revista hondureÑa de - cidbimenacidbimena.desastres.hn/rhn/pdf/1999/pdf/vol3-1-1999.pdfcongreso...
TRANSCRIPT


REVISTA HONDUREÑA DE NEUROCIENCIAS
Publicación oficial de la Asociación Hondureña de Neurología
ISSN 1029-1911
Asociada a la Federación de Revistas Neurológicas en Español
Director y Editor Dr. Marco Tulio Medina Hernández Editor Asociado Dr. Ricardo Madrid Asistente de Editor Dra. Reyna Durón Martínez Consejo Editorial Dr. Winston Mejía Dr. Salvador Moneada Dr. Javier Sánchez M. Dr. Nicolás Nazar Dr. Efraín Bu F. Dr. Elmer Mayes Dr. Nelson Chinchilla Dr. Inf. Richard K. Haré Dr. Hermán Corletto Dr. Francisco León Gómez Dr. Américo Reyes Dr. Rolando Machado Dra. Janeth Bu F. Dra. Heyke Hesse Dr. Edgardo Girón Lic. Rosa Palencia Dr. Olman Arguedas Dr. Rene Valladares Transcriptor Br. Eddy Amaya Espinal Diseño y Diagramación: SCANCOLOR PREPRESS Impresión: SCANCOLOR Honduras, C.A.

Dr. Salvador Moneada Inglaterra
Dr. Gustavo Cosenza Guatemala
Dr. Carlos Díaz Manzano El Salvador
Dr. Manuel Carvajal L. Costa Rica
Dr. Franz Chávez S. Costa Rica
Dr. Antonio V. Delgado-Escueta Estados Unidos
Dr. Henry Stokes Guatemala
Dr. Ramiro Sirias Nicaragua
Dr. Amín Hassan Nicaragua
Dr. Fernando Gracia Panamá
Dr. Pierre Genton Francia
Dr. Osear H. Del Brutto Ecuador
Dr. Sergio Córdova L. México
Dr. Francisco Rubio Donnadieu México
Dr. Gregory Quirk Estados Unidos
Dr. Luis Salguero Guatemala
Dr. Charlotte Dravet Francia
Dr. Michelle Bureau Francia
Dr. Joseph Roger Francia
Dr. Jorge Matías-Guiu España

Agradecemos a la empresa HOSPITEC y al Ingeniero José
Roberto Dheming por el apoyo en la publicación de esta Revista.
Agradecemos a todos aquellos dentro y fuera de Honduras que
de una u otra manera hacen realidad la vigencia este valioso
proyecto.

REVISTA HONDUREÑA DE NEUROCIENCIAS
Publicación oficial de la Asociación Hondureña de Neurología
Págs.
8 Editorial
9 ARTÍCULOS ESPECIALES SPECIAL ARTICLES
PRIMER CONGRESO VIRTUAL IBEROAMERICANO DE NEUROLOGÍA
FIRST VIRTUAL IBERO AMERICAN CONGRESS OF
NEUROLOGY.
14 Status epilepticus: epidemiología, clasificación y tratamiento
Status epilepticus: epidemiology, classification and treatment Antonio Delgado-Escueta, Ch.- Y.G. Fong, Marco Tulio Medina Reyna Durón Martínez
22 Neurocisticercosis Neurocysticercosis Osear H. Del Brutto.
37 Participación del sistema nervioso periférico en la enfermedad
de Chagas Participation of the periferic nervous system in Chagas disease. Roberto Sica

45 RESUMENES/ABSTRACTS
IX Congreso Centroamericano de Neurología y
II Congreso Centroamericano de Neurofisiología Clínica
IX Central American Congress of Neurology and
II Central American Congress of Clinical Neurophysiology
58 Instrucciones a los Autores/Instructions for Authors

La Federación Centroamericana de Neurología representa el reflejo del desarrollo organizativo de la neurología en nuestra región. El Congreso Panamericano de Neurología organizado brillantemente por el Dr. Luis Salguero y colaboradores en la Ciudad de Guatemala en 1995, marca un hito en Centro América, tanto por la magnificencia de este evento como por sus logros. Durante el mismo, tanto la delegación costarricense como la hondureña obtuvieron la membresía como capítulos de la Federación Mundial de Neurología y se tomó la decisión de organizar el Congreso Centroamericano de Neurología y de Neurofisiología Clínica en San José, Costa Rica en diciembre de 1996, naciendo con ello la Federación Centroamericana de Neurología, bajo la Presidencia del Dr. Manuel Carvajal Lizano, un entusiasta unionista centroamericano.
Es durante el Congreso de 1996 que se da un mandato a la actual Junta Directiva para fortalecer la neurología en nuestro istmo. Durante la gestión de la misma, se ha logrado que Nicaragua fuese aceptada como Capítulo de la World Federation of Neurology (WFN), la aceptación de la Federación Centroamericana de Neurología como Capítulo Regional de la WFN, la aceptación de la Sociedad Panameña de Neurología y Neurocirugía como miembro de nuestra Federación y la participación activa de la delegación centroamericana durante el Congreso Mundial de Neurología en Buenos Aires, Argentina en septiembre de 1997.
Asimismo se obtuvo la promoción a nivel mundial por parte de la
WFN, de nuestro Congreso Centroamericano, el apoyo de nuestra Federación al Primer Congreso Virtual Iberoamericano de Neurología, la creación de una Federación de Revistas Neurológicas en Español bajo el 'Acuerdo de Buenos Aires', la inclusión del Dr. Salvador Moneada, descubridor del óxido nítrico y candidato a premio Nobel de Medicina, como Miembro Honorario de nuestra Federación, el impulso por parte de la WFN para la creación de Programas de Residencia de Neurología en Centroamérica con el apoyo de Theodore Munsat, Chairman de Educación, del Dr. Frank Clifford Rose, Secretario de la WFN y del Dr. Alberto Porteras-Sánchez, así como otras muchas actividades que no es posible enumerar aquí.
Consideramos que todos estos hechos han fortalecido a la Neurología Centroamericana a pesar de los valladares que hemos tenido que superar. El vice presidente de la WFN, Dr. Jun Kimura y los neurólogos centroamericanos, seremos testigos en San Pedro Sula en junio del presente año durante el Congreso Centroamericano de Neurología, del paso de la antorcha a una nueva Junta Directiva, que sabemos la llevará muy alto.
Tenemos que agradecer a muchas personas e instituciones que creen en nosotros, no hay espacio para mencionarles a cada uno, pero nuestro aprecio por su contribución es grande. Deseamos tomar las palabras del Divino Maestro cuando dijo 'la verdad os hará libres1, para expresar a los centroamericanos que el trabajar nos hará libres, el ser positivos nos hará libres... de nuestras limitaciones.

Revista Hondureña de Neurociencia Vol. 3 No. 1,1999
PRIMER CONGRESO VIRTUAL IBEROAMERICANO DE NEUROLOGÍA
INTRODUCCIÓN
Internet ha derribado las barreras geográficas, de tal forma, que esta herramienta de formación, comunicación e información permite "encuentros" de profesionales en un determinado ámbito, aunque estén situados geográficamente a muchos kilómetros de distancia. Lo que hasta ayer era algo difícil y eventual, un encuentro de los interesados en un mismo lugar y tiempo, hoy día es algo mucho más accesible y con muchas más posibilidades: la comunicación a través de la Internet.
De esta manera, ya es posible organizar "reuniones" científicas de diversa índole para transmitir como en los congresos tradicionales actualizaciones, enseñanzas, experiencias, ideas de cada uno o de cada grupo de trabajo, para compartir con los demás colegas la propia ciencia, aprender de la experiencia ajena y cooperar en el progreso de la disciplina científica. Cuando esa reunión es de considerable envergadura, se le llama Congreso Virtual.
No es necesario ponderar las ventajas añadidas de comodidad, duración, facilidad de conservación de las comunicaciones científicas, mayor ámbito de influencia, y sobre todo, un encuentro a través de Internet que obvia muchas de las dificultades propias de la movilización que, como personas y como trabajadores sufrimos: "nadie" puede estar en dos sitios a la vez.
Un Congreso Virtual permite el encuentro con un amplio número de profesionales que de otra forma no podrían participar en la experiencia. Se trata de una actividad innovadora que, como en los encuentros tradicionales, tiene algunas limitaciones, como el idioma o la masificación, que no incluye por el momento las actividades "sociales" habituales de los congresos, tal como los hemos conocido hasta la fecha.
Esta herramienta, sin ser nueva, es aún poco conocida y está escasamente desarrollada. De esto no puede hacerse una lectura negativa, sino que este supuesto handicap debe ser tomado como un reto, y recordar que en materia de computación en el ámbito médico, la Neurología ha sido pionera. Y trataremos de seguir siéndolo. Contamos para ello, con el apoyo de muchas instituciones y hemos reunido toda la experiencia posible, pero sobre todo, contamos con el colectivo más emprendedor de especialistas médicos, fuertemente cohesionados a través de otras actividades como es nuestra Lista de Distribución, y con una media de edad que nos permite decir que somos un grupo de jóvenes pioneros.
Nuestra Comunidad Virtual de Neurociencias, va a disponer para este Primer Congreso Virtual Iberoamericano de Neurología, de cuantos recursos podamos capitalizar, empezando por nuestro afán de superación, sin excluir a nadie. Afortunadamente disponemos también de las herramientas de UniNet, de las experiencias que nos han brindado los organismos de un congreso anterior, el Iberoamericano de Anatomía Patológica -coordinado en español-, la colaboración instrumental de la red nacional de Redlris, y esperamos la ayuda de diferentes entidades públicas y privadas.
Un Congreso Virtual es una reunión científica celebrada en Internet, en la que las aportaciones y trabajos científicos son publicados en forma de páginas web, y a la que se asiste mediante la lectura de dicha páginas y la discusión a través de correo electrónico y charlas interactivas en tiempo real con los autores. Aunque novedosa, no es una experiencia nueva y sus características de economía en la organización, gratuidad en la asistencia, difusión universal y posibilidades de participación, hacen de esta herramienta un instrumento de utilidad incuestionable y futuro brillante.

SOCIEDADES CIENTÍFICAS QUE RECONOCEN Y APOYAN EL CONGRESO
Sociedad Valenciana de Neurología Sociedad Catalana de Neurología Federación Centroamericana de Neurología Asociación Hondureña de Neurología Sociedad Española de Informática de la Salud Liga Española Contra la Epilepsia Sociedad Andaluza de Neurología Sociedad Murciana de Neurología Sociedad de Neurología de Castilla la Mancha Listas Médicas de Redlris Sociedad Argentina de Neurología Sociedad Española de Neurología Pediátrica Instituto Nacional de Neurología y Neurocirugía de México Sociedad Mexicana de Neurología Pediátrica, etc.
OBJETIVOS
Los objetivos de este I Congreso Virtual Iberoamericano de Neurología son:
1) Difundir universalmente la experiencia de la Comunidad Científica de habla hispana, en el campo de las Neurociencias e intercambiar conocimientos con la comunidad internacional, en especial, la de habla portuguesa.
2) Propiciar un ámbito de comunicación y relaciones entre los profesionales de ambos lados de Atlántico, principalmente entre España, Portugal e Iberoamérica; superadora de la barrera impuesta por el dominio anglosajón de la comunicaciones, y las diferencias de medios económicos en los países americanos de cultura hispánica.
3) Enriquecimiento y formación continuada de los profesionales implicados en el campo de la Neurociencias, que padecen aislamiento geográfico, residen en lugares con escasas oportunidades de formación continuada, o carecen de posibilidad de desplazamiento por la causa que sea. Este punto es especialmente importante ya que podría ser un valioso medio de enlace de esto profesionales con los centros de referencia y los profesionales más favorecidos.
4) Establecer lazos permanentes de relación entre los profesionales, más allá del espacio temporal del Congreso, propiciadores de transmisión fluida de la información, y cooperación en la formación y la comunicación de los profesionales dedicados a Neurociencias. Ello conllevará frutos importantes en la asistencia, docencia e investigación en nuestro ámbito.
5) Un incremento de la presencia de los profesionales de la Neurología en Internet, la herramienta más potente de comunicación, cuya era no ha hecho más que comenzar.
6) Desarrollo de experiencia de utilización de Internet como medio asistencial, docente, e investigador, al servicio de la Ciencia y la Cultura universal.
DESARROLLO
Las actividades del Congreso se desarrollarán desde el 15 de octubre al 30 de noviembre de 1998, pero la exposición de los trabajos se prolongará al menos un año.
Los resúmenes de las publicaciones, cursos y conferencias invitadas serán publicados por la Revista de Neurología y a través de la Federación Iberoamericana de Revistas de Neurología, que permitirán darle a la participación en el Congreso la consistencia curricular que implica la participación en cualquier congreso convencional.
Se verterán los contenidos íntegros del Congreso en soporte informático permanente (tipo CD-ROM) que será distribuido gratuitamente entre todos los profesionales participantes en el evento.
PARTICIPACIÓN
La inscripción de los participantes es libre y gratuita. El plazo para el envío de resúmenes de comunicaciones libre y pósters electrónicos terminó el 15 de julio de 1998. A través de la lista de distribución de correo electrónico, en el que se incluirá a todos los participantes, y de información en formato web a través del Boletín del Congreso, se irá informando puntualmente de las incidencias precongreso.

La participación remedará en parte las actividades de las reuniones científicas tradicionales, incluidas las actividades sociales. Además de conferencias invitadas y ponencias oficiales, habrá comunicaciones científicas "orales" con su correspondiente discusión, (a través de una red de TRC-Científica) y en póster (en forma web), sin más limitaciones que las impuestas por el Comité Científico.
Durante la fase precongresual, se proporcionará a los participantes que lo requieran, de las enseñanzas teóricas y prácticas para adiestrarles en el manejo de las herramientas de Internet, incluido el programa de conversación en tiempo real y diseño de páginas web. Se emitirán certificación que acredita la participación de los profesionales en el Congreso, para que adquiera un valor curricular.
CONVOCATORIA
La Convocatoria partió del Equipo de Moderación de la Lista Neurología. Nuestro proyecto, incluido dentro de la acción más amplia de la Comunidad Virtual de Neurociencias presentada ante la Redlris, recoge las anteriores experiencias desarrolladas por la Sociedad Española de Informática de la Salud (SEIS), entidad que actúa como copatrocinador, y encuentra su primer aval en la Revista de Neurología de España que posibilita la difusión del Congreso en los formatos convencionales y permite que la participación obtenga desde un punto de vista curricular y científico un impacto parecido a un Congreso Ordinario.
UNINet la red de servicios universitarios IRC presta su infraestructura de servidores de web y de IRC para dotar al Congreso de una mayor velocidad de acceso y la capacidad de interacción entre participantes y asistentes.
La iniciativa ha encontrado su aprobación por las diferentes Sociedades Científicas neurológicas iberoamericanas y los organizadores continúan su labor de explicación y difusión del proyecto, esperando la gradual adhesión al proyecto de nuevas sociedades hispano-portuguesas en el campo de las Neurociencias.
COMITÉ ORGANIZADOR
Está formado por:
• SANTIAGO MOLA CABALLERO DE RODAS. Coordinador General del Congreso. Coordinador de la Comunidad Virtual de Neurociencias de Redlris, Unidad de Neurología, Hospital del SVS "Vega Baja". Alicante, España, [email protected]
• MARCO T. MEDINA. Coordinador del Congreso Iberoamérica, Profesor de Neurología de la Universidad Nacional Autónoma de Honduras, Editor de la Revista Hondurena de Neurología. [email protected]
• ÁNGEL PÉREZ SEMPERE. Coordinador del Comité Científico y Secretaría del Congreso, Equipo de Moderación de la Lista "Neurología" de Redlris, Unidad de Neurología, Hospital de SVS "Vega Baja". Alicante, España, [email protected]
• ENRIQUE BOTIA PANIAGUA. Coordinador Técnico del Congreso, Coordinador de la Web de la Comunidad Virtual de Neurociencias de Redlris, Unidad de Neurología del Hospital General del INSALUD "La Mancha-Centro", Alcázar de San Juan. Ciudad Real, España. [email protected]
• JORGE MATIAS-GUIU GUIA. Coordinador de Publicaciones y Presupuestos, Director de la "Revista de Neurología", Jefe de Servicio de Neurología, Hospital Universitario de Alicante. Alicante, España. [email protected]
JAUME COLL CANTI. Coordinador del Curso de Formación del Congreso, Coordinador de la Web de la Sociedad Catalana de Neurología, Servicio de Neurología, Hospital "Germans Trías i Pujol". Badalona. Barcelona, España, [email protected]
• JAUME MORERA GUITART. Coordinador de la Web de la Sociedad Valenciana de Neurología, Unidad de Neurología del Hospital del SVS "La Marina Alta". Denia. Alicante, España, [email protected]
• CRISTÓBAL CARNERO PARDO. Servicio de Neurología del Hospital del SAS "Virgen

de las Nieves". Granada, España. [email protected]
• JULIO ÓSCAR PROST. Jefe de Sección de Neurofisiología, Servicio de Neurología. Hospital Municipal B. Rivadavia, Buenos Aires, Argentina, [email protected]
• J GUSTAVO VEGA GAMA. Unidad de Diagnóstico del Centro, Servicio de Neurología. Celaya, Guanajuato, México. [email protected]
• MARCIAL GARCÍA ROJO. Representante de la Sociedad Española de la Informática de la Salud (SEIS), Servicio de Anatomía Patológica, Complejo Hospitalario de Ciudad Real, [email protected]
• MARÍA JESÚS COMA. Representante de UNINet, Responsable de la Unidad de Investigación del Hospital General Yaguc, Servicio de Anatomía Patológica. Burgos. [email protected]
• DAVID EZPELETA ECHAVARRI. Representante del Área de Información del Congreso. Neurólogo. Madrid. [email protected]
COMITÉ CIENTÍFICO
Presidente Dr. Antonio Culebras
Secretario
Dr. Ángel Pérez Sempere
Miembros
Dr. Román Alberca Dr. Gregorio R.P. Albiusi Dr. Fernando Barinagamenteria Dr. Félix Bermejo Dr. Enrique Botia Dr. Antonio Bové Dr. José Bueri Dr. Juan Burguera Hernández Dr. Cristóbal Carnero Dr. Manuel Carvajal (Costa Rica) Dr. Carlos Casas Fernández Dr. José Castillo Dr. Nelson Chinchilla (Honduras) Dr. Jaume Coll Cantí Dr. Josep Dalmau Obrador Dr. Osear Del Brutto Dr. Carmina Díaz Marín Dr. Exuperio Diez Tejedor
Dr. Jacinto Duarte Dr. Conrado Estol Dr. Isidro Ferrer Dr. Otto J. Fustes Dr. Jesús Gómez-Plascencia Dr. Fernando Gracia (Panamá) Dr. Francesc Graus Dr. Josep María Grau Veciana Dr. Javier Jiménez Jiménez Dr. Luis Javier López del Val Dr. José Maestre Moreno Dr. José Feliz Marti-Massó Dr. Eduardo Martínez Vila Dr. Jordi Matías-Guiu Dr. Marco Medina (Honduras) Dr. Adolfo Minguez Dr. Santiago Mola Dr. José Manuel Moltó Jordá Dr. Jaume Morera Dr. Luis Guillermo Palacios Dr. Julio Osear Prost Dr. Pedro J. Serrano Dr. Carlos Singer
LOGOTIPO DEL CONGRESO
En el logotipo están representados todos los colores de las banderas de los países iberoamericanos, que centran su interés y se organizan en tormo a la "neurona" (neurología). Su "potencial de acción" se propaga por el axón (Congreso Virtual) a la comunidad científica.
CONFERENCIAS INVITADAS
En esta sección la organización del Congreso pretende dar cabida, en un formato flexible, pero en el que valen de modelo las normas generales de publicación en las Revista Tradicionales, a: 1) Los autores de líneas de investigación y
publicación de reconocido prestigio. 2) Aquellos trabajos de carácter epidemiológico
que den a conocer la realidad de la Neurología Iberoamericana. Nuestro objetivo es componer un retrato transparente de los contextos de trabajo diferentes.
3) Los tópicos más candentes de la especialidad, agrupados bajo la forma de debates generales, dando cabida a las posiciones, muchas veces enfrentadas. Pretendemos romper las barreras, a veces artificiales, entre las diversas especialidades que tienen que ver con la asistencia a los pacientes con problemas neurológicos.
4) Todas las iniciativas que los autores consideren de interés.

Conferencias invitadas
Áreas temáticas Coordinadores
Neuropediatria Dr. Gómez-Plasencia Trastornos del movimiento Dr. Javier Jiménez Jiménez Epilepsias Dr. Marco T. Medina Neuropsicología Dr. Cristóbal Camero Cefalea Dr. Manuel Carvajal Vascular Dr. Conrado Estol,
Dr. Jorge-Guiu Esclerosis múltiple Dr. Miguel Ángel Hernández Neuromuscular Dr. Carmina Díaz Martínez Infecciosas Dr. Osear del Brutto Asistencia en neurología Dr. Jaume Morera Sueño Dr. Antonio Bové Terapéutica y neuroyatrogenia Dr. Otto JH Fustes Perspectivas tecnológicas Dr. Pedro J Serrano Neurogenética Dr. Luis Guillermo Palacios Epidemiología Dr. AP Sempere Neurología en el Hospital General Dr. Santiago Mola Internet y Neurología Dr. Enrique Botia
Coordinador: Dr. Ángel P. Sempere.
CURSOS DE FORMACIÓN
En este apartado se desarrollarán cursos de formación para residentes y médicos generales. Tienen cabida aquí todas las áreas temáticas de la neurología en las que la organización considere que
existen recursos suficientes para la exposición ordenada y extensivas de determinados tópicos de la especialidad. Estamos convencidos de que la experiencia adquirida nos permitirá desarrollar nuevos proyectos en el campo de la telemedicina.
Áreas temáticas de los Cursos de Formación
Áreas temáticas Coordinador
• Patología neuromuscular Dr. Jaume Coll i Canti
• Cefaleas Dr. Feliu Titus Albareda
• Neurología tropical Dr. Marco T. Medina
• Trastornos del sueño Dr. Joan Santamaría
• Vascular cerebral Dr. Domingo Escudero
• Neurooncología Dres. J. Dalmau y F. Graus
• Trastornos del movimiento Dr. Jaume Kulisevki
• Demencia Dr. Secundino López Pousa
• Esclerosis múltiple Dr. Txomin Arbizu
Coordinador: Dr. Jaume Coll i Cantí.

Revista Hondureña de Neurociencia Vol. 3 No. 1,1999
I CONGRESO VIRTUAL IBEROAMERICANO DE NEUROLOGÍA CURSO DE FORMACIÓN EN EPILEPSIA
STATUS EPILEPTICUS: EPIDEMIOLOGÍA, CLASIFICACIÓN Y TRATAMIENTO
Antonio Delgado-Escueta (1), Ch.-Y.G. Fong (1), Marco Tulio Medina (1,2), Reyna Durón Martínez (2)
(1) California Comprehensive Epilepsy Program (2) Asociación Hondureña de Neurología (3) Unidad de Investigación Científica, Facultad de Ciencias
Médicas, Universidad Nacional Autónoma de Honduras
La Comisión de Epidemiología y Pronóst ico de la Liga Internacional Contra la Epilepsia ha definido el status epilepticus (SE) como una actividad epiléptica que dura más de 30 minutos o la presencia de dos o más crisis secuénciales sin recu-peración de la conciencia entre las crisis. En este capítulo se revisarán la historia, epide-miología clínica y se discutirán los protocolos de tratamiento exis-tentes. Además, se considerarán los conceptos sobre el daño cerebral consecuente al SE.
HISTORIA DEL STATUS EPILEPTICUS Y LOS SIMPOSIOS
INTERNACIONALES DE MARSELLA SANTA MÓNICA CALIFORNIA
Las primeras descripciones de convulsiones continuas se pueden
encontrar en tablillas cuneiformes neo-babilónicas escritas entre los años 718-612 A.C. Fue L.F. Calmeil quien adoptó el témino état de mal en 1874. El escribió en su tesis que "Hay momentos que en cuanto una crisis termina otra empieza, una siguiendo a la otra en sucesión, de tal forma que uno puede contar hasta 40 ó 60 crisis s i n interrupción: los pacientes llaman a esto état de-mal. El peligro es inminente, muchos pacientes mueren". El término status epilepticus se utilizó por primera vez en la traducción de Bazire de las cátedras de Trousseau sobre medicina clínica en 1867.
La historia moderna del SE probablemente inició cuando Gastaut, Roger y Lob organizaron el Primer Simposio Internacional sobre SE ("la Conferencia de Marsella") en los años 60. Aquí se definió al SE como "crisis convulsivas que se repiten con tanta frecuencia o que son tan
prolongadas que ocasionan un estado fijo y duradero de la condición epiléptica". El concepto también incluyó crisis que duraban más de 30 minutos, aun sin alteración de la conciencia.
En 1977, se organizó el Segundo Simposium Internacional (la "Conferencia de Santa Mónica). Durante la misma se clasificó el SE según se muestra en la Tabla 1.
Desde el simposium de 1977, se han reportado más de 100 casos de status no convulsivo y SE parcial. Además, Treiman y colaboradores han descrito el SE sutil, una forma generalizada de status EEG con mínimas manifestaciones mo-toras, usual mente observado en anoxia cerebra l o paro car -diorrespiratorio.
Presentado durante Primer Congreso Virtual Iberoamericano de Neurología, 1998 Dirección Electrónica: http://cvneuro.org

Tabla 1. Clasificación del status epilepticus de la
Conferencia de Santa Mónica, 1979.
Status convulsivo • tónico-clónico • mioclónico • tónico • clónico
Status no convulsivo • de ausencias • parcial complejo* • parcial continuo**
• La presentación clínica es un estado "crepuscular" prolongado.
** epilepsia partialis continua, la conciencia está preservada.
Durante el 111 Simposio Internacional de Status Epilepticus en Santa Mónica, California (1997), David M. Treiman y colaboradores presentaron los resultados de un estudio nacional en EUA que duró cinco años y que incluyó 550 pacientes con SE convulsivo. El comparó cuatro diferentes esquemas de tratamiento. Se mencionarán datos más adelante.
INCIDENCIA Y PREVALENCIA
La incidencia anual en los Estados Unidos estimada por Hauser es entre 60,000 y 250,000 casos por año, mientras que Shorvon calcula que ocurren entre 3 80 a 280 casos por millón en el Reino Unido. De Lorenzo reportó que la mayor incidencia ocurre en la infancia (156 por 100,000).
En los países en vías de desarrollo, donde reside el 80% de los 40 millones de individuos con epilepsia en el mundo, parece ser muy frecuente, aunque sus características han sido poco estudiadas en estas regiones. Un estudio realizado en 1997 en el municipio rural de Salamá,
Honduras por Medina y colaboradores, reporta que 47% de los pacientes epilépticos habían desarrollado un SE, siendo el convulsivo el tipo más común, fue más frecuente en niños y en 42% de los casos estaba relacionado a la ausencia de tratamiento antiepiléptico.
CAUSAS DE STATUS EPILEPTICUS
El Estudio de Honduras reporta que 43% de los pacientes con SE presentaban epilepsia sintomática a neurocisticercosis y el resto se debía a causas genéticas, daño cerebral perinatal y malfor-maciones del SNC. La etiología era desconocida en un 28% de los casos.
En países desarrollados, las causas más comunes de epilepsia en hospitales generales son: enfermedad cerebro vascular (25%), supresión de terapia anticonvulsivante (18%), abuso o abstinencia de alcohol (12%), deprivación de sueño, infecciones y el inicio de nuevos anticonvulsivantes que disminuyen los niveles plasmáticos de antiepilépticos concomitantes (12%).
Otras causas incluyen la abstinencia de otras drogas sedantes como las benzo-diacepinas y el fenobarbital, alteraciones metabólicas, hipocal-cemia, hipo- e hiperglicemia, hiponatremia, insuficiencia hepática o renal, intoxicaciones (isoniazida, antidepresivos tricí-clicos, neurolépticos, estricnina), actividad física extenuante, radioterapia y el embarazo o parto. En centros de epilepsia, las causas más comunes son daños estructurales como neoplasias, abscesos cerebrales, infarto agudo cerebral embólico, encefalitis,
meningitis, epilepsias refractarias y epilepsias mioclónicas pro-gresivas.
Los infantes y niños representan los grupos más afectados por SE. El 40% de los casos ocurren antes de los dos años de edad. En la niñez, la causa mas común es la infección aguda del sistema nervioso central (SNC) (36%). Cerca de 20% se debe a cambios en la medicación y las alteraciones hidro-electrolíticas son etiologías y desencadenantes comunes.
En aproximadamente 50% de los casos no hay historia de crisis epilépticas. Medina y col. reportan que 4.4% de los epilépticos de su estudio presentaron SE como primera manifestación de la epilepsia. El riesgo de SE recurrente es de 26% y aumenta si el status dura más de 24 horas. Cuando el SE es la primera manifestación de una alteración del SNC, el 66% de los pacientes desarrolla epilepsia. En niños ne urológicamente normales, el riesgo de recurrencia es bajo.
MECANISMOS
Los mecanismos moleculares y celulares que transforman una crisis única en un estado epiléptico fijo y progresivo son desconocidos tanto en humanos como en animales con SE experimental. Los mecanismos básicos por los cuales un status convulsivo lleva a epileptogénesis y a epilepsia aún son motivo de estudio. La mayoría de la evidencia de que el SE debe controlarse en 20 a 30 minutos, procede de animales con status experimental. Sin embargo, las técnicas modernas de bioquímica in vivo, imagen de resonancia magnética funcional, espec-troscopia con resonancia

magnética v diffusion-weighted imaging (una forma especial de IRM), nos ofrecen ahora la posibilidad de estudiar ios mecanismos de daño y las posibilidades de tratamiento del SE en humanos.
TRATAMIENTO
En 1977, durante el Primer Simposio Internacional sobre Status Epiléptico, en Santa Mónica, California, Escueta y Col. propusieron los principios de tratamiento y recomendaron el diazepam intravenoso (I.V.) para el control agudo de SE y la fenitoína I.V. para prevenir su recurrencia. Estos principios de tratamiento para el SE han sido aceptados mundialmente y han pasado la prueba del tiempo y el reto de nuevos ensayos comparativos. Los fármacos que han sido aprobados recientemente por la FDA (EUA) y que han tenido impacto en el tratamiento del SE el diazepam en gel rectal (Diastat), la fosfeniíoína I.V. y el Valproato I.V.
Principios del manejo de urgencia Se sugiere seguir los principios de manejo racional presentados por el II Simposio Internacional de Status Epilepticus (1980) y recomendados por el grupo de trabajo de la Epilepsy Foundation of America: • Documentación clínica del
SE • Identificación de la causa del
SE • Seguimiento del ABC de
sostén vital • Control de las crisis lo antes
posible
Documentación clínica del status epilepticus
Se debe suponer la presencia de SE e instaurar un tratamiento inmediato si se da alguna de las siguientes condiciones:
• Dos convulsiones tónico- clónicas sin que el paciente recupere la conciencia entre ellas,
• Crisis clónicas continuas durante al menos cinco minutos sin recuperación de la conciencia,
• Dos crisis parciales complejas sin que el paciente recobre la conciencia entre ellas.
Durante el período de observación se debe identificar el tipo de SE. Existen tantos tipos de SE como tipos de crisis epilépticas. El de tipo convulsivo y el parcial complejo requieren tratamiento agresivo e inmediato. Se deben diferenciar los de tipo tónico, clónico y el tónico-clónico, pues las drogas de elección son diferentes. Se debe realizar un electroencefalograma (EEG) de urgencia cuando se duda del diagnóstico o, particularmente, si
Parcial complejo 4.8% Mioclónico2.4% De ausencia 2.4%
se sospecha un status no convulsivo.
Identificación
de la causa o desencadenante del SE
Si se presenta un status tónico-clónico, hay que realizar una búsqueda de una lesión aguda del SNC (infarto cerebral agudo, meningitis, encefalitis, trauma cerebral severo, una masa progresiva como un absceso o neoplasia). Si hay ausencia de una lesión cerebral, se deben considerar causas menos comunes como anormalidades electrolí-ticas, accidentes cerebrovascu-lares antiguos, abandono de drogas anticonvulsivantes, causas metabolicas y anoxia cerebral por falla cardiopulmonar.
El examen neurológico puede demostrar signos de focalización. Se deben realizar exámenes rápidos de sangre para investigar hiper- o hipoglicemia, hipoxia, hiponatremia, hipocalcemia y uremia, además de exámenes toxicológicos en orina y sangre, particularmente para cocaína. También pueden medirse niveles séricos de antiepilépticos.
Figura 1 Estudio de Salamá, Honduras 1997
TIPO DE STATUS EPILEPTICUS (n=42)
Sec. generalizado Tónico - clónico 90.4%

Seguimiento del ABC de sostén vital
El tratamiento del SE empieza con las medidas que deben realizarse en todo paciente inconciente: el ABC del sostén vital (mantener la vía aérea, mantener la presión arterial y la circulación y además, monitorear la temperatura, la presión arterial, electrocardio-grama y función respiratoria). En estudios experimentales con animales, la severidad del daño cerebral se correlaciona muy bien con la hipoxia, hipotensión e hiper-pirexia, así como con la duración del SE.
Control de las crisis lo antes posible
Las convulsiones deben controlarse lo antes posible para prevenir el daño neuronal selectivo y las secuelas neurológicas serias. Los pacientes con SE no deben recibir agentes paralizantes para controlar las convulsiones sin una
Falla energética, hipoxia/isquemia
Aumento de glutamato Aumento de
la despolarización Aumento del
calcio intracclular Aumento del
lactato Activación de lipasas y
proteasas
monitorización cercana del EEG que asegure que las descargas neuronales anormales han sido controladas. Si estas descargas continúan por un tiempo suficiente pueden causar daño cerebral significativo.
El objetivo del tratamiento es detener toda actividad epiléptica y no sólo las manifestaciones clínicas. Además se deben corregir las complicaciones metabólicas secundarias.
En casi todos los casos el control de las crisis se logra con la administración endovenosa de benzodiazepinas. Si el SE convulsivo dura más de 60 minutos a pesar del tratamiento, el paciente debe ser llevado a la anestesia general. Cuando las convulsiones ceden, se deben prevenir posteriormente con fenitoína I.V. Las razones para impedir el progreso del SE más allá de los 20 ó 30 minutos residen en el daño cerebral demostrado en cerebros humanos afectados por SE convulsivo y que se resumen en la Figura 2.
Neurotoxicidad Disfunción
mitocondrial
Daño al hipocampo
Patología cortical
Necrosis celular
Apoptosis
Un estado hiperadrenérgico resultante de catecolaminas circulantes ocurre 30 minutos después del inicio del SE. El 60% de los SE presenta taquicardia, disrritmias severas y disrritmias potencialmente fatales. La acidosis y la hipoglicemia ocurren con mucha frecuencia. Otra razón importante de terminar un SE, especialmente en lactantes y niños es el potencial del SE para inducir epileptogénensis y epilepsia crónica y déficit neurológico.
Ensayos Farmacológicos
Si bien la droga ideal para el manejo del SE no existe, casi todos, si no todos los estudios, están de acuerdo en el uso de las benzodiazepinas I.V. (diazepam. lorazepam) para terminar el SE. Sin embargo, el estudio controlado que comparó feno-barbital versus la combinación diazepam-fenitoína por Shanner y col. concluyó que el fenobarbital es rápidamente efectivo, compa-rativamente seguro y con alguna ventaja práctica sobre la combinación diazepam/fenitoína.
El estudio doble ciego realizado por Treiman y col. que comparó lorazepam con fenitoína en el SE convulsivo, mostró una tasa de éxito de 20/26 (77%) para lorazepam como primera droga y de 6/10 (60%) como segunda droga, mientras que la fenitoína tuvo tasas de 12/22 (54%) y 5/6 (83%), respectivamente. Los pacientes con SE convulsivo general izado sut i l no respondieron a ninguna de las drogas. El lorazepam fue tan seguro como el diazepam.
Leppik y col. mostraron que el lorazepam produjo depresión respiratoria en igual número de pacientes que el diazepam. Pueden ocurrir efectos
Figura 2. EVENTOS DURANTE EL S.E. QUE RESULTAN EN DAÑO CEREBRAL

secundarios como aletargamiento, ataxia- confusión, agitación y alucinaciones pero no son irreversibles. En los casos manejados con lorazepam, 3 pacientes con ausencias atípicas presentaron status tónico paradójico y uno presentó paro respiratorio transitorio. Aparentemente, el lorazepam no produce efectos secundarios cardíacos como la fenitofna.
Treiman y col. completaron en 1997 un ensayo comparativo nacional en Estados Unidos sobre el tratamiento I.V. del SE convulsivo con lorazepam vrs. diazepam/fenitoína vrs. fenobarbital vrs. fenitoína en 384 SE obvios y 134 SE sutiles. Los resultados del SE obvio no muestran diferencia entre los esquemas, excepto para la fenitoína sola, la cual fue estadísticamente menos efectiva que los otros esquemas. Un dato interesante fue que el fenobarbital resultó ser superior en el manejo de SE sutiles por anoxia cerebral o fallo cardiorrespiratorio. El éxito general del tratamiento fue de 55?5%, de 20 a 30% menos efectivo que en las series de SE en niños, adolescentes y adultos jóvenes. Sorprendentemente, no hubo diferencias en complicaciones como hipoventilación, hipotensión, arritmias cardíacas y paro cardíaco entre los cuatro esquemas de tratamiento.
Manejo inicial
Las tres opciones actuales en el manejo inicial del SE tónico-clónico generalizado son:
• Diazepam en bolo T.V. y una infusión lenta de fosfenitoína, o
• Lorazepam en bolo T.V., o • Fenobarbital I.V.
Estos deben acompañarse del uso intravenoso de dextrosa al 50% (bolo de 50cc), tiamina (bolo de lOOmg), piridoxina (bolo 100-200mg en niños menores de 18 meses) y 1 a 2 ampollas de bicarbonato de acuerdo al resultado de los gases arteriales.
ANTIEPILEPTICOS ESPECÍFICOS
Diazepam
Si la terapia se inicia con diazepam, éste cruza la barrera hematoencefálica inmediatamente y controla el SE en minutos, pero se redistribuye rápidamente a otros sitios del cuerpo, disminuyendo !a concentración cerebral y causando recurrencias en un tercio de los pacientes. Por lo tanto, casi siempre es necesario iniciar tratamiento concomitante con fosfenitoína y monitoreo estricto de los signos vitales.
El diazepam se administra a una velocidad de 2 mg/min para una dosis de 0.3 mg/kg (dosis total usual de 20 mg). Si es necesario, se puede dar una dosis adicional de 20 mg. Nuestra experiencia nos ha mostrado que cuando funciona, el diazepam controla el SE en 3 minutos en 33% de los pacientes y en 5 minutos en 80%. Esto significa que al término de 5 minutos (a una dosis de 10 mg), 80% de los pacientes se habrá controlado con diazepam. El 20% restante requerirán de los 20 mg completos o de dosis extras de 20mg.
En aquellos pacientes en los que no hay acceso a una vía I.V., el diazepam puede ser utilizado rectal a dosis de 0.5 mg/kg (dosis máxima de 20 mg) en forma de gel especialmente en pacientes pediátricos aun en esquemas extrahospitalarios de manejo del
SE. Las dosis recomendadas en niños de 2-5 años son de 0.5 mg/ kg y de 0.2-0.3 mg/kg en mayores de seis años. Los niveles séricos máximos se alcanzan en 15-30 min y duran 4 horas.
Lorazepam
Su vida biológica (15 horas) es más corta que la del diazepam y no contiene metabolitos activos, por lo que el riesgo de intoxicación es menor. Es más lipofílico y se cree que tiene menor redistribución tisular, la duración de la acción es mayor, pero alcanza concentraciones cerebrales máximas menos rápidamente (15-30 min). Sin embargo, controla el SE en 10 a 30 minutos.
En seis estudios realizados entre 1979 y 1981, el lorazepam controló la actividad convulsiva en 88% de 113 niños y adultos. Los status mioclónicos parecen ser los más refractarios, lográndose su control únicamente en 55% de los casos. El lorazepam se administra a 0.1 mg/ kg en bolo I.V. y se pude repetir esta dosis en 15 min si es necesario. Walker dio una dosis fija de 4 mg en dos minutos de lorazepam I.V., lográndose el control de las crisis durante 2 a 72 horas, tiempo mayor que el logrado usualmente con diazepam.
Clonazepam, midazolam, flunitrazepam
Tassinari y col. encontraron respuestas positivas en 60 a 100% de los casos en 11 reportes sobre clonazepam en SE. Es tan efectivo como el diazepam y tiene las mismas complicaciones. El midazolam y flunitrazepam I.V.

también son muy efectivos, pero su forma I.V. no se encuentra en los EUA. Aún no hay datos sobre el uso de midazolam en niños. El clonazepam I.V. fue utilizado en 17 niños de 2 semanas a 26 meses. Todos respondieron a dosis de 0.25-0.75 mg/kg en bolos de 0.25 mg, repitiendo cada 30 min si era necesario. Seis de estos niños tuvieron recurrencia 45 min a 46 horas después.
Fenitoína
La fenitoína está disuelta en propilenglicol, solvente en parte responsable de la hipotensión, el fallo cardíaco, las arritmias y el síndrome del guante púrpura observado durante su administración en infusión. No recomendamos que la fenitoína i. V. sea utilizada sola. Sus efectos anticonvulsivantes administrada a 50 mg/min, tienen su inicio en 10-20 minutos (la dosis total de 1000 mg se administra en 20 minutos).
La monoterapia con fenitoína detendrá el SE convulsivo en los primeros 20 min. en 80% de los pacientes que no tienen una lesión aguda del SNC. Los niveles plasmáticos efectivos por encima de 10 microgramos/ml pueden persistir por 12 horas para prevenir crisis. Después de una carga endovenosa de 10-12 mg/ mi, la vida media de la fenitoína I.V. es relativamente menor (10-15 horas) que la de la fenitoína oral (24 horas).
Fosfenitoína
La fosfenitoína es una prodroga hidrosoluble esterificada de la fenitoína, disuelta en tetraglicol, el cual tiene menos efectos tóxicos y aunque no se ha relacionado h a s t a a h o r a c o n l a s complicaciones mencionadas para
la fenitoína. siempre recomendamos el monitoreo del EKG y de la presión arterial durante su infusión. Se convierte rápida y completamente en fenitoína y puede ser administrada más rápidamente a 75 mg/min, a una velocidad de 150mg/min para una dosis total de 700 mg para una persona de 70 kg.
Recomendamos se utilice en la prevención de la recurrencia del SE concomitante a las benzodi-zepinas. Los estudios sobre la farmacocinética, seguridad y tolerancia de la fosfenitoína I.V. en adultos muestran que alcanza concentraciones sanguíneas similares a la fenitoína I.V. Actualmente se realizan estudios en niños y lactantes.
Acido valproico IV
Hasta que no se completen más ensayos, el valproato se debe reservar para el status tónico, status de ausencias atípicas y SE refractarios, se ha utilizado rectalmente en estas situaciones. Vajda y Vajda y col recomiendan una dosis media rectal de 2100 mg/día para controlar el status tónico, parcial motor o mioclónico. Viani ha reportado el uso del valproato en status infantil y neonatal. Devinski y col. valoraron la seguridad del valproato I.V. en 318 pacientes administrándoles 375 mg en una hora. Se observó cefalea, somnolencia, y vómitos, pero los signos vitales se mantuvieron estables.
Paraldehído IV o rectal
No se recomiendan, pues existen drogas más eficaces y sin los riesgos inherentes a esta droga, como falla de corazón derecho y hemorragia o edema pulmonar.
PROTOCOLO DE LA EPILEPSY FOUNDATION 0F AMERICA (EFA)
Inicio con diazepam (0.3 mg/kg a 2mg/min), o
lorazepam (0.1 mg/kg en bolo I.V.)
Seguir con fenitoína/fosfenitoína 20 mg/kg a 100 mg/min
Fenobarbital 20 mg/kg a 100 mg/min
Si el diazepam es utilizado como primera droga, la fosfenitoína debe ser utilizada como segunda droga por la corta vida media de la primera. Si el lorazepam es utilizada como primera droga y controla las crisis, la fosfenitoína no debe ser utilizada necesa-riamente, a menos que se vaya a usar como fármaco a largo plazo. Si posteriormente se administra fenobarbital sin éxito, se deben utilizar dosis anestésicas de barbitúricos, pues para ese entonces ya habrán transcurrido 30 minutos desde el inicio de la terapia para el SE. Se pueden utilizar el fenobarbital, pentobarbital o tiopentona para suprimir las descargas convulsivas.
Observaciones a las recomendaciones de la EFA
En la actualidad nuestra escuela todavía considera que el fenobarbital no debe ser la droga de primera elección para SE. El fenobarbital requiere frecuentemente del uso de dosis anestésicas para controlar las crisis. Cuando causa hipotensión o paro respiratorio, éstos duran más que los producidos por el diazepam. Además puede empeorar las convulsiones

primarias tipo gran mal y las ausencias, a menos que se utilicen dosis anestésicas. Sí estamos de acuerdo en utilizar el fenobarbital si la terapia con benzodiacepinas y fenitoína no ha dado resultado para controlar el SE en 30 min. La velocidad de infusión del diazepam debe ser de 2 mg/min. Las velocidades de 5-10 mg/min resultan en una mayor probabilidad de depresión respiratoria. Todavía considera-mos el diazepam como primera droga por las siguientes razones:
1. La mayor distribución tisular del lorazepam en comparación con el diazepam y su acción teóricamente más prolongada no se ha materializado en los estudios prospectivos de Leppik y col. y de Treiman y col. Las tasas de recurrencia fueron de 30% an ambos esquemas de tratamiento. Por lo tanto, no hay una ventaja práctica del lorazepam. Cuando ocurren hipotensión o depresión respiratoria, éstos persisten por mas tiempo que con el diazepam.
2. Si el manejo con diazepam/ lorazepam y fenitoína no da resultado, es necesario intubar el paciente y suministrar respiración artificial antes de usar los barbitúricos. La combinación de benzodiacepinas y barbitúricos aumenta el riesgo de hipotensión y depresión respiratoria.
STATUS EPILEPTICUS REFRACTARIO
Al presente, no existe un protocolo estandarizado para el tratamiento del SE refractario. Cuando el SE persiste por más de 30 a 60 minutos, requiere un
tratamiento más agresivo. Se requiere monitorización con EEG e intubación, siendo las opciones el uso de pentobarbital, fenobarbital o tiopentona I.V. o valproato I.V. Además del uso de vasopresores. Los barbitúricos I.V. parecen ser ideales, pues pueden reducir la hipertensión cerebral, el metabolismo y la hipoxia.
En estos casos, hemos utilizado diazepam para mantener concen-traciones séricas óptimas y prevenir recurrencias. Se debe evaluar la eficacia de combinar el diazepam I.V. y el rectal, que dura cuatro horas, lo que podría obviar el uso de fenitoína en el 30% de los SE que recurren. El uso en el hogar de diazepam rectal ha controlado efectivamente las convulsiones prolongadas o en racimo de 85% de los pacientes (81%), con la ventaja de prevenir la visita a las unidades de emergencia, mejorar la calidad de vida de las familias, y reducir los costos.
PERSPECTIVAS PARA LA EVALUACIONDELDAÑO CEREBRAL SECUNDARIO AL STATUS EPILEPTICUS
El SE es temido por la posibilidad de muerte súbita y por el daño cerebral debido a las depolarizaciones continuas. Muchos han sugerido la necesidad de terapias neuroprotectoras para status convulsivo y status parciales complejos de origen en el hipocampo, pero no se ha elaborado un protocolo específico para evaluar el daño cerebral in vivo en humanos.
Jackson y col. han demostrado que la IRM funcional (fIRM) puede detectar activación cortical
un minuto antes de cambios clínicos en el EEG durante las crisis parciales motoras, sugiriendo cambios vasculares y de oxigenación tempranos. Pritchard y otros seis investigadores, independiente-mente, realizaron espectroscopia de resonancia magnética en esclerosis del hipocampo y detectaron que la intensidad de la señal para el N-acetilaspartato disminuía en 22% en las regiones límbicas, señalando pérdida neuronal correlacionable con esclerosis mesial temporal.
Pritchard y col. también estudiaron el SE en el cerebro de conejos y notaron un descenso en la fosfocreatina y aumento del fosfato inorgánico Pi mientras el pH bajaba a 6.8 in vivo. El lactato aumenta aun después de terminadas las crisis. Zhongycol. midieron la longitud de la difusión de las moléculas de agua por medio de diffusion weighted imaging en SE de ratas y notaron una disminución de la longitud media de la difusión del agua.
Estos estudios deberían realizarse en humanos, debemos definir con espectroscopia de resonancia magnética los cambios bioquí-micos del SE que realmente preceden y llevan al daño cerebral definido por fIRM y espectros-copia. Solo después de realizar ese paso podremos valorar el uso de drogas neuroprotectoras en el status epilepticus.
PROBLEMÁTICA LATINOAMERICANA
Un reciente estudio sobre la experiencia y características del status epilepticus en Latinoamé-rica hace las siguientes conclusiones:-
• El tipo de S.E. más común es el convulsivo (secundaria-

mente generalizado tónico-clónico).
• Existe una alta tasa de mortalidad y secuelas neurológicas.
• El inicio del tratamiento está determinado por aspectos socioeconómicos y culturales.
• Los puntos de mayor preocupación son la prevención, educación y tratamiento temprano.
Estos son sin duda aspectos sobre los cuales debe trabajarse en la región.
BIBLIOGRAFÍA
Browne TR. Paral dehyde, chlormethiazole, and lidocaine for treatment of status epilepticus. Tn: Delgado-Escueta AV, Wasterlain CG, Treiman DM, Porter RJ, Eds. Status epilepticus: mechanisms of brain damage and treatment. New York: Raven Press, 1983:507-517. (Advances in Neurology, Vol 34.).
Calmeil JL. De l'épilepsíe, étudiéé sous le rapport de son siége et de son influence sur la productíon de l'aliénation mentale. Thesis, Université de Paris, 1824.
Commissíon on Epidemiology and Prognosis, International League Against Epilepsy. Guidelines for epidemiológical studies on epilepsy. Epilepsia, 1993;34(4):592-6.
Delgado-Escueta AV. Enrile-Bacsal F. Combination therapy for status epilepticus: intravenous diazepam and phenytoin. In: Delgado-Escueta AV, Wasterlain CG, Treiman DM, Porter RJ, Eds. Status epilepticus: mechanisms of brain damage and treatment. New York: Raven Press, 1983:15-35. (Advances in
Neurology, Vol 34.).
Delgado-Escueta AV, Wasterlain CG, Treiman DM, Porter RJ, Eds. Status epilepticus. New York: Raven Press, 1983. (Advances in Neurology, Vol 34.).
Gastaut H. Classification of status epilepticus. In: Delgado-Escueta AV, Wasterlain CG, Treiman DM, Porter RJ, Eds. Status epilepticus. New York: Raven Press, 1983:15-35. (Advances in Neurology, Vol 34.).
Gastaut H, RogerJ, Lob H. les étals de mal épileptiques. Paris; Masson, 1967.
Hauser WA. Status epilepticus: frequeney, etiology, and neurological sequelae. In: Delgado-Escueta AV, Wasterlain CG, Treiman DM, Porter RJ. Eds. Status epilepticus. New York: Raven Press, 1983:3-14. (Advances in Neurology, Vol 34.).
Hauser WA. Status epilepticus: cpidemiologic considerations. Neurology 1990;40(suppl2):9-13.
Leppik IE. Status epilepticus: the next decade. Neurology 1990;40(suppl 2):4-9.
Leppik IE, Derivan AT, Homan RW, Walker J, Ramsay RE, Patrick B. Double blind study of lorazepam and diazepam in status epilepticus. JAMA 1983;249:1452-4.
Lothman EW, Bertram EH. Epilcptogenic effeets of status epilepticus. Epilepsia 1993; 34(suppl 1):59-7Ü.
Lowestein DH, Aminoff MJ. Simón RP. Barbiturale anesthesía in the treatment of status epilepticus. Neurology 1988;38:395-40().
Marco T. Medina, Francisco Rubio-Donnadieu, Rey na Durón. Status epilepticus in Latin America. VIII Panamerican Epilepsy Congress. Sept. 12-13, 1997, Buenos Aires, Argentina. Memoria.
Ramsay E. Treatment of status epilepticus. Epilepsia 1993;34(suppl
Shanncr DM, McCurdy SA, Herring MO, Gabor AJ. Treatment of status epilepticus: a prospective comparison of diazepam and phenytoin versus phenobarbital and optional phenytoin. Neurology 1988;38:202-7.
Treiman DM. Status epileplicus. In: Resor SR, Kutt H, eds. The medical treatment of epilepsy. New York: Marcel Dekker, 1992:183-93.
Treiman DM, DeGiorgio CM, Salisbury SM, Wickboldt CL. Subtle generalizaed convulsive status epilepticus. Epilepsia 1984;25:653.
Treiman DM, Meyers PD. Coiling C. DVA Status Epilepticus Cooperative Study Group. Design of a large prospective double-blind trial to compare intravenous treatments of generalized convulsive status epilepticus. Epilepsia 1990a;31:635.
Vajda FJE, Mihaly GW. Miles JL, Donnnan GA, Bladin PF. Rectal administration of sodium valproate in status epilepticus. Neurology 1978;28:897-9.
Wasterlain CG, Fujikawa DG, Penix L, Sankar R. Pathophysiological mechanisms of brain damage from status epilepticus. Epilepsia 1993; 34(suppl l):37-53.
Wilson JVK, Trynolfd EH. Translation and analysis of a cuneiform text forming part of a babylonian treatisc on epilepsy. J MedHist 1990;34:185-98.
Working Group on Status Epilepticus. Treatment of convulsive status epilepticus. Epilepsy Foundation of America, 1995.

Revista Hondureña de Neurociencia Vol. 3 No. 1,1999
I CONGRESO VIRTUAL IBEROAMERICANO, DE NEUROLOGÍA CURSO DE FORMACIÓN EN NEUROLOGÍA TROPICAL
NEUROCISTICERCOSIS
Osear H. Del Brutto.
Senido de Neurología, Hospital "Luis Vernaza", Guayaquil, Ecuador.
La cisticercosis ocurre como consecuencia de la infección por el estadio larvario de la Taenia solium, la cual se produce cuando el hombre se convierte, en forma accidental, en el huésped intermediario de dicho céstodo [1]. El parásito tiene una predisposición particular por afectar el sistema nervioso, condicionando una enfermedad pleomórfica denominada neurocisticercosis [2-4]. A continuación revisaremos los aspectos más importantes de la neurocisticercosis, con especial atención a los avances recientes en diagnóstico y tratamiento.
EPIDEMIOLOGÍA
La neurocisticercosis es la enfermedad parasitaria más frecuente del sistema nervioso central, representando una patología neurológica común, así como un serio problema de salud pública en diferentes países de América Latina, África y Asia [5-12]. Por otra parte, el aumento reciente en el turismo, los grandes movimientos de refugiados y la imigración masiva de individuos
provenientes de aras endémicas, ha condicionado un aumento en la frecuencia de la neurocis-ticercosis en países desarrollados, donde esta entidad era considerada una rareza en las últimas décadas [13-17].
La prevalencia exacta de la neurocisticercosis es muy difícil de determinar en vista de la inespecificidad de sus manifes-taciones clínicas y de la falta de una prueba completamente confiable y segura, que pueda ser utilizada en estudios epidemio-lógicos a gran escala.
A finales del siglo pasado, la cisticercosis era prevalente en varios países Europeos; s in embargo, mejoras en los sistemas de salud pública produjeron una reducción considerable en su prevalencia [18]. Luego de la segunda guerra mundial, debido al regreso masivo de tropas, se notó un incremento transitorio en la prevalencia de la cisticercosis en Alemania y Francia. De igual manera, el regreso de soldados provenientes de la India condicionó un incremento en el
número de casos de cisticercosis en Inglaterra [19,20]. Nueva-mente, la implementación de medidas sanitarias redujo dicha prevalencia y en la actualidad, la cisticercosis es rara en dichas naciones [21-23]. Por el contrario, esta entidad continúa endémica en ciertos países de Europa del este [24,25], así como en España [26] y Portugal [27].
La cisticercosis era una enfermedad rara en Los Estados Unidos de América, con menos de 150 casos reportados hasta 1979. En la última década, la entrada masiva de imigrantes provenientes de América Latina ha condicionado un incremento importante de casos en dicho país especialmente en los estados del suroeste, incluyendo Texas y California [15-17]. Por otra parte, también se han reportado casos de neurocisticercosis en ciudadanos estadounidenses que nunca han estado en áreas endémicas [28-30]; en la mayoría de esos casos la fuente de infección ha sido un contacto portador asintomático de T. solium [29,30].
Presentado durante el primer Congreso Virtual Iberoamericano de Neurología, 1998 http://cvneuro.org
En África y Asia, la naturaleza endémica de la taeniosis/ cisticercosis se encuentra directamente relacionada con la tendencia religiosa y los hábitos alimenticios de sus habitantes. Debido a que el Koran prohibe el consumo de carne de cerdo, estas enfermedades son prácticamente inexistentes entre los Musulmanes. Por el contrario, la taeniosis y la cisticercosis son endémicas en países de África central, en la India y en el sudeste de Asia, donde la carne de cerdo es consumida s i n control higiénico adecuado [9-12,31-35].
De igual manera, la cisticercosis es endémica en varios países de América Latina. En México, estudios de autopsia han demostrado que el 2,5% al 3,6% de la población tiene neurocisticercosis [36,37]. La enfermedad es más prevalente en la zona geográfica denominada "El Bajío", lugar donde extensas plantaciones de frutas y vegetales alternan con grandes ranchos de ganado porcino [38]. Por otra parte, diversos estudios revelan que la neurocisticercosis es causa importante de admisiones hospitalarias y de procedimientos neuroquirúrgicos en México [5,39,40].
La cisticercosis también es endémica en Sudamérica, principalmente en Brasil [41,42], Colombia [8], Ecuador [6] y Perú [7,43]; en dichos países, la neurocisticercosis es causa importante de epilepsia de inicio tardío [44-46]. Al igual que en Asia y África, la endemia de la taeniosis/cisticercosis en América Latina se debe a las pobres condiciones socio-económicas de la mayoría de sus habitantes, así como al desconocimiento de la naturaleza de esta enfermedad y de su forma de adquisición [47].
Ciclo biológico de la Taenia solium: LaT. solium es una de las 30 especies de céstodos que pueden invadir al hombre. Dichas especies presentan ciclos biológicos complejos que requie-ren usualmente dos o más huéspedes para poder comple-tarse. En el caso de la T. solium, los humanos son los únicos huéspedes definitivos, mientras que tanto cerdos como humanos pueden actuar como huéspedes intermediarios.
La T. solium adulta está compuesta por una cabeza (escólex) armada con cuatro ventosas y una doble corona de ganchos, un cuello angosto y un cuerpo elongado que consiste en varios cientos de proglótides hermafroditas [48]. El parásito adulto habita en el tubo digestivo del ser humano, donde se mantiene firmemente adherido a la pared intestinal mediante sus ventosas y ganchos.
Cada día, varios proglótides grávidos se separan del extremo distal de la taenia y son expulsados con las heces.
Cada proglótide contiene miles de huevecillos que se liberan en el ambiente y que pueden permanecer viables durante largo tiempo. En lugares donde la eliminación de excretas es inadecuada, los cerdos se alimentan con heces humanas e ingieren los huevos de la T. solium.
Una vez ingeridos por el cerdo, los huevecillos pierden su cubierta y se liberan las oncosferas (embrio-nes hexacantos), los que atraviezan la pared intestinal y entran al flujo sanguíneo desde donde son transportados a los tejidos del cerdo, principalmente músculos estriados y cerebro.
En dichos tejidos, las oncosferas evolucionan y se transforman en larvas (cisticercos) [49]. Cuando el hombre ingiere carne de cerdo mal cocida y contaminada con cisticercos, las larvas se evaginan en el intestino delgado, el escólex se adhiere a la pared intestinal y el cuerpo del parásito comienza a crecer y a formar proglótides [50].
ETIOPATOGENIA
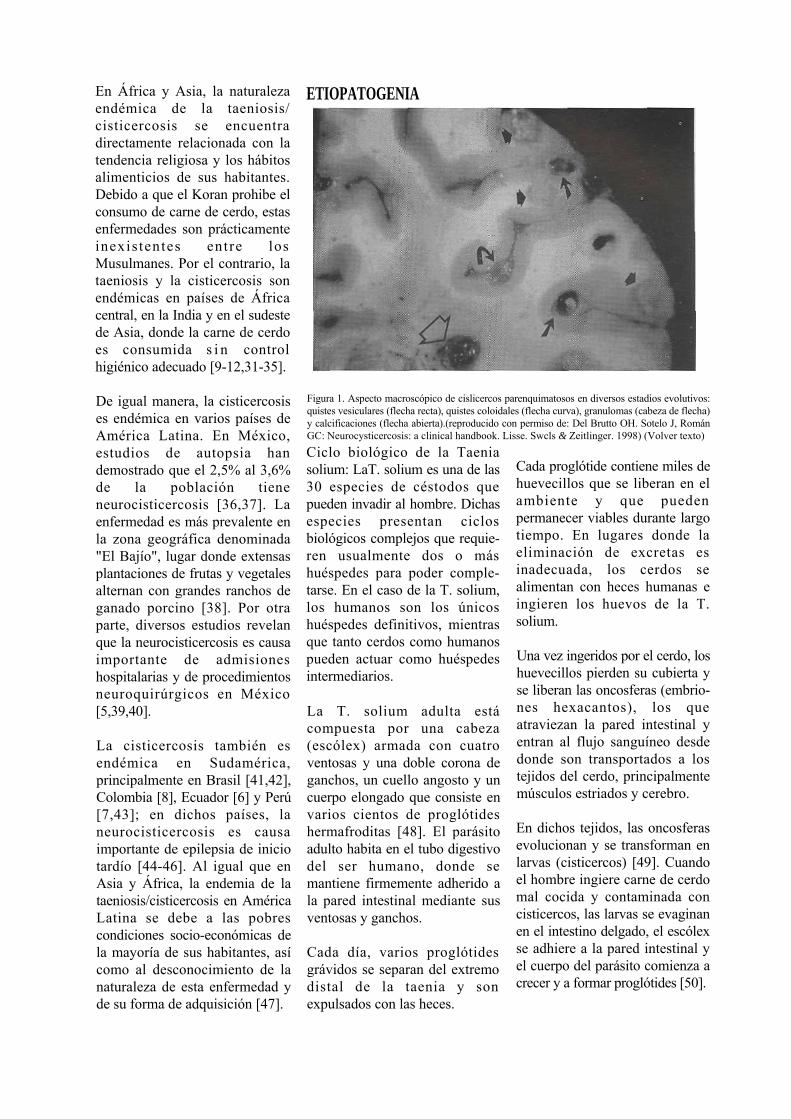
Figura 1. Aspecto macroscópico de cislicercos parenquimatosos en diversos estadios evolutivos: quistes vesiculares (flecha recta), quistes coloidales (flecha curva), granulomas (cabeza de flecha) y calcificaciones (flecha abierta).(reproducido con permiso de: Del Brutto OH. Sotelo J, Román GC: Neurocysticercosis: a clinical handbook. Lisse. Swcls & Zeitlinger. 1998) (Volver texto)

Por otra parte, el hombre puede también convertirse en huésped intermediario de la T. solium al ingerir sus huevecillos; bajo estas circunstancias, se desarrolla la cisticercosis humana [51]. El mecanismo por el cual los huevecillos entran al torrente sanguíneo y son distribuidos a los tejidos del hombre es similar al descrito en los cerdos. Las formas principales de contagio humano incluyen ingestión de comida contaminada con huevecillos de T. solium y contaminación ano-mano-boca en individuos portadores de la T. solium en su intestino, los que pueden auto-infectarse o infectar a otras personas, sobretodo a sus contactos domésticos.
La transmisión aérea de huevecillos de T. solium y la regurgitación de proglótides desde el intestino delgado hacia el estómago (auto-infección interna) no han sido adecuadamente demostrados como fuentes importantes de adquisición de la enfermedad. Como ejemplos anecdóticos de contagio, se encuentran la transmisión transplacentaria de cisticercos [52] y la ingestión voluntaria de proglótides de T. solium con propósitos medicinales [53],
Características de los cisticercos: Los cisticercos son vesículas llenas de líquido que contienen en su interior un escólex invaginado. La pared de la vesícula es una estructura membranosa com-puesta de tres capas, cuticular o externa, celular o media y reticular o interna [51 ]. El escólex presenta una estructura similar a la de la T. solium adulta, con una cabeza o róstelo que presenta ventosas y ganchos y un rudimento de cuerpo, que incluye al canal espiral. Algunas de las proteínas de los cisticercos tienen propiedades antigénicas y
estimulan la producción de antígenos específicos [54,55].
Sin embargo, estos antígenos no tienen mayor efecto en la protección contra la enfermedad ya que los cisticercos desarrollan una serie de mecanismo evasores que le permiten sobrevivir al ataque inmunológico del huésped [51,56]. Entre esos mecanismos destacan el mimetismo molecular y la depresión de la inmunidad celular, la cual puede condicionar una serie de complicaciones en enfermos con cisticercosis.
En algunos cisticercos el escólex no puede ser identificado. Estos parásitos están formados por membranas adheridas entre sí, las que tienden a agruparse en racimos [57]. Dichos cisticercos se localizan principalmente en las cisternas de LCR en la base del cráneo y en ellos, el escólex ha desaparecido como consecuencia de un proceso de degeneración hidrópica, condicionado por la entrada continua de LCR a la vesícula [58].
Es una práctica frecuente denominar Cysticercuscellulosae a aquellos parásitos que tienen escólex y Cysticercus racemosus a aquellos que no lo tienen; sin embargo, dicha terminología es inadecuada y crea confusión ya que se puede pensar que se trata de dos especies distintas de taenia [59]. En la actualidad se prefiere la terminología de "forma celulosa" y "forma racemosa" de cisticercos, respectivamente. De igual manera, dichos términos no deben ser escritos en itálicas ya que los cisticercos no son taxonómicamente independientes sino que representan formas larvarias de un céstodo adulto.
El aspecto macroscópico de los cisticercos varía dependiendo de su localización en el neuroeje
[52], Los cisticercos paren-quimatosos suelen ser pequeños y se localizan de preferencia en la corteza cerebral y en los ganglios básales debido a la gran vascularidad de estas áreas. Estos quistes rara vez miden más de 10mm de diámetro ya que la presión que ejerce el parénquima cerebral impide su crecimiento. Los cisticercos subaracnoideos pueden ser pequeños si se localizan en la profundidad de los surcos corticales o pueden alcanzar tamaños mayores de 5cm si están a nivel de las cisternas de LCR en la base del cráneo [581.
Los cisticercos ventriculares pueden ser pequeños o grandes, usualmente son únicos y se localizan e preferencia en el IV ventrículo; estos parásitos pueden estar adheridos a la capa ependimaria o encontrarse frotando libremente en las cavidades ventriculares [52, 58, 60]. Los cisticercos espinales se localizan en el espacio subaracnoideo o en el parénquima medular y su aspecto macros-cópico es similar al de los quistes localizados en el cerebro [61,62].
Estadios de involución de los cisticercos: Una vez que los cisticercos entran en el sistema nervioso, éstos se encuentran en un estado denominado vesicular, en el que los parásitos son viables y desencadenan cambios infla-matorios mínimos en el tejido cerebral adyacente. En algunos casos, los cisticercos permanecen durante décadas en este estadio ya que sus mecanismos evasores evitan que el sistema inmune del huésped los destruya [63].
Sin embargo, en otros casos los cisticercos entran, como resultado de un complejo ataque inmunológico del huésped, en un proceso degenerativo que termina con la muerte del parásito. Los
estadios por los que atraviezan los cisticercos hasta su destrucción comprenden: estadio coloidal, estadio granular y estadio calcificado [58].
Cada uno de estos estadios se caracteriza por cambios específicos en el interior de los parásitos, por cambios en el tejido cerebral vecino y como veremos posteriormente, por alteraciones específicas en los estudios de neuroimagen, lo cual permite su fácil reconocimiento. Es frecuente encontrar parásitos en diferentes estadios involutivos en el mismo paciente ; sin embargo, no se sabe si esto se debe a la presencia de infecciones recurrentes o a una sola infección en la que solamente algunos parásitos evaden la respuesta inmune mientras que otros son atacados intensamente.
Cambios estructurales en el s istema nervioso: El pleomorfismo clínico de la neurocisticercosis se debe, en gran parte, a la multiplicidad de lesiones que se producen en el sistema nervioso. Hemos descrito la diversidad de íocalización y aspecto de los cisticercos en el neuroeje. Por otra parte, la reacción inflamatoria desen-cadena una serie de cambios en el parénquima cerebral, el espacio subaracnoideo, las cavidades ventriculares y la médula espinal [52,58]. Dicha reacción inflamatoria se encuentra constituida principalmente por linfocitos, células plasmáticas y eosinófilos, se asocia con diversos grados de edema y gliosis reactiva y varía dependiendo del grado de involución de los cisticercos.
Los cisticercos localizados en el espacio subaracnoideo desenca-denan una intensa reacción inflamatoria perilesional, con formación de un denso exudado compuesto por fibras colágenas,
linfocitos, células gigantes rnultinucleadas, eosinófilos y membranas parasitarias hialini-zadas. Esto condiciona engrasa-miento anormal de las lepto-meninges en la base del cráneo, el cual puede extenderse desde la región optoquiasmática hasta el agujero magno.
El quiasma óptico, así como los demás nervios craneales que atraviezan el espacio suba-racnoideo, se encuentran atrapados en este denso exudado [58.64J. De igual manera, los agujeros de Luschka y Magendie pueden ocluirse con el subsecuente desarrollo de hidrocefalia [65]. Los vasos sanguíneos que forman el polígono de Willis también se afectan por esta reacción y las paredes de las pequeñas arterias son invadidas por células inflamatorias, lo cual induce el desarrollo de endarteritis proliferativa con oclusión de la luz arterial [66].
Los cisticercos ventriculares desencadenan una reacción inflamatoria localizada si se encuentran adheridos a los plexos coroideosoalapared ventricular.
En esos casos, las células ependimarias protruyen hacia el interior de las cavidades ventriculares y pueden bloquear la normal circulación de LCR a nivel del acueducto de Silvio [67] o de los agujeros de Monro [68]; este proceso, que suele acompañarse de hidrocefalia, se denomina ependimitis granular [65].
Los cisticercos espinales también pueden desencadenar cambios inflamatorios y desmielinizantes a nivel de las raíces nerviosas ventrales y dorsales al igual que los cisticercos intracraneales lo hacen con los nervios craneales. De igual manera, los cambios que se producen en el parénquima medular son similares a l o s observados en el parénquima cerebral [58].
MANIFESTACIONES CLÍNICAS
La cisticercosis es una enfermedad pleomórfica [2-4]. Dicho pleomorfismo se debe a diferencias individuales en el número y Íocalización de los parásitos, así como a la amplia
Figura
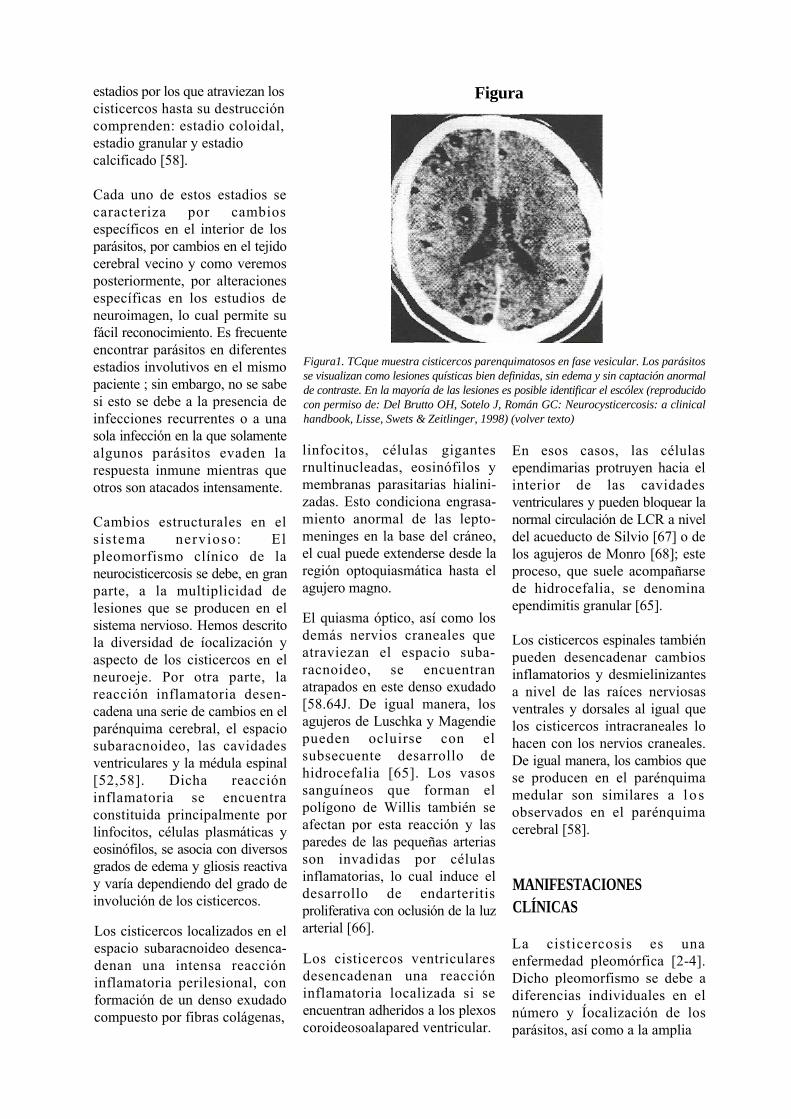
Figura1. TCque muestra cisticercos parenquimatosos en fase vesicular. Los parásitos se visualizan como lesiones quísticas bien definidas, sin edema y sin captación anormal de contraste. En la mayoría de las lesiones es posible identificar el escólex (reproducido con permiso de: Del Brutto OH, Sotelo J, Román GC: Neurocysticercosis: a clinical handbook, Lisse, Swets & Zeitlinger, 1998) (volver texto)

variación que existe en la respuesta inmune del huésped frente al parásito [69J. La epilepsia es la manifestación clínica más frecuente de la neurocisticercosis, observándo-sela en el 50% a 80% de los casos, particularmente en pacientes con compromiso de parénquima cerebral [70-73].
En regiones donde la cisticercosis es endémica, la presencia de crisis convulsivas de inicio reciente en sujetos mayores de 25 años de edad (epilepsia de inicio tardío), es altamente sugestiva de neurocisticercosis [44-46,74]. La mayoría de estos enfermos tienen un examen neurológico normal y se presentan con crisis convulsivas tónico-clónicas generalizadas [70,75]; por lo tanto, la práctica de estudios de neuroimagen es de fundamental importancia en todo paciente con epilepsia de inicio tardío con el objeto de confirmar o descartar la sospecha clínica de neurocisticercosis [75,76].
Se han descrito una gran variedad de signos neurológicos focales en enfermos con neurocisticercosis, particularmente en aquellos con quistes localizados en áreas cerebrales elocuentes [77-82]. Los signos más frecuentes incluyen: déficit motor, signos de liberación piramidal, ataxia cerebelosa, signos de disfunción de tallo cerebral y movimientos involuntarios [2,73]. Estas manifestaciones usualmente siguen un curso progresivo, por lo que es difícil el diagnóstico diferencial con neoplasias o con otros procesos infecciosos del sistema nervioso [76]. En algunos casos los signos focales aparecen en forma súbita, especialmente cuando se relacionan con infartos cerebrales secundarios a angeitis cisticercosa [66, 83-901.
Algunos enfermos presentan evidencia clínica de hipertensión endocraneal, la cual puede o no asociarse con crisis convulsivas, con signos focales o con alteraciones mentales [2]. La causa más frecuente de este síndrome es la hidrocefalia, la cual puede ser secundaria a aracnoiditis cisticercosa. ependimitis granular o quistes ventriculares [60,65,67,91-93]. En la mayoría de los casos la hipertensión endocraneal sigue un curso lentamente progresivo, el cual puede interrumpirse por episodios súbitos de pérdida de conciencia relacionados con movimientos de la cabeza (síndrome de Bruns), cuando la causa de la hidrocefalia es un cisticerco en el IV ventrículo [94,95].
Otras formas de neurocis-ticercosis que se asocian con hipertensión endocraneal son los quistes subaracnoideos gigantes y la encefalitis cisticercosa. Esta últ ima es una forma particularmente grave de neurocisticercosis que ocurre como resultado de la infección masiva de cisticercos al parénquima cerebral con la subsecuente intensa reacción inflamatoria del huésped [96]; la encefalitis cisticercosa es más frecuente en niños y mujeres jóvenes y se caracteriza por deterioro de conciencia, crisis convulsivas, disminución de agudeza visual, cefalea, vómitos y papiledema [72,96-99].
Las manifestaciones clínicas de la neurocisticercosis espinal también son inespecíficas. La aracnoiditis se manifiesta por dolor radicular asociado con debilidad muscular, la cual sigue un patrón de distribución sugestivo de afección de múltiples raíces nerviosas. Los quistes parenquimatosos suelen
condicionar un cuadro de mielitis transversa, con trastornos esfinterianos así como alteraciones motoras y sensitivas por debajo del sitio de la lesión [61,62,100,101].
DIAGNOSTICO
Únicamente la interpretación correcta de los exámenes de neuroimagen e inmunológicos permite el diagnóstico de la cisticercosis [76]. Por otra parte, la presencia de cisticercosis extracerebral, facilita conside-rablemente el diagnóstico de esta entidad en pacientes con manifestaciones neurológicas y hallazgos de neuroimagen sugestivos, mas no concluyentes.
Fuera del neuroeje, los cisticercos pueden localizarse en el globo ocular donde son visualizados mediante examen oftalmoscópico [102-104] o en los músculos esqueléticos o el tejido celular subcutáneo, donde pueden identificarse mediante radio-grafías simples o incluso a la palpación [105-1 10]. Es importante recordar, sin embargo, que no todos los nodulos subcutáneos palpables son cisticercos (aun en áreas donde esta enfermedad es endémica).
Estudios de neuroimagen: Tanto la TC como la IRM facilitan el diagnóstico de la neuro-cisticercosis ya que permiten visualizar el número y localización de los parásitos así como su estadio evolutivo [1 lili 7], De hecho, los hallazgos de TC e IRM en la neurocisticercosis parenquimatosa dependen fundamentalmente del grado de viabilidad de los cisticercos. De estos hallazgos, los más característicos son las lesiones quísticas bien definidas en las que es posible identificar el escólex en

su interior (Figura 2) y las calcificaciones punti formes múltiples [111,115]. Por el contrario, las lesiones anulares (únicas o múltiples) no son específicas y representan un problema diagnóstico).
Diversas entidades, incluyendo abscesos cerebrales, tuberculomas y neoplasias primarias o secundarías del sistema nervioso pueden cursar con lesiones similares en TC o IRM [118-121]. En algunos de estos casos, ni siquiera la práctica de otros exámenes complementarios tales como angiografía o estudio de LCR permiten un diagnóstico certero [2]. Como se describirá más adelante, un ensayo terapéutico con drogas anticis-ticercosas es de gran utilidad en estos casos [122-124] y representa una alternativa intermedia entre la actitud nihilística frente a estas lesions, propuesta por algunos autores, y la biopsia rutinaria, sugerida por otros [125-126].
La TC y la IRM en pacientes con neurocisticercosis meníngea suelen revelar hidrocefalia, captación anormal del contraste en las leptomeninges básales, quistes subaracnoideos e infartos cerebrales [113,116,127]. En los pacientes con infarto, la angiografía puede mostrar estenosis segmentaria u oclusión de arterias intracraneales de mediano calibre [84-90]. Con excepción de las lesiones quísticas, la mayoría de los hallazgos de neuroimagen en la neurocisticercosis meníngea no son específicos y pueden observarse en otro tipo de infecciones del sistema nervioso [121].
En estos casos, el análisis citoquímico del LCR es de fundamental importancia para el diagnóstico correcto [76,114]; los
niveles de glucosa en LCR suelen ser normales en pacientes con neurocisticercosis a diferencia de lo observado en pacientes con meningitis tuberculosa o micótica, en los que usualmente existe hipoglucorraquia [128]. De igual manera, las pruebas inmu-nológicas destinadas a la detección de anticuerpos anticisticerco son de gran utilidad en el diagnóstico de la neurocisticercosis meníngea [129,130].
Los cisticercos ventriculares se visualizan como lesiones quísticas que distorcionan el sistema ventricular y producen hidro-cefalia asimétrica [131-133]. Estos quistes suelen ser isodensos con el LCR y no se aprecian bien con TC, por lo que suele ser necesaria la administración de medio de contraste intratecal para confirmare! diagnóstico [134]. La IRM permite una mejor visualización de estas lesiones ya que el escólex puede ser iden-tificado y la señal del líquido vesicular suele ser distinta a la del LCR en los cortes potenciados en T2 [132,133,135].
En términos generales, la IRM es mejor que la TC para el diagnóstico de la neurocisti-cercosis, especialmente en pacientes con lesiones quísticas en la base del cráneo, tallo cerebral, cavidades ventriculares y médula espinal. Sin embargo, una limitación importante de la IRM es su mala resolución para detectar pequeñas calcificaciones parenquimatosas [136]. Debido a que muchos pacientes con epilepsia y neurocisticercosis presentan calcificaciones como única evidencia de la enfermedad, la práctica exclusiva de IRM puede condicionar errores diagnósticos. LaTC es el método de imagen de elección para el estudio de pacientes con probable
neurocisticercosis; la IRM debe reservarse para aquellos casos con TC normal o en los que el aspecto tomográfico de las lesiones no sea concluyente [76].
Pruebas inmunológicas: Existen varias pruebas destinadas a la detección de anticuerpos anticis-ticerco en sangre, saliva y LCR, entre las que destacan la reacción de fijación de complemento, el ensayo inmunoabsorbente ligado a enzimas (ELISA) y el inmunoblot [136-141]. Estas pruebas son un complemento importante de los estudios de neuroimagen, pero nunca deben ser utilizadas en forma aislada para confirmar o descartar el diagnóstico de neurocisticercosis debido al elevado porcentaje de resultados falso-positivos y falso-negativos.
De las pruebas serológicas, la más fidedigna es el inmunoblot. A pesar que se ha sugerido que el inmunoblot es 100% específico y 98% sensible para el diagnóstico de neurocisticercosis [142], es importante recordar que los pacientes con taeniosis o aquellos que tienen cisticercos musculares presentan resultados positivos en sangre, sin que eso signifique que tengan neurocisticercosis. Algo similar ocurre con el ELÍSA, cuya certeza diagnóstica en suero es decepcionante [143].
La práctica de pruebas inmunológicas en LCR suele ser más confiable que en suero; sin embargo, la positividad de dichas pruebas se relaciona directamente con la viabilidad y la localización de los cisticercos [ 144,145]. Tanto la reacción de fijación de complemento como el ELISA son muy sensibles en la neurocis-ticercosis meníngea [129,130]; esta sensibilidad declina considerablemente en la neurocisticercosis parenquima-

tosa, particularmente si las lesiones se encuentran calcificadas [144].
TRATAMIENTO
Debido al pleomorfismo clínico de la neurocisticercosis, no es posible que un solo esquema de tratamiento sea útil en todos los casos. Por lo tanto, la caracterización precisa de la enfermedad, en lo que respecta a viabilidad y localización de las lesiones, es de fundamental importancia con el objeto de planificar un tratamiento adecuado [146-148].
Drogas cestocidas: Tanto el praziquantel como el albendazol son potentes drogas cestocidas. El praziquantel es una isoquinolina que ha sido utilizada para el tratamiento de la neurocis-ticercosis humana desde 1979, luego de que Robles y Chavarrfa [149] reportaran el tratamiento satisfactorio de un niño con quistes parenquimatosos. Estu-dios posteriores demostraron que el praziquantel condiciona la desaparición del 60% a 70% de los cisticercos parenquimatosos luego de un curso de 15 días de tratamiento a dosis de 50 mg/kg/ día [150-152].
El albendazol es un imidazol con potente efecto cestocida. Esta droga inicialmente se utilizó en dosis de 15 mg/kg/día durante 30 días [153]; sin embargo, estudios posteriores demostraron que la duración del tratamiento podía ser reducida a 8 días con iguales resultados [154]. El albendazol destruye el 75% a 90% de los cisticercos parenquimatosos y ha probado ser superior al praziquantel en diversos estudios comparativos [155-158], no solamente por su mejor porcentaje de destrucción de quistes
parenquimatosos sino por su capacidad de destruir quistes subaracnoideos [ 159,160] y por su menor costo, un aspecto importante ya que la cisticercosis usualmente afecta a gente de bajos recursos económicos.
Si bien los primeros estudios de tratamiento de la neuro-cisticercosis fueron destinados a documentar el porcentaje de destrucción de quistes que tanto el praziquantel como el albendazol inducían en TC, estudios recientes han demostrado que el tratamiento con estas drogas también produce cambios considerables en el curso clínico de los enfermos [70,119,161-164].
Como se describirá más adelante, el control de crisis convulsivas en pacientes con epilepsia y neurocisticercosis es considerablemente mejor si los enfermos son manejados con drogas cestocidas [70,161,164]. Por otra parte, diversos estudios han documentado marcada mejoría en signos neurológicos de focalización en pacientes con quistes gigantes luego del tratamiento cestocida [160,163].
Otro potencial de las drogas cestocidas es su utilidad diagnóstica en pacientes con lesiones anulares únicas. Como se mencionó previamente, estas lesiones representan un reto diagnóstico ya que pueden ser producidas por un gran número de entidades; en caso de duda, la administración temprana de estas drogas facilitará el diagnóstico diferencial al acelerar la destrucción de las lesiones anulares causadas por cisticercos [123,124].
Existen formas de neurocis-ticercosis que no deben recibir
tratamiento cestocida. Entre ellas se encuentra la encefalitis cisticercosa ya que el uso de dichas drogas puede exacerbar el edema cerebral que acompaña esta forma de la enfermedad y condicionar aumento en la presión intracraneal [97,98]. Los enfermos con hidrocefalia y quistes parenquimatosos o subaracnoideos pueden recibir drogas cestocidas una vez que la hidrocefalia ha sido resuelta mediante la implantación de un sistema de derivación ventricular.
En los pacientes con quistes gigantes y en aquellos con quistes ventriculares, el tratamiento cestocida debe individualizarse y su beneficio debe ser comparado con los potenciales riesgos inherentes a su empleo [160]. Finalmente, los pacientes con cisticercos calcificados no deben recibir tratamiento cestocida ya que estas lesiones representan parásitos muertos que han sido previamente destruidos por el sistema inmune del individuo.
Tratamiento sintomático: Las drogas antiepilépticas son utilizadas en un gran número de enfermos con neurocisticercosis. En pacientes con epilepsia secundaria a calcificaciones, la administración de una droga antiepiléptica de primera línea (carbamazepina, fenitoína o fenobarbital) usualmente produce un control adecuado de las crisis. Sin embargo, los pacientes con quistes viables deben recibir primero un curso de tratamiento con drogas cestocidas para lograr un control posterior de crisis con drogas antiepilépticas.
Esto fue demostrado en una serie de 203 pacientes con epilepsia secundaria a neurocisticercosis, en la que se documentó control de crisis convulsivas en 83% de los

enfermos que fueron sometidos previamente a tratamiento con albendazol o praziquantel, a diferencia de aquellos que no recibieron dicho tratamiento, en los que únicamente el 27% logró un control adecuado de crisis [70]. Estos hallazgos fueron confirmados en dos estudios posteriores en los que el control de crisis fue definitivamente mejor en enfermos que recibieron tratamiento cestocida previo [161,162].
Recientemente se ha demostrado que la administración de praziquantel disminuye los niveles séricos de fenitoína y carbamazepina [165], hecho que puede condicionar descontrol de crisis convulsivas en pacientes con neurocisticercosis tratados con praziquantel. No hay estudios similares con el albendazol, sin embargo, es prudente el monitoreo de niveles séricos de drogas antiepiléticas en estos casos para evitar el desarrollo de crisis convulsivas.
Aún no se determina la duración óptima del tratamiento antiepilétíco en pacientes con neurocisticercosis. Un estudio retrospectivo reciente demostró recidiva de crisis en el 50% de los casos luego de la suspensión de drogas antiepilépticas, aun en enfermos que se encontraban por lo menos dos años libres de crisis [ 166]. Los factores que incidieron adversamente en el riesgo de recidiva de c r i s i s en dichos enfermos fueron la presencia de calcificaciones así como el antecedente de haber tenido crisis múltiples antes de alcanzar el control gracias al tratamiento antiepiléptico. Los resultados de dicho trabajo sugieren que el pronóstico de la epilepsia secundaria a neurocisticercosis no es del todo favorable ya que un
gran número de enfermos necesitan tratamiento antiepiléptico por tiempo indefinido.
Los corticosteroides son drogas frecuentemente utilizadas en pacientes con neurocisticercosis. Estos representan la forma principal de tratamiento de la encefalitis cisticercosa, donde usualmente son necesarias dosis altas de dexametasona (24-32 mg/ día) para reducir el edema cerebral que acompaña a esta forma de la enfermedad [147]; en estos casos suele ser necesaria la administración concomitante de diuréticos osmóticos (manitol, 2 mg/kg/día) para lograr un adecuado control de síntomas.
Los corticosteroides a dosis altas son también de utilidad en enfermos con angéitis cisticercosa con el objeto de reducir el riesgo de infartos recurrentes [66]. De igual manera, los corticosteroides se encuentran indicados en pacientes con aracnoiditis cisticercosa para reducir el riesgo de hidrocefalia o de lesiones irreversibles de nervios craneales.
La administración simultánea de corticosteroides durante el tratamiento cestocida es controversial. Se ha sugerido su empleo para disminuir los efectos adversos que pueden ocurrir durante el tratamiento con albendazol o praziquantel [167]. Dichos efectos no se deben a toxicidad de estas drogas sino a la destrucción de los cisticercos como resultado del tratamiento. En la mayoría de los casos, estos efectos secundarios pueden ser controlados adecuadamente con analgésicos comunes y drogas antieméticas, evitando el uso rutinario de corticosteroides.
Este aspecto es de especial importancia en enfermos tratados
con praziquantel ya que la administración simultánea de corticosteroides disminuye hasta en 50% los niveles plasmáticos de dicha droga [168]. Por el contrario, los niveles plasmáticos de albendazol aumentan con la administración de corticosteroides por lo que pueden ser utilizados libremente en estos casos [169].
Existen indicaciones absolutas para el uso simultáneo de corticosteroides y drogas cestocidas, incluyendo pacientes con quistes subaracnoideos gigantes, quistes ventriculares y quistes localizados en la médula espinal. En la mayoría de estos casos, los corticosteroides deben ser administrados antes, durante y después del tratamiento cestocida con el objeto de disminuir el riesgo de infartos cerebrales, hidrocefalia o edema medular, respectivamente.
Cirugía: Los pacientes con hidrocefalia secundaria a aracnoiditis cisticercosa usual-mente requieren la implantación de un sistema de derivación ventricular [ 170,171 ]. El principal problema en estos casos es el gran número de disfunciones de dicho sistema, lo cual aumenta considerablemente el riesgo de muerte en estos casos [39,172], Recientemente se ha desarrollado un nuevo sistema de derivación que aparentemente funciona mejor que los sistemas conven-cionales en pacientes con neuro-cisticercosis.
Este nuevo sistema permite el drenaje de LCR a una velocidad constante sin necesidad de un mecanismo de válvula, por lo que no se permite la entrada de LCR subaracnoideo al interior de las cavidades ventriculares [173]. En pacientes con neurocisticercosis, esta inversión del flujo de LCR es
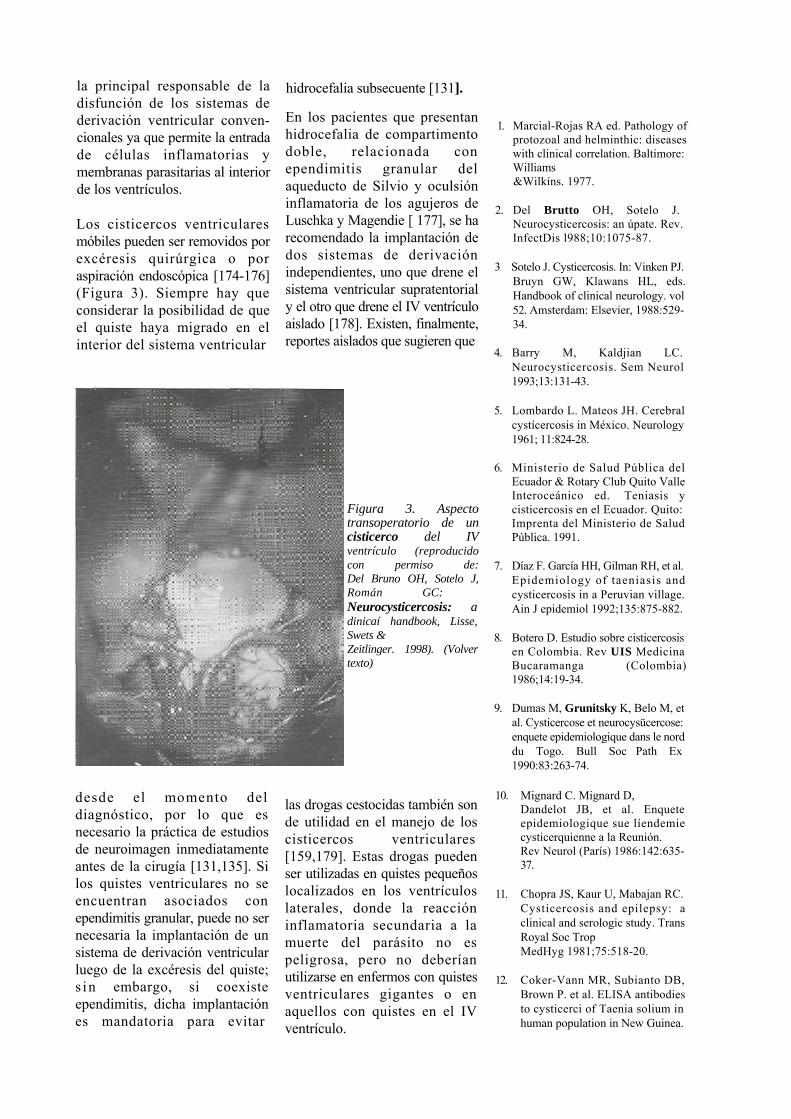
la principal responsable de la disfunción de los sistemas de derivación ventricular conven-cionales ya que permite la entrada de células inflamatorias y membranas parasitarias al interior de los ventrículos.
Los cisticercos ventriculares móbiles pueden ser removidos por excéresis quirúrgica o por aspiración endoscópica [174-176] (Figura 3). Siempre hay que considerar la posibilidad de que el quiste haya migrado en el interior del sistema ventricular
desde el momento del diagnóstico, por lo que es necesario la práctica de estudios de neuroimagen inmediatamente antes de la cirugía [131,135]. Si los quistes ventriculares no se encuentran asociados con ependimitis granular, puede no ser necesaria la implantación de un sistema de derivación ventricular luego de la excéresis del quiste; s in embargo, si coexiste ependimitis, dicha implantación es mandatoria para evitar
En los pacientes que presentan hidrocefalia de compartimento doble, relacionada con ependimitis granular del aqueducto de Silvio y oculsión inflamatoria de los agujeros de Luschka y Magendie [ 177], se ha recomendado la implantación de dos sistemas de derivación independientes, uno que drene el sistema ventricular supratentorial y el otro que drene el IV ventrículo aislado [178]. Existen, finalmente, reportes aislados que sugieren que
Figura 3. Aspecto transoperatorio de un cisticerco del IV ventrículo (reproducido con permiso de: Del Bruno OH, Sotelo J, Román GC: Neurocysticercosis: a dinicaí handbook, Lisse, Swets & Zeitlinger. 1998). (Volver texto)
las drogas cestocidas también son de utilidad en el manejo de los cisticercos ventriculares [159,179]. Estas drogas pueden ser utilizadas en quistes pequeños localizados en los ventrículos laterales, donde la reacción inflamatoria secundaria a la muerte del parásito no es peligrosa, pero no deberían utilizarse en enfermos con quistes ventriculares gigantes o en aquellos con quistes en el IV ventrículo.
1. Marcial-Rojas RA ed. Pathology of protozoal and helminthic: diseases with clinical correlation. Baltimore: Williams &Wilkíns. 1977.
2. Del Brutto OH, Sotelo J. Neurocysticercosis: an úpate. Rev. InfectDis l988;10:1075-87.
3 Sotelo J. Cysticercosis. In: Vinken PJ. Bruyn GW, Klawans HL, eds. Handbook of clinical neurology. vol 52. Amsterdam: Elsevier, 1988:529-34.
4. Barry M, Kaldjian LC. Neurocysticercosis. Sem Neurol 1993;13:131-43.
5. Lombardo L. Mateos JH. Cerebral cystícercosis in México. Neurology 1961; 11:824-28.
6. Ministerio de Salud Pública del Ecuador & Rotary Club Quito Valle Interoceánico ed. Teniasis y cisticercosis en el Ecuador. Quito: Imprenta del Ministerio de Salud Pública. 1991.
7. Díaz F. García HH, Gilman RH, et al. Epidemiology of taeniasis and cysticercosis in a Peruvian village. Ain J epidemiol 1992;135:875-882.
8. Botero D. Estudio sobre cisticercosis en Colombia. Rev UIS Medicina Bucaramanga (Colombia) 1986;14:19-34.
9. Dumas M, Grunitsky K, Belo M, et al. Cysticercose et neurocysücercose: enquete epidemiologique dans le nord du Togo. Bull Soc Path Ex 1990:83:263-74.
10. Mignard C. Mignard D, Dandelot JB, et al. Enquete epidemiologique sue líendemie cysticerquienne a la Reunión. Rev Neurol (París) 1986:142:635- 37.
11. Chopra JS, Kaur U, Mabajan RC. Cysticercosis and epilepsy: a clinical and serologic study. Trans Royal Soc Trop MedHyg 1981;75:518-20.
12. Coker-Vann MR, Subianto DB, Brown P. et al. ELISA antibodies to cysticerci of Taenia solium in human population in New Guinea.
hidrocefalia subsecuente [131].

Oceanía, and Southeast Asia. Southeast Asian J Trop Med Public Healthl981;12:499-505.
13. Loo L, Braude A. Cerebral cysticercosis in San Diego: a report of 23 cases and a review of the literature. Medicine 1982;61:341-59.
14. McCormick GF, Zee C-S, Heiden J. Cysticercosis cerebri: a review of 127 cases. Arch Neurol 1982;39:540-44.
15. Richards FO Jr, Schantz PM, Ruiz- Tiben E, Sorvillo FJ. Cysticercosis in Los Angeles County. JAMA 1985 ;254:3444-48.
16. Shandera WX, White CA Jr, Chen JC, Diaz P, Armstrong R. Neurocysticercosis in Houston, Texas. A report of 112 cases. Medicine 1994;73:37-52.
17. Cuetter AC, Guerra LG, Meza AD, Brower RD. Neurocysticercosis: a special problem in the soulhwerstern United States. J Trop Geogr Neurol 1992;2:172-76.
18. Henneberg R. Die tierischen parasiten des zentral nerven system. In: Bumke O, Foerster O, eds. Handbuch der neurologie. vol 14. Berlin: Julius Springer, 1936:286.
19. Dixon HBF, Hargreaves WH. Cysticercosis (Taenia solium): a further ten yearsí clínica! study covering 284 cases. Q J Med 1944;13:107-21.
20. Dixon HBF, Lipscomb FM. Cysticercosis: an analysis and follow-up of 450 cases. Med Res Council Spec Rep No 299. London: Her Majestyís Stationary Office, 1961:1-58.
21. Hitchcock ER. Cysticercosis in the UK. j Neurol Neurosurg Psychiat 1987;50:108O-81.
22. Raverdy PH, Gentilini M, Smagghe A, Aranud JP, Fouche Y. Cystícercose cerebrale. Trois cas observé dans la región parisienne chez des travailleur immigres. Rev Neurol (Paris) 1976;132:55-62.
23. Paolocci S, Francia A, DíAloja E, Argentino C. Intracranial cysticercosis: report of two cases. Acta Neurol (Napoli) 1984;6:152- 56.
24. Obrador S. Cysticercosis cerebri. Acta Neurochir 1972;3:320-64.
25. Stepién L. Cerebral cysticercosis in Poland: clinical symptoms and operative resuits in 132 cases. J Neurosurg 1962;19:505-13.
26. García-Albea E. Cisticercosis en España. Algunos datos epidemiológicos. Rev Clin Esp 1989; 184:3-6.
27. Monteiro L, Coelho T, Stocker A. Neurocysticercosis - A review of 231 cases. Infection 1992;20:61-65.
28. Tasker WG, Plotkin SA. Cerebral cysticercosis. Pediatrics 1979;63:761-63.
29. KruskaJ BA, Moths L, Teele DW. Neurocysticercosis in a child with no history of travel outside the continental United States. Clin InfectDis 1993:16:290-92.
30. Schantz PM, Moore AC, Muñoz J, et al. Neurocysticercosis in an orthodox Jewish community i n New York city. N Engl J Med 1992;327:692-95.
31. Dumas M, Ndiaye IP, Dumas JP, Gueye M. Cysticercosc cerebrales (deux nouveaux cas scncgalais). Bull Soc Med Afr Noire 1976;21:203-ll.
32. Michel R Callies P, Raharison H, Guyon P, Holvoet L, Genin C. Epidémiologie de la cysticercose a Madagascar. Bull SocPathEx 1993;86:62-67.
33. Mahajan RC, Chopra JS, Ganguly NK. Human cysticercosis and epilepsy: a serologic study. In: Flisser A, Willms Ks Láclete JP, Larralde C. Ridaura C, Beltrán F, eds. Cysticercosis: present state of knowledge and perspectives. New York: Academic Press, 1982:171- 78.
34. Gajdusek CD. Introduction of Taenia s o l i u m into West New Guinea with a note on an epidemic of. burns from cysticercus epilepsy in the Ekari pcople of the Wissel Lakes área. Papua New Guinea Med J 1978:21:329-42.
35. Kong Y, Cho SY, Cho MS, et al. Seroepidemiological observations of Taenia solium cysticercosis in
epileptic patients in Korea. J Korean MedScil993;8:145-52.
36. Costero I ed. Tratado de anatomía patológica. México: Editorial Atlante, 1946.
37. Briceño C, Biagi F, Martinez B. Cisticercosis: observaciones de 97 casos de autopsia. Prensa Med Mex 1961;26:193-97.
38. Mateos-Gómez JH, Zenteno-Alanís GH. Neurocisticercosis. Análisis de 1000 casos consecutivos. Neurología-Neurocirugía- Psiquiatría (México) 1987;27(Suppl):53-55.
39. Penagos P. Cirugía en neurocisticercosis: experiencia de 10 años en el Instituto Nacional de Neurología y Neurocirugía de México. México: Universidad Nacional Autónoma de México, 1988.
„ - ? • ■ ■ 40. Flisser A, Woodhouse E, Larralde
C. The epidemiology of human cysticercosis in México. I n : Palacios E, Rodriguez-Carbajal J, Taveras JM, eds. Cysticercosis of the central nervous system. Springfield: Charles C. Thomas, 1983:7-17.
41. Vianna LG, Macedo V, Mello P, Costa JM, Yoo JM. Estudo clínico e laboralorial da neurocisticercose em Brasilia. Rev Bras Neurol 1990;26:35-40.
42. Costa-Cruz JM, Rocha A, Silva AM, et al. Ocorrencia de cisticercose em necropsias realizadas em Uberlandia, Minas Geraís, Brasil. Arq Neuropsiquiat 1995;53:227-32.
43. García HH, Martinez M, Gilman R, et al. Diagnosis of cysticercosis in endemic regions. Lancet 1991;338:549-5L
44. Del Brutto OH, Noboa CA. Late- onsetepilepsy in Ecuador: aetíoiogy and clinical features in 225 patients. J Trop Geogr Neurol 1991 ;l:31-34.
45. Amida WO. Etiology of epilepsy: a prospective study of 210 cases. Arq Neuro Psiquiat (Sao Paolo) 1991;49:251-54.
46. García HH, Gilman R, Martinez M, et al. Cyslicercosis as a major cause

of epilepsy in Peni. Lancet 1993;341:197-200.
47. Velasco-Suárez M. Cysticercosis: personal impact and socioeconomic significancc. In: Palacios E, Rodriguez-Carbajal J, Taveras JM. eds. Cysticercosis of the central nervous system. Springfield: Charles C. Thomas, 1983:3-6.
48. Faust EC, Russell PF, June RC eds. Craig and Faustís clinical parasitology, 8th ed. Philadelphia: Lea & Febiger. 1970.
49. Corwin RM, DiMarco NK. McDowell AE, Pratt SE. Infernal parasites. In: Leman AD, Straw B, Glock RD, Mengeling WL Penny RHC, eds. Diseases of swine, 6th ed. Ames: Iowa State University Press, 1986:646-64.
50. Flisser A, Pcrez-Montfort R, Larralde C. The immunology of human and animal cysticercosis: a review. BulI WHO 1979;57:839-56.
51. Del Bruno OH, Sotelo J. Etiopatogenia de la neurocisticercosis. Rev Ecuat Neurol 1993;2:22-32.
52. Escobar A, Nieto D. Parasitic diseases. In: Minckler J. ed. Pathology of the nervous system. vol 3. New York: McGraw-Hill, 1972:2503-21.
53. Bourke GJ, Petana WB. Human Taenia cysticercosis: abizarremode of transmission. Trans Roya! Soc Trop Med Hyg 1994;88:680.
54. Grogl M, Estrada JJ, McDonald G, Kuhn RE. Anligen-antíbody analyses in neurocysticercosis. J parásito! 1985:71:433-42.
55. Khan NA, Sotelo J. Presentation of a membrane cysticercus antigen and i t s homology wilh excretory- secretory antigen. Acta Leidensia 1989;57:123-29.
56. Flisser A. Woodhouse E, Larralde C. Human cysticercosis: antigens. antibodies and non-responders. Clin Exp Immunol 1980;39:27-37.
57. Bickerstaff ER, Cloake PCP, Hughes B. Smith WT. The racemose form of cerebral cysticercosis. Brain 1952:75:1-18.
58. Escobar A. The pathology of neurocysticercosis. In: Palacios E, Rodriguez-Carbajal J, Taveras JM, eds. Cysticercosis of the central nervous systern. Springfield: Charles C.Thomas, 1983:27-54.
59. Rabiela MT, Rivas A. Flísser A. Morphological types of Tenia solium cysticerci. Parasitology Today 1989;5:357-59.
60. Madrazo I, García-Rentería JA, Sandoval M, López-Vega FJ. Intraventricular cysticercosis. Neurosurgery 1983;12:148-52.
61. Cabieses F, Vallenas M, Landa R. Cysticercosis of the dorsal spinal cord. J Neurosurg 1959;16:337-41.
62. Venkataramana NK, Jain VK, Das BS. Rao TV. Intramedullary cysticercosis. Clin Neurol Neurosurg 1989^1:337-41.
63. Flisser A. Taenia solium cysticercosis: some mechanisms of parasite survival in immunocompetent hosts. Acta Leidensia 1989:57:259-63.
64. Del Brutto OH, Guevara J. Sotelo J. Intraseilar cysticercosis. J Neurosurg 1988;69:58-60.
65. Estañol B, Kleríga E, Loyo M. et al. Mechanisms of hydrocephalus in cerebral cysticercosis: ímplications for therapy. Neurosurgery 1983:13:119-23.
66. Del Brutto OH. Cysticercosis and cerebrovascular disease: a review. J Neurol Neurosurg Psychiat 1992;55:252-4.
67. Salazar A, Sotelo J, Martínez H, Escobedo F. Diffcrential diagnosis betwen ventriculitis and fourth ventricle cysts in neurocysticercosis. J Neurosurg 1983;59:660-63.
68. Siqueíra EB, Richardson RR. Kranzler LI. Cysticercosis cerebri occluding the foramen of Monro. Surg Neurol 1980;13:429-31.
69. Del Brutto OH, García E, Talamas O. Sotelo J. Sex-related severity of inflammation in parenchymal brain cysticercosis. Arch Intern Med 1988:148:544-46.
70. Del Brutto OH, Santibañez R, Noboa CA, Aguirre R, Díaz E, Alarcón TA. Epilepsy due to neurocysticercosis:
analysis of 203 patients. Neurology 1992;42:389-92.
71. Arseni C, Cristescu A. Epilepsy due to cerebral cysticercosis. Epilepsia 1972:13:253-58.
72. López-Hernández A. Garaizar C. Childhood cerebral cysticercosis: clinical features and computed tomographic findings in 89 Mexican children. Can J Neurol Sci 1982;9:401-07.
73. Sotelo J, Guerrero V. Rubio F. Neurocysticercosis: a new cías sifi catión based on active and inatíve forms. Arch Intern Med 1985;145:442-45.
74. Medina MT, Rosas E. Rubio- Donnadieu F, Sotelo J. Neurocysticercosis as the main cause of late-onset epilepsy in México. Arch Intern Med 1990;l 50:323-25.
75. Del Brutto OH. CT findings in neurologically normal adults with a single generalized seizure. J Epilepsy 1994;7:38-40.
76. Del Brutto OH. Cysticercosis. In: Feldmann E, ed. Current diagnosis in neurology. St. Louis: Mosby, 1994:125-29.
77. Bhigjee AI, Kemp T. Cosnett JE. Cysticercosis presenting with hemichorea. J Neurol Neurosurg Psychiat 1987;50:1561-62.
78. Poon TP, Arida EJ, Tyschenko WP. Cerebral cysticercosis with aqueductal obstruction: case report. J Neurosurg 1980;53:252-55.
79. Barinagarrementeria F, De! Brutto OH. Ataxic hemiparesis from parenchymal brain cysticercosis. J Neurol 1988;235:325.
80. Ramina R, Hunhevicz SC. Cerebral cysticercosis presenting as mass lesión. Surg Neurol 1986:25:89-93.
81. Berman JD, BeaverPC, CheeverAW, Quindlen EA. Cysticercus of 60-milliliter in human brain. Am J Trop MedHygl981;30:616-19.
82. Keane JR. Neuro-ophthalmologic signs and symptomsof cysticercosis. Arch Ophthalmol 1982;100:1445- 48.

83. Barinagarrementeria F. Del Brutto OH. Neurocysticercosis and puré motor hemiparesis. Stroke 1988;19:1156-58.
84. Barinagarrementeria F, Del Brutto OH. Lacunar syndrome due to neurocysticercosis. Arch Neurol 1989;46:415-17.
85. Gauthier N, Sangla S, Stroh-Marcy A, Payen L. Neurocysticercose revelé par un accident vasculaire cerebral. J Radiol I995;76:l 19-23.
86. McCormick GF, Giannotta S, Zee C- S, Fisher M. Carotid occlusion in cysticercosis. Neurology 1983;33:1078-80.
87. Rodriguez-Carbajal J, Del Brutto OH, Penagos P, Huebe J, Escobar A. Occlusion of the middíe cerebral artery due to cysticercotic angiitis. Stroke 1989;20:1095-99.
88. ter-Penning B, Litchman C, HeierL. Bilateral middle cerebral artery occlusions in neurocysticercosis. Stroke 1992;23:280-83.
89. Monteiro L, Almeida-Pinto J, Leite I, Xavier J, Correia M. Cerebral cysticercus arteritis: fivc angiographic cases. Cerebrovasc Dis 1994;4:125-33.
90. Levy AS, Lillehei KO, Rubinstein D, Stears JC. Subarachnoid neurocysticercosis with occlusion of major intracranial arteries: case repon. Neurosurgery 1995;36:183-
91. Rangel-Guerra R, Martínez HR. Diagnóstico diferencial de la estenosis del acueducto de Silvio en el adulto. Rev Invest Clin (Mex) 1986;38:21-27.
92. Milenkovic Z, Penev G, Stojanovic D, Jovicic V, Antovic P. Cysticercosis cerebri involving the lateral ventricle. Surg Ncurol 1982;18:94-96.
93. Jankowski R, Zimmerman RD, Leeds NE. Cysticercosis presenting as a mass lesión at foramen of Monro. J ComputAssistTomogr 1979;3:694-96.
94. Bickerstaff ER, Small JM, Woolf AL. Cysticercosis of the posterior fossa. Brain 1956;79:622-34.
95. Arellano-Sanchez J, Aruffo C, Escobar A. Cisticercosis del IV ventrículo y síndrome de Bruns. Rev Fac Med UNAM (Mex) 1985;28:11- 19.
96. Madrazo I, Olhagaray B, Becerra M. Sandoval MA, Soto R. Acute cysticercotic encephalitis: description of a histologically confírmed case. Neurosurgery 1983;13:593-95.
97. Rangel R, Torres B, Del Brutto O, Sotelo J. Cysticercotic encephalitis. A severe form in young témales. Am J trop Med Hyg 1987;36:387-92.
98. Noboa CA. Encefalitis cisticercosa: análisis de 10 casos. Rev Ecuat Neurol 1992;1:61-71.
99. Del Brutto OH, Sotelo J. Neurocysticercosis simulating pseudotumor ce re bri (pseudopseudotumor). J Clin Neuro- ophthalmol 1988;8:87-91.
100. Akiguchi I, Fujiwara T. Matsuyama H, Muranaka H, Kameyama M. Intramedullary spinai cysticercosis. Neurology 1979;29:1531-34.
101. Firemark HM. Spinai cysticercosis. Arch Neurol 1978;35:25G-5L
102. Lozano-Elizondo D. Ophthalmic cysticercosis. In: Palacios E, Rodriguez-Carbajal J, Taveras JM, eds. Cysticercosis of the central nervous system. Springfield: Charles C.Thomas, 1983:84-100.
103. Kruger-Leite E. Jalkh AE, Quiroz H, Schepens CL. Intraocular cysticercosis. Am J Ophthalmol 1985;99:252-57.
104. Cárdenas F, Quiroz H, Planearte A, Meza A. Dalma A, Flisser A. Taenia solium ocular cysücercosis: findings in 30 cases. Ann Ophthalmol 1992;24:25-28.
105. OíGrady TC. Robbins BA. Subcutaneous cysticercosis simulating metastatic breast carcinoma. Int J Dermatol 1993;32:62-64.
106. Case Records of the Massachusetts General Hospitals. Case 26-1994. N Engl J Med 1994:330:1887-93.
107. Jacob JC, Mathew NT. Pseudohypertrophic myopathy in cysticercosis. Neurology 1968;18:767-71.
108. Jolly SS, Pallis C. Muscular pseudohypertrophy due to cysticercosis. J Neurol Sci 1971;12:155-62.
109. Sawhney BB, Chopra JS, Banerji AK, Wahi PL. Pseudohypertrophic myopathy in cysticercosis. Neurology 1976;26:270-72.
110. Wadia N, Desai S, Bhatt M. Disseminated cysticercosis. New observations including CT sean findings and experience with treatment by praziquantel. Brain 1988;111:597-614.
111. Carbajal JR, Paiacios E, Azar-Kia B, Churchill R. Radiology of cysticercosis of the central nervous system including computed tomography. Radiology 1977;125:127-31.
112. Mervis B. Lotz JW. Computed tomography (CT) in parenchymatous cerebral cysticercosis. Clinical Radiol 1980;31:521-28.
113. Rodacki MA, Detoni XA, Teixeira WR, Boer VH, Oliveira GC. CT features of Cellulosae and Racemosus neurocysticercosis. J Compu: Assist Tomogr 1989;13:1013-16.
114. Del Brutto OH. Diagnosis and management of cysticercosis. J Trop Geogr Neurol 1992:2:1-9.
115. Suss RA, Maravilla KR. Thompson J. MR imaging of intracranial cysticercosis: comparison with CT and anaíomopathologic features. AJNR 1986;7:235-42.
116. Martinez HR. Rangel-Guerra R, Elizondo G. et al. MR imaging in neurocysticercosis: a study of 56 cases. AJNR 1989; 10:1011 -19.
117. Creasy JL, Alarcon JJ. Magnetic resonance imaging of neurocysticercosis. Top Mag Res Imag 1994;6:59-68.
118. Rodriguez-Carbajal J, Salgado P. Gutierrez-Alvarado R, Escobar- Izquierdo A. Aruffo C. The acute encephalitic phase of

neurocysticercosis: computed tomographic manifestations. AJNR 1983;4:51-55.
119. Del Brutto OH. Single parenchymal brain cysticercus in the acute encephalitic phase: definition of a distinct forro of neurocysticercosis with benign prognosis. J Neurol Neurosurg Psychiat 1995;58:247-49.
120. Del Brutto OH, Zenteno MA, Salgado P, Sotelo J. MR imaging in cysticercotic encephalitis. AJNR1989;10:S18-S20.
121. Atlas SWed. Magneiicresonance imaging of the brain and spine. New York: Raven Press, 1991.
122. Rawlings D. Ferriero DM. Messing RO. Early CT evaluation after empirical praziquantel therapy in neurocysticercosis. Neurology 1989;39:739-41.
123. Del Brutto OH. The use of albendazole in patients with single lesions enhanced on contrast CT. NEngl JMed 1993;328:356-357.
124. Del Brutto OH, Quintero LA. Cysticercosis mimickíng brain tumor: the role of albendazole as a diagnostic tool. Clin Neurol Neurosurg (in press)
125. Rajshekhar V. Etíology and management of single small CT lesions in patients with seizures: understanding a controversy. Acta Neurol Scand 1991;84:465-70.
126. ChandyMJ, Rajshekhar V, Ghosh S, et al. Single small enhancing CT lesions in Indian patients with epilepsy: clinical radiological and pathological considerations. J Neurol Neurosurg Psychiat 1991;54;702-05.
127. Bandres JC, White AC Jr, Samo T, Murphy EC, Harris RL. E x t r a p a r e n c h y m a l neurocysticercosis: report of five cases and review of management. Clin Infect Dis 1992;15:799-811.
128. Leonard JM. Cerebrospinal fluid formula in patients with CNS infections. Neurol Clinics North Am 1986;4:3-12.
129. Rosas N, Sotelo J, Nieto D. ELISA in the diagnosis of
neurocysticercosis. Arch Neurol 1986;43:353-56.
130. Garcia E. Sotelo J. A new complement fixation test for the diagnosis of neurocysticercosis in cerebrospinal fluid. J Neurol 1991;238:379-92.
131. Zee C-S, Segail HD, Apuzzo MLL Ahmadi J, Dobkin WR. Intraventricuíar cysticercal cysts: further neuroradiological observations and neurosurgical implications. AJNR 1984;5:727- 30.
132. Ginier BL, Poirier V.MR imaging of intraventricuíar cysticer- cosis.AJNR 1992;13:1247-48
133. Rangel-Guerra R, Herrera J, Elizondo G, González-Morantes J. Neurocysticercosis. Arch Neurol 1988;45:492.
134. Madrazo í. Rentería JA, Paredes G, Olhagaray B. Diagnosis of intraventricuíar and cisternal cysticercosis by computerized tomography with positive intraventricuíar contrast medium. J Neurosurg 1981;55:947-51.
135. Kramer J, Carrazana E, Cosgrove GR, Kleefield J, Edelman RR. Transaqueductal migration of a neurocysticercus cyst. J Neurosurg 1992;77:956-58.
136. Richards F Jr, Schantz PM. Laboratory diagnosis of cysticercosis. Clin Lab Med 1991 ;11:1011-28.
137. Nieto D. Cysticercosis of the central nervous system: diagnosis by means of the spinal fluid complement fixation test. Neurology 1956;6:725-38.
138. Miller B. GoldbergMA, HeinerD, Myers A, Goldberg A. A new immunologic test for CNS cysticercosis. Neurology 1984;34:695-97.
139. Baily GG, Masón PR, Trijssenar FEJ, Lyons NF. Serological diagnosis of neurocysticercosis: evaluation of ELISA tests using cyst fluid and other components of Taenia solium cysticerci as antigens. Trans Roy Soc Trop Med Hyg 1988;82:295-99.
140. Flisser A, Planearte A, Correa D, et al. New approaches in the diagnosis of Taenia solium cysticercosis and taeniasís. Ann Parasitol Hum Comp 1990;65(Suppll):95-98.
141. Tsang VCW, Brand J, Boyer E. E n z y m e - l i n k e d immunoelectrotransferency blot assay and glycoprotein antigens for diagnosing human cysticercosis. J Infect Dis 1989;159:50-59.
142. Garcia HH, Herrera G, Gilman RH. et al. Discrepancies between cerebral computed tomography and western blot in the diagnosis of neurocysticercosis. Am J Trop Med Hyg 1994;50:152-57.
143. Ramos-Kuri M, Montoya RM, Padilla A, et al. Immunodiagnosis of neurocysticercosis: disappointing performance of serology (enzyme-linked immunosorbent assay) in an unbiased sample of neurological patients. Arch Neurol 1992;49:633-36.
144. Ziní D, Farrell VJR. Wadee AA. Therelationshipofantibody levéis to the clínica! spectrum of human neurocysticercosis. J Neurol Neurosurg Psychiat 1990;53:656- 61.
145. Chang KH, Kim WS, Cho SY, Han MC, Kim C-W. Comparative evaluation of brain CT and ELISA in the diagnosis of neurocysticercosis. AJNR 1988;9:125-130.
146. Sotelo. Cysticercosis. In: Johnson RT, ed. Current therapy in neurologic disease, vol 2. Philadelphia: Decken 1987:114- 17.
147. Del Brutto OH, Sotelo J, Román GC. Therapy for neurocysticercosis: a reappraisal. Clin Infec Dis 1993;17:730-35.
148. Del Brutto OH. Medical management of neurocysticercosis. ínt J Ant Ag 1993;3:133-37.
149. Robles C, Chavarria M. Presentación de un caso clínico de cisticercosis cerebral tratado médicamente con un nuevo

fármaco: praziquantel. Salud Pub Mex 1979;21:603-618.
150. Sotelo J, Escobedo F, Rodriguez- Carbajal J, Torres B, Rubio- Donnadieu F. Therapy of parenchymal brain cysticercosis wíth praziquantel. N Engl J Med 1984:301:1001-1007.
151. Sotelo J, Torres B, Rubio- Donnadieu F, Escobedo F, Rodriguez-Carbajal J. Praziquantel in the treatment of neurocysticercosis: a long-term follow-up. Neurology 1985;35:752-755.
152. Robles C, Sedano AM, Vargas- Tentori N, Gaiindo-Virgen S. Long-term results of praziquantel therapy in neurocysticercosis. J Neurosurg 1987 ;66:359-363.
153. Escobedo F, Penagos P, Rodríguez J, Sotelo J. Albendazole therapy for neurocysticercosis. Arch Intern Med 1987;147:738-741.
154. Sotelo J, Penagos P, Escobedo F. Del Brutto OH. Short course of albendazole therapy for neurocysticercosis. Arch Neurol 1988;45:1130-1133.
155. Sotelo J, Escobedo F, Penagos P. Albendazole vs praziquantel therapy of neurocysticercosis: a controllcd trial. Arch Neurol 1988;45:532-534.
156. Sotelo J. Del Brutto OH, Penagos P, et al. Comparison of therapeutic régimen of anticysticercal drugs for parenchymal brain cysticercosis. J Neurol 1990:237:69-72.
157. Takayanagui OM, Jardim E. Therapy for neurocysticercosis: comparison between albendazole and praziquantel. Arch Neurol 1992;49:290-294.
158. Cruz M, Cruz I, Horton J. Albendazole vs praziquantel in the treatment of cerebral cysticercosis: clinical evaluation. Trans Roy Soc TropMedHyg 1991;85:244-47.
159. Del Brutto OH, Sotelo J. Albendazole therapy for subarachnoid and ventricular cysticercosis. J Neurosurg 1990:72:816-817.
160. Del Brutto OH, Sotelo J, Aguirre R, Díaz-Calderón E, Alarcón TA. Albendazoie therapy for giant subarachnoid cysticerci. Arch Neurol 1992;49:535-538.
161. Vasquez N, Sotelo J. The course of seizures after treatment for cerebral cysticercosis. N Engl J Med 1992;327:696-701.
162. Medina MT, Genton P, Montoya MC, Cordova S, Dravet C. Sotelo J. Effect of anticysticercal treatment on the prognosis of epilepsy in neurocysticercosis: a pilot trial. Epilepsy 1993:34:1024- 27.
163. Saníoyo H, Corona R, Sotelo J. Total recovery of visual function after treatment for cerebral cysticercosis. N Engl J Med 1991;324:1137-n39.
164. Del Brutto OH. Medical treatment of cysticercosis—effective. Arch Neurol 1995;52:102-104.
165. Bittencourt PRM, Gracia CM, Martins R, Fernandes AG. Diekmann HW, Jung W. Phenytoin and carbamazepine decrease oral bioavailability of praziquantel. Neurology 1992;42:492-496.
166. Del Brutto OH. Prognostic factors for seizure recurrence after withdrawal of antíepileptic drugs in patients with neurocysticercosis. Neurology 1994;44:1706-1709.
167. deGhetaldi LD, Norman RM, Douville AW jr. Cerebral cysticercosis treated biphasically with dexamethasone and praziquantel. Ann Intern Med 1983;99:179-181-
168. Vázquez ML. Jung H, Sotelo J. Plasma levéis of praziquantel decrease when dexamethasone is given simultaneously. Neurology 1987;37:1561-1562.
169. Jung H, Hurtado M, Medina MT, Sánchez M, Sotelo J. Dexamethasone increases plasma levéis of aibendazole. J Neurol 1990;237:279-280.
170. Colli BO, Martelli N, Assirati JA Jr, Machado HR, Forjaz SV. Results of surgical treatment of neurocysticercosis in 69 cases. J Neurosurg 1986:65:309-15.
171. Rueda-Franco F. Surgical considerations in neurocyslicercosis. Childs Nerv Syst 1987;3:212.
172. Sotelo J, Marin C. Hydrocephalus secondary to cysticercotic arachnoiditis: a long-term floow- up review of 92 cases. J Neurosurg 1987:66:686-89.
173. Sotelo J. A new ventriculoperitoneal shunt for treatment of hydrocephalus. Experimental results.
174. Couldwell WT, Apuzzo MLJ. Management of cysticercosis cerebri. Contemporary Neurosurgery 1989:19:1-6.
175. Apuzzo MLJ, Dobkin WR, Zee C- S, Chan JC, Giannotta SL, Weiss MH. Surgical considerations in treatment of intraventricular cysticercosis: an analysis of 45 cases. J Neurosurg 1984;60:400- 07.
176. Neal JH. An endoscopic approach to cysticercosis cysts of the posterior third ventricle. Neurosurgery 1995;36:1040-43.
177. DeFeo DR, Foltz EL, Hamilton E. Double compartment hydrocephalus in a patient with cysticercotic meningitis. Surg Neurol 1975:4:247-51.
178. Colli BO, Pereira CU. Assirati JA Jr, Machado HR. Isolated fourth ventricle in neurocysticercosis: pathophysiology, diagnosis, and treatment. Surg Neurol 1993;39:305-10.
179. AIlcul DA. Coulthard A. Neurocysticercosis: regression of a fourth ventricular cyst with praziquantel. J Neurol Neurosurg Psychiat 1991;54:461-62.

JUNTA DIRECTIVA DE LA FEDERACIÓN CENTROAMERICANA DE NEUROLOGÍA
96-99
PRESIDENTE
Dr. Marco Tulio Medina H.
VICEPRESIDENTES
Dr. Jenner Velásquez Guatemala
Dr. Nelson Chinchilla Honduras
Dr. Alfredo Orellana El Salvador
Dr, Ramiro Sirias Nicaragua
Dr. Manuel Carvajal Lizano Costa Rica
Dr. Fernando Gracia Panamá
SECRETARIO
Dr. Ricardo Madrid
TESORERO
Dr. Rolando Machado
FISCAL
Dr. Edgardo Girón

Revista Hondureñ a de Neurociencia Vol 3 No. 1,1999
I CONGRESO VIRTUAL IBEROAMERICANO DE NEUROLOGÍA CURSO DE FORMACIÓN EN NEUROLOGÍA TROPICAL
PARTICIPACIÓN DEL SISTEMA NERVIOSO
PERIFÉRICO EN LA ENFERMEDAD DE CHAGAS
Roberto E.P. Sica
División de Neurología. Hospital Ramos Mejía. Buenos Aires. Cátedra de
Neurología. Facultad de Medicina. Universidad de Buenos Aires, Argentina
Desde las primeras descripciones la enfermedad de Chagas fue considerada responsable de alteraciones a nivel del sistema nervioso periférico (SNP). Ha habido descripciones clínicas y experimentales que han señalado ese daño en el curso de esta enfermedad (1,2,3,4,5).
El relato que sigue expondrá los resultados obtenidos por nosotros en el hombre. Cuando resulte oportuno, se darán datos experimentales que avalen lo encontrado en el humano. El material reunido permite aceptar que una proporción de pacientes, en el período crónico de la enfermedad, desarrolla lesiones en algún segmento de su SNP, a nivel o por debajo de la neurona motora del asta anterior de la médula y/o de aquella otra ubicada en el ganglio de la raíz posterior. Aunque más débiles y escasas, existen observaciones que sugieren la alteración del SNP en la etapa aguda de la infección.
En 1975 Sica y Sanz, en un estudio preliminar limitado a 30 pacientes en el estadio crónico de la infección y libres de todo padecimiento neurológico, buscaron, electromiográfica- mente, la presencia de denervación. Para ello seleccionaron sujetos que tuviesen al menos dos reacciones positivas de tres ( i n m u n o f l u o r e s c e n c i a , hemoaglutinación y fijación del complemento) y cuyo examen neurológico fuese normal. Encontraron que el 66% de ellos tenía evidencias de denervación seguida por reinervación en uno o más músculos (resultados no publicados).
Este hallazgo inicial justificó un estudio electromiográfico más extendido en el que participaron 80 personas con Enfermedad de Chagas en estadio crónico. Se halló que el 79% de los integrantes del grupo tenían manifestaciones de denervación antisua, más
frecuentes en miembros inferiores (6,7,8). En este trabajo se observó que entre el 17.5% y el 54.3% de los potenciales de unidad motora remanentes eran de aspecto polifásico y tenían amplitud elevada, hecho que sugería que uno de los sitios posibles de lesión fuese la neurona motora del asta anterior de médula.
Se cuantificó, entonces, el número y tamaño de las unidades motoras funcionantes en los músculos de las eminencias tenar e hipotenar, soleo y extensor corto de los dedos, empleando técnicas electrofisiológicas (9,10,11). En total fueron investigados 187 pacientes. Los resultados obtenidos fueron comparados con 263 controles sanos, sin positividad para las reacciones de Chagas (8,12,13). Se vio que la pérdida de unidades motoras comprometía al 51% de los enfermos al explorar los músculos de tenar, al 36% al hacerlo con el soleo, al 20% con el extensor
Presentado durante el primer Congreso Virtual Iberoamericano de Neurología, 1998 http://cvneuro.org

corto y al 18% con los músculos de hipotenar. Al mismo tiempo, De Faria y cols. (14) trabajando de manera independiente en Brasil, habían llegado a resultados similares.
En aquellos pacientes que tenían disminución del número de unidades motoras funcionantes y, por lo tanto, denervación parcial de alguno(s) de sus músculos, el tamaño de las unidades motoras remanentes estaba aumentado en relación a los controles (8); ese aumento se relacionaba negativamente con la pérdida de unidades en el músculo comprometido (r: -0.86 para tenar, -0.84 para el soleo, -0.92 para el extensor corto y -0.97 para hipotenar. p <0.001 para todos) (8). Esto permitía inferir que la denervación muscular, producida en algún momento de la historia natural de la infección, había sido adecuadamente compensada por reinervación de las fibras musculares denervadas mediante colateralización axonal a partir de axones indemnes; siendo esto así, era de esperar que no hubiese atrofia muscular clínica, pero sí agrupamiento de fibras musculares en la imagen histológica.
El hallazgo más habitual, en la histología del músculo, fue la existencia de agolpamientos de fibras musculares, tanto de tipo I como de tipo 11 (8, 15). Esto fortificó la convicción de la existencia de denervación en esta etapa de la infección humana.
Se cuantificó, luego, el ruido sináptico en la unión neuromuscular del extensor corto de los dedos (16). Los resultados obtenidos en 8 pacientes, que exhibían denervación en ese músculo, fueron comparados con controles y con otros 5 enfermos,
no chagásicos, denervados a causa de lesión probada del asta anterior de médula (8). Este procedimiento mide la frecuencia de los potenciales de placa registrados extracelularmente. La frecuencia de los potenciales recogidos en los pacientes chagásicos fue similar a la de aquellos que padecían compromiso de asta anterior de médula (8).
Esta otra observación señalaba la existencia de inervación doble de la fibra muscular, hallazgo que hacía suponer que la fibra muscular denervada había recibido una colateral axónica desde un axón no comprometido pero que, posteriormente, readquiría su primera inervación, sugiriendo que su axón original se había repuesto y logrado, nuevamente, capacidad de inervación. Esto insinuaba la presencia de "motoneuronas enfermas" (17) con capacidad para volver a obtener sus propiedades, haciendo funcionalmente útil su conexión al músculo nuevamente, contribuyendo, entonces, a la producción del ruido de placa.
Esto lleva a pensar que la alteración ocasionada por la enfermedad se produce en un momento determinado de la historia de la enfermedad y que no necesariamente lleva a la muerte neuronal en forma definitiva; es posible que una proporción de neuronas quede no funcionantes (en estado de enfermedad), con la capacidad de recuperarse en la medida en que las circunstancias se le tornen propicias, por ejemplo la limitación del tiempo de agresión de la enfermedad.
Quedaba, aún, por conocer como era el comportamiento del nervio. En el nervio cubital, por ejemplo,
se estudió la velocidad de conducción motora máxima, que involucra a los axones de mayor diámetro, tanto en sus segmentos distales como en los proximales; y la velocidad de conducción lenta en la que participan las fibras motoras de menor diámetro. Las técnicas empleadas fueron las convencionales (18,19,20). Del total de enfermos investigados el 14.5% mostró reducción de sus valores de conducción máxima distal, el 29.3% de los de la conducción máxima proximal y el 33.3% de aquellos que corresponden a la conducción de las fibras más lentas (21).
El estado de las fibras sensitivas fue explorado en los nervios mediano y safeno externo. Se halló que 47.8% de los pacientes mostraba reducción de los valores de conducción en el segmento más distal del nervio, en tanto que sólo unos pocos lo hacían en los segmentos medio o proximal (22). Al medir la amplitud del potencial sensitivo se encontró que algo más de un tercio de los enfermos mostraba reducido su valor en cualquiera de los niveles estudiados a lo largo del nervio (22). Se exploró, también, el número de ondas que componen esa deflexión y que es la expresión de la actividad eléctrica de axones de diferente diámetro. En el nervio mediano, en sujetos normales, el número de estas ondas varió entre 5 y 8 con una media de 6.4 +1-2.1. En el caso de los pacientes, 13 de 23 explorados exhibieron menos de 5 ondas con una media de 3.38 +/-0.96 (p:<0.001). Los resultados obtenidos en el safeno externo fueron similares a los del mediano (22).
Estas observaciones permitieron incluir al tronco nervioso como uno de los sitios de lesión. Los

datos electrofisiológicos eran suficientes como para suponer la existencia de dos tipos de alteración, por un lado la desmielinización del nervio, evidenciada por la disminución de los valores de conducción, y por el otro la pérdida axonal, puesta de manifiesto por la reducción de la amplitud del potencial de acción sensitivo y por la disminución del número de ondas que lo integraban.
El estudio histológico del nervio safeno externo mostró pérdida de fibras mielínicas, existencia de cubiertas mielínicas delgadas en relación con el diámetro axonal y presencia de grupos de axones de bajo diámetro. Las últimas dos observaciones corresponden a remielinización y regeneración axonal. En las fibras aisladas pudo hallarse retracción de la mielina, desmielinización segmentaria y paranodal y acortamiento de la distancia entre nodos (22). Nunca fueron vistos cambios inflama-torios ni nidos de parásitos.
El cuadro histológico dio evidencias de cambios dinámicos que aparentemente suceden en el nervio, ya que junto a la pérdida de algunos axones existe el intento de regeneración de otros que incluye su ulterior remieli-nización.
Se exploró, luego, la transmisión entre el nervio y el músculo. Para ello fueron estudiados 58 pacientes, sin neuropatía, en los que se procedió al estímulo repetitivo a intensidad supramáxima del nervio cubital registrando la respuesta motora en músculos de la eminecia hipotenar. Se encontró que el 48.3% de ellos mostraba algún tipo de alteración de la transmisión, traducido por una disminución o incremento de la
amplitud de la respuesta más allá de los límites controles o combinando ambas conductas (23). Esta observación señaló la alteración en la transmisión neuromuscular, pero no permitió individualizar el sitio del compromiso.
Junto con González Cappa y su grupo se desarrolló un modelo experimental de la infección en el ratón. Inicialmente los animales fueron infectados con distintas cepas de parásitos buscando, por un lado, la susceptibilidad del huésped y, por otro, el mayor tropismo por el sistema nevioso periférico del agente infectante. El primer modelo se obtuvo infectando ratones Rockland con tripomastigotes provenientes de la cepa CA-I. En esos experimentos fueron combinadas técnicas de detección electrofisiológicas con otras histológicas del músculo y del nervio.
Los estudios fueron hechos a los 4, 6, 9 y 12 meses post-infeccion (pi). A los 4 meses pi se hallo reducción del trazado interfe-rencial del electromiograma en aproximadamente la mitad de los ratones infectados junto a aumento de la duración y amplitud media de los potenciales de unidades motoras remanentes. A los 6 meses fueron vistos potenciales de fibrilación en el 50% de los animales tratados, reducción de la capacidad de reclutamiento voluntario de unidad motora en también el 50% y normalización de los valores medios de amplitud y duración del potencial de unidad motora. A los 9 meses pi se mantenía la presencia de actividad espontánea, ahora en una proporción menor de ratones infectados, y la reducción del trazado de interferencia del electromiograma, con simultánea
disminución de la duración y amplitud medias del potencial de unidad motora.
Por fin, a los 12 meses pi, la actividad espontánea podía aún verse en algunos animales, en tanto que el trazado de interferencia electromiográfico estaba reducido en la mayor parte de ellos junto a aumento de la amplitud y duración medias de los potenciales de las unidades motoras remanentes (24). El examen histológico del músculo y el nervio mostró miositis con compromiso perivascular, en forma de arteritis, especialmente a los 4 y 12 meses pi. Se vieron, además, fibras musculares atroncas y otras hipertróficas en todos los cortes y unos pocos nidos de amastigotes. En el nervio ciático se encontraron infiltrados inflamatorios, incluyendo imágenes de arteritis, fibras desmielinizadas y degeneración axonal en todos los ratones infectados. La pérdida de fibras mielínicas fue notoriamente mayor a los 12 meses/?; (24).
Estos hallazgos sugerían que los ratones infectados desarrollaban una denervación muscular bifásica con un período temprano que aparecía en algún momento antes de los 4 meses pi, seguido por reinervación y una segunda onda denervatoria acaecida alrededor del 6o. mes pi y continuada por una reinervacion lenta y tardía. Ello ocurría en el marco de una miositis, cuya existencia había sido ya descrita por otros autores (25).
Se hicieron, luego, nuevos experimentos con cortes en el tiempo más tempranos y frecuentes en el mismo modelo. Los estudios electrofisiológicos, que ahora incluían el registro de la transmisión neuromuscular

mediante el empleo de microelectrodos, y anatómicos fueron hechos a los 7, 15, 37, 60. 90, 120, 180, 270 y 360 días pi (26). Los hallazgos electro-miográficos fueron, con algunas variantes, similares a los del primer estudio, observándose que los cambios denervatorios aparecían entre los 15 y los 30 días pi (26). En el registro de la unión neuromuscular con microelec-trodos se vio que la amplitud de los pequeños potenciales de placa era menor que la de los controles a partir del día 37 pi. Su frecuencia y contenido cuántico de acetilcolina no diferían de las de los ratones normales, hecho que señalaba la ausencia de alteración de la secresión del transmisor en esa sinapsis.
Finalmente, se encontraron potenciales de placa dobles, en una proporción significativamente mayor que en los controles, a partir del día 90 pi (26). El examen histológico descubrió cambios inflamatorios tempranos en el músculo, presentes a partir del día 7 pi. Las alteraciones atribuibles al compromiso de su inervación se hicieron evidentes a partir del día 90 pi y consistieron en agolpamiento de fibras de tipo 11 y, luego del día 180, atrofia agrupada de fibras musculares (26). En el nervio ciático se detectaron infiltrados de células monucleadas a partir del día 15 pi. identificados como macrófagos la mayor parte de ellos. Junto a los infiltrados se observaron imágenes de desmielinización internodal y paranodal y pérdida de fibras mielínicas. En los cortes de médula se vieron densos infiltrados perivasculares com-puestos por mononucleares que comprometían las raíces y los ganglios espinales (26).
En síntesis, lo observado en el ratón fue la existencia de procesos de denervación y reinervación que se sucedían, a manera de ondas. Un dato más fue la participación del ganglio de la raíz posterior y de ambas raíces, ventral y dorsal, en el proceso patológico. En estos experimentos no fue encontrada lesión de la neurona motora del asta anterior de la médula. Sin embargo, Molina y cols. (27) hallaron pérdida del 21% de neuronas motoras medulares en ratones C3H/HeN infectados con tripomastigotes de la cepa Tulahuen. Posiblemente esta diferencia esté dada por el distinto tropismo de las diversas cepas para determinadas estructuras.
Estos resultados explicaban algunos de los hechos vistos en los humanos. Así, la disminución de la amplitud del potencial sensitivo que fue vista a lo largo de todo el nervio mediano, podía ser atribuida a la lesión del ganglio de la raíz posterior, cuya consecuencia es la despoblación neuronal en él y la pérdida axonal que sigue a la muerte de esas neuronas. Otra aclaración provino del registro de placa y pudo ser referida a las alteraciones vistas en el hombre cuando se estimuló en forma repetitiva el nervio cubital; en los experimentos animales pudo demostrarse que el aspecto pre-sináptico de la unión neuromuscular se mantenía indemne, ya que su contenido cuántico no estaba alterado ni tampoco lo estaba la frecuencia de presentación de los pequeños potenciales de placa, en tanto que la porción post-sináptica era la comprometida, hecho puesto en evidencia por la disminución de la amplitud de los pequeños potenciales de placa, que habla de probable reducción del número de receptores a la acetilcolina a partir del día 37 pi.
Estos últimos resultados parecen contradecir los hallazgos de Tanowitz y cois. (28) quienes encontraron aumento del número de receptores a la acetilcolina en el músculo de ratones infectados con Tripanosoma Cruzi empleando técnicas de marcación con alfa-bungarotoxina, hecho que favorece la presunción de hipersensibilidad de la fibra muscular por denervación; sin embargo, estos autores hicieron sus observaciones hasta el día 30 pi, en tanto que nuestros datos electrofisiológicos que señalan reducción de la cantidad de esos receptores aparecen después del día 37 pi. La presencia de potenciales de placa dobles en el modelo experimental dio también el sustento para la observación del aumento de frecuencia de los potenciales registrados en el extensor corto humano empleando el macroelectrodo de tungsteno. Finalmente, la demostración de Molina y cols. (27) dio el apoyo suficiente como para aceptar el compromiso de la neurona del asta anterior de la médula.
Lo descrito muestra la convergencia de los hallazgos humanos y experimentales y permite aceptar que el sistema nervioso periférico puede estar comprometido en el curso de la enfermedad de Chagas, en su etapa crónica.
Resta por decir si lo descrito en el hombre tiene correlato clínico o es la expresión subclínica de un daño cuya intensidad no es la suficiente como para transformarse en enfermedad clínica y también, cuál es el mecanismo que lleva a la participación del sistema nervioso periférico en esta parasitosis.

En respuesta al primer interrogante, nuestro grupo completó un estudio de rastreo de presencia de enfermedad del sistema nervioso periférico en 550 pacientes con Enfermedad de Chagas en estadio crónico o indeterminado, en los que se dejó de lado toda otra causa que hubiera podido lesionar su sistema nervioso periférico. Se halló que el 10.17% mostraba signos clínicos objetivos que señalaban la presencia de polineuropatía, con predominio del componente sensitivo (29).
En cuanto a la investigación del mecanismo de daño. En el modelo animal han sido identificadas algunas de las poblaciones linfocitarias que infiltran el músculo y el nervio. Así, en ratones Rockland infectados con tripomastigotes de la cepa CA-I el número absoluto de linfocitos L3T4+ y de T Lyt 2+ aumenta continuamente desde el día 15 pi y hasta los 9 meses pi e igual ocurre para los fagocitos MAC-1 + . Por otra parte, también pudieron ser identificados linfocitos B portadores de IgM de superficie en ambos tejidos y depósitos de IgG perivasculares a los 4 meses pi (30). En este aspecto los datos señalan que el fundamento de la alteración está en un desorden inmunológico que aparece una vez que el parásito ha penetrado a su huésped (30).
En favor de esta posibilidad están también las observaciones de Said y cols. (31) quienes pudieron desarrollar granulomas en ratones recipientes sanos mediante la inoculación de linfocitos T "helpers" provenientes de ratones crónicamente infectados con el parásito; este hallazgo sugiere que la inmunidad celular retardada puede tener papel en la patogenia del daño. Sin embargo, resulta útil
recordar los anteriores trabajos de Ribeiro Dos Santos en esta misma línea, quien describió la presencia de anticuerpos antineurona séricos, en pacientes crónicos, ubicados en las fracciones IgG e IgM (32).
En la etapa aguda de la enfermedad, es decir inmediatamente después de la primoinoculación, algunos pacientes muestran capacidad para lesionar su sistema nervioso periférico. Un estudio restringido, exclusivamente electrofisiológico y clínico, en enfermos de área de alta prevalencia de infección, se llevo a cabo en 35 pacientes cuyo tiempo de infección iba entre 5 y 30 días. En 20 de ellos se halló disminución de los valores de conducción nerviosa en uno o más de 4 nervios explorados, en 7 signos de afección muscular inflamatoria y en 9 compromiso de la transmisión neuromuscular (33,34). De Faria y cols. (35) exploraron 8 pacientes en igual período y en todos ellos encontraron signos de reciente denerv ación.
BIBLIOGRAFÍA
1.- Chagas, C. Les formes nerveuses d'un nouvelle trypanosomiase. T. Cruzi inocule par Triatoma Mcgista. Nouv.Iconogr.Salpetriere. 26:1,1913.
2.- Chagas, C. Vilella, E. Forma cardiaca de trypanosomiase americana. Mem. Inst. O. Cruz, 14:5, 1922.
3.- Villela, E. Torres, C.B.M. Estudo histo-patologico do systema nervoso central en paralysia experimental determinada pelo Schizotrypanum Cruzi. Mem. Insr. O.Cruz, 19:175, 1926.
4.- Vichi, F.L. Molestia de Chagas experimental: parasitismo na cadena ganglionar simpática, medul espinal, ganglio raquidiano e musculatura estriada. Hospital, 64:131, 1963.
5.- Vichi, F.L. Destruicao de neuronios motores de ratos na fase aguda da molestia de Chagas. Rev. Inst. Med. Trop. S. Paulo, 6:159, 1964.
6.- Pagano, M., O'Neil l , E. , Aristimuno. G. Electromiograma de de detección en la enfermedad de Chagas. Rev. Neurol. Arg. 3:464, 1977.
7.- Pagano, M., Aristimuno, G., Basso, S., Colombi, A., Sica, R.E.P. Electromyographical findings in human chronic Chagas' disease. Arq. Neuro-Psiquiat. 36:316,1978.
8.- Sica, R.E.P., Sanz, O.P., Aristimuno, G., Basso, S., Pagano, M.A., Taratuto, A., Fumo, T. Ratusnu, A., Colombi, A. Muscle denevation in chronic Chagas' disease. Medicina, 39:579, 1979.
9.- Me Comas, AJ, Fawcett, P., Campbell, M., Sica, R.E.P. Electrophysiological estimations of the number of motor units within a human muscle. J. Neurol. neurosurg. Psychiat. 34:121,1971.
10.- Sica. R.E.P., Me Comas, A.J., Upton, A. Longmire, D. Motor unit estimations in the small muscles of the hand. J. Neurol. Neurosurg. Psychiat. 33:55, 1974.
11.- Sica, R.E.p., Sanz, O.P., Colombi, A. The effeets of ageing upon the human soleus muscle Medicina, 36:443, 1976.
12.- Sanz, O.P. Sica, R.E.P., Colombi, A., O'Neill, E., Pagano, M., Aristimuno, G. Despoblación de neuronas motoras alfa en la enfermedad de Chagas crónica. Medicina, 37:551, 1977.
13.- Sanz, O.P., Ratusnu, A., Aritimunu, G., O'Neill, E., Sica, R.E.P. An electrophysiological investigation of skletal muscle in human chronic Chagas' disease. Arq. Neuro-Psiquiat. 36:319, 1978.
14.- De Faria, C.R., Melo-Souza, S., Rassi, A. Evidencias eletromiograficas de desnervacao motora en pacientes una fase crónica da doenca de Chagas. Rev. Goiana Med. 23:125, 1977.
15.- Taratuto, A., Pagano, M.. Fumo,T.. Sanz, O.P., Sica, R.E.P. Histológica! and histochemical

changes of ihe skeletal muscle in human chronic Chagas' disease. Arq. Neuro-Psiquiat. 36:327, 1978.
16.- O'Neill, E., Sica, R.E.P. Recording of neuromuscular synapticactivity in hcalthy subjccts and in patients with ncuromuscular diseases. Medicina, 38:389, 1978.
17.- Me Comas, A.J., Sica, R.E.P., Campbell, M. Sick motoncuronc. A unifying concept of muscle discases. Lancct, i:321, 1971.
18.- Kimura, J. F wave velocity in the central segments of the median and ulnar nerves. Neurology, 24:539, 1974.
19.- Fernamdez L.s N.A., García Erro, M., Pacagnini, G. Campos, C, Sica, R.E.P. Conducción nerviosa periférica proximal y distal en la insuficiencia renal crónica. Medicina, 47:477, 1987.
20.- Hopf, H.C. Untersuchungen uber die unterschiede in der leigeschwindigkeit motorischer nervenfasern beim menschen. Deuts. Zeits. Nervenheilk. 183:579, 1962.
21.- Sica, R.E.P., Genovese, O., Garcia Erro, M. Peripheral motor nerve conduction studies in patients with chronic Chagas' disease. Arq. Neuro-Psiquiat. 49:405, 1991.
22.- Sica, R.E.P., Füipini, D., Panizza, M., Fumo, T. Basso, S., Lazzari, J., Molina, H. Involvement of the peripheral sensory nervous system in human chronic Chagas' disease, Medicina, 46:662, 1986.
23.- Garcia Erro, M., Genovese, O., Corréale, J. , Sica, R.E.P. Neuromuscular transmission
studies in human chronic Chagas' disease. Arq. Neuro-Psiquiat. 47:279, 1989.
24.- González Cappa, S.M., Sanz, O.P., Muller, L., Molina, H., Fernandez, J., Rimoldi, M.T., Sica, R.E.P. Peripheral nervous system damage in experimental chronic Chagas' disease. Am. J. Trop. Med. Hyg. 36:41, 1987.
25.- Laguens, R., Cossio, P, Diez, C, Segal, A., Vázquez, C. Kreutzer, E., Khoury, E., Arana, R. Inmunopathologic and morphologic studies of skclctal muscle in Chagas' disease. Am. J. Pathol. 80:153, 1975.
26.- Losavio, A., Jones, M., Sanz, O.P., Mirkin, G., González Cappa, S.M., Muchnik. S., Sica R.E.P. A sequential study of the peripheral nervous system involvement in experimental Chagas' disease. Am.J. Trop. Med. Hyg. 41:539, 1989.
27.- Molina, H., Cardoni, R., Rimoldi, M.T. The neuromuscular pathology of experimental Chagas' disease. J. Neurol.Sci. 81:287, 1987.
28.- Tanowitz, H., Davies, R, WiUner, M. Alterations in acelylcholine receptors in experimental Chagas' disease. J. Infect. Diseases. 147:460, 1983.
29.- Genovese O., Ballario, C, Slorino, R., Segura, E.. Sica REP.Clinical manifestations of peripheral nervous system involvement in human chronic Chagas Disease. Arq. Neuropsiquiatr. 54:190,1996.
30.- Mirkin. GA., Jones, M., Sanz, OR, Rey, R., Sica, RER,González Cappa, SM. Experimental Chagas'
disease: eiectrophysiology and cell composition of neuromyopathic inflammatory lesions in mice infected with a myotropic and pantropic train of Trypanosoma Cruzi. Clin. Immunol. Immunopathol.73:69, 1994.
31.- Said, G. Joskowits, M. Barreira, A., Eisen H. Neuropathy associaled with experimental Chagas7 disease. Ann.Neurol. 18:676, 1985.
32.- Ribeiro Dos Santos, R., Márquez, J.O. Mecanismos inmunologicos comprometidos en la destrucción neuronal que ocurre en la enfermedad de Chagas. Rev. Neurol.Arg. 3:439, 1977.
33.- Bcnavcntc, O., Ledesma, P.O., Lugones, H., Kalala, E., Marteleur, A., Sica, R.E.R Compromiso del sistema nervioso periférico en la fase aguda de la enfermedad de Chagas. Medicina, 46:645, 1986.
34.- Benavente, O., Ledesma, P.O., Baez, R, Lugones, H., Kalala, E., Ribas, C, Genovese, O., Sica, R.E.R. Motor unit involvement in human acute Chagas' disease. Arq. Neuro-Psiquiat 47:283, 1989.
35.- De Faria, C, Melo-Souza, S., Rassi, A., Lima, A. Evidencias eletromiograficas de desnervacao motora en pacientes una fase aguda da doenca de Chagas. Rev. Goiana Med. 25:153, 1989.

RESÚMENES IX CONGRESO CENTROAMERICANO DE
NEUROLOGÍA YII CONGRESO CENTROAMERICANO DE
NEUROFISIOLOGIA CLÍNICA
ENFERMEDAD CEREBROVASCULAR
MANAGEMENT OFACÜTE STROKE, STRATEGIES FOR A NEW MILLENIUM
William 1. Brannon, MD, FACP University of South Carolina
The past two decades have brought a new understanding of the pathophysiology of cerebral infarction. With that understanding, new and exciting opportunities for treatment to minimize the adverse outcome of cerebral infarction have been and are being developed. Traditional methods of anticoagulant therapy folio wed by attempts at rehabilitation are no longer sufficient therapies. Physicians practícing in the new millenium will have the opportunity to use new treatments for patients with acute stroke. Preventive measures modifying risk factors for síroke wiíl remain a first line treatment approach.
For those patients who have an acute cerebral vascular event. the naíure of the event must be recognized as quickly as possible and the patient received for acute treatment. Patients who have a thromboembolic event as demostrated by examination and by an image of the brain do not have an intracerebral hemorrhage acute therapy should be instituted. Available therapies include:
1) anticoagulant drugs, 2) thrombolytic therapy, 3) neuroprotective drugs and 4) combinations of
neuroprotective drugs and thrombolytic or anticoagulaní therapies. Each of these therapies will be discussed in this presentation.
Key words: stroke, therapy, anticoagulant and neuroprotective drugs, thrombolytic therapy
HEMORRAGIA SUBARACNOIDEANO TRAUMÁTICA: CONTROVERSIAS
Dr. Roberto López Aguilar Hospital Nacional Rosales, Centro Médico Nacional, El Salvador
La hemorragia subaracnoidea es una entidad clínica conocida por primera vez en pasajes bíblicos, es producida por ruptura de aneurismas o malformaciones arteriovenosas, siendo el síntoma principal la cefalea intensa nunca antes conocida por el paciente. Su diagnóstico se realiza por tomografía cerebral o análisis del líquido cefalorraquídeo. Debe tenerse en cuenta que las dos grandes complicaciones de HSA son resangrado y vasoespasmo.
Hoy en día, las corrientes actuales más consistentes apoyan que sólo la cirugía del aneurisma en forma temprana previene el resangrado; así como la instauración de la triple terapia H (hipertensión, hipervolemia y hemodilución) y el uso de las 1,4 dihidropiridinas (nimodipina y nicardipina) en el manejo de vasoespasmo y la isquemia cerebral. La principal acción de la nimodipina es favorecer la apertura de pequeños vasos colaterales piales en el área isquémica; una acción similar presenta la nicardipina, con mejor efecto en la prevención del vasoespasmo y reducción de los síntomas isquémicos.
Se concluye que el manejo quirúrgico del aneurisma, previo estudio angiográfico lo más temprano posible, es la terapia definitiva para los aneurismas; así como instaurar en forma temprana el uso de bloqueadores de los canales de calcio dependientes de voltaje y la terapia triple H para el manejo del vasoespasmo arterial y la isquemia cerebral.
Palabras clave: hemorragia subaracnoidea, tratamiento, complicaciones

TRATAMIENTO DE LA HIPOTENSIÓN ORTOSTÁTICA
Dr. Eduardo Benarroch Mayo Clinic; Rochester, Minnesota
Los objetivos en el tratamiento de la hipotensión ortostática (HO) son mejorar la tolerancia ortostática. educar al paciente, corregir causas reversibles con uso de vasodilatadores y diuréticos, aumentar el volumen intravascular; mejorar el tono vasomotor; y evi tar la hipertensión en la posición supina. El paciente debe ingerir por lo menos 10-20 gramos de sal, y 2.0-2.5 litros de líquidos diarios.
Las maniobras posturales, medias compresivas y ejercicio moderado aumentan la tolerancia ortostática. El dormir con la cabecera de la cama elevada 15-30 cm puede evitar la hipertensión y poliuria nocturnas. La fluodrocortisona (0.1-1 mg) es la droga de primera línea para tratar la HO. El uso de cafeína o inhibidores de prostaglandina puede ayudar en casos de hipotensión postprandial.
Los vasoconstrictores ( f e n i l p r o p a n o l a m i n a , d i h i d r o e r g o t a m i n a ) , particularmente la midodrina (10-40 mg diarios) son el segundo paso. Drogas útiles en casos especiales son la eritropoyetina (anemia hipocrómica), la desmopresina (nicturia marcada) y el octreótido (hipertensión postprandial). La hipertensión en posición supina se evita con la elevación de la cabecera de la cama, abstinencia de drogas presoras por la tarde, y el uso de nifedipina, hidralazina o captopril al acostarse.
Palabras clave: hipotensión, tratamiento
NEUROLOGÍA DEL SINCOPE
Dr. Eduardo Benarroch Mayo Clinic; Rochester, Minnesota
El síncope es la pérdida brusca y transitoria de la conciencia debida a una disminución global del flujo sanguíneo cerebral. En el 90% de los casos, se debe a mecanismos hemodinámicos, tales como reflejos vasodepresores (síncope vasovagal o neurocardiogénico, del seno carotídeo o situacional); hipotensión ortostática; o cardiopatías (arritmias o cardiopatías estructurales). En un 10%, las causas son primariamente neurogénicas, incluyendo la epilepsia temporal, la enfermedad cerebrovascular (isquemia vertebrovasilar); la hipertensión intracraneal (hidrocéfalo agudo, malformación de Arnold-Chiari tipo 1) y, raramente, la hiperventilación o la hipoglucemia.
El neurólogo está involucrado en el diagnóstico diferencial entre síncope y crisis epiléptica; la evaluación de la posibilidad de enfermedad cerebrovascular o enfermedad estructural intracraneal como causa del síncope, y la evaluación de la hipotensión ortostática neurogénica. La historia clínica, el examen físico y el electrocardiograma son los elementos más importantes en el diagnóstico. El uso del electroencefalograma prolongado y video-E.E.G. pueden ser útiles en el diagnóstico diferencial entre síncope y epilepsia. En la ausencia de déficits focales, los estudios cerebrovasculares son de
poca utilidad. Las pruebas de función autonómica, incluyendo la prueba de basculación, son de utilidad en la valoración de la hipotensión ortostática y el síncope de causa desconocida.
Palabras clave: síncope, causas, diasnóstico diferencial
AVANCES EN EL MANEJO DE LA MIGRAÑA
Dr. Fernando Gracia Jefe Del Servicio De Neurología Hospital Santo Tomás, Panamá
La serotonina descubierta en 1948 causó gran impacto en la comunidad científica al ser relacionada con la patogénesis de la migraña. Su precursor es el triptófano de la dieta proteica, luego de la acción de la enzima triptófano-hidroxil as a es almacenada en vesículas y por exocitosis es liberada al espacio sináptico. Existen siete receptores 5HT-1-7, la estimulación selectiva de los receptores 5HT1B/5HT1D puede mejorar el ataque de la migraña. Los triptanes estimulan el 5HT1B produciendo vasocontricción de los vasos sanguíneos meníngeos de la dura y de la piamadre, al estimular el receptor 5HT1D produce inhibición de la inflamación neurogénica de la duramadre.
En 1991 se aprueba el uso del primer triptánico, el sumatriptán. En 1997 el zolmitriptán y el naratriptán y en 1998 el rizatriptán. En la actualidad se sintetizan tres nuevos triptánicos, el eletriptán, almitriptán y el frovatriptán, cuyos lanzamientos se estiman para el año 1999-2000. La denominada guerra de los triptánicos de la década de los 90's

surge a consecuencia de no haberse encontrado el medicamento ideal para el ataque agudo de la migraña, que actúe rápidamente para el alivio del dolor y síntomas asociados, mínimos efectos secundarios, que su eficacia se mantenga con el tiempo y recurrencias mínimas, con mejoría significativa de la calidad de vida.
La eficacia clínica, frecuencia de recurrencia y efectos adversos de los triptánicos disponibles no demuestran diferencias significativas, aunque los conocidos como triptánicos de segunda generación se observan algunas ventajas farmacocinéticas. Esperamos que la toma de decisión en el uso del mejor triptánico sea finalmente por su mejor eficacia clínica, característica de los pacientes y, experiencia del médico tratante y no por razones de mercadeo.
Palabras clave: triptánicos, serotonina
NIMODIPINAY NEUROPROTECCIÓN
Dr. Carlos Antonio Díaz Manzano El Salvador
La nimodipina es una dihidropiridina sintetizada por el Doctor N. Meyer en los Laboratorios de Investigación BAYER, es un calcio antagonista cuya principal indicación, inicial, es para la prevención del vasoespasmo en la HSA. Pero posteriormente al estudiar su mecanismo de acción, bloqueando los canales del Ca su indicación se desbordó ya que las patologías que causan con el
incremento del Ca2 intraneural son muy variadas en el SNC, desde una isquemia cerebral que resulta en demencias. Todos ellos conllevan un incremento de Ca, lo cual puede iniciar una cascada de eventos bioquímicos que nos pueden llevar a una lisis celular y muerte neuronal. Así la nimodipina es un fármaco que tiene propiedades de neuroprotección siendo de mucha utilidad en neurología.
Palabras clave: nimodipina, isquemia cerebral, muerte neuronal
ENFERMEDAD
NEUROMUSCULAR
YNEUROFISIOLOGIA
PRINCIPLESIN NEUROPHYSIOLOGY SHORTANDLONGOFNERVE CONDUCTIONSTUDIES
Jun Kimura, M.D. Department of Neurology Kyoto University Hospital
Electrical stimulation of the nerve initiates an impulse, which travels along motor, sensory, or mixed nerves. For the assessment of conduction characteristics, compound evoked potentials are recorded from the muscle in the study of the motor fibers and from the nerve itself in the case of the sensory fibers. With standardization the methods allows precise localization of a lesión and accurate characterization of peripheral nerve function. Conventional studies primarily deal with the evaluation of the fastest conducting fibers, based on the Iatency measured to the onset of
the evoked potential. In some clinical entities, other aspects of nerve conduction may help.
The routine conduction studies include those for the motor and sensory fibers of the median, ulnar, and radial nerves, the motor fibers of the peroneal and tibial nerves, the sensory fibers of the sural and superficial peroneal nerves, and the facial and accessory nerves. The basic principies apply to any nerves despite the anatomic peculiarities that dictate specific approaches to some of the commonly tested invividual nerves. Measuring the blink reflex, the F wave, or the H reflex supplements the limited information provided by the ordinary conduction studies on the central or most proximal nerve segment such as the radicular portion. Thisreview summarizes the fundamental concepts of nerve stimulation techniques, and discusses their proper application in the clinical domain.
Optimal application of the nerve conduction study depends on an understanding of the principies and recognition of the pitfalls of the technique. The conventional methods deal primarily with distal nerve segments in the extremity. Newer techniques allow one to assess nerve segments in less accessible anatómica! regions, to improve the accuracy in precisely localizing a focal lesión, and to increase sensitivity in detecting subclinical abnormalities. Despite certain limitations, these methods can provide diagnostically pertinent information if they are used judiciously in appropriate clinical contexts.
Key words: neurophysiology, nerve conduction, evoked potentials

DESNUTRICIÓN Y FISIOLOGÍA DE LA MÉDULA ESPINAL
Humberto Su, Heyke Hesse, Winston Mejía, María.Félix Rivera, GregoryJ., Quirk Laboratorio de Neurofisiología, Depto. de Ciencias Fisiológicas, Facultad de Ciencias Médicas, Ü.N.A.H..
Se ha estimado que más de 1/3 de los niños de Honduras sufren de retardo de crecimiento secundario a desnutrición crónica (Onis et al, 1993). Se indujo desnutrición postnatal temprana en cachorros lactantes desde el día del parto al destete (día 21). Después del destete, los cachorros de rata tuvieron acceso libre de alimentos hasta la adultez (día 75), tiempo en el cual fueron examinadas sus respuestas corticoespinales. Bajo anestesia con uretano, la corteza motora fue estimulada con electrodos de superficie para registrar las respuestas poblacionales del tracto corticoespinal a nivel de Cl (Patton y Amassian, 1954; Stewart et al, 1990). Además de reducir el peso corporal en 20 % y el peso cerebral en 9 % con respecto a los controles, la desnutrición temprana redujo la velocidad de conducción de las fibras corticoespinales más veloces de 20 m/s a 16.9 m/s (16%,p<0.001).
Además, el tamaño de la respuesta directa a la estimulación superficial fue significativamente reducida en las ratas desnutridas. Este efecto fue más pronunciado a menores intensidades de estímulo, las cuales activan las fibras de menor umbral y de mayor diámetro. Estos resultados sugieren que la desnutrición temprana reduce el número de fibras de mayor diámetro en la rata adulta.
En un estudio paralelo, comparamos los potenciales evocados somatosensoriales (SEP) en 20 niños con retardo severo del crecimiento y 20 niños controles de la misma edad (7-8 años). Los niños desnutridos fueron seleccionados por tener una talla para edad por debajo del tercer percentil. Los 2 grupos de niños fueron estadísticamente diferentes en término de hematócrito y desempeño en un test neurointegrativo (Bender). Los SEP se obtenían estimulando el nervio mediano y se registraba la respuesta cervical (NI3) y cortical (N20). A pesar de las diferencias en las otras variables, no hubo diferencia en el tiempo de conducción central (N13-N20) entre los dos grupos (Control= 6.30 ms y Desnutrido = 6.19 ms). No está claro que factores podrían explicar l a s diferencias encontradas. Las posibilidades incluyen la severidad y la duración de la desnutrición, susceptibilidad a la desnutrición específica a cada especie y, más probablemente, una diferencia entre los ciclos mielogenéticos de los sistemas motores y somatosensoriales.
Palabras clave: desnutrición, potenciales evocados, test de Bender
SÍNDROME DE GUILLAIN BARRÉ: EXPERIENCIA EN HONDURAS
Dr. Carlos A. Orellana, Dr. Marco A. Quiñónez Sala De Cuidados Intensivos, Hospital Escuela, Honduras
Antecedentes: El síndrome de Guillain Barré es la causa más frecuente de parálisis generalizada
aguda, su incidencia anual es de 0.75 a 2 casos por 100 mil habitantes. Son criterios clínicos para su diagnóstico: debilidadad progresiva en extremidades y arreflexia. Son características distintivas: simetría, disfunción autonómica, concentración aumentada de proteínas en LCR y el electrodiagnóstico. El tratamiento consiste en corticosteroides, plasmaféresis e inmunogl obulina.
Objetivos: Describir la evolución de pacientes con Sd. de. Guillain Barré que progresaron a insuficiencia respiratoria.
Métodos: Revisamos datos clínicos y epidemiológicos de los pacientes admitidos en UCI/HE, durante el período de enero/95 a diciembre/98.
Resultados: El análisis de distribución por sexo mostró mayor incidencia en hombres. El grupo de edad más afectado fue el de adulto joven (26-50 años). En más del 50% no se encontró ningún antecedente epidemiológico. En la mitad de los casos los pacientes buscaron atención médica temprana (1-5 días). Los miembros inferiores fue el grupo muscular más afectado. La concentración proteica del LCR a las 2 semanas fue elevada en el 50% de los casos. La mayoría tuvo parálisis ascendente y simétrica. Casi todos (14 pacientes) necesitaron ventilación mecánica, 10 de ellos sufrieron infecciones nosocomiales, ITU, traqueítis y neumonía en orden de frecuencia y todos más de una infección. En su mayoría fueron retirados del ventilador en las primeras tres semanas. El 79% presentó disautonomías que consistieron en HTA taquicardia y diaforesis. La permanencia en UC1 fue en promedio tres semanas. La

mortalidad fue de 25%.
Palabras clave: Guillain-Barré en Honduras, características clínicas, complicaciones
THE''CYCLAMEN HAND" IN INCLUSIÓN BODYMYOSITIS
Dn Parker A. Towle The Hitchcock Clinic-Littleton Neurology E-mail: Parker.A. Towle @ Hitchcock. org
The diagnosis of IBM (inclusión body mytosis) is often missed in spite of the fact that it is the most common primary muscle disease of middle and older age. The reasons for this are many. While it was defined in 1967 and named in 1970, most of the literature about the condition has been written only in the past 5 years. Laboratory studies aside from the creatine phosphokinase (CPK) assay may not be particularly helpful and electrodiagnosis studies in at least a third of cases may show neuropathic in addition to myopathic features. Associated diseases may obscure the clinical presentation.
The intent of this study was to define clinical characteristics that might be helpful in diagnosis, prognosis, and therapy. For this purpose I used cases from my own practice and reviewed the relevant literature. The pattem of weakness was distinctive being both próxima! and distal, occasionally asimetrical. Long finger flexors and, in particular, the flexor digitorum profundus muscles were often prefentially involved in such a manner that on attempting a fist the fingertips actually extended producing a hand posture resembling the petal
formation of the cyclamen flower bloom. This posture had been described by Dalakis and others but not previously so named.
Other usual distictive clinical features of IBM were quadriceps muscle weakness, older age of onset, male predominance (2:1). occasional dysphagia, less pain than other adult onset muscle diseases (polymyositis, dermatomyositis), and not inherited. In conclusión IBM may be suspected on the suspected on the basis of characteristic clinical features, including the "cyclamen hand", CPK may be helpful but other laboratory and electrodiagnostic testing may not. Muscle biopsy confirms the diagnosis. Histopathology, differential diagnosis, therapy, and prognosis will be briefly mentioned.
Key words: cyclamen hand, inclusión body myositis, diagnosis
EPILEPSIA
STATUS EP1LEPTWVS: MECHANISMS, TREATMENT
Antonio V. Delgado-Escueta, M.D (1) California Comprehensive
Epilepsy Program, UCLA School of Medicine and West Los Angeles DVA Medical Centén cind
(2) UCLA Brain Research Institute, Los Angeles, California, USA;
(3 GENetic Epilepsy Studies (GENES) International Consortium.
Status Epilepticus (SE) has remained a worldwide health problem ever since it was first
recognized by Calmeil in 1824. The yearly incidence of individuáis who sufferfrom status epilepticus in the USA ranges from 60,000 to 250,000. In this communication, we review four topics in detail: (1) The molecular events that set in motion the fine structural changes of cell damage induced by SE and the basic mechanisms by whích convulsive status leads to epileptogenesis and sets up chronic epilepsy. (2) The principies of SE management and the recently completed comparative triáis of AEDs in SE by Treiman et al. and the acute treatment of "refractory" SE. (3) Dmgs recently approved by the USA Food and Drug Administration, namely, (a) diazepam rectal gel, (b) intravenous fosphenytoin, and (c) intravenous valproate. (4) How the present tools for in vivo biochemistry, functional magnetic resonance imaging (fMRI), magnetic resonance spectroscopy (MRS), and diffusion-weighted imaging (a special form of MRI), offer special promise in allowing us to examine basic mechanisms and test our rationale for treating SE in human.
Key words: Status Epiiepticus, Mechanisms, Treatment
THE SEARCH FOR EPILEPSY GENES INJUVENILE MY0CL0NIC EPILEPSY (JME): DISCOVERIESALONGTHE WAY
Antonio V. Delgado-Escueta, MD (1,2,3), Maria E. Alonso, MD (3,4), Marco T. Medina, MD (1,3,5), Manyee N. Gee (1,3)
(1) California Comprehensive Epilepsy Program, UCLA School

DVA Medical Center, and (2) UCEA Brain Research Institute, Los Angeles, California, USA; (3) GENetic Epilepsy Studies (GENES) International Consortium; (4) National Institute of Nenrology and Neurosurgery, México City, México; (5) National University of Honduras, Tegucigalpa, Honduras
Of the 40 to 100 million persons with epilepsy, worldwide, the most common idiopathic epilepsy (10% to 30%) is Juvenile Myoclonic Epilepsy (JME), which is also known in Europe as the Janzsyndrome. Asaresultof our quest for epilepsy genes and our recruitment of families with myoclonic seizures, our GENES (GENetic Epilepsy Studies) International Consortium localized the gene for the Laforas progressive myoclonus epilepsy in chromosome 6q24 and helped to identify the mutation in the gene for the protein tyrosine phosphatase. Our consortium also identified (1) the 0.6 cM critical región for classic JME located some 30 cM below HLA in chromosome 6pll, (2) the 7 cM área for pyknoleptic childhood absences epilepsy (CAE) that evolves to or is mixed with JME in chromosome lp, and (3) the 0.5 cM área for pyknoleptic CAE with grand mal and irregular 3 to 4 Hz spike waves in chromosome 8q24. Now we also report on the progress in mapping the syndrome of photogenic CAE with eyelid myoclonia that evolves to JME. Separating the syndromes of pyknoleptic absences mixed with JME and the syndrome of typical childhood absence from classic JME facilitates the search for genes underlying JME.
Palabras clave: neurogenética, epilepsias idiopáticas, mapeo genético, ligamiento genético.
TERAPIAFARMACOLOGICA DE LAS EPILEPSIAS
Dr Franz Chaves Sell, Clínica Bíblica San José, Costa Rica
Es en el año de 1857 cuando Charles Locock descubre en forma indirecta las propiedades antiepilépticas de los bromuros. A pesar de los grandes limitantes en su uso: sedación, impotencia sexual, trastornos dermatológicos y gastrointestinales y la encefalopatía tóxica severa, fue la única droga disponible hasta 1912. En esta fecha, Hauptmann introduce el fenobarbital y los barbitúricos en general, medicamentos que vinieron a revolucionar el tratamiento de este mal gracias a su efectividad y seguridad. En 1937, Merritt y Putnam introducen la difenilhi-dantoína y prácticamente con estos dos medicamentos se manejó la mayoría de pacientes por largos años, restando soluciones efectivas para los pacientes con crisis de ausencias, mioclonías, crisis parciales complejas y espasmos infantiles.
En los años 60 a 70 aparecen las benzodiazepinas, la carbamazepina y el ácido valpróico, que junto a los dos primeros citados, constituyen el principal arsenal terapéutico en epileptología. También el mecanismo de acción de las drogas antiepilépticas se conoce hasta recientemente ya que el descubrimiento de neurotrasmisores y receptores específicos data de los últimos 30 años. Actualmente podemos definir los principales modelos de acción farmacológica a partir de los siguientes mecanismos: 1) Asentes modificadores de la
transmisión sináptica : a) aumentando la inhibición GABAérgica: benzodiazepinas, barbitúricos, vigabatrina, b) disminuyendo la transmisión exicitatoria : fenobarbital y D-CPPene. 2). Agentes que actúan a nivel de canales iónicos: a) prolongando la inactivación de los canales de sodio ( fenitoína, carbamazepina, valproato), b) bloqueando los canales lentos de calcio: etosuximida y trimethadiona.
A partir de la década de los 80 una nueva era de investigación da inicios con resultados poco satisfactorios y no es hasta los 90 en que se introducen nuevos productos con resultados prometedores como lamotrigina. vigabatrina, gabapentina, tiagabina, topiramato y la oxcarbazepina entre otros. Sin embargo a pesar del gran aporte que estos nuevos medicamentos han traído sobre todo en relación a una mejor tolerancia por menos efectos secundarios y una mayor especificidad en sus mecanismos de acción farmacológica, aún tenemos alrededor de un 20% de pacientes epilépticos que no responden a la terapia medicamentosa (refractarios) por lo que se hace necesario continuar avanzando en la búsqueda de soluciones como la cirugía y otras drogas que vendrán en un futuro próximo.
Palabras clave: antiepilépticos, historia, mecanismos
LENNOX-GASTAUT SYNDROME AND INFANTILE SPASMS
Edwin Trevathan, M.D., M.P.H., Pediatric Epilepsy Center, Departments of Neurology & Pediatrics, Washington University

School of Medicine, St. Louis Childrenís Hospital, st, Louis, Missouri, USA.
Lennox-Gastaut Syndrome (LGS) and Infanule Spasms (IS) are rare; in Atlanta LGS accounts for about 5% of childhood epüepsy and IS has an incidence of 3 per 10,000 births. Several therapies for IS and LGS have been introduced in the 1990s, challenging traditional methods of treatment. ACTH, benzodiazepines, and valproate (VPA) have been first-line drugs for IS. Recently vigabatrin (VGB) has been a first-line drug for TS in Europe. but retinal side effects may reduce the use of VGB. Tiagabine, topiramate (TPM), lamotrigine (LTG), and ganaxalone may be helpful in IS. Although VPA has been the drug of first choice for LGS, clinical triáis of VPA in LGS have never been published.
Felbamate (FBM), LTG, andTPM have all been effective in reducing múltiple seizure types among patients with LGS in clinical triáis. The efficacy of the ketogenic diet has been reproduced in open-label studies, and an NIH-sponsored blinded clinical trial of the ketogenic diet is in progress. Other clinical triáis of new drugs and vagus nerve stimulation in LGS are pending. A rational approach to the management of IS and LGS, based upon available efficacy and adverse event data will be presented with opportunity for discussion.
Key words: Lennox-Gastaut Syndrome, Infantile Spasms, treatment, drugs of choice
TRATAMIENTO
ANTIEPILÉPTICO: TOPIRAMATO
Dr. Henry B. Stokes, Neurólogo-Neu rofi siólogo Clínico, Guatemala
La epilepsia como desorden, enfermedad o síndrome se remonta desde los orígenes de la humanidad, y ha tenido una descripción como entidad médica hace más de dos mil años. Constituye junto con las cefaleas la causa más frecuente de dolencias neurológicas. Su tratamiento efectivo se remonta a 1857 cuando Charles Locock observó en un hospital psiquiátrico que los pacientes masturbadores y con conductas sexuales anormales, y que a veces tenían epilepsia, mejotraban su condición epiléptica al darles brumuros. Por 50 años este fue el único tratamiento antiepiléptico.
En 1912 Hauptman introduce los barbitúricos y luego en 1938 Merrit y Putman utilizando modelos experimentales de producción de crisis con pentiniltetrazol, pricrotoxina, biculina estricnina descubre la fenitoína, teniendo núcleos imidazoles similares al fenobarbital y modificando esta estructura básica con radicales en distintas áreas, surgieron los otros medicamentos con perfil antiepiléptico dependiendo de los sustitutos en el núcleo central o en sus cademas, por ejemplo las dionas succimidas para las crisis parciales o generalizadas convulsivas correspondiendo a este grupo el fenobarbital, las fenitoinas, las carbamazepinas y las acetil ureas.
En 1982, Goodman introduce el concepto de umbral y propagación de las descargas. Así es como surgen los modelos biológicos del tratamiento de las crisis, encontrando que la neuroexcitación neuronal era prevenida con estimulaciones del GABA mien t ras que estimulaciones de aspartato y el glutamato producían excitoxicidad y crisis epilépticas.
Surge un nuevo tipo de antiepilépticos distinto a base de ácidos valproico. Luego aparecen los nuevos antiepilépticos con blanco en los receptores y neurotransmisores con perfiles limitados y amplios. Mecanismos encontrados: a) Bloqueadores de canales de sodio: bajan excitación: fenitoinas, carbazepinas, b) bloqueadores de canales de calcio: efectivos en crisis de ausencia: valproato, succinamidas, efecto básicamente subcortical talámico, c) Tiagagabine, vigabatrin, gabapentina: aumentando la actividad gabaérgica y estimulando los canales de cloro para hiperpolizar la membrana, d) Efecto a varios niveles: valproatos, topitramato, ferbamato, estos actúan a nivel de canales de sodio, de calcio, y GABA además de que algunos también intervienen en disminuir la excitoxicidad inhibiendo los receptores que a su sobreactivación producen estimulación de receptores NMDA y no como NMDA como el AMPA y kainato.
El topiramato es un novedoso antiepiléptico, de los mejores por lo siguiente: a) Actúa sobre los canales de sodio y calcio, b) tiene efecto inhibitorio sobre los receptores NMDA y no NMDA como el AMPA, c) tienen efecto sobre la anhidrasa carbónica, lo

que favorece una acidificación local cerebral aumentando el umbral antiepiléptico, d) farmacocinecia y farmacodimamia que permite darlo una vez por día, e) buena tolerancia, f) poca toxicidad, g) una excreción no hepática, lo cual evita muchas de las interacciones medicamentosas, h) efecto de amplio espectro: crisis parciales, generalizadas convulsivas y no convulsivas, i) tolerancia gastrointestinal buena, algunos casos complicados con nefrolitiasis, j) potencialmente por su efecto sobre los receptores que median la excitoxicidad como son el NMDA y AMPA, puede tener utilidad como un neuroprotector contra enfermedades neurológicas crónicas.
Palabras clave: antiepilépticos, topiramato, mecanismos, usos
OXCARBACEPINA: MESA REDONDA
Dr. Jenner Velásquez (Guatemala), Dr. Franz Chávez (Costa Rica), Marco Tulio Medina (Honduras)
La oxcarbacepina (OCBZ) es el ceto-análogo de la carbamacepina que comparte su eficacia pero que no se metaboliza a epóxido, lo que la hace tener menos efectos secundarios e interacciones medicamentosas. Por ello, en algunos países ha sustituido a la carbamacepina (CBZ) como droga de elección en mono y politerapia para las epilepsias parciales en niños y adultos . Su similitud estructural y eficacia c l ín ica respecto a la carbamacepina sugieren que su mecanismo de acción involucra la inhibición de potenciales de acción anormales de sodio voltaje-dependientes.
Su absorción es rápida y completa alcanzando niveles terapéuticos después de 3 a 4 dosis en un régimen de dos veces al día. Su componente es rápida y extensamente metabolizado al metabolito 10,11-dihydro-10-hydroxy-carbamacepina, un derivado monohidróxido (MHD) que es responsable del efecto terapéutico y que tiene una vida media de 8 a 10 horas. El 27% de la dosis es excretada en orina sin transformación, 49% en forma de conjugado glucorónido. Estudios de farmacocinética indican que OCBZ no induce el grupo citocromo P-450 aunque sí lo hace con el grupo citocromo P-450 111A, responsable del metabolismo de los estrógenos y los bloqueadores de canales de calcio tipo dihidropiridinas.
En aquellos pacientes con crisis refractarias, la sustitución de CBZ por OCBZ ha mostrado disminución en la frecuencia de crisis y mejoría cognitiva. La sustitución puede hacerse cambiando la dosis inmediatamente de forma que el paciente toma la última dosis de CBZ un día y comienza con una dosis normal de OCBZ el siguiente. Aún pacientes con hipersensibilidad a CBZ pueden ser tratados con OCBZ. Otros usos que la oxcarbacepina comparte con la CBZ es el tratamiento de neuralgias y estudios controlados preliminares sugieren propiedades antimaníacas en algunos pacientes.
Palabras clave: oxcarbacepina, epilepsias parciales, farmacocinética
CYSTICERCOSISAND EPILEPSY
Dr. Marco Tulio Medina, Chairman, Research Unit, School of Medical Sciences, Honduras Dra. Reyna Durón-Martínez, Resident of Neurology, Hospital Escuela, Honduras
Cysticercosis is acquired by ingesting Taenia solium eggs (fecal contaminaron), and may occur in humans who neither eat pork ñor share environments with pigs. Neurocysticercosis (NCC) is the most frequent parasitosis of the central nervous system in several countries in Latin America, Asia and África and due to the high immigration from endemic to non endemic áreas, it is commonly seen in countries previously free of the disease. Acute symptomatic seizures and epilepsy are the most common presentation, however, there is a high percentage of the population in endemic countries harboring active or inactive NCC remaining asymptomatic.
In a recent case-control study in Honduras (Sánchez et al 1999). 480 persons were studied, the overal seroprevalence of antibodies for cysticercosis was 17% using Western Blot (ETTB) and the taeniasis prevalence was 2.5%, this studied community could be considered an hyperendemic for Taenia solium. Twenty one percent of seropositive persons had CT sean lesions compatible with NCC, only 6% had seizures or epilepsy, the majority was asymptomatic.
Tn other epidemiological study, we performed a house to house survey in the community of Salama Honduras (6.743 persons), finding and overall epilepsy prevalence of 23.1 per

1000, and 17 per 1000 of active epilepsy. Brain CT sean, video-EEG and serum EITB were done in ninety out of a hundred of the active epilepsy patients. Thirty seven of them had epilepsy due to NCC, the diagnosis was done according with Del Brutto et al and Sánchez et al diagnostic criteria. We found that epilepsy due to NCC has some characteristic features: a) the high frequeney of partial seizures with or without secondary generalization (mainly of frontal or temporal lobe), b) clinical manifestation depending on the location of the parasite in brain parenchyma, and c) high frequeney of calcified granulomas as sequelae of NCC, without other signs of active disease.
The epileptogenesis is related to: a) hight frequeney of gliosis and probable a sprouting mechanism, b) location of the lesions, c) probable genetic predisposition. The outeome of NCC after specific treatment (albendazole and praziquantel) has been judged mostly on the disappearance of cysts on repeat CT Sean (Sotelo etal 1984, 1988, 1990, Escobedo et al 1987). Controversy over the importance of specific anticysticercal treatment is ongoing, but Vasquez and Sotelo's study (1992) and Medina's et al study (1991, 1993), suggest a positive effect of the anticysticercar treatment on the prognosis of acute symptomatic seizures or epilepsy caused by active NCC.
Key words: cysticercosis, epidemiology, characteristics, treatment
MITOCONDRIOPATIAS
DOESTHEPATIENTHAVEA MIT0CH0NDRIAL DISEASE?
Dr. Salvatore Di Mauro Columbia University College Of Physicians And Surgeons New York, Ny. USA
This has become in recent years one of the most common questions asked by general pediatricians and specialist alike. In this context, "mitochondrial disease" refers to defeets in a restricted but vital área of mitochondrial metabolism, the respiratory chain, which presents the "business end" of oxidative metabolisms, wheremostcellular ATP is generated.
The ubiquitous nature of mitochondria, the dual genetic control of the respitatory chain, and the peculiar rules of mitochondrial genetics are all factors that contribute to the extraordinary spectrum of clinical presentations associated with defeets of oxidative phosphorylation. After a brief historical background, I will briefy review the principies of mitochondrial genetics, including ( i ) maternal inheritance; (ii) heteroplasmy and the threshold effect; and (iii) mitotic segregation, discussing their relevance to clinical practice.
In trying to give practitioners useful diagnostic clues, I will consider secuentially eight different tools: 1) Symptoms and signs; 2) inheritance; 3) laboratory data; 4) neuroradiology; 5) exercise physiology; 6) muscle biopsy; 7) biochemistry: and 8) molecular genetics. For each
approach, I will discuss which results should raise "red flags" suggesting mitochondrial dysfunction, and how different results can orient the diagnosis towards a defect in the nuclear or the mitochondrial genome.
Key words: mitochondrial genetics, oxidative phosphorylation, respiratory chain, diasnosis
MITOCHONDRIAL
ENCEPHALOMYOPATIES: WHERED0WEG0?
Dr. Salvatore Di Mauro Columbia University College Of Physician And Surgeons New York, Ny, USA
In few áreas of medicine has progress been more spectacular than in the field of mitochondrial diseases, especially trióse related to mitochondrial DNA (mtDNA) mutations. Much remains to be done, however, and this brief review will discuss the following áreas of research where progress has been more limited or data are stilí controversial:
1) mtDNA-related disorders:: a) mutations in structural genes: new findings and new puzzles, b) mutations in genes affecting protein synthesis; why is MELAS different from MERRF?
2) nuclear DNA (nDNA)-related disorders: a) mutations in genes encoding subunits of the respiratory cain: the opening of a new chapter, b) defeets in trans locases: mitochondrial cinderellas?, c) defeets of mitochondrial protein importation: which and how many?, d) defeets of intergenomic signaling: fascinating results are

appearing, e) from ragged-red fibers to genes and back again. Unexpected mitochondrial diseases.
Key words: mt-DNA disorders, nuclear DNA, research
ESCLEROSIS MÚLTIPLE
PREDOMINANCIA EN LA DISFUCIÓN DEL SISTEMA NERVIOSO AUTÓNOMO (SNA) SIMPÁTICO SOBRE EL PARASIMPÁTICOEN ESCLEROSIS MÚLTIPLE (EM)
L.E. Gomales-Sánchez, J. Valls-Sole, F.R. Graos. Servicio De Neurología. Hospital Clínico Y Provincia. Barcelona. Unidad De Neurología Y Neurofisiologia, Medicentro Plaza San Salvador, El Salvador.
Introducción y Objetivos: Actualmente se conoce el grado de participación simpática y parasimpática (S-PS) que media algunas de las respuestas de los test que estudian el SNA por métodos no invasivos; algunos son disponibles a laboratorios de EMG. Tal información no ha sido aplicada extensamente para catalogar la función autónoma que frecuentemente afecta en EM. Eí objetivo del estudio fue investigar el estado S-PS en EM.
Sujetos/Métodos: Fueron estudiados 14 pacientes con EM definida y 13 controles. Se exploraron cinco test autónomos (TA) que presumiblemente evalúan: 1) El brazo aferente o eferente (BA-E) paras impático; respiración profunda (AR-6), valsalva, bipedestación y 2) El
BA-E simpático: Handgrip y la respuesta sudomotora simpático cutánea (SSR). La SSR se consideró anormal cuando era ausente o presentaba un retraso de latencia superior a 3SD de la media de los sujetos sanos. La disfunción parasimpática (DFPS) si al menos uno de los 2 test era anormal; simpática (DFS) si al menos uno de los 2 test era anormal y mixta (DfM) si existía combinación de disfunciones.
Resultados: En los controles, los valores de la TA, no difieren de los reportados previamente. Ocho pacientes (51%) presentaron (DfS), 3 pacientes (21.3%) DfM, 1 paciente (7.1%) DFPS y 2 pacientes fueron normales (14.3%). La DFS predomina significativamente sobre la DFPS (P<0.01).
Conclusiones: La disfunción autónoma simpática predomina sobre la parasimpática en los pacientes con EM que hemos investigado.
Palabras clave: EM, esclerosis múltiple, sistema nervioso autónomo.
ESCLEROSIS MÚLTIPLE EN HONDURAS, UN ESTUDIO PRELIMINAR
Dr. Edgardo Girón Flores, Jefe Servicio de Neurología Dr. Tulio Pompeyo Murillo Alvarado, Médico Residente de Neurocirugía Hospital Escuela, Tegucigalpa, Honduras
Antecedentes: En Honduras, el Hospital Escuela es el centro asistencial más grande del país.
La Consulta Externa de Neurología atiende un promedio de 8.528 casos de patología neurológica al año. De ellas, un poco menos del 0.05% son casos de esclerosis múltiple.
Objetivos: Correlacionar las características clínicas y de la enfermedad en Honduras.
Métodos: Realizamos un estudio prospectivo utilizando el expediente clínico como fuente de información. Los datos se introdujeron al programa estadístico para su análisis.
Resultados: Trabajamos con 24 casos en quienes el rango de edad más afectado era el de 21 a 50 años, no encontrándose en estos sujetos ningún factor predisponente. El sexo femenino es el más afectado en una proporción de 3:1. Los síntomas o signos principales del debutó de la enfermedad fueron en 38% motores, 29.7% sensitivos y el resto distribuido entre trastornos psíquicos, autonómicos e indefinidos. El 54% de ellos no ha tenido recaídas aún. La resonancia magnética mostró una sensibilidad de 95% y los potenciales evocados 75% y el 66% de nuestros pacientes se diagnosticaron como esclerosis múltiple definitiva, el resto fueron probable esclerosis múltiple. Diecisiete de los 24 que fueron tratados en fase aguda mejoraron.
Conclusiones: La esclerosis múltiple en Honduras tiende a comportarse de igual manera como lo reporta la literatura internacional.
Palabras clave: Esclerosis múltiple en Honduras, características clínicas, diagnóstico

VARIOS
PARANEOPLASTIC SYNDROMES
Dr. Jorge Alberto Martínez Cerrata Hospital Bautista Managua, Nicaragua
The disorders of central nervous system (CNS) in patiens with cáncer is caused by metástasis, neurotoxicity, opportunist infections or paraneoplastic syndromes. These are less frequent than the metástasis and permit the diagnosis of a hidden cáncer by up to several years. We classified them as paraneoplastic syndromes of the central nervous system (CNS) and paraneoplastic syndromes of the periferic nervous system (PNS). The paraneoplastic syndromes of the PNS are motor neuronopathies, sensory neuronopathies, neuropathic miasthenic syndromes and myopathies. The paraneoplastic syndromes of the CNS are limbic encephalitis, cerebellar syndromes, necrotic myelopathy and retinal degeneration. The pathogenic is autoinmune. The tumor cells express antigen(s) identical or antigenically related to molecules normally expressed by neurons.
In the anti-Hu syndrome, the HuD antigen is found in the neuron nucleous and manifests as an encephalomyeloneuritis. In the Eaton Lambert syndrome, the Ac are directed against the presynaptic Ca++channel. That's why, everyone of the paraneoplastic syndromes have Ac, Ag and the associated cáncer more frequent. In the cerebellar
syndromes, you have to distingush the Yo syndrome, Hu syndrome, Tr syndrome cerebellar syndrome associated to Lambert- Eaton and CV-2 syndrome. The opsoclonus-myoclonus has different characteristics in children, adults and in the Ri syndrome. The limbic encephalitis can be part of the Hu syndrome, stem encephaliíis or puré limbic encephalitis.
The encephalomyeloneuritis (Hu syndrome) is associated to small-cell lung carcinoma (SCLC), Hogdkin's diseases and other carcinoma, while the stem encephalitis or puré limbic is associated to seminoma. The retinopathy associated to cáncer is the carcinoma of the female genital tract. The frequency of the assocíate neurophaties with carcinoma is 1.7 to3.5%by clinic and 30 to 40% by studies of the nervous conduction velocity. The subacute motor neuropathy is associated to Anti-Hu Ac and ANNA-1. The sensory-motor neuropahy is four times frequent than the sensory neuropathy. In relation to the paraneoplastic myopaties, you have to distinguish: necrotic myopathy, dermatomyositis, atrophy of type 2 muscle fibers and Anti-Decorin myopathy.
Key words: paraneoplastic syndromes, classification
CRITERIOS DE AUTISMO
Dr. Francisco León Gómez Asociación Hondureña De Neurología
El autismo es una patología psiquiátrica de la infancia temprana cuya prevalencia es de hasta 2 por cada 1,000 habitantes. Se manifiesta por déficit en la
socialización, comunicación y desarrollo neurológico del niño. El diagnóstico se basa en una historia minuciosa, pruebas neuro-psicológicas y estudios paraclínicos. Las causas son adquiridas y genéticas y el manejo incluye rehabilitación y fármaco terapia.
Palabras clave: autismo, desarrollo, terapia, rehabilitación.
AGONISTAS
DOPAMINÉRGICOSENLA ENFERMEDAD DE PARKINSON. AVANCES
Dr. Edgardo Girón Flores Jefe del Servicio de Neurología, Hospital Escuela Tegucigalpa, Honduras
Hay una reciente evidencia de los estudios clínicos que sugieren un rol amplio de los agonistas dopamínicos como monoterapia dopaminérgica inicial en el tratamiento de la enfermedad de Parkinson (EP). El uso racional de la monoterapia con agonistas dopaminérgicos en etapas tempranas de la EP se hace con el objeto de demorar la terapia con la levodopa o disminuir la exposición total a la misma con el objeto de reducir las complicaciones motoras de uso crónico de la misma Los agonistas dopaminérgicos cuando son usados solos, raramente promueven el desarrollo de diskinesias y de fluctuaciones motoras que complican seriamente la terapia con levodopa. Adicionalmente, los agonistas dopaminérgicos tienen un efecto potencial neuroprotectivo disminuyendo el metabolismo oxidativo de la dopamina y generación de radicales libres.

Debido a que ellos actúan en los receptores post-sinápticos dopaminérgicos del estriado, los agonistas dopamínicos actúan de forma independiente en su síntesis, la cual tampoco es dependiente de las neuronas presinápticas que están en proceso de degeneración a nivel de la sustancia negra. Esta presentación revisará el rol tradicional de los agonistas dopaminérgicos y enfocará las estrategias novedosas en el tratamiento de la EP incluyendo la monoterapia temprana con los agonistas dopaminérgicos o asociados con la levodopa.
Palabras clave: agonistas dopamínicos, monoterapia, ventajas
HIPO INTRATABLE. MANEJO CONGABAPENTINA. ¿NUEVA INDICACIÓN?
Dr. Nelson Chinchilla Cálix Centro De Diagnóstico Neuroíógico San Pedro Sula, Honduras, C.A.
Se presenta el caso de un paciente de 65 años de edad, con historia previa de hipertensión arterial sistémica e hipercolesterolemia sin control adecuado, quien sufrió un ictus isquémico en el territorio de las arterias tálamo perforantes izquierdas produciendo una lesión talamocapsular, cursando con hipo persistente e incapacitante por más de una semana, déficit sensitivo-motor derecho, trastorno afásico motor y Horner izquierdo. El paciente no
asoció afecciones sistémicas o periféricas que explicaran el hipo.
El tratamiento con clorpromazina le controlaba pero los efectos colaterales fueron suficientemente importantes como para interrumpir el medicamento. Tras tratamiento con gabapentina a dosis de 900 mgs diarios, el control completo ocurrió en menos de 48 horas. Dos semanas después, el medicamento fue retirado sin presentar nuevas recurrencias. La tolerancia a la gabapentina fue adecuada sin efectos colaterales permitiendo iniciar la rehabilitación física para su ictus.
Se presenta una revisión acerca de las vías y centros del hipo así como aspectos fisiopatológicos y terapéuticos. En vista de la dificultad en hacer estudios prospectivos con series amplias de hipo intratable por lo infrecuente de esta queja, se sugiere realizar estudios colaborativos con gabapentina en el manejo médico del hipo intratable dado su perfil farmacológico favorable.
Palabras clave: gabapentina, hipo intratable, presentación de caso.
VÉRTIGO: ORIENTACIONES CLÍNICAS
Dr. Juan Carlos Barrientes A., Hospital Escuela, Tegucigalpa Honduras
El vértigo podría definirse como una pérdida de la orientación espacial, una sesnación errónea y subjetiva de desplazamiento de los objetos en relación al sujeto y viceversa.
La palabra vértigo procede del término latino "verteré" que significa "dar vueltas", tiene su expresión más definida en la típica crisis aguda vertiginosa con una sensación giratoria intensa.
Existen otras alteraciones del equilibrio que no son propiamente giratorias, como pueden ser la sensación de desplazamiento de la base de sustentación del cuerpo, como movimientos hacia adelante o hacia atrás, sensación de caída en el vacío, "sensación de andar borracho" por movimientos en general bruscos de la cabeza; en la práctica se vé que son sensaciones muy difíciles de describir por el enfermo, pero en todas hay una característica en común: el movimiento es ireal.
Tanto en el vértigo como en los desequilibrios no giratorios, se provoca un defecto de la información que llega al tronco cerebral; el laberinto está informando que el movimiento está teniendo lugar, mientras que los ojos informan que el entorno está inmóvil y los receptores de la sensabilidad profunda infonnan que el cuerpo está tejido en el suelo.
Esta confusión de información da lugar a que el enfermo perciba un movimiento aparente pero que es irreal.
Las causas de estas alteraciones del equilibrio pueden ser muy variadas, pero las más frecuentes se localizan en el laberinto y en los núcleos vestibulares.
No obstante y considerando como premisa que el vértigo es puramente un síntoma, se describen a continuación algunos pasos para llegar al diagnóstico correcto.
El diagnóstico diferencial del vértigo exige la realización de una historia clínica muy detallada, un examen físico extenso y minucioso.

Idealmente se realizarán exámenes especiales de tipo audiológico, radiológico y laboratorial. El manejo en equipo de estos pacientes obliga a contar en cada caso con la opinión del especialista neurólogo, oftalmólogo, otorrinolaringólogo. internista y de otras según el caso.
Se harán consideraciones sobre lo que es el diagnóstico del vértigo agudo, las causas otológicas y extraotológicas en el diagnóstico diferencial, desde una perspectiva escencialmente clínica y desde el p un to de v i s t a de l otorrinolaringólogo.

INSTRUCCIONES A LOS AUTORES
La REVISTA HONDURENA DE NEÜROCIENCIAS invita a todos los profesionales médicos a enviar trabajos científicos para que sean considerados para publicación, previa calificación del Consejo Editorial de la Revista.
Los manuscritos deberán ser enviados a: Dr. Marco Tulio Medina, Editor, Revista Hondurena de Neurociencias, Centro de Neurodiagnóstico, Edificio Gómez Zúniga, Colonia La Alameda, Tegucigalpa, M.D.C., Honduras. Los trabajos que se acepten lo serán en ei entendimiento de que no han sido publicados previamente ni lo serán en otra revista s in el consentimiento escrito del editor y pasan a ser propiedad de la REVISTA HONDURENA DE NEÜROCIENCIAS.
La REV1STAH0NDUREÑA DE NEÜROCIENCIAS acepta para publicación trabajos científicos sobre todas las ramas de las neurociencias e incluye las siguientes secciones en forma regular: artículos originales de investigación clínica o experimental, trabajos de revisión, reporte de casos clínicos y cartas al editor. Además se publicarán las memorias de congresos o cursos de neurología realizados en el país e información general sobre las actividades de la Asociación Hondurena de Neurología.
En términos generales se recomienda que los autores se ajusten a las siguientes normas en la preparación de manuscritos:
a) Los manuscritos deberán ser escritos a máquina, en papel bond tamaño carta, a doble espacio, por una sola cara y con márgenes de 4 cm. por lo menos. Se enviará un original y una fotocopia de buena calidad, incluyendo tablas y figuras. Se recomienda que los autores guarden en su poder una copia del manuscrito.
b) La hoja frontal deberá contener el título del trabajo, el cual debe ser corto y específico, así como el nombre completo de l o s autores, su grado académico más importante, la institución a la que pertenecen y la dirección completa del autor que será encargado de la c o r r e s p o n d e n c i a concerniente a dicho artículo. Para evitar la confusión con el nombre de los autores a nivel internacional, el apellido paterno y materno (en caso que se desee incluirlo) serán unidos por un guión.
c) Los artículos originales deberán incluir, en la segunda, hoja, un resumen de 200 palabras o menos, en el que se incluya los antecedentes, el objetivo del trabajo, métodos, resultados y las conclusiones principales. El resumen deberá ser enviado en español e inglés. Los trabajos de revisión y los reportes de casos clínicos
también deben tener un resumen, cuya extensión dependerá del tamaño del manuscrito.
d) Los artículos originales deberán tener las siguientes secciones: introducción, material y métodos, resultados y conclusiones. Los reportes de casos clínicos deben incluir una breve introducción, la descripción completa del caso y un comentario final sobre los aspectos relevantes de dicho caso. El formato de los trabajos de revisión queda a libre criterio de los autores
e) Las cartas al editor son bienvenidas. Estas deberán ser escritas a doble espacio y no serán de más de 300 palabras. Pueden incluir una figura y hasta cinco referencias bibliográficas. Tratarán sobre temas neurológicos generales o sobre comentarios de artículos publicados en números previos de la revista.
f) Los cuadros deberán ser escritos en hoja aparte, n u m e r a d o s consecutivamente, de acuerdo a su ubicación en el texto y deberán llevar un pie explicativo conciso. Se evitará el exceso de tablas, evitando repetir en el texto lo que se exprese en ellas.
g) Las fotografías deberán ser

enviadas por duplicado, en blanco y negro, y deberán estar claramente identificadas con el número de la figura, el nombre de! autor principal y una flecha indicando la parte superior de la figura; estos datos deberán ser escritos sobre una etiqueta adhesiva pegada en el reverso de la figura o escritos directamente sobre el reverso de la figura con un lápiz. Se recomienda utilizar únicamente aquellas fotografías que contribuyan significativamente con el texto. Las fotografías a color serán aceptadas si aportan datos que no pueden ser revelados en fotografías en blanco y negro. Las fotografías en las que aparezcan datos que permitan la identificación personal de determinados pacientes deberán acompañarse del consentimiento escrito por parte de dichos pacientes. No es suficiente cubrir los ojos para ocultar una identidad.
h) Las referencias bibliográficas deberán ser escritas en hoja aparte, a doble espacio y serán ordenadas de acuerdo a su aparición en el texto (no por orden alfabético de los autores). Todas las referencias deberán estar citadas en el texto o en las tablas, con un número entre paréntesis, que i) corresponderá al de la lista final. Los autores son responsables de la veracidad de las referencias y de su correcta transcripción. Las referencias deberán seguir el estilo y puntuación de los siguientes ejemplos:
Referencias de revistas: Sommer C, Schroder JM. Hereditary motor and sensory neuropathy with optic atrophy. Arch Neurol 1989; 46:973-977.
Referencias de libros: Adams RD, Víctor M. Principies of Neurology, 3erd
Ed, New York: McGraw-Hill, 1986.
Referencias de capítulos de libros: Marshall J. Thomas DJ. Vascular Disease, in: Asbury AK, MacKham system, vol 2. Phíladelphia: Saunders, 1986: 1101-1135.
Los manuscritos que no se acepten para publicación no serán devueltos. Los autores únicamente recibirán una carta explicando los motivos de la no aceptación, así como las figuras de dicho manuscrito.
Los autores cuyos manuscritos sean aceptados para publicación deberán firmar un certificado de transferencia de derechos de autor, que será enviado por el Consejo Editorial de la Revista.