tema 18 patologÍa endocrina - uji
TRANSCRIPT

TEMA 18
PATOLOGÍA ENDOCRINA
Esther Roselló Sastre

GENERALIDADES• El sistema endocrino se encarga de la síntesis de
hormonas endocrinas• Está repartido por todo el cuerpo, como glándulas
macroscópicas o como grupos microscópicos de células secretoras (células neuroendocrinas)
• Regulado de forma compleja, con relaciones de activación y auto- control por feed-back
• La patología del sistema endocrino se clasifica como – Funcional:
• Hiperproducción hormonal• hipoproducción hormonal
– Tumoral• Efecto masa• Funcional/no funcional

GENERALIDADES
• El estudio de la patología endocrina requiere la integración de– Morfología– Bioquímica hormonal y
sus metabolitos– Bioquímica global
(efectos de la hiperactividad o insuficiencia hormonal)
Regulación de las hormonas hipofisarias sobre
resto de glándulas endocrinas

PATOLOGÍA DE LA HIPÓFISIS

PATOLOGÍA DE LA HIPÓFISISRecuerdo anatómico
• Adenohipófisis (80% de la glándula, restos epiteliales)– Seis tipos diferentes
celulares, secretores de • Hormona de crecimiento GH
y Prolactina• ACTH y Melatonina• Tirotropa• Gonadotropas: FSH, LH
• Neurohipófisis (20%, células gliales modificadas– Secreción de
• Oxitocina• ADH

PATOLOGÍA DE LA HIPÓFISISManifestaciones clínicas
• Efecto funcional– Hipopituitarismo
• Por isquemia (Síndrome de Sheehan): post-parto• Por traumatismos, RT• Por adenomas no funcionantes
– Hiperpituitarismo• Por tumores funcionantes (adenomas)
– Efecto masa• Compresión de quiasma óptico, con alteraciones de la visión• Destrucción de la silla turca

PATOLOGÍA DE LA HIPÓFISISADENOMA HIPOFISARIO• Es una enfermedad de adulto
joven (30-50 a) (14% de autopsias, con microadenomasno funcionantes
• Tipos– Microadenoma<1cm (lo más
frecuente)/Macroadenoma>1cm– No funcionante (lo más
frecuente)/Funcionante• El más frecuente, secretor de
Prolactina (30% de todos los adenomas)
• Causas– Activación continua de la vía de
las proteínas G: cAMP– Un pequeño grupo,
relacionados con cáncer familiar• MEN-1• Otros

PATOLOGÍA DE LA HIPÓFISISADENOMA HIPOFISARIO
• Morfología– Proliferación nodular de
células epiteliales de un único tipo, con pérdida parcial de la trama de reticulina
– Con o sin cápsula– Capacidad de infiltrar
quiasma óptico o tejido cerebral de hipotálamo (no es criterio de malignidad)
– Mayor agresividad en tumores con elevada actividad mitótica y expresión de P53

PATOLOGÍA DE LA HIPÓFISIS
Clasificación de los Adenoma funcionantes de hipófisis
Clínica dependiente de la hormona segregada, en los casos funcionantes

PATOLOGÍA TIROIDEA

PATOLOGÍA TIROIDEA NO NEOPLÁSICA
Folículo tiroideo
Células foliculares y células C
Fisiología del Tiroides

Patología Tiroidea no neoplásica
• Adenoma• Carcinoma
– Bien diferenciado• Papilar• Folicular
– oncocítico
– Poco diferenciado• Insular• anaplásico
– Neuroendocrino• medular
Patología Tiroides neoplásica
HIPERPLÁSICA• BOCIO COLOIDE
MULTINODULAR/BOCIO ADENOMATOSO
• HIPERPLASIA DIFUSA (ENFERMEDAD DE GRAVES)
INFLAMATORIA• TIROIDITIS DE
HASHIMOTO• TIROIDITIS DE DE
QUERVAIN• TIROIDITIS DE RIEDEL
PATOLOGÍA TIROIDEA

PATOLOGÍA TIROIDEA NO NEOPLÁSICA
• Crecimiento difuso
• BOCIO COLOIDE• DIFUSO• MULTINODULAR/BOCIO ADENOMATOSO
• HIPERPLASIA DIFUSA (ENFERMEDAD DE GRAVES)

BOCIO COLOIDE DIFUSO
• Bocio: agrandamiento de la glándula tiroides por hiperplasia folicular
• Causa: estímulo de la TSH– Déficit de Yodo– Agentes bociógenos (Bocio endémico)
• Crecimiento difuso y homogéneo glandular, que puede alcanzar un gran tamaño– Fenómenos de compresión (bocio endotorácico)
• Patología tiroidea más frecuente en nuestra zona– Predominio en mujeres de mediana edad– Relación con estado de hormonas femeninas

BOCIO COLOIDE DIFUSO
Dos fases de crecimientoFase de hiperplasia folicular, por estímulo de TSH inicialFase de quistificación, con atrofia epitelial y acúmulo de coloide

BOCIO COLOIDE MULTINODULAR
• Derivado de un Bocio difuso, por crecimiento irregular de los folículos, con fibrosis y atrofia de algunas zonas– Fibrosis estromal– Calcificaciones distróficas
• El crecimiento puede ser muy grande y adoptar morfología polinodular– Estado hormonal eutiroideo– Difícil diagnóstico diferencial con Carcinomas/adenomas

BOCIO COLOIDE MULTINODULAR
• Diagnóstico diferencial con neoplasia folicular:– Ausencia de cápsula alrededor de los nódulos
• Posibilidad de crecimiento autónomo de algún nódulo y de implantación de neoplasias– Seguimiento mediante ecografía y PAAF

BOCIO HIPERPLÁSICO DIFUSO-ENFERMEDAD DE GRAVES
• Hiperplasia e hipertrofia difusa de los folículos tiroideos, con induración de la glándula (consistencia carnosa)
• Causa autoinmune: – Anticuerpos anti-tiroideos (anti-Receptor de TSH---
-Hiperestimulación TSH)– Clínica de Hipertiroidismo
• Exoftalmos y Mixedema pretibiial• Causa más frecuente de hipertiroidismo endógeno
– Predisposición genética

BOCIO HIPERPLÁSICO DIFUSO
• Histología: – Folículos con epitelio
hiperplásico, alto, que puede conformar pequeñas papilas
– Morfología irregular, con rebordes pronunciados
– Infiltrado linfoide acompañante• Formación de folículos
linfoides• El tratamiento modifica de forma
importante la histología

PATOLOGÍA TIROIDEA NO NEOPLÁSICA
Inflamación de la glándula1.TIROIDITIS DE HASHIMOTO2.TIROIDITIS DE DE QUERVAIN3.TIROIDITIS DE RIEDEL

TIROIDITIS LINFOCITARIA-ENFERMEDAD DE HASHIMOTO
• Enfermedad autoinmune que destruye el tiroides– Anticuerpos anti-
tiroglobulina+Hipersensibilidad celular-----destrucción de folículos tiroideo
• Causa más frecuente de hipotiroidismo en nuestro medio– Mujeres 30-50 años– Inicio como hipertiroidismo
• Crecimiento homogéneo de la glándula, consistencia aumentada– Nódulos blanquecinos al
corte (Folículos linfoides)
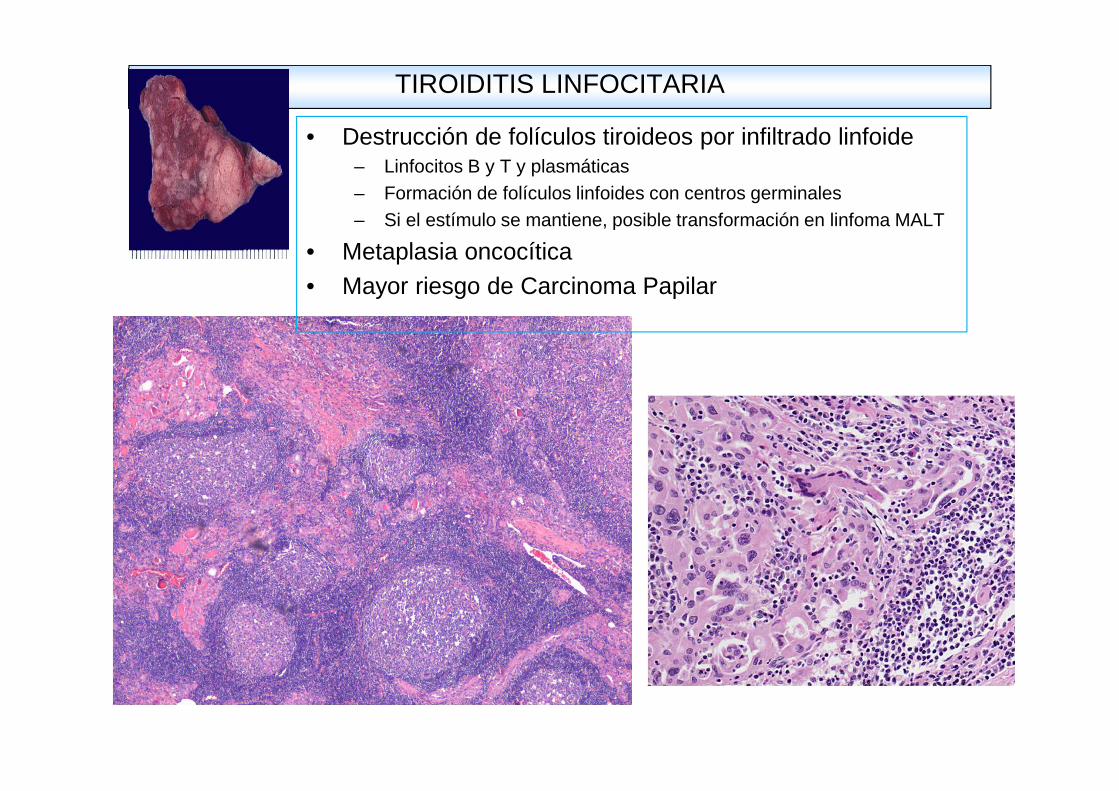
TIROIDITIS LINFOCITARIA
• Destrucción de folículos tiroideos por infiltrado linfoide– Linfocitos B y T y plasmáticas– Formación de folículos linfoides con centros germinales– Si el estímulo se mantiene, posible transformación en linfoma MALT
• Metaplasia oncocítica• Mayor riesgo de Carcinoma Papilar

TIROIDITIS GRANULOMATOSA-ENFERMEDAD DE DE QUERVAIN
• Inflamación autolimitada de glándula tiroides, secundaria a infección viral– Autoinmunidad inducida
por la infección viral• Poco frecuente
– Mujeres, 50 años– Dolor cervical y fiebre
• Histología– Destrucción del epitelio
por infiltrado inflamatorio granulomatoso
• Células gigantes multinucleadas
• Fibrosis, sin adherencia a otras estructuras

TIROIDITIS FIBROSANTE-ENFERMEDAD DE RIEDEL
• Inflamación de glándula tiroides, de etiología desconocida, que evoluciona rápido hacia la fibrosis glandular
– Probable etiología autoinmune– Relación con Enfermedad IgG4)
• Asociación a otros procesos de fibrosis idiopática (Fibrosis retroperitoneal) y Pancreatitis fibrosante
• Poco frecuente– Mujeres, 50 años– Clínica compresiva-disnea de rápida
evolución– Diagnóstico diferencial con Carcinoma
anaplásico por su rápida evolución
• Histología– Destrucción del epitelio por infiltrado
inflamatorio y fibrosis– Presencia de abundantes plasmáticas,
con predominio de formas IgG4– Adherencia a órganos adyacentes

NEOPLASIAS DE TIROIDES
• Benignas (LO MÁS FRECUENTE)• ADENOMA FOLICULAR
• Tumores foliculares de potencial maligno incierto
• Tumor papilar encapsulado• Tumor folicular con infiltración capsular cuestionable
• Malignas (LO MENOS FRECUENTE)• CARCINOMA TIROIDEO BIEN DIFERENCIADO
– Carcinoma papilar (>85%)– Carcinoma folicular (5-15%)
» Carcinoma oncocítico• CARCINOMA TIROIDEO POCO DIFERENCIADO
– Carcinoma insular o poco diferenciado – Carcinoma anaplásico o indiferenciado (<5%)
• Células parafoliculares– Carcinoma medular (5%)
• Linfoides– Linfoma primario de tiroides (<5%)

NEOPLASIA DE TIROIDES BENIGNA ADENOMA FOLICULAR
• Neoplasia encapsulada benigna de células foliculares, sin rasgos nucleares típicos de carcinoma papilar (SIN núcleos claros)
– Clonalidad (diag dif de nódulo adenomatosopor técnicas moleculares)
• Mutaciones en RAS
– Sin criterios de malignidad
• Sin infiltración capsular
• Sin invasión vascular
– Sin signos de hiperfuncionalidad en su mayoría
• Patogenia poco conocida
– Relación con exposición a la RT y al déficit de I
– Relación con alteraciones genéticas
• PTEN síndrome
• Poliposis múltiple
• Tratamiento quirúrgico y pronóstico excelente

NEOPLASIA DE TIROIDES BENIGNA - ADENOMA FOLICULAR
• Histología:
– Patrón folicular (macro o microfolículos) delimitado por una cápsula íntegra
– Con atipia ocasionales
– Con metaplasia oncocítica ocasional
ADENOMA ONCOCÍTICO

NEOPLASIA DE TIROIDES BENIGNA - ADENOMA FOLICULAR

CARCINOMAS DE TIROIDES
• Son carcinomas poco frecuente (1.5% de todos los cánceres)
– El más frecuente, el CARCINOMA PAPILAR (85% de todos)
– Seguido de CARCINOMA FOLICULAR (5% de todos)
– El CARCINOMA MEDULAR, generalmente familiar (5%)
– El CARCINOMA ANAPLÁSICO, el menos frecuente
• Patogenia común
– Mutaciones en la vía de los Receptores de Factores de Proliferación
• Mutaciones en RAS y BRAF
• Aumento de proliferación celular y de supervivencia celular
• Tratamiento quirúrgico (tiroidectomía completa) y ablación con Yodo radioactivo

CARCINOMA PAPILAR DE TIROIDES
• Neoplasia maligna de tiroides más frecuente
– 85% de todos los carcinomas
– Mujeres jóvenes, como nódulo solitario
– Extensión frecuente a adenopatías cervicales (palpables)
– Muy relacionado con radiaciones ionizantes (Chernobil)
• Patogenia
– Mutaciones en la vía de los Receptores de Factores de Proliferación
• Mutaciones puntuales en BRAF en variante papilar clásica
• Mutaciones en RAS en variantes foliculares
• Pronóstico excelente (supervivencias a los 5 años del 95%)
• Morfología
– Nódulo no encapsulado, blanco
– Fibrosis y calcificaciones
– Papilas, con núcleo claro e inclusiones intranucleares

CARCINOMA PAPILAR DE TIROIDES

CARCINOMA PAPILAR DE TIROIDES
– BUEN DIAGNÓSTICO POR PAAF
PAAF: formación de papilas, presencia de cuerpos de psamoma

CARCINOMA PAPILAR DE TIROIDESVARIANTES
Carcinoma papilar “Tall cell”•Mutaciones frecuentes en BRAF•Comportamiento agresivo•Pacientes añosos
Carcinoma papilar esclerosante difuso•Pacientes jóvenes•Mutaciones en RET•Buen pronóstico•Difícil diagnóstico diferencial con Tiroiditis
Carcinoma papilar folicular•Mutaciones similares a Carcinoma folicular (RAS)•Comportamiento más afín a Carcinoma Folicular microinvasor

CARCINOMA PAPILAR DE TIROIDESMICROCARCINOMA PAPILAR
Microcarcinoma papilar
• Histología
– Hallazgo incidental en otra patología tiroidea (bocio)
• Tumor<1cm
– Hallazgo frecuente, sin repercusión clínica. Precursor?
– Generalmente encapsulado

CARCINOMA FOLICULAR DE TIROIDES
• Segunda neoplasia maligna más frecuente (5% del total)
– Tumor folicular parcialmente encapsulado con ROTURA CAPSULAR O INFILTRACIÓN VASCULAR
• Mínimamente invasor
• Ampliamente invasor
• Encapsulado angioinvasor
– No diagnosticable por PAAF (sólo como neoplasia folicular, que requiere cirugía)
– No diagnosticable por estudio intraoperatorio (requiere estudio de toda la cápsula)
• Mujeres, de 50-60 años
• Clínica: nódulo frío de crecimiento progresivo
• Mutaciones génicas
– Mutaciones puntuales en RAS
– Mutaciones en PTEN
• Relación con síndrome de Cowden y otros

CARCINOMA FOLICULAR DE TIROIDES

CARCINOMA FOLICULAR DE TIROIDES
• Tumor folicular encapsulado con ROTURA CAPSULAR O INFILTRACIÓN VASCULAR– No diagnosticable por PAAF
(sólo como neoplasia folicular, que requiere cirugía)
– No diagnosticable por estudio intraoperatorio(requiere estudio de toda la cápsula)

CARCINOMA FOLICULAR DE TIROIDES
• Pronóstico (Dependiendo de la infiltración capsular-vascular)
– Microinfiltrante: supervivencia de 95% a los 5 años
– Macroinifltración: supervivencia del 50% a los 5 años

CARCINOMA DE CÉLULAS DE HURTHLE (ONCOCÍTICOS)
• Tumor folicular invasor con predominio de células de Hurthle
– Comportamiento más agresivo que la variante folicular no oncocítica
– Oncocito: acúmulo intracitoplásmico de megamitoconodrias
• Alteraciones genéticas en genes reguladores de mitocondrias
– Mal pronóstico
• Mutaciones en P53
• Aneuploidía y ganancias de DNA

CARCINOMA DE TIROIDES POBREMENTE DIFERENCIADO (CARCINOMA INSULAR)
• Carcinoma de células foliculares sin diferenciación de papilas o folículos, de comportamiento biológico intermedio entre Carcinoma diferenciado y Carcinoma anaplásico
– Comportamiento agresivo
– Extensión rápida ganglionar y a otros órganos
• Microscópico
– Islotes o cordones sólidos de células de tamaño intermedio
– Expresión de marcadores tiroideos (Tiroglobulina, TTF, PAX-8)
• Mutaciones génicas propias de carcinoma bien diferenciado + criterios de agresividad
– BRAF, RAS
– P53, AKT, ALK

CARCINOMA ANAPLÁSICO DE TIROIDES
• Muy poco frecuente, pero muy agresivo
– Pacientes>65 años
– Crecimiento rápido (días), con invasión de estructuras vecinas
– Pronóstico infausto (mortalidad 100% a los 5 años)
– Difícil diagnóstico diferencial con otros carcinomas y sarcomas (muy indiferenciado, células Tiroglobulina-, TTF1-, PAX-8-)
• Patogénesis
– Desdiferenciación de tumor previo (normalmente Carcinoma Papilar de larga evolución)
• Perfil genético de tumor agresivo+carcinoma papilar
– P53, PI3K+BRAF
Patrones de crecimiento•Fusocelular •Epiteloide•De células gigantes

CARCINOMA MEDULAR DE TIROIDES
• Único que deriva de células C, secretor de CALCITONINA
• Dos tipos
– ESPORÁDICO (70% de casos)
• Hombre, >40 años
• Nódulo único
– FAMILIAR (NEOPLASIAS ENDOCRINAS MÚLTIPLES MEN)
• Jóven, <10 años
• Nódulos múltiples, bilaterales
• Perfil genético
– Mutaciones en RET
• Familiar: línea germinal
• MEN 2, Carcinoma medular familiar
• Pronóstico agresivo

CARCINOMA MEDULAR DE TIROIDES
• Histología variada
– Células fusiformes, trabéculas, folículos
– Células secretoras de Cromogranina
– Identificación de Calcitonina mediante IHQ
– Islotes de Amiloide
Calcitonina

PATOLOGÍA de LA GLÁNDULA PARATIROIDES

CAUSAS DE HIPERPARATIROIDISMO

PARATIROIDES-NEOPLASIAS
• ADENOMA: 85-95% de casos– Nódulo único en una glándula paratiroides
• Peso: 0.5-5 g• Tamaño: 1-3 cm
– Atrofia del resto de glándulas– Población monoclonal de células paratiroideas (células principales)– Rodeado por cápsula– Baja actividad mitótica. Presencia frecuente de atipia celular
• Sobre-expresión de Ciclina-D1• Mutaciones esporádicas en MEN1

PARATIROIDES-NEOPLASIAS
• CARCINOMA: <1%– Difícil diagnóstico diferencial con Adenoma en fase inicial– Fibrosis e infiltración de tejidos adyacentes y/o metástasis
para su diagnóstico certero– Actividad proliferativa elevada– Suelen tener tamaño mayor que Adenoma
Carcinoma de paratiroides infiltrando músculo estriado cervical

PARATIROIDES-HIPERPLASIA
• HIPERPLASIA PRIMARIA : 5-10%– Crecimiento nodular o difuso de las 4 glándulas– Celularidad polimorfa, con formación de nódulos sólidos o microquistes– Actividad proliferativa baja
• Todos ellos, tanto la hiperplasia como el adenoma o carcinoma, pueden estar en relación con MEN 1 y 2 (mutaciones en línea germinal de MEN1 o RET)

PATOLOGÍA de LA GLÁNDULA SUPRARRENAL

PATOLOGÍA de LA GLÁNDULA SUPRARRENAL
• Hiperplasia de glándula SR– Hipercortisolismo (Síndrome de Cushing)– Síndrome adrenogenital
• Neoplasias de glándula SR– Neoplasias de la corteza SR
• Adenoma• Carcinoma
– Neoplasias de la médula SR• Feocromocitoma• Neuroblastoma

SÍNDROME DE CUSHING (HIPERCORTISOLISMO)
Patología más frecuente de hiperfuncionalidadadrenal, con hipersecreción de glucocorticoides, cuyo origen es diverso:
– Aporte exógeno de Corticoides (yatrogenia): lo más frecuente
• Atrofia de las SR– Adenoma hipofisario secretor de ACTH (lo
más frecuente endógeno) • Hiperplasia de SR
– Secreción de ACTH ectópica (síndrome paraneoplásico)
– Adenoma de suprarrenal, secretor de glucocorticoides (10% de casos)
• Tumor benigno de SR– Carcinoma de SR, secretor de
glucocorticoides (<5%)• Tumor maligno de SR

SÍNDROME DE CUSHING (HIPERCORTISOLISMO)
Etiopatogenia del Síndrome de Cushing

HIPERCORTISOLISMO CON HIPERPLASIA ADRENAL
Aumento de ambas glándulas (peso>30 g), secundario a estímulo externo de ACTH
• Patrón difuso o nodular, con macro o micronódulos• Representación de todas los estratos de la corteza suprarrenal, con
pigmentación endógena de algunas células (lipofucsina)

SÍNDROME ADRENO-GENITAL
Alteraciones de la diferenciación sexual, secundarias a hipersecreción de andrógenos corticales
• De causa neoplásica: generalmente por Carcinoma de SR funcionante
• De causa enzimática: HIPERPLASIA CONGÉNITA DE SR– Defecto congénito autosómico recesivo, con déficit parcial o total de
enzimas implicados en la síntesis de glucocorticoides, con incremento compensatorio de secreción de ACTH, que produce hiperplasia adrenal bilateral
• Déficit de 21-Hidroxilasa como patología más frecuente – Representación de todas los estratos de la corteza suprarrenal, con
pigmentación endógena de algunas células (lipofucsina)– Sospecha de Hiperplasia congénita de SR en cualquier neonato con
genitales ambiguos

SÍNDROME ADRENO-GENITAL
Hiperplasia congénita de SR
Genitales ambiguos en un neonato XX con déficit de 21-hidroxilasa e Hiperplasia de suprarrenales

NEOPLASIAS DE LA CORTEZA SR
ADENOMA DE SR:– Nódulo encapsulado, amarillo, productor de hormonas SR, autónomo
• Corticoides: Síndrome de Cushing• Aldosterona: Síndrome de Conn
– Incidentalomas no funcionantes en un 4% de la población. Cirugía sólo si clínica o gran tamaño.
– Micro: celularidad homogénea, con atipia ocasional y baja actividad mitótica
– Puede presentar áreas adiposas con células hematopoyéticas: MIELOLIPOMA

NEOPLASIAS DE LA CORTEZA SR
CARCINOMA DE SR:– Neoplasia muy infrecuente– Nódulo encapsulado de gran tamaño, normalmente funcionante– Difícil diagnóstico diferencial con Adenoma
• Fibrosis capsular• Necrosis, mitotis, metástasis
– Supervivencia media:2 años

NEOPLASIAS DE LA MEDULAR SR
• FEOCROMOCITOMA:– Tumor secretor de catecolaminas (céluls derivadas de la cresta
neural), con granos neuroendocrinos Cromogranina+– 25% ligados a síndromes familiares, con mutaciones en línea
germinal (MEN, VHL, Déficit de SDH)– Sin criterios histológicos de benignidad-malignidad: 10%
agresivos, malignos• Metástasis como mejor criterio
– Pequeño grupo de tumores no funcionantes– Histología: nidos celulares rodeados de células sustentaculares
(“zellballen”) S100+• Este tumor se denomina PARAGANGLIOMA en otras localizaciones
(tumor del cuerpo carotídeo)
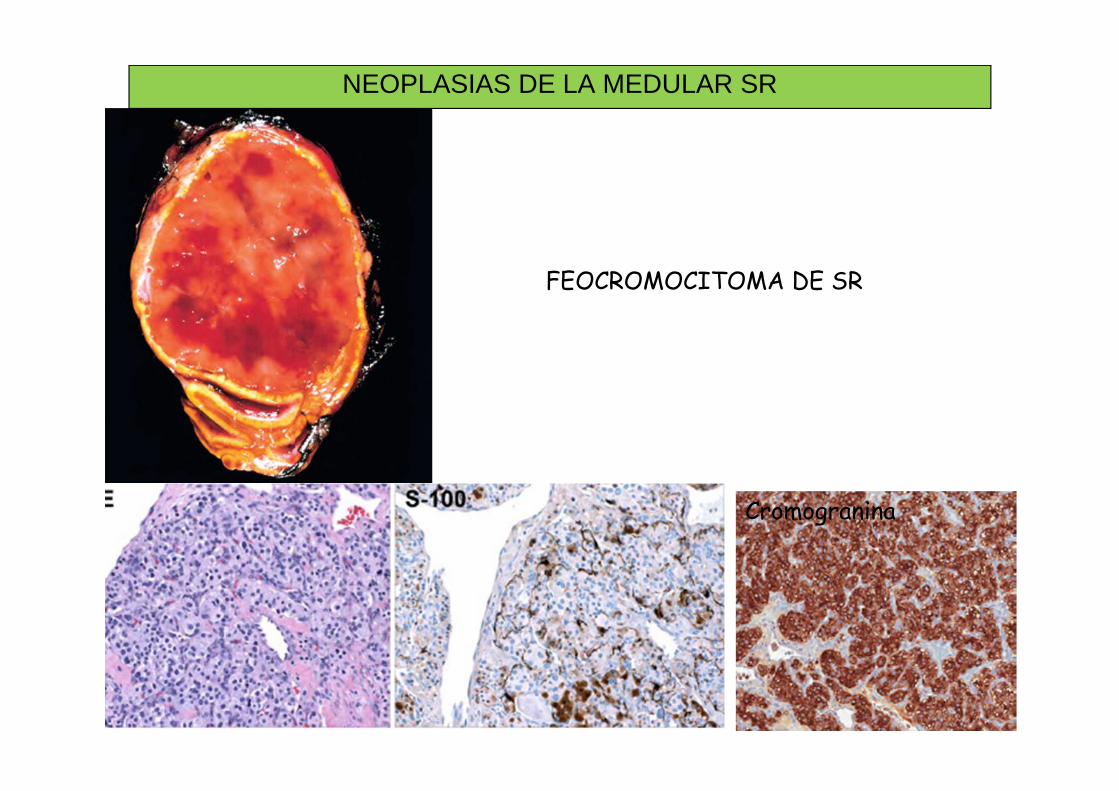
NEOPLASIAS DE LA MEDULAR SR
Cromogranina
FEOCROMOCITOMA DE SR

NEOPLASIAS DE LA MEDULAR SR
NEUROBLASTOMA:– Tumor maligno pediátrico, derivado de las células inmaduras de la
cresta neural– Localizado en un 40% de casos en Médula SR. El resto, en otros
paraganglios– Tamaño variable: cm-grandes masas– Crecimiento muy rápido, pero con capacidad de maduración
espontánea o inducida con QT• Buena respuesta al Tto en la mayoría de casos• Mal pronóstico si amplificación de N-MYC
– Células pequeñas, redondas, que conforman rosetas (Homer-Wright), con diferenciación neural-secreción de catecolaminas: NSE+. Cromogranina+

NEOPLASIAS DE LA MEDULAR SR
Amplificación de n-MYC (FISH)
Rosetas en un NEUROBLASTOMA

PATOLOGÍA del PÁNCREAS ENDOCRINO
Islotes pancréaticos productores de hormonas

PATOLOGÍA DEL PÁNCREAS ENDOCRINO
TUMORES NEUROENDOCRINOS– Tumores poco frecuente, generalmente no funcionantes
(Incidentalomas), constituidos por células neuroendocrinas (Cromogranina+)
– Los funcionantes: con clínica dependiente de la hormona liberada
• INSULINOMA• GASTRINOMA (Sind de Zollinger-Ellison)
– Hipersecrecion de ácido gástrico, con múltiples úlceras pépticas
• TUMOR SECRETOR DE MÚLTIPLES HORMONAS (VIP, Serotonina, Insulina…)
– Sin criterios histológicos bien definidos de malignidad– Frecuente asociación a Síndromes hereditarios (MEN 1, VHL)

PATOLOGÍA DEL PÁNCREAS ENDOCRINO
Tumor neuroendocrino en cuerpo pancreático
Tumor neuroendocrino de cabeza de páncreas, extendiéndose a hígado

TUMORES NEUROENDOCRINOS DE CUALQUIER LOCALIZACIÓN
• Todos los tumores neuroendocrinos pueden tener un comportamiento maligno– Difieren en velocidad de progresión y capacidad
metastástica• La clasificación está basada en el grado de
diferenciación PERO con peculiaridades topográficas• Estadiaje pronóstico basado en – Grado de diferenciación: según la actividad
proliferativa que tenga el tumor (número de mitosis; Ki-67)
– TNM: según el tamaño que tenga el tumor y la presencia o no de metástasis

TUMORES NEUROENDOCRINOS PANCREÁTICOS
Actividad proliferativa con Ki-67 y Clasificación

PATOLOGÍA DEL PÁNCREAS ENDOCRINO
TUMORES NEUROENDOCRINOS
Cromogranina Sinaptofisina
Octreoscan

PATOLOGÍA ENDOCRINA FAMILIAR

MEN-NEOPLASIAS ENDOCRINAS MÚLTIPLES
• Grupo de enfermedades hereditarias con lesiones proliferativas (hiperplasia-adenoma-carcinoma) en múltiples órganos endocrinos
• Mutaciones en línea germinal– MEN-1: mutación en gen supresor MEN1– MEN-2 A: mutaciones en gen RET, que
activan el receptor de RET– MEN-2B: mutaciones en RET, diferentes
a los de MEN-2 A • Las mutaciones en RET requieren tiroidectomía
profiláctica de la descendencia
Mutaciones del protooncogen RET presentes en MEN 2 y Carcinoma Medular Familiar

MEN

PATOLOGÍA ENDOCRINA FAMILIAR
SÍNDROME DE VON HIPPEL LINDAU:– Mutación autosómica dominante
en el gen supresor tumoral VHL– Tumores múltiples
• Hemangioblastoma en cerebelo
• Carcinoma renal y poliquistosis renal
• Tumores endocrinos pancreáticos y poliquistosispancreática
• Feocromocitoma bilateral– Difícil diagnóstico diferencial con
otras patologías congénitas hereditarias
• Fibrosis Quística• Poliquistosis renal
Páncreas

PATOLOGÍA ENDOCRINA FAMILIAR
Algunos síndromes familiares con afectación de órganos endocrinos

FINNN