u n i d a d i z t a p a l a p a - 148.206.53.84148.206.53.84/tesiuami/uami12537.pdf · con el uso...
TRANSCRIPT


1
U N I D A D I Z T A P A L A P A
PROYECTO TERMINAL
División de Ciencias Básicas e Ingeniería.
Licenciatura en Química
Área de Fisicoquímica Teórica
Análisis del efecto del tamaño del espacio activo en el método MCSCF para moléculas
aromáticas monosustituidas.
Alumno: Orlando Martinez Zapata (201319438)
Asesor: Dr. Andrés Cedillo Ortiz
Vo. Bo. Dra. Silvia Solís (Coordinadora de la Licenciatura en Química)
Julio de 2005

2
Introducción
En este trabajo se pretende estudiar teóricamente el efecto del sustituyente en los sitios de
reacción de una sustitución electrofílica aromática. Si bien este fenómeno se ha estudiando
ampliamente de manera experimental, en la actualidad no se cuenta con el estudio teórico que
apoye a estos resultados.
Las reacciones más características de los compuestos aromáticos bencenoides son las de
sustitución que se llevan a cabo al tratarlos con reactivos electrofílicos,
Areno-H + E+ ------> Areno-E + H+.
En todas estas reacciones una especie deficiente en electrones, un electrófilo, actúa sobre el
anillo bencénico. El benceno es susceptible de ataque electrofílico, principalmente a causa de
sus electrones π expuestos. En estas reacciones se encuentra que el electrófilo es un ion
positivo, una especie con una carga positiva parcial y en otras más es un ácido de Lewis.
Efecto de los sustituyentes: reactividad y orientación
Bencenos monosustituidos
Cuando los bencenos sustituidos sufren un ataque electrofílico, se observa que los grupos ya
presentes en el anillo afectan tanto la velocidad de la reacción como el punto en que se verifica
el ataque. Por tanto, puede decirse que, en la orientación aromática electrofílica, el grupo
sustituyente afecta tanto la reactivadad como la orientación.
Los grupos sustituyentes pueden dividirse en dos categorías dependiendo de la forma en que
influyen en la reactividad del anillo. Aquellos grupos que hacen que el anillo sea más reactivo
que el benceno mismo se llaman grupos activadores. Aquellos grupos que hacen que el anillo
sea menos reactivo que el benceno se llaman grupos inhibidores.
También se encuentra que los grupos sustituyentes pueden dividirse en dos categorías
dependiendo de cómo influyen en la orientación del ataque del electrófilo que entra. En una de
las categorías, los grupos tienden a producir una sustitución electrofílica principalmente en las
posiciones orto y para. Estos grupos se conocen como orientadores orto-para. En la segunda
categoría, los grupos tienden a dirigir al electrófilo hacia la sustitución en la posición meta.
Estos orientadores se llaman orientadores meta. La tabla 1 muestra una clasificación de los
sustituyentes más comunes

3
Tabla 1. Efecto de los sustituyentes sobre la sustitución aromática electrofílica
Orientadores orto-para Orientadores meta
Activadores potentes
—NH2, —NHR, —NR2
—OH
Inhibidores potentes
—NO2
—NR3+, —CF3, —CCl3
Activadores moderados
—NHCOCH3, —NHCOR
—OCH3,—OR
Inhibidores moderados
—C≡N, —CHO, —COR
—SO3H, —COOH, —COOR
Activadores débiles
—CH3, C2H5, —R
—C6H5
Inhibidores débiles
—F , —Cl , —Br , —I
En este trabajo se toma una serie de compuestos aromáticos, los cuales pertenecen a las
distintas clasificaciones según la tabla 1,
Anilina Fenol Tolueno Estireno Clorobencen
Fluorobenceno Benceno Nitrobenceno Triclorometilbenceno Trifluorometilbenceno
Benzonitrilo Benzaldehído Acetofenona Ácido benzoico Benzamida
.. .. ..
.. ..
.. ..
.. .. ..
..
.. .. .. ..
.. : : .. ..
.. : :

4
Degeneración en el benceno.
La geometría del benceno descrita con la hibridación sp2 de cada carbono le proporciona la
forma plana al anillo, una longitud de enlace C-C igual a 1.39 Å y ángulos equivalentes de 120°.
La particularidad de esta simetría genera que los orbitales moleculares tengan cierto orden de
degeneración como se observa en los orbitales de enlace y de antienlace, como se ve en la
Figua 1.
Figura 1. Combinación de seis orbitales
moleculares π, de los cuales tres son enlazantes y
tres son antienlazantes.
En el momento en que se quiere describir el catión del benceno con métodos de un solo
determinante, se presenta el problema de decidir cual es electrón que conviene ser removido
de uno de los dos orbitales degenerados que tiene el benceno debido a que no se cuenta con
un criterio que determine dicha preferencia. Resaltando que se esta tratando con partículas
indistinguibles, conduciendo a dos posibilidades vistas en la figura 3. Aquí, la necesidad de
utilizar métodos mas complejos y que no se limiten con el uso de un sólo determinante,
encontrando al método de multiconfiguraciones (MCSCF) [2] como una alternativa al problema
de la degeneración.
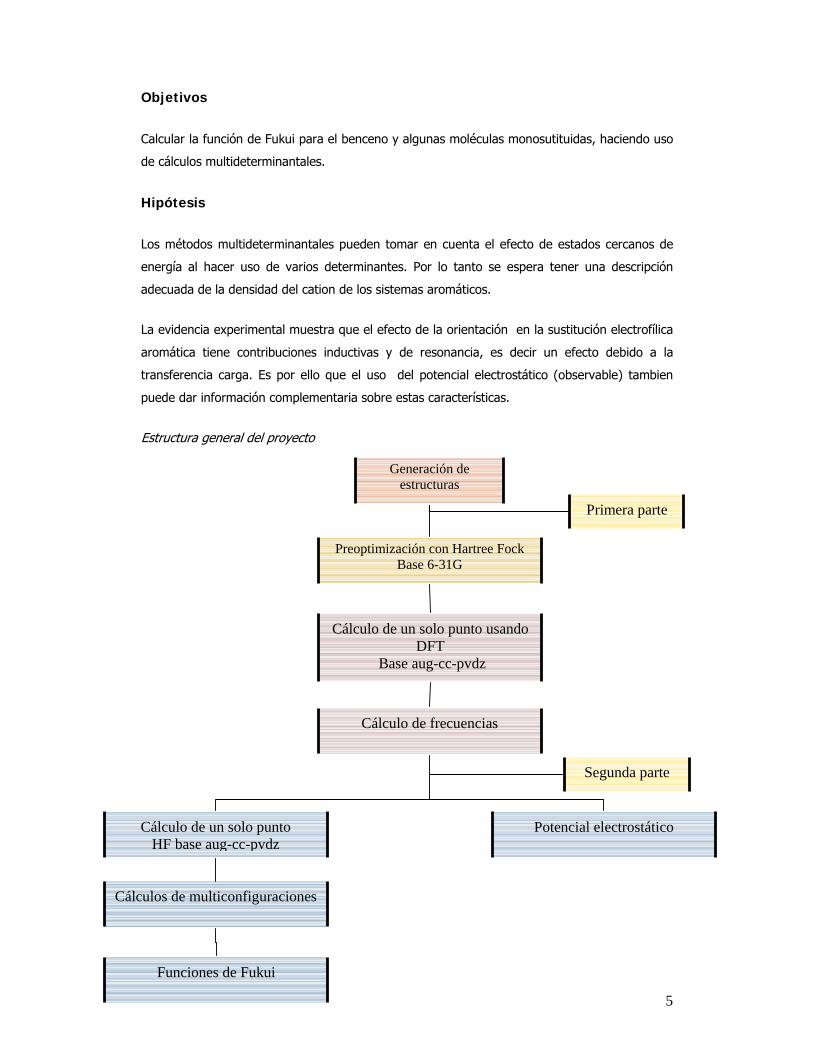
5
Generación de estructuras
Preoptimización con Hartree Fock Base 6-31G
Cálculo de un solo punto usandoDFT
Base aug-cc-pvdz
Cálculo de frecuencias
Potencial electrostático Cálculo de un solo punto HF base aug-cc-pvdz
Cálculos de multiconfiguraciones
Primera parte
Segunda parte
Funciones de Fukui
Objetivos
Calcular la función de Fukui para el benceno y algunas moléculas monosutituidas, haciendo uso
de cálculos multideterminantales.
Hipótesis
Los métodos multideterminantales pueden tomar en cuenta el efecto de estados cercanos de
energía al hacer uso de varios determinantes. Por lo tanto se espera tener una descripción
adecuada de la densidad del cation de los sistemas aromáticos.
La evidencia experimental muestra que el efecto de la orientación en la sustitución electrofílica
aromática tiene contribuciones inductivas y de resonancia, es decir un efecto debido a la
transferencia carga. Es por ello que el uso del potencial electrostático (observable) tambien
puede dar información complementaria sobre estas características.
Estructura general del proyecto

6
Generación de estructuras.
Previamente se realizo la calibración de la base, usando como molécula de trabajo al CO[1]. A
partir de este estudio se decidió usar el conjunto de base aug-cc-pvdz.
Con el uso del programa NWChem 4.5, en esta etapa se utilizaron los conjuntos de base 6-31G
y aug-cc-pvdz con el funcional BLYP para el estudio de las especies neutras.
El propósito de esta primera etapa es optimizar las geometrías de los compuestos aromáticos,
Figura 2. Se usa el funcional BLYP para incluir la correlación electrónica al hacer cálculos con la
teoría de funcionales de la densidad (DFT). Adicionalmente se hace un análisis de frecuencias
con el fin de verificar que las estructuras propuestas pertenecen a un mínimo en la superficie
de energía potencial.
Figura 2. Estructura básica empleada en la
construcción de las geometrías, en donde x es
cambiado por diferentes sustituyentes.
Estructura electrónica y análisis de la reactividad.
Método de MCSCF.
El benceno y algunas moléculas que orientan en la posición meta presentan los orbítales HOMO
y HOMO-1 casi degenerados, o degenerados como en el caso del benceno, como se ve en la
figura 3. Esto representa un problema en el momento de querer describir estos sistemas
empleando sólo un determinante. Este problema afecta tanto a los métodos basados en la
función de onda, Hartree-Fock (HF), como a los métodos DFT, Kohn-Sham (KS).
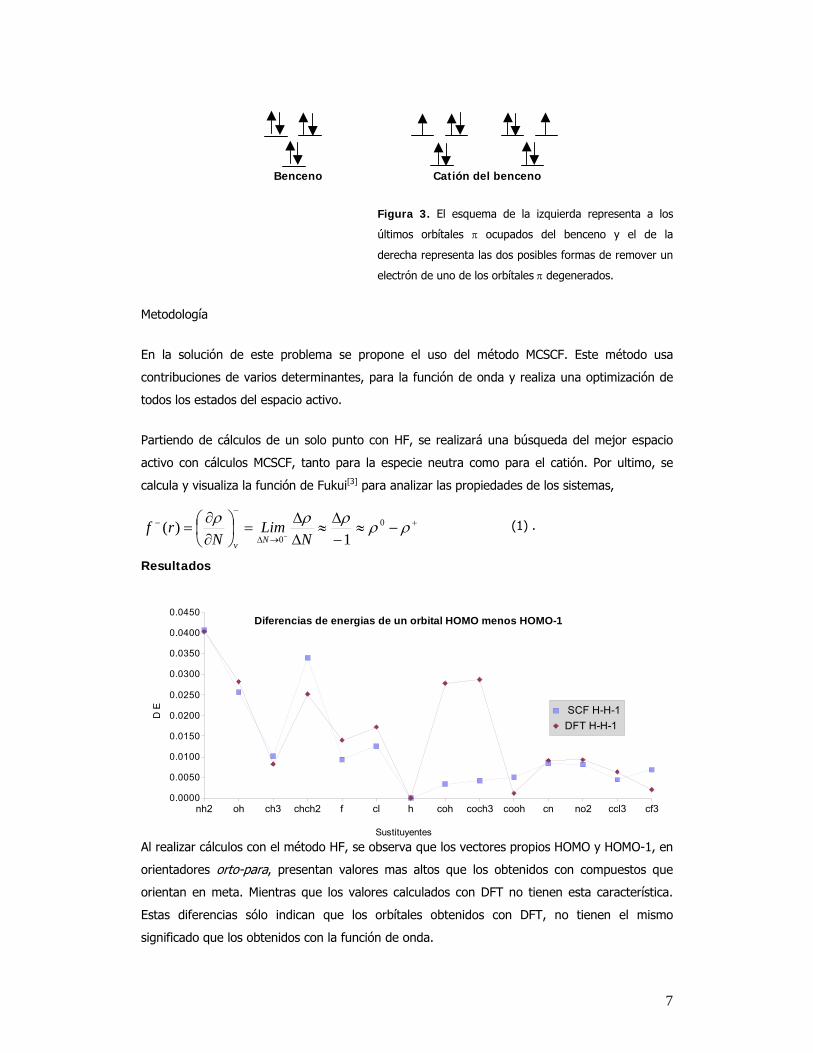
7
nh2 oh ch3 chch2 f cl h coh coch3 cooh cn no2 ccl3 cf30.0000
0.0050
0.0100
0.0150
0.0200
0.0250
0.0300
0.0350
0.0400
0.0450Diferencias de energias de un orbital HOMO menos HOMO-1
SCF H-H-1DFT H-H-1
Sustituyentes
D E
Benceno Catión del benceno
Figura 3. El esquema de la izquierda representa a los
últimos orbítales π ocupados del benceno y el de la
derecha representa las dos posibles formas de remover un
electrón de uno de los orbítales π degenerados.
Metodología
En la solución de este problema se propone el uso del método MCSCF. Este método usa
contribuciones de varios determinantes, para la función de onda y realiza una optimización de
todos los estados del espacio activo.
Partiendo de cálculos de un solo punto con HF, se realizará una búsqueda del mejor espacio
activo con cálculos MCSCF, tanto para la especie neutra como para el catión. Por ultimo, se
calcula y visualiza la función de Fukui[3] para analizar las propiedades de los sistemas,
(1) .
Resultados
Al realizar cálculos con el método HF, se observa que los vectores propios HOMO y HOMO-1, en
orientadores orto-para, presentan valores mas altos que los obtenidos con compuestos que
orientan en meta. Mientras que los valores calculados con DFT no tienen esta característica.
Estas diferencias sólo indican que los orbítales obtenidos con DFT, no tienen el mismo
significado que los obtenidos con la función de onda.
+
→∆
−− −≈
−∆
≈∆∆
=⎟⎠⎞
⎜⎝⎛
∂∂
=−
ρρρρρ 0
0 1)(
NLim
Nrf
Nv
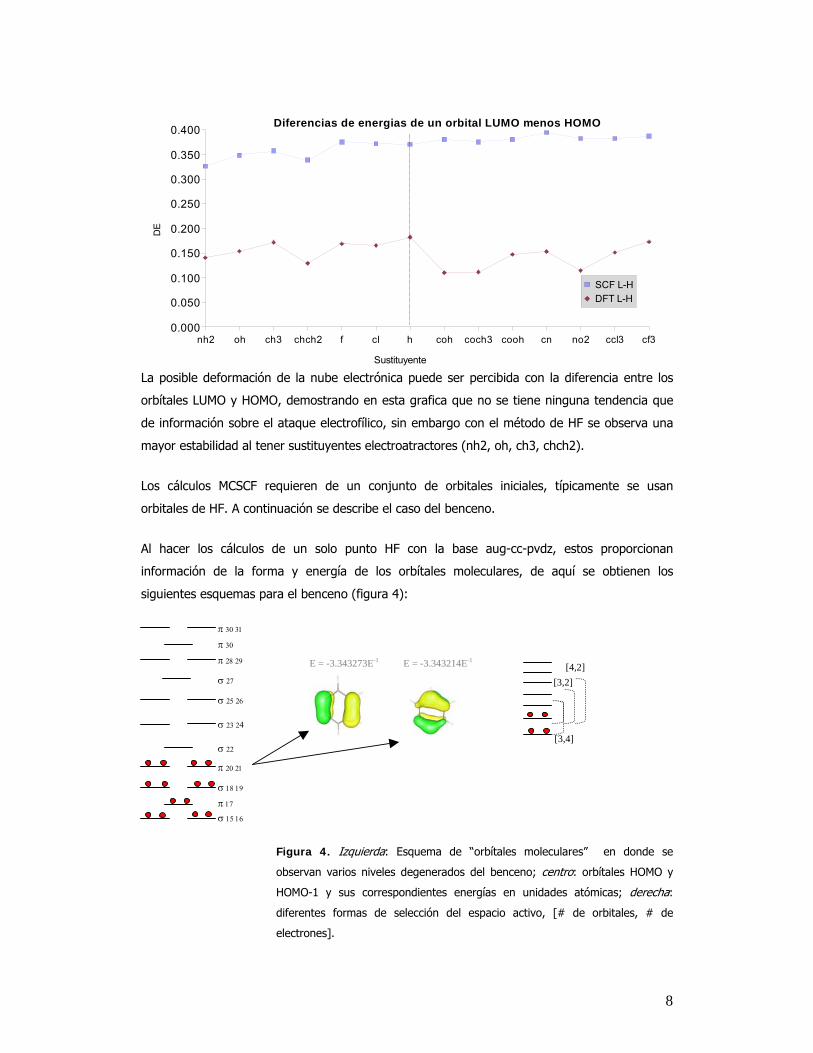
8
nh2 oh ch3 chch2 f cl h coh coch3 cooh cn no2 ccl3 cf30.000
0.050
0.100
0.150
0.200
0.250
0.300
0.350
0.400 Diferencias de energias de un orbital LUMO menos HOMO
SCF L-HDFT L-H
Sustituyente
DE
La posible deformación de la nube electrónica puede ser percibida con la diferencia entre los
orbítales LUMO y HOMO, demostrando en esta grafica que no se tiene ninguna tendencia que
de información sobre el ataque electrofílico, sin embargo con el método de HF se observa una
mayor estabilidad al tener sustituyentes electroatractores (nh2, oh, ch3, chch2).
Los cálculos MCSCF requieren de un conjunto de orbitales iniciales, típicamente se usan
orbitales de HF. A continuación se describe el caso del benceno.
Al hacer los cálculos de un solo punto HF con la base aug-cc-pvdz, estos proporcionan
información de la forma y energía de los orbítales moleculares, de aquí se obtienen los
siguientes esquemas para el benceno (figura 4):
Figura 4. Izquierda: Esquema de “orbítales moleculares” en donde se
observan varios niveles degenerados del benceno; centro: orbítales HOMO y
HOMO-1 y sus correspondientes energías en unidades atómicas; derecha:
diferentes formas de selección del espacio activo, [# de orbitales, # de
electrones].
σ 15 16 π 17
σ 18 19
π 20 21
σ 22
σ 23 24
σ 25 26
σ 27
π 28 29
π 30
π 30 31
[3,4]
[3,2] [4,2] E = -3.343273E-1 E = -3.343214E-1
9
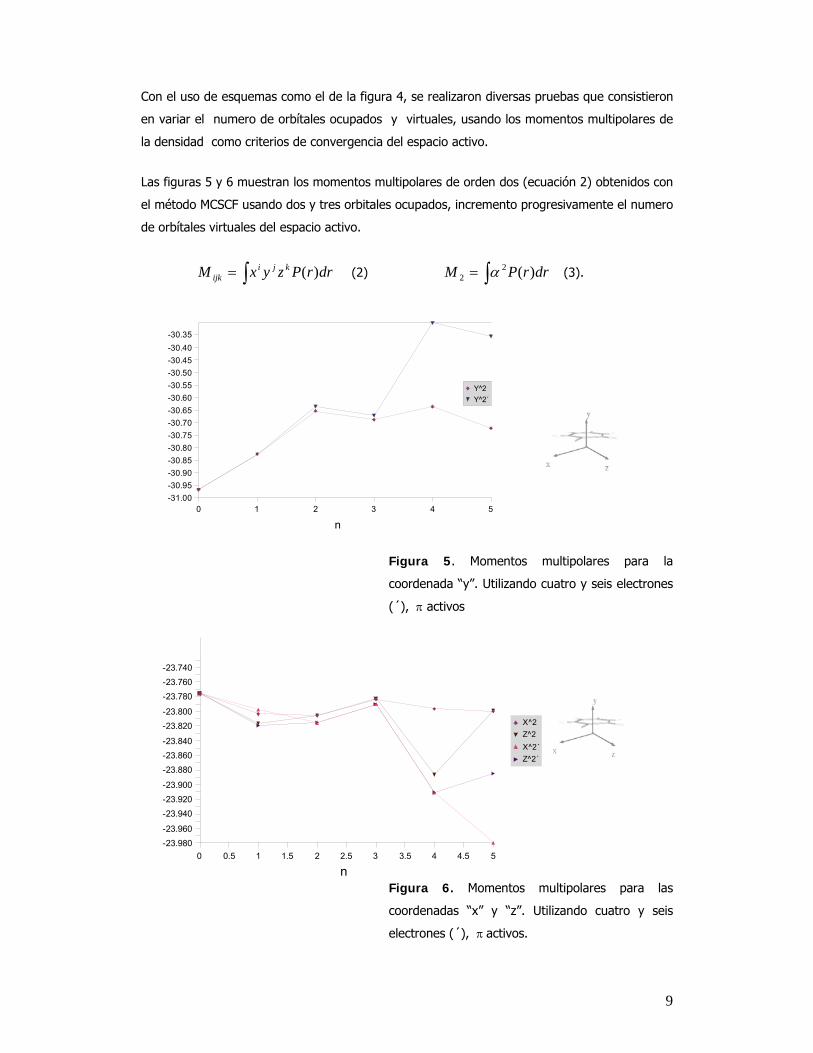
0 1 2 3 4 5-31.00-30.95-30.90-30.85-30.80-30.75-30.70-30.65-30.60-30.55-30.50-30.45-30.40-30.35
Y^2Y^2´
n
0 0.5 1 1.5 2 2.5 3 3.5 4 4.5 5-23.980-23.960
-23.940-23.920-23.900
-23.880-23.860-23.840
-23.820-23.800
-23.780-23.760-23.740
X^2Z^2X^2´Z^2´
n
Con el uso de esquemas como el de la figura 4, se realizaron diversas pruebas que consistieron
en variar el numero de orbítales ocupados y virtuales, usando los momentos multipolares de
la densidad como criterios de convergencia del espacio activo.
Las figuras 5 y 6 muestran los momentos multipolares de orden dos (ecuación 2) obtenidos con
el método MCSCF usando dos y tres orbitales ocupados, incremento progresivamente el numero
de orbítales virtuales del espacio activo.
∫= drrPzyxM kjiijk )( (2) drrPM )(2
2 ∫= α (3).
Figura 5. Momentos multipolares para la
coordenada “y”. Utilizando cuatro y seis electrones
(´), π activos
Figura 6. Momentos multipolares para las
coordenadas “x” y “z”. Utilizando cuatro y seis
electrones (´), π activos.

10
0 1 2 3 4 5-0.0050
-0.0045
-0.0040
-0.0035
-0.0030
-0.0025
-0.0020
-0.0015
-0.0010
-0.0005
0.0000
X´Y´Z ´
n
Figura 7. Componentes “x”, “y” y “z” del
momento dipolar empleando cuatro electrones (´)
π activos.
En las figuras 5 y 6 se observa que todos los datos parten de los valores correspondientes al
método HF. En estas propiedades al aumentar sucesivamente el espacio activo presentan
cambios que se encuentran en un intervalo pequeño, esto se debe a que estos valores ya están
prácticamente convergidos para el benceno neutro.
Al aumentar el tamaño del espacio activo es importante incluir juntos a los orbitales que están
degenerados. Cuando no se hace de esta forma, las propiedades obtenidas no son
representativas del sistema. Por ejemplo, si se coloca sólo un orbital π virtual dentro del espacio
activo, se provocan cambios, tanto en la forma de los nuevos orbítales HOMO y HOMO–1, en la
distribución de cargas vistas en el análisis de población de Mulliken y en los momentos
multipolares de la densidad. En la figura 7, el punto con un orbital virtual en el espacio activo
provoca un cambio brusco en la tendencia de los puntos. Algo similar se observa en la figura 6
cuando se usan cuatro orbitales virtuales. En este caso nuevamente se incluye sólo una parte
del grupo de orbitales degenerados.
También se hicieron pruebas empleando solo orbítales sigma en el espacio activo, obteniendo
contribuciones del orden de 10-7 en los coeficientes de estos determinantes. Las pruebas
realizadas utilizando bloques de orbítales π y σ tanto ocupados como virtuales, se muestran en
la figura 8. La nomenclatura [n,m] representa, n orbitales del espacio activo y m, el numero de
electrones colocados en dichos orbitales.
π π π π
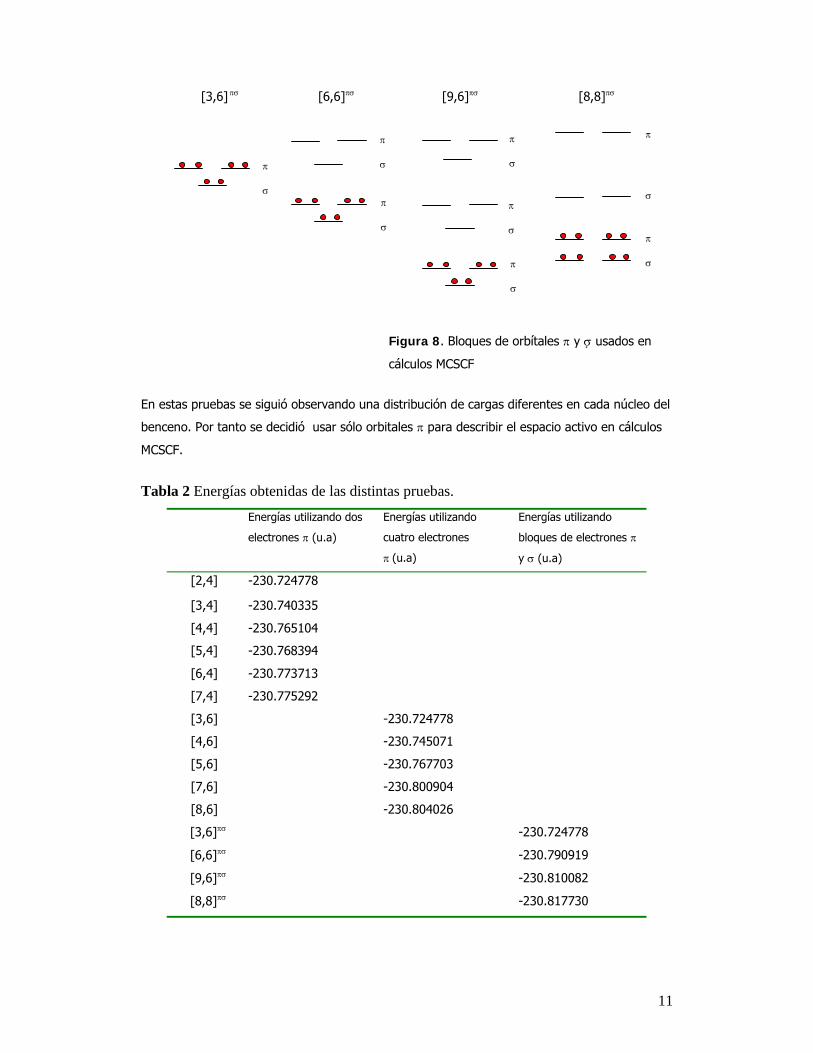
11
[3,6] πσ [6,6]πσ [9,6]πσ [8,8]πσ
Figura 8. Bloques de orbítales π y σ usados en
cálculos MCSCF
En estas pruebas se siguió observando una distribución de cargas diferentes en cada núcleo del
benceno. Por tanto se decidió usar sólo orbitales π para describir el espacio activo en cálculos
MCSCF.
Tabla 2 Energías obtenidas de las distintas pruebas. Energías utilizando dos
electrones π (u.a)
Energías utilizando
cuatro electrones
π (u.a)
Energías utilizando
bloques de electrones π
y σ (u.a)
[2,4] -230.724778
[3,4] -230.740335
[4,4] -230.765104
[5,4] -230.768394
[6,4] -230.773713
[7,4] -230.775292
[3,6] -230.724778
[4,6] -230.745071
[5,6] -230.767703
[7,6] -230.800904
[8,6] -230.804026
[3,6]πσ -230.724778
[6,6]πσ -230.790919
[9,6]πσ -230.810082
[8,8]πσ -230.817730
π σ
π σ
π σ
π σ
π σ
π σ
π σ
π σ

12
E =-0.552 E = -0.297 (20) (21)
En la tabla 2 se observan los valores de la energías en unidades atómicas, correspondientes a
los distintos cálculos y pruebas hechas. Observando la disminución paulatina de la energía con
forme se aumenta el numero de orbitales activos.
Figura 9. Densidad característica presentada
en los distintos cálculos hechos.
Al concluir con las pruebas realizadas con el benceno neutro, se inició con las visualizaciones
de la isosuperficie de la densidad total, encontrando una distribución uniforme en los casos
donde no se tenia un rompimiento de la simetría, figura 9.
Catión
Un calculo de multiconfiguraciones requiere de un conjunto de orbitales ortogonales. Para el
catión del benceno, en el estado doblete, se puede usar los orbitales de un calculo ROHF del
catión, o bien los orbitales del benceno neutro, RHF.
Utilizando información de cálculos obtenidos del benceno neutro, RHF, se obtuvo el estado
doblete del catión del benceno. Con la densidad de este sistema, calculada mediante el método
MCSCF se obtuvo la función de Fukui. Algunos resultados se observan en la figura 10.
(a) (b)
E = -0.552 E = -0.297 (20) (21)

13
E = -0.552 E = -0.297 (20) (21)
(c)
Figura 10. Funciones de Fukui empleando MCSCF
utilizando: (a) un electrón y un orbital activo, (b)
colocando un electrón y un orbital activo e
intercambiando los orbítales 20 y 21, (c) colocando
tres electrones y dos orbítales en el espacio
activo.
Al emplear orbitales ROHF se obtuvieron resultados similares. En este caso sólo se muestran los
análisis de población de Mulliken sobre los átomos de carbono del benceno, ver tabla 3.
Tabla 3. Análisis de Mulliken de distintos cálculos
Átomo
HF [3,3] π [4,3] π
[4,3] π
*
[4,3] π
**
[5,9] π [3,5] π [6,5]σ
C1 5.45 5.35 5.42 5.58 5.58 5.62 5.45 5.4
C2 5.45 5.59 5.54 5.54 5.54 5.45 5.62 5.56
C3 5.45 5.53 5.58 5.42 5.42 5.45 5.45 5.55
C4 5.45 5.44 5.37 5.54 5.54 5.62 5.45 5.4
C5 5.45 5.53 5.58 5.58 5.58 5.45 5.62 5.56
C6 5.45 5.59 5.54 5.37 5.37 5.45 5.45 5.56
π Utilizando de orbítales pi, σ Utilizando de orbítales sigma, * Un intercambio, * Dos intercambios
En la tabla 3 se observan las cargar obtenidas con el benceno neutro a nivel HF y las
resultantes con el catión del benceno a nivel MCSCF. Siendo desfavorable en el ultimo caso, ya
que molécula debe de seguir conservando una densidad de carga totalmente simétrica.
14
1 2 3 4 5-6.295
-6.293
-6.290
-6.288
-6.285
-6.283X^2Z^2
n
1 2 3 4 5-24.025-24.020-24.015-24.010-24.005-24.000-23.995-23.990-23.985-23.980-23.975
Y^2
n
22)(
20
0
+
→∆
−− −
≈−∆
≈∆∆
=⎟⎠⎞
⎜⎝⎛
∂∂
=−
ρρρρρN
LimN
rfNv
Debido a que los resultados del catión del benceno no corresponden con las propiedades
electrónicas, predichas teóricamente. Se decidió utilizar el bicatión del benceno para evaluar la
función de Fukui.
(4)
Siguiendo el mismo procedimiento empleado con el benceno neutro en el crecimiento del
espacio activo, obteniendo en esta ocasión distribuciones de carga equitativas sobre todos los
carbonos así como orbitales HOMO y HOMO-1 equivalentes a los orbitales obtenidos con la
especie neutra.
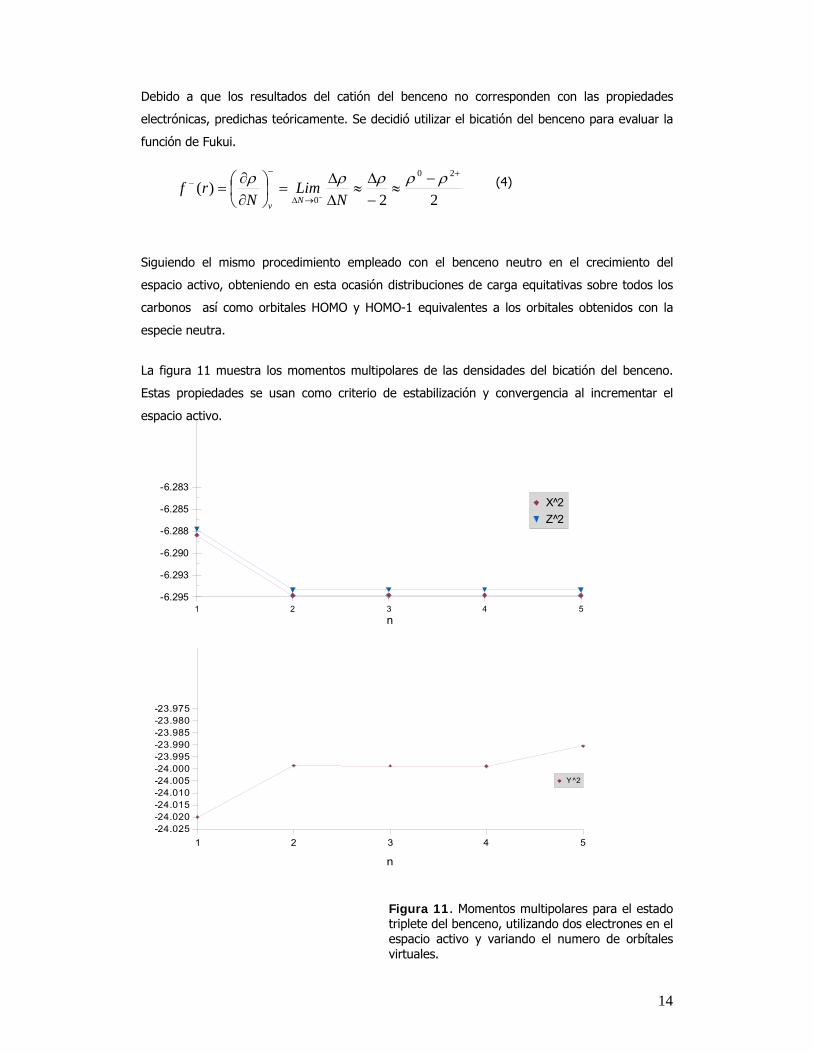
La figura 11 muestra los momentos multipolares de las densidades del bicatión del benceno.
Estas propiedades se usan como criterio de estabilización y convergencia al incrementar el
espacio activo.
Figura 11. Momentos multipolares para el estado triplete del benceno, utilizando dos electrones en el espacio activo y variando el numero de orbítales virtuales.

15
Con el uso del estado triplete del benceno, los valores de los momentos multipolares y dipolares tienen a estabilizarse rápidamente como se muestra en la figura 11.
En todos los cálculos hechos con el bicatión, la función de Fukui tiene simetría hexagonal. En la figura 12 se presenta la función de Fukui en donde todos los núcleos de carbono y hidrógeno, tienen contribuciones iguales.
Figura 12. Función de Fukui calculada con DFT, para el bicatión del benceno.
Debido a que no se encontraron diferencias al visualizar las funciones de Fukui obtenidas
mediante el método HF con orbitales RHF y UHF , así como en cálculos DFT y usando el método
MCSCF. Sólo se presenta en la figura 12, la función de Fukui obtenida con DFT.
Moléculas sustituidas
Las funciones de Fukui obtenidas con cálculos HF y DFT del estireno y nitrobenceno muestran
una similitud al orbital HOMO correspondiente, ver figura 13.
Figura 13 Funciones de Fukui del estireno, donde
se comparan con color amarillo la función usando
DFT y en verde con HF.

16
Potencial electrostático
En muchos casos, el electrófilo que reacciona con un compuesto aromático en la sustitución
electrofílica aromática es una especie positiva. Por esta razón se analiza el potencial
electrostático (ecuación 5) de las moléculas aromáticas. Para este análisis, se utilizo la base 6-
311++g(3df, 3pd).
⎥⎥⎦
⎤
⎢⎢⎣
⎡
−+
−−=Φ ∫ ∑
000
11)()(rR
Zdrrr
rqrαα
αρ (5)
Para cada benceno monosustituido se realizó una búsqueda de las regiones de valores mas
negativos, o bien del mínimo de potencial electrostático. Este representa los sitios donde hay
mayor acumulación de carga negativa (electrones), siendo zonas donde una carga positiva es
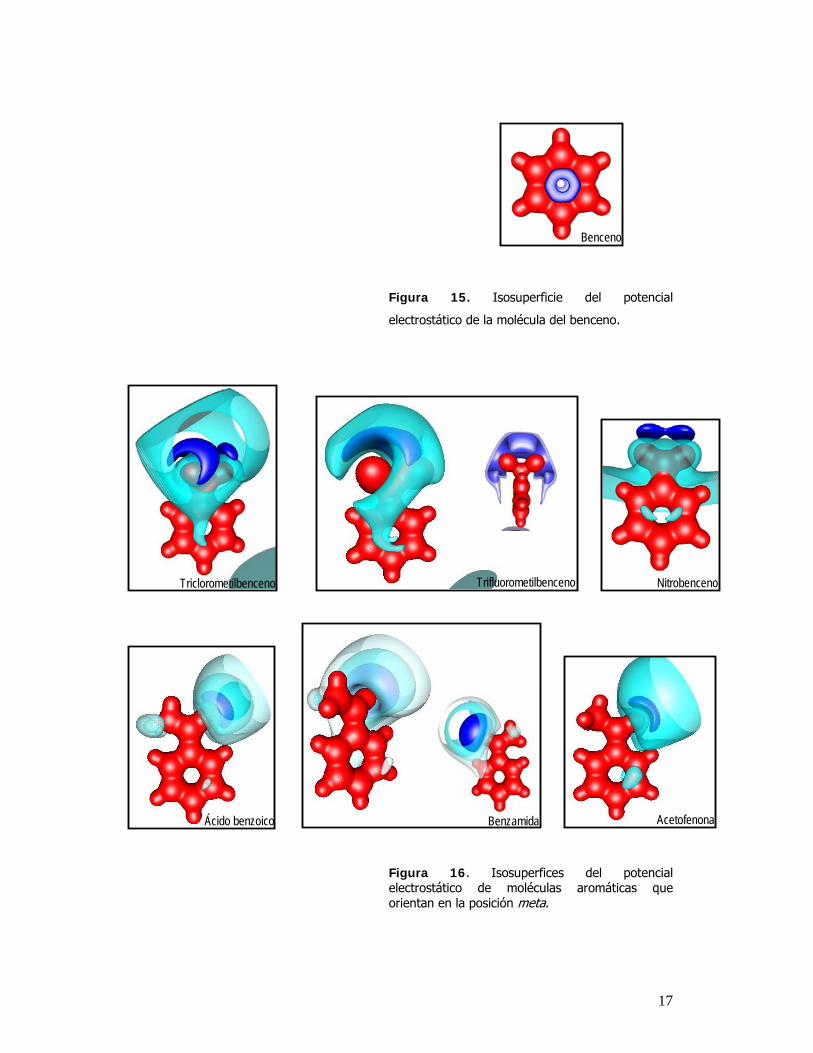
atraída más fuertemente. En la figura 14, 15 y 16 se muestran isosuperficies del potencial
electrostático, con valores cercanos al mínimo.
Figura 14. Isosuperficies del potencial
electrostático de moléculas aromáticas que
orientan en la posición orto-para.
Fluorobenceno Clorobenceno
Anilina Estireno
Tolueno
17
Figura 15. Isosuperficie del potencial
electrostático de la molécula del benceno.
Figura 16. Isosuperfices del potencial electrostático de moléculas aromáticas que orientan en la posición meta.
Benceno
Nitrobenceno Triclorometilbenceno Trifluorometilbenceno
Ácido benzoico Benzamida Acetofenona

18
En las figuras 14,15 y 16, el color rojo representa para todos los casos un valor positivo de 0.3
y se usa para obtener una superficie de la molécula; las isosuperficies de diferentes colores
azules representan las regiones más negativas de las moléculas como pares libres y enlaces
múltiples.
La figura 15 presenta al benceno con el valor del potencial mas negativo de manera simétrica
en el anillo.
En la mayoría de las moléculas estudiadas que orientan en posiciones orto-para se encontró
que el mínimo del potencial electrostático tiene valores negativos, y que el mínimo en el anillo
se encuentra en las posiciones orto o para (ver figura 14),
Se aplicó este mismo procedimiento de búsqueda en moléculas que tienden a orientar en meta.
Encontrando que los valores negativos están posicionados en sitios meta y en el caso especifico
de el nitrobenceno no se encontró ninguna región negativa en el anillo pero si una región
mínima positiva tambien en las posiciones meta de la molécula.
Conclusiones
Se obtuvo sin dificultades la densidad del benceno neutro, pero por deficiencias en el nivel de cálculo utilizado para la descripción del catión del benceno (MCSCF) no se obtuvo una función de Fukui confiable. Para solucionar este problema se uso el bicatión, obteniendo una función de Fukui que si representa al sistema.
El empleo de orbitales π y σ para describir el espacio no es una buena elección, ya que se no se tiene una población de electrones equitativa para los núcleos.
El uso de orbítales sigma en la construcción del espacio activo no proporcionaron contribución alguna, por tanto el uso de estos orbítales no ayuda a una mejorar la descripción de estos sistemas.
El incremento de bloques de orbítales π con la misma energía proporciona una estabilidad tanto en densidad electrónica obtenida, como en una tendencia suave en los momentos multipolares de la densidad. Concluyendo que sólo el uso de bloques de orbítales π es la manera mas adecuada al hacer cálculos de multiconfiguraciones para estos sistemas.
El uso de potenciales electrostáticos posibilita la identificación de los sitios mas reactivos en las posiciones meta y para, pero no en las posiciones orto. Esperando que en el futuro, el análisis de las funciones de Fukui permita estimar este efecto.
Adicionalmente se encontró que la diferencia de los vectores propios entre los orbitales HOMO y HOMO-1 en el método HF, permite diferenciar a las moléculas con respecto al efecto orientador de los sustituyentes. Es importante comentar que esto no ocurre con el método DFT.

19
Falta por analizar las distintas funciones de Fukui de las moléculas aromáticas estudiadas en este análisis.
Agregando por ultimo que queda inconclusa una comparación mas detallada de los distintos métodos utilizados y el conocer las verdaderas ventajas al utilizar los métodos MCSCF.
Bibliografía.
[1] Andrés Cedillo. Tercera Reunión Mexicana de Fisicoquímica Teórica, Puebla, Puebla 2004
[2] McWeeny, R. Methods of Molecular Quantum Mechanics. 2nd ed, Academia (1996)
[3] Robert G. Parr and Weitao Yang. Density-Functional Theory of Atoms and Molecules.1989
[4] Solomons, T. W. Graham. Quimica orgánica. 2da edición 1999.