u7 – diseÑo y anÁlisis de estudios genÉticos · herencia de la enfermedad y estimar la...
TRANSCRIPT

DISEÑO y ANÁLISIS de INVESTIGACIONES CLÍNICAS: MÓDULO IV
U7– Diseño y análisis de estudios genéticos
U7 – DISEÑO Y ANÁLISIS DE ESTUDIOS GENÉTICOS
Juan Ramón González Ruiz. Doctor en Estadística. Investigador. Centre de Recerca
en Epidemiologia Ambiental (CREAL). Unitat de Bioestadistica. Departament de Salut
Pública. Universitad de Barcelona (UB)
1. INTRODUCCIÓN
La publicación del primer borrador del genoma humano que se llevó a cabo en el año
2001 [ver lecturas recomendadas] ha permitido realizar numerosos estudios genéticos
en los que se han encontrado genes de susceptibilidad para más de 40 enfermedades
complejas (Alzheimer, cáncer, diabetes, …). Tradicionalmente los diseños de los
estudios genéticos se han basado en casos afectos con una mayor predisposición a
desarrollar la enfermedad. Por ejemplo, individuos con una aparición temprana de la
enfermedad o aquellos con varios familiares afectados. En estos casos los estudios se
basan en reclutar individuos relacionados (pedigríes o trios). Actualmente, sin
embargo, los estudios genéticos suelen disponer de un grupo control y son similares a
los que se utilizan epidemiología tradicional como los estudios de casos y controles y
los de cohortes (principalmente los primeros). Una ventaja de utilizar este tipo de
diseños es que se suele disponer de una amplia información recogida en los
cuestionarios, así como de bancos con muestras biológicas que permiten analizar el
ADN de los individuos. No obstante, antes de llevar a cabo un estudio de asociación,
debemos tener evidencias que los factores genéticos juegan algún papel en la
enfermedad que estamos estudiando. Para ello, los diseños tradicionales en genética
basados en estudios con familias resultan cruciales.
En esta unidad se empezará describiendo los marcadores genéticos que actualmente
se utilizan en los estudios de genética, así como las tecnologías existentes para
obtener la información sobre estos marcadores. Después se describirán los principales
diseños, no sólo para establecer asociación si no también para poder determinar la
existencia de algún gen de susceptibilidad para nuestra enfermedad de interés. Más
tarde se ilustrará cómo realizar el análisis estadístico, que incluirá: cómo evaluar
asociación, cómo corregir por estratificación poblacional (es decir, confusión por raza o
etnia) y cómo tener en cuenta las comparaciones múltiples. Estos métodos se

DISEÑO y ANÁLISIS de INVESTIGACIONES CLÍNICAS: MÓDULO IV
U7– Diseño y análisis de estudios genéticos
ilustrarán mediante un software libre basado en una aplicación Web que realiza todos
estos análisis de forma sencilla y rápida, utilizando además varios conjuntos de datos
reales.
2. DISEÑO DE ESTUDIOS GENÉTICOS
2.1 VARIABILIDAD GENÉTICA
Empezaremos describiendo los marcadores genéticos que comúnmente se utilizan en
estudios genéticos. Estos marcadores permitirán medir la variabilidad genética entre
individuos, así como poder establecer posibles diferencias entre individuos sanos y
enfermos. De esta forma, se podrán establecer qué regiones del genoma están
involucradas en el desarrollo, susceptibilidad o etiología de una enfermedad. Para
establecer una similitud con otros tipos de estudios, diremos que, estas variantes
genómicas juegan el mismo papel que los factores de riesgo en los estudios de
epidemiología tradicional.
Las variaciones genéticas heredables se llaman polimorfismos, que ocurren cuando el
DNA muta en las células germinales que se transmiten a los descendientes. La
mayoría de estos polimorfismos no tienen un impacto funcional (es decir, no hacen
que aparezca una enfermedad o un rasgo determinado), sólo algunos polimorfismos
tienen algún impacto y normalmente viene determinado por la evolución. Cuando uno
de estas variantes presenta una ventaja para el individuo, el polimorfismo aumenta en
frecuencia en la población.
Existen tres tipos de polimorfismos a nivel genético: Single Nucleotide Polimorphism
(SNP) (pronunciado “esnip”), Variable Number Tandem Repeats (VNTR) y Copy
Number Variations (CNV). Los SNPs son los polimorfismos más frecuentes y son los
más analizados en la actualidad. En este curso estudiaremos este tipo de variantes
genéticas.
Los SNPs son polimorfismos de un cambio en un nucleotido en una posición concreta
del genoma que afecta a una sola base (adenina (A), timina (T), citosina (C) o guanina
(G)). Por ejemplo, en el gen de la apolipoproteína E (ApoE) se han descrito varios
polimorfismos frecuentes que consisten en cambios de una única base. Uno de ellos,
denominado ApoE * -4, resulta en un cambio en el aminoácido C de la posición 112

DISEÑO y ANÁLISIS de INVESTIGACIONES CLÍNICAS: MÓDULO IV
U7– Diseño y análisis de estudios genéticos
por una A. Esta variante se asocia con la enfermedad de Alzheimer. El cambio de un
único nucleótido, si ocurre en una zona codificante como en el ejemplo de la ApoE,
puede provocar un cambio de aminoácido en la proteína resultante, y ello puede
resultar en una modificación de su actividad o función. Los cambios también pueden
ocurrir en zonas del promotor de un gen y modificar su expresión. Estas zonas
promotoras modulan el proceso de transcripción del ADN en ARN (la transcripción es
el primer paso de la decodificación de un gen a una proteína). Lo mismo puede ocurrir
si el cambio se produce en un intrón, como el ejemplo de la ataxia de Friedrich.
Aunque los intrones no se traducen a proteína, cambios en su estructura pueden
modular la expresión del gen. Otras veces, probablemente la mayoría, los cambios son
silentes y no tienen repercusiones funcionales. Mientras que sólo estudios moleculares
específicos pueden poner de manifiesto si los polimorfismos son funcionales, los
estudios epidemiológicos son fundamentales para valorar si hay efectos en la salud de
la población. En esta unidad se tratará principalmente cómo evaluar y cuantificar este
efecto. La información genética obtenida a partir de los SNPs (genotipos) se puede
obtener mediante distintas técnicas de genotipado que se describirán en la siguiente
sección.
Los VNTR aparecen con menos frecuencia que los SNPs y consisten en repeticiones
seriadas de una serie de nucleotidos con tamaño variable. Por ejemplo,
ATATATAT=(AT)4, ATATATATATAT=(AT)6. Las repeticiones pueden ser
mononucleotidas (AAAAA), dinucleotidas (AT) o repeticiones más largas. Estos
polimorfismos también se conocen como microsatélites y comúnmente son
multialélicos ya que el número de repeticiones puede variar enormemente. Los VNTRs
pueden tener un impacto funcional si están presentes en regiones codificantes. Un
ejemplo típico es la diabetes tipo I que se ha asociado a VNTR en el gen de la insulina.
Los individuos con un número pequeño de repeticiones (menos de 50) tiene el doble
de riesgo que los sujetos con más de 200 repeticiones.
Los CNVs se han identificado más recientemente como una fuente adicional de
variabilidad genética. La variación en el número de copias de un gen puede ser un
factor de riesgo para algunas enfermedades como por ejemplo osteoporosis o
psoriasis donde individuos con menos copias de los genes UGT2B17 y LCE3C,
respectivamente, presentan una mayor probabilidad de desarrollar dichas
enfermedades.

DISEÑO y ANÁLISIS de INVESTIGACIONES CLÍNICAS: MÓDULO IV
U7– Diseño y análisis de estudios genéticos
2.2 TECNOLOGÍAS DE GENOTIPADO
Los genotipos de un SNP, es decir la combinación de sus dos correspondientes alelos
(dos homocigotos AA y BB y un heterocigoto AB o BA, donde A y B puede ser
cualquiera de las 4 bases A,C,G ó T ) se obtienen a partir de las intensidades que se
obtienen de una imagen escaneada de un experimento de microarrays. Para cada uno
de los alelos también se dispone de información de una sonda para las dos
direcciones del DNA (sense y antisense), así finalmente se dispone de 4 valores de
intensidad. Los genotipos se determinan utilizando algoritmos que procesan estas
intensidades y que dependen del número de SNPs que se estén procesando cada vez.
Al inicio de realizarse estudios genéticos los genotipos solían obtenerse mediante
métodos basados en PCR (polymerase chain reaction). Estos métodos permiten
genotipar un número limitado de marcadores en cada ensayo, por lo que se
empezaron a diseñar otros métodos que permitieran obtener información de un
número mayor de SNPs en cada experimento. Estos métodos utilizan arrays de SNPs.
Las primeras tecnologías de SNP arrays eran capaces de determinar unos 1.500
SNPs en un único ensayo. Más adelante aparecieron los arrays 100K (K indica 1,000
por lo que estos arrays eran capaces de genotipar 100,000 SNPs), 300K, 500K,
1,000K y actualmente existen arrays incluso de 4,000K. Actualmente se dispone de
dos plataformas distintas para obtener estos genotipos: Affymetrix e Illumina [ver links
a Illumina y Affymetrix en lecturas recomendadas].
2.3 DISEÑOS DE ESTUDIOS GENÉTICOS
Encontrar genes que se asocien a enfermedades es un largo proceso y necesita
responder a varias preguntas antes de establecer esta relación (Tabla 7.01). Cada
pregunta normalmente requiere llevar a cabo un diseño de estudio específico y medir
la información genética con diferentes niveles de precisión. Sin embargo, actualmente
se puede obtener información para todo el genoma completo de cada individuo de
forma relativamente sencilla y barata, por lo que estos pasos previos no suelen
llevarse a cabo. En este curso nos basaremos en este tipo de estudios que pretenden
evaluar una posible asociación entre los genes y ciertas enfermedades o rasgos. Sin

DISEÑO y ANÁLISIS de INVESTIGACIONES CLÍNICAS: MÓDULO IV
U7– Diseño y análisis de estudios genéticos
embargo, es importante conocer en qué escenarios se pueden emplear este tipo de
estudios en relación a otros estudios de genética.
Ante de diseñar un estudio que pretenda establecer asociaciones entre SNPs y cierta
enfermedad, se debe contestar a una pregunta obvia importante: ¿Están los genes
relacionados con la enfermedad que estamos estudiando? Esta pregunta se puede
contestar utilizando información de estudios de casos y controles en el caso que
seamos capaces de demostrar que hay cierta agregación familiar. Un análisis que
incluya una variable que indique si hay familiares diagnosticados con nuestra
enfermedad de interés en los individuos analizados y que demuestre que la proporción
de afectos es superior en casos que en controles, puede ser concluyente. Sin
embargo, este es un mecanismo muy naive ya que puede haber factores que
confundan los resultados debido a exposiciones ambientales que comparten los
familiares. Otra posibilidad de obtener evidencias de la existencia de algún factor
genético es considerar estudios con inmigrantes. Esto ocurriría si las tasas de
enfermedad en las segundas generaciones de inmigrantes es más similar a las tasas
observadas en los países de origen que en los países de residencia. No obstante, los
estudios más concluyentes sobre la existencia de algún gen relacionado con la
enfermedad de estudio, vendría dado por los estudios de gemelos. Estos estudios son
los más potentes para estimar la heredabilidad (la proporción de casos atribuible a
factores genéticos). En estos estudios se trataría de comparar la concordancia entre
los gemelos monocigotos y dicigotos combinada con la información ambiental que
comparten los hermanos [ver artículo de Lichtenstein et al., en lecturas
recomendadas].
Cuando una enfermedad aparece de forma recurrente en algunas familias, se suelen
llevar a cabo estudios de segregación en pedigríes para poder determinar el modo de
herencia de la enfermedad y estimar la penetrancia. Los estudios de ligamiento,
también realizados en familias, nos dan una idea de en qué región del genoma se
encentran estos genes.
Cuando se dispone de suficiente evidencia sobre el hecho que existen factores
genéticos como causa de una enfermedad específica, la siguiente pregunta que se
debe realizar es: ¿qué genes son? Es en este punto donde se ubican los estudios de
asociación que vamos a tratar en esta asignatura. El hecho de que hagamos mayor
énfasis en los estudios de asociación viene dado por la mejora que se ha llevado a

DISEÑO y ANÁLISIS de INVESTIGACIONES CLÍNICAS: MÓDULO IV
U7– Diseño y análisis de estudios genéticos
cabo en los últimos años en la tecnología para medir marcadores genéticos. Como se
ha comentado anteriormente, la secuenciación del genoma ha permitido que se pueda
establecer qué regiones del genoma están asociadas a ciertas enfermedades, y en
última instancia cuáles son responsables. Los estudios de asociación utilizan SNPs
como marcadores genéticos y son la aproximación más potente para contestar a esta
pregunta. Los diseños en estudios de asociación son los que normalmente se llevan a
cabo en epidemiología tradicional ya que los investigadores aprovechan la información
de estos estudios y la combinan con la información genética que obtienen a partir de
material biológico que estos estudios suelen disponer. Así los estudios de asociación
genética se basan en diseños de casos y controles y diseños de cohortes. Estos
diseños se han descrito en una unidad anterior (unidad 5 sobre diseño y análisis de
estudios de factores de riesgo), por lo que no entraremos en detalle en ellos.
Los estudios de asociación comparan las frecuencias de los genotipos de una serie de
SNPs entre casos y controles no relacionados de una muestra de una población dada.
La posibilidad de poder incluir casos y controles no relacionados, permite utilizar
tamaños de muestra grandes para aumentar el poder de detección. La estrategia que
suelen llevara a cabo para encontrar genes responsables de una enfermedad
normalmente se basa en seleccionar SNPs pertenecientes a genes candidatos porque
se conozca que estos genes tienen una relación con el mecanismo de acción de la
enfermedad. Por ejemplo, los genes típicos que se estudian en cáncer están
relacionados con el control del ciclo celular, la inflamación, el metabolismo, o la
reparación de DNA.
Actualmente la tecnología es capaz de genotipar millones de SNPs a la vez. De esta
forma, en vez de realizar estudios de asociación con genes candidatos en los que se
incluye unos cientos de SNPs, también se puede llevar a cabo un análisis completo del
genoma en el que se intenta determinar qué SNPs se asocian con la enfermedad, sin
establecer ninguna hipótesis a priori. Estos estudios se conocen como GWAS (del
inglés Genome Wide Association Studies). Un ejemplo ilustrativo de estos estudios, en
los que no se establece ninguna hipótesis previa aparece en cáncer de próstata,
donde se ha identificado una región en la que no hay genes que puedan establecerse
como responsables de esta enfermedad.
El hecho de encontrar un SNP asociado con la enfermedad no quiere decir que sea el
responsable de la misma. Esto puede ocurrir por tres causas, principalmente. La

DISEÑO y ANÁLISIS de INVESTIGACIONES CLÍNICAS: MÓDULO IV
U7– Diseño y análisis de estudios genéticos
primera es que realmente sea el SNP causal, la segunda a que este SNP esté en
desequilibrio de ligamiento (LD), es decir, correlacionado con el verdadero SNP
causal, y la tercera a que los resultados estén confundidos por lo que se conoce como
estratificación poblacional (este problema se tratará en la sección de análisis). En
ocasiones, para encontrar el verdadero SNP causal tendremos que volver a re-
secuenciar llevando a cabo un genotipado más detallado (fine-mapping) de la región
de intereses. Finalmente, el SNP causal requerirá de estudios funcionales para
documentar el mecanismo de acción que determina el riesgo.
Para algunos genes, la variación genética no es probablemente suficiente para causar
una enfermedad, a menos que actúe un factor ambiental de forma simultánea. Por
ejemplo, el polimorfismo NAT2 se ha asociado con un riesgo aumentado de cáncer de
vejiga en los fumadores y este riesgo no se ha observado en fumadores [ver artículo
de García-Closas et al. en lecturas recomendadas]. Este es un ejemplo de interacción
gen-ambiente. Ignorar el factor ambiental puede hacer que no encontremos un riesgo
para ciertos genes puesto que éste se puede atenuar cuando incluimos individuos que
están expuestos a un factor ambiental (promediar el efecto en fumadores y no
fumadores). De la misma forma, también es probable que las interacciones gen-gen
puedan existir y que sólo los individuos portadores de múltiples variantes presente un
riesgo aumentado para ciertas enfermedades. Ambos tipos de interacciones pueden
estudiarse en estudios de asociación donde la dificultad radica en el hecho de que, sin
hipótesis a priori, el número de interacciones que podemos estudiar es muy grande,
requiriendo además un tamaño de muestra muy elevado.
2.4 ESTUDIOS DE ASOCIACIÖN COMPLETOS DEL GENOMA (GWAS)
La continua mejora llevada a cabo en la tecnología de genotipado y sobre todo su
abaratamiento, ha hecho posible que actualmente se lleven a cabo numerosos
estudios de asociación en los que se interroga el genoma completo. Los GWAS
utilizan tecnologías de genotipado a gran escala (en inglés high-throughput) para
analizar cientos de miles de SNPs y relacionarlos con variables clínicas, rasgos
cuantitativos o enfermedades. Una de las principales ventajas de los GWAS es que no
presuponen ninguna hipótesis a priori con respecto a una posible relación entre un gen
y la enfermedad de estudio, permitiendo así encontrar nuevos genes de

DISEÑO y ANÁLISIS de INVESTIGACIONES CLÍNICAS: MÓDULO IV
U7– Diseño y análisis de estudios genéticos
susceptibilidad para enfermedades comunes como por ejemplo Alzheimer, cáncer,
esquizofrenia, diabetes, Parkinson o esclerosis múltiple entre otras [ver WTCC en
lecturas recomendadas]. Por otro lado, uno de los principales problemas inherentes a
los GWAS es que pueden darse un número muy elevado de falsos positivos puesto
que se están llevando a cabo cientos de miles de tests estadísticos, haciéndose
necesario adoptar correcciones estadísticas o llevar a cabo estudios suplementarios
para replicar los resultados obtenidos. El tema de cómo tener en cuenta las
comparaciones múltiples se estudiará más adelante en la sección de análisis.
El diseño más empleado en los GWAS ha sido hasta ahora el de casos y controles.
Como se ha comentado anteriormente, la razón principal es que ya existen estudios
epidemiológicos que además disponen almacenado material biológico del que se
puede extraer información genética. Estos estudios, además, son menos costosos que
los estudios de cohortes, donde se requiere décadas de seguimiento para alcanzar el
número de casos requerido para detectar efectos genéticos moderados, y de ahí su
adopción en este tipo de estudios.
La mayoría de GWAS adoptan diseños multi-estado para reducir el número de falsos
positivos minimizando el coste de genotipado y manteniendo el poder estadístico [ver
Hirschhorn and Daly en lecturas recomendadas]. En la práctica, los GWAS se llevan a
cabo en 2 (o a veces más) pasos. En el primer paso se genotipan todos los SNPs
(dependiendo de la plataforma entre 300,000 y 1,000,000 de SNPs o más) en un
grupo inicial de casos y controles. Después, en el paso 2, sólo los marcadores más
“prometedores” (es decir, los SNPs que muestran una significación estadística más
importante) son re-genotipados en otro grupo de casos y controles. El número de
SNPs e individuos incluido en ambos pasos puede variar dependiendo del coste. Uno
de los principales problemas en estos diseños es establecer el punto de corte que
determine qué SNPs vale la pena re-genotipar. En la sección de análisis indicaremos
algunas aproximaciones que se están adoptando actualmente para tratar este
problema.

DISEÑO y ANÁLISIS de INVESTIGACIONES CLÍNICAS: MÓDULO IV
U7– Diseño y análisis de estudios genéticos
3. ANÁLISIS DE ESTUDIOS GENÉTICOS
Los análisis estadísticos que se llevan a cabo en los estudios de asociación en
genética no son complicados, pues principalmente se basan en analizar modelos de
regresión logística que son los que normalmente se usan en estudios de casos y
controles. El principal problema que aparece en este tipo de estudios es el coste
computacional, ya que además de evaluar la asociación entre la enfermedad y cada
SNP (recordemos que pueden ser cientos de miles) también se suelen probar
diferentes modelos de herencia como veremos más adelante. Afortunadamente,
existen varios programas estadísticos y aplicaciones web que permiten realizar estos
análisis de forma sencilla y rápida. En esta unidad ilustraremos cómo analizar los
estudios de asociación mediante el programa estadístico SNPstats [ver material
suplementario] que es muy útil cuando se dispone de un número moderado de SNPs
(decenas). En esta unidad mostraremos cómo interpretar los resultados que se
obtienen de SNPstats para cada tipo de análisis que debemos realizar en un estudio
de asociación. En el caso de disponer de una cantidad mayor de SNPs (miles) es
recomendable usar otros programas disponibles en R como por ejemplo SNPassoc
(González et al., Bioinformatics, 2008) que también tiene una implementación en Web
[ver material suplementario].
Para ilustrar cómo realizar un análisis de asociación usaremos unos datos reales que
están disponibles en la Web de SNPstats (para acceder a ellos, ir a
http://bioinfo.iconcologia.net/index.php?module=Snpstats, botón “Run SNPStats” y
enlace “Dataset 1”). Los datos corresponden a un estudio de casos y controles para
cáncer colorectal, donde se dispone de información genética y ambiental. La figura 1
ilustra cómo importar los datos a SNPstats. Como se puede observar, el formato
requerido es un archivo de texto (ASCII) donde se puede incluir información tanto de
los SNPs (cada alelo separado por “/”) como variables ambientales. El manual
suplementario describe cómo realizar el pre-proceso de los datos donde se indica
nuestra variable dependiente (caso-control o rasgo cuantitativo) así como otras
posibles variables confusoras.

DISEÑO y ANÁLISIS de INVESTIGACIONES CLÍNICAS: MÓDULO IV
U7– Diseño y análisis de estudios genéticos
3.1 EQUILIBRIO DE HARDY-WEINBERG
Antes de analizar la asociación entre un SNP y la enfermedad, debemos comprobar
que dado un SNP y su frecuencia alélica para una población específica se cumpe el
equilibrio de Hardy-Weimberg (HWE en inglés). Este principio determina qué
frecuencias genotípicas deberíamos observar, asumiendo que los alelos se transmiten
de forma independiente entre generaciones y siempre que no haya selección sobre
ellos. Así, si tenemos un SNP con dos alelos, A y B, con una frecuencia en la
población p y q = 1-p respectivamente, las probabilidades esperadas, f, para cada
genotipo vienen dadas por:
fAA=p2
fAB=2.p.q
fBB=q2
Para comprobar que un SNP cumple el HWE se suele utilizar una prueba de bondad
de ajuste de χ2. La hipótesis nula es que el SNP cumple con el HWE, por lo que
necesitamos obtener p-valores mayores a 0.05. Sin embargo, la prueba de ji-cuadrado
puede tener problemas cuando analizamos un SNP con valores pequeños para un
genotipo, en ese caso se suele utilizar un test exacto de Fisher.
Las razones para que un SNP no cumpla HWE pueden ser varias:
• Tener un tamaño de muestra pequeño
• Fallo en el genotipado
• El SNP está mapando varias regiones genómicas
• Haya habido una selección positiva de ciertos alelos (es decir, que un alelo se
asocie a una mayor longevidad)
En el contexto de un estudio de casos y controles, debemos tener en cuenta que HWE
sólo debe ser evaluado en la población control, que es donde debería cumplirse
este principio, ya que esperamos que en los casos algunas de las frecuencias
genotípicas observadas sean distintas a las esperadas ya que ese polimorfismo puede

DISEÑO y ANÁLISIS de INVESTIGACIONES CLÍNICAS: MÓDULO IV
U7– Diseño y análisis de estudios genéticos
ser responsable, en cierta forma, de la aparición de la enfermedad. Si no tenemos
claro el porqué un SNP no cumple HWE debemos excluirlo del análisis.
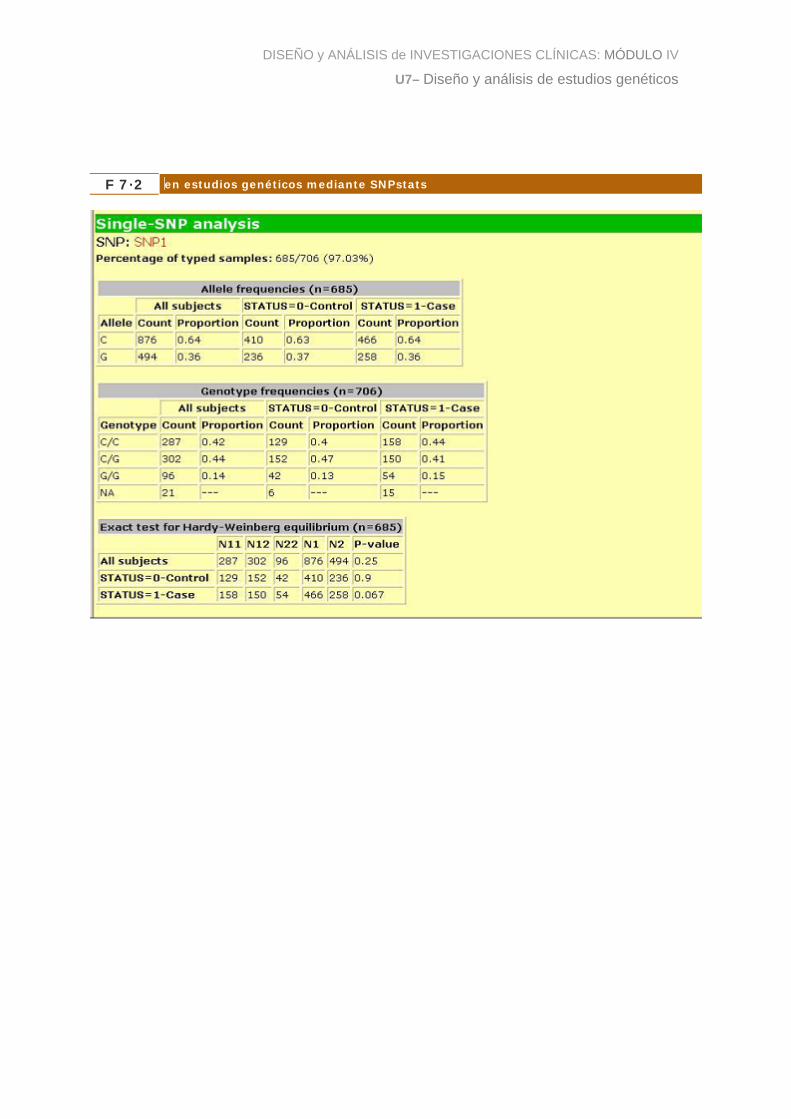
La figura 2 muestra los resultados de la prueba de HWE (junto con una descriptiva de
las frecuencias alélicas y genotípicas) para nuestro ejemplo sobre cáncer colorectal.
Se observa que para este SNP el p-valor es de 0.9 en los controles, por lo que no
tendríamos evidencias para rechazar la hipótesis nula. En otras palabras, podemos
aceptar que las frecuencias genotípicas observadas para este SNP son compatibles
con HWE.
3.2 ANÁLISIS SIMPLE DE SNPs: ASOCIACIÓN ENTRE UN SNP Y UN RASGO
El análisis simple de SNPs es el primer análisis que se lleva a cabo tras realizar un
control de calidad en nuestros datos (test de HWE). Esencialmente, el análisis consiste
en evaluar asociación entre los genotipos de un SNP y una variable respuesta. Este
análisis es sencillo y computacionalmente fácil de realizar en estudios de asociación
[ver Corden and Clayton en lecturas recomendadas]. Notemos sin embargo que, en el
contexto de un GWAS, éste tipo de análisis presenta algunas dificultades tanto desde
el punto de vista estadístico como bioinformático, especialmente para aquellos
estudios de asociación con grandes tamaños de muestra. La gran cantidad de datos
que generan estos estudios (miles de casos y controles y cientos de miles de SNPs)
demanda tener ciertas habilidades computacionales como estadísticas, así como una
infraestructura potente para llevar a cabo los análisis estadísticos. A continuación
detallamos cómo evaluar asociación entre un SNP y un rasgo específico. Dividiremos
los diferentes tipos de análisis dependiendo del tipo de variable respuesta que
tengamos: binaria o cuantitativa.
Rasgos binarios: El escenario con una variable respuesta categórica (binaria
generalmente) suele ser el más usual y suele encontrarse en estudios de asociación
poblacionales (casos y controles). En estos estudios se dispone de información de
individuos afectos (casos) y no afectos (controles). La manera de evaluar asociación
entre el SNP y la enfermedad se realiza mediante la tabla de contingencia 3x2 (tabla
2). Para testar la hipótesis nula de no asociación entre genotipos y la variable
respuesta, podemos llevar a cabo un test de χ2 con 2 grados de libertad. Cuando

DISEÑO y ANÁLISIS de INVESTIGACIONES CLÍNICAS: MÓDULO IV
U7– Diseño y análisis de estudios genéticos
tenemos unas frecuencias genotípicas pequeñas (menor de 5) en una o más de una
celda, es recomendable realizar un test exacto de Fisher.
El modelo que tiene en cuenta la tabla completa de los tres genotipos se conoce como
codominante o de 2 grados de libertad. Básicamente el modelo asume que puede
haber un riesgo distinto de los individuos heterocigotos y homocigotos variantes
respecto a los homocigotos normales. Para algunas enfermedades complejas se
puede asumir que el efecto para los individuos heterocigotos (AB) es la mitad que para
los homocigotos variantes (BB). Este modelo se conoce como modelo additivo y se
puede testar mediante un test de tendencia lineal (por ejemplo el de Mantel-Haenszel
que se utiliza en epidemiología tradicional) que se conoce en genética como test de
Cochran-Armitage. La hipótesis nula en este caso es que la pendiente para la recta
que relaciona el riesgo y los tres genotipos es cero. Este modelo también se conoce
como modelo multiplicativo ya que modela los efectos en la escala de odds. Una
ventaja importante de este modelo es que no necesita que se cumpla HWE, así que
puede ser una opción cuando esta hipótesis no se puede validar en nuestra población.
Sin embargo, algunas veces se espera que nuestro SNP siga otros patrones de
herencia como el dominante o el recesivo. En otras palabras, a veces se asume que
basta con tener un alelo variante para conferir riesgo (modelo dominante) o que es
necesario tener las 2 copias del alelo variante (modelo recesivo) para presentar la
enfermedad. Así pues, para llevar a cabo el análisis de asociación bajo esos modelos,
basta con reorganizar la tabla de contingencia 3x2 en una tabla 2x2 tal y como
podemos ver en las tablas 3 y 4, respectivamente. Cabe decir que también se suele
testar un modelo en el que se comparan los dos genotipos homocigotos contra el
heterocigoto. Este modelo se conoce con varios nombres, entre ellos sobredominante
(overdominant en inglés) o de heterocigosidad, aunque no suele usarse demasiado en
la práctica dado que desde un punto de vista biológico resulta difícil explicar este tipo
de efectos. El análisis de las tablas 3 y 4 puede realizarse de la misma forma con un
test de χ2 (con 1 grado de libertad) o con un test exacto de Fisher, dependiendo del
número de individuos en cada celda.
Puesto que normalmente los investigadores están interesados en analizar
enfermedades que no sólo están afectadas por factores genéticos si no que también
existen factores ambientales conocidos, muchas veces interesa realizar los análisis de
asociación entre el SNP y la enfermedad, ajustados por esos factores ambientales,

DISEÑO y ANÁLISIS de INVESTIGACIONES CLÍNICAS: MÓDULO IV
U7– Diseño y análisis de estudios genéticos
bien para tener más posibilidad de encontrar una asociación positiva entre el factor
genético y la enfermedad o bien para poder evitar algún tipo de confusión debido a
estos factores. En este caso, los modelos de regresión logística ofrecen una
posibilidad más flexible para evaluar asociación entre un SNP y el rasgo binario. Para
muestras de tamaño grandes, le test de razón de verosimilitud para el modelo logístico
contra la hipótesis nula βAA = βAB = βBB es equivalente al test de χ2 con 2 grados de
libertad (modelo codominante). La ventaja es que el modelo de regresión logística
permite ajustar por variables ambientales, así como evaluar interacciones gen-gen y
gen-ambiente. Es por ello que este modelo se usa para testar la asociación entre
SNPs y el rasgo binario de forma más general.
Para especificar los modelos de herencia en un modelo de regresión logística, tan sólo
necesitamos restringir los valores de los coeficientes β. Así, forzando que βAB = βBB o
βAA = βAB testaríamos el efecto dominante o recesivo, respectivamente. Si restringimos
que βAB sea la mitad entre βAA y βBB entonces el modelo logístico es equivalente al test
de Cochran-Armitage (tendencia lineal o modelo aditivo).
Rasgos cuantitativos: Un ejemplo típico de estudio de asociación entre un rasgo
cuantitativo es aquél en el que queremos testar si la expresión de un gen se ve
afectado por el genotipo de un SNP específico (que puede estar o no en el mismo
gen). Este tipo de asociación suele ser interesante en enfermedades con una base
genética importante como puede ser el cáncer. Otro ejemplo puede darse en estudios
en los que se quiera determinar si la inteligencia viene determinada o no por los
genes. En ese caso podríamos medir el cociente intelectual (variable continua) y
preguntarnos si este índice es mayor o menor en individuos que tengan ciertas
variantes genéticas.
Una primera aproximación para evaluar el grado de asociación entre un SNP y el
rasgo cuantitativo, podría ser mediante la categorización del la respuesta continua en
dos clases (por ejemplo “valores normales” vs “valores altos o anormales”) y luego
utilizar las aproximaciones que se han mencionado anteriormente. Sin embargo este
análisis no sería óptimo pues perderíamos mucho poder para detectar diferencias
estadísticamente significativas entre grupos. De esta forma, en vez de usar esta
aproximación, una forma más natural para testar la asociación en presencia de un
rasgo cuantitativo es usar un test como el ANOVA o un modelo de regresión lineal.

DISEÑO y ANÁLISIS de INVESTIGACIONES CLÍNICAS: MÓDULO IV
U7– Diseño y análisis de estudios genéticos
Mientras que un modelo ANOVA (para los tres genotipos) es equivalente a un test de
χ2 con 2 grados de libertad, la regresión lineal asume linealidad entre los genotipos y
las medias de la respuesta, por lo que los grados de libertad se reducen a 1. Además,
ambos tests necesitan que el rasgo se distribuya según una distribución normal y que
tengan una variabilidad similar en los tres genotipos. Estos problemas pueden
resolverse mediante trasformaciones de la variable respuesta tal y como se enseña en
los cursos de introducción a la estadística.
Al igual que se ha explicado para los rasgos binarios, los modelos de herencia también
se pueden especificar en este caso, colapsando los genotipos de forma adecuada
para generar un modelo dominante, recesivo o aditivo que contemple diferentes
patrones genéticos.
Otros tipos de rasgos: En estudios de cohortes, o en los que se estudian factores
pronóstico la variable analizada suele ser el tiempo hasta que se observa un evento de
interés. Desde un punto de vista estadístico, el estimador de Kaplan-Meier o el modelo
de Cox pueden usarse para estimar tanto las curvas de supervivencia o el riesgo en
función del genotipo. En otras ocasiones, el rasgo cuantitativo no es binario (por
ejemplo casos y controles donde los casos presentan diferentes sub-fenotipos). En
este caso otros modelos como la regresión multinomial pueden emplearse de la misma
forma que los modelos de regresión logística. En ambos casos, también se puede
forzar a que el SNP siga un patrón de herencia especificado.
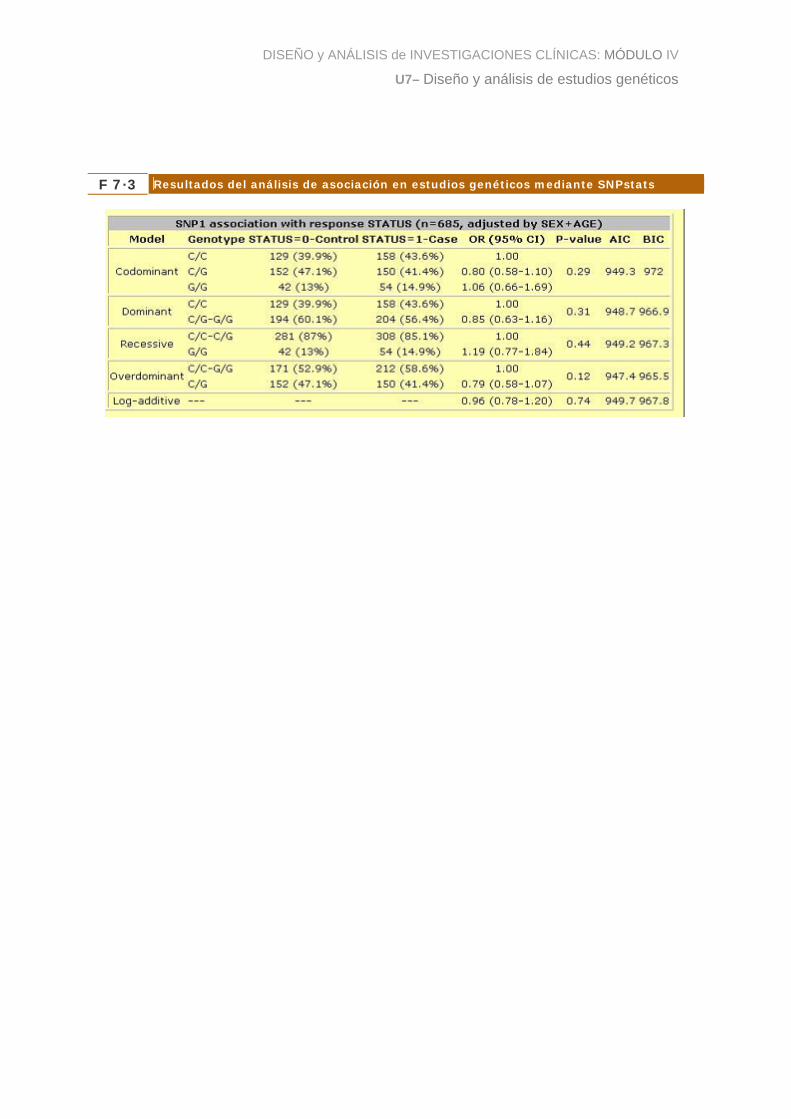
La figura 3 muestra los resultados donde se lleva a cabo un test para evaluar la
asociación entre el SNP1 y el cáncer colorectal. Para nuestro ejemplo, como se puede
leer en la figura, este análisis se ha realizado ajustado por edad y sexo, lo que supone
que el p-valor de asociación corresponde a un test de razón de verosimilitud donde se
ha comparado un modelo que incluye las variables edad y sexo como independientes,
con el modelo que incluye la edad, el sexo y el SNP como variables explicativas.
Observamos que ninguno de los p-valores es inferior a 0.05, por lo que no podemos
rechazar la hipótesis nula de igualdad de efectos. Las columnas AIC (Akaike
Information Criteria) y BIC (Bayesian Information Criteria) sirven para determinar qué
modelo de herencia es el que se ajusta mejor a los datos. En este caso el menor AIC y
BIC serían los correspondientes al modelo sobredominante indicando que ése es el
que mejor se ajusta. Notemos que también corresponde al modelo de herencia con un
p-valor menor, cosa que ocurren en la mayoría de ocasiones.

DISEÑO y ANÁLISIS de INVESTIGACIONES CLÍNICAS: MÓDULO IV
U7– Diseño y análisis de estudios genéticos
A modo de ilustración, y para aprender a interpretar los resultados, diríamos que, por
ejemplo, asumiendo un modelo de herencia recesivo, el riesgo de desarrollar cáncer
colorectal para los individuos homocigotos variantes (es decir, para los GG) es un 16%
superior que para los individuos que son homocigotos normales o heterocigotos
(CC+CG). El OR para el modelo dominante lo interpretaríamos como: los individuos
con el alelo variante, G, tienen un 15% menos de probabilidad de desarrollar la
enfermedad que los portadores del alelo normal. Notemos que ninguno de estos
resultados es estadísticamente significativo puesto que el p-valor no es menor que el
p-valor nominal del 5%. Sólo se pretende mostrar cómo se interpretan los resultados.
En cuanto a los análisis para un rasgo cuantitativo, las tablas son similares a las que
se obtienen de un estudio de casos y controles, pero en vez de mostrar OR se
presentan diferencias de medias de la variable respuesta para cada genotipo.
3.3 ANÁLISIS DE MULTIPLES SNPs
Los estudios de asociación raramente se restringen al análisis simple de un marcador.
Aunque son una buena estrategia para detectar posibles asociaciones con el rasgo, el
análisis simple de SNPs se han mostrado bastante ineficientes ya que no integran
información sobre marcadores que estén cercanos. Puesto que es bastante
improbable que se haya genotipado el verdadero SNP causal, el análisis de múltiples
SNPs puede proporcionar una ventaja adicional al análisis puntual de un único SNP.
Existen dos aproximaciones principales para evaluar asociación para múltiples
marcadores: los modelos de regresión y el análisis de haplotipos. Los modelos de
regresión se basan en la regresión logística o los modelos lineales (dependiendo del
tipo de respuesta que tengamos). Sin embargo, como el número de SNPs que se
suelen analizar es muy elevado, los modelos de regresión tradicionales suelen ser
ineficientes para el análisis de multiples SNPs. Es por ello que se han probado otros
modelos más complejos (árboles de regresión y de clasificación, redes neuronales,
random forest, boosting, regresión lógica, …) para intentar abordar este problema.
Estos modelos requieren un conocimiento avanzado tanto de métodos estadísticos
como de computación y quedarían emplazados para cursos más avanzados en

DISEÑO y ANÁLISIS de INVESTIGACIONES CLÍNICAS: MÓDULO IV
U7– Diseño y análisis de estudios genéticos
estudios genéticos. Esto hace que nos centremos en los métodos basados en
haplotipos ya que constituyen una alternativa muy atractiva. A continuación
introduciremos los conceptos básicos de la teoría de haplotipos y revisaremos los
métodos de asociación basados en ellos.
Un haplotipo es una combinación de alelos de múltiples loci polimórficos a lo largo de
un cromosoma. Aunque un cromosoma entero puede verse como un haplotipo,
normalmente sólo se consideran regiones no mayores a 100Kbp (100 pares de
kilobases) con polimorfismos altamente ligados (es decir, relacionados). Así, para un
conjunto de marcadores dado, cada persona tiene dos haplotipos, uno heredado del
padre y otro de la madre. Se puede calcular de forma sencilla que un conjunto de n
SNPs bialélicos generan 2n haplotipos potenciales. Sin embargo, las tasas de
recombinación hacen que el número de haplotipos que suelan generarse sean mucho
más pequeños que ese número máximo teórico.
Un problema serio que tiene los análisis que requieren información haplotípica es que
los estudios de genotipos normalmente generan datos sin conocer la fase. Es decir,
para un sujeto no conocemos realmente qué alelo proviene de cada uno de los
progenitores. Las técnicas de laboratorio que permiten conocer esta información son
muy caras y consumen mucho tiempo de análisis. Para resolver esta falta de
información se necesitan aproximaciones estadísticas que nos permita inferir los
haplotipos a para un conjunto de muestras no relacionadas y varios marcadores
genéticos. La inferencia de estos haplotipos y su asociación con un rasgo se basa en
métodos estadísticos muy complejos (basados en el algoritmo EM y máxima
verosimilitud) pero que afortunadamente están implementados en varios programas
informáticos. La figura 4 muestra los resultados del análisis de haplotipos que
obtendríamos con SNPstats. La primera tabla muestra una descriptiva de los
haplotipos estimados mediante el algoritmo EM. Las celdas en rojo significa que son
haplotipos con una frecuencia menor al 1% (esto es un parámetro del programa) por lo
que para el análisis de asociación, todos estos haplotipos se considerarán como una
categoría única llamada “rare” (haplotipos raros). También observamos que el
haplotipo más frecuente tanto en casos como en controles es el haplotipo CTCGG que
está presente en casi el 60% de la población. En la segunda tabla observamos, por
ejemplo, que los individuos que tienen el haplotipo CCAGG tienen casi 2.5 veces más
probabilidad de desarrollar cáncer colorectal que aquellos individuos con el haplotipo

DISEÑO y ANÁLISIS de INVESTIGACIONES CLÍNICAS: MÓDULO IV
U7– Diseño y análisis de estudios genéticos
más frecuente. Siendo esta OR estadísticamente significativa, con un nivel de
significación del 5% (p-valor 0.03).
3.4 ANÁLISIS GEN-GEN Y GEN-AMBIENTE
Existe evidencia empírica que sostiene la idea que tanto los factores genéticos como
ambientales afectan las enfermedades comunes. Es por ello que muchos estudios
genéticos, tras demostrar la relación de un gen con la enfermedad, suelen preguntase
si este efecto es el mismo entre subgrupos de pacientes definidos por alguna
característica clínica o que están expuestos a diferentes riesgos ambientales
(interacción gen-ambiente) o si existe otro gen que modifique el efecto de éste
(interacción gen-gen que se conoce en los estudios genéticos como epistasis). La
forma más sencilla de estudiar estas interacciones es mediante modelos estadísticos.
Al igual que en el caso del análisis de un SNP, se utilizan modelos de regresión
logística o modelos lineales dependiendo de la naturaleza de los datos. En este caso,
el test de razón de verosimilitud viene dado por la comparación de los modelos (nótese
que para el análisis de un rasgo cuantitativo es idéntico)
logit(p)=α+β·Gen+δ·Amb
logit(p)=α+β·Gen+δ·Amb+γ·Gen*Amb
donde p corresponde a la probabilidad de ser caso, Gen indica gen y Amb ambiente.
Este tipo de análisis también puede llevarse a cabo mediante SNPstats. La figura 5
muestra el resultado correspondiente a un análisis de interacción entre el SNP1 y el
sexo. La primera tabla muestra las ORs para la interacción entre el SNP y el sexo.
Como referencia se toma el genotipo homocigoto que tiene el alelo más frecuente y la
categoría de referencia para la variable sexo (en este caso las mujeres). En ocasiones,
para ayudar a interpretar una interacción, resulta útil presentar el análisis estratificado.
Las siguientes dos tablas muestran el análisis de asociación estratificado por el SNP y
luego por la variable sexo, respectivamente.
3.5 MÉTODOS DE ESTADÍSITICOS EN GWAS
Los GWAS utilizan tecnologías de genotipado a gran escala (Illumina o Affymetrix)
para evaluar la asociación de cientos de miles de SNPs con variables clínicas o

DISEÑO y ANÁLISIS de INVESTIGACIONES CLÍNICAS: MÓDULO IV
U7– Diseño y análisis de estudios genéticos
cualquier otro tipo de rasgos. Como ya hemos mencionado anteriormente, el principal
inconveniente de este tipo de estudios es que debemos realizar un número muy
grande de test estadísticos (tantos como SNPs hallamos genotipado). Esto puede
llevar a encontrar un número de falsos positivos no deseado, por lo que necesitamos
adoptar algún método de corrección por comparaciones múltiples o realizar algún tipo
de réplica de nuestros hallazgos.
El análisis estadístico para evaluar asociación entre un SNP y nuestro rasgo de interés
es el mismo que se ha descrito para el análisis simple de un SNP. El diseño más
utilizado es el de casos y controles, por lo que las limitaciones de este tipo de análisis
son las mismas que aparecen en este tipo de estudios. El sesgo de recuerdo aparece
cuando los casos reportan su historia de exposición de forma distinta que los
controles. Sin embargo en los estudios genéticos esto no es un problema ya que los
genotipos (que juega el papel de factor de exposición) se miden de forma exacta
utilizando el DNA de los individuos. Sin embargo, en algunas ocasiones este sesgo
puede aparecer si el DNA se recoge de forma distinta entre casos y controles. Por otro
lado, el sesgo de selección aparece cuando los controles no provienen de la misma
población que los casos. En este caso, la carga genética o ambiental puede diferir
como resultado del diseño del estudio y no por diferencias genéticas reales.
Finalmente, el sesgo por confusión aparece cuando un factor de riesgo también se
asocia con el marcador. En estudios genéticos este problema se conoce por
estratificación poblacional y aparece de forma más marcada en el contexto de GWAS
ya que estos estudios requieren de un número de muestra muy grande (miles de
casos y controles) y suelen llevarse a cabo en distintos países. Esta situación se
observa cuando tanto la enfermedad como la frecuencia alélica se correlacionan
mediante la etnia. En otras palabras, este problema puede aparecer cuando los casos
y controles de nuestra muestra provienen de poblaciones (razas) distintas. Este
problema se puede resolver en la fase de diseño si apareamos los casos y controles
por raza o seleccionamos a los controles de la misma familia que los casos (diseño
apareado). Sin embargo, puesto que los estudios de asociación genéticos suelen
utilizar individuos de estudios existentes, no podemos adoptar esta solución y
debemos tener en cuenta que los GWAS suelen incorporar individuos de varios países
para obtener un número elevado de casos y controles que les permita detectar efectos
pequeños (a menudo ORs de entre 1.1 y 1.6). De esta forma, sólo podemos controlar
esta confusión mediante métodos estadísticos. Existen varias aproximaciones, pero la

DISEÑO y ANÁLISIS de INVESTIGACIONES CLÍNICAS: MÓDULO IV
U7– Diseño y análisis de estudios genéticos
más usada en el contexto de los GWAS es un método conocido como EIGENSTRAT.
Básicamente este método realiza un análisis de componentes principales con los
genotipos (previa normalización) y usa los valores de la primera y segunda
componente principal para ajustar la asociación. En la figura 6 podemos ver un
análisis de este tipo para una muestra de 270 individuos europeos (CEU), africanos
(Yoruba, YRI) y chinos mas japoneses (CHB+JPT). En ella se puede ver como las dos
primeras componentes principales discriminan perfectamente a los individuos de cada
población. El eje 1 separa a los africanos de los asiáticos y europeos y la segunda
componente separa a los europeos de los asiáticos.
Una vez se dispone de esta información la asociación entre la enfermedad y el SNP se
realiza mediante el siguiente modelo de regresión logística:
logit(p)=α+β1·SNP+ β2·e1+ β3·e2
donde e1 y e2 son los valores que observamos en el eje X e Y de la figura 1 que
corresponde con los valores de la 1ª y 2ª componente principal y p es la probabilidad
de ser caso. Como se puede observar, este modelo no es más que un modelo de
asociación ajustado por una variable que tiene en cuenta las diferencias entre
poblaciones y que es un modelo similar al que se utiliza en epidemiología tradicional
para tener en cuenta la confusión cuando se evalúa la asociación entre un factor de
riesgo y la enfermedad.
Un punto crítico de los GWAS es el de comparaciones múltiples. Este problema
aparece porque testamos de forma simultánea un número muy elevado de hipótesis
(una para cada SNP). La corrección por comparaciones múltiples se realiza para
controlar el conjunto de hipótesis y para proteger al investigador a la hora de encontrar
falsas asociaciones que pudieran atribuirse al azar. En nuestro caso, la corrección por
comparaciones múltiples supondrá lo mismo que determinar un threshold para el cual
un p-valor se considere estadísticamente significativo y que garantice un error de tipo I
global igual a nuestro nivel nominal que suele ser del 5%.
La idea más simple se basa en corregir la tasa de error que se define como el hecho
de cometer al menos un error del tipo I. La forma más sencilla y conocida de realizar
esta corrección es mediante el método de Bonferroni. Este método requiere p-valores
inferiores a α/Μ donde α es el nivel de significación nominal (normalmente 0.05) y M
es el número de SNPs analizados. Utilizando esta corrección, en un GWAS que

DISEÑO y ANÁLISIS de INVESTIGACIONES CLÍNICAS: MÓDULO IV
U7– Diseño y análisis de estudios genéticos
incluya 1,000,000 SNPs tendríamos un valor de significación corregido de 5.0x10-8.
Sin embargo hay estudios que dicen que este valor es demasiado conservador y
sugieren otros valores menos extremos. También se suele utilizar otro método de
corrección que se conoce como false discovery rate (FDR) o métodos basados en
permutaciones que son capaces de estimar el verdadero número de hipótesis que se
testan en un GWAS [ver Dudbridge and Gustano en lecturas recomendadas].
Otra aproximación muy usada para proteger al investigador ante los falsos positivos
minimizando el coste del estudio y manteniendo el poder estadístico consiste en
adoptar un diseño multi-etapa (Figura 7). En la práctica los GWAS se llevan a cabo en
2-3 (o a veces más) etapas. En la primera etapa, se genotipa todo el conjunto de
marcadores (dado por la plataforma de análisis: Illumina, Affymetrix, …) en una
muestra inicial de individuos. En la segunda etapa, los marcadores más significativos
son re-genotipados en otro grupo de individuos utilizando una matriz de SNPs más
pequeña. El número de SNPs y de individuos suele depender del presupuesto del
investigador. De esta forma, se seleccionan aquellos SNPs que han mostrado una
asociación con la enfermedad con un significación estadística dada. En el primer paso,
no se suele ser muy restrictivo y se puede considerar un p-valor más liberal (es decir,
sin tener en cuenta las comparaciones múltiples). En la segunda etapa, si que se
consideran aquellos SNPs que pasan un p-valor más restrictivo que está corregido por
comparaciones múltiples. Finalmente, en la tercera etapa, aquel conjunto de SNPs que
muestran una asociación estadísticamente significativa tras corregir por
comparaciones múltiples, es validado en una muestra independiente, que
generalmente suele ser de otro pais o varios países [ver Hirschhorn and Daly en
lecturas recomendadas].
El Wellcome Trust Case Control Consortium (WTCC) [ver lecturas recomendadas]
publicó uno de los primeros GWAS en los que se analizaron siete enfermedades
comunes. En este artículo se discuten los temas mencionados anteriormente, entre
ellos el problema de estratificación poblacional, el de comparaciones múltiples y el
diseño multi-etapa.
En cuanto al software para analizar GWAS, podemos decir que existen varios
programas (PLINK, SNPTEST, snpMatrix, SNPassco,…) pero que ninguno de ellos
está implementado en una aplicación tan amigable como SNPstats. Además estos
programas requieren un conocimiento avanzado de programación en programas

DISEÑO y ANÁLISIS de INVESTIGACIONES CLÍNICAS: MÓDULO IV
U7– Diseño y análisis de estudios genéticos
compilados (C, C++, …) o en R. Existe un proyecto para usar SNPassoc (que está
implementado en R) en un entorno Web, que puede utilizarse en estos casos [ver
material suplementario].
4. LECTURAS RECOMENDABLES
• International human genome sequencing consortium. Initial sequencing and
analysis of the human genome. Nature, 2001, 409:860-921
• Venter, J.C. et al. The sequence of the human genome. Science, 2001, 291:1304-
1351.
Primer artículo (ambos se publicaron a la vez) en el que se describe la secuencia
del genoma humano. Útil para conocer cómo se obtuvo la información genética en
humanos.
• Información de cómo se obtienen los genotipos según las dos plataformas que
actualmente se utilizan en estudios de asociación, sobre todo en el caso de GWAS
o Illumina: www.illumina.com
o Affymetrix: www.affymetrix.com
• P. Lichtenstein, et al. Environmental and heritable factors in the causation of
cancer-analysis of cohorts of twins from Sweden, Denmark and Finland. N Engl J
Med, 2003;43:78-85.
Artículo donde se muestra cómo el cáncer puede estar debido a factores genéticos
basado en un estudio de gemelos
• M. García-Closas, et al. NAT2 slow acetylation, GSTM1 null genotype, and risk of
bladder cancer: results from the Spanish bladder cancer study and meta-analysis.
Lancet, 2005;366:649-659.
Estudio donde se muestra cómo el cáncer de vejiga puede estar debido a una
interacción gen-ambiente.

DISEÑO y ANÁLISIS de INVESTIGACIONES CLÍNICAS: MÓDULO IV
U7– Diseño y análisis de estudios genéticos
• H. J. Cordell and D. Clayton. Genetic Epidemiology 3: Genetic association studies.
Lancet, 2005;366:1121-1131.
• J. N. Hirschhorn and M. J. Daly. Genome-wide association studies for common
diseases and complex traits. Nature Revision Genetic, 2005;6:95:108
Estos dos artículos son dos buenas revisiones donde se discuten los métodos
estadísticos en estudios de asociación genética y los diseños en GWAS.
• The Wellcome Trust Case Control Consortium (WTCC). Genome-wide association
study of 14,000 cases of seven common diseases and 3,000 shared controls.
Nature, 2007;447:661-678.
Artículo en el que se lleva a cabo un GWAS para 14.000 casos de siete
enfermedades comunes y 3,000 controles. En este estudio se muestran todos los
problemas que aparecen en estudios de asociación (comparaciones múltiples,
estratificación, …) así como los diseños empleados en estudios completos del
genoma.

DISEÑO y ANÁLISIS de INVESTIGACIONES CLÍNICAS: MÓDULO IV
U7– Diseño y análisis de estudios genéticos
5. RESUMEN
En esta unidad se ha descrito los principales diseños y métodos estadísticos que se
utilizan en estudios genéticos. Los métodos estadísticos incluyen cómo evaluar
asociación entre un SNP y un rasgo (ya sea binario o cuantitativo), cómo evaluar
interacciones entre factores genéticos y ambientales, cómo analizar varios SNPs
utilizando haplotipos y cómo tener en cuenta las comparaciones múltiples. Este último
punto es uno de los principales problemas que se presentan en este tipo de estudios
puesto que se suelen analizar cientos de miles de SNPs. Esta metodología se ha
ilustrado con ejemplos pertenecientes a datos reales utilizando un software de libre
disposición que permite analizar los datos que se generan en este tipo de
investigaciones de forma sencilla y rápida.

DISEÑO y ANÁLISIS de INVESTIGACIONES CLÍNICAS: MÓDULO IV
U7– Diseño y análisis de estudios genéticos
6. EJERCICIOS
EJERCICIO 1
La base de datos “pulmon.txt” corresponde a un estudio de casos y controles, donde
los casos son pacientes diagnosticados de cáncer de pulmón, para los que se han
obtenido información genética para 8 SNPs. Además se dispone de información sobre
edad, sexo y hábito tabáquico (0:no fuma + exfumador, 1:fuma). Realiza los siguientes
análisis y contesta a las siguientes preguntas:
1. Realiza un análisis descriptivo de los genotipos para cada SNP
2. ¿Están todos los SNPs en HWE? ¿cómo lo has comprobado?
3. Evalúa la asociación entre la variable grupo (0: control, 1:caso) y cada SNP
ajustando por edad y sexo
a. ¿existe alguna asociación estadísticamente significativa? ¿bajo qué
modelo de herencia? ¿cómo interpretarías los resultados?
b. ¿es esta asociación estadísticamente significativa tras tener en cuenta
las comparaciones múltiples?
4. Realiza un análisis de interacción entre el SNP d9850 y el hábito tabáquico e
interpreta los resultados
Nota: Para ver y contestar la pregunta de este caso, debe acceder a la versión on line
del curso, que encontrará en el Campus del CEC.

DISEÑO y ANÁLISIS de INVESTIGACIONES CLÍNICAS: MÓDULO IV
U7– Diseño y análisis de estudios genéticos
EJERCICIO 2
Se conoce que el tratamiento con fármacos antidepresivos es la mejor opción para
tratar los episodios psicóticos que aparecen durante la evolución de los trastornos de
ánimo. Sin embargo, un tercio de los pacientes que reciben estos tratamientos no
muestran una buena respuesta. Existen varios factores que pueden contribuir a una
mala respuesta como por ejemplo: el sexo, la edad, el número de episodios previos, el
tipo de depresión (unipolar o bipolar) o la severidad de la enfermedad entre otros.
También se conoce que existen algunos factores genéticos que pueden influenciar en
una mejor repuesta al tratamiento. Algunos estudios han mostrado que la reducción de
los niveles de BDNF (grain-derived neurotrophic factor) pueden aumentar la
vulnerabilidad a sufrir depresión y es por ello que algunos autores se han planteado la
pregunta científica sobre si los genes del BDNF pueden asociarse o no a una mejor
respuesta al tratamiento farmacológico (ver artículo Gratacós et al. The
Pharmacogenomics Journal, 2008;8:110-112).
Para este ejercicio se dispone de la información contenida en la base de datos
“BDNF.txt” sobre 374 casos diagnosticados con algún trastorno del estado de ánimo
donde se tiene información sobre variables clínicas y genéticas (8 SNPs). Esta base
de datos corresponde a la información publicada en el artículo anteriormente citado.
Contesta a las siguientes preguntas.
1. Realiza un análisis descriptivo de los genotipos para cada SNP
2. ¿Están todos los SNPs en HWE? ¿existe algún problema en este caso?
3. Evalúa la asociación entre la respuesta al tratamiento y cada SNP
a. ¿existe alguna asociación estadísticamente significativa? ¿bajo qué
modelo de herencia? ¿cómo interpretarías los resultados?
b. ¿es esta asociación estadísticamente significativa tras tener en cuenta
las comparaciones múltiples?

DISEÑO y ANÁLISIS de INVESTIGACIONES CLÍNICAS: MÓDULO IV
U7– Diseño y análisis de estudios genéticos
4. Realiza un análisis de asociación entre los haplotipos formados por los SNPs
rs12273363 , rs908867, y rs1491850 y la respuesta al tratamiento ¿cómo
interpretarías los resultados?
5. Realiza un análisis de interacción entre el SNP rs12273363 y el estado
piscótico e interpreta los resultados
Nota: Para ver y contestar la pregunta de este caso, debe acceder a la versión on line
del curso, que encontrará en el Campus del CEC.

DISEÑO y ANÁLISIS de INVESTIGACIONES CLÍNICAS: MÓDULO IV
U7– Diseño y análisis de estudios genéticos
FIGURAS
F 7·1 Entrada de datos para el análisis de asociación en estudios genéticos mediante SNPstats
DISEÑO y ANÁLISIS de INVESTIGACIONES CLÍNICAS: MÓDULO IV
U7– Diseño y análisis de estudios genéticos
F 7·2 en estudios genéticos mediante SNPstats
DISEÑO y ANÁLISIS de INVESTIGACIONES CLÍNICAS: MÓDULO IV
U7– Diseño y análisis de estudios genéticos
F 7·3 Resultados del análisis de asociación en estudios genéticos mediante SNPstats

DISEÑO y ANÁLISIS de INVESTIGACIONES CLÍNICAS: MÓDULO IV
U7– Diseño y análisis de estudios genéticos
F 7·4 Resultados del análisis de haplotipos en estudios genéticos mediante SNPstats

DISEÑO y ANÁLISIS de INVESTIGACIONES CLÍNICAS: MÓDULO IV
U7– Diseño y análisis de estudios genéticos
F 7·5 Resultados del análisis de interacción en estudios genéticos mediante SNPstats

DISEÑO y ANÁLISIS de INVESTIGACIONES CLÍNICAS: MÓDULO IV
U7– Diseño y análisis de estudios genéticos
F 7·6 Análisis de componentes principales usando el método EIGENSTRAT para detectar estratificación poblacional en la muestra.

DISEÑO y ANÁLISIS de INVESTIGACIONES CLÍNICAS: MÓDULO IV
U7– Diseño y análisis de estudios genéticos
F 7·7 Diseño multi-etapa para un estudio completo del genoma (GWAS)

DISEÑO y ANÁLISIS de INVESTIGACIONES CLÍNICAS: MÓDULO IV
U7– Diseño y análisis de estudios genéticos
F 7·8 Definición de variables para el ejercicio 1

DISEÑO y ANÁLISIS de INVESTIGACIONES CLÍNICAS: MÓDULO IV
U7– Diseño y análisis de estudios genéticos
F 7·9 Solución a la pregunta 1 del ejercicio 1
F 7·10 Solución a la pregunta 2 del ejercicio 1

DISEÑO y ANÁLISIS de INVESTIGACIONES CLÍNICAS: MÓDULO IV
U7– Diseño y análisis de estudios genéticos
F 7·11 Solución a la pregunta 3 a) del ejercicio 1
F 7·12 Solución a la pregunta 4 del ejercicio 1

DISEÑO y ANÁLISIS de INVESTIGACIONES CLÍNICAS: MÓDULO IV
U7– Diseño y análisis de estudios genéticos
F 7·13 Definición de variables para el ejercicio 2

DISEÑO y ANÁLISIS de INVESTIGACIONES CLÍNICAS: MÓDULO IV
U7– Diseño y análisis de estudios genéticos
F 7·14 Solución a la pregunta 1 del ejercicio 2
F 7·15 Solución a la pregunta 2 del ejercicio 2

DISEÑO y ANÁLISIS de INVESTIGACIONES CLÍNICAS: MÓDULO IV
U7– Diseño y análisis de estudios genéticos
F 7·16 Solución a la pregunta 3 del ejercicio 2

DISEÑO y ANÁLISIS de INVESTIGACIONES CLÍNICAS: MÓDULO IV
U7– Diseño y análisis de estudios genéticos
F 7·17 Definición de variables para la pregunta 4 del ejercicio 2

DISEÑO y ANÁLISIS de INVESTIGACIONES CLÍNICAS: MÓDULO IV
U7– Diseño y análisis de estudios genéticos
F 7·18 Solución a la pregunta 4 del ejercicio 2

DISEÑO y ANÁLISIS de INVESTIGACIONES CLÍNICAS: MÓDULO IV
U7– Diseño y análisis de estudios genéticos
F 7·19 Solución a la pregunta 5 del ejercicio 2

DISEÑO y ANÁLISIS de INVESTIGACIONES CLÍNICAS: MÓDULO IV
U7– Diseño y análisis de estudios genéticos
TABLAS
T 7·1 Preguntas relevantes y diseños de estudios en epidemiología genética
Pregunta Diseño de estudio
¿Están los genes relacionados con la enfermedad?
Agregación familiar, estudios con gemelos
¿Cuál es el modo de herencia? Segregación
¿Dónde están los genes? Ligamiento
¿Qué genes son? Asociación
¿Cuál es la variante causal? Fine-mapping
¿Cuál es el mecanismo? Estudios funcionales
Interacciones Asociación Gen-Gen y Gen-Ambiente
T 7·2 Tabla de contingencia con el número de individuos para cada genotipo en casos y controles
Genotipo Controles Casos
AA nAAco nAAca
AB nABco nABca
BB nBBco nBBca

DISEÑO y ANÁLISIS de INVESTIGACIONES CLÍNICAS: MÓDULO IV
U7– Diseño y análisis de estudios genéticos
T 7·3 Tabla de contingencia para el modelo dominante donde el alelo B es el alelo de riesgo.
En este caso se espera que los individuos heterocigotos tengan el mismo riesgo que los individuos homocigotos BB, por ello ambas categorías se colapsan.
Genotipo Controles Casos
AA nAAco nAAca
AB + BB nABco + nBBco nABca + nBBca
T 7·4 Tabla de contingencia para el modelo recesivo donde el alelo B es el alelo de riesgo
En este caso se espera que los individuos heterocigotos BB tenga riesgo para desarrollar la enfermedad, por ello los individuos
con un alelo A se colapsan en una misma categoría.
Genotipo Controles Casos
AA + AB nABco + nAAco nABca + nAAca
BB nBBco nBBca