintamente el ánodo o el cátodo, encargado de recoger los
iones producidos.
Cuando el detector es atravesado solamente por gas portador, el campo eléctrico
entre ambos electrodos origina una corriente de fondo que aumenta o disminuye, según
los casos, cuando aparece un soluto. Todos estos detectores requieren para su correcto
funcionamiento mantener una geometría constante mediante una alineación cuidadosa de
los electrodos, y un aislamiento eléctrico eficaz, ya que los potenciales aplicados suelen
ser relativamente altos.

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
81
4.2.1. Detector de ionización de llama (FID)
Es el más utilizado por ser casi universal, tener un intervalo lineal amplio, una
sensibilidad elevada y un volumen muerto casi inexistente, así como un mantenimiento
sencillo. La figura muestra su esquema. La energía de ionización la suministra la llama
producida por una mezcla del efluyente de la columna con un gas combustible,
normalmente hidrógeno; a su alrededor fluye una corriente de aire, de tal manera que los
bordes de la llama son ricos en 02 y el centro en H2.
Figura V.14
Esquema de un detector de ionización de llama.
Cuando la llama se somete a una diferencia de potencial de ~100-300 V, se produce
una pequeña corriente de fondo debida a la formación de iones H+, O2- y OH-. Al aparecer
en la llama una sustancia orgánica, se produce un incremento notable de la corriente que
se atribuye a la ionización química de la sustancia en la parte más fría de la llama. Las
reacciones que se producen en la llama son muy complejas y del tipo
CH + 0 -> CHO + e-
La combustión prosigue hasta quemar totalmente los solutos, dando CO2 y H2O. La
mayor parte de las sustancias orgánicas tienen una respuesta muy similar, aunque los
compuestos con pocos enlaces C–H y ricos en O y heteroátomos dan respuestas
notablemente bajas; el ácido fórmico, por ejemplo, tiene una respuesta prácticamente
nula.

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
82
El FID es un detector de flujo másico muy sencillo de calibrar. La temperatura de la
llama se regula mediante los flujos de gas que forman parte de ella. Cuando se trabaja
con columnas capilares, que tienen un flujo de gas portador muy bajo, es frecuente
añadir una cantidad extra de gas inerte llamada supletorio a la salida de la columna para
enfriar ligeramente la llama y aumentar la respuesta. La mayor parte de los equipos
comerciales detectan 10-12 g de carbono/s, con un intervalo lineal de 107.
4.2.2. Detector termoiónico (TID).
También es conocido como NPD (detector de nitrógeno y fósforo). Son una serie de
dispositivos derivados del FID, que pueden funcionar con y sin llama. La fuente de
ionización es una superficie cerámica que contiene algún metal alcalino (como Rb o Cs)
que puede calentarse hasta 800 °C.
Esta superficie, en caliente, puede ceder fácilmente electrones a sustancias
electronegativas que entren en contacto con ella; los iones formados son recogidos por el
electrodo colector. Los distintos modos de trabajo se obtienen variando la composición
química de la superficie emisora, su temperatura y el tipo de gas portador.
• Con un gas inerte, tal como He o N2, y una superficie rica en Cs, se consigue una sensibilidad muy alta para grupos electronegativos como NO2, SH, halógenos, etc., que interaccionan directamente con la superficie, con factores de respuesta que pueden llegar a ser hasta 108 veces mayores que los de los hidrocarburos.
• Introduciendo gases reactivos se originan reacciones químicas en fase gaseosa que hacen variar la especificidad. Por ejemplo, introduciendo aire y un flujo de H2 reducido se forma una cuasi-llama junto a la superficie caliente, con radicales que descomponen químicamente a las moléculas orgánicas; este proceso forma selectivamente iones de productos que contengan nitrógeno y fósforo, con respuestas de 102 y 104 veces superiores a la del C respectivamente, consiguiéndose límites de detección del orden de 1-10 pg.
• Otra variante incluye un FID: los solutos se queman en la llama y los productos de combustión se ionizan, lo que hace que la respuesta de los compuestos con heteroátomos resulte independiente de la estructura original.

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
83
4.2.3. Detector de captura de electrones (DCE).
Es un detector muy popular en análisis medioambiental dada su especificidad y
sensibilidad hacia compuestos con grupos electronegativos. Un esquema de este detector
puede verse en la figura V.15.
Figura V.15
Detector de captura de electrones.
La ionización se consigue mediante una fuente radiactiva (de 3H o 63Ni) de baja
intensidad (unos 10 pCi) que emite radiación beta, constituida por electrones. Si se
establece una diferencia de potencial baja entre los electrodos (<20 V), los electrones
serán lentos, condición que se acentúa por sus choques elásticos con las moléculas del
gas portador. El N2 y la mezcla Ar/CH4 95:5 son gases adecuados para este detector,
mientras que el He y el Ar puros no lo son. Cuando un soluto con afinidad electrónica
llega a la cámara, encuentra un plasma de electrones lentos entre los dos electrodos; al
capturar algunos de estos electrones se forman iones negativos más pesados, que son
más lentos que los electrones, y la corriente de fondo disminuye.
La respuesta depende de factores propios del detector (Lovelock y Lipsky, 1960),
como la diferencia de potencial entre los electrodos (los equipos modernos pueden
trabajar a tensión continua o pulsada), el tipo de fuente y la temperatura; pero también
se ve afectada por factores como el flujo y la naturaleza del gas portador o el sangrado
de la columna.
La respuesta varía mucho de unos compuestos a otros (Pellizzari, 1974), y eso
favorece la detección de organoclorados en presencia de otros compuestos que
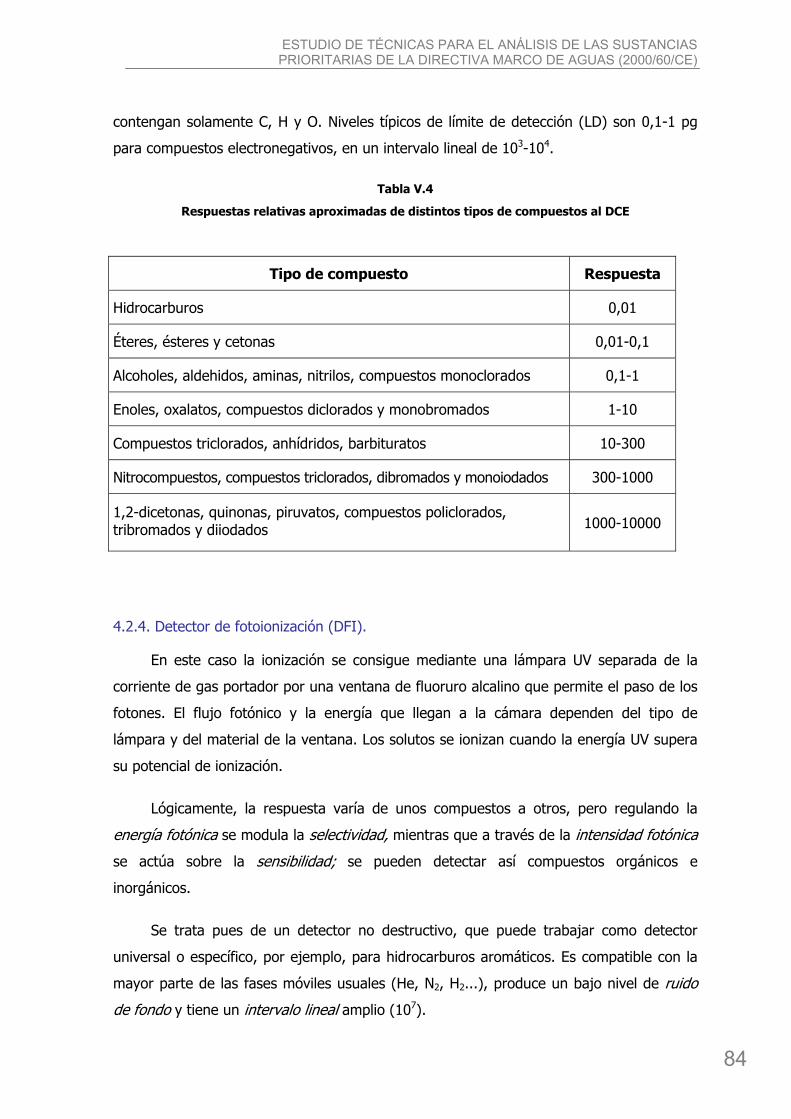
ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
84
contengan solamente C, H y O. Niveles típicos de límite de detección (LD) son 0,1-1 pg
para compuestos electronegativos, en un intervalo lineal de 103-104.
Tabla V.4
Respuestas relativas aproximadas de distintos tipos de compuestos al DCE
4.2.4. Detector de fotoionización (DFI).
En este caso la ionización se consigue mediante una lámpara UV separada de la
corriente de gas portador por una ventana de fluoruro alcalino que permite el paso de los
fotones. El flujo fotónico y la energía que llegan a la cámara dependen del tipo de
lámpara y del material de la ventana. Los solutos se ionizan cuando la energía UV supera
su potencial de ionización.
Lógicamente, la respuesta varía de unos compuestos a otros, pero regulando la
energía fotónica se modula la selectividad, mientras que a través de la intensidad fotónica
se actúa sobre la sensibilidad; se pueden detectar así compuestos orgánicos e
inorgánicos.
Se trata pues de un detector no destructivo, que puede trabajar como detector
universal o específico, por ejemplo, para hidrocarburos aromáticos. Es compatible con la
mayor parte de las fases móviles usuales (He, N2, H2...), produce un bajo nivel de ruido
de fondo y tiene un intervalo lineal amplio (107).
Tipo de compuesto Respuesta
Hidrocarburos 0,01
Éteres, ésteres y cetonas 0,01-0,1
Alcoholes, aldehidos, aminas, nitrilos, compuestos monoclorados 0,1-1
Enoles, oxalatos, compuestos diclorados y monobromados 1-10
Compuestos triclorados, anhídridos, barbituratos 10-300
Nitrocompuestos, compuestos triclorados, dibromados y monoiodados 300-1000
1,2-dicetonas, quinonas, piruvatos, compuestos policlorados, tribromados y diiodados 1000-10000

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
85
En cambio, su respuesta varía de unos compuestos a otros ya que los ioniza de
distinta forma. Otro inconveniente es que tiene cierto volumen muerto al producirse la
ionización en una cámara. El empleo de láseres puede hacer bajar su volumen a niveles
de subnanolitros y la sensibilidad al nivel del femtogramo. Se emplea en aplicaciones
ambientales (hidrocarburos aromáticos, tetraetilplomo, cloruro de vinilo, etc.).
4.3. Detectores electroquímicos.
La mayor parte de estos detectores se han diseñado para análisis ambientales de
sustancias con heteroátomos (halógenos, S, N).
4.3.1. Detector de conductividad electrolítica (DCEL).
El efluyente de la columna pasa por un reactor de Ni que puede calentarse hasta
1.000 °C, donde se mezcla con H2 (que forma haluros con los halógenos y NH3 con el
nitrógeno) o con 02 (que forma óxidos con el azufre). Los productos de reacción se llevan
a una célula electrolítica por la que circula una solución ácida o básica. La aparición de
compuestos ionizables hace variar la conductividad eléctrica, que es la propiedad que se
mide en continuo. Si se desea que sea realmente específico, el detector debe llevar
también un sistema de sustracción de compuestos para eliminar los posibles productos de
interferencia antes de que lleguen a la célula.
4.4. Espectógrafo de Masas.
Este método pertenece al grupo de las técnicas espectroscópicas que por la
importancia de los datos que suministran pueden considerarse en realidad métodos
analíticos por sí solos en vez de simples detectores. La espectrometría de masas es la más
importante en su acoplamiento con los métodos cromatográficos, aunque también
podemos nombrar a las espectrofotometrías IR y UV-visible, pero debido a la escasa
presencia de estas otras técnicas espectrométricas, vamos a comentar directamente la
espectrografía de masas.
La Espectrometría de Masas es una técnica analítica en la cual los átomos o
moléculas de una muestra son ionizados, separados de acuerdo con su relación masa /
carga (m/z) y detectados. Nace en 1886 cuando Golstein descubre los iones positivos en
un tubo de descarga eléctrica a baja presión. Wien (N. Física 1911) en 1898 muestra que

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
86
los rayos de esos iones podían ser desviados por campos eléctricos y magnéticos. Entre
1912 y 1919, Aston (N. Química 1922) y Thomson (N. Física 1906) realizaron
modificaciones a la técnica, anticipando este último el enorme potencial de esta técnica
dentro de la química analítica. En 1924, Aston fue capaz de determinar las abundancias
isotópicas de más de 50 elementos. Pero no fue sino hasta los años cuarenta que se
popularizó su uso en Química. Hoy en día existen diferentes tipos de espectrómetros de
masas comercialmente disponibles. Sus diferencias se encuentran en la forma de llevar a
cabo cada uno de los procesos necesarios para la detección de las sustancias.
En forma esquemática podemos decir que los espectrómetros de masas consisten
de cuatro secciones fundamentales: 1) Sistema de introducción de muestra; 2) Cámara de
ionización; 3) Analizador y 4) Detector. Las modificaciones instrumentales han sido de la
mayor trascendencia en la Espectrometría de Masas. Los analizadores, que comenzaron
siendo sectores magnéticos, han evolucionado a sectores de cuadrupolo, sextupolos,
octupolos, trampa de iones, analizadores por tiempo de vuelo, llegando a contener uno o
dos sectores para lograr una mayor precisión.
4.4.1. Sistema de introducción de muestra
Para poder llevar a cabo el análisis existen dos requisitos previos, que la muestra se
halle en estado gaseoso y que exista vacío.
Figura V.16
Esquema espectómetro de masas (http://www.asms.org/whatisms/index.html).

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
87
El primer requisito limitaba antiguamente la aplicación a muestras en estado
gaseoso, pero gracias al desarrollo de estos equipos, hoy en día se puede aplicar también
a muestras en estado líquido, e incluso embebidas sobre una matriz sólida.
La espectrometría de masas cumple satisfactoriamente la mayor parte de los
requisitos para poderse acoplar con la cromatografía de gases, al ser una técnica de
elevada sensibilidad, capaz de hacer barridos rápidos y de registrar los espectros de los
compuestos en fase gaseosa. Sin embargo, presenta el problema de operar a alto vacío,
lo que parece en principio incompatible con la elución de la muestra acompañada de una
cantidad elevada de gas portador.
Existen diversas razones para que se necesite un sistema de vacío en un
espectrómetro de masas:
• La cámara de ionización podría contaminarse, y oxidarse el filamento u otros elementos a alta temperatura.
• La presencia en la cámara de una gran proporción de un gas competiría con la muestra para la ionización y produciría fenómenos de ionización química.
• En la etapa de separación de masas las colisiones intermoleculares reducirían la resolución.
En el acoplamiento CG-EM, el uso como portador de un gas inerte altamente
purificado reduce el problema de la contaminación, pero sigue presente la necesidad de
eliminarlo en la cámara de ionización y en el analizador. Para ello existen dos
posibilidades que dependen del tipo de columna empleada, ya que ésta condiciona el flujo
de gas portador.
• Acoplamiento con columnas capilares de diámetro inferior a 0,3 mm.
Estas columnas son las más utilizadas en la actualidad, y su flujo no suele
sobrepasar 1 ml/min. Cuando se emplea helio como gas portador, los sistemas
de bombeo utilizados en los actuales espectrómetros de masas pueden
eliminar este flujo de gas manteniendo una presión inferior a 10-4 mmHg en la
cámara de ionización e incluso más reducida en el analizador. La columna
capilar puede introducirse, debidamente termostatizada, hasta la cámara de
ionización sin ningún requisito instrumental.

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
88
Figura V.17
Acoplamiento directo CG-EM.
Hay que tener en cuenta el cambio en las condiciones de operación del proceso cromatográfico, ya que debido a que la salida de la columna se encuentra a vacío en lugar de a presión atmosférica, se produce una dilución de las zonas cromatográficas. Otros problemas de este tipo de conexión son la necesaria interrupción del sistema de vacío cuando se cambia la columna, y las variaciones de la presión en cámara causadas por los cambios en las condiciones cromatográficas.
Para evitarlos se ha recurrido a la interfase denominada de división en régimen abierto, en la que la cámara de ionización y la salida de la columna se conectan a un dispositivo abierto a la atmósfera y barrido por un flujo de helio. Si las dimensiones del tubo de conexión cámara-dispositivo se calculan de forma que una caída de presión de 1 atm a su través corresponda a un flujo de helio que permita mantener un vacío adecuado, este vacío no variará cuando cambie el flujo de la columna cromatográfica, ni siquiera cuando ésta se sustituya.
Figura V.18
Divisor en régimen abierto.
• Interfases para columnas rellenas o capilares de elevado diámetro (>0,3 mm). Estas columnas dan lugar en su operación habitual a flujos de 5-25 ml/min, que no pueden eliminarse de forma satisfactoria con los sistemas de vacío propios de un espectrómetro de masas. El aumento de presión resultante puede dar lugar a pérdida de resolución en el analizador y a reacciones ión-molécula en la cámara de ionización.

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
89
La eficacia de una interfase se mide mediante el rendimiento, Y, definido como el porcentaje de muestra de la columna que alcanza el detector.
Otro parámetro del sistema es el enriquecimiento, E, dado por la relación entre las concentraciones de la muestra en la cámara de ionización y en el gas portador a la salida de la columna.
La interfase más sencilla es un divisor de flujo, que introduce en la cámara de ionización el flujo que ésta puede soportar (<1 ml/min) mientras que el resto se elimina o se desvía a otro detector para el registro del cromatograma. Sin embargo, tanto su rendimiento, próximo al 5% para un flujo de 20 ml/min, como su enriquecimiento, igual a la unidad, son muy poco satisfactorios. Por este motivo se han desarrollado diversos tipos de interfases que presentan distintas ventajas e inconvenientes según el tipo de muestras a analizar.
La más extendida es la interfase tipo haz molecular (Jet), en la que el efluyente de la columna se hace pasar por un pequeño orificio donde adquiere una gran velocidad. Enfrentado con dicho orificio, a una distancia entre 0,1-0,5 mm se encuentra otro, directamente conectado a la cámara de ionización. Mientras que las moléculas de muestra relativamente pesadas atraviesan con pérdidas mínimas el camino entre los dos orificios, las más ligeras del gas portador se desvían de él debido a su mayor facilidad de difusión y son atrapadas por el sistema de vacío de la interfase. Esta interfase presenta un rendimiento típico del 50-80% y un enriquecimiento próximo a 10, que utilizando sistemas de doble interfase puede aproximarse a 100. Sin embargo, la extensión del uso de las columnas capilares ha conducido a la práctica desaparición de estas interfases para el acoplamiento CG-EM.
4.4.2. Cámara de ionización
Como ya dijimos, el espectrómetro de masa mide la relación masa/carga de los
iones, por lo que antes de realizar su análisis necesitamos cargar eléctricamente las
moléculas generalmente neutras del analito, y esto se realiza en la cámara de ionización.
La ionización más común es la ionización por impacto electrónico, donde las
moléculas ya gaseosas del analito son sometidas a un bombardeo con electrones de alta
energía, los que provocan la fragmentación en iones. Los iones con la carga que nos
interesa (sólo se pueden analizar los iones de un determinado signo en cada vez) son
impulsados hasta el analizador por el campo eléctrico presente, mientras que los otros
iones van a parar al electrodo recolector y las moléculas neutras son expulsadas. Con esta

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
90
técnica de ionización se producen muchos menos iones negativos que positivos, por lo
que suele registrarse el espectro de masas positivo.
Figura V.19
Cámara de ionización por impacto electrónico (http://www.asms.org/whatisms/index.html).
En los casos en los que es importante determinar la masa molecular y la estructura
del analito, esta técnica presenta el gran inconveniente de producir una gran
fragmentación, siendo difícil o imposible relacionar el ión original con los resultantes de la
fragmentación. Existen otras posibilidades que sí producen una ionización “suave”; estas
técnicas están basadas en la ionización química y en la ionización desortiva.
La Ionización química suele producirse por una transferencia de protones al exponer
el analito a un exceso de gas ionizante, como puede ser el metano.
La otra técnica consiste en un proceso donde las moléculas son a la vez evaporadas
de una superficie e ionizadas, no conociéndose el proceso exacto por el que se produce.
Los distintos métodos englobados en esta técnica son:
• Bombardeo con átomos rápidos: la ionización se produce por impacto de átomos de alta velocidad en una muestra disuelta en un medio protector líquido no volátil.
• SIMS (Secundary Ion EM): consiste en el impacto de iones de alta velocidad sobre una fina lámina de muestra con un substrato metálico o disuelta en una matriz líquida.
• Desorción con plasma: impacto de fragmentos procedentes de una fisión nuclear, sobre una muestra sólida depositada en una lámina metálica.

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
91
• MALDI (Matrix Assisted Laser Desorption/Ionization): fotones de alta
energía impactan sobre una muestra embebida en una matriz sólida orgánica.
• Desorción de campo eléctrico: La muestra depositada sobre un soporte sólido especial es sometida a un alto gradiente eléctrico.
• Electrospray: Consiste en generar gotas cargadas muy pequeñas al nebulizar la muestra a través de una aguja sometida a un alto voltaje. El agua de la gota se irá evaporando gradualmente hasta un punto donde el número de cargas electrostáticas repulsivas en la superficie es tan grande con relación al tamaño de la gota que se produce una explosión, debida a la repulsión de las cargas en la gota. Esta explosión genera un gran número de gotas cargadas más. Esta técnica tiene la característica especial de que la muestra entra en la cámara de vacío ya en forma de iones.
4.4.3. Analizador
El analizador es el encargado de que solamente los iones con una determinada
relación masa/carga lleguen al detector, para lo cual se pueden usar distintos métodos:
con sectores magnéticos, con filtros de cuadrupolos, trampas de iones, etc.
Figura V.10
Esquema analizador Sector magnético.
Una de las formas más comunes consiste en usar un campo magnético para curvar
la trayectoria de los iones, el radio de esta curvatura dependerá de la relación m/z y sólo
aquellos iones con una trayectoria adecuada terminarán en el detector, el resto se
dispersarán. Una forma más avanzada consiste en combinar está técnica con un enfoque
por campo eléctrico. Este campo eléctrico se sitúa previamente al magnético y actúa de
forma que enfoca a los iones con una determinada energía cinética. Este equipo se llama
espectrómetro de masas de doble enfoque, y es capaz de diferenciar compuestos de la
misma masa nominal pero distinta composición química.

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
92
Figura V.2
Esquema analizador de cuadrupolos.
Los filtros de cuadrupolos tienen una resolución menor que los de doble enfoque,
pero su bajo coste hace que sean la opción elegida en muchos casos. Su nombre proviene
de estar formado por cuatro polos paralelos, los iones enfocados dependen de la
trayectoria inducida en los iones al ser sometidos simultáneamente a un campo constante
y una radiofrecuencia, de forma que solo aquellos que entren en resonancia llegarán al
detector.
Las trampas de iones operan con un principio parecido al anterior, pero tienen la
particularidad de que no opera como un filtro, ya que los iones que no son enfocados
permanecen retenidos, pudiéndose usar posteriormente. Consiste en una estructura tipo
sándwich donde los iones son seleccionados en función del voltaje aplicado en los
electrodos. La forma de operar con este EM suele ser haciendo un barrido, de forma que
los iones son seleccionados secuencialmente, analizándose todos.
Figura V. 22
Esquema analizador trampa de iones.
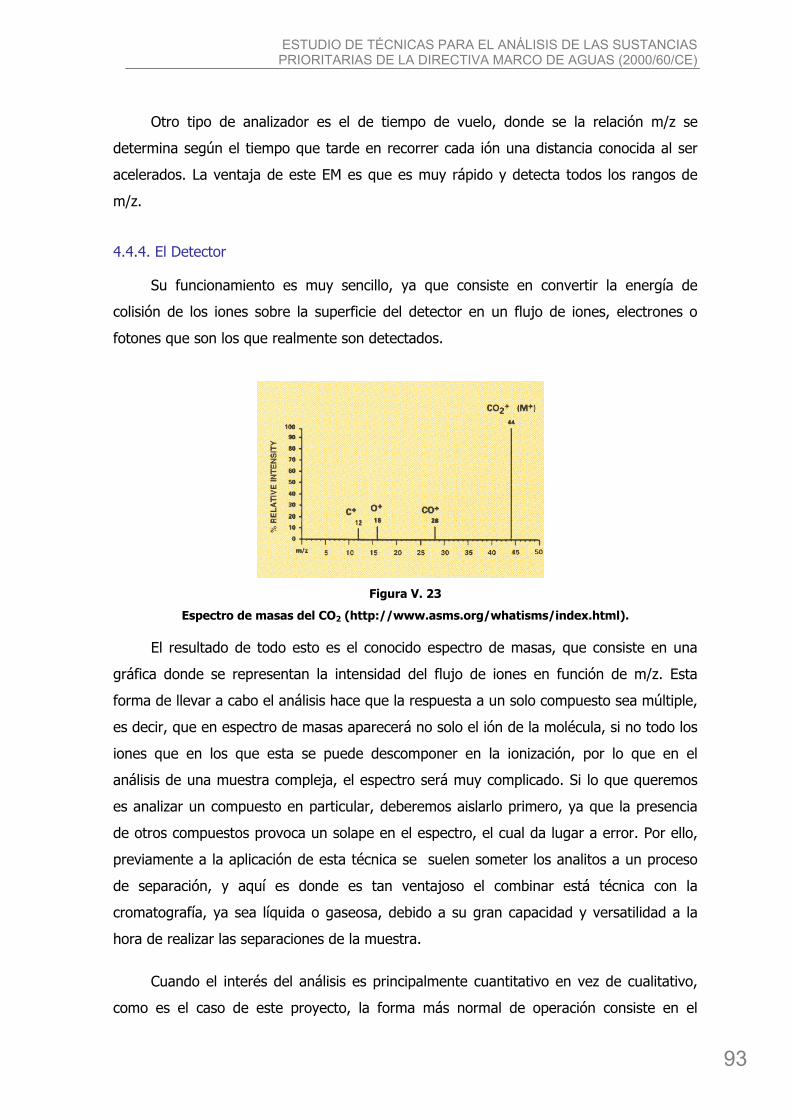
ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
93
Otro tipo de analizador es el de tiempo de vuelo, donde se la relación m/z se
determina según el tiempo que tarde en recorrer cada ión una distancia conocida al ser
acelerados. La ventaja de este EM es que es muy rápido y detecta todos los rangos de
m/z.
4.4.4. El Detector
Su funcionamiento es muy sencillo, ya que consiste en convertir la energía de
colisión de los iones sobre la superficie del detector en un flujo de iones, electrones o
fotones que son los que realmente son detectados.
Figura V. 23
Espectro de masas del CO2 (http://www.asms.org/whatisms/index.html).
El resultado de todo esto es el conocido espectro de masas, que consiste en una
gráfica donde se representan la intensidad del flujo de iones en función de m/z. Esta
forma de llevar a cabo el análisis hace que la respuesta a un solo compuesto sea múltiple,
es decir, que en espectro de masas aparecerá no solo el ión de la molécula, si no todo los
iones que en los que esta se puede descomponer en la ionización, por lo que en el
análisis de una muestra compleja, el espectro será muy complicado. Si lo que queremos
es analizar un compuesto en particular, deberemos aislarlo primero, ya que la presencia
de otros compuestos provoca un solape en el espectro, el cual da lugar a error. Por ello,
previamente a la aplicación de esta técnica se suelen someter los analitos a un proceso
de separación, y aquí es donde es tan ventajoso el combinar está técnica con la
cromatografía, ya sea líquida o gaseosa, debido a su gran capacidad y versatilidad a la
hora de realizar las separaciones de la muestra.
Cuando el interés del análisis es principalmente cuantitativo en vez de cualitativo,
como es el caso de este proyecto, la forma más normal de operación consiste en el
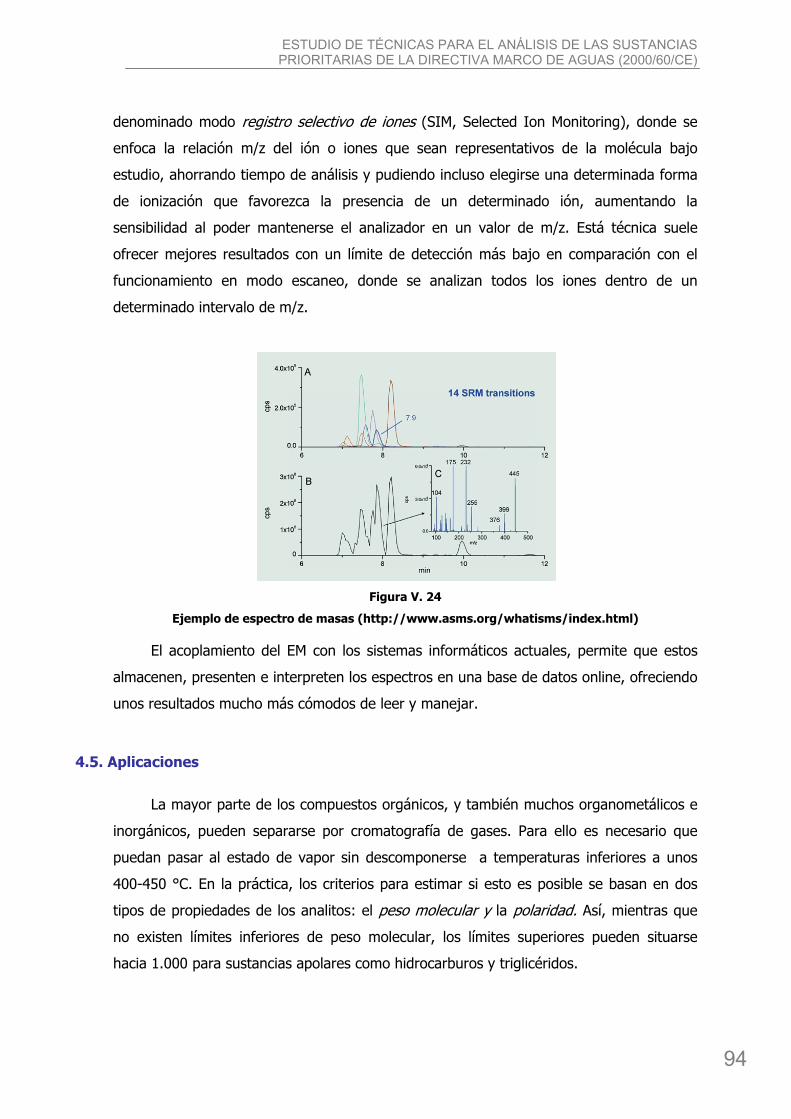
ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
94
denominado modo registro selectivo de iones (SIM, Selected Ion Monitoring), donde se
enfoca la relación m/z del ión o iones que sean representativos de la molécula bajo
estudio, ahorrando tiempo de análisis y pudiendo incluso elegirse una determinada forma
de ionización que favorezca la presencia de un determinado ión, aumentando la
sensibilidad al poder mantenerse el analizador en un valor de m/z. Está técnica suele
ofrecer mejores resultados con un límite de detección más bajo en comparación con el
funcionamiento en modo escaneo, donde se analizan todos los iones dentro de un
determinado intervalo de m/z.
Figura V. 24
Ejemplo de espectro de masas (http://www.asms.org/whatisms/index.html)
El acoplamiento del EM con los sistemas informáticos actuales, permite que estos
almacenen, presenten e interpreten los espectros en una base de datos online, ofreciendo
unos resultados mucho más cómodos de leer y manejar.
4.5. Aplicaciones
La mayor parte de los compuestos orgánicos, y también muchos organometálicos e
inorgánicos, pueden separarse por cromatografía de gases. Para ello es necesario que
puedan pasar al estado de vapor sin descomponerse a temperaturas inferiores a unos
400-450 °C. En la práctica, los criterios para estimar si esto es posible se basan en dos
tipos de propiedades de los analitos: el peso molecular y la polaridad. Así, mientras que
no existen límites inferiores de peso molecular, los límites superiores pueden situarse
hacia 1.000 para sustancias apolares como hidrocarburos y triglicéridos.

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
95
La polaridad, estimada por la cantidad y tipo de grupos funcionales, puede limitar
de manera drástica la volatilidad. Mientras que ácidos libres o aminas de peso molecular
medio pueden analizarse fácilmente, la presencia de ambas funciones en una misma
molécula (aminoácidos), aun con pesos moleculares inferiores a 200, impide la
volatilización y por tanto el análisis directo por esta técnica. Por otra parte, la formación
de derivados se emplea a veces para aumentar la sensibilidad o especificidad del método
analítico.
Los tipos de aplicaciones en cromatografía de gases se pueden clasificar en tres
categorías:
• Preparativas. Van dirigidas a la preparación de productos puros industriales y muy pocas veces se emplean en los laboratorios analíticos. Los problemas de escalado son similares a los que se pueden plantear en otras técnicas; pero al tratarse de una mezcla gaseosa, las dificultades (principalmente la formación de aerosoles) pueden surgir al provocar la condensación de los productos de interés.
• Químico-físicas. La técnica se utiliza, no para separar compuestos, sino para determinar ciertas propiedades del sistema como superficies específicas, isotermas de adsorción, cinética de reacciones, etc. (Laub y Pecsock, 1978; Conder y Young, 1979).
• Analíticas. Constituyen el grupo más numeroso. Aunque son casi ilimitadas, hay que destacar sus posibilidades frente a otras técnicas: se consiguen resoluciones muy elevadas en sustancias poco polares, así como sensibilidades muy altas mediante su acoplamiento con técnicas espectroscópicas. Pueden mencionarse una serie de campos en los que se utiliza preferencialmente, como gases permanentes, hidrocarburos (petroquímica, geoquímica), ambientales (aires, aguas, suelos, organismos), alimentos y cosméticos (aromas, lípidos, carbohidratos, aditivos), muestras biológicas (componentes de tejidos, metabolitos, drogas, fármacos, hormonas, feromonas), microorganismos (metabolitos, productos de fermentación), química forense (incendios provocados, drogas), etc.
5. Control del proceso cromatográfico
Las características geométricas de la columna así como el tipo y cantidad de fase
estacionaria que contienen, juegan un papel fundamental en el proceso cromatográfico.
Desde el punto de vista del cromatografista, es conveniente poder influir sobre este
proceso para adecuarlo a los requisitos de un análisis determinado, y no siempre es
posible disponer de la columna óptima para ello.

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
96
En el caso de la cromatografía de gases, existen dos magnitudes: el flujo de gas
portador y la temperatura de la columna, que son fáciles de controlar desde el
instrumento y pueden utilizarse para modificar el desplazamiento de los solutos a través
de la columna así como el respectivo cromatograma.
• Flujo de gas portador
El flujo a través de la columna puede controlarse mediante un regulador de la presión en cabeza de la columna o mediante un regulador de flujo. La relación entre presión y flujo, en el caso de las columnas abiertas de sección circular, podría calcularse mediante la ley de Poiseville.
La velocidad media del gas portador ū a lo largo de la columna está relacionada con el flujo, y éste se puede medir fácilmente de forma experimental, aunque en el caso de columnas capilares con flujos muy bajos puede ser difícil medirlo con exactitud.
La velocidad media del gas portador o la inversa del tiempo básico tM, se pueden utilizar para estimar el flujo en una misma columna con fines comparativos; siendo su ventaja que se pueden determinar con mayor exactitud a partir de medidas en el cromatograma.
El flujo interviene en la retención de forma que para un compuesto y fase estacionaria dados, el tiempo de retención es inversamente proporcional al flujo en la columna. Según esto, sería conveniente aumentar el flujo para reducir el tiempo de análisis, el problema es que como ya se vio, existe un valor óptimo de la velocidad, uop, para el que se produce el valor mínimo de la altura de plato. Es fácil concluir que, mientras que nunca se deben utilizar flujos inferiores al óptimo, puede ser conveniente emplear flujos superiores para reducir el tiempo de análisis si la pérdida de eficacia no degrada la separación de la muestra a resolver.
Es importante señalar que el flujo es capaz de modificar el tiempo de retención, tR, pero no tiene ninguna incidencia sobre el factor de retención, k, lo que en otras palabras significa, que afecta por igual a todos los componentes de la muestra.
• Temperatura de la columna
La temperatura de la columna se supone que es la misma del horno en el que está instalada, y esta última puede controlarse y medirse con exactitud. Cuando el gas portador se controla mediante un regulador de presión, su flujo a través de la columna varía con la temperatura de ésta debido al cambio de

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
97
viscosidad. Sin embargo, la influencia fundamental de la temperatura sobre la retención viene dada por su relación con el volumen de retención ya que cuando la temperatura aumenta, la retención disminuye de forma muy marcada.
En general, los valores de k muy bajos no son recomendables para llevar a cabo una separación, ya que la resolución tiende a cero cuando k también lo hace. Tampoco deben utilizarse valores muy altos, ya que el tiempo de análisis aumenta sin ventajas notables. Por estos motivos, dependiendo de la facilidad de separación de la muestra, se recomienda trabajar a una temperatura tal que los valores de k se encuentren entre 1 y 15.
Para intervalos de temperatura que no exceden los 30 °C, puede considerarse que la entalpía y entropía de tránsito permanecen constantes. Por tanto, cuando se mide la retención (K, Vg, k o tR) en un intervalo así y se representan sus logaritmos frente al inverso de la temperatura absoluta, se obtiene una recta para cada componente de la mezcla. Estas rectas tienen pendientes parecidas cuando corresponden a moléculas con grupos funcionales similares, ya que también lo son sus interacciones con la fase estacionaria y, por tanto, sus entalpías de tránsito. Las ordenadas en el origen, más relacionadas con el efecto entrópico, están influidas por otras características de la molécula, tales como su tamaño y forma. Se puede deducir que las rectas originadas por los miembros de una serie homóloga, que se diferencian en un grupo metileno, son paralelas y equidistantes. En cambio, cuando las sustancias poseen estructuras diferentes, se originan rectas con pendientes distintas, que pueden cruzarse (recta a con b y c en la figura). En este caso existe una temperatura en la que los picos coinciden, y a partir de ella se invierte el orden de elución.
Figura V.25
Variación de la retención con la temperatura (Dabrio et al., 2000)
Existen casos en los que se obtienen líneas quebradas en vez de rectas cuando se representa lnk frente a 1/T. Cuando este fenómeno se produce es debido a que en el intervalo experimental de temperaturas se ha producido algún tipo de transformación de la fase estacionaria, como cambio de estado de

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
98
agregación, sistema de cristalización u otro capaz de alterar el valor de la entalpía de tránsito. El resultado es que el punto en que cambia la pendiente corresponde a la temperatura de transformación.
La medida de esta temperatura por el procedimiento indicado se utiliza frecuentemente en el estudio de polímeros. Para ello se coloca el polímero como fase estacionaria en una columna y se inyectan patrones apropiados a varias temperaturas. Esta técnica se conoce inadecuadamente como cromatografía inversa.
6. Temperatura programada
En una muestra con numerosos y diversos componentes suele ser imposible la
elución de todos ellos en una sola operación a temperatura constante: una temperatura
baja, adecuada para la separación de los menos retenidos, implica un tiempo de análisis
muy alto para los últimos en eluir, mientras que si la temperatura se aumenta para que
éstos presenten una retención conveniente, los de menor retención se eluyen a valores de
k muy bajos, con una resolución insuficiente. En estos casos suele ser obligado recurrir a
la elución a temperatura programada.
El párrafo anterior justifica que en la mayor parte de los análisis por cromatografía
de gases de muestras complejas se recurra a la operación en el modo de temperatura
programada (Harris y Habgood, 1966), en el que la temperatura de la columna varía a lo
largo del proceso cromatográfico. Dicha variación suele consistir en un incremento por
unidad de tiempo (velocidad de calentamiento o de programación, expresada
generalmente en °C/min). Se denomina rampa a un periodo de operación con velocidad
de calentamiento constante; el desarrollo de un cromatograma puede incluir varias
rampas con distintas velocidades y periodos isotermos.
La variación de la temperatura provoca que el valor del factor de retención, k, de un
compuesto varíe a lo largo de la columna.
El cociente r/F, al que se llama programa, es el parámetro de operación más
significativo: si permanece constante, la temperatura de retención, TR, de un compuesto
no varía. En ese cociente, r representa la velocidad de calentamiento y F el flujo.
La temperatura influye directamente en la retención cromatográfica a través de la
relación entre Vg y T. Como la viscosidad del gas portador varía con la temperatura, si la

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
99
presión de entrada en la columna es constante, la reducción del flujo de fase móvil a lo
largo de una programación de temperatura puede llegar a ser muy marcada cuando el
incremento de temperatura alcanza los 100 °C. Si se utiliza un regulador de flujo, lo que
cambia es la presión de entrada.
La compleja intervención de la temperatura en las magnitudes que regulan la
velocidad de la zona en cromatografía de gases impide la resolución matemática, para lo
que sería necesaria una relación sencilla entre el volumen y la temperatura. Este es uno
de los inconvenientes de trabajar a temperatura programada, que complica la
interpretación teórica de los resultados así como el cálculo de magnitudes básicas de la
columna. Su empleo habitual pretende sólo conseguir que los compuestos atraviesen la
columna en las mejores condiciones para su separación.
Desde el punto de vista práctico, la programación de temperatura da lugar a
cromatogramas fácilmente distinguibles de los registrados en isoterma. Entre las
variaciones que implica en la respuesta cromatográfica se puede distinguir:
Figura V.26
Elución de una mezcla de compuestos: (a) en isoterma y (b) con temperatura programada. (Dabrio et al., 2000).

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
100
Anchura de pico. A diferencia del cromatograma isotermo, en que la anchura del
pico, w, aumenta de manera prácticamente lineal con k; en temperatura programada los
picos tienen una anchura que es similar a lo largo de una rampa, aunque crece en
periodos isotermos. Esto significa que pierden su sentido algunos parámetros basados en
la determinación de w, como la mayor parte de los relacionados con la eficacia.
• Tiempo y volumen de retención. Dado que la temperatura varía a lo largo del proceso cromatográfico, no puede calcularse el volumen de retención. Es muy utilizada la temperatura de retención, TR, que se define como aquella temperatura a la que se eluye el máximo del pico y que se relaciona fácilmente con el tiempo de retención ya que las rampas usadas tienen pendiente constante.
• Cuando se compara con un cromatograma isotermo, el cambio más llamativo en la retención es que, con programación lineal, los componentes de series homólogas se eluyen con un espaciado similar. Para la comparación de datos de retención procedentes de distintos laboratorios, se emplean los índices lineales en operación a temperatura programada en lugar de los índices de Kováts, aplicables a una programación lineal de la temperatura. Se pueden sustituir sus valores por los de los tiempos de retención.
• Otra característica suele ser la deriva de la línea base, que puede llegar a ser muy marcada si el sangrado de fase estacionaria es elevado. Según sea el equipo disponible, se puede compensar de dos formas: utilizando dos columnas iguales en paralelo, una de ellas para eluir la muestra y otra que sólo produce la señal de fondo y restando ambas señales en tiempo real, o con una sola columna, haciendo un programa en blanco sin muestra antes del análisis y efectuando la resta de ambos cromatogramas una vez almacenados en un ordenador.

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
101
VI. PRECONCENTRACIÓN Y PURIFICACIÓN DE MUESTRAS.
1. Introducción.
La etapa más delicada del análisis de estos compuestos es el fraccionamiento, que
es cuando se separan estos compuestos de la matriz de la muestra para introducirlos en
la columna cromatográfica. Las formas de llevarlo a cabo son diversas, y de usar una u
otra dependerán qué compuestos se aíslen y en qué medida. También tiene un gran coste
de tiempo, por lo que las mejoras en este sentido, como pueda ser la automatización del
proceso, son también muy importantes.
La extracción líquido-líquido (ELL) y la extracción en fase sólida (EFS) son las
técnicas de tratamiento de muestras más empleadas en el análisis cromatográfico de
aguas. La prevalencia de las técnicas de extracción líquido-líquido en el pasado se debía al
carácter hidrofóbico de los contaminantes considerados entonces como prioritarios. Aún
en la actualidad, la ELL es ampliamente usada en muchos métodos, especialmente aque-
llos orientados al enriquecimiento de trazas de contaminantes no polares en agua, aunque
su uso se encuentra actualmente desaconsejado por diversos los organismos
medioambientales que recomiendan el uso de las llamadas técnicas limpias (aquellas que
no usan disolvente).
Las técnicas limpias presentan dos ventajas:
• El disolvente no interfiere con compuestos de volatilidad similar en su análisis por Cromatografía Gaseosa.
• No es necesaria la etapa de preconcentración del disolvente, lo que evita la pérdida de compuestos de alta volatilidad así como la concentración de las impurezas existentes en los disolventes.
El objetivo del presente estudio es discutir el potencial y posibilidades de los
procesos de extracción líquido-líquido y de extracción en fase sólida, que son los dos
procedimientos de tratamiento de muestras previos a la determinación cromatográfica,
más empleados, en lo que se refiere a su aplicación en rutina en programas de vigilancia
de la calidad de aguas superficiales, centrándonos en el caso de los compuestos volátiles
ya que son el tipo de contaminantes más importante de este estudio.

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
102
El fundamento teórico de ambos procesos se basa en un principio similar, la
distribución de un soluto en la interfase de un sistema de dos fases. La distribución del
soluto entre ambas fases (líquido-líquido o líquido-sólido) viene gobernada por la afinidad
de este soluto por cada una de las fases, y viene caracterizada por una constante de
distribución. En la extracción líquido-líquido el transporte de los solutos de una fase
acuosa a un disolvente orgánico inmiscible con el agua, depende fundamentalmente de la
hidrofobicidad del soluto y de su solubilidad en el disolvente.
2. Técnicas de extracción con disolventes.
Se basan en la polaridad de los sustancias ya que la mayor o menor afinidad entre
los compuestos y el disolvente reside en la semejanza entre sus polaridades. Al tener una
gran cantidad de compuestos volátiles baja polaridad, un disolvente apolar dará un buen
resultado.
La extracción se puede realizar con matrices en estado sólido, líquido o gaseoso,
siendo asequibles y de fácil utilización, pero teniendo como inconveniente el tener que
usar disolventes de elevada pureza y los pobres resultados con compuestos que sean muy
volátiles.
En el caso de extracción líquido-líquido, la preconcentración se lleva a cabo en una
única etapa de transferencia del soluto de la fase acuosa a un disolvente orgánico. Una
vez alcanzado el equilibrio, una alícuota del disolvente orgánico es introducida en el
analizador. En ocasiones, sin embargo, puede ser necesario redisolver el analito en otro
disolvente que sea compatible con el sistema de análisis.
Existen técnicas más complejas que optimizan la extracción. Un ejemplo es la
Soxhlet, que al implicar un calentamiento, no se puede aplicar a compuestos termolábiles.
Otro ejemplo es la llamada Single-Drop, que consiste en poner en contacto la
muestra con una gota de disolvente que pende de una jeringa de inyección en
cromatografía gaseosa que, a pesar de su simplicidad conceptual, presenta problemas a
la hora de llevarla a la práctica como la inestabilidad de la gota y la complejidad
operacional (Jeannot y Cantwell, 1997; Jeannot y Cantwell, 1997; He y Lee, 1997).
La extracción líquido-líquido se emplea rutinariamente para la preconcentración de
PCBs, OCPs y HHAAPP. Para la extracción de plaguicidas organoclorados se puede

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
103
emplear hexano como disolvente de extracción debido a las buenas recuperaciones que
proporciona y a su compatibilidad con el sistema de análisis CG-DCE. Para la extracción
de hidrocarburos poliaromáticos se emplea freón dadas sus buenas recuperaciones y sus
ventajas de cara a su mayor inocuidad para el operador. Sin embargo el impacto adverso
que el freón provoca en la capa de ozono está haciendo que su uso se vaya reduciendo
cada vez más y se estudie su sustitución por hexano.
3. Extracción con fluidos supercríticos (McNally, 1996; King, 2002).
Su principio de funcionamiento es semejante al de los disolventes (de hecho se
comportan como auténticos disolventes), pero sin el impacto medioambiental de estos
últimos. Además actúan en parte como líquidos y en parte como gases, aunando las
ventajas de ambos. Por contra, tienen un alto coste de instrumentación (Kerrola, 1995).
4. Técnicas de destilación.
El fraccionamiento tiene lugar según la volatilidad de los compuestos, no siendo
apto para compuestos poco volátiles, pero tampoco para los compuestos muy volátiles
por el inconveniente que supone la etapa de preconcentración. Debido al calentamiento al
que se ven sometidos, los compuestos deben ser termoestables.
5. Extracción-destilación simultánea (EDS).
Conjuga tanto extracción con disolventes como destilación, por lo que su capacidad
de extracción depende tanto de la volatilidad como de la polaridad de los compuestos
(Chaintreau, 2001).
6. Inyección directa.
Es una técnica muy sencilla y rápida que consiste en inyectar el vapor contenido en
un recipiente, siendo el inconveniente la sensibilidad que se obtiene, que no es alta.
Han surgido otras que modifican algunos de sus aspectos, entre las que destaca el
arrastre con gas inerte, aplicable a muestras tanto sólidas (desorción térmica directa),
líquidas (burbujeo) y gaseosas. Las técnicas automáticas que se basan en estas técnicas
son la desorción térmica automática (ATD) y el “Purga&Trampa”, de las que no solo
destaca su automatización sino la ausencia de disolventes orgánicos y su alta sensibilidad.

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
104
7. Técnicas de fraccionamiento por absorción.
Estas técnicas han sido inspiradas en las cromatográficas, compartiendo sus mismos
principios y tecnología. Como ejemplo de esto se puede citar el PDMS, que es uno de los
materiales absorbentes más usados y también una de las fases estacionarias más
comunes para columnas capilares.
La creciente popularidad de las técnicas de extracción en fase sólida se debe no
sólo a sus evidentes ventajas sobre la ELL (menor consumo de disolventes orgánicos, no
formación de emulsiones, menor contacto del analista con sustancias potencialmente
tóxicas), sino también al progresivo perfeccionamiento de estos métodos que han
reducido las desventajas iniciales.
La aparición de los discos de extracción de membranas ha permitido aumentar la
velocidad del proceso de preconcentración, y la mayor producción de materiales para EFS
han abaratado sus precios. La amplia gama de materiales para EFS recientemente
desarrollados les ha permitido cubrir las exigencias de los nuevos contaminantes
(plaguicidas y otras sustancias químicas industriales y sus productos de degradación).
Actualmente, las principales características que hacen de la EFS una técnica pre-
ferente son: su facilidad de automatización, su capacidad de conservación y
almacenamiento de las muestras, y su gran potencial para ofrecer soluciones a complejos
problemas de preseparación y preconcentración en los estudios de vigilancia de la calidad
de las aguas superficiales.
El principio básico de este tipo de fraccionamiento es el uso de un material
extractante no solo como preconcentrador si no como elemento de fraccionamiento,
obteniéndose excelentes resultados con componentes de volatilidad alta y media.
Se pueden clasificar en dos tipos según el muestreo (Baltussen et al., 2002):
• Muestreo dinámico. La muestra se pone en contacto con el absorbente al hacerla circular a través de éste. Entre sus tipos se hayan trampas capilares abiertas, trampas capilares abiertas múltiples y tubos rellenos.
• Muestreo estático. En este caso tanto la muestra como el absorbente comparten el mismo volumen, no siendo la agitación o sonificación más que una forma de acelerar la llegada al equilibrio. En este tipo encontramos

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
105
microextracción en fibra capilar, extracción dinámica en fase sólida y extracción por absorción sobre barra agitadora.
La mayoría de los procedimientos de extracción en fase sólida comprenden la
transferencia del soluto en dos fases. Inicialmente, el soluto es adsorbido de la fase
acuosa, sobre la superficie sólida de un absorbente o en la fase estacionaria enlazada a la
misma, y, posteriormente, el soluto es eluido con un disolvente adecuado (o desorbido
térmicamente en un cromatógrafo de gases). En este caso, la selección del solvente
adecuado (es decir, la selección de la fuerza de las interacciones responsables de la unión
del soluto en la fase sólida) y la selección del disolvente de elución o de las condiciones
de desorción térmica (es decir, la energía necesaria para romper estas uniones) deben
realizarse adecuadamente a fin de obtener una preconcentración cuantitativa.
Cuando se emplean procedimientos de extracción en fase sólida, normalmente es
necesario filtrar las muestras a través de un filtro de membranas antes de la extracción.
De este modo, los resultados obtenidos tienen en cuenta únicamente el contenido de
analito en la fase acuosa. Aún en el caso de que no se aplicara ningún proceso de
filtración previo a la extracción, difícilmente los analitos adsorbidos en las partículas
sólidas serían retenidos en los adsorbentes de los cartuchos de extracción en fase sólida.
Existen numerosos estudios comparativos de ambas técnicas a nivel general (Junk
et al., 1987; Liska et al., 1989; Barceló, 1991), y ensayos experimentales de comparación
de ambas técnicas (Chiadek y Maraño, 1984; Fingler et al., 1987), que han demostrado la
superioridad de la extracción en fase sólida en numerosas aplicaciones, especialmente
cuando se pretende preconcentrar compuestos de carácter polar.
7.1. Trampas capilares abiertas (TCA).
Las trampas capilares abiertas consisten en una columna capilar recubierta de una
fina capa de fase estacionaria (10 - 17 μl), que es la encargada de atrapar los analitos
(Bicchi et al., 1987, 1988). La extracción de éstos se realizara generalmente por desorción
térmica (Burger y Munro, 1986; Blomberg y Roeraade, 1987), aunque también se pueden
emplear disolventes (Blomberg y Roeraade, 1987).
El inconveniente de esta técnica es la pequeña cantidad de fase estacionaria
presente, que hace que no sea muy sensible, no siendo demasiado factibles como

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
106
soluciones ni el aumentar el largo de la columna (Zhang y Pawliszyn, 1996), ni el espesor
de la capa estacionaria (Roeraade y Blomberg, 1989; Burger et al., 1990; Blomberg y
Roeraade, 1988). Otro inconveniente es el bajo flujo a través del capilar (< 10 ml / min),
que implica altos tiempos de muestreo.
7.2. Trampas capilares abiertas múltiples (TCAM).
Ortner y Rohwer (1996) mejoraron las trampas capilares abiertas colocando varios
TCAM en paralelo, que aunque aumenta notablemente la cantidad de fase estacionaria
absorbente, la limitación del flujo (15 ml / min) y la geometría de la trampa hace que no
se consiga una retención cuantitativa de los analitos.
7.3. Tubos Rellenos (Packed Beds).
Los tubos rellenos fueron introducidos por Baltussen et al. (1997) y supusieron otro
avance en lo que se refiere a la cantidad de fase estacionaria presente (hasta 300 μl),
aumentando la sensibilidad. Otra ventaja añadida es que permiten flujos mucho mayores
que las trampas capilares (Baltussen et al., 1999), lo que disminuye notablemente el
tiempo de muestreo.
Sin embargo, presentan inconvenientes en su difícil aclopamiento directo con CG o
CG/EM debido, entre otras cosas, a los altos caudales usados para la desorción y a la
imposibilidad de muestrear compuestos de alta volatilidad en muestras líquidas, ya que se
perderían en la necesaria etapa de secado.
El primer inconveniente se podría solucionar con un divisor de corrientes, pero iría
en detrimento de la sensibilidad; la otra opción sería usando complejos dispositivos de
desorción con dos puntos de división de flujo y una trampa fría entre ellos.
7.4. Microextracción en fibra capilar (MEFC).
Presentada por Arthur y Pawliszyn (1998) a principio de los noventa, consiste en un
dispositivo a modo de jeringa que posee una fibra capilar retráctil recubierta con una fase
extractante, ya sea adsorbente, absorbente o una combinación de ambas.
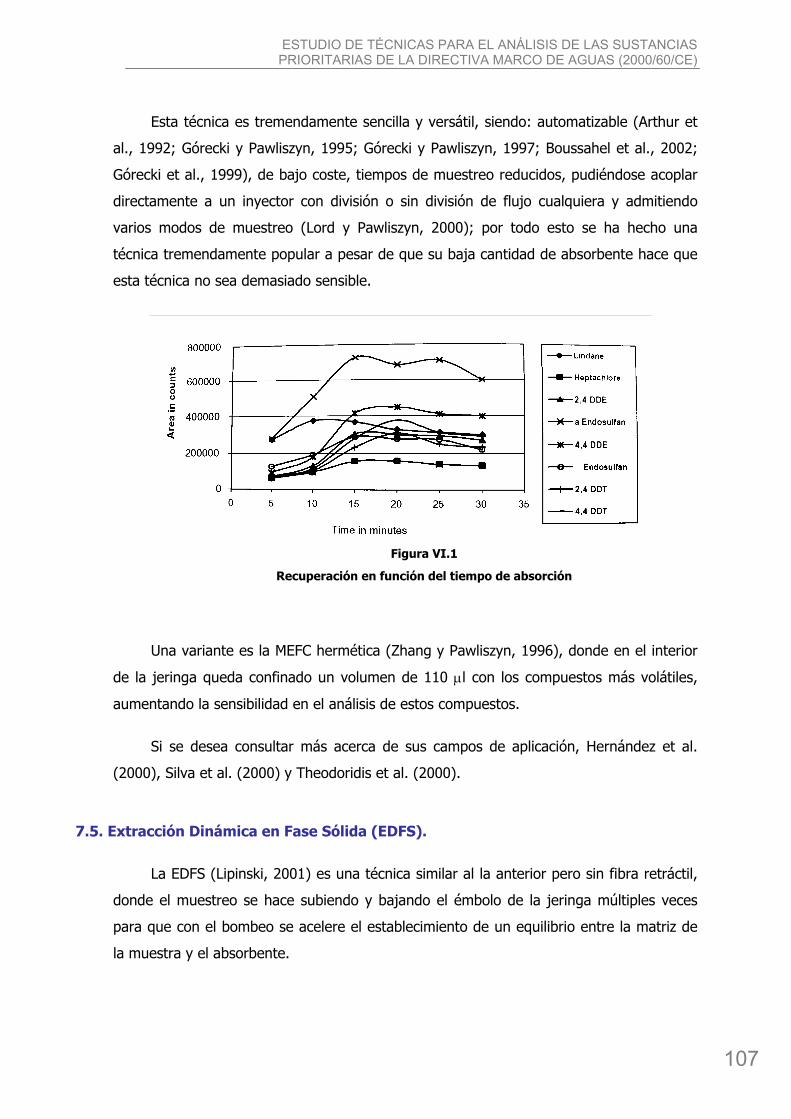
ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
107
Esta técnica es tremendamente sencilla y versátil, siendo: automatizable (Arthur et
al., 1992; Górecki y Pawliszyn, 1995; Górecki y Pawliszyn, 1997; Boussahel et al., 2002;
Górecki et al., 1999), de bajo coste, tiempos de muestreo reducidos, pudiéndose acoplar
directamente a un inyector con división o sin división de flujo cualquiera y admitiendo
varios modos de muestreo (Lord y Pawliszyn, 2000); por todo esto se ha hecho una
técnica tremendamente popular a pesar de que su baja cantidad de absorbente hace que
esta técnica no sea demasiado sensible.
Figura VI.1
Recuperación en función del tiempo de absorción
Una variante es la MEFC hermética (Zhang y Pawliszyn, 1996), donde en el interior
de la jeringa queda confinado un volumen de 110 μl con los compuestos más volátiles,
aumentando la sensibilidad en el análisis de estos compuestos.
Si se desea consultar más acerca de sus campos de aplicación, Hernández et al.
(2000), Silva et al. (2000) y Theodoridis et al. (2000).
7.5. Extracción Dinámica en Fase Sólida (EDFS).
La EDFS (Lipinski, 2001) es una técnica similar al la anterior pero sin fibra retráctil,
donde el muestreo se hace subiendo y bajando el émbolo de la jeringa múltiples veces
para que con el bombeo se acelere el establecimiento de un equilibrio entre la matriz de
la muestra y el absorbente.

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
108
7.6. Extracción por Absorción sobre Barra Agitadora (ESBA).
El ESBA es una técnica nueva (Baltussen et al., 1999), consistente en un imán
encapsulado en una funda de vidrio que a su vez está recubierta por una capa de PDMS
(la cantidad de absorbente varia según las dimensiones).
Las ventajas de este método son muchas, entre ellas que la cantidad de absorbente
que maneja es elevada (semejante a la de los tubos rellenos y las TCAM multicanal), lo
que hace que su sensibilidad sea elevada incluso para los compuestos más volátiles, ya
que el secado tras el muestreo de un líquido es sencillo y rápido.
La sencillez de la preparación de la muestra junto con la capacidad de ser
automatizado hacen de este método uno de los mejores para diversas aplicaciones (en
especial para el análisis de pesticidas en aguas), siendo su gran inconveniente el alto
coste, al venderse junto con el equipo de desorción térmica de la casa, que lo
comercializa bajo el nombre de “twister”.
Existe una nueva técnica desarrollada por Tienpont et al. (2000) llamada
“headspace sorptive extraction” (HSSE) con fundamentos muy similares a los de MEFC, y
consistente en recubrir con PDMS unas varillas de vidrio que se colocan en el espacio en
cabeza de un vial o Erlenmeyer que contenga la muestra a fraccionar en agitación
(Tienpot et al., 2000).
En general, el progreso en este campo ha sido grande, pero seguramente no
termine aquí si no que seguirá avanzando hacia técnicas aún más sofisticadas.

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
109
Tabla VI.1
Ventajas e inconvenientes de las técnicas de fraccionamiento de VOCs por Absorción (D. Estaban, 2003)
TÉCNICA VENTAJAS INCONVENIENTES
Bajos flujos de muestreo
Baja sensibilidad TCAM (Dinámica) Trampa fácil de adquirir
Desorción compleja
Sensibilidad Bajos flujos de muestreo TCAM Multicanal
(Dinámica) Desorción térmica directa en el inyector Trampa difícil de elaborar
Sensibilidad Análisis de volátiles en muestras líquidas Tubos rellenos
(Dinámica) Altos flujos de muestreo Complejo acoplamiento con CG por
los flujos de introducción de Muestra
Sencillez
Bajo coste
Versatilidad
Automatización MEFC (Estática)
Fibras que combinan absorbentes con adsorbentes
Baja sensibilidad
Sencillez EDFS (Estática)
Muestreo más eficaz que MEFC Técnica nueva con falta de
aplicaciones
Sensibilidad ESBA (Estática)
Automatización Instrumentación cara
Sensibilidad HSSE (Estática)
Automatización Instrumentación cara
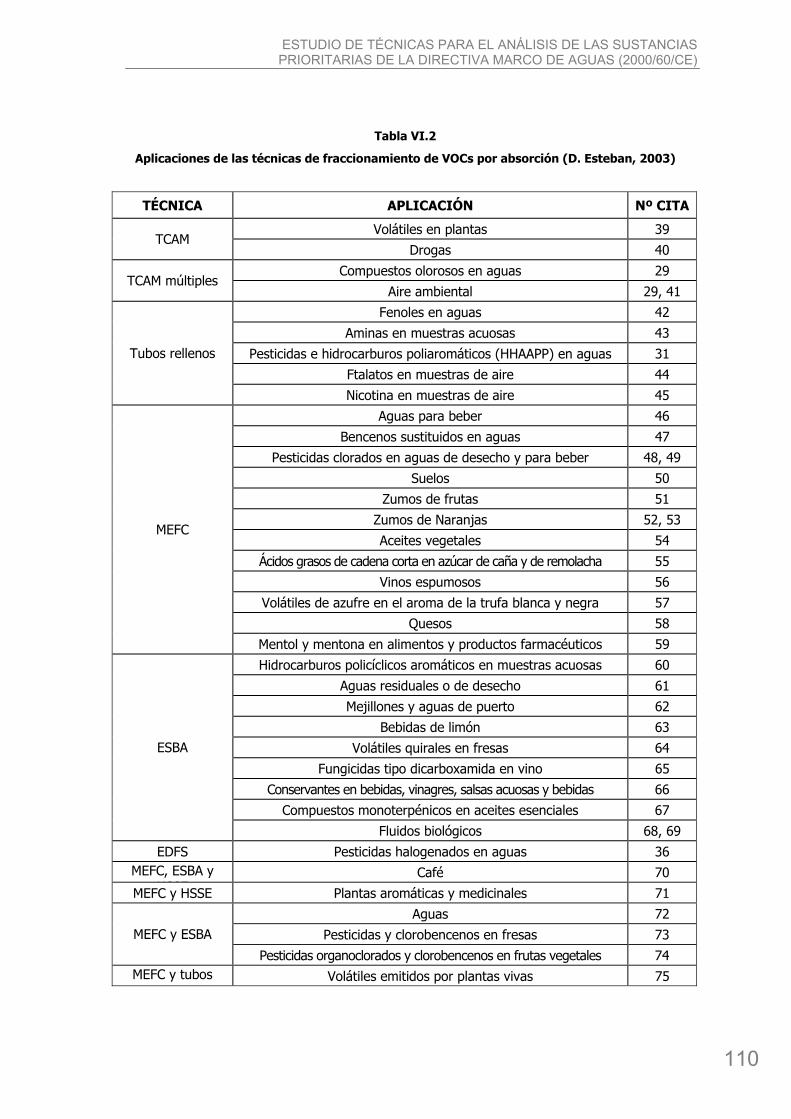
ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
110
Tabla VI.2
Aplicaciones de las técnicas de fraccionamiento de VOCs por absorción (D. Esteban, 2003)
TÉCNICA APLICACIÓN Nº CITA
Volátiles en plantas 39 TCAM
Drogas 40
Compuestos olorosos en aguas 29 TCAM múltiples
Aire ambiental 29, 41
Fenoles en aguas 42
Aminas en muestras acuosas 43
Pesticidas e hidrocarburos poliaromáticos (HHAAPP) en aguas 31
Ftalatos en muestras de aire 44
Tubos rellenos
Nicotina en muestras de aire 45
Aguas para beber 46
Bencenos sustituidos en aguas 47
Pesticidas clorados en aguas de desecho y para beber 48, 49
Suelos 50
Zumos de frutas 51
Zumos de Naranjas 52, 53
Aceites vegetales 54
Ácidos grasos de cadena corta en azúcar de caña y de remolacha 55
Vinos espumosos 56
Volátiles de azufre en el aroma de la trufa blanca y negra 57
Quesos 58
MEFC
Mentol y mentona en alimentos y productos farmacéuticos 59
Hidrocarburos policíclicos aromáticos en muestras acuosas 60
Aguas residuales o de desecho 61
Mejillones y aguas de puerto 62
Bebidas de limón 63
Volátiles quirales en fresas 64
Fungicidas tipo dicarboxamida en vino 65
Conservantes en bebidas, vinagres, salsas acuosas y bebidas 66
Compuestos monoterpénicos en aceites esenciales 67
ESBA
Fluidos biológicos 68, 69
EDFS Pesticidas halogenados en aguas 36 MEFC, ESBA y
HSSECafé 70
MEFC y HSSE Plantas aromáticas y medicinales 71
Aguas 72
Pesticidas y clorobencenos en fresas 73 MEFC y ESBA
Pesticidas organoclorados y clorobencenos en frutas vegetales 74 MEFC y tubos
llVolátiles emitidos por plantas vivas 75

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
111
7.7. Dispersión en Matriz de Fase Sólida (DMFS)
En esta técnica se produce simultáneamente una disrupción y extracción de
muestras sólidas y semisólidas mediante la dispersión de la muestra sobre un soporte
sólido, seguido por un lavado y elusión con una pequeña cantidad de solvente.
Su acoplamiento a cromatografía líquida es muy sencillo y, en esencia, se puede
considerar como un método que no usa disolventes.

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
112
VII. MÉTODOS PROPUESTOS.
1. Análisis.
1.1. Introducción
Las características más importantes que se han tenido en cuenta en la elección de
las técnicas analíticas son:
Alcanzar bajos límites de detección.
Sencillez de la preparación de la muestra.
Un amplio campo de aplicación.
Para los límites de detección se han tomado como referencia los valores dados en el
estudio encargado por la Unión Europea a la Sociedad Fraunhofer, siendo estos
provisionales (Lepper, 2002).
A la hora de comparar las técnicas de preconcentración se han agrupado los
espectrómetros de masas, ya que en la literatura se pueden encontrar distintos equipos,
como el CIPA (ionización a presión atmosférica) o el EMAR (espectrómetro de masas de
alta resolución) entre otros.
Las sustancias prioritarias se han agrupado en:
Pesticidas semivolátiles
Hidrocarburos policíclicos
Organoclorados volátiles
Otras sustancias.
En la bibliografía consultada han aparecido multitud de posibles técnicas para el
análisis de los distintos grupos, aunque muchas de ellas no se han considerado lo
suficientemente relevantes como para ser comentadas.
1.2. Pesticidas semivolátiles
El análisis medioambiental de estas sustancias se viene realizando desde hace
mucho tiempo, por lo que existe una gran variedad de bibliografía para su estudio. Este
grupo no posee propiedades semejantes, si no que contiene a aquellas sustancias útiles

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
113
en la lucha contra las plagas. Esto explica el porqué de la amplia variedad de técnicas
para su análisis y el reducido campo de acción de algunas de ellas.
Las sustancias incluidas dentro de los pesticidas volátiles y presentes en la directiva
son: Alacloro, Aldrina, Atracina, Clorofenvifos, Cloropirifos, o,p-DDT, p,p-DDT, Dieldrina,
Diurón, Endosulfán(alfa), Endrina, α-HCH, β-HCH, δ-HCH, Isodrina, Isoproturón, Lindano
(γ-HCH), Simacina, Trifluoralina.
Los pesticidas de mayor polaridad han sido excluidos de esta clasificación, ya que
los métodos aplicables a estas sustancias para obtener unos resultados aceptables, son
distintos de los usados para las sustancias aquí estudiadas, debido especialmente a la
inefectividad de la preconcentración por EFS, MEFS y ESBA, siendo en estos casos más
corriente la aplicación de P&T o EC. Tanto EFS como MEFS y ESBA pertenecen a la misma
familia de técnicas de fraccionamiento por absorción, siendo EFS el comienzo de esta
rama que ha caído ya en un notable desuso, aunque aún está presente en una parte
considerable de la bibliografía seleccionada, incluyendo interesantes artículos para ciertos
compuestos. La diferencia entre MEFS y ESBA es menor, siendo la principal distinción
entre ellos la mayor facilidad de aplicación del más moderno, que consiste simplemente
en utilizar un agitador magnético que está recubierto del material absorbente. En
resumen, el uso de una técnica u otra suele deberse a la antigüedad del estudio en
cuestión.
Entre las técnicas más importantes aparece la CG-EM, usándose como método de
preconcentración EFS, MEFS o ESBA. La CG es siempre una columna capilar de
cromatografía gaseosa con He por fase móvil, y que trabaja con un gradiente de
temperatura.
El detector es un elemento que varía, tanto en el método usado en la detección
(escaneo, registro selectivo de iones,…), como en el tipo (ionización a presión
atmosférica,...). La espectrometría de masas en tándem se considera una técnica distinta,
aunque podría englobarse como un tipo de detector de masas avanzado, ya que su
campo de aplicación es el mismo. Estos equipos están basados en los espectrómetros que
salieron a la luz hace unos 5 años, consistentes en un triple cuádruplo con fuentes de
ionización a presión atmosférica. Entre sus ventajas está la de aislar un determinado ión y
provocar su fragmentación mediante colisiones, permitiendo una mejor discriminación de
las posibles interferencias de la matriz de la muestra. Los requisitos especiales que debe

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
114
satisfacer este equipo son: buena selección del ión, un aislamiento y almacenamiento
eficiente del mismo y unas condiciones óptimas de la disociación inducida por colisión.
El detector de masas en tándem se distingue del modelo simple en que realiza la
detección en dos etapas. El modo de análisis más común es el llamado ESCANEO DE
IONES, donde primero se realiza un análisis de todos los iones dentro del rango m/z de
interés, y a continuación se seleccionan los iones de un determinado m/z, para pasar a la
segunda etapa, en que entran en la cámara de fragmentación y se analizan los
fragmentos resultantes de la misma.
La CG-EM es la técnica con mayor campo de aplicación y mejores resultados para
los pesticidas, siendo las excepciones el Diurón y el Isoproturón, para los que la
cromatografía líquida sigue siendo la técnica por excelencia, aunque el alto límite fijado
por la directiva para el Isoproturón hace que otros métodos menos aptos lo alcancen
fácilmente.
Carabias-Martínez et al. (2003), consigue analizar el Isoproturón mediante MEFS-
CG-EM, siendo esto lo más relevante de su artículo, ya que sus resultados son en general
pobres, alcanzando un LD de 0.02 μg/l y una precisión de 18.6 con una concentración de
0.3 μg/l para el Isoproturón. Sus resultados para los otros pesticidas no son mejores. En
otros estudios se pueden observar valores mucho mejores aplicando la misma técnica:
véase Estefani Morales (2004), y Gonçalves y Alpendurada (2004); por lo que es un
resultado extraño el que consiga tan buenos valores en el análisis de los dos pesticidas
más complicados en el análisis por CG, como son el Diurón e Isoproturón. Pese a que se
ha realizado una búsqueda, no se han encontrado estudios que puedan afianzar la idea
del análisis por CG de estos compuestos.
La utilización más general de esta técnica es la de Estefani Morales (2004), donde
aplica ESBA-CG-EM a todos los pesticidas del grupo, a excepción de los dos citados
anteriormente, con unos LLDD cercanos a 0.005 μg/l para la mayoría de los casos, y
obteniendo un buen coeficiente de correlación (r2>0.99) excepto en el DDT y la Simacina.
Cabe destacar que es el único estudio de los consultados, donde se realiza la detección de
la Isodrina, siendo posible que en otros, aunque no se hayan analizado, sea viable la
aplicación de sus métodos para el análisis de esta sustancia.
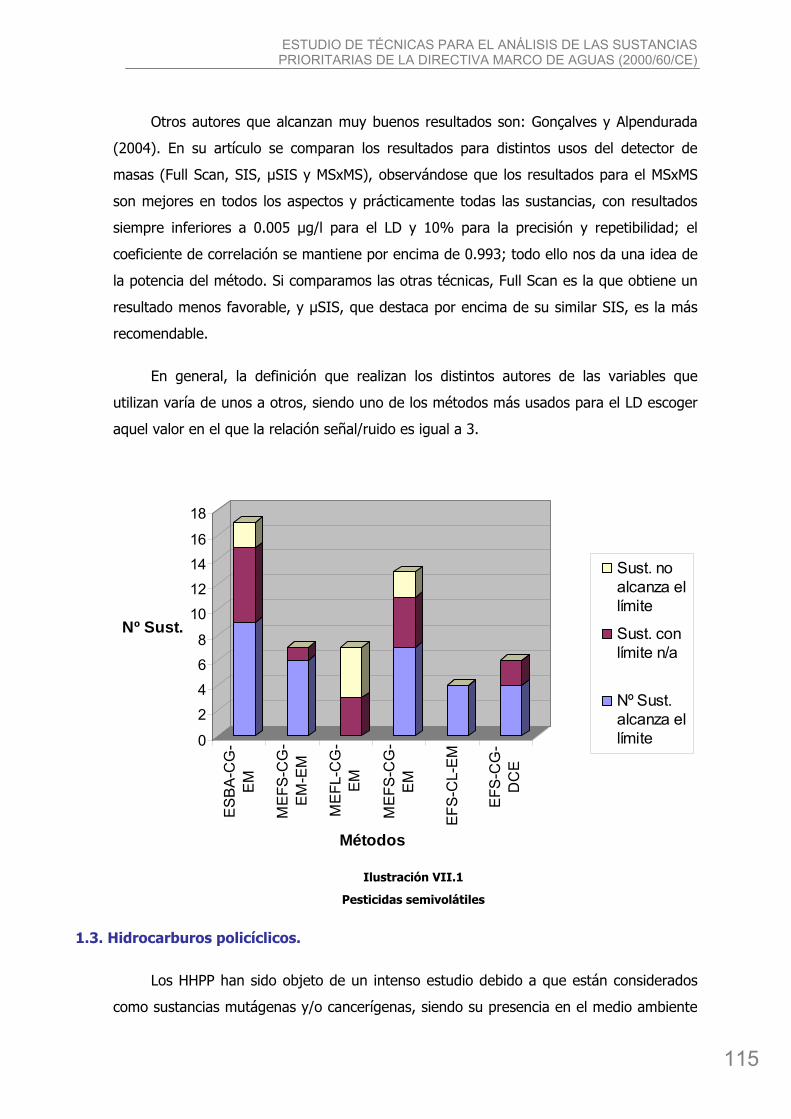
ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
115
Otros autores que alcanzan muy buenos resultados son: Gonçalves y Alpendurada
(2004). En su artículo se comparan los resultados para distintos usos del detector de
masas (Full Scan, SIS, μSIS y MSxMS), observándose que los resultados para el MSxMS
son mejores en todos los aspectos y prácticamente todas las sustancias, con resultados
siempre inferiores a 0.005 μg/l para el LD y 10% para la precisión y repetibilidad; el
coeficiente de correlación se mantiene por encima de 0.993; todo ello nos da una idea de
la potencia del método. Si comparamos las otras técnicas, Full Scan es la que obtiene un
resultado menos favorable, y μSIS, que destaca por encima de su similar SIS, es la más
recomendable.
En general, la definición que realizan los distintos autores de las variables que
utilizan varía de unos a otros, siendo uno de los métodos más usados para el LD escoger
aquel valor en el que la relación señal/ruido es igual a 3.
1.3. Hidrocarburos policíclicos.
Los HHPP han sido objeto de un intenso estudio debido a que están considerados
como sustancias mutágenas y/o cancerígenas, siendo su presencia en el medio ambiente
0
2
4
6
8
10
12
1416
18
Nº Sust.
ES
BA-
CG
-E
M
ME
FS-C
G-
EM
-EM
ME
FL-C
G-
EM
ME
FS-C
G-
EM
EFS-
CL-
EM
EFS
-CG
-D
CE
Métodos
Sust. noalcanza ellímite
Sust. conlímite n/a
Nº Sust.alcanza ellímite
Ilustración VII.1
Pesticidas semivolátiles

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
116
fruto de: combustiones incompletas de compuestos orgánicos, emisión de vehículos,
vertidos industriales, vertidos accidentales de petróleo, etc.
Las sustancias incluidas en este grupo son: Antraceno, Benzo(a)pireno,
Benzo(b)fluoranteno, Benzo(g,h,i)perileno, Benzo(k)fluranteno, Fluoranteno,
Indenol(1,2,3-cd)pireno y Naftaleno.
Las técnicas instrumentales de análisis más utilizadas para su determinación se
encuentran en la Cromatografía de Gases (CG) y la Cromatografía de Líquidos de Alta
Resolución (HPLC), así como la espectrofotometría de absorción UV-Visible y la
espectrofluorimetría, aún cuando las técnicas espectroscópicas convencionales requieren
de procesos de separación previos por las grandes interferencias espectrales presentes.
En la tabla puede observarse como los resultados obtenidos por Estefani Morales
(2004) cumplen los requisitos pedidos por la directiva, si bien siempre quedan por debajo
de los obtenidos por Kolahgar et al. (2002). Ambos autores utilizan la misma técnica
(ESBA-CG-EM), e incluso equipo de la misma marca, por lo que la diferencia entre los
resultados debe de hallarse en el procedimiento en sí o en la definición de los resultados
presentados.
Respecto a lo primero, Estefani Morales (2004) usa un Twister de 20 mm
adicionando un 20% de NaCl para aumentar las fuerzas iónicas, mientras que Kolahgar et
al. (2002) adiciona Hiomina hasta una concentración de 10 μg/l, para evitar la absorción
de los analitos sobre la pared de vidrio del material usado durante la preparación de la
muestra. Hay que recordar que según Gimeno et al. (2002), uno de los principales
problemas de los HHAAPP es su tendencia a ser absorbidos por las superficies con las que
entran en contacto.
Otro punto donde divergen ambos estudios es en el tiempo de desorción. En
Estefani Morales (2004) es de 14 h., para asegurarse de una completa extracción (el
procedimiento para este autor agrupa también al heterogéneo grupo de los pesticidas),
mientras que en Kolahgar et al. (2002) es de tan sólo 3.5 horas, ya que a partir de ese
momento aparecen extrañas bajadas y subidas en la recuperación.
La otra posible diferencia está en las definiciones de los valores aportados por cada
uno, que es uno de los problemas más importantes encontrados en la elaboración del
presente proyecto, ya que impide la comparación directa de los valores. Mientras que el

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
117
primer autor calcula los límites de detección según lo indicado en las normas “ISO 11843-
1” e “ISO 11843-2”, el segundo lo calcula como los valores estadísticos teóricos basados
en la fluctuación de la respuesta al Blanco y la sensibilidad del método (3σ entre la
sensibilidad), reconociendo que el límite cuantitativo práctico aumenta hasta 1 ng/l
aproximadamente, valor que sigue mejorando el obtenido por Estefani Morales (2004).
Decir cual de los métodos es más exacto es complicado, y recordamos lo dicho en el
apartado anterior acerca del tema.
En cualquier caso, los límites de detección alcanzados por ambos autores en el uso
de esta técnica, son satisfactorios para el análisis de los HHPP dentro de la directiva
MARCO, por lo que es una técnica recomendable al analizarse todas las sustancias de este
grupo con unos LLDD del orden de 1 ng/l para Kolahgar et al. (2002) y del orden de 10
ng/l para Estefani Morales (2004). Este último obtiene unos mejores coeficientes de
correlación, >0.99 para todos excepto el Naftaleno y Benzo(g,h,i)perileno para los que
son levemente inferiores; el otro autor consigue resultados semejantes en la mayoría de
los casos excepto para el Benzo(a)pireno, donde obtiene tan solo un 0,92543. La
precisión también es mejor en el estudio realizado por Estefani Morales (2004), donde
permanece por debajo del 10% en comparación del 15% que tiene como valor máximo
Kolahgar et al. (2002), quien sí alcanza unos muy buenos valores de recuperación, como
se puede ver en la tabla, la mayoría del 100%, valores que, aunque Estefani Morales
(2004) no proporcione, deberían ser similares.
012345678
Nº Sust.
ESBA-CG-EM EFS-CL-DF
Métodos
Sust. no alcanzael límite
Sust. con límiten/a
Nº Sust. alcanzael límite
Ilustración VII.2
Hidrocarburos Policíclicos.

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
118
El otro método es el de Gimeno et al. (2002), que usa EFS-CL-F, cuyos resultados
varían notablemente, siendo en general semejantes a los de Estefani Morales (2004) salvo
para el Indenol(1,2,3-cd)pireno, para el que alcanza un pobre LD de 0.1 (μg/l). Los
resultados de este autor se realizan sobre muestras reales de agua de mar y, arguye, que
los realizados con el detector de Fluorescencia deben superar a los obtenidos por EM
debido a la dificultad de los HHPP para ser ionizados; un contrapunto a este argumento
puede ser el uso de CIPA-EM, especialmente eficaz en la detección de elementos poco
ionizables; sin embargo, la comparación entre estos dos métodos realizada por el mismo
autor parece confirmarlo en todos los casos, salvo en el Indenol(1,2,3-cd)pireno. Es
también el que alcanza un mejor Coeficiente de Determinación, siempre superior a
0.9996. No analiza ni el Naftaleno ni el Antraceno.
1.4. Organoclorados volátiles.
En este grupo incluimos: Cloroformo (Triclorometano), Diclorometano, 1, 2-
dicloroetano, Tetracloroeteno, Tetraclorometano, 1,2,4-triclorobenceno y 1,1,2-
tricloroeteno.
Este grupo resulta problemático ya que la mayoría de las técnicas hasta ahora
vistas, estaban enfocadas a sustancias poco polares, no dando buenos resultados para los
compuestos más volátiles; por ello aparecen en este apartado métodos no presentes en
apartados anteriores.
De los tres procedimientos estudiados, P&T-CG-EM es el más potente respecto a sus
resultados, alcanzando unos límites realmente bajos y dejando sin analizar sólo un
compuesto, que seguramente, pueda ser determinado por este método sin mayor
problema. De los otros dos métodos, IDA-CG-DCE resulta más atractivo que ES-CG-DCE,
obteniendo unos mejores resultados y siendo su aplicabilidad muy buena, además, EC-
CG-DCE no alcanza el límite en una de las sustancias, mientras que IDA-CG-ECD deja dos
elementos sin analizar, siendo posible el que se pudiera aplicar. La diferencia básica entre
ambos métodos estriba en la forma de introducir la muestra en la columna.
Ekdahl y Abrahamsson (1997) hacen un interesante estudio, donde alcanzan unos
resultados muy por encima del resto de la bibliografía consultada, realizando además una
comparación entre dos métodos: P&T-CG-DCE y P&T-CG-EM. Con el primero analizan
Cloroformo, Tetracloroeteno y Tetraclorometano hasta unos niveles del orden de 10-12

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
119
μg/l, realizando una nueva comparación dentro de este método, entre una trampa de
gran volumen acoplada a una columna cromatográfica (0.53mm de diámetro) y una
trampa de pequeño volumen en combinación con una columna capilar (0.32mm de
diámetro); el resultado obtenido por esta última mejora los resultados en el límite de
detección en, aproximadamente, uno o dos ordenes de magnitud. Usando la segunda
técnica, los límites de detección aumentan tres órdenes de magnitud, pero analiza el
diclorometano además de las ya citadas. Las eficiencias alcanzadas por ambos métodos
rozan el 100%, siendo la repetibilidad muy buena, ya que la desviación estándar relativa
no supera el 3% para ninguno de los casos.
Los resultados antes vistos dan una idea de la potencia de los métodos que
combinen P&T con el DCE para el grupo de los organoclorados volátiles, siendo además
muy probable que se pueda aplicar al resto de sustancias, ya que todas ellas han sido
analizadas por otros autores usando el DCE, como Cristina Huertas, que usa EC-CG-DCE.
Huybrechts et al. (2000), que analiza todas las sustancias del grupo menos el
diclorometano y el 1,1,2-Tricloroeteno mediante P&T-CG-EM, obtiene unos resultados
que, aunque buenos (~ng/l), son más pobres que los anteriores. Lo que puede ser de
interés en el artículo, es que aumente el campo de aplicación del método sin pérdidas
relativas en sus resultados, lo que avala la idea de que se pueda aplicar el método para
0
1
2
3
4
5
6
7
Nº Sust.
IAD
-CG
-D
CE
EC
-CG
-D
CE
P&
T-C
G-
EM
P&
T-C
G-
DE
A
P&
T-C
G-
DC
E
Métodos
Sustanciasdonde noAlcanza elLímite
Nº sustanciasdonde alcanzael límite
Ilustración VII.3
Organoclorados Volátiles.

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
120
todas las sustancias.
El artículo de Wolska et al. (1998) ofrece unos resultados llamativos al compararlos
con los obtenidos por medio de EC-CG-DCE; usando un detector y columna similar y una
introducción de la muestra extremadamente simple, obtiene unos límites de detección
entre 1 y 4 ordenes de magnitud inferiores sobre muestras reales, estando todos por
debajo de los 0.1 μg/l excepto para el 1,2,4-triclorobenceno, para el cual obtiene 0.24. La
repetibilidad expresada en R.S.D permanece por debajo de 8.9%, valores obtenidos a
unas concentraciones cercanas al límite de detección, por lo que la precisión resulta
bastante satisfactoria. El coeficiente de determinación permanece por encima de 0.998,
por lo que indica una relación altamente lineal. La excepción en estos dos valores vuelve
a ser el 1,2,4-triclorobenceno, compuesto para el cual se obtienen unos valores algo
superiores.
La técnica usada para obtener estos valores es muy sencilla, y consiste en inyectar
la muestra acuosa de forma directa, lo que ahorra mucho tiempo de análisis al eliminar la
parte de la preparación previa, que es la más difícilmente automatizable. El procedimiento
recibe su nombre de inyección directa acuosa (“Direct Acuous Injection”), no siendo más
que eso, la inyección de una pequeña cantidad (1-2 μl) en una columna con una fase
estacionaria especialmente adaptada para separar los compuestos del agua. Esto elimina
el problema antes existente de separar en el proceso previo los analitos, ya que estos
poseen una gran afinidad con el agua. El inconveniente es que debido a la pequeña
cantidad de analitos inyectados, los límites de detección no son tan bajos como los
obtenidos por otros métodos en otras sustancias.
1.5. Otras sustancias.
En este grupo se encuentran las sustancias que no han podido incluirse en ninguno
de los anteriores. Como consecuencia inmediata de esto, nos encontramos con que el
número de métodos aplicables con buenos resultados aumenta con respecto a los otros
casos, siendo su aplicación mucho más específica. La excepción a esto es el ESBA-CG-EM,
donde Estefani Morales (2004) vuelve a aplicarlo con éxito a todas las sustancias.
Las sustancias incluidas son: Benceno, Hexaclorobenceno, Hexaclorobutadieno, 4-
Nonilfenoles, 4-tert-Octilfenol y Pentaclorobenceno.
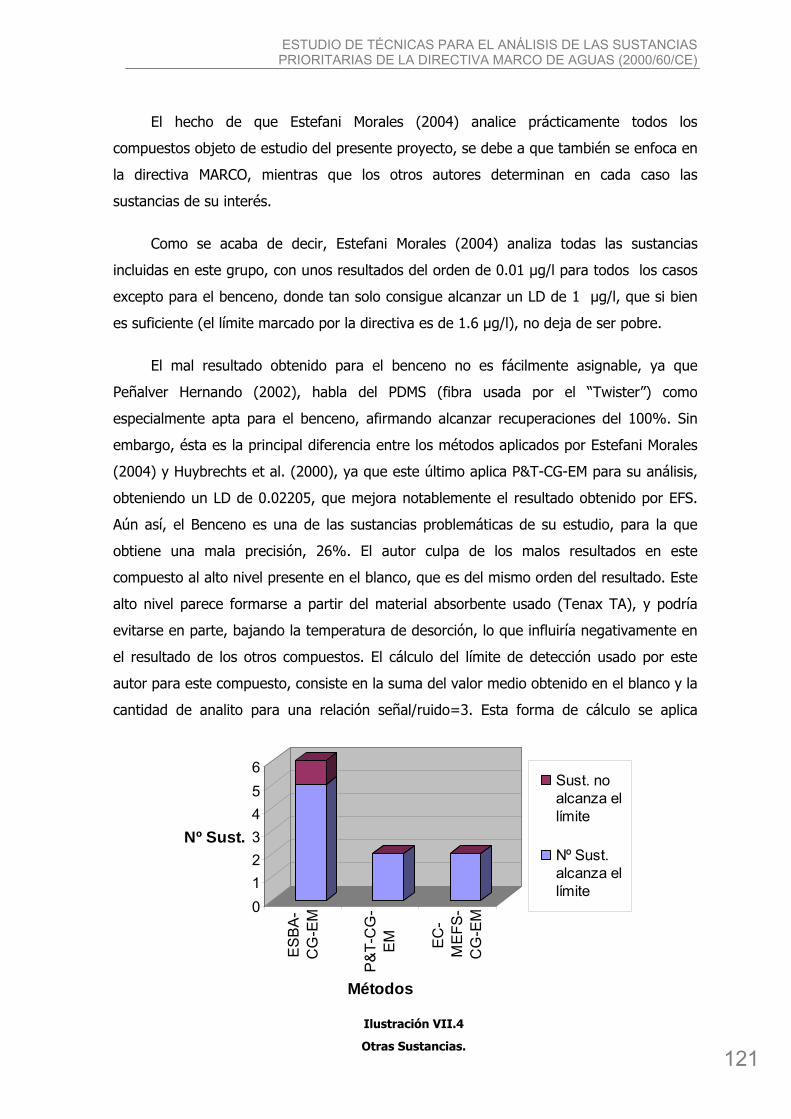
ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
121
El hecho de que Estefani Morales (2004) analice prácticamente todos los
compuestos objeto de estudio del presente proyecto, se debe a que también se enfoca en
la directiva MARCO, mientras que los otros autores determinan en cada caso las
sustancias de su interés.
Como se acaba de decir, Estefani Morales (2004) analiza todas las sustancias
incluidas en este grupo, con unos resultados del orden de 0.01 μg/l para todos los casos
excepto para el benceno, donde tan solo consigue alcanzar un LD de 1 μg/l, que si bien
es suficiente (el límite marcado por la directiva es de 1.6 μg/l), no deja de ser pobre.
El mal resultado obtenido para el benceno no es fácilmente asignable, ya que
Peñalver Hernando (2002), habla del PDMS (fibra usada por el “Twister”) como
especialmente apta para el benceno, afirmando alcanzar recuperaciones del 100%. Sin
embargo, ésta es la principal diferencia entre los métodos aplicados por Estefani Morales
(2004) y Huybrechts et al. (2000), ya que este último aplica P&T-CG-EM para su análisis,
obteniendo un LD de 0.02205, que mejora notablemente el resultado obtenido por EFS.
Aún así, el Benceno es una de las sustancias problemáticas de su estudio, para la que
obtiene una mala precisión, 26%. El autor culpa de los malos resultados en este
compuesto al alto nivel presente en el blanco, que es del mismo orden del resultado. Este
alto nivel parece formarse a partir del material absorbente usado (Tenax TA), y podría
evitarse en parte, bajando la temperatura de desorción, lo que influiría negativamente en
el resultado de los otros compuestos. El cálculo del límite de detección usado por este
autor para este compuesto, consiste en la suma del valor medio obtenido en el blanco y la
cantidad de analito para una relación señal/ruido=3. Esta forma de cálculo se aplica
0123456
Nº Sust.
ES
BA
-C
G-E
M
P&
T-C
G-
EM EC
-M
EFS
-C
G-E
M
Métodos
Sust. noalcanza ellímite
Nº Sust.alcanza ellímite
Ilustración VII.4
Otras Sustancias.

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
122
debido al alto valor del blanco y a que la variación de la concentración en el mismo no
fluctúa por encima del 30%.
Este mismo autor también analiza el Hexaclorobutadieno, ahora con mejores
resultados (LD: 0,00124 μg/l, Repetibilidad: 12.9%), alcanzando el límite marcado por la
directiva para esta sustancia (este límite no fue alcanzado por Estefani Morales (2004).
En dos estudios más se aplican la familia de técnicas de fraccionamiento por
absorción en combinación con CG-EM, son Gonçalves y Alpendurada (2004), ya
comentado en el apartado de pesticidas, y Mol et al. (2000), haciéndolo para el
Hexaclorobenceno y los 4-Nonilfenoles respectivamente. Ambos autores alcanzan un LD
aproximadamente de la mitad que Estefani Morales (2004), si bien la repetibilidad y
coeficiente de determinación del primero son relativamente malos. El análisis realizado
por Mol et al. (2000), encuentra diversos problemas en el análisis directo de los 4-
Nonilfenoles, entre ellos la adsorción y el ensanchamiento del pico cromatográfico, debido
a interacciones con otros compuestos, dependiendo esto de la matriz de la muestra y de
la antigüedad de la columna, lo que complica aun más la detección. Por ello decide
realizar una derivación del compuesto, lo que le permite evitar los problemas al hallar el
producto de esta reacción. La derivación se realizó añadiendo como reactivo derivador N-
metil-N-(tert.-butildimetiltrifluoroacetamida) (MTBSTFA), la solución se mantiene a 75º C
durante tres horas, y el detector se configuró en modo escaneo (esto sugiere una posible
Ilustración VII.1
Cromatogramas para el 4-n-octilfenol and 4-n-nonilfenol.

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
123
mejoría del LD usando otro método). De todas formas, el autor no recomienda esta
técnica para realizar análisis cuantitativos de esta sustancia, al ser determinada de forma
indirecta.
La otra técnica encontrada para el análisis del Hexaclorobenceno y
pentaclorobenceno, es la que consiste en acoplar el espacio en cabeza con la extracción
sobre fase sólida, y aparece desarrollada en el artículo de He et al. (2000). Este
acoplamiento entre ambas técnicas consiste en que la fibra está en contacto con la fase
gaseosa, disminuyendo el tiempo necesario para la extracción, al ser mayor la difusividad
en el gas que en el líquido. La extracción se realiza a 40º C, y en ella se obtienen
resultados que mejoran de forma importante los obtenidos a temperatura ambiente, para
los dos compuestos en cuestión. Esta técnica es especialmente apta para compuestos
volátiles o semivolátiles. Los LLDD son del orden de los conseguidos por los otros autores,
pero tiene una buena repetibilidad, 1.19% y 8.19% para ambos compuestos, obtenido
con el EM en modo RSI, y aporta una alternativa al análisis de estos compuestos.
1.6. Sustancias Prioritarias Orgánicas.
Tras el análisis de los distintos métodos, se puede llegar a la conclusión de que la
técnica más apta para el análisis de las sustancias prioritarias es el ESBA-CG-EM, que
analiza la mayoría de los compuestos, alcanzando los límites de detección marcados por el
estudio encargado para la directiva MARCO, éste sería, por tanto, el método base para el
análisis de las sustancias prioritarias orgánicas en aguas.
Habría que recordar de nuevo que el uso de la EM en tándem sería, en principio,
más recomendable que el uso de la EM simple, aunque la mejoría no esta muy
contrastada, y en algunos casos parece ser insuficiente para que compense el de este
equipo.
Para abarcar un número aún mayor de compuestos se podría complementar está
técnica con alguna otra. Los métodos más aptos para ello serían P&T-CG-EM como la que
mejor complementaría el resultado del EFS-CG-EM, ya que obtiene muy buenos
resultados con los organoclorados volátiles, además de ser capaz también de analizar el
hexaclorobutadieno; o IDA-CG-DCE, la cual complementaría a la EFS-CG-EM en el análisis
de las sustancias polares, obteniendo unos resultados algo peores que la anterior, pero de
aplicación más sencilla y obteniendo unos resultados muy satisfactorios.

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
124
La última alternativa del análisis de los organoclorados volátiles sería el uso de EC-
CG-DCE, pero teniendo en cuenta que esta técnica, aunque ofrece buenos resultados, no
alcanza el límite propuesto en el Diclorometano, pero consiguendo analizar el 1,1,2-
tricloroeteno, que no fue objeto de estudio en las otras técnicas mencionadas.
Con todo esto se analizarían la mayoría de las sustancias, y ya sólo quedarían el
Isoproturón y el Diurón, cuyo método propuesto sería la cromatografía líquida, y los
Cloropirifos, para los que no se encontró ninguna técnica capaz de alcanzar el límite propuesto.
2. Cromatografía de gases con espectrometría de masas (CG-EM).
En este apartado vamos a comentar en profundidad la técnica que hemos adoptado
en el apartado anterior como base, estudiando sus distintas variables para optimizar el
procedimiento lo máximo posible.
0
5
10
15
20
25
30
35Nº Sust.E
SB
A-C
G-E
M
P&
T-C
G-E
M
ME
FS-C
G-E
M-E
M
ME
FL-C
G-E
M
EFS
-CG
-DC
E
ME
FS-C
G-E
M
EFS
-CL-
EM
IAD
-CG
-DC
E
EC
-CG
-DC
E
P&
T-C
G-D
EA
P&
T-C
G-D
CE
Métodos
Sust. noalcanza ellímite
Sust. conlímite n/a
Nº Sust.alcanza ellímite
Ilustración VII.5 Sustancias Prioritarias.

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
125
El acoplamiento con la espectrometría de masas proporciona a la cromatografía de
gases la solución a su desventaja de su poca capacidad para el análisis cualitativo, por lo
que es usado principalmente en métodos de análisis que buscan la identificación de los
compuestos analizados. La complementariedad de ambas técnicas se basa en que la
cromatografía de gases diferencia claramente miembros de series homólogas con similar
espectro y permite la distinción de isómeros de posición, distinción difícil de llevar a cabo
por espectrometría de masas, mientras que dos sustancias de diferente estructura que se
solapan en un cromatograma pueden tener un espectro de masas completamente
distinto.
El uso práctico de este acoplamiento se ha extendido a partir de 1980 mediante una
serie de mejoras tanto en la cromatografía de gases (columnas capilares de sílice fundida,
fases inmovilizadas...) como en la espectrometría de masas (eficaces sistemas de vacío,
alta sensibilidad...).
Los avances informáticos han sido también fundamentales en el desarrollo de este
acoplamiento, permitiendo realizar el tratamiento de datos necesario para interpretar la
compleja información producida a un bajo coste. Un primer paso en el tratamiento es el
almacenamiento en forma digital de todos los espectros de masas registrados a lo largo
de la elución cromatográfica. A partir de estos datos, se reconstruye un perfil
cromatográfico similar a los producidos por cualquier detector a partir de la integración de
la señal de todos los fragmentos iónicos. Los espectros individuales se pueden interpretar
o comparar automáticamente con los almacenados en una biblioteca.
Las características mencionadas hacen del acoplamiento CG-EM el método analítico
más eficaz para el análisis cualitativo y cuantitativo de mezclas complejas de productos
volátiles.
2.1. Instrumentación
Desde el punto de vista instrumental, el acoplamiento CG-EM se presenta de
acuerdo con dos planteamientos distintos.
El primero de ellos es propio de grandes espectrómetros y considera que el
cromatógrafo de gases es un sistema de introducción de muestras en la cámara del

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
126
espectrómetro de masas equivalente a una sonda de introducción directa, un depósito
para introducción indirecta o a otras posibles modalidades.
El punto de vista opuesto considera al espectrómetro de masas como un detector
sofisticado del cromatógrafo de gases, e incluso lo denomina detector de masas o
detector de iones. Es propio de instrumentos sencillos y relativamente económicos, cuya
accesibilidad ha sido el origen del gran desarrollo de aplicaciones basadas en la
espectrometría de masas a finales de los años 80 y principios de los 90 (Wach, 1994).
Estos aparatos presentan prestaciones inferiores a las de los espectrómetros de
masas convencionales. Su intervalo de masas suele alcanzar sólo las 450-800 uma, lo que
impide el análisis de sustancias de alto peso molecular. El tamaño y la capacidad de
bombeo limitan el uso de distintos métodos de ionización de muestras, que generalmente
se reduce al impacto electrónico. El sistema de separación de masas suele ser un
cuadrupolo o una trampa de iones, que no permiten medidas de alta resolución. Estas
limitaciones, sin embargo, no son importantes en el uso típico de estos aparatos, que es
el análisis cualitativo de mezclas complejas separables por CG.
Si se fija el analizador en un valor m/z determinado en lugar de llevar a cabo un
barrido amplio, aparece a cambio una elevada sensibilidad hacia los compuestos que
presenten un fragmento de esa masa en su espectro, tanto mayor cuanto mayor sea la
intensidad relativa del fragmento y menor su presencia en el ruido de fondo del
instrumento. Este modo de operación, que combina la sensibilidad y selectividad propias
de la espectrometría de masas con la separación que proporciona la cromatografía de
gases, recibe el nombre de registro selectivo de iones (SIM, Selective Ion Monitoring) y
permite utilizar el acoplamiento CG-EM como un sistema de análisis cuantitativo de gran
sensibilidad e independiente de interferencias de compuestos que, aunque solapasen con
el pico correspondiente al compuesto buscado, no interferirían en su determinación si no
presentaran en su espectro el fragmento utilizado.
2.2. Procedimiento.
El procedimiento que se va a describir a continuación, está destinado al análisis de
compuestos semivolátiles con un equipo con extracción por absorción sobre barra
agitadora (ESBA), la entrada de muestras tiene lugar por una desorción térmica (DT) que

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
127
introduce la muestra en un cromatógrafo de gases de columna capilar (CG), con un
detector del tipo espectrómetro de masas (ESBA-DT-CG-EM).
El Procedimiento óptimo ha de permitir el análisis rutinario de un gran número de
muestras el mismo día del muestreo, de forma automatizada y con una sensibilidad lo
suficientemente elevada como para poder cumplir al menos con los criterios establecidos.
La absorción y fraccionamiento se realiza sobre polidimetilsiloxano (PDMS) que recubre
una barra magnética. Tras un cierto tiempo predeterminado de extracción, los analitos
atrapados sobre el polímero son desorbidos térmicamente en el inyector, produciéndose a
continuación un crioenfoque de la muestra que favorece un estrechamiento de las bandas
cromatográficas.
2.2.1. Material.
La extracción se realiza mediante la técnica de ESBA, en la que la cantidad de
polímero usado para la absorción es de 25-100 μl frente a los tan solo 0.5 μl que ofrece
MEFC; aparte de esta ventaja, el resto de mecanismos son similares entre ambas. En
especial vamos a realizar la extracción con “twisters” (nombre comercial de las barritas
agitadoras), usando el modelo de 10mm × 0.5mm (largo × espesor del recubrimiento),
siendo el proveedor Gerstel (Müllheim a/d Ruhr, Alemania).
Los “twisters” deben ser sometidos a una desorción térmica a 300ºC durante 4 h en
un flujo de helio de 50 ml / min antes del primer uso.
La extracción se realiza en Erlenmeyer de 100 ml de capacidad donde se introduce y
somete a agitación en un agitador magnético, también perteneciente a la casa Gerstel.
Para la elaboración de la matriz se usa cloruro de sodio y metanol CLAE-grade. Las
muestras se elaborarán con agua pura obtenida de un Mili-Q (Millipore, milford, MA, USA).
Los factores que afectan a la desorción son: la temperatura, el tiempo y el flujo de
helio en la desorción, así como la temperatura de crioenfoque en el inyector de muestras.
La optimización de estos factores se realizó comparando las áreas de los picos
obtenidos bajo las mismas condiciones: 20 ml de agua pura, y 20% NaCl, con barras de
10 mm × 0.5 mm PDMD absorbiendo durante 4 h a 1400 rpm.

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
128
2.2.2. Factores de influencia.
Según Baltussen et al. (1999), los factores que afectan a la extracción del analito en
equilibrio son solamente dos, el coeficiente de reparto octanol-agua (Kow) y el radio de
fase.
En la práctica es mayor el número de factores que afectan a la operación de
extracción: la adición de NaCl, la adición de metanol, el volumen de las muestras, el tipo
de barra agitadora usada, el perfil del equilibrio con el tiempo en la barra y la estabilidad
del analito en la misma; a los que hay que añadir los factores que determinan la
desorción térmica y el crioenfoque de la muestra en el sistema de inyección que son:
temperatura y tiempo de desorción, flujo de desorción, y temperatura de crioenfoque.
El estudio se realizó prestando especial atención a los analitos más polares ya que
es con estos con quienes demuestra una menor eficacia la extracción sobre PDMD
(apolar).
a. Temperatura de desorción (TDS):
Experimentalmente se puede comprobar que la temperatura de desorción no afecta a la calidad del cromatograma, obteniéndose áreas de picos similares; aunque si afecta al sangrado del recubrimiento de PDMS, siendo este sangrado menor a 280ºC por lo cual seleccionamos esta temperatura para la desorción térmica en el TDS.
b. Tiempo de desorción (TDS):
El suministrador de los twister recomienda usar flujos de desorción de 50 ml / min y, usando este flujo para nuestro procedimiento, se llega a la conclusión experimental de que un tiempo de desorción de 6 min es suficiente para extraer todos los compuestos de interés en nuestro caso, incluidos los menos volátiles, por lo que tomamos este tiempo para nuestro procedimiento de análisis (con 4min basta para simacina, lindano a-HCH; para p,p’.DDD, indenopireno, p,p’-DDE y benzo[k]fluoranteno, es necesario los 6 min).
c. Flujo de desorción (PTV):
Se puede comprobar que flujos pequeños de 20 ml / min son insuficientes para una adecuada transferencia de los compuestos menos volátiles como el benzo[k]fluoranteno, y que los flujos excesivos también resultan contraproducentes para el análisis, seguramente por impedir una correcta concentración de los compuestos más volátiles en el PTV. Un flujo de 75 ml/min de He puede resultar un buen compromiso entre ambos.

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
129
d. Temperatura de crioenfoque (PTV):
Las temperaturas relativamente elevadas como los 60ºC, resultan poco eficientes para atrapar los compuestos más volátiles como pueden ser el HCH y el lindano, que resultan tremendamente perjudicados.
Las temperaturas bajas para el resto de los compuestos dan lugar a áreas de pico inesperadamente pequeñas en el cromatograma, quizás por perdida de efectividad en la transferencia de los analitos atrapados a causa de la condensación del agua o la formación de hielo en el puerto de inyección, siendo el área del pico correspondiente al benzo[k]fluoranteno una de las que disminuye en mayor medida.
Se toma una temperatura de 20ºC como compromiso entre ambos efectos.
e. Adición de NaCl:
Debido a la naturaleza del material absorbente, la eficiencia de extracción será menor para los compuestos polares que para los no polares (Baltussen et al., 1999). La adición de sal al medio provocará un incremento de las fuerzas iónicas, mejorando la extracción de estos compuestos, especialmente para la simacina y la atracina, que pueden alcanzar 5 veces el área original con una adición del 20% de NaCl (Catherine et al., 1992; Eisert y Levsen, 1995; Valor et al., 1997).
En este caso también existe un efecto perjudicial para el otro grupo de sustancias, en este caso las apolares (Beltrán et al., 1998), aunque el uso de cantidades moderadas (5-10% NaCl) pueden incluso favorecer su análisis. Cantidades mayores (20-30%) pueden provocar pérdidas del área cromatográfica que superen ampliamente el 50% del valor original en el caso de indenolpireno, perileno, aldrin y p,p’-DDE.
Elegimos una adición de NaCl hasta el 20% como compromiso entre ambos efectos.
f. Efecto de la adición de metanol y la silanización en la absorción sobre las paredes de vidrio:
Los compuestos muy apolares, principalmente los HHAAPP de 5 ó 6 anillos, son adsorbidos por las paredes del instrumental de vidrio lo que limita en gran medida su recuperación (Baltussen et al., 1999; Heiden et al., 2001; Kicinski, 1993; Ackerman y Hurtubise, 2000; Benjits, 2001), recomendándose la adición de metanol para mantenerlos en solución. Esta adsorción depende del estado de la pared de vidrio, absorbiendo más aquellas dañadas por un limpiado abrasivo o ácidos fuertes.

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
130
Si bien la reproducibilidad no se ve afectada, no deja de ser un efecto adverso
a evitar, siendo la adición de un 5-10% de metanol una opción que disminuye la adsorción de los HHAAPP de 5 ó 6 anillos entre un 30 y un 100%, pero que disminuye la absorción de los compuestos más polares de un 30 a un 70%. Al ser nuestro análisis multielemental, esta opción resulta no muy recomendable.
Al contrario de lo que se pudiera pensar (Wennrich et al., 2001; Beltrán et al., 1998), la silanización de las paredes de vidrio no afecta a la adsorción una gran cantidad de compuesto como comprobó León et al., (2003). Sin embargo, esté problema afecta especialmente a los HHAAPP, compuestos para los que resulta conveniente la adición de Hiomina hasta una concentración de 10 μg/l (Kolahgar et al., 2002).
g. Volumen de muestra y cantidad de PDMS:
Para los compuestos más polares, principalmente la simacina y en menor grado la atracina, el incrementar el volumen de muestra tiene muy poco efecto sobre la cantidad extraída.
Para compuestos apolares, el aumento del volumen de muestra por encima de los 50 ml tiene un efecto limitado debido a que el tiempo para alcanzar el equilibrio es mayor que el tiempo que se mantiene la agitación. Además, la geometría del recipiente, la longitud del “twister” y el volumen de muestra son tres factores
Figura VII.2
Influencia del Metanol y la Hyamina en la recuperación de 100 ng/l PAH en soluciones acuosas Kolahgar et al. (2002).

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
131
interrelacionados que también afectan de manera sensible, siendo importante que busquemos la mayor eficiencia de agitado posible.
Elegimos un volumen de muestra de 100 ml y “twisters” de 20 mm × 0.5 mm para el análisis.
h. Tiempo de agitación:
La relación entre extracción y tiempo de agitación depende de la velocidad de agitación y la temperatura a la que se realiza, resultando que a temperatura ambiente, la absorción de los compuestos más volátiles tiene lugar en unas 4-6 horas, siendo necesarias 24 para los menos volátiles. Teniendo en cuenta lo importante de este tiempo para la aplicabilidad del método, seleccionamos 14 horas como compromiso entre ésta y un buen análisis.
Es muy importante que en condiciones donde no se alcance el equilibrio, se controlen los tiempos usados para realizar la absorción en orden de obtener una buena reproducibilidad del experimento.
i. Estabilidad de la muestra extraída:
El proceso de análisis dura unos 58 min, y teniendo en cuenta la posibilidad de introducir 20 muestras en el muestreados automático, hace un tiempo de unas 20 horas. Este tiempo no afecta prácticamente a la respuesta como se puede comprobar al ver que la respuesta tras 30 horas alcanza el 99% de la inicial. Para tiempos de hasta 63 horas si existe una disminución sensible de la recuperación (entre un 10 y un 25%). Estos resultados contradicen los previsibles teóricamente, ya que se podría esperar unas pérdidas de los compuestos más volátiles por evaporación.
j. Velocidad de agitación:
Una mayor velocidad de agitación disminuye el tiempo necesario para alcanzar el equilibrio, siendo el único inconveniente el obtener una rotación estable. Escogemos 900 rpm como velocidad alta y estable de agitación.

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
132
VIII. CONCLUSIONES.
En este apartado se muestran las conclusiones alcanzadas en la elaboración de
presente Proyecto Fin de Carrera acerca de los métodos para el análisis de las sustancias
prioritarias orgánicas en la directiva MARCO de aguas.
Para la realización del estudio se han consultado extensa bibliografía, usándose más
de 70 estudios como fuentes directas de datos en la elaboración de los resultados. Como
consecuencia de todo ello se puede llegar a la conclusión de que existe un método que
destaca sobre el resto en el análisis de las sustancias prioritarias, debido principalmente a
su gran campo de aplicación, pero también por su aplicabilidad y alto nivel de
automatización, este método es el ESBA-CG-EM, con el que se consiguen analizar a 29 de
las 40 sustancias prioritarias.
Los compuestos analizados dentro de los requerimientos por el método serían:
Alacloro, Aldrina, Atracina, Clorofenvifos, o,p-DDT, p,p-DDT, Dieldrina, Endosulfán(alfa),
Endrina, α-HCH, β-HCH, δ-HCH, Isodrina, Lindano (γ-HCH), Simacina, Trifluoralina,
Antraceno, Benzo(a)pireno, Benzo(b)fluoranteno, Benzo(g,h,i)perileno,
Benzo(k)fluranteno, Fluoranteno, Indenol(1,2,3-cd)pireno y Naftaleno, , Benceno,
Hexaclorobenceno, , 4-Nonilfenoles, 4-tert-Octilfenol y Pentaclorobenceno.
Tras esto nos quedarían 11 compuestos donde, o bien no se alcanzaba el límite de
detección requerido, o bien no se han encontrado referencias bibliográficas que usaran el
método en el análisis particular de los compuestos.
La técnica de P&T-CG-EM, aunque de elevado coste, sería la que mejor
complementaría el campo de aplicación de ESBA-CG-EM, consiguiendo analizar 7 de los
11 compuestos, 6 de ellos organoclorados volátiles, y siendo probable que pueda analizar
el organoclorado restante (1,1,2-tricloroeteno).
Las sustancias analizadas serían: Cloroformo (Triclorometano), Diclorometano, 1,2-
dicloroetano, Tetracloroeteno, Tetraclorometano, 1,2,4-triclorobenceno y
Hexaclorobutadieno.
Aplicándo ambos métodos, nos quedarían aún cuatro sustancias sin técnica
asignada para su análisis, las cuales pasamos a detallar a continuación:

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
133
o Isoproturón y diurón tendrían que ser analizadas por CL, técnica por
excelencia para el análisis de estas sustancias según la bibliografía
consultada.
o 1,1,2-tricloroeteno puede ser analizado por EC-CG-DCE.
o Los Cloropirifos no han podido ser analizados por ninguno de los métodos
presentes en la bibliografía dentro de los requisitos marcados, siendo por
tanto el único compuesto para el que no se han alcanzado los obtejivos
marcados.
Ésta sería la combinación más exhaustiva y con mejores posibilidades de análisis de
las sustancias prioritarias orgánicas, si bien existen otras alternativas que sustituyen a
P&T-CG-EM como método complementario de la ESBA-CG-EM:
o EC-CG-ECD: este método analizaría Cloroformo (Triclorometano), 1,2-
dicloroetano, Tetracloroeteno, Tetraclorometano, 1,2,4-triclorobenceno y
1,1,2-tricloroeteno, no alcanzando el límite en el Diclorometano ni
analizando el Hexaclorobutadieno. Su ventaja está en la mayor economía del
equipo.
o IAD-CG-DCE: este método, aunque con un campo de aplicación algo inferior,
es más simple y de más fácil aplicación que los anteriores; Cloroformo
(Triclorometano), 1,2-dicloroetano, Tetracloroeteno, Tetraclorometano, y
1,2,4-triclorobenceno, serían analizados satisfactoriamente, quedando sin
analizar Diclorometano, 1,1,2-tricloroeteno y Hexaclorobutadieno.
Con esto queda establecido para cada Sustancia Prioritaria, un método
recomendado de análisis, en función de las consideraciones tomadas en el Proyecto.

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
134
IX. ANEXOS

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
135
ABREVIATURAS
CG Cromatografía Gaseosa CL Cromatografía Líquida DCE Detector de Captura de Electrones DDL Detector de Diodos en Línea DF Detector de Fluorescencia EC Espacio en Cabeza EM Espectrómetro de Masas EM-EM Espectrómetro de Masas en Tándem EFL Extracción en Fase Líquida EFS Extracción en Fase Sóida ESBA Extracción Sobre Barra Agitadora IAD Inyección Directa Acuosa MEFL MicroExtracción en Fase Líquida MEFS MocroExtracción en Fase Sólida P&T Purga y Trampa

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
136
ANEXO 1.
PESTICIDAS SEMIVOLÁTILES
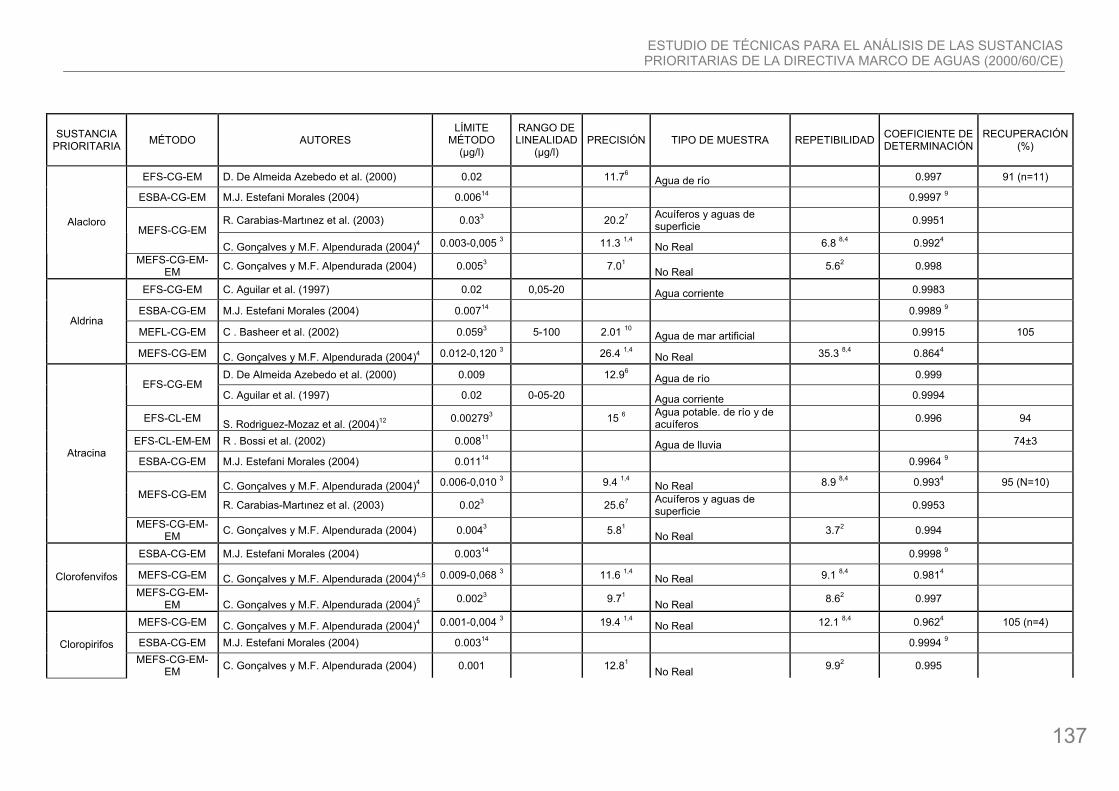
ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
137
SUSTANCIA PRIORITARIA MÉTODO AUTORES
LÍMITE MÉTODO
(μg/l)
RANGO DE LINEALIDAD
(μg/l) PRECISIÓN TIPO DE MUESTRA REPETIBILIDAD COEFICIENTE DE
DETERMINACIÓNRECUPERACIÓN
(%)
EFS-CG-EM D. De Almeida Azebedo et al. (2000) 0.02 11.76 Agua de río 0.997 91 (n=11)
ESBA-CG-EM M.J. Estefani Morales (2004) 0.00614 0.9997 9
R. Carabias-Martınez et al. (2003) 0.033 20.27 Acuíferos y aguas de superficie 0.9951
MEFS-CG-EM C. Gonçalves y M.F. Alpendurada (2004)4 0.003-0,005 3 11.3 1,4 No Real 6.8 8,4 0.9924
Alacloro
MEFS-CG-EM-EM C. Gonçalves y M.F. Alpendurada (2004) 0.0053 7.01 No Real 5.62 0.998
EFS-CG-EM C. Aguilar et al. (1997) 0.02 0,05-20 Agua corriente 0.9983
ESBA-CG-EM M.J. Estefani Morales (2004) 0.00714 0.9989 9
MEFL-CG-EM C . Basheer et al. (2002) 0.0593 5-100 2.01 10 Agua de mar artificial 0.9915 105 Aldrina
MEFS-CG-EM C. Gonçalves y M.F. Alpendurada (2004)4 0.012-0,120 3 26.4 1,4 No Real 35.3 8,4 0.8644
D. De Almeida Azebedo et al. (2000) 0.009 12.96 Agua de río 0.999 EFS-CG-EM
C. Aguilar et al. (1997) 0.02 0-05-20 Agua corriente 0.9994
EFS-CL-EM S. Rodriguez-Mozaz et al. (2004)12 0.002793 15 6 Agua potable. de río y de acuíferos 0.996 94
EFS-CL-EM-EM R . Bossi et al. (2002) 0.00811 Agua de lluvia 74±3
ESBA-CG-EM M.J. Estefani Morales (2004) 0.01114 0.9964 9
C. Gonçalves y M.F. Alpendurada (2004)4 0.006-0,010 3 9.4 1,4 No Real 8.9 8,4 0.9934 95 (N=10) MEFS-CG-EM
R. Carabias-Martınez et al. (2003) 0.023 25.67 Acuíferos y aguas de superficie 0.9953
Atracina
MEFS-CG-EM-EM C. Gonçalves y M.F. Alpendurada (2004) 0.0043 5.81 No Real 3.72 0.994
ESBA-CG-EM M.J. Estefani Morales (2004) 0.00314 0.9998 9
MEFS-CG-EM C. Gonçalves y M.F. Alpendurada (2004)4,5 0.009-0,068 3 11.6 1,4 No Real 9.1 8,4 0.9814 Clorofenvifos MEFS-CG-EM-
EM C. Gonçalves y M.F. Alpendurada (2004)5 0.0023 9.71 No Real 8.62 0.997
MEFS-CG-EM C. Gonçalves y M.F. Alpendurada (2004)4 0.001-0,004 3 19.4 1,4 No Real 12.1 8,4 0.9624 105 (n=4)
ESBA-CG-EM M.J. Estefani Morales (2004) 0.00314 0.9994 9 Cloropirifos MEFS-CG-EM-
EM C. Gonçalves y M.F. Alpendurada (2004) 0.001 12.81 No Real 9.92 0.995
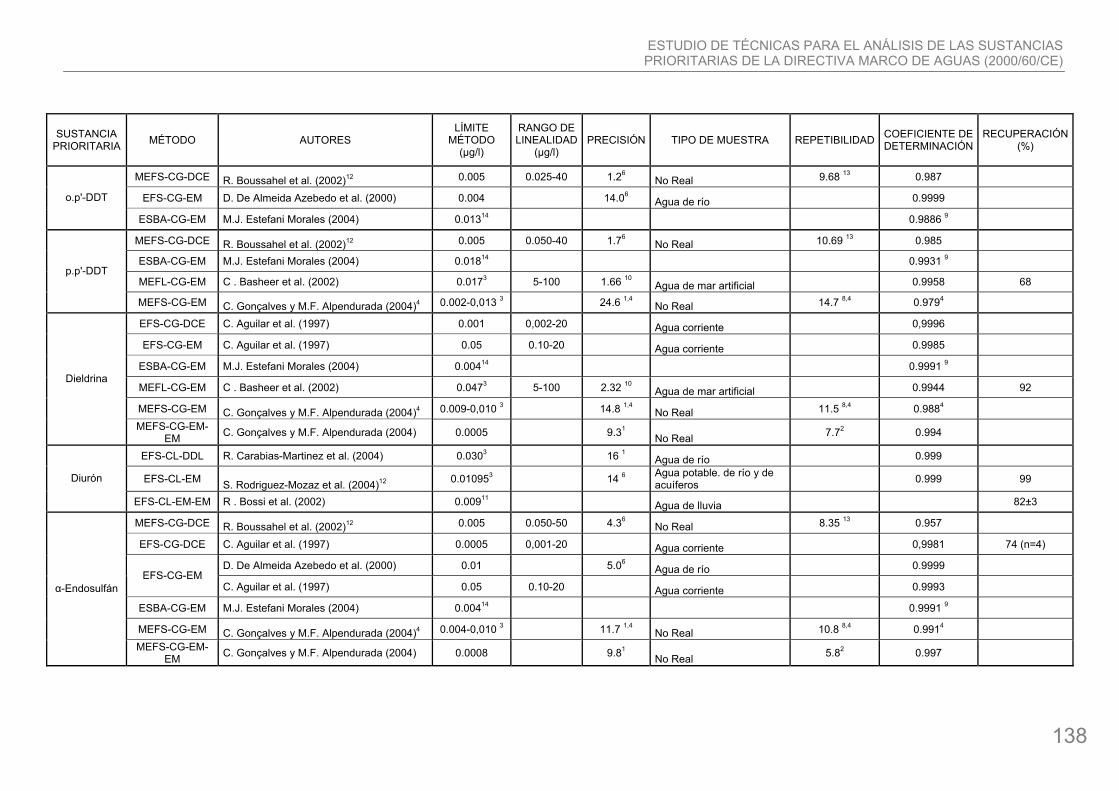
ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
138
SUSTANCIA PRIORITARIA MÉTODO AUTORES
LÍMITE MÉTODO
(μg/l)
RANGO DE LINEALIDAD
(μg/l) PRECISIÓN TIPO DE MUESTRA REPETIBILIDAD COEFICIENTE DE
DETERMINACIÓNRECUPERACIÓN
(%)
MEFS-CG-DCE R. Boussahel et al. (2002)12 0.005 0.025-40 1.26 No Real 9.68 13 0.987
EFS-CG-EM D. De Almeida Azebedo et al. (2000) 0.004 14.06 Agua de río 0.9999 o.p'-DDT
ESBA-CG-EM M.J. Estefani Morales (2004) 0.01314 0.9886 9
MEFS-CG-DCE R. Boussahel et al. (2002)12 0.005 0.050-40 1.76 No Real 10.69 13 0.985
ESBA-CG-EM M.J. Estefani Morales (2004) 0.01814 0.9931 9
MEFL-CG-EM C . Basheer et al. (2002) 0.0173 5-100 1.66 10 Agua de mar artificial 0.9958 68 p.p'-DDT
MEFS-CG-EM C. Gonçalves y M.F. Alpendurada (2004)4 0.002-0,013 3 24.6 1,4 No Real 14.7 8,4 0.9794
EFS-CG-DCE C. Aguilar et al. (1997) 0.001 0,002-20 Agua corriente 0,9996
EFS-CG-EM C. Aguilar et al. (1997) 0.05 0.10-20 Agua corriente 0.9985
ESBA-CG-EM M.J. Estefani Morales (2004) 0.00414 0.9991 9
MEFL-CG-EM C . Basheer et al. (2002) 0.0473 5-100 2.32 10 Agua de mar artificial 0.9944 92
MEFS-CG-EM C. Gonçalves y M.F. Alpendurada (2004)4 0.009-0,010 3 14.8 1,4 No Real 11.5 8,4 0.9884
Dieldrina
MEFS-CG-EM-EM C. Gonçalves y M.F. Alpendurada (2004) 0.0005 9.31 No Real 7.72 0.994
EFS-CL-DDL R. Carabias-Martinez et al. (2004) 0.0303 16 1 Agua de río 0.999
EFS-CL-EM S. Rodriguez-Mozaz et al. (2004)12 0.010953 14 6 Agua potable. de río y de acuíferos 0.999 99 Diurón
EFS-CL-EM-EM R . Bossi et al. (2002) 0.00911 Agua de lluvia 82±3
MEFS-CG-DCE R. Boussahel et al. (2002)12 0.005 0.050-50 4.36 No Real 8.35 13 0.957
EFS-CG-DCE C. Aguilar et al. (1997) 0.0005 0,001-20 Agua corriente 0,9981 74 (n=4)
D. De Almeida Azebedo et al. (2000) 0.01 5.06 Agua de río 0.9999 EFS-CG-EM
C. Aguilar et al. (1997) 0.05 0.10-20 Agua corriente 0.9993
ESBA-CG-EM M.J. Estefani Morales (2004) 0.00414 0.9991 9
MEFS-CG-EM C. Gonçalves y M.F. Alpendurada (2004)4 0.004-0,010 3 11.7 1,4 No Real 10.8 8,4 0.9914
α-Endosulfán
MEFS-CG-EM-EM C. Gonçalves y M.F. Alpendurada (2004) 0.0008 9.81 No Real 5.82 0.997

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
139
ESBA-CG-EM M.J. Estefani Morales (2004) 0.00614 0.9996 9
MEFL-CG-EM C . Basheer et al. (2002) 0.0333 5-100 1.93 10 Agua de mar artificial 0.9995 98 Endrina
MEFS-CG-EM C. Gonçalves y M.F. Alpendurada (2004)4 0.019-0,060 3 18.8 1,4 No Real 14.6 8,4 0.9844
EFS-CG-DCE C. Aguilar et al. (1997) 0.0002 0.0005-20 Agua de río 0.9993
D. De Almeida Azebedo et al. (2000) 0.02 16.06 Agua de río 0.999 EFS-CG-EM
C. Aguilar et al. (1997) 0.02 0.05-20 Agua corriente 0.9993
ESBA-CG-EM M.J. Estefani Morales (2004) 0.00714 0.9998 9
α-HCH
MEFL-CG-EM C . Basheer et al. (2002) 0.0173 5-100 13.72 10 Agua de mar artificial 0.9995 139
EFS-CG-EM D. De Almeida Azebedo et al. (2000) 0.02 13.06 Agua de río 0.9985
ESBA-CG-EM M.J. Estefani Morales (2004) 0.00614 0.9997 9 β-HCH
MEFL-CG-EM C . Basheer et al. (2002) 0.0293 5-100 10.29 10 Agua de mar artificial 0.9963 83
EFS-CG-DCE C. Aguilar et al. (1997) 0.0002 0,0005-20 Agua corriente 0.9996
D. De Almeida Azebedo et al. (2000) 0.01 12.06 Agua de río 0.9994 EFS-CG-EM
C. Aguilar et al. (1997) 0.02 0.05-20 Agua corriente 0.9999 δ-HCH
ESBA-CG-EM M.J. Estefani Morales (2004) 0.00514 0.9997 9
Isodrina ESBA-CG-EM M.J. Estefani Morales (2004) 0.00814 0.997 9
EFS-CL-DDL R. Carabias-Martinez et al. (2004) 0.0243 11 1 Agua de río 1.000
EFS-CL-EM S. Rodriguez-Mozaz et al. (2004)12 0.004893 12 6 Agua potable. de río y de acuíferos 0.999 99
EFS-CL-EM-EM R . Bossi et al. (2002) 0.00611 Agua de lluvia 79±4 Isoproturón
MEFS-CG-EM R. Carabias-Martınez et al. (2003) 0.023 18.67 Acuíferos y aguas de superficie
EFS-CG-DCE C. Aguilar et al. (1997) 0.0002 0,0005-20 Agua corriente 0,9971
D. De Almeida Azebedo et al. (2000) 0.02 31.06 Agua de río 0.999 EFS-CG-EM
C. Aguilar et al. (1997) 0.02 0.05-20 Agua corriente 0.9991
ESBA-CG-EM M.J. Estefani Morales (2004) 0.00814 0.9999 9
MEFL-CG-EM C . Basheer et al. (2002) 0.0133 5-100 14.00 10 Agua de mar artificial 0.9991 74
Lindano (γ-HCH)
MEFS-CG-DCE R. Boussahel et al. (2002)12 0.025 0.025-50 3.36 No Real 10.22 13 0.986
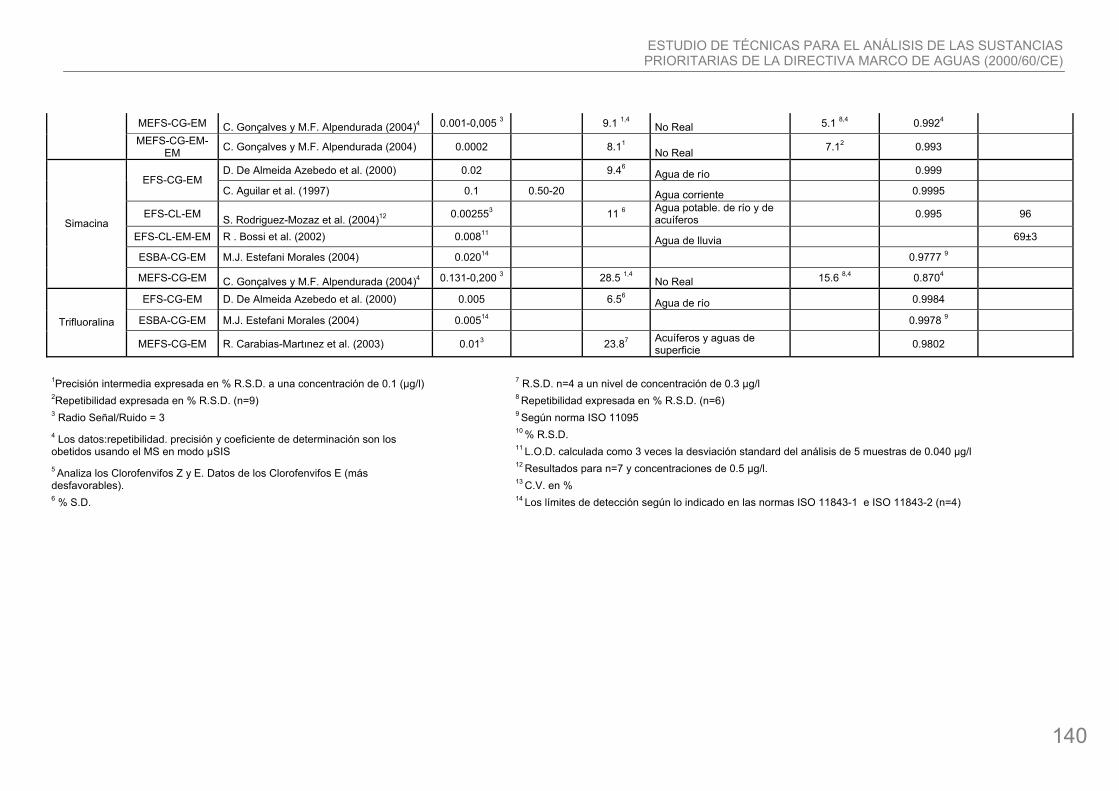
ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
140
MEFS-CG-EM C. Gonçalves y M.F. Alpendurada (2004)4 0.001-0,005 3 9.1 1,4 No Real 5.1 8,4 0.9924
MEFS-CG-EM-EM C. Gonçalves y M.F. Alpendurada (2004) 0.0002 8.11 No Real 7.12 0.993
D. De Almeida Azebedo et al. (2000) 0.02 9.46 Agua de río 0.999 EFS-CG-EM
C. Aguilar et al. (1997) 0.1 0.50-20 Agua corriente 0.9995
EFS-CL-EM S. Rodriguez-Mozaz et al. (2004)12 0.002553 11 6 Agua potable. de río y de acuíferos 0.995 96
EFS-CL-EM-EM R . Bossi et al. (2002) 0.00811 Agua de lluvia 69±3
ESBA-CG-EM M.J. Estefani Morales (2004) 0.02014 0.9777 9
Simacina
MEFS-CG-EM C. Gonçalves y M.F. Alpendurada (2004)4 0.131-0,200 3 28.5 1,4 No Real 15.6 8,4 0.8704
EFS-CG-EM D. De Almeida Azebedo et al. (2000) 0.005 6.56 Agua de río 0.9984
ESBA-CG-EM M.J. Estefani Morales (2004) 0.00514 0.9978 9 Trifluoralina
MEFS-CG-EM R. Carabias-Martınez et al. (2003) 0.013 23.87 Acuíferos y aguas de superficie 0.9802
1Precisión intermedia expresada en % R.S.D. a una concentración de 0.1 (μg/l) 7 R.S.D. n=4 a un nivel de concentración de 0.3 μg/l 2Repetibilidad expresada en % R.S.D. (n=9) 8 Repetibilidad expresada en % R.S.D. (n=6) 3 Radio Señal/Ruido = 3 9 Según norma ISO 11095
10 % R.S.D. 4 Los datos:repetibilidad. precisión y coeficiente de determinación son los obetidos usando el MS en modo μSIS 11 L.O.D. calculada como 3 veces la desviación standard del análisis de 5 muestras de 0.040 μg/l
12 Resultados para n=7 y concentraciones de 0.5 μg/l. 5 Analiza los Clorofenvifos Z y E. Datos de los Clorofenvifos E (más desfavorables). 13 C.V. en % 6 % S.D. 14 Los límites de detección según lo indicado en las normas ISO 11843-1 e ISO 11843-2 (n=4)

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
ANEXO 2.
HIDROCARBUROS POLICÍCLICOS

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
142
SUSTANCIA PRIORITARIA MÉTODO AUTORES LÍMITE MÉTODO (μg/l) RANGO DE LINEALIDAD (μg/l) TIPO DE MUESTRA REPETIBILIDAD
COEFICIENTE DE
DETERMINACIÓRECUPERACIÓN (%)
M.J. Estefani Morales (2004) 0.002 4 2 6 0,9999 5
B. Kolahgar et al. (2002) 0.0012 1,3 15 2 0,99865 99M.J. Estefani Morales
(2004) 0.008 4 8 6 0,9984 5
B. Kolahgar et al. (2002) 0.0012 1,3 10 2 0,92543 100
EFS-CL-DF R.A. Gimeno et al. (2002) 0.001 1 0,004-1 Agua de Mar >0,9996M.J. Estefani Morales
(2004) 0.014 4 7 6 0,9859 5
B. Kolahgar et al. (2002) 0.0003 1,3 10 2 0,92903 100
EFS-CL-DF R.A. Gimeno et al. (2002) 0.008 1 0,02-5 Agua de Mar >0,9996M.J. Estefani Morales
(2004) 0.012 4 6 6 0,9875 5
B. Kolahgar et al. (2002) 0.0002 1,3 14 2 0,99848 100
EFS-CL-DF R.A. Gimeno et al. (2002) 0.06 1 0,1-10 Agua de Mar >0,9996M.J. Estefani Morales
(2004) 0.011 4 10 6 0,9993 5
B. Kolahgar et al. (2002) 0.0005 1,3 6 2 0,97357 100
EFS-CL-DF R.A. Gimeno et al. (2002) 0.003 1 0,007-5 Agua de Mar >0,9996M.J. Estefani Morales
(2004) 0.003 4 2 6 0,9996 5
B. Kolahgar et al. (2002) 0.0001 1,3 10 2 0,99946 100
EFS-CL-DF R.A. Gimeno et al. (2002) 0.004 1 0,008-10 Agua de Mar >0,9996M.J. Estefani Morales
(2004) 0.007 4 6 6 0,9948 5
B. Kolahgar et al. (2002) 0.0014 1,3 12 2 0,99894 100
EFS-CL-DF R.A. Gimeno et al. (2002) 0.1 1 0,3-10 Agua de Mar >0,9996M.J. Estefani Morales
(2004) 0.011 4 7 6 0,9869 5
B. Kolahgar et al. (2002) 0.0005 1,3 12 2 0,98782 83
5 Según norma ISO 11095 6 sr
ESBA-CG-EM
ESBA-CG-EM
ESBA-CG-EM
ESBA-CG-EM
ESBA-CG-EM
ESBA-CG-EM
1 Límite para σ=32Repetibilidad expresada en % para una concentración de 10 ng/l3 Límite estadístico Teórico
Antraceno
Benzo(a)pireno
Benzo(b)fluoranteno
Benzo(g,h,i)perileno
Benzo(k)fluranteno
Fluoranteno
Indenol(1,2,3-cd)pirenoESBA-CG-
EM
ESBA-CG-EM
4 Los límites de detección según lo indicado en las normas ISO 11843-1 e ISO 11843-2 (n=4)
Naftaleno

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
ANEXO 3.
ORGANOCLORADOS VOLÁTILES
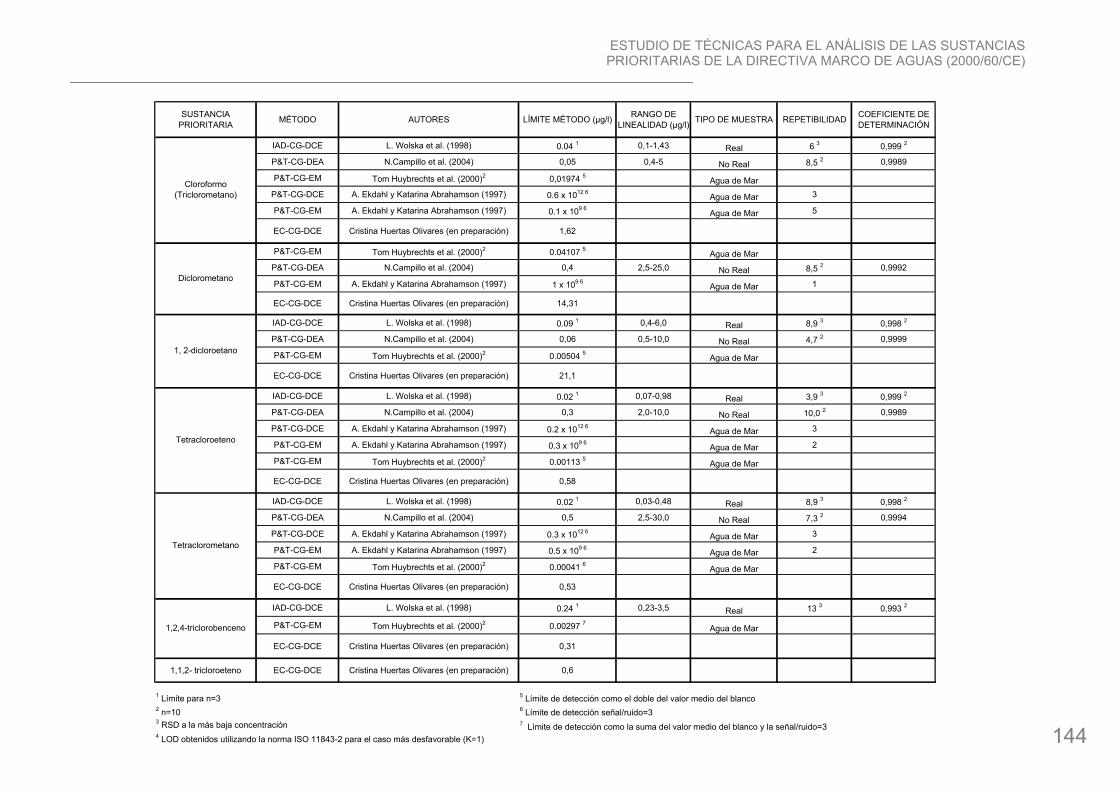
ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
144
SUSTANCIA
PRIORITARIA MÉTODO AUTORES LÍMITE MÉTODO (μg/l) RANGO DE LINEALIDAD (μg/l) TIPO DE MUESTRA REPETIBILIDAD COEFICIENTE DE
DETERMINACIÓN
IAD-CG-DCE L. Wolska et al. (1998) 0.04 1 0,1-1,43 Real 6 3 0,999 2
P&T-CG-DEA N.Campillo et al. (2004) 0,05 0,4-5 No Real 8,5 2 0,9989
P&T-CG-EM Tom Huybrechts et al. (2000)2 0,01974 5 Agua de MarP&T-CG-DCE A. Ekdahl y Katarina Abrahamson (1997) 0.6 x 1012 6
Agua de Mar 3
P&T-CG-EM A. Ekdahl y Katarina Abrahamson (1997) 0.1 x 109 6Agua de Mar 5
EC-CG-DCE Cristina Huertas Olivares (en preparación) 1,62
P&T-CG-EM Tom Huybrechts et al. (2000)2 0.04107 5 Agua de MarP&T-CG-DEA N.Campillo et al. (2004) 0,4 2,5-25,0 No Real 8,5 2 0,9992
P&T-CG-EM A. Ekdahl y Katarina Abrahamson (1997) 1 x 109 6Agua de Mar 1
EC-CG-DCE Cristina Huertas Olivares (en preparación) 14,31
IAD-CG-DCE L. Wolska et al. (1998) 0.09 1 0,4-6,0 Real 8,9 3 0,998 2
P&T-CG-DEA N.Campillo et al. (2004) 0,06 0,5-10,0 No Real 4,7 2 0,9999
P&T-CG-EM Tom Huybrechts et al. (2000)2 0.00504 5 Agua de Mar
EC-CG-DCE Cristina Huertas Olivares (en preparación) 21,1
IAD-CG-DCE L. Wolska et al. (1998) 0.02 1 0,07-0,98 Real 3,9 3 0,999 2
P&T-CG-DEA N.Campillo et al. (2004) 0,3 2,0-10,0 No Real 10,0 2 0,9989
P&T-CG-DCE A. Ekdahl y Katarina Abrahamson (1997) 0.2 x 1012 6Agua de Mar 3
P&T-CG-EM A. Ekdahl y Katarina Abrahamson (1997) 0.3 x 109 6Agua de Mar 2
P&T-CG-EM Tom Huybrechts et al. (2000)2 0.00113 5 Agua de Mar
EC-CG-DCE Cristina Huertas Olivares (en preparación) 0,58
IAD-CG-DCE L. Wolska et al. (1998) 0.02 1 0,03-0,48 Real 8,9 3 0,998 2
P&T-CG-DEA N.Campillo et al. (2004) 0,5 2,5-30,0 No Real 7,3 2 0,9994
P&T-CG-DCE A. Ekdahl y Katarina Abrahamson (1997) 0.3 x 1012 6Agua de Mar 3
P&T-CG-EM A. Ekdahl y Katarina Abrahamson (1997) 0.5 x 109 6Agua de Mar 2
P&T-CG-EM Tom Huybrechts et al. (2000)2 0.00041 6 Agua de Mar
EC-CG-DCE Cristina Huertas Olivares (en preparación) 0,53
IAD-CG-DCE L. Wolska et al. (1998) 0.24 1 0,23-3,5 Real 13 3 0,993 2
P&T-CG-EM Tom Huybrechts et al. (2000)2 0.00297 7 Agua de Mar
EC-CG-DCE Cristina Huertas Olivares (en preparación) 0,31
1,1,2- tricloroeteno EC-CG-DCE Cristina Huertas Olivares (en preparación) 0,6
1 Límite para n=32 n=10
4 LOD obtenidos utilizando la norma ISO 11843-2 para el caso más desfavorable (K=1)
5 Límite de detección como el doble del valor medio del blanco6 Límite de detección señal/ruido=37 Límite de detección como la suma del valor medio del blanco y la señal/ruido=3
Tetraclorometano
1,2,4-triclorobenceno
3 RSD a la más baja concentración
Cloroformo (Triclorometano)
1, 2-dicloroetano
Diclorometano
Tetracloroeteno

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
ANEXO 4.
OTRAS SUSTANCIAS
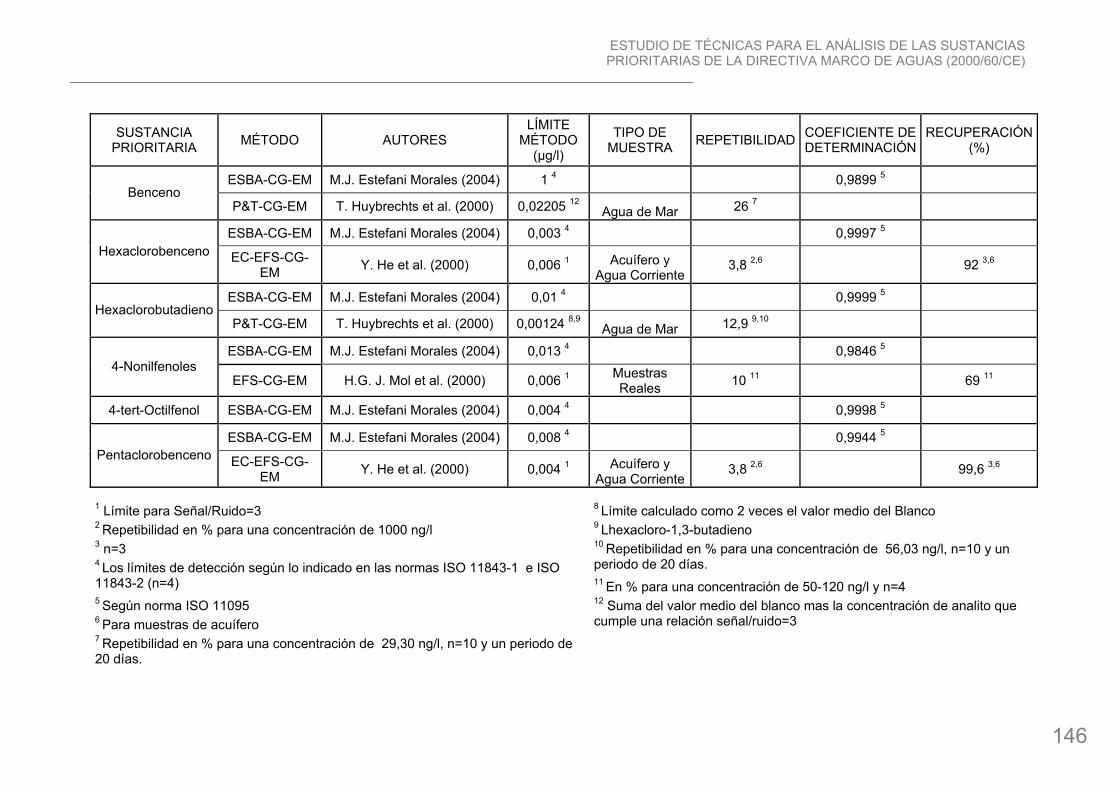
ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
146
SUSTANCIA PRIORITARIA MÉTODO AUTORES
LÍMITE MÉTODO
(μg/l)
TIPO DE MUESTRA REPETIBILIDAD COEFICIENTE DE
DETERMINACIÓNRECUPERACIÓN
(%)
ESBA-CG-EM M.J. Estefani Morales (2004) 1 4 0,9899 5 Benceno
P&T-CG-EM T. Huybrechts et al. (2000) 0,02205 12 Agua de Mar 26 7
ESBA-CG-EM M.J. Estefani Morales (2004) 0,003 4 0,9997 5 Hexaclorobenceno EC-EFS-CG-
EM Y. He et al. (2000) 0,006 1 Acuífero y Agua Corriente
3,8 2,6 92 3,6
ESBA-CG-EM M.J. Estefani Morales (2004) 0,01 4 0,9999 5 Hexaclorobutadieno
P&T-CG-EM T. Huybrechts et al. (2000) 0,00124 8,9 Agua de Mar 12,9 9,10 ESBA-CG-EM M.J. Estefani Morales (2004) 0,013 4 0,9846 5
4-Nonilfenoles EFS-CG-EM H.G. J. Mol et al. (2000) 0,006 1 Muestras
Reales 10 11 69 11
4-tert-Octilfenol ESBA-CG-EM M.J. Estefani Morales (2004) 0,004 4 0,9998 5
ESBA-CG-EM M.J. Estefani Morales (2004) 0,008 4 0,9944 5 Pentaclorobenceno EC-EFS-CG-
EM Y. He et al. (2000) 0,004 1 Acuífero y Agua Corriente
3,8 2,6 99,6 3,6
1 Límite para Señal/Ruido=3 8 Límite calculado como 2 veces el valor medio del Blanco 2 Repetibilidad en % para una concentración de 1000 ng/l 9 Lhexacloro-1,3-butadieno 3 n=3 10 Repetibilidad en % para una concentración de 56,03 ng/l, n=10 y un
periodo de 20 días. 4 Los límites de detección según lo indicado en las normas ISO 11843-1 e ISO 11843-2 (n=4) 11 En % para una concentración de 50-120 ng/l y n=4 5 Según norma ISO 11095 6 Para muestras de acuífero
12 Suma del valor medio del blanco mas la concentración de analito que cumple una relación señal/ruido=3
7 Repetibilidad en % para una concentración de 29,30 ng/l, n=10 y un periodo de 20 días.

ESTUDIO DE TÉCNICAS PARA EL ANÁLISIS DE LAS SUSTANCIAS PRIORITARIAS DE LA DIRECTIVA MARCO DE AGUAS (2000/60/CE)
X. BIBLIOGRAFÍA.

- Ackerman A.H., Hurtubise R.J., Talanta 52 (2000) 853. - Aguilar C., Borrull F., Marcé R.M., Journal of Chromatography A, 771 (1997) 221-231. - Andersson Ö., Blomkwist G., Chemosphere 10 (1981) 1051. - Anon. Report QUASIMEME Workshop on PCB analisys, Rodkilde, Denmark, June, FRS Marine
Laboratory, Aberdeen, UK, 1995. - Antle R.E., Goldberg A.P., Snyder L.R., Journal of Chromatography 321: 1 (1985). - Ariese F., Kok S.J.,Verkaik M., Gooijer C., Velthorst N.H., Hofstraat J.W., Aquat. Toxicol. 26 (1993)
273. - Arthur C., Killam L., Buchholz K., Berg J., Pawliszyn J., Anal. Chem. 64 (1992) 1960. - Arthur C.L., Pawliszyn J., Anal. Chem. 62 (1998) 1960. - Azebedo DA., Lacorte S., Vinhas T., Viana P., Barceló D., Journal of Chromatography A. 879
(2000) 13-26. - Bagheri H., Saber A., Mousavi S.R., Journal of Chromatography A, 1046 (2004) 27–33. - Baker B., Bull. Environ. Contam. Toxicol. 10 (1973) 279. - Baltussen B., Sandra P., David F., Cramers C., J. Microcol. Sep 11 (1999) 737. - Baltussen E., Cramers C.A., Sandra P.J.F., Anal. Bioanal. Chem. 373 (2002) 3. - Baltussen E., Janssen H.-G., Sandra P., Cramers C.A., J. High Resolut. Chromatogr. 20 (1997)
385. - Baltussen E., Janssen H.-G., Sandra P., Cramers C.A., J. High Resolut. Chromatogr. 20 (1997)
395. - Bamford H.A., Bezabeh D.Z., Schantz M.M., Wise S.A., Baker J.E., Chemosphere 50 (2003) 575. - Banks K.E., Hernandez S., Talanta 61 (2003) 257-265. - Barceló D., Analyst, 116 (1991) 681. - Basheer C., Lee H.K., Obbard J.P., Journal of Chromatography A, 968 (2002) 191–199. - Beliaeff B., 0'ConnorT.P., Munschy C., Raffin C., Claisse D., Environ. Toxicol. Chem. 21 (2002)
1783. - Beltrán J., López F.J., Cepria O., Hernandez F., Journal of Chromatography A 808 (1998) 257. - Bicchi C., D´Amato A., David F., Sandra P., Flavour Frag. J. 2 (1987) 49. - Bicchi C., D´Amato A., David F.,Sandra P., Flavour Frag. J. 3 (1988) 143. - Bij K.E., Horváth Cs., Melander W.R., Nahum A., Journal of Chromatography 203: 65 (1981). - Biziuk M., Namieśnik J., Czerwifiski J., Gorlo D., Makuch B., Janicki W., Polkowska Z., Wolska L.,
Journal of Chromatography A, 733 (1996) 171-183. - Björklund J., Gas Chromatography and mass spectrometry of polybrominated diphenyl ethers, PhD
thesis, Stockholm University, Stockholm, Sweden, 2003. - Bjørseth A. (Ed.), Handbook of Polycyclic Aromatic Hydrocarbons. Marcel Dekker, New York, 1983,
p. 397. - BlascoC., Font G., Pico Y., Journal of Chromatography A, 970 (2002) 201–212. - Blomberg S., Roeraade J., HRC&CC 11 (1988) 457. - Blomberg S., Roeraade J., Journal of Chromatography 394 (1987) 443. - Boer H.. Vapour Phase Chromatography H. Desty Ed., Butterworths, London 1957, p. 169. - Bossi R., Vejrup K.V., Mogensen B.B., Asman W.A.H., Journal of Chromatography A, 957 (2002)
27–36. - Boussahel R., Bouland S., Moussaoui K.M., Baudu M., Montiel A., Water Research 36 (2002) 1909-
1911. - Bøwadt S., Johansson B., Fruekilde P., Hansen M., Zilk D., Larsen B., de Boer J., Journal of
Chromatography A 675 (1994) 189. - Braun P., Moeder M., Schrader St., Popp P., Kuschk P., Engewald W., Journal of Chromatography
A, 988 (2003) 41–51. - Brilis G.M., Marsden P.J., Chemosphere 21 (1990) 91. - Burger B.V., LeRoux M., Burger W.J.G., J. High Resolut. Chromatogr. 12 (1990) 777. - Burger V., Munro Z., Journal of Chromatography 370 (1986) 449. - Caldas E.D., Coelho R., Souza L.C.K.R., Silva S.C., Bull. Environ. Contain. Toxicol. 62 (1999) 199. - Carabias-Martínez R., García-Hermida C., Rodríguez-Gonzalo E., Soriano-Bravo F.E., Hernández-
Méndez J., Journal of Chromatography A, 1002 (2003) 1–12.

- Carabias-Martínez R., Rodríguez-Gonzalo E., Herrero-Hernández E., Hernández-Méndez J., Analytica Chimica Acta 517 (2004) 71–79.
- Carabias-Martínez R., Rodríguez-Gonzalo E., Revilla-Ruiz P., Journal of Chromatography A, 1056 (2004) 131–138.
- Caro E., Marcé R.M., Cormack P.A.G., Sherrington D.C., Borrull F., Journal of Chromatography A, 1047 (2004) 175–180.
- Carpinteiro J., Quintana J.B., Rodríguez I., Carro A.M., Lorenzo R.A., Cela R., Journal of Chromatography A, 1056 (2004) 179–185.
- Castillo M., Puig D., Barceló D., Journal of Chromatography A, 778 (1997) 301-311. - Catherine L., Killam M., Buchholz D., Pawliszyn J., Anal. Chem. 64 (1992) 1960. - Clark J., Barber R.C., Boudreau C., Buchinger F., Crawford J.E., Gulick S., Hardy J.C., Heinz A.,
Lee J.K.P., Moore R.B., Savard G., Seweryniak D., Sharma K.S., Sprouse G., Vaz J., Wang J.C., Zhou Z., Nuclear Instruments and Methods in Physics Research B 204 (2003) 487–491.
- Collings B.A., Douglas D.J., International Journal of Mass Spentrometry and Ion Provesses 163 (1997) 121-127.
- ConderJ.R., Young C.L., Physicochemical Measurement by Gas Chromatography. J.Wiley & Sons. Chichester(1979).
- Covaci A., de Boer J., Ryan J.J., S. Voorspoels, S. Schepens, Anal. Chem. 74 (2002) 790. - Covaci A., Voorspoels S., de Boer J., Environ. Int. Volume 29, Issue 6, September 2003, Pages
735-756. - Chaintreau A., Flavour Frag. J. 16 (2001) 136. - Chiadek E., Maraño J.S., J. Chrom. ScL, 22 (1984) 313. - Dabrio M.V., Cromatografía y electroforesis en columna, Springer-Verlag Ibérica, Barcelona (2000). - Dandenau R.D., Zerrener E.H., J. High Resolut. Chromatogr. 6 (1979) 351. - de Boer J., de Boer K.. Boon J.P., J. Paasivirta (Ed.), Handbook of Environmental Chemistry, Vol. 3,
Springer, Heidelberg, 2000, p. 61, Part K. - de Boer J., Duinker J.C., Calder J.A., van der Meer J., J.Assoc. Off. Anal. Chem. 75 (1992) 1054. - de Boer J., Law R.J., Journal of Chromatography A, 1000 (2003) 223-251. - de Boer J.. Chemosphere 17 (1988) 1803. - de Boer J.. Chemosphere 17 (1988) 1811. - de BoerJ., Cofino W.P., Chemosphere 46 (2002) 625. - Di Corcia A., Marchetti M., Environ. Sci. Technol. 26 (1992) 66. - Díaz A., Vàzquez L., Ventura F., Galceran M.T., Analytica Chimica Acta 506 (2004) 71–80. - Díaz Moroles N.E., Alfaro Barbosa J.M., Garza Ulloa H., Método analítico para detectar
hidrocarburos aromáticos policíclicos en agua potable, Ciencia UANL Vol. IV, No. 4, Octubre-Diciembre 2001.
- Dill K.A., J.Phys.Chem. 91: 91 (1980). - Dill K.A., Naghizade J., Marqusee J.A., Ann.Rev.Phys.Chem. 39: 425 (1988). - Dorsey J.G., Cooper W.T., Anal.Chem. 66: 857A (1994). - Dorsey J.G., Dill K.A., Chem.Rev. 89: 331 (1989). - Ehrhardt M., Klungsøyr J., Law RJ., ICES Times Series 12 (1991) 1. - Eisert R., Levsen K., Fresenius J. Anal. Chem. 351 (1995) 555. - Ekdahl A., Abrahamsson K., Analytica Chimica Acta 357 (1997) 197-209. - El-Shahawi M.S., Journal of Chromatography A, 760 (1997) 179 - 192. - Esteban D., Técnicas de absorción de compuestos orgánicos volátiles, Cromatografía y Técnicas
Afines, Vol.24 (1), 2003. - Estefani Morales M.J., Análisis de hidrocarburos aromáticos policíclicos y pesticidas en aguas
mediante la técnica SBSE – GC/MS, Departamento de Ingeniería Química y Ambiental, Universidad de Sevilla (2004).
- Ettre L.S., Sakodynskii K.I., Chromatographia 35: 223 (1993). - Ettre L.S., Sakodynskii K.I., Chromatographia 35: 229 (1993). - Fernandez-Alba A.R.,Agüera A., Contreras M., Peñuela G., Ferrer I., Barceló D., Journal of
Chromatography A 823 (1998) 35. - Ferrer I., Pichon V., Hennion M.-C., Barceló D., Journal of Chromatography A. 777 (1997) 91-98.

- Flórez Menéndez J.C., Fernández Sánchez M.L., Fernández Martínez E., Sánchez Uría J.E., Sanz-Medel A., Talanta 63 (2004) 809-814.
- Fuoco R., Ceccarini A., Onor M., Marrara L., Journal of Chromatography A, 846 (1999) 387-393. - Fuoco R., Colombini M.P., Samcova E., Chromatographia 36 (1993) 65. - Futo I., Molnar M., Palcsu L., Svingor E., Szanto Zs., Vacuum 61 (2001) 441-445. - Gascón J., Salau J.S., Oubina A., Barceló D., Analyst 123 (1998) 941. - Gimeno R.A., Aguilar C., Marcé R.M., Borrull F., Journal of Chromatography A, 915 (2001) 139–
147. - Gimeno R.A., Altelaar A.F.M., Marcé R.M., Borrull F., Journal of Chromatography A, 958 (2002)
141–148. - Golay M.J.E., Gas Chromatography 1958. D.H. Desty Ed. Academic Press, New York (1959). - Gonçalves C., Alpendurada M.F., Journal of Chromatography A, Volume 1026, Issues 1-2, 13
February 2004, Pages 239-250 . - Górecki T., Pawliszyn J., Field Anal. Chem. Technol. 1(5) (1997) 277. - Górecki T., Pawliszyn J., J. High Resolut. Chromatogr. 18 (1995) 161. - Griffiths J.H., James D.H., Phillips G.S.G., Analyst 77:897 (1952). - Grob K., Classical split and splitless injection in capillary GC. Hüthig-Verlag, Heidelberg (1986). - Grob K., Making and manipulating capillary columns for GC. Hüthig-Verlag, Heidelberg (1986). - Grob K., On-column injection in capillary GC. Hüthig-Verlag, Heidelberg (1987). - Halasz I., Z. Anal. Chem. 236: 15 (1968). - Harris W.E., Habgood H.W., Temperature programmed gas chromatography, J.Wiley & Sons, New
York (1966). - Hauser B., Popp P., Bauer C., Kleine-Benne E., Semi-Automated Stir Bar Sorptive Extraction
(SBSE) in Combination with HPLC - Fluorescence Detection for the Determination of Polycyclic Aromatic Hydrocarbons in Water, Global Analytical Solutions, Gerstel, Appnote 1 (2002).
- Hauser B.,Popp P., Kleine-Benne E., Membrane-Assisted Solvent Extraction of Triazines and Other Semivolatile Contaminants Directly Coupled to Large-Volume Injection / GC-MS, Global Analytical Solutions, Gerstel, Appnote 12 (2002).
- He Y., Wang Y., Lee H.K., Journal of Chromatography A, 874 (2000) 149–154. - Heiden A.C., Hoffmann A., Kolahgar B., Comparison of the Sensitivity of Solid Phase
MicroExtraction (SPME) and Stir Bar Sorptive Extraction (SBSE) for the Determination of Polycyclic Aromatic Hydrocarbons (PAH) in Water and Soil Samples, Gerstel, Application Note 8, 2001.
- Hernández F., Beltrán J., López F.J., Gaspar J.V., Anal. Chem. 2000; 72:2313-22. - HeY., Lee H.K., Anal. Chem. 69 (1997) 4634. - Hinshaw J.V., LCGC, Volume 18, Number 9, pages 940-949, September 2000. - Hinshaw J.W., Ettre L.S., J.High Resol.Chromatogr. 20:471 (1997). - Horváth Cs., Melander W., Journal of Chromatography Sci. 15: 393 (1977). - Horváth Cs., Melander W., Molnar I., Anal.Chem. 49: 2295 (1977). - Horváth Cs., Melander W., Molnar I., Journal of Chromatography 125: 129 (1976). - http://www.asms.org/whatisms/index.html - http://www.tecnociencia.es/especiales/intercambio_ionico/introduccion.htm - http://www.uib.es/depart/dqu/dquo/pau/Cromatograf%92a/columnas.html - Hunt E.G., Bischoff A.I., Calif. Fish Game 46 (1960) 91. - Huybrechts T., Dewulf J., Moerman O., Van Langenhove H., Journal of Chromatography A, 893
(2000) 367–382. - Ikonomou M.G., Fernandez M.P., He T., Cullon D., J. of Chromatography A, 975 (2002) 319-333. - James A.T., Martin A.J.P., Biochem.J. 50: 679 (1952). - Janssen H.-G-, Sample Introduction Techniques For Capillary Gas Chromatography, Gerstel, 1998. - Jaouen-Madoulet A., Abarnou A., Le Guelleca A.M., Loizeaua V., Leboulenger F., Journal of
Chromatography A, 886 (2000) 153–173. - Jeannot M.A., Cantwell F.F., Anal. Chem. 69 (1997) 235. - Jeannot M.A., Cantwell F.F., Anal. Chem. 69 (1997) 2936. - Jeannot R., Sabik H., Sauvard E., Genin E., Journal of Chromatography A, 879 (2000) 51–71. - Jensen P., Renberg L., Reutergårdh L., Anal. Chem. 49 (1977) 316.

- Junk G.A., Suffet I.H., Malayiandi M.(Eds.), Organic Pollutants in Water, ACS Symposium Series No. 214, Washington D.Q, (1987), p. 201.
- Kerrola K., Food Rev. Int. 11 (1995) 547. - Kicinski H.G., GIT Fachz. Labor 11 (1993) 999. - Kiselev A.V., Yashin Y.I., Gas-adsorption chromatography. Plenum Press, New York (1969). - Knap A.H., Binkley K.S., Deuser W.G., Nature 319 (1986) 572. - Kolahgar B., Hoffmann A., Heiden A.C., Journal of Chromatography A, 963 (2002) 225–230. - König W.A., Gas chromatographic enantiomer separation with modified cyclodextrins, Hüthig.
Heidelberg(1992). - Lambropoulou D.A., Albanis T.A., Journal of Chromatography A, Volume 1072, Issue 1, 22 April
2005, Pages 55-61. - Langerfeld J.J., Hawthorne S.B., Miller D.J., Pawliszyn J., Anal. Chem. 6 (1993) 338. - Laub R.J., Pecsock R.L., Physicochemical applicatios of gas chromatography. J.Wiley & Sons. New
York (1978). - León V.M., Álvarez B., Cobollo M.A., Muñoz S., Valor I., Journal of Chromatography A, 999 (2003)
91–101. - Lepper P., Towards the Derivation of Quality Standards for Priority Substances in the Context of the
Water Framework Directive, Fraunhofer-Institute Molecular Biology and Applied Ecology, 2002. - Li H.-P., Li G.-V., Jen J.-F., Journal of Chromatography A, 1012 (2003) 129-137. - Lipinski J., Fresenius J. Anal. Chem. 369 (2001) 57. - Liska I., Krupcik J., Leclercq P.A., J. High Resolut. Chromatogr. Commu., 12 (1989) 577. - Liu R., Zhou J.L., Wilding A., Journal of Chromatography A, 1022 (2004) 179–189. - Lord H., Pawliszyn J., Journal of Chromatography A 885 (2000) 153. - Lovelock J.E., Journal of Chromatography 1 (1958) 35. - Lovelock J.E., Journal of Chromatography 1:25 (1958). - Lovelock J.E., Lipsky S.R., J.Am.Chem.Soc. 82: 431 (1960). - MajorsR.E., LC-GC International 8: 368 (1995). - MamyrinB.A., International Journal of Mass Spectrometry 206 (2001) 251–266. - Manoli E., Samara C., Chromatographia 43 (1996) 135. - Martf I., Ventura F., Journal of Chromatography A, 786 (1997) 135-144. - Martínez E., Lacorte S., Llobet I., Viana P., Barceló D., J. of cromatography A, 959 (2002) 181-190. - Marvin C.H., Smith R.W., Bryant D.W., McCarry B.E., Journal of Chromatography A 863 (1999) 13. - McReynolds O.W., Journal of ChromatographySci. 8: 685 (1970). - McWilliam I.G., Dewar R.A., Gas Chromatography Amsterdam 1958. G. butterworths Ed., Londres
(1958). - Melander W.R., Horváth Cs., High Performance Liquid Chromatography. Advances ana
Perspectives. Vol. 1, Cs. Horváth (ed). Academic Press, New York (1980). - Mohnke M., Saffert W., Gas Chromatography 1962. M.M.Swaay, Londres (1962). - Mol H.G.J., Sunarto S., Steijger O.M., Journal of Chromatography A, 879 (2000) 97–112. - Molera M.J., García Domínguez J.A., Fernández Biarge J., Journal of Chromatography Sci. 7: 305
(1969). - Moon H.M., Kim K., Lee S.C., Kim H., Microchemical Journal 63, 172–186 (1999). - Nahum A., Horváth Cs., Journal of Chromatography 203: 53 (1981). - Ngo T.T. (ed), Molecular Interactions in Bioseparations. Plenum Press, New York (1993). - Nikolaou A.D., Lekkas T.D., Golfinopoulos S.K., Kostopoulou M.N., Talanta 56 (2002) 717–726. - Ohlenbusch G., Zwiener C., Meckenstock R.U., Frimmel F.H., Journal of Chromatography A, 967
(2002) 201–207. - Oubina A., Martínez E., Gascón J., Barceló D., Alleluia I.B., Int. J. Environ. Anal. Chem. 70 (1998)
75. - Peaden P.A., Lee M.L., Hirata Y., Novotny M., Anal. Chem. 52 (1980) 2268. - Pellizzari E.D., Journal of Chromatography 98: 323 (1974). - Peñálver Hernando A.M., Aplicación de la Microextracción en Fase Sólida al Análisis
Medioambiental, Departament de Química Analítica i Química Orgànica, Universitat Rovira i Virgili (2002).
- Pereira W.F., Rostad C.E., Leoker T.J., Anal. Chim. Acta 228 (1990) 69.

- Picó Y., Blasco C., Font G, Environmental and Food Applications of Lc–Tandem Mass Spectrometry in Pesticide-Residue Analysis: An Overview, Laboratori de Bromatologia i Toxicologia, Facultat de Farmàcia, Universitat de València (2003).
- Pocurull E., Aguilar C., Alonso M.C., Barceló D., Borrull F., Marcé R.M., Journal of Chromatography A, 854 (1999) 187–195.
- Reutergårdh L., Identification and distribution of chlorinated organic pollutants en the environment, PhF thesis, Stockholm University, Stockholm, Sweden, 1988, p. 42.
- Roeraade J., Blomberg A., J. High Resolut. Chromatogr. 12 (1989) 139. - Sanders L.C., Wise S.A., LC-GC International 3 (6): 24 (1990). - Sara Rodriguez-Mozaz, Maria J. López de Alda, Damià Barceló, Journal of Chromatography A,
1045 (2004) 85–92. - Scott R.P.W., Chromatographic detectors. Marcel Dekker Inc., New York (1996). - Scott R.P.W., Liquid chromatograpphy Column Theory, J. Wiley & Sons, Chichester (1992). - Schaad R.E., Chromatogr. Rev. 13 (1970) 61. - Schomburg G., Sample introduction in capillary gas chromatography. Vol. 1. P. Sandra Ed. Hüthig-
Verlag, Heidelberg (1985). - Schurig V., Angewandte Chem.Int. 16: 110 (1977). - Sentel K.B., Dorsey J.G., Anal.Chem. 61: 930 (1989). - Shigun Z., Zhanxia Z., Guoying S., Yushun M., Jiamo F., Environ. Monit, Assess. 44 (1997) 563. - Silva F.C., de Carvalhot C.R., del Cardeal Z., Journal of Chromatography Sci. 2000; 38(7):315-8. - Sleight R.B., Journal of Chromatography 83 (1973) 31. - Smedes F., de Boer J., Trends Anal. Chem. 16 (1997) 503. - Snell R.P., Danielson J.W., Oxborrow G.S., Journal of Chromatography Sci. 25 (1987) 225. - Snyder L.R., Glajh J.L., Kirkland J.J., Practical HPLC Method Development. J.Wiley, New York
(1988). - Snyder L.R., High Performance Liquid Chromatography. Advances and Perspectives. Vol. 1. Cs.
Horváth (ed). Academic Press, New York (1980). - Snyder L.R., Principles of Adsorption Chromatography. Marcel Dekker, New York (1968). - Staniewski J., Jansen H.-G., Rijks J.A., Cramers C.A., J.Microcol. Sep. 5 (1993) 429. - sz Kováts E.. Adv. Chromatogr. 1: 230 (1965). - Tchapla A., Héron S., Lesellier E., Colin H.. Journal of Chromatography A 656: 81 (1993). - Theodoris G., Koster E.H.M., De Jong G.J., Journal of Chromatography B, Biomed. Sci. Appl. 2000;
745(1)49-82. - Tienpont B., David F., Bicchi V., Sandra P., J. microcol. Sep. 12(11) (200) 577. - Tswett M.S., Khromofilly v Rastitelínom i Zhivotnom Mire (Clorofilas en el mundo vegetal y animal),
Karbasnikov Publishers, Varsovia (1910). - Valor I., Moltó J.C., Apraiz D., Font G., Journal of Chromatography A 767 (1997) 195. - van Deemter J.J., Zuiderwerg F.J., Klinkenberg A., Chem.Eng.Sci. 5:271 (1956). - Wach F., Anal.Chem. 66: 927A (1994). - Webster L., Fryer R.J., Dalgamo E.J., Megginson C., Moffat C.F., J. Environ. Monit. 3 (2001) 591. - Wennrich L., Popp P., Köller G., Breuste J., J. AOAC Int. 84 (2001) 1194. - Wiencke W.W., Burke K.A.. J. Assoc. Off. Anal. Chem. 52 (1969) 1277. - Wilke C.R., Chang P., Am.Inst.Chem.Eng.J. 1: 264 (1955). - Wolska L., Galer K., Górecki T., Namieśnik J., Talanta 50 (1999) 985–991. - Wolska L., Olszewska C., Turska M., Zygmunt B., Namiesnik J., Chemospfere, Vol. 37, Nº 13, pp.
2645-2651, 1998. - Wolska L., Wiergowski M., GaIer K., Górecki T.‘, and Namieśnik J., Chemosphere, A25 - Woodhead R.J., Law R.J., Matthiessen P., Mar. Pollut. Bull. 38 (1999) 773. - Zhang Z., Pawliszyn J., J. High Resolut. Chromatogr. 19 (1996) 155.