memoria practicas quimica uned_2011
TRANSCRIPT

Página 1 de 27
Fundamentos Químicos de la Ingeniería Grado en Ingeniería Mecánica
Curso 2010-2011
PRÁCTICAS DE LABORATORIO

Página 2 de 27
ACTIVADAD DE LOS METALES
1.-INTRODUCCIÓN (Fundamento Teórico) La mayor o menor reactividad de los metales frente a los agentes químicos es muy variada dependiendo no sólo de sus propiedades, sino también, de la naturaleza del agente y de las condiciones del entorno. Los procesos de interacción metal-reactivo, son procesos de tipo redox, en los cuales existe un intercambio de electrones, uno pierde electrones y se oxida y el otro gana electrones y se reduce. El concepto de reactividad o de actividad de un, metal se refiere precisamente a la facilidad con que el metal cede electrones, obviamente el proceso es relativo ya que dependerá del reactivo al que se enfrente. Por ejemplo, la reactividad de los metales frente al ión hidronio (H3O+) del agua y de las disoluciones diluidas de ácidos no oxidantes, es muy variable, se clasifica en tres grandes grupos: Metales muy activos. Reaccionan con el agua pura (H3O+=10-7). Este es el caso de la mayoría de los metales alcalinos y algunos alcalinotérreos (Na, K, Ca). Todos ellos son muy reductores, tienen Potenciales Normales de Reducción muy negativos.
)(22)( 2/1 gs HOHNaOHNa
Metales activos. Reaccionan con el agua en soluciones ácidas (H3O+>10-7) son más reductores que el hidrógeno, por lo que tienen también Potenciales Normales de Reducción negativos. En este grupo se encuentran los metales de mayor interés industrial, entre otros: Mg, Zn, Al, Ti, Cr, Fe, Mn, Co, Ni, Sn y Pb.
OHHZnOHZn gaqaqs 2)(22
)()(3)( 22
Metales no activos. Están por debajo del hidrógeno en la Serie de Potenciales, tienen los Potenciales Normales de Reducción positivos, no reaccionan con el agua ni en soluciones ácidas (no oxidantes). Incluye entre otros: Cu, Ag, Hg, Pd, Os, Ir y Pt. Los más inactivos de este grupo se le nombra también como metales nobles (Ag, Au y Pt). En presencia de otros agentes, incluidos los agentes atmosféricos, la actividad puede cambiar, incluso, se pueden originar productos de oxidación que impiden que la reacción progrese, perdiendo así su actividad. Este es el caso del aluminio que a pesar de ser un metal muy activo, la capa de óxido que se forma en contacto con un agente oxidante, queda frecuentemente adherida al metal protegiéndole de su posterior oxidación. 2.- OBJETIVOS El objetivo fundamental es determinar de una forma experimental la actividad de diversos metales en diferentes medios más o menos reactivos, que nos permitan en cada caso ordenar la reactividad de los metales ensayados. 3.- MATERIALES -Gradilla con tubos de ensayo (16mm x 150mm). -Material auxiliar de laboratorio: Pipetas, probeta, vasos, espátulas, varilla agitadora de vidrio, vidrios de reloj, pinzas de acero inoxidable, lápiz rotulador para vidrio, papel de filtro, etc. -Metales en pequeñas piezas: Zn, Mg, Cu, Pb, Fe. -Guantes. -Papel rejilla. -Recipiente para depositar los residuos de metales.

Página 3 de 27
4.- REACTIVOS -Ácido Clorhídrico: HCl 6N. -Nitrato de Magnesio: Mg(NO3)2 0,1M. -Sulfato de Zinc: ZnSO4 0,1M -Nitrato de Plomo: Pb(NO3)2 0,1M -Nitrato de Cobre: Cu(NO3)2 0,1M -Nitrato de Plata: AgNO3 5.- PROCEDIMIENTO EXPERIMENTAL Experimento 1 Se toman dos piezas de cobre y otras dos de zinc se limpian con papel de lija para quitar la capa de óxido y se introducen en cuatro tubos de ensayo numerados, conteniendo respectivamente: Tubo 1: Zn(s) + 3 ml AgNO3 0,1M Tubo 2: Cu(s) + 3 ml ZnSO4 0,1M Tubo 3: Zn(s) + 3 ml Cu(NO3)2 0,1M Tubo 4: Cu(s) + 3 ml AgNO3 0,1M Se anota si ocurre alguna reacción en el intervalo de unos diez minutos. Debe observar especialmente si se produce algún cambio físico, como: desprendimiento de gases, formación de un precipitado, cambio de color, calentamiento del tubo, etc. Registrar sus observaciones en le cuaderno de laboratorio y emplearlas después para responder a las preguntas formuladas en el apartado 6 (Análisis de Resultados). Experimento 2 Pequeños trocitos de plomo, magnesio, zinc y cobre, convenientemente lijados, se colocan en 8 tubos de ensayo conteniendo soluciones de Pb(NO3)2, Mg(NO3)2 y Cu(NO3)2 con la siguiente distribución: Tubo 1: Mg + 3 ml Pb(NO3)2
Tubo 2: Mg + 3 ml Cu(NO3)2 Tubo 3: Pb + 3 ml Cu(NO3)2 Tubo 4: Pb + 3 ml Mg(NO3)2 Tubo 5: Cu + 3 ml Pb(NO3)2 Tubo 6: Cu + 3 ml Mg(NO3)2 Tubo 7: Zn + 3 ml Mg(NO3)2
Tubo 8: Zn + 3 ml Pb(NO3)2
Se anota si ocurre alguna reacción en el intervalo de unos diez minutos. Debe observar especialmente si se produce algún cambio físico, como: desprendimiento de gases, formación de un precipitado, cambio de color, calentamiento del tubo, etc. Registrar sus observaciones en le cuaderno de laboratorio y emplearlas después para responder a las preguntas formuladas en el apartado 6 (Análisis de Resultados). Experimento 3 Colocar piezas limpias de cobre, zinc, magnesio y hierro en sendos tubos de ensayo conteniendo en todos ellos 3 ml de HCl 6M, anotar como en casos anteriores, si se produce alguna reacción, así como la intensidad en el desprendimiento de gases. Utilice dichas observaciones para contestar a las preguntas del apartado 6.
Página 4 de 27
Precauciones de seguridad:
(1) Las soluciones concentradas de ácidos y bases son peligrosas, pueden producir quemaduras en contacto con la piel, para su manipulación deben usar guantes. En caso de una salpicadura accidental, lavar la piel afectada con agua abundante.
(2) Tanto los residuos de las soluciones metálicas, como los propios metales, son peligrosos para el medioambiente, por ello deben recogerse en contenedores especiales para su posterior gestión.
6.- ANÁLISIS DE LOS RESULTADOS OBTENIDOS Experimento 1 1.1.- Completar la siguiente TABLA I, con los resultados obtenidos en el experimento 1
TABLA I
Tubo Observaciones Elemento oxidado
Elemento reducido
1 Los compuestos reaccionan de manera que se forma un precipitado blanquecino de Ag y el Zn se disuelve. Zn Ag
2 No se observa ninguna reacción transcurrido el tiempo del experimento, el Cu se queda en el fondo del tubo.
3 La reacción es lenta, se descompone el Cu y hay un cambio de coloración en el líquido que pasa de ser azulado a verdoso. Zn Cu
4 La reacción en este tubo es más rápida que en los anteriores, en el fondo del tubo aparece un compuesto blanquecino que es Ag, el cobre desaparece. Cu Ag
1.2.- Escribir las reacciones, si ha lugar, que se producen en cada tubo:
Tubo 1: )(23
120
)(
1
)(3
1
)(
0)( lsls NOZnAgNOAgZn
Tubo 2:
)(4
262
)(
0
ls OSZnCu No hay reacción
Tubo 3: )(2
1
3
20
)()(2
1
3
2
)(
0)()( lsls NOZnCuNOCuZn
Tubo 4: )(2
1
3
2
)(
01
)(3
10
)( )( lsls NOCuAgNOAgCu
1.3.- Completar el orden de reactividad decreciente en los tubos.
Tubo 1 > Tubo 4 > Tubo 3 > Tubo 2 1.4.- Ordenar los metales por actividad creciente (incluida la plata).
Potenciales Normales de Reducción
Zn2+|Zn Zn2+ + 2e = Zn -0,763 Cu2+|Cu Cu2+ + 2e = Cu +0,336 Ag+|Ag Ag+ + e = Ag +0,799
Zn < Cu < Ag
Página 5 de 27
Experimento 2 2.1.- De acuerdo con las posibles reacciones que tienen lugar, completar la TABLA II.
TABLA II
Tubo Observaciones Reacción que tiene lugar Elemento oxidado
Elemento reducido
1 Los elementos reaccionan, reduce Pb y oxida Mg. )(2
1
3
20
)()(2
1
3
20
)( )()( lsls NOMgPbNOPbMg
Mg Pb
2 Reaccionan de manera que Mg oxida y Cu reduce. )(2
1
3
20
)()(2
1
3
2
)(
0)()( lsls NOMgCuNOCuMg
Mg Cu
3 Reacciona de manera que Pb oxida y Cu reduce.
)(2
1
3
20
)()(23
120
)( )()( lslS NOPbCuNOCuPb
Pb Cu
4 No existe reacción en el tubo, el Pb se queda en su estado original
)(2
1
3
20
)( )( ls NOMgPb No hay reacción
5 No hay reacción visible, el Cu se queda en su estado original.
)(2
1
3
20
)( )( ls NOPbCu No hay reacción
6 No hay reacción visible.
)(2
1
3
20
)( )( ls NOMgCu No hay reacción
7 No hay reacción entre los elementos.
)(2
1
3
20
)( )( ls NOMgZn No hay reacción
8 Los elementos reaccionan dando lugar a la precipitación de Pb
)(2
1
3
20
)()(23
120
)( )()( lslS NOZnPbNOPbZn
Zn Pb
2.2.- Ordenar los cinco metales (incluida la plata), por actividad decreciente.
Potenciales Normales de Reducción
Ag+|Ag Ag+ + e = Ag +0,799 Cu2+|Cu Cu2+ + 2e = Cu +0,336 Zn2+|Zn Zn2+ + 2e = Zn -0,763 Pb2+|Pb Pb2+ + 2e = Pb -0,126 Mg2+|Mg Mg2+ + 2e = Mg -2,363
Ag > Cu > Zn > Pb > Mg
Página 6 de 27
Experimento 3 3.1.- Completar las reacciones que tienen lugar en cada tubo. Tubo 1 )()(2)()( 22 gaclS HCuClHClCu Reacción inapreciable por la lentitud de la
misma.
Tubo 2 )()(2)()( 22 gacacs HZnClHClZn Reacción rápida con desprendimiento de gas y aumento de la temperatura.
Tubo 3 )()(2)()( 22 gacacs HMgClHClMg Reacción muy rápida con desprendimiento e gas y aumento de la temperatura.
Tubo 4 )()(2)()( 22 gacacs HFeClHClFe Reacción lenta, se aprecia desprendimiento de gas por el burbujeo en el tubo de ensayo.
3.2.- Comparar la velocidad de desprendimiento de gas en los 4 tubos.
Potenciales Normales de Reducción
Mg2+|Mg Mg2+ + 2e = Mg -2,363 Zn2+|Zn Zn2+ + 2e = Zn -0,763 Fe2+|Fe Fe2+ + 2e = Fe -0,44 Cu2+|Cu Cu2+ + 2e = Cu +0,336
Tubo 3 → Tubo 2 → Tubo 4 → Tubo 1
La diferencia que se aprecia en la velocidad de las reacciones es debido a las diferencias de Potencial Normal de Reducción de los metales. 3.3.- Ordenar por actividad creciente frente al ión H3O+ todos los metales de los experimento, incluida la plata y el hidrógeno.
Potenciales Normales de Reducción
Mg2+|Mg Mg2+ + 2e = Mg -2,363 Zn2+|Zn Zn2+ + 2e = Zn -0,763 Fe2+|Fe Fe2+ + 2e = Fe -0,44
H+|H2 (Pt) 2H+ + 2e = H2 0,000 Cu2+|Cu Cu2+ + 2e = Cu +0,336 Ag+|Ag Ag+ + e = Ag +0,799
Ag < Cu < H < Fe < Zn < Mg

Página 7 de 27
VOLUMETRÍA DE COMPLEJACIÓN: DETERMINACIÓN DE LA DUREZA DEL AGUA
Objetivo general Aplicar los principios básicos de las volumetrías de complejación en la determinación de la dureza de una muestra de agua. Objetivos específicos 1.- Determinar la dureza total, la dureza debida al magnesio y la dureza debida al calcio de una muestra de agua residual. 2.- Reforzar los conceptos relacionados con la formación de complejos metálicos. Duración del experimento 2-3 horas. 1. INTRODUCCIÓN TEÓRICA La dureza del agua se define como la suma de las concentraciones de calcio y magnesio, expresadas como CaCO3 en mg/L. El rango de dureza varía entre 0 y cientos de mg/L, dependiendo de la fuente de agua y el tratamiento a que haya sido sometida. Existen dos tipos de dureza: Dureza temporal: está determinada por el contenido de carbonatos y bicarbonatos de calcio y magnesio. Puede ser eliminada por ebullición del agua y posterior eliminación de precipitados por filtración. Dureza permanente: está determinada por todas las sales de calcio y magnesio excepto carbonatos y bicarbonatos. No puede ser eliminada por ebullición. Por tanto:
Dureza total= Dureza temporal + Dureza permanente
Según la dureza las aguas se clasifican según la tabla siguiente:
Dureza como CaCO3 Clasificación 0-75 mg/L Agua blanda
75-150 mg/L Agua poco dura 150-300 mg/L
(límite para agua potable) Agua dura
>300 mg/L Agua muy dura
En las volumetrías de complejación se mide el volumen de solución tipo, necesario para formar un complejo con un catión metálico del compuesto que se analiza. Muchos cationes metálicos reaccionan con especies dadoras de electrones llamadas ligandos, para formar compuestos de coordinación o complejos. El ligando debe de tener por lo menos un par de electrones sin compartir. Los complejos llamados quelatos, se producen por la coordinación de un catión y un ligando, en los que el catión (metálico) es parte de uno o varios anillos de cinco o seis miembros. Los compuestos orgánicos más conocidos que forman quelatos utilizables en análisis cuantitativo son el ácido nitrilotriacético, el ácido etildiaminotetracético (EDTA) y la sal disódica del EDTA; estos compuestos se conocen comercialmente con los nombres Titriplex I, II y III respectivamente, también se utilizan los nombres de Complexotas, Vercenos o Secuestrenos. El más empleado de los anteriores es la sal disódica del EDTA (Na2-EDTA), por la facilidad de disolución en agua; la solución se prepara por el método directo dado el carácter reactivo tipo primario de la sal disódica.

Página 8 de 27
El EDTA contiene hidrógenos ácidos, por esta razón se representa también como H4Y. El EDTA forma complejos estables con la mayoría de los cationes y entra siempre en relación molar 1:1 en la fórmula del complejo, independiente de la carga del catión, como se muestra en las siguientes reacciones:
Mg+2 + Y-4 → MgY-2
Al+3 + Y-4 → AlY-1
Ca+2 + Y-4 → CaY-2
Ag+ + Y-4 → AgY-3
Los iones formados en las reacciones anteriores son incoloros, de tal manera que para determinar el punto final se emplean indicadores llamados metalcrómicos. Estos tienen la propiedad de formar complejos con cationes como el Ca+2 y el Mg+2 de distinto color al que presenta el indicador libre. Estos indicadores son ácidos débiles que se representan como Hin. Determinación de la dureza total El colorante utilizado para determinar la dureza total del agua (debida al calcio y al magnesio), es eriocromo negro T (ENT). Este colorante es triprótico y existe inicialmente como anión divalente de color azul Hin-2 a pH 10. A la muestra se le adiciona solución buffer de pH 10 +0.1, para mantener la estabilidad de los complejos formados; no puede incrementarse el pH de este valor, por cuanto precipitan el CaCO3 o el Mg(OH)2, además porque el indicador cambia de color a pH elevado, obteniéndose In-3 de color naranja. La reacción del indicador con los iones Ca+2 y Mg+2 presentes en la solución que se valora es del siguiente tipo:
Ca+2 + Mg+2 + ENT + buffer (pH 10) → [Ca-Mg---ENT]
Color azul Complejo púrpura
[Ca-Mg---ENT] + EDTA → [Ca-Mg---EDTA]
Color azul
Al adicionar EDTA a la solución que contiene la muestra con el indicador, el EDTA se combina primero con el Ca+2 y luego con el Mg+2, ya que el complejo EDTA-Ca+2, es más estable que el complejo EDTA-Mg+2 mediante las siguientes reacciones:
EDTA + Ca+2 → [Ca---EDTA] K = 1010.7
EDTA + Mg+2 → [Mg---EDTA] K = 108.7
Determinación de la dureza debida al calcio
El calcio y el magnesio son ambos complejados por el EDTA a pH 10; la determinación de la dureza debida únicamente al calcio se hace a pH elevado (12-13); en este rango de pH , el magnesio precipita como Mg(OH)2 y no interviene en la reacción; además el indicador utilizado para esta determinación sólo se combina con el calcio. El indicador murexida se emplea para determinar la dureza debida al Ca+2, vira de rojo claro (cuando forma el complejo con el Ca+2) a violeta (cuando está libre).
Determinación de la dureza debida al magnesio
La diferencia entre la dureza total y la dureza cálcica (expresada ambas como mg/L de CaCO3), da directamente la dureza magnésica.
Interferencias de precisión
En la determinación de la dureza puede haber interferencias con otros elementos (Al, Cd, Co, Cu, Fe(2), Fe(3), Pb, Mn, Ni, Zn o polifosfátos) que si están presentes son titulados como dureza.

Página 9 de 27
Este método tiene un error relativo del 2% y una desviación estándar relativa de 9,2%.
2. MATERIAL Y PRODUCTOS Materiales - Una probeta de 100ml. - Una pipeta de 10ml. - Dos erlenmeyer de 250ml. - Una bureta de 25ml. - Un vidrio de reloj. - Un matraz aforado de 100ml. - Un matraz aforado de 250ml. - Un frasco lavador. - Un agitador de vidrio. - Un embudo de sólidos. - Papel pH. - Una balanza analítica. - Una espátula.
Productos
- Indicador preparado de negro de eriocromo T. - Murexida. - Trietanolamina, solución de trietanolamina al 30% (V/V). - Solución buffer de pH 10. - Titriplex III (Na2-EDTA) (mantenido en estufa durante dos horas a 105ºC enfriado en un desecador). - Solución de amoníaco, NH3 6N - Cloruro de ácido clorhídrico, HCl 6N - Indicador preparado de murexida. - Solución de hidróxido potásico, KOH al 20% (P/V). - Agua destilada. - Etanol. 3. REFERENCIAS BIBLIOGRÁFICAS 1.- American Society for testing materilas. Annual book of standars 1994. Determinación de dureza de agua. Método ASTM 1126-92. 2.- APHA, AWWA, WPCF. Métodos normalizados para el análisis de aguas potables y residuales. Ed. Díaz de Santos, S.A. Madrid 1992. 3.- RODIER, J. Análisis de las aguas. Ed. Omega. Barcelona. 1981. 4. PROCEDIMEINTO EXPERIMENTAL Preparación del indicador eriocromo negro T
Colocar en un vaso de precipitados de 100 mL 15 mL de trietanolamina y 5mL de etanol; adicione 0,1 g de negro de eriocromo T sólido y disuelva; almacenar en un recipiente.
Preparación de la solución buffer de pH 10

Página 10 de 27
Colocar en un vaso de precipitados de 100 mL 6,56 g de NH4Cl y disolver en 57 mL de amoníaco concentrado y completar a 100 mL con agua destilada en un matraz aforado; almacenar en un recipiente.
Preparación de la solución de Na2-EDTA 0,01 N
Pesar 0,93 g de Na2-EDTA en un matraz aforado de 250 mL, colocar 50 mL de agua destilada y transferir el EDTA al matraz con la ayuda de un embudo y de un frasco lavador, agitar hasta que el EDTA (el EDTA se disuelve lentamente); una vez disuelto completar hasta enrasar con agua destilada. (Si no se prepara la solución en el momento se procederá a estandarizar la solución).
Determinación de la dureza total de la muestra de agua
Medir 100 mL de la muestra en un matraz aforado de 100 mL y colocar en un erlenmeyer de 250 mL, ajustar el pH entre 7 y 10 mediante la adición de NH3 6N o HCl 6N. Adicionar 1 mL de la solución buffer de pH 10 y 2 gotas de indicador eriocromo negro T y agitar.
Adicionar lentamente desde una bureta la solución de EDTA, continuar la titulación hasta que el color de la solución cambie de rojo a azul. Calcular la dureza del agua como el promedio de dos valoraciones que no difieran más de 0,2 mL.
Determinación de la dureza cálcica de la muestra de agua Medir 100 mL de la muestra de agua en un matraz aforado de 100 mL y colocarlos en un erlenmeyer de 250 mL; agregar aproximadamente 4 mL de la solución KOH al 20% (P/V); agitar y ajustar el pH de la solución entre 12 y 13 con la solución de KOH; adicionar aproximadamente 0,1 g del indicador murexida (una punta de espátula). Titular con la solución EDTA hasta que el color de la solución cambie de rosa pálido a violeta. Calcular la dureza debida al calcio como el promedio de dos valoraciones que no difieran en más de 0,2 mL.

Página 11 de 27
5. CÁLCULOS Y RESULTADOS Completar la siguiente tabla y calcular la dureza total del agua (mg/L de CaCO3), la dureza cálcica (mg/L de CaCO3) y la dureza magnésica (mg/L de CaCO3), adicione los cálculos respectivos.
TABLA DE DATOS
Información requerida Ensayo 1 Ensayo 2 Volumen de la muestra de agua (mL) 100 mL 100 mL Normalidad de la solución de EDTA 0,01 N 0,01 N Volumen de EDTA gastado en la dureza total (mL) (VT) 2 mL 2 mL Volumen de EDTA gastado en la dureza de Ca+2 (VCA) 1,5 mL 1,5 mL
Ya que:
muestra de mL1000NV
)Mgy meq/L(Ca T22
total Dureza muestra de mL
100001,40NV )Mgy (Ca mg/L T22
2Ca2 Ca al debida Dureza muestra de mL
100001,40NV )(Ca mg/L
donde, VCa = mL EDTA requeridos usando el indicador para el calcio VT = mL EDTA requeridos usando el indicador de eriocromo negro T N = Normalidad del EDTA Dureza debida al Mg+2 = Dureza total - Dureza debida al Ca+2 Cálculos realizados con los datos obtenidos en el experimento
mg/L 8,002 mL 100
100040,010,01mL 2 )Mgy (Ca mg/L 22
mg/L 8,002 mL 100
100001,4001,0mL 2 )Mgy (Ca mg/L 22
mg/L 6,002 mL 100
100001,4001,0mL 1,5 )(Ca mg/L 2
mg/L 6,401 mL 100
100040,010,01mL 1,6 )(Ca mg/L 2
Dureza debida al Mg+2 = 8,002 mg/L - 6,002 mg/L = 2 mg/L Dureza debida al Mg+2 = 8,002 mg/L - 6,401 mg/L = 1,601 mg/L
Resultados Ensayo 1 Ensayo 2 Promedio Dureza total (mg/L de CaCO3) 8,002 mg/L 8,002 mg/L 8,002 mg/L Dureza debida al Ca+2 (mg/L de CaCO3) 6,002 mg/L 6,401 mg/L 6,201 mg/L Dureza debida al Mg+2 (mg/L de CaCO3) 2 mg/L 1,601 mg/L 1,801 mg/L

Página 12 de 27
6. EJERCICIOS 1.- ¿Cómo se clasifican las aguas en términos de dureza, expresadas como mg/L de CaCO3? ¿A qué clase de agua de acuerda con la dureza corresponde la muestra analizada?
Dureza como CaCO3 Clasificación
0-75 mg/L Agua blanda 75-150 mg/L Agua poco dura
150-300 mg/L (límite para agua potable) Agua dura
>300 mg/L Agua muy dura El agua analizada según la tabla de durezas se corresponde con un agua blanda 2.- ¿Qué problemas ocasiona la dureza en las aguas potables?
La dureza del agua se reconoció originalmente por la capacidad que tiene el agua para precipitar el jabón, esto es, las aguas requieren de grandes cantidades de jabón para producir espuma. Otra característica de suma importancia en la industria, reconocida posteriormente, es la producción de incrustaciones en los tubos de agua caliente, calentadores, boilers y algunas otras unidades en las que la temperatura del agua es alta.
La capacidad de consumo de jabón es de importancia desde el punto de vista económico y por la dificultad de obtener condiciones apropiadas para una limpieza óptima. Sin embargo, con los detergentes sintéticos este problema ha disminuido, por lo que, la demanda del público de aguas suavizadas en las plantas de tratamiento municipal también ha disminuido y la tendencia es hacia instalaciones de ablandamiento privadas e industriales excepto en aquellos lugares en los que la dureza es sumamente alta.
El problema de las incrustaciones no ha disminuido y es de consideración muy importante, principalmente en la industria, porque las incrustaciones pueden obstruir las tuberías a tal grado que se produzcan explosiones o que se inutilicen las unidades de los procesos industriales, resultando más económico darle a las aguas un tratamiento de ablandamiento, que sustituir tuberías, equipo, etc.
Las aguas duras no causan problemas al cuerpo humano y son tan satisfactorias como las aguas blandas sin embargo, la aceptación del público es variable de un lugar a otro, y su sensibilidad depende del grado de dureza al que las personas estén acostumbradas. Muchos consumidores ponen objeción cuando la dureza del agua excede de 150 mg/L CaCO3.
EXTRACCIÓN SIMPLE Experimento 5 Objetivo general Adquirir soltura en la realización práctica de la extracción, así como aprender el fundamento teórico y la utilidad de esta técnica. Objetivos específicos
1. Determinar cuando podrá separarse una sustancia de una mezcla mediante un proceso de extracción.
2. Elegir un disolvente apropiado para realizar la extracción de una sustancia disuelta en otro.

Página 13 de 27
3. Manejar con habilidad el embudo de decantación al realizar una extracción. 4. Tomar las precauciones necesarias durante un proceso de extracción. 5. Determinar en cualquier caso que se presente cuál es la capa del disolvente con el que
se realiza la extracción. 6. Aprender los métodos de romper una emulsión, una vez formada. 7. Separar un compuesto orgánico de carácter ácido de un compuesto neutro por una
extracción con un hidróxido alcalino. Duración del experimento Dos horas, aproximadamente. ESQUEMA 1. INTRODUCCIÓN TEÓRICA
1.A. Definición y utilización de esta técnica. 1.B. Fundamento. 1.C. Equipo y procedimiento operatorio
o Precauciones. o Emulsiones.
2. MATERIAL Y PRODUCTOS 3. TÉCNICAS UTILIZADAS 4. PROCEDIMIENTO EXPERIMENTAL
4.A. Elección de un disolvente para realizar la extracción de hidroquinona. 4.B .Extracción de hidroquinona con éter etílico 4.C. Separación de una mezcla de ácido benzóico y 1,3-dinitrobenceno.
5. EJERCICIOS

Página 14 de 27
1. INTRODUCCIÓN TEÓRICA 1.A. Definición y utilización de esta técnica La extracción consiste en un proceso mediante el cual una sustancia que se encuentra disuelta en un determinado disolvente es transferida a otro disolvente. Es, pues, una técnica que sirve para separar una sustancia de otras con las que se encuentre mezclada y que no sean solubles en un segundo disolvente. La sustancia a separar puede tratarse tanto de un sólido como de un líquido. Esta técnica se utiliza en muchas ramas de la Química, pero sobre todo en la Química Orgánica. Así, en “productos naturales”, para separar un determinado compuesto de otros que lo acompañan en los tejidos de un animal o de una planta, tejidos en los que hay un elevado contenido en agua. Otras veces, para aislar una sustancia orgánica del medio de reacción acuoso en que fue obtenida. Por esto generalmente –aunque no siempre- en las extracciones uno de los disolventes es el agua y el otro un disolvente orgánico (éter dietílico, pentano, hexano, cloroforma, tetracloruro de carbono, etc. son los más utilizados). En otros casos puede ocurrir que la disolución orgánica contenga restos de ácidos inorgánicos o de hidróxidos. Estos restos se eliminan lavando esta disolución orgánica con una solución acuosa diluida de una base o de un ácido inorgánicos, respectivamente. Cuando lo que se contiene son restos de sales, se lava simplemente con agua. Pues bien, estos procesos de lavado no son más que extracciones. Otro ejemplo muy importante es la extracción de un compuesto orgánico aprovechando su carácter ácido o su carácter básico. La disolución orgánica en que se encuentra se trata con una solución de un hidróxido alcalino, si el compuesto es ácido, o con una solución de un ácido inorgánico si fuera básico. De esta forma la sustancia pasa a la solución acuosa en forma de la correspondiente sal, que es insoluble en el disolvente orgánico. Si el compuesto orgánico era ácido se liberará tratando su sal, así formada, con un ácido inorgánico fuerte, y si era básico, tratándola con un hidróxido alcalino. 1.B. Fundamento El fundamento teórico que explica esta transferencia de un disolvente a otro es el siguiente: Tenemos la sustancia o soluto S disuelta en el disolvente. Si agitamos esta disolución con otro disolvente de esa sustancia –disolvente 2- que sea inmiscible con el disolvente 1, resultará que este soluto S se distribuirá o repartirá ahora entre ambos disolventes. Este sistema está constituido por dos fases líquidas, ya que los dos disolventes, como dijimos, son inmiscibles entre sí. Dejando el sistema en reposo las dos fases líquidas se separan en dos capas, llegándose así a una situación de equilibrio, en la que la relación de las concentraciones de la sustancia S en cada disolvente es constante. Esta constante, llamada coeficiente de distribución o también coeficiente de reparto, K, queda definida por la relación
constante) (T CCK
1
2
Donde C1 y C2 son las concentraciones de equilibrio del soluto S en los disolventes1 y 2, respectivamente (expresadas en gramos de S por volumen de disolvente). Esta constante K depende, como toda constante de equilibrio, de la temperatura y, además, del sistema considerado. Es decir, tiene un valor constante para cada soluto distribuido entre dos disolventes determinados a una cierta temperatura (K suele venir dado para la temperatura ambiente, 25ºC). Así, para una cierta sustancia, con que cambiemos uno solo de los dos disolventes, K variará. Sin embargo, K es independiente tanto de la cantidad de soluto como de las cantidades de los disolventes. Podrán separarse selectivamente los componentes de una mezcla simplemente jugando con la distinta solubilidad de cada uno de ellos en el par de disolventes considerado. Para esto será muy importante tener en cuenta la diferente polaridad de dichos componentes. El disolvente extractor habrá de ser, pues, inmiscible con el disolvente en que se encuentra la sustancia a extraer y deberá disolver a ésta mejor que esotro disolvente.

Página 15 de 27
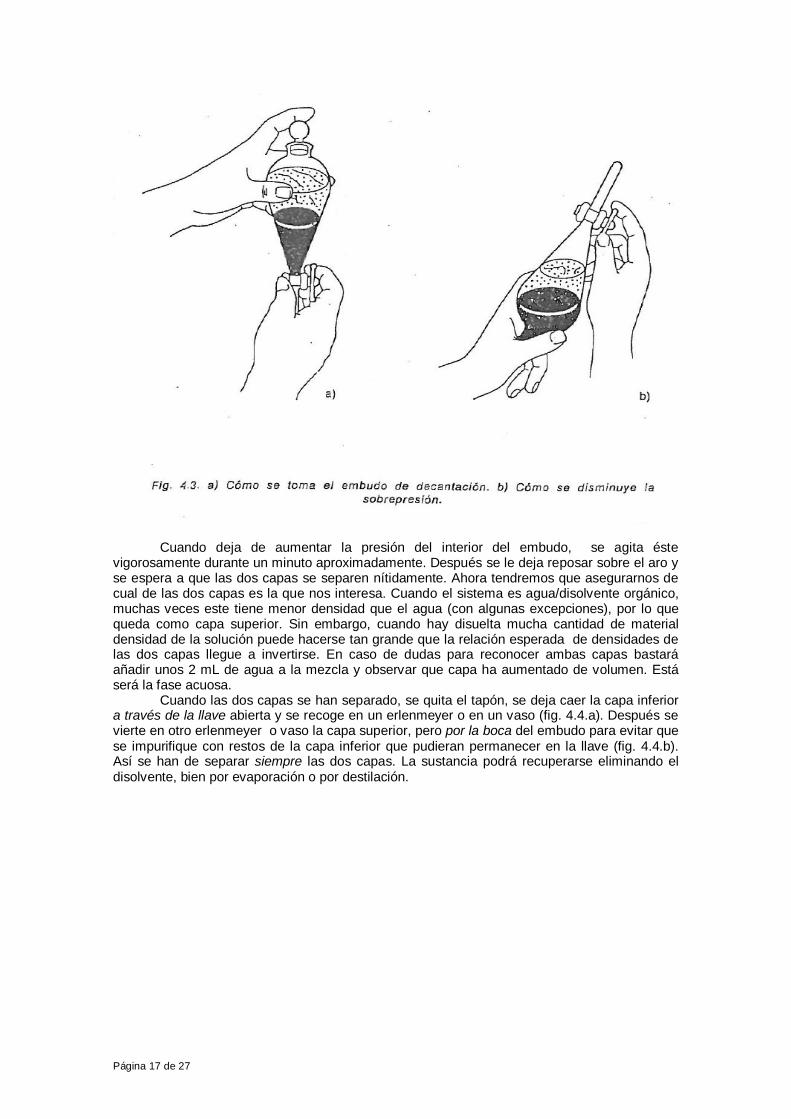
¿Qué cantidad del disolvente extractor será mejor utilizar para separar la mayor proporción de la sustancia S? Esta cantidad es variable, aunque para solutos bastante más en el disolvente extractor que en el agua generalmente es suficiente emplear del disolvente extractor un tercio y una mitad del otro disolvente. 1.C. Equipo y procedimiento operatorio La pieza fundamental utilizada en esta técnica es el embudo de decantación o de extracción (fig. 4.1.a), que esta provisto de una llave en su parte inferior y de un tapón en la superior. Ha de tener el tamaño suficiente para contener de dos a cuatro veces el volumen d el a solución que se vaya a extraer. Este embudo se coloca sobre un aro de hierro –en el que se habrán puesto unos trocitos de tubo de goma para evitar posibles roturas- , situado en un soporte (fig. 4.1.b).

Página 16 de 27
Primeramente, se introduce la solución en el embudo de decantación y, a continuación, el volumen requerido del disolvente extractor (fig. 4.2).
Debe tenerse cuidado de que la solución esté fría, ya que de lo contrario el disolvente orgánico podría hervir al estar en contacto con aquélla, con la consecuente pérdida del mismo (la mayoría de los disolventes orgánicos tienen puntos de ebullición bajos). Siempre debe colocarse un vaso debajo del embudo para que, en caso de rotura de éste, el líquido que contuviera caiga en el vaso. Seguidamente, se cierra el embudo con su tapón se le toma con ambas manos, sujetando con una la llave y con otra el tapón (fig. 4.3.a). Se invierte, sujetándolo siempre de la misma manera, y se agita suavemente. En este momento es muy importante tener en cuenta que, al mezclarse los dos disolventes, la presión de vapor de uno y de otro –presión de vapor parcial- se suman y dan lugar a una presión de vapor total de la mezcla. Esto ocasiona que se produzca en el interior del embudo una sobrepresión. Para disminuir esta presión interna, inmediatamente después de agitar –invirtiendo el embudo- se ha de abrir la llave para que los gases acumulados se vayan, cerrándola luego (fig. 4.3.b) Este proceso de agitar y de abrir la llave es muy importante y se repetirá varias veces hasta que no haya más salida de gases. Si no se eliminan estos gases, podrían hacer saltar el tapón, con el consiguiente derramamiento de líquido.
Página 17 de 27
Cuando deja de aumentar la presión del interior del embudo, se agita éste vigorosamente durante un minuto aproximadamente. Después se le deja reposar sobre el aro y se espera a que las dos capas se separen nítidamente. Ahora tendremos que asegurarnos de cual de las dos capas es la que nos interesa. Cuando el sistema es agua/disolvente orgánico, muchas veces este tiene menor densidad que el agua (con algunas excepciones), por lo que queda como capa superior. Sin embargo, cuando hay disuelta mucha cantidad de material densidad de la solución puede hacerse tan grande que la relación esperada de densidades de las dos capas llegue a invertirse. En caso de dudas para reconocer ambas capas bastará añadir unos 2 mL de agua a la mezcla y observar que capa ha aumentado de volumen. Está será la fase acuosa. Cuando las dos capas se han separado, se quita el tapón, se deja caer la capa inferior a través de la llave abierta y se recoge en un erlenmeyer o en un vaso (fig. 4.4.a). Después se vierte en otro erlenmeyer o vaso la capa superior, pero por la boca del embudo para evitar que se impurifique con restos de la capa inferior que pudieran permanecer en la llave (fig. 4.4.b). Así se han de separar siempre las dos capas. La sustancia podrá recuperarse eliminando el disolvente, bien por evaporación o por destilación.

Página 18 de 27
Este tipo de extracción, mediante la que se ha separado la sustancia en una única operación de extracción, con solo una porción de disolvente se denomina extracción simple.
Por último, acabada la extracción, hay que limpiar bien el embudo de decantación. Deben quitarse siempre la llave y el tapón, limpiarlos y engrasarlos con grasa especial para vidrio esmerilado, antes de colocarlos de nuevo en el embudo. De lo contrario podrían quedarse adheridos a éste muy fácilmente, resultando así inservible. De la misma manera, siempre que se vaya a utilizar el embudo de extracción se lubricarán antes llave y tapón con esta grasa para que queden bien ajustados (si el tapón no es de vidrio, sino de plástico, no hay que echar grasa). Emulsiones
Dentro de las precauciones a tomar en una extracción, el problema de la formación de
emulsiones merece una medición especial. Cuando se realiza una extracción entra agua y un disolvente orgánico de tipo
aromático o clorado, o buen cuando hay sustancias muy viscosas en la solución, es muy frecuente la formación de una emulsión. Una emulsión es la suspensión coloidal de un líquido en la seno de otro, con lo cual pequeñas gotas de la disolución orgánica quedan suspendidas en la disolución acuosa. En estos casos, aunque el sistema se deje en reposo, las dos capas no llegan a separarse. El hecho de que los dos disolventes –aunque les califiquemos de “inmiscibles”- sean siempre algo solubles mutuamente, colabora a la aparición de emulsiones. Por eso, a veces se pueden romper éstas añadiendo una solución saturada de cloruro sódico o de otra sal que, al provocar la aparición de iones en la fase acuosa, la hace más incompatible con la orgánica. Otras veces se consigue su ruptura dejando más tiempo en reposo la mezcla. De todas formas, la destrucción de una emulsión siempre requiere grandes dosis de paciencia

Página 19 de 27
y trabajo, por lo que es mejor evitarlas cuando se sospecha de que pueden formarse. Conviene así que, en lugar de agitar vigorosamente el embudo, girarlo suavemente alrededor de su posición vertical durante dos o tres minutos. O, también, invertirlo varias veces seguidas muy suavemente.
2. MATERIAL Y PRODUCTOS 2.A. Material - Tubos de ensayo. - Dos vidrios de reloj. - Tres vasos de 100 mL (o vasos erlenmeyer). - Una probeta de 25 mL. - Un embudo de decantación de 250 mL. - Un embudo normal cónico. - Un aro. - Un soporte metálico. - Una espátula. - Una varilla de vidrio para agitar. - Papel pH indicador universal. - Balanza. 2.B. Productos - Hidroquinona. C6H4(OH) - Éter etílico. C4H10O - Acetona. C3H6O - Etanol. C2H6O - Benceno. C6H6 - Ácido benzóico. C6H5-COOH - 1,3-dinitrobenceno. C6H4N2O4/C6H4(NO2)2 - Hidróxido sódico. NaOH - Ácido clorhídrico concentrado. HCl - Grasa para esmerilados. 3. TÉCNICAS UTILIZADAS 3.1 Técnicas que se introducen en este experimento - Extracción. - Prueba de solubilidad. - Medida del pH con papel indicador. 3.B. Técnicas introducidas en otros experimentos - Pesada con balanza. - Medida de volumen de líquidos con probeta. - Engrasado de esmerilados de vidrio. - Como agitar tubos de ensayo.

Página 20 de 27
4. PROCEDIMIENTO EXPERIMENTAL
4.A. Elección de un disolvente para realizar la extracción de hidroquinona La hidroquinona es un compuesto orgánico que por su carácter reductor se utiliza en el revelado fotográfico, ya que reduce los iones Ag+ de los haluros metálicos a plata metálica. Su fórmula es
es decir, es el 1,4-dihidroxibenceno. A continuación, probará la solubilidad de la hidroquinona en una serie de disolventes que se la indican seguidamente. Para ello, tome unos cristales de esta sustancia en la punta de su espátula, colóquelos en el fondo de un tubo de ensayo (t.e.) limpio y seco y añada a éste aproximadamente 1 mL (más o menos la altura de 1 cm en el t.e.) de agua destilada. Agite y vea si la sustancia disuelve o no. Repita la misma operación, en sendos tubos de ensayo limpios y secos, con: etanol, acetona, benceno y éter etílico. La hidroquinona (C6H4(OH)2) se ha disuelto en Agua (H2O), Etanol (C2H6O), Acetona (C3H6O) y Éter etílico (C4H10O).
DIS. 1 + S
DIS. 2
DIS. 2 + S
DIS. 1
DIS. 2 + S
DIS. 2 S
Destilar el disolvente
Fig. 4.5. Esquema de la realización de una extracción simple.
DIS. 1

Página 21 de 27
La hidroquinona no se ha disuelto en Benceno (C6H6). Si tuviera hidroquinona disuelta en agua, ¿con cuál/es de los disolventes orgánicos anteriores podría extraerla? Con éter etílico, etanol o acetona.
¿Por qué?
El éter etílico es más ligero que el agua su densidad es de 736 kg/m3, su solubilidad en agua es 6.9 g/100 ml H2O (20 ºC). Se puede separar físicamente sin problemas.
El etanol es más ligero que el agua su densidad es 789 kg/m3. El problema es que es miscible con agua en cualquier proporción; a la concentración de 95% en peso se forma una mezcla azeotrópica (es una mezcla líquida de dos o más componentes que posee un único punto de ebullición constante y fijo, y que al pasar al estado vapor (gaseoso) se comporta como un compuesto puro, o sea como si fuese un solo componente). Habría que sepáralos por destilación.
La acetona también es más ligera que el agua, su densidad es de 790 kg/m3, el problema es que es soluble en agua y al igual que con el etanol habría que proceder a una destilación para su separación.
Seguidamente, extraerá hidroquinona disuelta en agua con un disolvente químico apropiado. 4.B. Extracción de hidroquinona con éter etílico Tome un vidrio de reloj de unos diez cm de diámetro y péselo en la balanza (con una precisión de 0,01 gramos). Anote este peso en su cuaderno de laboratorio (37,37 g). A continuación, pese alrededor de 0,3 g de hidroquinona –anotando la pesada hasta su segunda cifra decimal- (0,30 g) y colóquelos en un vaso de 100 mL. Mida con una probeta 25 mL de agua y viértalos en el vaso. Agite con una varilla y espere hasta disolución total (en caso de que no se disolviera completamente añada otros 5 mL de agua). Coja un embudo de decantación de 125 mL y engrase llave y tapón. Para ello tome en la yema del dedo un poco de grasa de esmerilados (grasa de silicona), saque la llave del embudo y extienda una fina capa de grasa sobre su superficie, introdúzcala de nuevo en el embudo y gire suavemente. Compruebe si al girar no aparece resistencia al roce entre las paredes de esmerilado. Haga lo mismo con el tapón si es de vidrio (si es de plástico no es necesario darle grasa. Corte tres trozos de tubo de goma de unos dos cm de largo, haga una sección longitudinal e introdúzcalos equidistantes en el aro. Monte éste en un soporte y coloque el embudo encima. ADVERTENCIA: Cierre la llave del embudo y coloque debajo un vaso de 50 o de 100 mL (fig. 4.2.). Introduzca, a través de un embudo normal, la disolución de hidroquinona en el embudo de decantación. Después mida con una probeta 15 mL de éter etílico y añádalos al embudo de extracción. Póngale el tapón y tómelo en sus manos, según se ha indicado en el apartado 1.C (fig. 4.3.a).

Página 22 de 27
Invierta el embudo y agítelo un momento. Ahora abra la llave con el embudo invertido, como en la figura 4.3.b ¿Nota algún ruido especial al abrirla?
Sí, un soplido, sale gas del interior del embudo.
En caso afirmativo, ¿a que se debe?
Se libera aire debido al cambio de volumen del contenido porque al mezclar el éter etílico con la disolución de agua e hidroquinona se suman sus presiones de vapor y aumenta el volumen de los gases en el interior del embudo de extracción.
¿Por qué debe abrir la llave?
Para reducir la presión interna de los gases dentro del embudo.
Cierre la llave, agite de nuevo y vuelva a abrirla después. Repita este proceso hasta que no sienta salida de gases. Después, agite el embudo vigorosamente durante un minuto, aproximadamente. Coloque de nuevo el embudo en el aro y déjelo reposar hasta que las dos capas se separen nítidamente. ¿Cuál es la capa acuosa?
La inferior.
Compruébelo. ¿Qué ha hecho para comprobarlo? Como sabemos, la densidad del éter etílico es inferior a la del agua, por lo tanto
éste debería estar siempre en la capa superior; en caso de que al disolverse la hidroquinona hubiera aumentado su densidad nos bastaría con ver la diferencia de volúmenes, en el caso del agua teníamos 30 ml y el éter etílico sólo 15 ml. A la vista se puede apreciar que la solución de mayor volumen es la de la capa inferior. Seguidamente, quite o afloje el tapón, abra la llave del embudo y eje caer la capa inferior en el vaso (al principio puede caer rápidamente, pero cuando quede ya poco líquido cierre parcialmente la llave para que caiga más lentamente). Cuando haya pasado toda esta capa, cierre la llave. Después vierta la capa superior, a través de la boca del embudo, sobre otro vaso o sobre un erlenmeyer. ¿Por qué ha vertido esta capa a través de la boca del embudo?
Para evitar que el agua que haya quedado en llave del embudo se mezcle con la solución de éter e hidroquinona y la contamine.
Ahora -a ser posible en una vitrina- vierta con cuidado el extracto etéreo sobre el vidrio de reloj que antes había pesado. Hágalo poco a poco, a medida que el éter vaya evaporando, para que no se derramen (el éter es muy volátil y se evapora muy rápidamente). ADVERTENCIA: Insistimos: ¡mantenga el ambiente de éter lejos de toda llama! ¿Qué finalidad tienen el colocar el extracto etéreo sobre un vidrio de reloj?
Para facilitar la extracción de la hidroquinona por evaporación del éter etílico.
¿Por qué se utiliza un vidrio de reloj y no otro recipiente?
Porque al ser cóncavo puede contener los restos de hidroquinona y al no tener paredes la velocidad de evaporación del éter es mucho mayor puesto que la circulación de aire ayuda a la misma.

Página 23 de 27
Espere unos 5 ó 10 minutos. Cuando se haya evaporado todo el éter, pese el vidrio de reloj (anote la pesada hasta las centésimas de gramo).
Observe atentamente el contenido del vidrio. ¿Qué será el residuo sólido que aparece en él?
La hidroquinona. ¿Que cantidad de hidroquinona ha extraído?
Peso del vidrio vacío – Peso del vidrio con hidroquinona = Extracto de hidroquinona 37,37 g - 37,65 g = 0,28 g
Finalmente, limpie bien el embudo de decantación, engrase la llave y el tapón y
guárdelo. 4.C. Separación de una mezcla de ácido benzóico y 1,3-dinitrobenceno Tome con una espátula unos 0,3 g de ácido benzoico (no hace falta saber el peso exacto) y póngalos en un vaso de 100mL. Tome otros 0,3 g aproximadamente de 1,3-dinitrobenceno y colóquelos también en el vaso. ¿Qué aspecto tiene el ácido benzoico? Polvo blanco ¿Y el 1,3-dinitrobenceno? Polvo amarillo ocre Añada unos 20 mL de éter etílico y agite con una varilla hasta su disolución total. ¿Qué color tiene la disolución? Color amarillento ocre Prepare ahora el equipo de extracción, como se le indicó anteriormente. Vierta en el embudo de decantación –ayudándose de un embudo normal- la disolución orgánica anterior. Después mida con una probeta unos 15 mL de agua, viértalos en un vaso de 100 mL y añada con una espátula unos 1,5 g de hidróxido sódico (Cuidado: ¡no toque las escamas de hidróxido sódico con las manos, podría quemarse). Agite hasta que se disuelva todo el hidróxido y entonces eche la disolución en el embudo de extracción. ¿Haría falta medir con una pipeta los 20 mL de éter y los 15 mL de agua? No, sería suficiente con la probeta puesto además de venir calibrada, la cantidad de productos para el experimento no tiene que tener la exactitud que ofrece la pipeta, además, las pipetas no suelen ser de más de 5 mL y tendríamos que realizar varias cargas de producto. Tome el embudo de decantación y realice todas las operaciones necesarias para efectuar una extracción. Seguidamente separe ambas fases. Recoja la acuosa en un vaso (o en un erlenmeyer) de 100 mL –vertiéndola a través de la llave del embudo- y la etérea en otro vaso de 100 mL.
¿Por dónde ha vertido la fase etérea? Por ka boca del embudo para que no se contamine con los resto de agua que
pudieran quedar en la llave. En otro vaso coloque unos 5 mL de agua. Mida con una probeta otros 5 mL de HCl
concentrado (¡Cuidado con este ácido!) y vaya añadiéndolos lentamente sobre el agua. Agregue esta solución de HCl diluido sobre la fase acuosa resultante de la extracción poco a poco hasta llegar a pH ácido.

Página 24 de 27
NOTA: Para saber cuando ha llegado a pH ácido utilice papel pH. Este consiste en
unas tiras de papel impregnado en un indicador, sustancia que cambia de color según el pH del medio. El más utilizado es el papel indicador universal, que muestra distintos colores en un intervalo grande de pH. Para medir así el pH, tome un trozo del papel, y con una varilla de vidrio limpia ponga una gota de la solución en él. Compare el color que toma con los de la escala de colores del sobre del papel pH.
¿Qué observa en ese vaso después de acidular? Cambia de color amarillento ocre a color blanquecino. ¿A qué será debido? A la reacción entre el ácido benzoico, el hidróxido de sodio y el ácido clorhídrico. Explique brevemente lo ocurrido La reacción de los compuestos de la disolución es la siguiente:
C7H6O2 + NaOH → C7H5O2Na + H2O
C7H5O2Na + HCl + H2O → ClNa + C7H6O2 + H2O
Lo que ha sucedido según la reacción es que ha precipitado el ácido benzóico y queda una disolución salina en agua de cloruro sódico.
Por otra parte, tome un vidrio de reloj de 10 cm de diámetro y vaya echando en él la
capa etérea (poco a poco, para evitar que se derrame). Cuando se ha evaporado el éter, ¿qué aparece en el vidrio?
Un compuesto de color amarillo ocre. ¿Qué será? Extracto de 1,3-dinitrobenceno. ¿Por qué? Porque su disolución es mejor en éter que en agua. ¿Estará puro? A simple vista no lo sabemos, tendríamos que comprobarlo con algún
procedimiento. Al final limpie bien el embudo de decantación, engrase la llave y el tapón y guárdelo.
Limpie también el resto de material utilizado.

Página 25 de 27
DETERMINACIÓN DEL PUNTO DE FUSIÓN
1.- FUNDAMENTO TEÓRICO 1.1. INTRODUCCIÓN Los métodos y los aparatos descritos a continuación se utilizan para determinar el punto de fusión de los productos químicos, cualquiera que sea su grado de pureza. La selección del método dependerá de la naturaleza de la sustancia problema. En consecuencia, el factor limitante dependerá de que la sustancia sea fácil, difícilmente o no pulverizable. 1.2. DEFINICIONES Y UNIDADES El punto de fusión se define como la temperatura a la que se produce la transición de fase del estado sólido al líquido a presión atmosférica normal; esta temperatura corresponde idealmente a la temperatura de congelación. Dado que la transición de fase de numerosas sustancias se extiende en una amplia gama de temperaturas, ésta se designa muchas veces con el nombre de intervalo de fusión. Conversión de las unidades (K a °C) t = T - 273,15 t = temperatura Celsius, grado Celsius (°C) T = temperatura termodinámica, Kelvin (K)
1.3. SUSTANCIAS DE REFERENCIA No es necesario emplear sustancias de referencia cada vez que se estudie una nueva sustancia. Deberán servir, esencialmente, para comprobar el funcionamiento del método de vez en cuando y para comparar con los resultados obtenidos mediante otros métodos. 1.4. PRINCIPIO DEL MÉTODO DE ENSAYO Se determina la temperatura (o intervalo de temperatura) de transición de fase del estado sólido al líquido o viceversa. En la práctica, las temperaturas del inicio y del final del proceso de fusión/congelación se determinan al calentar/enfriar una muestra de la sustancia problema a presión atmosférica. Se describen dos tipos de métodos: el método de tubo capilar y el método de superficie caliente. 1.4.1. Método de tubo capilar 1.4.1.1. Dispositivos de temperatura de fusión con baño líquido Introducir en un tubo capilar una pequeña cantidad de sustancia finamente pulverizada y comprimirla firmemente. Calentar dicho tubo al mismo tiempo que un termómetro y ajustar el aumento de temperatura a poco menos de 1 K por minuto, durante la fusión real. Tomar nota de las temperaturas correspondientes al comienzo y al final de la fusión. 1.4.1.2. Dispositivos de temperatura de fusión con bloque metálico El fundamento es el mismo que el descrito en el apartado 1.4.1.1, con la diferencia de que el tubo capilar y el termómetro están colocados en un bloque de metal calentado y se observan a través de aberturas practicadas en este último. 1.4.1.3. Detección fotoeléctrica

Página 26 de 27
Calentar automáticamente en un cilindro metálico la muestra contenida en el tubo capilar. Por una abertura practicada en el cilindro, enviar un rayo de luz a través de la sustancia hacia una célula fotoeléctrica cuidadosamente calibrada. En el momento de la fusión, las propiedades ópticas de la mayor parte de las sustancias se modifican en el sentido de que la opacidad da paso a la transparencia. En consecuencia, la intensidad de la luz que llega a la célula fotoeléctrica aumenta y envía una señal de parada al indicador digital que registra la temperatura del termómetro de resistencia de platino colocado en la cámara de calentamiento. Este método no es aplicable a determinadas sustancias muy coloreadas. 1.4.2. Método de superficie caliente 1.4.2.1. Método de la placa caliente de Kofler La placa caliente de Kofler se compone de dos piezas de metal de conductividad térmica diferente, que se calientan eléctricamente. Está hecha de manera que el gradiente de temperatura sea casi lineal en toda su longitud. La temperatura de dicha placa puede variar de 283 a 573 K gracias a un dispositivo especial de lectura de la temperatura que tiene un cursor con un índice y una regleta graduada, especialmente concebido para dicha placa. Para determinar un punto de fusión se deposita una fina capa de sustancia directamente sobre la placa caliente. En unos segundos, se forma una fina línea de división entre la fase fluida y la fase sólida. Leer la temperatura a la altura de dicha línea, colocando el índice frente a esta última. 1.4.2.2. Microscopio de fusión Se utilizan diferentes microscopios de platina caliente para determinar puntos de fusión con cantidades de sustancia muy pequeñas. La temperatura se suele medir con un termopar sensible, pero a veces se usa un termómetro de mercurio. El dispositivo tipo tiene una carcasa de calor que contiene una platina de metal en la que se coloca una lámina de vidrio sobre la que se deposita la muestra. El centro de la platina metálica se atraviesa con un agujero que permite el paso de la luz procedente del espejo de iluminación del microscopio. Al utilizarlo, la carcasa se cierra con una placa de vidrio para impedir la entrada de aire a la zona de la muestra. El calentamiento de la muestra se regula con un reóstato. Para realizar mediciones muy precisas se puede utilizar luz polarizada en el análisis de las sustancias ópticamente anisótropas. 1.4.2.3. Método de menisco Este método se aplica específicamente a las poliamidas. Se determina la temperatura a la cual se observa, a simple vista, el desplazamiento de un menisco de aceite de silicona, atrapado entre una superficie caliente y un cubreobjetos colocado encima de la muestra de poliamida.

Página 27 de 27
Medición del punto de fusión mediante el tubo de Thiele
Equipo económico para determinación manual de los puntos de fusión y ebullición diseñado para laboratorios con rutinas y requerimientos moderados.
Nos ofrece:
Pantalla amplia y clara 4 puntos de calibración Tiempos de calentamiento y enfriamiento cortos Rango de temperatura hasta 400ºC Analiza 3 muestras simultáneamente