sustituciÓn del diclorometano por etanol … publicaciones qf-sintesis.pdf · sustituir el dcm y...
TRANSCRIPT

SUSTITUCIÓN DEL DICLOROMETANO POR ETANOL EN UNA ETAPA DEL PROCESO DE SÍNTESIS DE LA
OXIMETOLONA.
Autores: Alain Zarragoitia González, Yoanna M. Alvarez Ginarte, Mayra Reyes Moreno, Harold Curiel Hernández, Eddy
Castellanos Gil, Ulises J. Jáuregui Haza, Jose A. Ruiz García.
Centro de Química Farmacéutica. Calle 200 y 21, Atabey, Playa; P.O. Box 16042, Ciudad de La Habana, Cuba, C.P.11600 Telef.:
(537) 21 7822 / 217809 / 217925. Fax: (537) 33 6471. e-mail: [email protected]
RESUMENEn el CQF se desarrolló un procedimiento para la obtención de oximetolona, esteroide anabólico que se usa en el tratamiento de las
anemias aplásticas. Uno de los intermediarios en su proceso de obtención es la oxima del 5α-16-pregnen-3β-ol-20-ona, en cuya
purificación se emplea el diclorometano (disolvente clase 2)[2]. Teniendo en cuenta que las regulaciones internacionales
recomiendan la retirada paulatina de los disolventes clases 1 y 2 en los procesos productivos de principios activos farmacéuticos,
por su impacto en la salud del hombre y del medio ambiente, se hace necesario modificar la etapa de purificación de la oxima con
disolventes de menor toxicidad. El presente trabajo expone los resultados obtenidos en un nuevo proceso de purificación de la
oxima del 5α-16-pregnen-3β-ol-20-ona al sustituir el diclorometano por etanol de producción nacional. Con el fin de evaluar la
pureza de la oxima del 5α-16-pregnen-3β-ol-20-ona se desarrolló un procedimiento por cromatografía líquida de alta resolución. El
proceso de purificación con etanol garantiza iguales niveles de pureza que los obtenidos con diclorometano. Se realizó la
evaluación económica de la nueva variante y se demostró la viabilidad de su empleo en los estudios de escalado y ulterior
producción de la oximetolona, de manera que garantice un adecuado rendimiento y la calidad farmacéutica del producto final.
INTRODUCCIÓN
La oximetolona (C21H32O3: 17-βhidroxi-2-(hidroximetilen)-17-αmetil-5α-androstan-3-ona), es un esteroide anabólico empleado en
el tratamiento de las anemias aplásticas. En nuestro país, la oximetolona se importa como forma terminada y se utiliza,
actualmente, en el Instituto de Hematología. En el CQF se desarrolló un procedimiento para la síntesis química de la oximetolona a
partir de la hecogenina, a escala de laboratorio [1]. Uno de los intermediarios en el proceso de obtención de oximetolona es la
oxima del 5α-16-pregnen-3β-ol-20-ona, la cual, por requerimientos del proceso de síntesis, es sometida a una etapa de
purificación, donde se hace uso de diclorometano (DCM) y metanol, ambos disolventes clase 2 [2]. Estos deben ser usados con
mucha precaución en la obtención de sustancias activas debido a los niveles de toxicidad que presentan o a los daños que infringen
al medio ambiente [1]. Por ello, están estrictamente limitados en su uso según los valores de la Exposición Diaria Permitida (PDE)
y la Concentración Límite (CL). Los disolventes de interés en la etapa de purificación de la oxima de la oximetolona presentan
como restricciones: metanol: PDE = 30 mg/día, CL = 3000 ppm y diclorometano: PDE = 2 mg/día, CL = 3000 ppm. El objetivo de
este trabajo fue desarrollar una nueva variante de purificación que posibilitara la sustitución de los disolventes antes mencionados
por etanol, de producción nacional, el cual se obtiene a partir de una fuente natural. El etanol no está incluido en el listado de
sustancias tóxicas prohibidas, la oxima se solubiliza en él a su temperatura de reflujo (79 ºC) y cristaliza a 0 °C. Igualmente se
evaluó la factibilidad técnica y económica del nuevo procedimiento.
MATERIALES Y MÉTODOS
Atendiendo a que la oxima es soluble en compuestos polares, se comenzó el estudio de sustitución utilizando etanol 96%. Con el
fin de evaluar la factibilidad técnica del proceso se realizaron experimentos en dos escalas de laboratorio, a 500 mL y 1 L. Una de
las limitaciones principales para el uso del etanol es la menor solubilidad de la oxima en dicho disolvente que en DCM, por lo que

era de esperar que el volumen de etanol a usar sería mayor. Considerando los precios de compra de los disolventes se fijó un nivel
máximo de volumen de etanol a utilizar en la purificación, de tal forma que no afectara la economía del procedimiento global y
que, a la vez, permitiera asemejarr los rendimientos obtenidos con el DCM.
Descripción de la Instalación Experimental
Para la realización de las corridas experimentales, se utilizó un sistema que consistió en un matraz reactor de fondo redondo de 2
bocas (500 mL ó 1 L) con condensador. El calentamiento se realizó con una plancha AGIMATIC-N con agitación magnética. La
instalación experimental permitió operar con régimen de reflujo o de destilación según la disposición del condensador. El
procedimiento de purificación requiere de una filtración en caliente para la cual se utilizó un filtro Büchner con papel de filtro y
Celite como ayuda filtrante, y un segundo filtrado de la oxima cristalizada a 0 °C que se llevó a cabo en un embudo Büchner con
placa filtrante de vidrio aglomerado N° 3. El secado de los productos se realizó en un horno de aire recirculado a 70 °C. Todas las
pesadas se realizaron en balanza analítica.
Organización de los Experimentos
Para la escala de 500 mL se realizaron experimentos con incremento progresivo del volumen de etanol, utilizándose 115 mL, 230
mL, 345 mL, 400 mL y 460 mL del alcohol, que corresponden a una relación volumétrica de DCM:Etanol de 1:1, 1:2, 1:3, 1:3.5, y
1:4 respectivamente, considerándose la unidad como el volumen total de DCM utilizado en el procedimiento inicial para disolver la
oxima. Los experimentos se realizaron una sola vez, con excepción de la relación 1:4 a 500 mL de la que se desarrollaron 4
réplicas, y 3 réplicas en la escala de 1 L con el objetivo de comprobar la reproducibilidad del método. En la figura 1 se muestra el
nuevo esquema de purificación propuesto.
Determinación de la pureza de la Oxima por HPLC
Los análisis se realizaron utilizando una bomba inteligente L-6200A (Merck-Hitachi, Darmstadt, Alemania) equipada con un
inyector Rheodyne (USA) con volumen de inyección de 20 µL y un detector L-4250 UV-Vis (Merck-Hitachi) fijado a 245 nm
acoplado a una PC compatible con el programa BioChrom 1.2 (CIGB, La Habana, Cuba). Se utilizó una columna preempacada
LiChroCART 125x4 mm (LiChrospher 100 RP-18, 5 µm, Merck, Darmstadt, Alemania). La fase móvil fue una mezcla de
Figura. 1– Esquema de purificacíón de la Oxima del 5α-16-pregnen-3β-ol-20-ona con etanol
OXIMA CRUDA
AGITACION
FILTRACION
DESTILACION Etanol (Recuperación
de disolventes)
T= 00C
OXIMA PURIFICADA
Tref = 79 °C EtanolCarbón activado
Etanol (Recuperación dedisolventes)
FILTRACION
Etanol T= 70 °C
Torta (residual)
SECADO
ENFRIAMIENTO

metanol/agua (75:25, v/v) previamente filtrados y desgasificados y el caudal fue de 1,5 mL/min. Todos los reactivos empleados
fueron de calidad analítica.
RESULTADOS Y DISCUSIÓN
Al evaluar las cinco relaciones de volumen
estudiadas a escala de 500 mL, se observó que al
utilizar 460 mL de etanol se obtuvo una
recuperación en oxima de 77.24 ± 2.72 %, similar a
la obtenida cuando se utilizó DCM que fue de 76%
aproximadamente [1]. La relación de sustitución
DCM:Etanol 1:4 se verificó a la escala de 1 L con
resultados satisfactorios. La figura 2 muestra una
comparación de los cromatogramas obtenidos para
las oximas purificadas por ambos métodos. Siendo
el pico correspondiente a la oxima de magnitud
similar en los dos cromatogramas. Esto sugiere que la modificación en el procedimiento de purificación garantiza niveles similares
de pureza.
El análisis económico del proceso demostró que el cambio de
disolvente es viable (tabla 1). Considerando que el consumo
potencial de oximetolona es de 120 000 tabletas/año de 50 mg
la dosis [1], lo cual requiere una producción de 6 kg de
oximetolona al año, y conociendo que para satisfacer esa
necesidad es necesario contar anualmente con 15 kg de oxima
purificada [3], puede estimarse un ahorro potencial de más de
10700 $/año, únicamente por concepto de sustitución de DCM y
metanol por etanol. Esto influye de manera directa en los costos
globales del proceso de síntesis de la oximetolona y en un
incremento de la eficiencia del proceso.
CONCLUSIONES
Se desarrolló un procedimiento para la purificación de la oxima de la oximetolona con buenos rendimientos y pureza, que permite
sustituir el DCM y el metanol, disolventes clase 2, por etanol de producción nacional. El proceso es factible tanto desde el punto de
vista técnico como económico.
REFERENCIAS BIBLIOGRAFICAS
1- Ruiz J. A., Reyes M., Álvarez Y. Reglamento de laboratorio para la síntesis de oximetolona. JR-035-007-002-04. CQF 2000.
2- European agency for evaluation of medical products. Human medicines evaluation unit. Consensus guideline. 1998.
3- Jáuregui U., Zarragoitia A., Ruiz J. A. Factibilidad técnica para el escalado del proceso de obtención de oximetolona en la
planta piloto del CQF. UJ-035-010-005-01.CQF 2000.
Tabla 1- Resultados del ahorro introducido por elcambio de disolventes
GASTOS PARA CADA VARAINTE
GASTOS POR USO DE DCM Y METANOL
(Por cada 5.6 g de oxima sometida a purificación)5,87 $
GASTOS POR USO DE ETANOL
(Por cada 5.6 g de oxima sometida a purificación)1,84 $
AHORRO INTRODUCIDO
(Por cada 5.6 g de oxima sometida a purificación)
4, 02 $
Figura 2- Comparación de los cromatogramas
0 5 1 0 1 5 2 0 2 5 3 0
Im p u re z a
O x im a /E ta n o l (1 .0 0 3 m g /m L )O x im a /D C M (1 .0 0 7 m g /m L )
T ie m p o (m in .)

ESTUDIO DE LA REACCIÓN DE NITROSACIÓN EN FLAVONOIDES FRENTE A UNA MOLÉCULA DEAGUA
Autores: Elismary Rodríguez y Gerardo Gonzáles
Institución: Departamento de Química. Facultad de Ingeniería Química- Farmacia. Universidad de Camagüey, C.Cir,Km 5 ½ , C.P 74650, Camagüey, Cuba. e-mail: [email protected]
RESUMEN.
Los flavonoides son compuestos que aparecen en una gran variedad de vegetales, generalmente se cree que son
beneficiosos sobre un gran número y sistemas de órganos del cuerpo humano, sin embargo se ha encontrado que varios
de estos compuestos pueden presentar acción mutagénica en el ensayo de AMES cuando se someten a una reacción de
nitrosación, en este trabajo se hace una valoración de las características de los Complejos Intermediarios y Estados de
Transición formados en la reacción de nitrosación frente a una molécula de agua, particularmente de la Energía de
Activación encontrándose resultados similares a los alcanzados en fase gaseosa, es decir, un proceso libre sin Energía
de Activación que hace aconsejable el uso de un mayor número de moléculas de agua.
INTRODUCCIÓN
Los flavonoides son metabolitos de una amplia distribución en el reino vegetal y por este motivo se encuentran entre las
sustancias que más intervienen en la dieta del hombre desde los tiempos más remotos. Normalmente el hombre y otros
mamíferos los excretan tal y como los consumen pero también puede glicosilarlos o conjugarlos con el ácido
glucurónico e incluso transformarlos en alguna extensión para lograr una mejor excreción.
Se le atribuyen o se le han comprobado a los flavonoides, una multitud de efectos beneficiosos y curativos que van
desde antibacteriales hasta broncodilatadores y han sido objetos de manipulación química para mejorar sus propiedades
y quizás el ejemplo clásico sea la síntesis del Dicromoglicato Sódico (DCGS), fármaco usado en la prevención de los
ataques de asma, a partir de una modificación química de la estructura de la khelina.
En trabajos previos (González,2000 y Rodríguez,2001) han calculado el proceso de interacción de diversos flavonoides
con HNO2, reacción importante que puede ocurrir a nivel estomacal y que explican el carácter mutagénico de varios
flavonoides según (Rueff, Gaspar y Laires, 1995), habiéndose obtenido evidencias de que el proceso en fase gaseosa era
espontáneo, aunque con pequeñas diferencias no conclusivas apuntando a la mutagenisidad o no de estos compuestos.
En el presente trabajo pretendemos extender el estudio a un sistema que contenga una molécula de agua para modelar
en cierto sentido (aunque insuficiente) el efecto del solvente sobre esta reacción.
MATERIALES Y MÉTODOS

Nombre 3 5 7 2' 4' 5' Activ.1) Morina OH OH OH OH H OH 48002) 3-Oh Flavona OH H H H H H 03) Luteolina H OH OH H OH OH 46004) Flavona H H H H H H 0
O
O
A C
B12
345
6
78
1'2'
3'
4'5'
6'
Se utilizaron 4 de las estructuras de los flavonoides bajo estudio en el trabajo de Rueff, Gaspar y Laires,1995. Los datos
generales de estos compuestos se muestran en la Tabla I.
Tabla I. Datos estructurales y actividades para los flavonoides bajo estudio.
*La actividad está dada por el número de revertantes por placas en el ensayo de Ames (Rueff, Gaspar y Laires, 1995).
Las estructuras fueron generadas utilizando el programa Moby 1.50 3.92 de Udo Höeler, se fijaron parámetros
obtenidos del análisis estructural reportados por van Acker et al. 1996. Se realizó la optimización mediante el uso del
programa MOPAC 6.0 usando las palabras claves:AM1 o PM3 para identificar el método de cálculo, EF, el
procedimiento de optimización, VECTORS, salida de información de los coeficientes orbitálicos, GEO-OK, que no
chequee la geometría inicial
En la búsqueda de los posibles Estados de transición se utilizaron las siguientes palabras claves:SADDLE,
especificando el cálculo del estado de transición y POINT y STEP, cuando se hallaba el camino de reacción,
especificando por su parte la coordenada por estudiar con el valor de -1 en la matriz de entrada, XYZ, para especificar al
programa que realice la optimización de geometrías en coordenadas cartesianas cuando fue necesario.
RESULTADO Y DISCUSIÓN.
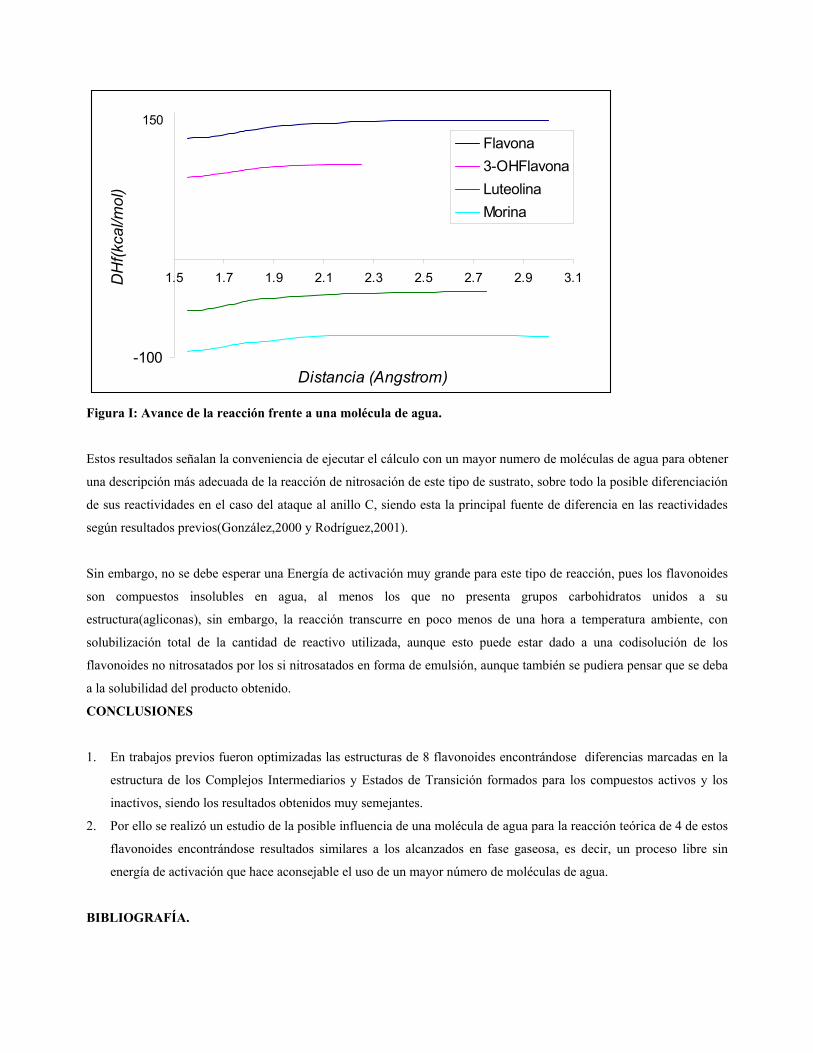
En la figura I se muestra los resultados obtenidos en nuestros cálculos, la Morina es la única molécula que presenta una
barrera de activación alta pero esta es de solo 2,4 Kcal/mol, que a temperatura ambiente no es de consideración. En
todos los demás casos se encontraron comportamientos idénticos a los descritos previamente por nuestro grupo de
trabajo, siendo la reacción un proceso libre sin energía de activación y con una caída continúa de energía hasta el
complejo intermediario tetrahédrico.
-100
1.5 1.7 1.9 2.1 2.3 2.5 2.7 2.9 3.1
Distancia (Angstrom)
DH
f(kc
al/m
ol)
Flavona
3-OHFlavona
Luteolina
Morina
150
Figura I: Avance de la reacción frente a una molécula de agua.
Estos resultados señalan la conveniencia de ejecutar el cálculo con un mayor numero de moléculas de agua para obtener
una descripción más adecuada de la reacción de nitrosación de este tipo de sustrato, sobre todo la posible diferenciación
de sus reactividades en el caso del ataque al anillo C, siendo esta la principal fuente de diferencia en las reactividades
según resultados previos(González,2000 y Rodríguez,2001).
Sin embargo, no se debe esperar una Energía de activación muy grande para este tipo de reacción, pues los flavonoides
son compuestos insolubles en agua, al menos los que no presenta grupos carbohidratos unidos a su
estructura(agliconas), sin embargo, la reacción transcurre en poco menos de una hora a temperatura ambiente, con
solubilización total de la cantidad de reactivo utilizada, aunque esto puede estar dado a una codisolución de los
flavonoides no nitrosatados por los si nitrosatados en forma de emulsión, aunque también se pudiera pensar que se deba
a la solubilidad del producto obtenido.
CONCLUSIONES
1. En trabajos previos fueron optimizadas las estructuras de 8 flavonoides encontrándose diferencias marcadas en la
estructura de los Complejos Intermediarios y Estados de Transición formados para los compuestos activos y los
inactivos, siendo los resultados obtenidos muy semejantes.
2. Por ello se realizó un estudio de la posible influencia de una molécula de agua para la reacción teórica de 4 de estos
flavonoides encontrándose resultados similares a los alcanzados en fase gaseosa, es decir, un proceso libre sin
energía de activación que hace aconsejable el uso de un mayor número de moléculas de agua.
BIBLIOGRAFÍA.

1. Höeler, U. Moby 1.50 3.92. 1992
2. Liehr, J.G., European Journal of. Cancer Prevention. 6:310, 1997.
3. Morrison, R.T. y Boyd, R.N. Química Orgánica Addison-Wesley Iberoamericana. Wilmington. 1990
4. Rueff, J.; Gaspar, J. Y Laires, A.; Mutagenesis. 10 (4):325. 1995.
5. Stewart, J Manual de usuuarios MOPAC v 6.0, 1989
6. Van Acker, S.A.B.E.; de Groot, M.J.; van den Berg, D.; Tromp, den Kelder, G.D.; G.R.M.M. van der Vijgh, W.J.Fy Bast, A.; Chem. Res. Toxicol. 9:1305. 1996.
7. Van Acker, S.A.B.E.; Tromp, M.N.J.L.; Haenen, G.R.M.M.; van der Vijgh, W.J.F y Bast, A.; Biochem andBiophys. Res. Comm. 214(3):755. 1995.
8. González,G. Tesis Presentada en Opción al Grado de Maestro en Química. Universidad de la Habana, 2000
9. Rodríguez,E. Tesis de Grado. Universidad de Camaguey,2001.
10. Avendaño, M. Principios de Química Farmacéutica. Ediciones Alhambra, Madrid. 1986
11. Foyé, W.O. Principios de Química Farmacéutica, Edición Revolucionaria, La Habana, 1985.

DESARROLLO DE UN MODELO ALTERNATIVO (QSAR) DE
REEMPLAZAMIENTO PARA PREDECIR RIESGO INMUNOTOXICO
AUTORES: MSc Esvieta Tenorio*, Lic Alfredo Peña**,Dr Antonio Perez*, Lic
Edisleidy Aguila*,Msc Rafael Sosa*, Dr Mv Osmany Marrero*, Dr Remigio
Cortes*,Dr Mv Armando Morales*.Osvaldo Norman*
*Centro de Bioactivos Químicos
** Universidad de Granma
RESUMEN
El QSAR es un método alternativo desarrollado actualmente y que por su
aplicabilidad fue introducido en los estudios toxicologicos con la finalidad de reducir el
tiempo y los costos de estos ensayos además de reducir el empleo de animales de
laboratorio, la naturaleza de una acción tóxica esta relacionada con la estructura molecular
del químico. Nuestro trabajo esta encaminado a predecir de forma teórico práctica
empleando un diseño molecular con una base grafo-teórica la actividad inmunotóxica de la
molécula de G-1 por lo que desarrollamos un modelo basándonos en las técnicas
bioinformaticas capaz de discriminar adecuadamente entre fármacos inmunosupresores y
aquellos que son inactivos, este modelo posee un porciento de clasificación de 98.6 % por
lo que se considera adecuado.
Palabras Claves: QSAR, Métodos alternativos
Introducción
La actividad de un químico sobre un organismo viviente depende de la acción física
o química en los tejidos biológicos y la naturaleza de tal acción esta relacionada con la
estructura molecular del químico, este fundamento fue denominado Relación Estructura
Actividad y se estableció desde hace 100 años atrás pero especialmente en las dos última
décadas se han efectuado muchos intentos para comprobar lo anterior planteado empleando
para ello varias vías cualitativas y cuantitativas. [1,2,3]

En el caso del compuesto objeto de estudio el cual pertenece a los vinilfuranos, uno
de los aspectos que más polémica ha provocado entre los especialistas es la confusión de
estos con los nitrofuranos los cuales son susceptibles a generar especies radicalarias
durante el proceso de reducción que sufre el grupo nitro por la acción de las nitroreductasas
causando daños genético y contribuyendo al desarrollo de tumores.
Las técnicas en que se basa la relación estructura actividad son variadas, y la
actividad biológica de interés puede ser un efecto o una acción tóxica específica
usualmente bioquímica relacionada con la toxicidad[4]
Nuestro trabajo esta encaminado a predecir de forma teórico práctica empleando
un diseño molecular con una base grafo-teórica la actividad inmunotóxica de la molécula
de G-1 por lo que desarrollamos un modelo capaz de discriminar adecuadamente entre
fármacos inmunosupresores y aquellos que son inactivos.
MATERIALES Y MÉTODOS
En el desarrollo del modelo para predecir la actividad inmunosupresora se tomaron
más de 8000 fármacos representantes típicos con esta propiedad farmacológica
(compuestos activos) y un número elevado de fármacos que no poseen esta actividad
(compuestos inactivos), con estos dos conjuntos activos e inactivos se construyó la serie de
entrenamiento y la de aprendizaje con la cual se construyó el modelo luego de un análisis
estadístico. Para el cálculo de los momentos espectrales se empleo el software TOSMODE.
Calculándose los momentos espectrales ponderando los grafos moleculares con el momento
dipolo estándar de enlace.
El modelo de clasificación se obtuvo empleando el análisis discriminante, para el
que se empleo el Statistica Ver. 5.0, el mismo fue validado empleando una serie de

predicción con moléculas de fármacos con acción inmunosupresora y un conjunto de
fármacos inactivos, para la evaluación de las moléculas con el modelo encontrado y
validado se empleo el Microsoft Excel 200
RESULTADOS
En la tabla # 1 que se muestra a continuación se puede apreciar las características
del modelo encontrado, el mismo es capaz de discriminar adecuadamente entre fármacos
inmunosupresores y aquellos que son inactivos.
Tabla #1 Matriz de clasificación del modelo
Matriz de clasificación del modeloPorcentaje declasificación correcto
G-1:-1 G-2:1
G-1:-1 100 152 0G-2:1 95.01 2 38Total 98. 9 154 38
En la tabla numero 2 se observa el comportamiento de la familia G la cual posee un
amplio espectro terapéuticos con una doble acción anibacteriana y antifungica.
Tabla # 2 Comportamiento de los porcientos de probabilidades calculado por
el modelo TOSSMoDe de la Actividad IM. (inmunosupresora)
Estructura Activ. IM %Base1 6.34G-0 39.35G-1 46.99MBr, (5) 42.90MBr, (C) 43.44
Leyenda:
mBr (5) monobromado en posición 5 del furano
mBr (C) monobromado en cadena.

DISCUSION
Se nota que la clasificación para los inactivos es superior a los activos con un 100%
y un 95.01 respectivamente, esto pudiera parecer una desventaja cuando se analiza que
algunos fármacos activos pudieran ser clasificados como inactivos (falsos inactivos)
perdiéndose la posibilidad de identificarlos, sin embargo es una ventaja al asegurar que la
mayoría de los compuestos que son clasificados como activos realmente lo sean,
eliminando o reduciendo al mínimo los posibles falsos activos[5,6].
Se entiende por un falso activo aquel compuesto que siendo inactivo el modelo lo
reconoce como activo y falso inactivo es aquel que el modelo lo clasifica como inactivo
siendo realmente activo. La función discriminante fue validada con una serie de predicción
en la que se comprobó que el mismo reconoció correctamente el 100% de los compuestos
tanto activos como inactivos. Con el modelo validado fue posible calcular la contribución
del la estructura básica de los vinilfuranos, evaluándose tres moléculas de la serie G, los
resultados obtenidos se muestran a continuación en la tabla 2 se muestra que el porciento de
probabilidad de una actividad inmunosupresora relativa en la serie fue calculado por el
modelo y que su valor es de 46.99 % para la molécula objeto de estudio, y siguiendo los
criterios emitidos por la bibliografía especializada donde solo los valores mayores a un 75
% son considerados significativos valoramos estos resultados y consideramos que es baja
la probabilidad de que esta molécula exprese la actividad inmunosupresora.
Debe notarse además que el porcentaje de clasificación total del modelo es de
98.9%, este resultado es muy importante toda vez que en la literatura un modelo se

considera adecuado si su porcentaje de clasificación es superior a 85%. [3]
Como se puede observar la estructura base de los vinilfuranos Fig 2
O
tiene una contribución relativa importante en la actividad inmunosupresora con un valor de
contribución de 6.24%, sin embargo la presencia del grupo nitro es quizás el hecho más
importante debido a la gran contribución a la actividad Inmunosupresora que este confiere a
la estructura, como se muestra en la tabla anterior el valor de esta propiedad es de 39.35 %.
La presencia de un átomo de bromo tanto en la cadena como en la posición 5 del
anillo furánico producen un ligero incremento de esta actividad, aunque se puede notar que
la presencia de un átomo de bromo en la cadena produce in incremento mayor. Esto
explica el hecho de que en el caso del G-1 se observa el mayor valor calculado de la
actividad Inmunosupresora en esta serie de compuestos.
CONCLUSIONES
1. El porcentaje de clasificación total del modelo desarrollado es de 98.9%
considerándose adecuado para la predicción
2. El porciento de predicción de la actividad inmunosupresora de la molécula G-1
empleando el TOSMODE fue de 46.99% considerándose no significativa.
BIBLOGRAFIA
1. Turner L.,Choplin P. Structure- Activity Relationsships in Toxicology and Ecotoxicology: an
Assessment. Toxic in vitro Vol (1): 143-171.1987.
2. Estrada, RE. Spectral Moment of edge adyacency matrix. No. I, J. Chem. Inf. and Comp. Sci.

31-32 1995.
3. Gálvez. J., García R. Diseño de fármacos por conectividad molecular. (Monografía). p.355-
384 Ed. Farmaindustria. Madrid 1994.
4. Randic, M., J. Math.Chem. 7 155 1991.
5. Gálvez, AJ. Diseño de Fármacos por Conectividad Molecular. (Monografía) p 357. Ed.
Farmaindustria. 1994.
6. Balaban, AT. J. Chem Inf. and Comp. Sci. 25, 334. 1985.

Diseño, síntesis y evaluación biológica “in vivo” de nuevos compuestos Antitumorales. Parte I.
Dr. Enrique Molina Pérez.
Universidad de Camagüey.
Resumen
En el presente trabajo se utiliza una serie de nuevos descriptores moleculares, denominados momentos espectrales, los que
fueron correlacionados con la actividad biológica de los compuestos seleccionados. El estudio permitió la obtención de un
modelo capaz de clasificar y predecir correctamente la actividad antitumoral de los compuestos a partir de la utilización de
la técnica estadística ADL, implementada en el paquete de programas STATISTICA. Las contribuciones de los diferentes
fragmentos y grupos sustituyentes son calculadas por el programa TOSS MODE, resultado que permitió el diseño de los
nuevos compuestos antitumorales.
Se sintetizaron los compuestos diseñados y se caracterizaron inequívocamente con el empleo de las técnicas
espectroscópicas más modernas, lográndose obtener un total de 15 nuevas benzofurocumarinas. La construcción del
esqueleto tetracíclico se realizó mediante la vía retrosintética.
Los compuestos sintetizados fueron evaluados biológicamente, estudio que permitió corroborar los resultados de la
predicción realizada a partir del modelo antitumoral, observándose una relación mayor del 80 % entre lo predicho
teóricamente y lo obtenido experimentalmente, resultado que es considerado de muy alta correspondencia.
Los estudios de actividad biológica “in vivo” y de toxicidad, mostraron que las benzofurocumarinas son más activas y
menos tóxicas a diferencia del 8-MOP, fármaco utilizado en la terapia actual contra el Cáncer. Las benzofurocumarinas 1 y
2 se corresponden con los compuestos más prometedores, presentando una buena capacidad de inhibición del crecimiento
celular aún en ausencia de radiación y no manifiestan fototoxicidad cutánea a altas dosis, convirtiéndolas en compuestos
con un gran perfil farmacológico.
Introducción
En décadas pasadas, la búsqueda de nuevos fármacos estuvo basada en los métodos de prueba y error que necesitaban
ensayar sobre 10 000 compuestos, de los cuales 10 superaban todos los ensayos y sólo uno lograba convertirse en un
medicamento de prescripción,1 lo que condujo a grandes pérdidas de recursos.
En los últimos años se han desarrollado los métodos de Diseño Racional de Fármacos, los que se basan en el estudio de las
relaciones entre la estructura química de los fármacos y la actividad biológica, conocido con las siglas QSAR.
El paso más crítico en el descubrimiento de un fármaco sigue siendo la identificación y optimización de compuestos líderes
de forma rápida y costeable, que se logra a través del enfoque conocido por CADD (Computer-Aided Drug Discovery) que
ofrece una alternativa al mundo real de la búsqueda y la síntesis2 y comprende “todas las técnicas asistidas por computación
usadas para descubrir, diseñar y optimizar compuestos con propiedades y estructura deseadas”.3
La obtención de un nuevo fármaco se puede lograr a partir de una fuente natural, por síntesis orgánica o biotecnológica. En
este sentido, el uso de la síntesis orgánica constituye la vía que aporta el mayor número de nuevos productos al cada vez
más próspero mercado farmacéutico. 4
Las cumarinas, que constituyen las materias primas de las estructuras tetracíclicas obtenidas, son compuestos de origen
natural y sintético con interesantes propiedades farmacológicas; estas son usadas en el tratamiento de enfermedades severas
de la piel,5 como antioxidantes y anticoagulantes,6 como agentes fotoquimioterápicos,7 entre otras.
Este trabajo esta basado en la utilización de una nueva metodología desarrollada con el objetivo de diseñar, sintetizar y
evaluar biológicamente nuevos compuestos con posible actividad antitumoral con el empleo de descriptores moleculares
novedosos.
Materiales y métodos
Los momentos espectrales se calcularon con el uso del paquete informático TOSS MODE.8
El análisis estadístico se efectuó utilizando el paquete de programas para el procesamiento estadístico STATISTICA9 para
WINDOWS, versión 4.3. El modelo cualitativo de clasificación y predicción se obtuvo con la utilización de la técnica del
Análisis Discriminante Lineal (ADL) conjuntamente con los estadígrafos generados, como son: número de casos (N), la
lambda de Wilk (λ), el cuadrado de la distancia de Mahalanobis (D2) y la razón de Fisher (F).10,11
La síntesis del esqueleto tetracíclico se realizó mediante la vía retrosintética, destacándose cada uno de los etapas y
condiciones de reacción.12
Los compuestos fueron objeto de estudio rutinario por espectrofotometría IR, empleando un espectrofotómetro PERKIN-
ELMER 1640 FTIR. Las muestras se prepararon en pastillas de KBr, utilizando la técnica estándar.

1
El ensayo de proliferación celular se realizó sobre células HL-60 mantenidas en incubación durante 24 h. La sustancia a
examen fue añadida disuelta en etanol. Después de 3 h se irradiaron las células 10 min. y se dejaron durante 21 h incubando.
Las células se contaron con Trypan Blu en cámara de Burker y los resultados se expresan en DI50.
Los experimentos de fototoxicidad cutánea se realizaron sobre piel depilada de cobaya albina, aplicando tópicamente 50
µg/cm2 de las sustancias estudiadas disueltas en etanol. Los animales se mantuvieron 45 min. en la oscuridad y
posteriormente fueron irradiados con un a dosis total UV equivalente 9.9x10-4 J/m2 para ser absorbidos 48 h más tarde.
Resultados y discusión
Previamente se obtuvo un modelo teórico antitumoral capaz de clasificar y predecir correctamente los nuevos compuestos
con actividad deseada. Para ello, se partió de una muestra de entrenamiento (ME) seleccionada, la que contiene tanto
compuestos activos (antitumorales) como inactivos (otras acciones farmacológicas), con gran diversidad estructural. El
modelo obtenido, conjuntamente con sus estadígrafos, se muestra a continuación:13, 14
7061095,610040,015
81078,61471011,4
841027,77022,0 6125,0 4202,0 3242,0 2552,0 1234,1 998,9 .
µµµµµµ
µµµµµµµ
×−⋅+×−×−⋅+×−⋅−
×−⋅+×+×−×+×−×−×+−=Act
N = 222 λ = 0,357 D2 = 5,90 F = 55,33
Se seleccionó una muestra de predicción (MP) con características semejantes a la ME, pero conteniendo otras estructuras no
incluidas en la ME. El porciento de buena clasificación para la ME y la MP fue de 91,9 y 94,4, respectivamente.
Con el modelo antitumoral obtenido, se calcularon las contribuciones de los fragmentos y grupos a la actividad deseada.15, 16
A partir de los resultados de este estudio, se tomaron en cuenta las siguientes consideraciones para el diseño de las nuevas
benzofurocumarinas:
- Al aumentar la longitud de la cadena R, se produce un incremento gradual de la contribución a la actividad antitumoral
- El anillo bencénico A y el anillo furánico C, contribuyen positivamente a la actividad.
- El anillo de lactona B presenta una contribución negativa.
- Cuando el anillo D tiene una insaturación contribuye positivamente y cuando es aromático lo hace negativamente.
Todo lo anterior, permitió el diseño de las nuevas benzofurocumarinas no reportadas en la bibliografía consultada con este
tipo de acción.
La síntesis de las nuevas benzofurocumarinas se realizó mediante la vía retrosintética, las que fueron caracterizadas
inequívocamente, observándose como evidencia espectroscópica significativa la desaparición de la banda del grupo OH en
el espectro IR del compuesto precursor.12
Los compuestos sintetizados fueron evaluados experimentalmente como antitumorales frente a las líneas celulares HeLa
(Human Cervix Adenocarcinoma) y HL-60 (Human Promyelocytic Leukemia) utilizando como patrón de referencia el 8-
MOP, fármaco que se utiliza actualmente en la terapia contra el Cáncer. 12 Los resultados más significativos se recogen en la
Tabla 1.
Tabla 1 Inhibición del crecimiento de las células en presencia de los compuestos sintetizados y del principio activo de
referencia 8-MOP. Clasificación y probabilidad obtenidos a partir del modelo antitumoral para los compuestos examinados.
CI50 (µM)a
Comp. Estructura HeLa HL-60 Clasif. Prob.
oscuro UV oscuro UV
1
O O
CH3
O
R
A BCD
>20 1.1±0.3 5.3±2.1 0.5±0.3 + 98.33
2
O O
CH3R
O
A B
C
D
>20 2.8±0.3 9.2±1.1 0.7±0.2 + 90.87
8-MOP
O O
O
H3CO
>20 10±3.0 >20 5.4±0.7 + 85.12
R = -O(CH2)3N(CH3)2; a Concentración requerida para la reducción proliferativa de las células tumorales al 50 %.

2
La capacidad de inhibición del crecimiento en las células HL-60 es también muy buena; los resultados observados para los
compuestos 1 y 2 son comparables con los obtenidos para el 8-MOP después de la irradiación (ver Tabla 2); pero con las
ventajas, sobre este último compuesto, de: 12
- Ser activos hasta en la oscuridad (ver Tabla 2), que no lo es el 8-MOP y
- No manifestar aparición de eritema cutáneo (característico del 8-MOP), aún a dosis cinco veces superiores (ver Tabla 3).
Tabla 2 Ensayo de inhibición de la proliferación celular.
Compuesto DI50 en ausencia de DI50 en presencia de
radiación (µM) radiación (µM)
1 1.80 1.10
2 2.07 1.15
8-MOP no activo 0.75
Tabla 3 Estudios de fototoxicidad cutánea.
Compuesto Formación de eritema (50 µg/cm2)
1 -
2 -
Compuesto de referencia Formación de eritema (10 µg/cm2)
8-MOP +++
- ausencia de eritema;
+++ evidente formación de eritema con edema.
Bibliografía1 Johnson, L., IUCr. Newsletter, 2 (1994) 5.2 Hann, M. and Green, R., Curr. Opin. Chem. Biol., 3 (1999) 411.3 van de Waterbeemd, H., et. al., Ann. Rep. Med. Chem., 33 (1998) 397.4 Evolución y perspectivas de la Industria Farmacéutica mundial en: El mundo en cifras. Farmacia y Biotecnología.
BIOMUNDI. La Habana, 2 (1996) 6.5 Kornhauser, A., et. al., Science, 217 (1982) 733.6 Foti, M., et. al., J. Agric. Food Chm., 2 (1996) 497.7 Gia, O., et. al., J. Med. Chem., 40 (1998) 352.8 Gutierrez, Y., et. al., TOSS MODE versión 2.5 for Windows. Universidad Central de Las Villas, Santa Clara, Cuba. 1995.9 STATISTICA for Windows versión 4.3 Copyright StatSoft Inc. 1993.10 Krishnaiah, P. R., Handbook of Statistic 2 Classification Patter Recognition and reduction of the dimensionality. Ed.
North Holland. 61, 1982.11 Alzina, R. B., Introducción conceptual al análisis multivariable. Ed. Ppu. Barcelona, 1989.12 Molina, E., Desarrollo de nuevos compuestos antitumorales y modelación de la actividad antimicrobiana a través del
empleo de descriptores moleculares novedosos. Tesis de Doctorado, 2002.13 Estrada, E. and Molina, E., In the book QSAR/QSPR Studies by Molecular Descriptors. M. Diudea (Ed.), Nova Science,
New York. 91, 2001.14 Estrada, E. and Molina, E., SAR & QSAR in Environm. Res., 12 (2001) 445.15 Molina, E. et. al., J. Mol. Graphics and Mod., 20 (2001) 54.16 Estrada, E. and Molina, E., J. Chem. Information and Comput. Sci., 41 (2001) 1015.

SINTESIS DEL 1-O-DODECILGLICEROL.
León J.L., Merchán F., Bilbao M., Nils A.Instituto de Farmacia y Alimentos. Universidad de La Habana.
Introducción. En los últimos años, los éteres de glicerilo han recibido gran atención debido a su granactividad biológica, sus mecanismos de acción constituyen temas actuales deinvestigación. Los efectos de la actividad farmacológica y fisiológica se explican porqueéstos presentan la estructura base de los más complejos éteres de glicerolípidos1
Los 1-o-alquilgliceroles exhiben actividad antimicrobiana, insecticida y herbicida2,3
inmunosupresoras4, antitumorales, promotoras de la absorción5 ,etc. La actividad antimicrobiana de los ésteres del glicerol ha sido ampliamente estudiada yel efecto de la cadena y el grado de esterificación han sido reportados. La actividad antimicrobiana no solamente puede estar asociada a los ésteres del glicerol,sino también a los éteres alquílicos del glicerol, como se reporta en el trabajo de Osanai ycol.6, que plantea que el 1-o-dodecilglicerol, presenta un marcado efecto bacteriostático. Para el 1-O-dodecilglicerol se ha reportado actividad antitumoral por activación demacrófagos7,8
La síntesis de los éteres de glicerilo ha sido abordada con menor o mayor éxito pordiversos autores, en la medida en que se han aplicado diferentes variantes a lo largo delproceso. Un método interesante se reporta por M.M. Ponpipom y col9, para la síntesis del1-o-hexadecilglicerol, preparando “in situ” el 1-o-isopropilidenoxihexadecano a partir deisopropilidenglicerol, hidruro de sodio y bromuro de hexadecilo, utilizando comodisolvente dimetilformamida y posterior hidrólisis del mismo.
Materiales y métodos. Para la síntesis del 1-o-dodecilglicerol se ha utilizado n-dodecanol (alcohol laurílico),dimetilformamida, cloruro de tionilo, todos de la BDH Chemical Ltd. Piridina (paraanálisis) Merck, 1,2-o-isopropilidenglicerol racémico de la Sigma Chemical Co. Con un97% de pureza. Todos los disolventes fueron de calidad analítica. El punto de fusión fue determinado en un equipo ELECTROTHERMAL 9100 defabricación inglesa, realizándose tres determinaciones. Para la espectroscopia infrarroja fue utilizado un equipo SPECOL-80 de la Carl Zeiss. La espectroscopia RMN en un equipo Brucker Ac-250F . Las determinaciones cromatográficas se realizaron por TLC utilizando como soporteSilica Gel 60G y como reveladores cámara de yodo y solución al 10% de ácido sulfúrico yposteriormente solución de permanganato de potasio al 3%.
Parte experimental.Preparación del 1 clorododecano. 20g de n-dodecanol, 8,48g de piridina, se agitan atemperatura ambiente y se añaden 12,76 g de cloruro de tionilo gota a gota. Culminada laadición se calienta en baño de parafina durante 6 horas, se deja reposar durante l2 horas yposteriormente se añaden 5 g de cloruro de tionilo, se refluja durante 8 horas más.Posteriormente se lava con agua, solución de bicarbonato de sodio y agua hasta pH 7. Seseca con sulfato de sodio anhidro durante 24 horas. Rendimiento 95% de 1-clorododecanocon 94% de pureza, determinada por cromatografía gaseosa.Preparación del 1-o-dodecilglicerol. Se mezclan 18,34g de isopropiliden glicerol, 18,0gde 1-clorododecano y 30ml de dimetilformamida, se agita vigorosamente a temperatura de

50ºC . Se añade lentamente 3g de sodio metálico fragmentado en pequeñas porciones. Sedeja a temperatura ambiente durante 12h y consumido todo el metal se calienta a 70ºCdurante 8h . La mezcla de reacción se vierte sobre agua helada y se extrae con 40ml de n-hexano. Se evapora todo el disolvente y el aceite remanente se disuelve en metanol, y seañade 30ml de metanol/HCl al 10% y se refluja durante 30 minutos. Se enfría a 0ºC y seextrae con 20ml de n-hexano frio. La fase alcohólica se evapora totalmente hasta obtenerun aceite amarillo viscoso, el mismo se disuelve en cloroformo se lava con agua , soluciónde bicarbonato de sodio al 5% hasta pH 7. Se evapora todo el cloroformo y se cristaliza den-hexano frio. Se obtienen cristales de punto de fusión 49-50ºC con un rendimiento del48,7%.
Resultados y discusión. La síntesis de 1-o-alquilgliceroles “in situ” es un método práctico que elimina lapurificación de los intermediarios durante la síntesis y sobre todo evita la resinificación dela sal sódica del isopropilidenglicerol , por oxidación en un medio fuertemente alcalino. En el presente trabajo se propone la síntesis “in situ” del 1-o-dodecilglicerol a partir delderivado clorado (menos reactivo que los yoduros y bromuros) y sodio metálico utilizadocomunmente en la preparación de alcóxidos , para hacerlo reaccionar posteriormente conel derivado halogenado y formar el éter. La reacción “in situ” en estas condiciones puedeprovocar si no se toman precauciones, la formación de una parafina , la cual se favorece enhaluros de alquilo de alto peso molecular y número par de átomos de carbono. El método“in situ”descrito por M.M.Ponpipom y col. Para la preparación del 1-o-hexadecilglicerol ,evita la reacción de Wurtz , pero el hidruro de sodio se inflama con facilidad en airehúmedo, lo que dificulta su manipulación. La síntesis propuesta en el presente trabajo limita la reacción de Wurtz, al añadirse elsodio lentamente a la temperatura de 50º C. Por otra parte el 1- clorododecano está enexceso con relación al alcóxido que se va formando, lo que no favorece la reacción deeliminación. Al efectuar la síntesis con el halogenuro de alquilo menos reactivo sedemuestra que aun en condiciones adversas el rendimiento del éter es satisfactorio y conun alto grado de pureza. La temperatura de fusión del éter de glicerilo obtenido por estavía se corresponde con lo reportado en la literatura (49-50ºC) La cromatografía en placadelgada empleando diferentes sistemas de solventes revela la presencia de una solamancha. El espectro infrarrojo presenta las bandas características del enlace C-O-C a 1,010 cm-1
la banda ancha característica del grupo hidroxilo asociado en la región de los 3,200-3,500cm-1 y las bandas características de los grupos metilenos de valencia simétrica 2,850-3,000 cm-1. El espectro de H´ RMN 250 MHz muestra las siguientes señales (CDCl3, δ ppm): 0.86 t3H, 1.26 s 18H, 1.58 t 2H, 3.50 m 4H, 3.66 2H, 3.85 m 1H.
Conclusiones. Se ha sintetizado el 1-o-dodecilglicerol utilizando el método “in situ” en la formación deléter intermediario, limitando las reacciones colaterales que se producen. Se utiliza el 1-clorododecano en lugar del yoduro o bromuro de dodecilo comúnmemte utilizados en lasíntesis de los éteres de glicerilo por ser más reactivos, pero más complejos en supreparación. En consideración al derivado halogenado de partida el rendimiento del 1-o-dodecilglicerol es satisfactorio y se alcanza con facilidad un producto de elevada pureza.

Referencias.1. Andreeseu,R. y Munder,P.G. "Ether-Lipophospholipids and celular inmunity. A
potencial role for antitumor activity. New trends lipids Mediators Res.; 1: 16-20;(1988).
2. Weber,N. "Biologycal effects of alkylglycerols". Prog. Biochem. Pharmacol., 22:48-50; (1988).
3. Osmond, D.G., et al.: The Action of Batyl Alcohol and Selachyl Alcohol on theBone Marrow of the Guinea Pig. Acta Hematol. Quie, P.G.: Antimicrobialdefenses in the neonate. Semin Perinatol, 14: 2-9, 1990.
4. Valdés,Y. y col. "1-o-alquilglicerols induced supression of human lymphocyteresponse to mitogens". I.M.L.: 242; (1992).
5. Werbach, M.R.: Alkylglycerols in Cancer. 1 Orth Molec Med, 9:71; 1994.6. Osanai,S.; Wakisaka,T. and Inove,H. "Synthesis of alcoxyl and fenoxyl sustitute
glycerides and their antimicrobial properties". J. Antibact. Antifung. Agents.VOL.14(3): 109-116; (1986).
7. Werbach, M.R.: Alkylglycerols in Cancer. 1 Orth Molec Med, 9:71; 1994.8. Ngwenya BZ, Fiavey NP, Mogashoa MM, Anti-neoplastic action of peritoneal
macrophages following oral administration of ether analogues oflysophospholipids. Eur J Cancer 1992;28A(10):1637-42.
9. Ponpipom,M. Y col. Síntesis of azide and amide analogs of platelet-activantingfactor and related derivatives. Chemestry and Physics of Lipids, 35 (1984), 29-37.

SISTEMA ACETATO DE VANADILO / CLOROFORMO. POTENCIAL CATALÍTICO PARA PROCESOS DE
ACETILACIÓN DE ALCOHOLES SECUNDARIOS.
Lic. J. L. Leyva 1 *, Dr. J. E. Tacoronte 2, M. Sc. F. A. Verdecia 1, Lic. L. G. Pérez 1, Lic. M. T. Cabrera 2.
1.- Departamento de Investigaciones, Empresa Laboratorio Farmacéutico "Mario Muñoz". QUIMEFA, MINBAS. Hacendado No.
1, Habana Vieja. C. Habana. CP 10200. Cuba. Tef: 863-4269. Fax: 863-1295. E-mail: [email protected];
2.- Laboratorio de Producto Naturales, Fac. de Química. U/H. Zapata y G, Plaza. C. Habana. CP 10400. Cuba.
RESUMEN
La utilización de un sistema heterogéneo tipo VO(AcO)2 / CHCl3 para la obtención de ésteres, acetatos (alcohol secundario +
anhídrido acético, a reflujo, 12-36 h) permitió llevar a cabo una reacción ecológicamente limpia, alcanzándose rendimientos
>70%, minimizando los riesgos de contaminación ambiental al no ser utilizada la piridina o bases amínicas tóxicas, disminuyendo
la carga energética y optimizando el proceso operativo al disminuir el número de etapas y la elaboración de la mezcla reaccionante.
De gran significación metodológica fue la posibilidad de reutilizar el catalizador acetato de vanadilo (V2O5 + (AcO)2O, reflujo, 3
h), que es simplemente filtrado y activado a 50°C durante 1 hora antes de iniciar otro ciclo catalítico.
INTRODUCCION
La acetilación de esteroides y monoterpenos constituye en muchos casos una etapa de síntesis necesaria en la búsqueda de
intermediarios y/o substancias biológicamente activas tipo ecdiesteroides, promotores del crecimiento vegetal y control de
metamorfosis de insectos y derivados hormonales de uso clínico-terapéutico 1.
El uso de la piridina como catalizador y solvente y un anhídrido de ácido o cloruro de ácido para la acetilación de alcoholes es el
método más antiguo conocido. Aunque la acetilación de alcoholes primarios y secundarios presenta pocos problemas, los alcoholes
impedidos estéricamente son resistentes a la acetilación. Es por esto que se desarrollaron otros métodos para la acilación
empleando como catalizadores la 4 -dimetilaminopiridina (DAP) o 4 -pirrolidinopiridina (Ppy) 2, así como otras substancias:
tributilfosfina (Bu3P) 3, cloruro de cobalto (CoCl2) 4, trimetilsililtrifluorometano sulfonato 5, trifluorometano sulfonato de escandio 6,
Sc(NTf)3 7, mediante sílica gel impregnada con Ce(SO4)2 , Ti(SO4)2 , Fe2(SO4)3 , NaHSO4
8, Tl(OEt) 9.
El objetivo de este trabajo fue sustituir la piridina en reacciones de acetilación de
alcoholes por el sistema heterogéneo tipo acetato de vanadilo/cloroformo como potencial
catalítico y anhídrido acético a reflujo para lograr un proceso ecológicamente limpio.
El acetato de vanadilo es una substancia sólida; química y térmicamente estable. No produce alteraciones metabólico-
funcionales del tracto gastro-intestinal en seres humanos, afecciones renales, trastornos hemohepáticos, ni a nivel del
sistema nervioso central 10.
MATERIALES Y METODOS
El control de las reacciones se realizó mediante cromatografía de capa fina, empleándose cromatoplacas de sílica gel 60F254
MERCK de 5 cm de ancho por 10 cm de largo y 0.25 mm de espesor. Como fase móvil se utilizaron diferentes mezclas de
solventes.
El revelado se realizó utilizando una solución de H2SO4(c)/ETOH (96%) (1:1 v/v) y una lámpara UV a λ =365 nm.
El aislamiento y la purificación de los acetoderivados se realizó mediante cromatografía de columna, empleando como fase
estacionaria sílica gel 60 (70-230 mesh ASTM) y diferentes mezclas de solventes como fase móvil.

Las temperaturas de fusión se determinaron en un Electrothermal modelo IA6304, sin corrección. Los espectros infrarrojos se
registraron en espectrofotómetro PHILIPS FTIR PU-9600, las muestras se prepararon en pastillas de bromuro de potasio. Los
espectros RMN-1H y 13C se registraron en un equipo BRUKER ACF-250, utilizando como solventes cloroformo y dimetilsulfóxido
deuterados. Como referencia interna se usó tetrametilsilano (TMS).
Síntesis del acetato de vanadilo.
En un matraz de fondo redondo se colocó 18.2 g (0.1mol) de V2O5 y de 150 ml (1.6 mol) de (AcO)2O. La mezcla reaccionante se
reflujó 3 h, se filtró a vacío y el producto (1, ver figura) se lavó con 105 ml de CHCl3. Luego el mismo se secó a vacío a 60°C.
Método general para la síntesis de acetoderivados.
En un matraz de fondo redondo se colocó 1 mmol de los alcoholes secundarios (2, 3, 4, 5, 6, 7, 8, ver figura) y 6 ml de cloroformo,
este sistema se agitó durante diez minutos. Posteriormente se adicionó 5mmol (0.45 ml) de anhídrido acético y 0.5 mmol
(76.45mg) de acetato de vanadilo (1) previamente activado. La mezcla reaccionante se reflujó durante 12-36 h. Luego se filtró a
vacío, el catalizador se lavó con 1.5 ml de cloroformo (en dos porciones), para ser utilizado en otro ciclo catalítico y el filtrado
resultante se retoevaporó a sequedad. De la mezcla resultante se aislaron los acetoderivados de interés (9, 10, 11, 12, 13, 14, 15, ver
figura) mediante cromatografía de columna.
Figura: Estructuras de los substratos y acetoderivados.
VAcO
AcOO
1- Acetato de Vanadilo
O
O
O
H
HecogeninaAcetato de Hecogenina
RO
2- R=H
9- R=Ac 3- R=H (25R)-2α,3α-dihidroxi-espirostan-6-ona 10- R=Ac (25R)-2α-acetoxi-3α-hidroxi-espirostan-6-ona
RO
O
HO
O
OH
4- R=H (25R)-3β,5α-dihidroxi-espirostan-6-ona11- R=Ac (25R)-3β-acetoxi-5α-hidroxi-espirostan-6-ona
O
O
OOHRO
O
OR
TestosteronaAcetato de Testosterona12- R=Ac
5- R=H EstronaRO
O
Acetato de Estrona6- R=H
13- R=Ac
Diosgenina
O
O
RO
Acetato de Diosgenina14- R=Ac7- R=H Borneol
H3C CH3
CH3
H
OR
Acetato de Bornilo8- R=H
15- R=Ac

RESULTADOS Y DISCUSION
La conversión de alcoholes secundarios en sus correspondientes acetoderivados transcurrió en condiciones ecológicas y
metodológicas sencillas y limpias sin el uso de parámetros experimentales extremos. A continuación se presenta una tabla con los
rendimientos y la temperatura de fusión de los acetoderivados obtenidos.
Tabla I. Rendimientos y temperaturas de fusión del acetato de vanadilo y los acetoderivados.
COMPUESTOS RENDIMIENTOS (%) Tf (°C)
1 91 % (16.56 g) > 360 °C9 78.18 % (369 mg) 243-244 °C
10 74 % (330 mg) 260-262 °C11 70 % (311.7 mg) 280-282 °C12 78.58 % (259.3 mg) 154-155 °C13 76.11 % (235.8 mg) 126-128°C14 72 % (299.52 mg) 194-195°C15 70 % (107.98 mg) 206-207 °C
Como se observa en la tabla I, con el empleo de este sistema catalítico se logró alcanzar rendimientos mayores del 70% para los
acetoderivados obtenidos, coincidiendo sus temperaturas de fusión con las reportadas en la literatura.
En la tabla II se presentan algunas características espectroscópicas (IR, RMN1H, RMN13C) de los compuestos sintetizados, lo cual
permitió una adecuada caracterización.
Para los acetoderivados obtenidos en el espectro FTIR se observaron bandas de 1205 a 1249 cm-1 asociadas a νasc-c-o; así como
también en RMN-1H las señales pertenecientes a los protones Hα-3, Hβ-2 y Hα-17 entre 4.30-5.02 ppm todas como multipletes y
en RMN-13 C las correspondientes a los carbonilos de ésteres detectadas entre 168.46-172.34 ppm, corroborándose con todo ello la
formación de los acetoderivados obtenidos.
En el espectro FTIR para productos espirostánicos (9, 10, 11 y 14) se observó bandas entre 987-860 cm-1, típicas para sistemas
espirocetálicos asociados a νC-C y νC-O y entre 890-925 cm-1 que indican la existencia de la serie esteroidal 25R; lo que demostró
que este sistema catalítico no afectó estructuralmente dichos fragmentos moleculares (anillo E y F).
Tabla II. Algunas características espectroscópica de los acetoderivados.
SEÑALES
COMPUESTOS IR (cm-1) RMN-1H (ppm) RMN-13C (ppm)
9 1237.90; 955, 920, 882 4.65 (Hα, m, H3) 172.34 (CH3-CO-O)10 1242; 975, 920, 890, 860 4.60(Hβ, m, H2) 168.46 (CH3-CO-O)
11 1240; 920, 980, 903, 866 5.02(Hα, m, H3) 171.20 (CH3-CO-O)
12 1248.99 4.66(Hα, m, H17) 171.15 (CH3-CO-O)
13 1205.38 - 169.20 (CH3-CO-O)
14 1239.98; 987, 960, 890, 888 4.30(Hα, m, H3) 170.44 (CH3-CO-O)
15 1245.20 4.95(Hβ, m, H2) 171.50 (CH3-CO-O)
Los datos espectroscópicos demostraron que los alcoholes secundarios se acetilaron en estas condiciones empleadas. Derivado
cíclico monoterpénico como el Borneol (8) se acetiló (15), en las mismas condiciones. Alcohol espirostánico terciario α-
carbonílico (4) no se acetiló, lo que permitió utilizar el sistema catalítico selectivamente para alcoholes secundarios.
CONCLUSIONES

1.- El acetato de vanadilo, un ácido de Lewis sui géneris, cataliza la reacción de acetilación de alcoholes secundarios no
impedidos estéricamente, de procedencia natural y derivado cíclico monoterpénico, como el Borneol, en condiciones
heterogéneas y ecológicamente tolerantes en ausencia de piridina y derivados sin generar procesos colaterales.
2.- Se obtienen rendimientos mayores del 70 % en el proceso de obtención de acetoderivados sin la utilización de parámetros
experimentales extremos (atmósfera inerte, elevadas temperaturas o agentes corrosivos).
3.- El sistema desarrollado permite la acetilación selectiva de substratos con sustituyentes OH secundarios en presencia de OH
terciarios α-carbonílicos.
R E F E R E N C I A S
1. XIIIth EDCYZONE Workshop, Jena, July 27-31, ed. Jena Universitat. 1998, 1-112.
2. E. F. Vscriven Chem. Soc. Rev. 1983, 12, 129.
3. E. Verdej, N. S. Bennett, L. M. Conn, S. T. Diver, M. Gingras, S. Lin, P. A. Oliver, M. J. Peterson. J. Org. Chem. 1993, 58,
7286.
4. J. Iqbal, R. R. Srivastava J. Org. Chem., 1992, 57, 2001.
5. P. A. Procopion, S. D. P. Baugh, S. S. Flack, G. G. A. Iglis, J. Chem. Soc. Chem. Commun. 1996, 2625.
6. K. Ishijara, M. Kubota, H. Kurihara, H. Yamamota. J. Org. Chem. 1996, 61, 4560.
7. I. Shina, T. Mukayama. Synlett, 1996, 265.
8. T. Nishiguchi, H. Taya. J. Chem. Soc. Perkin Trans. 1990, 1, 172.
9. J. E. Herz, S. Cruz, J. V. Torres, A. Murillo. Synth. Commun. 1977, 7(6), 383-385.
10. N. Irving Sax. Dangerous Properties of Industrial Materials. Ed. Rev. ICL. 1968, 1067, 1223.

SÍNTESIS DEL [2,3-d ]-ISOXAZOL-17α-ETINIL-17β-HIDROXI-4-ANDROSTENO
Mayra Reyes; Yoanna Ma. Alvarez; José A. Ruiz; Harold Curiel y Hermán Vélez
Centro de Química Farmacéutica.
RESUMENEl [2,3-d]-isoxazol-17α-etinil-17β-hidroxi-4-androsteno (danazol) es un esteroide sintético obtenido a partir de 4-androsten-3,17-diona (AD). Su uso más frecuente es en el tratamiento de la displacía benigna e hipertrofia pubertal demamas, además de desórdenes como la ginecomastia. En el presente trabajo se presentan los resultados logrados en lasíntesis, así como la elucidación estructural y las temperaturas de fusión de cada uno de los productos.
INTRODUCCIONHoy en día el término esteroide se ha reservado sólo para aquellos compuestos que presentan una marcada acción sobre elorganismo humano, es decir, los esteroides terapéuticamente activos, como las hormonas sexuales, los corticosteroides,anabólicos, todos de gran valor terapéutico. Muchos de estos compuestos se encuentran en los animales (incluyendo alhombre) en muy pequeñas cantidades, por lo que su obtención es muy compleja y costosa a partir de dichas fuentes.El hombre se ha visto precisado a desarrollar otras vías de obtención de dichos compuestos, tales como la síntesis químicay la transformación microbiológica a partir de fuentes vegetales.1
El danazol, es un esteroide de amplio uso en el tratamiento de la endometriosis, la hipertrofia de los senos, el angioedemahereditario, la pubertad precoz y el síndrome premenstrual.2
El propósito de este trabajo es la síntesis de este medicamento y su comparación con lo reportado.4-8
El procedimiento diseñado en este trabajo (figura 1) consta de cuatro etapas de síntesis:
O
O
C2H5O
O OH
C C H
O
OH
C C H
O
HC
OH
OH
C C H
ON
HC(C2H5O)3
Na/MeOHHCOOC2H5/Py
NH2OH.HCl/AcONa
EtOH
MSA/EtOH abs.K/IspOH/C2H2/THF
LiC CH.H2NCH2CH2NH2
ó
1 2
3
5 4 Figura 1 Esquema de obtención del Danazol
MATERIALES Y METODOS
3-etoxi-3,5-androstadien-17-ona.(enol éter de androstendiona) 2: En un matraz de 250 mL, de una boca provisto deagitación se introducen 150 mL de etanol absoluto, 25 g de 1 y 20 mL de ortoformiato de etilo, se agita 5 minutos. A estasuspensión se le adicionan 0,075 mL de ácido metanosulfónico (MSA) y se agita 30 minutos. Se realiza cromatografía encapa delgada empleando como mezcla de disolventes tolueno : acetato de etilo 4:1. Terminada la reacción, se añaden 4,5mL de piridina, se agita 5 minutos y enfría a 0 0C por espacio de 2 horas. Se filtra y seca el producto en el filtro a vacío atemperatura ambiente. Rend. 24,5 g (90%). IR (cm-1) 2900 (f), 1722(f), 1620 (m), 1375 (m), 1360 (m), 1218 (f), 1160 (f),1040 (m). RMN 1H (CHCl3, ppm) 0,92 (3H,s,CH3-18); 1,0 (3H,s,CH3-19); 1,25 (3H,t,CH3-CH2-O); 3,75 (2H,c,R-CH2-O); 5,1(1H, s, H-4); 5,2 (1H,m,H-6). 17α-etinil-17β-hidroxi-4-androsten-3-ona (etisterona) 3.En un matraz de 250 mL de 3 bocas, provisto de agitación magnética y embudo goteador, se adicionan 25 g de acetiluro delitio en etilendiamina y se enfría a 0oC. Se adiciona en 10 min una disolución que contiene 18 g de 2 en 100 mL detetrahidrofurano. Se agita la reacción durante 24 horas cuidando que la temperatura se mantenga entre 5 y 10 ºC.Transcurrido este tiempo se controla la reacción por CCD. Culminada la misma se añaden 60 ml de agua y agita 15 min. Seseparan las fases, el extracto acuoso se extrae con 40 ml de acetato de etilo, se separan las fases y el extracto orgánico seune a la fracción orgánica principal. La fracción orgánica principal se lava con 3 x 40 mL de una disolución saturada de

cloruro de sodio y se concentra a sequedad con vacío. Al residuo obtenido se le adicionan 70 mL de metanol y se agita 30min. Posteriormente, se adicionan 20 mL de agua y 0,5 mL de. ácido metanosulfónico. La mezcla se calienta a reflujo por 1hora, se enfría a 00C obteniéndose un sólido que se filtra y se lava con pequeñas porciones de metanol frío. Se seca enhorno de aire recirculado a 60 ºC. Rend. 16 g (90%). t.f. 255-58 oC. IR (cm-1) 3400 (f), 3250 (f), 2950 (f), 2900 (f), 1640 (f),1370 (m), 1225(m), 1045 (f). RMN 1H (CHCl3, ppm) 0,9 (3H,s,CH3-18); 1,2 (3H,s,CH3-19); 2,15 (1H,s, OH-17); 2,55 (1H, s,H-Alquino); 5,7 (1H,s,H-4). 17α-etinil-17β-hidroxi-2-hidroximetilen-4-androsten-3-ona 4.En un matraz de fondo redondo de dos bocas de 250 ml, provisto de agitador magnético, condensador y tubo de cloruro decalcio, se introducen 40 ml de etanol absoluto y se añaden lentamente 2,25 g de sodio hasta disolución total. Se pasanitrógeno y se adiciona una disolución de 5 g de 3 en 25 mL de piridina seca (KF< 0,7 %). Se agita 30 min. Se adicionan8,5 mL de etilformiato y se continúa agitando por 24 horas a temperatura ambiente. Se realiza el control de la reacción porCCD. La mezcla de reacción se vierte en un vaso de precipitados de 1 L que contiene 400 mL de ácido clorhídrico 6N y seobserva la formación de un precipitado amarillo. La suspensión se extrae con 3 x 50 mL de diclorometano y los extractosorgánicos son extraídos con 4 x 50 mL de una disolución de hidróxido de potasio al 10 %. Los extractos acuosos se enfríanentre 8 y 10 0C y se precipitan con 40 a 45 mL de ácido clorhídrico 6 N hasta lograr un pH igual a 2. Se agita 30 min y sevuelve a controlar el pH. Se filtra y se lava el sólido con agua hasta pH neutro. Se seca por succión y después en horno deaire recirculado a 50 0C. Rend. 3,27 g (60 %). t.f 193-5 oC. IR (cm-1) 3415 (f), 3300 (f), 2938 (f), 2876 (f), 1646 (f), 1192 (f),1061(f). RMN 1H (CHCL3, ppm) 0,88 (3H,s,CH3-18); 1,2 (3H,s,CH3-19); 2,15 (1H,s,OH-17); 2,33 (1H,s,OH-hidroximetileno);2,55 (1H,s,H-Alquino); 5,75 (1H,s,H-4); 7,35 (1H,s,HC=R).[2,3-d ]-isoxazol-17α-etinil-17β-hidroxi-4-androsteno 5.En un matraz de fondo redondo de dos bocas de 100 ml, provisto de agitador y condensador, se adicionan 45 ml de etanolabsoluto y 3 g de 4. Después de disuelto el esteroide se añade una disolución que contiene 0,675 g de hidroxilamina, 0,86g de acetato de sodio en 1,8 mL de agua. La reacción se calienta a reflujo durante 1 hora, se deja enfriar y se viertelentamente y con agitación sobre un vaso de precipitado de 500 mL que contiene 250 mL de agua. El sólido resultante sefiltra, se lava con agua y se seca en vacío a 50 0C. Rend. 2,37 (80%), t.f 225-7 oC. IR (cm-1) 3516 (f), 3262 (f), 2941 (f),2877 (f), 1600 (f), 1470 (f), 1435 (f), 1062 (f), 947 (f), 858 (m). RMN 1H (CHCl3, ppm) 0,95 (3H,s,CH3-18); 0,83 (3H,s,CH3-19); 0,98 (1H,H-6a); 1,3 (1H,H-11a); 1,4 (1H,H-15a); 1,45 (1H,H-14); 1,55 (1H,H-15e);1,6 (1H,H-12a); 1,7 (1H,H-12e); 1,67(1H,H-11e); 1,94 (1H,H-16e); 2,25 (1H,H-16a); 2,35 (1H,H-6e); 2,32 (1H,H-7a); 2,38 (1H,H-7e); 2,43 (1H,H-1a); 2,65 (1H,H-1e); 2,5 (1H,s,H-Alquilo); 6,10 (1H,s,H-4); 7,93 (1H,s,H-vinílico del isoxazol). RMN 13C (CHCl3, ppm) C5, 164,9; C3,154,4; C22, 148,6; C4, 108,7; C2, 107,6; C20, 87,3; C17, 79,7; C21, 74,1; C9, 53,8; C14, 49,8; C13, 46,7; C10, 41,0;C16, 38,9; C8, 36,9; C1, 33,3; C12, 32,6; C7, 32,2; C6, 30,6; C11, 23,2; C15, 21,2; C18, 18,7; C19, 12,7;
RESULTADOS Y DISCUSIÓN
La obtención de 2 consiste en la protección del grupo carbonilo de la posición 3 del compuesto 1 para realizar laintroducción del grupo etinilo en la posición 17. Muchos son los ácidos de Lewis que se utilizan para realizar estaprotección. Después de haber evaluado diferentes catalizadores como ácido p-toluensulfónico, ácido metanosulfónico,ácido sulfúrico, ácido clorhídrico y disolventes como benceno, tolueno, tetrahidrofurano, dioxano y etanol absoluto, seseleccionó la combinación MSA con etanol absoluto por ser el etanol el disolvente menos tóxico y el MSA un ácido fácil demanipular y cuantificar, además de obtenerse muy buenos resultados con estos reactivos en experiencias anteriores.3
Se comprobó la formación del enol éter por el análisis del espectro IR de 2, donde se mostró la desaparición de la banda de1680 cm-1, perteneciente a las vibraciones de valencia del grupo carbonilo de la cetona α,β-insaturada que se encuentra enel AD de partida.. En el espectro RMN 1H de 2 se observó una señal de multiplicidad cuadruplete, a 3,75 ppm,característica del grupo metileno perteneciente al éter de enol. También, una señal a 1,25 ppm, perteneciente al metilo delmismo grupo que aparece como un triplete por el acoplamiento con el metileno vecino. Todo esto confirmó que laprotección fue completa. Además, aparecieron señales de protones vinílicos de las posiciones 4 y 6 a 5,1 y 5,2 ppm,respectivamente Para la obtención de 3 se ensayaron dos métodos:• La etinilación usando potasio e isopropanol.• El empleo del acetiluro de litio en etilendiamina.El primero de estos métodos consistió en preparar un alcóxido de potasio "in situ", o sea, a partir de isopropanol y potasio.Para esto se empleó 1 mol de isopropanol por mol de potasio. Esta cantidad está en una relación de 1:16,5 con respecto alesteroide. Este exceso se debe a que después de formado el isopropóxido de potasio se pasa corriente de acetileno paraintercambiar las sales y obtener el acetiluro de potasio que es quién finalmente ataca a la molécula de esteroide. Ladificultad de este método radica en la peligrosidad que existe al manipular potasio y además garantizar que el acetilenoutilizado sea filtrado.

En el segundo procedimiento se trabajó con un reactivo comercial el acetiluro de litio en etilendiamina4 y la relación molarfue de 1:5 con respecto al esteroide, todo en atmósfera de nitrógeno y usando como disolvente el tetrahidrofurano. La introducción del grupo etinilo quedó demostrada del análisis del espectro IR de 3 donde se observan dos bandas en 3400y 3250 cm-1 pertenecientes a las vibraciones de valencia del grupo hidroxilo y el grupo etinilo formados. Además,desapareció la banda de 1722 cm-1, que pertenecía al carbonilo de la posición 17 que fue modificado. También se observa labanda en 1640 cm-1, perteneciente a las vibraciones de valencia del grupo carbonilo de la cetona α,β-insaturada de laposición 3. En el espectro de RMN 1H de 3 se observaron señales a 2,15 y 2,55 ppm como singuletes, pertenecientes a losprotones de los grupos hidroxilo y etinilo respectivamente. Además, la señal del protón vinílico del C4, como singulete, a5,7 ppm.La obtención de 4 consistió en la introducción del grupo hidroximetileno en la posición 2 de 35. Por la insolubilidad quepresenta la etisterona en los disolventes que se emplean en este tipo de reacción, se decide hacer la misma en piridina enrelación de 1:25 y emplear etóxido de sodio (que se prepara "in situ" con sodio y etanol), como base fuerte para eliminar elhidrógeno α al grupo carbonilo en una relación con el esteroide de 1:6. El etilformiato se empleó como agente acilante enrelación con el esteroide de 1:6,6. En el caso del espectro IR de 4 la única diferencia significativa además de las presentes en la región dactilar, es la que sepresenta en la banda que aparece en 3415 cm-1 que la misma se ancha de forma apreciable debido a que surge un nuevogrupo hidroxilo en la molécula. En el espectro de RMN 1H de 4, se observaron señales a 2,15 y 2,55 ppm como singuletes,pertenecientes a los protones de los grupos hidroxilo y etinilo, respectivamente. Además, la señal del protón vinílico del C4,como singulete, a 5,7 ppm.La última etapa de obtención del danazol 5 consistió en la formación del anillo isoxazólico para lo que se empleó unarelación esteroide:clorhidrato de hidroxilamina de 1:1,1 y una relación esteroide:acetato de sodio de 1:0,25. El acetato desodio garantiza el tampón para evitar que se deshidrate el grupo hidroxilo de la posición 176,7,8. Se comparó el espectro IR de 5 con el de un patrón internacional Steraloid, existiendo una perfecta coincidencia.Evidencias de que la reacción de obtención de 5 tuvo lugar lo mostró el análisis del espectro de RMN 1H, en donde seobservaron señales a 0,88 y 1,2 ppm como singuletes, correspondientes a los metilos 18 y 19, respectivamente. Un singuletea 2,15 ppm, que corresponde al protón del grupo hidroxilo de la posición 17, otro a 2,33 ppm, perteneciente al protón delgrupo hidroxilo del hidroximetileno y uno a 2,55 ppm, perteneciente al protón del grupo alquino. Además, un singulete a5,75 ppm, producto del protón vinílico en el C4 y por último, la señal a 7,35 ppm como singulete, del protón vinílico en elgrupo hidroximetileno.
Bibliografía
1. Applezweig, N. Steroid Drugs. New York, Mc. Graw-Hill Book, Inc. Vol 1, p. 20-25, (1962).2. Pharmaceutical manufacturing Encyclopedie 2nd Ed. Vol1, p.427-428, 1988. Marshal s Sittig United States of
America by Noyes Publications.3. Reyes, M., Alvarez, Y.Ma., Ruiz, J.A., Díaz, O., Acosta, J., Jáuregui, U.J. Optimización de la reacción de
formación del enol éter de androstendiona. Revista CENIC de Ciencias Químicas, en prensa.4. Stabilised Monolitium Acetylide and the ethynilation of steroids. EUROPEAN PATENT APPLICATION.
Number: 0148616. 1997.5. Ringold H.J., Batres E., Halpen O., Necoechea E. Steroids cv. 2-methyl and 2-hidoxymethylene-androstane
derivatives. J. Am. Chem. Soc. 81, p. 427, 1959.6. Clinton R.O., Manson A.J., Stonner F.W., et al. Steroidal [3,2-c] pyrazoles.II. Androstanes, 19-Norandrostanes and
their Unsaturated Analogs. J.Am.Chem.Soc., 83, 1478, 1961.7. Clinton R.O., Clarke R.L., Stonner F.W., Manson A.J., Jennings K.F., Phillips D.K. Steroidal Heterocycles.VI.
Formylation of A/B-cis-3-Ketosteroids. Preparation of 5β-Steroidal [3,2-c] pyrazoles. J.Org.Chem. 27, 2800, 1962.8. Manson, A.J., Stonner, F.W., Neumann, H.C., y col. Steroidal Heterocycles. VII. Androstano [2,3-d) isoxazoles
and related compounds. Journal of Medicinal Chemistry, Vol. 6, (1), p. 1-9, (1963).

NUEVOS PROCEDIMIENTOS PARA LA FORMACIÓN DEL ANILLO DE 2-AMINOTIAZOL DURANTE LA
SÍNTESIS DEL 2-(2-AMINOTIAZOL-4-IL)-(Z)-2-(HIDROXIMINO) ACETATO DE ETILO
M. González, Z. Rodríguez, B. Tolón, R. Avila, O. Díaz, H. Vélez, M. A. López.(Centro de Química Farmacéutica).
RESUMEN
El 2-(2-aminotiazol-4-il)-(Z)-2-(hidroximino) acetato de etilo (I) es un importante intermediario utilizado durante la síntesis
de antibióticos cefalosporánicos de amplio espectro. Durante el presente trabajo se desarrollaron 2 procedimientos para la
formación del anillo de 2-aminotiazol, fragmento molecular característico de este compuesto, mediante el uso de una
mezcla de THF-agua como disolvente y acetato de sodio como catalizador básico o empleando N,N-dimetilacetamida con
este doble propósito. Ambos métodos permitieron alcanzar rendimientos del intermediario superiores en un casi un 9 % y un
20 % respectivamente, a los alcanzados cuando se utilizan los procedimientos publicados en la literatura.
INTRODUCCION
El 2-(2-aminotiazol-4-il)-(Z)-2-(hidroximino) acetato de etilo (I) es un importante intermediario utilizado durante la síntesis
de antibióticos cefalosporánicos de amplio espectro, el cual se obtiene a partir del acetoacetato de etilo, por formación de la
oxima, halogenación del grupo metilo α-cetónico y formación del anillo de 2-aminotiazol mediante el método de Hantschz.
Los rendimientos que se obtienen, a través de los procedimientos publicados en la literatura1-4 son muy bajos, hecho que
influye negativamente, desde el punto de vista del balance económico del proceso, sobre la preparación de estos
antibióticos. El objetivo del presente trabajo consistió en desarrollar procedimientos para la formación del anillo de 2-
aminotiazol durante la preparación de I que permitieran obtener rendimientos más elevados de este intermediario.
MATERIALES Y METODOS
Las temperaturas de fusión (Tf) se determinaron en un equipo Gallenkamp y no fueron corregidas. Los espectros 1H RMN
fueron registrados en un Equipo Bruker AC 250F utilizando dimetilsulfóxido deuterado (DMSO-d6) como disolvente y TMS
como referencia.
Síntesis del 2-(2-aminotiazol-4-il)-(Z)-2-(hidroximino) acetato de etilo (I)
a) Formación de la oxima. Se disuelven 29,2 g (225 mmol) de acetoacetato de etilo en 29,6 mL de ácido acético glacial, se
enfría hasta 0 OC y se adicionan 18 g (308 mmol) de nitrito de sodio en 40 mL de agua manteniendo la temperatura por
debajo de 10 OC. Se agita 30 minutos a 10 OC y se eliminan el agua y el ácido acético por destilación al vacío. El residuo se
disuelve en 50 mL de acetato de etilo y se lava con una disolución de NaHCO3 al 5 % (2 x 50 mL). Después de separar las
fases, la fase orgánica se seca sobre Na2SO4 anhidro, se filtra y el filtrado se evapora hasta sequedad bajo presión reducida.
b) Halogenación de la oxima. El producto del proceso anterior se disuelve en 23 mL de ácido acético glacial, se calienta
hasta 58-60 OC, se adicionan 23,1 g (171 mmol) de cloruro de sulfurilo, se agita por 1 h a 58-60 OC y se evapora hasta
sequedad al vacío. El residuo se disuelve en 90 mL de acetato de etilo y se lava con disolución saturada de NaCl
(3 x 30 mL). La fase orgánica se separa, se seca sobre Na2SO4 anhidro, se filtra y el filtrado se evapora a sequedad al vacío.
c) Formación del anillo de 2-aminotiazol.
Uso de etanol como disolvente en presencia de N,N-dimetilanilina como base (Método 1, reportado)3.
El residuo obtenido en el paso anterior se disuelve en 50 mL de etanol, se adicionan 7,7 mL (59,3 mmol) de N,N-
dimetilanilina y se incorporan 4,2 g (55,2 mmol) de tiourea. Se agita por 2 h a temperatura ambiente y el precipitado que se
forma se separa por filtración. El sólido obtenido se lava con etanol (20 mL), acetona (20 mL), éter dietílico (20 mL) y se
seca a 40 OC durante 1 h.

Uso de etanol-agua como disolvente en ausencia de una base (Método 2, reportado)1,2,4.
Sobre una disolución de 14,0 g (184 mmol) de tiourea en una mezcla de etanol (42 mL) y agua (84 mL) se adiciona una
disolución de la oxima halogenada (36 g, 186 mmol) en 42 mL de agua. La mezcla se agita por 1 h a temperatura ambiente,
se concentra hasta la mitad del volumen inicial y se ajusta a pH 6 por adición de una disolución acuosa al 5 % de NaHCO3.
El sólido precipitado se colecta por filtración, se lava con éter dietílico (50 mL), acetona (50 mL) y se seca a 40 OC por 1 h.
Uso de THF-agua como disolvente en presencia de acetato de sodio como base (Método 3, desarrollado).
Se disuelven 18 g (93 mmol) de la oxima halogenada en 48 ml de THF, se añaden 48 ml de agua y a continuación de forma
sucesiva se adicionan 8,5 g (111,6 mmol) de tiourea y 15,0 g (182,8 mmol) de acetato de sodio anhidro. Se agita por 4 h a
temperatura ambiente, se ajusta a pH 6,7-6,8 con 5,2 g (61,8 mmol) de NaHCO3 y se extrae con acetato de etilo (2 x 100
mL). Después de eliminar la fase orgánica, el precipitado formado en la fase acuosa se separa por filtración, se lava con una
mezcla de acetato de etilo-agua (1:1) (2 x 20 ml) y se seca a 40 OC durante 3 h al vacío.
Uso de N,N-dimetilacetamida como disolvente y como base (Método 4, desarrollado).
Se disuelven 18 g (93 mmol) de la oxima halogenada en 40 ml de N,N-dimetilacetamida (DMA), se adicionan 4,3 g (56,5
mmol) de tiourea y se agita por 3 h a temperatura ambiente. La mezcla se vierte sobre 200 mL de agua fría, se añaden
200 mL de acetato de etilo y se ajusta hasta pH 6,1-6,2 por adición de 4,3 g (51,1mmol) de NaHCO3. Se elimina la fase
orgánica, el sólido precipitado en la fase acuosa se separa por filtración, se lava con acetato de etilo-agua (1:1) (2 x 20 ml) y
se seca a 40 OC durante 3 h al vacío.
RESULTADOS Y DISCUSION
En la Figura 1 se muestra el esquema para la síntesis de I y en la Tabla 1 los resultados obtenidos mediante los métodos
desarrollados en comparación con el procedimientos reportados en la literatura para la síntesis de este intermediario:
acetoacetatode etilo
CH3OH3C
O O
O CH3N
O
N
SH2N
OH
ClO CH3
O
N
O
OH
CH3OH3C
O O
N
OH
NaNO2HAc / H2O
SO2Cl2 HAc tiourea
oxima oxima halogenada IFigura 1. Esquema general para la síntesis del compuesto I
Tabla 1. Rendimientos obtenidos durante la preparación del compuesto I
Método Disolvente Catalizador Rendimiento de I (%)1 etanol N,N-dimetilanilina 13,92 etanol/agua - 17,23 THF/agua acetato de sodio 26,14 DMA - 36,9
La Temperatura de fusión y las constantes espectroscópicas de los productos obtenidos según las 4 técnicas utilizadas
fueron iguales y coincidieron con las reportadas en la literatura. Tf: 191-193 OC; 1H-RMN (δ ppm): 1,25 (3H,t,CH3); 4,24
(2H,q,CH2); 6,83 (1H,s,H, aminotiazol); 7,20 (2H,s,NH2); 11,65 (1H,s,NOH).
El proceso de obtención de I consta de 3 pasos que implican la formación de la oxima a partir del acetoacetato de etilo por
tratamiento con nitrito de sodio, la halogenación del grupo metilo α-cetónico con cloruro de sulfurilo y la formación del
anillo de 2-aminotiazol por reacción del derivado halogenado con tiourea. En el presente trabajo I se obtuvo de acuerdo al
método reportado con respecto a la ejecución de los 2 primeros pasos de síntesis y sin introducir modificación alguna. La

formación del anillo de 2-aminotiazol inicialmente se realizó por los métodos descritos1-4, basados en el tratamiento de la
oxima halogenada con tiourea utilizando etanol como disolvente y en presencia de N,N-dimetilanilina como base (Método
1)3 o en disolución hidroalcohólica y en ausencia de catalizador básico (Método 2)1,2,4. Aunque en ambos casos, estas
condiciones de reacción permiten preparar el isómero syn, que es el que despliega mayor actividad biológica y por lo tanto
el isómero de interés, también propician la formación de cantidades notables del isómero anti, lo cual se reflejó en los bajos
rendimientos obtenidos y que fueron similares a los reportados (13,9 % y 17,2 % respectivamente con relación al
acetoacetato de etilo).
Como consecuencia, en el presente trabajo se desarrollaron 2 procedimientos para la formación del anillo de 2-aminotiazol
con tiourea, el primero mediante el uso de una mezcla de THF-agua como disolvente y acetato de sodio como aceptor del
cloruro de hidrógeno formado (Método 3) y el segundo utilizando DMA con la doble función de disolvente y catalizador del
proceso (Método 4). Como se observa, con los métodos desarrollados se obtuvieron rendimientos significativamente
mayores a los reportados en la literatura. El mejor procedimiento fue aquel donde se utilizó DMA como disolvente y
catalizador de la reacción. En este caso se alcanzó un rendimiento superior en casi un 20 % (2,14 veces) al reportado en la
literatura. Sin lugar a dudas, en ambos procedimientos se logró un balance más favorable en la proporción isómero syn /
isómero anti. Los resultados obtenidos se pueden explicar si se considera que los isómeros syn y anti se encuentran en
equilibrio, el cual se conoce que está favorecido hacia la formación del isómero anti en medio ácido. De acuerdo al
mecanismo de formación del anillo de 2-aminotiazol por el método de Hantschz, durante el paso del ataque nucleofílico de
la tiourea al átomo de carbono halogenado adyacente al carbonilo cetónico, se genera como subproducto cloruro de
hidrógeno el cual incrementa la acidez del medio de reacción. Por esta causa, el uso de una sal básica como el acetato de
sodio (cuando se utiliza THF-agua como disolvente) o de un disolvente como la DMA, el cual posee propiedades básicas,
contribuyen a reducir la acidez del medio de reacción y por lo tanto a desplazar el equilibrio isomérico hacia la formación
del isómero syn. El uso de la DMA conduce a rendimientos más elevados, debido a que en este caso la reacción transcurre
en un medio homogéneo. Este disolvente es capaz de disolver tanto a la tiourea como al compuesto halogenado de partida,
propiciando una mejor interacción entre los reaccionantes. Cuando se utiliza como disolvente la mezcla THF-agua y el
acetato de sodio como catalizador, la reacción transcurre en un medio heterogéneo constituido por 2 fases: la orgánica
donde está disuelto el derivado halogenado de partida y la acuosa donde se encuentra la tiourea y el acetato de sodio, lo cual
resulta más desfavorable (rendimiento inferior en casi un 11 %). En el procedimiento reportado en la literatura donde se
utiliza etanol como disolvente, a pesar de la presencia de la N,N-dimetilanilina como aceptor del ácido formado, los
rendimientos obtenidos son muy bajos, inclusive inferiores a los que se alcanzan en medio hidroalcohólico en ausencia de
catalizador básico, hecho que ha sido reportado con anterioridad1,4.
BIBLIOGRAFIA
1. Bucourt, R., Heymes, R., Lutz, A., Pénasse, L., Perronet, J. Cephalosporines a chaines amino-2-thiazolyl-4-acetyles.
Tetraedron, Vol. 34, 2233-2243, (1978).
2. Heymes, R., Lutz, A. Akyloxymes of 7-amino-thiazolyl-acetamido-cephalosporanic acids. US Pat. 4 396 618 (1983).
3. Takeda Chemical Industries. Ltd. Thiazoles substitués et leur procedé de préparation. Fr. Pat. 2 357 552 (1978).
4. Ochiai, M., Aki, O., Morimoto, A., Matsushita, Y., Okada, T. New cephalosporin derivatives with high antibacterial
activities. Chem. Pharm. Bull., Vol. 25 (11), 3115-3117, (1977).


SINTESIS DEL HETEROCICLO 11H-PIRIDO[2,1-b]QUINAZOLIN-11-ONA Y DERIVADOS
EMPLEANDO IRRADIACION ULTRASONICA
Maite L. Docampo Palacios y Rolando F. Pellón Comdom
Centro de Química Farmacéutica Apartado 16042, La Habana, Cuba
Telef. (537)217809 Fax (537) 336471 E-mail: [email protected]
RESUMEN
Se reporta un método ventajoso para la síntesis del heterociclo 11H-pirido[2,1-b]quinazolin-11-ona y
derivados, mediante la condensación de derivados del ácido 2-clorobenzoico y derivados de la 2-
aminopiridina con el empleo de N,N-dimetilformamida como disolvente en presencia de irradiación
ultrasónica. Los derivados fueron obtenidos con altos rendimientos en tiempos cortos de reacción.
INTRODUCCIÓN
En una comunicación previa1 reportamos el empleo de la N,N-dimetilformamida como disolvente en la
condensación de Ullmann- Goldberg entre el ácido 2-clorobenzoico (I) con la 2-aminopiridina (II) para la
síntesis del heterociclo de la 11H-pirido[2,1-b]quinazolin-11-ona (IV). Esta reacción transcurre a través de la
formación del correspondiente ácido 2-(2-piridilamino) benzoico (III) el cual cicla en el medio de reacción.
N
NCO
NH
N
COOH
COOH
Cl
+N
NH2
I II III IV
Recientemente una serie de derivados del heterociclo de la 11H-pirido[2,1-b] quinazolin-11-ona se han
estudiado como agentes antialérgicos 2, protectores celulares 3 y como agentes hipolipémicos 4.
En el presente trabajo con la finalidad de reducir los tiempos de reacción estudiamos el empleo de la
irradiación ultrasónica en la síntesis del heterociclo de la 11H-pirido[2,1-b]quinazolin-11-ona y sus
derivados.
MATERIALES Y METODOS
PROCEDIMIENTO GENERAL:
Síntesis de 11H-pirido[2,1-b]quinazolin-11-ona empleando N,N-dimetilformamida como disolvente.
Una mezcla de ácido 2-clorobenzoico (0.04mol), carbonato de potasio anhidro (0.02mol), 2-aminopiridina
sustituída (0.08mol), cobre en polvo (0.2g) en N,N-dimetilformamida (25mL) fue tratada durante 25 minutos
con irradiación ultrasónica usando una aguja ultrasónica de 20 kHz. La mezcla es lentamente añadida con
agitación en agua (100 mL) y se deja toda la noche en reposo. Precipita el correspondiente derivado del
heterociclo 11H-pirido[2,1-b]quinazolin-11-ona, el cual es purificado mediante disolución en el disolvente
correspondiente y tratado con carbón a ebullición. Los valores de RF y el disolvente empleado en cada
derivado se presentan en la tabla 1 donde también se reporta el rendimiento, punto de fusión (sin corregir) y
el análisis elemental para cada derivado del heterociclo obtenido.

Tabla 1 Derivados del heterociclo 11H-pirido[2,1-b]quinazolin-11-ona y resultados analíticos.
R5
N
NCR1
R2
R4
O
R3
No Sustituyente Rend. (%) P.F. (oC)(no corregido)
P.F .(oC)Reportado
Rf
1 R1=R2=R3=R4=R5=H 75 211-12a 2105,6 0,602 R4=CH3 82 151-52b 152-535,7 0,633 R5=CH3 69 90-2a 92-35 0,614 R1=OCH3 79 158-60b 1575,6 0,715 R2=Cl 65 190-91b 191-925,6 0,676 R3=NO2 67 199c 1985 0,697 R1=NO2 78 257-58c 2585 0,708 R1=R3=NO2 89 287-89c 288-895 0,739 R2=NO2 63 207-09b 2085 0,72
Valores del análisis elementalCalculado (%) Experimental (%)
No Fórmula C H C H1 C12H8N2O 73,47 4,08 73,40 4,232 C13H10N2O 74,28 4,76 74,53 5,113 C13H10N2O 74,28 4,76 74,35 4.684 C13H10N2O2 69,03 4,42 68,90 4,255 C12H7ClN2O 62,47 3,04 62,61 3,196 C12H7N3O3 59,75 2,90 59,55 2,847 C12H7N3O3 59,75 2,90 59,88 2,678 C12H6N4O5 50,35 2,10 50,49 1,919 C12H7N3O3 59,75 2,90 59,63 2,63
aCristalizado de etanol/agua; bCristalizado de etanol ; cCristalizado de dioxano/agua.
RESULTADOS Y DISCUSION
En la primera parte de este trabajo estudiamos el tiempo de reacción en presencia de irradiación ultrasónica,
tomamos como modelo la síntesis del heterociclo 11-H pirido[2,1-b]quinazolin-11-ona, mediante la
condensación del ácido 2-clorobenzoico y la 2-aminopiridina en N,N-dimetilformamida como disolvente. El
tiempo se varió entre 35 y 20 minutos, realizando mediciones cada 5 minutos. Se observó que con 25 minutos
de reacción se obtienen los mejores rendimientos, tiempos superiores permanecen constantes los
rendimientos. Todos los experimentos realizados en este trabajo fueron repetidos 5 veces y los rendimientos
que se reportan representan una media de los rendimientos obtenidos para cada reacción.
En la tabla 2 comparamos los resultados empleando irradiación ultrasónica en 25 minutos de reacción y
empleando reflujo durante 6 horas, en todos los casos los rendimientos obtenidos empleando ultrasonido son
superiores a aquellos que se obtienen usando reflujo de la reacción. Es interesante señalar que en le caso del
compuesto 3 el ácido 2-(2-piridilamino-6-metil)benzoico fue obtenido empleando condiciones de reflujo, este
ácido fue ciclado usando ácido sulfúrico a 1000C a la correspondiente 9-metil-11H-pirido[2,1-b]quinazolin-
11-ona, pero en condiciones donde se emplea la irradiación ultrasónica el derivado de la piridoquinazolona
fue obtenido directamente probablemente debido a las altas temperaturas que se generan por las implosiones
durante el proceso de irradiación ultrasónica.
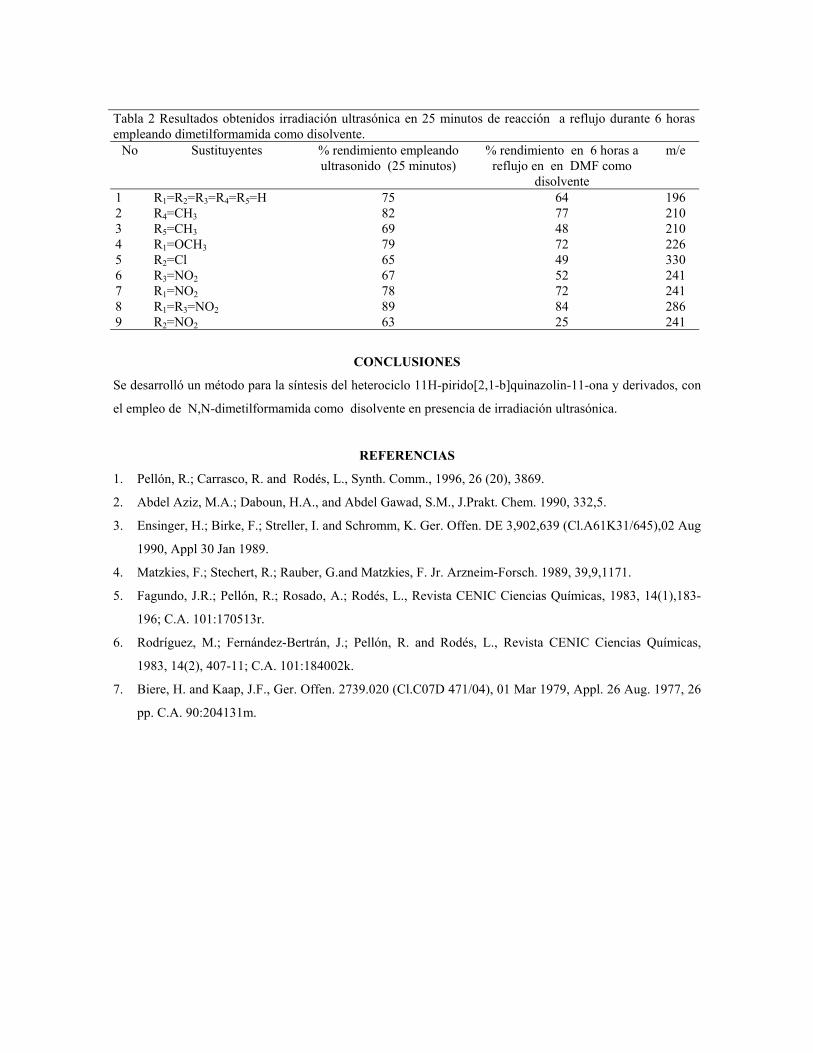
Tabla 2 Resultados obtenidos irradiación ultrasónica en 25 minutos de reacción a reflujo durante 6 horasempleando dimetilformamida como disolvente.
No Sustituyentes % rendimiento empleandoultrasonido (25 minutos)
% rendimiento en 6 horas areflujo en en DMF como
disolvente
m/e
1 R1=R2=R3=R4=R5=H 75 64 1962 R4=CH3 82 77 2103 R5=CH3 69 48 2104 R1=OCH3 79 72 2265 R2=Cl 65 49 3306 R3=NO2 67 52 2417 R1=NO2 78 72 2418 R1=R3=NO2 89 84 2869 R2=NO2 63 25 241
CONCLUSIONES
Se desarrolló un método para la síntesis del heterociclo 11H-pirido[2,1-b]quinazolin-11-ona y derivados, con
el empleo de N,N-dimetilformamida como disolvente en presencia de irradiación ultrasónica.
REFERENCIAS
1. Pellón, R.; Carrasco, R. and Rodés, L., Synth. Comm., 1996, 26 (20), 3869.
2. Abdel Aziz, M.A.; Daboun, H.A., and Abdel Gawad, S.M., J.Prakt. Chem. 1990, 332,5.
3. Ensinger, H.; Birke, F.; Streller, I. and Schromm, K. Ger. Offen. DE 3,902,639 (Cl.A61K31/645),02 Aug
1990, Appl 30 Jan 1989.
4. Matzkies, F.; Stechert, R.; Rauber, G.and Matzkies, F. Jr. Arzneim-Forsch. 1989, 39,9,1171.
5. Fagundo, J.R.; Pellón, R.; Rosado, A.; Rodés, L., Revista CENIC Ciencias Químicas, 1983, 14(1),183-
196; C.A. 101:170513r.
6. Rodríguez, M.; Fernández-Bertrán, J.; Pellón, R. and Rodés, L., Revista CENIC Ciencias Químicas,
1983, 14(2), 407-11; C.A. 101:184002k.
7. Biere, H. and Kaap, J.F., Ger. Offen. 2739.020 (Cl.C07D 471/04), 01 Mar 1979, Appl. 26 Aug. 1977, 26
pp. C.A. 90:204131m.

SINTESIS DE DERIVADOS DE ACIDO SALICILICO EN PRESENCIA DE IRRADIACION
ULTRASONICA EMPLEANDO AGUA COMO DISOLVENTE
Maite L. Docampo Palacios y Rolando F. Pellón Comdom
Centro de Química Farmacéutica Apartado 16042, La Habana, Cuba
Teléfonos (537)217809 Fax (537) 336471 E-mail: [email protected]
RESUMEN
Empleando cobre y piridina como catalizadores se pudo realizar la síntesis de derivados de ácido salicílico empleando
agua como disolvente en presencia de irradiación ultrasónica con buenos rendimientos en tiempos muy cortos de
reacción. Con el empleo de las condiciones de reacción desarrolladas en este trabajo se logró la síntesis de varios
derivados del ácido salicílico.
INTRODUCCION
El desarrollo de una nueva forma de síntesis de derivados del ácido salicílico es interesante, ya que estos
derivados son ampliamente utilizados en la síntesis orgánica y en trabajos de laboratorio 1.
El aumento de la velocidad de las reacciones orgánicas por el empleo de la irradiación ultrasónica ha sido
ampliamente ejemplificada2 y ha ganado popularidad sobre las reacciones usuales, tanto homogéneas como
heterogéneneas3 estas reacciones pueden ser llevadas a cabo de forma rápida y en muchos casos obteniéndose
productos puros con rendimientos casi cuantitativos, con procedimientos limpios, seguros y operacionalmente
sencillos.
Existen pocas referencias en la literatura relacionadas con la hidrólisis de los ácidos 2-halogenobenzóicos para
la obtención de ácidos salicílicos. La hidrólisis del ácido 2-bromobenzoico se reportó por Listsyn 4 en presencia
de acetato cúprico y piperidina acuosa.
Recientemente, nosotros hemos estudiado el empleo del cobre y la piridina como catalizadores para la hidrólisis
de los derivados del ácido 2-clorobenzoico empleando agua como disolvente5. En la presente comunicación, con
la finalidad de reducir el tiempo de reacción, estudiamos el empleo de la irradiación ultrasónica en la síntesis de
derivados del ácido salicílico.
PARTE EXPERIMENTAL
Procedimiento general:
Síntesis de derivados del acido salicílico empleando agua como disolvente.
Una mezcla de ácido 2-clorobenzoico sustituido, (0.04 mol), carbonato de potasio anhidro (0.06 mol), piridina
(0.02 mol), cobre en polvo (0.2g) y 25 mL de agua como disolvente fue tratada durante 15 minutos con
irradiación ultrasónica. La mezcla de reacción fue enfriada, vertida sobre agua y acidificada con ácido
clorhídrico (1:1) a pH 3, el sólido se filtra y se recristaliza de etanol/agua (1:2). El rendimiento y el punto de
fusión (sin corregir) de cada derivado de ácido salicílico obtenido se reporta en la tabla 3
RESULTADOS Y DISCUSION
Los mejores rendimientos obtenidos en nuestra investigación previa para la hidrólisis del ácido 2-clorobenzoico
en presencia de agua como disolvente se lograron con el empleo de 3 equivalentes de carbonato de potasio, 1

equivalente de piridina y 3% (en peso) de cobre por mol de ácido 2-clorobenzoico en 2 horas de reflujo. Estas
condiciones fueron empleadas para a síntesis de varios derivados del ácido salicílico.
Primeramente estudiamos el tiempo de reacción en presencia de irradiación ultrasónica para lo cual tomamos
como modelo la hidrólisis del ácido 2-clorobenzoico en las condiciones previamente reportadas. Como
podemos apreciar en la Tabla 1 en sólo 15 minutos de reacción se obtuvieron los mejores rendimientos. Todas
las experiencias realizadas en este trabajo fueron repetidas 5 veces. El rendimiento reportado representa una
media de los valores obtenidos para cada reacción.
Tabla 1 Efecto del tiempo de reacción sobre los rendimientos de la reacción empleandoirradiación ultrasónica en presencia de agua como disolvente.
R1
R2
R3
Cl
COOH COOHR3
R2
R1
OH
K2CO3/CuH2O/Pird/)))
Tiempo de IrradiaciónUltrasónica (minutos)
% de rendimiento de ácidosalicílico
Desviación estandard (S)
30 95 2.025 96 1.520 95 1.015 94 2.010 57 2.0
La Tabla 2 muestra los resultados para varios experimentos de la hidrólisis del ácido 2-clorobenzoico con
diferentes cantidades de piridina empleando polvo de cobre (3% en peso) y tres equivalentes de carbonato de
potasio por mol de ácido 2-clorobenzoico, el tiempo de reacción en todos los casos fue de 15 minutos. En estas
condiciones de reacción los mejores resultados fueron obtenidos empleando 0.5 equivalentes de piridina.
Tabla 2 Efecto de la piridina sobre los rendimientos de la reacción empleando irradiaciónultrasónica en presencia de agua como disolvente.
Equivalentes de piridina % de rendimiento de ácidosalicílico
Desviación estandard (S)
1.00 94 1.50.75 95 1.00.50 95 1.00.25 61 1.5
Una vez que se estableció la cantidad de piridina necesaria para la reacción se obtuvo una serie de derivados de
ácido salicílico empleando las condiciones de reacción estudiadas: 3 equivalentes de carbonato de potasio, 1
equivalente de piridina y 3% (en peso) de cobre por mol de ácido 2-clorobenzoico y 15 minutos de irradiación
ultrasónica.
En la Tabla 3 se muestran los derivados del ácido salicílico sintetizados de los correspondientes ácidos 2-
clorobenzoicos, los puntos de fusión (sin corregir) y los rendimientos obtenidos se compararon con aquellos
logrados empleando agua como disolvente a reflujo durante 2 horas de reacción.

Tabla 3 Derivados del ácido salicílico sintetizados a partir de los correspondientes derivados del 2-
clorobenzoico.
COOH
OH
R1
R2
R3
No R1 R2 R3 % de rendimientoempleando ultrasonido
% de rendimiento en 2horas de reflujo
P.F.Sin corregir
1 H H H 95 90 1596
2 H Cl H 95 88 2127
3 H NO2 H 96 90 2258
4 H H Cl 92 84 1719
5 H H NO2 97 95 23410
6 NO2 H NO2 96 93 17011
7 H H CH3 92 87 14112
8 Cl H H 94 85 18713
CONCLUSIONES
Se elaboró un método para la síntesis de derivados de ácido salicílico empleando agua como disolvente en presencia
de irradiación ultrasónica con buenos rendimientos en tiempos muy cortos de reacción.
REFERENCIAS
1. Merk Index Eighth Edition 930
2. Mason, T.J., Chem. Ind., 1993, 47.
3. Einhorn, C.; Einhorn, J. and Luche, J. L., Synthesis, 1989, 787.
4. Logosvskaya, E. K.; Lisetsyn, V. N., Tr. Mosk. Khim.Tekhnol. Inst., 1972, 70, 87.
5. Synthesis of salicylic acid from the corresponding o-chlorobenzoic acids derivatives using water as solvent.
Pellón, R. F.; Docampo M. L., Synth. Comm., (ENVIADA).
6. Korner, B., Ber., 1982, 25, 2189.
7. Ullmann, F. Kopetschni, R.,Ber., 1911, 44, 1911, 428.
8. Hirwe, N.H., Proc Indian Acad. Sci., 1938, 8A, 208.
9. Ullmann, F. and Wagner C., Ann., 1907, 355, 359.
10. Hirsch, M., Ber., 1900,33, 3239.
11. Ullmann, F. and Liebigs, A.,Der. Chemie, 1909,366, 85.
12. Ullmann, F. and Kipper, H., Ber., 1905, 32, 2120.
13. Varnholt, A., J. Practice, ,1945, 36 (2), 22.

LAS REDES DE NEURONAS COMO BATERÍA DE TAMIZAJE BIOLÓGICO VIRTUAL. UN EJEMPLO EN
ANTIBACTERIALES.
Ramón Carrasco1 y Julio C. Escalona2
1Centro de Química Farmacéutica Ave. 21 y calle 200 Atabey, La Habana Apdo. 16040
Teléfono: 271 7925; FAX: 33 6471; Email: [email protected]
2Facultad de Farmacia, Universidad de Oriente, Patricio Lumumba s/n Santiago de Cuba
RESUMEN
En el mundo informático de hoy es necesario enfocar la solución de los problemas con esa óptica. Para la búsqueda de
nuevos agentes antibacterianos, es práctica común el disponer de baterías de ensayo microbiológico para la evaluación de
los compuestos frente a la mayor variedad de bacterias. Es lo que se conoce como batería de screening. Por otra parte, las
redes de neuronas (RN) son una herramienta matemática de introducción relativamente reciente en la modelación de
actividad biológica de sustancias activas. Su utilización en este campo se centró en gran medida en la clasificación de
compuestos en clases de actividad (activo-inactivo). A diferencia del Análisis Multivariado tradicional, ellas permiten
obtener simultáneamente más de una respuesta en cada modelo. Esta capacidad se ha aprovechado para proponer un
enfoque de aplicación que permita, a partir de un mismo juego de descriptores estructurales, crear un modelo matemático
que sirva para evaluar teóricamente el potencial antibacteriano de compuestos no incluidos en la muestra, en más de una
actividad biológica. Esta forma de aplicación brinda un modelo de screening virtual de la actividad antibacteriana. Se
presenta un modelo de RN en que las dos neuronas de salida son la actividad frente a S. aureus y E. cloacae de una familia
de cefalosporinas. La correlación entre valores predichos y observados es mayor de 0.98 en ambos casos y el % de error
menor de 3. Se determinó la potencia predictiva Q2 del modelo y resultó mayor de 0.96.
INTRODUCCIÓN
Los modelos de redes neuronales artificiales (RNA) se basan en la interacción colectiva y en paralelo de una gran
cantidad de procesadores simples que simulan la conducta de las neuronas biológicas. Una RNA la podemos considerar una
caja negra que acepta un conjunto de datos, los procesa y brinda una o más salidas. Las RNA’s han sido aplicadas en
química tanto para estudios de relación estructura-propiedad como estructura-actividad biológica, de forma cualitativa y
cuantitativa. En los estudios QSAR y QSPR es de uso común los perceptrones multicapa para ambos tipos de
procesamiento. La selección del tipo de red y la definición de su topología se establece dependiendo del trabajo a realizar.
Una RNA consiste de un número de unidades de procesamiento conectadas entre sí usualmente, pero no siempre en
diferentes capas. En este trabajo se empleó el tipo de red conocida como perceptrón multicapa (PMC) por ser la más
ampliamente aplicada en estudios QSAR y QSPR, en el que la arquitectura es la de una capa de entrada, una oculta y una de
salida.
Las redes de neuronas se han utilizado en estudios de relación estructura química-actividad biológica para la clasificación
en clases de actividad (activos-inactivos). También se han utilizado con más de una neurona en la capa de salida para fijar
diferentes clases de actividad, en estudios vida media-selectividad1 y actividad-selectividad.2 Sin embargo, no han aparecido
reportes del empleo de las redes de neuronas para predecir más de una actividad biológica simultáneamente, aunque si para
propiedades químico-físicas3.
Por otra parte, en el caso particular de los antibióticos del tipo de los β-lactámicos, se plantea que los estudios
cuantitativos de relación estructura-actividad son un sueño imposible4. El objetivo del presente trabajo es demostrar la

posibilidad de realizar estudios QSAR en antibióticos β-lactámicos y a la vez, predecir cuantitativamente la actividad de
estos frente a más de una bacteria de forma simultánea, simulando una pequeña batería de ensayos in vitro.
PROCEDIMIENTO
Se trabajó con una muestra de 65 cefalosporinas con actividad reportada frente a S. aureus y E. cloacae. Para la
descripción estructural de los compuestos se dividieron las moléculas en tres regiones (sustituyente en R7, núcleo central β-
lactámico y sustituyente en R3, fig. 1). A los fragmentos correspondientes a los sustituyentes en 7 y 3 se les calcularon los
índices topológicos y topográficos de Estrada (ε, ερ, Ω, Ωq, ΩqC) y los topológicos de Randi (χ, χV). con el programa
MODEST1. Los descriptores de los sustituyentes en la posición 3 se calcularon a partir de dicho fragmento unido a un grupo
vinilo. Los correspondientes al sustituyente R7 se calcularon en forma de la amida N-metil sustituida. La actividad biológica
se trató como log[1/CMI] en la que CMI es la concentración mínima inhibitoria en términos de concentración µM.
El método de cálculo utilizado para los descriptores
químico-cuánticos fue el PM35,6 incluido dentro del
programa MOPAC 6.07,8. Como entrada de datos a este
programa se utilizó la matriz Z de salida de cada
estructura optimizada por mecánica molecular con el
método MM+ incluido en el programa HyperChem9. Para
la selección de las variables se empleó el método de
análisis cluster empleando RN reportado por Cruz10. Se
seleccionaron las variables 1Ωq, 2Ω, Ω3 y 1Ωq
3C para R7;
la distancia C-N, y 2Ω, 4Ω, Ωq3 y 1χV
3 para R3. El
algoritmo empleado para el establecimiento del modelo
fue el de backpropagation. El número de neuronas en la capa oculta de la red de tres capas fue seleccionada por
minimización del error relativo en el rango de 3 a 13 neuronas en dicha capa, por lo que la arquitectura final es 9:13:2. Para
el trabajo con los perceptrones multicapas (PMC) se empleó el programa profesional NeuroShell 211
RESULTADOS Y DISCUSIÓN
Se entrenaron PMC de arquitectura 9-x-2 en las que x tomó valores entre 3 y 13 para determinar el número de neuronas en la
capa oculta. Para el entrenamiento se seleccionaron al azar 5 compuestos para la muestra de validación. Se utilizó como criterio
de parada del entrenamiento, el menor valor del error relativo en forma de porciento. Este se calcula con la ecuación
Error Relativo (%)= (| residual | / | valor experimental |) • 100.
Se seleccionó la red con 13 neuronas en la capa
oculta por ser la que mejor resultado brindó en ambas
actividades (fig. 2). Con arquitectura 9-13-2 se utilizó la
técnica del leave one out para formar un conjunto de 65
redes cuyos valores de predicción se promediaron para
dar el valor definitivo de la misma. Se obtuvieron
coeficientes de correlación entre predichos y
observados de 0.983 para el S. aureus y de 0.979 para el
E. cloacae utilizando los valores promedio de la
N
S
COOH
R3O
R7CONH
Figura 1. Fórmula general de las cefalosporinas. Se señalan
los fragmentos en que se dividieron las moléculas para la
realización de este estudio.
0.000
0.020
0.040
0.060
0.080
0.100
5 6 7 8 9 10 11 12 13
E.(s.aureus)
V.(S.aureus)
E.(E.cloacae)
V.(E.cloacae)
Fig. 2 Resultados en la búsqueda de la mejor arquitectura de red

predicción. Se encontraron bajos % de error en la predicción de cada bacteria los cuales son 2,6 y 2,4% para el S. aureus y E
cloacae respectivamente. Estos errores se consideran muy pequeños para este tipo de estudios. Al modelo se le calculó la
potencia predictiva12 Q2 por la ecuación: Q2=1-Σ(real-predicho)2 / Σ(real-promedio)2
Para cada una de las neuronas de salida se encontraron valores de Q2 de 0.966 para el S. aureus y de 0.958 para el E. cloacae.
Estos altos valores de Q2 le confieren una elevada capacidad predictiva al modelo.
Las redes de neuronas han sido catalogadas como cajas
negras de difícil interpretación. Eso ha llevado al
desarrollo de métodos alternativos y no convencionales
para poder interpretar los resultados, que del entrenamiento
y establecimiento de un modelo se deriven. Es así como se
calculan y grafican las contribuciones relativas de las
diferentes variables al modelo. De la figura 3 se observa
que la variable 5 (Distancia C-N en el anillo β-lactámico)
tiene relativamente poca influencia en el modelo. El
entrenamiento de una red con una muestra de validación de
5 compuestos indica que para el S. aureus no hay
modificación en el valor del % de error promedio de la
predicción. Sin embargo, para el E. cloacae aumenta dicho error. Este resultado preliminar sugiere la existencia de diferencias
pequeñas en el nivel de participación de cada variable en cada una de las respuestas biológicas.
CONCLUSIONES
Se presenta un modelo de screening virtual de cefalosporinas empleando redes de neuronas para la predicción cuantitativa
simultánea de actividad antibacterial de estos compuestos. Los magníficos resultados en cuanto a la potencia predictiva del
modelo confirman la múltiple aplicabilidad de las RNA’s en modelos de relación estructura-actividad.
BIBLIOGRAFÍA
1 Quiñones, C.; Cáceres, J.; Stud, M. y Martínez, A.; Quant Struct.-Act. Relat, 19, 448-454, 20002 Martinez, A.; Castro, A.; Stud, M.; Rodriguez, J.; Cardelus, I.; Llenas, J.; Fernandez, A. y Palacios, J. M.; Med.Chem. Res., 3, 171-180, (1998)3 Gakh, A.A.; Gakh, E.G.; Sumpter, B.G.; Noid, D.W. Neural Network-Graph Theory Approach to the Prediction of thePhysical Proprties of Organic Compounds. J. Chem. Inf. Comput. Sci., 34, 4, 832-839(1994)4 Frere, J. M.; Joris, B.; Varetto, L.; Crine, M. Structure-activity Relationships in the β-lactam family: An ImpossibleDream. Molecular Pharmacology, 125-132(1989)1 Rodríguez L., Estrada E., Muñoz I. y Gutiérrez Y., MODEST (Molecular DESign Tools) 2.0. Universidad Central de LasVillas: Santa Clara, (1994)5 J.J.P. Stewart, J.Comp.Chem.,10, 209(1989)6 J.J.P. Stewart, J.Am.Chem.Soc., 10 221(1989)7 J. J. P. Stewart, Manual del MOPAC 6.0. QCPE # 5818 J. J. P. Stewart, J. Comp. Aid. Mol. Design, 4, 1, 1(1990)9 HyperChem. Versión 3 para WINDOWS. Molecular Modeling System. Hypercube, Inc y Autodesk, Inc. 199310 Cruz R., López N., Quintero M, Rojas G. Cluster Analysis from molecular similarity matrices using a Non-linear NeuralNetwork., J. Math. Chem. 20 (1997) 385-39411 NeuroShell 2, Release 1.1, WSG copyright@1993 by Ward Systems Group, Inc.12 Wailzer, B.;Klocker, J.; Buchbauer, G.; Ecker, G.; Wolschann, P.; Prediction of the aroma quality and the thresholdvalues of some pyrazines using artificial neural networks. J. Med. Chem. 44, 17, 2805—13(2001)
Figura 3. Contribución relativa de cada variable al modelo

ESTUDIO DE LA REACCIÓN DE ALQUILACIÓN DE DERIVADOS DE ACRIDONA MEDIANTE EL
EMPLEO LA CATÁLISIS POR TRANSFERENCIA DE FASES LÍQUIDO-LÍQUIDO
Autores: Rolando F. Pellón Comdom y Lisbet Xuárez Marill
Centro de Química Farmacéutica, Departamento de Química Orgánica.
Calle 200 y 21, Atabey, Playa, Aptdo. 16042, La Habana 11600. Cuba.
RESUMEN
Recientemente ha existido un desarrollo en las investigaciones biológicas de las acridonas, familia heterocíclica que
tiene actividad antitumoral y antiviral. En la estructura base de las acridonas existe un centro duro que es el oxígeno
(átomo más electronegativo) y un centro blando que es el nitrógeno. Por la importancia de propiedades de las
acridonas en este trabajo, se realizó la obtención y el estudio de derivados N/O alquilados de esta familia, con el
empleo de la catálisis por transferencia de fases líquido-líquido como técnica no convencional. En este trabajo se
demuestra que cuando la alquilación se realizó con sulfato de dimetilo se obtuvo el derivado O-alquilado y cuando
se realizó con bromuro de etilo se obtuvo el derivado N-alquilado.
INTRODUCCIÓN
Las acridonas son compuestos heterocíclicos que pueden ser aislados como principio activo de fuentes naturales o
pueden ser obtenidas por síntesis química. En los últimos años varios investigadores han llevado a cabo estudios
relacionados con sus propiedades biológicas y han encontrado derivados con actividad antitumoral (Abadi (1), 1999;
Cholody (2), 1999; Kawaii (3), 1999; Tabarrini (4), 1999) y antiviral (Takemura (5), 1995; Turpin (6), 1998; Vance(7),
1999). Con la finalidad de obtener nuevos derivados de esta familia, en este trabajo se propuso realizar el estudio de
alquilación de la 9(10-H) acridona, la 2-metil-6-cloro-9(10-H) acridona, la 2-carboxi-7-cloro-9(10-H) acridona y la
4-carboxi-1,6 dicloro-9(10-H) acridona con sulfato de dimetilo (electrófilo duro) y bromuro de etilo (electrófilo
blando) con el empleo de la técnica de catálisis por transferencia de fases líquido-líquido para comprobar si en la
reacción de alquilación de acridonas se cumple el Principio de Simbiosis
MATERIALES Y MÉTODOS
Obtención de derivados de la-9-metoxi acridina (1)
En un balón de 100 mL de capacidad provisto de un agitador mecánico se adicionan 1mmol la acridona
correspondiente, 50 mL de una disolución de hidróxido de potasio al 50%, se agita fuertemente durante 20 min. y se
añaden 0,3mmol de bromuro de trietilbencilamonio, 15mmoles de sulfato de dimetilo y 75 mL de butanona. La
mezcla de reacción se calienta a reflujo por tres horas. Al concluir este tiempo se separan las fases. De la fase
orgánica se obtiene por eliminación del disolvente la acridona alquilada, la cual se recristaliza de etanol. 2-metil-6-
cloro-9-metoxi acridina (1) Sólido amarillo claro. Se obtuvo un 89,2% de rendimiento con una Tf (0C): 235-6.
RMN-1H (ppm): 8,30 (d, H-C8); 8,10 (d, H-C1); 7,86 (d, H-C5); 7,65 (dd, H-C3); 7,30 (dd, H-C7); 7,74 (d, H-C4),
3,90 (s, H-OCH3), 2,40 (s, H-CH3). RMN-13C (ppm): 165,50 (COCH3). 142,73 (C4a); 140,29 (C5a); 138,58 (C6);
135,22 (C3); 130,67 (C2); 128,36 (C4); 128,36 (C1); 121,96 (C7); 121,48 (C1a); 120,84 (C5); 119,89 (C8a); 115,96
(C7); 115,21 (C8); 62,05 (OCH3); 19,86 (CH3). De igual forma se obtuvo la 2-carboxi-7-cloro-9-metoxi acridina

(2) con un 85,8% de rendimiento. Tf (0C): 245-6 y la 4-carboxi-1,6-dicloro-9-metoxiacridina (3) con un 80,6% de
rendimiento. Tf (0C): 300
Obtención de la 9-(10-etil) acridona (4) y de la 2-metil-6-cloro-9-(10-etil) acridona (5)
En un balón de 100 mL de capacidad provisto de un agitador mecánico se adicionan (0,01 mol) del derivado
correspondiente de la 9(10-H) acridona, 50 mL de una disolución de hidróxido de potasio al 50%, se agita
fuertemente durante 20 min. y se añaden (0,3.10-3) de bromuro de trietilbencilamonio, (0,015 moles) de bromuro de
etilo y 75 mL de butanona. La mezcla de reacción se calienta a reflujo por tres horas. Al concluir este tiempo se
separan las fases. De la fase orgánica se obtiene por eliminación del disolvente la acridona alquilada la se recristaliza
de etanol Se obtuvo la 9-(10-etil) acridona con un 89,3% de rendimiento. Tf (0C): 235-237. RMN-1H (ppm): 8,35
(d, H-C3); 7,88 (d, H-C4 y H-C5); 7,84 (m, H-C3 y H-C6); 7,34 (m, H-C2 y H-C7); 4,60 (q, H-CH2) y 1,45 (t, H-CH3).
RMN-13C (ppm): 176,40 (C=O); 141,15 (C4a y C5a); 134,22 (C3 y C6); 126,73 (C1 y C8); 121,55 (C1a y C8a); 121,16
(C2 y C7); 115,66 (C4 y C5); 40,12 (NCH2) y 12,30 (NCH3). De igual forma se obtuvo la 2-metil-6-cloro-9-(10-etil)
acridona con un 90,7% de rendimiento. Tf (0C): 187-89 y la 4-carboxi-1,6-dicloro-9-(10-etil) acridona (7) como
sólido amarillo claro. Rendimiento: 65,2 % Tf (0C): 162-164.
DISCUSIÓN DE RESULTADOS
En la estructura base de las acridonas existe un centro duro que es el oxígeno (átomo más electronegativo) y un
centro blando que es el nitrógeno. Teniendo en cuenta que existen varios factores que influyen en la reactividad dual
de un compuesto ambidentado como son: la polarizabilidad (carácter duro-blando) del nucleófilo, los efectos de
solvatación y el grupo saliente entre otros, y conociendo el principio de Pearson de clasificación de los ácidos y
bases en duros y blandos se puede clasificar a los nucleófilos y a los electrófilos de acuerdo a esta misma fortaleza.
Según el Principio de Simbiosis que plantea que las reacciones ocurren más rápidamente entre sistemas duro-duro o
blando-blando, esta reacción de alquilación debe ocurrir por el centro del nucleófilo de igual dureza que el
electrófilo.
O
N
C
N
OCH3
(CH3)2SO4
50( % )KOHCTF
1
2
3
45
6
7
8
9
105
8 1
4
a
a aa
R R R RH
1
2
3
45
6
7
8
9
10
(1): R2= CH3, R6= Cl y R7= H; 2): R2 = CO2H, R6= H y R7= Cl; (3): R1 = Cl , R6 = Cl, R4 =CO2H
Esquema 1: Secuencia de síntesis para la obtención de las 9-metoxi acridinas.
En la reacción de alquilación de acridonas el disolvente juega un papel importante, por su influencia sobre la
reactividad del sustrato. En la literatura existen algunos estudios de alquilación de acridonas bajo condiciones de
transferencia de fases(9-12).
En las reacciones de alquilación con sulfato de metilo bajo Catálisis por Transferencia de Fases, se obtuvieron los
productos de la O-alquilación como único derivado, ya que de existir mezcla de isómeros aparecería una señal en el
espectro de RMN1H adicional a δ=2,5ppm correspondiente a los protones del grupo metilo unido al átomo de
nitrógeno, además la señal a 2,40 ppm es característicos de protones metilos unidos a un átomo de oxígeno. Esta

reacción es regioespecífica ya que aunque es posible obtener dos isómeros estructurales (N-alquilado y O-alquilado)
sólo se obtiene el O-alquilado.
Cuando la alquilación se realiza empleando como agente alquilante bromuro de etilo en condiciones de catálisis por
transferencia de fases esta reacción es regioespecífica ya que aunque es posible obtener dos isómeros estructurales
(N-alquilado y O-alquilado) sólo se obtiene el N-alquilado. Esto se comprueba en el espectro RMN1H, la señal de
los protones metilenos aparece en 4,60 ppm y la de los protones metilos a 1,40 ppm lo cual es típico de los
productos N-CH2-CH3, en el espectro RMN 13C, la señales de los carbonos CH2 y CH3 aparecen a 40,12 y 12,30
ppm respectivamente característicos de carbonos unidos a un átomo de nitrógeno. Estos resultados permiten
corroborar la formación del producto N-alquilado.
Al comparar el valor de la temperatura de fusión de estas acridonas con las no alquiladas se observa una disminución
del valor de la temperatura de fusión que se debe a la ruptura de los puentes de hidrógeno intermolecular como
consecuencia de la formación del producto N-alquilado.
O
N
C
N50( % )KOH
CTF
1
2
3
45
6
7
8
9
105
8 1
4
a
a aa
R R R RH
1
2
3
45
6
7
8
9
10
C2H5Br
O
C2H5
(4): R = H; (5): R2 =CH3, R6= Cl, R7= H; (6): R2 = CO2H, R6= H y R7= Cl; (7): R1 = Cl , R6 = Cl, R4 =CO2H
Esquema 2: Secuencia de síntesis para la obtención de las 9-(10-etil) acridonas.
CONCLUSIONES
Se comprobó que en la reacción de alquilación estudiada para las acridonas, con el empleo de la catálisis por
transferencia de fases se cumple el principio de simbiosis de reactividad. Se establece las condiciones de reacción
para la obtención regioespecífica de acridonas N/O alquiladas.
BIBLIOGRAFÍA
1. Abadi, A. et. al, Arzn. Forsch. Drug Res. 49, 259-66 (1999).
2. Cholody, W. et al.. J. Med. Chem. 38, 3043-52 (1995).
3. Kawaii, S. et. al. Leukemia Research, 23, 263-69 (1999).
4. Tabarrini, O. et. al.. J. of Medicinal Chem. 42, 2136-44 (1999).
5. Takemura, Y. et. al.. Planta Med. 61, 366-8 (1995).
6. Turpin, J. A. et al.. Antimicrobial Agent and Chemotherapy 42 (3), 487-94 (1998).
7. Vance, J. et. al. Bochemical Pharm. 58, 703-8 (1999).
8. Xuárez, L. Tesis de Maestría UH (1996).
9. 11. Galy, J.P., Elguero, J. And E.J. Vicent.. Síntesis, 944, (1979).
10. 12. Berny, H., Bsiri, N., Clarbit, J., Galy, A. M, et al. Arzneim Forsch-Drug Res. 42 (I), Nr.5, 674-9 (1992).
11. 13. Ngady, L., Galy, A. M, Barbe, J. . Arzneim Forsch-Drug Res. 43 (I), Nr.4, 480-83 (1993).
12. 14. Papadopulus, K., J. Fur Praktiche Chem. Zet. 335, 633-36 (1993)

Synthesis of pharmaceuticals and intermediates by Supported Aqueous Phase Catalysis (SAPC).
Ulises J. Jáuregui-Haza1*, Anne Marie Wilhelm2 and Henri Delmas2
1 Centro de Química Farmacéutica, Apdo. 16042, C. Habana, Cuba. Tel: 271-5106, Fax: 336471, e-mail:
[email protected] Ecole Nationale Supérieure d'Ingénieurs en Arts Chimiques et Technologuiques, 118 route de Narbonne, 31077
Toulouse, France
AbstractSupported Aqueous Phase Catalysis (SAPC) becomes attractive for manufacture of pharmaceuticals and intermediates.
An overview about the synthesis of some pharmaceuticals and intermediates by SAPC is presented. This method has
been successfully used in the asymmetric reduction of 2-(6'-methoxy-2'-naphtyl)acrylic acid to the commercially anti-
inflammatory agent naproxen and in the hydrogenation of retinal. The hydroformylation of α, β-unsaturated esters by
SAPC has been studied also. Several 2-formylpropanoate esters, which are extensively used as intermediates in the
synthesis of pharmaceuticals like rifamicine and vitamin E were obtained using the hydrosoluble complexes. The
perspectives of the SAPC in the hydroformylation reaction for obtaining molecules having a broadspectrum therapeutic
activity, such pheniramines, milverine, lidoflazine and penfluridol are shown.
Introduction.
One of the challenges of the pharmaceutical industry is to find new ways of synthesis for active principles and
intermediates. Among these methods the Supported Aqueous Phase Catalysis (SAPC) seems to be advantageous from
an economical and ecological point of view1. SAPC is a special case of the supported liquid phase catalysis whose
development began according to proposals of Moravec2. Two recent reviews on SAPC show that this process has been
mainly used in the synthesis of bulk chemicals3, 4. The first report of the use of SAPC for obtaining a pharmaceutical
appeared in 19935. In this work, an overview about the synthesis of some pharmaceuticals and intermediates by SAPC
is shown.
Technique of SAPC
In SAPC1, the catalytic phase, immiscible with the organic phase containing the reactant and products, consists of an
aqueous solution of a water-soluble organometallic catalyst. To provide for the necessary interfacial area required for
the reaction to proceed at a reasonable rate, the catalytic phase is immobilized in the pores of a high surface area
hydrophilic support, like controlled porous glass, silica, alumina and phosphate1, 3-6. Reactions of liquid phase, water-
insoluble organic reactants take place at the film-organic interface.
The concept of SAPC is fairly general. It has been proved that supported aqueous phase catalysts can conduct a broad
spectrum of reactions (hydroformylation, hydrogenation, oxidation), and that the organometallic complexes remain
immobilized when the catalysts are appropriately synthesized3-6. Despite promising results, there is not any industrial
process on SAPC. The activity of SAPC is strongly dependent on the water content of the solid particle. The optimum
range is between 3 and 7 wt-%. Decreased activity is observed at higher or lower water-loadings and not due to metal
loss through leaching. Crucial measurement and control of the water content are impossible during continuous
operation modes. Additionally, the water-soluble ligands used, like TPPTS, are to some extent degraded under reaction
conditions. This loss can not be compensated for by simply adding ligand6. Our recent report6,7 about the possibility of
SAPC to take place in the external surface of the support solved above mentioned problems. Therefore, this
modification opens the way to apply SAPC at a commercial scale.

Synthesis of pharmaceuticals and intermediates by SAPC.
Wan and Davis8 used a supported-phase, asymmetric, hydrogenation catalyst (SAP-Ru-BINAP-4SO3Na) for the
reduction of 2-(6'-methoxy-2'-naphtyl)acrylic acid to the commercially anti-inflammatory agent naproxen, using as
support a controlled-pore glass CPG-240. An enantiomeric excess of 96 % was obtained. The supported aqueous phase
catalyst was found to be 50 times more active than its two-phase counterpart and only seven times less active than its
homogeneous analogue. Both the catalytic activity and the enantioselectivity were dependent on the water content of
the support, with the hydrated SAP catalyst being at least 2000 times more active than the dried catalyst. Recycling of
the SAP catalyst was easily achieved without any leaching of ruthenium into the organic phase.
MeO
COOH COOH
MeO+ H2 Me*
Figure 1. Asymmetric synthesis of naproxen
Another example of hydrogenation by SAPC is the synthesis of retinol from retinal using the complex RuH2(TPPTS)4
on silica5. The reaction was carried out in hexane (conversion of 70 %) and methanol (99 %). Although the high
conversions were obtained, the supported aqueous phase catalyst was not stable to be recycled. However, for the high
valuable products the process in one cycle with the regeneration of catalytic system can be considered.
The hydroformylation of α, β-unsaturated esters by SAPC has been studied also9. Several 2-formylpropanoate esters,
which are extensively used as intermediates in the synthesis of pharmaceuticals like rifamicine and vitamin E, were
obtained using the hydrosoluble Rh-TPPTS on silica gel with an exceptionally high initial turnover frequency of 4300
h-1. As it was reported for other SAP systems, the results obtained in this case with silica gel supported catalyst were
strikingly dependent on the water content. The maximum initial activity was obtained for a water content of 37 % by
weight; even slight deviation from this value led to decreases activities.
The synthesis of many pharmaceutical agents and complex molecules from natural sources is strongly dependent on the
availability of intermediate compounds liable to further structural elaboration. In this context, the hydroformylation by
SAPC, especially when catalyzed by rhodium carbonyl complexes, which ensure higher chemo- and regioselectivity
with respect to other metal derivatives under comparable reaction conditions, offers a concrete possibility to obtain a
wide variety of molecules endowed with therapeutic activity. As an example the hydroformylations of styrene and
related vinylaromatics to yield 2-arylpropanoic acids (anti-inflammatory and analgesic agents)10 can be accomplished
by SAPC (Fig. 2).
CO H2 + +Rh o Pt
cat.
CHOCHO
+
Figure 2. Styrene hydroformylation reaction.

On the other hand, hydroformilation is the key step in the synthesis of pheniramine and several structurally related
compounds. For example, N, N-dimethylcinnamylamines would produce by SAPC 2-aryl-4-(dimethyamino)butanals by
Rh complexe-catalyzed reaction; these amino-aldehydes can be converted further in two steps into pheniramines, a well
known generation family of H1 antihistaminic agents. Figure 3 shows the obtention of pheniramines' intermediates
starting from cinnamaldehyde acetals.
CH CH CHO
X
CH CH CH(OR)2
X
CH
X
CH2 CH(OR)2
CHO
CO, H2
Rh cat.
ROH
cat. ácido
Figure 3. Synthesis of pheniramines' intermediates starting from cinnamaldehyde acetals.
These results show the perspectives of the SAPC for obtaining molecules having a broadspectrum therapeutic activity,
such milverine fluspirilene, pimozide and penfluridol (neuroleptics); lidoflazine (vasodilator) and azinothricins
(antitumor antibiotics).
References.
1. Arhancet, J.P.; Davis, M.E.; Merola, S.S.; Hanson, B.E. Nature, 399, 454-455, 1989.
2. Moravec, R.Z.; Schelling, W.T.; Oldershaw, C..F. Brit. Patent 511,556, 1939.
3. Anson, M.S.; Leese, M.P.; Tonks, L.; Willians, J.M.J. J. Chem. Soc., Dalton Trans., 21, 3529-3538, 1998.
4. Davis, M. E. Aqueous-phase organometallic catalysis. Wiley-VCH Verlag GmbH: Weinheim, Germany, 241-251,
1998.
5. Fache, E.; Mercier, C.; Pagnier, N.; Despeyroux, B.; Panster, P. J. Mol. Catal., 79, 117-131, 1993.
6. Jáuregui-Haza, U. J. Hidroformilación del octeno por catálisis en fase acuosa soportada. Cinética, parámetros de
ingeniería y propiedades físico-químicas del sistema. Tesis de Doctorado. C. Habana-Toulouse, 2002.
7. Jáuregui-Haza, U. J.; Dessoudeix, M.; Kalck, Ph.; Wilhelm, A. M.; Delmas, H. Catalysis Today, 66, 297-302, 2001.
8. Wan, K.T.; Davis, M.E. J. Catal., 148, 1-8, 1994.
9. Fremy, G.; Monflier, E.; Carpentier, J.F.; Castanet, Y.; Mortreux, A. Angew. Chem. Int. Ed. Engl., 34(12-13), 1474-
1476, 1995.
10. Botteghi, C.; Marchetti, M.; Paganelli, S. Transition Met. Org. Synth., 1, 25-46, Wiley-VCH Verlag GmbH:
Weinheim, Germany, 1998.

Síntesis de oxazolonas bajo irradiación de microondas. (Poster)
Autores: Taimirys Mamposo Pérez, Miriam Mesa Hernandez, Rolando Pellón Comdom, Miguel López López.Centro de Química Farmacéutica. Calle 21 esq. 200, Atabey, Playa, Apdo. 16042, La Habana 11600, Ciudad de laHabana.Teléfono: 271-7809, Fax: 33-6471 Email: [email protected]
Resumen:
La búsqueda de compuestos o sustancias químicas con propiedades farmacólogicas ha sido siempre de gran interés. Esto ha
llevado a los químicos orgánicos al desarrollo de nuevas condiciones de reacción sin disolventes y bajo irradiación de
microondas en busca de mayores velocidades de reacción, rendimientos, selectividad y bajos costos, con fáciles
procedimientos de aislamiento y purificación, y una menor contaminación del entorno. Este trabajo se refiere a la síntesis
bajo irradiación de microondas y en ausencia de disolventes, de una serie de oxazolonas derivadas de N-acilglicina,
importantes intermediarios para moléculas con actividad farmacológica reconocida, con el objetivo de estudiar el sistema en
las condiciones seleccionadas.
Introducción.
En los últimos años ha crecido el interés sobre el empleo de la irradiación con microondas en la síntesis orgánica1-3. Se
afirma que en estas condiciones, las reacciones son efectuadas en cortos tiempos de reacción (en pocos minutos) con un
incremento de la pureza y la mejora del rendimiento de los productos orgánicos resultantes, además de que las mismas
pueden realizarse en ausencia de disolventes4.
Las oxazolonas comunmente conocidas como azlactonas, son importantes síntones de moléculas biológicamente activas. En
la literatura se describe la preparación de estos compuestos por condensación de derivados de N-acilglicina con aldehídos
aromáticos en presencia de diferentes catalizadores5-8. En este trabajo se reporta un nuevo método de síntesis de oxazolonas
derivadas de acetilglicina y benzoilglicina utilizando irradiación de microondas y soportes sólidos minerales. Los
rendimientos y pureza de los productos obtenidos son superiores a los reportados por métodos convencionales.
Materiales y Métodos.
Las materias primas empleadas en la síntesis fueron de calidad reactivos. Los puntos de fusión fueron determinados en un
equipo Gallenkamp con calentamiento variable y registro de temperatura, los mismos no fueron corregidos.
En las corridas cromátograficas se utilizaron placas de gel de sílice 60 con indicador de fluorescencia (Merck). Para el
desarrollo de los cromatogramas se utilizó como fase móvil Acetato de Etilo: N-Hexano: Acido acético Glacial (8:6:1
v/v/v). Como revelador se empleo una lámpara UV-Visible a 254 o 366 nm. Las reacciones por el método no clásico se
realizaron en un horno doméstico de microondas a 2450 MHz, en reactores abiertos de 25 mL (PYREX) ocupando un 10 %
del volumen total del recipiente. El disolvente utilizado para el aislamiento y purificación de los productos fue acetona.

RESULTADOS Y DISCUSIÓN
En el presente trabajo reportamos la síntesis de una serie de oxazolonas derivadas de acetilglicina y benzoilglicina, bajo
irradiacion de microondas y con el empleo de soportes sólidos inorgánicos (Fig. # 1)
Fig. # 1: Esquema de síntesis.
La primera parte de ste trabajo estuvó encaminada a realizar estudios preliminares con el objetivo de seleccionar el soporte
adecuado, optimizar la cantidad relativa de sustrato/soporte y el tiempo de reacción. Para la obtención de las oxazolonas se
escogió como reacción modelo la condensación de benzoilglicina y benzaldehido. Los resultados obtenidos con los
soportes ensayados, se muestran en la tabla I.
Tabla I: Síntesis de 6a bajo irradiación de microondas.
Modo de activación
Microondas Calentamiento clásico*
Soporte (1g) Tiempo de reacción(min.)
Rendimiento 3a (%) Tiempo de reacción(min.)
Rendimiento 3a (%)
Al2O3 básica 1,5 64 60 ≤ 5
120 8
Al2O3 – NH4OAc 1,5 67 60 10
120 13
Al2O3-KF 1 75 60 10
120 15*La temperatura en el calentamiento convencional fue prefijada de 80º - 100º C previendo la descomposición de los productos.
Los resultados obtenidos por este método no convencional de síntesis se compararon con los alcanzados por el método
tradicional, existiendo una diferencia marcada entre los rendimientos de la reacción. El experimento se realizó
reproduciendo las condiciones de reacción utilizadas cuando se usa las radiaciones de microondas, pero empleando
calentamiento clásico (baño de aceite) y en ausencia de disolventes. Todas las temperaturas fueron fijadas tomando cuidado
de que no ocurriera descomposición de los reactivos de partida. Se comprobó que a mayor bacisidad del soporte empleado,
mayor es el porciento de conversión del producto deseado y menor tiempo de reacción.
En la tabla II se muestran los resultados alcanzados para una serie de oxazolonas derivadas de acetilglicina (4) y
benzoilglina (6) respectivamente con las condiciones de reacción establecidas anteriormente.
Tabla II: Resultados obtenidos para oxaolonas derivadas de acetilglicina y benzoilglicina.
R C
O
NH CH2
COOH +
R1
CHO
Al 2O 3/KF
MW 1-3 min.
R = CH 3 (4) C 6H 5 (6)
CH
N O
R
R1
R = H (a), 2-Cl (b), 4-Cl (c), 3-NO 2 (d), 4-NO 2 (e), 4-CH 3(f), 3-OCH 3 (g), 4-N(CH 3)2 (h), 3-OCH 3 4-OH (i), 3,4-(OCH 3)2(j)
1
4(a-j)6(a-j)
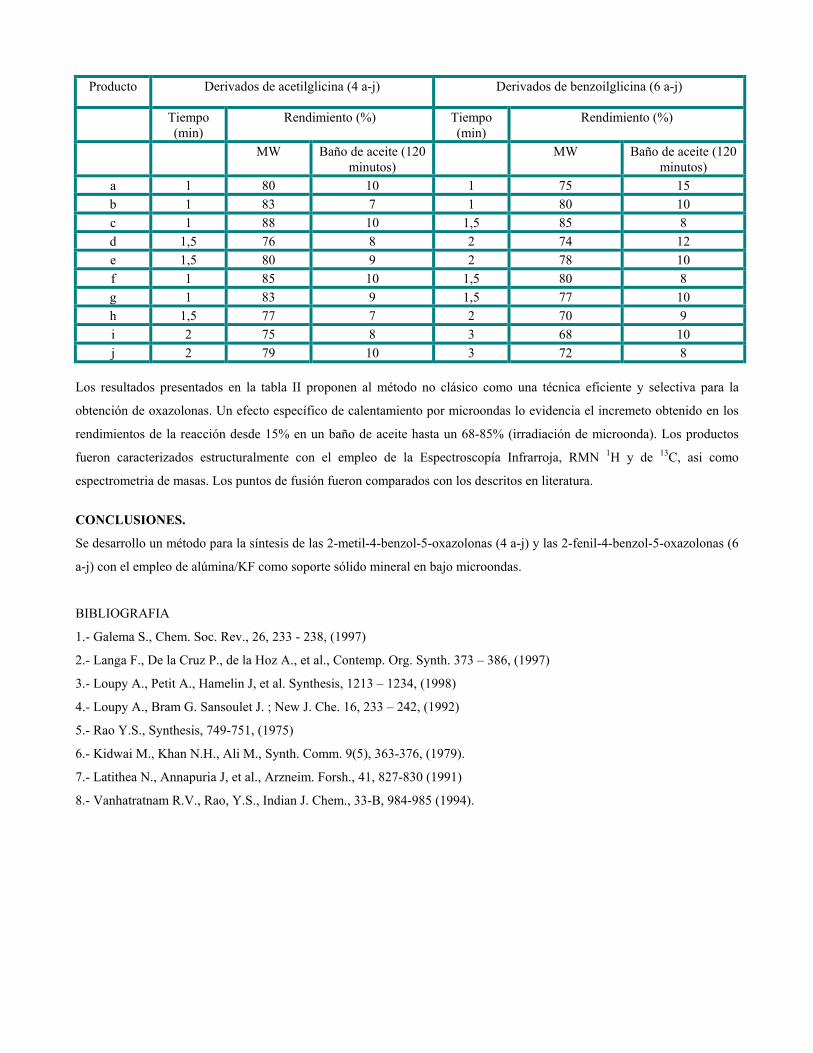
Producto Derivados de acetilglicina (4 a-j) Derivados de benzoilglicina (6 a-j)
Tiempo(min)
Rendimiento (%) Tiempo(min)
Rendimiento (%)
MW Baño de aceite (120minutos)
MW Baño de aceite (120minutos)
a 1 80 10 1 75 15
b 1 83 7 1 80 10
c 1 88 10 1,5 85 8
d 1,5 76 8 2 74 12
e 1,5 80 9 2 78 10
f 1 85 10 1,5 80 8
g 1 83 9 1,5 77 10
h 1,5 77 7 2 70 9
i 2 75 8 3 68 10
j 2 79 10 3 72 8
Los resultados presentados en la tabla II proponen al método no clásico como una técnica eficiente y selectiva para la
obtención de oxazolonas. Un efecto específico de calentamiento por microondas lo evidencia el incremeto obtenido en los
rendimientos de la reacción desde 15% en un baño de aceite hasta un 68-85% (irradiación de microonda). Los productos
fueron caracterizados estructuralmente con el empleo de la Espectroscopía Infrarroja, RMN 1H y de 13C, asi como
espectrometria de masas. Los puntos de fusión fueron comparados con los descritos en literatura.
CONCLUSIONES.
Se desarrollo un método para la síntesis de las 2-metil-4-benzol-5-oxazolonas (4 a-j) y las 2-fenil-4-benzol-5-oxazolonas (6
a-j) con el empleo de alúmina/KF como soporte sólido mineral en bajo microondas.
BIBLIOGRAFIA
1.- Galema S., Chem. Soc. Rev., 26, 233 - 238, (1997)
2.- Langa F., De la Cruz P., de la Hoz A., et al., Contemp. Org. Synth. 373 – 386, (1997)
3.- Loupy A., Petit A., Hamelin J, et al. Synthesis, 1213 – 1234, (1998)
4.- Loupy A., Bram G. Sansoulet J. ; New J. Che. 16, 233 – 242, (1992)
5.- Rao Y.S., Synthesis, 749-751, (1975)
6.- Kidwai M., Khan N.H., Ali M., Synth. Comm. 9(5), 363-376, (1979).
7.- Latithea N., Annapuria J, et al., Arzneim. Forsh., 41, 827-830 (1991)
8.- Vanhatratnam R.V., Rao, Y.S., Indian J. Chem., 33-B, 984-985 (1994).

Hidrólisis de oxazolonas bajo irradiación de microondas.
Autores: Taimirys Mamposo Pérez, Miriam Mesa Hdez, Rolando Pellón Comdom, Miguel López López.
Centro de Química Farmacéutica, calle 21, esq 200, Atabey, Playa, Apdo 16042, La Habana 11600, Ciudad de la Habana.Teléfono: 271-7809, Fax: 33-6471. Email: [email protected].
Resumen:
En la última década se ha llevado a cabo un gran número de reacciones de hidrólisis utilizando la energía de microondas,
lográndose una mayor selectividad y un aumento considerable del rendimiento de la reacción. Generalmente estas
reacciones se realizan en presencia de soportes inorgánicos como alúminas y sílica. Con el objetivo de aportar nuevos datos
a las investigaciones sobre este tipo de reacción, se dirigió este trabajo, al estudio de la hidrolisis de oxazolonas utilizando
irradiación de microondas en ausencia de disolventes, no descrito con anterioridad en la literatura. Los experimentos se
realizaron utilizando diferentes soportes sólidos, evitando la descomposición de los sustratos en un horno doméstico a
2450mhz. Se obtuvieron rendimientos cuantitativos con el uso de la alumina impregnada en fluoruro de potasio como
soporte sólido y a una potencia de 800 watts.
Introducción.
El empleo de las microondas como fuente de enrgía ha comenzado a desarrollarse en la última década1-3. El ambito de
aplicación de la radiación de microondas esta aún por delimitar, sin embargo pueden ya indicarse un número de
aplicaciones particularmente importantes como son: equilibrios que suponen la pérdida de moléculas pequeñas4, reacciones
que requieren de condiciones drásticas5, reacciones que utilizan reactivos poco estables en condicones energicas6. Las
oxazolonas son hidrólizadas a sus correspondientes ácidos, con reactivos ácidos o básicos, dependiendo las condiciones de
esta reacción, del sustituyente presente en el anillo de oxazolona7. Con el objetivo de aportar nuevos datos a las
investigaciones a este tipo de reacción, se dirigió este trabajo al estudio de la hidrólisis de oxazolonas derivadas de
acetilglicina y benzoilglicina utilizando irradiación de microondas, no descrito con anterioridad..
Materiales y Métodos.
Las materia primas empleadas en la síntesis fueron de calidad reactivos. Los puntos de fusión fueron determinados en un
equipo Gallenkamp con calentamiento variable y registro de temperatura, los mismos no fueron corregidos.En las corridas
cromátograficas se utilizaron placas de gel de sílice 60 con indicador de fluorescencia (Merck). Para el desarrollo de los
cromatogramas se utilizó como fase móvil Acetato de Etilo: N-Hexanol (8:6 v/v). Como revelador se empleo una lámpara
UV-Visible a 254 o 366 nm. Las reacciones por el método no clásico se realizaron en un horno doméstico de microondas a
2450 MHz, en reactores abiertos de 25 mL (PYREX) ocupando un 10 % del volumen total del recipiente. El disolvente
utilizado para el aislamiento y purificación de los productos fue acetona.
Resultados y Discusión.
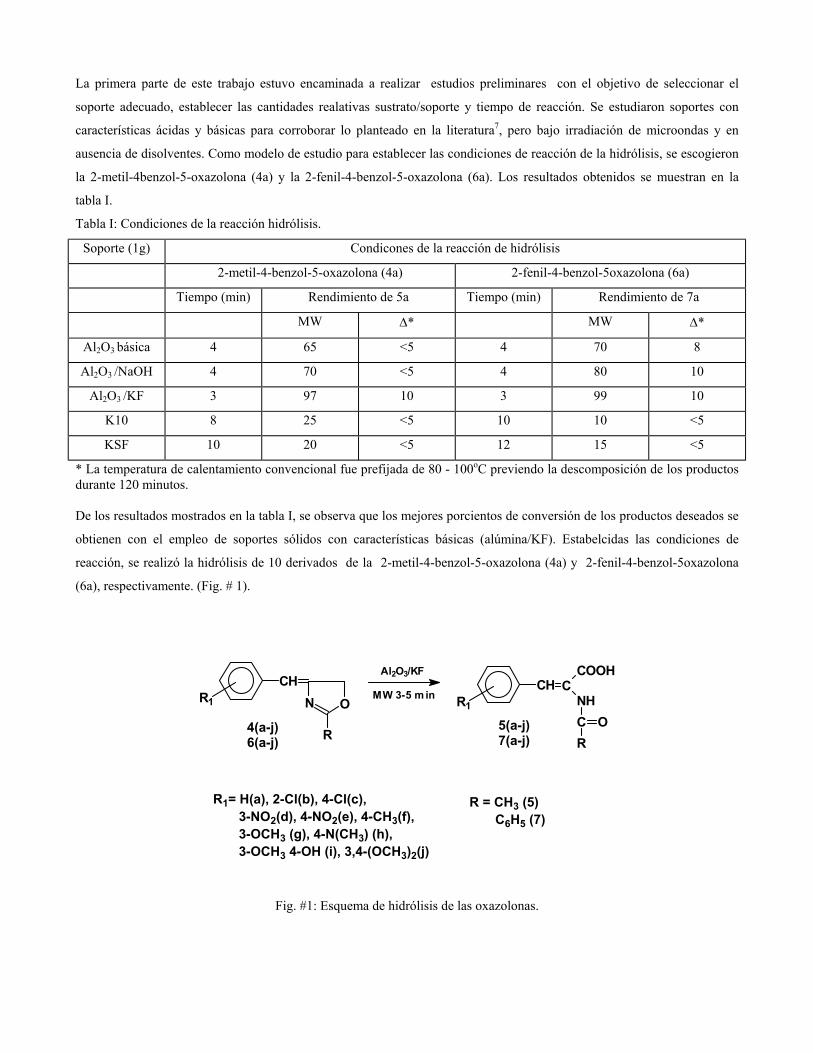
La primera parte de este trabajo estuvo encaminada a realizar estudios preliminares con el objetivo de seleccionar el
soporte adecuado, establecer las cantidades realativas sustrato/soporte y tiempo de reacción. Se estudiaron soportes con
características ácidas y básicas para corroborar lo planteado en la literatura7, pero bajo irradiación de microondas y en
ausencia de disolventes. Como modelo de estudio para establecer las condiciones de reacción de la hidrólisis, se escogieron
la 2-metil-4benzol-5-oxazolona (4a) y la 2-fenil-4-benzol-5-oxazolona (6a). Los resultados obtenidos se muestran en la
tabla I.
Tabla I: Condiciones de la reacción hidrólisis.
Soporte (1g) Condicones de la reacción de hidrólisis
2-metil-4-benzol-5-oxazolona (4a) 2-fenil-4-benzol-5oxazolona (6a)
Tiempo (min) Rendimiento de 5a Tiempo (min) Rendimiento de 7a
MW ∆* MW ∆*
Al2O3 básica 4 65 <5 4 70 8
Al2O3 /NaOH 4 70 <5 4 80 10
Al2O3 /KF 3 97 10 3 99 10
K10 8 25 <5 10 10 <5
KSF 10 20 <5 12 15 <5
* La temperatura de calentamiento convencional fue prefijada de 80 - 100oC previendo la descomposición de los productosdurante 120 minutos.
De los resultados mostrados en la tabla I, se observa que los mejores porcientos de conversión de los productos deseados se
obtienen con el empleo de soportes sólidos con características básicas (alúmina/KF). Estabelcidas las condiciones de
reacción, se realizó la hidrólisis de 10 derivados de la 2-metil-4-benzol-5-oxazolona (4a) y 2-fenil-4-benzol-5oxazolona
(6a), respectivamente. (Fig. # 1).
Al2O3/KF
MW 3-5 m inCH
N O
R
R1 R1
CH CCOOH
NH
C O
R4(a-j)6(a-j)
R1= H(a), 2-Cl(b), 4-Cl(c), 3-NO2(d), 4-NO2(e), 4-CH3(f), 3-OCH3 (g), 4-N(CH3) (h), 3-OCH3 4-OH (i), 3,4-(OCH3)2(j)
5(a-j)7(a-j)
R = CH3 (5) C6H5 (7)
Fig. #1: Esquema de hidrólisis de las oxazolonas.
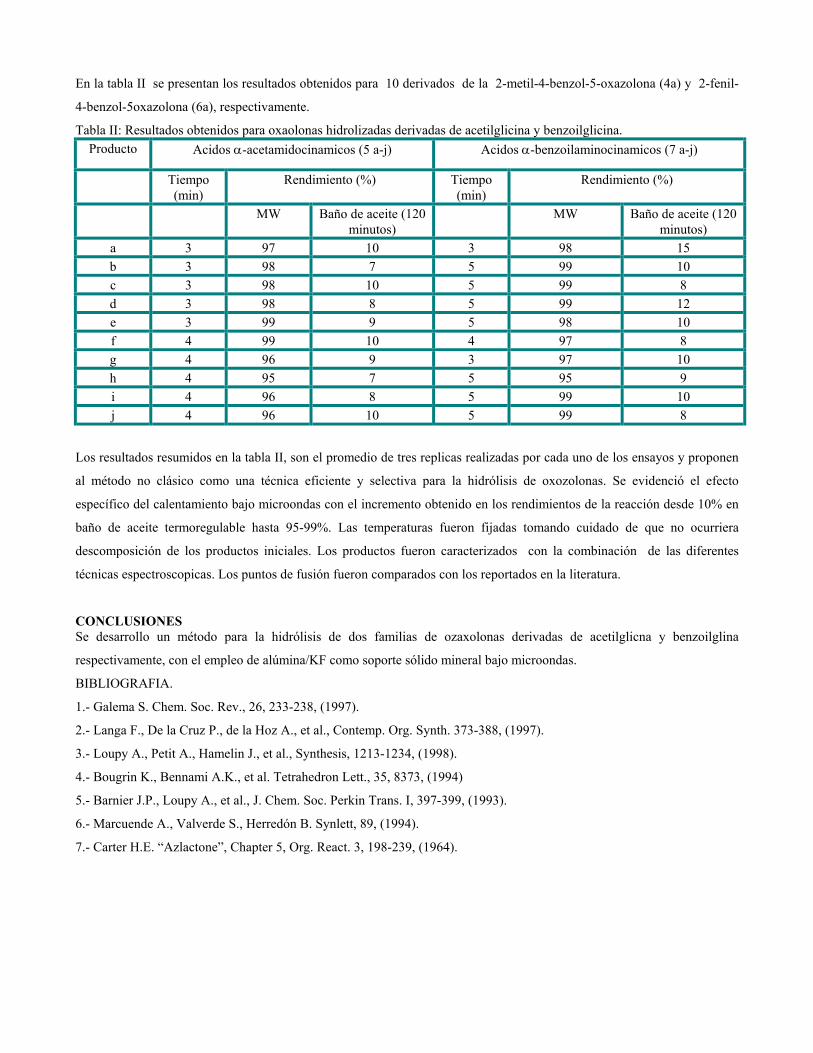
En la tabla II se presentan los resultados obtenidos para 10 derivados de la 2-metil-4-benzol-5-oxazolona (4a) y 2-fenil-
4-benzol-5oxazolona (6a), respectivamente.
Tabla II: Resultados obtenidos para oxaolonas hidrolizadas derivadas de acetilglicina y benzoilglicina.
Producto Acidos α-acetamidocinamicos (5 a-j) Acidos α-benzoilaminocinamicos (7 a-j)
Tiempo(min)
Rendimiento (%) Tiempo(min)
Rendimiento (%)
MW Baño de aceite (120minutos)
MW Baño de aceite (120minutos)
a 3 97 10 3 98 15
b 3 98 7 5 99 10
c 3 98 10 5 99 8
d 3 98 8 5 99 12
e 3 99 9 5 98 10
f 4 99 10 4 97 8
g 4 96 9 3 97 10
h 4 95 7 5 95 9
i 4 96 8 5 99 10
j 4 96 10 5 99 8
Los resultados resumidos en la tabla II, son el promedio de tres replicas realizadas por cada uno de los ensayos y proponen
al método no clásico como una técnica eficiente y selectiva para la hidrólisis de oxozolonas. Se evidenció el efecto
específico del calentamiento bajo microondas con el incremento obtenido en los rendimientos de la reacción desde 10% en
baño de aceite termoregulable hasta 95-99%. Las temperaturas fueron fijadas tomando cuidado de que no ocurriera
descomposición de los productos iniciales. Los productos fueron caracterizados con la combinación de las diferentes
técnicas espectroscopicas. Los puntos de fusión fueron comparados con los reportados en la literatura.
CONCLUSIONESSe desarrollo un método para la hidrólisis de dos familias de ozaxolonas derivadas de acetilglicna y benzoilglina
respectivamente, con el empleo de alúmina/KF como soporte sólido mineral bajo microondas.
BIBLIOGRAFIA.
1.- Galema S. Chem. Soc. Rev., 26, 233-238, (1997).
2.- Langa F., De la Cruz P., de la Hoz A., et al., Contemp. Org. Synth. 373-388, (1997).
3.- Loupy A., Petit A., Hamelin J., et al., Synthesis, 1213-1234, (1998).
4.- Bougrin K., Bennami A.K., et al. Tetrahedron Lett., 35, 8373, (1994)
5.- Barnier J.P., Loupy A., et al., J. Chem. Soc. Perkin Trans. I, 397-399, (1993).
6.- Marcuende A., Valverde S., Herredón B. Synlett, 89, (1994).
7.- Carter H.E. “Azlactone”, Chapter 5, Org. React. 3, 198-239, (1964).

SNTESIS DE ÁCIDOS 2-CARBOXIDIFENILAMINA EMPLEANDO AGUA COMO DISOLVENTE CON
EL EMPLEO DE IRRADIACIÓN ULTRASONICA.
Rolando F. Pellón Comdom y Maite L. Docampo Palacios
Centro de Química Farmacéutica Apartado 16042, La Habana, Cuba
Telef. (537)217809 Fax (537) 336471 E-mail: [email protected]
RESUMEN
Se reporta un método ventajoso para la síntesis del ácido 2-carboxidifenilamina a partir del ácido 2-clorobenzoico y
anilina, usando agua como disolvente y cobre como catalizador en presencia de irradiación ultrasónica. Se estudia el
efecto del uso de la piridina sobre el tiempo de reacción. Se sintetizaron varios derivados del ácido 2-
carboxidifenilamina con muy buenos rendimientos en tiempos cortos de reacción.
INTRODUCCION
Una inspección a las nuevas y tecnologías ya existentes en la síntesis química nos permite concluir que el uso de
ondas ultrasónicas es un campo reconocido en esta rama con estudios teóricos y prácticos que revelan la complejidad
del fenómeno químico subyacente(1).
El ultrasonido es un método adecuado para reacciones en medio heterogéneo y en algunos casos en sistemas
homogéneos, pero es especialmente efectivo cuando la reacción involucra la presencia de un metal disuelto, pues las
ondas ultrasónicas limpian la superficie del mismo haciéndolo más reactivo y por tanto aumenta la velocidad de la
reacción y los rendimientos de los productos(2-5).
En trabajos anteriores se ha estudiado la influencia de varios parámetros de la reacción de Ullmann-Goldberg para la
obtención del ácido 2-carboxidifenilamina a partir de ácido 2-clorobenzoico y anilina. Se demostró que estos ácidos
se podían obtener satisfactoriamente empleando agua como disolvente, en presencia de cobre como catalizador y
carbonato de potasio como base(6).
El uso de la piridina como cocatalizador ha sido reportado para la obtención de ácidos 2-carboxidifenilamina
empleando como disolventes agua o alcohol amílico con el objetivo de reducir los tiempos de reacción(7).
El presente trabajo tiene como objetivo estudiar el efecto del ultrasonido en la condensación de Ullmann-Goldberg
para la síntesis de ácidos 2-carboxidifenilamina a partir de ácidos 2-clorobenzoicos y anilinas sustituidas empleando
agua como disolvente, en presencia de cubre y con o sin el empleo de piridina como catalizadores de la reacción.
MATERIALES Y METODOS
Procedimiento General
Una mezcla del ácido 2-clorobenzoico sustituido correspondiente (0,04mol), el derivado de la anilina
correspondiente (0,08mol), carbonato de potasio anhidro (0,02mol), cobre en polvo (0,2g), piridina (1 mL) y 25 mL
de agua se trata durante 20 minutos con irradiación ultrasónica en un sonicador a 20 kHz. La mezcla de reacción se
enfría, se vierte sobre agua y se acidula con ácido clorhídrico (1:1). El sólido se filtra, se lava con agua y se extrae
con agua hirviendo, obteniéndose el ácido 2-carboxidifenilamina correspondiente, el cual se recristaliza de
etanol/agua (1:1).

RESULTADOS Y DISCUSION
En trabajos anteriores se optimizó la síntesis del ácido ácido 2-carboxidifenilamina usando la reacción de Ullmann-
Goldberg con el empleo de agua como disolvente. Las condiciones óptimas obtenidas fueron: 1 equivalente de
carbonato de potasio, 3% en peso de cobre, 2 equivalentes de anilina por mol de ácido 2-clorobenzoico y 5 horas
como tiempo de reacción(6).
En el presente trabajo estudiamos el uso del agua como disolvente en la obtención de estos ácidos con el empleo del
ultrasonido. Como modelo de la reacción se tomó la condensación del ácido 2-clorobenzoico con anilina empleando
las concentraciones de reaccionantes y catalizador previamente optimizadas (fig. 1).
NH
COOH
R1 R2
COOH
Cl
R1 +H2N
R2
Cu
H20 / )))
Figura 1: Obtención de ácidos 2-carboxidifenilamina.
Con la finalidad de ajustar el tiempo de reacción al emplear ultrasonido, se realizaron varias experiencias a
diferentes tiempos de irradiación, demostrándose que con 20 minutos se obtienen los mejores rendimientos del ácido
2-carboxidifenilamina como se muestra en la tabla 1, para tiempos mayores los rendimientos permanecen constantes.
Todos los experimentos se realizaron por triplicado y se reportan los valores de la desviación estándar en cada
punto.
En estudios anteriores, con el objetivo de reducir los tiempos de reacción, se demostró que la piridina podía ser
usada como cocatalizador en la síntesis del ácido 2-carboxidifenilamina ntranílico empleando agua o alcohol amílico
como disolvente, reduciéndose los tiempos de reacción de 5 a sólo 2 horas(7). Cuando se empleó la piridina en una
concentración de 0,5 equivalentes por mol de ácido 2-clorobenzoico y las concentraciones estudiadas anteriormente
en la síntesis del ácido 2-carboxidifenilamina con el empleo de irradiación ultrasónica, el tiempo de exposición de la
mezcla de reacción se redujo a sólo 15 minutos y el rendimiento del ácido es semejante a cuando no se utiliza la
piridina (tabla 2). Este hecho demuestra, que en presencia de irradiación ultrasónica, la piridina cataliza la reacción.
Todas las experiencias se realizaron por triplicado reportándose la desviación estándar en cada punto. En todos los
casos se obtuvo como subproducto de la reacción el ácido salicílico.
% de rendimiento del ácido 2-carboxidifenilaminaTiempo de irradiaciónultrasónica (minutos)
Sin piridina Desviación estándar(S)
Con piridina Desviación estándar(S)
30 80 1.0 80 2,125 81 1.5 81 2,120 81 2.0 80 2,015 58 1.5 82 1,010 - - 45 2,0
Tabla 1: Estudio del tiempo de irradiación ultrasónica en la síntesis del ácido 2-carboxidifenilamina con y sin el
empleo de la piridina.
Una vez establecido el tiempo de irradiación, se sintetizaron varios derivados del ácido 2-carboxidifenilamina con y
sin piridina en presencia de ultrasonido (tabla 3). Los resultados obtenidos empleando irradiación ultrasónica se
compararon con los obtenidos empleando 5 horas como tiempo de reacción y sin el empleo de piridina (tabla 3). En

todos los casos los rendimientos de los ácidos con el empleo del ultrasonido son semejantes a los obtenidos con el
método a reflujo, pero la pureza de los productos es mayor al emplear irradiación ultrasónica.
N
H R1
R2
R3
R4
COOH
R % de rendimiento de los ácidos 2-carboxidifenilamina usandoagua como disolvente
No R1 R2 R3 R4 5 horas dereflujo sinpiridina
20 minutos conultrasonido y sin
piridina
15 minutos conultrasonido y con
piridina
Punto de fusiónsin corregir (OC)
1 H H H H 86 88 89 184(2)
2 H H COOH H 63 65 64 289-90(9)
3 COOH H H H 79 81 82 293-94(10)
4 H H NO2 Cl 58 61 60 234-35(11)
5 CH3 CH3 H H 78 79 80 231(12)
6 H OCH3 H H 80 82 83 131-32(13)
7 H H OCH3 H 86 90 92 181-82(14)
Tabla 3: Resultados de la síntesis de los derivados del ácido N-fenilantranílico usando agua como disolvente.
CONCLUSIONES
Se desarrolló un método para la síntesis de derivados del ácido 2-carboxidifenilamina usando agua como disolvente
en presencia de irradiación ultrasónica con o sin el empleo de piridina como catalizador.
REFERENCIAS
1. Pestman M. J.; Engberts, J. B. and de Jong , F. Recueil des Travaux Chimiques des Pays-Bas. 1994, 113/12.
2. Haiza, M. ; Lee, J.; Snyder, J.K. J. Org Chem. 1990, 55, 5008.
3. Lee, J.; Mei, H. S. ; Snyder, J. K. J. Org Chem. 1990, 55, 5013.
4. Abdulla, R. F. ; Aldrichimica Acta. 1988, 21, 31.
5. Goldberg, Y. Sturkovich, R. and Lukevics, E. Heterocycles. 1989, 29, 597.
6. Pellón, R. F. ; Carrasco, R.; Rodés, L. Synthetic Communications, 1993, 23 (10) , 1447-53.
7. Pellón, R. F. ; Mamposo, T.; Carrasco, R.; Rodés, L. Synthetic Communications, 1996, 26 (20) , 3877-83
8. Allen, C.F.H. and McKee, G. H. W. Organic Syntheses 1939, 19, 6.
9. Ullmann, F. and Hoz, B., Ann., 1907, 355, 352.
10. Drozdov, N. and Bekhli, A., J. Gen. Chem. (USSR), 1938, 8, 1505.
11. Aggarwal, M.L., Sen-Gupta, I. and Ahmad, B., J. Indian Chem. Soc., 1945, 22, 41.
12. Sherrer, R.A., U.S. Patent, 1964, 3, 138, 636.
13. Lehmstedt, K. and Schrader, K., Ber. 1937, 70, 838.
14. Bauer, Chem. Ber. 1950, 83, 10.

Predicción de las interacciones de derivados de ácido barbitúrico con hydroxipropil--ciclodextrina
utilizando una aproximación TOMO-COMD.
Yovani Marrero*,§, †, Leysi N. Reyes§, Mirtha M. Gonzáles†, & Arnaldo García§.§Departmento de farmacia, Facultad de Química-Farmacia. Universidad Central de las Villas, Santa Clara,
54830, Villa Clara, Cuba.†Centro de Bioactivos Químicos, Universidad Central de las Villas, Santa Clara, 54830, Villa Clara, Cuba.*Author to whom correspondence should be addressed. E-mail: [email protected]
RESUMEN
La interacción entre 33 derivados de ácido barbitúrico con hydroxipropil--ciclodextrina (HPCD) fue
estudiada utilizando un novedoso enfoque topológico-molecular (TOMO-COMD). Usando la regresión
lineal múltiple se desarrollaron modelos cuantitativos para la explicación de la fuerza de la interacción
fármaco-CD. Así, se obtuvieron modelos que describen la hidrofobicidad de los compuestos en estudio
(RMO), el área de superficie hidrofóbica (bE) y la capacidad para la formación del complejo de inclusión con
CD (bH). Los resultados mostraron que la intensidad de la interacción se incrementa con el aumento de la
lipofilia de las moléculas huésped, demostrándose el rol preponderante de las interacciones hidrofóbicas en
la formación de los complejos de inclusión.
Introducción
Las ciclodextrinas forman complejos de inclusión con muchas fármacos de variada estructura química,
modificando sus propiedades. Asi por ejemplo, si se añade ciclodextrina a la fase móbil de un proceso de
separación por HPLC (en fase reversa), se puede observar que decrece el tiempo de retención de las
moléculas que se acomplejan, dependiendo de forma proporcional a la concentración de ciclodextrina y a la
magnitud de la kC [1]. Wang, M et al. establecieron un método cromatográfico sencillo que permite
determinar las constantes de estabilidad para algunos complejos con ß-CD [2]. Frecuentemete se ha
empleado la cromatografía de capa delgada (TLC) para el estudio de interacciones moleculares [3]. Este
método fue usado para la investigación de la formación de los complejos de inclusión de algunos derivados
de ácidos barbitúricos con β-CD polimérica [4] y con HP-b-CD [5] y con Taxol y otras drogas
anticancerigenas [6]. La teoría y los métodos para calcular la fuerza de la interacciín a partir de los datos de
retensión en TLC han sido publicados en la referencia [7].
Materiales y metodos
Los datos de la interacción de compuestos químicos con CDs medidos por TLC de 39 derivados de ácidos
barbitúricos utilizados en el estudio fueron tomados de la referencia [5]. Los índices cuadráticos fueron
calculados con el paquete computacional TOMO-COMD [8] y la regresión lineal multiple con el programa
STATISTICA [9]

Resultados y discusión
Utilizando el procedimiento stepwise se encontraron modelos QSPR que relacionan los índices cuadráticos
totales y locales con los valores de RMO, BE, BH y BBH. Los modelos obtenidos junto con los parámetros
estadísticos se dan a continuación:
RMO=-0.5686+99.122x10-3.eq1(x)–37.88x10-3.eq2(x)+3.19x10-5.eq7(x)-7x10-7.eq9L(x,F1-2) [1]
N=29 R=0.969 F(4.24)=92.707 S=0.344 p<0.00000
RMO = - 2.1565 +28.86x10-3.eq0(x) [2]
N=29 R=0.94 F(1.27)=224.62 S=0.43 p<0.00000
BE = -0.03844 + 12.55x10-3 .eq1(x) [3]
N=29 R=0.953 F(1.27)=269.25 S=0.537 p<0.00000
BH = 3.567 + 1.18x10-7.eq15L(x, F4) – 0.3047.eq2L(x, F4) +043.603x10-3.eq1L(x,F1-2) [4]
–2.1x10-5.eq7L(x,F1-2)–0.258.Xeq0L(x)
N=28 R=0.95 F(5.22)=42.449 S=0.68 p<0.00000
BEH = -0.3016 + 18.542x10-3.eq0L(x, F1-2) + 3.4x10-9.eq14L(x, F1-2)+29.17x10-4.Xeq2L(x) [5]
N=28 R=0.85 F(3.24)=21.272 S=0.28 p<0.00000
Las variables incluidas en la ecuación 1 son los índices cuadráticos totales de orden 1, 2 y 7 y local de
orden 9 para el fragmento 1-2, es decir, en las posiciones 5 del ácido barbitúrico. Como se puede apreciar el
modelo presenta un buen poder predictivo. No obstante, hemos obtenido otro modelo para describir RMO
con una sola variable. La variable incluida es el índice cuadrático total de orden cero, que presenta una
contribución positiva lo cual es lógico si tenemos en cuenta que este descritor es indicativo del tamaño
molécular. El parámetro BE es el coeficiente de la variable concentracioón de alcohol. Los valores de RM de
los compuestos decrecen en cada instante con el aumento en la concentración de alcohol. En la mayoría de
los casos, un incremento en la concentración HP-β-CD puede decrecer en valor RM, indicando la formación
del complejo. Esto presenta gran importancia ya que sugiere que las propiedades biológicas (adsorción,
tiempo de vida media, etc) del complejo fármaco-HP-β-CD puede ser diferente a las de los fármacos
desacomplejados, resultando una modificación efectiva. En esta ecuación, el índice cuadrático local de
orden cero, que tiene en cuenta los heteroátomos en la molécula, contribuye de forma negativa, lo cual es
lógico, debido a que estos átomos aumentan la hidrofilía del compuesto y con esto disminuye la
contribución del efecto hidrofóbico y las interacciones de Van der Waals en el interior de la cavidad de la
CD. BEH es indicativo del impacto de la interacción ternaria etanol-HP-β -CD-Guest en el RM.. Los valores
observados y calculados utilizando las ecuación encontradas para las series de predicción se presentan en
las Tablas 1-5.
Tabla 1. Valores de RMO experimentales y calculados utilizando la ecuacion 1 para la serie de predicción.
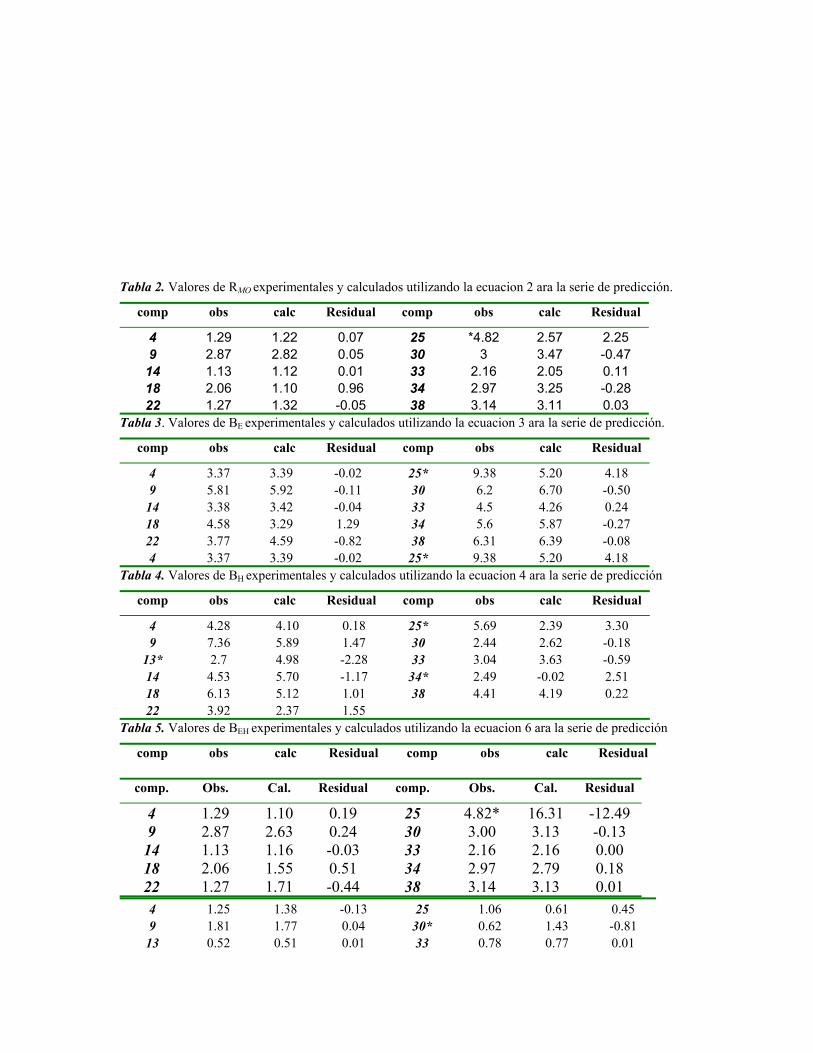
Tabla 2. Valores de RMO experimentales y calculados utilizando la ecuacion 2 ara la serie de predicción.
comp obs calc Residual comp obs calc Residual
4 1.29 1.22 0.07 25 *4.82 2.57 2.259 2.87 2.82 0.05 30 3 3.47 -0.4714 1.13 1.12 0.01 33 2.16 2.05 0.1118 2.06 1.10 0.96 34 2.97 3.25 -0.2822 1.27 1.32 -0.05 38 3.14 3.11 0.03
Tabla 3. Valores de BE experimentales y calculados utilizando la ecuacion 3 ara la serie de predicción.
comp obs calc Residual comp obs calc Residual
4 3.37 3.39 -0.02 25* 9.38 5.20 4.189 5.81 5.92 -0.11 30 6.2 6.70 -0.50
14 3.38 3.42 -0.04 33 4.5 4.26 0.2418 4.58 3.29 1.29 34 5.6 5.87 -0.2722 3.77 4.59 -0.82 38 6.31 6.39 -0.084 3.37 3.39 -0.02 25* 9.38 5.20 4.18
Tabla 4. Valores de BH experimentales y calculados utilizando la ecuacion 4 ara la serie de predicción
comp obs calc Residual comp obs calc Residual
4 4.28 4.10 0.18 25* 5.69 2.39 3.309 7.36 5.89 1.47 30 2.44 2.62 -0.18
13* 2.7 4.98 -2.28 33 3.04 3.63 -0.5914 4.53 5.70 -1.17 34* 2.49 -0.02 2.5118 6.13 5.12 1.01 38 4.41 4.19 0.2222 3.92 2.37 1.55
Tabla 5. Valores de BEH experimentales y calculados utilizando la ecuacion 6 ara la serie de predicción
comp obs calc Residual comp obs calc Residual
4 1.25 1.38 -0.13 25 1.06 0.61 0.459 1.81 1.77 0.04 30* 0.62 1.43 -0.81
13 0.52 0.51 0.01 33 0.78 0.77 0.01
comp. Obs. Cal. Residual comp. Obs. Cal. Residual
4 1.29 1.10 0.19 25 4.82* 16.31 -12.499 2.87 2.63 0.24 30 3.00 3.13 -0.1314 1.13 1.16 -0.03 33 2.16 2.16 0.0018 2.06 1.55 0.51 34 2.97 2.79 0.1822 1.27 1.71 -0.44 38 3.14 3.13 0.01

14 1.3 1.25 0.05 34 0.52 0.24 0.2818 1.63 1.38 0.25 38 1.09 1.12 -0.0322 1.44 0.61 0.83
Bibliografía
1. Zarzycki, P. K., Wierzbowska, H., y Lamparczyk, H. Journal of Pharmaceutical and Biomedical
Analysis. 1997.15, 1281 - 1287.
2. Wang, M., Ueda, H, y Nagai, T. Drug.Dee.Ind.Pharm. 1990. 16, 571.
3. Cserháti, T., Valkó,K., CRC Press, FL, 1993. 1-120.
4. Cserháti, T., Bojarski, J.; Fenyvesi, E, Szejetli, J. J chomatogr. 1986. 351. 356-362.
5. Csabai, K., Cserháti, T., Szejtli, J. International Journal of Pharmaceutics. 1993. 91, 15-22
6. Cserháti, T., Holló, J. International Journal of Pharmaceutics. 1994. 108, 69-75
7. Cserháti, T.,Szögyi, M. J. Biochem. Biophys. Methods, 14. 1987. 101-108.
8. Marrero, Y., Romero, V. TOMOCOND program. Universidad central de las villas. 2002.
9. STATISTICA ver. 5.5, Statsoft, Inc. 1999.

Predicción de la temperatura de ebullición de alcoholes alifáticos utilizando índices cuadráticos
locales.
Yovani Marrero*, †& Vicente Romero.§
* Department of Pharmacy, Faculty of Chemical-Pharmacy. Universidad Central de las Villas, Santa Clara,
54830, Villa Clara, Cuba.†Centro de Bioactivos Químicos, Universidad Central de las Villas, Santa Clara, 54830, Villa Clara, Cuba.§ Faculty of Informatics. Universidad de Cienfuegos, Cienfuegos, Cuba.*Author to whom correspondence should be addressed. e-mail:[email protected]
Resumen
En este trabajo se establece un enfoque local (invariante local) de los índices cuadráticos. Este enfoque está
basado, en la utilización de una matriz local [Mk(G, Fi)] como matriz de la forma. Esta matriz local es
obtenida a partir de la k-ésima potencia de la matriz M [Mk(G)] de adyacencia entre vértices de un
seudografo molecular (G). Mk(G, Fi) tiene en cuenta los elementos del fragmento de interés y a los que
están enlazados con este, mediante caminos de longitud k. Los índices locales junto con sus análogos
totales han sido utilizados para la obtención de modelos QSPR que relacionan la temperatura de ebullición
(b.p) de 58 alcoholes con su estructura molecular. La comparación con otras aproximaciones topológicas
revela un muy buen comportamiento de la metodología propuesta.
Palabras claves: Indice cuadrático local, QSPR, alcoholes alifáticos, temperatura de ebullición.
1. Introducción.
Hasta el momento existen cientos de descriptores moleculares de variada base teórica1. Los índices
topológicos han sido de los más difundidos demostrando gran eficacia en diferentes aplicaciones QSPR y
QSAR2. En un trabajo reciente, Randic3, propuso una lista de atributos deseables para un índice topológico.
Uno de los criterios importantes es la posibilidad de definir localmente los descriptores. En ocasiones, las
propiedades de un grupo de moléculas esta más relacionado con un fragmento que con la molécula como
un todo, por lo que la definición global no satisface los requerimientos estructurales. Por tal motivo, hemos
tenido como objetivo fundamental, proponer una definición local de las formas cuadráticas moleculares
qk(x), manteniendo la generalización a “análogos superiores” (como una secuencia de números3).
2. Materiales y Métodos
Las estructuras y los datos de la b.p de los 58 alkyl alcoholes fueron tomados de la referencia4 y se
muestran en la Tabla 2. Los índices cuadráticos fueron calculadas con el programa TOMO-COMD5
(TOpological MOlecular COMputer Desing). La regresión lineal fue realizada con STATISTICA6.
3. Enfoque Teórico. índices cuadráticos locales. La definición de este descriptor, invariantes grafo-
teóricas para un fragmento Fi dado dentro de un seudografo específico (G) es la siguiente:
qkL(x)= ∑∑==
m
j
km
i
aijLXiXj11
[1]

Donde m es el número de átomos del fragmento de interés y kaijL es el elemento de la fila “i” y columna “j”
de la matriz MkLMk(G, Fi). Esta matríz se extrae de la matríz k-ésima potencia de M y contiene la
información referida a los vértices del fragmento Fi de interés y también del entorno molecular.
La matriz MkL=[kaijL] y los elementos kaijL son definidos como se muestra a continuación:
kaijL= kaij si tanto vi como vj son vértices contenidos en el fragmento de interés.
=1/2 kaij si vi o vj están contenidos en el fragmento de interés pero no ambos
=0 de otra forma
siendo kaij los elementos de la k-ésima potencia de M. Estos análogos locales también pueden ser
expresados de forma matricial por la expresión: qkL(x) =Xt MkL X. Cada orden de los índices cuadráticos
locales tienen un significado particular, especialmente para los primeros valores de k, contiene información
sobre la estructura del fragmento Fi en sí, para valores mayores, contiene información sobre el entorno del
fragmento Fi considerado dentro del seudografo molecular (G).
4. APLICACION QSPR.
En orden de aplicar esta nueva aproximación se ha escogido un conjunto de moléculas empleadas por
Randic8 y posteriormente por Krenkel et. al.4 de las cuales se han computado la b.p de 58 alcoholes
alifáticos. Utilizando el análisis de RLM se han obtenido dos ecuaciones QSPR.
Bp(oC)=2.8042+4.1936.eq0(x)–0.1167.eq4(x) +2.026.10 -3. eq8(x) – 6.7.10-3. eHq8L(x) [2]
Bp(oC)=26.655+2.56.eq1(x)–0.55.eq2(x) –10-4. eq13(x) + 4.73x10-5 . eq14(x) [3]
+1.4262 . Heq2L(x) –0.0512 . Heq6L(x)
Los parámetros estadísticos (tabla I), demuestran la calidad estadística de los modelos obtenidos, incluso
superiores a los obtenidos con anterioridad. Además con el objetivo de reconocer la mejor calidad de
nuestra predicción se ha considerado el número de predicciones con una desviación mayor que 5oC. En este
sentido, utilizando la ecuación 2 se registraron 7 casos, mientras que la data de Randic presentaron 10. Por
otra parte, la ecuación 3 registra para la serie de predicción un solo caso, mientras que para la serie de
predicción de 29 compuestos propuesta por Krenkel et. al. se registraron 4 casos utilizando la ecuación 114.
Tabla I. Parámetros estadísticos correspondientes a las ecuaciones de regresión.
Equation
Set Regression
Coefficient (R)
Standar Fischer Average
Error (S) ratio (F) Deviation
Eq. 2
Complete 0.9944
3.90 11752.71 2.05
Randic and Basak /8/
Complete 0.9938

4.039 2193 2.90
Eq. 3
Training 0.9981
Test 0.9944
2.8937 980 2.067
3.0198 2405 2.40
Eq. 11 /4/
Training 0.9953
Test 0.9948
2.903 5733 2.20
3.025 2529 2.50
En la tabla II se presentan la Bp Experimental y calculada de alcoholes alifáticos incluidos en la misma
serie de predicción externa utilizada en la referencia 4
Tabla II. Bp Experimental y calculada de alcoholes alifáticos incluidos en la serie de predicción
Alkyl alcohol bp exp bp calc. (eq. 2) % bp calc. (eq. 11)
5. 1-butanol 117.7 116.31 1.39 1.18 116.25 (1.45)
7. 2-methyl-1-propanol 107.9 107.84 0.06 0.06 109.79 (-1.89)
10. 2-pentanol 119 120.54 -1.54 -1.29 117.13 (1.87)
12. 2-methyl-1-butanol 128.7 126.63 2.07 1.61 129.34 (-0.64)
13. 3-methyl-1-butanol 131.2 128.13 3.07 2.34 129.23 (1.97)
15. 3-methyl-2-butanol 111.5 111.34 0.16 0.14 110.67 (0.83)
17. 1-hexanol 157.13 155.75 1.38 0.88 155.13 (1.87)
19. 3-hexanol 135.4 138.75 -3.35 -2.47 136.57 (-1.17)
21.3-methyl-1-pentanol 152.4 147.57 4.83 3.17 148.68 (3.72)
23. 2-methyl-2-pentanol 121.4 126.11 -4.71 -3.88 123.86 (-2.46)
24. 3-methyl-2-pentanol 134.2 131.35 2.85 2.13 130.11 (4.09)
25. 4-methyl-2-pentanol 131.7 134.36 -2.66 -2.02 130.11 (1.59)
28. 2-ethyl-1-butanol 146.5 145.87 0.63 0.43 148.68 (-2.18)
30. 2,3-dimethyl-1-butanol 149 147.00 2.00 1.34 142.22 (6.78)
31. 3.3-dimethyl-1-butanol 143 140.47 2.53 1.77 136.55 (6.45)
32. 2,3-dimethyl-2-butanol 118.6 116.56 2.04 1.72 117.40 (1.20)
33. 3,3-dimethyl-2-butanol 120 121.60 -1.60 -1.33 117.99 (2.01)
36. 4-heptanol 155 158.70 -3.70 -2.38 156.01 (-1.01)
38. 3-methyl-3-hexanol 142.4 146.23 -3.83 -2.69 143.30 (-0.90)
40. 2,3-dimethyl-2-pentanol 139.7 141.51 -1.81 -1.30 136.84 (2.86)
42. 2.2-dimethyl-3-pentanol 136 140.00 -4.00 -2.94 137.43 (-1.43)
43. 2,3-dimethyl-3-pentanol 139 138.85 0.15 0.11 136.84 (2.16)
46. 2-octanol 179.8 179.88 -0.08 -0.04 175.45 (4.35)
47. 2-ethyl-1-hexanol 184.6 186.16 -1.56 -0.84 187.56 (-2.96)
50. 2-nonanol 198.5 199.92 -1.42 -0.72 194.89 (3.61)
51. 3-nonanol 194.7 198.43 -3.73 -1.92 194.89 (-0.19)
54. 7-methyl-1-octanol 206 208.16 -2.16 -1.05 207.00 (1.00)
55. 2,6-dimethyl-4-heptanol 178 189.44 -11.44 -6.43 181.99 (-3.99)57. 3,3,5-trimethyl-1-hexanol 193 195.00 -2.00 -1.04 188.43 (4.57)
5. Conclusiones
Los indices cuadráticos totales junto con los análogos locales, constituyen una importante serie que pueden
ser empleados en estudios QSAR y QSPR. Estos índices locales son de gran importancia en la modelación
de propiedades de moléculas con heteroátomos. Además, utilizando estos descriptores se obtuvieron 2
modelos QSPR que relacionan la b.p de 58 alcoholes con su estructura molecular. La comparación con
otras aproximaciones revela un muy buen comportamiento de la metodología propuesta.
6. Referencias
1. Needham, D., E., Wei, I-Chien, Seybold, P., G. J. Amer. Chem. Soc. 1998, 110, 4186-4194.
2. L.B. Kier and L. H. Hall. Molecular Conectivity in Chemistry and Drug Research; Academic Press:
New YorK. 1976
3. Randic, M. Generalized molecular descriptors. J. Math. Chem. 1991, 7, 155-168.
4. Castro, E., A; Krendel, G.. International Journal of Molecular Sciences. 2001, 2, 57-65
5. Marrero, Y., Romero, V. TOMOCOND program. Universidad central de las villas. 2002.
6. STATISTICA ver. 4.13, Statsoft, Inc. 1993.

Screening virtual y Modelación Sub-Estructural (ToSS-MoDe) de Efectos Nocivos Provocados por
Medicamentos sobre el Sistema Sanguíneo e Inmunológico: Trombocitopenia.
Yovani Marreroa,b Humberto González, Luis A. Torres a,b, José A. Marrerob y Mariuchy Mayónb.a Centro de Bioactivos Químicos, Universidad Central de Las Villas, 54830, Cuba.b Departamento de Farmacia, Universidad Central de Las Villas, 54830, Cuba.
* Enviar correspondencia a: [email protected]
Resumen:
Se realizó un estudio de “Relación Cuantitativa Estructura- Reacción Adversa al Medicamento (RCE-
RAM)” para la Trombocitopenia; mediante el uso de la metodología ToSS MoDe. Se desarrolló un modelo
para la predicción de esta RAM a nuevas entidades moleculares (NEM) y el cálculo de las contribuciones a
la trombocitopenia de grupos químicos frecuentes en los fármacos. Estas contribuciones fueron
interpretadas en términos químico-físicos mediante el Análisis de Componentes Principales (ACP). Por
último pudieron postularse algunos de estos grupos como candidatos a toxicóforos.
Palabras Claves: Trombocitopenia, ToSS MoDe, ACP, Hematología, Reacción Adversa.
1. Introducción.
Una de las reacciones adversas de mayor frecuencia son las alteraciones hematológicas producidas por
los fármacos en especial la trombocitopenia1. Las plaquetas juegan un importante papel en la hemostasia al
formar tampones temporales y participando en la reacción de la coagulación. La trombocitopenia es una
depleción del número de plaquetas circulantes. Entre las causas principales de trombocitopenia están los
fármacos; actuando de forma directa en la medula ósea, comprometiendo la función o el número de
megacariocitos. De lo anterior se deriva la importancia de obtener modelos predictivos, que puedan ser
interpretados en términos estructurales, permitiendo un eficiente diseño de NEM menos tóxicas sin perder
la eficacia farmacológica.
El presente trabajo tuvo como objetivo, con el uso de la aproximación ToSS MoDe2, encontrar una
función que nos permita establecer relaciones lineales entre los momentos espectrales (µk) y la
trombocitopenia. Calcular las contribuciones de diferentes grupos químicos. Interpretar desde el punto de
vista químico-físico la propiedad estudiada y proponer los posibles grupos toxicóforos.
2. Materiales y Métodos.
Los datos de los 133 compuestos utilizados en el trabajo fueron obtenidos del INTERCOM3 y
corroborados en el Diccionario de Especialidades Farmacéuticas4 y Bases Farmacológicas de la
Terapéutica5. Los momentos espectrales (µk) fueron calculados con el programa TOSS-MODE6. El análisis
Discriminante Lineal y el ACP fueron realizados con el programa STATISTICA7.
3. Resultados y Discusión.
En este trabajo se ha contado con una amplia data de 133 compuestos. Esta data fue dividida en dos
subseries, una conteniendo 100 compuestos (serie de entrenamiento) y otra 33 compuestos (serie de
predicción).
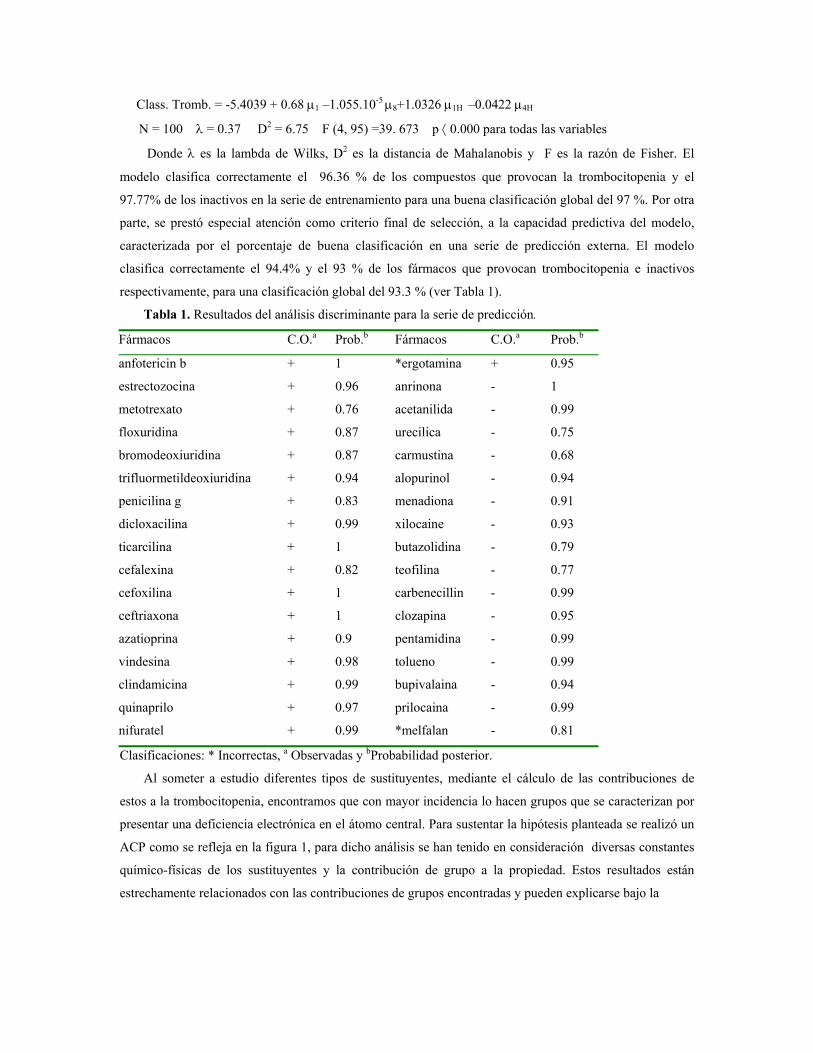
Class. Tromb. = -5.4039 + 0.68 µ1 –1.055.10-5 µ8+1.0326 µ1H –0.0422 µ4H
N = 100 λ = 0.37 D2 = 6.75 F (4, 95) =39. 673 p ⟨ 0.000 para todas las variables
Donde λ es la lambda de Wilks, D2 es la distancia de Mahalanobis y F es la razón de Fisher. El
modelo clasifica correctamente el 96.36 % de los compuestos que provocan la trombocitopenia y el
97.77% de los inactivos en la serie de entrenamiento para una buena clasificación global del 97 %. Por otra
parte, se prestó especial atención como criterio final de selección, a la capacidad predictiva del modelo,
caracterizada por el porcentaje de buena clasificación en una serie de predicción externa. El modelo
clasifica correctamente el 94.4% y el 93 % de los fármacos que provocan trombocitopenia e inactivos
respectivamente, para una clasificación global del 93.3 % (ver Tabla 1).
Tabla 1. Resultados del análisis discriminante para la serie de predicción.
Fármacos C.O.a Prob.b Fármacos C.O.a Prob.b
anfotericin b + 1 *ergotamina + 0.95
estrectozocina + 0.96 anrinona - 1
metotrexato + 0.76 acetanilida - 0.99
floxuridina + 0.87 urecilica - 0.75
bromodeoxiuridina + 0.87 carmustina - 0.68
trifluormetildeoxiuridina + 0.94 alopurinol - 0.94
penicilina g + 0.83 menadiona - 0.91
dicloxacilina + 0.99 xilocaine - 0.93
ticarcilina + 1 butazolidina - 0.79
cefalexina + 0.82 teofilina - 0.77
cefoxilina + 1 carbenecillin - 0.99
ceftriaxona + 1 clozapina - 0.95
azatioprina + 0.9 pentamidina - 0.99
vindesina + 0.98 tolueno - 0.99
clindamicina + 0.99 bupivalaina - 0.94
quinaprilo + 0.97 prilocaina - 0.99
nifuratel + 0.99 *melfalan - 0.81
Clasificaciones: * Incorrectas, a Observadas y bProbabilidad posterior.
Al someter a estudio diferentes tipos de sustituyentes, mediante el cálculo de las contribuciones de
estos a la trombocitopenia, encontramos que con mayor incidencia lo hacen grupos que se caracterizan por
presentar una deficiencia electrónica en el átomo central. Para sustentar la hipótesis planteada se realizó un
ACP como se refleja en la figura 1, para dicho análisis se han tenido en consideración diversas constantes
químico-físicas de los sustituyentes y la contribución de grupo a la propiedad. Estos resultados están
estrechamente relacionados con las contribuciones de grupos encontradas y pueden explicarse bajo la
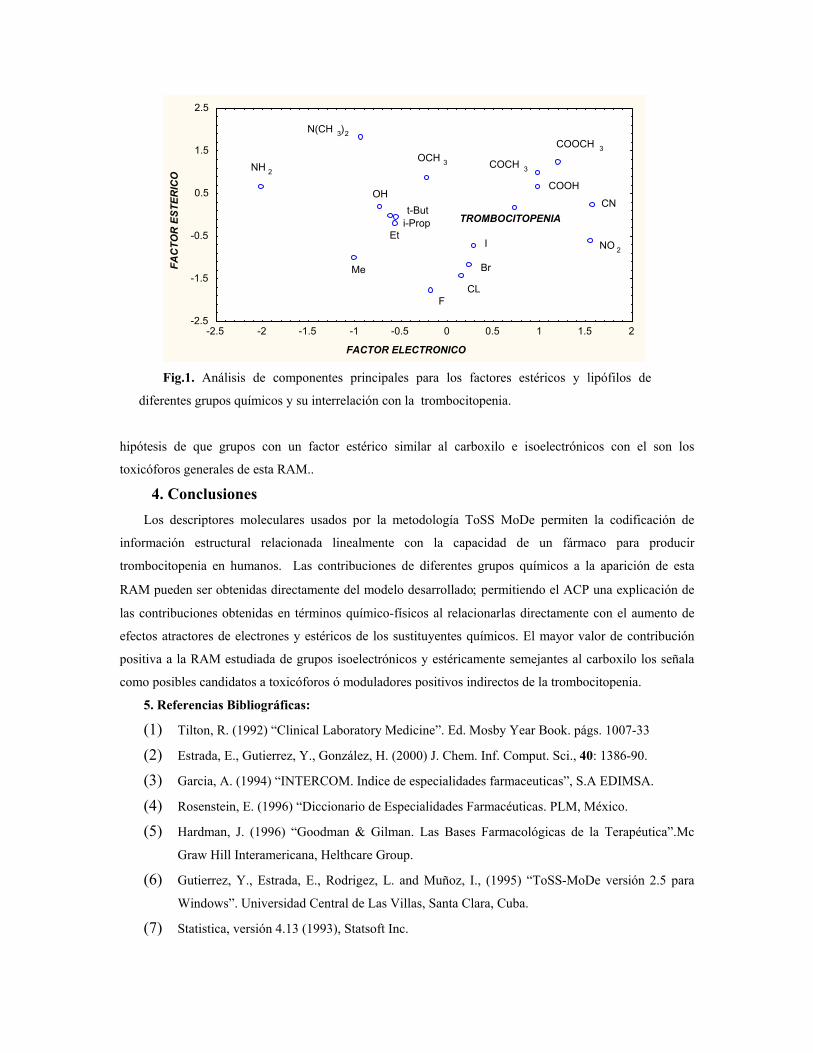
FACTOR ELECTRONICO
FA
CT
OR
ES
TE
RIC
O
-2.5
-1.5
-0.5
0.5
1.5
2.5
-2.5 -2 -1.5 -1 -0.5 0 0.5 1 1.5 2
TROMBOCITOPENIA
NH 2
NO 2
CN
COOCH 3
N(CH 3)2
F
OCH 3
CL
Br
I
COCH 3
COOH
t-But
Me
Eti-Prop
OH
Fig.1. Análisis de componentes principales para los factores estéricos y lipófilos de
diferentes grupos químicos y su interrelación con la trombocitopenia.
hipótesis de que grupos con un factor estérico similar al carboxilo e isoelectrónicos con el son los
toxicóforos generales de esta RAM..
4. Conclusiones
Los descriptores moleculares usados por la metodología ToSS MoDe permiten la codificación de
información estructural relacionada linealmente con la capacidad de un fármaco para producir
trombocitopenia en humanos. Las contribuciones de diferentes grupos químicos a la aparición de esta
RAM pueden ser obtenidas directamente del modelo desarrollado; permitiendo el ACP una explicación de
las contribuciones obtenidas en términos químico-físicos al relacionarlas directamente con el aumento de
efectos atractores de electrones y estéricos de los sustituyentes químicos. El mayor valor de contribución
positiva a la RAM estudiada de grupos isoelectrónicos y estéricamente semejantes al carboxilo los señala
como posibles candidatos a toxicóforos ó moduladores positivos indirectos de la trombocitopenia.
5. Referencias Bibliográficas:
(1) Tilton, R. (1992) “Clinical Laboratory Medicine”. Ed. Mosby Year Book. págs. 1007-33
(2) Estrada, E., Gutierrez, Y., González, H. (2000) J. Chem. Inf. Comput. Sci., 40: 1386-90.
(3) Garcia, A. (1994) “INTERCOM. Indice de especialidades farmaceuticas”, S.A EDIMSA.
(4) Rosenstein, E. (1996) “Diccionario de Especialidades Farmacéuticas. PLM, México.
(5) Hardman, J. (1996) “Goodman & Gilman. Las Bases Farmacológicas de la Terapéutica”.Mc
Graw Hill Interamericana, Helthcare Group.
(6) Gutierrez, Y., Estrada, E., Rodrigez, L. and Muñoz, I., (1995) “ToSS-MoDe versión 2.5 para
Windows”. Universidad Central de Las Villas, Santa Clara, Cuba.
(7) Statistica, versión 4.13 (1993), Statsoft Inc.

Predicción de las interacciones del taxol y otros fármacos anticancerígenos con hydroxipropil--
ciclodextrina utilizando una aproximación TOMO-COMD.
Yovani Marrero*,§, †, Leysi N. Reyes§ , Mirtha M. Gonzáles†, & Arnaldo García§.§Departmento de farmacia, Facultad de Química-Farmacia. Universidad Central de las Villas, Santa Clara,
54830, Villa Clara, Cuba.†Centro de Bioactivos Químicos, Universidad Central de las Villas, Santa Clara, 54830, Villa Clara, Cuba.*Author to whom correspondence should be addressed. E-mail: [email protected]
RESUMEN
La interacción de 23 fármacos anticancerígenos y hydroxipropil--ciclodextrina (HPCD) fue estudiada
utilizando un novedoso enfoque topológico-molecular. Se desarrollaron, utilizando los índices cuadráticos
como descriptores moleculares, modelos cuantitativos para la explicación de la fuerza de la interacción
fármaco-CD. Los resultados corroboraron que la capacidad de formación de los complejos con fármacos
difiere considerablemente acorde a la estructura química y mostraron además, que las interacciones
aumentan significativamente con el incremento de la hidrofobicidad de las moléculas huésped,
demostrándose el rol preponderante de las interacciones hidrofóbicas en la formación de los complejos de
inclusión.
INTRODUCCIÓN
El taxol es una promisoria droga anticancerígena aislada de varias especies Taxus tales como Taxus
baccata L. [1], Taxol ha sido usado satisfactoriamente en el tratamiento de cáncer que hace metástasis en
los senos [2] y carcinoma de ovario [3]. Debido a su hidrofobicidad [4] la administracion del taxol presenta
considerables dificultades [5]. Muchos esfuerzos se han realizado para desarrollar un derivado
semisintético del taxol que presente mejores parámetros de aplicación [6]. El objetivo de este trabajo fue el
estudio de la interacción del taxol y otros fármacos anticancerígenos con hidroxipropil-β-ciclodextrina
(HP-β-CD), mediante la obtención de modelos QSPR que permiten predecir, interpretar y ayudar a explicar
los resultados experimentales de varios parámetros de la interacción fármaco-CD obtenidos por
cromatografia de capa delgada (TLC)
RESULTADOS Y DISCUSIÓN
El enfoque TOMOCOMD. En este software se han implementado computacionalmente 10 nuevas
familias de descriptores moleculares. En específico, hemos utilizado los índices cuadráticos “de la matriz
de adyacencia entre vértices de un seudografo molecular”. Estos descriptores se calculan según la ecuación:
Aplicaciones QSPR. Los parámetros TLC (RMO, BE y BH) que describen la interacción entre 23 fármacos
anticancerígenos (ver Tabla I) y HP-β-CD fueron tomados de la referencia [7]. Los modelos obtenidos para
qkL(x)= ∑∑==
m
j
km
i
aijLXiXj11
[1]

la lipofilia (RMO), el área específica hidrofóbica (BE) y la capacidad de formación del complejo o también
denominado fuerza de la interacción (BH) se dan a continuación, junto con sus parámetros estadísticos.
Tabla I.. Fármacos anticancerígenos utilizados en el estudio
No Nombre No Nombre No Nombre
1 ftoraful 9 paraplatin 17 Estracyt
2 bicnu 10 zitazonium 18 Deticene
3 leukeran 11 farmorubicin 19 methotrexate
4 vincristine 12 adriblastine 20 Myelobromol
5 vinblastine 13 natulan 21 Zytostop
6 vumon 14 alexan 22 Elobromol
7 provera 15 Mytomicin C kyowa 23 taxol
8 bleogin 16 cytoxan
RMO = - 0.46868 + 0.012188. eq0H(x) –9x10-9. eq12
H(x) + 1.55x10-9. eq13(x) [2]
N=14 R=0.96 F(3.10)=41.424 S=0.24 p<0.00000
BE= -0.28247+0.014419. eq0(x)–2.2x10-10.Aeq15LH(x)+0.014726.eq3L
H(x)– 4.2x10-9 .Eeq13LH(x) [3]
N=14 R=0.95 F(4.9)=24.352 S=0.35 p<0.00008
BH = 2.128569+1.83x10-6 .Aeq8L(x)–0.01703. Eeq0L(x)+0.020723. eq2LH(x) –1.3x10-8.Eeq13L(x) [4]
N=11 R=0.95 F(4.6)=15.229 S=0.19 p<0.00269
Los valores de estos parámetros experimentales y calculados por las ecuaciones 2 y 3 para la serie de
entrenamiento y predicción son dados en las Tablas II, III.
La capacidad de los fármacos de incluirse difiere considerablemente acorde a su estructura química.
Además, la capacidad de la interacción se incrementa significativamente con el incremento de la
hidrofobicidad de la molécula huésped, demostrándose, una vez más, el rol de las interacciones
hidrofóbicas en la formación de los complejos de inclusión..
Tabla II. Valores de RMO experimentales y calculados utilizando la ecuación 2 .
comp
obs
calc
Residual
comp
obs
calc
Residual

Serie entrenamiento
2
1.04
1.04
0.00
13
0.86
0.68
0.18
3
0.93
1.04
-0.11
15
1.11
1.09
0.02
4
2.44
2.32
0.12
16
1.18
1.49
-0.31
5
2.18
2.38
-0.20

17
1.67
1.84
-0.17
6
1.96
2.04
-0.08
18
0.94
0.59
0.35
7
2.34
2.10
0.24
19
0.76
1.14
-0.38
11
1.67
1.51
0.16
23
3.5
3.32
0.18
Serie prediccón

1
0.36
-0.47
0.83
10
2.15
2.10
0.05
8
1.43
1.38
0.05
12
1.58
-0.47
2.05
La variable eq0(x) [índice cuadrático total de orden cero], fue incluida en los 2 primeras ecuaciones con una
contribución positiva, lo cual es lógico si tenemos en cuenta que este descritor es indicativo del tamaño
molécular y del tipo de átomos que conforman la molécula.
Tabla III. Valores de BE experimentales y calculados utilizando la ecuación 3.
comp
obs
calc
Residual
comp
obs
calc
Residual

Serie entrenamiento
2
1.9
1.67
0.23
13
2.26
2.07
0.19
3
0.75
1.21
-0.46
15
2.81
2.83
-0.02
4
3.2
2.95
0.25
16
2.18
1.84
0.34
5
2.9
2.90
0.00

17
1.47
1.88
-0.41
6
2.41
2.26
0.15
18
2.11
1.72
0.39
7
2.86
3.19
-0.33
19
2.26
2.72
-0.46
11
2.12
2.07
0.05
23
5.27
5.19
0.08
Serie predicción

1
1.75
1.80
-0.05
10
2.01
1.99
0.02
8
2.25
2.21
0.04
12
1.83
2.33
-0.50
Los compuestos 9, 14, y 20-22 fueron cercanos al frente en el sistema eluyente, indicando que estos
fármacos son altamente hidrofílicos y su interacción con HP-β-CD no puede ser determinado bajo las
condiciones experimentales empleadas
CONCLUSIONES
En el trabajo se realizó el estudio de la interacción del taxol y otros fármacos anticancerígenos con
hidroxipropil-β-ciclodextrina (HP-β-CD), desarrollando modelos QSPR que permiten predecir, interpretar
y explicar los resultados experiemtales de parámetros de la interacción fármaco-CD obtenidos por TLC.
REFERENCIAS
1. Senilh, V., Blechert, S.; Colin, M., Guenard, D., Picot, F., Potier, P. J. NaturalProd., 1984. 47. 134-139.
2. Holmes, F.A., Walters, R.S. Theriault, R.L., Forman, A. D., Newton, L. K, Raber,M.N., Buzdar, A. U., Fye, D.K., Hortobagy, G.N.. J. Nat. Cancer Inst., 1991. 83.1797-1805.
3. Markman, M.. Yale J. Biol. Med., 1991. 64, 583-590
4. Kingston, D. G. I., The chemistry of taxol. Pharmacol. Ther., 1991. 52, 1-34.
5. Gurite-Voegelein, F.,Guenard, D., Potier, P. C. R. Soc. Biol., 1992, 186, 433-440.

6. Mathew, A. E., Mejillano, M. R., Nath, J. P., Himes, R. H., Stella, V. J. J. Med.Chem., 1992, 35, 145-151.
7. Cserháti, T., Holló, J. International Journal of Pharmaceutics. 1994. 108, 69-75

Quadratic Index for the “Adjacency Matrix of the vertexes of a Molecular Pseudograph”.
Yovani Marrero*, † & Vicente Romero.§
*Department of Pharmacy, Faculty of Chemical-Pharmacy. Universidad Central de las Villas, Santa Clara,
54830, Villa Clara, Cuba.†Centro de Bioactivos Químicos, Universidad Central de las Villas, Santa Clara, 54830, Villa Clara, Cuba.§ Faculty of Informatics. Universidad “Carlos Rafael Rodriguez” de Cienfuegos, Cienfuegos, Cuba.*Author to whom correspondence should be addressed. E-mail: [email protected]
Summary
A novel topologic-molecular approach to obtain a family of molecular descriptors is proposed. For that, a
set of all the organic molecules is defined, as a vector space E, where by E is a vector space in the form of
a direct sum of subspaces. This way, we can express the molecules with i-atoms as elements (vectors) of
the vector spaces (product spaces) ℜ i (i=1, 2, 3,..., n; where n is number of atoms in the molecule) and
the components of the vectors are atomic properties that characterize each atom in particular. The
“Quadratic Indexes” are based on the calculation of quadratic forms, which uses the Matrixes (M) of
adjacency between vertexes of molecular pseudograph and the canonical bases, as matrix and bases of the
forms, respectively. This index was generalized for “higher analogues”. The results obtained are valid to
establish that this new index has the ideal requirements as proposed by Randic for a new descriptor.
Keywords: Molecular vector space, Quadratic index, QSPR, Hydrocarbons, Boiling points.
1. INTRODUCTION
The graph theory has great applications in the field of economics, sociology, technology, biology and other
branches [1]. In the area of the organic chemistry we can represent compounds and basing on the so-called
"molecular graph", several matrixes can be obtained that characterize it [2]. The most commonly used
matrixes have been those of incidence, those of adjacency between vertexes, or of topological distances
between these. Using any of the last two or a combination of both, most of the topological indexes
described in the literature have been obtained [3]. Therefore, these topological descriptors (indexes) are
calculated using the graph theory applied to the organic chemistry and they constitute numeric descriptors
of the molecular structure. This work is based on the most fundamental thing in the definition of a new
topological index, related to the adjacency matrix (M) between vertexes of a molecular pseudograph. This
new approach is also applied to the prediction of boiling point of saturated and unsaturated non-cyclic
aliphatic hydrocarbons.
2. MATERIALS AND METHODS
The structures and the data of the boiling point of the hydrocarbons were taken from the literatures [4]. The
Quadratic Indexes [eqk(x)] were calculated with the TOMO-COMD (TOpological MOlecular COMputer
Design) program [5]. The multiple linear regression was carried out with the package of statistical
programs STATISTICA [6].
2. THEORETICAL APPROACH

A. Molecular Vectorial space. Each element of the periodic table presents inherent atomic properties, such
as the electro-negativity, density, atomic radio, etc. Each one of these properties characterizes numerically
each atom, taking values in the real set (ℜ ). Recently, these authors [7-8] defined the E set; which
includes all the possible molecules with i-atoms, as vector space in the set of real space (ℜ -space). ℜ i
was defined as direct sum of all sub vector spaces on ℜ , where the dimension of E is the sum of the
dimensions of each one of the sub-spaces. Therefore the dimension is n(n+1)/2. By this way, a molecule
with n atoms can be “represented” by means of vectors with 2, 3, 4,...., n components belonging to the sub-
spaces ℜ 2, ℜ 3, ℜ 4,..., ℜ n respectively.
B. Matrix representation. The adjacency M matrix of a molecular pseudograph G = < V,E > will be the
nxn matrix M(G) = [aij] , where n is the number of vertexes, and the elements aij are defined as:
where Pij is the number of edges that satisfies ek ∼ vi,vj between the vertexes vi and vj. Lij is the number of
loop in vi.The elements aij of M represent the bonds between an atom "i" and other "j". Which vertexes can
be reached using path of length 2 (P2)? An interesting property of M is that Mk gives the number of paths
of length k that unite the vertexes vi and vj.
C. Mathematical definition of the descriptor. The quadratic index ( q(x)) is calculated using the matrix
M as in eq. 1.
qk(x)= ∑∑==
n
j
kn
i
aijXiXj11
[1]
where aij = aji and n is the number of atoms in the molecule. The coefficients kaij are the elements aij of the
k-th power of M From the expression of qk(x). Then q(x) can be written in the form of a matrix product
XtMkX = qk(x), k ≥ 10q(x). So if we use the canonic bases, the coordinates of any vector X coincide with
the components of that vector.
4. QSRP APPLICATION.
To prove the possibilities of the application of the Quadratic Index in studies QSRP, we have selected the
boiling point of non-cyclic aliphatic hydrocarbons. The equations obtained, are given below, together with
their statistical parameters:
b. p (oC)= -100.834 + 7.2605 eq0(x) - 1.5649 eq2(x) + 9.8724 x10-2 eq5(x) - 5.92 x10-3 eq7(x)
N=53 R=0.9837 F(4.48)=359.75 S=6.29 p<0.0000
The statistics show the quality of the model. So for example, the coefficient of multiple linear correlations
has a value above 0.98 for the training set. Also, a relationship between the experimental values and those
calculated through the model for the compounds included in the prediction set was established. The
statistical coefficients for these correlations are given as follows: N=29, R=0.986, F(1.27)=930.85, S=4.386
aij = Pij if i≠j y ∃ ek ∈ E / ek ∼ vi,vj
= Lij if i = j
= 0 otherwise
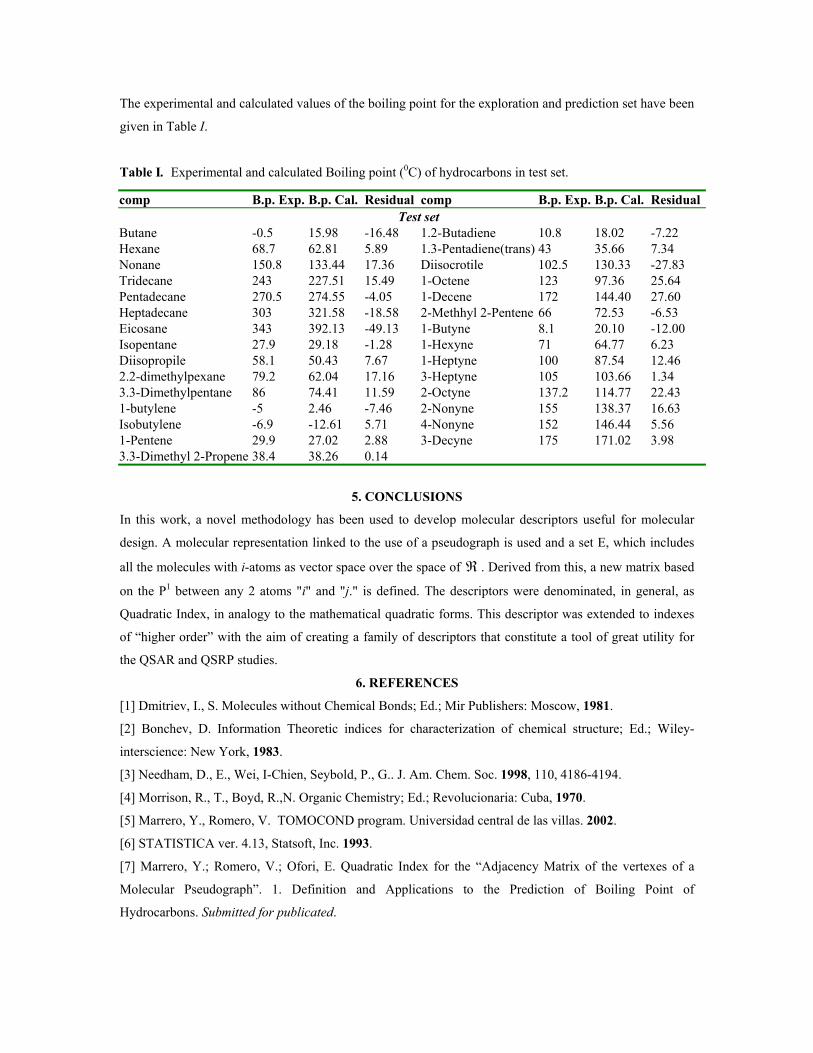
The experimental and calculated values of the boiling point for the exploration and prediction set have been
given in Table I.
Table I. Experimental and calculated Boiling point (0C) of hydrocarbons in test set.
5. CONCLUSIONS
In this work, a novel methodology has been used to develop molecular descriptors useful for molecular
design. A molecular representation linked to the use of a pseudograph is used and a set E, which includes
all the molecules with i-atoms as vector space over the space of ℜ . Derived from this, a new matrix based
on the P1 between any 2 atoms "i" and "j." is defined. The descriptors were denominated, in general, as
Quadratic Index, in analogy to the mathematical quadratic forms. This descriptor was extended to indexes
of “higher order” with the aim of creating a family of descriptors that constitute a tool of great utility for
the QSAR and QSRP studies.
6. REFERENCES
[1] Dmitriev, I., S. Molecules without Chemical Bonds; Ed.; Mir Publishers: Moscow, 1981.
[2] Bonchev, D. Information Theoretic indices for characterization of chemical structure; Ed.; Wiley-
interscience: New York, 1983.
[3] Needham, D., E., Wei, I-Chien, Seybold, P., G.. J. Am. Chem. Soc. 1998, 110, 4186-4194.
[4] Morrison, R., T., Boyd, R.,N. Organic Chemistry; Ed.; Revolucionaria: Cuba, 1970.
[5] Marrero, Y., Romero, V. TOMOCOND program. Universidad central de las villas. 2002.
[6] STATISTICA ver. 4.13, Statsoft, Inc. 1993.
[7] Marrero, Y.; Romero, V.; Ofori, E. Quadratic Index for the “Adjacency Matrix of the vertexes of a
Molecular Pseudograph”. 1. Definition and Applications to the Prediction of Boiling Point of
Hydrocarbons. Submitted for publicated.
comp B.p. Exp. B.p. Cal. Residual comp B.p. Exp. B.p. Cal. ResidualTest set
Butane -0.5 15.98 -16.48 1.2-Butadiene 10.8 18.02 -7.22Hexane 68.7 62.81 5.89 1.3-Pentadiene(trans) 43 35.66 7.34Nonane 150.8 133.44 17.36 Diisocrotile 102.5 130.33 -27.83Tridecane 243 227.51 15.49 1-Octene 123 97.36 25.64Pentadecane 270.5 274.55 -4.05 1-Decene 172 144.40 27.60Heptadecane 303 321.58 -18.58 2-Methhyl 2-Pentene 66 72.53 -6.53Eicosane 343 392.13 -49.13 1-Butyne 8.1 20.10 -12.00Isopentane 27.9 29.18 -1.28 1-Hexyne 71 64.77 6.23Diisopropile 58.1 50.43 7.67 1-Heptyne 100 87.54 12.462.2-dimethylpexane 79.2 62.04 17.16 3-Heptyne 105 103.66 1.343.3-Dimethylpentane 86 74.41 11.59 2-Octyne 137.2 114.77 22.431-butylene -5 2.46 -7.46 2-Nonyne 155 138.37 16.63Isobutylene -6.9 -12.61 5.71 4-Nonyne 152 146.44 5.561-Pentene 29.9 27.02 2.88 3-Decyne 175 171.02 3.983.3-Dimethyl 2-Propene 38.4 38.26 0.14

[8] Marrero, Y.; Romero, V.; Ofori, E. Quadratic Index for the “Adjacency Matrix of the vertexes of a
Molecular Pseudograph”. 2. Local Definition and Applications to the Prediction of Boiling Point of Alkyl
alcohols. Submitted for publicated.

Índices Cuadráticos y predicción de la temperatura de ebullición de moléculas que contienen ciclos.
Yovani Marrero,*, δ, † Vicente Romero. §
δDepartment of Pharmacy, Faculty of Chemical-Pharmacy. Universidad Central de las Villas, Santa Clara,
54830, Villa Clara, Cuba.†Centro de Bioactivos Químicos, Universidad Central de las Villas, Santa Clara, 54830, Villa Clara, Cuba.§Faculty of Informatics. Universidad “Carlos Rafael Rodriguez” de Cienfuegos, Cienfuegos, Cuba.*Author to whom correspondence should be addressed. E-mail: [email protected]
Resumen
En el presente trabajo los índices cuadráticos “de la matriz de adyacencia entre vértices de un seudografo
molecular” han sido utililizados en un estudio QSPR. Esta nueva aproximación es aplicada a moléculas que
contienen ciclos. Utilizando este enfoque, se obtuvieron dos modelos que describen adecuadamente la
temperatura de ebullición (b.p) de los 80 compuestos incluidos en la serie de entrenamiento. El poder
predictivo de estos modelos fue evaluado utilizando una serie de predicción de 26 cicloalcanos. La
comparación con otras aproximaciones revela un buen comportamiento de la metodología propuesta.
Palabras claves: QSPR, índices cuadráticos, moléculas que contienen ciclos, temperatura de ebullición.
1. Introducción.
En la actualidad, el enfoque grafo-teórico es una importante alternativa para los métodos de diseño
molecular asistido por computadoras [1]. Los más usados y conocidos son los llamados índices topológicos
[2]. Estos índices son obtenidos a partir de una representación grafo-teórica de la molécula, en forma de
invariantes [3]. Estas invariantes constituyen un número o un conjunto de números que describen de forma
cuantitativa la estructura molecular, por lo que pueden ser usados en estudios QSPR y QSAR [2-3]. En dos
artículos previos [4.5], estos autores han propuesto una nueva invariante grafo-teórica. En el presente
trabajo, nosotros aplicamos los índices cuadráticos a la descripción de la b.p de moléculas que contienen
ciclos alifáticos. Este tipo de moléculas son muy importantes ya que representan una gran parte del campo
de la química y muchos de los fármacos contienen ciclos en su estructura.
2. Materiales y Métodos.
Las b.p de los 106 cicloalcanos fueron tomados de la referencia [6]. Los índices cuadráticos fueron
calculadas con el programa TOMO-COMD [7]. La regresión lineal fue realizada con STATISTICA [8].
3. Enfoque teórico:Índices Cuadráticos. Tomemos una molécula con n átomos (vector de ℜ n). Se
definen los k-ésimos índices cuadráticos totales [ec. 1] y locales [ec. 2] a las aplicaciones q: ℜ n ℜ , si
para X=x1a1+...+xnan, donde (ai)1in es una base de ℜ n se cumple:
qk(x)= ∑∑==
n
j
kn
i
aijXiXj11
[1] qkL(x)= ∑∑==
m
j
km
i
aijLXiXj11
[2]

donde n es el número de átomos de la molécula. Los coeficientes kaij son los elementos aij de la k-ésima
potencia de la matríz M del seudografo molecular. Luego, M=[aij], y los elementos aij se definen
donde Pij es el número de aristas que cumplen ek ∼ vi,vj entre los vértices vi y vj. Lij es el número de lazos
en vi. En ec. 2 m es el número de átomos del fragmento de interés y kaijL es el elemento de la fila “i” y
columna “j” de la matriz MkLMk(G, Fi). Esta matríz se extrae de la matriz k-ésima potencia de M.
kaijL= kaij si tanto vi como vj son vértices contenidos en el fragmento de interés.
=1/2 kaij si vi o vj están contenidos en el fragmento de interés pero no ambos
=0 de otra forma
4. QSPR Aplicación.
Se obtuvieron 2 modelos de RLM que describen la b.p de los compuestos en la serie de entrenamiento.
Bp (oC)=-105.146+3.1629 eq1(x) –0.4933 eq2(x) [4]
Bp (oC)=-108.197+1.6358 eq0(x)+2.038 eq1(x)–0.3016 eq2(x)-1.75x10-5 eq14(x)
+6.42x10-6 eq15(x) [5]
Los estadígrafos de estas 2 ecuaciones y los valores reportados en [6] se presentan en la tabla I.
Tabla I. Parámetros estadísticos correspondientes a las ecuaciones de regresión.
Equation
Set Regression
Coefficient (R)
Standar Fischer
Error (S) ratio (F)
Eq. [4] two descriptors
Training 0.9823
Test 0.9726
7.8211 1058.2 10.245 421.21
Eq. [5] Five descriptors
Training 0.9927
Test 0.9938
5.0145 5257.9
4.7865 2025.4
Eq. [1] /6/. Six descriptors
Training 0.9937
Test 0.9943
aij = Pij if i≠j y ∃ ek ∈ E / ek ∼ vi,vj
= Lij if i = j
= 0 otherwise
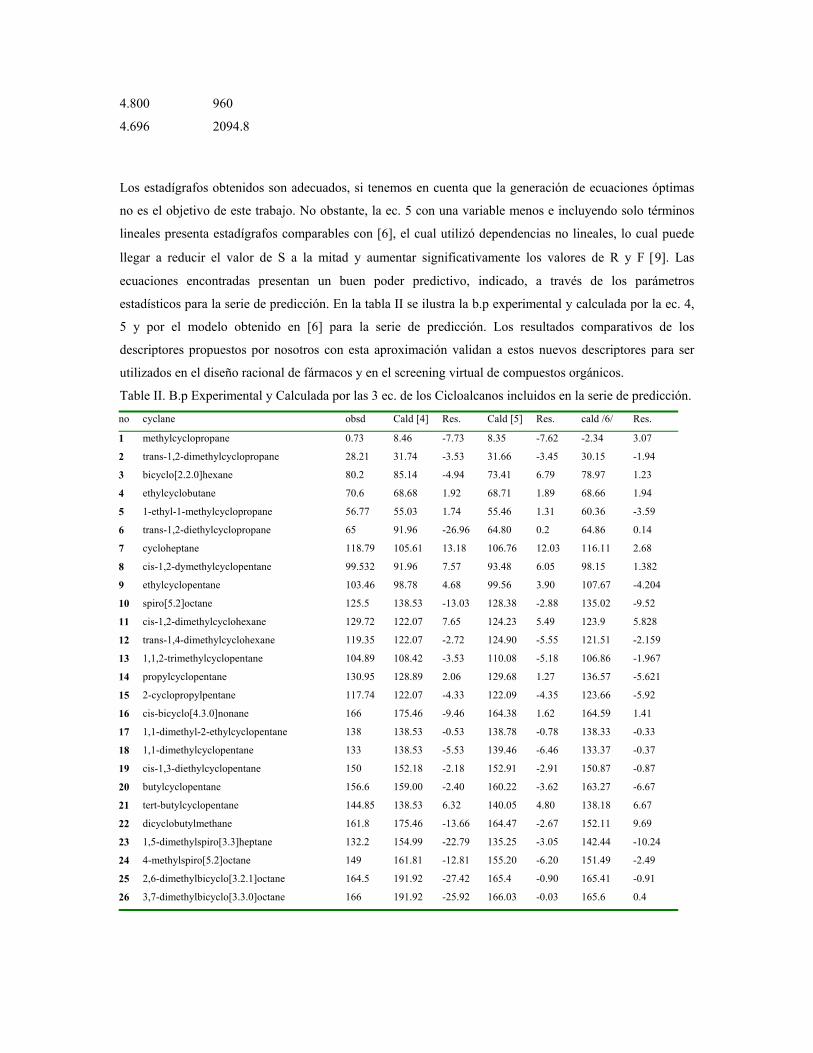
4.800 960
4.696 2094.8
Los estadígrafos obtenidos son adecuados, si tenemos en cuenta que la generación de ecuaciones óptimas
no es el objetivo de este trabajo. No obstante, la ec. 5 con una variable menos e incluyendo solo términos
lineales presenta estadígrafos comparables con [6], el cual utilizó dependencias no lineales, lo cual puede
llegar a reducir el valor de S a la mitad y aumentar significativamente los valores de R y F [9]. Las
ecuaciones encontradas presentan un buen poder predictivo, indicado, a través de los parámetros
estadísticos para la serie de predicción. En la tabla II se ilustra la b.p experimental y calculada por la ec. 4,
5 y por el modelo obtenido en [6] para la serie de predicción. Los resultados comparativos de los
descriptores propuestos por nosotros con esta aproximación validan a estos nuevos descriptores para ser
utilizados en el diseño racional de fármacos y en el screening virtual de compuestos orgánicos.
Table II. B.p Experimental y Calculada por las 3 ec. de los Cicloalcanos incluidos en la serie de predicción.
no cyclane obsd Cald [4] Res. Cald [5] Res. cald /6/ Res.
1 methylcyclopropane 0.73 8.46 -7.73 8.35 -7.62 -2.34 3.07
2 trans-1,2-dimethylcyclopropane 28.21 31.74 -3.53 31.66 -3.45 30.15 -1.94
3 bicyclo[2.2.0]hexane 80.2 85.14 -4.94 73.41 6.79 78.97 1.23
4 ethylcyclobutane 70.6 68.68 1.92 68.71 1.89 68.66 1.94
5 1-ethyl-1-methylcyclopropane 56.77 55.03 1.74 55.46 1.31 60.36 -3.59
6 trans-1,2-diethylcyclopropane 65 91.96 -26.96 64.80 0.2 64.86 0.14
7 cycloheptane 118.79 105.61 13.18 106.76 12.03 116.11 2.68
8 cis-1,2-dymethylcyclopentane 99.532 91.96 7.57 93.48 6.05 98.15 1.382
9 ethylcyclopentane 103.46 98.78 4.68 99.56 3.90 107.67 -4.204
10 spiro[5.2]octane 125.5 138.53 -13.03 128.38 -2.88 135.02 -9.52
11 cis-1,2-dimethylcyclohexane 129.72 122.07 7.65 124.23 5.49 123.9 5.828
12 trans-1,4-dimethylcyclohexane 119.35 122.07 -2.72 124.90 -5.55 121.51 -2.159
13 1,1,2-trimethylcyclopentane 104.89 108.42 -3.53 110.08 -5.18 106.86 -1.967
14 propylcyclopentane 130.95 128.89 2.06 129.68 1.27 136.57 -5.621
15 2-cyclopropylpentane 117.74 122.07 -4.33 122.09 -4.35 123.66 -5.92
16 cis-bicyclo[4.3.0]nonane 166 175.46 -9.46 164.38 1.62 164.59 1.41
17 1,1-dimethyl-2-ethylcyclopentane 138 138.53 -0.53 138.78 -0.78 138.33 -0.33
18 1,1-dimethylcyclopentane 133 138.53 -5.53 139.46 -6.46 133.37 -0.37
19 cis-1,3-diethylcyclopentane 150 152.18 -2.18 152.91 -2.91 150.87 -0.87
20 butylcyclopentane 156.6 159.00 -2.40 160.22 -3.62 163.27 -6.67
21 tert-butylcyclopentane 144.85 138.53 6.32 140.05 4.80 138.18 6.67
22 dicyclobutylmethane 161.8 175.46 -13.66 164.47 -2.67 152.11 9.69
23 1,5-dimethylspiro[3.3]heptane 132.2 154.99 -22.79 135.25 -3.05 142.44 -10.24
24 4-methylspiro[5.2]octane 149 161.81 -12.81 155.20 -6.20 151.49 -2.49
25 2,6-dimethylbicyclo[3.2.1]octane 164.5 191.92 -27.42 165.4 -0.90 165.41 -0.91
26 3,7-dimethylbicyclo[3.3.0]octane 166 191.92 -25.92 166.03 -0.03 165.6 0.4

5. Conclusiones
En el presente trabajo, esta nueva aproximación topológica-molecular es aplicada a moléculas que
contienen ciclos. Los modelos QSPR obtenidos utilizando este enfoque pueden ser considerados
estadísticamente significativos y describen adecuadamente la b.p de los compuestos incluidos, tanto en la
serie de entrenamiento como de predicción. Las ecuaciones obtenidas emplean solo términos lineales.
6. References
1. Rouvray, D. New Scientist 1993, May 29, 35-38.
2. Balaban, A. T. J. Chem. Inf. Compt. Sci. 1985, 25, 334-343.
3. Hansen, P. J.; Jurs, P. C. J. Cem. Educ. 1988, 65, 574-580.
4. Quadratic Index for the “Adjacency Matrix of the vertexes of a Molecular Pseudograph”. 1. Definition
and Applications to the Prediction of Boiling Point of Hydrocarbons. Submitted for publicate
5. Quadratic Index for the “Adjacency Matrix of the vertexes of a Molecular Pseudograph”. 2. Local
Definition and Applications to the Prediction of Boiling Point of alkyl alcohols. Submitted for publicate
6. Estrada, E. J. Chem. Inf. Compt. Sci. 1998, 38, 23-27.
7. Marrero, Y., Romero, V. TOMOCOND program. Universidad central de las villas. 2002.
8. STATISTICA ver. 5.5, Statsoft, Inc. 1999.
9. Estrada, E. J. Chem. Inf. Comput. Sci., 1996, 36, 844.

Un modelo estructural para el cálculo de propiedades químico-físicas de compuestos orgánicos. La presión crítica
como función de índices topológicos y topográficos.
Z. Kunakbaeva y R. Carrasco
Centro de Química Farmacéutica, Ave. 21 y calle 200 Atabey, Ciudad de La Habana
Email: [email protected]
RESUMEN
En muchos casos prácticos, el mejor método de estimación de una propiedad químico-física puede no resultar el mejor para
los objetivos que el investigador persigue. Por eso, resulta útil disponer de diferentes métodos de estimación de la
propiedad o propiedades en cuestión. Por otra parte, los índices topológicos y topográficos son descriptores estructurales
que han demostrado su versatilidad en la química medicinal. Son de cálculo fácil y bajos requerimientos computacionales.
Dado que todas las propiedades macroscópicas de las sustancias están relacionadas con la estructura molecular, es posible
asociar los índices mencionados con propiedades químico-físicas. La temperatura crítica es una propiedad de uso frecuente
en trabajos de química fina. Existen diferentes modelos de estimación de dicha propiedad. El método de Ambrose calcula la
temperatura crítica por una técnica de contribución de grupos y en función de la temperatura de ebullición experimental. En
el presente trabajo se propone un método basado en índices topológicos y topográficos para la estimación de la temperatura
crítica. Se parte de una función topológico-topográfica de la temperatura de ebullición de los compuestos la cual se incluye
como una variable más. Por el método de regresión múltiple a una muestra de más de 400 compuestos orgánicos de tres ó
más átomos pesados se desarrolla un modelo predictivo de esta propiedad con un coeficiente de correlación superior a 0.9 y
un error relativo del 5%. La ventaja principal del método es que no requiere de ningún tipo de medición experimental y los
descriptores son de fácil implementación computacional.
INTRODUCCIÓN.
Uno de los aspectos más relevantes de la química es su capacidad para crear nuevas sustancias que eran desconocidas
previamente y quizás insospechadas por el hombre. Muchos de los estudios encaminados a racionalizar el proceso de
búsqueda de nuevas moléculas con propiedades deseadas están basados en el desarrollo de relaciones cuantitativas entre la
estructura química y las propiedades en cuestión. El problema fundamental de estas relaciones radica en el hecho de que las
propiedades son por lo general expresadas por medidas cuantitativas, o sea por números, mientras que la estructura química
es un concepto de naturaleza no numérica. El problema entonces consiste en desarrollar métodos que permitan expresar
numéricamente la estructura química y convertir las relaciones estructura-propiedades en relaciones entre dos series de
números, unos representando la estructura química y los otros las propiedades.
Desde el siglo XIX Arthur Cayley y James Sylvester plantearon la idea de que los grafos no son sólo una herramienta
válida para la representación de las estructuras químicas, sino que estos pueden brindar invariantes que son características
de las especies representadas. Las aplicaciones de la teoría de grafos a los estudios de relaciones cuantitativas estructura-
propiedades están basadas fundamentalmente en la utilización de descriptores grafo-teóricos, llamados índices topológicos,
para codificar información estructural.
Un índice topológico es aquel índice numérico derivado de una representación grafo teórica que caracteriza la molécula
como un todo y que es calculado a partir de la matriz de conectividad entre los átomos pesados de la molécula. La
ponderación de estas matrices por valores como las distancias de enlace o las densidades de carga calculadas por un método

teórico les confiere características tridimensionales y se les considera topográficos. Tanto los unos como los otros son
siempre correlacionados con alguna propiedad químico-física y sus aplicaciones principales han estado enfocadas a los
estudios QSAR. Eso hace que muchos especialistas en química medicinal cuenten con descriptores estructurales de este tipo
dentro de su arsenal de variables aplicables a los respectivos problemas. No resulta entonces desacertado tratar de establecer
modelos cuantitativos de relaciones estructura–propiedades químico-físicas con estas variables de fácil manejo, que le
permita a estos especialistas emplear dichos modelos en la predicción de algunas de ellas a sus compuestos objetos de
estudio en una primera aproximación. En el presente trabajo se establecen modelos de regresión lineal múltiple de la
temperatura de ebullición y la temperatura crítica de una amplia muestra de compuestos orgánicos de diferente naturaleza.
En el caso de la temperatura crítica se establecerá un modelo de regresión en función de la temperatura de ebullición
calculada previamente con el modelo correspondiente.
MATERIALES Y MÉTODOS.
Se trabajó con una muestra total de 433 compuestos orgánicos1 con valores experimentales de la temperatura de
ebullición (422 comp.) y la temperatura crítica (431 comp.). Para la descripción estructural de los compuestos se les
calcularon los índices topológicos y topográficos de Estrada (ε, ερ, Ω, Ωq, ΩqC) y los topológicos de Randi (χ, χV) con el
programa MODEST2. El método de cálculo químico-cuántico utilizado para la ponderación de las matrices de conectividad
correspondiente fue el PM33,4 incluido dentro del programa MOPAC 6.05,6. Como entrada de datos a este programa se
utilizó la matriz Z de salida de cada estructura optimizada por mecánica molecular con el método MM+ incluido en el
programa HyperChem7. Además de estos índices, se emplearon como variables independientes el peso molecular (M) y el
Area de Superficie Accesible al Disolvente calculada por el método DelPhi (ADELP). El tratamiento estadístico se realizó
con el paquete Statistica8 empleando la técnica de regresión por pasos. Como criterios para la exclusión de outliers se
determinó que si un compuesto resultaba outlier en tres criterios entre el residual estándar, la distancia de Mahalanobis, la
distancia de Cook, el residual borrado y el por ciento de error, el compuesto era excluido y se reiniciaba el análisis.
RESULTADOS Y DISCUSIÓN.
Modelo de regresión de la temperatura de ebullición. Por el método de Análisis de Regresión por pasos se obtiene la
ecuación 1 después de la exclusión de 10 outliers. Es una ecuación en función de diferentes parámetros que incluyen un
descriptor desarrollado recientemente9. En la tabla 1 se presenta un resumen del resultado alcanzado.
TB= 211.28(±5.80) - 141.42(±27.38) 2MRχ3 + 0.97(±0.09) M + 80.67(±36.97) 3MRχ3 - 83.00(±9.25) 1Ωqc3 +
130.79(±11.36) 1ε 3 - 344.92(±37.06) MRχ3 - 0.09(±0.02)W3D + 38.05(±3.60) 2χ - 51.70(±9.46) Ωq3
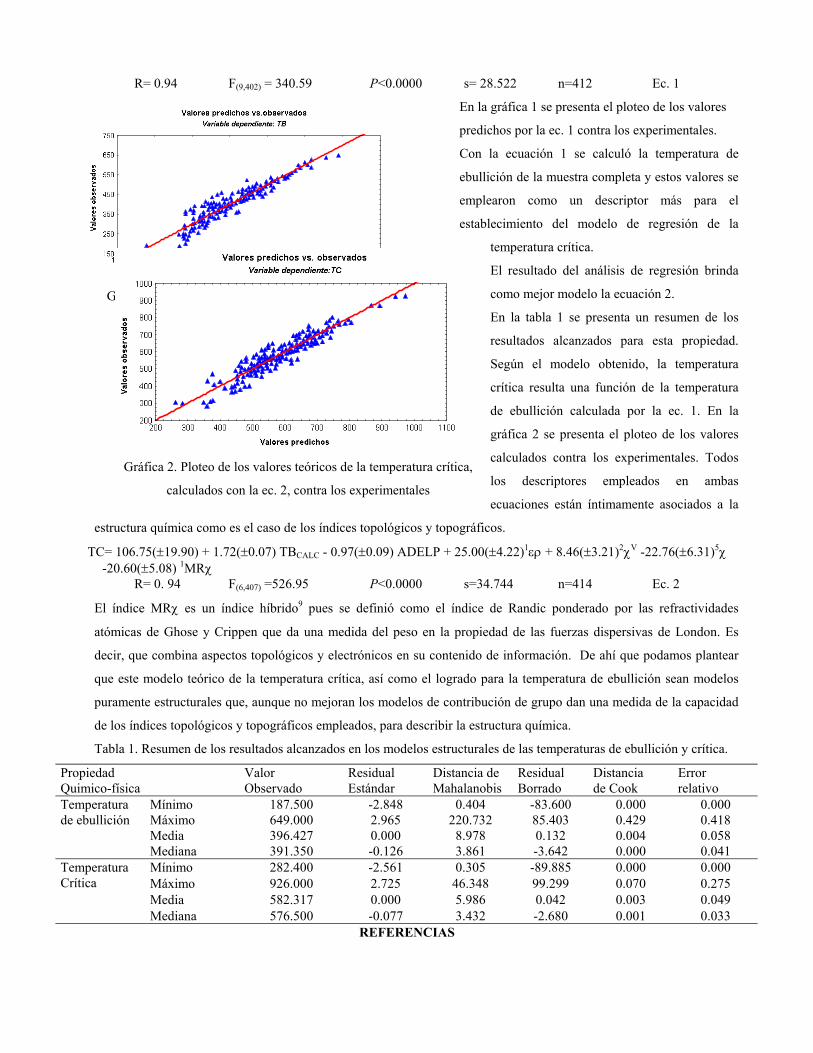
R= 0.94 F(9,402) = 340.59 P<0.0000 s= 28.522 n=412 Ec. 1
En la gráfica 1 se presenta el ploteo de los valores
predichos por la ec. 1 contra los experimentales.
Con la ecuación 1 se calculó la temperatura de
ebullición de la muestra completa y estos valores se
emplearon como un descriptor más para el
establecimiento del modelo de regresión de la
temperatura crítica.
El resultado del análisis de regresión brinda
como mejor modelo la ecuación 2.
En la tabla 1 se presenta un resumen de los
resultados alcanzados para esta propiedad.
Según el modelo obtenido, la temperatura
crítica resulta una función de la temperatura
de ebullición calculada por la ec. 1. En la
gráfica 2 se presenta el ploteo de los valores
calculados contra los experimentales. Todos
los descriptores empleados en ambas
ecuaciones están íntimamente asociados a la
estructura química como es el caso de los índices topológicos y topográficos.
TC= 106.75(±19.90) + 1.72(±0.07) TBCALC - 0.97(±0.09) ADELP + 25.00(±4.22)1ερ + 8.46(±3.21)2χV -22.76(±6.31)5χ -20.60(±5.08) 1MRχ
R= 0. 94 F(6,407) =526.95 P<0.0000 s=34.744 n=414 Ec. 2
El índice MRχ es un índice híbrido9 pues se definió como el índice de Randic ponderado por las refractividades
atómicas de Ghose y Crippen que da una medida del peso en la propiedad de las fuerzas dispersivas de London. Es
decir, que combina aspectos topológicos y electrónicos en su contenido de información. De ahí que podamos plantear
que este modelo teórico de la temperatura crítica, así como el logrado para la temperatura de ebullición sean modelos
puramente estructurales que, aunque no mejoran los modelos de contribución de grupo dan una medida de la capacidad
de los índices topológicos y topográficos empleados, para describir la estructura química.
Tabla 1. Resumen de los resultados alcanzados en los modelos estructurales de las temperaturas de ebullición y crítica.
Propiedad Valor Residual Distancia de Residual Distancia ErrorQuimico-física Observado Estándar Mahalanobis Borrado de Cook relativo
Mínimo 187.500 -2.848 0.404 -83.600 0.000 0.000Máximo 649.000 2.965 220.732 85.403 0.429 0.418Media 396.427 0.000 8.978 0.132 0.004 0.058
Temperaturade ebullición
Mediana 391.350 -0.126 3.861 -3.642 0.000 0.041Mínimo 282.400 -2.561 0.305 -89.885 0.000 0.000Máximo 926.000 2.725 46.348 99.299 0.070 0.275Media 582.317 0.000 5.986 0.042 0.003 0.049
TemperaturaCrítica
Mediana 576.500 -0.077 3.432 -2.680 0.001 0.033REFERENCIAS
Gráfica 1. Ploteo de valores predichos vs observados para la
temperatura de ebullición
Gráfica 2. Ploteo de los valores teóricos de la temperatura crítica,
calculados con la ec. 2, contra los experimentales

1 Reid, R. C.; Prausnitz, J. M y Poling, B. E.; “The Properties of Gases and Liquids”, McGraw-Hill Book Co., New York,4th Ed. (1987)2 Rodríguez L., Estrada E., Muñoz I. y Gutiérrez Y., MODEST (Molecular DESign Tools) 2.0. Universidad Central deLas Villas: Santa Clara, (1994)3 J.J.P. Stewart, J.Comp.Chem.,10, 209(1989)4 J.J.P. Stewart, J.Am.Chem.Soc., 10 221(1989)5 J. J. P. Stewart, Manual del MOPAC 6.0. QCPE # 5816 J. J. P. Stewart, J. Comp. Aid. Mol. Design, 4, 1, 1(1990)7 HyperChem. Versión 3 para WINDOWS. Molecular Modeling System. Hypercube, Inc y Autodesk, Inc. 19938 Statistica for Windows, Release 5.0, Copyright StatSoft, Inc. 1984-19859 Padrón, J.A.; Carrasco, R. y Pellón, R.F.; J. Pharm. & Pharmac. Sci., Aprobada

NUEVOS MÉTODOS PARA LA SÍNTESIS DEL 7β-FENILACETAMIDO-3-HIDROXIMETIL-3-CEFEM-4-
CARBOXILATO DE DIFENILMETILO.
Z. Rodríguez, M. González, B. Tolón, R. Avila, O. Díaz, H. Vélez, M. A. López.(Centro de Química Farmacéutica).
RESUMEN
El 7β-fenilacetamido-3-hidroximetil-3-cefem-4-carboxilato de difenilmetilo (III) es un intermediario utilizado durante la
síntesis de antibióticos cefalosporánicos. En este trabajo se desarrollaron 2 procedimientos para la síntesis de III que
implicaron el aislamiento del derivado 3-hidroximetilo, luego de la hidrólisis básica del ácido 7β-aminocefalosporánico
(7-ACA), seguido por la fenilacetilación de la función amino en medio orgánico utilizando N,O-bis(trimetilsilil)acetamida
(BSA) para disolver el núcleo y cloruro de fenilacetilo o el ácido fenilacético activado por el Reactivo de Vilsmeier como
agentes acilantes. Ambos métodos permitieron alcanzar rendimientos del intermediario superiores en un 11 % y un 18 %
respectivamente, al que se logra cuando se utiliza el procedimiento previamente publicado en la literatura.
INTRODUCCIONEl 7β-fenilacetamido-3-hidroximetil-3-cefem-4-carboxilato de difenilmetilo (III) es un intermediario utilizado durante la
síntesis de antibióticos cefalosporánicos, el cual se obtiene a partir del 7-ACA por hidrólisis básica del grupo acetoximetilo,
protección de la función amino por fenilacetilación y bloqueo de la función ácida por formación del éster de difenilmetilo
con difenildiazometano. Los rendimientos que se obtienen son muy bajos, hecho que influye negativamente sobre los
rendimientos que se alcanzan durante la preparación de estos antibióticos. El objetivo del trabajo fue desarrollar métodos
para la preparación de III que permitan obtener rendimientos más elevados de este importante intermediario.
MATERIALES Y METODOS
La cromatografía de capa delgada (CCD) se realizó en placas de gel de sílice GF-254 (Merck), acetato de
etilo/etanol/agua/ácido fórmico (60:25:15:1) como fase móvil y luz UV (254 nm) para visualizar los cromatogramas. Las
temperaturas de fusión (Tf) se determinaron en un equipo Gallenkamp y no fueron corregidas. Los espectros 1H RMN fueron
registrados en un equipo Bruker AC 250F con dimetilsulfóxido deuterado (DMSO-d6) como disolvente y TMS como referencia.
Difenildiazometano (I). Se obtuvo por oxidación de benzofenona hidrazona con cloramina T, según métodos reportados1,2
Acido 7β-amino-3-hidroximetil-3-cefem-4-carboxílico (II). Se suspenden 9,0 g (33 mmol) de 7-ACA en 60 mL de agua y
60 mL de metanol (MeOH), se enfría hasta -20 OC y se adicionan 7 mL de una disolución de NaOH 10 mol/L. Se agita 25
min. entre -20 y -10 OC y se ajusta a pH 3 con HCl al 37 %. El sólido formado se separa por filtración, se lava con MeOH,
acetona y éter dietílico y se seca a 40-50 OC durante 1 h.
7β-fenilacetamido-3-hidroximetil-3-cefem-4-carboxilato de difenilmetilo (III). (Método reportado). Se suspenden 16,0
g (59 mmol) de 7-ACA en 64 mL de agua,. se enfría hasta 2-5 OC y se añaden 5,2 g (130 mmol) de NaOH en 26 mL de
agua. Se agita 5 min., se ajusta a pH 8,5 con HAc y se añaden 48 mL de acetona. Se enfría a 0-5 OC y se adicionan 11,0 g
(71 mmol) de cloruro de fenilacetilo en 11 mL de acetona, mientras el pH se mantiene entre 7,5-8,5 con trietilamina (TEA).
Se agita 1 h a 0-5 OC, la acetona se evapora al vacío. y la mezcla se extrae con acetato de etilo (AcOEt) (1 x 220 mL y 1 x
100 mL) previo ajuste del pH a 3,5 con HCl 6 mol/L. Los extractos orgánicos se combinan, se lavan con disolución saturada
de NaCl (2 x 80 mL), se secan sobre Na2SO4,y se filtra. Sobre el filtrado se adicionan 13,77 g (71 mmol) de I en 50 mL de
AcOEt y se agita 1 h a temperatura ambiente. Se concentra hasta 50 mL y se enfría a 0-5 OC por 12 h. El precipitado que se
obtiene se separa por filtración, se lava con AcOEt (3 x 25 mL) y se seca a 40 OC durante 2 h.

Acilación con cloruro de fenilacetilo en medio orgánico. Se suspenden 5,0 g (21,7 mmol) de II en 50 mL de N,N-
dimetilacetamida (DMA), se añaden 16 mL (57,8 mmol) de N,O-bis(trimetilsilil)acetamida (BSA) y se agita 30 min. Se
enfría hasta -30 OC, se adicionan 3,5 mL (26,4 mmol) de cloruro de fenilacetilo y se agita 90 min. entre -20 y -10 OC. La
disolución se vierte sobre 200 mL de agua-hielo y se extrae con AcOEt (1 x 100 mL y 1 x 45 mL). Los extractos orgánicos
se combinan, se lavan con disolución saturada de NaCl (2 x 35 mL), se secan sobre Na2SO4 y se filtra. Sobre el filtrado se
adicionan 5,12 g (26,4 mmol) de I en 25 mL de AcOEt y se agita 1 h a temperatura ambiente. Se concentra hasta 25 mL y se
enfría a 0-5 OC por 12 h. El precipitado formado se filtra, se lava con AcOEt (3 x 25 mL) y se seca a 40 OC por 2 h.
Acilación con ácido fenilacético activado con el Reactivo de Vilsmeier.
a) Preparación del Reactivo de Vilsmeier y activación del ácido fenilacético. Se disuelven 2,7 mL (35 mmol) de DMF
en 30 mL de THF, se enfría hasta 0-5 OC, se adicionan 3,3 mL (36 mmol) de oxicloruro de fósforo y se agita durante 30
min. a 0-5 OC. Sobre el Reactivo de Vilsmeier se adicionan 4,33 g (31,8 mmol) de ácido fenilacético y se agita 1 h a 0-5 OC.
b) Acilación. Se suspenden 7,33 g (31,87 mmol) de II en 70 mL de THF, se calienta hasta 40 OC, se adicionan 24 mL
(98,16 mmol) de BSA y se agita 5 min. Se enfría hasta -30 OC y se añade la disolución del ácido fenilacético activado con el
Reactivo de Vilsmeier. Se agita 1 h entre -10 y -20 OC, se vierte sobre 200 mL de agua y se extrae con AcOEt (1 x 145 mL y
1 x 66 mL). Los extractos orgánicos se combinan, se lavan con agua (3 x 50 mL), seguido por 50 mL de disolución saturada
de NaCl, se seca sobre Na2SO4 y se filtra. Sobre el filtrado se adicionan 7,42 g de I en 35 mL de AcOEt y se agita 1 h a
temperatura ambiente. Se concentra hasta 37 mL y se enfría a 0-5 OC durante 12 h. El precipitado formado se filtra, se lava
con AcOEt (3 x 25 mL) y se seca a 40 OC durante 2 h.
RESULTADOS
En la Figura 1 se muestra el esquema para la síntesis de III y en Tabla 1 los resultados obtenidos:
N
SH2N
O
O CH3
O
O OH
N
SH2N
O
O OH
OH N
SNH
O
O OCHPh2
OH
O
7-ACA II III
2
3
45
67
8
910
11
12
Figura 1. Esquema general para la síntesis del compuesto III
Tabla 1. Rendimientos obtenidos durante la preparación del compuesto III
Agente acilante Catalizador Disolvente Respecto II (%) Respecto 7-ACA (%)cloruro de fenilacetilo (método reportado) TEA acetona-agua - 32cloruro de fenilacetilo BSA DMA 51,9 42,6ácido fenilacético / Reactivo Vilsmeier BSA THF 60,4 49,6
Las constantes físicas y espectroscópicas de los productos obtenidos según la 3 técnicas utilizadas fueron iguales y
coincidieron con las reportadas en la literatura. Tf: 178-180 OC; 1H-RMN (δ ppm): 3,52 y 3,57 (2H,ABq,H-10); 3,63 (2H,s,H-
2); 4,23 (2H,d,H-12); 5,10 (1H,d,H-6); 5,18 (1H,t,OH-12); 5,73 (1H,dd,H-7); 6,91 (1H,s,H-11); 7,20-7,55 (15H,m,aromáticos);
9,14 (1H,d,NH-7)
DISCUSION
Acilación con cloruro de fenilacetilo en acetona acuosa (técnica reportada)

En primer lugar se ensayó el procedimiento reportado en la literatura3, donde como paso inicial se efectuó la hidrólisis
básica del grupo acetoximetilo por tratamiento del 7-ACA con NaOH en agua a 0-5 OC para obtener II, el cual no se aisló
del medio de reacción. A continuación, se protegió el grupo amino por acilación con cloruro de fenilacetilo en acetona
acuosa y mediante el uso de TEA como aceptor del ácido formado y como último paso se bloqueó la función ácida por
tratamiento con I. El rendimiento que se obtiene no supera el 32 %, hecho que puede tener varias causas, la primera de las
cuales podría estar relacionada con la hidrólisis básica del 7-ACA, ya que las condiciones bajo las que se realiza dificultan
el control de la reacción. El uso de un medio de reacción acuoso, obliga a utilizar temperaturas de reacción superiores a
0 OC e implica tiempos de reacción muy cortos (5 min.) para evitar al máximo la degradación del núcleo cefalosporánico.
Por otra parte, el análisis por CCD de la mezcla al concluir la fenilacetilación, permitió comprobar la presencia de 3
manchas, una asignable al derivado fenilacetilado de interés y las dos restantes debidas al compuesto que resulta de la
esterificación del grupo hidroxilo de II y al derivado difenilacetilado, por la función amino y por el grupo hidroxilo. La
posible descomposición parcial del cloruro de fenilacetilo en el medio de reacción acuoso, parece ser otra causa del bajo
rendimiento que se obtiene mediante esta técnica. Como consecuencia, se desarrollaron 2 procedimientos para la síntesis de
III que posibilitaran mejorar los rendimientos de este intermediario de reacción y que reunieran los siguientes requisitos:
condiciones de reacción durante la hidrólisis básica del 7-ACA que permitan ejercer un mejor control durante la preparación
de II; efectuar la fenilacetilación en un medio orgánico para reducir la descomposición de los agentes acilantes empleados y
proteger por sililación el grupo hidroxilo de II para de evitar reacciones colaterales indeseables.
La hidrólisis básica del 7-ACA se realizó por tratamiento con NaOH en MeOH acuoso, lo que permitió emplear
temperaturas más bajas (- 10 y - 20 OC) y extender el tiempo de reacción hasta 25-30 min. De esta forma se logró ejercer un
mejor control durante el proceso y aislar II (Rendimiento: 82 %), paso necesario para implementar los restantes requisitos.
Acilación con cloruro de fenilacetilo en medio orgánico. En este método, II se trató con BSA en DMA a temperatura
ambiente. El núcleo disuelto se trató con cloruro de fenilacetilo y se efectuó la protección final de la función ácida con I.
Con respecto al 7-ACA se alcanzó, un rendimiento de 42,6 %, superior en casi un 11 % a cuando se empleó acetona acuosa.
Acilación con ácido fenilacético activado por el Reactivo de Vilsmeier. Como en el caso anterior se partió de II, el cual
se disolvió en THF por reacción con BSA a 40-45 oC. La acilación se realizó con ácido fenilacético activado por el Reactivo
de Vilsmeier y se obtuvieron rendimientos, respecto al 7-ACA, de 49,6 %, es decir superior casi en un 18 % a cuando se
utilizó el procedimiento reportado. Esta técnica fue la más eficaz, ya que en este caso se genera in situ el cloruro de
fenilacetilo4 y se reducen sus posibilidades de descomposición. La CCD efectuada durante fenilacetilación en los 2 métodos
desarrollados, permitió comprobar la presencia de una sola mancha en el cromatograma, asignable al derivado fenilacetilado
de interés, ya que la sililación de II bloquea al grupo hidroxilo y evita la formación de derivados fenilacetilados colaterales,
hecho que contribuye decisivamente al incremento de los rendimientos que se observó en ambos casos.
BIBLIOGRAFIA
1. Adamson, J. R., Bywood, R., Easlick, D. T., Gallagher, G., Walker, D., Wilson, E. M. Amino-acids and peptides. Part II.
A new method for preparation of diphenildiazomethane and related compounds. J. C. S. Perkin I, 2030-2033, (1975).
2. López, M. A., Rodríguez, Z., González, M., Valdés, B., Vélez, H., Agüero, J., Fini, A. Improvements of the síntesis of
diphenylmethyl 7β-(o-hydroxy)benzylidene amino-3-hydroxymethyl-3-cephem-4-carboxylate. Il Fármaco, 000, 1-3, (2001).
3. Takasugi, H., Takaya, T., Murakawa, T., Nakano, H. Studies on β-lactam antibiotics. III. Synthesis and enzymatic
stability of 3-acyloxymethyl-7β-[(Z)-2-(2-amino-4-thiazolyl)-2-(methoxymino)acetamido]-3-cephem-4-carboxylic acids. J.
Antibiotics, Vol. 34 (10), 1300-1318, (1981).

4. Just, C., in Iminium Salts in Organic Chemistry, H. Böhme and H. G. Viehe (Eds.), Vol. 9, in Advances in Organic
Chemistry: Methods and Results, Wiley-Interscience, New York, 225-342, (1976).