tecnolologÍa de alto vacÍo - guzlop editoras … · 2015-04-03 · tecnolologÍa de alto vacÍo...
TRANSCRIPT

TECNOLOLOGÍA DE ALTO VACÍO
HERRAMIENTA BÁSICA PARA EL
DESARROLLO INDUSTRIAL
Arturo Talledo

TECNOLOLOGÍA DE ALTO VACÍO
HERRAMIENTA BÁSICA PARA EL
DESARROLLO INDUSTRIAL
Primera edición digital
Octubre, 2013
Lima - Perú
© Arturo Talledo
PROYECTO LIBRO DIGITAL
PLD 0639
Editor: Víctor López Guzmán
http://www.guzlop-editoras.com/[email protected] facebook.com/guzlop twitter.com/guzlopster731 2457 - 999 921 348Lima - Perú

PROYECTO LIBRO DIGITAL (PLD)
El proyecto libro digital propone que los apuntes de clases, las tesis y los avances en investigación (papers) de las profesoras y profesores de las universidades peruanas sean convertidos en libro digital y difundidos por internet en forma gratuita a través de nuestra página web. Los recursos económicos disponibles para este proyecto provienen de las utilidades nuestras por los trabajos de edición y publicación a terceros, por lo tanto, son limitados.
Un libro digital, también conocido como e-book, eBook, ecolibro o libro electrónico, es una versión electrónica de la digitalización y diagramación de un libro que originariamente es editado para ser impreso en papel y que puede encontrarse en internet o en CD-ROM. Por, lo tanto, no reemplaza al libro impreso.
Entre las ventajas del libro digital se tienen:• su accesibilidad (se puede leer en cualquier parte que tenga electricidad),• su difusión globalizada (mediante internet nos da una gran independencia geográfica),• su incorporación a la carrera tecnológica y la posibilidad de disminuir la brecha digital (inseparable de la competición por la influencia cultural),• su aprovechamiento a los cambios de hábitos de los estudiantes asociados al internet y a las redes sociales (siendo la oportunidad de difundir, de una forma diferente, el conocimiento),• su realización permitirá disminuir o anular la percepción de nuestras élites políticas frente a la supuesta incompetencia de nuestras profesoras y profesores de producir libros, ponencias y trabajos de investiga-ción de alta calidad en los contenidos, y, que su existencia no está circunscrita solo a las letras.
Algunos objetivos que esperamos alcanzar:• Que el estudiante, como usuario final, tenga el curso que está llevando desarrollado como un libro (con todas las características de un libro impreso) en formato digital.• Que las profesoras y profesores actualicen la información dada a los estudiantes, mejorando sus contenidos, aplicaciones y ejemplos; pudiendo evaluar sus aportes y coherencia en los cursos que dicta.• Que las profesoras y profesores, y estudiantes logren una familiaridad con el uso de estas nuevas tecnologías.• El libro digital bien elaborado, permitirá dar un buen nivel de conocimientos a las alumnas y alumnos de las universidades nacionales y, especialmente, a los del interior del país donde la calidad de la educación actualmente es muy deficiente tanto por la infraestructura física como por el personal docente.• E l pe r sona l docente jugará un r o l de tu to r, f ac i l i t ador y conductor de p r oyec tos

de investigación de las alumnas y alumnos tomando como base el libro digital y las direcciones electró-nicas recomendadas.• Que este proyecto ayude a las universidades nacionales en las acreditaciones internacionales y mejorar la sustentación de sus presupuestos anuales en el Congreso.
En el aspecto legal:• Las autoras o autores ceden sus derechos para esta edición digital, sin perder su autoría, permitiendo que su obra sea puesta en internet como descarga gratuita.• Las autoras o autores pueden hacer nuevas ediciones basadas o no en esta versión digital.
Lima - Perú, enero del 2011
“El conocimiento es útil solo si se difunde y aplica” Víctor López Guzmán Editor

PRÓLOGO
El concepto de “industria” es muy amplio, el número de bienes y servicios
que se pueden producir industrialmente es prácticamente infinito. La tecnología de alto
vacío es fundamental en muchas industrias que usan tecnología de punta como la
microelectrónica, la microscopía electrónica, la tecnología de láseres, etc. Sin embargo,
entre la producción sólo de materia prima y la tecnología de punta hay un gran número
de aplicaciones industriales de “tecnología intermedia” en la que nuestros jóvenes
pueden ser preparados con una razonable inversión en equipamiento de laboratorios de
nuestras universidades.
En este libro se lanza la propuesta de desarrollar este campo de la
tecnología moderna en las universidades del país como una manera de entregar a los
jóvenes profesionales una herramienta que puede ser aplicada a diferentes ramas de la
industria. Aquí se trata de explicar en qué consiste la tecnología de alto vacío y mostrar
con ejemplos provenientes de tesis profesionales desarrolladas en la UNI y sustentadas
tanto en esta universidad como otras universidades del país cómo esta tecnología se
puede aplicar para producir bienes con valor agregado. Los campos de aplicación que se
muestran son: (i) Recubrimientos Duros para prolongar el tiempo de vida de
herramientas de corte y otras piezas mecánicas de máquinas industriales, (ii)
Recubrimientos Anticorrosivos, (iii) Producción de baterías recargables de litio y (iv)
Producción de filtros ópticos.
Este libro puede ser de interés para jóvenes que hacen post grado o
antegrado en Ciencia de los Materiales así como para jóvenes estudiantes de Ingeniería
Industrial, Ingeniería Metalúrgica, Ingeniería Mecánica y profesionales en Ciencias
Básicas como Física y Química.

Autor: Arturo Fernando Talledo Coronado
Coautores de algunos capítulos
Capítulo III: Héctor Valdivia
Capitulo IV: Bertha Ponce, Manuel Cruz y Robinson Figueroa
Capítulo V: Elizabeth Chávez
Capítulo VI: Bertha Ponce y Héctor Valdivia
Capítulo VII: Giovanna Huamán y Gelacio Tafur

CONTENIDO
I. ALTO VACIO
I.1 Conceptos básicos
I.2 Bombas de vacío
I.3 Vacuómetros
II. TÉCNICAS DE PRODUCCION
II.1 Evaporación Térmica
II.2 Evaporación Iónica
II.3 Sistemas de alto vacío en la UNI
III TECNICAS DE CARACTERIZACION
III.1 Microscopía electrónica de barrido
III.2 Espectroscopía por dispersión de energía
III.3 Retrodispersión Rutheford
III.4 Espectroscopía XPS
III.5 Difracción de rayos X
III.6 Espectroscopía de Absorción en el Infrarrojo
IV RECUBRIMIENTOS DUROS
IV.1 Dureza
IV.2 Normas para medir dureza
IV.3 Microdureza Vickers del nitruro de titanio
IV.4 Rapidez de deposición
IV.5 Adhesión
IV.6 Resistencia la desgaste por abrasión
IV.7 Coeficiente de fricción

IV.8 Nanoindentación
IV.9 Análisis químico y estructural.
V. RECUBRIMIENTOS ANTICORROSIVOS
V.1 Corrosión Electroquímica
V.2 Corrosión Seca
VI. MICROBATERÍAS DE LITIO
VI.1 Conceptos Básicos
VI.2 Baterías de Litio
VI.3 Películas de Pentóxido de Vanadio Como Cátodos
VI.4 Prototipos de Microbaterías Desarrolladas en la UNI
VII. FILTROS OPTICOS FABRY PROT
VII.1 El Interferómetro Fabry Perot
VII.2 Películas Delgadas de Oxido de Titanio
VII.3 Prototipos de Filtros Fabry Perot

Capítulo I Alto Vacío
1
CAPITULO I
ALTO VACÍO
La Física del alto vacío es en realidad la Física de los gases a bajas
presiones. En efecto, en 1958, la American Vacuum Society, definió vacío como “un
determinado espacio lleno con gas a presiones menores que la presión atmosférica”.
Naturalmente, cualquier región del espacio a unos kilómetros por encima de la
superficie terrestre puede considerarse un sistema de vacío. Sin embargo, en este libro
nos interesa el vacío obtenido artificialmente al evacuar un recipiente (una cámara)
mediante un sistema de bombas. Visto así, podemos decir que la ciencia y tecnología
del vacío nace en 1654 cuando Otto von Guerick logra evacuar los célebres hemisferios
de Magdeburgo. Los avances de esta rama del conocimiento se desarrollan en la primera
mitad del siglo XX con los siguientes inventos: la bomba de paletas rotatorias por
Wolfgang Gaede en 1905, el vacuómetro de conductividad térmica por Marcelo Pirani
en 1906, la bomba de difusión de aceite por Kenneth Hickman en 1936, el vacuómetro
de ionización en frío por F.M. Penning en 1937 y la bomba turbomolecular por W.
Becker en 1958.
En este capítulo precisamos los órdenes de magnitud de presión
correspondientes, revisamos algunos conceptos básicos de la teoría cinética de gases y
dinámica de fluidos. También describimos los fundamentos de operación de las bombas
de vacío y de los vacuómetros, es decir, las máquinas necesarias para conseguir tales
bajas presiones y los instrumentos para medirlas

Capítulo I Alto Vacío
2
I.1. CONCEPTOS BÁSICOS
Nos referimos aquí a los conceptos de la teoría cinética de gases que
describen el comportamiento hidrostático de los sistemas de alto vacío, así como, a
conceptos provenientes de la dinámica de fluidos tales como flujo y conductancia.
También a conceptos más propios de la tecnología de alto vacío como degasificación y
velocidad de bombeo.
I.1.1. PRESION, UNIDADES Y ORDENES DE MAGNITUD
La presión, P, que una fuerza ejerce sobre una superficie se define como la
razón
AFP n (1.1)
donde Fn es la componente de la fuerza perpendicular a la superficie y A es el área de la
superficie.
Las paredes de un recipiente que contiene un líquido o un gas (un fluido) en
reposo, así como las superficies de cualquier cuerpo sumergido en un fluido,
experimentan una presión llamada presión hidrostática. En los líquidos la fuerza que
origina la presión hidrostática es el peso de los líquidos y en los gases es una
consecuencia del cambio de momentum que experimentan las moléculas al chocar con
la superficie de los cuerpos.
La unidad de presión en el sistema internacional (SI) es el Pascal (Pa)
1 Pa = 1 N/m2
Una buena referencia para comprender las definiciones de las unidades de
presión y los órdenes de magnitud en sistemas de vacío es la presión que el aire que
rodea a la tierra ejerce sobre la superficie de los cuerpos sobre ella. La presión

Capítulo I Alto Vacío
3
atmosférica a 273 K a nivel del mar es 101.3 kPa, es decir, aproximadamente, 105 Pa.
Un múltiplo importante del pascal es, entonces, el bar ( 1 bar = 105 Pa).
La presión atmosférica fue medida por primera vez por Evangelista
Torricelli, en 1643, usando un instrumento llamado barómetro, el cual está basado en el
resultado de que la diferencia de presión hidrostática, ΔP, entre dos puntos dentro de un
líquido está dada por
hgP , (1.2)
donde es la densidad del líquido usado, generalmente mercurio, g es la aceleración de
la gravedad en el punto de la tierra donde se hace el experimento y h es la diferencia de
altura. Se demostró experimentalmente que la presión atmosférica es equivalente a la
presión que ejerce una columna de mercurio de 760 mm de altura. La presión
correspondiente a una diferencia de altura de 1 mm de Hg constituye una unidad de
presión, más bien histórica, denominada Torr. El siguiente cuadro muestra la relación
entre las diferentes unidades de presión. En este libro tratamos de usar la unidad del SI.
1 atmósfera = 1.013 x 105 Pa
bar = 105 Pa
1 atmósfera 1 bar = 1000 mbar
1 atmósfera = 760 Torr
1 mbar 0.76 Torr
1 Torr = 131 Pa
1 mbar = 100 Pa
Cuando decimos alto vacío nos referimos a sistemas cuya presión está en el
orden de 10-5 a 10-7 mbar, es decir, de 10-3 a 10-5 Pa. También puede decirse, sistemas
de alto vacío son aquellos con valores de presión entre 10-8 y 10-10 veces el valor de la
presión atmosférica. De los sistemas a presiones menores que 10-8 mbar se dice que son

Capítulo I Alto Vacío
4
sistemas de ultra alto vacío, y podemos decir de los sistemas a presiones mayores que
10-4 mbar, sistemas de vacío pobre.
I.1.2. VELOCIDADES DE LAS MOLÉCULAS DE UN GAS IDEAL
Un gas real está constituido por un gran número N de moléculas, del orden
de 1020, que se reparten una energía total U. Hay muchas maneras de repartirse la
energía respetando las leyes de conservación:
N = n1 + n2 + n3 + . . .
U = n1E1 + n2E2 + n3E3 + . . . ,
donde E1, E2, E3, ...son los valores de energía que individualmente puede tomar cada
molécula y n1, n2, n3, . . . son las poblaciones (número de moléculas) con los
respectivos valores de energía. La teoría estadística de Maxwell Boltzmann [ref. 1]
demuestra que la partición más probable es aquella en la cual el número de moléculas
con valores de energía entre E y E + dE está dado por:
dE TE/keAdE n(E)dN B , (1.3)
siendo A una constante de normalización ( N dE n(E) ).
Un gas real a bajas presiones y densidades pequeñas puede ser considerado
como un conjunto de N partículas de volumen despreciable que no interaccionan entre
sí salvo, eventualmente, por colisiones elásticas. En estas condiciones, la energía de
cada molécula es sólo energía cinética. A un sistema como éste se le llama gas ideal.
La teoría estadística de Maxwell Boltzmann permite deducir directamente que el
número de moléculas con velocidades entre v y v + dv está dado por:
dv Tk 2 vm -
e vTk 2
m 4 N dv v)(n dN B
2
2
B
(1.4)

Capítulo I Alto Vacío
5
donde m es la masa de cada molécula y kB = 1, 38 10-23JK-1 es la constante de
Boltzmann.
El valor medio del cuadrado de la velocidad, que se define por la ecuación:
0
02
2
dv (v)ndv (v)nv
v (1.5)
resulta, en el caso del gas ideal:
m
Tk 3v B2 (1.6)
y teniendo en cuenta el resultado experimental de que para un gas ideal las variables de
estado presión (P) , volumen (V) y temperatura (T) se relacionan por la ecuación del
gas ideal:
PV = NkBT (1.7)
podemos escribir también:
P 3
N m
V P 32v (1.8)
Una cantidad muy usada para tener una idea del movimiento de las
moléculas de un gas es la velocidad cuadrática media, vrms,
P 3
M
T R 3
m
T Bk 3
2v rmsv (1.9)
donde R = 8,31 J/mol K es la constante universal de los gases y M es la masa
molecular.
En un recipiente que contiene oxígeno a temperatura ambiente (300 K) la
velocidad cuadrática media de las moléculas es 483 m/s y si el gas es nitrógeno la
velocidad cuadrática media es 516 m/s. El aire contiene aproximadamente 80% de
nitrógeno y 20% de oxígeno y puede establecerse que la velocidad cuadrática media es
490 m/s.

Capítulo I Alto Vacío
6
Experimentalmente se pueden hacer medidas de las distribuciones de
velocidades moleculares. Dos ejemplos de estos experimentos son: el realizado por
Esterman, Simpson y Stern, y el de Zartman y Ko, por otro lado, [ref. 2]. Estos
experimentos confirman la hipótesis de la distribución Maxwell Boltzmann.
I.1.3. TRAYECTORIA LIBRE MEDIA
La trayectoria libre media ( ) es la longitud que en promedio recorre una
molécula entre dos choques sucesivos. Este es un parámetro importante en el diseño de
sistemas tales como aceleradores de partículas o sistemas de deposición de películas
delgadas donde se necesita que partículas microscópicas tales como electrones, iones o
moléculas viajen distancias considerables con una cantidad mínima de obstáculos.
Para hacer una estimación del valor de podemos suponer que las
moléculas son esféricas de diámetro d. Dos moléculas chocarán si el centro de una
molécula se aproxima una distancia menor que d de la otra. Para simplificar el cálculo
podemos suponer que una molécula tiene diámetro 2d y que todas las otras moléculas
tienen diámetro despreciable (partículas puntuales).
Si es la trayectoria libre media podemos decir que en promedio, en el
volumen 2d , hay encerrada una partícula, es decir:
1 d 2
VN
o, usando la ley universal de los gases ideales,
P1 d
T k2
B (1.10)
A temperatura ambiente (300 K) y considerando que d es aproximadamente
5Å podemos estimar la trayectoria libre media usando la siguiente fórmula:

Capítulo I Alto Vacío
7
P
10 3.5 3
(1.10 a)
donde resultará en metros si P se expresa en Pascales.
Así, a la presión atmosférica ( P = 1 bar = 105 Pa ), una molécula recorre
en promedio una distancia = 530 Å antes de experimentar un choque con otra
molécula. En un vacío pobre (P = 10-3 mbar = 0.1 Pa), recorre = 5.3 cm. En alto vacío
( P = 10-6 mbar = 10-4 Pa), recorre = 53 m; y en ultra alto vacío ( P = 10-9 mbar = 10-7
Pa), = 53 km.
Considerando una velocidad cuadrática media vrms = 500 m/s, podemos
estimar que a la presión atmosférica una molécula experimenta un choque cada 10-10 s y
en alto vacío (10-6 mbar) una molécula experimenta un choque cada 0.1 segundo.
I.1.4. RAPIDEZ DE INCIDENCIA MOLECULAR
Conocer el número de moléculas que inciden sobre una superficie en la
unidad de tiempo es un tema muy importante tanto en la tecnología de alto vacío como
en la producción de películas delgadas. Aquí deduciremos una expresión para calcular
el número de moléculas de un gas ideal en equilibrio que inciden sobre la unidad
de área de una superficie en la unidad de tiempo.
Sea dNx el número de moléculas con velocidades entre v y v + dv . Las
moléculas que pasan a través de la superficie de área A entre los instantes t y t + t son
las que en el instante t están encerradas en el volumen Av t, es decir,

Capítulo I Alto Vacío
8
t xvAV
xdN
La contribución al flujo de estas moléculas es
xx
VdNΦd v (1.11)
Por otro lado, usando la ec. (1.4) puede demostrarse que
xB
x
/
Bx dT k
m - e )
T πm N ( dN v2
v
k2
2
21 (1.4a)
y reemplazando el valor de dNx de la ec (1.4a) en la ec (1.11) e integrando, obtenemos:
sm
moléculas MTP Φ x 2
231009.2 (1.12)
donde P es la presión expresada en pascales y M es la masa molecular del gas.
Por ejemplo para gas nitrógeno a presión atmosférica y 300 K
smmoléculas Φ 2
27108.2
y a una presión de 10-4 Pa
smmoléculas Φ 2
19108.2
A vx
Vx Δt
Fig. 1.1 Esquema para obtener una relación entre
el flujo de moléculas y la velocidad de las
moléculas.

Capítulo I Alto Vacío
9
I.1.5. REGÍMENES DE FLUJO ESTACIONARIO
La discusión anterior sobre la teoría cinética de gases es válida en
condiciones hidrostáticas, es decir, no hay flujo neto de gases. Cuando hay un flujo
neto, es conveniente distinguir entre regímenes diferentes de flujo. Cuando un sistema
de vacío como el de la figura 2, por ejemplo, comienza a funcionar, el flujo neto de
moléculas a través de una sección transversal en general varía con el tiempo pero
después de unos segundos se alcanza una situación tal que el flujo neto de moléculas a
través de cualquier sección transversal permanece constante. Decimos entonces que
estamos en una situación de régimen estacionario.
El criterio para distinguir entre los regímenes estacionarios está basado en la
magnitud del número de Knudsen, Kn, que se define como la razón del diámetro de la
cámara o tubería, tD , a la trayectoria libre media,
tn
DK (1.13)
El régimen molecular ocurre a muy bajas presiones, cuando la trayectoria libre
media es grande comparada con las dimensiones de la cámara ( > tD ). Las
moléculas se comportan independientemente una de otra. Esta es la situación en
equipos de análisis tales como espectrómetros Auger, en los microscopios electrónicos,
Régimen Número de Knudsen
Flujo molecular Kn < 1
Fujo intermedio 1 < Kn < 110
Flujo viscoso Kn > 110

Capítulo I Alto Vacío
10
en equipos de deposición por evaporación térmica y otras formas de deposición donde
intervienen vapores obtenidos por medios físicos (PVD).
En el otro extremo, cuando los valores de presión son del orden de la
presión atmosférica, la trayectoria libre media es pequeña ( 110/tD ) y las
moléculas chocan más frecuentemente entre sí que con las paredes del recipiente. En
este caso se dice que tenemos un régimen viscoso. Esta es la situación, por ejemplo, en
muchos de los procesos de deposición por reacciones químicas de vapores (CVD).
El régimen viscoso puede ser turbulento, cuando el flujo es irregular, o
laminar cuando el flujo es regular, sin remolinos, con velocidad nula cerca de las
paredes y máxima en el eje de la tubería.
Si el número de Knudsen toma los valores intermedios entre 1 y 110 se dice
que el régimen es intermedio.
Las bombas mecánicas no son efectivas en el régimen molecular, mientras
que las bombas de difusión y las turbomoleculares sólo pueden trabajar en este régimen.
I.1.6. GASTO, CAUDAL Y THROUGHPUT
La masa de gas por unidad de tiempo que pasa a través de una superficie se
llama gasto
ΔtΔmG (1.14)
Haciendo un esquema como el de la figura 1.1, puede demostrarse que
ΔtΔVρvAρG ,
donde v es la velocidad del fluido, la densidad y V es el volumen de gas que pasa a
través de la sección transversal de área A.

Capítulo I Alto Vacío
11
Caudal se define como el volumen de gas que pasa a través de una sección
transversal
tΔVΔS , (1.15)
es decir, la relación entre gasto y caudal queda establecida por la siguiente ecuación.
SρG (1.16)
Puesto que es más sencillo medir la presión que la densidad del gas en un
punto dado del sistema y puesto que hay una relación de proporcionalidad directa entre
densidad y presión, es conveniente definir una nueva cantidad para analizar los sistemas
de fluidos en movimiento. Se define Throughput, Q, a través de una superficie por la
expresión:
PSQ , (1.17)
donde S es el caudal y P es la presión del gas en los puntos de la superficie. Throughput
es una medida indirecta del gasto, es decir, de la masa por unidad de volumen que pasa
a través de una sección transversal.
I.1.7. CONDUCTANCIA
Consideremos el sistema básico de vacío de la figura 1.2. En régimen
estacionario se establece una diferencia de presión entre la entrada de la bomba (b) y la
salida de la cámara (c). El throughput constante tiene el mismo valor a través de
cualquier sección transversal del tubo de conexión. Puede establecerse
experimentalmente la siguiente relación entre el throughput y la diferencia de presión:
Q = C (Pc - Pb) (1.18)
donde C queda definida como la conductancia del tubo de conexión, y Pc y Pb son las
presiones medidas en los puntos b y c.

Capítulo I Alto Vacío
12
Fig. 1.2. Sistema básico de vacío
Si comparamos un sistema de vacío como el de la figura 1.2 con un circuito
eléctrico, el throughput (Q) es equivalente a la corriente, la diferencia de presión (Pc -
Pb) es equivalente a la diferencia de potencial eléctrico y la conductancia (C) vendría a
ser el análogo del inverso de la resistencia eléctrica.
En régimen viscoso, que es el caso de sistemas de vacío pobre o algunas
líneas de alto vacío conectada a bomba mecánica (vacío previo), la conductancia
depende de los valores de presión involucrados y de la geometría del elemento
conductor. En régimen molecular, en el cual operan las bombas de difusión y
turbomoleculares, la conductancia de un elemento conductor depende sólo de la
geometría del elemento, no depende de los valores de presión.
En algunos libros especializados [ref. 3, 4] se usan los métodos de Hagen y
Poiseuille para calcular la conductancia de diferentes elementos en el régimen viscoso,
así como los métodos usados originalmente por Smoluchowsky [ref. 5], Knudsen [ref.
6] y Loeb [ref. 7] para calcular conductancias en regímenes moleculares. A
continuación presentamos los resultados para tres elementos conductores:
cámara
bombaa laatmósfera
c
b

Capítulo I Alto Vacío
13
i) Conductancia de una abertura, para aire a 20ºC:
a) En régimen viscoso, en el rango 0,525 < 1
2
PP < 1,
C =
1
2
PP1
20A
b) En régimen molecular:
C = 11,6 A
ii) Conductancia de un tubo cilíndrico de diámetro D
a) En régimen viscoso
C = 91 L
D4 (P1 + P2)
b) En régimen molecular
C = 12,1 L
D3
iii) Conductancia de tubo cilíndrico de radios interno R1, externo R2
y
longitud L
a) En régimen viscoso:
C = 1450 (L
2 1 PP ) )/log(/ 1022
120
41
40 RRRRRR
b) En régimen molecular:
C = 12,1 ( D2 – D1)2 (D1 + D2) (K0 /L)
donde los valores de K0 dependen de D1/D2 según la siguiente tabla:
D1/D2 0 0,259 0,5 0.707 0.866 0,966
K0 1 1,072 1,154 1,254 1,430 1,675

Capítulo I Alto Vacío
14
Cuando las conductancias se asocian en serie la conductancia del sistema es:
...CCCsis 21
111
y cuando se asocian en paralelo es:
. . .CCCsis 21
Es importante observar que aun cuando C, la conductancia de un tubo, y el
caudal S a través de una sección transversal tienen las mismas unidades, e inclusive
pueden tomar valores del mismo orden de magnitud, son dos conceptos diferentes. La
conductancia se refiere a un tubo o a un componente tubular del sistema, mientras que el
caudal se refiere a cualquier sección transversal.
I.1.8. FUGAS, DEGASIFICACION Y RETROFLUJO
Cuando se quiere instalar un sistema de alto vacío lo más importante es
tener cuidado en lograr un cerrado hermético de la cámara y todas las partes que limitan
el sistema de vacío con la atmósfera (excepto, por supuesto, la abertura de salida de la
bomba mecánica, en la figura 1.2). Cualquier agujero, rendija o defecto en el sellado
entre dos partes del sistema dará lugar a la penetración de aire de la atmósfera hacia el
interior del sistema. Esto es lo que llamamos fugas.
Pero, aún cuando el sistema esté herméticamente cerrado, es imposible
obtener presión cero. A parte de las fugas hay dos fenómenos que se oponen al vacío
perfecto: la degasificación y el retroflujo.
La degasificación es la emanación de gases de las superficies de los
materiales dentro del sistema. Estos gases emanados pueden ser vapor de agua, oxígeno
u otros gases adheridos a las superficies internas de las paredes del sistema de vacío o

Capítulo I Alto Vacío
15
también átomos de las superficies sólidas que se liberan debido a las condiciones de
presión y temperatura.
Este fenómeno es más importante a presiones del orden de 1 Pa o menores.
La degasificación es el principal factor con respecto a la presión última que un sistema
puede alcanzar, asumiendo que las fugas están ausentes. El nivel de degasificación es
reducido manteniendo el sistema limpio y seco y con una apropiada selección de
materiales para la construcción de la cámara. El diseño de un sistema de ultra alto vacío
incluye la posibilidad de eliminar capas de vapor de agua y otros gases adheridas a las
paredes de la cámara y tuberías del sistema por calentamiento a temperaturas de al
menos 150ºC.
Para las aplicaciones que describimos en este libro este nivel de limpieza no
es necesario. Sin embargo, las componentes del sistema deben ser conservadas limpias
(no huellas digitales u otros), secas y aisladas del ambiente (la mayor fuente de
humedad) tanto como sea posible.
Un concepto físico que caracteriza la degasificación de los materiales es la
presión de vapor. Todos los materiales emiten vapores de sus partes constituyentes y
estos vapores se suman al gas carga en el sistema. El agua es el más común de los
materiales encontrados y es un buen ejemplo de lo que significa presión de vapor. A
100ºC, la presión de vapor del agua es 1 atm (760 Torr). Bajo estas circunstancias
cuando la presión del ambiente que rodea al agua es igual a su presión de vapor todos
sabemos lo que sucede – el agua hierve. A temperatura ambiente la presión de vapor de
agua disminuirá a 17,5 Torr y hervirá a esa presión. No es recomendable mantener agua
en el interior de un sistema vacío. Otros materiales con alta presión de vapor son los
plásticos y metales tales como mercurio, zinc, plomo y cadmio. Entre los materiales
con baja presión de vapor están los vidrios, cobre, aluminio, acero inoxidable, plata y

Capítulo I Alto Vacío
16
algunos cauchos sintéticos. Puesto que la presión de vapor es una función de la
temperatura, es posible usar en sistemas de vacío materiales como zinc y bronce
siempre y cuando no se utilicen a alta temperatura.
Normalmente el flujo de gas y vapor en un sistema de vacío es del interior
de la cámara hacia la atmósfera a través de las bombas. Sin embargo en el régimen
molecular algunas moléculas se comportan individualmente y pueden ir en la dirección
opuesta al flujo principal. Este fenómeno conocido como retroflujo (backstreaming) es
particularmente nocivo en un sistema de alto vacío cuando hay elementos indeseables
en este flujo; por ejemplo el aceite de las bombas difusoras puede ir hacia la cámara de
vacío en vez de ir siempre hacia la atmósfera. Es necesario diseñar elementos especiales
como trampas para disminuir este efecto, o usar preferentemente bombas
turbomoleculares en vez de difusoras.
I.1.9. VELOCIDAD DE BOMBEO
Bombeo es el proceso de extraer moléculas de gas del sistema a través de la
acción de bombas. La velocidad de bombeo, bS , es definida como el volumen de gas
por unidad de tiempo que pasa a través del plano de la abertura de entrada de la bomba,
es decir, el caudal en la abertura de entrada de una bomba de vacío.
b
b PQS (1.19)

Capítulo I Alto Vacío
17
Fig. 1.3 Cámara, bomba y tubo de conexión (a) sin considerar la degasificación, (b) con
degasificación (adaptada de ref. [8]).
Cuando un extremo de un tubo es conectado a una bomba, el caudal en ese
extremo del tubo tendrá un valor mayor que en el otro, en la figura 1.3, bS > S. Para
obtener una relación entre el caudal S en la salida de la cámara y la velocidad de
bombeo bS , combinemos la ecuación Q = C ( P – Pb) con la ec. (1.19). Se obtendrá:
CS
SS
b
b
/1 , (1.20)
El caudal S en la base de la cámara nunca excede los valores numéricos de
C ni de bS . Cuando bS = C, el caudal en la base de la cámara (o velocidad de bombeo
efectiva ) será S/2.
Ejemplo: Si un tubo de 2 cm de diámetro y 60 cm de longitud se conecta
por un lado a una bomba mecánica de velocidad de bombeo nominal S = 5,0 l/s (a 100
mTorr) y por otro lado a una bomba de difusión. Durante la operación del sistema se
obtendrá una velocidad de bombeo experimental S = 4,8 l/s, es decir, este tubo de
conexión no limita el rendimiento de la bomba mecánica. Si el mismo tubo es conectado
entre una bomba difusora, que tiene una velocidad de bombeo especificada de 100 l/s , y
cámara
bomba
cámara
bomba a la atmósfera
a la atmósfera
P , S , Q
C
Pp, Sp
Q
Qp
(a) (b)

Capítulo I Alto Vacío
18
una pequeña cámara se obtendrá una velocidad de bombeo efectiva de sólo 3,1 l/s, es
decir, un tubo de conductancia pobre puede limitar fuertemente la bomba el
funcionamiento eficiente de la bomba; la ahorca. Si se usara un tubo de 5 cm de
diámetro, aún se tendría una velocidad de bombeo de sólo 25 l/s.
En un sistema real debemos considerar la degasificación y el retroflujo
como se muestra en la figura 3b, por un término de throughput inverso Qd. En tal caso,
el throughput neto quedará expresado por:
db QPSQ . (1.21)
)1(PS
QPSQ
b
db
Cuando el sistema se acerca a la presión final, Pf , se tendrá Qd proporcional
a Pf y por lo tanto, la velocidad de bombeo efectiva será:
)PP
α(SPQS f
b 1 . (1.22)
TIEMPO DE BOMBEO
Un tema importante en la tecnología de alto vacío es el tiempo requerido
para alcanzar una determinada presión, Pf. Una fórmula para estimar el tiempo de
bombeo, Tb, puede obtenerse rápidamente asumiendo que el gas es ideal y el proceso
isotérmico, es decir, PV = constante y por lo tanto:
dtdPV
dtdVP
Esto nos permite escribir el throughput en la forma:
dtdPVQ

Capítulo I Alto Vacío
19
db QPSdtP(t)dV (1.23)
donde Qd incluye el efecto de la degasificación y el retroflujo. Después de integrar se
obtiene
)V
tS(PPPP(t)
/SQP/SQ(t)P b
fi
f
bdi
bd exp (1.23a)
lo cual permite decir que una buena estimación del tiempo de bombeo está dada por la
expresión:
f
i
bb P
PSVT ln (1.24)
Ejemplo: Una bomba mecánica cuya velocidad de bombeo es 16 m3/h ( 4,44 l/s )
tardará 5 minutos para evacuar una cámara de 100 litros desde la presión atmosférica
hasta una presión de 10 Pa.
I.2. BOMBAS DE VACIO
En un sistema de alto vacío, el elemento fundamental son las bombas de
vacío, máquinas que reducen la presión de un recipiente cerrado, es decir, evacuan una
cámara. Los principios básicos para el funcionamiento de las bombas de vacío son dos:
(i) transferencia de gas del interior al exterior de la cámara y (ii) adherencia de
moléculas de gas a una superficie sólida dentro de la cámara. Las bombas mecánicas,
bombas de difusión y turbomoleculares se basan en el principio de transferencia de gas.
Ejemplos de bombas que se basan en la captura de las moléculas por una superficie son
las bombas criogénicas y las bombas de adsorción. En esta sección sólo describiremos
los principios básicos de funcionamiento de las bombas comúnmente usadas en
procesos de producción de materiales, que son precisamente las que funcionan por
transporte de gas.

Capítulo I Alto Vacío
20
Entre las bombas que funcionan por transferencia de gas es importante
distinguir entre las bombas mecánicas que funcionan en el régimen viscoso (presiones
entre 100 y 105 Pa) y las bombas de difusión y turbomoleculares que trabajan en el
régimen molecular (P < 100 Pa). Las bombas de difusión y turbomoleculares siempre
trabajan en serie con una bomba mecánica.
I.2.1. BOMBAS MECANICAS
El principio de las bombas mecánicas consiste en aislar una cantidad de gas
de un recipiente, comprimirlo y trasladarlo al exterior. Existen varios tipos de bombas
mecánicas, según la geometría del sistema compresor: de paletas rotatorias, de pistón
rotatorio y las llamadas bombas de raíces. Discutiremos aquí sólo las bombas de paletas
rotatorias.
BOMBAS DE PALETAS ROTATORIAS
La figura 1.4 muestra el esquema de una bomba de paletas rotatorias. Consiste de dos
cilindros eccéntricos. El externo o estator, que es hueco, y el interno o rotor. A lo largo
diámetro del rotor hay una ranura en la cual se han insertado dos paletas radiales
separadas en el centro por un resorte. La cavidad del cilindro estator está conectado por
una abertura a la cámara de vacío (entrada) y por otra abertura al ambiente externo
(salida). Las aberturas de entrada y salida están a uno y otro lado de la línea de contacto
entre estator y rotor separados por una pequeña distancia angular.

Capítulo I Alto Vacío
21
Fig. 1.4 Esquema de una bomba de paletas rotatorias, [ref. 3 ].
El funcionamiento de este tipo de bombas puede entenderse con ayuda de la
figura 1.5. Cuando la paleta A pasa por la abertura de entrada la cámara de vacío entra
en contacto con el espacio entre rotor y estator (Fig. 1.5a). El volumen de este espacio
aumenta cuando la paleta continúa su barrido produciendo así una disminución en la
presión de la cámara. Esto continúa hasta que la paleta B pasa por la abertura de
entrada, instante en el cual una porción de gas queda aislada en el espacio entre rotor,
estator y paletas (Fig. 1.5b). Puesto que el barrido continua, el gas aislado se comprime
(Fig. 1.5c) y es expulsado cuando la paleta B llega a la abertura de salida (fig. 1.5d). La
velocidad angular del rotor es usualmente entre 400 y 1500 rpm y la temperatura del
sistema en funcionamiento normal es entre 60 y 80ºC.

Capítulo I Alto Vacío
22
Fig. 1.5 Funcionamiento de una bomba mecánica de paletas rotatorias, [ref. 3 ].
En la figura 1.6 se muestra la sección transversal de una bomba mecánica
comercial. Un detalle que aparece en la figura 1.6 pero no en las figuras 1.4 y 1.5 es la
conexión para el lastre de gas. Algunas veces es necesario introducir en la cámara de
vacío algunos gases que se pueden condensar (o licuar) en las etapas de compresión y
expulsión. Estos líquidos son nocivos para el funcionamiento de la bomba porque
disminuyen la efectividad del lubricante que sella el estator con el rotor. Si éste es el
caso, es conveniente inyectar en la etapa de compresión una porción de gas inerte desde
el exterior para evitar que los gases expelidos desde la cámara se conviertan en líquidos
antes de ser expulsados. Esta porción de gas inerte es la que se llama lastre de gas (gas
ballast).

Capítulo I Alto Vacío
23
Fig. 1.6 Sección transversal de una bomba mecánica de paletas rotatorias: (1) Entrada o
conexión a la cámara, (2) filtro, (3) válvula antisuccionadora, (4) canal de entrada, (5) paletas,
(6) cilindro estator, (7) cilindro rotor, (8) conexión para el lastre de gas inerte (opcional) , (9)
canal de salida, (10) válvula de salida, (11) demistor interno, (12) bucle de resortes, (13)
conexión para el filtro de aceite, [ref 9.]
Las características más importantes de las bombas son dos: (i) la presión
última o presión mínima obtenida cuando la bomba sólo se evacua a sí misma (ii) la
velocidad de bombeo. Para mejorar estas características algunas veces se usan bombas
de paletas rotatorias de dos etapas. La figura 1.7 muestra un esquema de este tipo de
bombas. Con bombas de paletas rotatorias es posible alcanzar presiones del orden de 10-
2 Pa. La figura 1.8 muestra la variación de la velocidad de bombeo en función de la
presión para bombas de paletas rotatorias de una y de dos etapas con dispositivos para
lastre de aire y sin ese dispositivo.

Capítulo I Alto Vacío
24
Fig. 1.7 Esquema de una bomba mecánica de paletas rotatorias de de dos etapas, [ref. 4].
presión en la entrada de la bomba (Pa)
10-2 100 102
104
bomba de paletas rotatoriasde una etapa
bomba de paletas rotatorias de dos etapas
con lastre de gas
sin lastre de gas
6
8
10
Fig. 1.8 Gráfico típicos de velocidad de bombeo vs presión para bombas mecánicas de
una y dos etapas con y sin dispositivo de lastre de gas.
I.2.2. BOMBAS DE DIFUSIÓN
Las bombas de difusión siempre funcionan entre la cámara a evacuar y una
bomba mecánica. Cuando una bomba mecánica ha evacuado la cámara y la bomba de
difusión hasta una presión de 100 Pa o menos puede ponerse en funcionamiento la
bomba de difusión para obtener presiones de hasta 10-8 Pa.
El principio de las bombas de difusión es la compresión de un gas por
transferencia de momentum en una dirección determinada desde un chorro de vapor.
La figura 1.9 muestra la sección transversal de una bomba de difusión. En
el fondo, un líquido de baja presión de vapor (mercurio o aceite) es calentado. Un

Capítulo I Alto Vacío
25
chorro de ese vapor sube por la chimenea y regresa al chocar en el techo de ésta y en
las paredes de las boquillas. Los chorros reflejados arrastran las moléculas de gas a
baja presión que se encuentran alrededor de la chimenea. El vapor, de aceite o mercurio,
se condensa en las paredes refrigeradas de la bomba y regresa al fondo de la bomba,
mientras que las moléculas de gas comprimidas salen a través de la bomba mecánica. El
gas residual de la cámara por encima del techo de la chimenea reduce su presión porque
sus moléculas entran por difusión a la región donde actúa el chorro de vapor.
Las bombas de difusión funcionan comprimiendo gases a presiones del
orden de 10-8 Pa a presiones del orden de 100 Pa. Por esta razón las bombas de difusión
siempre funcionan en serie con una bomba mecánica. La acción de bombeo cesa a
presiones mayores de 100 Pa porque el chorro de vapor reflejado ya no se extiende
hasta la pared sino que termina en un frente de choque cerca al chorro principal.
Usualmente, la presión obtenida por la bomba mecánica que respalda a la bomba de
difusión es del orden de 25 a 75 Pa. Las bombas de difusión modernas tienen varias
etapas de compresión – usualmente 3 a 5 para bombas pequeñas y hasta 7 para bombas
grandes. La bomba de difusión mostrada en el esquema de la figura 1.9 es de tres
etapas. Cada etapa comprime el gas a presiones sucesivamente más altas que la
precedente hasta llegar a la salida donde el gas comienza a ser evacuado por la bomba
mecánica.

Capítulo I Alto Vacío
26
Fig. 1.9 Sección transversal de una bomba de difusión. (1) Sombrerete enfriado para evitar
que el vapor entre a la cámara, (2) heater para compensar la pérdida de calor en la parte superior
de la chimenea, (3) superficie aerodinámica para evitar turbulencia, (4) etapas múltiples
para obtener bajas presiones, (5) estuche alargado, (6) obstáculo para impedir que entre líquido
al chorro de vapor, (7) heater para supercalentar el vapor, (8) etapa de ejector lateral, (9)
obstáculo cónico, (10) purificador de aceite (11) drenaje de aceite altamente volátil, (12)
obstáculos para evitar pérdidas de aceite, (13) chimeneas concéntricas que permiten el
fraccionamiento del chorro de vapor de aceite, [ref. 4]
La velocidad de bombeo está determinada por el área de la puerta de
conexión a la cámara y por el factor de Ho. De acuerdo con la ec (1.12) para la rapidez
de incidencia molecular, podemos calcular el caudal máximo que puede pasar a través
de la abertura de conexión entre la cámara y la bomba.
Smax = 11. 6 A l s-1,
y si consideramos como área efectiva de la abertura A = 4 ( D2 – t2), donde D es el
diámetro de la abertura y t el de la tapa de la chimenea, obtendremos:
S = H Smax = 3,64 (T/M)1/2 H ( 4/ ) t (2D – t) (1.25)

Capítulo I Alto Vacío
27
Si asumimos el factor de Ho, H = 0,4 y t = D/3, entonces, una bomba con
D ≈ 9 pulgadas tiene una velocidad de bombeo de 1000 l/s. Las bombas de difusión
comerciales generalmente están disponibles en el rango de 1 a 40 pulgadas de diámetro
de puerta de conexión a la cámara y sus respectivas velocidades de bombeo están en el
rango de 10 a 105 l/s.
Según la ec. (1.25) la velocidad de bombeo no depende de la presión. Esto
es cierto en un rango de presiones de varios órdenes de magnitud, digamos de 10-1 a 10-4
Pa. En la práctica, como se muestra en la figura 1.10, pueden distinguirse tres regiones
en un gráfico S vs. P: (i) Si comenzamos a disminuir la presión a partir de la presión
máxima de funcionamiento de la bomba, el throughput (Q = SP) se mantiene constante
y por lo tanto S aumenta al disminuir la presión (región AB en figura 1.11a), (ii) la
región BC donde S = Sm es independiente de P, y (iii) cerca de la presión última Pu la
velocidad de bombeo S tiende a cero debido a que la razón de compresión no puede ser
infinita.
S = Sm [ 1- ( Pu / P) ]
La dependencia de S respecto de M-1/2, observada en la ec. (1.25), se verifica
experimentalmente como lo muestran las curvas en la figura 1.11.
A
C B
presión en la entrada de la bomba P (Pa)
10 -9 10 0 10 -1
100
1000
Fig. 1.10. Velocidad de bombeo en función de la presión mostrando tres regiones de
comportamiento diferente.

Capítulo I Alto Vacío
28
presión en la entrada de la bomba P (Pa)
10-410010-3 10-2
10-1
100
1000 N2
Ar
H2
Fig. 1.11.Velocidad de bombeo en función de la presión para gases diferentes, [ref. 3].
I.2.3. BOMBAS TURBOMOLECULARES
Una turbina es una rueda con paletas en su periferia especialmente
diseñadas para diferentes aplicaciones. En la tecnología de máquinas térmicas las
turbinas se usan para transformar la cantidad de movimiento de las moléculas de un
fluido que chocan con las paletas en movimiento del eje de la turbina. Por el contrario,
en una bomba turbomolecular el eje de la turbina es impulsada por un motor eléctrico
(generalmente trifásico) a velocidades angulares de 30000 rev/min, y las paletas están
especialmente diseñadas para transferir cantidad de movimiento a las moléculas de un
gas, empujándolas siempre en una dirección determinada, logrando de este modo la
compresión de una porción de gas. La figura 1.12 es la sección transversal de una típica
bomba turbomolecular (Turbovac 151 de Leybold) y la figura 1.13 es la fotografía de
una turbina típica de una bomba turbomolecular.
La función que cumplen las bombas turbomoleculares es la misma que las
bombas de difusión, es decir, colocadas en serie entre la cámara a evacuar y la bomba
mecánica de prevacío deben transportar moléculas del gas residual desde la cámara
hasta la bomba mecánica. Mientras que en las bombas de difusión las moléculas del gas
residual son arrastradas por un chorro de vapor de aceite, en las bombas

Capítulo I Alto Vacío
29
turbomoleculares son arrastradas por las paletas de una turbina, obteniéndose por lo
tanto la ventaja de un vacío más limpio. En las bombas de difusión es imposible evitar
que algunas pequeñas cantidades de gases tales como CO2, CO, y algunos hidrocarburos
provenientes de la descomposición del aceite entren en la cámara.
La velocidad de bombeo y valores de presión última para las bombas
turbomoleculares son similares a los de las bombas de difusión.
Fig. 1.12 Sección transversal de una bomba turbomolecular: (1) Brida para conexión a cámara,
(2) estator, (3) brida para conexión a ventilación, (4) brida para conexión a bomba mecánica de
pre-vacío, (5) malla protectora, (6) rotor, (7) cubierta o chasis de la bomba, (8) rodaje, (9)
conexión para agua de enfriamiento, (10) motor trifásico, (11) rodaje, [ref. 9]

Capítulo I Alto Vacío
30
Fig. 1.13 Fotografía del rotor de una bomba turbomolecular: serie de turbinas. Si la velocidad
angular de las turbinas es de 30000 rev/min. A 10 cm del eje la velocidad lineal de un punto de
las paletas es de 314 m/s. Del mismo orden de magnitud que las velocidades de las moléculas de
O2, Ar, o N2 a temperatura ambiente, [ref. 4]
Para comprender el principio de funcionamiento de las bombas
turbomoleculares consideremos las figuras 1.14. La figura 1.14a es una vista lateral de
una de las turbinas que componen el rotor de la bomba turbomolecular. Las paletas
hacen un ángulo con el plano de rotación de la rueda. Los puntos de una paleta tienen
una velocidad lineal u. La turbina separa las regiones del espacio 1 y 2 y transportará
moléculas de la región 1 a la región 2 porque de acuerdo a su geometría la probabilidad
de transmisión de la región 1 a la región 2 es mayor que de 2 a 1. En efecto, si
asumimos que la velocidad de las moléculas es igual a la velocidad de las paletas, la
parte delantera (izquierda) de la paleta es alcanzada sólo por aquellas moléculas dentro
del ángulo 1 . Del punto A las moléculas pueden rebotar directamente a la región 2
dentro del ángulo 1 o hacia la paleta opuesta dentro del ángulo 1 . De manera análoga
las moléculas que chocan con la paleta en el punto B (fig 1.14b), bajo el ángulo 2 ,
pueden rebotar dentro de un ángulo 2 a la región 1; puesto que el diseño es tal que:
2
2
1
1

Capítulo I Alto Vacío
31
se tendrá que la transmisión de la región 1 a la región 2 es mayor que en la dirección
opuesta.
u
A
B
u
u C
vw
u
Fig. 1.14 . (a,b) Vista lateral de una turbina de una bomba turbomolecular mostrando que,
debido a la inclinación de las paletas y la velocidad de éstas que es del orden de la velocidad de
las moléculas, existe un flujo neto de la región 1 a la región 2. (c) Cinemática del choque de una
molécula con una paleta de una bomba turbomolecular. [ref. 9]
La cinemática de una simple colisión es explicada con ayuda de la figura
1.14c. Una partícula de la región 1 que incide con velocidad v en el punto C en un
borde de la paleta se adhiere a ésta por un tiempo muy corto y cuando es desorbida "se
ha olvidado" de la dirección de incidencia. Asumiendo que la temperatura de la paleta
es igual a la del gas la partícula dejará el punto C de la paleta con velocidad v en la
dirección más probable, es decir, perpendicular a la superficie. La suma del vector
a)
b)
c)

Capítulo I Alto Vacío
32
velocidad v con la velocidad u de la paleta da la velocidad resultante w. Debido al
ángulo de inclinación de la paleta respecto al plano de rotación los vectores w son
preferencialmente dirigidos a la región 2.
La velocidad de bombeo de una bomba turbomolecular está principalmente
determinada por la geometría de las turbinas y el área de conexión a la cámara que se
desea evacuar. Las curvas de velocidad de bombeo vs. presión son similares a las de
una bomba de difusión, es decir, se tiene una velocidad de bombeo constante para
presiones entre 10-8 y 10-1 Pa y para presiones mayores, donde comienza el régimen
viscoso laminar, se tiene una disminución de la velocidad de bombeo. La velocidad de
bombeo es mayor para gases pesados como argón, nitrógeno e hidrocarburos que para
gases livianos, como helio o hidrógeno, esto se debe a que la condición: velocidad lineal
de las paletas es aproximadamente igual a la velocidad de las moléculas, se cumple
mejor para gases de medio o alto peso molecular.
Fig. 1.15 Curvas velocidad de bombeo vs presión obtenidas con una bomba turbomolecular
para diferentes tipos de gas residual.
La presión última de una bomba turbomolecular o difusora es del orden de
10-7 a 10-9 Pa pero puede variar dependiendo de la bomba mecánica de vacío previo. Es
presión en la entrada de la bomba P (Pa)
10 -8 10 0 10 -6 10 -4 10 -2
100
1000 N 2
Ar
H 2

Capítulo I Alto Vacío
33
importante observar la diferencia entre presión última de un sistema de bombas y la
presión de fondo en un sistema de vacío. Cuando se habla de presión última de una
bomba mecánica o de un sistema de bombas no se incluye la cámara, es decir, las
bombas sólo se evacuan a sí mismas. La siguiente ecuación resume los parámetros que
influyen en la presión de fondo o presión mínima en la cámara de un sistema de vacío.
ef
Du
ef
c S
Q P
tV
S-
e P P 0 (1.26)
Pc = Presión de fondo o presión mínima en la cámara
P0 = Presión en la cámara al principio de la evacuación (usualmente presión
atmosférica)
Sef = Velocidad de bombeo efectiva dentro del rango de presión
concerniente
V = Volumen de la cámara
Pu = Presión última de la bomba turbomolecular
QD = Throughput debido a fugas y degasificación
I.3. VACUOMETROS
Existen un buen número de instrumentos para medir presiones de gases
enrarecidos, entre ellos podemos mencionar a: (i) El vacuómetro McLeod, que es una
variante del simple tubo en U, se usa para medir presiones por encima de la atmosférica,
(ii) el vacuómetro de Bourdon, que mide la deformación de un tubo en forma de hélice
cuando se conecta a un sistema de vacío, (iii) el vacuómetro de diafragma, que se basa
en una cápsula herméticamente cerrada en la cual se ha hecho el vacío. La variación de
la presión externa produce deformación de las paredes de la cápsula, y (iv) el

Capítulo I Alto Vacío
34
manómetro de capacitancia, que es un vacuómetro de diafragma en el cual se mide la
capacitancia eléctrica del condensador formado por una de las paredes de la cápsula y
un contraelectrodo.
El principio de funcionamiento y los diseños de estos tipos de vacuómetros
son ampliamente discutidos en las referencias [3] y [4]. Aquí sólo discutimos el
principio de funcionamiento de los dos tipos de vacuómetros más ampliamente usados
en los laboratorios que utilizan los sistemas de alto vacío para producir materiales de
alta pureza; los vacuómetros térmicos, que miden la presión indirectamente a través de
la conductividad térmica del gas residual, y los vacuómetros de ionización, que lo hacen
a través de la corriente de ionización de una fracción del gas residual.
I.3.1. VACUÓMETROS TÉRMICOS
Miden indirectamente la presión a través de mediciones de la conductividad
térmica del gas residual. El esquema de la figura 1.16 muestra su principio de
operación: Un filamento de tungsteno o de otro metal adecuado es encerrado en un tubo
metálico o de vidrio y éste a su vez es conectado a la cámara de vacío. Para simplificar
la explicación digamos que el filamento es conectado a una fuente que le proporciona
una corriente eléctrica constante y que lo calienta por efecto Joule. Si la presión del gas
residual fuera constante se establecería rápidamente un flujo estacionario de calor del
filamento a las paredes del tubo y el filamento mantendría su temperatura constante.
Cualquier variación en la presión del gas residual varía su conductividad térmica y por
lo tanto la temperatura del filamento. Así pues, midiendo la temperatura del filamento
se mide indirectamente la conductividad térmica del gas y la presión del gas en la
cámara.

Capítulo I Alto Vacío
35
1
2
3
4
5
Fig. 1.16. Esquema de un vacuómetro térmico: (1) filamento de tungsteno, (2) soporte para el
filamento, (3) tubo de vidrio o de acero inoxidable, (4) conexiones eléctricas, (5) conexión a la
cámara de vacío.
Los vacuómetros térmicos son útiles en el rango de 1 a 100 Pa porque se
basan en el fenómeno de conducción de calor. Por encima de este rango, la transferencia
de calor por convección es tan importante como la conducción y por debajo de este
rango predomina la radiación.
Si la temperatura del filamento se mide indirectamente a través de la
resistencia eléctrica del filamento, se dice que nuestro vacuómetro térmico es un Pirani
y si la temperatura del filamento se mide con una termocupla tendremos un vacuómetro
TC.
Vacuómetro Pirani
La característica de este tipo de vacuómetros térmicos es que el filamento
de tungsteno es una de las cuatro resistencias de un puente Wheastone. Para una presión
determinada se obtiene una resistencia del filamento que mantiene el puente balanceado.
Cualquier cambio de presión hace que la temperatura del filamento, y por lo tanto su
resistencia eléctrica, cambie y que el puente se desbalancee.

Capítulo I Alto Vacío
36
G
Fig. 1.17 Esquema mostrando como el filamento del Pirani forma parte de un puente
Wheastone
Si un circuito de control actúa rápidamente para adaptar la potencia del
calentamiento aplicado al filamento y mantener balanceado el puente se dice que el
vacuómetro Pirani está siendo operado en el modo de temperatura constante.
También se puede operar el vacuómetro alimentándolo con voltaje constante
o corriente constante y calibrar la presión en función de la corriente a través de G (Fig.
1.17) que indica el desbalance del puente Wheastone. El modo de temperatura constante
es más preciso porque las pérdidas por efecto de bordes o por radiación son constantes,
sin embargo, el modo de voltaje constante es más sencillo porque una vez que el puente
es balanceado a una presión conocida no se necesita volver a balancearlo para cada
valor de presión.
Vacuómetro TC
En un vacuómetro térmico TC una termocupla es soldada en el centro del filamento. El
filamento se calienta con corriente constante y la variación de la temperatura es una
medida de la conductividad térmica del gas residual y por lo tanto de la presión de la
cámara. Usualmente un microamperímetro de resistencia pequeña funciona como
voltímetro.

Capítulo I Alto Vacío
37
I.3.2. VACUOMETROS DE IONIZACION
El principio de operación de estos instrumentos consiste en inducir la
ionización de una fracción de las moléculas del gas residual y establecer una corriente
de moléculas ionizadas. La presión de la cámara se mide entonces indirectamente a
través de la corriente iónica. Discutiremos dos tipos de vacuómetros de ionización: El
vacuómetro de cátodo caliente y el vacuómetro de cátodo frío o Penning.
Vacuómetro de ionización de cátodo caliente
Un filamento de tungsteno se calienta para lograr la emisión de electrones
por efecto termoiónico. El filamento está rodeado por una rejilla metálica a potencial
positivo (100 - 200 V) respecto al filamento de modo que los electrones emitidos por
éste son acelerados radialmente alejándose del filamento. Los electrones chocan con
moléculas del gas residual y lo ionizan. Se establece entonces una corriente de iones
positivos hacia el filamento. Algunos electrones pasan a través de la rejilla pero son
acelerados por el colector, un tercer electrodo a potencial más negativo que el cátodo
(fig. 1.18).
Este tipo de instrumentos es útil en el rango de 1 a 10-12 Pa. Tiene que
hacerse una calibración para cada tipo de gas.

Capítulo I Alto Vacío
38
Fig. 1.18 a) esquema de un vacuómetro de cátodo caliente : 1. Filamento, 2. rejilla, 3. colector,
4. conexión a fuente para calentamiento del cátodo. b) Circuito de control y medida.
Vacuómetro de ionización de cátodo frío (Penning)
El proceso de ionización en una región de gas enrarecido se puede iniciar
sin necesidad de un filamento caliente si se establece un campo eléctrico
suficientemente alto. El proceso se realza colocando un campo magnético paralelo al
campo eléctrico. Este es el principio de funcionamiento de los vacuómetros de cátodo
frío más conocidos como Penning. La figura 1.19 es un esquema del vacuómetro
Penning. Estos instrumentos pueden medir presiones tan bajas como 10-10 Pa.
2000 V
1
2
3
3
mA
4
a la cámara de vacío
Fig. 1.19. Esquema de un vacuómetro Penning: (1) ánodo en forma de anillo, (2) cátodo
cilíndrico, (3) imanes, (4) chasis de material aislante.
mA
amplificador
A
30 V
200 V
1
2 3
( a )
( b )

Capítulo I Alto Vacío
39
REFERENCIAS
[1] “Fundamental University Physics”, Vol. III, M. Alonso y E. J. Finn, Addison-
Wesley, 1968.
[2] “Mechanics, wave motion and heat”, F. W. Sears, Addison-Wesley, 1958.
[3] “Vacuum Technology”, A. Roth, North Holland, 1976.
[4] “A User´s guide to vacuum technology. J. F. O`Haulon.
[5] M. von Smolochowsky, Ann. Phys., 33, 386 (1911).
[6] M. Knudsen, Ann. Phys., 28, 75 (1909); 35, 389 (1911).
[7] “The kinetic theory of gases”, L. B. Loeb, Mc. Graw-Hill, 1934.
[8] “The material science of thin films” M. Ohring, Academic Press, Inc., 1991.
[9] Manual Leybold

Capítulo II Técnicas de producción
40
CAPITULO II
TECNICAS DE PRODUCCION
Los materiales producidos por técnicas de alto vacío son obtenidos en
forma de películas delgadas sobre superficies de cuerpos sólidos. Estas películas tienen
espesores del orden de 100 Ǻ a 100 m . Los dispositivos que se producen por estas
técnicas están constituidos generalmente por sucesivas capas de diferentes materiales.
El principio básico para producir materiales por técnicas de alto vacío
consiste en la vaporización de un sólido en un ambiente enrarecido. Según que la
vaporización sea causada por medios físicos o por medios químicos se habla de
técnicas PVD (Physical Vapor Deposition) o técnicas CVD (Chemical Vapor
Deposition), respectivamente. En este libro tratamos sólo las técnicas en que la
vaporización que da origen a los nuevos materiales es producido por medios físicos.
Las técnicas PVD son básicamente dos: (1) Evaporación Térmica, en la cual la
vaporización es producida por transferencia de calor al sólido fuente y (2) Evaporación
Iónica, en la cual la vaporización es producida por transferencia de momentum de un
flujo de iones de alta energía al material fuente.
La obtención de películas delgadas por evaporación iónica fue reportada
por primera vez por W.R. Grover en 1852 [ref. 1] y por evaporación térmica por
Faraday en 1857 [ref. 2]. Hasta 1960 la evaporación térmica era ampliamente más
usada, pero en la actualidad debido a la gran variedad de materiales que se necesitan

Capítulo II Técnicas de producción
41
para aplicaciones en óptica, magnetismo, microelectrónica y otras disciplinas
científicas, las técnicas de vaporización iónica se han hecho más populares.
II.1. EVAPORACION TERMICA
La producción de películas delgadas por evaporación térmica en alto vacío
puede ser mejor explicada refiriéndonos al esquema de la figura 2.1. En una cámara de
alto vacío (Presión menor que 10-3 Pa) se colocan en forma adecuada: el material fuente
y el sólido (substrato) que se quiere cubrir con una película delgada. El material fuente
se coloca sobre un crisol que a su vez funciona como un resistor conectado a una fuente
eléctrica de alta corriente (del orden de 100 A) y bajo voltaje (10V). Cuando el material
fuente empieza a calentarse pero todavía no alcanza la temperatura de vaporización se
desprenden algunos vapores no deseados, por lo cual, es necesario mantener una placa
(puerta) que separe el substrato de la fuente. Cuando se alcanza la temperatura de
vaporización, se abre la puerta y se permite que el material se deposite sobre el
substrato.
A continuación discutimos, en las secciones II.1.1 y II.1.2 aspectos
relacionados a la vaporización de la fuente y en las secciones II.1.3 y II.1.4 aspectos
relacionados al depósito del material en el substrato. Luego, en la sección II.1.5
describimos las características de los crisoles, material auxiliar que se requiere para
vaporizar el material fuente, y en la sección II.1.6 presentamos la evaporación por haz
de electrones como una variante de la técnica de evaporación térmica.

Capítulo II Técnicas de producción
42
Fuente dealta corriente (100 A)
bombamecánica
bomba dedifusión
crisol
materialfuente
substrato
puerta
control dela puerta
Fig. 2.1 Esquema de un sistema de alto vacío para producir materiales de alta pureza por evaporación
térmica.
II.1.1. RAPIDEZ DE EVAPORACIÓN DE LA FUENTE
Imaginemos un trozo de material condensado en equilibrio con su vapor. En
estas condiciones la rapidez con que se produce el vapor es igual al número de
partículas que se condensan sobre la superficie del material. Podemos admitir que el
número de partículas que se condensan por unidad de tiempo en una unidad de área está
dado aproximadamente por la rapidez de incidencia molecular (ec. 1.12). Si
bruscamente se extrajera el vapor que rodea a la fuente, reduciendo la presión
hidrostática a cero entonces la rapidez neta con que la fuente está produciendo vapor
estaría dada también por la ec. (1.12). Estos argumentos originales de Hertz permiten
entender la ecuación básica para la rapidez de vaporización de un sólido o líquido.
TM
PP heqe
)(1003,2 23
sm
moléculas2 ( 2.1)
donde α es un factor de eficiencia, Ph es la presión hidrostática que rodea a la fuente y
Peq es la presión de vapor del material, es decir, la presión del vapor (en Pa) a la cual se
mantiene en equilibrio termodinámico con el material condensado.

Capítulo II Técnicas de producción
43
Una ecuación equivalente a la anterior es la siguiente:
)(34.0 heqe PPTM
smgramos
2 (2.2)
Desde el punto de vista práctico, para determinar la presión de la cámara, Ph y la
temperatura T a la cual debe realizarse el proceso de evaporación térmica para un
material específico, es importante comprender la relación entre la presión de vapor de
cada material y la temperatura.
II.1.2 PRESION DE VAPOR Y TEMPERATURA
Para comprender cómo es que a una temperatura dada un determinado
material puede estar en equilibrio con su respectivo vapor a una presión específica (Peq)
es conveniente recordar los siguientes conceptos termodinámicos:
(a) Energía libre de Gibbs, que se define en términos de la entalpía, la entropía y la
temperatura, como:
TSHG . (2.3)
Su respectivo diferencial completo es:
VdPSdTdG (2.3a)
(b) Dos fases en equilibrio termodinámico tienen el mismo valor para la energía de
Gibbs:
ggcc TSHTSH (2.4)
dPVdTSdPVdTS ggcc (2.4a)
Estas ideas conducen a la siguiente ecuación
)( cg
cg
cg
cg
VVTHH
VVSS
dTdP

Capítulo II Técnicas de producción
44
conocida como la ecuación de Clausius Clapeyron y que se puede escribir en forma
aproximada:
g
eq
TVH
dTdP )(
Usando la ecuación del gas ideal para reemplazar Vg, tendremos:
2
)(TdT
RH
PdP qe (2.5)
cuya integración conduce a
constante)(
lnRTH
P qeeq (2.6)
Es necesario hacer algunas correcciones a esta ecuación para tener en cuenta
que la entalpía depende a su vez de la temperatura. Por ejemplo para el aluminio se
obtiene:
T ,- T , -, /T , P -eq
610 10523log99905261499315log .
La figura 2.2 muestra esta dependencia de la presión de vapor respecto de la
temperatura para algunos materiales.
Log
10P
resió
n d
e v
apor
(Pa)
104/T (K -1)
5 10 15 20
5
-5
3
1
-1
-3
1000 5002000
Temperatura (K)
AlW Pb Li Zn Hg
Fig. 2.2 Relación de presión de vapor (Peq) vs. temperatura para algunos materiales
[adaptada de ref. 3].

Capítulo II Técnicas de producción
45
Para ilustrar la importancia práctica de estas gráficas consideremos el caso del aluminio.
Si quisiéramos efectuar el proceso de evaporación a la presión atmosférica (105 Pa)
necesitaríamos calentar el material fuente a temperaturas mayores que 2200 K, pero si
la presión es de 10-5 Pa, la temperatura sería sólo del orden de 1000 K. Sin embargo a
estas presiones tan bajas, la rapidez de depósito es demasiado baja. Por lo cual es usual
depositar Al a presiones del orden de 1 Pa y temperaturas del orden de 1100 K.
II.1.3. GEOMETRIA Y CANTIDAD DE MATERIAL DEPOSITADO
La masa que se evapora del material fuente, a una temperatura determinada,
depende de la geometría de su superficie y del tiempo que dura el proceso, esto se puede
expresar en la forma:
see dAdtM
La masa depositada sobre una porción de área dAs del substrato depende de
la geometría de la fuente y de la posición relativa fuente-substrato. Aquí consideramos
dos fuentes posibles: (i) una fuente puntual como se muestra en la figura (2.3a) y
(ii) una fuente que llamaremos superficial o fuente de Knudsen, que consiste en un
recipiente cerrado de superficie isotérmica sobre la cual se ha hecho un pequeño
agujero, como se muestra en la figura 2.3b.
Para una fuente como la de la fig. 2.3a se supone que la masa Me evaporada
de la fuente se deposita uniformemente en una esfera de radio r. Por lo tanto sobre un
elemento de área dAc sobre la esfera se tendrá
24 rM
dAdM e
c
s .
La cantidad de material que se deposita sobre una superficie dAs que no
necesariamente se encuentra sobre la esfera es entonces:

Capítulo II Técnicas de producción
46
24 rCosM
dAdM e
s
s (2.7)
Para una fuente superficial, la teoría cinética de gases permite establecer:
2rCosCosM
dAdM e
s
s (2.8)
donde θ es el ángulo que la normal al substrato hace con la recta fuente-substrato y es
el ángulo que la normal a la fuente hace con la recta fuente substrato.
Para fuentes en crisoles que contienen una piscina de material fundido
podemos usar en forma aproximada la ec. (2.8).
θ
(a) (b)
dAs
dAsdAc
θ
Fig. 2.3 Esquemas para discutir la cantidad de material depositada en función de la disposición
geométrica de la fuente y substrato: (a) fuente puntual, (b) fuente superficial.
II.1.4. ESPESOR Y UNIFORMIDAD DE LAS PELICULAS DELGADAS
En algunas aplicaciones, como en el caso de recubrimientos duros para
prolongar el tiempo de vida de herramientas de corte, la uniformidad es deseable pero
no es necesaria. En cambio, en otras aplicaciones, como por ejemplo en la fabricación
de filtros ópticos Fabry Perot, se requiere que el espesor de las películas se mantenga
constante con un error máximo de 1% sobre toda la superficie del substrato. En esta

Capítulo II Técnicas de producción
47
sección discutimos las consideraciones geométricas que deben ser tenidas en cuenta
para obtener espesores uniformes.
La masa depositada, dMs, sobre un elemento infinitesimal de área, dAs,
sobre el substrato se puede escribir en la forma
tdAdM ss (2.9)
donde es la densidad del nuevo material y t es el espesor de la película. Combinando
esta ecuación con la ec. (2.7) y teniendo en cuenta la geometría de la figura 2.4 se
obtiene:
2/32232 )(444 hhM
rhM
rCosMt eee .
Observamos que el espesor es máximo cuando = 0 y su valor es:
20 4 hMt e
Tomando este valor como referencia, obtenemos la ec. (2.10) para el espesor
en cualquier otro punto del substrato.
hr
substrato
Fig.2.4 Espesor vs. parámetros y h para fuentes puntuales.
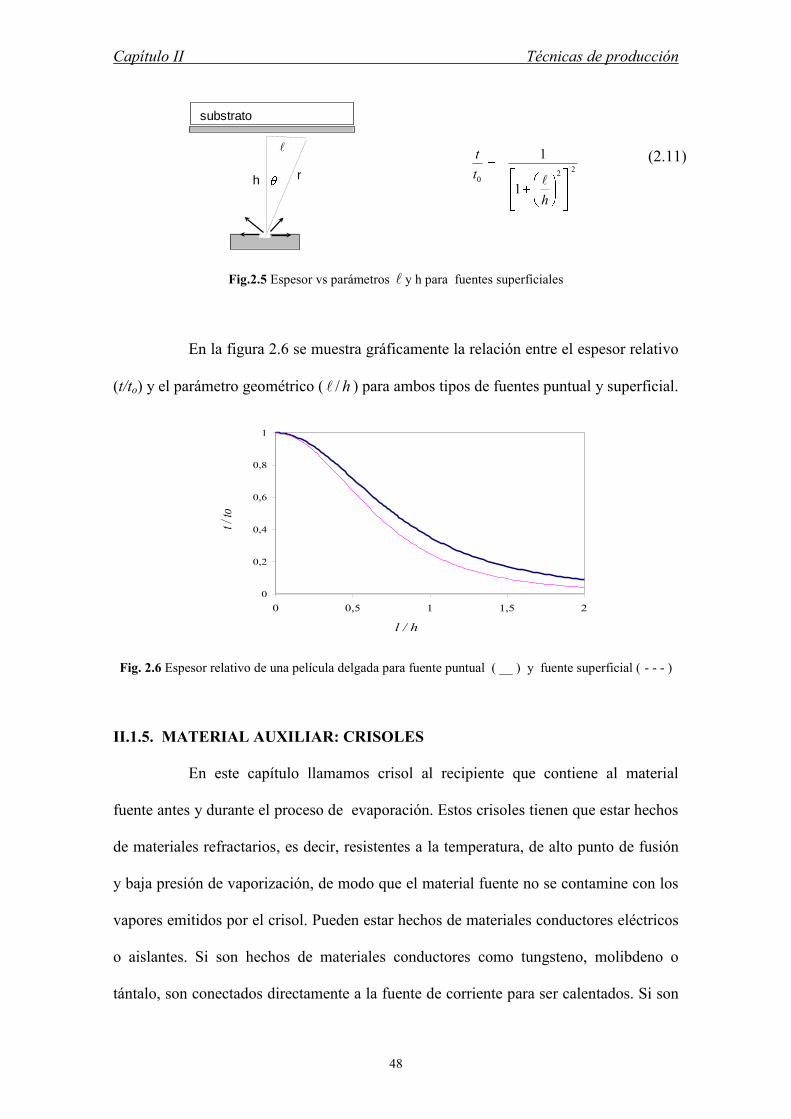
Similarmente, para una fuente superficial puede obtenerse la ec. (2.11) que
relaciona el espesor de la película y los parámetros geométricos y h de la figura 2.5.
2/320
1
1
h
tt
(2.10)
Capítulo II Técnicas de producción
48
substrato
h
r
Fig.2.5 Espesor vs parámetros y h para fuentes superficiales
En la figura 2.6 se muestra gráficamente la relación entre el espesor relativo
(t/to) y el parámetro geométrico ( h/ ) para ambos tipos de fuentes puntual y superficial.
0
0,2
0,4
0,6
0,8
1
0 0,5 1 1,5 2
l / h
t / to
Fig. 2.6 Espesor relativo de una película delgada para fuente puntual ( __ ) y fuente superficial ( - - - )
II.1.5. MATERIAL AUXILIAR: CRISOLES
En este capítulo llamamos crisol al recipiente que contiene al material
fuente antes y durante el proceso de evaporación. Estos crisoles tienen que estar hechos
de materiales refractarios, es decir, resistentes a la temperatura, de alto punto de fusión
y baja presión de vaporización, de modo que el material fuente no se contamine con los
vapores emitidos por el crisol. Pueden estar hechos de materiales conductores eléctricos
o aislantes. Si son hechos de materiales conductores como tungsteno, molibdeno o
tántalo, son conectados directamente a la fuente de corriente para ser calentados. Si son
220
1
1
h
tt
(2.11)

Capítulo II Técnicas de producción
49
hechos de materiales aislantes como alúmina, por ejemplo, son calentados externamente
por un filamento conductor. La figura 2.7 muestra diferentes formas de crisoles
requeridos en la práctica y que pueden ser obtenidos comercialmente.
Fig 2.7 Diferentes tipos de crisoles o fuentes de evaporación térmica disponibles comercialmente [ref. 5].
II.1.6. EVAPORACIÓN POR HAZ DE ELECTRONES
Cuando se desea evaporar materiales de alto punto de fusión como por
ejemplo tungsteno, es imposible transferir el calor necesario desde un crisol sin que el
material del crisol mismo se evapore y contamine al material que se quiere producir.
Una alternativa para evitar esta desventaja de la evaporación térmica es calentar al
material fuente con un haz de electrones. El principio de funcionamiento de la
evaporación por haz de electrones se ilustra en la figura 2.8. Un filamento de tungsteno,
o LaB6, emite electrones por efecto termoiónico, los cuales son acelerados por un alto

Capítulo II Técnicas de producción
50
potencial y enfocados con ayuda de lentes magnéticas a un punto escogido dentro del
material fuente.
filamento caliente
20 kV
ánodosubstrato
haz de electrones
recipiente del material fuente
material fuente lente
magnética
Fig. 2.8 Principio de funcionamiento de la técnica de evaporación por haz de electrones.
El material fuente se calienta sólo puntualmente. Por ejemplo si el material
fuente es tungsteno puntualmente puede alcanzar 3000ºC, pero el recipiente que lo
contiene puede estar a 35ºC.
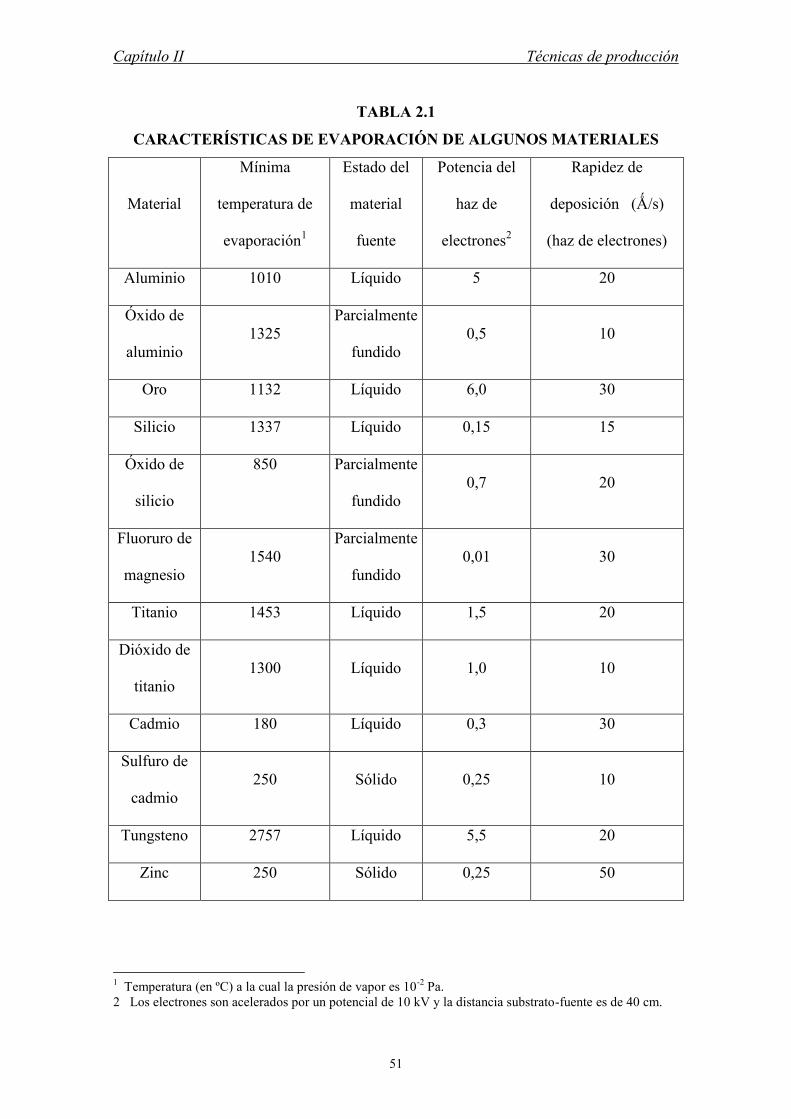
En la tabla 2.1 mostramos algunas características de evaporación de algunos
materiales por ambas técnicas evaporación térmica y por haz de electrones. Los datos
de evaporación por haz de electrones se refieren a una fuente de 10 kV.
Capítulo II Técnicas de producción
51
TABLA 2.1
CARACTERÍSTICAS DE EVAPORACIÓN DE ALGUNOS MATERIALES
Material
Mínima
temperatura de
evaporación1
Estado del
material
fuente
Potencia del
haz de
electrones2
Rapidez de
deposición (Ǻ/s)
(haz de electrones)
Aluminio 1010 Líquido 5 20
Óxido de
aluminio 1325
Parcialmente
fundido 0,5 10
Oro 1132 Líquido 6,0 30
Silicio 1337 Líquido 0,15 15
Óxido de
silicio
850
Parcialmente
fundido 0,7 20
Fluoruro de
magnesio 1540
Parcialmente
fundido 0,01 30
Titanio 1453 Líquido 1,5 20
Dióxido de
titanio 1300 Líquido 1,0 10
Cadmio 180 Líquido 0,3 30
Sulfuro de
cadmio 250 Sólido 0,25 10
Tungsteno 2757 Líquido 5,5 20
Zinc 250 Sólido 0,25 50
1 Temperatura (en ºC) a la cual la presión de vapor es 10-2 Pa. 2 Los electrones son acelerados por un potencial de 10 kV y la distancia substrato-fuente es de 40 cm.

Capítulo II Técnicas de producción
52
II.2. EVAPORACION IONICA (*)
Para explicar el fundamento de la técnica de evaporación iónica nos
referimos al esquema de la figura 2.9. En una cámara de vacío donde previamente se
han obtenido presiones del orden de 10-3 Pa o menos se introduce controladamente un
gas inerte (generalmente argón) hasta una presión del orden de 10-1 a 1 Pa. Se establece
luego un campo eléctrico con ayuda de una fuente de alto voltaje de 500 a 3000 V y
baja corriente (100 a 500 mA). El campo eléctrico establecido ioniza los átomos de
argón y estos iones son acelerados hacia el cátodo (blanco o target). Como consecuencia
de este impacto los átomos del blanco son extraídos y el vapor obtenido se mezcla con
el gas inerte. El vapor proveniente del blanco se condensa en cualquier superficie sólida
dentro de la cámara, en particular, en la superficie del sólido (substrato) que se quiere
recubrir.
+++
ARGON
bomba mecánica
bombaturbo-molecular
blanco
substrato fuente0 a 1000 V
-
Fig. 2. 9 Esquema para explicar el principio de la técnica de evaporación iónica para producir materiales
y dispositivos modernos.
(*) En la literatura internacional, en inglés, esta técnica es conocida como sputtering, sin embargo, he creído conveniente traducirlo de esta manera por dos razones: (i) porque sputtering no tiene traducción literal en español y (ii) porque el nombre evaporación iónica es más indicativo de lo que sucede en esta técnica.

Capítulo II Técnicas de producción
53
En la sección II.2.1 describimos brevemente los procesos que ocurren
cuando un ion interacciona con la superficie del blanco o target. En la sección II.2.2
discutimos el concepto de eficiencia iónica .En la sección II.2.3 describimos el ambiente
en el cual se realiza el proceso, esto es, la descarga luminosa. En las secciones II.2.4 a
II.2.7 discutimos las diversas variantes de esta técnica tales como evaporación iónica dc,
evaporación iónica rf , magneto evaporación iónica y evaporación iónica reactiva.
II.2.1. INTERACCION ION – SUPERFICIE
Muchos fenómenos pueden ocurrir cuando un ion incide sobre la superficie
de un sólido (el blanco). La figura 2.10 ilustra algunos de estos fenómenos. Todos no
ocurren a la vez. ¿Cuáles suceden? depende de la energía y la masa de los iones así
como de los átomos y la estructura del sólido. Desde el punto de vista de producción de
películas delgadas el fenómeno que nos interesa es la extracción de átomos del blanco.
Un ion incidente de alta energía al chocar con el sólido produce una colisión en cascada
y el resultado es que uno o más átomos (o iones) del blanco son liberados de la
superficie. Algunos de estos átomos forman parte del vapor que luego se va a condensar
en el substrato. Otros son retroacelerados y se vuelven a depositar sobre el blanco.
fotón
ion incidentede alta energía
iones reflejados
colisión en cascada
átomos extraídos
átomos retro- acelerados
implantación de iones
superficie del blanco
Fig. 2.10 Esquema para explicar algunos de los resultados de la interacción de un ión con la superficie
del blanco.

Capítulo II Técnicas de producción
54
Respecto a los iones incidentes puede suceder que estos sean reflejados o
que queden incrustados en el target. A este último fenómeno se le conoce como
implantación iónica. Ambos fenómenos tienen aplicaciones en otros campos de la
tecnología de producción de materiales. Por ejemplo la implantación iónica es muy
usado para introducir impurezas tipo p o tipo n en cristales de silicio. El cambio de
energía cuando un haz de iones de helio incide y se refleja en un sólido da información
sobre la masa atómica de los átomos que constituyen el sólido. Esto constituye la
técnica de análisis de materiales conocida como Rutherford Back Scattering.
Electrones y fotones son también obtenidos como sub-productos de la
interacción ion superficie. Los electrones son conocidos como electrones secundarios y
los fotones generalmente son rayos X. Defectos de la red puede ser otra consecuencia
de la interacción ion-superficie.
II.2.2 EFICIENCIA IONICA (*)
La eficiencia iónica (Y) se define como el número de átomos del blanco
que en promedio son extraídos por cada ion incidente. Su valor depende de la energía
del ion incidente (E1) , de la energía de enlace de los átomos del blanco (Eb), de la masa
atómica del ion incidente (M1) y de la masa atómica de los átomos del blanco (M2).
Sigmund en 1969 [ref. 5] desarrolló dos fórmulas para el cálculo de la eficiencia
iónica:
bE
EMMMMY 1
221
212 )(
443 ( )11 keVE (2.12a)
b
n
EES
MMM
ZZZZY
)(56,3
21
13/2
23/2
1
21 keVE 11 (2.12b)
(*) Esta es una traducción de la cantidad conocida en la literatura internacional, en inglés, como sputter yield

Capítulo II Técnicas de producción
55
Estas ecuaciones dependen de dos parámetros complejos y Sn (E). El
parámetro es una medida de la eficiencia de la transferencia de momentum en las
colisiones. Aumenta monótonamente de 0.17 a 1.4 cuando M1/ M2 aumenta de 0,1 a 10.
El parámetro Sn(E) es una medida de la pérdida de energía por unidad de longitud
debido a las colisiones nucleares.
En la tabla 2.2 se muestra la eficiencia iónica de iones de He con energía de
0,5 keV y iones de Ar con 0,5 y 1 keV para extraer iones de superficies sólidas de
algunos metales.
TABLA 2.2
Eficiencia iónica (número de átomos / ion) de He+ y Ar+ para extraer algunos metales
[ref. 4]
Metal He +
(0,5 keV)
Ar +
(0,5 keV)
Ar +
(1 keV)
Ag 0,20 3,12 3,8
Al 0,16 1,05 1,0
Au 0,07 2,40 3,6
Ti 0,07 0,51
Si 0,13 0,50 0,6
C 0,07 0,12
Cu 0,24 2,35 2,85
W 0,01 0,57
II.2.3 CONCEPTOS DE DESCARGA LUMINOSA Y PLASMA
Descarga luminosa es el fenómeno que se produce en la cámara
simultáneamente al proceso de evaporación iónica. Cuando los átomos del gas inerte
(argón) son sometidos a un campo eléctrico, se ionizan y son acelerados hacia el
blanco del cual extraen átomos, obteniéndose así, una mezcla de: iones y átomos
neutros de argón, electrones, átomos neutros e iones del material del blanco. Además,

Capítulo II Técnicas de producción
56
en esta situación se producen choques entre las diferentes partículas mencionadas, lo
cual hace que se produzcan una serie de transiciones electrónicas entre diferentes
niveles atómicos, razón por la cual (asociada con la corriente iónica) hay radiación
electromagnética en casi todo el espectro electromagnético desde infrarrojo hasta
rayos X, en particular en el visible. Esta mezcla de átomos, iones y electrones que
emite radiación electromagnética en un amplio rango del espectro con un máximo de
intensidad en una determinada frecuencia del visible es lo que se conoce como descarga
lumiosa (glow discharge).
Por otro lado, B. Chapman [ref. 4] define plasma de la siguiente manera:
“Un gas parcialmente ionizado que consiste de igual número de cargas positivas y
negativas, y un número diferente de moléculas neutras no ionizadas”. Podemos decir
entonces que el fenómeno de descarga luminosa es un caso particular de plasma y que el
ambiente en el cual se realiza el proceso de evaporación iónica es un plasma o una
descarga luminosa. En este tipo de plasmas, el grado de ionización es típicamente de
10-4, de modo que el plasma consiste mayoritariamente de partículas neutras. La figura
2.11 muestra una vista del plasma, a través de una ventana de la cámara, durante la
producción de materiales por la técnica de evaporación iónica, en el laboratorio de
Física de la Universidad Nacional de Ingeniería.
Fig. 2.11 Vista del plasma, a través de una ventana en la cámara, durante la producción
de materiales por técnica de evaporación iónica, en la UNI.

Capítulo II Técnicas de producción
57
II.2.4. EVAPORACION IONICA DC
Es el proceso que se obtiene cuando la fuente que produce el campo
eléctrico que ioniza el gas inerte y sostiene el plasma es una fuente de corriente contínua
(corriente directa).
Nos proponemos discutir dos aspectos de este proceso: (1) cuáles son las
características básicas de la descarga luminosa durante este proceso y (2) cómo escoger
los parámetros del plasma: presión y corriente para obtener una rapidez de deposición
apreciable.
En general durante una descarga luminosa dc, cuando sólo intervienen dos
electrodos en un ambiente de gas enrarecido, se distinguen siete regiones: (i) brillo del
cátodo, (ii) zona obscura de Crookes, (iii) zona brillante negativa, (iv) zona obscura de
Faraday, (v) zona brillante positiva, (vi) espacio obscuro del ánodo y (vi) brillo del
ánodo. La figura 2.12a muestra un esquema de estas regiones y la figura 2.12 b muestra
como varía el potencial dentro del plasma.
cátodo brillo del cátodo
zona de Crookes
brillo negativo
zona de Faraday
2000 V
brillo positivo brillo del ánodo
zona obscura del ánodo
V
-2000 V
(a)
(b)
0
substrato
Fig 2.12 . (a) Esquema para mostrar las regiones dentro de una descarga luminosa dc. (b) Variación del
potencial con la posición dentro del plasma. Obsérvese que sólo hay campo eléctrico apreciable cerca al
cátodo y cerca al ánodo. [adaptada de la ref. 4]

Capítulo II Técnicas de producción
58
En un experimento de evaporación iónica dc el blanco es el cátodo y el
substrato se coloca en la zona brillante negativa, cerca de la zona obscura de Faraday.
Para tener una idea de cómo escoger los parámetros de corriente y presión
durante un experimento de evaporación iónica conviene considerar la figura 2.13 y los
siguientes argumentos: La rapidez de deposición depende de la potencia disipada por el
plasma, es decir, es proporcional al cuadrado de la corriente. Para un voltaje fijo, y a
bajas presiones la corriente es pequeña por que la trayectoria libre media de los
electrones antes de ionizar un átomo de argón es grande. La corriente aumenta al
aumentar la presión. Sin embargo, al aumentar la presión demasiado también aumenta el
número de colisiones que experimenta un átomo que sale del blanco antes de llegar al
substrato, razón por la cual la eficiencia iónica disminuye. Estas tendencias opuestas
son mostradas en la figura 2.13 y se muestran las condiciones óptimas de operación.
corr
iente
(m
A)
20
40
60
80
100
120
140
3 6 9 12 15 18 21
Presión de argón ( Pa)
efi
cie
ncia
ió
nic
a y
rap
idez d
e d
ep
osic
ión
rela
tiva
0,2
0,4
0,6
0,8
1,0
típicas condiciones de
evaporación iónica
curv
a A
curva B
curva C
Fig. 2.13 Influencia de la presión de trabajo y de la corriente de la descarga luminosa sobre la rapidez de
deposición en un proceso de evaporación iónica dc. Curva A: presión vs corriente. Curva B: presión vs.
efisiencia iónica relativa. Curva C: rapidez de deposición relativa vs presión. [adaptada de la ref. 4]

Capítulo II Técnicas de producción
59
II.2.5. EVAPORACION IONICA RF
En este proceso la única variante con respecto a la evaporación iónica dc es
que la fuente que sostiene el plasma es una fuente de corriente alterna (ver figura 2.14).
La frecuencia convencional para este tipo de aplicaciones está en el rango de
radiofrecuencia, 13,56 MHz. Es especialmente adecuado para producir evaporación de
materiales no conductores eléctricos, pero también se usa para materiales conductores.
Si intentáramos efectuar la evaporación iónica dc a un material aislante, la incidencia
de iones positivos hace que rápidamente el blanco se cargue positivamente y el proceso
de extracción de átomos del blanco dura sólo algunos segundos. En un proceso normal
dc las corrientes son del orden de 1 mA/cm2, el espesor del blanco es del orden de
0,1 cm y el voltaje a través del blanco del orden de 100 V. Un cálculo sencillo, 100 V =
J1,0 , permite concluir que es imposible el proceso de evaporación iónica dc de
materiales con resistividades eléctricas del orden de 106 cm o mayor.
La figura 2.14 muestra la comparación de un sistema de evaporación iónica
dc y uno de rf.
Fig. 2.14 Comparación entre un sistema de evaporación iónica dc y uno rf.

Capítulo II Técnicas de producción
60
Una pregunta interesante es ¿en un plasma alimentado con corriente alterna,
los iones positivos son acelerados al blanco sólo durante un semiperíodo? La respuesta
es no. Los iones positivos son continuamente acelerados hacia la superficie del blanco.
La razón es que debido a la diferencia entre las velocidades promedios de los electrones
(portadores ligeros) y de los iones (portadores pesados), el blanco se polariza
negativamente y el sistema se comporta como en la evaporación iónica dc. Para
comprender mejor esta polarización del blanco consideremos el circuito de la descarga
luminosa en la figura 2.15a. Sea Va el potencial alterno de la fuente y veamos como
varía el potencial Vb en la superficie del blanco. Comencemos considerando que el
plasma es alimentada con una fuente de onda cuadrada y que en t = 0 el capacitor está
descargado. Cuando Va va a –1000 V, Vb lo sigue. Empieza la descarga, el blanco se
comienza a cargar positivamente, su potencial aumenta a –800 V. Cuando Va aumenta
en 2 kV, Vb aumenta a 1200 V. El blanco positivamente cargado atrae electrones muy
rápidamente de modo que Vb llega por ejemplo a –100 V. Cuando Va cambia de signo
Vb cambia a – 1900V. Después de unos pocos ciclos se puede decir que el potencial de
Vb, en la superficie del blanco es siempre negativa. En las figuras 2.15 queda ilustrada
esta situación.

Capítulo II Técnicas de producción
61
Va
Vb
(a)
Fig. 2.15 (a) Esquema de uncircuito de una descarga luminosa rf, (b) Potencial de una fuente de onda
cuadrada, (c) Potencial en la superficie del target del cuando la fuente es de onda cuadrada, (d) voltaje
alterno (e) Potencial en lasuperficie del target cuando la fuente es sinusoidal. [adaptada de la ref. 4]

Capítulo II Técnicas de producción
62
II.2.6. MAGNETO EVAPORACION IONICA
Si a los sistemas de evaporación iónica representados en la figura 2.14
agregamos detrás del cátodo un sistema de imanes que nos permitan obtener un campo
magnético como se representa en la figura 2.16, diremos que tenemos un sistema de
magneto evaporación iónica dc o magneto evaporación iónica rf, respectivamente.
entrada de argón
bombas de vacío
V (DC)
entrada de argón
bombas de vacío
blanco
substratos
aislante
DC RF
SN N SN N
B B
imanes
Fig. 2.16 Magneto evaporación iónica dc y magneto evaporación iónica rf.
El efecto del campo magnético es cambiar la trayectoria de los electrones
del plasma, aumentar su recorrido libre medio y tiempo de permanencia en el plasma.
Ya no irán en línea recta hacia el ánodo (substratos) sino que se moverán en trayectorias
helicoidales muy cerca al blanco, lo cual trae dos consecuencias prácticas positivas: (1)
Disminuye el calentamiento del substrato porque será menos golpeado por los
electrones, (2) aumenta la rapidez de depósito, porque al permanecer más tiempo los
electrones en el plasma experimentan más choques con átomos neutros de argón,
multiplicando tanto el número de iones positivos como el número de electrones. Es
posible entonces obtener corrientes más altas a menores presiones, lo cual implica
menos choques de los átomos extraídos del blanco con iones o átomos de argón.

Capítulo II Técnicas de producción
63
II.2.7. EVAPORACION IONICA REACTIVA
Si en los sistemas de evaporación iónica 2.14 ó 2.16 permitimos que junto
con el argón ingrese a la cámara un gas reactivo como oxígeno o nitrógeno de modo que
el material que se deposite sobre el substrato no es el mismo del blanco sino el
resultante de una reacción química entre los átomos del blanco y las moléculas del gas
reactivo entonces diremos que estamos usando la técnica de evaporación iónica reactiva
dc o rf, o magneto evaporación iónica dc o rf.
Los materiales más cómunes que se producen por evaporación iónica
reactiva son óxidos y nitruros. En nuestro laboratrio por ejemplo, hemos producido
nitruro de titanio usando un blanco de titanio e introduciendo en la cámara una mezcla
de gases de 15% de N2 y 85 % de Ar. También hemos producido óxido de vanadio y
óxido de titanio, introduciendo en la cámara 10% de O2 y 90 % de Ar. También es
posible obtener carburos (TiC, WC, SiC) introduciendo gases tales como metano,
propano o acetileno junto con el argón. Se ha reportado también la producción de
sulfuros (CdS, CuS, ZnS) introduciendo junto con el argón gas H2S (ácido sulfhídrico).
Otros materiales posibles de obtener con esta técnica son oxinitruros y oxicarburos de
titanio, aluminio, silicio, etc.
En todos los casos es posible obtener materiales con estequiometría
variable, por ejemplo es posible obtener Ta Nx con valores de x desde 0,01 hasta 1. Esto
se logra controlando el flujo de gas reactivo hacia la cámara.
II.2.8 EVAPORACION IONICA CON POLARIZACION DEL SUSTRATO
Usualmente los substratos están en contacto eléctrico con el chasis de la
cámara, es decir, a tierra. Sin embargo se ha observado que, para muchas aplicaciones,
se obtienen películas de mejor calidad cuando el substrato también es conectado a un

Capítulo II Técnicas de producción
64
potencial negativo. Esto lo hemos observado en nuestro laboratorio en el caso de
recubrimientos duros de nitruro de titanio sobre acero. La dureza del material obtenido
mejora notablemente cuando se polariza el substrato. Si se trata sólo de evaporación
iónica dc es usual usar - 2000 V como potencial del blanco y –100 V como potencial de
los substratos. Sin embargo en magneto evaporación iónica –50 V es un buen valor de
voltaje polarización para el substrato. La explicación cualitativa para esta mejora es que
el voltaje de polarización del substrato acelera algunos iones de argón hacia el substrato
y extrae los átomos más débilmente adheridos, quedando sólo los que fueron adheridos
con mayor energía cinética.
Hay dos maneras de conectar un voltaje de polarización al substrato como se
muestra en las figura 2.17. Se puede usar un divisor de voltaje y la misma fuente que
polariza el blanco o una segunda fuente especial para polarizar los substratos.
entrada de gas
conexión a las bombas de vacío
entrada de gas
conexión a las bombas de vacío
(a) (b)
2 kV 2 kV
100 V
Fig. 2.17 Dos formas de polarizar el substrato: (a) con divisor de voltaje, (b) con otra fuente.

Capítulo II Técnicas de producción
65
TABLA 2.3
ALGUNAS APLICACIONES DE MATERIALES PRODUCIDOS POR
EVAPORACIÓN IONICA
AREA DE APLICACION USO ESPECÍFICO MATERIALES USADOS
Recubrimientos protectores
Antidesgaste
Anticorrosión
lubricación solida
barrera térmica
TiN, TiC, TiCN, TiAlN,…
TiN, BN, CrN, TiB,..
TiN, TiC, ....
ZrO2
Medicina
biocompatibilidad para
insertos quirúrgicos
lentes de contacto e
intraoculares
TiN,TiO2
Polímeros
Recubrimientos decorativos
Utensilios de cocina
Pulseras para relojes
Bijouterie/joyeria
TiN, TiCN, TiO2…
TiN, TiCN…
TiN, TiCN…
Recubrimientos ópticos
Antirraya transparente
para lentes orgánicos
Filtros ópticos
Decorativos para vidrios
de edificios
SiO2
Dieléctricos (MgF, Al2O3)
Matales. Dieléctricos
Microelectrónica
Contactos eléctricos
para circuitos integrados
Memorias magnéticas
Circuitos integrados
(Pd-Si)
aleaciones de tierras raras
ataque químico seco.
Microbaterías
Cátodos
Ánodos
Sólidos electrolitos
LiCoO2, V2O5
Grafito
LiPON
Electrocromismo Materiales
electrocrómicos
WO3

Capítulo II Técnicas de producción
66
II.3. SISTEMAS DE ALTO VACIÓ EN LA UNI
En el Laboratorio de Física de la Universidad Nacional de Ingeniería
contamos con tres sistemas de alto vacío que son utilizados para la producción a nivel
experimental de materiales por la técnica de evaporación iónica. Aunque fácilmente
pueden ser acondicionados para producir materiales por evaporación térmica o por
técnicas híbridas.
SISTEMA 1 Se puede ver una fotografía en la figura 2.18. Es un sistema comercial
UNIVEX 300 de Leybold. Está constituido por:
- Bomba mecánica de paletas rotatorias modelo Trivac D8B
- Bomba turbomolecular modelo Turbovac 151
- Fuente para bomba turbomolecular Turbotronik NT 150
- Cámara de acero inoxidable de 30 cm de diámetro por 30 cm de altura
- Fuente de radiofrecuencia de 0 a 300W, modelo Cesar
- Vacuómetros Pirani y Penning controlados por un solo panel electrónico
- Cátodo con imanes incorporados para magneto evaporación iónica con blancos de
75 mm de diámetro
Fig 2.18 Sistema de alto vacío UNIVEX 300.

Capítulo II Técnicas de producción
67
SISTEMA 2 Es un sistema diseñado en la Universidad de Hamburgo, Alemania, y
donado recientemente a nuestra Universidad, donde le hemos hecho algunas
modificaciones. Esta constituido por:
- Bomba mecánica de paletas rotatorias modelo Trivac D16B de Leybold
- Bomba turbomolecular modelo Turbovac 151
- Fuente para bomba turbomolecular Turbotronik NT 150
- Cámara de acero inoxidable de 30 cm de diámetro por 30 cm de altura
- Fuente de radiofrecuencia de 0 a 500W, Hüttinger IS 0,5 / 13560
- Vacuómetros Pirani y Penning con sus respectivas fuentes y controles electrónicos
- 2 Cátodos para evaporación iónica dc o rf con blancos de 75 mm de diámetro.
Con este sistema es posible hacer evaporación iónica desde dos blancos
simultáneamente para producir materiales compuestos. O, también es posible producir
multicapas de materiales deferentes sin abrir la cámara, lo cual es importante en la
producción de dispositivos. Una vista de este sistema se muestra en la figura 2.19.
Fig. 2.19 Sistema de alto vacío Nro. 2 del Laboratorio de Física de la UNI.

Capítulo II Técnicas de producción
68
SISTEMA 3. Cronológicamente es el primer sistema que se montó en la UNI para
producir materiales por la técnica evaporación iónica dc (figura 2.20). Muchas de las
piezas que lo constituyen fueron construidos o adaptadas en nuestros talleres. Consta de:
- Bomba mecánica de paletas rotatorias modelo Trivac D4B de Leybold
- Bomba turbomolecular modelo Pfeiffer y su respectiva fuente
- Cámara de acero inoxidable de 30 cm de diámetro por 30 cm de altura
- Fuente de radiofrecuencia de 0 a 500W, Hüttinger IS 0,5 / 13560
- Vacuómetros Pirani y Penning con sus respectivas fuentes y controles electrónicos
- Cátodo para evaporación iónica dc o rf con blancos de 75 mm de diámetro.
Fig. 2.20 Sistema de alto vacío No. 3 del Laboratorio de Física de la UNI.

Capítulo II Técnicas de producción
69
REFERENCIAS
[1] W. R. Grove, Phil. Trans. Roy. Soc., London A 142, 87 (1852).
[2] M. Faraday, Phil. Trans. 147, 145 (1857)
[3] “The material Science of the thin films”, M. Ohring, Academic Press, Inc., 1991.
[4] “Glow Discharge Processes”, B. Chapman, Jhon Wiley & sons, 1980.
[5] Manual de compañía, R & D Mathis Company

70
CAPÍTULO III
TÉCNICAS DE CARACTERIZACIÓN
Cada vez que se produce un material nuevo, es conveniente verificar sus
características generales, concretamente, su morfología, su composición química y la
estructura cristalina. Dependiendo de la aplicación en algunos casos es conveniente evaluar
además propiedades más específicas como por ejemplo propiedades mecánicas tales como
microdureza, resistencia al desgaste por abrasión etc, así como también propiedades
electroquímicas o propiedades ópticas. En este capítulo sólo describimos los fundamentos de
las técnicas que nos permiten obtener las propiedades generales de los materiales. Estas son:
Microscopía electrónica de barrido que nos permite observar la morfología de los
materiales producidos, tamaño de grano, espesor etc.
EDS o espectroscopía por dispersión de energía, XPS o Espectroscopía por
fotoelectrones de rayos X, RBS o Retrodispersión Rutherford Para estudiar la composición
química de un material,
Para el estudio de la estructura cristalina hemos usado la Difracción de rayos X y
Para el estudio de las vibraciones de la red cristalina hemos usado algunas veces técnicas de
reflexión de radiación infrarroja.
3.1 MICROSCOPIA ELECTRONICA DE BARRIDO (MEB)
Esta técnica la hemos usado para obtener imágenes amplificadas de pequeñas
áreas, tanto de la superficie como de la sección transversal.
En el MEB, la imagen es formada por un haz muy fino de electrones que es
enfocada en la superficie del espécimen. El haz recorre la muestra en una serie de líneas
paralelas dentro de una área seleccionada (hace un barrido). El control para que el haz de
electrones ejecute dicho barrido es realizado por unas pequeñas bobinas. La figura 3.1
muestra un esquema del microscopio electrónico de barrido.

71
A los electrones que en un instante dado bombardean un espécimen pueden ocurrirle varias
cosas: Pueden ser elásticamente reflejados sin pérdida de energía; pueden ser absorbidos por
la muestra y dar lugar a electrones secundarios de muy baja energía y rayos X; también
pueden ser absorbidos y dar lugar a la emisión de luz visible (efecto conocido como cátodo
luminiscencia) y finalmente, pueden dar lugar a corrientes eléctricas dentro del espécimen.
Todos estos efectos pueden ser usados para producir una imagen. Por mucho, el fenómeno
más usado para producir la formación de imágenes es la producción de electrones
secundarios de baja energía.
Fig. 3.1 Esquema de un microscopio electrónico de barrido
Los electrones secundarios son selectivamente atraídos a una rejilla mantenida a
bajo voltaje positivo (50 voltios) con respecto a la muestra. Detrás de la rejilla hay un disco
mantenido a aproximadamente 10 kV positivos con respecto a la muestra. El disco consiste
de una capa de material centelleante cubierto con una capa delgada de aluminio. Los
electrones secundarios pasan a través de la grilla y golpean el disco causando la emisión de
luz del material centelleante. La luz es conducida a un tubo fotomultiplicador que convierte
los fotones en voltaje. La intensidad de este voltaje depende del número de electrones
secundarios que han golpeado el disco centelleante. Así, los electrones secundarios

72
producidos desde determinada área de la muestra dan lugar a una señal de voltaje de
intensidad particular. Esta señal es conducida fuera de la columna del microscopio a una
consola electrónica donde es procesada y amplificada para prodcir un punto brillante en la
pantalla de un tubo de rayos catódicos (o televisor). Una imagen es producida barriendo el
haz de electrones a través del espécimen en sincronía exacta con el barrido del haz de
electrones en el tubo de rayos catódicos.
El microscopio electrónico de barrido no tiene lentes objetivo, intermedia y
proyector para magnificar la imagen como en un microscopio óptico. En vez de esto, la
magnificación resulta de la razón del área barrida el área de la pantalla de televisión.
Aumentar la magnificación en un microscopio electrónico de barrido es entonces realizado en
forma muy simple, barriendo el haz de electrones sobre un área más pequeña del espécimen.
En las figuras 3.2 mostramos ejemplo de imágenes de una fractura producida en una película
de nitruro de titanio durante el proceso de indentación de bido a una falla en la adherencia
entre substrato y película.
Fig. 3.2 Imágenes de una fractura obtenidas por microscopía electrónica de barrido (micrografías
obtenidas por el autor en Laboratorio Åmstrong, Uppsala, Suecia).

73
3.2 ESPECTROSCOPIA POR DISPERSION DE ENERGIA (EDS)
Las siglas EDS que identifican esta técnica vienen de su nombre en inglés:
Energy Dispersive Spectroscopy. Esta técnica permite determinar la composición química de
los materiales en la superficie de una muestra determinando la energía de los fotones de rayos
X que son emitidos por la muestra durante el bombardeo con electrones en un microscopio
electrónico de barrido o de transmisión.
Determinando las energías de los rayos X emitidos por el área bombardeada se
puede saber cuáles son los elementos químicos presentes en la muestra (análisis cualitativo) y
la razón de intensidades de estos rayos característicos nos permite obtener las cantidades de
cada elemento (análisis cuantitativo). Un sistema EDS mide simultáneamente la intensidad de los rayos X en un
amplio rango de energías y proporciona información sobre la composición química exacta de
la muestra en segundos. Las componentes más importantes del sistema son: un detector de
rayos X, un procesador de pulsos para medir exactamente la energía de los rayos X
detectados y una interface para controlar el sistema con su respectivo software para analizar
los resultados.
En el capítulo IV sección 10 se muestra un ejemplo de la aplicación de esta
técnica para estudiar la composición química de nuestras muestras de nitruro de titanio (TiN)
así como de TiVN.
3.3 RETRODISPERSION RUTHERFORD
La espectroscopía de retrodispersión Rutherford (Rutherford Back Scattering ó
RBS) es una técnica de caracterización de películas delgadas muy conocida. Da información
acerca de la composición atómica de las películas. En general, cuando es aplicable, RBS
puede detectar niveles de composición del orden del 1 %. Cuando es aplicada a compuestos
binarios la exactitud composicional es del orden de ± 1 %.
3.1 El principio de la técnica
La figura 3.1 muestra esquemáticamente la geometría y notación para explicar el
principio físico de la técnica RBS. Un haz de iones de muy alta energía (MeV) y poca masa
incide sobre una muestra. Ocurre una colisión elástica entre un ion incidente (masa M0 y
energía E0) y un átomo de la muestra (masa M). Después del choque la energía del ion

74
desviado un ángulo θ es E1. De los principios de conservación de la energía y del momentum,
se obtiene:
E}M+M
)M+)senM-M({=E 02
0
01/22
022
1cos
, (3.1)
Para una combinación particular de M0, M y θ , la simple fórmula:
EK=E 0M1 , (3.2)
donde KM es una constante, relaciona la energía de los iones emergentes con la de los iones
incidentes.
Fig. 3.3 Geometría y notación para explicar el principio físico de la técnica RBS.
M0, E
0
espécimen
detector
M0, M1

Capítulo III Técnicas de caracterización
75
Cuando los iones (usualmente partículas alfa, i.e, iones 4He+) penetran en la película,
ellos pierden parte de su energía por su interacción con las nubes electrónicas de los
átomos de la muestra. Estas "colisiones electrónicas" son tan numerosas que la
perdida de energía puede ser considerada de dependencia continua respecto de la
profundidad en la muestra. Consecuentemente, los iones desviados de diferentes
profundidades (d) son desviados con diferentes valores de energía:
E(d)-E=E 1 (3.3) En el caso de átomos con peso atómico M 50 ó inferior, este efecto es favorable
porque permite a uno determinar la estequiometría de la película a diferentes
profundidades debajo de la superficie de la película. Sin embargo, para muestras que
contienen átomos más pesados, los intervalos de energía correspondientes a los
isótopos vecinos se translapan. A valores M 200, solamente ΔM > 20 puede ser
resuelto. Por ejemplo 209Bi y 190Os serían indistinguibles. Para mayor información
sobre la técnica RBS, ver Ziegler, [1].
Fig. 3.4 Espectro RBS para una película de V2O5 policristalino de 100 nm de espesor.

Capítulo III Técnicas de caracterización
76
Tabla 3.3
Composición atómica de una película de V2O5 policristalino según la técnica RBS
Elemento
Porcentje de átomos
Vanadio
28.6
Oxígeno
71.4
III.4 Espectroscopia de emisión de electrones por absorción de rayos X (XPS)
También es llamada espectroscopia de fotoelectrones X (XPS) es una
técnica de análisis de superficies que permite la identificación de los elementos
presentes con número de carga Z>2, y cuantificarlos (con error relativo del orden del
5%). Permite la identificación del compuesto químico del que forma parte el
elemento analizado, determinación del estado de valencia y el estado químico de los
átomos. Esta técnica puede destruir la superficie del material.
Cuando fotones X de baja energía, de 1 keV a 10 keV (por ejemplo
radiación K 1 y K 2 de Aluminio; ver figura 3.3; ó K de Magnesio, etc.) inciden
sobre la superficie de un material se produce una serie de eventos que resulta en la
emisión de electrones, los mismos que se analizan para obtener información de la
superficie. El efecto fotoeléctrico es el fundamento físico de esta espectroscopia. Los
fotoelectrones que provienen de capas internas del átomo (verbigracia la capa K)
emergen, dejando vacancias que son llenadas luego de aproximadamente 10-14 s, por
otros electrones de niveles energéticos más altos dando lugar a fotones X
fluorescentes y además con mucha mayor probabilidad electrones Auger.
El aparato que se emplea para esta técnica se conoce como espectrómetro
XPS. Usualmente el espectrómetro está unido eléctricamente a la sustancia que se
está analizando, de modo que su nivel de energía de fermi Ef y el de una muestra

Capítulo III Técnicas de caracterización
77
metálica sean iguales (ver figura 3.3). Esta se utiliza como referencia para la
evaluación de la energía de ligadura EB del fotoelectrón eyectado en función de la
energía cinética Ek, medida por el aparato.
La expresión matemática que relaciona estas últimas energías es [ref 2]:
EB = h - Ek - sp – ER - E
Donde h es la energía del fotón X incidente, sp es la función trabajo del
espectrómetro y es constante que depende únicamente del aparato, ER es la energía
de retroceso del fotoelectrón (término despreciable pues es del orden 0,1 eV a 0,01
eV) y el término E refleja la presencia de cargas electrostáticas sobre la superficie,
que debe tomarse en cuenta en el caso de aislantes; sin embargo, experimentalmente
se calibra el espectrómetro para evitar la acumulación de carga en la muestra
(desplazaría el nivel de Fermi), eliminando así la contribución de dicho efecto a la
ecuación. La energía de ligadura EB es típica para cada elemento y permite
identificar a los que están presentes en la superficie de la sustancia analizada.
Fig. 3.5 Esquema de los niveles de energía relevantes para la medida de la energía de enlace o de
ligadura; es la función trabajo de la muestra.

Capítulo III Técnicas de caracterización
78
En el esquema del espectrómetro de XPS que se muestra en la figura 3.4, se aprecia
la instrumentación básica del equipo. La fuente de rayos X, que tiene como ánodo al
aluminio, cuyos fotones X de 1486,6 eV y ancho de línea de 1,0 eV, (también se usa
el magnesio que emite fotones X de 1253,6 eV y 0,8 eV de ancho de línea) inciden
sobre la muestra produciendo electrones (fotoelectrones, electrones Auger) que son
captados por el analizador de energía, que los selecciona según su cantidad y energía
cinética Ek y posteriormente son contados en el detector de electrones. No se muestra
la cámara de ultra alto vacío (UHV), necesaria para que la superficie de la muestra
no se contamine. Debe anotarse que en XPS se utilizan preferentemente analizadores
hemisféricos (CHA: Concentric Hemispherical Analyzer), y su resolución disminuye
con el aumento de la energía cinética de los fotoelectrones.
Fig 3.6 Esquema básico de un espectrómetro XPS: fuente de rayos X (de Al); analizador de energía,
y; detector de electrones.
En los espectros XPS se puede apreciar un fondo continuo en el que se distinguen
dos regiones: una es la región de alta energía de enlace que se debe
fundamentalmente a las colisiones inelásticas de los electrones, otra es la región de

Capítulo III Técnicas de caracterización
79
baja energía de enlace donde es básicamente responsable la radiación continua de
frenado inherente a la no monocromaticidad de la fuente de rayos X.
La información relevante de los espectros se obtiene de los picos que aparecen
encima del fondo, los cuales son de varios tipos, fundamentalmente los de interés
son:
(a) Los picos debido a los fotoelectrones, que son agudos e intensos y aparecen
como singletes (s) o dobletes (p,d,f) debido a los fenómenos de acople espín –
órbita (L – S) electrónico, que dan lugar a la notación del tipo “nlj” y a la
relación de intensidades (que son proporcionales a la degeneración de
slj ) en el caso de doblete. El desplazamiento en energía de estos picos
dan información del entorno químico del átomo del cual proviene el
fotoelectrón, cuya magnitud depende del tipo y número de enlaces, valencia,
diferencia de electronegatividad, etc. y varía de fracciones a unas pocas
unidades de eV.
(b) Los picos debido a los electrones Auger, que son intensos no dependen de la
energía de los fotones incidentes, pero nos proporciona información adicional
sobre la naturaleza físico – química de los átomos en la superficie.
Los electrones Auger se originan según el siguiente proceso: un fotón
incidente es absorbido por un electrón de una capa interna que abandona el
átomo (fotoelectrón), dejando un hueco que luego es ocupado por otro
electrón que proviene de un nivel de mayor energía emitiendo energía (de
energía igual a la diferencia de energía de los niveles involucrados) que se
transfiere a un segundo electrón, que es el llamado electrón Auger, para
extraerlo del átomo.

Capítulo III Técnicas de caracterización
80
(c) Los picos menos intensos, llamados secundarios, como son los satélites,
plasmones y desdoblamiento de multiplete, aparecen en algunos espectros.
Como por ejemplo, si la muestra es metálica es posible la aparición de
excitaciones colectivas de los átomos de la banda de conducción (plasmones),
que son excitados por los fotoelectrones salientes. Los satélites resultan de la
interacción de los fotoelectrones con los electrones de valencia. Los
desdoblamientos de multiplete de un pico fotoelectrónico, ocurre en átomos
con electrones desapareados en la banda de valencia y consiste de distintas
configuraciones espín-órbita de los electrones internos.
Un método de análisis cuantitativo para determinar las concentraciones
relativas de los diversos constituyentes de una muestra homogénea, consiste en
utilizar el área de los picos. El número de fotoelectrones por segundo I en un pico es
dado por:
TAλyθσfnΙ
donde n es el número de átomos del elemento por cm3 de la muestra, f es el flujo de
rayos X en fotones cm-2 s-1, es la sección eficaz fotoeléctrica para el orbital
atómico de interés en cm2, es un factor de eficiencia angular para el espectrómetro
basado en el ángulo que forma el fotón incidente y el electrón detectado, y es la
eficiencia en el proceso fotoeléctrico, es el camino libre medio de los
fotoelectrones de la muestra, A es el área de la muestra, y T es la eficiencia para los
electrones emitidos desde la muestra. De la ecuación anterior: SΙ
TAλyθσfΙn
donde se ha definido S como el factor de sensibilidad atómica. Al considerar un pico
agudo de cada uno de dos elementos, entonces:

Capítulo III Técnicas de caracterización
81
22
11
SΙ
SΙ
2
1
nn
El parámetro S, para un determinado espectrómetro se encuentra en
tablas para cada elemento y depende del ángulo de incidencia del fotón incidente
relativa al analizador. Si generalizamos la expresión anterior, la fracción atómica de
cualquier constituyente en la muestra, Mx, por ejemplo, es:
ii
xx
SΙ
SΙ
i
xx
nn
M
III.5 DIFRACCIÓN DE RAYOS X
Puesto que la distancia entre átomos de un sólido es del orden de 10-10 m, es posible
estudiar su estructura microscópica utilizando radiación electromagnética cuya
longitud de onda sea del mismo orden de magnitud, y la energía que le corresponde
es dada por la ecuación: eV300121010
hcλhcω que es del rango de los rayos X
en el espectro electromagnético.
W. L. Bragg asumió que un cristal está constituido por planos cristalográficos, y
encontró que cuando un haz monocromático de rayos X (de longitud de onda )
incide sobre un cristal, con ángulo de incidencia , es reflejado especularmente (es
decir el ángulo de incidencia es igual al ángulo de reflexión) por planos paralelos
separados una distancia d, de tal manera que interfieren constructivamente cuando se
cumple la condición, llamada de Bragg: [ref 3]
λnsenθd2 ,
donde el número entero n es el orden de difracción, se mide desde el plano de
reflexión.

Capítulo III Técnicas de caracterización
82
La difracción de rayos X permite obtener difractogramas o espectros, en los cuales,
la condición de Bragg produce picos agudos, de los cuales es posible deducir el valor
de uno o más parámetros de la red cristalina, es decir permite conocer la celda
unitaria, comparando los valores de medidos para los picos con los patrones
internacionalmente aceptados [ref 4]. También es posible determinar la posición de
los átomos en la celda unitaria y la orientación de los planos cristalinos en la película
delgada, así como la posible identificación de la composición del material debido a la
existencia de la gran cantidad de información en las tablas o patrones
internacionalmente aceptados.
Las películas fabricadas por sputtering son, generalmente, estructuras policristalinas
de grano muy pequeño, tanto que de ser más pequeños de 0,1 m se produce un
ensanchamiento de los picos de difracción, y es difícil obtener picos cuando el
tamaño del grano es menor de 0.01 m. El tamaño medio del grano tg se estima
utilizando la ecuación de Scherrer:
cosθΓλ0,9
tg
Donde (medido en radianes) es el ancho del pico de difracción a la mitad de la
intensidad máxima.
Esta técnica es no destructiva y la muestra no necesita preparación especial alguna.
Los materiales amorfos no producen patrones de difracción con picos agudos
definidos.
III.6 ESPECTROSCOPIA DE ABSORCION EN EL INFRARROJO (IR)
Esta espectroscopia es útil para medir las vibraciones y rotaciones
moleculares que se pueden producir por la absorción de radiación usualmente en la
región del infrarrojo medio del espectro electromagnético, también es posible obtener

Capítulo III Técnicas de caracterización
83
información útil acerca de la estructura. Se puede considerar la región IR de interés
en función de la longitud de onda , desde = 2,5 m hasta = 25 m o
equivalentemente en número de onda ν , que es el inverso de la longitud de onda,
desde ν = 4000 cm-1 hasta ν = 400 cm-1. Para explicar la absorción de radiación por
las moléculas debe utilizarse la mecánica cuántica. La apariencia y la intensidad de
las bandas de absorción depende, además de las condiciones cuánticas, del momento
dipolar de la molécula. La onda infrarroja sólo se absorbe cuando el momento
dipolar de la molécula interacciona con el vector eléctrico de la onda incidente.
Para tratar el movimiento rotacional, se puede partir de un modelo clásico para una
molécula, por ejemplo diatómica: dos masas puntuales unidas por una varilla rígida
sin peso que rotan alrededor de un eje perpendicular a la varilla y que pasa por su
centro de masa (C.M.). La energía rotacional Erot en función del operador momentum
angular P es: 2IPE
2
rot , donde I es el momento de inercia de la molécula respecto del
C.M.. Para transformarlo a mecánica cuántica reemplazamos P por el operador
cuántico J, de modo que el hamiltoniano Hrot es 2IJH
22
rot , luego la ecuación de
Schrödinger es:
ΨEΨH rotrot
siendo la función de onda y Erot los autovalores correspondientes que se expresan
en la forma 1)J(J)2I
(E2
rot donde J = 0, 1, 2, ... Las reglas de selección para los
niveles rotacionales en un nivel de vibración dado es de acuerdo a las transiciones
dipolares eléctricas dado por 10,ΔJ .
En el caso del movimiento de vibración, consideremos nuevamente una molécula
diatómica como ejemplo, el modelo clásico sería dos masas puntuales unidas por un

Capítulo III Técnicas de caracterización
84
resorte, la única posibilidad de vibración es a lo largo del resorte y alrededor del
C.M. que no se mueve. La amplitud del movimiento de un átomo se aproxima a la
mitad del promedio de la distancia interatómica. Este problema de dos cuerpos es
equivalente al movimiento de una partícula de masa reducida en el sistema centro
de masa y cuya coordenada de posición, en cualquier instante, es igual al
desplazamiento total q de las masas o lo que es lo mismo decir q = longitud de
enlace.
La energía mecánica del sistema es dada por Eclas = Ecin + Epot =
22
kq21
2μp , siendo qμp el momentum lineal y k es la constante de fuerza para todo
el enlace. En cuántica se utiliza los operadores del oscilador armónico simple para
reemplazar pq
ip y qq y obtener el hamiltoniano que aplicado a la
función de onda se construye la ecuación de Schrödinger:
ΨEΨkq21
dq2μΨd
n2
2
22
En es la energía asociada a los niveles de energía cuantizados. Los autovalores que
satisfacen la ecuación de Schrödinger son
hν21nEn con n = 0, 1, 2, ...
y donde μk
2π1ν es la frecuencia de oscilación. La regla de selección dominante
es n = +1, +2. Los niveles de energía obtenidos están igualmente separados por
En = h , lo que no representa una molécula real. La energía potencial que las
describe mejor es un potencial anarmónico, que puede conseguirse perturbando el

Capítulo III Técnicas de caracterización
85
potencial armónico, pero con validez sólo en el entorno de la distancia interatómica
de equilibrio.
Fig. 3.7 Potencial de Morse para la molécula HCl. Los niveles de energía no están a escala. E = h
es el cambio de nivel de energía y E0 = energía del punto cero, n = 0. r0 es la distancia de equilibrio
para el enlace.
Sin embargo, los autovalores de la molécula real pueden visualizarse (ver
figura 3.1) empleando la energía potencial de Morse: 2)rβ(re )e(1DU(r) 0 , aquí De
se llama energía de disociación del enlace, es una constante que depende de la
molécula (tiene que ver con el ancho de la curva cerca de la distancia de equilibrio
r0) y r es la distancia entre los átomos de la molécula. Se puede colegir rápidamente
que el oscilador tiene comportamiento armónico en la vecindad de r0 con constante
de fuerza k = 2De2.
Resolviendo la ecuación de Schrödinger usando el potencial de Morse los
autovalores de la energía son [ref 5].
e2
vib xhν)21(n)hν
21(nE
donde h xe es llamada constante de vibración anarmónica, xe es adimensional.

Capítulo III Técnicas de caracterización
86
El espectrómetro IR más utilizado es FTIR [ref 6], es decir, el de
transformada de Fourier, cuyo fundamento consiste en una fuente de ondas IR que
emite radiación de todas las frecuencias, radiación que disminuye la intensidad al
pasar a través de la muestra. Las vibraciones moleculares producidas por la
excitación corresponde a las frecuencias cuya intensidad disminuye. La radiación
residual se mide con un detector y se transforma electrónicamente en un espectro.
La fuente debe radiar continuamente la banda completa de frecuencias de
interés, usualmente se emplea el llamado Globar de carburo de silicio que opera a
una temperatura de 1500 K. El detector registra la radiación emergente y transforma
las señales ópticas en eléctricas, el más corriente es el detector DTGS (sulfato de
triglicina deuterado). La señal eléctrica que sale del detector es almacenada en un
ordenador para aplicarle la transformada de Fourier, es decir para obtener la señal
registrada en función del número de onda incidente, al que se suele llamar espectro
infrarrojo, IR.
Fig. 3.8 Espectro de emisión de rayos X del Aluminio (a) esquema de las transiciones atómicas, y (b)
espectro de ambas componentes juntas.
En la interpretación de espectros, son esencialmente útiles aquellas
vibraciones que se limitan inicialmente a enlaces aislados o a grupos funcionales de
una molécula, es decir, a los modos localizados. Estas vibraciones localizadas sirven

Capítulo III Técnicas de caracterización
87
para la identificación de los grupos funcionales. El espectro IR se compone de dos
regiones: para números de onda mayores que 1500 cm-1 se encuentran las bandas de
absorción asignables a grupos funcionales aislados, mientras que la región de
números de onda menores que 1500 cm-1 contiene muchas bandas que caracterizan a
la molécula como tal. Esta zona se denomina región de la huella dactilar. La
utilización de la zona de la huella dactilar en la determinación estructural de una
sustancia por comparación con una muestra de catálogo es por lo general bastante
fiable, aún cuando es necesario verificar estos resultados utilizando otras técnicas de
espectroscopia.
REFERENCIAS:
[1] Ziegler J.F. (1975), in New Uses of Ion Accelerators, Ed. by Ziegler J.F. (Plenum
Press, New York).
[2] Handbook of X-Ray Photoelectron Spectroscopy, Perkin-Elmer, Physical Electronics
Division, Dec. 1978.
[3] Ashcroft, N. W. y Mermin, N. D. Solid State Physics. USA: Holt, Rinehart and Winston.
cap 6, 1975.
[4 ] Manual XRD, Tablas de datos del espectrómetro de rayos X. Phillips. X´Pert.
[ 5 ] Hollas, J. M. Modern Spectroscopy 3rd Ed. John Wiley & Sons. Cap. 04 y 05, 1996.
[6] Manual IR, Manual del usuario del espectrómetro infrarrojo. Shimatzu. FTIR – 8300.
.

Capítulo IV Recubrimientos duros
88
CAPITULO IV
RECUBRIMIENTOS DUROS
Los materiales de alta dureza son ampliamente investigados en la actualidad
[ref . 1, 2, 3 ] por sus propiedades de alta resistencia a temperaturas altas, al ataque
químico, al desgaste por abrasión, al desgaste por fricción. Sin embargo, son también
frágiles o quebradizos, razón por la cual en aplicaciones donde se necesitan de
materiales duros y tenaces, una solución es usar materiales tenaces, aceros por ejemplo,
recubiertos con capas de algunos micrómetros de espesor de materiales duros. Uno de
los materiales más usados e importantes es el nitruro de titanio, que además de poseer
una alta dureza tiene una apariencia muy atractiva, similar a la del oro.
Los recubrimienos duros de nitruro de titanio son usados en la actualidad
para prolongar el tiempo de vida de herramientas de corte tales como brocas, fresas,
cuchillas de uso industrial; también se usan para prolongar el tiempo de vida útil de
moldes en fábricas de producción de artículos de plástico por inyección. Por su
presentación atractiva agregada a su resistencia al desgaste se usa también en utensilios
domésticos. En la industria textil, algunas piezas de la maquinaria son recubiertas con
nitruro de titanio para disminuir el coeficiente de fricción entre la maquinaria y las
fibras tratadas con partículas de dióxido de titanio.
En las figuras 4.1 mostramos algunas de las aplicaciones más generalizadas
de los recubrimientos de nitruro de titanio. Las herramientas mostradas en las figuras
4.1a y 4.1b son producidas industrialmente. Las de la figura 4.1c fueron producidas
experimentalmente en nuestro laboratorio.

Capítulo IV Recubrimientos duros
89
(a)
(b)
(c)
Fig. 4.1 Diferentes tipos de herramientas de acero de alta velocidad recubiertas con nitruro de titanio (a) y
(b) comerciales, (c) recubiertas a nivel experimental en nuestro Laboratorio de Física de la UNI.

Capítulo IV Recubrimientos duros
90
La relación costo beneficio depende de cada aplicación, sin embargo, como
regla general podemos decir que recubrir una pieza con nitruro de titano duplica su
costo y sextuplica su tiempo de vida útil. A continuación algunos ejemplos prácticos.
Con una broca de 8 mm de diámetro, 117 mm de longitud, hecha de acero
de alta velocidad puede hacer un promedio de 34 huecos a través de una placa de acero
inoxidable de 30 mm a una velocidad de 25 mm/min. La misma broca recubierta con
una capa de 3,5 m de espesor nitruro de titanio, en las mismas condiciones, puede
hacer en promedio 219 huecos.
Un molde de plástico de 25 25 25 mm3 hecho de bakelita puede ser usado
para hacer 48 000 piezas, mientras que el mismo molde recubierto con una capa de
4 m de espesor de nitruro de titanio es útil para hacer 300 000 piezas.
En este capítulo definimos el concepto de dureza, discutimos cómo se mide
esta cantidad física, discutimos la importancia tecnológica de los materiales de alta
dureza, mostramos los resultados obtenidos en nuestro laboratorio en relación a la
producción de recubrimientos duros. Puesto que la dureza está intimamente ligada a los
conceptos de desgaste por abrasión y rozamiento, incluimos en este capítulo también
los resultados de las pruebas de desgaste y de coeficiente de fricción a las que fueron
sometidas nuestras muestras de TiN y TiVN. Incluimos también los resultados de los
análisis químico y estructural, realizados por las técnicas de espectroscopía de
fotoelectrones emitidos por rayos X y difracción de rayos X, respectivamente. Al final
del capítulo adjuntamos una tabla de las propiedades físicas de una gran variedad de
recubrimientos duros que se usan alternativa o complementariamente al nitruro de
titanio.

Capítulo IV Recubrimientos duros
91
IV.2. DUREZA. La dureza es la propiedad física de los materiales relacionada a la
resistencia de un cuerpo a ser rayado o penetrado por un cuerpo puntiagudo de otro
material. Según esta definición Mohs hizo una clasificación de los materiales de 1 a 10
según su dureza.
Escala de Mohs
1. Talco, 2. yeso, 3. calcita, 4. fluorita, 5. apatita, 6. ortosa, 7. cuarzo,
8.topacio, 9. corindón, 10. diamante.
En términos modernos se dice que la dureza es una medida de la resistencia
de los materiales a la deformación plástica localizada. Los físicos teóricos del estado
sólido asocian la dureza con el módulo de compresibilidad, B, de la siguiente manera
dVdE
VB 1 (4.1)
donde V es el volumen y E es la energía interna total del cuerpo.
IV. 3. NORMAS PARA MEDIR DUREZA
Existen varias normas para medir la dureza [ref. 4]. Todas ellas se basan en
el uso de un objeto penetrador, indentador (generalmente de forma esférica, cónica o
piramidal), el cual es sometido a un determinado peso o carga, para luego ser
presionado durante cierto tiempo sobre el cuerpo cuya dureza interesa conocer. La
dureza así determinada será función de la carga aplicada y del tamaño de la huella
dejada, indentación, sobre la superficie del cuerpo.
Dureza Brinell, se usa como indentador una esfera de 10 mm de diámetro de acero
endurecido o de carburo de tungsteno. Las cargas normalizadas están entre 500 y 3 000
kg en incrementos de 500 kg; durante el ensayo la carga se mantiene constante durante

Capítulo IV Recubrimientos duros
92
un tiempo especificado (entre 10 y 30 s). El número de dureza Brinell, HB, es una
función tanto de la carga como del diámetro de la huella resultante.
Dureza Rockwell y Rocwell superficial, los indentadores son conos de diamante y
esferas de acero de 1/16, 1/8, 1/4 y 1/2 pulgada de diámetro. Las cargas varían entre
10 kg y 150 kg.
Dureza Knoop, según esta norma el indentador es una pirámide de base rombohedral,
los valores de carga van desde 1 gf a 2 kgf, y se define como
222,14mmkg
dLHK (4.2)
donde L es el valor de la carga aplicada en gf y d es el valor de la diagonal mayor de la
indentación en m.
Dureza Vickers, es la norma que nosotros aplicamos en nuestro laboratorio. El
indentador es una pirámide de diamante de base cuadrada. La geometría exacta está
indicada en la figura 4.2. Los valores de las cargas van desde 1 gf a 2 kgf. El valor de
dureza viene dado por el número que se obtiene de la siguiente fórmula:
22 /1840 mmkg
dLHV (4.3)
donde L es el valor de la carga aplicada en gf y d es el valor de la diagonal promedio de
la indentación en m.
Fig. 4.2 Esquema de indentador usado para medir dureza según norma Vickers.

Capítulo IV Recubrimientos duros
93
Todos los resultados de dureza en este capítulo han sido obtenidos según la
norma Vickers, usando el microdurómetro Buehler, serie Micromet® 2100.
La figura 4.3 muestra la indentación dejada por la aplicación de una carga
de 500 gf sobre una muestra de acero de alta velocidad (hss) recubierta con nitruro de
titanio (TiN). Esta fotografía que muestra una vista tridimensional no ha sido hecha para
tener una información cuantitativa de la dureza, sino para resaltar la forma piramidal de
la indentación. En la práctica, para hacer medidas lo que interesa es la diagonal del
cuadrado, intersección entre la pirámide y la superficie de la muestra.
Fig. 4.3 Micrografía con microscopio electrónico de barrido de la huella o impronta dejada por el
indentador Vickers sobre una muestra de nitruro de titanio depositada sobre acero de alta velocidad. IV.4. MICRODUREZA VICKERS DEL NITRURO DE TITANIO
En esta sección mostramos los resultados de las medidas de dureza Vickers
de acero hss y de acero hss recubierto con una capa de 4.0 μm de TiN. Los
recubrimientos de TiN fueron producidos en nuestro laboratorio de Física de la UNI, en
1999, por la técnica magneto evaporación iónica dc. En la actualidad podemos producir

Capítulo IV Recubrimientos duros
94
también nitruro de titanio por la técnica magneto evaporación iónica rf, siendo los
resultados muy similares.
Los substratos fueron trozos de cuchilla de 30 10 6 mm3 hechas de acero
hss pulidas hasta darles una terminación especular. Una lámina de silicio de 5 5 mm2
cubría parcialmente la superficie de la cuchilla durante la deposición, a manera de
máscara, de modo que fuera posible comparar la dureza del material recubierto con la
del no recubierto. Las indentaciones fueron observadas en un microscopio electrónico
de barrido, el instrumento usado fue Zeiss D5M de la Universidad de Uppsala, Suecia.
Los valores de carga usados para la comparación de la dureza son de 500,
25 y 10 gf. El propósito es hacer notar que para valores de carga grandes, la dureza del
sistema substrato más recubrimiento es prácticamente la misma que la del substrato
solo, mientras que para valores de carga pequeños se observa un incremento en el valor
de la dureza hasta por un factor de 8. Esto se explica porque debido a la geometría del
indentador la penetración de éste es sólo 1/7 del valor de la diagonal, así que para cargas
de 500 gf el indentador penetra unas 7 μm, profundidad mayor que el espesor del
recubrimiento, es decir, si la carga es grande lo que se está midiendo básicamente es la
dureza del substrato.
Un detalle a ser observado en las micrografías mostradas es la presencia de
granos de carburo de tungsteno en las substratos no recubiertos del acero hss, los cuales
no se observan en el substrato recubierto.

Capítulo IV Recubrimientos duros
95
(a) (b)
Fig. 4.4 Micrografías de indentaciones Vickers sobre: (a) acero hss, (b) acero hss recubierto con TiN. La
carga usada en ambos casos fue de 500 gf. La dureza Vickers correspondiente al substrato es 402 gf/mm2
y al substrato recubierto con TiN es de 397 kgf/ mm2.
a)
(a) (b)
Fig. 4. 5 Micrografías de indentaciones Vickers sobre: (a) acero hss, (b) acero hss recubierto con TiN.
La carga usada en ambos casos fue de 25 gf. La dureza Vickers correspondiente al substrato es
400 kgf/mm2 y al substrato recubierto con TiN es de 1600 kgf/ mm2.

Capítulo IV Recubrimientos duros
96
(a) (b) Fig. 4.6 Micrografías de indentaciones Vickers sobre: (a) acero hss, (b) acero hss recubierto con TiN. La
carga usada en ambos casos fue de 10 gf. La dureza Vickers correspondiente al substrato es 400 kgf/mm2
y al substrato recubierto con TiN es de 2000 kgf/ mm2
IV.5. RAPIDEZ DE DEPOSICION
La rapidez de deposición se calcula dividiendo el espesor del recubrimiento
por el tiempo de deposición. El espesor de los recubrimientos se mide haciendo un corte
transversal al sistema recubrimiento más substrato y observando la sección transversal
en el microscopio electrónico de barrido. El corte transversal se puede hacer de dos
maneras: (a) cortando el sustrato por la cara opuesta al recubrimiento hasta llegar casi al
recubrimiento y forzando el material para obtener una fractura. Este método es conocido
como “fracture cross section”, (b) haciendo una probeta con la muestra recubierta y
resina conductora, cortándola transversalmente y puliendo la sección transversal antes
de hacer la observación en el microscopio, éste es el método “polished cross section”.
En las figuras 4.7a y 4.7b mostramos micrografías de las secciones
transversales de acero hss recubierto con TiN. En ambos casos el espesor que se observa
es de alrededor de 4 m, y teniendo en cuenta que el tiempo de deposición fue de
2 horas, significa que la rapidez de deposición de TiN en nuestro equipo es de 2 m/ h.

Capítulo IV Recubrimientos duros
97
Fig. 4.7 Micrografías con microscopio electrónico de barrido mostrando el espesor de un recubrimiento
de TiN sobre acero hss. (a) fracture cross section, (b) polished cross section. El espesor observado es de
4, 11 y 4,06 m respectivamente.
IV. 6. ADHESION
En general, para las diferentes aplicaciones, es muy importante la adhesión
entre el recubrimiento y el respectivo substrato, pero es particularmente importante en el
caso de aplicaciones mecánicas como en las herramientas de corte donde la pieza
recubierta va a tener contacto y va estar sujetas a tensiones y compresiones con otros
cuerpos.
Aquí consideramos la siguiente definición: Adhesión es la fuerza o trabajo
requerido para desprender una capa del substrato. En realidad se trata de una fuerza de
adhesión por unidad de superficie adherida o desprendida [ref 5].
Hay varias técnicas para determinar la adhesión. Aquí solo mencionamos
algunas de ellas:
(a) Pull-off test, que consiste en enganchar un cilindro metálico con un adhesivo (epoxy
generalmente) a la superficie del sistema a estudiar. Una vez seco el adhesivo, se
procede a ejercer sobre el cilindro metálico una fuerza de tracción para separarlo del
sistema.

Capítulo IV Recubrimientos duros
98
(b) Blister test o Delaminación por gas a presión, que consiste en hacer un pequeño
orificio en el substrato por el que se puede introducir aire hasta la interface. La capa se
desprenderá por efecto de la presión ejercida por el aire.
(c) Flexión cóncava o convexa del substrato (three point flexure test). Se trata de que la
flexión del substrato y, por consiguiente de la capa, provoque el fallo adhesivo en la
interface.
(d) Rayado con indentador Rockwell (scrtach test) , consiste en efectuar lo mismo que
en una indentación Rockwell pero desplazando la muestra en una dirección
determinada.
(e) Ensayo Estático con indentador rockwell, en este caso se trata de presionar la
muestra recubierta con un indentador Rockwell con cargas del orden de 20 a 60 kgf y
después observar al microscopio electrónico las partículas en forma de escamas (flakes)
que se desprenden.
Nuestras muestras fueron sometidas a este ensayo estático con cargas de 20
y 50 kgf. Las micrografías obtenidas (figura 4.8) muestran características similares a las
reportadas en otros trabajos [ref. 6].
(a) (b) Fig. 4.8 Micrografías con microscopio electrónico de barrido de muestras de TiN/HSS después de haber
sido indentadas con indentador Rockwel (a) con carga de 20 kg, (b) con carga de 50 kg.

Capítulo IV Recubrimientos duros
99
IV.7. RESISTENCIA AL DESGASTE POR ABRASION
La prueba del cráter podría ser el nombre en español para la técnica
desarrollada en la Universidad de Uppsala, Suecia, [ref. 7, 8, 9], para evaluar la
resistencia a la abrasión de películas delgadas así como de material voluminoso
homogéneo.
Esta técnica utiliza un cavador de hoyuelos comercial (figura 4.9)
normalmente usada para pre-adelgazamiento de especímenes de microscopía electrónica
de transmisión y de espectroscopía Auger.
Fig 4.9 Esquema del arreglo experimental para la prueba del cráter.
Los movimientos combinados de la muestra rotando en un plano horizontal y el disco
rotando en un plano vertical, conducen a la formación de un cráter en forma de casquete
esférico en la muestra. Cualquier medio abrasivo puede ser usado, en nuestros ensayos
utilizamos pasta de diamante.
Para materiales voluminosos homogéneos, la ecuación de Archard establece
que
LSV , (4.4)

Capítulo IV Recubrimientos duros
100
donde V es el volumen del crater, S es la distancia recorrida por un punto del disco, L es
la carga y es la rapidez de desgaste específica. Para películas delgadas la ecuación
propuesta es:
)/( sscc VSLV (4.5)
donde Vc y Vs son los volúmenes del crater; c y s son las rapideces específicas de
desgaste para el recubrimiento y sustrato, respectivamente, L es la carga y S la distancia
recorrida por un punto del disco.
Los volúmenes son determinados de los radios del crater y del disco, la
carga es decidida por el operador, y las distancias S dependen del número de vueltas y
el diámetro del disco. Los resultados mostrados a continuación fueron obtenidos con
una carga de 5 mN, el diámetro del disco fue 20 mm y la pasta de diamante tenía 2,5 µm
de tamaño de grano. Los cráteres hechos fueron con 50 a 400 revoluciones del disco, lo
que equivale a valores de S entre 3.14 y 12.56 m.
El disco usado es de acero inoxidable, por lo tanto la técnica es aplicable
sólo para materiales de mayor dureza que este tipo de acero. Para recubrimientos duros
depositados sobre acero inoxidable hemos usado esta técnica con la condición que la
profundidad del crater sea menor que el espesor del recubrimiento. En la práctica esto
significa que el número de vueltas del disco debe ser menor que 50. Materiales
voluminosos de silicio y acero hss fueron también analizados como referencias.
En la tabla 4.1 se muestran los resultados de las medidas de desgaste por
abrasión para recubrimientos de TiN sobre substratos de acero inoxidable y sobre acero
de alta velocidad. También se muestran resultados para recubrimientos de TiVN sobre
acero inoxidable y materiales voluminosos de acero hss y silicio.

Capítulo IV Recubrimientos duros
101
TABLA 4.1
Rapidez de desgaste específico para diferentes materiales en bulto y en forma de
recubrimientos
Material Número de
revoluciones
Rapidez de
desgaste
(µm3/mmN)
Si (bulk) 50-400 5600
Hss (bulk) 50-400 1280
TiN/hss 50-400 216
TiN/inox 50 364
(Ti,V)N/inox 50 242
Los valores obtenidos para TiN/hss están en buen acuerdo con resultados
publicados en trabajos anteriores [ref. 9]. La resistencia al desgaste para el acero de alta
velocidad está también en buen acuerdo con resultados en la literatura [ref. 9 y 10]. El
valor de la rapidez de desgaste para TiVN/inox es menor que para TiN/inox, indicando
que es posible mejorar la resistencia al desgaste del nitruro de titanio introduciendo un
porcentaje de vanadio dentro de la estructura del TiN. En este caso particular el TiVN
fue obtenido por la técnica magneto evaporación iónica dc, insertando en el blanco de
titanio de 75 mm de diámetro, dos discos de vanadio de 10 mm de diámetro
simétricamente dispuestos. La composición química resultante evaluada por la técnica
XPS fue 40% Ti, 10% V, 50% N.
Las figuras 4.10 muestran una vista usando un microscopio óptico de los
cráteres hechos a recubrimientos de nitruro de titanio sobre sustratos de acero de alta
velocidad con 50 y 200 vueltas del disco. En la figura 4.10b puede observarse una capa
intermedia entre el sustrato y la capa de nitruro de titano. Se trata de una capa de titanio
metálico colocada allí para mejorar la adherencia.

Capítulo IV Recubrimientos duros
102
(a) (b)
Fig. 4.10 Micrografías ópticas de cráteres hechos en TiN/hss para medir resistencia a abrasión (a) con
50 vueltas, (b) con 200 vueltas del disco vertical.
IV.8. COEFICIENTE DE FRICCION
La técnica billa sobre disco (ball on disk) es ampliamente usada para medir
el coeficiente de fricción cinético [ref. 10, 11 y 12]. Una billa de 5 a 10 mm de diámetro
se mantiene fija en la parte inferior de un eje sometida a cargas de 1 a 10 N. La muestra
gira respecto a un eje paralelo al que sostiene a la billa, obteniéndose una huella de la
trayectoria como se muestra en la figura 4.11. Un sensor de fuerza nos permite
determinar la fuerza de fricción y puesto que la normal es conocida, podemos
determinar el coeficiente de fricción μk.
Fig. 4.11 Esquema que muestra el principio de funcionamiento del método billa sobre disco para medir el
coeficiente de fricción, [ref. 18]. .

Capítulo IV Recubrimientos duros
103
En este trabajo tres tipos de billas fueron usadas: alumina, acero 100Cr6 y
nitruro de silicio para medir el coeficiente de fricción de recubrimientos duros de nitruro
de titanio y TiVN. El diámetro de las bolas fue 5 mm y la longitud de la trayectoria
15.70 mm. Las muestras fueron sometidas a limpieza con ultrasonido en alcohol y
acetona antes de la prueba. Todos los experimentos fueron realizados bajo las siguientes
condiciones:
Fuerza normal: 5N
Velocidad de deslizamiento: 0.005 m/s
Tiempo: 30 min
Temperatura: 22°C
Humedad relativa: 40%
Sin lubricar.
Los datos experimentales de fuerza de fricción y tiempo fueron adquiridos
electrónicamente durante el experimento y transformados automáticamente en archivos
de coeficiente de fricción versus distancia de deslizamiento.
El comportamiento del coeficiente de fricción en función de la distancia
deslizada obtenido por la técnica billa sobre disco es generalmente semiperiódica con
período igual a la longitud de la circunferencia de la trayectoria. Esto significa que cada
cierta longitud deslizada ocurren máximos y mínimos aun cuando los valores del
coeficiente de fricción no se repiten exactamente. Esto está ilustrado en la figura 4.12.
Cuando cientos de períodos son considerados el gráfico deja tene la
apariencia de una curva y luce más bién como una franja más o menos homogénea [ref.
11]. Hasta ahora no se ha reportado ninguna explicación cuantitativa para este
comportamiento, sin embargo la situación puede ser cualitativamente comprendida si
consideramos que la fuerza medida tiene dos contribuciones [ref. 13]. Una es la fuerza de

Capítulo IV Recubrimientos duros
104
deformación debido a las asperezas en la superficie opuesta y que son responsables de
desgaste abrasivo, y la otra es la fuerza de rozamiento por deslizamiento que da origen a
desgaste adhesivo, es decir, intercambio de material. Estas fuerzas dependen una de la
otra; por ejemplo, el intercambio de material por adhesión causa asperezas y estas a su
vez causan abrasión. Estas fuerzas varían al azar dentro de cierto rango dependiendo del
material intercambiado en cada período.
0
0,1
0,2
0,3
0,4
0,5
0,6
0,7
0 0,05 0,1 0,15 0,2 0,25
distancia, m
Coef
icien
te de
fricc
ión
Fig 4.12 Coeficiente de fricción vs. distancia deslizada para una muestra de acero inoxidable recubierta
con TiN, mostrando el carácter semiperiódico de la curva.
Se ha obtenido también un coeficiente de fricción promedio vs distancia,
tomando el promedio cada 100 valores, es decir, cada 7 cm de deslizamiento. Es la línea
blanca dentro de la franja obscura de cada gráfico coeficiente de fricción vs distancia.
Fricción contra la billa de alúmina
Las figuras 4.13 a y b muestran respectivamente el comportamiento del
coeficiente de fricción en función de la distancia deslizada cuando se usa una billa de
alúmina sobre nitruro de titanio y TiVN. Las figuras 4.14 a y b muestran las huellas
dejadas por la billa en las películas de TiN/inox y TiVN/inox. No se observa mucha
diferencia entre los valores del coeficiente de fricción para ambos tipos de
recubrimientos, sin embargo nos gustaría enfatizar dos cosas: (a) los bajos valores del

Capítulo IV Recubrimientos duros
105
coeficiente de fricción del TiN concuerdan con los resultados obtenidos de
experimentos similares reportados en la ref. 10, y (b) las huellas muestran que el TiVN
sufre menos desgaste por fricción que el TiN.
(a) (b) Fig 4.13 Variación del coeficiente de fricción con la distancia recorrida por una billa de alúmina sobre
(a) TiN/inox, (b) TiVN/inox.
(a) (b)
Fig. 4.14 Huellas de desgaste dejada por una billa de alúmina sobre (a) TiN/inox, (b) TiVN/ inox.
Fricción contra la billa de acero Cr6
Las figuras 4.15 a y b muestran la evolución del coeficiente de fricción en
función de la distancia de deslizamiento para recubrimientos TiN y TiVN sobre acero
inoxidable, respectivamente. Los valores de para el TiN están en buen acuerdo con
0
0,1
0,2
0,3
0,4
0,5
0 2 4 6 8
distancia, mco
efic
ient
e de
fric
ción
0
0,1
0,2
0,3
0,4
0,5
0 1 2 3 4 5 6 7 8 9distancia, m
coef
icie
nte
de fr
icci
ón

Capítulo IV Recubrimientos duros
106
los reportados en anteriores publicaciones [ref. 12]. Los valores de para el TiVN son,
sin embargo mucho mas bajos.
Las figuras 4.16 a y b muestran micrografías ópticas para las respectivas
huellas. El color más obscuro de la huella (figura 4.16b) respecto al TiN es atribuida a la
relativamente alta transferencia de material de la billa a la película. Tal transferencia ha
sido reportada en varios estudios [ref. 10, 14 y 15]. Ha sido demostrado también que la
dureza del recubrimiento no tiene mucho efecto en el desgaste de la bola.
(a) (b) Fig. 4.15 Coeficiente de fricción vs. distancia recorrida para una billa de acero deslizándose sobre
(a) TiN/inox y (b) TiVN/inox.
(a) (b) Fig 4.16 Huellas de desgaste dejada por una billa de acero sobre (a) TiN/inox y (b) TiVN/ inox.
Fricción contra una billa de nitruro de silicio
De las figuras 4.17 a y b puede observarse que el comportamiento del
coeficiente de fricción es similar para TiN y TiVN, sin embargo la apariencia en las
0
0,1
0,2
0,3
0,4
0,5
0 1 2 3 4 5 6 7 8 9
distancia, m
coef
icie
nte
de fr
icci
ón

Capítulo IV Recubrimientos duros
107
huellas en las figuras 4.18 a y b es muy diferente, mientras que la película de TiN es
notoriamente destruida por la bola de nitruro de silicio, la figura 4.18b evidencia
transferencia de material de la billa al recubrimiento.
(b)
Fig. 4.17 Coeficiente de fricción vs. distancia recorrida para una billa de nitruro de silicio
deslizándose sobre (a) TiN/inox y b) TiVN/inox.
(a) (b)
Fig 4.18. Huellas de desgaste dejada por una billa de nitruro de silicio sobre (a) TiN/inox y
(b) TiVN/ inox
IV.9. NANOINDENTACION
La microdureza y el módulo de Young fueron medidos usando el
instrumento Nanoindenter XP de acuerdo al procedimiento seguido por Oliver and
Pharr [ref ].
La microdureza H y el módulo de Young E de tres tipos de muestras fueron
determinados usando la técnica de nanoindentación. Los especímenes fueron TiN/hss,
0
0,2
0,4
0,6
0,8
1
0 1 2 3 4 5 6 7 8 9
distancia, m
coef
icie
nte
de fr
icci
ón
0
0,2
0,4
0,6
0,8
1
0 1 2 3 4 5 6 7 8 9
distancia, m
coef
icie
nte
de fr
icci
ón
(a)
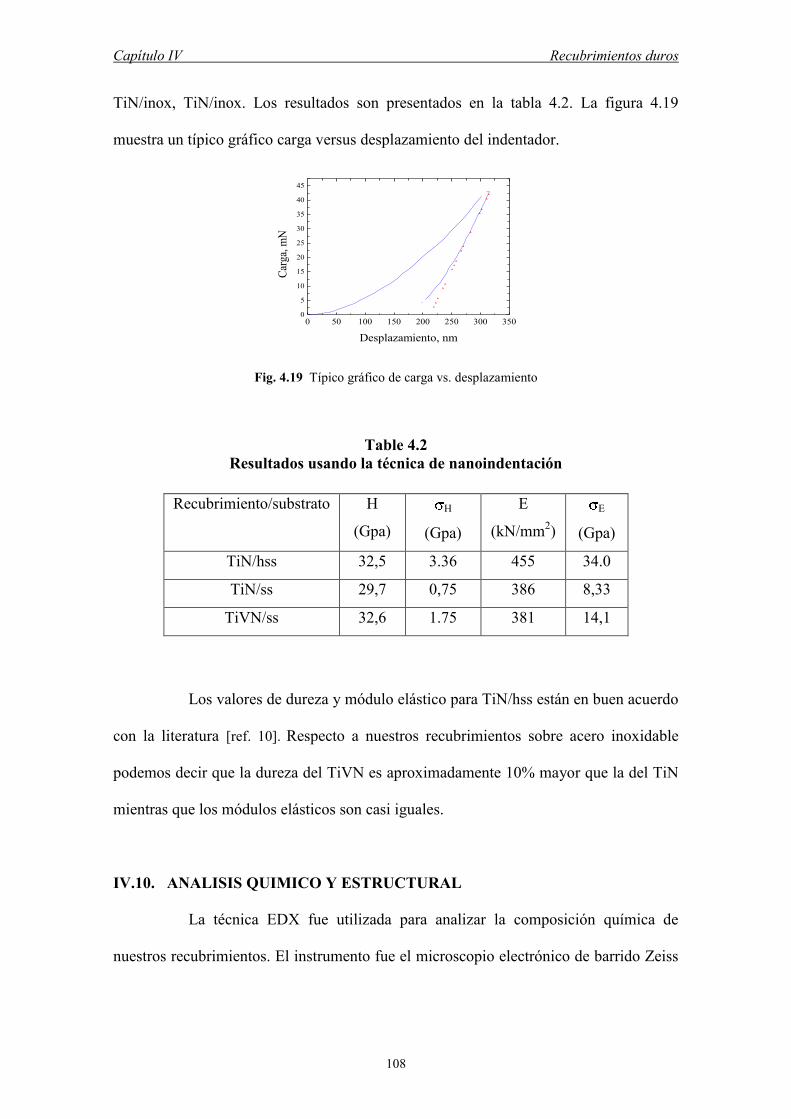
Capítulo IV Recubrimientos duros
108
TiN/inox, TiN/inox. Los resultados son presentados en la tabla 4.2. La figura 4.19
muestra un típico gráfico carga versus desplazamiento del indentador.
0 50 100 150 200 250 300 3500
5
10
15
20
25
30
35
40
45
Carg
a, m
N
Desplazamiento, nm
Fig. 4.19 Típico gráfico de carga vs. desplazamiento
Table 4.2 Resultados usando la técnica de nanoindentación
Recubrimiento/substrato H
(Gpa) H
(Gpa)
E
(kN/mm2) E
(Gpa)
TiN/hss 32,5 3.36 455 34.0
TiN/ss 29,7 0,75 386 8,33
TiVN/ss 32,6 1.75 381 14,1
Los valores de dureza y módulo elástico para TiN/hss están en buen acuerdo
con la literatura [ref. 10]. Respecto a nuestros recubrimientos sobre acero inoxidable
podemos decir que la dureza del TiVN es aproximadamente 10% mayor que la del TiN
mientras que los módulos elásticos son casi iguales.
IV.10. ANALISIS QUIMICO Y ESTRUCTURAL
La técnica EDX fue utilizada para analizar la composición química de
nuestros recubrimientos. El instrumento fue el microscopio electrónico de barrido Zeiss

Capítulo IV Recubrimientos duros
109
D5M. La energía de los electrones incidentes fue 20 keV y el tiempo de medida fue
usualmente 300 segundos.
La figura 4.20a muestra el espectro EDX para una muestra de acero
inoxidable recubierta con una capa de 4 m de TiN. Se observan los picos K del
nitrógeno y del titanio a 0.34 y 567 eV, respectivamente. El pico K del titanio aparece
a 590 eV. La figura 4.20b muestra el respectivo espectro para TiVN; es posible observar
que aparentemente el pico K correspondiente al titanio se ha incrementado respecto
al K , sin embargo, lo que sucede realmente es que el pico K del vanadio aparece
casi exactamente al mismo valor de energía. Puede verse además el pico K del
vanadio en 553 eV. La composición química calculada para el TiN es N 50,5% y Ti
49,5% mientras que para el TiVN es N 50.4%, Ti 40% y V 9,6%.
(a) (b)
Fig. 4.20. Espectros EDX de (a) nitruro de titanio y (b)nitruro de titanio y vanadio.
En las figuras 4.21 a, b y c se muestran los resultados de análisis químicos
hechos por otra técnica, específicamente por la técnica XPS.
La técnica de difracción de rayos X fue usada para analizar la estructura
cristalina de nuestros recubrimientos. El instrumento fue Siemens D5000, operado a 40
kV. El blanco para producir los rayos X fue de cobre y la longitud de onda filtrada fue
= 0, 15418 nm. El arreglo 2 fue usado.
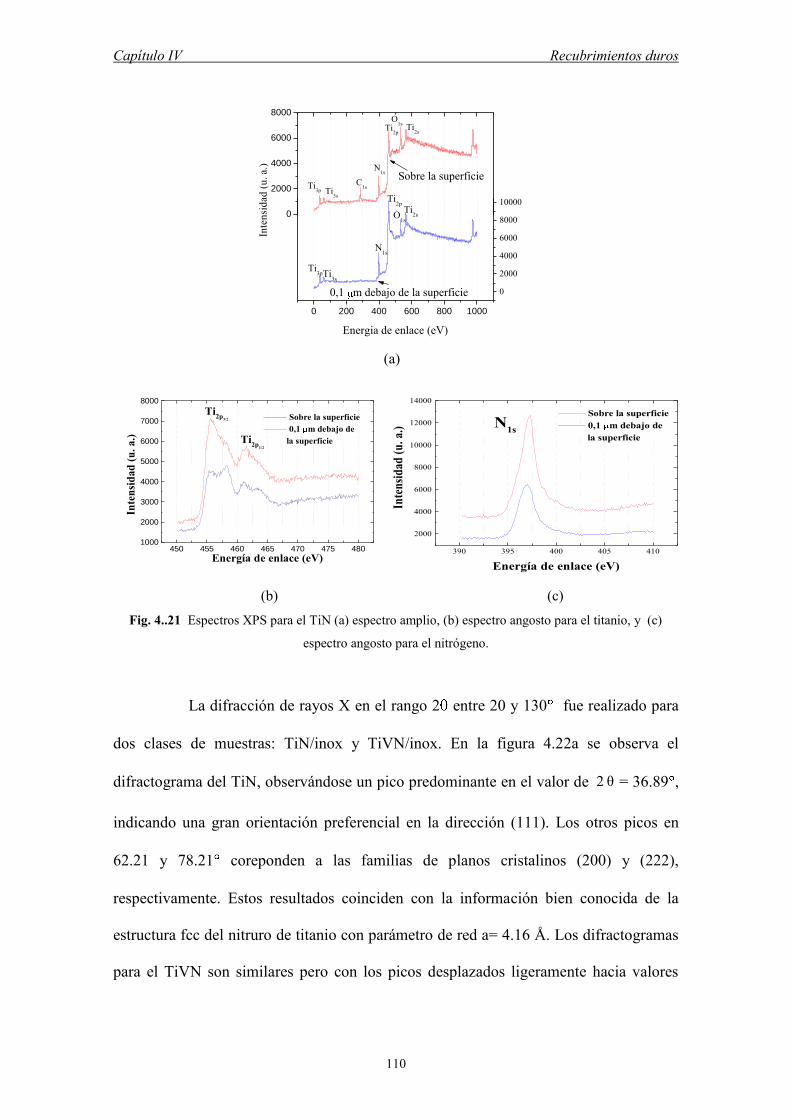
Capítulo IV Recubrimientos duros
110
0 200 400 600 800 1000
0
2000
4000
6000
8000
10000
Ti3sTi3p
N1s
Ti2sO1s
Ti2p
0,1 m debajo de la superficie
Inte
nsid
ad (u
. a.)
Energia de enlace (eV)
0
2000
4000
6000
8000
Ti3s
Ti3p
Ti2s
O1sTi
2p
N1s
C1s
Sobre la superficie
(a)
(b) (c) Fig. 4..21 Espectros XPS para el TiN (a) espectro amplio, (b) espectro angosto para el titanio, y (c)
espectro angosto para el nitrógeno.
La difracción de rayos X en el rango 2 entre 20 y 130 fue realizado para
dos clases de muestras: TiN/inox y TiVN/inox. En la figura 4.22a se observa el
difractograma del TiN, observándose un pico predominante en el valor de θ2 = 36.89 ,
indicando una gran orientación preferencial en la dirección (111). Los otros picos en
62.21 y 78.21 coreponden a las familias de planos cristalinos (200) y (222),
respectivamente. Estos resultados coinciden con la información bien conocida de la
estructura fcc del nitruro de titanio con parámetro de red a= 4.16 Å. Los difractogramas
para el TiVN son similares pero con los picos desplazados ligeramente hacia valores
450 455 460 465 470 475 4801000
2000
3000
4000
5000
6000
7000
8000
Sobre la superficie 0,1 m debajo de
la superficieTi2p1/2
Ti2p3/2
Inte
nsid
ad (u
. a.)
Energía de enlace (eV)
390 395 400 405 410
2000
4000
6000
8000
10000
12000
14000
Sobre la superficie 0,1 m debajo de
la superficieN1s
Inte
nsid
ad (u
. a.)
Energía de enlace (eV)

Capítulo IV Recubrimientos duros
111
mayores de 2 , lo que indica una disminución del parámetro de red. la figura 4.22b
muestra el corrimiento del pico (111) del TiVN respecto del de TiN.
(a) (b) Fig. 4.22 Difractograma de rayos X (a) de nitruro de titanio, (b) desplazamiento del pico (111) del TiVN
con respecto al TiN.
IV.11. OTROS MATERIALES DE ALTA DUREZA
En la siguiente tabla 4.3 [ref 17] se muestran las propiedades físicas de
otros materiales de alta dureza que se pueden usar alternativamente o
complementariamente con el nitruro de titanio.
20 40 60 80 100 120 140
0
5000
10000
15000
20000
25000
30000
TiN (222)
TiN (220)
TiN (111)
Inten
sidad
(u.
a.)
2 (grados)34 35 36 37 38 39 40
0
10000
20000
30000
40000
50000
60000
TiN (111)
TiVN (111)
Inte
nsid
ad (u
. a.)
2 (grades)

Capítulo IV Recubrimientos duros
112
TABLA 4.3
Propiedades físicas de algunos materiales de alta dureza
Material Dureza Vickers
(kgmm-2)
Punto de fusión
( C)
Módulo de Young
(kgmm-2)
TiN 2100 2950 590
TiC 2800 3067 460
TiO2 110 1867 200
TiCN 2200 (*) (*)
TiAl 600 (*) (*)
TiAlN 2200 (*) (*)
TiB2 3000 3225 560
ZrO2 1200 2710 200
SiO2 1100 1700 151
HfC 2700 3928 460
TaC 1600 3985 560
WC 2300 2776 720
WC-6% Co (*) 1500 640
BN 2730 440
(*) Información no disponible

Capítulo IV Recubrimientos duros
113
HSS 900 1400 250
REFERENCIAS
[1] L. Karlsson, Arc Evaporated titanium Carbonitride Coatings, Ph.D. Thesis, Linköping
University, 1999.
[2] S. Rudenja, P.Kulu, E.Tallimets, V.Mikli, C.A. Straede, Surface and Coatings
Technology, 100-101 (1998) 247-250.
[3] H. Ljungcrantz, L. Hultman, and J.-E. Sundgren J. Appl. Phys. 78 (2), 1995, 832-837.
[4] William D. Callister, Jr. Introducción a la Ciencia e Ingeniería de los Materiales, Editorial
reverté S.A. 1995
[5] J. Esteve Pujol, Elena Martínez, Medidas de Adhesión y Fricción mediante el método de
scatch test, Conferencia, Ensenada B.C. , Mexico, 1998.
[6] M. Larsson, Deposition and Evaluation of Thin Hard PVD Coatings, Ph.D. Thesis,
Uppsala University, 1996.
[7] U. Wiklund, P. Hedenqvist, S. Hogmark, Surface and Coatings Technology 97 (1997)
773-778.
[8] Å. Kassman, S. Jacobson, L. Erickson, P. Hedenqvist and M. Olsson, Surface and
Coatings Technology, 50 (1991) 75-84
[9] R. Gåhlin, M. Larsson, P. Hedenqvist, S. Jacobson, S. Hogmark, Surface and Coatings
Technology, 90 (1997) 107-114.
[10] U.Wiklund, Mechanics and Tribology of Micro-and Nanolayered PVD coatings, Ph. D.
Thesis, Uppsala University 1999.
[11] G.A. Garnizo-Demo, F.L. Lama, Surface and Coatings Technology, 76-77 (1995) 487-
493.
[12] Takadoum, H. Houmid Bennani, M. Allouard, Surface and Coatings Technology, 88
(1996) 232-238.
[13] I.M. Hutchings, Tribology, CRC Press, Boca Raton, FL, 1992, 26-35.
[14] D. Klaffke, W. Wasche, H. Czichoz, Wear, 153 (1992) 149
[15] H. Ronkainen, J. Likonen and J. Koskinenen, Surface and Coatings Technology 54/55
(1992) 570.
[16] W.C. Oliver, and G.M. Pharr, J. Mater. Res., Vol. 7 No. 6, 1992, 1564-1583.
[17] Milton Ohring, The Materials Science of Thin Films, Academic Press, INC, 1991
[18] Micro Photonics Surface Test

Capítulo IV Recubrimientos duros
114

Capítulo V Recubrimientos anticorrosivos
114
CAPITULO V
RECUBRIMIENTOS ANTICORROSIVOS
La corrosión, en el sentido más amplio, es la destrucción o el
deterioro de un material a causa de su reacción con el medio ambiente [ref 1]. En
este capítulo nos referimos a la corrosión de metales. Esta puede considerarse
como un proceso inverso al proceso metalúrgico extractivo. En su estado natural
los metales se encuentran formando compuestos y el ingeniero metalurgista tiene
que usar diferentes métodos físicos y químicos para purificarlos, sin embargo, la
tendencia natural de los metales es combinarse con otros elementos químicos para
formar compuestos, y en cierto modo, regresar a su estado natural. Esta tendencia
se explica desde el punto de vista termodinámico por la necesidad que tiene
cualquier sistema de minimizar su energía libre de Gibbs.
La importancia del fenómeno de corrosión desde el punto de vista
económico puede quedar establecida con los siguientes datos estadísticos: Causa
pérdidas del orden del 3,5 % del producto bruto interno en los países desarrollados
y en vías de desarrollo [ref 2] . Un cuarto a un tercio de la producción mundial de
acero se dedica a la reposición de las estructuras metálicas deterioradas [ref 3].
Según el mecanismo de reacción los procesos de corrosión pueden
clasificarse en: (i) Corrosión seca (oxidación directa), aquella en la que el metal
reacciona con un medio no iónico, por ejemplo, oxidación en aire a altas
temperaturas y (ii) Corrosión electroquímica es la que ocurre en presencia de un
electrolito.

Capítulo V Recubrimientos anticorrosivos
115
En este capítulo exploramos las posibilidades de disminuir la
velocidad de corrosión de algunos metales de uso generalizado en la industria
recubriendo su superficie con capas de materiales producidos por la técnica de
evaporación iónica. En concreto, estudiaremos dos casos: (i) Los efectos de
recubrimientos de titanio y nitruro de titanio sobre acero y cobre y (ii) los efectos
de la aleación TiAl para disminuir la velocidad de oxidación del cobre a altas
temperaturas.
V.1 CORROSION ELECTROQUIMICA
Debido a las imperfecciones microscópicas de un metal ( impurezas, dislocaciones, etc.)
las superficies metálicas no son homogéneas y es posible obtener naturalmente
acumulaciones de cargas positiva y negativa que actúan como ánodos y cátodos locales.
Si estas superficies están expuestas a medios donde hay compuestos iónicos se
establecen entonces corrientes iónicas similares a las que se producen en una pila. Estas
corrientes iónicas están relacionadas a un conjunto de reacciones químicas que se
producen en lo electrodos. Las reacciones más comunes que se producen en un
fenómeno de corrosión son:
Reacción anódica (oxidación)
Me Mez+ + ze- (disolución del metal)
Reacción catódica (reducción)
½ O2 + H2 O + 2 e- 2OH- (en medios neutros o alcalinos)
2 H+ + 2 e- H2 (en medios ácidos)
Me z + + z e- Me (reducción de cationes metálicos)

Capítulo V Recubrimientos anticorrosivos
116
Para comprender el significado de los parámetros que aparecen al explicar
un fenómeno de corrosión electroquímica comparemos una fuente de fuerza
electromotriz, batería o pila con las micropilas que se obtienen en el proceso de
corrosión. Una diferencia de potencial característica se establece entre cada electrodo y
el medio electrolítico de una batería. Análogamente una diferencia de potencial aparece
entre el medio corrosivo y cada uno de los microelectrodos que se forman en la
superficie metálica Una corriente estacionaria se establece cuando una resistencia de
carga RL se conecta entre los electrodos de la pila. Esta corriente es iónica dentro del
medio electrolítico y electrónica a través de la carga. Sucede exactamente lo mismo en
las microbaterías de corrosión, Una corriente Icorr circula entre los microelectrodos, el
medio corrosivo y una parte neutra del metal, con la diferencia que la resistencia de
carga es parte misma del material y no es removible.
a)
Pb
O=
SO4=
carga
-
-
-
-
b)
Fig 5.1 Analogía entre una pila o fuente de fuerza electromotriz y las micropilas que se
forman en un proceso de corrosión electroquímica de metales.
El parámetro más importante es la corriente de corrosión por unidad de área, icorr , la
cual está directamente relacionado a la velocidad de corrosión del metal. El problema es
que Icorr no se puede medir como la corriente en el circuito de la figura 5.1a, porque en

Capítulo V Recubrimientos anticorrosivos
117
los circuitos de la figura 5.1b no podemos insertar un amperímetro. Los métodos de
análisis que discutimos en la próxima sección tienen el propósito de determinar la
corriente de corrosión, icorr. Otro parámetro importante es Ecorr que es el potencial
común, mixto o potencia l de corrosión de un metal en medio con respecto a un
electrodo de referencia. Este parámetro sí se puede medir directamente y da información
del peligro real de los pares galvánicos formados por acoplamiento de materiales
metálicos diferentes.
V.1.1 METODOS DE ANÁLISIS DE CORROSION ELECTROQUIMICA
Curvas de polarización
Un método para estimar la corriente de corrosión Icorr consiste en
introducir la muestra en estudio en una solución electrolítica y construir una celda
electrolítica como se muestra en la figura 5.2. Se mide el potencial de equilibrio (Ecorr)
entre la muestra (electrodo de trabajo, ET) y el electrodo de referencia (ER). Con ayuda
de un potenciostato y un contraelectrodo inerte se aplica la corriente necesaria para que
el potencial del electrodo de trabajo vaya alcanzando sucesivos potenciales más
positivos respecto al electrodo de referencia. El respectivo gráfico corriente vs potencial
constituye la curva de polarización anódica. Similarmente se aplica la corriente
necesaria para que ET vaya disminuyendo su potencial respecto a ER y se obtiene la
curva de polarización catódica.
La extrapolación de los valores de corriente de ambas curvas (catódica y
anódica) para el valor de E = Ecorr es la corriente de corrosión Icorr. Este método de
estimación de la corriente de corrosión fue propuesto por U. R. Evans en 1932 (ref 27),
razón por la cual al conjunto de curvas de polarización catódica y anódica se llama
diagrama de Evans. En la figura 3 se muestra un típico diagrama de Evans. Se usa
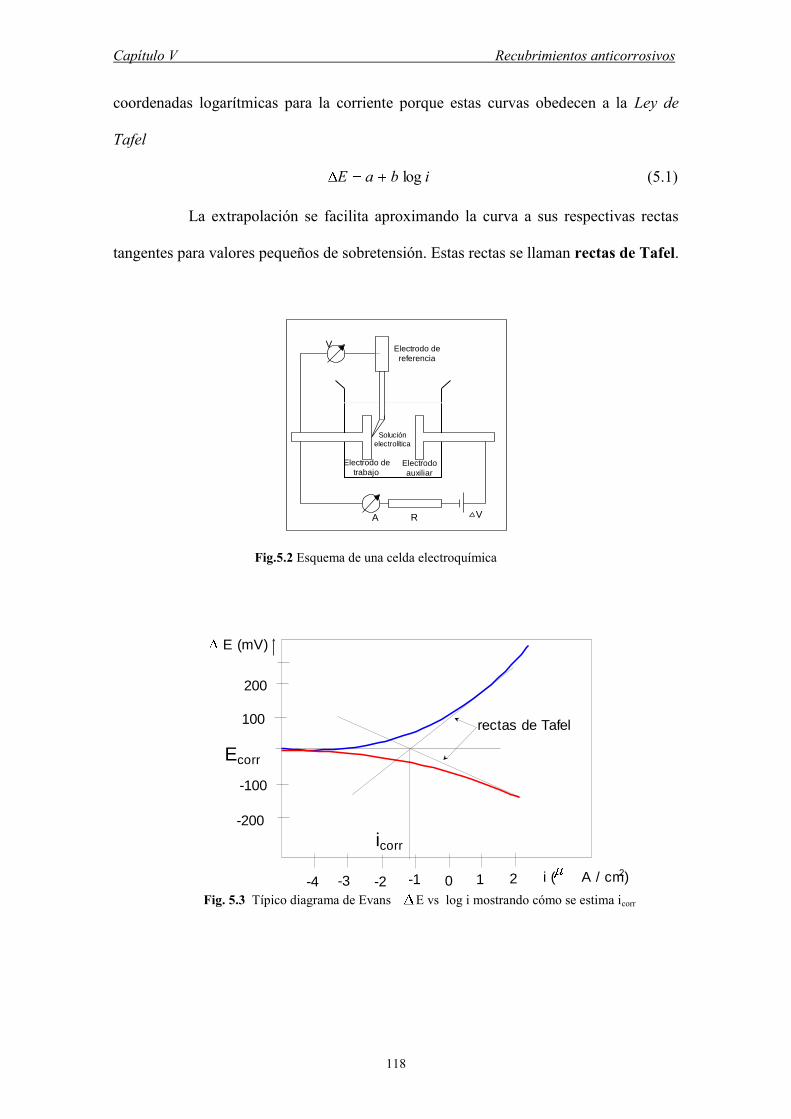
Capítulo V Recubrimientos anticorrosivos
118
coordenadas logarítmicas para la corriente porque estas curvas obedecen a la Ley de
Tafel
ibaE log (5.1)
La extrapolación se facilita aproximando la curva a sus respectivas rectas
tangentes para valores pequeños de sobretensión. Estas rectas se llaman rectas de Tafel.
Solución
electrolítica
Electrodo de
referencia
Electrodo de
trabajoElectrodo
auxiliar
V
A R V
Fig.5.2 Esquema de una celda electroquímica
Ecorr
icorr
rectas de Tafel100
200
-100
-200
-4 -3 -2 10-1 2 i ( A / cm2)
E (mV)
Fig. 5.3 Típico diagrama de Evans E vs log i mostrando cómo se estima icorr

Capítulo V Recubrimientos anticorrosivos
119
Método de resistencia a la polarización lineal
Un método alternativo para obtener la corriente de corrosión icorr consiste en
obtener la resistencia a la polarización de la celda electrolítica. Esto significa hacer
pequeñas variaciones del potencial alrededor del Ecorr, ajustar estas curvas a una recta y
obtener la pendiente.
IE
ILím
pR0 (5.2)
La corriente de corrosión se estima entonces por la ecuación:
pR
Bcorri (5.3)
donde Rp es la resistencia a la polarización y B es una constante de proporcionalidad.
Una variedad de métodos son usados para determinar la constante B [ref
14], siendo el más utilizado el uso de las pendientes anódica ( a) y catódica ( b) de las
rectas de Tafel en las curvas de polarización; en este trabajo calculamos B por la
siguiente relación:
cacaB
3..21
(5. 4)
donde a y c son respectivamente las pendientes de las rectas de Tafel anódica y
catódica respectivamente.

Capítulo V Recubrimientos anticorrosivos
120
El potenciostato
En la figura 5.4 se muestra el circuito potenciométrico utilizado para obtener
las curvas de polarización, cuyo principio de funcionamiento es el siguiente: el voltaje
V2 medido entre el electrodo de referencia y el electrodo de trabajo es registrado por el
amplificador A2 y llevado hacia el amplificador operacional A1. El usuario selecciona
el valor de entrada POL V, en condiciones de equilibrio, no hay salida de corriente a
través de A1 debido a que los potenciales de entrada son iguales, se prefija la velocidad
de barrido del potencial de entrada POL V, a polarizaciones positivas o negativas,
forzando al sistema a salir del equilibrio, midiéndose una corriente resultante que es la
que envía el amplificador operacional A1 hacia el interior de la celda vía el
contraelectrodo (CE), si en el curso del ensayo V1 y V2 difieren, el circuito ajusta
electrónicamente la corriente de salida hasta que V2 - V1 = 0, quedando restituida la
diferencia de potencial programada entre el electrodo de trabajo y el electrodo de
referencia.
- A1
+
A2
R1
R2
-POLV
CE
ER
ETCelda
Rs
V2
V1
Fig. 5.4 Esquema del circuito potenciostático

Capítulo V Recubrimientos anticorrosivos
121
V.1.2 PRODUCCION DE RECUBRMIENTOS ANTICORROSIVOS
Condiciones de deposición
Los recubrimientos producidos por la técnica magneto evaporación iónica
dc están constituidos por dos materiales: titanio y nitruro de titanio. Ambos materiales
fueron producidos casi bajo las mismas condiciones, con las siguientes diferencias: (i)
que durante la deposición del titanio metálico no se introduce nitrógeno en la cámara,
(ii) la deposición del titanio metálico fue hecha a menor temperatura del substrato que
en el caso del TiN y (iii) generalmente los tiempos de deposición para el titanio fueron
menores.
TABLA 5.1
Condiciones de deposición para la deposición de Ti y TiN
Titanio Nitruro de titanio
Blanco Disco de titanio 76 mm Disco de titanio 76 mm
Pureza del blanco 99.97% 99.97%
Pureza del Ar 99.999% 99.999%
Pureza del N2 99.997% 99.997%
temperatura de substrato 100 a 2000C 300 a 4500C
Presión de fondo 5 10-6 mbar 5 10-6 mbar
Presión parcial de N2 - 4 10-4 mbar
Presión total 4 10-3 mbar 4 10-3 mbar
Corriente 300 mA 400 mA
Voltaje 600 V 800 V
tiempo de deposición 8 a 10 minutos 8 a 240 minutos
Los espesores de las diferentes capas fueron medidas haciendo un cráter en la
superficie de la película con la misma técnica que para estudiar el desgaste por
abrasión y midiendo el diámetro luego con ayuda de un microscopio óptico.

Capítulo V Recubrimientos anticorrosivos
122
a)
b)
Fig. 5.5 Micrografías ópticas donde se puede observar cráteres hechos sobre muestras de acero de
alta velocidad. a) muestra tipo 1, 10 minutos de titanio y 2 horas de nitruro de titanio b) muestra
tipo 4a donde cada capa de titanio fue depositada por 10 minutos y cada capa de nitruro de titanio
por 10 minutos.
V.1.3 INFLUENCIA DE LOS RECUBRIMIENTOS SOBRE LA CORROSION
La hipótesis del presente trabajo es la siguiente: siendo la causa más
frecuente de la destrucción de metales la corrosión atmosférica y existiendo factores que
aceleran dicho proceso; tal es el caso de la contaminación atmosférica originada por
iones Cl- y SO4=, hemos elegido como solución electrolítica NaCl 0.01M y Na2SO4 0.1
M . En esta sección describimos los experimentos realizados para investigar el proceso
de corrosión de cobre electrolítico y acero martensítico revenido sin recubrir y
recubiertos.
Las muestras investigadas fueron las siguientes:
1. Discos de cobre electrolítico
2. Muestras tipo 2a : Discos de cobre recubiertos con una capa de 0,5 m de titanio
y 2 m de nitruro de titanio.

Capítulo V Recubrimientos anticorrosivos
123
3. Muestras tipo 2b : Discos de cobre recubiertos con una capa de 0,5 m de titanio
y 4 m de nitruro de titanio.
4. Cuchillas de acero martensítico revenido.
5. Muestras tipo 3a : Secciones de las cuchillas anteriores recubiertos con una capa
de 0,5 m de titanio y 2 m de nitruro de titanio.
6. Muestras tipo 3b : Secciones de las mismas cuchillas recubiertos con una capa
de 0,5 m de titanio y 4 m de nitruro de titanio.
7. Muestras tipo 4a : cuchillas recubiertas con tres capas de Ti y TiN intercaladas
siendo el espesor de la capa de titanio de 0,4 m y de la capa de TiN 0,4 m .
8. Muestras tipo 4b : cuchillas recubiertas con tres capas de Ti y TiN intercaladas
siendo el espesor de la capa de titanio de 0,4 m y de la capa de TiN 0,8 m .
9. Muestras tipo 4c : cuchillas recubiertas con tres capas de Ti y TiN intercaladas
siendo el espesor de la capa de titanio de 0,4 m y de la capa de TiN 1,2 m .
Las pruebas realizadas fueron tres: Potencial de corrosión, Curvas de
polarización y resistencias a la polarización.:
POTENCIALES DE CORROSION LIBRE (Ecorr)
Para obtener el valor del potencial de corrosión libre, Ecorr, no se induce
corriente alguna, sólo hay que colocar un voltímetro entre el electrodo de trabajo (ET) y
el electrodo de referencia (ER) equilibrio. En la figura 5.6 Se representa
esquemáticamente el sistema empleado para la obtención del Ecorr. Los datos obtenidos
del Ecorr fueron tomados una hora después de introducir la muestra en la solución y
posteriormente cada 24 horas para todas las muestras en estudio y en ambiente
controlado a una temperatura de 220 C aproximadamente.
El sistema electródico utilizado fue:

Capítulo V Recubrimientos anticorrosivos
124
(i) Electrodos de trabajo (cobre y acero rápido no recubiertos y recubiertos con
TiN/Ti).
(ii) Electrodo de referencia de Plata- Cloruro de plata 3M (Ag/AgCl).
(iii) Electrolito: NaCl 0,01M más Na2SO4 0,1 M, dicho electrolito fue elegido a
fin de simular la corrosión atmosférica del material en estudio en presencia
de iones cloruro (Cl-) y sulfatos (SO4=) como elementos contaminantes.
V
Fig. 5.6 Representación del sistema para la obtención del Ecorr
CURVAS DE POLARIZACION
El dispositivo experimental utilizado para obtener las curvas de polarización
consta de una celda electrolítica, conteniendo en ella los siguientes componentes: un
electrodo de trabajo (material a estudiar), electrodo de referencia (Ag/AgCl 3M) y un
contraelectrodo de platino, estos asociados a un circuito potenciostático, que a su vez
está conectado a una computadora.
Los pasos a seguir para la obtención de las curvas de polarización son:
(1) Se prefija el potencial a circuito abierto (Ecorr) entre el electrodo de trabajo
respecto al electrodo de referencia, el tiempo de estabilización para Ecorr fue de
15 minutos.

Capítulo V Recubrimientos anticorrosivos
125
(2) Luego el circuito es cerrado, mediante el programa M352 [26] existente en el
computador, se prefijo el potencial obtenido a circuito abierto Ecorr dando un
barrido desde -250 mV a + 250 mV a partir del Ecorr.
(3) La velocidad de barrido fue de 0,4 mV/seg y el área expuesta del electrodo de
trabajo fue de 0,5 cm2 para todas las muestras en estudio. El electrolito utilizado
fue una solución de NaCl 0.01M y Na2SO4 0.1 M.
Sistema electródico
El dispositivo electródico utilizado está conformado por tres electrodos
dispuestos en una célula electrólitica (ver Fig.2.8), conteniendo en ella una solución de
NaCl a una concentración de 0.01M más Na2SO4 a 0.1 M.
Electrodo de trabajo: en el presente trabajo se utilizó
(4) El cobre no recubierto y recubierto con TiN/Ti.
(5) El acero martensítico revenido no recubierto y recubierto con TiN/Ti, así como
también con multicapas de TiN/Ti..
Electrodo de referencia: fue empleado el electrodo de Ag-AgCl 3M, este electrodo se
encuentra conectado al electrodo de trabajo a través de un puente salino (capilar de
Luggin)
Electrodo auxiliar o contra electrodo: este electrodo tiene la propiedad de no corroerse,
es decir es inerte, en este caso se utilizó como contraelectrodo el platino.
El equipo utilizado:
Potenciostato-glavanostato Marca EG& G, Modelo 273A

Capítulo V Recubrimientos anticorrosivos
126
AV
ELECTRODO AUXILIAR
ELECTRODO DEREFERENCIA
ELECTRODODE
TRABAJO
Fig. 5.7 Esquema del montaje de una celda electrolítica para la
obtención de las curvas de polarización.
CURVAS DE POLARIZACION LINEAL
Las curvas de polarización lineal se obtienen en forma similar a las curvas
de polarización descritas anteriormente pero el barrido de potencial está sólo en el rango
Ecorr 20 mV. Fue realizado en 30 segundos y los datos de potencial E, y la corriente
I, fueron almacenados automáticamente con ayuda de una computadora. Las curvas E vs
I, obtenidas son ajustadas a rectas dentro del intervalo de voltajes Ecorr 5 mV. La
pendiente de cada curva proporciona el correspondiente valor de Rp. Los valores de Rp
fueron medidas de esta manera para las nueve muestras reportadas en las tablas 5.6 y
5.7
V.1.4 RESULTADOS EXPERIMENTALES Y DISCUSION
A) Potenciales de corrosión libre
En la tabla 5.2 se presenta la evolución en el tiempo del parámetro Ecorr
para no recubierto y recubierto con bicapas de titanio y nitruro de titanio con diferentes
espesores (muestras 2a y 2b). Los datos presentados son para tiempos de 1h, 24h, 48h y
100h de permanencia de las muestras en la solución.

Capítulo V Recubrimientos anticorrosivos
127
La tabla 5.3 se refiere al acero martensítico revenido de las cuchillas y al
mismo material recubierto con bicapas de titanio y nitruro de titanio de 2 y de 4
m (muestras 3a y 3b). En este caso presentamos datos para 1h, 24h y 100h de
permanencia en la solución.
En la tabla 5.4 se presenta los resultados referidos a las cuchillas recubiertas
con multicapas de titanio y de nitruro de titanio intercaladas. Las capas de titanio de 0,5
m y las de nitruro de titanio tuvieron un espesor de 0,4, 0,8 y 1,2 m ,
respectivamente.
Comentarios acerca de los recubrimientos sobre cobre
El cobre es uno de los metales más nobles (menos activos), por lo tanto
menos proclives a la corrosión electroquímica. Es importante observar que los
recubrimientos de titanio y nitruro de titanio refuerzan esta propiedad del cobre
haciéndolo aún menos activo. Esta tendencia aumenta con el transcurrir del tiempo.
Comentarios acerca de los recubrimientos sobre las cuchillas
Los valores de potencial que presenta el acero matensítico revenido indica la
gran tendencia a la corrosión que presenta dicho material. Esto fue evaluado sólo hasta
las 48 horas, debido a la rápida disolución del material lo cual alteraba la composición
de la solución no permitiendo realizar una toma real de datos del material en estudio.
Al recubrir el acero de las cuchillas con bicapas de titanio y nitruro de
titanio, se observó que la tendencia a la oxidación (potenciales menos negativos)
disminuye pero a las 48 horas ésta experimenta un salto acercándose a la del material no
recubierto. Los resultados obtenidos para el Ecorr y el comportamiento que presentan los
recubrimientos de TiN en el tiempo están en concordancia con los resultados obtenidos
por Ries y colaboradores [ref 17]; quienes mostraron, que este fenómeno está asociado
con la aparición de picaduras (pitting) en el recubrimiento.
Capítulo V Recubrimientos anticorrosivos
128
TABLA 5.2
Evolución del ECORR, obtenido para el cobre y cobre recubierto, en una solución de
NaCl 0.01M y Na2SO4 0.1 M, en función del tiempo
Material estudiado 01 hora
Ecorr (mV)
24 horas
Ecorr (mV)
48 horas
Ecorr (mV)
100 horas
Ecorr (mV)
Cobre -261 -190 -187 -168
muestra 2a -236 -144 -91 -41
muestra 2b -200 -165 -170 -51
TABLA 5.3
Evolución del ECORR, obtenido para el acero rápido y acero recubierto, en una
solución de NaCl 0.01M y Na2SO40.1 M, en función del tiempo
Material estudiado 01 hora
Ecorr (mV)
24 horas
Ecorr (mV)
48 horas
Ecorr (mV)
cuchilla -721 -731 -754
muestra 3a -565 -502 -601
muestra 3b -532 -524 -591
TABLA 5.4
Evolución del ECORR, obtenido para el acero rápido recubierto con una multicapa
de TiN/Ti en una solución de NaCl 0.01M y Na2SO4 0.1 M, en función del tiempo
Multicapas de
TiN/Ti/Acero martensítico
revenido
01 hora
Ecorr (mV)
24 horas
Ecorr (mV)
100 horas
Ecorr (mV)
muestra 4ª -568 -581 -637
muestra 4b -497 -565 -607
muestra 4c +42 +15.7 +14

Capítulo V Recubrimientos anticorrosivos
129
Como puede observarse en la tabla 5.4 los valores de Ecorr para las cuchillas
recubiertas con multicapas relativamente delgadas (muestras 4a y 4b) tuvieron valores
parecidos a los mostrados en la tabla 5.3 para cuchillas recubiertas con bicapas. Se
observó que el salto en el Ecorr es atribuido a las picaduras que aparecieron a las 5 horas
de estar inmersa la cuchilla en la solución.
El acero recubierto con las multicapas cuyas capas de TiN son mas
gruesas, 1,2 m de espesor (muestra 4c), muestra un comportamiento mucho más noble
que el substrato y todas las muestras descritas anteriormente. El Ecorr para las muestras
tipo 4c varía de + 42 a +14 mV al cabo de 100 horas indicando una mínima tendencia a
la corrosión. Esta notable mejora es atribuida a una mayor compacidad del
recubrimiento, es decir, una disminución de la porosidad. Es de resaltar que al cabo de 5
días, no se formaron picaduras en la superficie del recubrimiento.
B) Curvas de polarización
En las figuras 5.8, 5.9, y 5.10 se presentan las curvas de polarización
experimentales para los diferentes materiales en estudio.
En la Fig.5.8 se muestran las curvas de polarización obtenidas para el cobre
sin recubrimiento y recubierto con TiN/Ti (muestra 2b). El Ecorr para el cobre fue -219
mV, luego de permanecer en la solución 1 hora. Para la muestra 2b el Ecorr fue de –96
mV, luego de permanecer en la solución 15 minutos.
Se puede observar de la figura 5.8 un corrimiento hacia la izquierda de la
curva de polarización para el TiN/Ti/Cobre, indicando una disminución en un orden de
magnitud en la densidad de corriente de corrosión, icorr, respecto a la del cobre sin
recubrimiento.

Capítulo V Recubrimientos anticorrosivos
130
La figura 5.9 muestra las curvas de polarización para el acero sin
recubrimiento y recubierto con TiN/Ti (muestra 3b); el Ecorr para el acero fue -767 mV,
luego de permanecer en la solución 2 horas y para el TiN/Ti/acero fue de –491 mV,
luego de permanecer en la solución 1 hora.
En la figura 5.10 se muestran las curvas de polarización para el acero
recubierto con la multicapa TiN/Ti. Las tres muestras fueron evaluadas luego de
encontrarse sumergidas en el medio agresivo (NaCl 0.01M y Na2SO4 0.1 M) durante
un tiempo de 15 minutos. El Ecorr para la muestra 4a fue de -421 mV, para la muestra 4b
fue de –493 mV y para la muestra 4c de -326 mV.
En la figura 5.10 se observa que la muestra 4c tiene mejores propiedades
anticorrosivas que las muestras 4a y 4b. Pues el potencial de corrosión Ecorr es menos
negativo para la muestra 4c y la intersección de las pendientes (no están dibujadas) en
esta muestra está más corrida hacia la izquierda, indicando un menor icorr. Sin embargo,
estas primeras observaciones deberian ser verificadas cuantitativamente, para lo cual se
utilizará el valor de la constante B que nos servirá para obtener los valores de icorr para
cada muestra estudiada a partir de los ensayos de Rp.
Capítulo V Recubrimientos anticorrosivos
131
-4 -3 -2 -1 0 1 2
-500
-400
-300
-200
-100
0
100
200
TiN/Ti/Cu, muestra 2b
Cobre sin recubrimiento
E v
s.A
g/ A
gC
l (m
V)
Log i ( A/cm2)
Fig.5.8 Curvas de polarización para el Cobre y cobre recubierto con TiN/Ti.
-4 -3 -2 -1 0 1 2
-1000
-900
-800
-700
-600
-500
-400
-300
-200
Acero sin recubrimiento
TiN/Ti/Acero, muestra 3b
E v
s. A
g/ A
gC
l (m
V)
Log i ( A/cm2)
Fig.5.9 Curvas de polarización para el acero y acero recubierto.
-3 -2 -1 0 1 2 3-800
-700
-600
-500
-400
-300
-200
-100
muestra 4c
muestra 4a
muestra 4b
E v
s.
Ag/A
gC
l (m
V)
Log i ( A/cm2)
Fig.5.10 Curvas de polarización para acero recubierto con multicapas
de titanio y nitruro de titanio.

Capítulo V Recubrimientos anticorrosivos
132
Constante de proporcionalidad B
En la tabla 5.5 se muestran los valores de las pendientes anódica y catódica
obtenidas de las curvas de polarización, extrapolando las rectas de Tafel.
Tabla 5.5
Constantes ba, bc y B obtenidas a partir de las curvas de polarización (Tafel)
Material en estudio a (V) c (V) B (V)
Cobre 0.283 0.162 0.045
TiN/Ti/Cu, muestra 2b 0.251 0.220 0.051
Acero martensítico revenido 0.111 0.217 0.032
TiN/Ti/acero, muestra 3b 0.136 0.199 0.035
Multicapa/acero muestra 4c 0.164 0.121 0.030
Comentario sobre los valores de las constantes:
Según Stern y Weisert los valores de a y c varían por lo general entre 0,03 V y
0,18 V, estando comprendidas en la mayoría de las veces entre 0,06 V y 0,12 V.
Los valores en esta tesis varían entre 0,11V y 0,28V. Los resultados
obtenidos se encuentran fuera de los márgenes según la referencia bibliográfica [ ref 5 ].
Sin embargo como hace constar Mansfeld, las pendientes de Tafel varían con el tiempo
para muchos sistemas, pero la constante de proporcionalidad es menos sensible a estos
cambios, al quedar afectados tanto el numerador como el denominador de B (ver
ecuación 5.4).
La precisión con la que se realizó el ajuste lineal para la obtención de las
pendientes anódica y catódica fue del 99%.

Capítulo V Recubrimientos anticorrosivos
133
C) Resistencia de polarización lineal (Rp)
En las figuras 5.11, 5.12 y 5.13 se muestran los gráficos E vs I obtenidos
para el cobre sin recubrimiento y recubierto con TiN/Ti (muestras 2a y 2b).
En las figuras 5.14, 5.15 y 5.16 se muestran los gráficos E vs I obtenidos
para el acero de las cuchillas sin recubrimiento y recubierto con TiN/Ti (muestras 3a y
3b).
En las figuras 5.17, 5.18 y 5.19 se muestran los gráficos E vs I obtenidos
para el acero de las cuchillas sin recubrimiento y recubierto con multicapas de titanio y
TiN descritas como muestras 4a, 4b y 4c.

Capítulo V Recubrimientos anticorrosivos
134
-0.2 -0.1 0.0 0.1 0.2 0.3
-280
-260
-240
-220
-200
-180
-160
100 horas
48 horas
24 horas
01hora
E v
s. A
g/A
gC
l (m
V)
I ( A)
Fig.5.11 Curvas de polarización lineal para el cobre sin recubrimiento
0.00 0.01 0.02 0.03-250
-200
-150
-100
-50
0
50
01 hora
100 horas
48 horas
24 horasE v
s.
Ag/A
gC
l (m
V)
I ( A)
Fig.5.12 Curvas de polarización lineal para el TiN/Ti/Cu, muestra tipo 2ª
-0.005 0.000 0.005 0.010
-200
-150
-100
-50
0
100 horas
24 horas
01 hora
48 horas
E v
s.
Ag/A
gC
l (m
V)
I ( A)
Fig. 5.13 Curvas de polarización lineal para el TiN/Ti/Cu, muestra tipo 2b

Capítulo V Recubrimientos anticorrosivos
135
0 2-800
-780
-760
-740
-720
-700
-680
48 horas
24 horas
01 hora
E v
s.A
g/
AgC
l (m
V)
I ( A)
Fig. 5.14 Curvas de polarización lineal para el acero sin recubrimiento
-0.10 -0.05 0.00 0.05 0.10-680
-660
-640
-620
-600
-580
-560
-540
-520
100 horas
48 horas
01 hora
24 horas
E v
s.A
g/ A
gC
l (m
V)
I ( A)
Fig. 5.15 Curvas de polarización lineal para el TiN/Ti/acero, muestras 3ª
-0.15 -0.10 -0.05 0.00 0.05
-660
-640
-620
-600
-580
-560
-540
-520
100 horas
48 horas
24 horas
01hora
E v
s. A
g/A
gC
l (m
V)
I ( A)
Fig. 5.16 Curvas de polarización lineal para el TiN/Ti/acero, muestras 3b
Capítulo V Recubrimientos anticorrosivos
136
-1 0 1-660
-650
-640
-630
-620
-610
-600
-590
-580
-570
-560
-550
-540
100 horas
28 horas5 horas
E v
s
Ag/A
gC
l (m
V)
I ( A)
Fig. 5.17 Curvas de polarización lineal para muestras tipo 4ª
-1 0 1-640
-620
-600
-580
-560
-540
-520
-500
-480
28 horas
100 horas
5 horas
E v
s A
g/A
gC
l (m
V)
I ( A)
Fig. 5.18 Curvas de polarización lineal para muestras tipo 4b
-0.3 -0.2 -0.1 0.0 0.1
0
20
40
60
80
100
120
140
160
180
100 horas
28 horas
05 horas
E v
s. A
g/A
gC
l (m
V)
I ( A)
Fig. 5.19 Curvas de polarización lineal para muestras tipo 4c

Capítulo V Recubrimientos anticorrosivos
137
En la tabla 5.6 se resumen los valores de resistencia a la polarización, Rp,
para el cobre y para el cobre recubierto con bicapas de titanio y nitruro de titanio. Se
observa un aumento en la resistencia a la polarización desde 51 k cm2 para el cobre
sin recubrir a 2,2 M cm2 para el cobre recubierto con capas de 0,5 m de titanio y 4
m de nitruro de titanio. Estos valores corresponden a las muestras después de haber
permanecido 100 horas en la solución electrolítica (NaCl 0.01M y Na2SO4 0.1 M).
En la tabla 5.7 se resumen los resultados obtenidos para el ensayo de
resistencia de polarización Rp del acero martensítico revenido y de éste recubierto con
bicapas TiN/Ti (muestras 3a y 3b), y de muestras recubiertas con multicapas (muestras
4a, 4b y 4c).
Si bien es cierto que la resistencia a la polarización para el acero recubierto
con la bicapa (muestras 3a y 3b) mejoró, incrementandose en 5 veces aproximadamente
para ambas, respecto al acero no recubierto. Estas medidas fueron superadas por la
multicapa de TiN/Ti/ Acero rápido (muestra 4c) para la cual Rp resultó quedar
multiplicada por un factor de 4000 respecto a la del acero no recubierto y por un factor
900 respecto al acero recubierto con bicapas (muestras 3a y 3b).
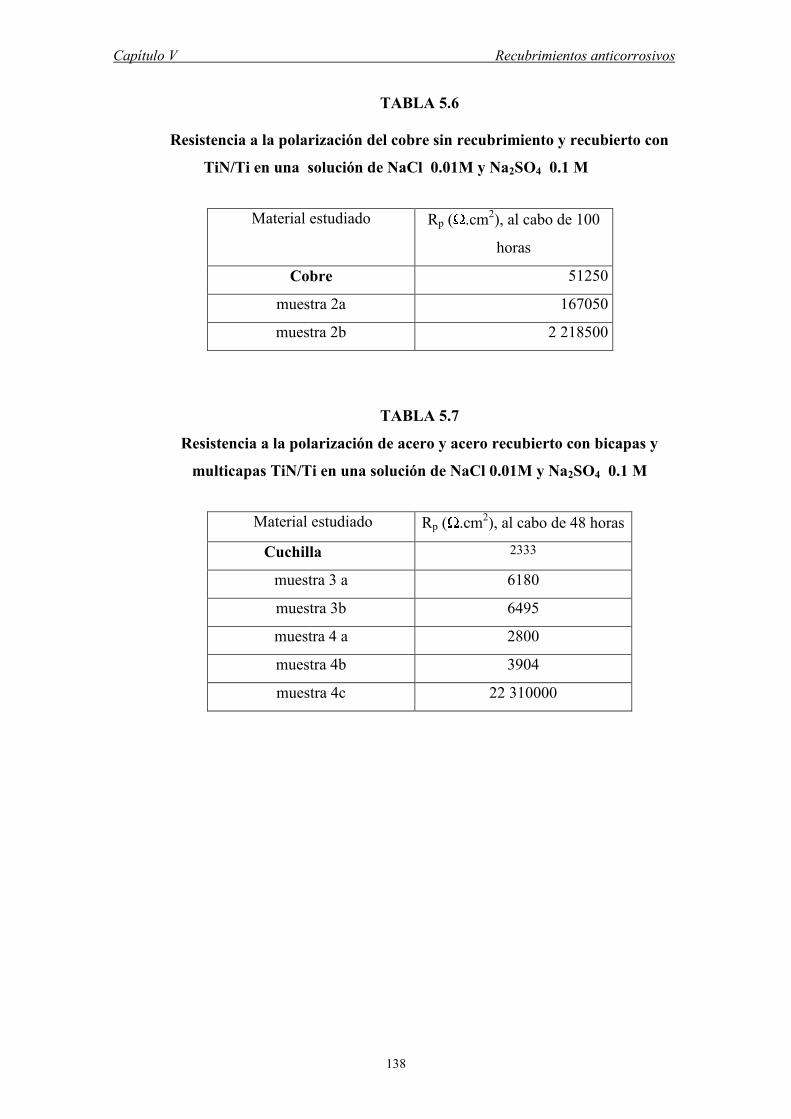
Capítulo V Recubrimientos anticorrosivos
138
TABLA 5.6
Resistencia a la polarización del cobre sin recubrimiento y recubierto con
TiN/Ti en una solución de NaCl 0.01M y Na2SO4 0.1 M
Material estudiado Rp ( .cm2), al cabo de 100
horas
Cobre 51250
muestra 2a 167050
muestra 2b 2 218500
TABLA 5.7
Resistencia a la polarización de acero y acero recubierto con bicapas y
multicapas TiN/Ti en una solución de NaCl 0.01M y Na2SO4 0.1 M
Material estudiado Rp ( .cm2), al cabo de 48 horas
Cuchilla 2333
muestra 3 a 6180
muestra 3b 6495
muestra 4 a 2800
muestra 4b 3904
muestra 4c 22 310000

Capítulo V Recubrimientos anticorrosivos
139
Evolución en el tiempo de la resistencia a la polarización lineal
Se efectuó un seguimiento a lo largo del tiempo del valor de Rp hasta
aproximadamente 100 horas de permanencia de cada muestra en el medio agresivo
(solución de NaCl 0.01M y Na2SO4 0.1 M).
En la figura 5.20 se muestra la evolución en el tiempo de Rp para el cobre y
para el cobre recubierto con bicapas TiN/Ti (muestra 2a y 2b). Observándose que el
metal sin recubrimiento muestra una resistencia a la polarización bastante menor (mayor
velocidad de corrosión) respecto al material recubierto. Asimismo se observa que el
metal recubierto con la capa de mayor espesor de TiN (0,4µm) mantiene un valor
elevado de Rp (menor corrosión) durante las 100h de ensayo.
La disminución de la resistencia de polarización en la muestra 2a al cabo de
80 horas a valores comparables con los del cobre sin recubrir puede ser atribuida a
fenómenos de corrosión por picadura. El mantenimiento de valores altos de Rp para la
muestra 2b, aún después de 100 horas de ensayo, indica que la corrosión por picaduras
del recubrimiento ha sido disminuidad con recubrimientos de mayor espesor.
En la figura 5.21 se muestra la evolución en el tiempo de la resistencia a la
polarización para el acero con bicapas de TiN/Ti (muestras 3a y 3b).
En la figura 5.22 se presenta la evolución en el tiempo de Rp para el acero
recubierto con multicapas (muestras 4a , 4b y 4c).
De la figura 5.21 se observa que el acero recubierto con bicapas TiN/Ti,
muestra 3b, presenta una resistencia a la polarización muy superior respecto al acero no
recubierto y a la muestra 3a, durante las primeras 24 horas aproximadamente.
Asimismo, se observa que las muestras recubiertas mencionadas mantienen un Rp
ligeramente superior a la del material sin recubrir, al cabo 100 horas, con lo cual se
verifica la mejora de la resistencia a la corrosión. La caída de la resistencia a la

Capítulo V Recubrimientos anticorrosivos
140
polarización (aumento de la corrosión) para las bicapas con respecto a la cuchilla puede
deberse probablemente a la porosidad y defectos existentes en dichos recubrimientos
[ref 18].
En la figura 5.22 se observa que la resistencia a la polarización lineal del
acero recubierto con multicapas de titanio y nitruro de titanio depende del espesor de
las capas de nitruro de titanio. En este tipo de probetas las capas de titanio fueron de
aproximadamente 0,5 m mientras que el espesor de las capas de nitruro de titanio fue
variable: 0,4 m (muestras 4a), 0,8 (muestras 4b) y 1,2 m (muestars 4c). No se
observa diferencia apreciable entre los valores Rp para las muestras 4a y 4b. Esta
elevada resistencia a la polarización se mantiene alta aún después de 100 horas de
permanencia de la muestra en la solución electrolítica, lo cual permite afirmar que en
este tipo de muestras se ha eliminado el problema de la corrosión por picaduras,
probablemente por ser más compacta y menos porosa.
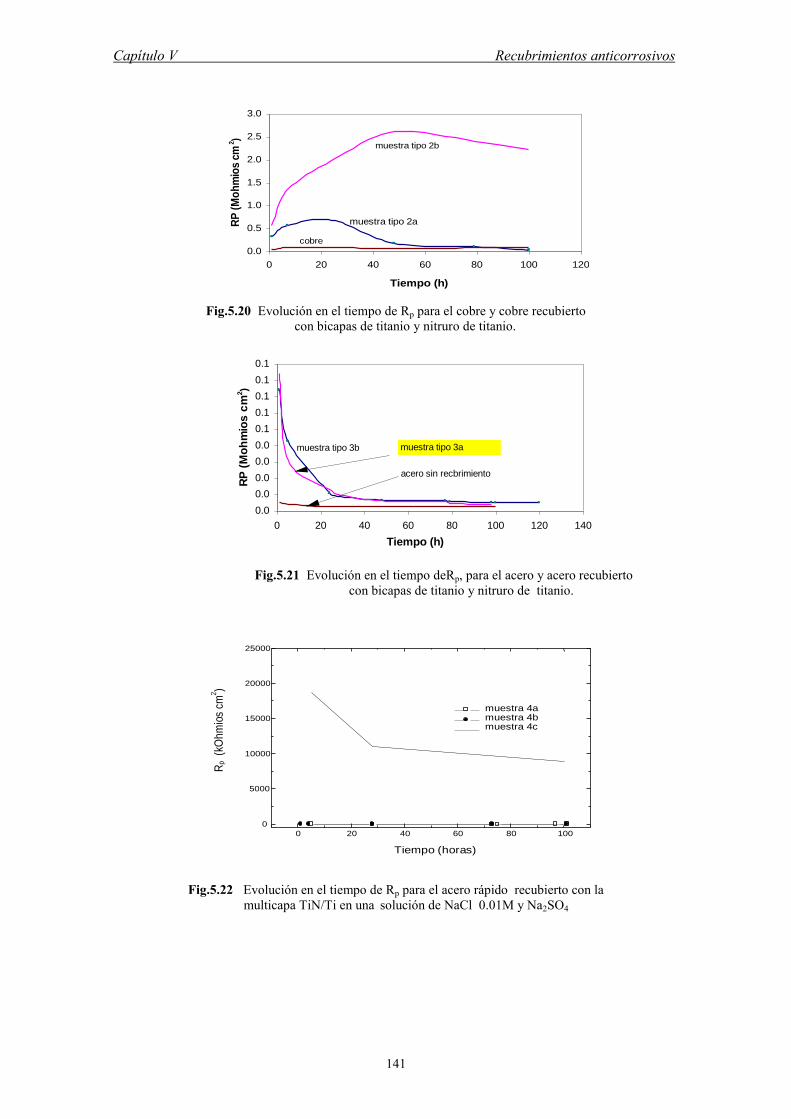
Capítulo V Recubrimientos anticorrosivos
141
0.0
0.5
1.0
1.5
2.0
2.5
3.0
0 20 40 60 80 100 120
Tiempo (h)
RP
(M
oh
mio
s cm
2)
muestra tipo 2b
muestra tipo 2a
cobre
Fig.5.20 Evolución en el tiempo de Rp para el cobre y cobre recubierto
con bicapas de titanio y nitruro de titanio.
0.0
0.0
0.0
0.0
0.0
0.1
0.1
0.1
0.1
0.1
0 20 40 60 80 100 120 140
Tiempo (h)
RP
(M
oh
mio
s c
m2)
muestra tipo 3b muestra tipo 3a
acero sin recbrimiento
Fig.5.21 Evolución en el tiempo deRp, para el acero y acero recubierto
con bicapas de titanio y nitruro de titanio.
0 20 40 60 80 100
0
5000
10000
15000
20000
25000
muestra 4amuestra 4bmuestra 4c
Rp
(kO
hm
ios
cm2)
Tiempo (horas)
Fig.5.22 Evolución en el tiempo de Rp para el acero rápido recubierto con la multicapa TiN/Ti en una solución de NaCl 0.01M y Na2SO4

Capítulo V Recubrimientos anticorrosivos
142
Densidad de corriente de corrosión (icorr)
En las figuras 5.23 y 5.24 se muestra la evolución en el tiempo del icorr para
las diferentes probetas estudiadas, calculadas según la ec. (5.4) y con ayuda de los
resultados experimentales de la tabla (5.5) y en las figuras 5 .20, 5.21, y 5.22.
En la figura 5.23 se puede observar que el cobre sin recubrimiento muestra
una densidad de corriente de corrosión muy superior respecto al material recubierto,
durante las primeras 100 horas aproximadamente. Así mismo, se observa que el metal
recubierto con capas de TiN de 4 m , muestra 2b , mantiene su densidad de corrosión
constante a partir de las 24 horas aproximadamente, disminuyendo en 83 veces
aproximadamente su densidad de corrosión. Podemos afirmar que el recubrimiento de
TiN/Ti , disminuye notablemente la velocidad de corrosión, es decir, mejora la
resistencia a la corrosión del cobre.
En la figura 5.24 se observa que el acero recubierto con la bicapa de titanio
y TiN, muestra 3b, presenta una velocidad de corrosión menor por un factor de 1,4 con
respecto al material no recubierto. Pero el resultado más importante de este trabajo es la
observación de que el metal recubierto con la multicapa, muestra 4c, presenta una
densidad de corriente de corrosión muy inferior respecto al material recubierto con la
bicapa y el material sin recubrimiento, dicho comportamiento se mantiene a lo largo del
tiempo. Este recubrimiento disminuye en 22000 veces aproximadamente respecto del no
recubierto.
En ambos casos los recubrimientos mejoran la resistencia a la corrosión del
acero rápido. Sin embargo se ha comprobado experimentalmente que las multicapas
mejoran notablemente la resistencia a la corrosión, protegiendo al material base del
medio agresivo al que estuvo expuesto (NaCl a 0.01M y Na2SO4 a 0.1 M). Los
resultados obtenidos para las bicapas están en concordancia con los presentados por

Capítulo V Recubrimientos anticorrosivos
143
Ries y colaboradores [ref 7], siendo nuestro aporte el mostrar evidencias de que el
problema de la corrosión por picadura puede ser resuelto alterando capas de Ti y TiN
con los espesores apropiados. En nuestro caso las multicapas más protectoras resultaron
ser aquellas con 500 nm de titanio y 1,2 m de nitruro de titanio.
0 20 40 60 80 100
0.00
0.05
0.10
0.15
0.20
0.25
TiN/Ti/cobre, muestra 2b
Cobre sin recubrimiento
i corr
(µA
/cm
2 )(
A/c
m2 )
Tiempo (h)
Fig.5..23 icorr para el cobre sin recubrimiento y TiN/Ti/cobre.
0 10 20 30 40 50 60 70-1
0
1
2
3
4
5
6
7
8
Multicapa TiN/Ti/acero, muestra 4c
TiN/Ti/acero, muestra 3b
Acero sin recubrimiento
i corr (µ
A/c
m2 )
Tiempo (h)
Fig.5.24 icorr para el acero sin recubrimiento y TiN/Ti/acero.
En la tabla 5.8 se muestra los resultados obtenidos de la densidad de
corriente de corrosión icorr para la muestra 2b, respecto al cobre no recubierto.

Capítulo V Recubrimientos anticorrosivos
144
En la tabla 5.9 se muestra la evolución en el tiempo de la densidad de
corriente de corrosión para las muestras 3b y 3b respecto al acero martensítico revenido
no recubierto.
TABLA 5.8
Densidades de corriente de corrosión para el cobre no recubierto y
recubierto con bicapas de titanio y nitruro de titanio para un tiempo de
100 horas, en una solución de: NaCl 0.01M y Na2SO4 0.1 M
Materiales estudiados icorr ( A/cm2)
Cobre 0.22173
TiN/Ti/Cu, muestra 2b 0.00268
TABLA 5.9 Densidades de corrosión para el acero martensítico revenido no recubierto y recubierto
con bicapas de titanio y nitruro de titanio y multicapas para un tiempo de 100 horas, en
una solución de: NaCl 0.01M y Na2SO4 0.1 M.
0.01M y Na2SO4 0.1 Mmateriales
estudiados
icorr ( A/cm2)
Acero martensítico revenido 5.72482
TiN/Ti/acero, muestra 3b 4.02466
Acero recubiertos con multicapa,
muestra 4c
0.00025

Capítulo V Recubrimientos anticorrosivos
145
V.2 CORROSION SECA
El cobre es uno de los recursos naturales más importantes de la minería en
nuestro país. Sin embargo sus aplicaciones tecnológicas en procesos a altas
temperaturas están muy limitadas por su tendencia a la oxidación. El proceso de
oxidación del cobre a altas temperaturas ha sido ampliamente estudiado [ref 9]. El
principal producto de oxidación es el óxido cuproso Cu2O, aunque después de un
tiempo de oxidación suficientemente largo se obtiene también óxido cúprico CuO. La
aleación intermetálica TiAl, en cambio, es conocida [ref 10,11] por su excelente
resistencia a la oxidación hasta alrededor de 7000C. Se conoce también que esta
aleación tiene la importante propiedad de aumentar su resistencia a la tracción con la
temperatura hasta 7000C.
Existen algunos estudios previos que muestran la posibilidad de obtener
recubrimientos de TiAl usando la técnica de evaporación iónica. Takakazu et al. [ref 10]
han producido este tipo de recubrimientos usando la técnica evaporación iónica rf con
blancos múltiples de titanio y aluminio; por otro lado, Hardwick y Cordi [ref 11] lo han
hecho usando blancos de composición Ti-53Al-3Nb y TiB2.
En esta sección se describe la producción de recubrimientos TiAl por la
técnica evaporación iónica dc, se muestran los resultados del análisis estructural de este
material. Por difracción de rayos X. Usando una técnica termogravimétrica
demostramos que la rapidez de oxidación de una superficie de cobre recubierta con TiAl
a 600 y 7000 C disminuye notablemente con respecto a una superficie de cobre no
recubierta.

Capítulo V Recubrimientos anticorrosivos
146
V.2.1 PRODUCCION DE LOS RECUBRIMIENTOS
El equipo para la producción de recubrimientos por la técnica evaporación iónica dc es
el sistema No. 3 mostrado en el capítulo 2 de este libro. El blanco fue construido
incrustando un disco de titanio 99.5% puro, de 35 mm de diámetro y 3 mm de espesor
en un disco de aluminio de 100 mm de diámetro y 6 mm de espesor, 99% puro, de modo
que la superficie de ambos discos estaba expuesta al bombardeo de iones de argón. Las
condiciones de deposición se muestran en la tabla 1.
TABLA 5.10
Condiciones de operación durante el proceso evaporación iónica dc
Presión de fondo 2 x 10-5 mbar
Presión durante deposición 8 x 10-3 mbar
Temperatura durante deposición 3500C
Voltaje 2700 V
Corriente 50 mA
Tiempo de deposición 2 a 3 horas
Tipos de substratos Láminas de cobre de 1 mm de espesor
DIFRACCION DE RAYOS X
La estructura cristalina de nuestros recubrimientos fue analizada usando un
difractómetro Philips PW 1130/00 operado a 40 kV y 20 mA . La radiación usada fue

Capítulo V Recubrimientos anticorrosivos
147
Cu K de 0.15418 nm de longitud de onda. La disposición -2 fue usada. El paso fue
de 0.010 y el análisis fue hecho entre 20 y 700.
TERMOGRAVIMETRIA
En la figura 2 se puede ver el equipo usado para las medidas
termogravimétricas. La técnica consiste en operar el horno hasta alcanzar una
temperatura determinada, se introduce la muestra y se espera 5 minutos para que ésta
alcance el equilibrio térmico; el cambio de peso de la muestra se mide usando una
balanza analítica con resolución de 0.001g. Los datos obtenidos son luego tabulados y
expresados en una gráfica de cambio de peso ( P) en función del tiempo (t). El cambio
de peso está directamente relacionado con el incremento del espesor de la capa de óxido
en la superficie del metal. Este equipo tiene facilidades para efectuar medidas en
atmósferas controladas conteniendo diferentes tipos de gases, pero en el presente
estudio las medidas fueron hechas sólo en presencia de aire.
Fig 5.25. Esquema del equipo para medidas termogravimétricas.

Capítulo V Recubrimientos anticorrosivos
148
V.2.2 RESULTADOS Y DISCUSIÓN
a) DIFRACCION DE RAYOS X
La estructura cristalina [ref 12] de la aleación TiAl en la fase es tetragonal con
parámetros de red a = 0.399 nm y c = 0.407 nm. En la figura 5 mostramos un
difractograma de un recubrimiento de 2.0 m de aleación TiAl sobre una placa de
cobre. Se observan claramente los picos en los valores 2 = 38.8, 44.5 y 65.0 que
corresponden a las familias de planos cristalinos (111), (002) y (202) respectivamente.
0
1000
2000
3000
4000
5000
6000
20 30 40 50 60 70
2 THETA (Angulo)
TiAl
TiAlTiAl
(002)
(111)(202)
Fig 5.26 Difractograma de rayos X de aleación TiAl producido por la técnica
evaporación iónica dc
B) TERMOGRAVIMETRIA
Placas de cobre de 7 cm de largo, 15 mm de ancho y 1 mm de espesor fueron usadas
para el estudio comparativo de la cinética de corrosión en superficies de cobre sin
recubrimiento y en superficies con recubrimiento TiAl de 2.0 m de espesor sobre cada
una de las dos caras de mayor área de las placas.
En la figura 6 se muestra el aumento de peso ( P) en función del tiempo para una placa
de cobre sin recubrimiento y otra de exactamente las mismas dimensiones pero con
recubrimiento, cuando la temperatura de las placas era de 6000C en atmósfera de aire.

Capítulo V Recubrimientos anticorrosivos
149
En la figura 7 se reportan resultados similares pero cuando la temperatura de las placas
era estable a 7000C.
0
10
20
30
40
50
60
70
80
90
100
0 5 10 15 20 25 30 40 50 60 70 80 90 100
Tiempo (minutos)
P (
milig
ram
os)
a
a
b
Fig. 5.27 Variación del peso en función del tiempo para una lámina de cobre a) sin recubrimiento y b)
con recubrimiento de aleación TiAl de 2.0 m de espesor. La temperatura de ambas placas fue de 6000C
0
20
40
60
80
100
120
140
0 5 10 15 20 25 30 40 50 60 70 80 90 100
Tiempo (minutos)
P (
milig
ram
os)
a
b
Fig. 5.28 Variación del peso en función del tiempo para una lámina de cobre a) sin recubrimiento y b)
con recubrimiento de aleación TiAl de 2.0 m de espesor. La temperatura de ambas placas fue de 7000C .

Capítulo V Recubrimientos anticorrosivos
150
Referencias
[ 1 ] J.A. Gonzales , “Control de la Corrosión: estudio y medida por técnicas
experimentales”, Editorial CENIM-CSIC, Madrid, 1984.
[ 2 ] Report of the committee on Corrosion and Protection, Dept of Trade and
Industry . HMSO Londres, 1971
[ 3 ] M. Pourbaix Lecciones de corrosión electroquímica. Ed. Instituto Español de la
corrosión, Madrid, 1987.
[ 4 ] U.R. Evans y T.P Hoar, Proc. Royal Soc., A 137 (1932) p 343
[ 5 ] R. Grauer, P.J. Moreland and G. Pini, “A Literature Review of Polarization
Resistance Constant (B) Values for the Measurements of Corrosion Rate” (1982)
[ 6 ] Corrosion Measurement and Analysis Software , Model 352 SoftcorrTM II,
Copyright 19991 EG y G Instruments Corporation.
[ 7 ] L.A.S. Ries, D.S. Azambuja, I.J.R. Baumvol, Surface and Coatings Technology,
89 (1997) 114-120.
[ 8 ] B.F. Chen, W.L. Pan, G.P.Yu, J. Hwang, J.H. Huang; Surface and Coatings
Technology, 111 (1999) 16-21.
[ 9] Arne Ross, Teddy Chibuye and Bjorn Karlsson in Preparation and Characterization
of Selective Surfaces on Cu prepared by chemical and thermal oxidation, Uppsala
University, 1986.
[ 10 ] Takakazu Suzuki et al. J. Mater. Res., Vol 9. No 4, p 1028 ,Apr 1994
[ 11 ] D.A. Hardwick and R.C. Cordi, en Intermetallic Matrix Composites, edited by
D.L. Anton, P.L. Martin, D.B. Miracle, and R. McMeeking (Mater. Res. Soc. Symp.
Proc. 194, Pittsburgh. PA 1990), p. 65
[ 12 ] Metals Handbook, Vol. 8 editado por ASM, 1985.

Capítulo VI Microbaterías recargables de litio
151
CAPÍTULO VI
MICROBATERÍAS RECARGABLES DE LITIO
La explosión del consumo de productos electrónicos junto con la toma de
conciencia a nivel mundial de la necesidad de conservar el medio ambiente incentiva la
búsqueda de nuevas y eficientes fuentes de energía.
Entre las diferentes fuentes de energía, las de origen electroquímico, ocupan
un lugar muy importante. Por un lado, se investigan baterías recargables de alta energía
como una nueva solución al desarrollo de vehículos eléctricos, y por otro lado, baterías
de pequeñas dimensiones, son cada vez más necesarias para satisfacer una demanda
creada por el crecimiento del mercado de dispositivos electrónicos tales como teléfonos
celulares, microcomputadoras portátiles, cámaras digitales, alarmas, etc.
Como muestra del interés económico y académico en las fuentes
electroquímicas de energía mencionamos aquí [ref 1] sólo dos datos estadísticos: (a) El
mercado global de baterías en 1997 era de 22 billones de dólares y en el 2002 de 41
billones de dólares, (b) El número de publicaciones utilizando las palabras claves
battery o batteries entre 1974 y 1998 fue de 14 973.
En este capítulo describimos brevemente los conceptos básicos sobre
baterías, mostramos los resultados obtenidos en nuestro laboratorio en la UNI respecto
a la producción de prototipos de microbaterías recargables de litio con electrolito
líquido, y describimos los experimentos que estamos realizando en este momento
(2004) para la elaboración de prototipos de microbaterías recargables de iones de litio
con la totalidad de materiales en estado sólido.

Capítulo VI Microbaterías recargables de litio
152
VI.1. CONCEPTOS BASICOS
Una celda o célula electroquímica (batería) es un dispositivo en el cual se
desarrollan reacciones químicas de oxidación y reducción, espontáneas o forzadas, en
cada uno de los electrodos, los cuales están separados por un electrolito iónicamente
conductor. Si la reacción química es espontánea, parte de la energía de reacción se
transforma en energía eléctrica y se conoce como celda galvánica. Si la reacción se
fuerza mediante la adición de energía eléctrica se llama celda electrolítica.
Las celdas galvánicas se subdividen usualmente según su operación: celdas
de combustible, en las que se dosifican los reactivos en forma continua, y de los que se
extraen los productos también de forma continua; y, celdas primarias, se llama así a los
dispositivos de estado no uniforme que contienen cantidades iniciales fijas de reactivos,
verbigracia las pilas secas.
Hay un tercer tipo de celdas electroquímicas conocidas como secundarias,
que funcionan como las galvánicas cuando están en uso, pero que se pueden regenerar
(recargarse) invirtiendo la reacción de la celda mediante la aplicación de energía
eléctrica, tal como las baterías de automóvil, en las que se encuentran conectadas en
serie varias celdas. La figura 6.1 ilustra el proceso de descarga y recarga de una típica
batería de automóvil Pb/H2SO4 /PbO2.
La celda electroquímica más simple consta de tres fases al menos,
incluyendo dos electrodos y uno o más electrolitos1 [ref 4]. Durante su funcionamiento,
operación llamada de descarga, los electrodos de la batería se conectan a una carga
externa, y los electrones fluyen a través de ella, desde el ánodo (electrodo negativo)
hacia el cátodo (electrodo positivo), para mantener el equilibrio de carga los iones
1 Los electrodos deben ser buenos conductores de electrones y los electrolitos deben ser buenos conductores de iones pero pobres conductores de electrones para evitar el corto circuito interno en la batería convencional

Capítulo VI Microbaterías recargables de litio
153
negativos (aniones) y los iones positivos (cationes), contenidos en el electrolito, se
desplazan hacia el ánodo y el cátodo, respectivamente. En las baterías recargables, el
proceso de recarga ocurre en el sentido inverso, debido a que la batería es conectada a
una fuente de energía eléctrica.
resistencia de carga
PbPbO2
SO4=
-
(cátodo) (ánodo)
I
(ele
ctro
nes)
PbPbO2
SO4=
-
(ánodo) (cátodo)
I
(ele
ctrones)
(a)(b)
H+
H+
Fig. 6.1 Esquema de una batería recargable Pb/H2SO4/PbO2 mostrando los procesos de (a) descarga y (b)
recarga.
La capacidad de una celda electroquímica se expresa en términos de la
cantidad de carga involucrada en la reacción electroquímica. Usualmente se expresa en
coulombs (o ampere hora, (Ah) ). Por la ley de Faraday es posible calcular la carga Q
involucrada en la reacción de una mol:
Q = z n F , ( 6.1 )
donde z es el número de electrones intercambiados en la reacción, n es el número de
moles que participan en la reacción y F es la constante de Faraday (96487 C o 26,8 Ah).
Es también importante expresar la capacidad de una batería en términos de
carga por unidad de masa (en Ah/g) o carga por unidad de volumen (Ah/cm3). Para esto
se usan las siguintes dos ecuaciones:

Capítulo VI Microbaterías recargables de litio
154
M
n 26.8C (6.2)
o:
M
n 26.8V
n 26.8C (6.3)
donde M es la masa atómica o molecular del material, V es el volumen y es la
densidad. A capacidade teórica de uma bateria é baseada apenas nos materiais ativos
participantes da reação eletroquímica.
La calidad de una bateria puede también ser expresada en términos de la
energia entregada. Esta es usualmente expresa em Watts horas, y puede ser calculada
por la ecuación:
(Ah) Capacidad (V) VoltajeE(Wh) (6.4)
VI.2 BATERIAS DE LITIO
Utilizan el litio metálico como ánodo, un electrolito conductor de iones de
litio y un óxido de metales de transición como cátodo o receptor de iones de litio
durante el proceso de descarga o de trabajo útil de la batería. Este último, es
generalmente un compuesto de estructura abierta capaz de aceptar y liberar iones de
litio interna y externamente a su red cristalina.
Las baterías de litio en principio pueden tener una larga vida y los ciclos
pueden repetirse una gran cantidad de veces, siempre que se mantenga las
características estructurales del material catódico. La figura 6.2 muestra un esquema del
proceso de descarga y de recarga de una batería de litio que usa pentóxido de vanadio
como cátodo y perclorato de litio disuelto en carbonato de propileno como electrolito
líquido.

Capítulo VI Microbaterías recargables de litio
155
resistencia de carga
LiV2O5
ClO4=
-
(cátodo) (ánodo)
I
(ele
ctro
nes)
LiV2O5
ClO4=
-
(ánodo) (cátodo)
I
(ele
ctrones)
(a)(b)
LiClO4 + CPLiClO4 + CP
colector colector
Li+ Li+
Fig. 6.2 Esquema de una batería de litio mostrando el proceso de (a) descarga y (b) recarga.
Puesto que el pentóxido de vanadio es un semiconductor (no es buen
conductor electrónico) debe estar en contacto con una barra metálica que actúa como
colector de electrones. Baterías de este tipo dan un voltaje de 3,5 V y tienen una
densidad de energía de 460 a 700 Wh/ kg [ref 5] que superan notablemente a las de
plomo-acido que proporcionan un voltaje de 1,69 V y tienen una densidad de energía de
25 a 40 Wh/kg.. Oxidos de otros metales de transición tales como MnO2 pueden
también ser usados.
Si las dimensiones de los electrodos y de la solución electrolítica son
pequeñas como en el caso de que se usen películas delgadas del orden de un micrómetro
de espesor, se habla de microbaterías. Es posible construir microbaterías usando
electrolito líquido, en efecto, en la sección 6.4 describimos la elaboración de un tipo de
estas microbaterías, sin embargo, las aplicaciones requieren microbaterías de estado
sólido, es decir aquellas en que tanto los electrodos como la solución electrolítica estén
constituidas por películas delgadas en estado sólido. En particular, el electrolito puede
ser un polímero, un gel, o simplemente un sólido electrolítico como el LiPON.

Capítulo VI Microbaterías recargables de litio
156
Fig. 6.3 Esquema de una microbatería de estado sólido, [ref 8].
Una de las ventajas de las microbateías de litio de estado sólido es que al
reducir las dimensiones del electrodo se reduce la resistencia interna de la batería. Otra
ventaja es la posibilidad de integrarla a los circuitos electrónicos. Existen en la
literatura varios ejemplos de películas delgadas integradas a dispositivos electrónicos
[ref 6 y ref 7] como sensores de gas, cronómetros, microcélulas combustibles, capacitores,
dispositivos electrocrómicos, etc. La figura 6.3 muestra un esquema de microbaterías de
estado sólido [ref 8]
Una de las desventajas de las baterías de litio es la dificultad para trabajar
con el litio metálico, ya que éste no puede estar en contacto con la atmósfera por que
produce una explosión violenta. Una solución a esta dificultad es el uso de
microbaterías de iones de litio. Estas baterías están constituidas por un óxido de metal
transición que contenga litio (ejemplo, LiCoO2), un sólido electrolito como LiPON y un
ánodo de estructura abierta como el grafito [ref 9, 10 y 11].

Capítulo VI Microbaterías recargables de litio
157
VI.3 PELÍCULAS DE PENTOXIDO DE VANADIO COMO CATODOS
En esta sección describimos los experimentos realizados en el Laboratorio
de Física de la UNI para producir las películas delgadas de pentóxido de vanadio.
También mostramos los resultados de análisis químico y estructural hechos por
Rutherford Back Scattering, Difracción de rayos X, Espectroscopía de Reflexion en el
Infrarrojo y X-ray Photoelectron Spectroscopy, tanto a muestras de pentóxido de
vanadio como a muestras de pentóxido de vanadio a las cuales se les ha intercalado litio
electrolíticamente y se han constituido en LiyV2O5.
Para la elaboración de las películas delgadas de V2O5, se utilizó la técnica
evaporación iónica rf En la cámara de vacío se utilizó como blanco (cátodo) un disco
de vanadio metálico de 7,5 cm de diámetro y 3 mm de espesor, en un ambiente de gas
argón de 99,99% de pureza y oxígeno de 99,99% de pureza. La disposición de la
cámara es de tipo diodo asimétrico donde el blanco o target, en este caso vanadio
metálico es el electrodo de voltaje variable conectado a la fuente de radio frecuencia (rf)
y donde el sustrato y la campana metálica es parte del electrodo conectado a tierra.
Después de hacer vacío en la cámara hasta aproximadamente 1 mPa (10-5
mbar) se introduce el oxígeno hasta una presión de 81 mPa (8 x 10-4 mbar), luego se
introduce el Argón hasta la presión total de 0,52 Pa (5,1 x 10-3 mbar). La relación entre
la presión parcial de oxígeno, PO2, y la presión parcial de argón, PAr, es %6,182
Ar
O
PP
,
aproximadamente.
El tiempo de deposición para cada muestra fue de 0,5h, ó de1h, ó de1,5h y
hasta de 2h, para obtener diversos espesores de la película de V2O5, pues de la literatura
se conoce que el espesor es proporcional al tiempo de deposición, cuyo orden de

Capítulo VI Microbaterías recargables de litio
158
magnitud es de una décima parte de micra. Se observó que el color característico del
pentóxido de vanadio es amarillo intenso.
Se debe precisar que el término a temperatura ambiente es utilizado en este
trabajo para hacer notar que no se le da al sustrato un calentamiento directo in situ, es
decir mientras dura la deposición. Sin embargo, a través del cátodo pasa corriente
eléctrica, de modo que se calienta y por radiación calienta el ambiente al interior de la
cámara de vacío en forma irregular, particularmente en la posición del sustrato se estima
que la temperatura está en el rango de los 50 °C - 100 °C. Este amplio rango depende
del tiempo de deposición y en nuestro caso este tiempo varía hasta en cuatro veces el
tiempo mínimo de deposición.
El tratamiento térmico consiste en calentar cada muestra hasta una
temperatura de 350 °C durante el intervalo de 3h a 4h, y luego se deja que enfríe
lentamente al contacto térmico con el medio ambiente. Las muestras fueron analizadas
antes y después del tratamiento térmico, para observar si hay cambios, que
posteriormente se reportará.
A temperatura ambiente bajo las mismas condiciones para la cámara de
vacío como el vacío inicial y la presión total; parámetros de la fuente de radio
frecuencia, el tiempo de deposición y variando solamente la presión del oxígeno hasta
aproximadamente 40 mPa (4 x 10-4 mbar) de modo que la relación de presiones de los
gases sea %8,51P
P
Ar
O2 , se fabricaron algunas películas delgadas de pentóxido de vanadio
sobre vidrio. A estas muestras también se les dio el mismo tratamiento térmico para
analizarlas y evaluar la influencia de la relación de presiones en la estructura de la
película delgada.

Capítulo VI Microbaterías recargables de litio
159
DIFRACCIÓN DE RAYOS X DE PELÍCULAS DELGADAS DE V2O5
La estructura cristalina de nuestras películas delgadas de pentóxido de
vanadio fue estudiada usando difracción de rayos X . El instrumento PW11300 Philips
de la Universidad de Chalmers en Gotemburgo, Suecia fue operado a 40 kV, 20 mA. La
radiación utilizada fue la proveniente de la transición no resuelta K del cobre con =
0,154 nm . El ángulo de difracción 2 vs intensidad fue almacenado automáticamente.
La figura 6.4 muestra los difractogramas de rayos X de tres películas
delgadas de pentóxido de vanadio producidas en las mismas condiciones de evaporación
iónica pero con diferentes tiempos de depósito y por lo tanto diferente espesor. Estos
difractogramas están de acuerdo con la estructura ortorrómbica del pentóxido de
vanadio reportada en la literatura [ref 12] con parámetros de red :
ao = 11.519 Å bo= 3.564 Å co = 4.373 Å
Fig. 6.4 Difractogramas de rayos X de tres películas delgadas preparadas a las mismas condiciones,
a 300 0C con diferentes espesores.
0
2000
4000
6000
8000
10000
12000
14000
INT
EN
SID
AD
D
IFR
AC
TA
DA
(c
p s)
s)
13 18 23 28 33 38 43
600 nm thick
ANGULO DE DIFRACCION, 2 (grados)
200 nm thick
100 nm thick
(200) V2O5
(400
) V
2O5
V2O
5
(001) V2O5
(002) V2O5
(2
22)
ITO
(2
22)
ITO
(2
22)
IT0
(002) V2O5
(002) V2O5
(40
0) V
2O5
(200) V2O5
(321) ITO
(321) ITO
(321) ITO
(211
) IT
O
ITO
(21
1) I
TO
(2
11)
ITO

Capítulo VI Microbaterías recargables de litio
160
INSERSION ELECTROLITICA DE LITIO EN PENTOXIDO DE VANADIO
Durante el proceso normal de descarga de una batería iones de litio se van
introduciendo en la estructura del cátodo de pentóxido de vanadio. Nosotros hemos
introducido en forma controlada iones de litio a la estructura del pentóxido de vanadio y
hemos interrumpido el proceso para estudiar los cambios estructurales.
El proceso de intercalación de iones de litio en la estructura del V2O5 se
realiza utilizando una celda electrolítica como se muestra en la figura y con ayuda de un
galvanostato establecer una corriente constante entre el electrodo de trabajo (V2O5) y el
contraelectrodo (litio metálico) Durante la intercalación (descarga de la batería) es
como si una resistencia se estuviera colocando entre el electrodo de trabajo y el
contraelectrodo, y durante la des-intercalación es como si una fuerza electromotriz
externa se estuviera conectando a la batería. Este proceso tiene que realizarse en una
atmósfera inerte, en una caja de guantes (glove box)
Fig. 6. 5 Esquema de celda electrolítica para introducir iones de litio en V2O5
Los experimentos de intercalación de litio fueron realizados de la siguiente manera:
(1) La película de V2O5 fue colocada en la glove box en la posición ET de la celda
electrolítica.
(2) Se esperó por 10 minutos para que la muestra alcance un estado de equilibrio

Capítulo VI Microbaterías recargables de litio
161
(3) El potenciostato-galvanostato fue programado para que funcione en el modo
galvanostático para producir una corriente constante durante un tiempo t. Fue
programado también para medir la diferencia de potencial ( V) entre ET y ER
(litio metálico también).
Después del proceso de intercalación la muestra fue extraída de la glove
box, limpiada con alcohol y nitrógeno gaseoso y estaba lista para los diferentes análisis.
Los valores de corriente constante (I) y el tiempo de intercalación (t) fueron
escogidos considerando las dimensiones de la muestra (área y espesor) y el valor
deseado de y en la fórmula LiyV2O5. Por ejemplo, para obtener y = 1,5 en una película
de 600 nm de espesor y 2,8 cm2 de área se programó una corriente de 250 A durante
1800 segundos y para obtener y = 0,04 en una película de 100 nm de espesor se usó
una corriente de 3,33 A/cm2 durante 220 segundos.
Los valores de y fueron estimados usando los siguientes datos
Espesor de la película d (Å)
Area de la película A (cm2)
Masa molecular de V2O5 (M) 181.88
Corriente I ( A)
Tiempo de intercalación t (s)
Densidad del V2O5 3.357 g/cm3
Y considerando la siguiente fórmula
y = n / N , (6.26)
donde n es el número de iones de litio por cm3 en la película y está dado por
e tI n (6.27a)
y N es el número de moléculas de V2O5 por cm3 . Está dado por:

Capítulo VI Microbaterías recargables de litio
162
M
dA N N Avog (6.27b)
La siguiente fórmula práctica, deducida de las ecuaciones (6. 27) puede ser usada:
nm) ( d )(cmA 176
(s) t A I y 2 . (6.28)
De este modo es posible obtener valores de y entre 0,0 y 3,0.
DIFRACCIÓN DE RAYOS X DE LiyV2O5
Uno de los resultados más espectaculares desde el punto de vista de la Física
del Estado Sólido es la variación de la estructura cristalina de las películas delgadas de
pentóxido de vanadio cuando se les intercala iones de litio. El parámetro a de la
estructura ortorrómbica varía desde 0,44 nm hasta 0,47 nm cuando y varía entre 0,0 y
1,0. Puede variar hasta a = 0,49 nm si y varía hasta 3,0. La figura 6,6 muestra los
difractograma de rayos X que sustentan lo dicho.
Fig 6.7 Difractogramas de rayos X de LiyV 2O5

Capítulo VI Microbaterías recargables de litio
163
REFELECTANCIA EN EL INFRARROJO
Estos análisis buscan averiguar en qué frecuencias los materiales tienen
picos de absorción. Estas son también las frecuencias de los movimientos oscilatorios
de los átomos dentro del cristal respecto a sus posiciones de equilibrio. Aquí
reportamos los resultados obtenidos con el espectrofotómetro Perkin Elmer 580B de la
Universidad de Chalmers, Gotemburgo, Suecia. Las muestras fueron películas delgadas
de pentóxido de vanadio depositadas sobre vidrio previamente recubierto con un
electrodo transparente (óxido de estaño e indio, ITO) , así como muestras a las que se
les ha introducido litio electrolíticamente para valores de y = 0, 5 y 1,5.
20 o
R
0
20
40
60
80
10 0
400 600 800
Refl
ectan
ce (
%)
1000 1200 1400
y = 1.5 0y = 0.5 0
su bstrate
Wavenumber (cm -1 )
V 2 O 5 -b ased film
as depo sited
I T O film
Co rnin g glass
Fig 6.8 Espectros de reflectancia en el infrarrojo para tres muestras de Liy V2O5
La figura 6.8 muestra los espectros de reflectancia en el infrarrojo para las
tres muestras ya descritas. La importancia de estos resultados puede apreciarse
observado primero en el espectro del V2O5 los tres picos de absorción para los valores

Capítulo VI Microbaterías recargables de litio
164
1035, 980 y 950 cm-1. Estos picos han sido previamente estudiados [ref 13] y se asignan
a las vibraciones de un átomo de vanadio y uno de oxígeno que tienen un doble enlace y
que oscilan en la dirección cristalográfica c. Estos picos notoriamente se desplazan
hacia frecuencias menores cuando el litio es intercalado en la red del V2O5.
VI.4 PROTOTIPOS DE MICROBATERÍAS DESARROLLADOS EN LA UNI
Se han producido un prototipo de microbaterías de litio basado en litio
metálico como ánodo, pentóxido de vanadio como cátodo y perclorato de litio (LiClO4)
disuelto en propileno carbonato como electrolito líquido. También, actualmente se
vienen desarrollando prototipo de baterías de iones de litio basadas en películas
delgadas de LiCoO2, como cátodo, LiPON, como electrolito sólido y como ánodo
películas delgadas de grafito o películas delgadas de una aleación de aluminio y litio.
PROTOTIPO Li / LiClO4 (PC) / V2O5 (CON ELECTROLITO LIQUIDO)
En la figura (6.9) mostramos un esquema del prototipo de microbaterías de
litio desarrollado en la UNI. El cátodo está constituido por una película delgada de
V2O5 de 200 nm de espesor depositada sobre un sustrato de vidrio Corning previamente
recubierto con óxido de estaño (SnO). El electrolito líquido perclorato de litio (LiClO4)
1M disuelto en carbonato de propileno fue adherido a papel filtro y colocado sobre la
película delgada de pentóxido de vanadio. Una lámina delgada de litio metálico fue
colocada sobre el papel filtro. Después de colocar los contactos eléctricos de cobre,
todo el sistema fue empaquetado y cerrado herméticamente con cera en una atmósfera
de argón, para lo cual se hizo de una glove box. El área de los electrodos de este
prototipo fue de 4 cm2.

Capítulo VI Microbaterías recargables de litio
165
Fig 6.9 Esquema del prototipo de microbaterías recargables de litio desarrollado en la UNI
CURVAS DE CARGA Y DESCARGA DEL PROTOTIPO DE BATERIA
Haciendo uso de un potenciostato operado en el modo galvanostato, se
descargó la batería a corriente constante de 5 A. Mientras ocurría la descarga, los
valores de diferencia de potencial entre el electrodo de litio y el de pentóxido de
vanadio se acumulaban en función del tiempo. Estos datos están graficados como curva
de descarga en la figura 6.10a. Inicialmente el voltaje dado por nuestro prototipo fue de
3,4 V, el cual se mantiene por 100 minutos, luego baja a 3,2 V y se mantiene en ese
voltaje constante por 250 minutos. Después de lo cual baja a 2,4 V. La capacidad de
esta microbatería fue de 34,5 mC/cm2.
El proceso de recarga fue realizado también a corriente constante de 5 A.
La figura 6.10b muestra la curva de recarga. Se puede calcular que después de recibir
una carga de 0,24 C la batería regresa a un potencial de 3,0 V muy cercano al inicial.
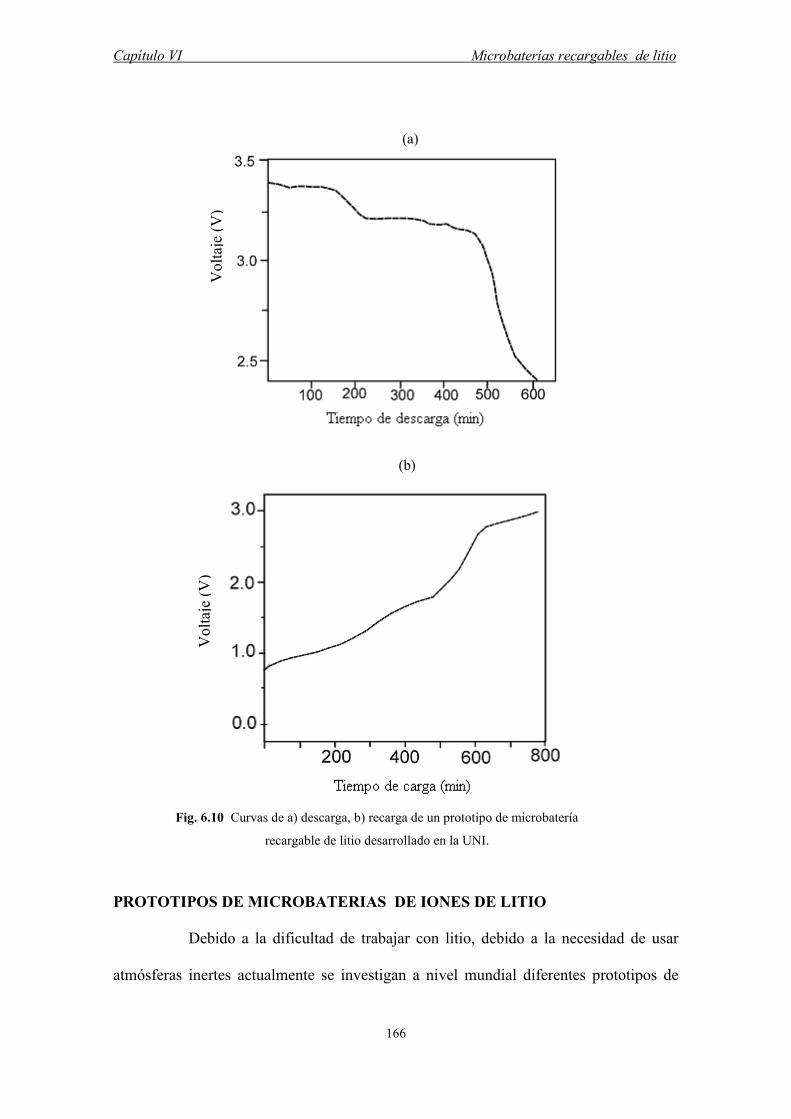
Capítulo VI Microbaterías recargables de litio
166
(a)
(b)
Fig. 6.10 Curvas de a) descarga, b) recarga de un prototipo de microbatería
recargable de litio desarrollado en la UNI.
PROTOTIPOS DE MICROBATERIAS DE IONES DE LITIO
Debido a la dificultad de trabajar con litio, debido a la necesidad de usar
atmósferas inertes actualmente se investigan a nivel mundial diferentes prototipos de
Vol
taje
(V)
Vol
taje
(V)

Capítulo VI Microbaterías recargables de litio
167
microbaterías recargables de litio. En nuestro laboratorio, en la UNI, estamos
desarrollando dos prototipos de microbaterías de iones de litio con electrolito sólido:
Prototipo 1: LiCoO2/LiPON/C (grafito)
Prototipo2: Li CoO2/ LiPON/ Al-Li
Ambos prototipos están basados en la producción de películas delgadas por
la técnica de evaporación iónica rf.
Para producir películas delgadas de LiCoO2 se usa un blanco de 75
mm de diámetro, 4 mm de espesor, 99.99% puro. El proceso de evaporación
iónica se realiza en una atmósfera de 90% de argón y 10% de oxígeno.
La producción de nuestras películas delgadas de electrolito sólido se
hace a partir de un blanco de fosfato de litio (Li3PO4) de 75 mm de diámetro, 4
mm de espesor y 99,97 % de pureza. El proceso de evaporación iónica se realiza
en una atmósfera de nitrógeno de pureza 99.999%. El material obtenido es
llamado LiPON.
Para nuestro prototipo 1 usamos películas delgadas de garfito, las
cuales son obtenidas por evaporación iónica a partir de un blanco de grafito en
una atmósfera de argón.
Para nuestro prototipo 2 usamos películas delgadas de una aleación de
litio y aluminio que se obtiene haciendo evaporación iónica de aluminio y
evaporación térmica de perclorato de litio simultáneamente. La evaporación
térmica del perclorato de litio se logra por el calentamiento natural del target de
aluminio durante el proceso de evaporación iónica del aluminio.
Nuestro prototipo 2 es más prometedor puesto que se han logrado
fuerzas electromotrices de 1,6 V, mientras que con electrodo de grafito hemos
llegado a 0,9 V.

Capítulo VI Microbaterías recargables de litio
168
REFERENCIAS
[1] Airton Lourenço, tesis de doctorado: Filmes Finos de Óxido de Vanádio como
Catodos de Microbaterias Recarregáveis de Lítio, 1999, Facultad de Ingeniería
Mecánica , Universidad de Campinas, Brasil.
[2] Arturo Talledo, tesis de doctorado, Vanadium Pentoxide-based thin films:
Optical, Structural, and Electrophysical characterization (applications to
electrochromic devices and microbatteries) 1992 Facultad de Ciencias UNI
[3] Arturo Talledo, Héctor Valdivia y Carsten Benndorf, J. Vac. Sci. Technol.
A, Vol. 21 No. 4 , 2003.
[4] Perry, R., Chilton, C. Manual del Ingeniero Químico, 5ta ed. Inglés, 2da
ed. Español, Vol I, McGraw Hill, p 4-76. 4-81, 1982
[5] West K. (1989) in High Conductivity ionic Conductors: Recent Trends and
Applications, Ed by Takahashi, (World Scientific Publishing, Company,
Singapore)
[6] Julien, C., Technological Applications of Solid-State Ionics, Mat Sci Eng
B-Solid 6: (1) 9-28 May 1990
[7] Kennedy, J.H., ThinSolid Films 43: (1-2) 41-92
[8] Ribes 1990, in Solid State Microbatteries, Ed. By Akridge J.R. and Balkansky M.
(Plenum Press, New York ) p 41
[9] N.J. Dudney y B.J. Neudecker, Current opinión in solid state and Materials
Science 4 (1999) 479-482
[10] . J.B. Bates et al Solid State Ionics 135 (2000) 33-45
[11] Bruno Scrosati et al Journal of Power Sources 105 (2002) 161-168
[12] Wyckoff R. W. G. (1964) Crystal Structures (Interscience Publishers, New York)
Second edition, Vol 2 p 186-187.
[13] Clauws et al, (1985), Phys Stat. Sol. (b), Vol. 131, p 459.

Capítulo VII Filtros ópticos Fabry Perot
169
CAPITULO VII
FILTROS OPTICOS FABRY PEROT
Una de las más antiguas y más importantes aplicaciones de la tecnología de
alto vacío es la producción de dispositivos ópticos. Desde espejos simples, espejos
semitransparentes (beam spliters) hasta las modernas guías de onda.
Un dispositivo óptico muy importante y que conserva actualidad a pesar de
haber sido desarrollado desde 1899 es el interferómetro Fabry Perot [ref 1]. Consiste de
dos láminas de vidrio recubiertas con películas semitransparentes de aluminio u otro
metal separadas por una pequeña distancia.
En la actualidad se investigan diferentes modelos de filtros Fabry Perot
sintonizables [ref 2] para aplicaciones en telecomunicaciones por fibras ópticas.
Generalmente están constituidos por superposición de tres capas: metálica, transparente
y metálica, donde la capa transparente del centro puede ser un cristal líquido o cristal
opto-eléctrico de modo que su índice de refracción pueda modularse por la aplicación
de una diferencia de potencial.
En este capítulo mostramos los avances desarrollados en la Universidad
Nacional de Ingeniería para la producción de Filtros Fabry Perot no sintonizables.
Nuestros prototipos están basados en películas delgadas de óxido de titanio y de
aluminio. Describimos aquí la producción de películas delgadas de óxido de titanio,
verificamos su estructura por difracción de rayos X, analizamos sus propiedades ópticas
por espectroscopia UV visible y obtenemos el índice de refracción Finalmente
describimos la producción de prototipos de filtros Fabry Perot y mostramos sus
características ópticas.

Capítulo VII Filtros ópticos Fabry Perot
170
VI.1 EL INTERFEROMETRO FABRY PEROT
El arreglo de las capas metálicas sobre superficies de vidrio y el espacio
transparente entre ellas se llama etalón. La figura 7 muestra un etalón en forma
esquemática y la definición de los coeficientes de amplitud de transmisión y reflexión.
capas
reflectantes
espaciador
placas transparentes
del etalon
aire o medio
transparente
dirección de la
luz incidente
ds
ta+
tb+
rb+
ra-
Fig. 7.1 a) Esquema de un etalón Fabry Perot b) Definición de los índices de amplitud que dan lugar a
los coeficientes de intensidad usados en las ecuaciones 7.1 a 7.4.
La teoría electromagnética [ref 3]. muestra que la transmisión para una
onda plana esta dada por:
1
222/122/1 2)(1
41
)(1ba
ba
ba
ba
ba senRR
RR
RR
TTT (7.1)
donde /cos2 sssdn . El espesor y el índice de refracción están designados por
d y n respectivamente.Haciendo que la transmitancias y reflectancias de las dos
superficies sean iguales, es decir que 0ba , y que ns sea igual a la unidad, la
expresión para T se convierte en
222 )1/(411
1 senRRRT
Tsss
s (7.2)

Capítulo VII Filtros ópticos Fabry Perot
171
y, definiendo
214
s
s
RR
F (7.3)
entonces:
22 11
1 FsenRT
Ts
s (7.4)
VII-2 PELICULAS DELGADAS DE OXIDO DE TITANIO
El óxido de titanio TiO2, anatasa, es un semiconductor con un ancho de banda
prohibida de 3,2 eV, razón por la cual es transparente a la luz visible. Esto lo hace un
buen candidato como electrodo de celdas fotoelectroquímicas [ref 4]. En este trabajo
aprovechamos sólo la propiedad de transparencia del TiO2 y lo usamos como espaciador
entre dos capas metálicas semitransparentes.
La técnica magneto evaporación iónica reactiva fue usada para producir
películas delgadas de óxido de titanio entre 100 y 250 nm sobre substratos de vidrio
Corning. El blanco usado fue un disco de 75 mm de diámetro y 4 mm de espesor. La
mezcla de gases usada fue 90% de argón y 10% de oxígeno. Las dimensiones de los
substratos fueron 71 26 1 mm3. El tiempo de depósito fue de entre 10 y 30 minutos
con una potencia de la fuente de radio frecuencia de 200 W. El voltaje de polarización
fue de 600 V. De estas muestras hemos estudiado la estructura, usando difracción de
rayos X, y las propiedades ópticas, usando espectroscopia UV visible de transmisión.
ANÁLISIS ESTRUCTURAL:
La estructura cristalina de nuestras películas fue estudaiada por difracción
de rayos X. El instrumento Philips X´ Pert de la Universidad Nacional de Ingeniería fue

Capítulo VII Filtros ópticos Fabry Perot
172
usado. La radiación utilizada es la proveniente de la transición K no resuelta del
cobre con = 0,154 nm.
La figura 3.2 muestra el Difractograma de rayos X de una muestra de óxido
de titanio de aproximadamente 220 nm de espesor. Se observa un pico bien definido
para el valor de 2 = 25,3020. Este pico corresponde a la familia de planos (101) de la
estructura tetragonal del TiO2 (anatasa) , con parámetros de red a = b = 3,78 A , c =
9,51 A ref 5 . El hecho de que sólo aparezca un pico significa que hay un
crecimiento preferencial del TiO2 en la dirección (101).
0
200
400
600
800
1000
1200
0 10 20 30 40 50 60 70
Ángulos de Difracción 2ø
Inte
ns
ida
d d
e D
ifra
cc
ión
(u
.a)
Fig 7.2 Difractograma de rayos X de una muestra de TiO2 sobre vidrio
.

Capítulo VII Filtros ópticos Fabry Perot
173
PROPIEDADES ÓPTICAS:
La transmitancia óptica de las películas delgadas de óxido de titanio sobre
vidrio Corning, en el rango de longitudes de onda comprendido entre 300 y 800 nm , fue
obtenido usando el espectrofotómetro UV Visible SHIMADZU modelo UV 1601 de la
Universidad Nacional de Ingeniería. Estos espectros fueron digitalizados y a partir de
ellos fueron obtenidos el espesor y el índice de refracción de las películas.
Las figuras 7.3, 7.4 y 7.5 muestran los espectros de transmitancia para
tres muestras.
300 400 500 600 700 800
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1.0
Tra
nsm
ita
ncia
Longitud de onda (nm)
Fig. 7.3: Espectro de transmitancia de una película delgada de 82 nm de TiO2 sobre vidrio. El tiempo de
deposición fue de 10 minutos, el índice de refracción calculado fue de 2.58

Capítulo VII Filtros ópticos Fabry Perot
174
300 400 500 600 700 800
0.0
0.2
0.4
0.6
0.8
1.0
Tra
nsm
ita
ncia
Longitud de onda (nm)
Fig. 7.4 Espectro de transmitancia de una película delgada de 135 nm de TiO2 sobre vidrio. El tiempo de
deposición fue de 20 minutos, el índice de refracción calculado fue 2.59
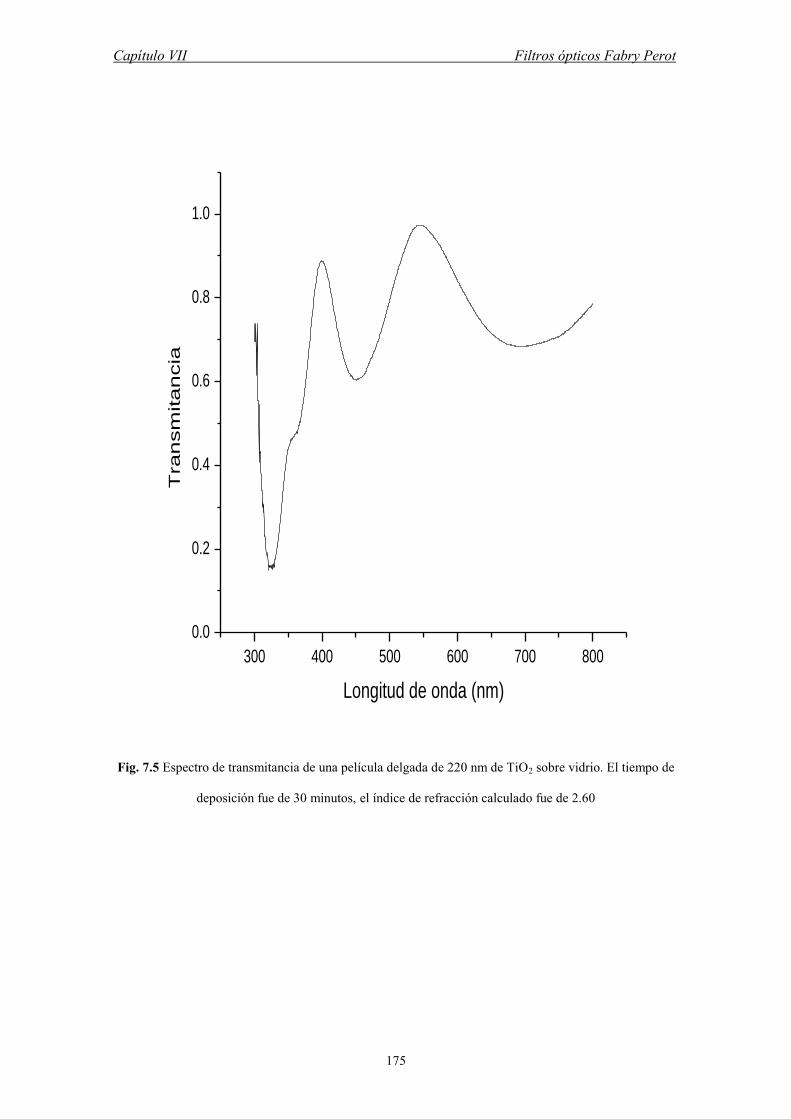
Capítulo VII Filtros ópticos Fabry Perot
175
300 400 500 600 700 800
0.0
0.2
0.4
0.6
0.8
1.0
Tra
nsm
ita
ncia
Longitud de onda (nm)
Fig. 7.5 Espectro de transmitancia de una película delgada de 220 nm de TiO2 sobre vidrio. El tiempo de
deposición fue de 30 minutos, el índice de refracción calculado fue de 2.60

Capítulo VII Filtros ópticos Fabry Perot
176
VII.3 PROTOTIPOS DE FILTROS OPTICOS FABRY PEROT
Dos tipos de filtros ópticos Fabry Perot vienen desarrollándose en la
Universidad Nacional de Ingeniería, usando la técnica magneto evaporación iónica rf
reactiva. La figura 7.6 muestra un esquema de estos prototipos. El filtro simple de la
figura 7.6a consiste de dos películas de aluminio de aproximadamente 80 nm de
espesor, separadas por una película de óxido de titanio de aproximadamente 200nm de
espesor. El sustrato es una lámina portaobjeto de vidrio corning. El filtro doble de la
figura 7.6b tiene una capa adicional de óxido de titanio de 200 nm de espesor y una
película adicional de aluminio de 80 nm de espesor.
substrato
Al
Al
AlTiO2
TiO2
substrato
Al
AlTiO2
(a) (b)
Fig. 7.6 Esquemas de filtros ópticos fabry Perot desarrollados en la UNI usando técnicas de alto vacío
(a)
(b)
Fig. 7.7 Micrografías por microscopía electrónica de barrido de filtros ópticos Fabry Perot desarrollados
en la UNI usando técnicas de alto vacío, a) filtro simple, b) filtro doble.

Capítulo VII Filtros ópticos Fabry Perot
177
Las figuras 7.7 muestran micrografías tomadas con el microscopio
electrónico de barrido Philips 600 de la Universidad Nacional de Ingeniería de un filtro
simple y de un filro doble. Se aprecia claramente las interfaces entre las películas
metálicas de aluminio y las aislantes de óxido de titanio.
ESPECTROS DE TRANSMITANCIA DE FILTROS ÓPTICOS
En las figuras 7.8 y 7.9 muestran los espectros de transmitancia de nuestros
prototipos de filtros ópticos fabry Perot en el rango de 300 a 800 nm.
El espectro de la figura 7.8 corresponde a un filtro simple. Se observa que
básicamente este filtro permite el paso sólo de longitudes de onda de 460 nm y el ancho
a media altura es de = 93. Cálculos simples nos permiten calcular la finesa de este
dispositivo: F ~ 10.
El espectro de la figura 7.9 corresponde a un filtro doble. La longitud de
onda seleccionada es 488 nm y el ancho a media altura es 53, lo ual nos permite estimar
una finesa F ~ 34 para este dispositivo.

Capítulo VII Filtros ópticos Fabry Perot
178
0.00E+00
2.00E-02
4.00E-02
6.00E-02
8.00E-02
1.00E-01
1.20E-01
1.40E-01
0 200 400 600 800 1000
Longitud de Onda (nm)
Tra
ns
mit
an
cia
Fig.7.8 Espectro de transmitancia de filtro Fabry Perot simple Al / TiO2 / Al. Obsérvese que la longitud
de onda = 460,06 nm, el ancho a media altura = 93,09 nm y la transmitancia máxima T = 11,2 .
Tiempo de 40 minutos de TiO2.
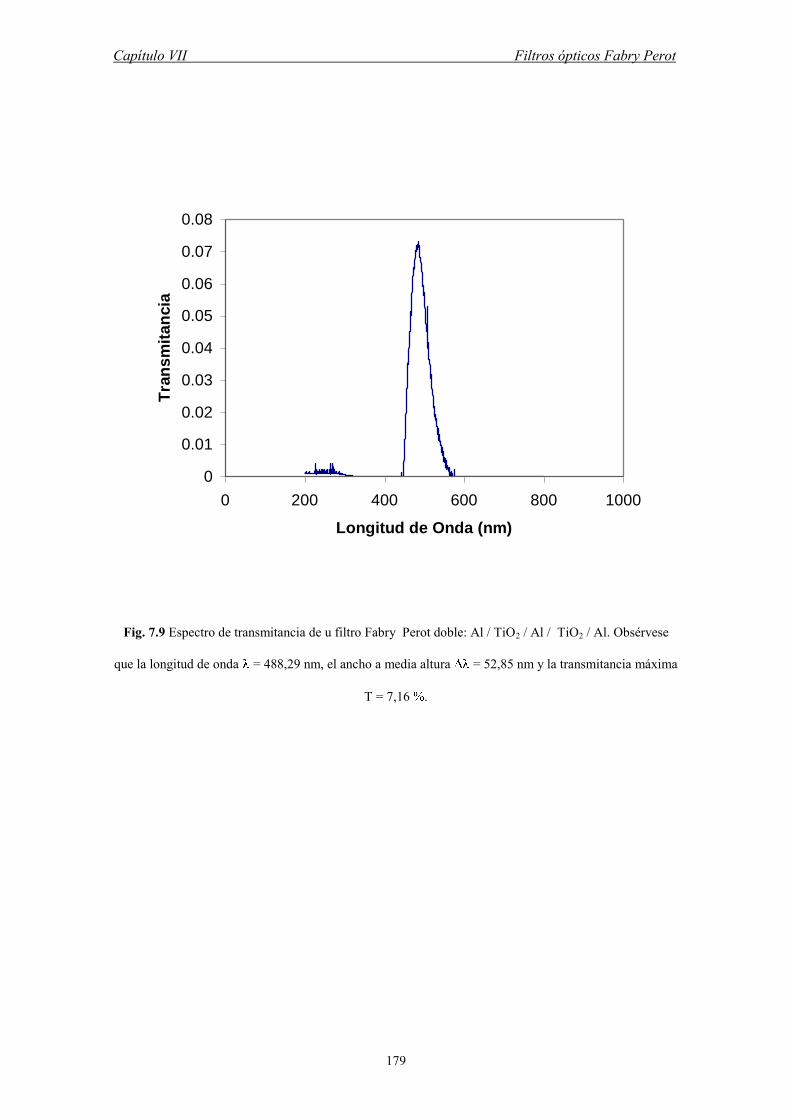
Capítulo VII Filtros ópticos Fabry Perot
179
0
0.01
0.02
0.03
0.04
0.05
0.06
0.07
0.08
0 200 400 600 800 1000
Longitud de Onda (nm)
Tra
ns
mit
an
cia
Fig. 7.9 Espectro de transmitancia de u filtro Fabry Perot doble: Al / TiO2 / Al / TiO2 / Al. Obsérvese
que la longitud de onda = 488,29 nm, el ancho a media altura = 52,85 nm y la transmitancia máxima
T = 7,16 .

Capítulo VII Filtros ópticos Fabry Perot
180
REFERENCIAS 1. Fabry C. and Perot A, 1899 Théorie et Applications d´ une nouvelle methode de
spectroscopie interferentielle, Ann. Chim. Phys., Paris 16, p 115-144. 2. Dan Sadot and Efraim Boimovich, IEEE Communications Magazine., Dic 1998
3. H.A Macleod, Thin Film Optical Filtres, Adam Hilger, Ltd, Bristol 1986
4. Mónica Marcela Gómez León, Photoelectrochemical and Physical Properties of
sputter deposited titanium oxide electrodes, Tesis de Doctorado, UNI, Lima,
Perú, 2001.
5. Wyckoff R. W. G. (1964) Crystal Structures (Interscience Publishers, New York)
Second edition, Vol 2.
