manual qir · alquenos y dienos 329 tema 5. alquinos 343 ... los átomos de o, n, y los halógenos...
TRANSCRIPT

Manual QIR
VOLUMEN VB: MANUAL DE QUÍMICA ORGÁNICA
Autor: Dr. Melvin M. Morales Aguilera
Editora: Dra. Iliana Perdomo López

Manual QIR. VOLUMEN V: MANUAL DE QUÍMICA
INORGÁNICA Y ORGÁNICA
Autor: Dr. Melvin M. Morales Aguilera
© Iliana Perdomo López (editora). Depósito legal: DLC 616-2012
Reservado todos los derechos. Está prohibido, bajo las sanciones penales y el resarcimiento civil previsto en las leyes, reproducir, registrar o transmitir esta publicación, íntegra o parcialmente por cualquier sistema de recuperación y por cualquier medio, sea mecánico, electrónico, magnético, por fotocopia o por cualquier otro, sin la autorización previa por escrito de la editora.

UNIDAD II ÍNDICE DE QUÍMICA ORGÁNICA
TEMA 1.INTRODUCCIÓN. CONCEPTOS GENERALES 293
TEMA 2. HIDROCARBUROS ALIFÁTICOS. ALCANOS 315
TEMA 3. HALOALCANOS ( DERIVADOS HALOGENADOS) 321
TEMA 4. ALQUENOS Y DIENOS 329
TEMA 5. ALQUINOS 343
TEMA 6. COMPUESTOS ORGANOMETÁLICOS 351
TEMA 7. ALCOHOLES, FENOLES Y TIOLES 365
TEMA 8. ÉTERES Y EPÓXIDOS. TIOÉTERES 377
TEMA 9. BENCENO. SISTEMAS AROMÁTICOS 385
TEMA 10. ALDEHÍDOS Y CETONAS 393
TEMA 11. ÁCIDOS CARBOXÍLICOS Y DERIVADOS 411
TEMA 12. COMPUESTOS DIFUNCIONALIZADOS 425
TEMA 13. CARBOHIDRATOS 431
TEMA 14. SÍNTESIS DE PÉPTIDOS 443
TEMA 15. AMINAS 449
TEMA 16. HETEROCICLOS 457
TEMA 17. REACCIONES PERICÍCLICAS 479
TEMA 18. PRODUCTOS NATURALES 489
TEMA 19. POLÍMEROS 501
ANEXOS 507
BIBLIOGRAFÍA 521
Química Orgánica InspiracleQIR/2015
293
TEMA 1. INTRODUCCIÓN. CONCEPTOS GENERALES. (2003) 114, 122, 135;
(2004) 116, 121, 126, 130; (2005) 98, 99, 102, 103, 105, 106, 107, 118, 119, 121, 123, 127; (2006) 93, 95, 96,
103, 106, 109, 119; (2007) 127, 132, 133; (2008) 92, 97, 101, 103, 123, 131; (2009) 13, 36, 39, 40, 140, 255;
(2010) 88, 97, 98, 115, 116, 254; (2011) 94, 103, 109, 120, 128; (2012) 105, 226, 230, 232; (2013) 77, 115;
(2014) 76.
QUÍMICA ORGÁNICA: rama de la Química que estudia la estructura, propiedades,
aplicaciones, síntesis y reactividad de los compuestos cuyo elemento básico de constitución es el
átomo de carbono; por ello, con frecuencia, esta parte de la ciencia es denominada QUÍMICA
DEL CARBONO.
ENLACE EN LOS COMPUESTOS DEL CARBONO (MOLÉCULAS ORGÁNICAS):
El átomo de C presenta una extraordinaria capacidad para combinarse consigo mismo y con otros
elementos, frecuentemente con los átomos de H, O y N, y con kenor asiduidad los átomos de S, P
y halógenos.
En estas uniones, el átomo de C forma enlace covalente. Su estructura electrónica es 1s2 2s2 2p2 y
tiende a adquirir una estructura más estable (la estructura del gas noble Ne, con 8 electrones en la
capa de valencia 1s2 2s2 2p6) compartiendo sus 4 electrones más externos.
La teoría de LEWIS constituye una visón simplificada y clásica, y muy útil en Química Orgánica,
del enlace covalente. La descripción mecánico-cuántica del enlace basada en la HIBRIDACIÓN
DE ORBITALES. También permite explicar la tetravalencia del átomo de C. Según esta teoría el
átomo de carbono forma enlaces covalentes por solapamiento de orbitales atómicos híbridos. Son
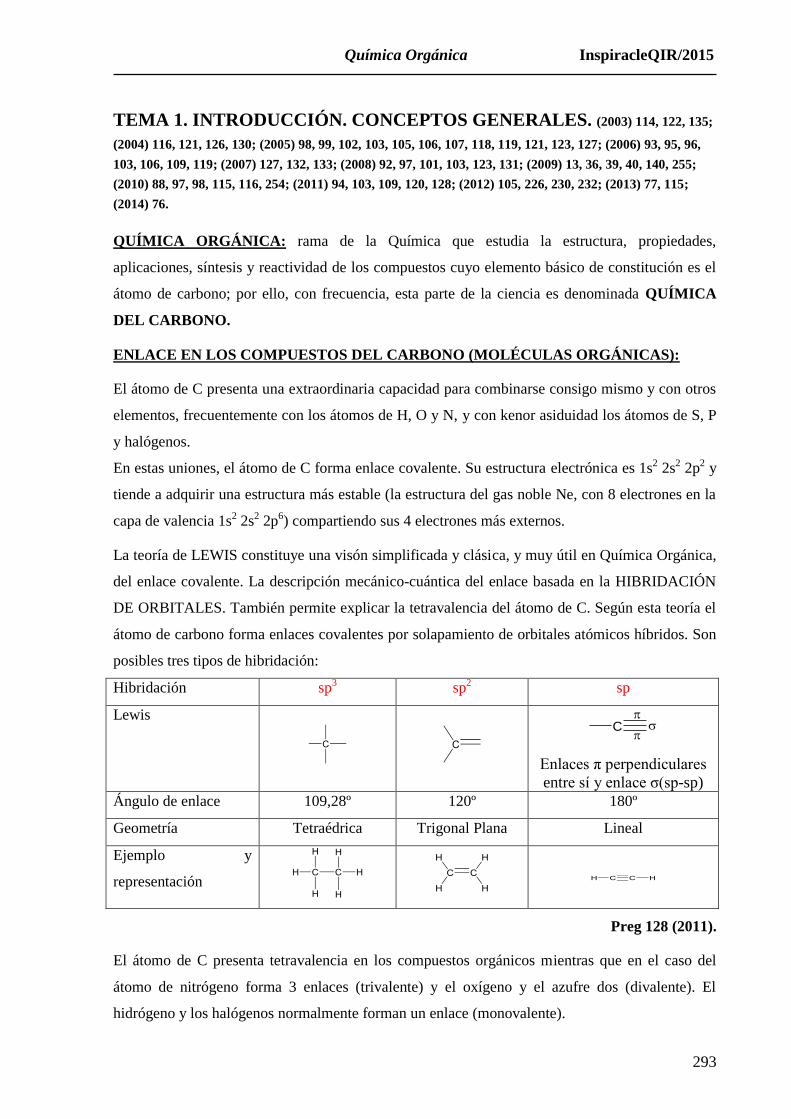
posibles tres tipos de hibridación:
Hibridación sp3 sp2 sp
Lewis
C
Enlaces π perpendiculares
entre sí y enlace σ(sp-sp)
Ángulo de enlace 109,28º 120º 180º
Geometría Tetraédrica Trigonal Plana Lineal
Ejemplo y
representación
Preg 128 (2011).
El átomo de C presenta tetravalencia en los compuestos orgánicos mientras que en el caso del
átomo de nitrógeno forma 3 enlaces (trivalente) y el oxígeno y el azufre dos (divalente). El
hidrógeno y los halógenos normalmente forman un enlace (monovalente).
InspiracleQIR/2015 Química Orgánica
294
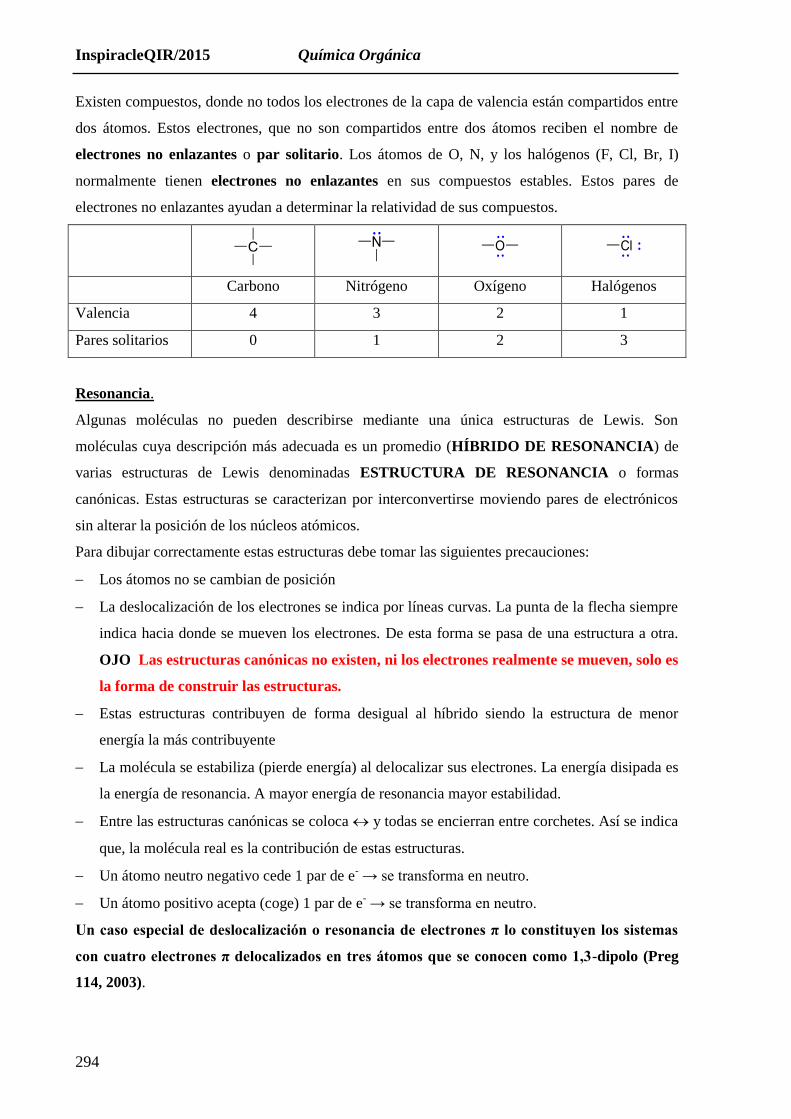
Existen compuestos, donde no todos los electrones de la capa de valencia están compartidos entre
dos átomos. Estos electrones, que no son compartidos entre dos átomos reciben el nombre de
electrones no enlazantes o par solitario. Los átomos de O, N, y los halógenos (F, Cl, Br, I)
normalmente tienen electrones no enlazantes en sus compuestos estables. Estos pares de
electrones no enlazantes ayudan a determinar la relatividad de sus compuestos.
Carbono Nitrógeno Oxígeno Halógenos
Valencia 4 3 2 1
Pares solitarios 0 1 2 3
Resonancia.
Algunas moléculas no pueden describirse mediante una única estructuras de Lewis. Son
moléculas cuya descripción más adecuada es un promedio (HÍBRIDO DE RESONANCIA) de
varias estructuras de Lewis denominadas ESTRUCTURA DE RESONANCIA o formas
canónicas. Estas estructuras se caracterizan por interconvertirse moviendo pares de electrónicos
sin alterar la posición de los núcleos atómicos.
Para dibujar correctamente estas estructuras debe tomar las siguientes precauciones:
Los átomos no se cambian de posición
La deslocalización de los electrones se indica por líneas curvas. La punta de la flecha siempre
indica hacia donde se mueven los electrones. De esta forma se pasa de una estructura a otra.
OJO Las estructuras canónicas no existen, ni los electrones realmente se mueven, solo es
la forma de construir las estructuras.
Estas estructuras contribuyen de forma desigual al híbrido siendo la estructura de menor
energía la más contribuyente
La molécula se estabiliza (pierde energía) al delocalizar sus electrones. La energía disipada es
la energía de resonancia. A mayor energía de resonancia mayor estabilidad.
Entre las estructuras canónicas se coloca y todas se encierran entre corchetes. Así se indica
que, la molécula real es la contribución de estas estructuras.
Un átomo neutro negativo cede 1 par de e- → se transforma en neutro.
Un átomo positivo acepta (coge) 1 par de e- → se transforma en neutro.
Un caso especial de deslocalización o resonancia de electrones π lo constituyen los sistemas
con cuatro electrones π delocalizados en tres átomos que se conocen como 1,3-dipolo (Preg
114, 2003).

Química Orgánica InspiracleQIR/2015
299
Están favorecidas por una débil diferencia de polaridad entre A y B.
Tienen lugar en fase gaseosa o en disolventes no polares; muchas sólo transcurren a
temperaturas elevadas.
Se ven afectadas por catalizadores que son compuestos de los metales de transición.
Se forman radicales que atacan a otras moléculas, con lo que el proceso se propaga dando
lugar a una reacción en cadena, cuya terminación tiene lugar al desaparecer los radicales por
unión entre sí.
La activación fotoquímica favorece este tipo de rupturas. Es un ejemplo característico la
halogenación (bromación, cloración) fotoquímica de alquenos.
RUPTURA HETEROLÍTICA: sólo uno de los dos átomos enlazados retiene los dos electrones
del enlace. Se forman CATIONES y ANIONES.
Están favorecidas por una fuerte diferencia de polaridad entre A y B.
Tienen lugar en solución, y sólo muy raramente en fase gaseosa, estando favorecidas por los
disolventes de constante dieléctrica elevada.
Se ven afectadas por catalizadores de tipo electrolito, como los ácidos y las bases.
La velocidad de reacción aumenta al elevarse la temperatura.
Su cinética es simple, y son frecuentes las reacciones de segundo orden.
Carbocatión o ión carbonio:
Cuando los dos electrones del enlace se separan con el sustituyente X, el fragmento orgánico
resultante se denomina ión carbonio o carbocatión, y el átomo de carbono procedente del enlace
soporta una carga positiva. Los carbocationes son iones positivos que contienen un átomo de
carbono con solo seis electrones en tres enlaces. Son planos y tienen hibridación sp2.
Debido a su deficiencia electrónica, los carbocationes tienen gran tendencia a captar electrones y
completar así su octete. Los sustituyentes que ceden electrones estabilizan los carbocationes por
efecto +I (disminuyen densidad de carga positiva), mientras que los aceptores de electrones lo
desestabilizan por efecto –I (incrementan densidad de carga positiva). En el caso de los
carbocationes bencilo y alilo, estos son más estables (Preg 149, 2007) por estar estabilizados
por resonancia.

InspiracleQIR/2015 Química Orgánica
300
Un catión con una carga positiva en uno de los dos átomos de carbono de un doble enlace
C=C se conoce como catión vinilo (Preg 127, 2005).
C
- Plano (sp2)- orbital p vacío
Estabilidad: Bencilo alilo > 3º > 2º > 1º > CH3+
Preg 105 (2012)
Carbanión:
Cuando los dos electrones del enlace quedan con el fragmento orgánico resultante, se origina un
carbanión, y el átomo de carbono soporta carga negativa. Los carbaniones son iones
negativamente cargados que contienen un átomo de carbono con tres enlaces y un par electrónico
sin compartir.
Tienden a ceder su par de electrones no compartido para formar un enlace covalente coordinado.
La estabilidad de los carbaniones también está muy influenciada por la naturaleza de los átomos o
sustituyentes unidos al átomo de carbono deficitario en electrones. Así, los sustituyentes que
ceden electrones desestabilizan los carbaniones por efecto +I (aumentan la densidad de carga
negativa), mientras que los aceptores de electrones lo estabilizan por efecto –I (reducen la
densidad de carga negativa.
C
- Carbono sp3, piramidal, con 2 e- en orbital no enlazante
Estabilidad: CH3- > 1º > 2º > 3º
Las reacciones que suceden en una sola etapa, la formación de los nuevos enlaces se produce al
mismo tiempo que la ruptura de los enlaces, se denominan concertadas y transcurren sin
formación de intermedios. Las reacciones en varias etapas sí originan intermedios (y el número de
intermedios es el número de etapas menos uno). Preg 115 (2013); Preg 76 (2014).
De particular interés resultan los carbaniones alílicos y bencílicos puesto que están estabilizados
por resonancia.
Carbenos:
En ciertas reacciones se forman especies altamente reactivas como los carbenos, cuyo átomo
de carbono presenta 6 electrones de valencia. Preg 101 (2008); Preg 13 (2009); Preg 115 (2010).
El carbeno más sencillo es el metileno (CH2:) el cual se forma por el calentamiento (exposición a
la luz o en presencia de Cu metálico) del diazometano, liberándose nitrógeno.

InspiracleQIR/2015 Química Orgánica
312
Racemización: es la transformación de un compuesto ópticamente activo por combinación
con su enantiómero (opuesto óptico) o por conversión en racémico. Es la pérdida de
actividad óptica que ocurre cuando una reacción no muestra total retención de la
configuración ni total inversión de la configuración (Preg 121, 2005).
8) Resolución: separación de los dos enantiómeros de una mezcla. Preg 100 (2011). Métodos:
Método Pasteur: separación mecánica de cristales de los dos enantiómeros.
Conversión en diastereoisómeros.
Separación o resolución cinética: los enantiómeros se hacen reaccionar con reactivo
quiral no racémico. Se separan porque las velocidades de los enantiómeros son
diferentes (Preg 126, 2004; Preg 94, 2005).
9) Rendimiento óptico: exceso enantiomérico de los productos en relación a los materiales de
partida (Preg 108, 2004; Preg 107, 2005).
10) Reacciones estereoselectivas: reacciones que conducen a la formación preferente de un
estereoisómero sobre otros que podrían formarse. Preg 88 (2010).
11) Nomenclatura eritro y treo:
Los términos eritro y treo se utilizan en moléculas disimétricas, cuyos extremos son
diferentes. El diastereómero eritro es el que tiene los grupos similares al mismo lado en la
proyección de Fischer, mientras que el diastereómero treo tiene grupos similares en lados
opuestos de la proyección de Fischer.
12) Molécula Proquiral: Compuestos aquirales que se convierten en quirales a través de una
sencilla transformación.

Química Orgánica InspiracleQIR/2015
313
Ejemplos de Casos posibles. P reg 232 (2012).
1 centro asimétrico 2n = 21 = 2 estereoisómeros R y S son enantiómeros
2 centros asimétrico (A - B) 2n = 22 = 4 estreoisómeros 2 pares de enantiómeros
Ejemplo: 2-bromo-3-clorobutanodiferentes
CH
CH3
C
Br
ClH
CH3
CH
CH3
C
Br
HCl
CH3
CH3
BrH
Cl
CH3
H
CH3
BrH
H
CH3
Cl
No son imágenesespeculares
1
2
3
4
1 1 1
2 2 2
33 3
4 4 4
R
S
R S S
S S S
(2S,3R) (2S,3R) (2S,3S)(2R,3R)
enantiómeros enantiómeros
CH3
HBr
OH
CH3
Cl
1
2
3
4
S
R
(2R,3S)
CH3
HBr
Cl
CH3
H
1
2
3
4
R
R
(2S,3S)
diastereómeros entre sí
Dos compuestos en que todos sus carbonos asimétricos tienen configuraciones opuestas son enantiómeros
Dos compuestos en que no todos sus carbonos asimétricos tienen configuraciones opuestas son enantiómeros
2 centros asimétricos3 estereoisómeros 1 par de enantiómeros y una forma MESO
de igual composición(A - A)
CH
COOH
C
OH
OHH
COOH
CH
COOH
C
OH
HHO
COOH
COOH
OHH
OH
COOH
H
COOH
OHH
H
COOH
HO
No son imágenesespeculares
1
2
3
4
1 1 1
2 2 2
33 3
4 4 4
R
S
R
R RS
R R
(2R,3S) (2S,3R) (2S,3S)(2R,3R)
mismo estereoisómero enantiómeros
Ej: ácido tartárico (ácido 2,3-hidroxi-butanodioico)
COOH
HHO
OH
COOH
HO
1
2
3
4
S
R
(2R,3S)
COOH
HHO
OH
COOH
H
1
2
3
4
S
S
(2S,3S)
forma MESO
diastereómeros entre sí
La forma MESO es un diastereoisómero respecto de cualquiera de los dos enantiómeros. Es un compuesto simétrico (presenta plano de simetría y un centro de simetría), y por ello no es ópticamente activa. Es un estreoisómero que noes quiral a pesar de presentar C*

InspiracleQIR/2015 Química Orgánica
314
Topicidad
En Química, la topicidad es la relación estereoquímica que se establece entre dos grupos que están
unidos a un mismo átomo. Desde el puto de vista estereoquímico, y dependiendo de esa relación,
esos grupos pueden ser homotópicos, enantiotópicos o diasterotópicos.
Los grupos homotópicos son aquellos donde todos los grupos son equivalentes. Dos átomos A y B
son homotópicos si la molécula permanece igual (estereoquímicamente) cuando estos grupos
dichos grupos pueden intercambiarse con el resto de los grupos y la estructura de la molécula no
se afecta. Por ejemplo, los cuatro átomos de hidrógeno del metano (CH4) son homotópicos. Lo
mismo pasa con los átomos de hidrógeno del cloruro de metileno (CH2Cl2).
El término enantiotópico se refiere a la relación que se establece entre dos átomos, los cuales, si
uno de ellos es reemplazado, se generará un compuesto quiral, dando una mezcla de
enantiómeros. Así, si un átomo de carbono está unido a dos hidrógenos y a dos grupos
diferentes, los dos hidrógenos se denominan enantiotópicos (Preg 133, 2007).
butano (R)-2-bromobutano (S)-2-bromobutano
El término diastereotópico se refiere a la relación que se establece entre dos átomos, los cuales, si
uno de ellos es reemplazado, una mezcla de diastereoisómeros:
(S)-2-bromobutano (2S,3R)-2,3-dibromobutano (2S,3S)-2,3-dibromobutano

Química Orgánica InspiracleQIR/2015
321
TEMA 3. HALOALCANOS (DERIVADOS HALOGENADOS). (2005) 91, 96;
(2008) 90; (2009) 7, 9; (2010) 130); (2012) 88; (2013) 95, 108; (2014) 104.
Los haloalcanos representan una de las familias de compuestos orgánicos que han encontrado
gran aplicación industrial. Por ejemplo, un grupo de compuestos clorofluorcarbonados
llamados freones han sido utilizados como refrigerantes, propelentes y disolventes (Preg 128,
2007; Preg 110, 2008).
Propiedades físicas: Preg 65 (2012)
F Cl Br I
Tamaño
Electronegatividad
Longitud de enlace: aumenta al aumentar X
Polaridad de enlace: enlaces polares
Fuerza de enlace: disminuye al aumentar X (Preg 257, 2005; Preg 153, 2007)
C X
Cuanto más polar es X mayor polarización del enlace
Puntos de ebullición: más elevados que en alcanos debido a que las fuerzas de interacción entre moléculas son mayores. Además, mayores al aumentar el tamaño de X
Preg 116 (2010)
REACTIVIDAD CARACTERÍSTICA
Reacciones de Sustitución:
Mecanismo SN2 (sustitución nucleófila bimolecular): Preg 253, 259 (2011); Preg 87 (2012).
- Reacción concertada (1 etapa). Preg 129 (2010).
- Reacción bimolecular y sigue una cinética de orden 2 [ ][ ]v k RX Nu
- Estereoquímica Inversión de la configuración del centro atacado. (Estereoespecífica).
(Preg 104 (2014).
- Ataque dorsal

Química Orgánica InspiracleQIR/2015
327
El ión -OH (hidróxido), RO- (alcóxido), -NH2 y otras bases fuertes son grupos salientes malos en
las reacciones SN2. Por ejemplo, el grupo OH de un alcohol es un grupo saliente malo porque
tendría que liberarse como ión hidróxido.
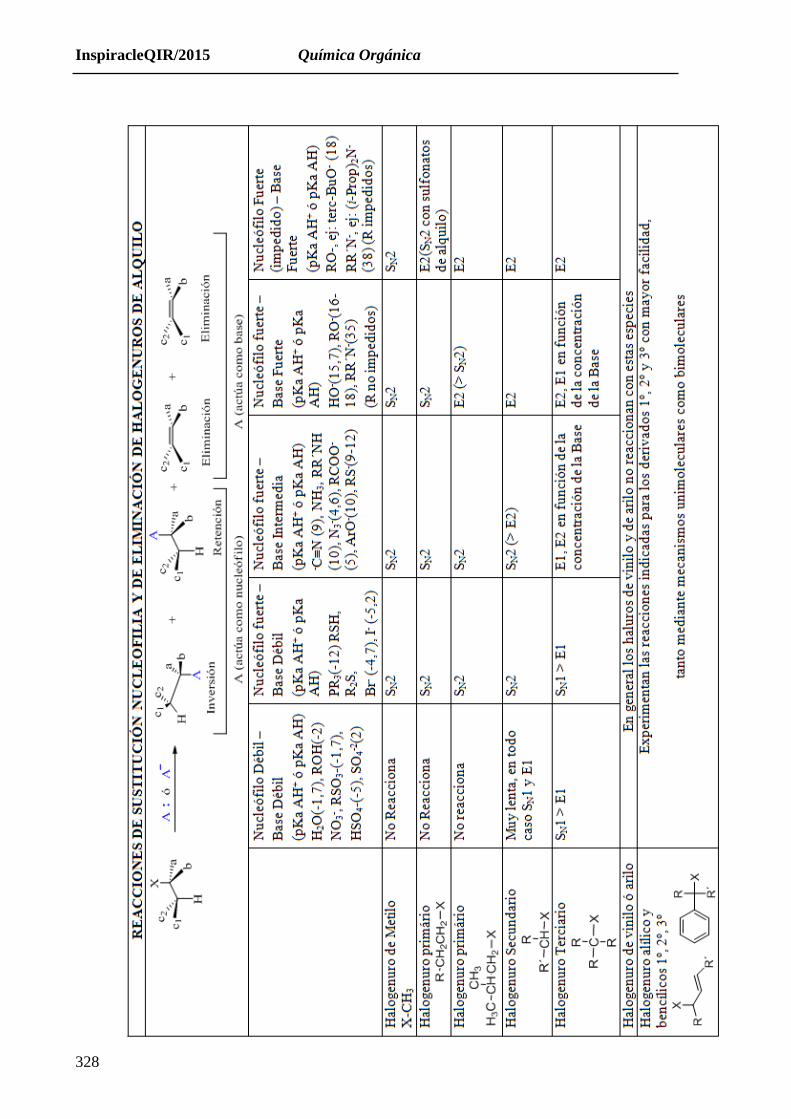
En la siguiente tabla resumimos como pueden ocurrir los procesos de sustitución o eliminación en
función del tipo de R-X o Nucleófilo o base (A:; A¯) que reaccionan entre sí
InspiracleQIR/2015 Química Orgánica
328

Química Orgánica InspiracleQIR/2015
329
TEMA 4. ALQUENOS Y DIENOS. (2003) 92, 93, 95, 99, 123, 124; (2004) 82, 95, 98, 109,
118; (2005) 110, 120, 134; (2006) 100, 112, 113, 127; (2007) 145, 151, 167; (2008) 81, 122, 126; (2009) 10, 15,
16, 24, 25, 29, 42, 45, 46, 47; (2010) 85, 89, 91, 94, 95, 114, 123; (2011) 84, 101; (2012) 61, 62, 72, 77, 90,95,
96; (2013) 72, 80, 82, 89, 110; (2014) 81, 90, 102, 115.
Alquenos: hidrocarburos que contienen dobles enlaces C=C.
Fórmula CnH2n.
Son hidrocarburos que también reciben el nombre de olefinas.
Nomenclatura E-Z: (YA VISTA ANTEIORMENTE EN ESTEREOISOMERÍA: Regla
Cahn-Ingold-Prelog).
Estructura electrónica y enlace:
- Hibridación sp2 de los átomos de C.
- Doble enlace = σ + π Preg 127 (2006) σ = Csp2-Csp2 π = C2p-C2p
- Geometría trigonal plana para cada átomo de C del doble enlace.
- El doble enlace C=C es más fuerte y de menor longitud que el enlace C-C en alcanos.
- El enlace σ es más fuerte (108 Kcal/mol) que el enlace π (65 Kcal/mol).
- Los alquenos son planos. No existe posibilidad de libre giro alrededor de los dobles
enlaces.
- El doble enlace de los alquenos constituye una región rica en electrones. De esta forma
los alquenos son nucleófilos y reaccionan con electrófilos (Preg 92, 2003).
Estabilidad relativa de alquenos:
La estabilidad de los alquenos es mayor cuanto más sustituidos están y el alqueno trans es más
estable que el cis. Preg 91 (2010).
La variable que permite establecer esta estabilidad es el CALOR DE HIDROGENACIÓN (Preg
95, 2004). Mientras menor calor de hidrogenación mayor estabilidad.
¿Por qué el más sustituido es el más estable? Preg 72 (2012).
Efectos inductivos de los grupos alquilos unidos satisfacen las características aceptoras de e-
de los Csp2 del doble enlace. Además efecto de hiperconjugación. Preg 102 (2014).
¿Por qué el trans es más estable que el cis?

InspiracleQIR/2015 Química Orgánica
330
En el isómero cis los dos sustituyentes voluminosos del doble enlace interfieren entre ellos, se
produce impedimento estérico y se eleva la energía de tensión de van der Waals; en el isómero
trans, los sustituyentes están más alejados, interferencia estérica menor y por tanto menor energía
(más estable). Preg 47 (2009).
Teniendo en cuenta esto se puede predecir dado una serie de alquenos, cuál de ellos es más
estable (Preg 167, 2007):
2-metil-2-buteno 2-metil-1-buteno (E)-2-buteno
H HH
H(Z)-2-buteno 1-buteno
MÁS ESTABLE
Y en esta otra serie (Preg 101, 2005):
2-metil-1-buteno2-metil-2-buteno 3-metil-1-buteno (E)-2-buteno
H HH
H(Z)-2-buteno 1-buteno
MÁS ESTABLE
¡CUIDADO!: En cicloalquenos de pequeño o mediano tamaño el isómero cis es el más estable.
El cicloocteno trans es el cicloalqueno más pequeño que puede aislarse (los anteriores obligados
a ser cis).
Propiedades físicas de alquenos.
Algunos son débilmente polares (pero aún así son muchos menos polares que haloalcanos y
alcoholes). Estas propiedades se deben a la electronegatividad del Csp2 del doble enlace (recordar
que la electronegatividad de Csp > Csp2 > Csp3; aumenta con el carácter s).
El carácter atractor de e- del Csp2 explica:
1- Los H vinílicos (unidos directamente al C del doble
enlace) son más ácidos que los alquílicos.
2- El momento dipolar neto de los isómeros cis frente a los trans:
Consecuencia: la mayor polaridad del isómero cis hace que las fuerzas intermoleculares
(dipolo-dipolo) sean mayores que en el trans los isómeros cis tienen mayores puntos
de ebullición que los trans.

Química Orgánica InspiracleQIR/2015
479
TEMA 17. REACCIONES PERICÍCLICAS. (2003) 104; (2004) 101, 117; (2006) 102,
104, 111; (2007) 138, 160; (2008) 100; (2009) 42; (2011) 120; (2012) 62, 67, 95; (2013) 98; (2014) 100, 112.
Se entiende por reacciones pericíclicas la transformación concertada y estereoespecífica
que sucede a través de un estado de transcición cíclico respecto a núcleos y electrones. Preg 160
(2007); Preg 120 (2011).
Los tres tipos de reacciones pericíclicas más importantes son las cicloadiciones, los
reordenamientos sigmatrópicos y las reacciones electrocíclicas. Preg 138 (2007).
16.1. Reacciones de cicloadición (Cicloadiciones).
Una cicloadición es una reacción pericíclica (y, por lo tanto, concertada y
estereoespecífica) en la que dos enlaces π se pierden para formar dos nuevos enlaces σ. Las
cicloadicioens se describen en función del número de electrones (e-) π de cada uno de los
reactivos. Las reacciones que involucran 4n+2 e- π suelen ser térmicas y las cicloadiciones 4n e- π
son activadas fotoquímicamente.
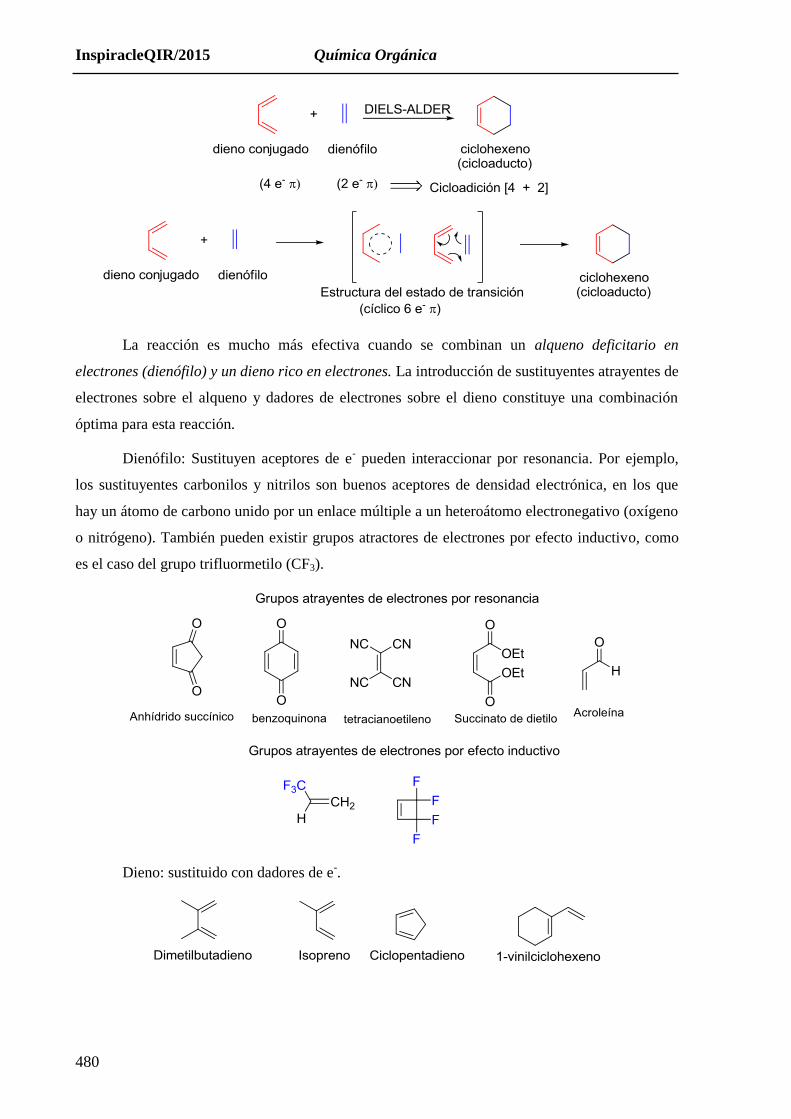
16.1.1. Reacción de Diels-Alder. Preg 62, 95 (2012).
Cicloadición concertada (única etapa) y estereoespecífica que sigue la regla endo y tiene
lugar entre un dieno s-cis y un dienófilo para formar un anillo de 6 miembros. Preg 42 (2009);
Preg 111 (2006); Preg 117 (2004).
InspiracleQIR/2015 Química Orgánica
480
La reacción es mucho más efectiva cuando se combinan un alqueno deficitario en
electrones (dienófilo) y un dieno rico en electrones. La introducción de sustituyentes atrayentes de
electrones sobre el alqueno y dadores de electrones sobre el dieno constituye una combinación
óptima para esta reacción.
Dienófilo: Sustituyen aceptores de e- pueden interaccionar por resonancia. Por ejemplo,
los sustituyentes carbonilos y nitrilos son buenos aceptores de densidad electrónica, en los que
hay un átomo de carbono unido por un enlace múltiple a un heteroátomo electronegativo (oxígeno
o nitrógeno). También pueden existir grupos atractores de electrones por efecto inductivo, como
es el caso del grupo trifluormetilo (CF3).
Dieno: sustituido con dadores de e-.

Química Orgánica InspiracleQIR/2015
489
TEMA 18. PRODUCTOS NATURALES. (2004) 107; (2005) 109; (2009) 38; (2010) 126;
(2013) 103, 114.
17.1. ALCALOIDES.
La palabra alcaloide significa semejante a la sosa y hace alusión a una de sus propiedades
más comunes: su carácter básico.
Los alcaloides son compuestos nitrogenados de origen natural —excluidos los de
naturaleza proteica—, con carácter básico y dotados de propiedades farmacológicas notables.
Preg 38 (2009); Preg 126 (2010).
En muy pocos vegetales se conocen la función de estas moléculas. Sin embargo en los
animales, pueden presentar distintas actividades farmacológicas: Un ejemplo, lo constituyen los
alcaloides de la amapola del opio: morfina (analgésico central), codeía (analgésico, antitusivo),
papaverina (espasmolítico). En el caso del alcaloide quinina, aislada de la corteza de la cicona, es
el más antiguo agente antimalárico efectivo conocido.
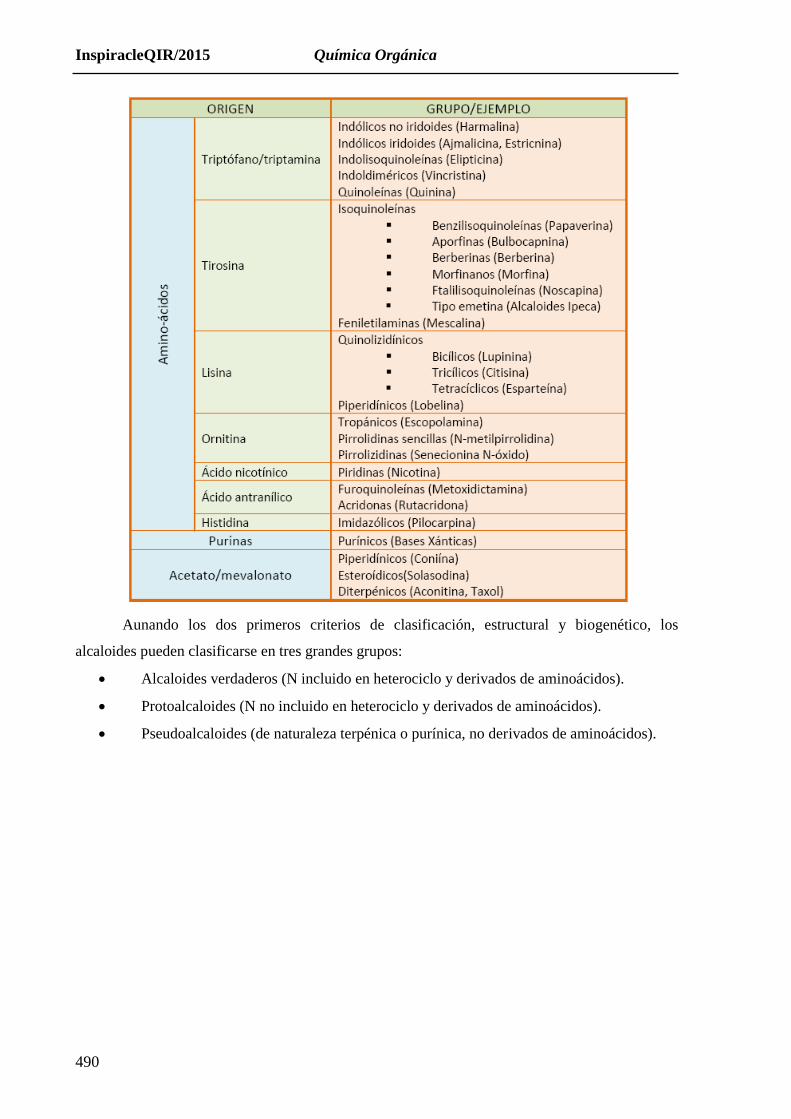
17.1.1. Clasificación:
La variedad y el grado de complejidad estructural de los alcaloides es tal, que según el
autor y el contexto en el que se presente, la clasificación de esta clase de compuestos puede
establecerse desde diferentes puntos de vista: estructural, biogenético, farmacológico…
Atendiendo a la situación que ocupa el nitrógeno en la molécula, los alcaloides podrían
catalogarse en los siguientes grupos:
Aminas 2as y 3as (protonadas o no, según el pH), en las que se incluyen la gran mayoría
de los alcaloides.
Aminas 4as, con carga a todos los pH (tubocurarina, por ejemplo).
Amidas (aminas neutras), donde se agruparían alcaloides del tipo de la colchicina.
En forma de N-óxidos, típicos de los derivados pirrolizidínicos.
Biogenéticamente, la gran mayoría de los alcaloides son construidos a partir de aminoácidos,
pero otros se originan por rutas biosintéticas muy diferentes, pudiendo así derivar de la
purina, de terpenos aminados o incluso proceder de la vía de los poliacetatos.
InspiracleQIR/2015 Química Orgánica
490
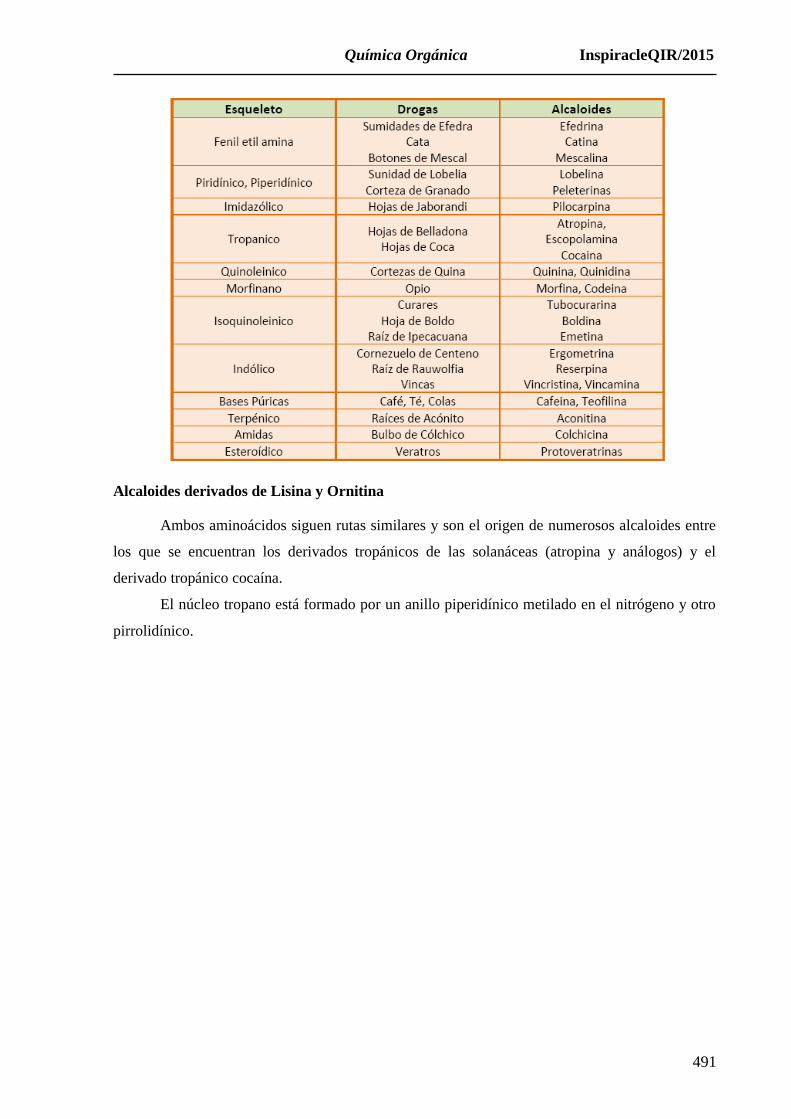
Aunando los dos primeros criterios de clasificación, estructural y biogenético, los
alcaloides pueden clasificarse en tres grandes grupos:
Alcaloides verdaderos (N incluido en heterociclo y derivados de aminoácidos).
Protoalcaloides (N no incluido en heterociclo y derivados de aminoácidos).
Pseudoalcaloides (de naturaleza terpénica o purínica, no derivados de aminoácidos).
Química Orgánica InspiracleQIR/2015
491
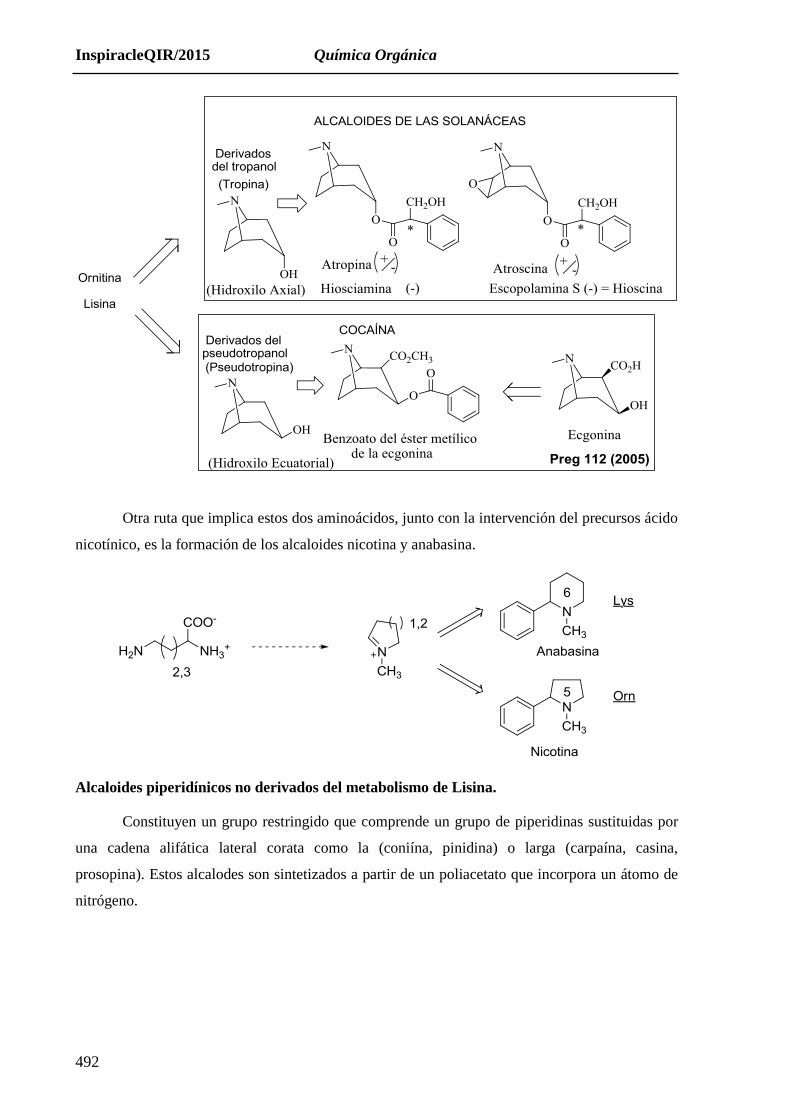
Alcaloides derivados de Lisina y Ornitina
Ambos aminoácidos siguen rutas similares y son el origen de numerosos alcaloides entre
los que se encuentran los derivados tropánicos de las solanáceas (atropina y análogos) y el
derivado tropánico cocaína.
El núcleo tropano está formado por un anillo piperidínico metilado en el nitrógeno y otro
pirrolidínico.
InspiracleQIR/2015 Química Orgánica
492
Otra ruta que implica estos dos aminoácidos, junto con la intervención del precursos ácido
nicotínico, es la formación de los alcaloides nicotina y anabasina.
Alcaloides piperidínicos no derivados del metabolismo de Lisina.
Constituyen un grupo restringido que comprende un grupo de piperidinas sustituidas por
una cadena alifática lateral corata como la (coniína, pinidina) o larga (carpaína, casina,
prosopina). Estos alcalodes son sintetizados a partir de un poliacetato que incorpora un átomo de
nitrógeno.
Química Orgánica InspiracleQIR/2015
493
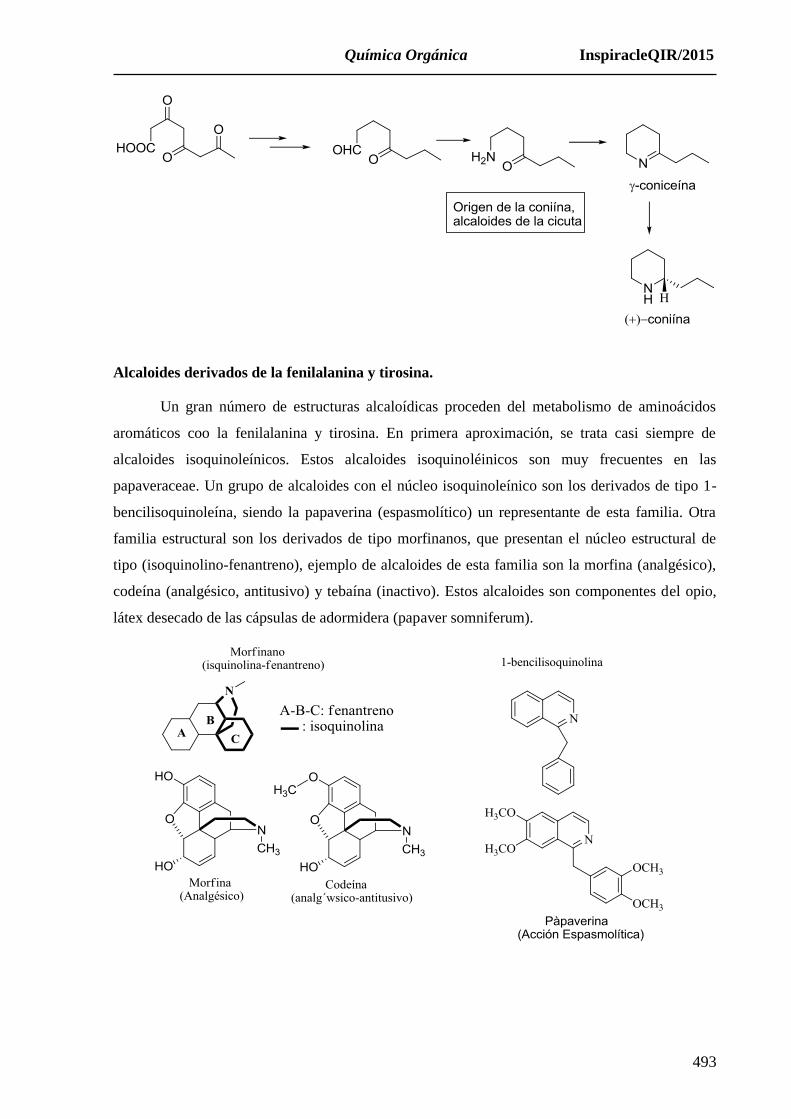
Alcaloides derivados de la fenilalanina y tirosina.
Un gran número de estructuras alcaloídicas proceden del metabolismo de aminoácidos
aromáticos coo la fenilalanina y tirosina. En primera aproximación, se trata casi siempre de
alcaloides isoquinoleínicos. Estos alcaloides isoquinoléinicos son muy frecuentes en las
papaveraceae. Un grupo de alcaloides con el núcleo isoquinoleínico son los derivados de tipo 1-
bencilisoquinoleína, siendo la papaverina (espasmolítico) un representante de esta familia. Otra
familia estructural son los derivados de tipo morfinanos, que presentan el núcleo estructural de
tipo (isoquinolino-fenantreno), ejemplo de alcaloides de esta familia son la morfina (analgésico),
codeína (analgésico, antitusivo) y tebaína (inactivo). Estos alcaloides son componentes del opio,
látex desecado de las cápsulas de adormidera (papaver somniferum).

InspiracleQIR/2015 Química Orgánica
496
17.3. TERPENOS Y ESTEROIDES.
Los terpenos y esteroides son elaborados a partir de los mismos precursores. La inmensa
mayoría de los terpenos son específicos del reino vegetal, pero se pueden encontrar en el los
animales: feromonas y hormonas juveniles sesquiterpénicasde los insectos, diterpenos de
organismos marinos (Celentéreos, Espongiarios). Igualmente los triterpenos son específicos del
reino vegetal.
17.3.1. TERPENOS.
Los terpenos están formados por la unión de un número entero de unidades
pentacarbonadas ramificadas derivadas del 2-metil-1,3-butadieno “unidades de isopreno”. Preg
103 (2013).
Biosíntesis.
En la naturaleza son biosintetizados no a partir del isopreno: su
precursor natural es el ácido mevalónico.
Clasificación: Preg 114 (2013).
Se clasifican según su tamaño en:
Monoterpenos: C10 (2xC5) Sesquiterpenos: C15 (3xC5)
Diterpenos: C20 (4xC5) Triterpenos: C30 (6xC5)
Monoterpenos: son el resultado del acoplamiento de dos unidades isoprénicas. Son los
constituyentes más frecuentes de los aceites esenciales.
Sesquiterpenos: El precursor común de los sesquiterpenos es el 2E,6E-pirofosfato de farnesilo
(FPP). Son terpenos constituidos por 3 unidades de isopreno. Preg 109 (2005). Existe un grupo de
sesquiterpenos se caracteriza por la presencia de un γ-lactona. Numerosas lactonas son
antibacterianas, sobre todo frente a bacterias gram (+), un ejemplo es la cnicina del cardo santo.
En el caso de la artemisinina, posee efecto antimalárico.

Química Orgánica InspiracleQIR/2015
497
17.3.2. ESTEROIDES
Tienen un origen biosintético común con los triterpenos, pues parten del escualeno
fosforilado (farnesil-farnesil pirofosfato), que sufre una oxidación, con formación de un epoxi que
conduce a la biosíntesis de triterpenos o esteroides, según el siguiente esquema:
La formación de esteroides, supone la desaparición de los metilos situados en los Carbono
C4; C8 y C14 y la presencia de un metilo en C13
Estructura:
Son triterpenos. Presentan una estructura tetracíclica, que consta
de tres anillos ciclohexano y uno de ciclopenteno. Son derivados del
sistema ciclopentanoperhidrofenantreno o gonano. Estos cuatro ciclos se
designan con las letras A, B, C, D. Pueden contener metilos en C10, C13 y
funciones oxigenadas en C3 y C17. Según el tipo de cadena carbonada que
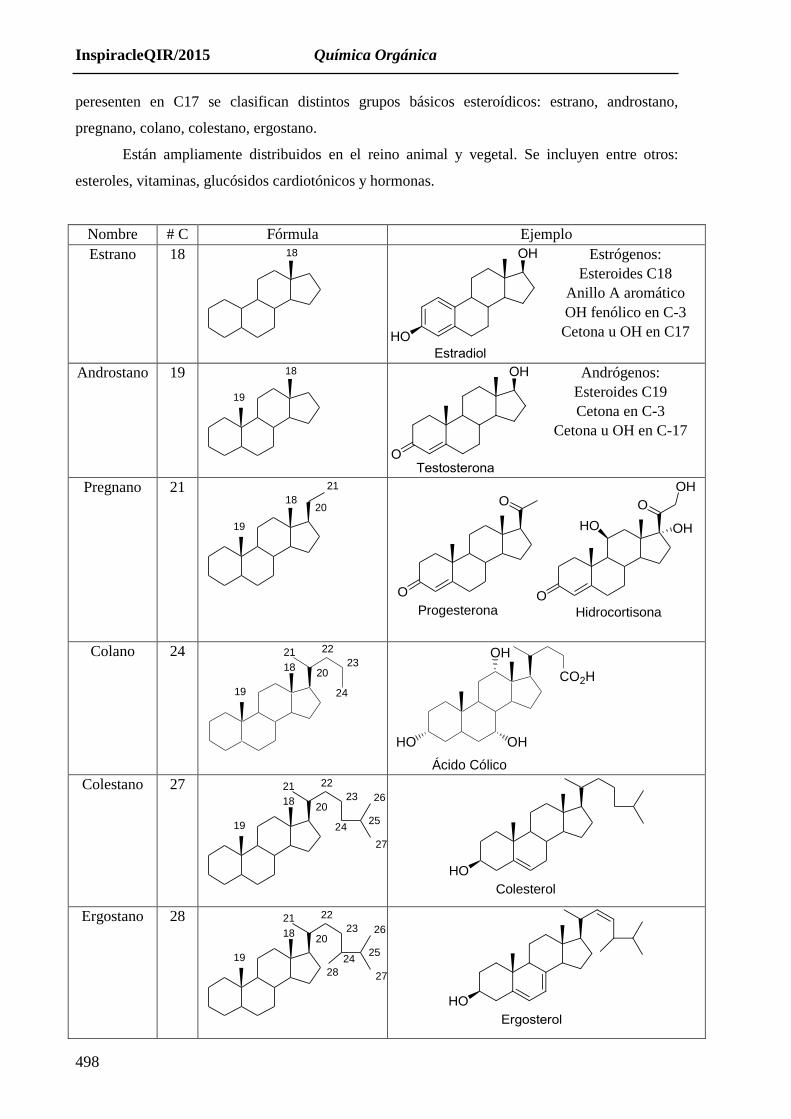
InspiracleQIR/2015 Química Orgánica
498
peresenten en C17 se clasifican distintos grupos básicos esteroídicos: estrano, androstano,
pregnano, colano, colestano, ergostano.
Están ampliamente distribuidos en el reino animal y vegetal. Se incluyen entre otros:
esteroles, vitaminas, glucósidos cardiotónicos y hormonas.
Nombre # C Fórmula Ejemplo
Estrano 18 18
Estrógenos:
Esteroides C18
Anillo A aromático
OH fenólico en C-3
Cetona u OH en C17
Androstano 19
19
18
Andrógenos:
Esteroides C19
Cetona en C-3
Cetona u OH en C-17
Pregnano 21
20
21
19
18
O
O
O
O
OH
HO OH
Progesterona Hidrocortisona
Colano 24 22
23
24
20
21
19
18
CO2H
HO OH
OH
Ácido Cólico Colestano 27
26
27
22
23
2425
20
21
19
18
HO
Colesterol
Ergostano 28
28
26
27
22
23
2425
20
21
19
18

Química Orgánica InspiracleQIR/2015
499
HETEROSIDOS CARDIOTONICOS.
La digital y otros glucósidos cardíacos tienen en común una poderosa acción sobre el
miocardio de gran utilidad en el tratamiento de la insuficiencia cardiaca. Estos glucósidos se
encuentran presentes en numerosas plantas (digital, estrofanto, escila) y algunos se hallan también
en el veneno de algunos sapos.
Las moléculas de digitálicos están formadas por la unión de una genina con una o cuatro
moléculas de azúcar. La actividad farmacológica reside en el aglicón o genina, pero los azucares
unidos a él modifican la hidro y liposolubilidad y la potencia del glucósido resultante. El aglicón
presenta una estructura básica de un núcleo de ciclopentanoperhidrofenantreno al que se le une, en
posición C-17, un anillo de lactona insaturada que presenta una estructura Δ, y que puede tener 5
o 6 miembros.

Química Orgánica InspiracleQIR/2015
507
ANEXOS. (2003) 120; (2004) 88, 89, 119; (2005) 116, 135; (2006) 135; (2007) 140, 154, 165, 170; (2008)
98, 99, 105; (2009) 3, 4, 11, 26, 31, 32; (2010) 106; (2011) 92; (2012) 97; (2013) 83; (2014) 84, 105.
NOMENCLATURA
ORDEN DE PRIORIDAD PARA ELEGIR GRUPO FUNCIONAL. Preg 88 (2004); 11, (2009);
105 (2014)
- Cationes
- Acidos:
Carboxílicos
Tioácidos
Sulfónicos
Sulfínicos
Sulfénicos
- Derivados de ácido:
Anhídridos
Ésteres
Haluros
Amidas
Hidrazidas
Imidas
- Nitrilos e isocianuros
- Aldehídos
Tioaldehídos
Derivados de los aldehídos
- Cetonas
Tiocetonas
Derivados de las cetonas
- Alcoholes y fenoles
Derivados del azufre (tioles)
- Hidroperóxidos
- Aminas, iminas, hidracinas
- Éteres
Sulfuros
- Peróxidos

Química Orgánica InspiracleQIR/2015
515
biciclo y por tanto, inestabilidad. La regla que indica, que un compuesto biciclico con puente no
puede tener un doble enlace en la posicion del puente a menos que uno de los anillos contenga
como minimo ocho atomos de carbono se conoce como regla de Bredt (Preg 3, 2009).
Sistemas cíclicos espiránicos.
Se dice que dos anillos están unidos formando un espirano (contienen un carbono espiro)
cuando comparten un solo átomo y ésta es la unión, directa o indirecta, que existe entre ellos.
Preg 99 (2008); Preg 92 (2011). El átomo común, que casi siempre es un carbono, se denomina
“átomo espiránico”. Cuando hay uno, dos, tres, etc. Átomos espiránicos en un sistema, éste se
designa coo monoespirano, diespirano, triespirano, etc. Respectivamente.
Un ejemplo de compuesto natural con sistema espiránico lo constituye el antibiótico
griseofulvina, el cual presenta efectos antifúngico.
InspiracleQIR/2015 Química Orgánica
518
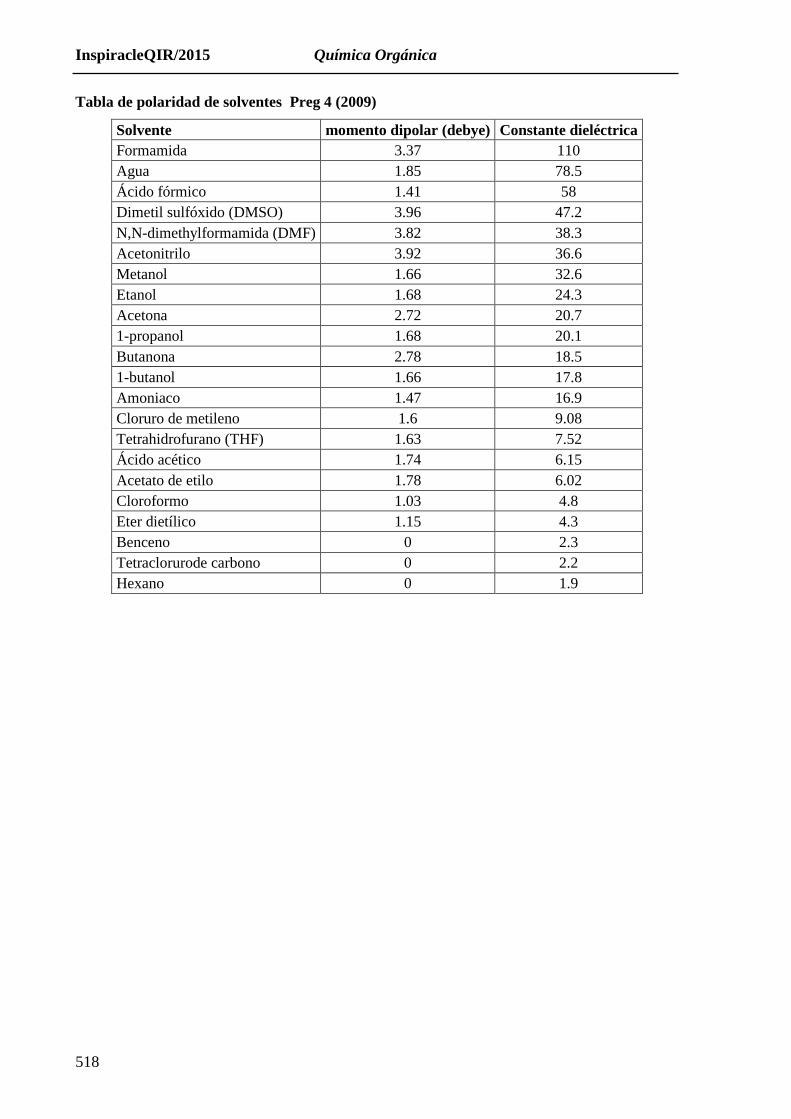
Tabla de polaridad de solventes Preg 4 (2009)
Solvente momento dipolar (debye) Constante dieléctrica
Formamida 3.37 110
Agua 1.85 78.5
Ácido fórmico 1.41 58
Dimetil sulfóxido (DMSO) 3.96 47.2
N,N-dimethylformamida (DMF) 3.82 38.3
Acetonitrilo 3.92 36.6
Metanol 1.66 32.6
Etanol 1.68 24.3
Acetona 2.72 20.7
1-propanol 1.68 20.1
Butanona 2.78 18.5
1-butanol 1.66 17.8
Amoniaco 1.47 16.9
Cloruro de metileno 1.6 9.08
Tetrahidrofurano (THF) 1.63 7.52
Ácido acético 1.74 6.15
Acetato de etilo 1.78 6.02
Cloroformo 1.03 4.8
Eter dietílico 1.15 4.3
Benceno 0 2.3
Tetraclorurode carbono 0 2.2
Hexano 0 1.9

Química Orgánica InspiracleQIR/2015
519
Preg 26 (2009).

Química Orgánica InspiracleQIR/2015
521
BIBLIOGRAFÍA
1. Química Orgánica. L. G. Wade. Editorial Pearson. Quinta Edición. 2004.
2. Química Orgánica Moderna. R.W. Griffin Jr. Editorial reverté, s.a. 1981.
3. Química Orgánica. Tercera edición. Volhardt, K. P.C. Omega, Barcelona, 2001