introducción a la química orgánica · web viewpentanal ch3 cho...
TRANSCRIPT

www.monografías.com
Introducción a la Química Orgánica1. Introducción. 2. Sustitucion electrófila aromática 3. Compuestos organometálicos. 4. Alcoholes y fenoles. 5. Compuestos 1,3-dicarbonílicos. 6. Reacciones. 7. Conformaciones. 8. Proyecciones de Newman. 9. Estereoisomería geométrica debida al doble enlace.
Introducción.
Estructuras de Lewis.
Las estructuras de Lewis pueden dibujarse para todos los elementos y compuestos representativos de una molécula unidos mediante enlaces covalentes. Un enlace covalente se produce cuando dos átomos comparten electrones. Si los dos átomos del enlace son iguales o tienen electronegatividad similar, los electrones son compartidos por igual entre los dos átomos y el enlace es considerado no polar. Si los dos átomos tienen electronegatividad significativamente diferente, los electrones no son compartidos por igual entre los dos átomos y el enlace es considerado como polar. En un enlace polar, el elemento más electronegativo adquiere una carga parcial negativa, y el elemento menos electronegativo adquiere una carga parcial positiva. Las cargas parciales se denotan comúnmente con la letra griega “delta” ().
La electronegatividad es la medida de la capacidad de un átomo para atraer electrones. En la tabla periódica la electronegatividad aumenta de izquierda a derecha en un período. En un grupo aumenta de abajo a arriba. La mayor electronegatividad corresponde al Flúor (3.9) y la menor al Francio (0.7).
Una regla para determinar si un enlace covalente va a ser polar o no polar es como se explica a continuación:
Si las casillas de la tabla periódica en las que se encuentran los elementos que forman el enlace covalente tienen un lado común (boro y carbono, por ejemplo), la diferencia de electronegatividades es, en general, lo suficientemente pequeña como para que el enlace entre estos dos elementos sea considerado no polar. Si las casillas de los átomos no tienen ningún lado en común (carbono y oxígeno, por ejemplo), entonces cualquier enlace entre estos elementos es considerado como polar. Las cargas parciales en este último enlace pueden representase por:
C – O
Cuando se aplica esta aproximación, el hidrógeno (electronegatividad 2.1) debería situarse entre el boro y el carbono.
Método para el dibujo de las estructuras de Lewis.
1. Se cuenta el número de electrones que tiene cada átomo en su capa exterior y se obtiene el número total de electrones que tiene la molécula en su capa exterior.
El hidrógeno tiene 1 electrón en su capa exterior, el carbono 4, el nitrógeno y el fósforo 5, el oxígeno y el azufre 6, los halógenos 7.
2. Se determina el número de enlaces que tiende a formar cada átomo considerando la regla del octeto.
Los átomos se unen adquiriendo la estructura del gas noble más próximo en la tabla periódica, que para la mayoría de los elementos que intervienen en los compuestos orgánicos es el Neón, el cual posee 8 electrones en su capa más externa.

3. Se coloca el átomo que tiende a formar más enlaces en el centro.
4. Se coloca el hidrógeno, flúor, bromo, cloro en el exterior.
5. Una vez colocados los átomos, conectarlos mediante enlaces simples.
6. Se colocan los electrones en los átomos exteriores para satisfacer la regla del octeto para cada uno de estos átomos.
7. Si hay electrones disponibles, se añaden al átomo central para que se cumpla la regla del octeto.
8. Se crean los enlaces múltiples (dobles y triples) necesarios entre los átomos exteriores y centrales para satisfacer la regla del octeto moviendo los electrones no compartidos para formar pares de enlace. Pongamos el ejemplo del cianuro de hidrógeno (HCN):
ÁTOMO ELECTRONES EN CAPA EXTERNA
ENLACES A FORMAR
H 1 1
C 4 4
N 5 3
Se dibuja el esqueleto con los átomos conectados por enlaces simples. Puesto que el carbono es el que más enlaces requiere, se coloca en el centro.
H – C - N
Se añaden los electrones restantes a los átomos más exteriores hasta llegar a los 10 electrones totales.
H – C - N
El carbono no cumple la regla del octeto (pero el hidrógeno y el nitrógeno sí). Se mueven los electrones de valencia desde el nitrógeno para formar un enlace múltiple entre el carbono y el nitrógeno (triple).
H – C N
Carga formal.
Los átomos más comunes en los compuestos suelen formar un número determinado de enlaces (el carbono 4 enlaces, el nitrógeno 3, el oxígeno 2, el hidrógeno y los halógenos 1 enlace). Cuando dichos átomos forman un número de enlaces que no es el indicado, para que la contabilidad de electrones sea coherente, es necesario colocar en ellos una carga formal que se indica mediante un signo + o un signo – en las cercanías del átomo correspondiente.
Se calcula tomando el número del grupo del átomo en la tabla periódica, o lo que es lo mismo, número de electrones de valencia en un átomo libre neutro, y restando el número de electrones asociados con él mediante la fórmula:
Carga formal = número de grupo o nº de electrones de valencia – ½ (nº de electrones compartidos) – (nº de electrones no compartidos)
La suma aritmética de todas las cargas formales debe ser igual a la carga total en la molécula o ion. Por ejemplo:
H + Para el Hidrógeno: CARGA FORMAL =
H N H electrones libres (1) - electrones asignados (1) = 0
H Para el Nitrógeno: CARGA FORMAL =
electrones libres (5) – electrones asignados (4) = + 1
CARGA EN EL ION = (4) x (0) + 1 = +1
Página Página 11

Resonancia.Existen diversas moléculas cuyos electrones no parecen estar localizados en posiciones fijas (dadas por las estructuras de Lewis) sino dispuestos en diferentes posiciones. La teoría de la resonancia explica esto suponiendo que dichas moléculas son un compuesto intermedio entre una serie de estructuras moleculares llamadas formas resonantes. Cada una de ellas, por sí misma, no existe, existe el conjunto llamado híbrido de resonancia.Por ejemplo, los electrones en el ion acetato (CH3CO2
-) pueden considerarse como localizados en dos diferentes disposiciones:
H O H O
H – C – C H – C – C
H O H OEl híbrido de resonancia posee una menor energía (y por tanto es más estable) que cada una de las formas resonantes.Las distintas formas canónicas o resonantes deben tener el mismo número y la misma clase de núcleos de átomos y el mismo número de electrones. La diferencia entre unas formas canónicas y otras se encuentra exclusivamente en la posición de los electrones.Para que exista resonancia deben existir al menos dos formas resonantes que tengan energía similar si no la molécula perfectamente representada por la de menor energía que sería la predominante.Cuanto mayor sea el número de formas resonantes la molécula será más estable.Toda molécula que sólo posea enlaces simples no tiene formas resonantes. Las moléculas con enlaces múltiples pueden tener formas resonantes o no.La resonancia estabiliza las moléculas e iones, lo cual puede explicar diversas tendencias de las reacciones químicas. De dichas tendencias se induce lo siguiente:
1. Si un reactivo (material de partida) está estabilizado por resonancia, las reacciones químicas de esta molécula estarán menos favorecidas que en ausencia de resonancia.
2. Si un producto (material resultante) está estabilizado por resonancia, las reacciones de las que se obtiene estarán más favorecidas.
Acidos y bases.
Según la Teoría de Bronsted-Lowry:ÁCIDO es todo compuesto capaz de ceder un protón.
BASE es todo compuesto capaz de aceptar un protón.
Según la Teoría de Lewis: ÁCIDO es toda sustancia capaz de aceptar un par de electrones.
BASE es toda sustancia capaz de ceder un par de electrones.
Aunque la Teoría de Lewis parece diferente, es coherente con otras teorías. La teoría de Bronsted-Lowry se refiere al ion H+. La definición de Lewis se refiere a los pares de electrones (que conllevan una carga negativa). En la teoría de Bronsted-Lowry es el protón H (H+) el que se mueve. En la teoría de Lewis, los electrones forman enlaces, que “tiran” de los átomos para llevarlos a sus nuevos posiciones.
Ácido Base
Bronsted-Lowry Donante de H+ Receptor de H+
Lewis Receptor de e- Donante de e-
NOMENCLATURA.
Notación y convenciones.
Líneas ZIG-ZAG.
En numerosas ocasiones los compuestos orgánicos se representan mediante líneas zig-zag que siguen las siguientes reglas:
Página Página 22

Cada extremo y vértice de la línea representa un átomo de carbono. Los hidrógenos se omiten salvo que estén unidos a heteroátomos (átomos que no son ni
carbono ni hidrógeno). Los heteroátomos se representan todos. Por ejemplo, las dos representaciones siguientes son equivalentes:
Br CH3
CH – CH2 – CHBr - CH3
CH3
Convenciones.
Una flecha de este tipo significa el cambio de posición de un par de electrones.
Una flecha de este tipo significa el cambio de posición de un solo electrón.
Una flecha bicefálica de este tipo significa formas resonantes
Dos flechas significan equilibrio reversible al 50 por ciento.
Dos flechas significan equilibrio reversible desplazado hacia la derecha.
Dos flechas significan equilibrio reversible desplazado hacia la izquierda.
Alcanos.
Alcanos lineales.
Se nombran mediante un prefijo que indica el número de átomos de carbono de la cadena y el sufijo –ano.
Número de
carbonos
Prefijo Número de
carbonos
Prefijo Número de
carbonos
Prefijo
1 Met 6 Hex 11 Undec
2 Et 7 Hept 12 Dodec
3 Prop 8 Oct 13 Tridec
4 But 9 Non 14 Tetradec
5 Pent 10 dec 15 Pentadec
CH3 – CH2 – CH3 CH3 – CH2 – CH2 – CH2 – CH3
PROPANO PENTANO
Grupos alquilo.
Son el resultado que un alcano pierda un átomo de hidrógeno. Se nombran sustituyendo, en el nombre del alcano correspondiente, el sufijo –ano por –ilo.
CH3 – CH2 – CH2 - CH3 – CH2 – CH2 – CH2 – CH2 -
PROPILO PENTILO
Alcanos ramificados.Se localiza la cadena continua más larga de átomos de carbono. Esta cadena determina el nombre base del alcano.Si una molécula tiene dos o más cadenas de igual longitud se selecciona como cadena base o principal aquella que tiene un mayor número de sustituyentes.Se nombran todos los grupos unidos la cadena más larga como sustituyentes alquilo.Se numera la cadena principal comenzando por el extremo más próximo a uno de los sustituyentes. Si tenemos dos sustituyentes a igual distancia de los extremos se utiliza el orden alfabético para determinar la numeración. En una cadena lateral el carbono 1 es siempre el que está unido a la cadena principal.
Página Página 33

Para nombrar el compuesto se colocan los nombres de los sustituyentes por orden alfabético precedidos del número del carbono al que están unidos y de un guión, y a continuación se añade el nombre de la cadena principal.En el caso de cicloalcanos se antepone el prefijo ciclo- al nombre del alcano de igual número de átomos de carbono.En caso de cicloalcanos monosustituidos si el sustituyente tiene más átomos de carbono, entonces ese sustituyente es la cadena principal. Si el sustituyente tiene igual o menor número de átomos de carbono entonces la cadena principal es el cicloalcano y no es necesario numerar la posición de aquel. En caso de cicloalcanos multisustituidos se ordenan alfabéticamente los sustituyentes y se indica su posición relativa con un número asignándoles los localizadores más bajos posibles.
Me
Me
Me
1,1,2-trimetilciclopentano
4-etil-5-isopropil-3-metil-8-propilundecano
Alquenos.Se busca la cadena más larga que contenga el doble enlace y tomando como base ese número de carbonos se nombra utilizando el sufijo –eno.Se numera la cadena principal de forma que se asigne el número más bajo posible al doble enlace.La posición del doble enlace se indica mediante el localizador del primero de los átomos que intervienen en el doble enlace. Si hay más de un doble enlace se indica la posición de cada uno de ellos y se emplean los sufijos –dieno, -trieno, -tetraeno, etc.
CH2 = CH – CH = CH – CH3
1,3 - Pentadieno
3 - etil - 6 - metil - 2 - heptenoLos cicloalquenos se nombran de manera similar, al no existir ningún extremo en la cadena, el doble enlace se numera de forma que esté situado entre los carbonos 1 y 2.Los bencenos monosustituidos se nombran anteponiendo el nombre del sustituyente a la palabra benceno.Los bencenos disustituidos se nombran anteponiendo el prefijo orto, meta o para y los nombres de los sustituyentes a la palabra benceno.
S1
S1 S1
S2
S2 S2
Orto- Meta- Para-o- m- p-
Página Página 44

En los bencenos trisustituidos o más se numeran los carbonos de forma que tenga los localizadores más bajos posibles y se nombran teniendo en cuenta el orden alfabético.Alquinos.Se busca la cadena más larga que contenga el triple enlace y tomando como base ese número de carbonos se nombra utilizando el sufijo –ino.Se numera la cadena principal de forma que se asigne el número más bajo posible al triple enlace.La posición del triple se indica mediante el localizador del primero de los átomos que intervienen en el triple enlace. Si hay más de un triple enlace se indica la posición de cada uno de ellos y se emplean los sufijos –dieno, trieno, tetraeno, etc.Si en una molécula existen dobles y triples enlaces se les asigna los localizadores más bajos posibles. Al nombrarlos se indican primero los dobles enlaces y después los triples.Si un doble y triple enlace están en posiciones equivalentes se empieza a numerar por el extremo que da el localizador más bajo al doble enlace.
CH3 – C C – CH2 – CH – C CH CH2 = CH – C CH1- buten – 3 - ino
CH2 – CH2 – CH3
3 – propil –1,5- heptadiino
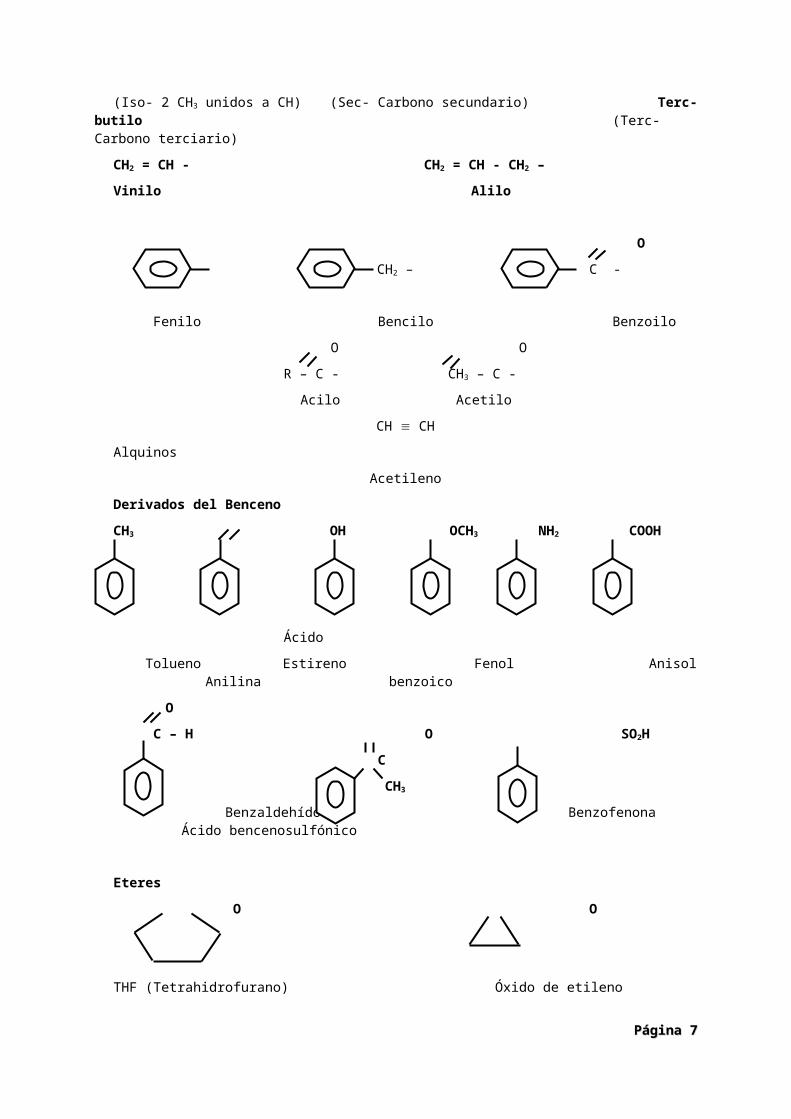
Nombres no normalizados.Radicales
CH3 CH3
CH – CH2 – CH3 – CH2 - CH - CH3 CH3 – C –CH3
Isobutilo sec-butilo CH3 (Iso- 2 CH3 unidos a CH) (Sec- Carbono secundario) Terc-butilo
(Terc- Carbono terciario)
CH2 = CH - CH2 = CH - CH2 –
Vinilo Alilo
O
CH2 – C -
Fenilo Bencilo Benzoilo
O O
R – C - CH3 – C -
Acilo Acetilo
CH CH
Alquinos
Acetileno
Derivados del Benceno
CH3 OH OCH3 NH2 COOH
Ácido
Tolueno Estireno Fenol Anisol Anilina benzoico
O
Página Página 55

C – H O SO2H
C
CH3
Benzaldehído Benzofenona Ácido bencenosulfónico
Eteres
O O
THF (Tetrahidrofurano) Óxido de etileno
Aldehidos y cetonas
O O O
Formaldehído Acetaldehído Acetona
Acidos carboxilicos
O O COOH COOH HOOC
OH OH COOH COOH COOHÁcido fórmico Ácido acético Ácido oxálico Ácido malónico Ácido succínico
Alcanos y cicloalcanos.Los alcanos son hidrocarburos en los cuales todos los enlaces carbono-carbono son enlaces simples. Su fórmula molecular es CnH2n+2.Los cicloalcanos son alcanos en los cuales los átomos de carbono están unidos formando un anillo.Propiedades físicas. Punto de ebullición:Los puntos de ebullición de los alcanos no ramificados aumentan al aumentar el número de átomos de carbono. Para los isómeros, el que tenga la cadena más ramificada, tendrá un punto de ebullición menor.
Solubilidad:
Los alcanos son casi totalmente insolubles en agua debido a su baja polaridad y a su incapacidad para formar enlaces con el hidrógeno. Los alcanos líquidos son miscibles entre sí y generalmente se disuelven en disolventes de baja polaridad. Los buenos disolventes para los alcanos son el benceno, tetracloruro de carbono, cloroformo y otros alcanos.
Síntesis.
El principal método para la obtención de alcanos es la hidrogenación de alquenos. H H
Catalizador C = C + H2 C C
El catalizador puede ser platino, paladio o níquel.
Página Página 66
Reacciones.
Las reacciones más importantes de los alcanos son:
Pirólisis: Se produce cuando se calientan alcanos a altas temperaturas en ausencia de oxígeno. Se rompen enlaces C-C y C-H, formando radicales, que se combinan entre sí formando otros alcanos de mayor número de carbonos.
Ruptura 1,2
CH3 + CH2 – (CH2)3 – CH3
Ruptura 2,3
CH3 – CH2 + CH2 – (CH2)2 – CH3
Ruptura 3,4
2 CH3 – CH2 – CH2
1 + 3 = CH3 – CH2 – CH3
2 + 4 =
Combustión:
2 CnH2n+2 + (3n+1) O2 2nCO2 + (2n+2) H2O + calor
Halogenación: Clluz
CH3 – CH2 – CH3 + Cl2 CH3 – CH2 – CH2 – Cl + CH3 – CH – CH3
25ºC
45% 55% Br
luzCH3 – CH2 – CH3 + Br2 CH3 – CH2 – CH2 – Br + CH3 – CH – CH3
35ºC
3% 97%
El bromo es muy selectivo y con las condiciones adecuadas, prácticamente, se obtiene un solo producto, que será aquel que resulte de la adición del bromo al carbono más sustituido.
El flúor es muy poco selectivo y puede reaccionar violentamente, incluso explosionar, por lo que apenas se utiliza para la halogenación de alcanos.
La halogenación de alcanos mediante el yodo no se lleva a cabo.
Alquenos.
Los alquenos son hidrocarburos cuyas moléculas contienen el doble enlace carbono – carbono.
Propiedades físicas.
Punto de ebullición:Los puntos de ebullición de los alquenos no ramificados aumentan al aumentar la longitud de la cadena. Para los isómeros, el que tenga la cadena más ramificada tendrá un punto de ebullición más bajo.Solubilidad:
Página Página 77
1 2
3 4
5

Los alquenos son casi totalmente insolubles en agua debido a su baja polaridad y a su incapacidad para formar enlaces con el hidrógeno.Estabilidad:Cuanto mayor es el número de grupos alquilo enlazados a los carbonos del doble enlace (más sustituido esté el doble enlace) mayor será la estabilidad del alqueno.
R R R R R H H R H H R H H H
C = C > C = C > C = C > C = C C = C > C = C > C = C
R R R H H R H R R R H H H H
Tetrasustituido Trisustituido Monosustituido No sustituido Disustituido
Síntesis.
Los métodos más utilizados para la síntesis de los alquenos son la deshidrogenación, deshalogenación, deshidratación y deshidrohalogenación, siendo estos dos últimos los más importantes. Todos ellos se basan en reacciones de eliminación que siguen el siguiente esquema general:
Y Z Eliminación
C – C C = C
(-YZ)
Y, Z pueden ser iguales, por ejemplo en la deshidrogenación y en la deshalogenación. Y, Z también pueden ser diferentes como en la deshidratación y en la deshidrohalogenación.
Deshidrogenación.
R1 R2 catalizador R1 R2
CH – CH calor C = C H H (-H2) H H
Deshalogenación.
R1 R2 acetona R1 R2
CH – CH Zn C = C(X=Halógeno)
X X (-ZnX2) H H
Deshidratación.
R1 R2 Ácido R1 R2
CH – CH C = C
H OH (-H2O) H H
Deshidrohalogenación.
R1 R2 Base R1 R2
CH – CH C = C (X=Halógeno)
H X (-HX) H H
Mediante la deshidratación y la deshidrohalogenación se puede obtener más de un producto. Para determinar cual será el mayoritario se utiliza la REGLA DE ZAITSEV: “Cuando se produce una deshidratación o una deshidrohalogenación el doble enlace se produce preferentemente hacia el carbono más sustituido”.
No todas las reacciones de deshidrohalogenación siguen la regla de Zaitsev. Cuando se utiliza una base muy voluminosa (Terc-butóxido de sodio, LDA) siguen la regla de Hoffman, el doble enlace se produce preferentemente hacia el carbono menos sustituido.
Página Página 88

Reacciones.
Las reacciones más comunes de los alquenos son las reacciones de adición. En la adición puede intervenir un agente simétrico.
X X
C = C + X - X C - C
O un agente asimétrico:
X Y
C = C + X - Y C - C
Cuando el agente asimétrico es un haluro de hidrógeno la adición cumple la REGLA DE MARKOVNIKOV: ”El halógeno se adiciona al átomo de carbono menos hidrogenado”, o lo que es lo mismo “el hidrógeno se adiciona al carbono más hidrogenado”.La excepción ocurre en la adición del bromuro de hidrógeno en presencia de peróxidos, en este caso la reacción se produce de forma ANTIMARKOVNIKOV.En la siguiente tabla se exponen las distintas reacciones de los alquenos:
TIPO REACCIÓN SIN / ANTI
Hidrogenación catalítica
Me H H2 H H
C = C Catalizador Me – C – C – H
Me H (Pt, Pd o Ni) Me H
SIN
Halogenación
(X = Cl, Br)
Me H X2 X H
C = C Me – C – C – H
Me H Me X
ANTI
Hidrohalogenación
(X= Cl, Br, I)
(Markovnikov)
Me H HX X H
C = C Me – C – C – H
Me H Me H
Adición de HBr con peróxidos
(Antimarkovnikov)
Me H HBr H H
C = C Peróxidos Me – C – C – H
Me H Me Br
Hidratación
(Markovnikov)
Me H H2O OH H
C = C H2SO4 Me – C – C – H
Me H Me H
Hidroboración – Oxidación
(Antimarkovnikov)
Me H 1) BH3 OH H
C = C 2) H2O2, OH- Me – C – C – H
Me H Me H
Ozonólisis
Me H 1) O3 Me H
C = C 2) Zn, H3O+ C = O O = C
Me H Me H
Tratamiento con KMnO4
Página Página 99

KMnO4 en caliente (1)
Concentrado y en caliente O
R – CH = CH2 H+ R – C – OH + CO2 + H2O
Tratamiento con KMnO4 en caliente
(2)
R1 KMnO4
C = CH2 Concentrado y en caliente O
R2 H+ R1 – C – R2 + CO2 + H2O
Tratamiento con KMnO4 en caliente
(3)
KMnO4
Concentrado y en caliente O O
R1 – CH = CH – R2 H+ R1 – C – OH + R2 – C – OH
Tratamiento con KMnO4 en caliente
(4)
R1 KMnO4 Concentrado
C = CH – R3 y en caliente O O
R2 H+ R1 – C – R2 + R1 – C – OH
Tratamiento con KMnO4 en caliente
(5)
R1 R3 KMnO4 Concentrado
C = C y en caliente O O
R2 R4 H+ R1 – C – R2 + R3 – C – R4
Epoxidación
R1 R3 O R1 O R3
C = C R – C – O – O – H C – C
R2 R4 Perácido R2 R4
Alquenos conjugados.
Los alquenos conjugados son aquellos en los que se alternan enlaces dobles y enlaces simples. Estos alquenos dan lugar a sistemas insaturados conjugados formados por los radicales libres, carbocationes y carbaniones.
Sistema alílico.
El sistema alílico es el sistema insaturado conjugado más sencillo. Está formado por:
Radical libre alilo.
CH2 = CH – CH2 CH2 – CH = CH2
Carbocatión alilo.
CH2 = CH – CH2+ +CH2 – CH = CH2
Carbanión alilo.
CH2 = CH – -CH2 -CH2 – CH = CH2
Dienos conjugados.
Dienos conjugados son aquellos compuestos que presentan dos enlaces dobles separados por un enlace simple.
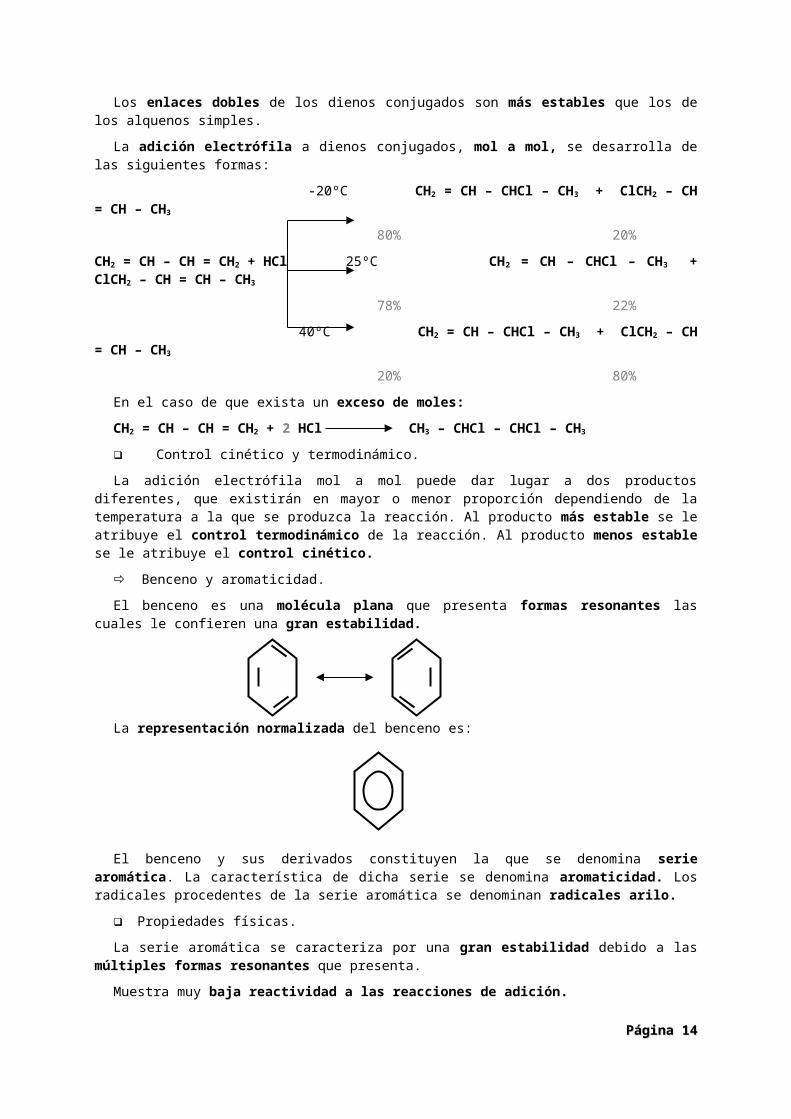
Los enlaces dobles de los dienos conjugados son más estables que los de los alquenos simples.
La adición electrófila a dienos conjugados, mol a mol, se desarrolla de las siguientes formas:
-20ºC CH2 = CH – CHCl – CH3 + ClCH2 – CH = CH – CH3
80% 20%
CH2 = CH – CH = CH2 + HCl 25ºC CH2 = CH – CHCl – CH3 + ClCH2 – CH = CH – CH3
78% 22%
40ºC CH2 = CH – CHCl – CH3 + ClCH2 – CH = CH – CH3
Página Página 1010

20% 80%
En el caso de que exista un exceso de moles:
CH2 = CH – CH = CH2 + 2 HCl CH3 – CHCl – CHCl – CH3
Control cinético y termodinámico.
La adición electrófila mol a mol puede dar lugar a dos productos diferentes, que existirán en mayor o menor proporción dependiendo de la temperatura a la que se produzca la reacción. Al producto más estable se le atribuye el control termodinámico de la reacción. Al producto menos estable se le atribuye el control cinético.
Benceno y aromaticidad.
El benceno es una molécula plana que presenta formas resonantes las cuales le confieren una gran estabilidad.
La representación normalizada del benceno es:
El benceno y sus derivados constituyen la que se denomina serie aromática. La característica de dicha serie se denomina aromaticidad. Los radicales procedentes de la serie aromática se denominan radicales arilo.
Propiedades físicas.
La serie aromática se caracteriza por una gran estabilidad debido a las múltiples formas resonantes que presenta.
Muestra muy baja reactividad a las reacciones de adición.
El benceno es una molécula plana con un alto grado de saturación lo cual favorece las reacciones de sustitución. Es un líquido menos denso que el agua y poco soluble en ella. Es muy soluble en otros hidrocarburos. Es un compuesto muy tóxico.
Reacciones.
Las reacciones más comunes del benceno son las reacciones de sustitución. Los sustituyentes producen diversos efectos sobre la reactividad y orientación de una nueva sustitución. Considerando estos efectos los sustituyente se clasifican en:
Reactividad:
ACTIVADORES: Aumentan la reactividad.
INHIBIDORES: Disminuyen la reactividad.
Orientación:
ORTOPARADIRIGENTES: El nuevo sustituyente se sitúa en posición ORTO o PARA respecto a él (pero no meta).
METADIRIGENTES: El nuevo sustituyente se sitúa en posición META respecto a él.
En general, los ACTIVADORES SON ORTOPARADIRIGENTES y los INHIBIDORES SON METADIRIGENTES.
SUSTITUCION ELECTRÓFILA AROMÁTICA
Página Página 1111

Las reacciones más características del benceno son las de sustitución electrófila. También son de destacar las reacciones de la cadena lateral de los compuestos aromáticos.
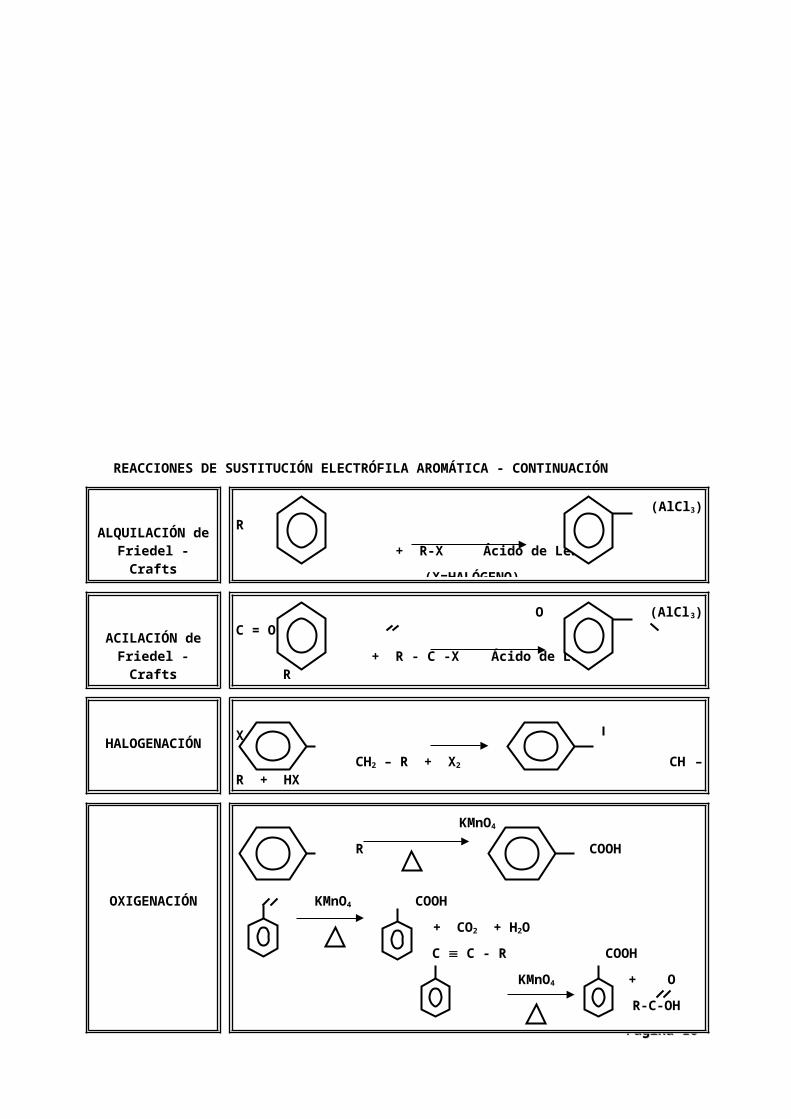
REACCIONES DE SUSTITUCIÓN ELECTRÓFILA AROMÁTICA
REACCIONES DE SUSTITUCIÓN ELECTRÓFILA AROMÁTICA - CONTINUACIÓN
Página Página 1212
ORIENTADORES ORTO-PARA
ORIENTADORES META
Activadores potentes
Activadores moderados
Activadores débiles
Inhibidores débiles
Inhibidores potentes
Inhibidores moderados
- N ; - OH
- O – R ; - NHCOR
-R (Radical alquilo); (Radical arilo)
-F ; -Cl ; -Br ; -I
-NO2 ; -CF3 ; -N+ -
-C N; -COOH; -COOR;-SO3H; -COH; -COR;
BROMACIÓN FeBr3 Br + Br2
CLORACIÓN AlCl3 Cl + Cl2
NITRACIÓN H2SO4 NO2
+ HNO3
SULFONACIÓN H2SO4 SO3H + SO3 (REVERSIBLE)
ALQUILACIÓN de Friedel - Crafts
(AlCl3) R + R-X Ácido de Lewis
(X=HALÓGENO)
ACILACIÓN de Friedel - Crafts
O (AlCl3) C = O + R - C -X Ácido de Lewis R
(X=HALÓGENO)HALOGENACIÓN
X CH2 – R + X2 CH – R + HX
OXIGENACIÓN
KMnO4
R COOH
KMnO4 COOH + CO2 + H2O
C C - R COOHKMnO4 + O
R-C-OH

Alquinos.
Los alquinos son hidrocarburos cuyas moléculas contienen el triple enlace carbono-carbono.
Propiedades físicas.
Como podría esperarse, las propiedades físicas de los alquinos son muy similares a las de los alquenos y los alcanos. Los alquinos son ligeramente solubles en agua aunque son algo más solubles que los alquenos y los alcanos. A semejanza de los alquenos y alcanos, los alquinos son solubles en disolventes de baja polaridad, como tetracloruro de carbono, éter y alcanos. Los alquinos, al igual que los alquenos y los alcanos son menos densos que el agua.
Los tres primeros alquinos son gases a temperatura ambiente.
Síntesis.
Existen tres procedimientos para la obtención de alquinos:
Deshidrohalogenación de halogenuros de alquilo vecinales (vec-dihalogenuros).
Br Br Base
R1 – C – C – R2 R1 – C C – R2
H H
Deshidrohalogenación de halogenuros de alquilo geminales (gem-dihalogenuros).
H Br Base
R1 – C – C – R2 R1 – C C – R2
H Br
Alquilación de alquinos.
Se produce debido a la acidez del hidrógeno en los alquinos terminales. Mediante esta reacción se sintetizan alquinos internos a partir de alquinos terminales. Tiene lugar en dos etapas:
1) R – C CH + NaNH2 R – C CNa + NH3
2) R – C CNa + R1 – X R – C C – R1 + NaX
Página Página 1313
Para que se produzca esta última reacción es necesario utilizar haloalcanos primarios.
Reacciones.
Muchas de las reacciones de los alquinos son reacciones de adición que siguen el siguiente esquema:
Y – Z Y – Z Y Z Z Y
– C C - C = C - C – C - ó - C – C -
Y Z Y Z Y Z
Otras de las reacciones de los alquinos se deben a la acidez del hidrógeno de los alquinos terminales.
TIPO REACCION
Página Página 1414
Hidrogenación
H H H2 (Pt ó Pd) R1 R2 H2 (Pt ó Pd)
R1 – C C – R2 C = C R1 – C – C – R2
H H H H
H2 R1 R2
R1 – C C – R2 C = C (CIS) Catalizador de Lindlar H H
Li ó Na, NH3 R1 HR1 – C C – R2 C = C (TRANS)
H R2
Hidrohalogenación
(X= Cl, Br, I)(Markovnikov)
X H HX R H HXR – C C – H C = C R – C – C - H
X H
X H
Hidrobromación
con peróxidos(Antimarkovnikov
)
H Br HBr R H HBrR – C C – H C = C R – C – C - H
H Br
H Br
Hidratación(Markovnikov)
OH H O H H2SO4, H2O R – C C – H R - C = C - H R – C – C - H
HgSO4 H
Hidroboración -
oxidación (Antimarkovni
kov)
H O 1) BH3 R – C C – H R – C – C - H
2) H2O2, OH-
H Halogenación
(X= Cl, Br)
X X X2 R1
X X2R1 – C C – R2 C = C R1 – C – C – R2
X R2 (Trans) X X
Ozonólisis
1) O3 O OR1 – C C – R2 R1 – C – OH + R2 – C - OH
2) H3O+
1) O3 O R – C C – H R – C – OH + CO2 + H2
2) H3O+
Tratamiento con KMnO4 en caliente
KMnO4 O OR1 – C C – R2 R1 – C – OH + R2 – C - OH
KMnO4 O R – C C – H R – C – OH + CO2 + H2
Acidez de alquinos
terminales
H NaNH2 RCH2X CH3 – C C – H CH3 – C C:- CH3 – C C – C - R NH3
H

Grupos funcionales.
Orden de prioridad.
Cuando en un compuesto existen varios grupos funcionales es necesario establecer un orden de prioridad con el fin de numerar la cadena, de manera que el grupo funcional de mayor prioridad se le asigne el localizador más bajo posible y sirva de base para nombrar el compuesto. Este orden de prioridad es el siguiente:
1. Sales.2. Ácidos carboxílicos.3. Derivados de ácidos carboxílicos.4. Nitrilos.5. Aldehídos.6. Cetonas.7. Alcoholes.8. Aminas.9. Haloalcanos.10. Nitroderivados.11. Éteres.12. Peróxidos.
Los grupos funcionales tienen prioridad respecto a los enlaces múltiples.
Haluros orgánicos.
Los haluros orgánicos presentan enlaces C – X donde X es un halógeno (F, Cl, Br, I). Como el halógeno es más electronegativo que el carbono, éste adquirirá una carga parcial positiva mientras que el halógeno adquirirá una carga parcial negativa:
C - X
Nomenclatura.
Los haluros orgánicos se nombran considerándolos como sustituyentes anteponiendo al nombre del hidrocarburo correspondiente los siguientes prefijos: Fluoro, Cloro, Bromo, Yodo.
CH3 – CHCl – CHCl - CH3 Br Cl
2,3 - Diclorobutano
1-bromo-3-clorociclopentano
Propiedades físicas.Los haluros orgánicos son moléculas polares con fuerzas intermoleculares mayores que los hidrocarburos de los que proceden y, por tanto, con puntos de ebullición más altos que éstos. Para un mismo sustituyente alquílico, el punto de ebullición aumenta al aumentar el tamaño del halógeno.
Página Página 1515
Al igual que en los alcanos, los puntos de ebullición disminuyen con el aumento de las ramificaciones. Son sustancias solubles en disolventes no polares e insolubles en agua.
Síntesis.
Halogenación de alcanos. Sólo en casos muy concretos se utiliza la halogenación de alcanos en la síntesis de halogenuros de alquilo, debido a que en la mayoría de las veces se obtienen mezclas de productos.
R – H + X2 h R – X + HX
Halogenación de alquenos. X
R1CH = CHR2 X2 R1CH – CHR2
X
Hidrohalogenación de alquenos. X H
R1CH = CHR2 HX R1CH – CHR2
Halogenación de alquinos. X X
R1CH CHR2 2X2 R1C – CR2
X X
Hidrohalogenación de alquinos. X H
R1CH CHR2 2HX R1C – CR2
X H
Halogenación aromática.
FeBr3 o AlCl3 X+ X2 (X= Cl o Br)
A partir de alcoholes:
R – OH R – X (Mecanismo carbocatiónico) HX
R – OH R – X PBr3 o PI3
R – OH R - X PCl5
R – OH R – X (En presencia de trietilamina (CH3CH2)3N) SOCl2
Reacciones.1. Deshalogenación.2. Deshidrohalogenación.
COMPUESTOS ORGANOMETÁLICOS.
Los compuestos organometálicos presentan enlaces C – M donde M es un metal. En este documento se van a tratar aquellos compuestos en los que el metal es litio (organolíticos) o magnesio (organomagnesianos).
MgCl
Li Li CH3 – CH2 – CH – CH3
Butil-litio Isopropil-litio Cloruro de sec-butilmagnesio
Página Página 1616

Síntesis.
Siendo X es un halógeno.
R – X + 2Li RLi+ XLi
R – X + Mg RMgX
La reacción se produce cuando el halógeno es yodo puesto que es el mejor grupo saliente.
Reacciones.
En los compuestos organolíticos y organomagnesianos el carbono que está enlazado al metal se puede considerar a todos los efectos como un carbanión, el cual puede tener dos comporta-mientos: base o nucleófilo.
Comportamiento como bases:
R Li + R’ – OH R – H + R’ – O - Li
R – MgX R – H + HO - MgX H2O
Los compuestos organometálicos reaccionan como bases con aquellos sustratos que tengan un pKa menor que el de ellos.
Comportamiento como nucleófilos:
O OLi H3O+ OH
R - Li + R R
O OLi H3O+ OH
R - Li + H H R R
O OLi H3O+ OH
R1 - Li + R2 H R1 R1
R2 R2
O OLi H3O+ OH
R1 - Li + R2 R3 R1 R1
R2 R3 R2 R3
O O- -OR3 O
R1 - Li + R2 - C - R3 R1 - C – OR3 R2 – C – R1
R1 R1Li H3O+
OH
R2
R1 R1
ALCOHOLES Y FENOLES.
Los alcoholes presentan el grupo funcional –OH unido a un átomo de carbono saturado. Si el átomo de carbono saturado es un grupo alquilo, alquenilo o alquinilo, hablamos de alcoholes en general pero si se une a un anillo bencénico hablamos de fenoles.
Página Página 1717

Nomenclatura.
Para nombrar los alcoholes tenemos dos alternativas:
1ª. Añadir el sufijo –OL al nombre del hidrocarburo de referencia (por ejemplo: Propanol).
2ª. Citar primero la función (alcohol) y luego el radical (por ejemplo: alcohol propílico).
En compuestos ramificados el nombre del alcohol deriva de la cadena más larga que contenga el grupo –OH.
Al numerar la cadena se asigna al C unido al –OH el localizador más bajo posible.
Cuando el grupo –OH interviene como sustituyente se utiliza el prefijo –hidroxi.
En alcoholes cíclicos el carbono unido al –OH ocupa siempre la posición 1.
CH3 – CH2 – CH2OH Cl OH
1-propanol OH
(Alcohol propílico) alcohol sec-butílico 3-clorociclopentanol
(2-butanol)
Para los fenoles, indicamos los sustituyentes en sus posiciones respecto al anillo bencénico y terminamos indicando la palabra fenol. Al hidroxibenceno se le conoce como fenol.
OH OH OH
Cl
Br
m-bromofenol o-clorofenol NO2 p-nitrofenol
Propiedades físicas.Los alcoholes pueden formar enlaces mediante puentes de hidrógeno, lo que causa que estos compuestos tengan puntos de ebullición más altos que los correspondientes haloalcanos. La solubilidad de los alcoholes en agua disminuye gradualmente a medida que se agranda la porción de la molécula correspondiente al hidrocarburo; los alcoholes de cadena larga son semejantes a los correspondientes alcanos y, por lo tanto, son menos semejantes al agua.Los fenoles son también capaces de formar enlaces de hidrógeno intermoleculares fuertes, lo que ocasiona que los fenoles se asocien y, por tanto, que posean temperaturas de ebullición más altas que los hidrocarburos del mismo peso molecular. Son algo solubles en agua.
Síntesis.
Hidratación de alquenos.La adición de agua al enlace doble de un alqueno, catalizada por ácidos es un método para la preparación de alcoholes de bajo peso molecular que tiene su mayor utilidad en los procesos industriales a gran escala. Los catalizadores típicos son el ácido sulfúrico (H2SO4) y el fosfórico (H3PO4). La reacción es reversible y el mecanismo para la hidratación de un alqueno es, simplemente, el inverso del de la deshidratación de un alcohol.
C = C - C – C+ - - C – C - - C – C -
H H +OH2 H OH
Hidroboración-oxidación de alquenos.
THF:BH3 H2O2/OH-
3CH3CH = CH2 (CH3CH2CH2)3B 3 CH3CH2CH2OH
Propeno (Hidroboración) Tripropilborano Alcohol propílico
Página Página 1818
+H+
-H+
-H+
+H+
+H2O
-H2O

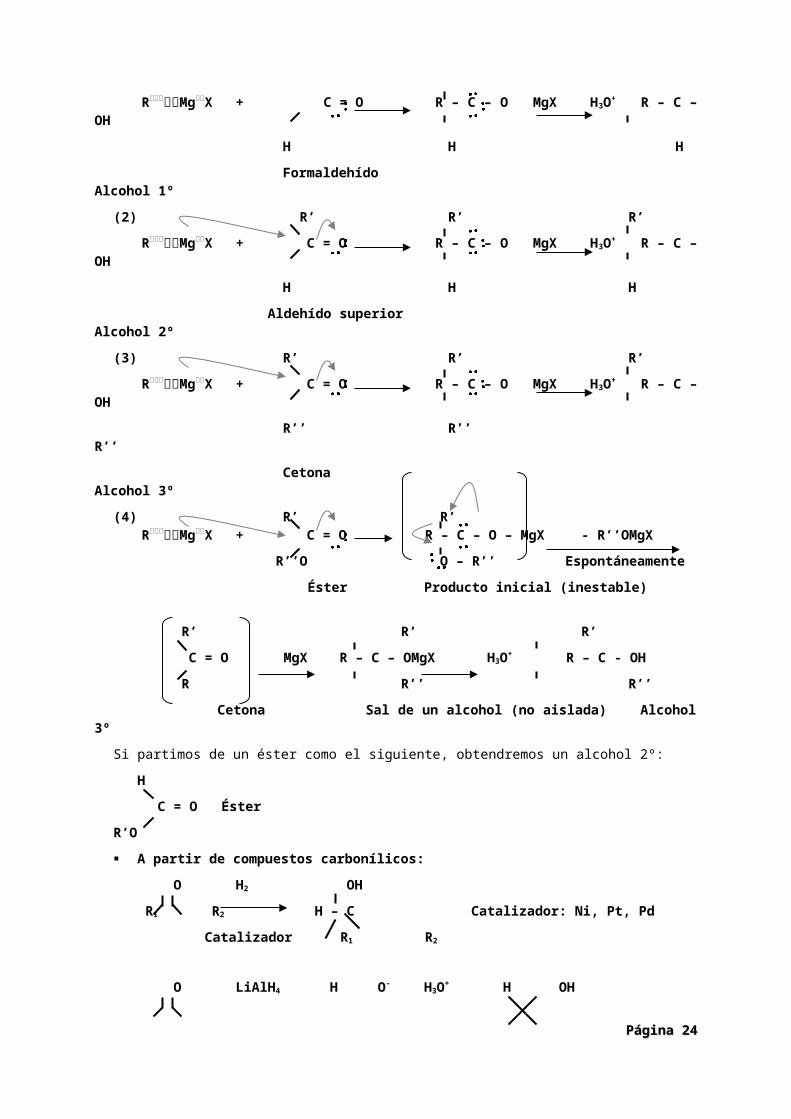
A partir de compuestos organometálicos. La adición de reactivos organometálicos a aldehídos y cetonas va a proporcionar un método más para la obtención de alcoholes. El tipo de alcohol sintetizado dependerá de los reactivos empleados.
1. La reacción con metanal (formaldehído) da lugar a alcoholes primarios.2. El resto de aldehídos da lugar a alcoholes secundarios.3. La reacción con cetonas da lugar a alcoholes terciarios.4. La reacción de ésteres carboxílicos con dos equivalentes del compuesto organometálicos da
alcoholes secundarios y terciarios.
(1) H H H
RMgX + C = O R – C – O MgX H3O+ R – C – OH
H H H
Formaldehído Alcohol 1º
(2) R’ R’ R’
RMgX + C = O R – C – O MgX H3O+ R – C – OH
H H H
Aldehído superior Alcohol 2º
(3) R’ R’ R’
RMgX + C = O R – C – O MgX H3O+ R – C – OH
R’’ R’’ R’’
Cetona Alcohol 3º
(4) R’ R’RMgX + C = O R – C – O – MgX - R’’OMgX
R’’O O – R’’ Espontáneamente
Éster Producto inicial (inestable)
R’ R’ R’
C = O MgX R – C – OMgX H3O+ R – C - OH
R R’’ R’’
Cetona Sal de un alcohol (no aislada) Alcohol 3º
Si partimos de un éster como el siguiente, obtendremos un alcohol 2º:
H
C = O Éster
R’O
A partir de compuestos carbonílicos:
O H2 OH
R1 R2 H – C Catalizador: Ni, Pt, Pd
Catalizador R1 R2
O LiAlH4 H O- H3O+ H OH
R1 R2
ó NaBH4 R1 R2 R1 R2
Página Página 1919

El borohidruro sódico (NaBH4) no presenta problemas en cuanto a su manipulación y puede disolverse en agua o en alcohol. El hidruro de litio y aluminio (LiAlH4) es más reactivo y, también, más peligroso, ya que reacciona violentamente con el agua, debiéndose utilizar disuelto en éter o tetrahidrofurano (THF) anhidros.La reducción de un aldehído da lugar a un alcohol primario. La reducción de una cetona da lugar a un alcohol secundario. Los ésteres y ácidos carboxílicos pueden reducirse para formar alcoholes primarios, aunque con mayor dificultad que los aldehídos y las cetonas. Por ello, las reducciones se realizan con LiAlH4, y no con NaBH4 pues éste reduce a los ésteres lentamente y no reduce a los ácidos.
O OH
CH3CH2CH2C – H CH3CH2CH2C – H
Butanal 1-butanol(85%) H
O OH
CH3CH2CH2COCH3 CH3CH2CH2C – H + CH3OH
Butanoato de metilo H
Reacciones.
Las reacciones de los alcoholes son, esencialmente, de tres tipos:
Ruptura del enlace C – O.
Deshidratación de alquenos.
Síntesis de haluros orgánicos a partir de alcoholes.
Ruptura del enlace O – H.
Síntesis de éteres.
R – OH + R – OH R – O – R
Esta reacción no conduce necesariamente al éter como producto mayoritario, puesto que existe competencia con la eliminación.
Tratamiento con metales alcalinos.
R – OH + Na R – O Na
Síntesis de éteres de Williamson.
R - O Na + R’ – L R – O – R’
Siendo L un buen grupo saliente (I, Br, Cl)
Esta reacción da excelentes resultados cuando R’ es un metilo o radical primario pues no hay competencia con la eliminación.
Síntesis de ésteres.
O O
R – OH + R’ – C – OH R’ – C – OR
O O
R – OH + R’ – C – Cl R’ – C – OR
Oxidación.
Alcohol primario.
O
Página Página 2020
NaBH4
EtOH
1) LiAlH4, éter
2) H+, H2O
-H2O
H3O+
BASE
CrO3 / Py
Piridina:N

R – CH2 – OH R – C - H
Se utiliza la piridina (Py) para detener la reacción en el aldehído. A este reactivo se le conoce como reactivo de Sarrett.
O
R – CH2 – OH R – C - H
A este reactivo se le conoce como reactivo de Jones.También se utiliza como reactivo el permanganato potásico, (KMnO4).
Alcohol secundario.
OH O
R – CH – R’ R – C – R’
Alcohol terciario.
No se da la reacción ya que los alcoholes terciarios no tienen hidrógenos en posición alfa ().
FENOLES.
Síntesis.
+
Fenol
Reacciones.
Las reacciones de los fenoles son de sustitución electrófila aromática teniendo en cuenta que ya existe el sustituyente –OH.
Éteres.
Los éteres presentan el grupo funcional – O -.
Nomenclatura.
Para nombrar los éteres tenemos dos alternativas:
(1) Considerar el grupo alcoxi R – O – como un sustituyente (siendo R el radical más sencillo).
(2) Citar los dos radicales que están unidos al O por orden alfabético y, a continuación, la palabra éter.
CH3 – O – CH2 – CH3 CH3 – CH2 – O – CH2 – CH3 CH3 – CH2 – O – CH = CH2
Etil metil éter Dietil éter Etil vinil éter
(Metoxietano) (Etoxietano) (Etoxietileno)
Un caso particular de éteres son los epóxidos, (también llamados oxiranos) cuyo esquema es:
O
C - C
Para nombrarlos se utiliza el prefijo epoxi- seguido del nombre del hidrocarburo correspondiente, indicando los carbonos a los que está unido el oxígeno, con dos localizadores lo más bajos posibles, en caso de que sea necesario.
O O
H2C – CH – CH3 CH3 – CH – CH – CH2 –CH3
Epoxipropano 2,3 – epoxipentano
El epóxido más simple tiene el nombre de óxido de etileno.
Página Página 2121
CrO3 / H+
acetona
KMnO4
Alquilación de Friedel-Crafts
O2 O – O - H
H3O+
OH O

O
H2C – CH2
Propiedades.
Los éteres no pueden formar enlaces mediante puentes de hidrógeno, lo que hace que estos compuestos tengan puntos de ebullición más bajos que los alcoholes de mismo peso molecular.
Los éteres de cadena corta son solubles en agua. A medida que aumenta la longitud de la cadena disminuye la solubilidad.
Los éteres son muy poco reactivos, a excepción de los epóxidos, por lo que se utilizan como disolventes. Ejemplo de estos disolventes es:
Tetrahidrofurano o THF
O (Oxaciclopentano)
Síntesis.
A partir de alcoholes primarios, por deshidratación intermolecular.
R – OH + HO – R R – O - R
Epoxidación de alquenos.
O O O
RCH = CHR + R’C – O – OH RHC – CHR + R’C – OH
Alqueno Peroxiácido Epóxido Ácido
Reacciones.
Los éteres no son muy reactivos a excepción de los epóxidos. Al calentar los dialquil éteres con ácidos muy fuertes (HI, HBr y H2SO4), se hace que éstos intervengan en reacciones en las que se rompe el enlace carbono – oxígeno. Por ejemplo, el dietil éter reacciona con ácido bromhídrico concentrado caliente para formar dos equivalentes molares de bromuro de etilo.
Las reacciones de los epóxidos pasan por la apertura del ciclo. Dicha apertura puede ser catalizada por ácido o apertura mediante un nucleófilo.
Apertura catalizada por ácido.
O HO OH
R R R
Nu
El nucleófilo ataca al carbono más sustituido.
Apertura por nucleófilo.
O O-
R R OH
Nu R Nu
El nucleófilo ataca al carbono menos sustituido.
Página Página 2222
H+( -H2O)
H3O+ Nu -
H3O+ Nu -

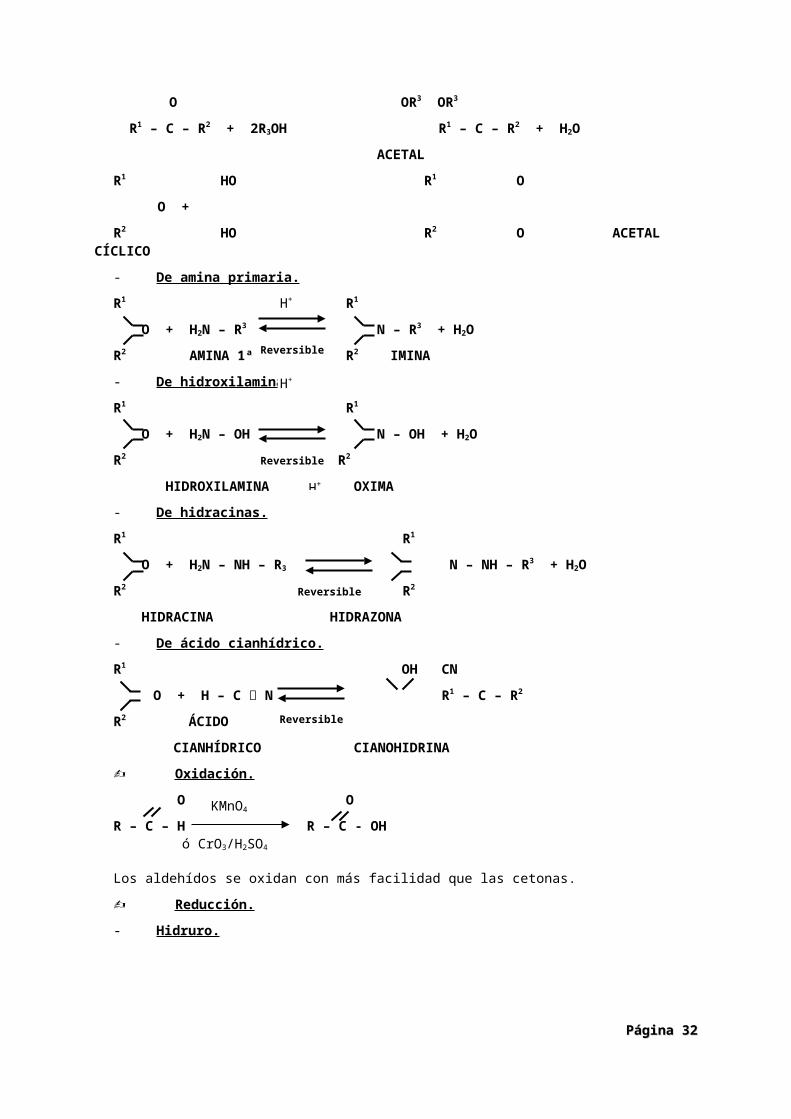
Aldehídos y cetonas.
Tanto aldehídos como cetonas se caracterizan por tener el grupo carbonilo C = O
O
La fórmula general de los aldehídos es R – C - H
O
La fórmula general de las cetonas es R – C – R’
Nomenclatura.
Aldehídos.
El sistema de nomenclatura corriente consiste en emplear el nombre del alcano correspondiente terminado en –al. Cuando el grupo CHO es sustituyente se utiliza el prefijo formil-. También se utiliza el prefijo formal- cuando hay tres o más funciones aldehídos sobre el mismo compuesto. En esos casos se puede utilizar otro sistema de nomenclatura que consiste en dar el nombre de carbaldehído a los grupos CHO (los carbonos de esos CHO no se numeran, sino que se consideran que no forman parte de la cadena). Este último sistema es el idóneo para compuestos con grupos CHO unidos directamente a ciclos.
O
CH3 – CH2 - CH2 - CH2 – C – H CHO CHO – CH - CH2 - CH2 – CHO
Pentanal CH3 CHO
2-formil-4-metilpentanodial
CHO
1,3-ciclohexanodicarbaldehido
Cetonas.
Para nombrar las cetonas tenemos dos alternativas:
1ª) El nombre del hidrocarburo del que procede terminado en –ona. Como sustituyente debe emplearse el prefijo oxo-.
2ª) Citar los dos radicales que están unidos al grupo carbonilo por orden alfabético y a continuación la palabra cetona.
O O
CH3 – C - CH2 - CH2 – CH3
2-Pentanona
(Metil propil cetona) 2,5-ciclohexanodienona
Algunas cetonas poseen nombres comunes que se conservan en el sistema IUPAC:
O O O
CH3 – C – CH3 C – CH3 C
Acetona Acetofenona Benzofenona
Propiedades físicas.
Los compuestos carbonílicos presentan puntos de ebullición más bajos que los alcoholes de su mismo peso molecular debido a que no pueden forman enlaces de puente de hidrógeno como lo hacen los alcoholes. Pero sus puntos de ebullición son mayores que los de los correspondientes alcanos. No hay grandes diferencias entre los puntos de ebullición de aldehídos y cetonas de igual peso molecular.
Los compuestos carbonílicos de cadena corta son solubles en agua ya que el átomo de oxígeno del carbonilo permite que las moléculas de aldehídos y cetonas forman enlaces de hidrógeno fuertes
Página Página 2323
con las moléculas de agua. A medida que aumenta la longitud de la cadena disminuye la solubilidad. La acetona y el acetaldehído se disuelven en agua en cualquier proporción.
Algunos aldehídos aromáticos que se obtienen de fuentes naturales poseen fragancias muy agradables. Ejemplo de ello son:
CHO CHO CHO
OH
OCH3
OH
Síntesis.
Ozonólisis de alquenos.
R R’’ 1) O3 R R’’
C = C 2) Zn, H3O+ C = O + O = C
R’ H R’ H
Cetona + Aldehído
Tratamiento con KMnO 4 en caliente de alquenos.
Este método sólo es válido para la preparación de cetonas.R1 KMnO4
C = CH2 Concentrado y en caliente O
R2 H+ R1 – C – R2 + CO2 + H2O
Hidratación de alquinos. OH H O H
H2SO4, H2O R – C C – H R - C = C - H R – C – C - H
HgSO4 HEn este caso da una cetona pero si en vez de R tuviésemos H, obtendríamos un aldehído.
Hidroboración-oxidación de alquinos. H O
1) BH3
R – C C – H R – C – C - H 2) H2O2, OH-
H Acilación de Friedel-Crafts del Benceno.
O (AlCl3) C = O + R - C - X R Ácido de Lewis
(X=Halógeno) Oxidación de alcoholes:
- Primarios.
O
R – CH2 – OH R – C - H
Página Página 2424
Benzaldehído(de almendras amargas)
Salicilaldehído (de la ulmaria)
Vainillina(de las vainas de la vainilla)
CrO3 / Py

- Secundarios.
OH O
R – CH – R’ R – C – R’
Con alcoholes terciarios no se da la reacción.
Reacciones.
Las reacciones de los alcoholes son esencialmente de tres tipos:
Adición nucleófila.
Debido a la resonancia del grupo carbonilo:
La reacción más importante de aldehídos y cetonas es la reacción de adición nucleofílica cuyo mecanismo es el siguiente:
- De alcoholes.
O OR3 OR3
R1 – C – R2 + 2R3OH R1 – C – R2 + H2O
ACETAL
R1 HO R1 O
O +
R2 HO R2 O ACETAL CÍCLICO
- De amina primaria.
R1 R1
O + H2N – R3 N – R3 + H2O
R2 AMINA 1ª R2 IMINA
- De hidroxilamina.
R1 R1
O + H2N – OH N – OH + H2O
R2 R2
HIDROXILAMINA OXIMA
- De hidracinas.
R1 R1
O + H2N – NH – R3 N – NH – R3 + H2O
R2 R2
HIDRACINA HIDRAZONA
- De ácido cianhídrico.
R1 OH CN
O + H – C N R1 – C – R2
R2 ÁCIDO
Página Página 2525
KMnO4
C = O C+ - O-
C = O C - O- C – OHNu- Nu H3O+ Nu
H+
Reversible
H+
Reversible
H+
Reversible
Reversible
H+
Reversible
H
Reversible

CIANHÍDRICO CIANOHIDRINA
Oxidación.
O O
R – C – H R – C - OH
Los aldehídos se oxidan con más facilidad que las cetonas.
Reducción.
- Hidruro.
O OH
R1 – C – R2 R1 – CH – R2 + H2O
- Hidrogenación.
O OH
R1 – C – R2 R1 – CH – R2
- Reducción de Clemmensen.
O
R1 – C – R2 R1 – CH – R2
- Reacción de Wolff-Kishner.
O
R1 – C – R2 R1 – CH – R2
Anión enolato.
Cuando los compuestos carbonílicos tienen hidrógenos en el carbono adyacente al grupo carbonilo estos hidrógenos son ácidos, es decir, tienen tendencia a desprenderse propiciando la formación de un anión. El anión así formado recibe el nombre de anión enolato. Por ejemplo:
O O
+ H+
_
ANIÓN ENOLATO
El anión enolato está estabilizado por resonancia. Por ejemplo:
O O-
_
Si una vez obtenido el anión enolato efectuamos una hidrólisis:
O O
H3O+ FORMA CETO
O- OH
H3O+ FORMA ENOLPágina Página 2626
KMnO4
ó CrO3/H2SO4
1) NaBH4 ó LiAlH4
2) H3O+
H2
Zn (Hg) / HCl
1) NH2 –NH2
2) OH- /

Al equilibrio entre la FORMA CETO y la FORMA ENOL se denomina tautomería ceto-enólica. Dicho equilibrio suele estar muy desplazado hacia la FORMA CETO.
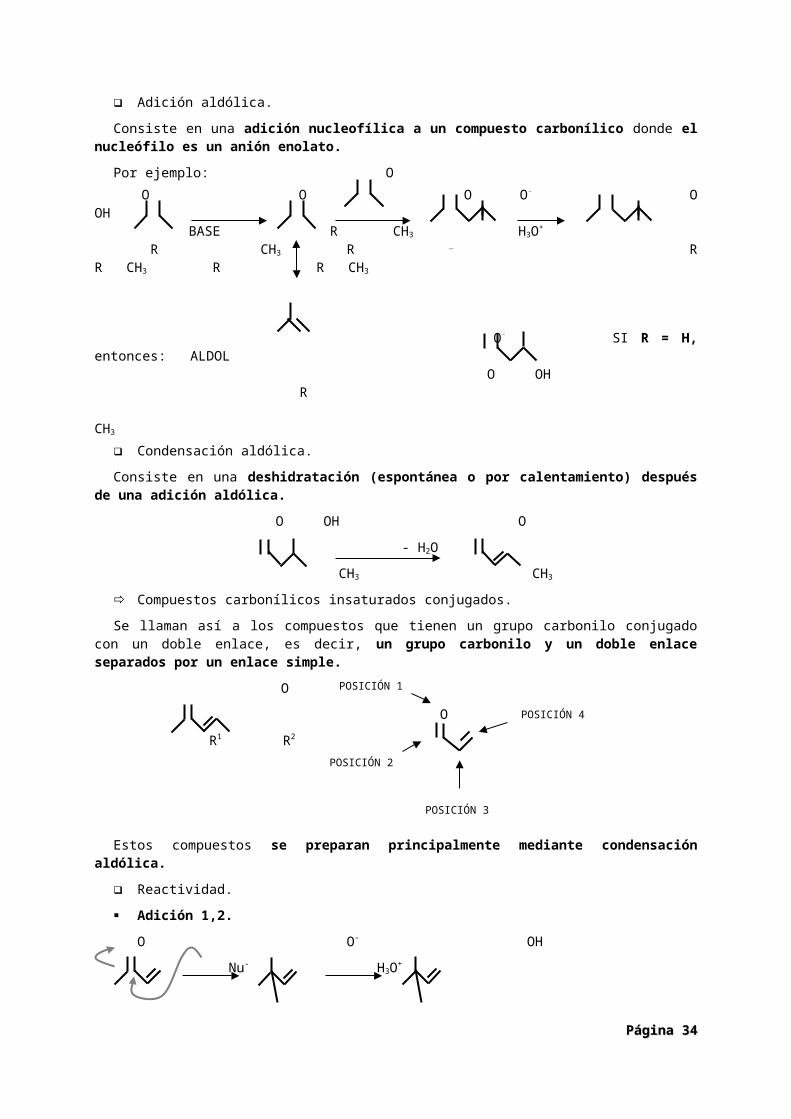
Adición aldólica.
Consiste en una adición nucleofílica a un compuesto carbonílico donde el nucleófilo es un anión enolato.
Por ejemplo: OO O O O- O OH
BASE R CH3 H3O+
R CH3 R _ R R CH3 R R CH3
O- SI R = H, entonces: ALDOL O OH
R CH3
Condensación aldólica.
Consiste en una deshidratación (espontánea o por calentamiento) después de una adición aldólica.
O OH O
- H2O
CH3 CH3
Compuestos carbonílicos insaturados conjugados.
Se llaman así a los compuestos que tienen un grupo carbonilo conjugado con un doble enlace, es decir, un grupo carbonilo y un doble enlace separados por un enlace simple.
O
O
R1 R2
Estos compuestos se preparan principalmente mediante condensación aldólica.
Reactividad.
Adición 1,2.
O O- OH
Nu- H3O+
Nu Nu
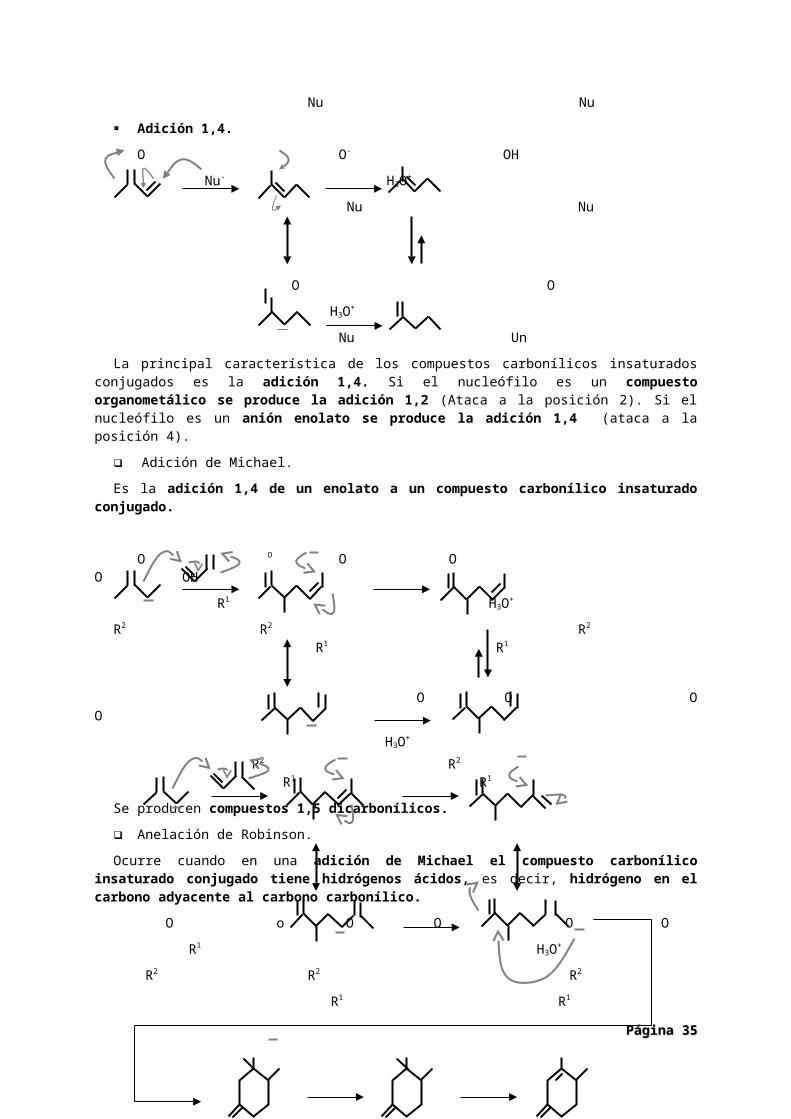
Adición 1,4.
O O- OH
Nu- H3O+
Nu Nu
Página Página 2727
POSICIÓN 1
POSICIÓN 2
POSICIÓN 3
POSICIÓN 4

O O
H3O+
Nu Un
La principal característica de los compuestos carbonílicos insaturados conjugados es la adición 1,4. Si el nucleófilo es un compuesto organometálico se produce la adición 1,2 (Ataca a la posición 2). Si el nucleófilo es un anión enolato se produce la adición 1,4 (ataca a la posición 4).
Adición de Michael.
Es la adición 1,4 de un enolato a un compuesto carbonílico insaturado conjugado.
O O O O O OH
R1 H3O+
R2 R2 R2
R1 R1
O O O O
H3O+
R2 R2
R1 R1
Se producen compuestos 1,5 dicarbonílicos.
Anelación de Robinson.
Ocurre cuando en una adición de Michael el compuesto carbonílico insaturado conjugado tiene hidrógenos ácidos, es decir, hidrógeno en el carbono adyacente al carbono carbonílico.
O o O O O O
R1 H3O+
R2 R2 R2
R1 R1
O O O O
H3O+
R2 R2
R1 R1
R2 O R2 OH R2
R1 H3O+ R1 -H2O R1
O O O
Tiene lugar una anillación intramolecular (Anelación tiene la misma raíz etimológica que anilla). Se producen ciclohexenonas.
Página Página 2828

Ácidos carboxílicos y derivados.
Los ácidos carboxílicos presentan el grupo:
O
- C - OH
Derivados.
Consideramos como derivados de ácidos carboxílicos los siguientes compuestos:
Ésteres. O
R1 O – R2 Anhídridos. O O
R1 O R2
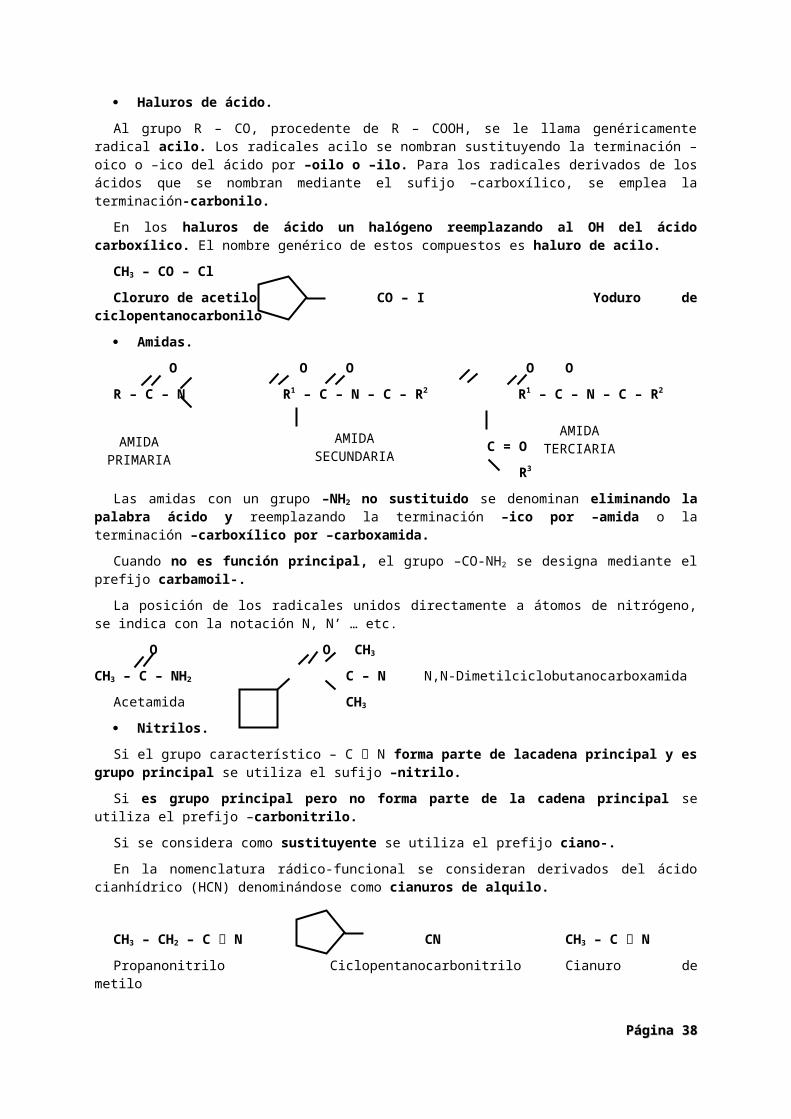
Haluros de ácido. O
X= Halógeno.
R1 X
Amidas. O
R1 N
Nitrilos. R – C N
Nomenclatura.
Ácidos carboxílicos.
Cuando el grupo carboxílico es la función principal se antepone la palabra ácido al nombre del hidrocarburo correspondiente acabado en –oico.
CH3 – CH2 - CH2 – COOH CH3 - C C – CH2 – CH = CH – COOH
Ácido butanoico ácido 2-hepten-5-inoico
Cuando en un compuesto hay tres o más grupos COOH y en caso de ácidos cíclicos se utiliza el sufijo –carboxílico.
COOH Ácido ciclopentanocarboxílico
Cuando el grupo COOH se considera como sustituyente se utiliza el prefijo carboxi-.
Sales.
Se sustituye la terminación –ico del ácido por laterminación –ato. En caso de que se haya utilizado el sufijo –carboxílico para nombrar el ácido se sustituye por –carboxilato. A continuación el nombre del metal correspondiente.
CH3
CH3 – COONa CH3 – CH – CH2 – COOK
Acetato de sodio 3-metilbutanoato de potasio
Ésteres.
Página Página 2929

Se utiliza el mismo procedimiento que para las sales poniendo el nombre del radical correspondiente en vez del metal.
Cuando el grupo característico es sustituyente frente a otro principal o frente a otros grupos carboxilato, se emplean los prefijos alcoxicarbonil-, arilocarbonil-, o en su caso se utiliza el preifjo aciloxi-.
O
CH3 – COO – CH2 – CH3 OMe
Acetato de etilo Butanoato de metilo
Anhídridos de ácido.
Se antepone la palabra anhídrido al nombre del ácido del que provienen.
O O O
2 x -H2O
CH3 OH CH3 O CH3
Anhídrido acético
Haluros de ácido.
Al grupo R – CO, procedente de R – COOH, se le llama genéricamente radical acilo. Los radicales acilo se nombran sustituyendo la terminación –oico o –ico del ácido por –oilo o –ilo. Para los radicales derivados de los ácidos que se nombran mediante el sufijo –carboxílico, se emplea la terminación-carbonilo.
En los haluros de ácido un halógeno reemplazando al OH del ácido carboxílico. El nombre genérico de estos compuestos es haluro de acilo.
CH3 – CO – Cl
Cloruro de acetilo CO – I Yoduro de ciclopentanocarbonilo
Amidas.
O O O O O
R – C – N R1 – C – N – C – R2 R1 – C – N – C – R2
C = O
R3
Las amidas con un grupo –NH2 no sustituido se denominan eliminando la palabra ácido y reemplazando la terminación –ico por –amida o la terminación –carboxílico por –carboxamida.
Cuando no es función principal, el grupo –CO-NH2 se designa mediante el prefijo carbamoil-.
La posición de los radicales unidos directamente a átomos de nitrógeno, se indica con la notación N, N’ … etc.
O O CH3
CH3 – C – NH2 C – N N,N-Dimetilciclobutanocarboxamida
Acetamida CH3
Nitrilos.
Si el grupo característico – C N forma parte de lacadena principal y es grupo principal se utiliza el sufijo –nitrilo.
Página Página 3030
AMIDA PRIMARIA
AMIDA SECUNDARIA
AMIDA TERCIARIA

Si es grupo principal pero no forma parte de la cadena principal se utiliza el prefijo –carbonitrilo.
Si se considera como sustituyente se utiliza el prefijo ciano-.
En la nomenclatura rádico-funcional se consideran derivados del ácido cianhídrico (HCN) denominándose como cianuros de alquilo.
CH3 – CH2 – C N CN CH3 – C N
Propanonitrilo Ciclopentanocarbonitrilo Cianuro de metilo
Propiedades.
Los compuestos carboxílicos que tengan enlaces O – H ó N – H (pueden formar enlaces mediante puentes de hidrógeno) tendrán un punto de ebullición más elevado que aquellos que no posean esos enlaces.
La principal característica de los ácidos carboxílicos, como su propio nombre indica, es la acidez.
O O
R – C – O – H R – C – O- + H+
Por lo que forman sales con gran facilidad.
Síntesis y reacciones.
Las reacciones de ácidos carboxílicos y derivados tienen lugar mediante un proceso de sustitución por adición-eliminación.
O O- O
R – C – X R – C – X R – C - Nu
Donde X puede ser un haluro, un anhidrido, un éster, una amida o un ácido. El orden de reactividad de los compuestos carboxílicos es el siguiente:
Haluros de ácido > Anhidridos > Esteres > Amidas > Acidos carboxílico
Es sencillo obtener un compuesto menos reactivo a partir de otro más reactivo, lo contrario, en general, no.
Ácidos carboxílicos.
Síntesis.
a) Oxidación de alquenos con KMnO4 en caliente.
b) Ozonólisis de alquinos.
c) Oxidación de alquinos con KMnO4 en caliente.
d) Oxidación de cadena lateral del Benceno con KMnO4 en caliente.
e) Oxidación de alcoholes primarios.
f) Oxidación de aldehídos.
g) Hidrólisis de nitrilos: Esta reacción sólo es válida para nitrilos que proceden de haloalcanos primarios.
H3O+ /
R – CN R - COOH
Página Página 3131
Nu-
Adición
-X-
Eliminación
1) OH-
2) H3O+
h) A partir de compuestos organometálicos:
R – Li R - COOH
Reacciones.
a) Reducción.
O
R – C – OH R – CH2 - OH
Haluros de ácido.
Síntesis.
SOCl2
O
R – C - Cl
O
R – C – OH
PBr3 O
R – C – Br
Reacciones.
O O R1 – C – O – C – R2
O R1 – C – O – R2
R1 – C – Cl O R1 – C – N
O R1 – C – OH
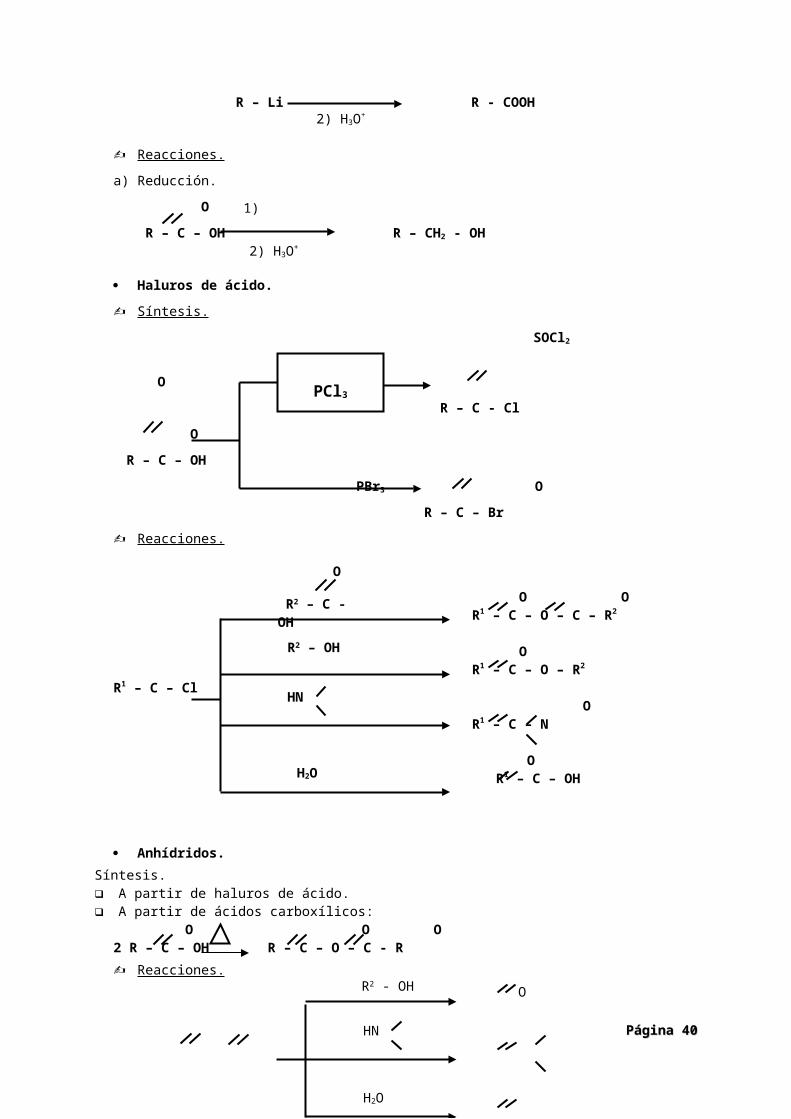
Anhídridos.Síntesis. A partir de haluros de ácido. A partir de ácidos carboxílicos:
O O O2 R – C – OH R – C – O – C - R Reacciones.
OR1 – C – O – R2
Página Página 3232
1) CO2
2) H3O+
1) LiAlH4
2) H3O+
PCl3
O
R2 – C - OH
R2 – OH
HN
H2O
R2 - OH
HN
H2O

O O O
R – C – O – C – R R1 – C – N
OR1 – C - OH
Ésteres.
Síntesis.
a) A partir de haluros de ácido.
b) A partir de anhídridos.
c) A partir de ácidos carboxílicos (esterificación):
O O
R1 – C – OH + R2 – OH R1 – C – O – R2
d) A partir de otros ésteres (transesterificación).
O O
R1 – C – O – R2 + R3 – OH R1 – C – O – R3
Reacciones.
O O
R1 – C – O – R2 R1 – C – N
O O
R1 – C – O – R2 R1 – C – O- + R2 – OH (Saponificación)
O
R1 – C – O – R2 R1 – CH2OH
También reacciona con compuestos organometálicos para formar alcoholes.
Amidas.Síntesis.
1. A partir de haluros de ácido.2. A partir de anhídridos.3. A partir de ésteres.4. A partir de ácidos carboxílicos:
O O
R – C – OH + NH R – C – N
Reacciones.
O
R – C – N R – CH2 – N
Página Página 3333
H3O+
H3O+
NH
OH-
1) LiAlH4
2) H3O+
1) LiAlH4
2) H3O+
Nitrilos.
Síntesis.
R – CH2 – Br R – CH2 - CN
(Primario)
Reacciones.
a) Obtención de ácidos carboxílicos mediante hidrólisis de nitrilos.
b) Reducción:
R – CN R – CH2 – NH2
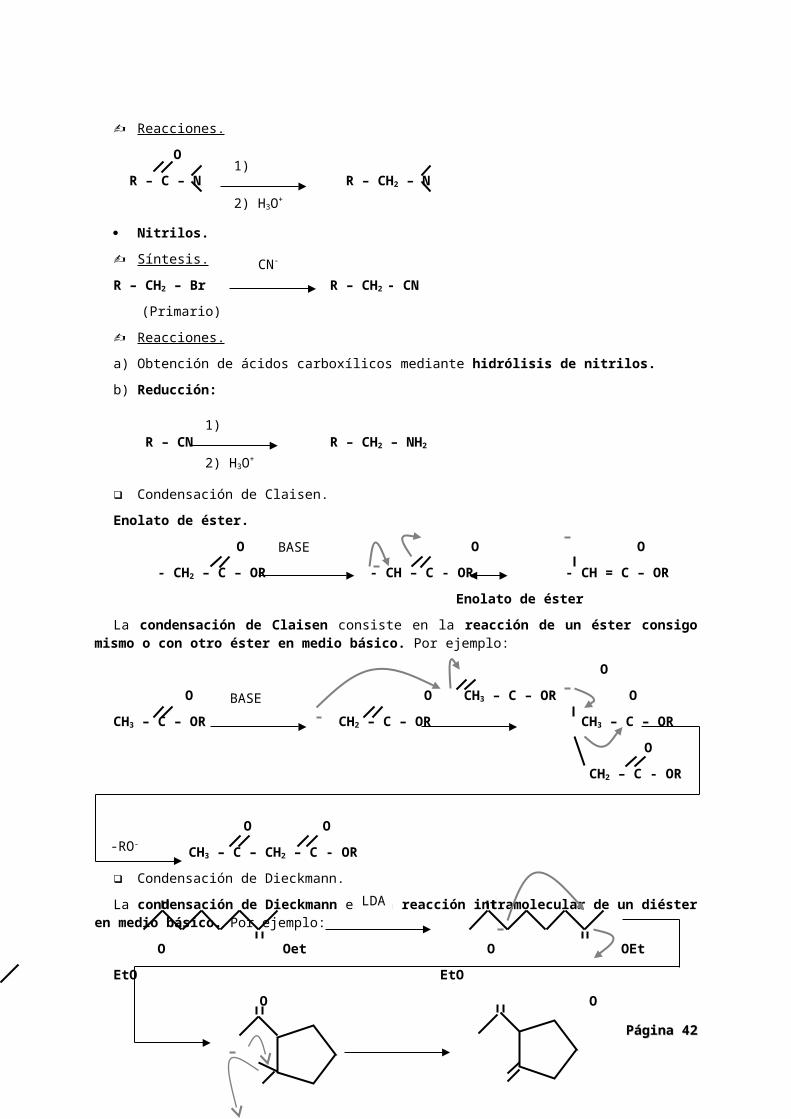
Condensación de Claisen.
Enolato de éster.
O O O
- CH2 – C – OR - CH – C - OR - CH = C – OR
Enolato de éster
La condensación de Claisen consiste en la reacción de un éster consigo mismo o con otro éster en medio básico. Por ejemplo:
O
O O CH3 – C – OR O
CH3 – C – OR CH2 – C – OR CH3 – C – OR
O
CH2 – C - OR
O O
CH3 – C – CH2 – C - OR
Condensación de Dieckmann.
La condensación de Dieckmann es una reacción intramolecular de un diéster en medio básico. Por ejemplo:
O Oet O OEt
EtO EtO
O O
O O
EtO EtO
O
OEt O
COMPUESTOS 1,3-DICARBONÍLICOS.
Generalidades.
Los compuestos 1,3-dicarbonílicos se caracterizan por poseen el siguiente grupo:
Página Página 3434
CN-
1) LiAlH4
2) H3O+
BASE
BASE
-RO-
LDA
O O
Al tratar este tipo de compuestos con una base se obtiene un obtiene un anión que presenta las siguientes formas resonantes:
O O O O O O O O
Existen dos compuestos 1,3-dicarbonílicos que tienen especial importancia:
Acetoacetato de etilo Malonato de dietilo
O O O O
OEt EtO OEt
Síntesis acetil-acética.
O O O O O O
OEt OEt R OEt
O O O O O
R O- R OH R
Síntesis malónica.
O O O O O O
EtO OEt EtO OEt EtO OEt R
O O O O O
-O R O- HO R OH HO R
Nótese que tanto en la síntesis acetilacética como en la síntesis malónica intervienen las siguientes secuencias características:
1) OH-
2) H3O+
3)
Procesos de dialquilación.
Consisten en dos reacciones de alquilación sucesivas.Página Página 3535
BASE
EtONa R - X
X = Halógeno
OH- H3O+
-CO2
EtONa R - X
X = Halógeno
H3O+
-CO2
EtONa R1 - X
X = Halógeno
R2 - XBase FuerteOH-
H3O+
HO

O O O O O O
EtO OEt EtO OEt EtO OEt R1
O O O O O
EtO R1 OEt EtO R1 R2 OEt 3) R1 R2
Para la síntesis acetilacética el proceso es análogo.
Obtención de ciclos.
Es posible la obtención de ciclos en la síntesis malónica y acetilacética utilizando dihaloalcanos terminales.
O O O O O O
EtO OEt EtO OEt EtO OEt
O O O O O
EtO X EtO OEt 3)
Para la síntesis acetilacética el proceso es análogo.
Aminas.
Las aminas pueden considerarse como derivados del amoníaco.
NH3 -NH2 NH N - N+
Amoniaco aminaamina amina sal de
primaria secundaria terciaria tetraalquilamonio
Nomenclatura.
El método más extendido para nombrar las aminas es el radicofuncional que consiste en tomar como base el radical más complejo y añadirle el sufijo –amina. Los otros radicales se nombran como sustituyentes sobre el nitrógeno.
CH3 CH2 – CH3
CH3 – CH2 – CH2 – NH – CH3 CH3 – N - CH2 – CH2 – CH2 – N - CH2 – CH3
N – metilpropilamina N,N – dietil – N’,N’ – dimetil – 1,3 - propanodiamina
Cuando la función amina no es principal se utiliza el prefijo –amino.
CH3 – CH – COOH COOH
NH2
Ácido 2-aminopropanoico Ácido m-aminobenzoico
NH2
Página Página 3636
EtONa X X
Base Fuerte
HO
X
OEt

Propiedades.
Las aminas primarias y secundarias (pueden formar puentes de hidrógeno) tienen puntos de ebullición más altos que las terciarias de igual peso molecular. Las aminas son compuestos eminentemente básicos.
Síntesis.
Procesos de reducción.
R – C N R – CH2 – NH2
2) H+
(Reducción)
NO2 NH2
R1 R1
N – R3 CH – NHR3
R2 IMINA R2
R1 R1
N – OH CH – NH2
R2 OXIMA R2
O
R1 – C – N R1 – CH2 – N
AMIDA
Reacciones.Activante en reacciones de sustitución electrófila aromática (Orientador Orto-Para).Base.
N: + H+ N+H
Nucleófilo en procesos de alquilación.
N: + Br – CH2 - R N+- CH2 - R
Nucleófilo en procesos de acilación.
O O
N: + R – C – Cl R – C - N
Oxidación de aminas aromáticas.
NH2 NO2
Reacción de Sandmeyer.
Consiste en la sustitución del grupo diazonio (N2+), en una sal de arenodiazonio, por Cl, Br o
CN.
Ar – Cl
Página Página 3737
H2 / Catalizador1) LiAlH4
H2 / CatalizadorFe /HCl
REDUCCIÓN
REDUCCIÓN
REDUCCIÓN
-HCl
KMnO4 ó Perácido
HNO2
H2SO4Sal de arenodiazonio
HCl
CuClHBr
CuBr
KCN
CuCN
Ar – NH2 Ar – N+ N H2SO4- Ar - Br
Ar = Radical arilo Ar – C N
Eliminación de Hoffman
NH2
R
+ N - R
R
REACCIONES.
Clasificación.
Las reacciones orgánicas más importantes son:
Reacciones de sustitución. Un átomo o grupo de átomos de una molécula son reemplazados por un átomo o grupo de átomos de otra molécula. Ejemplo.
CH3OH N + HBr CH3 – Br + H2O
En una reacción de sustitución podemos distinguir cuatro partes:Sustrato, recibe este nombre la molécula en la cual tiene lugar la sustitución.Reactivo o grupo entrante. Es el átomo o grupo de átomos que ataca al sustrato.Grupo saliente. Es el átomo o grupo de átomos que es expulsado del sustrato.Producto. Es el resultado de la sustitución del grupo saliente por el nucleófilo.
Cuando el reactivo es un nucleófilo se produce una sustitución nucleofílica. Ejemplo:
R – C C:- + R’ – CH2 – Br R – C C – CH2 – R’ + Br -
El ataque se produce en el carbono indicado por la flecha puesto que el bromo es más electronegativo que el carbono, por lo que el bromo “tira” de los electrones del enlace (adquiriendo una carga parcial negativa) y llevándose dichos electrones al producirse el ataque del nucleófilo.El nucleófilo puede tener carga negativa o neutra. El sustrato puede ser neutro o tener carga positiva. Existen, pues, cuatro posibilidades:
1. Nu:- + R – L Nu – R + :L-
2. Nu: + R – L [Nu – R] + + :L-
3. Nu:- + R – L+ Nu – R + :L-
4. Nu: + R – L+ [Nu – R] + + :L-
Si el nucleófilo es negativo, el producto es neutro.
Si el nucleófilo es neutro, el producto es positivo.
Estas reacciones pueden tener lugar según dos mecanismos diferentes:1. Sustitución nucleofílica monomolecular (Sn1). En este caso la reacción procede por dos
etapas, disociándose primero los compuestos en sus iones y reaccionando después estos iones entre sí. Se produce por medio de carbocationes.
2. Sustitución nucleofílica bimolecular (Sn2). En este caso la reacción transcurre en una sola etapa, produciéndose simultáneamente el ataque del reactivo y la expulsión del grupo saliente. En este caso, si el ataque tiene lugar sobre un carbono quiral se produce una inversión en la configuración, aunque puede no pasar de R a S o viceversa, puesto que el sustituyente puede alterar el orden de prioridades.
Página Página 3838
1) RI / HO- 2) Ag2O/H2O
3)

Reacciones de eliminación. A partir de una molécula grande se obtiene una molécula pequeña. Aumenta el grado de multiplicidad del enlace. Ejemplo.
CH3CH2OH CH2 = CH2 + H2O
Estas reacciones pueden ser de dos tipos:
1) Eliminación monomolecular (E1). En este caso la reacción transcurre en dos etapas, para dar lugar a un carbocatión en una primera etapa y a un doble enlace en una segunda etapa; Es una reacción competitiva con la Sn1.
2) Eliminación bimolecular (E2). En este caso la reacción transcurre en una sola etapa a través de un agente nucleofílico. Si el sustrato es un haloalcano, para que se produzca esta reacción el halógeno y el hidrógeno adyacente tienen que estar en disposición anti.
CRITERIOS DE REACTIVIDAD.Orden del sustrato
Nucleófilo débil (H2O)
Nucleófilo Base débil (I)
Nucleófilo No IMPEDIDO Base
fuerte (EtO-)
Nucleófilo IMPEDIDO Base
fuerte (tBuO-)
Metilo NO HAY REACCIÓN
Sn2 Sn2 Sn2
Primario NO HAY REACCIÓN
Sn2 Sn2 (mayoritaria) E2
Secundario Sn1 + E1 (Muy lenta)
Sn2 E2 E2
Terciario Sn1 (mayoritaria) + E1
Sn1 + E1 E2 E2
La reacción Sn2 está muy influenciada por efectos estéricos.Al aumentar la temperatura se favorece la eliminación.Los sustratos más usuales son haloalcanos.Bases fuertes impedidas estéricamente: LDA, tBuO-Na+.Bases fuertes no impedidas estéricamente: EtO-Na+, MeO-Na+, NH2
-Na+.Nucleófilos débiles: H2O, ROH (Alcoholes).
Reacciones de adición. Una molécula grande asimila una molécula pequeña. Disminuye el grado de multiplicidad del enlace. Ejemplo.
CH2 = CH2 + H – Br CH3CH2 – Br
HC CH + H2 CH2 = CH2
Intermedios de reacción.
Las reacciones químicas transcurren por ruptura de determinados enlaces y formación de otros nuevos. Existe un momento en el proceso de reacción en el que se ha roto el enlace del reactivo y no se ha formado el enlace del producto. La sustancia en este estado se denomina intermedio de reacción.
Se corresponden con un mínimo relativo en la curva Energía – Enlace de la reacción. Son especies químicas de estructura definida y representables según la teoría de Lewis. Están presentes en la reacción durante un tiempo determinado y en ocasiones se pueden aislar y almacenar. La ruptura de los enlaces puede ser de dos tipos:
Homolítica: Cada uno de los grupos en que se divide la molécula se lleva un electrón del enlace. Este tipo de ruptura da lugar a los radicales libres.
H H
H - C – H H – C * + H *
H H
Página Página 3939
Radical libre Metilo
Heterolítica: Uno de los grupos en que se divide la molécula se lleva los dos electrones. Este tipo de ruptura da lugar a los carbocationes y carbaniones.
H H
H - C – H H – C + + : H -
H H
Carbocatión Metilo
H H
H - C – H H – C :- + H +
H H
Carbanión Metilo
Estabilidad de radicales libres, carbocationes y carbaniones.
Radical libre. Será tanto más estable cuantos más grupos alquilo rodeen al átomo de carbono. Además su estabilidad si presenta formas resonantes.
H H R R
H - C R – C R – C R - C
H H H R
Carbocatión. Será tanto más estable cuantos más grupos alquilo rodeen al átomo de carbono. Además, su estabilidad aumentará si presenta formas resonantes, es decir, lo mismo que para los radicales libres.
H H R R
H - C + R – C + R – C + R – C +
H H H R
Carbanión. Será tanto más estable cuantos menos grupos alquilo rodeen al átomo de carbono (lo contrario a radicales libres y carbocationes). Su estabilidad aumentará si presenta formas resonantes.
R R H H
R – C - R – C - R – C - H – C -
R H H H
Isomería.
La existencia de moléculas que poseen la misma fórmula molecular y propiedades distintas se conoce como isomería. Los compuestos que presentan esta característica reciben el nombre de isómeros. La isomería puede ser de dos tipos:
Isomería constitucional.
Las moléculas que presentan este tipo de isomería se diferencian en la conectividad, es decir, tienen los mismos átomos conectados de forma diferente (distinta fórmula estructural). La isomería constitucional se clasifica en:
Isomería de cadena u ordenación. Presentan isomería e cadena u ordenación aquellos compuestos que tienen distribuidos los átomos de carbono de la molécula de forma diferente.
CH3CH2CH2CH3 CH3CHCH3
Página Página 4040
Los carbocationes se comportan como nucleófilos (bases de Lewis).
Los carbaniones se comportan como electrófilos (ácidos de Lewis).
Estabilidad
Estabilidad
Estabilidad

CH3
Isomería de posición. La presentan aquellos compuestos que teniendo las mismas funciones químicas están enlazadas a átomos de carbono que tienen localizadores diferentes.
CH3 – CH2 – CH2 – CO - CH3 CH3 – CH2 – CO - CH2 - CH3
Isomería de función. La presentan aquellos compuestos que tienen distinta función química.
CH3 – CH2 – CH2OH CH3 – CH2 – O - CH3
Metámeros. Tienen el mismo grupo funcional sustituido de formas distintas.
CH3 - CH2 - CO - NH2 CH3 - CO - NH - CH3
Isomería en el espacio o estereoisomería.
La isomería en el espacio se clasifica en:
Isomería conformacional.
Aparece en los compuestos que presentan conformaciones diferentes. Se denomina análisis conformacional al estudio de dichas conformaciones.
CONFORMACIONES.
Se denominan conformaciones las diferentes disposiciones espaciales de los átomos cuando la cadena realiza un giro cuyo eje es un enlace simple C – C.
H H
H H
H HPROYECCIONES DE NEWMAN.
Para representar las distintas conformaciones se utilizan las proyecciones de Newman.
En estas proyecciones, el grupo más cercano
al observador se representa como:
El grupo más lejano al observador se representa como:
Entonces:
representa
En los alcanos pueden distinguirse tres conformaciones:
Alternada. Es la más estable (menor energía) pues sus átomos están lo más separados posible y, por tanto, la interacción es mínima.
Eclipsada. Es la menos estable (mayor energía).
Sesgada o desviada. Se llaman así a las infinitas conformaciones que existen entre la alternada y la eclipsada. Su energía es mayor que la alternada y menor que la eclipsada.
ANÁLISIS CONFORMACIONAL DEL CICLOHEXANO.
Las conformaciones más importantes que puede presentar el ciclohexano son la silla y el bote. Estas conformaciones experimentan una interconversión continua. La forma de silla es más
Página Página 4141
a
bc
df
e
a
c
b
f
d
e
estable que la de bote, por lo que la forma de silla es mayoritaria. Se estima que, en un momento dado, más del 99% de las moléculas se encuentran en forma de silla. Entre las conformaciones de silla y de bote destaca la conformación de bote torsionado de mayor estabilidad que el bote.
En la conformación silla existen dos tipos de enlaces: axiales y ecuatoriales.
Los enlaces axiales en la silla 1 pasan a ser ecuatoriales en la silla 2.Los enlaces ecuatoriales en la silla 1 pasan a ser axiales en la silla 2.Las dos sillas son indistinguibles si no poseen algún sustituyente.
Isomería configuracional.La isomería configuracional se clasifica a su vez en:
ESTEREOISOMERÍA GEOMÉTRICA.
La presentan los compuestos que se diferencian únicamente en la disposición de sus átomos en el espacio. Moléculas con fórmulas moleculares idénticas pueden presentar estructuras espaciales diferentes. Estas moléculas pueden ser:
AQUELLAS CUYAS CADENAS TIENEN DOBLES ENLACES. Una característica del doble enlace es su rigidez, que impide la libre rotación y reduce los posibles intercambios de posición que pueden experimentar los átomos de una molécula.
SISTEMAS CÍCLICOS PLANOS. Página Página 4242
RR R SILLA I
BOTE TORSIONADO I BOTE
R
BOTE TORSIONADO II
R
SILLA II
ENERGÍA
SILLA IBOTE
TORSIONADO I
BOTE TORSIONADO
II
BOTE
SILLA II
10.8 Kcal/mol
5.5 Kcal/mol
1.6 Kcal/mol
CC
C
CCC
H H
H H
H
H
SILLA IGris oscuro: Ecuatorial
Gris claro: Axial
H
HH
H
HH
C
C
C
C
CC
H H
H
H
HH
SILLA IIGris oscuro: Axial
Gris claro: Ecuatorial
H
H
H HH
H

SISTEMAS CÍCLICOS PLEGADOS.
ESTEREOISOMERÍA ÓPTICA.
Las moléculas que presentan este tipo de isomería se diferencian únicamente en el efecto que tienen sobre la luz. Recibe el nombre de MOLÉCULA QUIRAL aquella que no se puede superponer con su imagen especular. Toda molécula no quiral recibe el nombre de AQUIRAL. Si una molécula posee un plano de simetría es aquiral. Una molécula quiral puede presentar, al menos, dos configuraciones diferentes, una imagen especular de la otra, que constituyen una pareja de ENANTIÓMEROS. Uno de ellos gira el plano de polarización de la luz hacia la derecha (dextrógiro) y se identifica con la letra R; el otro gira el plano de polarización de la luz hacia la izquierda (levógiro) y se identifica con la letra S. Este tipo de nomenclatura recibe el nombre de NOMENCLATURA R,S.
CONFIGURACIÓN.
Aparece en los compuestos que presentan configuraciones diferentes.
Para pasar de una configuración a otra es necesario, al menos, romper un enlace y formar otro nuevo. Dos configuraciones son diferentes si no son superponibles.
Las configuraciones se representan mediante las proyecciones de Fischer. En una proyección de Fischer, un átomo de carbono tetraédrico se representa por dos líneas cruzadas. Por convención, las líneas horizontales representan enlaces que salen de la página hacia el lector y las líneas verticales representan enlaces que entran en la página y se alejan del lector.
R1 R1 R1
C R2 R3 C R4 R3 R4 R3 R4
R2 R2
Si en una proyección de Fischer se realiza un número par de permutaciones entre los sustituyentes se obtiene la configuración original. Si se realiza un número impar de permutaciones se obtiene la imagen especular de la configuración original.
R1 R2 R4 R4 R4 R2
R3 R1 R3 R1 R2 R1
R3 R4 R2 R3
A B C D A y B son configuraciones diferentes. B y C son configuraciones diferentes. C y D son configuraciones diferentes. A y D son configuraciones diferentes. A y C son la misma configuración. B y D son la misma configuración.Se denomina carbono asimétrico o estereocentro aquel carbono que tiene los 4 sustituyentes distintos. Si una molécula tiene un único carbono asimétrico, solo puede existir un par de enantiómeros. Si tiene dos carbonos asimétricos da por resultado un máximo de cuatro estereoisómeros (dos pares de enantiómeros). En general, una molécula con n carbonos asimétricos hace posibles 2n estereoisómeros (2n-1 pares de enantiómeros). Los estereoisómeros que no son imágenes especulares se denominan DIASTEREISÓMEROS. Esta generalización implica que los estereoisómeros CIS-TRANS son un tipo de diastereoisómeros.Se denominan compuesto meso a aquellos que, conteniendo carbonos asimétricos, son aquirales (existe un plano de simetría).Se denomina mezcla racémica a aquella que contiene un par de enantiómeros en una proporción del 50% de cada uno. La desviación de la luz polarizada producida por dicha mezcla es nula.
Página Página 4343
IsomeríaMisma fórmula
molecular y distintas
propiedades
Isómeros constitucionalesMismos átomos con distinta conectividad. (Diferente fórmula estructural).EstereoisómerosMismos átomos
con diferente disposición
espacial
Isomería conformacionalDiferentes conformaciones
Isomería configuracionalDiferentes configuraciones
Enantiómeros: Estereoisómeros que son imágenes especulares

ESTEREOISOMERÍA GEOMÉTRICA DEBIDA AL DOBLE ENLACE.
El doble enlace C = C no permite el giro cuyo eje sea dicho enlace. Supongamos un doble enlace C = C, disustituido, siendo ambos sustituyentes idénticos. Si los dos sustituyentes están del mismo lado el compuesto es CIS. Si están en distinto lado es TRANS.
R R H R
RCH = CHR C = C C = C
H H R H
CIS TRANS
Aunque los sustituyentes no sean iguales también ocurre este tipo de isomería si el tamaño de dos de los sustituyentes es grande con respecto a los otros dos:
R R’ H R’
RCH = CHR’ C = C C = C
H H R H
CIS TRANS
En el caso de doble enlace trisustituido o tetrasustituido con los 3 ó 4 sustituyentes diferentes se emplea la denominación E,Z basada en las reglas de secuencia de Cahn, Ingold y Prelog:
1. Se observan los átomos que están directamente unidos a cada carbono del doble enlace y se establece un orden de prioridad. Un átomo de mayor número atómico recibe prioridad más alta.
2. Si no es posible tomar una decisión considerando los primeros átomos del sustituyente, se recurre a los segundos, terceros, y así sucesivamente hasta que se encuentre una diferencia.
3. Si los átomos de mayor prioridad están del mismo lado el compuesto es Z. Si están del lado contrario el compuesto es E.
CH3CH2 CH3 CH3 CH3
C = C C = C
CH3 H CH3CH2 H
ESTEREOISOMERÍA GEOMÉTRICA EN SISTEMAS CÍCLICOS PLANOS.
En este caso el plano de simetría para determinar si una molécula es CIS o TRANS es el plano que contiene el esqueleto carbonado de dicha molécula.
CH3 H CH3 H
H CH3
CH3 H
ESTEREOISOMERÍA GEOMÉTRICA EN SISTEMAS CÍCLICOS PLEGADOS. Página Página 4444
Diastereoisómeros: Estereoisómeros que no son
imágenes especulares
(Z)–3–metil–2-penteno
(E)–3–metil–2-penteno
Cis-1,2-dimetilciclopentano
Trans-1,2-dimetilciclopentano

Para determinar si un sistema cíclico plegado es CIS o TRANS consideramos, como planos de referencia, planos ortogonales a los enlaces axiales que contengan a los carbonos. Si los sustituyentes prioritarios están del mismo lado de dichos planos la molécula es CIS. Si están en distinto lado es TRANS.En el caso del ciclohexano por cada estereoisómero CIS-TRANS habrá dos confórmeros, uno ecuatorial y otro axial.
Nomenclatura R,S.
La nomenclatura R,S se utiliza para determinar la configuración absoluta de los carbonos quirales. Para determinar si una molécula es R ó S se actúa de la siguiente forma:
1. Se observan los átomos que están directamente unidos a cada carbono del doble enlace y se establece un orden de prioridad siguiendo las reglas de Cahn, Ingold y Prelog. El átomo de mayor número atómico recibe la prioridad más alta.
2. Si no es posible tomar una decisión considerando los primeros átomos del sustituyente, se recurre a los segundos, terceros, y así sucesivamente hasta encontrar un punto de discordancia.
3. Se considera que los átomos unidos con enlaces múltiples son equivalentes al mismo número de átomos unidos con enlace sencillo. H – C = O equivale a H – C – O – C
O 1. cuando la única diferencia entre los sustituyentes sea isotópica tendrá prioridad el átomo de
mayor masa atómica.2. Una vez determinadas las prioridades, se recorren los átomos por orden de prioridad (de
mayor a menor) sin tener en cuenta el de menor prioridad. El sentido de las agujas del reloj es R, el sentido contrario a las agujas de reloj es S.
3. Si el átomo de menor prioridad se encuentra hacia atrás (posición vertical en la proyección de Fischer) se mantiene el sentido, es decir, si el sentido de recorrido de los átomos era R la molécula será R, y si el sentido de recorrido de los átomos era S la molécula será S.
4. Si el átomo de menor prioridad se encuentra hacia delante (posición horizontal en la proyección de Fischer) se invierte el sentido, es decir, si el sentido de recorrido de los átomos era R la molécula será S, y si el sentido de recorrido de los átomos era S la molécula será R.
Luz María Pastor Vá[email protected] en ciencias químicas
Página Página 4545
Br
CH3
H
H
Br
HCH3
H
Cis-1-bromo-3-metilciclohexanoAxial-axial
Trans-1-bromo-3-metilciclohexanoEcuatorial-ecuatorial