jornada sobre déficit de alfa-1 y...
TRANSCRIPT

Jornada sobre déficit de alfa-1 y EPOCLas enfermedades minoritarias como modelos de patologías de gran prevalencia
Aula Magna-Aula CajalFacultad de Medicina de la Universitat de València.Avda. Blasco Ibáñez nº 1546010 - Valencia.
Valencia, 12 de diciembre de 2013

2 /Valencia, 12 de diciembre de 2013
Jornada sobre déficit de alfa-1 y EPOC. Las enfermedades minoritarias como modelos de patologías de gran prevalencia.
PRESENTACIÓN
La jornada está organizada por el Instituto de Investigación Sanitaria del Hospital Clínico de Valencia INCLIVA, con la colaboración de la Universitat de València y el VLC Campus de Excelencia.
La Facultad de Medicina de Valencia acogerá una jornada monográfica sobre la enfermedad minoritaria déficit de alfa-1 antitripsina (DAAT) destinada a profesionales sanitarios, pacientes y familiares.
La jornada que ha sido avalada por diversas sociedades científicas, contará con la participación de expertos clínicos y científicos de ámbito nacional e internacional que han contribuido al conocimiento de la enfermedad.
El encuentro albergará tres actos diferentes, pero complementarios entre sí. La mañana se dedicará a sesiones de información básica sobre el DAAT. La tarde la ocupará la parte clínica de la enfermedad, en la que además de las charlas de los correspondientes especialistas en la materia, se incluirá una sesión de casos clínicos especialmente dirigida a los médicos residentes. De manera paralela, la tarde se centrará en la divulgación social de la enfermedad a través de un debate entre sociólogos, periodistas y asociaciones de pacientes y familiares.
DirectorDr. Francisco DasíInvestigador INCLIVA-Prof. asociado UVEG
Comité OrganizadorDra. Amparo EscribanoPediatra HCUV-Profª UVEGDra. Pilar CodoñerPediatra H. Dr. Peset. Valencia-Profª UVEGDra. Beatriz LaraNeumóloga REDAATDr. Francisco SanzNeumólogo HGUVDra. Mónica AmorPediatra HCUVDra. Silvia CastilloPediatra HCUVDr. Federico PallardóDecano Facultad Medicina UVEGDr. José Luis GarcíaInvestigador CIBERERDr. Felipe SerranoInvestigador Cambridge Institute for Medical ResearchDª. María Mercedes NavarroEstudiante periodismo UVEG
CARACTERÍSTICAS
¿A quién va dirigida?
Profesionales sanitarios, científicos y pacientes juntos ante la enfermedad.
La jornada se compone de charlas magistrales y una sesión de casos clínicos relacionados con el DAAT, y va dirigida a investigadores, pacientes y fundamentalmente a profesionales sanitarios: pediatras, neumólogos, hepatólogos, médicos de atención primaria, personal de enfermería, estudiantes de ciencias de la Salud, etc. Asimismo se realizará una sesión paralela destinada a debatir sobre los aspectos sociales relacionados con la enfermedad.
OBJETIVO
El objetivo fundamental de esta jornada es colaborar en la creación de una conciencia global de lo que supone el DAAT en la vida de los pacientes y en el conjunto de la sociedad. La jornada dará a conocer la enfermedad tanto en el ámbito médico como en el social. La interrelación investigador-médico-paciente-sociedad es una oportunidad para debatir sobre el impacto y el estado de la enfermedad y para identificar necesidades aún no satisfechas. Del mismo modo, la jornada permitirá generar una cultura de la ciencia que refuerce las labores de investigación que se realizan en universidades e institutos de investigación sanitaria.
Avales Sociedades Científicos
Sociedad Valenciana de Neumología

Valencia, 12 de diciembre de 2013 /3
Jornada sobre déficit de alfa-1 y EPOC. Las enfermedades minoritarias como modelos de patologías de gran prevalencia.
C.O. 01. AATD: 50 YEARS LATER
C.O. 02. AATD: CLINICAL MANIFESTATIONS/ THE LUNG
C.O. 03. AATD: CLINICAL MANIFESTATIONS/ THE LIVER
Encuentro de Excelencia Internacional en Enfermedades Raras. La investigación en DAATModeradores: Dra. Martínez, Dr. Pallardó, Dr Dasí
La investigación en DAATModeradores: Dr. Pallardó, Dr. Blanco, Dr Dasí.
C.O. 04. EPIDEMIOLOGÍA DEL DAAT
C.O. 05. EL DESAFIO DE DIAGNOSTICAR DAAT
C.O. 06. REDAAT: LA IMPORTANCIA DE LOS REGISTROS
C.O. 07. MESA REDONDA: LAS ENFERMEDADES MINORITARIAS COMO MODELOS DE PATOLOGÍAS DE GRAN PREVALENCIA.
Aspectos clínicos del DAATModeradores: Dra. Escribano Dr. Blanco.
C.O. 08. QUÉ DEBE SABER UN MÉDICO SOBRE DAAT
C.O. 09. DIAGNÓSTICO CLÍNICO Y DE LABORATORIO
C.O. 010. AAT: DEL PLASMA AL PRODUCTO FINAL
C.O. 011. EXPERIENCIA DE LAS COMUNIDADES AUTÓNOMAS EN DAAT: GALICIA-CV
C.O. 012. CASOS CLÍNICOS DAAT
Aspectos sociales del DAATModerador: Dr. Antonio Ariño
C.O. 013. EL DAAT EN LOS MEDIOS DE COMUNICACIÓN: ANÁLISIS Y RETOS
C.O. 014. MESA REDONDA: EL PAPEL DE LAS ASOCIACIONES DE PACIENTES
C.O. 015. HERRAMIENTAS WEB PARA LA COMUNICACIÓN. EJ.: FEDER
C.O. 016. LA INVESTIGACIÓN TRASLACIONAL: SINERGIA ENTRE LA INVESTIGACIÓN ACADÉMICA Y LOS HOSPITALES PÚBLICOS. EJ.: INCLIVA
C.O. 017. QUÉ DEBE SABER UN PACIENTE SOBRE DAAT

4 /Valencia, 12 de diciembre de 2013
Jornada sobre déficit de alfa-1 y EPOC. Las enfermedades minoritarias como modelos de patologías de gran prevalencia.
ABSTRACT
Alpha-1-antitrypsin deficiency (AATD) discovered 50 years ago, is a common hereditary conditions associated with liver and lung diseases occurring most frequently among Caucasians of northern European ancestry. It is believed that the Z (Glu342Lys) mutation causing severe AATD was spread by the Vikings between 66 and 120 generations ago (2000-4000 years ago).
The Z mutation causes AAT protein to assemble into polymers and retain intracellular. Because hepatocytes are the main source for AAT, polymerization and accumulation of AAT within the hepatocytes can cause liver damage with a variable clinical presentation, from neonatal hepatitis to liver cirrhosis and hepatocellular carcinoma in adults. On the other hand, low secreted levels of Z AAT protein, together with its increased propensity for extracellular polymerization and oxidation, results in dramatically reduced functional levels of AAT and increased risk for early-onset emphysema. Therefore, plasma purified AAT is used as a therapeutic for AATD-related emphysema.
A well-described function of AAT is to inhibit neutrophil elastase and proteinase 3. However, novel findings suggest that AAT possess broad anti-inflammatory and immuno-modulatory activities which are independent on elastase inhibition. Hence, accumulating knowledge allows for us and other researchers in the field to propose that AAT can be used as potential treatment for a broad spectrum of inflammatory and immune-mediated diseases, not necessary related to inherited AATD. Among the group of disorders that are considered to be linked to the reduced levels of AAT are panniculitis, vasculitis, HIV type 1 infection, hepatitis C infection and diabetes mellitus. The putative expansion of the use of AAT therapy points towards a need for more detailed investigations into the relationships between the concentration, structure and function of AAT protein.
Encuentro de Excelencia Internacional en Enfermedades Raras. La investigación en DAATModeradores: Dra. Martínez, Dr. Pallardó, Dr Dasí
C.O. 01 AATD: 50 YEAR LATER
RESUMEN
El déficit de alfa-1 antitripsina (DAAT) descubierto hace 50 años, es una de las enfermedades heredita-rias más frecuentes asociada con enfermedad hepática y pulmonar. La enfermedad es muy común entre los caucásicos de ascendencia norte-europea. Se cree que la mutación Z (Glu342Lys), que causa DAAT severa, la extendieron los vikingos y se remonta a 66-120 genera-ciones atrás (2000 a 4000 años).
La mutación Z causa que la alfa-1 antitripsina (AAT) polimerice y quede retenida intracelularmente. Debido a que los hepatocitos son la fuente principal de AAT, la polimerización y acumulación de AAT en los hepatoci-tos pueden causar daños en el hígado con una presenta-ción clínica variable, que va desde hepatitis neonatal a cirrosis hepática y carcinoma hepatocelular en adultos. Por otro lado, los bajos niveles séricos de la forma Z de la proteína, junto con su mayor propensión para la polimerización extracelular y la oxidación, resultan en una reducción drástica de los niveles AAT funcional, y en un mayor riesgo de desarrollo de enfisema a eda-des tempranas. Por lo tanto, la proteína AAT purificada a partir del plasma se utiliza como agente terapéutico para el tratamiento del enfisema relacionado con el DAAT.
Una de las funciones de la AAT es la de inhibir la elastasa de los neutrófilos y la proteinasa 3. Sin embar-go, nuevos descubrimientos sugieren que la AAT posee amplias actividades anti-inflamatorias e inmuno-mo-duladoras que son independientes de la inhibición de la elastasa. Por lo tanto, la acumulación de conoci-mientos nos permite a nosotros y otros investigadores en el campo proponer que la AAT pueda ser utilizada como potencial tratamiento para un amplio espectro de enfermedades inflamatorias y autoinmunes, no ne-cesariamente relacionadas con DAAT. Entre el grupo de trastornos que se considera que pueden estar relacio-nados con los niveles reducidos de AAT se encuentran la paniculitis , la vasculitis, la infección VIH tipo 1, la hepatitis C y la diabetes mellitus. La posible expansión de la utilización de puntos de terapia AAT hacia una necesidad de investigaciones más detalladas sobre las relaciones entre la concentración, la estructura y fun-ción de la proteína AAT.
DRA. SABINA JANCIAUSKIENE HANNOVER MEDICAL SCHOOL HANNOVER, GERMANY.

Valencia, 12 de diciembre de 2013 /5
Jornada sobre déficit de alfa-1 y EPOC. Las enfermedades minoritarias como modelos de patologías de gran prevalencia.
Encuentro de Excelencia Internacional en Enfermedades Raras. La investigación en DAATModeradores: Dra. Martínez, Dr. Pallardó, Dr Dasí
C.O. 02. AATD: CLINICAL MANIFESTATIONS/ THE LUNG
RESUMEN
La descripción original del déficit de alfa-1-antitrip-sina (DAAT) puso de manifiesto el desarrollo temprano de enfisema panacinar basal grave. Este se reconoció como el fenotipo DAAT clásico y los pacientes con patologías similares eran seleccionados para determi-nar en ellos los niveles de AAT, con lo que se asoció el DAAT con este cuadro clínico.
La progresión y la indicación para el empleo de la terapia sustitutiva se ha centrado en el FEV1, espe-cialmente en los pacientes dentro del rango de función pulmonar entre 30 - 60% del valor teórico. Más recien-temente, se ha reconocido que el DAAT es una carac-terística común a muchos fenotipos clínicos distintos, y estos afectan, no sólo a diferentes regiones del pulmón, sino también al deterioro fisiológico y a la sensibilidad de la monitorización. La TAC se ha mostrado eficaz para señalar la validez de la terapia de reemplazo, pero su relación con la fisiología y la progresión de la en-fermedad no es mayoritariamente aceptada. El control de los pacientes con DAAT requiere un nuevo enfoque basado en la fisiopatología de la enfermedad.
ABSTRACT
The original description of alpha-1-antitrypsin deficiency (aatd) noted the early onset of severe basal panacinar emphysema. This became recognised as the classical aatd phenotype and similar patients were subsequently targeted for testing, endorsing the association of aatd with this clinical presentation.
Progression and the indication for augmentation therapy has focussed on the fev1 especially in the range pf 30-60% predicted. More recently it has become recognised that aatd is a feature of many different clinical phenotypes and these affect, not only different regions of the lung but also the physiological impairment and sensitivity of monitoring. The role of ct scan has proven effective in demonstrating efficacy of augmentation therapy but its’ relationship to physiology and disease progression is not widely appreciated. Monitoring patients with aatd requires a new approach based on the pathophysiology of the disease.
PROFESSOR ROBERT A. STOCKLEYQUEEN ELIZABETH HOSPITALBIRMINGHAM, UK.

6 /Valencia, 12 de diciembre de 2013
Jornada sobre déficit de alfa-1 y EPOC. Las enfermedades minoritarias como modelos de patologías de gran prevalencia.
ABSTRACT
In patients with homozygous Alpha-1-Antitrypsin Deficiency (AATD) a defective AAT protein leads to accumulation of the protein in the hepatocytes. Interestingly, the majority of patients do not develop a severe hepatic manifestation and therefore, other co-factors must be relevant with regards to the clinical manifestation . On the other hand, a rapid progression to liver cirrhosis and the need for liver transplantation may occur, even in childhood. Moreover, liver fibrosis and liver cirrhosis have an impact on hepatic carcinogenesis.
There is evidence that the majority of homozygous AATD patients present in the neonatal period with a prolonged icterus and/or elevated transaminases but in clinical practice only 10 to 20 percent of the patients are beeing diagnosed at an early stage of the disease. Consequently, Pediatricians should increase their awareness for the disease and they should be attempt to diagnose more patients in the neonatal period.
Given the fact that 10% of homozygous patients will statistically die from liver cirrhosis, there is a strong need to establish statregies to identify affected patients as early as possible. A screening program is beeing controversially discussed in some countries.
Encuentro de Excelencia Internacional en Enfermedades Raras. La investigación en DAATModeradores: Dra. Martínez, Dr. Pallardó, Dr Dasí.
C.O. 03. AATD: CLINICAL MANIFESTATIONS/ THE LIVER
RESUMEN
En los pacientes homocigotos para el déficit de alfa-1 antitripsina (DAAT), la proteína AAT defectuosa se acumula en el interior de los hepatocitos. Curiosamen-te, la mayoría de los pacientes no desarrollan una ma-nifestación hepática grave y por lo tanto, deben existir otros cofactores implicados en la manifestación clínica del déficit. Por otro lado, se puede producir una rápida progresión a cirrosis hepática y la necesidad de un tras-plante de hígado, incluso en la infancia. Por otra parte, la fibrosis y la cirrosis hepática tienen un impacto en la carcinogénesis hepática.
Hay pruebas de que la mayoría de los pacientes ho-mocigotos DAAT debutan en el período neonatal con una ictericia prolongada y/o elevación de las transami-nasas, pero en la práctica clínica sólo del 10-20 % de los pacientes se encuentran diagnosticados en los estadios iniciales de la enfermedad. En consecuencia, los pedia-tras deben incrementar su conciencia de la enfermedad y se debería intentar diagnosticar a un mayor número de pacientes en el período neonatal.
Dado el hecho de que el 10% de los pacientes homo-cigotos fallecerá estadísticamente por cirrosis hepática, existe una gran necesidad de establecer estrategias para identificar los pacientes afectados lo antes posible. Los programas de cribado están siendo discutidos en varios países.
DR. RAINER GANSCHOW UNIVERSITY CHILDREN’S HOSPITAL BONN, GERMANY

Valencia, 12 de diciembre de 2013 /7
Jornada sobre déficit de alfa-1 y EPOC. Las enfermedades minoritarias como modelos de patologías de gran prevalencia.
ABSTRACT
Severe alpha-1 antitrypsin (AAT) deficiency, defined by AAT serum levels below 35% of the mean expected value, or 50 mg/dL (measured by nephelometry), is generally related to Pi*Z alleles, and much less frequently with combinations of Z, S, “rare” and null alleles. In fact, in clinical practice, 96% of AAT deficiency-related pathologies occur in Pi*ZZ genotypes, and 4% remaining in subjects with SZ, Z-null, Z-rare, null-null, rare-rare, or rare-null genotypes.
The highest prevalence of Pi*ZZ (1:1.500-2.000 individuals) are found in the Baltic Republics, south of the Scandinavian Peninsula and Denmark. It declines, but still remains high (1:2.500-1:4.000 individuals), in Belarus, Ukraine, Poland, Germany, the Netherlands, France, Britain Islands, and the Iberian Peninsula. Values significantly decline (1:10.000-90.000) in the outermost regions of Northern, Southern, and Western Europe, to practically disappearing in Lapland, Southern Italy and the Balkan Peninsula (Fig. 1.). Its prevalence is moderate in Caucasian populations from Canada and the US (1:5,000-6,000), and is low in Mexico, Caribbean, and Central and South America. In Anglo-Saxon populations from New Zealand and Australia, Pi*Z prevalence is similar to that reported in the UK (1:2.500 y 1:4.000). The Pi*ZZ genotype is practically absent in most regions of Asia and Africa.
Among an estimated total of 5.264.150.044 of humans in 97 out of 193 countries in the World whit available data, there might be 181.894 Pi*ZZ at high risk for development of AAT deficiency-related diseases, most of them Caucasians living in Western and Central Europe (74.000, 41% of the total), 20% in the US (34.000), and 16% en South-America. In addition, there might be 1.269.054 Pi*SZ, with potential, risk of developing AAT deficiency-related diseases, 615.193 living in Europe, and the remaining in North America, South America, Australia and New Zealand.
RESUMEN
El déficit grave de alfa-1 antitripsina (DAAT), defini-do por niveles séricos de AAT por debajo del 35% del valor medio esperado, o 50 mg/dl (determinado por ne-felometría), se encuentra generalmente relacionado con alelos PI*Z, y con mucha menos frecuencia con combi-naciones de alelos Z , S , “raros” y nulos. De hecho, en la práctica clínica, el 96% de las patologías relacionadas con el déficit se producen en los genotipos Pi*ZZ , y el 4% restante en los sujetos con genotipos SZ, Z-nulo, Z-raro, nulo-nulo, raro-raro o raro-nulo.
La mayor prevalencia del Pi*ZZ (1:1.500-2.000 per-sonas) se encuentra en las repúblicas bálticas, al sur de la península escandinava y Dinamarca. Disminuye, pero continúa siendo alta (1:2.500-1:4.000 individuos), en Bielorrusia, Ucrania, Polonia, Alemania, Holanda, Francia, Las Islas Británicas y la Península Ibérica. Los valores disminuyen significativamente (1:10,000-90,000) en las regiones periféricas del Norte, Sur, y Europa Occidental, para desaparecer prácticamente en Laponia, el sur de Italia y la península de los Balcanes (Fig. 1). Su prevalencia es moderada en poblaciones caucásicas de Canadá y los EE.UU. (1:5.000-6.000) , y es baja en México, el Caribe, América del Sur y Cen-tral. En las poblaciones anglosajonas de Nueva Zelanda y Australia, la prevalencia del Pi*Z es similar a la del Reino Unido (1:2.500 y 1:4.000). El genotipo Pi*ZZ está prácticamente ausente en la mayoría de las regiones de Asia y África.
Entre un total estimado de 5.264.150.044 de seres humanos de 97 países de los 193 que hay en el mundo, podría haber 181.894 Pi*ZZ con un riesgo elevado de desarrollo de enfermedades relacionadas con DAAT, la mayoría de ellos de raza blanca que viven en Euro-pa occidental y central (74.000, el 41% del total), 20% en los EE.UU. (34.000), y el 16% en América del Sur. Además, podría haber 1.269.054 Pi*SZ, con riesgo po-tencial de desarrollo de enfermedades relacionadas con las deficiencias de AAT, 615.193 viven en Europa, y el resto de América del Norte , América del Sur , Austra-lia y Nueva Zelanda.
La investigación en DAATModeradores: Dr. Pallardó, Dr Dasí.
C.O. 04. EPIDEMIOLOGÍA DEL DAAT
DR. IGNACIO BLANCONEUMÓLOGOCOORDINADOR REDAAT

8 /Valencia, 12 de diciembre de 2013
Jornada sobre déficit de alfa-1 y EPOC. Las enfermedades minoritarias como modelos de patologías de gran prevalencia.
Figu
re 1
. Map
of
Pi*Z
freq
uenc
ies
in E
urop
e, e
stim
ated
by
mea
ns o
f th
e Ar
cMap
[an
info
rmat
ics
mat
hem
atic
al a
ppro
ach
for
Mic
roso
ft W
indo
ws]
, bas
ed o
n th
e in
vers
e di
stan
ce w
eigh
ting
(ID
W)
mul
tivar
iate
inte
rpol
atio
n m
etho
d, w
hich
cre
ates
new
num
eric
al p
oint
s fr
om k
now
n da
ta, u
sing
a s
impl
e lo
garit
hm b
ased
in t
he d
ista
nce
exis
ting
betw
een
them
. B
lanc
o I,
de
Serr
es F
J, C
árca
ba V
, Lar
a B
, Fer
nánd
ez-B
usti
llo E
. Alp
ha-1
ant
itryp
sin
defic
ienc
y PI
*Z a
nd P
I*S
gene
freq
uenc
y di
strib
utio
n us
ing
on m
aps
of t
he w
orld
by
an in
vers
e di
stan
ce
wei
ghtin
g (I
DW
) m
ultiv
aria
te in
terp
olat
ion
met
hod.
Hep
at M
on. 2
012;
12(
10 H
CC):
e743
4.

Valencia, 12 de diciembre de 2013 /9
Jornada sobre déficit de alfa-1 y EPOC. Las enfermedades minoritarias como modelos de patologías de gran prevalencia.
La investigación en DAATModeradores: Dr. Pallardó, Dr Blanco.
C.O. 05. EL DESAFIO DE DIAGNOSTICAR DAAT
RESUMEN
El déficit de alfa-1 antitripsina (DAAT) es una enfer-medad infradiagnosticada. Los diversos programas de detección han puesto de manifiesto que únicamente se diagnostican entre el 2-10% de los individuos con DAAT.
Por otra parte, existe, desde que aparecen los prime-ros síntomas, un retraso medio de 8 años en el diag-nóstico de la enfermedad y los pacientes visitan varios especialistas hasta obtener un diagnóstico definitivo.
El diagnóstico precoz de la enfermedad cobra par-ticular importancia en esta patología, ya que existen una serie de estrategias (evitar fumar, consejo genético, terapia sustitutiva), que, utilizadas en el momento ade-cuado, pueden retrasar el daño hepático o pulmonar y en algunos casos incluso evitarlo.
En esta charla se discuten las diversas estrategias empleadas para mejorar el diagnóstico del DAAT. Aun-que los resultados obtenidos varían entre los diferentes estudios, en general, indican un aumento en el número de pacientes diagnosticados.
A pesar de todo lo anterior, la enfermedad sigue es-tando infradiagnosticada, lo que indica que se deben di-señar nuevas estrategias que conduzcan al diagnóstico precoz de la enfermedad, especialmente en un contexto en el que van apareciendo nuevos recursos terapéuti-cos de las que podrían beneficiarse los pacientes con DAAT.
ABSTRACT
Alpha-1 antitrypsin deficiency (AATD) is an underdiagnosed disease. Screening programs have shown that only 2-10 % of individuals with AATD have been diagnosed.
Moreover, there is a mean diagnostic delay period of 8 years since the first symptoms appeared and patients visit multiple physicians until a definitive diagnosis is accomplished.
Early diagnosis is particularly important in this disease, because a number of strategies (avoid smoke exposure, genetic counselling, replacement therapy) are available, which used at the right time, could delay (and in some cases avoid) the liver or lung damage observed in AATD patients.
In this talk various strategies employed to improve the diagnosis of AATD are discussed. While results vary across studies, they generally indicate an increase in the number of diagnosed patients.
In spite of the above, the disease remains underdiagnosed, indicating the need for new strategies that lead to early diagnosis of the disease, especially in a context in which new therapeutic strategies are on the horizon, which could be of benefit for AATD patients.
DR. FRANCISCO DASÍINVESTIGADORINCLIVA

10 /Valencia, 12 de diciembre de 2013
Jornada sobre déficit de alfa-1 y EPOC. Las enfermedades minoritarias como modelos de patologías de gran prevalencia.
La investigación en DAATModeradores: Dr. Dasí, Dr. Blanco.
C.O. 06 REDAAT: LA IMPORTANCIA DE LOS REGISTROS
DRA. BEATRIZ LARANEUMÓLOGAGESTORA REDAAT
ABSTRACT
Patients’ registries are a crucial tool in rare disease field, not only from a scientific point of view even though from health related institutions. Their importance has been reported in European Health Comission documents and in the Spanish Strategy for Rare Diseases published by the Ministry of Health.
In Spain, the Spanish Registry of Patients afected of alpha-1 antitrypsin deficiency (REDAAT) from the Spanish Society of Pulmonology is available. Its main goal is the improvement of knowledge about AATD in order to a better management of afected individuals.
REDAAT has an advisory board including pulmonologist, pediatricians and basic researchers and a reference laboratory. More than 300 physicians from all over the country collaborate reporting their cases. The REDAAT’s website: www.redaat.es has public contents related with AATD and a restricted area where database is located and also real-time information about the registered population is shown.
REDAAT advisory board collaborate with International Alpha-1 Registry and other registries and it shares information about AATD with the National Registry of Rare Diseases.
Nowadays, 505 individuals have been registered (ZZ, SZ and very rare variants). Most of them are afected by respiratory diseases and only a few suffered from liver disease.
Guidelines for physicians, patient’s guides and other materials have been published thanks to the shared experience collected in the Registry.
RESUMEN
Los registros de pacientes dentro del campo de las en-fermedades minoritarias son un elemento fundamental, no sólo desde el punto de vista científico, sino también institucional como queda recogido tanto en los docu-mentos de la Comisión Europea de Salud, como en la Estrategia Nacional de Enfermedades Raras del Minis-terio entre otras.
En el caso del déficit de alfa 1 antitripsina (DAAT) en España contamos con el Registro Español de pacientes con DAAT (REDAAT) que forma parte de la SEPAR. Su finalidad es profundizar en el conocimiento sobre el DAAT, estimular la investigación, contribuir a su difu-sión y mejorar el tratamiento de las personas afectadas. Cuenta con un comité asesor (neumólogos, pediatras, investigadores básicos) y un laboratorio de referencia. Colaboran en el registro de casos más de 300 médicos de toda España. El principal recurso técnico es www.redaat.es incluye un área de acceso público con infor-mación general, y un área de acceso restringido para profesionales sanitarios que incluye la ficha de recogida de datos de los pacientes, así como información a tiem-po real de los casos registrados y características globa-les y diagnóstico y tratamiento. Colabora activamente en el registro internacional AIR desde su formación y con algunos registros nacionales europeos y comparte información con el Registro Nacional de enfermedades raras.
Actualmente, recoge 505 individuos con déficit gra-ve (ZZ, SZ y otras variantes menos frecuentes) prin-cipalmente, afectados por patología respiratoria y una minoría hepática.
Gracias a la experiencia recogida en él se han publi-cado diversos trabajos, tanto normativas de diagnóstico y tratamiento como información para profesionales y pacientes e investigación básica (nuevas variantes).

Valencia, 12 de diciembre de 2013 /11
Jornada sobre déficit de alfa-1 y EPOC. Las enfermedades minoritarias como modelos de patologías de gran prevalencia.
La investigación en DAATModeradores: Dr. Dasí, Dr. Blanco.
C.O. 07 MESA REDONDA: LAS ENFERMEDADES MINORITARIAS COMO MODELOS DE PATOLOGÍAS DE GRAN PREVALENCIA.
RESUMEN
El DAAT es una entidad incluida dentro de ese amplio y variado grupo de enfermedades raras, que no figu-ran en el listado de diagnósticos comunes que el clínico maneja día a día, de ahí que se trate de una enfermedad infradiagnosticada, incluso entre los adultos con mani-festaciones clínicas de enfermedad pulmonar obstructi-va crónica (EPOC), que llegan a esperar una media de 7 años y a acudir a 5 médicos diferentes antes de poder ser diagnosticados.
Si esto ocurre en la edad adulta, cuando el DAAT tiene una forma clara de expresión, ¿qué podemos es-perar en la infancia, cuando sus manifestaciones clíni-cas son limitadas o están ausentes? Aún así, sabemos que el diagnóstico precoz logra, en la mayor parte de los procesos crónicos, incluso en los letales, retrasar su progresión, mejorar el pronóstico y aumentar la espe-ranza de vida, por lo que debe ser el pediatra el encarga-do de sospecharlos y diagnosticarlos. Si bien el DAAT debería sospecharse en niños con hepatopatía crónica, hipertensión portal o cirrosis, como la afectación pul-monar no se expresa durante la infancia, los pediatras o los neumólogos infantiles no suelen plantear este diag-nóstico.
Sin embargo, sólo se encuentra aquello que se bus-ca, por lo que si la alfa-1 antitripsina se incluyera entre los parámetros a valorar cuando se solicita un análisis de sangre, aumentaría la probabilidad de detectar pre-cozmente un DAAT. Ésta es nuestra experiencia y así hemos podido diagnosticar a un grupo relativamente numeroso de niños alfa y, a partir de ellos, de padres afectos, en los que unos hábitos de vida saludables y un control de los factores de riesgo quizás eviten una EPOC futura.
DRA. AMPARO ESCRIBANOJEFA UNIDAD NEUMOLOGÍA PEDIÁTRICAHOSPITAL CLÍNICO UNIVERSITARIO DE VALENCIA
ABSTRACT
The AATD is an entity included within that large and varied group of rare diseases, not listed in the list of common diagnoses that the physician handles daily, hence it is an under-diagnosed disease, even among adults with clinical manifestations of COPD, reaching can wait for an average of seven years and visit up to 5 different doctors before being diagnosed.
If this occurs during adulthood, when the AATD has a clear form of expression, what can we expect during childhood, when clinical manifestations are limited or absent? Still, we know that early diagnosis can, in most chronic processes, even lethal, slow its progression, improve prognosis and increase life expectancy, so it must be the pediatrician responsible for its suspicion and diagnosis. While the AATD should be suspected in children with chronic liver disease, cirrhosis or portal hypertension, as pulmonary involvement is not expressed during childhood, pediatricians or pediatric pulmonologists do not usually pose this diagnosis.
However, only what is sought can be detected, so if alpha-1 antitrypsin was included among the parameters to be evaluated when a blood test is requested, the likelihood of detecting early AATD would increase. This is our experience and thus we have been able to diagnose a relatively large group of alpha children and, from them, affected parents in which healthy lifestyle and control of risk factors could perhaps prevent a future COPD.

12 /Valencia, 12 de diciembre de 2013
Jornada sobre déficit de alfa-1 y EPOC. Las enfermedades minoritarias como modelos de patologías de gran prevalencia.
DRA. CRUZ GONZÁLEZNEUMÓLOGAHOSPITAL CLÍNICO UNIVERSITARIO DE VALENCIA
ABSTRACT
Guidelines for treatment of Chronic Obstructive Pulmonary Disease (COPD) apply equally to alpha-1-antitrypsin deficiency (AATD), except for lung volume reduction surgery, which is generally not recommended. The most severely affected individuals should be considered for lung transplantation. Cigarette smoke and environmental pollution avoidance is determinant in preventing the development of all forms of COPD. Also is possible the correction of the plasma deficiency with intravenous augmentation of pooled plasma purified alpha-1-antitrypsin (AAT).
Based on the understanding of the pathogens of the disease as a deficiency in liver production of AAT resulting from inherited genetic variation in both parental AAT genes, the knowledge that AAT functions primarily to inhibit neutrophil elastase (NE), and the observation that NE instilled into the lung of experimental animals resulted in emphysema, the concept evolved that the pulmonary manifestations of the disease could be halted by intermittent intravenous infusions of AAT purified from pooled human plasma. Following preliminary clinical and pharmaceuticals studies the FDA approved the use of weekly AAT augmentation therapy with AAT from human plasma for specific treatment of AATD. Intravenous augmentation therapy has demonstrated to be safe and weekly infusions of AAT have demonstrated to result in plasma AAT concentration above those considered protective for the lungs. Randomized, placebo-controlled clinical trials have confirmed a reduction in the decline in lung density in patients receiving augmentation therapy. This is the first example of an antiprotease effective in restoring the protease/antiprotease imbalance in the lungs and changing the natural history of congenital emphysema.
RESUMEN
Las directrices para el tratamiento de la Enfermedad Pulmonar Obstructiva Crónica (EPOC) se aplican por igual al déficit de alfa-1 antitripsina (DAAT), a excep-ción de la cirugía de reducción de volumen pulmonar, que en general no se recomienda. Las personas con mayor afectación pulmonar deben ser consideradas para trasplante pulmonar. Evitar tanto el humo del cigarrillo como la contaminación medio ambiental es determinante para prevenir el desarrollo de todas las formas de EPOC. Por otra parte, es posible la correc-ción de los niveles plasmáticos deficientes mediante el empleo de alfa-1 antitripsina (AAT) purificada a partir de plasma humano.
Sobre la base de la comprensión de los diversos factores implicados en el desarrollo de la enfermedad, como la deficiencia en la producción hepática de AAT como consecuencia de la variación genética heredada en ambos genes AAT parentales, el conocimiento de que la principal función de la AAT es inhibir la elasta-sa producida por los neutrófilos (NE), y la observación de que la instilación de elastasa en los pulmones de los animales de experimentación produjo en estos enfise-ma, dio origen al concepto de que se podrían detener las manifestaciones pulmonares de la enfermedad me-diante el empleo de infusiones intravenosas intermi-tentes de AAT purificada a partir de plasma humano.
Tras los estudios clínicos y farmacéuticos prelimi-nares, la FDA aprobó el uso de la terapia de sustitución semanal con AAT purificada a partir de plasma huma-no para el tratamiento específico del DAAT. La terapia de sustitución endovenosa ha demostrado ser segura e infusiones semanales de AAT producen concentracio-nes plasmáticas de AAT superiores a las consideradas protectoras para los pulmones. Los ensayos clínicos aleatorizados y controlados con placebo han confirma-do una reducción en la disminución de la densidad pul-monar en pacientes que reciben terapia sustitutiva. Este es el primer ejemplo de una antiproteasa eficaz en la reparación del desequilibrio proteasa/antiproteasa pul-monar lo que contribuirá a cambiar la historia natural de enfisema congénito.
La investigación en DAATModeradores: Dr. Dasí, Dr. Blanco.
C.O. 07 MESA REDONDA: LAS ENFERMEDADES MINORITARIAS COMO MODELOS DE PATOLOGÍAS DE GRAN PREVALENCIA.

Valencia, 12 de diciembre de 2013 /13
Jornada sobre déficit de alfa-1 y EPOC. Las enfermedades minoritarias como modelos de patologías de gran prevalencia.
DR. FRANCISCO SANZNEUMÓLOGOHOSPITAL GENERAL UNIVERSITARIO DE VALENCIA
ABSTRACT
Chronic obstructive pulmonary disease (COPD) is a leading cause of morbidity and mortality worldwide. Tobacco smoking is an essential etiopathogenic factor in this disease and the fight against smoking is one of the cornerstones in the management of this disease. However it has been detected that up to 25-45 % of COPD patients have never smoked. Exogenous factors play an important role in COPD developing. Those factors include the exposure to biomass smoke, occupational exposures, respiratory infections and socioeconomical status. Other endogenous conditions exist which can facilitate the development of COPD in never-smoking patients with no exposure to exogenous risk factors. Alpha-1 antitrypsin deficiency (AATD) and other genetic diseases are responsible for the development of COPD in that group of patients. The alpha-1 antitrypsin deficiency causes emphysema in earlier stages of life and it presents with a histological pattern which is different from tobacco smoke causes. At the present time, replacement therapy is available for those AATD patients and this treatment has demonstrated an increase in survival from COPD. However, despite the availability of these therapeutic tools, a huge underdiagnosis of this disease is detected. Estimates show that AATD is identified in less than 10% of cases, and it is related to a delay in the diagnosis of this deficiency and thus to the progression of emphysema. Education and training of physicians and clinical suspicion is essential, especially in young patients with pulmonary emphysema and no history of tobacco smoking in which AATD is mandatory to be ruled out.
La investigación en DAATModeradores: Dr. Dasí, Dr. Blanco.
C.O. 07 MESA REDONDA: LAS ENFERMEDADES MINORITARIAS COMO MODELOS DE PATOLOGÍAS DE GRAN PREVALENCIA.
RESUMEN
La enfermedad pulmonar obstructiva crónica (EPOC) es una causa principal de morbilidad y mortalidad en todo el mundo. El consumo de tabaco es un factor etiopatogénico fundamental en esta enfermedad y la lucha contra el tabaquismo es uno de los pilares del tratamiento. Sin embargo, se ha detectado que hasta un 25-45% de los pacientes con EPOC nunca han fumado. Los factores exógenos desempeñan un papel importante en el desarrollo de la EPOC. Esos factores incluyen la exposición al humo de biomasa, las exposiciones laborales, las infecciones respiratorias y el estatus socioeconómico. Existen otras condiciones endógenas que pueden facilitar el desarrollo de la EPOC en pacientes nunca fumadores sin exposición a factores de riesgo exógenos. El déficit deAlfa-1 antitripsina (DAAT) y otras enfermedades genéticas son responsables del desarrollo de la EPOC en ese grupo de pacientes. El DAAT causa enfisema en las primeras etapas de la vida y se presenta con un patrón histológico que es diferente de causas de humo de tabaco. En la actualidad, la terapia de reemplazo está disponible para aquellos pacientes DAAT, y este tratamiento ha demostrado un aumento en la supervivencia de la EPOC. Sin embargo, a pesar de la disponibilidad de estas herramientas terapéuticas, se detecta un gran infradiagnóstico de esta enfermedad. Las estimaciones muestran que DAAT se identifica en menos del 10% de los casos, y se relacionan con un retraso en el diagnóstico de esta deficiencia y así la progresión del enfisema. La formación de los médicos y la sospecha clínica es fundamental, sobre todo, en pacientes jóvenes con enfisema pulmonar y sin antecedentes de consumo de tabaco en los que es obligatorio descartar DAAT.

14 /Valencia, 12 de diciembre de 2013
Jornada sobre déficit de alfa-1 y EPOC. Las enfermedades minoritarias como modelos de patologías de gran prevalencia.
DRA. PILAR DE LUCASPRESIDENTA SEPAR
ABSTRACT
Alpha-1 antitrypsin deficiency (AATD) is the main genetic cause of COPD. It is estimated that this etiology is responsible for 1% of COPD cases and 4% of cases of emphysema. No doubt that AATD is a rare disease, however, according to the EPISCAN epidemiological study, in Spain, there are about 2.000.000 people with COPD, meaning that between 10.000 to 20.000 cases are related to the presence of AATD, which in most of the cases remain undiagnosed. An under-diagnosed disease with a high morbidity and mortality rates is certainly very worrying.
SEPAR’s main purpose is not only to advance on the knowledge and research in respiratory diseases, but also to foster measures to promote respiratory health and to participate in the training and information of citizens, directly and through collaboration with patients’ associations, with the ultimate goal of preventing respiratory diseases and improve care for patients and their families once the respiratory problem has occurred. In this overall framework, attention to AATD plays an important role.
Focusing on COPD, the COPD SEPAR-ALAT guide and the Spanish guidelines for the management of COPD, GESEPOC, recommend alpha-1 antitrypsin screening for all patients with COPD at least once in patient’s life. Undoubtedly, this measure will help to solve the problem of under-diagnosis. In addition, SEPAR’s experts group on AATD, published in 2006, the SEPAR regulations on AATD, directed to the diagnosis and treatment of disease, still in force today. Besides to these fundamental policy documents, SEPAR has performed a great task of research and dissemination of knowledge about the disease, and has given support to patients and families which has been articulated around the work done by this group of experts forming the REDAAT: Working Group on AATD registry. Dr. Marc Miravitlles, who became its first director, created the AATD registry in 1993 and although its characteristics, objectives and outcomes will be discussed in another talk of this meeting, it has to be mentioned at this point, since it is the core of the SEPAR activities for the diagnosis and monitoring of the disease.
Aspectos clínicos del DAATModeradores: Dr. Dasí, Dr. Blanco.
C.O. 07 MESA REDONDA: LAS ENFERMEDADES MINORITARIAS COMO MODELOS DE PATOLOGÍAS DE GRAN PREVALENCIA.
RESUMEN
El déficit de alfa-1 antitripsina constituye la principal cau-sa genética de enfermedad pulmonar obstructiva cróni-ca, estimándose que esta etiología es la responsable del 1% de casos de EPOC y del 4% de casos de enfisema. Sin duda se trata de una enfermedad minoritaria, pero si, de acuerdo con el estudio epidemiológico EPISCAN en España existen en torno a 2.000.000 de personas que pa-decen EPOC, es posible que entre 10.000 y 20.000 casos estén relacionados con la presencia de un déficit de alfa-1 antitripsina, en la mayor parte de los mismos no diagnos-ticado. El infradiagnóstico de una enfermedad de elevado impacto en morbilidad y mortalidad resulta sin duda muy preocupante.
SEPAR tiene como objetivos fundamentales no solo avanzar en el conocimiento y la investigación de las enfer-medades respiratorias, sino también impulsar medidas de promoción de la salud respiratoria, participar en la formación e información de los ciudadanos, de forma directa y a través de la colaboración con las asociaciones de pacientes y todo ello con el fin último de prevenir la enfermedad respiratoria y mejorar la atención a los enfermos y sus familiares una vez que aquella se produce. En este marco global, la atención al déficit de alfa-1 antitripsina ocupa un importante lugar.
De una forma más genérica, enfocada en la EPOC, tan-to la guia de EPOC SEPAR-ALAT como la guía española para el manejo de la EPOC, GESEPOC, recogen la indi-cación de que en todo paciente con EPOC se debe realizar una determinación alfa-1-antitripsina al menos una vez en la vida. Esta medida serviria sin ninguna duda para mejo-rar las tasas de infradiagnóstico. Además, el grupo experto de la sociedad, publico el año 2006 una Normativa SEPAR encaminada de forma especifica al diagnóstico y tratamien-to de la enfermedad, normativa actualmente vigente. Pero además de estos fundamentales documentos de actuación, la sociedad ha desarrollado una gran labor de investigación y difusión del conocimiento en torno a la enfermedad, así como de apoyo a pacientes y familiares, que se han articu-lado en torno al trabajo realizado por ese grupo de expertos que conforma el REDAAT: grupo de trabajo del registro de alfa-1-antitripsina. El registro fue creado en 1993 por el Dr. Marc Miravitlles, quien se convirtió en su primer direc-tor, y aunque sus caracteristicas, objetivos y resultados en permanente movimiento corresponden a otra sesión de esta jornada, hay que mencionarlo aquí pues es el núcleo de las actividades SEPAR para el diagnóstico y control de la en-fermedad. Estas actividades pueden ser clasificadas por el colectivo al que van dirigidas y por su contenido.

Valencia, 12 de diciembre de 2013 /15
Jornada sobre déficit de alfa-1 y EPOC. Las enfermedades minoritarias como modelos de patologías de gran prevalencia.
DRA. PILAR CODOÑERJEFA DE SERVICIO PEDIATRÍAHOSPITAL UNIVERSITARIO DR. PESET DE VALENCIA
ABSTRACT
Alpha-1-antitrypsin deficiency (AATD) is a genetic disorder characterized by low serum levels of AAT, a blood protein produced in the liver. In subjects with AAT deficiency, the protein is still produced but the genetic defect means that the molecule configuration is changed. As a result, it is retained inside the liver and so cannot pass to the lungs and widespread. In normal lungs, alpha-1 antitrypsin protects lung tissue by trapping and destroying neutrophil elastase, an enzyme that digest damaged or aging cells and bacteria. However in AATD this enzyme is free to destroy tissue. As a consequence lung disease (emphysema) develops. Liver disease associated with AATD is not caused by the protein deficiency, but by excessive amounts of AAT stuck in the liver cells.
Genetic defect consists in a mutation in the SERPINA1 gene (previously known as the Pi gene) on chromosome 14. M alleles are the normal variants of the gene. Other common deficient variants are S and Z. Someone who is homozygous MM, will produce normal amounts of AAT. Common genotypes for people with AAT deficiency are ZZ, SS, SZ, MS, MZ. However, not everyone with AAT deficiency develops clinically significant disease. The different genotypes will lead to different serum levels of AAT. It is the serum level of AAT that will determine the likelihood of developing clinically significant disease.
Recommendation exists to test virtually all patients with chronic obstructive pulmonary disease for AATD, and those with asthma with incomplete reversibility on bronchodilator testing. Patients with liver disease of unrecognized origin should also be tested.
Aspectos clínicos del DAATModeradores: Dra. Escribano Dr. Blanco.
C.O. 08 QUÉ DEBE SABER UN MÉDICO SOBRE DAAT
RESUMEN
La deficiencia de alfa-1 antitripsina (DAAT) es un tras-torno genético que se caracteriza por los bajos niveles de AAT en suero, una proteína sanguínea producida en el hígado. Los pacientes con DAAT, sí producen la proteína, pero la configuración molecular ha cambiado. Como resultado, la proteína se retiene en el interior del hígado, por lo que no puede pasar a los pulmones y di-fundirse. En condiciones normales, la alfa-1 antitripsina protege el tejido pulmonar, atrapando y destruyendo la elastasa de los neutrófilos. La elastasa es una enzima que digiere células dañadas o envejecidas y bacterias. Sin embargo, en la DAAT, esta enzima está libre para destruir el tejido. Como consecuencia se desarrolla en-fermedad pulmonar (enfisema). La enfermedad hepá-tica asociada con DAAT no está causada por la defi-ciencia de proteína, sino por las excesivas cantidades de proteínas atrapadas en los hepatocitos.
El defecto genético consiste en una mutación en el gen SERPINA1 (anteriormente conocido como el gen Pi) en el cromosoma 14. Los alelos M son las variantes normales del gen. Otras variantes deficientes comunes son S y Z. Los homocigotos MM produce cantidades normales de AAT. Los genotipos más comunes para personas con deficiencia de AAT son: ZZ, SS, SZ, MS, MZ. Sin embargo, no todas las personas con deficien-cia de AAT desarrollan clínicamente la enfermedad. Los diversos genotipos producirán diferentes niveles de AAT en suero. Es el nivel sérico de AAT lo que va a determinar la probabilidad de desarrollar clínicamente la enfermedad.
Se recomenienda comprobar la DAAT a todos los pacientes con enfermedad pulmonar obstructiva cróni-ca, y a las personas con asma de reversibilidad incom-pleta en las pruebas de broncodilatador, así como a los pacientes con enfermedad hepática de origen desco-nocido.

16 /Valencia, 12 de diciembre de 2013
Jornada sobre déficit de alfa-1 y EPOC. Las enfermedades minoritarias como modelos de patologías de gran prevalencia.
DRA. BEATRIZ MARTÍNEZ UNIDAD DE GENÉTICA MOLECULAR. ÁREA DE GENÉTICA HUMANAINSTITUTO DE INVESTIGACIÓN DE ENFERMEDADES RARAS (IIER). INSTITUTO DE SALUD CARLOS III (ISCIII)
ABSTRACT
Alpha-1 Antitrypsin deficiency (AATD) is an autosomal-codominant genetic disorder that predisposes individuals to the development of liver and lung disease. Although it has been estimated a relatively high prevalence of the ZZ form in different populations, the AATD is a significantly underrecognized and underdiagnosed disease. Many AATD individuals, although at risk of developing symptoms of disease during life, can remain asymptomatic for a long time, and makes it difficult to determine the true prevalence of AATD.
The diagnosis of AAT deficiency is generally made after the identification of COPD or liver disease or after the deficiency has been diagnosed in a family member. Three strategies are commonly used to diagnose AAT deficiency: measurement of the serum or plasma protein level, AAT protein phenotyping of serum or plasma, and AAT genotyping. Each one of them has its own limitations and then to overcome these limitations, the testing of levels or protein phenotyping should be performed with genotype testing. Although there are different methodology and diagnostic algorithms currently in use, measurement of the level of AAT in the blood and the determination of the genotype are nowadays the two major diagnostic techniques.
Early identification will allow preventive measures to be taken, as could be the avoidance of smoking and exposure to environmental pollutants.
Aspectos clínicos del DAATModeradores: Dra. Escribano Dr. Blanco.
C.O. 09 DIAGNÓSTICO CLÍNICO Y DE LABORATORIO
RESUMEN
El déficit de alfa-1 antitripsina (DAAT) es una enferme-dad genética autosómica codominante, que predispone a los individuos para desarrollar enfermedad hepática y pulmonar. Aunque se ha estimado una prevalencia rela-tivamente alta del genotipo ZZ en diferentes poblacio-nes, el DAAT es significativamente poco reconocido y está infradiagnosticado. Existen muchos individuos en los que el déficit puede permanecer asintomático du-rante mucho tiempo, aunque el riesgo de desarrollar los síntomas de la enfermedad dure toda la vida, esto hace que sea difícil determinar la verdadera prevalencia de DAAT.
Generalmente, el diagnóstico de la deficiencia de AAT se realiza después de la identificación de EPOC o enfermedad hepática o después del diagnóstico de DAAT de algún familiar. Tres estrategias se utilizan comúnmente para diagnosticar DAAT: medición del nivel de proteína en suero o plasma, fenotipado de la proteína AAT en suero o plasma y genotipado de AAT. Cada uno de ellos tiene sus propias limitaciones, para superarlas se deben realizar las pruebas de los niveles de proteína o fenotipado de esta junto con la determina-ción del genotipo. Aunque hay diferentes metodologías y algoritmos de diagnóstico actualmente en uso, la me-dición del nivel de AAT en sangre y la determinación del genotipo son hoy en día las dos principales técnicas de diagnóstico.
La identificación temprana permitirá medidas pre-ventivas, como podría ser evitar el consumo de tabaco y la exposición a contaminantes ambientales.

Valencia, 12 de diciembre de 2013 /17
Jornada sobre déficit de alfa-1 y EPOC. Las enfermedades minoritarias como modelos de patologías de gran prevalencia.
DR. PERE RISTOL ÁREA DE INVESTIGACIÓN Y DESARROLLO INSTITUTO GRIFOLS
ABSTRACT
AAT is a protein found in human plasma with inhibitory activity of serine proteases, and more specifically of elastase, whose deficit has been associated with pulmonary disease (emphysema).
Currently the therapeutic AAT comes exclusively from human plasma for fractionation because its molecular characteristics and its concentration in the starting plasma makes it suitable for industrial production by conventional methods.
The AAT concentrates safety is ensured by screening plasma from the donor to the final product through serological assays (specific antibodies) and NAT (nucleic acids) of the transmissible infectious agents known. Also, production methods add security to the intrinsic plasma for fractionation in case of unlikely pathogens presence. The plasma fractionation industry deliberately includes specific steps to remove or destroy infectious agents. Thus, all production processes are validated complying with EU guidelines.
At present, industrial production methods allow to process several thousand liters of plasma per batch and thus to obtain marketable quantities of AAT with a high level of purity (> 60%) and functionality (activity/antigen ratio > 0.7). Lyophilized final products (Prolastina®, Trypsone®) are stable at room temperature for at least 2 years.
AAT worldwide consumption and its availability from human plasma, proving the potential capacity to meet this concentrate future needs in case of an demand increase, for example because of new indications or an increasing number of emphysema diagnosis.
In the near future new developing AAT concentrates will provide greater ease of administration (dose, concentration, ready to use, route), but replacing the plasma by recombinant technology will not be set up over the long term.
RESUMEN
La AAT es una proteína que se encuentra en el plasma humano con actividad inhibitoria de las serina protea-sas, y más específicamente de la elastasa, éste déficit se ha asociado con enfermedad pulmonar (enfisema).
Actualmente la AAT terapéutica proviene exclusiva-mente del fraccionamiento del plasma humano, debido a sus características moleculares y su concentración plasmática, en el punto de partida, hace que sea ade-cuado para la producción industrial por métodos con-vencionales.
En los concentrados de AAT la seguridad está garan-tizada gracias al cribado plasmático desde el donante hasta el producto final a través ensayos serológicos (an-ticuerpos específicos) y NAT (ácidos nucleicos) de los agentes infecciosos transmisibles conocidos. Además, los métodos de producción añaden seguridad al fraccio-namiento intrínseco del plasma en caso de presencia de patógenos no deseados. La industria de fraccionamien-to del plasma incluye deliberadamente medidas con-cretas para eliminar o destruir agentes infecciosos. Por tanto, todos los procesos de producción están validados mediante cumplimiento de las directrices de la UE.
En la actualidad, los métodos de producción indus-trial permiten procesar varios miles de litros de plasma por lote y así obtener cantidades comercializables de AAT con un alto nivel de pureza (> 60 %) y funcionali-dad (actividad / antígeno relación > 0,7). Los productos finales liofilizado (Prolastina®, Trypsone®) son estables a temperatura ambiente durante al menos 2 años.
El consumo de AAT en todo el mundo y su disponi-bilidad a través de plasma humano, prueban la capaci-dad potencial para satisfacer las necesidades futuras de este concentrado en caso de que la demanda aumente, como consecuencia de nuevas indicaciones o por un au-mento del número de diagnósticos de enfisema.
En un futuro próximo, nuevas vías de concentrado de AAT proporcionarán una mayor facilidad de admi-nistración (dosis, concentración, listo para usar, ruta), pero la sustitución del plasma por tecnología recombi-nante es una visión a largo plazo.
Aspectos clínicos del DAATModeradores: Dra. Escribano Dr. Blanco.
C.O. 010 AAT: DEL PLASMA AL PRODUCTO FINAL

18 /Valencia, 12 de diciembre de 2013
Jornada sobre déficit de alfa-1 y EPOC. Las enfermedades minoritarias como modelos de patologías de gran prevalencia.
DRA. MARÍA TORRES NEUMÓLOGACOMPLEXO HOSPITALARIO UNIVERSITARIO DE VIGO.
Aspectos clínicos del DAATModeradores: Dra. Escribano Dr. Blanco.
C.O. 011 EXPERIENCIA DE LAS COMUNIDADES AUTÓNOMAS EN DAAT: GALICIA
RESUMEN
El déficit de alfa-1 antitripsina es una enfermedad he-reditaria autosómica co-dominante, su principal mani-festación clínica en adultos es el desarrollo temprano de enfisema y EPOC. Desde hace 20 años tenemos un tratamiento específico para la enfermedad a través de una infusión intravenosa de concentrados purificados de AAT . A pesar de la experiencia, aún persiste cierta controversia acerca de la eficacia del tratamiento, debi-do a la falta de grandes estudios (ECA).
En 2003, se creó en Galicia una “Comisión Far-macoterapéutica para el tratamiento de pacientes con DAAT” con la finalidad de asesorar a los médicos que tratan a estos pacientes. El Registro del Paciente fue creado inspirado en otros existentes como el AIR o RE-DAAT. Los médicos deben incluir en el Registro todos los pacientes con fenotipos ZZ, SZ u otras variantes del déficit severo. Es obligatorio hacerlo cuando se requiere tratamiento sustitutivo para un paciente.
La Comisión evalúa y aprueba todas las solicitudes de tratamiento sustitutivo de todos los hospitales galle-gos. Se han establecido unos criterios de tratamiento que incluyen: edad > 18 años, PiZZ u otras variantes del déficit, los niveles de A1AT < 11 mM / L, enfisema pulmonar, FEV1: 30-65 % , y ser no fumador o ex -fu-mador (más de 6 meses).
En 10 años de actividad, se evaluaron 81 solicitudes de tratamiento, se aprobaron 71 tratamientos , 7 fueron rechazados (6 por no cumplir con los criterios y una pe-tición reiterada) y actualmente están siendo evaluadas 3 peticiones.
Los pacientes en tratamiento reciben una dosis de 180mg/kg de A1AT cada 21 días en el “Hospital de Día”. Se recomienda seguimiento clínico, con pruebas de función pulmonar, cada 6 meses.
ABSTRACT
Alpha 1 Antitrypsin deficiency is an autosomal co-do-minant hereditary disease which main clinical manifes-tation in adults is the early development of emphysema and COPD. For over 20 years we have specific treat-ment for the disease through intravenous infusion of purified concentrates of AAT. Despite the experience, there remains some controversy about the efficacy of treatment, due to the lack of large studies (RCT).
In 2003 a “Pharmacologic-Therapeutic Commission for the treatment of patients with AATD” was created in Galicia to serve as advisory board for physicians treating these patients. A Patient́ s Registry was created inspired in other existing like AIR or REDAAT. Physi-cians should include in the Registry all patients with ZZ phenotype or other severe deficiency variants and SZ phenotype. It is mandatory to do it when augmentation therapy is required for a patient.
The Commission evaluates and approves all requests for augmentation therapy from all Galician hospitals. Treatment criteria are established and includes: age > 18 years, PiZZ phenotype or other deficient variants, levels of AAT <11mM/L, pulmonary emphysema, FEV1: 30-65%, and be non-smoker or ex-smoker (more than 6 months).
In 10 years of activity, 81 applications for treatment were assessed, 71 treatments were approved, 7 were re-jected (6 for not meet criteria and one repeated request) and 3 are currently being evaluated.
Patients on treatment receive a dose of 180mg/kg of A1AT every 21 days at “Day Hospital”. Clinical follow up with lung function test every 6 months is recommen-ded.

Valencia, 12 de diciembre de 2013 /19
Jornada sobre déficit de alfa-1 y EPOC. Las enfermedades minoritarias como modelos de patologías de gran prevalencia.
ABSTRACT
Since 2011, the Valencia Health Agency is developing a specific strategy for Chronic Obstructive Pulmonary Disease (COPD), an ambitious program aimed to reduce the incidence, decrease the underdiagnosis and improve morbidity, mortality and quality of care in patients with COPD. Additionality this program also promotes training of profesionals and quality research. One of the priority strategic line is focused on the fight against the underdiagnosis (line 1). For this area we have designed a research program in the field of primary care, intended to assess whether the early diagnosis through case-finding approach and early therapeutic intervention could reduce the adverse long term consequences of the disease. The study will be developed in a large number of primary care centers, near 16.000 patients at risk of COPD (without previous diagnosis) will be selected and a longitudinal follow-up using electronic health history during several years will be included. The design of this case-finding program is an excellent oportunity to obtain information about epidemiological distribution of the alpha-1-antitrypsin deficiency (AAT). The first cross-sectional phase of the study will be intended to determine the prevalence of AAT deficiency in those newly diagnosed COPD patients. The posterior longitudinal follow-up also will add information on the longitudinal development of intermediate deficits and the natural history of both ZZ and the other phenotypes.
RESUMEN
Desde 2011, la Agencia Valenciana de Salud está desa-rrollando una estrategia específica para la Enfermedad Pulmonar Obstructiva Crónica (EPOC), un ambicioso programa destinado a reducir la incidencia, disminuir el infradiagnóstico y mejorar la morbilidad, mortalidad y calidad de la atención en los pacientes con EPOC. Ade-más, este programa también promueve la formación de profesionales e investigación de calidad. Una línea de prioridad estratégica se centra en la lucha contra el in-fradiagnóstico (línea 1). En esta área hemos diseñado un programa de investigación en el campo de la aten-ción primaria, con la intención de evaluar si el diagnós-tico precoz a través del método de detección de casos y la intervención terapéutica temprana podrían reducir las consecuencias adversas de la enfermedad a largo plazo. El estudio se desarrollará en un gran número de centros de atención primaria, se seleccionarán cerca de 16.000 pacientes con riesgo de EPOC (sin diagnóstico previo) y se incluirá un seguimiento longitudinal me-diante historia clínica electrónica durante varios años. El diseño de este programa de detección de casos es una excelente oportunidad para obtener información acerca de la distribución epidemiológica de la deficiencia de alfa-1-antitripsina (AAT). En la primera fase transver-sal del estudio, se pretende determinar la prevalencia de deficiencia de AAT en los nuevos pacientes diagnos-ticados con EPOC. El seguimiento longitudinal pos-terior, también, tendrá información sobre el desarrollo longitudinal de déficits intermedios y la historia natural de ZZ y otros fenotipos.
Aspectos clínicos del DAATModeradores: Dra. Escribano Dr. Blanco.
C.O. 011 EXPERIENCIA DE LAS COMUNIDADES AUTÓNOMAS EN DAAT: COMUNITAT VALENCIANA
DR. JUAN JOSÉ SOLERCOORDINADOR PROGRAMA EPOC-PLAN DE SALUD 2010-14COMUNITAT VALENCIANA

20 /Valencia, 12 de diciembre de 2013
Jornada sobre déficit de alfa-1 y EPOC. Las enfermedades minoritarias como modelos de patologías de gran prevalencia.
DR. FRANCISCO CASAS NEUMÓLOGOHOSPITAL UNIVERSITARIO SAN CECILIO DE GRANADA PRESIDENTE DE NEUMOSUR
ABSTRACT
Clinical case 1. Female 47 years old. Primary school teacher. Nine years ex-smoker (15 pack/year) ago. Married without children. Eight brothers, one sister died of breast cancer and other with respiratory problems. Sent to outpatient primary care from frequent respiratory infections. Now presents with purulent bronchial infection process again. Clinical history of productive mucoid cough morning and dyspnoea grade 1-2 MRC from 5-6 years. Frequent respiratory infections winter stores: cough, expectoration abundant mucopurulent, wheezing and increased dyspnoea. He denies chest pain or fever. In the last 6 months has made successive treatments with antibiotics does not remember. Physical examination: good general condition. SpO2= 95%. BMI: 25,6. Cardiac auscultation normal. Respiratory auscultation with light widespread hypophonesis.
Clinical case 2. Student girl 15 years of age. Proper vaccination schedule. No family history of asthma or other respiratory problems. Acute bronchiolitis newborn aspiration of gastric contents. Pneumonia in 2002 and in february 2012 (hospital admissions). Menarche at 11 years of age. Passive smoking (home and car). Father active smoker (40 pack/year) diagnosed ischemic heart disease. Mother ex-smoker since 2 years (20 pack/year) ago. Clinical history of productive mucoid cough and usual dyspnoea grade 1 MRC in the last year. He denies symptoms with pets. He denies symptoms of bronchial hyperreactivity. Good appetite and weight stability. No digestive discomfort. Physical examination: Good general condition. SpO2= 95%. Height: 149 cm, weight: 45 kg, BMI: 20,3. Cardiac auscultation normal. Respiratory auscultation with mild hypophonesis right base.
Aspectos clínicos del DAATModeradores: Dr. Blanco, Dr. Dasí.
CO 012. CASOS CLÍNICOS DAAT
RESUMEN
Caso clínico 1. Mujer de 47 años. Maestra de escue-la primaria. Exfumadora desde hace nueve años (15 paquetes/año). Casada sin hijos. Ocho hermanos, una hermana murió de cáncer de mama y otro con proble-mas respiratorios. Enviada a Atención Primaria por pa-cientes con infecciones respiratorias frecuentes. En la actualidad, presenta otro proceso infeccioso bronquial purulento. Historia clínica de tos matutina, expectora-ción mucoide y disnea de medianos-grandes esfuerzos (grado 1-2 MRC) desde hace 5 -6 años. Infecciones respiratorias frecuentes en invierno: tos, expectoración abundante mucopurulenta, pitos y aumento de la disnea. Niega dolor torácico o fiebre. En los últimos 6 meses su-cesivos tratamientos con antibióticos que no recuerda. Exploración física: buen estado general. SpO2 = 95%. IMC: 25,6. Auscultación cardiaca normal. Auscultación respiratoria con leve hipofonesis generalizada.
Caso clínico 2. Chica estudiante de 15 años. Calendario de vacunación adecuado. Sin antecedentes familiares de asma u otros problemas respiratorios. Bronquiolitis aguda de recién nacida por aspiración de contenido gástrico. Neumonía en 2002 y en febrero de 2012 (ingresos hospitalarios). Menarquia a los 11 años. Tabaquismo pasivo (casa y coche). Padre fumador activo (40 paquetes/año) diagnosticado de enfermedad cardíaca isquémica. Madre de exfumadora desde hace 2 años (20 paquetes/año). Historia clínica de tos con expectoración mucoide y disnea de grandes esfuerzos (grado 1 MRC) en el último año. Niega síntomas con mascotas. Niega síntomas de hiperreactividad bronquial. Buen apetito y estabilidad de peso. Sin molestias digestivas. Examen físico: buen estado general. SpO2 = 95%. Altura: 149 cm, peso: 45 kg, IMC: 20,3. Auscultación cardiaca normal. Auscultación respiratoria con leve hipofonesis en la base derecha.

Valencia, 12 de diciembre de 2013 /21
Jornada sobre déficit de alfa-1 y EPOC. Las enfermedades minoritarias como modelos de patologías de gran prevalencia.
DÑA. MERCEDES NAVARROESTUDIANTE DE PERIODISMOFACULTAD DE FILOLOGÍA, TRADUCCIÓN Y COMUNICACIÓN UNIVERSITAT DE VALÈNCIA
ABSTRACT
On the 50th anniversary of the discovery of AATD, a rare disease that affects many individuals around the world, putting them at risk of having lung and liver disease, an study to assess the coverage of the disease by the press was performed. Methods: News in four EU newspapers were analyzed. Two Spanish (El País; El Mundo) and two UK newspapers (The Sun; Sunday Express). The most relevant and the only AATD-related news covered by the four newspapers was “Michael Jackson needs lung transplant”. Formal components of that new were registered and the informative coverage given by the four newspapers was analyzed. Results: The analysis of the sources used by the four newspapers highlighted some differences. The Spanish newspapers, El Pais y El Mundo, used the information provided by the The Sun and Sunday Express. Although the Spanish newspapers have enough resources to carry out a complete coverage, minor coverage was preferred, virtually echoing a rumor. The coverage given by the UK newspapers was quite similar to that provided by the Spanish ones. The Sunday Express (which claimed exclusivity on the news) briefly mentioned the disease and gave their readers more information on the disease.The four newspapers used the same criteria of newsworthiness to convert fact into news. All used expressions highlighted the event/drama. The treatment of the news was tabloid-type, bordering on hearsay.
Conclusions. We emphasize that press media could have been used to provide information on one condition that affects hundreds of thousands of individuals around the world. Analysis of the news showed lack of informative rigor in the editorial lines of the main newspapers of two EU countries, which have great influence in the public opinion, and the withdrawal of the social function attributed to rigorous journalism oriented to the information of health-related issues.
RESUMEN
En el 50 aniversario del descubrimiento de la enfer-medad minoritaria déficit de alfa-1 antitripsina, estu-diamos cómo los medios de comunicación de prensa generalista dan cobertura a esta enfermedad. Tras se-leccionar la noticia más significativa, presentamos el análisis del tratamiento periodístico. Métodos. Se han analizado cuatro periódicos: dos de ámbito nacional, El País y El Mundo, y dos de otro país europeo, The Sun y Sunday Express (Reino Unido). Tras una búsqueda de noticias relacionadas con la enfermedad déficit de alfa 1 antitripsina, se seleccionó la noticia “Michael Jackson necesita un trasplante de pulmón”, única no-ticia relacionada con la enfermedad que fue tratada en los cuatro periódicos. Se registraron los componentes formales de presentación, y se analizó el tratamiento informativo que dichos periódicos daban a la noticia.Resultados. Al analizar las fuentes, destacó la diferen-cia de recursos entre ellos, siendo El País y El Mundo los que emplearon menor cobertura en la elaboración de la noticia, ya que solo recogieron las informaciones dadas por The Sun y Sunday Express. Hemos obser-vado que los periódicos nacionales, aunque disponen de suficientes recursos para realizar una cobertura más completa, optaron por un tratamiento menor, práctica-mente se hicieron eco de un rumor. Asimismo, los dos periódicos de ámbito internacional utilizaron recursos de cobertura similares. Aunque Sunday Express, que se atribuye la exclusividad, mencionó brevemente la en-fermedad dando mayor cobertura e información a sus lectores. Los cuatro utilizaron los mismos criterios de noticiabilidad para convertir el hecho en noticia. Todos utilizaron expresiones que remarcan el suceso/drama. El tratamiento de la noticia fue sensacionalista, rozando la rumorología.
Conclusiones. Destacamos que los medios genera-listas podrían haber utilizado la noticia para dar difu-sión a una de las enfermedades poco frecuente que es desconocida por los lectores. Del análisis de la noticia se desprende la falta de rigor en las líneas editoriales de cabeceras influyentes en sus respectivos países y el alejamiento de la función social que se atribuye al pe-riodismo de rigor, orientado a la divulgación de temas de salud.
Aspectos sociales del DAATModerador: Dr. Antonio Ariño
CO 013. EL DAAT EN LOS MEDIOS DE COMUNICACIÓN: ANÁLISIS Y RETOS

22 /Valencia, 12 de diciembre de 2013
Jornada sobre déficit de alfa-1 y EPOC. Las enfermedades minoritarias como modelos de patologías de gran prevalencia.
DÑA. SHANE FITCH PRESIDENTAFUNDACIÓN LOVEXAIR
ABSTRACT
RESUMEN
Actualmente el papel de la asociación de pacientes tiene nuevas oportunidades no explotables a este nivel ante-riormente. Sin embargo, requiere una mayor profesio-nalización de las organizaciones para saber aprovechar mejor esta nueva etapa.
Si la asociación tiene dificultad entre sus afiliados para encontrar recursos profesionales para acompa-ñarles en este proceso, tiene que identificar recursos externos, aliados que les podrían prestar este servicio y también deben ser más pro-activos en conseguir sus objetivos.
El entorno es competitivo para obtener recursos eco-nómicos y es fundamental que la asociación aprenda a hacer frente a ello igual que hacen las demás organiza-ciones y empresas.
Hay que preparar los recursos y la red de relaciones que podrían contribuir a su misión, objetivos y soste-nibilidad. Conseguir visibilidad y credibilidad es fun-damental, actuando con una acción coordinada y en un sentido realista acorde a los tiempos y la situación actual
Encontrar medios y recursos apoyándose en las nue-vas tecnologías es el vía al alcance de todos. Lo impor-tante es planificar los pasos y la estrategía con el obje-tivo de crear valor para sus afiliados y la comunidad afectada y relacionada en su entorno.
Aspectos sociales del DAATModerador: Dr. Antonio Ariño
C.O. 014. MESA REDONDA: EL PAPEL DE LAS ASOCIACIONES DE PACIENTES
In our current environment patient associations can be-nefit from new opportunities not previously available. However this requires a more professional approach to be made by the organizations to take advantage of this situation.
If the association has difficulty finding professional skills amongst its members in this process, they need to identify external resources, allies who can deliver this service and they need to be more pro-active in achie-ving their objectives.
The environment is competitive in identifying eco-nomic resources and its essential for the association to find ways of confronting this situation the same as other organizations and businesses.
Both resources and the network of key relationships that contribute to their mission, goals and sustainabli-ty need to be prepared. Gain visibility, and credibility through cooordinated action and a realistic approach to the current times and situation are vital.
Identifying the necessary means and resources through new technology is within everyone’s reach the-se days. What is important is to plan the strategy and steps with the goal of creating value for the members and the community who are affected and related to their environment. This necessarily includes all stakeholders.

Valencia, 12 de diciembre de 2013 /23
Jornada sobre déficit de alfa-1 y EPOC. Las enfermedades minoritarias como modelos de patologías de gran prevalencia.
D. ALAN HEYWOOD-JONESPRESIDENTEASOCIACIÓN ALFA 1 EUROPA
ABSTRACT
Patient Organisations, especially those concerned with rare diseases, have an important part to play in relieving the burden of illness from the patient, his family, the health services and society at large.
Rare disease patients live with infirmities which often limit what they can do in life and this can also affect their families. Patients, working together in association, can provide support and encouragement using their collective experience. One patient talking to another is an effective way of communicating how to cope with an illness and reduce the consequential stress.
In addition to support given to individuals, associations can collaborate with doctors in developing better models of care and with researchers in helping increase our knowledge of the disease process. Patients often want to do more to help and joining registries, especially those for rare diseases, is a major contribution that they can make.
Finally, patient organisations need to inform society of the burden that illness places on their members and how this is also a burden on society. Many small patient associations find it difficult to make their voice heard in places where decisions about health care are made. For this reason it is necessary to work with other associations as members of EURORDIS, EPPOSI, FEDER, etc.
In the modern world patients are seem as equal partners in designing the best health care that we can provide. We measure the quality of our society by how we care for those with infirmities.
Aspectos sociales del DAATModerador: Dr. Antonio Ariño
C.O. 014. MESA REDONDA: EL PAPEL DE LAS ASOCIACIONES DE PACIENTES
RESUMEN
Las organizaciones de pacientes, especialmente, los re-lacionados con las enfermedades raras, tienen un papel importante que desempeñar en el alivio de la carga de la enfermedad por parte del paciente, su familia, los servi-cios de salud y la sociedad en general.
Los pacientes de enfermedades raras viven muy li-mitados, y esto puede afectar también a sus familias. Los pacientes, que trabajan juntos en asociación, pue-den proporcionar apoyo y aliento a través de su expe-riencia colectiva. Un paciente que habla con otro es una forma efectiva de comunicar cómo hacer frente a una enfermedad y reducir el estrés.
Además del apoyo dado a los individuos, las asocia-ciones pueden colaborar con los médicos en el desarro-llo de mejores modelos de atención, y con los investiga-dores para ayudar a aumentar nuestro conocimiento del proceso de la enfermedad. Los pacientes quieren hacer más para ayudar y unirse a los registros, en especial pa-cientes con enfermedades infrecuentes, ya que es im-portante la contribución que pueden hacer.
Por último, las organizaciones de pacientes deben informar a la sociedad de la carga que supone la enfer-medad en sus miembros y cómo esta es también una carga para la sociedad. Muchas asociaciones con pocos pacientes tienen dificultades para hacer oír su voz en los lugares donde se toman las decisiones sobre el cui-dado de la salud. Por esta razón, es necesario trabajar con otras asociaciones como miembros: EURORDIS, EPPOSI, FEDER , etc
En el mundo moderno, los pacientes deben verse como socios iguales que participan en el diseño de la mejor atención de la salud que podamos ofrecer. Medi-mos la calidad de nuestra sociedad por la forma en que cuidamos de las personas con enfermedades.

24 /Valencia, 12 de diciembre de 2013
Jornada sobre déficit de alfa-1 y EPOC. Las enfermedades minoritarias como modelos de patologías de gran prevalencia.
DÑA. ALMUDENA AMAYADELEGADAFEDER- COMUNIDAD VALENCIANA
RESUMEN
El aumento del acceso a internet y el uso de las redes sociales ha llevado a la Federación Española de Enfer-medades Raras (FEDER) a plantearse la utilización de éstas como herramienta de comunicación en salud.
Además, ha favorecido realizar una reflexión sobre como internet y las redes sociales pueden funcionar de motor, en la búsqueda de información relativa a la sa-lud. Deteniéndose, en el posicionamiento de las Orga-nizaciones sanitarias frente a este hecho y en conocer cuál es la actual presencia de centros sanitarios en la red y redes sociales.
También ha profundizado en las consideraciones a tener en cuenta a la hora de trabajar o gestionar una red social desde una asociación de pacientes, las expectati-vas del colectivo y los beneficios que aportan las redes en centros sanitarios entre otras cuestiones.
En la actualidad, los pacientes buscan a través de internet y las redes sociales, no sólo información sani-taria, si no también, apoyo de otros pacientes o profe-sionales. Se habla de un paciente informado como un colaborador activo.
Sin duda, FEDER apuesta por la utilización de estas herramientas para favorecer la interrelación entre los profesionales sanitarios y los pacientes, al mismo tiem-po que, se favorece la mejora de los servicios.
Aspectos sociales del DAATModerador: Dr. Antonio Ariño
C.O. 015. HERRAMIENTAS WEB PARA LA COMUNICACIÓNEJ.: FEDER
ABSTRACT
The increase in internet access and use of social ne-tworks has led to the Spanish Federation for Rare Di-seases (FEDER) to consider using them as a tool for health communication.
It has also helped make a reflection on how the In-ternet and social networks can operate motor in search of information on health. Stopping in positioning health organizations face this fact and know what the current presence of health centers in the network and social ne-tworks.
He has also delved into the considerations to keep in mind when working or managing a social network from a patient organization, the expectations of the group and the benefits of networks in health centers among others.
Currently, patients seek through internet and social networks, not just health information , but also support from other patients or professionals. There is talk of an informed patient as an active partner.
Surely FEDER commitment to using these tools to encourage interaction between healthcare professionals and patients, while the improvement of services is en-couraged.
DÑA. PILAR HERNÁNDEZ TRABAJADORA SOCIAL

Valencia, 12 de diciembre de 2013 /25
Jornada sobre déficit de alfa-1 y EPOC. Las enfermedades minoritarias como modelos de patologías de gran prevalencia.
DRA. MARTA PEIRÓCOORDINADORA ÁREA CIENTÍFICAINCLIVA
ABSTRACT
The Foundation for the Research of the Hospital Clínico Universitario of Valencia was constituted in the year 2000, being the first Valencian research foundation attached to a public hospital. In 2010, certain excellence groups in biomedical research from the University of Valencia and IUIVI (Valencian Infertility Institute) joined the Foundation through the establishment of assignment agreements, thus creating the INCLIVA Health Research Institute.
INCLIVA manages biomedical research performed and encourages teaching and scientific activities, thus improving patient treatment and knowledge sharing.
After being accredited, in 2011, as a Health Research Institute by the Ministry of Science and Innovation, INCLIVA obtained preferential by the Carlos III Health Institute being recognized its excellent research.
INCLIVA is organized around four priority areas of research, based on a selection of internal scientific excellence, health demands and funding requirements of the R+D+i system, which are: Cardiovascular, Oncological, Metabolism and Organic Damage Research and Reproductive Medicine Research. INCLIVA has produced over 500 international publications with a cumulated impact factor of 2,000 in the last year. Besides, during 2011, its attached research groups have carried out more than 100 granted competitive projects, including four European Framework Programme major cooperation projects.
Finally, the ultimate mission of INCLIVA is to transfer the knowledge generated by research to clinical practice and to the productive sector. Thus, the research groups have participated in the generation of 34 practice guidelines published in international journals and up to 9 patent requests have been submitted in the last three years.
Aspectos sociales del DAATModerador: Dr. Antonio Ariño
C.O. 016. LA INVESTIGACIÓN TRASLACIONAL: SINERGIA ENTRE LA INVESTIGACIÓN ACADÉMICA Y LOS HOSPITALES PÚBLICOSEJ.: INCLIVA
RESUMEN
La Fundación para la Investigación del Hospital Clínico Universitario de Valencia se constituyó en el año 2000, siendo la primera fundación de investigación valen-ciana asociada a un hospital público. En 2010, varios grupos de excelencia en investigación biomédica de la Universidad de Valencia y el IVI (Instituto Valenciano de Infertilidad) se unieron a la Fundación a través del establecimiento de acuerdos de cesión, creando el Insti-tuto de Investigación Sanitaria- INCLIVA.
El INCLIVA gestiona la investigación biomédica y fomenta actividades docentes y científicas, mejorando así el tratamiento de los pacientes y el intercambio de conocimiento.
Después de ser acreditado, en 2011, como un Institu-to de Investigación Sanitaria por el Ministerio de Cien-cia e Innovación, el INCLIVA obtuvo la acreditación del Instituto de la Salud Carlos III reconociendo su ex-celente investigadora.
El INCLIVA está organizado en cuatro áreas priori-tarias de investigación, basado en una selección de ex-celencia científica interna, las demandas de salud y los requisitos de financiación del sistema I+D+i, que son: Cardiovascular, Oncología, Metabolismo y Daño Or-gánico y Medicina Reproductiva. El INCLIVA ha pro-ducido más de 500 publicaciones internacionales con un factor de impacto acumulado de 2.000 en el último año. Además, durante 2011, sus grupos de investigación han llevado a cabo más de 100 proyectos competitivos, incluyendo cuatro proyectos de cooperación dentro del marco europeo. Finalmente, la última misión del IN-CLIVA es transferir el conocimiento generado por la investigación a la práctica clínica y al sector productivo. Así, los grupos de investigación han participado en la generación de 34 guías publicados en revistas interna-cionales y hasta 9 solicitudes de patentes se han presen-tado en los últimos tres años.

26 /Valencia, 12 de diciembre de 2013
Jornada sobre déficit de alfa-1 y EPOC. Las enfermedades minoritarias como modelos de patologías de gran prevalencia.
DRA. BEATRIZ LARANEUMÓLOGAGESTORA REDAAT
ABSTRACTRESUMEN
En esta sesión se realizará una aproximación básica a la molécula de la AAT y las alteraciones sufridas por las diferentes mutaciones que ocasionan déficit, cómo se hereda, cómo favorece el desarrollo de enfermedades y se hará hincapié en los cuidados y tratamientos reco-mendados para los afectados en un formato que facilite la participación de los asistentes.
Asimismo, se presentará la nueva guía para familias de niños con déficit mediante la descripción en la sesión de los contenidos más destacados.
Además de los aspectos mencionados se comentarán las asociaciones y otras iniciativas de apoyo a los pa-cientes y sus familias y los recursos on line existentes.
Aspectos sociales del DAATModerador: Dr. Antonio Ariño
C.O. 017. QUÉ DEBE SABER UN PACIENTE SOBRE DAAT
In this session a basic approach to the AAT molecu-le will be done as well as the alterations caused by the different mutations that produce AATD. How it is in-herited, how favors disease development and will em-phasize care and recommended treatments for those affected in a format that facilitates the participation of the attendees.
Also, the new guide for families of children with AATD will be presented by describing some of the most important contents.
Besides the above mentioned aspects, the role of pa-tients’ associations and other initiatives to support pa-tients and their families and existing online resources will be discussed.
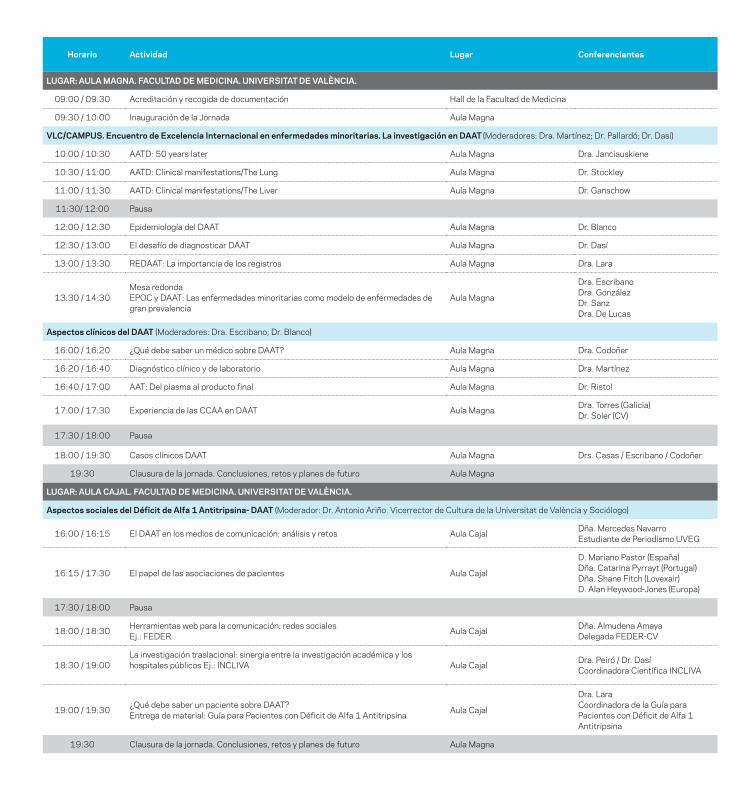
PRESENTACIÓN
La jornada está organizada por el Instituto de Investigación Sanitaria del Hospital Clínico de Valencia INCLIVA, con la colaboración de la Universitat de València y el VLC Campus de Excelencia.
La Facultad de Medicina de Valencia acogerá una jornada monográfica sobre la enfermedad minoritaria déficit de alfa-1 antitripsina (DAAT) destinada a profesionales sanitarios, pacientes y familiares.
La jornada que ha sido avalada por diversas sociedades científicas, contará con la participación de expertos clínicos y científicos de ámbito nacional e internacional que han contribuido al conocimiento de la enfermedad.
El encuentro albergará tres actos diferentes, pero complementarios entre sí. La mañana se dedicará a sesiones de información básica sobre el DAAT. La tarde la ocupará la parte clínica de la enfermedad, en la que además de las charlas de los correspondientes especialistas en la materia, se incluirá una sesión de casos clínicos especialmente dirigida a los médicos residentes. De manera paralela, la tarde se centrará en la divulgación social de la enfermedad a través de un debate entre sociólogos, periodistas y asociaciones de pacientes y familiares.
OBJETIVO
El objetivo fundamental de esta jornada es colaborar en la creación de una conciencia global de lo que supone el DAAT en la vida de los pacientes y en el conjunto de la sociedad. La jornada dará a conocer la enfermedad tanto en el ámbito médico como en el social. La interrelación investigador-médico-paciente-sociedad es una oportunidad para debatir sobre el impacto y el estado de la enfermedad y para identificar necesidades aún no satisfechas. Del mismo modo, la jornada permitirá generar una cultura de la ciencia que refuerce las labores de investigación que se realizan en universidades e institutos de investigación sanitaria.
CARACTERÍSTICAS
¿A quién va dirigida?Profesionales sanitarios, científicos y pacientes juntos ante la enfermedad.La jornada se compone de charlas magistrales y una sesión de casos clínicos relacionados con el DAAT, y va dirigida a investigadores, pacientes y fundamentalmente a profesionales sanitarios: pediatras, neumólogos, hepatólogos, médicos de atención primaria, personal de enfermería, estudiantes de ciencias de la Salud, etc. Asimismo se realizará una sesión paralela destinada a debatir sobre los aspectos sociales relacionados con la enfermedad.
Horario Actividad Lugar Conferenciantes
LugAR: AuLA MAgNA. FACuLTAD DE MEDICINA. uNIVERSITAT DE VALèNCIA.
09:00 / 09:30 Acreditación y recogida de documentación Hall de la Facultad de Medicina
09:30 / 10:00 Inauguración de la Jornada Aula Magna
VLC/CAMPuS. Encuentro de Excelencia Internacional en enfermedades minoritarias. La investigación en DAAT (Moderadores: Dra. Martínez; Dr. Pallardó; Dr. Dasí)
10:00 / 10:30 AATD: 50 years later Aula Magna Dra. Janciauskiene
10:30 / 11:00 AATD: Clinical manifestations/The Lung Aula Magna Dr. Stockley
11:00 / 11:30 AATD: Clinical manifestations/The Liver Aula Magna Dr. Ganschow
11:30/ 12:00 Pausa
12:00 / 12:30 Epidemiología del DAAT Aula Magna Dr. Blanco
12:30 / 13:00 El desafío de diagnosticar DAAT Aula Magna Dr. Dasí
13:00 / 13:30 REDAAT: La importancia de los registros Aula Magna Dra. Lara
13:30 / 14:30Mesa redondaEPOC y DAAT: Las enfermedades minoritarias como modelo de enfermedades de gran prevalencia
Aula Magna
Dra. EscribanoDra. GonzálezDr. SanzDra. De Lucas
Aspectos clínicos del DAAT (Moderadores: Dra. Escribano; Dr. Blanco)
16:00 / 16:20 ¿Qué debe saber un médico sobre DAAT? Aula Magna Dra. Codoñer
16:20 / 16:40 Diagnóstico clínico y de laboratorio Aula Magna Dra. Martínez
16:40 / 17:00 AAT: Del plasma al producto final Aula Magna Dr. Ristol
17:00 / 17:30 Experiencia de las CCAA en DAAT Aula Magna Dra. Torres (Galicia)Dr. Soler (CV)
17:30 / 18:00 Pausa
18:00 / 19:30 Casos clínicos DAAT Aula Magna Drs. Casas / Escribano / Codoñer
19:30 Clausura de la jornada. Conclusiones, retos y planes de futuro Aula Magna
LugAR: AuLA CAJAL. FACuLTAD DE MEDICINA. uNIVERSITAT DE VALèNCIA.
Aspectos sociales del Déficit de Alfa 1 Antitripsina- DAAT (Moderador: Dr. Antonio Ariño. Vicerrector de Cultura de la Universitat de València y Sociólogo)
16:00 / 16:15 El DAAT en los medios de comunicación: análisis y retos Aula Cajal Dña. Mercedes NavarroEstudiante de Periodismo UVEG
16:15 / 17:30 El papel de las asociaciones de pacientes Aula Cajal
D. Mariano Pastor (España)Dña. Catarina Pyrrayt (Portugal)Dña. Shane Fitch (Lovexair)D. Alan Heywood-Jones (Europa)
17:30 / 18:00 Pausa
18:00 / 18:30 Herramientas web para la comunicación: redes socialesEj.: FEDER Aula Cajal Dña. Almudena Amaya
Delegada FEDER-CV
18:30 / 19:00La investigación traslacional: sinergia entre la investigación académica y los hospitales públicos Ej.: INCLIVA Aula Cajal Dra. Peiró / Dr. Dasí
Coordinadora Científica INCLIVA
19:00 / 19:30 ¿Qué debe saber un paciente sobre DAAT?Entrega de material: Guía para Pacientes con Déficit de Alfa 1 Antitripsina Aula Cajal
Dra. LaraCoordinadora de la Guía para Pacientes con Déficit de Alfa 1 Antitripsina
19:30 Clausura de la jornada. Conclusiones, retos y planes de futuro Aula Magna

Jornada sobre déficit de alfa-1 antitripsina y EPOCLas enfermedades minoritarias como modelos de patologías de gran prevalencia
Valencia, 12 diciembre 2013
Lugar de celebración: Facultad de Medicina de la Universitat de València.Avda. Blasco Ibáñez nº 15 - 46010 - Valencia
Derechos de inscripción: Investigadores, profesionales sanitarios: 60€Estudiantes de Ciencias de la Salud: 30€ Reconocido con 1 crédito de libre opción por la Universitat de València (Para obtener el reconocimiento de los créditos, el estudiante deberá abonar en su centro el 25% del valor de los créditos)Pacientes y familiares: 10€
Más información e inscripciones:Fundación Universidad-Empresa de Valencia, ADEITTeléfono: 962057923/25Página web: congresos.adeituv.es/alfa-1
DirectorDr. Francisco Dasí Investigador INCLIVA-Prof. asociado UVEG
Comité OrganizadorDra. Amparo EscribanoPediatra HCUV-Profª UVEGDra. Pilar Codoñer Pediatra H. Dr. Peset. Valencia-Profª UVEGDra. Beatriz Lara Neumóloga REDAATDr. Francisco SanzNeumólogo HGUVDra. Mónica AmorPediatra HCUVDra. Silvia CastilloPediatra HCUVDr. Federico PallardóDecano Facultad Medicina UVEGDr. José Luis García Investigador CIBERERDr. Felipe SerranoInvestigador Cambridge Institute for Medical ResearchDª. María Mercedes NavarroEstudiante periodismo UVEG
Organiza:
Avalada por:
Colaboran: