udigestiÓn de lÍpidos - fmed.uba.ar y tp de lipoprote+¡nas... · e qm, vldl, idl, hdl ligando de...
TRANSCRIPT

UDIGESTIÓN DE LÍPIDOS
La mayor reserva de lípidos en los alimentos tanto de origen vegetal como animal está
constituida por los triacilgliceroles (TG). La ruta de digestión de los TG depende en cierta
manera del largo de la cadena de los ácidos grasos que los constituyen. Los TG que contienen
ácidos grasos de cadena corta y media (no más de 12 átomos de carbono) son hidrolizados en
la boca y en el estómago a 2-monoacilglicerol (2-MAG) y ácidos grasos (AG) libres por acción
de las enzimas denominadas lipasas sintetizadas en la lengua (lingual) y en el estómago
(gástrica). Estas dos enzimas presentan mayor actividad en los niños que ingieren grandes
cantidades de leche de vaca que contiene alto contenido en TG con ácidos grasos de cadena
corta. La lipasa lingual es importante en el recién nacido porque en ellos no está bien
establecida la secreción de lipasa pancreática. Los ácidos grasos de cadena corta y media son
absorbidos directamente por las células epiteliales intestinales. Luego entran a la sangre portal
y son transportados al hígado unidos a la albúmina.
El resto de la grasa dietaria ingresa al intestino donde es emulsionada en pequeñas
partículas, que son estabilizadas por acción de compuestos anfipáticos llamados sales biliares.
Figura 1: Colato (una sal biliar)
Las sales biliares son sintetizadas en el hígado y almacenadas en la vesícula biliar para
luego se secretadas en el lumen del intestino. Estos compuestos actúan como detergentes,
uniéndose a las gotas lipídicas que son disgregadas por la acción peristáltica del músculo
intestinal. Esta grasa emulsionada es atacada por enzimas digestivas provenientes del
páncreas entre las que se encuentran la lipasa y la colipasa. La lipasa pancreática es la
responsable de separar los ácidos grasos de todos los largos de cadena de los TG
produciendo ácidos grasos libres y 2-MAG. La colipasa es secretada como pro-colipasa y
convertida en colipasa por acción de la tripsina. El complejo lipasa-colipasa es el que tiene
actividad hidrolítica. Además se secreta bicarbonato (cuya secreción es estimulada por la
hormona intestinal secretina) que neutraliza el ácido que entra al intestino desde el estómago
junto con la comida parcialmente digerida, incrementando el pH a aproximadamente 6,
condiciones óptimas para la acción de las enzimas digestivas del intestino. La secreción de
sales biliares y de enzimas pancreáticas está estimulada por la hormona intestinal
colecistoquinina. El páncreas también produce esterasas y fosfolipasa A2 que remueven el
colesterol de los ésteres de colesterol y el ácido graso en posición 2 de los fosfolípidos
(resultando en lisofosfolípidos) respectivamente. Los ácidos grasos y 2-MAGs producidos por
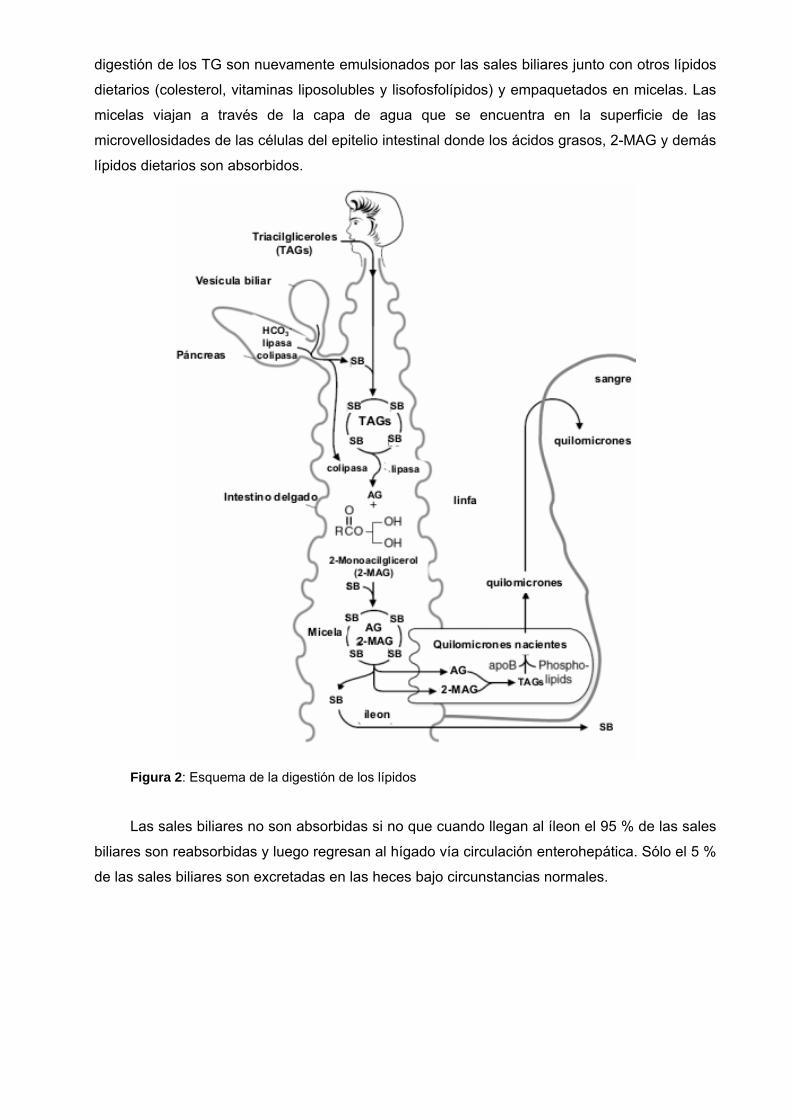
digestión de los TG son nuevamente emulsionados por las sales biliares junto con otros lípidos
dietarios (colesterol, vitaminas liposolubles y lisofosfolípidos) y empaquetados en micelas. Las
micelas viajan a través de la capa de agua que se encuentra en la superficie de las
microvellosidades de las células del epitelio intestinal donde los ácidos grasos, 2-MAG y demás
lípidos dietarios son absorbidos.
Figura 2: Esquema de la digestión de los lípidos
Las sales biliares no son absorbidas si no que cuando llegan al íleon el 95 % de las sales
biliares son reabsorbidas y luego regresan al hígado vía circulación enterohepática. Sólo el 5 %
de las sales biliares son excretadas en las heces bajo circunstancias normales.

Figura 3: Recirculación de las sales biliares
Una vez dentro de las células del intestino los ácidos grasos son reesterificados en el
retículo endoplásmico liso con el 2-MAG para formar TG. En este proceso los ácidos grasos
son activados a acil-CoA de la misma forma en la cual se activan para sufrir β-oxidación. Los
TG son insolubles en agua y no pueden entrar directamente en el torrente sanguíneo ya que
coalescerían impidiendo el flujo sanguíneo por lo cual necesitan ser transportados en partículas
que reciben el nombre de lipoproteínas. Las células intestinales empaquetan los TG junto con
proteínas, colesterol, vitaminas liposolubles y fosfolípidos en lipoproteínas llamadas
Quilomicrones. Más adelante se describirá en profundidad el metabolismo de esta lipoproteína.
ULIPOPROTEÍNAS PLASMÁTICAS
Debido a su carácter hidrofóbico, los lípidos son transportados en el plasma asociados a
proteínas. Las lipoproteínas son complejos moleculares de lípidos y proteínas específicas,
denominadas apoproteínas. Los triglicéridos (TG) y los ésteres de colesterol se ubican en el
centro hidrofóbico de las lipoproteínas mientras que los grupos polares de los fosfolípidos,
colesterol y apoproteínas se ubican en la parte externa de la misma, en contacto con la fase
acuosa. Estas partículas son dinámicas y están en un estado constante de síntesis,
degradación y remoción del plasma. Las lipoproteínas permiten tanto el transporte de los
lípidos como su liberación en los tejidos.

Fosfolípidos
Ésteres de colesterol
ApoproteínaTriglicéridos
Colesterol
Quilomicrones
Estructura de una lipoproteína Lipoproteínas visualizadas por microscopía electrónica
Fosfolípidos
Ésteres de colesterol
ApoproteínaTriglicéridos
Colesterol
Quilomicrones
Estructura de una lipoproteína Lipoproteínas visualizadas por microscopía electrónica
Fosfolípidos
TriglicéridosFosfolípidos
Ésteres de colesterol
ApoproteínaTriglicéridos
Colesterol
Quilomicrones
Estructura de una lipoproteína Lipoproteínas visualizadas por microscopía electrónica
Fosfolípidos
Ésteres de colesterol
ApoproteínaTriglicéridos
Colesterol
Quilomicrones
Estructura de una lipoproteína Lipoproteínas visualizadas por microscopía electrónica
Fosfolípidos
Triglicéridos
Figura 1: Representación de la estructura de una lipoproteína (a) y tamaño relativo de las
lipoproteínas observadas por microscopía electrónica (b).
Las diferentes lipoproteínas presentan una composición relativa de lípidos y proteínas
característica (Tablas 1 y 2), lo que les otorga una densidad diferencial. A medida que aumenta
la proporción de lípidos en una lipoproteína su densidad disminuye y cuanto mayor es su
proporción de proteínas su densidad aumenta.
Cuando se somete a las lipoproteínas a un proceso de ultracentrifugación, éstas se
distribuyen de acuerdo a su densidad. Este método permite separar las lipoproteínas en cinco
fracciones, dando lugar a la nomenclatura más utilizada para estas partículas:
• Quilomicrones (Qm), que son las lipoproteínas menos densas y de mayor tamaño.
• Lipoproteínas de muy baja densidad (VLDL)
• Lipoproteínas de densidad intermedia (IDL)
• Lipoproteínas de baja densidad (LDL)
• Lipoproteínas de alta densidad (HDL), que son las lipoproteínas más densas y
pequeñas.
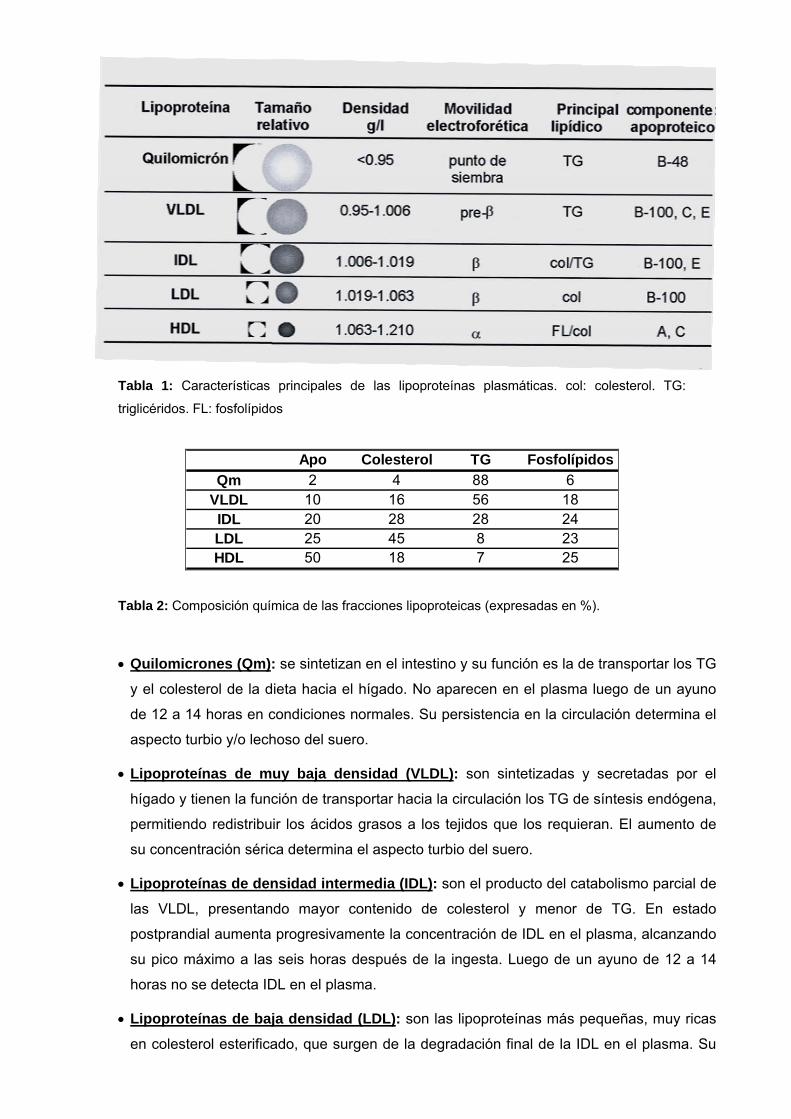
Tabla 1: Características principales de las lipoproteínas plasmáticas. col: colesterol. TG:
triglicéridos. FL: fosfolípidos
Apo Colesterol TG FosfolípidosQm 2 4 88 6
VLDL 10 16 56 18IDL 20 28 28 24LDL 25 45 8 23HDL 50 18 7 25
Tabla 2: Composición química de las fracciones lipoproteicas (expresadas en %).
• UQuilomicrones (Qm)U: se sintetizan en el intestino y su función es la de transportar los TG
y el colesterol de la dieta hacia el hígado. No aparecen en el plasma luego de un ayuno
de 12 a 14 horas en condiciones normales. Su persistencia en la circulación determina el
aspecto turbio y/o lechoso del suero.
• ULipoproteínas de muy baja densidad (VLDL)U: son sintetizadas y secretadas por el
hígado y tienen la función de transportar hacia la circulación los TG de síntesis endógena,
permitiendo redistribuir los ácidos grasos a los tejidos que los requieran. El aumento de
su concentración sérica determina el aspecto turbio del suero.
• ULipoproteínas de densidad intermedia (IDL)U: son el producto del catabolismo parcial de
las VLDL, presentando mayor contenido de colesterol y menor de TG. En estado
postprandial aumenta progresivamente la concentración de IDL en el plasma, alcanzando
su pico máximo a las seis horas después de la ingesta. Luego de un ayuno de 12 a 14
horas no se detecta IDL en el plasma.
• ULipoproteínas de baja densidad (LDL)U: son las lipoproteínas más pequeñas, muy ricas
en colesterol esterificado, que surgen de la degradación final de la IDL en el plasma. Su

función es la de distribuir colesterol a los tejidos que lo requieren para la reposición de
sus componentes de las membranas celulares o para la síntesis de hormonas esteroides
o de sales biliares.
• ULipoproteínas de alta densidad (HDL)U: las HDL pueden provenir de la síntesis hepática
e intestinal. Las HDL recién sintetizadas o nacientes son discoidales y se las conoce
como pre-β HDL. Luego, se convierten en HDL maduras, proceso en el cual interviene el
catabolismo de las lipoproteínas ricas en TG. La función de las HDL es vehiculizar el
colesterol, desde los tejidos periféricos hacia el hígado, proceso conocido como
transporte reverso del colesterol.
UAPOPROTEÍNAS
Las apoproteínas no sólo son parte estructural de las lipoproteínas sino que también tienen
un rol activo en su metabolismo ya que les confieren la capacidad de llevar los lípidos a los
tejidos específicos evitando en su itinerario su dispersión por intercambio o difusión. Esto
ocurre por mecanismos en los cuales las apoproteínas actúan como ligandos de receptores de
superficie o como cofactores para lipasas de la superficie celular. Así los componentes
proteicos de las lipoproteínas determinan cómo los lípidos son metabolizados en una
determinada partícula lipoproteica.
En la siguiente tabla se resume la distribución y función de las apoproteínas más conocidas.
Apoproteína Lipoproteína Funciones
A-I HDL y Qm Estructura de HDL
Activador de LCAT
Ligando del Receptor SR-BI
B-48 Qm Ensamble y secreción de Qm en intestino
B-100 VLDL, LDL, IDL Ensamble y secreción de VLDL en hígado
Ligando del Receptor de LDL
C-II Qm, VLDL, HDL Cofactor/activador de LPL
E Qm, VLDL, IDL, HDL Ligando de Receptores (de LDL y E)
Tabla 3: Distribución y función de las principales apoproteínas.
UENZIMAS que intervienen en el metabolismo lipoproteico
• ULipoproteínlipasa (LPL) U:
Es una enzima que se localiza en la superficie de las células endoteliales de los capilares
de los tejidos extrahepáticos principalmente adiposo y muscular (esquelético y cardíaco). Es

responsable de la hidrólisis de los TG de los Qm y de las VLDL produciendo así Qm
remanentes e IDL, respectivamente. Por su actividad triglicérido hidrolasa produce ácidos
grasos libres y 2-monoacilglicéridos. Es activada por Apo C-II (cofactor) e inhibida por Apo C-III.
La expresión de la LPL en los diferentes tejidos es regulada de manera tal de dirigir los ácidos
grasos en función de la demanda metabólica. Por ejemplo, existe un marcado incremento en la
actividad de LPL en la glándula mamaria durante la lactancia con una correspondiente
disminución en la actividad de esta enzima en el tejido adiposo. Además, el ejercicio
incrementa la actividad de la LPL en músculo y la disminuyen en tejido adiposo. En cambio,
luego de una ingesta se incrementa la actividad de la LPL en tejido adiposo y se disminuye en
músculo. Tales cambios son mediados por la acción de hormonas, tales como insulina y
glucocorticoides que inducen su expresión.
• ULipasa Hepática (HL) U:
Esta enzima se localiza principalmente en la membrana de los hepatocitos, pero también de
células esteroidogénicas. Actúa hidrolizando los TG de las IDL y de las HDL. Los ácidos grasos
liberados son tomados por estos tejidos y degradados produciendo acetil-CoA precursor en la
síntesis de colesterol necesario para la síntesis de sales biliares (en el hígado) y hormonas
esteroides (en gónadas y glándula adrenal). Es por ello que la actividad de la HL se regula en
función de la demanda de colesterol celular.
• ULecitina-colesterol aciltransferasa (LCAT) U:
Es una enzima de síntesis hepática que circula en el plasma asociada a HDL. Es la
responsable de la esterificación del colesterol en las HDL. Actúa transfiriendo ácidos grasos de
la posición 2 de la lecitina al colesterol libre, resultando la formación de lisolecitina y colesterol
esterificado. Es activada por la Apo A-I y por la Apo E (menos eficiente). Una vez que actúa
LCAT esterificando el colesterol libre de las HDL, éste es transferido a las otras lipoproteínas
con Apo B-100 por medio de una Uproteína transportadora de colesterol esterificado (CETP) U.
CETP circula asociada a HDL y también transporta TG, pero en sentido inverso es decir desde
las lipoproteínas con Apo B-100 hacia las HDL. Se considera que el conjunto LCAT/APO A-
I/CETP es el complejo esterificante y de transferencia del colesterol plasmático.
Figura 2: Reacción catalizada por la enzima LCAT.

URECEPTORES que intervienen en el metabolismo lipoproteico
Las células presentan dos mecanismos distintos para la captación de lípidos mediada por
receptor a partir de las lipoproteínas: endocitosis mediada por receptor de toda la lipoproteína y
la captación selectiva de lípidos de ciertos componentes de las partículas.
• Receptor de LDL (o Receptor Apo B-100/E)
Es una glicoproteína que se localiza en la membrana celular en zonas especiales
denominadas cavidades revestidas de clatrina. Estos receptores están ampliamente
distribuidos en varios tipos de células como fibroblastos, músculo liso, hepatocitos y células que
utilizan colesterol para la síntesis de hormonas esteroides (glándulas suprarrenales, testículos,
ovarios), entre otros tejidos.
A este receptor pueden unirse las lipoproteínas que contengan Apo B-100 y/o Apo E, que
son principalmente LDL, IDL y remanentes de Qm. Luego el complejo lipoproteína-receptor es
endocitado.
La síntesis de este receptor es regulada por los niveles intracelulares de colesterol. Un
aumento de colesterol reprime la síntesis de los receptores.
• Receptor E
Se localiza en el hígado y une Apo E, por lo que interactúa principalmente con los Qm
remanentes, pero también es capaz de unir VLDL e IDL, pero no LDL. Luego el complejo
lipoproteína-receptor es endocitado.
• Receptor SR-BI
Es una glicoproteína que se localiza principalmente en hígado y tejidos esteroidogénicos.
Reconoce a la Apo A-I de la HDL, permitiendo la captación selectiva de ésteres de colesterol
de estas lipoproteínas por las células que expresan el receptor, pero sin internalizar por
endocitosis toda la partícula.
• Transportador ABC-AI (ATP binding cassette transporter A1)
Se localiza en tejidos extrahepáticos y reconoce a la Apo A-I de HDL. Utiliza la hidrólisis de
ATP para transportar el colesterol desde la cara interna a la externa de la membrana
plasmática de células ricas en colesterol. De esta manera, las HDL pueden captar el colesterol
de las células.
• Receptores Scavenger (SR-A)
Se localizan en los macrófagos y cobran importancia porque están relacionados con el
desarrollo de lesiones ateromatosas. Pueden unir las siguientes lipoproteínas: LDL (normales y
modificadas), IDL y VLDL anómalas. La síntesis de este receptor no es regulada por el nivel de

colesterol intracelular, por lo que este lípido se acumula progresivamente en las células que
poseen receptores scavenger, convirtiéndose en células espumosas.
METABOLISMO DE LAS LIPOPROTEÍNAS
Metabolismo exógeno de las lipoproteínas
Tanto los ácidos grasos provenientes de la hidrólisis de los TG como el colesterol ingerido
en la dieta son reesterificados en el retículo endoplásmico de las células de la mucosa
intestinal, produciéndose nuevamente triglicéridos y colesterol esterificado.
Estos lípidos se ensamblan con apoproteínas, principalmente B-48 y A-I, además de unirse
con fosfolípidos y colesterol libre, constituyendo de esta manera el Quilomicrón naciente. Éste
es vertido al sistema linfático y desde allí al sistema sanguíneo donde recibe otras
apoproteínas como las C-II y E, cedidas por las HDL, transformándose en Quilomicrón maduro.
Debido a la presencia de Apo C-II, el Qm maduro interactúa con la enzima LPL en la
superficie del endotelio capilar de los tejidos adiposo y muscular produciendo la hidrólisis de los
TG. Al mismo tiempo se desprenden de la estructura del Qm moléculas de colesterol,
fosfolípidos y Apo A y C, que son transferidas a las HDL. La partícula resultante es llamada
Quilomicrón remanente, y contiene menos TG y más colesterol que el Qm maduro, carece de
Apo C-II, pero es muy rica en Apo E. La presencia de esta última Apo permite la unión del Qm
remanente con los receptores E hepáticos, para su internalización y degradación. El contenido
de colesterol de estos remanentes puede ser excretado por vía biliar, o incorporarse a las
lipoproteínas de síntesis hepática.
Entonces, la principal función de los Qm es el transporte de los lípidos provenientes de la
dieta (exógenos). La mayor parte del colesterol procedente de la dieta llega hasta el hígado y
se incorpora al reservorio hepático de colesterol, interviniendo en la regulación de la síntesis de
mismo. Mientras, los ácidos grasos libres provenientes de los TG transportados han sido
liberados en el tejido muscular para su consumo o en el tejido adiposo para su
almacenamiento.

Figura 3: Representación del metabolismo exógeno de las lipoproteínas
Metabolismo endógeno de las lipoproteínas
Los triglicéridos sintetizados en el hígado (provenientes fundamentalmente de los hidratos
de carbono de la dieta) son ensamblados en forma de VLDL nacientes junto con fosfolípidos,
colesterol y Apo B-100 y E, principalmente. Las VLDL son secretadas y en el plasma, maduran
adquiriendo Apo C-II procedente de las HDL, convirtiéndose de esta manera en sustrato de la
LPL que produce la hidrólisis de los triglicéridos. Al mismo tiempo que las VLDL van perdiendo
los TG, aumenta su contenido de ésteres de colesterol gracias a la acción de la CTEP. Como
se mencionó anteriormente, esta proteína tiene la doble misión de transportar los TG desde las
partículas ricas en ellos hacia las HDL y de trasportar los ésteres de colesterol desde las HDL
hacia las VLDL. A medida que las VLDL pierden sus TG, van transfiriendo la Apo CII a las HDL
perdiendo así la capacidad de ser metabolizadas por la LPL, convirtiéndose en partículas ricas
en ésteres de colesterol, Apo B-100 y E, recibiendo el nombre de VLDL residuales o IDL.
Las IDL pueden ser tomadas por receptores E hepáticos, sin embargo éste es el camino
minoritario ya que la mayor parte de las IDL continúan su degradación por la enzima HL. Esta
enzima actúa sobre estas lipoproteínas, completando la hidrólisis de TG, al tiempo que pierde
todas las apoproteínas excepto la Apo B-100, produciendo finalmente lipoproteínas de baja
densidad (LDL).
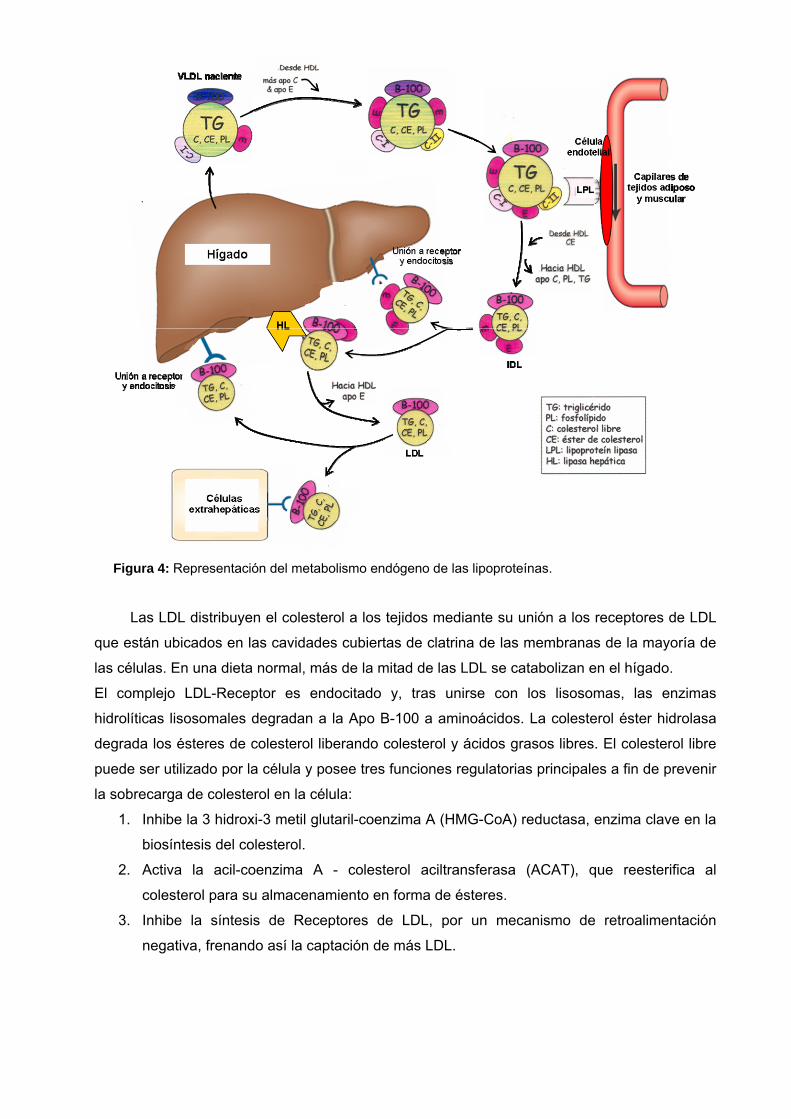
Figura 4: Representación del metabolismo endógeno de las lipoproteínas.
Las LDL distribuyen el colesterol a los tejidos mediante su unión a los receptores de LDL
que están ubicados en las cavidades cubiertas de clatrina de las membranas de la mayoría de
las células. En una dieta normal, más de la mitad de las LDL se catabolizan en el hígado.
El complejo LDL-Receptor es endocitado y, tras unirse con los lisosomas, las enzimas
hidrolíticas lisosomales degradan a la Apo B-100 a aminoácidos. La colesterol éster hidrolasa
degrada los ésteres de colesterol liberando colesterol y ácidos grasos libres. El colesterol libre
puede ser utilizado por la célula y posee tres funciones regulatorias principales a fin de prevenir
la sobrecarga de colesterol en la célula:
1. Inhibe la 3 hidroxi-3 metil glutaril-coenzima A (HMG-CoA) reductasa, enzima clave en la
biosíntesis del colesterol.
2. Activa la acil-coenzima A - colesterol aciltransferasa (ACAT), que reesterifica al
colesterol para su almacenamiento en forma de ésteres.
3. Inhibe la síntesis de Receptores de LDL, por un mecanismo de retroalimentación
negativa, frenando así la captación de más LDL.

Figura 5: Unión de LDL a su receptor y captación de colesterol.
El 15% de las LDL se degradan por una vía alternativa denominada camino de barrido
(scavenger). Los macrófagos del sistema retículo-endotelial son capaces de unir LDL por medio
de receptores de baja afinidad, de digerir la partícula y de depositar el colesterol en el
citoplasma en forma de oleato de colesterilo. El nivel intracelular de colesterol no regula la
síntesis de estos receptores ni la síntesis de colesterol intracelular, lo cual puede conducir a
que estas células se sobrecarguen de colesterol, adquiriendo un aspecto de células
espumosas.
Transporte Reverso del Colesterol (Metabolismo de HDL)
Este es el proceso mediante el cual las HDL transportan el colesterol excedente de los
tejidos periféricos hacia el hígado para su posterior reciclado (síntesis de VLDL) o eliminación
(síntesis de ácidos biliares), razón por la cual es considerado un mecanismo anti-aterogénico.
Las HDL conforman una familia de lipoproteínas que difieren entre sí en la composición relativa
de lípidos en relación a las proteínas y en su perfil de apoproteínas, cambios que surgen por el
constante intercambio con otras lipoproteínas (Qm y VLDL).
Las HDL nacientes generadas en intestino, hígado o incluso la circulación plasmática tienen
forma discoidal y contienen fosfolípidos y Apo A-I, C y E, principalmente.
Las HDL nacientes captan el colesterol libre proveniente de las células, transformándose en
una partícula de mayor tamaño. Por acción de la LCAT, el colesterol libre es esterificado y
migra hacia el interior de las partículas lipoproteicas, generando una estructura esférica (HDL maduras). Durante este proceso se incorporan además de colesterol libre, fosfolípidos y
apoproteínas provenientes de la hidrólisis de Qm y VLDL.
El transporte reverso del colesterol puede dividirse en cuatro etapas que se suceden en forma
consecutiva:

1. Eflujo del colesterol libre desde las células periféricas hacia el espacio extracelular: el
colesterol esterificado en el citoplasma celular es hidrolizado por la enzima colesterol
éster hidrolasa. Luego, el colesterol libre es translocado a la membrana plasmática y
expuesto hacia el espacio extracelular. En este proceso participa un transportador
activo de membrana ABC-A1 que actúa como mediador entre la secreción de
colesterol desde las células hacia las HDL utilizando Apo A-I como ligando.
2. Esterificación del colesterol libre por la enzima LCAT en la circulación plasmática: la
enzima LCAT circula en el plasma asociada a HDL y esterifica el colesterol libre
presente en su superficie. El colesterol recién esterificado migra hacia el interior de
las partículas lipoproteicas debido a su carácter altamente hidrofóbico, generando un
estructura esférica (HDL maduras).
3. Transferencia del colesterol esterificado desde las HDL hacia las lipoproteínas con
Apo B-100 circulantes: mediante la CETP, que circula asociada a la HDL. El
colesterol esterificado de las HDL es transferido a las lipoproteínas con Apo B-100 a
la vez que se transportan TG en sentido inverso, es decir desde las lipoproteínas con
Apo B-100 hacia las HDL. Se considera que el conjunto LCAT / APO A-I / CETP es el
complejo esterificante y de transferencia del colesterol plasmático. Como
consecuencia de estos intercambios, se origina una HDL con mayor contenido en TG
que es susceptible a la acción de la HL, la cual la convierte en una partícula más
pequeña, liberando al medio parte de ciertos componentes de la superficie como la
Apo A-I y fosfolípidos. La Apo A-I liberada, ávida por lípidos, se asocia nuevamente
con fosfolípidos y se regeneran así partículas de HDL naciente, reiniciándose de esta
manera el ciclo metabólico de las HDL.
4. Depuración hepática del colesterol esterificado: existen diferentes vías de llegada del
colesterol esterificado al hígado:
- una vía indirecta, por medio de las lipoproteínas con Apo B-100 que aceptaron el
colesterol esterificado de las HDL y que se unen a los receptores de LDL (Apo B-
100/E) hepáticos.
- una vía directa, por medio de la captación hepática de las HDL mediante su unión a
receptores SR-BI, que reconocen Apo A-I.
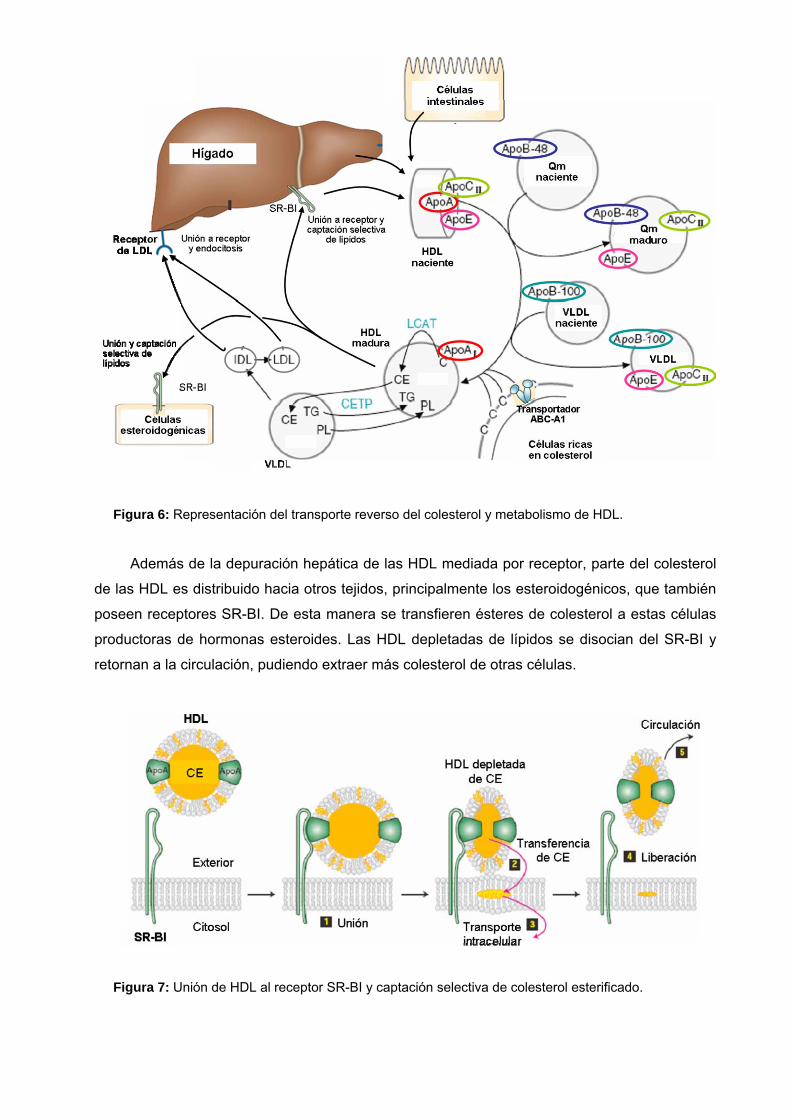
Figura 6: Representación del transporte reverso del colesterol y metabolismo de HDL.
Además de la depuración hepática de las HDL mediada por receptor, parte del colesterol
de las HDL es distribuido hacia otros tejidos, principalmente los esteroidogénicos, que también
poseen receptores SR-BI. De esta manera se transfieren ésteres de colesterol a estas células
productoras de hormonas esteroides. Las HDL depletadas de lípidos se disocian del SR-BI y
retornan a la circulación, pudiendo extraer más colesterol de otras células.
Figura 7: Unión de HDL al receptor SR-BI y captación selectiva de colesterol esterificado.

LIPOPROTEÍNAS-ATEROGÉNESIS
La enfermedad coronaria es la principal causa de muerte en los países industrializados de
Occidente. A su vez, la mayoría de los casos de enfermedad coronaria están asociados a otra
patología: la aterosclerosis (acumulación de depósitos de lípidos en las paredes arteriales). El
crecimiento de estos depósitos o placas ateroscleróticas provoca la formación de coágulos que
impiden el flujo sanguíneo. Si un coágulo llega a ocluir una arteria se produce la isquemia del
tejido por falta de irrigación. Si el proceso es sostenido y se acompaña de necrosis (muerte
celular) se produce un infarto.
Se conocen algunos factores que pueden predisponer a la aterosclerosis y posterior
enfermedad coronaria. La hipertensión arterial (HTA), la diabetes, el hábito de fumar y otros
factores parecen aumentar la probabilidad de una enfermedad coronaria prematura. Las dietas
ricas en colesterol y en ácidos grasos saturados contribuyen a la elevación de los niveles de
lípidos en la sangre y a la progresión de la aterosclerosis. La constitución genética de un
individuo desempeña también cierto papel: algunas personas pueden ingerir cantidades
enormes de alimentos con colesterol en su dieta durante períodos prolongados sin que se
produzca una elevación de la colesterolemia. Abundan sin embargo, quienes con valores de
colesterol extraordinariamente elevados, jamás padecerán de enfermedad coronaria y por
último sí pueden sufrirla aquellos que tienen un bajo perfil de riesgo.
Debido al riesgo cardiovascular que representa la hipercolesterolemia, es de suma
importancia la medición de distintas lipoproteínas que transportan colesterol. Como las HDL
remueven el colesterol desde los tejidos periféricos, mientras que las LDL lo depositan en ellos,
el riesgo cardiovascular está en relación inversa a las cifras de colesterol unido a las HDL y en
relación directa con el colesterol unido a las LDL.
La acción pro-aterogénica de las lipoproteínas no solamente depende de su concentración
plasmática sino también de su heterogeneidad (tamaño, densidad y composición). La vida
media de las LDL en plasma es de 3 días. Durante este tiempo, estas lipoproteínas pueden
sufrir modificaciones como glicosilación (considerar en los pacientes diabéticos), oxidación y
carbamilación (importante en los pacientes con insuficiencia renal). Si la vida media aumenta,
aumentan aún más las modificaciones de las mismas, lo que disminuye la capacidad de
interacción con sus receptores fisiológicos en los tejidos, lo cual resulta en un metabolismo
incrementado a través de vías metabólicas alternativas que aceleran el proceso aterogénico.

Figura 8: Modificaciones químicas de las LDL
Lipoproteínas modificadas:
Entre las subfracciones de mayor importancia fisiopatológica se destacan:
- LDL pequeña y densa: Las LDL pueden interactuar con las VLDL, tomando TG y
entregando colesterol esterificado. Por acción de la lipasa hepática, los TG son hidrolizados,
disminuyendo el tamaño de las LDL y transformándolas en "LDL muy pequeñas y densas"
(small dense LDL o sLDL), que presentan gran capacidad para atravesar el endotelio y
alcanzar a las células musculares lisas, y mayor capacidad de unión a los proteoglicanos
favoreciendo su retención en la pared arterial. Estas sLDL son particularmente sensibles a los
procesos de oxidación y glicosilación. Pierden afinidad por el receptor de Apo B-100 y son
atrapadas por los macrófagos que se cargan de colesterol esterificado, modifican su
metabolismo y se convierten en células espumosas.
Se ha encontrado que la presencia de partículas sLDL se asocia con un aumento de riesgo
de enfermedad coronaria. El aumento de sLDL ocurre raramente como un desorden aislado, se
acompaña frecuentemente de hipertrigliceridemia, niveles de HDL disminuidos, obesidad
abdominal, resistencia a insulina, y otras alteraciones metabólicas.
Figura 9: Formación de LDL pequeñas y densas (sLDL).

- LDL ricas en triglicéridos: Son partículas de LDL con un ligero aumento en la proporción
de TG, lo que distorsiona el reconocimiento de la lipoproteína por el receptor fisiológico. Su
presencia se asocia con disminución o ausencia de la actividad de la lipasa hepática (diabetes
tipo 1 o en insuficiencia renal crónica) o por un aumento en la actividad de la CETP (común en
las hipertrigliceridemias) dado que facilita los intercambios de TG por colesterol entre
lipoproteínas y por lo tanto el enriquecimiento en TG de las LDL.
- LDL glicosiladas: Se forma como parte del proceso de glicosilación no-enzimática de las
proteínas estructurales y circulantes, de manera proporcional a Ia magnitud de la
hiperglucemia, mediante la unión covalente de la glucosa a la Apo B-100 de la LDL.
- LDL oxidadas: La oxidación de LDL ocurre en el subendotelio. Si bien la producción de
especies reactivas del oxígeno es inherente al metabolismo celular normal, los diversos
factores de riesgo cardiovascular producen un incremento del estrés oxidativo que acelera
marcadamente este proceso. Por ejemplo; oxidantes como el superóxido, peróxido de
hidrogeno, óxido nítrico, favorecen la oxidación de las LDL mientras que compuestos como la
vitamina E, ácido ascórbico, beta-caroteno protegen contra la oxidación.
Cabe destacar, que la sLDL, la LDL rica en TG y la LDL glicosilada, presentan una mayor
tendencia a oxidarse.
- Lipoproteína (a): Es sintetizada por el hígado y sus cifras plasmáticas están
genéticamente determinadas puesto que no dependen de factores dietarios (ingesta rica en
colesterol o alteraciones metabólicas de los lípidos). La Lp(a) es comparable en tamaño y
contenido de colesterol esterificado a la LDL, pero posee mayor densidad. Se trata de una Apo
B-100 enlazada covalentemente por puentes disulfuro a una molécula proteica símil
plasminógeno (Apo(a)).
El papel fisiológico de la Lp(a) aún no se conoce, aunque se encuentra abundantemente en
sangre de muchas personas cuya vulnerabilidad a la enfermedad cardíaca no podía atribuirse a
otros factores. El metabolismo es independiente de la LDL y la forma de eliminación es poco
comprendida puesto que se une escasamente a los receptores para B-100.
La Apo(a) es muy parecida al plasminógeno, que es el componente principal del sistema
fibrinolítico o de disolución del coagulo sanguíneo, pero a diferencia de éste carece de
actividad enzimática.
La Lp(a) incrementa el riesgo cardiovascular porque suma el depósito de colesterol
endotelial a su acción procoagulante, dado que compite con el plasminógeno en su unión al
endotelio.
Algunas hormonas pueden modificar su concentración plasmática: hormonas tiroides, los
estrógenos y los esteroides anabólicos la reducen en tanto que la hormona de crecimiento la
incrementa pero no se modifica por medicamentos hipolipemiantes o por la dieta.

ATEROGÉNESIS
La aterogénesis es la formación de placas de ateroma (génesis del ateroma o
ateromatosis) o depósito lípido-celular en el subendotelio de las grandes arterias. El proceso se
desencadena como consecuencia de la desregulación del metabolismo del colesterol en la
íntima de las arterias. Normalmente el colesterol que llega a la pared arterial es consumido por
fibroblastos y macrófagos. Además, el endotelio produce eicosanoides y otros factores
antiaterogénicos que promueven la remoción de colesterol a través de las HDL.
Sin embargo, bajo circunstancias no bien precisadas tales como:
- el excesivo aporte de LDL (como se ve en las hiperliloproteinemias secundarias)
- la alteración de los receptores de la Apo-B100 y el clearence anómalo de las LDL (como
se ve en la hiperlipoproteinemias primarias).
- las modificaciones de la secuencia de aminoácidos de la Apo-B100 (que media
interacciones entre ésta y su receptor).
- los microtraumas en la pared vascular provocados por la HTA,
- el envejecimiento celular (a partir del efecto deletéreo de los radicales libres del oxígeno),
... las células endoteliales responden a la agresión produciendo factores de crecimiento,
citoquinas y factores proinflamatorios que atraen macrófagos y desencadenan la proliferación
de las células musculares lisas. Tanto los macrófagos como los miocitos se cargan de
colesterol esterificado por fagocitosis de las LDL extracelulares y por la alteración de su
metabolismo lipídico se convierten en células espumosas o foam cells. El acúmulo celular y de
colesterol subendotelial levanta esta capa de tejido generando la placa incipiente. La
intervención macrofágica subendotelial aumenta la reacción inflamatoria local que extiende la
lesión, altera a los proteoglicanos (el contenido de heparán-sulfato en el tejido conectivo de la
íntima arterial es antiaterogénico) y permite el depósito de calcio el cual lleva a la calcificación
de la placa.
Finalmente la placa se autoperpetúa no sólo por el aporte excesivo de LDL, sino porque en
segunda instancia las plaquetas y las citoquinas contribuyen al crecimiento del ateroma.
Cuando la obstrucción de la luz arterial es importante, los tejidos que son nutridos por ese vaso
se quedan sin aporte de sangre (isquemia) y sobreviene la necrosis (infarto) (ver Figura 10).
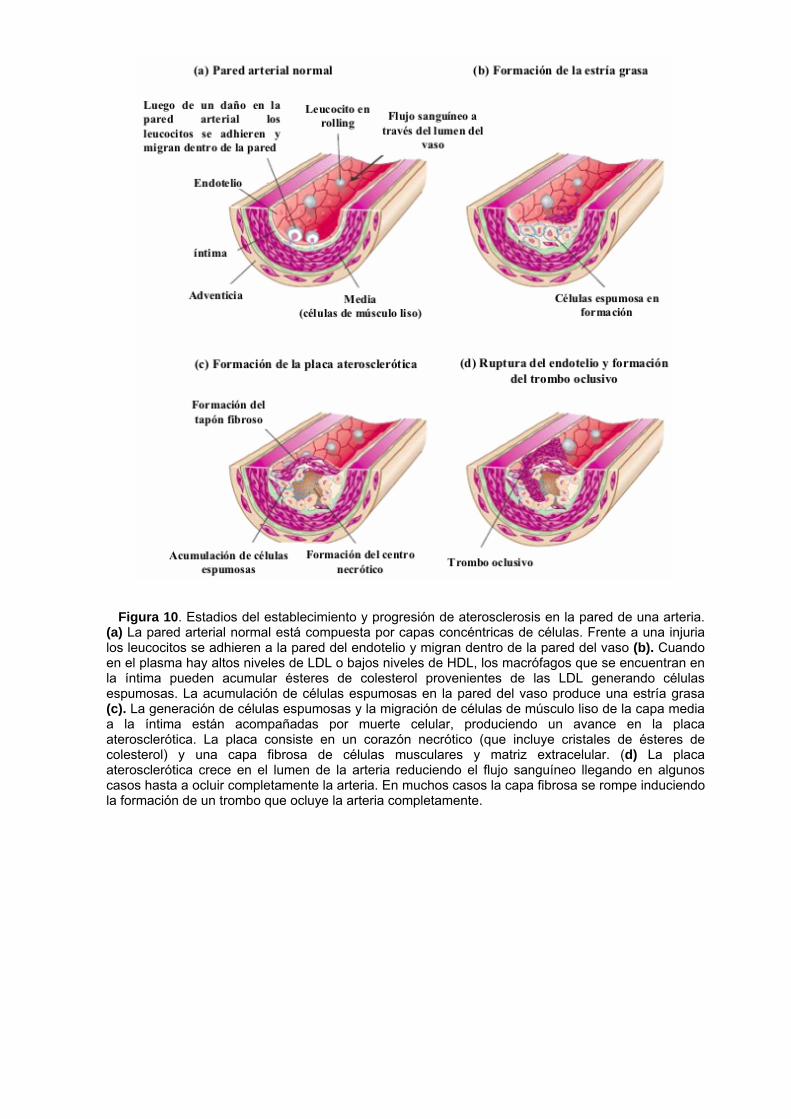
Figura 10. Estadios del establecimiento y progresión de aterosclerosis en la pared de una arteria.
(a) La pared arterial normal está compuesta por capas concéntricas de células. Frente a una injuria los leucocitos se adhieren a la pared del endotelio y migran dentro de la pared del vaso (b). Cuando en el plasma hay altos niveles de LDL o bajos niveles de HDL, los macrófagos que se encuentran en la íntima pueden acumular ésteres de colesterol provenientes de las LDL generando células espumosas. La acumulación de células espumosas en la pared del vaso produce una estría grasa (c). La generación de células espumosas y la migración de células de músculo liso de la capa media a la íntima están acompañadas por muerte celular, produciendo un avance en la placa aterosclerótica. La placa consiste en un corazón necrótico (que incluye cristales de ésteres de colesterol) y una capa fibrosa de células musculares y matriz extracelular. (d) La placa aterosclerótica crece en el lumen de la arteria reduciendo el flujo sanguíneo llegando en algunos casos hasta a ocluir completamente la arteria. En muchos casos la capa fibrosa se rompe induciendo la formación de un trombo que ocluye la arteria completamente.

(a)
(b)
Figura 11. Generación de células espumosas en la pared arterial (a) En el sitio de infección o daño
(1) los monocitos se adhieren y migran a través de la pared del endotelio activado hacia la íntima (2), donde se diferencian a macrófagos. Cuando los niveles de LDL plasmáticos son altos la concentración de LDL en la íntima es alta y parte es oxidado (LDLox) (3). Los receptores scavenger expresados por los macrófagos unen y endocitan las LDLox, que son degradadas. Su colesterol se acumula como ésteres de colesterol en gotas lipídicas citosólicas conduciendo a la acumulación de colesterol y a la formación de células espumosas (4). Los macrófagos también expresan ABC-A1 y SR-BI que pueden mediar la salida del exceso de colesterol de las células en forma de HDL hacia la intima (5). (b) Micrografía de una arteria coronaria con una placa aterosclerótica conteniendo muchas células espumosas llenas de cristales esféricos de ésteres de colesterol. También están presentes algunas células de músculo liso que también contienen gotas lipídicas (flecha).

DIAGNÓSTICO DE DISLIPOPROTEINEMIAS Y EVALUACIÓN DEL RIESGO CARDIOVASCULAR
La presencia de un defecto en algún paso en el metabolismo de las lipoproteínas trae
aparejadas alteraciones en la concentración y calidad de las lipoproteínas plasmáticas,
expresadas como dislipoproteinemias.
Se demostró que existe una relación directa y estrecha entre el nivel de colesterol y la
incidencia de la enfermedad coronaria, más aún, el descenso del colesterol plasmático detiene
la progresión de la aterosclerosis y sus complicaciones.
Numerosos estudios demuestran que no es suficiente la medida del colesterol total, sino
que es necesario conocer su distribución en las diferentes lipoproteínas, en especial HDL y
LDL, y la relación que existe entre ellas.
El estudio de los lípidos plasmáticos debe realizarse no solamente para el diagnóstico de
las dislipoproteinemias y el control de su tratamiento, sino también en una actitud preventiva,
ya que el inicio de las lesiones ateroscleróticas se produce desde edad temprana,
acelerándose con la presencia de otros factores de riesgo.
Es importante tener en cuenta algunas características del paciente como su edad y sexo,
antecedentes personales y familiares de dislipemias o de enfermedad cardiovascular
manifestada en edad temprana, y presencia de otros factores de riesgo concomitantes
(hipertensión, tabaquismo, obesidad, diabetes, sedentarismo, hipercoagulabilidad,
hiperuricemia, etc.).
De acuerdo a estos antecedentes y al objetivo del estudio, se decide cuáles serán los
parámetros que van a conformar el perfil de lípidos adecuados para cada individuo.
Se sugiere un perfil lipídico básico como una primera aproximación al conocimiento del
estado del metabolismo lipoproteico que comprende:
a) observación del aspecto del suero
b) colesterol total (CT)
c) triglicéridos (TG)
d) colesterol de HDL (c-HDL)
e) colesterol de LDL (c-LDL)
f) índice CT /c-HDL
Para realizar un estudio de lípidos se requiere un ayuno de 12 a 14 horas. Si el ayuno es
menor, no logran metabolizarse completamente los Qm de la dieta.
a- En condiciones normales y en ayunas, el aspecto del suero es límpido. Cuando se
incrementan las VLDL y/o aparecen los Qm, el suero se enturbia debido al gran tamaño de

estas partículas. Las LDL no alteran el aspecto del suero dado su pequeño tamaño. Si el
tiempo de ayuno no es el adecuado, puede apreciarse opalescencia en el suero, debido a la
presencia de algunos Qm.
b- El valor de CT aislado, salvo que se encuentre francamente aumentado, aporta poca
información en cuanto a la evaluación del riesgo cardiovascular. Es necesario conocer su
distribución entre las dos lipoproteínas que principalmente lo transportan: la LDL (aterogénica)
y la HDL (antiaterogénica).
No deben considerarse los valores normales de colesterol de una población, ya que se aprecia
que los valores ideales no coinciden con los valores reales. Más importante que obtener un
dato de CT dentro de los valores normales, es mantenerlo cerca del rango ideal
correspondiente a la edad y al sexo, además de relacionar ese dato con las demás
lipoproteínas. Así, según estudios poblacionales realizados, se recomienda que el CT no
supere los 200 mg/dl.
c- Existe una discrepancia en la existencia de una asociación entre la trigliceridemia y la
enfermedad coronaria, por lo que deben evaluarse los niveles de TG junto a alteraciones
cuanti/cualitativas de lipoproteínas. En general se considera menor a 150 mg/dl de plasma
como valor deseable.
d- Se ha demostrado una correlación negativa entre el colesterol de las HDL y la incidencia
de enfermedades ateroscleróticas y se puede afirmar que el c-HDL tiene un alto valor
predictivo.
El valor medio de c-HDL es de 45 mg/dl para los varones y de 55 mg/dl para las mujeres
premenopáusicas.
La concentración de HDL es regulada por un conjunto de factores moduladores. El ejercicio
físico, el consumo moderado de alcohol, los estrógenos, entre otros, elevan los niveles de HDL.
En cambio el tabaquismo, el sedentarismo y el sobrepeso, son algunas de las circunstancias
que producen el descenso en los niveles de c-HDL. El conocimiento de estos factores
moduladores sirve para explicar las alteraciones del nivel de HDL y corregirlos en beneficio del
paciente.
e- Con respecto al c-LDL, en general su valor se calcula por la fórmula de Friedwald
(siempre que la concentración de TG no supere los 300 mg/dl):
c-LDL = CT - ( TG + c-HDL)
5
donde TG/5 sería una estimación del colesterol correspondiente a las VLDL. Con este cálculo
no siempre se obtienen buenos resultados, dado que la relación TG/CT en las VLDL varía, aún
más en los casos patológicos.
Los valores deseables de c-LDL dependerán de cada paciente, en virtud de la presencia de
otros factores de riesgo. Así, el c-LDL podría considerarse normal hasta 160 mg/dl. En caso de

presentar dos o más factores de riesgo, el valor deseable de c-LDL debe ser menor a 130
mg/dl. En tanto que en aquellos pacientes con enfermedad coronaria o equivalentes, el c-LDL
no debe superar los 100 mg/dl.
f- Por otro lado, el valor de los índices CT/c-HDL (índice aterogénico o de Castelli) y de
c-LDL/c-HDL, aportan más información que los datos aislados.
La relación c-LDL/c-HDL tiene la misma utilidad que CT/c-HDL, sin que hasta ahora se
hayan demostrado mayores ventajas sobre esta última. Ambas otorgan un alto poder
discriminador de enfermedad coronaria y una gran capacidad predictiva.
El valor esperado para un individuo sano de la relación CT/c-HDL se considera hasta 4,5 y el
de la relación c-LDL/c-HDL no debe superar 2,9.
Estudios complementarios
1) Determinación de los valores plasmáticos de Apo B-100 y Apo A-I.
A diferencia de las mediciones de c-HDL y c-LDL, las cuales son indirectas, los niveles de
apoproteínas pueden medirse directamente. Por lo tanto, la medida de las apoproteínas B-100
y A-I contribuyen con mayor sensibilidad y exactitud a la detección y clasificación de individuos
con riesgo o con enfermedad cardiovascular aterosclerótica.
Dado que sólo una molécula de Apo B-100 está presente en cada partícula de lipoproteínas,
el valor de Apo B-100 indica el número total de lipoproteínas potencialmente aterogénicas.
La Apo B-100 fue evidenciada como el mejor marcador de aterosclerosis en comparación a
otros parámetros lipídicos y/o lipoproteicos dado que puede encontrarse aumentada aún
cuando el colesterol total y c-LDL sean normales.
Por otro lado, la Apo A-I es el constituyente proteico mayoritario de las HDL, cuya función
está relacionada con los procesos antiaterogénicos.
De lo expuesto se deduce la necesidad de tener en cuenta en algunos casos la
determinación de las apoproteínas A-I y B-100 en el estudio de los lípidos. Ella debe realizarse
especialmente en aquellos casos donde los demás parámetros lipídicos se encuentren dentro
de los rangos normales y existan signos sospechosos de aterosclerosis o antecedentes
familiares importantes.
2) Lipidograma electroforético.
Para la tipificación de las dislipoproteinemias, en algunos casos, es de utilidad la realización
de un lipidograma electroforético. Para ello, el plasma de un paciente es sometido a
electroforesis utilizando un soporte de acetato de celulosa o gel de agarosa a pH 8,6, de forma
similar a un proteinograma electróforético. Posteriormente, se realiza una tinción de los lípidos
utilizando un colorante especial de lípidos, observándose entonces solamente las bandas de

lipoproteínas. Se obtiene así un lipidograma electroforético en el cual se pueden observar
cuatro bandas.
• Una primera banda, que migra con las α- globulinas y contiene a las HDL.
• Una segunda banda, que migra hacia una región inmediatamente anterior a la región β y
se llama pre- β, que contiene a las VLDL
• Una tercera banda, que migra con las β-globulinas y corresponde a las LDL e IDL. Estas
últimas se encuentran en muy baja proporción luego de un ayuno de 12 hs.
• Una última banda, que permanece en el origen, corresponde a los Qm, y que sólo
aparece en estados patológicos o después de la ingestión de alimentos.
El lipidograma electroforético de un individuo normal con un ayuno de 12 horas contiene
una banda β prominente (LDL) y una leve banda α (HDL). Apenas se observa la banda pre- β
(VLDL).
Lipidograma electroforético Proteinograma electroforético
Figura 12: Representación de un lipidograma y un proteinograma de un individuo normal.
Valores de perfil lipídico y riesgo de enfermedad coronaria
Los valores deseables de CT, c-HDL, c-LDL y TG dependerán de cada paciente, en virtud
de la presencia de otros factores de riesgo. La American Heart Association (2007,
Hwww.americanheart.org H) proporciona un conjunto de guías de los valores de dichas
determinaciones en relación al riesgo de padecer enfermedad coronaria.
0BColesterol total < 200 mg/dL = valores deseados
200 – 239 mg/dL = límite de riesgo elevado
≥ 240 mg/dL = alto riesgo
1BColesterol - HDL < 40 mg/dL para hombres riesgo < 50 mg/dL para mujeres alto

40-50 mg/dL para hombres valores esperados en individuos 50-60 mg/dL para mujeres sanos (es mejor cuanto mayor es su nivel) ≥ 60 mg/dL = se asocia a un cierto nivel de protección contra enfermedad cardíaca
2BColesterol – LDL
< 70 mg/dL = valores óptimos si posee un muy alto riesgo de padecer enfermedad cardíaca
< 100 mg/dL = valores óptimos si padece enfermedad cardíaca o diabetes
100 mg/dL - 129mg/dL = óptimo a casi óptimo
130 mg/dL - 159mg/dL = límite de alto riesgo
160 mg/dL - 189mg/dL = alto riesgo
≥ 190 mg/dL y más arriba = muy alto riesgo
3BTriglicéridos
< 150 mg/dL = normal
150 mg/dL – 199 mg/dL = límite de alto riesgo
200 mg/dL – 499 mg/dL = alto riesgo
≥ 500 mg/dL = muy alto riesgo
TRASTORNOS ENDÓGENOS DEL METABOLISMO DE LAS LlPOPROTEÍNAS
Los trastornos endógenos del metabolismo de las lipoproteínas comprenden los
trastornos primarios o hereditarios y los trastornos secundarios o adquiridos que son
secundarios a otra enfermedad o tratamiento farmacológico. Las dislipoproteinemias
comprenden las hiperlipoproteinemias y las hipolipoproteinemias.
Una clasificación clásica de las hiperlipoproteinemias es la clasificación fenotípica de
Fredrickson, asumida por la OMS. En la actualidad esta clasificación tiene una importancia
fundamentalmente descriptiva, pero es necesario tener en cuenta que no considera la causa ni
la entidad nosológica, no considera a las HDL y se basa en la interpretación de la electroforesis
de las lipoproteínas del plasma. Por otro lado, un paciente puede evolucionar cambiando la
manifestación fenotípica y además, una hiperlipoproteinemia endógena puede tener
manifestaciones fenotípicas diferentes.

Tabla 4: Clasificación de hiperlipoproteinemias según la OMS.
Referencias: P: presente, A: ausente, IR: dentro de los intervalos de referencia, Prev: prevalencia
Aspecto del plasma: (+) indica presencia de sobrenadante cremoso. Con distinto número de (+) se indica la cremosidad relativa.
Figura 13: Representación de lipidogramas patológicos, correspondientes a la clasificación de
Fredrickson de las hiperlipoproteinemias.
Hiperlipoproteinemias primarias
A- Hipercolesterolemia
Corresponden al fenotipo IIa de la clasificación de Fredrickson y las causas más frecuentes
son:
1- Deficiencia del receptor de LDL
Se caracteriza por la acumulación de las LDL en el plasma debido a mutaciones que afectan
a los receptores de LDL (carencia de receptores, afinidad disminuida o dificultades en el
proceso de endocitosis mediada por receptor). El aumento de las LDL en el plasma favorece su
depósito en la piel y en los tendones formando xantomas tendinosos y/o xantelasmas,

presentan arco corneal y ateromas al depositarse en las arterias, razón por la cual estos
pacientes presentan un riesgo aterogénico elevado.
La enfermedad se expresa con el fenotipo IIa de hiperlipoproteinemia, sin embargo, con
menor frecuencia algunos pacientes pueden presentar fenotipo IIb dado que las partículas de
VLDL e IDL también son catabolizadas por el receptor LDL.
Se hereda en forma autosómica dominante, siendo la forma homocigota la más severa con
valores de colesterol que pueden superar los 1.000 mg/dl de plasma. Esta forma puede ocurrir
con una frecuencia de 1 en 1.000.000 entre la población norteamericana y europea. Estos
pacientes presentan hipercolesterolemia grave desde el nacimiento y mueren por cardiopatía
isquémica en la adolescencia o juventud. En cambio, la forma heterocigota, con valores de
colesterol alrededor de 500 mg/dl de plasma, tiene una incidencia de 1 en 500.
El tratamiento de los pacientes heterocigotas se basa en reducir los niveles de c-LDL a
través de medidas dietéticas y con fármacos hipocolesteromiantes, como las estatinas, de por
vida. En los pacientes homocigotas, se recurre a la disminución de los niveles de c-LDL y, en
algunos casos, al trasplante hepático.
2- Defecto familiar de Apo B-100
Otra mutación que altera el metabolismo de LDL es la presencia de una Apo B-100 mutada
en la zona que interviene en el reconocimiento y unión al receptor de LDL. Esta patología se
hereda en forma autosómica dominante dando lugar a una hipercolesterolemia semejante a la
observada en pacientes con mutación del receptor de LDL.
3- Hipercolesterolemia poligénica
Es la más frecuente dentro de las hipercolesterolemias primarias con fenotipo Ila y dentro de
este grupo se debe incluir a los individuos con colesterol total y c-LDL elevados en los que no
se haya demostrado la presencia de las hipercolesterolemias previamente descritas. Se han
descrito mutaciones que afectan distintos pasos del metabolismo lipídico que junto con factores
ambientales desencadenan esta enfermedad. Se expresa a partir de los 20 años de edad, con
valores variables de c-LDL siendo la aterosclerosis coronaria la manifestación clínica más
importante con ausencia de las manifestaciones cutáneas.
B- Hipertrigliceridemia
1- Hiperquilomicronemia
a) Deficiencia familiar de LPL (Fenotipo l de la Clasificación de Fredrickson)
Tiene una incidencia muy baja, se transmite en forma autosómica recesiva y las
manifestaciones clínicas, como los xantomas eruptivos, comienzan en la primera década de
vida. Existe un marcado aumento de los Qm (con la consiguiente elevación de los TG) que
otorga un característico aspecto lechoso al suero. Los niveles de HDL se encuentran
disminuidos. Si bien la LPL cataboliza tanto Qm como VLDL, la deficiencia familiar de LPL

debería traducirse en un aumento de ambas partículas. Sin embargo las VLDL no se
encuentran elevadas sino que sus niveles suelen ser normales o disminuidos, debido quizás a
que su síntesis se encuentra relativamente disminuida.
b) Deficiencia familiar de Apo C-II (Fenotipo I o V de la clasificación de Fredrickson).
La Apo C-II, cofactor de la LPL, genera una hipertrigliceridemia semejante al déficit de dicha
enzima. Los pacientes con esta deficiencia genética pueden presentar un fenotipo I o V según
que la elevación de Qm se acompañe o no de aumento de VLDL.
La complicación más grave de la hiperquilomicronemia es la pancreatitis aguda que se
generaría por la lenta circulación inducida por Ios Qm en los capilares pancreáticos. Son
frecuentes los dolores abdominales difusos y con frecuencia hay hepato y esplenomegalia, y
presencia de xantomas eruptivos en nalgas y extremidades. Los pacientes por lo general son
obesos y el metabolismo de glúcidos es normal.
Como primera medida en el tratamiento de los pacientes portadores de esta dislipemia se
debe restringir la ingesta de grasas, principalmente de ácidos grasos de cadena larga. Se
recomienda la ingesta de ácidos grasos de menos de 12 átomos de carbono, que pueden
pasar directamente a la sangre para ser utilizados por el hígado.
2- Hipertrigliceridemia familiar (Fenotipo IV de la clasificación de Fredrickson).
Su prevalencia se estima entre 0,5-1 % de la población general. Se hereda en forma
autosómica dominante y se manifiesta en la segunda década de vida. Se caracteriza por
aumento de VLDL y puede originarse por un trastorno en el metabolismo de carbohidratos, una
ingestión excesiva de carbohidratos o un aumento en la producción de VLDL. Se observa la
presencia de partículas de VLDL con mayor contenido de TG, que las convierte en un sustrato
menos adecuado para la LPL.
C- Hiperlipemia mixta
Se caracterizan por cursar con aumento tanto de colesterol como de TG. Dentro de las
hiperlipoproteinemias primarias se distinguen:
1. Hiperlipemia familiar combinada (Fenotipos Ila, Ilb, IV y V de la clasificación de
Fredrickson).
Es la forma familiar más común de hiperlipemia en sobrevivientes de infarto de miocardio
jóvenes. Su prevalencia es del 1% de la población general. El defecto genético y la
fisiopatología en detalle aún se desconocen. La alteración fundamental consiste en un aumento
en la síntesis hepática de Apo B-100 y VLDL asociado frecuentemente a TG elevados. Los
individuos afectados presentan aumento de LDL, de VLDL o ambos frecuentemente
acompañados de descenso de HDL y prevalencia de LDL pequeñas y densas. Se observa un

aumento en la concentración plasmática de Apo B-100, que refleja un mayor número de
partículas.
Es característico que se detecten distintos fenotipos lipoproteicos entre los miembros de una
misma familia y que puedan modificarse a lo largo de la vida.
2. Disbetalipoproteinemia (Hiperlipoproteinemia tipo III de Fredrickson)
Esta enfermedad se caracteriza por la acumulación de lipoproteínas resultantes de la
acción de la LPL, las IDL, los Qm remanentes y eventualmente VLDL. Estas lipoproteínas
presentan cambios en sus características físico-químicas (movilidad electroforética,
enriquecimiento en colesterol). Este conjunto heterogéneo de lipoproteínas es denominado β-
VLDL, que presenta movilidad β – preβ y se visualiza como una banda ancha en la
electroforesis. Una de las causas por la que se produce una eliminación disminuida de estas
lipoproteínas residuales es debida a una alteración estructural de la Apo E (presencia de la
isoforma E2) que le impide ser reconocida adecuadamente por el receptor hepático.
Como resultado se acumulan estas lipoproteínas en plasma, aunque pueden ser captadas
por los macrófagos. Las LDL están reducidas pero como todas las partículas remanentes son
ricas en ésteres de colesterol, la concentración de colesterol total está aumentada. Los adultos
muestran la presencia de xantomas tuberosos o planares y pliegues cutáneos color naranja por
el depósito de carotenoides y otros lípidos.
Las manifestaciones clínicas se observan en 1 cada 10.000 individuos. Se propone que la
expresión completa de la enfermedad requiere un factor disparador como el hipotiroidismo, la
diabetes u otra hiperlipoproteinemia familiar.
Hiperlipoproteinemias Secundarias
Algunas enfermedades, estados fisiológicos, ingesta de alcohol o tratamientos
farmacológicos pueden alterar el metabolismo lipídico y presentar como manifestación
secundaria una hiperlipoproteinemia. Debe realizarse el diagnóstico o exclusión de una
hiperlipoproteinemia secundaria antes de la investigación de una hiperlipoproteinemia primaria.
Diabetes mellitus.
Los trastornos del metabolismo de los lípidos que se observan en los pacientes diabéticos
están relacionados con el tipo de diabetes, el tratamiento aplicado y el control de la
enfermedad. Si bien es posible encontrar valores de lípidos basales normales, se puede afirmar
la existencia de alteraciones en el metabolismo intermedio de las lipoproteínas, que se
traducen en acumulación de VLDL, disminución de los niveles de HDL, persistencia de IDL en
el plasma y/o formación de LDL de composición alterada. Estas situaciones contribuyen, junto
a otros factores de riesgo, a que los pacientes presenten una aterosclerosis acelerada y una
morbi-mortalidad cardiovascular de dos a tres veces mayor que la población normal. Hoy en día

se considera a la diabetes, especialmente la de tipo 2, como un factor de altísimo riesgo para el
desarrollo de enfermedades vasculares (infarto agudo de miocardio, accidente cerebro-
vascular, etc.). Los pacientes diabéticos presentan el mismo riesgo de padecer un infarto de
miocardio que los pacientes que ya lo han sufrido.
Los mecanismos que producen las alteraciones lipoproteicas van a diferir según el tipo de
diabetes.
-Diabetes tipo 1: la dislipoproteinemia ocurre como consecuencia de la deficiencia de
insulina. La actividad de la LPL está disminuida por lo tanto el catabolismo de las lipoproteínas
ricas en TG (Qm y VLDL) se encuentra reducido (aumentan los niveles de Qm y VLDL). Un
mecanismo adicional que contribuiría a la hipertrigliceridemia, sería la falta de inhibición de la
lipasa hormono-sensible ubicada en el tejido adiposo. Esta enzima en condiciones normales es
inhibida por la insulina e hidroliza los TG almacenados aportando así ácidos grasos libres que
son utilizados por el hígado como fuente de energía mediante la beta-oxidación, a fin de poder
realizar la síntesis de glucosa por gluconeogénesis. En la diabetes tipo 1 descontrolada, el
destino principal del acetil-CoA formado será la síntesis de cuerpos cetónicos.
Los niveles de HDL en el paciente diabético tipo 1 pobremente controlado, están
disminuidos como consecuencia del escaso catabolismo de las VLDL y los Qm, lo cual impide
la maduración de las HDL. Otra característica del cuadro lipoproteico en la diabetes tipo 1, es la
presencia de IDL varias horas después de la ingesta. Esta lipoproteína se acumula al no poder
ser degradada por la lipasa hepática, ya que esta enzima también es estimulada por la insulina.
Además, se debe tener en cuenta que estas lipoproteínas tienen alteraciones, como un mayor
contenido en Apo C, que impide su captación por los receptores.
-Diabetes tipo 2: este tipo de diabetes se halla frecuentemente asociada a la obesidad y a la
hiperinsulinemia. En respuesta a un estado de resistencia a la insulina, existe una mayor
producción de precursores para la síntesis hepática de TG y la formación y secreción de VLDL.
En estos pacientes, cuando la remoción de VLDL del plasma es incompleta, se acumulan
remanentes, persistiendo IDL aumentada en ayunas.
Es frecuente encontrar niveles elevados de LDL, debidos principalmente al aumento de su
precursor (VLDL) que se cataboliza por la LPL, cuya actividad es normal en los primeros
estadíos de la enfermedad. Otra razón que contribuye a elevar los niveles de LDL, consiste en
la disminución de la capacidad de interacción con los receptores de LDL dado que se altera la
estructura de esta lipoproteína por glicosilación. La presencia de LDL glicosilada es común a
ambos tipos de diabetes dado que la glicosilación de LDL es consecuencia de la hiperglucemia.
Un subtipo de LDL asociado con la diabetes tipo 2 y otros estados de resistencia a la insulina,
es la aparición de LDL pequeña y densa, asociada a alto riesgo aterogénico. Los niveles de
HDL en la diabetes tipo 2 se encuentran disminuidos, en estrecha relación con la
hipertrigliceridemia, la obesidad asociada y el grado de control de la diabetes. Además, la
lipasa hepática, como consecuencia de la hiperinsulinemia, acelera la remoción de la HDL.

Hipotiroidismo
La hipercolesterolemia es un hallazgo muy frecuente en el hipotiroidismo, así como los
niveles bajos de colesterol en el hipertiroidismo. Si bien la hipofunción tiroidea puede cursar
con trigliceridemia normal, también se han encontrado valores de TG moderadamente
elevados, dependiendo fundamentalmente del grado de disfunción tiroidea.
Las hormonas tiroideas estimulan la síntesis de los receptores-LDL, de la enzima lipasa
hepática y en menor medida de la LPL. La hipercolesterolemia que frecuentemente se observa
en los pacientes hipotiroideos, se debe a una disminución en el catabolismo de LDL, como
consecuencia de la disminución del número de receptores. Por otro lado, como consecuencia
de la disminución en la actividad de la lipasa hepática, se observa permanencia de
lipoproteínas intermedias en la circulación, con gran dificultad para continuar su camino
metabólico. Esto explica los hallazgos de una lipoproteína rica en colesterol, con características
de β-VLDL, en el suero de algunos pacientes hipotiroideos. Finalmente, ante un descenso en la
actividad de LPL, se manifiesta una hipertrigliceridemia con aumento de VLDL.
Los niveles de HDL no disminuyen dado que la actividad baja de la lipasa hepática se asocia
con un menor catabolismo hepático de la HDL2.
Los pacientes presentan frecuentemente el fenotipo IIa aunque también pueden presentar
un fenotipo IIb, III y IV.
Síndrome nefrótico
El aumento en la concentración de lipoproteínas plasmáticas es una característica del
síndrome nefrótico.
La síntesis hepática de proteínas y lipoproteínas se encuentra incrementada, en respuesta a
la disminución de la presión oncótica causada por la hipoalbuminemia. Esto incluye una
sobreproducción de VLDL y, por lo tanto, de IDL y LDL. Suele observarse también aumento de
las concentraciones de Lp(a). Además existe una disminución del catabolismo de las
lipoproteínas ricas en TG, supuestamente por pérdida de cofactores o activadores de la LPL
(principalmente Apo C-II), por la orina.
Las HDL, por su menor tamaño, son las únicas lipoproteínas que pueden atravesar el riñón,
por lo tanto es frecuente encontrar los niveles de HDL descendidos. Dado que también se
produce aumento de su síntesis y de Apo A-I, lo que compensaría rápidamente la pérdida, los
valores de los niveles de HDL frecuentemente se encuentran normales.
Por otro lado, si la pérdida de albúmina es grande, disminuye la hidrólisis de las VLDL
puesto que la albúmina es la que acepta los ácidos grasos libres. Estos ácidos grasos libres
ante la falta de albúmina se unen a las lipoproteínas, alterándolas y disminuyendo la capacidad

de hidrólisis. Aumenta entonces la concentración de VLDL y disminuye la concentración de
LDL.
Cabe destacar que además del aumento del colesterol en sus fracciones más aterogénicas
(LDL, IDL y Lp(a)) la composición de las lipoproteínas en el síndrome nefrótico es anormal,
tanto en su contenido lipídico como lipoproteico generando en estos pacientes un riesgo
cardiovascular aumentado.
Obstrucción biliar
Dado que el hígado es el lugar de síntesis y catabolismo de gran parte de las lipoproteínas y
de enzimas fundamentales para el metabolismo lipoproteico, cualquier enfermedad hepatobiliar
traería aparejada anormalidades en la síntesis lipoproteica y/o su catabolismo.
La colestasis intra o extra hepática cursa con concentraciones muy aumentadas de
colesterol y fosfolípidos. Los ácidos biliares se acumulan e inhiben la degradación del colesterol
por retroalimentación negativa. Esto unido a un aumento de la síntesis de colesterol hepático,
explicarían la hipercolesterolemia.
La anormalidad lipoproteica característica de la colestasis es la aparición de la lipoproteína x
(LPx). Esta es de estructura discoidal, contiene fosfolípidos y colesterol libre en igual relación,
mientras que la albúmina y apoproteínas del grupo C conforman su contenido proteico. Esta
lipoproteína se formaría ante la necesidad de transportar el exceso de fosfolípidos y colesterol
acumulados.
La actividad de la enzima LCAT es variable, dependiendo en gran parte del grado de
obstrucción, de la injuria hepática, y del aporte de su cofactor Apo A-l. La disminución de la
enzima determina que la hipercolesterolemia se produzca a expensas del colesterol libre.
En la colestasis, las lipoproteínas presentan alteraciones relacionadas, tal vez, con la
deficiencia de LCAT. Las VLDL tienen una composición apoproteica y lipídica alterada. Las
HDL se presentan en forma discoidal o naciente y son ricas en colesterol libre y fosfolípidos.
Ambas lipoproteínas, al igual que la LPx, tienen movilidad beta en la electroforesis, coincidente
con la LDL que, a su vez, es rica en TG.
Inqesta de Alcohol
Existe una relación inversa entre el consumo moderado de alcohol y la incidencia de
enfermedad cardiovascular. El alcohol es oxidado a acetaldehído y por acción de la aldehído
deshidrogenasa se forman acetato y NADH. Así, el aumento del poder reductor que se genera,
es el responsable de la mayoría de los efectos del etanol sobre el hígado. Los ácidos grasos
libres aumentan, ya sea por incremento de su síntesis como por disminución de su
catabolismo, conduciendo a un aumento de la síntesis de TG y de VLDL.
Para interpretar el efecto del alcohol, se debe tener presente la cantidad y forma de
consumo. La ingesta de más de 170 g de alcohol en un período menor a 6 hs, lleva a una

hipertrigliceridemia aguda, por disminución de la LPL. Esta hipertrigliceridemia es más severa si
el consumo agudo de alcohol ocurre conjuntamente con una dieta rica en grasas.
El alcoholismo crónico moderado, considerado como la ingesta de 15 a 40 g/día durante 2
años, logra mantener un perfil lipoproteico favorable con TG-VLDL normales, LDL normal o
disminuida y HDL aumentada. Esto último responde a la inducción hepática de la síntesis de
Apo A-I y Apo A-II y por ende de HDL.
Cuando la ingesta de alcohol es más severa aumentando a 60-80 g/día durante más de 2
años, incrementa la síntesis y secreción de TG, pero también la actividad de LPL, por lo tanto
su nivel puede mantenerse normal. La concentración de HDL y Apo A-I va a depender de la
funcionalidad hepática. Cuando comienza el daño hepático, decae la secreción de TG-VLDL y
se instala el hígado graso, que puede conducir a la hepatitis alcohólica, y/o a la cirrosis. Este
cuadro se acompaña de una frecuente malnutrición del individuo alcohólico, disminuyendo aún
más el aporte proteico necesario para la funcionalidad hepática.
El consumo moderado de alcohol protege de la enfermedad cardiovascular no sólo por el
aumento de Apo A-I y de HDL sino también, por tener un efecto antiagregante plaquetario y de
vasodilatación, así como la presencia de antioxidantes adjudicados a componentes no-
alcohólicos contenidos en vinos, cervezas y licores que poseen polifenoles y flavonoides.
Hormonas sexuales
Es bien conocida la menor incidencia de enfermedad cardiovascular en la mujer
premenopáusica con respecto al hombre. Las mujeres presentan niveles más bajos de LDL y
más elevados de HDL que los varones. Esto se debe al papel de los estrógenos que estimulan
la síntesis de los receptores LDL y de Apo A-I, principal constituyente de HDL. Además los
estrógenos inhiben la actividad de la lipasa hepática contribuyendo al aumento de HDL.
Como consecuencia de la disminución estrogénica en la menopausia se produce un
marcado deterioro del perfil lipoproteico, asociado con un elevado riesgo cardiovascular
característico de esta etapa. La dislipemia más frecuentemente observada en la mujer
postmenopáusica es la hipercolesterolemia, presentando: aumento de colesterol-total, aumento
de c-LDL, c-IDL y aumento de Apo B-100, sin grandes variaciones en el c-HDL.
La progesterona y los andrógenos tienen un efecto opuesto al de los estrógenos ya que
producen disminución de las HDL.
Otra situación fisiológica donde se producen alteraciones hormonales es durante el
embarazo. El marcado aumento estrogénico característico del tercer trimestre conduce a un
aumento en la síntesis de TG que lleva a una mayor secreción de VLDL. Por lo tanto, es
frecuente observar hipertrigliceridemia en este trimestre, que no es considerada de riesgo,
salvo que la mujer embarazada presente antecedentes de dislipemia o diabetes, que agraven
el cuadro. En estos casos la hipertrigliceridemia debe ser controlada cuidadosamente para
prevenir el desarrollo de una pancreatitis aguda. En la mayoría de las mujeres, el colesterol y

los TG se mantienen elevados hasta la cuarta semana del postparto y el descenso posterior de
los niveles lipídicos es más marcado en las mujeres que amamantan.
A B
C D
AA BB
CC DD
Figura 14: Imágenes de algunos signos y síntomas de pacientes con hiperlipoproteinemias. A: Arco corneal (banda clara) en hombre con Hipercolesterolemia familiar homocigota (flechas). B: Múltiples xantomas tendinosos y planos en manos del mismo paciente. C: Xantomatosis eruptiva en un paciente con Hiperquilomicronemia. D: Xantomas tuboeruptivos en codos de un varón con Disbetalipoproteinemia.
Hipolipoproteinemias
Las hipolipoproteinemias son mucho menos frecuentes que las hiperlipoproteinemias. Su
presencia puede responder a causas primarias o secundarias.
Hipolipemias primarias
1. Abetalipoproteinemia: es una enfermedad de transmisión autosómica recesiva de muy
baja incidencia que se manifiesta desde la infancia. El defecto radica en una modificación post-
traduccional de Apo B-100 que impide su normal incorporación a las lipoproteínas. Se
caracteriza por total ausencia de producción de Qm, VLDL, IDL y LDL, con concentraciones
plasmáticas de TG y colesterol extremadamente bajas. Se produce acumulación de TG en
hígado e intestino. Las manifestaciones clínicas consisten en esteatorrea, mala absorción de
vitaminas liposolubles y graves alteraciones neurológicas, oculares, musculares y
hematológicas.
2. Hipoalfalipoproteinemia: esta patología es poco frecuente y se han descrito distintas
mutaciones que causan hipoalfalipoproteinemia. Entre las más conocidas se encuentran la
enfermedad de Tangier, donde se produce un defecto en la síntesis de HDL a partir de una
síntesis defectuosa de Apo A-I que es destruida rápidamente de forma tal que la vida media de

la HDL es muy corta. Esta carencia se traduce en una alteración del metabolismo de otras
lipoproteínas fundamentalmente los Qm y las VLDL debido a la disponibilidad reducida de Apo
C-II. Su riesgo aterogénico no es claro.
3. Deficiencia de LCAT: Otro trastorno relacionado con una disminución de las HDL es la
deficiencia en la lecitina colesterol acil transferasa. Los pacientes tienen TG plasmáticos
elevados, el colesterol es normal o está aumentado pero en mayor proporción el colesterol no
esterificado. Las VLDL y Qm son muy ricas en colesterol no esterificado y parecen tener
monocapas anormales en la superficie. Las LDL poseen TG. Los depósitos de colesterol no
esterificado y fosfolípidos en los vasos sanguíneos llevan a la destrucción de las nefronas y a la
insuficiencia renal. También se produce anemia, los glóbulos rojos presentan un aumento del
colesterol no esterificado y lecitina en sus membranas.
Hipolipoproteinemias secundarias
Las hipolipemias secundarias se relacionan principalmente con situaciones de alteraciones
hepáticas graves, neoplasias, desnutrición, infecciones agudas, anemias graves,
hipertiroidismo, talasemia mayor, lupus eritematoso sistémico, artritis reumatoidea, etc.
CASOS CLÍNICOS
1- A un hombre de 36 años de edad se le detectó hipercolesterolemia. Sus niveles de
colesterol plasmáticos tras un ayuno de 14 horas, determinado en dos ocasiones distintas eran
de 330 mg/dl aproximadamente. Fue tratado durante 3 meses con una fórmula dietética exenta
de colesterol, pero los niveles de colesterol plasmático en ayunas tan sólo descendieron a 300
mg/dl. Posteriormente fue tratado con colestiramina, una resina que adsorbe sales biliares.
Esta resina no es adsorbida y permanece en el intestino, donde va captando sales biliares, que
son eliminadas en grandes cantidades por las heces. Este tratamiento farmacológico tuvo éxito
y el colesterol plasmático en ayuno disminuyó a valores entre 200-220 mg/dl.
a- ¿Por qué se efectuaron las determinaciones en plasma tras un ayuno de 14 horas?
b- ¿Cómo es posible que el paciente siga presentando hipercolesterolemia tras seguir una
dieta exenta de colesterol?
c- ¿Qué relación existe entre las sales biliares y el colesterol?
d- ¿Por qué una excreción aumentada de sales biliares conlleva en ciertos casos un
descenso en la concentración plasmática de colesterol?
e- ¿Por qué el descenso en la concentración plasmática de colesterol beneficiaría a este
paciente?
f- ¿Qué otros estudios lipídicos podría realizarle a este paciente además del colesterol total?
¿Qué información le brindarían?

2- Un hombre de 75 años de edad, presentaba cefaleas, debilidad muscular y calambres en
miembros inferiores. Se le registró hipertensión arterial, moderada (200/100 mm de Hg) y
soplos en las arterias carótidas y femorales. Presentaba además deterioro cognitivo,
alteraciones mnésicas, fallas en la denominación de objetos e ideas delirantes. Súbitamente
sufrió hemiparesia facio-braquio-crural derecha y afasia. Llevado de urgencia a un instituto
especializado se le practicó una angiografía de la arteria carótida izquierda que demostró un
estrechamiento de la luz arterial. Una resonancia magnética de cerebro demostró un infarto
extenso correspondiente al territorio vascular de la arteria silviana izquierda, además de
múltiples focos isquémicos e infartos lacunares en ambos hemisferios cerebrales. El nivel de
colesterol en ayunas era de 385 mg/dl y la trigliceridemia era de 150 mg/dl. Se le practicó una
endarterectomía de urgencia de la arteria carótida izquierda. Fue dado de alta y se le prescribió
lovastatin, fármaco que inhibe a la enzima HMGCoA reductasa.
a- ¿Cómo esperaría encontrar los valores de c-HDL y c-LDL en este paciente?
a-¿Qué relación existe entre la hipercolesterolemia y aterosclerosis?
b- Indique como se relaciona el colesterol con las lipoproteínas plasmáticas.
c- ¿Qué beneficio puede obtenerse con el uso del lovastatin? ¿Sería útil también en este
paciente utilizar una dieta pobre en colesterol?
d- ¿Cómo captan LDL las células humanas?
e- ¿Cuáles pueden ser las causas de esta dislipoproteinemia?
3- En un exámen de rutina se encontró que un hombre de 36 años presentaba una
concentración plasmática de colesterol en ayunas de 250 mg/dl (valor normal < 200 mg/ dl.) y
una concentración plasmática de triglicéridos de 500 mg/dl (valor normal <150 mg/dl). No
presentaba ninguna manifestación clínica. Se le recomendó primero una dieta normocalórica
que contenía 15 % de proteínas, 15% de grasas, 70 % de glúcidos y 300 mg de colesterol
diarios. Posteriormente se sugirió una dieta sin grasas. En ninguno de los dos casos se
alteraron los resultados de laboratorio, por el contrario los valores de triglicéridos aumentaron.
Como este tratamiento no dio resultado, se cambió la dieta, se redujeron los glúcidos y se
reemplazaron por alimentos de alto contenido en grasas. En poco tiempo se observó una
notable disminución de los lípidos (colesterol 200 mg/dl y triglicéridos 170 mg/dl).
a- ¿Qué importancia tiene en el diagnóstico en este caso el lipidograma electroforético?
¿Qué lipoproteína espera encontrar elevada?
b- ¿Cuál es el lípido mas aumentado en plasma? ¿Qué le sugiere este resultado en relación
al diagnóstico?
c- ¿Por qué la dieta pobre en grasas no fue efectiva? ¿Cuáles son a largo plazo los peligros
de una dieta sin lípidos?

d- Explique la mejoría después de la dieta pobre en glúcidos.
f- Indique cuál es la función y la composición de la lipoproteína aumentada. Relacione su
composición con su densidad y movilidad electroforética.
e- ¿A qué fenotipo corresponde esta patología de acuerdo al lipidograma?
4- Un joven de 15 años tenía antecedentes de dolores abdominales, incluyendo accesos de
dolor abdominal tan severos que requirieron narcóticos para aliviarlo. Estas situaciones
ocurrían en promedio cada seis meses. El paciente se había sentido bien recientemente hasta
que comenzó con dolor abdominal. Su madre afirmó que el dolor había comenzado 4 horas
después de haber comido costillas de cerdo, papas fritas, leche y helado con crema. Nadie
más en la familia se había enfermado por la comida. A su llegada se tomó una muestra de
sangre. A los 15 minutos el técnico de laboratorio reportó que no podían obtenerse resultados
válidos porque el plasma estaba cargado con grasa. Era lechoso pero luego de centrifugarlo se
aclaró considerablemente y había una banda compacta de crema en la parte superior de la
muestra.
a) ¿Qué tipo de anormalidad lipídica sospecha en este paciente?
b) ¿Cuál sería el resultado del lipidograma? ¿A qué fenotipo corresponde?
c) ¿Qué tipo de dieta sería útil para aliviar la enfermedad?
d) ¿Recomendarían una dieta suplementada con triacilglicéridos que contengan ácidos
grasos de cadena media? ¿Por qué?
e) ¿Por qué esta enfermedad puede provocar pancreatitits?
f) ¿Cuál podría ser una causa de la enfermedad?