retos en de medicamentos en la aempsaemps:: actual y sus … · 2016-02-15 · ensayos clínicos de...
TRANSCRIPT

IX Santiago de Chile Septiembre 2012
“Desafíos Regulatorios de la Globalización y el Acceso a Medicamentos de Calidad”
““ óó““Retos en regulación de medicamentos Retos en regulación de medicamentos en la en la AEMPSAEMPS: Situación actual y sus : Situación actual y sus
ii ””proyeccionesproyecciones” ”
Belén Crespo SánchezBelén Crespo Sánchez‐‐ EznarriagaEznarriagaDirectora de la AEMPSDirectora de la AEMPSDirectora de la AEMPSDirectora de la AEMPS

IX Santiago de Chile Septiembre 2012
MISIÓNGarantizar a la sociedad, desde la perspectiva , p pde servicio público, la calidad, seguridad, eficacia y correcta información de los medicamentos y productos sanitarios en el más amplio sentido, desde su investigación p , ghasta su utilización, en interés de la protección y promoción de la salud de las personas y de los animales.

IX Santiago de Chile Septiembre 2012
Cooperación Europea
2012 Implementar la nueva legislación en FarmacovigilanciaDirectiva 2010/84/UE y Reglamento 1235/2010 sobre Farmacovigilancia
RETOS 1235/2010 sobre Farmacovigilancia
2013 Implementar la nueva legislación 2013 Implementar la nueva legislación en medicamentos falsificadosen medicamentos falsificadosDirectiva 2011/62/UEDirectiva 2011/62/UE
Work programme2012 Adopted by the Management B d 15 D b 2011
Road map to 2015Board on 15 December 2011 ______________
The European Medicines Agency’s contribution toscience, medicines and Health
Adopted by the Agency’s Management Board on 16 December
Globalización de:Ensayos clínicos Producción de ingredientesProducción de ingredientes
farmacéuticos activos (APIs) y productos terminados
Colaboración internacional Redes Área de Buena Practica ClínicaInspecciones de Buena Práctica
de Manufactura
A Strategy for theHeads of Medicines Agencies, 2011-15 En revisión la Directiva de Ensayos Clínicos
Propuesta de revisión de la legislación de productos sanitarios orientada a la interacción de productos sanitarios, productos p , pdiagnósticos

45 años de45 años dearmonizaciónarmonizaciónContextoContexto
•• Sistema altamente garantistaSistema altamente garantista
europeaeuropeaContextoContexto
Sistema altamente garantistaSistema altamente garantista
•• Basado en laBasado en la permanente evaluación de la relación del beneficio riesgo que se que se inicia en la investigación y se prolongainicia en la investigación y se prolongainicia en la investigación y se prolonga inicia en la investigación y se prolonga todo el ciclo de vida del medicamento todo el ciclo de vida del medicamento
• Utiliza los mismos criterios de evaluación
H d d lH d d l l ió i di id l l•• Ha pasado de laHa pasado de la evaluación individual a la evaluación en red
• Identifica necesidades de mejora y difi ió á fi imodificación para ser más eficiente
•• Es másEs más trasparente
•• EstáEstá más abierto a la sociedadEstá Está más abierto a la sociedad
““““Nuevo sistema de Nuevo sistema de farmacovigilanciafarmacovigilanciapara Europapara Europa” ”

Directiva 2010/84/UE y Reglamento 1235/2010 sobre Directiva 2010/84/UE y Reglamento 1235/2010 sobre Farmacovigilancia Farmacovigilancia
Textos Aprobados bajo Presidencia española de la UE del año 2010Textos Aprobados bajo Presidencia española de la UE del año 2010Fecha de Publicación: 31 de diciembre de 2010Fecha de Publicación: 31 de diciembre de 2010
gg
Fecha de Publicación: 31 de diciembre de 2010 Fecha de Publicación: 31 de diciembre de 2010 Fecha de entrada en vigor Reglamento: 2 de julio de 2012; Directiva: 21 de julio de 2012Fecha de entrada en vigor Reglamento: 2 de julio de 2012; Directiva: 21 de julio de 2012
ObjetivoObjetivo
Promover y proteger la salud pública fortaleciendo el sistema europeo de Promover y proteger la salud pública fortaleciendo el sistema europeo de monitori ación de la seg ridad el balance beneficiomonitori ación de la seg ridad el balance beneficio riesgo de los medicamentosriesgo de los medicamentosmonitorización de la seguridad y el balance beneficiomonitorización de la seguridad y el balance beneficio‐‐riesgo de los medicamentosriesgo de los medicamentos
Reforzar la vigilancia, transparencia y comunicaciónReforzar la vigilancia, transparencia y comunicación de la seguridad de los medicamentos de la seguridad de los medicamentos una vez comercializados así como mejorar una vez comercializados así como mejorar los actuales procedimientos y coordinarlos actuales procedimientos y coordinar mejor las mejor las actuaciones entre los distintos Estados miembros.actuaciones entre los distintos Estados miembros.
Se basa, por un lado, en sistemas y estructuras ya existentes que refuerza y, por otro, en la Se basa, por un lado, en sistemas y estructuras ya existentes que refuerza y, por otro, en la imposición de nuevas tareas que se añaden a las ya existentesimposición de nuevas tareas que se añaden a las ya existentes

Nuevo sistema de farmacovigilanciaNuevo sistema de farmacovigilancia
11. . EstablecerEstablecer la la Vigilancia proactivaVigilancia proactiva. . Mejores mecanismos para conseguir garantizar la Mejores mecanismos para conseguir garantizar la seguridad de los medicamentosseguridad de los medicamentos (agencias y compañías farmacéuticas)(agencias y compañías farmacéuticas)
gg
seguridad de los medicamentos seguridad de los medicamentos (agencias y compañías farmacéuticas)(agencias y compañías farmacéuticas)
2. 2. IncrementarIncrementar la la ParticipaciónParticipación de lde los pacientesos pacientes. . sospechas de reacciones adversas a las sospechas de reacciones adversas a las autoridades competentes de los Estados Miembros y a acceder a la informaciónautoridades competentes de los Estados Miembros y a acceder a la información
33.. Incrementar la Incrementar la Transparencia Transparencia y acceso a la información en todas las actividades de FVy acceso a la información en todas las actividades de FV
55 MejorarMejorar CoordinaciónCoordinación Procedimientos coordinados para la evaluación de aspectosProcedimientos coordinados para la evaluación de aspectos
44.. Mejorar la Mejorar la comunicación de riesgos comunicación de riesgos (decisiones reguladoras y su justificación) (decisiones reguladoras y su justificación)
55. . MejorarMejorar CoordinaciónCoordinación.. Procedimientos coordinados para la evaluación de aspectos Procedimientos coordinados para la evaluación de aspectos relacionados con la seguridad de los medicamentos. Comité Europeo de Farmacovigilancia con relacionados con la seguridad de los medicamentos. Comité Europeo de Farmacovigilancia con representación de todas las agencias de medicamentos nacionales y también en comunicaciónrepresentación de todas las agencias de medicamentos nacionales y también en comunicación
6.6. Mejorar la eficiencia del sistema europeo Mejorar la eficiencia del sistema europeo (vigilancia proporcional al riesgo, reparto de tareas (vigilancia proporcional al riesgo, reparto de tareas entre los Estados Miembros, simplificación de los procedimientos)entre los Estados Miembros, simplificación de los procedimientos)

Nuevo sistema de farmacovigilanciaNuevo sistema de farmacovigilancia
11. . Se amplía definición de Se amplía definición de reacción adversareacción adversa, que engloba las reacciones adversas , que engloba las reacciones adversas
gg
derivadas de cualquier uso, abuso y errores de medicación.derivadas de cualquier uso, abuso y errores de medicación.
22 Se establecen criterios claros acerca de lasSe establecen criterios claros acerca de las obligaciones y funcionesobligaciones y funciones de las partesde las partes22. . Se establecen criterios claros acerca de las Se establecen criterios claros acerca de las obligaciones y funcionesobligaciones y funciones de las partes de las partes responsables implicadas.responsables implicadas.
33 Se refuerzan las obligaciones de losSe refuerzan las obligaciones de los titulares de la autorización detitulares de la autorización de33. . Se refuerzan las obligaciones de los Se refuerzan las obligaciones de los titulares de la autorización de titulares de la autorización de comercializacióncomercialización, , debiendo identificar potenciales problemas de seguridad de forma debiendo identificar potenciales problemas de seguridad de forma proactiva,enproactiva,en un un plan de gestión de riesgosplan de gestión de riesgos que forma parte de la autorización de que forma parte de la autorización de comercializacióncomercialización
44. . Se refuerza la vigilancia de los nuevos medicamentos y de aquellos en los que Se refuerza la vigilancia de los nuevos medicamentos y de aquellos en los que surja un potencial problema de seguridad Estos medicamentos bajo seguimientosurja un potencial problema de seguridad Estos medicamentos bajo seguimientosurja un potencial problema de seguridad. Estos medicamentos bajo seguimiento surja un potencial problema de seguridad. Estos medicamentos bajo seguimiento adicional adicional tendrán un distintivo en la ficha técnica y prospecto, para priorizar la tendrán un distintivo en la ficha técnica y prospecto, para priorizar la notificación de sospechas de reacciones adversas. La lista de estos medicamentos notificación de sospechas de reacciones adversas. La lista de estos medicamentos será pública.será pública.

Nuevo sistema de farmacovigilanciaNuevo sistema de farmacovigilanciagg
55.. Se refuerza la toma de decisionesSe refuerza la toma de decisiones sobre la seguridad de los medicamentossobre la seguridad de los medicamentos55.. Se refuerza la toma de decisiones Se refuerza la toma de decisiones sobre la seguridad de los medicamentos sobre la seguridad de los medicamentos autorizados. Se crea el autorizados. Se crea el Comité para la Evaluación de Riesgos en FarmacovigiComité para la Evaluación de Riesgos en Farmacovigilancialanciadel que forman parte las agencias nacionales de medicamentos. del que forman parte las agencias nacionales de medicamentos.
66. . Se racionaliza y Se racionaliza y se armoniza la toma de decisiones tras la evaluación de los se armoniza la toma de decisiones tras la evaluación de los riesgosriesgos asociados a los medicamentos, implementándose de forma equitativa, asociados a los medicamentos, implementándose de forma equitativa, completa y simultánea las decisiones en todos los países de la Unión Europeacompleta y simultánea las decisiones en todos los países de la Unión Europea
77. . Se Se estrecha la colaboración de las agencias de medicamentos nacionalesestrecha la colaboración de las agencias de medicamentos nacionales en la en la l ó l l l ll ó l l l l
completa y simultánea las decisiones en todos los países de la Unión Europea. completa y simultánea las decisiones en todos los países de la Unión Europea.
evaluación de los riesgos de medicamentos y la toma de decisiones y se aclara el evaluación de los riesgos de medicamentos y la toma de decisiones y se aclara el papel coordinador de la EMA.papel coordinador de la EMA.
88. . Se incorpora la necesidad de Se incorpora la necesidad de evaluar el impacto de las medidas que se adoptan evaluar el impacto de las medidas que se adoptan para minimizar los riesgos de los medicamentospara minimizar los riesgos de los medicamentos..

Nuevo sistema de farmacovigilanciaNuevo sistema de farmacovigilanciagg
9.9. La legislación establece una nueva aproximación a los Estudios PostLa legislación establece una nueva aproximación a los Estudios Post‐‐Autorización de Autorización de Seguridad y Eficacia (PASS/PAES) para apoyar la toma de decisiones de los reguladores sobre el Seguridad y Eficacia (PASS/PAES) para apoyar la toma de decisiones de los reguladores sobre el perfil de seguridad y beneficio/riesgo de un medicamento.perfil de seguridad y beneficio/riesgo de un medicamento.
66. . Se Se estrecha la colaboración de las agencias de medicamentos nacionalesestrecha la colaboración de las agencias de medicamentos nacionales en la en la evaluación de los riesgos de medicamentos y la toma de decisiones y se aclara el evaluación de los riesgos de medicamentos y la toma de decisiones y se aclara el
l di d d l EMAl di d d l EMA
perfil de seguridad y beneficio/riesgo de un medicamento.perfil de seguridad y beneficio/riesgo de un medicamento.
••PASS es un estudio en un medicamento autorizado quePASS es un estudio en un medicamento autorizado que ::
•• identifica, caracteriza o cuantifica un riesgo de seguridad, identifica, caracteriza o cuantifica un riesgo de seguridad,
papel coordinador de la EMA.papel coordinador de la EMA.•• confirma el perfil de seguridad de un medicamento o confirma el perfil de seguridad de un medicamento o
•• evalúa la efectividad de las medidas de minimización de riesgos aplicadas evalúa la efectividad de las medidas de minimización de riesgos aplicadas
••PAES es un estudio en un medicamento autorizado que está dirigido aPAES es un estudio en un medicamento autorizado que está dirigido a: clarificar la eficacia de: clarificar la eficacia dePAES es un estudio en un medicamento autorizado que está dirigido aPAES es un estudio en un medicamento autorizado que está dirigido a: clarificar la eficacia de : clarificar la eficacia de un medicamento en el mercado utilizado en condiciones de práctica clínica. un medicamento en el mercado utilizado en condiciones de práctica clínica.
Estos estudios tienen que ser evaluados y autorizados por las autoridades competentes en el Estos estudios tienen que ser evaluados y autorizados por las autoridades competentes en el contexto del nuevo Comité de Farmacovigilancia (PRAC) que, de nuevo, tienen que evaluar sus contexto del nuevo Comité de Farmacovigilancia (PRAC) que, de nuevo, tienen que evaluar sus resultados una vez realizados.resultados una vez realizados.
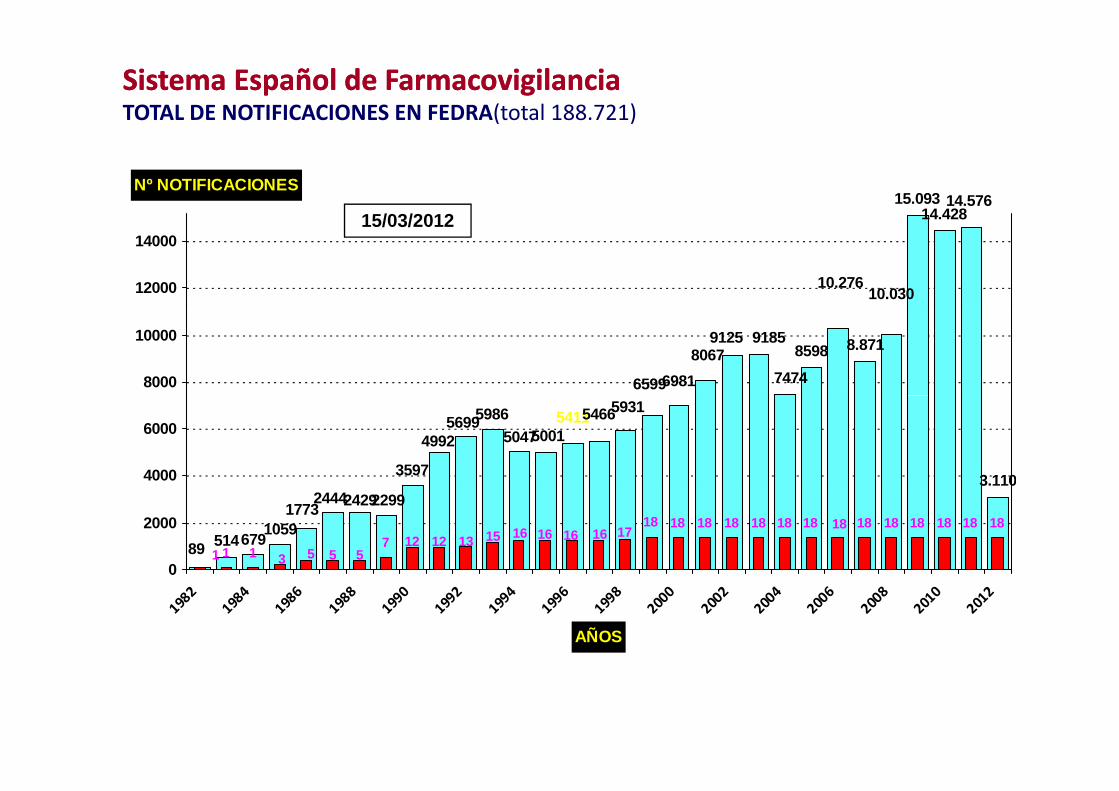
Sistema Español de FarmacovigilanciaSistema Español de FarmacovigilanciaTOTAL DE NOTIFICACIONES EN FEDRA(total 188.721)
15.09314 428
14.576Nº NOTIFICACIONES
15/03/2012 14.428
10.27610.03012000
1400015/03/2012
8598 8.8718067
65996981 7474
9125 9185
8000
10000
3.110
5931
5047 5001541159865699
4992
3597
5466
4000
6000
3.110229924292444
1773105967951489 1 1
718 18 18 18 18 18 18 18 18 18 18 1818
3 55512 12 13 16 16 16
1817
11615
0
2000
1982
1984
1986
1988
1990
1992
1994
1996
1998
2000
2002
2004
2006
2008
2010
2012
AÑOS

Sistema Español de FarmacovigilanciaSistema Español de Farmacovigilancia2011 14.886 SRA2011 14.886 SRA
EudravigilanceEudravigilance
8 041 EMA
Prof Sanit: 77,48%77,48%
8.041 EMA
FEDRAFEDRA == Farmacovigilancia Española, Datos De
TAC: 22,52%22,52%
Reacciones Adversa
.••15.47315.473 enviadas a la OMS
••3.759 3.759 enviadas a la industria.
• 272 272 certificaciones a petición de los TAC

Sistema Español de FarmacovigilanciaSistema Español de Farmacovigilancia
Algunos de los problemas de seguridad más conocidos detectados Algunos de los problemas de seguridad más conocidos detectados
por el Sistema Español de Farmacovigilanciapor el Sistema Español de Farmacovigilanciapor el Sistema Español de Farmacovigilanciapor el Sistema Español de Farmacovigilancia
Medicamento ProblemaMedicamento Problema seguridad
Benzbromarona and Benziodarona HepatotoxicidadBenzbromarona and Benziodarona
(Urinorm ®, Dilafurane®, Acifugan ®)
Hepatotoxicidad
Cerivastatina (Lipobay ®) Rabdomiolisis
Nimesulida (Antifloxil ®, Guaxan ®) Hepatotoxicidad
Extracto té verde (Exolise ®) HepatotoxicidadExtracto té verde (Exolise ®) Hepatotoxicidad
Veraliprida (Agreal ®) Síndrome de retirada
Infliximab (Remicade ®) Tuberculosis

44.. Incremento de la transparencia de la EMA relacionada con la Incremento de la transparencia de la EMA relacionada con la vigilancia de los fármacosvigilancia de los fármacos
•• EnEn lala comunicacióncomunicación compromisocompromiso dede tenertener enen lala WebWeb dede cadacada EEMMEEMM• Informes públicos de evaluación de todos los medicamentos• Prospectos y fichas técnicas de todos los medicamentosProspectos y fichas técnicas de todos los medicamentos• Resúmenes de los PGR entendible por la sociedad,• Información sobre como notificar y herramientas de notificación
Li t d di t j t it i ió di i l• Lista de medicamentos sujetos a monitorización adicional• Acceso a las agendas y resúmenes de los Comités relacionados con la
seguridad de los medicamentos• Todas las notas informativas de seguridad.
Consecuencias GlobalesConsecuencias Globales
Gestión y Comunicación de riesgosGestión y Comunicación de riesgosGest ó y Co u cac ó de esgosGest ó y Co u cac ó de esgos

IX Santiago de Chile Septiembre 2012
Revisión de Revisión de la la l i l ió dl i l ió dlegislación de legislación de
Ensayos ClínicosEnsayos Clínicos

CompetenciasCompetencias

PropuestaPropuesta de Reglamento del Parlamento Europeo y del de Reglamento del Parlamento Europeo y del Consejo sobre los ensayos clínicos de medicamentos de Consejo sobre los ensayos clínicos de medicamentos de j yj y
uso humano, y por el que se deroga la Directiva uso humano, y por el que se deroga la Directiva 2001/20/CE2001/20/CE
Fecha de publicación de la Propuesta 17 julio 2012 BruselasFecha de publicación de la Propuesta 17 julio 2012 BruselasFecha de publicación de la Propuesta: 17 julio 2012 BruselasFecha de publicación de la Propuesta: 17 julio 2012 Bruselas
ObjetivoObjetivojj
l ll l i l li ió d lí ii l li ió d lí iQue la UE resulte Que la UE resulte atractiva para la realización de ensayos clínicosatractiva para la realización de ensayos clínicos, y para , y para ello se plantea simplificar los requisitos aplicables a los ensayos ello se plantea simplificar los requisitos aplicables a los ensayos
clínicos clínicos sin disminuir las garantías de los sujetossin disminuir las garantías de los sujetos, de forma coherente , de forma coherente c cosc cos s d s u as ga a t as de os sujetoss d s u as ga a t as de os sujetos, de o a co e e te, de o a co e e tecon la armonización de los ensayos clínicos a nivel internacional y con la armonización de los ensayos clínicos a nivel internacional y
buscando una reducción en los costes para el promotor. buscando una reducción en los costes para el promotor.

Propuesta de Reglamento del Parlamento Europeo y del Consejo sobre los Propuesta de Reglamento del Parlamento Europeo y del Consejo sobre los ensayos clínicos de medicamentos de uso humano, y por el que se deroga la ensayos clínicos de medicamentos de uso humano, y por el que se deroga la
Directiva 2001/20/CEDirectiva 2001/20/CE
La Directiva 2001/20/ECLa Directiva 2001/20/EC supuso una mejora importante en la garantíasupuso una mejora importante en la garantíaLa Directiva 2001/20/EC La Directiva 2001/20/EC supuso una mejora importante en la garantía supuso una mejora importante en la garantía
de seguridad, consideraciones éticas y eficacia de los ensayosde seguridad, consideraciones éticas y eficacia de los ensayos; sin ; sin
b hb h d d íd l lí i l UE
Pérdida de atractivo de la investigación orientada al paciente y de susPérdida de atractivo de la investigación orientada al paciente y de sus
embargo, se ha embargo, se ha detectado una caída en los ensayos clínicos en la UE.
Pérdida de atractivo de la investigación orientada al paciente y de sus Pérdida de atractivo de la investigación orientada al paciente y de sus correspondientes estudios en la UEcorrespondientes estudios en la UE
El número de solicitudes de autorización de ensayos clínicos en la UE pasó El número de solicitudes de autorización de ensayos clínicos en la UE pasó de 5de 5 028 en 2007 a 4028 en 2007 a 4 400 en 2010400 en 2010de 5de 5 028 en 2007 a 4028 en 2007 a 4 400 en 2010400 en 2010
Entre los Entre los aspectos mas criticados aspectos mas criticados se encuentran: se encuentran:
el incremento de costes asociados, fundamentalmente administrativos el incremento de costes asociados, fundamentalmente administrativos y de cobertura de riesgos, y y de cobertura de riesgos, y
el retraso en el periodo necesario para el lanzamiento de los ensayosel retraso en el periodo necesario para el lanzamiento de los ensayosel retraso en el periodo necesario para el lanzamiento de los ensayos el retraso en el periodo necesario para el lanzamiento de los ensayos hasta 152 días, un 90% superior al existente previamentehasta 152 días, un 90% superior al existente previamente

Propuesta de Reglamento del Parlamento Europeo y del Consejo sobre los Propuesta de Reglamento del Parlamento Europeo y del Consejo sobre los ensayos clínicos de medicamentos de uso humano, y por el que se deroga la ensayos clínicos de medicamentos de uso humano, y por el que se deroga la
Directiva Directiva 2001/20/CE2001/20/CE
Presentación por separado, con evaluación y seguimiento reglamentario divergentes, de las solicitudes de ensayos clínicos
Mayor dificultad para realizar EC debido a disposiciones reglamentarias no adaptadas a consideraciones y necesidades prácticas
Fiabilidad de los datos procedentes de ensayos clínicos en un entorno de i ti ió di li dinvestigación mundializado

Propuesta de Reglamento del Parlamento Europeo y del Consejo sobre los Propuesta de Reglamento del Parlamento Europeo y del Consejo sobre los ensayos clínicos de medicamentos de uso humano, y por el que se deroga la ensayos clínicos de medicamentos de uso humano, y por el que se deroga la
Directiva 2001/20/CEDirectiva 2001/20/CE
Objetivo nº 1: un marco regulador moderno para la presentación, la evaluación y j g p p , yel seguimiento reglamentario de las solicitudes de ensayos clínicos, teniendo en cuenta el entorno de investigación multinacional. El objetivo operativo es reducir las cargas administrativas, los gastos de funcionamiento y los retrasos en comenzar el ensayo clínico que dependan del marco regulador.
Objetivo nº 2: disposiciones reglamentarias adaptadas a consideraciones, condicionantes y necesidades prácticas sin comprometer la seguridad, el y p p g ,bienestar y los derechos de los participantes en ensayos clínicos ni la consistencia de los datos. El objetivo operativo es reducir las cargas administrativas y los gastos de funcionamiento relacionados con dos di i i l t i f d t l l i f l d id d ldisposiciones reglamentarias fundamentales: el informe anual de seguridad y el seguro o indemnización obligatorios.
Objetivo nº 3: abordar la dimensión mundial de los ensayos clínicos, garantizandoal mismo tiempo el cumplimiento de las BPC. El objetivo operativo es garantizar elcumplimiento de las BPC en los ensayos clínicos efectuados en países exteriores ala UEla UE.
Globalización de ensayos clínicos y sus retos
Cuando se discuten las pautas éticas que deben regir la
Globalización de ensayos clínicos y sus retos
investigación en los países en vías de desarrollo hay varios temas que siguen siendo motivo de discusión:
imperialismo ético, y universalismo o relativismo moral;moral; tratamiento estándar en los países en vías de desarrollo y concepto de vulnerabilidad; balance riesgo‐beneficio; proceso del consentimiento debidamente informado;la capacidad para realizar los EC y la revisión ético‐científica; la interpretación de lo que exige la aplicación ella interpretación de lo que exige la aplicación el principio de justicia en investigación; las situaciones en la que los investigadores tienen un conflicto de intereses los desafíos especiales que plantea la investigación en genética humana, transferencia genética y células madre

Santiago de Chile Septiembre 2012
“P d“P d“Propuesta de “Propuesta de revisión de la revisión de la legislaciónlegislación de de
productosproductosproductos productos sanitariossanitarios” ”
23

Santiago de Chile Septiembre 2012
PROPUESTAS REVISIÓN LEGISLACIÓN UE
Afecta a TODOS los productos: PS, PSIA y IVDCambio de 3 Directivas a 2 Reglamentos (PS PSIA y IVD)Cambio de 3 Directivas a 2 Reglamentos (PS, PSIA y IVD)Incorporación principios Reglamento Nuevo Enfoque sobre comercialización de los productoscomercialización de los productos. Incorporación nuevos procedimientos legislativos comunitarios: actos delegados actos de ejecucióncomunitarios: actos delegados, actos de ejecución.Objetivo: reforzar las garantías de los productos (caso PIP) y conseguir mayor armonizacióny conseguir mayor armonización.
24

Santiago de Chile Septiembre 2012
CALENDARIOCALENDARIO
Comienzo de discusiones en Consejo de la UE en mes de joctubre.
2 años para finalizar discusiones
5 años para aplicación
25

Santiago de Chile Septiembre 2012
Puntos de contacto Direcciones de Productos Sanitarios en EUhtt // /h lth/ di l d i /li k / t t i t hthttp://ec.europa.eu/health/medical‐devices/links/contact_points_en.htm
INFORMACIÓN SOBRE REGULACIÓN EN PRODUCTOS SANITARIOS
A través de INTERNET, en la página web de la Agencia Española de Medicamentos y Productos Sanitarios: http://www.aemps.gob.es
A través de INTERNET en la página web de la Comisión Europea:A través de INTERNET, en la página web de la Comisión Europea:http://ec.europa.eu/health/medical‐devices/index_en.htm
26

Gracias por su atención!!!!