guía de laboratorio vi i propiedades coligativas y ... · centro interdisciplinario de líquidos...
TRANSCRIPT

Capacitación de docentes en
de la Química desde la perspectiva de la
Guía de LaboratorioPropiedades Coligativas y
Capacitación de docentes en la enseñanza
de la Química desde la perspectiva de la
Química Verde
Guía de Laboratorio VIPropiedades Coligativas y
Química Verde
Santiago, 2015
la enseñanza
de la Química desde la perspectiva de la
II Propiedades Coligativas y

Centro Interdisciplinario de Líquidos Iónicos Página 1

Centro Interdisciplinario de Líquidos Iónicos Página 2
Tabla de contenidos.
Parte I. Propiedades coligativas de las Soluciones.
1. Introducción.
2. Aumento Ebulloscópico.
3. Descenso crioscópico.
4. Presión Osmótica.
5. Presión de vapor.
Parte II. Propiedades coligativas de las Soluciones y Química Verde.
1. Introducción.
2. Principios de la Química Verde.
3. Compuestos orgánicos volátiles.
4. COV’s y la salud humana.
5. ¿De dónde provienen los COV’s?
6. Proceso de refinación de petróleo.
Parte III. Trabajo Práctico
1. Objetivos de la actividad.
2. Conceptos asociados en la actividad.
3. Materiales y reactivos.
4. Metodología.
Parte IV. Ficha de trabajo.
1. Observaciones sustraídas de la actividad.
2. Explicación de las observaciones.
3. Principio de la Química Verde abordado.
4. Aprendizajes obtenidos. Conclusiones preliminares.
5. Preguntas y Cuestionamientos.
Referencias.

Centro Interdisciplinario de Líquidos Iónicos Página 3
Parte I. Propiedades de las Soluciones.
1. Introducción Las propiedades coligativas
permiten explicar fenómenos que
ocurren en la vida cotidiana. Tal
como cuando se agrega sal al agua
hirviendo, ésta deja de hervir. Pues
bien, este fenómenos se explicar
mediante una de las propiedades
coligativas. La imagen, representa
un vaso de precipitado, que
contiene agua y sal (NaCl), cuando
el cloruro de sodio se disuelve en
agua, modifica las propiedades originales que tiene el agua a una determinada temperatura. En
esta unidad, estudiarás como afecta la presencia de un soluto en las propiedades de las
soluciones. Las propiedades que dependen de la concentración de las partículas de soluto, pero no
de su naturaleza, son las propiedades coligativas.
Año Suceso
1758 Joseph Black formula el concepto de calor latente para explicar fenómenos de cambios de
estado.
1803
Louis Joseph Gay-Lussac, descubre propiedades físicas y químicas del aire y otros gases y
realiza las pruebas experimentales de las leyes de Boyle y de Charles, así como las
relaciones entre la densidad y la composición de los gases.
1848 Lord Kelvin, establece el concepto de cero absoluto, que es la temperatura a la cual
teóricamente todo movimiento molecular se detiene.
1866 James Maxwell construyó la teoría cinética sobre un modelo para los gases.
1883 William Nicol propone teoría de interacción entre las moléculas de soluto y disolvente.
1901
Jacobus Herricus Van`t Hoff, de los países bajos, recibe el premio nobel, por su
descubrimiento sobre las leyes de la dinámica química y de la presión osmótica en
disolución.
1945 Se descubre el fenómeno de resonancia nuclear magnética (RMN), por E.M Purcell y F.
Bloch, quienes lo logran de manera separada.
1948 Se reemplaza el nombre de grado centígrado para la unidad de temperatura y se establece
el grado celcius (°C)
2011 Daniel Shechtman, Israelí, gana el premio nobel de química por su descubrimiento de los
“cuasicristales”
Centro Interdisciplinario de Líquidos Iónicos Página 4
2. Aumento ebulloscópico.
Los autos tienen un motor el que debe ser protegido, sobre todo en la época de verano,
donde se usa una sustancia llamada anticongelante. Su principal ingrediente es una sustancia
llamada etilenglicol. El radiador del auto y su sistema de enfriamiento están sellados para
mantener el anticongelante a presión, de modo que no se evapore a las temperaturas normales de
funcionamiento del motor. Sin embargo, cuando la temperatura atmosférica se eleva en el verano,
el radiador podría “llegar a la ebullición” si no estuviera protegido con “anticongelante”. De esta
manera al agregarle este líquido no volátil, la solución del radiador adquiere un punto de
ebullición más alto que del agua pura.
El punto de ebullición de una disolución es la temperatura a la cual su vapor de presión
iguala a la presión atmosférica externa. Por ejemplo la temperatura de ebullición del agua sobre el
nivel del mar es 100 °C. Debido a la presencia de un soluto no volátil, disminuye la presión de
vapor de una disolución y por lo tanto el punto de ebullición de la disolución se afectado.
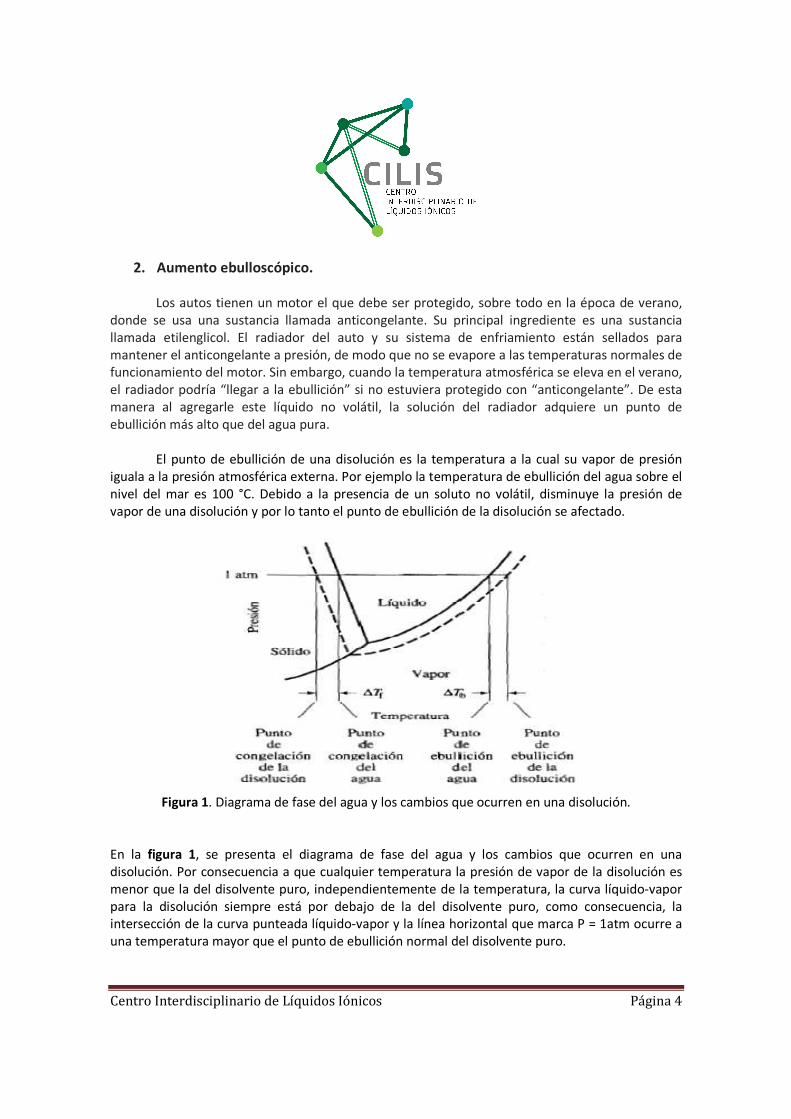
Figura 1. Diagrama de fase del agua y los cambios que ocurren en una disolución.
En la figura 1, se presenta el diagrama de fase del agua y los cambios que ocurren en una
disolución. Por consecuencia a que cualquier temperatura la presión de vapor de la disolución es
menor que la del disolvente puro, independientemente de la temperatura, la curva líquido-vapor
para la disolución siempre está por debajo de la del disolvente puro, como consecuencia, la
intersección de la curva punteada líquido-vapor y la línea horizontal que marca P = 1atm ocurre a
una temperatura mayor que el punto de ebullición normal del disolvente puro.

Centro Interdisciplinario de Líquidos Iónicos Página 5
El análisis de esta gráfica muestra que el punto de ebullición de la disolución es mayor
que el del agua. La elevación del punto de ebullición (∆Tb), se define como el punto de ebullición
de la disolución (Tb) menos el punto de ebullición del disolvente puro (Tb0): ΔTb = Tb – T°b
Debido a que ΔTb = (Tb – T°b) > 0, es una cantidad positiva que depende de la cantidad de masa de
soluto, entonces, ΔTb α m. Por lo tanto, ΔTb = Kb*m
El valor de ∆Tb, es proporcional a la disminución de la presión de vapor y también es proporcional
a la concentración (molalidad) de la disolución, es decir, donde m es la molalidad de la disolución y
Kb es la constante molal de elevación del punto de ebullición (Constante ebulloscópica). Las
unidades de Kb son 0C/m. Es importante entender la selección de las unidades de concentración en
este caso, y la razón de optar por la molalidad en vez de la molaridad, es que ésta última depende
de la temperatura, y el sistema con el que se trabaja la temperatura no se mantiene constante.
En la siguiente tabla se encontrará las constantes ebulloscópicas de varios líquidos comunes:
Tabla1. Puntos de ebullición y constante ebulloscópica, de disolventes comunes.
Ejemplo práctico.
Cuál es el asenso ebulloscópico, de una solución formada por 20,1 g de una sustancia cuya masa
molar es 62,0 g, esta cantidad se encuentra en 400 g de agua.
Paso 1. Organizar la información entregada.
Masa de soluto: 20,1 g
Masas molar del soluto: 62 g
Masa del solvente: 400 g (agua)
Determinar: elevación del punto de ebullición

Centro Interdisciplinario de Líquidos Iónicos Página 6
Paso 2. Escoger la expresión matemática
La expresión matemática que permite desarrollar este ejercicio es: ∆Tb = Kb m
La constante ebulloscópica para el agua, la podrás encontrar en la Tabla 1 de esta lección.
Para utilizar esta expresión matemática, se debe determinar la molalidad de la solución, con
los datos que se han entregado.
Masa de soluto (g) x 1 mol de soluto = cantidad de soluto (mol)
Masa molar de soluto (g)
Cantidad de soluto (mol) x 1000 g de agua = molalidad
Masa de solvente (g) 1 kg de agua
∆Tb = Kb m
Paso 3. Sustituir los datos en la expresión matemática y resolver el problema:
20, 1 g de soluto x 1 mol de soluto = 0,324 mol de soluto
62, 0 g de soluto
0,324 mol de soluto x 1000 g de agua = 0,810 mol de soluto = 0,810 m
400 g de agua 1 kg de agua kg de agua
∆Tb = Kb m = 0,52 °C/m x 0,810 m = 0,41 °C
El aumento ebulloscópico es de 0,41 °C
3. Descenso crioscópico
Imagina que necesitas mantener frías unas bebidas. Lo normal es meterlas en hielo con agua de
manera que se mantuviesen frías. Pero como la temperatura del agua es mayor que la del hielo,
tenderá a estabilizarse y, por tanto, tardará relativamente poco en calentarse, por lo tanto no se
mantendrán mucho tiempo a la temperatura que deseas. El truco usado es usar agua salada muy
fría con hielos, de esa manera, conseguirás que las bebidas estén en un medio frío durante más
tiempo, ya que los hielos se disolverán por razones coligativas de descenso crioscópico no por
razones relacionadas con la temperatura. El agua salada aguantará temperaturas menores que
cero grados y tardará más en calentarse, manteniéndolas frías más tiempo. Esto lo podrás
entender de mejor manera en la siguiente lección.
La disminución del punto de congelación (∆Tf) se define como el punto de congelación del
disolvente puro (Tf0) menos el punto de congelación de la disolución (Tf): ΔTf = Tf – T°f. Debido a
que ΔTf = (Tf – T°f) > 0, es una cantidad positiva. De nuevo, ∆Tf es proporcional a la concentración
de la disolución: ΔTf α m. Por lo tanto, ΔTf = Kf*m

Centro Interdisciplinario de Líquidos Iónicos Página 7
Donde m es la concentración de soluto en unidades de molalidad y Kf es la constante molal
de la disminución del punto de congelación (Constante crioscópica). Al igual que para Kb (Tabla 1)
las unidades de Kf son 0C/m. La explicación cualitativa de la disminución del punto de congelación,
se debe a que la congelación implica la transición de un estado desordenado a un estado
ordenado. Para que esto ocurra, el sistema debe liberar energía, y debido a que en una disolución
hay mayor desorden que en el disolvente, es necesario que libere más energía para generar orden
que en el caso de un disolvente puro. Por lo tanto, la disolución tiene menor punto de congelación
que el disolvente (Tabla 2).
Tabla 2. Puntos de congelación y constante crioscópica, de disolventes comunes.
Figura 2. Descenso del punto de congelación de una solución compuesta por agua y azúcar.
Ejemplo práctico.
¿Cuál es el descenso crioscópico, de una solución formada por 17,1 g de sucralosa, C12H22O11, en
200 g de agua? ¿Cuál es el punto de ebullición de la disolución?

Centro Interdisciplinario de Líquidos Iónicos Página 8
Paso 1. Organizar la información entregada.
Masa de soluto y fórmula química: 17,1 g C12H22O11
Masa del solvente: 200 g (agua)
Determinar: Descenso del punto de congelación
Punto de congelación de la solución
Paso 2. Escoger la expresión matemática
La expresión matemática que permite desarrollar este ejercicio es: ∆Tf = Kf m
La constante crioscópica para el agua, la podrás encontrar en la Tabla 2 de esta lección.
Para utilizar esta expresión matemática, se debe determinar la molalidad de la solución, con
los datos que se han entregado.
Masa de soluto (g) x 1 mol de soluto = cantidad de soluto (mol)
Masa molar de soluto (g)
Cantidad de soluto (mol) x 1000 g de agua = molalidad
Masa de solvente (g) 1 kg de agua
∆Tf = Kf m
Por otro lado:
Punto de congelación de la solución = Punto de congelación del solvente + ∆Tf
Paso 3. Sustituir los datos en la expresión matemática y resolver el problema:
17,1 g C12H22O11 x 1 mol de C12H22O11 = 0,0500 mol de C12H22O11
342, 34 g C12H22O11
0,0500 mol de C12H22O11 x 1000 g de agua = 0,250 mol de C12H22O11= 0,250 m
200 g de agua 1 kg de agua kg de agua
a. ∆Tf = Kf m = (-1,86 °C/m) x 0,250 m = -0,465 °C
b. Punto de congelación de la solución = 0,0 °C + (-0,465 °C) = -0,465 °C

Centro Interdisciplinario de Líquidos Iónicos Página 9
Seres vivos con anticongelantes
Algunos animales evitan la congelación
usando anticongelantes fisiológicos. Se trata de
solutos elaborados o incorporados a sus líquidos
corporales durante las estaciones frías, con los
que incrementan la concentración total de solutos
y reducen la temperatura de congelación. Muchos
insectos, por ejemplo, producen concentraciones
elevadas de glicerol, sorbitol o manitol durante el
invierno. Es el caso, por ejemplo, de las larvas de
invierno de Bracon cephi que tienen una
temperatura de congelación que puede alcanzar
los -17 °C durante el invierno, mientras que los
individuos de verano sólo pueden alcanzar los -1 °C. También los huevos de invierno de la oruga
Malacosoma contienen glicerol en una cantidad del orden del 35% de su peso en seco.
La Rana sylvatica, que vive en la mitad
septentrional de Norteamérica, es capaz de
prosperar en un clima subártico. Este animal puede
hibernar, sobreviviendo a la congelación de entre
el 35 y el 45% de su cuerpo. Para ello se sirve de
proteínas especiales, urea acumulada en los tejidos
y glucosa almacenada en el hígado para limitar la
cantidad de hielo que se forma en su cuerpo y
reducir así la contracción osmótica de las células.
En el océano Antártico viven un centenar de
especies del género Notothenioidea que
sintetizan un tipo de proteínas con
características anticongelantes que se
disuelven en la sangre y que les permiten
sobrevivir a temperaturas de hasta -1,8 °C.
Estas proteínas tienen un mecanismo de
funcionamiento que no se basa en el
descenso crioscópico y una efectividad unas 500 veces mayor. Al parecer, estas se adhieren a los
núcleos de cristalización del hielo que se forman inicialmente e impiden su crecimiento posterior.

Centro Interdisciplinario de Líquidos Iónicos Página 10
¿Por qué no usar sales como anticongelantes?
La disminución de la temperatura de congelación de un disolvente debido a la presencia de
un soluto se usa para evitar la solidificación del agua de refrigeración en los motores de
combustión. En las regiones frías, donde la temperatura puede bajar de los 0 °C, se añaden
sustancias al agua de refrigeración para rebajar su temperatura de congelación y evitar así que
esta se congele, ya que, de producirse, el aumento del volumen del hielo podría romper el sistema
de refrigeración. Las sales muy solubles en agua, como el cloruro de calcio, podrían ser apropiadas,
ya que una disolución con un 30,5 % de CaCl2 se congela a -50 °C. Sin embargo, no pueden usarse
ya que corroen los metales.
Como anticongelantes se usan disoluciones de etanol, etilenglicol o glicerina, ya que sus
disoluciones al 36,5 % en masa, 39 % en volumen y 44,4 % en masa, respectivamente, congelan a
partir de -25 °C. Si se desea disminuir más la temperatura de congelación se usan disoluciones de
glicerina al 58 % que congelan a -50 °C. El más usado es el etilenglicol. En los circuitos de
refrigeración de paneles de energía solar el anticongelante que se utiliza es el propilenglicol.
El descenso crioscópico también se aprovecha para eliminar capas de hielo de las carreteras,
autopistas y pistas de aeropuertos. Para ellos se lanza cloruro de sodio (NaCl) o de calcio (CaCl2)
sobre las placas de hielo, con lo que se disminuye la temperatura de congelación y se funden las
placas de hielo. Una ventaja del cloruro de calcio es que, cuando este se disuelve, libera gran
cantidad de calor que ayuda a fundir más el hielo. Para eliminar las capas de hielo que se forman
sobre los aviones también se usa el etilenglicol.
En la construcción se usan anticongelantes para los hormigones en lugares donde las
temperaturas son muy bajas y se congelaría el agua. No es posible usar grandes cantidades de
anticongelante ya que provocaría problemas de corrosión. Los anticongelantes que se usan son
sales, normalmente cloruro de calcio, CaCl2, que en una proporción del 2 % reduce la temperatura
de congelación a -5 °C.

Centro Interdisciplinario de Líquidos Iónicos Página 11
4. Presión Osmótica.
Osmosis
Existen muchos procesos químicos y biológicos que dependen de la osmosis, ¿pero qué es la
osmosis?, se entiende por osmosis, al paso selectivo de las moléculas del disolvente a través de
una membrana porosa desde una disolución diluida hacia una de mayor concentración. La difusión
del líquido de un medio a otro, a través de membranas, fue observada por primera vez en 1758
por el padre francés Jean Antoine Nollet. La figura 1 muestra el proceso de la osmosis.
Figura 3. Las esferas amarillas representan moléculas
de soluto, y las esferas celestes, moléculas de agua. La
osmosis (paso selectivo de las moléculas del disolvente
a través de una membrana porosa desde una disolución
diluida hacia una de mayor concentración) es el paso de
moléculas de solvente, a través de una membrana
semipermeable, desde una zona diluida hacia una zona
concentrada.
La ósmosis es realizada con el auxilio de una membrana semipermeable que permite el paso del
solvente y no permite el paso del soluto. Existen muchos tipos de esas membranas. Como
ejemplo, podemos citar el papel celofán, la vejiga animal, pared de las células, porcelana,
zanahoria hueca, etc. El flujo de agua es más intenso en el sentido de la solución. Cuando los flujos
se igualan, no existirán alteraciones en los niveles de los líquidos.
Presión osmótica
Si quisiéramos interrumpir la ósmosis, basta ejercer sobre el sistema formado por dos soluciones o
una solución y un solvente, separados por una membrana semipermeable, una presión en el
sentido inverso al de la ósmosis, con la misma intensidad de aquella presión que ejerce el solvente
para atravesar la membrana semipermeable. A esa presión, capaz de impedir el fenómeno de la
ósmosis, damos el nombre de presión osmótica. O sea, es definida como el equivalente a la
presión que es necesaria aplicar sobre un recipiente conteniendo solvente puro de modo de
impedir la ósmosis. Lo anterior se puede esquematizar en la figura 4.

Centro Interdisciplinario de Líquidos Iónicos Página 12
Figura 4. Se muestra un experimento donde se
sumerge una solución concentrada en solvente puro,
separados por una semipermeable al solvente. La
solución comienza a ascender por el tubo, lo que
genera una diferencia de altura, esto es generado por
la presión ejercida por el solvente para ingresar a la
solución concentrada, presión que se conoce como
presión osmótica (A mayor presión osmótica, mayor es
la tendencia del solvente para entrar en la solución).
Fenómenos relacionados con la presión osmótica
Es muy importante conocer la naturaleza de las soluciones que se ponen en contacto a través
de membranas semipermeables al solvente, ya que si hay diferencias de concentración, el
solvente migrará de una solución a la otra de acuerdo a lo explicado en los párrafos anteriores.
¿Existirá diferencia si dejamos en contacto disoluciones de diferente concentración entre sí, y
cuando ponemos en contacto soluciones de igual concentración? Esta respuesta es de suma
importancia en los seres vivos, ya que gracias a eso podemos realizar la migración de solvente
en los diferentes sistemas de los organismos. Para esto revisemos tres situaciones, las cuales
se resumen en la figura 5. Si ponemos en contacto a través de una membrana semipermeable
al solvente, dos disoluciones con igual concentración, no existirá migración de solvente ya que
existirá un equilibrio osmótico. A esta solución de igual concentración a la estudiada se
llamara solución isotónica (iso: igual - tónica: tensión), es decir, ambas soluciones provocan la
misma presión del solvente por lo que existe un equilibrio de las fuerzas por unidad de área.
En el caso de tener una disolución dada, la cual se pone en contacto con una disolución
externa de mayor concentración, el solvente migrará de la disolución hacia el exterior. Esta
solución externa se llamara solución hipertónica porque posee mayor concentración que la
solución en estudio. En el caso de la disolución más diluida se llamará disolución hipotónica.
Figura 5. Una solución en
contacto con otra en estudio,
puede tener una
concentración igual (solución
isotónica), mayor (solución
hipertónica) o menor
(solución hipotónica) que la
solución estudiada.

Centro Interdisciplinario de Líquidos Iónicos Página 13
Estos conceptos se aplican en medicina,
ya que a las personas enfermas deben
agregar suero que sea isotónico a la
presión osmótica ejercida por lo glóbulos
de la sangre. Lo mismo ocurre en las
plantas, ya que las vacuolas pueden
llenarse o vaciarse de agua dependiendo
del medio externo que se agregue. Esto se
puede ilustrar de mejor forma en la figura
del lado.
Conexión con el lenguaje. Investiga los
nombres de los fenómenos ocurridos
entre soluciones isotónicas, hipertónicas e
hipotónicas en células animales y
vegetales, confecciona un tríptico y
preséntalo a tu curso.
A continuación, trabajaremos los
conceptos necesarios para calcular la
presión osmótica en un sistema.
Cálculos de presión osmótica
Es muy importante cuantificar la presión osmótica, ya que debemos saber si estamos en
presencia de soluciones isotónicas, hipertónicas o hipotónicas.
Para esto debemos hacer uso de la ecuación de Van´t Hoff, quien recibió el premio nobel de
Química en 1901 por su descubrimiento de las leyes de la dinámica química y de la presión
osmótica en disoluciones.
La presión osmótica (π), de una disolución es la presión que se requiere para detener la
osmosis. La presión osmótica de una disolución está dada por: π = M R T
Donde M corresponde a la molaridad de la disolución, R es la constante de los gases (0,082 L
atm/mol K) y T la temperatura absoluta (K). La presión osmótica, π se expresa en atm. En el
caso de esta propiedad, se trabaja con molaridad, ya que las mediciones se realizan a
temperatura constante, por lo que no es necesario utilizar la molalidad. La presión osmótica
puede ser medida aplicándose una presión externa que bloquee la ósmosis.

Centro Interdisciplinario de Líquidos Iónicos Página 14
Ejercicio práctico.
Presión osmótica en la sangre.
La presión osmótica en la sangre es de 7,7 atm a 25 ºC. ¿Qué concentración de glucosa será
isotónica a la sangre?
Paso 1. Organizar la información entregada. Como nos dan la presión osmótica y la
temperatura, podemos despejar la concentración de la ecuación de Van´t Hoff.
Paso 2. Escoger la expresión matemática. π = M R T
Paso 3. Sustituir los datos en la expresión matemática y resolver el problema:
M = π / R T = 7,7 atm /(0,082 L atm K-1 mol-1) (298 K) = 0,31 M
Comentario: En situaciones clínicas, las concentraciones de las disoluciones generalmente se
expresan en términos de porcentaje en masa. El porcentaje en masa de una disolución 0,31 M
de glucosa es 5,3 % m/m.
5. Presión de vapor
Si tienes un líquido al aire libre podrías observar dos fenómenos, por un lado el líquido podría
evaporarse hasta pasar todo al estado gaseoso, o bien, se mantiene en el estado líquido. Esto
se puede observar claramente con los siguientes líquidos a temperatura ambiente, si derramas
acetona, ésta se evaporará luego de un tiempo, pero si derramas bencina, ésta se mantendrá
sin evaporarse. ¿A qué se deben estas diferencias?
Se plantea que algunos líquidos, llamados volátiles, sufren un fenómeno en la superficie de
éstos. Este fenómeno consiste en establecer un equilibrio entre el estado líquido y gaseoso, es
decir, algunas moléculas se evaporan y otras se condensan. Esto ocurre ya que las moléculas
de la superficie ejercen una presión para pasar al estado gaseoso, esta presión se llama
presión de vapor (presión a la cual un líquido en estado puro y su vapor están en equilibrio a
una determinada temperatura. Se incrementa cuando la temperatura aumenta, y viceversa).
La presión de vapor es una medida del número de moléculas que escapan de la superficie del
líquido por unidad de área. Por lo tanto, diferentes líquidos con distintas fuerzas de atracción
entre sus moléculas, tendrán una presión de vapor característica.
De acuerdo a la presión de vapor, se habla de líquidos volátiles y no volátiles. Los líquidos
volátiles pasan con facilidad al estado de vapor debido a que las fuerzas de atracción entre sus
moléculas son muy débiles; por lo tanto, estos tienen una alta presión de vapor. Ejemplos de
líquidos volátiles son el alcohol y la acetona. De esta información deducimos que la presión de
vapor de un líquido es inversamente proporcional a la fuerza de atracción entre las moléculas
que conforman. En la figura 6, se muestra un diagrama de la presión de vapor de un líquido
volátil.

Centro Interdisciplinario de Líquidos Iónicos Página 15
Figura 6. Las moléculas de un líquido en la superficie tienden
a escapar al estado gaseoso. Mientras mayor sea la presión
de vapor de un líquido, mayor tendencia a evaporarse tendrá.
La presión de vapor tiene directa relación con la temperatura, ya que al aumentar la
temperatura, aumentamos la energía cinética de las moléculas, por lo tanto la cantidad de
colisiones, entonces la presión de vapor aumenta. Cuando la presión de vapor de un líquido
alcanza la presión atmosférica, se establece que el líquido comienza a ebullir, es decir, todas
las moléculas del líquido, las de la superficie y del seno del líquido, pasan al estado gaseoso.
La presión de vapor se mide en unidades de presión, es decir, en atm, mmHg, torr o Pa, donde
la equivalencia entre cada una de estas unidades es la siguiente: 1 atm = 760 mmHg = 760 torr
= 101.325 Pa
Disminución de la presión de vapor
¿Qué ocurre si se agrega un soluto no
volátil a este líquido volátil?
Figura 7. En la imagen de la izquierda
se muestra el equilibrio entre el
estado líquido-gaseoso del solvente
puro, y a la derecha se observa el
mismo solvente pero ahora con un
soluto disuelto.
Al observar la figura 7, podemos ver que la diferencia entre ambas imágenes es el número de
moléculas del solvente por unidad de área en la superficie. Es lógico pensar que, a una misma
temperatura, un solvente puro deja escapar mayor número de moléculas por unidad de área
que una disolución formada por este solvente y un soluto no volátil, tal como se muestra en la
figura 6.
Por lo tanto una menor cantidad de moléculas de solvente podrá pasar al estado gaseoso, en
presencia de un soluto. Entonces, la presión de vapor de un líquido puro es siempre mayor
que la de su disolución, con un soluto no volátil, ambos a la misma temperatura.

Centro Interdisciplinario de Líquidos Iónicos Página 16
Ley de Raoult
Cuando se habla de un soluto cuya presión de vapor no se puede medir, es decir, es no volátil, la
presión de vapor de sus disoluciones siempre es menor que la del disolvente puro, por lo que la
relación entre la presión de vapor de la disolución y la presión de vapor del disolvente puro
depende de la concentración del soluto de la disolución. La relación que se genera se expresa
mediante la Ley de Raoult, planteada por Francois Raoult en 1866, la que establece lo siguiente:
“La presión parcial de un disolvente puro, P0
1, multiplicada por la fracción molar del disolvente en la
disolución, X1”: P1 = X1 P0
1
En una disolución que contenga sólo un soluto X1 = 1 – X2, donde X2 es la fracción molar del soluto.
Por lo tanto, la ecuación anterior se puede reescribir como: P1 = (1 - X2) P0
1
o bien, P1 = P0
1 - X2 P0
1
Se tiene finalmente: P0
1 - P1 = ∆P =X2 P0
1
De la última ecuación se puede observar que la disminución de la presión de vapor, ∆P, es
directamente proporcional a la concentración del soluto (la que es medida en fracción molar).
Si ambos componentes de una disolución son volátiles (es decir sus presiones de vapor se pueden
medir), la presión de vapor de la disolución es la suma de las presiones parciales individuales. La
ley de Raoult, también se cumple en este caso:
PA = XA P0
A
PB = XB P0
B
Recordar que por definición:
Donde n1 y n2 son los números de moles del disolvente y del soluto, respectivamente.
Debido a que la fracción molar de la urea en esta disolución es sólo de 0,033, la disolución
es diluida y se supone que n1 es mucho mayor que n2, por tanto se escribe:

Centro Interdisciplinario de Líquidos Iónicos Página 17
Donde PA y PB son las presiones parciales de los componentes A y B de la disolución; P0
A y P0
B son
las presiones de vapor de las sustancias puras XA y XB son sus fracciones molares. La presión total
está dada por la Ley de Dalton de las presiones parciales:
PT = PA + PB
PT = XA P0
A + XB P0
B
La Historia de la Osmosis Inversa
El fenómeno de la ósmosis inversa fue descrito por vez primera por Sir Charles E. Reid en el año
1953 para obtener agua potable procedente del agua del mar y constituir una fuente de agua
potable. La propuesta de Sir Reid fue sometida a consideración por la Oficina de Aguas Salinas de
EEUU y aparecieron algunas objeciones a la idea. En una de ellas se consideraba a la ósmosis
inversa como un proceso no práctico o de difícil logro y que en caso de funcionar, sería tan solo
una curiosidad a modo de experimento didáctico de laboratorio.
La controversia más dura a la propuesta de Reid fue cuando se demostró que se carecía, por aquel
entonces, de una membrana de características y calidad adecuada para efectuar de forma
eficiente el proceso de ósmosis inversa tal como se había descrito. Efectivamente, al aprobarse el
proyecto de Sir Reid se determinó que era un problema enorme el hecho de conseguir una
membrana osmótica que tuviera las capacidades físicas y químicas de realizar ese proceso sin
sufrir alteraciones en su naturaleza ni interferir químicamente con el disolvente. Las dificultades
técnicas aludidas y las objeciones básicamente fueron las siguientes:

Centro Interdisciplinario de Líquidos Iónicos Página 18
1) La carencia de una membrana lograda que resistiera de manera estable químicamente las
soluciones salinas sin degradarse.
2) Las membranas logradas en la época eran muy poco porosas o inconsistentes como para
permitir el tránsito del solvente, que se trataba de agua pura y demasiado abierta en el poro para
tener un bajo rechazo de sales disueltas.
3) La rápida saturación de la membrana osmótica requería de una limpieza constante o lavado
tangencial periódico o lo que resultaba más grave, su sustitución.
La solución al problema de la separación del agua pura a partir de agua marina o aguas salobres,
fue resuelta con éxito por el descubrimiento de una membrana de acetato de celulosa en el año
1959 y su subsiguiente producción en serie.
Uno de los mayores logros en el terreno de los polímeros de la década de los años sesenta, fue
hacer económica y rentable la aplicación de la ósmosis inversa en la obtención de agua potable a
partir de aguas salobres o para mejorar el agua potable doméstica y entrar en competencia con
otros sistemas de purificación como era la destilación y otras filtraciones mecánicas. Esta
posibilidad fue consecuencia directa del perfeccionamiento de la membrana de acetato de
celulosa y una comprensión más clara de los procesos físicos en una solución salina con presencia
de dichas membranas.
Uso de solventes orgánicos en adhesivos
El uso de adhesivos en base disolvente orgánico en la industria del calzado, a pesar de los
beneficios que aporta, conlleva una serie de riesgos, entre los que se pueden destacar sus
repercusiones medioambientales y los efectos perniciosos para la salud humana. Los
inconvenientes más importantes que presentan estos adhesivos derivan de la inflamabilidad y
nocividad de los disolventes orgánicos, fundamentalmente el n-hexano, causante de una
polineuropatía desmielinizante, la cual ha llegado a la opinión pública creando una fuerte alarma
social.
Dicha polineuropatía ha sido considerada enfermedad profesional y se caracteriza por una pérdida
de fuerza progresiva en las extremidades. Entre los factores que favorecen la aparición de esta
enfermedad destaca la ausencia de medidas de protección en el lugar de trabajo, ni individual
(mascarillas, guantes, etc.), ni colectiva (cabinas de extracción), alta temperatura en el local de
trabajo, mala ventilación y posiblemente exceso de fuentes de emisión de disolventes orgánicos
nocivos.

Centro Interdisciplinario de Líquidos Iónicos Página 19
El uso de adhesivos con disolventes orgánicos no sólo origina problemas para la salud humana sino
que también repercute en el medio ambiente. Los compuestos orgánicos volátiles (COVs)
presentes en la atmósfera intervienen en el proceso de formación de niebla contaminante (smog),
procediendo algunos de ellos de la emisión de disolventes orgánicos al medio ambiente en
determinadas actividades industriales.
En la actualidad, los adhesivos en base acuosa junto con los adhesivos 100% sólidos o
termofusibles, son las principales alternativas a la tecnología de los adhesivos basados en
disolventes orgánicos.

Centro Interdisciplinario de Líquidos Iónicos Página 20
Parte II. Propiedades Coligativas y Química Verde.
1. Introducción.
Alrededor del 70% de las fuentes de los compuestos orgánicos volátiles (COV) es natural, siendo la
mayor parte alcanos naturales. El resto es producido por el hombre, correspondiendo las mayores
proporciones a productos aromáticos y alcanos antropogénicos.
El tratamiento apropiado de las emisiones de COV es crucial para comprender las reacciones
químicas en la atmósfera. Por ejemplo, los COV contribuyen a la formación de ozono fotoquímico
en escala regional y urbana. Ellos también juegan un rol en la depositación ácida, ya que
contribuyen a la generación de los radicales responsables de la conversión de los óxidos de azufre
en ácido sulfúrico. Similar situación se da con respecto a los óxidos de nitrógeno y el ácido nítrico,
así como en la formación de peróxidos que influyen en las formaciones ácidas de las nubes.
Algunos COV llegan incluso a fases de transformación en aerosoles, con su consiguiente
disminución de visibilidad.
Los COV más frecuentes son el metano, etano, propano, acetileno, alcanos, bencenos y solventes,
entre otros. El hombre es responsable de las emisiones de COV cuando produce, refina o
distribuye masivamente el petróleo y el gas natural. También genera emisiones cuando utiliza
carbón o leña, solventes, transportes y quema basura.
Las fuentes de emisión de amoníaco son los suelos naturales, los animales salvajes y domésticos,
la quema de biomasa, la combustión de carbón, la aplicación y producción de fertilizantes, los
océanos y mares, así como la excreta humana, el lodo de basural, los automóviles y los cultivos
agrícolas.
2. Principios de la Química Verde.
La Química Verde o Sostenible corresponde al diseño de productos químicos y procesos
que reducen o eliminan el uso y generación de sustancias nocivas o contaminantes. Este término
introducido por Anastas (Anastas, 1998) describe los esfuerzos de los químicos para desarrollar
procesos y productos que prevengan la contaminación y que sean seguros tanto para los seres
humanos como para el medio ambiente. El diseño de productos amigables con el medioambiente
se guía por los doce principios de la química sostenible enunciados por Anastas y Warner.

Centro Interdisciplinario de Líquidos Iónicos Página 21
Los 12 Principios de la Química Verde.
1. Es mejor prevenir la formación de residuos que limpiarlos una vez formados. (Prevención).
2. Los métodos sintéticos deben diseñarse para maximizar la incorporación en el producto
final de todos los materiales usados en el proceso. (Economía atómica).
3. Siempre que sea posible, deben diseñarse metodologías sintéticas que usen y generen
sustancias que no sean tóxicas para la salud y el medio ambiente. (Métodos de síntesis
menos peligrosos).
4. Los productos químicos deben diseñarse para mantener la eficacia de su función, pero
reduciendo la toxicidad. (Diseño de productos más seguros).
5. El uso de sustancias auxiliares (por ejemplo, disolventes, agentes de separación, etc)
debería ser innecesario en la medida de lo posible e inocuo cuando sean necesarios.
(Disolventes y auxiliares más seguros).
6. Los requerimientos energéticos deben ser tenidos en cuenta debido a su impacto
medioambiental y económico, y deben ser minimizados. Los métodos sintéticos deben
realizarse a temperatura ambiente cuando sea posible. (Eficacia energética).
7. Las materias primas deben ser renovables cuando sea posible técnica y económicamente.
(Uso de materias primas renovables).
8. Debe evitarse el uso y generación de derivados (grupos bloqueantes,
protección/desprotección, modificación temporal de las condiciones físicas/químicas)
cuando sea posible. (Reducir el uso de derivados).
9. Los reactivos catalíticos (tan selectivos como sea posible) son mejores que los reactivos
estequiométricos. (Catálisis).
10. Los productos químicos deben diseñarse de manera que su función no persista en el
medio ambiente y degradarse a productos inocuos. (Diseño para la degradación).
11. Necesidad de desarrollo de metodologías analíticas que permitan analizar, monitorear y
controlar previamente a la formación de sustancias peligrosas. (Análisis en tiempo real).
12. Deben escogerse las sustancias y la forma de una sustancia utilizada en un proceso
químico de manera que se minimice el potencial de accidentes químicos, incluyendo
escapes, explosiones e incendios. (Síntesis químicas más seguras).

Centro Interdisciplinario de Líquidos Iónicos Página 22
De los principios de la Química Verde, se observa que el N° 5 sugiere evaluar el uso de
sustancias auxiliares (disolventes, reactivos de separación, agentes secantes, etc.) que se emplean
en las reacciones y las síntesis. De la experiencia empírica, se ha visto que los residuos generados
por estas sustancias auxiliares son significativos y en ocasiones supera la cantidad de desperdicio
generado directamente por la reacción. Es por ello que se sugiere no usar sustancias auxiliares.
Muchas reacciones orgánicas utilizan grandes cantidades de disolventes que con
frecuencia son tóxicos. Muchos de ellos finalizan en el agua, suelo y aire dando como resultado
una contaminación significativa del medio ambiente. En ese sentido, los esfuerzos se dirigen a
reemplazar los disolventes orgánicos por otros menos peligrosos, o definitivamente, no utilizar
disolventes sino que reemplazar “el medio” por otro, por ejemplo, energía.
El principio 6 considera las necesidades energéticas de una reacción. Es preferible llevar a
cabo reacciones a presión y temperatura ambientes, aunque muchas reacciones químicas
requieren calor y/o frío y presiones distintas a las ambientales. Esto supone el uso de una fuente
de energía y la mayoría de las veces esta energía procede de un combustible fósil.
El principio 9, los reactivos usados en cantidades catalíticas son preferibles a los usados en
cantidades estequiométricas. Sin embargo, aunque se usen cantidades estequiométricas, siempre
que sea posible, se deben realizar procesos de recuperación, reciclado y reutilización de los
productos no deseados. En ese sentido, recientemente se han logrado grandes progresos
desarrollando reacciones que son promovidas por catalizadores no tóxicos y recuperables.
Muchos de los principios antes destacados están en directa relación con los conceptos que
se tratarán a continuación, Principalmente el uso de sustancias auxiliares como solventes,
incremento de presión o temperatura.
3. Compuestos Orgánicos Volátiles (COV)
Los compuestos orgánicos volátiles (COV) son todos aquellos hidrocarburos que se presentan en
estado gaseoso a la temperatura ambiente normal o que son muy volátiles a dicha temperatura.
Se puede considerar como COV aquel compuesto orgánico que a 20ºC tenga una presión de vapor
de 0.01 kPa o más, o una volatilidad equivalente en las condiciones particulares de uso.
Suelen presentar una cadena con un número de carbonos inferior a doce y contienen otros
elementos como oxígeno, flúor, cloro, bromo, azufre o nitrógeno. Su número supera el millar, pero
los más abundantes en el aire son metano, tolueno, n-butano, i-pentano, etano, benceno, n-
pentano, propano y etileno. Tienen un origen tanto natural (COV biogénicos) como antropogénico
(debido a la evaporación de disolventes orgánicos, a la quema de combustibles, al transporte,
etc.).

Centro Interdisciplinario de Líquidos Iónicos Página 23
Con respecto a su peligrosidad los COV pueden clasificarse en 3 grupos:
Compuestos extremadamente peligrosos para la salud: Benceno, cloruro de vinilo y 1,2
dicloroetano.
Compuestos clase A: los que pueden causar daños significativos al medio ambiente, como por
ejemplo: acetaldehído, anilina, tricloroetileno, etc.
Compuestos clase B: tienen menor impacto en el medio ambiente. Pertenecen a este grupo, entre
otros, acetona y etanol.
La presencia de los COV está fundamentalmente influenciada por actividades en las que se
empleen disolventes orgánicos. Algunas de las actividades donde es posible que se den emisiones
de COV son:
Pinturas y barnices (e industrias donde se usen éstos)
Industria siderúrgica
Industria de la madera
Industria cosmética
Industria farmacéutica
Los COV afectan tanto de manera medioambiental como directamente sobre la salud del ser
humano. En primer lugar, algunos COV son destructores del ozono, como el tetracloruro de
carbono, por tanto son compuestos que afectan al fenómeno de disminución de la capa de ozono.
Además, los COV en conjunto con los óxidos de nitrógeno y la luz solar, son precursores del ozono
a nivel de suelo (ozono tropósferico) que es perjudicial para la salud provocando daños
respiratorios. Se puede producir el llamado smog fotoquímico que es una niebla de color marrón-
rojizo.
Con respecto a daños directos sobre la salud, estos se producen principalmente por vía
respiratoria aunque también pueden entrar a través de la piel. Además estos compuestos son
liposolubles por lo que se bioacumulan en las grasas de los organismos vivos.
Como efectos que pueden producir están problemas respiratorios, irritación de ojos y garganta,
mareos, etc. También se pueden dar efectos psiquiátricos (irritabilidad, dificultad de
concentración, etc.). Además a largo plazo pueden causar daños renales, al hígado o al sistema
nervioso central o algunos COV tienen efecto cancerígeno como por ejemplo el benceno.

Centro Interdisciplinario de Líquidos Iónicos Página 24
4. ¿Qué efectos pueden provocar los COV en la Salud humana?
Los disolventes clorados se absorben fácilmente a través del tracto digestivo (por ingestión) y los
pulmones (por inhalación). Una vez absorbidos, se mueven por todo el organismo a través de la
sangre. Durante un corto periodo de tiempo, pueden acumularse en el hígado, los riñones, el
cerebro y los tejidos adiposos. En el hígado, los disolventes clorados se transforman en otras
sustancias y finalmente se eliminan del organismo. Generalmente, la mayoría de estas sustancias
se eliminan del organismo en pocos días tras el fin de la exposición.
Se comprobó que niveles elevados de disolventes clorados provocan mareos, disminuyen la
capacidad de concentrarse y recordar, dañan el sistema nervioso, y producen un pulso irregular en
las personas que están expuestas a estas sustancias en el lugar de trabajo y en los animales de
laboratorio. Algunos disolventes clorados (tricloroetileno, tetracloroetileno, cloruro de metileno,
tetracloruro de carbono, cloruro de vinilo y 1,2-dicloroetano) han provocado cáncer en animales
de laboratorio expuestos a dosis elevadas. El cloruro de vinilo también ha provocado cáncer de
hígado en personas que han utilizado esta sustancia química en el trabajo.
Los componentes de combustibles se absorben fácilmente a través de los pulmones (por
inhalación) y del tracto digestivo (por ingestión). Estas sustancias químicas se transportan
rápidamente por todo el organismo a través de la sangre, principalmente al cerebro y al sistema
nervioso. Los componentes de combustibles también pueden acumularse temporalmente en los
tejidos adiposos, la médula ósea, el hígado y los riñones. El hígado transforma estas sustancias
químicas en otras sustancias (por ejemplo, el benceno se transforma en fenol) para que puedan
eliminarse mediante la orina.
En dosis elevadas, los componentes de combustibles pueden provocar somnolencia, mareos y
cefaleas. La exposición durante un largo periodo de tiempo a niveles elevados de tolueno o xileno
puede provocar daños en el hígado y los riñones. El benceno es el componente de combustible
más tóxico y puede afectar gravemente a los glóbulos rojos. Los trabajadores industriales
expuestos a niveles elevados de benceno en el aire corrían un mayor riesgo de padecer anemia y
de tener una cantidad inferior de leucocitos que otros trabajadores que no estaban expuestos.
Los trabajadores industriales tenían más probabilidades de padecer leucemia, un tipo de cáncer de
los leucocitos, en comparación con otros trabajadores. También se dispone de información
limitada que sugiere que el benceno puede dañar los fetos o provocar un aborto. Algunas
personas expuestas a niveles elevados de MTBE en el aire han reportado irritación en la nariz y la
garganta. Se detectaron daños en los riñones y el hígado en animales de laboratorio expuestos a
niveles elevados de MTBE. La exposición de animales a niveles muy elevados de MTBE ha
provocado tumores en diversos órganos del cuerpo.

Centro Interdisciplinario de Líquidos Iónicos Página 25
Puesto que muchas de estas sustancias químicas aún no se han testeado adecuadamente en
niveles de exposición bajos, no se dispone de información suficiente acerca de los efectos
perjudiciales de los COV en el agua potable y en el aire. Normalmente, la exposición a los COV en
el agua potable contaminada ha sido muy inferior a las cantidades a las que se han expuesto los
trabajadores y los animales de laboratorio.
Según cifras entregadas por la Organización Internacional del Trabajo (OIT) se estima que el 22%
de los 2 millones de muertes laborales que ocurren al año en el mundo se produce por la
exposición de trabajadores a productos químicos. Una clase de estos son los Compuestos
Orgánicos Volátiles (COVs), los cuales son incorporados por los trabajadores a través de las vías
aéreas, es decir, el aparato respiratorio. En Chile el Decreto Supremo Nº 594 regula la
concentración ambiental de 213 compuestos químicos en ambientes laborales, de los cuales el
30% corresponden a COVs. Estos compuestos presentan un alto nivel de riesgo para la salud, y se
les ha asociado como precursores de enfermedades neurotóxicas, nefrotóxicas y hepatotóxicas,
así como de la aparición de cáncer en las personas expuestas.
Diversos estudios indican que entre un 4 y un 40% de los casos de cáncer detectados tienen su
origen en las condiciones laborales del enfermo (por ejemplo la asociación entre exposición a
benceno y leucemia) y entre un 15 a 30% de las licencias médicas que se presentan en Chile
pueden ser asociadas a distintos niveles de intoxicación por compuestos químicos, incluidos COVs.
Respecto del problema de la exposición de COV en ambientes laborales, científicos indican que en
las empresas se hacen algunas mediciones, pero los métodos utilizados son caros, las técnicas de
monitoreo son rudimentarias, poco específicas y la periodicidad es muy espaciada (una vez al
año), por lo que la información que se puede obtener de eso es bastante pobre y no permite
realizar las medidas de prevención y mucho menos de mitigación necesarias para proteger la salud
de los trabajadores.
5. ¿De dónde se producen los COV?
Los Compuestos orgánicos volátiles, al ser principalmente cadenas hidrocarbonadas, el origen de
estos se debe principalmente a la industria del petróleo. Razón por la cual se hace necesario
comprender el proceso por medio del cual es posible obtener diversos compuestos a partir de la
destilación de los distintos componentes del petróleo.
La destilación del petróleo se realiza mediante las llamadas, torres de fraccionamiento. En esta, el
petróleo asciende por la torre aumentando su temperatura, obteniéndose los derivados de este
en el siguiente orden:
1. Residuos sólidos / 2. Aceites y lubricantes / 3. Gasóleo y fuel / 4. Querosen / 5. Naftas /
6. Gasolinas / 7. Disolventes / 8. GLP (Gases licuados del petróleo)

Centro Interdisciplinario de Líquidos Iónicos Página 26
Si hay un excedente de un derivado del petróleo de alto peso molecular, pueden romperse las
cadenas de hidrocarburos para obtener hidrocarburos más ligeros mediante un proceso
denominado craqueo.
6. Proceso de refinación del Petróleo
El petróleo finalmente llega a las refinerías en su estado natural para su procesamiento. Aquí
prácticamente lo que se hace es cocinarlo. Por tal razón es que al petróleo también se le denomina
"crudo".
Una refinería es un enorme complejo donde ese petróleo crudo se somete en primer lugar a un
proceso de destilación o separación física y luego a procesos químicos que permiten extraerle
buena parte de la gran variedad de componentes que contiene. El petróleo tiene una gran
variedad de compuestos, al punto que de él se pueden obtener por encima de los 2.000
productos. El petróleo se puede igualmente clasificar en cuatro categorías: parafínico, nafténico,
asfáltico o mixto y aromático.
Los productos que se sacan del proceso de refinación se llaman derivados y los hay de dos tipos:
los combustibles, como la gasolina, ACPM, etc.; y los petroquímicos, tales como polietileno,
benceno, etc. Las refinerías son muy distintas unas de otras, según las tecnologías y los esquemas
de proceso que se utilicen, así como su capacidad. Las hay para procesar petróleos suaves,
petróleos pesados o mezclas de ambos. Por consiguiente, los productos que se obtienen varían de
una a otra.
La refinación se cumple en varias etapas. Es por esto que una refinería tiene numerosas torres,
unidades, equipos y tuberías. Es algo así como una ciudad de plantas de proceso. En términos
sencillos, el funcionamiento de una refinería se cumple de la siguiente manera: El primer paso de
la refinación del petróleo crudo se cumple en las torres de "destilación primaria" o "destilación
atmosférica". En su interior, estas torres operan a una presión cercana a la atmosférica y están
divididas en numerosos compartimientos a los que se denominan "bandejas" o "platos". Cada
bandeja tiene una temperatura diferente y cumple la función de fraccionar los componentes del
petróleo. El crudo llega a estas torres después de pasar por un horno, donde se "cocina" a
temperaturas de hasta 400 grados centígrados que lo convierten en vapor. Esos vapores entran
por la parte inferior de la torre de destilación y ascienden por entre las bandejas.
A medida que suben pierden calor y se enfrían. Cuando cada componente vaporizado encuentra
su propia temperatura, se condensa y se deposita en su respectiva bandeja, a la cual están
conectados ductos por los que se recogen las distintas corrientes que se separaron en esta etapa.
Al fondo de la torre cae el "crudo reducido", es decir, aquel que no alcanzó a evaporarse en esta
primera etapa. Se cumple así el primer paso de la refinación.

Centro Interdisciplinario de Líquidos Iónicos Página 27
De abajo hacia arriba se han obtenido, en su orden: gasóleos, acpm, queroseno, turbosina, nafta y
gases ricos en butano y propano. Algunos de estos, como la turbosina, queroseno y acpm, son
productos ya finales. Las demás corrientes se envían a otras torres y unidades para someterlas a
nuevos procesos, al final de los cuales se obtendrán los demás derivados del petróleo.
Así, por ejemplo, la torre de "destilación al vacío" recibe el crudo reducido de la primera etapa y
saca gasóleos pesados, bases parafínicas y residuos. La Unidad de Craqueo Catalítico o Cracking
recibe gasóleos y crudos reducidos para producir fundamentalmente gasolina y gas propano. Las
unidades de Recuperación de Vapores reciben los gases ricos de las demás plantas y sacan gas
combustible, gas propano, propileno y butanos.
La planta de mezclas es en últimas la que recibe las distintas corrientes de naftas para obtener la
gasolina motor, extra y corriente. La unidad de aromáticos produce a partir de la nafta: tolueno,
xilenos, benceno, ciclohexano y otros petroquímicos. La de Parafinas recibe destilados parafínicos
y nafténicos para sacar parafinas y bases lubricantes.
De todo este proceso también se obtienen azufre y combustóleo. El combustóleo es lo último que
sale del petróleo. Es algo así como el fondo del barril. En resumen, el principal producto que sale
de la refinación del petróleo es la gasolina motor. El volumen de gasolina que cada refinería
obtiene es el resultado del esquema que utilice. En promedio, por cada barril de petróleo que
entra a una refinería se obtiene 40 y 50 por ciento de gasolina. El gas natural rico en gases
petroquímicos también se puede procesar en las refinerías para obtener diversos productos de
uso en la industria petroquímica.
La destilación del petróleo se realiza mediante las llamadas, torres de fraccionamiento. En esta, el
petróleo asciende por la torre aumentando su temperatura, obteniéndose los derivados de este
en el siguiente orden:
1. Residuos sólidos
2. Aceites y lubricantes
3. Gasóleo y fuel
4. Querosen
5. Naftas
6. Gasolinas
7. Disolventes
8. GLP (Gases licuados del petróleo)

Centro Interdisciplinario de Líquidos Iónicos Página 28

Centro Interdisciplinario de Líquidos Iónicos Página 29
Parte III. Trabajo Práctico.
1. Objetivos de la actividad.
• Verificar experimentalmente las relaciones existentes entre la temperatura y la
concentración de las soluciones.
• Analizar la relación de la temperatura y la concentración de las soluciones,
contrastándolos con problemas cotidianos.
• Comprender las relaciones existentes las interacciones moleculares y la presión de vapor.
2. Conceptos asociados en la actividad.
• Concentración molal de Soluciones.
• Propiedades coligativas de Soluciones y la Química Verde.
3. Materiales y reactivos.
Este trabajo práctico se debe realizar en grupos de 3 ó 4 personas. Cada uno de los cuales
contará con una bandeja de materiales necesarios para el desarrollo de la experiencia. Los
reactivos e insumos serán entregados por los docentes, esto con el fin de que dentro de lo posible
que no se generen gastos excesivos para no tener mayores desechos.
Es conveniente tener un mesón con todos los reactivos e insumos, y con los materiales
necesarios para que puedan retirar los insumos del práctico. A su vez, es necesario recalcar que
este trabajo práctico cuenta con tres partes independientes entre sí, razón por la cual se sugiere
comenzar por la que reviste mayor tiempo de preparación inicial.
Materiales y Reactivos.
Bandejas 1 y 2.
- 1 vaso de precipitados de 250 mL.
- 1 Probeta de 100 mL
- Trípode, rejilla y mechero.
- Balanza granataria.
- Bagueta.

Centro Interdisciplinario de Líquidos Iónicos Página 30
Bandejas 3, 4 y 5.
- Sistema de destilación simple.
- 1 Probeta de 100 mL.
Materiales.
- AlK(SO4)3.
- NaCl
- Agua Destilada.
- Metanol.
4. Metodología.
Experimento 1. Preparando una curva de Temperatura v/s de la molalidad.
En este capítulo has estudiado la relación existente entre la temperatura y la concentración de las
disoluciones. Es así como aprendiste en qué consiste la temperatura de fusión y ebullición de un
solvente. Estos temas son importantes en nuestra vida cotidiana. En este laboratorio, deberás
aplicar lo aprendido para verificar experimentalmente la relación entre la temperatura y la
concentración de las disoluciones.
Experimento 1 y 2. Temperatura de ebullición de una disolución.
1. Agregue la cantidad de NaCl sólido que se detalla en la siguiente tabla, en un vaso de
precipitado.
2. En una probeta agregue 100 mL de agua destilada y viértalo en el vaso de precipitado.
3. Comience a calentar el agua destilada del punto 2 hasta la ebullición. Registre la
temperatura cuando esté en ebullición.
4. Repita el mismo procedimiento del punto 3 con cada una de las cantidades mostradas en
la tabla. Tanto para el NaCl y el ALK(SO4)3.
Molalidad Masa de NaCl Masa de AlK(SO4)3
0.0 0 0
0.6 3.51 28.5
0.8 4.68 38.0
1.0 5.58 47.4
1.2 7.02 57.0
2.4 14.0 113.9

Centro Interdisciplinario de Líquidos Iónicos Página 31
Experimento 2. Separación de mezclas puras.
1. Instale el sistema de destilación simple.
2. Agregue 100 mL de agua destilada al balón de destilación.
3. Agregue 100 mL de metanol al balón de destilación.
4. Agregue perlas de ebullición.
5. Comience a calentar la mezcla.
6. Cuando llegue a 50° C, registre la temperatura cada 3 minutos.
Experimento 4. Separación de soluciones.
1. Instale el sistema de destilación simple.
2. Agregue 100 mL de agua destilada al balón de destilación.
3. Agregue 100 mL de metanol al balón de destilación.
4. Agregue 7 g de NaCl a la mezcla del balón.
5. Agregue perlas de ebullición.
6. Comience a calentar la mezcla.
7. Cuando llegue a 50° C, registre la temperatura cada 3 minutos.
Experimento 5. Separación de soluciones.
1. Instale el sistema de destilación simple.
2. Agregue 100 mL de agua destilada al balón de destilación.
3. Agregue 100 mL de metanol al balón de destilación.
4. Agregue 57 g de ALK(SO4)3 a la mezcla del balón.
5. Agregue perlas de ebullición.
6. Comience a calentar la mezcla.
7. Cuando llegue a 50° C, registre la temperatura cada 3 minutos.
Analiza los resultados
Experimento 1 y 2.
Analizar observaciones. Compara los resultados obtenidos en las disoluciones de NaCl, y el agua
pura. Ordena los datos en una tabla.
Tratar datos. Con los datos obtenidos experimentalmente, elabore una grafica de temperatura de
ebullición de la disolución de NaCl en función de la molalidad. A partir de la gráfica y la ecuación

Centro Interdisciplinario de Líquidos Iónicos Página 32
de la recta verifique el valor de la constante de ebullición del agua y la temperatura de ebullición
del agua pura. Recuerde dividir por 2 (para el NaCl) ó 5 (para el ALK(SO4)3) la pendiente de la
gráfica.
Saca conclusiones
Evaluar resultados. ¿Cómo varía la temperatura de ebullición y fusión del solvente, al agregar un
soluto?
Analiza los resultados
Experimento 3, 4 y 5.
Analizar observaciones. Compara los resultados obtenidos entre las tres experiencias de
destilación. Ordena los datos en una tabla.
Tratar datos. Con los datos obtenidos experimentalmente, elabore una grafica de temperatura en
función del tiempo.
Saca conclusiones
Evaluar resultados. ¿Cómo varía la temperatura de ebullición y fusión del solvente, al agregar un
soluto?

Centro Interdisciplinario de Líquidos Iónicos Página 33
Parte IV. Ficha de trabajo.
1. Observaciones sustraídas de la actividad.
2. Explicación de las observaciones.
3. Principio de la Química Verde abordado.
4. Aprendizajes obtenidos. Conclusiones preliminares.
5. Preguntas y Cuestionamientos.

Centro Interdisciplinario de Líquidos Iónicos Página 34
Referencias.
1. Química. La ciencia central T.L. Brown, H.E. LeMay Jr., B.E. Bursten, C.J. Murphy, P. Woodward .
Ed. Pearson, 11ª Edicion, 2009.
2. Química, R. Chang, Ed. Mc Graw Hill, 10ª Edición, 2010.
3. D.M.P. Mingos, D.R. Baghurst, Applications of Microwave Dielectric Heating Effects to Synthetic
Problems in Chemistry, Microwave-Enhanced Chemistry, American Chemical Society,Washington,
DC, USA, 1997.
4. D. R. Baghurst, D. M. P. Mingos, Chem. Soc. Rev., 20, 1991, 1.
5. C. Gabriel, S. Gabriel, E. H. Grant, B. S. Halstead, D. M. P. Mingos, Chem. Soc. Rev., 27, 1998,
213.
6. A. Díaz-Ortiz, A. de la Hoz, A. Moreno,Microwave in Organic Synthesis, (Ed.: A. Loupy, 2nd
Edition), Wiley-VCH, Weinheim, 2006.
7. E. Van der Eycken, C. O. Kappe (Eds), Microwave-Assisted Synthesis of Heterocycles, Series:
Topics in Heterocyclic Chemistry, Springer: New York, 2006.
8. P. Walla, C. O. Kappe, Chem. Commun.,564, 2004.
9. D. M. P. Mingos, Microwave-Asssisted Organic Synthesis, Eds.: P. Lidström, J. P. Tierney,
Blackwell, Oxford, 2005.
10. Z. Zhao, D. D. Wisnoski, S. E. Wolkenberg, W. H. Leister, Y. Wang, C. W. Lindsley, Tetrahedron
Lett., 45,2004, 4873
11. E. Van der Eycken, P. Appukkuttan, W. De Borggraeve, W. Dehaen, D. Dallinger, C. O. Kappe, J.
Org. Chem., 67,2002, 7904.
12. Loupy, A. Spectra Anal, 22, 1993, 175.
13. C. O. Kappe, D. Dallinger, S. S. Murphree, Practical Microwave Synthesis for Organic Chemists—
Strategies, Instruments, and Protocols, Wiley-VCH, Weinheim, 2008,
14. P. T. Anastas, J. C. Warner, Green Chemistry: Theory and Practice; Oxford University Press, New
York, 1988.
15. M. Lancester, Green Chemistry: An Introductory Text; Royal Society of Chemistry, Cambridge,
2002.
16. D. Dallinger, C. O. Kappe, Chem. Rev., 107, 2007, 2563.
17. Ciencias Naturales. Química 2° Medio; Contreras M., Letelier R., Rojas M., Von Marttens H.; Ed.
Mc Graw Hill; 2003.
18. Ciencia Químicas, Educación Media” III, IV; López J.T., Martínez M, Jorquera J., Muñoz M.; Ed.
Santillana; 1995.