responsabilidades del investigador segÚn...
TRANSCRIPT

RESPONSABILIDADES DEL
INVESTIGADOR SEGÚN GCP-ICH
Lic. Eugenia Farré
Phoenix Cuyo - 7 y 8nov13

2
Calificaciones y acuerdos del investigador
• Formación académica
• CV – Matrícula Profesional
• Conoce y cumple
GCP GCP
• Conoce la droga del estudio
• Experiencia
• comprobable

3
Cumplir el protocolo
§ Analiza, estudia, discute y luego Firma el protocolo y un acuerdo con el patrocinante
§ No implementar desviaciones al protocolo
§ Documentar, explicar y reportar toda desviación involuntaria del protocolo

4
Responsabilidad del investigador
§ Asegurar la confidencialidad de la información (preparación, ejecución y finalización del estudio), así
como de la identidad de las personas incorporadas
§ Comunicar reacciones adversas serias (OMS)
§ - Muerte
§ - Amenaza la vida
§ - Hospitalización o prolongación de hospitalización
§ - Discapacidad o Incapacidad persistente o
significativa
§ - Anomalía congénita/defecto de nacimiento

5
Cuidado médico de los voluntarios del estudio
• Información al paciente de un tratamiento médico para
una enfermedad intercurrente (complicación) Informar al
médico de cabecera
§ Respetar los derechos del paciente. Aunque el sujeto
puede salir del estudio en forma prematura, deberá indagar
acerca de las razones del abandono
• Brindar tratamiento médico adecuado.
• Responsable de todas las decisiones médicas del estudio.

6
Consentimiento Informado § Obtener el consentimiento escrito por parte del
paciente el cual debe estar fechado y firmado. Se le debe entregar una copia
§ Previo a la firma del CI el investigador deberá
brindar tiempo suficiente para que el paciente/rep. legal formule las preguntas necesarias y se aclaren todas las dudas
§ Asegurarse que el paciente entendió y aclaró todas sus dudas.

7
§ Seguir los procedimientos de aleatorización
§ Mantener el ciego del estudio
§ Explicar y documentar la ruptura prematura del ciego
Patient Number 43
contains treatment allocation
Aleatorización y ciego del estudio

8
Garantizar: Recursos adecuados • Cantidad de pacientes prometidos (datos retrospectivos y actuales)
• Instalaciones adecuadas
• Personal calificado y correctamente entrenado (protocolo, fármaco del estudio GCP-ICH, regulaciones y normas): TODOS

9
- Mantener bajo custodia la totalidad del material utilizado en el estudio, siendo responsables del uso correcto de los mismos - Conocer las responsabilidades del patrocinante
Responsabilidad del investigador
Permitirá el monitoreo y auditorías del patrocinante e inspecciones de las
autoridades regulatorias

10
§ Aceptar monitoreos regulares
§ Identificar y seleccionar pacientes
§ Atender pacientes del ensayo (visitas)
§ Completar y analizar documentos fuente
§ Completar documentos regulatorios y del protocolo
§ Para entrenar y supervisar a su equipo
§ FIRMAR CRF: ojo!!!! Requiere revisión!
¡¡ Tiempo suficiente !!
Recursos adecuados

11
Trascripción al CRF correcta, precisa, completa, legible,
actualizada, consistente con documento fuente
Mantenimiento de documentos fuente y esenciales hasta 2 años luego de la interrupción
del desarrollo de la droga (15 años) o última solicitud de aprobación en algún país ICH
Documentar aspectos financieros (contrato)
Registros y reportes

12
Eventos adversos serios hasta su resolución
§ Al patrocinante antes de las 24 horas
§ A los comités
§ A las autoridades regulatorias
Reportes de seguridad
Notificaciones al investigador
§ A los comités
Eventos adversos no serios
§ Al patrocinante en el CRF

13
Investigador Principal
Asume total responsabilidad por la conducción del estudio
Delega ciertas tareas en su personal.
No puede delegar su responsabilidad por el total ensayo

14
El investigador principal de un proyecto delega funciones NUNCA
responsabilidades
IMPORTANTE concepto regulatorio

15
Sub-investigador
Miembro del equipo a quien se le delegan ciertas responsabilidades

16
Coordinador / Asistente del Estudio
§ Screening de pacientes/enrrolamiento • Logística de muestras biológicas • CRF • Mantenimiento de la carpeta regulatoria/obtencion de documentos • Documentos fuente • Atención al paciente
Todas deben estar previamente delegadas en forma documentada por el investigador

17
Reportes escritos sobre la evolución del estudio
§ A la institución
§ A los comités al menos una vez al año
Reportes
Reportes escritos sobre cambios en la conducción del estudio y
aumento del riesgo de los pacientes
§ A la institución
§ A los comités
§ Al patrocinante
Reportes escritos sobre nueva información de la
droga
§ A la institución
§ A los comités

18
Registro detallado de la droga del estudio
§ Cantidades recibidas del Patrocinante
§ Cantidades disponibles en el Centro
§ Cantidades entregadas a cada Paciente
§ Cantidades devueltas por cada Paciente
§ Cantidades devueltas al Patrocinante
§ Número de lote
§ Fecha de vencimiento
§ Código o número de identificación
Farmacia

19
Recordar al paciente el uso de la medicación y la devolución de envases vacíos o medicación no
utilizada
Almacenar la medicación del estudio de acuerdo a lo
indicado (humedad, temperatura) con acceso
restringido
Asegurar que la medicación se utilice de acuerdo a lo especificado
en el protocolo
Farmacia
Trazabilidad

n Temperature Log n Protocolo: …………………………………………
n Site: n PI
n Fecha
n T° Max/Min n Responsable****
n Observaciones n
n n
n n
n
n n
n n
n n
n n
n
n n
n n
n n
n
n n
n
20
Farmacia

n Coordinar retiro de muestras según Tº n Pautas de recolección de muestras
n Pedido de materiales n Embalaje/refrigerantes
n Guía Aerea
n Alertas/sitemas n Resolución de discordancias
21
Bioquimico
22
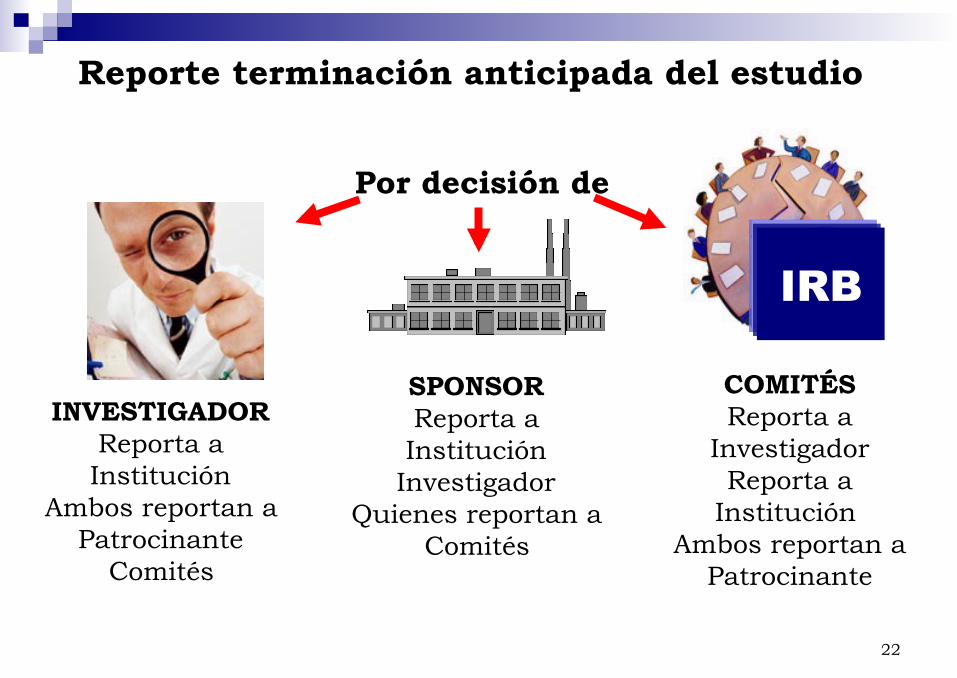
Reporte terminación anticipada del estudio
INVESTIGADOR Reporta a Institución
Ambos reportan a Patrocinante
Comités
SPONSOR Reporta a Institución
Investigador Quienes reportan a
Comités
Por decisión de
COMITÉS Reporta a
Investigador Reporta a
Institución Ambos reportan a
Patrocinante
IRB

23
Reporte terminación anticipada del estudio
§ Informar a los voluntarios del estudio
§ Asegurar su seguimiento, atención, No al
abandono de pacientes
§ Indicarles el tratamiento adecuado
§ Informar a las autoridades regulatorias si corresponde

24
Cumplimiento de las BPC
Documentar,
Documentar,
Y por último…

25
Recordar
Lo que no está escrito...

Estudios Preclínicos

¿Qué se busca con los ensayos clínicos?
Información científica sobre las
propiedades de las drogas en seres humanos
Eficacia, seguridad, biodisponibilidad de las drogas


Investigational New Drug Application (IND)
Incluye: :
• Farmacología animal • Experiencia previa en humanos
• Información de Manufactura • Los protocolos clínicos planificados

Investigational New Drug Application (IND)
Se renueva cada año � Nueva información
� Resultados de estudios � Plan Clínico para el próximo año
Enmiendas
� Al protocolo � Otros cambios

Ensayos Clínicos en Humanos
9 En sujetos voluntarios sanos o enfermos
9 Comienzan cuando se ha
demostrado seguridad en animales
9 Las Fases no son siempre
distintivas pero sirven como marcadores

Desarrollo de la Droga
Estudios Clínicos Fase I
Objetivo principal: SEGURIDAD
Busca definir: - Efectos secundarios
- Dosis toleradas
- Farmacodinamia
Duración media: ~ 1 - 1,5 años
N° de sujetos: ~ 20 - 80 voluntarios sanos
- Farmacocinética básica humana

Desarrollo de la Droga
Estudios Clínicos Fase II (IIa y IIb)
Objetivo principal: EFICACIA
Busca definir: - Eficacia
- Tolerabilidad a corto plazo - Dosis óptima
- Farmacodinamia
Duración media: ~ 1 a 2 años
N° de sujetos: ~ 100 a 300 pacientes

Desarrollo de la Droga
Estudios Clínicos Fase II (IIa y IIb)
¾ Búsqueda de dosis
¾ Datos cinéticos (PK) que correlacionen niveles de droga con efecto
farmacológico
¾ Al finalizar Meeting con FDA

Desarrollo de la Droga
Estudios Clínicos Fase III (IIIa)
Objetivo principal: EFICACIA Y SEGURIDAD
Busca definir: - Efectos de la droga en un grupo
- Ampliado de pacientes
Duración media: ~ 4 a 6 años
~ 1000 a 3000 pacientes

Desarrollo de la Droga
Estudios Clínicos Fase III (IIIa) Estudios Multicéntricos
(con similar diseño en diferentes centros) que aportan :
¾Datos e información concluyente para la seguridad y eficacia del fármaco en un número alto de pacientes.
¾Grupos de población especiales.
¾Presencia de múltiples enfermedades. �
¾Uso simultaneo de múltiples fármacos. �
¾Edad, Genero, Raza, Peso.

37
Farmacología Experimental Farmacocinética Farmacodinamia Toxicidad
20/80 sanos Seguridad Dosis tolerable Farmacodinamia 1-1.5 años
20/200 sanos Eficacia Tolerabilidad a corto plazo Dosis optima 1-2 años Roll out
300/3000 Seguridad Eficacia Edad/Raza/Género 4-6 años
Post Mkt Info adicional Riesgos/beneficios Uso optimo Diferentes Presentaciones Farmacovigilancia

New Drug Application (NDA)
• Requerimiento formal de aprobación a FDA
• Necesariamente tiene que proveerse datos de seguridad y eficacia completos
• Incluye la información del prospecto

Nueva Indicación
Si el Patrocinador quiere estudiar una nueva indicación, los estudios se consideran desde la Fase II, y
requiere de nueva solicitud de NDA.

Calidad en todas las fases
E. Preclínica Etapa Clínica
FI FII FIII FIV Marketing
IFPMA Code
Estándar de GLP
Calidad
GMP
GCP
Ciclo de vida
de un producto