universidad de chile facultad de ciencias quÍmicas y...
TRANSCRIPT

UNIVERSIDAD DE CHILE
FACULTAD DE CIENCIAS QUÍMICAS Y FARMACÉUTICAS
DEPARTAMENTO DE QUÍMICA FARMACOLÓGICA Y TOXICOLÓGICA
LABORATORIO DE FARMACOLOGÍA
ESTUDIO DE BIODISPONIBILIDAD RELATIVA DE UNA FORMULACION ORAL DE IMATINIB RESPECTO AL PRODUCTO INNOVADOR
PATROCINANTE y CO-DIRECTOR
Prof. María Eugenia Letelier M.
DIRECTOR DE MEMORIA
Prof. Iván Saavedra S.
Laboratorio de Farmacología. Dpto.
Química Farmacológica y Toxicológica.
Facultad Ciencias Químicas y
Farmacéuticas. Universidad de Chile
Laboratorio de Farmacocinética y
Biodisponibilidad. Centro de
Investigaciones Farmacológicas y
Toxicológicas (IFT). Facultad de
Medicina. Universidad de Chile
Memoria para optar al título de Químico Farmacéutico
Jaime Alfredo Sasso Aguirre
SANTIAGO DE CHILE
2008

II
A mis padres.

III
Agradecimientos
Ante todo, agradecer a mis pilares, mi familia, a mis padres Estrella y Jaime por
su sacrificio y dedicación, a mi hermana Daniela. Gracias a mi Lela por ser la imagen
de fortaleza frente a los momentos adversos.
Agredecer a la Profesora María Eugenia Letelier por confiar en mi y enseñarme
que todo puede ser posible, al Profesor Iván Saavedra por enseñarme a ser un
profesional íntegro, con una visión mas igualitaria y juzta, y al Profesor Luis Quiñones
por su amistad y por creer en mis capacidades.
Al Centro de Investigaciones Farmacológicas y Toxicológicas, mi segundo
hogar, a cada una de las personas que construyen este lugar y creen en él como un
aporte a la comunidad.
A mis amigos de la Universidad, Fabiola, Cristian, Pablo y Francisco, por estar
siempre a mi lado, en los momentos dificiles y de máxima diversión.

IV
INDICE GENERAL
Página
DEDICATORIA .......................................................................................... ........... II
AGRADECIMIENTOS ........................................................................................... III
INDICE GENERAL ................................................................................................IV
INDICE DE FIGURAS ........................................................................................... VI
INDICE DE TABLAS ............................................................................................. VI
RESUMEN ............................................................................................................ VII
SUMMARY ........................................................................................................... VIII
I. INTRODUCCIÓN ............................................................................................... 1
Hipótesis ............................................................................................................... 5
Objetivos generales .............................................................................................. 6
Objetivos especificos ............................................................................................ 6
II. VOLUNTARIOS, MATERIALES Y MÉTODOS..................................................7
Selección de Voluntarios.……………………………………………………………… 7
Medicamentos en estudio, adquisición y manejo de los productos a ensayar...... 9
Metodología.……………………………………………………………………. ……… 12
III. RESULTADOS.……………………………………………………………………. 17
Adaptación de la metodología bioanalítica…………………………………………..17
Validación de la metodología bioanalítica……………………………………………18
Características antropométricas de los voluntarios. …….......................................24
Análisis físico-químico del producto test y el producto referencia………………… 25
Exámenes clínicos y de laboratorio clínico………………………………………….. 25
Consentimiento informado ……………………………………………………………. 26

V
Dieta administrada a los voluntarios ………………………………………………….26
Análisis cromatográfico de las muestras plasmáticas ……………………………...26
Análisis Farmacocinético.……………………………………………………………… 31
Análisis estadístico de las muestras …………………………………………………. 35
Evaluación de Bioequivalencia.………………………………………………………..37
Encuesta de reacciones Adversas …………………………………………………… 39
IV. DISCUSIÓN.…………………………………………………………………………40
V. REFERENCIAS ............................................................................................... 42
ANEXOS................................................................................................................ 45

VI
ÍNDICE DE FIGURAS
Figura Nº 1 Fórmula química de Imatinib. Pág. 2
Figura Nº 2 Cromatogramas de separación y cuantificación de Imatinib
y Clozapina
Pág. 20
Figura Nº 3 Curva de Calibración de Imatinib Pág. 21
Figura Nº 4 Curvas promedio de concentración plasmática de Imatinib Pág. 30
ÍNDICE DE TABLAS
Tabla 1 Validación Analítica: Determinación de exactitud Pág. 21
Tabla 2 Validación Analítica: Determinación de precisión intradía. Pág. 21
Tabla 3 Validación Analítica: Determinación de precisión interdía. Pág. 22
Tabla 4 Validación Analítica: Determinación del porcentaje de
recuperación.
Pág. 23
Tabla 5 Características Antropométricas de los voluntarios. Pág. 24
Tabla 6 Concentraciones plasmáticas (ηg/mL) vs el tiempo (h) en 17
voluntarios sanos para A y B
Pág. 27
Tabla 7 Concentraciones promedio de Imatinib (ng/mL). Pág. 29
Tabla 8 Parámetros farmacocinéticas ABC0→24, ABC 0→∞, Cmáx y tmáx. Pág. 32
Tabla 9 Otros parámetros farmacocinéticos de Imatinib. Pág. 33
Tabla 10 Análisis de varianza para los parámetros farmacocinéticos. Pág. 36
Tabla 11 Intervalos de Confianza al 90%. Pág. 38
Tabla 12 Prueba de dos hipótesis de Schuirmann para los
parámetros farmacocinéticos.
Pág. 39

VII
RESUMEN
En este estudio se determinó la biodisponibilidad relativa para la bioequivalencia
del producto similar Zeite® de Laboratorios Recalcine S.A., producto de prueba, con
Glivec® fabricado por Laboratorios Novartis AG, producto de referencia; ambas
formulaciones orales de Imatinib de 400 mg. Para este estudio doble ciego, cruzado y
randomizado se seleccionaron 24 voluntarios sanos; de los cuales, 17 voluntarios
finalizaron el estudio. Los niveles plasmáticos de Imatinib, después de la administración
de una sola dosis, se determinaron esencialmente por la técnica HPLC descrita por
Velpandian y colaboradores, 2004, modificada en el Laboratorio. Para este efecto la
técnica fue validada de acuerdo a los parámetros siguientes: especificidad, exactitud,
precisión y linealidad. Para evaluar la biodisponibilidad se compararon los siguientes
parámetros farmacocinéticos de los productos Zeite® y Glivec®: área bajo la curva ABC
(concentración plasmática versus tiempo) y concentración máxima (Cmáx).
Los resultados de los parámetros farmacocinéticos fueron: ABC0→∞ 18,5154 ±
6,4942 y 20,8107 ± 6,1045 µg/h/mL; ABC0→24 12,9506 ± 4,4824 µg/h/mL y 14,6367 ±
3,6799 µg/h/mL; Cmáx 1215,12 ± 410,96 ηg/mL y 1189,42 ± 293,70 ηg/mL, para A y B,
respectivamente. Todas estas diferencias resultaron ser estadísticamente no
significativas, p>0.05. Los intervalos de confianza para el cuociente A/B fueron:
ABC0→∞ 91,27 % a 99,40 %; ABC0→24, 89,31 % a 98,73 % y Cmáx 97,64 % a 102,03 %.
El rango de confianza recomendado por la FDA es 80% a 125 % con una
probabilidad de bioequivalencia del 100%. Todos los parámetros cinéticos para la
bioequivalencia (Cmáx, ABC0→24, ABC0→∞) ensayados se encontraron dentro de este
rango; por lo tanto, se concluye que Zeite® 400 mg, A (test), es un genérico
bioequivalente e intercambiable con Glivec® 400 mg, B (referencia).

VIII
SUMMARY
RELATIVE STUDY OF BIOAVAILAILITY OF AN ORAL FORMULATION OF IMATINIB
WITH RESPECT TO THE INNOVATOR PRODUCT.
This study determined the relative bioavailability to demonstrate the
bioequivalence of the similar product Zeite® of Laboratories Recalcine S.A., test
product, and Glivec® of Laboratories Novartis AG, reference product. For this double
blind, crossed and randomizaded study which include 24 healthy volunteers, which
received one dose of 400 mg Imatinib.
The Imatinib plasmatic levels, was determinated essentially for the HPLC
technique described by Velpandian et al., 2004. This technique was validated respect to
its specificity, accuracy, precision and linearity. The pharmacokinetics parameters
compared were: area under the curve AUC, (plasmatic concentration vs time) and
maximum concentrations (Cmax).
The results of the pharmacokinetics parameters of samples A and B were: AUC0→∞
18,5154 ± 6,4942 y 20,8107 ± 6,1045 µg/h/mL; AUC0→24 12,9506 ± 4,4824 µg/h/mL y
14,6367 ± 3,6799 µg/h/mL; Cmáx 1215,12 ± 410,96 ηg/mL and 1189,42 ± 293,70 ηg/mL,
respectively. Significant differences between the kinetic parameters values of both
products, p>0.05 were not observed. The confidence intervals for the rate A/B were:
AUC0→∞ 91,27 % to 99,40 %; AUC0→24, 89,31 % to 98,73 % and Cmáx 97,64 % to 102,03
%. The range recommended by FDA is 80 % to 125 % with a bioequivalence probability
of 100 %. All the kinetic parameters for the bioequivalence (Cmáx, AUC0→24, AUC0→∞)
were included in this range; therefore, Zeite® 400 mg, (A test), is a generic product
bioequivalent and interchangeable with Glivec®, (B reference).

IX
I. INTRODUCCIÓN
Imatinib (mesilato de Imatinib), agente antineoplásico, es un derivado de la
fenilaminopiridina designado químicamente como 4-[(4-metil-3-[{4-(3-piridinil)-2-
pirimidil}amino]fenil]-benzimida (Figura Nº 1). Imatinib es miembro de una nueva clase
de fármacos conocida como inhibidores de las señales de transducción. En el año
2001 este medicamento fue aprobado por la FDA para el tratamiento de la Leucemia
Mieloide Crónica (LMC). El principio activo interfiere con la proteína que provoca una
proliferación más rápida de las células leucémicas en comparación con las células
sanguíneas normales, actúa inhibiendo selectivamente la BCR-ABL tirosina-quinasa,
responsable esencial en la aparición de la LMC; esta proteína proviene de la expresión
del cromosoma Filadelfia, el cual es el resultado de la translocación entre el gen BCR
del cromosoma 22 y el gen ABL del cromosoma 9 [1, 2]. Esta enzima cataliza la
fosforilación de múltiples substratos generando un aumento de la transducción de
señales y una desregulación del crecimiento celular. La característica que hace
interesante a Imatinib es que es una droga selectiva cuyo efecto se ejerce
exclusivamente en las células leucémicas ya que se ha diseñado considerando la
biología molecular de la enfermedad, por lo que provoca pocos efectos secundarios al
no afectar a las células normales [3].
Un estudio multicéntrico realizado en 177 hospitales de todo el mundo con 1.106
pacientes, ha permitido determinar que este nuevo fármaco es la mejor opción
terapéutica para tratar a los enfermos afectados por leucemia mieloide crónica. Cabe
señalar que el tratamiento por muchos años fue el Interferón α con Citarabina; el
Interferón α se liga a receptores específicos de la superficie celular, lo que
desencadena una serie de fenómenos intracelulares, entre ellos una inhibición de la

X
N
NH
NH
N
N
O
CH3
N
N
CH3
proliferación celular y una intensificación de la actividad inmunitaria; la Citarabina es un
análogo de la citidina, por lo que actúa como un antimetabolito y es así como produce
inhibición de la síntesis de ADN. Los datos de la investigación publicados en "The New
England Journal of Medicine", son concluyentes, dejando al Interferón α combinado
con Citarabina como opción de segunda línea para el tratamiento de la enfermedad [3].
Figura Nº 1. Fórmula química de Imatinib.
La literatura consultada señala que la farmacocinética clínica de Imatinib
administrado en tabletas por vía oral, ha sido ampliamente estudiada. A dosis de 1 x
400 mg los parámetros farmacocinéticos obtenidos fueron; concentración máxima
(Cmáx): 1,606 ± 647 ηg/mL; tiempo al cual se llega a la Cmáx o tiempo máximo promedio
(tmáx): 2,5 h (1,5 a 4,0 h); área bajo la curva desde tiempo cero a 24 horas (ABC0-24
h):18,658 ± 8,016 µg/h/mL; área bajo la curva desde tiempo cero a infinito (ABC0-inf):
25,5 ± 11,85 µg/h/mL; volumen de distribución (Vd/F): 404 ± 144 L; tiempo de vida
media de eliminación (t1/2): 15,7 ± 2,8 h; clearence (Cl/F): 18,3 ± 5,7 L/h [4]. La droga
posee una biodisponibilidad absoluta del 98 % y su absorción no es afectada
significativamente por la comida. Su metabolización se produce en forma extensa en el

XI
hígado, siendo la CYP3A4 la isoenzima mayoritariamente involucrada en su
metabolismo microsómico. Es importante tener en cuenta el posible polimorfismo
genético de esta enzima ya que se ha demostrado que tanto el metabolismo como los
niveles plasmáticos de Imatinib son alterados por los fármacos que compiten por el
sitio de metabolización, como por ejemplo el ketoconazol, un inhibidor de la CYP3A4
[5, 6].
La selección de principios activos para exigencia de estudios de bioequivalencia
(BE), es un criterio que debe estar incluido en las políticas que regulan la salud pública
de cada nación y como tal, se debe evaluar para cada fármaco, la relación
riesgo/beneficio. La biodisponibilidad de los principios activos, es decir aquella cantidad
proveniente de una forma farmacéutica que llega a la circulación sistémica y la
velocidad a la cual esto ocurre, puede variar entre dos equivalentes farmacéuticos.
Esto porque existen varios factores que pueden afectarla, entre ellos, las
características fisicoquímicas del principio activo, la forma farmacéutica (comprimidos,
grageas, cápsulas, comprimido recubierto) y la calidad de ésta, que incluye: el tipo y la
calidad de los excipientes, el proceso de fabricación, la estabilidad del producto, etc.
Por lo tanto, demostrar que una formulación es intercambiable por otra, involucra el
desarrollo de un estudio de BE en un Centro que cuente con acceso a voluntarios
sanos, personal médico, enfermeros, analistas químicos, analistas estadísticos y
experiencia en el tema.
Al respecto, cada país confecciona una lista de principios activos que, por sus
características farmacodinámicas y farmacocinéticas, deben someterse a estudios
comparativos “in vivo” para demostrar su biodisponibilidad y certificar así, su
equivalencia terapéutica. De esta manera se pueden seleccionar aquellos fármacos

XII
que son más riesgosos para la salud de la población, bajo una clasificación de riesgo
sanitario alto. En Chile en la actualidad existe una lista de 16 principios activos a los
cuales se les debe realizar estudios de BE; Imatinib no está incluido dentro de esta lista
de fármacos; sin embargo, internacionalmente las drogas anti-neoplásicas deben
cumplir con los criterios de equivalencia terapéutica por sus características de drogas
de gran toxicidad y utilizadas en una enfermedad catastrófica. La India es hoy uno de
los mayores productores de medicamentos similares de calidad, a un precio accesible
para los enfermos graves, entre ellos, enfermos de cáncer y SIDA. La industria
farmacéutica India proporciona más del 50 % de los medicamentos similares que se
distribuyen a muchos países en vías de desarrollo a través de organismos mundiales
como UNICEF, de los cuales se necesita conocer la BE con el medicamento innovador.
Este es un problema que afecta a los medicamentos que la India exporta. Un ejemplo
de esto es el juicio que actualmente está en proceso entre Novartis, empresa suiza
productora del fármaco innovador Imatinib, y los medicamentos similares formulados
en la India. En 1993 Novartis (laboratorio inventor de la molécula) patentó el Imatinib;
en 1997 esta empresa solicitó la patente para un polimorfo del cristal de mesilato de
Imatinib en varios países y en 1998 Novartis solicitó en la India la patente de Imatinib
bajo la marca comercial Glivec®. India no concedió esta patente como medicamento
innovador, lo cual llevó a un juicio internacional de patentes [7]. La resolución de este
juicio es muy importante para la salud de la población india y también, de aquellas
naciones a las cuales la India exporta este medicamento, dado el significativo menor
costo de los medicamentos formulados en la India comparado con aquellos de
Novartis.

XIII
Los antecedentes presentados muestran que Imatinib es una droga incluida en
medicamentos cuyas formas farmacéuticas sólidas provienen de fuentes múltiples; por
lo tanto, deben ser sometidas a estudios de equivalencia terapéutica, entre ellos,
estudios de biodisponibilidad. La realización de estos estudios requiere la utilización de
un estándar, el cual es el fármaco innovador del mercado farmacéutico mundial; esto
permite intercambiar el producto innovador por el medicamento similar, ya que si
ambos poseen la misma biodisponibilidad, la concentración plasmática será la misma y
se asegura así, el éxito del producto similar en la terapia farmacológica.
Existen diferentes modalidades para demostrar la equivalencia terapéutica, entre
ellos: a) estudios de biodisponibilidad comparativa; b) estudios clínicos controlados.
Estos estudios son regulados por la OPS/OMS, a través de “Multisource (generic)
pharmaceutical products: Guidelines on registration requirements to establish
interchangeability”, WHO Tecnical Report Series, Nº 937, 2006 (WHO-06), quienes
proponen los aspectos y criterios científico-técnicos que deben ser considerados en
una norma de estudios de BE [8, 9]. De acuerdo a esta norma y respecto a los estudios
de biodisponibilidad comparativa, se entiende que cuando una formulación sólida es
similar a otra en cuanto a la velocidad de entrega del principio activo y de la cantidad
de droga que se absorbe a partir de ella, pueden considerarse ambas bioequivalentes
e intercambiables [10].
Hipótesis
Se postula que las dos formulaciones orales de Imatinib, el producto innovador
Glivec® y el producto similar Zeite®, son bioequivalentes.

XIV
OBJETIVOS
Objetivo general
Determinar, en un diseño de doble ciego, cruzado y randomizado la
biodisponibilidad relativa de una formulación farmacéutica de Imatinib respecto del
producto innovador.
Objetivos específicos
1. Validación de la técnica HPLC para medir las concentraciones plasmáticas de
Imatinib descrita por Velpandian y colaboradores, 2004.
2. Seleccionar 24 voluntarios sanos mediante exámenes clínicos y de laboratorio.
3. Someter los productos comerciales Glivec® y Zeite® a análisis fisicoquímicos de la
farmacopea.
4. Medir las concentraciones plasmáticas de Imatinib en el plasma de los voluntarios
sanos.
5. Realizar el análisis farmacocinético de los resultados obtenidos de la determinación
de las concentraciones plasmáticas en el tiempo.
6. Realizar el análisis estadístico de los resultados de los parámetros
farmacocinéticos de ambos productos similares.

XV
II. VOLUNTARIOS, MATERIALES Y MÉTODOS.
A. Selección de Voluntarios
El número de sujetos que participan en el estudio debe realizarse mediante un
método adecuado que nos permita garantizar la confiabilidad de los resultados; este
número no debe ser menor a 12 individuos, pero normalmente es de 18 a 24 personas
[8].
En la primera semana del estudio un grupo de adultos, hombres y mujeres,
sanos(as), con edades que fluctuaron entre los 18 y 55 años, con apellidos
hispanoamericanos, fueron citados a las dependencias del Centro de Investigación, en
ayunas. Los sujetos se entrevistaron con un médico cirujano quien les realizó un
examen médico completo y una interrogación sobre su historia médica. La entrevista
con el médico incluyó los siguientes puntos:
- Identificación: nombre, edad, sexo.
- Examen físico general: presión sanguínea, pulso, ritmo respiratorio, medición de
altura y peso en ropa interior. Aspecto de la piel, cabeza, cuello, ojos, nariz,
oídos, boca, garganta, pecho, abdomen, espina dorsal y nódulos linfáticos.
- Anamnesis: enfermedades existentes y preexistentes, historia familiar de
patologías, historia quirúrgica y hábitos (tabaco, drogas, alcohol).
- Anamnesis farmacológica: medicación, RAM, alergia a medicamentos.
Posteriormente, a aquellas personas que cumplían con los criterios de selección
(inclusión y exclusión) se les extrajo una muestra de sangre y se les solicitó una
muestra de orina para análisis de Laboratorio Clínico. Los análisis que se realizaron
antes y después del estudio, comprendieron: hemograma y VHS, orina completa, VIH,

XVI
screening de drogas de abuso, glicemia, uremia, proteinemia, fosfatasas alcalinas,
bilirubinemia, transaminasas oxálica y pirúvica y creatinemia. En el caso de las mujeres
se les solicitó además, un test de embarazo. Los exámenes de VIH y test de embarazo
se realizaron con el consentimiento informado de los voluntarios.
Luego de obtener los resultados de los exámenes clínicos, el médico seleccionó
los 24 voluntarios completamente sanos y el Químico Farmacéutico, coordinador del
estudio, los instruyó acerca del propósito del estudio, les explicó los posibles riesgos y
luego de esto cada voluntario seleccionado certificó con su firma el consentimiento
informado para participar en el estudio.
A.1. Criterios de selección de voluntarios para el estudio:
A.1.1. Criterios de Inclusión:
• Grupo de hombres y mujeres sanos de edades entre 18 y 55 años con apellidos
hispanoamericanos e índice de masa corporal (IMC) entre 19 y 30.
• No fumadores, no consumidores de drogas de abuso ni de alcohol.
• Sin alergias a medicamentos.
• Sin terapias concomitantes y no haber estado en tratamiento farmacológico a lo
menos dos meses antes del estudio.
• Sin presencia en la historia clínica de enfermedades que hayan afectado la
absorción, distribución y eliminación de drogas desde el organismo.
• Dar negativo en el análisis de drogas, VIH y/o test de embarazo.
• Con resultados de los exámenes de laboratorio en rangos normales y declarados
aptos para el estudio por el médico.

XVII
A.1.2. Criterios de Exclusión:
• Historia clínica de hipersensibilidad a cualquier medicamento.
• Presencia en la historia clínica de problemas gastrointestinales, de hígado, riñón,
pulmón, hematológicos, neurológicos, psiquiátricos, endocrinos, inmunológicos o
dermatológicos significativos.
• Mantención de una terapia medicamentosa o bien consumo de alcohol, tabaco,
marihuana u otras drogas de abuso.
• Haber tenido cualquier enfermedad de importancia en los 28 días previos al
estudio.
• Haber usado en los 28 días previos al estudio, fármacos que interactúen con la
biotransformación a través del sistema oxidativo del citocromo P450 (todos los
barbitúricos, corticoesteroides, fenilhidantoínas, etcétera).
• Haber usado cualquier medicamento en los 7 días antes del estudio, incluyendo
medicamentos de venta directa o sin receta médica.
• Haber participado en otro estudio similar en 90 días previos al estudio actual.
• Presencia de historia de desmayos o miedo a la extracción de sangre.
B. Materiales: Medicamentos en estudio, adquisición y manejo de los productos
a ensayar
B.1. Medicamentos en estudio. Los medicamentos fueron comprimidos recubiertos
que contenían 400 mg de Imatinib como Mesilato.
Medicamento Test: El producto farmacéutico Zeite®, fue administrado como
comprimidos de 400 mg envasados en blister; ellos fueron fabricados por Laboratorios

XVIII
Recalcine S.A. con el número de serie A-07108 y fecha de vencimiento Enero del
2009.
Medicamento Referencia: El producto farmacéutico considerado como referencia,
Glivec®, fueron administrados como comprimidos de 400 mg envasados en blister;
ellos fueron fabricados por Laboratorios Novartis AG., con el número de serie S0080 y
fecha de vencimiento Agosto del 2008.
B.2. Adquisición, obtención y manejo de los productos a ensayar
B.2.1. Adquisición de los productos a ensayar. El producto farmacéutico a evaluar
Zeite®, fue proporcionado por Laboratorios Recalcine S.A. y el producto considerado
como referencia Glivec®, fue adquirido en el mercado norteamericano.
B.2.2. Preparación de los medicamentos en estudio. Se solicitó a un profesional
Químico Farmacéutico ajeno al Centro que en confidencia confeccionara el ciego,
colocando 24 comprimidos de cada producto en un frasco ámbar, rotulando las
muestras para el estudio como “Producto A” y “Producto B”. Este procedimiento se
llevó a cabo en el momento previo a la administración del medicamento a los sujetos
de manera de asegurar la estabilidad del comprimido hasta el momento de su
administración. Su decisión quedó escrita en sobre sellado junto a los datos de número
de serie, fechas de elaboración y expiración, esta carta fue entregada en duplicado al
investigador principal y a Laboratorios Recalcine S.A. Los investigadores no conocieron
la identidad de los productos hasta el momento en que se realizó el análisis de
equivalencia, ya que en este procedimiento se debe conocer si el producto A o el

XIX
producto B corresponde al medicamento test o medicamento referencia. Previamente
al estudio de BE ambos productos, de ahora en adelante, producto A y producto B,
fueron sometidos a análisis de identidad de principio activo, valoración de principio
activo, variación de peso del comprimido y perfil cinético de disolución por Laboratorio
Recalcine S.A.
B.2.3. Administración de los productos farmacéuticos. Los productos
farmacéuticos se administraron a través de un diseño experimental randomizado,
cruzado y ciego. El profesional a cargo de la realización del ciego, realizó la
randomización de los voluntarios seleccionados, asignándole un número al azar de 1 a
24 a cada voluntario, además incluyó la secuencia para la administración de los
medicamentos, creando en cada período dos bloques equilibrados con la secuencia AB
o BA. De esta manera, en la secuencia AB, el voluntario recibió el fármaco A en el
primer período y el fármaco B en el segundo período, y en la secuencia BA, el
voluntario recibió el fármaco B en el primer período y el fármaco A en el segundo
período.
Diseño
Periodo 1 Periodo 2
Secuencia 1 A B
Secuencia 2 B A
En la segunda semana de estudio se cito a los 24 voluntarios seleccionados,
del total de voluntarios citados un número de 20 voluntarios, asistieron al Período 1, de

XX
manera tal que diez de ellos recibieron el producto A y los otros diez recibieron el
producto B. Los voluntarios que participaron realizaron un ayuno de 12 horas, con la
indicación de consumir solo agua hasta 1 hora previa a la administración del fármaco.
El fármaco se administró en una dosis oral única con 250 mL de agua potable estando
de pie. Luego de transcurridos 14 días (transcurridas más de 7 vidas medias del
fármaco), los voluntarios se cruzaron de manera tal de recibir el otro producto.
C. Metodología
C.1. Cuidado de los voluntarios. Durante el estudio los voluntarios estuvieron en
contacto con los médicos y fueron atendidos por todo el equipo profesional del Centro.
Su alimentación, que incluyó desayuno, almuerzo, meriendas y cena fue diseñada por
un profesional Nutricionista sobre la base de lo aconsejado por el Médico. La dieta
proporcionada a los voluntarios durante el estudio se encuentra en el Anexo 1.
Frente a la carencia de Seguros para este tipo de investigación clínica en el país
y tal como se acostumbra en este tipo de trabajos, la Institución que solicitó el trabajo
se comprometió a pagar el tratamiento de cualquier problema de salud que surja a
causa del estudio. Se estimó prudente financiar el costo de hospitalización y
tratamientos.
En cada uno de los procedimientos empleados, los voluntarios se sometiron
rigurosamente respecto a las normas de investigación de fármacos en seres humanos
[11]. Así para realizar este estudio se solicitó la autorización de la comisión de bioética
de la Facultad de Medicina de la Universidad de Chile. La aprobación del estudio por
parte del comité de ética se encuentra en el Anexo 2.

XXI
Las reacciones adversas a medicamentos (RAM), fueron debidamente abordadas
por el Médico. De acuerdo a lo descrito en la literatura científica del tema, la
administración de una sola dosis de 400 mg podía producir problemas de reacciones
adversas al medicamento (RAM) en el voluntario, tales como náuseas, vómitos y dolor
de cabeza, de intensidad moderada a suave [4].
C.2. Recolección de muestras. Para la extracción seriada de sangre las Enfermeras
Universitarias instalaron a primera hora y a cada voluntario una bránula ante-braquial
(Beckton & Dickinson, 18 G) provista de llave de tres pasos, la que se mantuvo en
forma permeable con heparina sódica como anticoagulante hasta la obtención de la
última muestra del primer día de cada período. Se recolectaron muestras de 5 mL de
sangre en tubos de ensayo de vidrio que contenían 10 µL de heparina sódica como
anticoagulante. Las muestras se recolectaron a tiempo 0 (antes de administrar la
droga), y a las 0,5; 1,0; 1,5; 2,0; 2,5; 4,0; 6,0; 8,0; 12,0 y 24,0 h. El plasma se obtuvo
mediante centrifugación a 3.000 rpm en una centrífuga Jouan® modelo GR412 y 10 °C.
Los plasmas obtenidos se separaron en dos tubos (muestra y contramuestra) y se
almacenaron a –20 ºC. Sólo las muestras fueron utilizadas en este estudio; las contra-
muestras se mantendrán almacenadas durante un período de 5 años.
C.3. Técnica analítica: Método para la determinación del principio activo en
sangre. Se utilizó esencialmente el método de Cromatografía Líquida de Alta
Resolución (HPLC) descrito por Velpandian y colaboradores, 2004 [12]. La validación
de este método fue definida respecto a su especificidad, linealidad, recuperación,
límites de detección, limites de cuantificación, exactitud, precisión y estabilidad.

XXII
El sistema cromatográfico utilizado consistió en un equipo HPLC marca Elite La
Cherom, Merck – Hitachi compuesto por un detector UV – visible L 2420, una bomba L
2130, un muestrador automático L 2200, un sistema organizador computacional Merck.
Las condiciones cromatográficas fueron las siguientes, una columna Shimp-pack
CLC C8 (150 mm x 4,6 mm y tamaño de partículas de 5 µm), un sistema isocrático de
fase móvil compuesto por una solución acuosa de KH2PO4 0,02 M y acetonitrilo (67:33
v/v), una velocidad de flujo de 1 mL/min. El Imatinib y Clozapina (estándar interno)
fueron detectados midiendo su absorbancia a 265 nm
Las muestras, fueron sometidas a un proceso de extracción, para este efecto, se
descongelaron los tubos lentamente, pasando de una temperatura de -20 °C a 8 °C y
luego a temperatura ambiente; luego, una alícuota de 100 µL de plasma, 15 µL de
Clozapina (200 ηg/mL) y 200 µL de alcohol metílico fueron mezclados y agitados por 3
minutos. Las proteínas precipitadas se separaron por centrifugación a 4.000 rpm a 10
°C durante 10 minutos. Una alícuota de 50 µL de cada muestra del sobrenadante se
inyectó directamente al HPLC.
Los estándares de referencia USP (Imatinib y Clozapina) fueron donados por
Laboratorio Recalcine S.A. Los reactivos y solventes que se emplearon, fueron
adquiridos en Merck Química Chilena.
C.4. Análisis Farmacocinético. Los parámetros farmacocinéticos correspondientes a
la concentración plasmática máxima (Cmáx) y el tiempo máximo (tmáx) se obtuvieron por
inspección directa de las curvas de concentración plasmática en el tiempo. Utilizando el
programa computacional Microsoft Office Excel se graficaron las curvas de
concentración plasmática de Imatinib versus tiempo para cada voluntario.

XXIII
Los parámetros farmacocinéticos tales como la constante de velocidad de
absorción (Ka), el tiempo de vida media (t½), el área bajo la curva de las
concentraciones sanguíneas en el tiempo [ABC (0 – 24) y ABC (0 - ∞)], el volumen de
distribución aparente/F (Vd/F) y el clearance/F (Cl/F), fueron determinados a partir de
los datos que arrojaron las curvas de niveles plasmáticos de la droga en el tiempo pos-
administración. Para ello se utilizó el programa computacional AUC-RPP disponible en
el Laboratorio, donado por el Profesor Ritschel [13].
C.5. Análisis estadístico. Se utilizó el test de análisis de varianza multifactorial
(ANOVA) para establecer las posibles diferencias entre los parámetros determinados
para cada producto farmacéutico en cada voluntario, estimándose una diferencia
estadísticamente significativa para valores de p≤0,05. Se utilizaron las
recomendaciones de las Guías del FDA, tanto para el diseño del estudio como para el
análisis farmacocinético y estadístico. Como fuentes de variación se consideraron el
producto administrado, el período de administración y la secuencia. Además, se
calcularon los intervalos de confianza (IC) de 90% para la razón de las medias
obtenidas con los productos A y B [14].
Schuirmann OST/TOST o la prueba de las dos hipótesis monocaudales (Two One
Side Test Procedure) permite determinar la condición de bioequivalencia entre el
producto farmacéutico en estudio y el innovador. Esta prueba estadística consiste en
descomponer el intervalo de confianza de las hipótesis en dos grupos de hipótesis de
una cola, H0 y H1:
H01: µT - µR ≤ θinf H02: µT - µR ≥ θsup Bioinequivalencia
H11: µT - µR > θinf H02: µT - µR < θsup Bioequivalencia

XXIV
Donde µT - µR es la diferencia entre las medias poblacionales de los productos
en estudio y el innovador.
La hipótesis nula H0 comprende H01 y H02 y establece que T y R no son
bioequivalentes, es decir, bioinequivalentes. La hipótesis alterna H1 comprende H11 y
H12 y establece que T y R son bioequivalentes. Estos datos nos permiten rechazar o no
la hipótesis nula H0, y afirmar si la hipótesis alterna H1 es la verdadera. Sin embargo, si
no se rechaza la hipótesis nula H0, no significa necesariamente que H0 sea verdadera,
sino que no tenemos evidencia suficiente que nos permita concluir que H1 es verdadera
[15]. La evaluación estadística de los resultados se realizó utilizando el programa
computacional STATA versión 10.
C.6. Criterios de Bioequivalencia. Para establecer la bioequivalencia promedio de
fármacos que no poseen características farmacocinéticas y clínicas complejas, se
tomaron los valores del intervalo de confianza de 90%, estos valores deben estar
dentro de los límites de bioequivalencia de 80 % a 125 %, para la razón de las medias
del ABC y Cmáx del producto test (T) respecto del producto de referencia (R). El rango
de aceptación para tmáx se determina para cada fármaco, el cual debe ser clínicamente
relevante. Para establecer la BE, los resultados del estudio deben ser aceptables para
los parámetros farmacocinético ABC y Cmáx [17].

XXV
III. RESULTADOS
A. Adaptación de la metodología bioanalítica. La metodología fue adaptada y
modificada de acuerdo a las condiciones presentes en el laboratorio. Debido a que el
estándar interno utilizado en la bibliografía consultada no estaba disponible en el
laboratorio (Imatinib deuterizado), se procedió a realizar una búsqueda de un estándar
interno, mediante ensayos cromatográficos se realizaron pruebas con distintos
estándares que debían cumplir con un grado de similitud estructural, tener un rango de
absorción y un tiempo de retención similar a Imatinib; se hicieron pruebas con
Olanzapina, Pramiprexol, Quetiapina, Escitalopram y Clozapina. Una vez elegido el
estándar interno (Clozapina) se procedió a variar la fase móvil de manera de obtener
una mejor resolución de los analitos; la fase móvil utilizada por Velpandian y
colaboradores, 2004 [12], era KH2PO4 0,02 M y acetonitrilo (70:30 v/v), para obtener el
objetivo planteado se varió gradualmente la proporción de los componentes, hasta
obtener una mezcla final de KH2PO4 0,02 M y acetonitrilo (67:33 v/v).
Un factor crítico en el proceso de extracción del fármaco desde la muestra
plasmática, correspondió al tiempo de agitación. Las muestras utilizadas para la
instalación de la técnica se sometieron a diferentes tiempos de agitación para
establecer la mayor eficiencia del proceso, estos tiempos fueron 1, 2, 3 y 4 minutos. A
los 1 y 2 minutos se obtuvieron % de recuperación menor a un 50 %, con 3 minutos se
obtuvo un % de extracción promedio de 88 %, este porcentaje de extracción no fue
superior a mayor tiempo de agitación, por lo tanto la variable se estableció en tres
minutos. Lo ideal sería obtener una recuperación de un 100 %, sin embargo la FDA

XXVI
acepta que ésta sea hasta un 50 % a 60 %, siempre que los resultados sean precisos,
exactos y reproducibles.
B. Validación de la metodología bioanalítica
B.1. Especificidad. Según la FDA no deben existir interferencias en las muestras
blanco de plasma a los tiempos de retención de los analitos. Esta prueba se realizó
demostrando que en los cromatogramas, a los tiempos de retención de los compuestos
de interés (Imatinib y Clozapina) no existen interferencias de las señales endógenas,
para esto se utilizaron seis fuentes diferentes de plasma. En el cromatograma
experimental (Figura N° 2-A) se observa que no hay interferencias tanto para Imatinib
como para Clozapina, esto se repitió en las seis muestras diferentes de plasma.
B.2. Linealidad. La linealidad del método se obtuvo en un rango de concentración de
Imatinib de 100 a 2000 ηg/mL. Para establecer linealidad se realizaron curvas de
calibración que incluían 6 puntos (100, 200, 500, 1000, 1600 y 2000 ηg/mL). En la
Figura N° 3 se observa una de las curvas experimentales de calibración utilizadas para
la validación, el valor obtenido para el coeficiente de correlación fue 0,9988 y la FDA
exige un coeficiente de correlación mayor a 0,95.
B.3. Exactitud. Se determinó la exactitud del método, realizando una cuantificación de
analito en plasma fortificado con Imatinib y Clozapina en concentraciones conocidas; la
relación de área obtenida se ingresó a la curva de calibración de manera de obtener el
valor de concentración el cual se comparó con el valor real de concentración. Se

XXVII
realizó la medición en tres niveles de concentración (bajo, medio, alto), cada nivel se
repitió 5 veces. Los valores obtenidos en % mostraron una diferencia menor al 15%
que es lo exigido por la FDA (Tabla 1).
B.4. Precisión. Se determinó la precisión del método, realizando una cuantificación de
analito en plasma fortificado con Imatinib y Clozapina en concentraciones conocidas.
Se realizó la medición en tres niveles de concentración (bajo, medio, alto) y se
realizaron ensayos intradía e interdía. Para los ensayos intradía cada medición se
repitió 5 veces y para los ensayos interdía se realizaron un total de 10 mediciones en 3
días distintos. Para determinar la precisión de la metodología se calculó el promedio, la
desviación estándar y el coeficiente de variación para cada punto. Los valores
obtenidos para CV mostraron una diferencia menor al 15% que es lo exigido por la
FDA (Tabla 2 y 3).
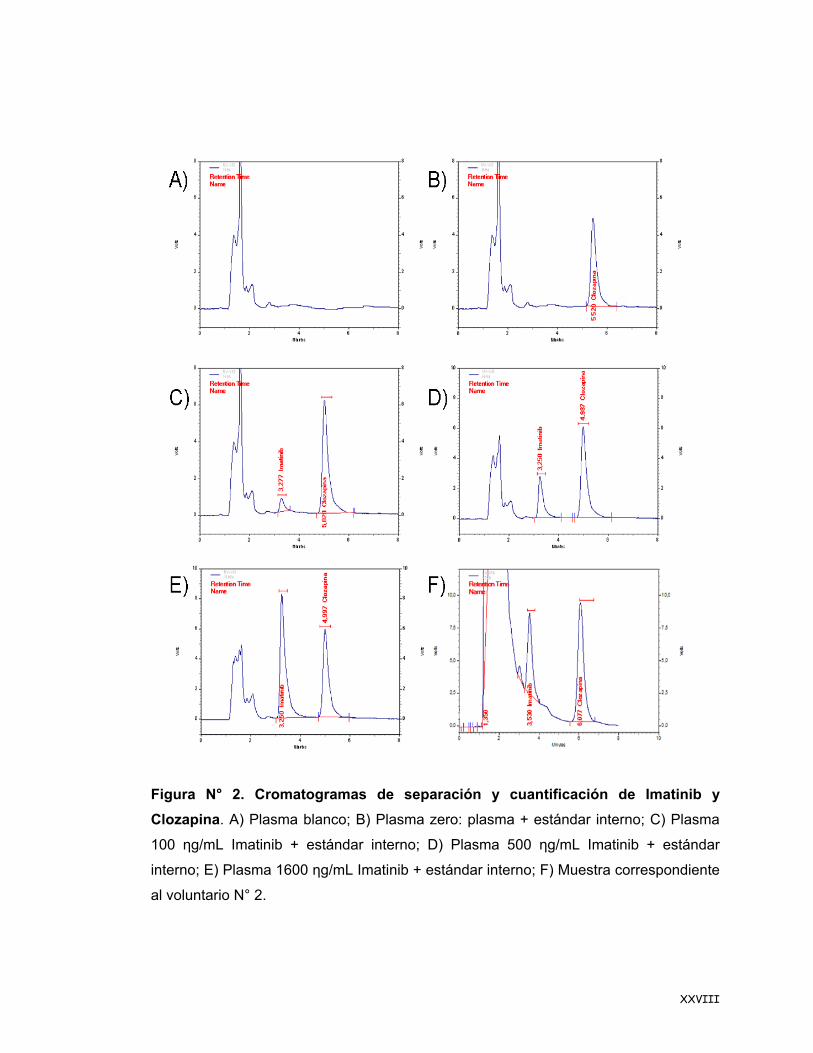
XXVIII
Figura N° 2. Cromatogramas de separación y cuantificación de Imatinib y Clozapina. A) Plasma blanco; B) Plasma zero: plasma + estándar interno; C) Plasma
100 ηg/mL Imatinib + estándar interno; D) Plasma 500 ηg/mL Imatinib + estándar
interno; E) Plasma 1600 ηg/mL Imatinib + estándar interno; F) Muestra correspondiente
al voluntario N° 2.

XXIX
Curva Calibración Imatinib
0
0,2
0,4
0,6
0,8
1
1,2
0 500 1000 1500 2000 2500
Concentración (ng/mL)
Rel
ació
n de
Are
a
Figura N° 3. Curva de Calibración de Imatinib. Eje X= Concentración de Imatinib. Eje
Y= Relación de área: Área de Imatinib / Área de Clozapina.
Tabla 1. Validación Analítica: Determinación de exactitud.
Conc. real (ng/mL) n RA promedio Ecuación
Concentración (ng/mL) (%)
100 5 0,01284 y = 0,0005195x - 0,0330285 88,29 88,294500 5 0,24789 y = 0,0005195x - 0,0330286 477,17 95,434
1600 5 0,80982 y = 0,0005195x - 0,0330287 1558,85 97,428 RA = Relación de Área = Área Imatinib/Área Clozapina
n = número de repeticiones
Tabla 2. Validación Analítica: Determinación de precisión intradía.
Concentración(ng/mL) n RA promedio S.D. C.V.
100 5 0,038012 0,0025157 6,62500 5 0,207811 0,0138739 6,68
1600 5 0,780372 0,0200576 2,57

XXX
Tabla 3. Validación Analítica: Determinación de precisión interdía.
Concentración(ng/mL) n RA promedio S.D. C.V.
100 10 0,03669 0,0049544 13,50500 10 0,204964 0,0114516 5,59
1600 10 0,800178 0,0231695 2,90 RA = Relación de Área = Área Imatinib/Área Clozapina
S.D. = Desviación estándar
CV. = Coeficiente de variación
n = número de repeticiones
B.5. Limite de Detección y Cuantificación. El límite de detección corresponde a 3
veces la señal ruido de la muestra blanco y el límite de cuantificación corresponde a 5
veces la señal ruido de la muestra blanco. Los límites fueron 18 ηg/mL y 60 ηg/mL,
respectivamente.
B.6. Recuperación. Se determinó la recuperación del proceso de extracción,
realizando una cuantificación de analito en plasma y agua, ambos fortificados con
Imatinib y Clozapina en concentraciones conocidas. Las muestras de plasma se
sometieron al proceso de extracción descrito para el método. Se realizó la medición en
tres niveles de concentración (bajo, medio, alto), cada nivel se repitió 5 veces. Se
comparó la relación de área obtenida en la muestras de plasma respecto a las
obtenidas en las muestras preparadas en agua. Los resultados se muestran en la
Tabla 4, los valores obtenidos para % de recuperación muestran un valor menor al
100%, valor aceptado por la FDA.

XXXI
Tabla 4. Validación Analítica: Determinación del porcentaje de recuperación.
Concentración (ng/mL) n
RA promedio plasma
RA promedio agua % Recuperación
100 5 0,035390 0,040716 86,92500 5 0,214944 0,244588 87,88
1600 5 0,806179 0,903687 89,21
RA = Relación de Área = Área Imatinib/Área Clozapina
n = número de repeticiones
B.7. Estabilidad. Se determinó la estabilidad de las muestras en tres niveles de
concentración, previamente a los ensayos de estabilidad, cada una de las muestras se
analizó.
a) Estabilidad por ciclos de congelamiento y descongelamiento. Las muestras
sometidas a los ciclos de congelamiento/descongelamiento fueron estables luego de
tres ciclos.
b) Estabilidad de corta duración. Las muestras se mantuvieron a temperatura
ambiente durante un período de 24 horas; todas ellas fueron estables en estas
condiciones.
c) Estabilidad de larga duración. Las muestras se congelaron durante dos semanas,
tiempo que transcurrió entre la primera toma de muestra y la segunda toma de
muestra; luego se descongelaron y se midió la concentración del fármaco. Todas las
muestras fueron estables en estas condiciones.
d) Estabilidad de solución stock. Las soluciones stock de Imatinib y Clozapina fueron
almacenadas a 4 °C y -20 °C, respectivamente durante un mes; debido a que las
muestras no eran estables luego de este período se procedió a preparar soluciones
stock nuevas.

XXXII
e) Estabilidad post-preparación. Terminado el proceso de extracción, las muestras
se mantuvieron durante 6 horas a 20 ºC. También en estas condiciones las muestras
permanecieron estables.
C. Características antropométricas de los voluntarios. La Tabla 5 muestra el
promedio, la desviación estándar y el coeficiente de variación para los parámetros de
edad, peso, altura y el índice de masa corporal (IMC) de los voluntarios.
Tabla 5. Características Antropométricas de los voluntarios.
Iniciales Peso (kg) Talla (cm) Edad IMC 1 AJB 79 164 46 29,37 2 CSP 67 160 22 26,17 3 CNT 82 179 21 25,59 5 DCV 52 167 167 18,65 6 FAL 63 160 39 24,61 8 JLB 70 176 22 22,6 9 JAR 59 179 27 18,41
10 JGB 105 188 26 29,71 11 LYS 56 154 38 23,61 12 MGG 76 174 174 25,1 13 MCJ 63 162 28 24,01 14 MVV 53 152 31 22,94 15 MLV 65 163 21 24,46 16 MRP 68 151 51 29,82 17 NRS 66 160 34 25,78 19 XNG 52 164 28 19,33 20 YLP 59,9 167 167 21,48 21 FMA 74,4 179 21 23,22 22 FCA 76,5 175 21 24,98 23 NNV 48 156 25 19,72
PROMEDIO. 67,04 166,5 50,45 23,98 D.E. 13,33 10,36 51,95 3,39
C.V. (%) 19,88 6,22 102,97 14,15

XXXIII
Los voluntarios 4, 7, 18 y 24 no se presentaron al estudio, a pesar de haber sido
evaluados y seleccionados por el Médico durante la primera semana. Ellos no dieron
excusa alguna, lo que no es exigible, debido a las normas éticas, expresamente
señaladas en el consentimiento informado (Anexo 3).
D. Análisis físico-químico del producto test Zeite® y el producto referencia
Glivec®. Estos análisis fueron realizados en el “Laboratorios Recalcine S.A”. El
resultado de los análisis se muestra en el Anexo 4. En este análisis se demostró que
ambas formulaciones cumplían con las especificaciones de la United States
Pharmacopeia (USP), en cuanto a identidad, valoración y test de disolución, de manera
que ambas productos cumplen con los criterios de calidad.
E. Exámenes clínicos y de laboratorio clínico. En el Anexo 5 se muestra a modo de
ejemplo un informe de laboratorio con los resultados clínicos de uno de los voluntarios
que fue seleccionado por el equipo médico para participar en este estudio. Los
resultados de los exámenes clínicos se adjuntan a la ficha clínica, la cual fue utilizada
exclusivamente por el médico del estudio (Anexo 6). Tanto los resultados de los
exámenes como la información registrada en la ficha son confidenciales. El conjunto de
antecedentes médicos fueron entregados, una vez finalizado el estudio al Director de
este proyecto, Dr. Q.F. Iván Saavedra.

XXXIV
F. Consentimiento informado. En el Anexo 3 se muestra la copia del consentimiento
informado entregada a cada uno de los voluntarios participantes en el estudio. Tanto
este documento como el proyecto fueron aprobados por la Comisión de Ética de
Investigación en Seres Humanos de la Facultad de Medicina de la Universidad de
Chile.
G. Dieta administrada a los voluntarios. La dieta fue controlada, estrictamente
estandarizada y especificada. Los alimentos administrados a cada voluntario fueron
determinados por una Nutricionista y aparecen en el Anexo 1.
H. Análisis cromatográfico de las muestras plasmáticas. De los 20 voluntarios que
iniciaron el estudio, un número de 19 finalizó el estudio, ya que 1 de ellos fue retirado
por el médico a cargo debido a que presentó vómitos 1 hora post-administración del
fármaco.
De los 19 voluntarios que completaron ambos períodos de estudio, 2 de ellos
presentaron interferencias en el análisis cromatográfico para el tiempo de retención de
Imatinib, esta interferencia se presentó a partir de la muestra correspondiente al tiempo
0. Los datos de concentración plasmática en el tiempo, para los 17 voluntarios que
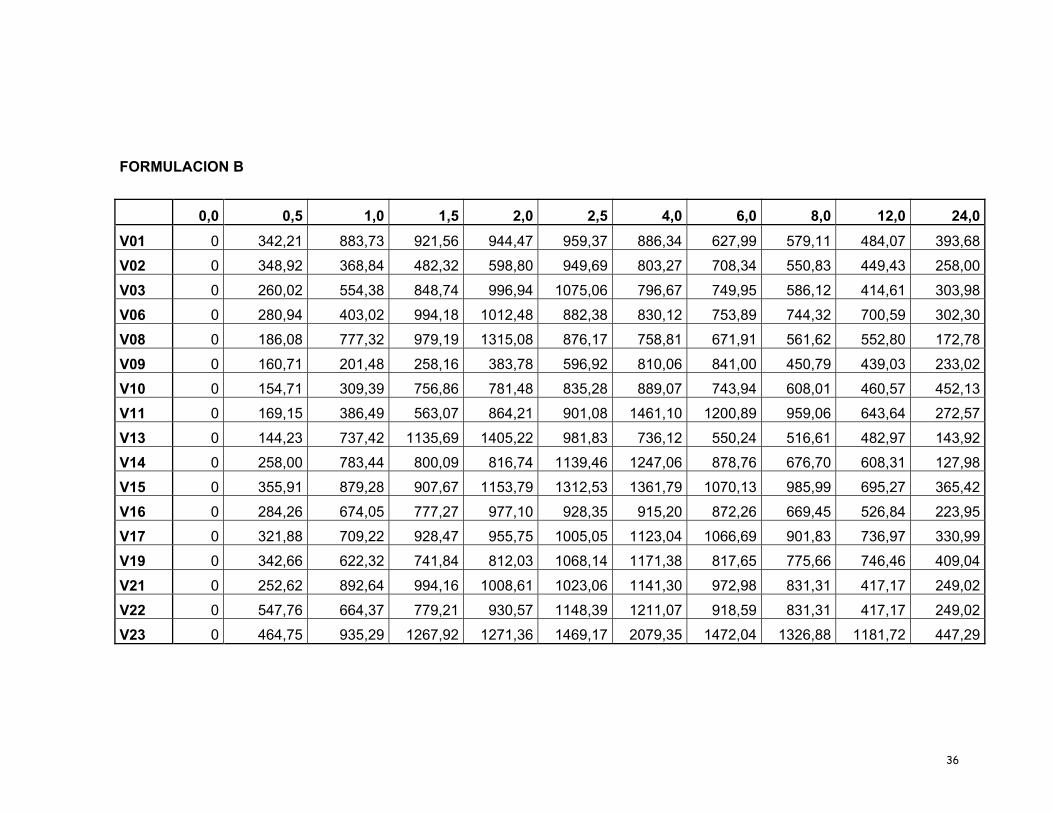
participaron en el estudio completo se presentan en la Tabla 6.
Las concentraciones promedio de Imatinib y la desviación estándar (ηg/mL) de
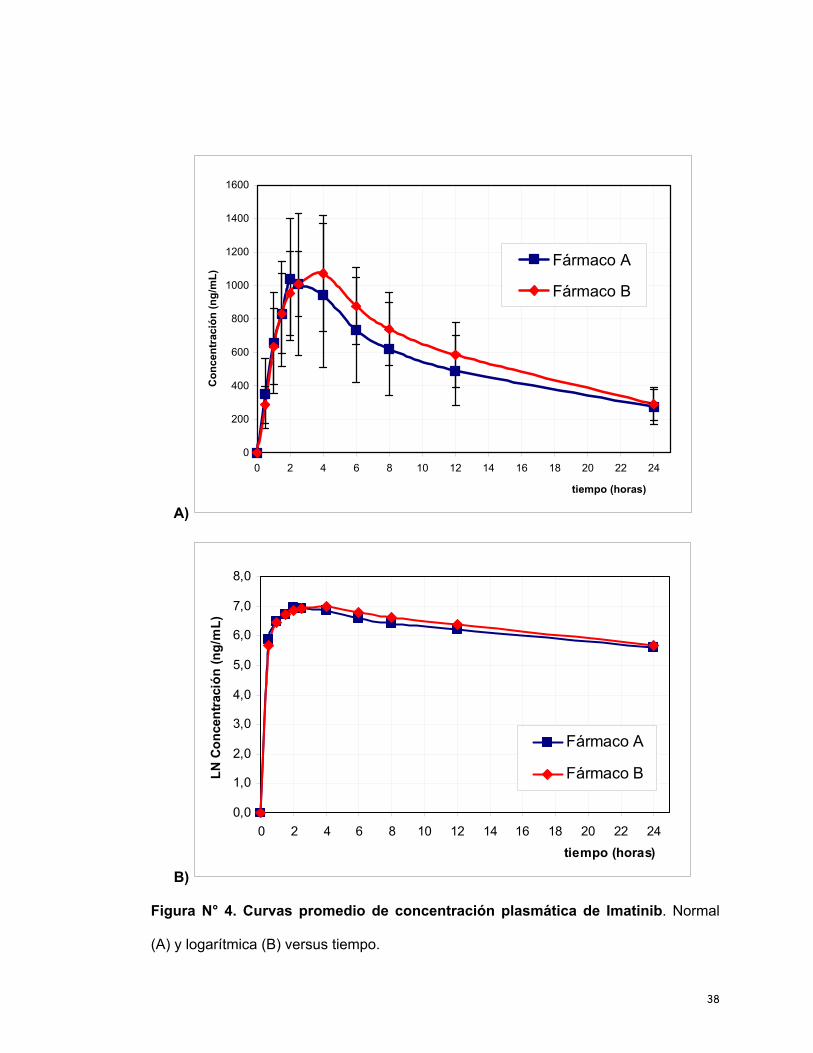
ambos productos aparecen en la Tabla 7. A partir de estos valores se construyeron las
curvas promedio de concentraciones plasmáticas de Imatinib (A y B) vs el tiempo
transcurrido desde la administración del fármaco hasta las 24 horas; estos resultados
se muestran en la Figura N° 4.

35
Tabla 6. Concentraciones plasmáticas (ηg/mL) vs el tiempo (h) en 17 voluntarios sanos para A y B.
FORMULACION A
0,0 0,5 1,0 1,5 2,0 2,5 4,0 6,0 8,0 12,0 24,0V01 0 294,75 544,65 688,56 871,73 842,25 774,48 745,23 697,73 642,00 284,81
V02 0 417,97 796,27 818,71 1019,48 1045,45 1200,44 1121,72 1043,26 783,38 453,31
V03 0 455,03 986,95 1034,41 1317,43 916,58 784,07 651,56 488,87 422,48 257,99
V06 0 251,17 531,02 725,04 838,01 780,30 772,05 668,90 605,75 476,67 270,01
V08 0 133,81 262,86 428,37 510,71 438,03 405,78 388,16 335,20 244,28 169,51
V09 0 139,88 274,16 376,61 767,60 430,51 367,32 361,99 302,00 298,60 153,60
V10 0 187,98 474,74 1292,47 1702,15 1701,67 484,34 351,74 319,42 287,10 221,94
V11 0 651,17 1201,87 1530,56 1859,25 2079,78 1614,12 773,89 522,85 380,95 253,67
V13 0 220,26 439,57 768,50 939,98 779,62 745,61 711,57 699,49 654,32 213,48
V14 0 140,61 432,80 603,62 1165,14 985,65 806,16 585,72 461,21 335,58 175,46
V15 0 929,46 1137,42 1161,68 1436,73 1363,31 1233,54 1045,86 727,98 603,73 364,16
V16 0 185,57 442,81 490,56 759,18 696,36 511,06 325,76 272,32 272,09 159,46
V17 0 289,40 449,78 734,05 818,15 1160,73 1441,38 1100,50 760,99 747,04 264,36
V19 0 346,88 606,49 807,82 876,16 944,50 1113,39 1376,49 1319,11 745,23 493,79
V21 0 495,29 604,97 608,82 623,82 677,10 1316,05 589,08 565,71 542,33 256,29
V22 0 418,43 1079,79 1096,47 1151,53 982,24 671,33 596,98 522,62 140,29 118,76
V23 0 443,37 884,85 934,99 985,13 1285,11 1783,50 1089,95 895,46 758,66 437,99
36
FORMULACION B
0,0 0,5 1,0 1,5 2,0 2,5 4,0 6,0 8,0 12,0 24,0V01 0 342,21 883,73 921,56 944,47 959,37 886,34 627,99 579,11 484,07 393,68
V02 0 348,92 368,84 482,32 598,80 949,69 803,27 708,34 550,83 449,43 258,00
V03 0 260,02 554,38 848,74 996,94 1075,06 796,67 749,95 586,12 414,61 303,98
V06 0 280,94 403,02 994,18 1012,48 882,38 830,12 753,89 744,32 700,59 302,30
V08 0 186,08 777,32 979,19 1315,08 876,17 758,81 671,91 561,62 552,80 172,78
V09 0 160,71 201,48 258,16 383,78 596,92 810,06 841,00 450,79 439,03 233,02
V10 0 154,71 309,39 756,86 781,48 835,28 889,07 743,94 608,01 460,57 452,13
V11 0 169,15 386,49 563,07 864,21 901,08 1461,10 1200,89 959,06 643,64 272,57
V13 0 144,23 737,42 1135,69 1405,22 981,83 736,12 550,24 516,61 482,97 143,92
V14 0 258,00 783,44 800,09 816,74 1139,46 1247,06 878,76 676,70 608,31 127,98
V15 0 355,91 879,28 907,67 1153,79 1312,53 1361,79 1070,13 985,99 695,27 365,42
V16 0 284,26 674,05 777,27 977,10 928,35 915,20 872,26 669,45 526,84 223,95
V17 0 321,88 709,22 928,47 955,75 1005,05 1123,04 1066,69 901,83 736,97 330,99
V19 0 342,66 622,32 741,84 812,03 1068,14 1171,38 817,65 775,66 746,46 409,04
V21 0 252,62 892,64 994,16 1008,61 1023,06 1141,30 972,98 831,31 417,17 249,02
V22 0 547,76 664,37 779,21 930,57 1148,39 1211,07 918,59 831,31 417,17 249,02
V23 0 464,75 935,29 1267,92 1271,36 1469,17 2079,35 1472,04 1326,88 1181,72 447,29

37
Tabla 7. Concentraciones promedio de Imatinib (ηg/mL)
Tiempo (h) Producto A Producto B
PROMEDIO D.E. PROMEDIO D.E.
0,0 0,00 0,00 0,00 0,00
0,5 353,00 208,84 286,75 110,75
1,0 655,94 300,19 634,27 227,18
1,5 829,49 312,79 831,55 240,35
2,0 1037,77 365,46 954,61 251,52
2,5 1006,42 423,35 1008,94 194,77
4,0 942,62 429,78 1071,87 344,24
6,0 734,42 312,97 877,49 228,63
8,0 620,00 279,02 738,57 219,97
12,0 490,28 209,71 585,74 193,69
24,0 267,56 106,80 290,30 99,15
38
A)
0
200
400
600
800
1000
1200
1400
1600
0 2 4 6 8 10 12 14 16 18 20 22 24
tiempo (horas)
Con
cent
raci
ón (n
g/m
L)
Fármaco A
Fármaco B
B)
0,0
1,0
2,0
3,0
4,0
5,0
6,0
7,0
8,0
0 2 4 6 8 10 12 14 16 18 20 22 24tiempo (horas)
LN C
once
ntra
ción
(ng/
mL)
Fármaco A
Fármaco B
Figura N° 4. Curvas promedio de concentración plasmática de Imatinib. Normal
(A) y logarítmica (B) versus tiempo.

39
I. Análisis Farmacocinético. Los datos correspondientes a Cmáx y tmáx fueron
obtenidos por observación directa de las tablas y curvas de concentración plasmática
de Imatinib vs tiempo. Para realizar el análisis, y así obtener los otros parámetros
farmacocinéticos: área bajo la curva (ABC0-24, ABC0-∞), tiempo de vida media (t1/2),
constante de absorción (ka), tiempo medio de residencia (MRT), clearence (CL/f) y
volumen de distribución (Vd/f), se utilizó el programa computacional AUC-RPP. El
informe de estos parámetros para cada voluntario y formulación se encuentran en el
Anexo 7.
Los parámetros farmacocinéticos para establecer bioequivalencia son Cmáx, ABC0-
24 y ABC0-∞, el parámetro tmáx es utilizado para establecer equivalencia cuando este es
clínicamente importante. Así los parámetros obtenidos de cada voluntario para cada
formulación, fueron expresados en números naturales y logaritmo natural. Los
resultados se presentan como el promedio de los valores y su desviación estándar
(Tabla 8).
Los otros parámetros: Intercepto, tiempo de vida media (t1/2), constante de
velocidad de absorción (ka), tiempo medio de residencia (MRT), clearence (CL/f) y
volumen de distribución (Vd/f), se calcularon de manera de caracterizar cinéticamente
de forma más completa al fármaco, estos parámetros no son necesarios para
determinar BE. El informe para estos parámetros se detalla en la Tabla 9.

40
Tabla 8. Parámetros farmacocinéticas ABC 0→24, ABC 0→∞, Cmáx y tmáx.
A B A B A B A B Vol. N° Cmáx (ng/mL) Cmáx (ng/mL) ABC 0a24 (ugxh/mL) ABC 0a24 (ugxh/mL) ABC 0a∞ (ugxh/mL) ABC 0a∞ (ugxh/mL) tmáx tmáx Ln Ln Ln Ln Ln Ln (h) (h)
1 871,72 6,7705 959,37 6,8663 13,8260 2,6265 13,2834 2,58651 18,6773 2,9273 29,0288 3,3683 2,0 2,5
2 1200,44 7,0904 949,69 6,8561 19,0326 2,9462 11,4675 2,43952 27,9491 3,3304 16,9408 2,8297 4,0 2,5
3 1317,42 7,1834 1075,06 6,9801 11,8831 2,4751 12,1983 2,50130 18,2985 2,9068 20,2970 3,0105 2,0 2,5
6 838,01 6,7310 1012,48 6,9202 11,8925 2,4759 14,8397 2,69731 17,2559 2,8482 19,9218 2,9918 2,0 2,0
8 510,71 6,2358 1315,08 7,1817 6,5693 1,8824 12,3207 2,51128 10,2861 2,3308 14,6975 2,6877 2,0 2,0
9 767,60 6,6433 841,00 6,7346 6,7928 1,9159 10,4613 2,34769 10,1913 2,3215 15,7685 2,7580 2,0 6,0
10 1702,15 7,4396 889,06 6,7902 9,6681 2,2688 13,1017 2,57274 18,7596 2,9317 32,3552 3,4768 2,0 4,0
11 2079,78 7,6400 1461,10 7,2869 15,2119 2,7221 16,5130 2,80415 21,1677 3,0525 20,0525 2,9984 2,5 4,0
13 939,18 6,8450 1405,22 7,2479 13,3057 2,5882 11,3589 2,43000 16,0218 2,7739 13,0576 2,5694 2,0 2,0
14 1165,14 7,0606 1247,06 7,1285 9,8600 2,2885 14,0730 2,64425 12,8373 2,5524 15,2730 2,7261 2,0 4,0
15 1436,73 7,2701 1361,79 7,2166 17,1451 2,8417 18,1969 2,90125 25,6093 3,2430 24,2498 3,1884 2,0 4,0
16 759,18 6,6322 977,10 6,8846 7,1318 1,9646 13,1976 2,58004 11,5605 2,4476 16,4387 2,7996 2,0 2,0
17 1441,38 7,2734 1123,04 7,0238 16,8753 2,8259 17,1486 2,84192 20,4189 3,0165 22,3050 3,1048 4,0 4,0
19 1376,49 7,2273 1171,38 7,0659 19,8465 2,9880 16,7657 2,81934 28,7992 3,3603 26,4283 3,2744 6,0 4,0
21 1316,05 7,1824 1141,30 7,0399 12,8983 2,5571 13,8657 2,62942 17,7819 2,8782 17,2052 2,8452 4,0 4,0
22 1151,52 7,0488 1211,06 7,0993 8,6266 2,1549 13,8913 2,63127 9,8084 2,2832 17,3189 2,8518 4,0 4,0
23 1783,50 7,4863 2079,35 7,6398 19,5939 2,9752 26,1399 3,26346 29,3387 3,3789 32,4433 3,4795 4,0 4,0
Prom. 1215,12 7,0447 1189,42 7,0566 12,9506 2,4998 14,6367 2,65891 18,5154 2,8578 20,8107 2,9977 2,9 3,4
DE. 410,96 0,3607 293,70 0,2214 4,4824 0,3700 3,6799 0,21948 6,4942 0,3654 6,1045 0,2791 1,2 1,1

41
Tabla 9. Otros parámetros farmacocinéticos de Imatinib.
Fármaco A Cltot/f Cltot/f Vol. N° Lambda Z
(1/H) t½ (h) ka (1/h) MRT (h)
(mL/min) (mL/min/Kg)Vdbeta/f
(mL) Vdbeta/f (L/Kg)
1 0,0587 11,81 3,0667 18,03 356,93 4,3529 364797,80 4,44882 0,0508 13,63 1,8400 20,81 238,53 3,5601 281508,90 4,20163 0,0402 17,24 3,0667 23,02 364,33 4,4430 543576,80 6,62906 0,0503 13,76 3,0667 20,37 386,34 6,1324 460451,30 7,30888 0,0456 15,20 3,0667 22,96 648,14 9,2591 852651,10 12,18079 0,0452 15,33 3,0667 21,98 654,24 11,0888 868528,40 14,7208
10 0,0244 28,39 3,0667 35,85 355,37 3,3845 873443,40 8,318511 0,0426 16,27 2,3000 18,80 314,95 5,6240 443668,00 7,922613 0,0786 8,82 3,0667 14,20 416,10 6,6048 317643,50 5,042014 0,0589 11,76 3,0667 16,17 519,32 9,7985 528717,90 9,975815 0,0430 16,11 3,0667 21,69 260,32 4,0050 363039,70 5,585216 0,0360 19,25 3,0667 25,56 576,68 7,7929 960972,80 12,986117 0,0746 9,29 1,8400 14,13 326,48 4,9467 262604,50 3,978819 0,0516 12,57 1,1500 20,14 231,48 4,4516 251827,90 4,842821 0,0525 13,21 1,8400 18,69 374,91 5,0664 428631,40 5,792322 0,1005 6,90 3,0667 10,21 679,69 8,9433 405710,30 5,338323 0,0449 15,42 2,5000 21,78 227,23 4,7340 303336,90 6,3195
Promedio 0,0529 14,41 2,6590 20,26 407,71 6,1287 500653,56 7,3877DE. 0,0179 4,80 0,6261 5,57 152,39 2,3843 238251,72 3,2600CV. 33,8434 33,34 23,5459 27,47 37,38 38,9040 47,59 44,1277Máximo 0,1005 28,39 3,0667 35,85 679,69 11,0888 960972,80 14,7208Mínimo 0,0244 6,90 1,1500 10,21 227,23 3,3845 251827,90 3,9788Mediana 0,0503 13,76 3,0667 20,37 364,33 5,0664 428631,40 6,3195

42
Fármaco B Cltot/f Cltot/f Vol. N° Lambda Z
(1/H) t ½ (h) ka (1/h) MRT (h)
(mL/min) (mL/min/Kg)Vdbeta/f
(mL) Vdbeta/f (L/Kg)
1 0,0250 27,72 2,3000 39,37 229,65 2,8006 551095,10 6,72072 0,0471 14,70 2,3000 21,17 393,53 5,8736 500903,60 7,47623 0,0375 18,47 2,3000 25,97 328,46 4,0056 525047,80 6,40306 0,0595 11,65 3,0667 17,83 334,64 5,3118 337549,00 5,35798 0,0727 9,54 3,0667 13,56 453,58 6,4797 374394,60 5,34849 0.043906 15,79 2,3000 22,27 422,78 7,1658 577750,00 9,7924
10 0,0235 29,51 2,3000 44,06 206,24 1,9642 526897,80 5,018111 0,0770 9,00 1,8400 14,13 332,46 5,9368 259034,80 4,625613 0,0843 8,18 3,0667 12,03 510,56 8,1041 361558,50 5,739014 0,1066 6,50 1,8400 10,29 436,50 8,2359 245571,60 4,633415 0,0604 11,48 1,8400 17,18 274,92 4,2296 273217,60 4,203316 0,0691 10,03 3,0667 14,89 405,56 5,4806 352006,70 4,756817 0,0642 10,80 1,8400 16,56 298,89 4,5286 279373,30 4,232919 0,0423 16,37 1,8400 23,91 252,25 4,8511 357535,00 6,875721 0,0746 9,29 1,8400 14,26 387,49 5,2363 311758,50 4,213022 0,0727 9,54 1,8400 14,41 384,94 5,0650 317885,60 4,182823 0,0710 9,77 1,8400 15,05 205,49 4,2810 173746,70 3,6197
Promedio 0,0617 13,43 2,2639 19,82 344,58 5,2676 372078,01 5,4823DE. 0,0221 6,55 0,5014 9,30 90,57 1,6620 120751,92 1,5640CV. 35,7502 48,74 22,1461 46,93 26,28 31,5509 32,45 28,5279Máximo 0,1066 29,51 3,0667 44,06 510,56 8,2359 577750,00 9,7924Mínimo 0,0235 6,50 1,8400 10,29 205,49 1,9642 173746,70 3,6197Mediana 0,0667 10,80 2,3000 16,56 334,64 5,2363 352006,70 5,0181
Lambda Z: Intercepto; t ½: tiempo de vida media; ka: constante de absorción; MRT: tiempo medio de residencia;
Cl/f: clearence; Vdbeta/f: volumen de distribución; f: fármaco.

43
J. Análisis estadístico de las muestras. Como sugiere la FDA, se realizó un análisis
de varianza (ANOVA). En la Tabla 10, se muestra el resultado final del análisis
aplicado a los datos del estudio. Dicho análisis se realizó introduciendo en el programa
estadístico STATA versión 10 las variables: voluntario, secuencia, período, formulación
y parámetro farmacocinético, y se analizaron, donde la variable dependiente fue el
parámetro farmacocinético (ABC, Cmáx y tmáx) y los efectos principales fueron las
variables secuencia, período y formulación.
Los resultados muestran que no existen efectos de período, formulación y
secuencia significativos (columna p). No se observaron diferencias estadísticamente
significativas entre los parámetros farmacocinéticos Cmáx, tmáx, ABC0-24 y ABC0-∞ de
ambas formulaciones farmacéuticas, ya que los valores de p obtenidos son mayores
que 0,05.

44
Tabla 10. Análisis de varianza para los parámetros farmacocinéticos.
A) Cmáx
N° de Obs: 34 R-cuadrado: 0,0078MSE : 0,309295 R-cuadrado corregido: -0,0915Fuente Parcial SS MS df F pModelo 0,02242 0,00748 3 0,08 0,9713tratamiento 0,00002 0,00023 1 0,00 0,9877secuencia 0,00471 0,00471 1 0,05 0,8259periodo 0,01694 0,01694 1 0,18 0,6769Residual 2,86990 0,09566 30Total 2,89236 0,08765 33
B) tmáx
N° de Obs: 34 R-cuadrado: 0,0928MSE : 1,186 R-cuadrado corregido: 0,0021Fuente Parcial SS MS df F pModelo 4,319 1,440 3 1,02 0,3963tratamiento 2,860 2,860 1 2,03 0,1643secuencia 0,988 0,988 1 0,7 0,4086periodo 0,948 0,948 1 0,67 0,4182Residual 42,211 1,407 30
C) ABC0-24
N° de Obs: 34 R-cuadrado: 0,1432MSE : 0,301173 R-cuadrado corregido: 0,0576Fuente Parcial SS MS df F pModelo 0,45496 0,15165 3 1,67 0,1940tratamiento 0,25897 0,25897 1 2,86 0,1015secuencia 0,15182 0,15182 1 1,67 0,2056periodo 0,08801 0,08801 1 0,97 0,3325Residual 2,72115 0,90705 30Total 3,17611 0,09625 33
D) ABC 0-∞
N° de Obs: 34 R-cuadrado: 0,0865MSE : 0,328715 R-cuadrado corregido: -0,0048Fuente Parcial SS MS df F pModelo 0,30709 0,10237 3 0,95 0,4302tratamiento 0,19727 0,19727 1 1,83 0,1867secuencia 0,08190 0,15182 1 0,76 0,3909periodo 0,05898 0,08801 1 0,55 0,4657Residual 3,24160 0,10805 30Total 3,54870 0,10754 33
SS: Suma de los cuadrados; MS: varianza; df: Grados de libertad.

45
Una vez descartada la existencias de los efectos de secuencia, período y
formulación se analizó si el valor medio (de las ABC y de la Cmáx) del medicamento test
(T) era equivalente con el medicamento referencia (R) dentro de los límites fijados. Se
calcularon los intervalos de confianza al 90% para la razón de las medias
logarítmicamente transformadas para los parámetros farmacocinéticos ABC0-24, ABC0-∞
y Cmáx y no logarítmicamente transformados para tmáx.
La agencia reguladora FDA requiere que los datos utilizados para estudiar la
bioequivalencia se sometan a una transformación logarítmica antes de realizar
cualquier análisis comparativo. Existe un acuerdo general acerca de la comparación
primaria de interés en un estudio de bioequivalencia es la razón entre la formulación
test (T) y la formulación referencia (R) T/R, mas que la diferencia entre las medias. Los
modelos estadísticos disponibles más desarrollados no evalúan cuocientes sino
diferencias, por lo que la transformación logarítmica permite aplicar los modelos
estadísticos óptimos a la razón T/R, expresada como lnT-lnR [18].
K. Evaluación de Bioequivalencia. La Tabla 11 muestra los intervalos de confianza al
90% para Cmáx, ABC0-24, ABC0-∞ y tmáx. Los resultados obtenidos muestran que los
límites del intervalo de confianza se encuentran dentro de los límites de equivalencia
(80 % a 125 %) para los parámetros Cmáx, ABC0-24 y ABC0-∞. El límite inferior para tmáx
se encuentra por fuera del límite de equivalencia, pero este dato es considerado como
parámetro necesario para la equivalencia cuando tiene una importancia clínica
relevante, para el caso de Imatinib no es así ya que el esquema posológico es de una
dosis diaria y donde los resultados clínicos se miden a largo plazo.

46
Tabla 11. Intervalos de Confianza al 90%.
inferior superior inferior superior
diferencia -1,411 1,411 -0,167 0,143radio % 80,0 125,0 97,64 102,03
diferencia -0,532 0,532 -0,284 -0,034radio % 80,0 125,0 89,31 98,73
Ln ABC 0a∞diferencia -0,600 0,600 -0,262 -0,018radio % 80,0 125,0 91,27 99,40
diferencia -0,653 0,653 -1,144 0,085radio % 80,0 125,0 66,19 102,51
tmax
Limites de Limites del Test
Ln Cmax
Ln ABC 0a24
Tratamiento A: medicamento test
Tratamiento B: medicamento referencia
La Tabla 12 muestra el análisis estadístico utilizando el método de Schuirmann.
Este método indica los valores inferiores y superiores para la hipótesis nula. Los
resultados muestran que la diferencias entre los parámetros cinéticos ABC0-24, ABC0-∞ y
Cmáx fueron estadísticamente significativas, rechazando la hipótesis nula (p<0,05). Por
lo tanto, ambas formulaciones farmacéuticas son equivalentes. Para el parámetro tmáx
se obtuvieron valores mayores a 0,05, lo que no permite rechazar la hipótesis nula,
pero tal como se mencionó anteriormente este parámetro no es necesario para
demostrar bioequivalencia.

47
Tabla 12. Prueba de dos hipótesis de Schuirmann para los parámetros
farmacocinéticos.
Upper sideLower side
Upper sideLower side
Ln ABC 0a∞Upper sideLower side
Upper sideLower side 0,420 0,681
6,616 <0.0001tmáx
-3,441 0,004
5,217 <0.0001
-10,642 <0.0001
15,818 <0.0001Ln ABC 0a24
-9,671 <0.0001
Two One Side Test pLn Cmáx
-16,117 <0.0001
Ambas metodologías cumplen con los requisitos de la FDA de EEUU para probar
bioequivalencia, cuyas regulaciones establecen que para ABC0-24, ABC0-∞ y Cmáx los
intervalos de confianza deben estar dentro de los limites de 80 % a 125 % para la
razón A/B y además de lo anterior la hipótesis nula planteada se debe rechazar.
L. Encuesta de reacciones Adversas. La encuesta fue realizada por el Médico y
estuvo orientada a detectar los efectos adversos. Se presentaron reacciones adversas
relacionadas con el sistema gastrointestinal (vómitos) y relacionadas con el sistema
nervioso central (cefalea y nausea). Todas las reacciones adversas medicamentosas
fueron leves y tratadas no farmacológicamente con reposo y observación.

48
IV. DISCUSIÓN
La metodología analítica fue adaptada y modificada en dos aspectos, primero se
utilizó Clozapina como estándar interno a diferencia de Imatinib deuterizado que es el
utilizado por Velpandian y colaboradores, 2004 [12]. La segunda modificación realizada
consistió en variar la relación de los componentes de la fase móvil de manera de
aumentar la resolución de los picos de interés, Imatinib y Clozapina.
Los resultados obtenidos para la validación de la técnica analítica fueron los
requeridos por la guía de la FDA [21] para este tipo de análisis. Las curvas de
calibración poseían un coeficiente de relación superior a 0,95; la metodología posee
una linealidad entre 100 y 2000 ηg/mL. La exactitud y precisión de la metodología
cumplió con lo exigido. El error estándar de los valores de las mediciones de exactitud
no superó el 15 %. Asimismo, el coeficiente de variación para los valores de precisión
tampoco superó el 15 %. El rendimiento del proceso de extracción fue 88% en
promedio. Los ensayos de estabilidad mostraron que las muestras eran estables en
todas las condiciones testeadas.
Por el método estadístico ANOVA, se analizó el efecto de secuencia, período y
formulación. Este análisis estadístico es útil cuando se aplica en situaciones complejas
ya que permite, mediante una prueba única, observar si los datos de una población son
diferentes entre si; además, asigna diferencias a otras fuentes de error como la
formulación, los grupos, los períodos y otras causas aleatorias que se definen como
errores experimentales.
Para que dos formulaciones sean declaradas bioequivalentes, la FDA ha
establecido que los intervalos de confianza de la razón de las medias deben caer en un
intervalo de 80% a 125%. Los resultados obtenidos para los parámetros ABC 0-24, ABC

49
0-∞ y Cmáx cumplen con esta condición, por lo tanto ambas formulaciones pueden ser
intercambiables. El intervalo de confianza para tmáx en su límite inferior escapa del
rango de aceptación, ya que en el caso de este fármaco no es clínicamente relevante.
El número de sujetos que participan en un estudio de biodisponibilidad relativa
para la bioequivalencia debe ser calculado sobre la base de estimaciones estadísticas.
Para este efecto es necesario conocer la variabilidad de los parámetros
farmacocinéticos tanto intraindividual como interindividual. Con respecto a esto, se
presenta el problema de la escasa información bibliográfica de la cual se dispone y lo
segundo, que es poco viable realizar un estudio para determinar esta variabilidad. Por
lo tanto, la elección del número de sujetos a incluir en este tipo de estudio se realiza
sobre lo aconsejado por las entidades regulatorias, que indican un número ideal de 18
a 24 voluntarios.
Finalmente, los antecedentes presentados nos permiten aseverar que los
productos Zeite® y Glivec® son equivalentes farmacéuticos y terapéuticos. Esto
implica que ambos productos son desarrollados en la misma forma farmacéutica y
poseen el mismo principio activo, en la misma concentración y que sus perfiles de
disolución se encuentran dentro de lo aceptado por la farmacopea; más aún, no se
observaron diferencias estadísticamente significativas entre los valores de las pruebas
farmacocinéticas utilizadas para determinar biodisponibilidad.

50
V. REFERENCIAS
1. Deininger M, Druker B. (2003). Specific targeted therapy of chronic myelogenous
leukemia with Imatinib. Pharmacol. Rev. 55:401-423.
2. Zhang Z, Li M, Rayburn ER, Hill DL, Zhang R, Wang H. (2005). Oncogenes as
novel targets for cancer therapy (part I): growth factors and protein tyrosine
kinases. Am. J. Pharmacogenomics. 5(3):173-190.
3. O’Brien S, Guilhot F, Larson R, Gathmann I, Baccarani M, Cervantes F,
Cornelissen J, Fischer T, Hochhaus A, Hughes T, Lechner K, Nielsen J,
Rousselot P, Reiffers J, Saglio G, Shepherd J, Simonsson B, Gratwohl A,
Goldman J, Kantarjian H, Taylor K, Verhoef G, Bolton A, Capdeville R, Druker B.
(2003). Imatinib Compared with Interferon and Low-Dose Cytarabine for Newly
Diagnosed Chronic-Phase Chronic Myeloid Leukemia. New Engl. J. Med. 348:
994-1004.
4. Nikolova Z, Peng B, Hubert M, Seiberling M, Keller U, Ho Y, Schan H, Capdeville
R. (2004). Bioequivalence, safety, and tolerability of Imatinib tablets compared
with capsules. Cancer Chemother. Pharmacol. 53: 433-438.
5. Peng B, Lloyd P, Schan H. (2005). Clinical pharmacokinetics of Imatinib. Clin.
Pharmacokinet. 44 (9): 879-94.
6. Dutreix C, Peng B, Mehing G, Hayes M, Capdeville R, Pokorny R, Seiberling M.
(2004). Pharmacokinetic interaction between ketoconazole and Imatinib mesylate
(glivec) in healthy subjects. Cancer Chemother. Pharmacol. 54: 290-294.
7. Novartis contra la India: el caso Glivec. FARMAMUNDI. Documento de posición
Nº1(Vol. 1). 1-7, 2007.

51
8. Guidelines on registration requirements to establish interchangeability, WHO
Technical Report Series, Nº 937, 2006 (WHO-06).
9. Guidance for Industry. “BA and BE Studies for Orally Administered Drug Products-
General Considerations” Draft Guidance, US. Department of Health and Human
Services, FDA, Center for Drug Evaluation and Research, 1999.
10. Saavedra I. Conceptos de farmacodinamia y farmacocinética en la perspectiva
clínica. En, Rosselot E. y Biagini L. (Eds.). Farmacología Clínica en Medicina
Interna. Series Clínicas Sociedad Médica de Santiago, Editorial Mediterráneo,
Vol. VII (1): 15-27, 1988
11. Declaración de Helsinki. Recomendaciones para guiar a los médicos en la
investigación biomédica en seres humanos, hecha por la Asociación Médica
Mundial en la Asamblea del Helsinki, Finlandia, en 1964. En Ética Médica,
normas y documentos. Colegio Médico de Chile. Graphos Comunicaciones Ltda;
pp. 91- 95, 1986.
12. Velpandian T, Mathur R, Agarwal N, Arora B, Kumar L, Gupta S. (2004).
Development and validation of a simple liquid chromatographic method with
ultraviolet detection for the determination of Imatinib in biological samples. J.
Chromatogr. B 804: 431-434.
13. Ritschel, WA. (1986). “AUC-RPP Basic computer program for compartment model
independent pharmacokinetic analyisis”. Meth. Find. Exptl. Clin. Pharmacol. 8:
633-640.
14. Norma que define criterios para establecer equivalencia terapéutica a productos
farmacéuticos en Chile. Unidad de Asuntos Farmacéuticos. Ministerio de Salud.
Diario Oficial. Resol. Exenta N° 727, 29 Noviembre 2005.

52
15. Schuirmann DJ. (1987). “A comparison of the two one–sided tests procedure and
the power. Approach for assessing the bioequivalence of average bioavailability”.
J Pharmacikinet Biopharm 15: 657-680.
16. Saavedra I, Saldaña A, Ruminot C. (2006). Medicamentos Genéricos. Cuadernos
Médicos Sociales. 46 (3): 205-211.
17. Bioavailability and Bioequivalence Studies for Orally Administered Drug Products -
General Considerations. U.S. Department of Health and Human Services Food
and Drug Administration. Center for Drug Evaluation and Research (CDER).
March, 2003.
18. Guidance for Industry Statistical Approaches to Establishing Bioequivalence. U.S.
Department of Health and Human Services Food and Drug Administration. Center
for Drug Evaluation and Research (CDER). January, 2001.
19. Peng B, Hayes M, Resta D. (2004). Pharmacokinetics and Pharmacodynamics of
Imatinib in a Phase I Trial with Chonic Myeloid Leukemia Patients. J. Clin. Oncol.
22 (5): 935-942.
20. Guetens G, Prenen H, De Boeck G, van Oosterom A, Schöffski P, Highley M, de
Bruijn E. (2007). Simultaneous determination of AMN107 and Imatinib (Gleevec®,
Glivec®, STI571) in cultured tumour cells using an isocratic high-performance
liquid chomatography procedure with UV detection. J. Chromatogr. B 846: 341-
345.
21. Bioanalytical Methods Validation for Human Studies, Guidance for Industry, U.S.
Department of Health and Human Services, FDA, Center for Drug Evaluation and
Research, 2001.

53
ANEXOS Anexo 1: Dieta proporcionada a los voluntarios durante el estudio.
Anexo 2: Aprobación ética de la comisión de bioética de la Facultad de Medicina de la
Universidad de Chile.
Anexo 3: Consentimiento Informado.
Anexo 4: Informe del análisis físico-químico de los productos Zeite® y Glivec®.
Anexo 5: Exámenes realizados a uno de los voluntarios.
Anexo 6: Ficha clínica de estudio de Biodisponibilidad relativa de Imatinib.
Anexo 7: Informe de parámetros farmacocinéticos de uno de los voluntarios.