formas no clasica de herencia
TRANSCRIPT

Integrantes:
Davila Sasturain Karla Judith
Duarte Siqueiros Diana Laura
Jara Rendon Dulce Alejandra
López Ramírez Esther Margarita
Valdez Ahumada Jennifer
Zambrano Moreno Carlos

*
*La impronta genética o "imprinting" es un fenómeno genético por el que ciertos genes son expresados de un modo específico que depende del sexo del progenitor. Es un proceso biológico por el cual un gen o dominio genómico se encuentra marcado bioquímicamente, indicando su origen parental. Las improntas pueden deberse a uniones covalentes (metilación de ADN) o no covalentes (interacciones ADN-ARN). El proceso de impronta requiere una maquinaria enzimática nuclear

*
*La impronta parental se establece durante la gametogénesis, en la que un cromosoma de cada pareja de homólogos es segregado al espermatozoide o al óvulo; posteriormente, durante la embriogénesis y el desarrollo a adulto, los alelos de los genes improntados se mantienen en sus dos estados epigenéticos"conformacionales": materno o paterno.De esta manera, las improntas genómicas hacen de molde en su propia replicación, son heredables, y pueden ser identificadas mediante análisis molecular, sirviendo como marcadores del origen parental de las regiones genómicas.

** Es consecuencia de una
alteración genética originada por la falla
en la expresión de genes del cromosoma
15. En la etapa de lactancia se caracteriza
por hipotonía y dificultad para succionar, lo
que ocasiona un retraso en el
crecimiento. Posteriormente, durante la
infancia, se produce un retraso en
el desarrollo psicomotor junto
con discapacidad intelectual y problemas
en el comportamiento. La enfermedad
cursa con una deficiencia en la producción
de hormonas del eje hipotalámico-
hipofisario-adrenal,
del crecimiento, gonadotrofinas y tiroideas
, ocasionando obesidad, apetito excesivo,
tendencia a padecer diabetes, alteraciones
en el control de la temperatura, capacidad
baja de sentir dolor, trastornos de la
respiración al dormir, alteraciones
del sueño, junto con otros problemas-


**Un ejemplo clásico de enfermedad genética
cuyo origen y herencia dependen del
mecanismo de impronta genética. Ambas
enfermedades se deben a la ausencia de
expresión de genes que se encuentran en
el mismo locus del cromosoma 15. Una
persona sana recibe dos copias del
cromosoma 15: Una de la madre y otra del
padre. La expresión de los genes que se
encuentran en el locus relacionado con
ambas enfermedades es diferente según se
trate del cromosoma materno o paterno
debido a la epigenética.
* Por tanto, en un individuo sano se expresan
unos genes en el cromosoma materno y
otros en el paterno. Si el locus materno se
pierde o está mutado, se produce el
síndrome de Angelman

*Reacción:
*Retraso en el desarrollo
* Una capacidad lingüística
reducida o nula
*Escasa receptividad
comunicativa
*Escasa coordinación motriz
*Con problemas de equilibrio y
movimiento
*Ataxia
*Estado aparente de alegría
permanente, con risas y sonrisas
en todo momento.
*También se pueden mostrar
fácilmente excitables, con
hipermotricidad y déficit de
atención

*
*Un mosaico genético o mosaicismo es una alteración genética en la que, en un mismo individuo, coexisten dos o más poblaciones de células con distinto genotipo supuestamente originadas a partir de un mismo cigoto. Para ilustrar este fenómeno se suele recurrir al ejemplo de las mujeres, dado que al tener uno de sus cromosomas X inactivados pueden ser consideradas como mosaicos. Este fenómeno de inactivación ocurre en la embriogénesis temprana (alrededor del décimo día de desarrollo) y, a partir de ese momento, todas las células heredan el patrón de cromosoma X inactivado. Las células tumorales son también un tipo de mosaicismo, en este caso patológico.

**Se trata de un síndrome que afecta a la piel y en muchos de los casos al
sistema nervioso, presentando manchas con poco pigmento distribuidas en el
cuerpo y con frecuencia manifestaciones neurológicas como crisis convulsivas,
retraso mental, entre otras.
*¿QUE LA CAUSA ? Aunque permanece bajo estudio, se considera que la
Hipomelanosis de Ito se presenta a consecuencia de la presencia de
mosaicos cromosómicas en personas de piel oscura. Se llama mosaico
cuando no todas las células del organismo presentan la misma configuración de
cromosomas, por lo que unas células pueden presentar la configuración para
una piel oscura y otras para una piel de un tono más claro.


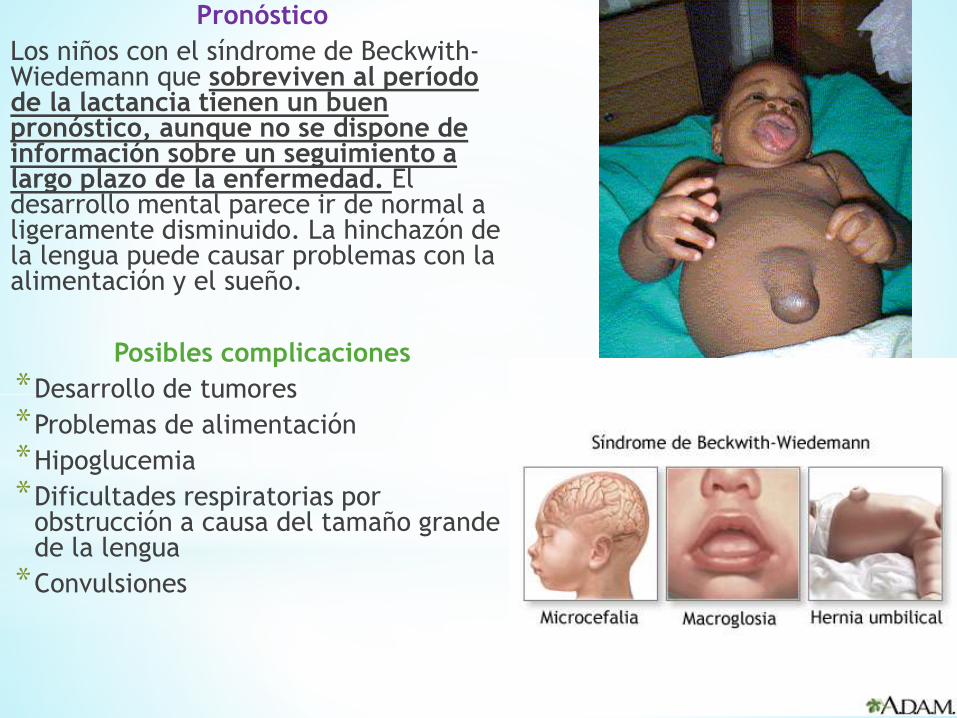
*Síndrome de Beckwith-
WiedemannEs un trastorno congénito (presente al
nacer) del crecimiento que provoca un
tamaño corporal grande, órganos
grandes y otros síntomas
Causas
La causa del síndrome de Beckwith-
Wiedemann se desconoce, pero parece
tener origen genético. La mayoría de los
casos pueden estar asociados con un
defecto en el cromosoma número 11.
La lactancia puede ser un período crítico
debido al azúcar bajo en la sangre
(hipoglucemia ), onfalocele y a un aumento
en la tasa de desarrollo de tumores. El
tumor de Wilms y el carcinoma suprarrenal
son los tumores más comunes en pacientes
con este síndrome.

Pronóstico
Los niños con el síndrome de Beckwith-Wiedemann que sobreviven al período de la lactancia tienen un buen pronóstico, aunque no se dispone de información sobre un seguimiento a largo plazo de la enfermedad. El desarrollo mental parece ir de normal a ligeramente disminuido. La hinchazón de la lengua puede causar problemas con la alimentación y el sueño.
Posibles complicaciones
*Desarrollo de tumores
*Problemas de alimentación
*Hipoglucemia
*Dificultades respiratorias por obstrucción a causa del tamaño grande de la lengua
*Convulsiones

*
*La mayor parte del material genético se encuentra en los cromosomas en el interior del núcleo de la célula, pero las mitocondrias, unos orgánulos del interior celular que producen la energía que se utiliza en el metabolismo, también contienen una pequeña cantidad de ADN denominado ADN mitocondrial. Las alteraciones del material genético de las mitocondrias son la causa de algunas enfermedades que se transmiten con un patrón característico debido a que las mitocondrias solo se heredan de la madre. Todos los hijos e hijas de una mujer afectada heredarán las mitocondrias con la mutación y serán afectados por la enfermedad(Figura 1) mientras que ninguno de los hijos e hijas de un hombre afectado heredaran la alteración ni desarrollaran la enfermedad.(Figura 2).
Figura 1
Figura 2


*
*La Neuropatía óptica hereditaria de Leber (NOHL) o Atrofia óptica de Leber es una degeneración de los gangliocitos de la retina y sus axones, heredada mitocondrialmente (de la madre a todos sus hijos), que conlleva una pérdida aguda o subaguda de visión central. Esto afecta predominantemente a varones adultos jóvenes. Sin embargo la NOHL sólo se transmite a través de la madre ya que se debe principalmente a mutaciones en el genoma mitocondrial (no el nuclear) y sólo el óvulo aporta mitocondrias al embrión. La NOHL se debe habitualmente a una de tres posibles mutaciones puntuales patogénicas en el ADN mitocondrial. Estas mutaciones afectan a los nucleótidos de las posiciones 11.778, 3.460 y 14.484, respectivamente en los genes de las subunidades ND4, ND1 y ND6, del complejo I de la cadena de transporte de electrones en las mitocondrias. Los hombres no pueden transmitir esta enfermedad a sus hijos.


*
*En función de la tasa de mutación -
que el genoma humano es muy
variable- se pueden distinguir 2 tipos
de mutación: mutación estática y
mutación dinámicas. En la secuencia
originada tras la mutación es la misma
tanto para la secuencia originada tras
la mutación dinámicas, que ocurren
en el ADN repetitivo, se caracteriza
por un tipo de mutación: la variación
es el numero de copias. En este caso,
la tasa de mutación depende del
numero de copias, por lo que la
mutabilidad de una secuencia
organizada por mutación es diferente
de la secuencia de la que proviene.
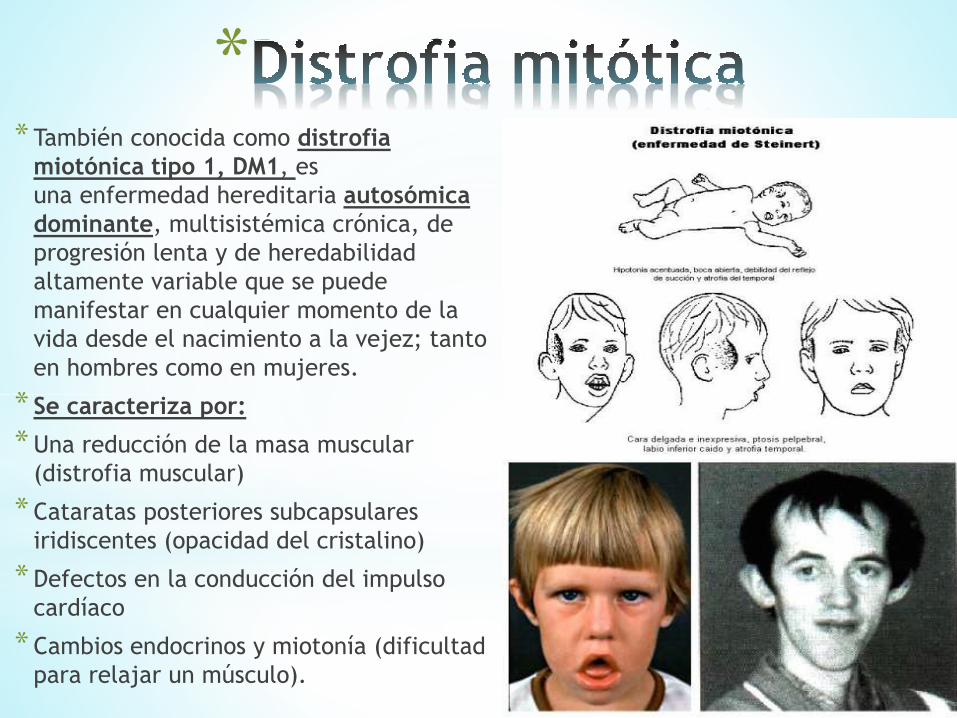
** También conocida como distrofia
miotónica tipo 1, DM1, es
una enfermedad hereditaria autosómica
dominante, multisistémica crónica, de
progresión lenta y de heredabilidad
altamente variable que se puede
manifestar en cualquier momento de la
vida desde el nacimiento a la vejez; tanto
en hombres como en mujeres.
* Se caracteriza por:
*Una reducción de la masa muscular
(distrofia muscular)
* Cataratas posteriores subcapsulares
iridiscentes (opacidad del cristalino)
* Defectos en la conducción del impulso
cardíaco
* Cambios endocrinos y miotonía (dificultad
para relajar un músculo).

*La DM es una enfermedad de
transmisión genética donde está
involucrado un patrón de dominancia
autosómica, significando que hay un gen
mutante de uno de los padres que
resultará en esta condición. Hay un
50% de probabilidad de recibir DM del
progenitor afectado. En la DM1,
el gen afectado es el DMPK (proteína
quinasa de distrofia miotónica), que
codifica la kinasa miosina, expresada en
los músculos esqueléticos. Este gen se
localiza en el brazo largo
del cromosoma 19. La DM2 es
similarmente causada por un defecto
del gen ZNF9 en el cromosoma 3 q21. Patrón de herencia de una
enfermedad autosómica
dominante cuando uno sólo de
los padres es portador
heterocigota


**La enfermedad de Huntington (llamada
también corea de Huntington y conocida
antiguamente como baile de San Vito o
mal de San Vito, al igual que otras
coreas como la corea de Sydenham) es
un trastorno genético hereditario cuya
consideración clínica se puede resumir
en que es un trastorno
neuropsiquiátrico. Sus síntomas suelen
aparecer hacia la mitad de la vida de
la persona que lo padece (unos 30 o
50 años de media) aunque pueden
aparecer antes y los pacientes muestran
degeneración neuronal constante,
progresiva e ininterrumpida hasta el
final de la enfermedad que suele
coincidir con el final de su vida por
demencia y muerte o suicidio.

*Esta enfermedad genética presenta una herencia autosómica dominante, lo cual significa que cualquier niño en una familia en la cual uno de los progenitores esté afectado, tiene un 50% de probabilidades de heredar la mutación que causa la enfermedad.
*Causas:
*La enfermedad de Huntington es causada por un defecto genético en el cromosoma N.° 4. El defecto hace que una parte del ADN, llamada repetición CAG, ocurra muchas más veces de lo que se supone que debe ser. Normalmente, esta sección del ADN se repite de 10 a 28 veces, pero en una persona con la enfermedad de Huntington, se repite de 36 a 120 veces.
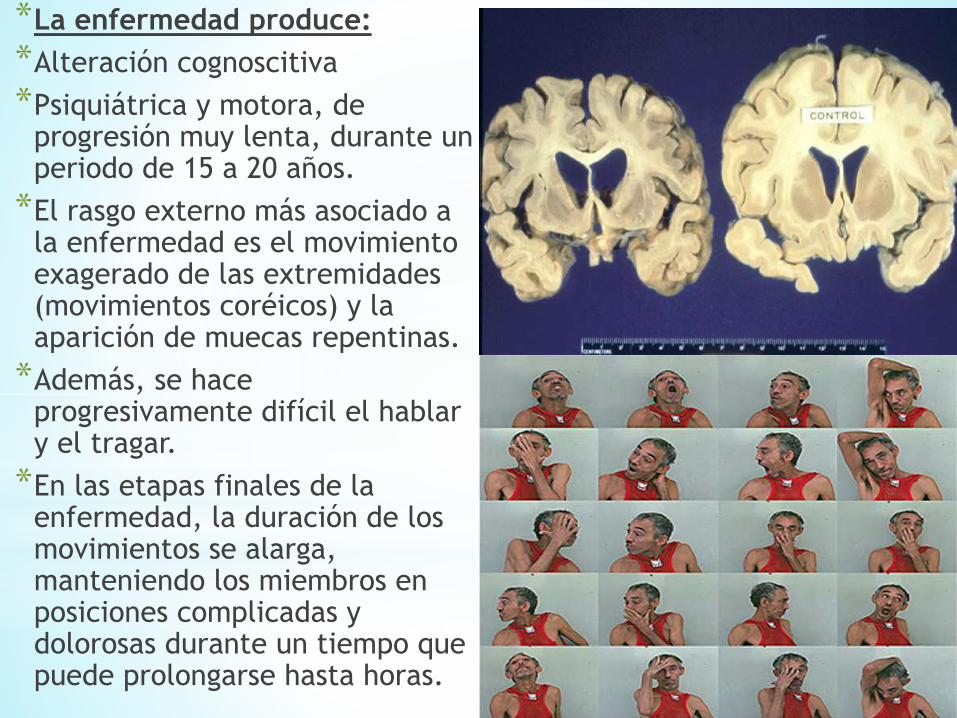
*La enfermedad produce:
*Alteración cognoscitiva
*Psiquiátrica y motora, de progresión muy lenta, durante un periodo de 15 a 20 años.
*El rasgo externo más asociado a la enfermedad es el movimiento exagerado de las extremidades (movimientos coréicos) y la aparición de muecas repentinas.
*Además, se hace progresivamente difícil el hablar y el tragar.
*En las etapas finales de la enfermedad, la duración de los movimientos se alarga, manteniendo los miembros en posiciones complicadas y dolorosas durante un tiempo que puede prolongarse hasta horas.

*