proyecto fin de carrera - unizar.eszaguan.unizar.es/record/13614/files/taz-pfc-2014-166.pdf ·...
TRANSCRIPT

Repositorio de la Universidad de Zaragoza – Zaguan
http://zaguan.unizar.es
Proyecto Fin de Carrera
CARACTERIZACIÓN, INTEGRACIÓN Y
COMPORTAMIENTO DE NUEVOS MATERIALES
TIPO OXIAPATITA EN UNA PILA DE
COMBUSTIBLE DE ÓXIDO SÓLIDO
MICROTUBULAR
Autor
Samuel Poblador Cester
Directores
Alodia Orera Utrilla
Miguel Ángel Laguna Bercero
Ponente
Patricia Beatriz Oliete Terraz
Departamento de Ciencia y Tecnología de Materiales y Fluidos
Escuela de Ingeniería y Arquitectura. Universidad de Zaragoza
2014


ProyectoFindeCarreraIngenieríaIndustrial
CARACTERIZACIÓN,INTEGRACIÓNYCOMPORTAMIENTODENUEVOS
MATERIALESTIPOOXIAPATITAENUNAPILADECOMBUSTIBLEDEÓXIDO
SÓLIDOMICROTUBULAR
Autor
SamuelPobladorCester
Directores
AlodiaOreraUtrillaMiguelÁngelLagunaBercero
Ponente
PatriciaBeatrizOlieteTerraz
InstitutodeCienciadeMaterialesdeAragónDepartamentodeCienciayTecnologíadeMaterialesyFluidos
ÁreadeCienciadeMaterialeseIngenieríaMetalúrgica


Caracterización, integración y comportamientodenuevosmateriales tipooxiapatita enunapiladecombustibledeóxidosólidomicrotubular
RESUMEN
Enesteproyectosehafabricadounapiladecombustibledeóxidosólido(SOFC)detipomicrotubular soportada sobre ánodo con dos nuevos materiales de tipo oxiapatita,La9.67Si6O26.5 y La9.67Si3Ge3O26.5, sustituyendo el material de electrolito que se utilizaactualmente, la YSZ (circona estabilizada con itria), lo que permite bajar el punto deoperación del sistema de los 1000°C al rango de los 700 ‐ 800°C debido a losmejoresvaloresdeconducciónde los ionesdeoxígenoa temperaturas intermediasquepresentaestetipodemateriales.Paraellosehatenidoque:
1. Sintetizar las oxiapatitas, realizando las mezclas, moliendas y tratamientos térmicosadecuados,comprobandodespuéssucristalinidadypurezamediantedifracciónderayosX y espectroscopia Raman así como sus propiedades de conducción iónica medianteespectroscopiadeimpedancias(EIS).
2.Caracterizaryoptimizar lamorfologíadelpolvode loscompuestosobtenidosparasuposteriorutilizaciónenlafabricacióndelaspilas,diferenciandoaquepartedelapilaestédestinadocadapolvo,yaseaparaánodo,electrolitoocátodo.Paraello,sehaestudiadoladistribución del tamaño de partícula, se han realizado ensayos de dilatometría paradeterminarlastemperaturasdesinterizacióndelelectrolitoyporúltimo,sehanllevadoacabo ensayos de reología de las suspensiones para optimizar el proceso de dip‐coating(recubrimientopor inmersión) por el que se deposita el electrolito y el cátodo sobre elánodo.
3.Unavezoptimizados losparámetros, sehan fabricado losánodos (conunamezcladeNiO y oxiapatita)mediante CIP (prensado isostático en frío) y un posterior tratamientotérmicodepresinterización.Acontinuación,sehanhechomúltiplespruebasdedeposiciónde electrolito sobre el ánodo mediante la técnica de dip‐coating con un posteriortratamiento térmico de sinterización. Por último, después de obtenerse una correctaintegraciónentreelánodoylacapadeelectrolito,sehadepositadoelcátodo(mezcladePr2NiO4 y oxiapatita) sobre el electrolito. La integración entre las diferentes capas quecomponen la pila, así como los espesores de cada una de las fases, se ha estudiadomediantelastécnicasdemicroscopiaóptica(OM)yelectrónicadebarrido(SEM).
4. Finalmente, como se ha logrado con éxito la integración de las distintas capas, se hareducidoelNiOaNimetálicoenelánododejando lapilaensu forma funcional,yseharealizado la caracterización electroquímica mediante el control del voltaje a circuitoabierto(OCV)yconmedidasdelascurvasintensidad‐voltaje(I‐V).


Índice1.Introducción.......................................................................................................................................................11.1Ámbitodedesarrollodelproyecto....................................................................................................11.2Objetivoyalcancedelproyecto..........................................................................................................11.3Pilasdecombustible................................................................................................................................21.4Pilasdecombustibledeóxidosólido(SOFC)................................................................................2
1.4.1Conceptosgenerales.....................................................................................................................21.4.2Requisitoscomunesdeloscomponentes............................................................................31.4.3Ánodo.................................................................................................................................................31.4.4Electrolito.........................................................................................................................................31.4.5Cátodo................................................................................................................................................41.4.6Problemáticaactualyvíasdesolución.................................................................................4
1.5Propiedadesdelasoxiapatitas............................................................................................................51.5.1Estructura.........................................................................................................................................51.5.2Mecanismodeconducción.........................................................................................................5
1.6Procesogeneraldelfabricadodelapila...........................................................................................62.Síntesisdelasoxiapatitas..............................................................................................................................72.1Introducción...............................................................................................................................................72.2Mezclaestequiométrica.........................................................................................................................72.3Moliendadebolas.....................................................................................................................................82.4Compactado................................................................................................................................................92.5Tratamientotérmico(síntesis)...........................................................................................................92.6Comprobacióndelapurezadelasíntesis......................................................................................9
2.6.1EspectroscopiaRaman................................................................................................................92.6.2DifractometríaporrayosX.......................................................................................................10
2.7Medidadeconductividaddelasoxiapatitas(EIS)....................................................................123.Procesado,caracterizaciónyoptimizacióndelospolvosdeoxiapatita...................................143.1Tamañoymorfologíadelpolvodeseados....................................................................................14
3.1.1Polvoparaánodo.........................................................................................................................143.1.2Polvoparaelectrolito................................................................................................................143.1.3Polvoparacátodo........................................................................................................................14
3.2Métodosdecaracterización...............................................................................................................143.2.1Microscopíaelectrónicadebarrido(SEM).......................................................................143.2.2EspectrometríadeCorrelaciónFotónica(DLS)...............................................................15
3.3Procesosdetransformaciónaplicadosalospolvos.................................................................163.3.1Moliendaconmortero...............................................................................................................163.3.2Moliendadebolas.......................................................................................................................163.3.3Atrición............................................................................................................................................163.3.4Ultrasonidosdealtaintensidad..............................................................................................17
3.4Dilatometríadeelectrolito...................................................................................................................173.5Densimetría...............................................................................................................................................183.6Pastasdeelectrolitoycátodo.Caracterizaciónreológica......................................................19
4.Fabricacióndepilas......................................................................................................................................204.1Ánodo.Fabricaciónycaracterización............................................................................................20
4.1.1Mezclaparaánodo......................................................................................................................204.1.2Preformatubular.........................................................................................................................20

4.1.3Prensadoisostáticoenfrío(CIP)..........................................................................................204.1.4Presinterizado..............................................................................................................................214.1.5Dilatometríadeánodo.................................................................................................................21
4.2Deposicióndelacapadeelectrolito................................................................................................214.2.1Dip‐coating.....................................................................................................................................214.2.2Resultadosyoptimizacióndeparámetros.......................................................................22
4.3Cosinterizado(ánodo+electrolito)................................................................................................234.3.1Estudiodilatométrico................................................................................................................234.3.2Estudiodereactividad...............................................................................................................25
4.4Integracióndelcátodo..........................................................................................................................264.4.1Dip‐coating.....................................................................................................................................264.4.2Sinterizado.....................................................................................................................................26
5.CaracterizaciónelectroquímicadelaSOFC.........................................................................................275.1Preparacióndelaspilas.......................................................................................................................27
5.1.1Muestrasapreparar...................................................................................................................275.1.2Montajeentubos.........................................................................................................................275.1.3Montajedecontactoseléctricos............................................................................................27
5.2Montajedeequipoycaracterizaciónelectroquímica..............................................................285.2.1Montajedeequipo......................................................................................................................285.2.2Comprobacióndelaintegridadestructural.....................................................................285.2.3TratamientodereducciónymedidaOCV.........................................................................295.2.4Curvasintensidad‐voltaje.........................................................................................................30
6.Conclusiones....................................................................................................................................................316.1Visiónglobal.............................................................................................................................................316.2Conclusionesporfasesdedesarrollo.............................................................................................316.3Líneasfuturas...........................................................................................................................................33
Bibliografía............................................................................................................................................................34Tabladefiguras...................................................................................................................................................35Listadeabreviaturas.........................................................................................................................................38A1.Pilasdecombustible..................................................................................................................................40A1.1Reseñahistórica...................................................................................................................................40A1.2Tiposdepilasdecombustible........................................................................................................41
A2.Oxiapatitas.....................................................................................................................................................43A2.1Elmineraldeapatita..........................................................................................................................43A2.2Silicatos(germanatos).......................................................................................................................43
A2.2.1Concepto......................................................................................................................................43A2.2.2Clasificaciónestructural........................................................................................................44
A2.3SistemabinarioLa2O3−SiO2.............................................................................................................45Anexo3.Elaboracióndemezclasypastas...............................................................................................46A3.1Aditivosquímicosutilizados...........................................................................................................46
A3.1.1Alcoholdepolivinilo(PVA)..................................................................................................46A3.1.2Butiraldepolivinilo(PVB)...................................................................................................46A3.1.3BeycostatC213..........................................................................................................................46
A3.2Mezclasparaánodo.............................................................................................................................47A3.2.1Condicionesycálculosprevios...........................................................................................47A3.2.2Procesodefabricacióndelamezcla.................................................................................49A3.2.3Mezclasrealizadas...................................................................................................................50

A3.3Pastadeelectrolito.............................................................................................................................51A3.3.1Condicionesycálculo..............................................................................................................51A3.3.2Procesodefabricacióndelapasta....................................................................................51A3.3.3Pastasdeelectrolitorealizadas..........................................................................................52
A3.4Pastadecátodo.....................................................................................................................................53A3.4.1Condicionesycálculo..............................................................................................................53A3.4.2Procesodefabricacióndelapasta....................................................................................53A3.4.3Pastadecátodorealizada.....................................................................................................54
Anexo4.Técnicasdeprocesado...................................................................................................................55A4.1Moliendadebolas................................................................................................................................55A4.2Atrición....................................................................................................................................................56A4.3Compactado............................................................................................................................................56
A4.3.1Prensadouniaxial.....................................................................................................................56A4.3.2Prensadoisostático(CIP)......................................................................................................56
A4.4Sinterizado.............................................................................................................................................57A4.5Dip‐coating..............................................................................................................................................58
Anexo5.Técnicasdecaracterización.........................................................................................................59A5.1EspectroscopiaRaman......................................................................................................................59A5.2DifracciónderayosX.........................................................................................................................60
A5.2.1Concepto......................................................................................................................................60A5.2.2Equipoutilizado........................................................................................................................60A5.2.3Medidasrealizadas..................................................................................................................60
A5.3Microscopíaelectrónica....................................................................................................................61A5.4EspectrometríadeCorrelaciónFotónica...................................................................................62A5.5Dilatometría...........................................................................................................................................63
A5.5.1Concepto......................................................................................................................................63A5.5.2Dilatómetro.................................................................................................................................63A5.5.3Tratamientodedatos.............................................................................................................63A5.5.4Ejemplodedilatometríasanalizadas...............................................................................64
A5.7Reometría...............................................................................................................................................65A5.7.1Reología.Conceptoyaplicaciónenelreómetro..........................................................65A5.7.2Equipodemedida....................................................................................................................66A5.7.3Procesodemedida...................................................................................................................66
A5.8Espectroscopiadeimpedanciacompleja...................................................................................67

1.Introducción
‒1‒
1.Introducción
1.1ÁmbitodedesarrollodelproyectoEsteproyectoseenmarcadentrodeuna líneade trabajode investigaciónenmateriadepilasdecombustibledeóxidossólidosenelInstitutodeCienciadeMaterialesdeAragón(ICMA, instituto mixto CSIC‐Universidad de Zaragoza), en concreto, en el grupo deProcesadoyCaracterizacióndeCerámicasEstructuralesyFuncionales(ProCaCef).
1.2ObjetivoyalcancedelproyectoElprincipalobjetivodeesteproyectoeslafabricacióndeunapiladecombustibledeóxidosólido (SOFC) de tipo microtubular soportada sobre ánodo cambiando los materialesconvencionales que se utilizan actualmente de electrolito, como la YSZ (circonaestabilizada con itria),por dos nuevos materiales de tipo oxiapatita, La9.67Si6O26.5 yLa9.67Si3Ge3O26.5 (abreviadamente LSO y LSGO), los cuales presentanmejores valores deconducción de los iones de oxígeno a temperaturas intermedias. Sin embargo, estosmateriales presentan importantes limitaciones en su procesado, debidofundamentalmenteasudifícildensificación, loquehacenecesariounestudiominuciosodesuprocesodesinterización.Paraellosehatenidoque:
1. Sintetizar las oxiapatitas, realizando las mezclas, moliendas y tratamientos térmicosadecuados, comprobando después su cristalinidad mediante difracción de rayos X yespectroscopia Raman así como sus propiedades de conducción iónica medianteespescroscopiadeimpedancias(EIS).
2.Caracterizaryoptimizar lamorfologíadelpolvode loscompuestosobtenidosparasuposteriorutilizaciónenlafabricacióndelaspilas.Paraello,sehaestudiadoladistribucióndel tamaño de partícula, se han realizado ensayos de dilatometría para determinar lastemperaturasdesinterizacióndelelectrolitoyporúltimo,sehanllevadoacaboensayosdereologíadelassuspensionesparaoptimizarelprocesodedip‐coating(recubrimientoporinmersión)porelquesedepositaelelectrolitoyelcátodosobreelánodo.
3.Unavezoptimizados losparámetros, sehan fabricado losánodosdeNiOyoxiapatita,mediante CIP (prensado isostático en frío). Posteriormente, se han hecho múltiplespruebasdedeposicióndeelectrolitosobreelánodomediante latécnicadedip‐coatingyporúltimo,sehadepositadoelcátodo(mezcladePr2NiO4yoxiapatita)sobreelelectrolito.Laintegraciónentrelasdiferentescapasquecomponenlapila,asícomolosespesoresdecadaunade las fases, seha estudiadomediante las técnicas demicroscopia óptica y debarrido(SEM).
4. Finalmente, como se ha logrado con éxito la integración de las multicapas, se harealizado la caracterización electroquímica mediante el control del voltaje a circuitoabierto(OCV)ymedidasdelascurvasintensidad‐voltaje(I‐V).

1.Introducción
‒2‒
1.3PilasdecombustibleUna pila de combustible, al igual que cualquier otro tipo de pila, es un dispositivoelectroquímicoquepermiteobtenerunacorrienteeléctricaapartirdeunareacciónredoxespontánea entredos agentesquímicos, unoqueoxida (agenteoxidante) yotroque esoxidado(agentereductor).Enunapiladecombustible,porlogeneral,elagenteoxidanteeseloxígenodelaatmósferayeloxidadosueleserhidrógenoounhidrocarburo,elcualesalimentado,puntualocontinuamente,deahíeltérminopila“decombustible”.
Comoestosdispositivospermitenlageneracióndirectadeenergíaeléctricaapartirdelaoxidaciónelectroquímicadeuncombustible, evitan las limitacionesdictadasporel ciclode Carnot que rigen el tradicional proceso de combustión. Con lo cual, se tratan dedispositivosquepermitenunusomáseficientedelaenergíaquímicadeuncombustible.Otradelasventajasdeestossistemasessuimpactoambiental,yaquecomoenlamayoríadecasosseutilizahidrógenocomocombustible,noemitenCO2,siendovapordeaguaelúnicoresiduo.
Losdistintostiposdepilasdecombustibleexistentes,comosuscaracterísticasyunabrevereseñahistóricaseañadenenelanexoA1.
1.4Pilasdecombustibledeóxidosólido(SOFC)
1.4.1ConceptosgeneralesLaspilasdecombustibledeóxidosólido,oSOFC(SolidOxideFuelCell),sonunaclasedepilas de combustible caracterizadas por el uso de un material de óxido sólido comoelectrolito,porelcual los ionesóxido(O2‐)viajandesdeelcátodoalánodo,enelcualseforma el vapor de agua. Son dispositivos enteramente sólidos (tanto electrodo comoelectrolitos),adiferenciadeotrostiposcuyoselectrolitossonlíquidos[1].Enlafigura3semuestranlasreaccionesylosmaterialesquevaahaberenlasdistintaspartesdelaSOFCquesevaafabricarenesteproyecto.
Estas pilas son muyversátilesenlaeleccióndecombustibles, puedenutilizar tanto hidrógenocomo algún hidrocarburo(principalmentemetano).
Como el rango detemperaturas al queoperan es muy elevado,entre los 600°C y 1000°C(condición necesaria paralabuenaconductividadiónicadelelectrolito),lascinéticasdelasreaccionesdeoxidaciónyreducción del hidrógeno y oxígeno que ocurren en el interior sonmuy rápidas, con loquenoesindispensablelapresenciadeplatinocomoelectrocatalizador,suponiendounaventajaeconómicayaquesepuedenutilizarotrosmuchomásbaratoscomoelníquel.
Figura1.ReaccionesquímicasymaterialesquevaahaberenlasdistintaspartesdelaSOFCconstruidaenesteproyecto.

1.Introducción
‒3‒
1.4.2RequisitoscomunesdeloscomponentesTodosloscomponentesdeunaSOFCdebentenerunaseriederequisitoscomunes: Los componentes no deben reaccionar entre sí en las condiciones de fabricación y
operación(temperaturaypresiónparcialdeoxígeno),paraevitarlaposibleformacióndefasessecundariasenlasinterfases,quepuedenproducircaídasóhmicasenlapila.
Lamicroestructuranodebeevolucionarconeltiempoalatemperaturadetrabajo,yaquede lo contrario seproduciríaunavariaciónde la respuestade lapiladurante sufuncionamiento.
Los materiales deben presentar coeficientes de expansión térmica semejantes paraminimizarroturasoseparacióndelosmismosporfatigamecánica.
Esdeseablequeloscomponentesseanbaratosyquesuprocesadoseasencillodecaraasuproducciónagranescalaybajocoste.
1.4.3ÁnodoEn él se produce la oxidación electroquímica del combustible, concretamente en losllamadosTPB(TriplePhaseBoundary)quesonlospuntosdondecoincidenporo,metalycerámica. En la figura 5 se explica de unamaneramuysimpleelprocesoenunTPB.
El ánodo debe de ser poroso, para permitir eltransportegaseosodelasmoléculasdeH2alossitiosdereacción(TPBs),yunconductormixto,paraconducirtantoelflujodeelectronescomoel de iones O2−. En este proyecto se van aconstruir los ánodos con un cermet de níquel(que aportará la conductividad electrónica) yoxiapatita (que aportará la conductividadiónica)enunarelacióndevolumen1:1.
1.4.4ElectrolitoDebedeactuarcomo: Conductor iónico: Cuantamayor sea el flujode ionesO2−mayor será la potenciaque
proporcionarálapila. Barreraelectrónica:Elelectrolitodebeaislareléctricamenteelánododelcátodo,sino
habrácorrientesinternasdefugaquedisminuiránlapotencia. Barrerafísicaparagases:Eltránsitodegasesentreambosladosdelapilaimplicauna
pérdida de rendimiento, y en caso de fugas sustanciales, una situación de riesgo porunaposibleexplosión.
Portanto,lascaracterísticasquedebetenerelmaterialqueloconstituyeson: Altaconductividadiónica. Conductividadelectrónicadespreciable. Buenasinterabilidad:Sielmaterialdelelectrolitonodensificaprácticamenteal100%
puedequehayaalgunazonaporosa.
El material más comúnmente utilizado y extendido es la YSZ (circona estabilizada conitria).Enesteproyectosevanautilizarloscompuestosdeoxiapatitayamencionados.Enlasección1.5sedescribenmásampliamente.
Figura2.ReaccionesquímicasenunTBP.ElH2sedisociaendose−ydosprotones.Lose−sonconducidosporelNihastaelcontactoeléctricoylosprotonessecombinanconeliónO2−enlasuperficiedelLSOformandounamoléculadeH2O.

1.Introducción
‒4‒
1.4.5CátodoEsdonde tiene lugar la reduccióndel oxígeno.En la figura 7 se explicademaneramuysimpleelprocesoquímicoquetienelugar.
El cátodo debe de ser poroso con el fin depermitir la llegada de flujo del oxidante a lossitios activos y el material que lo compongadebetenerunamuybuenaconductividadtantoelectrónica como iónica. Además debe poseeractividadcatalíticaparapromoverlareduccióndelO2 ygenerarlosionesO2−. El niquelato depraseodimio(Pr2NiO4abreviadamentePNO)esun material perteneciente al tipo de losconductoresmixtosque cumpleperfectamentecon los requisitosdescritos [2].EnelproyectoelcátodoestarácompuestoporunamezcladePr2NiO4yoxiapatita.
1.4.6ProblemáticaactualyvíasdesoluciónComo se puede prever, la gran problemática que presentan las SOFC convencionalesactuales es el rango tan elevado de temperatura al que funcionan, lo cual conlleva unproblema de estabilidad termoquímica de los componentes y sus interfases que acabaderivando en una disminución de la efectividad y duración de la celda, además de laimposibilidaddeusarmetalescomocolectoresdecorriente.Loqueseintentaesdisminuiresta elevada temperatura de funcionamiento (evitandoasí sus problemas derivados) sinqueellorepercutaenunadisminucióndelosvaloresdepotenciadelapila.Lasdosvíasdeinvestigaciónmásrepresentativasqueseestánestudiandoactualmenteson:
o Cambiar la geometría soportada sobre electrolito, de las actuales SOFC, por unageometría soportada sobre ánodo. Esto permite la construcción de una capa deelectrolitomuyfina,lacual,amenortemperatura,puedetenerunvalorderesistenciaglobal igualomenorque lacapagruesadeelectrolitoquetienen laspilassoportadassobreelectrolito.[2]
o Sustituir los tradicionales materiales empleados en el electrolito, como la YSZ, pornuevosmaterialesquepresenten,amenor temperatura, igualesomejoresvaloresdeconductividadiónica.
Hay otros materiales, como el CeO2 y elBi2O3 que tienen mayor conductividadaniónica que la YSZ, pero son menosestables a presiones parciales bajas deoxígeno, originando la formación dedefectos en el óxido que provocan laaparicióndecorrienteseléctricasdefugainternas. Entre los nuevos materialesdestaca el LSGM (LaSrGaMgO) que a800°C tiene los mismos valores deconductividadaniónicaquelaYSZa1000°C,comosepuedeapreciarenlafigura5,aunqueeste compuesto es 5 veces más caro que la YSZ[3], con lo que no es un material taneficientedesdeunpuntodevistaeconómico‐energéticoglobaldelapila.
Figura3.Reaccionesquímicasenelcátodo.Una molécula de O2 (gas) es adsorbida por lasuperfice del PNO, donándole cuatro e− yconvirtiéndolaendos ionesO2−queviajan tantoporelPNOcomoporelLSOhastaelelectrolito.
Figura 4.Valores de conductividad para electrolitos deóxidosólidoydiversasconfiguracionesdepila.[1]

1.Introducción
‒5‒
1.5PropiedadesdelasoxiapatitasLapublicacióndeestudiosdelapresenciadeunaaltísimaconductividaddeionesóxidoensilicatos de lantánidos con la estructura de la apatita provocó el interés por lainvestigacióndeestetipodematerialescomoposibleselectrolitosdepilasdecombustiblede óxido sólido. Posteriormente, germanatos con idéntica estructura que estos silicatos,tambiénfueronidentificadoscomoexcelentesconductoresdeionesóxidos.
En términos económicos, los silicatos de lantano son bastante baratos, aunque losgermantos de lantano, por el contrario, son caros. Por esta razón, se va a utilizar uncompuestomixtodeambos(La9.67Si3Ge3O26.5),sobreelqueapenashayunlimitadonúmerodeestudiosenlaliteratura.
1.5.1EstructuraLafórmulageneraldelasoxiapatitasesM10(XO4)6O2,dondeMesunmetalpertenecientealastierrasrarasoalcalino,yXesunelementodelbloquep(electronesdevalenciaenelorbital p) tales como P, Si o Ge. Laestructura cristalina típica de apatitaestá compuesta portetraedros aislados(XO4)4−, con el catiónM situado en doscavidades, una con un número decoordinación7(esferasrojasenlafigura5) y el otro con un número decoordinación 9 (esferas verdes). Losátomos de oxígeno adicionales (esferasvioletas) ocupan canales en toda laestructura.[4]
1.5.2MecanismodeconducciónElmecanismodeconduccióniónicaenlasoxiapatitasesdetipointersticial.Unexcesodeionesóxidoenlaestructura(dentrodeunrango)mejoralosvaloresdeconducción.Enlosestudios iniciales que se realizaron se obtuvieron valores de conductividad a 500°C de1.1x10−4S·cm−1 para el La9.33(SiO4)6O2y de 1.3x10−3S·cm−1 para el La9.67(SiO4)6O2.5 (elcompuesto que se va a utilizar en este proyecto). En la figura 7 se puede observar unasimulaciónatomísticadondesepredicequeelcaminodeconducciónintersticialquevaaseguir un ión oxido va a tener una forma sinusoidal compleja. Una característicaimportanteesqueelprocesodeconducciónestáayudadoporlosdesplazamientosdelostetraedros silicato (SiO4)4−, lo que sugiere que la flexibilidad de estas subestructuras esesencialparalasaltasconductividadesiónicasobservadas.[5]
Figura5.Estructuradelcristaldeoxiapatitavistadelejez.
Figura7.SimulaciónatomísticadelavíademigracióndeliónO2−intersticialenlaoxiapatitaLa9.33(SiO4)6O2Vistafrontaldelejec(a)yvistaperpendicularalejec(b).
Figura 6.Comparación de la conductividad deoxiapatitasconotrosconductoresdeionesóxido.

1.Introducción
‒6‒
1.6ProcesogeneraldelfabricadodelapilaEn el siguiente diagrama se plasma el proceso general de fabricación de una pila degeometría tubular soportada sobre ánodo con el material que más se estudia en esteproyecto,elLSO.Esteesquemaservirádeíndicevisualparaunamejorubicaciónalolargode la lecturade lamemoria, cuyosapartados correspondenadivisiones secuencialesdeesteesquema.Despuésdefabricarlapilahayqueacondicionarla(reduccióndeNiOaNi)pararealizarsucaracterizaciónelectroquímica.
Relaciónentreapartadosdelamemoriaybloquesenelprocesogeneraldefabricadodelapila:
2.Síntesisdelasoxiapatitas(bloqueazul)
3.Procesado,caracterizaciónyoptimizacióndelospolvosdeoxiapatita(bloquenaranja)
4.Fabricacióndepilas(bloqueverde)

2.Síntesisdelasoxiapatitas
‒7‒
2.Síntesisdelasoxiapatitas
2.1IntroducciónAlolargodeesteproyectoentotalsehanrealizadotressíntesisdeLSO(referidascomoLSO.1, LSO.2 y LSO.3) y una síntesis de LSGO. Elmétodo de síntesis empleado es el de"reacciónenestadosólidoaaltatemperatura".
2.2MezclaestequiométricaLoscompuestosdeoxiapatitaquesevanasintetizarsondos,elLSOyelLSGO.Larelacióndereactivosnecesariaparalaobtencióndeamboscompuestoseslaqueseobtienesegúnlassiguientesrelacionesestequiométricas:
4.833 ⋅ La O 6 ⋅ SiO ⟶ La . SiO O .
4.833 ⋅ La O 3 ⋅ SiO 3 ⋅ GeO ⟶ La . SiO GeO O .
De la apatita con germanio sequierenobtener 10gdepolvo,mientrasquede la que esenteramentesilicatodelantanosequierenobtener15g.Parahallarlascantidadesquesehan de mezclar se efectúan los correspondientes cálculos. En el siguiente cuadro semuestran la fracción molar que tiene cada óxido con su respectivo peso molecular, lafracciónmásicaquesedeberíamezclardecadaunosisetuvieranenestadopuroal100%,lapurezadelosóxidosquesehanutilizadoenellaboratorioylacantidaddecadaunodeestos productos comerciales que se tiene que mezclar para cumplir la relaciónestequiométricaquesehaindicadoanteriormente:
LSO LSGOLa2O3 SiO2 La2O3 SiO2 GeO2
fracciónmolar 0.4462 0.5538 0.4462 0.2769 0.2769pesomolecular[g/mol] 325.82 60.09 325.82 60.09 104.59fracciónmásicapura 0.8137 0.1863 0.7612 0.0871 0.1517purezadelpolvocomercial 0.9999 0.998 0.9999 0.998 0.9998fracciónmásica 0.8134 0.1866 0.7611 0.0873 0.1517masadelamezcla[g] 15 10masadecadaóxido[g] 12.2013 2.7987 7.6106 0.8729 1.5165Anteriormentealamezclasedebenprepararlospolvoscomercialesquesevanautilizar.Entodaslassíntesis,elóxidodelantanose hasometidoauntratamientotérmico,enelquesecalientahasta980°Cyselemantienedurantedoshoras,conelfindedeshidratarydescarbonatar, yaqueesuncompuestoaltamentehigroscópicoyqueatrapadióxidodecarbono del aire con cierta facilidad. Adicionalmente, en la tercera síntesis de LSO, sesometióalóxidodesilicioalmismotratamiento.
Elpesadodeloscomponentesserealizaenunabalanzadeprecisión(deunadiezmilésimade gramo). Inmediatamente después semuelen juntos, excepto en la tercera síntesis deLSO,enlacualestuvieronpremezclándosedurante12horasenunrecipientecolocadoenunagitadorderodillosantesdelamolienda.

2.Síntesisdelasoxiapatitas
‒8‒
2.3MoliendadebolasElmolinodebolaseselmétodomásutilizadoparalamezcladepolvoscerámicosyparalareduccióndetamañodesuspartículas.Entresusventajassedestacanlasencillezyelbajoprecio del equipo. No obstante presenta algunos problemas, el más importante es lacontaminaciónquesepuedeproducirpordesgastedelmediodemolienda(bolas),obienporelrecipienteenelcualseintroduceelmaterialquesevaamoler.
Esta técnica permite obtener una mezcla muy homogénea con un tamaño de granoreducido,locualfavoreceenormementeelprocesodesíntesisposterior.
Entodaslassíntesisrealizadas, lamezcladelpolvoseharealizadoconunamoliendadebolas a 300 rpm usando como medio líquido etanol. En las primeras síntesis (LSGO yLSO.1)conunaduraciónde5horasyenlasotrasdos(LSO.2yLSO.3)conunaduraciónde2.5horas.
ExceptoenlaprimerasíntesisdeLSO,queseutilizóunboldecircona(ZrO2)con90bolasdecirconadediámetro10mm,enelrestode lassíntesisseutilizóunboldeágata(SiO2)con 90 bolas de ágata de diámetro 10mm.Esto corresponde a la relación general (envolumen)quesesueleemplear:1/3dematerial,1/3demediolíquidoy1/3debolas.
El molino que se ha utilizado durante todo el proyecto es un Planetary Mono MillPULVERISETTE6classiclinedelacompañíaFritsch[4].
Despuésderealizarselamoliendaseprocedealarecuperacióndelpolvo.Elcontenidodelbol se transvasaa una bandeja. Tanto el interior de bol, como las bolas, se limpian conetanol para recuperar la mayor cantidadposibledepolvo,quetambiénseverteráalamisma bandeja, la cual, se introduce en unhorno a unos 85°C durante unas 3‐4 horasparasusecado.
Unavezevaporadotodoeletanol,elpolvoseencuentra formando una lámina seca en elfondodelabandeja.Seprocedeaunraspadopara obtener el polvo suelto y se guarda enunrecipienteherméticoparadificultarquelamezcladeóxidossepuedahidratar.
Figura 8.Bol y bolas de ágata con polvo antes deañadiretanolparaprocederasumezcla‐molienda.
Figura9.Boldecirconacolocadoyaseguradoenelmolinoplanetarioantesdeempezarlamolienda.
Figura10.Transvasedemoliendaabandeja.

2.Síntesisdelasoxiapatitas
‒9‒
2.4CompactadoAlpolvoqueseharecuperadodelamezcla‐moliendadebolas,selerealizauncompactado,elcualaportaunaaproximaciónmáselevadaentrelaspartículas,aumentandoelcontactosuperficialentreellas,favoreciendo,portanto,elprocesodereacciónenestadosólidoquetendrálugardespués.
En lasdosprimerassíntesis (LSO.1yLSGO)elpolvose compactó en forma de pastillas con una prensauniaxial. Cada una fue prensada con un troquelcilíndricode15mmdediámetroyunapresiónde5toneladasdurante 5minutos. Laspastillas teníanunespesorde1.5mmyunpesoaproximadode1.2g.
Enlasdossiguientesmezclasparaobtenerlosnuevoscompuestos (LSO.2 y LSO.3) no se utilizó la prensa.Simplemente, en el crisol que se introduce al hornopara el tratamiento térmico, el polvo fue depositado en capas finas, y entre cadadeposición, conunmacillo (manodemortero)deágata, se fueprensandomanualmentehastaobtenerunamasacompacta.
2.5Tratamientotérmico(síntesis)Despuésde tener lamezcladeóxidosenuna formacompactadepolvohomogéneo contamañodegranoadecuadoselesometealtratamientotérmicoqueproducelareacciónenestado sólido la cual sintetiza los óxidos iniciales en el compuesto de oxiapatitabuscado.Para todas las síntesis ha sido elmismo, un calentamiento a razón de 6°C porminuto,hastaunatemperaturade1500°C,enlaquesehamantenidodurante4horas,yundescensohastatemperaturaambiente,tambiénaunavelocidadde6°C/min.
2.6ComprobacióndelapurezadelasíntesisHansidodoslastécnicasutilizadasparacomprobarlapurezadelasmuestrasobtenidasylaexistenciadeotrasfasesnodeseadas:laespectroscopiaRamanyladifracciónporrayosX.EnelanexoA5seincluyemásinformaciónsobreestastécnicas.
2.6.1EspectroscopiaRamanEstatécnicaesmuyflexibleporquenohacefaltaprepararunamuestraenpolvo,puesesposiblemedirdirectamentesobrelasuperficiedelaspastillascompactadasysintetizadas.Conestatécnicaseobtieneunaresoluciónespacialdelamuestra,porloquesecompruebasuhomogeneidad.Ademáselgrupo(ProCaCef)disponedeesteequipodemedida,conlacorrespondienterapidezenquesepuedeobteneresta.Sinembargo,elproblemaprácticoque supone esta técnica es que, debido a su extrema sensibilidad, se magnifica en losresultados de medida las pequeñas impurezas que pueda haber en la muestra. En lasoxiapatitasesmuycomúnencontrarpequeñassegundasfasesdeLa2SiO5(oxiortosilicatode lantano)yLa2Si2O7 (sorosilicatode lantano),yaqueenel sistemabinarioLa2O3−SiO2(veranexoA2.3)estoscompuestoscoexisten[5].
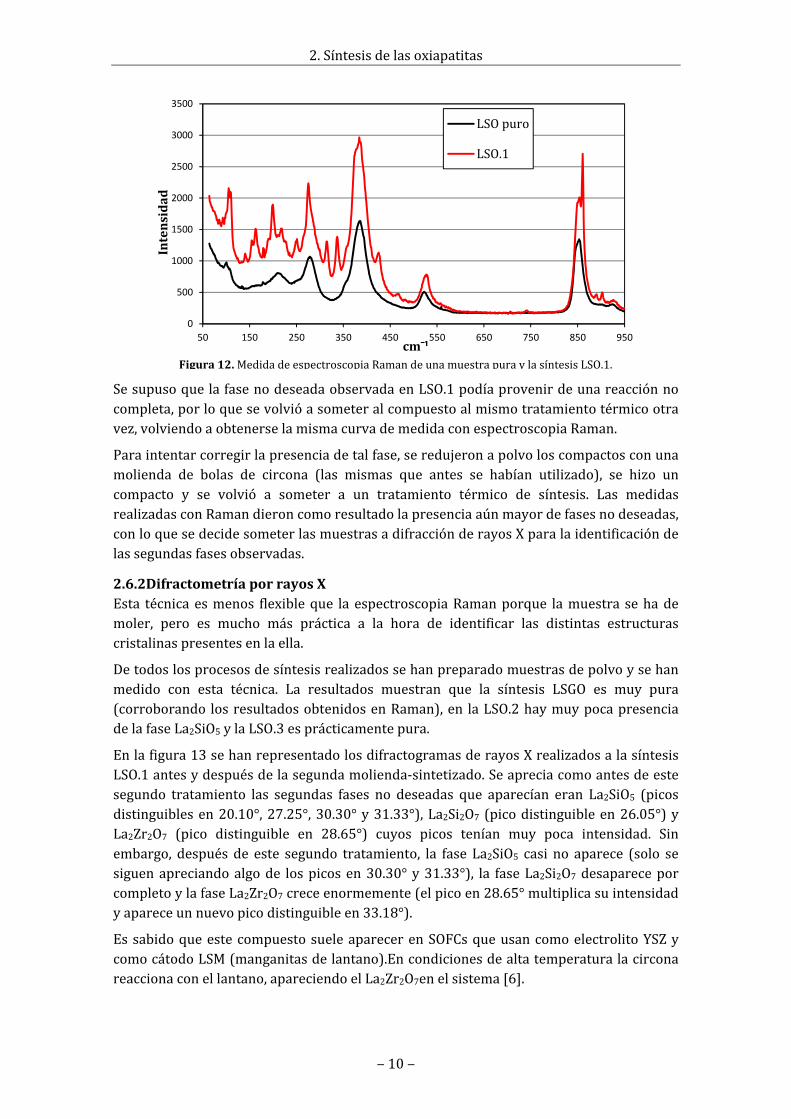
Con esta técnica se observó que la pureza en la muestra de LSGO sintetizado eramuyelevada, es decir, la curva medida con el espectrómetro era muy ajustada a la de unamuestrapura,mientrasqueenlamuestraquesemidiódeLSO.1,aparecíareflejadaenlacurvalaexistenciadeunafasenodeseada,comosepuedeobservarenlafigura12.
Figura11.Pastillareciénprensadaadheridaaltroquellistaparasuextracción.
2.Síntesisdelasoxiapatitas
‒10‒
SesupusoquelafasenodeseadaobservadaenLSO.1podíaprovenirdeunareacciónnocompleta,porloquesevolvióasometeralcompuestoalmismotratamientotérmicootravez,volviendoaobtenerselamismacurvademedidaconespectroscopiaRaman.
Paraintentarcorregirlapresenciadetalfase,seredujeronapolvoloscompactosconunamolienda de bolas de circona (las mismas que antes se habían utilizado), se hizo uncompacto y se volvió a someter a un tratamiento térmico de síntesis. Las medidasrealizadasconRamandieroncomoresultadolapresenciaaúnmayordefasesnodeseadas,conloquesedecidesometerlasmuestrasadifracciónderayosXparalaidentificacióndelassegundasfasesobservadas.
2.6.2DifractometríaporrayosXEsta técnica esmenos flexibleque la espectroscopiaRamanporque lamuestra sehademoler, pero es mucho más práctica a la hora de identificar las distintas estructurascristalinaspresentesenlaella.
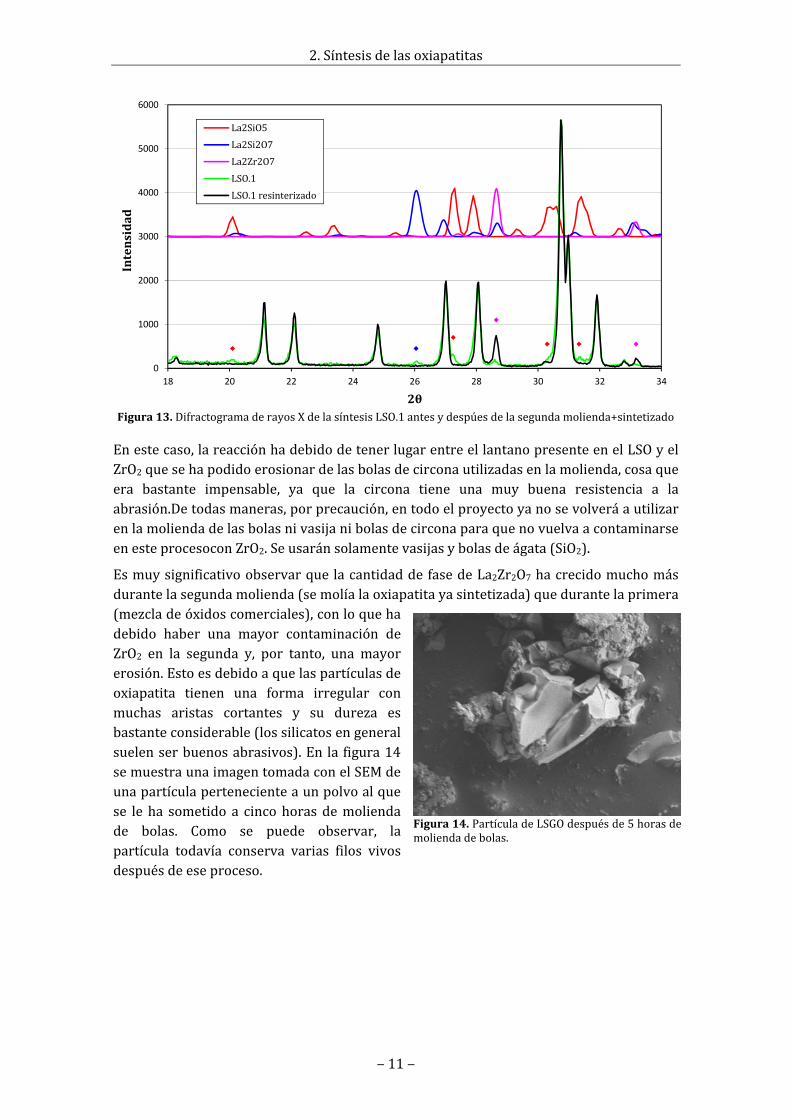
Detodoslosprocesosdesíntesisrealizadossehanpreparadomuestrasdepolvoysehanmedido con esta técnica. La resultados muestran que la síntesis LSGO es muy pura(corroborando losresultadosobtenidosenRaman),en laLSO.2haymuypocapresenciadelafaseLa2SiO5ylaLSO.3esprácticamentepura.
Enlafigura13sehanrepresentadolosdifractogramasderayosXrealizadosalasíntesisLSO.1antesydespuésdelasegundamolienda‐sintetizado.Seapreciacomoantesdeestesegundo tratamiento las segundas fases no deseadas que aparecían eran La2SiO5 (picosdistinguiblesen20.10°,27.25°,30.30°y31.33°),La2Si2O7 (picodistinguibleen26.05°)yLa2Zr2O7 (pico distinguible en 28.65°) cuyos picos tenían muy poca intensidad. Sinembargo, después de este segundo tratamiento, la fase La2SiO5 casi no aparece (solo sesiguenapreciandoalgode lospicosen30.30°y31.33°), la faseLa2Si2O7desapareceporcompletoylafaseLa2Zr2O7creceenormemente(elpicoen28.65°multiplicasuintensidadyapareceunnuevopicodistinguibleen33.18°).
EssabidoqueestecompuestosueleaparecerenSOFCsqueusancomoelectrolitoYSZycomocátodoLSM(manganitasdelantano).Encondicionesdealtatemperaturalacirconareaccionaconellantano,apareciendoelLa2Zr2O7enelsistema[6].
Figura12.Medidadeespectroscopia Raman deunamuestrapurayla síntesisLSO.1.
0
500
1000
1500
2000
2500
3000
3500
50 150 250 350 450 550 650 750 850 950
Intensidad
cm¯¹
LSOpuro
LSO.1
2.Síntesisdelasoxiapatitas
‒11‒
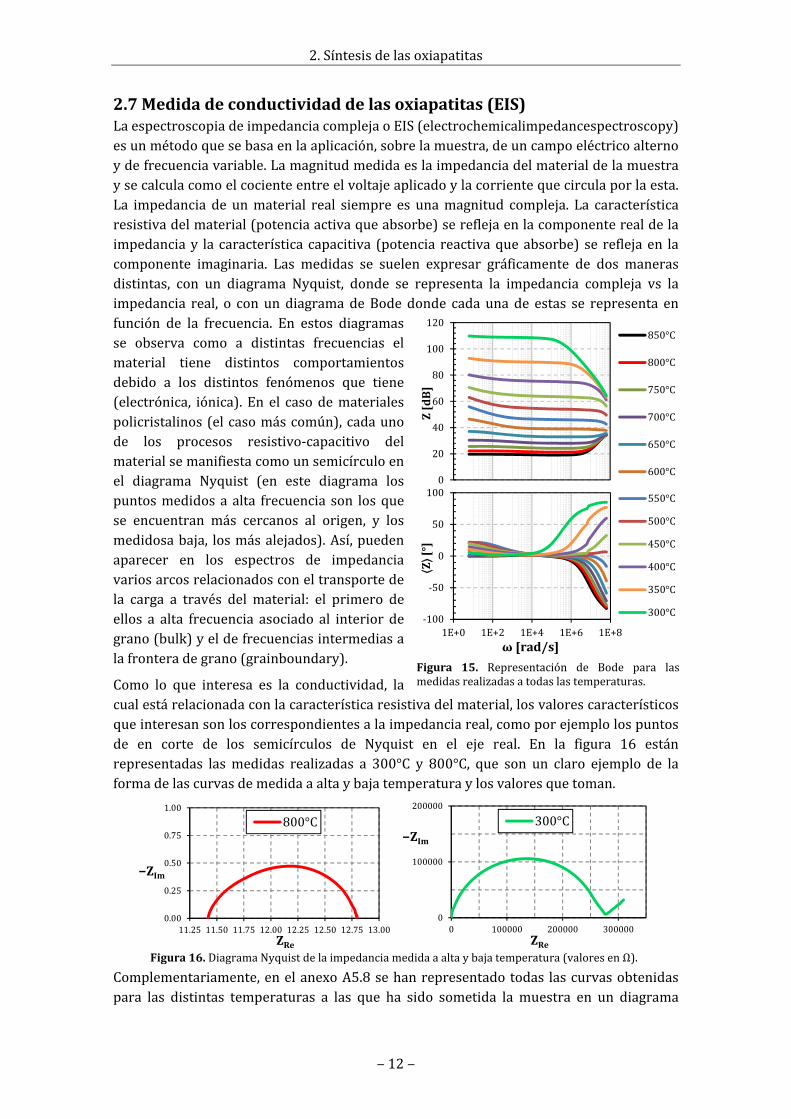
Enestecaso,lareacciónhadebidodetenerlugarentreellantanopresenteenelLSOyelZrO2quesehapodidoerosionardelasbolasdecirconautilizadasenlamolienda,cosaqueera bastante impensable, ya que la circona tiene una muy buena resistencia a laabrasión.Detodasmaneras,porprecaución,entodoelproyectoyanosevolveráautilizarenlamoliendadelasbolasnivasijanibolasdecirconaparaquenovuelvaacontaminarseenesteprocesoconZrO2.Seusaránsolamentevasijasybolasdeágata(SiO2).
EsmuysignificativoobservarquelacantidaddefasedeLa2Zr2O7hacrecidomuchomásdurantelasegundamolienda(semolíalaoxiapatitayasintetizada)quedurantelaprimera(mezcladeóxidoscomerciales),conloquehadebido haber una mayor contaminación deZrO2 en la segunda y, por tanto, una mayorerosión.Estoesdebidoaquelaspartículasdeoxiapatita tienen una forma irregular conmuchas aristas cortantes y su dureza esbastanteconsiderable(lossilicatosengeneralsuelenserbuenosabrasivos).En la figura14semuestraunaimagentomadaconelSEMdeunapartículapertenecienteaunpolvoalquese le ha sometido a cincohorasdemoliendade bolas. Como se puede observar, lapartícula todavía conserva varias filos vivosdespuésdeeseproceso.
Figura13.DifractogramaderayosXdelasíntesisLSO.1antesydespúesdelasegundamolienda+sintetizado
0
1000
2000
3000
4000
5000
6000
18 20 22 24 26 28 30 32 34
Intensidad
2θ
La2SiO5
La2Si2O7
La2Zr2O7
LSO.1
LSO.1resinterizado
Figura14.PartículadeLSGOdespuésde5horasdemoliendadebolas.
2.Síntesisdelasoxiapatitas
‒12‒
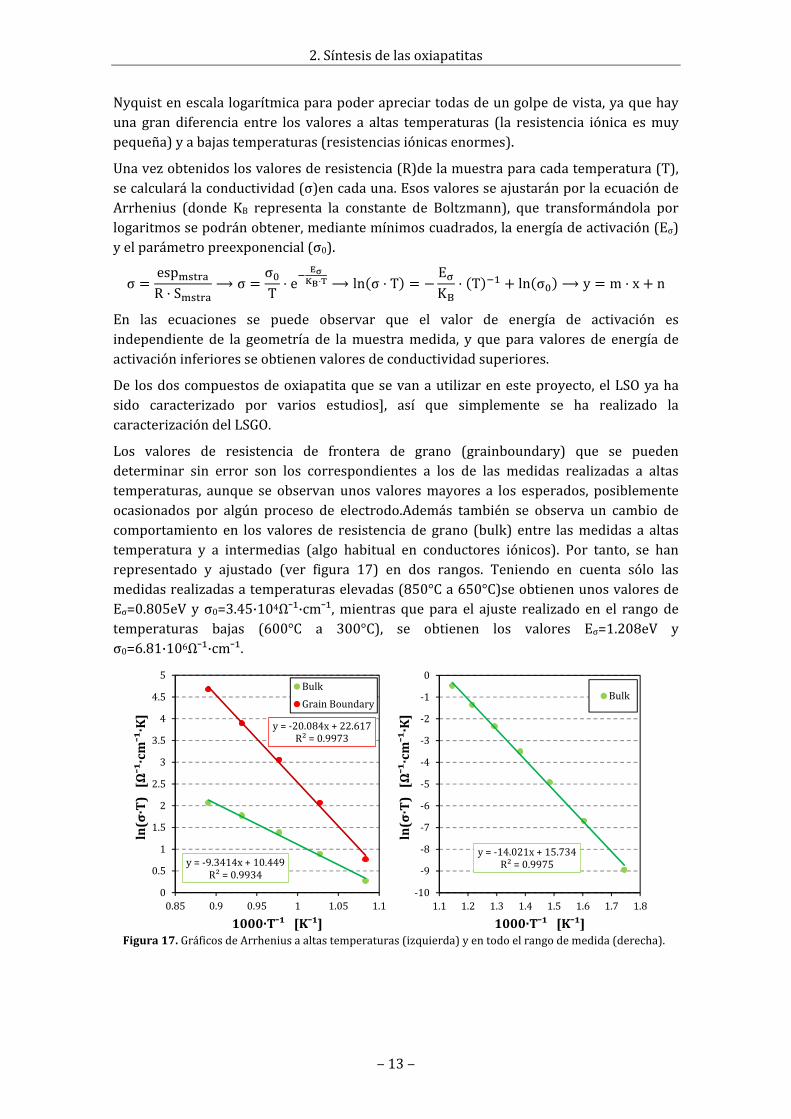
2.7Medidadeconductividaddelasoxiapatitas(EIS)LaespectroscopiadeimpedanciacomplejaoEIS(electrochemicalimpedancespectroscopy)esunmétodoquesebasaenlaaplicación,sobrelamuestra,deuncampoeléctricoalternoydefrecuenciavariable.Lamagnitudmedidaeslaimpedanciadelmaterialdelamuestraysecalculacomoelcocienteentreelvoltajeaplicadoylacorrientequecirculaporlaesta.La impedancia de unmaterial real siempre es unamagnitud compleja. La característicaresistivadelmaterial(potenciaactivaqueabsorbe)sereflejaenlacomponenterealdelaimpedanciay la característicacapacitiva (potencia reactivaqueabsorbe) se reflejaen lacomponente imaginaria. Las medidas se suelen expresar gráficamente de dos manerasdistintas, con un diagrama Nyquist, donde se representa la impedancia compleja vs laimpedancia real, o conundiagramadeBodedonde cadaunade estas se representa enfunción de la frecuencia. En estos diagramasse observa como a distintas frecuencias elmaterial tiene distintos comportamientosdebido a los distintos fenómenos que tiene(electrónica, iónica).En el casodematerialespolicristalinos(elcasomáscomún),cadaunode los procesos resistivo‐capacitivo delmaterialsemanifiestacomounsemicírculoenel diagrama Nyquist (en este diagrama lospuntosmedidosa alta frecuencia son losquese encuentran más cercanos al origen, y losmedidosabaja, losmásalejados).Así,puedenaparecer en los espectros de impedanciavariosarcosrelacionadosconeltransportedela carga a través del material: el primero deellos a alta frecuencia asociado al interior degrano(bulk)yeldefrecuenciasintermediasalafronteradegrano(grainboundary).
Como lo que interesa es la conductividad, lacualestárelacionadaconlacaracterísticaresistivadelmaterial,losvalorescaracterísticosqueinteresansonloscorrespondientesalaimpedanciareal,comoporejemplolospuntosde en corte de los semicírculos de Nyquist en el eje real. En la figura 16 estánrepresentadas lasmedidas realizadas a 300°C y 800°C, que son un claro ejemplo de laformadelascurvasdemedidaaaltaybajatemperaturaylosvaloresquetoman.
Complementariamente,enelanexoA5.8sehanrepresentadotodas lascurvasobtenidaspara las distintas temperaturas a las que ha sido sometida la muestra en un diagrama
Figura16.DiagramaNyquistdelaimpedanciamedidaaaltaybajatemperatura(valoresenΩ).
0.00
0.25
0.50
0.75
1.00
11.25 11.50 11.75 12.00 12.25 12.50 12.75 13.00
−ZIm
ZRe
800°C
0
100000
200000
0 100000 200000 300000
−ZIm
ZRe
300°C
Figura 15. Representación de Bode para lasmedidasrealizadasatodaslastemperaturas.
0
20
40
60
80
100
120
Z[dB]
850°C
800°C
750°C
700°C
650°C
600°C
‐100
‐50
0
50
100
1E+0 1E+2 1E+4 1E+6 1E+8
Z[°]
ω [rad/s]
550°C
500°C
450°C
400°C
350°C
300°C
2.Síntesisdelasoxiapatitas
‒13‒
Nyquistenescalalogarítmicaparapoderapreciartodasdeungolpedevista,yaquehayuna gran diferencia entre los valores a altas temperaturas (la resistencia iónica esmuypequeña)yabajastemperaturas(resistenciasiónicasenormes).
Unavezobtenidoslosvaloresderesistencia(R)delamuestraparacadatemperatura(T),secalcularálaconductividad(σ)encadauna.EsosvaloresseajustaránporlaecuacióndeArrhenius (donde KB representa la constante de Boltzmann), que transformándola porlogaritmossepodránobtener,mediantemínimoscuadrados,laenergíadeactivación(Eσ)yelparámetropreexponencial(σ0).
σespR S
⟶ σσT⋅ e ⟶ ln σ T
EK
T ln σ ⟶ y m x n
En las ecuaciones se puede observar que el valor de energía de activación esindependiente de la geometría de lamuestramedida, y que para valores de energía deactivacióninferioresseobtienenvaloresdeconductividadsuperiores.
De losdoscompuestosdeoxiapatitaquesevanautilizarenesteproyecto,elLSOyahasido caracterizado por varios estudios], así que simplemente se ha realizado lacaracterizacióndelLSGO.
Los valores de resistencia de frontera de grano (grainboundary) que se puedendeterminar sin error son los correspondientes a los de las medidas realizadas a altastemperaturas, aunque se observanunos valoresmayores a los esperados, posiblementeocasionados por algún proceso de electrodo.Además también se observa un cambio decomportamiento en los valores de resistencia de grano (bulk) entre lasmedidas a altastemperatura y a intermedias (algo habitual en conductores iónicos). Por tanto, se hanrepresentado y ajustado (ver figura 17) en dos rangos. Teniendo en cuenta sólo lasmedidasrealizadasatemperaturaselevadas(850°Ca650°C)seobtienenunosvaloresdeEσ=0.805eV yσ0=3.45·104Ω¯¹·cm¯¹,mientras quepara el ajuste realizado en el rangodetemperaturas bajas (600°C a 300°C), se obtienen los valores Eσ=1.208eV yσ0=6.81·106Ω¯¹·cm¯¹.
Figura17.GráficosdeArrheniusaaltastemperaturas(izquierda)yentodoelrangodemedida(derecha).
y=‐9.3414x+10.449R²=0.9934
y=‐20.084x+22.617R²=0.9973
0
0.5
1
1.5
2
2.5
3
3.5
4
4.5
5
0.85 0.9 0.95 1 1.05 1.1
ln(σ·T)[Ω¯¹·cm¯¹·K]
1000·T¯¹[K¯¹]
Bulk
GrainBoundary
y=‐14.021x+15.734R²=0.9975
‐10
‐9
‐8
‐7
‐6
‐5
‐4
‐3
‐2
‐1
0
1.1 1.2 1.3 1.4 1.5 1.6 1.7 1.8
ln(σ·T)[Ω¯¹·cm¯¹·K]
1000·T¯¹[K¯¹]
Bulk

3.Procesado,caracterizaciónyoptimizacióndelospolvosdeoxiapatita
‒14‒
3. Procesado, caracterización y optimizacióndelospolvosdeoxiapatitaDependiendodesielpolvocerámicovaaserutilizadoparafabricaruntubodeánodoosise va a utilizar en forma de pasta para la fabricación del electrolito o del cátodo, lascaracterísticasquedebepresentarsondistintas.Paraelloseleaplicarándistintosmétodosdeprocesado(moliendadebolas,atriciónyultrasonidos).Paraverificarsisehalogradoadecuarelpolvocorrectamenteydecidirsisehadevolveraprocesarono,semediráladistribución de tamaños de partícula con un espectrómetro de correlación fotónicay seobservarátantoladimensióncomolamorfologíadelosgranosdepolvoenunmicroscopioelectrónico de barrido. Además, en el caso del polvo procesado para electrolito, serealizarándilatometríasydensimetríasparacaracterizarsudensificación.
3.1Tamañoymorfologíadelpolvodeseados
3.1.1PolvoparaánodoPara fabricar un tubo de ánodo se van a utilizar dos polvos cerámicos, uno será laoxiapatita sintetizada y el otro seráóxido de níquel (además de almidón demaíz comoformadordeporo).Laspartículasdeamboscomponenteshandeserdesimilar tamañoporquesino,unodelosdospodríadejaraisladoreticularmenteapartesdelotro.Todalaestructura de grano de la apatita debe estar interconectada para que los iones O2−producidosentodoslosTPB(triplephaseboundary,puntostriplesdondetienelareacciónquímica)delánodopuedanllegarhastaelelectrolito.Igualmente,sialgunazonadeníquelestuvieraincomunicada,nolellegaríaflujodeelectrones,conloquelosTPBdeesazonanointervendríanenelmecanismo,reduciendolaeficienciaypotenciadelapila.
3.1.2PolvoparaelectrolitoAdiferenciadelánodo,quevaaserunaestructuraporosa,elelectrolitodebetenerunaporosidad prácticamente nula. Para favorecer su densificación durante el proceso desinterizadosuspartículasdebendeserpequeñas(veranexoA4.4)demodoquepresentenunasinterizacióncaracterísticacompatibleconeltuboquehacedesoporte.
3.1.3PolvoparacátodoEl cátodo será una mezcla de niquelato de praseodimio con oxiapatita. Igual que en elánodo,todoslosgranosdeniquelatodelaestructuradelcátododeberánestarconectadosparaquenohayaningunazonaquesequedesinflujoelectrónico.
3.2Métodosdecaracterización
3.2.1Microscopíaelectrónicadebarrido(SEM)El microscopio electrónico de barrido o SEM (ScanningElectronMicroscopy) permiteobservarladimensiónylamorfologíadelosgranosdepolvo.Ambascaracterísticassonimportantes tanto a la hora de conformar el tubo de ánodo como a la hora de lasinterización.Conestatécnicapodemosobservarademáslapresenciadeaglomeradosysilas partículas tienenuna forma cercana a la esfera (muy recomendable para unabuenadensificación)oporsilocontrariosonirregulares.
3.Procesado,caracterizaciónyoptimizacióndelospolvosdeoxiapatita
‒15‒
3.2.2EspectrometríadeCorrelaciónFotónica(DLS)Esta técnica, también conocida como dispersión dinámica de luz o DLS (Dynamic lightscattering)seutilizaparamedirladistribucióndeltamañodepartículaenunasuspensión.
Como medio dispersante en las suspensiones se ha utilizado etanol con un 1% dedefloculante Beycostat C213 (ver anexo A3.1.3) en peso del polvo utilizado. Para evitarproblemasdeestabilidadestassuspensionesdemedidasehicieronmuydiluidas(siempredentrodelosparámetrosrecomendadosporelfabricantedelaparato).
Como las oxiapatitas son materiales nuevos que pocas veces se han medido en elespectrómetroquesevaautilizar,laincertidumbreenelprocesodemedidaesunfactoratener en cuenta (respecto a su valor absoluto; aunque sirven perfectamente paracomparar muestras).Para minimizar tal problema, se ha procedido midiendo siempretodaslassuspensionesconidénticaconcentración,paraqueesteparámetronointroduzcaunadisonanciaenlosresultados.Además,siempresehanrealizadovariasrepeticionesdemedidadecadamuestrahastaqueelsoftwarequeanalizalasmedidasdelespectrómetrodaunvalordeconfianzaqueseconsidereaceptable.
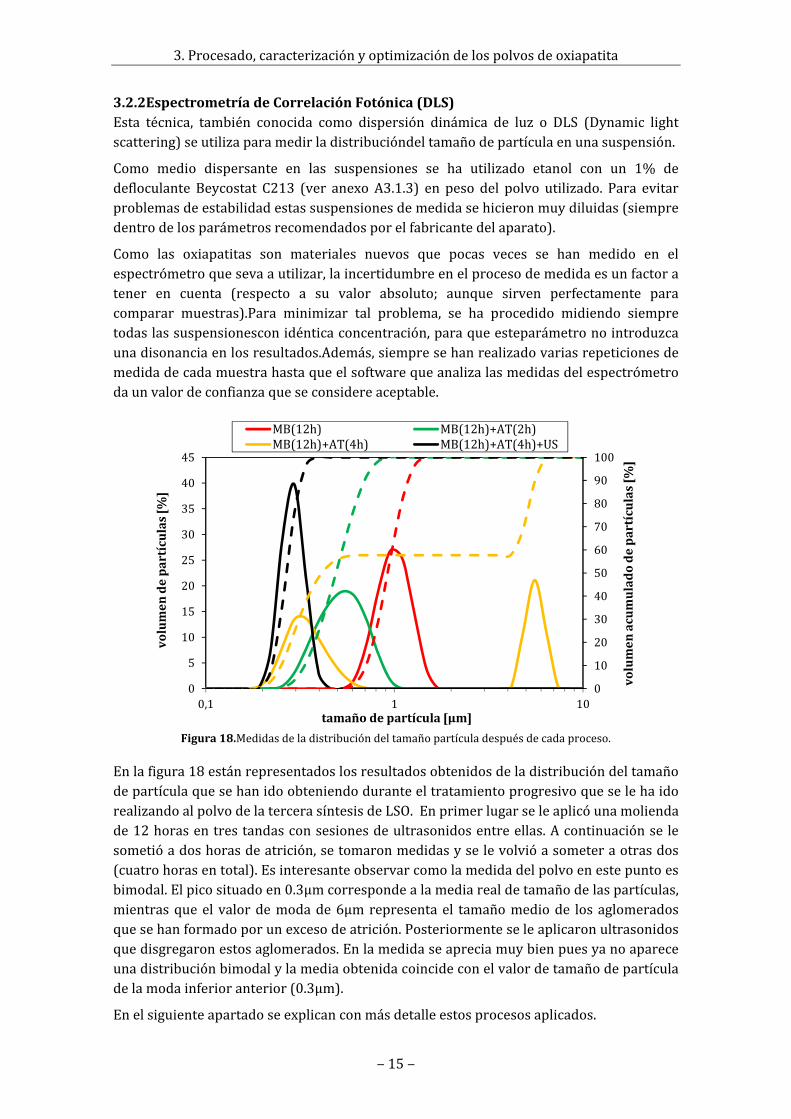
Enlafigura18estánrepresentadoslosresultadosobtenidosdeladistribucióndeltamañodepartículaquesehanidoobteniendoduranteeltratamientoprogresivoqueselehaidorealizandoalpolvodelatercerasíntesisdeLSO.Enprimerlugarseleaplicóunamoliendade12horasentrestandasconsesionesdeultrasonidosentreellas.Acontinuaciónse lesometióadoshorasdeatrición,setomaronmedidasyselevolvióasometeraotrasdos(cuatrohorasentotal).Esinteresanteobservarcomolamedidadelpolvoenestepuntoesbimodal.Elpicosituadoen0.3μmcorrespondealamediarealdetamañodelaspartículas,mientrasqueel valordemodade6μmrepresentael tamañomediode losaglomeradosquesehanformadoporunexcesodeatrición.Posteriormenteseleaplicaronultrasonidosquedisgregaronestosaglomerados.Enlamedidaseapreciamuybienpuesyanoapareceunadistribuciónbimodalylamediaobtenidacoincideconelvalordetamañodepartículadelamodainferioranterior(0.3μm).
Enelsiguienteapartadoseexplicanconmásdetalleestosprocesosaplicados.
Figura18.Medidasdeladistribucióndeltamañopartículadespuésdecadaproceso.
0
10
20
30
40
50
60
70
80
90
100
0
5
10
15
20
25
30
35
40
45
0,1 1 10
volumenacumuladodepartículas[%
]
volumendepartículas[%
]
tamañodepartícula[μm]
MB(12h) MB(12h)+AT(2h)MB(12h)+AT(4h) MB(12h)+AT(4h)+US

3.Procesado,caracterizaciónyoptimizacióndelospolvosdeoxiapatita
‒16‒
3.3Procesosdetransformaciónaplicadosalospolvos
3.3.1MoliendaconmorteroDespuésdelprocesodesíntesis seobtieneuncompactodelcompuesto, el cual ha de ser premolido paraposteriormenteaplicarle molienda de bolas. Estepreprocesadodelpolvoserealizaconunmorteroyunpilónde ágata. Al ser un proceso completamente manual, losresultados obtenidos suelen ser variables. El principalproblemaesquesinoesrealizadoadecuadamente,aunqueasimple vista el polvo parezcahomogéneo y fino, se puedenhaber quedado partículas de gran tamaño (~50μm), lascuales, con bastante probabilidad, no se reduzcan
adecuadamente en la molienda de bolas,pudiendocausarproblemasdespuéscuandoelpolvoseutiliceenlafabricacióndelapila.Gracias al SEM se puede observar quedespués de este proceso, el tamaño departícula más grande encontrado estáentorno los 10μm, siendo las 3‐5μm ladimensión más común, aunque se hanllegado a visualizar partículas de hasta200nm. En la figura 21 se aprecia unaglomeradocompuestoporunapartículade5μm,otrade3μmyvariasmáspequeñas.
3.3.2MoliendadebolasSe utiliza para reducir el tamaño de polvo (ver punto 2.3). Para todos los polvos se haempleadounboldeágatacon90bolasdeágatadediámetro10mmymediolíquidoetanol.Despuésdelosresultadosobtenidos(medidasdedistribucióndetamañodepartículasymicrografíasSEM)conlasdistintassíntesisylasdiversastandasdemoliendarealizadas,se ha determinadoque el proceso óptimo es unamoliendade12horas,entrestandas,entrelasquese realizará sesiones de ultrasonidos de altaintensidada la suspensión de la molienda paradeshacer los aglomerados que se hayan idoagregando. El diámetro medio de partícula seencuentra entorno a la micra, aunque se puedeencontrar algunas de hasta 3μm. El polvoprocesado con esta técnica es el que se va aemplear para realizar la mezcla cerámica (juntoconelóxidodeníquel)conlaquesefabricaránlostubosdeánodo.
3.3.3AtriciónEsunmétodosimilaralamoliendadebolas.Enlugardeservirsedelasfuerzascentrífugasdelplatorotatorioyelgiropropiodelcontenedordelamoliendaparaimpulsarlasbolas,estas son impulsadas directamente por unos brazos sujetos a un eje rotatorio. Con este
Figura 20.Aglomerado de partículas de LSOdespuésdeunamoliendaconmortero.
Figura21.Partículas de LSO después de 12horasdemoliendaconbolas.
Figura 19.Mortero utilizado enlamoliendadelospolvos.

3.Procesado,caracterizaciónyoptimizacióndelospolvosdeoxiapatita
‒17‒
proceso lasdimensionesde laspartículassereducen mucho máshasta cierto momento,en el que el proceso no puede disminuir eltamaño de partícula y comienza a formaraglomerados.Porlasmedidasrealizadas(verfigura 22) se determinó que esto sueleocurrirentrelasdosylascuatrohoras.Enlamicrografía de la figura 24 se aprecia comomuchaspartículaspequeñassehanagregadohastaformarunaglomeradodeunos12μm.
Todos losprocesosdeatriciónrealizadosenesteproyectohansidoutilizandounmediolíquidodispersanteformadoporetanolmásundefloculante(BeycostatC‐213)en1%delamasadelpolvo.Sehanempleado1410bolasdecircona de diámetro 3mm y un bol de 100ml. La velocidad de la atrición ha sido de500rpm. Tanto el bol como los brazos rotatorios son de polímero para minimizar losefectosdeloschoquesentreestosylasbolas.
3.3.4UltrasonidosdealtaintensidadComo anteriormente se la ha referido, estatécnicaselaempleapararomperlosposiblesaglomeradosque sehayan formadodurantelas distintas moliendas. Consiste enintroducir una sonda que generaultrasonidos en la suspensión en la que seencuentra el polvo. Las vibraciones de altaenergía queproduce la sonda se transmitenpor el medio líquido rompiendo losaglomerados. Siempre se realiza con lasuspensiónsumergidaenunbañofrío,tantoparaevitarlaevaporacióndelmediodelasuspensión(sueleseretanoloacetona),comoevitarlanuevaformacióndeaglomeradosencuantosedejedeaplicarultrasonidos,yaquesonfavorecidosporelincrementodetemperaturaproducidoporlosultrasonidos.
3.4DilatometríadeelectrolitoEnunensayodedilatometríaseaplicaunprocesotérmicoaunmaterialconelfindemedirla deformación que presenta en función de la temperatura. En esta deformación estánincluidostantoelfenómenodecontracciónporlacoalescenciaentrelaspartículascomoeldedilatacióndelmaterial(muypequeñoenestecasoportratarsedecerámicas).
Para preparar una muestra se coge una pequeñacantidaddepolvo(unos0.5gaproximadamente)alaque se le añade un aglutinante (PVA) paraproporcionarleunamayorresistenciaenverde.Coneste se rellena un pequeño tubo fino de látex quedespuésseembridayseestiradentrodeuncuerpolargoyagujereadodelatón.Asíseconsiguequeestetubito flexible rellenodelpolvode electrolito tenga
Figura 22.Aglomerado presente en el polvoformadoporunexcesodeatrición.
Figura23.Aparienciadelpolvodespuésde aplicarultrasonidosalamuestradeaglomerados.
Figura 24.Compacto de LSO después deCIPparamuestradedilatometría.

3.Procesado,caracterizaciónyoptimizacióndelospolvosdeoxiapatita
‒18‒
una forma alargada y cilíndrica en su mayor parte, la cual conservará después delprensado isostático en frío (CIP), que se le aplica a continuación. Una vez obtenido elcompacto(verfigura24),de lasecciónmásregularsecortauncilindrodeunos7mmalqueselerealizaunrefrentadoconunalijasuaveparaobtenerdosbasesparalelas.
Enlafigura25semuestranlosresultadosdelasdilatometríasrealizadasalosdospolvosdeLSOquesehanutilizadoparaelectrolito.ElpolvodeLSO.2hasidoprocesadocon12horasdemoliendadebolas.ElpolvoLSO.3,adicionalmenteaesamismamolienda, selehaaplicadounaatriciónde4horasmásultrasonidos.SeobservacomoelLSO.3,alteneruntamañodepartículainferiorqueelLSO.2,hafavorecidoelprocesodesinterizado,porlo que la contracciónha comenzado a temperaturasmás bajas.Como se puede observarambos polvos comparten el mismo máximo global en la velocidad de contracción a1480°C,aunqueadicionalmenteelLSO.3poseeunmáximolocalen1380°C.
3.5DensimetríaUno de los principales problemas de estos materiales deoxiapatita es su dificultad para su completa densificación.Para estimar los valores de esta simplemente semedirá ladensidaddeunamuestradespuésdeuntratamientotérmicode sinterización y se comparará con las densidadescristalinas teóricas de los compuestos de lasoxiapatitasutilizados (ρLSO=5.457g/cm3, ρLSGO=5.834g/cm3) paraaveriguar su proporción porosa. Si la muestra es bastanteregularsepuedemedirporparámetrosgeométricosysino,hayqueutilizarunkitdedeterminacióndedensidadcomoeldelafigura26.
SemidióunamuestradeLSO.2(12horasmoliendadebolas,2horasa1600°C)cuyadensidadalcanzóel90%delateóricay otra de LSGO (12 horas demolienda de bolas + 3 horas de atrición + ultrasonidos, 2horasa1440°C)lacualdensificócasiporcompleto(porosidadnula).
Figura25.Dilatometríasdedospolvosdeoxiapatitaprocesadosparareducirsutamañodepartícula:AlLSO.2selehaaplicado12hdemoliendadebolasyalLSO.3,además4hdeatrición.
‐0.07
‐0.06
‐0.05
‐0.04
‐0.03
‐0.02
‐0.01
0
0.01
0.02
‐16
‐14
‐12
‐10
‐8
‐6
‐4
‐2
0
0 200 400 600 800 1000 1200 1400 1600
velocidaddeε(%
)[°C−
1]
ε(%
)
T[°C]
LSO.2‐εLSO.3‐εLSO.2‐Δε/ΔTLSO.3‐Δε/ΔT
Figura26.Kitdedensidad.
3.Procesado,caracterizaciónyoptimizacióndelospolvosdeoxiapatita
‒19‒
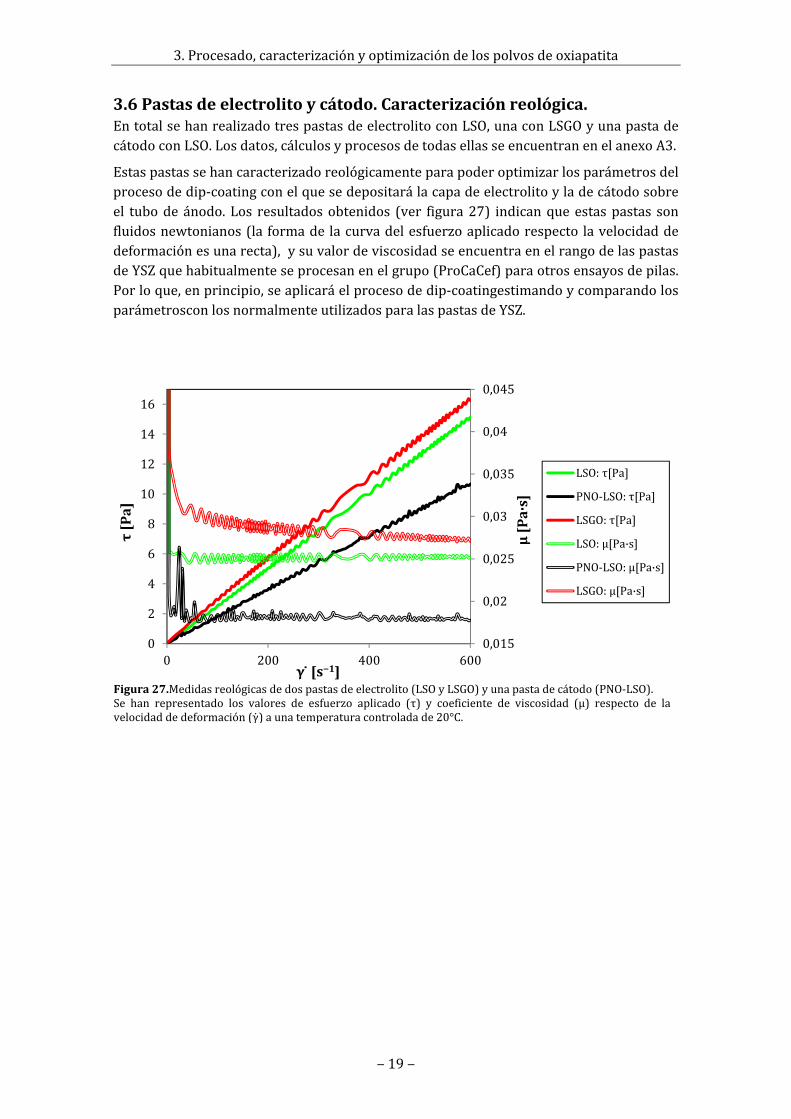
3.6Pastasdeelectrolitoycátodo.Caracterizaciónreológica.EntotalsehanrealizadotrespastasdeelectrolitoconLSO,unaconLSGOyunapastadecátodoconLSO.Losdatos,cálculosyprocesosdetodasellasseencuentranenelanexoA3.
Estaspastassehancaracterizadoreológicamenteparapoderoptimizarlosparámetrosdelprocesodedip‐coatingconelquesedepositarálacapadeelectrolitoyladecátodosobreel tubodeánodo.Los resultadosobtenidos (ver figura27) indicanqueestaspastas sonfluidosnewtonianos(la formade lacurvadelesfuerzoaplicadorespecto lavelocidaddedeformaciónesunarecta),ysuvalordeviscosidadseencuentraenelrangodelaspastasdeYSZquehabitualmenteseprocesanenelgrupo(ProCaCef)paraotrosensayosdepilas.Porloque,enprincipio,seaplicaráelprocesodedip‐coatingestimandoycomparandolosparámetrosconlosnormalmenteutilizadosparalaspastasdeYSZ.
Figura27.Medidasreológicasdedospastasdeelectrolito(LSOyLSGO)yunapastadecátodo(PNO‐LSO).Se han representado los valores de esfuerzo aplicado (τ) y coeficiente de viscosidad (μ) respecto de lavelocidaddedeformación(γ)aunatemperaturacontroladade20°C.
0,015
0,02
0,025
0,03
0,035
0,04
0,045
0
2
4
6
8
10
12
14
16
0 200 400 600
μ[Pa·s]
τ[Pa]
γ ̇ [s−1]
LSO:τ[Pa]
PNO‐LSO:τ[Pa]
LSGO:τ[Pa]
LSO:μ[Pa·s]
PNO‐LSO:μ[Pa·s]
LSGO:μ[Pa·s]

4.Fabricacióndepilas
‒20‒
4.Fabricacióndepilas
4.1Ánodo.Fabricaciónycaracterización.
4.1.1MezclaparaánodoUna relación 1:1 en volumen cermet‐poro (después de la reducción de NiO a Ni) es ladeseadaaobtener en los tubosdeánodoque se fabriquenenesteproyecto.Además sedeseaqueelcermet(Ni‐LSO)tengaelmismovolumendemetalquedecerámica.ApartirdeesarelaciónsecalculalaproporciónmásicadeNiO,LSOyformadordeporoquedebehaberenlamezclayseprocedearealizarla.Unavezpreparada,seleañadeunaglutinante(PVA) para que aumente la resistencia en verde del tubo compactado. Los cálculos, lospasos realizados en el proceso y las características de todas las mezclas realizadas seencuentrandetalladaselanexoA3.2.
4.1.2PreformatubularElmolde cilíndrico, que va a ser rellenado con lamezcla, se obtiene combinando un tubo de gomaflexible (necesario si luego se quiere aplicar CIP)con un macho cilíndrico en el interior(varilla deacero nitrurado de 3mm de diámetro). Se ha deprocederconbastantecuidadoparaqueelllenadotenga un buen grado de homogeneidad yuniformidadporque la intrusión de cualquierdefectoenestapartedelprocesopuedellegaraserarrastradohastaelconformadofinaldelapila.Unavez lleno el molde, el extremo abierto del tuboflexible es embridado para lograr una buenaestanqueidad que evitará la posible introduccióndeaceitemientrasserealizaelCIP.
4.1.3Prensadoisostáticoenfrío(CIP)TambiénconocidoporcoldisostatingpressingoCIP,esunmétododecompactadoen laqueseejerceunapresiónidénticasobretodalasuperficieexteriordelcompacto,locualloconvierte en un método ideal para fabricar un tubo por su simetría cilíndrica. Lasdensidadesobtenidasconestemétodosonsuperioresquelasobtenidasconelprensadouniaxial,ademásdequelosgradientesdedensidadessonmenores,cosaqueimplicaunamenor probabilidad de aparición de grietas en el proceso.La preforma es sometida a200MPadurante5minutos.Seguidamentesedesmoldacondelicadezapueslaresistenciamecánicaquetieneenverdeesmuypequeñayselesometeaunprocesodeobservaciónparadescubrirsihayalgunagrietaodefectoquepuedacomprometerposteriormente laintegridadestructuraldeltubo.
Figura28.Elementosempleadosenelmoldedeltubodeánodo.

4.Fabricacióndepilas
‒21‒
4.1.4PresinterizadoEsun tratamiento térmicoenelquesimplementese llevaacabo laprimeraetapade lasinterización,enlaquesecreanunionesenformadecuelloentrelaspartículasqueestánen contacto. Se le suele someter a 950°C durante 4 horas. Este tratamiento permiteobtener un tubo de ánodo más resistenteque en verde, pero sin estar sinterizado(este debe hacerse junto con el electrolitoparaqueamboscontraiganjuntosyselogreunabuena integración), lo que asegura unamejor manipulación del tubo, algocompletamentenecesarioparacuandoselevayaaaplicarelrecubrimientodepastadeelectrolito. Además, durante estepresinterizado todos los compuestosorgánicospresenteseneltuboprocedentesdel aglutinante y el formador de poro soneliminados.
En la figura 29 se observa una grieta en el extremo de uno de los dos tubos. En eldesmoldadoyasehabíaobservadoqueaparecíaunadeuntamañoaproximadodeunos2mmpero ha crecido durante el presinterizado alcanzando algomás del centímetro delongitud. En etapas posteriores tal defecto se puede ir agravando por los tratamientostérmicosalosqueserásometido.
4.1.5DilatometríadeánodoSe realiza para caracterizarel comportamiento de contracción quesufrirá el ánodo durante el sinterizado.La muestra medida es unasección cortada del tubo de unos 6‐7mm que posteriormente hasidorefrentada hasta lograr un buen paralelismoentre las bases(indispensableparalograrunamedidafiable).Comosepuedeapreciarenlafigura30,semontaentredosláminasdeplatinoydospastillasdealúminaparaevitar lacontaminaciónde loselementoscerámicosdeldilatómetro. Los resultados obtenidos se muestran en el punto 4.3dondesecomparancon lasdilatometríasdeelectrolitoparaestudiarelprocesodesinterizacióncomún.
4.2DeposicióndelacapadeelectrolitoPrimeroserealizaladeposicióndelelectrolitosobreelánodomediantede este la técnica de dip‐coating. Después se procederá al sinterizado.Unavezsinterizado,sepodráobservar,tantoconelmicroscopioópticocomoconelelectrónico,sielespesordecapadeelectrolitoobtenidoeselbuscado,alrededorde10‐15μm.
4.2.1Dip‐coatingOrecubrimientoporinmersión,esunatécnicaenlaquebásicamente,eltubodeánodo,previamentealquesehacubiertosusextremosconcinta
Figura29.Tubosdeánododespuésdepresinterizarydetalledegrietaenelextremodeunodelostubos.
Figura30.Muestradedeánodocolocadaendilatómetro.
Figura 31.Muestrareciénimpregnada.

4.Fabricacióndepilas
‒22‒
de teflón para que no pueda introducirse nada en suinterior,es sumergidoverticalmenteenunaprobetaenla que se encuentra la pasta de electrolito. Acontinuación es ascendido a una velocidad controlada.Unavezqueeletanoldelapastaimpregnadaalapareddel ánodo se ha evaporado, se puede volver a efectuar
otrainmersión.Esimportanteagitarvigorosamentelapastaantesdecadainmersiónparahomogeneizarla, ya que estando en reposo, parte de las partículas de la suspensión sepuededepositar en el fondo. Para alcanzar el espesor de capa requerido se efectuarántantasrepeticionescomosecreanecesario.Tanto lavelocidaddeextracción,elacabadosuperficialdelánodoyelcomportamientoviscosodelapastainfluyenenelespesordelacapaadherida,por loquetanto lavelocidaddeascensocomoelnúmerode inmersionesaplicadasseestimaconlosresultadosobtenidosdelapruebadereologíaqueserealizóalapastaqueseestáutilizando.
4.2.2ResultadosyoptimizacióndeparámetrosEnlasprimeraspruebasdedeposiciónseprocedióa realizar tres inmersiones a una velocidad de3mm/s.Losresultadosobtenidosmostrabanquesehabía obtenido un espesor de capa de 80μm (verfigura33)traselsinterizado,unvalormuydistintoalesperado.
Enconsecuenciaseactúaendosparámetros.Porunladosereduceelnúmerodeinmersionesasólouna.Por otro lado se hace una pasta nueva de la queparte se deja con las condiciones de la anterior(13%envolumendesólido)yotrapartesediluye(10%envolumendesólido).Conestadiferencia de concentración se consigue una viscosidad distinta que proporcionará unaadherenciadiferente.Serealizanvariaspruebasdedip‐coatingenvariasmuestrasdetubodeánodoyseapreciaqueconlapastadiluidanosealcanzaelespesordeseadomientrasqueconlanodiluidasí(verfigura34).
Figura 34.Imágenes de microscopía óptica en donde se aprecia la diferencia de espesor de capa deelectrolitosegúnlaconcentracióndepastautilizada.Izquierda:pastaconcentradaal10%desólido.Derecha:pasta concentradaal 13% desólido.
Figura33.Micrografía SEMde una capa deelectrolitodeespesordeunos80μm.
Figura 32.Muestra después derealizardip‐coating(teflónretirado).
4.Fabricacióndepilas
‒23‒
4.3Cosinterizado(ánodo+electrolito)Coneste sinterizado común sebusca logrardosobjetivos: la completa densificacióndelelectrolitoysucorrectaintegraciónconelánodo.
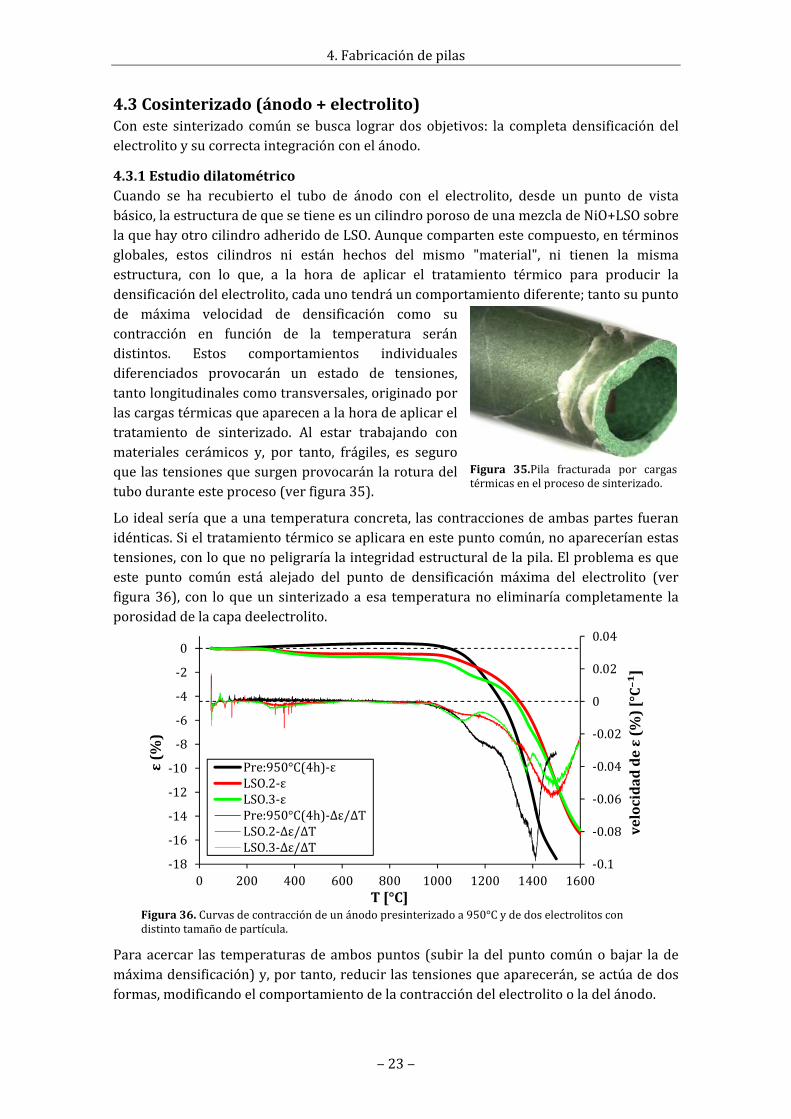
4.3.1EstudiodilatométricoCuando se ha recubierto el tubo de ánodo con el electrolito, desde un punto de vistabásico,laestructuradequesetieneesuncilindroporosodeunamezcladeNiO+LSOsobrelaquehayotrocilindroadheridodeLSO.Aunquecompartenestecompuesto,entérminosglobales, estos cilindros ni están hechos del mismo "material", ni tienen la mismaestructura, con lo que, a la hora de aplicar el tratamiento térmico para producir ladensificacióndelelectrolito,cadaunotendráuncomportamientodiferente;tantosupuntode máxima velocidad de densificación como sucontracción en función de la temperatura serándistintos. Estos comportamientos individualesdiferenciados provocarán un estado de tensiones,tantolongitudinalescomotransversales,originadoporlascargastérmicasqueaparecenalahoradeaplicareltratamiento de sinterizado. Al estar trabajando conmateriales cerámicos y, por tanto, frágiles, es seguroquelastensionesquesurgenprovocaránlaroturadeltuboduranteesteproceso(verfigura35).
Loidealseríaqueaunatemperaturaconcreta, lascontraccionesdeambaspartesfueranidénticas.Sieltratamientotérmicoseaplicaraenestepuntocomún,noapareceríanestastensiones,conloquenopeligraríalaintegridadestructuraldelapila.Elproblemaesqueeste punto común está alejado del punto de densificación máxima del electrolito (verfigura36), con loqueunsinterizadoaesa temperaturanoeliminaría completamente laporosidaddelacapadeelectrolito.
Paraacercar las temperaturasdeambospuntos (subir ladelpuntocomúnobajar lademáximadensificación)y,portanto,reducirlastensionesqueaparecerán,seactúadedosformas,modificandoelcomportamientodelacontraccióndelelectrolitooladelánodo.
Figura36.Curvasdecontraccióndeunánodopresinterizadoa950°Cydedoselectrolitoscondistintotamañodepartícula.
‐0.1
‐0.08
‐0.06
‐0.04
‐0.02
0
0.02
0.04
‐18
‐16
‐14
‐12
‐10
‐8
‐6
‐4
‐2
0
0 200 400 600 800 1000 1200 1400 1600
velocidaddeε(%
)[°C−¹]
ε(%
)
T[°C]
Pre:950°C(4h)‐εLSO.2‐εLSO.3‐εPre:950°C(4h)‐Δε/ΔTLSO.2‐Δε/ΔTLSO.3‐Δε/ΔT
Figura 35.Pila fracturada por cargastérmicasenelprocesodesinterizado.
4.Fabricacióndepilas
‒24‒
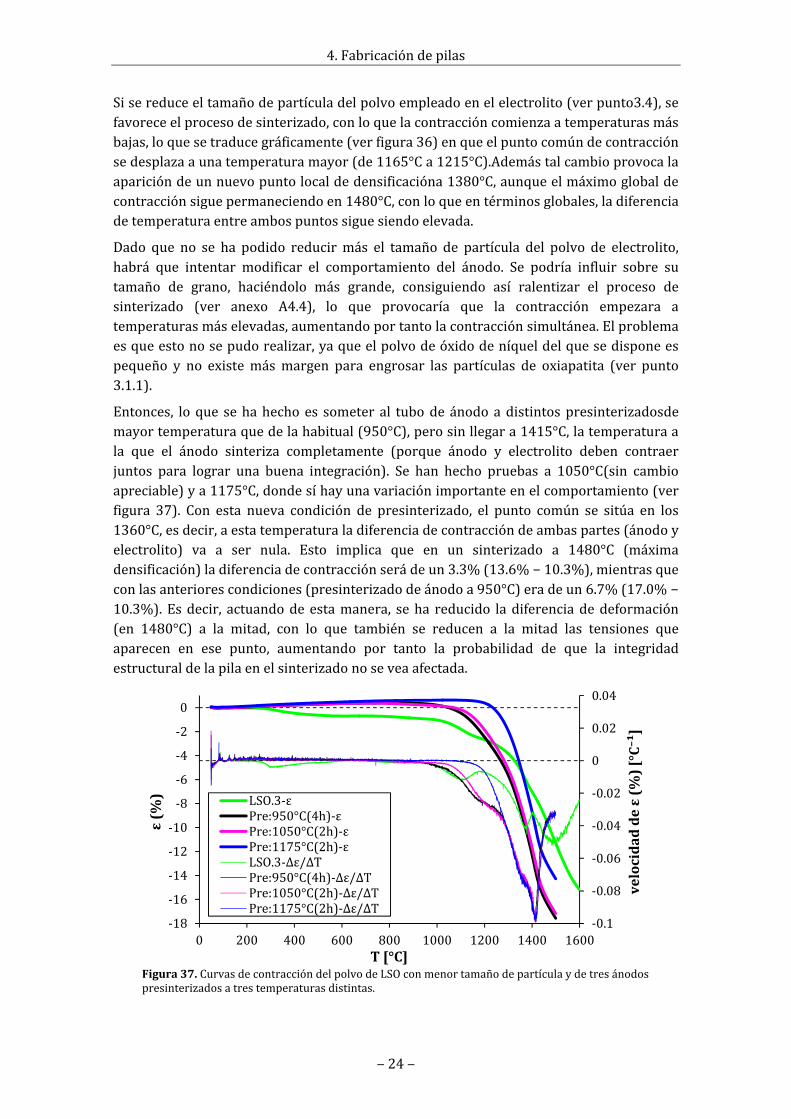
Sisereduceeltamañodepartículadelpolvoempleadoenelelectrolito(verpunto3.4),sefavoreceelprocesodesinterizado,conloquelacontraccióncomienzaatemperaturasmásbajas,loquesetraducegráficamente(verfigura36)enqueelpuntocomúndecontracciónsedesplazaaunatemperaturamayor(de1165°Ca1215°C).Ademástalcambioprovocalaaparicióndeunnuevopuntolocaldedensificacióna1380°C,aunqueelmáximoglobaldecontracciónsiguepermaneciendoen1480°C,conloqueentérminosglobales,ladiferenciadetemperaturaentreambospuntossiguesiendoelevada.
Dado que no se ha podido reducirmás el tamaño de partícula del polvo de electrolito,habrá que intentar modificar el comportamiento del ánodo. Se podría influir sobre sutamaño de grano, haciéndolo más grande, consiguiendo así ralentizar el proceso desinterizado (ver anexo A4.4), lo que provocaría que la contracción empezara atemperaturasmáselevadas,aumentandoportantolacontracciónsimultánea.Elproblemaesqueestonosepudorealizar,yaqueelpolvodeóxidodeníqueldelquesedisponeespequeño y no existemásmargen para engrosar las partículas de oxiapatita (ver punto3.1.1).
Entonces, loque sehahechoes someter al tubodeánodoadistintospresinterizadosdemayortemperaturaquedelahabitual(950°C),perosinllegara1415°C,latemperaturaala que el ánodo sinteriza completamente (porque ánodo y electrolito deben contraerjuntos para lograr una buena integración). Se han hecho pruebas a 1050°C(sin cambioapreciable)ya1175°C,dondesíhayunavariaciónimportanteenelcomportamiento(verfigura 37). Con esta nueva condición de presinterizado, el punto común se sitúa en los1360°C,esdecir,aestatemperaturaladiferenciadecontraccióndeambaspartes(ánodoyelectrolito) va a ser nula. Esto implica que en un sinterizado a 1480°C (máximadensificación)ladiferenciadecontracciónserádeun3.3%(13.6%−10.3%),mientrasqueconlasanteriorescondiciones(presinterizadodeánodoa950°C)eradeun6.7%(17.0%−10.3%).Esdecir, actuandodeestamanera, seha reducido ladiferenciadedeformación(en 1480°C) a la mitad, con lo que también se reducen a la mitad las tensiones queaparecen en ese punto, aumentando por tanto la probabilidad de que la integridadestructuraldelapilaenelsinterizadonoseveaafectada.
Figura37.CurvasdecontraccióndelpolvodeLSOconmenortamañodepartículaydetresánodospresinterizadosatrestemperaturasdistintas.
‐0.1
‐0.08
‐0.06
‐0.04
‐0.02
0
0.02
0.04
‐18
‐16
‐14
‐12
‐10
‐8
‐6
‐4
‐2
0
0 200 400 600 800 1000 1200 1400 1600
velocidaddeε(%
)[°C−¹]
ε(%
)
T[°C]
LSO.3‐εPre:950°C(4h)‐εPre:1050°C(2h)‐εPre:1175°C(2h)‐εLSO.3‐Δε/ΔTPre:950°C(4h)‐Δε/ΔTPre:1050°C(2h)‐Δε/ΔTPre:1175°C(2h)‐Δε/ΔT
4.Fabricacióndepilas
‒25‒
Una vez que se han optimizado los parámetros del proceso con el compuesto LSO, seaplicanalotrocompuestoLSGO,cuyotamañodepartículafuereducidohastaeladecuadocon12horasdemoliendadebolasy3horasdeatrición.
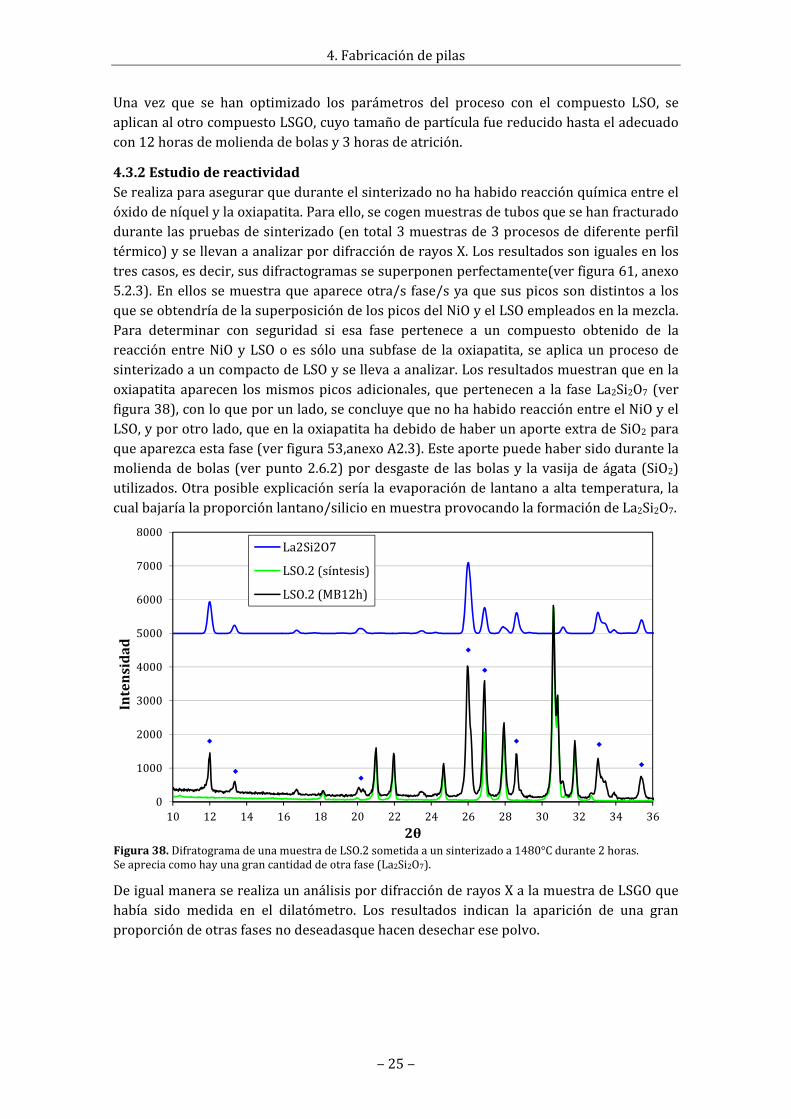
4.3.2EstudiodereactividadSerealizaparaasegurarqueduranteelsinterizadonohahabidoreacciónquímicaentreelóxidodeníquelylaoxiapatita.Paraello,secogenmuestrasdetubosquesehanfracturadodurantelaspruebasdesinterizado(entotal3muestrasde3procesosdediferenteperfiltérmico)ysellevanaanalizarpordifracciónderayosX.Losresultadossonigualesenlostrescasos,esdecir,susdifractogramassesuperponenperfectamente(verfigura61,anexo5.2.3).Enellossemuestraqueapareceotra/sfase/syaquesuspicossondistintosa losqueseobtendríadelasuperposicióndelospicosdelNiOyelLSOempleadosenlamezcla.Para determinar con seguridad si esa fase pertenece a un compuesto obtenido de lareacciónentreNiOyLSOoes sólounasubfasede laoxiapatita, seaplicaunprocesodesinterizadoauncompactodeLSOysellevaaanalizar.Losresultadosmuestranqueenlaoxiapatitaaparecen losmismospicosadicionales,quepertenecena la faseLa2Si2O7 (verfigura38),conloqueporunlado,seconcluyequenohahabidoreacciónentreelNiOyelLSO,yporotrolado,queenlaoxiapatitahadebidodehaberunaporteextradeSiO2paraqueaparezcaestafase(verfigura53,anexoA2.3).Esteaportepuedehabersidodurantelamoliendadebolas (verpunto2.6.2)pordesgastede lasbolasy lavasijadeágata (SiO2)utilizados.Otraposibleexplicaciónseríalaevaporacióndelantanoaaltatemperatura, lacualbajaríalaproporciónlantano/silicioenmuestraprovocandolaformacióndeLa2Si2O7.
DeigualmaneraserealizaunanálisispordifracciónderayosXalamuestradeLSGOquehabía sido medida en el dilatómetro. Los resultados indican la aparición de una granproporcióndeotrasfasesnodeseadasquehacendesecharesepolvo.
Figura38.DifratogramadeunamuestradeLSO.2sometidaaunsinterizadoa1480°Cdurante2horas.Seapreciacomohayunagrancantidaddeotrafase(La2Si2O7).
0
1000
2000
3000
4000
5000
6000
7000
8000
10 12 14 16 18 20 22 24 26 28 30 32 34 36
Intensidad
2θ
La2Si2O7
LSO.2(síntesis)
LSO.2(MB12h)

4.Fabricacióndepilas
‒26‒
4.4IntegracióndelcátodoUna vez que se consiguen fabricar muestras sinterizadas de ánodo y electrolito cuyaintegridad estructural no ha resultado dañada, se procede a añadirles el cátodo. Elprocedimiento es exactamente igual que con el electrolito. Primero se realiza unadeposición de pasta de cátodo mediante la técnica de dip‐coating para posteriormenterealizarunsinterizado.Unavezobtenidalapilacompleta,laintegraciónentresuscapasesobservadaalmicroscopioelectrónico.
4.4.1Dip‐coatingEs exactamente el mismo procedimiento que el realizado con el electrolito (ver punto4.2.1) con la salvedad de que en el anterior caso el tubo de ánodo era cubiertoenteramente por una capa de electrolito (excepto los extremos que estaban cubiertos),mientras que ahora simplemente se va a depositar 1 cm2 de área, para normalizar laposteriorcaracterizaciónelectroquímicadelapila.Paraellosecubriráconcintadeteflóntodoeltubomenosunaseccióncentralde9.1mm(⇐radiodelapila=3.5mm).
La pasta de cátodo utilizada, mezcla de Pr2NiO4 y LSO (abreviadamente PNO‐LSO)previamente ha sido caracterizadareológicamente (ver punto 3.6). De los resultadosobtenidossedeterminaqueserealizarán4inmersionesa4.5mm/s.
4.4.2SinterizadoEste sinterizado se realiza a baja temperatura, similar al presinterizado realizado a losánodos.Eltratamientotérmicoaplicadoesde2horasa1100°Cconunasvelocidadesdesubidaybajadade2°C/min.Enestesinterizadosólosebuscalauniónentrepartículas,yno un densificado, ya que el cátodo debe de ser una estructura porosa.Deanteriorestrabajosdelgrupo[7] (ProCaCef)sesabequeenesteprocesonovaaexistir reactividadquímicaentreelPNOyelLSO.Enlasfiguras39y40seapreciaelresultadoconseguido.
Figura39.Aspectodeunapiladespuésdelaintegracióndelcátodo.
Figura40.MicrografíasSEMdondeseaprecialacorrectaintegraciónentrelascapasdelapilafabricada.Enunapilaacabadalosespesoresdecapason:ánodo400μm,electrolito10~15μm,cátodo30μm.

5.CaracterizaciónelectroquímicadelaSOFC
‒27‒
5.CaracterizaciónelectroquímicadelaSOFCComo se ha observado al SEM que se ha logrado una correcta integración entra lasdistintascapas,seprocedearealizarunacaracterizaciónelectroquímicaparaobservarsidurante su funcionamiento lapila respondeelectroquímicamente comodeberíaoporelcontrario aparecen determinados fenómenos que indiquen que la pila realmente tienedefectosinternos.
5.1Preparacióndelaspilas
5.1.1MuestrasaprepararLaspilasfabricadasquesevanamedirsondos,unacuyacosinterizadoseharealizadoa1480°Cdurante2horasyotraqueentotal(seharealizadoen3tandasdesinterizado)sehacosinterizadodurante12horasa1380°C.
5.1.2MontajeentubosLa pila se va a medir dentro de un horno tubular en el cual se alcanzará una elevadatemperatura.Parasuministrarun flujo internodehidrógenoyevacuarelvapordeaguagenerado en el proceso electroquímico la pila hace faltadisponer de un conducto queaguantetalestemperaturas,porloquelapilasemontaentredostubosdealúmina(aloscualesestosflujosdegasesllegaránoseevacuaránmediantetubosdegomaqueestaránconectados en el extremo contrario donde la temperatura de tubo de alúmina será laambiente).Senecesitaunselladoqueaguantetambiéntales condiciones. Para ello se utiliza una pastacerámicaqueseaplicaendosveces(cubrirelhuecoyluego un repaso superficial) con posteriores secadosbajouna lámparade infrarrojos.Porúltimo,paraquelapastasolidifique,esnecesariosometeralconjuntoaun tratamiento térmico a 371°C. Durante estetratamientounade laspilas(sinterizadaa1480°C)seharotoporsegundavez(verfigura41),conloquenosevanaobtenermedidasdeésta.
5.1.3MontajedecontactoseléctricosUnavezlauniónconlostubosdealúminaestáselladaysolidificadaseprocedeamontarloscontactoseléctricos(hilosdeplatinoparaaguantarlasaltastemperaturassinoxidarseconlaatmósferaexterior).Porelexteriorunhilovaaserenrolladosobreelcátodo(verfigura42),aplicandodespuésunapinturadeplatinoquemejoraráeléctricamentelaunióny por el interior, se colocará un enrollamiento que presione ligeramente la pared delánodoalamismaalturadondeexternamenteseencuentraelcátodo.
Figura42.Contactoseléctricosmontadosenlapila
Figura41.Pilarotaduranteelmontaje.Teníaunamicrogrietainternaquesehaidoextendiendohastasurotura.
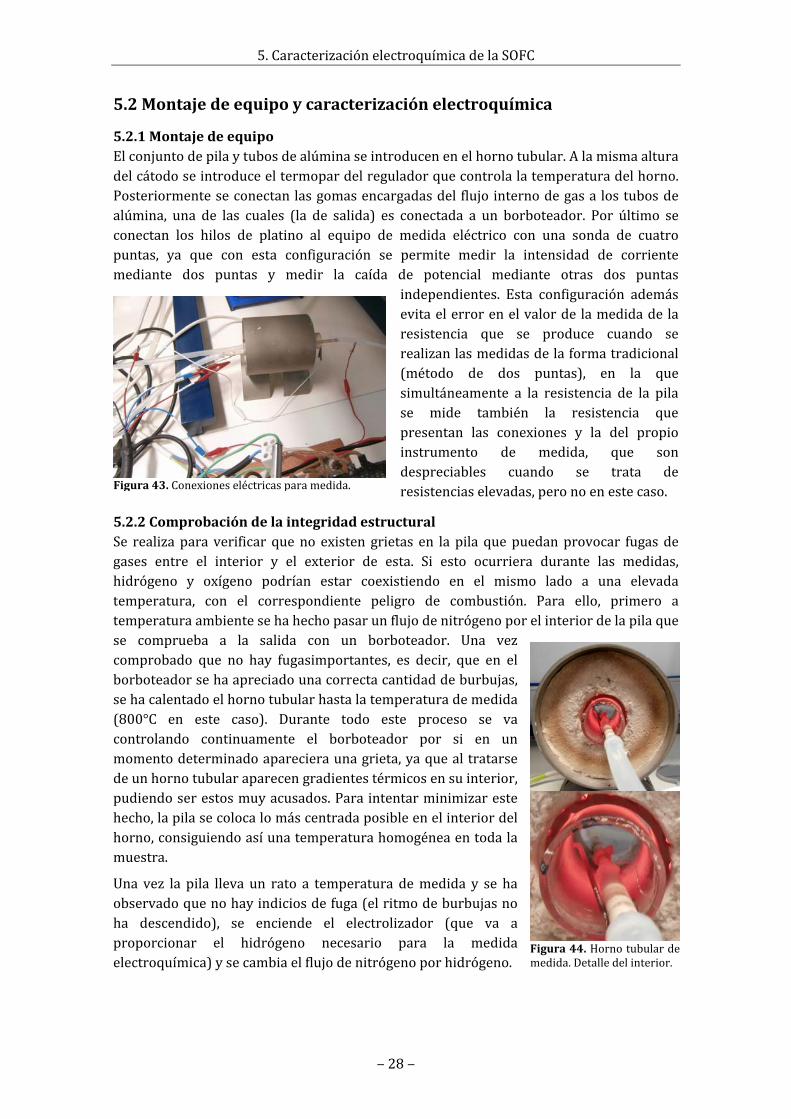
5.CaracterizaciónelectroquímicadelaSOFC
‒28‒
5.2Montajedeequipoycaracterizaciónelectroquímica
5.2.1MontajedeequipoElconjuntodepilaytubosdealúminaseintroducenenelhornotubular.Alamismaalturadelcátodoseintroduceeltermopardelreguladorquecontrolalatemperaturadelhorno.Posteriormenteseconectanlasgomasencargadasdel flujo internodegasa lostubosdealúmina, una de las cuales (la de salida) es conectada a un borboteador. Por último seconectan los hilos de platino al equipo de medida eléctrico con una sonda de cuatropuntas, ya que con esta configuración se permite medir la intensidad de corrientemediante dos puntas y medir la caída de potencial mediante otras dos puntas
independientes. Esta configuración ademásevitaelerrorenelvalordelamedidadelaresistencia que se produce cuando serealizanlasmedidasdelaformatradicional(método de dos puntas), en la quesimultáneamente a la resistencia de la pilase mide también la resistencia quepresentan las conexiones y la del propioinstrumento de medida, que sondespreciables cuando se trata deresistenciaselevadas,peronoenestecaso.
5.2.2ComprobacióndelaintegridadestructuralSe realizapara verificarqueno existen grietas en lapila quepuedanprovocar fugasdegases entre el interior y el exterior de esta. Si esto ocurriera durante las medidas,hidrógeno y oxígeno podrían estar coexistiendo en el mismo lado a una elevadatemperatura, con el correspondiente peligro de combustión. Para ello, primero atemperaturaambientesehahechopasarunflujodenitrógenoporelinteriordelapilaquese comprueba a la salida con un borboteador. Una vezcomprobado que no hay fugasimportantes, es decir, que en elborboteadorsehaapreciadounacorrectacantidaddeburbujas,sehacalentadoelhornotubularhastalatemperaturademedida(800°C en este caso). Durante todo este proceso se vacontrolando continuamente el borboteador por si en unmomentodeterminadoaparecieraunagrieta,yaquealtratarsedeunhornotubularaparecengradientestérmicosensuinterior,pudiendoserestosmuyacusados.Paraintentarminimizarestehecho,lapilasecolocalomáscentradaposibleenelinteriordelhorno,consiguiendoasíunatemperaturahomogéneaentodalamuestra.
Una vez la pila lleva un rato a temperatura demedida y se haobservadoquenohayindiciosdefuga(elritmodeburbujasnoha descendido), se enciende el electrolizador (que va aproporcionar el hidrógeno necesario para la medidaelectroquímica)ysecambiaelflujodenitrógenoporhidrógeno.
Figura44.Hornotubulardemedida.Detalledelinterior.
Figura43.Conexioneseléctricasparamedida.
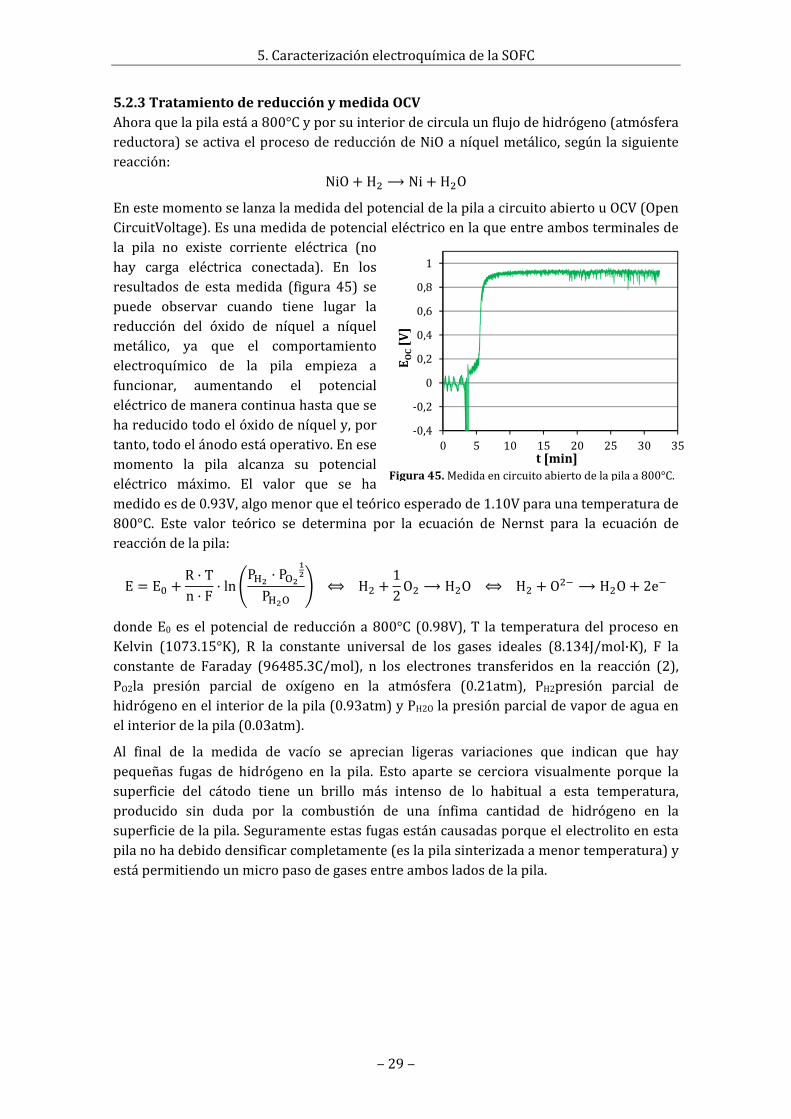
5.CaracterizaciónelectroquímicadelaSOFC
‒29‒
5.2.3TratamientodereducciónymedidaOCVAhoraquelapilaestáa800°Cyporsuinteriordecirculaunflujodehidrógeno(atmósferareductora)seactivaelprocesodereduccióndeNiOaníquelmetálico,segúnlasiguientereacción:
NiO H ⟶ Ni H O
EnestemomentoselanzalamedidadelpotencialdelapilaacircuitoabiertouOCV(OpenCircuitVoltage).Esunamedidadepotencialeléctricoenlaqueentreambosterminalesdela pila no existe corriente eléctrica (nohay carga eléctrica conectada). En losresultados de estamedida (figura 45) sepuede observar cuando tiene lugar lareducción del óxido de níquel a níquelmetálico, ya que el comportamientoelectroquímico de la pila empieza afuncionar, aumentando el potencialeléctricodemaneracontinuahastaquesehareducidotodoelóxidodeníquely,portanto,todoelánodoestáoperativo.Enesemomento la pila alcanza su potencialeléctrico máximo. El valor que se hamedidoesde0.93V,algomenorqueelteóricoesperadode1.10Vparaunatemperaturade800°C. Este valor teórico se determina por la ecuación de Nernst para la ecuación dereaccióndelapila:
E ER Tn F
lnP P
P⟺ H
12O ⟶ H O ⟺H O ⟶ H O 2e
dondeE0es elpotencialde reduccióna800°C (0.98V),T la temperaturadelprocesoenKelvin (1073.15°K), R la constante universal de los gases ideales (8.134J/mol·K), F laconstante de Faraday (96485.3C/mol), n los electrones transferidos en la reacción (2),PO2la presión parcial de oxígeno en la atmósfera (0.21atm), PH2presión parcial dehidrógenoenelinteriordelapila(0.93atm)yPH2Olapresiónparcialdevapordeaguaenelinteriordelapila(0.03atm).
Al final de la medida de vacío se aprecian ligeras variaciones que indican que haypequeñas fugas de hidrógeno en la pila. Esto aparte se cerciora visualmente porque lasuperficie del cátodo tiene un brillo más intenso de lo habitual a esta temperatura,producido sin duda por la combustión de una ínfima cantidad de hidrógeno en lasuperficiedelapila.Seguramenteestasfugasestáncausadasporqueelelectrolitoenestapilanohadebidodensificarcompletamente(eslapilasinterizadaamenortemperatura)yestápermitiendounmicropasodegasesentreambosladosdelapila.
Figura45.Medidaencircuitoabiertodelapilaa800°C.
‐0,4
‐0,2
0
0,2
0,4
0,6
0,8
1
0 5 10 15 20 25 30 35
E OC[V]
t[min]
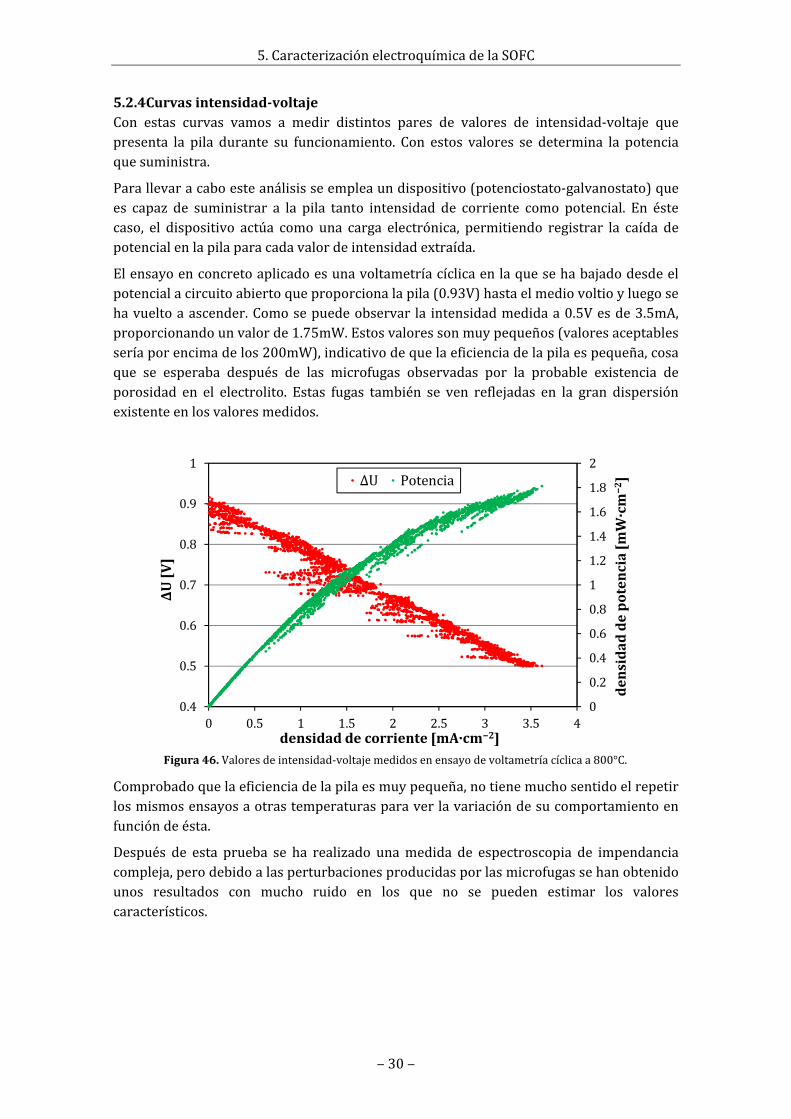
5.CaracterizaciónelectroquímicadelaSOFC
‒30‒
5.2.4Curvasintensidad‐voltajeCon estas curvas vamos a medir distintos pares de valores de intensidad‐voltaje quepresenta la pila durante su funcionamiento. Con estos valores se determina la potenciaquesuministra.
Parallevaracaboesteanálisisseempleaundispositivo(potenciostato‐galvanostato)quees capaz de suministrar a la pila tanto intensidad de corriente como potencial. En éstecaso, el dispositivo actúa como una carga electrónica, permitiendo registrar la caída depotencialenlapilaparacadavalordeintensidadextraída.
Elensayoenconcretoaplicadoesunavoltametríacíclicaenlaquesehabajadodesdeelpotencialacircuitoabiertoqueproporcionalapila(0.93V)hastaelmediovoltioyluegosehavueltoaascender.Comosepuedeobservar la intensidadmedidaa0.5Vesde3.5mA,proporcionandounvalorde1.75mW.Estosvaloressonmuypequeños(valoresaceptablesseríaporencimadelos200mW),indicativodequelaeficienciadelapilaespequeña,cosaque se esperaba después de las microfugas observadas por la probable existencia deporosidad en el electrolito. Estas fugas también se ven reflejadas en la gran dispersiónexistenteenlosvaloresmedidos.
Comprobadoquelaeficienciadelapilaesmuypequeña,notienemuchosentidoelrepetirlosmismosensayosaotrastemperaturasparaverlavariacióndesucomportamientoenfuncióndeésta.
Después de esta prueba se ha realizado unamedida de espectroscopia de impendanciacompleja,perodebidoalasperturbacionesproducidasporlasmicrofugassehanobtenidounos resultados con mucho ruido en los que no se pueden estimar los valorescaracterísticos.
Figura46.Valoresdeintensidad‐voltajemedidosenensayodevoltametríacíclicaa800°C.
0
0.2
0.4
0.6
0.8
1
1.2
1.4
1.6
1.8
2
0.4
0.5
0.6
0.7
0.8
0.9
1
0 0.5 1 1.5 2 2.5 3 3.5 4
densidaddepotencia[mW·cm
−2]
ΔU[V]
densidaddecorriente[mA·cm−2]
ΔU Potencia

6.Conclusiones
‒31‒
6.Conclusiones
6.1VisiónglobalElprincipalobjetivodeesteproyectoeralograrlafabricacióndeunapiladecombustibledeóxido sólido (SOFC) de tipo microtubular soportada sobre ánodo con dos nuevosmaterialesdetipooxiapatita,LSO yLSGO,sustituyendoelmaterialdeelectrolitoqueseutilizaactualmente,laYSZ,loquepermitebajarelpuntodeoperacióndelsistemadelos1000°C al rango de los 700 ‐ 800°C debido a losmejores valores de conducción de losionesdeoxígenoatemperaturasintermediasquepresentaestetipodemateriales.
Esteobjetivosehacumplidoyaquesehalogradoobtenerpilasconunabuenaintegraciónentre lasdistintas capasque la componen (ánodo, electrolitoy cátodo)y ademásunadeellassehapodidocaracterizarelectroquímicamente.Paralograrestehecho,sehantenidoque ir determinando los parámetros necesarios y adecuadospara la fabricaciónde estapilamediantenumerosaspruebas,medidasyobservaciones.Hayciertosparámetrosquepuedensermejoradosydeberíanserestudiadosconmásdetalle,peroesocorrespondeaunfuturoprocesodemejora.
Como cuestión relevante, mencionar que parte de los resultados conseguidos en esteproyectosehanutilizadoenunproyectodeinvestigaciónamayorescalaypresentadosenuncongresosobreestosnuevosmaterialesdeoxiapatitaysuutilizaciónenSOFC:
Proyecto:NUEVOSPROCEDIMIENTOSDEPROCESADODEMATERIALESPARALAFABRICACIONDEPILASDECOMBUSTIBLEYELECTROLIZADORESDEOXIDOSOLIDO(MAT2012‐30763)Congreso:AUTHORS:A.Orera,J.Silva,M.A.Laguna‐Bercero,S.Poblador,R.I.MerinoTITLE: Symmetrical cells of Pr2NiO4+d on electrolytes with the apatite‐type structure.FabricationofMicrotubularApatite‐basedSOFCTYPEOFPRESENTATION:PosterCONGRESS:H2FCSUPERGENMEETINGPLACE:Birmingham(UK)YEAR:2013(December16‐18)Para estructurar la memoria de este proyecto en distintos apartados se ha hecho unadivisiónporbloquessecuencialessegúnelprocesodefabricacióndeunapila,perocomoenesteproyecto,loqueserealizaesunadeterminacióndelosparámetrosycondicionesparasufabricación,todaslaspartesestáninterrelacionadas,yaquedefectosqueaparecenen el últimoestadiode fabricaciónpueden estar relacionados con lospasos iniciales deesta. En el siguiente apartado se exponen las conclusiones obtenidas en cada fase dedesarrollodelprocesodefabricacióndelapilaysusposiblesimplicacionesfuturas.
6.2Conclusionesporfasesdedesarrollo Sehallevadoacabolasíntesisdedoscomposicionesdeapatita,LSOyLSGO,con
elnecesarioniveldepurezadecaraasuaplicación

6.Conclusiones
‒32‒
o Durante las pruebas de síntesis se observó que el resultado era igual desatisfactorio si los polvos de los óxidos reactivos se compactabanmanualmenteenelcrisolqueibaaserintroducidoenelhornoenlugardeser prensados en pastillas uniaxialmente, lo cual supone un ahorrosignificativoeneltiempoinvertido(devariashorasaunos30‐40minutos).
o También durante esta fase se observó el carácter abrasivo de lasoxiapatitas, descubriendo que existe desgaste de los elementos demolienda usando bolas y vasija de circona (materialmuy resistente a laabrasión). Asímismo, la observación al microscopio electrónico nosmostraba una considerable cantidad de partículas que después de variashorasdemoliendaseguíanconservandofilosvivos,loquehacepatentelanecesidaddeprocesosdemoliendaexhaustivos.
Sehaconseguidolaadecuacióndelpolvocerámicofabricadoenlafaseanteriordeacuerdoconlaaplicaciónalaqueestádestinado.Así,
o Sehadeterminadoqueel tamañodepartículadepolvodeoxiapatitaquemejorseadaptaalpolvocomercialdisponibledeNiO(paralafabricaciónde tubos de ánodo), se logra procesándolo con 12 horas demolienda entres tandas, entre las que se realizan sesiones de ultrasonidos de altaintensidad que deshacen los agregados de la tanda anterior, impidiendoquecrezcanenlatandasiguiente.Enelpolvoqueserealizólamoliendasinultrasonidos intermedios se observaron agregados más grandes quepodíanocasionardefectospuntualesconsiderables.
o Decaraalafabricacióndelelectrolito,sehacomprobadoquelareducciónmáximadetamañodegranoquesepuederealizaraunpolvodeoxiapatita(con los métodos mecánicos empleados), al cual previamente se le haaplicadounamoliendade12horas,esconunprocesodeatricióndetreshoras. A partir de ese momento, las partículas no se reducen y pasan aformar agregados. Hay que recordar que un polvo fino es indispensablepara lograrunabuenadensificacióndel electrolitoduranteel sinterizadoya que las dimensiones de una pila son reducidas, y la aparición de unagregadode10μm,quepuedeseralgoposible, tiene lamismadimensiónqueel espesorde la capadeelectrolito (10‐15μm), con las implicacionesqueelloconlleva.
Sehallevadoacaboconéxitoladeposicióndecapasdelgadasdeelectrolitosobreánodosmicrotubulares.
o Mediante el estudio reológico de las distintas pastas de electrolitorealizadas, diversas pruebas de impregnación por inmersión (con susinterizado posterior) y la observación de los resultados tanto almicroscopioópticocomoelectrónico,sehacomprobadoquelatécnicadedip‐coating es adecuada para este tipo de polvo, y que realizar una solainmersión con la extracción de la muestra a una velocidad de 3mm/sproporcionaelespesordecapadeelectrolitodeseado.
Sehaestudiado lacosinterizaciónde lasdistintascapasde lapila, alcanzándoseunacorrectaintegracióndelasmismas.

6.Conclusiones
‒33‒
o Los estudios de dilatometría, junto con una serie de pruebas decosinterizadoqueacabaronen fractura,mostraronquecomonosepodíamejorarelcomportamientodecontraccióndelacapadeelectrolito,habíaque presinterizar el tubo de ánodo a temperaturas más altas de lashabitualesempleadasconotrosmateriales,paraquesucomportamientoala temperatura de densificación del electrolito, se acercara a la del este,consiguiendo disminuir suficientemente el valor de las tensiones queaparecen entre ambas partes (por diferencias de contracción), quepermitiólograrobteneruncosinterizadosinfractura.
SehaverificadoquenohayningúntipodereacciónentreelNiOylasoxiapatitas.
o Como contrapartida,mencionar que se halló una importante cantidaddesegundafasenodeseadaLa2Si2O7enelelectrolitoqueindicaunexcesodeproporción de silicio respecto lantano, que puede provenir tanto de unacontaminación de SiO2 por desgaste de las bolas y la vasija de ágatautilizados durante la molienda de bolas, como por una evaporación delantano a alta temperatura durante el sinterizado. Esta fase no deseadapuede haber contribuido a los bajos valores de potencia obtenidos en lacaracterizaciónelectroquímica.
Al final se fabricaron dos pilas con el compuesto LSO a dos temperaturas decosinterizado distintas para observar su diferencia de comportamientoelectroquímico.Esunalástimaquedeestasdosmuestras,lamásprometedora(lasinterizada a temperaturamás elevada) se rompió en la fase demontaje por lapresencia de unamicrogrieta interna que no se había apreciado. Los resultadosobtenidosconlapilasinterizadaamenortemperaturacorroboranlasospechadequeaunqueexisteunmáximodecontraccióna1380°C,noessuficientesinterizara esta temperatura, teniendo que subir a 1480°C, que es la que corresponde almáximodecontracciónglobal.
6.3LíneasfuturasAlcanzadoelobjetivoprincipaldeesteproyecto,queera la integraciónde lasdiferentescapas delgadas cerámicas en una pila de combustible microtubular, un posible trabajofuturosecentraríaenlaoptimizacióndelcomportamientoeléctricodelsistema.Paraellohabríaque limitar laapariciónde fasessecundariasenelelectrolito,quedisminuyensuconductividad total. Para ello se podría determinar la causa realizando un análisis dedifracciónde rayosX aunamuestradepolvoprocesada conunamoliendade similarescaracterísticas. Si se corroborara esta sospecha se podría actuar midiendo eldesgaste/aportación de SiO2 en varios intérvalos de molienda (tomando muestrasintermediasyanalizándolas)paraobtenerunatasadedesgasteeintentarcompensarconunexcesodeLa2O3añadidoalaúltimatandademolienda,elcualpodríareaccionarconelSiO2 durante el sinterizado, conservando la estequiometría de la oxiapatita en el polvofinal.

Bibliografía
‒34‒
Bibliografía
[1] J.LarminieyA.Dicks,FuelCellSystemsExplained,JohnWiley&Sons,Ltd.,2003.
[2] D.Parfitt,A.ChroneosyJ.A.a.G.R.W.Kilner,«MoleculardynamicsstudyofoxygendiffusioninPr2NiO4+d,»PhysicalChemistryChemicalPhysics,vol.12,nº25,p.6834–6836,2010.
[3] A. Larrea, R. Campana, R. Merino, I. Villarreal y V. Orera, «SOFC mini‐tubularesbasadasenYSZ,»Boletínde la sociedadespañoladeCerámicayVidrio,pp.189‐195,2007.
[4] NexTech Materials, «Fuel Cell Materials,» [En línea]. Available:www.fuelcellmaterials.com.
[5] S.Guillot,S.Beaudet‐SavignatyS.Lambert,«Evidenceof localdefects intheoxygenexcessapatiteLa9.67(SiO4)6O2.5fromhighresolutionneutronpowderdiffraction,»JournalofSolidStateChemistry,nº182,p.3358–3364,2009.
[6] E.Kendrick,M.S.IslambyP.R.Slater,«Developingapatitesforsolidoxidefuelcells:into structural, transport and doping properties,» Journal of Materials Chemistry,2007.
[7] Fritsch,«FritschMilling,»[Enlínea].Available:http://www.fritsch‐milling.com/.
[8] F. Koichiro, A. Toru y U. Tomohiro, «Thermal expansion of lanthanum silicateoxyapatite (La9.33+2x(SiO4)6O2+3x), lanthanum oxyorthosilicate (La2SiO5) andlanthanum sorosilicate (La2Si2O7),» Journal of Solid State Chemistry, vol. 194, pp.157‐161,2012.
[9] M. A. Schubert, S. Senz y D. Hesse, «Formation of La2Zr2O7 on yttria‐stabilizedZrO2(110)singlecrystalsduringvapour–solidreaction,»SolidState Ionics,vol.179,pp.453‐457,2008.
[10]T.BallouyA.Orera,«Formulationand IntegrationofApatite‐typeMaterialsforSolidOxideFuelCells».
[11]G.Hoogers,FuelCellTechnologyHandbook,CRCPressLLC,2003.
[12]H.‐H. Möbius, «On the history of solid electrolyte fuel cells,» Journal of Solid StateElectrochemistry,1997.
[13]M. Warshay y P. R. Prokopius, «The Fuel Cell in Space: Yesterday, Today andTomorrow,»deGroveAnniversaryFuelCellSymposium,London,1989.
[14]E.E. Jay,P.M.Mallinson, S.K. Fong,B. L.Metcalfe yR.W.Grimes, «Divalent cationdiffusionincalciumfluorapatite,»JournalofMaterialsScience,2011.
[15]C.Umbach,«ApplicationsofRamanSpectroscopy,»CornellUniversity.
[16]K.Fukuda,T.IwatayE.Champion,«Crystalstructureoflanthanumoxyorthosilicate,La2SiO5,»PowderDiffraction,vol.21,pp.300‐303,2006.
[17]E. M. Levin, C. R. Robbins y H. F. McMurdie, Phase Diagrams for Ceramists: 1969Supplement,Columbus,Ohio:TheAmericanCeramicSociety,1969.

Tabladefiguras
‒35‒
TabladefigurasFigura 1. Reacciones químicas ymateriales que va a haber en las distintas partes de laSOFCconstruidaenesteproyecto.................................................................................................................2Figura2.ReaccionesquímicasenunTBP..................................................................................................3Figura3.Reaccionesquímicasenelcátodo...............................................................................................4Figura 4.Valores de conductividad para electrolitos de óxido sólido y diversasconfiguracionesdepila.[1]................................................................................................................................4Figura5.Estructuradelcristaldeoxiapatitavistadelejez.................................................................5Figura6.Comparaciónde laconductividaddeoxiapatitasconotrosconductoresde ionesóxido...........................................................................................................................................................................5Figura 7. Simulación atomística de la vía de migración del iónO2−intersticial en laoxiapatitaLa9.33(SiO4)6O2...................................................................................................................................5Figura8.Bolybolasdeágataconpolvoantesdeañadiretanolparaprocederasumezcla‐molienda...................................................................................................................................................................8Figura9.Boldecirconacolocadoyaseguradoenelmolinoplanetarioantesdeempezarlamolienda...................................................................................................................................................................8Figura10.Transvasedemoliendaabandeja............................................................................................8Figura11.Pastillareciénprensadaadheridaaltroquellistaparasuextracción......................9Figura12.MedidadeespectroscopiaRamandeunamuestrapuraylasíntesisLSO.1........10Figura 13.Difractogramade rayosXde la síntesis LSO.1 antes y despúesde la segundamolienda+sintetizado.......................................................................................................................................11Figura14.PartículadeLSGOdespuésde5horasdemoliendadebolas.....................................11Figura15.RepresentacióndeBodeparalasmedidasrealizadasatodaslastemperaturas.....................................................................................................................................................................................12Figura16.DiagramaNyquistdelaimpedanciamedidaaaltaybajatemperatura(valoresenΩ).........................................................................................................................................................................12Figura17.GráficosdeArrhenius a altas temperaturas (izquierda)y en todoel rangodemedida(derecha)................................................................................................................................................13Figura18.Medidasdeladistribucióndeltamañopartículadespuésdecadaproceso.........15Figura19.Morteroutilizadoenlamoliendadelospolvos................................................................16Figura20.AglomeradodepartículasdeLSOdespuésdeunamoliendaconmortero............16Figura21.PartículasdeLSOdespuésde12horasdemoliendaconbolas.................................16Figura22.Aglomeradopresenteenelpolvoformadoporunexcesodeatrición....................17Figura 23.Apariencia del polvo después de aplicar ultrasonidos a la muestra deaglomerados..........................................................................................................................................................17Figura24.CompactodeLSOdespuésdeCIPparamuestradedilatometría..............................17Figura25.Dilatometríasdedospolvosdeoxiapatitaprocesadosparareducirsu tamañodepartícula:..........................................................................................................................................................18Figura26.Kitdedensidad...............................................................................................................................18Figura 27.Medidas reológicas de dos pastas de electrolito (LSO y LSGO) y una pasta decátodo(PNO‐LSO)...............................................................................................................................................19Figura28.Elementosempleadosenelmoldedeltubodeánodo....................................................20Figura29.Tubosdeánododespuésdepresinterizarydetalledegrietaenel extremodeunodelostubos...................................................................................................................................................21

Tabladefiguras
‒36‒
Figura30.Muestradedeánodocolocadaendilatómetro.................................................................21Figura31.Muestrareciénimpregnada.......................................................................................................21Figura32.Muestradespuésderealizardip‐coating(teflónretirado)..........................................22Figura33.MicrografíaSEMdeunacapadeelectrolitodeespesordeunos80μm...................22Figura34.Imágenesdemicroscopíaópticaendondeseaprecialadiferenciadeespesordecapadeelectrolitosegúnlaconcentracióndepastautilizada.........................................................22Figura35.Pilafracturadaporcargastérmicasenelprocesodesinterizado.............................23Figura36.Curvasdecontraccióndeunánodopresinterizadoa950°Cydedoselectrolitoscondistintotamañodepartícula..................................................................................................................23Figura37.Curvasde contraccióndelpolvodeLSOconmenor tamañodepartículaydetresánodospresinterizadosatrestemperaturasdistintas..............................................................24Figura 38. Difratograma de una muestra de LSO.2 sometida a un sinterizado a 1480°Cdurante2horas...................................................................................................................................................25Figura39.Aspectodeunapiladespuésdelaintegracióndelcátodo...........................................26Figura40.MicrografíasSEMdondeseaprecialacorrectaintegraciónentrelascapasdelapilafabricada........................................................................................................................................................26Figura41.Pilarotaduranteelmontaje.....................................................................................................27Figura42.Contactoseléctricosmontadosenlapila............................................................................27Figura43.Conexioneseléctricasparamedida.......................................................................................28Figura44.Hornotubulardemedida.Detalledelinterior.................................................................28Figura45.Medidaencircuitoabiertodelapilaa800°C....................................................................29Figura46.Valoresdeintensidad‐voltajemedidosenensayodevoltametríacíclicaa800°C.....................................................................................................................................................................................30Figura47.IlustracióndecuatrodelasceldasdelexperimentodeGrove...................................40Figura48. PropuestadeBaur yPreispara la realizaciónunapilade combustible con la“masaNernst”comoelectrolitosólido.......................................................................................................40Figura49.ApolloCSM‐102.............................................................................................................................40Figura50.Estructuracristalinayceldaunidaddelmineraldefluroapatita.............................43Figura51.Estructuracristalinadeloxiortosilicatodelantano(La2SiO5)vistaalolargodelejeb...........................................................................................................................................................................44Figura52.Clasificaciónestructuraldelossilicatos..............................................................................44Figura53.DiagramadeequilibriodefasesdelsistemabinarioLa2O3−SiO2.[13]....................45Figura54.Molinoutilizado.............................................................................................................................55Figura55.Dinámicadelasfuerzasqueintervienenenlamoliendadebolas...........................55Figura56.Diagramadelprocesodeatrición..........................................................................................56Figura57.Prensauniaxialhidraúlicayelementosdeltroquelutilizado.....................................56Figura58.Servoelectromecánico................................................................................................................58Figura59.Momentosanterioryposterioralainmersióndedosmuestras..............................58Figura60.DifracciónderayosXporplanosatómicos........................................................................60Figura 61. Difractograma de tres túbos de ánodo sometidos a diferentes tratamientostérmicos..................................................................................................................................................................60Figura62.EsquematécnicodeunSEM.....................................................................................................61Figura 63.Microscopio electrónico de barrido de emisión de campo (FESEM) Carl ZeissMERLIN™................................................................................................................................................................61Figura64.ZetasizerNano‐Zs,MalvernInstruments.............................................................................62Figura65.Equipoutilizadoyesquemainternodelacámaradeldilatómetro.........................63

Tabladefiguras
‒37‒
Figura66.CurvasdecontraccióndelpolvodeLSGO(12horasdemolienda+3horasdeatrición)ydeánodopresinterizadoa1175°C........................................................................................64Figura67.Deformacióndeunlíquidosometidoaunesfuerzocortanteenelreómetro......65Figura68.ReómetroHaakeRheostressMarsII,ThermoScientific..................................................66Figura69.CiclocompletodelareometríadeunapastadeLSO......................................................66Figura70.DiagramaNyquistenescaladoblelogarítmicadelascurvasmedidasconEIS..67Figura71.DiagramaNyquistconlascurvasnormalizadasalaresistenciade300°C...........67

Listadeabrebiaturas
ListadeabreviaturasCIP ColdIsostaticPressing(prensadoisostáticoenfrío)EIS ElectrochemicalImpedanceSpectroscopy(espectroscopiadeimpedancias)LSO La9.67Si6O26.5=La9.67(SiO4)6O2.5LSGO La9.67Si3Ge3O26.5=La9.67(SiO4)3(GeO4)3O2.5
NiO óxidodeníquelNPD NeutronPowderDiffraction(Difraccióndeneutrones)OCV OpenCircuitVoltage(voltajedecircuitoabierto)PNO Pr2NiO4(niquelatodepraseodimio)PVA PolyvinylAlcohol(alcoholdepolivinilo)PVB PolyvinylButyral(butiraldepolivinilo)SEM ScanningElectronMicroscopy(microscopioelectrónicodebarrido)SOFC SolidOxideFuelCell(piladecombustibledeóxidosólido)TPB TriplePhaseBoundary(triplefronteradefase)XRD XRayDiffraction(difracciónderayosX)YSZ YttriaStabilizedZirconia(circonaestabilizadaconitria)

ANEXOS

Anexo1.Pilasdecombustible
‒40‒
A1.Pilasdecombustible
A1.1ReseñahistóricaEn1838enSuiza,ChristianFriedrichSchönbeinpublicabaelprincipiodefuncionamientode la célula de combustible, la propiedad catalítica del platino en la reacción entre elhidrógenoyeloxígeno.En1839,SirWilliamGrovediseñóunexperimentoqueconsistíaenunirenserievariasceldaselectroquímicas,cadaunade lascualesestabacompuestapor un electrodo con hidrógeno y otro con oxígeno,separados por un electrolito. Grove comprobó que lareaccióndelhidrógenoenelelectrodonegativocombinadacon la del oxígeno en el positivo generaba una corrienteeléctrica, aunque para que esto fuera posible tuvo queconstruir una pila con 26 celdas para tener la suficientesuperficiedecontacto.[7]
BaurandPreis,en1937,empezaronadesarrollarlaspilasdecombustibledeóxidosólidoporlanecesidaddeteneruntipodepilasdecombustibleconunelectrolitomásmanejable
quelasdeelectrolitodesalesfundidas,queeranlasque se habían estado desarrollando a principio delsigloXX.Sebasaronenlallamada“masaNernst”,uncompuesto de 85%de circona (ZrO2) y un 15%deitria (Y2O3), que Wilhelm Nernst había estadoinvestigandoen1899.En1951,HundconfirmóconcristalografíaderayosXlaexistenciadevacantesdelionóxidoenelmaterialdeNerst[8].Enlaactualidad,75añosdespuésdeaquelprimerprototipo,laspilasde combustible de óxido sólido siguen teniendo elmismocompuesto,lacirconaestabilizadaconitriaoYSZ (Yttria Stabilized Zirconia), como el material
másutilizadoenlacomposicióndesuselectrolitos.
Eldesarrollo industrialefectivodelaspilasdecombustiblenosedio hasta 1959, cuando el ingeniero británico Francis ThomasBacon desarrolló con éxito una de tipo alcalino con unfuncionamiento estacionarioque permitía accionar una máquinade soldadura. Esta suministraba 5 kilowatios, estaba compuestade cuarenta celdas y tenía una eficiencia del 60%. En 1960 elprogramaespacialdelaNASAdecidióemplearlasenlasmisionesGéminis y Apolo[9]. A pesar del éxito en estos programasespaciales, estos sistemas se limitaron a aplicaciones especiales,dondeelcostenoesunproblema.
Nofuehastaelfinaldelosaños80yprincipiosdelos90quelaspilasde combustible se convirtieronenunaopción realparaunusomásamplio.Variasinnovaciones que permitieron la reducción de la cantidad de platino presente en el
Figura47. Ilustraciónde cuatrodelasceldasdelexperimentodeGrove.
Figura48.PropuestadeBauryPreisparala realizaciónunapilade combustible conla“masaNernst”comoelectrolitosólido.
Figura49.ApolloCSM‐102

Anexo1.Pilasdecombustible
‒41‒
catalizador y electrodos de película fina, hicieron bajar su coste de fabricación,permitiendo que el desarrollo de ciertos tipos de sistemas, como los de membranapolimérica,conaplicacionescomercialescomoparaautomóviles,comenzaraaserrealista.
Hoy en día nos podemos encontrar con una gran variedad comercial de pilas decombustiblequealimentanordenadoresportátiles,bicicletaseléctricas,coches,autobuses,sistemasgeneradoresparaedificios…
A1.2TiposdepilasdecombustibleApesardequetodassebasanenunatecnologíasimilar,existengranvariedaddepilasdecombustible.ElproyectosehacentradoenlasSOFCperoacontinuaciónsedetallanotrostipos:
Pilas de combustible alcalinas (AFC): Este tipo de pila de combustible opera atemperaturas típicamente entre 60 y 100 ⁰C. Se caracterizan por tener un electrolitolíquido, generalmente formado por una disolución de KOH y pueden utilizar comocatalizador una amplia variedad de metales no muy sofisticados. Como desventajaspresentansualtocosteyunaelevadasensibilidadalenvenenamientoporCO2.
Pilasdecombustibledeácidofosfórico(PAFC):Secaracterizanportenerunelectrolitolíquido de ácido fosfórico y electrodos porosos de carbono con platino. Operan atemperatura relativamente baja (150 – 200 ⁰C), pero son pesadas y voluminosascomparadas con otros tipos, por lo que se las utiliza generalmente como dispositivosestacionarios o para propulsar barcos. Presentan una sensibilidad menor al CO2 encomparaciónconelrestodepilas.
Pilas de combustible de membrana de intercambio protónico (PEMFC): Secaracterizan por tener un electrolito polimérico a través del cual se intercambian losprotones.Setratadeunapiladecombustibledebajatemperatura,entornoa80⁰C,porloquelasreaccionessonmuylentas,requiriendoelusodecatalizadoresmuysofisticadosyde alto precio como el platino. Adicionalmente este tipo de pilas presenta una especialsensibilidad al envenenamiento por CO, por lo que requieren H2 de elevada pureza.Existendiseñosenlosquesealimentaconmetanolyéstesereformadirectamenteenelinteriordelapilaparaproducirelhidrógenoquevaaserconsumido.Éstaspilasrecibentambién el nombre de pilas de combustible de metanol directo (DMFC) y tienen graninterésparaaplicacionesportátiles.
Pilasdecombustibledecarbonatosfundidos(MCFC):Estetipodepilasdecombustiblesecaracterizaportenerunelectrolito formadoporsalesdecarbonato fundidas.Setratade pilas de combustible de alta temperatura 600 – 700 ⁰C lo que permite trabajar concatalizadoresbaratosenlugardeplatino,lesconfiereunaaltaeficienciaylashacemenossusceptibles al envenenamiento por CO. Por otra parte, tienen una durabilidad limitadadebidoalacorrosióndesuscomponentesytienentiemposdepuestaenmarchalargos.
En la siguiente tabla se resumen las características de los distintos tipos de pilasexplicadosysecomparanconlascaracterísticasdelasSOFC:
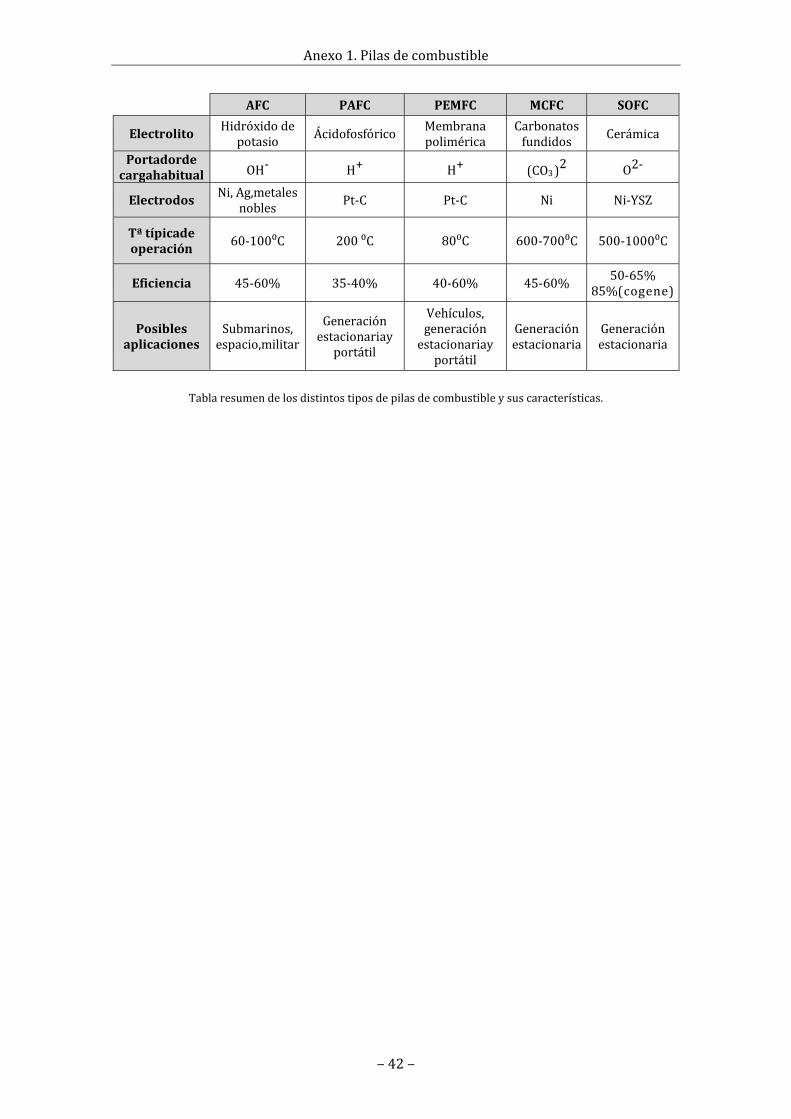
Anexo1.Pilasdecombustible
‒42‒
AFC PAFC PEMFC MCFC SOFC
ElectrolitoHidróxidodepotasio
ÁcidofosfóricoMembranapolimérica
Carbonatosfundidos
Cerámica
Portadordecargahabitual OH‐ H+ H+ (CO3)
2 O2‐
ElectrodosNi,Ag,metales
noblesPt‐C Pt‐C Ni Ni‐YSZ
Tªtípicadeoperación
60‐100⁰C 200⁰C 80⁰C 600‐700⁰C 500‐1000⁰C
Eficiencia 45‐60% 35‐40% 40‐60% 45‐60%50‐65%
85%(cogene)
Posiblesaplicaciones
Submarinos,espacio,militar
Generaciónestacionariay
portátil
Vehículos,generaciónestacionariay
portátil
Generaciónestacionaria
Generaciónestacionaria
Tablaresumendelosdistintostiposdepilasdecombustibleysuscaracterísticas.

Anexo2.Oxiapatitas
‒43‒
A2.Oxiapatitas
A2.1ElmineraldeapatitaLas apatitas convencionales pertenecen al grupo de los minerales fosfato, másconcretamente al de los ortofosfatos. Las formas más habituales son: hidroxiapatita,fluorapatita y cloroapatita, denominadas así por las altas concentraciones de los ionesOH‾,Cl‾yF‾,respectivamente,enelcristal.Lafórmuladelaceldaunidaddelcristales:
Ca PO X ⟶ X OH, F, CL
En la figura 50[10] se muestra la coordinación de todos los átomos en la estructuracristalina de la fluorapatita, la cual es idéntica a la de las oxiapatitas utilizadas en esteproyecto(conelcorrespondientecambioatómico:PO4→SiO4,Ca→La,F→O).
A2.2Silicatos(germanatos)Los germanatos,mineralogísticamente, están incluidos dentro del grupo de los silicatospor formar estructuras cristalinas muy parecidas a estos, ya que su unidad básica, eltetraedro(GeO4)4−tienepropiedadesmuysimilaresconlaunidadbásicadelossilicatos.
A2.2.1ConceptoLos silicatos son materiales compuestos principalmente por silicio y oxígeno, los doselementosmásabundantesenlacortezaterrestre.Launidadbásicadelossilicatoseseltetraedro(SiO4)4−,elcualestáformadoporunátomodesilicio(Si4+)enelcentrounidoacuatroátomosdeoxígeno(O2−)localizadosenlasesquinasdeltetraedro.LosenlacesSi−Otienen un significativo carácter covalente, siendo direccionales y relativamente fuertes,característica por la cual a menudo los silicatos no son considerados iónicos.
Figura50.Estructuracristalinayceldaunidaddelmineraldefluroapatita.

Anexo2.Oxiapatitas
‒44‒
Independientementedel carácterdel enlaceSi−O, existeuna carga eléctrica−4 asociadacon cada tetraedro, puesto que cada uno de los cuatro átomos de oxígeno requiere unelectrónextraparaalcanzarunaestructuraelectrónicaestable.
Estaspropiedadesprovocanquelaunidadbásicadelossilicatos,eltetraedro(SiO4)4−,seconsidereunaentidadcargadanegativamente,yen lugardecaracterizar lasestructurascristalinas de estos materiales en términos de cedillas unidad, es más convenienteclasificarlossegúnlostiposdecombinacionesgeométricasqueformanestostetraedros.
A2.2.2Clasificaciónestructural Ortosilicatos o nesosilicatos. Tienen todos sus tetraedros (SiO4)4− aislados (no
enlazados). La carga estos tetraedros aislados se equilibra con otros cationes. A estegrupopertenecentantolasoxiapatitas,comounadelasfasesnodeseadasqueapareceenlasíntesisdeestas,elLa2SiO5,cuyaestructurasepuedeapreciarenlafigura51.
Disilicatososorosilicatos.Estánformadosporgrupostetraédricosdobles,esdecir,pordostetraedrosquecompartenunoxígeno.Suunidadestructuralesel(Si2O7)6−.AestegrupoperteneceelLa2Si2O7(otradelasfasesnodeseadasqueaparecenenlasíntesisdelasoxiapatitas).
Ciclosilicatos. Están formados por anillos de tetraedros enlazados. Hay tres tiposposiblesdeconfiguraciones:(Si3O9)6−,(Si4O12)8−y(Si6O18)12−.
Inosilicatos o silicatos de cadena. Existen dos grupos importantes, los piroxenos,formadosporcadenassimplesdetetraedroscuyaunidadesel(SiO3)n2n−ylosanfíboles,formadospordoblescadenasdetetraedroscuyaunidadesel(Si4O11)n6n−.
Filosilicatososilicatoslaminares.Presentanunaestructurabidimensionalenformadecapasoláminasproducidaalcompartirtresdeloscuatroionesdeoxígenodecadaunodelostetraedros.Lafórmulaunidadqueserepiteesdeltipo(Si2O5)2−.
Tectosilicatos.Todos los tetraedros comparten todos susoxígenos con los tetraedrosadyacentes formandouna red tridimensional.Esel casode la síliceuóxidode silicio(SiO2),unodelosreactivosqueseutilizanenlasíntesisdelasoxiapatitas.
Figura51. Estructura cristalina deloxiortosilicatodelantano(La2SiO5)vistaalolargodelejeb.Está compuesta por tetraedros (SiO4)4− aislados,con dos tipos de cationes de La: uno con unnúmero de coordinación 9 (La1) y el otro con unnúmerodecoordinación7(La2).[12] Figura52.Clasificaciónestructuraldelossilicatos.

Anexo2.Oxiapatitas
‒45‒
A2.3SistemabinarioLa2O3−SiO2LosreactivosutilizadosparaobtenerelcompuestodeoxiapatitaLSOdeesteproyectosonelóxidodelantano(La2O3)yelóxidodesilicio(SiO2).Paraverdeunamaneramásvisualeintuitiva la relación entrela estructura y la composición y las cantidades de fases enequilibrio,sehaanexado(figura53)eldiagramadefasesdelsistemabinarioLa2O3−SiO2.
LasoxiapatitasseencuentranenlaregióncomprendidaentreelLa2SiO5(oxiortosilicatodelantano)yelLa2Si2O7(sorosilicatodelantano).Susecuacionesdereacciónson:
La O SiO ⟶ La SiO La O 2 SiO ⟶ La Si O
La relación molar del La2SiO5 es 50% de La2O3 y 50% de SiO2 (relación 1:1) y la delLa2Si2O7es33.33%deLa2O3y66.67%deSiO2(relación1:2).
ElcompuestodeoxiapatitautilizadoenesteproyectoeselLa9.67(SiO4)6O2.5óLa9.67Si6O26.5sise expresa en relación estequiométrica. Su ecuación de reacción, expresada de dosmanerasdistintas,eslasiguiente:
4.833 ⋅ La O 6 ⋅ SiO ⟶ La . Si O . La O 1.241 ⋅ SiO ⟶ La Si . O .
Surelaciónmolares44.62%deLa2O3y55.38%deSiO2(relación1:1.241).Estáseñaladaconunamarcarojaeneldiagramadefases.
Hay que señalar que hasta el 60% en mol de SiO2 (relación 2:3) donde se forma elLa4Si3O12,compuesto en el que todos los oxígenos están localizados en los tetraedros desilicato(SiO4)4−,loscompuestosformadospertenecenalgrupodelosortosilicatos,esdecir,tienen todos sus tetraedros (SiO4)4− aislados. A concentraciones más elevadas de SiO2,estostetraedrosdesilicatoempiezanaenlazarse(compartensusoxígenos).
Figura53.DiagramadeequilibriodefasesdelsistemabinarioLa2O3−SiO2.[13]

Anexo3.Elaboracióndemezclasypastas
‒46‒
Anexo3.ElaboracióndemezclasypastasEnesteanexosevaaexponerdeunamaneramásdetalladalascondicionesdeseadas,loscálculos de composición y los procesos utilizados en la realización de lasmezclas paraánodoylaspastasdeelectrolitoycátodo,ademásdelosaditivosquímicosempleadosenellas.
A3.1Aditivosquímicosutilizados
A3.1.1Alcoholdepolivinilo(PVA)El alcohol de polivinilo o PVA (PolyVinyl Alcohol), también llamado polietenol opolialcohol vinílico, es un polímero sintético soluble en agua, cuya fórmula químicaes[CH2CH(OH)]n.
ElPVAqueseutilizaenellaboratorioseencuentradisueltoenH2Oenunaconcentracióndeun5%enmasa.
Sehautilizadocomoaglutinanteparaproporcionarunamayorresistenciaenverdealostubosdeánodoymuestrasdemedidasdedilatometría.
A3.1.2Butiraldepolivinilo(PVB)Elbutiral de polivinilo o PVB (PolyVinylButyral), también conocido como Butiral, es uncompuestoquímicoresultadodemezclaralcoholdepolivinilo(PVA)conbutiraldehído.Elmaterial resultante es un polímero de gran adherencia y durabilidad, utilizadoprincipalmenteenlaindustriadelvidrio.
ElPVButilizadoseencuentraenunadisolución20gdePVB+100gdeetanol.
Enellaboratoriosehaempleadoenlarealizacióndelaspastasdeelectrolitoycátodoparadotarlesdeunamayorviscosidadyadherencia.
A3.1.3BeycostatC213El defloculante éster de fosfato Beycostat‐C213 (CECA) se utiliza para proporcionar laestabilizaciónelectroestérica(estabilizaciónelectrostática+estabilizaciónestérica).
En este proyecto se ha utilizado junto con etanol para obtener un medio líquidodispersante para las suspensiones de polvo, evitando agregados y sedimentaciones. Enconcreto se ha utilizado en lasmedidas de tamañodepartículas, en la atrición y en lasfasesdemezcladodelospolvosensuspensiónenlaspastasdeelectrolitoycátodo.
Estecompuesto,deunamanerasimple,puededecirsequecubrelaspartículasdelpolvocerámico en suspensión, reduciendo las energías de interacción existentes entre ellas.Estas energías están formadas por fuerzas atractivas (generalmente fenómenos deLondon‐VanderWaals)yfuerzasrepulsivas(electrostáticasyestéricas).

Anexo3.Elaboracióndemezclasypastas
‒47‒
A3.2Mezclasparaánodo
A3.2.1CondicionesycálculospreviosSequiereobtenerunánododondelafraccióndevolumendeporoseaigualaladecermet,yenel cermet, laproporcióndeníquelyoxiapatitadebenser iguales también, esdecir,50%deporo,25%deníquely25%deoxiapatita.Apartirdelasfracciónvolumétricassedebencalcularlafraccionesmásicasdeloscompuestos(NiO,apatitayformadordeporo)quesevanamezclar.
Separtedelareaccióndereduccióndelóxidodelníquelaníquelmetálico:
2 NiO ⟶ 2 Ni O
2mol 74.7094gmol
NiO ⟶ 2mol 58.71gmol
Ni 1mol 15.9994gmol
O
149.4188g NiO ⟶ 117.42g Ni 15.9994g O
V149.4188g
6.6722.4016 cm V
117.42 g
8.913.1933cm
Después de que el NiO se reduzca a Ni quedarán los huecos del oxígeno.El resto devolumendeporoquequeremosloconseguiremosconformadordeporo(maicena):
V V V 22.4016cm NiO 13.1933cm Ni 9.2084cm porosO
V V V
Lasfraccionesdevolumenparaelvolumentotalfinal(despuésdeNireducido)son:
FV 0.25FV 0.25FV 0.5
→V V FVV V FVV V FV
→V
VFV
13.1933cm0.25
52.7730cm
V 52.7730cm 0.25 13.1933cm
V 52.7730cm 0.25 26.3865cm
Lacantidaddeformadordeporonecesariavaaser:
V V V 26.3865cm 9.2084cm 17.1782cm
m 17.1782cm 1.9gcm
32.6385g fp
Lamasadeapatita:
m 13.1933cm 5.3066gcm
70.0113g apa
Secalcula lamasa totaldemezcla (antesdereduccióndelNiO)yconella las fraccionesmásicas:
m m m m 149.4188g 70.0113g 32.6385g 252.0686g
FMmm
149.4188g252.0686g
0.5928
FMm
m70.0113g252.0686g
0.2777
FMm
m32.6385g252.0686g
0.1295

Anexo3.Elaboracióndemezclasypastas
‒48‒
Si, por ejemplo, sequiere tenerunamasademezcla inicialde5g las cantidadesdeNi0,apatitaymaicenaserán:
m FM m 0.5928 5g 2.9639g NiOm FM m 0.2777 5g 1.3887g apam FM m 0.1295 5g 0.6474g fp
Siseprocededeigualformadecálculo,perodeunamaneraalgebraica,esdecir,utilizandosólo parámetros y ninguna cantidad numérica, se obtienen las siguientes expresionesgenerales, que serán de utilidad si se desea cambiar de proporción volumétrica en loscomponentesdelánodo:
FMmm
M
M 1
FMm
m
ρ
M 1
FMm
m
ρ
M 1
FM 1 FM FM

Anexo3.Elaboracióndemezclasypastas
‒49‒
A3.2.2Procesodefabricacióndelamezcla1.SepesanlascantidadescalculadasdeNiO,LSOymaicena.
2.EnunvasodeprecipitadosemezclaelNiOconelLSOconunaespátulahastaobtenerunpolvo homogéneo. Se añade la maicena y se mezcla hasta obtener otra vez un polvohomogéneo.
3.Seañadeacetonaalpolvoparatenerloensuspensiónyuniformizarlamezcla.Seañadeuna cantidad adecuada que permita la agitación magnética en el vaso de precipitado;normalmentequecubra1‐2mmelimán.
4.Agitaciónmagnética (durante15minutos). Se adecuará la velocidadde giro según sevealascondicionesexistentes.
5.Ultrasonidos(4x15segundos)enbaño frío.Elvasodeprecipitadoestaráenbaño frío(bandeja con agua y cubitos) para que la mezcla esté fría mientras se aplican losultrasonidosconlasonda.Estocontrarrestaelcalorqueseproduceporlosultrasonidosevitando que se evapore la acetona y se formen aglomerados en la mezcla. Entre cadatandade15segundosdeultrasonidossedeberáremoverelvasodeprecipitadoenelbañofríounos15segundosparafacilitarqueellamezclalibereelcalorproducidoporlasondadeultrasonidos.
Serepitenlospasos4y5tresvecesmásparaqueentotalsehayaaplicadoalamezclauntotalde60minutosdeagitaciónmagnéticay4minutosdeultrasonidos.Sidespuésdeunatandade ultrasonidos (exceptodespuésde la tanda final) se aprecia que el volumendeacetona ha disminuido, se repondrá su nivel para que sea adecuado para la siguienteagitaciónmagnética.
6.Seintroduceelvasodeprecipitado,conlamezclayacompletamentehomogeneizada,enlaestufaaunos70°Cparaqueseevaporelaacetona.
7.Unavezevaporadalaacetona,serecuperaelpolvodelvasodeprecipitado.
8. Se añade un aglutinante, alcohol de polivinilo (PVA), que se encuentra en forma dedisoluciónal5%demasaenH2O.Semezclaenunarelaciónde10gotasporcada3gdepolvodeánodo.Elaglutinanteaumentalaresistenciaenverdedeloscompactosparaquepuedansermanipuladosenverde.LadisolucióndePVAseañadealpolvoanódicoenelmortero;seañadeunaodosgotascadavezysemezclahastaquenohayningunapartedelpolvoquepresenteunestadopegajoso.UnavezquesehaañadidoymezcladoeltotaldedisolucióndePVAconelpolvo,estesecolocadebajodeunalámparapotenteparaqueelaguadeladisoluciónseevaporeysolopermanezcaelPVA.
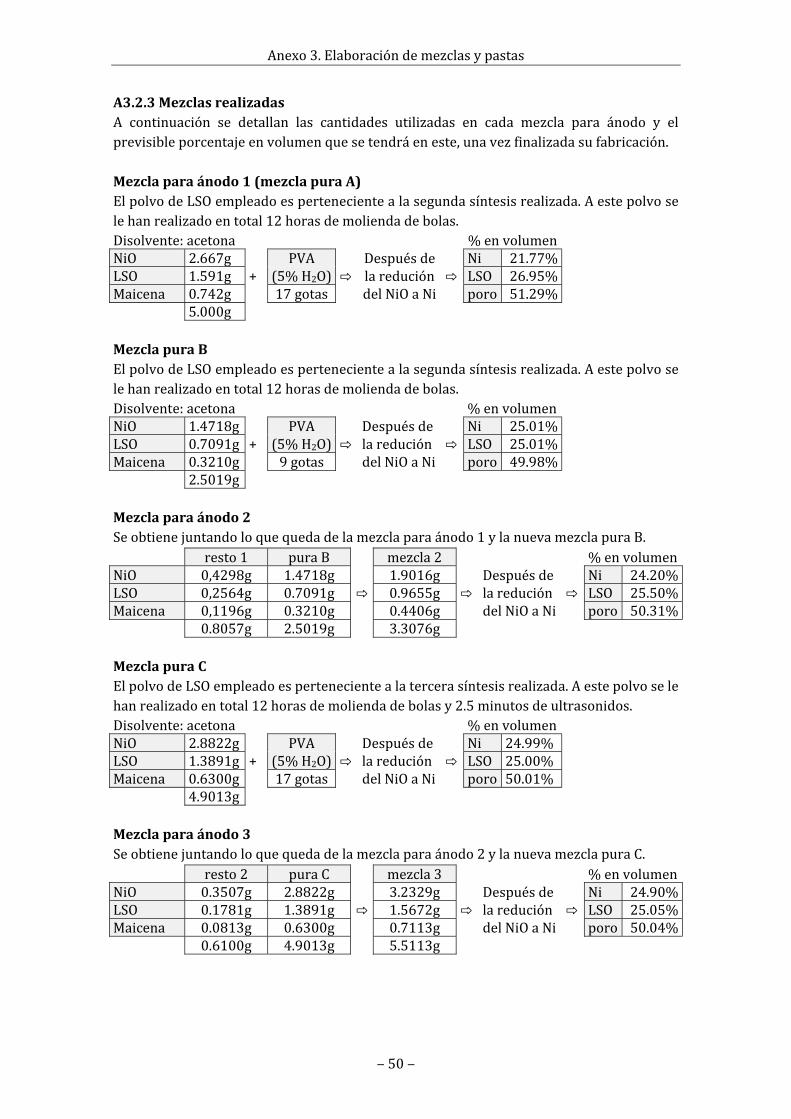
Anexo3.Elaboracióndemezclasypastas
‒50‒
A3.2.3MezclasrealizadasA continuación se detallan las cantidades utilizadas en cada mezcla para ánodo y elprevisibleporcentajeenvolumenquesetendráeneste,unavezfinalizadasufabricación.Mezclaparaánodo1(mezclapuraA)ElpolvodeLSOempleadoespertenecientealasegundasíntesisrealizada.Aestepolvoselehanrealizadoentotal12horasdemoliendadebolas.Disolvente:acetona %envolumenNiO 2.667g PVA Despuésde Ni 21.77%LSO 1.591g + (5%H2O) ⇨ laredución ⇨ LSO 26.95%Maicena 0.742g 17gotas delNiOaNi poro 51.29% 5.000g MezclapuraBElpolvodeLSOempleadoespertenecientealasegundasíntesisrealizada.Aestepolvoselehanrealizadoentotal12horasdemoliendadebolas.Disolvente:acetona %envolumenNiO 1.4718g PVA Despuésde Ni 25.01%LSO 0.7091g + (5%H2O) ⇨ laredución ⇨ LSO 25.01%Maicena 0.3210g 9gotas delNiOaNi poro 49.98% 2.5019g Mezclaparaánodo2Seobtienejuntandoloquequedadelamezclaparaánodo1ylanuevamezclapuraB. resto1 puraB mezcla2 %envolumenNiO 0,4298g 1.4718g 1.9016g Despuésde Ni 24.20%LSO 0,2564g 0.7091g ⇨ 0.9655g ⇨ laredución ⇨ LSO 25.50%Maicena 0,1196g 0.3210g 0.4406g delNiOaNi poro 50.31% 0.8057g 2.5019g 3.3076g MezclapuraCElpolvodeLSOempleadoespertenecientealatercerasíntesisrealizada.Aestepolvoselehanrealizadoentotal12horasdemoliendadebolasy2.5minutosdeultrasonidos.Disolvente:acetona %envolumenNiO 2.8822g PVA Despuésde Ni 24.99%LSO 1.3891g + (5%H2O) ⇨ laredución ⇨ LSO 25.00%Maicena 0.6300g 17gotas delNiOaNi poro 50.01% 4.9013g Mezclaparaánodo3Seobtienejuntandoloquequedadelamezclaparaánodo2ylanuevamezclapuraC. resto2 puraC mezcla3 %envolumenNiO 0.3507g 2.8822g 3.2329g Despuésde Ni 24.90%LSO 0.1781g 1.3891g ⇨ 1.5672g ⇨ laredución ⇨ LSO 25.05%Maicena 0.0813g 0.6300g 0.7113g delNiOaNi poro 50.04% 0.6100g 4.9013g 5.5113g

Anexo3.Elaboracióndemezclasypastas
‒51‒
A3.3Pastadeelectrolito
A3.3.1CondicionesycálculoCondicionesdelapasta:
apatitaal50%demasa(restoetanol) Beycostat:1‐3%enmasadelaapatita PVB(Butiraldepolivinilo):5%enmasadelaapatita⟶seobtienedeunadisolución
20gdePVB+100gdeetanol.
Aclaracionessobrelascantidadesdemezcla:
1. La cantidad de Beycostat y de PVB no se tienen en cuenta para el cálculo de laproporciónmásicatotal,sólolaapatitayeletanol,conloquelamasadeambosseráigual.
2.ElPVBseobtienedeunadisolución20gdePVB+100gdeetanol⇒paraobtener20gdePVBhacenfalta120gdedisolución:
m _12020
m 6 0.05 m 0.3 m
3.Eletanolseañadeendosformas,juntoconelPVB(porqueesteestáendisoluciónconetanol) e independientemente. La cantidad de etanol que se añade con la disolución dePVB‐etanoles:
m añadidocondisoluciónPVB10020
m 5 0.05 m 0.25 m
Lacantidadqueseañadeparacompletaraltotales:m añadidosolo m Totalenpasta m añadidocondisoluciónPVB m añadidosolo m 0.25 m 0.75 m
A3.3.2Procesodefabricacióndelapasta1.Sepesalacantidaddeapatitaenunabandejita.
2. En el bote que se va a realizar lamezcla se pesa el Beycostat (un 2% enmasa de laapatita).
3.Seañadeetanol(un75%enmasadelaapatita).
4.Agitaciónmagnética(seponeparafilmparareducirlaevaporacióndeletanol).
5.Seañadelaapatita(pesadaen1).
6.Agitaciónmagnética(conparafilm).
7.Ultrasonidosenbañofrío:3minutosentandasde15‐20segundos.
8.SeañadeelPVB(cantidaddedisolución20/100dePVB=30%enmasadelaapatita).
9.Agitaciónmagnética.
10.Transvasealbotecontenedordondepermanecerá.Secierraherméticamenteysedejaenlabandejaderodillosparaquelapastanoseapelmace.

Anexo3.Elaboracióndemezclasypastas
‒52‒
A3.3.3PastasdeelectrolitorealizadasAcontinuaciónsedetallanlascantidadesrealesutilizadasen lasmezclasparalaspastasdeelectrolito,laadecuacióndeestasconlascondicionesteóricasinicialesylaproporciónde polvo respecto el total de pasta, con la que se puede estimarmás intuitivamente elresultadodeladeposicióndelacapadeelectrolitoporlatécnicadedip‐coating:
Pastadeelectrolito1ElpolvodeLSOempleadoespertenecientealasegundasíntesisrealizada.Aestepolvoselehanrealizadoentotal12horasdemoliendadebolas.apatita 6.9896g polvoBeycostat 0.1403g %masadelpolvo (respectototaldepasta)etanol 5.2790g ⇨ Beycostat 2.00% %enmasa 49.87%PVB(disolución) 2.0960g PVB 5.00% %envolumen 12.80%Seobtieneunvolumentotaldepastade10.0083cm3.
Pastadeelectrolito2ElpolvodeLSOempleadoespertenecientealasegundasíntesisrealizada.Aestepolvoselehanrealizadoentotal12horasdemoliendadebolas.apatita 4.8358g polvoBeycostat 0.1005g %masadelpolvo (respectototaldepasta)etanol 3.6432g ⇨ Beycostat 2.08% %enmasa 49.92%PVB(disolución) 1.4507g PVB 5.00% %envolumen 12.82%Seobtieneunvolumentotaldepastade6.9136cm3
Pastadeelectrolito2diluidaPara contrastar la relación de concentración de apatita en la pasta y el espesor deelectrolitodepositadoporelprocesodedip‐coating,sehadiluidoestapastahastaobtenerun valor de un 10% en volumen de apatita aproximadamente. Para ello se han cogido2.47gdepastayseleshaañadido0.4410gdeetanol:cantidadesutilizadas polvo(respectototaldepasta)pastaelectrolito2 2.4700g ⇨ %enmasa 42.35%etanolañadido 0.4410g %envolumen 9.78%Seobtieneunvolumentotaldepastade2.3105cm3
Pastadeelectrolito3ElpolvodeLSOempleadoespertenecientealatercerasíntesisrealizada.Aestepolvoselehanrealizadoentotal12horasdemoliendadebolas,4horasdeatricióny3minutosdeultrasonidosdespuésdelaatrición.apatita 2.9954g polvoBeycostat 0.0642g %masadelpolvo (respectototaldepasta)etanol 2.2582g ⇨ Beycostat 2.14% %enmasa 49.88%PVB(disolución) 0.9015g PVB 5.02% %envolumen 12.80%Seobtieneunvolumentotaldepastade4.2873cm3
Pastadeelectrolito4AlpolvodeLSGOempleadoenestapastaselehanrealizadoentotal12horasdemoliendadebolasy2horasdeatrición.apatita 3.2997g polvoBeycostat 0.0683g %masadelpolvo (respectototaldepasta)etanol 2.5210g ⇨ Beycostat 2.07% %enmasa 49.59%PVB(disolución) 0.9994g PVB 5.05% %envolumen 11.95%Seobtieneunvolumentotaldepastade4.7319cm3

Anexo3.Elaboracióndemezclasypastas
‒53‒
A3.4Pastadecátodo
A3.4.1CondicionesycálculoCondicionesdelapasta:
laapatitaesun20%deltotaldelpolvo⇒PNOesun80%delpolvo elpolvo(apatita+PNO)seráel45%delamasatotaldelapasta(restoetanol) Beycostat:masaañadida=1%delamasadepolvo PVB(Butiraldepolivinilo):masaañadida=5%de lamasadepolvo⟶seobtienede
unadisolución20gdePVB+100gdeetanol.
Aclaracionessobrelascantidadesdemezcla:
1. La cantidad de Beycostat y de PVB no se tienen en cuenta para el cálculo de laproporciónmásicatotal,sólolaapatita,elPNOyeletanol,conloqueeletanolsuponeel55%enmasadelapasta.
2.ElPVBseobtienedeunadisolución20gdePVB+100gdeetanol⇒paraobtener20gdePVBhacenfalta120gdedisolución:
m _12020
m 6 0.05 m 0.3 m
3.Eletanolseañadeendosformas,juntoconelPVB(porqueesteestáendisoluciónconetanol) e independientemente. La cantidad de etanol que se añade con la disolución dePVB‐etanoles:
m añadidocondisoluciónPVB10020
m 5 0.05 m 0.25 m
Lacantidadqueseañadeparacompletaraltotales:m añadidosolo m Totalenlapasta m añadidocondisoluciónPVB
m añadidosolo0.550.45
m 0.25 m3536
m 0.9722 m
A3.4.2Procesodefabricacióndelapasta1.Sepesalacantidaddeapatitaenunabandejita.
2.SepesalacantidaddePNO(4veceslamasadeapatita)enunabandejita.
3. Se mezclan la apatita y el PNO en seco con una espátula hasta obtener un polvohomogéneo.
4. En el bote que se va a realizar lamezcla se pesa el Beycostat (un 1%de lamasa depolvo).
5.Seañadeetanol(un97.22%delamasadepolvo).
6.Agitaciónmagnética(seponeparafilmparareducirlaevaporacióndeetanol).
7.Seañadeelpolvomezcladoen3).
8.Agitaciónmagnética(conparafilm).
9.Ultrasonidosenbañofrío:3minutosentandasde15‐20segundos.
10.Siseobservaqueenelfondodelvasosehaapelmazadoelpolvodelasuspensión,estaseremueveconunasaspasaunas500rpm.

Anexo3.Elaboracióndemezclasypastas
‒54‒
11. Se añade el PVB (cantidad de disolución 20/100 de PVB = 30% en de la masa depolvo).
12.Agitaciónmagnética.
13.Transvasealbotecontenedordondepermanecerá.Secierraherméticamenteysedejaenlabandejaderodillosparaquelapastanoseapelmace,anoserquevayaserutilizadoinmediatamente,momentohastaelqueseseguiráteniendoenagitaciónmagnética.
A3.4.3PastadecátodorealizadaAcontinuaciónsedetallan las cantidades realesutilizadasen lamezclapara lapastadecátodo, la adecuación de estas con las condiciones teóricas iniciales y la proporción depolvo respecto el total de pasta, con la que se puede estimar más intuitivamente elresultadodeladeposicióndelacapadecátodoporlatécnicadedip‐coating.
ElpolvodeLSOempleadoespertenecientealatercerasíntesisrealizada.Aestepolvoselehanrealizadoentotal12horasdemoliendadebolas,4horasdeatricióny3minutosdeultrasonidosdespuésdelaatrición.LSO 2.2497g %masadelpolvo polvoPNO 8.9961g LSO 20.00% (respectototaldepasta)Beycostat 0.1126g ⇨ PNO 80.00% %enmasa 44.78%etanol 11.018g Beycostat 1.00% %envolumen 14.25%PVB(disolución) 3.4400g PVB 5.10% Seobtieneunvolumentotaldepasta20.0907cm3

Anexo4.Técnicasdeprocesado
‒55‒
Anexo4.Técnicasdeprocesado
A4.1MoliendadebolasLa molienda de bolas es el método más utilizado para lamezcladepolvoscerámicosyparalareduccióndetamañodesuspartículas.Entre sus ventajas se destacan la sencillez y elbajo precio del equipo. No obstante presenta algunosproblemas, el más importante es la contaminación que sepuedeproducirpordesgastedelmediodemolienda(bolas),obienporelrecipienteenelcualseintroduceelmaterialquesevaamoler.
Elmolinoque sehautilizadodurante todoelproyectoesunPlanetary Mono Mill PULVERISETTE 6 classic line de lacompañíaFritsch(figura54).
Las fuerzas centrífugas ocasionadas por las rotaciones, tantodelrecipientedemoliendasobresupropioeje,comodeldiscodelsoportederotación,sonlasqueactúansobrelasbolasdemolienda y el material en el recipiente de molienda.Elrecipiente de molienda y el disco de soporte giran endirecciones opuestas, de manera que las fuerzas centrífugasactúan alternativamente en las mismas y en direccionesopuestas.(verfigura55)
Estodacomoresultado, comounefectode fricción, lasbolasdemoliendaa lo largodelinterior de la pared del recipiente demolienda, y por efecto de impacto, que las bolasimpactencontralaparedopuestadelrecipientedemolienda.
Lamáquinaesaccionadaporunmotordecorrientealternatrifásicosinmantenimiento,queesoperadoconunconvertidordefrecuencia.
Figura55.Dinámicadelasfuerzasqueintervienenenlamoliendadebolas
Figura54.Molinoutilizado

Anexo4.Técnicasdeprocesado
‒56‒
A4.2AtriciónEsunmétodosimilaralamoliendadebolas.Enlugarde servirse de las fuerzas centrífugas del platorotatorio y el giro propio del contenedor de lamolienda para impulsar las bolas, estas sonimpulsadas directamente por unos brazos sujetos aunejerotatorio.(figura56)
Tanto el recipiente, como los brazos rotatorios, sonde polímero para minimizar los efectos de loschoquesentreestosylasbolas.
Enelproyectosehausadoparareducireltamañodegrano a la dimensión más pequeña posible que sepuedaobtenerporunprocesomecánico.
A4.3CompactadoParaobtenercompactossehanutilizadodosmétodos,elprensadouniaxialyelprensadoisostáticoenfrío,utilizandounaprensauniaxialyunaprensaisostáticarespectivamente.Con la prensauniaxial se han obtenidopastillas compactas demezcla de óxidos sólidosparaluegosometerlasaltratamientotérmicodesíntesis,yconlaprensaisostáticasehanobtenidolostubosdeánodoenverde.
A4.3.1PrensadouniaxialEnunaprensauniaxial(figura57)lapresiónseaplicaalpolvo en una sola dirección. Mediante compactaciónuniaxial pueden obtenerse piezas en verde condimensiones y acabados precisos, obteniéndose unaalta productividad en la industria mediante estatécnica. Un inconveniente de la compactación uniaxiales la baja relación longitud/diámetro que puedeobtenerseenlaspiezasdebidoalgradientededensidadque seproduceentreel centrode lapiezay las zonasmás próximas al punzón. Para obtener un compactocon mayor densidad se emplean prensas de dobleémbolo(dobleacción).
A4.3.2Prensadoisostático(CIP)El prensado isostático en frío o CIP (ColdIsostaticPressing)esunmétododecompactaciónqueserealizaencerrando herméticamente el polvo en moldeselásticostípicamentedegoma,látexoPVC,aplicándolespresiónhidrostáticamedianteunfluidoquepuedeseraguaoaceite.Laspiezasenverdeobtenidasporestesistematienenpropiedades uniformes e isótropas. Una de las principales ventajas de este método decompactacióneslaaltarelaciónlongitud/diámetroquepuedeobtenerseenlaspiezasconrespecto a la compactación uniaxial. Es uno de los métodos más utilizados para lacompactacióndepiezascerámicas.
Figura56.Diagramadelprocesodeatrición.
Figura 57. Prensa uniaxialhidraúlica yelementosdeltroquelutilizado.

Anexo4.Técnicasdeprocesado
‒57‒
A4.4SinterizadoDuranteelprocesodecompactaciónseconsiguequeunpolvosueltoadquirieraunaformadeterminada, y que pueda ser manejado sin que el compacto se desmorone. Laconsolidación o densificación de estos compactos en verde aporta una serie depropiedadesfísicasymecánicasque loshaceninteresantesparasuposteriorutilización.Engeneral, alcanzandounaaltadensidadsemejoran laspropiedadesdeunmaterial, espor esta razón que uno de los objetivos durante la sinterización sea, en determinadoscasos (como en el de la capa de electrolito), alcanzar la densidad teórica, es decir, laeliminacióntotaldelaporosidadresidual.
Lasinterizacióneselprocesoporelcualsecreanunionesentrepartículasatemperaturaspor debajo de la temperatura de fusión mediante fenómenos de difusión atómica. Lasinterización lleva asociada normalmente la densificación del compacto, aunque puedeocurrirquelaspartículascreenunionessinqueseproduzcacontracción.
Lafuerzaimpulsoradelasinterizaciónvienedadaporlareduccióndelaenergíalibredelsistema,locualpuedeproducirseporunadisminucióndelacurvaturadelassuperficiesoporlareduccióndelaenergíaasociadaaláreasuperficial,yaseapordisminucióndeéstaoporlasustitucióndesuperficiesólido‐vaporporsuperficiesólido‐sólidodemenorenergía.
ETAPASENLASINTERIZACIÓNENESTADOSÓLIDOElmodelobásicodesinterizaciónenestadosólidopuedeexplicarseapartirdeunmodelogeométricamente sencillo consistente en dos partículas inicialmente esféricas. Existenvariasetapasdentrodelproceso.Aunquenoexisteunaclaradistinciónentrelasdiversasetapasqueocurrendurante la sinterización,puedeaceptarse ladivisiónde lamismaentresetapas.
LaprimeraetapadelasinterizaciónsedefinecomoaquellaqueocurremientrasX/R˂0.3parapolvosnocompactados(Xeseldiámetrode lapartículasyRelradiodelcuellodeuniónqueseformaentreambaspartículas).Lafuerzaconductoraenestaetapaesdebidaa los gradientes de curvatura existentes en los cuellos generados entre las partículas.Duranteestaetapalaporosidadesabiertaytotalmenteinterconectada,aunquelaformapuedenosermuylisa.Enlaetapaintermedia,losporossonmuchomáslisos,tienenunaestructuracilíndricayestáninterconectados.Laatencióncambiadelcrecimientodecuelloentrepartículasalaestructuragranos‐poros.Lafuerzaconductoravieneoriginadaporladisminución en el área superficial y el descenso de la energía superficial libre poreliminacióndelainterfacessólido‐vapor.Laspropiedadesdelproductofinaldependeránen granmanerade esta segunda etapa.Al final de esta segunda etapa suele ocurrir uncrecimiento de grano que puede motivar el movimiento o aislamiento de la porosidadexistente.Durantelasinterizaciónlainteracciónentreporosylímitesdegranospuedeserdedosformas:1.Los poros puede ser arrastrados por elmovimiento de las juntas de grano durante elcrecimientodegrano.2.Las juntasdegranopuedensepararsede losporosdejandoaestosenel interior (losporossemuevenmáslentamentequelasjuntasdegrano).
Cuando la porosidad residual llega a ser aproximadamente un 8% los poros cilíndricoscolapsan en poros esféricos y éstos no son tan efectivos deteniendo el crecimiento degrano. La estructura de poros asociada a las juntas de grano es energéticamente másfavorable,yaqueelpororeduceeláreacorrespondientealajuntadegrano.Noobstante,

Anexo4.Técnicasdeprocesado
‒58‒
bajociertascondicioneslafuerzaconductoradelcrecimientodegranosuperaestafuerzarestrictivadebidaalosporos,conloqueseproducelaseparaciónentreéstosylasjuntasdegrano.Elaislamientodeporosenelinteriordelosgranosoriginaunacaídaimportanteenlavelocidaddedensificación.Lacantidaddeporosidaddecrececoneltiempo,aunqueeltamañodelosporospuedeaumentar.Enestaetapalafuerzaconductoravienedadaporlaeliminacióndelainterfasevapor‐sólido.Lapresenciadeungasinsolubleenelinteriordelporolimitarálacantidaddedensificaciónenestaúltimaetapa.
A4.5Dip‐coatingLa técnicadeldipcoatingseutiliza tantopararecubrirlostubosdeánodoconunacapadepastadeelectrolito,como para recubrir el tubo cosinterizado de ánodo yelectrolitoconunacapadepastadecátodo.Paraello,se ha utilizado un servo electromecánico (figura 58)que, con ayuda de un motor con regulador develocidad,hacedescendery ascender lasplacas en lavertical permitiendo que estas se introduzcan y seextraigandelapastarespectivamente.
Se requiere un control de la viscosidad, carga desólidos y velocidad de inmersión de lamuestra en lapasta para garantizar un espesor del recubrimientocorrecto. Para evitar aglomeraciones y decantacionesen la suspensión es necesaria una agitación vigorosaantesde cada inmersión,pudiendohaceruso si fueranecesariodesondadeultrasonidos.
Figura59.Momentosanterioryposterioralainmersióndedosmuestras.
Figura58. Servoelectromecánico.

Anexo5.Técnicasdecaracterización
‒59‒
Anexo5.Técnicasdecaracterización
A5.1EspectroscopiaRamanLa Espectroscopia Raman es una técnica fotónica de alta resolución que proporcionaenpocossegundosinformaciónquímicayestructuraldecasicualquiermaterialocompuestoorgánicoy/oinorgánicopermitiendoasísuidentificación.Elanálisismedianteestatécnicasebasaenelexamendelaluzdispersadaporunmaterialalincidirsobreélunhazdeluzmonocromático.Unapequeñaporcióndelaluzesdispersadainelásticamente(dispersiónRaman)experimentandoligeroscambiosdefrecuenciaquesoncaracterísticosdelmaterialanalizadoeindependientesdelafrecuenciadelaluzincidente.Setratadeunatécnicadeanálisisqueserealizadirectamentesobreelmaterialaanalizarsinnecesitarésteningúntipodepreparaciónespecialyquenoconllevaningunaalteracióndelasuperficiesobrelaqueserealizaelanálisis,esdecir,esno‐destructiva.
En lasiguientetablaserefleja la informaciónqueproporcionan lascaracterísticasdeunpicoenunamedidadeespectrometríaRaman[11]:
frecuenciascaracterísticas
composicióndelmaterial
cambiosenlafrecuenciadelpico
estadodecargasydeformaciones
polarizacióndelpico simetríayorientacióndelcristal
anchuradelpico calidaddelcristal
intensidaddelpico cantidaddematerial
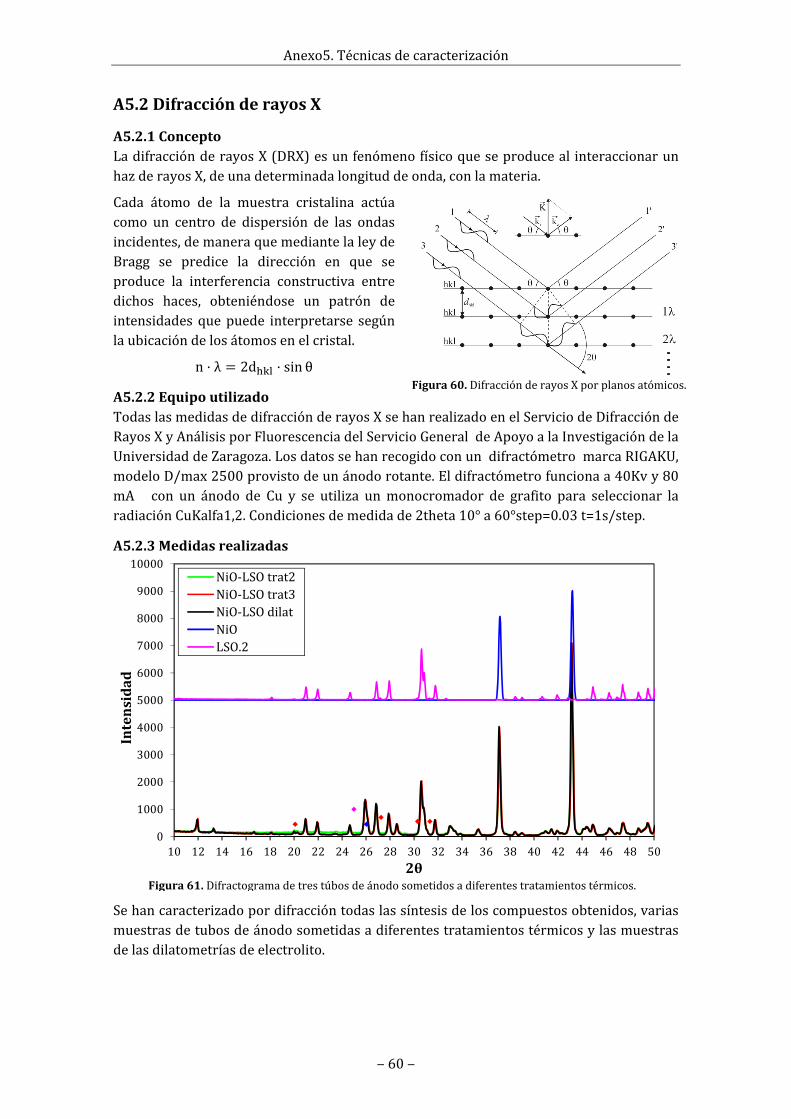
Anexo5.Técnicasdecaracterización
‒60‒
A5.2DifracciónderayosX
A5.2.1ConceptoLadifracciónderayosX(DRX)esunfenómenofísicoqueseproduceal interaccionarunhazderayosX,deunadeterminadalongituddeonda,conlamateria.
Cada átomo de la muestra cristalina actúacomo un centro de dispersión de las ondasincidentes,demaneraquemediantelaleydeBragg se predice la dirección en que seproduce la interferencia constructiva entredichos haces, obteniéndose un patrón deintensidadesquepuede interpretarse segúnlaubicacióndelosátomosenelcristal.
n λ 2d sin θ
A5.2.2EquipoutilizadoTodaslasmedidasdedifracciónderayosXsehanrealizadoenelServiciodeDifraccióndeRayosXyAnálisisporFluorescenciadelServicioGeneraldeApoyoalaInvestigacióndelaUniversidaddeZaragoza.LosdatossehanrecogidoconundifractómetromarcaRIGAKU,modeloD/max2500provistodeunánodorotante.Eldifractómetrofuncionaa40Kvy80mA con un ánodo de Cu y se utiliza unmonocromador de grafito para seleccionar laradiaciónCuKalfa1,2.Condicionesdemedidade2theta10°a60°step=0.03t=1s/step.
A5.2.3Medidasrealizadas
Sehancaracterizadopordifraccióntodaslassíntesisdeloscompuestosobtenidos,variasmuestrasdetubosdeánodosometidasadiferentestratamientostérmicosylasmuestrasdelasdilatometríasdeelectrolito.
Figura61.Difractogramadetrestúbosdeánodosometidosadiferentestratamientostérmicos.
0
1000
2000
3000
4000
5000
6000
7000
8000
9000
10000
10 12 14 16 18 20 22 24 26 28 30 32 34 36 38 40 42 44 46 48 50
Intensidad
2θ
NiO‐LSOtrat2NiO‐LSOtrat3NiO‐LSOdilatNiOLSO.2
Figura60.DifracciónderayosXporplanosatómicos.

Anexo5.Técnicasdecaracterización
‒61‒
A5.3MicroscopíaelectrónicaEstatécnicaestábasadaenlaformacióndeunaimagenapartirdelaproyeccióndeunhazdeelectronesfocalizadossobrelamuestramedianteunsistemaformadoporuncañóndeelectronesyunaseriedelenteselectromagnéticas.
Este haz interactúa con la región de la muestrapróximaalasuperficieysegeneranunaseriedeseñales que pueden ser recogidas mediantedetectores,principalmentetres.
Entre ellas están los electrones dispersadosinelásticamente, o electrones secundarios, queconstituyen una señal que nos proporcionainformación de la morfología de la muestramediante una imagen con gran profundidad decampo. El punto será más brillante cuanto másorientado hacia el detector se encuentre, ya queunafracciónmayordeloselectronesdispersadosincidirásobreeldetector.
Otrapartedeloselectronessufreunadispersiónelástica y son retrodispersados, este fenómenoocurre más cuanto más alto sea el númeroatómico en la región afectada, por tantoobtenemosunaimagenconcontrasteentrezonascondiferentenúmeroatómico.
Porúltimoelhazdeelectronesimpulsaenlosátomossobrelosqueincideunmecanismode excitación/relajación que genera a su vez una emisión de rayos X. Esta emisión escaracterísticadecadaátomo,porloqueestaseñalnosproporcionainformacióndetalladasobrelacomposicióndelamuestra.
El equipo utilizado es un microscopioelectrónico de barrido de emisión de campo(FESEM) Carl Zeiss MERLIN™ cuyascaracterísticassonlassiguientes:
•Cañóndeemisióndeelectronesporemisióndecampodepuntacaliente.•Permite observaciones de hasta 0.8nm deresoluciónespacial.•Voltajesdeaceleraciónentre0.02y30kV.•Detectores de electrones secundarios yretrodispersadosenlacámarayenlacolumna(in‐lens).•DetectorEDSparaAnálisisdelaEnergíadelosRayosXdispersadosINCA350deOxfordInstrumentsconresoluciónenenergíade127eVa5.9KeV.•DetectorEBSD(ElectronBackScatterDiffraction)paraelregistroyanálisisdediagramasdeDifraccióndeelectronesretrodispersadosymapasdeorientacióncristalográfica.•DetectorSTEM.
Figura62.EsquematécnicodeunSEM.
Figura63.Microscopioelectrónicodebarridodeemisióndecampo(FESEM)CarlZeissMERLIN™.

Anexo5.Técnicasdecaracterización
‒62‒
A5.4EspectrometríadeCorrelaciónFotónicaLa espectrometría de correlación fotónica o dispersión dinámica de luz o máscomúnmenteconocidaporDLS(Dynamiclightscattering)esunatécnicaempleadaparaladeterminacióndeladistribucióndetamañosdepartículasensuspensión
ElespectrómetroutilizadoesunZetasizerNano‐ZSdelacompañíaMalvernInstrumentsquepermitedeterminartamañosdepartículaentre0.5y800μm.
Para medir la distribución de tamaño departícula de un polvo cerámico en primerlugar hay que acondicionarlo. El manual delespectrómetro nos indica los rangos devalores de concentración que han de serusadossegúnelrangodetamañoenelqueseencuentran las partículas de la muestra depolvoquevaasercaracterizada.Enelcasodeesteproyecto,entretalesindicacionesyvariasmedidasdepruebasehadeterminadoqueunaconcentración de 0.025g/l podría ser laidónea. Para llegar hasta tal valor deconcentración, el cual es muy pequeño, setienen que realizar varias disoluciones intermedias. El polvo se va a suspender en unmediodispersante compuestopor etanol yun defloculante ésterde fosfato (Beycostat‐C213,CECA,Francia)queseutilizaparaproporcionarlaestabilizaciónelectroestéricadelasuspensión.Estedefloculanteseañadiráenunaproporciónde1%delamasadelpolvo.Después de tener la suspensión realizada se le aplicará agitaciónmagnética, y si fueranecesario, una sesión de ultrasonidos. De esta manera, ya está en condiciones de sermedida.
La suspensiónque sedeseamedir se introduceenunas celdaso cubetasespecialesqueluegoseintroduciránenelaparato.Seajustanlosparámetrosdelprogramaquecontrolael espectrómetro y se lanza la medida. Después, un haz láser de He‐Ne (λ=632.8nm)interaccionaráconlaspartículassuspendidasyseprocederáamensurarlaintensidaddela dispersión dinámica de luz que se produzca. Para partículas por debajo de 0.5μm elequiporealizaladeterminaciónmediantedispersióndiferencialdeluzpolarizada.
Figura64.ZetasizerNano‐Zs,MalvernInstruments

Anexo5.Técnicasdecaracterización
‒63‒
A5.5Dilatometría
A5.5.1ConceptoEn un ensayo de dilatometría se aplica un proceso térmico a unmaterial con el fin demedir la deformación que presenta en función de la temperatura. En esta deformaciónestánincluidostantoel fenómenodecontracciónpor lacoalescenciaentre laspartículascomoeldedilatacióndelmaterial(muypequeñoenestecasoportratarsedecerámicas).
A5.5.2DilatómetroLos ensayos se llevan a cabo en un equipo SetaramSetsys 2000. Consiste en un hornoverticalquedetectaloscambioslinealesenlongitudquesufrelamuestraenfuncióndelatemperatura.
A5.5.3TratamientodedatosDuranteelproceso térmicoque tiene lugar en la cámaradehornodeldilatómetro, esteproporciona computerizadamente los datos de tiempo transcurrido y temperaturadeproceso(T)eincrementodelongitud(ΔL)delamuestra.Elvalorqueinteresaparacadamuestraesladeformación(ε)enfuncióndelatemperatura(ysuvelocidaddevariaciónoderivada respecto de la temperatura), ya que es unamedida normalizada con la que sepuedecomparardemaneracorrectalosresultadosentrelasdistintasmuestrasalasquesehasometidoaunprocesodedilatometría.Losdatospuntualeshansidotratadossegúnlassiguientesfórmulas:
ε TΔL TL
dε TdT
≃Δε TΔT
⇒dε TdT
≃ε T ε T
T T
Figura65.Equipoutilizadoyesquemainternodelacámaradeldilatómetro.
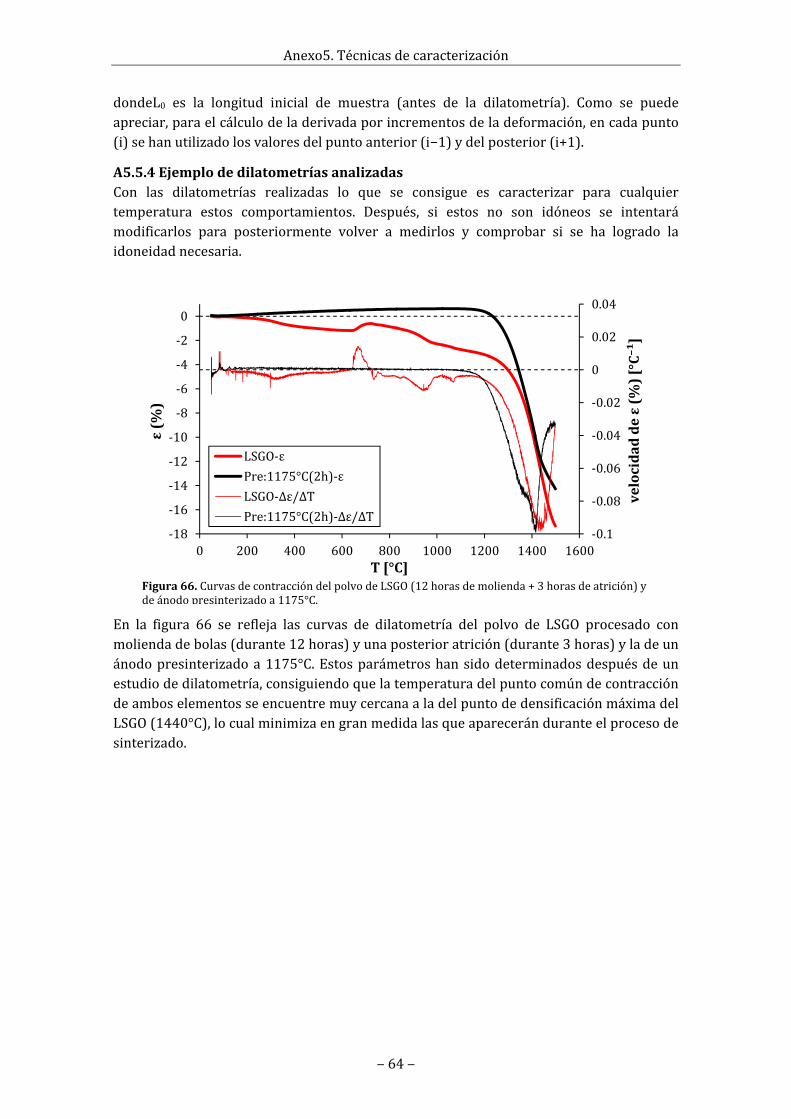
Anexo5.Técnicasdecaracterización
‒64‒
dondeL0 es la longitud inicial de muestra (antes de la dilatometría). Como se puedeapreciar,paraelcálculodeladerivadaporincrementosdeladeformación,encadapunto(i)sehanutilizadolosvaloresdelpuntoanterior(i−1)ydelposterior(i+1).
A5.5.4EjemplodedilatometríasanalizadasCon las dilatometrías realizadas lo que se consigue es caracterizar para cualquiertemperatura estos comportamientos. Después, si estos no son idóneos se intentarámodificarlos para posteriormente volver a medirlos y comprobar si se ha logrado laidoneidadnecesaria.
En la figura 66 se refleja las curvas de dilatometría del polvo de LSGO procesado conmoliendadebolas(durante12horas)yunaposterioratrición(durante3horas)yladeunánodopresinterizadoa1175°C.Estosparámetroshansidodeterminadosdespuésdeunestudiodedilatometría,consiguiendoquelatemperaturadelpuntocomúndecontraccióndeamboselementosseencuentremuycercanaaladelpuntodedensificaciónmáximadelLSGO(1440°C),locualminimizaengranmedidalasqueapareceránduranteelprocesodesinterizado.
Figura66.CurvasdecontraccióndelpolvodeLSGO(12horasdemolienda+3horasdeatrición)ydeánodopresinterizadoa1175°C.
‐0.1
‐0.08
‐0.06
‐0.04
‐0.02
0
0.02
0.04
‐18
‐16
‐14
‐12
‐10
‐8
‐6
‐4
‐2
0
0 200 400 600 800 1000 1200 1400 1600
velocidaddeε(%
)[°C−¹]
ε(%
)
T[°C]
LSGO‐ε
Pre:1175°C(2h)‐ε
LSGO‐Δε/ΔT
Pre:1175°C(2h)‐Δε/ΔT

Anexo5.Técnicasdecaracterización
‒65‒
A5.7Reometría
A5.7.1Reología.Conceptoyaplicaciónenelreómetro.Lareologíaeslapartedelafísicaqueestudialarelaciónentreelesfuerzoyladeformaciónen losmaterialesquesoncapacesde fluirsiendopor tantounapartede lamecánicademedioscontinuos.Laspropiedadesmecánicasestudiadasporlareologíasepuedenmedirmediante reómetros, aparatos que permiten someter al fluido a diferentes tipos dedeformacionescontroladasymedirlosesfuerzosoviceversa.Elreómetro,portanto,nosvaapermitirmedirlaformaenquefluyeunlíquido,mezclaosuspensiónbajolaaccióndefuerzasexternas. Seempleapara fluidosquenopuedendefinirse conunúnicovalordeviscosidad y por tanto requieren más parámetros que los que puede proporcionar unviscosímetro.Estostiposdefluidosepuedencaraterizarconunmodelomatemáticosegúnlasmedidasdelreómetro.Algunostiposson:
Fluidosnewtonianos τ μ γPlásticodeBingham τ τ μ γOstwald‐de‐Waele τ K γHerschel‐Bulkley τ τ K γ
En lamedida, el reómetro proporciona unmomento (M) que establece un giro relativoentredoscilindrosconcéntricos,elcualproduceunesfuerzodecizalla(τ)sobreelfluidocontenidoenelplatoportamuestras.Ellíquidosedeformaunánguloγ:
FMR
⟹ τFA⟹ γ
Porsimetríacilíndricaladeformaciónsolamentedependedelacoordenadar,conloquelavelocidad de deformación y el gradiente de velocidades, creado entre las placasconcéntricas,esequivalente:
γdγdt
du rdr
Cuando el esfuerzo de cizalla es linealmente dependiente del gradiente de velocidad sedice que el fluido es newtoniano (sólo depende de la temperatura). La constante deproporcionalidadeselcoeficientedeviscosidadμ,queindicalaresistenciaalflujodebidoalasfriccionesinternasentrelasmoléculasdellíquido:
τ μ γ
Figura67.Deformacióndeunlíquidosometidoaunesfuerzocortanteenelreómetro.
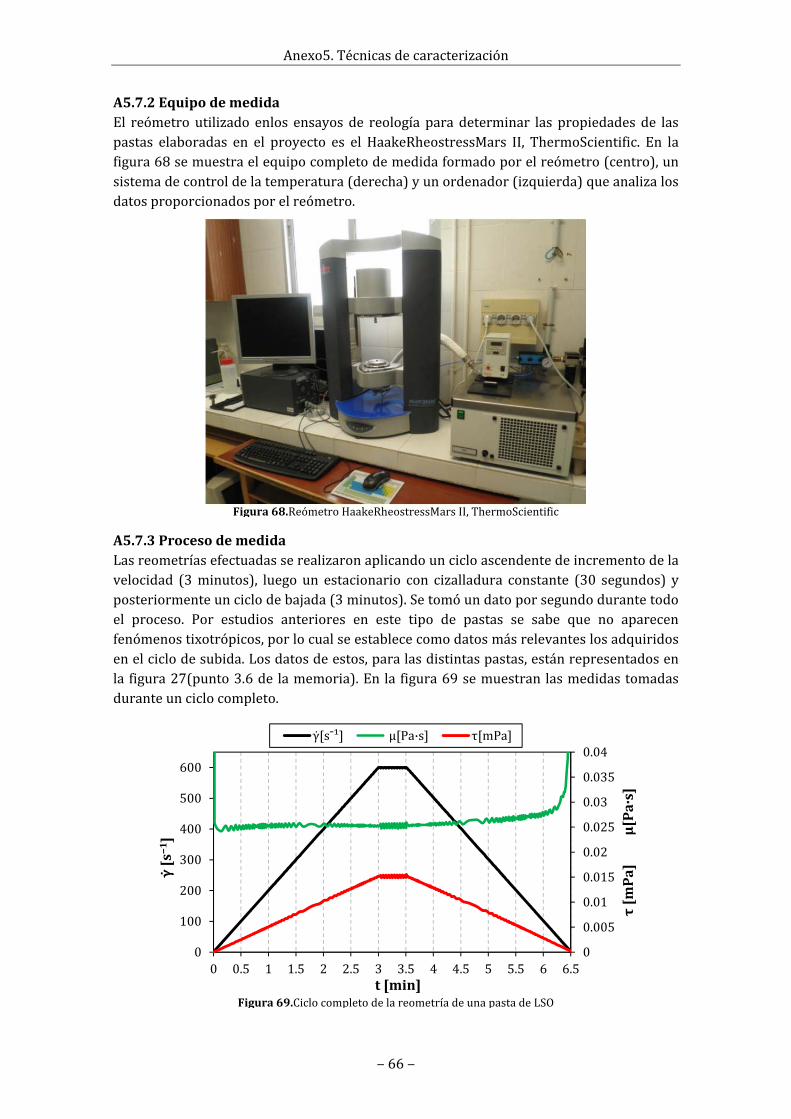
Anexo5.Técnicasdecaracterización
‒66‒
A5.7.2EquipodemedidaEl reómetroutilizado enlos ensayos de reología para determinar las propiedades de laspastas elaboradas en el proyecto es el HaakeRheostressMars II, ThermoScientific. En lafigura68semuestraelequipocompletodemedidaformadoporelreómetro(centro),unsistemadecontroldelatemperatura(derecha)yunordenador(izquierda)queanalizalosdatosproporcionadosporelreómetro.
A5.7.3ProcesodemedidaLasreometríasefectuadasserealizaronaplicandouncicloascendentedeincrementodelavelocidad (3minutos), luego un estacionario con cizalladura constante (30 segundos) yposteriormenteunciclodebajada(3minutos).Setomóundatoporsegundodurantetodoel proceso. Por estudios anteriores en este tipo de pastas se sabe que no aparecenfenómenostixotrópicos,porlocualseestablececomodatosmásrelevanteslosadquiridosenelciclodesubida.Losdatosdeestos,paralasdistintaspastas,estánrepresentadosenla figura27(punto3.6de lamemoria).En la figura69semuestran lasmedidastomadasduranteunciclocompleto.
Figura68.ReómetroHaakeRheostressMarsII,ThermoScientific
Figura69.CiclocompletodelareometríadeunapastadeLSO
0
0.005
0.01
0.015
0.02
0.025
0.03
0.035
0.04
0
100
200
300
400
500
600
0 0.5 1 1.5 2 2.5 3 3.5 4 4.5 5 5.5 6 6.5
τ[mPa]μ[Pa·s]
γ̇[s−1]
t[min]
γ̇[s¯¹] μ[Pa·s] τ[mPa]
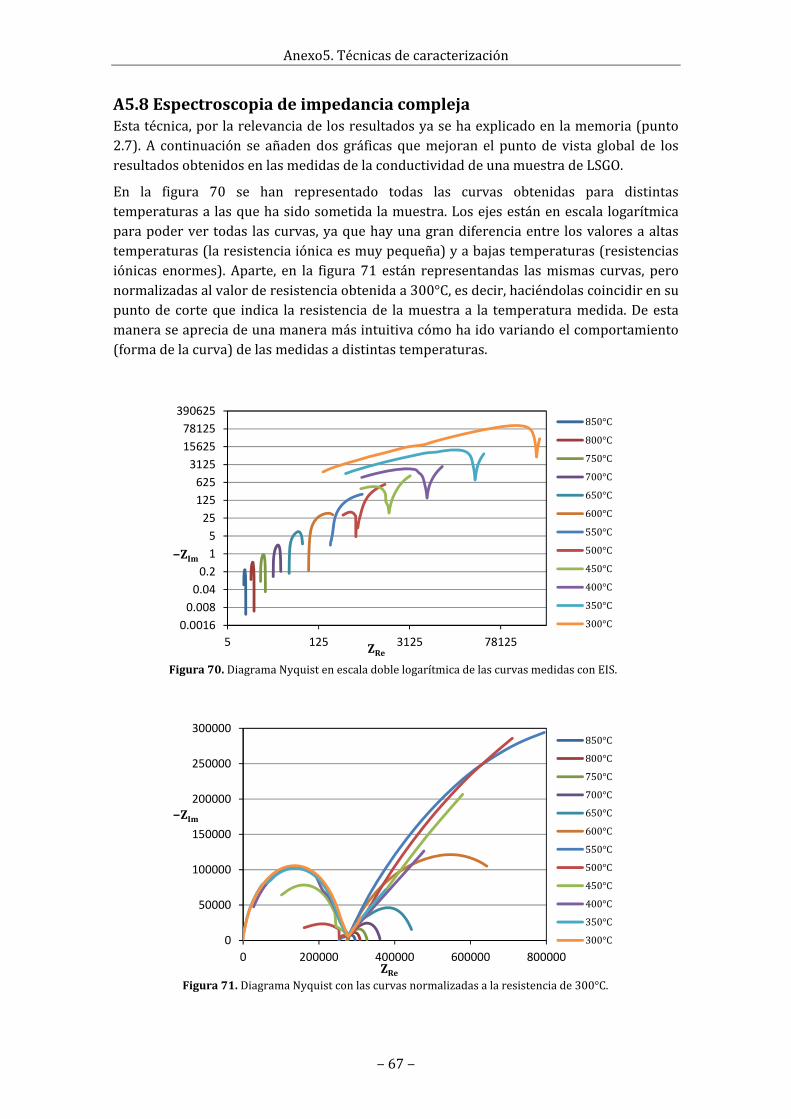
Anexo5.Técnicasdecaracterización
‒67‒
A5.8EspectroscopiadeimpedanciacomplejaEstatécnica,porlarelevanciadelosresultadosyasehaexplicadoenlamemoria(punto2.7).A continuación se añadendos gráficasquemejoran el puntode vista global de losresultadosobtenidosenlasmedidasdelaconductividaddeunamuestradeLSGO.
En la figura 70 se han representado todas las curvas obtenidas para distintastemperaturasalasquehasidosometidalamuestra.Losejesestánenescalalogarítmicaparapodervertodas lascurvas,yaquehayunagrandiferenciaentre losvaloresaaltastemperaturas(laresistenciaiónicaesmuypequeña)yabajastemperaturas(resistenciasiónicas enormes). Aparte, en la figura 71 están representandas lasmismas curvas, peronormalizadasalvalorderesistenciaobtenidaa300°C,esdecir,haciéndolascoincidirensupuntodecorteque indica la resistenciade lamuestraa la temperaturamedida.Deestamaneraseapreciadeunamaneramásintuitivacómohaidovariandoelcomportamiento(formadelacurva)delasmedidasadistintastemperaturas.
Figura70.DiagramaNyquistenescaladoblelogarítmicadelascurvasmedidasconEIS.
0.0016
0.008
0.04
0.2
1
5
25
125
625
3125
15625
78125
390625
5 125 3125 78125
−ZIm
ZRe
850°C
800°C
750°C
700°C
650°C
600°C
550°C
500°C
450°C
400°C
350°C
300°C
Figura71.DiagramaNyquistconlascurvasnormalizadasalaresistenciade300°C.
0
50000
100000
150000
200000
250000
300000
0 200000 400000 600000 800000
−ZIm
ZRe
850°C
800°C
750°C
700°C
650°C
600°C
550°C
500°C
450°C
400°C
350°C
300°C