modificación y síntesis de polímeros de isopreno vía ... · tesis para obtener el grado de...
TRANSCRIPT

Tesis para obtener el grado de
Doctor en Tecnología de Polímeros
“Modificación y Síntesis de Polímeros de
Isopreno vía Polimerización Radicálica
Controlada”
Por: M. C. David Contreras López
Asesor:
Dr. Enrique Saldívar Guerra
Dr. Octavio Manero Brito
Saltillo, Coahuila Septiembre 2014




II
Agradecimientos
Al Centro de Investigación en Química Aplicada (CIQA) por las facilidades de ser becado por la institución para lograr la elaboración y culminación de este proyecto de doctorado, aunado por el soporte técnico que me brindo durante la realización de esta Tesis.
Al Dr. Enrique Saldívar Guerra por la confianza depositada en mí desde que le solicite nuevamente ser parte de su equipo de trabajo. Su apoyo académico y moral fueron y han sido valiosos hacia mi persona que los tendré en cuenta como un aprendizaje constante. Es un orgullo haberlo conocido y tenerlo por enésima ocasión como mi mentor.
Al Dr. Octavio Manero Brito, por su amistad y apoyo durante mi desarrollo doctoral. En particular por participar como coasesor de esta tesis. Gracias por permitirme entrar de nuevo a su grupo de trabajo y la confianza depositada en mi persona.
Al Dr. Robin Hutchinson y al Dr. Scott Parent por darme la oportunidad de colaborar dentro de sus grupos de trabajo en la universidad de Queen en Kingston, Canadá. Al Dr. Parent, por compartir sus conocimientos en el área de bioreactores y la aplicación de los hidrogeles, un campo que bien se puede aplicar en el país. Al Dr. Hutchinson y en especial a Jan Schier por apoyarme en la realización de los experimentos en PLP.
A mis sinodales Dra. Catalina Pérez, Dra. Martha Albores, Dr. Gregorio Cadenas y Dr.
Yibran Perera por sus acertados comentarios y discusiones, los cuales enriquecieron el desarrollo de este proyecto de doctorado.
Al Dr. Francisco Ramos por compartir algunos aspectos de su experiencia profesional y discusiones sobre algunos resultados de esta tesis.
Al Dr. Ernesto Elizalde por confiar en mí e interceder para que obtuviera la beca de CIQA. Sin ese apoyo, difícilmente hubiera logrado este logro académico.
Al personal del CIQA, en particular a Patricia Siller y José Luis por su apoyo en las búsquedas bibliográficas. A Rosario Rangel por su apoyo en la capacitación recibida para el uso del equipo de IR, al Dr. Román Torres por su apoyo en el equipo de RMN. En especial a Ricardo
Mendoza y Almendra Ordaz por su paciencia y apoyo en planta 2, sin ustedes este trabajo de doctorado se hubiera complicado. Mi estimado Richard, muchas gracias por brindarme tu amistad y por compartir experiencias personales, créeme, extrañare esos momentos.
A mis amigos y compañeros Edgar, Sergio (a los dos), Esperanza, Cristina (a las dos), Ángel, Nidia, Claude, Omar, Olga, Quique y a los que me faltaron, gracias por dedicar parte de su tiempo haciendo más ameno el trabajo.

III
Dedicatorias
Esta tesis de doctorado se la dedico especialmente a mi suegra Sra. Jovita Nativitas
García (QEPD). Una excelente persona que tuve el honor y gusto de conocerla. La extrañamos mucho pero siempre la vemos presente en su nieto.
A mi madre Sra. María de la Luz López Frontana, por ser mi gran ejemplo y maestra de la vida. Gracias por enseñarme como alcanzar los objetivos que me trazo en la vida. Te quiero mucho y aunque lejos andamos, siempre estás en mi corazón.
A mi bella esposa Martha, por que siempre estás en todo momento al pendiente, por disfrutar cada instante bueno y malo, por creer en mí, por hacer mágico cada momento, por todo eso y más… gracias vida mía. Te amo y es para toda la vida.
A quien en los momentos más oscuros que vivimos hace poco, llegaste como un bello haz de luz a darle alegría a nuestros corazones embriagados por la tristeza, por que tu alegría nos invita a recordar que la vida es para disfrutarse, a ti mi bello niño te dedico este esfuerzo. A David Tadeo.
A Tino, Cristina, Alina y Peque… saben lo que siento por ustedes.
Al Dr. Carlos Rius Alonso; Dr. Carlos, sigo en pie de lucha, su ejemplo me sigue y no me detendré, gracias por todo.
"En la vida terminamos siendo los libros que leemos y los amigos de los que nos rodeamos."
Ikram Antaki (Damasco 1948 – Mexico, DF 2000)

IV
Índice General
Agradecimientos ................................................................................................................................. II
Dedicatorias ...................................................................................................................................... III
Índice General ........................................................................................................................................ IV
Acrónimos ......................................................................................................................................... IX
Lista de reactivos .............................................................................................................................. XI
Lista de disolventes ......................................................................................................................... XII
Índice de figuras .............................................................................................................................XIII
Índice de tablas .............................................................................................................................. XVI
Índice de esquemas ....................................................................................................................... XVII
Resumen ......................................................................................................................................... XIX
Capítulo 1: Enfoque de la Tesis ........................................................................................................... 1
1.1. Hipótesis .................................................................................................................................. 1
1.2. Justificación ............................................................................................................................ 3
1.3. Objetivo general ..................................................................................................................... 4
1.4. Objetivos particulares ............................................................................................................ 4
Capítulo 2: Antecedentes ...................................................................................................................... 6
2.1 Polimerización por radicales libres ............................................................................................ 8

V
2.2 Polimerización controlada-viviente por radicales libres (CRP). ............................................ 11
2.2.1 Fundamentos de la CRP. Consideraciones generales .......................................................... 12
2.2.2 Polimerización radicálica por transferencia de átomo (ATRP) .......................................... 16
2.2.3 Polimerización radicálica controlada por radicales libres estables (SFRP)-Polimerización
radicálica mediada por nitróxidos (NMP) ..................................................................................... 18
2.2.4 Polimerización radicálica por transferencia de cadena de adición-fragmentación
reversible (RAFT) ............................................................................................................................ 23
2.2.5 Comparación de los métodos más representativos de la CRP ............................................ 30
2.3 Síntesis de copolímeros de tipo injerto ..................................................................................... 30
2.3.1 Mecanismo del proceso de injerto .......................................................................................... 33
2.4 Uso de compatibilizantes en mezclas de polímeros inmiscibles .............................................. 38
2.5 Índices de reactividad ................................................................................................................ 42
2.6 Método de PLP-SEC .................................................................................................................. 49
2.7 Polimerización de 1,3-dienos. Isopreno .................................................................................... 53
Capítulo 3: Parte Experimental .......................................................................................................... 56
3.1 Materiales y reactivos ................................................................................................................ 57
3.2 Purificación de monómeros y polímeros .................................................................................. 57
3.3 Técnicas instrumentales de análisis y métodos de caracterización. ....................................... 57
3.3.1 Resonancia Magnética Nuclear (RMN) ................................................................................. 57
3.3.2 Espectroscopia infrarroja con transformada de Fourier (FT-IR) ...................................... 58
3.3.3 Cromatografía de Permeación en Gel (GPC) ....................................................................... 58
3.3.4 Calorimetría Diferencial de Barrido (DSC) .......................................................................... 59
3.3.5 Análisis mecánico diferencial (DMA) .................................................................................... 59
3.4 Procedimiento general de la cinética y copolimerización del IP/MAH. Índices de
reactividad ........................................................................................................................................ 59

VI
3.5 Procedimiento general de la cinética y copolimerización del isopreno del IP/GMA. Índices
de reactividad ................................................................................................................................... 61
3.6 Procedimiento general de la cinética de homo y copolimerización del isopreno.
Polimerización mediada por nitróxidos (NMP). Proceso bimolecular ........................................ 62
3.7 Procedimiento general de la cinética de la homo y copolimerización del isopreno.
Polimerización RAFT ...................................................................................................................... 63
3.8 Procedimiento general de la cinética de la homo y copolimerización del isopreno por NMP.
Proceso unimolecular ....................................................................................................................... 64
3.9 Purificación del poliisopreno (PIP) ........................................................................................... 64
3.10 Funcionalización del PIP con el nitróxido .............................................................................. 64
3.11 Reacción del proceso de injerto de IP/GMA en el PIP funcionalizado................................ 65
3.12 Reacción de la homo y copolimerización del isopreno por PLP-SEC ................................. 65
Capítulo 4: Resultados y discusión. Índices de reactividad. ............................................................ 67
4.1 Copolímero de isopreno-metacrilato de glicidilo (IP/GMA). Consideraciones generales ... 67
4.2 Análisis espectroscópico ............................................................................................................. 68
4.3 Determinación de los índices de reactividad del sistema IP/GMA ........................................ 70
4.3.1 Método de Finemann-Ross (FR) y Finemann-Ross Invertida (FRI) .................................. 73
4.3.2 Método de Kelen-Tüdos (KT) y Kelen-Tüdos extendido (KTE) ......................................... 75
4.3.3 Método de Tidwell-Mortimer (TM) ....................................................................................... 78
4.4 Propiedades térmicas del copolímero de IP/GMA .................................................................. 83
4.5 Copolímero de isopreno-anhídrido maleico (IP-MAH). Consideraciones generales ........... 85
4.6 Análisis espectroscópico ............................................................................................................. 86
4.7 Determinación de los índices de reactividad para el sistema IP/MAH. ................................. 89
4.8 Uso de los métodos de cálculo para la determinación de los índices de reactividad ............ 90
4.9 Propiedades térmicas del copolímero IP/MAH ....................................................................... 94

VII
4.10 Determinación de la kp del isopreno por PLP-SEC ............................................................... 95
Capítulo 5: Resultados y discusión. Homo y copolimerización del isopreno mediada por
nitróxidos ............................................................................................................................................... 99
5.1 Cinéticas de isopreno-metacrilato de glicidilo (IP-GMA) en solución .................................. 99
5.2 Cálculos semiempíricos (AM1-RHF) ...................................................................................... 102
5.3 Efecto de la concentración del nitróxido ................................................................................ 105
5.4 Efecto del disolvente en el medio de reacción. ....................................................................... 106
5.5 Reacciones de copolimerización IP-GMA .............................................................................. 107
5.6 Uso de aditivos en la NMP. Incremento en la velocidad de polimerización ........................ 110
5.7 Crecimiento de la cadena polimérica (carácter viviente). Uso del alcoxiaminas ................ 114
5.8 Análisis espectroscópico ........................................................................................................... 118
Capítulo 6: Resultados y discusión. Homo y copolimerización del isopreno por el proceso RAFT
.............................................................................................................................................................. 121
6.1 Cinéticas de isopreno-metacrilato de glicidilo (IP-GMA) en solución ................................ 121
6.2 Cálculos semiempíricos (AM1-RHF) ...................................................................................... 124
6.3 Reacciones de homopolimerización del isopreno .................................................................. 128
6.4 Reacciones de copolimerización IP-GMA .............................................................................. 129
6.5 Crecimiento de la cadena polimérica (carácter viviente del proceso RAFT) ..................... 132
6.6 Análisis espectroscópico ........................................................................................................... 135

VIII
Capítulo 7: Resultados y discusión. Copolimerización de injerto del isopreno mediada por
nitróxidos ............................................................................................................................................. 138
7.1 Copolimerización de injerto del isopreno .............................................................................. 138
7.2 Análisis espectroscópico ........................................................................................................... 140
7.3 Propuesta mecanística de la incorporación del radical nitróxido ........................................ 144
7.4 Análisis por cromatografía de permeación en gel (GPC) ..................................................... 149
Capítulo 8: Conclusiones y trabajo a futuro .................................................................................... 151
8.1 Utilización de los copolímeros de IP sintetizados. ................................................................. 151
8.2 Conclusiones. ............................................................................................................................ 152
8.3 Trabajo a futuro ....................................................................................................................... 155
8.4 Logros académicos ................................................................................................................... 156
Referencias .......................................................................................................................................... 158

IX
Acrónimos
% w/w Porcentaje en peso 13C RMN Resonancia magnética nuclear de carbono 1H RMN Resonancia magnética nuclear de protón
AA Anhídrido acético
ABS Terpolímero de acrilonitrilo-butadieno-estireno
Ac-d6 Acetona deuterada
AIBN Azo-bis-isobutironitrilo
AN Acrilonitrilo
ATRP Polimerización radicálica por transferencia de átomo
BCTC Bis(carboximetil) tritiocarbonato
BPO Peróxido de benzoilo
CDCl3 Cloroformo deuterado
CDSPA Ácido 4-ciano-4-[(dodecilsulfaniltiocarbonil)sulfanil) pentanoico
CM Concentración del monómero
CRP Polimerización por radicales libres controlada-viviente
CSA Ácido canforsulfónico
CTA Agente de transferencia de cadena
ᴆ Dispersidad (de la distribución de los pesos moleculares, ᴆ = Mw/Mn)
DMA Análisis mecánico diferencial
DMF N,N’-dimetil formamida
DMPA 2,2-dimetoxi-2-fenilacetofenona
DMSO Dimetil sulfóxido
DPn Grado de polimerización promedio en número
DQF-COSY Espectroscopía correlacionada-doble cuántica filtrada
DSC Calorimetría diferencial de barrido
fCTA Eficiencia del CTA (=Mn exp/Mn teo)
fi Fracción molar del comonómero en la alimentación
Fi Fracción molar del comonómero en el copolímero
FR Método de Finemann-Ross
FRI Método de Finemann-Ross invertido
FRP Polimerización por radicales libres convencionales
FT-IR Espectroscopía de infrarrojo por transformada de Fourier
Glu D-glucosa
GMA Metacrilato de glicidilo
GPC Cromatografía de permeación en gel
HETCOR Espectroscopía de correlación heteronuclear
HO-TEMPO 4-hidroxi-2,2,6,6- tetrametil piperidina-N-oxil
I Iniciador
IP Isopreno
ki Constante de rapidez de iniciación

X
kp Constante de rapidez de propagación
kt Constante de rapidez de terminación
ktr Constante de rapidez de transferencia de cadena
kz Constante de rapidez de inhibición
KT Método de Kelen-Tüdos
KTE Método de Kelen-Tüdos extendido
Li Grado de polimerización en PLP-SEC
LLS Técnica de cálculo de mínimos cuadrados lineales
M Monómero
MAH Anhídrido maleico
MCTA Macroagente de control
MHS Mark-Houwink-Sakurada
MMA Metacrilato de metilo
Mn Peso molecular del polímero promedio en número
Mw Peso molecular del polímero promedio en peso
MWD Distribución de pesos moleculares
NLLS Técnica de cálculo de mínimos cuadrados no lineales
NMP o NMRP Polimerización radicálica mediada por nitróxidos
PB Polibutadieno
PIP Poliisopreno
PLP-SEC Polimerización de pulso de láser/cromatografía de exclusión de tamaño
PP Polipropileno
PRE Efecto del radical persistente
PS Poliestireno
RAFT Polimerización radicálica de transferencia de adición-fragmentación reversible
ri Índice de reactividad
S Estireno
SFRP Polimerización radicálica controlada por radicales libres estables
SG1/DEPN N-(2-metilpropil)-N-(1-dietilfosfono-2,2-dimetilpropil)-N-oxil
TBP Peróxido de tert-butilo (Luperox DI)
TEMPO 2, 2, 6, 6-tetrametil, piperidin–N-oxil
Tg Temperatura de transición vítrea
THF Tetrahidrofurano
TIPAL N-tert-butil-N-(2-metil-1-fenilpropil)-O-(1-feniletil) hidroxilamina
TIPNO 2, 2, 5-trimetil-4-fenil-3-azahexan-3-nitróxido
TM Método de Tidwell-Mortimer
tp Periodo de tiempo de pulso
vrep Velocidad de repetición del pulso (Hz)
δ Desplazamiento químico (ppm)
XI
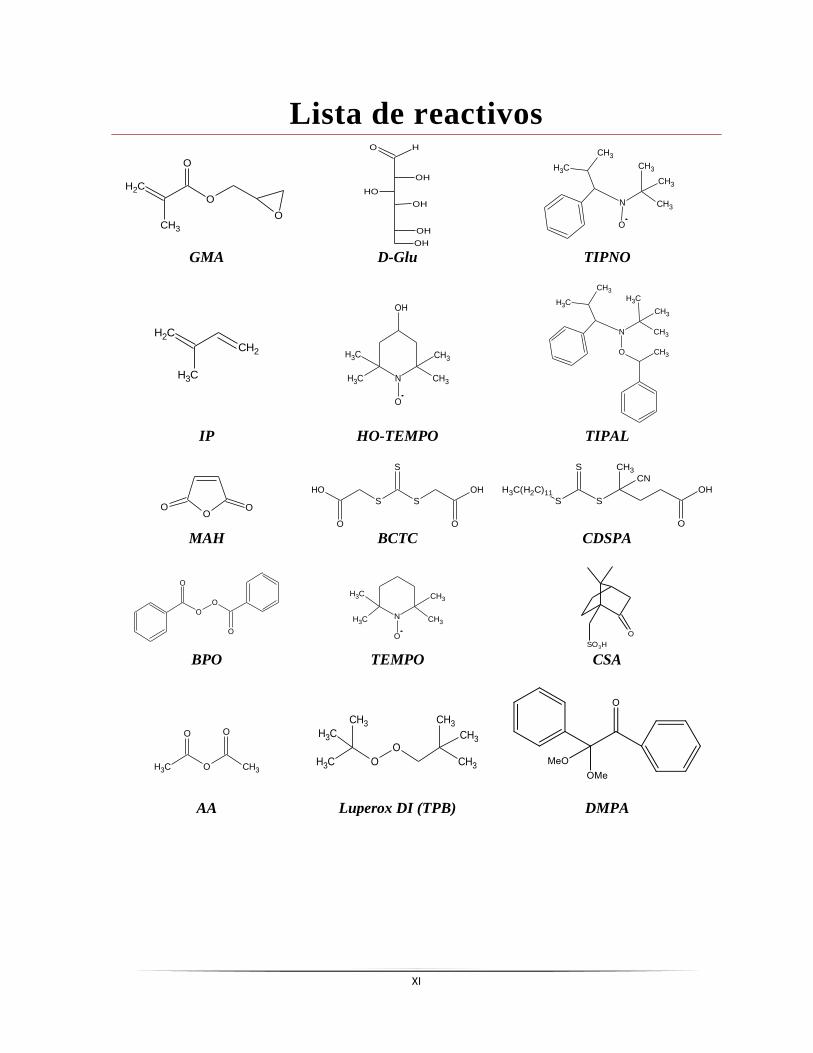
Lista de reactivos
O
O
CH2
CH3
O
OH
OH
OH
HO
OH
OH
N
O
CH3
CH3
CH3
CH3
CH3
GMA D-Glu TIPNO
CH2
CH2
CH3 N
OH
CH3 CH3
CH3 CH3
O
N
O
CH3
CH3
CH3
CH3
CH3
CH3
IP HO-TEMPO TIPAL
OO O
OH
S S
OH
O
S
O
H3C(H2C)11
S S
S CH3
CN
OH
O MAH BCTC CDSPA
O
O
O
O
N
CH3
CH3
CH3
CH3
O O
SO3H BPO TEMPO CSA
O
CH3 O CH3
O
O
O
CH3
CH3
CH3
CH3
CH3
CH3
AA Luperox DI (TPB) DMPA

XII
Lista de disolventes
H
Cl
Cl Cl O
N
O
H
CH3
CH3
Cloroformo THF DMF
CH3 OH CH3
O
CH3
CH3
Metanol Acetona Tolueno
CH3CH3
n-Hexano

XIII
Índice de figuras
Figura 1: Materiales poliméricos que se pueden obtener por medio de la CRP………..…... 12
Figura 2: Comportamiento lineal de la conversión con respecto al tiempo de reacción…….. 14
Figura 3: Comportamiento del grado de polimerización (DPn) con respecto a la conversión 14
Figura 4: Registro histórico de publicaciones de ATRP, SFRP/NMP y RAFT de acuerdo a
SciFinder Scholar..…………………………...………………………………....….. 16
Figura 5: Demostración del efecto del radical persistente……………………...…………….. 20
Figura 6: Efectos de inhibición y retardación en la polimerización del acrilato de metilo con
ditiobenzoato de cumilo a 80 ……………………………………………………..…. 26
Figura 7: Intervalo de aplicación de los diferentes tipos de agentes RAFT……….……..…… 28
Figura 8: Intervalo de acción de los agentes RAFT de acuerdo a la naturaleza del grupo R.... 29
Figura 9: Mecanismo de reacción para el proceso de injerto mediado por radicales libres..... 35
Figura 10: Terminación radical-radical del macroradical en ausencia de monómero...……... 36
Figura 11: Representación esquemática del proceso de compatibilización en mezclas de
polímeros inmiscibles………………………………………………………………. 40
Figura 12: Curvas de composición vs composición de alimentación del comonómero a
diferentes ri..……………………………………….…………………………..…… 48
Figura 13: Distribución del peso molecular del poliacrilato de dodecilo para la
determinación de la kp usando la técnica de PLP-SEC……………....…….…..…… 52
Figura 14: Sistema del equipo de PLP……………………………..…….………………..…... 66
Figura 15: Espectro de FT-IR del copolímero de IP/GMA en diferentes relaciones molares
en la alimentación………………….………………………………………...…... 68
Figura 16: Espectro de 1H RMN del copolímero de IP/GMA en diferentes relaciones molares
en la alimentación…………………………………………………………..…..... 69
Figura 17: Espectro de 1H RMN del copolímero de IP/GMA con una relación molar en la
alimentación 50:50…………………………….………………………....….….... 71
Figura 18: Curva de composición (composición del copolímero vs composición en la
alimentación del comonómero) para el sistema IP/GMA………...…………...…. 74
Figura 19: Gráfica de Finemann-Ross para el sistema IP/GMA…………….……………..…. 75
Figura 20: Gráfica de Finemann-Ross Invertida para el sistema IP/GMA..………...……..…. 75
Figura 21: Gráfica de Kelen-Tüdos para el sistema IP/GMA……………….…….………..…. 76

XIV
Figura 22: Gráfica de Kelen-Tüdos Extendido para el sistema IP/GMA………..…..……..…. 77
Figura 23: JCR de 95% para los valores evaluados de r1 y r2 de los métodos de KT, KTE y
TM para el sistema IP/GMA………………………….………………………….. 81
Figura 24: Comportamiento de la Tg del copolímero de IP/GMA como una función de la
concentración en peso del IP………………………….…………………………. 84
Figura 25: DMA del copolímero de IP/GMA 90:10…………...………………………………. 85
Figura 26: Espectro de FT-IR del copolímero de IP/MAH en diferentes relaciones molares
en la alimentación….………………….……………………………………....…. 87
Figura 27: Espectro de 1H RMN del copolímero de IP/MAH en diferentes relaciones molares
en la alimentación….………………………………………………………….…. 88
Figura 28: Espectro de 1H RMN del copolímero de IP/MAH con una relación molar en la
alimentación 50:50….………….…………………………………………….…... 89
Figura 29: Curva de composición (composición del copolímero vs composición en la
alimentación del comonómero) para el sistema IP/MAH…..……….………..….. 91
Figura 30: Gráfica de FT y FTI para el sistema IP/MAH…………...…….………………..…. 91
Figura 31: Gráfica de KT y KTE para el sistema IP/MAH………...………………...……..…. 92
Figura 32: JCR de 95% para los valores evaluados de r1 y r2 de los métodos de KT, KTE y
TM para el sistema IP/MAH………………..……………………………………. 93
Figura 33: Comportamiento de la Tg del copolímero de IP/MAH como una función de la
concentración en peso del IP………………..………………………………….... 94
Figura 34: DMA del copolímero de IP/MAH 90:10…………………………………...………. 95
Figura 35: Distribuciones de pesos moleculares para los homopolímeros de GMA y del IP,
producidos en masa por PLP a 25 °C en diferentes tiempos de pulsado……..…. 97
Figura 36: Constante de propagación del copolímero IP-GMA vs fracción mol del IP,
obtenido por PLP-SEC a 25 °C………………………...………………………… 97
Figura 37: Efecto del disolvente en la velocidad de polimerización del isopreno por NMP a
diferentes relaciones de N/I………………………………………………………. 105
Figura 38: Efecto del disolvente en la velocidad de polimerización del isopreno por NMP a
N/I=1.8……………………………………………………………………………
………… 107
Figura 39: DSC del copolímero IP/GMA con una composición molar IP/GMA 98:2 ..………. 108
Figura 40: Perfil de la cinética de la formación del copolímero IP/GMA y del homopolímero
de IP a 120 °C y 1170 kPa iniciado por 14…………..……………………….…. 109
Figura 41: Cromatogramas de GPC del homo- y copolímero de IP por NMP………………... 110
Figura 42: Efecto de la concentración aditivo acelerante en la polimerización del IP usando
13 y BPO…..…………………………………………………………………..…. 112
Figura 43: Efecto del aditivo acelerante en el perfil cinético de la polimerización del IP por
NMP..………………………………………………………...………………..…. 114
Figura 44: Perfil de la cinética de la polimerización del IP con 14 y 18 en solución con
tolueno.….….…………………………………………………………………..…
. 116
Figura 45: Perfil cinético de la formación del copolímero IP/GMA con 18 en solución con
tolueno.….…………………………………………………………….………..…
. 117
Figura 46: Cromatograma de GPC antes y después de la extensión de cadena del copolímero
de IP con GMA …………………………………………………………..…....…. 118

XV
Figura 47: Espectro de FT-IR del copolímero de IP por NMP……………………………..…. 119
Figura 48: Espectro de 1H RMN del copolímero de IP por NMP…………………..….…...…. 120
Figura 49: Correlación de la energía LUMO y el calor de reacción relativo de algunos
agentes RAFT……………………………………………………………...…..…. 126
Figura 50: Correlación de la energía LUMO y la longitud de enlace de algunos agentes
RAFT…………………………………………………………….………………..
…. 127
Figura 51: Perfil de la cinética de la polimerización iniciada por 19 y 20 en solución
(conversión vs tiempo)…………………………………………………..……..…. 128
Figura 52: Perfil de la cinética de la polimerización iniciada por 19 y 20 en solución (Mn vs
conversión)………………………………………………………………………..
….. 129
Figura 53: DSC del copolímero de IP/GMA con una composición molar de 9:1……………... 130
Figura 54: Perfil de la cinética de la formación del copolímero de IP en solución
(conversión vs tiempo) .…………………………………………………..…..….. 131
Figura 55: Perfil de la cinética de la formación del copolímero de IP en solución (Mn vs
conversión)…..…………………………………………………………….…..….
.….. 131
Figura 56: Cromatogramas de GPC del copolímero de IP por el proceso RAFT…………….. 132
Figura 57: Perfil cinético de la formación del homopolímero de IP con 20 en solución con
tolueno (conversión vs tiempo y Mn vs conversión)…..…………..……………… 134
Figura 58: Cromatogramas de GPC en la extensión de la cadena del copolímero de IP por el
proceso RAFT……………………………………………………………….……. 135
Figura 59: Espectro de FT-IR del copolímero de IP/GMA generado por 20…………………. 135
Figura 60: Espectro de 1H RMN del copolímero de IP/GMA generado por 20………..…..…. 136
Figura 61: Espectro de 1H RMN del CDSPA (20)……………..………….………………..…. 137
Figura 62: Espectro de FT-IR del PIP funcionalizado e injertado por NMP…………..…..…. 141
Figura 63: Espectro de 1H RMN del PIP funcionalizado e injertado por NMP…………....…. 142
Figura 64: Espectro DQF-COSY del PIP funcionalizado e injertado por NMP……………… 143
Figura 65: Cromatograma de GPC del PIP funcionalizado e injertado por NMP…………… 149

XVI
Índice de tablas
Tabla 1: Clasificación de agentes RAFT de acuerdo a la naturaleza química de Z…………...…. 28
Tabla 2: Comparativo de la técnicas de polimerización NMP, ATRP y RAFT………………….... 30
Tabla 3: Energías de disociación para C-H……………….………………….………………..…. 34
Tabla 4: Relación de las constantes de desproporción - combinación en solución………….....… 36
Tabla 5: Propiedades físico-térmicas de los copolímeros dependiendo de su composición….…... 45
Tabla 6: Índices de reactividad para el sistema estireno(1)-metacrilato de metilo(2) en distintas
técnicas de polimerización…………………………………….………………….…. 48
Tabla 7: Resultados de la copolimerización del IP con GMA……………………...…………..…. 72
Tabla 8: Resultados de los índices de reactividad usando el método de TM…………………..…. 80
Tabla 9: Resultados de los índices de reactividad usando diversos métodos de cálculo para el
sistema IP-GMA………………………………………………………………......…. 83
Tabla 10: Resumen de los valores de Tg por DSC/DMA para el sistema IP/GMA……………….. 85
Tabla 11: Resultados de la copolimerización del IP con MAH………………………………….... 90
Tabla 12: Resultados de los índices de reactividad usando el método de TM……………………. 92
Tabla 13: Resultados de los índices de reactividad usando diversos métodos de cálculo para el
sistema IP-MAH……………………………………………………………….….…. 93
Tabla 14: Resumen de los valores de Tg por DSC/DMA para el sistema IP/MAH……………….. 95
Tabla 15: Condiciones experimentales y resultados de PLP para el sistema IP-GMA a 25 °C.…. 96
Tabla 16: Resumen de la (co)polimerización del isopreno por NMP……………………...…..…. 100
Tabla 17: Propiedades predichas de alcoxiaminas con cálculos semiempíricos (AM1-RHF).…... 102
Tabla 18: Propiedades físicas de algunos disolventes………….....………….………………..…. 106
Tabla 19: Resumen de la homopolimerización del IP con el uso de aditivos acelerantes en la
NMP.….…………………………………………………………………..…………..
…. 111
Tabla 20: Resumen de la copolimerización del IP por NMP. Crecimiento de la cadena......…….. 115
Tabla 21: Resumen de la (co)polimerización del IP por RAFT…………………………………... 122

XVII
Tabla 22: Propiedades predichas de alcoxiaminas con cálculos semiempíricos (AM1-RHF).…... 125
Tabla 23: Resumen de la (co)polimerización del IP por RAFT. Crecimiento de la cadena…...…. 133
Tabla 24: Resumen de la copolimerización de injerto del IP por NMP…...….………………..…. 139
Tabla 25: Desplazamientos químicos observados para el experimento 55…………………….…. 143
Índice de esquemas
Esquema 1: Reacciones reversibles de activación-desactivación que experimenta el radical
propagante Pi en los sistemas de CRP……………..……………….……………….. 13
Esquema 2: Mecanismo de polimerización por ATRP………………………....……………….. 17
Esquema 3: Desactivación temporal del radical propagante por el radical nitróxido.………… 18
Esquema 4: Equilibrio de activación-desactivación en la NMP…………………….………….. 19
Esquema 5: Representación de la polimerización del estireno mediada por nitróxidos...……… 21
Esquema 6: Derivados de nitróxidos empleados exitosamente como agentes controladores en
la NMP………………………………………………………………..……………. 22
Esquema 7: Mecanismo de reacción para macromonómeros de MMA como agentes de
transferencia………….……………………………………………………….……
…….. 23
Esquema 8: Mecanismo de reacción de la polimerización radicálica controlada tipo RAFT….. 25
Esquema 9: Reacción propuesta para la terminación cruzada entre radicales libres
propagantes y radicales libres intermediarios……………………...……………... 27
Esquema 10: Terminación radical-radical del macroradical en ausencia de monómero......................... 36
Esquema 11: Isopreno y las microestructuras del poliisopreno…………………..…………….. 55
Esquema 12: Reacción de copolimerización del IP-GMA…………………………………...….. 67
Esquema 13: Resumen de los picos presentados en el espectro de 1H RMN de la Figura 16….. 70
Esquema 14: Reacción de copolimerización del IP-MAH……………..……….……………….. 86

XVIII
Esquema 15: Resumen de los picos presentados en el espectro de 1H RMN de la Figura 27….. 88
Esquema 16: Estructuras químicas de los nitróxidos HO-TEMPO, TIPNO y TEMPO………… 99
Esquema 17: Formación del (co)polímero de isopreno con grupos nitróxido………………….. 101
Esquema 18: Ángulos y enlaces involucrados en el cálculo semiempírico AM1-RHF…………. 103
Esquema 19: Estructuras de los aditivos acelerantes utilizados en la polimerización del IP por
NMP…….…………………………………………………………………..………
….. 113
Esquema 20: Acilación de la alcoxiamina con el grupo terminal que conlleva la incremento de
su reactividad………………………………………………………………...…….. 113
Esquema 21: Estructura del TIPAL…………………...……………….……….……………….. 115
Esquema 22: Estructuras químicas de los agentes RAFT 19, 20, 21, 22 y 23………………….. 121
Esquema 23: Formación del copolímero de isopreno con grupos RAFT...………….………….. 122
Esquema 24: Funcionalización del IP con grupos nitróxidos y la etapa de injerto
controlada/viviente del copolímero IP/GMA………………………...…………….. 139
Esquema 25: Mecanismo de reacción para el proceso de funcionalización de cadenas
olefínicas…………………………………………………………...……………….
.
144
Esquema 26: Mecanismo de reacción para el caso de la funcionalización del PIP-trans con
nitróxidos….……………………………………………………………….……….
. 145
Esquema 27: Descomposición unimolecular del radical t-butoxido (eliminación )…………... 148

XIX
Resumen
Los polímeros funcionalizados son de gran interés en las áreas de tipo científico e
industrial por la diversidad de usos en los que se pueden aplicar; tales como, materiales
compatibilizantes en mezclas de polímeros inmiscibles, agentes acoplantes en compositos de
una matriz polimérica reforzada con alguna fibra o en la incorporación de nanoarcillas. En
particular, los materiales elastoméricos son muy preciados por su alta potencialidad en diversas
aplicaciones (en particular, los derivados de monómeros de tipo dieno). El interés radica en
poder emplearlos como compatibilizantes y/o agentes acoplantes, debido a que esto abriría una
amplia gama de posibles usos; sin embargo, cuando se realiza una investigación en la literatura
especializada acerca de la síntesis de elastómeros del tipo dieno, y en el caso específico de la
polimerización por radicales libres, ésta es muy pobre y más aún en el campo de la
polimerización radicálica controlada viviente (CRP).
Es por ello que en este estudio doctoral, se llevó a cabo la síntesis de copolímeros en
bloque de isopreno (IP) con metacrilato de glicidilo (GMA) por medio de la polimerización
mediada por nitróxidos (NMRP) y por el proceso de transferencia de cadena de adición -
fragmentación reversible (RAFT), dos de la técnicas más exitosas que tiene la CRP;
adicionalmente, también se obtuvo la funcionalización de polímeros de isopreno (tanto en la
conformación trans como en la conformación cis) con grupos nitróxido y sus correspondientes
macroalcoxiaminas, las que fueron utilizadas para la obtención de injertos controlados en
presencia de los monómeros de isopreno y GMA para llevar a cabo el crecimiento de la cadena
del copolímero correspondiente.
Las condiciones de operación para llevar a cabo dichas reacciones de copolimerización,
(tanto de cadena lineal como de tipo injerto) fueron llevadas a cabo en un reactor de acero
inoxidable a una presión no menor de 1170 kPa (170 psi), 125 °C y en solución (el disolvente
fue tolueno grado reactivo).

XX
Preliminarmente, se efectuaron las copolimerizaciones de los sistemas isopreno-
metacrilato de glicidilo (IP/GMA) e isopreno-anhídrido maleico (IP/MAH) por radicales libres
convencionales (FRP) a diferentes relaciones molares en la alimentación del par de monómeros,
esto fue con el fin de determinar los índices de reactividad (ri) correspondientes para cada
sistema de comonómeros.
Para obtener dichos índices de reactividad se utilizaron diferentes métodos de cálculo
(tanto lineales como no lineales), indicando que existe una fuerte tendencia en estos sistemas a
formar copolímeros alternados; para conocer las composiciones de los monómeros en el
copolímero, éstos se midieron por medio de resonancia magnética nuclear de protón (1H RMN),
aunque existen claras diferencias entre ambos sistemas tales como los valores de ri y los aspectos
espectroscópicos que determinan las características de cada sistema.
Ambos sistemas poliméricos presentan un comportamiento térmico (observado por DSC
y DMA), que se ajusta adecuadamente al modelo de Couchman con respecto a la determinación
de la temperatura de transición vítrea (Tg) correspondiente.
Dado que muchos de los datos cinéticos de la polimerización del isopreno vía radicales
libres se desconocen, se tuvo la oportunidad de determinar la constante de propagación para este
monómero tipo dieno por medio de la técnica de polimerización de láser pulsado/cromatografía
de exclusión de tamaño (PLP-SEC). Adicionalmente, también se logró determinar la constante
de propagación del copolímero de IP/GMA en diferentes concentraciones molares de isopreno
en la alimentación.

Capítulo 1| Enfoque de la Tesis
1
1.
Enfoque de la Tesis
La motivación para realizar este estudio doctoral parte de la intención de preparar
poliolefinas que contengan grupos polares que se puedan utilizar como agentes
compatibilizantes mediante la síntesis directa en cadena lineal o mediante la modificación de
polímeros existentes para formar copolímeros de tipo injerto en forma controlada, utilizando la
polimerización radicálica controlada viviente (CRP, por sus siglas en inglés).
Esta idea nació sabiendo que la elaboración de mezclas de polímeros compatibles presenta
ventajas económicas en la manufactura de un material que cumple con las especificaciones que
requieren los clientes a un menor costo.1 Sin embargo, cuando se tratan de mezclar especies
poliméricas, la mayoría de los polímeros son inmiscibles y la mezcla resultante tiene
propiedades físicas que son inferiores que la de sus componentes iniciales. Se sabe que cuando
se mezclan dos polímeros inmiscibles, se forma un producto heterogéneo cuya morfología
dependerá de las tensiones interfaciales y acompañadas de la adhesión interfacial. Una alta
tensión interfacial da lugar a una falta de estabilidad en la interfase y por lo consiguiente se
presenta una separación brusca de las fases durante el procesamiento o durante el uso, mientras
que una mala adhesión conduce a un comportamiento mecánico frágil en la mezcla, cuyas
propiedades mecánicas llegan a ser peores que las de los componentes individuales.2
Los copolímeros que llegan a ser miscibles (pero no necesariamente de composición
similar) con cada fase en la mezcla, mitigan la inestabilidad interfacial y mejoran las
propiedades mecánicas de la mezcla. Esta propiedad de los copolímeros es debida a su
propensión a segregarse completamente en las dos fases, reduce la tensión interfacial y mejora
la adhesión entre las dos fases.3

Capítulo 1| Enfoque de la Tesis 1.1 Hipótesis
1
De acuerdo a los argumentos anteriores, la idea es obtener copolímeros de isopreno (IP)
con un monómero polar (en este caso, metacrilato de glicidilo) ya sea en cadena lineal o de tipo
injerto por medio de la polimerización controlada-viviente por radicales libres. Para el caso de
la síntesis de copolímeros en cadena lineal, se copolimerizaron los monómeros antes
mencionado para generar la estructura poli (isopreno-b-(isopreno-co-metacrilato de glicidilo))
con una relación especifica al 1 % w/w del sustrato polar con respecto al IP (ver Esquema 17,
pp. 101 ó Esquema 23, pp. 122). Al mismo tiempo, a este material polimérico se le hizo crecer
la cadena polimérica de manera sucesiva con la adición de isopreno al medio de reacción
(recordando que este material polimérico es ya un macroagente de control, y con ello se
demostró el carácter viviente que tuvo el sistema) hasta alcanzar la longitud de cadena deseada.
Alternativamente, en la síntesis de los copolímeros de tipo injerto, se requirió en un primer
paso, de la utilización de un sustrato polimérico (poliisopreno comercial tanto en una
conformación cis como en una conformación trans), el cual se funcionalizó con un grupo
nitróxido a lo largo de la cadena principal. Como segundo paso, ya funcionalizado el polímero
(el cual ya es una macroalcoxiamina), se le hizo reaccionar en presencia de una mezcla de
monómeros (isopreno y GMA), los cuales crecieron como injerto de copolímero para dar la
estructura final de poli (isopreno-g-(isopreno-co-metacrilato de glicidilo)) (ver Esquema 24,
pp. 139).
1.1. Hipótesis
Es viable la sintesis de polímeros de isopreno conteniendo grupos polares con estructuras
de copolímeros en bloque por medio de dos de las técnicas de CRP (la polimerización mediada
por nitróxidos, NMP, y por el proceso de transferencia de cadena de adición-fragmentación
reversible, RAFT) o lograr la modificación de polímeros de isopreno funcionalizados con
grupos nitróxido y a partir de estos últimos, en presencia de isopreno con monómeros polares,
se plantea la posibilidad de generar injertos de manera controlada.

Capítulo 1| Enfoque de la Tesis 1.2 Justificación
3
Se espera que al funcionalizar los polímeros portadores con los grupos polares o reactivos,
éstos presentarán propiedades como agentes acoplantes en compositos de una matriz polimérica
y/o agentes compatibilizantes en mezclas de polímeros inmiscibles.
1.2. Justificación
Actualmente existen muy pocos trabajos científicos y/o patentes en la síntesis de
(co)polímeros de isopreno por radicales libres convencionales (a pesar de que la polimerización
por radicales libres no requiere de condiciones tan estrictas como lo es su contraparte iónica y
de coordinación), y mucho menos existe información disponible en el área de la polimerización
radicálica controlada-viviente de este monómero; las posibles causas a la falta de información
científica en la polimerización del isopreno por radicales libres se deban tal vez al uso de
sistemas presurizados, y temperaturas mayores a 115 °C con el fin de obtener una velocidad de
reacción razonable en la polimerización de 1,3-dienos por CRP y finalmente, como punto a
resaltar, la posibilidad de formarse un ciclo aducto derivado por la reacción de Diels-Alder
durante la síntesis de los copolímeros de isopreno con monómeros polares vinílicos.
El reto que presenta este proyecto doctoral es el compromiso de hallar una ruta de síntesis
para obtener copolímeros de isopreno con monómeros polares tales como metacrilato de
glicidilo (GMA, ver Esquema 12, pp. 67) y anhídrido maleico (MAH, ver Esquema 14, pp.
86), así como la determinación de los índices de reactividad respectivos para cada sistema de
copolímero, estudiar los mecanismos cinéticos para la generación de los copolímeros
correspondientes, empleando rutas de síntesis científicamente viables y tecnológicamente
atractivas que permitan realizar de un modo sistemático los estudios de copolimerización.
Aunada a las aportaciones científicas de índole básica, el presente trabajo de tesis doctoral
también involucra una motivación tecnológica ya que desde el punto de vista económico, los
compatibilizantes promueven la miscibilidad de mezclas de polímeros, por lo que éstos
representan una forma de convertir materiales de bajo costo ya con valor agregado en materiales
de especialidad con gran valía comercial. Asimismo, al obtener polímeros de isopreno con una
fracción polar en su cadena principal, se ofrece la posibilidad de conseguir materiales

Capítulo 2/ Antecedentes 1.3 Objetivo general.
4
poliméricos con cadenas hidrocarbonadas saturadas por medio de la hidrogenación de los dobles
enlaces que presenta el poliisopreno en su estructura (lo cual, le daría un agregado potencial a
sus posibles aplicaciones tecnológicas para este tipo de materiales). Demostrar la síntesis del
copolímero de isopreno (tanto en cadena lineal como en injerto) con estructuras controladas
poseyendo grupos polares o reactivos, abre la posibilidad de elaborar con el sector industrial el
desarrollo de materiales de ingeniería.
1.3. Objetivo general
El objetivo de este trabajo de tesis, es obtener copolímeros en cadena lineal (por medio de
la polimerización NMP y RAFT) que tengan grupos polares o reactivos (tal como el GMA);
adicionalmente se desea modificar una serie de polímeros comerciales (poliisopreno en
conformación trans y cis) con grupos nitróxido, para posteriormente hacerle crecer
controladamente, injertos de copolímero poseyendo grupos polares o reactivos (GMA).
1.4. Objetivos particulares
a. Sintetizar copolímeros de cadena lineal de isopreno con algún monómero funcional, tal
como el metacrilato de glicidilo o el anhídrido maleico mediante polimerización por
radicales libres en masa para determinar sus índices de reactividad (ri) para cada sistema
por medio de diferentes métodos de cálculo auxiliados con la técnica espectroscópica de
resonancia magnética nuclear protónica (1H RMN).
b. Determinación de la constante de propagación, kp, del isopreno por medio de la técnica
de polimerización de láser por pulso-cromatografía de exclusión de tamaño (PLP-SEC).
Adicionalmente, determinar la kp del copolímero de IP-GMA a diferentes
concentraciones del par de monómeros por PLP-SEC.
c. Sintetizar copolímeros de cadena lineal de isopreno con algún monómero funcional, tal
como el metacrilato de glicidilo, para formar estructuras del tipo poli-(isopreno-b-

Capítulo 2/ Antecedentes 1.4 Objetivos particulares.
5
(isopreno-co-monómero funcional)) vía las técnicas de polimerización de NMP y RAFT
en solución (tolueno).
d. Sintetizar copolímeros de injerto a partir de poliisopreno comercial (en conformación cis
y trans individualmente), con ramas que contengan un monómero funcional tal como el
metacrilato de glicidilo, para formar estructuras del tipo poli-(isopreno-g-(isopreno-co-
monómero funcional)) vía NMP en solución (en tolueno).
e. Caracterizar los copolímeros descritos en los puntos a-d por espectroscopía de infrarrojo
por transformada de Fourier (FT-IR), resonancia magnética nuclear (RMN),
cromatografía de permeación en gel (GPC) y calorimetría diferencial de barrido (DSC).

Capítulo 2/ Antecedentes
6
2.
Antecedentes
La industria de los plásticos está situada como una de las áreas más importantes que hay
en la actualidad por la creciente demanda de sus productos, y para ello se han desarrollado
tecnologías que van desde la polimerización o copolimerización hasta llegar a la mezcla de
polímeros.1 Hoy en día, los polímeros sintéticos son ahora una parte esencial de numerosos y
diversos materiales dentro de nuestra vida cotidiana. En las últimas décadas, la ciencia de los
polímeros se ha desarrollado dentro de un campo de investigación multidisciplinario, como en
las áreas relacionadas a la electrónica, óptica, biomédica, entre otras; obteniéndose un
incremento de materiales funcionales de alta valía. Esto explica por qué la ciencia de los
polímeros es considerada actualmente como un campo de investigación esencial e innovativo
tanto en la academia como en la industria. Con respecto a la mezcla de polímeros o materiales
poliméricos compuestos, hay un gran auge tecnológico ya que ofrecen la posibilidad de obtener
nuevos materiales poliméricos con propiedades muy diferentes y características mejoradas; y
para conseguirlo, se combinan dos o más polímeros que son miscibles entre sí o se mezclan por
medio de la compatibilización de polímeros inmiscibles.4 Alternativamente, se pueden utilizar
copolímeros con ciertas características específicas como agentes acoplantes para la dispersión
y exfoliación de nanoarcillas en matrices poliméricas no polares.
El uso de polímeros funcionalizados como compatibilizantes o acoplantes dentro de la
industria del plástico es un área de gran interés científico y comercial y está bien documentado
para algunos sistemas. Un ejemplo en particular es el polipropileno (PP) maleatado (modificado
con anhídrido maleico (MAH)),4b, 5 que incluso se produce comercialmente (EXXON, Uniroyal)
y se utiliza como compatibilizante en mezclas de PP con poliamidas para aplicaciones en partes

Capítulo 2/ Antecedentes
7
de automóviles, aparatos eléctricos o electrónicos, etc. Otro ejemplo, es el poliisobutileno
bromado (hule butilo), que se ha utilizado por EXXON para exfoliación de nanoarcillas en una
matriz de este hule, a fin de incrementar la impermeabilidad a gases de este material en
aplicaciones de la industria llantera.5c Otros polímeros que se funcionalizan industrialmente con
MAH son el EPDM (abreviatura en inglés de poli(etilen-propilen-monómero diénico)) y el
SEBS (abreviatura en inglés para el poli(estireno-etileno-butileno-estireno) en bloques) los
cuales también se utilizan como compatibilizantes en mezclas de polímeros en las que se busca
mejorar las propiedades de la mezcla resultante en comparación con las propiedades de los
polímeros individuales.4a
Por otra parte, la utilización de polímeros como agentes acoplantes se puede ejemplificar
en el proceso de la incorporación de nanoarcillas en elastómeros, tales como en el terpolímero
EPDM (poli(etilen-propilen-monómero diénico))6 y en elastómeros termoplásticos como el SIS
(estireno-isopreno-estireno)7 por citar algunos ejemplos; con esto se entiende que es un campo
de estudio que recientemente ha cobrado importancia en la academia y en la industria. En
conclusión, existe un gran interés tecnológico por el desarrollo de materiales nanocompuestos
con matriz polimérica, porque esta alteración confiere al material resultante un refuerzo en sus
propiedades mecánicas,4a, 8 una mayor retardancia a la flama,5c, 9 y/o una mejora en la
transparencia con respecto a la exhibida por el polímero original.7, 10 Para lograr esto, es
necesario sintetizar un agente compatibilizante o acoplante para obtener una mayor afinidad
entre los materiales a mezclar. Sin embargo, el procedimiento de funcionalización que se usa
industrialmente, tanto en el caso de poliolefinas (PE, PP o EPDM) como en el caso del SEBS,
es la extrusión reactiva del polímero base (p. ej. PP) con MAH en presencia de un peróxido.1
No obstante, en este proceso se tiene muy poco control sobre la estructura resultante, dado que
puede conducir a reacciones de ruptura que afectan de forma incontrolada al peso molecular del
polímero funcionalizado, como en el caso del PP maleatado, donde se ha reportado que el
número de grupos funcionales de MAH en el PP es difícil de controlar.11
En los últimos años se han realizado enormes esfuerzos por mejorar la síntesis precisa de
los compatibilizantes y/o acoplantes, dado que existen varios sistemas poliméricos de interés
comercial cuyas mezclas con otros polímeros o nanoarcillas han sido poco estudiados. Tal es el

Capítulo 2/ Antecedentes 2.1 Polimerización por radicales libres
8
caso de las poliolefinas y los elastómeros. Las poliolefinas cuentan con una excelente
procesabilidad a bajo costo y con buenas propiedades mecánicas, y por su parte, los elastómeros
son materiales poliméricos muy atractivos desde el punto de vista científico e industrial por sus
aplicaciones potenciales como reforzantes,12 espesantes,13 adhesivos,14 compatibilizantes,15
elastómeros termoplásticos,16 etc., siendo así, los materiales poliméricos a base de estos
sustratos conteniendo bloques o injertos controlados de polímero con grupos polares como
metacrilato de glicidilo (GMA), anhídrido maleico (MAH), o acrilonitrilo (AN) son
considerados materiales de gran valía e impacto comercial.17 Finalmente, hasta hace 20 años,
las polimerizaciones vivientes iónicas eran los únicos métodos existentes para lograr un alto
grado de homogeneidad estructural y composicional de los polímeros; sin embargo, con el
advenimiento de nuevas herramientas sintéticas fáciles (como la polimerización por radicales
libres controlada-viviente o CRP), se ha logrado llevar a cabo arquitecturas macromoleculares
sintéticas más complejas con objetivos deseados y en condiciones experimentales más suaves.
Existen diferentes métodos para producir copolímeros de bloques, la mayoría de ellos caen
en una de estas tres categorías: a) polimerización secuencial de monómeros presentes en el
medio de reacción, b) polimerización de un nuevo bloque sobre un polímero y c) acoplamiento
de dos polímeros producido previamente. El método más común para la síntesis de copolímeros
en bloque es la polimerización viviente. La característica particular de este tipo de
polimerización es la de poseer un grado muy limitado de cadenas terminadas irreversiblemente,
permitiendo que la adición secuencial de monómero(s) forme un copolímero tipo multibloque
con estructuras moleculares bien definidas. Los métodos para la obtención de copolímeros de
bloques a los que se recurre son: polimerizaciones aniónicas,18 catiónicas,19 de transferencia de
grupo,20 metátesis de apertura de anillo21 y los basados en radicales libres controlados. Dado
que el enfoque de la presente tesis de doctorado es en la polimerización radicálica controlada,
ésta es la que se discutirá a continuación dejando a un lado las demás técnicas de polimerización.
2.1 Polimerización por radicales libres
La polimerización por radicales libres es un proceso comercial muy importante para la
producción de polímeros de alto peso molecular. Hoy por hoy, cerca del 50% de la producción
mundial de los polímeros sintéticos se obtiene mediante este proceso.22 Las principales ventajas

Capítulo 2/ Antecedentes 2.1 Polimerización por radicales libres
9
que ofrece la polimerización por adición (tradicionalmente denominada polimerización
radicálica) es la capacidad de polimerizar una amplia gama de monómeros tipo acrilatos,
acrilamidas, acrilonitrilo, estirénicos, dienos y vinílicos, presenta una alta tolerancia a grupos
funcionales no protegidos en el monómero y/o en el disolvente (p. ej. OH, NR2, COOH, CONR2
y SO3H), se puede llevar a cabo en medios acuosos o próticos, y presenta una amplia
compatibilidad con los diversos procesos de polimerización que comúnmente se manejan (en
masa, solución, emulsión, miniemulsión y suspensión) dentro de un amplio intervalo de
temperaturas (que van desde -100 °C hasta por encima de 200 °C).23
Sin embargo, la principal desventaja de la polimerización por radicales libres
convencionales (FRP, por sus siglas en inglés) es el bajo control sobre algunos parámetros
estructurales como: el peso molecular, la dispersión de la distribución de los pesos moleculares
(ᴆ), funcionalidad, composición, etc. Este tipo de polimerización es inherente a la química que
rige el mecanismo;23 es decir, el tiempo de vida promedio efectivo que tiene un radical
propagante es del orden de un segundo, por lo que esto impide lograr manipular la estructura
del material polimérico resultante durante ese corto tiempo. Esto implica, que en un pequeño
intervalo de tiempo se lleven a cabo las reacciones (etapas) de iniciación, propagación y de
terminación, generando cadenas de polímero muerto con un grado de polimerización (DP por
sus siglas en inglés) de alrededor de 103 – 104 unidades.24
Las cadenas de polímero formadas se acumulan a lo largo de la polimerización y no
participan más en las reacciones de propagación (incrementando con ello la dispersidad
molecular del polímero). Esta desventaja inhabilita a la FRP como una práctica de síntesis para
la obtención de polímeros con estructura específica, (p. ej. materiales con arreglos de dos o más
monómeros en forma de bloques (copolímeros)). No obstante, en las tres últimas décadas, ha
existido un gran volumen de trabajos sobre el desarrollo y uso de diversos artificios que permiten
controlar las etapas de terminación, intercambiando activamente las especies propagantes por
especies temporalmente inactivas. Como resultado, esto ha permitido acceder a copolímeros en
bloque como se hace comúnmente en las polimerizaciones vivientes de tipo aniónica.

Capítulo 2/ Antecedentes 2.1 Polimerización por radicales libres
10
En 1956, Szwarc18 reportó la obtención de un copolímero en bloque por adición secuencial
de monómeros por medio de una polimerización iniciada con un sistema dual de sodio-naftaleno
(polimerización aniónica). Esta polimerización se denominó como viviente debido a que las
cadenas de polímero crecían sin experimentar reacciones de transferencia y/o de terminación,
lo que dio como resultado una uniformidad en el tamaño de las cadenas (baja dispersidad) y
como consecuencia, todas las cadenas tuvieron la misma longitud debido a que cada una de ellas
creció con la misma velocidad en un periodo de tiempo muy corto. En estos casos, la conversión
total y la longitud de la cadena están dadas por la relación de monómero (M) e iniciador (I):
𝐷𝑃𝑛 = [𝑀] [𝐼]⁄ (1)
Este descubrimiento dio pie al diseño estructural de las macromoléculas; empero, los
sistemas iónicos poseen la gran desventaja de la rigurosidad en la pureza de los reactivos y los
disolventes empleados, aunada con el uso de condiciones inertes para evitar la desactivación del
iniciador. Esto trajo como consecuencia, que numerosos grupos de investigación tuvieran interés
por mejorar y ampliar las condiciones de reacciones de este tipo de sistemas de polimerización,25
pero fue hasta la década de los 80’s en que Otsu et al.26 acuñaron un nuevo concepto para la
polimerización, el iniferter (iniciador-agente de trasferencia-terminador).
Los iniferters son moléculas que tienen la capacidad de ser un iniciador de la
polimerización por radicales libres a través de la iniciación, propagación y terminación de los
radicales libres generados y actuar como un agente de transferencia durante todo el proceso. El
prototipo de este tipo de agentes de control, son sustancias azufradas que actúan como
terminadores efectivos en la propagación de la polimerización por radicales libres pero con la
diferencia de que la terminación es reversible. En este tipo de mecanismo de reacción, la
terminación bimolecular y de otras reacciones de transferencia son despreciables. Gracias a este
procedimiento, es como se tiene el origen de lo que ahora se conoce como polimerización
controlada-viviente por radicales libres o CRP y aun cuando existen algunas controversias con
el nombre que debe recibir una polimerización radicálica con características vivientes,27 la
realidad es que el desarrollo de esta área permitió obtener estructuras macromoleculares que
nunca se habían obtenido por los métodos hasta entonces conocidos.

Capítulo 2/ Antecedentes 2.2. Polimerización controlada-viviente por
radicales libres (CRP)
11
2.2 Polimerización controlada-viviente por radicales libres (CRP).
Una de las técnicas de síntesis o modificación de polímeros que existe en la actualidad es
la polimerización controlada-viviente por radicales libres o CRP, esta técnica permite
funcionalizar sustratos poliméricos con grupos reactivos y controlar el crecimiento de los
bloques o injertos poliméricos; sin embargo, antes de hablar de la CRP, es esencial señalar que
los términos controlado y viviente no se deben interpretar como sinónimos.
La polimerización controlada, permite determinar el resultado de la polimerización; esto
es, cuando se va a realizar la estrategia de síntesis del material polimérico se puede manipular
la arquitectura final de éste, así como la presencia de un grupo funcional terminal, la
composición y disposición de los (co)monómeros del material resultante; además de estimar el
peso molecular promedio a partir de la relación de la concentración de monómero con respecto
al iniciador. Por otra parte, en la polimerización viviente, las etapas de terminación y de
transferencia de cadena se encuentran ausentes (o minimizadas), por lo que existe el potencial
de reiniciar el crecimiento de la cadena polimérica siempre y cuando exista la presencia de
monómero(s) en el medio de reacción.28
Una de las principales características de la CRP, es combinar lo mejor de la química
convencional de los radicales libres y de las polimerizaciones vivientes sin las desventajas de
ambos, de tal manera que, la CRP ofrece una robustez y flexibilidad que son características de
los radicales libres aunadas a un carácter viviente para producir estructuras controladas y
versátiles. Por lo tanto, es válido realizar una comparación de la polimerización por radicales
libres con su contraparte iónica en el tema del control (tanto en reactividad como en selectividad
de las especies en crecimiento, las cuales se regulan por medio de las interacciones electrónicas
de su contraión en el caso de la polimerización iónica). Debido a los atributos propios que
ofrecen las técnicas de CRP, sobresalen la polimerización mediada por nitróxidos (NMRP o
NMP),29 la polimerización radicálica de transferencia de átomo (ATRP)30 y el proceso de
transferencia de cadena de adición-fragmentación reversible (RAFT).31

Capítulo 2/ Antecedentes 2.2.1 Fundamentos de la CRP. Consideraciones generales
12
Con estas técnicas, se ha logrado la síntesis de materiales poliméricos bien definidos como
homopolímero, copolímero de tipo injerto, dibloque, tribloque, estrella; entre otras arquitecturas
mucho más complejas, incluyendo microgeles y polímeros de tipo peine, por lo que existe un
potencial de revolucionar gran parte de la industria de los polímeros.28-29, 32 Las diferentes
técnicas de CRP han abierto nuevas expectativas para el diseño y desarrollo de nuevos
materiales, siendo factible sintetizar materiales poliméricos en función de su composición,
topología y grupos funcionales en particular (Figura 1). Para lograr la síntesis de estos nuevos
materiales, se puede recurrir al uso combinado de los métodos de CRP con otros procedimientos
de polimerización (que pueden ser o no vivientes-controlados) como son las polimerizaciones
por apertura de anillo, iónicas, por coordinación o por procesos radicálicos convencionales en
diferentes medios de reacción. 28-29, 32
Figura 1: Materiales poliméricos que se pueden obtener por medio de la CRP.30
2.2.1 Fundamentos de la CRP. Consideraciones generales
Desde la publicación del trabajo de Otsu,26 muchas de las investigaciones se orientaron en
proveer un mejor carácter viviente a la polimerización iniciada por radicales libres; en la década
de los 90’s arribó con éxito el desarrollo de una de las tres de las estrategias más empleadas: la
polimerización controlada por radicales libres estables (SFRP, por sus siglas en inglés, en este
caso entra la NMP).29 Las otras dos técnicas mayormente recurridas son la transferencia de
átomo (ATRP, por sus siglas en inglés)30 y por transferencia degenerativa, en este caso con

Capítulo 2/ Antecedentes 2.2.1 Fundamentos de la CRP. Consideraciones generales
13
compuestos de yodo,33 porfirinas de cobalto34 o por compuestos de tiocarboniltio mediante
reacciones reversibles de adición-fragmentación (RAFT).31-32 Mecanisticamente, una de las
características de la CRP que lo distingue de la FRP es la presencia de un proceso de activación
reversible (Esquema 1).35 En esta etapa, la cadena dormida Pi-X está propensa a ser activada
por un radical polimérico Pi•. En presencia de monómero M en el medio de reacción, Pi• pasará
por un proceso de propagación hasta que sea desactivado nuevamente a Pi-X.
Pi
X Pi
.+
kact kp
X.
kdact
M
(activa)(durmiente)
Esquema 1: Reacciones reversibles de activación-desactivación que experimenta el radical propagante Pi• en
los sistemas de CRP
Desde el punto de vista de un intervalo de tiempo largo, las cadenas propagantes crecen
lentamente, por lo que el tiempo que existe entre la activación y la subsiguiente desactivación
de una misma cadena es típicamente de 0.1 a 10 ms.24 En el caso en que la cadena polimérica
viviente experimente estos ciclos de activación y desactivación sobre el periodo de
polimerización, todas las cadenas vivientes tendrán una posibilidad prácticamente igual de
crecer y se generará un producto de baja dispersidad. La CRP también se distingue por la (casi)
ausencia de las etapas de terminación semejante a la polimerización viviente aniónica (en su
forma ideal) a diferencia de la FRP que involucra etapas de terminación bimolecular,
transferencia de cadena y otras reacciones elementales.
Puesto que en una CRP se reduce marcadamente la etapa de terminación irreversible, se
espera que la concentración de radicales libres no varíe a lo largo de la reacción. Es por ello, de
acuerdo a la deducción presentada en el recuadro superior derecho de la Figura 2, que la
representación gráfica de la conversión de monómero (expresada como ln ([M]o/[M]) con
respecto al tiempo se ajusta a un comportamiento lineal lo cual indica que la reacción tiene un
comportamiento de primer orden con respecto a la concentración de monómero y que la
concentración de radicales libres [M•] permanece constante a lo largo de la reacción.30b, 36 El
valor de ln ([M]o/[M]) se determina fácilmente a partir del valor de la conversión experimental
p (ver recuadro inferior derecho de la Figura 2). Una posible desviación a este comportamiento
lineal es la existencia de una iniciación lenta o una reacción de terminación, las cuales modifican
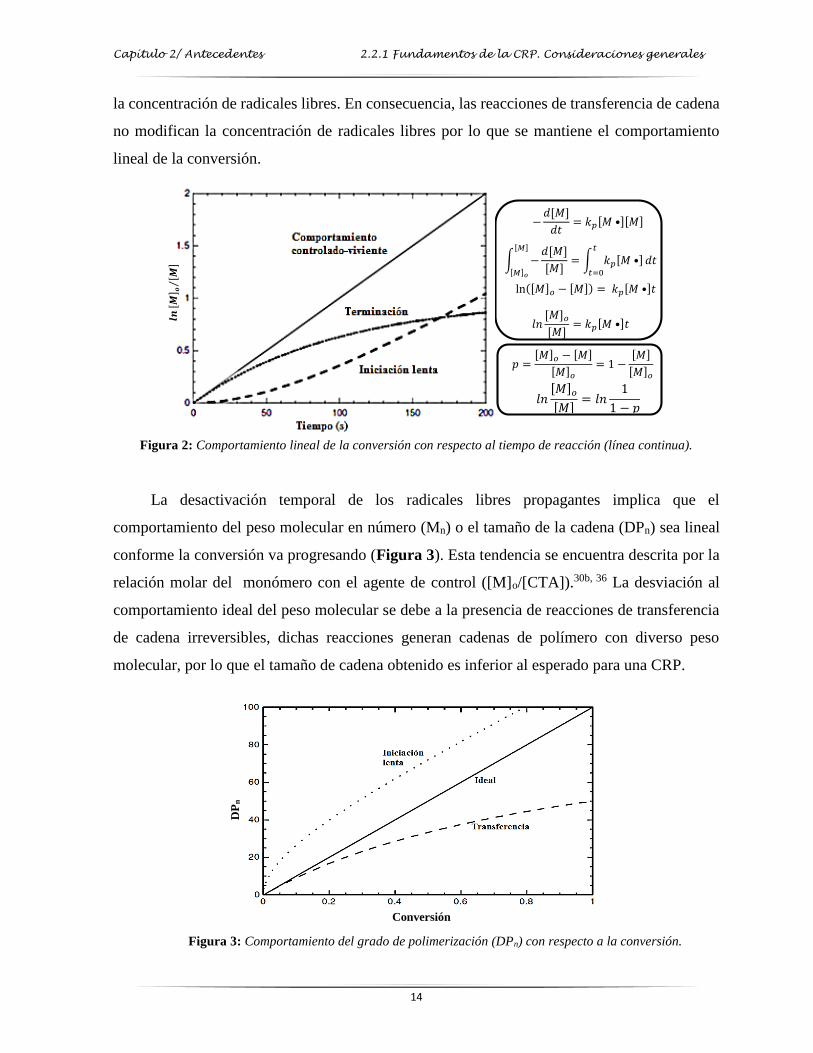
Capítulo 2/ Antecedentes 2.2.1 Fundamentos de la CRP. Consideraciones generales
14
la concentración de radicales libres. En consecuencia, las reacciones de transferencia de cadena
no modifican la concentración de radicales libres por lo que se mantiene el comportamiento
lineal de la conversión.
Figura 2: Comportamiento lineal de la conversión con respecto al tiempo de reacción (línea continua).
La desactivación temporal de los radicales libres propagantes implica que el
comportamiento del peso molecular en número (Mn) o el tamaño de la cadena (DPn) sea lineal
conforme la conversión va progresando (Figura 3). Esta tendencia se encuentra descrita por la
relación molar del monómero con el agente de control ([M]o/[CTA]).30b, 36 La desviación al
comportamiento ideal del peso molecular se debe a la presencia de reacciones de transferencia
de cadena irreversibles, dichas reacciones generan cadenas de polímero con diverso peso
molecular, por lo que el tamaño de cadena obtenido es inferior al esperado para una CRP.
Figura 3: Comportamiento del grado de polimerización (DPn) con respecto a la conversión.
−𝑑[𝑀]
𝑑𝑡= 𝑘𝑝[𝑀 •][𝑀]
−𝑑[𝑀]
[𝑀]= 𝑘𝑝[𝑀 •]
𝑡
𝑡=0
𝑑𝑡[𝑀]
[𝑀]𝑜
ln [𝑀]𝑜 − [𝑀] = 𝑘𝑝[𝑀 •]𝑡
𝑙𝑛[𝑀]𝑜[𝑀]
= 𝑘𝑝[𝑀 •]𝑡
𝑝 =[𝑀]𝑜 − [𝑀]
[𝑀]𝑜= 1 −
[𝑀]
[𝑀]𝑜
𝑙𝑛[𝑀]𝑜[𝑀]
= 𝑙𝑛1
1 − 𝑝
𝒍𝒏[ 𝑴
] 𝒐[ 𝑴
]⁄
Conversión
DP
n

Capítulo 2/ Antecedentes 2.2.1 Fundamentos de la CRP. Consideraciones generales
15
Adicionalmente, una iniciación lenta de las cadenas también produce una desviación al
comportamiento viviente-controlado de la polimerización, tal escenario se tiene cuando el
agente de control experimenta una lenta adición sobre el radical propagante, dando como
resultado una polimerización aproximándose a un proceso por FRP. Si el control en el
crecimiento de las cadenas es efectivo, se espera que la dispersión de la distribución de peso
molecular (Ɖ) sea estrecha; esto es, que ésta se encuentre constituida por cadenas del mismo
tamaño.
No obstante, a conversiones bajas, diferencias ligeras en el tamaño de las cadenas tiene
un mayor impacto sobre la dispersidad de la distribución de los pesos moleculares, la cual se
define como la razón que existe entre los promedios del peso molecular en peso y en número
(Ɖ=Mw/Mn). Es así que este parámetro tiende a disminuir a valores cercanos a la unidad a
medida que avanza la conversión. Por convención, se ha tomado el valor de 1.5 como límite
superior para considerar a una polimerización radicálica como controlada ya que una FRP
convencional no puede tener valores menores a este límite. Finalmente, la funcionalización con
el agente de control en al menos una de las extremidades de la cadena polimérica permite que
en una etapa posterior sea posible una extensión de cadena o formación de un segundo bloque,
al mismo tiempo, esto permite evaluar el carácter viviente de la CRP. No obstante, el que alguna
de las características descritas en los enunciados anteriores no se cumpla, no implica que una
polimerización radicálica no llegue a tener características controladas-vivientes.
El gran auge que han tenido las técnicas de CRP se refleja en el número de reportes
científicos publicados en revistas con arbitraje internacional (Figura 4); estas publicaciones
comprenden desde trabajos hechos en la síntesis de controladores, el entendimiento del
mecanismo involucrado, hasta la obtención de diversas arquitecturas macromoleculares. Aun
cuando se tiene que el número de trabajos en ATRP supera en conjunto los reportes realizados
en SFRP y RAFT, no significa que estas dos últimas técnicas sean menos efectivas en proveer
características vivientes a la polimerización. De hecho, cada técnica tiene ventajas y
limitaciones que generan su desarrollo continuo. Actualmente en México, diversos grupos de
investigación han dirigido sus trabajos hacia el modelamiento de la cinética de polimerización,37
la obtención de copolímeros y/o nuevos materiales con estructuras definidas38 o la síntesis de
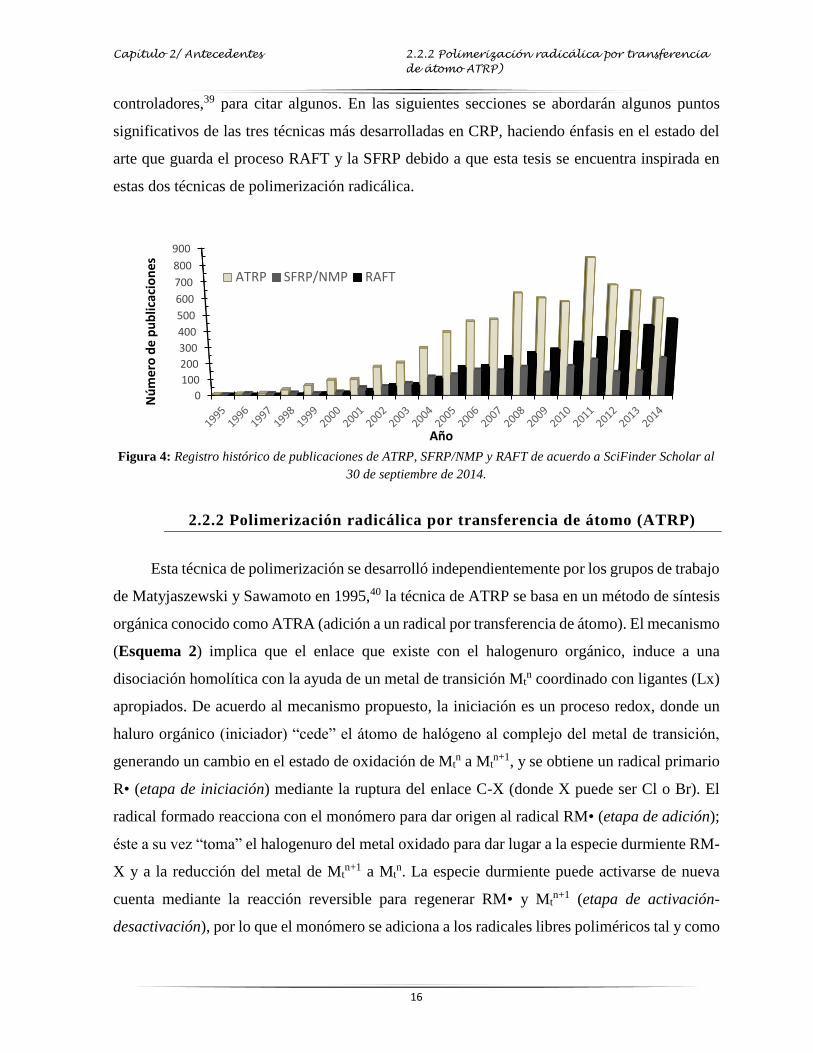
Capítulo 2/ Antecedentes 2.2.2 Polimerización radicálica por transferencia
de átomo ATRP)
16
controladores,39 para citar algunos. En las siguientes secciones se abordarán algunos puntos
significativos de las tres técnicas más desarrolladas en CRP, haciendo énfasis en el estado del
arte que guarda el proceso RAFT y la SFRP debido a que esta tesis se encuentra inspirada en
estas dos técnicas de polimerización radicálica.
Figura 4: Registro histórico de publicaciones de ATRP, SFRP/NMP y RAFT de acuerdo a SciFinder Scholar al
30 de septiembre de 2014.
2.2.2 Polimerización radicálica por transferencia de átomo (ATRP)
Esta técnica de polimerización se desarrolló independientemente por los grupos de trabajo
de Matyjaszewski y Sawamoto en 1995,40 la técnica de ATRP se basa en un método de síntesis
orgánica conocido como ATRA (adición a un radical por transferencia de átomo). El mecanismo
(Esquema 2) implica que el enlace que existe con el halogenuro orgánico, induce a una
disociación homolítica con la ayuda de un metal de transición Mtn coordinado con ligantes (Lx)
apropiados. De acuerdo al mecanismo propuesto, la iniciación es un proceso redox, donde un
haluro orgánico (iniciador) “cede” el átomo de halógeno al complejo del metal de transición,
generando un cambio en el estado de oxidación de Mtn a Mt
n+1, y se obtiene un radical primario
R• (etapa de iniciación) mediante la ruptura del enlace C-X (donde X puede ser Cl o Br). El
radical formado reacciona con el monómero para dar origen al radical RM• (etapa de adición);
éste a su vez “toma” el halogenuro del metal oxidado para dar lugar a la especie durmiente RM-
X y a la reducción del metal de Mtn+1 a Mt
n. La especie durmiente puede activarse de nueva
cuenta mediante la reacción reversible para regenerar RM• y Mtn+1 (etapa de activación-
desactivación), por lo que el monómero se adiciona a los radicales libres poliméricos tal y como
0
100
200
300
400
500
600
700
800
900
Nú
me
ro d
e p
ub
licac
ion
es
Año
ATRP SFRP/NMP RAFT

Capítulo 2/ Antecedentes 2.2.2 Polimerización radicálica por transferencia
de átomo ATRP)
17
es en una polimerización convencional por radicales libres, con la salvedad de que
posteriormente puede desactivarse (estado durmiente). La técnica de ATRP requiere una
iniciación rápida y una desactivación reversible de las especies activas para permitir el
crecimiento homogéneo y uniforme a través del sistema de reacción y por consiguiente la
obtención de distribuciones moleculares estrechas (Ɖ<1.5). Existen un buen número de haluros
iniciadores disponibles comercialmente.
Esquema 2: Mecanismo de polimerización por ATRP
Al comparar ATRP con los otros dos métodos importantes de CRP se observa que esta
técnica de polimerización demanda cuidado al definir el sistema multicomponente para que este
logre ser eficiente: haluro iniciador, catalizador de metal de transición y el ligando soluble. Se
han establecido rutas sintéticas que van desde pequeñas moléculas hasta macro iniciadores para
la obtención de haluros iniciadores. El complejo del metal de transición utilizado en el proceso
de iniciación es de vital importancia y juega un papel fundamental en la determinación de la
posición de equilibrio de la transferencia del átomo y éste determina la dinámica del intercambio
entre las especies activa y durmiente.
Los complejos de metales de transición más exitosos están basados en cobre debido a su
bajo costo y a su gran versatilidad; sin embargo, otros complejos de metales de transición han
sido utilizados exitosamente para ATRP tales como Pd, Re, Fe, Ru, Mo y Ni.40b, 41 Algunas
clases de monómeros que pueden ser polimerizados por esta técnica incluyen derivados
estirénicos, acrilatos, acrilonitrilos, metacrilatos y raramente con ácidos metacrílicos y
acrilamidas.42

Capítulo 2/ Antecedentes 2.2.3 Polimerización radicálica controlada por radicales
libres estables (SFRP)-Polimerización radicálica mediada por nitróxidos (NMP)
18
Adicionalmente, debido a su amplia aplicabilidad y al número de grupos de investigación
que se han enfocado en ATRP, se han obtenido materiales poliméricos con funcionalidades
novedosas como copolímeros alternados, en bloque, en injerto, copolímeros dendríticos y de
tipo estrella, así como también esquemas de polimerización funcionales para preparar polímeros
telequélicos y macromonómeros.43 Con la finalidad de minimizar la oxidación del catalizador y
maximizar su desempeño, se han desarrollado recientemente diferentes estrategias tales como:
AGET, ICR y ARGET.44 No obstante, la principal desventaja de ATRP es la necesidad de
remover el complejo del metal de transición del polímero.
2.2.3 Polimerización radicálica controlada por radicales libres estables
(SFRP)-Polimerización radicálica mediada por nitróxidos (NMP)
El primer ejemplo de polimerización radicálica viviente con nitróxidos fue reportado por
Moad et al.45 A principios de los años 80, demostraron que el uso de los nitróxidos (tal como el
2, 2, 6, 6-tetrametilpiperidiniloxi (TEMPO)) como alcoxiaminas, funcionaban como atrapador
de radicales libres en la polimerización de estireno y metacrilato de metilo. Este proceso
permitió un control limitado sobre el proceso de polimerización; posteriormente Georges et
al.,46 introdujeron mejoras en el proceso de obtención de resinas de poliestireno con
dispersidades estrechas, usando TEMPO con peróxido de benzoilo como iniciador a 130°C. Los
nitróxidos, como el TEMPO y sus derivados, se recombinan fácilmente con radicales libres
efímeros mediante la formación de un enlace C-O, tal y como sucede en una reacción de
polimerización radicálica convencional, con la diferencia que los radicales libres nitróxidos
actúan como un terminador que atrapa reversiblemente los macro radicales libres formados,
generando así, un polímero durmiente; es decir, un polímero con una alcoxiamina como grupo
final (Esquema 3).
Esquema 3: Desactivación temporal del radical propagante por el radical nitróxido.

Capítulo 2/ Antecedentes 2.2.3 Polimerización radicálica controlada por radicales
libres estables (SFRP)-Polimerización radicálica mediada por nitróxidos (NMP)
19
La técnica de polimerización SFRP (a la que pertenece la polimerización radicálica
mediada por nitróxidos, NMP), ofrece un crecimiento controlado de las cadenas poliméricas a
través del concepto de terminación reversible como se puede apreciar en el Esquema 4. La
NMP se basa en el mecanismo de terminación reversible entre los macro radicales libres
propagantes en crecimiento y el nitróxido (que actúa como agente de control de la
polimerización) para obtener una macroalcoxiamina como especie predominante. Esta
funcionalidad durmiente (especie inactiva) se puede regenerar a un radical propagante y a un
nitróxido por medio de un rompimiento homolítico si se trabaja a una temperatura adecuada.
Esquema 4. Equilibrio de activación-desactivación en la NMP. Iniciación por un proceso a) unimolecular, b)
bimolecular.
Es así como se conforma un equilibrio dinámico de activación-desactivación entre las
especies participantes y este equilibrio presenta la ventaja de ser un proceso puramente térmico
donde no es necesaria la participación de un catalizador o algún otro medio de intercambio
bimolecular para llevar a cabo la polimerización. La cinética de polimerización está gobernada
tanto por dicho equilibrio dinámico (con una constante de equilibrio K=kd/kc) y por el efecto de
radical persistente (PRE, por sus siglas en inglés).47 El concepto de PRE fue elucidado por
Fischer y Fukuda,24 quienes explicaron teóricamente la minimización de las reacciones
terminales irreversibles. Este concepto consiste en lo siguiente (Figura 5): considerando un
compuesto RY que se descompone en un radical efímero (transitorio) R• y en un radical
persistente (estable) Y• con una concentración inicial de radicales libres igual a cero; al inicio
de la reacción, las concentraciones de ambas especies radicálicas aumentan linealmente con el
tiempo, y se rigen por un coeficiente de velocidad de descomposición kd. Este período (que es
un régimen de pre-equilibrio) dura hasta que la concentración de radicales libres total se
convierte en lo suficientemente grande como para que las especies de radicales libres puedan
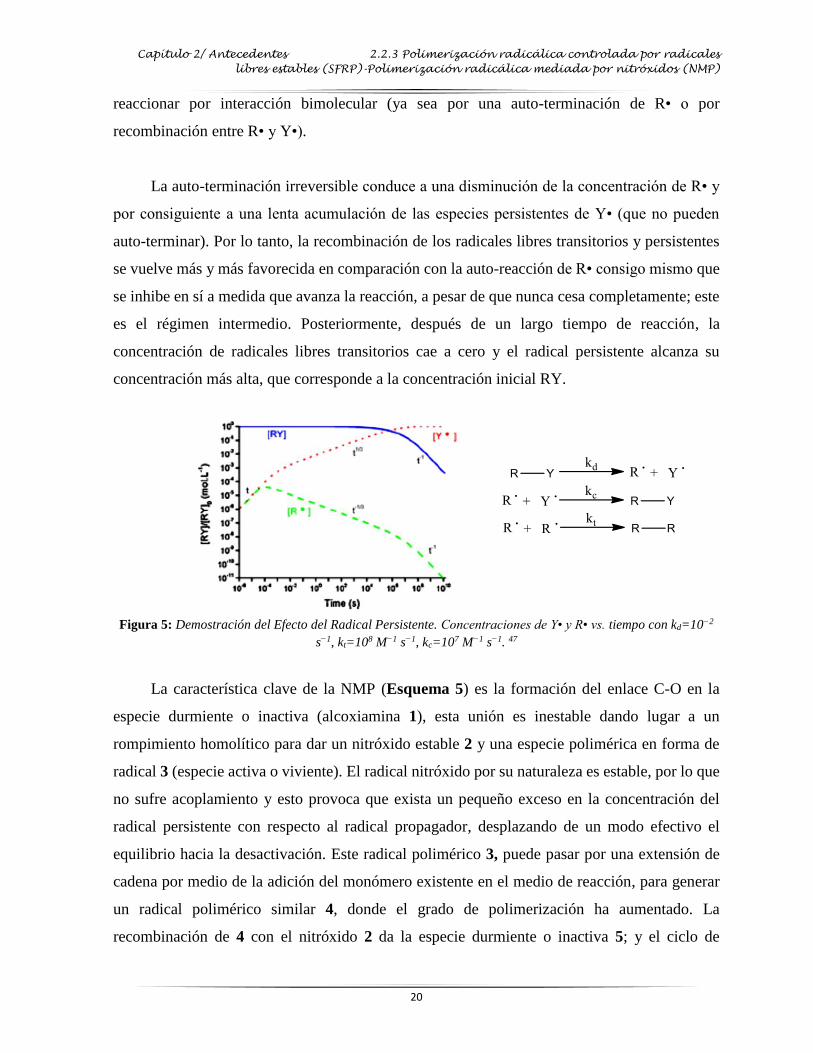
Capítulo 2/ Antecedentes 2.2.3 Polimerización radicálica controlada por radicales
libres estables (SFRP)-Polimerización radicálica mediada por nitróxidos (NMP)
20
reaccionar por interacción bimolecular (ya sea por una auto-terminación de R• o por
recombinación entre R• y Y•).
La auto-terminación irreversible conduce a una disminución de la concentración de R• y
por consiguiente a una lenta acumulación de las especies persistentes de Y• (que no pueden
auto-terminar). Por lo tanto, la recombinación de los radicales libres transitorios y persistentes
se vuelve más y más favorecida en comparación con la auto-reacción de R• consigo mismo que
se inhibe en sí a medida que avanza la reacción, a pesar de que nunca cesa completamente; este
es el régimen intermedio. Posteriormente, después de un largo tiempo de reacción, la
concentración de radicales libres transitorios cae a cero y el radical persistente alcanza su
concentración más alta, que corresponde a la concentración inicial RY.
Figura 5: Demostración del Efecto del Radical Persistente. Concentraciones de Y• y R• vs. tiempo con kd=10−2
s−1, kt=108 M−1 s−1, kc=107 M−1 s−1. 47
La característica clave de la NMP (Esquema 5) es la formación del enlace C-O en la
especie durmiente o inactiva (alcoxiamina 1), esta unión es inestable dando lugar a un
rompimiento homolítico para dar un nitróxido estable 2 y una especie polimérica en forma de
radical 3 (especie activa o viviente). El radical nitróxido por su naturaleza es estable, por lo que
no sufre acoplamiento y esto provoca que exista un pequeño exceso en la concentración del
radical persistente con respecto al radical propagador, desplazando de un modo efectivo el
equilibrio hacia la desactivación. Este radical polimérico 3, puede pasar por una extensión de
cadena por medio de la adición del monómero existente en el medio de reacción, para generar
un radical polimérico similar 4, donde el grado de polimerización ha aumentado. La
recombinación de 4 con el nitróxido 2 da la especie durmiente o inactiva 5; y el ciclo de

Capítulo 2/ Antecedentes 2.2.3 Polimerización radicálica controlada por radicales
libres estables (SFRP)-Polimerización radicálica mediada por nitróxidos (NMP)
21
homólisis-adición de monómero-recombinación se repite hasta terminar con todo el monómero
presente.
Esquema 5: Representación de la polimerización del estireno mediada por nitróxidos.
Numerosas revisiones están disponibles en la literatura para este tipo de proceso de
polimerización por radicales libres.29b, 29d, 48 Cabe señalar que en el punto de la disociación
inicial, las concentraciones del radical persistente y de la especie viviente son prácticamente
iguales en las etapas tempranas de la reacción, por lo que ocurre una terminación irreversible
entre los radicales libres propagadores y los iniciadores generando pequeñas cantidades de las
moléculas de bajo peso molecular en el principio de la polimerización. La NMP se puede iniciar
empleando el proceso bimolecular, donde se hace uso de un iniciador de radicales libres
convencionales (como el peróxido de benzoilo, BPO o el 2, 2’-azobisisobutironitrilo, AIBN por
ejemplo) en combinación con el radical nitróxido.46 Este sistema tiene la ventaja de usar el
proceso de polimerización por radicales libres convencionales con la diferencia de solo
adicionar radicales libres nitróxido al medio de reacción (esto puede ser altamente deseable
desde el punto de vista económico y práctico). La relación molar de [nitróxido]o/[iniciador]o es
de gran importancia dentro de la NMP, debido a que la cinética de esta polimerización está
gobernada por la cantidad de nitróxido en exceso presente después de la etapa de iniciación.49
Una posible consecuencia de la relación molar nitróxido/iniciador de alimentación a la
polimerización se presenta con el exceso de nitróxido libre que se da en el equilibrio de
activación-desactivación, ocasionando a que éste se desplace hacia las especies durmientes, y

Capítulo 2/ Antecedentes 2.2.3 Polimerización radicálica controlada por radicales
libres estables (SFRP)-Polimerización radicálica mediada por nitróxidos (NMP)
22
trayendo como consecuencia una disminución de la velocidad de polimerización. Por otra parte,
todos los iniciadores térmicos sufren de la dificultad para determinar precisamente la eficiencia
de los radicales libres primarios que se producen por su la descomposición térmica para inducir
la polimerización (esto se debe al efecto de “jaula” –cage en inglés- y a la descomposición
inducida), dependiendo de la naturaleza de los grupos iniciantes, la mayoría de estos radicales
libres primarios pasan por reacciones de reordenamiento o de fragmentación.50 Es por ello, que
la relación [nitróxido]o/[iniciador]o originalmente fue de 1.3; empero, Dollin et al.,51
investigaron la influencia que tiene esta relación en la cinética y demostraron que si este valor
es finamente optimizado y puede disminuirse hasta 0.95 en ciertas condiciones de reacción con
lo que la cinética del sistema podría verse apreciablemente acelerada.
La gran limitante que tiene la NMP cuando se hace uso del TEMPO y sus derivados, es el
número reducido de monómeros polimerizables por esta técnica, los cuales se restringen a
estirenos sustituidos principalmente. Recientemente, el desarrollo de nuevos tipos de nitróxidos
(que son considerados nitróxidos de segunda generación, Esquema 6);52 o el uso combinado
del TEMPO y sus derivados con compuestos α-hidroxicarbonilos como aditivos53 han permitido
el control de la polimerización de otros tipos de especies monoméricas tales como acrilato de
butilo (BA), por citar un ejemplo. No obstante, a la fecha no se ha logrado la polimerización de
monómeros metacrílicos debido a factores estéricos y a que este tipo de nitróxidos tienen la
posibilidad de abstraer protones β en lugar de formar alcoxiaminas con los radicales libres
propagantes de la polimerización.44
Esquema 6: Derivados de nitróxidos empleados exitosamente como controladores en la NMP: N-tert-butil-
dietil-fósforo-2,2-dimetil-propil N-propil N-oxido (DEPN, también conocido como SG-1) y el 2, 2, 5-trimetil-4-
fenil-3-azahexano N-oxido (TIPNO)

Capítulo 2/ Antecedentes 2.2.4 Polimerización radicálica por transferencia de cadena
de adición-fragmentación reversible (RAFT)
23
Otra forma de llevar a cabo una polimerización controlada por nitróxidos es mediante la
iniciación unimolecular,48a, 54 donde la molécula iniciante se descompone en el radical iniciante
de la polimerización y el nitróxido correspondiente. A este compuesto (originalmente
denominado unímero) se le llama alcoxiamina iniciante. Debido a su particular estructura, se
obtiene una relación molar 1:1 del radical iniciante: nitróxido después de la disociación
homolítica. Experimentalmente, se ha observado que los sistemas unimoleculares llevan a un
mejor control del peso molecular y sus correspondientes dispersidades comparados con los
sistemas bimoleculares.54a
2.2.4 Polimerización radicálica por transferencia de cadena de adición -
fragmentación reversible (RAFT)
En 1998, Rizzardo et al.,55 reportaron un nuevo tipo de polimerización radicálica
controlada, a la cual designaron con el nombre de proceso de transferencia de cadena por
reacciones reversibles de adición-fragmentación (RAFT). Esta técnica se fundamenta en la
transferencia degenerativa y no en el efecto de radical persistente como en el caso de la NMP y
ATRP. El proceso RAFT tiene sus orígenes en trabajos previos realizados con macromonómeros
metacrílicos.56 Dichos macromonómeros funcionaron como agentes de transferencia en las
reacciones de polimerización con monómeros metacrílicos (Esquema 7).
Esquema 7: Mecanismo de reacción para macro monómeros de MMA como agente de transferencia.
Para hacer más eficiente la reacción de transferencia fue necesario desarrollar agentes más
reactivos, el éxito de esta técnica de polimerización se dio con el uso de compuestos tipo
tiocarboniltio como: ditioésteres, ditiocarbamatos, xantatos y tritiocarbonatos, los cuales
mostraron ser adecuados como agentes de transferencia reversibles. La gran cantidad de reportes

Capítulo 2/ Antecedentes 2.2.4 Polimerización radicálica por transferencia de cadena
de adición-fragmentación reversible (RAFT)
24
sobre esta invención muestran su aplicabilidad, tanto en sistemas homogéneos como
heterogéneos, para una amplia gama de monómeros y de condiciones de reacción. La
característica habitual de los agentes RAFT es que estos cuentan con un doble enlace C=S, que
es propenso a sufrir una adición por parte del radical propagante debido a su baja energía de
enlace, comparada con la energía de enlace C-S y un grupo R, que se libera fácilmente en forma
de un radical que reinicia el proceso de la polimerización.
En el Esquema 8 se presenta el mecanismo propuesto por el grupo de CSIRO.55 La etapa
I corresponde al proceso de iniciación, en el que se generan los radicales libres que inician el
crecimiento de cadenas macromoleculares Pm•. El pre-equilibrio del proceso RAFT mostrado en
la etapa II, se lleva a cabo en los primeros instantes de la reacción de los macro radicales libres
propagantes -generalmente oligoméricos- adicionándose al doble enlace C=S del agente RAFT
6, resultando un radical intermediario centrado sobre el carbono en 7. En la etapa III, la adición
de los radicales libres primarios generados por la descomposición del iniciador (I•) sobre el
agente RAFT inicial, generalmente no se considera; sin embargo, puede llegar a tener un efecto
importante en el caso de altas concentraciones del radical aducto 7 formado en el pre-equilibrio
que experimenta una ruptura β, ya sea en el sentido de los reactivos o liberando un radical re-
iniciante R•, de tal manera que se forma un polímero funcionalizado con el grupo –SC(S)Z. En
la etapa IV ocurre una reacción similar a la sucedida en la etapa II, salvo que en este caso, un
macro radical reacciona con el agente RAFT polimérico, conocido como especie durmiente,
para establecer reacciones periódicas en equilibrio entre las cadenas “durmientes” y “vivientes”
con coeficientes de reacción kβ y k-β.
Es claro que el esquema de reacción de la polimerización RAFT se sobrepone al de una
polimerización FRP incluyendo la reacción de terminación (etapa V). El efecto que tiene el
agente RAFT en la polimerización es notorio cuando la constante de transferencia es alta
(definida como Ctr=kp/ktr).

Capítulo 2/ Antecedentes 2.2.4 Polimerización radicálica por transferencia de cadena
de adición-fragmentación reversible (RAFT)
25
Esquema 8: Mecanismo de reacción de la polimerización radicálica controlada tipo RAFT.
En este caso, el equilibrio dinámico entre las especies propagantes y las especies
funcionalizadas con el grupo carbonilo produce un crecimiento de las cadenas involucradas de
un modo uniforme, debido a que la polimerización RAFT se lleva a cabo generalmente con una
gran cantidad de agente de transferencia con respecto a la cantidad de radicales libres primarios
generados por el iniciador. Es importante resaltar que la relación molar de monómero-agente
RAFT, determina el peso molecular en número, como se puede apreciar en la ecuación 2 (esta
aproximación teórica también se puede aplicar para la polimerización mediada por nitróxidos)
debido a que durante la reacción, cada cadena de polímero creada consume una molécula de
agente RAFT (o de iniciador, cuya cantidad es muy pequeña con respecto al agente RAFT).
𝑀𝑛,𝑡𝑒𝑜̅̅ ̅̅ ̅̅ ̅̅ =
[𝑀]𝑜∗𝑀𝑚∗𝑐𝑜𝑛𝑣𝑒𝑟𝑠𝑖ó𝑛
[𝑅𝐴𝐹𝑇]𝑜+ 𝑀𝑅𝐴𝐹𝑇 (2)
[M]o es la concentración molar inicial del monómero, Mm es el peso molecular del monómero,
conversión es la conversión en peso del polímero, [RAFT]o y MRAFT son la concentración inicial
y peso molecular del agente RAFT respectivamente.
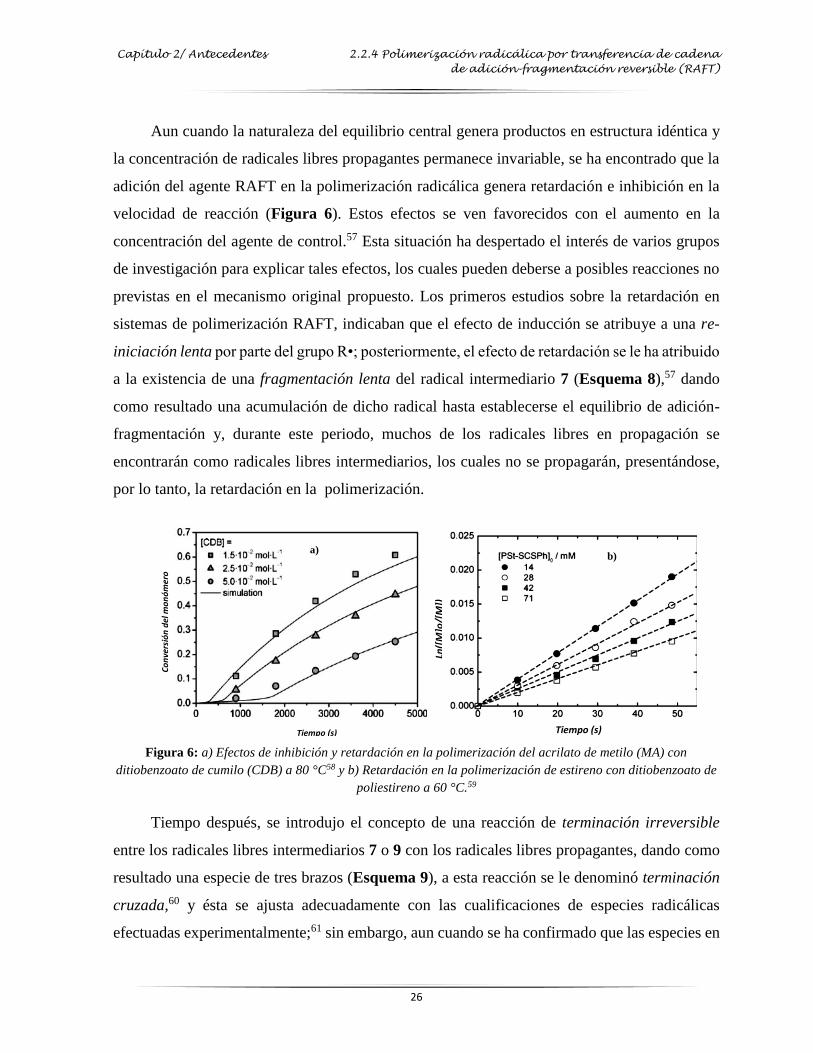
Capítulo 2/ Antecedentes 2.2.4 Polimerización radicálica por transferencia de cadena
de adición-fragmentación reversible (RAFT)
26
Aun cuando la naturaleza del equilibrio central genera productos en estructura idéntica y
la concentración de radicales libres propagantes permanece invariable, se ha encontrado que la
adición del agente RAFT en la polimerización radicálica genera retardación e inhibición en la
velocidad de reacción (Figura 6). Estos efectos se ven favorecidos con el aumento en la
concentración del agente de control.57 Esta situación ha despertado el interés de varios grupos
de investigación para explicar tales efectos, los cuales pueden deberse a posibles reacciones no
previstas en el mecanismo original propuesto. Los primeros estudios sobre la retardación en
sistemas de polimerización RAFT, indicaban que el efecto de inducción se atribuye a una re-
iniciación lenta por parte del grupo R•; posteriormente, el efecto de retardación se le ha atribuido
a la existencia de una fragmentación lenta del radical intermediario 7 (Esquema 8),57 dando
como resultado una acumulación de dicho radical hasta establecerse el equilibrio de adición-
fragmentación y, durante este periodo, muchos de los radicales libres en propagación se
encontrarán como radicales libres intermediarios, los cuales no se propagarán, presentándose,
por lo tanto, la retardación en la polimerización.
Figura 6: a) Efectos de inhibición y retardación en la polimerización del acrilato de metilo (MA) con
ditiobenzoato de cumilo (CDB) a 80 °C58 y b) Retardación en la polimerización de estireno con ditiobenzoato de
poliestireno a 60 °C.59
Tiempo después, se introdujo el concepto de una reacción de terminación irreversible
entre los radicales libres intermediarios 7 o 9 con los radicales libres propagantes, dando como
resultado una especie de tres brazos (Esquema 9), a esta reacción se le denominó terminación
cruzada,60 y ésta se ajusta adecuadamente con las cualificaciones de especies radicálicas
efectuadas experimentalmente;61 sin embargo, aun cuando se ha confirmado que las especies en
Tiempo (s)
Co
nve
rsió
n d
el m
on
óm
ero
Tiempo (s)
Ln([
M]o
/[M
])
a) b)

Capítulo 2/ Antecedentes 2.2.4 Polimerización radicálica por transferencia de cadena
de adición-fragmentación reversible (RAFT)
27
forma de estrella de tres brazos son estables a las temperaturas típicas de polimerización,59 no
se han aislado productos derivados de este tipo de reacción en los sistemas que presentan
retardación.62 Recientemente se ha sugerido un modelo hibrido o compuesto que combina
ambas propuestas,63 adicionalmente, el grupo de trabajo de Buback at al.64 se han enfocado en
las determinaciones de las constantes de equilibrio, Keq, lo que permite comprender el
comportamiento cinético del mecanismo de reacción que se lleva a cabo en el proceso RAFT.
Ellos han observado que existe una lenta adición de los radicales propagantes al agente RAFT
y una lenta fragmentación del radical intermediario.
Esquema 9: Reacción propuesta para la terminación cruzada entre radicales libres propagantes y radicales
libres intermediarios.
La capacidad que tiene un agente RAFT para controlar la polimerización está gobernada
por dos grupos químicos conocidos como Z y R. El grupo Z determina en gran medida la
velocidad de adición de los radicales libres a las especies durmientes, mientras que R debe ser
un buen grupo saliente que al mismo tiempo determina la velocidad de fragmentación del
intermediario; por lo tanto, la efectividad del agente RAFT en proveer un carácter viviente se
asocia a una alta constante de transferencia que garantiza una rápida velocidad de intercambio
entre las cadenas activas y durmientes. Los factores que afectan la velocidad de adición en la
polimerización RAFT dependen de la constante de adición (kβ) la que está en función de la
reactividad del radical propagante y del agente RAFT, aquí se involucran los efectos de
resonancia, polares y estéricos de las especies participantes.
Debido a que los radicales libres se adicionan sobre el átomo de azufre del grupo
tiocarbonilo, la velocidad de adición aumenta con la activación del doble enlace por los
sustituyentes Z y R. De acuerdo a la literatura, los sustituyentes que contienen orbitales π (p. ej.
fenilo), inducen un aumento en el valor de kβ por resonancia, en comparación con sustituyentes
que contienen un átomo con electrones (p. ej. halógenos, -OR, -NR2, -SR).50 Considerando los
efectos polares, los sustituyentes electroatractores (halógenos, -COOR, CN) permiten un

Capítulo 2/ Antecedentes 2.2.4 Polimerización radicálica por transferencia de cadena
de adición-fragmentación reversible (RAFT)
28
incremento en el valor de kβ, ya que los radicales libres atacantes son más nucleófilos. La
influencia que los sustituyentes ejercen individualmente por dichos efectos sobre la activación
del doble enlace, es difícil de cuantificar debido a que éstos actúan simultáneamente y en
algunos casos en forma antagónica. El sustituyente R juega un papel limitado en la activación
del doble enlace C=S (contrario al fuerte impacto que exhibe el sustituyente Z), por lo que hasta
el día de hoy se tienen cinco clasificaciones generales de agentes RAFT (Tabla 1).
Tabla 1: Clasificación de agentes RAFT de acuerdo a la naturaleza química de Z
Clasificación Z
Ditioésteres -R1 (alquil o aril)
Tritiocarbonatos -SR1
Ditiocarbamatos -NR1R2
Ditiocarbonatos
(xantatos)
-OR1
En la Figura 7 se presenta el intervalo de aplicación de cada tipo de agente RAFT. Como
regla general, se asume que la constante de transferencia de los agentes RAFT disminuye de la
siguiente manera: ditiobenzoatos > tritiocarbonatos ≈ Ditiocarbonatos (xantatos) >
ditiocarbamatos.50
Figura 7: Intervalo de aplicación de los diferentes tipos de agentes RAFT.65
Por otra parte, algunos de los factores que afectan principalmente a la velocidad de
fragmentación son:

Capítulo 2/ Antecedentes 2.2.4 Polimerización radicálica por transferencia de cadena
de adición-fragmentación reversible (RAFT)
29
Efecto cinético. La constante de fragmentación (k-β) depende tanto de la estabilización
del radical intermediario como de la fragilidad del enlace C-S.
Efecto de selectividad. La fragmentación preferencial resulta más favorecida por los
efectos estéricos, electrónicos y polares para los grupos R y Pn del radical
intermediario. Como regla general, se considera un buen grupo saliente al radical
que una vez liberado se estabiliza fácilmente y está impedido estéricamente.66 La
habilidad de fragmentación se relaciona al grado de sustitución del radical que se
fragmenta y la naturaleza de sus sustituyentes. Siendo así se tiene que: carbono
primario < secundario < terciario, y con la presencia del sustituyente estabilizante
aumenta esta capacidad (por efectos de resonancia o polares, por ejemplo para los
grupos –Ph, -CN).
Entre los grupos R más empleados están los siguientes sustituyentes: cianoisopropilo (-
C(CH3)2CN), cumilo (-C(CH3)2Ph), feniletilo (-CH(CH3)Ph), bencilo (-CH2Ph). En la Figura 8
se presenta el intervalo de aplicación para cada tipo de grupo saliente R. En este caso, la
constante de transferencia para diversos grupos R disminuye de la siguiente manera:
C(CH3)(CH2(CH2)2OH)CN > C(CH3)CN ≈ C(CH3)2Ph > C(CH3)2CO2Et > C(CH3)2CONH-
alquilo > C(CH3)2CH2C(CH3)3 ≥ CH(CH3)Ph > C(CH3)3 ≈ CH2Ph.66a
Figura 8: Intervalo de acción de los agentes RAFT de acuerdo a la naturaleza del grupo R (con igual o diferente
grupo Z).65 La línea punteada indica poca influencia en la actividad del monómero correspondiente.

Capítulo 2/ Antecedentes 2.2.5 Comparación de los métodos más representativos de la CRP
30
2.2.5 Comparación de los métodos más representativos de la CRP
A fin de resumen, y dado que cada uno de los métodos de CRP antes mencionados
presenta ventajas y desventajas entre ellos, algunas de ellas se presentan en la Tabla 2
comparando cuatro características típicas que incluyen: tipo de monómeros a polimerizar,
condiciones de reacción (esto es, temperatura, sensibilidad a impurezas, etc.), la naturaleza de
los grupos funcionales y la existencia de aditivos tales como catalizadores y/o aceleradores.
Tabla 2: Comparativo de las técnicas de polimerización NMP, ATRP y RAFT
NMP ATRP RAFT
Monómeros Estireno con TEMPO y
derivados. Acrilatos y
acrilamidas en
combinación TEMPO y
derivados con aditivos
acelerantes, nitróxidos de
2da generación. No hay
para metacrilatos
Casi todos los
monómeros vinílicos
con grupos
activadores. No existe
para el acetato de
vinilo
Casi todos los
monómeros vinílicos.
Condiciones
de reacción
T elevadas (> 120 para
TEMPO). Sensible a la
presencia de oxígeno
Amplio intervalo de T
(-30 a 150 °C).
Relativa tolerancia a
O2 y a los inhibidores
con Mtn
Amplio intervalo de T
(Tamb a 200 °C) aunque
T elevadas a monómeros
menos reactivos.
Sensible al O2
Iniciadores/
grupos
terminales
Alcoxiaminas.
Térmicamente inestables.
Relativamente caros.
(Pseudo) halogenuros.
Térmica y
fotoquímicamente
estables. Barata y
disponible.
Ditioésteres, yoduros
poco estables térmicos y
fotoquímicamente.
Relativamente caro. El
polímero adquiere color
y olor.
Aditivos No son necesarios. La
NMP puede acelerarse con
compuestos acilos.
Los catalizadores
deben removerse y
reciclarse. Puede o no
usar aditivos.
Iniciadores de RL
convencionales.
2.3 Síntesis de copolímeros de tipo injerto
Las poliolefinas que contienen diferentes grados de polietileno (PE) y polipropileno (PP)
dentro de su composición estructural tienen un gran valor comercial en el mercado. En 2006, el
PE y el PP conformaron el 69% del tonelaje de los termoplásticos producidos en los Estados
Unidos y Canadá.67 Cabe recordar que las poliolefinas son materiales de bajo costo, que

Capítulo 2/ Antecedentes 2.3 Síntesis de copolímeros de tipo injerto
31
presentan una excelente resistencia química aunada con buenas propiedades mecánicas y fácil
procesabilidad.68 Sin embargo, carecen de la funcionalidad reactiva o polar, lo que limita su
potencial para la adhesión o la compatibilidad con otros polímeros. Por lo tanto, una mezcla de
poliolefinas con polímeros que contengan una funcionalidad reactiva o polar, da la oportunidad
a ser una posible ruta para mejorar las propiedades finales de las resinas debido a que la mayoría
de las mezclas de poliolefinas con polímeros polares son completamente inmiscibles. La
búsqueda para obtener un copolímero que sirva como un puente de unión entre dichas mezclas
inmiscibles y que logre estabilizar la composición deseada, es uno de los objetivos prioritarios
de la ciencia y tecnología de polímeros.
Muchos polímeros son candidatos idóneos a ser modificados químicamente para aumentar
sus aplicaciones; destacan de este grupo en particular, las poliolefinas y los elastómeros. Los
elastómeros son materiales poliméricos muy atractivos desde el punto de vista científico e
industrial por sus diversas aplicaciones potenciales como compatibilizantes,69 reforzantes,12
adhesivos,14 entre otras aplicaciones, por lo que los productos a base de estos sustratos
conteniendo injertos controlados de polímero con grupos polares tales como anhídrido maleico
(MAH), metacrilato de glicidilo (GMA), acrilonitrilo (AN), etc.; se pueden considerar
materiales de gran valía y de un buen impacto comercial. El injerto de grupos polares, cadenas
de copolímero con grupos polares, o bien la síntesis “in situ” de ramas de homopolímeros o
copolímeros sobre un sustrato polimérico, es también conocida como síntesis de copolímeros
de injerto. Muchos de los métodos para obtener los copolímeros de injerto se pueden realizar
por medio de radicales libres; sin embargo, también es posible hacerlo por métodos iónicos.23
Existen tres técnicas básicas para sintetizar este tipo de copolímeros: injerto sobre (“grafting
onto”), injerto desde (“grafting from”) e injerto a través de (“grafting through”).
La copolimerización de injerto sobre,
involucra la reacción entre los grupos funcionales
de dos tipos diferentes de polímeros; por lo que
se requiere de un sustrato con grupos capaces de
modificarse por medios químicos; esto es, la presencia de una cadena polimérica con un grupo
funcional terminal, y la presencia de un grupo funcional complementario afín al grupo funcional
A
B+
A B
Injerto sobre "Grafting onto"

Capítulo 2/ Antecedentes 2.3 Síntesis de copolímeros de tipo injerto
32
del injerto, en el esqueleto del sustrato a modificar. Este grupo funcional adicional servirá de
“puente” enlazando químicamente al injerto con el sustrato:23, 70
En el injerto desde, participa un polímero con
grupos funcionales a lo largo de la cadena principal que
inician la polimerización del monómero; este tipo de
técnica es muy útil, debido a que se puede usar con un
extenso intervalo de condiciones de reacción (tanto radicálica como iónica).
Finalmente, el injerto a través de involucra la
polimerización de un macromonómero, por lo que es
necesario efectuar la síntesis de dicho
macromonómero, el cual tiene como característica la
presencia de cadenas poliméricas de bajo peso
molecular (oligómeros), conteniendo en sus extremos especies polimerizables, específicamente
extremos de tipo vinílico.
Posteriormente, con la adición de un segundo monómero e iniciador en presencia del
macromonómero mencionado anteriormente, se tienen como resultado homopolímeros y/o
copolímeros ramificados. La síntesis de elastómeros y poliolefinas modificadas con injertos
poliméricos o con grupos polares, se ha dado principalmente mediante la polimerización por
radicales libres;71 sin embargo, como ya se ha explicado en las primeras secciones, la FRP no
permite obtener un control en el número y la longitud de las cadenas injertadas. Típicamente,
en un proceso por FRP, las cadenas son iniciadas, propagadas y terminadas en cuestión de
segundos. En este corto periodo de tiempo se presentan eventos de terminación prematura en la
cadena creciente46 por reacciones de combinación, desproporción y transferencia, evitando la
obtención de cadenas poliméricas de longitud homogénea. La falta de control produce como
consecuencia amplias distribuciones de pesos moleculares reflejadas en altos índices de
dispersidad (Ð> 3).
De los muchos métodos potenciales que existen para la producción de copolímeros de tipo
injerto, el proceso por radicales libres llega a ser un método conveniente para introducir un
Injerto desde "Grafting from"
A
+ nM
A(M)n
Injerto a través de "Grafting through"
CH2 CH + CH2 CHY * CH2 CH CH2 CHY *

Capítulo 2/ Antecedentes 2.3.1 Mecanismo del proceso de injerto
33
grupo reactivo o polar en poliolefinas. Esta técnica es usada en escala industrial como muchas
reacciones de modificación de polímeros y pueden llevarse a cabo en equipos de procesamiento
convencional.72 Los beneficios económicos que ofrecen los radicales libres en las técnicas de
injerto se deben a sus características particulares; es decir, debido a su naturaleza robusta, se
pueden llevar a cabo las reacciones en solución, en fase fundida o como un proceso en estado
sólido. Este tipo de reacciones (como se ha mencionado anteriormente) son relativamente
insensibles a la presencia de humedad o al aire, y en ciertos casos la reacción se mejora aún en
presencia de oxígeno.50 Existen reportes que discuten con profundidad la química del proceso
de injerto,50, 72-73 que a continuación se explicará brevemente.
2.3.1 Mecanismo del proceso de injerto
Las principales etapas de reacción implicadas en el proceso de funcionalización (y
posterior proceso de injerto) mediado por radicales libres se representan en las Figuras 9 y 10.
Al ser un proceso por radicales libres, la Figura 9 presenta las etapas de iniciación y de
propagación para el injerto de grupos sobre una cadena principal de polímero saturado. Los
modos de terminación para el macrorradical se presentan en la Figura 10. Este ejemplo fue
seleccionado con el fin de que ayuda a ilustrar el proceso de la obtención de un copolímero tipo
injerto sobre una poliolefina (que en nuestro caso de estudio se produjo sobre poliisopreno (PIP)
tanto en conformación cis y trans funcionalizado con grupos nitróxido). Alternativamente,
permite este ejemplo entender las etapas básicas de las reacciones que se presentan durante la
copolimerización, aunado con la factibilidad que tienen ciertos átomos de hidrógeno a ser
extraídos de la cadena principal del polímero bajo ciertas condiciones de reacción.
Iniciación: Los iniciadores de radicales libres más comunes para la modificación de
poliolefinas son los peróxidos de dialquilo, ya que proporcionan un tiempo de vida media
de descomposición razonable a las temperaturas de fusión del injerto, siendo la
temperatura un parámetro operacional importante en el control de la selectividad de los
radicales libres formados. Otro factor a considerar es el efecto de la viscosidad en el estado
fundido ya que a temperaturas más bajas, aumenta la viscosidad y se limita una buena
dispersión tanto del iniciador como del polímero a modificar. La Figura 9 ilustra las
principales vías de descomposición del peróxido de dicumilo (DCP) y el radical cumiloxi

Capítulo 2/ Antecedentes 2.3.1 Mecanismo del proceso de injerto
34
resultante. Tras la exposición del DCP al calor, el enlace peróxido se rompe para formar
dos radicales libres cumiloxi, cuya eficacia se rige por el efecto “jaula” (cage effect en
inglés), una medida del efecto “jaula” es la competencia entre la velocidad de
recombinación del radical y la velocidad a la que los radicales libres recién formados se
pueden difundir. En ausencia de los efectos “jaula”, el radical cumiloxi abstrae un
hidrógeno para formar el alcohol de cumilo, ésta especie también se puede descomponer
a través de un β-rompimiento para formar acetofenona y un radical metilo para añadirse a
una fracción del radical nitróxido.73b
Energías de enlace de disociación: La preferencia de un radical de retirar un átomo de
hidrógeno en la adición del nitróxido está controlada por la labilidad de los donantes de
hidrógeno disponibles y la naturaleza de los tipos de radicales libres. En referencia a la Figura
9, la extracción de hidrógeno desde el polímero (kH, kHi) lleva a la formación del macrorradical
polimérico. Las energías de disociación de enlace de hidrógeno para varias moléculas relevantes
se muestran en la Tabla 3. Estas energías de disociación proporcionan una idea de la factibilidad
que se puede tener en la etapa de la extracción del hidrógeno. Por ejemplo, la energía de
disociación de enlace del ciclohexano puede ser representativa para la abstracción de hidrógeno
en una molécula de polietileno por los grupos metileno internos del polímero, donde todos son
equivalentes. En el caso de la energía de disociación de enlace para el benceno, justifica el uso
de disolventes aromáticos en las reacciones de radicales libres, ya que es relativamente inerte a
la abstracción de hidrógeno. Finalmente, la presencia del TEMPO es susceptible a la extracción
de un hidrógeno secundario, generándose un biciclo por medio de una reacción intramolecular
(kH, NO).
Tabla 3: Energías de disociación de enlace para C-H comunes en las reacciones de injerto
mediante radicales libres.74
Enlace Energía de disociación
(kJ/mol)
CH3-H 439
(CH3)2CH-H 413
Ciclohexilo-H 400
(CH3)3C-H 404
CH2=CH(CH3)CH-H 345
Tetrahidrofurano-2-il-H 385
CH3CH(OH)-H 389
C6H5-H 365

Capítulo 2/ Antecedentes 2.3.1 Mecanismo del proceso de injerto
35
Figura 9: Mecanismo de reacción para el proceso de injerto mediado por radicales libres del TEMPO en
poliolefinas saturadas.75 P: polímero, PhH: benceno.
Incorporación del grupo nitróxido: La abstracción de hidrógeno del polímero forma el
macrorradical correspondiente (P⋅), el cual puede participar en la secuencia de
propagación como se muestra en la Figura 9. Estos macrorradicales libres poliméricos
tienden a añadirse en el extremo menos sustituido del enlace carbono-carbono debido a
los efectos estéricos. Los grupos nitróxido son por excelencia buenos atrapadores de
radicales libres,45b, 54b de tal manera que por su naturaleza, estas moléculas radicálicas se
adicionan donde se haya establecido la extracción del hidrógeno. Con esto se asegura un
polímero funcionalizado con grupos nitróxido, el cual funciona como una

Capítulo 2/ Antecedentes 2.3.1 Mecanismo del proceso de injerto
36
macroalcoxiamina multifuncional y en el momento de agregar algún monómero al medio
de reacción, se obtiene el copolímero de tipo injerto correspondiente.
Terminación: La modificación de injerto mediada por radicales libres en poliolefinas,
conduce a la formación de una amplia variedad de especies de radicales libres que están
sujetos al proceso de terminación. La terminación requiere de la interacción de dos
especies radicálicas, y la velocidad de terminación es un proceso controlado por
difusión.50 Un esquema generalizado para la terminación de los radicales libres se muestra
en la Figura 10.
Figura 10: Terminación radical-radical del macroradical en ausencia de monómero (superior: combinación;
inferior: desproporcionación).
Las velocidades relativas del proceso de desproporción sobre la combinación
(kdespr/kc) han sido ampliamente descritos en la literatura para la terminación de radicales
libres alquilo y alilo.74 Algunos valores se presentan en la Tabla 4. La tendencia de un par
radical para terminar por desproporción en lugar de combinación, es altamente
dependiente de la estructura de las especies de radicales libres. La disponibilidad de un
hidrógeno lábil en la posición β favorece a la desproporción.
Tabla 4: Relación de las constantes de desproporcionación-combinación en solución.74
Radical Tipo kdespr/kc
2 CH3CH2• 1° 0.15
2 (CH3)2CH• 2° 1.20
2 C6H11• 2° 1.10
2 (CH3)3C• 3° 4.50
2 CH2=CHC• (CH3)2 alílico 0.00

Capítulo 2/ Antecedentes 2.3.1 Mecanismo del proceso de injerto
37
La introducción de un reactante como es en el caso de la Figura 9, complica los
productos formados debido a la terminación. Pocos datos han sido reportados en la
literatura sobre la selectividad de los modos de terminación para las reacciones de
radicales libres mediadas por injerto, especialmente a las temperaturas usadas en este
estudio de investigación. Aunque el macrorradical y su aducto pueden terminar por
combinación y desproporción, el radical alílico formado debido a la transferencia de la
cadena terminará casi exclusivamente a través del proceso de combinación.
El método “grafting from” vía NMP para la modificación de poliolefinas y polidienos
particularmente se ha discutido en patentes; dentro de nuestro grupo de trabajo, se han
desarrollado estudios en el área de la modificación de polidienos por el método de “grafting
from”, elucidando el mecanismo de reacción que se lleva a cabo para realizar la síntesis de este
tipo de materiales poliméricos.38f, 76 Esto ilustra que el desarrollo de nuevas técnicas (que es una
de las ventajas que ofrece la polimerización radicálica controlada-viviente) para incorporar
injertos poliméricos o grupos polares en sustratos con insaturaciones como: polibutadieno (PB),
poliisopreno (PIP), poli(etilenpropilen-monómero diénico), (EPDM), poli(estireno-b-isopreno-
b-estireno), (SIS), hule butilo (BR) y materiales sin insaturaciones en la cadena principal tales
como polipropileno (PP), polietileno (PE), elastómeros hidrogenados (SEBS) y sustratos no
poliméricos han sido y siguen siendo uno de los retos a largo plazo de la ciencia y la tecnología
de polímeros.
Con el anterior argumento, es como nace la idea en la presente tesis de doctorado, lograr
la síntesis de copolímeros de injerto controlado (además, de la síntesis de los copolímeros de
tipo bloque) dado que los materiales resultantes pueden ser considerados materiales de
ingeniería, pudiendo presentar nuevas y/o mejoradas propiedades físicas y/o aplicaciones, por
ejemplo: permeabilidad selectiva a nitrógeno y oxígeno para embalaje de alimentos,
mejoramiento en el desempeño de propiedades mecánicas en mezclas de polímeros inmiscibles,
nano dispersiones en matrices poliméricas para aplicaciones biomédicas, materiales híbridos
para aplicaciones en celdas solares, etc., o bien ser usados como agentes compatibilizantes en
mezclas de polímeros inmiscibles o acoplantes en materiales poliméricos.

Capítulo 2/ Antecedentes 2.4 Uso de compatibilizantes en mezclas de polímeros inmiscibles
38
2.4 Uso de compatibilizantes en mezclas de polímeros inmiscibles
El uso de la extrusión reactiva ha contribuido a la funcionalización de poliolefinas, pero
esta técnica no ofrece un control de la estructura final de los polímeros. El número de grupos
funcionales injertados es difícil de controlar por este tipo de técnica; lo que sería deseable dado
que el número y distribución de grupos funcionales injertados en un polímero puede afectar la
morfología y sus propiedades.2 La utilización de copolímeros como agentes compatibilizantes,
promueve la miscibilidad en las mezclas poliméricas; esto es, la función del compatibilizante es
reducir la tensión interfacial entre las fases cuando se dispersa un polímero en otro, con el fin
de mejorar las propiedades de la mezcla final. Sin embargo, el diseño de moléculas
compatibilizantes y su uso en mezclas, no se ha extendido para el uso industrial, debido a la
resistencia al cambio pues permanece en buena medida como un arte semiempírico el mezclado
polimérico, donde las moléculas se diseñan y se seleccionan por prueba y error, con un uso
limitado de principios científicos. La incorporación de un compatibilizante en una mezcla de
polímeros tiene un efecto importante sobre la morfología y el grado de dispersión de las
partículas de un polímero en otro.2, 4b
Diversos trabajos han estudiado los factores que influyen en la mejora de propiedades en
mezclas cuando se utilizan compatibilizantes de tipo injerto y/o funcionalizados. Sin embargo,
los estudios no han sido sistemáticos debido al poco control que se ha tenido en la síntesis de
esos compatibilizantes al emplear métodos tradicionales de síntesis y estos métodos no han
cambiado mucho en los últimos 35 años. El uso de mezclas de polímeros no es reciente, se
considera que las primeras mezclas reconocidas comercialmente tuvieron sus inicios en los años
ochenta y a partir de esta fecha tuvieron un crecimiento rápido. El mezclado no se limitó a los
polímeros sino que también se comenzaron a usar cargas o rellenos para reforzar los polímeros
existentes. En la actualidad, la complejidad de las mezclas se ha incrementado
considerablemente, es por ello que el interés por las mezclas de polímeros comenzó por la
necesidad de abaratar los polímeros de ingeniería diluyendo estos compuestos en polímeros de
bajo costo, para intentar reciclar plásticos y para ajustar las propiedades de los polímeros a los
requerimientos del consumidor, entre otros.1 La mayoría de las parejas de polímeros tomadas al
azar son termodinámicamente inmiscibles, lo que imposibilita la formación de una sola fase

Capítulo 2/ Antecedentes 2.4 Uso de compatibilizantes en mezclas de polímeros inmiscibles
39
homogénea en la mezcla final. Adicionalmente, las altas tensiones interfaciales impiden lograr
el grado suficiente de dispersión en la mezcla para alcanzar las propiedades deseadas, y
trasfieren además una pobre estabilidad a la dispersión, trayendo como consecuencia la
delaminación o separación de las fases en etapas posteriores de transformación o durante el uso
del material en la aplicación final. En síntesis, una pobre adhesión impide alcanzar propiedades
mecánicas necesarias para usar la mezcla como material en diversas aplicaciones.77
La miscibilidad en las mezclas de polímeros está relacionada con el equilibrio
termodinámico, tomando en cuenta variables como la temperatura, presión, peso molecular, etc.,
donde la energía libre de mezclado debe ser negativa Gm<0. El hecho de que una mezcla sea
miscible no asegura que sus propiedades físicas y mecánicas sean las esperadas.1 La tensión y
la adhesión interfacial influyen sobre la mezcla modificando sus propiedades, es por ello que en
una mezcla inmiscible, la adhesión entre las fases es muy débil; y como consecuencia, la
transmisión de esfuerzos a la fase dispersa es pobre, lo que genera una disminución de las
propiedades mecánicas del material polimérico formado. Por eso, es deseable al tener una
mezcla de polímeros inmiscible y transformarlo en un material útil modificando su adhesión
interfacial.77
A modo de conclusión, el término compatibilidad se utiliza en la literatura científica como
sinónimo de miscibilidad en el sentido termodinámico; sin embargo, en la literatura técnica o
tecnológica se usa en un sentido menos estricto para caracterizar la facilidad relativa de
obtención de las propiedades deseadas en una mezcla de polímeros; por lo tanto, los polímeros
que al mezclarse resisten la separación macroscópica de fases y resultan en mezclas con
propiedades aceptables, se dice que son “compatibles”, aunque termodinámicamente no sean
miscibles.
Ciertos copolímeros funcionalizados o de injerto pueden contribuir a disminuir la alta
tensión interfacial, a ayudar en la dispersión y, por lo consiguiente, a mejorar las propiedades
finales de la mezcla polimérica. A este tipo de materiales se les conoce como compatibilizantes
y tienen una función análoga al que ofrece un surfactante dentro de una dispersión coloidal,
debiendo contener segmentos afines termodinámicamente o reactivos con cada uno de los

Capítulo 2/ Antecedentes 2.4 Uso de compatibilizantes en mezclas de polímeros inmiscibles
40
polímeros a mezclar.2 Dichos materiales se ubican en la interfase como se observa en la Figura
11; los segmentos (azules) afines al polímero A se orientan hacia la matriz polimérica y los
segmentos (rojos) afines al polímero B se orientan hacia la fase dispersa. El efecto neto provoca
la disminución de la tensión interfacial proporcionando los resultados benéficos buscados en las
propiedades mecánicas del material polimérico.
Para que una compatibilización sea efectiva, se necesita optimizar la arquitectura y el peso
molecular de los copolímeros funcionalizados en cadena lineal o de injerto que se usarán como
compatibilizantes. Los compatibilizantes de pesos moleculares muy bajos se difundirán más
rápidamente a la interfase, pero no facilitarán estabilidad en la morfología, mientras que los de
peso molecular muy alto tienden a ser poco efectivos ya que no se difunden fácilmente en la
interfase y poseen una concentración micelar crítica muy baja. Evidentemente, aquellos métodos
de síntesis de compatibilizantes que permitan controlar su arquitectura molecular, facilitarán la
labor de optimización; esta es una de las razones del porqué en este estudio de doctorado, se
desea aplicar la técnica de CRP para lograr tal objetivo.
Figura 11: Representación esquemática del proceso de la compatibilización en mezcla de polímeros inmiscibles;
a) copolímeros de cadena lineal y b) copolímeros de injerto. Los compatibilizantes se ubican en la interfase entre
los polímeros I y II de la mezcla. Los segmentos azules representan unidades afines al polímero I y los segmentos
rojos son afines al polímero II.
Cuando se utilizan copolímeros de cadena lineal, cada segmento del mismo debe ser
compatible con uno de los integrantes de la mezcla y al ser mezclados los copolímeros deben

Capítulo 2/ Antecedentes 2.4 Uso de compatibilizantes en mezclas de polímeros inmiscibles
41
situarse en la interfase de los dos polímeros o componentes de la mezcla para promover la
estabilidad y evitar la coalescencia. Cabe señalar, que la coalescencia es la propiedad que
presenta algunos polímeros que al estar en estado fundido o en solución, comienzan a unirse.
Este tipo de copolímeros han demostrado cambios en la morfología de mezclas al usar solo el
2% en peso del mismo, promoviendo la resistencia a la tensión y la resistencia al impacto. La
morfología de una mezcla está relacionada con sus propiedades reológicas, peso molecular,
composición, etc., correspondientes al agente acoplante usado durante el mezclado.
La concentración del compatibilizante también puede producir efectos diversos en la
mezcla, donde se ha encontrado que a altas concentraciones de compatibilizante se incrementa
el tamaño de partícula promedio debido a la formación de micelas de copolímero en la matriz.78
Así mismo, una baja concentración de compatibilizante no permite la reducción del tamaño de
partícula, debido a una saturación interfacial del mismo en una de las fases.
En conclusión, cuando se usa un compatibilizante, la tensión interfacial disminuye pero
tiene un límite de efectividad. Se han encontrado reportes79 donde mencionan que la
composición y la estructura de los copolímeros multibloque influyen considerablemente en las
capas interfaciales en partículas de poliolefina dispersas en polímeros de estireno; a diferencia
de copolímeros dibloques los cuales muestran eficiencia de compatibilización menor comparada
con los multibloques. Se ha encontrado que el injerto del copolímero de estireno-anhídrido
maleico en una poliolefina (p. ej. PP) se ha llevado a cabo con éxito y se logra obtener una
cadena larga de polímero con grupos reactivos colgantes.80
En cuanto a los efectos de compatibilización, los copolímeros pueden influir en la
cristalización de la mezcla, especialmente cuando se trata de compatibilizantes que tienden a
formar estructuras cristalinas (p. ej. copolímero de PP). El utilizar este tipo de copolímeros
puede cambiar la cristalinidad de la mezcla, el tamaño de los cristales existentes en los
homopolímeros, la orientación de los cristales, etc.6

Capítulo 2/ Antecedentes 2.5 Índices de reactividad
42
2.5 Índices de reactividad
Si en una reacción química intervienen dos o más monómeros distintos, se forma un
copolímero y estas unidades monoméricas se pueden unir de diferentes formas, las cuales son
clasificadas en función de cómo estén dispuestas estas unidades a lo largo de la cadena (ver
Figura 1, en la columna composición). Los copolímeros industriales más conocidos son: el ABS
(acrilonitrilo-butadieno-estireno), el SBR (caucho de estireno-butadieno), el caucho de nitrilo,
el copolímero de estireno acrilonitrilo (SAN), el elastómero de estireno-isopreno-estireno (SIS)
y el copolímero de etileno-acetato de vinilo (más conocido como goma EVA). Los hules
termoplásticos son también ejemplos de copolímeros, como el copolímero de etileno-propileno
(EPR). La determinación precisa de los índices de reactividad (ri), es una herramienta útil para
la estimación cuantitativa de la composición del copolímero, de la cual se pueden inferir sus
propiedades. Es común que los índices de reactividad se calculen utilizando la ecuación de
composición instantánea del copolímero (también conocida, como modelo de Mayo-Lewis,
ecuación 3) para un par de monómeros 1 y 2, dado que esta composición está solamente en
función de la alimentación instantánea.81 Para entender en qué consiste el modelo de Mayo-Lewis,
es importante comprender cómo se desarrolla la cinética de la homopolimerización. Una cadena
macromolecular es creada por la síntesis de un homopolímero durante la etapa de propagación
considerando un proceso de polimerización por radicales libres, es en esta etapa donde la mayor cantidad
de monómero se consume y el mayor número de enlaces químicos (M-M) se forman:23
Donde –M• es el macrorradical libre y n es el grado de polimerización promedio; por lo tanto,
se puede deducir que la copolimerización binaria es una ampliación del proceso de
homopolimerización vía la introducción de un segundo monómero (en este caso Mj), por lo que
un cambio significativo sucede en la etapa de propagación; donde se incluye la
homopolimerización de los comonómeros y la propagación cruzada de estos:

Capítulo 2/ Antecedentes 2.5 Índices de reactividad
43
Donde i≠j, y se generan también enlaces -Mi-Mj-, lo que hace la diferencia entre un
homopolímero y un copolímero. La estructura de un copolímero depende de cada momento en
la reacción y de las concentraciones relativas del comonómero y de su reactividad (esto es, de
la polaridad, resonancia y factores estéricos).82
La reacción decisiva en la configuración de las moléculas obtenidas en la
copolimerización binaria es la propagación, donde las cadenas activas –M1• y –M2• incorporan
a su estructura, nuevas moléculas de monómero M1 y M2, creando con ello una nueva molécula
activada con un monómero más. Como se mencionó anteriormente, en la etapa de propagación
se pueden considerar cuatro posibles reacciones de adición de los monómeros:
Siendo k11, k12, k21 y k22 las respectivas constantes de rapidez para la propagación de la
cadena terminal en Mi. A las reacciones 3 y 6 frecuentemente se les refiere como
homopropagación o autopropagación de un centro reactivo por adición al otro monómero,
mientras que a las reacciones 4 y 5, se les hace referencia como propagación cruzada o reacción
cruzada. Todas las reacciones se consideran como irreversibles en el modo más simple.
Desde el punto de vista de la arquitectura de la cadena, los copolímeros se pueden
clasificar de acuerdo a la distribución de las uniones Mi-Mi y de Mi-Mj en:23
I. Copolímeros estadísticos (aleatorio o al azar). Es el resultado de las cadenas poliméricas
en la que ambos monómeros se ubican de manera aleatoria.
II. Copolímeros alternados. La macromolécula es formada por la sucesión rigurosa de -Mi-
Mj-Mi-Mj-.

Capítulo 2/ Antecedentes 2.5 Índices de reactividad
44
III. Copolímeros en bloque. El número de enlaces –Mi-Mi- (los cuales se forman de las
secuencias de homopolímero llamados “bloques”) excede por mucho el número de
enlaces generados a través de la propagación entrecruzada.
IV. Copolímeros de injerto. Exhiben solo una cadena principal y varias ramificaciones.
Mayo y Lewis81 introdujeron el primer modelo cinético de una copolimerización ,
denominado modelo cinético terminal donde se asume que la reactividad de las especies
propagantes es dependiente solamente de la unidad monomérica al final de la cadena (referido
como la unidad última o terminal, tal y como se presenta en las expresiones 3-6, donde cuatro
reacciones de propagación son solamente posibles para un par de monómeros a reaccionar). Este
modelo describe el cambio relativo de las concentraciones del monómero (dM1/dM2) en función
únicamente de las concentraciones instantáneas de los monómeros M1 y M2 y de los índices de
reactividad, ri. Esta expresión fue establecida en relación a la etapa de propagación (que es la
más significativa, pues es ahí donde los comonómeros se consumen) y toma en cuenta la
contribución de cuatro posibilidades de reacción por separado en la copolimerización;
obteniéndose la ecuación de copolimerización (o ecuación de composición del copolímero):
𝑑𝑀1
𝑑𝑀2=
𝑚1
𝑚2=
𝑀1
𝑀2
𝑟1𝑀1+𝑀2
𝑀1+𝑟2𝑀2 (7)
La composición del copolímero, dM1/dM2, es la relación molar de las dos unidades
monoméricas que se incorporan instantáneamente en el copolímero y de acuerdo a la ecuación
de copolimerización, está relacionada con las concentraciones de los dos monómeros en la
alimentación inicial M1 y M2 y con los parámetros r1 y r2 (donde r1=k11/k12 y r2=k22/k21, estos
son los cuales son los índices de reactividad de los monómeros 1 y 2). Los índices de reactividad
son constantes, independientes de la alimentación y característicos para cada par de monómeros
en estudio.
Cuando las reactividades son muy bajas, las reacciones 3 y 6 son muy lentas y se produce
un copolímero alternado. Por el contrario, si las reactividades son muy grandes, las reacciones
3 y 6 son más rápidas y entonces se obtiene una mezcla de homopolímeros. Por lo tanto, la
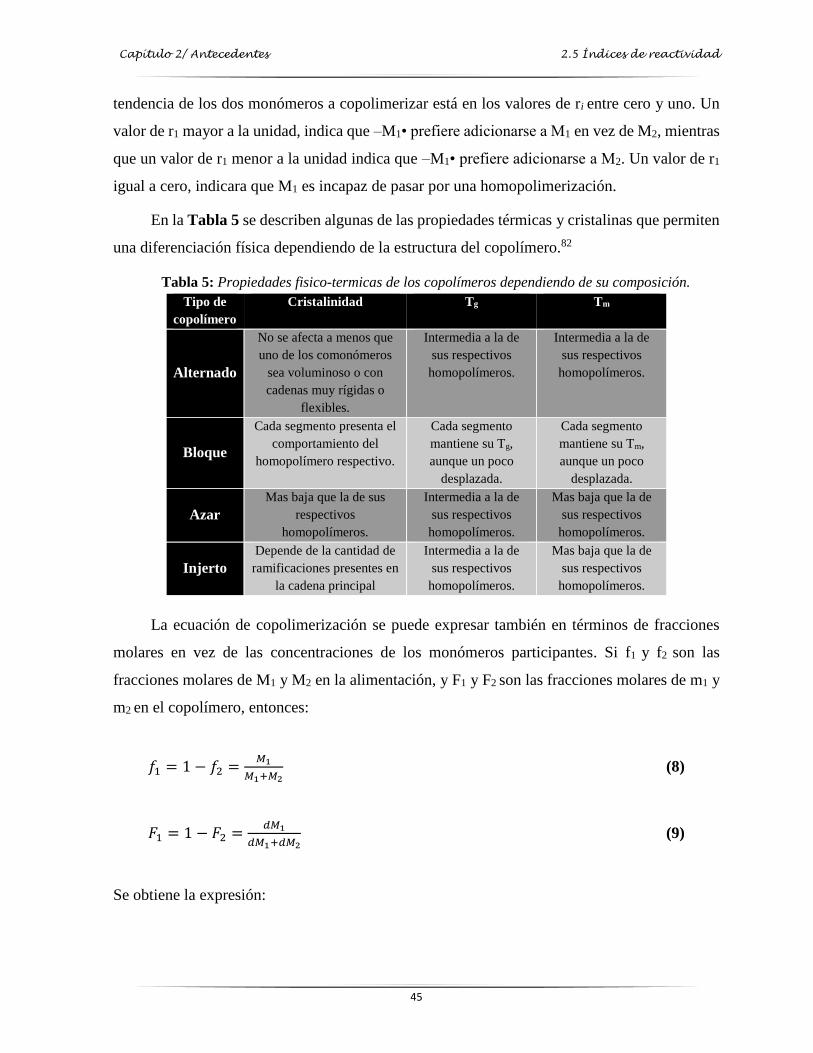
Capítulo 2/ Antecedentes 2.5 Índices de reactividad
45
tendencia de los dos monómeros a copolimerizar está en los valores de ri entre cero y uno. Un
valor de r1 mayor a la unidad, indica que –M1• prefiere adicionarse a M1 en vez de M2, mientras
que un valor de r1 menor a la unidad indica que –M1• prefiere adicionarse a M2. Un valor de r1
igual a cero, indicara que M1 es incapaz de pasar por una homopolimerización.
En la Tabla 5 se describen algunas de las propiedades térmicas y cristalinas que permiten
una diferenciación física dependiendo de la estructura del copolímero.82
Tabla 5: Propiedades fisico-termicas de los copolímeros dependiendo de su composición.
Tipo de
copolímero
Cristalinidad Tg Tm
Alternado
No se afecta a menos que
uno de los comonómeros
sea voluminoso o con
cadenas muy rígidas o
flexibles.
Intermedia a la de
sus respectivos
homopolímeros.
Intermedia a la de
sus respectivos
homopolímeros.
Bloque
Cada segmento presenta el
comportamiento del
homopolímero respectivo.
Cada segmento
mantiene su Tg,
aunque un poco
desplazada.
Cada segmento
mantiene su Tm,
aunque un poco
desplazada.
Azar
Mas baja que la de sus
respectivos
homopolímeros.
Intermedia a la de
sus respectivos
homopolímeros.
Mas baja que la de
sus respectivos
homopolímeros.
Injerto
Depende de la cantidad de
ramificaciones presentes en
la cadena principal
Intermedia a la de
sus respectivos
homopolímeros.
Mas baja que la de
sus respectivos
homopolímeros.
La ecuación de copolimerización se puede expresar también en términos de fracciones
molares en vez de las concentraciones de los monómeros participantes. Si f1 y f2 son las
fracciones molares de M1 y M2 en la alimentación, y F1 y F2 son las fracciones molares de m1 y
m2 en el copolímero, entonces:
𝑓1 = 1 − 𝑓2 =𝑀1
𝑀1+𝑀2 (8)
𝐹1 = 1 − 𝐹2 =𝑑𝑀1
𝑑𝑀1+𝑑𝑀2 (9)
Se obtiene la expresión:

Capítulo 2/ Antecedentes 2.5 Índices de reactividad
46
𝐹1 =𝑟1𝑓1
2+𝑓1𝑓2
𝑟1𝑓12+2𝑓1𝑓2+𝑟2𝑓2
2 (10)
ó
𝐹1
𝐹2=
𝑓1
𝑓2 𝑟1𝑓1+𝑓2
𝑟2𝑓2+𝑓1 (11)
Las expresiones 10 u 11, se usan para efectuar gráficos de la fracción molar contenida
en el copolímero con respecto a la fracción molar en la alimentación (ver Figura 12) para
expresar el comportamiento a diferentes composiciones. Los índices de reactividad no son sólo
parámetros adecuados para la estimación de las reactividades relativas de los monómeros,
también proporcionan información valiosa y precisa para la determinación de parámetros micro
estructurales, tales como la distribución de las unidades y longitudes de secuencia a lo largo de
las cadenas macromoleculares.23 Por lo tanto, los procedimientos de cálculo empleados para la
determinación de ri, deben ser ajustados los más cuantitativamente posible al comportamiento
experimental del sistema de copolimerización. Adicionalmente, la importancia que tienen los
índices de reactividad en los monómeros, es que son valores cuantitativos para predecir la
composición del copolímero para cualquier alimentación inicial en reactores por lotes, semilotes
o continuos y para entender la cinética y los aspectos mecanísticos de la copolimerización.
En general, se observan dos tipos de comportamiento diferentes: a. ambos índices de
reactividad están debajo de 1.0 (ri<1.0 y rj<1.0) y b. solo uno de los dos está debajo de 1.0 (ri>1.0
y rj<1.0). El modelo de Mayo-Lewis permite el estudio de varios casos, uno de ellos es cuando
ambas reactividades son iguales hacia ambos radicales libres;83 otro caso es cuando los radicales
libres reaccionan exclusivamente con el monómero del otro tipo (r1r2≈0). Cuando r1=r2=1.0, se
tiene el caso especial en que m1=M1 (definición de azeotropía). Por otra parte, si r1r2≈1.0, se
tiene la aproximación de la copolimerización “ideal” y en el caso de tener r1r2<0.03, existe una
fuerte tendencia a formar copolímeros alternados.84 Para el tipo en que r1>1.0 (o r2>1.0), una
composición azeotrópica no llega a ser posible. El copolímero es más rico en M1 cuando r1>1 y
es más pobre en M1 cuando r1<1.
Las copolimerizaciones tienen como resultado que: un monómero es más reactivo que el
otro. Por lo general, los índices de reactividad son determinados a baja conversión.82 La Figura

Capítulo 2/ Antecedentes 2.5 Índices de reactividad
47
12, ilustra las curvas de composición del copolímero generado por las composiciones de
alimentación del comonómero correspondiente.
Casos especiales de ri
I. r1r2=0 Formación de copolímero alternado, donde ni r1 o r2 es mayor que la
unidad. Con este comportamiento, existen dos tipos de alternado,
comportamiento alternado extremo y el moderado. En el primer caso,
r1 y r2 son cero, donde los dos monómeros ingresan en el copolímero
en cantidades equimolares a lo largo de la cadena. Cada uno de los
tipos de especies propagantes se adicionará preferentemente al otro
monómero; esto es, –M1• se adiciona solo con M2 y viceversa. El
comportamiento alternado moderado ocurre cuando a) r1 y r2 son muy
pequeños (r1r2= muy pequeño, cercano a cero) o b) uno de los valores
de r es pequeño y el otro es cero (r1r2=0). La composición del
copolímero tiende hacia la alternancia pero no es una estructura
perfectamente alternante.
II. r1=r2= Solo se producen homopolímeros.
III. r1=r2=1 Formación de copolímero aleatorio, la composición en el copolímero
es la misma que la concentración inicial de la alimentación.
IV. r1r2=1 Copolimerización “ideal”, la composición en el copolímero no
necesariamente es la misma que la concentración inicial de la
alimentación (los monómeros muestran la misma preferencia por
adicionarse con el otro par). Se puede apreciar que un comportamiento
ideal extremo ocurre cuando r1 y r2 son muy diferentes (p. ej. 10 y 1)
y se presenta un comportamiento ideal moderado cuando r1 y r2 no son
muy diferentes (p. ej. 0.5 y 2).
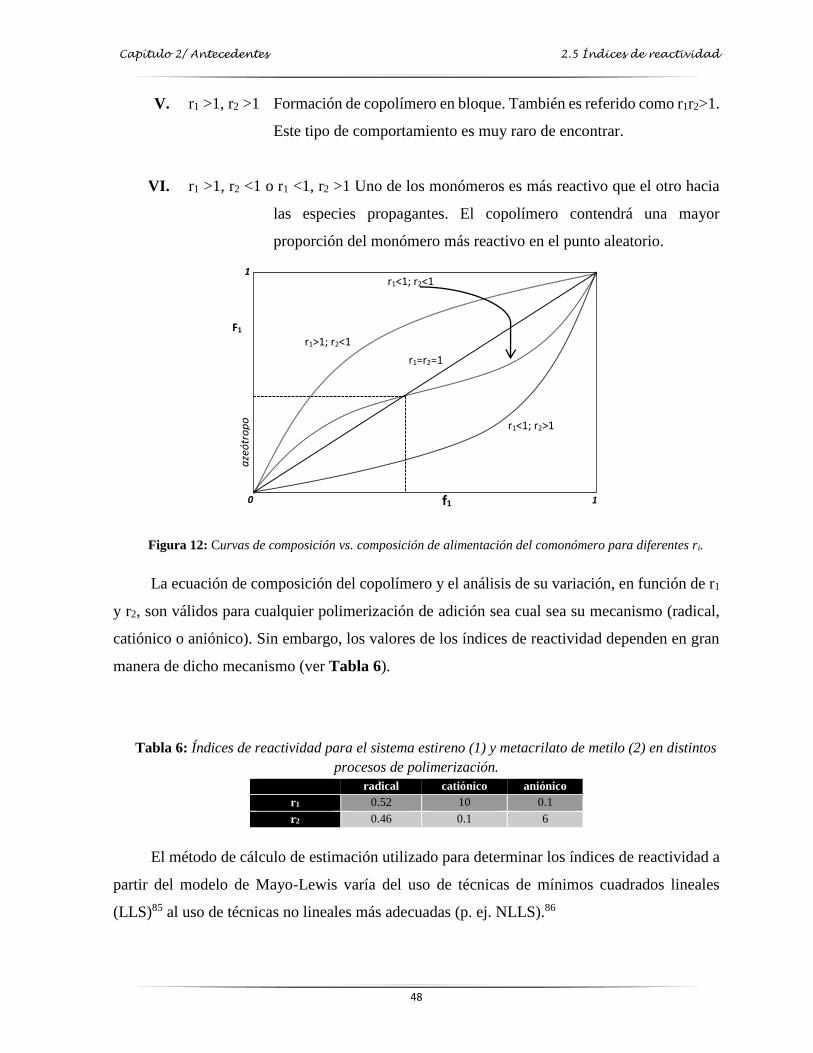
Capítulo 2/ Antecedentes 2.5 Índices de reactividad
48
V. r1 >1, r2 >1 Formación de copolímero en bloque. También es referido como r1r2>1.
Este tipo de comportamiento es muy raro de encontrar.
VI. r1 >1, r2 <1 o r1 <1, r2 >1 Uno de los monómeros es más reactivo que el otro hacia
las especies propagantes. El copolímero contendrá una mayor
proporción del monómero más reactivo en el punto aleatorio.
Figura 12: Curvas de composición vs. composición de alimentación del comonómero para diferentes ri.
La ecuación de composición del copolímero y el análisis de su variación, en función de r1
y r2, son válidos para cualquier polimerización de adición sea cual sea su mecanismo (radical,
catiónico o aniónico). Sin embargo, los valores de los índices de reactividad dependen en gran
manera de dicho mecanismo (ver Tabla 6).
Tabla 6: Índices de reactividad para el sistema estireno (1) y metacrilato de metilo (2) en distintos
procesos de polimerización.
radical catiónico aniónico
r1 0.52 10 0.1
r2 0.46 0.1 6
El método de cálculo de estimación utilizado para determinar los índices de reactividad a
partir del modelo de Mayo-Lewis varía del uso de técnicas de mínimos cuadrados lineales
(LLS)85 al uso de técnicas no lineales más adecuadas (p. ej. NLLS).86
0 1
1
f1
F1
azeótropo
r1>1; r2<1
r1<1; r2>1
r1=r2=1
r1<1; r2<1

Capítulo 2/ Antecedentes 2.6 Método de PLP-SEC
49
2.6 Método de PLP-SEC
A pesar de la enorme importancia que ofrece la polimerización por radicales libres, el
entendimiento detallado de la cinética involucrada en la polimerización llega a ser incompleta
por la falta de información que se tiene de las constantes de rapidez implicadas durante el
proceso. Las constantes de rapidez de propagación y terminación, aunadas con la cinética de
iniciación y las concentraciones iniciales de los radicales libres primarios, son las que ayudan a
determinar la conversión del monómero con respecto al tiempo involucrado en dicha reacción
de polimerización; sin embargo, lamentablemente en su gran mayoría no son propiamente
conocidas. Esto puede verse fácilmente en el Polymer Handbook,87 donde aún para las mismas
condiciones de reacción, los valores de las constantes de rapidez están muy variados dentro de
la misma tabla de información. El conocimiento de éstas constantes cinéticas de rapidez también
se necesitan con el fin de describir y simular la distribución del peso molecular del polímero,
dado que ésta afecta a las propiedades del material polimérico.
Tal y como se ha descrito anteriormente, la polimerización mediada por radicales libres,
además de presentar las etapas básicas en su mecanismo de reacción, puede presentar las etapas
de transferencia de cadena e inhibición; de tal manera que se presentan cinco diferentes tipos de
constantes de rapidez para dichas etapas de reacción: iniciación (ki), propagación (kp),
terminación (kt), transferencia de cadena (ktr) e inhibición (kz). Algunos cocientes de constantes
asociados a la velocidad de propagación (kp/kt1/2), a la de transferencia (ktr/kp) y a la de inhibición
(kz/kp), se pueden obtener por métodos descritos en la literatura especializada.23, 30b, 87 Sin
embargo, la evaluación individual de estas constantes cinéticas en condiciones de régimen
permanente necesita de la determinación aproximada de la concentración del radical propagante,
facilitando con ello encontrar el valor de la kp a partir de la expresión:
𝑅𝑝 = 𝑘𝑝|M ∙||𝑀| (12)
Ya obtenida la kp, se pueden calcular kt, ktr y kz de las relaciones kp/kt1/2, ktr/kp y de kz/kp.
Muchos de los datos que existen en la literatura especializada fueron determinados por el método

Capítulo 2/ Antecedentes 2.6 Método de PLP-SEC
50
del sector rotante hasta finales de los 80’s; en la actualidad, con la tecnología de pulso de láser
combinado con la cromatografía de exclusión de tamaño (PLP-SEC)88 se ofrece una mucho
mejor alternativa para la determinación directa de kp. Cabe señalar que ambos métodos
involucran polimerizaciones bajo condiciones de régimen no permanente. En el caso del método
del sector rotante, se usan las velocidades de polimerización para obtener kp; mientras que en el
método de PLP-SEC, se utilizan los pesos moleculares del polímero para determinar dicha
constante cinética. Esta técnica experimental combina la polimerización iniciada por pulso de
láser (PLP) con un subsecuente análisis del polímero obtenido por cromatografía de exclusión
de tamaño (SEC). La IUPAC recomienda la técnica de PLP-SEC como uno de los métodos
directos y robustos para la determinación de la kp.88d
A diferencia del método del sector rotante, el método de PLP-SEC permite la medición
directa de kp sin la necesidad del acoplamiento con la constante de rapidez de terminación. El
método de PLP-SEC involucra una fotopolimerización en régimen no permanente, bajo la
irradiación del pulso de láser, por lo tanto, los radicales libres primarios son formados en un
corto tiempo (≈10 ns de ancho de pulso) comparado con el tiempo de ciclo (≈1 s). El ancho del
pulso de láser es también muy corto comparado con los tiempos de vida de los radicales libres
propagantes y los tiempos para la conversión de los radicales libres primarios a radicales libres
propagantes. El método de PLP-SEC para medir la kp requiere que las condiciones de reacción
seleccionadas no sean propicias para que ocurra la transferencia de cadena.
Una manera breve de describir este procedimiento es del modo siguiente: Los primeros
pulsos de láser generan un racimo casi instantáneo de radicales libres primarios en una alta
concentración (≈10-6 mol/L). Durante el periodo oscuro (ausencia de la presencia del haz del
láser) que sigue al pulso, muchos de los radicales libres primarios forman radicales libres
propagantes, los cuales se propagan pero algunos de los radicales libres primarios terminan por
acoplamiento. El acoplamiento terminal y la desproporción ocurren durante el periodo oscuro,
pero una gran fracción de cadenas propagantes no terminan, debido a que el ciclo de tiempo es
muy corto. El segundo pulso de láser produce nuevamente un racimo de radicales libres
primarios y muchas de las cadenas propagantes sobrevivientes (provenientes del primer periodo

Capítulo 2/ Antecedentes 2.6 Método de PLP-SEC
51
oscuro entre el primer y segundo pulso de láser) son casi instantáneamente terminadas, por la
alta concentración de radicales libres primarios formados en el segundo pulso láser.
Sin embargo, algunos de los radicales libres propagantes sobreviven en el segundo pulso
y continúan propagándose durante el segundo periodo oscuro. Algunos de los radicales libres
primarios en el segundo pulso láser inician la polimerización que procede en el segundo periodo
oscuro, al mismo tiempo que las cadenas propagantes del primer periodo oscuro son propagadas
en el segundo periodo oscuro. El proceso continua de modo similar en cada pulso de láser y
subsecuente periodo oscuro. Un número suficiente de pulsos de láser son usados de este modo
con tal de lograr la cantidad del polímero necesario para el análisis por cromatografía de
exclusión de tamaño. La cantidad de conversión es de solo 2 – 3 %, lo que indica que la
concentración del monómero es esencialmente constante en el transcurso del experimento.23
Algunas de las cadenas propagantes terminan por acoplamiento normal y/o desproporcionación
durante los periodos oscuros.
La mayoría del polímero producido está conformado de cadenas propagantes que se han
extendido por diferentes intervalos de tiempo del periodo oscuro tp. La tp es esencialmente el
mismo ciclo de tiempo, debido a que el ancho del pulso es mucho más corto que tp. Las cadenas
se propagan para 1tp, 2tp, 3tp, etc. El grado de polimerización para las cadenas terminadas por
radicales libres primarios está dada por:
𝐿𝑖 = 𝑖𝑘𝑝𝐶𝑀𝑡𝑝 (13)
Donde CM es la concentración del monómero, tp es el tiempo entre dos pulsos sucesivos de pulso
láser y Li es el número de etapas de propagación entre dos pulsos subsecuentes. Li es calculado
de acuerdo a Li =Mi/MM, donde MM es el peso molecular del monómero y Mi es el peso
molecular para las cadenas que se han propagado a través de i ciclos en la distribución de pesos
moleculares. Li es obtenido de la medición de la distribución del peso molecular del polímero
en el SEC (ver Figura 13).88d

Capítulo 2/ Antecedentes 2.6 Método de PLP-SEC
52
Figura 13: Distribución del peso molecular del poliacrilato de dodecilo para la determinación de Kp usando la
técnica de PLP-SEC.88d
Li es obtenido del primer punto de inflexión en el lado de bajo peso molecular del pico
mayor de la gráfica de la distribución de pesos moleculares. En la práctica, es más fácil
determinar el máximo en una gráfica diferencial de la distribución de pesos moleculares
dw(logM)/dlogM con respecto a log M. Para casos en que existen varios Li, los valores de kp
calculado para L1, L2 y L3 están dentro del 2% uno con respecto al otro, el valor de L4 está dentro
del 6 al 7% de los otros valores. En la práctica, los valores de kp mas aceptados son los
determinados a partir de L1 y L2, considerados lo suficientemente validos por el método de PLP-
SEC.
Por otra parte, kt se puede obtener también por el método de PLP-SEC por medio de la
expresión:
𝑘𝑡 =𝑘𝑝2|𝑀| 3−𝛿
𝑅𝑝𝐿𝑤 (14)
donde Rp y Lw son la velocidad de polimerización y el grado de polimerización en peso
promedio y δ es la contribución relativa de desproporción con respecto a la terminación, definido
como ktd/(ktd+ktc).88d

Capítulo 2/ Antecedentes 2.7 Polimerización de 1,3-dienos. Isopreno
53
2.7 Polimerización de 1,3-dienos. Isopreno
El isopreno (IP) es un monómero es muy atractivo e interesante debido a que está
disponible a bajo costo, aunado a que el polímero correspondiente es muy similar al hule natural
tanto en estructura como en sus propiedades. Actualmente, el poliisopreno sintético se ocupa en
una amplia variedad de aplicaciones en donde se requiere un bajo hinchamiento en presencia de
agua, buena elasticidad, alta resistencia en caliente y alta adherencia, por lo que es común ver
los derivados de este elastómero en bandas de goma, tetinas de biberones, mangueras, en
neumáticos, soportes para motor y bujes de los amortiguadores, entre otros productos
moldeados. También en la industria del calzado es muy socorrido este material polimérico así
como en el área médica.89 Adicionalmente, el isopreno es la base estructural de moléculas de
origen natural como los terpenos, por lo que se puede encontrar en moléculas como el escualeno,
retinol o vitamina E, que son compuestos biocompatibles, sugiriendo que el poliisopreno
sintético con una estructura controlada puede tener interesantes aplicaciones biomédicas
análogas a las recientemente desarrolladas para el escualeno.90
La polimerización de 1,3-dienos (p. ej. isopreno, butadieno entre otros), es un proceso
laborioso tanto en los laboratorios industriales como en los académicos;91 aun así, los polímeros
de este tipo cuentan con una gran importancia comercial como ya se mencionó. Los copolímeros
de este tipo de elastómeros han sido típicamente generados por técnicas de polimerización
aniónicas,92 catiónicas93 y de coordinación,94 técnicas que son difíciles de aplicarse para la
síntesis de estos materiales poliméricos y presentan dificultades para hacerlas compatibles con
un amplio intervalo de grupos funcionales o comonómeros. En el caso de la polimerización por
radicales libres, la homo- y copolimerización de 1,3-dienos tiene como objetivo expandir la
importancia de su campo comercial.95 Los materiales producidos por esta técnica, son de gran
interés en la vida cotidiana dado que ofrecen el potencial de poderse usar para la producción de
hule o como compatibilizantes en mezclas de caucho natural con polímeros de acrílico, para
citar un ejemplo, o para aplicaciones médicas,89 ente otros tantos usos. Es sabido que la
polimerización del isopreno tiene como resultado, la formación de polímeros que mantienen
dobles enlaces reactivos a lo largo de su cadena principal, lo que le confiere ser un material
funcional. Por las propiedades interesantes que ofrece, tales como la degradabilidad química por

Capítulo 2/ Antecedentes 2.7 Polimerización de 1,3-dienos. Isopreno
54
ozonólisis,96 puede ser entrecruzado por una gran variedad de rutas químicas,95, 97 y ofrece
muchas oportunidades para la funcionalización (p. e. vía epoxidación,98 hidrogenación,99 etc.).
Es un candidato ideal para la hidrogenación pues el producto generado es un material excelente
como poliolefina funcionalizada, el que por otras vías es muy complicado de obtener. El
poliisopreno, es también interesante por su baja Tg.
Siendo así, entre los polímeros que son accesibles por CRP, los poli (1,3-dienos) tal como
el poliisopreno (PIP) son interesantes por la inclusión de la funcionalidad del doble enlace en la
cadena principal del polímero. Esta funcionalidad le imparte propiedades atractivas debido a
que presenta un potencial para una subsecuente funcionalización.100 Otro de los puntos a
destacar del PIP es que ofrece una estructura hidrocarbonada insaturada y presenta una baja Tg,
lo que le permite ser atractivo para diversas aplicaciones tal como crear surfactantes de cadena
larga (creando copolímeros de IP con una fracción de un comonómero polar) para que sean
directamente disueltas en agua.101 Por lo tanto, este tipo de polímeros son una selección natural
para usarse como componentes lipofílicos de los surfactantes.
A pesar de todas estas propiedades destacables, la CRP de 1-3 dienos presentan numerosos
retos experimentales; la polimerización aniónica es una de las técnicas más destacables para
obtener estos materiales poliméricos con un control estructural altamente deseado, pero también
es muy sensible a la presencia de aire o impurezas electrofílicas o ácidas, dándole una apreciable
ventaja a la polimerización controlada-vivientes por radicales libres, debido a que ésta es más
tolerante a una amplia gama de funcionalidades; sin embargo, se requiere el uso de equipo
presurizado para polimerizar al butadieno o al isopreno con elevadas temperaturas, a fin de
obtener una velocidad de reacción razonable. Esta es una de las posibles razones del por qué los
reportes relacionados a la CRP del IP sean muy escasas52a, 53b, 95, 102
De las técnicas comunes de CRP, ATRP no puede ser aplicado a la polimerización del
isopreno debido a la quelación del catalizador de cobre con el IP.102d Existen varios reportes de
la polimerización por el proceso RAFT del IP102c-e pero es más común la técnica por NMP.52a,
53b, 95, 96b, 102a, b Los estudios recientes en NMP del IP reportan la producción de polímeros con
dispersidades de 1.36 – 1.53. El mayor avance se dio en el trabajo reportado por Benoit et al.52a

Capítulo 2/ Antecedentes 2.7 Polimerización de 1,3-dienos. Isopreno
55
en el 2000 con el uso de una alcoxiamina con funcionalidad TIPNO y en el 2011 por Harrisson
et al.102a con el uso del SG1 y sus derivados como alcoxiaminas en lugar del TEMPO empleado
por investigadores previos. Esto permitió la obtención de poliisopreno y polibutadieno con pesos
moleculares arriba de 20 000 g/mol y dispersidades típicas de 1.1 – 1.2. La polimerización del
isopreno procede en un modo tal que, debido a la presencia del grupo metilo que tiene la
molécula, genera distintas microestructuras (Esquema 11) que muestran diferentes propiedades
químicas, físicas y mecánicas.103 Por ejemplo, el cis-1,4-PIP es el componente mayoritario del
hule natural y funciona como un excelente elastómero; por otra parte, el trans-1,4-PIP y el 3,4-
PIP son polímeros cristalinos y con una Tg mayor que el cis-1,4-PIP.
Esquema 11: Isopreno y las microestructuras del poliisopreno.

Capítulo 3/ Parte Experimental
56
3.
Parte Experimental
En este capítulo se describen los materiales, métodos de purificación de reactivos y
disolventes, además del detalle de los procedimientos de síntesis implementados en este
proyecto de doctorado a fin de cumplir con los siguientes puntos:
a. Determinar los índices de reactividad para los sistemas de isopreno-metacrilato de
glicidilo (IP/GMA) e isopreno-anhídrido maleico (IP/MAH).
b. Determinar las constantes de propagación involucradas en los sistemas de IP/GMA por
PLP-SEC.
c. Sintetizar (co)polímeros lineales de IP/GMA por la técnica de polimerización mediada
por nitróxidos.
d. Sintetizar (co)polímeros lineales de IP/GMA por la técnica de polimerización RAFT.
e. De los copolímeros sintetizados en los puntos c. y d., demostrar el carácter viviente de
estas técnicas de polimerización al adicionar más isopreno al medio de reacción
(extensión de cadena).
f. Sintetizar diversas macroalcoxiaminas multifuncionales, modificando químicamente la
cadena principal de polímeros de isopreno en conformación cis y trans en solución en
presencia de un tipo de iniciador peroxídico y de algunos nitróxidos, empleando técnicas
estándar ya reportadas.76b
g. Polimerizar injertos de copolímeros de IP/GMA de longitud controlada sobre los
polímeros funcionalizados en solución empleando técnicas estándar ya reportadas.76b
h. Diseñar y escalar un compatibilizante en base a poliisopreno controlando el nivel de
injerto o el tamaño de la cadena lineal del copolímero funcional en solución.

Capítulo 3/ Parte Experimental 3.1 Materiales y reactivos
57
3.1 Materiales y reactivos
Los reactivos y disolventes fueron adquiridos en Sigma-Aldrich Co. y en J. F. Baker (a menos
que se indique otra cosa). Los reactivos y materiales utilizados fueron: ácido canforsulfónico (CSA),
ácido 4-ciano-4-[(dodecilsulfaniltiocarbonil) sulfanil] pentanoico (CDSPA), anhídrido acético (AA),
hidróxido de sodio (NaOH), peróxido de benzoilo (BPO), 2, 2, 5-trimetil-4-fenil-3-azahexan-3-
nitróxido (TIPNO), N-t-butil-N-(2-metil-1-fenilpropil)-O-(1-feniletil) hidroxilamina (TIPAL), 4-
hidroxi-2, 2, 6, 6-tetrametil piperidin-1-oxil (HO-TEMPO, de CIBA), 2,2-dimetoxi-2-fenilacetofenona
(DMPA), Bis(carboximetil) tritiocarbonato (BCTC), peróxido de t-butilo (TBP o Luperox DI), D-
glucosa (Glu), PIP- alto cis (código SIR 3L de Indo Java Rubber Planting Co).
Los disolventes empleados son de grado reactivo y fueron: acetona, metanol, hexano, tolueno,
tetrahidrofurano (THF), N, N-dimetil formamida (DMF). Los disolventes deuterados para los estudios
de resonancia magnética nuclear (RMN) fueron adquiridos por Sigma-Aldrich Co., utilizados tal como
se recibieron: cloroformo (CDCl3) y acetona (Ac-d6).
3.2 Purificación de monómeros y polímeros
El monómero isopreno se destiló previamente a una polimerización a presión atmosférica y el
metacrilato de glicidilo se destiló a presión reducida. El anhídrido maleico (MAH, Fluka) fue
recristalizado en cloroformo.
El poliisopreno-cis (PIP-cis, Sigma-Aldrich, Mn = 6150 Da, Ð = 1.18) y el poliisopreno-trans
(PIP-trans, Sigma-Aldrich, Mn = 29550 Da, Ð = 1.15) se purificaron para retirar todo el posible
antioxidante o aditivos que pudieran contener, disolviéndolos en tolueno y precipitándolos en metanol
(dos veces se repitió el proceso).
3.3 Técnicas instrumentales de análisis y métodos de caracterización.
3.3.1 Resonancia Magnética Nuclear (RMN)
La determinación de los espectros de 1H, 13C, DQF-COSY y HETCOR se llevaron a cabo en un
equipo Jeol Eclipse 300 Instrument a 300 MHz de 7.07 Teslas de intensidad de campo magnético en

Capítulo 3/ Parte Experimental 3.3.2 Espectroscopia infrarroja con transformada
de Fourier (FT-IR)
58
tubos de 5 mm de diámetro externo, usando cloroformo deuterado (CDCl3) a temperatura ambiente para
los copolímeros de IP-GMA y acetona deuterada (Ac-d6) a temperatura ambiente para los copolímeros
de IP-MAH. Para el caso de 1H se usaron 16 barridos con muestras de 25 mg en 0.6 mL del disolvente
deuterado, mientras que para el caso de 13C fueron 1500 barridos con muestras de 40 mg en 0.6 mL del
disolvente deuterado.
Los desplazamientos químicos (δ) fueron medidos en partes por millón (ppm). Se utilizó
tetrametilsilano (TMS) como referencia interna para los espectros de RMN.
3.3.2 Espectroscopia infrarroja con transformada de Fourier (FT-IR)
El espectrofotómetro utilizado para obtener los espectros de FT-IR fue un equipo “Thermo-
Nicolet Nexus 470 Spectrometer”. Las muestras fueron analizadas en forma de película depositada
sobre un cristal de KBr puro para el caso de los copolímeros de IP/GMA y en forma de pastilla para el
sistema IP/MAH, obtenidas mediante una mezcla con KBr; el intervalo de frecuencia de trabajo fue de
4000-400 cm-1 usando 32 barridos y 4 cm-1 de resolución. Mediante esta técnica se analizaron las bandas
de absorción presentes en los diversos compuestos poliméricos obtenidos.
3.3.3 Cromatografía de Permeación en Gel (GPC)
Los pesos moleculares promedio en número (Mn), promedio en peso (Mw) y los índices de
dispersidad (Ð =Mw/Mn) fueron obtenidos en un cromatógrafo de permeación en gel HPLC 1100
Hewlett Packard. Los pesos moleculares relativos a estándares de poliestireno fueron determinados
usando columnas “Ultrastyragel” en serie PLGel (1 – 40 K para 10-3Å, 40 K – 4 M para 10-5Å y 400 K
– 40 M para 10-6Å) en THF a 40 °C y un flujo de 1 mL/min acoplado a un detector de índice de
refracción y ultravioleta “Waters 410”.
Se realizó una curva de calibración de poliisopreno para convertir los picos de los pesos
moleculares relativos a estándares del PS, MPS, a los pesos moleculares del PIP, MPIP, usando
las constantes de Mark-Houwink-Sakurada (MHS) para ambos polímeros en CCl4 a 25 °C
(Ecuación 16).104 Para el poliisopreno, las constantes de MHS que se usaron son: KPIP=2.44 x

Capítulo 3/ Parte Experimental 3.3.4 Calorimetría Diferencial de Barrido (DSC).
59
10-4 y PIP=0.712. Para el PS, KPS=7.1 x 10-4 y PS=0.54 (PM<16700 g/mol) o KPS=1.44 x 10-4
y PS=0.713 (PM16700 g/mol).
𝐾𝑃𝐼𝑃𝑀𝑃𝐼𝑃∝𝑃𝐼𝑃 = 𝐾𝑃𝑆𝑀𝑃𝑆
∝𝑃𝑆 (15)
3.3.4 Calorimetría Diferencial de Barrido (DSC)
El análisis térmico de los materiales poliméricos se llevó a cabo usando un instrumento Perkin Elmer
Serie 7. La velocidad de calentamiento fue de 10 °C/min (primer y segundo calentamiento), con
atmósfera de nitrógeno a un flujo de 50 mL/min con un intervalo de temperatura fue de -100 a 130 °C.
3.3.5 Análisis mecánico diferencial (DMA)
El análisis mecánico diferencial se realizó en un equipo TA Instrument DMAQ800 con
una tensión de la abrazadera a una frecuencia de 1 Hz y una tensión de amplitud de 20 µm. La
velocidad de calentamiento fue de 5 ºC/min y el intervalo de temperatura fue de -100 a 90 °C.
Nota: Todas las reacciones de (co)polimerización se llevaron a cabo dentro de una
campana de extracción.
3.4 Procedimiento general de la cinética y copolimerización del IP/MAH.
Índices de reactividad
El procedimiento para llevar a cabo la copolimerización del IP/MAH se realizó a diferentes
composiciones del isopreno con respecto al MAH. En muchos casos, la polimerización se completó tan
rápido como la adición del dieno terminó.

Capítulo 3/ Parte Experimental 3.4 Procedimiento general de la cinetica y
copolimerización del IP/GMA. Indices de Reactividad.
60
En un matraz enchaquetado de dos bocas equipado con agitación magnética y un condensador,
se cargó una solución de MAH recristalizado y DMF (usando una relación del 5% w/w con respecto al
anhídrido maleico, el DMF se empleó con el fin de llevar a cabo una disolución completa del MAH en
el medio de reacción; sin la presencia de este disolvente la solubilidad del MAH en el IP está limitada
alrededor de 3-5% en peso). i Se eliminó el oxígeno de la solución con nitrógeno (grado UAP) por
espacio de 20 minutos.
Por otra parte, se preparó una solución de isopreno recién destilado y BPO (en una relación al
1.5% en mol con respecto al IP), ésta fue desoxigenada con nitrógeno (grado UAP) por 20 minutos.
Durante el periodo de desoxigenación de la solución, el recipiente que contuvo la mezcla de reacción
estuvo sumergida dentro de un baño de hielo para prevenir la pérdida del isopreno por evaporación. La
mezcla se adicionó cuidadosamente al matraz de dos bocas que contenía el MAH, por medio de un
embudo de adición por un periodo aproximado de 5 a 20 minutos (dependiendo de la relación IP/MAH
en cuestión).
El tiempo de reacción fue registrado a partir de la adición inicial de la mezcla de IP/BPO; este
procedimiento permite favorecer la polimerización sobre la cicloadición de Diels-Alder.105 Previamente,
el matraz que contiene al anhídrido maleico fue precalentado a 70 °C ± 0.1°C con una agitación continua
por medio de un baño recirculador de aceite. El inicio de la polimerización fue instantáneo y la
formación del copolímero fue visible en la superficie de la mezcla de reacción.
Las muestras fueron tomadas a intervalos de tiempo regulados (típicamente cada hora) para
determinar la conversión por gravimetría. Una vez alcanzada la conversión deseada (< 15%), se detuvo
la reacción agregando una solución de hidroquinona en THF al 2% y se sumergió la mezcla de reacción
en un baño de hielo, posteriormente se agregaron aproximadamente 20 mL de acetona para disolver el
copolímero y lentamente se vertió la mezcla en 100 mL de hexano en agitación para precipitar el
material polimérico, el exceso de disolvente se retiró por decantación. Este procedimiento se repitió
cinco veces; el copolímero precipitó en forma fibrosa. Finalmente se filtró y se lavó nuevamente con
hexano. Todas las muestras se secaron a vacío a 40 °C por 24 horas y se analizaron por FT-IR, 1H RMN,
i Dado que la cantidad de DMF que se usa en los experimentos de copolimerización es muy pequeña, se puede considerar en términos prácticos que es muy similar
a una polimerización en masa y por lo tanto, se estará refiriendo a estas reacción de polimerización como experimentos en masa).104

Capítulo 3/ Parte Experimental 3.5 Procedimiento general de la cinética y
copolimerización del isopreno del IP/GMA. Índices de reactividad.
61
GPC y DSC. El producto seco presentó una tonalidad desde amarillenta a café claro (dependiendo de la
relación IP-MAH). El análisis composicional de los copolímeros con respecto a la fracción molar del
isopreno en el copolímero fue determinado por 1H RMN.
FT-IR (cm-1) 3440, 3100, 3000, 2924, 1854, 1780, 1666, 1645, 1450, 1361, 1245, 1059, 889, 837,
692
1H RMN (300 MHz, Ac-d6): δ (ppm) = 5.9, 5.6, 4.9, 4.6, 3.1, 2.8, 2.6, 2.0, 1.7, 1.4, 1.0
3.5 Procedimiento general de la cinética y copolimerización del isopreno
del IP/GMA. Índices de reactividad
Las reacciones de copolimerización se realizaron en viales de vidrio tipo Pyrex sellados con una
gárgola de aluminio y septum de hule. Los viales tienen una longitud de 8.5 cm de largo con un diámetro
externo de 23 mm.
Los monómeros y el iniciador (previamente pesados a la relación molar deseada) se mezclaron y
se dividieron en alícuotas de 2 mL de volumen. Los viales fueron sellados, sumergidos en un baño de
hielo por 20 min y se les hizo burbujear una corriente de nitrógeno (grado UAP) para promover la
desoxigenación en la mezcla de reacción. Esta operación se repitió tres veces y, posteriormente, los
viales se sumergieron dentro de un baño de aceite a 70 °C ± 0.1°C para iniciar la copolimerización,
retirando un vial en cada intervalo de tiempo especificado (cada hora en promedio) hasta alcanzar la
conversión deseada (<15%). Para detener la reacción de polimerización, se les adicionó una solución de
hidroquinona en THF al 2%, sumergiendo el vial en un baño de hielo. Posteriormente, se agregaron 15
mL de cloroformo y lentamente se vertió el contenido en 60 mL de metanol para precipitar el
copolímero. Este procedimiento de repitió cinco veces.
El copolímero precipitó inmediatamente en forma fibrosa, finalmente se filtró y se lavó con
hexano. Todas las muestras se secaron a vacío a 40 °C por 24 horas y se analizaron por IR-FT, 1H RMN,
GPC y DSC. El producto seco presentó una tonalidad blanca. El análisis composicional de los
copolímeros con respecto a la fracción molar del isopreno en el copolímero se determinó por 1H RMN.

Capítulo 3/ Parte Experimental 3.6 Procedimiento general de la cinética de
(co)polimerización del isopreno. Polimerización mediada por nitróxidos.
62
FT-IR (cm-1) 3440, 3100, 3000, 2924, 1733, 1666, 1645, 1485, 1361, 1256, 1147, 1078, 908, 889,
837, 725
1H RMN (300 MHz, CDCl3): δ (ppm) = 5.6, 4.9, 4.8-4.6, 4.3, 3.1, 2.7, 2.2, 2.0, 1.9-1.7, 1.4, 1.0, 0.9
3.6 Procedimiento general de la cinética de homo y copolimerización del
isopreno. Polimerización mediada por nitróxidos (NMP). Proceso
bimolecular
En un procedimiento típico para la cinética de la (co)polimerización del isopreno por
NMP, se utilizó un reactor de acero inoxidable con una capacidad de 250 mL acondicionado
con una válvula alimentadora y una válvula de purga, donde se cargó una mezcla de nitróxido
con BPO, un aditivo acelerante de la velocidad de polimerización (en el caso de los
experimentos que así lo requirieron) a una relación molar especificada e isopreno (30 mL, 300
mmol) en una cantidad requerida de disolvente (o-diclorobenceno o tolueno según se especificó)
con la condición de cumplir la relación de 30 % w/w de sólidos en el disolvente. Se burbujeó en
el interior de la mezcla de reacción nitrógeno (grado UAP), manteniéndola fría dentro de un
baño de hielo (para poder disminuir la presión de vapor del IP y con ello evitar pérdidas del
monómero) por espacio de 40 minutos, después se procedió a presurizar el reactor con nitrógeno
(grado UAP) a 1170 kPa. Realizado lo anterior, se sumergió el sistema en un baño de aceite a
120 ± 1 °C para tomar alícuotas (de 6±2 mL) cada intervalo de tiempo requerido.
A cada alícuota se le adicionó 0.5 mL de una solución de hidroquinona en THF al 2 %
para detener completamente la reacción y fue sumergida en un baño de hielo. La conversión de
cada muestra fue determinada por gravimetría. Posteriormente, a cada alícuota se le evaporó el
disolvente por medio de un extractor de aire y la mezcla viscosa resultante fue redisuelta en
tolueno (5 mL) y precipitada (2 veces) en metanol (60 mL). El polímero precipitó
inmediatamente en forma chiclosa y el exceso de disolvente fue evaporado dentro de una estufa
a 40 °C a vacío hasta que un peso constante fue obtenido. Las muestras se mandaron a
caracterizar por 1H RMN, FT-IR, GPC y DSC.

Capítulo 3/ Parte Experimental 3.7 Procedimiento general de la cinética de
(co)polimerización del isopreno. Polimerización RAFT.
63
En el caso de la copolimerización aleatoria, se utilizaron las mismas condiciones antes
mencionadas (sin la utilización del reactivo acelerante de la velocidad de polimerización) pero
se adicionó a la mezcla de reacción metacrilato de glicidilo (0.2043 g, que representa el 1% w/w
con respecto al IP). El proceso de la purificación del material polimérico procedió de similar
manera.
3.7 Procedimiento general de la cinética de la homo y copolimerización
del isopreno. Polimerización RAFT
La cinética de polimerización se llevó a cabo de acuerdo al procedimiento descrito en la
polimerización del isopreno mediada por nitróxidos (apartado 3.6), con la diferencia de que la
temperatura de reacción fue de 125 °C o 65 °C dependiendo del caso de estudio y sin la
utilización de aditivo acelerante.
Un procedimiento típico para efectuar las reacciones mediadas por RAFT fue el siguiente;
una mezcla de reacción se preparó con el agente de control CDSPA, peróxido de t-butilo o BPO
(dependiendo de la temperatura de operación) e isopreno (30 mL, 20.43 g, 300 mmol) con una
relación molar de agente RAFT/iniciador de radicales libres de 5.3.
En el caso de la copolimerización aleatoria del isopreno, se utilizaron las mismas
condiciones que las utilizadas para la homopolimerización, adicionando a la mezcla de reacción
metacrilato de glicidilo (0.2043 g, que representa el 1% w/w con respecto al IP).
Previamente para cada extensión de cadena, se procedió a purificar el copolímero para
realizar el siguiente experimento. Para la extensión de la cadena el procedimiento fue similar al
anteriormente descrito, pero en la condiciones iniciales para realizar la copolimerización, se
manejó 1.0215 g de GMA (que representa el 5% w/w con respecto al IP), posteriormente se
realizó una mezcla de reacción con isopreno y el macroagente de control resultante (MCTA),
manteniendo una relación molar de isopreno/MCTA de 1400 en una solución de tolueno por 24
horas. El procedimiento del crecimiento de la cadena se repitió hasta alcanzar un Mn de al menos
1x105 Da.

Capítulo 3/ Parte Experimental 3.8 Procedimiento general de la cinética de
(co)polimerización del isopreno por NMP. Proceso unimolecular.
64
3.8 Procedimiento general de la cinética de la homo y copolimerización
del isopreno por NMP. Proceso unimolecular
Para llevar a cabo el proceso unimolecular, esta parte experimental fue similar al
procedimiento llevado a cabo en la homopolimerización del isopreno mediada por nitróxidos
descrito en el apartado 3.6 (sin la utilización de BPO y el aditivo acelerante), por lo cual se
realizó una mezcla de reacción con la alcoxiamina TIPAL (0.0976 g, 0.3 mmol) e isopreno (30
mL, 300 mmol).
Para el caso de la copolimerización del isopreno, las condiciones de reacción son similares
a lo mencionado, agregando metacrilato de glicidilo en la mezcla de reacción original (0.2043
g, 1.437 mmol, que representa el 1% w/w con respecto al IP).
La extensión de la cadena polimérica se llevó a cabo de igual forma a como se describe
en el apartado 3.8, utilizando las condiciones correspondientes para la polimerización mediada
por nitróxidos en un proceso unimolecular.
3.9 Purificación del poliisopreno (PIP)
10 g de PIP (ya sea en la conformación cis o trans) se disolvieron en tolueno (150 mL).
La solución resultante se adicionó gota a gota en metanol (400 mL) para precipitarlo en medio
de una agitación vigorosa (este procedimiento se repitió dos veces). El PIP purificado (sin
antioxidantes) fue recuperado por decantación y secado en una estufa de vacío a 40 °C hasta
obtener un peso constante. La muestra se caracterizó por 1H RMN y por FT-IR.
3.10 Funcionalización del PIP con el nitróxido
En un procedimiento típico para realizar la funcionalización del PIP, 5 g de poliisopreno
purificado-cis y 0.56 mmol del nitróxido (2.38% w/w con respecto al PIP) fueron disueltos en

Capítulo 3/ Parte Experimental 3.11 Reacción de copolimerización de injerto de
IP/GMA en el PIP funcionalizado.
65
70 mL de tolueno en un matraz de dos bocas de 250 mL equipado con un condensador, agitación
magnética y un embudo de adición sumergidos dentro de un baño de aceite. Cuando la mezcla
de reacción alcanzó la temperatura de 125 °C, una solución de TBP (0.63 mmol en 20 mL de
tolueno) se adicionó gota a gota con el fin de prevenir la reacción de entrecruzamiento. La
cantidad de nitróxido utilizada fue calculada considerando que el 10% en mol de las unidades
de PIP en el sustrato se unirán con el nitróxido en la cadena principal. Una relación molar
nitróxido: iniciador de radicales libres (N/I) de 2.7 fue usada para llevar a cabo la reacción de
funcionalización, bajo una atmósfera de nitrógeno (grado UAP) por 6 horas. El polímero
funcionalizado fue purificado tres veces con metanol y disuelto en tolueno con el fin de retirar
el iniciador y el nitróxido libre que no hubiera reaccionado. El material fue secado en una estufa
de vacío a temperatura ambiente hasta obtener un peso constante. La muestra fue caracterizada
por 1H RMN, DQF-COSY y FT-IR.
La síntesis para el PIP-trans fue similar al procedimiento descrito anteriormente, salvo
con la excepción de manejar 0.38 mmol del radical nitróxido.
3.11 Reacción del proceso de injerto de IP/GMA en el PIP
funcionalizado
El proceso de injerto se llevó a cabo bajo condiciones diluidas para minimizar los efectos
viscosos. En conjunto, se prepararon 5 g de la macroalcoxiamina preparada en el procedimiento
3.10 fueron disueltos en 15 g (22 mL, 0.22 mol) de isopreno y 3.48 g de GMA (3.24 mL, 0.025
mol) en una cantidad suficiente de tolueno hasta alcanzar una concentración de 30% w/w de
sólidos. La polimerización se condujo de acuerdo a la técnica descrita en el procedimiento 3.6
(sin la participación del BPO y el aditivo acelerante de la velocidad de polimerización).
3.12 Reacción de la homo y copolimerización del isopreno por PLP-SEC
Las homopolimerizaciones de baja conversión del IP y copolimerizaciones de IP/GMA se
llevaron a cabo en una configuración de pulso de láser que consiste en un “Spectra-Physics

Capítulo 3/ Parte Experimental 3.12 Reacción de la (co)polimerización del
isopreno por PLP-SEC.
66
Quanta-Ray” con un láser Nd:YAG de 100 Hz que es capaz de producir un pulso de láser de
355 nm de duración de 7-10 ns de ancho de pulso y una energía de 1-50 mJ por pulso. El haz de
láser se refleja dos veces (180°) para resplandecer en una celda de muestra cúbica de cuarzo
utilizada como reactor de PLP. Un generador de retardo digital (DDG, Stanford Instruments)
está unido al láser para regular la velocidad de repetición de los pulsos de salida a un valor entre
1 y 100 Hz.
La (co)polimerización de los monómeros fue en masa, usando DMPA (5 mmol) como
fotoiniciador, los cuales se añadieron a la celda de cuarzo para exponerlos a la energía láser, con
temperatura controlada por un baño de aceite en recirculación como lo muestra la Figura 14.
Los experimentos se llevaron a cabo a una temperatura de 25 °C a 1, 2, 10, 25 y 50 Hz, la
fracción molar de GMA en la mezcla de monómeros varió entre 0-100 %. La temperatura se
controló durante el pulso de láser, y nunca aumentó más de 0.5 ° C durante la polimerización.
Las conversiones se controlaron por debajo de 3.0% para evitar una desviación de composición
significativa en la mezcla copolimérica. Después de realizar la purificación del material
polimérico, se mandó a caracterizar (y correlacionar) por GPC.ii
Figura 14: Sistema del equipo de PLP. Parte superior; interior del equipo donde se colocan las celdas de
cuarzo para efectuar la polimerizacion. Parte inferior izquierda; baño de recirculacion que controla la
temperatura del interior del equipo. Parte inferior derecha; tarjeta electronica para la conexión con el equipo de
computo
ii Desarrollo experimental realizado en el Laboratorio del Departamento de Ingeniería Química, Dupuis Hall, Queen’s University, Kingston,
Ontario, Canadá en colaboración con el Dr. Robin Hutchinson y el Dr. Scott Parent.

Capítulo 4/ Índices de reactividad 4.1 Copolimero de IP/GMA. Consideraciones
generales.
67
4.
Resultados y discusión. Índices de reactividad.
4.1 Copolímero de isopreno-metacrilato de glicidilo (IP/GMA).
Consideraciones generales
La copolimerización en masa del isopreno (10) con metacrilato de glicidilo (11, Esquema 12) se
estudió variando las fracciones mol del dieno desde 0.0 hasta 1.0 en la alimentación y el tiempo de
reacción estuvo en función de generar suficiente cantidad de material polimérico para mandarlo a
caracterizar para cada punto experimental, pero cumpliendo la condicionante de lograr conversiones
<15% en peso (requisito vital para la validez de la ecuación de copolimerización de Mayo-Lewis a bajas
conversiones81). Como iniciador de radicales libres se utilizó el BPO en exceso (la relación utilizada fue
de 1.5 % w/w con respecto al dieno), para garantizar la polimerización con respecto a la reacción
secundaria de cicloadición de Diels-Alder.105-106 Las composiciones de la alimentación y en los
copolímeros son presentadas en la Tabla 7. Los copolímeros fueron solubles en DMF, THF, DMSO,
acetona, tolueno y cloroformo pero fueron insolubles en metanol, etanol, agua y hexano.
Esquema 12: Reacción de copolimerización del IP-GMA.
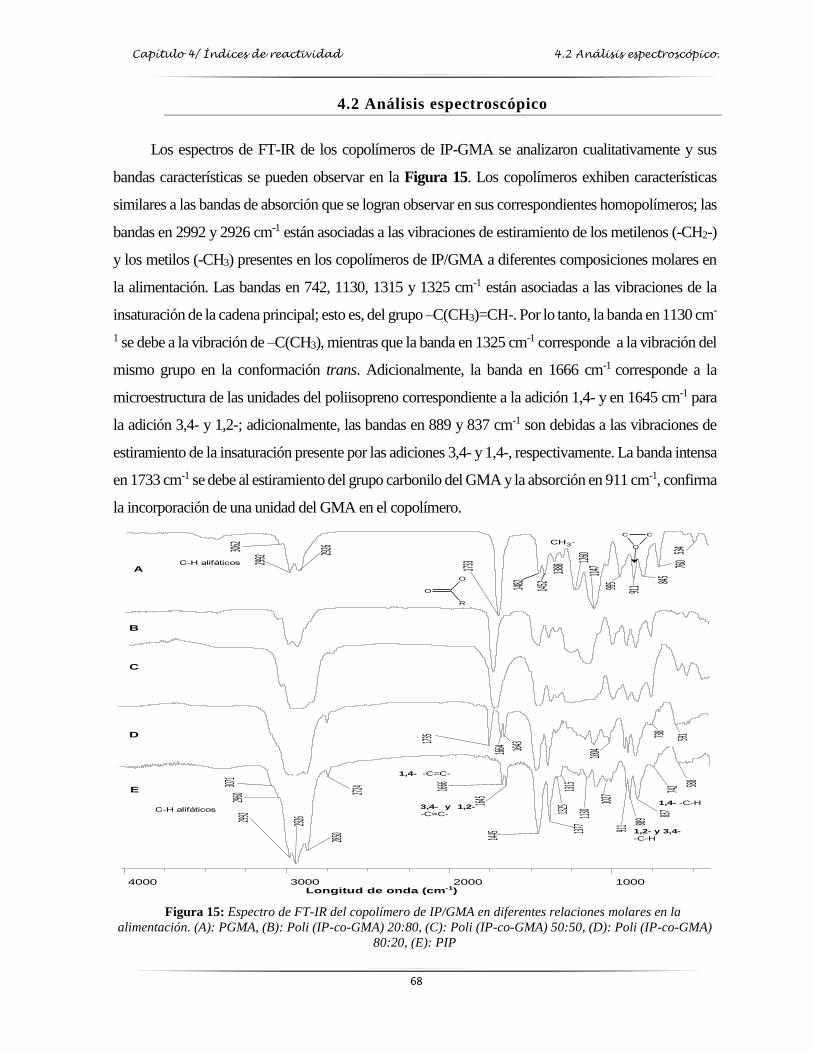
Capítulo 4/ Índices de reactividad 4.2 Análisis espectroscópico.
68
4.2 Análisis espectroscópico
Los espectros de FT-IR de los copolímeros de IP-GMA se analizaron cualitativamente y sus
bandas características se pueden observar en la Figura 15. Los copolímeros exhiben características
similares a las bandas de absorción que se logran observar en sus correspondientes homopolímeros; las
bandas en 2992 y 2926 cm-1 están asociadas a las vibraciones de estiramiento de los metilenos (-CH2-)
y los metilos (-CH3) presentes en los copolímeros de IP/GMA a diferentes composiciones molares en
la alimentación. Las bandas en 742, 1130, 1315 y 1325 cm-1 están asociadas a las vibraciones de la
insaturación de la cadena principal; esto es, del grupo –C(CH3)=CH-. Por lo tanto, la banda en 1130 cm-
1 se debe a la vibración de –C(CH3), mientras que la banda en 1325 cm-1 corresponde a la vibración del
mismo grupo en la conformación trans. Adicionalmente, la banda en 1666 cm-1 corresponde a la
microestructura de las unidades del poliisopreno correspondiente a la adición 1,4- y en 1645 cm-1 para
la adición 3,4- y 1,2-; adicionalmente, las bandas en 889 y 837 cm-1 son debidas a las vibraciones de
estiramiento de la insaturación presente por las adiciones 3,4- y 1,4-, respectivamente. La banda intensa
en 1733 cm-1 se debe al estiramiento del grupo carbonilo del GMA y la absorción en 911 cm-1, confirma
la incorporación de una unidad del GMA en el copolímero.
534760
845
911995114
71260
14521482
1733
2926
2992306
2
591738
1004164
3
1664173
5
588742837
889911102
7
1130
1315
1325
1377
1445
1645166
6
2724
2850
2926
2960
2992
3071
1000 2000 3000 4000 Longitud de onda (cm-1)
O
R
O
1388
CH3-
CC
O
C-H alifáticos
C-H alifáticos
1,4- -C=C-
3,4- y 1,2- -C=C-
1,2- y 3,4- -C-H
1,4- -C-H
A
B
C
D
E
Figura 15: Espectro de FT-IR del copolímero de IP/GMA en diferentes relaciones molares en la
alimentación. (A): PGMA, (B): Poli (IP-co-GMA) 20:80, (C): Poli (IP-co-GMA) 50:50, (D): Poli (IP-co-GMA)
80:20, (E): PIP

Capítulo 4/ Índices de reactividad 4.2 Análisis espectroscópico.
69
La composición del copolímero se analizó cuantitativamente por medio de la técnica
espectroscópica de resonancia magnética nuclear de protón (1H RMN). El espectro de 1H RMN del
poli(IP-co-GMA) se muestra en la Figura 16, en dicha figura se efectúa la comparación con los
homopolímeros respectivos y la variación en diferentes relaciones molares (% mol en la alimentación)
de IP-GMA. En el espectro de 1H RMN del PGMA (Figura 16A) existen picos (protones h e i) en 3.1
y 2.6 ppm atribuidos a los protones metileno y metino del grupo epóxido respectivamente, y un grupo
de multipletes en 4.3 y 3.8 ppm asignados debido al metileno (g) que existe entre el grupo éster y los
protones epóxicos. La señal del grupo metilo (f) aparece en 1.3 ppm. En el caso del espectro de 1H RMN
del PIP (Figura 16E), se observan en la región de 4.9-5.4 ppm los protones (b) correspondientes a la
adición 1,4-, el pico ubicado en 5.7 ppm es asignado al protón vinílico (l) correspondiente a la adición
1.2- y en la región de 4.5-4.9 ppm, se muestran los protones (m y q) que corresponden a las adiciones
1,2- y 3,4-. Las diferencias claras se pueden observar en las Figuras 16C y D.
El espectro de 1H RMN del poli(IP-co-GMA) (Figuras 16B-D) muestra picos en 4.9 ppm
asignados al protón (b) de la insaturación proveniente del isopreno. Los protones de los metilenos (d)
de la cadena principal dan picos en la región de 2.0-2.2 ppm. Los protones del –CH3 (c) perteneciente
al isopreno, se encuentran asignados en 1.7 ppm.
(ppm)5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 1.0 0.5
B
A
C
D
E
CH3
*
O O
O
CH3
*ba
c
d
e
f
g
h
i
bg
ia
d
c
e
f
*CH3
CH3 CH2
CH2
*j
kn
o
lm
p q
l q, m
o
pn
jk
Figura 16: Espectro de 1H RMN de copolímeros de IP/GMA en diferentes relaciones molares en la
alimentación. (A): PGMA, (B): Poli (IP-co-GMA) 20:80, (C): Poli (IP-co-GMA) 50:50, (D): Poli (IP-co-GMA)
80:20, (E): PIP.

Capítulo 4/ Índices de reactividad 4.3 Determinación de los índices de reactividad del
sistema IP/GMA.
70
Como una forma de resumen, en el Esquema 13 se presentan las siguientes unidades
estructurales que conforman la cadena polimérica del sistema IP/GMA (los números adjuntos a dichas
unidades estructurales son los desplazamientos químicos (en ppm)):
Esquema 13: Resumen de los picos presentados en el espectro de 1H RMN de la Figura 16.
4.3 Determinación de los índices de reactividad del sistema IP/GMA
Con el fin de determinar la cantidad de cada comonómero que fue incorporado en el copolímero
formado, la integración de los picos seleccionados del espectro de 1H RMN se manejó para tal propósito.
Esta técnica es ampliamente aceptada y usada tanto en el ámbito industrial como académico.107 La
asignación de los picos estuvo de acuerdo a datos disponibles en la literatura;108 algunos de ellos fueron
también confirmados vía el software ACD/ChemSketch Version 4.55.
Para ilustrar los cálculos involucrados en la determinación de la composición del copolímero,
tomaremos como ejemplo el espectro de 1H RMN del copolímero formado de IP-GMA con una relación
molar en la alimentación 50:50 (Figura 17). Es posible identificar tres regiones en el espectro: a) los
picos correspondientes a los protones ubicados en los grupos -CH-, -CH2- y –CH3 de las unidades de
GMA e isopreno en la región de δ=0.9-2.3 ppm; b) los picos asignados en el grupo glicidilo de unidades
del GMA en la región de δ=2.4-4.5 ppm y c) los picos de los protones adyacentes al doble enlace, –
HC= y =CH2 de unidades del isopreno en la región de δ=4.6-5.3 ppm.

Capítulo 4/ Índices de reactividad 4.3 Determinación de los índices de reactividad del
sistema IP/GMA.
71
Figura 17: Espectro de 1H RMN del copolímero de IP-GMA con una relación molar en la alimentación
50:50.
Siguiendo la técnica de cálculo de Rusakova et al.,109 se definen las fracciones mol del GMA y
del isopreno presente en el copolímero como m2 y (1 - m2) respectivamente y dado que la unidad de
GMA contiene 10 protones y la de isopreno tiene 8, el número de protones correspondiente al área total
del espectro (ST) es:
𝑆𝑇 ∝ 10𝑚2 + 8 1 − 𝑚2 = 8 + 2𝑚2 (16)
Por otra parte, los picos en el rango de 2.4-4.5 ppm corresponden a los cinco protones del
grupo glicidilo, cuya área de integración es denotada como SG, resultando en:
𝑆𝐺
𝑆𝑇=
5𝑚2
8+2𝑚2; 𝑚2 =
8𝑆𝐺
5𝑆𝑇−2𝑆𝐺 (17)
La Tabla 7 presenta las composiciones molares del copolímero obtenidas por medio de las
expresiones 16 y 17 para varias series de copolímeros de IP-GMA a baja conversión, donde la relación
molar en la alimentación fue variada.
Los pesos moleculares promedio en número (Mn) y en peso (Mw) de los copolímeros (los cuales
fueron determinados por GPC) también se presentan en la Tabla 7; los índices de dispersidad (Đ) están
entre 1.9 hasta 3.5, se aprecia que el peso molecular promedio en peso disminuye con el contenido de

Capítulo 4/ Índices de reactividad 4.3 Determinación de los índices de reactividad del
sistema IP/GMA.
72
isopreno en el copolímero. Esto puede deberse a dos factores; a) la baja constante de propagación
conforme aumenta el contenido de IP en la alimentación, b) una posible alta rapidez de transferencia de
cadena al monómero conforme se incrementa el contenido de IP. Aunque no existe suficiente
información acerca de los parámetros cinéticos en la literatura especializada para este par de monómeros
(IP y GMA), se pueden realizar algunas estimaciones aproximadas para los valores de las constantes
cinéticas relevantes, los cuales se pueden obtener de datos ya publicados o de monómeros similares.
Tabla 7. Resultados de la copolimerización del IP con GMA.
fracción
mol IPa
fracción
mol
GMAa
Composición
del monómero
inicial (M1)a, c
Conversión SG ST
Composición
del copolímero
(m1)b, c
Mn Mw Đ
10 90 0.11 9.9 2.95 7.28 0.23 93000 230000 2.5
20 80 0.21 11.12 2.63 7.20 0.32 79000 210000 2.7
30 70 0.29 7.98 2.31 7.46 0.43 57000 170000 3.0
40 60 0.38 11.54 1.87 6.19 0.45 39000 76000 1.9
50 50 0.50 8.66 1.24 4.65 0.52 10200 28000 2.7
60 40 0.61 13.31 0.97 3.63 0.52/0.54d 8900 26000 2.9
70 30 0.72 12.11 0.79 3.07 0.54 9900 33000 3.3
80 20 0.80 14.05 0.58 2.44 0.58 3400 12000 3.5
90 10 0.93 14.65 0.33 2.00 0.72 5200 18000 3.5
Condiciones de operación: Temperatura: 70 °C; iniciador: BPO. 1: IP, 2: GMA Para fines prácticos, los valores de la composición
inicial de la alimentación (% mol) de las dos primeras columnas son valores nominales. Los valores reales se hallan en la columna 3
(solo el monómero 1 se muestra) ligeramente diferente de los nominales debido al error experimental en el momento del pesaje de las
muestras. a Composición en la alimentación. b Determinado por 1H RMN. c Composición molar. d Este experimento se replicó.
El único valor que se tiene reportado para la constante de propagación (kp) del isopreno es de 2.8
L/(mol s) a 5 °C, mientras que para el butadieno (un dieno similar sin la presencia del grupo metilo que
tiene el isopreno) a la misma temperatura de operación es de 5.69 L/(mol s); esto es, el doble del valor
del isopreno; ambos valores reportados en una antigua referencia.110 El valor de kp para el GMA a 5 °C
es reportado como 251.05 L/(mol s),88c alrededor de 44 veces más grande para el butadieno a la misma
temperatura y casi 90 veces más grande para el isopreno si se usan todos los datos anteriores y se hacen
simples extrapolaciones. Estos valores son consistentes con la tendencia mostrada por los resultados
obtenidos de las reacciones de copolimerización efectuando simples cálculos lineales basados en la
conversión vs tiempo de la Tabla 7; esto es, la velocidad de polimerización para la copolimerización
10/90 del IP/GMA es nueve veces más grande que para la reacción 90/10 del IP/GMA.
El cambio en la kp y en la velocidad de polimerización con la composición del monómero

Capítulo 4/ Índices de reactividad 4.3.1 Método de FR y FRI.
73
contribuirá a obtener un Mn más bajo del copolímero conforme se incrementa el contenido del IP, pero
no es suficiente para explicar el cambio dramático sobre el peso molecular con la composición. Por otra
parte, para las constantes de rapidez de transferencia al monómero (ktr), se halló el valor de 1.32 L/(mol
s) para el butadieno a 70 °C,111 pero no existe un valor para el GMA; sin embargo, para otros
metacrilatos (como el metacrilato de metilo, MMA), el valor de la ktr reportado a 50 °C es de 0.0334
L/(mol s).112
Basado en la kp del MMA a 50 °C de 1157 L/(mol s), la constante de transferencia de CM (ktr/kp)
para el MMA a 50 °C es de 2.9x10-5 L/(mol s). Similarmente, el valor estimado para el butadieno a 70
°C sería de 4.5x10-3 L/(mol s), alrededor de dos órdenes de magnitud más grande que para el MMA.
Esto sugiere que el valor de la CM para el isopreno podría ser significativamente más grande que la del
GMA, lo que también contribuiría a un Mn más bajo de los copolímeros con el incremento del IP en la
alimentación.
4.3.1 Método de Finemann-Ross (FR) y Finemann-Ross Invertida (FRI)
Los resultados experimentales presentados en la Tabla 7 fueron tratados por diversos métodos
de cálculo para el sistema conformado por el IP y GMA, de tal manera que los índices de reactividad y
las composiciones del copolímero se determinaron por las técnicas de cálculo de mínimos cuadrados
lineales (LLS), como el método de Finemann-Ross (FR),85a Finemann-Ross Invertida (FRI),85a Kelen-
Tüdos (KT),85b Kelen-Tüdos extendida (KTE)85c y por una técnica de mínimos cuadrados no lineales
(NLLS), el método de Tidwell-Mortimer (TM).86a
Todos estos métodos están basados en el modelo cinético terminal de la ecuación de Mayo-Lewis
(ecuación 7). La Figura 18 muestra la curva de composición del copolímero generada por las
correspondientes composiciones de alimentación expresadas como fracción mol de comonómero
arrojados por la Tabla 7 (en este caso se utilizará la siguiente nomenclatura: 1 para designar al IP y 2
para el GMA). En dicha gráfica se muestra que el par de monómeros forman una mezcla azeotrópica
con un contenido de 50.7 fracción mol.
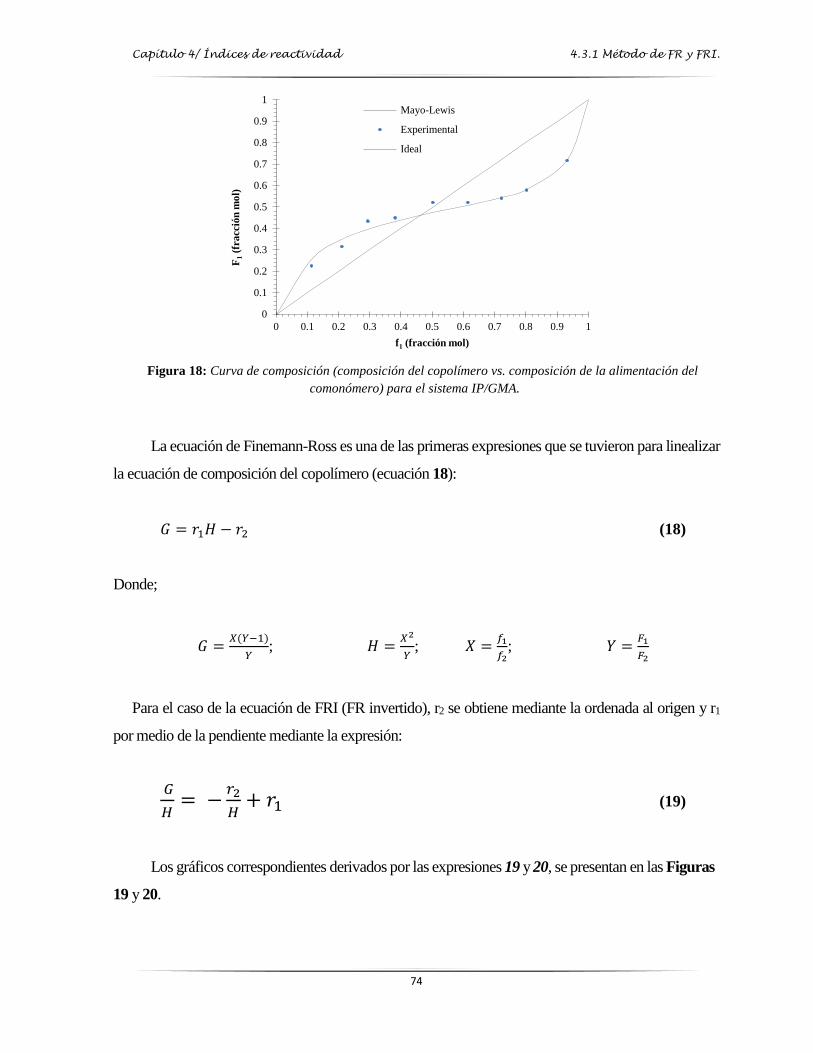
Capítulo 4/ Índices de reactividad 4.3.1 Método de FR y FRI.
74
Figura 18: Curva de composición (composición del copolímero vs. composición de la alimentación del
comonómero) para el sistema IP/GMA.
La ecuación de Finemann-Ross es una de las primeras expresiones que se tuvieron para linealizar
la ecuación de composición del copolímero (ecuación 18):
𝐺 = 𝑟1𝐻 − 𝑟2 (18)
Donde;
𝐺 =𝑋 𝑌−1
𝑌; 𝐻 =
𝑋2
𝑌; 𝑋 =
𝑓1
𝑓2; 𝑌 =
𝐹1
𝐹2
Para el caso de la ecuación de FRI (FR invertido), r2 se obtiene mediante la ordenada al origen y r1
por medio de la pendiente mediante la expresión:
𝐺
𝐻= −
𝑟2
𝐻+ 𝑟1 (19)
Los gráficos correspondientes derivados por las expresiones 19 y 20, se presentan en las Figuras
19 y 20.
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1
0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1
F1
(fra
cció
n m
ol)
f1 (fracción mol)
Mayo-Lewis
Experimental
Ideal
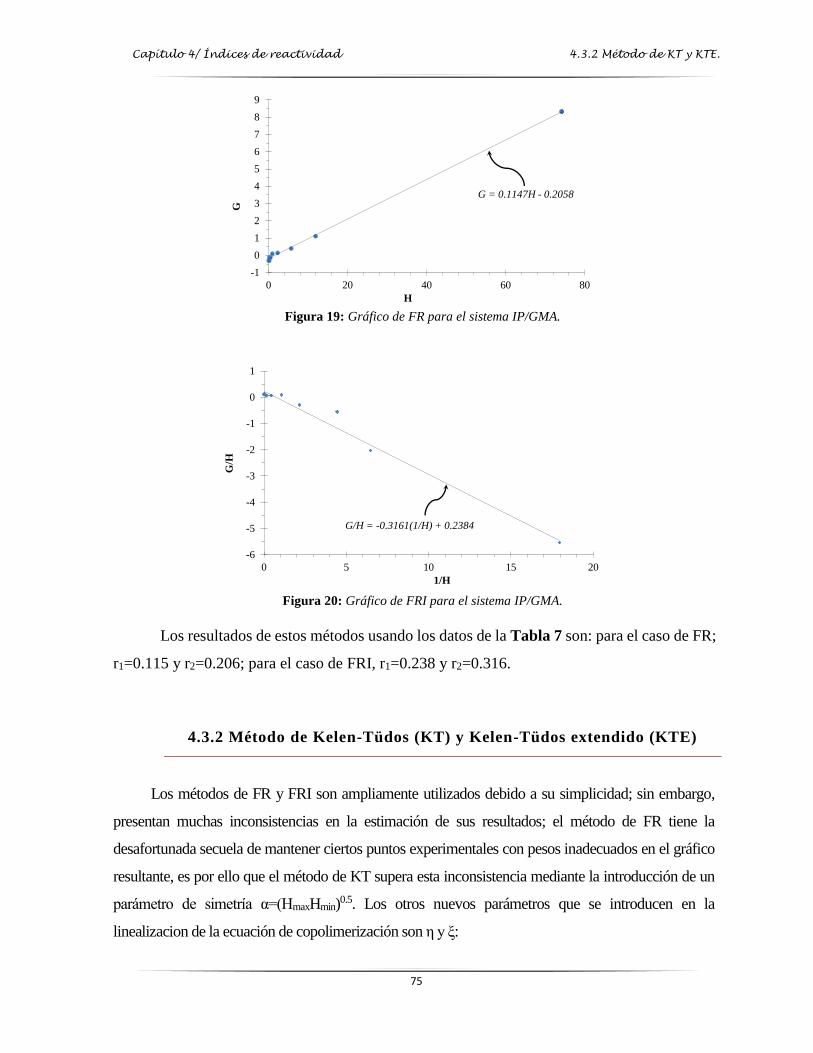
Capítulo 4/ Índices de reactividad 4.3.2 Método de KT y KTE.
75
Figura 19: Gráfico de FR para el sistema IP/GMA.
Figura 20: Gráfico de FRI para el sistema IP/GMA.
Los resultados de estos métodos usando los datos de la Tabla 7 son: para el caso de FR;
r1=0.115 y r2=0.206; para el caso de FRI, r1=0.238 y r2=0.316.
4.3.2 Método de Kelen-Tüdos (KT) y Kelen-Tüdos extendido (KTE)
Los métodos de FR y FRI son ampliamente utilizados debido a su simplicidad; sin embargo,
presentan muchas inconsistencias en la estimación de sus resultados; el método de FR tiene la
desafortunada secuela de mantener ciertos puntos experimentales con pesos inadecuados en el gráfico
resultante, es por ello que el método de KT supera esta inconsistencia mediante la introducción de un
parámetro de simetría α=(HmaxHmin)0.5. Los otros nuevos parámetros que se introducen en la
linealizacion de la ecuación de copolimerización son η y ξ:
G = 0.1147H - 0.2058
-1
0
1
2
3
4
5
6
7
8
9
0 20 40 60 80
G
H
G/H = -0.3161(1/H) + 0.2384
-6
-5
-4
-3
-2
-1
0
1
0 5 10 15 20
G/H
1/H
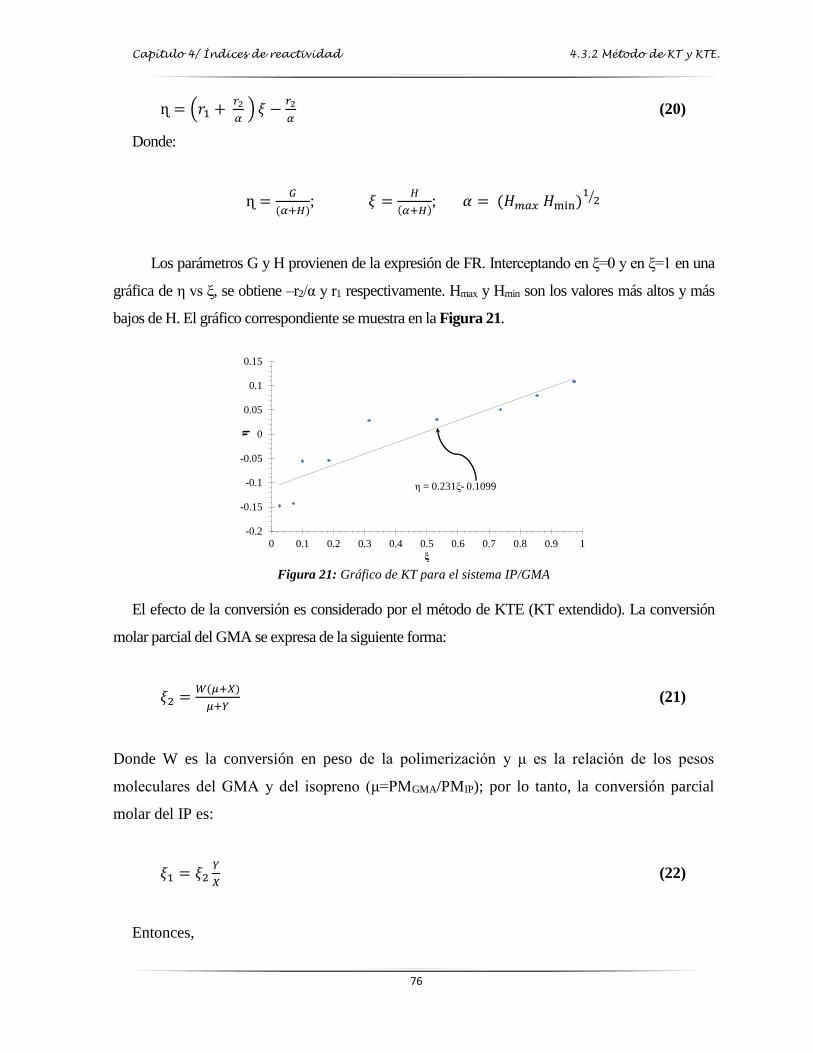
Capítulo 4/ Índices de reactividad 4.3.2 Método de KT y KTE.
76
ɳ = (𝑟1 + 𝑟2
𝛼 ) 𝜉 −
𝑟2
𝛼 (20)
Donde:
ɳ =𝐺
𝛼+𝐻 ; 𝜉 =
𝐻
𝛼+𝐻 ; 𝛼 = 𝐻𝑚𝑎𝑥 𝐻min
12⁄
Los parámetros G y H provienen de la expresión de FR. Interceptando en ξ=0 y en ξ=1 en una
gráfica de η vs ξ, se obtiene –r2/α y r1 respectivamente. Hmax y Hmin son los valores más altos y más
bajos de H. El gráfico correspondiente se muestra en la Figura 21.
Figura 21: Gráfico de KT para el sistema IP/GMA
El efecto de la conversión es considerado por el método de KTE (KT extendido). La conversión
molar parcial del GMA se expresa de la siguiente forma:
𝜉2 =𝑊 𝜇+𝑋
𝜇+𝑌 (21)
Donde W es la conversión en peso de la polimerización y μ es la relación de los pesos
moleculares del GMA y del isopreno (μ=PMGMA/PMIP); por lo tanto, la conversión parcial
molar del IP es:
𝜉1 = 𝜉2𝑌
𝑋 (22)
Entonces,
η = 0.231ξ- 0.1099
-0.2
-0.15
-0.1
-0.05
0
0.05
0.1
0.15
0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1
η
ξ
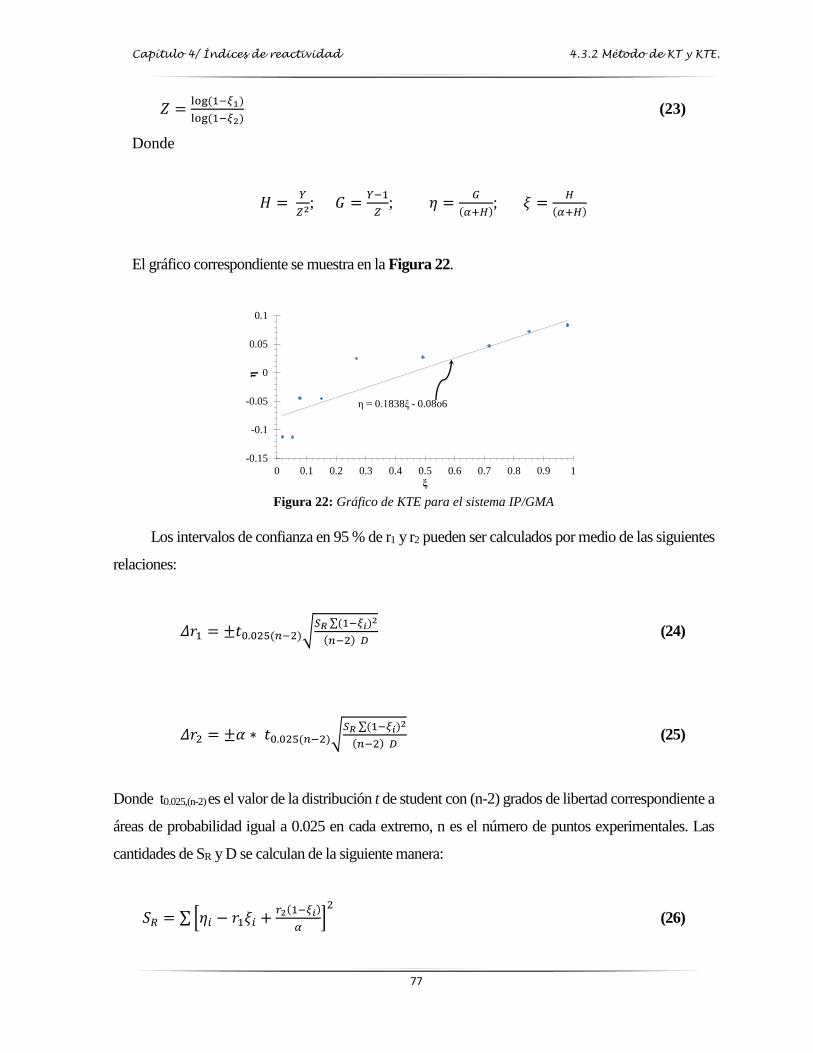
Capítulo 4/ Índices de reactividad 4.3.2 Método de KT y KTE.
77
𝑍 =log 1−𝜉1
log 1−𝜉2 (23)
Donde
𝐻 = 𝑌
𝑍2; 𝐺 =𝑌−1
𝑍; 𝜂 =
𝐺
𝛼+𝐻 ; 𝜉 =
𝐻
𝛼+𝐻
El gráfico correspondiente se muestra en la Figura 22.
Figura 22: Gráfico de KTE para el sistema IP/GMA
Los intervalos de confianza en 95 % de r1 y r2 pueden ser calculados por medio de las siguientes
relaciones:
𝛥𝑟1 = ±𝑡0.025 𝑛−2 √𝑆𝑅 ∑ 1−𝜉𝑖
2
𝑛−2 𝐷 (24)
𝛥𝑟2 = ±𝛼 ∗ 𝑡0.025 𝑛−2 √𝑆𝑅 ∑ 1−𝜉𝑖
2
𝑛−2 𝐷 (25)
Donde t0.025,(n-2) es el valor de la distribución t de student con (n-2) grados de libertad correspondiente a
áreas de probabilidad igual a 0.025 en cada extremo, n es el número de puntos experimentales. Las
cantidades de SR y D se calculan de la siguiente manera:
𝑆𝑅 = ∑[𝜂𝑖 − 𝑟1𝜉𝑖 +𝑟2 1−𝜉𝑖
𝛼]2
(26)
η = 0.1838ξ - 0.08o6
-0.15
-0.1
-0.05
0
0.05
0.1
0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1
η
ξ

Capítulo 4/ Índices de reactividad 4.3.3 Método de Tidwell-Mortimer.
78
𝐷 = ∑𝜉𝑖2 ∑ 1 − 𝜉𝑖
2 − [∑𝜉𝑖 1 − 𝜉𝑖 ]2 (27)
Los resultados de los índices de reactividad por estos métodos, usando los datos de la Tabla 7
son los siguientes: para el caso de KT r1=0.121±0.060 y r2=0.224±0.121; en el caso de KTE,
r1=0.103±0.053 y r2=0.203±0.133. Dado que r1 y r2 no son independientes uno del otro, es apropiado
utilizar una región de confianza conjunta (JCR) del 95%, de acuerdo a la siguiente expresión:
𝑅1 − 𝑟1 2 ∑𝜉𝑖
2 +2
𝛼 𝑅1 − 𝑟1 𝑅2 − 𝑟2 ∑ 𝜉𝑖 𝜉𝑖 − 1 + (
𝑅2−𝑟2
𝛼)2∑ 𝜉𝑖 − 1 2 =
2𝑆𝑅
𝑛−2𝐹0.05,2, 𝑛−2 (28)
Donde F0.05, 2, (n-2) es la distribución F teniendo 2 y n-2 grados de libertad; r1 y r2 es la estimación
de mínimos cuadrados de los índices de reactividad respectivamente. El JCR para ambos métodos de
cálculo, se muestran en la Figura 23. Los índices de reactividad generalmente se determinan a
bajas conversiones debido a la consideración en que el corrimiento en la composición del
comonómero es despreciable a bajas conversiones. Sin embargo, para una aproximación más
detallada de los índices de reactividad, se pueden calcular los efectos de la conversión. Es por
ello que el método KTE considera este aspecto en su proceso de cálculo.
4.3.3 Método de Tidwell-Mortimer (TM)
El método de Tidwell-Mortimer es una de las técnicas más comunes a utilizar cuando se desea
aplicar las más apropiadas técnicas de estimación no lineales (NLLS), dado que es considerado uno de
los procedimientos más exactos para la determinación de los valores de los índices de reactividad de
monómeros.86a El método es una modificación del procedimiento de ajuste de curva tal que la suma de
los cuadrados de la diferencia entre las composiciones observadas y las calculadas son minimizadas.
Una breve descripción del método consiste en estimar los valores iniciales de r1 y r2, un conjunto
de cálculos se realizan mediante la suma de los cuadrados de las diferencias entre las composiciones

Capítulo 4/ Índices de reactividad 4.3.3 Método de Tidwell-Mortimer.
79
observadas y las calculadas. La suma es minimizada por iteración, obteniéndose por lo tanto los índices
de reactividad. Este procedimiento evalúa las ri’s que se han estimado y verifica que los datos sean
consistentes con la ecuación de composición del copolímero por métodos de mínimos cuadrados no
lineales. El procedimiento TM consiste en lo siguiente: se estiman los parámetros de ri’s iniciales
(obtenidos por algún otro método o suponiendo los valores), Gi se calcula por medio de la expresión:
𝐺𝑖 = 𝑟2𝑓2𝑖
2+ 𝑓1𝑖𝑓2𝑖
𝑟2𝑓2𝑖2+2𝑓1𝑖 𝑓2𝑖 +𝑟1 𝑓1𝑖
2 (29)
La diferencia di, entre las composiciones del polímero observadas y calculadas (en este
caso, el objetivo es minimizar la suma de los cuadrados de las diferencias por iteración), las
derivadas parciales δGi/δr1, δGi/δr2 y el estimado de mínimos cuadrados b1 y b2 se determinan
por medio de:
𝑑𝑖 = 𝐹2𝑖 − 𝐺𝑖 (30)
𝜕𝐺𝑖
𝜕𝑟1=
−𝑓1𝑖 2 𝑟2𝑓2𝑖
2+𝑓1𝑖𝑓2𝑖
𝑃2 (31)
𝜕𝐺𝑖
𝜕𝑟2= 𝑓2𝑖
2 𝑟1𝑓1𝑖
2+𝑓1𝑖𝑓2𝑖
𝑃2 (32)
Donde:
𝑃 = 𝑟2𝑓2𝑖2 + 2𝑓1𝑖𝑓2𝑖 + 𝑟1𝑓1𝑖
2 (33)
𝑏1 = [𝛴
𝜕𝐺𝑖𝜕𝑟2
2
𝛴(𝜕𝐺𝑖𝜕𝑟1
)2 𝛴𝑑𝑖
𝜕𝐺𝑖
𝜕𝑟1−
𝛴 𝜕𝐺𝑖𝜕𝑟2
𝜕𝐺𝑖𝜕𝑟1
𝛴𝑑𝑖𝜕𝐺𝑖𝜕𝑟2
𝛴(𝜕𝐺𝑖𝜕𝑟1
)2 ] /𝐶 (34)
𝑏2 = [𝑛𝛴𝑑𝑖
𝜕𝐺𝑖𝜕𝑟2
𝛴(𝜕𝐺𝑖𝜕𝑟1
)2 −
𝛴 𝜕𝐺𝑖𝜕𝑟2
2
𝛴(𝜕𝐺𝑖𝜕𝑟1
)2 𝛴 𝑑𝑖 (
𝜕𝐺𝑖
𝜕𝑟1 )] /𝐶 (35)

Capítulo 4/ Índices de reactividad 4.3.3 Método de Tidwell-Mortimer.
80
𝐶 = 𝑛 [𝛴
𝜕𝐺𝑖𝜕𝑟2
2
𝛴(𝜕𝐺𝑖𝜕𝑟1
)2] − (
∑𝜕𝐺𝑖𝜕𝑟2
∑𝜕𝐺𝑖𝜕𝑟1
)
2
(36)
Los valores de Sk=[Σ(di)2]k para r2=r2
anterior+[(k-1)/2]b1 y r1=r1anterior+[(k-1)/2]b2 son
determinados para valores de k=1, 2 y 3; para S4=[Σ(di)2]4 para r2=r2
anterior+Vb1 y
r1=r1anterior+Vb2, donde:
𝑉 = 0.5 + 𝑆1−𝑆3
4 𝑆1−2𝑆2+𝑆3 (37)
Si S4<S1 se repite el proceso usando las nuevas ri’s obtenidas. Si S4>S1, entonces se
reevalúa V después de haber divido en dos b1 y b2. El procedimiento se repite hasta que Σ(di)2
se reduce a su mínimo valor. Para el sistema que se está estudiando, se tomó a r1=r2=10 como
estimado inicial y después de varias iteraciones, los valores generados se muestran en la Tabla
8.
Tabla 8: Resultados de las iteraciones para el cálculo de los índices de reactividad usando el método
de TM.
r1 r2 (di)2
0.12 0.22 0.0070020
0.12224 0.27935 0.0069320
0.12223 0.28017 0.0069672
0.12222 0.28104 0.0070053
0.12221 0.28196 0.0070467
0.12219 0.28294 0.0070918
0.11395 0.23417 0.0067476
0.11782 0.24615 0.0063839
0.11802 0.24645 0.0063349
0.11836 0.24691 0.0063349
0.11895 0.24764 0.0063349
r1: índice de reactividad para el isopreno, r2: índice de reactividad para el GMA.
El JCR del 95% para el método de TM se presenta en la Figura 23 el cual está definido
por la expresión:
𝑎11 𝑅1 − 𝑟1 2 + 2𝑎12 𝑅1 − 𝑟1 𝑅2 − 𝑟2 + 𝑎22 𝑅2 − 𝑟2 = 2𝐹0.05 2, 𝑛−2
∑ 𝑑𝑖 2
𝑛−2 2 (38)
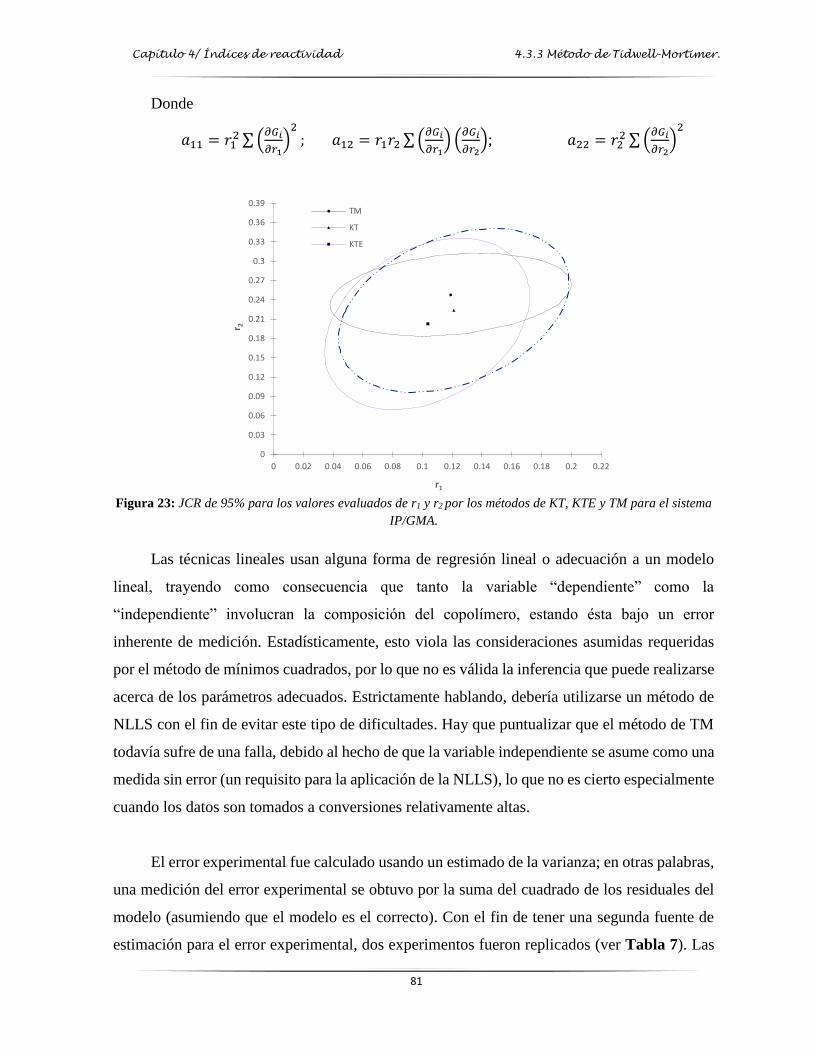
Capítulo 4/ Índices de reactividad 4.3.3 Método de Tidwell-Mortimer.
81
Donde
𝑎11 = 𝑟12 ∑(
𝜕𝐺𝑖
𝜕𝑟1)2
; 𝑎12 = 𝑟1𝑟2 ∑(𝜕𝐺𝑖
𝜕𝑟1) (
𝜕𝐺𝑖
𝜕𝑟2); 𝑎22 = 𝑟2
2 ∑(𝜕𝐺𝑖
𝜕𝑟2)2
Figura 23: JCR de 95% para los valores evaluados de r1 y r2 por los métodos de KT, KTE y TM para el sistema
IP/GMA.
Las técnicas lineales usan alguna forma de regresión lineal o adecuación a un modelo
lineal, trayendo como consecuencia que tanto la variable “dependiente” como la
“independiente” involucran la composición del copolímero, estando ésta bajo un error
inherente de medición. Estadísticamente, esto viola las consideraciones asumidas requeridas
por el método de mínimos cuadrados, por lo que no es válida la inferencia que puede realizarse
acerca de los parámetros adecuados. Estrictamente hablando, debería utilizarse un método de
NLLS con el fin de evitar este tipo de dificultades. Hay que puntualizar que el método de TM
todavía sufre de una falla, debido al hecho de que la variable independiente se asume como una
medida sin error (un requisito para la aplicación de la NLLS), lo que no es cierto especialmente
cuando los datos son tomados a conversiones relativamente altas.
El error experimental fue calculado usando un estimado de la varianza; en otras palabras,
una medición del error experimental se obtuvo por la suma del cuadrado de los residuales del
modelo (asumiendo que el modelo es el correcto). Con el fin de tener una segunda fuente de
estimación para el error experimental, dos experimentos fueron replicados (ver Tabla 7). Las
0
0.03
0.06
0.09
0.12
0.15
0.18
0.21
0.24
0.27
0.3
0.33
0.36
0.39
0 0.02 0.04 0.06 0.08 0.1 0.12 0.14 0.16 0.18 0.2 0.22
r 2
r1
TM
KT
KTE

Capítulo 4/ Índices de reactividad 4.3.3 Método de Tidwell-Mortimer.
82
dos fuentes fueron combinadas para proveer una estimación del error experimental (desviación
estándar), la cual para el sistema IP-GMA, fue de σ=0.001 con ocho grados de libertad. Como
resultado, el estimado del intervalo de confianza del 95% para los índices de reactividad (por
la técnica de TM) son r1=0.119±0.049 y r2=0.248±0.081. Los intervalos de confianza de 95% de
r1 y r2 pueden ser calculados por medio de las siguientes relaciones, obteniendo primero la fracción
molar teórica en el copolímero (F1,teo) y comparándola con la experimental (F1,exp):
𝛼 =𝑓1
𝑓2 𝑟1𝑓1+𝑓2
𝑓1+𝑟2𝑓2 (39)
𝐹1,𝑡𝑒𝑜 =𝛼
1+𝛼 (40)
𝜎2 =∑(𝐹1,𝑒𝑥𝑝−𝐹1,𝑡𝑒𝑜)
2
𝑛−1 (41)
𝐽𝑇𝐽 = (∑(
𝜕𝐺𝑖
𝜕𝑟1)2
∑𝜕𝐺𝑖
𝜕𝑟1
𝜕𝐺𝑖
𝜕𝑟2
∑𝜕𝐺𝑖
𝜕𝑟1
𝜕𝐺𝑖
𝜕𝑟2∑(
𝜕𝐺𝑖
𝜕𝑟2)2) (42)
𝑉𝑖𝑖 = 𝜎2 𝐽𝑇𝐽 −1 (43)
𝛥𝑟1 = ±𝑡0.025 𝑛−1 √𝑉11 (44)
𝛥𝑟2 = ±𝑡0.025 𝑛−1 √𝑉22 (45)
Donde t0.025,(n-2) es el valor de la distribución t de student con (n-1) grados de libertad
correspondiente a áreas de probabilidad igual a 0.025 en cada extremo, n es el número de puntos
experimentales. Los índices de reactividad del par de monómeros obtenidos por diversos
métodos de cálculo se presentan en la Tabla 9.
De acuerdo a los valores que se pueden apreciar en la Tabla 9 a excepción del método
de FRI, el producto r1r2 es menor que 0.03, indicando que el sistema IP/GMA copolimeriza con
una fuerte tendencia a alternar.82 Para este sistema de comonómeros, los únicos valores

Capítulo 4/ Índices de reactividad 4.4 Propiedades térmicas del copolímero de IP/GMA.
83
reportados en la literatura son los de Rusakova et al.,109 donde obtuvieron los valores: r1=0.135
y r2=0.195 (confirmando que el copolímero es aproximadamente alternado); sin embargo, no
reportan los detalles del método usados para su estimación. Estos valores son muy cercanos a
los calculados por el método de KTE en este trabajo de investigación.
Tabla 9: Resultados de los índices de reactividad usando diversos métodos de cálculo para el sistema
IP/GMA.
Método r1 r2 r1* r2
FR 0.115 0.206 0.024
FRI 0.238 0.316 0.075
KT 0.121 0.224 0.027
KTE 0.125 0.198 0.025
TM 0.119 0.248 0.030
r1: relación de reactividad para el IP, r2: relación de reactividad para el GMA.
Es también interesante notar que los valores reportados de los índices de reactividad para
la copolimerización del isopreno con algún otro metacrilato (IP=1, metacrilato de metilo,
MMA=2), r1=0.65 y r2=0.26, significativamente difieren del sistema estudiado de IP/GMA en
el valor de r1. Esto parece estar relacionado con una relativa mayor afinidad termodinámica del
GMA que la del MMA hacia el isopreno, medido por la diferencia al cuadrado de los
parámetros de solubilidad: 0.49 cal/cm3 para GMA/IP comparado con 1.69 cal/cm3 para
MMA/IP;iii estas estimaciones están basados en la referencia.113
4.4 Propiedades térmicas del copolímero de IP/GMA
La Figura 24 muestra las temperaturas de transición vítrea (Tg) obtenidas por DSC como
una función de la concentración del comonómero. Se probaron diversos modelos populares de
predicción de Tg, los cuales incluyen el modelo de Fox, Pochan, Wood y Couchman. La mejor
correlación con los experimentos tratados en la copolimerización del IP se obtuvo con el
modelo basado en la teoría de Couchman:114
iii Los parámetros de solubilidad se estimaron usando la contribución de grupo por medio de la formula (ρΣGi/M), donde Gi es la constante de
atracción molar del grupo i, ρΣGi es la suma de todos los átomos y grupos en la molécula, ρ es la densidad del polímero y M es el peso
molecular del polímero.
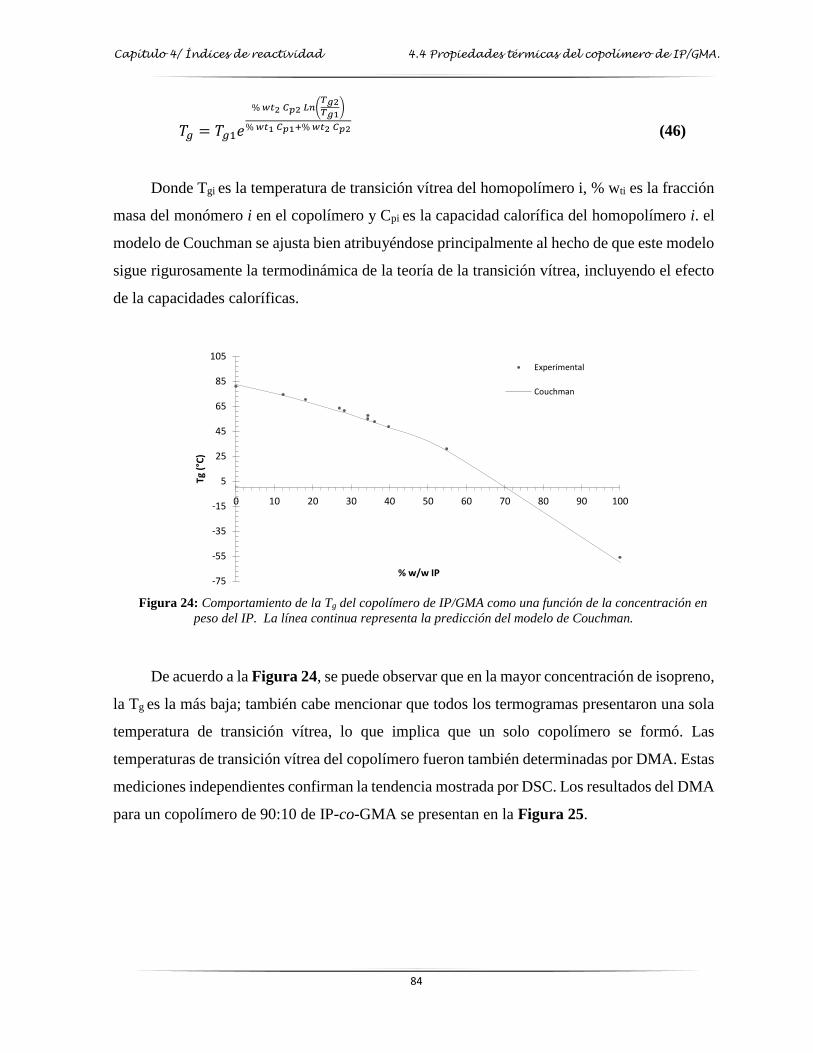
Capítulo 4/ Índices de reactividad 4.4 Propiedades térmicas del copolímero de IP/GMA.
84
𝑇𝑔 = 𝑇𝑔1𝑒
% 𝑤𝑡2 𝐶𝑝2 𝐿𝑛(𝑇𝑔2𝑇𝑔1
)
% 𝑤𝑡1 𝐶𝑝1+% 𝑤𝑡2 𝐶𝑝2 (46)
Donde Tgi es la temperatura de transición vítrea del homopolímero i, % wti es la fracción
masa del monómero i en el copolímero y Cpi es la capacidad calorífica del homopolímero i. el
modelo de Couchman se ajusta bien atribuyéndose principalmente al hecho de que este modelo
sigue rigurosamente la termodinámica de la teoría de la transición vítrea, incluyendo el efecto
de la capacidades caloríficas.
Figura 24: Comportamiento de la Tg del copolímero de IP/GMA como una función de la concentración en
peso del IP. La línea continua representa la predicción del modelo de Couchman.
De acuerdo a la Figura 24, se puede observar que en la mayor concentración de isopreno,
la Tg es la más baja; también cabe mencionar que todos los termogramas presentaron una sola
temperatura de transición vítrea, lo que implica que un solo copolímero se formó. Las
temperaturas de transición vítrea del copolímero fueron también determinadas por DMA. Estas
mediciones independientes confirman la tendencia mostrada por DSC. Los resultados del DMA
para un copolímero de 90:10 de IP-co-GMA se presentan en la Figura 25.
-75
-55
-35
-15
5
25
45
65
85
105
0 10 20 30 40 50 60 70 80 90 100
Tg (
°C)
% w/w IP
Experimental
Couchman

Capítulo 4/ Índices de reactividad 4.4 Propiedades térmicas del copolímero de IP/GMA.
85
Figura 25: DMA del copolímero de IP/GMA 90:10
La Tabla 10 muestra un comparativo de los valores de la Tg obtenidos por las técnicas
de DSC y el DMA; es notorio que el incremento de la concentración del GMA en el copolímero,
el valor de su Tg se incrementa. Este efecto es principalmente debido a la presencia del
comonómero polar del GMA.
Tabla 10: Resumen de los valores de Tg por DSC/DMA para el sistema IP/GMA.
% w/w
IP
DSC
Tg (°C)
DMA
Tg (°C)
12 74.4 71.3
40 48.7 52.6
55 30.3 33.7
4.5 Copolímero de isopreno-anhídrido maleico (IP-MAH).
Consideraciones generales
La copolimerización del IP (10) con anhídrido maleico (12) en masa (Esquema 14) se estudió
variando las fracciones mol del dieno desde 0.4 hasta 0.9 en la alimentación. Este intervalo de
composición fue seleccionado debido a que debajo de 0.4, existe la tendencia a la formación del ciclo
aducto de Diels-Alder, impidiendo conseguir las condiciones para la copolimerización. Para satisfacer
la ecuación de copolimerización de Mayo-Lewis, las conversiones estuvieron abajo de 15%.81 La
reacción de copolimerización se llevó a cabo en masa y como iniciador de radicales libres se utilizó
BPO en exceso (la relación utilizada fue de 1.5 % en peso con respecto al dieno), para favorecer la
polimerización con respecto a la reacción colateral.105-106 Las composiciones de la alimentación y de los
0
0.02
0.04
0.06
0.08
0.1
0.12
0.14
0
2000
4000
6000
8000
10000
12000
14000
16000
0 20 40 60 80 100
Tan
De
lta
Mó
du
lo (
MP
a)
Temperatura (°C)
Storage Modulus
Loss Modulus
Tan Delta
40.7 °C
33.7 °C

Capítulo 4/ Índices de reactividad 4.5 Copolímero de IP/MAH. Consideraciones
generales.
86
copolímeros se presentan en la Tabla 11. Los copolímeros fueron solubles en DMF, THF, DMSO y
acetona, pero fueron insolubles en metanol, etanol, cloroformo, tolueno agua y hexano.
Esquema 14: Reacción de copolimerización del IP-MAH.
4.6 Análisis espectroscópico
Los espectros de FT-IR de los copolímeros de IP-MAH fueron analizados cualitativamente y sus
estructuras son presentadas en la Figura 26. Puede observarse que los copolímeros tienen bandas de
absorción características similares a las de sus respectivas unidades homopoliméricas; bandas amplias
en 3077, 1666, 1642, 888 y 842 cm-1 se deben a las vibraciones de estiramiento del isopreno presente
en sus diferentes microestructuras, tal como la correspondiente adición 1,4- en 1666 cm-1
correlacionándose con la banda de absorción en 842 cm-1; mientras que la banda de absorción en 1642
cm-1 corresponde a las adiciones 1,2- y 3,4-; las cuales están correlacionadas con la banda ubicada en
888 cm-1. Los grupos metilos (CH3-), que pertenecen a estas adiciones, se encuentran en 1445 y 1378
cm-1, respectivamente. Las fuertes bandas de absorción tanto en 1850 como en 1778 cm-1 confirman la
presencia de los dos grupos carbonilos presentes en la unidad del MAH; así como las absorciones en las
bandas de 1242 y 1082 cm1 son un indicativo de la presencia del grupo C-O-C, el cual está asociado al
anillo del MAH.
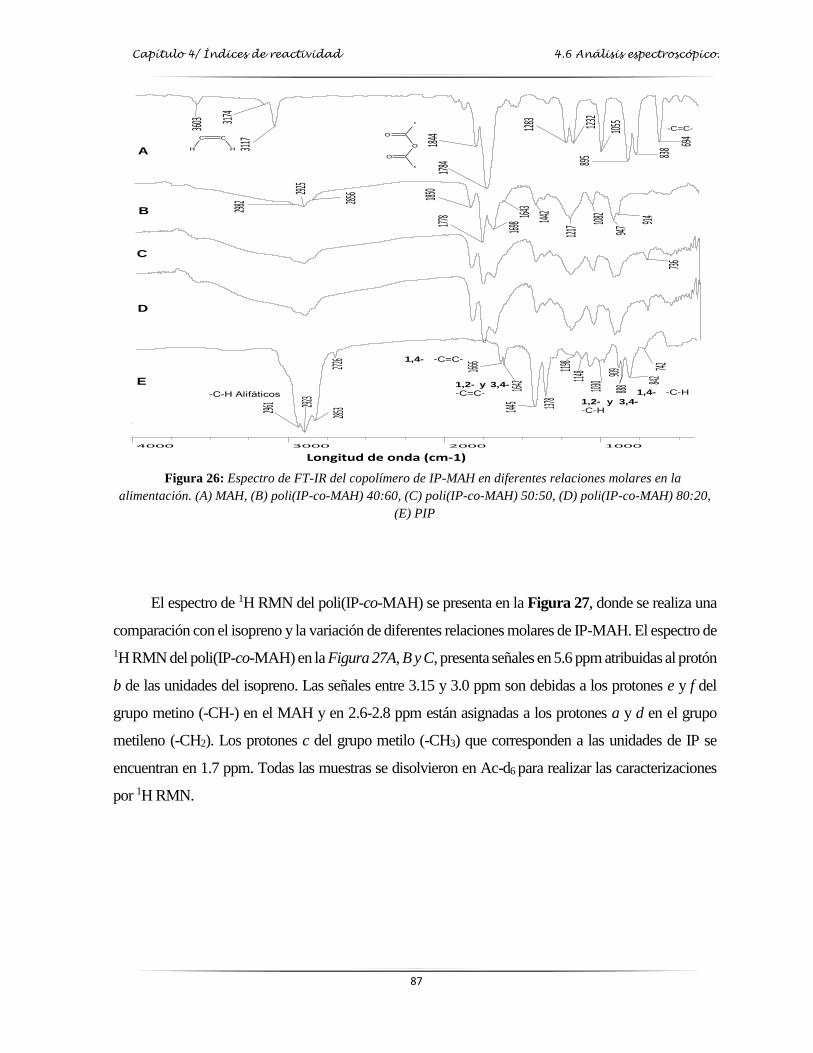
Capítulo 4/ Índices de reactividad 4.6 Análisis espectroscópico.
87
694
838
895
1055123
2
1283
1784
1844
3117
3174
3603
914
947
1082
1217144
21643
1698177
8185
0
2856292
5
2982
736742
842
888909
1030114
81198
1378
1445
1642
1666272
6285
32923
2961
1000 2000 3000 4000
Longitud de onda (cm-1)
O
*
O
*
O-C=C-
C C
HH
-C-H Alifáticos
1,4- -C=C-
1,2- y 3,4- -C=C-
1,2- y 3,4- -C-H
1,4- -C-H
A
B
C
D
E
Figura 26: Espectro de FT-IR del copolímero de IP-MAH en diferentes relaciones molares en la
alimentación. (A) MAH, (B) poli(IP-co-MAH) 40:60, (C) poli(IP-co-MAH) 50:50, (D) poli(IP-co-MAH) 80:20,
(E) PIP
El espectro de 1H RMN del poli(IP-co-MAH) se presenta en la Figura 27, donde se realiza una
comparación con el isopreno y la variación de diferentes relaciones molares de IP-MAH. El espectro de
1H RMN del poli(IP-co-MAH) en la Figura 27A, B y C, presenta señales en 5.6 ppm atribuidas al protón
b de las unidades del isopreno. Las señales entre 3.15 y 3.0 ppm son debidas a los protones e y f del
grupo metino (-CH-) en el MAH y en 2.6-2.8 ppm están asignadas a los protones a y d en el grupo
metileno (-CH2). Los protones c del grupo metilo (-CH3) que corresponden a las unidades de IP se
encuentran en 1.7 ppm. Todas las muestras se disolvieron en Ac-d6 para realizar las caracterizaciones
por 1H RMN.

Capítulo 4/ Índices de reactividad 4.6 Análisis espectroscópico.
88
6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 1.0 0.5
(ppm)
O
*
O
O
CH3
*
CH3
CH3 CH2
CH2
*
*g
hk
l
ij
m n
a
c
b
d
e f
bf
e da
c
i j nl
k
m
g h
A
B
C
D
Figura 27: Espectro de 1H RMN de los copolímeros de IP/MAH en diferentes relaciones molares en la
alimentación. (A) poli(IP-co-MAH) 40:60, (B) poli(IP-co-MAH) 50:50, (C) poli(IP-co-MAH) 80:20, (D) PIP
Como resumen, en el Esquema 15 se presentan las unidades estructurales que están contenidas
en la cadena polimérica del sistema IP/MAH (los números adjuntos a dichas unidades estructurales
denotan los desplazamientos químicos (en ppm)):
----H2C C CH
CH3
CH2----
2.8
1.75
5.6 2.6CH3 C
CH3
HC
.
CH2
1.4
1.0
5.9
4.9
. CH
CCH2 CH3
CH2------
4.6 1.7
2.0 1.4C
O
CH
C
HC. .
O O
3.1 3.1
Adición 1,4- Adición 1,2-
Adición 3,4-
Esquema 15: Resumen de los picos presentados en el espectro de 1H RMN de la Figura 27.

Capítulo 4/ Índices de reactividad 4.7 Determinación de los índices de reactividad del
sistema IP/MAH.
89
4.7 Determinación de los índices de reactividad para el sistema IP/MAH.
Como en el caso del espectro para el copolímero de IP-GMA es posible localizar tres regiones de
señales en el espectro del copolímero IP-MAH (ver Figura 28): i) las señales de los protones en la
región de δ = 1.0-2.8 ppm que pertenecen a los grupos -CH-, -CH2- y -CH3 de las unidades de isopreno;
ii) los picos de los protones en la región de δ = 3.0-3.3 ppm de los grupos metileno del MAH y iii) los
picos en la región de δ = 4.5-5.9 ppm de los grupos -CH= y =CH2 de las unidades de isopreno.
Figura 28: Espectro de 1H RMN del copolímero de IP/MAH con una relación molar en la alimentación
50:50.
Si las fracciones molares de MAH e isopreno en el copolímero son designadas como m2 y (1-
m2), respectivamente y dado que el número de protones en las unidades son 2 y 8 para el MAH y el
isopreno respectivamente, el área total de los picos en el espectro es ST, el cual es proporcional a:
𝑆𝑇 ∝ 2𝑚2 + 8 1 − 𝑚2 = 8 − 6𝑚2 (47)
El área SM correspondiente a los protones del grupo del MAH se halla en el rango de 3.0-3.3
ppm, el cual es proporcional a 2m2, de la relación anterior se tiene que:
𝑆𝑀
𝑆𝑇=
2𝑚2
8−6𝑚2; 𝑚2 =
8𝑆𝑀
2𝑆𝑇+6𝑆𝑀 (48)
De la ecuación 48 es posible estimar la composición del copolímero. En la Tabla 11 se presentan
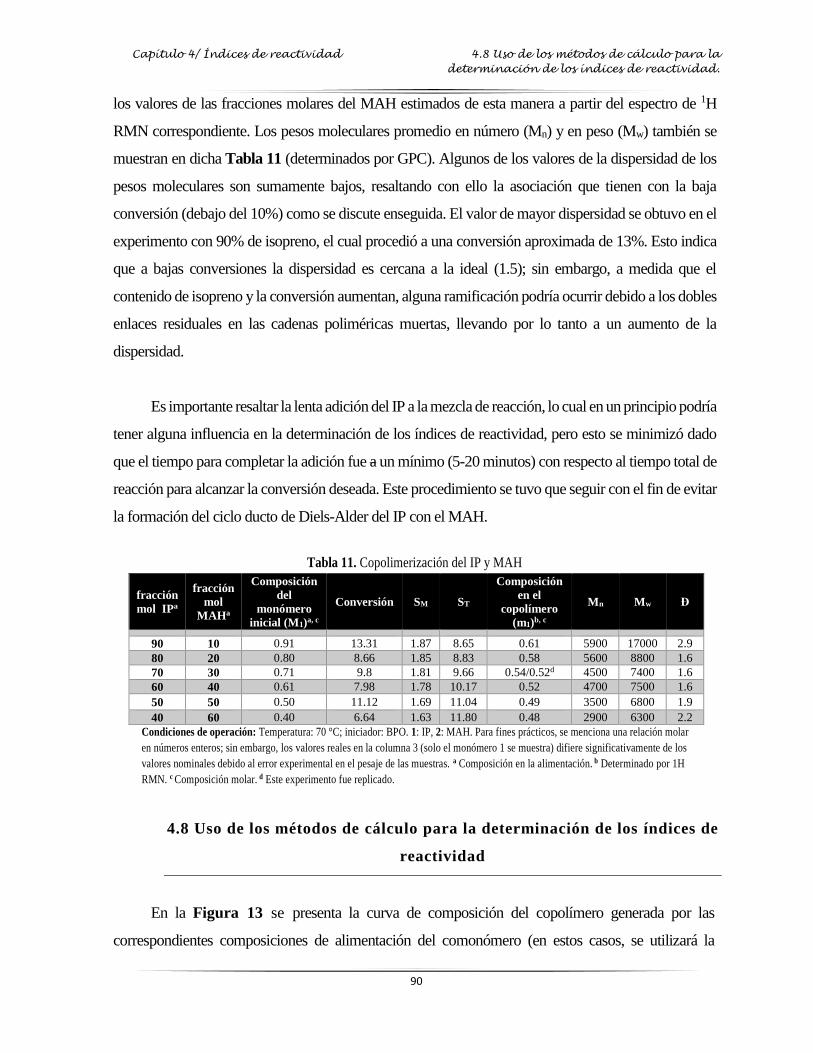
Capítulo 4/ Índices de reactividad 4.8 Uso de los métodos de cálculo para la
determinación de los índices de reactividad.
90
los valores de las fracciones molares del MAH estimados de esta manera a partir del espectro de 1H
RMN correspondiente. Los pesos moleculares promedio en número (Mn) y en peso (Mw) también se
muestran en dicha Tabla 11 (determinados por GPC). Algunos de los valores de la dispersidad de los
pesos moleculares son sumamente bajos, resaltando con ello la asociación que tienen con la baja
conversión (debajo del 10%) como se discute enseguida. El valor de mayor dispersidad se obtuvo en el
experimento con 90% de isopreno, el cual procedió a una conversión aproximada de 13%. Esto indica
que a bajas conversiones la dispersidad es cercana a la ideal (1.5); sin embargo, a medida que el
contenido de isopreno y la conversión aumentan, alguna ramificación podría ocurrir debido a los dobles
enlaces residuales en las cadenas poliméricas muertas, llevando por lo tanto a un aumento de la
dispersidad.
Es importante resaltar la lenta adición del IP a la mezcla de reacción, lo cual en un principio podría
tener alguna influencia en la determinación de los índices de reactividad, pero esto se minimizó dado
que el tiempo para completar la adición fue a un mínimo (5-20 minutos) con respecto al tiempo total de
reacción para alcanzar la conversión deseada. Este procedimiento se tuvo que seguir con el fin de evitar
la formación del ciclo ducto de Diels-Alder del IP con el MAH.
Tabla 11. Copolimerización del IP y MAH
fracción
mol IPa
fracción
mol
MAHa
Composición
del
monómero
inicial (M1)a, c
Conversión SM ST
Composición
en el
copolímero
(m1)b, c
Mn Mw Đ
90 10 0.91 13.31 1.87 8.65 0.61 5900 17000 2.9
80 20 0.80 8.66 1.85 8.83 0.58 5600 8800 1.6
70 30 0.71 9.8 1.81 9.66 0.54/0.52d 4500 7400 1.6
60 40 0.61 7.98 1.78 10.17 0.52 4700 7500 1.6
50 50 0.50 11.12 1.69 11.04 0.49 3500 6800 1.9
40 60 0.40 6.64 1.63 11.80 0.48 2900 6300 2.2
Condiciones de operación: Temperatura: 70 °C; iniciador: BPO. 1: IP, 2: MAH. Para fines prácticos, se menciona una relación molar
en números enteros; sin embargo, los valores reales en la columna 3 (solo el monómero 1 se muestra) difiere significativamente de los
valores nominales debido al error experimental en el pesaje de las muestras. a Composición en la alimentación. b Determinado por 1H
RMN. c Composición molar. d Este experimento fue replicado.
4.8 Uso de los métodos de cálculo para la determinación de los índices de
reactividad
En la Figura 13 se presenta la curva de composición del copolímero generada por las
correspondientes composiciones de alimentación del comonómero (en estos casos, se utilizará la
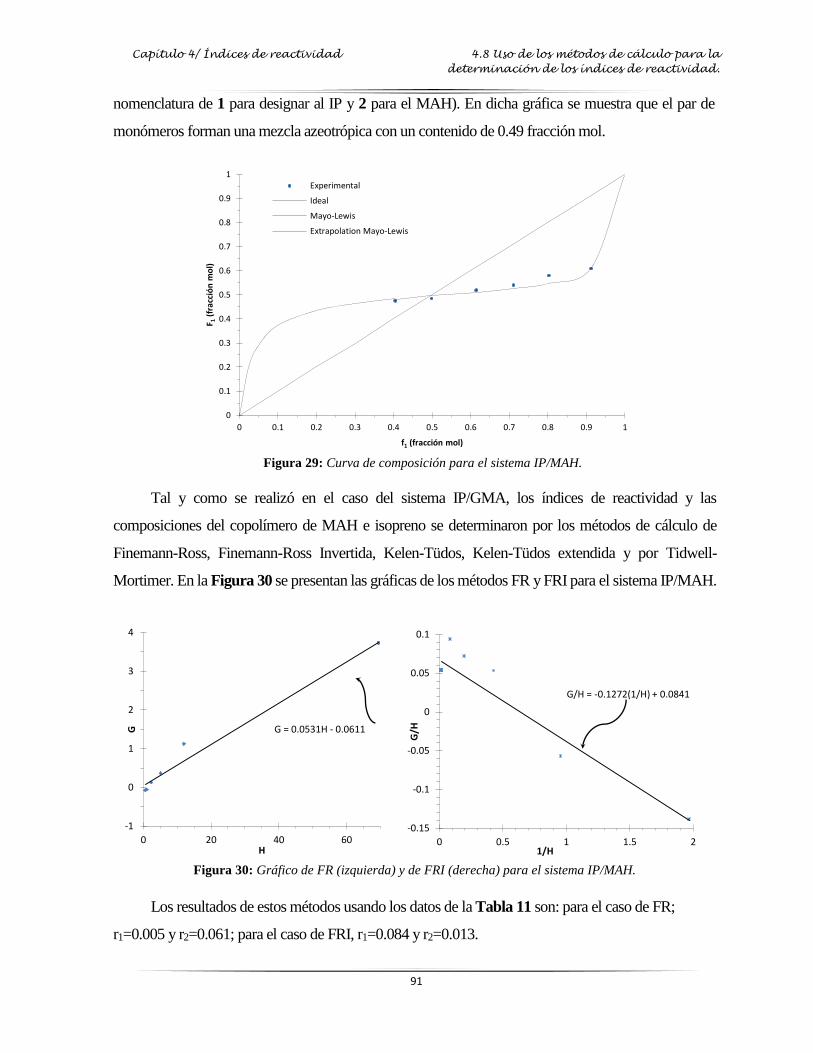
Capítulo 4/ Índices de reactividad 4.8 Uso de los métodos de cálculo para la
determinación de los índices de reactividad.
91
nomenclatura de 1 para designar al IP y 2 para el MAH). En dicha gráfica se muestra que el par de
monómeros forman una mezcla azeotrópica con un contenido de 0.49 fracción mol.
Figura 29: Curva de composición para el sistema IP/MAH.
Tal y como se realizó en el caso del sistema IP/GMA, los índices de reactividad y las
composiciones del copolímero de MAH e isopreno se determinaron por los métodos de cálculo de
Finemann-Ross, Finemann-Ross Invertida, Kelen-Tüdos, Kelen-Tüdos extendida y por Tidwell-
Mortimer. En la Figura 30 se presentan las gráficas de los métodos FR y FRI para el sistema IP/MAH.
Figura 30: Gráfico de FR (izquierda) y de FRI (derecha) para el sistema IP/MAH.
Los resultados de estos métodos usando los datos de la Tabla 11 son: para el caso de FR;
r1=0.005 y r2=0.061; para el caso de FRI, r1=0.084 y r2=0.013.
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1
0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1
F 1(f
racc
ión
mo
l)
f1 (fracción mol)
Experimental
Ideal
Mayo-Lewis
Extrapolation Mayo-Lewis
G = 0.0531H - 0.0611
-1
0
1
2
3
4
0 20 40 60
G
H
G/H = -0.1272(1/H) + 0.0841
-0.15
-0.1
-0.05
0
0.05
0.1
0 0.5 1 1.5 2
G/H
1/H
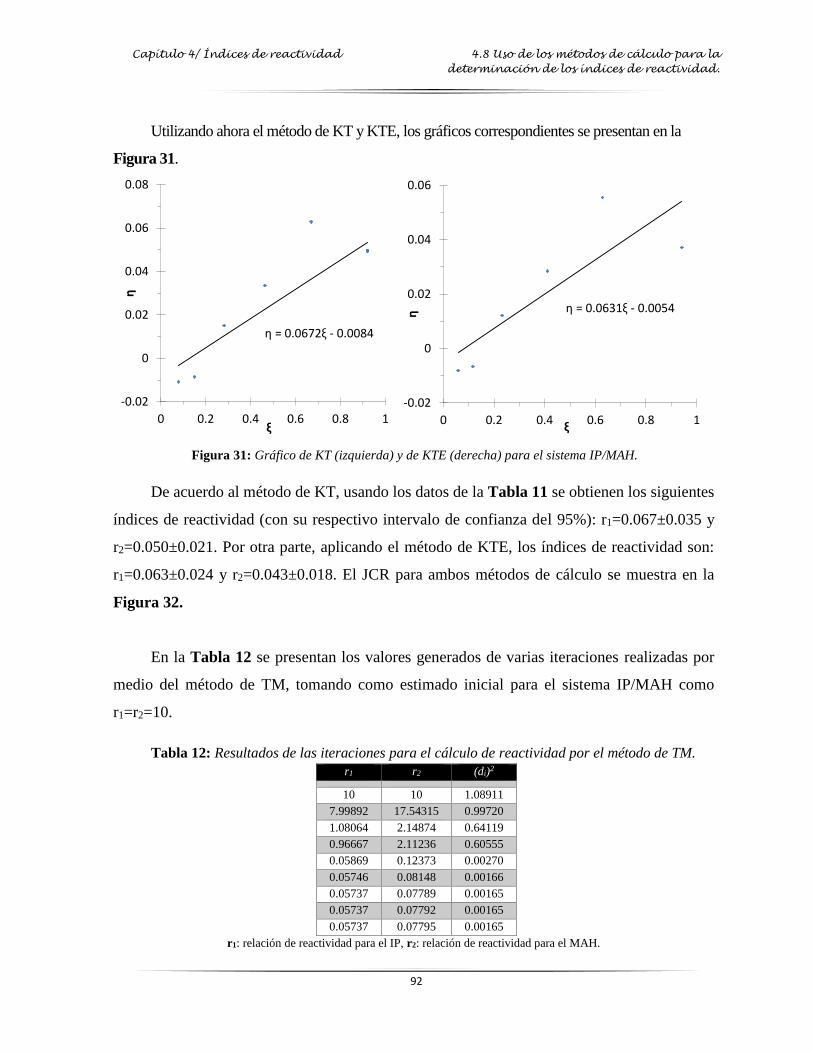
Capítulo 4/ Índices de reactividad 4.8 Uso de los métodos de cálculo para la
determinación de los índices de reactividad.
92
Utilizando ahora el método de KT y KTE, los gráficos correspondientes se presentan en la
Figura 31.
Figura 31: Gráfico de KT (izquierda) y de KTE (derecha) para el sistema IP/MAH.
De acuerdo al método de KT, usando los datos de la Tabla 11 se obtienen los siguientes
índices de reactividad (con su respectivo intervalo de confianza del 95%): r1=0.067±0.035 y
r2=0.050±0.021. Por otra parte, aplicando el método de KTE, los índices de reactividad son:
r1=0.063±0.024 y r2=0.043±0.018. El JCR para ambos métodos de cálculo se muestra en la
Figura 32.
En la Tabla 12 se presentan los valores generados de varias iteraciones realizadas por
medio del método de TM, tomando como estimado inicial para el sistema IP/MAH como
r1=r2=10.
Tabla 12: Resultados de las iteraciones para el cálculo de reactividad por el método de TM.
r1 r2 (di)2
10 10 1.08911
7.99892 17.54315 0.99720
1.08064 2.14874 0.64119
0.96667 2.11236 0.60555
0.05869 0.12373 0.00270
0.05746 0.08148 0.00166
0.05737 0.07789 0.00165
0.05737 0.07792 0.00165
0.05737 0.07795 0.00165
r1: relación de reactividad para el IP, r2: relación de reactividad para el MAH.
η = 0.0672ξ - 0.0084
-0.02
0
0.02
0.04
0.06
0.08
0 0.2 0.4 0.6 0.8 1
η
ξ
η = 0.0631ξ - 0.0054
-0.02
0
0.02
0.04
0.06
0 0.2 0.4 0.6 0.8 1η
ξ
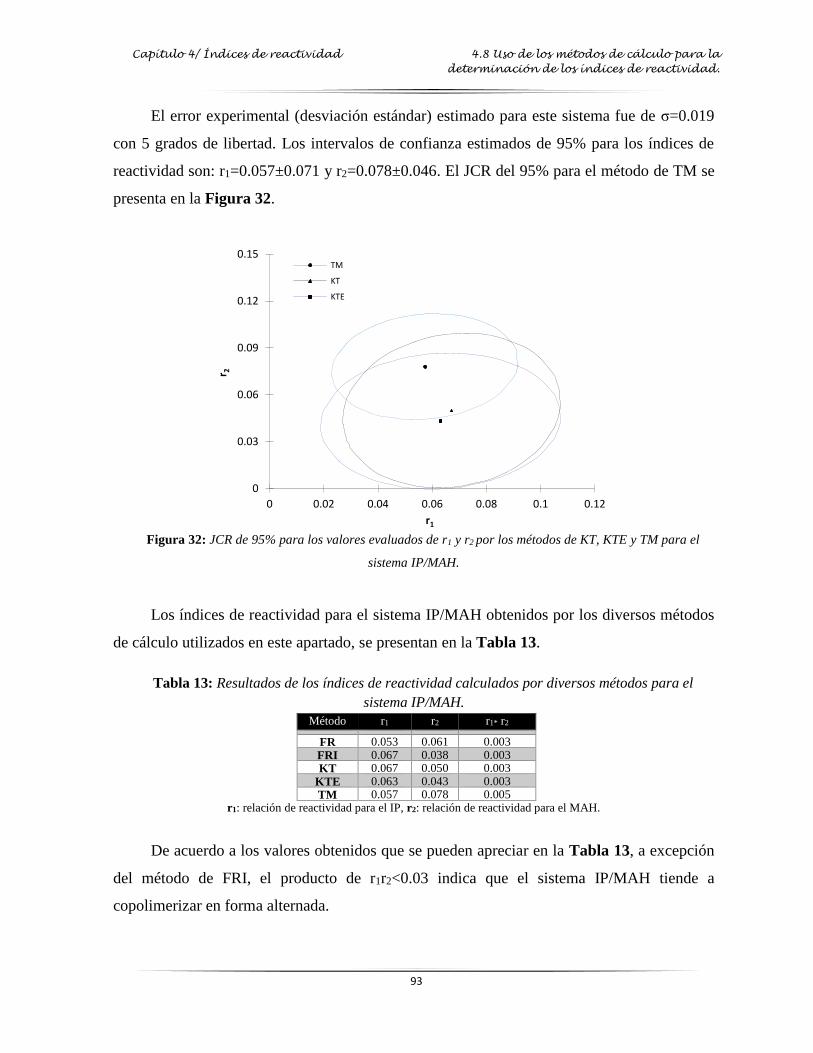
Capítulo 4/ Índices de reactividad 4.8 Uso de los métodos de cálculo para la
determinación de los índices de reactividad.
93
El error experimental (desviación estándar) estimado para este sistema fue de σ=0.019
con 5 grados de libertad. Los intervalos de confianza estimados de 95% para los índices de
reactividad son: r1=0.057±0.071 y r2=0.078±0.046. El JCR del 95% para el método de TM se
presenta en la Figura 32.
Figura 32: JCR de 95% para los valores evaluados de r1 y r2 por los métodos de KT, KTE y TM para el
sistema IP/MAH.
Los índices de reactividad para el sistema IP/MAH obtenidos por los diversos métodos
de cálculo utilizados en este apartado, se presentan en la Tabla 13.
Tabla 13: Resultados de los índices de reactividad calculados por diversos métodos para el
sistema IP/MAH.
Método r1 r2 r1* r2
FR 0.053 0.061 0.003 FRI 0.067 0.038 0.003 KT 0.067 0.050 0.003
KTE 0.063 0.043 0.003 TM 0.057 0.078 0.005
r1: relación de reactividad para el IP, r2: relación de reactividad para el MAH.
De acuerdo a los valores obtenidos que se pueden apreciar en la Tabla 13, a excepción
del método de FRI, el producto de r1r2<0.03 indica que el sistema IP/MAH tiende a
copolimerizar en forma alternada.
0
0.03
0.06
0.09
0.12
0.15
0 0.02 0.04 0.06 0.08 0.1 0.12
r 2
r1
TM
KT
KTE

Capítulo 4/ Índices de reactividad 4.9 Propiedades térmicas del copolímero IP/MAH.
94
4.9 Propiedades térmicas del copolímero IP/MAH
En la Figura 33 se muestran las temperaturas de transición vítrea (Tg) obtenidas de los
termogramas de DSC y se exhibe la tendencia que hay con respecto a la concentración del
comonómero presente en el material polimérico; se puede observar que a mayor concentración
de isopreno, se presenta la menor Tg. Cabe mencionar que todos los termogramas presentaron
una sola temperatura de transición vítrea, indicando con ello claramente la naturaleza de la
formación de un copolímero de naturaleza aleatoria (si bien con tendencia a alternar).
Figura 33: Grafico del comportamiento de la Tg con respecto a la concentración molar de isopreno.
Los valores de Tg obtenidos por DSC fueron confirmados por técnicas de DMA. La
Figura 34 ilustra el comportamiento exhibido para el copolímero de IP-MAH 90:10; mientras
que la Tabla 14 muestra un resumen comparativo de las temperaturas de transición vítrea
obtenidas por las técnicas de DSC y DMA. En dicha tabla se muestra que a una disminución
de la concentración del isopreno en el copolímero, la Tg se incrementa debido a la presencia
del monómero polar de anhídrido maleico.
-70
-50
-30
-10
10
30
50
70
90
110
30 40 50 60 70 80 90 100
T g(°
C)
% w/w IP
EXPE
Couchman

Capítulo 4/ Índices de reactividad 4.10 Determinación de la kp del isopreno por PLP-SEC
95
Figura 34: DMA del copolímero IP/MAH 90:10.
Tabla 14: Resumen de los valores de Tg por DSC/DMA para el sistema IP/MAH.
% w/w
IP
DSC
Tg (°C)
DMA
Tg (°C)
40 99.2 100.9
49 72.2 75.0
52 65.9 69.8
4.10 Determinación de la kp del isopreno por PLP-SEC
Los coeficientes de rapidez de propagación, kp, para la homopolimerización del GMA y
del IP en masa fueron obtenidos de acuerdo a la ecuación 12. Los pesos moleculares absolutos
del primer punto y del segundo punto de inflexión, M1 y M2, fueron calculados usando las
constantes de MHS, teniéndose que para el GMA: α=0.537 y k=0.0278 mL g-1 y para el IP:
α=0.712, k=0.0244 mL g-1. En la Tabla 15 se resumen las condiciones experimentales y los
datos arrojados por éstos, donde los pesos moleculares absolutos y el primer y segundo punto
de inflexión, M1(abs) y M2(abs), con los valores de kp para todos los experimentos de PLP
llevados a cabo, se presentan en dicha tabla. En todos los casos se consiguió una excelente
reproducibilidad de los datos experimentales.
0
0.02
0.04
0.06
0.08
0.1
0.12
0.14
0
200
400
600
800
1000
1200
1400
1600
50 60 70 80 90 100 110 120
Tan
De
lta
Mó
du
lo (
MP
a)
Temperatura (°C)
Storage Modulus
Loss Modulus
Tan Delta
74.5 °C
69.8 °C
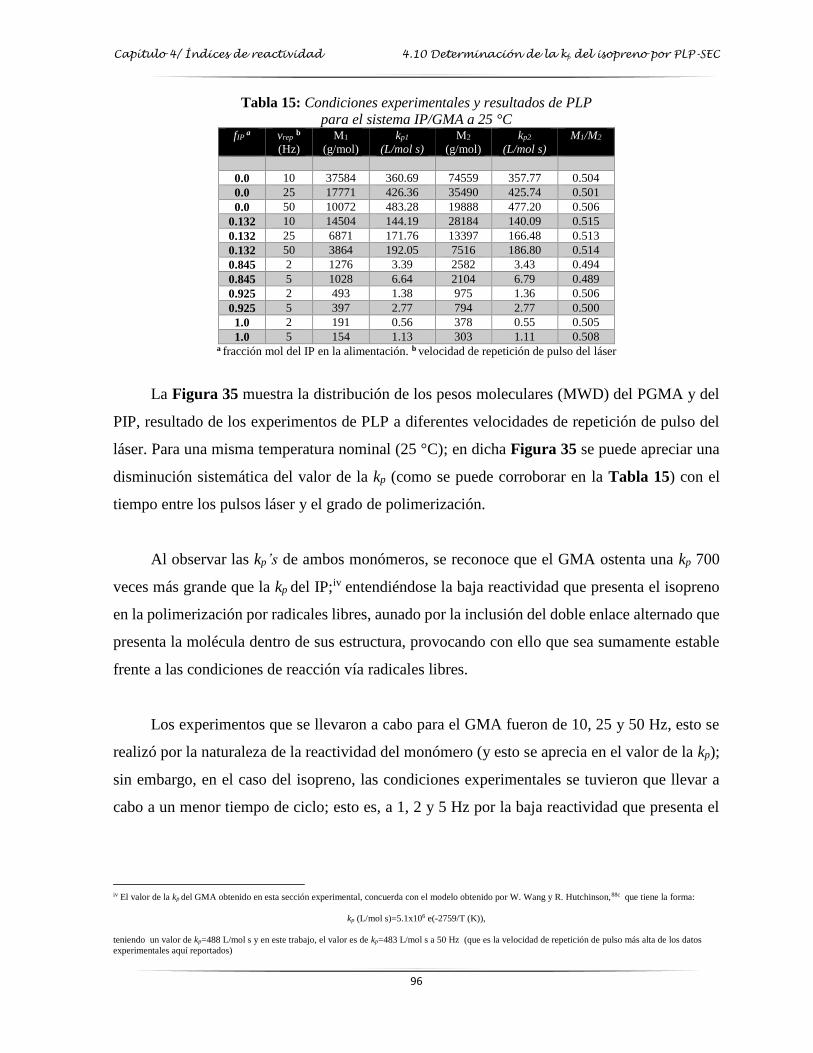
Capítulo 4/ Índices de reactividad 4.10 Determinación de la kp del isopreno por PLP-SEC
96
Tabla 15: Condiciones experimentales y resultados de PLP
para el sistema IP/GMA a 25 °C fIP
a vrep b
(Hz)
M1
(g/mol)
kp1
(L/mol s)
M2
(g/mol)
kp2
(L/mol s)
M1/M2
0.0 10 37584 360.69 74559 357.77 0.504
0.0 25 17771 426.36 35490 425.74 0.501
0.0 50 10072 483.28 19888 477.20 0.506
0.132 10 14504 144.19 28184 140.09 0.515
0.132 25 6871 171.76 13397 166.48 0.513
0.132 50 3864 192.05 7516 186.80 0.514
0.845 2 1276 3.39 2582 3.43 0.494
0.845 5 1028 6.64 2104 6.79 0.489
0.925 2 493 1.38 975 1.36 0.506
0.925 5 397 2.77 794 2.77 0.500
1.0 2 191 0.56 378 0.55 0.505
1.0 5 154 1.13 303 1.11 0.508 a fracción mol del IP en la alimentación. b velocidad de repetición de pulso del láser
La Figura 35 muestra la distribución de los pesos moleculares (MWD) del PGMA y del
PIP, resultado de los experimentos de PLP a diferentes velocidades de repetición de pulso del
láser. Para una misma temperatura nominal (25 °C); en dicha Figura 35 se puede apreciar una
disminución sistemática del valor de la kp (como se puede corroborar en la Tabla 15) con el
tiempo entre los pulsos láser y el grado de polimerización.
Al observar las kp’s de ambos monómeros, se reconoce que el GMA ostenta una kp 700
veces más grande que la kp del IP;iv entendiéndose la baja reactividad que presenta el isopreno
en la polimerización por radicales libres, aunado por la inclusión del doble enlace alternado que
presenta la molécula dentro de sus estructura, provocando con ello que sea sumamente estable
frente a las condiciones de reacción vía radicales libres.
Los experimentos que se llevaron a cabo para el GMA fueron de 10, 25 y 50 Hz, esto se
realizó por la naturaleza de la reactividad del monómero (y esto se aprecia en el valor de la kp);
sin embargo, en el caso del isopreno, las condiciones experimentales se tuvieron que llevar a
cabo a un menor tiempo de ciclo; esto es, a 1, 2 y 5 Hz por la baja reactividad que presenta el
iv El valor de la kp del GMA obtenido en esta sección experimental, concuerda con el modelo obtenido por W. Wang y R. Hutchinson,88c que tiene la forma:
kp (L/mol s)=5.1x106 e(-2759/T (K)),
teniendo un valor de kp=488 L/mol s y en este trabajo, el valor es de kp=483 L/mol s a 50 Hz (que es la velocidad de repetición de pulso más alta de los datos
experimentales aquí reportados)
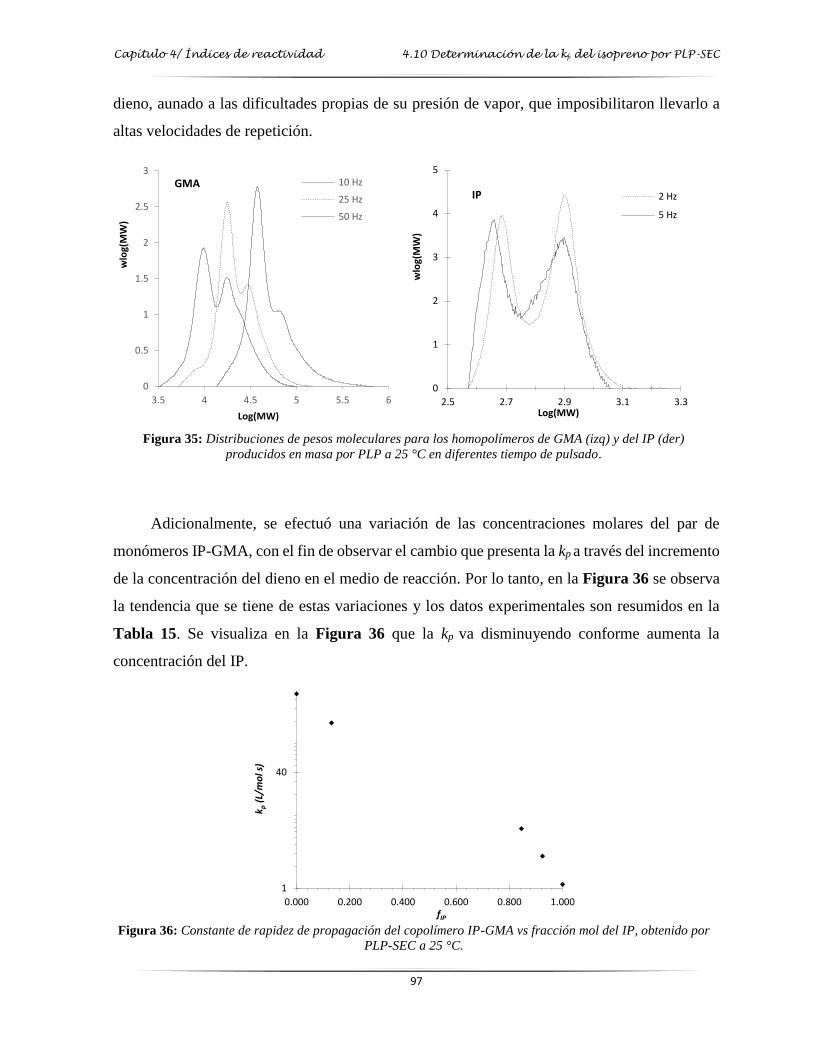
Capítulo 4/ Índices de reactividad 4.10 Determinación de la kp del isopreno por PLP-SEC
97
dieno, aunado a las dificultades propias de su presión de vapor, que imposibilitaron llevarlo a
altas velocidades de repetición.
Figura 35: Distribuciones de pesos moleculares para los homopolímeros de GMA (izq) y del IP (der)
producidos en masa por PLP a 25 °C en diferentes tiempo de pulsado.
Adicionalmente, se efectuó una variación de las concentraciones molares del par de
monómeros IP-GMA, con el fin de observar el cambio que presenta la kp a través del incremento
de la concentración del dieno en el medio de reacción. Por lo tanto, en la Figura 36 se observa
la tendencia que se tiene de estas variaciones y los datos experimentales son resumidos en la
Tabla 15. Se visualiza en la Figura 36 que la kp va disminuyendo conforme aumenta la
concentración del IP.
Figura 36: Constante de rapidez de propagación del copolímero IP-GMA vs fracción mol del IP, obtenido por
PLP-SEC a 25 °C.
0
0.5
1
1.5
2
2.5
3
3.5 4 4.5 5 5.5 6
wlo
g(M
W)
Log(MW)
10 Hz
25 Hz
50 Hz
GMA
0
1
2
3
4
5
2.5 2.7 2.9 3.1 3.3
wlo
g(M
W)
Log(MW)
2 Hz
5 Hz
IP
1
40
0.000 0.200 0.400 0.600 0.800 1.000
k p(L
/mo
l s)
fIP

Capítulo 4/ Índices de reactividad 4.10 Determinación de la kp del isopreno por PLP-SEC
98
Por lo tanto, lo que se observa es que la kp del isopreno es 1.13 L mol-1 s-1 a 25 °C,
mientras que la kp del GMA es de 483 L mol-1 s-1 a la misma temperatura de operación. Es entre
estos dos valores que se encuentra las diferentes relaciones molares de estos dos comonómeros.
Por ejemplo, para la relación molar de IP/GMA de 0.13/0.87 tiene un valor de 192 L mol-1 s-1
y se observa, como el comonómero de IP influye apreciablemente en la kp de este par de
monómeros. Esto se puede apreciar, por ejemplo en la relación de IP/GMA de 0.84/0.16 que
tiene un valor de 6.64 L mol-1 s-1.

Capítulo 5/ (Co) polimerización del IP por NMRP 5.1 Cinéticas de IP-GMA en solución.
99
5.
Resultados y discusión. Homo y copolimerización del
isopreno mediada por nitróxidos
5.1 Cinéticas de isopreno-metacrilato de glicidilo (IP-GMA) en solución
Todas las condiciones experimentales utilizadas en este apartado se reúnen en la Tabla
16; por lo tanto, se procederá a describir la cinética de la (co)polimerización del isopreno por
medio de la técnica de polimerización mediada por nitróxidos (NMP), utilizando dos tipos de
relación molar de nitróxido/iniciador (N/I). La polimerización fue llevada a cabo con los
nitróxidos HO-TEMPO (13) y TIPNO (14), (Ver Esquema 16) usando BPO como iniciador de
radicales libres en solución con o-diclorobenceno o tolueno (al 30 % w/w en sólidos) en periodos
de tiempo requerido de 50–75 h (según sea el caso).
Esquema 16: Estructuras de los nitróxidos HO-TEMPO (13), TIPNO (14) y TEMPO (15).

Capítulo 5/ (Co) polimerización del IP por NMRP 5.1 Cinéticas de IP-GMA en solución.
100
Tabla 16: Resumen de la (co)polimerización del isopreno por NMP a 120 °C.
a Abreviaciones: BPO, peróxido de benzoilo; GMA, metacrilato de metilo; IP, isopreno; o-ClBz, o-diclorobenceno; TBP,
peróxido de t-butilo; Tol, tolueno. b Condiciones de reacción: 120 °C @ 1170 kPa (170 psig). Isopreno en la cantidad
requerida del disolvente (con el fin de obtener un 30 % w/w de sólidos). Relación molar IP:N, 490:1 c Experimento
llevado en condiciones de FRP. d Relación molar entre monómeros; 99.5:0.5 (para obtener un 1 % w/w del GMA con
respecto al IP). e Relación molar nitróxido/iniciador. f Mn, teo se calculó a partir de la expresión Mn, teo=conversión
|M|/|N| PMM + PMN + PMI/2; donde |M|: concentración del monómero (mol); |N|: concentración del nitróxido (mol);
PMM, PMN, PMI: peso molecular del monómero, nitróxido e iniciador (en g/mol) respectivamente. g 𝑀𝑛𝐺𝑃𝐶 se determinó
por GPC, calibrado con estándares de PS y convertidos a PIP usando las constantes de MHS de la referencia104 h
Constante de rapidez aparente; k’= -ln(1-conversión)/t, t:tiempo de reacción. i Eficiencia del nitróxido; fNO=Mn, exp/Mn, teo
El poliisopreno (PIP) es insoluble en disolventes polares tales como el DMSO, DMF,
alcoholes y acetona, pero soluble en 1,4-dioxano, piridina, tolueno y benceno.87 En el proceso
de la (co)polimerización del isopreno, la ruta sintética que se tiene propuesta para obtener el
material polimérico controlado se presenta en el Esquema 17. La macroalcoxiamina resultante
se obtiene calentando al monómero isopreno (o la mezcla de monómeros IP/GMA), en presencia
de un iniciador de radicales libres (proceso bimolecular), en solución (ya sea en o-
diclorobenceno (o-ClBz) o tolueno (Tol)). La relación molar del nitróxido: iniciador empleado
en la (co)polimerización del isopreno fue de 1.8 y 0.9.
Experi-
mento Monómero(s)
(disolvente,
ml) a
Nitróxido
(N)b
Iniciador
(I) N/I e
Tiempo
de
reacción
(h)
Conversión Mn, teof 𝑴𝒏
𝑮𝑷𝑪 g fNO i Ð
k’x10-3
(h-1) h
1
IP (o-ClBz, 36.5)
13 BPO 1.8 75 0.2648 9090 6200 0.68 1.53 4.10
2 IP
(Tol, 60) 13 BPO 1.8 75 0.1854 6440 4500 0.70 1.58 2.73
3 IP
(Tol, 60) 13 BPO 0.9 75 0.2176 7520 5100 0.68 1.61 3.27
4 IP
(o-ClBz, 36.5) 14 BPO 1.8 50 0.1968 6790 4800 0.71 1.24 4.38
5 IP
(Tol, 60) 14 BPO 1.8 50 0.1531 5840 4300 0.74 1.31 3.32
6 IP/GMAd
(Tol, 60) 14 BPO 1.8 50 0.1735 6130 4400 0.72 1.29 3.81
7 IP/GMAd
(o-ClBz, 36.5) 14 BPO 1.8 50 0.1992 6990 5100 0.73 1.24 4.44
8 IP/GMAd
(o-ClBz, 36.5) 14 BPO 0.9 50 0.2641 9170 6100 0.67 1.30 6.13
9 IP/GMAd
(Tol, 60) 14 BPO 0.9 50 0.2286 7980 5200 0.65 1.34 5.19
10 IP
(Tol, 60)c c TBP ----- 36 0.3187 12760 15400 ---- 2.37 10.66
11 IP/GMAd
(Tol, 60)c c TBP ----- 36 0.3561 12000 17000 ---- 2.16 12.23

Capítulo 5/ (Co) polimerización del IP por NMRP 5.1 Cinéticas de IP-GMA en solución.
101
Esquema 17: Formación del homo y copolímero de isopreno con grupos nitróxido.
Realizando un comparativo de estos dos agentes de control y de acuerdo a los datos
arrojados en la Tabla 17, se observa que el nitróxido 13 exhibe un pobre control en la
polimerización del isopreno con respecto a 14, y esto puede ser atribuido a la constante de
rapidez de disociación de la macroalcoxiamina formada in-situ (Esquemas 3 y 4) y a la de
recombinación. Otra característica a destacar, es la diferencia que hay en las estructuras
químicas de los nitróxidos presentes, pues en el caso de 13, ésta es una molécula cíclica y posee
en la posición al nitróxido un carbono cuaternario, mientras que 14 es acíclica y tiene un
hidrógeno en la posición , dando como resultado una constante de disociación más alta que
13.52a
Es importante señalar que el éxito de una polimerización radicálica por NMP (tanto en
dispersidad como en la velocidad de polimerización), es dependiente de la constante de rapidez
de la homólisis del enlace C-O de la alcoxiamina, y ésta depende de la estructura del nitróxido
formado (y del radical centrado en el carbono); por otra parte, la constante de rapidez de
acoplamiento (reacción inversa) muestra una dependencia de los factores estéricos de las
moléculas involucradas, que son los que influyen principalmente, mientras que las
características termodinámicas y polares, son contribuyentes secundarios. Por lo tanto, para que
la velocidad de polimerización sea rápida con obtención de dispersidades (Ð) estrechas, es
esencial que la porción del nitróxido de la alcoxiamina formada deba poseer ciertas
características, para que la homólisis del enlace C-O sea rápida y con pocas reacciones
secundarias a la temperatura de reacción.

Capítulo 5/ (Co)polimerización del IP por NMRP 5.2 Cálculos semiempíricos (AM1-RHF).
102
5.2 Cálculos semiempíricos (AM1-RHF)
Existen diversos estudios donde se utilizan cálculos semiempíricos con el fin de apoyarse
y/o establecer los factores que influyen en los valores de las energías de disociación del enlace
C-O (Dc-o); además de poder lograr una visión clara de la relación estructura-reactividad del
nitróxido involucrado. Diferentes autores han observado una diferencia significante entre los
valores estimados de la entalpia de formación (Hf) (y Dc-o) para los radicales libres
involucrados en la polimerización mediada por nitróxidos, por medio de los métodos de cálculo
semiempíricos AM1 y PM3, encontrando que los valores de AM1 generalmente son los más
cercanos a los valores experimentales.115 Para demostrar y comprender más el comportamiento
de los nitróxidos utilizados en esta sección (agregando para el cálculo al TEMPO (15), como un
punto de referencia), una serie de cálculos semiempíricos se llevaron a cabo con optimización
de la geometría con el método de cálculo AM1-RHF, tanto para la serie de alcoxiaminas
propuestas como para sus reacciones de homólisis correspondientes (ver Tabla 17). Los
radicales libres planteados que componen la parte alquílica de las alcoxiaminas a analizar son
los siguientes:
Tabla 17: Propiedades predichas (orden decreciente en Dc-o) de alcoxiaminas con cálculos semiempíricos (AM1-
RHF)d Hf (kJ/mol) DC-O
b C-O c N-O c C-N c C-N-C c N-O-C c
Alcoxiaminaa radical nitróxido alcoxiamina (kJ/mol) (Å) (Å) (Å) (°) (°)
13-Me 130.57 -256.42 -294.67 168.82 1.440 1.355 2.341 116.13 113.72
15-Me 130.57 -76.31 -112.97 167.23 1.439 1.355 2.341 116.04 113.72
14-Me 130.57 46.21 13.78 162.99 1.439 1.354 2.350 112.33 114.52
13-IP 80.45 -256.42 -283.62 107.65 1.456 1.353 2.344 116.14 113.15
15-IP 80.45 -76.31 -101.89 106.03 1.455 1.353 2.344 116.07 113.13
13-iPr 28.50 -256.42 -332.62 104.70 1.460 1.354 2.371 115.41 114.84
15-iPr 28.50 -76.31 -151.75 103.95 1.460 1.353 2.371 115.23 114.80
13-S 172.32 -256.42 -185.88 101.78 1.459 1.355 2.372 116.13 114.85
15-S 172.32 -76.31 -4.10 100.11 1.459 1.355 2.372 116.13 114.85
14-iPr 28.50 46.21 -13.58 88.29 1.460 1.349 2.398 113.29 117.16
14-IP 80.45 46.21 41.67 84.99 1.445 1.356 2.382 114.18 116.51
14-S 172.32 46.21 134.30 84.23 1.462 1.349 2.386 116.99 116.04
13-tBu -10.79 -256.42 -331.69 64.48 1.470 1.350 2.422 115.13 118.31
15-tBu -10.79 -76.31 -149.93 62.84 1.469 1.351 2.422 115.09 118.30
14-tBu -10.79 46.21 -17.19 52.60 1.472 1.350 2.414 112.15 118.58 a Las alcoxiaminas comprenden al nitróxido y el radical indicado. b Calor de reacción para la
homólisis del enlace C-O. c Consultar el Esquema 18. d Hyperchem 5.01 para Windows.
Molecular Model System. Hypercube, Inc. Gainesville, Florida.

Capítulo 5/ (Co)polimerización del IP por NMRP 5.2 Cálculos semiempíricos (AM1-RHF).
103
Los cálculos confirman lo que experimentalmente se observa, un incremento en el tamaño
de los sustituyentes α al nitrógeno del nitróxido genera una mejora en la rapidez de la
homólisis;115a esto es, se percibe en la disminución de la fuerza de enlace C-O al pasar de un
nitróxido con estructura cerrada a uno de cadena abierta o simplemente con el incremento del
ángulo C-O-N. Las velocidades relativas de la homólisis del enlace C-O para dichas
alcoxiaminas, también se debe a factores estéricos. Los cálculos predicen los valores de DC-O y,
de acuerdo a ellos, este disminuye en la serie del nitróxido 13>15>14. En el Esquema 18 se
especifican los enlaces calculados y los ángulos involucrados en cuestión. Con respecto a la
participación de los efectos estéricos en la determinación de las energías de disociación, se
puede anticipar que existirá una mayor longitud para el enlace C-O y con ello facilitará su
rompimiento; adicionalmente, la presencia del grupo HO- en 13, influye moderadamente en la
fragmentación homolítica de dicho enlace.
De acuerdo con los anteriores argumentos, se observa que existe una correlación entre las
longitudes de enlace C-O calculados con el tamaño del radical formado. Por ejemplo, la longitud
de enlace C-O para las t-butoxiaminas (1.470 ± 0.002 Å) es significativamente más grande que
para las correspondientes metoxiaminas (1.400 ± 0.001 Å).
O
N C
*
*
*
C
C
R2 *
*
*
R1
*
Esquema 18: Ángulos y enlaces involucrados en el cálculo semiempírico AM1-RHF.
Una mejor medida de la influencia de los factores estéricos involucrados, es la distancia
C-N dentro del fragmento C-O-N (Esquema 18). Esta distancia comprende los cambios en las
longitudes de los enlaces C-O y N-O; al mismo tiempo, del ángulo de enlace C-O-N. Se puede
deducir que un incremento en alguno de estos parámetros tendrá como resultado una reducción
de las interacciones estéricas entre los fragmentos alquílicos y el nitróxido. La variación en la
distancia C-N con la estructura del nitróxido, aumenta con el incremento del tamaño del

Capítulo 5/ (Co)polimerización del IP por NMRP 5.2 Cálculos semiempíricos (AM1-RHF).
104
fragmento del alquilo; para el grupo metoxi, por ejemplo, no hay mucha variación, indicando
que los factores estéricos no son relativamente importante para dicho sistema en el que se crean
radicales libres alquilo primarios.
Es importante también señalar que otros factores pueden influir en los valores de DC-O en
esta serie de alcoxiaminas. La tendencia en DC-O y en las velocidades de homólisis relativas, se
correlacionan con una disposición hacia el incremento en la estabilidad termodinámica de los
nitróxidos con el incremento del ángulo de enlace C-N-C; sin embargo, no existen datos
experimentales termoquímicos de los nitróxidos como una función del tamaño del anillo o de
cadena abierta, pero existen datos disponibles sobre los efectos de la estructura en la
conformación, propiedades y reactividades de nitróxidos y sus derivados. Se ha reportado116 que
la fuerza del enlace -NO-H en las hidroxilaminas aumenta con la disminución del ángulo C-N-
C en el fragmento del nitróxido y esto es atribuido a una variación en la estabilidad del nitróxido.
Se ha afirmado que la reactividad de los nitróxidos disminuye (y por ende, la estabilidad de
estos aumenta) con el incremento del ángulo de enlace C-N-C; sin embargo, no existen ejemplos
específicos de estos compuestos químicos.
Es pertinente también considerar los datos para la reacción inversa (acoplamiento) de los
radicales libres centrados en el carbono con los nitróxidos. La reacción de acoplamiento entre
C- y N-O se sabe que ocurre con constantes de rapidez altamente extremas (ca. 109 M-1 s-1).
Bowry et al.,117 encontraron que las constantes de rapidez para la reacción de acoplamiento de
radicales libres centrados en el carbono con nitróxidos despliegan una tendencia con la
estructura del nitróxido, la cual es inversa a lo hallado para la reacción de homólisis; esto es,
anillo de 5 miembros> anillo de 6 miembros> cadena abierta. Este comportamiento es
consistente con la tendencia que se observa en la presente serie de los cálculos de AM1. Se
atribuye esta tendencia a las constantes de rapidez para la reacción de acoplamiento por la
influencia de factores estéricos. Estos cálculos, ayudan a comprender porqué el nitróxido 14
llega a ser más eficiente en la polimerización del isopreno por NMP que 13 (Figuras 37 y 38).
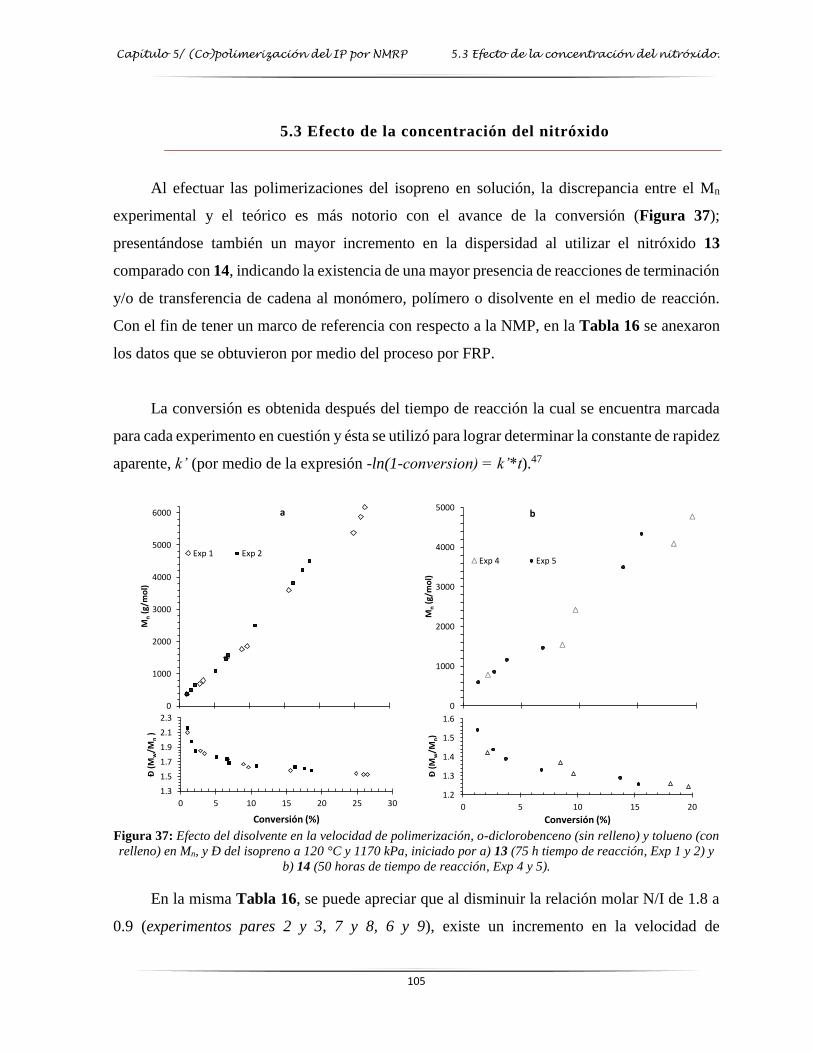
Capítulo 5/ (Co)polimerización del IP por NMRP 5.3 Efecto de la concentración del nitróxido.
105
5.3 Efecto de la concentración del nitróxido
Al efectuar las polimerizaciones del isopreno en solución, la discrepancia entre el Mn
experimental y el teórico es más notorio con el avance de la conversión (Figura 37);
presentándose también un mayor incremento en la dispersidad al utilizar el nitróxido 13
comparado con 14, indicando la existencia de una mayor presencia de reacciones de terminación
y/o de transferencia de cadena al monómero, polímero o disolvente en el medio de reacción.
Con el fin de tener un marco de referencia con respecto a la NMP, en la Tabla 16 se anexaron
los datos que se obtuvieron por medio del proceso por FRP.
La conversión es obtenida después del tiempo de reacción la cual se encuentra marcada
para cada experimento en cuestión y ésta se utilizó para lograr determinar la constante de rapidez
aparente, k’ (por medio de la expresión -ln(1-conversion) = k’*t).47
Figura 37: Efecto del disolvente en la velocidad de polimerización, o-diclorobenceno (sin relleno) y tolueno (con
relleno) en Mn, y Ð del isopreno a 120 °C y 1170 kPa, iniciado por a) 13 (75 h tiempo de reacción, Exp 1 y 2) y
b) 14 (50 horas de tiempo de reacción, Exp 4 y 5).
En la misma Tabla 16, se puede apreciar que al disminuir la relación molar N/I de 1.8 a
0.9 (experimentos pares 2 y 3, 7 y 8, 6 y 9), existe un incremento en la velocidad de
0
1000
2000
3000
4000
5000
6000
Mn
(g/m
ol)
Exp 1 Exp 2
a
0
1000
2000
3000
4000
5000
Mn
(g/m
ol)
Exp 4 Exp 5
b
1.3
1.5
1.7
1.9
2.1
2.3
0 5 10 15 20 25 30
Ɖ (
Mw
/Mn
)
Conversión (%)
1.2
1.3
1.4
1.5
1.6
0 5 10 15 20
Ɖ (
Mw
/Mn)
Conversión (%)

Capítulo 5/ (Co)polimerización del IP por NMRP 5.5 Reacciones de copolimerización IP/GMA.
106
polimerización (un modo de apreciarlo cuantitativamente, es con el valor numérico de k’),
teniéndose una mayor amplitud en la distribución de pesos moleculares (Đ); esto se debe a la
disminución del nitróxido presente en el medio de reacción, induciendo a una iniciación lenta
en la velocidad de la polimerización (presencia de una desactivación temporal de los radicales
libres propagantes). Adicionalmente, los pesos moleculares promedio en número Mn, se
encuentran en un intervalo del 65 al 74% con respecto al Mn teórico (esto se puede apreciar
mejor en la eficiencia del nitróxido).
5.4 Efecto del disolvente en el medio de reacción.
La Tabla 18 muestra las propiedades físicas de algunos disolventes propuestos, con el fin
de comprender la relación que pueda existir con la polaridad del disolvente en la velocidad de
polimerización del IP por NMP.
Tabla 18: Propiedades físicas de algunos disolventes.118
Disolvente Pea
(°C) ea
ma
(D20 °C)
1,4-dioxano 101 2.25 0.45
Benceno 80 2.27 0
Tolueno 111 2.38 0.43
Xileno 144 2.57 0.62
Anisol 154 4.33 1.38
o-dicloro
benceno 181 9.93 2.27
Piridina 115 12.4 2.37
Acetona 56 20.7 2.85
Metanol 65 32.7 1.70
DMF 152 36.7 3.86
DMSO 189 46.7 3.9
a e: constante dieléctrica; m: momento dipolar; Pe: punto de ebullición
En el presente estudio de investigación, se eligieron dos disolventes de la serie presentada
en la Tabla 18, el tolueno que es relativamente no polar con un momento dipolar de 0.43 D
(constante dieléctrica de 2.38) y el o-diclorobenceno, moderadamente polar (momento dipolar
de 2.27 D y constante dieléctrica de 9.93); cabe mencionar que ambos disolventes tienen puntos
de ebullición arriba de 100 °C (punto importante, debido a que la reacción se lleva a 120 °C y
1170 kPa). Así mismo, se eligieron dos relaciones molares de N/I (1.8 y 0.9) para observar el
efecto que tienen en la velocidad de polimerización. El comportamiento de la cinética es
aparentemente de primer orden con respecto al monómero como se puede observar en la Figura

Capítulo 5/ (Co)polimerización del IP por NMRP 5.5 Reacciones de copolimerización IP/GMA.
107
38 (resultado de una rápida disociación de la macroalcoxiamina formada a la temperatura de
reacción).
Figura 38: Efecto del disolvente en la velocidad de polimerización, o-diclorobenceno (sin relleno) y tolueno (con
relleno) en la polimerización del isopreno a 120 °C y 1170 kPa, iniciado por 13 (Experimentos 1 y 2) y 14
(Experimentos 4 y 5).
Como se aprecia en la Figura 38, existe una mejora en la evolución de la conversión
(velocidad de polimerización) de la polimerización del isopreno; así mismo, también se aprecia
una mejora en la distribución de los pesos moleculares con el o-diclorobenceno comparándolo
con tolueno (ver Figura 37). Estas observaciones son coincidentes con reportes ya publicados,
donde muestran que la homólisis del enlace C-O de la macroalcoxiamina formada es acelerada
por disolventes polares,115a, 119 debido a que estabilizan al radical (carácter viviente); mientras
que la reacción inversa (recombinación o transición a carácter durmiente), es desacelerada.120
Adicionalmente, estas observaciones se confirman con los valores de la constante de rapidez
aparente para cada experimento involucrado.
5.5 Reacciones de copolimerización IP-GMA
A pesar de que las polimerizaciones del isopreno llevadas a cabo con el disolvente o-
diclorobenceno muestran una apreciable velocidad de polimerización con respecto a las
efectuadas con tolueno; el efecto no es tan significativo, por lo que de aquí en adelante se
procedió a realizar las polimerizaciones en solución con tolueno como disolvente, lo que está
0
5
10
15
20
25
30
0
0.05
0.1
0.15
0.2
0.25
0.3
0.35
0 10 20 30 40 50 60 70 80
con
versión
(%)
-Ln
(1-c
on
vers
ion
)
Tiempo (h)
Exp 1 Exp 2 Exp 4 Exp 5
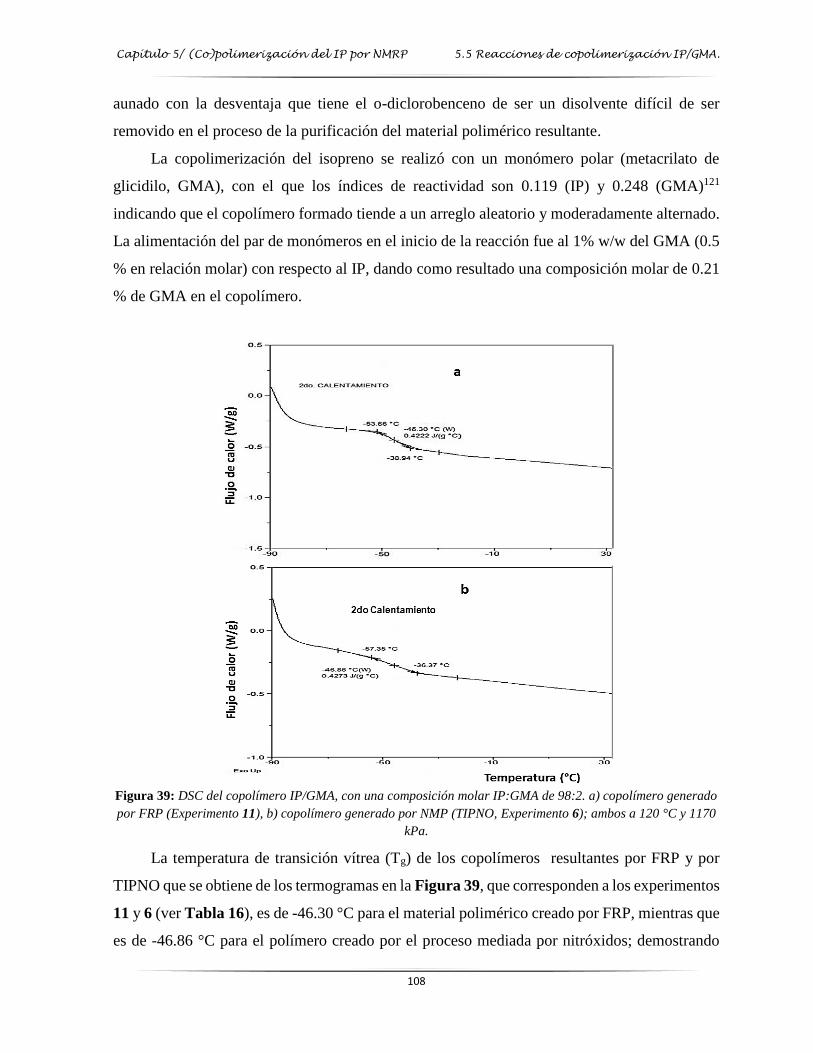
Capítulo 5/ (Co)polimerización del IP por NMRP 5.5 Reacciones de copolimerización IP/GMA.
108
aunado con la desventaja que tiene el o-diclorobenceno de ser un disolvente difícil de ser
removido en el proceso de la purificación del material polimérico resultante.
La copolimerización del isopreno se realizó con un monómero polar (metacrilato de
glicidilo, GMA), con el que los índices de reactividad son 0.119 (IP) y 0.248 (GMA)121
indicando que el copolímero formado tiende a un arreglo aleatorio y moderadamente alternado.
La alimentación del par de monómeros en el inicio de la reacción fue al 1% w/w del GMA (0.5
% en relación molar) con respecto al IP, dando como resultado una composición molar de 0.21
% de GMA en el copolímero.
Figura 39: DSC del copolímero IP/GMA, con una composición molar IP:GMA de 98:2. a) copolímero generado
por FRP (Experimento 11), b) copolímero generado por NMP (TIPNO, Experimento 6); ambos a 120 °C y 1170
kPa.
La temperatura de transición vítrea (Tg) de los copolímeros resultantes por FRP y por
TIPNO que se obtiene de los termogramas en la Figura 39, que corresponden a los experimentos
11 y 6 (ver Tabla 16), es de -46.30 °C para el material polimérico creado por FRP, mientras que
es de -46.86 °C para el polímero creado por el proceso mediada por nitróxidos; demostrando
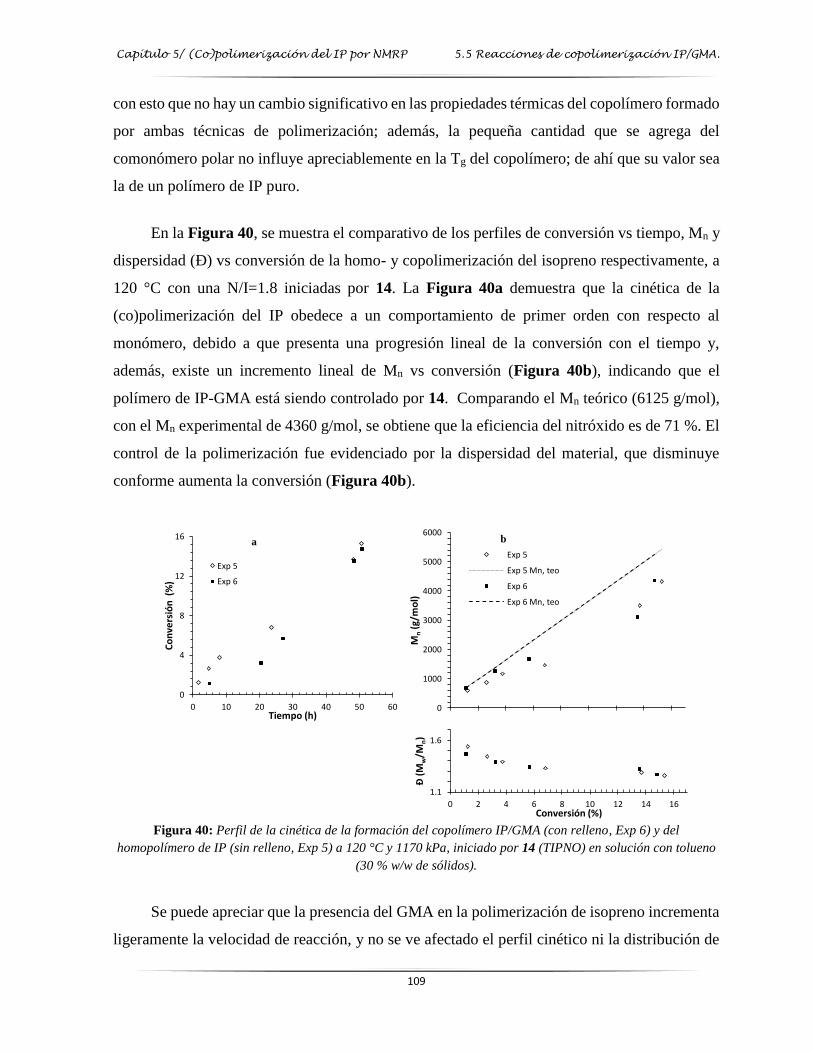
Capítulo 5/ (Co)polimerización del IP por NMRP 5.5 Reacciones de copolimerización IP/GMA.
109
con esto que no hay un cambio significativo en las propiedades térmicas del copolímero formado
por ambas técnicas de polimerización; además, la pequeña cantidad que se agrega del
comonómero polar no influye apreciablemente en la Tg del copolímero; de ahí que su valor sea
la de un polímero de IP puro.
En la Figura 40, se muestra el comparativo de los perfiles de conversión vs tiempo, Mn y
dispersidad (Ð) vs conversión de la homo- y copolimerización del isopreno respectivamente, a
120 °C con una N/I=1.8 iniciadas por 14. La Figura 40a demuestra que la cinética de la
(co)polimerización del IP obedece a un comportamiento de primer orden con respecto al
monómero, debido a que presenta una progresión lineal de la conversión con el tiempo y,
además, existe un incremento lineal de Mn vs conversión (Figura 40b), indicando que el
polímero de IP-GMA está siendo controlado por 14. Comparando el Mn teórico (6125 g/mol),
con el Mn experimental de 4360 g/mol, se obtiene que la eficiencia del nitróxido es de 71 %. El
control de la polimerización fue evidenciado por la dispersidad del material, que disminuye
conforme aumenta la conversión (Figura 40b).
Figura 40: Perfil de la cinética de la formación del copolímero IP/GMA (con relleno, Exp 6) y del
homopolímero de IP (sin relleno, Exp 5) a 120 °C y 1170 kPa, iniciado por 14 (TIPNO) en solución con tolueno
(30 % w/w de sólidos).
Se puede apreciar que la presencia del GMA en la polimerización de isopreno incrementa
ligeramente la velocidad de reacción, y no se ve afectado el perfil cinético ni la distribución de
0
4
8
12
16
0 10 20 30 40 50 60
Co
nve
rsió
n (
%)
Tiempo (h)
Exp 5
Exp 6
0
1000
2000
3000
4000
5000
6000
Mn
(g/m
ol)
Exp 5
Exp 5 Mn, teo
Exp 6
Exp 6 Mn, teo
1.1
1.6
0 2 4 6 8 10 12 14 16
Ɖ (
Mw
/Mn)
Conversión (%)
a b

Capítulo 5/ (Co)polimerización del IP por NMRP 5.6 Uso de aditivos en la NMP.
Incremento en la velocidad de polimerización.
110
los pesos moleculares. En la Figura 41 se muestra una comparación de las distribuciones de los
pesos moleculares del homopolímero y copolímero por medio del nitróxido 14 y por
polimerización radicálica convencional, donde se puede observar la tendencia hacia pesos
moleculares altos con la utilización de la técnica por FRP y la presencia en este caso también de
un hombro debido a las reacciones de terminación o de transferencia de cadena durante el
transcurso de la reacción.
Figura 41: Cromatogramas de GPC del homo- y copolímero del IP por NMP (Exp 5 y 6) y por FRP (Exp 10 y
11).
5.6 Uso de aditivos en la NMP. Incremento en la velocidad de
polimerización
El interés en esta sección es incrementar la conversión (y consecuentemente, el peso
molecular) del polímero mediante el uso de aditivos acelerantes en la velocidad de
polimerización del isopreno mediante nitróxidos. Este interés nace con la intención de obtener
un Mn aproximado de 1x105 Da, debido a su posible aplicación como compatibilizante en
mezclas poliméricas no miscibles. Siendo así, todas las condiciones experimentales utilizadas
se reúnen en la Tabla 19. En los párrafos siguientes se describirá la cinética involucrada en la
hopolimerización del IP utilizando como agente de control a los radicales 13 y 14 (por medio
del proceso bimolecular) con el uso de tres aceleradores de la polimerización en nitróxidos:
anhídrido acético (AA), glucosa (Glu) y ácido canforsulfónico (CSA) a diferentes relaciones
molares de acelerador:nitróxido (Acel/N).
2.5 3 3.5 4 4.5 5 5.5
Log MW (g/mol)
Exp 6
Exp 5
Exp 11
Exp 10

Capítulo 5/ (Co)polimerización del IP por NMRP 5.6 Uso de aditivos en la NMP.
Incremento en la velocidad de polimerización.
111
Tabla 19. Resumen de la homopolimerización del IP con el uso de aceleradores en la NMP.
Experi-
mento
Acelera-
dora Nitróxido (N)b
Acel/
N c
Tiempo
de
reacción
(h)
Conver-
sión Mn, teo
d MnRMN e 𝑴𝒏
𝑮𝑷𝑪 f fNO h Ð k’x10-3
(h-1) g
12 CSA HO-TEMPO 2.0 48 0.2793 9700 9300 9100 0.96 1.62 6.82
13 CSA HO-TEMPO 1.0 48 0.2564 9000 9900 9850 1.10 1.64 6.17
14 CSA HO-TEMPO 0.2 48 0.1667 6000 3800 3950 0.63 1.69 3.80
15 GLU HO-TEMPO 2.0 48 0.2918 10100 10500 10700 1.04 1.58 7.19
16 GLU HO-TEMPO 1.0 48 0.1426 5200 5400 5350 1.04 1.60 3.20
17 GLU HO-TEMPO 0.2 48 0.2370 8300 5800 5950 0.70 1.63 5.64
18 AA HO-TEMPO 2.0 48 0.4602 15800 18100 18300 1.15 1.54 12.85
19 AA HO-TEMPO 1.0 48 0.4285 14700 18000 17850 1.22 1.59 11.66
20 AA HO-TEMPO 0.2 48 0.1876 6700 7800 7850 1.16 1.61 4.33
21 CSA TIPNO 2.0 24 0.1639 5900 6300 6300 1.07 3.10 7.46
22 GLU TIPNO 2.0 24 0.2260 8000 7400 7250 0.93 2.45 10.67
23 AA TIPNO 2.0 24 0.3326 11600 12400 12600 1.07 3.34 16.86
24 ---- TIPNO 0.0 24 0.0761 2600 2400 2450 0.92 1.34 3.30
25 ---- HO-TEMPO 0.0 48 0.1237 4100 3100 3000 0.76 1.60 2.75
a Abreviaciones: AA, anhídrido acético; BPO, peróxido de benzoilo; CSA, ácido canforsulfónico; GLU, glucosa;
HO-TEMPO, 4-hidroxi-TEMPO; IP, isopreno; TIPNO: 2, 2, 5-trimetil-4-fenil-3-azahexano-3-nitroxido; Tol,
tolueno. b Condiciones de reacción: 1170 kPa. 30 ml IP en la cantidad requerida del disolvente (con el fin de obtener
un 30 % w/w de sólidos). Relación molar IP:N:I, 490:1:0.55; iniciador: BPO en solución con tolueno (60 mL); para
el HO-TEMPO 135 °C, para el TIPNO 120 °C. c Relación molar del nitróxido/acelerador. d Mn, Teo se calculó a partir
de la expresión Mn, Teo=conversión|M|/|N| PMM + PMN + PMI/2; donde |M|: concentración del monómero (mol); |N|:
concentración del nitróxido (mol); PMM, PMN, PMI: peso molecular del monómero, nitróxido (g/mol) e iniciador
respectivamente. e Se determinó por RMN122 f Se determinó por GPC, calibrado con estándares de PS y convertidos
a PIP usando las constantes de MHS según la referencia104 g Constante de rapidez aparente; k’= -ln(1-conversión)/t,
t:tiempo de reacción. i Eficiencia del nitróxido; fNO=Mn, exp/Mn, teo
Como se puede apreciar en la Tabla 19 hay un incremento considerable en la velocidad
de la polimerización al agregar el aditivo en la reacción (la modificación de la velocidad depende
de la relación molar que se tenga de Acel/N, la velocidad de polimerización se puede observar
cualitativamente con el valor numérico de la constante aparente, k’). En la misma Tabla 19 se
anexaron datos que se obtuvieron sin la presencia del aditivo para tener un marco de referencia.
La polimerización del isopreno por NMP fue llevada a cabo por 13 y por 14 (Esquema 16) a
135 °C y 120 °C respectivamente, usando BPO como iniciador de radicales en solución con
tolueno (30 % w/w en sólidos) en periodos de tiempo de 24–48 h. En todos los casos
experimentales, la relación molar de isopreno al nitróxido fue de 490:1; se formaron series
experimentales con los aditivos (aceleradores de la polimerización) manejando una relación
molar del nitróxido al aditivo de 2.0, 1.0 y 0.2.
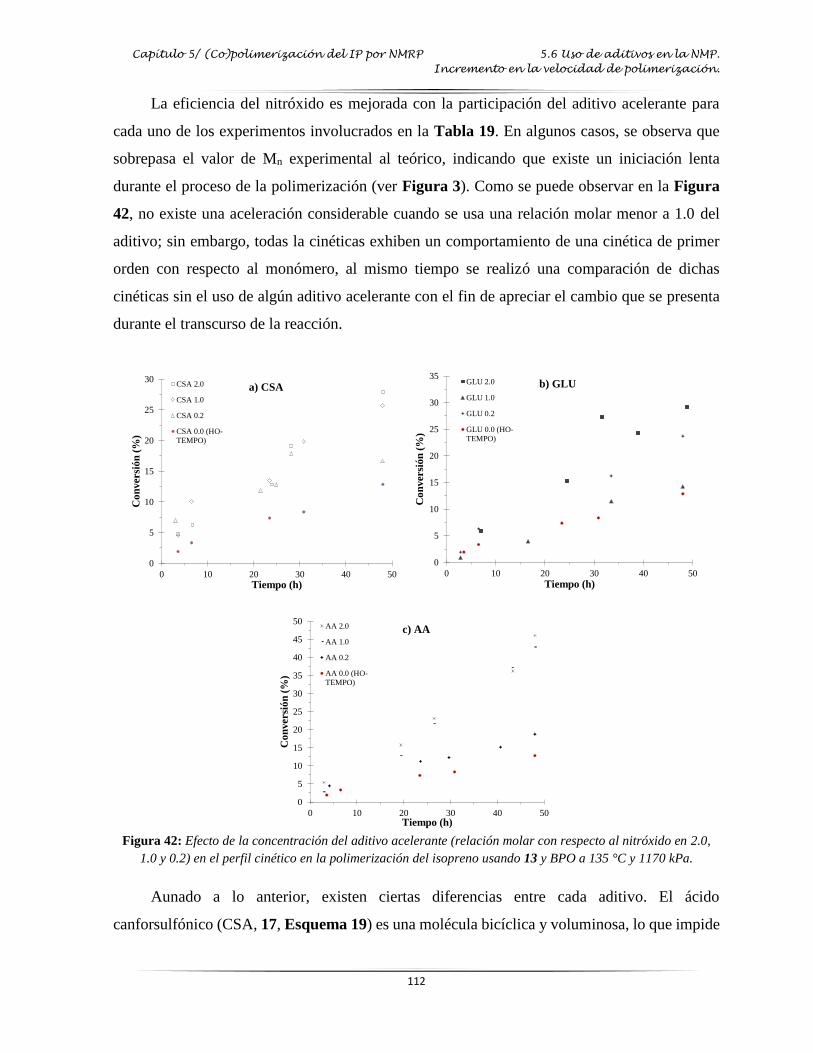
Capítulo 5/ (Co)polimerización del IP por NMRP 5.6 Uso de aditivos en la NMP.
Incremento en la velocidad de polimerización.
112
La eficiencia del nitróxido es mejorada con la participación del aditivo acelerante para
cada uno de los experimentos involucrados en la Tabla 19. En algunos casos, se observa que
sobrepasa el valor de Mn experimental al teórico, indicando que existe un iniciación lenta
durante el proceso de la polimerización (ver Figura 3). Como se puede observar en la Figura
42, no existe una aceleración considerable cuando se usa una relación molar menor a 1.0 del
aditivo; sin embargo, todas la cinéticas exhiben un comportamiento de una cinética de primer
orden con respecto al monómero, al mismo tiempo se realizó una comparación de dichas
cinéticas sin el uso de algún aditivo acelerante con el fin de apreciar el cambio que se presenta
durante el transcurso de la reacción.
Figura 42: Efecto de la concentración del aditivo acelerante (relación molar con respecto al nitróxido en 2.0,
1.0 y 0.2) en el perfil cinético en la polimerización del isopreno usando 13 y BPO a 135 °C y 1170 kPa.
Aunado a lo anterior, existen ciertas diferencias entre cada aditivo. El ácido
canforsulfónico (CSA, 17, Esquema 19) es una molécula bicíclica y voluminosa, lo que impide
0
5
10
15
20
25
30
0 10 20 30 40 50
Co
nversi
ón
(%
)
Tiempo (h)
a) CSA CSA 2.0
CSA 1.0
CSA 0.2
CSA 0.0 (HO-
TEMPO)
0
5
10
15
20
25
30
35
0 10 20 30 40 50
Co
nversi
ón
(%
)
Tiempo (h)
b) GLUGLU 2.0
GLU 1.0
GLU 0.2
GLU 0.0 (HO-
TEMPO)
0
5
10
15
20
25
30
35
40
45
50
0 10 20 30 40 50
Co
nversi
ón
(%
)
Tiempo (h)
c) AAAA 2.0
AA 1.0
AA 0.2
AA 0.0 (HO-
TEMPO)

Capítulo 5/ (Co)polimerización del IP por NMRP 5.6 Uso de aditivos en la NMP.
Incremento en la velocidad de polimerización.
113
tener la suficiente capacidad de acelerar la reacción por cuestiones estéricas, esto le imposibilita
mucho acercarse adecuadamente a la molécula objetivo y lograr interaccionar con el grupo
nitróxido; por otra parte, con el uso de una molécula más pequeña como es el anhídrido acético
(AA, 16), ésta tiene la ventaja de ser fácilmente removida dentro de la mezcla de reacción por
su alta volatilidad, es muy soluble en la mezcla de reacción e incrementa considerablemente la
velocidad de polimerización. En un punto intermedio queda la glucosa (GLU, 18) dentro de este
análisis experimental.
Esquema 19: Estructuras de los aditivos utilizados como aditivos acelerantes en la polimerización del IP por
NMP: 16 (ácido acético, AA), 17 (ácido canforsulfónico, CSA) y 18 (glucosa, GLU).
Dicho lo anterior y de acuerdo con Malmström et al.,53a el efecto de la aceleración en la
velocidad de polimerización, se presenta por la participación del par de electrones que tiene el
nitrógeno de la macroalcoxiamina con el aditivo acelerante, pasando por una reacción de
acilación reversible (Esquema 20), y disminuyendo el exceso de nitróxido presente en el medio
de reacción. La coordinación del grupo acilo con el átomo de nitrógeno induce a una carga
positiva parcial, teniendo como resultado una polarización del enlace N-O hacia el nitrógeno,
generando un debilitamiento al enlace C-O y promoviendo la disociación térmica de dicha
unión; creando así un incremento en la velocidad de polimerización. Al mismo tiempo, los
agentes de acilación pueden reaccionar con el nitróxido libre presente, disminuyendo por lo
tanto, la concentración y/o exceso del agente de control.
N
CH3
CH3
CH3CH3
OR
Ac2ON
+
CH3
CH3
CH3CH3
OR
CH3
O
Esquema 20: Acilación de la alcoxiamina con el grupo terminal que conlleva al incremento de su reactividad
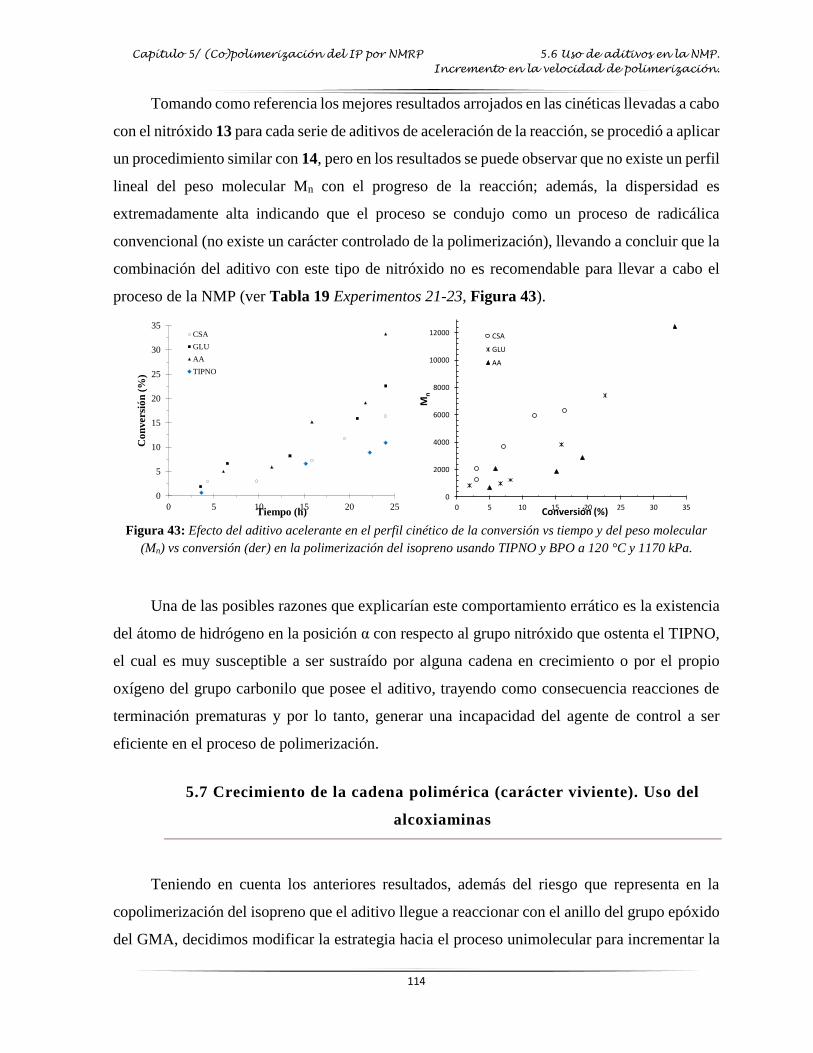
Capítulo 5/ (Co)polimerización del IP por NMRP 5.6 Uso de aditivos en la NMP.
Incremento en la velocidad de polimerización.
114
Tomando como referencia los mejores resultados arrojados en las cinéticas llevadas a cabo
con el nitróxido 13 para cada serie de aditivos de aceleración de la reacción, se procedió a aplicar
un procedimiento similar con 14, pero en los resultados se puede observar que no existe un perfil
lineal del peso molecular Mn con el progreso de la reacción; además, la dispersidad es
extremadamente alta indicando que el proceso se condujo como un proceso de radicálica
convencional (no existe un carácter controlado de la polimerización), llevando a concluir que la
combinación del aditivo con este tipo de nitróxido no es recomendable para llevar a cabo el
proceso de la NMP (ver Tabla 19 Experimentos 21-23, Figura 43).
Figura 43: Efecto del aditivo acelerante en el perfil cinético de la conversión vs tiempo y del peso molecular
(Mn) vs conversión (der) en la polimerización del isopreno usando TIPNO y BPO a 120 °C y 1170 kPa.
Una de las posibles razones que explicarían este comportamiento errático es la existencia
del átomo de hidrógeno en la posición α con respecto al grupo nitróxido que ostenta el TIPNO,
el cual es muy susceptible a ser sustraído por alguna cadena en crecimiento o por el propio
oxígeno del grupo carbonilo que posee el aditivo, trayendo como consecuencia reacciones de
terminación prematuras y por lo tanto, generar una incapacidad del agente de control a ser
eficiente en el proceso de polimerización.
5.7 Crecimiento de la cadena polimérica (carácter viviente). Uso del
alcoxiaminas
Teniendo en cuenta los anteriores resultados, además del riesgo que representa en la
copolimerización del isopreno que el aditivo llegue a reaccionar con el anillo del grupo epóxido
del GMA, decidimos modificar la estrategia hacia el proceso unimolecular para incrementar la
0
5
10
15
20
25
30
35
0 5 10 15 20 25
Co
nversi
ón
(%
)
Tiempo (h)
CSA
GLU
AA
TIPNO
0
2000
4000
6000
8000
10000
12000
0 5 10 15 20 25 30 35
Mn
Conversión (%)
CSA
GLU
AA

Capítulo 5/ (Co)polimerización del IP por NMRP 5.7 Crecimiento de la cadena polimérica
(carácter viviente). Uso de alcoxiaminas.
115
conversión y el peso molecular del producto polimérico mediante la utilización de una
alcoxiamina conteniendo el fragmento del nitróxido TIPNO (TIPAL, 18, Esquema 21),
manejando diferentes relaciones molares de monómero/nitróxido (M/N) de 490 a 1600. Todas
las condiciones experimentales se resumen en la Tabla 20 manteniendo la misma concentración
de sólidos en tolueno (30 % w/w).
Esquema 21: Estructura del TIPAL (18).
Tabla 20. Resumen de la homo y copolimerización del IP por NMP. Experi-
mento
Monóme-
ro(s)a CTAb M/N c
Conver-
sión Mn, teo
e MnRMN d 𝑴𝒏
𝑮𝑷𝑪 f fNO n Ð k’x10-3
(h-1) g
26 IP TIPAL 490 0.1514 5600 7800 7900 1.01 1.19 6.84
27 IP TIPAL 750 0.2395 12800 17100 17150 1.36 1.23 11.41
28 IP TIPAL 1000 0.2927 20500 26800 26300 1.31 1.26 14.43
29 IP TIPAL 1300 0.3419 30800 38200 38550 1.24 1.37 17.43
30 IP TIPAL 1600 0.5122 56400 67500 67450 1.20 1.44 29.91
31 IP/GMA TIPAL 1000 0.4329 33500 37000 36850 1.11 1.21 23.77
32 IP Exp 31j 1400 0.4348 53100 56700 56950 1.07 1.26 24.42
33 IP Exp 32k 1400 0.4439 83300 89100 90100 1.07 1.33 24.45
34 IP Exp 33l 1400 0.4216 112000 120500 119800 1.08 1.37 22.81
35 IPm TIPNO 1000 0.1652 11300 8500 8450 0.75 1.29 7.52
a Abreviaciones: GMA: metacrilato de glicidilo; IP, isopreno; TIPNO: 2, 2, 5-trimetil-4-fenil-3-azahexano-3-nitroxido;
TIPAL: N-tert-butil-N-(2-metil-1-fenilpropil)-O-(1-feniletil) hidroxilamina. b Condiciones de reacción: 24 h, 1170 kPa a
120 °C en 30 ml IP con 60 mL de tolueno (con el fin de obtener un 30 % w/w de sólidos). c Relación molar
Monómero/Nitróxido. d Se determinó por RMN.122 e Mn, Teo se calculó a partir de la expresión Mn, Teo=conversion |M|/|N|
PMM + PMN ; donde |M|: concentración del monómero (mol); |N|: concentración del nitróxido (mol); PMM, PMN, PMI:
peso molecular del monómero, nitróxido (g/mol) e iniciador respectivamente. f Se determinó por GPC, calibrado con
estándares de PS y convertidos a PIP usando las constantes de MHS según la referencia104 g Constante de rapidez aparente;
k’= -ln(1-conversión)/t, t:tiempo de reacción. i Relación molar IP:GMA=9:1 j Macroalcoxiamina obtenida en el
experimento 31 k Macroalcoxiamina obtenida en el experimento 32 l Macroalcoxiamina obtenida en el experimento 33 m
Relación molar Nitróxido/Iniciador= 1.8. n Eficiencia del nitróxido; fNO=Mn, exp/Mn, teo
Tal y como se puede observar en los Experimentos 26-30 (Tabla 20) y en concordancia a
lo esperado, al aumentar M/N de 490 a 1600, se presenta un incremento en la velocidad de
polimerización pero con una mayor amplitud en la distribución de pesos moleculares, debido a
que la concentración del nitróxido en el medio va disminuyendo en esta serie experimental,
trayendo como consecuencia un menor grado de control en la polimerización. A partir de estas
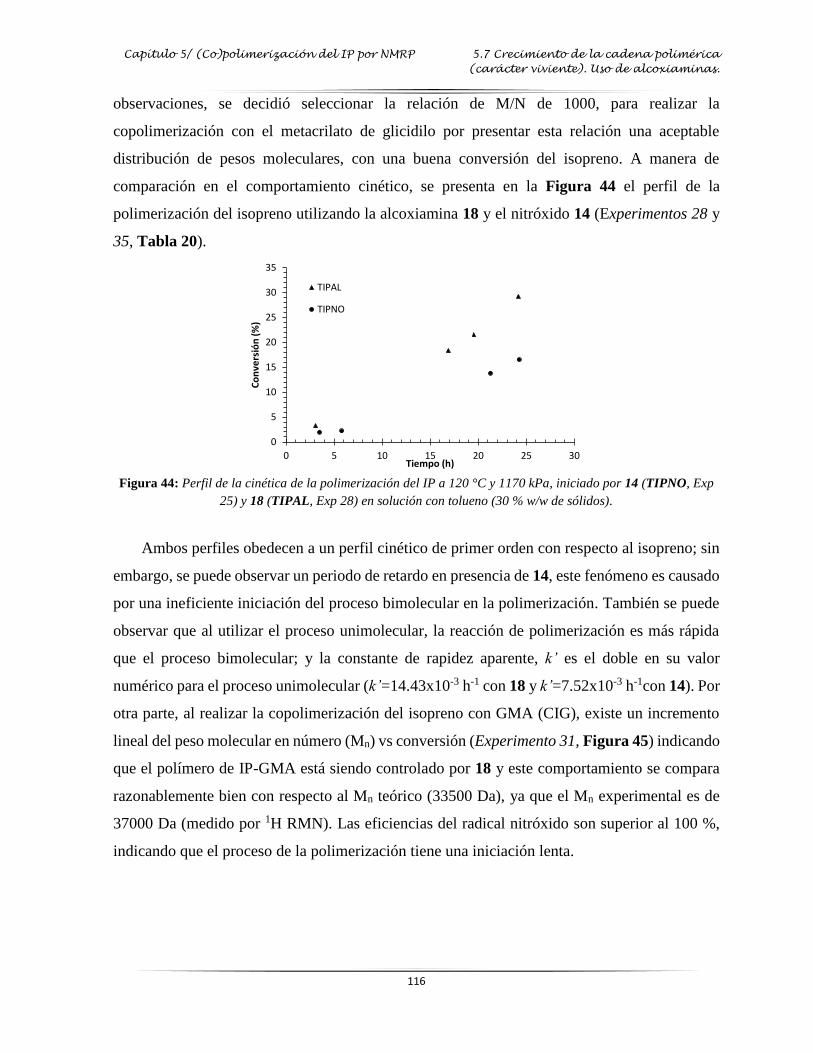
Capítulo 5/ (Co)polimerización del IP por NMRP 5.7 Crecimiento de la cadena polimérica
(carácter viviente). Uso de alcoxiaminas.
116
observaciones, se decidió seleccionar la relación de M/N de 1000, para realizar la
copolimerización con el metacrilato de glicidilo por presentar esta relación una aceptable
distribución de pesos moleculares, con una buena conversión del isopreno. A manera de
comparación en el comportamiento cinético, se presenta en la Figura 44 el perfil de la
polimerización del isopreno utilizando la alcoxiamina 18 y el nitróxido 14 (Experimentos 28 y
35, Tabla 20).
Figura 44: Perfil de la cinética de la polimerización del IP a 120 °C y 1170 kPa, iniciado por 14 (TIPNO, Exp
25) y 18 (TIPAL, Exp 28) en solución con tolueno (30 % w/w de sólidos).
Ambos perfiles obedecen a un perfil cinético de primer orden con respecto al isopreno; sin
embargo, se puede observar un periodo de retardo en presencia de 14, este fenómeno es causado
por una ineficiente iniciación del proceso bimolecular en la polimerización. También se puede
observar que al utilizar el proceso unimolecular, la reacción de polimerización es más rápida
que el proceso bimolecular; y la constante de rapidez aparente, k’ es el doble en su valor
numérico para el proceso unimolecular (k’=14.43x10-3 h-1 con 18 y k’=7.52x10-3 h-1con 14). Por
otra parte, al realizar la copolimerización del isopreno con GMA (CIG), existe un incremento
lineal del peso molecular en número (Mn) vs conversión (Experimento 31, Figura 45) indicando
que el polímero de IP-GMA está siendo controlado por 18 y este comportamiento se compara
razonablemente bien con respecto al Mn teórico (33500 Da), ya que el Mn experimental es de
37000 Da (medido por 1H RMN). Las eficiencias del radical nitróxido son superior al 100 %,
indicando que el proceso de la polimerización tiene una iniciación lenta.
0
5
10
15
20
25
30
35
0 5 10 15 20 25 30
Co
nve
rsió
n (
%)
Tiempo (h)
TIPAL
TIPNO
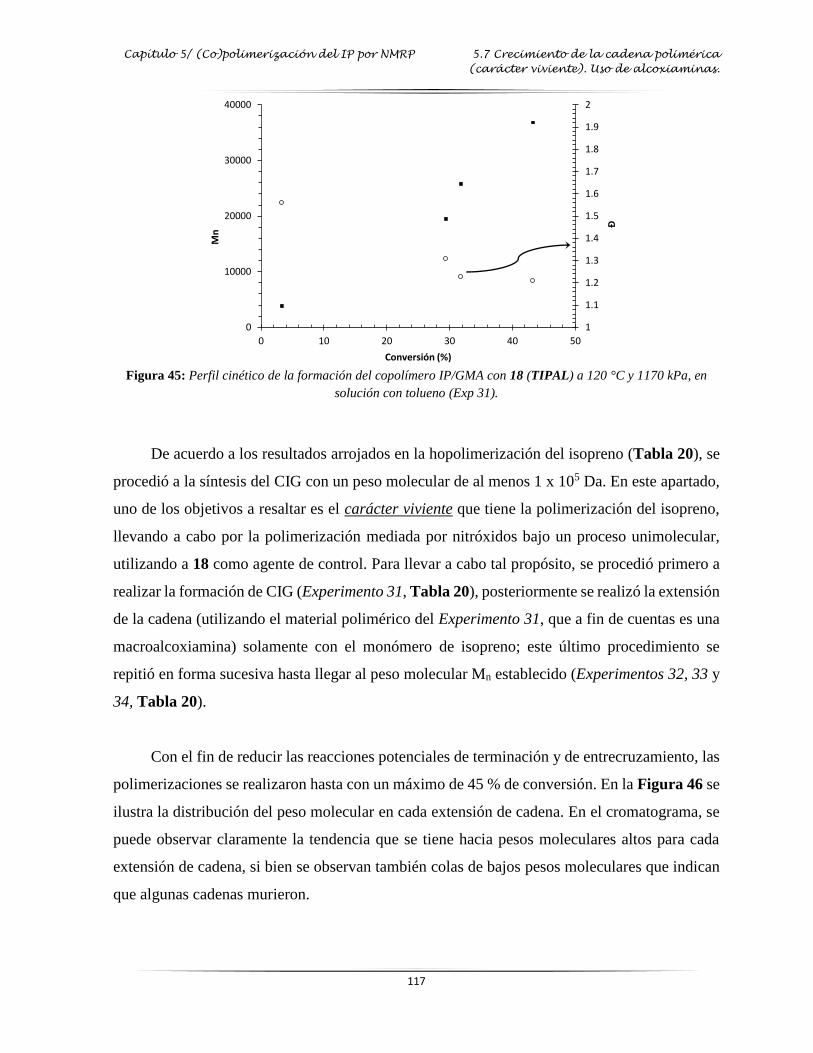
Capítulo 5/ (Co)polimerización del IP por NMRP 5.7 Crecimiento de la cadena polimérica
(carácter viviente). Uso de alcoxiaminas.
117
Figura 45: Perfil cinético de la formación del copolímero IP/GMA con 18 (TIPAL) a 120 °C y 1170 kPa, en
solución con tolueno (Exp 31).
De acuerdo a los resultados arrojados en la hopolimerización del isopreno (Tabla 20), se
procedió a la síntesis del CIG con un peso molecular de al menos 1 x 105 Da. En este apartado,
uno de los objetivos a resaltar es el carácter viviente que tiene la polimerización del isopreno,
llevando a cabo por la polimerización mediada por nitróxidos bajo un proceso unimolecular,
utilizando a 18 como agente de control. Para llevar a cabo tal propósito, se procedió primero a
realizar la formación de CIG (Experimento 31, Tabla 20), posteriormente se realizó la extensión
de la cadena (utilizando el material polimérico del Experimento 31, que a fin de cuentas es una
macroalcoxiamina) solamente con el monómero de isopreno; este último procedimiento se
repitió en forma sucesiva hasta llegar al peso molecular Mn establecido (Experimentos 32, 33 y
34, Tabla 20).
Con el fin de reducir las reacciones potenciales de terminación y de entrecruzamiento, las
polimerizaciones se realizaron hasta con un máximo de 45 % de conversión. En la Figura 46 se
ilustra la distribución del peso molecular en cada extensión de cadena. En el cromatograma, se
puede observar claramente la tendencia que se tiene hacia pesos moleculares altos para cada
extensión de cadena, si bien se observan también colas de bajos pesos moleculares que indican
que algunas cadenas murieron.
1
1.1
1.2
1.3
1.4
1.5
1.6
1.7
1.8
1.9
2
0
10000
20000
30000
40000
0 10 20 30 40 50
Ð
Mn
Conversión (%)
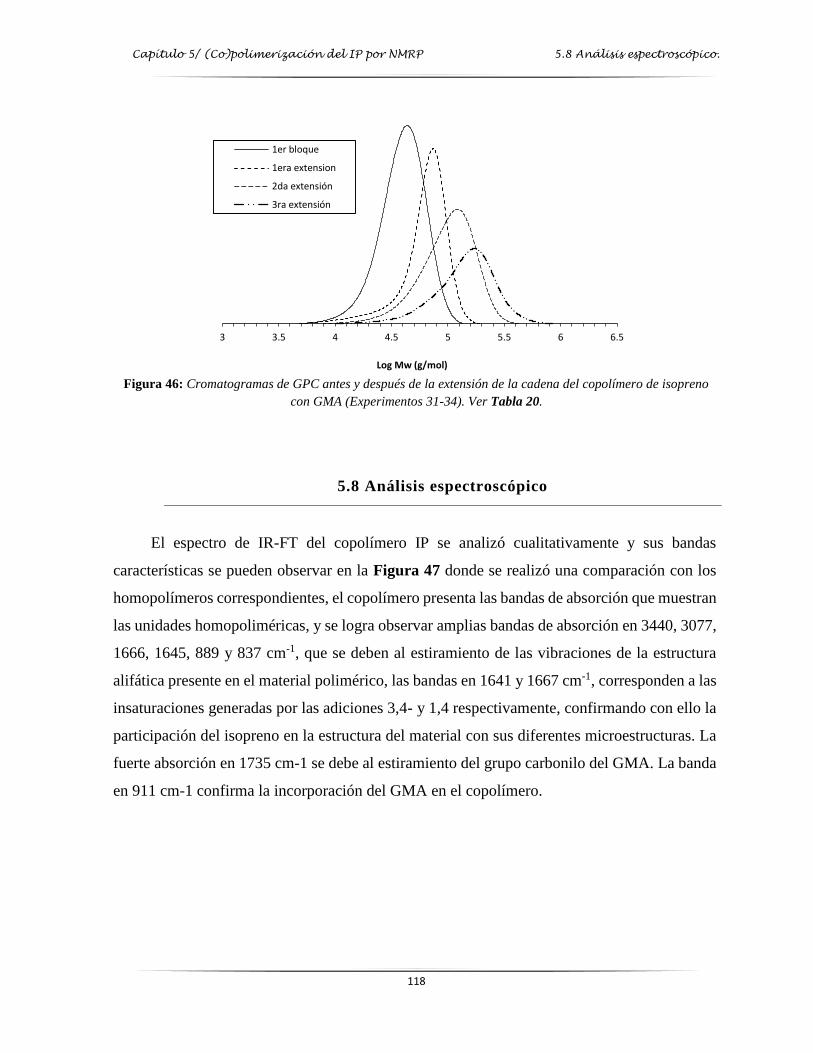
Capítulo 5/ (Co)polimerización del IP por NMRP 5.8 Análisis espectroscópico.
118
Figura 46: Cromatogramas de GPC antes y después de la extensión de la cadena del copolímero de isopreno
con GMA (Experimentos 31-34). Ver Tabla 20.
5.8 Análisis espectroscópico
El espectro de IR-FT del copolímero IP se analizó cualitativamente y sus bandas
características se pueden observar en la Figura 47 donde se realizó una comparación con los
homopolímeros correspondientes, el copolímero presenta las bandas de absorción que muestran
las unidades homopoliméricas, y se logra observar amplias bandas de absorción en 3440, 3077,
1666, 1645, 889 y 837 cm-1, que se deben al estiramiento de las vibraciones de la estructura
alifática presente en el material polimérico, las bandas en 1641 y 1667 cm-1, corresponden a las
insaturaciones generadas por las adiciones 3,4- y 1,4 respectivamente, confirmando con ello la
participación del isopreno en la estructura del material con sus diferentes microestructuras. La
fuerte absorción en 1735 cm-1 se debe al estiramiento del grupo carbonilo del GMA. La banda
en 911 cm-1 confirma la incorporación del GMA en el copolímero.
3 3.5 4 4.5 5 5.5 6 6.5
Log Mw (g/mol)
1er bloque
1era extension
2da extensión
3ra extensión

Capítulo 5/ (Co)polimerización del IP por NMRP 5.8 Análisis espectroscópico.
119
Figura 47: Espectro de FT-IR del homopolímero de IP, GMA y el respectivo copolímero, con una relación
molar en el copolímero de 9:1 (IP-GMA). a) PGMA, b) Poli (IP-ran-GMA, experimento 31), c) PIP
(experimento 28).
El espectro de 1H RMN del poli(IP-ran-GMA) se presenta en la Figura 48, donde se
realizó una comparación con el respectivo homopolímero (Experimento 28); en dicha figura se
muestra el copolímero correspondiente al Experimento 31 y la primera extensión de la cadena
polimérica (Experimento 32), donde se puede observar con ello el carácter viviente que ostenta
el sistema. Como se aprecia en la Figura 48b y c, en la región entre 2.5 a 4.5 ppm, están las
contribuciones más notorias del GMA en el copolímero, donde se observa que se encuentran
en 2.83 y 3.2 ppm los protones que se deben al epóxido y un grupo de multipletes en 4.38 y
3.89 ppm que son debidos a los protones diasterotópicos del metileno (-CH2-) que existe entre
el grupo éster y los protones epóxicos. Con respecto a la contribución de PIP, las señales en
4.7, 4.9 y 5.8 ppm se deben al protón de la insaturación de las diferentes microestructuras que
presenta el PIP. Los protones de los metilenos de la cadena principal dan señales en 2.0 - 2.2
ppm. El grupo metilo (–CH3) de la unidad del isopreno se encuentra en 1.7 y en 1. 6 ppm
mientras que el del GMA está en 1.3 ppm.
Longitud de onda (cm-1)

Capítulo 5/ (Co)polimerización del IP por NMRP 5.8 Análisis espectroscópico.
120
La composición de las microestructuras que conforman al PIP mostrado en la Figura 48
son: 3.1% para la microestructura correspondiente a la adición 1,2-, 6.5% para la adición 3,4-
33.4% para la adición 1,4- en conformación cis y 57.0% para la adición 1.4% en conformación
trans. Estos valores están muy cercanos a los reportado en la literatura (página 690 de la
referencia 23).
DPIPHRX10-2 1H EN CDCL3-1.ESP
7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 1.0 0.5
Chemical Shift (ppm)
a
b
c
1,2-
1,4-
1,2- + 3,4-
(ppm)
Figura 48: Espectro de 1H RMN del polímero de a) PIP (Experimento 28), b) y c) IP-GMA (Experimentos 31
y 32) mediada por 18 (TIPAL) en solución con tolueno (30 % w/w de sólidos) a 120 °C y 1170 kPa.
Como resumen, en el Esquema 13 (pp. 70) se muestran las unidades que conforman la
cadena polimérica de isopreno con sus respectivos desplazamientos químicos.

Capítulo 6/ (Co)polimerización del isopreno por RAFT 6.1 Cinéticas de IP/GMA en solución.
121
6.
Resultados y discusión. Homo y copolimerización del
isopreno por el proceso RAFT
6.1 Cinéticas de isopreno-metacrilato de glicidilo (IP-GMA) en solución
Siguiendo la dinámica llevada a cabo en el capítulo anterior, todas las condiciones
experimentales para la polimerización del isopreno por el proceso RAFT en solución con
tolueno se resumen en la Tabla 21. En esta sección, se describe la cinética involucrada en la
(co)polimerización del IP vía RAFT con una relación molar de CTA/I (I: iniciador) de 5.3 en
solución con tolueno. En dicha Tabla 21 se anexaron también los datos obtenidos por medio de
una polimerización por radicales libres convencionales, esto con el fin de ofrecer un marco de
referencia con respecto al proceso RAFT. La polimerización RAFT del isopreno fue llevada a
cabo con los agentes de control 19 y 20 (Esquema 22) a 125 °C usando TBP como iniciador de
radicales libres en solución con tolueno (30 % w/w en sólidos) con periodos de tiempo de
reacción de 46–75 h (excepto para los experimentos 40 y 41, donde se utilizaron las condiciones
a 65 °C con AIBN como iniciador de radicales libres en un tiempo de reacción de 96 h).
Esquema 22: Estructuras de los agentes RAFT; BCTC (19), CDSPA (20), ETSPE (21),102e DDTAL (22)102d y
CPDB (23).102e

Capítulo 6/ (Co)polimerización del isopreno por RAFT 6.1 Cinéticas de IP/GMA en solución.
122
Tabla 21. Resumen de la (co)polimerización del IP por RAFT. Experi-
mento
Monóme-
ro(s)a
RAFT
(CTA)b
Inicia-
dor (I)
CTA
/Ie
Tiempo
de
reacción
(h)
Conver-
sión
Mn, teof 𝑴𝒏
𝑮𝑷𝑪 g fRAFT
k
Ð k’x10-3
(h-1)j
36 IP 20 TBP 5.3 75 0.2632 9278 6432 0.69 1.20 4.18
37 IP/GMAd 20 TBP 5.3 46 0.1785 6283 4712 0.75 1.22 4.27
38 IP 19i TBP 5.3 75 0.1187 5947 3746 0.63 1.52 1.69
39 IP/GMAd 19i TBP 5.3 46 0.0858 3041 1932 0.64 1.53 1.95
40 IP/GMAd 20 AIBNh 5.3 96 0.2019 8267 6200 0.75 1.21 2.35
41 IP/GMAd 19i AIBNh 5.3 96 0.0982 5281 3324 0.64 1.52 1.08
42 IP c TBP ----- 96 0.6423 21400 15416 ----- 2.77 10.71
43 IP/GMAd c TBP ----- 96 0.6936 23100 16957 ----- 3.18 12.32
a Abreviaciones: AIBN, azobisisobutironitrilo; GMA, metacrilato de glicidilo; IP, isopreno; TBP, peróxido de tert-butilo;
Tol, tolueno. b Condiciones de reacción: 125 °C @ 1170 kPa, 30 ml IP en la cantidad requerida del disolvente (con el fin
de obtener un 30 % w/w de sólidos). Relación molar IP:CTA, 490:1 c Experimento llevado a cabo en condiciones de
polimerización por radicales libres convencionales. d Relación molar entre monómeros; 99.5:0.5 (para obtener un 1 % w/w
del GMA con respecto al IP). e Relación molar del agente RAFT/iniciador. f El Mn, teo se calculó a partir de la expresión Mn,
teo=conversión |M|/|RAFT| * PMM + PMN + PMI; donde |M|: concentración del monómero (mol); |RAFT|: concentración
del agente RAFT (mol); PMM, PMRAFT, PMI: peso molecular del monómero, agente RAFT (g/mol) e iniciador
respectivamente. g El 𝑀𝑛𝐺𝑃𝐶 se determinó por GPC, calibrado con estándares de PS y convertidos a PIP usando las
constantes de MHS de la referencia104 h Polimerización llevada a cabo a 65 °C @ 1170 kPa . i Para llevar a cabo la
polimerización, a la mezcla de reacción se agregó DMSO para poder disolver al BCTC (dado que éste es insoluble en
tolueno y en IP), usando una relación de 5% w/w con respecto al CTA, Dado que la cantidad de DMSO que se usa en los
experimentos es muy pequeña, se puede considerar en términos prácticos que es muy similar a una polimerización en
solución y por lo tanto, se estará refiriendo a las reacciones como experimentos en solución de tolueno. j Constante de
rapidez aparente; k’= -ln(1-conversión)/t, t:tiempo de reacción. k Eficiencia del agente RAFT; fRAFT=Mn, exp/Mn, teo
El proceso de la (co)polimerización del isopreno sigue la ruta sintética que se exhibe en
el Esquema 23. La macroalcoxiamina resultante se obtiene calentando al monómero isopreno
o, en su defecto, la mezcla de monómeros IP/GMA, en presencia de un iniciador de radicales
libres y el agente RAFT, en solución con tolueno (30 % w/w de sólidos).
Esquema 23: Formación del (co)polímero de isopreno con grupos RAFT.

Capítulo 6/ (Co)polimerización del isopreno por RAFT 6.1 Cinéticas de IP/GMA en solución.
123
Observando los datos que arroja la Tabla 21, se tiene que la especie 19 presenta una mayor
dispersidad en la polimerización con respecto a 20, esto puede ser debido a que este agente de
transferencia posee una constante de transferencia más baja que 20,102e (ver Esquema 8). Es
importante recordar que el equilibrio dinámico que existe entre las especies propagantes
(especies vivientes) y las especies funcionalizadas con el agente RAFT (especies durmientes),
deben producir un crecimiento uniforme de las cadenas involucradas; es por ello que, la
selección de los grupos Z y R en el agente RAFT es crucial y punto clave para tener éxito en la
polimerización por medio de esta técnica, ya que esta combinación es la que determinara la
constante de transferencia que ostentara dicho agente de control tipo RAFT.
Dicho lo anterior y analizando 19, se observa que es una molécula simétrica, lo que hace
pensar que al presentar dos grupos salientes idénticos ofrecerá dos rutas viables para la
fragmentación del aducto intermediario y la partición del aducto radical (7 ó 9 Esquema 8)
entre los materiales iniciantes y los productos, los cuales estarían determinados por la capacidad
relativa del grupo saliente, tanto el grupo R (como etapa inicial) y el radical poliisopreno (como
radical propagante). Sin embargo, los factores estéricos y polares influyen para que Ctr sea más
baja que 20, y este último, es el que llega a ser más efectivo en la polimerización del isopreno
bajo las condiciones experimentales presentadas en la Tabla 21.
Por otra parte, el efecto del grupo Z es de consideración dentro de la funcionalidad del
agente RAFT, ya que tiene una influencia en el grupo ditiocarbonilo y en la rapidez de adición
del radical propagante al doble enlace C=S. Diversos factores deben ser considerados para la
determinación de la rapidez de adición del radical en los compuestos ditiocarbonilos, la
capacidad del grupo Z para estabilizar un radical adyacente es crucial para el proceso de la
polimerización. Es por ello que la utilización de herramientas de cómputo como son los cálculos
semiempíricos, han demostrado ser un apoyo crucial en este tipo de estudios con el fin de
entender la relación que existe en estructura y reactividad para este tipo de reacciones de
polimerización.

Capítulo 6/ (Co)polimerización del IP por RAFT 6.2 Cálculos semiempíricos (AM1-RHF).
124
6.2 Cálculos semiempíricos (AM1-RHF)
Tal y como se hizo con los nitróxidos presentados en el capítulo anterior, se procedió a
realizar cálculos por el método de cálculo semiempírico AM1-RHF, con el fin de elucidar la
relación que existe entre la estructura del agente RAFT y su reactividad frente a la
polimerización del isopreno. Los grupos R y Z que se proponen a analizar son los siguientes
(además de los agentes RAFT incluidos en el Esquema 22):
Con el fin de observar una dimensión cualitativa del efecto que existe en los grupos R y
Z hacia la reactividad del C=S en la adición del radical (y con ello, tener una idea de la magnitud
de la Ctr), se procedió a calcular los calores de reacción (∆Hr) para la adición de varios radicales
libres al doble enlace C=S, las energías LUMO y HOMO de estos agentes de control, además
las entalpias de formación (AHf), las cargas atómicas y la distancia del doble enlace C=S. Las
optimizaciones de la geometría para el agente RAFT y su correspondiente aducto, se realizaron
con el método de cálculo AM1-RHF. Estos datos se presentan en la Tabla 22. Se ha reportado
que la constante de rapidez para la adición de un radical dado llega a ser más alta para
compuestos que poseen la energía LUMO más baja.66b Dentro de esta serie de cálculos, también
se añadieron tres agentes RAFT que fueron reportados por otros investigadores para efectuar la
polimerización del isopreno, obteniéndose aceptables resultados con el uso de 22.
Estos cálculos semiempíricos, confirman la influencia que tienen los grupos Z y R en el
agente RAFT. La participación del grupo Z se puede observar en la Tabla 22 donde la tendencia
de la serie involucrada en los valores de la energía LUMO es hacia los radicales estirenilo e

Capítulo 6/ (Co)polimerización del IP por RAFT 6.2 Cálculos semiempíricos (AM1-RHF).
125
isoprenilo, ya que éstos presentan los índices más altos en cada serie, esto se debe a que ambos
radicales libres son estabilizados por efectos de resonancia y estérico; mientras que, el radical
CN1 mantiene la energía LUMO más baja (dado que sólo es estabilizado por el efecto inductivo
que presenta el grupo ciano).
Tabla 22: Propiedades predichas de agentes RAFT por cálculos semiempíricos (AM1-RHF) en
orden decreciente de la energía de LUMO para cada bloque del grupo Z.e
AHf (kJ/mol) ∆Hra Energía (eV) Carga atómica =S
Especie b Aductob
Agente
RAFT (kJ/mol) HOMO LUMO =S -S (Å)
23 c (BzS-CN3) -54.78 311.15 365.93 -8.718 -1.271 0.007 0.254 1.540
21c -687.80 -339.53 348.27 -8.768 -1.288 0.078 0.279 1.523
22d -1010.10 -635.59 374.52 -8.939 -1.416 0.014 0.271 1.535
20 (TS-CN4) -916.21 -540.05 376.16 -9.007 -1.458 0.044 0.257 1.530
19 (AcS-Ac) -1045.46 -679.06 366.41 -9.344 -1.692 0.077 0.324 1.527
BzS-IP -167.27 193.53 360.80 -8.632 -1.138 -0.044 0.270 1.550
BzS-S -70.21 286.85 357.06 -8.670 -1.144 -0.050 0.262 1.550
BzS-CN4 -459.61 -88.93 370.68 -8.822 -1.371 0.011 0.260 1.540
BzS-Ac -539.31 -176.84 362.47 -8.987 -1.404 0.008 0.304 1.541
BzS-CN2 -43.17 310.81 353.98 -9.017 -1.457 -0.011 0.279 1.545
BzS-CN1 -33.36 327.84 361.20 -9.069 -1.494 -0.006 0.294 1.544
TS-IP -607.52 -257.60 349.92 -8.773 -1.089 -0.055 0.266 1.550
TS-S -522.98 -164.08 358.90 -8.827 -1.101 -0.059 0.254 1.550
TS-CN3 -510.07 -142.60 367.47 -8.880 -1.363 0.037 0.241 1.531
TS-Ac -1077.18 -627.65 449.53 -9.106 -1.397 0.012 0.294 1.538
TS-CN2 -496.46 -139.64 356.82 -9.151 -1.440 -0.020 0.270 1.544
TS-CN1 -481.75 -121.99 359.75 -9.202 -1.472 -0.021 0.282 1.545
EtS-S -528.20 131.32 659.52 -8.676 -1.120 -0.032 0.242 1.542
EtS-IP -670.03 -180.30 489.73 -8.999 -1.386 0.046 0.251 1.535
EtS-CN2 -218.28 156.23 374.51 -8.926 -1.415 0.038 0.248 1.531
EtS-Ac -709.57 -339.17 370.40 -9.131 -1.434 0.009 0.302 1.539
EtS-CN3 -225.15 149.09 374.24 -8.993 -1.453 -0.005 0.262 1.539
EtS-CN1 -204.82 165.78 370.61 -9.206 -1.476 -0.023 0.283 1.545
EtS-CN4 -615.93 -251.76 364.17 -9.087 -1.561 0.005 0.268 1.537
AcS-S -572.55 -206.91 365.65 -8.775 -1.195 -0.013 0.265 1.544
AcS-IP -670.03 -180.30 489.73 -8.999 -1.386 0.046 0.251 1.535
AcS-CN3 -552.75 -190.16 362.59 -9.020 -1.406 0.044 0.258 1.353
AcS-CN2 -553.79 -180.17 373.62 -9.076 -1.463 0.052 0.269 1.534
AcS-CN4 -948.44 -589.77 358.67 -9.128 -1.511 0.050 0.266 1.534
AcS-CN1 -534.58 -171.53 363.05 -9.379 -1.622 0.040 0.305 1.536
a Calor de reacción relativo = ∆Hf (6) – ∆Hf (7) (ver Esquema 8). b Pn se
consideró con un DP=5, iniciado con TBP c Agente RAFT utilizado en la
referencia.102e d Agente RAFT utilizado en la referencia.102d e Hyperchem 5.01
para Windows. Molecular Model System. Hypercube, Inc. Gainesville, Florida.
El comportamiento del grupo Z en el carácter del doble enlace C=S se puede apreciar al
realizar una gráfica de energía LUMO vs AHr (Figura 49). Para el caso específico del agente
RAFT 20, éste ostenta el mayor calor de reacción, pero su energía LUMO se encuentra
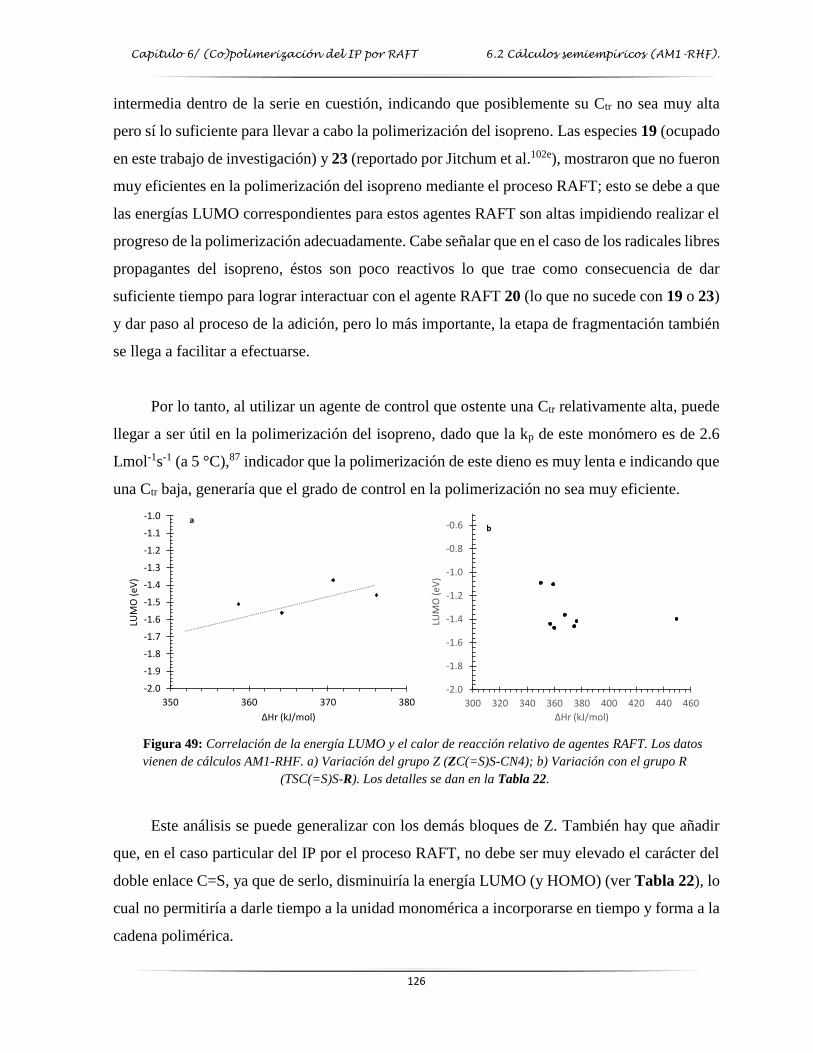
Capítulo 6/ (Co)polimerización del IP por RAFT 6.2 Cálculos semiempíricos (AM1-RHF).
126
intermedia dentro de la serie en cuestión, indicando que posiblemente su Ctr no sea muy alta
pero sí lo suficiente para llevar a cabo la polimerización del isopreno. Las especies 19 (ocupado
en este trabajo de investigación) y 23 (reportado por Jitchum et al.102e), mostraron que no fueron
muy eficientes en la polimerización del isopreno mediante el proceso RAFT; esto se debe a que
las energías LUMO correspondientes para estos agentes RAFT son altas impidiendo realizar el
progreso de la polimerización adecuadamente. Cabe señalar que en el caso de los radicales libres
propagantes del isopreno, éstos son poco reactivos lo que trae como consecuencia de dar
suficiente tiempo para lograr interactuar con el agente RAFT 20 (lo que no sucede con 19 o 23)
y dar paso al proceso de la adición, pero lo más importante, la etapa de fragmentación también
se llega a facilitar a efectuarse.
Por lo tanto, al utilizar un agente de control que ostente una Ctr relativamente alta, puede
llegar a ser útil en la polimerización del isopreno, dado que la kp de este monómero es de 2.6
Lmol-1s-1 (a 5 °C),87 indicador que la polimerización de este dieno es muy lenta e indicando que
una Ctr baja, generaría que el grado de control en la polimerización no sea muy eficiente.
Figura 49: Correlación de la energía LUMO y el calor de reacción relativo de agentes RAFT. Los datos
vienen de cálculos AM1-RHF. a) Variación del grupo Z (ZC(=S)S-CN4); b) Variación con el grupo R
(TSC(=S)S-R). Los detalles se dan en la Tabla 22.
Este análisis se puede generalizar con los demás bloques de Z. También hay que añadir
que, en el caso particular del IP por el proceso RAFT, no debe ser muy elevado el carácter del
doble enlace C=S, ya que de serlo, disminuiría la energía LUMO (y HOMO) (ver Tabla 22), lo
cual no permitiría a darle tiempo a la unidad monomérica a incorporarse en tiempo y forma a la
cadena polimérica.
-2.0
-1.9
-1.8
-1.7
-1.6
-1.5
-1.4
-1.3
-1.2
-1.1
-1.0
350 360 370 380
LUM
O (
eV)
∆Hr (kJ/mol)
a
-2.0
-1.8
-1.6
-1.4
-1.2
-1.0
-0.8
-0.6
300 320 340 360 380 400 420 440 460
LUM
O (
eV)
∆Hr (kJ/mol)
b
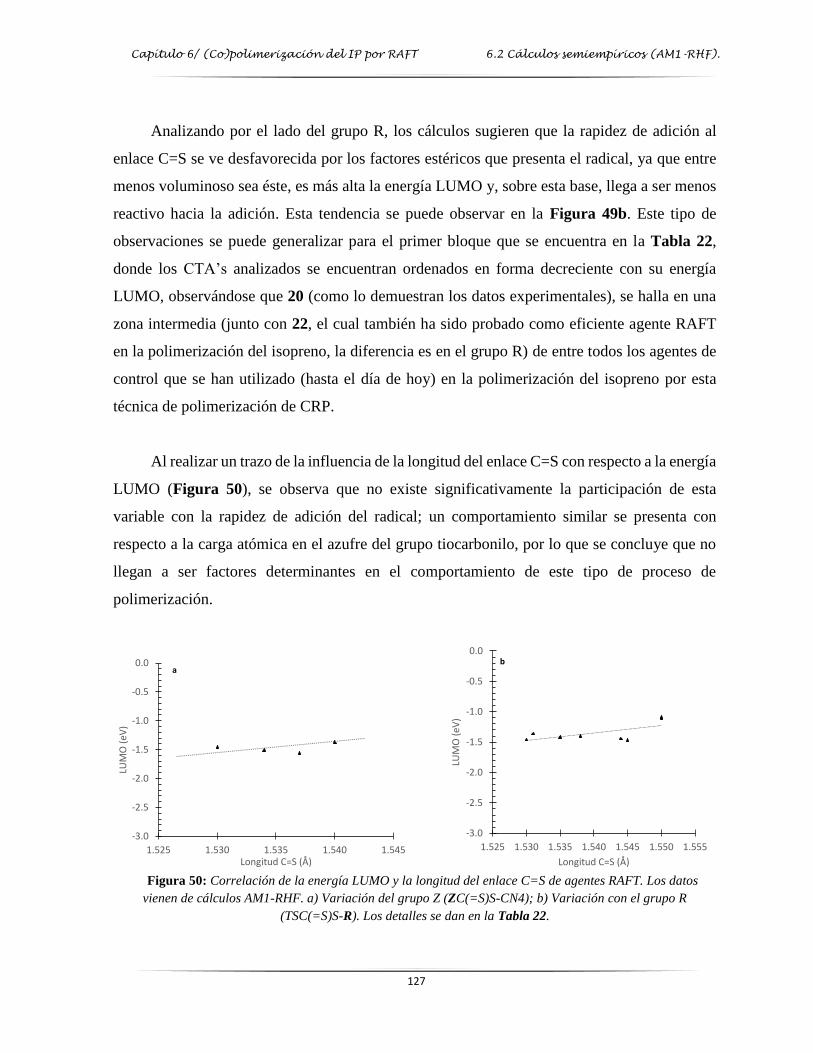
Capítulo 6/ (Co)polimerización del IP por RAFT 6.2 Cálculos semiempíricos (AM1-RHF).
127
Analizando por el lado del grupo R, los cálculos sugieren que la rapidez de adición al
enlace C=S se ve desfavorecida por los factores estéricos que presenta el radical, ya que entre
menos voluminoso sea éste, es más alta la energía LUMO y, sobre esta base, llega a ser menos
reactivo hacia la adición. Esta tendencia se puede observar en la Figura 49b. Este tipo de
observaciones se puede generalizar para el primer bloque que se encuentra en la Tabla 22,
donde los CTA’s analizados se encuentran ordenados en forma decreciente con su energía
LUMO, observándose que 20 (como lo demuestran los datos experimentales), se halla en una
zona intermedia (junto con 22, el cual también ha sido probado como eficiente agente RAFT
en la polimerización del isopreno, la diferencia es en el grupo R) de entre todos los agentes de
control que se han utilizado (hasta el día de hoy) en la polimerización del isopreno por esta
técnica de polimerización de CRP.
Al realizar un trazo de la influencia de la longitud del enlace C=S con respecto a la energía
LUMO (Figura 50), se observa que no existe significativamente la participación de esta
variable con la rapidez de adición del radical; un comportamiento similar se presenta con
respecto a la carga atómica en el azufre del grupo tiocarbonilo, por lo que se concluye que no
llegan a ser factores determinantes en el comportamiento de este tipo de proceso de
polimerización.
Figura 50: Correlación de la energía LUMO y la longitud del enlace C=S de agentes RAFT. Los datos
vienen de cálculos AM1-RHF. a) Variación del grupo Z (ZC(=S)S-CN4); b) Variación con el grupo R
(TSC(=S)S-R). Los detalles se dan en la Tabla 22.
-3.0
-2.5
-2.0
-1.5
-1.0
-0.5
0.0
1.525 1.530 1.535 1.540 1.545
LUM
O (
eV)
Longitud C=S (Å)
a
-3.0
-2.5
-2.0
-1.5
-1.0
-0.5
0.0
1.525 1.530 1.535 1.540 1.545 1.550 1.555
LUM
O (
eV)
Longitud C=S (Å)
b

Capítulo 6/ (Co)polimerización del IP por RAFT 6.2 Cálculos semiempíricos (AM1-RHF).
128
6.3 Reacciones de homopolimerización del isopreno
Cuando se realizan las reacciones de homopolimerización del isopreno, los mejores
resultados se obtienen (en términos de la evolución de la conversión, velocidad de
polimerización y en la distribución de los pesos moleculares) cuando se utiliza el agente RAFT
20. Las Figuras 51 y 52 muestran los perfiles comparativos de conversión vs tiempo, así como
de Mn y dispersidad (Ð) vs conversión, respectivamente a 125 °C y 1170 kPa utilizando los
agentes 19 y 20. La Figura 51 indica que la cinética obedece a un comportamiento de primer
orden con respecto al monómero debido a que presenta una aparente progresión lineal de la
conversión con el tiempo cuando la polimerización es llevada a cabo con 20 mientras que 19
muestra un comportamiento errático durante el proceso; además, existe un incremento lineal del
peso molecular en número (Mn) vs conversión (Figura 52) indicando que el polímero está
siendo mejor controlado por 20.
Figura 51: Perfil de la cinética de la polimerización del IP iniciado con 19 (con relleno, Exp 38) y con 20 (sin
relleno, Exp 36) a 125 °C y 1170 kPa en solución con tolueno (30 % w/w de sólidos).
La naturaleza del control de la polimerización del isopreno fue evidenciada por la
dispersidad decreciente del polímero conforme aumenta Mn (Figura 52). Cabe mencionar que
la polimerización del IP llevada a cabo por el agente RAFT 20, procede con una velocidad
mayor que su contraparte 19, alcanzando una dispersidad final de 1.20. La eficiencia final del
agente de transferencia 20 alcanzó un valor del 69%.
0
5
10
15
20
25
30
0 20 40 60 80
Co
nve
rsió
n (
%)
Tiempo (h)
Exp 36
Exp 38
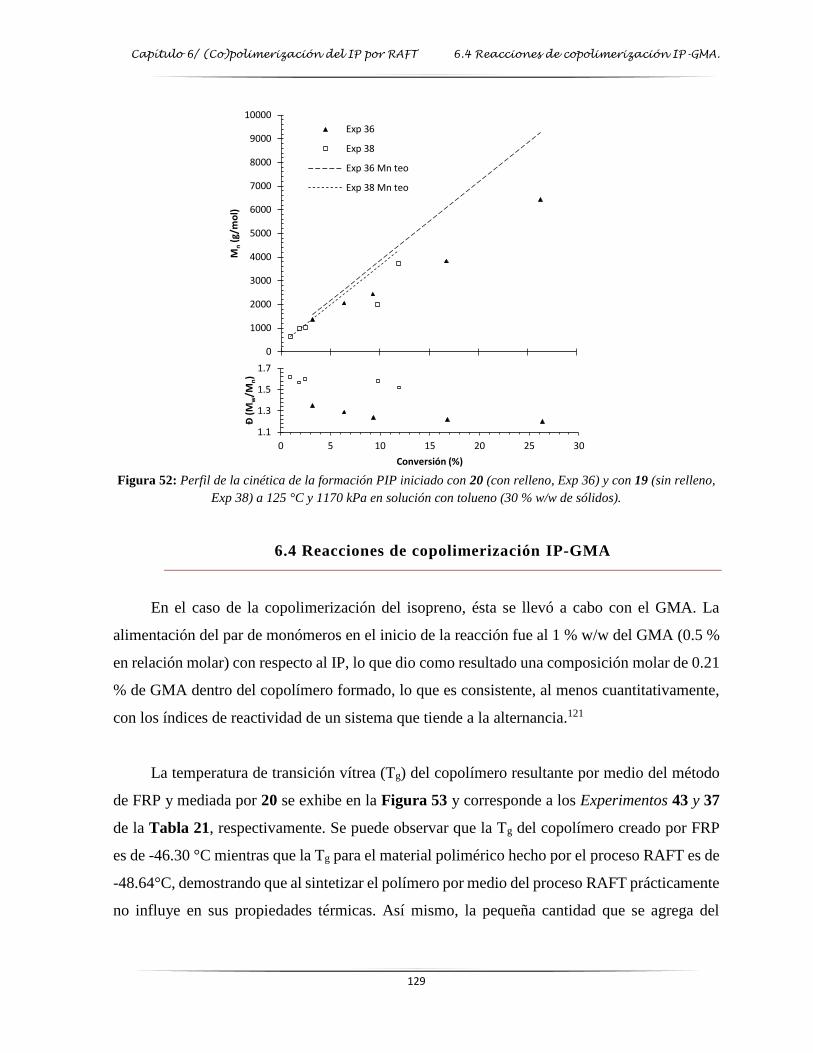
Capítulo 6/ (Co)polimerización del IP por RAFT 6.4 Reacciones de copolimerización IP-GMA.
129
Figura 52: Perfil de la cinética de la formación PIP iniciado con 20 (con relleno, Exp 36) y con 19 (sin relleno,
Exp 38) a 125 °C y 1170 kPa en solución con tolueno (30 % w/w de sólidos).
6.4 Reacciones de copolimerización IP-GMA
En el caso de la copolimerización del isopreno, ésta se llevó a cabo con el GMA. La
alimentación del par de monómeros en el inicio de la reacción fue al 1 % w/w del GMA (0.5 %
en relación molar) con respecto al IP, lo que dio como resultado una composición molar de 0.21
% de GMA dentro del copolímero formado, lo que es consistente, al menos cuantitativamente,
con los índices de reactividad de un sistema que tiende a la alternancia.121
La temperatura de transición vítrea (Tg) del copolímero resultante por medio del método
de FRP y mediada por 20 se exhibe en la Figura 53 y corresponde a los Experimentos 43 y 37
de la Tabla 21, respectivamente. Se puede observar que la Tg del copolímero creado por FRP
es de -46.30 °C mientras que la Tg para el material polimérico hecho por el proceso RAFT es de
-48.64°C, demostrando que al sintetizar el polímero por medio del proceso RAFT prácticamente
no influye en sus propiedades térmicas. Así mismo, la pequeña cantidad que se agrega del
0
1000
2000
3000
4000
5000
6000
7000
8000
9000
10000
Mn
(g/m
ol)
Exp 36
Exp 38
Exp 36 Mn teo
Exp 38 Mn teo
1.1
1.3
1.5
1.7
0 5 10 15 20 25 30
Ɖ (
Mw
/Mn)
Conversión (%)
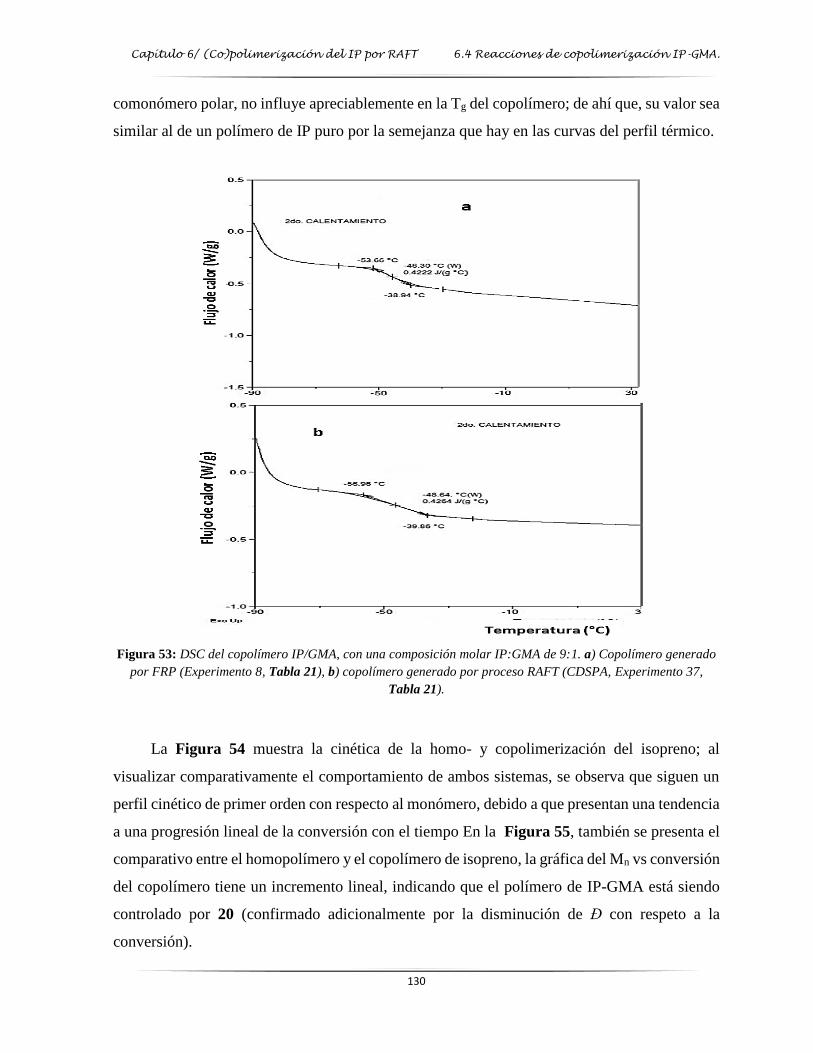
Capítulo 6/ (Co)polimerización del IP por RAFT 6.4 Reacciones de copolimerización IP-GMA.
130
comonómero polar, no influye apreciablemente en la Tg del copolímero; de ahí que, su valor sea
similar al de un polímero de IP puro por la semejanza que hay en las curvas del perfil térmico.
Figura 53: DSC del copolímero IP/GMA, con una composición molar IP:GMA de 9:1. a) Copolímero generado
por FRP (Experimento 8, Tabla 21), b) copolímero generado por proceso RAFT (CDSPA, Experimento 37,
Tabla 21).
La Figura 54 muestra la cinética de la homo- y copolimerización del isopreno; al
visualizar comparativamente el comportamiento de ambos sistemas, se observa que siguen un
perfil cinético de primer orden con respecto al monómero, debido a que presentan una tendencia
a una progresión lineal de la conversión con el tiempo En la Figura 55, también se presenta el
comparativo entre el homopolímero y el copolímero de isopreno, la gráfica del Mn vs conversión
del copolímero tiene un incremento lineal, indicando que el polímero de IP-GMA está siendo
controlado por 20 (confirmado adicionalmente por la disminución de Đ con respeto a la
conversión).

Capítulo 6/ (Co)polimerización del IP por RAFT 6.4 Reacciones de copolimerización IP-GMA.
131
Figura 54 Perfil de la cinética de la formación homopolímero de IP (con relleno, Exp 36) y el correspondiente
copolímero, IP-GMA (sin relleno, Exp 37), iniciado con el CTA 20 (CDSPA) a 125 °C y 1170 kPa en solución
con tolueno (30 % w/w de sólidos).
Figura 55: Progreso de Mn y Đ vs conversión en la formación del homopolímero de IP (con relleno, Exp 36) y el
correspondiente copolímero, IP-GMA (sin relleno, Exp 37), iniciado con el CTA 20 (CDSPA) a 125 °C y 1170
kPa en solución con tolueno (30 % w/w de sólidos).
En la Figura 56, el efectuar la comparación de las distribuciones de los pesos moleculares
del homopolímero y del copolímero por el CTA 20 y por FRP, se muestran claramente las
diferencias marcadas por ambas técnicas de polimerización, donde se observa que existe un
0
5
10
15
20
25
30
0 20 40 60 80
Co
nve
rsió
n (
%)
Tiempo (h)
Exp 36 Exp 37
0
2000
4000
6000
8000
10000
Mn
(g/
mo
l)
Exp 36
Exp 37
Mn teo Exp 36
Mn teo 37
1.1
1.3
1.5
0 5 10 15 20 25 30
Ɖ (
Mn
/Mw
)
Conversión (%)

Capítulo 6/ (Co)polimerización del IP por RAFT 6.5 Crecimiento de la cadena polimérica
(carácter viviente del proceso RAFT).
132
control en la polimerización por el agente de control bajo las condiciones ya especificadas
anteriormente en los Experimentos 36 y 37. Los experimentos no controlados (42 y 43)
presentan una cola en bajos pesos moleculares, una distribución más abierta y, en general, pesos
moleculares más altos, consistentes con un mecanismo convencional de radicales libres y
posiblemente con reacciones de transferencia al polímero.
Figura 56: Cromatogramas de GPC del homopolímero y copolímero del IP por RAFT (Exp 36 y 37) y por FRP
(Exp 42 y 43). Exp 37: Experimento 37, Exp 38: Experimento 38, Exp 42: Experimento 42, Exp 43: Experimento
43. Ver Tabla 21
6.5 Crecimiento de la cadena polimérica (carácter viviente del proceso
RAFT)
Dado que el agente de control 20 demostró dar una aceptable eficiencia en la ejecución de
la (co)polimerización del isopreno mediante el proceso RAFT, y aunado con la técnica
experimental utilizada en este trabajo de investigación en la extensión de la cadena polimérica
mediante nitróxidos (sección 5.7, página 79), se utilizó este agente RAFT para generar la
extensión de la cadena polimérica y con ello demostrar el carácter viviente de esta técnica de
polimerización. Las condiciones experimentales de las que se partió fueron con una relación
molar M/CTA de 1000, con un tiempo de reacción de 24 h a 125 °C y 1170 kPa, usando como
iniciador de radicales al peróxido de t-butilo (TBP). Este último compuesto orgánico fue
seleccionado por su prolongado tiempo de vida (10 h a 125 °C) y por la simetría que presenta
2.5 3 3.5 4 4.5 5 5.5
Log MW (g/mol)
Exp 37
Exp 36
Exp 43
Exp 42

Capítulo 6/ (Co)polimerización del IP por RAFT 6.5 Crecimiento de la cadena polimérica
(carácter viviente del proceso RAFT).
133
como peróxido; la relación molar entre el CDSPA (20) y el iniciador, CTA/I, fue de 4. La
reacción de polimerización se efectuó en una solución con tolueno a 30 % w/w de sólidos. Con
el fin de reducir las reacciones potenciales de terminación y de entrecruzamiento, las
polimerizaciones de extensión de cadena se realizaron hasta un máximo de 65 % de conversión.
Todas las condiciones experimentales se reúnen en la Tabla 23.
Tabla 23. Resumen de la (co)polimerización del IP por RAFT. Crecimiento de cadena.
Experimento Monóme-
ro(s) a CTA a, b M/CTA d
Conver-
sión Mn, teo
e 𝑴𝒏𝑹𝑴𝑵 f 𝑴𝒏
𝑮𝑷𝑪 g fRAFT k Ð
k’x10-3
(h-1) h
44 IP/GMAc CDSPA 1000 0.5753 39600 29700 29300 0.75 1.25 35.68
45 IP Exp 44i 1200 0.6378 82500 68800 68400 0.83 1.31 42.32
46 IP Exp 45j 1200 0.6287 120900 109600 107850 0.91 1.34 41.28
47 IP CDSPA 1000 0.5061 68200 61500 61150 0.90 1.22 29.39
a Abreviaciones: CDSPA: ácido 4-ciano-4-[(dodecilsulfaniltiocarbonil) sulfanil] pentanoico; GMA: metacrilato de
glicidilo; IP, isopreno; TBP: Peróxido de tert-butilo. b Condiciones de reacción: 125 °C @ 160 psig con 24 h como tiempo
de reacción en 30 ml IP con 60 mL de tolueno (con el fin de obtener un 30 % w/w de sólidos). Relación molar CTA/I de
4. c Relación molar entre monómeros; 90:10 (para obtener un 10 % w/w del GMA con respecto al IP). d Relación molar
del agente RAFT/iniciador. e Mn, teo se determinó a partir de Mn, teo = conversión*|M|/|CTA|*PMM+PMCTA+PMI; donde
|M|: concentración del monómero (mol); |CTA|: concentración del agente de transferencia (mol); PMM, PMCTA, PMI:
peso molecular del monómero, CDSPA e iniciador (g/mol) respectivamente. f Se determinó por RMN.122 g 𝑀𝑛𝐺𝑃𝐶 se
determinó por GPC, calibrado con estándares de PS y convertidos a PIP usando las constantes de MHS según la
referencia104 h Constante de rapidez aparente; k’= -ln(1-conversión)/t, t:tiempo de reacción. i Macroagente de control
obtenido en el experimento 44. j Macroagente de control obtenido en el experimento 45. k Eficiencia del agente RAFT;
fRAFT=Mn, exp/Mn, teo
Se puede apreciar en la Tabla 23 que se anexaron los datos obtenidos de
homopolimerización (Experimento 47) con el fin de tener un marco de referencia. En el caso de
la copolimerización, se tiene el uso de macroagentes de control (experimentos 45 y 46) obtenidos
para la extensión de la cadena polimérica. Así mismo, se incorporó en dicha Tabla 23 la rapidez
aparente k’ que tiene cada experimento, esto con el fin de tener una idea cuantitativa acerca de
la velocidad de polimerización para cada experimento. Se puede apreciar que la influencia del
comonómero GMA en la polimerización del isopreno (experimento 44), logra incrementar
discretamente, en un 21%, la velocidad de polimerización cuando se compara con su respetiva
homopolimerización (experimento 47).
Inicialmente, se realizó la cinética de la homopolimerización del isopreno y la Figura 57
muestra los correspondientes perfiles de conversión vs tiempo, Mn y Đ vs conversión
respectivamente, con una relación molar de M/CTA de 1000. Al comparar los perfiles cinéticos
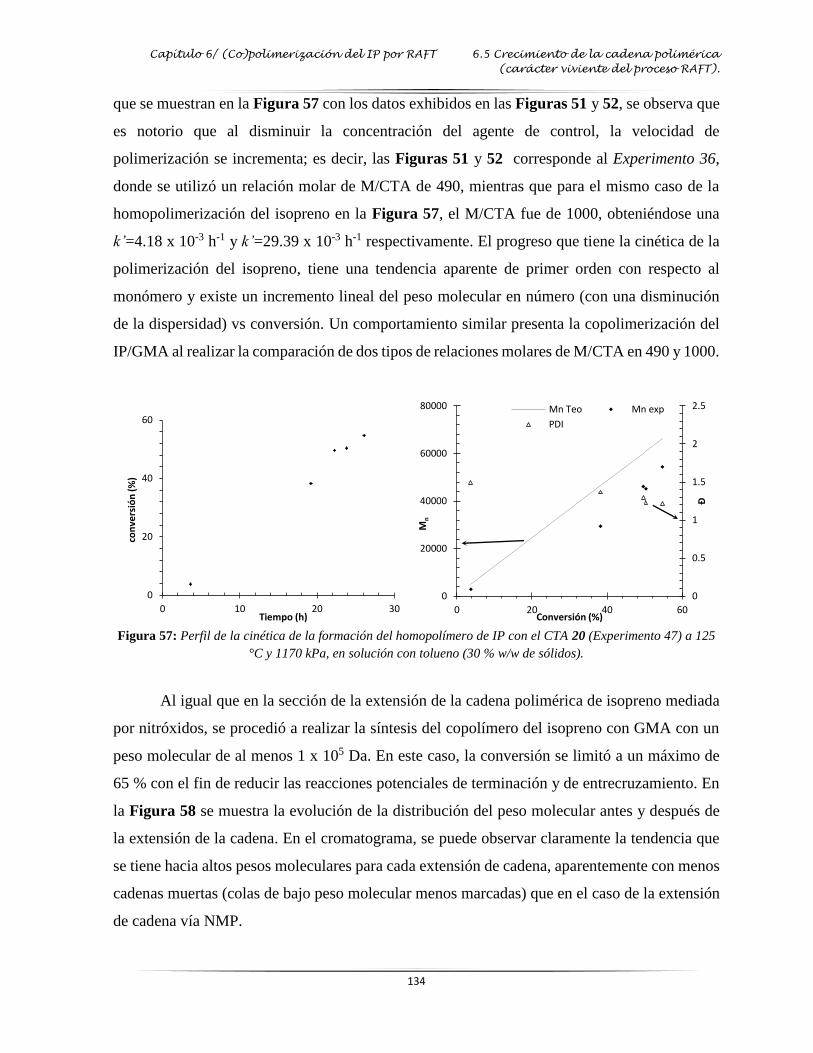
Capítulo 6/ (Co)polimerización del IP por RAFT 6.5 Crecimiento de la cadena polimérica
(carácter viviente del proceso RAFT).
134
que se muestran en la Figura 57 con los datos exhibidos en las Figuras 51 y 52, se observa que
es notorio que al disminuir la concentración del agente de control, la velocidad de
polimerización se incrementa; es decir, las Figuras 51 y 52 corresponde al Experimento 36,
donde se utilizó un relación molar de M/CTA de 490, mientras que para el mismo caso de la
homopolimerización del isopreno en la Figura 57, el M/CTA fue de 1000, obteniéndose una
k’=4.18 x 10-3 h-1 y k’=29.39 x 10-3 h-1 respectivamente. El progreso que tiene la cinética de la
polimerización del isopreno, tiene una tendencia aparente de primer orden con respecto al
monómero y existe un incremento lineal del peso molecular en número (con una disminución
de la dispersidad) vs conversión. Un comportamiento similar presenta la copolimerización del
IP/GMA al realizar la comparación de dos tipos de relaciones molares de M/CTA en 490 y 1000.
Figura 57: Perfil de la cinética de la formación del homopolímero de IP con el CTA 20 (Experimento 47) a 125
°C y 1170 kPa, en solución con tolueno (30 % w/w de sólidos).
Al igual que en la sección de la extensión de la cadena polimérica de isopreno mediada
por nitróxidos, se procedió a realizar la síntesis del copolímero del isopreno con GMA con un
peso molecular de al menos 1 x 105 Da. En este caso, la conversión se limitó a un máximo de
65 % con el fin de reducir las reacciones potenciales de terminación y de entrecruzamiento. En
la Figura 58 se muestra la evolución de la distribución del peso molecular antes y después de
la extensión de la cadena. En el cromatograma, se puede observar claramente la tendencia que
se tiene hacia altos pesos moleculares para cada extensión de cadena, aparentemente con menos
cadenas muertas (colas de bajo peso molecular menos marcadas) que en el caso de la extensión
de cadena vía NMP.
0
20
40
60
0 10 20 30
con
vers
ión
(%
)
Tiempo (h)
0
0.5
1
1.5
2
2.5
0
20000
40000
60000
80000
0 20 40 60
Ð
Mn
Conversión (%)
Mn Teo Mn exp
PDI

Capítulo 6/ (Co)polimerización del IP por RAFT 6.6 Análisis espectroscópico.
135
Figura 58: Cromatogramas de GPC antes y después de la extensión de la cadena del copolímero de isopreno
con GMA (experimentos 44-46). Ver Tabla 23.
6.6 Análisis espectroscópico
El espectro de IR del (co)polímero de isopreno obtenido por el proceso RAFT, se analizó
cualitativamente y sus bandas características se pueden observar en la Figura 59. El copolímero
presenta las mismas bandas de absorción características que muestran las unidades
homopoliméricas. La asignación de las bandas para las unidades del isopreno y del GMA se
describió en la sección correspondiente a la copolimerización mediada por nitróxidos, por lo
que nos enfocaremos a las bandas del agente de control. 73
781
284
2
887
909
1002
1033
1069
1151
1376
1448
1639
1664
1735
2043
2725
2853
2918
2923
.59
2961
3032
3449
1000 2000 3000 4000
Longitud de onda (cm-1)
OH -
NC -
s c
s c
Figura 59: Espectro de FT-IR del copolímero del IP/GMA generado por el CTA 20 (Experimento 44).
3 3.5 4 4.5 5 5.5 6
Log Mw (g/mol)
1er bloque
1era extension
2da extensión

Capítulo 6/ (Co)polimerización del IP por RAFT 6.6 Análisis espectroscópico.
136
La banda en 1069 cm-1 se debe a la vibración del grupo tiocarbonilo (C=S) y aquélla en
la región de 812 cm-1 a la unión C-S, adicionalmente, el HO- del ácido carboxílico se presenta
como una banda ancha en 3449 cm-1 y el grupo ciano está en 2043 cm-1, lo que confirma que el
material polimérico está funcionalizado con la porción tritioéster en la cadena principal.
Similarmente al análisis efectuado a partir del espectro de FT-IR, el espectro de 1H RMN
del poli(IP-ran-GMA) se presenta en la Figura 60, y solo se discutirán las señales del agente
de control que está unido a la cadena polimérica, aclarando que la relación molar de isopreno-
GMA en el copolímero en la segunda extensión es de 98:2. Las unidades del agente de control
son las siguientes; por el lado del grupo Z, la señal del metileno (CH2) en la posición α con
respecto al tritiocarbonilo en la cadena hidrocarbonada se encuentra en 3.31 ppm, mientras que
el resto de las metilenos están entre 1.37 a 1.14 ppm; el grupo metilo (CH3-), que está en la
posición ω se encuentra en 0.90 ppm.
Para el caso del grupo R que originalmente ostentaba el agente de control, se debe
observar el grupo metilo en 1.85 ppm, pero este se encuentra traslapado con las señales del
copolímero, mientas que los metilenos que están en la posición α y β al grupo ácido carboxílico
están en 2.84 y 2.65 ppm respectivamente. (Ver Figura 61 como referencia de las señales para
el CDSPA).
DPIPRX-31 1H EN CDCL3-1.JDF
5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 1.0Chemical Shift (ppm) (ppm)
Figura 60: Espectro de 1H RMN FT-IR del copolímero del IP/GMA generado por el CTA 20 (Experimento 44).

Capítulo 6/ (Co)polimerización del IP por RAFT 6.6 Análisis espectroscópico.
137
Figura 61: Espectro de 1H RMN del CDSPA (20).
Adicionalmente, a manera de resumen, en el Esquema 13 (página 70) se muestran las
unidades que conforman la cadena polimérica de isopreno con sus respectivos desplazamientos
químicos.
δ (ppm)

Capítulo 7/ Copolimerización de injerto del IP por NMRP 7.1 Copolimerización de injerto del IP.
138
7.
Resultados y discusión. Copolimerización de injerto
del isopreno mediada por nitróxidos
7.1 Copolimerización de injerto del isopreno
En este capítulo se estudia la funcionalización del poliisopreno (PIP) en presencia de los
radicales nitróxido HO-TEMPO (13) y TIPNO (14); tal y como se mencionó en el apartado de
Antecedentes y de acuerdo a la experiencia con la que cuenta nuestro grupo de trabajo,38f, 76 se
utilizó el método de injerto desde para llevar a cabo el proceso de la copolimerización,
manejando el TBP como iniciador de radicales con una relación molar de 2.7 (se mantuvo un
exceso de nitróxido con el fin de asegurar el proceso de injerto en la cadena polimérica del
elastómero) y utilizando poliisopreno como sustrato a funcionalizar (tanto en una conformación
cis como en otra en trans). Las condiciones experimentales se resumen en la Tabla 24.
El proceso de funcionalización se dio a 125 °C y presión atmosférica con un tiempo de
reacción de 8 horas. En el proceso de la funcionalización del poliisopreno, la ruta de síntesis
que se propuso para obtener la macroalcoxiamina multifuncional se presenta en el Esquema 24.
Esta macroalcoxiamina multifuncional se obtiene calentando al poliisopreno comercial en
solución con tolueno, en presencia del nitróxido y el TBP como iniciador de radicales; el TBP
se seleccionó por que los radicales formados (radicales t-butoxi) están centrados en el átomo de
oxígeno y esto tiene como resultado:
a. Que muestran relativamente altas regioespecificidades en reacciones en donde un
enlace C-C este presente,50
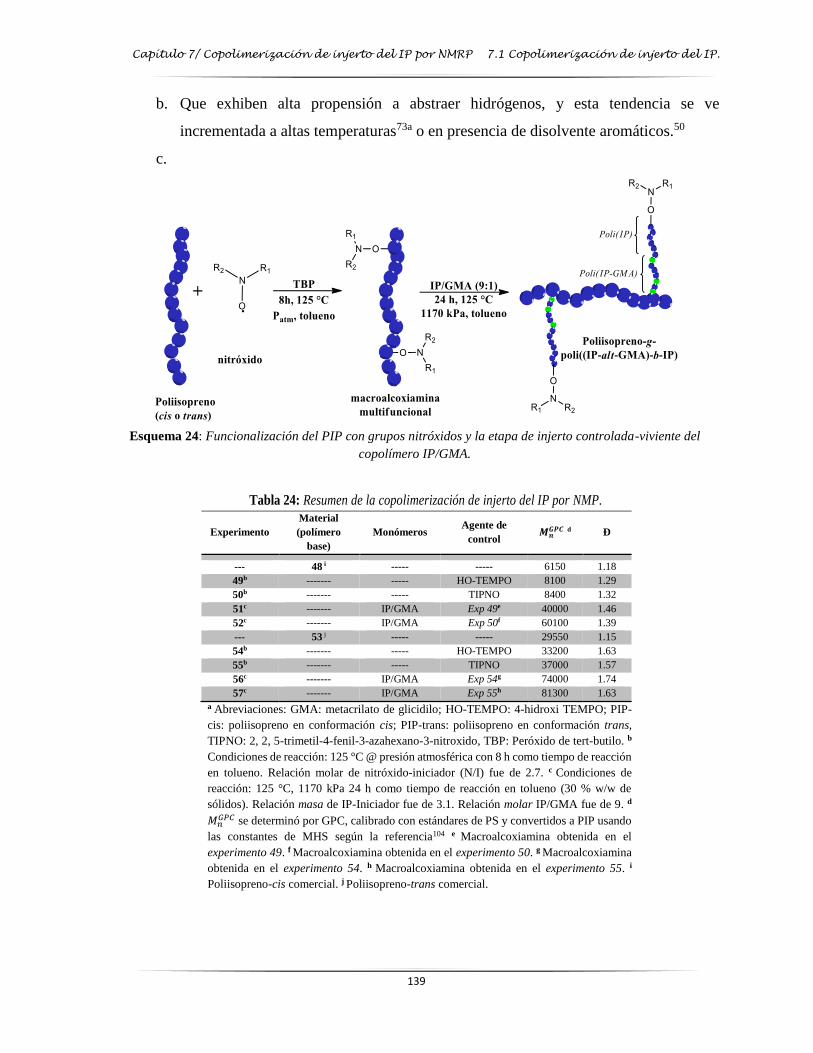
Capítulo 7/ Copolimerización de injerto del IP por NMRP 7.1 Copolimerización de injerto del IP.
139
b. Que exhiben alta propensión a abstraer hidrógenos, y esta tendencia se ve
incrementada a altas temperaturas73a o en presencia de disolvente aromáticos.50
c.
Esquema 24: Funcionalización del PIP con grupos nitróxidos y la etapa de injerto controlada-viviente del
copolímero IP/GMA.
Tabla 24: Resumen de la copolimerización de injerto del IP por NMP.
Experimento
Material
(polímero
base)
Monómeros Agente de
control 𝑴𝒏
𝑮𝑷𝑪 d Ð
--- 48 i ----- ----- 6150 1.18
49b ------- ----- HO-TEMPO 8100 1.29
50b ------- ----- TIPNO 8400 1.32
51c ------- IP/GMA Exp 49e 40000 1.46
52c ------- IP/GMA Exp 50f 60100 1.39
--- 53 j ----- ----- 29550 1.15
54b ------- ----- HO-TEMPO 33200 1.63
55b ------- ----- TIPNO 37000 1.57
56c ------- IP/GMA Exp 54g 74000 1.74
57c ------- IP/GMA Exp 55h 81300 1.63
a Abreviaciones: GMA: metacrilato de glicidilo; HO-TEMPO: 4-hidroxi TEMPO; PIP-
cis: poliisopreno en conformación cis; PIP-trans: poliisopreno en conformación trans,
TIPNO: 2, 2, 5-trimetil-4-fenil-3-azahexano-3-nitroxido, TBP: Peróxido de tert-butilo. b
Condiciones de reacción: 125 °C @ presión atmosférica con 8 h como tiempo de reacción
en tolueno. Relación molar de nitróxido-iniciador (N/I) fue de 2.7. c Condiciones de
reacción: 125 °C, 1170 kPa 24 h como tiempo de reacción en tolueno (30 % w/w de
sólidos). Relación masa de IP-Iniciador fue de 3.1. Relación molar IP/GMA fue de 9. d
𝑀𝑛𝐺𝑃𝐶 se determinó por GPC, calibrado con estándares de PS y convertidos a PIP usando
las constantes de MHS según la referencia104 e Macroalcoxiamina obtenida en el
experimento 49. f Macroalcoxiamina obtenida en el experimento 50. g Macroalcoxiamina
obtenida en el experimento 54. h Macroalcoxiamina obtenida en el experimento 55. i
Poliisopreno-cis comercial. j Poliisopreno-trans comercial.

Capítulo 7/ Copolimerización de injerto del IP por NMRP 7.3 Propuesta mecanística de la
incorporación del radical nitróxido.
140
Los radicales nitróxido además de ser usados como capturadores de radicales en altas
temperaturas (aproximadamente a 125 °C), tienen la habilidad de extraer átomos de hidrógeno
de moléculas hidrocarbonadas5b, 32 o adicionarse a dobles enlaces C=C a moderadas
temperaturas.123 La relación molar nitróxido: iniciador empleada en la funcionalidad del
poliisopreno fue de 2.7, y esta fue deducida considerando: i. Dos radicales t-butoxi generados
por la descomposición del TBP; ii. El mantener un exceso del 100% del iniciador, lo que asegura
la presencia de los radicales en el medio de reacción y; iii. Una eficiencia global del iniciador
(f) de 0.67; esto es por el hecho de que, 2*2*f = 2.7. Finalmente, la macroalcoxiamina
multifuncional sintetizada se calentó en presencia de una mezcla de monómeros (IP-GMA), con
una relación molar de 9:1 a 125 °C y 1170 kPa en solución con tolueno (30% w/w de sólidos),
produciendo injertos controlados del copolímero de isopreno, sobre la cadena principal del
material polimérico. El TBP se utilizó como iniciador de radicales por su prolongado tiempo de
vida media (10 h a 125 ° C).
7.2 Análisis espectroscópico
En la Figura 62, se presenta el espectro de FT-IR del PIP-trans (sin antioxidantes), PIP-
trans funcionalizado con TIPNO y PIP-trans injertado con isopreno-metacrilato de glicidilo
(Material 53, Experimento 55 y 57 respectivamente). Las estructuras fueron confirmadas por
esta técnica espectroscópica y fueron cualitativamente analizadas. Se pueden observar las
bandas características para el caso del Material 53 (Figura 62a), donde aquéllas en las regiones
de 730-665 cm-1, 960-980 cm-1 corresponden a las vibraciones de tensión (C-H) fuera del plano
del isómero trans. Cerca de 3000 cm-1 está la presencia de una banda intensa atribuida a las
vibraciones de estiramiento (C-H) correspondiente a átomos de hidrógeno del doble enlace. En
la región de 1666 cm-1, se observa una banda de absorción que corresponde a la vibración de
estiramiento simétrico del doble enlace (C=C, trans) y en 1430 cm-1, hay una banda de absorción
correspondiente a vibraciones de flexión asimétrica en el plano del enlace. C-H. en la Figura
62b (Experimento 55), se aprecian nuevas bandas; en la región 1933-1820 corresponde a las
vibraciones de tensión del anillo aromático monosustituido que corresponde al nitróxido TIPNO
y de acuerdo a Bonilla et al.,76b las bandas en la región de 807 y en 729 cm-1 corresponden a la
vibración del grupo nitróxido (-N-O).
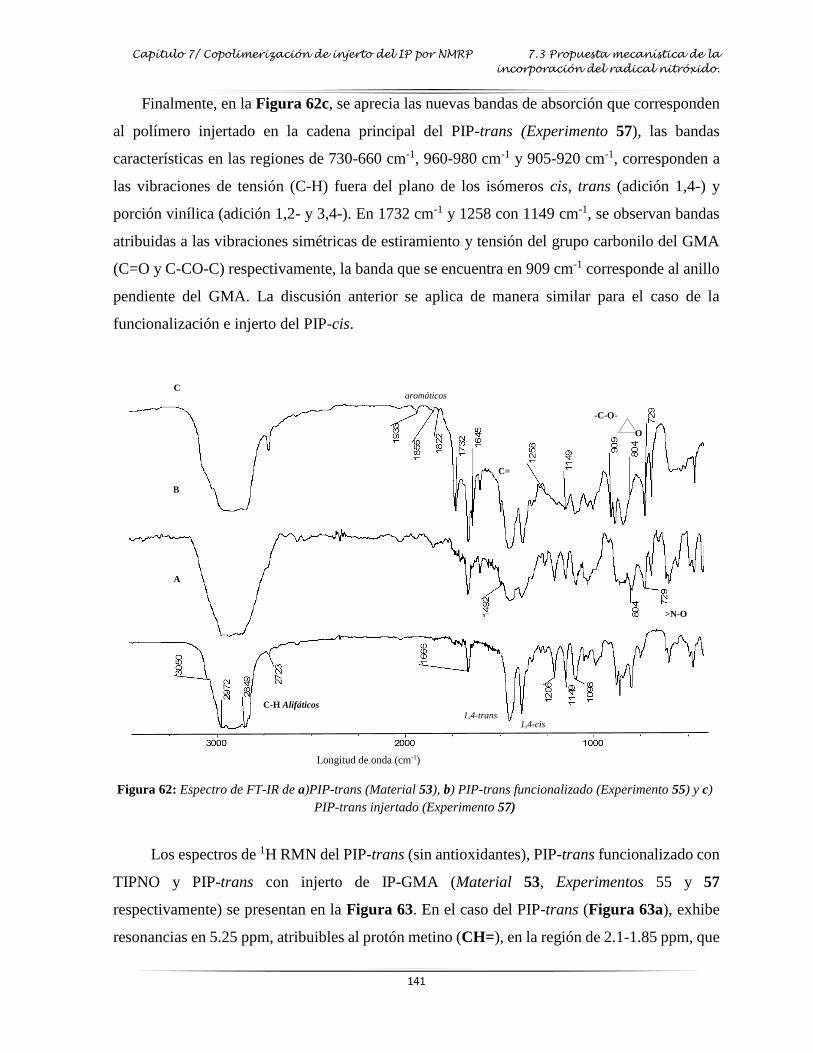
Capítulo 7/ Copolimerización de injerto del IP por NMRP 7.3 Propuesta mecanística de la
incorporación del radical nitróxido.
141
Finalmente, en la Figura 62c, se aprecia las nuevas bandas de absorción que corresponden
al polímero injertado en la cadena principal del PIP-trans (Experimento 57), las bandas
características en las regiones de 730-660 cm-1, 960-980 cm-1 y 905-920 cm-1, corresponden a
las vibraciones de tensión (C-H) fuera del plano de los isómeros cis, trans (adición 1,4-) y
porción vinílica (adición 1,2- y 3,4-). En 1732 cm-1 y 1258 con 1149 cm-1, se observan bandas
atribuidas a las vibraciones simétricas de estiramiento y tensión del grupo carbonilo del GMA
(C=O y C-CO-C) respectivamente, la banda que se encuentra en 909 cm-1 corresponde al anillo
pendiente del GMA. La discusión anterior se aplica de manera similar para el caso de la
funcionalización e injerto del PIP-cis.
Figura 62: Espectro de FT-IR de a)PIP-trans (Material 53), b) PIP-trans funcionalizado (Experimento 55) y c)
PIP-trans injertado (Experimento 57)
Los espectros de 1H RMN del PIP-trans (sin antioxidantes), PIP-trans funcionalizado con
TIPNO y PIP-trans con injerto de IP-GMA (Material 53, Experimentos 55 y 57
respectivamente) se presentan en la Figura 63. En el caso del PIP-trans (Figura 63a), exhibe
resonancias en 5.25 ppm, atribuibles al protón metino (CH=), en la región de 2.1-1.85 ppm, que
Longitud de onda (cm-1)
aromáticos
C=
O
1,4-cis
-C-O-
C O
>N-O
C-H Alifáticos
1,4-trans
C
B
A

Capítulo 7/ Copolimerización de injerto del IP por NMRP 7.3 Propuesta mecanística de la
incorporación del radical nitróxido.
142
corresponden a los dos pares de protones de metileno (-CH2-) que conforman la cadena principal
y en 1.6 ppm, asignadas a los protones metilo (CH3-). Los dos desplazamientos centrados en
1.5 y 0.5 ppm son atribuidas a los protones de metilenos y metilos terminales. En la Figura 63b,
correspondiente al Experimento 55, se puede observar un nuevo desplazamiento químico en
3.35 ppm, el cual fue atribuido al metino correspondiente al hidrógeno α al grupo nitróxido
respectivamente.
Los desplazamientos químicos en 1.3 ppm y 1.9 ppm corresponden a los metilos del t-butilo
y del isopropilo presentes en el TIPNO (aunque estos están traslapados con las señales
correspondientes de los protones del poliisopreno). También se observaron señales en 2.3 ppm
y fueron atribuidas a los protones vecinos en donde el nitróxido se enlazó químicamente. La
Figura 63c (que corresponde al Experimento 57), muestra las señales idénticas correspondiente
a las observadas en la Figura 63a, pero adicionalmente se presentan las señales
correspondientes al poliisopreno con las respectivas adiciones 1,4- cis/trans, 1,2- y 3,4-; aunado
con las correspondientes al comonómero de metacrilato de glicidilo. (Ver Esquema 13, página
46).
Figura 63: Espectro de 1H RMN de a)PIP-trans (Material 53), b) PIP-trans funcionalizado (Experimento 55) y
c) PIP-trans con injerto (Experimento 57)
c
a
b

Capítulo 7/ Copolimerización de injerto del IP por NMRP 7.3 Propuesta mecanística de la
incorporación del radical nitróxido.
143
Los polímeros funcionalizados y con injerto fueron analizados por DQF-COSY, sus
espectros se presentan en la Figura 64, donde se pueden apreciar las correlaciones protón-
protón. Estas evidencias se resumen en la Tabla 25.
7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 1.0 0.5 0
F2 Chemical Shift (ppm)
0
1
2
3
4
5
6
7
F1 C
hem
ical S
hift (ppm
)
7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 1.0 0.5
F2 Chemical Shift (ppm)
1
2
3
4
5
6
7
F1 C
hem
ical S
hift (ppm
)
Figura 64: DQF-COSY: lado izquierdo PIP-trans funcionalizado (Experimento 55) y lado derecho PIP-trans
injertado (Experimento 57).
Tabla 25: Desplazamientos químicos observados para el Experimento 55.
Análisis por 1H RMN y DQF-COSY
Exp
55
d (ppm) 1.5b 1.8c 1.5b 5.11
Especie CH3- -CH2- CH3- -HC=
Vecindad -HC=a,d -HC=CH-a -CH2-a,c =CH-
d (ppm) 5.12 5.12 1.8 5.26 a acoplamiento de largo alcance. b asignado al metilo del carbono donde se unió químicamente
el nitróxido. c asignado a la posición alfa del carbono donde se unió químicamente el nitróxido. d asignado a la posición alfa del carbono donde se unió químicamente el nitróxido.

Capítulo 7/ Copolimerización de injerto del IP por NMRP 7.3 Propuesta mecanística de la
incorporación del radical nitróxido.
144
Bonilla et al.,76b proponen que el mecanismo de reacción para la funcionalización de
cadenas olefínicas (Esquema 25), en presencia de una mezcla de nitróxido/iniciador de
radicales a 125 °C es el siguiente:
Esquema 25: Mecanismo de reacción para el proceso de funcionalización de cadenas olefínicas.76b
7.3 Propuesta mecanística de la incorporación del radical nitróxido
Como se puede observar en el mecanismo de reacción, al ser extraído el hidrógeno α a
la posición del doble enlace C=C por el radical proveniente del iniciador, se forma un radical
alílico y posteriormente se presenta la incorporación del nitróxido en la cadena alifática;
logrando finalmente la funcionalización del polímero hidrocarbonado. El análisis de DQF-
COSY (Tabla 25), reveló que los desplazamientos químicos en 5.11 ppm y 1.8 ppm, tienen
vecindad con la unión química del grupo nitróxido y, de acuerdo a diversos autores,76b, 102f, 124
indican que el nitróxido se une químicamente a una cadena hidrocarbonada en la posición α a
un doble enlace C=C, dando lugar a un desplazamiento químico que se encuentra centrado entre
4.1 ppm y 4.4 ppm en 1H RMN; sin embargo, dichos autores trabajaron con estructuras que no
presentaban grupos colgantes en su cadena principal.
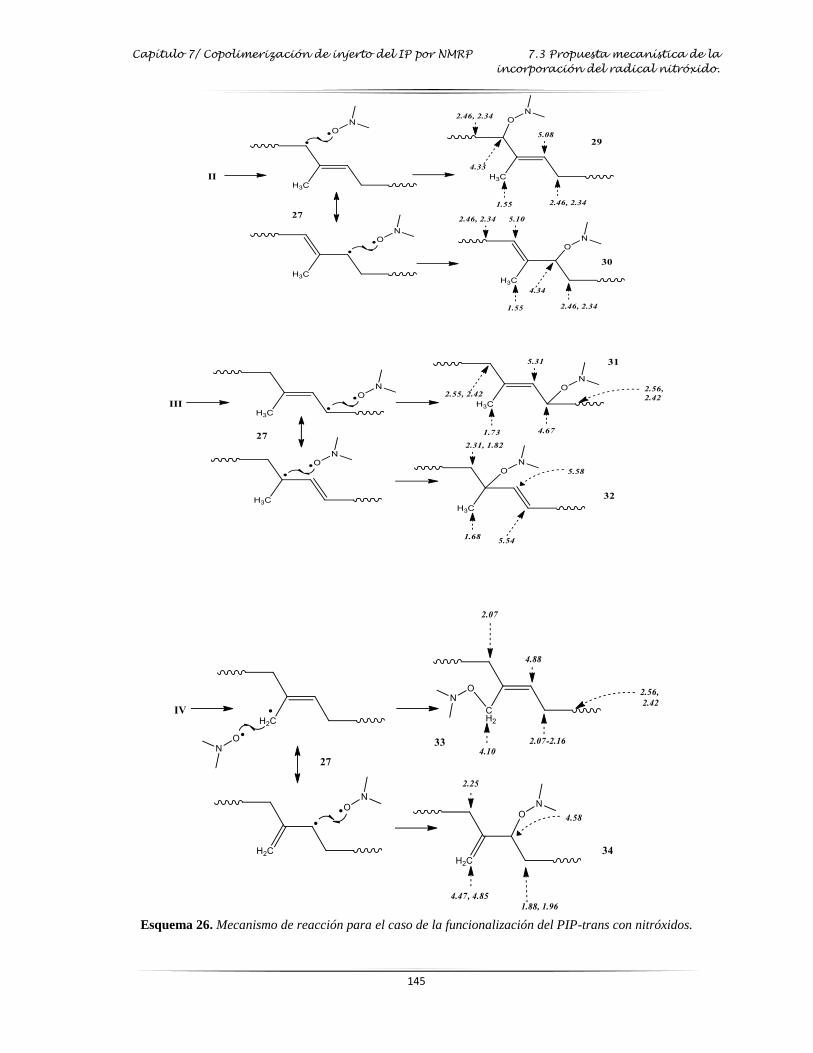
Capítulo 7/ Copolimerización de injerto del IP por NMRP 7.3 Propuesta mecanística de la
incorporación del radical nitróxido.
145
Esquema 26. Mecanismo de reacción para el caso de la funcionalización del PIP-trans con nitróxidos.

Capítulo 7/ Copolimerización de injerto del IP por NMRP 7.3 Propuesta mecanística de la
incorporación del radical nitróxido.
146
En nuestro caso de estudio, la estructura del poliisopreno-trans ostenta un grupo metilo
(CH3-) como especie colgante en la cadena principal, confiriéndole con ello, que en el momento
de la reacción de extracción del átomo de hidrógeno se generen diversos tipos de radicales. El
nitróxido tiende a incorporarse en el radical más estable. Siendo así, y como un medio de
explicación en función de las evidencias mostradas anteriormente, se decidió proponer
estructuras posibles teóricas para el PIP-trans funcionalizado (Experimentos 55 y 54) con
grupos nitróxido y simuladas, usando el paquete “1H NMR predictor” del software ACDLabsTM,
con el fin de apoyarse a predecir los desplazamientos químicos teóricos correspondientes (los
cuales fueron comparados con los desplazamientos químicos experimentales). Las estructuras
propuestas que se consideraron en el mecanismo de reacción de la extracción del átomo de
hidrógeno (existiendo otras opciones de propuestas tal como a) ramificación y b)
entrecruzamiento, que no se consideraron, dado que ya se presentaron en la referencia 77b). Por
lo tanto, el Esquema 26 muestra las estructuras funcionalizadas con el radical nitróxido más
probables.
De acuerdo a lo mostrado en dicho Esquema 26, existen cuatro rutas posibles para obtener
la funcionalización de la cadena polimérica; la estructura 28 es resultado de la ruta I, donde se
tiene lugar la extracción del átomo de hidrógeno en el doble enlace C=C, y por lo tanto, no se
genera alguna señal en 1H RMN en dicha unión química pero se pueden observar las señales
correspondientes de los protones de los grupos vecinos. Por ejemplo, el grupo metilo (CH3-)
que está como sustituyente en dicho doble enlace, muestra un desplazamiento químico (δ) en
1.61 ppm y los metilenos (-CH2-)vecinos, presentan un desplazamiento químico en 1.91 ppm y
2.25 ppm respectivamente.
Para el caso de la estructura 29, resultado de la ruta II donde se extrae un átomo de
hidrógeno del metileno α al doble enlace C=C, se pueden observar los siguientes
desplazamientos químicos: el grupo metilo tiene un δ=1.55 ppm, los protones diasterotópicos
del CH2 vecino al punto de unión con el grupo nitróxido presentan dos señales (δ=2.46 ppm y
2.34 ppm); el protón del carbono donde el nitróxido se enlazó químicamente está en δ=4.33
ppm. La estructura 30, es un derivado de la ruta II debido al efecto de la conjugación que se
forma con el radical alilo. Con ello, se genera un sitio alterno para la incorporación química del

Capítulo 7/ Copolimerización de injerto del IP por NMRP 7.3 Propuesta mecanística de la
incorporación del radical nitróxido.
147
nitróxido; siendo así, se pueden observar los siguientes desplazamientos químicos: los
hidrógenos del metilo se encuentran en δ=1.55 ppm, el protón (del metino, –HC=)
correspondiente al doble enlace C=C en δ=5.10 ppm, los hidrógenos diasterotópicos del
metileno vecino al punto de unión y el protón del carbono donde el nitróxido se unió
químicamente, están en δ=2.46, 2.34 ppm y 4.34 ppm, respectivamente.
Con respecto a la estructura 31, resultado de la ruta III, donde se abstrae el átomo de
hidrógeno del metileno α al doble enlace C=C, se observan los siguientes desplazamientos
químicos: los protones del grupo metilo están en δ=1.73 ppm, los hidrógenos diasterotópicos
del metileno vecinos al doble enlace C=C y el protón del carbono donde el nitróxido se enlazo
químicamente, están en δ=2.55, 2.42 ppm y 4.67 ppm, respectivamente. Finalmente, la
estructura 32, al igual que la estructura 30, es resultado de la conjugación del radical alilo
formado por la abstracción del átomo de hidrógeno, pero a diferencia de la estructura 30 (donde
ambos radicales son secundarios), aquí se tiene una forma canónica, donde el radical reside en
un carbono terciario que es más estable que el radical secundario originado en la estructura 30.
Finalmente, la estructura 33, resultado de la ruta IV, donde se abstrae el átomo de
hidrógeno del metileno colgante de la cadena principal del PIP, se observan los siguientes
desplazamientos químicos: los protones de ambos metilenos vecinos al doble enlace C=C están
en δ=2.07-2.16 ppm, mientras que el metileno donde se encuentra enlazado el grupo nitróxido
está en δ=4.10 ppm y el protón que pertenece al doble enlace C=C tiene un desplazamiento
químico de 4.88 ppm. Por otra parte, la estructura 34, al igual que la estructura 30, es resultado
de la conjugación del radical alilo formado por la abstracción del átomo de hidrógeno en el
grupo metilo, se forma un radical secundario (más estable que el radical primario original de la
extracción del átomo de hidrógeno). Las señales teóricas que se forman son las siguientes: el
grupo metileno en la posición α al doble enlace C=C tiene un δ=2.25 ppm, mientras que el otro
grupo metileno (donde existen dos hidrógenos diasterotópicos) presenta un desplazamiento
químico de δ=1.88 y 1.96 ppm. El grupo metino que ostenta la unión química con el grupo
nitróxido tiene un δ=4.58 ppm.

Capítulo 7/ Copolimerización de injerto del IP por NMRP 7.3 Propuesta mecanística de la
incorporación del radical nitróxido.
148
De tal manera que en los desplazamientos químicos que se observan, no figura un protón
obvio en el punto de unión con el grupo nitróxido, por lo que los desplazamientos que se
interpretan son acoplamientos de largo alcance; como es el caso de los protones del metilo que
están en 1.68 ppm junto con los protones del doble enlace C=C que están centrados en δ=5.54
y 5.58 ppm y los protones diasterotópicos del metileno en δ=1.82 y 2.31 ppm.
De acuerdo a lo observado en las estructuras teóricas sugeridas, se puede concluir que las
estructura 32, 33 y 34 son la más propensas a generarse (como se puede comprobar con los datos
experimentales antes mencionados, donde no se aprecian visiblemente las otras posibles
estructuras en el análisis espectroscópico), debido a la formación de un radical estable, cuya
formación es más probable en el momento de la incorporación del radical nitróxido a la cadena
principal del PIP-trans. Con esta conclusión, también se demuestra porque no existe evidencia
de la señal en la región de 4.1 – 4.3 ppm en 1H RMN como ya se ha reportado anteriormente. 23,
76b, 102f
En el caso específico de las estructuras 33 y 34 es posible que se presenta ésta extracción
del átomo de hidrógeno al grupo metilo debido a que la reacción se realizó en solución con un
disolvente relativamente polar. De acuerdo a Finn et al.,125 el uso del peróxido de t-butilo como
iniciador de radicales libres presenta una descomposición unimolecular (Esquema 27) y la
extracción de hidrógeno se incrementa en disolventes polares, lo que sugiere que la velocidad
del proceso de eliminación se incrementa con respecto a la polaridad del disolvente (debido
al incremento de la polaridad en el estado de transición asociado con el desarrollo de la parte
del carbonilo que se forma). Por lo que, la constante de rapidez para la extracción del hidrógeno
son generalmente esperadas a incrementarse con la disminución de la fuerza de enlace.
Esquema 27. Descomposición unimolecular del radical t-butóxido (eliminación ).

Capítulo 7/ Copolimerización de injerto del IP por NMRP 7.4 Análisis por GPC.
149
Un análisis de los parámetros de activación revelaron que a temperatura ambiente, la
barrera de energía libre (ΔG╪) para la reacción del tBuO∙ con aminas terciarias dominó más los
factores entrópicos que los entálpicos (es decir, que la magnitud de TΔS╪ fue mayor que ΔH╪).
El alcance de este estudio ha sido significativamente expandido a otras especies. Estos
resultados y en conjunto con valores reportados en la literatura para otros sustratos, se midieron
las fuerzas de enlace C-H, encontrándose dichos valores en el intervalo de 79 a 101 kcal/mol;
con ello se tiene una idea acerca de las extracciones del átomo de hidrogeno del carbono por
medio del tBuO∙.
El anterior razonamiento, también se aplica de manera similar para el caso de la
funcionalización del PIP-cis.
7.4 Análisis por cromatografía de permeación en gel (GPC)
Las anteriores evidencias presentadas y el mecanismo de reacción propuesto, proveen
información valiosa para interpretar los cromatogramas de GPC en la determinación de sus
pesos moleculares para concluir con las estructuras resultantes de los polímeros funcionalizados.
La Figura 65 muestra comparativamente los cromatogramas del GPC para el PIP-trans
(Material 53), PIP-trans funcionalizado con TIPNO (Experimento 55) y PIP-trans con injerto
de un copolímero de IP-GMA (Experimento 57).
Figura 65: Cromatogramas de GPC del PIP-trans (Material 53), PIP-trans funcionalizado con TIPNO (Exp 55)
y PIP-trans injertado (Exp 57).
2 3 4 5 6 7
Log MW (g/mol)
Exp 57
Exp 53
Exp 55
Mn = 29550Ɖ = 1.15
Mn = 37000Ɖ = 1.57
Mn = 81300Ɖ = 1.63

Capítulo 7/ Copolimerización de injerto del IP por NMRP 7.4 Análisis por GPC.
150
Se puede apreciar en dicha Figura 65, el comparativo entre el Material 53 (PIP-trans), y
los Experimentos 55 (PIP-trans funcionalizado) y 57 (PIP-trans con injerto), donde existe un
desplazamiento del PIP-trans funcionalizado hacia altos pesos moleculares. Este
desplazamiento es atribuido principalmente a la modificación del volumen hidrodinámico del
PIP-trans virgen por el grupo TIPNO, indicando que se llevó a cabo la funcionalización. Esta
modificación tiende a ser uniforme en todas las cadenas del pico principal de la distribución de
los pesos moleculares; sin embargo, el hombro hacia pesos moleculares altos del Material 53 se
incrementó, provocando un aumento en la dispersidad. El peso molecular promedio, Mn, del
PIP-trans funcionalizado se incrementó en un 25.2% con respecto al Mn del PIP-trans, lo cual
es atribuible a la aparición de nuevas cadenas de alto peso molecular.
Cuando el polímero tiene injerto, es más notorio el desplazamiento hacia altos pesos
moleculares, considerando que se ha incrementado apreciablemente el volumen hidrodinámico
del material polimérico, dado que la dispersidad pasó del Material 53 al Experimento 57 de 1.15
a 1.63; principalmente atribuido a la aparición de cadenas de alto peso molecular, las cuales
principalmente se deben al injerto mismo. Esto puede ser explicado, debido a la presencia de
los radicales t-butoxi; ya que alguno de estos radicales extraen radicales hidrógeno de cadenas
adyacentes o en la misma cadena, originando el entrecruzamiento. Estas son más notorias en el
grafico correspondiente y señalado en el cromatograma respectivo.

Capítulo 8/ Conclusiones y trabajo a futuro 8.1 Utilización de los copolimeros de IP
sintetizados
151
8.
Conclusiones y trabajo a futuro
8.1 Utilización de los copolímeros de IP sintetizados.
Los copolímeros de IP que se obtuvieron en este proyecto de doctorado se tiene pensado
usarlos como compatibilizantes en mezclas poliméricas de isopreno con Nylon 66 y/o como
agentes de acoplamiento con nanoarcillas tipo montmorillonita para obtener nanocompositos de
estos materiales dentro de una matraz polimérica de isopreno. Este tipo de experimentos se
llevarán a cabo en el Instituto de Investigación de Materiales de la UNAM (IIM-UNAM) en
colaboración con el Dr. Octavio Manero Brito.
En el caso de la mezcla de polímeros, se llevará a cabo dentro de un extrusor doble husillo
y en el caso de la dispersión de las nanoarcillas, también se llevará a cabo en el extrusor de doble
husillo pero en la salida se tendrá un dispositivo de ultrasonido para amplificar dicha dispersión-
exfoliación. La intención es estudiar la influencia de la estructura y composición de los
compatibilizantes con sus propiedades finales, en particular se probará la influencia que tiene la
estructura en bloques o de injerto en composiciones globales similares. Posteriormente, las
mezclas resultantes se caracterizarán para obtener sus propiedades mecánicas y reológicas. La
caracterización se hará en términos de morfología (vía TEM), de propiedades mecánicas
(Instron) y resistencia al impacto (Impactómetro Izod), además de la pruebas reológicas
pertinentes.

Capítulo 8/ Conclusiones y trabajo a futuro 8.2 Conclusiones.
152
8.2 Conclusiones.
Los índices de reactividad para el isopreno (IP) con dos monómeros polares: metacrilato
de glicidilo (GMA) y anhídrido maleico (MAH) se lograron determinar. En el caso del
copolímero de IP-GMA, existía solo una publicación en la literatura que reportaba los índices
de reactividad respectivo.109 Para el caso del copolímero de IP-MAH, los procedimientos
experimentales sólo se detallaba la relación experimental para una composición molar en la
alimentación de 50:50, siendo así, sus índices de reactividad no se proveen del todo.105
La serie de copolímeros de poli(IP-co-GMA) por una parte y otra serie de copolímeros de
poli(IP-co-MAH) fueron preparados usando BPO como iniciador de radicales, a 70±1 °C. La
espectroscopia de 1H RMN reveló la presencia de ambos constituyentes monoméricos dentro de
la estructura del copolímero. Las composiciones fueron determinadas por 1H RMN y los índices
de reactividad fueron obtenidos por los métodos de cálculo de Finemann-Ross (FR), Finemann-
Ross Invertido (FRI), Kelen-Tüdos (KT) y Kelen-Tüdos Extendido (KTE); como técnicas tipo
de mínimos cuadrados lineales (LLS)85 y por Tidwell-Mortimer (TM), como técnica de mínimos
cuadrados no lineales (NLLS).86 De todos los métodos antes mencionados, el algoritmo de TM
provee el estimado de los índices de reactividad más confiable, debido a que es el único
rigurosamente correcto en su metodología (estimación no lineal) y a que presenta el área más
pequeña de JCR (lo que indica que presenta la más alta confianza en el estimado de estos
puntos). Los valores de r1 y r2 son menores a la unidad, indicando que el sistema presenta un
punto azeotrópico y una fuerte tendencia a formas copolímeros tipo alternado.
Adicionalmente, un incremento del contenido del monómero polar en el copolímero
(MAH o GMA según sea el caso), incrementa la Tg del material polimérico. El modelo de
Couchman para predecir los valores de Tg, da un excelente aproximado con los datos
experimentales usando las técnicas de caracterización de DSC y DMA para ambos sistemas de
IP/GMA y de IP/MAH.
Fue posible elucidar las condiciones de operación que son necesarias para llevar a cabo
la (co)polimerización del isopreno por medio de dos de las técnicas de CRP; RAFT

Capítulo 8/ Conclusiones y trabajo a futuro 8.2 Conclusiones.
153
(polimerización radicálica por transferencia de cadena de adición-fragmentación reversible) y
NMRP (polimerización radicálica mediada por nitróxidos). En el caso de la NMRP, se usaron
como agentes de control al HO-TEMPO y al TIPNO como nitróxidos. Éste último presentó un
mejor control en la distribución de los pesos moleculares; sin embargo, al manejar una
alcoxiamina con funcionalidad TIPNO (TIPAL), se obtuvo una mejora extra en dicha
distribución de los pesos moleculares, alcanzando un valor de 1.19.
Para el caso del proceso RAFT, se usaron como agentes de transferencia dos moléculas
tipo tritioésteres: BCTC y CDSPA. El CDSPA presentó un mejor comportamiento en la (co)
polimerización del dieno, alcanzando un valor en la distribución de pesos moleculares de 1.25.
Ambas técnicas de polimerización se llevaron a cabo en solución con tolueno a 30 % w/w de
solidos a 1170 kPa (160 psig) y 115 – 125 °C (dependiendo de la técnica de polimerización
utilizada).
En el caso de la polimerización mediada por nitróxidos, se confirma que la
polimerización del isopreno es posible usando HO-TEMPO y también la formación de sus
copolímeros; sin embargo, existen todavía algunos detalles que hay que investigar tal como la
presión o temperatura a la que se deben de realizar dichas polimerizaciones. También se
deberán investigar otras proporciones del monómero polar.
Fue posible obtener los copolímeros de cadena lineal por medio de las técnicas de
polimerización mediada por nitróxidos (NMP) utilizando TIPAL y por el proceso RAFT
(utilizando al CDSPA), con un peso molecular Mn de al menos 100 000 Da por medio del
crecimiento sucesivo (extensión) de la cadena polimérica funcionalizada (comprobando
implícitamente, el comportamiento viviente del sistema), todo esto en un medio de solución
con tolueno (30 % w/w en sólidos) a 125 °C y 160 psig con una relación molar de
monómero:CTA de 1000:1.
Se mostraron evidencias de que con el uso de nitróxidos de segunda generación; esto es,
por medio del uso del TIPAL, es posible llevar a cabo la (co)polimerización del isopreno a 115
°C y 1170 kPa (170 psig), demostrándose al mismo tiempo el carácter viviente que tiene esta

Capítulo 8/ Conclusiones y trabajo a futuro 8.2 Conclusiones.
154
técnica de polimerización cuando se adiciona más monómero al medio de reacción. Este
estudio de doctorado, complementa lo que se ha publicado en la literatura acerca de la
polimerización del IP por NMP.52a, 126 La caracterización de los materiales poliméricos fue
llevada a cabo por 1H RMN, FT-IR y GPC para corroborar tanto cuantitativamente como
cualitativamente la estructura del homo y copolímero resultante.
Los datos de cálculos semiempíricos soportan las evidencias encontradas y ofrecen una
visión muy clara del por qué funcionan mejor los nitróxidos de cadena abierta en la
polimerización del IP con respecto a los nitróxidos de cadena cerrada, y al mismo tiempo, se
puede llegar a poder predecir (o sugerir) el tipo de estructuras que pueden ser útiles para llevar
a cabo la polimerización del dieno bajo las condiciones de NMP.
Se estudió la cinética de la (co)polimerización del IP por NMP, encontrándose que se
mantiene un comportamiento de primer orden con respecto al monómero y una constante de
rapidez aparente, k’, más alta cuando se utiliza un nitróxido de segunda generación en lugar de
uno de primera generación.
Similarmente, se realizó la homo y copolimerización del isopreno por el proceso RAFT
a 125 °C y 1170 kPa (170 psig), encontrándose que el agente de control que mejores resultados
presenta como CTA, es con un grupo Z S-alifático de cadena larga (12 carbonos de longitud)
y un grupo R que presenta en la posición α un elemento electroatractor (como es el caso del
grupo ciano), el cual ofrece la estabilización requerida para llevar a cabo el proceso de la
polimerización controlada. Siguiendo la misma técnica descrita para la NMP, se procedió con
la extensión de la cadena polimérica y con ello se demostró también, el carácter viviente que
tiene el proceso. Esta información, complementa a los estudios publicados que existen en la
literatura acerca de la polimerización del IP por el proceso RAFT.102e, 127 La caracterización de
los materiales poliméricos obtenidos por el proceso RAFT se efectuó por 1H RMN, FT-IR y
GPC, los cuales confirmaron cuantitativa y cualitativamente la estructura del (co)polímero
resultante bajo estas condiciones de polimerización.
Los cálculos semiempíricos confirmaron que un agente RAFT tipo tritioéster de cadena

Capítulo 8/ Conclusiones y trabajo a futuro 8.3 Trabajo a futuro.
155
alifática larga es el que mejores resultados ofrece en la polimerización del IP con respecto a un
tritioéster simétrico; esto se puede apreciar con las energía LUMO, las cuales son un importante
parámetro durante el mecanismo de reacción que se lleva a cabo durante la polimerización tipo
RAFT.
Se pudo determinar la constante de propagación del isopreno, kp, y la de una serie de
copolímeros de IP/GMA por medio de la técnica de PLP-SEC, contribuyendo con ello a la
actualización de este dato cinético, dado que el único dato que se tenía hasta el día de hoy, es
un referencia de 1952.110 Esta contribución permite entender las necesidades de condiciones de
reacción altas para obtener aceptables resultados en la polimerización de dicho monómero.
En el caso de la funcionalización del poliisopreno, se manejaron las dos conformaciones
del elastómero (cis y trans individualmente), logrando la realización de la funcionalización y
comprobándose por las espectroscopías de FT-IR y por RMN. También se logró el posterior
crecimiento de las cadenas poliméricas de injertos sobre el sustrato.
8.3 Trabajo a futuro
Se propone continuar con los experimentos de (co)polimerización del IP, buscando las
condiciones experimentales adecuadas para llevarlas a cabo en condiciones de emulsión.
Se propone efectuar la copolimerización del IP/MAH en solución, estudiando a detalle las
condiciones experimentales necesarias para llevar a cabo esta reacción de polimerización.
Emplear otro método de cálculo tipo NLLS (tal como método de error de variables, EVM)
para afinar los resultados de los índices de reactividad del par de sistemas estudiados en este
estudio doctoral.

Capítulo 8/ Conclusiones y trabajo a futuro 8.4 Logros académicos.
156
Con los copolímeros de IP-GMA obtenidos en este trabajo doctoral, realizar mezclas
poliméricas en matrices de nylon o algún otra matriz polimérica semejante, con el fin de probar
su funcionalidad como materiales compatibilizantes y/o acoplantes para nanoarcillas.
Se propone (como un avance potencial), la realización de la hidrogenación de los
materiales poliméricos de IP-GMA y de IP-MAH, dado que es muy difícil obtener este tipo de
compuestos con una parte polar.
Con los cálculos llevados a cabo por los métodos semiempíricos y en función de las
características que se obtengan, se puede proponer la síntesis de posibles agentes de control para
llevar a cabo la (co)polimerización del isopreno tanto NMP como por el proceso RAFT.
8.4 Logros académicos
En esta sección se presentan los logros científicos producidos durante el desarrollo del
presente estudio doctoral:
Contreras-López, D., Luna-Bárcena, G., Saldívar-Guerra, E. Eur Polym J 2013, 49
(7), 1760. “Copolymerization of isoprene with polar vinyl monomers: Reactivity
ratios, characterization and thermal properties.”
XXVI Congreso Nacional de la Sociedad Polimérica de México “Copolimerización
de isopreno con monómeros vinílicos polares: Índices de reactividad.”; SPM. 6 –
9 noviembre 2013. Coatzacoalcos, Veracruz, México.
3ra reunión de la Red de Polímeros/2do simposio de radicálica controlada
“Copolímeros de isopreno con monómeros polares vía CRP”; CIQA-CIMAV
(Mty) 28-31 octubre 2013. Saltillo/Monterrey, México.
Estancia Doctoral en Queen’s University, Kingston, Ontario, Canadá; Enero– Julio
2014.

Capítulo 8/ Conclusiones y trabajo a futuro 8.4 Logros académicos.
157
Artículos en escritura:
“Copolymerization of isoprene with glycidyl methacrylate: RAFT and NMRP”
Contreras-López, D., Saldívar-Guerra, E.
“PLP-SEC Study of free radical copolymerization of Isoprene and Glycidyl
Methacrylate” Contreras-López, D., Saldívar-Guerra, E., Schier, J., Hutchinson, R.
“Synthesis of functional polyamines for use in two-phase partitioning bioreactors
(TPPBs)” Peterson, E., Contreras-López, D., Parent, S.

Referencias
158
Referencias
1. Utracki, L. A., Introduction to polymer blends Polymer Blends Handbook: New York, 2003; Vol. Capítulo 1.
2. Paul, D. R., Polymer Blends. Elsevier Science: 2012.
3. (a) Die Makrom Chem 1967, 101 (1), 58. Riess, V. G., et al., Über die verträglichkeit von copolymeren mit den
entsprechenden homopolymeren; (b) Polym Eng Sci 1996, 36 (12), 1574. Ajji, A., et al., Interphase and compatibilization of
polymer blends.
4. (a) Majumdar, et al., Polymer Blends. In Encyclopedia of Polymer Science and Technology, Horák, Z., et al., Eds.
John Wiley & Sons, Inc.: 2002; Vol. 1; (b) Datta, S., et al., Polymeric Compatibilizers. Hanser Publishers Münich, 1996; Vol.
Capítulo 1.
5. (a) Polymer 2007, 48 (20), 5960. Hoon Kim, D., et al., Effect of the ratio of maleated polypropylene to organoclay
on the structure and properties of TPO-based nanocomposites. Part I: Morphology and mechanical properties; (b) J Appl
Polym Sci 2008, 109 (2), 1048. Nguyen, Q. T., et al., Dispersion of nanoclay into polypropylene with carbon dioxide in the
presence of maleated polypropylene; (c) Polymer 2008, 49 (10), 2492. Kim, D. H., et al., Effect of the ratio of maleated
polypropylene to organoclay on the structure and properties of TPO-based nanocomposites. Part II: Thermal expansion
behavior.
6. J Comp Mater 2005, 39 (8), 745. Ahmadi, S. J., et al., Morphology and Characterization of Clay-reinforced EPDM
Nanocomposites.
7. (a) Macromolecules 2003, 36 (3), 804. Lee, K. M., et al., Linear Dynamic Viscoelastic Properties of Functionalized
Block Copolymer/Organoclay Nanocomposites; (b) Macromolecules 2004, 37 (20), 7649. Choi, S., et al., Effects of Triblock
Copolymer Architecture and the Degree of Functionalization on the Organoclay Dispersion and Rheology of Nanocomposites.
8. Horák, Z., et al., Polymer Blends. In Encyclopedia of Polymer Science and Technology, John Wiley & Sons, Inc.:
2002.
9. Polym Adv Tech 2011, 22 (4), 395. Joseph, P., et al., Reactive modifications of some chain- and step-growth polymers
with phosphorus-containing compounds: effects on flame retardance—a review.
10. Arrighi, V., et al., Miscibility. In Encyclopedia of Polymer Science and Technology, John Wiley & Sons, Inc.: 2002.
11. (a) J Polym Sci Part B 1995, 33 (12), 1751. González-Montiel, A., et al., Morphology of nylon 6/polypropylene blends
compatibilized with maleated polypropylene; (b) Polymer 1995, 36 (24), 4605. González-Montiel, A., et al., Impact-modified
nylon 6/polypropylene blends: 2. Effect of reactive functionality on morphology and mechanical properties; (c) Polymer 1995,
36 (24), 4587. González-Montiel, A., et al., Impact-modified nylon 6/polypropylene blends: 1. Morphology-property
relationships; (d) Polymer 1995, 36 (24), 4621. González-Montiel, A., et al., Impact-modified nylon 6/polypropylene blends:
3. Deformation mechanisms.
12. Adv Polym Tech 2013, 32 (S1), E399. Mujal-Rosas, R., et al., Dielectric, Thermal, and Mechanical Properties of
Acrylonitrile Butadiene Styrene Reinforced with Used Tires.
13. J Appl Polym Sci 1977, 21 (1), 113. Taylor, N. W., et al., Tailoring closely packed gel–particle systems for use as
thickening agents.
14. J Appl Polym Sci 2012, 124 (2), 1031. Poh, B. T., et al., Adhesion behavior of natural rubber-based adhesives
crosslinked by benzoyl peroxide.
15. (a) J Appl Polym Sci 2003, 88 (2), 266. Raghu, P., et al., Effect of styrene–isoprene–styrene, styrene–butadiene–
styrene, and styrene–butadiene–rubber on the mechanical, thermal, rheological, and morphological properties of
polypropylene/polystyrene blends; (b) J Appl Polym Sci 2013, 127 (2), 1164. Ismail, H., et al., Compatibilization of bentonite
filled ethylene-propylene-diene monomer composites: Effect of maleic anhydride grafted EPDM; (c) J Appl Polym Sci 2013,
127 (2), 1148. Sadik, T., et al., Polyolefins/Poly(3-hydroxybutyrate-co-hydroxyvalerate) blends compatibilization: Morphology,
rheological, and mechanical properties.
16. Macromolecules 1996, 29 (7), 2498. Fischer, M., et al., On the evolution of phase patterns during the high-impact-
modified polystyrene process.
17. (a) Macromolecules 2005, 38 (22), 8966. Wang, Z. M., et al., Synthesis of Maleic Anhydride Grafted Polypropylene
with High Molecular Weight Using Borane/O2 Radical Initiator and Commercial PP Polymers; (b) Polym Eng Sci 2013, n/a.
Gallego, R., et al., Synthesis of new compatibilizers to poly(lactic acid) blends.
18. J Amer Chem Soc 1956, 78 (11), 2656. Szwarc, M., et al., Polymerization initiated by electron transfer to monomer.
A new method of formation of block polymers.
19. Chem Rev 2009, 109 (11), 5245. Aoshima, S., et al., A Renaissance in Living Cationic Polymerization.
20. Science 1991, 251 (4996), 887. Webster, O. W., Living Polymerization Methods.

Referencias
159
21. Macromolecules 1989, 22 (4), 1558. Risse, W., et al., A novel route to block copolymers by changing the mechanism
from living ring-opening metathesis polymerization of cyclic olefins to aldol condensation polymerization of silyl vinyl ethers.
22. Mater Today 2005, 8 (3), 26. Matyjaszewski, K., et al., Controlled/living radical polymerization.
23. Odian, G., Principles of Polymerization. 4th ed.; Wiley: 2004.
24. Prog Polym Sci 2004, 29 (4), 329. Goto, A., et al., Kinetics of living radical polymerization.
25. (a) Prog Polym Sci 2006, 31 (12), 1068. Hadjichristidis, N., et al., Macromolecular architectures by living and
controlled/living polymerizations; (b) Prog Polym Sci 2006, 31 (12), 1041. Smid, J., et al., Perspectives on the contributions
of Michael Szwarc to living polymerization; (c) Prog Polym Sci 2007, 32 (2), 173. Baskaran, D., et al., Anionic vinyl
polymerization—50 years after Michael Szwarc.
26. Die Makrom Chem, Rap Commun 1982, 3 (2), 127. Otsu, T., et al., Role of initiator-transfer agent-terminator
(iniferter) in radical polymerizations: Polymer design by organic disulfides as iniferters.
27. (a) J Polym Sci Part A 2000, 38 (10), 1706. Darling, T. R., et al., Living polymerization: Rationale for uniform
terminology; (b) Matyjaszewski, K., et al., Naming of controlled, living and ‘living’ polymerizations, Macromolecular
Nomenclature Note 12. Nomenclature Committee of the ACS Division of Polymer Chemistry guest presentation. Disponible
en: http://www.polyacs.org/725.html
28. Account Chem Res 2008, 41 (9), 1133. Moad, G., et al., Toward Living Radical Polymerization.
29. (a) Angew Chem Int Ed 2011, 50 (22), 5034. Tebben, L., et al., Nitroxides: Applications in Synthesis and in Polymer
Chemistry; (b) Chem Rev 2001, 101 (12), 3661. Hawker, C. J., et al., New Polymer Synthesis by Nitroxide Mediated Living
Radical Polymerizations; (c) Polymer Reviews 2011, 51 (2), 104. Grubbs, R. B., Nitroxide-Mediated Radical Polymerization:
Limitations and Versatility; (d) Prog Polym Sci 2013, 38 (1), 63. Nicolas, J., et al., Nitroxide-mediated polymerization.
30. (a) Matyjaszewski, K., Radical Polymerization. In Controlled and Living Polymerizations, Wiley: 2010; pp 103; (b) Matyjaszewski, K., et al., Handbook of radical polymerization. Wiley-Interscience: 2002.
31. (a) Aust J Chem 2005, 58 (6), 379. Moad, G., et al., Living Radical Polymerization by the RAFT Process; (b) Aust
J Chem 2006, 59 (10), 669. Moad, G., et al., Living Radical Polymerization by the RAFT Process—A First Update; (c) Barner-
Kowollik, C., Handbook of RAFT Polymerization. Wiley: 2008.
32. Polymer 2008, 49 (5), 1079. Moad, G., et al., Radical addition–fragmentation chemistry in polymer synthesis.
33. Chem Rev 2006, 106 (9), 3936. David, G., et al., Use of Iodocompounds in Radical Polymerization.
34. (a) J Polym Sci Part A 2000, 38 (10), 1753. Gridnev, A., The 25th anniversary of catalytic chain transfer; (b) Chem
Rev 2001, 101 (12), 3611. Gridnev, A. A., et al., Catalytic Chain Transfer in Free-Radical Polymerizations.
35. Macromolecules 1994, 27 (3), 638. Greszta, D., et al., "Living" radical polymerization. 1. Possibilities and limitations.
36. J Macrom Sci Part C 2001, 41 (3), 139. Bisht, H. S., et al., Living free-radical polymerization -A review
37. (a) J Molec Model 2010, 16 (1), 95. Rodríguez-Sanchez, I., et al., Theoretical evaluation of the order of reactivity of
transfer agents utilized in RAFT polymerization; (b) Can J Chem Eng 2012, 90 (4), 804. Zapata-González, I., et al., Efficient
numerical integration of stiff differential equations in polymerisation reaction engineering: Computational aspects and
applications; (c) J Macrom Sci Part A 2006, 43 (7), 995. Tuinman, E., et al., Controlled Free‐Radical Copolymerization
Kinetics of Styrene and Divinylbenzene by Bimolecular NMRP using TEMPO and Dibenzoyl Peroxide; (d) Flores-Tlacuahuac,
A., et al., Dynamic modelling, nonlinear parameter fitting and sensitivity analysis of a living free-radical polymerization reactor.
In Computer Aided Chemical Engineering, Asprey, S. P.; Macchietto, S., Eds. Elsevier: 2003; Vol. Volume 16, pp 21; (e) J Macrom Sci Part A 2006, 43 (9), 1293. Pallares, J., et al., A Comparison of Reaction Mechanisms for Reversible Addition‐Fragmentation Chain Transfer Polymerization Using Modeling Tools.
38. (a) Appl Catal A 2011, 397 (1–2), 225. Barrientos-Ramírez, S., et al., Influence of the surface chemistry of activated
carbons on the ATRP catalysis of methyl methacrylate polymerization; (b) Polym Adv Tech 2012, 23 (3), 375. Enríquez-
Medrano, F. J., et al., Super impact strength of blends prepared from regular HIPS and poly(butyl acrylate)-block-poly(styrene)
obtained by RAFT polymerization; (c) J Appl Polym Sci 2011, 119 (4), 2476. Enríquez-Medrano, F. J., et al., Synthesis of
diblock and triblock copolymers from butyl acrylate and styrene by reverse iodine transfer polymerization; (d) Macrom Chem
Phys 2013, 214 (12), 1396. García-Valdez, O., et al., Grafting of Chitosan with Styrene and Maleic Anhydride via Nitroxide-
Mediated Radical Polymerization in Supercritical Carbon Dioxide; (e) J Mater Sci 2010, 45 (7), 1878. Nogueira, T. R., et al.,
Nitroxide-mediated radical copolymerization of styrene and divinylbenzene: increased polymerization rate by using TBEC as
initiator; (f) Eur Polym J 2010, 46 (2), 298. Bonilla-Cruz, J., et al., Controlled “Grafting-from” of poly[styrene-co-maleic
anhydride] onto polydienes using nitroxide chemistry.
39. (a) Polymer 2004, 45 (3), 815. Cuatepotzo-Dı́Az, R., et al., Nitroxide mediated polymerization using diphenyl
azabutane N-oxides. A study of electronic effects and of the [nitroxide]/[initiator] ratio on the polymerization control; (b) J Polym Res 2011, 18 (4), 559. Percino, M. J., et al., Atom transfer radical polymerization of monomers containing amide and
ester moieties monitored by dilatometric method; (c) Polymer 2009, 50 (21), 5024. Jaramillo-Soto, G., et al., Effect of stabilizer

Referencias
160
concentration and controller structure and composition on polymerization rate and molecular weight development in RAFT
polymerization of styrene in supercritical carbon dioxide.
40. (a) J Amer Chem Soc 1995, 117 (20), 5614. Wang, J.-S., et al., Controlled/"living" radical polymerization. atom
transfer radical polymerization in the presence of transition-metal complexes; (b) Macromolecules 1995, 28 (5), 1721. Kato,
M., et al., Polymerization of Methyl Methacrylate with the Carbon Tetrachloride/Dichlorotris-
(triphenylphosphine)ruthenium(II)/Methylaluminum Bis(2,6-di-tert-butylphenoxide) Initiating System: Possibility of Living
Radical Polymerization.
41. (a) Macromolecules 1997, 30 (24), 7631. Lecomte, P., et al., Controlled Radical Polymerization of Methyl
Methacrylate in the Presence of Palladium Acetate, Triphenylphosphine, and Carbon Tetrachloride; (b) Macromolecules
1996, 29 (27), 8576. Granel, C., et al., Controlled Radical Polymerization of Methacrylic Monomers in the Presence of a
Bis(ortho-chelated) Arylnickel(II) Complex and Different Activated Alkyl Halides; (c) J Organom Chem 1999, 584 (2), 246.
Brandts, J. a. M., et al., Controlled radical polymerization of styrene in the presence of lithium molybdate(V) complexes and
benzylic halides; (d) Macromolecules 1999, 32 (8), 2420. Kotani, Y., et al., Re(V)-Mediated Living Radical Polymerization of
Styrene:1 ReO2I(PPh3)2/R−I Initiating Systems; (e) Macromolecules 1997, 30 (26), 8161. Matyjaszewski, K., et al.,
Controlled/“Living” Radical Polymerization of Styrene and Methyl Methacrylate Catalyzed by Iron Complexes1.
42. (a) Chem Rev 2001, 101 (12), 3689. Kamigaito, M., et al., Metal-Catalyzed Living Radical Polymerization; (b) Macromolecules 1999, 32 (20), 6431. Matyjaszewski, K., et al., An Investigation into the CuX/2,2‘-Bipyridine (X = Br or Cl)
Mediated Atom Transfer Radical Polymerization of Acrylonitrile; (c) Macromol Rap Commun 2000, 21 (4), 190. Teodorescu,
M., et al., Controlled polymerization of (meth)acrylamides by atom transfer radical polymerization; (d) Chem Commun 1999,
(14), 1285. J. Ashford, E., et al., First example of the atom transfer radical polymerisation of an acidic monomer: direct
synthesis of methacrylic acid copolymers in aqueous media.
43. (a) Soft Matter 2009, 5 (23), 4788. Gao, C., et al., Facile synthesis and self-assembly of multihetero-arm
hyperbranched polymer brushes; (b) Aust J Chem 2009, 62 (10), 1371. Xiong, X. Q., Efficient Synthesis of Dendritic
Architectures by One-Pot Double Click Reactions; (c) Biomacromolecules 2008, 9 (11), 3231. Yin, M., et al., Dendritic Star
Polymers for Efficient DNA Binding and Stimulus-Dependent DNA Release.
44. Prog Polym Sci 2007, 32 (1), 93. Braunecker, W. A., et al., Controlled/living radical polymerization: Features,
developments, and perspectives.
45. (a) Polymer Bulletin 1982, 6 (11-12), 589. Moad, G., et al., A product study of the nitroxide inhibited thermal
polymerization of styrene; (b) Macromolecules 1982, 15 (3), 909. Moad, G., et al., Selectivity of the reaction of free radicals
with styrene; (c) Die Makrom Chem, Rap Commun 1982, 3 (8), 533. Moad, G., et al., Reactions of benzoyloxyl radicals with
some common vinyl monomers.
46. Macromol Symp 1994, 88 (1), 89. Georges, M. K., et al., Breathing new life into the free radical polymerization
process.
47. Chem Rev 2001, 101 (12), 3581. Fischer, H., The Persistent Radical Effect: A Principle for Selective Radical
Reactions and Living Radical Polymerizations.
48. (a) J Amer Chem Soc 1994, 116 (24), 11185. Hawker, C. J., Molecular Weight Control by a "Living" Free-Radical
Polymerization Process; (b) Account Chem Res 1997, 30 (9), 373. Hawker, C. J., “Living” Free Radical Polymerization: A
Unique Technique for the Preparation of Controlled Macromolecular Architectures; (c) Polym Rev 2011, 51 (2), 104. Grubbs,
R. B., Nitroxide-Mediated Radical Polymerization: Limitations and Versatility.
49. Macromolecules 1995, 28 (24), 8453. Odell, P. G., et al., Rate Enhancement of Living Free-Radical Polymerizations
by an Organic Acid Salt.
50. Moad, G., et al., The Chemistry of Radical Polymerization. 2nd ed.; Elsevier Science: Oxford, U. K., 2006.
51. J Polym Sci Part A 2007, 45 (23), 5487. Dollin, M., et al., Rapid additive-free TEMPO-mediated stable free radical
polymerizations of styrene.
52. (a) Macromolecules 2000, 33 (2), 363. Benoit, D., et al., Accurate Structural Control and Block Formation in the
Living Polymerization of 1,3-Dienes by Nitroxide-Mediated Procedures; (b) Macromolecules 1996, 29 (27), 8992. Listigovers,
N. A., et al., Narrow-Polydispersity Diblock and Triblock Copolymers of Alkyl Acrylates by a “Living” Stable Free Radical
Polymerization; (c) Macromolecules 2000, 33 (4), 1141. Grimaldi, S., et al., Acyclic β-Phosphonylated Nitroxides: A New
Series of Counter-Radicals for “Living”/Controlled Free Radical Polymerization.
53. (a) Tetrahedron 1997, 53 (45), 15225. Malmström, E., et al., Development of a new class of rate-accelerating
additives for nitroxide-mediated ‘living’ free radical polymerization; (b) Macromolecules 1998, 31 (21), 7559. Keoshkerian,
B., et al., Polyacrylates and Polydienes to High Conversion by a Stable Free Radical Polymerization Process: Use of Reducing
Agents; (c) Macromolecules 2006, 39 (16), 5359. Debuigne, A., et al., Stable Free Radical Polymerization of Acrylates
Promoted by α-Hydroxycarbonyl Compounds.

Referencias
161
54. (a) Macromolecules 1996, 29 (16), 5245. Hawker, C. J., et al., Initiating Systems for Nitroxide-Mediated “Living”
Free Radical Polymerizations: Synthesis and Evaluation; (b) Solomon, D. H., et al. Polymerization process and polymers
produced thereby. US Patent 4581429, 1986.
55. Macromolecules 1998, 31 (16), 5559. Chiefari, J., et al., Living Free-Radical Polymerization by Reversible
Addition−Fragmentation Chain Transfer: The RAFT Process.
56. Macromolecules 1995, 28 (15), 5381. Krstina, J., et al., Narrow Polydispersity Block Copolymers by Free-Radical
Polymerization in the Presence of Macromonomers.
57. Polym Intern 2000, 49 (9), 993. Moad, G., et al., Living free radical polymerization with reversible addition –
fragmentation chain transfer (the life of RAFT).
58. Polymer 2005, 46 (19), 8483. Drache, M., et al., Modeling RAFT polymerization kinetics via Monte Carlo methods:
cumyl dithiobenzoate mediated methyl acrylate polymerization.
59. (a) Macromolecules 2004, 37 (4), 1219. Kwak, Y., et al., Rate Retardation in Reversible Addition−Fragmentation
Chain Transfer (RAFT) Polymerization: Further Evidence for Cross-Termination Producing 3-Arm Star Chain; (b) Aust J
Chem 2002, 55 (7), 425. Vana, P., et al., Recent Advances in the Kinetics of Reversible Addition Fragmentation Chain-Transfer
Polymerization.
60. Macromolecules 2001, 34 (3), 349. Monteiro, M. J., et al., Intermediate Radical Termination as the Mechanism for
Retardation in Reversible Addition−Fragmentation Chain Transfer Polymerization.
61. Macromolecules 1999, 32 (16), 5457. Hawthorne, D. G., et al., Living Radical Polymerization with Reversible
Addition−Fragmentation Chain Transfer (RAFT): Direct ESR Observation of Intermediate Radicals.
62. (a) Macromolecules 2004, 37 (3), 744. Ah Toy, A., et al., Reversible Addition Fragmentation Chain Transfer (RAFT)
Polymerization of Methyl Acrylate: Detailed Structural Investigation via Coupled Size Exclusion
Chromatography−Electrospray Ionization Mass Spectrometry (SEC−ESI-MS); (b) Polymer 2005, 46 (19), 8448. Feldermann,
A., et al., An in-depth analytical approach to the mechanism of the RAFT process in acrylate free radical polymerizations via
coupled size exclusion chromatography–electrospray ionization mass spectrometry (SEC–ESI-MS).
63. J Polym Sci Part A 2006, 44 (20), 5809. Barner-Kowollik, C., et al., Mechanism and kinetics of dithiobenzoate-
mediated RAFT polymerization. I. The current situation.
64. (a) Macromol Rap Commun 2012, 33 (15), 1273. Meiser, W., et al., “Assessing the RAFT Equilibrium Constant via
Model Systems: An EPR Study”—Response to a Comment; (b) Macromol Rap Commun 2011, 32 (18), 1490. Meiser, W., et
al., Assessing the RAFT Equilibrium Constant via Model Systems: An EPR Study.
65. Macromol Rap Commun 2006, 27 (9), 653. Favier, A., et al., Experimental Requirements for an Efficient Control of
Free-Radical Polymerizations via the Reversible Addition-Fragmentation Chain Transfer (RAFT) Process.
66. (a) Macromolecules 2003, 36 (7), 2256. Chong, Y. K., et al., Thiocarbonylthio Compounds [SC(Ph)S−R] in Free
Radical Polymerization with Reversible Addition-Fragmentation Chain Transfer (RAFT Polymerization). Role of the Free-
Radical Leaving Group (R); (b) Macromolecules 2003, 36 (7), 2273. Chiefari, J., et al., Thiocarbonylthio Compounds
(SC(Z)S−R) in Free Radical Polymerization with Reversible Addition-Fragmentation Chain Transfer (RAFT Polymerization).
Effect of the Activating Group Z.
67. Chem & Eng News July 2, 2007, 85 (27), 55. Facts & figures of the chemical industry.
68. Peters, E. N., Plastics: Thermoplastics, Thermosets, and Elastomers. In Handbook of Materials Selection, John Wiley
& Sons, Inc.: 2007; pp 335.
69. (a) Polym Eng Sci 2011, 51 (1), 1. Patham, B., et al., Thermoplastic vibration welding: Review of process
phenomenology and processing–structure–property interrelationships; (b) J Appl Polym Sci 2013, n/a. Yan, G., et al.,
Development of lightweight thermoplastic composites based on polycarbonate/acrylonitrile–butadiene–styrene copolymer
alloys and recycled carbon fiber: Preparation, morphology, and properties; (c) J Appl Polym Sci 2012, 125 (1), 745. Juntuek,
P., et al., Effect of glycidyl methacrylate-grafted natural rubber on physical properties of polylactic acid and natural rubber
blends.
70. Datta, S., et al., Polymeric Compatibilizers: Uses and Benefits in Polymer Blends. Carl Hanser Verlag: 1996.
71. (a) J Polym Sci Part A 2006, 44 (9), 2972. Tsuchida, A., et al., Grafting of polymers onto and/or from silica surface
during the polymerization of vinyl monomers in the presence of γ-ray-irradiated silica; (b) Eur Polym J 2013, 49 (5), 1007.
Songsing, K., et al., Kinetics and mechanism of grafting styrene onto natural rubber in emulsion polymerization using cumene
hydroperoxide–tetraethylenepentamine as redox initiator; (c) Ind Crops & Prod 2013, 46 (0), 191. Panesar, S. S., et al.,
Functionalization of lignin: Fundamental studies on aqueous graft copolymerization with vinyl acetate.
72. Xanthos, M., Reactive Extrusion: Principles and Practice, Chapter 2 "Process analysis from reaction fundamentals:
Examples of polymerization and controlled degradation in extruders". Hanser Publishers: 1992.

Referencias
162
73. (a) Prog Polym Sci 1999, 24 (1), 81. Moad, G., The synthesis of polyolefin graft copolymers by reactive extrusion;
(b) Prog Polym Sci 2002, 27 (6), 1007. Russell, K. E., Free radical graft polymerization and copolymerization at higher
temperatures.
74. Chem Rev 1973, 73 (5), 441. Gibian, M. J., et al., Organic radical-radical reactions. Disproportionation vs.
combination.
75. Polym Eng Sci 2006, 46 (12), 1754. Parent, J. S., et al., Radical mediated graft modification of polyolefins:
Vinyltriethoxysilane addition dynamics and yields.
76. (a) Macromol Rap Commun 2007, 28 (13), 1397. Bonilla-Cruz, J., et al., Towards Controlled Graft Polymerization
of Poly[styrene-co-(maleic anhydride)] on Functionalized Silica Mediated by Oxoaminium Bromide Salt. Facile Synthetic
Pathway Using Nitroxide Chemistry; (b) Macrom Chem Phys 2008, 209 (21), 2268. Bonilla-Cruz, J., et al., Controlled
Grafting-From of Polystyrene on Polybutadiene: Mechanism and Spectroscopic Evidence of the Functionalization of
Polybutadiene with 4-Oxo-TEMPO; (c) Macromol Symp 2009, 283–284 (1), 110. Saldívar-Guerra, E., et al., Progress in
Controlled Grafting-From by Nitroxide Chemistry.
77. (a) J Appl Polym Sci 1994, 54 (3), 339. Majumdar, B., et al., Effect of extruder type on the properties and morphology
of reactive blends based on polyamides; (b) J Appl Polym Sci 1998, 69 (13), 2625. Radonjič, G., et al., Compatibilization of
polypropylene/polystyrene blends with poly(styrene-b-butadiene-b-styrene) block copolymer.
78. Polymer 1994, 35 (7), 1552. Favis, B. D., Phase size/interface relationships in polymer blends: the emulsification
curve.
79. Eur Polym J 1994, 30 (5), 597. Hlavatá, D., et al., Waxs and saxs investigation of polypropylene crystalline phase in
blends with high-impact polystyrene and compatibilizers.
80. J Macrom Sci Part C 2000, 40 (2-3), 167. Schellekens, M. a. J., et al., Synthesis of Polyolefin Block and Graft
Copolymers.
81. J Amer Chem Soc 1944, 66 (9), 1594. Mayo, F. R., et al., Copolymerization. I. A Basis for Comparing the Behavior
of Monomers in Copolymerization; The Copolymerization of Styrene and Methyl Methacrylate.
82. Hagiopol, C., Copolymerization. Toward a systematic approach. Kluwer Academic/Plenum Publisher: New York,
1999.
83. Chem Rev 1950, 46 (2), 191. Mayo, F. R., et al., Copolymerization.
84. Macromolecules 1993, 26 (19), 5249. Kathmann, E. E. L., et al., Water-soluble copolymers. 48. Reactivity ratios of
N-vinylformamide with acrylamide, sodium acrylate, and n-butyl acrylate.
85. (a) J Polym Sci 1950, 5 (2), 259. Finemann, M., et al., Linear method for determining monomer reactivity ratios in
copolymerization; (b) J Macrom Sci Part A 1975, 9 (1), 1 Kelen, T., et al., Analysis of the Linear Methods for Determining
Copolymerization Reactivity Ratios. I. A New Improved Linear Graphic Method; (c) J Macrom Sci Part A 1976, 10 (8), 1513
Tüdos, F., et al., Analysis of Linear Methods for Determining Copolymerization Reactivity Ratios. III. Linear Graphic Method
for Evaluating Data Obtained at High Conversion Levels.
86. (a) J Polym Sci Part A 1965, 3 (1), 369. Tidwell, P. W., et al., An improved method of calculating copolymerization
reactivity ratios; (b) J Royal Stat Soc. Serie C 1993, 42 (4), 693. Park M. Reilly, et al., Algorithm AS 286: Parameter Estimation
in the Error-in-Variables Model.
87. Brandrup, J., et al., Polymer Handbook. 4th ed.; John Wiley & Sons, Inc.: N. Y., USA, 1999.
88. (a) Eur Polym J 1989, 25 (7–8), 635. Olaj, O. F., et al., Laser-flash-initiated polymerization as a tool for evaluating
(individual) kinetic constants of free-radical polymerization—5. Complete analysis by means of a single experiment; (b) Macromolecules 2002, 35 (25), 9300. Beuermann, S., Requirements Associated with Studies into a Chain-Length Dependence
of Propagation Rate Coefficients via PLP−SEC Experiments; (c) Macromolecules 2008, 41 (23), 9011. Wang, W., et al.,
PLP/SEC/NMR Study of Free Radical Copolymerization of Styrene and Glycidyl Methacrylate; (d) Prog Polym Sci 2002, 27
(2), 191. Beuermann, S., et al., Rate coefficients of free-radical polymerization deduced from pulsed laser experiments.
89. (a) Biomaterials 2001, 22 (17), 2411. Nazhat, S. N., et al., Isoprene-styrene copolymer elastomer and
tetrahydrofurfuryl methacrylate mixtures for soft prosthetic applications; (b) J Appl Polym Sci 2003, 88 (4), 921.
Wootthikanokkhan, J., et al., Compatibilization efficacy of poly(isoprene–butyl acrylate) block copolymers in natural/acrylic
rubber blends.
90. (a) ACS Nano 2011, 5 (2), 1513. Arias, J. L., et al., Squalene Based Nanocomposites: A New Platform for the Design
of Multifunctional Pharmaceutical Theragnostics; (b) Molecular Pharmaceutics 2009, 6 (5), 1526. Reddy, L. H., et al.,
Anticancer Efficacy of Squalenoyl Gemcitabine Nanomedicine on 60 Human Tumor Cell Panel and on Experimental Tumor;
(c) Nano Letters 2006, 6 (11), 2544. Couvreur, P., et al., Squalenoyl Nanomedicines as Potential Therapeutics.
91. (a) Macromolecules 1999, 32 (8), 2806. Schöps, M., et al., Salt-Induced Switching of Microdomain Morphology of
Ionically Functionalized Diblock Copolymers; (b) Macromolecules 1999, 32 (9), 3017. Sen, T. Z., et al., Local Dynamics of
cis-1,4-Polybutadiene and cis-1,4-Polyisoprene. A Comparative Study Based on Cooperative Kinematics Theory and NMR

Referencias
163
Experiments; (c) Macromolecules 1997, 30 (6), 1582. Allgaier, J., et al., Synthesis and Characterization of Poly[1,4-isoprene-
b-(ethylene oxide)] and Poly[ethylene-co-propylene-b-(ethylene oxide)] Block Copolymers.
92. (a) Macromolecules 2005, 38 (10), 4124. Yuan, Z., et al., Synthesis of Arborescent Isoprene Homopolymers; (b) J Polym Sci Part A 2000, 38 (6), 931. Paraskeva, S., et al., Synthesis of an exact graft copolymer of isoprene and styrene with two
branches.
93. Polymer Bulletin 2002, 47 (6), 509. Donderer, M., et al., A combined radical/cationic synthetic route for poly(styrene-
g-isobutylene) and poly(styrene-g-isobutylene-co-isoprene).
94. (a) Macromol Rap Commun 2001, 22 (18), 1493. Zhang, Q., et al., Homopolymerization and Copolymerization of
Isoprene and Styrene with a Neodymium Catalyst Using an Alkylmagnesium Cocatalyst; (b) Macromolecules 2000, 33 (23),
8541. Markel, E. J., et al., Metallocene-Based Branch−Block Thermoplastic Elastomers.
95. J Polym Sci Part A 2005, 43 (14), 2977. Wegrzyn, J. K., et al., Preparation of poly(ethylene oxide)-block-
poly(isoprene) by nitroxide-mediated free radical polymerization from PEO macroinitiators.
96. (a) J Polym Sci Part A 2008, 46 (6), 1964. Chen, S.-Y., et al., Ozonolysis efficiency of PS-b-PI block copolymers for
forming nanoporous polystyrene; (b) J Amer Chem Soc 2006, 128 (21), 6808. Cheng, C., et al., Tandem Synthesis of
Core−Shell Brush Copolymers and Their Transformation to Peripherally Cross-Linked and Hollowed Nanostructures.
97. (a) J Amer Chem Soc 2007, 129 (17), 5630. Wang, X., et al., Shell-Cross-Linked Cylindrical Polyisoprene-b-
Polyferrocenylsilane (PI-b-PFS) Block Copolymer Micelles: One-Dimensional (1D) Organometallic Nanocylinders; (b) J
Appl Polym Sci 2003, 90 (10), 2785. Lu, Z., et al., Water-dispersible porous polyisoprene-block-poly(acrylic acid)
microspheres; (c) J Polym Sci Part A 1989, 27 (11), 3721. Ishizu, K., et al., Core-shell type polymer microspheres prepared
by domain fixing of block copolymer films.
98. (a) Macromolecules 2000, 33 (7), 2308. Grubbs, R. B., et al., Selectively Epoxidized Polyisoprene−Polybutadiene
Block Copolymers; (b) Macromolecules 2006, 39 (25), 8772. Bailey, T. S., et al., Routes to Alkene and Epoxide Functionalized
Nanoporous Materials from Poly(styrene-b-isoprene-b-lactide) Triblock Copolymers.
99. Macromolecules 2007, 40 (5), 1585. Mahanthappa, M. K., et al., Control of Mechanical Behavior in Polyolefin
Composites: Integration of Glassy, Rubbery, and Semicrystalline Components.
100. (a) J Polym Sci Part A 2010, 48 (17), 3797. Wang, G., et al., Investigation of thiol-ene addition reaction on
poly(isoprene) under UV irradiation: Synthesis of graft copolymers with “V”-shaped side chains; (b) Macromolecules 2002,
35 (19), 7182. Uhrig, D., et al., Synthesis of Combs, Centipedes, and Barbwires: Poly(isoprene-graft-styrene) Regular
Multigraft Copolymers with Trifunctional, Tetrafunctional, and Hexafunctional Branch Points.
101. Macromolecules 1999, 32 (8), 2783. Förster, S., et al., Synthesis of PB−PEO and PI−PEO Block Copolymers with
Alkyllithium Initiators and the Phosphazene Base t-BuP4.
102. (a) Macromolecules 2011, 44 (23), 9230. Harrisson, S., et al., SG1 Nitroxide-Mediated Polymerization of Isoprene:
Alkoxyamine Structure/Control Relationship and α,ω–Chain-End Functionalization; (b) Macromol Rap Commun 2012, n/a.
Harrisson, S., et al., Use of Solvent Effects to Improve Control Over Nitroxide-Mediated Polymerization of Isoprene; (c) Macrom Chem Phys 2007, 208 (23), 2481. Germack, D. S., et al., RAFT-Based Synthesis and Characterization of ABC versus
ACB Triblock Copolymers Containing tert-Butyl Acrylate, Isoprene, and Styrene Blocks; (d) J Polym Sci Part A 2007, 45 (17),
4100. Germack, D. S., et al., Isoprene polymerization via reversible addition fragmentation chain transfer polymerization; (e)
Macromolecules 2007, 40 (5), 1408. Jitchum, V., et al., Living Radical Polymerization of Isoprene via the RAFT Process; (f) Macromolecules 1998, 31 (25), 9087. Georges, M. K., et al., Block Copolymer Synthesis by a Nitroxide-Mediated Living Free
Radical Polymerization Process.
103. (a) Macromolecules 2007, 40 (16), 5638. Milione, S., et al., Stereoselective Polymerization of Conjugated Dienes
and Styrene−Butadiene Copolymerization Promoted by Octahedral Titanium Catalyst; (b) Macromolecules 2008, 41 (6), 1983.
Wang, B., et al., Highly 3,4-Selective Living Polymerization of Isoprene with Rare Earth Metal Fluorenyl N-Heterocyclic
Carbene Precursors; (c) Chem Rev 2006, 106 (6), 2404. Zeimentz, P. M., et al., Cationic Organometallic Complexes of
Scandium, Yttrium, and the Lanthanoids; (d) Macromolecules 2004, 37 (16), 5860. Kaita, S., et al., An Efficient Gadolinium
Metallocene-Based Catalyst for the Synthesis of Isoprene Rubber with Perfect 1,4-Cis Microstructure and Marked Reactivity
Difference between Lanthanide Metallocenes toward Dienes As Probed by Butadiene−Isoprene Copolymerization Catalysis.
104. J Polym Sci Part B 1995, 33 (16), 2229. Allorio, S., et al., Hydrodynamic behavior of anionically prepared linear
polyisoprenes and polystyrenes in carbon tetrachloride.
105. J Macrom Sci Part A 1971, 5 (5), 867 Gaylord, N. G., et al., Donor-Acceptor Complexes in Copolymerization. XVIII.
Alternating Diene–Dienophile Copolymers. 1. Conjugated Diene–Maleic Anhydride Copolymers through Radical-Catalyzed
Polymerization.
106. J Polym Sci Part A 1989, 27 (10), 3353. Sipos, A., et al., Kinetic studies of grafting of maleic anhydride to hydrocarbon
substrates.

Referencias
164
107. (a) Brandolin, A. J., et al., NMR spectra of polymer and polymer additives. Marcel Dekker, Inc: New York, 2000;
(b) Macomber, R. S., A complete introduction to modern NMR spectroscopy John Wiley and Sons, Inc.: New York, 1998.
108. Robert M. Silverstein, F. X. W., David Kiemle, Spectrometric Identification of Organic Compounds. 7th ed.; 2005; p
512.
109. Polymer Science U.S.S.R. 1974, 16 (12), 3279. Rusakova, K. A., et al., Study of copolymerization of isoprene with
glycidyl methacrylate.
110. J Polym Sci 1952, 8 (2), 215. Morton, M., et al., Absolute propagation rates in emulsion polymerization. II. Butadiene
in hydroperoxide-polyamine systems.
111. Die Makrom Chem 1993, 194 (12), 3287. Deibert, S., et al., Rate coefficients of the free-radical polymerization of
1,3-butadiene.
112. Macromolecules 1998, 31 (4), 994. Kukulj, D., et al., Chain Transfer to Monomer in the Free-Radical Polymerizations
of Methyl Methacrylate, Styrene, and α-Methylstyrene.
113. Polymer J 1970, 1 (6), 669. Oikawa, E., et al., Alternating Copolymerization of Isoprene and Methyl Methacrylate.
114. Sperling, L. H., Introduction to Physical Polymer Science. 4th ed.; 2006; p 880.
115. (a) Macromolecules 1995, 28 (26), 8722. Moad, G., et al., Alkoxyamine-Initiated Living Radical Polymerization:
Factors Affecting Alkoxyamine Homolysis Rates; (b) Macromolecules 1995, 28 (6), 1841. Kazmaier, P. M., et al., Free-Radical
Polymerization for Narrow-Polydispersity Resins. Semiempirical Molecular Orbital Calculations as a Criterion for Selecting
Stable Free-Radical Reversible Terminators; (c) J Comput Chem 1989, 10 (2), 221. Stewart, J. J. P., Optimization of
parameters for semiempirical methods II. Applications; (d) J Molec Struc 1991, 236 (3–4), 211. Tupper, K. J., et al.,
Semiempirical computation of homolytic O-H bond dissociation energies of alcohols: comparison of the AM1 and PM3
methods; (e) J Org Chem 1991, 56 (1), 134. Karelson, M., et al., AM1, PM3, and MNDO calculations of radical formation
energies in the gas phase and in solution; (f) J Molec Struc 1994, 314 (3), 261. Djennane-Bousmaha, S., et al., A PM3 versus
AM1 and RHF versus UHF study of the radical reactivity of vinyl monomers.
116. (a) J Org Chem 2010, 76 (2), 631. Billone, P. S., et al., Accurate O−H Bond Dissociation Energy Differences of
Hydroxylamines Determined by EPR Spectroscopy: Computational Insight into Stereoelectronic Effects on BDEs and EPR
Spectral Parameters; (b) J Amer Chem Soc 2012, 134 (24), 10156. Sajid, M., et al., N,N-Addition of Frustrated Lewis Pairs
to Nitric Oxide: An Easy Entry to a Unique Family of Aminoxyl Radicals.
117. J Amer Chem Soc 1992, 114 (13), 4992. Bowry, V. W., et al., Kinetics of nitroxide radical trapping. 2. Structural
effects.
118. Dean, J. A., Lange's Handbook of Chemistry (15th Edition). McGraw-Hill: 1999.
119. J Org Chem 2001, 66 (4), 1146. Marque, S., et al., Factors Influencing the C−O Bond Homolysis of Alkoxyamines:
Effects of H−Bonding and Polar Substituents.
120. J Amer Chem Soc 1992, 114 (13), 4983. Beckwith, A. L. J., et al., Kinetics of nitroxide radical trapping. 1. Solvent
effects.
121. Eur Polym J 2013, 49 (7), 1760. Contreras-López, D., et al., Copolymerization of isoprene with polar vinyl monomers:
Reactivity ratios, characterization and thermal properties.
122. J Chem Educ 2011, 88 (8), 1098. Izunobi, J. U., et al., Polymer Molecular Weight Analysis by 1H NMR Spectroscopy.
123. J Org Chem 2002, 67 (19), 6831. Babiarz, J. E., et al., The Thermal Reaction of Sterically Hindered Nitroxyl Radicals
with Allylic and Benzylic Substrates: Experimental and Computational Evidence for Divergent Mechanisms.
124. Ind & Eng Chem Res 2003, 42 (16), 3662. Scott, M. E., et al., Controlled Radical Grafting: Nitroxyl-Mediated
Maleation of Model Hydrocarbons.
125. J Amer Chem Soc 2004, 126 (24), 7578. Finn, M., et al., Chemistry of the t-Butoxyl Radical: Evidence that Most
Hydrogen Abstractions from Carbon are Entropy-Controlled.
126. Macromolecules 2011. Harrisson, S., et al., SG1 Nitroxide-Mediated Polymerization of Isoprene: Alkoxyamine
Structure/Control Relationship and α,ω–Chain-End Functionalization.
127. J Polym Sci Pol Chem 2007, 45 (17), 4100. Germack, D. S., et al., Isoprene polymerization via reversible addition
fragmentation chain transfer polymerization.