presentación1.pptx
TRANSCRIPT

• Yesika lizeth Ochoa
• II semestre • Enfermería

Síndrome de alport

Síndrome de alport
• El síndrome de Alport COL4A5 base de datos de variante.
Síndrome de Alport es una enfermedad renal progresiva con cocleares y afectación ocular. La forma más común (aproximadamente el 80%) se hereda en un patrón X-ligada. Alport ligado al cromosoma X Síndrome (XLAS) es causada por mutaciones en la cadena alfa del colágeno tipo IV 5 (COL4A5). Hemos desarrollado una base de datos específica de la enfermedad curada contengan informó variantes de secuencia en COL4A5.

• Actualmente la base de datos de archivos de un total de 520 variantes de la secuencia, para verificar su posición en el gen COL4A5 y nombrado siguiendo la nomenclatura estándar. variantes de secuencia se comunicarán con la información adjunta sobre el efecto de la proteína, la clasificación de mutación vs polimorfismo, tipo de mutación sobre la base de la primera descripción en la literatura, y enlaces a las publicaciones pertinentes.

Definición y estudio
• El síndrome de Alport es una enfermedad de tipo hereditario del colágeno IV conduce a la fibrosis renal progresiva, pérdida de audición y cambios oculares. insuficiencia renal terminal se desarrolla generalmente durante la adolescencia. COL4A3-/ - ratones sirven como modelo animal para la cicatrización renal progresiva en el síndrome de Alport. El presente estudio evalúa el papel de Discoidin dominio del receptor 1 (DDR1) en la interacción célula-matriz implicados en la patogénesis del síndrome de Alport, incluyendo la inflamación y la fibrosis renal

. DDR1/COL4A3 doble knock-outs fueron comparados con COL4A3-/ - ratones con 50% o 100% expresión de DDR1, tipo salvaje controles y DDR1-/ - + COL4A3 / controles por más de 6 años +. Haga doble nocauts vivió 47% más de tiempo, los ratones con un 50% DDR1 vivió 29% más tiempo y mostraron una mejoría de la función renal (reducción de la proteinuria y el nitrógeno ureico en sangre) en comparación con los animales con un 100% DDR1 expresión. La pérdida de células proinflamatorias DDR1 reducida, a través de la señalización de profibróticos TGFbeta, CTGF, NFkappaB e IL-6 y la disminución de la deposición de matriz extracelular.

Esto apoya nuestra hipótesis de que la interacción podocito de matriz a través de receptores de colágeno desempeña un papel importante en la PROGREION de la fibrosis renal en la enfermedad de Alport. El bloqueo de los receptores de colágeno DDR1 podría servir como un importante nuevo concepto terapéutico en enfermedades progresivas e inflamatorias fibróticas en el futuro. Copyright (c) 2010 Elsevier BV Todos los derechos reservados.

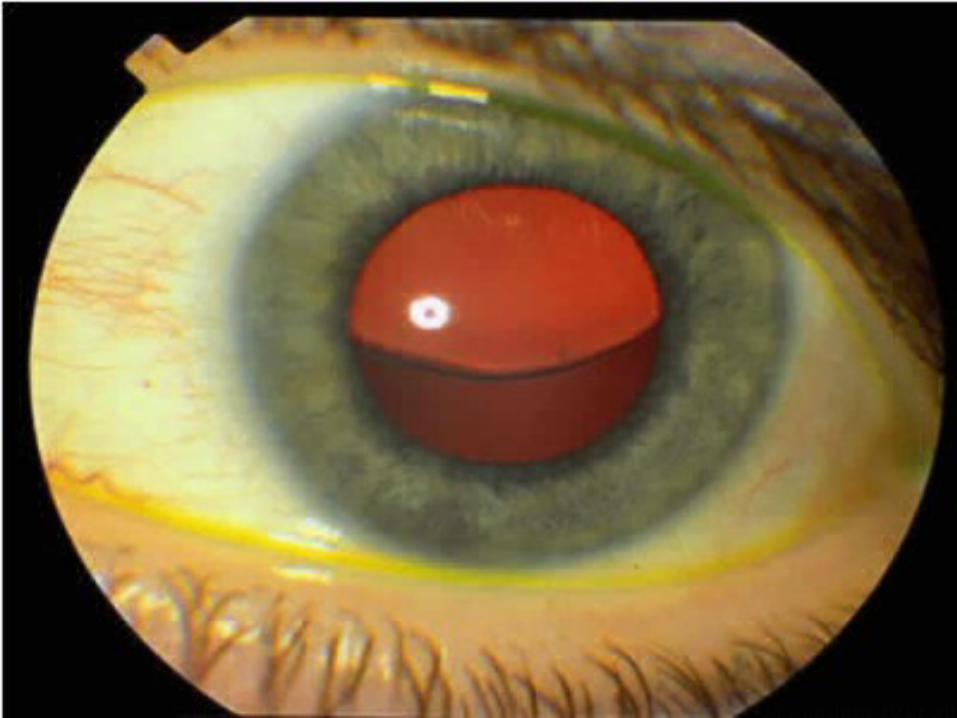
Pacientes y métodos:aberrometría Tracey frente de onda se utilizó para tratar a un paciente con lenticono bilaterales anteriores que tenían una historia de síndrome de Alport. Para el examen microscópico electrónico de transmisión, cápsulas anterior objetivo se obtuvieron durante la facoemulsificación del cristalino transparente e implante de lente intraocular. RESULTADOS: las aberraciones esféricas fueron predominantes las aberraciones de alto orden en los elementos ópticos internos de ambos ojos. El frente de onda aberrómetro Tracey mostró que la mayoría de los astigmatismos irregulares proceden de la parte lenticular. microscopía electrónica de transmisión de las muestras mostraron cápsulas anterior lente con menor grosor y dehiscencias múltiples.

CONCLUSIÓN: aberrometría Tracey frente de onda y de microscopía electrónica de transmisión son herramientas eficaces para la evaluación de lenticono anterior en el síndrome de Alport.

ANOMALÍAS AÓRTICA EN LOS VARONES CON SÍNDROME DE ALPORT.
ANTECEDENTES: Se han producido casos aislados de la enfermedad arterial en varones con síndrome de Alport (AS), una enfermedad sistémica del colágeno tipo IV. En este trabajo se describen cinco casos nuevos de AS significativos asociados con la enfermedad aórtica incluyendo la disección y aneurisma.

MÉTODOS: Se presentan breves descripciones clínicas de cinco varones con EA y la enfermedad aórtica. Se realizó el análisis inmunohistoquímico de la expresión de la cadena alpha5 de colágeno tipo IV en las membranas basales de piel de una familia, notificadas previamente con AS y las enfermedades de la aorta y en la media de la aorta de ratones machos con síndrome de Alport ligado al cromosoma X (XLAS) debido a una mutación sin sentido en el gen COL4A5.

RESULTADOS: Tres de los cinco pacientes mostraron un aneurisma y disección de la aorta torácica, que ocurren en 25-32 años de edad, mientras que uno había dilatación aórtica y otro tenía una insuficiencia aórtica. Los cinco hombres necesitaron tratamiento sustitutivo renal a los 20 años. La inmunohistoquímica de muestras de biopsia de piel en hermanos varones se informó anteriormente con enfermedad aórtica confirmaron que habían XLAS. Tenemos además que la cadena alpha5 de colágeno tipo IV es anormalmente ausente de media de la aorta de ratones transgénicos con XLAS.

CONCLUSIONES: La primera aparición de la enfermedad aórtica puede ser una característica inusual de la EA. Proyección de los hombres con AS para las anomalías del aórtica puede ser clínicamente indicado en algunas familias.

GENETICA
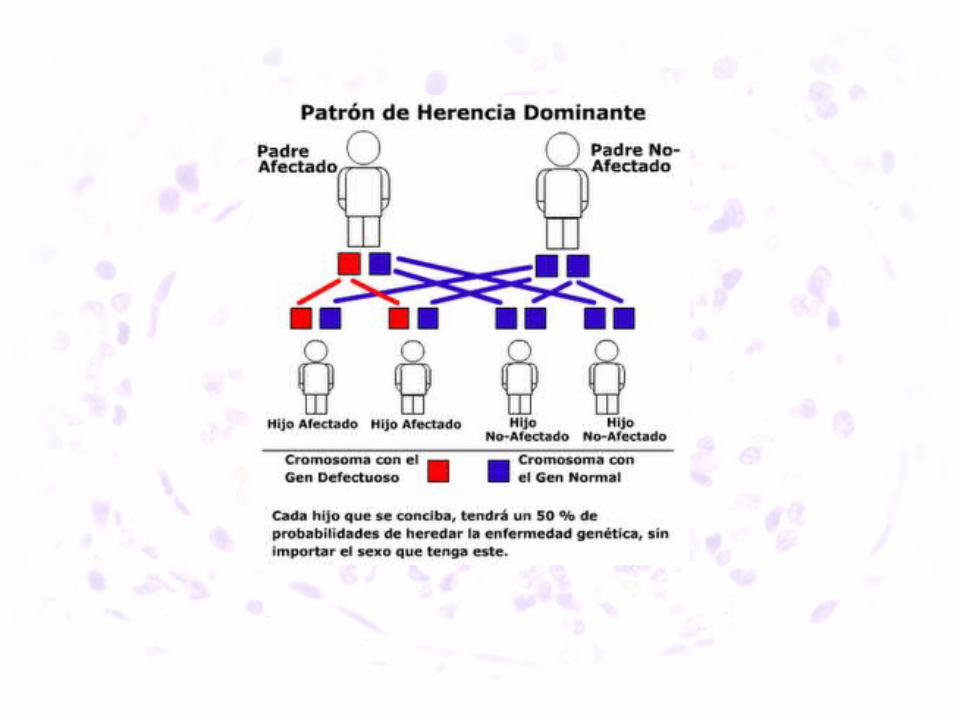
• Mutaciones en los genes que codifican las cadenas del colágeno IV.
• - FORMA CLASICA (80 %), ligada a X: gen COL4A5 en Xq22
• - Autosómicas (D/R): genes COL4A3 y COL4A4 en 2q34.

CASO CLINICO
• ANTECEDENTES: Se han producido casos aislados de la enfermedad arterial en varones con síndrome de Alport (AS), una enfermedad sistémica del colágeno tipo IV. En este trabajo se describen cinco casos nuevos de AS significativos asociados con la enfermedad aórtica incluyendo la disección y aneurisma. MÉTODOS: Se presentan breves descripciones clínicas de cinco varones con EA y la enfermedad aórtica. Se realizó el análisis inmunohistoquímico de la expresión de la cadena alpha5 de colágeno tipo IV en las membranas basales de piel de una familia, notificadas previamente con AS y las enfermedades de la aorta y en la media de la aorta de ratones machos con síndrome de Alport ligado al cromosoma X (XLAS) debido a una mutación sin sentido en el gen <en algunas familias.

• . RESULTADOS: Tres de los cZinco pacientes mostraron un aneurisma y disección de la aorta torácica, que ocurren en 25-32 años de edad, mientras que uno había dilatación aórtica y otro tenía una insuficiencia aórtica. Los cinco hombres necesitaron tratamiento sustitutivo renal a los 20 años. La inmunohistoquímica de muestras de biopsia de piel en hermanos varones se informó anteriormente con enfermedad aórtica confirmaron que habían XLAS. Tenemos además que la cadena alpha5 de colágeno tipo IV es anormalmente ausente de media de la aorta de ratones transgénicos con XLAS. CONCLUSIONES: La primera aparición de la enfermedad aórtica puede ser una característica inusual de la EA. Proyección de los hombres con AS para las anomalías del aórtica puede ser clínicamente

SINDROME DE ALPORT CLINICO
• El curso de la afectación renal y pérdida de audición es mucho más suave en los síndromes de Alport mayoría de las mujeres ligado al cromosoma X que en los pacientes masculinos.

FENOTIPO-GENOTIPO• aquellos con mutaciones del sitio de empalLas mutaciones en el
gen del síndrome de Alport COL4A5 causa ligada al cromosoma X (XLAS). Comprender la correlación entre las manifestaciones clínicas y las mutaciones que subyacen agrega valor pronóstico de la prueba genética, que es cada vez más disponibles. Nuestro objetivo fue determinar la asociación entre el genotipo y el fenotipo en 681 participantes masculinos afectados con XLAS de 175 familias de los EE.UU.. La pérdida de audición y cambios oculares estaban presentes en el 67 y el 30% de los participantes, respectivamente. El promedio de edad de los participantes al inicio de la enfermedad renal terminal fue de 37 años para aquellos con mutaciones sin sentido, 28 años para me, y 25 años para aquellos con mutaciones truncar (P <0,0001).

• Hemos demostrado una fuerte relación entre la posición de la mutación y la edad de inicio de la enfermedad renal terminal, con una menor edad de inicio de la enfermedad renal terminal asociado a mutaciones en el extremo 5 'del gen (índice de riesgo 0,766 [95% intervalo de confianza 0,694 a 0,846] por cada 1000 pb hacia el extremo 3 ', p <0,0001). participantes a los que afecta con mutaciones de empalme o truncar las mutaciones cada uno tenía probabilidades de dos veces mayor de desarrollar problemas en los ojos que aquellos con mutaciones sin sentido, el desarrollo de la discapacidad auditiva mostró una tendencia similar. La pérdida de audición y cambios oculares asociadas con mutaciones situada más cerca del 5; final del gen. Estos fuertes correlaciones genotipo-fenotipo podría ayudar en la evaluación y el asesoramiento de las familias de EE.UU. con XLAS.

SINDROME DE ALPORT





TRATAMIENTO
• Trasplante de riñón

http://www.ncbi.nlm.nih.gov/pubmed/20574986
http://www.ncbi.nlm.nih.gov/pubmed/20494893
http://www.ncbi.nlm.nih.gov/pubmed/20386926
http://www.ncbi.nlm.nih.gov/pubmed/20307660
BLIBIOGRAFIA
http://www.ncbi.nlm.nih.gov/pubmed/20594039