flavonoides con actividad antitumoral: identificación y ... · activates the sphingomyelin cycle...
TRANSCRIPT

DEPARTAMENTO DE PATOLOGÍA ANIMAL, PRODUCCIÓN ANIMAL,
CIENCIA Y TECNOLOGÍA DE LOS ALIMENTOS
Y
DEPARTAMENTO DE CIENCIAS CLÍNICAS
FLAVONOIDES CON ACTIVIDAD ANTITUMORAL:
IDENTIFICACIÓN Y ESTUDIO DEL MECANISMO DE ACCIÓN
Memoria para optar al Grado de DOCTOR
SARA RUBIO SÁNCHEZ
Las Palmas de Gran Canaria
2009






DEPARTAMENTO BIOQUÍMICA Y BIOLOGÍA MOLECULAR, FISIOLOGÍA,
GENÉTICA E INMUNOLOGÍA
El trabajo experimental de la presente memoria fue desarrollado por la doctoranda en el
laboratorio de Bioquímica y Biología Molecular, Fisiología, Genética e Inmunología de
la Facultad de Ciencias de la Salud.


“UNA ESTRELLA FUGAZ,
UNA LUZ EN LA OSCURIDAD”
Ángel Rubio Sánchez
A mis padres: ANGEL Y DELIA
A mis tesoros: FREDDY & BENJAMÍN


PUBLICACIONES A LAS QUE HA DADO ORIGEN ESTA TESIS • Rubio, S., J. Quintana, M. López, J. L. Eiroa, J. Triana, and F. Estévez. 2006.
Phenylbenzopyrones structure-activity studies identify betuletol derivatives as
potential antitumoral agents. Eur J Pharmacol 548:9-20.
• Rubio, S., J. Quintana, J. L. Eiroa, J. Triana, and F. Estévez. 2007. Acetyl
derivative of quercetin 3-methyl ether-induced cell death in human leukemia cells
is amplified by the inhibition of ERK. Carcinogenesis 28:2105-13.
• Rubio, S., J. Quintana, J. L. Eiroa, J. Triana, and F. Estévez. 2009, Betuletol 3-
methyl ether induces G2-M phase arrest and activates the sphingomyelin and
MAPK pathways in human leukemia cells. Aceptado con revisión en Molecular
Carcinogenesis.
COMUNICACIONES A CONGRESOS
• Rubio S., Quintana J, López M., Eiroa JL, Triana J, Estévez F. 2005. Induction
of G2/ M phase arrest and apoptosis by Betuletol derivates on leukaemia cells.
Póster. Comunicación Oral. Seleccionada por el Comité Organizador. XXVII
Congreso de la Sociedad Española de Farmacología. Methods and Findings in
Experimental and Clinical Pharmacology Vol. 27, Supl. 2 (ISSN: 0379-
0355).Girona,
• Rubio S, Quintana J, Eiroa JL, Triana J, Estévez F. 2006. Quercetin-derivative-
induced apoptosis in human leukaemia HL-60 cells does not require JNK but is
amplified by the inhibition of ERK. Póster, XXVIII Congreso de la Sociedad
Española de Farmacología. Methods and Findings in Experimental and Clinical
Pharmacology Vol 28, Suppl.2, 2006. ISSN: 0379-0355. Santiago de Compostela.

• Rubio S, Quintana J, Eiroa JL, Triana J, Estévez F. 2007. Betuletol 3-methyl
ether-induces apoptosis in human leukaemia cells overexpressing Bcl-2 and
activates the sphingomyelin cycle pathway, Póster. XXIX Congreso de la Sociedad
Española de Farmacologia. Methods and Findings in Experimental and Clinical
Pharmacology Vol 29, Suppl. 1, 2007 ISSN 0379-0355, Alcala de Henares.
• Rubio S., Quintana J, Eiroa JL, Triana J, Estévez F. 2008. Perturbation of
microtubule polymerization by Betuletol 3-methyl ether in human leukaemia cells
(HL-60).Póster. Comunicación Oral. Seleccionada por el Comité organizador.
XXX Congreso de la Sociedad Española de Farmacología. Methods and Findings in
Experimental and Clinical Pharmacology Vol. 30, Supl. 2, 2008, ISSN 0379-0355.
Bilbao.
ESTE TRABAJO HA SIDO FINANCIADO POR:
• Ministerio de Educación y Ciencia de España y FEDER (SAF 2004 -07928).
• Consejería de Educación, Cultura y Deportes (Gobierno de Canarias) y FEDER
(GRUP 2004 /44) y (SAF2007-62536).
• Instituto Canario de Investigación del Cáncer (08/2004), (RED
PRODNATCANCER-08).
• Fundación Canaria Universitaria de las Palmas (Clínica San Roque y La Caja de
Canarias).

ÍÍÍNNNDDDIIICCCEEE

Ilustración de portada: Fotomicrografía de una célula HL-60 obtenida mediante microscopia electrónica de transmisión. Ilustración de contraportada: Foto de izquierda: Allagopappus dichotomus; Foto derecha: Allagopappus viscosissimus.

Índice
3
ÍNDICE 1
AGRADECIMIENTOS 9
ABREVIATURAS 13
RELACIÓN DE FIGURAS Y TABLA 19
1
INTRODUCCIÓN 23
1.2 Flavonoides 28
1.2.1 Origen 28
1.2.2 Estructura química 28
1.2.3 Aislamiento y análisis 29
1.2.4 Biosíntesis y funciones 30
1.2.5 Fuentes de flavonoides 32
1.2.6 Dieta y flavonoides 33
1.2.7 Absorción y metabolización 33
1.2.8 Efectos adversos o mutagénicos 34
1.2.9 Actividad antitumoral 34
1.2.9.1 Inhibición de la iniciación del proceso cancerígeno 35
1.2.9.2 Inhibición de la proliferación 37
1.2.9.3 Inhibición de la fase invasiva 42
2
APOPTOSIS 42
2.1 El proceso apoptótico 44
2.1.1 Vía extrínseca: receptores de la muerte celular 45
2.1.2 Vía intrínseca: inicio de la señal de apoptosis en la mitocondria 49
2.1.3 Proteínas reguladoras de la señal de apoptosis 52
2.1.3.1 Proteínas de la familia Bcl-2 52
2.1.3.2 Proteínas de la familia de las caspasas 57

Indice
4
2.1.4 Vía de la MAPK quinasas 63
2.1.4.1 ERK1/2 (p42/44 MAPK) 65
2.1.4.2 Familia JNK (SAPK/JUN) 69
2.1.4.3 Familia MAPK/p38 71
2.1.5 Vía de supervivencia : vía pI3K/AKT 73
2.1.6 Esfingolípidos y apoptosis 74
2.1.6.1 Ceramida 77
3
OBJETIVOS 81
4
MATERIAL Y MÉTODOS 85
4.1
Productos y material utilizado 87
4.1.1 Agentes farmacológicos 87
4.1.2.1 Cultivos celulares 88
4.1.2.2 Fragmentación del ADN 88
4.1.2.2 Microscopía de fluorescencia (tinción con bisbencimida) 88
4.1.2.4 Citometría de flujo 89
4.1.2.5 Determinación de actividad caspasa 89
4.1.2.6 Determinación de actividad esfingomielinasa y determinación de
ceramidas 89
4.1.2.7 Inmunodetección de proteínas (“western blot”) 90
4.1.2.8 Microscopía inmunofluorescencia 91
4.1.3 Reactivos generales de laboratorio 91
4.2
Modelo experimental 91
4.2.1 Cultivo de células leucémicas promielocíticas humanas HL-60 y
células de linfoma histiocítico humano U937 91
4.2.2 Cultivo de células A431 (carcinoma epidermoide humano) y SK-
OV-3 (adenocarcinoma de ovario humano) 92
4.2.3 Cultivo de células HeLa (carcinoma de cervix humano) y HOS 92

Índice
5
(osteosarcoma)
4.2.4 Cultivo de células HL-60 transfectadas con el gen humano Bcl-xL,
HL-60 que sobre-expresan Bcl-xL, HL-60 con su vector control HL-
60 Bcl-neo y U937 (transfectadas con el gen humano de Bcl-2), que
sobre-expresan Bcl-2 92
4.2.5 Células mononucleares de sangre periférica de origen humano
(PBMC) 93
4.2.6 Tratamiento con los compuestos 93
4.3
Métodos 94
4.3.1 Evaluación de la citotoxicidad in vitro y estudios de la proliferación
celular 94
4.3.2 Estudio de la apoptosis celular 94
4.3.2.1 Tinción con el fluorocromo bisbencimida 94
4.3.2.2 Fragmentación del ADN 95
4.3.2.3 Cuantificación de las células hipodiploides por citometría de flujo 96
4.3.2.4 Determinación de la externalización de fosfatidilserina 97
4.3.2.5 Microscopía electrónica de transmisión 97
4.3.3 Determinación de actividad caspasa 98
4.3.4 Inmunodetección de proteínas (“western blot”) 98
4.3.5 Análisis de la tubulina 101
4.3.6 Determinación de la generación de EROs intracelulares 101
4.3.7 Análisis de la despolarización de la membrana mitocondrial (ΔΨm) 102
4.3.8 Análisis de la red de microtúbulos por microscopía de fluorescencia 103
4.3.9 Determinación de la actividad esfingomielinasa ácida y neutra 103
4.3.10 Determinación de la cantidad de ceramidas intracelular 105
4.4 Métodos estadísticos 106
5
RESULTADOS 107
5.1 Relación entre la estructura química de los flavonoides y su 109

Indice
6
capacidad citotóxica.
5.2 Los derivados del betuletol inducen apoptosis en células de leucemia
mieloide HL-60 120
5.3 BME induce apoptosis a través de un mecanismo dependiente de
caspasas 122
5.4 La apoptosis inducida por los derivados del betuletol implica la
activación de caspasa-3 125
5.5 Los derivados del betuletol inducen liberación de citocromo c
mitocondrial 125
5.6 BME induce cambios en el potencial de transmenbrana mitocondrial
(ΔΨm) 128
5.7 Caspasa-8 es activada en la apoptosis inducida por los derivados del
betuletol 130
5.8 La inducción de apoptosis por BME implica la hidrólisis de Bid a
través de un mecanismo independiente de Fas. 132
5.9 El inhibidor de la caspasa-8, bloquea la activación de la caspasa-3 en
la apoptosis inducida por BME 132
5.10 BME activa la vía MAPKs 134
5.11 Las especies reactivas de oxigeno (EROs) no son necesarias para la
muerte celular inducida por BME 137
5.12 BME induce generación de ceramida y activación de SMasa ácida 139
5.13 BME inhibe la proliferación celular de células de leucemia mieloide
humana mediante la inducción de la parada del ciclo celular en fase
G2-M 141
5.14 BME induce cambios en la expresión y el estado de fosforilación de
las proteínas reguladoras de la fase G2-M 141
5.15 BME induce un incremento de la población celular en fase M 143
5.16 BME desestabiliza la polimerización de los microtúbulos 143
5.17 BME inhibe el crecimiento e induce apoptosis en líneas celulares de
leucemia humana independientemente de los niveles de expresión de 146

Índice
7
Bcl-2 y Bcl-xL
5.18 Los derivados del betuletol no son citotóxicos para los linfocitos
normales de origen humano 148
5.19 QD inhibe la viabilidad de células de leucemia mieloide humana 150
5.20 QD bloquea el ciclo celular en la fase G2-M e induce apoptosis
independientemente de la biosíntesis de ARN y de proteínas 150
5.21 QD induce muerte celular vía caspasa-dependiente 153
5.22 El inhibidor específico de caspasa-3 es incapaz de inhibir la
apoptosis inducida por QD 156
5.23 QD activa MAPKs 158
5.24 La apoptosis inducida por QD en células leucémicas humanas se
potencia mediante el uso de inhibidores específicos de ERK 1/2. 160
5.25 QD inhibe el crecimiento, la viabilidad celular e induce apoptosis en
líneas celulares de leucemia humana, que expresan niveles elevados
de Bcl-2 y Bcl-xL 160
5.26 QD no es citotóxico para los linfocitos normales de origen humano 161
6
DISCUSIÓN 163
7
CONCLUSIONES 183
8
BIBLIOGRAFÍA 187
9
ANEXO: Publicaciones 211


AAAGGGRRRAAADDDEEECCCIIIMMMIIIEEENNNTTTOOOSSS


Agradecimientos
11
En primer lugar, quiero agradecer al Dr. Francisco Estévez Rosas y al Dr. José
Quintana Aguiar, Directores de esta tesis, su dedicación y entusiasmo en el trabajo
diario, por haber realizado una parte importante de esta tesis, por haber compartido
conmigo sus conocimientos y su pasión por la investigación. Aunque más importante si
cabe, por haberme dado la confianza necesaria para enfrentarme a algo impensable para
mi “una tesis y la lectura de la misma”.
Debo manifestar, mi agradecimiento a mis compañeros de laboratorio, cuya amistad ha
creado el ambiente propicio para poder realizar este trabajo de investigación. Hago
especial mención a Fernando Torres porque ha llegado a ser parte de los que considero
mi familia y que gracias a su compañía y a su ayuda esta tesis ha tomado forma a lo
largo de estos 5 años, a Gleddy por las infinidad de momentos compartidos tanto buenos
como malos, a Olga, Cristina, Mayte, Dionisio y Juan, quienes siempre estuvieron ahí
cuando los necesitaba.
Quiero agradecer al Dr. Javier Cabrera, y al Dr. Juan Francisco Loro Ferrer su
inestimable ayuda, a la Dra. Mª del Pino Santana, a la Dra. Inmaculada Servanda
Hernández, al Dr. Ignacio Javier González, por haber sido un gran apoyo en estos años
tanto a nivel del laboratorio como a nivel personal. A mis asesores técnicos Dr. Germán
Gallardo, Dr. Enrique Castro y Dr. Carlos Tabraue por haber aguantado mi ignorancia
acerca de la informática y haberme ayudado a resolver todos mis problemas con el
ordenador.
En especial quiero agradecer todo el apoyo, cariño y aliento en los momentos malos a
Freddy mi tesoro, que ha soportado como un campeón el peso de mi pequeño Benjamín
para que yo tuviera dedicación casi exclusiva a este trabajo, sin él la tesis nunca hubiera
sido una realidad; También se lo quiero dedicar a Benjamín, mi vida, por que en los días
mas cargantes su sonrisa y su amor ha disipado mi cansancio.
No podría concluir los agradecimientos sin dedicar este trabajo a mi familia que
incesantemente, me han dado muestras de su apoyo y afecto incondicional, aún desde la
distancia. Y… especialmente a ti que esté donde esté, siempre estás conmigo.
¡GRACIAS A TODOS!


AAABBBRRREEEVVVIIIAAATTTUUURRRAAASSS


Abreviaturas
15
ABREVIATURAS.
14-3-3 Proteínas adaptadoras y de andamiaje Ac-DEVD-Pna N-Acetil-Asp-Glu-Val-Asp-p-nitroanilina Ac-IETD-pNA Ac-lleGlu-Thr-Asp-p-nitroanilina ADN Ácido desoxirribonucleico AIF Factor inductor de apoptosis APAF-1 Factor activador de proteasas apoptóticas Ara-C 1-β-D-arabino-furanosil-citosina ARN Ácido ribonucleico BAD Proteína pro-apoptótica. Inductor de muerte asociado a Bcl-2 BAX Proteína X asociada a Bcl-2 BCL-2 Proteína anti-apoptótica BID Proteína pro-apoptótica BIR Dominio de interacción proteína- proteína presente en las
proteínas IAPS BME 3-metil éter de betuletol BME(A) Diacetato de 3- metil éter de betuletol CAK Proteína quinasa constitutiva activadora de quinasa tipo CDK CARD Dominio de reclutamiento de caspasa CAPPs Fosfatasas activadas por ceramidas. Cdc25 Proteína fosfatasa que participa en la activación del complejo
Cdk1/Cyc B CDK Proteína quinasa dependiente de ciclina CDK1 Proteína quinasa dependiente de ciclina 1 CDK2 Proteína quinasa dependiente de ciclina 2 CDK4 Proteína quinasa dependiente de ciclina 4 CDK6 Proteína quinasa dependiente de ciclina 6 CIP Proteínas inhibidoras de CDKs CKI Inhibidores de las quinasas dependientes de ciclinas CREB Factor de transcripción Cyc Ciclina DAG Diacilglicerol DCF Diacetato 2´,7´-diclorofluoresceína DD Dominio (de) muerte DED Dominio efector (de) muerte DETAPAC Ácido dietilen-triamina-penta-acético DGK Diacilglicerol quinasa DISC Complejo inductor de muerte DMSO Dimetilsulfóxido DNA-PK Proteína quinasa dependiente de ADN

Abreviaturas
16
DTT Ditiotreitol EDTA Ácido etilen-diamin-tetra-acético
EGTA Ácido etilenglicol-bis-N,N'-tetraacético Elk-1 Factor de transcripción nuclear ERK1/2 Proteína quinasa regulada por señales extracelulares 1 y 2 EROs Especies reactivas de oxígeno ETO Etopósido FADD Dominio (de) muerte asociado a Fas Fas Receptor de muerte, también llamado Apo1 o CD95 FasL Ligando del receptor (de) muerte Fas FBS Suero bovino fetal H2DCF-DA Diacetato dihidro-diclorofluoresceína HEPES N-[2-hidroxietil] piperazino N'-[2-etanosulfanílico] Hsp27 Proteína de choque térmico de 27 kDa Hsp40 Proteína de choque térmico de 40 kDa Hsp70 Proteína de choque térmico de 70 kDa IAP Proteína inhibidora de apoptosis IC50 Concentración de producto que inhibe el crecimiento celular a la
mitad ICAD/DFF45 Inhibidor de la DNasa activada por caspasa. IP Yoduro de propidio JC-1 Yodurode5,5',6,6'–tetracloro-1,1',3,3'-
tetraetilbenzimidazolocarbocianina JNK Proteína quinasa N-terminal de jun JNKK Proteína quinasa de las JNK Jun Factor de transcripcion de la familia de AP-1 MAPK Proteina quinasa activada por mitógenos MEK Proteína quinasa MAPK/ERK MEKK Proteína quinasa de las MEKs MOM Membrana mitocondrial externa m-TOR Proteína quinasa de la familia de la PI3K MTT Bromuro de 3-[4,5- Dimetiltiazol-2-il]-2,5-difeniltetrazolio Myc Factor de transcripción nuclear Na3VO4 Ortovanadato sódico NaCl Cloruro de sodio NaOH Hidróxido de sodio NF-ΚB Factor nuclear –κB (nuclear factor κB) P53 Proteína supresora de tumores PARP-1 Poli (ADPribosa) polimerasa-1 PBS Tampón fosfato salino PC Fosfatidilcolina

Abreviaturas
17
PI3K Fosfatidil inositol 3quinasa PIP2 PI 4,5-bifosfato PKA Proteína quinasa A PKB Proteína quinasa B PKC Proteína quinasa C PLA2 Fosfolipasa A2 PLC Fosfolipasa C PMSF Fluoruro de metilsulfonilfenilo PP1 Proteina fosfatasa 1 PP2A Proteina fosfatasa 2A PTP Poro de transición de permeabilidad PVDF Fluoruro de Poli-vinilideno QD Tetraacetato de 3- metil eter de quercetina Raf-1 MAPKKK Ras Proteína G pequeña RING Dominio interacción proteína-proteína SAPK MAPK activada por estrés SDS Dodecil sulfato sódico SK Esfingosina quinasa Smac Segundo activador de caspasas derivado de la mitocondria Sos Factor de intercambio de nucleótidos de guanina TBST 20 mM Tris-HCl (pH 7,5), 137 mM NaCl, 0,1 % Tween-20 TEMED N,N,N,N,-tetrametil-etilendiamina TLC cromatografia en capa fina. TNF Factor de necrosis tumoral TNFR Receptor de muerte de TNF- α TNFR1 Receptor 1 del factor necrótico tumoral TNF-α Factor de necrosis tumoral- α TRADD Dominio de muerte asociado al TNFR TRAF2 Factor asociado a al receptor de TNF TRAIL Ligando inductor de apoptosis relacionado con TNF VDAC Proteína de canal aniónico dependiente de voltaje VEGF Factor de crecimiento endotelial vascular Wee1 Tirosina quinasa Wee1


FFFIIIGGGUUURRRAAASSS YYY TTTAAABBBLLLAAASSS


Relación de Figuras y Tablas
21
INTRODUCCIÓN
Figura 1 Estructura básica de los flavonoides 29 Figura 2 Clasificación de los flavonoides 29 Figura 3 Biosíntesis de los flavonoides 31 Tabla 1 Actividades anticancerigenas de flavonoides en varias líneas de
células cancerigenas 35 Figura 4 Regulación del ciclo celular por CDKs y ciclinas 39 Tabla 2 Receptores de muerte y sus ligandos 45 Figura 5 Esquema de los mecanismos de activación de la apoptosis a partir
de los receptores de muerte 47 Figura 6 Esquema de los diferentes receptores de muerte. Activación de
caspasa 8/10 por receptores de muerte. 49 Figura 7 Vía intrínseca o mitocondrial 51 Figura 8 Miembros de la familia Bcl-2 52 Figura 9 Regulación de la apoptosis por la familia Bcl-2 56 Figura 10 Vías de activación de las quinasas MAPKs 64 Figura 11 Vía de señalización activada por PI3K/AKT 74 Figura 12 Balance ceramida-esfingosina 77 MATERIAL Y MÉTODOS Figura 13 Histograma de la distribución del ciclo celular 96 Figura 14 Método para determinar la despolarización de la membrana
mitocondrial 102 RESULTADOS Tabla 3 Estructura de los 22 flavonoides 110 Tabla 4 Efectos de los derivados fenil-benzo-γ-pirona sobre el crecimiento
de células tumorales humanas 111 Figura 15 Estructura química de los flavonoides 1-2-3, 1-12 112 Figura 16 Estructura química de los flavonoides 4-5, 9-5, 6-8, 11-12 114 Figura 17 Estructura química de los flavonoides 5-11, 7-10, 5-6, 7-8 115 Figura 18 Estructura química de los flavonoides 12-13, 13-14-15 118 Figura 19 Estructura química de los flavonoides 21-19-20, 21-18, 21-22 119 Figura 20 Estructura química del 3-metil éter de betuletol (BME) 120 Figura 21 Efecto del 3-metil éter de betuletol (1) sobre la inducción de
apoptosis en células HL-60 121 Figura 22 Efecto de los derivados del betuletol sobre la apoptosis en células
HL-60 123 Figura 23 Evaluación de la participación de las caspasas en la inducción de la
apoptosis por BME y BME(A) 124

Relación de Figuras y Tablas
22
Figura 24 Activación de caspasa-3 en células HL-60 por BME 126 Figura 25 Evaluación de la vía intrínseca de la apoptosis inducida por BME y
BME(A) 127 Figura 26 BME disminuye el potencial de membrana mitocondrial (ΔΨm) 129 Figura 27 Activación de la caspasa-8 por BME 131 Figura 28 BME induce apoptosis e hidrólisis de Bid a través de un
mecanismo independiente de Fas 133 Figura 29 La supresión de la actividad de la caspasa-8 bloquea la activación
de la caspasa-3 135 Figura 30 Efecto de BME sobre la fosforilación de las MAPKs y evaluación
de los inhibidores de esta vía en la línea celular HL-60 136 Figura 31 BME induce generación de EROs 138 Figura 32 Efecto de BME sobre la vía de la esfingomielina en células HL-60 140 Figura 33 Efecto de BME sobre el ciclo celular y sobre las proteínas que lo
regulan en células HL-60 142 Figura 34 BME induce arresto mitótico 144 Figura 35 BME inhibe la polimerización de la tubulina en células HL-60 145 Figura 36 BME inhibe el crecimiento e induce apoptosis en líneas celulares
de leucemia humana independientemente de los niveles de Bcl-2 y Bcl-xL expresados 147
Figura 37 Efecto de los derivados del betuletol BME y BME(A) sobre la viabilidad de los linfocitos humanos (PBMC) 149
Figura 38 Estructura química de QD y efecto de QD sobre la viabilidad de las células U937 151
Figura 39 Efectos de QD sobre la inducción de apoptosis en células de leucemia mieloide humana HL-60 152
TABLA 5 Efecto de la QD en la distribución de las fases del ciclo celular en células HL-60 y U937 a diferentes tiempos 154
Figura 40 Efecto de QD sobre la activación de las caspasas en células HL-60 y U937 155
Figura 41 Efecto de los inhibidores de las caspasas sobre la apoptosis inducida por QD en HL-60 157
Figura 42 Efecto sobre la fosforilación de las MAPKs e impacto de los inhibidores de esta vía en la apoptosis 159
Figura 43 QD induce apoptosis en líneas celulares de leucemia humana que expresan niveles altos de Bcl-2 y Bcl-xL y no es citotóxico para los linfocitos humanos normales (PBMC) 162

IIINNNTTTRRROOODDDUUUCCCCCCIIIÓÓÓNNN


Intoducción
La búsqueda de nuevos fármacos con fines terapéuticos se ha incrementado de forma
considerable. En los últimos años la existencia de enfermedades para las cuales no
existen medicamentos efectivos, como el cáncer, la resistencia de los agentes patógenos,
los avances en las técnicas de biología molecular que han llevado a la identificación de
un número cada vez mayor de moléculas “diana” junto con el conocimiento de la
relación entre anomalías funcionales y estructurales con ciertas patologías, son algunos
de los factores que han aumentado el interés en la búsqueda de fármacos que sean
capaces de ejercer acciones específicas y que sean potentes. Los productos naturales
facilitan dichas bases actuando como sustrato, modelo o estructura base para generar
nuevas moléculas antitumorales por química combinatoria con comprobación inmediata
de su actividad. Estudios recientes sobre compuestos antitumorales de origen vegetal
están proporcionando nuevas estructuras, muchas de ellas sumamente complejas por lo
que resulta difícil pensar que pudieran haber sido sintetizados de forma empírica de
hallar nuevos fármacos.
El cáncer es uno de los principales problemas de salud pública en el mundo y una de las
enfermedades que están más ligadas al fenómeno del progreso humano. Se encuentra en
general en todos los animales superiores, y forma parte de una patología compleja ya
que no existe un cáncer sino una multitud de cánceres (abarca un grupo heterogéneo con
más de 200 tipos de tumores malignos). Cada tipo de cáncer posee unas características
particulares, pudiendo considerarse enfermedades independientes, con sus causas, su
evolución y su tratamiento específico.
El cáncer es la principal causa de mortalidad y a nivel mundial, se le atribuyen 7,9
millones de defunciones en el 2007. La Organización Mundial de la Salud (OMS) ha
estimado que alrededor de 84 millones de personas morirán a causa de esta enfermedad
entre 2005 y 2015 [1]. La OMS señala que el consumo de tabaco es el principal factor
de riesgo para desarrollar la enfermedad, así como el sobrepeso u obesidad, una dieta
baja en frutas y hortalizas, el sedentarismo, el consumo de alcohol, la presencia de las
enfermedades de transmisión sexual tales como el Virus de Inmunodeficiencia Humana
(VIH) y el Virus del Papiloma Humano (VPH), la contaminación del aire urbano y el

Intoducción
26
humo por la utilización doméstica de combustibles sólidos como la leña [1]. En el
mundo occidental el riesgo de cáncer es mayor debido a los hábitos de vida poco
saludables como el tabaquismo, el sedentarismo o el consumo de dietas hipercalóricas
ricas en grasas saturadas que son factores que predisponen al cáncer de pulmón, al
cáncer de mama o al cáncer colon. En los países en vías de desarrollo el número de
fallecimientos por cáncer es menor que en los países desarrollados debido a la mayor
mortalidad infantil y a la mayor mortalidad por enfermedades infecciosas. En este
sentido no se debería olvidar que el cáncer tiene mayor incidencia en los grupos de
mayor edad y que, por tanto, la mayor esperanza de vida lograda en los países
desarrollados tiene como contrapartida mayor incidencia de enfermedades ligadas al
envejecimiento gradual de la población.
El cáncer se caracteriza por la multiplicación incontrolada de las propias células
corporales (resistiendo a las señales exógenas de inhibición del crecimiento,
proliferando sin control) y la diseminación dentro del organismo (invasión y
metástasis), que es capaz de comprometer la funcionalidad del cuerpo y llegar a
producir la muerte. Se produce como consecuencia de la acumulación de múltiples
alteraciones o errores en el sistema que la célula usa para regular la proliferación. Así la
célula adquiere la capacidad de propagarse de forma indefinida (capacidad de las células
para generar sus propias señales mitógenas), en su sitio o bien en zonas diferentes de su
localización normal como consecuencia de una pérdida de control de su proliferación,
diferenciación o de la respuesta a las señales que ordenan su eliminación controlada por
apoptosis (evadiendo la apoptosis) [2,3].
El conocimiento del cáncer es tan antiguo como la utilización de determinadas plantas
medicinales en su tratamiento [4]. Los principios activos de algunas plantas con
actividad antitumoral han podido ser identificados:
• El podofilino: utilizado desde hace mas de 2000 años por los chinos como
fármaco antitumoral; las resinas de esta planta Phodophyllum hexandrum han dado
lugar al aislamiento de lignanos y heterósidos que poseen actividad antitumoral [5].
Aunque los componentes mayoritarios no son utilizables como fármacos, si son

Intoducción
27
utilizados sus derivados semisintéticos como el etopósido que se utiliza en el
tratamiento de tumores germinales, cáncer microcítico de pulmón, neoplasias
hematológicas, cáncer de ovario, cáncer de mama, cáncer gástrico y tumores de origen
desconocido y el tenopósido que se utiliza en leucemias agudas infantiles
mielomonocítica y linfoide. También es activo en linfomas, tumores uroteliales y
neuroblastomas [6,7].
• La vinblastina y vincristina, alcaloides indólicos dímeros del Catharantus
roseus constituyen el mayor logro en la quimioterapia del cáncer con actividad
antileucémica, se emplean solos o en combinación con otras formas terapéuticas para el
tratamiento del cáncer. La vincristina se utiliza en numerosos tumores infantiles y
adultos (linfomas y leucemias, tumor de Wilms, sarcoma de Edwing, rabdomiosarcoma,
mieloma múltiple y carcinoma microcítico de pulmón). La vinblastina se utiliza en la
enfermedad de Hodgkin y cáncer de vejiga, también es activa en el sarcoma de Kaposi,
neuroblastoma, cáncer de mama y de riñón [8,9].
El estudio de la actividad de determinados compuestos naturales ha ayudado a
establecer mejor los mecanismos de acción de nuevos antitumorales. Así, los estudios
sobre el Taxol® (registrado por la compañía Bristol-Myers Squibb, obtenido de la
corteza del árbol Taxus brevifolia [10]) y derivados como el Taxotere® interaccionan
con los microtúbulos generando uniones estables que favorecen la polimerización de los
microtúbulos y los estudios sobre la camptotecina obtenida a partir del árbol nativo de
China Camptotheca acuminata (inhibición selectiva e irreversible de la ADN
topoisomerasa I [11] que impide la replicación del ADN) han permitido potenciar la
investigación en este campo, buscando nuevos antitumorales que actúen de forma
similar.
Los flavonoides representan un amplio grupo de compuestos de origen natural que
tienen gran valor como prometedores agentes antitumorales (de ahí el interés mostrado a
través de estos años y el fruto de esta tesis). Su amplia bioactividad y su elevada
presencia en la dieta humana los hace merecedores de la atención de la investigación

Intoducción
28
farmacológica. Actualmente, los campos en los que más estudios se realizan con
flavonoides son el cáncer y las enfermedades cardiovasculares (dos de los principales
problemas sanitarios del mundo occidental). La búsqueda de principios activos dentro
de los flavonoides tiene, desde el punto de vista farmacológico, algunas ventajas
respecto a otros grupos de compuestos naturales. La más importante es la uniformidad
de la configuración química de toda la familia, de modo que las relaciones entre
estructura y actividad son más fáciles de establecer. Por otro lado, la disponibilidad de
las moléculas flavónicas y la relativa facilidad de su obtención por síntesis favorecen la
evaluación de sus propiedades.
1.2 FLAVONOIDES.
El primer flavonoide fue identificado en 1930 por el premio Nobel de Fisiología y
Medicina Szent-Györgyi, quien aisló de la cáscara de limón una sustancia, la citrina,
que probó regular la permeabilidad de los capilares al ser consumida, aunque
probablemente la primera vez que se describió a los flavonoides fue cuando Robert
Boyle en 1664 hizo una primera descripción de los efectos de los pigmentos de las
flores en medio ácido y en medio básico [12].
1.2.1 ORIGEN.
Los flavonoides pueden haber existido en la naturaleza desde hace un billón de años y
haber interaccionado con la evolución de los organismos vegetales. La importancia del
papel que desempeñan en la naturaleza ha permitido que hayan permanecido durante la
evolución, haciendo posible el extraordinario rango de actividades farmacológicas y
bioquímicas que manifiestan en mamíferos y otros sistemas celulares.
1.2.2 ESTRUCTURA QUÍMICA.
Los flavonoides son compuestos polifenólicos que se encuentran omnipresentes en las
plantas, como producto del metabolismo secundario, y que abundan en los generos
Poligonaceae, Rutaceae, Leguminosae, Umbelliferae y Compositae. Son un grupo de

Intoducción
29
compuestos de bajo peso molecular con una estructura común fenil benzo–γ-pirona (C6-
C3-C6) (Figura 1).
Figura 1. Estructura básica de los flavonoides
En función del grado de saturación y de la apertura o no del anillo central pirano, los
flavonoides se clasifican en flavanoles, flavonas, isoflavonas, antocianidinas, chalconas,
flavanonas y flavonoles (Figura 2).
Figura 2. Clasificación de los flavonoides.
1.2.3 AISLAMIENTO Y ANÁLISIS.
En las plantas, los flavonoides se encuentran en estado libre o unido a azúcares
formando heterósidos, que es lo más frecuente, debido a que les confiere mayor
O
A
B
C
1
2
345
6
7
8 1'
2'
3'
4'
5'
6'
O
OH
A
B
C
Flavanol(Flavan-3-ol)
Antocianidina
O+
OHA
B
C
O
O
A
B
C
Flavanona
Flavona
O
O
A
B
C
OH
O
B
AChalcona
O
O
OHA
B
C
Flavonol(Flavon-3-ol)
A
B
CO
O
Isoflavona

Intoducción
30
estabilidad química. Los azúcares que aparecen unidos con más frecuencia a las geninas
son la D-glucosa, la D-galactosa, la L-ramnosa, la L-arabinosa, la D-xilosa y el D-ácido
glucurónico. Pueden aparecer como O-heterósidos (con los carbohidratos ligados a
través de átomos de oxígeno) o como C-heterósidos (con los carbohidratos ligados a
través de enlaces C-C). De todas estas formas naturales, los O-glicósidos son los más
comunes.
Estos heterósidos son polares y por lo tanto son generalmente hidrosolubles, se extraen
y se solubilizan en disolventes polares; mientras que sus geninas o agliconas son solo
ligeramente por lo que se extraen en disolventes orgánicos.
Para la identificación de los flavonoides se utilizan un gran número de métodos
analíticos: la resonancia magnética nuclear (RMN), espectrometría de masas,
epectofotometria UV o IR (suele ir acoplada a una separación cromatografica como por
ejemplo HPLC).
1.2.4 BIOSINTESIS Y FUNCIONES.
Los flavonoides se sintetizan en todas las plantas (taxón Embryophyta) y también en
algunas algas (Charophyta), que aunque comparten la vía biosintética central, poseen
una gran variabilidad en la composición química de sus productos finales y en los
mecanismos de regulación de su biosíntesis, por lo que la composición y concentración
de flavonoides es muy variable en función de la especie y en respuesta al ambiente. Los
flavonoides son sintetizados en el citoplasma y luego migran hacia su destino final en
las vacuolas celulares. Están disueltos como glicósidos en el jugo vacuolar, cloroplastos
y membranas. La luz no es esencial para su formación, pero influye de forma
cuantitativa.
La biosíntesis de los flavonoides se realiza a través de la vía del ácido shikímico y la
vía del acido malónico (Figura 3). La vía del ácido shikímico participa en la biosíntesis
de la mayoría de los fenoles de las plantas superiores y que utiliza como sustratos la
eritrosa-4-fosfato (de la vía de las pentosas fosfato) y el ácido fosfo-enol-pirúvico
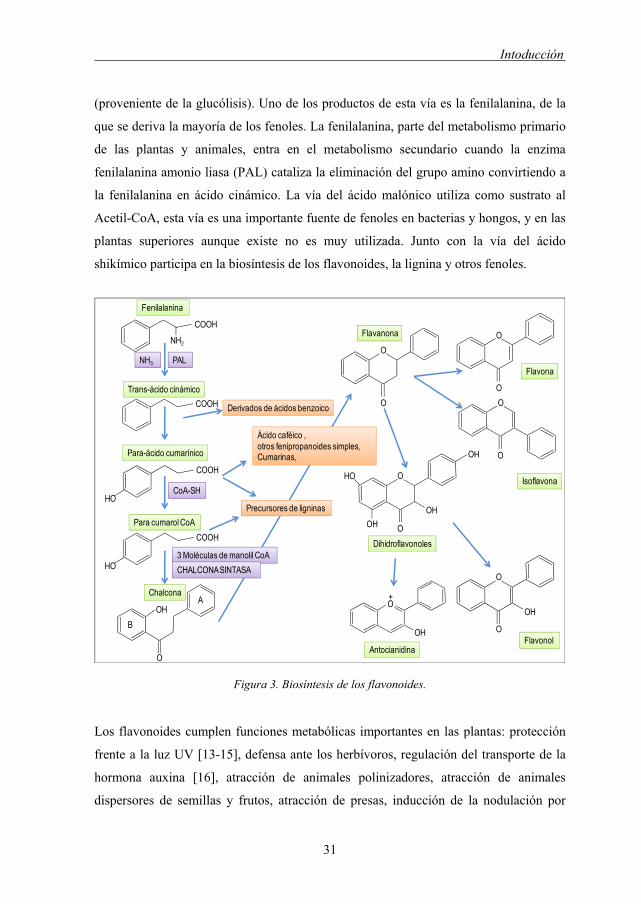
Intoducción
31
(proveniente de la glucólisis). Uno de los productos de esta vía es la fenilalanina, de la
que se deriva la mayoría de los fenoles. La fenilalanina, parte del metabolismo primario
de las plantas y animales, entra en el metabolismo secundario cuando la enzima
fenilalanina amonio liasa (PAL) cataliza la eliminación del grupo amino convirtiendo a
la fenilalanina en ácido cinámico. La vía del ácido malónico utiliza como sustrato al
Acetil-CoA, esta vía es una importante fuente de fenoles en bacterias y hongos, y en las
plantas superiores aunque existe no es muy utilizada. Junto con la vía del ácido
shikímico participa en la biosíntesis de los flavonoides, la lignina y otros fenoles.
Antocianidina
O+
OH
O
O
Flavanona
Flavona
O
O
OH
O
B
AChalcona
O
O
OH
Flavonol
O
O
Isoflavona
OH
O
ODihidroflavonoles
OH
HO
OH
OH
COOH
OH
COOH
COOH
Para cumarol CoA
Para-ácido cumarínico
Trans-ácido cinámico
COOHNH2
Fenilalanina
Ácido caféico ,otros fenipropanoides simples,Cumarinas,
Derivados de ácidos benzoico
Precursores de ligninas
NH3 PAL
CoA-SH
3 Moléculas de manolil CoACHALCONA SINTASA
Figura 3. Biosíntesis de los flavonoides.
Los flavonoides cumplen funciones metabólicas importantes en las plantas: protección
frente a la luz UV [13-15], defensa ante los herbívoros, regulación del transporte de la
hormona auxina [16], atracción de animales polinizadores, atracción de animales
dispersores de semillas y frutos, atracción de presas, inducción de la nodulación por

Intoducción
32
parte de las bacterias fijadoras de nitrógeno, protección contra los hongos etc. Algunas
funciones son comunes a todas las plantas y otras son específicas de algunos taxones.
Estos compuestos polifenólicos muestran un gran espectro de actividades biológicas en
los seres humanos: aumentan la resistencia capilar, propiedades cardiotónicas y
antitrombóticas, propiedades hepatoprotectoras, disminución del colesterol circulante,
protectores gástricos, propiedades antioxidantes, antiinflamatorios y analgésicos,
antivíricos, antibacterianos y antifúngicos y prevención de enfermedades
neurodegenerativas [17-24].
1.2.5 FUENTES DE FLAVONOIDES.
Los flavonoides se encuentran en frutas, verduras, semillas y flores, así como en
cerveza, vino, té verde, té negro y soja, los cuales se consumen en la dieta humana de
forma habitual y también se utilizan en forma de suplementos nutricionales, junto con
ciertas vitaminas y minerales. Los flavonoides se encuentran también en extractos de
plantas como arándano, gingko biloba, cardo-mariano o crataegus. Se ubican
principalmente en las hojas y, apareciendo sólo rastros de ellos en las partes de la planta
por encima de la superficie del suelo, a excepción de los tubérculos de cebolla, que
contienen una gran cantidad de quercetina. Se han identificado alrededor de 9.000
flavonoides [25]:
Las flavonas no suelen encontrarse en frutas pero sí en cereales y en muchas plantas
herbáceas. Los flavonoles se encuentran repartidos abundantemente en todos los
alimentos de origen vegetal, especialmente en las frutas y raramente se encuentran en
legumbres y hortalizas. Los flavanoles se encuentran en el cacao, la uva, el té, la
manzana y diversas bayas. Las flavanonas como son la hesperidina y la naringenina se
encuentran prácticamente restringidas a frutos cítricos. Las chalconas son compuestos
minoritarios, que los encontramos en manzana y fresa principalmente (floretina). Las
isoflavonas son compuestas con actividad estrogénica que se encuentran principalmente
en legumbres y más concretamente en la soja y los productos derivados de esta como las
judías, el tofu, el tempeh (alimento que resulta de la fermentación controlada de soja
con un hongo de Rhizopus), la leche de soja, la proteína vegetal texturizada, la harina y

Intoducción
33
el miso. Las proantocianidinas y las antocianidinas se localizan en las semillas de las
uvas y en las cerezas respectivamente, a las que confieren el color rojo y rojo azulado.
En general son pigmentos naturales de coloración comprendida entre el naranja y el
violeta, pasando por el rojo.
1.2.6 DIETA Y FLAVONOIDES.
Los hábitos alimenticios son muy diversos en el mundo, el valor medio de ingesta de
flavonoides se estima en torno al 23 mg/día [26] siendo predominantes los flavonoles,
especialmente la quercetina. Las fuentes alimenticias principales de los flavonoles son,
entre otras, el té negro, las cebollas, las manzanas, la pimienta negra —que contiene
cerca de 4 g/kg de quercitina y bebidas alcohólicas como el vino (donde los compuestos
fenólicos se sitúan entre 1,8- 4,0 g/L) y la cerveza (29 nmol/L). Entre los alimentos, el
té es una de las fuentes principales de quercetina, principalmente en Japón y los Países
Bajos, el vino tinto lo es en Italia y las cebollas en los Estados Unidos y Grecia.
La ingesta promedio de flavonoles y flavonas se sitúa entre los 20 y 26 mg/día [27-29],
excede a la de otros antioxidantes en la dieta, como el beta-caroteno (2-3 mg/día) y la
vitamina E (7-10 mg/día) mientras que es aproximadamente un tercio de la vitamina C
(70-100 mg/día). Así, pues los flavonoides representan una contribución importante al
potencial antioxidante de la dieta humana.
1.2. 7 ABSORCIÓN Y METABOLIZACIÓN.
Los diferentes grupos de flavonoides poseen distintas propiedades farmacocinéticas.
Los compuestos solubles son metabolizados en el tracto gastrointestinal. Las agliconas
libres son absorbidas a través de la mucosa del intestino delgado. La transformación de
los flavonoides tiene lugar en dos localizaciones: en primer lugar en el hígado, por
medio de reacciones de biotransformación de fase I en las que se introducen o exponen
grupos polares; en segundo lugar en el colon mediante reacciones de biotransformación
de fase II, en las que los microorganismos degradan los flavonoides no absorbidos [27].

Intoducción
34
La conjugación con el ácido glucurónico, sulfatos, o glicina, parecen tener lugar tanto
para los flavonoides como para sus metabolitos procedentes del colon. Los conjugados,
solubles en agua, pueden excretarse por la orina [30]. La biodisponibilidad es uno de los
factores más importantes a tener en cuenta para evaluar los efectos biológicos de los
flavonoides, así como de cualquier fármaco o componente alimenticio [30], en la que
van a influir factores como la estructura química, la absorción, la distribución y la
eliminación.
1.2.8 EFECTOS MUTAGÉNICOS.
Los estudios sobre la capacidad de alteración genética de los flavonoides en células de
mamíferos son limitados y ciertamente confusos. Es posible que las flavonas interfieran
indirectamente en la replicación celular y no directamente en el daño del ADN. Los
procesos de oxidación y reducción en los que toman parte los polifenoles en el interior
celular pueden desencadenar la generación de especies reactivas de oxigeno (EROs),
que serían las últimas responsables de la rotura de las cadenas de ADN, atribuyendo,
por tanto, un papel crucial a las EROs en el proceso. Algunos estudios sobre capacidad
mutagénica realizados con cepas microbianas indican la capacidad mutagénica de
algunos flavonoides.
En muchas de las ocasiones la actividad genotóxica observada, se produce a unas
concentraciones del flavonoide que difícilmente se podrían alcanzar de manera natural
en un tejido. En cualquier caso, los resultados de mutagenicidad obtenidos con algunos
flavonoides en experimentos in vitro no se han correspondido con experimentos in vivo,
con lo que la capacidad carcinogénica de estos compuestos esta todavía sin dilucidar.
Intoducción
35
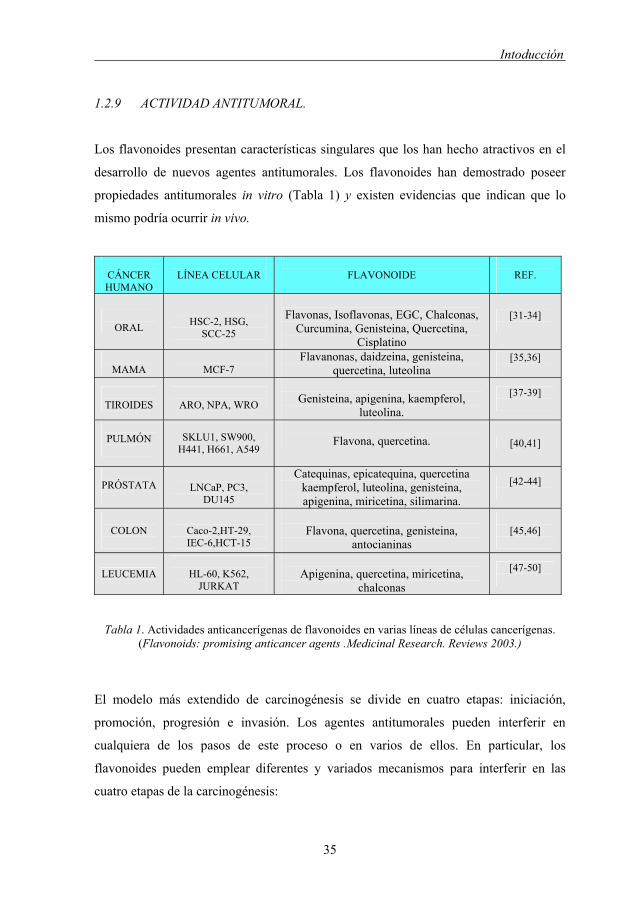
1.2.9 ACTIVIDAD ANTITUMORAL.
Los flavonoides presentan características singulares que los han hecho atractivos en el
desarrollo de nuevos agentes antitumorales. Los flavonoides han demostrado poseer
propiedades antitumorales in vitro (Tabla 1) y existen evidencias que indican que lo
mismo podría ocurrir in vivo.
CÁNCER HUMANO
LÍNEA CELULAR
FLAVONOIDE
REF.
ORAL
HSC-2, HSG,
SCC-25
Flavonas, Isoflavonas, EGC, Chalconas,
Curcumina, Genisteina, Quercetina, Cisplatino
[31-34]
MAMA
MCF-7
Flavanonas, daidzeina, genisteina, quercetina, luteolina
[35,36]
TIROIDES
ARO, NPA, WRO
Genisteina, apigenina, kaempferol,
luteolina.
[37-39]
PULMÓN
SKLU1, SW900,
H441, H661, A549
Flavona, quercetina. [40,41]
PRÓSTATA
LNCaP, PC3,
DU145
Catequinas, epicatequina, quercetina kaempferol, luteolina, genisteina, apigenina, miricetina, silimarina.
[42-44]
COLON
Caco-2,HT-29, IEC-6,HCT-15
Flavona, quercetina, genisteina,
antocianinas
[45,46]
LEUCEMIA
HL-60, K562, JURKAT
Apigenina, quercetina, miricetina,
chalconas
[47-50]
Tabla 1. Actividades anticancerígenas de flavonoides en varias líneas de células cancerígenas. (Flavonoids: promising anticancer agents .Medicinal Research. Reviews 2003.)
El modelo más extendido de carcinogénesis se divide en cuatro etapas: iniciación,
promoción, progresión e invasión. Los agentes antitumorales pueden interferir en
cualquiera de los pasos de este proceso o en varios de ellos. En particular, los
flavonoides pueden emplear diferentes y variados mecanismos para interferir en las
cuatro etapas de la carcinogénesis:

Intoducción
36
1.2.9.1 INHIBICIÓN DE LA INICIACIÓN DEL PROCESO CANCERÍGENO.
Modulación del metabolismo y disponibilidad de agentes carcinógenos. En estudios
realizados in vivo e in vitro se ha observado que algunos flavonoides son capaces de
inhibir el paso del agente precursor a mutágeno durante la elaboración de los alimentos,
mientras que otros dificultan la absorción del promotor tumoral en el tubo digestivo. Sin
embargo, muchos de los efectos antitumorales de los flavonoides podrían ser debidos a
la modulación de las enzimas metabólicas hepáticas [51]. Los flavonoides pueden
intervenir en la metabolización de los agentes mutagénicos, o de sus precursores, de dos
formas. Por un lado, pueden inducir la síntesis de enzimas metabólicas, y por otro lado,
pueden interferir en la actividad enzimática directamente.
Protección frente a mutágenos activos. Algunos flavonoides también pueden actuar
capturando el mutágeno o interponiéndose entre éste y su diana de actuación. La
eficacia inhibidora varía según el tipo de mutágeno frente al que actúan y dependerá de
la estructura química del flavonoide y de la polaridad de la molécula.
Actividad antioxidante. Los flavonoides pueden prevenir el inicio del cáncer al actuar
como antioxidantes naturales que evitan el daño del ADN celular causados por especies
reactivas de oxígeno (EROs) o por agentes cancerígenos y de esta forma previenen el
inicio de cáncer. Los mecanismos a través de los cuales ejercen su acción antioxidante
es el resultado de una combinación de sus propiedades quelantes de metales de
transición y secuestradoras de radicales libres (RL), así como de la inhibición de
actidades oxidasas como la lipooxigenasa (curcumina, silimarina), la ciclooxigenasa
(curcumina), la mieloperoxidasa, la NADPH oxidasa (quercetina y la rutina) y xantina
oxidasa, evitando la generación de especies reactivas de oxigeno (EROs) in vivo y la
acción sobre enzimas que están involucradas indirectamente en los procesos oxidativos,
como la fosfolipasa A2 (PLA2) [52-61].
Existen evidencias de una cierta relación entre la estructura y la actividad antioxidante
de los flavonoides. Los flavonoides con sustituyentes dihidroxílicos en posiciones 3' y 4'
en el anillo B son antioxidantes más efectivos, efecto que es potenciado por la presencia

Intoducción
37
de un doble enlace entre los carbonos 2 y 3, un grupo OH libre en la posición 3 y 5, y un
grupo carbonilo en la posición 4 [62-64].
1.2.9.2 INHIBICIÓN DE LA PROLIFERACIÓN.
Los procesos neoplásicos, en sus fases de promoción y progresión, se caracterizan por
alteraciones de la regulación de la proliferación y de la diferenciación celular. Algunos
flavonoides poseen acción sobre estas etapas en muy diferentes tipos celulares, lo que
puede conducir al uso de estos compuestos como agentes citostáticos en las últimas
etapas de la carcinogénesis más que como elementos preventivos de las primeras fases.
Inhibición de las vías de transducción de la señal. La maquinaria de transducción de las
señales que regulan el crecimiento, la proliferación y la diferenciación celular es
siempre una posible diana de las sustacias anticancerígenos. Desde este punto de vista
puede tener importancia la capacidad de determinados flavonoides para inhibir quinasas
como son las proteínas tirosina quinasas (PTK) (quercetina y la genisteina), la proteína
quinasa C (PKC) (apigenina y la curcumina) y las enzimas de la ruta de metabolización
del ácido araquidónico, las cuales están implicadas en la regulación de la proliferación
celular [65-69]
Inducción de la parada del ciclo celular. Las perturbaciones en la progresión del ciclo
celular se encuentran entre los efectos anticancerígenos de los flavonoides. La célula
puede encontrarse en: el estado de quiescencia o en el estado de división. Cuando la
célula decide dividirse entra en el llamado ciclo celular, proceso ordenado y repetitivo
en el que la célula se divide en dos células hijas. En el ciclo celular se distinguen varias
fases diferenciadas (G1 S, G2 y M) y su duración varía en función del tipo celular.
Para garantizar que el ciclo celular progrese la célula dispone de un complejo sistema de
regulación a lo largo del ciclo [70] (Figura 4) que consta de una amplia gama de
moléculas, entre los que se encuentran:

Intoducción
38
• Proteínas quinasas dependientes de ciclina (CDKs): CDK G1 (4,6 y 2), CDK de la
fase S (CDK 2), CDK mitóticas (CDKs 2 y 1) que fosforilan elementos claves en la
progresión del ciclo. Estas moléculas requieren unirse a las ciclinas para ser activas y
sus niveles siempre son los mismos a lo largo del todo el ciclo [71,72].
• Ciclinas: las ciclinas D (D1, D2, D3) se unen a la CDK 4 y a la CDK 6 para formar el
complejo necesario para entrar en la fase G1, la ciclina E se une a la CDK 2 para
regular la progresión del ciclo de G1 a S, la ciclina A se une a CDK 1, este complejo
se requiere durante la fase S. En la fase G2 y la fase temprana de M el complejo
ciclina A–CDK 1 promueve la entrada a la mitosis y es regulado por la formación del
complejo ciclina B-CDK 1. Las ciclinas se sintetizan y se degradan durante el ciclo
celular y actúan regulando positivamente el ciclo [73-78].
• Moléculas inhibidoras de CDKs: se pueden dividir en dos familias, la familia del
inhibidor de la cdk 4 (INK4) donde se encuentran p15, p16, p18, p19 que inhiben
específicamente a la CDK4 y a la CDK6 [79] y la familia inhibidora de CDK
(CIP/KIP) donde encontramos a p21, p27, p57 que son capaces de inhibir a todas las
CDKs [80] en todas las fases del ciclo. Las moléculas inhibidoras se unen a las CDKs
e inhiben su actividad, participan regulando negativamente al ciclo [81,82]. Sin
embargo, cuando se produce cualquier error, la célula dispone de puntos de
verificación que permiten detectar y evitar que continúe el ciclo celular hasta que se
reparen. En el caso de no producirse la reparación la célula induce su propia muerte
mediante apoptosis.
Un número amplio de cánceres esta asociado con hiperactividad de las CDKS (su
expresión suele estar aumentada en el 25-30 % de los cánceres humanos[83,84]) como
resultado de una mutación en los genes de CDKs o bien por mutaciones en los genes de
los inhibidores de CDKs (alteración y perdida de regulación de la actividad de las CDKs
son marcas inequívocas de neoplasia). Los inhibidores o moduladores de las CDKs
representan un nuevo arsenal de agentes terapéuticos contra el cáncer por lo que en los
últimos años, se ha llevado a cabo una intensa búsqueda de nuevos agentes con estas
propiedades [72].

Intoducción
39
Figura 4. Regulación del ciclo celular por CDKs y ciclinas.
Los flavonoides como la quercetina, la silimarina, la daidzeina, la luteolina, kaempferol,
la apigenina y la genisteina entre otros tienen actividad inhibitoria sobre varias quinasas
relacionadas con el ciclo celular [85,86].
Inducción de diferenciación celular. Ciertos flavonoides son capaces de provocar la
transformación de líneas celulares cancerígenas en células diferenciadas con
características fenotípicas de madurez. Así, la genisteína, la apigenina, la luteolina y la
quercetina inducen la diferenciacion de células de leucemia HL-60 en granulocitos y
monocitos [87,88].
Inhibición de la proliferación celular mediante interacción con los microtúbulos: Los
microtúbulos son polímeros dinámicos de dímeros de tubulina, en continua
polimerización y despolimerización, que juegan un papel crucial en un gran número de
funciones celulares: crecimiento, división celular, movilidad, fagocitosis, transporte o

Intoducción
40
localización intracelular de orgánulos, etc. La capacidad que tienen los micotúbulos del
citoesqueleto de interaccionar con un gran número de proteínas hace que module
muchos procesos de señalización intracelular.
Su papel vital en la mitosis los convierte en una diana muy atractiva para el desarrollo
de fármacos contra el cáncer. Los microtúbulos son las únicas dianas celulares que
interaccionan directamente con los agentes que afectan a los microtúbulos. Los agentes
que afectan a los microtúbulos inhiben la proliferación de células cancerosas por
imposibilidad de las mismas de formar el huso mitotico. Se produce parada mitótica en
la transición de la metafase /anafase y frecuentemente se desencadena la muerte por
apoptosis.
Existen una gran cantidad de compuestos capaces de unirse a la tubulina, modular su
estado de activación y de este modo interferir con la dinámica microtubular a
concentraciones intracelulares mucho más bajas que la de tubulina. Los compuestos que
modulan la actividad de tubulina pueden dividirse de forma general en dos grandes
grupos:
• Los inhibidores de la polimerización o desestabilizantes, como la colchicina,
dolastatina, nocodazol y los alcaloides de la Vinca (vincristina y vimblastina) [89,90]
que se unen a ésta impidiendo que forme microtúbulos.
• Los potenciadores de la polimerización o estabilizantes, como el paclitaxel (conocido
comercialmente como taxol®) y el docetaxel® [91,92].
La alteración de la dinámica de los microtúbulos induce una parada del ciclo celular en
la fase G2/M debido al daño del uso mitótico, seguida de la inducción de apoptosis. La
apoptosis puede activarse además por factores liberados como resultado de los cambios
en la polimerización microtubular ya que los microtubulos interacionan con un gran
número de proteínas. Los microtubulos en condiciones normales podrían estar
secuestrando estas moléculas e inhibiendo su papel señalizador. La liberación de estos

Intoducción
41
factores proteícos, les permitiría alcanzar sus dianas celulares y desencadenar una
respuesta, como por ejemplo, la apoptosis [93].
El mecanismo preciso por el cual se produce la inducción de apoptosis una vez que se
ha producido la parada mitótica no está muy claro. Se ha observado inactivación de Bcl-
2 por fosforilación y activación de Bax o Bad las cuales a su vez inactivan Bcl-2 o Bcl-
xL, en este último caso Bax translocaría a la mitocondria que independientemente de si
interacciona con Bcl-2 o con Bcl-xL dispararía la apoptosis por la via mitocondrial.
Además el aumento de la expresión de Bcl-2 inhibe la apoptosis inducida por los
agentes que se unen a los microtúbulos sin afectar su acción sobre los microtúbulos o
sobre la parada del ciclo celular en G2/M, lo cual indicaría que la apoptosis inducida por
los estos agentes depende de la vía mitocondrial [94-96].
El daño de los microtúbulos conduce al bloqueo de la degradación de la ciclina B1 por
el proteasoma y a una inducción de la expresión de CDK1, dando como resultado la
activación de la quinasa CDK1/ciclina B1 y parada del ciclo celular en G2/M. La
inhibición de la CDK1 por olomoucina o la inhibición de CDKs por medio del inhibidor
endógeno p21, previene la apoptosis inducida por taxol, sugiriendo que CDK1/ciclina-
B1 juega un papel importante en la apoptosis disparada por taxol. El complejo
CDK1/ciclina B1 puede fosforilar Bcl-2 en sistemas libres de células y se ha visto que
la activación de CDK1/ciclina B1 es necesaria para la fosforilación de Bcl-2, poniendo
de manifiesto la vinculación entre la alteración de la dinámica de los microtúbulos, la
activación de CDKs y las proteínas reguladoras de la apoptosis.
El tratamiento con agentes que alteran los microtúbulos provoca el aumento de la
expresión de numerosas proteínas como c-Mos, c-Jun, ciclina B1, CDK1, Mcl-1, DR4 y
DR5, Bak, Bax, p21, COX-2, TNF-α, así como la disminución de los niveles de Bcl-xL,
FLIP, o XIAP. Todos estos cambios en proteínas implicadas en la apoptosis estarían
también relacionados con la capacidad de inducir apoptosis o de sensibilizar a las
células tumorales a otras drogas anticancerígenas. La parada del ciclo podría
desencadenar la liberación de la survivina de los microtúbulos, disminuyendo así su
actividad antiapoptótica [96-108].

Intoducción
42
Algunos flavonoides han demostrado interferir la polimerización de la tubulina. Las
flavonas que contienen un grupo hidroxilo en C-3´y C-5 y grupos metoxi en C-3 y C-
4´, son capaces de alterar la dinámica de los microtúbulos. Se ha descrito que la
quercetina, se comporta como la colchicina, se une al mismo sitio que lo hace la
colchicina, induce la acividad GTPasa de la tubulina soluble y perturba la
polimerización de microtúbulos por inducción de cambios conformacionales en la
tubulina [109]. La casticina también se comporta como inhibidor de la polimerización
promueve parada en G2/M y activa la apoptosis.
1.2.10.3 INHIBICIÓN DE LA FASE INVASIVA.
La posibilidad de bloquear el crecimiento tumoral por inhibición de la angiogénesis
representa uno de los objetivos primordiales en el tratamiento de tumores sólidos. Las
metaloproteinasas de la matriz extracelular (MMPs), los factores de crecimiento
angiogénicos y sus receptores son los principales objetivos para la inhibición de la
angiogénesis. El galato de epigalocatequina (EGCG), uno de los flavonoides del té
verde, es un potente inhibidor de la MMP-2 y MMP-9 (metaloproteinasas de la matriz
extracelular) y de la quinasa de adhesión focal (FAK) inhibiendo la invasión y
metástasis de las células cancerígenas [110].
2. APOPTOSIS.
La supervivencia de un organismo multicelular depende del balance entre la vida y la
muerte de sus células. La apoptosis o "muerte celular programada" se puede definir
como el conjunto de reacciones bioquímicas que tienen lugar en la célula, que
concluyen con la muerte de la célula de una forma ordenada y silenciosa, sin producir
ningún tipo de reacción en los tejidos. La apoptosis es un proceso de autodestrucción
celular controlada que permite al organismo su correcta morfogénesis y la eliminación
de las células que amenacen su supervivencia además de ser necesaria para evitar la
sobreproducción celular. Este tipo de muerte celular es de vital importancia, tanto
durante el desarrollo embrionario como durante la vida adulta [111,112].

Intoducción
43
La muerte celular programada es parte integral del desarrollo de los tejidos tanto de
plantas como de animales pluricelulares. La célula apoptótica rápidamente es fagocitada
por macrófagos o por células vecinas debido a la exposición de marcadores de
fagocitosis en la superficie celular como la fosfatidilserina, evitando la exposición del
material intracelular al sistema inmune que podría desencadenar una respuesta
inflamatoria. La célula apoptótica sufre una serie de cambios morfológicos y
bioquímicos característicos [111,113]:
• Cambios morfológicos: la membrana plasmática se altera, existe una pérdida de
estructura de la superficie celular, disminuye el volumen celular y se produce la
condensación del citoplasma. Además se produce la pérdida de orgánulos
principalmente mitocondrias, el engrosamiento del retículo endoplasmático,
alteraciones en la membrana nuclear, así como condensación y fragmentación nuclear dando lugar a la formación de los llamados cuerpos picnóticos ó apoptóticos.
• Cambios bioquímicos: aumento de la concentración del calcio iónico libre,
interacción Bcl-2/Bax, deshidratación celular, perdida del potencial de membrana
mitocondrial, externalización de fosfatidilserina, proteólisis de laminina b,
desnaturalización e hidrólisis internucleosómica del ADN en 50-30 Kb.
La perdida de regulación de la apoptosis es un factor crítico en la oncogénesis. La
inducción de la apoptosis de células tumorales o el impedimento de su inhibición son
hipotéticos mecanismos antitumorales de algunos flavonoides, entre los mecanismos
que se barajan se encuentra: la disminución de las especies reactivas de oxigeno
(EROs), la regulación de la expresión de proteínas de choque térmico, la modulación de
las vías de señalización, la inhibición de la actividad de la topoisomerasa I/II, la
liberación de citocromo c con la subsiguiente activación de las caspasas-3 y -9, la
represión de proteínas antiapoptóticas como Bcl-2 y Bcl-xL y aumento de la expresión
de Bax y Bak (proteínas proapoptóticas), activación de la endonucleasa G y la supresión
de la proteína Mcl-1. La inducción de la rotura del ADN y la fragmentación proteíca es
otro posible mecanismo.

Intoducción
44
Diversas enfermedades están asociadas con la pérdida de la regulación del proceso
apotótico, algunas presentan un aumento de la supervivencia celular (asociado a la
inhibición de la apoptosis) y otras un exceso de muerte celular (asociado a un exceso de
activación de la apoptosis) [114]. Entre las enfermedades en las que hay una inhibición
de la apoptosis se incluye el cáncer, las infecciones virales y las enfermedades
autoinmunes [115]. Los trastornos neuro-degenerativos como el Alzheimer o el
Parkinson y el síndrome de inmunodeficiencia adquirida son ejemplos claros de un
exceso de apoptosis.
Dado que en muchos tumores malignos la apoptosis esta inhibida esta se acepta como
un mecanismo clave en todas las facetas del cáncer, incluyendo la hiperplasia,
transformación neoplásica, la expansión tumoral, neo- vascularización y la metástasis.
Los flavonoides son capaces de actuar como agentes anticancerígenos mediante la
inducción de apoptosis de ahí el estudio exhaustivo de esta forma de muerte
programada.
2.1 EL PROCESO APOPTÓTICO.
Existen varios factores por los cuales se puede desencadenar la apoptosis en las células.
La sensibilidad de la célula a un estímulo dependerá de la expresión de proteínas pro- y
anti-apoptóticas y de proteínas inhibidoras de la apoptosis, de la fuerza del estímulo y el
estado del ciclo celular en el que se encuentre.
El inicio de la señal apoptótica puede ocurrir tanto fuera de la célula, a través de los
receptores de muerte, como dentro de ella como consecuencia de estímulos que inducen
estrés celular (radiación, fármacos, infección viral, deficiencia de factores de
crecimiento, aumento de radicales libres que induzcan un estrés oxidativo) o daño en el
ADN. En general las señales intrínsecas desencadenan la apoptosis vía mitocondrial o
también conocida como vía intrínseca de la apoptosis mientras que las que se inician a
nivel de receptores de muerte desencadenan la vía extrínseca.
Intoducción
45
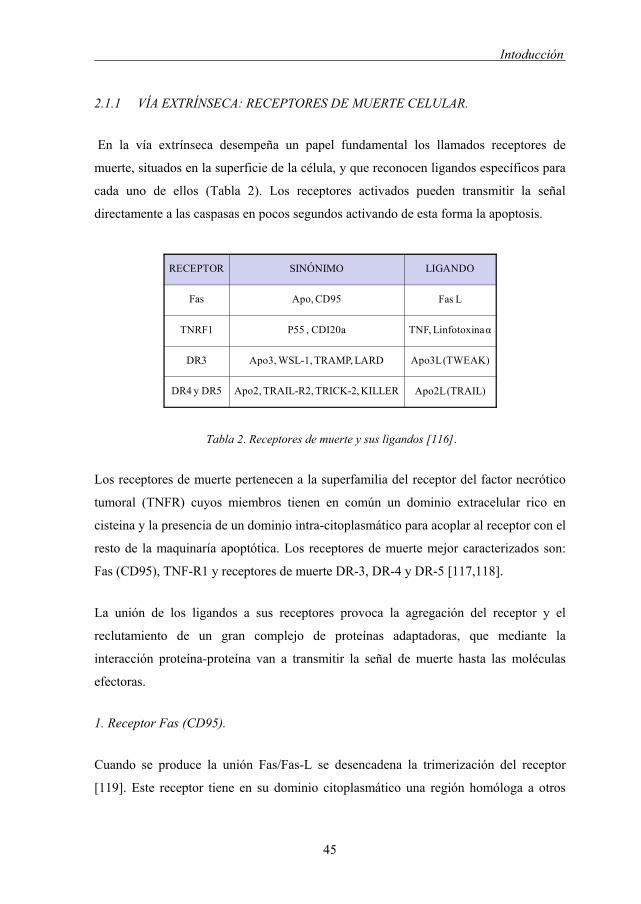
2.1.1 VÍA EXTRÍNSECA: RECEPTORES DE MUERTE CELULAR.
En la vía extrínseca desempeña un papel fundamental los llamados receptores de
muerte, situados en la superficie de la célula, y que reconocen ligandos específicos para
cada uno de ellos (Tabla 2). Los receptores activados pueden transmitir la señal
directamente a las caspasas en pocos segundos activando de esta forma la apoptosis.
Tabla 2. Receptores de muerte y sus ligandos [116].
Los receptores de muerte pertenecen a la superfamilia del receptor del factor necrótico
tumoral (TNFR) cuyos miembros tienen en común un dominio extracelular rico en
cisteina y la presencia de un dominio intra-citoplasmático para acoplar al receptor con el
resto de la maquinaría apoptótica. Los receptores de muerte mejor caracterizados son:
Fas (CD95), TNF-R1 y receptores de muerte DR-3, DR-4 y DR-5 [117,118].
La unión de los ligandos a sus receptores provoca la agregación del receptor y el
reclutamiento de un gran complejo de proteínas adaptadoras, que mediante la
interacción proteína-proteína van a transmitir la señal de muerte hasta las moléculas
efectoras.
1. Receptor Fas (CD95).
Cuando se produce la unión Fas/Fas-L se desencadena la trimerización del receptor
[119]. Este receptor tiene en su dominio citoplasmático una región homóloga a otros
RECEPTOR SINÓNIMO LIGANDO
Fas Apo, CD95 Fas L
TNRF1 P55 , CDI20a TNF, Linfotoxinaα
DR3 Apo3, WSL-1, TRAMP, LARD Apo3L (TWEAK)
DR4 y DR5 Apo2, TRAIL-R2, TRICK-2, KILLER Apo2L (TRAIL)

Intoducción
46
receptores capaces de inducir apoptosis. Al dominio implicado en la transducción de la
señal, se le denomina dominio de muerte, (DD). La activación de este receptor provoca
el reclutamiento y unión de otras moléculas adaptadoras como son FADD y RIP
[119,120].
• FADD (Proteína con dominio de muerte asociado a Fas) contiene dos dominios
importantes, que son el dominio DD, con el cual interacciona Fas, y el dominio DED
(dominio efector de muerte), que es el encargado de transmitir la señal apoptótica a
los efectores, ya que poseen también un dominio DED (caspasa 8/10).
• RIP (Proteína de interacción con receptor), es una proteína Ser/Thr quinasa que tiene
dos formas de actuación dependiendo de que su unión sea a Fas o a TNFR1. En el
caso de unirse a Fas, lo hace también por su dominio DD al DD de Fas. En este
momento está en condiciones de interaccionar con RAIDD (proteína homóloga
asociada a RIP con dominio de muerte), el cual también tiene un dominio DD y otro
dominio homólogo a caspasa-2, aunque esta vía no suele ser mayoritaria.
Dentro de esta activación existen dos mecanismos muy importantes, que dependen del
tipo celular [119,121]. Así algunos autores diferencian dos tipos de células (Figura 5):
Tipo I: células en las que la expresión de caspasa-8 es abundante y podrían activar
directamente a caspasa-3, aunque también a caspasa-6 y -7 a través de las cascadas de
las caspasas. Este tipo de inducción no requiere de la mitocondria y por tanto no es
inhibida por factores como Bcl-2 o Bcl-xL, ya que no requiere amplificación de la señal
por factores pro-apoptóticos derivados de la mitocondria [122].
Tipo II: células en las que la expresión de caspasa-8 no es abundante y no puede activar
eficientemente a caspasa-3, ni a otras caspasas en la cascada de caspasas. En estas
células la activación de caspasa-8 provoca la activación proteolítica de un miembro de
la familia Bcl-2, como es Bid. Esta activación provoca la translocación de Bid a la
mitocondria, donde interacciona con moléculas antiapoptóticas de la familia Bcl-2 como
son Bax y Bak que se encuentran como componentes de la membrana mitocondrial e
Intoducción
47
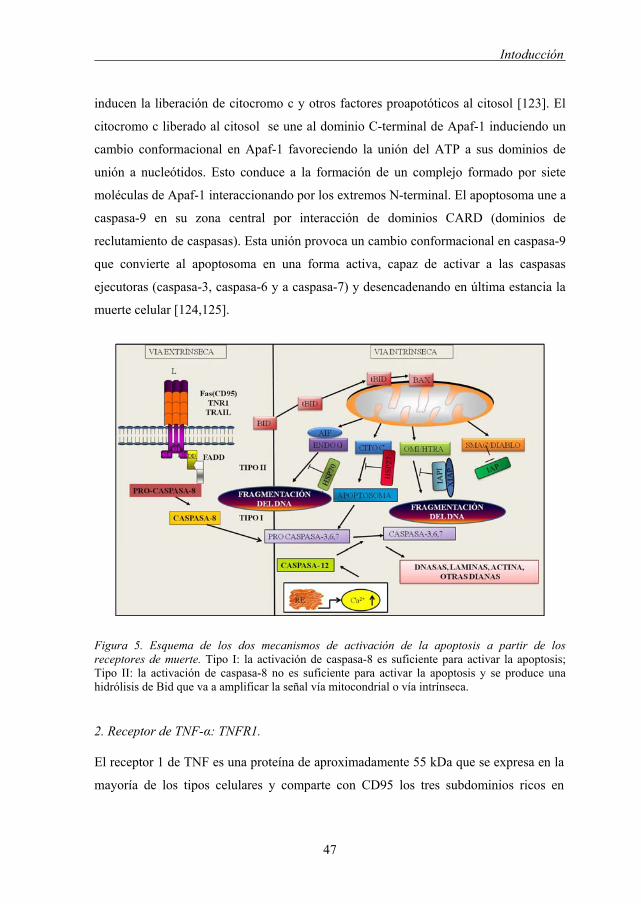
inducen la liberación de citocromo c y otros factores proapotóticos al citosol [123]. El
citocromo c liberado al citosol se une al dominio C-terminal de Apaf-1 induciendo un
cambio conformacional en Apaf-1 favoreciendo la unión del ATP a sus dominios de
unión a nucleótidos. Esto conduce a la formación de un complejo formado por siete
moléculas de Apaf-1 interaccionando por los extremos N-terminal. El apoptosoma une a
caspasa-9 en su zona central por interacción de dominios CARD (dominios de
reclutamiento de caspasas). Esta unión provoca un cambio conformacional en caspasa-9
que convierte al apoptosoma en una forma activa, capaz de activar a las caspasas
ejecutoras (caspasa-3, caspasa-6 y a caspasa-7) y desencadenando en última estancia la
muerte celular [124,125].
Figura 5. Esquema de los dos mecanismos de activación de la apoptosis a partir de los receptores de muerte. Tipo I: la activación de caspasa-8 es suficiente para activar la apoptosis; Tipo II: la activación de caspasa-8 no es suficiente para activar la apoptosis y se produce una hidrólisis de Bid que va a amplificar la señal vía mitocondrial o vía intrínseca.
2. Receptor de TNF-α: TNFR1.
El receptor 1 de TNF es una proteína de aproximadamente 55 kDa que se expresa en la
mayoría de los tipos celulares y comparte con CD95 los tres subdominios ricos en

Intoducción
48
cisteinas situados en la zona extracelular. El ligando de TNFR1 es TNF-α. A diferencia
de la pareja formada por CD95/ CD95L, el par TNFR1/TNF es capaz de trasmitir a la
célula dos tipos de señales muy distintas. La unión de TNF-α a su receptor promueve
que el dominio intracelular de TNFR1 sea reconocido por proteína (TRADD) que lleva
un dominio de muerte asociado y que puede unirse a RIP, TRAF-2 (factor 2 asociado a
TNF-R) o a FADD. Si TRADD se une a FADD esta última se unira a caspasa-8
activándola mediante autoproteólisis e inducirá la apoptosis. Si TRADD se une a
TRAF- 2 recluta proteínas inhibidoras de la apoptosis como cIAP-1 y cIAP-2, aunque
tambien puede activar MAPKKKs, activando la cascada de quinasas dando lugar a la
activación de JNK. Si TRADD se une a la proteina RIP se induce la activación de la
transcripción de NF-kB y AP-1 dando lugar a la inducción de genes de carácter
proinflamatorio e inmunomodulador y de esta manera, se opondría a la acción
apoptótica del TNF-α. La unión del ligando al receptor puede dar lugar a dos tipos de
complejos: Complejo I formado por TRADD unido a RIP y a TRAF2 que dará lugar a
la señalizacion de NF-kB y Complejo II: formado por TRADD unido a FADD que va a
inducir la autoproteólisis de caspasa-8/10 y por tanto va a inducir la apoptosis (Figura
6).
Los niveles de FLIP van a determinar que prevalezca una vía u otra. La señalización de
apoptosis por medio de TNFR1 es mucho más limitada que la mediada por CD95. En el
caso de TNFR1, la unión de su ligando induce apoptosis en algunos tipos celulares y
solo cuando la síntesis de proteínas ha sido bloqueada. De este hecho se deduce que
debe existir en las células algún factor que bloquee las señales de apoptóticas derivadas
de TNFR1 (existe la hipótesis de que el complejo I podría regular los niveles de FLIP),
por lo que se puede concluir que la expresión de este factor estará probablemente
controlada a través de NFkB y JNK/AP-1 [126].
Además TNF-R1 puede activar dos tipos de esfingomielinasas, una de ellas, localizada
en la membrana plasmática y denominada esfingomielinasa neutra, provocaría la
acumulación de ceramida al romper el enlace fosfodiester entre la fosfocolina y la
ceramida [127] activando una serie MAP-quinasas que llevan a la activación de la ruta
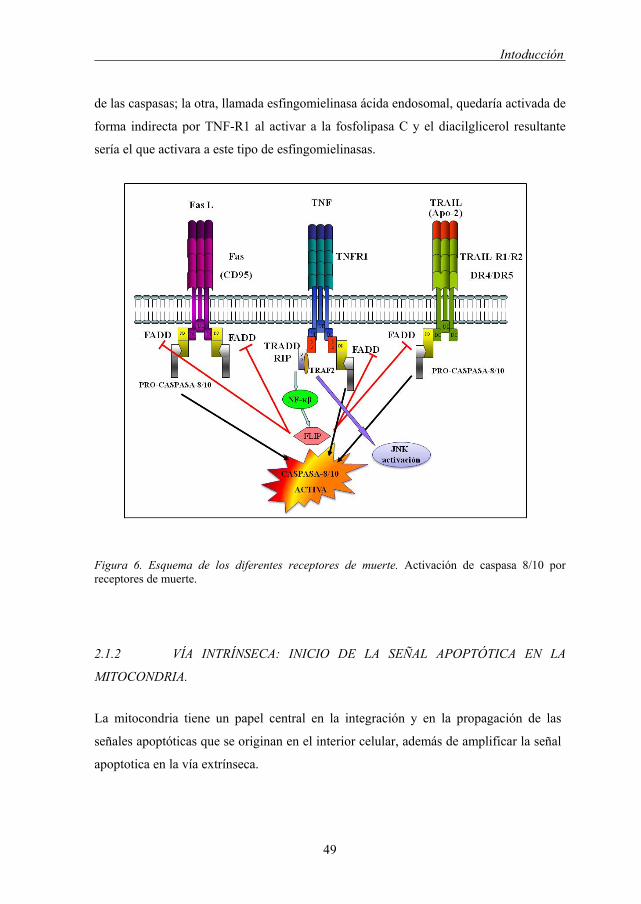
Intoducción
49
de las caspasas; la otra, llamada esfingomielinasa ácida endosomal, quedaría activada de
forma indirecta por TNF-R1 al activar a la fosfolipasa C y el diacilglicerol resultante
sería el que activara a este tipo de esfingomielinasas.
Figura 6. Esquema de los diferentes receptores de muerte. Activación de caspasa 8/10 por receptores de muerte.
2.1.2 VÍA INTRÍNSECA: INICIO DE LA SEÑAL APOPTÓTICA EN LA
MITOCONDRIA.
La mitocondria tiene un papel central en la integración y en la propagación de las
señales apoptóticas que se originan en el interior celular, además de amplificar la señal
apoptotica en la vía extrínseca.

Intoducción
50
Las señales de muerte incluyen una gran variedad de estímulos que implican estrés
celular como son fármacos, radiaciones, agentes oxidantes, sobrecarga de Ca+2, etc.
Algunos de estos estímulos actúan directamente sobre la mitocondria y otros lo hacen a
través de moléculas mediadoras como las ceramidas y el factor proapoptótico Bax. Los
efectos de estas señales se traducen en el incremento de la permeabilidad mitocondrial
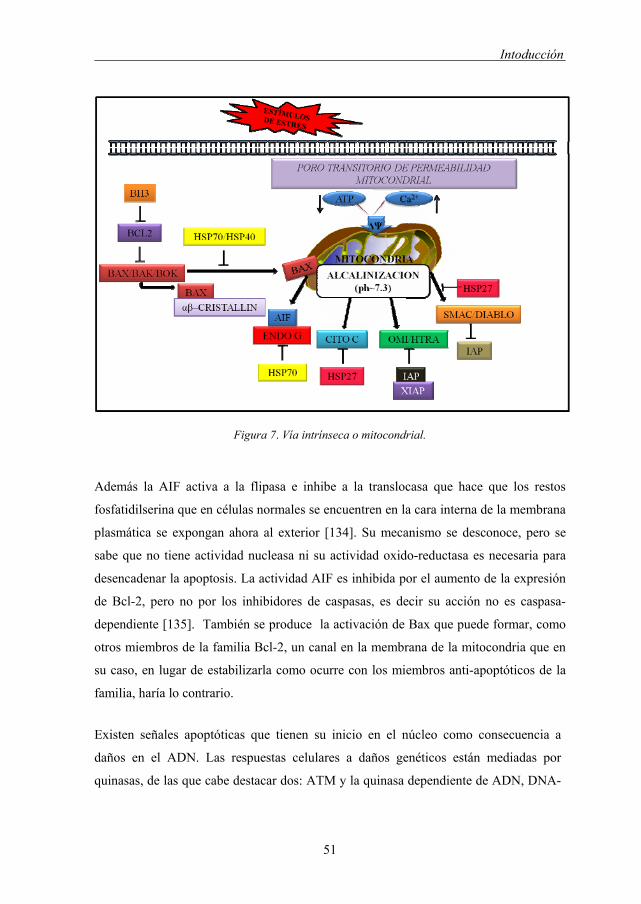
de modo que se produce una liberación de (Figura 7):
• Citocromo c: proteína que funciona como transportador electrónico mitocondrial y
que una vez liberada al citosol va a participar en la activación de caspasa-9 a través de
la formación del apoptosoma.
• Endonucleasa G: se trasloca al núcleo tras la liberación y está involucrada en la
fragmentación internucleosómica del ADN, produciendose un patrón en escalera
típico [128].
• Smac/Diablo: inhibidor de las proteínas inhibidoras de apoptosis (IAPs) las cuales
poseen la capacidad de inhibir a las caspasas activas (por tanto Smac/Diablo previene
la inactivación de las caspasas. La unión de las IAPs a caspasas y a Smac/Diablo se
produce en el mismo dominio, por tanto se produce una competencia de unión
[129,130]).
• Htr/Omi: es una serín proteasa que hidroliza e inactiva a IAPs, mediante la
inactivación por corte [131].
Como resultado se produce la ruptura de la cadena de transporte de electrones, la
liberación de iones superóxido [132,133] y la hiperpolarización de la membrana
mitocondrial interna (que puede terminar con una expansión de la matriz y ruptura de la
membrana mitocondrial externa de la mitocondria). La formación de poros en la
mitocondria conduce a la entrada de agua y solutos en la matriz con el consiguiente
choque osmótico, y liberación del factor inductor de apoptosis AIF, una flavoproteína
que transloca al núcleo donde provoca la condensación y rotura de ADN en fragmentos
de alto peso molecular (50Kb).
Intoducción
51
Figura 7. Vía intrínseca o mitocondrial.
Además la AIF activa a la flipasa e inhibe a la translocasa que hace que los restos
fosfatidilserina que en células normales se encuentren en la cara interna de la membrana
plasmática se expongan ahora al exterior [134]. Su mecanismo se desconoce, pero se
sabe que no tiene actividad nucleasa ni su actividad oxido-reductasa es necesaria para
desencadenar la apoptosis. La actividad AIF es inhibida por el aumento de la expresión
de Bcl-2, pero no por los inhibidores de caspasas, es decir su acción no es caspasa-
dependiente [135]. También se produce la activación de Bax que puede formar, como
otros miembros de la familia Bcl-2, un canal en la membrana de la mitocondria que en
su caso, en lugar de estabilizarla como ocurre con los miembros anti-apoptóticos de la
familia, haría lo contrario.
Existen señales apoptóticas que tienen su inicio en el núcleo como consecuencia a
daños en el ADN. Las respuestas celulares a daños genéticos están mediadas por
quinasas, de las que cabe destacar dos: ATM y la quinasa dependiente de ADN, DNA-

Intoducción
52
PK. Ambas dirigen una serie de respuestas entre las que se encuentran la detención del
crecimiento celular, la reparación y la apoptosis.
2.1.3 PROTEÍNAS REGULADORAS DE LA SEÑAL DE APOPTÓTICA:
2.1.3.1 PROTEÍNAS DE LA FAMILIA BCL-2
Una de las vías más importantes en la regulación de la apoptosis se lleva a cabo por la
familia de proteínas Bcl-2. Los miembros de esta familia tienen en común la presencia
en su estructura de al menos una de cuatro secuencias consecutivas posibles, BH1 a
BH4. De estos dominios, BH3 está directamente relacionado con una función pro-
apoptótica mientras que BH1, BH2 y BH4 desempeñan una función anti-apoptótica.
La familia Bcl-2 se clasifica en dos grandes grupos (Figura 8). Los miembros del
primer grupo comparten alta homología estructural y funcional con Bcl-2 y tienen
actividad anti-apoptótica (Bcl-xL, Bcl-w, A1/BFL-1, y Mcl-1). Los miembros del
segundo grupo son proteínas que comparten una homología menor con Bcl-2 y tiene
actividad pro-apoptótica (Bax, Bak, Bid).
Figura 8. Miembros de la familia Bcl-2.

Intoducción
53
Los factores pro-apoptóticos se dividen en dos grupos. Los que poseen los dominios
BH1, BH2, BH3 (Bax, Bak y Bok) y los que contienen solo el dominio BH3 necesario
y suficiente para realizar la acción de muerte celular (Bid, Bim, Bik, Bad, Bmf, Hrk,
Noxa, Puma, B1k, BNIP3 y Spike) [136]. La hetero-dimerización no es necesaria para
la actividad de los miembros anti-apoptóticos de la familia Bcl-2 y para los pro-
apoptóticos del grupo de Bax. Sin embargo, es muy importante para los miembros pro-
apoptóticos del grupo BH3 que basan gran parte de su funcionamiento en la unión a las
proteínas anti-apoptóticas alterando su actividad. Los miembros de la familia Bcl-2
interactúan de una forma dinámica para regular la apoptosis. Los miembros pro-
apoptóticos como Bax pueden hetero-dimerizar con los anti-apoptóticos como Bcl-2 y
bloquear su acción anti-apoptótica. Por tanto la relación en los niveles de expresión
entre anti-apoptóticos/pro-apoptóticos determina el destino celular. Cuando Bcl-2 está
en exceso, las células están protegidas de la muerte celular, sin embargo cuando Bax
está en exceso la célula está avocada a entrar en apoptosis. Bax también tiene la
habilidad de hetero-dimerizar con Bcl-xL, por lo cual un balance entre los dos también
es necesario para la inducción o represión del estimulo apoptótico.
Los miembros de la familia Bcl-2 parecen tener localizaciones subcelulares diferentes.
Los miembros anti-apoptóticos suelen estar como proteínas integrales en las membranas
del núcleo, del retículo endoplásmico y fundamentalmente de la mitocondria. En este
último orgánulo su principal función es la de estabilizar la membrana mitocondrial
externa, evitando la liberación del citocromo c y su posterior unión al factor de
activación de la apoptosis-1 (Apaf-1). En cambio los miembros pro-apoptóticos se
encuentran en el citosol o asociados con el citoesqueleto [137,138] donde a raíz de un
estímulo de muerte sufren un cambio conformacional que les permite integrarse también
en las membranas, especialmente en la membrana mitocondrial externa. Esto se ha
podido comprobar incluso con Bim, el cual sólo posee un dominio BH3 [138]. En la
mitocondria interactúan con los miembros anti-apoptóticos, inhibiendolos e induciendo
la activación de la maquinaria apoptótica. La regulación de la actividad de los miembros
de la familia Bcl-2 se lleva a cabo mediante homo/hetero-dimerización o mediante
fosforilación y des-fosforilación.

Intoducción
54
Factores anti-apoptóticos de la familia Bcl-2.
Entre los más representativos se encuentran:
• Bcl-2: es la proteína prototipo de esta familia. Su peso molecular es de 26 KDa y
posee los cuatro dominios que la definen (BH1-BH4). Es una proteína integral
localizada en la cara citoplasmática de la membrana externa de la mitocondria, en la
membrana del retículo endoplásmico y en la envuelta nuclear. Forma una estructura
similar a un poro que le permite modificar el flujo de moléculas o pequeñas proteínas
a través de la membrana, interviniendo en la estabilidad de orgánulos como la
mitocondria ante la existencia de posibles daños [136]. Puede evitar, o al menos
retrasar, la muerte celular inducida por retirada de factores de crecimiento,
irradiación, glucocorticoides y múltiples drogas quimioterápicas [139]. Promueve la
tumorogénesis mediante la prevención de la muerte de la célula, incrementando la
tasa de división celular.
• Bcl-xL: es una de las proteínas más estrechamente relacionada con Bcl-2, tanto en su
estructura como en su función. Posee los cuatro dominios BH y su peso molecular es
de 30 KDa. Puede mediar una resistencia significativa a la muerte celular por
apoptosis dependiente de la falta de factores de crecimiento. Bcl-xL ejerce su acción
anti-apoptótica mediante varios mecanismos: es capaz de interaccionar con las
moléculas pro-apoptóticas como Bax y bloquear de esta forma su acción pro-
apoptótica. Por otro lado regula la permeabilidad de la membrana mitocondrial
previniendo la liberación del citocromo c y su posterior unión al factor de activación
de la apoptosis-1 (Apaf-1). Bcl-xL es también capaz de unirse a Apaf-1, formando un
complejo que impide la activación de la caspasa-9.
Factores pro-apoptóticos de la familia Bcl-2.
• Subfamilia Bax: Bax es uno de los miembros pro-apoptóticos más importantes de la
familia de Bcl-2 y le da nombre a la subfamilia Bax. Su peso molecular es de 21
KDa y posee los dominios BH1, BH2 y BH3 aunque son BH1 y BH2 los que

Intoducción
55
guardan una estrecha homología con Bcl-2. Bax está presente en los distintos tejidos
y el aumento de su expresión acelera la muerte en respuesta a distintas señales. P53
regula los niveles de transcripción de Bax. En condiciones normales Bax se
encuentra en el citosol o en el citoesqueleto en forma monomérica. La exposición a
estímulos apoptóticos promueve la translocación de Bax a la mitocondria donde
puede integrarse tanto en la membrana externa como en la membrana interna de la
mitocondria, formando homo oligómeros o hetero oligómeros con Bak. Existe
controversia en cuanto a si Bax interacciona con la cardiolipina y si esta interacción
sería la única necesaria para alterar el potencial de membrana mitocondrial (MMP) o
si interacciona directamente con proteínas de la mitocondria como son el canal de
iones dependiente de voltaje (VDAC) o la translocasa de nucleótidos de adenina
(AWT). En cualquier caso la oligomerización y la asociación con la membrana
mitocondrial externa afecta al potencial de membrana (MMP), induciendo
homodimerización de la proteína Bax, que dará como consecuencia liberación del
citocromo c al citosol que se unirá a Apaf-1 formando el complejo que unirá a pro-
caspasa-9, activándola y dando lugar a la cascada proteolítica que conducirá a la
muerte celular [140,141].
La regulación de Bax se lleva a cabo por fosforilación, que inhibe su efecto pro-
apoptótico.
• Subfamilia BH3.
Esta subfamilia está compuesta solo por miembros pro-apoptóticos que, excepto por el
dominio BH3, no muestran homología con Bcl-2. Para ejercer su actividad, estas
proteínas pueden formar heterodímeros con miembros anti-apoptóticos de la familia:
como Bcl-2 y Bcl-xL [142]. Para ello, el dominio BH3 de los miembros de este grupo
puede introducirse en el hueco hidrofóbico formado por la asociación de las regiones
BH1, BH2 y BH3 de los miembros anti-apoptóticos. En esta subfamilia se encuentran
Bad, Bid y Bik. Bid y Bik actúan sobre la mitocondria, donde inducen la liberación de
citocromo c (Figura 9) [143].

Intoducción
56
Figura 9. Regulación de la apoptosis por la familia Bcl-2.
Bad es regulado por fosforilación/de-fosforilación (la de-fosforilación de Bad induce su
activación y la unión a las proteínas anti-apoptóticas de la familia Bcl-2 en la
mitocondria). Cuando Bad está en la forma fosforilada se encuentra inactiva
localizándose en el citosol, pudiendo estar unida a la proteína 14-3-3. La hidrólisis de la
proteína 14-3-3 promueve la muerte celular mediante la liberación de Bad, su
translocación a la mitocondria y su interacción con Bcl-xL. Bid es la proteína que inter-
relaciona las dos vías apoptóticas (vía extrínseca y vía intrínseca). La activación de
caspasa-8 induce la hidrólisis de Bid formando un fragmento truncado, tBid, que se
transloca a la membrana mitocondrial e indúce la liberación de citocromo c. Bid y Bad
cooperan para provocar la disfunción de la membrana mitocondrial. Un modelo
propuesto asume que Bid una vez en la membrana mitocondrial inactiva Bcl-2 o activa a
Bax.

Intoducción
57
La perdida de la regulación sobre los miembros de Bcl-2 está estrechamente vinculada a
la tumorogénesis. Los miembros anti-apoptóticos de la familia Bcl-2 parecen funcionar
como onco-proteínas y los pro-apoptóticos como supresores de tumores.
2.1.3.2 PROTEÍNAS DE LA FAMILIA DE LAS CASPASAS.
Los miembros ejecutores de la apoptosis son una serie de cisteín-proteasas dirigidas a
aspartato, englobadas bajo el nombre de caspasas. La familia de las caspasas se
compone de 15 miembros en mamíferos, que se agrupan en dos grandes sub-familias:
las caspasas relacionadas con la inflamación y las caspasas apoptóticas. Se han descrito
11 caspasas en humanos, caspasa-1, -2, -3, -4, -5, -6, -7, -8, -9, -10 y -14. La enzima
denominada inicialmente caspasa-13 fue posteriormente identificada como un
homólogo bovino de la caspasa-4 humana y las caspasas-11 y -12 son enzimas murinas
que representan los homólogos de las caspasas-4 y -5 humanas. Todas ellas tienen en
común que se encuentran en forma de zimógenos o proenzimas con una estructura bien
definida: a) un dominio N-terminal muy variable tanto en su secuencia como en su
longitud, con funciones de regulación y activación, b) y una región catalítica formada
por dos dominios, uno grande (20 KDa) y otro pequeño (10 KDa), que darán lugar a las
dos subunidades del enzima una vez activada. Las caspasas se dividen en dos grupos
según la longitud de su región reguladora N-terminal o prodominio. Las caspasas con
un prodominio largo como son las caspasa-1, -2, -4, -5, -8, -9 y -10 parecen estar
involucradas en funciones de regulación de la activación de la cascada. Ejemplos bien
conocidos de este grupo son las pro-caspasa-8 y -10 que contienen en sus largos
prodominios repeticiones de una secuencia de interacción proteina-proteina llamada
dominio efector de muerte o DED y las pro-caspasa-1, -2, -4, -5 y -9, que contienen
dominios de reclutamiento de caspasas o CARDs. La presencia en su estructura de estas
secuencias unido a su localización cercana a la membrana plasmática hacen posible su
reclutamiento hacia el complejo formado en torno a receptores de superficie
señalizadores de apoptosis como CD95 y TNF, activándose allí y dando lugar al
comienzo de la cascada de proteólisis. Por esta situación en la cascada se las denomina
caspasas iniciadoras. El otro grupo denominadas caspasas efectoras, está compuesto por

Intoducción
58
las caspasas con prodominio corto como son las caspasas-3, -6 y -7 que son activadas
por alguna de las caspasas iniciadoras. Actúan al final de la cascada hidrolizando
numerosas proteínas celulares. Las caspasas realizan su función enzimática de forma
específica y eficaz.
El mecanismo de acción de las caspasas tiene importancia a la hora de explicar tanto su
papel en la apoptosis como su activación. Todas las caspasas se activan por proteólisis
y cumplen todas las condiciones para que esta activación sea llevada a cabo por otras
caspasas. Una de estas moléculas activa a la otra cortando entre sus dominios. Este
proceso de activación puede permitir a las caspasas realizar su función con un efecto en
cascada que se amplifica con el estímulo. Se han descrito diferentes mecanismos para
la activación de las caspasas:
• Activación por otra caspasa: las caspasas son proteasas que cortan después de un
residuo de ácido aspártico. Un punto de corte de aspártico separa el prodominio de
p20 y uno o dos separan p20 de p10. Esto sugiere la posibilidad de activación
autocatalítica. Un modo simple de activar una procaspasa es exponerla a otra
previamente activada. Esta estrategia de activación, denominada cascada de
caspasas, es muy utilizada por las células para la activación de las tres caspasas que
tienen un prodominio corto (caspasa-3, -6 y -7). Estas tres caspasas son más
abundantes y activas que las otras caspasas con un prodominio más largo. La cascada
de caspasas amplifica las señales proapoptóticas, pero no pueden explicar cómo se
inicia la activación de la primera caspasa. Existen al menos dos aproximaciones que
explican dicha activación.
• Activación inducida por proximidad: las concentraciones elevadas de determinadas
pro-caspasas en la célula hace que estas se agrupen y se autoactiven. Esto sugiere
que las caspasas iniciadoras podrían encontrarse como monómeros a bajas
concentraciones y que la molécula adaptadora unida al receptor de muerte serviría
para unirlas propiciando su autocatálisis. La caspasa-8 es la caspasa iniciadora clave
en la vía de los receptores de muerte. Después de la unión del ligando, los receptores
de muerte como CD95 (Apo-1/Fas) se agregan y forman un complejo de
señalización en la membrana. Estos complejos reclutan, a través de sus proteínas

Intoducción
59
adaptadoras, varias moléculas de pro-caspasa-8 aumentando la concentración local
de zimógeno. En estas condiciones la actividad proteasa de la pro-caspasa-8 es
suficiente para permitir que varias moléculas de proenzima se corten mutuamente y
se activen unas a otras.
• Asociación con una subunidad reguladora: se denomina también autocatálisis
facilitada. Las caspasas se encuentran en la célula formando complejos o con una
conformación tal que previene su autocatálisis. En este estado un cofactor actuaría
facilitando la activación, cambiando la conformación, o liberando un inhibidor del
complejo, permitiendo la autocatálisis de la proteína. Un ejemplo de esta activación
es el utilizado por la caspasa-9. A diferencia de otras caspasas, el procesamiento
proteolítico de la procaspasa-9 tiene un efecto mínimo en su activación. El
requerimiento clave para la activación de la caspasa-9 es su asociación con un
cofactor de proteínas, Apaf-1. También es necesario el citocromo c liberado por la
mitocondria. El citocromo c y Apaf-1 se asocian en un proceso dependiente de ATP.
La oligomerización de Apaf-1 recluta procaspasas-9 formando el apoptosoma. La
activación de la caspasa-9 es debida a un cambio conformacional, no a proteólisis.
La señal de las caspasas como ya se mencionó anteriormente depende del tipo de
activación. Si la activación es via receptor (vía extrínseca) o bien si es vía mitocondrial
(vía intrínseca). Si es vía receptor existen dos vías de activación de caspasas: tipo 1
caracterizada por altos niveles de DISC y por tanto grandes cantidades de caspasa-8
capaz de activar a las caspasa efectoras y, tipo 2 donde la cantidad de caspasa-8 no es
suficiente para activar las caspasas efectoras y la señalización requiere la amplificación
mediante la via mitocondrial por hidrólisis de Bid, generacion de tBid y liberación de
citocromo c de la mitocondria dando lugar mediante su union a Apaf-1 y pro-caspasa-9
a la formación del apoptosoma, activación de la caspasa-9 y activación en última
estancia de las caspasas efectoras [144].
Si es por vía mitocondrial factores como UV, radiaciones, déficit de factores de
crecimiento, estrés, dan lugar a la liberación de citocromo c de la mitocondria.

Intoducción
60
Existen otras dos vías de activación de las caspasas:
• El retículo endoplasmático (RE): es otro sitio de regulación de muerte celular debido
a agentes que provocan estrés. La perturbación de la homeostasis del ion calcio
provoca la translocación de la caspasa-7 a la superficie del retículo endoplasmático
activando la caspasa-12 que a su vez activaría la caspasa-9 y ésta a su vez activaría a
las caspasas efectoras -3, -6 y -7 [145]. En este caso el citocromo c no es liberado de
la mitocondria.
• Activación vía Granzima B: Es una vía de apoptosis utilizada por los linfocitos T
citotóxicos y las células NK, para eliminar células infectadas por virus.
SUSTRATOS DE LAS CASPASAS.
Los sustratos celulares sobre los que actúan las caspasas, son un número determinado
de proteínas que son cortadas de manera coordinada con la finalidad de hacerles perder
su función o modificársela, de tal manera, que la organización celular resulte
desmantelada. Entre los sustratos de caspasas y algunos de sus efectos están:
• Moléculas implicadas en proteger a la célula del proceso de apoptosis como es el
caso de iCAD/DFF45, la molécula que mantiene inhibida a CAD, la nucleasa
responsable de la degradación del DNA durante la apoptosis. En condiciones
normales CAD esta formando un complejo inactivo con iCAD, durante la apoptosis
este inhibidor se degrada permitiendo a la nucleasa degradar el ADN. Otras
moléculas protectoras de la célula y que son degradadas por las caspasas son algunos
de los miembros anti-apoptóticos de la familia Bcl-2, (caspasa-8 fragmenta a Bid que
es un miembro pro-apoptótico, generando un fragmento de 15 KDa que se transloca
a la membrana mitocondrial, provocando la liberación de citocromo c). En el caso de
estas proteínas, la proteólisis libera un fragmento que tiene poder pro-apoptótico por
sí mismo. De esta forma, las caspasas realizan una retro-alimentación positiva de su
propio efecto [144].

Intoducción
61
• Moléculas implicadas directamente en la estructura celular como son la quinasa de
adhesión focal o FAK, actina y la quinasa 2 activada por p21, modificando su
actividad. En otras ocasiones, como en el caso de la gelsolina, la rotura por caspasa
le hace perder la región reguladora pasando a expresarse de forma constitutiva.
También degradan la fodrina que disocia la membrana plasmatica del citoesqueleto
[144].
• Proteínas relacionadas con el metabolismo, la reparación, replicación y transcripción
del ADN: DNA-PKCS, ADN-topoisomerasas, RNA-polimerasas o la poli (ADP-
ribosa) polimerasa (PARP).
• Proteínas del ciclo celular: p21, p27, pRB, proteínas quinasas: PKC y sus isoformas;
MAPK, ERK, Akt, Wee1, proteínas de la vía de transdución de señales: citoquinas
pro-interleukinas, fosfolipasas.
INHIBIDORES DE CASPASAS.
La activación o inactivación de las caspasas están controladas por distintos
mecanismos a través de diferentes inhibidores y activadores. La inhibición de las
caspasas se lleva a cabo por [144]:
• IAPs: inhibidores de la apoptosis liberados por la mitocondria que ejercen su acción
inhibiendo a las caspasas. Se han identificado ocho IAPs en mamíferos, entre las que
se encuentran en humanos NAIP, XIAP, c-IAP1, c-IAP2, ML-IAP y survivina. Las
IAPs son inhibidores directos de las caspasas. XIAP, c-IAP1 y c-IAP2 inhiben las
caspasas –3, -7 y –9. Estas proteínas inhiben a las caspasas mediante la unión a ellas
por dominios BIR. Las IAPs pueden presentar: a) uno o más dominios BIR, b)
dominios RING, que actúan como una ubiquitín-ligasa induciendo su
autodegradación y la degradación de la caspasa que va unida a él, a través del
proteasoma y c) dominios CARD que pueden permitir la regulación de la degradación
de caspasas por interacción entre dominios de este tipo. Dentro de esta familia de
proteínas, la mejor estudiada es la XIAP. Su estructura está formada por tres dominios
BIR y un dominio RING. El dominio BIR2 inhibe las caspasas-3 y 7, mientras que el

Intoducción
62
dominio BIR3 inhibe a la caspasa-9. Los antagonistas de XIAP pueden bloquear su
efecto inhibidor e inducir la activación proteolítica directa de la caspasa-3, activando
posteriormente a las caspasas-8 y -9 como consecuencia del bucle de amplificación de
la señal apoptótica. Esta primera activación de caspasa-3 es debida a que estos
antagonistas de XIAP se unen a BIR2 impidiendo el bloqueo de la caspasa. Además,
el efecto apoptótico del antagonista de XIAP no se observa en células normales y
posiblemente debido a que los niveles de expresión son menores que en las células
tumorales. Así, el tratamiento con antagonistas de XIAP podría tener valor
terapéutico en los casos con elevados niveles de expresión de la proteína.
• FLIP/FLICE: posee dos regiones homólogas de dominios efectores de muerte (DED)
en su extremo amino, asemejándose a la estructura de las caspasas-8 y -10. c-FLIP es
también llamado Casper/ CASH/ CLARP/ FLAME/ I-FLICE/ MRIT o usurpina. Se
han identificado dos variantes: c-FLIPS y c-FLIPL. Debido a su similitud estructural
con la caspasa-8, las proteínas FLIP interfieren en su activación a nivel de formación
del DISC. c-FLIPS se une al adaptador FADD y a la caspasa-8 a través de sus
dominios DED, inhibiendo la unión de la caspasa-8 a su receptor y su posterior
activación. c-FLIPL se une también a la caspasa-8 mediante sus dominios DED y a
una región homóloga a la caspasa, formandose un hetero-dímero caspasa-8/c-FLIPL,
que promueve la proteólisis parcial de ambos. Esta proteólisis incompleta mantiene a
las dos proteínas unidas al receptor, impidiendo la liberación de la forma activa de la
caspasa-8. c-FLIPL parece ser un inhibidor más potente que c-FLIPS para los
distintos receptores de muerte (Fas, TRAIL-R1, TRAIL-R2, TNF, etc.) [146].
• El inhibidor ARC, contiene un dominio CARD y también antagoniza la apoptosis
mediada por receptor, ya que es capaz de unirse e inactivar a la caspasa-2 y a la
caspasa-8 [147]. ARC es un inhibidor general de la apoptosis, tanto de la vía
extrínseca como de la vía intrínseca.
• Otros mecanismos de inhibición de caspasas son: a) Mediante fosforilación, por
ejemplo Akt, quinasa involucrada en la supervivencia celular y mediador de PI3K,
fosforila e inhibe a la caspasa-9 [148], b) Mediante nitrosilación reversible del óxido
nítrico [149] y c) Las proteínas Bcl-2 y Bcl-xL evitan la liberación del citocromo c y

Intoducción
63
por tanto la activación de la caspasa-9 y -3. Bcl-xL puede unirse a Apaf-1 impidiendo
la asociación entre Apaf-1 y pro-caspasa-9.
2.1.4 VÍA DE LA MAPK QUINASAS.
Las vías de señalización de las proteínas quinasas activadas por mitógenos (MAPKs)
juegan un papel fundamental en la transducción de señales en la célula a través de
cascadas de fosforilación. Las MAPKs son una gran familia de Ser/Thr quinasas,
activadas por un gran número de estímulos celulares y/o externos, que modulan eventos
importantísimos en la célula eucariota como son: la progresión del ciclo celular, la
migración celular, la diferenciación celular o la apoptosis.
Las MAPKs se encuentran en el citoplasma de las células y existen 4 subfamilias, con
una estructura básica similar: MAPK p38/RK/CSBP (p38), ERK (quinasa reguladora de
señales extracelulares), JNK/SAPK (c-Jun NH2 terminal quinasa), BMK1/ERK5. La
cascada de señalización de las MAPKs se encuentra organizada de forma jerárquica en
tres etapas, a través de la fosforilación de las proteínas específicas. La fosforilación de
un determinado sustrato por las MAPKs hace que la actividad de dicha proteína se
induzca o reprima. Los sustratos de las MAPKs son otras proteínas quinasas,
fosfolipasas, factores de trascripción y proteínas del citoesqueleto.
En la cascada de señalización las MAPKKK se activan interaccionando con una familia
de pequeñas GTP-asas y/o con otras quinasas que conectan las MAPKs con los
receptores de superficie o con los estímulos externos. Los receptores acoplados a
proteínas G (GPCR) son activados por una gran variedad de estímulos externos. Al
realizarse la activación se produce el cambio de GDP por GTP y desencadenan la
activación de diversas cascadas de señalización (Figura10). Para poder activar las
pequeñas cascadas de G-proteína/MAPK, se emplean tres tipos de tirosina quinasas, con
la posterior consecuencia de estimulación o inhibición de las rutas de MAPK. Las
señales dirigidas hacia el citoesqueleto acopladas a GPCR, integrinas y receptores de
tirosina quinasas (RKT), tienen distintos efectos en la actividad celular, produciendo
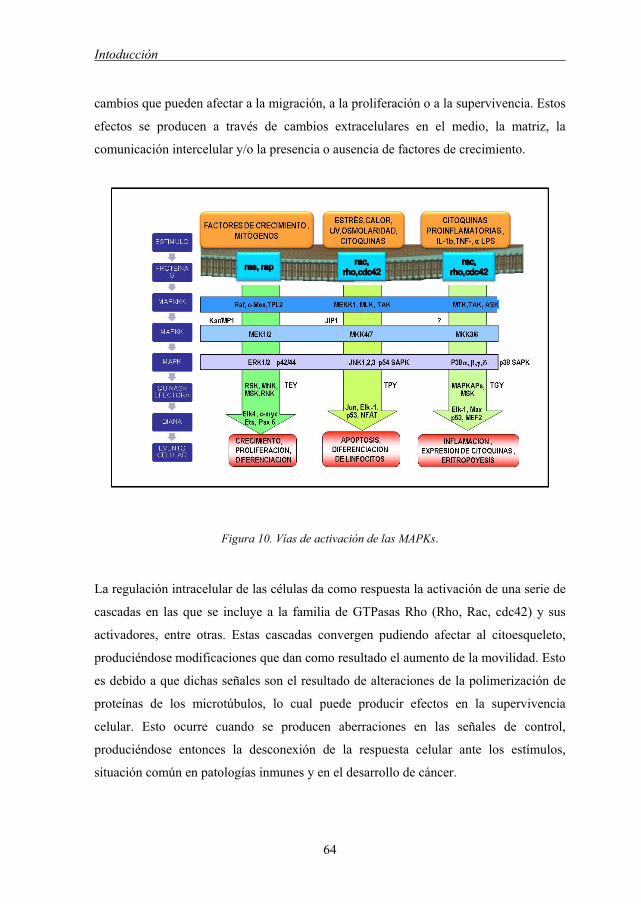
Intoducción
64
cambios que pueden afectar a la migración, a la proliferación o a la supervivencia. Estos
efectos se producen a través de cambios extracelulares en el medio, la matriz, la
comunicación intercelular y/o la presencia o ausencia de factores de crecimiento.
Figura 10. Vías de activación de las MAPKs.
La regulación intracelular de las células da como respuesta la activación de una serie de
cascadas en las que se incluye a la familia de GTPasas Rho (Rho, Rac, cdc42) y sus
activadores, entre otras. Estas cascadas convergen pudiendo afectar al citoesqueleto,
produciéndose modificaciones que dan como resultado el aumento de la movilidad. Esto
es debido a que dichas señales son el resultado de alteraciones de la polimerización de
proteínas de los microtúbulos, lo cual puede producir efectos en la supervivencia
celular. Esto ocurre cuando se producen aberraciones en las señales de control,
produciéndose entonces la desconexión de la respuesta celular ante los estímulos,
situación común en patologías inmunes y en el desarrollo de cáncer.

Intoducción
65
La activación de las MAPKs determina la inducción de varias respuestas celulares muy
importantes como activación de factores de transcripción, regulación de apoptosis,
regulaciones transcripcionales, remodelación de la cromatina nuclear, producción de
citoquinas, y progresión del ciclo celular. Cada actividad biológica es inducida por una
subfamilia MAPK y cada estímulo determinará la actividad producida. En general, la
vía Ras/ERK regula principalmente el crecimiento y supervivencia celular, pero en
algunas circunstancias determina también la diferenciación celular. Las vías de JNK y
p38 regulan principalmente la apoptosis, la respuesta inflamatoria y transmiten señales
inhibidoras del crecimiento. Se ha descrito, sin embargo, que la vía de MAPK/p38
puede inducir efectos antiapoptóticos o proliferativos, transmitiendo señales de
supervivencia celular en determinadas condiciones, en función del tejido y de la
isoforma de quinasa expresada.
La capacidad de las MAPKs para transmitir diferentes señales e incluso opuestas, en las
mismas células plantea la cuestión de cómo se regula la especificidad de las señales
transmitidas por las MAPKs. Se han propuesto varios mecanismos: la duración y la
fuerza de la señal, la interacción con diferentes proteínas adaptadoras, la localización
subcelular, la presencia de varias isoformas, en cada nivel de la cascada, conexiones
entre la vía de las MAPKs y otras vías y las modificaciones post-traduccionales distintas
a la fosforilación [150].
2.1.4.1 ERK1/2 (p42/44 MAPK).
La vía de activación ERK1/2 (p42/44 MAPK) está bien caracterizada y se activa por un
gran número de estímulos, tanto intracelulares como extracelulares: factores de
crecimiento, el suero, los ésteres de forbol, los ligandos de receptores acoplados a
proteína G, las citoquinas, el estrés osmótico y la desorganización de los microtúbulos.
La señalización de ERK1/2 está implicada en la proliferación celular y, por esta razón,
los inhibidores de esta vía están en fase de ensayos clínicos como potenciales fármacos
antitumorales [150].

Intoducción
66
Se conocen dos isoformas, ERK1 y ERK2 con niveles de expresión diferentes en los
tejidos. La cascada de señalización suele iniciarse mediante la activación de receptores
de superficie celular como los receptores tirosina quinasas (RTKs) y los GPCRs. La
señal es transmitida a las pequeñas proteínas G (por ejemplo, Ras), que transmiten la
señal mediante la unión a MAP3K (quinasas Raf: A-Raf, B-Raf y Raf-1) en la
membrana plasmática donde se activa a través del reclutamiento de SOS (un factor de
intercambio de nucleótidos de guanina). Así, SOS estimula Ras permitiendo la
interacción con un amplio rango de efectores en la ruta, incluyendo las isoformas de
Raf. El mecanismo de la activación de Raf parece que requiere la unión a Ras y
múltiples fosforilaciones en la membrana. Tanto la regulación de Ras como la de Raf
son cruciales para el mantenimiento de la proliferación celular y la mutación activadora
en estos genes lleva a la oncogénesis.
Otros componentes MAP3K que pueden activar la vía ERK1/2 bajo condiciones
específicas son el c-Mos, TPL2 y MEKK1/2/3, que actúan principalmente durante la
meiosis, la proliferación y la respuesta a estrés, respectivamente. Raf activado (u otra
MAP3Ks) se une y fosforila a las quinasas MEK1 y 2, que a su vez fosforilan y activan
a ERK 1/2. La activación de ERK1/2 determina la fosforilación de un gran número de
sustratos en todos los compartimentos celulares, incluyendo proteínas citosólicas y de
membrana (PLA2, CD120a, Syk y calnexin), sustratos nucleares (SRC-1, Pax6, NFAT,
Elk-1, MEF-2, c-fos, c-jun, c-myc y STAT3) y proteínas del citoesqueleto
(neurofilamentos y paxillin). Una proporción significativa de ERK1/2 se acumula en el
núcleo.
Además ERKs puede fosforilar y activar proteínas quinasas (MKs). Se conocen más de
150 sustratos de ERK1/2 hasta la fecha. Entre las dianas de ERK1/2 citoplasmáticas se
encuentra la fosfolipasa A2, que regula la producción de ácido araquidónico en células
estimuladas. Entre MKs, los miembros de la familia RSK son exclusivamente activados
por ERK1/2. Después de su activación se trasladan al núcleo y fosforilan otros sustratos.
Sin embargo, RSKs activados también se han encontrado en el citoplasma, lo que
sugiere que también existen sustratos citosólicos. Muchos de los sustratos de ERK1/2

Intoducción
67
son compartidos por RSKs y MSKs (quinasas mitógeno-estrés activadas), como factores
de transcripción y coactivadores como SRF, el CBP, Elk-1 y CREB que participan en la
respuesta temprana. Como en el caso de ERK1/2, existen genes que son fosforilados por
RSKs, por ejemplo, c-fos y c-jun [150].
El control sobre la vía ERK1/2 se ha perdido en aproximadamente un tercio de todos los
cánceres humanos. La participación de ras en el cáncer se conoce desde hace mucho
tiempo. El punto de aberración en la mayoria de los cánceres parece ser ras/raf. La
aberración abarca a toda la cascada de ERK1/2, desde un receptor mutado como EGFR
y sobreexpresión ErBB2, la mutación de Ras y Raf hasta la amplificación de dianas
nucleares como myc y AP-1. La heterodimerización de raf1 con B-Raf parece que
contribuye de forma considerable en la actividad de ERK y los estudios llevados a cabo
para dilucidar el mecanismo de acción de oncogén B-Raf señalan su participación en el
cáncer. Los MKs, RSKs, MNKs y MSKs, pueden trasladarse de forma independiente al
núcleo y fosforilar diversos factores de transcripción implicados en la proliferación
celular, contribuyendo de esta forma a la inducción de genes inmediatos y participan de
esta forma en la proliferación celular.
En algunos estudios también se ha llegado a la conclusión de que algunos inhibidores de
la vía ERK no se encuentran regulados en el cáncer entre ellos se incluyen: MAP
quinasa fosfatasa (MKPs) y los miembros de la familia Sprouty. Además de la
proliferación celular, otros sustratos de ERK juegan un papel clave en la angiogénesis,
la migración celular, la invasión y la metástasis. La señalización de ERK puede
promover más el fenotipo maligno del cáncer interrumpiendo las vías de señalización
Rho, fosforilando un gran número de proteínas implicadas en la migración celular
incluyendo MLCK, calpaína, FAK y paxillina, así como mediante la regulación de la
expresión de proteasas implicadas en la degradación de la membrana basal.
La vía ERK1/2 desempeña un papel central de la proliferación celular, el crecimiento y
la progresión del ciclo celular. Se han propuesto diversos mecanismos para explicar el
papel de la vía en la progresión del ciclo celular. Los sustratos más importantes son

Intoducción
68
ciclinas tipo D y c-myc. La activación de la vía ERK induce las ciclinas tipo D por
aumento y activación de los factores de transcripción implicados en su síntesis, por
ejemplo las proteínas de la familia AP1. También puede controlar su expresión a nivel
post-transcripcional. La activación de MEK1 también parece facilitar la formación de
complejos ciclina D1-CDK4 en fibroblastos [150]. La activación de ERK estabiliza
mediante fosforilación a c-myc, un factor de transcripción, que desempeña un papel
importante en la regulación del crecimiento celular, en la progresión celular y la
apoptosis [151].
Los mecanismos mediante los cuales la vía ERK1/2 promueve apoptosis no se conoce
con detalle y parece ocurrir a diferentes niveles. ERK1/2 puede actuar antes de la
liberación del citocromo c y de la activación de la caspasa-3 como se ha observado en la
apoptosis inducida por cisplatino. Varios estudios han demostrado una disminución de
los niveles de Bax y p53 después de la inhibición de ERK1/2. Esto indica que ERK1/2
pueden actuar a través de la regulación (aumentando los niveles) de Bax y p53,
inhibiendo la acción de las proteínas anti-apoptóticas Bcl-2 y Bcl-xL en la mitocondria.
ERK1/2 parece fosforilar directamente p53, contribuyendo a su papel apoptótico [152-
155]. Se ha sugerido que la vía ERK1/2 podría tener un papel relevante en la inducción
de la vía extrínseca por TNF-α y en la activación de la caspasa-8. La promoción de la
muerte celular por esta vía también puede ser el resultado de la represión de la señal de
supervivencia mediada por Akt [150].
La vía de ERK1/2 se encuentra relacionada con la oncogénesis, y la magnitud de la
actividad de ERK1/2 influye en la supervivencia del carcinoma. Una alta actividad de
ERK1/2 reduce la tasa de apoptosis en el carcinoma de colon e induce la detención del
ciclo celular por aumento de los niveles de los inhibidores de CDK, p21 y p27. Sin
embargo, también se han descito dianas de ERK1/2 implicadas en la apoptosis, se ha
demostrado que ERK1/2, activando B-Raf, inhibe la activación de la caspasa citosólica
y la liberación del citocromo c de la mitocondria. Una segunda diana de la vía ERK son
los miembros de la familia Bcl-2 pro-apoptóticos; Bad influye en la integridad de la
membrana y libera citocromo c de manera indirecta por asociación con Bcl-2 y Bcl-xL

Intoducción
69
inhibiendo el efecto antiapoptótico de los mismos. ERK fosforila a Bad, lo secuestra en
el citosol, y lo mantiene fuera de la mitocondria previniendo de esta forma la apoptosis
[150].
2.1.4.2 FAMILIA JNK (SAPKs).
Las vías JNKs son activadas en respuesta a las citoquinas, radiación ultravioleta,
ausencia de factores de crecimiento, agentes que dañan el ADN y, en menor medida, en
respuesta a receptores acoplados a proteína G, suero y factores de crecimiento
[156,157]. Las MAPKKs que activan JNK son MKK4 y MKK7. La diversidad de
activadores de MKK4 y MKK7 permite que la activación de la vía JNK se produzca
como consecuencia de un número elevado de estímulos externos. Entre los activadores
de las MAPKKs se encuentran las MAPKKK: MEKK1-4, MLK2 y -3, TPL-2, DLK,
TAO1 y 2, TAK1 y ASK 1/2. Las diferentes MAPKKKs son específicas para los
diferentes estímulos. Por ejemplo, TAK1 ha demostrado ser fundamental para la
activación de JNK en respuesta a citoquinas inflamatorias (IL-1, TNF-α, TGF-β y
linfotoxina -β) y a la activación de receptores tipo Toll (TLR-3, -4 y -9) [158,159]. En
estímulos de estrés, tales como la retirada de los factores de crecimiento, estrés del
retículo endoplasmático, radiación, etc, la vía JNK es muy activa. En estas
circunstancias, la activación de JNK puede inducir cambios en la expresión de los
genes, muerte celular o alteración de la proliferación celular [150].
La activación de la vía puede producir translocación de SAPK/JNK del citoplasma al
núcleo pero no en una proporción tan alta como ERK1/2. Entre los sustratos de JNK se
encuentran una amplia variedad de proteínas nucleares, principalmente factores de
transcripción y receptores nucleares de hormonas. Estos van a tener un efecto directo
sobre la expresión génica de:
• proteínas nucleares : c-jun, JunD, junB, familia ATF (ATF-2), familia FOS, Elk-1, c-
myc, p53, familia NFAT, STAT1 y -3, y proteínas de la familia Pax.

Intoducción
70
• receptores nucleares de hormonas: PPARγ1, receptor de glucocorticoides y receptores
de ácido retinoico (RXR y RARα).
• proteínas no nucleares como IRS-1, JIP1, JIP3 y 14-3-3 permiten la modulación de
otros eventos de señalización en la célula.
• proteínas mitocondriales de la familia de Bcl-2 (Bcl-2, Bcl-xL, Bad, Bim y Bax).
• Proteínas del citoesqueleto: paxillina, proteínas asociadas a microtúbulos tipo2 y 1B
(MAP-2, 1B) y Tau.
• Akt.
Un aspecto muy importante de JNK es su papel en la degradación de proteínas y en la
tumorogénesis ya que se aprecian altos niveles de actividad en diferentes líneas
celulares cancerígenas. La actividad de JNK y la fosforilación de c-jun desempeñan un
papel fundamental en tumores inducidos por Ras; Ras y c-jun cooperan en la
transformación celular. Nateri et al. [160] demostró que la perdida de c-jun o la
mutación de alguno de sus sitios de fosforilación para JNK reduce el número de tumores
y el tamaño de los mismos. Una función importante de c-jun parece que es la represión
transcripcional de p53.
Aunque el papel de JNK en la apoptosis está bien establecido, el mecanismo de acción
es controvertido y parece ser dependiente del estímulo y del tipo celular. Un objetivo en
la señalización pro-apoptótica de JNK es p53, la unión de ambos provoca que JNK
desestabilice p53 mediante la inducción de la degradación a través del proceso de
ubiquitinización. Por el contrario, la activación de JNK por estrés inhibe la degradación
dependiente de ubiquitina debido a la formación de un complejo estable.
Los miembros pro-y anti-apoptóticos de la familia Bcl-2 son también sustratos de JNK.
Proteínas anti-apoptóticos: Bcl-2 y Bcl-xL son inhibidas a través de su fosforilación por
JNK. Bax y 14-3-3 ha demostrado que son sustratos de JNK in vivo, los cuales se
pueden considerar pro-apoptóticos. Estudios recientes sugieren que JNK está
involucrado en la degradación del inhibidor de la caspasa-8, cFLIPL, y en la activación
de la variante de histona 2 (H2AX) que parece ser esencial para la fragmentación del

Intoducción
71
ADN. La caspasa 3 puede amplificar la activación de JNK, ya que es capaz de activar
MEKK1 y por tanto activar JNK. Si bien el papel de JNK en la apoptosis está bien
establecido, también se ha demostrado que JNK puede contribuir a la supervivencia.
Estos efectos opuestos podrían deberse en parte a la duración o la magnitud de la
activación de la vía y en parte debido a la activación de otros vías de supervivencia. Así
la activación prolongada de JNK parece que induce apoptosis, mientras que la
activación transitoria promueve la supervivencia celular [150].
2.1.4.3 FAMILIA MAPK/P38. La vía p38 esta implicada en respuestas a estímulos de estrés extracelular, en la
regulación del ciclo celular y en la osmorregulación. La vía p38 se activa por estrés
ambiental y citoquinas inflamatorias, y activado de forma contraria en menor medida
por la insulina y factores de crecimiento. Los mismos estímulos que activan JNKs
también activan p38 excepto en la isquemia por reperfusión donde sólo se activa p38
[161]. Hay cuatro isoformas conocidas de p38: α, β, γ y δ. Entre estos, p38 α es la
isoforma más estudiada. Al igual que todos las MAPKs, p38 son activadas por las
MKKs: MKK3 y MKK6. MKK6 puede activar las cuatro isoformas, mientras que
MKK3 no puede activar p38β. MKK4 y MKK7 (quinasas que activan JNK) también
puede activar isoformas p38, lo que indica que existe una vía de unión entre ambas vías
(JNK y p38). Los activadores de la vía p38 son un grupo más diverso que en la vía de
JNK. Esto explica su activación con una amplia variedad de estímulos. Varias MKKKs
(MAP3Ks) inducen la activación de p38, entre ellos: MTK1, MLK2/MST, MLK3,
DLK, ASK1 y TAK1. Estos confieren especificidad de la respuesta a diferentes
estímulos [150].
Las isoformas de p38 estan presentes tanto en el núcleo como en el citoplasma de las
células quiescentes. Aunque existen evidencias que sugieren que, a raíz de la activación,
p38 se transloca del citoplasma al núcleo, también están presentes en el citoplasma. La
activación de p38α en diferentes sistemas celulares ha sido descrita en respuesta a:
factores de crecimiento (FGF, NGF, IGF, PDGF, TGF-β, eritropoyetina etc.) agonistas

Intoducción
72
de GPCR, un agonista muscarínico, péptidos vasoactivos y choque térmico [162-166].
Los sustratos proteícos de p38 son: factores de transcripción (alterando la expresión de
determinados genes), MKs y proteínas citosólicas como fosfolipasa A2, Tau (proteína
asociada a microtúbulos) y queratina 8.
La participación de p38 en la ralentización de la proliferación celular se observó en
levaduras transfectadas con p38α. En células NIH3T3 se producía detención del ciclo
celular en G1 cuando se inyectaba p38α y esta detención desaparecía en MKK3 nulas,
mostrando la participación activa de p38α. El punto de unión entre el p38 y el control
del ciclo celular parece ser que es mediante la regulación de los sustratos como HBP1 y
p21. Muchos de los efectos de p38 sobre el ciclo celular están mediados vía MSKs, las
cuales fosforilan diversos sustratos como: CREB e influyen en la expresión génica.
Muchos agentes quimioterapéuticos requieren la activación de p38 para la inducción de
la apoptosis. La supresión de la activación de p38 parece aumentar la apoptosis en
respuesta al daño del ADN por agentes como la doxorrubicina y el cisplatino Existen
estudios que muestran la relación entre la vía de p38 con la vía de las caspasas: al
inhibir su actividad por inhibidores de las caspasas [150]. Así, niveles elevados de
MKK6 podría inducir la actividad de caspasa e incrementar la muerte celular. Otro
mecanismo de la inducción de apoptosis por p38 (por ejemplo en células endoteliales)
se produce a través de la fosforilación y disminución de los niveles de Bcl-xL y por
aumento de la expresión de p53.
La pérdida de MKK3 y MKK6 promueve la proliferación y aumenta la probabilidad de
formación de tumores. Otros estudios indican que p38 puede funcionar como un
supresor de tumores. Estos efectos supresores de tumor parece ser mediado por la
activación de p53 e inducción de apoptosis dependiente de p53 [150].

Intoducción
73
2.1.5 VÍA DE SUPERVIVENCIA.
La necesidad de la supervivencia celular requiere que se activen los mecanismos de
inhibici�n de la apoptosis, promoviendose la inhibici�n de la expresi�n de los factores
pro-apopt�ticos y el incremento en la expresi�n de los factores anti-apopt�ticos o de
supervivencia.
VÍA PI3K/AKT.
La activaci�n de la ruta PI3K/Akt se inicia mediante el reclutamiento de PI3K a la
membrana plasm�tica a trav�s de la uni�n de su dominio SH2 a prote�nas fosforiladas
en tirosina. PI3K es un heterod�mero que consiste en una subunidad reguladora (p85) y
una subunidad catal�tica (p110) encargada de transferir el grupo fosfato γ del ATP al
fosfatidil inositol, 4,5-bifosfato (PIP2), generando el fosfatidil inositol, 3, 4,5-trifosfato
(PIP3) y ADP. La actividad quinasa est� regulada por fosfatasas que act�an eliminando
los fosfatos de las prote�nas diana. PIP3 sirve como ligando para reclutar la
serina/treonina quinasa Akt (c-Akt, tambi�n llamada prote�na-quinasa B, PKB) a la
membrana plasm�tica a trav�s de la interacci�n directa con el dominio con homolog�a
con plecstrina (PH) de Akt. Una vez en la cara interna de la membrana, Akt es
fosforilado por una serina/treonina quinasa, la quinasa 1, dependiente de fosfatidil
inositol-3 (PKD1), resultando en la activaci�n de Akt.
Las c�lulas de mam�fero expresan tres isoformas de Akt: Akt1 (PKB), Akt2 (PKB) y
Akt3 (PKB), codificadas por diferentes genes. Akt1 se expresa en todos los tejidos
mientras que Akt2 se expresa en los tejidos sensibles a la insulina incluyendo el h�gado,
m�sculo esquel�tico y tejido adiposo. La expresi�n de Akt2 aumenta de forma dr�stica
durante la diferenciaci�n del tejido adiposo y del m�sculo esquel�tico. Akt3 es
abundante en el cerebro y en los test�culos mientras que en los �rganos intestinales y en
el tejido muscular su expresi�n es menor [167-174].

Intoducción
74
Figura 11. Representación esquemática de la vía de señalización activada por PI3K/AKT.
La activación de Akt controla la supervivencia celular a través de la fosforilación de las
dianas que dependen de ella, con el resultado neto del incremento en la supervivencia
celular, proliferación, crecimiento y metabolismo (Figura 11).
2.1.6 ESFINGOLÍPIDOS Y APOPTOSIS
Los esfingolípidos desempeñan un papel importante en la señalización intracelular,
particularmente la ceramida, la esfingosina y la esfingosina-1-fosfato (S1P). La
ceramida y la esfingosina se consideran mediadores celulares que desempeñan
importantes funciones celulares que afectan a los procesos de diferenciación,
senescencia, apoptosis y la parada del ciclo celular. La S1P en cambio, promueve la
supervivencia y la proliferación celular. (Figura12). El delicado equilibrio entre los
niveles intracelulares de cada uno de estos esfingolípidos está controlado por las
enzimas que los producen. La esfingosina quinasa (SK) es reguladora esencial de este
balance ya que, produce el metabolito pro-supervivencia esfingosina 1-fosfato y reduce
el contenido de los metabolitos pro-apoptóticos.

Intoducción
75
Existen varias rutas de entrada a la v�a de los esfingol�pidos que convergen en la
formaci�n de ceramida, que es clave en la generaci�n de otros esfingol�pidos activos.
La primera ruta de entrada es la bios�ntesis de novo donde la serina y el palmitoil
CoA se condensan y a trav�s de una serie de reacciones se forma ceramida en el
ret�culo endopl�smico y aparato de Golgi. La ceramida puede ser convertida en otros
esfingol�pidos como la esfingosina y la esfingosina 1-fosfato o bien formar
esfingomielina y glicoesfingol�pidos complejos. La eliminaci�n celular de todos los
esfingol�pidos requiere la transformaci�n final en S1P. Por lo tanto, la actividad de la
SK, enzima responsable de la fosforilaci�n de esfingosina para formar S1P, inicia la
�nica v�a bioqu�mica degradativa de eliminaci�n de ceramida de la c�lula. La
degradaci�n de S1P est� mediada por la S1P liasa y este mecanismo le otorga a la SK
una importancia indirecta adicional, como herramienta de salida celular de todos los
esfingol�pidos. Esta v�a es activada por agentes quimioter�picos, choque t�rmico,
LDL oxidadas y canabinoides. Diversos estudios sugieren que la ceramida formada
de novo esta involucrada en los efectos inducidos a respuestas de estr�s, y apoptosis.
La segunda ruta es la v�a que involucra la hidr�lisis de esfingomielina por acci�n de
esfingomielinasas. La hidr�lisis de la esfinomielina est� considerada como la
principal v�a para la producci�n de ceramida como transductor de se�ales que regulan
la muerte celular. En primer t�rmino se forma ceramida, la cual a su vez es
hidrolizada por la acci�n de ceramidasas para formar esfingosina. La esfingosina
sirve como sustrato de la esfingosina quinasa para formar S1P. Esta v�a es estimulada
en respuesta al tratamiento celular con TNF-α, el ligando Fas, o estr�s oxidativo
[175,176].
Recientemente se ha identificado un tercer mecanismo m�s complejo de formaci�n y
acumulaci�n de ceramida denominada v�a de salvamento. Este mecanismo se
relaciona con el catabolismo de esfingol�pidos complejos que se transformar�an en
esfingosina, que a trav�s de reacilaci�n ser�an reutilizados para producir ceramida. En
esta v�a hay una serie de enzimas claves entre las que se incluyen: esfingomielinasas,

Intoducción
76
posiblemente glucocerebrosidasas (�cido β-glucosidasa), ceramidasas, y (dihidro)
ceramida sintasas. La ceramida generada por esta v�a es transformada por la
ceramidasa �cida para formar esfingosina y �cidos grasos libres, que son capaces de
dejar el lisosoma en contraste con la ceramida que no parece salir del lisosoma. Una
vez liberados de los lisosomas pueden entrar en la v�a de la s�ntesis de ceramida o en
la s�ntesis de esfingosina-1 fosfato (mediante esfingosina quinasas).
En los �ltimos estudios realizados se apunta a esta v�a como responsable de las
funciones de la ceramida generada de muchas respuestas biol�gicas tales como la
detenci�n del crecimiento, la apoptosis, y la se�alizaci�n celular [177]. La activaci�n
diferente de las distintas v�as de formaci�n de ceramida es posible gracias a la
separaci�n espacial de las enzimas que contribuyen a la formaci�n de la misma.
.
CERAMIDA ESFINGOSINA ESFINGOSINA -1P
DETENCION DEL CRECIMIENTOMUERTE
DETENCION DEL CRECIMIENTOMUERTE
SENESCENCIADIFERENCIACION
PROLIFERACIONSUPERVIVENCIA
CERAMIDA ESFINGOSINA ESFINGOSINA -1P
DETENCION DEL CRECIMIENTOMUERTE
DETENCION DEL CRECIMIENTOMUERTE
SENESCENCIADIFERENCIACION
PROLIFERACIONSUPERVIVENCIA
Figura 12. Balance ceramida -esfingosina.
El metabolismo de los esfingol�pidos es complejo y puede ser regulado a m�ltiples
niveles, a trav�s del control de la expresi�n de enzimas, modificaciones post-
traduccionales y mecanismos alost�ricos. Algunos de estos mecanismos son espec�ficos
del tipo celular, tanto para controlar el tipo de esfingol�pido que se sintetiza durante
distintas etapas del desarrollo o en repuesta a se�ales espec�ficas.

Intoducción
77
2.1.6.1 CERAMIDA
La ceramida tiene una importancia fundamental, como mediadora de la respuesta al
estrés en las células eucariotas. Los efectos de la ceramida en la mayoría de los casos
son inhibidores de la proliferación celular. La ceramida induce parada del ciclo celular
mediante diversos mecanismos tales como: la inducción de la desfosforilación de la
proteína retinoblastoma Rb, la activación del inhibidor de la quinasa dependiente de
ciclina p21, y la inhibición de la quinasa dependiente de ciclina CDK2. Los niveles
elevados de la concentración de ceramida también se encuentran presentes en la
senescencia. El fenotipo senescente es atribuido a fallas en la vía fosfolipasa C/proteína
quinasa C, enzimas que son inhibidas por ceramida [178-182].
El papel de la ceramida más estudiado es su función como molécula pro-apoptótica. La
acumulación de ceramida que sigue al tratamiento de células con agentes pro-
apoptóticos constituye una evidencia de su implicación en la respuesta biológica a estos
agentes. Debido al efecto inductor de apoptosis, la ceramida se la denomina lípido
supresor de tumores [183].
Como ya se ha mencionado anteriormente la hidrólisis de esfingomielina por
esfingomielinasas da lugar a la liberación de ceramida. Se han identificado al menos
cinco subtipos diferentes de esfingomielinasas dependiendo de su pH óptimo, su
localización subcelular y su dependencia de cationes: SMasa neutra dependiente de
magnesio y unida a membrana (SMasa neutra), una SMasa neutra independiente de
magnesio, la SMasa ácida presente en los lisosomas (SMasa ácida), una forma secretada
y soluble de SMasa ácida dependiente de zinc y una SMasa alcalina [184]. A pesar de
los estudios basados para dilucidar el mecanismo de la activación de estas SMasas, su
papel específico no está todavía claro, siendo la SMasa neutra y la SMasa ácida las más
estudiadas. Ambas enzimas pueden ser activadas por el receptor de 55 kDa del factor
necrótico tumoral, TNF, así como por otros estímulos de estrés. La SMasa neutra es
activada por el receptor de TNF a través de una proteína adaptadora denominada FAN
que se asocia al dominio NSD del receptor [175]. La proteína FAN a su vez puede

Intoducción
78
asociarse con otras proteínas ensambladoras como el receptor de PKC activada RACK1,
que sirve de atracción y activación de numerosas proteínas implicadas en rutas de
señalización. La SMasa ácida, puede ser también activada por el receptor de TNF a
través de otras proteínas adaptadoras como FADD y TRADD que se unen al dominio de
muerte (DED) del receptor [185]. La SMasa ácida es activada por muchos estímulos
incluyendo CD95, CD40, DR5/TRAIL, CD20, FccRII, CD5, LFA-1, CD28, TNF, los
receptores de interleuquina-1, los receptores FAP, la infección por Pseudomonas
aeruginosa, Staphylococcus aureus, Neisseria gonorrhoeae, Sindbis-Virus, Rhinovirus,
-irradiación, luz ultravioleta, doxorrubicina, cisplatino, gemcitabina, paclitaxel,
alteración de la señalización por integrina y péptidos amiloides.
En la mayoría de los estímulos mencionados se produce una translocación de la SMasa
ácida (la enzima se encuentra en vesículas intracelulares) a la membrana celular donde
se fusiona con la misma y se produce una liberación de ceramida. Las moléculas de
ceramida forman dominios de membrana o balsas lipídicas enriquecidas en ceramida
(raft) que sirven para agregar receptores de membrana activados. La formación de estos
dominios de membrana enriquecidos en ceramida se debe a la tendencia de las
moléculas de ceramida a asociarse entre si. En estos casos la ceramida parece sustituir el
colesterol en estos dominios de la membrana dando lugar a cambios en las propiedades
de la membrana. Los dominios de membrana enriquecidos de ceramida sirven para una
reorganización de la señal celular y en particular para agrupar las moléculas de
receptores (alta densidad de receptores en un área pequeña de la membrana). Se ha
demostrado in vitro e in vivo el agrupamiento de CD95 o DR5 mediado por ASM. La
función de la agrupación de los receptores es amplificar la señal mediada por DR5 o
CD95. Esta agrupación es necesaria para el inicio de la señalización y por último, para
la inducción de la apoptosis. Este papel de amplificación de la señal no parece ser un
evento específico en la cascada de señalización iniciada por receptores [186-190].
Algunos agentes externos, tales como la luz ultravioleta la irradiación, y el estrés
activan múltiples vías de acumulación de la ceramida que juega un papel importante en
la muerte inducida por estos agentes [191-195].

Intoducción
79
Los estudios realizados por Fuks y Kolesnick demostraron que la deficiencia de
esfingomielinasa �cida no s�lo imped�a la liberaci�n de ceramida en respuesta a la
irradiaci�n, sino que tambi�n que proteg�a a la c�lula de la muerte inducida por la
radiaci�n. Los resultados de varios estudios concluyeron que para la inducci�n de
muerte celular por UV se requiere la expresi�n de la esfingomielinasa �cida y la
liberaci�n de ceramida. Sin embargo actualmente, los mecanismos de muerte celular
inducida por la generaci�n de ceramida en la membrana de la c�lula son desconocidos.
Los estudios realizados por Schutze et al. [196,197] demostraron que cuando el receptor
TNF se activaba, �ste se internalizaba r�pidamente, los endosomas se fusionaban con
los lisosomas donde se pon�a en contacto la ASM con el receptor de TNF dando lugar
a la activaci�n de la enzima y por consiguiente a la liberaci�n de ceramida intra
vesicular. La ceramida se une a catepsina D y la activa, la cual a trav�s de mecanismos
desconocidos transloca al citoplasma y ejecuta la muerte celular v�a bid, bax, y bad
[198,199].
Estudios recientes indican que la ceramida tambi�n est� presente en las mitocondrias y
que la ceramida mitocondrial puede desempe�ar un papel en la apoptosis, sin embargo,
los mecanismos de c�mo la ceramida mitocondrial media en la apoptosis est�n por
definir. La ceramida mitocondrial parece generarse a trav�s de la v�a de s�ntesis de
novo, y/o a trav�s de la actividad de la esfingomielinasa acida, que ha demostrado que
se encuentra en el espacio que hay entre la membrana externa e interna de la
mitocondria [200-204]. La ceramida parece estar implicada en la formaci�n de de poros
mitocondriales que favorecen la salida de factores pro-apopt�ticos como el citocromo c,
AIF, Smac/Diablo y la Endonucleasa G [205]. La ceramida, por tanto, act�a como
segundo mensajero activando numerosas v�as de transducci�n de se�ales: entre las
prote�nas que interaccionan con la ceramida se incluyen la prote�na quinasa activada por
ceramida (CAPK), la prote�na quinasa supresora de Ras (KSR), las fosfatasas PP2A y
PP1B, catepsina D, cPLA2 y PKCζ [177].
Se ha demostrado que la ceramida y sus an�logos inducen la translocaci�n de la PKC
at�pica PKCζ, desde el citosol a la membrana perinuclear y que tambi�n aumentan la
fosforilaci�n de esta quinasa tanto in vitro como in vivo. La uni�n de PKCζ a ceramida,

Intoducción
80
produce un cambio conformacional y la activaci�n de PKCζ por fosforilaci�n, lo cual
activa otras dianas, en particular la v�a de supervivencia de NF-Kb. Adem�s, la
ceramida activa m�ltiples cascadas de se�alizaci�n que involucran a JNK. Se ha
sugerido que la ceramida puede activar JNK via Rac-1, PKCζ y MEKK1 (miembro de
la familia de las quinasas activadas por mit�genos), la cual es an�loga de c-Raf en la
cascada de JNK [206-208].
Las fosfatasas activadas por ceramidas son la PP2A y PP1 (tambi�n llamadas fosfatasas
activadas por ceramidas, CAPPs). Entre los sustratos de CAPPs identificados in vitro se
encuentran Bcl-2, PKC, c-jun, prote�nas Rb, prote�nas SR y Akt [209]. La
fosforilaci�n y defosforilaci�n por quinasas y fosfatasas es el principal mecanismo por
el que se regula la funci�n de miembros de la familia de Bcl-2 tanto con actividad anti
apopt�tica (Bcl-2, Bcl-xL, Mcl-1) como pro-apopt�tica (Bad, Bax, Bim). Los factores
de supervivencia inducen la fosforilaci�n de Bad, inactivando su funci�n pro-
apopt�tica. La desfosforilaci�n de Bad por PP1 por lo tanto, induce apoptosis. Por el
contrario, PP2A podr�a desfosforilar Bcl-2 convirti�ndolo en una mol�cula pro-
apopt�tica [210].
Aunque muchos estudios indican un papel importante de la ceramida en la inducci�n de
la apoptosis tras un est�mulo agudo, existen pocos datos que demuestren una
contribuci�n de ceramida a una p�rdida de regulaci�n de la apoptosis en los trastornos
humanos. Entre las enfermedades que son debidas a la p�rdida de regulaci�n de la
ceramida celular se incluyen s�ndromes neurol�gicos, en particular la neuropat�a
sensorial hereditaria Tipo I causada por una mutaci�n de la prote�na serina
palmitoiltransferasa 1 y un aumento de la concentraci�n celular ceramida, que parece
estar directamente vinculadas a la degeneraci�n de neuronas sensoriales perif�ricas. Los
niveles celulares de ceramida est�n aumentados en el s�ndrome de Batten, el cual es
debido a una mutaci�n de palmitoil-tioesterasa y de CLN3 dando lugar a la muerte de
las c�lulas neuronales [211-213].

OOOBBBJJJEEETTTIIIVVVOOOSSS


Objetivos
83
OBJETIVOS.
1. Analizar la citotoxicidad de 22 compuestos con estructura fenilbenzo–-pirona en
cinco l�neas tumorales humanas: HL-60 (células leucémicas promielocíticas), A431
(carcinoma epidermoide), SK-OV-3 (adenocarcinoma de ovario), HeLa
(carcinoma de cervix) y HOS (células de osteosarcoma).
2. Establecer una posible relaci�n entre la estructura de los compuestos y su actividad
citot�xica.
3. Seleccionar los compuestos m�s potentes y estudiar su mecanismo de acci�n en la
l�nea celular m�s sensible.


MMMAAATTTEEERRRIIIAAALLL YYY MMMÉÉÉTTTOOODDDOOOSSS


Material y métodos
87
4.1 Productos y material utilizado:
4.1.1 Agentes farmacológicos.
Los estudios correspondientes al aislamiento, elucidación estructural y derivatizaciones
de los productos naturales se llevaron a cabo en el departamento de Química de la
Universidad de Las Palmas de Gran Canaria bajo la dirección del Dr. Jorge Triana
Muñoz. La mayoría de los compuestos utilizados en este estudio se aislaron de plantas
endémicas de las Islas Canarias. Los compuestos se obtuvieron mediante métodos de
aislamiento ya publicados. La identificación de las estructuras se realizó por resonancia
magnética nuclear (protón y 13C), espectroscopía de infrarrojos y espectrometría de
masas: Los compuestos 3-metil éter de betuletol (1), 3,3´-dimetil éter de quercetina (7),
3-metil éter de kaempferol (9), 5,7-dihidroxi-3,3´,4´-trimetoxiflavona (10), 4´,5,7-
trihidroxi-3,6-dimetoxi-flavona (12) y eridictiol (19) se aislaron de las partes aéreas de
Allagopappus dichotomus [214]. Los compuestos 3-metil éter de quercetina (5),
naringenina (21), eriodictiol (19) y 3-metil éter de kaempferol (9) se aislaron de
Allagopappus viscosissimus [215]. La axillarina (11) y la apigenina (22) se aislaron de
las partes aéreas de Tanacetum ferulaceum y de Tanacetum ptarmiciflorum
respectivamente [216,217]. Los derivados acetilados de 3-metil éter de betuletol (2), 3
metil éter de quercetina (6), 3,3´-dimetil éter de la quercetina (8) y de eriodictiol (20) se
obtuvieron mediante tratamiento de los correspondientes alcoholes con anhídrido
acético (Ac2 O) en piridina durante 12 horas a temperatura ambiente. El 3, 5,7-trimetil
éter de betuletol (3) fue obtenido por metilación de 4´,5,7-trihidroxi-3, 6-
dimetoxiflavona (12). Los mono (13), di (14) y triacetil (15) derivados de 4´,5,7
trihidroxi-3,6 dimetoxiflavona (12) se obtuvieron por acetilación de este último con
anhídrido acético en piridina. Los derivados acetilados fueron purificados mediante
cromatografía en columna de silicagel, eluidos con hexano y mezclas de hexano: acetato
de etilo (8:2). Los mono y diacetil derivados 7-acetoxi-4´,5-dihidroxi-3,6-
dimetoxiflavona (13) y 5,7-diacetoxi-4´-hidroxi-3,6-dimetoxiflavona (14) fueron
eluidos con metanol.
El compuesto 3,3´4´,5, 7-pentahidroxiflavona (4), más conocido como quercetina, fue
obtenido de Sigma. La criptostrobina (18) se aisló de Rumohra adiantiformis. La

Material y métodos
88
estructura de este compuesto fue determinada mediante análisis de espectroscopía y
comparación de bases de datos [218].
El 7,4´dimetileter de la dihidroquercetina (16) y el 7,4´-dimetileter del
dihidrokaempferol (17) fueron aislados de las partes aéreas de Pulicaria canariensis
[219]. La pureza de todos los compuestos fue del 99%.
4.1.2 Productos y material utilizado en:
4.1.2.1 Cultivos celulares.
El medio de cultivo RPMI 1640, DMEM, el suero bovino fetal (FBS), la tripsina,
HEPES (N-[2-hidroxietil] piperazino N'-[2-etanosulfanílico]), la L-glutamina y el azul
de tripán se obtuvieron de Gibco (Grand Island, NY, USA). El bicarbonato sódico y los
antibióticos (estreptomicina, gentamicina y penicilina G) fueron de Sigma Chemical Co.
(St. Louis, MO, USA). Las botellas de cultivo de 75 cm2 y las placas de 48 y 96 pocillos
estériles, así como el resto del material estéril utilizado fueron de Falcon (Becton y
Dickinson).
4.1.2.2 Técnica de fragmentación del ADN.
La RNasa A, la proteinasa K y el fenol fueron suministradas por Sigma Chemical Co.
(ST. Louis, MO, USA). El cloroformo y alcohol isoamílico se obtuvieron de BDH
(Poole, Inglaterra). La agarosa se obtuvo de Bio-Rad (Madrid, España) y el bromuro de
etidio de Sigma/Aldrich (España).
4.1.2.3 Microscopía de fluorescencia (tinción con bisbencimida).
El paraformaldehído y la bisbencimida (Hoechst nº 33258) se obtuvieron de Sigma
Chemical Co. (St. Louis, MO, USA). El microscopio invertido de contraste de fases
(Axiovert 135), los objetivos Plan-NEOFLUAR 20X y 63X y la lámpara de
fluorescencia (HBO 50) se adquirieron a Zeiss (Alemania). Las Fotos TMAX 100 pro
fueron de Kodak.

Material y métodos
89
4.1.2.4 Citometría de flujo.
El yoduro de propidio se obtuvo de Sigma/Aldrich. La anexina V se obtuvo de BD
PharmigenTH (Annexin V-FITC Apoptosis Detection Kit). Los fluorocromos H2DCF-
DA (dihidro-diclorofluoresce�na-diacetato) y JC-1 (yoduro de 5,5', 6,6’–tetracloro-1,1',
3,3’-tetraetil-benzimidazolo-carbocianina) se obtuvieron de Molecular Probes
(Invitrogen Corporation Carlsbad, CA). Los inhibidores de caspasas fueron obtenidos
de Sigma/Aldrich (Espa�a) a excepci�n del inhibidor de la caspasa-1 que fue obtenido
de Calbiochem y la caspasa 4 que fue obtenido de MP Biomedicals, (LLC). El
anticuerpo anti-Fas (human, neutralizing), clone ZB4, fue obtenido de Millipore
(Billerica, MA, USA).
El nocodazol, SP600125, PD98059, SB203580, los inhibidores de catepsina B y
catepsina L, Mu-Phe-HPh-fmk (MeOsuc-Phe-Homo-Phe-fluoromethylketone) y el
E64d [(2S, 3S)-trans-epoxysuccinyl-L-leucylamido-3-methylbutane ethyl ester], U0126
y el resto de inhibidores fueron obtenidos de Sigma (Saint Louis, MO, USA).
4.1.2.5 Determinación de la actividad caspasa.
Los substratos espec�ficos utilizados en el ensayo para determinar la actividad de
caspasa-3, -8 y -9 fueron N-acetil-Asp-Glu-Val-Asp-p-nitroanilina (DEVD-pNA), N-
acetil-Ile-Glu-Thr-Asp-p-nitroanilina (IETD-pNA) obtenidos de la casa Sigma y el N-
acetil-Leu-Glu-His-Asp-p-nitroanilina (LEHD-pNA) fu� obtenido de AnaSpec,
respectivamente. La paranitroanilina (pNA) fue obtenida de Calbiochem.
4.1.2.6 Determinación de actividad esfingomielinasa y determinación de ceramidas.
La N-metil-14C-esfingomielina (actividad especifica 56.6 mCi/mmol) y el [-32P] 5�-
trifosfato de adenosina (actividad especifica 3000 Ci/mmol) fueron obtenidos de Perkin
Elmer (Madrid, Spain) y sn-1,2-Diacilglicerol quinasa fue obtenido de Calbiochem
(Darmstadt, Germany). Las placas de TLC (placas de silica gel de 250 m de espesor)
fueron obtenidos de Whatman (Maidstone, UK).

Material y m�todos
90
4.1.2.7 Inmunodetecci�n de prote�nas (“Western Blot”).
Las membranas (PVDF) se obtuvieron de Millipore (Billerica, MA, USA). El sistema
de revelado por quimioluminiscencia, Supersignal � West Pico Chemiluminiscent
Substrate, fue proporcionado por Pierce (Rockford, USA). Las placas para
autorradiografia se adquirieron de Kodak (Sigma).
Los productos utilizados en la obtenci�n del lisado celular, fueron: Aprotinina
(inhibidor de ser�nproteasas que inhibe tripsina y quimiotripsina), Proteinasa K (enzima
para la digesti�n de prote�nas y nucleasas durante la purificaci�n del ADN, Leupeptina
(inhibidor de ciste�n y ser�nproteasas. Inhibe tripsina, papa�na y catepsina B), Pepstatina
A (inhibidor potente de proteasas �cidas), PMSF (inhibe ser�nproteasas, ciste�nproteasas
y acetilcolinesterasa), Ditiotreitol (DTT: agente reductor estereoselectivo para puentes
disulfuro en complejos moleculares), ortovanadato s�dico (inhibidor de fosfatasas
alcalinas) y el detergente trit�n X-100 fueron obtenidos de Sigma Chemical Co. (ST.
Louis, MO, USA).
Los anticuerpos para poli-(ADP-ribosa)-polimerasa (PARP), caspasa-3, caspasa-9,
caspasa-8, se obtuvieron de Stressgen (Victoria, British Columbia, Canada). Los
anticuerpos para citocromo c, caspasa-7 y Bid se obtuvo de BD PharMingen (San
Diego, CA, USA).
Los anticuerpos para α-tubulina y caspasa-6 se obtuvieron de Sigma (Saint Louis, MO,
USA) y de MBL respectivamente. Los anticuerpos para fosfo-p38MAPK (Thr180/Tyr182),
p38MAPK, fosfo-JNK/SAPK (Thr183/Tyr185), JNK/SAPK, fosfo-ERK1/2 (Thr202/Tyr204) y
ERK1/2 fueron obtenidos de New England Biolabs (Cell Signaling Technology,
Beverly, MA, USA). Los anticuerpos anti-ciclina B1, anti-fosfo-ciclina B1, anti-cox IV,
anti-cdk 1, anti-fosfo-cdk1 y anti-p21, se obtuvieron de Abcam (Cambridge, UK). El
anticuerpo monoclonal anti-β-actina se obtuvo de Sigma/Aldrich. Los anticuerpos
secundarios fueron obtenidos de GE Healthcare Bio-Sciences AB (Uppsala, Sweden).

Material y métodos
91
4.1.2.8 Microscopía inmunofluorescencia.
El anticuerpo anti-α-tubulina, la colchicina y el paclitaxel fueron obtenidos de Sigma
(Saint Louis, MO, USA), el BSA (albumina de suero bovino) de Sigma/ Aldrich y el
Anti –IgG de rat�n (H+L)-Cy2TM (cabra) de Jackson Immuno Research Laboratories,
Inc. (West Grove, PA, USA). El suero normal de cabra (NGS) fue obtenido de Sigma.
El VECTASHIELD� con DAPI (Vector Laboratories, INC. Burlingame, CA 94010) y
el microscopio que se utiliz� fue un Microscopio de fluorescencia LSM 5 PASCAL de
ZEISS.
4.1.3 Reactivos generales de laboratorio.
El agua desionizada y bidestilada se obtuvo con un equipo Mili-Q (Water Purification
System, Millipore Ib�rica SA, Madrid, Espa�a). El dimetilsulf�xido (DMSO), EDTA,
EGTA, NaCl, glicerol, trizma base, trizma-HCl, azida s�dica, bromuro de 3-[4,5-
dimetiltiazol-2-il]-2,5-difeniltetrazolio (MTT), la sacarosa, N-acetil-L-ciste�na (NAC),
tween 20, hidroxido de sodio (NaOH), �cido clorh�drico (HCL) y otros compuestos
utilizados en la preparaci�n de reactivos y tampones se adquirieron a Sigma Chemical
Co. (ST. Louis, MO, USA) o a BDH (Carlo Erba, E Merck y Fluka). El dodecil sulfato
s�dico (SDS) se obtuvo de Bio-Rad (Madrid, Espa�a).
4.2 Modelo Experimental.
Las c�lulas tumorales utilizadas se obtuvieron de la colecci�n europea de c�lulas
(Salisbury, UK). Las c�lulas se contaron en un hematocit�metro y la viabilidad siempre
fue superior al 95% utilizando el m�todo de exclusi�n de azul de trip�n.
4.2.1 Cultivo de células leucémicas promielocíticas humanas HL-60 y células de
linfoma histiocítico humano U937.
Las c�lulas HL-60 y U937 se cultivaron en medio RPMI 1640 suplementado con 10%
(v/v) de suero bovino fetal (FBS) inactivado por calor y antibi�tico (100 unidades /ml
de penicilina y 100 g/ml estreptomicina) en una atm�sfera humidificada (37 �C y 5%

Material y métodos
92
CO2) y a una densidad no superior a 0.5 x 10 6 células/ml [220,221]. A las células se les
cambió el medio dos veces por semana y se dividieron aproximadamente cada 24 horas.
4.2.2 Cultivo de células A431 (carcinoma epidermoide humano) y SK-OV-3
(adenocarcinoma de ovario humano).
Las células A431 y SK-OV-3 se cultivaron en monocapa en medio DMEM
suplementado con 10% (v/v) de suero bovino fetal (FBS) inactivado por calor y
antibiótico (100 unidades /ml de penicilina y 100 g/ml estreptomicina) en una
atmósfera humidificada (37 ºC y 5% CO2). Las células se pasaron dos veces por semana
y se duplicaron cada 24 horas [220,221].
4.2.3 Cultivo de células HeLA (carcinoma de cervix humano) y HOS (osteosarcoma).
Las células HeLa y HOS se crecieron en monocapa en medio EMEM suplementado con
10% (v/v) de suero bovino fetal (FBS) inactivado por calor, antibiótico (100 unidades
/ml de penicilina y 100 g/ml estreptomicina), 2 mM de glutamina y 1% de
aminoácidos no esenciales. Para su recuento se incubaron con 0,25 % tripsina-EDTA
durante 5 minutos y las células obtenidas por centrifugación (500 g durante 5 minutos)
se resuspendieron a una densidad de 1104 células /ml. Se mantuvieron en una
atmósfera humidificada (37 ºC y 5% CO2) [220,221].
4.2.4 Cultivo de células HL-60 transfectadas con el gen humano Bcl-xL (HL-60 que
sobre expresan bxl), HL-60 con su vector control HL-60 bcl-neo y U937 que sobre
expresan el gen humano de bcl-2 (denominadas U937/Bcl-2, obtenidas por donacion de
Dr. Jacqueline Bréard, INSERM U749, Faculté de Pharmacie Paris-Sud, Châtenay-
Malabry, France). HL-60 fueron transfectadas con el plasmido pSFFV-neo (HL-
60/neo) o con el plasmido pSFFV-bcl-xL (HL-60/Bcl-xL) que fueron establecidos por el
Dr. Kapil N Bhalla (Medical College of Georgia Cancer Center, GA, USA) y fueron
obtenidas por donación de Dr. Angelika Vollmar (Department of Pharmacy, University
of Munich, Germany).

Material y métodos
93
Las c�lulas HL-60/Bcl-xL y HL-60/neo se crecieron en medio RPMI 1640 advance con
suplementado con 2 mM de glutamina, 10 ml de geneticina y 10% de suero bovino fetal
inactivado por calor. El d�a previo a los experimentos se les cambi� el medio y se
a�adi� RPMI que conten�a 10% de suero bovino fetal inactivado por calor y antibi�tico
(100 unidades /ml de penicilina y 100 g/ml estreptomicina) en una atm�sfera
humidificada (37 �C y 5% CO2) [220,221].
U937/Bcl-2 se crecieron en medio RPMI 1640 suplementado con 10% (v/v) de suero
bovino fetal (FBS) inactivado por calor y antibi�tico (100 unidades /ml de penicilina y
100 g/ml estreptomicina) en una atm�sfera humidificada a 37 �C y 5% CO2, y a una
densidad que no excediera de 0.5 x 10 6 c�lulas/ml.
A las c�lulas se les cambi� el medio dos veces por semana y se duplicaron cada 24
horas.
4.2.5 Células mononucleares de sangre periférica de origen humano (PBMC).
Las c�lulas mononucleares de sangre perif�rica de origen humano (PBMC) fueron
aisladas de sangre obtenida de voluntarios sanos, recogidas en tubos heparinizados para
evitar la coagulaci�n y se aislaron mediante centrifugaci�n con Ficoll-Paque Plus
(Amersham Biosciences). Una parte de las PBMCs obtenidas se estimularon con
fitohemaglutinina (PHA, 2 mg / ml) durante 48 h antes del tratamiento experimental.
4.2.6 Tratamiento con los compuestos.
Los productos se prepararon a una concentraci�n de 10-100 mM en dimetilsulf�xido y
las al�cuotas se mantuvieron a –20� C. Las diluciones a partir de las anteriores se
hicieron en medio de cultivo justo antes del experimento. En todos los experimentos la
concentraci�n de DMSO no excedi� del 0,3%, concentraci�n no t�xica para las c�lulas.
La misma concentraci�n de DMSO fue a�adida a las c�lulas control.

Material y métodos
94
4.3 Métodos:
4.3.1 Evaluación de la citotoxicidad in vitro y estudios de la proliferación celular:
MTT.
La valoración de la proliferación celular se realizó, mediante el ensayo de la reducción
metabólica del bromuro de 3-[4,5-dimetiltiazol-2-il]-2,5-difeniltetrazolio (MTT) por la
actividad deshidrogenasa mitocondrial [222].
Las células se cultivaron en placas de 96 pocitos, por triplicado, a una densidad
aproximada de 1x104 células/ pocito en 0,2 ml de medio de crecimiento completo y en
presencia de diferentes concentraciones de los flavonoides durante 72 h. Las placas se
centrifugaron (3000 x rpm, 10 min) a temperatura ambiente y el medio se eliminó por
aspiración. A cada pocito se le añadió 100 l de MTT (0,5mg/ml en medio de
crecimiento suplementado con antibiótico pero sin suero) y las placas se incubaron
durante 4 h a 37 oC. La reacción se paró añadiendo 100 l de SDS (20%) en HCl 0.02N
e incubando la mezcla hasta la mañana siguiente. La cuantificación de la conversión del
MTT (amarillo) en su forma reducida (púrpura) por la deshidrogenasa mitocondrial se
determinó a 570 nm en un lector ELISA. Los datos se analizaron con el programa
informático Prism 2.0 (GraphPad) y se determinó la IC50 (concentración de producto
que inhibe el crecimiento celular a la mitad) [220,223].
4.3.2 Estudio de la apoptosis celular:
La apoptosis celular se determinó mediante:
Tinción con el fluorocromo bisbencimida, método cuantitativo, basado en la
capacidad del fluorocromo para unirse al ADN, permite visualizar la existencia de
cambios morfológicos del núcleo como son la condensación de la cromatina y su
compactación a lo largo de la periferia del núcleo y la segmentación del núcleo. Una vez
finalizados los tratamientos, las células (~5x105) se lavaron con PBS y se fijaron con 70
l de paraformaldehído (3%, en PBS) durante 10 minutos a temperatura ambiente. A

Material y métodos
95
continuación se centrifugó (16.000 x g 1 minuto), se eliminó el paraformaldehído y las
células se tiñeron con 30-50 l de una disolución 20 g/mL de bis-benzimida (Hoechst
33258) en PBS durante 15 minutos a temperatura ambiente y en la oscuridad. Una
alícuota (10 l) se fijó en un porta y se analizó la morfología nuclear de 500 células con
un microscopio de fluorescencia (Zeiss-Axiovert). Se consideraron células apoptóticas
aquellas cuyos núcleos presentaron condensación de la cromatina, su compactación a lo
largo de la periferia y la fragmentación nuclear en tres o más cuerpos apoptóticos [220].
Fragmentación del ADN, método cualitativo, basado en la detección de la
fragmentación internucleosómica del ADN, visualizado como una escalera discontinua
de bandas multimétricas de 185-200 pares de bases o múltiplos, características del
proceso apoptótico.
Las células a una densidad de 5 x 105 células/ml se trataron con diferentes
concentraciones de los distintos flavonoides durante períodos variables de tiempo, se
recolectaron por centrifugación (16.000 x g 1 minuto) y se lavaron dos veces con
tampón fosfato-salino frío (10 mM fosfato sódico, 150 mM NaCl, pH 7,4). Las células
se resuspendieron con 20 l de tampón de lisis [50 mM Tris-HCl (pH 8,0) 10 mM
EDTA, 0,5% SDS] y se incubaron sucesivamente con 1 g/ l de RNasa A (1 hora a 37
ºC) y con 1 g/ l de Proteinasa K (más de 1h a 50ºC). Se añadió a cada muestra 2 l de
azul bromofenol (0,25%) y el ADN se extrajo con 100 l de una mezcla fenol:
cloroformo: alcohol isoamílico (24:24:1). Se centrifugó 1 minuto a temperatura
ambiente y se recogió la fase acuosa de color azul. Se añadió 100 l de cloroformo a las
muestras, se agitaron, se centrifugaron y se extrajo la fase acuosa. Se añadió 5 l de
tampón de carga (10 mM EDTA, pH 8,0, conteniendo 1% (p/v) de agarosa de bajo
punto de fusión y 40% de sacarosa) y las muestras se incubaron a 70ºC durante 5-10
minutos. Finalmente 30 l de cada muestra se sometieron a electroforesis (5 V/cm)
durante 3-4 h en geles de agarosa al 2 %. Los geles se tiñeron con bromuro de etidio
(0,5 g/ml durante 30 min), los fragmentos de DNA se visualizaron a 260 nm en un
transiluminador y la imagen se captó con una cámara digital (DC290, Kodak) y se
analizó con el software Quantity One (Bio-Rad).
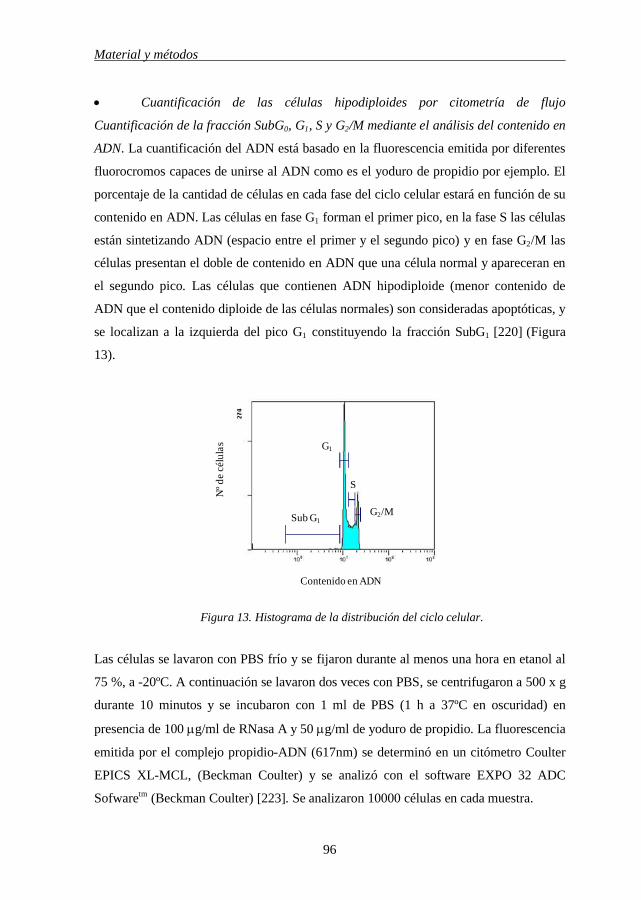
Material y métodos
96
Cuantificación de las células hipodiploides por citometría de flujo
Cuantificación de la fracción SubG0, G1, S y G2/M mediante el análisis del contenido en
ADN. La cuantificación del ADN está basado en la fluorescencia emitida por diferentes
fluorocromos capaces de unirse al ADN como es el yoduro de propidio por ejemplo. El
porcentaje de la cantidad de células en cada fase del ciclo celular estará en función de su
contenido en ADN. Las células en fase G1 forman el primer pico, en la fase S las células
están sintetizando ADN (espacio entre el primer y el segundo pico) y en fase G2/M las
células presentan el doble de contenido en ADN que una célula normal y apareceran en
el segundo pico. Las células que contienen ADN hipodiploide (menor contenido de
ADN que el contenido diploide de las células normales) son consideradas apoptóticas, y
se localizan a la izquierda del pico G1 constituyendo la fracción SubG1 [220] (Figura
13).
Figura 13. Histograma de la distribución del ciclo celular.
Las células se lavaron con PBS frío y se fijaron durante al menos una hora en etanol al
75 %, a -20ºC. A continuación se lavaron dos veces con PBS, se centrifugaron a 500 x g
durante 10 minutos y se incubaron con 1 ml de PBS (1 h a 37ºC en oscuridad) en
presencia de 100 g/ml de RNasa A y 50 g/ml de yoduro de propidio. La fluorescencia
emitida por el complejo propidio-ADN (617nm) se determinó en un citómetro Coulter
EPICS XL-MCL, (Beckman Coulter) y se analizó con el software EXPO 32 ADC
Sofwaretm (Beckman Coulter) [223]. Se analizaron 10000 células en cada muestra.
Contenido en ADN
Nºd
ecé
lula
s
Sub G1
G1
S
G2/M

Material y métodos
97
Determinación de las células apoptóticas mediante la determinación de la
externalización de fosfatidilserina por citometría de flujo. La fosfatidilserina es un
fosfolípido que normalmente se encuentra en la cara interna de la membrana plasmática
y que se transloca a la cara externa de la membrana durantelos estadíos más tempranos
de la apoptosis. La proteína Anexina V se une a fosfatidilserina en presencia de Ca2+ y
permite detectar las células apoptóticas por citometría de flujo previa incubación de las
células con Anexina V unida a un fluorocromo (FITC). Incubando las células
simultaneamente con yoduro de propidio (entra sólo en las células muertas) y Anexina
V podremos distinguir entre células necróticas (a las cuales se unirá también la Anexina,
por tener la membrana permeabilizada) y células apoptóticas (excluyen el yoduro de
propidio y unen Anexina V).
Las células (1x106 por tratamiento) una vez tratadas se lavaron con PBS, y se
resuspendieron en el tampón de incubación (10 mM Hepes-NaOH, pH 7,4, 140 mM
NaCl, 5 mM CaCl2), en presencia de 5 M de yoduro de propidio y Anexina-V- FITC
(BD Pharmingen).
La externalización de fosfatidilserina se determinó mediante citometría de flujo. Hay
que tener en cuenta en este punto que cuando las células en cultivo sufren apoptosis, al
no poder ser fagocitadas, sufren necrosis secundaria al proceso apoptótico, puede
aumentar el número de células que dan positivo a yoduro de propidio. De esta manera
distinguimos: a) Células viables: no unen anexina V y excluyen yoduro de propidio, b)
Células en apoptosis temprana: unen anexina V y excluyen yoduro de propidio, c)
Células en apoptosis tardía: unen anexina V e incorporan yoduro de propidio y d)
Células necróticas: unen anexina V e incorporan yoduro de propidio, o sólo incorporan
yoduro de propidio.
Microscopía Electrónica de Transmisión (MET).
Para el procesado de muestras en Microscopía Electrónica de Transmisión, las células
se centrifugaron, se resuspendieron y se fijaron en glutaraldehído al 2.5 % en tampón
fosfato (0.1M, pH 7.2) durante 24 horas. La post-fijación se realizó en OsO4
al 1% en
tampón fosfato. Las células se deshidrataron con una serie de concentraciones crecientes

Material y m�todos
98
de etanol. El sedimento se incluy� en resina EMBed 812 que polimeriz� a 70� C. Los
cortes fueron realizados con un ultramicrotomo Reichert Ultracut s (Leica). Los cortes
semifinos fueron te�idos con azul de toluidina, mientras que los ultrafinos fueron
contrastados con acetato de uranilo y plomo. Las fotograf�as fueron tomadas en un
Microscopio Electr�nico de Transmisi�n Zeiss EM 910 (Carl ZEISS, Germany)
equipado con C�mara Digital Proscan Slow-scan CCD-Camera for TEM (Fa. Proscan
Elektronische Systeme GmbH, Germany) y software Soft Imaging System (Germany) del
Servicio de Microscop�a Electr�nica (SME) de la Universidad de Las Palmas de Gran
Canaria (ULPGC).
4.3.3 Determinaci�n de la actividad caspasa.
Las c�lulas tratadas se centrifugaron a 1000 �g durante 5 min at 4 �C se lavaron con
PBS y se incubaron en hielo. Las c�lulas se resuspendieron en tamp�n de lisis (50 mM
HEPES, pH 7,4, 1 mM dithiothreitol, 0,1 mM EDTA, 0.1% Chaps) y se dejaron 5 min
en hielo. Se centrifugaron durante 10 min at 17,000 �g a 4�C, se determin� la
concentraci�n de prote�nas en los sobrenadantes mediante el m�todo de Bradford (Bio-
RAD) y se almacenaron a −20 �C hasta que se usaron para el estudio de la actividad
enzim�tica de las caspasas por colorimetr�a. Se utiliz� una cantidad equivalente de
prote�nas de los diferentes tratamientos (~20 �g) y el ensayo se realiz� en hielo. El
incremento en la absorbancia a 405 nm despu�s de la incubaci�n a 37 �C durante 1 hora
fue indicativo de la actividad enzim�tica de las caspasas. Los substratos colorim�tricos
espec�ficos utilizados en el ensayo para la actividad de caspasa-3, -8 y -9 fueron N-
acetil-Asp-Glu-Val-Asp-p-nitroanilina (DEVD-pNA), N-acetil-Ile-Glu-Thr-Asp-p-
nitroanilina (IETD-pNA) y N-acetil-Leu-Glu-His-Asp-p-nitroanilina (LEHD-pNA),
respectivamente.
4.3.4 Inmunodetecci�n de prote�nas (“Western blot”).
Las c�lulas (1-10 x 106, dependiendo del ensayo) se incubaron en medio de crecimiento
libre de suero con los productos de inter�s durante distintos tiempos a 37�C. A
continuaci�n se recolectaron por centrifugaci�n (500 x g, 10 minutos, 4�C) y se lavaron

Material y métodos
99
dos veces con PBS. En este punto, las células se procesaron de distinta forma: en
función de si se necesitaba el lisado celular o las diferentes fracciones subcelulares
(fracción nuclear, citosólica y mitocondrial).
Obtención del lisado celular:
El precipitado celular ( 7x106 células) se resuspendió en 100 l de tampón Tris [Tris-
HCl (pH 7,4), 2 mM EDTA, 137 mM cloruro sódico,10% glicerol,1% Triton X-100,
2mM de pirofosfato de sodio, 20 mM glicerofosfato de sodio, 10 mM fluoruro sódico, 2
mM ortovanadato sódico, 1mM PMSF, leupeptina (5 g/ml), aprotinina (5 g/ml) y
pepstatina A (5 g/ml)] e incubó durante 15 min a 4ºC. Los lisados se homogeneizaron
mediante sonicación (cinco ciclos de cinco segundos) y se centrifugaron a 11,000 x g
durante 10 min a 4ºC. Se determinó la concentración de proteínas mediante el método
de Bradford [222] y se ajustaron todas las muestras a la misma concentración utilizando
el mismo tampón anterior. Los lisados celulares se hirvieron en tampón de
electroforesis-SDS con colorante [50 mM Tris-HCl (pH 6,8), 15% sacarosa, 2mM
EDTA, 3% SDS, 5 mM -mercaptoetanol y 0,01% azul de bromofenol] a 100ºC durante
5 minutos [220].
Fraccionamiento subcelular
El precipitado celular ( 107 células) se resuspendió en 100 l de tampón de
homogeneización [20 mM HEPES-KOH (pH 7,5), 10 mM KCl, 1,5 mM MgCl2, 1 mM
EDTA, 1mM EGTA, 1 mM DTT, 250 mM sacarosa] con inhibidores de proteasas (0,1
mM PMSF, 10 g/ml de leupeptina, 10 g/ml aprotinina y 10 g/ml pepstatina A). Las
células se lisaron usando una aguja de 21G y el extracto resultante se centrifugó a 1.000
x g durante 10 min a 4 ºC. El precipitado (fracción nuclear) se resuspendió en 100 l de
tampón de homogeneización, mientras el sobrenadante resultante se centrifugó a
105.000 x g durante 45 minutos a 4ºC. El precipitado resultante (fracción mitocondrial)
se resuspendió en 40 l de tampón de homogeneización mientras que el sobrenadante
representa la fracción citosólica. Se determinó la concentración de proteínas en cada una
de las fracciones obtenidas mediante el método de Bradford y se igualaron

Material y métodos
100
posteriormente las concentraciones de todas las muestras. Cantidades equivalentes de
cada muestra se mezclaron con tampón de electroforesis-SDS con colorante [50 mM
Tris-HCl (pH 6,8), 15% sacarosa, 2mM EDTA, 3% SDS, 5 mM -mercaptoetanol y
0,01% azul de bromofenol] y se hirvieron a 100ºC durante 3 minutos [220].
Las muestras se sometieron a electroforesis en un gel de poliacrilamida (del 7,5% al
15%, dependiendo de la proteína bajo estudio: 7,5% para PARP, 10% para MAPKs y
tubulina, 12,5% para caspasas, 15% para citocromo c y Bid) conteniendo 0,1% SDS y
se transfirieron a membranas de PVDF.
Las membranas se bloquearon con 10% de leche desnatada en tampón TBST [20 mM
Tris-HCl (pH 7,5), 137 mM NaCl, 0,1 % Tween-20] durante 1 h a temperatura ambiente
(o toda la noche a 4ºC), seguido por incubación con el anticuerpo específico.
En todos los casos, los anticuerpos se diluyeron en TBST conteniendo 3% de leche
desnatada y las membranas se incubaron en presencia del anticuerpo de interés, durante
24 horas con agitación suave y a 4 ºC. Las membranas se lavaron con TBST tres veces
durante 15 minutos cada vez y se incubaron con el anticuerpo secundario apropiado,
acoplado a la actividad peroxidasa, durante 1 h a temperatura ambiente. Finalmente las
membranas se lavaron nuevamente con TBST en las mismas condiciones anteriores y la
detección de las proteínas específicas se determinó por emisión de quimioluminiscencia
utilizando un kit comercial (ECL Western Blotting System, Amersham Pharmacia
Biotech) y posterior exposición de las membranas sobre películas de autorradiografía
(Hyperfilm ECL, Amersham Pharmacia Biotech). Las imágenes fueron capturadas con
un escáner y analizadas con el programa Adobe Photoshop 7.0.
Como control de que se ha cargado y transferido la misma cantidad de proteínas, se
realizó la determinación de -actina mediante el uso de un anticuerpo específico anti--
actina.

Material y métodos
101
4.3.5 Análisis de la tubulina.
Las células ( 0,5x106 células) se incubaron a las concentraciones indicadas de BME,
paclitaxel y colchicina durante el tiempo seleccionado. Las células se recolectaron por
centrifugación (500 x g, 10 min, 4ºC) y se lavaron tres veces con PBS. Los precipitados
celulares se resuspendieron en 100 l de tampón de homogeneización (20 mM Tris-HCl
(pH 6,8), 1 mM MgCl2, 2 mM EGTA, 5 g/ml leupeptina, 5 g/ml aprotinina, 1 mM
PMSF, 1 mM ortovanadato y 0,5% Nonidet), se incubaron a 37º C durante 5 minutos y
se centrifugaron a 15.000 x g durante 10 minutos a 4ºC. El sobrenadante obtenido
constituye la fracción donde se encuentra la tubulina soluble y el precipitado es la que
contiene la tubulina insoluble. La fracción insoluble se solubilizó con 100 l de tampón
de homogeneización mediante sonicación. Se determinó la concentración de proteínas
en cada una de las fracciones obtenidas mediante el metodo de Bradford y se igualaron
posteriormente las concentraciones de todas las muestras. Cantidades equivalentes de
cada muestra se mezclaron con tampón de electroforesis-SDS con colorante [50 mM
Tris-HCl (pH 6,8), 15% sacarosa, 2mM EDTA, 3% SDS, 5 mM -mercaptoetanol y
0,01% azul de bromofenol] y se hirvieron a 100ºC durante 3 minutos. Las muestras se
sometieron a electroforesis en gel de SDS-poliacrilamida al 10% y las proteínas se
electrotransferieron a membranas de PVDF. Las membranas se bloquearon con 5%
leche desnatada en tampón TBST [50 mM Tris-HCl (pH 7,4), 150 mM NaCl] con 1%
Tween 20, y incubadas con un anticuerpo monoclonal anti--tubulina (Sigma) y
posteriormente incubadas con un anticuerpo secundario (GE Healthcare Bio-Sciences
AB, Uppsala, Sweden). El complejo antigeno-anticuerpo se visulizó mediante
quimiluminiscencia (Pierce) [220].
4.3.6 Determinación de la generación de EROs intracelular.
La producción de EROs intracelular se midió fluorimétricamente usando la sonda
diacetato 2´,7´-diclorofluoresceína (DCF). Las células fueron expuestas a los
tratamientos correspondientes y se incubaron con 5 M DCF durante los últimos 30
minutos a 37ºC. Posteriormente las células se lavaron y resuspendieron en RPMI libre
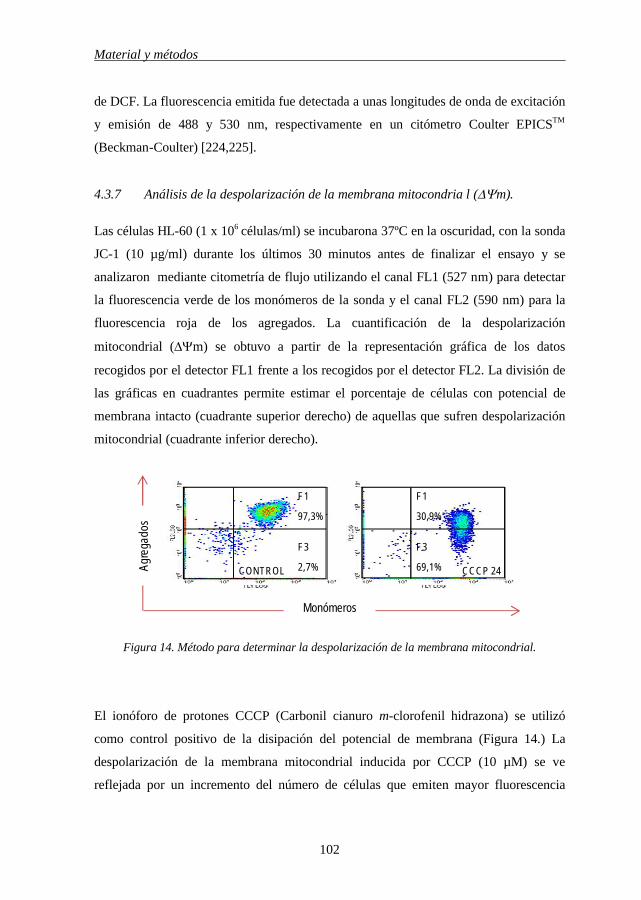
Material y métodos
102
de DCF. La fluorescencia emitida fue detectada a unas longitudes de onda de excitación
y emisión de 488 y 530 nm, respectivamente en un citómetro Coulter EPICSTM
(Beckman-Coulter) [224,225].
4.3.7 Análisis de la despolarización de la membrana mitocondria l (m).
Las células HL-60 (1 x 106 células/ml) se incubarona 37ºC en la oscuridad, con la sonda
JC-1 (10 µg/ml) durante los últimos 30 minutos antes de finalizar el ensayo y se
analizaron mediante citometría de flujo utilizando el canal FL1 (527 nm) para detectar
la fluorescencia verde de los monómeros de la sonda y el canal FL2 (590 nm) para la
fluorescencia roja de los agregados. La cuantificación de la despolarización
mitocondrial (m) se obtuvo a partir de la representación gráfica de los datos
recogidos por el detector FL1 frente a los recogidos por el detector FL2. La división de
las gráficas en cuadrantes permite estimar el porcentaje de células con potencial de
membrana intacto (cuadrante superior derecho) de aquellas que sufren despolarización
mitocondrial (cuadrante inferior derecho).
F1
97,3%
F3
2,7%
F1
30,9%
F3
69,1% CCCP 24CONTROL
Monómeros
Agre
gado
s
Figura 14. Método para determinar la despolarización de la membrana mitocondrial.
El ionóforo de protones CCCP (Carbonil cianuro m-clorofenil hidrazona) se utilizó
como control positivo de la disipación del potencial de membrana (Figura 14.) La
despolarización de la membrana mitocondrial inducida por CCCP (10 µM) se ve
reflejada por un incremento del número de células que emiten mayor fluorescencia

Material y métodos
103
verde y menos roja respecto a las c�lulas control y que se sit�an en el cuadrante inferior
derecho. Para el an�lisis se us� cit�metro Coulter EPICSTM (Beckman Coulter).
4.3.8 Análisis de la red de microtúbulos por microscopía de fluorescencia.
Una vez finalizados los tratamientos, las c�lulas (~1x106) se lavaron con PBS y se
fijaron con 70 l de paraformaldeh�do (3%, en PBS) durante 10 minutos a temperatura
ambiente. A continuaci�n se centrifug� (16.000 x g, 1 minuto), las c�lulas se lavaron
con PBS y se bloquearon con una disoluci�n de PBS conteniendo un 3 % de BSA
(alb�mina de suero bovino) y 0,1% de triton X-100 durante 1 hora a 4�C. Las c�lulas
se lavaron dos veces durante 5 minutos con PBS y se incubaron (24 h, 4�C) con anti-α-
tubulina (1/2000) en PBS, 3% BSA, 2% NGS (suero normal de cabra). Las c�lulas se
lavaron con PBS, se a�adi� el anticuerpo secundario (1/200), Anti–IgG de rat�n (H+L)-
Cy2TM, (de cabra)) en PBS, 3% BSA, 2% NGS e incubaron durante 1 hora. Se
realizaron tres lavados con PBS durante 10 minutos y se a�adi� 10 �L
VECTASHIELD�. Tras una incubaci�n de 15 minutos se fijaron las c�lulas en un
portaobjetos y se analiz� la morfolog�a nuclear (en azul se ti�eron los n�cleos) y la red
de microt�bulos (en verde se visualiz� la tubulina) en un microscopio de fluorescencia
LSM 5 PASCAL de ZEISS.
4.3.9. Determinación de la actividad esfingomielinasa ácida y neutra.
Los ensayos de determinaci�n de SMasa se realizaron utilizando [colina-metil- C14]
esfingomielina [226]. Las c�lulas HL-60 se trataron por triplicado en ausencia o en
presencia del compuesto. TNF- fue incluido como control positivo. A los tiempos
indicados de tratamiento se par� por inmersi�n en un ba�o de hielo seco con metanol,
las c�lulas se centrifugaron a 1500 rpm durante 5 min a 4�C y se lavaron dos veces con
PBS.

Material y métodos
104
- Determinación de la actividad de la esfingomielinasa neutra.
Las células se resuspendieron en un tampón que contenía 20 mM Hepes (PH 7,4), 10
mM MgCl2, 2 mM EDTA, 5 mM DTT, 0,1 mM ortovanadato sódico, 0,1 mM
molibdato sódico, 30 mM p-nitrofenilfosfato, 10 mM glicerofosfato, 750 µM ATP,
1mM PMSF, 10 M de leupeptina, 10 M pepstatina y 0,2% Triton X-100. Después de
incubar durante 5 minutos a 4ºC, las células se homogeneizaron y se obtuvo la fracción
citosólica mediante centrifugación. Se determinó la concentración de proteínas mediante
el método de Bradford y se ajustaron todas las muestras a la misma concentración.
Aproximadamente se incubaron 30 µg de proteínas de cada muestra se incubaron
durante 2 horas a 37ºC en un tampón que contenía 20 mM HEPES (PH 7,4), 1 mM
MgCl2 y 2,25 µl de [N-metil-14C] esfingomielina (0,2 µCi/ml, con una actividad
específica de 56,6 mCi/mmol). La cantidad de [14C] esfingomielina hidrolizada no
excedió del 10% de la cantidad de esfingomielina radiactiva añadida. La fosforilcolina
fue extraída con 800 µl de cloroformo: metanol 2:1 (v/v) y 250 µl de agua. La
fosforilcolina radiactiva producida a partir de [14C] esfingomielina fue determinada en
la fase acuosa en un contador de centelleo líquido.
- Determinación de la actividad de la esfingomielinasa ácida.
Las células tratadas se recolectaron mediante centrifugación a 500 x g a 4ºC durante 10
minutos, se lavaron dos veces con PBS frío y se resuspendieron en tampón de
homogeneización (0,2% Triton X-100) en la proporción 1x106 células por 0,3 ml. Las
células se lisaron, se centrifugó a 800 x g durante 5 minutos. La concentración de
proteínas se determinó en los sobrenadantes mediante el método de Bradford y 30 µg de
proteínas de cada muestra fueron incubadas durante 2 horas a 37ºC en un tampón que
contenía 250 mM acetato de sodio (PH 5,0), 1 mM EDTA, y 2,25 µl de [N-metil-14C]
esfingomielina. La cantidad de fosforilcolina producida a partir de [14C] esfingomielina
fue determinada en la fase acuosa en un contador de centelleo líquido.

Material y m�todos
105
4.3.10 Determinaci�n de la cantidad de ceramidas intracelular.
Para la determinaci�n de la cantidad de ceramida intracelular se utiliz� el ensayo de
diacilglicerol quinasa de Escherichia coli (DGK), en el cual la cantidad de ceramida se
determin� mediante cromatograf�a en capa fina (TLC) despu�s de su conversi�n a [32P]
ceramida-1-fosfato por la diacilglicerol quinasa [227]. Las c�lulas se incubaron a 37�C
durante diferentes tiempos con BME y el contenido lip�dico (3�106 c�lulas por tubo) se
extrajo con 1ml de cloroformo:metanol:1M HCL (100:100:1,v/v/v),270 �l de tamp�n
salino (135mM NaCl, 4,5mM KCL, 1,5 mM CaCL2, 0,5 mM MgCl2, 5,6 mM glucosa y
10 mM HEPES PH 7,2 ).
Al�cuotas de la fase org�nica (400 �l) se transferieron a tubos, se secaron bajo nitr�geno
y se sometieron a una hidr�lisis alcalina (0,1 M KOH en metanol durante 1 hora a 37
�C) para eliminar los glicerofosfol�pidos.
Los l�pidos se extrajeron con cloroformo, tamp�n salino, EDTA y se secaron bajo
nitr�geno. Se a�adi� 20 �l de una disoluci�n acuosa de 7,5% n-octyl β-
Dglucopiran�sido, 5 mM cardiolipina y 1 mM DETAPAC (�cido dietilenetriamina-
pentaac�tico) y los l�pidos se solubilizaron mediante sonicaci�n durante 2 minutos en un
ba�o. Para solubilizar los l�pidos se a�adi� 50 �l de 2 x de tamp�n de reacci�n (10 mM
imidazol, 100 mM NaCl, 2 mM EDTA, 25 mM MgCl2, pH 6,5), 2 �l 100 mM
ditiotreitol, 3,5 �l de diacilglicerol quinasa (1 mg/ml), 1 �l [γ-32P] ATP, y agua hasta un
volumen de 100 �l en total y se incub� a temperatura ambiente durante 30 minutos.La
reacci�n se par� mediante la adici�n de 1ml de cloroformo / metanol/ 1 M HCl
(100:100:1, v/v/v), 170 �l de 10 mM HEPES (PH 7,2), y 30 �l de 100 mM EDTA. La
fase org�nica se extrajo y se sec� bajo nitr�geno. Los l�pidos se separaron mediante
cromatograf�a en capa fina (TLC) desarrollada en cloroformo/metanol/acido ac�tico
glacial (65:15:5, v/v/v). Las placas cromatogr�ficas fueron revelados por
autorradiograf�a y las bandas correspondientes a ceramida se extrajeron y la
radiactividad se determin� mediante un contador de centelleo l�quido.

Material y métodos
106
4.4 Métodos estadísticos.
En todos los casos las determinaciones para cada grupo experimental se realizaron por
triplicado o cuadruplicado, y los valores representados se corresponden a datos de tres
experimentos como mínimo (Media ± S.E.M). La comparación entre los distintos
tratamientos se realizó por el método de la t de Student o análisis de la varianza.

RRREEESSSUUULLLTTTAAADDDOOOSSS


Resultados
109
5.1. Relación entre la estructura química de los flavonoides y su capacidad citotóxica:
En �sta Tesis hemos evaluado la actividad antitumoral de 22 flavonoides (tabla 3) en
cinco l�neas celulares tumorales de origen humano. Los estudios antiproliferativos (tabla
4) indican que el 3-metil �ter de betuletol (1) presenta propiedades citot�xicas en todas
las l�neas celulares analizadas, aunque las c�lulas HL-60 son las m�s sensibles (IC50=1,8
�M).
El 3-metil �ter de betuletol (1) posee dos grupos hidroxilos en C5 y C7,
respectivamente. Para estudiar como afectan dichos grupos a la capacidad
antiproliferativa del compuesto obtuvimos el derivado diacetilado, diacetato de 3-metil
�ter de betuletol (2) y el derivado metilado, dimetil-�ter de 3-metil �ter de betuletol (3)
(Figura 15A). Los resultados obtenidos con el derivado diacetilado no mostraron
grandes diferencias. As�, en HL-60, la IC50 pas� de 1,8 μM a 1,3 μM). Sin embargo, en
el caso del derivado metilado, el cambio de los dos grupos hidroxilo por los grupos
metoxi tuvo como consecuencia la supresi�n de la actividad antiproliferativa (IC50 >
100μM) en todas las l�neas celulares.
Dado que el grupo metoxilo es m�s peque�o que el grupo acetilo esta respuesta no
parece deberse a una consecuencia de impedimento est�rico. Es posible que los grupos
hidroxilo del 3-metil �ter de betuletol (1) est�n interactuando mediante puentes de
hidr�geno con grupos importantes de las dianas biol�gicas, funci�n que podr�an realizar
los grupos ceto en el derivado acetilado (2).
Un grupo que parece desempe�ar un papel clave en la determinaci�n de la potencia de
estos compuestos sobre la viabilidad celular es el grupo metilo en posici�n 4' del anillo
B dado que el 3-metil �ter de betuletol (1) es significativamente m�s potente que la 4',
5,7-trihidroxi-3 ,6-dimetoxiflavona (12) en todas las l�neas celulares estudiadas (Figura
15B).
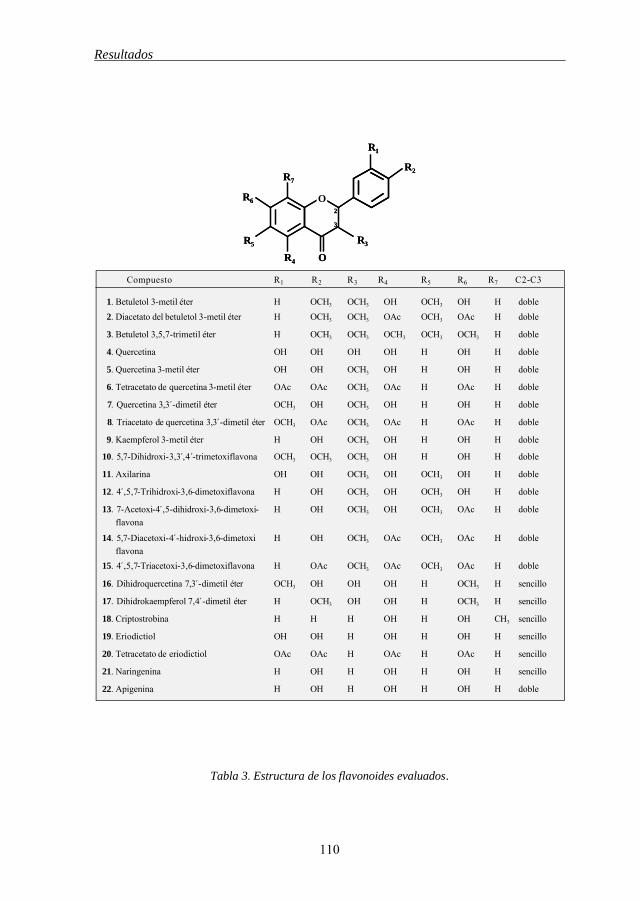
Resultados
110
Tabla 3. Estructura de los flavonoides evaluados.
Compuesto R1 R2 R3 R4 R5 R6 R7 C2-C3
1. Betuletol 3-metil éter H OCH3 OCH3 OH OCH3 OH H doble2. Diacetato del betuletol 3-metil éter H OCH3 OCH3 OAc OCH3 OAc H doble
3. Betuletol 3,5,7-trimetil éter H OCH3 OCH3 OCH3 OCH3 OCH3 H doble
4. Quercetina OH OH OH OH H OH H doble
5. Quercetina 3-metil éter OH OH OCH3 OH H OH H doble
6. Tetracetato de quercetina 3-metil éter OAc OAc OCH3 OAc H OAc H doble
7. Quercetina 3,3´-dimetil éter OCH3 OH OCH3 OH H OH H doble
8. Triacetato de quercetina 3,3 -dimetil éter OCH3 OAc OCH3 OAc H OAc H doble
9. Kaempferol 3-metil éter H OH OCH3 OH H OH H doble
10. 5,7-Dihidroxi-3,3 ,4 -trimetoxiflavona OCH3 OCH3 OCH3 OH H OH H doble
11. Axilarina OH OH OCH3 OH OCH3 OH H doble
12. 4 ,5,7-Trihidroxi-3,6-dimetoxiflavona H OH OCH3 OH OCH3 OH H doble
13. 7-Acetoxi-4 ,5-dihidroxi-3,6-dimetoxi- H OH OCH3 OH OCH3 OAc H dobleflavona
14. 5,7-Diacetoxi-4 -hidroxi-3,6-dimetoxi H OH OCH3 OAc OCH3 OAc H dobleflavona
15. 4 ,5,7-Triacetoxi-3,6-dimetoxiflavona H OAc OCH3 OAc OCH3 OAc H doble
16. Dihidroquercetina 7,3 -dimetil éter OCH3 OH OH OH H OCH3 H sencillo
17. Dihidrokaempferol 7,4 -dimetil éter H OCH3 OH OH H OCH3 H sencillo
18. Criptostrobina H H H OH H OH CH3 sencillo
19. Eriodictiol OH OH H OH H OH H sencillo
20. Tetracetato de eriodictiol OAc OAc H OAc H OAc H sencillo
21. Naringenina H OH H OH H OH H sencillo
22. Apigenina H OH H OH H OH H doble
O
OR4
R6
R2
R3R5
R1
R7
2
3
O
OR4
R6
R2
R3R5
R1
R7
2
3
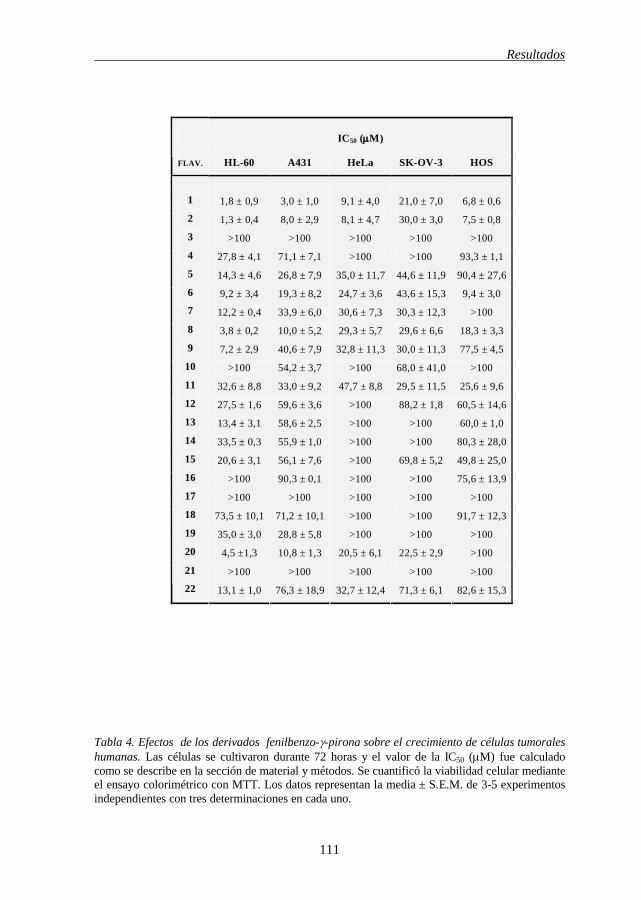
Resultados
111
IC50 (M)
FLAV. HL-60 A431 HeLa SK-OV-3 HOS
1,8 ± 0,9 3,0 ± 1,0 9,1 ± 4,0 21,0 ± 7,0 6,8 ± 0,6
1,3 ± 0,4 8,0 ± 2,9 8,1 ± 4,7 30,0 ± 3,0 7,5 ± 0,8
>100 >100 >100 >100 >100
27,8 ± 4,1 71,1 ± 7,1 >100 >100 93,3 ± 1,1
14,3 ± 4,6 26,8 ± 7,9 35,0 ± 11,7 44,6 ± 11,9 90,4 ± 27,6
9,2 ± 3,4 19,3 ± 8,2 24,7 ± 3,6 43,6 ± 15,3 9,4 ± 3,0
12,2 ± 0,4 33,9 ± 6,0 30,6 ± 7,3 30,3 ± 12,3 >100
3,8 ± 0,2 10,0 ± 5,2 29,3 ± 5,7 29,6 ± 6,6 18,3 ± 3,3
7,2 ± 2,9 40,6 ± 7,9 32,8 ± 11,3 30,0 ± 11,3 77,5 ± 4,5
>100 54,2 ± 3,7 >100 68,0 ± 41,0 >100
32,6 ± 8,8 33,0 ± 9,2 47,7 ± 8,8 29,5 ± 11,5 25,6 ± 9,6
27,5 ± 1,6 59,6 ± 3,6 >100 88,2 ± 1,8 60,5 ± 14,6
13,4 ± 3,1 58,6 ± 2,5 >100 >100 60,0 ± 1,0
33,5 ± 0,3 55,9 ± 1,0 >100 >100 80,3 ± 28,0
20,6 ± 3,1 56,1 ± 7,6 >100 69,8 ± 5,2 49,8 ± 25,0
>100 90,3 ± 0,1 >100 >100 75,6 ± 13,9
>100 >100 >100 >100 >100
73,5 ± 10,1 71,2 ± 10,1 >100 >100 91,7 ± 12,3
35,0 ± 3,0 28,8 ± 5,8 >100 >100 >100
4,5 ±1,3 10,8 ± 1,3 20,5 ± 6,1 22,5 ± 2,9 >100
>100 >100 >100 >100 >100
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22 13,1 ± 1,0 76,3 ± 18,9 32,7 ± 12,4 71,3 ± 6,1 82,6 ± 15,3
Tabla 4. Efectos de los derivados fenilbenzo--pirona sobre el crecimiento de células tumorales humanas. Las células se cultivaron durante 72 horas y el valor de la IC50 (M) fue calculado como se describe en la sección de material y métodos. Se cuantificó la viabilidad celular mediante el ensayo colorimétrico con MTT. Los datos representan la media ± S.E.M. de 3-5 experimentos independientes con tres determinaciones en cada uno.
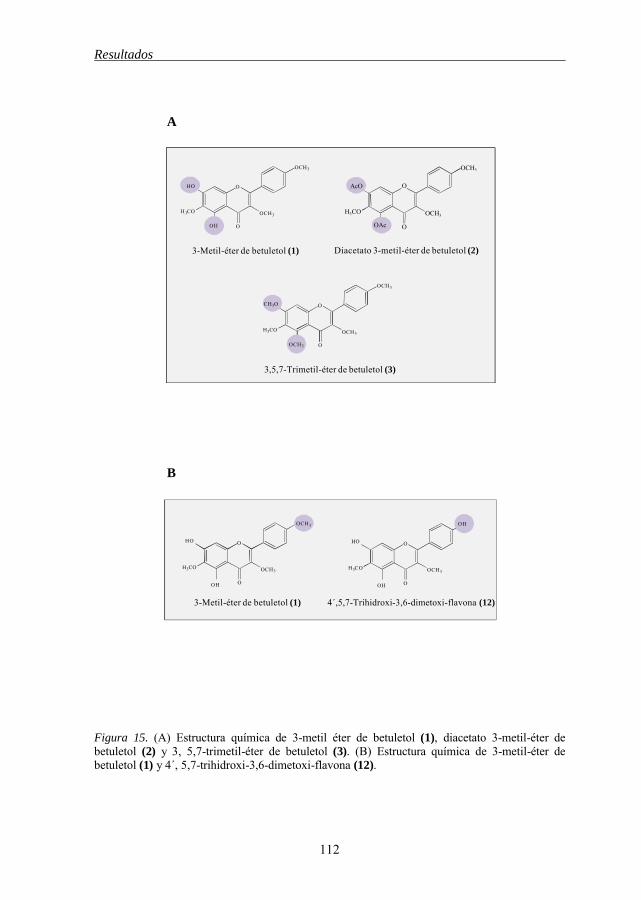
Resultados
112
Figura 15. (A) Estructura química de 3-metil éter de betuletol (1), diacetato 3-metil-éter de betuletol (2) y 3, 5,7-trimetil-éter de betuletol (3). (B) Estructura química de 3-metil-éter de betuletol (1) y 4´, 5,7-trihidroxi-3,6-dimetoxi-flavona (12).
O
O
OCH3
OCH3H3CO
CH3O
OCH3
3,5,7-Trimetil-éter de betuletol (3)
O
O
OCH3
OCH3H3CO
HO
OH
3-Metil-éter de betuletol (1)
O
O
OCH3
OCH3H3CO
AcO
OAc
Diacetato 3-metil-éter de betuletol (2)
3-Metil-éter de betuletol (1) 4´,5,7-Trihidroxi-3,6-dimetoxi-flavona (12)
O
O
OCH3H3CO
HO
OH
OH
OH
O
O
OCH3H3CO
HO
OCH3
A
B

Resultados
113
Se estudiaron también una serie de flavonoides que se encuentran estructuralmente
relacionados con la quercetina. El interés de este estudio se debe a que la quercetina (4)
es el flavonoide más abundante de la dieta humana. Los resultados indican que la
metilación del grupo hidroxilo en la posición C3 de la quercetina (4) da lugar a un
compuesto con un mayor actividad antiproliferativa el 3-metil éter quercetina (5)
(Figura 16A), en todas las líneas celulares, excepto en las células HOS, lo que sugiere
que el cambio afecta específicamente a determinados tipos de células.
Sin embargo, el grupo hidroxilo situado en la posición 3´ parece tener poco influencia
en la inhibición del crecimiento celular ya que la metilación de 3-metil éter quercetina
(5) para rendir el derivado dimetilado 3,3'-dimetil éter de quercetina (7) no alteró
significativamente el valor de IC50.
La presencia de un grupo hidroxilo en la posición 3' del anillo B tiene consecuencias
diferentes en función de la configuración de la molécula. Así el, 3-metil éter de
kaempferol (9) carece del mismo y la IC50 obtenida fue similar a su homologo 3-metil
éter quercetina (5) en todas las líneas ensayadas (Figura 16B). Del mismo modo, el
tetracetato de 3-metil éter de quercetina (6) y el triacetato del 3,3'-dimetil éter de
quercetina (8) (Figura 16C) que se diferencian sólo por la presencia de un grupo acetil o
un grupo metil éter, respectivamente, en posición 3', muestran una potencia similar. Por
el contrario, la presencia de un 3'-hidroxilo mejora significativamente la actividad
antiproliferativa de axillarina (11), en comparación con el 4´,5,7-trihidroxi-3,6-
dimetoxiflavona (12) que carece de este grupo químico (Figura 16D), en todos los tipos
celulares salvo en HL-60.
Cuando en el 3-metil éter de quercetina (5) un grupo metil éter sustituye al hidrogeno
en C6, el efecto sobre la IC50 depende del tipo celular. Así, el compuesto obtenido
axillarina (11) (Figura 17A) es menos citotóxico en HL-60 pero significativamente más
citotóxico en células HOS comparado con el 3-metil éter de quercetina (5). Sin embargo
la presencia del grupo metil éter no influyó en la citotóxidad del compuesto en las
restantes líneas celulares, A431, HeLa y SK-OV-3.
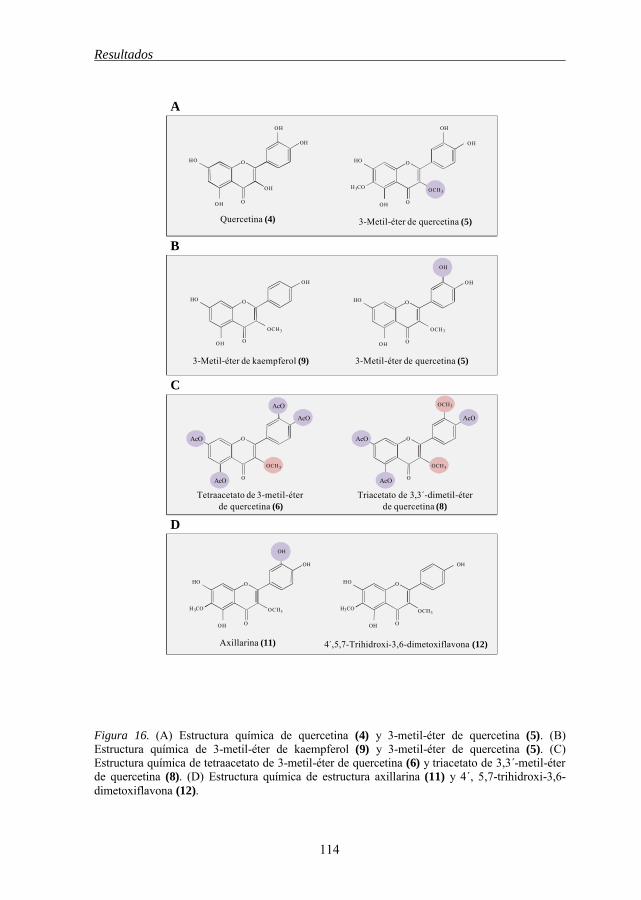
Resultados
114
Figura 16. (A) Estructura química de quercetina (4) y 3-metil-éter de quercetina (5). (B)Estructura química de 3-metil-éter de kaempferol (9) y 3-metil-éter de quercetina (5). (C)Estructura química de tetraacetato de 3-metil-éter de quercetina (6) y triacetato de 3,3´-metil-éter de quercetina (8). (D) Estructura química de estructura axillarina (11) y 4´, 5,7-trihidroxi-3,6-dimetoxiflavona (12).
Quercetina (4)
O
O
OH
HO
OH
OH
OH
3-Metil-éter de quercetina (5)
O
O
H3CO
HO
OH
OH
OH
OCH3
3-Metil-éter de quercetina (5)
O
O
HO
OH
OH
OH
OCH3
3-Metil-éter de kaempferol (9)
O
O
HO
OH
OH
OCH3
Triacetato de 3,3´-dimetil-éter de quercetina (8)
Tetraacetato de 3-metil-éterde quercetina (6)
O
O
OCH3
AcO
AcO
AcO
AcO
O
O
OCH3
AcO
AcO
AcO
OCH3
Axillarina (11) 4´,5,7-Trihidroxi-3,6-dimetoxiflavona (12)
O
O
H3CO
HO
OH
OH
OCH3
O
O
HO
OH
OH
H3CO OCH3
OH
A
B
C
D
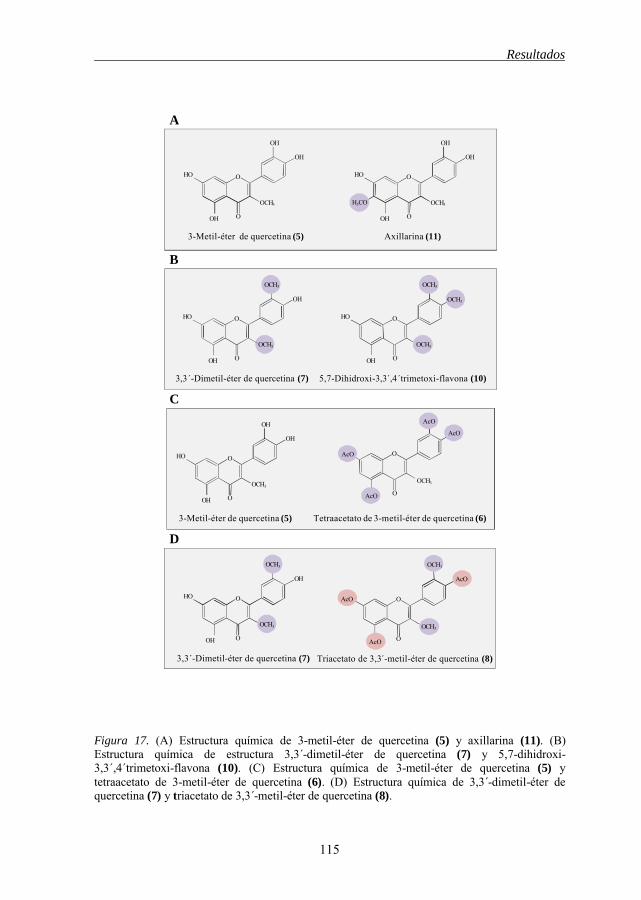
Resultados
115
Figura 17. (A) Estructura química de 3-metil-éter de quercetina (5) y axillarina (11). (B)Estructura química de estructura 3,3´-dimetil-éter de quercetina (7) y 5,7-dihidroxi-3,3´,4´trimetoxi-flavona (10). (C) Estructura química de 3-metil-éter de quercetina (5) y tetraacetato de 3-metil-éter de quercetina (6). (D) Estructura química de 3,3´-dimetil-éter de quercetina (7) y triacetato de 3,3´-metil-éter de quercetina (8).
3-Metil-éter de quercetina (5) Axillarina (11)
O
O
HO
OH
OH
OH
H3CO OCH3
O
O
HO
OH
OH
OH
OCH3
5,7-Dihidroxi-3,3´,4´trimetoxi-flavona (10)3,3´-Dimetil-éter de quercetina (7)
O
O
HO
OH
OH
OCH3
OCH3
O
O
HO
OH
OCH3
OCH3
OCH3
Triacetato de 3,3´-metil-éter de quercetina (8)
O
O
OCH3
AcO
AcO
AcO
OCH3
3,3´-Dimetil-éter de quercetina (7)
OH
O
O
HO
OH
OCH3
OCH3
A
B
C
D3-Metil-éter de quercetina (5) Tetraacetato de 3-metil-éter de quercetina (6)
O
O
HO
OH
OH
OHOCH3
O
O
OCH3
AcO
AcO
AcO
AcO

Resultados
116
Se ensay� tambi�n la influencia del grupo hidroxilo en la posici�n 4' del anillo B sobre
la viabilidad celular. Cuando el 3,3'-dimetil �ter de quercetina (7) es metilado en esa
posici�n el compuesto resultante, el 5,7-dihidroxi-3, 3', 4'-trimetoxiflavona (10) (Figura
17B) es significativamente menos citot�xico. Los valores de IC50 aumentaron de 12,2
μM y 30,6 μM a >100 μM, en HL-60 y c�lulas HeLa, respectivamente. Como en el
caso de otras sustituciones, estos cambios afectan de diferente forma a las distintas
l�neas celulares analizadas. La l�nea celular HOS es resistente a ambos compuestos.
La acetilaci�n de todos los grupos hidroxilo libres localizados en las posiciones 3', 4', 5
y 7 de 3-metil �ter de quercetina (5) para obtener el tetracetato 3-metil �ter de
quercetina (6) (Figura 17C) mejora considerablemente la actividad citot�xica en la l�nea
celular HOS, pasando de una IC50=90 μM a 9,4 μM. A diferencia de la l�nea celular
HOS, el resto de las l�neas celulares son resistentes a estos cambios qu�micos.
Cuando el 3,3'-dimetil �ter quercetina (7) es acetilado en las posiciones 4, 5 y 7
obteniendo el triacetato 3,3�-dimetil �ter de quercetina (8) (Figura 17D) se observa un
notable aumento en la actividad inhibidora del crecimiento en HL-60, A431 y, en
particular, en la l�nea celular HOS que es la m�s sensible a estas sustituciones.
Se realizaron adem�s acetilaciones sobre los grupos hidroxilo libres de la 4',5,7-
trihidroxi-3,6-dimetoxiflavona (12). La acetilaci�n del grupo hidroxilo en posici�n 7
rindi� un derivado monoacetilado, el 7-acetoxi-4',5-dihidroxi-3,6-dimetoxiflavona (13)
(Figura 18A), m�s activo que el compuesto de origen unicamente en la l�nea celular HL-
60. Este efecto se pierde cuando un segundo y un tercer grupo acetilo se introducen de
forma secuencial en las posiciones 5 y 4' para producir los compuestos 5,7-diacetoxi-4'-
hidroxi-3,6-dimetoxiflavona (14) y 4',5,7-triacetoxi-3,6-dimetoxiflavona (15) (Figura
18B), respectivamente.

Resultados
117
Otro conjunto de flavonoides son los relacionados con la estructura de la naringenina
(21), un compuesto con muy baja actividad citot�xica (IC50>100 μM), que tras la
introducci�n de un grupo hidroxilo en la posici�n 3' (eriodictiol (19)) aumenta
notablemente su actividad antiproliferativa en HL-60 (IC50= 35,0 μM) y en c�lulas
A431 (IC50= 28,8 μM). El derivado tetracetilado, de �ste �ltimo, el 3',4',5,7 tetracetato
de eriodictiol (20) (Figura 19A), es a�n m�s activo, con valores de IC50= 4,5 μM y 10,8
μM en HL-60 y A431, respectivamente.
Un incremento moderado de la citotoxicidad se observ� tambi�n en HL-60 y A431
cuando se elimina el grupo hidroxilo situado en la posici�n 4� en el anillo B de la
naringenina (21) y se introduce un grupo metilo en C8 para generar criptostrobina (18),
(Figura 19B). Otro requisito estructural que incrementa la actividad citot�xica de la
naringenina (21) es la introducci�n de un doble enlace entre C2 y C3. La flavona
resultante, apigenina (22) (Figura 19C) mostr� actividad antiproliferativa en todas las
l�neas celulares evaluadas, aunque HL-60 fue la m�s susceptible a este cambio qu�mico
(IC50= 13,1 μM).
As�, pues, podemos concluir que de las cinco l�neas celulares utilizadas la l�nea celular
promieloc�tica HL-60, es en general, la m�s sensible a la acci�n citot�xica promovida
por estos flavonoides.
La quercetina, el flavonoide m�s abundante en frutas y hortalizas, que generalmente es
consumido con mayor abundancia en la dieta, muestra propiedades citot�xicas en HL-
60.

Resultados
118
Figura 18. (A) Estructura química de 4',5,7-trihidroxi-3,6-dimetoxiflavona (12) y 7-acetoxi-4',5-dihidroxi-3,6-dimetoxiflavona (13). (B) Estructura química de 7-acetoxi-4',5-dihidroxi-3,6-dimetoxiflavona (13), 5,7- diacetoxi-4'-hidroxi-3 ,6-dimetoxiflavona (14) y 4',5,7-triacetoxi-3,6-dimetoxiflavona (15).
4´,5,7-Trihidroxi-3,6-dimetoxi-flavona (12)
O
O
OCH3H3CO
HO
OH
OH
O
O
OCH3H3CO
AcO
OH
OH
7 Acetoxi 4´,5-dihidroxi-3,6-dimetoxi-flavona (13)
O
O
OCH3H3CO
AcO
OH
OH
7 Acetoxi 4´,5-dihidroxi-3,6-dimetoxiflavona (13)
5,7 Diacetoxi 4´ hidroxi-3,6-dimetoxiflavona (14)
O
O
OCH3H3CO
AcO
OH
OH
AcO
4´,5,7-Triacetoxi-3,6-dimetoxi-flavona (15)
O
O
OCH3H3CO
AcO
OH
OH
AcO
AcO
A
B
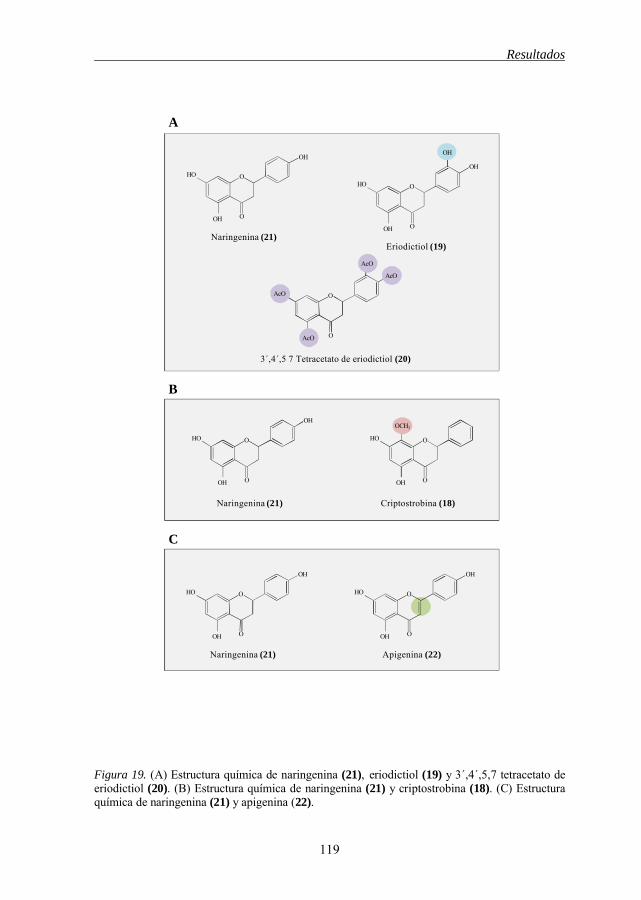
Resultados
119
Figura 19. (A) Estructura química de naringenina (21), eriodictiol (19) y 3´,4´,5,7 tetracetato de eriodictiol (20). (B) Estructura química de naringenina (21) y criptostrobina (18). (C) Estructura química de naringenina (21) y apigenina (22).
Naringenina (21) Criptostrobina (18)
O
O
HO
OH
OH
O
O
HO
OH
OCH3
Eriodictiol (19)
O
O
HO
OH
OH
OH
Naringenina (21)
O
O
HO
OH
OH
3´,4´,5 7 Tetracetato de eriodictiol (20)
O
O
HO
OH
OHAcO
AcO
AcO
AcO
Naringenina (21)
O
O
HO
OH
OH
Apigenina (22)
O
O
HO
OH
OH
A
B
C

Resultados
120
5.2. Los derivados del betuletol inducen apoptosis en células de leucemia mieloide
HL-60.
Los estudios antiproliferativos indican que el 3-metil éter de betuletol (1) posee notables
propiedades citotóxicas en todas las líneas celulares aunque HL-60 (IC50 de 1,8 µM) y
U937 (IC50= 2.0 M) demostraron ser las más sensibles (Tabla 4).
O
O
OCH3
HO
OHOCH3H3CO
Figura 20. Estructura química de 3-metil éter de betuletol (BME).
Con el fin de evaluar si la disminución de la viabilidad de los derivados del betuletol se
produce por apoptosis, se analizaron los cambios morfológicos característicos de las
células apoptóticas (condensación y fragmentación de la cromatina) por microscopia de
fluorescencia (Figura 21B) y por microscopía de transmisión electrónica (Figura 21C).
La evaluación de la morfología nuclear, indica que el porcentaje de células apoptóticas
aumentó de un 6 ± 1% a 24 ± 2% (4 veces más) en células tratadas con 3-metil éter de
betuletol durante 12 h a una concentración de 10 µM (Figura 21A).
Tambien se determinó el porcentaje de las células apoptóticas mediante el análisis de la
externalización de la fosfatidilserina por citometría de flujo en presencia de Anexina-V-
FITC. Los resultados indican un aumento de las células Anexina-V positivas en
respuesta al compuesto 1 (BME) (Figura 22A).
Se examinó si estos flavonoides derivados del betuletol eran capaces de inducir la
hidrólisis de la poli (ADP-ribosa) polimerasa, un marcador inequívoco que indica
activación de las caspasas.
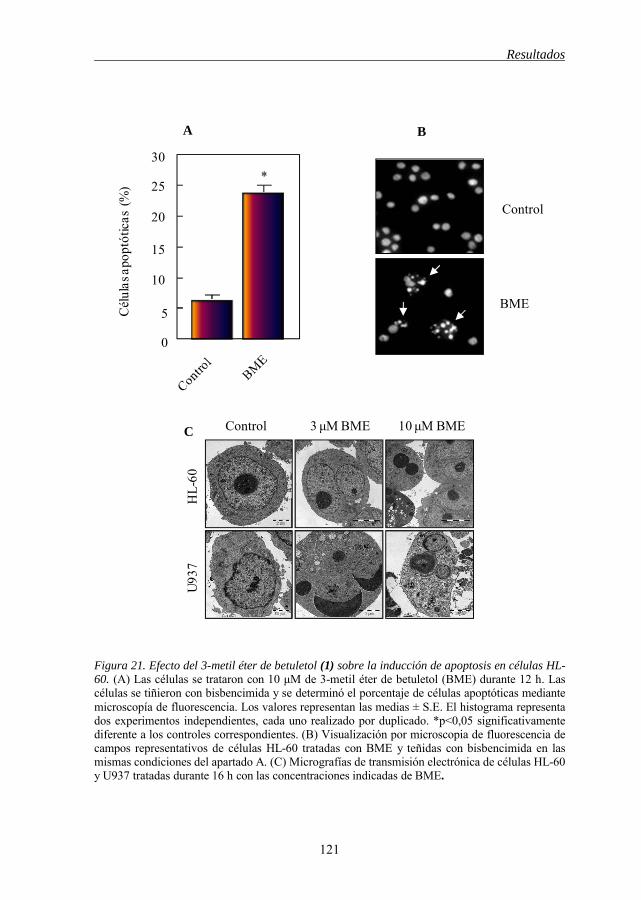
Resultados
121
Figura 21. Efecto del 3-metil éter de betuletol (1) sobre la inducción de apoptosis en células HL-60. (A) Las c�lulas se trataron con 10 μM de 3-metil �ter de betuletol (BME) durante 12 h. Las c�lulas se ti�ieron con bisbencimida y se determin� el porcentaje de c�lulas apopt�ticas mediante microscop�a de fluorescencia. Los valores representan las medias � S.E. El histograma representa dos experimentos independientes, cada uno realizado por duplicado. *p<0,05 significativamente diferente a los controles correspondientes. (B) Visualizaci�n por microscopia de fluorescencia de campos representativos de c�lulas HL-60 tratadas con BME y te�idas con bisbencimida en las mismas condiciones del apartado A. (C) Micrograf�as de transmisi�n electr�nica de c�lulas HL-60 y U937 tratadas durante 16 h con las concentraciones indicadas de BME.
A B
Control
BME
*
C�l
ulas
apop
t�tic
as(%
)
0
5
20
30
10
15
25
C Control 3 μM BME 10 μM BME
HL-
60U
937

Resultados
122
Despu�s de tratar las c�lulas con el 3-metil �ter de betuletol (1), el diacetato 3-metil �ter
de betuletol (2) y el derivado trimetilado 3, 5,7-trimetil �ter de betuletol (3) durante 12 h
a una concentraci�n de 10 μM se observ� la hidr�lisis de poli (ADP-ribosa) polimerasa
(116 kDa) mediante la detecci�n del fragmento de 85 kDa (Figura 22B), excepto en el
tratamiento con el 3, 5,7-trimetil �ter de betuletol (3). Estos resultados indican que la
hidr�lisis de poli (ADP-ribosa) polimerasa participa en la apoptosis inducida por estos
compuestos. El derivado trimetilado 3, 5,7-trimetil �ter de betuletol (3) no tuvo efecto
sobre la hidr�lisis de poli (ADP-ribosa) polimerasa consistente con los an�lisis de
citot�xicidad (tabla4).
El tratamiento con los flavonoides derivados del betuletol induce tambi�n la
fragmentaci�n del ADN en escalera, t�pico en las c�lulas apopt�ticas, y como era de
esperar, el 3,5,7-trimetil �ter de betuletol (3) no indujo la fragmentaci�n del ADN
(Figura 22C).
En conjunto, estos resultados indican que tanto el 3-metil �ter betuletol como el
diacetato, 3-metil �ter betuletol son potentes inductores de apoptosis en la l�nea celular
de leucemia mieloide humana HL-60 y que ambos compuestos son igualmente activos.
5.3. BME induce apoptosis por un mecanismo dependiente de caspasas: El
tratamiento previo con un inhibidor general de caspasas impide la hidrólisis de poli
(ADP-ribosa) polimerasa y la fragmentación del ADN, producida por el tratamiento de
las células con los derivados del betuletol.
Para determinar si los derivados del betuletol requieren la activaci�n de las caspasas
para la inducci�n de la apoptosis, las c�lulas HL-60 fueron pre-tratadas con un inhibidor
de caspasas de amplio espectro, z-VAD-fmk. Los resultados demuestran claramente que
el inhibidor bloquea totalmente la fragmentaci�n del ADN (Figura 23A) y la hidr�lisis
de poli (ADP-ribosa) polimerasa (Figura 23B) inducidas por el 3-metil �ter betuletol.
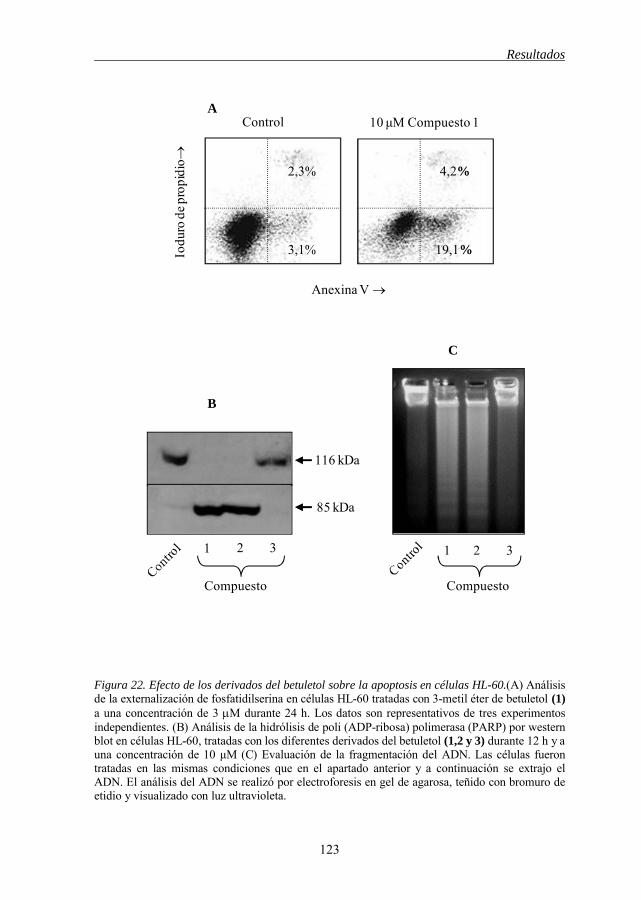
Resultados
123
Figura 22. Efecto de los derivados del betuletol sobre la apoptosis en células HL-60.(A) An�lisis de la externalizaci�n de fosfatidilserina en c�lulas HL-60 tratadas con 3-metil �ter de betuletol (1)a una concentraci�n de 3 M durante 24 h. Los datos son representativos de tres experimentos independientes. (B) An�lisis de la hidr�lisis de poli (ADP-ribosa) polimerasa (PARP) por western blot en c�lulas HL-60, tratadas con los diferentes derivados del betuletol (1,2 y 3) durante 12 h y a una concentraci�n de 10 μM (C) Evaluaci�n de la fragmentaci�n del ADN. Las c�lulas fueron tratadas en las mismas condiciones que en el apartado anterior y a continuaci�n se extrajo el ADN. El an�lisis del ADN se realiz� por electroforesis en gel de agarosa, te�ido con bromuro de etidio y visualizado con luz ultravioleta.
1 2 3
Compuesto
C
116 kDa
B
1 2
85 kDa
3
Compuesto
Control 10 μM Compuesto 1
3,1%
2,3%
19,1%
4,2%Io
duro
depr
opid
io
Anexina V
A
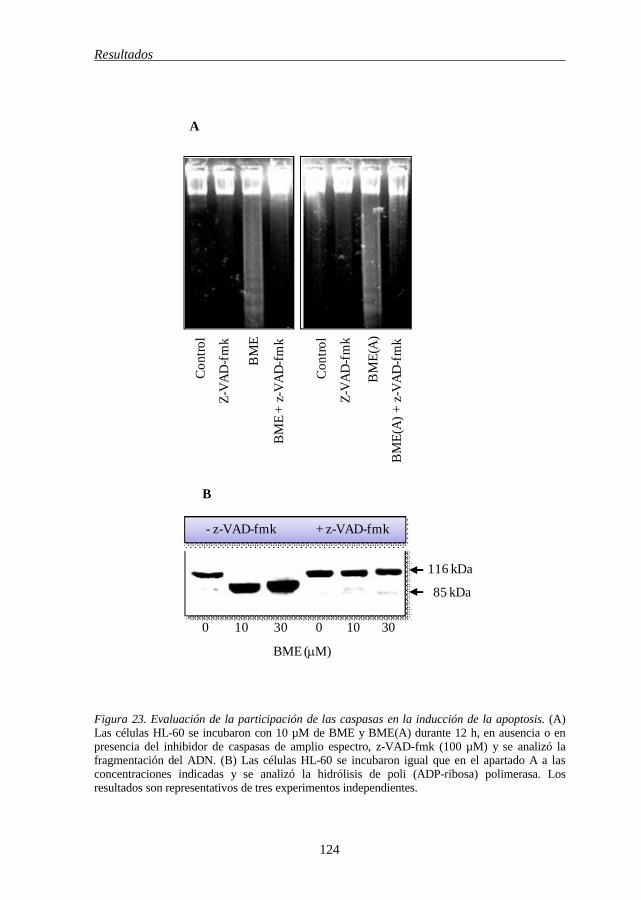
Resultados
124
Figura 23. Evaluación de la participación de las caspasas en la inducción de la apoptosis. (A) Las células HL-60 se incubaron con 10 µM de BME y BME(A) durante 12 h, en ausencia o en presencia del inhibidor de caspasas de amplio espectro, z-VAD-fmk (100 µM) y se analizó la fragmentación del ADN. (B) Las células HL-60 se incubaron igual que en el apartado A a las concentraciones indicadas y se analizó la hidrólisis de poli (ADP-ribosa) polimerasa. Los resultados son representativos de tres experimentos independientes.
B
0 10 30 0 10 30
- z-VAD-fmk + z-VAD-fmk
BME (M)
85 kDa
116 kDa
A
Con
trol
Z-V
AD
-fm
k
BM
E+
z-V
AD
-fm
k
BM
E
Con
trol
Z-V
AD
-fm
k
BM
E(A
)+z-
VA
D-f
mk
BM
E(A
)

Resultados
125
En conjunto, estos resultados indican que estos acontecimientos son dependientes de la
activaci�n de caspasa.
5.4. La apoptosis inducida por los derivados del betuletol implica la activación de
caspasa-3.
Para determinar si la caspasa-3 est� involucrada en la inducci�n de apoptosis por los
compuestos, las c�lulas HL-60 se incubaron con concentraciones crecientes de 3-metil
�ter de betuletol, los lisados celulares se analizaron en presencia del sustrato espec�fico
para caspasa-3, DEVD-pNA. Los resultados (Figura 24A), demuestran que la actividad
caspasa-3 aumenta 1,5 y 2,3 veces en las c�lulas HL-60 tratadas con 3 μM � con 10 μM
de 3-metil �ter de betuletol, respectivamente, durante 12 h.
La activaci�n de la caspasa-3 se evalu� adem�s en respuesta al 3-metil �ter de betuletol
y al diacetato 3-metil �ter de betuletol mediante western blot. Los resultados indican que
concentraciones bajas (3-10 μM) de estos compuestos son capaces de inducir la
hidr�lisis de pro-caspasa-3 e inducir la forma activa del enzima (Figura 24B). Si
previamente tratamos a las c�lulas con el inhibidor especifico de la caspasa-3, z-DEVD-
fmk (100 μM), el porcentaje de c�lulas apopt�ticas y la fragmentaci�n de ADN inducida
por el 3-metil �ter de betuletol disminuye de forma significativa.
5.5. Los derivados del betuletol inducen liberación de citocromo c mitocondrial.
Para determinar si la apoptosis inducida por el 3-metil �ter de betuletol y el diacetato del
3-metil �ter de betuletol en las c�lulas HL-60 implica la translocaci�n del citocromo c al
citosol, se realizaron experimentos con concentraciones crecientes de los compuestos y
los extractos citos�licos fueron analizados mediante western blot. Como se muestra en
la Figura 25A, concentraciones bajas de ambos compuestos (3 �M) promueven la
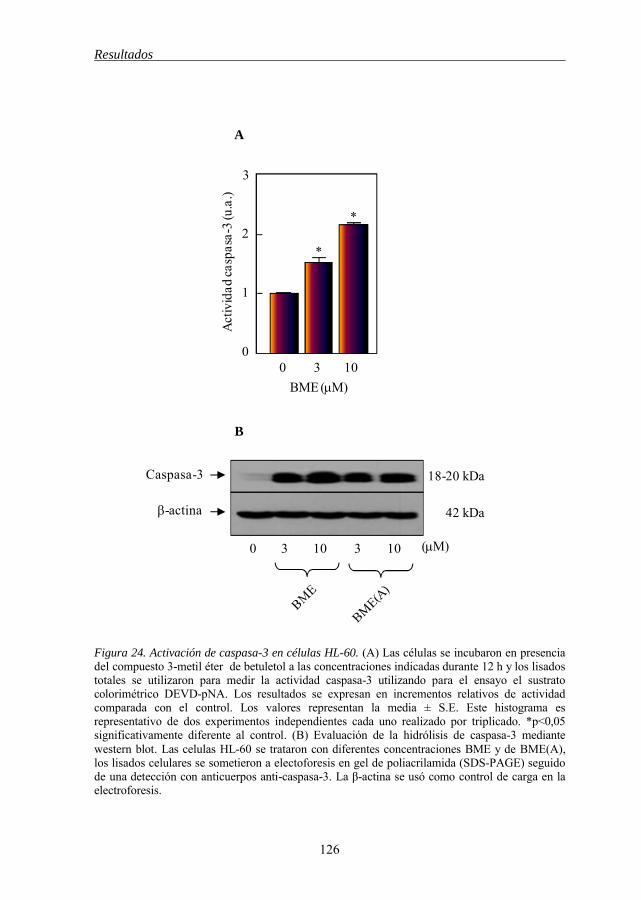
Resultados
126
Figura 24. Activación de caspasa-3 en células HL-60. (A) Las c�lulas se incubaron en presencia del compuesto 3-metil �ter de betuletol a las concentraciones indicadas durante 12 h y los lisados totales se utilizaron para medir la actividad caspasa-3 utilizando para el ensayo el sustrato colorim�trico DEVD-pNA. Los resultados se expresan en incrementos relativos de actividad comparada con el control. Los valores representan la media � S.E. Este histograma es representativo de dos experimentos independientes cada uno realizado por triplicado. *p<0,05 significativamente diferente al control. (B) Evaluaci�n de la hidr�lisis de caspasa-3 mediante western blot. Las celulas HL-60 se trataron con diferentes concentraciones BME y de BME(A), los lisados celulares se sometieron a electoforesis en gel de poliacrilamida (SDS-PAGE) seguido de una detecci�n con anticuerpos anti-caspasa-3. La β-actina se us� como control de carga en la electroforesis.
18-20 kDa
B
-actina
(M)3 10 3 100
Caspasa-3
A
Act
ivid
adca
spas
a-3
(u.a
.)
3 100BME (M)
0
1
2
3
*
*
42 kDa
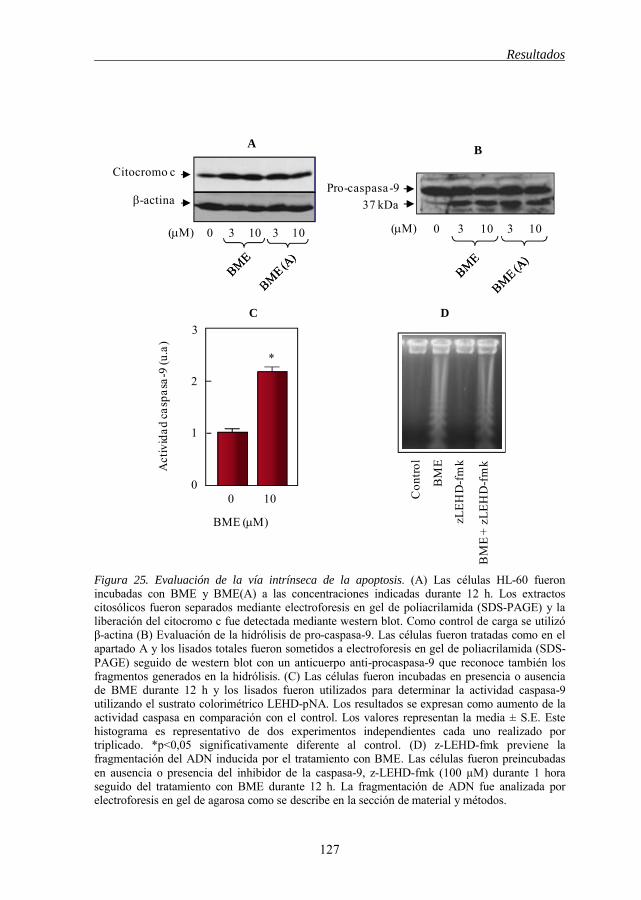
Resultados
127
Figura 25. Evaluación de la vía intrínseca de la apoptosis. (A) Las c�lulas HL-60 fueron incubadas con BME y BME(A) a las concentraciones indicadas durante 12 h. Los extractos citos�licos fueron separados mediante electroforesis en gel de poliacrilamida (SDS-PAGE) y la liberaci�n del citocromo c fue detectada mediante western blot. Como control de carga se utiliz� β-actina (B) Evaluaci�n de la hidr�lisis de pro-caspasa-9. Las c�lulas fueron tratadas como en el apartado A y los lisados totales fueron sometidos a electroforesis en gel de poliacrilamida (SDS-PAGE) seguido de western blot con un anticuerpo anti-procaspasa-9 que reconoce tambi�n los fragmentos generados en la hidr�lisis. (C) Las c�lulas fueron incubadas en presencia o ausencia de BME durante 12 h y los lisados fueron utilizados para determinar la actividad caspasa-9 utilizando el sustrato colorim�trico LEHD-pNA. Los resultados se expresan como aumento de la actividad caspasa en comparaci�n con el control. Los valores representan la media � S.E. Este histograma es representativo de dos experimentos independientes cada uno realizado por triplicado. *p<0,05 significativamente diferente al control. (D) z-LEHD-fmk previene la fragmentaci�n del ADN inducida por el tratamiento con BME. Las c�lulas fueron preincubadas en ausencia o presencia del inhibidor de la caspasa-9, z-LEHD-fmk (100 �M) durante 1 hora seguido del tratamiento con BME durante 12 h. La fragmentaci�n de ADN fue analizada por electroforesis en gel de agarosa como se describe en la secci�n de material y m�todos.
Con
trol
BM
E
zLEH
D-f
mk
BM
E+
zLEH
D-f
mk
D
Act
ivid
adca
spas
a-9
(u.a
)
BME (M)
0 100
1
2
3
*
C
A
Citocromo c
-actina
(M) 3 10 3 100
B
37 kDaPro-caspasa-9
(M) 3 10 3 100

Resultados
128
liberaci�n de citocromo c en la misma proporci�n. No se observaron diferencias con el
uso de concentraciones superiores de los dos compuestos (10 �M).
El efecto del 3-metil �ter de betuletol y el diacetato del 3-metil �ter de betuletol sobre la
caspasa-9 tambi�n se analiz�. Los resultados demuestran que ambos derivados del
betuletol inducen hidr�lisis de la procaspasa-9, generando un fragmento de 37 kDa
(Figura 25B). Para determinar si la hidr�lisis est� asociada con un aumento de la
actividad caspasa-9, los lisados de c�lulas tratadas se incubaron en presencia del
sustrato espec�fico para caspasa-9, LEHD-pNA. Los resultados indican que el
tratamiento con BME (10 μM durante 12 h) induce un aumento significativo de la
actividad de la caspasa-9 (Figura 25C).
Con el fin de determinar la contribuci�n de la caspasa-9 en el proceso apopt�tico
inducido por los derivados del betuletol, examinamos los efectos del inhibidor
espec�fico de caspasa-9, z-LEHD-fmk. Como se muestra en la Figura 25D, el inhibidor
es incapaz de impedir la la fragmentaci�n del ADN inducida por el 3-metil �ter de
betuletol. Este resultado sugiere que la caspasa-9 no parece desempe�ar un papel
relevante en el mecanismo de citot�xicidad de los derivados del betuletol.
5.6. BME induce cambios en el potencial de transmenbrana mitocondrial (m).
Nuestros resultados demuestran que BME induce la liberaci�n de citocromo c despu�s
de 12 h de tratamiento en c�lulas HL-60 (Figura 25A). Para determinar si este efecto
est� asociado con la p�rdida del potencial de membrana de la mitocondria, se realizaron
experimentos a diferentes tiempos. Los resultados muestran una p�rdida significativa
del m entre las 9 y las l2 h de tratamiento (Figura 26), esto sugiere que la disipaci�n
del potencial de membrana podr�a estar implicada en la inducci�n de la muerte celular
por BME.
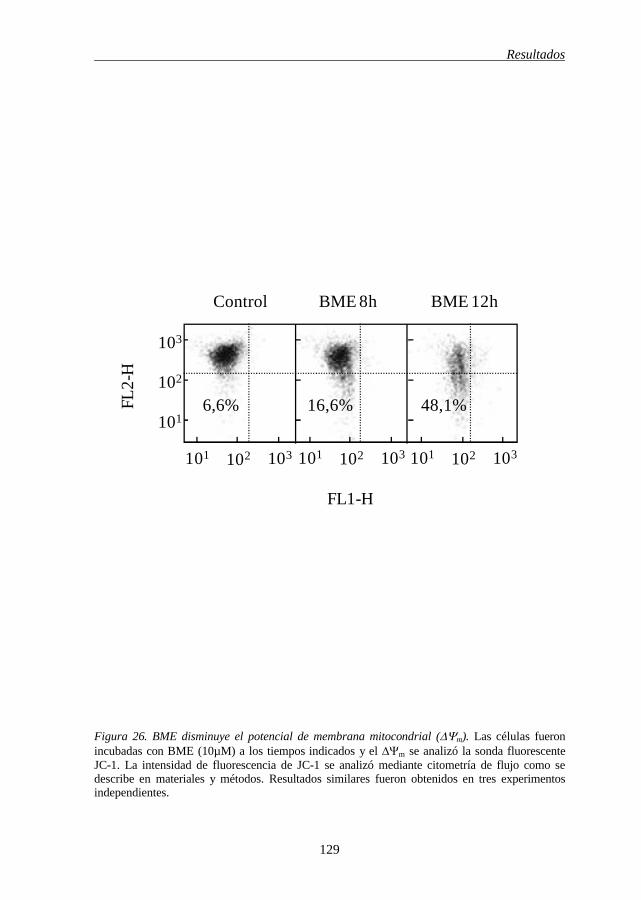
Resultados
129
Figura 26. BME disminuye el potencial de membrana mitocondrial (m). Las células fueron incubadas con BME (10µM) a los tiempos indicados y el m se analizó la sonda fluorescenteJC-1. La intensidad de fluorescencia de JC-1 se analizó mediante citometría de flujo como se describe en materiales y métodos. Resultados similares fueron obtenidos en tres experimentos independientes.
FL1-H
FL2-
H
101
102
103
Control BME 8h BME 12h
6,6%
101 103102 101 103102 101 103102
48,1%16,6%

Resultados
130
5.7. La caspasa-8 es activada por los derivados de betuletol.
Dado que la v�a intr�nseca no parece contribuir de forma significativa a la apoptosis
inducida por los derivados del betuletol se analiz� la contribuci�n de la v�a extr�nseca.
Las c�lulas HL-60 se trataron con concentraciones crecientes de 3-metil �ter de
betuletol (BME) y los lisados se incubaron con IETD-pNA, sustrato colorim�trico
espec�fico para la caspasa-8. Como se muestra en la Figura 27A, este compuesto induce
la activaci�n de la caspasa-8 de una forma claramente dependiente de la concentraci�n
empleada. La actividad enzim�tica aument� 2,4 y 3,5 veces respecto al control, con 3
�M y 10 μM de BME, respectivamente. Adem�s, los lisados celulares (Figura 27B)
fueron sometidos a an�lisis por western blot mediante el uso de anticuerpos anti-
caspasa-8 que reconocen tanto a la forma inactiva (pro-caspasa) como a la forma activa
(fragmentos de 32 kDa y 42-44 kDa, obtenidos tras la hidr�lisis de la pro-caspasa). Los
resultados demuestran que 3 �M de BME induce hidr�lisis significativa de la pro-
caspasa-8. A la concentraci�n de 10 μM la hidr�lisis de pro-caspasa-8 es m�s intensa
mientras que la concentraci�n m�s baja utilizada (1 μM) no tuvo ning�n impacto.
La activaci�n por prote�lisis de la caspasa iniciadora caspasa-8 suele ocurrir en
respuesta a la activaci�n de receptores de muerte que se encuentran en la membrana
celular, como el CD95 [228,229]. Para determinar el grado de implicaci�n de la
caspasa-8 en la inducci�n de apoptosis mediada por el derivado del betuletol BME, se
examinaron los efectos del inhibidor espec�fico de caspasa-8, z-IETD-fmk. Mediante el
an�lisis por citometr�a de flujo se observ� un aumento del porcentaje de c�lulas
apopt�ticas que pas� de un 6% (control) al 24% y al 30% con 10 �M y 30 �M de BME,
respectivamente (Figura 27C), que fue completamente suprimido en c�lulas pre-tratadas
con z-IETD-fmk. El inhibidor selectivo de caspasa-8 no afect� al nivel basal de
apoptosis. Su efecto tambi�n se analiz� mediante la visualizaci�n del ADN, y como
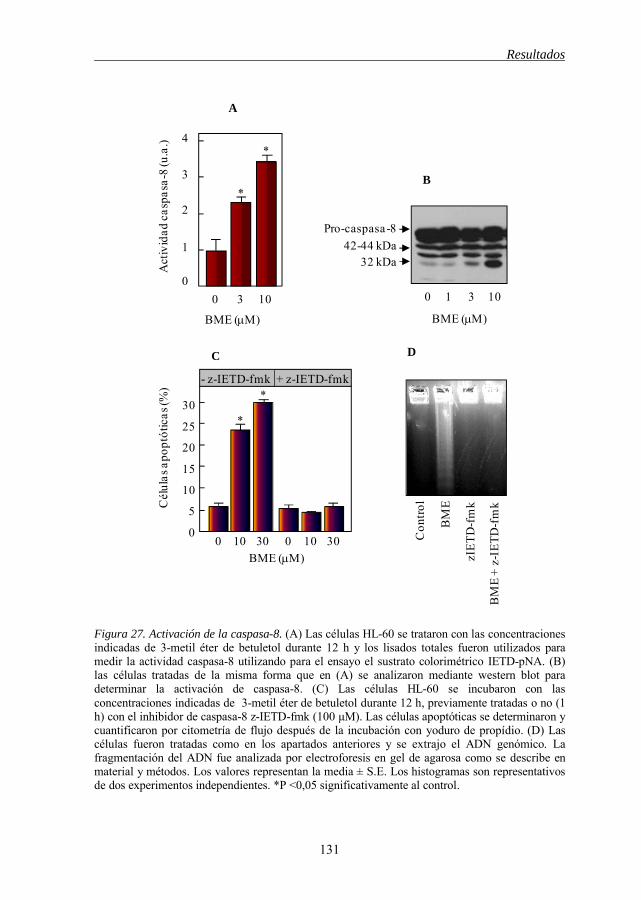
Resultados
131
Figura 27. Activación de la caspasa-8. (A) Las c�lulas HL-60 se trataron con las concentraciones indicadas de 3-metil �ter de betuletol durante 12 h y los lisados totales fueron utilizados para medir la actividad caspasa-8 utilizando para el ensayo el sustrato colorim�trico IETD-pNA. (B) las c�lulas tratadas de la misma forma que en (A) se analizaron mediante western blot para determinar la activaci�n de caspasa-8. (C) Las c�lulas HL-60 se incubaron con las concentraciones indicadas de 3-metil �ter de betuletol durante 12 h, previamente tratadas o no (1 h) con el inhibidor de caspasa-8 z-IETD-fmk (100 μM). Las c�lulas apopt�ticas se determinaron y cuantificaron por citometr�a de flujo despu�s de la incubaci�n con yoduro de prop�dio. (D) Las c�lulas fueron tratadas como en los apartados anteriores y se extrajo el ADN gen�mico. La fragmentaci�n del ADN fue analizada por electroforesis en gel de agarosa como se describe en material y m�todos. Los valores representan la media � S.E. Los histogramas son representativosde dos experimentos independientes. *P <0,05 significativamente al control.
D
Con
trol
zIET
D-f
mk
BM
E
BM
E+
z-IE
TD-f
mk
C
0
10
20
30
0 10 30 0 10 30BME (M)
- z-IETD-fmk + z-IETD-fmk
C�l
ulas
apop
t�tic
as(%
)
5
15
25
*
*
BME (M)
Act
ivid
adca
spas
a-8
(u.a
.)
A
0
1
2
3
4
0 10
*
3
*B
Pro-caspasa-842-44 kDa
32 kDa
3 100 1
BME (M)

Resultados
132
podemos observar en la Figura 27D inhibi� la fragmentaci�n de ADN en c�lulas
tratadas con 10 μM de 3-metil �ter de betuletol.
Globalmente, estos resultados demuestran que la caspasa-8 desempe�a un papel muy
importante en la apoptosis inducida por el compuesto BME.
5.8. La inducción de apoptosis por BME implica la hidrólisis de Bid a través de un
mecanismo independiente de Fas.
Dado que BME induce apoptosis en c�lulas HL-60 mediante un mecanismo que activa
la caspasa-8 y Bid es sustrato de �sta, se sometieron los lisados celulares a western blot
para analizar la posible hidr�lisis de Bid mediante el uso de un anticuerpo anti-Bid. Los
resultados demuestran que Bid, en c�lulas tratadas con BME, se hidroliza en fragmentos
de 13/15 kDa, los cuales se translocan a la mitocondria (Figura 28A).
La caspasa-8 (caspasa iniciadora) es esencial para la apoptosis inducida en respuesta a
la activaci�n del receptor de muerte Fas. Para determinar si la muerte celular inducida
por el tratamiento con BME es dependiente del receptor de Fas, se utiliz� un anticuerpo
antagonista anti-Fas (clon ZB4). Con este fin, las c�lulas fueron preincubadas durante 1
h con el anticuerpo ZB4 en ausencia y en presencia de BME. Los resultados indican que
ZB4 es incapaz de proteger a las c�lulas de la muerte inducida por BME. Esto sugiere
que la inducci�n de apoptosis por BME ocurre de forma independiente del receptor de
muerte Fas (Figura 28B).
5.9. El inhibidor de la caspasa-8 bloquea la activación de la caspasa-3 en la
apoptosis inducida por BME.
Para profundizar m�s en el papel que desempe�a la caspasa-8 en la inducci�n de
apoptosis por BME se realizaron experimentos adicionales. Para ello las c�lulas HL-60
se pre-trataron con el inhibidor de caspasa 8, z-IETD-fmk. Los resultados muestran que
la hidr�lisis de pro-caspasa-3 y la hidr�lisis de poli (ADP-ribosa) polimerasa (t�pico
sustrato de caspasa-3) est� bloqueada por el tratamiento con el inhibidor de caspasa-8
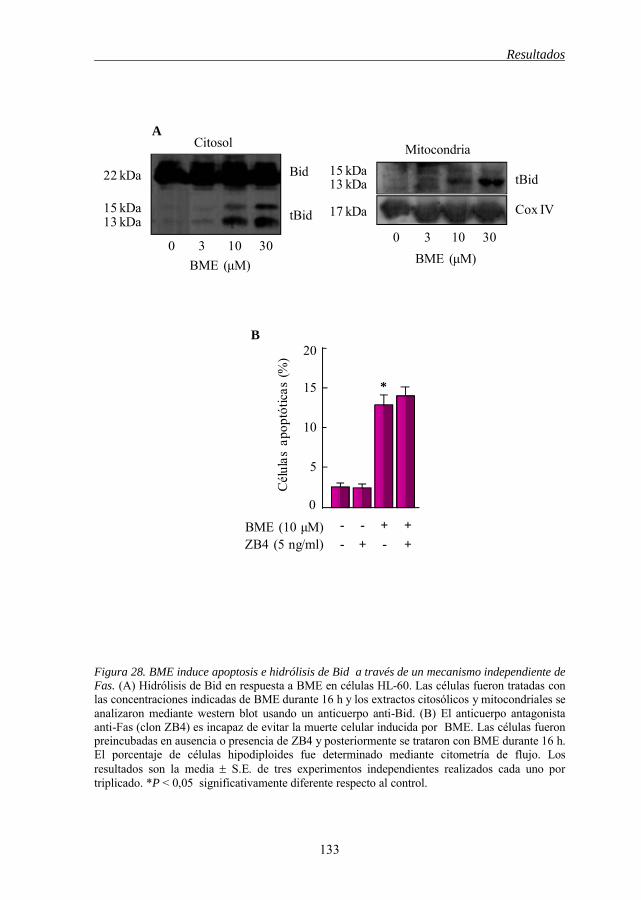
Resultados
133
Figura 28. BME induce apoptosis e hidrólisis de Bid a través de un mecanismo independiente de Fas. (A) Hidr�lisis de Bid en respuesta a BME en c�lulas HL-60. Las c�lulas fueron tratadas con las concentraciones indicadas de BME durante 16 h y los extractos citos�licos y mitocondriales se analizaron mediante western blot usando un anticuerpo anti-Bid. (B) El anticuerpo antagonista anti-Fas (clon ZB4) es incapaz de evitar la muerte celular inducida por BME. Las c�lulas fueron preincubadas en ausencia o presencia de ZB4 y posteriormente se trataron con BME durante 16 h. El porcentaje de c�lulas hipodiploides fue determinado mediante citometr�a de flujo. Los resultados son la media S.E. de tres experimentos independientes realizados cada uno por triplicado. *P < 0,05 significativamente diferente respecto al control.
AMitocondria
tBid
Cox IV
15 kDa13 kDa
17 kDa
0 3 10BME (μM)
30
BME (μM)
Citosol
Bid
tBid 15 kDa13 kDa
22 kDa
0 3 10 30
B
BME (10 μM)ZB4 (5 ng/ml)
--
++-
- ++
C�l
ulas
apop
t�tic
as(%
) 20
10
0
5
15 *

Resultados
134
(Figura 29A). Sin embargo, el tratamiento con el inhibidor espec�fico de la caspasa-9 (z-
LEHD-fmk) no muestra el mismo efecto. Bajas concentraciones (10 μM) de cada
inhibidor espec�fico de caspasas (z-LEHD-fmk para caspasa-9 y z-IETD–fmk para
caspasa- 8) fueron ineficaces en reducir el porcentaje de apoptosis y el bloqueo de la
fragmentaci�n del ADN inducida por BME (10�M).
Para definir los tiempos de activaci�n de las diferentes caspasas, determinamos las
actividades enzim�ticas de la caspasa-3, caspasa-8 y caspasa-9 en extractos celulares de
HL-60 tratadas con BME recolectadas a diferentes tiempos.
Como se muestra en la Figura 29B la inducci�n de la actividad de la caspasa-8 (3 veces)
y la caspasa-3 (3,8 veces) son detectables a las 8 h de tratamiento. En cambio, el
aumento de la actividad caspasa-9 (3 veces) no se detecta hasta las 12 h.
De estos resultados se deduce que el BME induce la activaci�n de la caspasa-8 y, por
tanto, directamente la activaci�n de caspasa-3 (hidr�lisis de caspasa-3).
5.10. BME activa la vía MAPKs.
El balance entre las v�as ERK activadas por factores de crecimiento y las v�as activadas
por estr�s JNK/SAPK y p38MAPK determina la supervivencia o la muerte celular. Para
explorar los mecanismos por los que BME es tan efectivo induciendo la muerte celular,
se determin� si BME induce la fosforilaci�n y la activaci�n de MAPKs en la l�nea
celular HL-60. Los resultados muestran que el tratamiento de las c�lulas con BME (10
M) induce fosforilaci�n de ERK1/2, JNK/SAPK y p38MAPK. La activaci�n de ERK 1/2
y p38 MAPK se detecta a los 30 minutos y 1 h respectivamente, despu�s de la adici�n de
BME y se mantienen elevadas al menos hasta 4 h (Figura 30A). Sin embargo la
activaci�n de JNK/SAPK no se observa hasta las 4 h. Los resultados sugieren que BME
induce la fosforilaci�n de las v�as MAPK siguiendo diferentes cin�ticas.
Se evalu� si la fosforilaci�n de las MAPKs jugaba un papel importante en la apoptosis
inducida por BME mediante el uso de inhibidores espec�ficos. El pre-tratamiento de las
c�lulas HL-60 con SB 203580 (2 M), un inhibidor especifico de p38MAPK, no afect� a
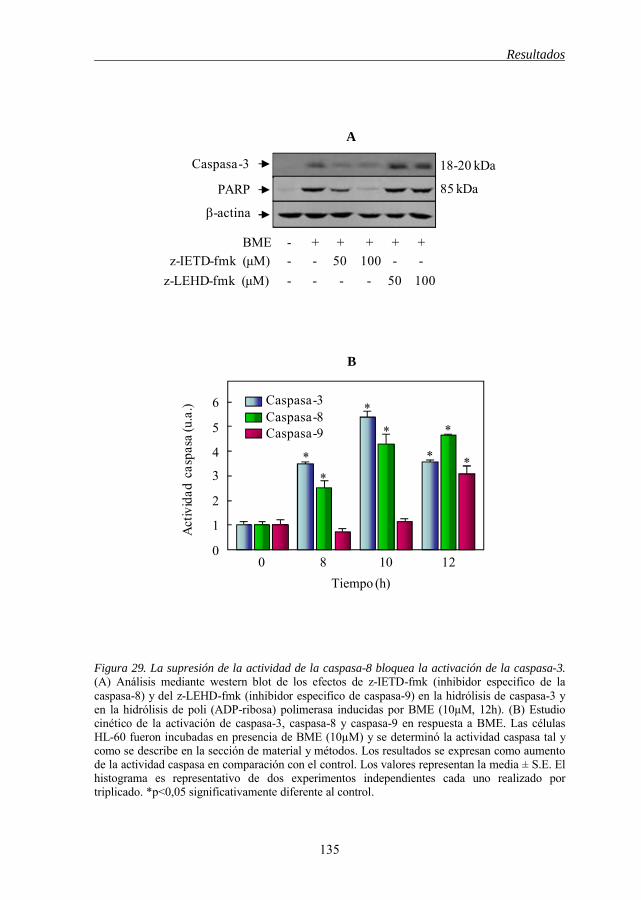
Resultados
135
Figura 29. La supresión de la actividad de la caspasa-8 bloquea la activación de la caspasa-3.(A) An�lisis mediante western blot de los efectos de z-IETD-fmk (inhibidor especifico de la caspasa-8) y del z-LEHD-fmk (inhibidor especifico de caspasa-9) en la hidr�lisis de caspasa-3 y en la hidr�lisis de poli (ADP-ribosa) polimerasa inducidas por BME (10�M, 12h). (B) Estudio cin�tico de la activaci�n de caspasa-3, caspasa-8 y caspasa-9 en respuesta a BME. Las c�lulas HL-60 fueron incubadas en presencia de BME (10�M) y se determin� la actividad caspasa tal y como se describe en la secci�n de material y m�todos. Los resultados se expresan como aumento de la actividad caspasa en comparaci�n con el control. Los valores representan la media � S.E. El histograma es representativo de dos experimentos independientes cada uno realizado por triplicado. *p<0,05 significativamente diferente al control.
A
BME - + + +z-IETD-fmk (μM)
z-LEHD-fmk (μM)
+ +- - 50 100 - -- - - - 50 100
PARP
-actina
Caspasa-3
85 kDa
18-20 kDa
B
*
* *
*
*
*
*
0
2
4
6
0 8 10 12
Act
ivid
adca
spas
a(u
.a.)
Tiempo (h)
Caspasa-8Caspasa-9
Caspasa-3
1
3
5
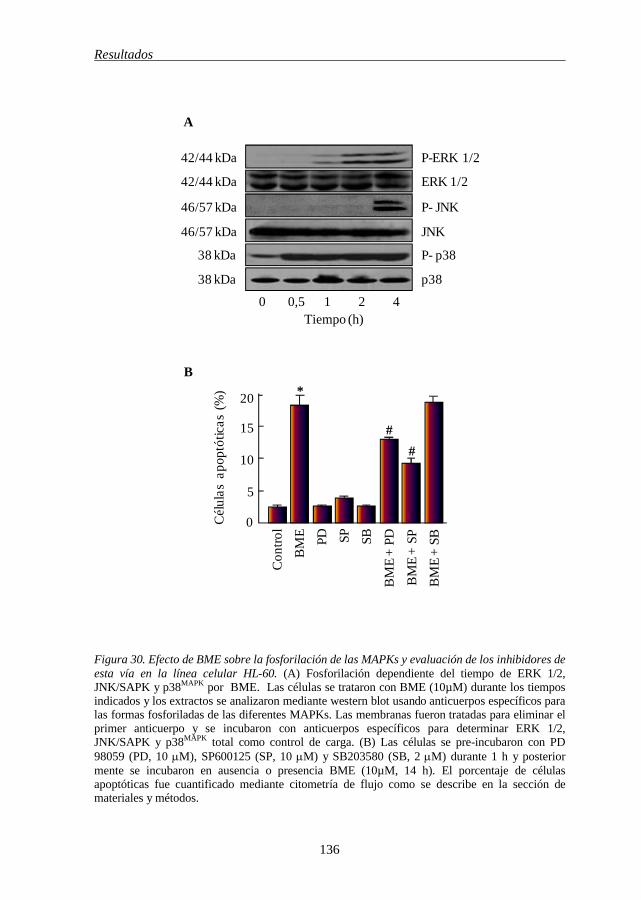
Resultados
136
Figura 30. Efecto de BME sobre la fosforilación de las MAPKs y evaluación de los inhibidores de esta vía en la línea celular HL-60. (A) Fosforilación dependiente del tiempo de ERK 1/2, JNK/SAPK y p38MAPK por BME. Las células se trataron con BME (10µM) durante los tiempos indicados y los extractos se analizaron mediante western blot usando anticuerpos específicos para las formas fosforiladas de las diferentes MAPKs. Las membranas fueron tratadas para eliminar el primer anticuerpo y se incubaron con anticuerpos específicos para determinar ERK 1/2, JNK/SAPK y p38MAPK total como control de carga. (B) Las células se pre-incubaron con PD 98059 (PD, 10 M), SP600125 (SP, 10 M) y SB203580 (SB, 2 M) durante 1 h y posteriormente se incubaron en ausencia o presencia BME (10µM, 14 h). El porcentaje de células apoptóticas fue cuantificado mediante citometría de flujo como se describe en la sección de materiales y métodos.
A
Tiempo (h)
P-ERK 1/242/44 kDa
ERK 1/242/44 kDa
JNK46/57 kDa
P- JNK46/57 kDa
38 kDa P- p38
0 0,5 1 2 4
p3838 kDa
B
Cél
ulas
apop
tótic
as(%
) 20
10
0
5
15
Con
trol
BM
E SPPD SB
BM
E+
PD
BM
E+
SP
BM
E+
SB
*
##

Resultados
137
la apoptosis mediada por BME (Figura 30B). Estos datos sugieren que la activación de
p38MAPK no está involucrada en la apoptosis inducida por BME. Sin embargo, el pre-
tratamiento de las células con SP600125, un inhibidor de JNK/SAPK, indujo una
disminución en la apoptosis inducida por BME de 18 2 % a 8 2 % (Figura 30B).
Se evaluó el impacto de ERK 1/2 en la vía de transducción de señal que lleva a la
muerte celular por BME. Para este fin, se usó PD98059, un inhibidor específico de la
ERK 1/2 con el fin de bloquear su activación. Los resultados indican que PD98059
atenúa de forma significativa la apoptosis mediada por BME (reducción de 18 2 % al
12 0.5 %).
El conjunto de los resultados por tanto indican que la muerte celular inducida por BME
está asociado con la activación de ERK 1/2, p38MAPK y JNK/SAPK pero solo se
encuentran involucrados en la activación de la apoptosis JNK/SAPK y ERK 1/2.
5.11. Las especies reactivas de oxígeno (EROs) no son necesarias para la muerte
celular inducida por BME.
Dado que la producción de especies reactivas de oxígeno (EROs) puede dar lugar a la
activación de MAPKs e inducir muerte celular [24-27], se estudió mediante el uso de la
sonda fluorescente sensible a la oxidación diacetato 2´,7´-dicloro-dihidro-fluoresceína
(H2-DCF-DA) si las EROs estaban implicadas en la apoptosis inducida por BME. Los
resultados muestran un incremento de la fluorescencia de 2´,7´-diclorofluoresceína
después de 12 h de tratamiento (Figura 31A y 31B) con 10µM de BME.
Para determinar si la generación de EROs está implicada en la muerte celular inducida
por BME se estudió el efecto del antioxidante trolox (2 mM). En la Figura 31C, como
se puede apreciar, el trolox inhibe casi completamente la generación de EROs, pero es
incapaz de bloquear la muerte celular Figura 31D. Estos resultados sugieren que la
apoptosis inducida por BME es independiente de la producción de EROs.
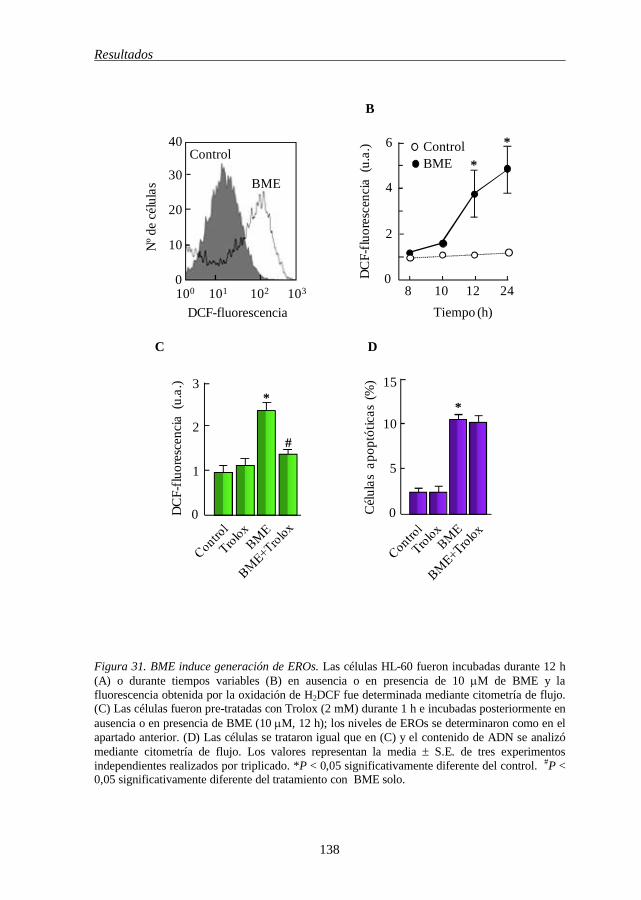
Resultados
138
Figura 31. BME induce generación de EROs. Las células HL-60 fueron incubadas durante 12 h(A) o durante tiempos variables (B) en ausencia o en presencia de 10 M de BME y la fluorescencia obtenida por la oxidación de H2DCF fue determinada mediante citometría de flujo. (C) Las células fueron pre-tratadas con Trolox (2 mM) durante 1 h e incubadas posteriormente en ausencia o en presencia de BME (10 M, 12 h); los niveles de EROs se determinaron como en el apartado anterior. (D) Las células se trataron igual que en (C) y el contenido de ADN se analizómediante citometría de flujo. Los valores representan la media S.E. de tres experimentos independientes realizados por triplicado. *P < 0,05 significativamente diferente del control. #P < 0,05 significativamente diferente del tratamiento con BME solo.
Control
BME
Nºd
ecé
lula
s
DCF-fluorescencia
40
0101 103102100
10
20
30
D
Cél
ulas
apop
tótic
as(%
) 15
0
10
5
*
B
DC
F-flu
ores
cenc
ia(u
.a.)
4
0
6
2
8 10 12Tiempo (h)
24
ControlBME *
*
C
2
0
3
1
DC
F-flu
ores
cenc
ia(u
.a.)
#
*

Resultados
139
5.12. BME induce la generación de ceramida y la activación de SMasa ácida
Se evaluó si este compuesto induce cambios en los niveles endógenos de ceramida en
células HL-60. El ensayo de la diacilglicerol quinasa fue utilizado para determinar el
contenido de ceramida en los extractos lipídicos. Con este fin, las células se trataron con
BME a diferentes tiempos, y los lípidos celulares se extrajeron y la ceramida fue
cuantificada. Cualquier aumento de ceramida en los extractos lipídicos refleja la
liberación celular de ceramida. Como se muestra en la Figura 32A, se observó un
incremento significativo de ceramida intracelular a tiempos cortos (2 h) después del
tratamiento con 10µM BME y los niveles intracelulares de ceramida se mantuvieron al
menos hasta las 4 h de tratamiento.
La generación de ceramida por el tratamiento con BME nos llevó a investigar la posible
participación de las SMasas. Tanto la SMasa ácida como la neutra están implicadas en
la generación de ceramida en respuesta a estrés ambiental. Las células HL-60 se trataron
con BME (10µM) a diferentes tiempos y los lisados (libres de núcleos) se analizaron a
pH 7,4 o pH 5,0 para determinar la actividad de la SMasa neutra y la actividad de la
SMasa ácida, respectivamente. Como se muestra en Figura 32B, los resultados indican
que BME induce la activación de la SMasa ácida obteniendo la máxima actividad a las
2 h de tratamiento. No se apreciaron cambios en la actividad total de la esfingomielinasa
neutra a los tiempos analizados.
Puesto que la ceramida se considera una molécula reguladora de la apoptosis en células
tumorales, se utilizó fumonisina B1 (100 M) para inhibir la dihidro-ceramida sintasa,
una enzima implicada no solo en la biosíntesis de novo de ceramida sino también en la
vía de salvamento [28]. La fumonisina B1 no bloquea la apoptosis inducida por BME en
HL-60 (Figura 32C). Ya que SMasa ácida se puede activar por sn-1,2-diacilglicerol
(DAG) se evaluó el impacto de los inhibidores de la generación de DAG. D609 es un
inhibidor específico de la PC-PLC. Este inhibidor no es capaz de bloquear la apoptosis
inducida por BME, indicando que la PC-PLC no está implicada en este evento (Figura
32D).
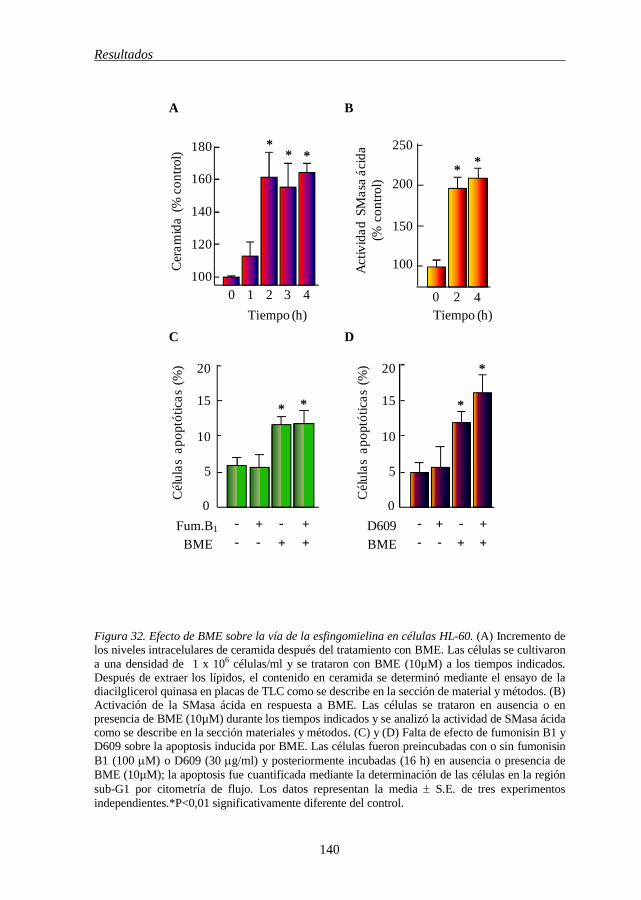
Resultados
140
Figura 32. Efecto de BME sobre la vía de la esfingomielina en células HL-60. (A) Incremento de los niveles intracelulares de ceramida después del tratamiento con BME. Las células se cultivarona una densidad de 1 x 106 células/ml y se trataron con BME (10µM) a los tiempos indicados. Después de extraer los lípidos, el contenido en ceramida se determinó mediante el ensayo de la diacilglicerol quinasa en placas de TLC como se describe en la sección de material y métodos. (B) Activación de la SMasa ácida en respuesta a BME. Las células se trataron en ausencia o en presencia de BME (10µM) durante los tiempos indicados y se analizó la actividad de SMasa ácidacomo se describe en la sección materiales y métodos. (C) y (D) Falta de efecto de fumonisin B1 y D609 sobre la apoptosis inducida por BME. Las células fueron preincubadas con o sin fumonisin B1 (100 M) o D609 (30 g/ml) y posteriormente incubadas (16 h) en ausencia o presencia de BME (10µM); la apoptosis fue cuantificada mediante la determinación de las células en la región sub-G1 por citometría de flujo. Los datos representan la media S.E. de tres experimentos independientes.*P<0,01 significativamente diferente del control.
A
0 1 2 3 4Tiempo (h)
100
120
140
160
180
Cer
amid
a(%
cont
rol)
** *
C
Cél
ulas
apop
tótic
as(%
) 20
10
0
BMEFum.B1 -
-++
-- ++
15
5
* *
D
Cél
ulas
apop
tótic
as(%
) 20
10
0
BMED609 -
-++
-- ++
15
5
*
*
B
Tiempo (h)0 2 4
100
200
250
150
Act
ivid
adSM
asa
ácid
a(%
cont
rol)
* *

Resultados
141
Los resultados sugieren que esta enzima no está implicada en la apoptosis inducida por
BME en células HL-60.
Los resultados nos permiten concluir que la generación de ceramida en células HL-60
en respuesta a BME es un evento que se produce antes de la activación de caspasas y
que la generación de ceramidas está mediada principalmente por la activación de la
esfingomielinasa ácida.
5.13. BME inhibe la proliferación celular de células de leucemia mieloide humana
mediante la inducción de la parada del ciclo celular en fase G2-M.
Para estudiar el efecto de BME sobre el ciclo celular, las células HL-60 se incubaron a
diferentes tiempos en presencia 3 M del compuesto y se analizaron mediante
citometría de flujo. La Figura 33A muestra el típico histograma de ADN y los datos
obtenidos relativos se refieren a la distribución del ciclo celular. Las células tratadas
con BME (10 µM), después de 4 h de incubación, comienzan a acumularse en la fase
G2-M del ciclo celular, y se alcanza un máximo después de 8-12 h de incubación. De
forma paralela se observa un descenso en las células en la fase G0/G1 del ciclo celular.
A las 12 h de tratamiento se observa un porcentaje significativo de células hipodiploides
(sub-G1). Los resultados indican que BME induce parada del ciclo celular en la fase
G2-M.
5.14. Efectos de BME en la expresión y el estado de fosforilación de las proteínas
reguladoras de la fase G2-M.
Para caracterizar el mecanismo molecular de la detención del ciclo celular en G2-M
inducido por BME en células de leucemia se examinó la expresión de las proteínas
asociadas a esta fase del ciclo celular. Los resultados muestran un incremento
significativo en los niveles de ciclina B1 y fosfo-ciclina B1 (fosfo S126) a las 3 h de
tratamiento mientras que un incremento de p21Cip1 se observa después de 6 h de
tratamiento con BME (Figura 33B).
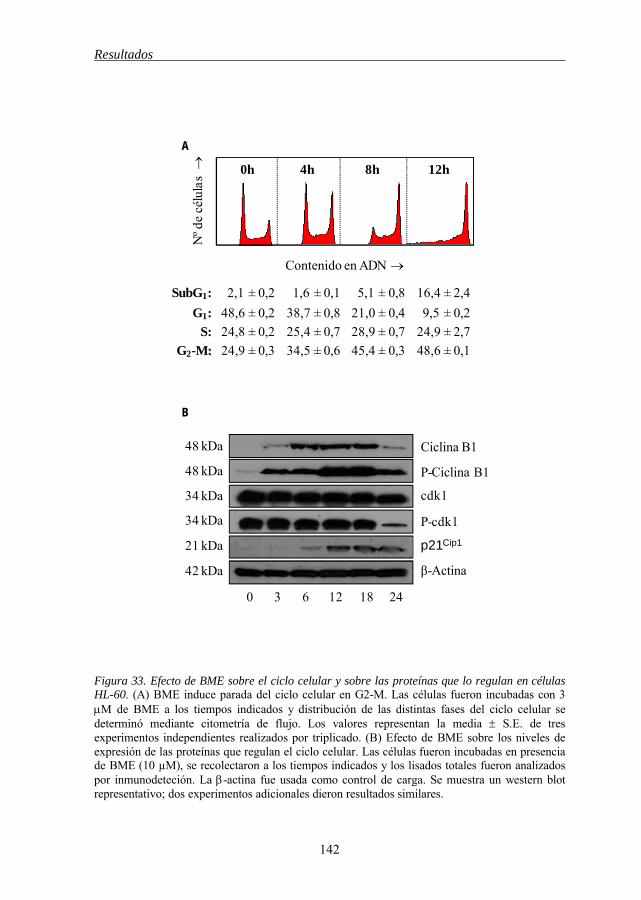
Resultados
142
Figura 33. Efecto de BME sobre el ciclo celular y sobre las proteínas que lo regulan en células HL-60. (A) BME induce parada del ciclo celular en G2-M. Las células fueron incubadas con 3 M de BME a los tiempos indicados y distribución de las distintas fases del ciclo celular sedeterminó mediante citometría de flujo. Los valores representan la media S.E. de tres experimentos independientes realizados por triplicado. (B) Efecto de BME sobre los niveles de expresión de las proteínas que regulan el ciclo celular. Las células fueron incubadas en presencia de BME (10 µM), se recolectaron a los tiempos indicados y los lisados totales fueron analizados por inmunodeteción. La -actina fue usada como control de carga. Se muestra un western blot representativo; dos experimentos adicionales dieron resultados similares.
A
0h 4h 8h 12h
2,1 ± 0,248,6 ± 0,224,8 ± 0,224,9 ± 0,3
1,6 ± 0,138,7 ± 0,825,4 ± 0,734,5 ± 0,6
5,1 ± 0,821,0 ± 0,428,9 ± 0,745,4 ± 0,3
16,4 ± 2,49,5 ± 0,2
24,9 ± 2,748,6 ± 0,1
SubG1:G1:
S:G2-M:
Nºd
ecé
lula
s
Contenido en ADN
B
Ciclina B148 kDa
P-Ciclina B148 kDa
cdk134 kDa
P-cdk134 kDa
p21Cip121 kDa
β-Actina
0 3 6 12 18 24
42 kDa

Resultados
143
La activación de cdk1 está controlada por múltiples pasos que incluyen la unión a la
ciclina y la fosforilación de la treonina 161. Sin embargo la etapa reguladora crítica en
la activación de cdk1 durante la progresión de la mitosis parece ser la des-fosforilación
de T14 y Y15. Esta doble des-fosforilación, la cual activa la cdk1, de los residuos está
catalizada por la fosfatasa Cdc25. Por ello se estudió si BME induce fosforilación de
cdk1 usando un anticuerpo que reconocía la forma fosforilda de cdk1 (pTpY 14/15).
Los resultados ponen de manifiesto que el tratamiento con BME induce la
desfosforilación y por lo tanto la activación de cdk1 (Figura 33B).
5.15. BME induce un incremento de la población celular en fase M: Comparación
del perfil del ciclo celular de los tratamientos con BME y nocodazol en HL-60.
Para determinar si el tratamiento celular con BME induce la parada del ciclo celular en
la fase M, las células fueron incubadas con Hoechst 33258 para el análisis cuantitativo
de la parada mitótica.
Se incluyó el nocodazol como control positivo, ya que bloquea el ciclo celular en la
transición de la metafase a la anafase e induce un incremento de la población celular en
la fase M del ciclo como control positivo. Los cambios morfológicos característicos de
las células mitóticas fueron analizados y cuantificados por microscopía de
fluorescencia. Los resultados indican que el porcentaje de células mitóticas incrementa
de 2,5 1% a 20 4% (8 veces el control) en células tratadas con BME después de 8 h
a una concentración de 10 M (Figura 34 A y 34 B).
5.16. BME desestabiliza la polimerización de los microtúbulos
Dado que BME causa acumulación de las células en la fase M del ciclo celular, se
estudió si el compuesto podría afectar a la polimerización de la tubulina en la línea
celular HL-60. Los resultados indican que BME inhibe la polimerización de la tubulina
de forma dependiente de la concentración. La colchicina y el paclitaxel fueron
utilizados como controles positivos de inhibición y promoción de la polimerización de
la tubulina, respectivamente (Figura 35A).
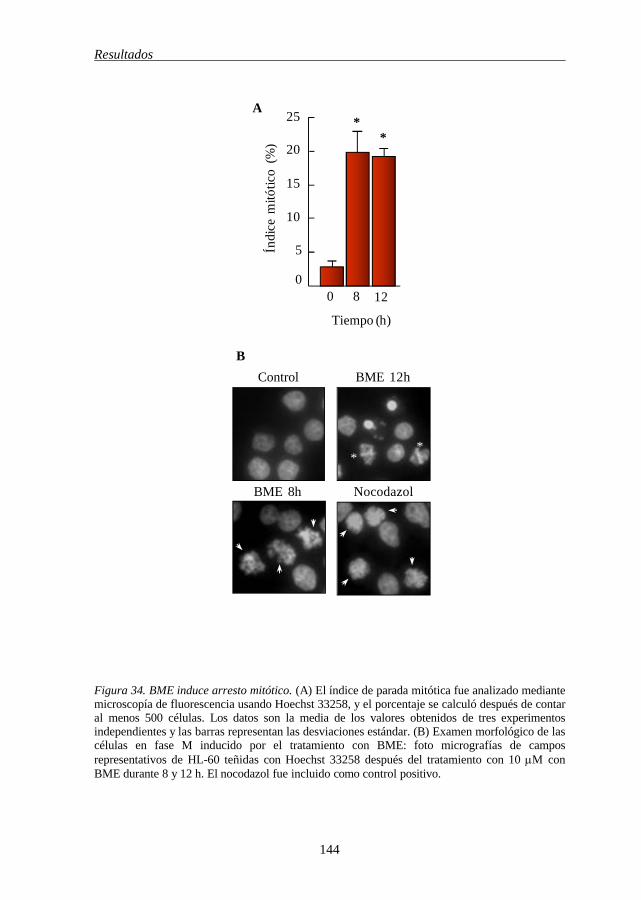
Resultados
144
Figura 34. BME induce arresto mitótico. (A) El índice de parada mitótica fue analizado mediante microscopía de fluorescencia usando Hoechst 33258, y el porcentaje se calculó después de contaral menos 500 células. Los datos son la media de los valores obtenidos de tres experimentos independientes y las barras representan las desviaciones estándar. (B) Examen morfológico de las células en fase M inducido por el tratamiento con BME: foto micrografías de campos representativos de HL-60 teñidas con Hoechst 33258 después del tratamiento con 10 M con BME durante 8 y 12 h. El nocodazol fue incluido como control positivo.
BControl
BME 8h
BME 12h
Nocodazol
**
A
Tiempo (h)
0 8 12
5
15
25
10
Índi
cem
itótic
o(%
) 20
0
**
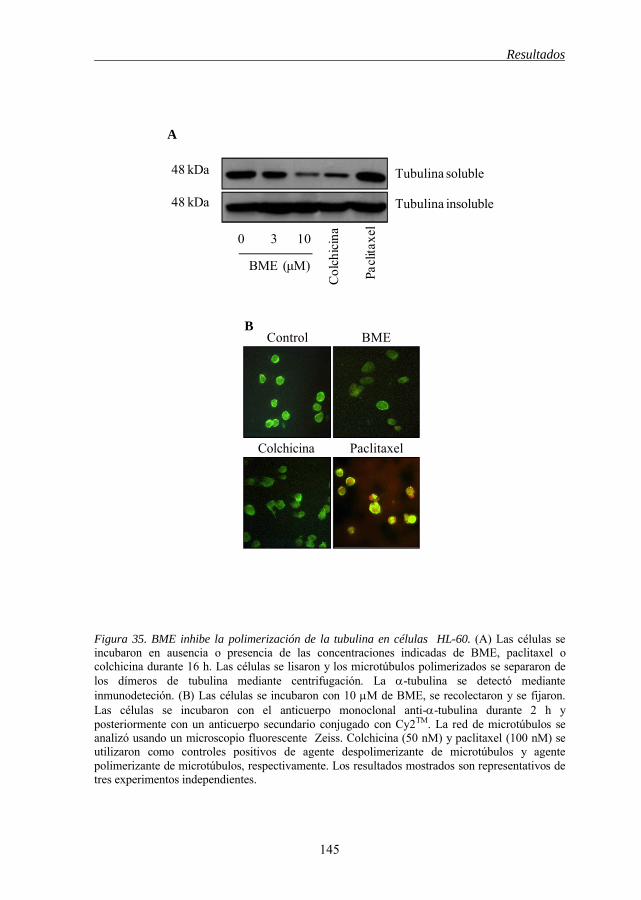
Resultados
145
Figura 35. BME inhibe la polimerización de la tubulina en células HL-60. (A) Las c�lulas se incubaron en ausencia o presencia de las concentraciones indicadas de BME, paclitaxel o colchicina durante 16 h. Las c�lulas se lisaron y los microt�bulos polimerizados se separaron de los d�meros de tubulina mediante centrifugaci�n. La -tubulina se detect� mediante inmunodeteci�n. (B) Las c�lulas se incubaron con 10 M de BME, se recolectaron y se fijaron. Las c�lulas se incubaron con el anticuerpo monoclonal anti--tubulina durante 2 h y posteriormente con un anticuerpo secundario conjugado con Cy2TM. La red de microt�bulos se analiz� usando un microscopio fluorescente Zeiss. Colchicina (50 nM) y paclitaxel (100 nM) se utilizaron como controles positivos de agente despolimerizante de microt�bulos y agente polimerizante de microt�bulos, respectivamente. Los resultados mostrados son representativos de tres experimentos independientes.
BControl BME
Colchicina Paclitaxel
A
Tubulina insoluble
Tubulina soluble
0 3 10
Col
chic
ina
BME (μM)
Pacl
itaxe
l
48 kDa
48 kDa

Resultados
146
Estudiamos el efecto de BME sobre la red de microtúbulos mediante técnicas de
inmunofluorescencia. Como se puede apreciar en la Figura 35B, el tratamiento con
BME causa una pérdida casi completa de los microtúbulos de la misma forma que la
colchicina. En cambio, el tratamiento con paclitaxel induce un incremento de la
densidad de los microtúbulos.
5.17. BME inhibe el crecimiento e induce apoptosis en líneas celulares de leucemia
humana independientemente de los niveles de expresión de Bcl-2 y Bcl-xL.
Estudiamos el efecto de BME sobre el crecimiento de células de leucemia mieloide
U937 y sobre células que expresan niveles elevados de proteínas anti-apoptóticas Bcl-2
(U937/Bcl-2) y Bcl-xL (HL-60/Bcl-xL). Los resultados indican que este compuesto tiene
una gran actividad citotóxica en todas las células de leucemia ensayadas. Los valores de
IC50 fueron de 2,0 0,7 M y 1,5 0,6 M en U937 y U937/Bcl-2, respectivamente,
mientras que en HL-60/neo y HL-60/Bcl-xL fueron 1,2 0,3 M y 2,6 0,6 M,
respectivamente. Estos resultados sugieren que el aumento de la expresión tanto de Bcl-
2 como de su homólogo Bcl-xL no confiere resistencia a las células frente a la
citotoxicidad inducida por BME.
También evaluamos el efecto de la inducción de apoptosis en células de leucemia que
expresan niveles elevados de las proteínas Bcl-2 y Bcl-xL. El análisis del número de
células hipodiploides muestra que el porcentaje de células apoptóticas incrementa no
solo en U937 y HL-60/neo, sino también en U937/Bcl-2 y HL-60/Bcl-xL de una forma
similar (Figura 36). Estos resultados confirman que BME también induce muerte celular
mediante apoptosis en células que expresan niveles elevados de las proteínas anti-
apoptóticas Bcl-2 y Bcl-xL.
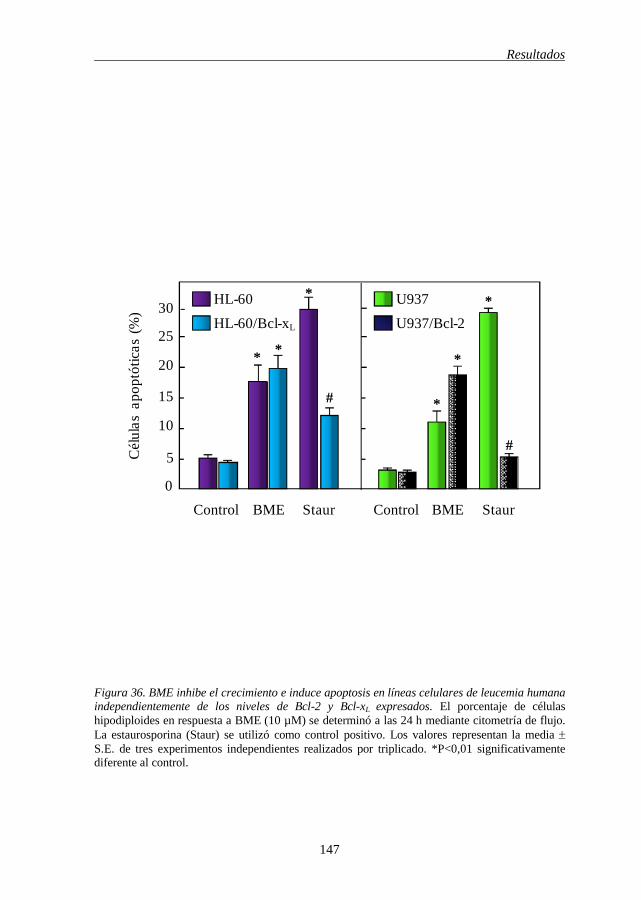
Resultados
147
Figura 36. BME inhibe el crecimiento e induce apoptosis en líneas celulares de leucemia humana independientemente de los niveles de Bcl-2 y Bcl-xL expresados. El porcentaje de células hipodiploides en respuesta a BME (10 µM) se determinó a las 24 h mediante citometría de flujo. La estaurosporina (Staur) se utilizó como control positivo. Los valores representan la media S.E. de tres experimentos independientes realizados por triplicado. *P<0,01 significativamente diferente al control.
*
Cél
ulas
apop
tótic
as(%
)
20
10
0
5
15
25
Control BME Staur
30
Control BME Staur
* *
# *
*
#
HL-60
HL-60/Bcl-xL
U937
U937/Bcl-2*

Resultados
148
La estaurosporina, un agente citot�xico, se utiliz� como control positivo. La
estaurosporina induce apoptosis en U937 y HL-60/neo, mientras que el aumento de la
expresi�n de las prote�nas anti-apopt�ticas Bcl-2 y Bcl-xL protegen a la c�lula de forma
significativa de la apoptosis inducida por este agente citot�xico [230,231].
5.18. Los derivados del betuletol no son citotóxicos para los linfocitos normales de
origen humano.
Partiendo de la premisa de que el mejor agente antitumoral ser�a aquel que tuviera
efecto en las c�lulas tumorales pero no en las c�lulas normales sanas estudiamos el
efecto de BME y su derivado diacetilado (BME(A)) en linfocitos humanos de
voluntarios sanos (c�lulas humanas mononucleares de sangre perif�rica, PBMC).
Los resultados obtenidos muestran que los compuestos BME y BME(A) no son
citot�xicos hasta una concentraci�n de 10 μM en los linfocitos humanos quiescentes o
proliferantes. Como control positivo, las c�lulas HL-60 tambi�n se incluyeron en el
experimento y, como era de esperar, hubo una reducci�n importante en la proliferaci�n
de estas c�lulas (Figura 37).
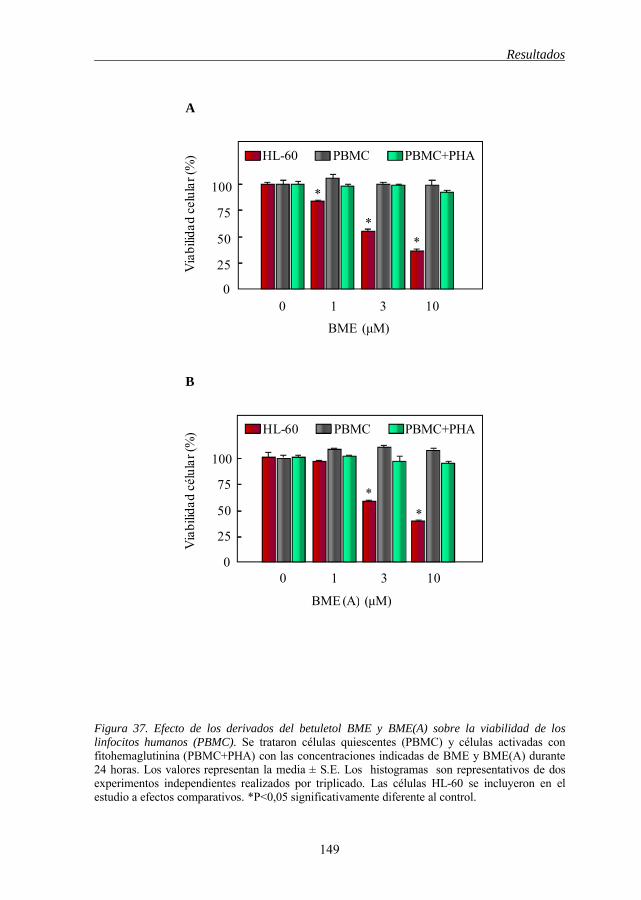
Resultados
149
Figura 37. Efecto de los derivados del betuletol BME y BME(A) sobre la viabilidad de loslinfocitos humanos (PBMC). Se trataron c�lulas quiescentes (PBMC) y c�lulas activadas con fitohemaglutinina (PBMC+PHA) con las concentraciones indicadas de BME y BME(A) durante 24 horas. Los valores representan la media � S.E. Los histogramas son representativos de dos experimentos independientes realizados por triplicado. Las c�lulas HL-60 se incluyeron en el estudio a efectos comparativos. *P<0,05 significativamente diferente al control.
BME (μM)
0
25
50
75
100V
iabi
lidad
celu
lar(
%)
0 1 3 10
*
**
PBMC+PHAPBMCHL-60
A
BME (A) (μM)
0
25
50
75
100
Via
bilid
adc�
lula
r(%
)
**
0 1 3 10
PBMC+PHAPBMCHL-60
B

Resultados
150
5.19. El Tetracetato de 3´-metil-éter de quercetina (QD) inhibe la viabilidad de
células de leucemia mieloide humana.
Los estudios antiproliferativos indicaron que las líneas celulares de leucemia mieloide
humana HL-60 y U937 son altamente sensibles a los efectos anti-proliferativos de QD
(Figura 38A) con una IC50= 9,2 ± 3,4 μM y de 5,4 ± 0,1 µM respectivamente.
5.20. QD bloquea el ciclo celular en la fase G2-M e induce apoptosis
independientemente de la biosíntesis de ARN y de proteínas.
Cuando las células son incubadas con QD, el análisis del ADN por electroforesis en gel
de agarosa, muestra un patrón de fragmentación típico (hidrólisis de cromatina inter-
nucleosomal), confirmando los efectos sobre la inducción de la apoptosis (Figura 39A).
Los cambios morfológicos característicos de las células apoptóticas (cromatina
condensada y fragmentada), se detectaron mediante microscopía fluorescente (Figura
39B).
El análisis por citometría de flujo muestra que el porcentaje de células apoptóticas
(hipodiploides) aumentó seis veces en células HL-60 tratadas con QD (Figura 39D).
Aunque el crecimiento de las células U937 disminuyó (Figura 39B), se obtuvo un índice
menor de inducción de apoptosis en comparación con las HL-60 como se muestra en los
experimentos de citometría de flujo (Figura 39D). Además, el pre-tratamiento con
actinomicina D o cicloheximida en células HL-60 a concentraciones que inhiben la
biosíntesis de ARN-m y de proteínas respectivamente, no afecta a la respuesta
apoptótica inducida por QD (Figura 39E).
Para evaluar si la inhibición del crecimiento inducido por QD está mediada por
alteraciones en la progresión del ciclo celular, se evaluó el efecto de este compuesto en
la distribución de las distintas fases del ciclo celular mediante estudios de citometría de
flujo.
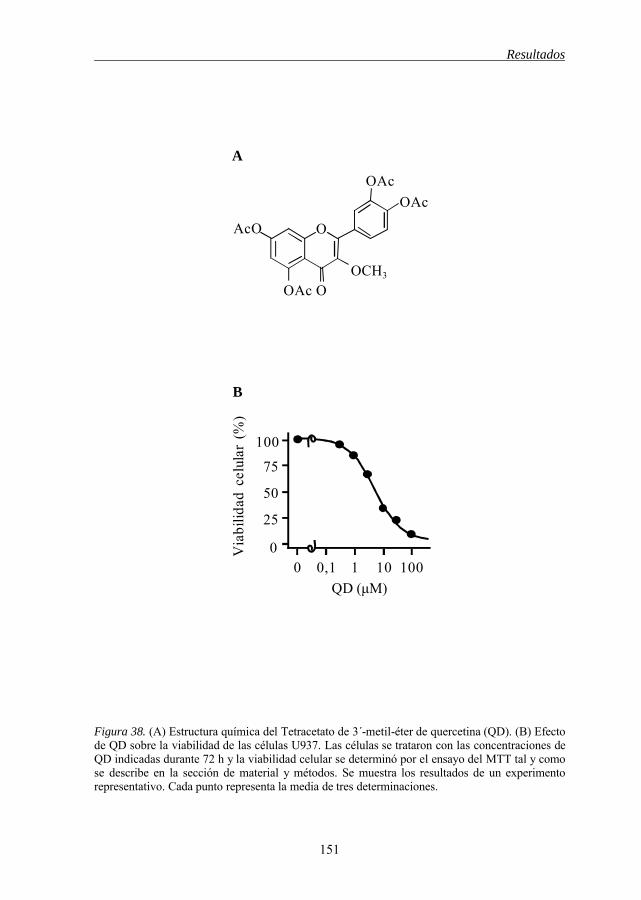
Resultados
151
Figura 38. (A) Estructura qu�mica del Tetracetato de 3�-metil-�ter de quercetina (QD). (B) Efecto de QD sobre la viabilidad de las c�lulas U937. Las c�lulas se trataron con las concentraciones de QD indicadas durante 72 h y la viabilidad celular se determin� por el ensayo del MTT tal y como se describe en la secci�n de material y m�todos. Se muestra los resultados de un experimento representativo. Cada punto representa la media de tres determinaciones.
O
O
OAcOAc
AcO
OAcOCH3
A
QD (μM)
0
25
50
75
100
10 10 100
Via
bilid
adce
lula
r(%
)
0,1
B
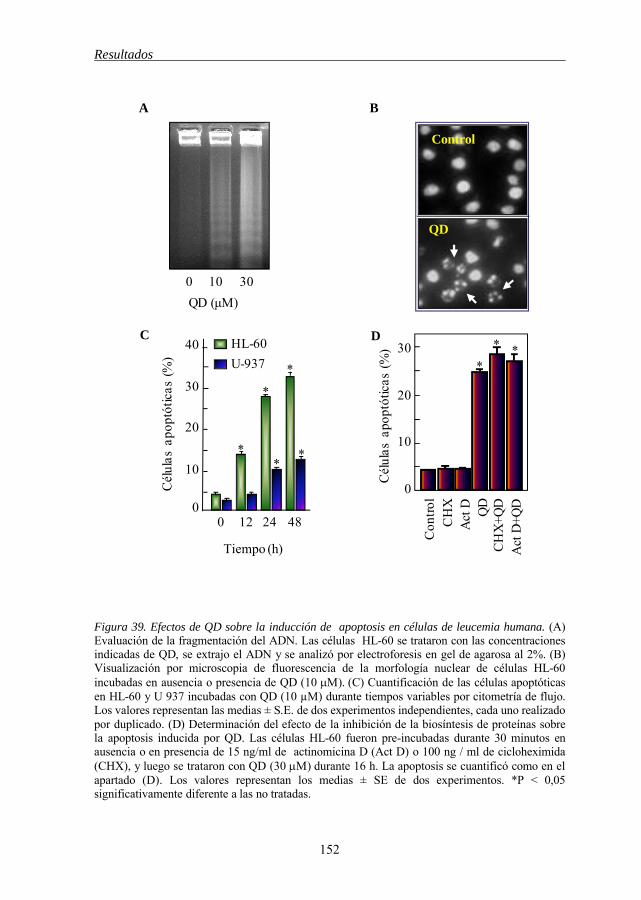
Resultados
152
Figura 39. Efectos de QD sobre la inducción de apoptosis en células de leucemia humana. (A) Evaluaci�n de la fragmentaci�n del ADN. Las c�lulas HL-60 se trataron con las concentraciones indicadas de QD, se extrajo el ADN y se analiz� por electroforesis en gel de agarosa al 2%. (B) Visualizaci�n por microscopia de fluorescencia de la morfolog�a nuclear de c�lulas HL-60 incubadas en ausencia o presencia de QD (10 ).(C) Cuantificaci�n de las c�lulas apopt�ticas en HL-60 y U 937 incubadas con QD (10 �M) durante tiempos variables por citometr�a de flujo.Los valores representan las medias � S.E. de dos experimentos independientes, cada uno realizado por duplicado. (D) Determinaci�n del efecto de la inhibici�n de la bios�ntesis de prote�nas sobrela apoptosis inducida por QD. Las c�lulas HL-60 fueron pre-incubadas durante 30 minutos en ausencia o en presencia de 15 ng/ml de actinomicina D (Act D) o 100 ng / ml de cicloheximida (CHX), y luego se trataron con QD (30 durante 16 h. La apoptosis se cuantific� como en el apartado (D). Los valores representan los medias � SE de dos experimentos. *P < 0,05 significativamente diferente a las no tratadas.
B
QD
Control
A
0 10 30QD (μM)
D *
C�l
ulas
apop
t�tic
as(%
)
Con
trol
CH
XA
ctD QD
CH
X+Q
DA
ctD
+QD
0
10
20
30*
*C
Tiempo (h)
120 24
C�l
ulas
apop
t�tic
as(%
)
0
20
30
10
48
HL-60U-937
40
*
*
*
**

Resultados
153
Como se muestra en la tabla 5, y en consonancia con los efectos inhibidores del
crecimiento, QD indujo la acumulación de células en la fase G2-M a expensas de la
población de células en la fase G1, en células HL-60 y U937.
5.21. QD induce muerte celular vía caspasa-dependiente.
Para determinar si las caspasas están involucradas, se examinó si QD induce hidrólisis
de PARP, un marcador apoptótico que indica activación de las caspasas (Figura 40A).
La hidrólisis de la proteína PARP de 116 kDa generando un fragmento de 85 kDa se
detectó en células tratadas con QD después de 12 horas de tratamiento a una
concentración de 3 µM , incrementándose de forma dosis-dependiente.
Para averiguar si hay liberación del citocromo c desde la mitocondria al citosol durante
la apoptosis inducida por la QD en células de HL-60, los extractos citosólicos se
analizaron por western blot. Como se observa en la Figura 40B se detectó un
incremento significativo en la cantidad total de citocromo c citosólico a las 12 horas de
tratamiento. Así mismo, se estudió el efecto de QD sobre el procesamiento de las
caspasas. Con este propósito las células HL-60 y U937 se trataron con concentraciones
crecientes de QD (3-10 µM) durante diferentes tiempos, y las caspasas iniciadoras
(caspasa-9 y caspasa-8) y ejecutoras (caspasa-7, -6 y -3) se determinaron por western
blot usando anticuerpos que se unen tanto a las pro-enzimas (precursoras de las
caspasas) como a los fragmentos activados. Primero se analizó el efecto de este
compuesto en la formación de caspasa-9.
El resultado muestra que QD estimula la hidrólisis de pro-caspasa-9 (inactiva) en los
correspondientes fragmentos activos de 35-37 kDa (Figura 40C). En células U937
también se observó procesamiento de pro-caspasa-9 aunque se requirió un tratamiento
de 24 horas (Figura 40C).
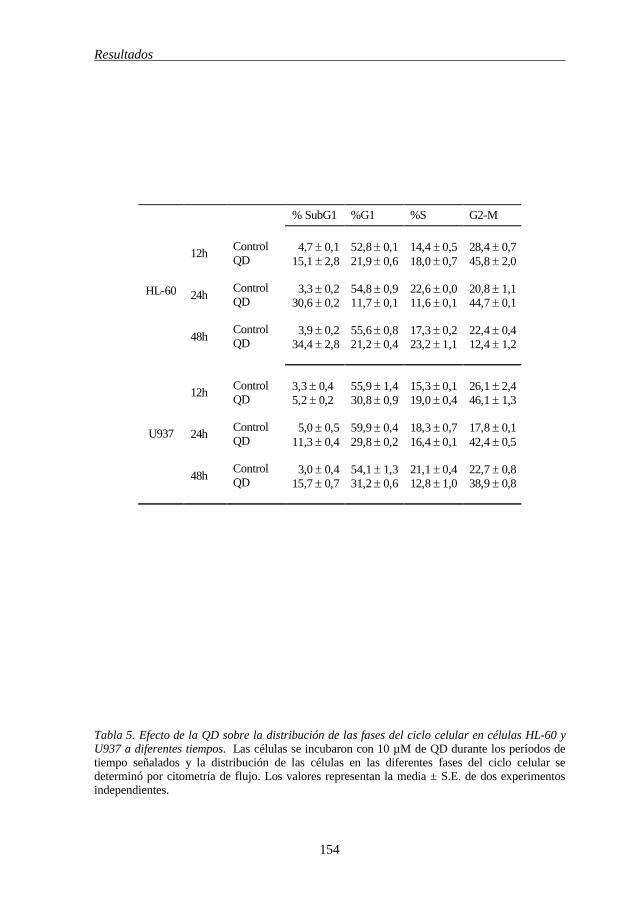
Resultados
154
Tabla 5. Efecto de la QD sobre la distribución de las fases del ciclo celular en células HL-60 y U937 a diferentes tiempos. Las células se incubaron con 10 µM de QD durante los períodos de tiempo señalados y la distribución de las células en las diferentes fases del ciclo celular se determinó por citometría de flujo. Los valores representan la media ± S.E. de dos experimentos independientes.
% SubG1 %G1 %S G2-M
Control 4,7 0,1 52,8 0,1 14,4 0,5 28,4 0,712h QD 15,1 2,8 21,9 0,6 18,0 0,7 45,8 2,0
Control 3,3 0,2 54,8 0,9 22,6 0,0 20,8 1,124h QD 30,6 0,2 11,7 0,1 11,6 0,1 44,7 0,1
Control 3,9 0,2 55,6 0,8 17,3 0,2 22,4 0,4
HL-60
48h QD 34,4 2,8 21,2 0,4 23,2 1,1 12,4 1,2
Control 3,3 0,4 55,9 1,4 15,3 0,1 26,1 2,412h QD 5,2 0,2 30,8 0,9 19,0 0,4 46,1 1,3
Control 5,0 0,5 59,9 0,4 18,3 0,7 17,8 0,124h QD 11,3 0,4 29,8 0,2 16,4 0,1 42,4 0,5
Control 3,0 0,4 54,1 1,3 21,1 0,4 22,7 0,8
U937
48h QD 15,7 0,7 31,2 0,6 12,8 1,0 38,9 0,8
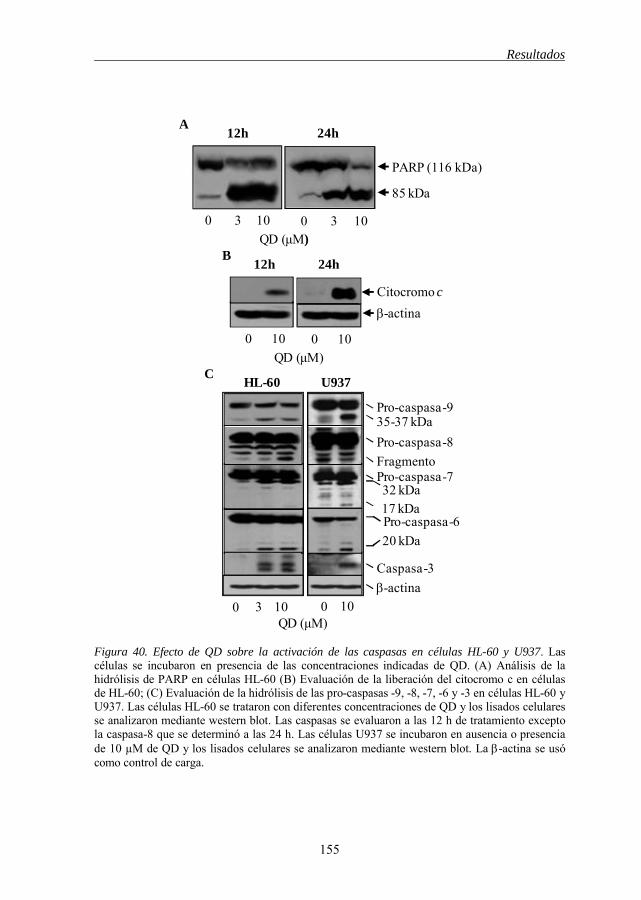
Resultados
155
Figura 40. Efecto de QD sobre la activación de las caspasas en células HL-60 y U937. Las c�lulas se incubaron en presencia de las concentraciones indicadas de QD. (A) An�lisis de la hidr�lisis de PARP en c�lulas HL-60 (B) Evaluaci�n de la liberaci�n del citocromo c en c�lulas de HL-60; (C) Evaluaci�n de la hidr�lisis de las pro-caspasas -9, -8, -7, -6 y -3 en c�lulas HL-60 y U937. Las c�lulas HL-60 se trataron con diferentes concentraciones de QD y los lisados celulares se analizaron mediante western blot. Las caspasas se evaluaron a las 12 h de tratamiento excepto la caspasa-8 que se determin� a las 24 h. Las c�lulas U937 se incubaron en ausencia o presencia de 10 �M de QD y los lisados celulares se analizaron mediante western blot. La -actina se us�como control de carga.
B
-actinaCitocromo c
12h 24h
0 10QD (μM)
0 10
85 kDa
PARP (116 kDa)
0 3 10QD (μM)
0 3 10
12h 24hA
HL-60 U937
QD (μM)0 3 10
C
35-37 kDaPro-caspasa-9
Caspasa-3-actina
0 10
Pro-caspasa-732 kDa17 kDa
Pro-caspasa-8Fragmento
Pro-caspasa-620 kDa

Resultados
156
Para determinar la contribución de la vía extrínseca en la apoptosis inducida por QD, las
células HL-60 se trataron con concentraciones crecientes de este compuesto y los
lisados celulares se sometieron a análisis por western blot. Los resultados demuestran
que QD (10 µM) induce de forma significativa la hidrólisis de la pro-caspasa-8, si bien
esta respuesta no se detectó hasta las 24 horas de tratamiento (Figura 40C). En cuanto a
las células U937, no hubo hidrólisis de pro-caspasa-8 a los tiempos ensayados. Como se
muestra en la figura 40C, QD estimula a su vez el procesamiento proteolítico de las
caspasas ejecutoras -7, -6 y -3. En células HL-60, concentraciones bajas de QD (3 µM)
inducen la hidrólisis de las pro-caspasas -7, -6 y -3 a las 12 horas. Por el contrario, en
células U937 se requieren concentraciones más altas (10 µM) y tratamientos más
prolongados (24 horas).
Para confirmar que la inducción de apoptosis por QD requiere de la activación de las
caspasas, las células HL-60 se pre-trataron con un inhibidor general de caspasas (z-
VAD-fmk). La inhibición de la apoptosis inducida por QD en presencia de z-VAD-fmk
demuestra que la apoptosis es caspasa dependiente.
5.22. El inhibidor específico de caspasa-3, z-DEVD-fmk, es incapaz de inhibir la
apoptosis inducida por QD.
Para identificar qué caspasas son importantes en la apoptosis inducida por QD se
estudió el efecto de los inhibidores permeables de caspasas. Ninguno de los inhibidores
utilizados como z-IETD-fmk (inhibidor específico de caspasa-8), z-LEHD-fmk
(inhibidor específico de la caspasa-9) o z-DEVD-fmk (inhibidor específico de las
caspasas -3 y -7) tuvieron efecto significativo en la muerte celular inducida por QD
(Figura 41A).
Sorprendentemente, el inhibidor específico de caspasa-3 (z-DEVD-fmk) fue incapaz de
inhibir la apoptosis inducida por QD en células de HL-60 (Figura 41A). Este resultado
sugiere que in vivo se requiere probablemente una segunda caspasa, posiblemente
caspasa-6 (Figura 41C), que es sensible a z-VAD-fmk, pero no a z-DEVD-fmk (26).
Sin embargo, la caspasa-3 activa se encuentra presente en niveles altos en células HL-
60 tratadas con QD (Figura 41C).
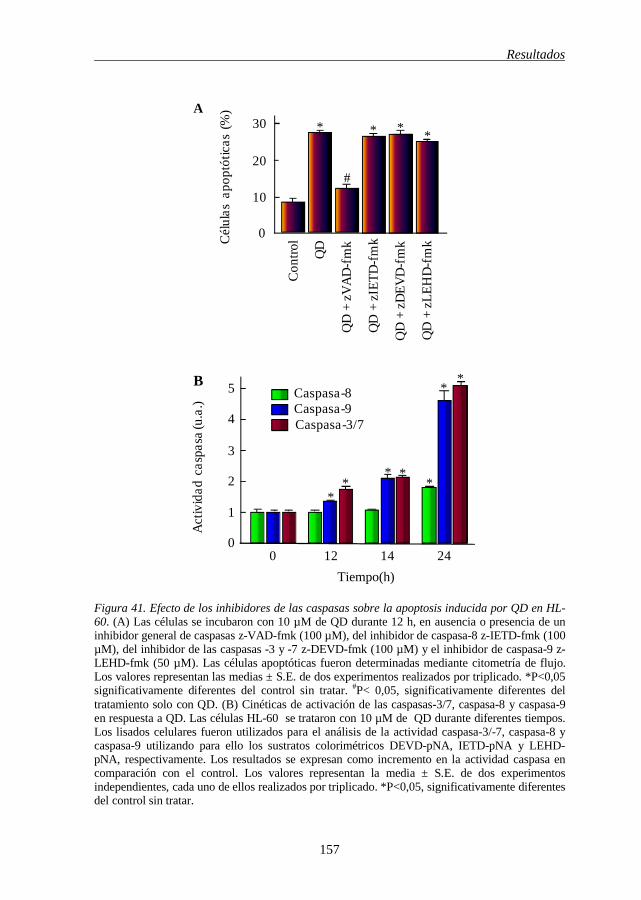
Resultados
157
Figura 41. Efecto de los inhibidores de las caspasas sobre la apoptosis inducida por QD en HL-60. (A) Las células se incubaron con 10 µM de QD durante 12 h, en ausencia o presencia de un inhibidor general de caspasas z-VAD-fmk (100 µM), del inhibidor de caspasa-8 z-IETD-fmk (100µM), del inhibidor de las caspasas -3 y -7 z-DEVD-fmk (100 µM) y el inhibidor de caspasa-9 z-LEHD-fmk (50 µM). Las células apoptóticas fueron determinadas mediante citometría de flujo.Los valores representan las medias ± S.E. de dos experimentos realizados por triplicado. *P<0,05 significativamente diferentes del control sin tratar. #P< 0,05, significativamente diferentes del tratamiento solo con QD. (B) Cinéticas de activación de las caspasas-3/7, caspasa-8 y caspasa-9 en respuesta a QD. Las células HL-60 se trataron con 10 µM de QD durante diferentes tiempos. Los lisados celulares fueron utilizados para el análisis de la actividad caspasa-3/-7, caspasa-8 y caspasa-9 utilizando para ello los sustratos colorimétricos DEVD-pNA, IETD-pNA y LEHD-pNA, respectivamente. Los resultados se expresan como incremento en la actividad caspasa en comparación con el control. Los valores representan la media ± S.E. de dos experimentos independientes, cada uno de ellos realizados por triplicado. *P<0,05, significativamente diferentes del control sin tratar.
A
Cél
ulas
apop
tótic
as(%
)
20
10
30
0
Con
trol
QD
QD
+zV
AD
-fm
k
QD
+zI
ETD
-fm
k
QD
+zD
EVD
-fm
k
QD
+zL
EHD
-fm
k
#
* * * *
B
Tiempo(h)0 12 14
0
1
2
3
4
5
Act
ivid
adca
spas
a(u
.a.)
24
Caspasa-8Caspasa-9Caspasa-3/7
***
**
**

Resultados
158
Puesto que el procesamiento de las caspasas no siempre se corresponde con un aumento
de la actividad, las actividades enzimáticas de la caspasa-3 y de las caspasas -8 y -9 se
determinaron en extractos de células HL-60 tratadas con QD a diferentes tiempos.
Como se muestra en la Figura 41B, las actividades de las caspasas-3 y -9 aumentan
significativamente a las 12-14 horas. Mientras que la actividad de la caspasa -8 se
detectó a las 24 horas del tratamiento.
5.23. QD activa MAPKs.
Puesto que las MAPKs como ERK, JNK/SAPK y p38 MAPK juegan un papel crucial en
el destino de la célula, se evaluó el efecto de QD sobre la activación de MAPKs. Los
resultados indican que la incubación de células HL-60 y U937 con QD estimuló la
fosforilación de JNK/SAPK, de p38 MAPK y de ERK 1/2 (Figuras 42A y B). La
fosforilación de p38 MAPK y de ERK 1/2 se detectó a los 30 minutos y a la hora de
añadir QD, respectivamente, y ambos valores permanecieron elevados hasta al menos
240 minutos (Figura 42A). Sin embargo, y en las mismas condiciones de
experimentación, no se observó la activación de la JNK/SAPK hasta 4 horas después de
añadir QD (Figura 42B). Estos resultados indican que QD induce la activación de
JNK/SAPK, de p38 MAPK y de ERK 1/2 siguiendo diferentes cinéticas. Para establecer si
la fosforilación de las MAPKs juega un papel esencial en la apoptosis inducida por QD,
se examinaron los efectos de los inhibidores de JNK/SAPK, de p38 MAPK y de ERK 1/2
(Figura 42C y F).
El pre-tratamiento de células HL-60 con inhibidores específicos de JNK/SAPK y de p38MAPK no alteró el porcentaje de apoptosis inducido por QD; Tanto SP600125 como
SB203580, no tuvieron influencia en los niveles basales de apoptosis (Figura 42D y E).
Estos resultados sugieren que la activación de JNK/SAPK y de p38 MAPK no está
involucrada en la apoptosis mediada por QD.
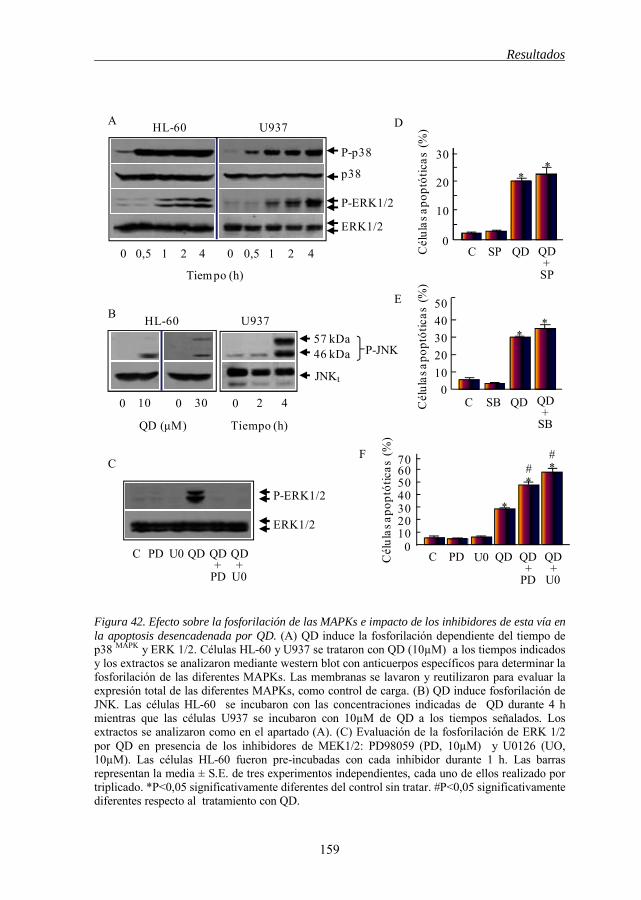
Resultados
159
Figura 42. Efecto sobre la fosforilación de las MAPKs e impacto de los inhibidores de esta vía en la apoptosis desencadenada por QD. (A) QD induce la fosforilaci�n dependiente del tiempo de p38 MAPK y ERK 1/2. C�lulas HL-60 y U937 se trataron con QD (10�M) a los tiempos indicados y los extractos se analizaron mediante western blot con anticuerpos espec�ficos para determinar la fosforilaci�n de las diferentes MAPKs. Las membranas se lavaron y reutilizaron para evaluar la expresi�n total de las diferentes MAPKs, como control de carga. (B) QD induce fosforilaci�n de JNK. Las c�lulas HL-60 se incubaron con las concentraciones indicadas de QD durante 4 hmientras que las c�lulas U937 se incubaron con 10�M de QD a los tiempos se�alados. Los extractos se analizaron como en el apartado (A). (C) Evaluaci�n de la fosforilaci�n de ERK 1/2 por QD en presencia de los inhibidores de MEK1/2: PD98059 (PD, 10�M) y U0126 (UO, 10�M). Las c�lulas HL-60 fueron pre-incubadas con cada inhibidor durante 1 h. Las barras representan la media � S.E. de tres experimentos independientes, cada uno de ellos realizado por triplicado. *P<0,05 significativamente diferentes del control sin tratar. #P<0,05 significativamente diferentes respecto al tratamiento con QD.
P-ERK1/2
ERK1/2
C PD U0 QD+
PD
QD+
U0
QD
C
D
C�l
ulas
apop
t�tic
as(%
)
010203040506070
C PD U0 QD+
PD
QD+
U0
QD
F
*
#*
#*
B
0 30
QD (μM)
0 10
JNKt
46 kDa57 kDa
P-JNK
HL-60
Tiempo (h)
20 4
U93750
0
20
40
10
30
C SB QD QD+
SBC
�lul
asap
opt�
ticas
(%)
E
**
C�l
ulas
apop
t�tic
as(%
)
10
20
30
0C SP QD QD
+SP
**
A HL-60
Tiempo (h)
1 20 0,5 4
P-p38
P-ERK1/2
U937
1 20 0,5 4
ERK1/2
p38

Resultados
160
5.24. La apoptosis inducida por QD en células leucémicas humanas se potencia
mediante el uso de inhibidores específicos de ERK 1/2.
Para investigar el impacto de ERK 1/2 en la muerte celular mediada por QD, se usaron
los inhibidores de la MEK (PD98059 y U0126) para bloquear la activación de ERK 1/2.
Los resultados indican que PD98059 y U0126 potencian la apoptosis mediada por QD
(Figura 42F). El porcentaje de células apoptóticas aumentó desde un 30 % en células
tratadas con QD hasta un 45 % y 55% con PD98059 y U0126, respectivamente. Para
confirmar el efecto de los inhibidores de MEK1/2, se determinó a su vez el estado de
fosforilación de ERK 1/2 con los diferentes tratamientos. Como se muestra en la Figura
42C, PD98059 y U0126 suprimieron completamente la fosforilación de ERK 1/2
inducida por QD.
La combinación de QD con U0126 duplicó el porcentaje de muerte celular respecto a
las células tratadas con QD y aumentó diez veces respecto a U0126 (Figura 42F). Estos
resultados sugieren que los inhibidores específicos de ERK 1/2 pueden ser útiles para
sensibilizar a las células leucémicas humanas frente a QD.
5.25. QD inhibe el crecimiento, la viabilidad celular e induce apoptosis en líneas
celulares de leucemia humana, que expresan niveles elevados de Bcl-2 y Bcl-xL.
Para dilucidar si la vía intrínseca es determinante en la inducción de apoptosis por QD,
comparamos los efectos en la inducción de apoptosis en la línea celular U937 que
expresan niveles elevados Bcl-2 (U937/Bcl-2) con la línea celular parental (U937).
Como se muestra en la Figura 43A, no hay diferencias en la inducción de apoptosis a
diferentes tiempos de tratamiento (12, 24 y 48 horas).
Estos resultados sugieren que Bcl-2 no está involucrado en la apoptosis inducida por
QD.

Resultados
161
5.26. QD no es citotóxico para los linfocitos normales de origen humano.
Se estudió el efecto de QD en linfocitos humanos (células humanas mononucleares de
sangre periférica, PBMC). Los resultados obtenidos muestran que el compuesto QD no
es citotóxico hasta una concentración de 10 μM tanto en PBMC quiescentes o
proliferantes (como control positivo, las células HL-60 también se incluyeron en el
experimento y, como era de esperar, hubo una reducción importante en la proliferación
de estas células (Figura 43B y C).
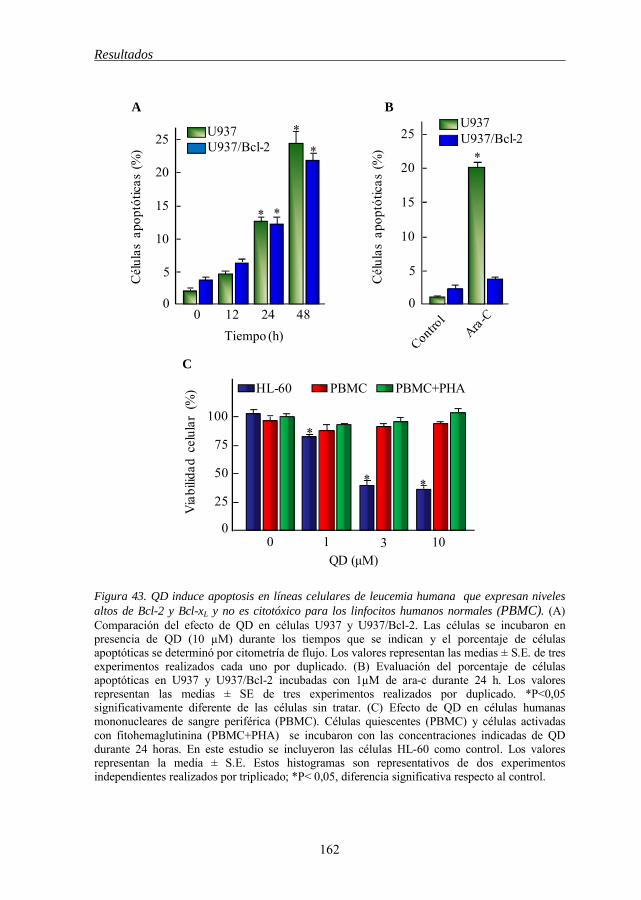
Resultados
162
Figura 43. QD induce apoptosis en líneas celulares de leucemia humana que expresan niveles altos de Bcl-2 y Bcl-xL y no es citotóxico para los linfocitos humanos normales (PBMC). (A)Comparaci�n del efecto de QD en c�lulas U937 y U937/Bcl-2. Las c�lulas se incubaron enpresencia de QD (10 �M) durante los tiempos que se indican y el porcentaje de c�lulas apopt�ticas se determin� por citometr�a de flujo. Los valores representan las medias � S.E. de tres experimentos realizados cada uno por duplicado. (B) Evaluaci�n del porcentaje de c�lulas apopt�ticas en U937 y U937/Bcl-2 incubadas con 1�M de ara-c durante 24 h. Los valores representan las medias � SE de tres experimentos realizados por duplicado. *P<0,05significativamente diferente de las c�lulas sin tratar. (C) Efecto de QD en c�lulas humanas mononucleares de sangre perif�rica (PBMC). C�lulas quiescentes (PBMC) y c�lulas activadas con fitohemaglutinina (PBMC+PHA) se incubaron con las concentraciones indicadas de QD durante 24 horas. En este estudio se incluyeron las c�lulas HL-60 como control. Los valores representan la media � S.E. Estos histogramas son representativos de dos experimentos independientes realizados por triplicado; *P< 0,05, diferencia significativa respecto al control.
B
C�l
ulas
apop
t�tic
as(%
)
20
10
15
25
0
5
U937U937/Bcl-2
*
A
Tiempo (h)
C�l
ulas
apop
t�tic
as(%
)
0
20
10
24120 48
5
15
25 U937U937/Bcl-2
*
**
*
C
QD (μM)
Via
bilid
adce
lula
r(%
)
0 1 3 100
25
50
75
100
HL-60 PBMC PBMC+PHA
* *
*

DDDIIISSSCCCUUUSSSIIIÓÓÓNNN


Discusión
165
En el ámbito de la quimioterapia contra el cáncer el 67% de los medicamentos eficaces
son de origen natural [232]. La mayoría de los fármacos actualmente disponibles para el
tratamiento del cáncer son muy caros, tóxicos y poco eficaces en el tratamiento de la
enfermedad. Esto sugiere que, los agentes procedentes de fuentes naturales deberían ser
investigados más en profundidad para la prevención y el tratamiento del cáncer.
Los flavonoides son compuestos con estructura fenilbenzo-γ-pirona de origen natural
que se encuentran en la dieta, en frutas, verduras y bebidas derivadas de las plantas [52].
Estos compuestos polifenólicos son de gran interés debido a que entre sus actividades
biológicas en el ser humano se encuentra la posibilidad de actuar como agentes
antitumorales [52].
En este estudio, se ha evaluado la sensibilidad de cinco líneas de células tumorales
humanas a veintidós compuestos naturales y semi-sintéticos que contienen el núcleo de
fenilbenzo-γ-pirona. Los resultados indican que el 3-metil éter de betuletol (1) y su
derivado diacetilado (2) son potentes inhibidores de la proliferación y tienen actividad
citotóxica en la línea celular de leucemia mieloide humana (HL-60) y en otras líneas
celulares, tales como las células de carcinoma epidermoide humano (A431) y las células
de osteosarcoma humano (HOS).
La baja sensibilidad de las células SK-OV-3 a estos compuestos podría explicarse por el
alto nivel de proteína HER-2/neu y a la alta actividad de Akt (una serina / treonina
quinasa antiapoptótica que media señales de supervivencia) en esta línea celular [233]
así como, estudios previos han demostrado que las células de adenocarcinoma de ovario
humano (SK-OV-3) son resistentes a TRAIL (ligando inductor de apoptosis relacionado
con TNF).
Sin embargo, las células HL-60 son especialmente sensibles al 3-metil éter de betuletol
(1) y a su derivado diacetilado (2). La introducción de un grupo metilo en la posición 4'
del anillo B parece contribuir a la citotoxicidad, ya que el 3-metil éter de betuletol (1) es

Discusión
166
mucho más potente que la 4', 5,7 trihidroxi-3 ,6-dimetoxiflavona (12) en todas las líneas
celulares estudiadas.
Un estudio previo sobre la estructura y la actividad antiproliferativa de los flavonoides
polimetoxilados demostró que el doble enlace en C2-C3 y el grupo 3-hidroxilo del
anillo C son factores estructurales importantes [234]. En función de los datos obtenidos
podemos afirmar que la naringenina (21) y la 7,3´-dimetil éter dihidro-quercetina (16)
no son eficaces como agentes antiproliferativos confirmando que el doble enlace en C2-
C3 es importante para dicha actividad antiproliferativa.
El papel del grupo 3'-hidroxilo se deduce al comparar las actividades antiproliferativas
de la naringenina (21) y el eriodictiol (19), ya que la única diferencia estructural entre
ambos flavonoides es la presencia de un grupo 3´-hidroxilo en la molécula de eriodictiol
(19), resultando en un aumento de su capacidad citotóxica. Al interpretar los resultados
hay que tener en cuenta que estos compuestos ejercen una actividad antitumoral que es
dependiente del tipo celular, ya que sólo la línea celular HL-60 y la línea celular A431
fueron susceptibles al eriodictiol (19).
Algunos autores han sugerido que la actividad antiproliferativa de los flavonoides
polimetoxilados puede deberse, en parte, a su estabilidad química [235]. Se ha
demostrado que la quercetina (4) puede sufrir autoxidación y degradarse, mientras que
la metilación de los hidroxilos fenólicos, como en el caso de la 3,3' dimetil éter de
quercetina (7), le confiere mayor estabilidad a este flavonoide. Por otra parte, la adición
de bajas concentraciones de ácido ascórbico aumenta la actividad antiproliferativa de la
quercetina (4) en carcinoma de células escamosas (HTB 43). Esto parece estar
relacionado con la capacidad del acido ascórbico de inhibir la degradación oxidativa de
los flavonoides polihidroxilados [235].
El hallazgo de una mayor actividad citotóxica del 3,3'-dimetil éter de quercetina (7)
respecto a la quercetina (4) en la mayoría de las líneas celulares estudiadas, concuerda
con lo anteriormente mencionado. Sin embargo la metilación de la quercetina (4) para

Discusión
167
obtener el 3,3',4'-trimetil éter de quercetina (10) provoca la supresión de la actividad
citotóxica. Del mismo modo, cuando el 3-metil éter de betuletol (1) es metilado a
betuletol 3,5,7-trimetil éter de betuletol (3), la actividad citotóxica se pierde. Aunque no
existen estudios sobre la estabilidad química del betuletol y de sus derivados
metoxilados, este resultado no puede ser explicado por un aumento de la estabilidad,
sino más bien por la pérdida de grupos hidroxilos esenciales. Aunque hay muchas
pruebas de que la posición y número de los grupos hidroxilos de los anillos A y B
parecen influir firmemente en la conformación de la molécula y modular su efecto
inhibitorio del crecimiento, se acepta de forma general que la actividad antiproliferativa
de los flavonoides no se puede predecir en función de su composición química y
estructura [3].
La citotoxicidad de la quercetina (4) mejora notablemente mediante la acetilación y la
metilación de los grupos hidroxilos libres en todas las líneas celulares del estudio. El
aumento de la actividad citotóxica en la línea celular HL-60 fue de diez veces. Una
posible explicación de la diferencia de IC50 podría ser que se alcance una concentración
intracelular más alta debido a que tanto los derivados acetilados como los metilados
pueden atravesar las membranas biológicas con más facilidad que la quercetina. Sin
embargo, probablemente existan factores adicionales que desempeñen un papel
importante en la respuesta biológica final de una línea celular tumoral. Por ejemplo,
aunque los derivados acetilados de eriodictiol (20) presentan mejor actividad
antiproliferativa en la mayoría de las líneas celulares (HL-60, A431, HeLa y SK-OV-3),
el aumento de la hidrofobicidad no puede explicar su ineficacia en las células HOS.
Entre los 22 compuestos que se presentan en la Tabla 3, el 3-metil éter de betuletol (1) y
su derivado diacetilado (2), son los que exhiben la actividad citotóxica más alta en todas
las líneas celulares tumorales evaluadas. En la mayoría de las líneas celulares los
valores de IC50 fueron ligeramente inferiores a 10 μM, mientras que en células HL-60
estos se sitúan alrededor de 1 μM.

Discusión
168
La selectividad hacia las células tumorales es un criterio muy importante a la hora de
utilizar o desarrollar agentes para el tratamiento contra el cáncer. En este estudio se
compararon los efectos de los compuestos (1) y (2) en las células HL-60 y en linfocitos
humanos (PBMC). Los experimentos de dosis-respuesta revelaron que tanto las PBMC
quiescentes como las PBMC en proliferación son resistentes a ambos derivados del
betuletol.
Entre las distintas estrategias que pueden ser utilizadas para la prevención o para el
tratamiento del cáncer están aquellas que afectan a la apoptosis, que es un proceso
fisiológico por el cual se eliminan las células cuando un agente químico o físico
produce daño en el ADN. Por lo tanto los compuestos que pueden inducir apoptosis
pueden ser útiles en el control y en la terapia contra el cáncer.
Aunque existen estudios previos que describen la muerte celular por apoptosis y
fragmentación del ADN por derivados de fenilbenzo-γ-pirona en células de leucemia
mieloide humanas HL-60 [236], hasta ahora los derivados del betuletol no se han
evaluado como potenciales agentes antitumorales.
El 3-metil éter de betuletol (BME) y diacetato de 3-metil éter de betuletol (BME(A)).
En este trabajo, se demuestra que BME y BME(A) tienen una alta actividad
antiproliferativa y similar efectividad (muestran valores similares IC50), en las células
HL-60, U937, K-562, THP-1, Jurkat y Molt-3. Ambos compuestos inducen en la línea
celular HL-60 la formación de cuerpos apoptóticos, la degradación inter-nucleosomal
del ADN y la hidrólisis de poli (ADP-ribosa) polimerasa, características típicas de la
apoptosis [237]. Con el uso de z-VAD-fmk, un inhibidor general de caspasas, se inhibe
los cambios morfológicos relacionados con la apoptosis, la fragmentación del ADN y la
hidrólisis de la poli (ADP-ribosa) polimerasa, poniendo de manifiesto que las caspasas
son componentes esenciales en la apoptosis inducida por BME.

Discusión
169
La caspasa-3 está considerada como la caspasa efectora más importante, tanto si la
inducción de la apoptosis es mediante la vía intrínseca como por la vía extrínseca. Se ha
observado que la caspasa-3 participa en la apoptosis inducida por agentes citotóxicos
como 1-β-D-arabino-furanosil-citosina, mitoxantrona, etopósido y CPT-11 [238] en
células leucémicas. En el presente estudio se encontró un alto nivel de activación y
procesamiento de la caspasa-3 después del tratamiento de las células HL-60 con ambos
derivados del betuletol.
La mayoría de agentes citotóxicos conocidos inducen liberación de citocromo c
mitocondrial [239], proteína que desencadena la activación de caspasa-9 mediante un
proceso dependiente de la formación del apoptosoma [240]. Aunque los derivados del
betuletol inducen liberación de citocromo c en el citosol y activación de la caspasa-9, la
vía intrínseca no parece estar involucrada en la inducción de apoptosis por estos
compuestos. La liberación de citocromo c y la correspondiente activación de caspasa-9
es posterior (12 h) a los eventos apoptóticos tempranos observados a las 8 h tales como
aumento de la actividad de la caspasa-8 y caspasa-3. Además, el uso de un inhibidor de
caspasa-9 (z-LEHD-fmk) fue incapaz de impedir el efecto de BME sobre la apoptosis
apoyando un modelo en el que BME induce apoptosis por un mecanismo mediado por
caspasa-8.
Numerosos estudios indican que la vía iniciada a través de los receptores de muerte y
que desencadena en la activación de la caspasa-8 (vía extrínseca), pueden interaccionar
con la vía intrínseca mediante hidrólisis de Bid, perturbación de la permeabilidad
mitocondrial, liberación del citocromo c al citosol y formación del apoptosoma. La
caspasa-8, caspasa iniciadora, tiene dos sustratos preferentes: la caspasa ejecutora
caspasa-3, que una vez que es activada causa directamente la muerte celular, y la
proteína Bid, factor pro-apoptótico de la familia de proteínas Bcl-2. La hidrólisis de Bid
por la caspasa-8 genera una proteína truncada, tBid, que produce perturbación en la
mitocondria que conduce a la liberación de citocromo c al citosol, activación de la
caspasa-9 y activación de la caspasa-3 en última instancia. En el presente estudio se

Discusión
170
demuestra que BME induce activación de caspasa-8 e hidrólisis de Bid, que está
asociado a la pérdida del potencial de membrana mitocondrial.
Estudios previos han demostrado que la apoptosis causada por algunos principios
activos o medicamentos contra el cáncer puede estar mediada a través de receptores de
muerte como Fas (CD95) [241], aunque muchos fármacos parecen iniciar la vía
intrínseca directamente. Se han descrito una amplia variedad de mecanismos de
resistencia a la apoptosis que interfieren en los diferentes niveles de la señalización
[230,242]. Dado que la caspasa-8 es esencial en la cascada apoptótica iniciada en la
activación del receptor de muerte Fas, se evaluó si este receptor estaba involucrado en la
apoptosis inducida por BME. Los resultados demuestran que la inducción de la
apoptosis es independiente de la unión de FasL a su receptor. Estos resultados sugieren
que otros receptores de muerte diferentes a Fas como son TNFR1 (receptores de TNF) y
los receptores de TRAIL (ligando inductor de apoptosis relacionado con TNF), TRAIL-
R1 (receptor de muerte 4, DR4) y TRAIL-R2 (receptor de muerte 5, DR5) pueden estar
implicados. Otra posibilidad podría ser que BME promueva un cambio conformacional
en la membrana plasmática seguido de agregación de receptores de Fas, ya que estudios
recientes con este receptor han puesto de manifiesto que puede ser activado
intracelularmente, en ausencia de su ligando. Por ejemplo el éter sintético, edelfosina,
induce apoptosis de forma selectiva en células tumorales sin dañar a los tejidos sanos,
activando Fas intracelularmente mediante un mecanismo independiente de la
interacción con su ligando natural FasL [243].
Ya que las catepsinas B y L pueden hidrolizar Bid [244] evaluamos la participación de
estas proteasas en la muerte inducida por el 3-metil éter de betuletol. Sin embargo, los
inhibidores de catepsinas B y L (Mu-Phe-HPh-fmk y E64d) y el inhibidor de la
catepsina lisosomal D (pepstatina A) no impidieron la muerte celular inducida por
BME. Estos resultados indican que las cisteín-proteasas catepsina B y L y la aspartil-
proteasa catepsina D no están implicadas en el mecanismo desencadenado por BME.

Discusión
171
Es ampliamente conocido que la vía intrínseca de la apoptosis está controlada por
miembros de la familia de Bcl-2 [179,245], que son reguladores cruciales de la
apoptosis en células de mamíferos. En este sentido los resultados obtenidos indican que
BME induce muerte celular en células de leucemia que expresan niveles elevados de
Bcl-2 y Bcl-xL. Estos resultados son de gran importancia dado que Bcl-2 y su homólogo
Bcl-xL confieren resistencia a la apoptosis evitando la permeabilidad mitocondrial, la
acumulación del citocromo c en el citosol y la activación de las caspasas ejecutoras de
la apoptosis [230,246-248]. Por tanto este compuesto podría ser útil en el tratamiento
del cáncer, ya que la alta expresión de Bcl-2 y Bcl-xL está asociada con la
quimioresistencia, especialmente en las neoplasias hematológicas malignas [249] en las
que el aumento de expresión de Bcl-xL tiene un peor pronóstico [250]. Por tanto, la
búsqueda de nuevos agentes que inducen la apoptosis por nuevas vías alternativas es un
objetivo primordial para superar la resistencia al tratamiento contra el cáncer.
Dado que las MAPKs juegan un papel importante en la apoptosis, se estudió su
implicación en la apoptosis inducida por el 3-metil éter de betuletol. Los resultados
ponen de manifiesto que este compuesto induce la fosforilación y la activación de ERK
1/2, p38MAPK y JNK/SAPK siguiendo diferentes cinéticas. La fosforilación de ERK1/2
y de p38MAPK aumentó inmediatamente después del tratamiento con el 3-metil éter de
betuletol (< 30 min.) y se mantuvo elevada al menos hasta las 4 h. Sin embargo, la
actividad de JNK/SAPK se detectó a un tiempo más prolongado de incubación (4 h).
Para evaluar si la fosforilación y activación de las MAPKs desempeñan un papel
relevante en la inducción de la apoptosis por BME se utilizaron inhibidores específicos
de ERK 1/2, p38MAPK y JNK/SAPK. Así, mientras el inhibidor de JNK/SAPK,
SP600125, disminuyó significativamente el porcentaje de células apoptóticas inducidas
por BME, el inhibidor de p38MAPK, SB203580, careció de efecto. Este resultado sugiere
que JNK/SAPK pero no p38MAPK desempeña un papel activo en el proceso. La función
pro-apoptótica de JNK/SAPK se ha demostrado en la apoptosis inducida por diferentes
agentes quimioterapeuticos, como la vinblastina, la doxorubicina y el etopósido
[251,252].

Discusión
172
Los datos también indican que el inhibidor PD98059 disminuye los efectos apoptóticos
del 3-metil éter de betuletol, lo que sugiere que la vía ERK 1/2 está implicada en la
señal de muerte celular. Estos resultados están en consonancia con los obtenidos por
otros autores en células A549 en los que la inhibición de la vía MEK-ERK con U0126 o
PD98059 bloquea la apoptosis inducida por la quercetina [253]. Además, la supresión
de la vía MEK-ERK por el inhibidor de MEK, PD98059, induce un incremento de
resistencia a cis-platino en células de carcinoma cervical humano (SiHa) y en células de
hepatoblastoma (HepG2) [254]. Por otro lado, la acroleína, un aldehído α,β-insaturado
altamente reactivo generado por peroxidación lipídica, induce la fosforilación de ERK
1/2 y la inhibición de su activación mediante PD98059 y U0126 bloquea la apoptosis
inducida por la acroleína [255]. Recientemente, se ha demostrado que la inhibición de
ERK 1/2 disminuye la muerte celular inducida por un flavonoide, el acetil derivado de
trifolina [256].
Las especies reactivas de oxígeno (EROs) han sido implicadas como segundos
mensajeros en múltiples vías de señalización y se han asociado con la activación de las
vías MAPKs. Aunque en el estudio llevado a cabo se ha observado un aumento en la
generación de EROs, éstos no parecen jugar ningún papel importante en la muerte
celular inducida por BME.
Los agentes antitumorales que afectan a la dinámica de los microtúbulos son de gran
interés y se utilizan en la quimioterapia actual. Diversos flavonoides que contienen un
grupo metoxilo en C-3 ejercen su actividad anti-proliferativa mediante la unión a los
microtúbulos de tubulina [257]. El bioflavonoide mejor estudiado, la quercetina, ejerce
su actividad inhibitoria del crecimiento celular afectando a los microtúbulos, inhibiendo
la polimerización mediante la unión de la misma con la tubulina [109]. Dado que BME
es un análogo de la quercetina y también contiene un grupo metoxilo en C-3, la parada
en la fase G2-M del ciclo celular inducida por BME podría ser explicada por la
inhibición de la formación de los microtúbulos. En el estudio llevado a cabo, hemos
demostrado efectivamente que BME inhibe la polimerización de la tubulina y promueve
cambios en la red de microtúbulos celular siguiendo un patrón similar a la inducida por

Discusión
173
la colchicina. La parada del ciclo celular inducida por BME se produce en la fase M y
está asociada con la inducción y la fosforilación de ciclina B1, la acumulación de
p21Cip1, y la activación de Cdk1 vía desfosforilación de fosfo T14-Y15.
Diversos estudios llevados a cabo por otros autores indican que la desorganización de la
estructura de los microtúbulos mediante agentes que se unen a la tubulina promueve un
aumento de la expresión de diferentes proteínas entre las que se encuentra p21Cip1 [258].
El aumento de p21Cip1 parece ser un evento importante en el efecto anti-proliferativo
inducido por BME y supone una vía independiente de p53, dado que las células HL-60
carecen de p53 funcional. Estos resultados tienen implicaciones importantes para el
desarrollo de BME como fármaco para la prevención del cáncer o bien como fármaco
quimioterapéutico, ya que aproximadamente el 50% de los cánceres humanos
manifiestan mutaciones en p53 [259,260].
Se ha descrito que el agente quimioterapéutico etopósido induce acumulación de
ceramida en células humanas de leucemia Molt-4 mediante la activación de la serina-
palmitoil-transferasa la cual es responsable de la generación de ceramida por la vía de
biosíntesis de novo [261]. En este estudio se demuestra que BME activa la SMasa ácida
e induce un aumento en los niveles de ceramida en células HL-60. En este sentido es
importante resaltar que la ceramida puede que esté implicada en el reclutamiento de los
receptores de Fas en balsas lipídicas [262]. A pesar de que múltiples vías metabólicas
convergen en la generación de ceramida, hasta ahora la vía de novo y la vía de las
esfingomielinasas han sido las más estudiadas. La fumonisina B1 no inhibe la muerte
inducida por el 3-metil éter de betuletol sugiriendo un papel poco importante o nulo de
la enzima ceramida sintasa en este proceso. La activación de la SMasa ácida es la
principal vía para la generación de ceramida en respuesta a agentes quimioterápicos
[175,209]. La SMasa ácida se encuentra principalmente en los lisosomas y en
microdominios ricos en esfingolípidos de la membrana plasmática que participan en la
señalización mediada por receptores [263]. La unión de la caveola a la SMasa ácida, o
la asociación de la parte interior de la misma a la SMasa ácida, parece ser responsable
de la generación de ceramida [264].

Discusión
174
Estudios recientes apuntan a la mitocondria como lugar de acción y generación de
ceramida [200,265,266]. En la mitocondria, la apoptosis inducida por ceramida puede
ser bloqueada por Bcl-2, indicando que la producción de ceramida está controlada a ese
nivel [200]. Curiosamente, BME induce apoptosis en células U937 que expresan niveles
elevados de Bcl-2. Estos resultados apoyan un papel crucial de la vía extrínseca en la
muerte celular inducida por BME en células con niveles marcadamente elevados de Bcl-
2. Sin embargo, podría ser también que BME fuera capaz de suprimir la protección
contra la apoptosis que confiere Bcl-2. En este sentido, diversos estudios han descrito la
inactivación de Bcl-2 a través de distintos mecanismos, entre los que se incluyen su
hidrólisis mediante la activación de caspasas y la hiperfosforilación [267,268]. Es
interesante resaltar que la ceramida puede ejercer sus efectos antes que la activación de
la vía JNK/SAPK, ya que esta vía se activa por esfingomielinasas exógenas, N-acetil-
ceramida y /o por el TNF-α [269].
Entre otros sistemas de señalización, el sn-1,2-diacilglicerol (DAG) desempeña un papel
anti-apoptótico y estimulador de la proliferación celular [270]. Estudios previos
demuestran que la fosfolipasa C de fosfatidilcolina (PC-PLC) modula la actividad de la
SMasa ácida a través de la generación de DAG [271] y que D609, un potente inhibidor
de la PC-PLC, inhibe selectivamente la activación SMasa ácida desencadenada por el
TNF-α en células U937 [226]. El inhibidor D609 también inhibe a la esfingomielina
sintasa, que participa en la regulación de los niveles de DAG y ceramida [272].
Además, el D609 aumenta los niveles de ceramida y mejora la capacidad de la
daunorubicina y mitoxantrona para producir apoptosis [272,273]. Se ha descrito que
DAG es también generado por la hidrólisis de fosfatidil inositol 4,5-bifosfato (PIP2) por
una fosfolipasa C específica. Sin embargo, la inhibición de las enzimas que intervienen
en la generación de DAG mediante el D609 y U73122 (inhibidor de fosfolipasa C de
PIP2) no tuvo ningún impacto en la muerte celular inducida por BME. Así, el
mecanismo de muerte celular provocada por BME parece ser independiente de la
generación de DAG por estas dos fosfolipasas C.

Discusión
175
Recientemente se ha demostrado que la SMasa ácida puede ser fosforilada por PKCδ y
que la fosforilación parece ser fundamental no solamente para la activación de la
enzima sino también para su translocación a la membrana plasmática [274]. Se
requieren más estudios para determinar el efecto de BME sobre la fosforilación de
SMasa ácida y la participación de PKCδ.
En resumen, el presente estudio proporciona evidencias acerca del mecanismo por el
cual BME induce muerte celular en células de leucemia humana y sugiere que éste
compuesto puede ser de utilidad en el futuro desarrollo de fármacos para tratamiento del
cáncer, ya sea como agente único o asociado a otros fármacos anticancerígenos.
Tetracetato de 3-metil éter quercetina (QD).
En la evaluación citotóxica de los 22 compuestos naturales y semi-sintéticos con
estructura de fenilbenzo-pirona, demostramos que la citotoxicidad de la quercetina se
aumentaba significativamente mediante la acetilación y la metilación de los grupos
hidroxilo libres. El 3-metil éter quercetina y el tetracetato de 3-metil éter de quercetina
(QD) presentan una potencia similar en células HL-60 con una IC50 de 14,3 μM y 9,2
μM, respectivamente. En consonancia con estos resultados los estudios de citometría de
flujo indican que QD y su precursor, el 3-metil éter quercetina, inducen apoptosis y
detención del ciclo celular en G2-M de una forma similar. Dado que los flavonoides son
compuestos fenólicos y, por tanto, susceptibles de oxidarse a quinonas, llevamos a cabo
la protección de los grupos 3', 4'-dihidroxi para generar un compuesto químicamente
más estable. Por lo tanto, los experimentos se llevaron a cabo con el tetracetato de 3-
metil éter quercetina (QD) en lugar del 3-metil éter de quercetina de origen natural.
También demostramos que QD presenta un efecto anti-proliferativo en una segunda
línea de células de la leucemia, U937, con un valor de IC50 similar al obtenido en la
línea celular HL-60. Dado que la selectividad hacia las células tumorales es un criterio
muy importante a la hora de buscar agentes para tratamiento del cáncer se estudió los
efectos de QD en linfocitos humanos obtenidos de sangre periférica (PBMCs). Los

Discusión
176
estudios dosis-respuesta revelaron que tanto las PBMC quiescentes como las
proliferantes son resistentes al tetracetato de 3-metil éter de quercetina.
En estudios anteriores se había demostrado que derivados de la estructura fenilbenzo-
pirona como wogonina y fisetina son capaces de inducir la muerte celular apoptótica en
la línea celular de leucemia mieloide humana HL-60 [236]. Estos flavonoides parecen
desencadenar la muerte celular por apoptosis a través de un mecanismo mediado por
caspasa-3 y asociado a una disminución de las especies reactivas de oxígeno (EROs).
Sin embargo, el mecanismo desencadenado por QD para disminuir el crecimiento de las
células en HL-60 y U937 no se ha evaluado hasta el momento. El análisis de la
distribución de las distintas fases del ciclo celular indica que QD promueve la detención
de las células en la fase G2-M y el aumento del porcentaje de células hipodiploides
(células apoptóticas).
Como se ha comentado anteriormente, muchos flavonoides parecen exhibir una
actividad anti-proliferativa a través de la unión a tubulina y por tanto ejercen su
actividad afectando a la actividad de los microtúbulos de la célula [257]. Uno de los
requisitos estructurales que encontramos habitualmente en las flavonas que
interaccionan con la tubulina es la presencia de un grupo metoxilo en C-3. Sin embargo,
esto no representa un requisito absoluto ya que la quercetina, que carece del grupo 3-
metoxilo, se une a la tubulina en el sitio de la colchicina, perturba la polimerización de
los microtúbulos e induce cambios conformacionales en el monómero [109]. Dado que
QD es un análogo de la quercetina y también contiene un grupo metoxilo en C-3, la
detención en G2-M inducida por el tetracetato de 3-metil éter de quercetina podría
explicarse por la inhibición de la formación de los microtúbulos. No obstante, los
cambios en la expresión y /o actividad de reguladores de G2-M del ciclo celular también
podrían estar involucrados.
Algunos aspectos celulares que caracterizan a la apoptosis como la fragmentación del
ADN, la formación de cuerpos apoptóticos y la hidrólisis de PARP [237] se distinguen
también en las células tratadas con QD.

Discusión
177
También se han llevado a cabo experimentos para comparar QD y quercetina. No hubo
evidencias de fragmentación de ADN o hidrólisis de PARP en las células HL-60
tratadas con quercetina a los tiempos y a las dosis ensayadas (hasta 10 μM, 24 h). Sin
embargo, QD y 3-metil éter quercetina inducen fragmentación del ADN e hidrólisis de
PARP a 3µM.
En un trabajo previo descrito por Nguyen et al. [253] se evaluaron las concentraciones
de quercetina y sus metabolitos en ratas. Las concentraciones fueron 25,1, 43,3 y 54,3
μM después de 10 días de tratamiento con 50, 100 y 150 microgramos de quercetina por
kg de peso corporal, respectivamente, lo que sugiere la posibilidad de que las
concentraciones de QD empleadas en el presente estudio se podrían alcanzar in vivo. En
este sentido, se han utilizado concentraciones 5 veces inferiores (10 μM frente a 54,3
μM) e incluso casi 20 veces inferiores (3 μM frente a 54,3 μM) a las empleadas por
Nguyen et al. Los estudios in vitro han demostrado que la quercetina se une a las
proteínas plasmáticas humanas y la albúmina es la principal proteína de unión. Sin
embargo, la sustitución del 3-OH en la quercetina debilita notablemente la unión a la
albúmina [275]. Esto sugiere que la introducción de un grupo metilo en C-3 podría
permitir una débil unión a la albúmina y, por tanto, aumentar su distribución a las
células. Estudios recientes han indicado también que la quercetina suministrada en
liposomas puede mejorar significativamente su solubilidad y biodisponibilidad de forma
que se podría mejorar la eficacia antitumoral [276]. Estos resultados podrían ser
aplicables a otros flavonoides y especialmente a QD.
Es interesante señalar que una concentración baja (3 μM), de QD fue suficiente para
inducir la liberación del citocromo c, la hidrólisis de las caspasas (caspasas-3, -6, -7, y -
9) y también la hidrólisis de PARP en células HL-60. Sin embargo, la hidrólisis de la
caspasa-8 no se detectó hasta 24 h después del tratamiento y sólo con la mayor
concentración utilizada (10 µM). Los resultados indican que la muerte celular inducida
por QD depende de la activación de caspasas dado que la apoptosis se inhibe
farmacologicamente por el inhibidor general de caspasas z-VAD-fmk.

Discusión
178
Aunque QD induce la perturbación del ciclo celular de forma similar en HL-60 y en
U937, es importante señalar que la línea celular U937 ha demostrado ser más resistente
a la apoptosis lo que pone de manifiesto la existencia de heterogeneidad en la respuesta
a QD. La inducción de la apoptosis por QD no implica la biosíntesis de proteínas y por
lo tanto, las células tumorales ya contienen el conjunto completo de las proteínas
necesarias para la apoptosis inducida por QD.
Es importante señalar que aunque la apoptosis inducida por QD fue suprimida por un
inhibidor de caspasa de amplio espectro (z-VAD-fmk), los inhibidores selectivos de
caspasa-8 (z-IETD-fmk), caspasa-3 (z-DEVD-fmk) y caspasa-9 (z-LEHD-fmk) fueron
incapaces de bloquear la muerte celular. En este trabajo hemos demostrado que la
apoptosis inducida por BME en células HL-60 es inhibida por z-IETD-fmk pero no por
z-LEHD-fmk, apoyando la idea de que la caspasa-8 desempeña un papel central en el
mecanismo de citotoxicidad. Por lo tanto, diferentes vías apoptóticas se activan en esta
línea celular en respuesta a las fenilbenzo-pironas QD y BME. Se puede descartar un
posible papel de Bid en la apoptosis inducida por QD, ya que requiere la activación
proteolítica de la caspasa-8 y la inhibición de esta caspasa con z-IETD-fmk no impide la
muerte celular.
Los datos demuestran que las caspasas-3 y -7 se activan en células HL-60 en respuesta a
QD. Sin embargo, la falta de efecto de z-DEVD-fmk, un inhibidor selectivo de las
caspasas-3 y -7, sobre la apoptosis inducida por QD indica que la muerte celular puede
ocurrir en ausencia de actividad de estas caspasas. Por tanto, es posible que otros
factores puedan compensar la falta de actividad de caspasa-3 y caspasa-7. Dado que z-
DEVD-fmk es un inhibidor más potente para la caspasa-3 que para la caspasa-7 [243],
es posible también que una actividad residual de esta última caspasa junto con la posible
participación de otra caspasa efectora como es la caspasa-6 contribuyan a la apoptosis
causada por QD.
La falta de efecto de z-LEHD-fmk sobre la muerte celular inducida por QD sugiere que
es posible que no esté involucrada en la muerte celular mediada por QD. No obstante,
este resultado es sorprendente dado que se produce proteólisis de la caspasa-9. Puesto

Discusión
179
que la activación de la caspasa-9 puede ocurrir sin procesamiento proteolítico [277,278]
también se realizaron estudios cinéticos. Los resultados demuestran claramente que la
caspasa-9 se activa por QD, lo que sugiere que la apoptosis inducida por este compuesto
está relacionada con el citocromo c, pero no con un mecanismo mediado por receptores.
La mayor protección ofrecida por z-VAD-fmk respecto a z-LEHD-fmk es debida
probablemente a su mejor accesibilidad a los espacios intracelulares donde se encuentra
la caspasa-9 [279]. Aunque a menudo es muy difícil determinar el orden de activación
de las caspasas, los estudios cinéticos indican que la inducción de las actividades de
caspasa-9 y -3 aumentaron significativamente a las 12-14 horas de tratamiento. En
contraste, el aumento de la actividad caspasa-8 no fue detectable hasta las 24 horas de
tratamiento.
Como se mencionó anteriormente el aumento de expresión de Bcl-2 se ha asociado con
quimioresistencia, especialmente en el caso de las neoplasias hematológicas [280]. Esto
determina que la búsqueda y el desarrollo de compuestos capaces de inducir la muerte
celular en células tumorales que expresan concentraciones elevadas de Bcl-2 sean de
gran importancia terapéutica. Los presentes resultados sugieren que el tratamiento con
QD induce muerte celular en células leucémicas que expresan altos niveles de Bcl-2. En
este sentido, se ha demostrado que el flavopiridol induce daño mitocondrial en diversas
células neoplásicas, incluídas las que presentan niveles de expresión de Bcl-2 elevados
[281]. Así pues, aunque se podría especular que la activación de la vía extrínseca puede
ser responsable de la muerte celular en células que presentan altas concentraciones de
Bcl-2, otra posibilidad podría ser que QD fuera capaz de neutralizar la resistencia al
daño mitocondrial y a la liberación de citocromo c que confiere este factor.
Las MAPKs son parte fundamental de la maquinaria de transducción de señales y
desempeñan un papel fundamental en el crecimiento celular, la diferenciación y en la
muerte celular. Así, JNK/SAPK y p38MAPK en general han sido relacionadas con
acciones pro-apoptóticas, mientras que ERK 1/2 en la mayoría de los casos ejerce
efectos citoprotectores [282]. La acción de los inhibidores de ERK 1/2 potencia los

Discusión
180
efectos antitumorales de los agentes citotóxicos como el 1-β-D-arabino-furanosil-
citosina (ara-C) [283].
La activación de la vía JNK /SAPK puede desencadenarse por señales de estrés en el
retículo endoplasmático en las células Jurkat, causando liberación del citocromo c y
activación de la caspasa-3 [284]. Aunque la mayoría de los estudios apoyan la idea de
que la vía JNK / SAPK contribuye a la apoptosis inducida por el estrés, su efecto parece
depender del tipo celular y del conjunto de otras señales recibidas por la célula [284].
En este sentido, QD induce activación de JNK/SAPK en células HL-60 y U937, aunque
el inhibidor selectivo, SP600125, es incapaz de suprimir su efecto sobre la apoptosis.
Este resultado sugiere que la vía JNK/SAPK no está involucrada en la muerte celular
inducida por QD.
La vía de señalización p38MAPK se ha relacionado no sólo con la muerte celular en
algunas líneas celulares, sino también con la supervivencia, el crecimiento y la
diferenciación celular. Por lo tanto, el papel de la p38MAPK depende del tipo celular del
que se trate y del tipo de estímulo [285]. En el presente estudio la fosforilación de
p38MAPK se produjo antes (30 min.) que la activación de las caspasas. Sin embargo, la
inhibición de la vía por SB203580 no atenuó la apoptosis inducida por QD lo que
sugiere que la vía p38MAPK no está implicada. La vía p38MAPK está relacionada con la
inducción de apoptosis en una gran variedad de tipos celulares como en U937 por el
cadmio [286] y células B malignas humanas por el resveratrol [287]. Sin embargo, su
activación no está implicada en la inducción de apoptosis en células U937 tratadas con
radiación UV [288] y en células Jurkat T tratadas con Fas y/o UV [289].
Diversos estudios han demostrado que la quercetina induce activación sostenida de
ERK 1/2 en células epiteliales de cáncer de pulmón A549 [253] y que la inhibición de
la activación de esta vía con U0126 o PD98059 suprime la apoptosis [253]. El
tratamiento del carcinoma de células de ovario [290,291] y de la línea celular de
melanoma C8161 [292] con cisplatino induce la activación de ERK 1/2 y muerte
celular. Este último efecto fue potenciado por el uso de los inhibidores de ERK 1/2 lo

Discusión
181
que indica que estas enzimas se comportan como quinasas de supervivencia en estas
células.
Aunque otros autores han demostrado que el aumento de la producción de EROs en
células leucémicas conduce a la activación de MAPKs y a la muerte celular [293-295],
esto no ha podido ser demostrado con QD. Así, el mecanismo de la muerte celular
provocada por QD parece ser diferente al de wogonina y fisetina que implica una
disminución de EROs [236].
En resumen, aunque QD induce apoptosis también aumenta la activación de la vía ERK
1/2 de modo que un tratamiento hipotético con este compuesto podría verse
adversamente afectado debido a un aumento de la proliferación celular y la
supervivencia. Sin embargo, el uso de inhibidores específicos de ERK 1/2 como
PD98059 y U0126 ha demostrado poseer un valor farmacológico adicional ya que
potencian significativamente el efecto citotóxico de QD. El uso de bajas dosis en
quimioterapia es importante, ya que son más fáciles de alcanzar durante el tratamiento y
menos tóxicas para los pacientes. Estos resultados tienen importantes implicaciones
clínicas para el uso de QD en combinación con los inhibidores de ERK 1/2 como
posibles agentes terapéuticos.


CCCOOONNNCCCLLLUUUSSSIIIOOONNNEEESSS


Conclusiones
185
1. Se ha evaluado la actividad antitumoral de 22 compuestos en cinco líneas
tumorales humanas, y se han seleccionado los más citotóxicos (valores de IC50
menores). La línea celular promielocítica HL- 60, fue, en general la que mostró
mayor sensibilidad a los efectos citotóxicos de estos compuestos.
2. El 3-metil éter de betuletol y su derivado diacetilado así como el tetraacetato de
3-metil éter de quercetina exhiben propiedades citotóxicas selectivas frente a
células tumorales a concentraciones que se podrían alcanzar in vivo.
3. Estos compuestos, 3-metil éter de betuletol y tetraacetato de 3-metil éter de
quercetina, inducen la detención del ciclo celular en la fase G2-M y apoptosis a
través de un mecanismo independiente de p53, en las líneas celulares de
leucemia mieloide humana HL-60 y U937.
4. El 3-metil éter de betuletol (BME) inhibe la polimerización de la tubulina y
promueve cambios en la red de microtúbulos siguiendo un patrón similar al de
la colchicina e induce la expresión y fosforilación de ciclina B1, así como la
acumulación de p21Cip1 y la activación de cdk1.
5. La apoptosis inducida por el 3-metil éter de betuletol y el tetraacetato de 3-
metil éter de quercetina está asociado con la activación de múltiples caspasas,
la liberación mitocondrial del citocromo c, la hidrólisis de PARP y la
fragmentación del ADN.
6. El 3-metil éter de betuletol induce apoptosis por un mecanismo mediado por
caspasa-8 caracterizado por activación de la SMasa ácida, la generación de
ceramida, la hidrólisis de Bid y la activación de las MAPKs.

Conclusiones
186
7. El tetraacetato de 3-metil éter de quercetina induce hidrólisis de las pro-
caspasas-8, -9, -3 y -7 aunque éstas no parecen ser totalmente responsables de
la muerte celular.
8. El tetraacetato de 3-metil éter de quercetina induce fosforilación y activación
de ERK 1/2, p38MAPK y JNK/SAPK. El tratamiento combinado de QD y los
inhibidores de ERK 1/2 potencia la muerte celular.
9. La protección de las mitocondrias por el aumento de expresión de proteínas
Bcl-2 (Bcl-2 y Bcl-xL) no impide la apoptosis inducida por los derivados del
betuletol y por el tetraacetato de 3-metil éter de quercetina.
Los datos del presente estudio proporcionan evidencias acerca del mecanismo por el que
el 3-metil éter de betuletol, el diacetato de 3-metil eter de betuletol y el tetraacetato de
3-metil éter de quercetina inhiben el crecimiento de las células tumorales mieloides
humanas e inducen apoptosis y sugieren que estos compuestos podrían ser útiles en el
desarrollo de nuevos fármacos antitumorales.

BBBIIIBBBLLLIIIOOOGGGRRRAAAFFFÍÍÍAAA


Bibliografía
189
[1] OMS (Enero de 2009.) Cáncer. Nota descriptiva N°297. [2] Hanahan, D. and Weinberg, R.A. (2000) The hallmarks of cancer. Cell 100,
57-70. [3] Kuntz, S., Wenzel, U. and Daniel, H. (1999) Comparative analysis of the
effects of flavonoids on proliferation, cytotoxicity, and apoptosis in human colon cancer cell lines. Eur J Nutr 38, 133-42.
[4] Cragg, G.M., Newman, D.J. and Snader, K.M. (1997) Natural products in drug discovery and development. J Nat Prod 60, 52-60.
[5] Stahelin, H. (1973) Activity of a new glycosidic lignan derivative (VP 16-213) related to podophyllotoxin in experimental tumors. Eur J Cancer 9, 215-21.
[6] Williams, S.D., Birch, R., Einhorn, L.H., Irwin, L., Greco, F.A. and Loehrer, P.J. (1987) Treatment of disseminated germ-cell tumors with cisplatin, bleomycin, and either vinblastine or etoposide. N Engl J Med 316, 1435-40.
[7] Harvey, A.L. (1999) Medicines from nature: are natural products still relevant to drug discovery? Trends Pharmacol Sci 20, 196-8.
[8] Noble, R.L. (1990) The discovery of the vinca alkaloids--chemotherapeutic agents against cancer. Biochem Cell Biol 68, 1344-51.
[9] Devita, V.T., Jr., Serpick, A.A. and Carbone, P.P. (1970) Combination chemotherapy in the treatment of advanced Hodgkin's disease. Ann Intern Med 73, 881-95.
[10] Wani, M.C., Taylor, H.L., Wall, M.E., Coggon, P. and McPhail, A.T. (1971) Plant antitumor agents. VI. The isolation and structure of taxol, a novel antileukemic and antitumor agent from Taxus brevifolia. J Am Chem Soc 93, 2325-7.
[11] Liu, L.F., Desai, S.D., Li, T.K., Mao, Y., Sun, M. and Sim, S.P. (2000) Mechanism of action of camptothecin. Ann N Y Acad Sci 922, 1-10.
[12] Winkel-Shirley, B. (2001) Flavonoid biosynthesis. A colorful model for genetics, biochemistry, cell biology, and biotechnology. Plant Physiol 126, 485-93.
[13] Middleton, E.M. and Teramura, A.H. (1993) The Role of Flavonol Glycosides and Carotenoids in Protecting Soybean from Ultraviolet-B Damage. Plant Physiol 103, 741-752.
[14] Hess, S., Alvarez, L., Iturra, G., Romero, M. (2002) Evidence of UV B differential response in Sophora microphylla from shady and sunny places. . Soc. Chil. Quim. 47, 501-510.
[15] Taiz, L., Zeiger, E. (2002) Plant physiology, Sunderland. Sinaver Associates. , 690 p. 3ª edición
[16] Brown, D.E., Rashotte, A.M., Murphy, A.S., Normanly, J., Tague, B.W., Peer,
W.A., Taiz, L. and Muday, G.K. (2001) Flavonoids act as negative regulators of auxin transport in vivo in arabidopsis. Plant Physiol 126, 524-35.
[17] Hertog, M.G., Kromhout, D., Aravanis, C., Blackburn, H., Buzina, R., Fidanza, F., Giampaoli, S., Jansen, A., Menotti, A., Nedeljkovic, S. and et al. (1995) Flavonoid intake and long-term risk of coronary heart disease and cancer in the seven countries study. Arch Intern Med 155, 381-6.

Bibliografía
190
[18] Yochum, L., Kushi, L.H., Meyer, K. and Folsom, A.R. (1999) Dietary flavonoid intake and risk of cardiovascular disease in postmenopausal women. Am J Epidemiol 149, 943-9.
[19] Garcia-Alonso, M., Rimbach, G., Rivas-Gonzalo, J.C. and De Pascual-Teresa, S. (2004) Antioxidant and cellular activities of anthocyanins and their corresponding vitisins A--studies in platelets, monocytes, and human endothelial cells. J Agric Food Chem 52, 3378-84.
[20] Bergan, J.J., Schmid-Schonbein, G.W. and Takase, S. (2001) Therapeutic approach to chronic venous insufficiency and its complications: place of Daflon 500 mg. Angiology 52 Suppl 1, S43-7.
[21] Labrid, C. (1994) Pharmacologic properties of Daflon 500 mg. Angiology 45, 524-30.
[22] Selway, J.W. (1986) Antiviral activity of flavones and flavans. Prog Clin Biol Res 213, 521-36.
[23] Hocman, G. (1989) Prevention of cancer: vegetables and plants. Comp Biochem Physiol B 93, 201-12.
[24] Plumb, G.W., De Pascual-Teresa, S., Santos-Buelga, C., Cheynier, V. and Williamson, G. (1998) Antioxidant properties of catechins and proanthocyanidins: effect of polymerisation, galloylation and glycosylation. Free Radic Res 29, 351-8.
[25] Williams, C.A. and Grayer, R.J. (2004) Anthocyanins and other flavonoids. Nat Prod Rep 21, 539-73.
[26] Hollman, P.C., Hertog, M.G. and Katan, M.B. (1996) Role of dietary flavonoids in protection against cancer and coronary heart disease. Biochem Soc Trans 24, 785-9.
[27] Rimm, E.B., Katan, M.B., Ascherio, A., Stampfer, M.J. and Willett, W.C. (1996) Relation between intake of flavonoids and risk for coronary heart disease in male health professionals. Ann Intern Med 125, 384-9.
[28] Hollman, P.C. and Katan, M.B. (1998) Bioavailability and health effects of dietary flavonols in man. Arch Toxicol Suppl 20, 237-48.
[29] Knekt, P., Jarvinen, R., Reunanen, A. and Maatela, J. (1996) Flavonoid intake and coronary mortality in Finland: a cohort study. Bmj 312, 478-81.
[30] Rowland, M. (1986) Scheele lecture. Pharmacokinetics: concepts, insights and applications. Acta Pharm Suec 23, 173-90.
[31] Shi, Y.Q., Fukai, T., Sakagami, H., Chang, W.J., Yang, P.Q., Wang, F.P. and Nomura, T. (2001) Cytotoxic flavonoids with isoprenoid groups from Morus mongolica. J Nat Prod 64, 181-8.
[32] Sakagami, H., Jiang, Y., Kusama, K., Atsumi, T., Ueha, T., Toguchi, M., Iwakura, I., Satoh, K., Fukai, T. and Nomura, T. (2000) Induction of apoptosis by flavones, flavonols (3-hydroxyflavones) and isoprenoid-substituted flavonoids in human oral tumor cell lines. Anticancer Res 20, 271-7.
[33] Fukai, T., Sakagami, H., Toguchi, M., Takayama, F., Iwakura, I., Atsumi, T., Ueha, T., Nakashima, H. and Nomura, T. (2000) Cytotoxic activity of low molecular weight polyphenols against human oral tumor cell lines. Anticancer Res 20, 2525-36.

Bibliografía
191
[34] Elattar, T.M. and Virji, A.S. (2000) Effect of tea polyphenols on growth of oral squamous carcinoma cells in vitro. Anticancer Res 20, 3459-65.
[35] Pouget, C., Lauthier, F., Simon, A., Fagnere, C., Basly, J.P., Delage, C. and Chulia, A.J. (2001) Flavonoids: structural requirements for antiproliferative activity on breast cancer cells. Bioorg Med Chem Lett 11, 3095-7.
[36] Elattar, T.M. and Virji, A.S. (2000) The inhibitory effect of curcumin, genistein, quercetin and cisplatin on the growth of oral cancer cells in vitro. Anticancer Res 20, 1733-8.
[37] Yin, F., Giuliano, A.E. and Van Herle, A.J. (1999) Growth inhibitory effects of flavonoids in human thyroid cancer cell lines. Thyroid 9, 369-76.
[38] Yin, F., Giuliano, A.E. and Van Herle, A.J. (1999) Signal pathways involved in apigenin inhibition of growth and induction of apoptosis of human anaplastic thyroid cancer cells (ARO). Anticancer Res 19, 4297-303.
[39] Kuo, S.M., Morehouse, H.F., Jr. and Lin, C.P. (1997) Effect of antiproliferative flavonoids on ascorbic acid accumulation in human colon adenocarcinoma cells. Cancer Lett 116, 131-7.
[40] Caltagirone, S., Ranelletti, F.O., Rinelli, A., Maggiano, N., Colasante, A., Musiani, P., Aiello, F.B. and Piantelli, M. (1997) Interaction with type II estrogen binding sites and antiproliferative activity of tamoxifen and quercetin in human non-small-cell lung cancer. Am J Respir Cell Mol Biol 17, 51-9.
[41] Bai, F., Matsui, T., Ohtani-Fujita, N., Matsukawa, Y., Ding, Y. and Sakai, T. (1998) Promoter activation and following induction of the p21/WAF1 gene by flavone is involved in G1 phase arrest in A549 lung adenocarcinoma cells. FEBS Lett 437, 61-4.
[42] Kampa, M., Hatzoglou, A., Notas, G., Damianaki, A., Bakogeorgou, E., Gemetzi, C., Kouroumalis, E., Martin, P.M. and Castanas, E. (2000) Wine antioxidant polyphenols inhibit the proliferation of human prostate cancer cell lines. Nutr Cancer 37, 223-33.
[43] Bhatia, N. and Agarwal, R. (2001) Detrimental effect of cancer preventive phytochemicals silymarin, genistein and epigallocatechin 3-gallate on epigenetic events in human prostate carcinoma DU145 cells. Prostate 46, 98-107.
[44] Agarwal, G., Bhatia, V., Cook, S. and Thomas, P.Q. (2000) Adrenocorticotropin deficiency in combined pituitary hormone deficiency patients homozygous for a novel PROP1 deletion. J Clin Endocrinol Metab 85, 4556-61.
[45] Wenzel, U., Kuntz, S., Brendel, M.D. and Daniel, H. (2000) Dietary flavone is a potent apoptosis inducer in human colon carcinoma cells. Cancer Res 60, 3823-31.
[46] Kamei, H., Hashimoto, Y., Koide, T., Kojima, T. and Hasegawa, M. (1998) Anti-tumor effect of methanol extracts from red and white wines. Cancer Biother Radiopharm 13, 447-52.
[47] Wang, I.K., Lin-Shiau, S.Y. and Lin, J.K. (1999) Induction of apoptosis by apigenin and related flavonoids through cytochrome c release and activation of caspase-9 and caspase-3 in leukaemia HL-60 cells. Eur J Cancer 35, 1517-25.

Bibliografía
192
[48] De Vincenzo, R., Ferlini, C., Distefano, M., Gaggini, C., Riva, A., Bombardelli, E., Morazzoni, P., Valenti, P., Belluti, F., Ranelletti, F.O., Mancuso, S. and Scambia, G. (2000) In vitro evaluation of newly developed chalcone analogues in human cancer cells. Cancer Chemother Pharmacol 46, 305-12.
[49] Csokay, B., Prajda, N., Weber, G. and Olah, E. (1997) Molecular mechanisms in the antiproliferative action of quercetin. Life Sci 60, 2157-63.
[50] Chung, H.S., Chang, L.C., Lee, S.K., Shamon, L.A., van Breemen, R.B., Mehta, R.G., Farnsworth, N.R., Pezzuto, J.M. and Kinghorn, A.D. (1999) Flavonoid constituents of Chorizanthe diffusa with potential cancer chemopreventive activity. J Agric Food Chem 47, 36-41.
[51] Conney, A.H. (2003) Enzyme induction and dietary chemicals as approaches to cancer chemoprevention: the Seventh DeWitt S. Goodman Lecture. Cancer Res 63, 7005-31.
[52] Middleton, E., Jr., Kandaswami, C. and Theoharides, T.C. (2000) The effects of plant flavonoids on mammalian cells: implications for inflammation, heart disease, and cancer. Pharmacol Rev 52, 673-751.
[53] Alia, M., Mateos, R., Ramos, S., Lecumberri, E., Bravo, L. and Goya, L. (2006) Influence of quercetin and rutin on growth and antioxidant defense system of a human hepatoma cell line (HepG2). Eur J Nutr 45, 19-28.
[54] Watson, W.H., Cai, J. and Jones, D.P. (2000) Diet and apoptosis. Annu Rev Nutr 20, 485-505.
[55] Russo, A., Acquaviva, R., Campisi, A., Sorrenti, V., Di Giacomo, C., Virgata, G., Barcellona, M.L. and Vanella, A. (2000) Bioflavonoids as antiradicals, antioxidants and DNA cleavage protectors. Cell Biol Toxicol 16, 91-8.
[56] Bravo, L. (1998) Polyphenols: chemistry, dietary sources, metabolism, and nutritional significance. Nutr Rev 56, 317-33.
[57] Bohm, H., Boeing, H., Hempel, J., Raab, B. and Kroke, A. (1998) [Flavonols, flavone and anthocyanins as natural antioxidants of food and their possible role in the prevention of chronic diseases]. Z Ernahrungswiss 37, 147-63.
[58] Kostiuk, V.A., Potapovich, A.I., Tereshchenko, S.M. and Afanas'ev, I.B. (1988) [Antioxidative activity of flavonoids in various systems of lipid peroxidation]. Biokhimiia 53, 1365-70.
[59] Ferrandiz, M.L. and Alcaraz, M.J. (1991) Anti-inflammatory activity and inhibition of arachidonic acid metabolism by flavonoids. Agents Actions 32, 283-8.
[60] de Groot, H. and Rauen, U. (1998) Tissue injury by reactive oxygen species and the protective effects of flavonoids. Fundam Clin Pharmacol 12, 249-55.
[61] Lindahl, M. and Tagesson, C. (1997) Flavonoids as phospholipase A2 inhibitors: importance of their structure for selective inhibition of group II phospholipase A2. Inflammation 21, 347-56.
[62] Ratty, A.K. and Das, N.P. (1988) Effects of flavonoids on nonenzymatic lipid peroxidation: structure-activity relationship. Biochem Med Metab Biol 39, 69-79.

Bibliografía
193
[63] Yoshino, M. and Murakami, K. (1998) Interaction of iron with polyphenolic compounds: application to antioxidant characterization. Anal Biochem 257, 40-4.
[64] Pietrangelo, A., Borella, F., Casalgrandi, G., Montosi, G., Ceccarelli, D., Gallesi, D., Giovannini, F., Gasparetto, A. and Masini, A. (1995) Antioxidant activity of silybin in vivo during long-term iron overload in rats. Gastroenterology 109, 1941-9.
[65] Ferry, D.R., Smith, A., Malkhandi, J., Fyfe, D.W., deTakats, P.G., Anderson, D., Baker, J. and Kerr, D.J. (1996) Phase I clinical trial of the flavonoid quercetin: pharmacokinetics and evidence for in vivo tyrosine kinase inhibition. Clin Cancer Res 2, 659-68.
[66] Markovits, J., Linassier, C., Fosse, P., Couprie, J., Pierre, J., Jacquemin-Sablon, A., Saucier, J.M., Le Pecq, J.B. and Larsen, A.K. (1989) Inhibitory effects of the tyrosine kinase inhibitor genistein on mammalian DNA topoisomerase II. Cancer Res 49, 5111-7.
[67] Lin, J.K. (2004) Suppression of protein kinase C and nuclear oncogene expression as possible action mechanisms of cancer chemoprevention by Curcumin. Arch Pharm Res 27, 683-92.
[68] Lin, J.K., Chen, Y.C., Huang, Y.T. and Lin-Shiau, S.Y. (1997) Suppression of protein kinase C and nuclear oncogene expression as possible molecular mechanisms of cancer chemoprevention by apigenin and curcumin. J Cell Biochem Suppl 28-29, 39-48.
[69] Surette, M.E., Fonteh, A.N., Bernatchez, C. and Chilton, F.H. (1999) Perturbations in the control of cellular arachidonic acid levels block cell growth and induce apoptosis in HL-60 cells. Carcinogenesis 20, 757-63.
[70] DeWolf, W.C. and Gaston, S.M. (2004) The cell cycle and its relevance to the urologist. J Urol 171, 1674-81.
[71] Sherr, C.J. and Roberts, J.M. (1999) CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev 13, 1501-12.
[72] Malumbres, M. and Barbacid, M. (2001) To cycle or not to cycle: a critical decision in cancer. Nat Rev Cancer 1, 222-31.
[73] Sherr, C.J. (1994) G1 phase progression: cycling on cue. Cell 79, 551-5. [74] Ohtsubo, M., Theodoras, A.M., Schumacher, J., Roberts, J.M. and Pagano, M.
(1995) Human cyclin E, a nuclear protein essential for the G1-to-S phase transition. Mol Cell Biol 15, 2612-24.
[75] Girard, F., Strausfeld, U., Fernandez, A. and Lamb, N.J. (1991) Cyclin A is required for the onset of DNA replication in mammalian fibroblasts. Cell 67, 1169-79.
[76] Walker, D.H. and Maller, J.L. (1991) Role for cyclin A in the dependence of mitosis on completion of DNA replication. Nature 354, 314-7.
[77] Arellano, M. and Moreno, S. (1997) Regulation of CDK/cyclin complexes during the cell cycle. Int J Biochem Cell Biol 29, 559-73.
[78] King, R.W., Jackson, P.K. and Kirschner, M.W. (1994) Mitosis in transition. Cell 79, 563-71.
[79] Carnero, A. and Hannon, G.J. (1998) The INK4 family of CDK inhibitors. Curr Top Microbiol Immunol 227, 43-55.

Bibliografía
194
[80] Pavletich, N.P. (1999) Mechanisms of cyclin-dependent kinase regulation: structures of Cdks, their cyclin activators, and Cip and INK4 inhibitors. J Mol Biol 287, 821-8.
[81] Harper, J.W., Elledge, S.J., Keyomarsi, K., Dynlacht, B., Tsai, L.H., Zhang, P., Dobrowolski, S., Bai, C., Connell-Crowley, L., Swindell, E. and et al. (1995) Inhibition of cyclin-dependent kinases by p21. Mol Biol Cell 6, 387-400.
[82] Roberts, J.M., Koff, A., Polyak, K., Firpo, E., Collins, S., Ohtsubo, M. and Massague, J. (1994) Cyclins, Cdks, and cyclin kinase inhibitors. Cold Spring Harb Symp Quant Biol 59, 31-8.
[83] Iida, H., Towatari, M., Tanimoto, M., Morishita, Y., Kodera, Y. and Saito, H. (1997) Overexpression of cyclin E in acute myelogenous leukemia. Blood 90, 3707-13.
[84] Scuderi, R., Palucka, K.A., Pokrovskaja, K., Bjorkholm, M., Wiman, K.G. and Pisa, P. (1996) Cyclin E overexpression in relapsed adult acute lymphoblastic leukemias of B-cell lineage. Blood 87, 3360-7.
[85] Zi, X., Feyes, D.K. and Agarwal, R. (1998) Anticarcinogenic effect of a flavonoid antioxidant, silymarin, in human breast cancer cells MDA-MB 468: induction of G1 arrest through an increase in Cip1/p21 concomitant with a decrease in kinase activity of cyclin-dependent kinases and associated cyclins. Clin Cancer Res 4, 1055-64.
[86] Choi, J.A., Kim, J.Y., Lee, J.Y., Kang, C.M., Kwon, H.J., Yoo, Y.D., Kim, T.W., Lee, Y.S. and Lee, S.J. (2001) Induction of cell cycle arrest and apoptosis in human breast cancer cells by quercetin. Int J Oncol 19, 837-44.
[87] Kim, S.Y., Gao, J.J. and Kang, H.K. (2000) Two flavonoids from the leaves of Morus alba induce differentiation of the human promyelocytic leukemia (HL-60) cell line. Biol Pharm Bull 23, 451-5.
[88] Mata-Greenwood, E., Ito, A., Westenburg, H., Cui, B., Mehta, R.G., Kinghorn, A.D. and Pezzuto, J.M. (2001) Discovery of novel inducers of cellular differentiation using HL-60 promyelocytic cells. Anticancer Res 21, 1763-70.
[89] Margolis, R.L. and Wilson, L. (1977) Addition of colchicine--tubulin complex to microtubule ends: the mechanism of substoichiometric colchicine poisoning. Proc Natl Acad Sci U S A 74, 3466-70.
[90] Himes, R.H., Kersey, R.N., Heller-Bettinger, I. and Samson, F.E. (1976) Action of the vinca alkaloids vincristine, vinblastine, and desacetyl vinblastine amide on microtubules in vitro. Cancer Res 36, 3798-802.
[91] Rowinsky, E.K. and Donehower, R.C. (1995) Paclitaxel (taxol). N Engl J Med 332, 1004-14.
[92] Cortes, J.E. and Pazdur, R. (1995) Docetaxel. J Clin Oncol 13, 2643-55. [93] Mollinedo, F. and Gajate, C. (2003) Microtubules, microtubule-interfering
agents and apoptosis. Apoptosis 8, 413-50. [94] Haldar, S., Jena, N. and Croce, C.M. (1995) Inactivation of Bcl-2 by
phosphorylation. Proc Natl Acad Sci U S A 92, 4507-11. [95] Tang, C., Willingham, M.C., Reed, J.C., Miyashita, T., Ray, S., Ponnathpur,
V., Huang, Y., Mahoney, M.E., Bullock, G. and Bhalla, K. (1994) High levels of p26BCL-2 oncoprotein retard taxol-induced apoptosis in human pre-B leukemia cells. Leukemia 8, 1960-9.

Bibliografía
195
[96] Gajate, C., Barasoain, I., Andreu, J.M. and Mollinedo, F. (2000) Induction of apoptosis in leukemic cells by the reversible microtubule-disrupting agent 2-methoxy-5-(2',3',4'-trimethoxyphenyl)-2,4,6-cycloheptatrien-1 -one: protection by Bcl-2 and Bcl-X(L) and cell cycle arrest. Cancer Res 60, 2651-9.
[97] Shankar, S., Chen, X. and Srivastava, R.K. (2005) Effects of sequential treatments with chemotherapeutic drugs followed by TRAIL on prostate cancer in vitro and in vivo. Prostate 62, 165-86.
[98] Manthey, C.L., Brandes, M.E., Perera, P.Y. and Vogel, S.N. (1992) Taxol increases steady-state levels of lipopolysaccharide-inducible genes and protein-tyrosine phosphorylation in murine macrophages. J Immunol 149, 2459-65.
[99] Koistinaho, J., Hicks, K.J. and Sagar, S.M. (1993) Long-term induction of c-jun mRNA and Jun protein in rabbit retinal ganglion cells following axotomy or colchicine treatment. J Neurosci Res 34, 250-5.
[100] Shibuya, E.K. and Ruderman, J.V. (1993) Mos induces the in vitro activation of mitogen-activated protein kinases in lysates of frog oocytes and mammalian somatic cells. Mol Biol Cell 4, 781-90.
[101] Blagosklonny, M.V., Schulte, T.W., Nguyen, P., Mimnaugh, E.G., Trepel, J. and Neckers, L. (1995) Taxol induction of p21WAF1 and p53 requires c-raf-1. Cancer Res 55, 4623-6.
[102] Liu, Q.Y. and Stein, C.A. (1997) Taxol and estramustine-induced modulation of human prostate cancer cell apoptosis via alteration in bcl-xL and bak expression. Clin Cancer Res 3, 2039-46.
[103] Townsend, K.J., Trusty, J.L., Traupman, M.A., Eastman, A. and Craig, R.W. (1998) Expression of the antiapoptotic MCL1 gene product is regulated by a mitogen activated protein kinase-mediated pathway triggered through microtubule disruption and protein kinase C. Oncogene 17, 1223-34.
[104] Chadebech, P., Truchet, I., Brichese, L. and Valette, A. (2000) Up-regulation of cdc2 protein during paclitaxel-induced apoptosis. Int J Cancer 87, 779-86.
[105] Subbaramaiah, K., Hart, J.C., Norton, L. and Dannenberg, A.J. (2000) Microtubule-interfering agents stimulate the transcription of cyclooxygenase-2. Evidence for involvement of ERK1/2 AND p38 mitogen-activated protein kinase pathways. J Biol Chem 275, 14838-45.
[106] Li, F., Ambrosini, G., Chu, E.Y., Plescia, J., Tognin, S., Marchisio, P.C. and Altieri, D.C. (1998) Control of apoptosis and mitotic spindle checkpoint by survivin. Nature 396, 580-4.
[107] Huisman, C., Ferreira, C.G., Broker, L.E., Rodriguez, J.A., Smit, E.F., Postmus, P.E., Kruyt, F.A. and Giaccone, G. (2002) Paclitaxel triggers cell death primarily via caspase-independent routes in the non-small cell lung cancer cell line NCI-H460. Clin Cancer Res 8, 596-606.
[108] Park, S.J., Wu, C.H., Gordon, J.D., Zhong, X., Emami, A. and Safa, A.R. (2004) Taxol induces caspase-10-dependent apoptosis. J Biol Chem 279, 51057-67.
[109] Gupta, K. and Panda, D. (2002) Perturbation of microtubule polymerization by quercetin through tubulin binding: a novel mechanism of its antiproliferative activity. Biochemistry 41, 13029-38.

Bibliografía
196
[110] Zhen, M.C., Huang, X.H., Wang, Q., Sun, K., Liu, Y.J., Li, W., Zhang, L.J., Cao, L.Q. and Chen, X.L. (2006) Green tea polyphenol epigallocatechin-3-gallate suppresses rat hepatic stellate cell invasion by inhibition of MMP-2 expression and its activation. Acta Pharmacol Sin 27, 1600-7.
[111] Kerr, J.F., Wyllie, A.H. and Currie, A.R. (1972) Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer 26, 239-57.
[112] Jacobson, M.D., Weil, M. and Raff, M.C. (1997) Programmed cell death in animal development. Cell 88, 347-54.
[113] Wyllie, A.H. (1980) Glucocorticoid-induced thymocyte apoptosis is associated with endogenous endonuclease activation. Nature 284, 555-6.
[114] Lopez-Hoyos, M., Carrio, R., Merino, J. and Merino, R. (1998) Regulation of B cell apoptosis by Bcl-2 and Bcl-XL and its role in the development of autoimmune diseases (Review). Int J Mol Med 1, 475-83.
[115] Thatte, U. and Dahanukar, S. (1997) Apoptosis: clinical relevance and pharmacological manipulation. Drugs 54, 511-32.
[116] Ashkenazi, A. and Dixit, V.M. (1998) Death receptors: signaling and modulation. Science 281, 1305-8.
[117] Nagata, S. (1997) Apoptosis by death factor. Cell 88, 355-65. [118] Krammer, P.H. (1998) The CD95(APO-1/Fas)/CD95L system. Toxicol Lett
102-103, 131-7. [119] Schmitz, I., Kirchhoff, S. and Krammer, P.H. (2000) Regulation of death
receptor-mediated apoptosis pathways. Int J Biochem Cell Biol 32, 1123-36. [120] Vincenz, C. (2001) Death receptors and apoptosis. Deadly signaling and
evasive tactics. Cardiol Clin 19, 31-43. [121] Thorburn, A. (2004) Death receptor-induced cell killing. Cell Signal 16, 139-
44. [122] Scaffidi, C., Fulda, S., Srinivasan, A., Friesen, C., Li, F., Tomaselli, K.J.,
Debatin, K.M., Krammer, P.H. and Peter, M.E. (1998) Two CD95 (APO-1/Fas) signaling pathways. Embo J 17, 1675-87.
[123] Luo, X., Budihardjo, I., Zou, H., Slaughter, C. and Wang, X. (1998) Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell 94, 481-90.
[124] Acehan, D., Jiang, X., Morgan, D.G., Heuser, J.E., Wang, X. and Akey, C.W. (2002) Three-dimensional structure of the apoptosome: implications for assembly, procaspase-9 binding, and activation. Mol Cell 9, 423-32.
[125] Slee, E.A., Harte, M.T., Kluck, R.M., Wolf, B.B., Casiano, C.A., Newmeyer, D.D., Wang, H.G., Reed, J.C., Nicholson, D.W., Alnemri, E.S., Green, D.R. and Martin, S.J. (1999) Ordering the cytochrome c-initiated caspase cascade: hierarchical activation of caspases-2, -3, -6, -7, -8, and -10 in a caspase-9-dependent manner. J Cell Biol 144, 281-92.
[126] Micheau, O. and Tschopp, J. (2003) Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell 114, 181-90.
[127] Obeid, L.M., Linardic, C.M., Karolak, L.A. and Hannun, Y.A. (1993) Programmed cell death induced by ceramide. Science 259, 1769-71.

Bibliografía
197
[128] Li, L.Y., Luo, X. and Wang, X. (2001) Endonuclease G is an apoptotic DNase when released from mitochondria. Nature 412, 95-9.
[129] Verhagen, A.M., Ekert, P.G., Pakusch, M., Silke, J., Connolly, L.M., Reid, G.E., Moritz, R.L., Simpson, R.J. and Vaux, D.L. (2000) Identification of DIABLO, a mammalian protein that promotes apoptosis by binding to and antagonizing IAP proteins. Cell 102, 43-53.
[130] Roberts, D.L., Merrison, W., MacFarlane, M. and Cohen, G.M. (2001) The inhibitor of apoptosis protein-binding domain of Smac is not essential for its proapoptotic activity. J Cell Biol 153, 221-8.
[131] Gray, C.W., Ward, R.V., Karran, E., Turconi, S., Rowles, A., Viglienghi, D., Southan, C., Barton, A., Fantom, K.G., West, A., Savopoulos, J., Hassan, N.J., Clinkenbeard, H., Hanning, C., Amegadzie, B., Davis, J.B., Dingwall, C., Livi, G.P. and Creasy, C.L. (2000) Characterization of human HtrA2, a novel serine protease involved in the mammalian cellular stress response. Eur J Biochem 267, 5699-710.
[132] Kroemer, G. and Reed, J.C. (2000) Mitochondrial control of cell death. Nat Med 6, 513-9.
[133] Kroemer, G., Zamzami, N. and Susin, S.A. (1997) Mitochondrial control of apoptosis. Immunol Today 18, 44-51.
[134] Susin, S.A., Daugas, E., Ravagnan, L., Samejima, K., Zamzami, N., Loeffler, M., Costantini, P., Ferri, K.F., Irinopoulou, T., Prevost, M.C., Brothers, G., Mak, T.W., Penninger, J., Earnshaw, W.C. and Kroemer, G. (2000) Two distinct pathways leading to nuclear apoptosis. J Exp Med 192, 571-80.
[135] Susin, S.A., Lorenzo, H.K., Zamzami, N., Marzo, I., Snow, B.E., Brothers, G.M., Mangion, J., Jacotot, E., Costantini, P., Loeffler, M., Larochette, N., Goodlett, D.R., Aebersold, R., Siderovski, D.P., Penninger, J.M. and Kroemer, G. (1999) Molecular characterization of mitochondrial apoptosis-inducing factor. Nature 397, 441-6.
[136] Cory, S. and Adams, J.M. (2002) The Bcl2 family: regulators of the cellular life-or-death switch. Nat Rev Cancer 2, 647-56.
[137] Hsu, Y.T., Wolter, K.G. and Youle, R.J. (1997) Cytosol-to-membrane redistribution of Bax and Bcl-X(L) during apoptosis. Proc Natl Acad Sci U S A 94, 3668-72.
[138] Puthalakath, H., Huang, D.C., O'Reilly, L.A., King, S.M. and Strasser, A. (1999) The proapoptotic activity of the Bcl-2 family member Bim is regulated by interaction with the dynein motor complex. Mol Cell 3, 287-96.
[139] Reed, J.C. (1998) Bcl-2 family proteins. Oncogene 17, 3225-36. [140] Antonsson, B., Montessuit, S., Lauper, S., Eskes, R. and Martinou, J.C. (2000)
Bax oligomerization is required for channel-forming activity in liposomes and to trigger cytochrome c release from mitochondria. Biochem J 345 Pt 2, 271-8.
[141] Tsujimoto, Y. and Shimizu, S. (2000) VDAC regulation by the Bcl-2 family of proteins. Cell Death Differ 7, 1174-81.
[142] Scorrano, L. and Korsmeyer, S.J. (2003) Mechanisms of cytochrome c release by proapoptotic BCL-2 family members. Biochem Biophys Res Commun 304, 437-44.

Bibliografía
198
[143] Shimizu, S. and Tsujimoto, Y. (2000) Proapoptotic BH3-only Bcl-2 family members induce cytochrome c release, but not mitochondrial membrane potential loss, and do not directly modulate voltage-dependent anion channel activity. Proc Natl Acad Sci U S A 97, 577-82.
[144] Chowdhury, I., Tharakan, B. and Bhat, G.K. (2008) Caspases - an update. Comp Biochem Physiol B Biochem Mol Biol 151, 10-27.
[145] Lamkanfi, M., Declercq, W., Kalai, M., Saelens, X. and Vandenabeele, P. (2002) Alice in caspase land. A phylogenetic analysis of caspases from worm to man. Cell Death Differ 9, 358-61.
[146] Thome, M., Schneider, P., Hofmann, K., Fickenscher, H., Meinl, E., Neipel, F., Mattmann, C., Burns, K., Bodmer, J.L., Schroter, M., Scaffidi, C., Krammer, P.H., Peter, M.E. and Tschopp, J. (1997) Viral FLICE-inhibitory proteins (FLIPs) prevent apoptosis induced by death receptors. Nature 386, 517-21.
[147] Koseki, T., Inohara, N., Chen, S. and Nunez, G. (1998) ARC, an inhibitor of apoptosis expressed in skeletal muscle and heart that interacts selectively with caspases. Proc Natl Acad Sci U S A 95, 5156-60.
[148] Hermann, C., Assmus, B., Urbich, C., Zeiher, A.M. and Dimmeler, S. (2000) Insulin-mediated stimulation of protein kinase Akt: A potent survival signaling cascade for endothelial cells. Arterioscler Thromb Vasc Biol 20, 402-9.
[149] Mannick, J.B., Hausladen, A., Liu, L., Hess, D.T., Zeng, M., Miao, Q.X., Kane, L.S., Gow, A.J. and Stamler, J.S. (1999) Fas-induced caspase denitrosylation. Science 284, 651-4.
[150] Krishna, M. and Narang, H. (2008) The complexity of mitogen-activated protein kinases (MAPKs) made simple. Cell Mol Life Sci 65, 3525-44.
[151] Pelengaris, S., Khan, M. and Evan, G. (2002) c-MYC: more than just a matter of life and death. Nat Rev Cancer 2, 764-76.
[152] Kim, G.S., Hong, J.S., Kim, S.W., Koh, J.M., An, C.S., Choi, J.Y. and Cheng, S.L. (2003) Leptin induces apoptosis via ERK/cPLA2/cytochrome c pathway in human bone marrow stromal cells. J Biol Chem 278, 21920-9.
[153] Park, B.G., Yoo, C.I., Kim, H.T., Kwon, C.H. and Kim, Y.K. (2005) Role of mitogen-activated protein kinases in hydrogen peroxide-induced cell death in osteoblastic cells. Toxicology 215, 115-25.
[154] Wu, Z., Wu, L.J., Tashiro, S., Onodera, S. and Ikejima, T. (2005) Phosphorylated extracellular signal-regulated kinase up-regulated p53 expression in shikonin-induced HeLa cell apoptosis. Chin Med J (Engl) 118, 671-7.
[155] Brown, L. and Benchimol, S. (2006) The involvement of MAPK signaling pathways in determining the cellular response to p53 activation: cell cycle arrest or apoptosis. J Biol Chem 281, 3832-40.
[156] Leppa, S. and Bohmann, D. (1999) Diverse functions of JNK signaling and c-Jun in stress response and apoptosis. Oncogene 18, 6158-62.
[157] Behrens, A., Sibilia, M. and Wagner, E.F. (1999) Amino-terminal phosphorylation of c-Jun regulates stress-induced apoptosis and cellular proliferation. Nat Genet 21, 326-9.

Bibliografía
199
[158] Wan, Y.Y., Chi, H., Xie, M., Schneider, M.D. and Flavell, R.A. (2006) The kinase TAK1 integrates antigen and cytokine receptor signaling for T cell development, survival and function. Nat Immunol 7, 851-8.
[159] Sato, S., Sanjo, H., Takeda, K., Ninomiya-Tsuji, J., Yamamoto, M., Kawai, T., Matsumoto, K., Takeuchi, O. and Akira, S. (2005) Essential function for the kinase TAK1 in innate and adaptive immune responses. Nat Immunol 6, 1087-95.
[160] Nateri, A.S., Spencer-Dene, B. and Behrens, A. (2005) Interaction of phosphorylated c-Jun with TCF4 regulates intestinal cancer development. Nature 437, 281-5.
[161] Kyriakis, J.M. and Avruch, J. (2001) Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol Rev 81, 807-69.
[162] Foltz, I.N., Lee, J.C., Young, P.R. and Schrader, J.W. (1997) Hemopoietic growth factors with the exception of interleukin-4 activate the p38 mitogen-activated protein kinase pathway. J Biol Chem 272, 3296-301.
[163] Xing, J., Kornhauser, J.M., Xia, Z., Thiele, E.A. and Greenberg, M.E. (1998) Nerve growth factor activates extracellular signal-regulated kinase and p38 mitogen-activated protein kinase pathways to stimulate CREB serine 133 phosphorylation. Mol Cell Biol 18, 1946-55.
[164] Cheng, H.L. and Feldman, E.L. (1998) Bidirectional regulation of p38 kinase and c-Jun N-terminal protein kinase by insulin-like growth factor-I. J Biol Chem 273, 14560-5.
[165] Yamauchi, J., Nagao, M., Kaziro, Y. and Itoh, H. (1997) Activation of p38 mitogen-activated protein kinase by signaling through G protein-coupled receptors. Involvement of Gbetagamma and Galphaq/11 subunits. J Biol Chem 272, 27771-7.
[166] Clerk, A., Michael, A. and Sugden, P.H. (1998) Stimulation of the p38 mitogen-activated protein kinase pathway in neonatal rat ventricular myocytes by the G protein-coupled receptor agonists, endothelin-1 and phenylephrine: a role in cardiac myocyte hypertrophy? J Cell Biol 142, 523-35.
[167] Coffer, P.J. and Woodgett, J.R. (1991) Molecular cloning and characterisation of a novel putative protein-serine kinase related to the cAMP-dependent and protein kinase C families. Eur J Biochem 201, 475-81.
[168] Jones, P.F., Jakubowicz, T., Pitossi, F.J., Maurer, F. and Hemmings, B.A. (1991) Molecular cloning and identification of a serine/threonine protein kinase of the second-messenger subfamily. Proc Natl Acad Sci U S A 88, 4171-5.
[169] Bellacosa, A., Franke, T.F., Gonzalez-Portal, M.E., Datta, K., Taguchi, T., Gardner, J., Cheng, J.Q., Testa, J.R. and Tsichlis, P.N. (1993) Structure, expression and chromosomal mapping of c-akt: relationship to v-akt and its implications. Oncogene 8, 745-54.
[170] Jones, P.F., Jakubowicz, T. and Hemmings, B.A. (1991) Molecular cloning of a second form of rac protein kinase. Cell Regul 2, 1001-9.
[171] Konishi, H., Shinomura, T., Kuroda, S., Ono, Y. and Kikkawa, U. (1994) Molecular cloning of rat RAC protein kinase alpha and beta and their

Bibliografía
200
association with protein kinase C zeta. Biochem Biophys Res Commun 205, 817-25.
[172] Hill, M.M., Clark, S.F., Tucker, D.F., Birnbaum, M.J., James, D.E. and Macaulay, S.L. (1999) A role for protein kinase Bbeta/Akt2 in insulin-stimulated GLUT4 translocation in adipocytes. Mol Cell Biol 19, 7771-81.
[173] Vandromme, M., Rochat, A., Meier, R., Carnac, G., Besser, D., Hemmings, B.A., Fernandez, A. and Lamb, N.J. (2001) Protein kinase B beta/Akt2 plays a specific role in muscle differentiation. J Biol Chem 276, 8173-9.
[174] Nakatani, K., Thompson, D.A., Barthel, A., Sakaue, H., Liu, W., Weigel, R.J. and Roth, R.A. (1999) Up-regulation of Akt3 in estrogen receptor-deficient breast cancers and androgen-independent prostate cancer lines. J Biol Chem 274, 21528-32.
[175] Andrieu-Abadie, N. and Levade, T. (2002) Sphingomyelin hydrolysis during apoptosis. Biochim Biophys Acta 1585, 126-34.
[176] Futerman, A.H. and Hannun, Y.A. (2004) The complex life of simple sphingolipids. EMBO Rep 5, 777-82.
[177] Kitatani, K., Idkowiak-Baldys, J. and Hannun, Y.A. (2008) The sphingolipid salvage pathway in ceramide metabolism and signaling. Cell Signal 20, 1010-8.
[178] Dbaibo, G.S., Pushkareva, M.Y., Jayadev, S., Schwarz, J.K., Horowitz, J.M., Obeid, L.M. and Hannun, Y.A. (1995) Retinoblastoma gene product as a downstream target for a ceramide-dependent pathway of growth arrest. Proc Natl Acad Sci U S A 92, 1347-51.
[179] Borner, C. (2003) The Bcl-2 protein family: sensors and checkpoints for life-or-death decisions. Mol Immunol 39, 615-47.
[180] Radin, N.S. (2003) Killing tumours by ceramide-induced apoptosis: a critique of available drugs. Biochem J 371, 243-56.
[181] Lee, J.Y., Leonhardt, L.G. and Obeid, L.M. (1998) Cell-cycle-dependent changes in ceramide levels preceding retinoblastoma protein dephosphorylation in G2/M. Biochem J 334 ( Pt 2), 457-61.
[182] Uchida, Y., Nardo, A.D., Collins, V., Elias, P.M. and Holleran, W.M. (2003) De novo ceramide synthesis participates in the ultraviolet B irradiation-induced apoptosis in undifferentiated cultured human keratinocytes. J Invest Dermatol 120, 662-9.
[183] Hannun, Y.A. (1996) Functions of ceramide in coordinating cellular responses to stress. Science 274, 1855-9.
[184] Marchesini, N. and Hannun, Y.A. (2004) Acid and neutral sphingomyelinases: roles and mechanisms of regulation. Biochem Cell Biol 82, 27-44.
[185] Schwandner, R., Wiegmann, K., Bernardo, K., Kreder, D. and Kronke, M. (1998) TNF receptor death domain-associated proteins TRADD and FADD signal activation of acid sphingomyelinase. J Biol Chem 273, 5916-22.
[186] Dumitru, C.A. and Gulbins, E. (2006) TRAIL activates acid sphingomyelinase via a redox mechanism and releases ceramide to trigger apoptosis. Oncogene 25, 5612-25.

Bibliografía
201
[187] Grassme, H., Jekle, A., Riehle, A., Schwarz, H., Berger, J., Sandhoff, K., Kolesnick, R. and Gulbins, E. (2001) CD95 signaling via ceramide-rich membrane rafts. J Biol Chem 276, 20589-96.
[188] Grassme, H., Schwarz, H. and Gulbins, E. (2001) Molecular mechanisms of ceramide-mediated CD95 clustering. Biochem Biophys Res Commun 284, 1016-30.
[189] Holopainen, J.M., Subramanian, M. and Kinnunen, P.K. (1998) Sphingomyelinase induces lipid microdomain formation in a fluid phosphatidylcholine/sphingomyelin membrane. Biochemistry 37, 17562-70.
[190] Kolesnick, R.N., Goni, F.M. and Alonso, A. (2000) Compartmentalization of ceramide signaling: physical foundations and biological effects. J Cell Physiol 184, 285-300.
[191] Santana, P., Pena, L.A., Haimovitz-Friedman, A., Martin, S., Green, D., McLoughlin, M., Cordon-Cardo, C., Schuchman, E.H., Fuks, Z. and Kolesnick, R. (1996) Acid sphingomyelinase-deficient human lymphoblasts and mice are defective in radiation-induced apoptosis. Cell 86, 189-99.
[192] Zhang, Y., Mattjus, P., Schmid, P.C., Dong, Z., Zhong, S., Ma, W.Y., Brown, R.E., Bode, A.M., Schmid, H.H. and Dong, Z. (2001) Involvement of the acid sphingomyelinase pathway in uva-induced apoptosis. J Biol Chem 276, 11775-82.
[193] Rotolo, J.A., Zhang, J., Donepudi, M., Lee, H., Fuks, Z. and Kolesnick, R. (2005) Caspase-dependent and -independent activation of acid sphingomyelinase signaling. J Biol Chem 280, 26425-34.
[194] Kashkar, H., Wiegmann, K., Yazdanpanah, B., Haubert, D. and Kronke, M. (2005) Acid sphingomyelinase is indispensable for UV light-induced Bax conformational change at the mitochondrial membrane. J Biol Chem 280, 20804-13.
[195] Paris, F., Fuks, Z., Kang, A., Capodieci, P., Juan, G., Ehleiter, D., Haimovitz-Friedman, A., Cordon-Cardo, C. and Kolesnick, R. (2001) Endothelial apoptosis as the primary lesion initiating intestinal radiation damage in mice. Science 293, 293-7.
[196] Heinrich, M., Neumeyer, J., Jakob, M., Hallas, C., Tchikov, V., Winoto-Morbach, S., Wickel, M., Schneider-Brachert, W., Trauzold, A., Hethke, A. and Schutze, S. (2004) Cathepsin D links TNF-induced acid sphingomyelinase to Bid-mediated caspase-9 and -3 activation. Cell Death Differ 11, 550-63.
[197] Schneider-Brachert, W., Tchikov, V., Neumeyer, J., Jakob, M., Winoto-Morbach, S., Held-Feindt, J., Heinrich, M., Merkel, O., Ehrenschwender, M., Adam, D., Mentlein, R., Kabelitz, D. and Schutze, S. (2004) Compartmentalization of TNF receptor 1 signaling: internalized TNF receptosomes as death signaling vesicles. Immunity 21, 415-28.
[198] Grassme, H., Jin, J., Wilker, B., von Kurthy, G., Wick, W., Weller, M., Moroy, T. and Gulbins, E. (2006) Regulation of pulmonary Pseudomonas aeruginosa infection by the transcriptional repressor Gfi1. Cell Microbiol 8, 1096-105.
[199] Utermohlen, O., Karow, U., Lohler, J. and Kronke, M. (2003) Severe impairment in early host defense against Listeria monocytogenes in mice deficient in acid sphingomyelinase. J Immunol 170, 2621-8.

Bibliografía
202
[200] Birbes, H., El Bawab, S., Hannun, Y.A. and Obeid, L.M. (2001) Selective hydrolysis of a mitochondrial pool of sphingomyelin induces apoptosis. Faseb J 15, 2669-79.
[201] Dai, Q., Liu, J., Chen, J., Durrant, D., McIntyre, T.M. and Lee, R.M. (2004) Mitochondrial ceramide increases in UV-irradiated HeLa cells and is mainly derived from hydrolysis of sphingomyelin. Oncogene 23, 3650-8.
[202] Birbes, H., Luberto, C., Hsu, Y.T., El Bawab, S., Hannun, Y.A. and Obeid, L.M. (2005) A mitochondrial pool of sphingomyelin is involved in TNFalpha-induced Bax translocation to mitochondria. Biochem J 386, 445-51.
[203] Bionda, C., Portoukalian, J., Schmitt, D., Rodriguez-Lafrasse, C. and Ardail, D. (2004) Subcellular compartmentalization of ceramide metabolism: MAM (mitochondria-associated membrane) and/or mitochondria? Biochem J 382, 527-33.
[204] Matsumoto, A., Comatas, K.E., Liu, L. and Stamler, J.S. (2003) Screening for nitric oxide-dependent protein-protein interactions. Science 301, 657-61.
[205] Anishkin, A., Sukharev, S. and Colombini, M. (2006) Searching for the molecular arrangement of transmembrane ceramide channels. Biophys J 90, 2414-26.
[206] Galve-Roperh, I., Haro, A. and Diaz-Laviada, I. (1997) Ceramide-induced translocation of protein kinase C zeta in primary cultures of astrocytes. FEBS Lett 415, 271-4.
[207] Kajita, K., Mune, T., Kanoh, Y., Natsume, Y., Ishizawa, M., Kawai, Y., Yasuda, K., Sugiyama, C. and Ishizuka, T. (2004) TNFalpha reduces the expression of peroxisome proliferator-activated receptor gamma (PPARgamma) via the production of ceramide and activation of atypical PKC. Diabetes Res Clin Pract 66 Suppl 1, S79-83.
[208] Shirakabe, K., Yamaguchi, K., Shibuya, H., Irie, K., Matsuda, S., Moriguchi, T., Gotoh, Y., Matsumoto, K. and Nishida, E. (1997) TAK1 mediates the ceramide signaling to stress-activated protein kinase/c-Jun N-terminal kinase. J Biol Chem 272, 8141-4.
[209] Pettus, B.J., Chalfant, C.E. and Hannun, Y.A. (2002) Ceramide in apoptosis: an overview and current perspectives. Biochim Biophys Acta 1585, 114-25.
[210] Garcia, A., Cayla, X., Guergnon, J., Dessauge, F., Hospital, V., Rebollo, M.P., Fleischer, A. and Rebollo, A. (2003) Serine/threonine protein phosphatases PP1 and PP2A are key players in apoptosis. Biochimie 85, 721-6.
[211] Dawkins, J.L., Hulme, D.J., Brahmbhatt, S.B., Auer-Grumbach, M. and Nicholson, G.A. (2001) Mutations in SPTLC1, encoding serine palmitoyltransferase, long chain base subunit-1, cause hereditary sensory neuropathy type I. Nat Genet 27, 309-12.
[212] Puranam, K.L., Guo, W.X., Qian, W.H., Nikbakht, K. and Boustany, R.M. (1999) CLN3 defines a novel antiapoptotic pathway operative in neurodegeneration and mediated by ceramide. Mol Genet Metab 66, 294-308.
[213] Puranam, K., Qian, W.H., Nikbakht, K., Venable, M., Obeid, L., Hannun, Y. and Boustany, R.M. (1997) Upregulation of Bcl-2 and elevation of ceramide in Batten disease. Neuropediatrics 28, 37-41.

Bibliografía
203
[214] González, A.G., Bermejo,J.,Triana J.,,Eiroa,J.L.,López,M. (1995) Sesquiterpene lactones and other constituents of Allagopappus species. J.Nat.Prod. 58.
[215] González, A.G., Bermejo,J.,Triana J.,,Eiroa,J.L.,López,M. (1992a) Germacrolides from allagopappus viscosissimus. Phytochemistry 31.
[216] González, A.G., Bermejo,J.,Triana J.,López,M.,Eiroa,J.L. (1990) Sesquiterpene lactones from tanacetum ferulaceum. Phytochemistry 29.
[217] González, A.G., Bermejo,J.,Triana J.,López,M.,Eiroa,J.L. (1992b) Sesquiterpene lactones and other constituents of tanacetum species. Phytochemistry
31. [218] Byrne, C., Gawad,Joshi,Skelton,Toia,White (1982) The crystal structure of
(S)-(-)-6-bromo-5,7-dihydroxy-8-methyl-2-phenyl-2,3-dihidro-4H-1benzopyran 4-one[(-)-6-bromocryptostrobin] and a 13C N.M.R. Aus.J.Chem. 35, 1851-1858.
[219] Triana, J., Lopez, M., Perez, F.J., Gonzalez-Platas, J., Quintana, J., Estevez, F., Leon, F. and Bermejo, J. (2005) Sesquiterpenoids from Pulicaria canariensis and their cytotoxic activities. J Nat Prod 68, 523-31.
[220] Rivero, A., Quintana, J., Eiroa, J.L., Lopez, M., Triana, J., Bermejo, J. and Estevez, F. (2003) Potent induction of apoptosis by germacranolide sesquiterpene lactones on human myeloid leukemia cells. Eur J Pharmacol 482, 77-84.
[221] Cabrera, J., Quintana, J., Reiter, R.J., Loro, J., Cabrera, F. and Estevez, F. (2003) Melatonin prevents apoptosis and enhances HSP27 mRNA expression induced by heat shock in HL-60 cells: possible involvement of the MT2 receptor. J Pineal Res 35, 231-8.
[222] Bradford, M.M. (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72, 248-54.
[223] Rubio, S., Quintana, J., Lopez, M., Eiroa, J.L., Triana, J. and Estevez, F. (2006) Phenylbenzopyrones structure-activity studies identify betuletol derivatives as potential antitumoral agents. Eur J Pharmacol 548, 9-20.
[224] Haridas, V., Higuchi, M., Jayatilake, G.S., Bailey, D., Mujoo, K., Blake, M.E., Arntzen, C.J. and Gutterman, J.U. (2001) Avicins: triterpenoid saponins from Acacia victoriae (Bentham) induce apoptosis by mitochondrial perturbation. Proc Natl Acad Sci U S A 98, 5821-6.
[225] Rubio, S., Quintana, J., Eiroa, J.L., Triana, J. and Estevez, F. (2007) Acetyl derivative of quercetin 3-methyl ether-induced cell death in human leukemia cells is amplified by the inhibition of ERK. Carcinogenesis 28, 2105-13.
[226] Wiegmann, K., Schutze, S., Machleidt, T., Witte, D. and Kronke, M. (1994) Functional dichotomy of neutral and acidic sphingomyelinases in tumor necrosis factor signaling. Cell 78, 1005-15.
[227] Okazaki, T., Bell, R.M. and Hannun, Y.A. (1989) Sphingomyelin turnover induced by vitamin D3 in HL-60 cells. Role in cell differentiation. J Biol Chem 264, 19076-80.

Bibliografía
204
[228] Budihardjo, I., Oliver, H., Lutter, M., Luo, X. and Wang, X. (1999) Biochemical pathways of caspase activation during apoptosis. Annu Rev Cell Dev Biol 15, 269-90.
[229] Krammer, P.H. (2000) CD95's deadly mission in the immune system. Nature 407, 789-95.
[230] Yang, J., Liu, X., Bhalla, K., Kim, C.N., Ibrado, A.M., Cai, J., Peng, T.I., Jones, D.P. and Wang, X. (1997) Prevention of apoptosis by Bcl-2: release of cytochrome c from mitochondria blocked. Science 275, 1129-32.
[231] Kim, C.N., Wang, X., Huang, Y., Ibrado, A.M., Liu, L., Fang, G. and Bhalla, K. (1997) Overexpression of Bcl-X(L) inhibits Ara-C-induced mitochondrial loss of cytochrome c and other perturbations that activate the molecular cascade of apoptosis. Cancer Res 57, 3115-20.
[232] Cragg, G.M. and Newman, D.J. (2005) Plants as a source of anti-cancer agents. J Ethnopharmacol 100, 72-9.
[233] Kim, K.M. and Lee, Y.J. (2005) Role of HER-2/neu signaling in sensitivity to tumor necrosis factor-related apoptosis-inducing ligand: enhancement of TRAIL-mediated apoptosis by amiloride. J Cell Biochem 96, 376-89.
[234] Kawaii, S., Tomono, Y., Katase, E., Ogawa, K. and Yano, M. (1999) Antiproliferative activity of flavonoids on several cancer cell lines. Biosci Biotechnol Biochem 63, 896-9.
[235] Kandaswami, C., Perkins, E., Soloniuk, D.S., Drzewiecki, G. and Middleton, E., Jr. (1993) Ascorbic acid-enhanced antiproliferative effect of flavonoids on squamous cell carcinoma in vitro. Anticancer Drugs 4, 91-6.
[236] Lee, W.R., Shen, S.C., Lin, H.Y., Hou, W.C., Yang, L.L. and Chen, Y.C. (2002) Wogonin and fisetin induce apoptosis in human promyeloleukemic cells, accompanied by a decrease of reactive oxygen species, and activation of caspase 3 and Ca(2+)-dependent endonuclease. Biochem Pharmacol 63, 225-36.
[237] Nicholson, D.W. and Thornberry, N.A. (1997) Caspases: killer proteases. Trends Biochem Sci 22, 299-306.
[238] Motwani, M., Delohery, T.M. and Schwartz, G.K. (1999) Sequential dependent enhancement of caspase activation and apoptosis by flavopiridol on paclitaxel-treated human gastric and breast cancer cells. Clin Cancer Res 5, 1876-83.
[239] Szewczyk, A. and Wojtczak, L. (2002) Mitochondria as a pharmacological target. Pharmacol Rev 54, 101-27.
[240] van Gurp, M., Festjens, N., van Loo, G., Saelens, X. and Vandenabeele, P. (2003) Mitochondrial intermembrane proteins in cell death. Biochem Biophys Res Commun 304, 487-97.
[241] Friesen, C., Herr, I., Krammer, P.H. and Debatin, K.M. (1996) Involvement of the CD95 (APO-1/FAS) receptor/ligand system in drug-induced apoptosis in leukemia cells. Nat Med 2, 574-7.
[242] Igney, F.H. and Krammer, P.H. (2002) Death and anti-death: tumour resistance to apoptosis. Nat Rev Cancer 2, 277-88.
[243] Margolin, N., Raybuck, S.A., Wilson, K.P., Chen, W., Fox, T., Gu, Y. and Livingston, D.J. (1997) Substrate and inhibitor specificity of interleukin-1 beta-converting enzyme and related caspases. J Biol Chem 272, 7223-8.

Bibliografía
205
[244] Cirman, T., Oresic, K., Mazovec, G.D., Turk, V., Reed, J.C., Myers, R.M., Salvesen, G.S. and Turk, B. (2004) Selective disruption of lysosomes in HeLa cells triggers apoptosis mediated by cleavage of Bid by multiple papain-like lysosomal cathepsins. J Biol Chem 279, 3578-87.
[245] Marsden, V.S. and Strasser, A. (2003) Control of apoptosis in the immune system: Bcl-2, BH3-only proteins and more. Annu Rev Immunol 21, 71-105.
[246] Adams, J.M. and Cory, S. (1998) The Bcl-2 protein family: arbiters of cell survival. Science 281, 1322-6.
[247] Perkins, C., Kim, C.N., Fang, G. and Bhalla, K.N. (1998) Overexpression of Apaf-1 promotes apoptosis of untreated and paclitaxel- or etoposide-treated HL-60 cells. Cancer Res 58, 4561-6.
[248] Green, D.R. and Reed, J.C. (1998) Mitochondria and apoptosis. Science 281, 1309-12.
[249] Amundson, S.A., Myers, T.G., Scudiero, D., Kitada, S., Reed, J.C. and Fornace, A.J., Jr. (2000) An informatics approach identifying markers of chemosensitivity in human cancer cell lines. Cancer Res 60, 6101-10.
[250] Addeo, R., Caraglia, M., Baldi, A., D'Angelo, V., Casale, F., Crisci, S., Abbruzzese, A., Vincenze, B., Campioni, M., Di Tullio, M.T. and Indolfi, P. (2005) Prognostic role of bcl-xL and p53 in childhood acute lymphoblastic leukemia (ALL). Cancer Biol Ther 4, 32-8.
[251] Brantley-Finley, C., Lyle, C.S., Du, L., Goodwin, M.E., Hall, T., Szwedo, D., Kaushal, G.P. and Chambers, T.C. (2003) The JNK, ERK and p53 pathways play distinct roles in apoptosis mediated by the antitumor agents vinblastine, doxorubicin, and etoposide. Biochem Pharmacol 66, 459-69.
[252] Mingo-Sion, A.M., Marietta, P.M., Koller, E., Wolf, D.M. and Van Den Berg, C.L. (2004) Inhibition of JNK reduces G2/M transit independent of p53, leading to endoreduplication, decreased proliferation, and apoptosis in breast cancer cells. Oncogene 23, 596-604.
[253] Nguyen, T.T., Tran, E., Nguyen, T.H., Do, P.T., Huynh, T.H. and Huynh, H. (2004) The role of activated MEK-ERK pathway in quercetin-induced growth inhibition and apoptosis in A549 lung cancer cells. Carcinogenesis 25, 647-59.
[254] Yeh, P.Y., Chuang, S.E., Yeh, K.H., Song, Y.C., Ea, C.K. and Cheng, A.L. (2002) Increase of the resistance of human cervical carcinoma cells to cisplatin by inhibition of the MEK to ERK signaling pathway partly via enhancement of anticancer drug-induced NF kappa B activation. Biochem Pharmacol 63, 1423-30.
[255] Tanel, A. and Averill-Bates, D.A. (2007) P38 and ERK mitogen-activated protein kinases mediate acrolein-induced apoptosis in Chinese hamster ovary cells. Cell Signal 19, 968-77.
[256] Torres, F., Quintana, J., Diaz, J.G., Carmona, A.J. and Estevez, F. (2008) Trifolin acetate-induced cell death in human leukemia cells is dependent on caspase-6 and activates the MAPK pathway. Apoptosis 13, 716-28.
[257] Beutler, J.A., Hamel, E., Vlietinck, A.J., Haemers, A., Rajan, P., Roitman, J.N., Cardellina, J.H., 2nd and Boyd, M.R. (1998) Structure-activity requirements for flavone cytotoxicity and binding to tubulin. J Med Chem 41, 2333-8.

Bibliografía
206
[258] Wang, L.G., Liu, X.M., Kreis, W. and Budman, D.R. (1999) The effect of antimicrotubule agents on signal transduction pathways of apoptosis: a review. Cancer Chemother Pharmacol 44, 355-61.
[259] Hollstein, M., Rice, K., Greenblatt, M.S., Soussi, T., Fuchs, R., Sorlie, T., Hovig, E., Smith-Sorensen, B., Montesano, R. and Harris, C.C. (1994) Database of p53 gene somatic mutations in human tumors and cell lines. Nucleic Acids Res 22, 3551-5.
[260] Hollstein, M., Sidransky, D., Vogelstein, B. and Harris, C.C. (1991) p53 mutations in human cancers. Science 253, 49-53.
[261] Perry, D.K., Carton, J., Shah, A.K., Meredith, F., Uhlinger, D.J. and Hannun, Y.A. (2000) Serine palmitoyltransferase regulates de novo ceramide generation during etoposide-induced apoptosis. J Biol Chem 275, 9078-84.
[262] Grassme, H., Cremesti, A., Kolesnick, R. and Gulbins, E. (2003) Ceramide-mediated clustering is required for CD95-DISC formation. Oncogene 22, 5457-70.
[263] Liu, P. and Anderson, R.G. (1995) Compartmentalized production of ceramide at the cell surface. J Biol Chem 270, 27179-85.
[264] Veldman, R.J., Maestre, N., Aduib, O.M., Medin, J.A., Salvayre, R. and Levade, T. (2001) A neutral sphingomyelinase resides in sphingolipid-enriched microdomains and is inhibited by the caveolin-scaffolding domain: potential implications in tumour necrosis factor signalling. Biochem J 355, 859-68.
[265] Birbes, H., El Bawab, S., Obeid, L.M. and Hannun, Y.A. (2002) Mitochondria and ceramide: intertwined roles in regulation of apoptosis. Adv Enzyme Regul 42, 113-29.
[266] Decaudin, D., Marzo, I., Brenner, C. and Kroemer, G. (1998) Mitochondria in chemotherapy-induced apoptosis: a prospective novel target of cancer therapy (review). Int J Oncol 12, 141-52.
[267] Yamamoto, K., Ichijo, H. and Korsmeyer, S.J. (1999) BCL-2 is phosphorylated and inactivated by an ASK1/Jun N-terminal protein kinase pathway normally activated at G(2)/M. Mol Cell Biol 19, 8469-78.
[268] Fadeel, B., Hassan, Z., Hellstrom-Lindberg, E., Henter, J.I., Orrenius, S. and Zhivotovsky, B. (1999) Cleavage of Bcl-2 is an early event in chemotherapy-induced apoptosis of human myeloid leukemia cells. Leukemia 13, 719-28.
[269] Westwick, J.K., Bielawska, A.E., Dbaibo, G., Hannun, Y.A. and Brenner, D.A. (1995) Ceramide activates the stress-activated protein kinases. J Biol Chem 270, 22689-92.
[270] Exton, J.H. (1994) Phosphatidylcholine breakdown and signal transduction. Biochim Biophys Acta 1212, 26-42.
[271] Schutze, S., Potthoff, K., Machleidt, T., Berkovic, D., Wiegmann, K. and Kronke, M. (1992) TNF activates NF-kappa B by phosphatidylcholine-specific phospholipase C-induced "acidic" sphingomyelin breakdown. Cell 71, 765-76.
[272] Luberto, C. and Hannun, Y.A. (1998) Sphingomyelin synthase, a potential regulator of intracellular levels of ceramide and diacylglycerol during SV40 transformation. Does sphingomyelin synthase account for the putative phosphatidylcholine-specific phospholipase C? J Biol Chem 273, 14550-9.

Bibliografía
207
[273] Bettaieb, A., Plo, I., Mansat-De Mas, V., Quillet-Mary, A., Levade, T., Laurent, G. and Jaffrezou, J.P. (1999) Daunorubicin- and mitoxantrone-triggered phosphatidylcholine hydrolysis: implication in drug-induced ceramide generation and apoptosis. Mol Pharmacol 55, 118-25.
[274] Zeidan, Y.H. and Hannun, Y.A. (2007) Activation of acid sphingomyelinase by protein kinase Cdelta-mediated phosphorylation. J Biol Chem 282, 11549-61.
[275] Dangles, O., Dufour, C., Manach, C., Morand, C. and Remesy, C. (2001) Binding of flavonoids to plasma proteins. Methods Enzymol 335, 319-33.
[276] Yuan, Z.P., Chen, L.J., Fan, L.Y., Tang, M.H., Yang, G.L., Yang, H.S., Du, X.B., Wang, G.Q., Yao, W.X., Zhao, Q.M., Ye, B., Wang, R., Diao, P., Zhang, W., Wu, H.B., Zhao, X. and Wei, Y.Q. (2006) Liposomal quercetin efficiently suppresses growth of solid tumors in murine models. Clin Cancer Res 12, 3193-9.
[277] Rodriguez, J. and Lazebnik, Y. (1999) Caspase-9 and APAF-1 form an active holoenzyme. Genes Dev 13, 3179-84.
[278] Stennicke, H.R., Deveraux, Q.L., Humke, E.W., Reed, J.C., Dixit, V.M. and Salvesen, G.S. (1999) Caspase-9 can be activated without proteolytic processing. J Biol Chem 274, 8359-62.
[279] Scoltock, A.B. and Cidlowski, J.A. (2004) Activation of intrinsic and extrinsic pathways in apoptotic signaling during UV-C-induced death of Jurkat cells: the role of caspase inhibition. Exp Cell Res 297, 212-23.
[280] Campos, L., Rouault, J.P., Sabido, O., Oriol, P., Roubi, N., Vasselon, C., Archimbaud, E., Magaud, J.P. and Guyotat, D. (1993) High expression of bcl-2 protein in acute myeloid leukemia cells is associated with poor response to chemotherapy. Blood 81, 3091-6.
[281] Achenbach, T.V., Muller, R. and Slater, E.P. (2000) Bcl-2 independence of flavopiridol-induced apoptosis. Mitochondrial depolarization in the absence of cytochrome c release. J Biol Chem 275, 32089-97.
[282] Cross, T.G., Scheel-Toellner, D., Henriquez, N.V., Deacon, E., Salmon, M. and Lord, J.M. (2000) Serine/threonine protein kinases and apoptosis. Exp Cell Res 256, 34-41.
[283] Yu, C., Wang, Z., Dent, P. and Grant, S. (2001) MEK1/2 inhibitors promote Ara-C-induced apoptosis but not loss of Deltapsi(m) in HL-60 cells. Biochem Biophys Res Commun 286, 1011-8.
[284] Herr, I. and Debatin, K.M. (2001) Cellular stress response and apoptosis in cancer therapy. Blood 98, 2603-14.
[285] Zarubin, T. and Han, J. (2005) Activation and signaling of the p38 MAP kinase pathway. Cell Res 15, 11-8.
[286] Galan, A., Garcia-Bermejo, M.L., Troyano, A., Vilaboa, N.E., de Blas, E., Kazanietz, M.G. and Aller, P. (2000) Stimulation of p38 mitogen-activated protein kinase is an early regulatory event for the cadmium-induced apoptosis in human promonocytic cells. J Biol Chem 275, 11418-24.
[287] Shimizu, T., Nakazato, T., Xian, M.J., Sagawa, M., Ikeda, Y. and Kizaki, M. (2006) Resveratrol induces apoptosis of human malignant B cells by activation of caspase-3 and p38 MAP kinase pathways. Biochem Pharmacol 71, 742-50.

Bibliografía
208
[288] Franklin, C.C., Srikanth, S. and Kraft, A.S. (1998) Conditional expression of mitogen-activated protein kinase phosphatase-1, MKP-1, is cytoprotective against UV-induced apoptosis. Proc Natl Acad Sci U S A 95, 3014-9.
[289] Juo, P., Kuo, C.J., Reynolds, S.E., Konz, R.F., Raingeaud, J., Davis, R.J., Biemann, H.P. and Blenis, J. (1997) Fas activation of the p38 mitogen-activated protein kinase signalling pathway requires ICE/CED-3 family proteases. Mol Cell Biol 17, 24-35.
[290] Hayakawa, J., Ohmichi, M., Kurachi, H., Ikegami, H., Kimura, A., Matsuoka, T., Jikihara, H., Mercola, D. and Murata, Y. (1999) Inhibition of extracellular signal-regulated protein kinase or c-Jun N-terminal protein kinase cascade, differentially activated by cisplatin, sensitizes human ovarian cancer cell line. J Biol Chem 274, 31648-54.
[291] Persons, D.L., Yazlovitskaya, E.M., Cui, W. and Pelling, J.C. (1999) Cisplatin-induced activation of mitogen-activated protein kinases in ovarian carcinoma cells: inhibition of extracellular signal-regulated kinase activity increases sensitivity to cisplatin. Clin Cancer Res 5, 1007-14.
[292] Mandic, A., Viktorsson, K., Heiden, T., Hansson, J. and Shoshan, M.C. (2001) The MEK1 inhibitor PD98059 sensitizes C8161 melanoma cells to cisplatin-induced apoptosis. Melanoma Res 11, 11-9.
[293] Chen, Y.R., Wang, W., Kong, A.N. and Tan, T.H. (1998) Molecular mechanisms of c-Jun N-terminal kinase-mediated apoptosis induced by anticarcinogenic isothiocyanates. J Biol Chem 273, 1769-75.
[294] Watabe, M., Kakeya, H. and Osada, H. (1999) Requirement of protein kinase (Krs/MST) activation for MT-21-induced apoptosis. Oncogene 18, 5211-20.
[295] Zhuang, S., Demirs, J.T. and Kochevar, I.E. (2000) p38 mitogen-activated protein kinase mediates bid cleavage, mitochondrial dysfunction, and caspase-3 activation during apoptosis induced by singlet oxygen but not by hydrogen peroxide. J Biol Chem 275, 25939-48.

AAANNNEEEXXXOOO


Phenylbenzopyrones structure-activity studies identify betuletol derivativesas potential antitumoral agents
Sara Rubio a, José Quintana a, Mariana López b, José Luis Eiroa b, Jorge Triana b,Francisco Estévez a,⁎
a Department of Biochemistry, I.C.I.C., University of Las Palmas de Gran Canaria, Plaza Dr. Pasteur s/n, 35016 Las Palmas de Gran Canaria, Spainb Department of Chemistry, I.C.I.C., University of Las Palmas de Gran Canaria, Campus Universitario de Tafira, 35017 Las Palmas de Gran Canaria, Spain
Received 10 March 2006; received in revised form 20 June 2006; accepted 13 July 2006Available online 22 July 2006
Abstract
We have analyzed the cytotoxicity of 22 compounds with a phenylbenzo-γ-pirone core structure, most of them obtained from natural sources,in five human tumor cell lines (HL-60, A431, SK-OV-3, HeLa and HOS). Betuletol 3-methyl ether and its diacetate were the most cytotoxiccompounds. The HL-60 cell line was especially sensitive to these compounds, with IC50 values of approximately 1 μM. Treatment of HL-60 cellswith betuletol 3-methyl ether was associated with apoptosis induction which was prevented by a non-specific caspase inhibitor (z-VAD-fmk) andalso by a specific inhibitor of caspase-8 (z-IETD-fmk) indicating activation of the extrinsic apoptotic pathway. The results suggest that betuletol 3-methyl ether has potential as new anticancer agent.© 2006 Elsevier B.V. All rights reserved.
Keywords: Apoptosis; Flavonoids; Cytotoxic activity; Caspases; DNA fragmentation; poly(ADP-ribose) polymerase
1. Introduction
Many anticancer drugs have been shown to cause the deathof sensitive cells through the induction of apoptosis. Thiscellular suicide program is initiated by ligation of deathreceptors, growth factor deprivation or various environmentalstresses which are mediated by molecular pathways thatculminate in the activation of a conserved family of aspartate-specific cysteine proteases, known as the caspases, whichorchestrate the dismantling and clearance of the dying cell.Caspases themselves are present as proenzymes that are readilycleaved and activated during apoptosis, providing the cell with ameans to rapidly amplify its apoptotic response (Cohen, 1997;Thornberry and Lazebnik, 1998). Two major pathways havebeen identified for the induction of apoptosis. The intrinsicpathway is activated by release of mitochondrial cytochrome c(Kluck et al., 1997; Liu et al., 1996; Yang et al., 1997) whichforms a complex with Apaf-1 and activates caspase-9 leading to
activation of caspase-3 (Li et al., 1997; Srinivasula et al., 1998).The extrinsic pathway is initiated by activation of deathreceptors and thereby cleavage of procaspase-8 (Boldin et al.,1996; Muzio et al., 1996). Caspase-8 directly activates caspase-3 (Stennicke et al., 1998). Caspase-8 also cleaves Bid-a Bcl-2family member— and thereby plays a role in the release ofcytochrome c. However, it is becoming increasingly clear thatapoptosis can also occur independently of caspase activation(Chipuk and Green, 2005).Flavonoids comprise a vast array of biologically active
compounds ubiquitous in plants, many of which have been usedin traditional Eastern medicine for thousands of years. Thesepolyphenolic compounds exert several biological functions suchas apoptosis-inducing activity, free radical scavenging activity,and anti-tumorigenic activity (Ko et al., 2002; Lee et al., 2002).Here we have studied the effect of twenty two flavonoids, mostof them isolated from endemic plants to the Canary Islands, oncell viability of five human tumor cell lines. The betuletolderivatives, betuletol 3-methyl ether and betuletol 3-methylether diacetate were the most potent cytotoxic compounds in allcell lines studied, but also the most potent apoptotic inducers on
European Journal of Pharmacology 548 (2006) 9–20www.elsevier.com/locate/ejphar
⁎ Corresponding author. Tel.: +34 928 451443; fax: +34 928 451441.E-mail address: [email protected] (F. Estévez).
0014-2999/$ - see front matter © 2006 Elsevier B.V. All rights reserved.doi:10.1016/j.ejphar.2006.07.020

human myeloid leukemia HL-60 cells. Increased levels of anti-apoptotic or decreased levels of pro-apoptotic proteins can causeacquired or intrinsic resistance against apoptosis induction byantineoplastic drugs thereby contributing to treatment failureand poor clinical prognosis. Thus, novel agents targetingaberrant apoptosis pathways or inducing alternative deathpathways may be suited to overcome treatment resistance(Igney and Krammer, 2002). The present studies demonstratethat betuletol 3-methyl ether induces apoptosis, at least in largepart, by activation of the extrinsic caspase-8 pathway of death.
2. Materials and methods
2.1. Materials
Most of the flavonoids used in this study were obtained fromthe Canary Islands' endemic plants and were isolated accordingto published methods with minor modifications. Structuralidentities of flavonoids were determined spectroscopically(proton nuclear magnetic resonance and 13C nuclear magneticresonance, infrared and UV/Visible spectroscopy and massspectrometry) as described previously: Betuletol 3-methyl ether1, quercetin 3,3′-dimethyl ether 7, kaempferol 3-methyl ether 9,5,7-dihydroxy-3,3′,4′-trimethoxyflavone 10, 4′, 5, 7-trihy-droxy-3,6-dimethoxyflavone 12 and eriodictyol 19 wereisolated from the aerial parts of Allagopappus dichotomus asdescribed (González et al., 1995). Quercetin 3-methyl ether 5,naringenin 21, eriodictyol 19 and kaempferol 3-methyl ether 9were isolated from Allagopappus viscosissimus as described(González et al., 1992a). Axillarin 11 and apigenin 22 wereisolated from the aerial parts of Tanacetum ferulaceum and Ta-nacetum ptarmiciflorumas described (González et al., 1990,1992b). Acetyl derivatives of betuletol 3-methyl ether (com-pound 2), quercetin 3-methyl ether (compound 6), quercetin3,3′-dimethyl ether (compound 8) and eriodictyol (compound20) were obtained by treatment of the corresponding alcoholswith acetic anhydride (Ac2O) in pyridine for 12 h at roomtemperature. Betuletol 3, 5, 7-trimethyl ether 3 was obtained bymethylation of 4′,5,7-trihydroxy-3, 6-dimethoxyflavone 12.Mono-di- and triacetyl derivatives (compounds 13, 14 and 15)of 4′,5,7-trihydroxy-3, 6-dimethoxyflavone 12were obtained byacetylation of this compound with acetic anhydride in pyridineas above. The acetyl derivatives were purified by chromatog-raphy on a silica gel column, eluted with hexane, and hexane-ethyl acetate mixtures (8:2). The mono and diacetyl derivatives,7-Acetoxy-4′,5-dihydroxy-3,6-dimethoxyflavone 13 and 5,7-diacetoxy-4′-hydroxy-3,6-dimethoxyflavone 14, are describedfor the first time.7-Acetoxy-4′,5-dihydroxy-3,6-dimethoxyflavone 13: UV/
Vis λmax nm, (MeOH): 289, 316,330; (+NaOMe): 286, 325,398; (+AlCl3): 294, 350, 396; (+AlCl3+ HCl): same as AlCl3;(+NaOAc): 290, 376; (+NaOAc+H3BO3): same as NaOAc.PMR (300 MHz, CDCl3): δ 8.11 (d, J=8.8 Hz, H-2′, H-6′),7.23 (d, J=8.8 Hz, H-3′, H-5′), 6.76 (s, H-8), 4.04 (s, OMe),3.87 (s, OMe), 2.35 (s, OAc). EIMS m/z (rel. int.): 372 [M]+
(100), 357 (15.6), 330 (41.1), 329 (45.8), 287 (39.1), 269 (17.8),121 (22.9), 69 (66.0).
5,7-diacetoxy-4′-hydroxy-3,6-dimethoxyflavone 14: UV/Vis λmax nm, (MeOH): 287, 323,329; (+NaOMe): 284, 325,393; (+AlCl3): same as MeOH; (+AlCl3+ HCl): same as AlCl3;(+NaOAc): 287, 360; (+NaOAc+H3BO3): 287,325. PMR(300 MHz, CDCl3): δ 8.06 (d, J=8.8, H-2′, H-6′), 7.21 (d,J=8.5, H-3′, H-5′), 6.89 (s, H-8), 3.94 (s, OMe), 3.76 (s, OMe),2.50 (s, OAc), 2.33 (s, OAc). EIMS m/z (rel. int.): 413 [M]+
(36.6), 372 (100), 330 (49.2), 315 (46.8), 312 (42.8), 287 (22.5),269 (10.6), 121 (14.9), 69 (15.6).Quercetin 4 (3,3′,4′,5,7-pentahydroxyflavone) was obtained
from Sigma. Cryptostrobin 18 was isolated from Rumohraadiantiformis. The structure of this compound was determinedby a combination of spectroscopic analysis and comparisonwith reported data (Byrne et al., 1982).Dihydroquercetin 7,3′-dimethyl ether 16 and dihydrokaemp-
ferol 7,4′-dimethyl ether 17 were isolated from the aerial partsof Pulicaria canariensis as recently described (Triana et al.,2005). All other reagents were of analytical grade. Purity of allcompounds was 99.0% as judged by high-performance liquidchromatography. Stock solutions of 10 mM flavonoids weremade in dimethyl sulfoxide (DMSO), and aliquots were frozenat −20 °C. Polyvinylidene difluoride (PVDF) membranes werepurchased from Millipore. Antibodies for poly(ADP-ribose)polymerase (PARP), caspase-3, caspase-8 and caspase-9 werepurchased from Stressgen. Antibody for cytochrome c waspurchased from BD PharMingen. Secondary antibodies werefrom Amersham Pharmacia Biotech. The caspase inhibitorswere from Sigma. The caspase-3 and caspase-8 substrates werefrom Sigma. The caspase-9 substrate was from AnaSpec.
2.2. Cell culture
HL-60 cells were cultured in RPMI 1640 medium containing10% (v/v) heat-inactivated fetal bovine serum and 100 units/mlpenicillin and 100 μg/ml streptomycin at 37 °C in a humidifiedatmosphere containing 5% CO2. The cultures were passed twiceweekly exhibiting characteristic doubling times of∼24 h. The cellnumbers were counted by a hematocytometer, and the viability wasalways greater that 95% in all experiments as assayed by the0.025% trypan blue exclusion method. Further dilutions of stocksolutions of flavonoids were made in culture media just before use.In all experiments, the final concentration of DMSOdid not exceed0.3% (v/v), a concentration which is non-toxic to the cells. Thesame concentration was present in control experiments. The A431cell linewas grown asmonolayers in plastic tissue flasks containingDulbecco's modified Eagle's medium (DMEM) supplementedwith 10% fetal bovine serum, 100 units/ml penicillin and 100μg/mlstreptomycin. SK-OV-3 was grown as monolayers in DMEM. Thehuman cervix carcinoma HeLa and human osteosarcoma HOScells were grown in Eagle's minimum essential medium (EMEM)supplementedwith 2mMglutamine, 1%Non essential amino acidsand 10% fetal bovine serum. Cells at logarithmic growth phasewere harvested using 0.25% trypsin-EDTA solution for 5 min,pelleted by centrifugation at 500 ×g for 5 min, washed once withmedium and resuspended at a concentration of 1×104 cells/ml.These cell lineswere obtained from theEuropeanCollection ofCellCultures (Salisbury, UK).
10 S. Rubio et al. / European Journal of Pharmacology 548 (2006) 9–20

Human peripheral blood mononuclear cells (PBMC) wereisolated from heparin-anticoagulated blood of healthy volunteersby centrifugation with Ficoll–Paque Plus (Amersham Bios-ciences). PBMCs were also stimulated with phytohemagglutinine(PHA, 2 μg/ml) for 48 h before experimental treatment.
2.3. Cytotoxicity of flavonoids on HL-60 cells, A431 cells,HeLa cells, SK-OV-3 cells and HOS cells
The cytotoxicity of flavonoids in HL-60 cells was analyzedby colorimetric 3-(4,5-dimethyl-2-thiazolyl-)-2,5-diphenyl-2H-tetrazolium bromide (MTT) assay. Briefly, 1×104 exponential-ly growing cells were seeded in 96-well microculture plateswith various flavonoids concentrations. After the addition ofMTT (0.5 mg/ml) cells were incubated at 37 °C for 4 h. Sodiumdodecyl sulfate (SDS) (10% w/v) in 0.05 M HCl was added tothe wells and then incubated at room temperature overnightunder dark conditions. The absorbance was measured at570 nm. Concentrations inducing a 50% inhibition of cellgrowth (IC50) were determined graphically for each experimentas described (Rivero et al., 2003). A431, SK-OV-3, HeLa andHOS cells (5×103−1×104) were inoculated in 96-well platesand were then incubated at 37 °C for 24 h to allow theattachment to plates after which the culture media were changedand flavonoids were added and provided different finalconcentrations. Cells were then incubated at 37 °C in a 5%CO2–95% air atmosphere for 72 h. Cell viability wasdetermined as above using a colorimetric assay with thereduction of MTT.
2.4. Quantitative fluorescent microscopy
Cells were harvested and fixed in 3% paraformaldehyde andincubated at room temperature for 10 min. The fixative wasremoved and the cells werewashedwith PBS, resuspended in 30–50 μl of PBS containing 20 μg/ml bis-benzimide trihydrochloride(Hoechst 33258), and incubated at room temperature for 15 min.Ten-microliter aliquots of the cells were placed on glass slides,and triplicate samples of 500 cells each were counted and scoredfor the incidence of apoptotic chromatin condensation using aZeiss fluorescent microscopy. Stained nuclei with condensedchromatin (supercondensed chromatin at the nuclear periphery),or nuclei that were fragmented into multiple smaller dense bodieswere considered as apoptotic. Nuclei with uncondensed anddispersed chromatin were considered as not apoptotic.
2.5. Quantification of apoptosis by flow cytometry
To study changes in the cell DNA content, histogrammeasurements of hypodiploid DNA formation was performedby flow cytometry using a Coulter EPICS™ cytometer (Beck-man Coulter). Histograms were analyzed with the Expo 32 ADCSoftware™. Cells were collected and centrifuged at 500 ×g,washed with PBS and resuspended in 50 μl of PBS. Followingdropwise addition of 1 ml of ice-cold 75% ethanol, fixed cellswere stored at −20 °C for 1 h. Samples were then centrifuged at500 ×g andwashedwith PBS before resuspension in 1ml of PBS
containing 50 μg/ml propidium iodide and 100 μg/ml RNase Aand incubation for 1 h at 37 °C in the dark. The percentage ofcells with decreased DNA staining, composed of apoptotic cellsresulting from either fragmentation or decreased chromatin, of aminimum of 10,000 cells per experimental condition wascounted. Cell debris was excluded from analysis by selectivegating based on anterior and right angle scattering.
2.6. Analysis of DNA fragmentation
HL-60 cells were washed with PBS and incubated in 20 μl of50 mM Tris-HCl (pH 8.0), 10 mM EDTA, 0.5% SDS and 1 μg/μl RNase A (Sigma) at 37 °C for 1 h. Then, 10 μg/μl proteinaseK (Sigma) (2 μl) was added, and the mixture was incubated at50 °C for 2 h more. DNAwas extracted with 100 μl of phenol-chloroform-isoamyl alcohol (24:24:1) and mixed with 5 μl ofloading solution (10 mM EDTA, pH 8.0, containing 1% (w/v)low melting-point agarose, 0.25% bromophenol blue and 40%sucrose). Samples were separated by electrophoresis in 2%agarose gels and visualized by UV illumination after ethidiumbromide staining.
2.7. Immunoblotting of caspase-3
HL-60 cells (1×106) were treated with flavonoids at theindicated concentrations in RPMI 1640 medium. Cell pelletswere lysed in lysis buffer containing 125 mM Tris-HCl pH 6.8,2% SDS, 5% glycerol, and 1% β-mercaptoethanol, and boiledfor 5 min. The samples were separated on 15% SDS-polyacrylamide gel, electrotransferred to a PVDF membrane,immunoblotted with anti-caspase-3 antibody [Stressgen; 1:1000dilution in Tris-buffered saline containing 0.1% Tween-20(TBST) supplemented with 3% nonfat milk] overnight. Afterwashing and incubation with horseradish peroxidase-conjugat-ed anti-rabbit (Amersham Pharmacia Biotech), the antigen–antibody complexes were visualized by enhanced chemilumi-nescence (ECL, Amersham Pharmacia Biotech) using themanufacturer's protocol.
2.8. Immunoblot analysis of poly(ADP-ribose) polymerasedegradation
Induction of apoptosis was also examined by proteolyticcleavage of poly(ADP-ribose) polymerase. After treatments, cellswere pelleted by centrifugation, and resuspended in lysis buffercontaining 25 mM PBS, 0.1 mM phenylmethylsufonylfluorideand protease inhibitors leupeptin, aprotinin and pepstatin A (5 μg/ml each). After centrifugation, the pellet was resuspended in theloading buffer containing 125 mM Tris-HCl, pH 6.8, 2% SDS,5% glycerol, and 1% β-mercaptoethanol. The mixture wassonicated for 30 s at 4 °C and then boiled to 100 °C for 3 min. Thecell lysates were fractionated on a 7.5% polyacrylamide gelcontaining 0.1% SDS and proteins were electrophoreticallytransferred onto a PVDFmembrane.Membranewas blockedwith5%nonfat milk in Tris-buffered saline containing 0.1%Tween-20for 1 h, followed by incubation with anti-poly(ADP-ribose)polymerase polyclonal antibody (Stressgen; 1:3000 dilution in
11S. Rubio et al. / European Journal of Pharmacology 548 (2006) 9–20

TBST supplemented with 3% nonfat milk) overnight. Afterwashing and incubation with anti-rabbit antibody conjugated tohorseradish peroxidase (Amersham Pharmacia Biotech), theantigen–antibody complexes were visualized by enhancedchemiluminiscence (ECL, Amersham Pharmacia Biotech) usingthe manufacturer's protocol. The appearance of an 85 kDacleavage product was used as a measure of apoptosis.
2.9. Detection of cytochrome c release from mitochondria
Release of cytochrome c from mitochondria was detected byWestern blot analysis. After treatments, HL-60 cells werewashed twice with PBS and then suspended in ice-cold buffer[20 mM HEPES (pH 7.5), 1.5 mM MgCl2, 10 mM KCl, 1 mMEDTA, 1 mM EGTA, 1 mM dithiothreitol, 0.1 mM phenyl-methylsulfonylfluoride, and 5 μg/ml leupeptin, aprotinin, andpepstatin A] containing 250 mM sucrose. After 15 minincubation on ice, cells were lysed by pushing them severaltimes through a 22-gauge needle and the lysate spun down at1000 ×g for 5 min at 4 °C. The supernatant fraction wascentrifuged at 105,000×g for 45 min at 4 °C and the resultingsupernatant was used as the soluble cytosolic fraction. Cytosolicproteins (50 μg) were resolved on an SDS/15% polyacrylamidegel and electrotransferred onto a PVDF membrane. Themembrane was probed with monoclonal anti-cytochrome cantibody (BD Transduction Laboratories) (1:250 dilution) andthen with secondary antibody conjugated to horseradishperoxidase. Proteins bands were detected by chemiluminescence(ECL, Amersham Pharmacia Biotech) as described above.
2.10. Assay of caspase activity
After treatments, cells were harvested by centrifugation at1000 ×g for 5 min at 4 °C and washed with PBS, and the cellpellets were kept on ice. The cells were resuspended in cell lysisbuffer (50 mM HEPES, pH 7.4, 1 mM dithiothreitol, 0.1 mMEDTA, 0.1% Chaps) and held on ice for 5 min. Aftercentrifugation for 10 min at 17,000 ×g at 4 °C, the supernatantswere analyzed for protein concentration by the Bradford dye-binding assay and stored at −20 °C until used to study caspasecolorimetric enzymatic activity. Equal amounts of protein(∼20 μg) from different treatments were used, and the assayswere set up on ice. The net increase of absorbance at 405 nmafter incubation at 37 °C was indicative of enzyme activity.Specific labelled substrates for caspase-3, -8 and 9 activitieswere N-acetyl-Asp-Glu-Val-Asp-p-nitroaniline (DEVD-pNA), N-acetyl-Ile-Glu-Thr-Asp-p-nitroaniline (IETD-pNA)and N-acetyl-Leu-Glu-His-Asp-p-nitroaniline (LEHD-pNA)respectively.
3. Results
3.1. Betuletol derivatives inhibit the growth and cell viability ofhuman tumor cell lines
In the present study, we examined the effects of 22 flavonoids(Fig. 1), on the growth of five human tumor cell lines.
Antiproliferative studies on betuletol 3-methyl ether 1 indicatethat this compound displays strong cytotoxic properties in allcell lines assayed, although HL-60 cells was especially sensitiveto this substance with an IC50 as low as 1.8±0.9 μM (Table 1).Since betuletol 3-methyl ether 1 has two hydroxyl groups on C5and C7, respectively, we next obtained the diacetate 2- anddimethyl ether 3-derivatives of betuletol 3-methyl ether 1 inorder to know more about the structural requirement for theantiproliferative action of this compound. Therefore, thecytotoxic studies performed with the di-acetylated compoundindicate that theses changes have no impact on the IC50 in HL-60cells (1.8 ± 0.9 μM vs 1.3 ± 0.4 μM). However, in thedimethylated derivative a dramatic consequence on the growthinhibitory activity is observed. Contrary to betuletol 3-methylether 1 and betuletol 3-methyl ether diacetate 2, betuletol 3, 5, 7-trimethyl ether 3 is ineffective as an antiproliferative agent in allcell lines tested. Since the methoxy group is smaller than theacetyl group, it seems clear that this is not a consequence of sterichindrance. Moreover, if the hydroxyl groups in the betuletol 3-methyl ether 1 interact by hydrogen bridges with vicinity groupson the biological targets, then the presence of the keto groups onthe di-acetyl derivative could be important. One group thatseems to play a key role in determining the potency of thesecompounds on cell viability is the methyl group on position 4′ofthe B ring (2-phenyl group) since betuletol 3-methyl ether 1 issignificantly more potent than 4′,5,7-trihydroxy-3,6-dimethox-yflavone 12 in all cell lines studied.The critical relationship of fruit and vegetable intake and
cancer prevention has been thoroughly documented (Block et al.,1992). Since quercetin 4 is the most abundant flavonoid in thehuman diet we have investigated the effect of a series ofstructurally related flavonoids. The results indicate that methyl-ation of hydroxyl group at position C3 of quercetin 4 yieldsquercetin 3-methyl ether 5, a compound with a higherantiproliferative activity. Cytotoxic effects were observed in allcancer cell lines tested except in HOS cells, which indicates thatthe above chemical change in the quercetin molecule 4 affects cellproliferation in a cell-type specific manner. However, thehydroxyl group located at the 3′ position seems to have littleimportance for the growth inhibitory activity of the abovequercetin derivative since the additional methylation at this levelto yield quercetin 3,3′-dimethyl ether 7 does not significantlychange the IC50 value. The presence of a hydroxyl group in the 3′position of ring B has different consequences depending on theconfiguration of molecule. For example, kaempferol 3-methylether 9 lacks the 3′-hydroxyl group and displays similar IC50 to its3′-hydroxylated counterpart, quercetin 3-methyl ether 5 in all celllines assayed. Similarly, quercetin 3-methyl ether tetracetate 6 andquercetin 3,3′-dimethyl ether triacetate 8 differ only by thepresence of an acetyl- ormethylether-group, respectively, at the 3′position, but they showed similar potency. On the other hand, thepresence of the 3′ hydroxyl group improves the antiproliferativeactivity of axillarin 11 as compared with the 3′-dehydroxylatedcounterpart 12 in all cell types tested except HL-60 cells. When amethoxy group is placed at C6 instead of hydrogen in quercetin 3-methyl ether 5, the impact on the IC50 is dependent on the cell typeunder study. The resulting compound (axillarin 11) is significantly
12 S. Rubio et al. / European Journal of Pharmacology 548 (2006) 9–20

less cytotoxic on HL-60 (IC50=14.3±4.6 μM versus 32.6±8.8 μM) but significantly more cytotoxic on HOS cells(IC50=90.4±27.6 μM versus 25.6±9.6 μM). In contrast, thepresence of this methyl ether group does not change its cytotoxicpotency on A431, HeLa and SK-OV-3 cells.The influence of the hydroxyl group at position 4′ of the B ring
(2-phenyl group) on cell viability was also tested. A dramaticdecrease in the growth inhibitory activity was observed whenquercetin 3,3′-dimethyl ether 7was methylated in that position toproduce 5,7-dihydroxy-3,3′,4′-trimethoxyflavone 10. The IC50increased from 12.2±0.4 μM and 30.6±7.3 μM to N100 μM, inHL-60 and HeLa cells respectively. As occurs with otherssubstitutions, this chemical change affected the cell lines understudy to different degrees. The HOS cell line was resistant to bothquercetin 3,3′-dimethyl ether 7 and 5,7-dihydroxy-3,3′,4′-trimethoxyflavone 10. Thus, acetylation of all free hydroxylgroups locating at positions 3′, 4′, 5 and 7 in the quercetin 3-methyl ether 5 molecule to produce quercetin 3-methyl ethertetracetate 6 greatly improved the cytotoxic activity onHOS cells.In this cell line the IC50 decreased about one order of magnitude
(90.4±27.6 μM versus 9.4±3.0). Unlike the HOS cells, theremaining cell lines were resistant to these chemical changes.Also, an increase in the growth inhibitory activity was clearlyobserved when quercetin 3,3′-dimethyl ether 7 was acetylated atpositions 4′, 5 and 7 to produce quercetin 3,3′-dimethyl ethertriacetate 8. HL-60 cells, A431 cells and particularly HOS cellswere susceptible to these chemicals substitutions.Selected acetylations were also performed on the free
hydroxyl groups of the 4′, 5, 7-trihydroxy-3, 6-dimethoxy-flavone molecule 12. Acetylation of the hydroxyl group atposition 7 yielded a monoacetylated derivative, 7-acetoxy-4′,5-dihydroxy-3,6-dimethoxyflavone 13, that was significantlymore active on HL-60 cells. This effect is abolished when asecond and a third acetyl group are sequentially introduced atposition 5 and at position 4′ to produce the derivativecompounds 5,7-diacetoxy-4′-hydroxy-3,6-dimethoxyflavone14 and 4′,5,7-triacetoxy-3,6-dimethoxyflavone 15 respectively.Another set of flavonoids are those related to the structure of
the low cytotoxic compound naringenin 21. This flavanonedecreased the proliferation rate of the cell lines studied only at
Fig. 1. Structures of the investigated flavonoids.
13S. Rubio et al. / European Journal of Pharmacology 548 (2006) 9–20
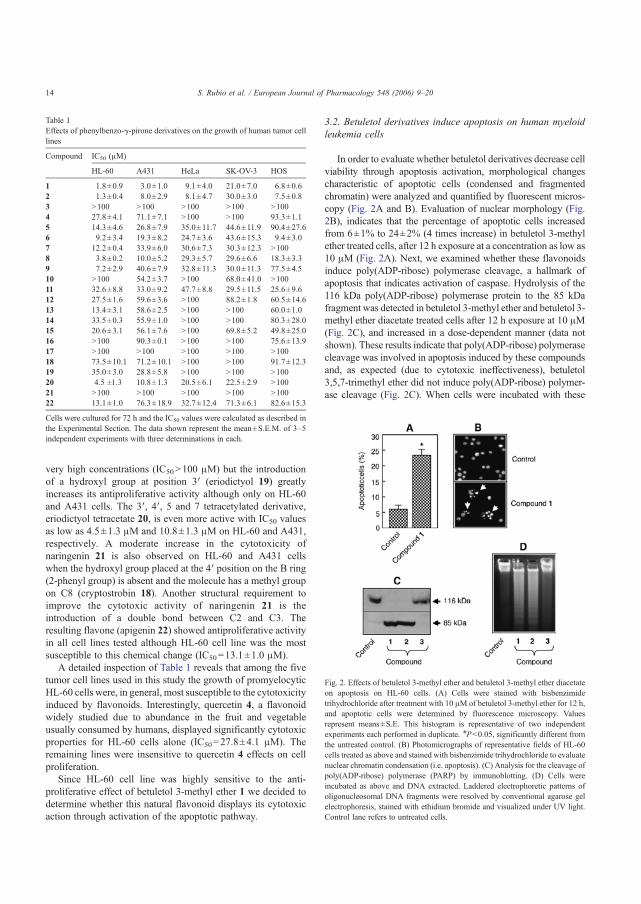
very high concentrations (IC50N100 μM) but the introductionof a hydroxyl group at position 3′ (eriodictyol 19) greatlyincreases its antiproliferative activity although only on HL-60and A431 cells. The 3′, 4′, 5 and 7 tetracetylated derivative,eriodictyol tetracetate 20, is even more active with IC50 valuesas low as 4.5±1.3 μM and 10.8±1.3 μM on HL-60 and A431,respectively. A moderate increase in the cytotoxicity ofnaringenin 21 is also observed on HL-60 and A431 cellswhen the hydroxyl group placed at the 4′ position on the B ring(2-phenyl group) is absent and the molecule has a methyl groupon C8 (cryptostrobin 18). Another structural requirement toimprove the cytotoxic activity of naringenin 21 is theintroduction of a double bond between C2 and C3. Theresulting flavone (apigenin 22) showed antiproliferative activityin all cell lines tested although HL-60 cell line was the mostsusceptible to this chemical change (IC50=13.1±1.0 μM).A detailed inspection of Table 1 reveals that among the five
tumor cell lines used in this study the growth of promyelocyticHL-60 cells were, in general, most susceptible to the cytotoxicityinduced by flavonoids. Interestingly, quercetin 4, a flavonoidwidely studied due to abundance in the fruit and vegetableusually consumed by humans, displayed significantly cytotoxicproperties for HL-60 cells alone (IC50=27.8±4.1 μM). Theremaining lines were insensitive to quercetin 4 effects on cellproliferation.Since HL-60 cell line was highly sensitive to the anti-
proliferative effect of betuletol 3-methyl ether 1 we decided todetermine whether this natural flavonoid displays its cytotoxicaction through activation of the apoptotic pathway.
3.2. Betuletol derivatives induce apoptosis on human myeloidleukemia cells
In order to evaluate whether betuletol derivatives decrease cellviability through apoptosis activation, morphological changescharacteristic of apoptotic cells (condensed and fragmentedchromatin) were analyzed and quantified by fluorescent micros-copy (Fig. 2A and B). Evaluation of nuclear morphology (Fig.2B), indicates that the percentage of apoptotic cells increasedfrom 6±1% to 24±2% (4 times increase) in betuletol 3-methylether treated cells, after 12 h exposure at a concentration as low as10 μM (Fig. 2A). Next, we examined whether these flavonoidsinduce poly(ADP-ribose) polymerase cleavage, a hallmark ofapoptosis that indicates activation of caspase. Hydrolysis of the116 kDa poly(ADP-ribose) polymerase protein to the 85 kDafragment was detected in betuletol 3-methyl ether and betuletol 3-methyl ether diacetate treated cells after 12 h exposure at 10 μM(Fig. 2C), and increased in a dose-dependent manner (data notshown). These results indicate that poly(ADP-ribose) polymerasecleavage was involved in apoptosis induced by these compoundsand, as expected (due to cytotoxic ineffectiveness), betuletol3,5,7-trimethyl ether did not induce poly(ADP-ribose) polymer-ase cleavage (Fig. 2C). When cells were incubated with these
Fig. 2. Effects of betuletol 3-methyl ether and betuletol 3-methyl ether diacetateon apoptosis on HL-60 cells. (A) Cells were stained with bisbenzimidetrihydrochloride after treatment with 10 μM of betuletol 3-methyl ether for 12 h,and apoptotic cells were determined by fluorescence microscopy. Valuesrepresent means±S.E. This histogram is representative of two independentexperiments each performed in duplicate. ⁎Pb0.05, significantly different fromthe untreated control. (B) Photomicrographs of representative fields of HL-60cells treated as above and stained with bisbenzimide trihydrochloride to evaluatenuclear chromatin condensation (i.e. apoptosis). (C) Analysis for the cleavage ofpoly(ADP-ribose) polymerase (PARP) by immunoblotting. (D) Cells wereincubated as above and DNA extracted. Laddered electrophoretic patterns ofoligonucleosomal DNA fragments were resolved by conventional agarose gelelectrophoresis, stained with ethidium bromide and visualized under UV light.Control lane refers to untreated cells.
Table 1Effects of phenylbenzo-γ-pirone derivatives on the growth of human tumor celllines
Compound IC50 (μM)
HL-60 A431 HeLa SK-OV-3 HOS
1 1.8±0.9 3.0±1.0 9.1±4.0 21.0±7.0 6.8±0.62 1.3±0.4 8.0±2.9 8.1±4.7 30.0±3.0 7.5±0.83 N100 N100 N100 N100 N1004 27.8±4.1 71.1±7.1 N100 N100 93.3±1.15 14.3±4.6 26.8±7.9 35.0±11.7 44.6±11.9 90.4±27.66 9.2±3.4 19.3±8.2 24.7±3.6 43.6±15.3 9.4±3.07 12.2±0.4 33.9±6.0 30.6±7.3 30.3±12.3 N1008 3.8±0.2 10.0±5.2 29.3±5.7 29.6±6.6 18.3±3.39 7.2±2.9 40.6±7.9 32.8±11.3 30.0±11.3 77.5±4.510 N100 54.2±3.7 N100 68.0±41.0 N10011 32.6±8.8 33.0±9.2 47.7±8.8 29.5±11.5 25.6±9.612 27.5±1.6 59.6±3.6 N100 88.2±1.8 60.5±14.613 13.4±3.1 58.6±2.5 N100 N100 60.0±1.014 33.5±0.3 55.9±1.0 N100 N100 80.3±28.015 20.6±3.1 56.1±7.6 N100 69.8±5.2 49.8±25.016 N100 90.3±0.1 N100 N100 75.6±13.917 N100 N100 N100 N100 N10018 73.5±10.1 71.2±10.1 N100 N100 91.7±12.319 35.0±3.0 28.8±5.8 N100 N100 N10020 4.5 ±1.3 10.8±1.3 20.5±6.1 22.5±2.9 N10021 N100 N100 N100 N100 N10022 13.1±1.0 76.3±18.9 32.7±12.4 71.3±6.1 82.6±15.3
Cells were cultured for 72 h and the IC50 values were calculated as described inthe Experimental Section. The data shown represent the mean±S.E.M. of 3–5independent experiments with three determinations in each.
14 S. Rubio et al. / European Journal of Pharmacology 548 (2006) 9–20
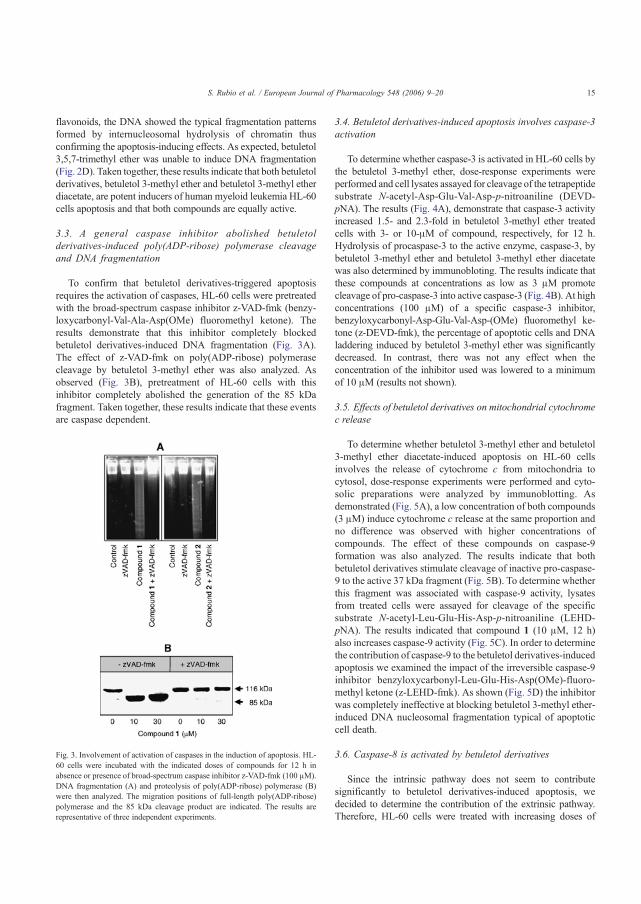
flavonoids, the DNA showed the typical fragmentation patternsformed by internucleosomal hydrolysis of chromatin thusconfirming the apoptosis-inducing effects. As expected, betuletol3,5,7-trimethyl ether was unable to induce DNA fragmentation(Fig. 2D). Taken together, these results indicate that both betuletolderivatives, betuletol 3-methyl ether and betuletol 3-methyl etherdiacetate, are potent inducers of human myeloid leukemia HL-60cells apoptosis and that both compounds are equally active.
3.3. A general caspase inhibitor abolished betuletolderivatives-induced poly(ADP-ribose) polymerase cleavageand DNA fragmentation
To confirm that betuletol derivatives-triggered apoptosisrequires the activation of caspases, HL-60 cells were pretreatedwith the broad-spectrum caspase inhibitor z-VAD-fmk (benzy-loxycarbonyl-Val-Ala-Asp(OMe) fluoromethyl ketone). Theresults demonstrate that this inhibitor completely blockedbetuletol derivatives-induced DNA fragmentation (Fig. 3A).The effect of z-VAD-fmk on poly(ADP-ribose) polymerasecleavage by betuletol 3-methyl ether was also analyzed. Asobserved (Fig. 3B), pretreatment of HL-60 cells with thisinhibitor completely abolished the generation of the 85 kDafragment. Taken together, these results indicate that these eventsare caspase dependent.
3.4. Betuletol derivatives-induced apoptosis involves caspase-3activation
To determine whether caspase-3 is activated in HL-60 cells bythe betuletol 3-methyl ether, dose-response experiments wereperformed and cell lysates assayed for cleavage of the tetrapeptidesubstrate N-acetyl-Asp-Glu-Val-Asp-p-nitroaniline (DEVD-pNA). The results (Fig. 4A), demonstrate that caspase-3 activityincreased 1.5- and 2.3-fold in betuletol 3-methyl ether treatedcells with 3- or 10-μM of compound, respectively, for 12 h.Hydrolysis of procaspase-3 to the active enzyme, caspase-3, bybetuletol 3-methyl ether and betuletol 3-methyl ether diacetatewas also determined by immunobloting. The results indicate thatthese compounds at concentrations as low as 3 μM promotecleavage of pro-caspase-3 into active caspase-3 (Fig. 4B). At highconcentrations (100 μM) of a specific caspase-3 inhibitor,benzyloxycarbonyl-Asp-Glu-Val-Asp-(OMe) fluoromethyl ke-tone (z-DEVD-fmk), the percentage of apoptotic cells and DNAladdering induced by betuletol 3-methyl ether was significantlydecreased. In contrast, there was not any effect when theconcentration of the inhibitor used was lowered to a minimumof 10 μM (results not shown).
3.5. Effects of betuletol derivatives on mitochondrial cytochromec release
To determine whether betuletol 3-methyl ether and betuletol3-methyl ether diacetate-induced apoptosis on HL-60 cellsinvolves the release of cytochrome c from mitochondria tocytosol, dose-response experiments were performed and cyto-solic preparations were analyzed by immunoblotting. Asdemonstrated (Fig. 5A), a low concentration of both compounds(3 μM) induce cytochrome c release at the same proportion andno difference was observed with higher concentrations ofcompounds. The effect of these compounds on caspase-9formation was also analyzed. The results indicate that bothbetuletol derivatives stimulate cleavage of inactive pro-caspase-9 to the active 37 kDa fragment (Fig. 5B). To determine whetherthis fragment was associated with caspase-9 activity, lysatesfrom treated cells were assayed for cleavage of the specificsubstrate N-acetyl-Leu-Glu-His-Asp-p-nitroaniline (LEHD-pNA). The results indicated that compound 1 (10 μM, 12 h)also increases caspase-9 activity (Fig. 5C). In order to determinethe contribution of caspase-9 to the betuletol derivatives-inducedapoptosis we examined the impact of the irreversible caspase-9inhibitor benzyloxycarbonyl-Leu-Glu-His-Asp(OMe)-fluoro-methyl ketone (z-LEHD-fmk). As shown (Fig. 5D) the inhibitorwas completely ineffective at blocking betuletol 3-methyl ether-induced DNA nucleosomal fragmentation typical of apoptoticcell death.
3.6. Caspase-8 is activated by betuletol derivatives
Since the intrinsic pathway does not seem to contributesignificantly to betuletol derivatives-induced apoptosis, wedecided to determine the contribution of the extrinsic pathway.Therefore, HL-60 cells were treated with increasing doses of
Fig. 3. Involvement of activation of caspases in the induction of apoptosis. HL-60 cells were incubated with the indicated doses of compounds for 12 h inabsence or presence of broad-spectrum caspase inhibitor z-VAD-fmk (100 μM).DNA fragmentation (A) and proteolysis of poly(ADP-ribose) polymerase (B)were then analyzed. The migration positions of full-length poly(ADP-ribose)polymerase and the 85 kDa cleavage product are indicated. The results arerepresentative of three independent experiments.
15S. Rubio et al. / European Journal of Pharmacology 548 (2006) 9–20
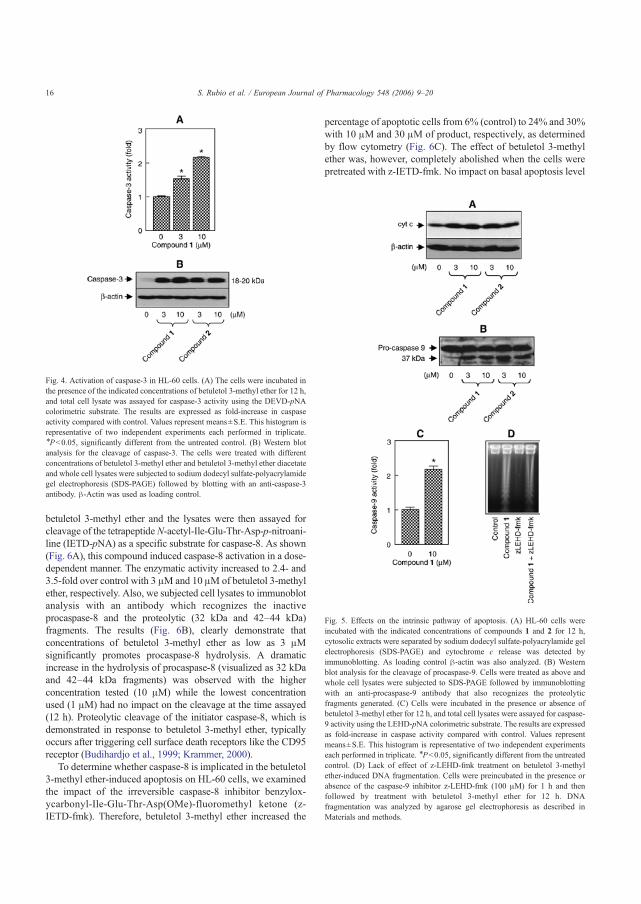
betuletol 3-methyl ether and the lysates were then assayed forcleavage of the tetrapeptideN-acetyl-Ile-Glu-Thr-Asp-p-nitroani-line (IETD-pNA) as a specific substrate for caspase-8. As shown(Fig. 6A), this compound induced caspase-8 activation in a dose-dependent manner. The enzymatic activity increased to 2.4- and3.5-fold over control with 3 μM and 10 μM of betuletol 3-methylether, respectively. Also, we subjected cell lysates to immunoblotanalysis with an antibody which recognizes the inactiveprocaspase-8 and the proteolytic (32 kDa and 42–44 kDa)fragments. The results (Fig. 6B), clearly demonstrate thatconcentrations of betuletol 3-methyl ether as low as 3 μMsignificantly promotes procaspase-8 hydrolysis. A dramaticincrease in the hydrolysis of procaspase-8 (visualized as 32 kDaand 42–44 kDa fragments) was observed with the higherconcentration tested (10 μM) while the lowest concentrationused (1 μM) had no impact on the cleavage at the time assayed(12 h). Proteolytic cleavage of the initiator caspase-8, which isdemonstrated in response to betuletol 3-methyl ether, typicallyoccurs after triggering cell surface death receptors like the CD95receptor (Budihardjo et al., 1999; Krammer, 2000).To determine whether caspase-8 is implicated in the betuletol
3-methyl ether-induced apoptosis on HL-60 cells, we examinedthe impact of the irreversible caspase-8 inhibitor benzylox-ycarbonyl-Ile-Glu-Thr-Asp(OMe)-fluoromethyl ketone (z-IETD-fmk). Therefore, betuletol 3-methyl ether increased the
percentage of apoptotic cells from 6% (control) to 24% and 30%with 10 μM and 30 μM of product, respectively, as determinedby flow cytometry (Fig. 6C). The effect of betuletol 3-methylether was, however, completely abolished when the cells werepretreated with z-IETD-fmk. No impact on basal apoptosis level
Fig. 5. Effects on the intrinsic pathway of apoptosis. (A) HL-60 cells wereincubated with the indicated concentrations of compounds 1 and 2 for 12 h,cytosolic extracts were separated by sodium dodecyl sulfate-polyacrylamide gelelectrophoresis (SDS-PAGE) and cytochrome c release was detected byimmunoblotting. As loading control β-actin was also analyzed. (B) Westernblot analysis for the cleavage of procaspase-9. Cells were treated as above andwhole cell lysates were subjected to SDS-PAGE followed by immunoblottingwith an anti-procaspase-9 antibody that also recognizes the proteolyticfragments generated. (C) Cells were incubated in the presence or absence ofbetuletol 3-methyl ether for 12 h, and total cell lysates were assayed for caspase-9 activity using the LEHD-pNA colorimetric substrate. The results are expressedas fold-increase in caspase activity compared with control. Values representmeans±S.E. This histogram is representative of two independent experimentseach performed in triplicate. ⁎Pb0.05, significantly different from the untreatedcontrol. (D) Lack of effect of z-LEHD-fmk treatment on betuletol 3-methylether-induced DNA fragmentation. Cells were preincubated in the presence orabsence of the caspase-9 inhibitor z-LEHD-fmk (100 μM) for 1 h and thenfollowed by treatment with betuletol 3-methyl ether for 12 h. DNAfragmentation was analyzed by agarose gel electrophoresis as described inMaterials and methods.
Fig. 4. Activation of caspase-3 in HL-60 cells. (A) The cells were incubated inthe presence of the indicated concentrations of betuletol 3-methyl ether for 12 h,and total cell lysate was assayed for caspase-3 activity using the DEVD-pNAcolorimetric substrate. The results are expressed as fold-increase in caspaseactivity compared with control. Values represent means±S.E. This histogram isrepresentative of two independent experiments each performed in triplicate.⁎Pb0.05, significantly different from the untreated control. (B) Western blotanalysis for the cleavage of caspase-3. The cells were treated with differentconcentrations of betuletol 3-methyl ether and betuletol 3-methyl ether diacetateand whole cell lysates were subjected to sodium dodecyl sulfate-polyacrylamidegel electrophoresis (SDS-PAGE) followed by blotting with an anti-caspase-3antibody. β-Actin was used as loading control.
16 S. Rubio et al. / European Journal of Pharmacology 548 (2006) 9–20
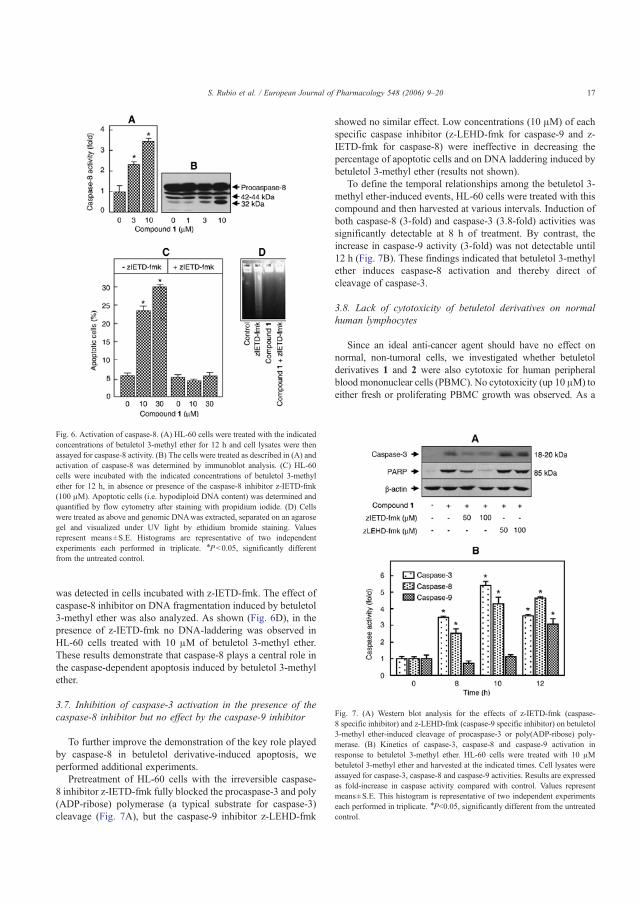
was detected in cells incubated with z-IETD-fmk. The effect ofcaspase-8 inhibitor on DNA fragmentation induced by betuletol3-methyl ether was also analyzed. As shown (Fig. 6D), in thepresence of z-IETD-fmk no DNA-laddering was observed inHL-60 cells treated with 10 μM of betuletol 3-methyl ether.These results demonstrate that caspase-8 plays a central role inthe caspase-dependent apoptosis induced by betuletol 3-methylether.
3.7. Inhibition of caspase-3 activation in the presence of thecaspase-8 inhibitor but no effect by the caspase-9 inhibitor
To further improve the demonstration of the key role playedby caspase-8 in betuletol derivative-induced apoptosis, weperformed additional experiments.Pretreatment of HL-60 cells with the irreversible caspase-
8 inhibitor z-IETD-fmk fully blocked the procaspase-3 and poly(ADP-ribose) polymerase (a typical substrate for caspase-3)cleavage (Fig. 7A), but the caspase-9 inhibitor z-LEHD-fmk
showed no similar effect. Low concentrations (10 μM) of eachspecific caspase inhibitor (z-LEHD-fmk for caspase-9 and z-IETD-fmk for caspase-8) were ineffective in decreasing thepercentage of apoptotic cells and on DNA laddering induced bybetuletol 3-methyl ether (results not shown).To define the temporal relationships among the betuletol 3-
methyl ether-induced events, HL-60 cells were treated with thiscompound and then harvested at various intervals. Induction ofboth caspase-8 (3-fold) and caspase-3 (3.8-fold) activities wassignificantly detectable at 8 h of treatment. By contrast, theincrease in caspase-9 activity (3-fold) was not detectable until12 h (Fig. 7B). These findings indicated that betuletol 3-methylether induces caspase-8 activation and thereby direct ofcleavage of caspase-3.
3.8. Lack of cytotoxicity of betuletol derivatives on normalhuman lymphocytes
Since an ideal anti-cancer agent should have no effect onnormal, non-tumoral cells, we investigated whether betuletolderivatives 1 and 2 were also cytotoxic for human peripheralblood mononuclear cells (PBMC). No cytotoxicity (up 10 μM) toeither fresh or proliferating PBMC growth was observed. As a
Fig. 7. (A) Western blot analysis for the effects of z-IETD-fmk (caspase-8 specific inhibitor) and z-LEHD-fmk (caspase-9 specific inhibitor) on betuletol3-methyl ether-induced cleavage of procaspase-3 or poly(ADP-ribose) poly-merase. (B) Kinetics of caspase-3, caspase-8 and caspase-9 activation inresponse to betuletol 3-methyl ether. HL-60 cells were treated with 10 μMbetuletol 3-methyl ether and harvested at the indicated times. Cell lysates wereassayed for caspase-3, caspase-8 and caspase-9 activities. Results are expressedas fold-increase in caspase activity compared with control. Values representmeans±S.E. This histogram is representative of two independent experimentseach performed in triplicate. ⁎Pb0.05, significantly different from the untreatedcontrol.
Fig. 6. Activation of caspase-8. (A) HL-60 cells were treated with the indicatedconcentrations of betuletol 3-methyl ether for 12 h and cell lysates were thenassayed for caspase-8 activity. (B) The cells were treated as described in (A) andactivation of caspase-8 was determined by immunoblot analysis. (C) HL-60cells were incubated with the indicated concentrations of betuletol 3-methylether for 12 h, in absence or presence of the caspase-8 inhibitor z-IETD-fmk(100 μM). Apoptotic cells (i.e. hypodiploid DNA content) was determined andquantified by flow cytometry after staining with propidium iodide. (D) Cellswere treated as above and genomic DNAwas extracted, separated on an agarosegel and visualized under UV light by ethidium bromide staining. Valuesrepresent means±S.E. Histograms are representative of two independentexperiments each performed in triplicate. ⁎Pb0.05, significantly differentfrom the untreated control.
17S. Rubio et al. / European Journal of Pharmacology 548 (2006) 9–20
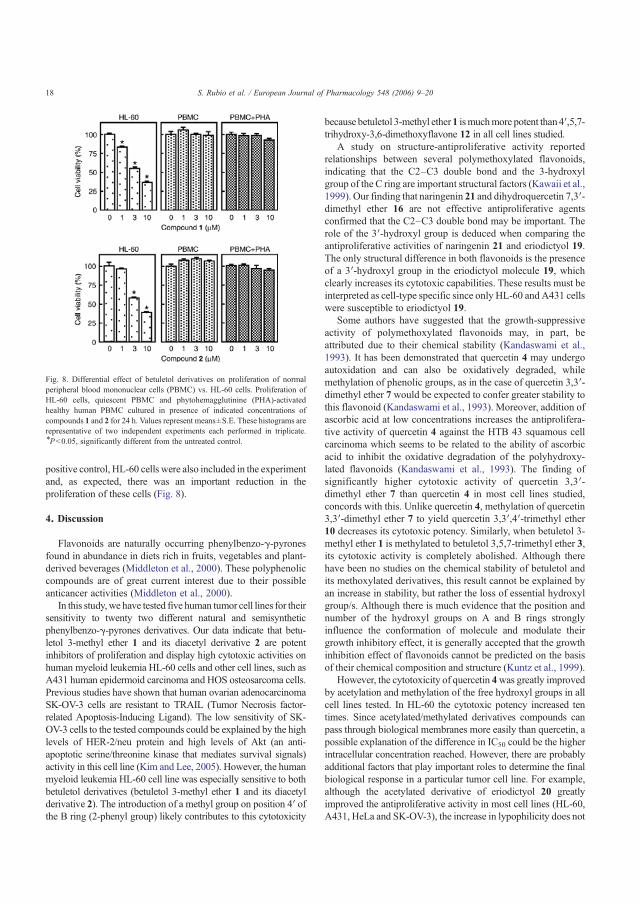
positive control, HL-60 cells were also included in the experimentand, as expected, there was an important reduction in theproliferation of these cells (Fig. 8).
4. Discussion
Flavonoids are naturally occurring phenylbenzo-γ-pyronesfound in abundance in diets rich in fruits, vegetables and plant-derived beverages (Middleton et al., 2000). These polyphenoliccompounds are of great current interest due to their possibleanticancer activities (Middleton et al., 2000).In this study,we have tested five human tumor cell lines for their
sensitivity to twenty two different natural and semisyntheticphenylbenzo-γ-pyrones derivatives. Our data indicate that betu-letol 3-methyl ether 1 and its diacetyl derivative 2 are potentinhibitors of proliferation and display high cytotoxic activities onhuman myeloid leukemia HL-60 cells and other cell lines, such asA431 human epidermoid carcinoma and HOS osteosarcoma cells.Previous studies have shown that human ovarian adenocarcinomaSK-OV-3 cells are resistant to TRAIL (Tumor Necrosis factor-related Apoptosis-Inducing Ligand). The low sensitivity of SK-OV-3 cells to the tested compounds could be explained by the highlevels of HER-2/neu protein and high levels of Akt (an anti-apoptotic serine/threonine kinase that mediates survival signals)activity in this cell line (Kim and Lee, 2005). However, the humanmyeloid leukemia HL-60 cell line was especially sensitive to bothbetuletol derivatives (betuletol 3-methyl ether 1 and its diacetylderivative 2). The introduction of a methyl group on position 4′ ofthe B ring (2-phenyl group) likely contributes to this cytotoxicity
because betuletol 3-methyl ether1 ismuchmore potent than 4′,5,7-trihydroxy-3,6-dimethoxyflavone 12 in all cell lines studied.A study on structure-antiproliferative activity reported
relationships between several polymethoxylated flavonoids,indicating that the C2–C3 double bond and the 3-hydroxylgroup of the C ring are important structural factors (Kawaii et al.,1999). Our finding that naringenin 21 and dihydroquercetin 7,3′-dimethyl ether 16 are not effective antiproliferative agentsconfirmed that the C2–C3 double bond may be important. Therole of the 3′-hydroxyl group is deduced when comparing theantiproliferative activities of naringenin 21 and eriodictyol 19.The only structural difference in both flavonoids is the presenceof a 3′-hydroxyl group in the eriodictyol molecule 19, whichclearly increases its cytotoxic capabilities. These results must beinterpreted as cell-type specific since only HL-60 and A431 cellswere susceptible to eriodictyol 19.Some authors have suggested that the growth-suppressive
activity of polymethoxylated flavonoids may, in part, beattributed due to their chemical stability (Kandaswami et al.,1993). It has been demonstrated that quercetin 4 may undergoautoxidation and can also be oxidatively degraded, whilemethylation of phenolic groups, as in the case of quercetin 3,3′-dimethyl ether 7 would be expected to confer greater stability tothis flavonoid (Kandaswami et al., 1993). Moreover, addition ofascorbic acid at low concentrations increases the antiprolifera-tive activity of quercetin 4 against the HTB 43 squamous cellcarcinoma which seems to be related to the ability of ascorbicacid to inhibit the oxidative degradation of the polyhydroxy-lated flavonoids (Kandaswami et al., 1993). The finding ofsignificantly higher cytotoxic activity of quercetin 3,3′-dimethyl ether 7 than quercetin 4 in most cell lines studied,concords with this. Unlike quercetin 4, methylation of quercetin3,3′-dimethyl ether 7 to yield quercetin 3,3′,4′-trimethyl ether10 decreases its cytotoxic potency. Similarly, when betuletol 3-methyl ether 1 is methylated to betuletol 3,5,7-trimethyl ether 3,its cytotoxic activity is completely abolished. Although therehave been no studies on the chemical stability of betuletol andits methoxylated derivatives, this result cannot be explained byan increase in stability, but rather the loss of essential hydroxylgroup/s. Although there is much evidence that the position andnumber of the hydroxyl groups on A and B rings stronglyinfluence the conformation of molecule and modulate theirgrowth inhibitory effect, it is generally accepted that the growthinhibition effect of flavonoids cannot be predicted on the basisof their chemical composition and structure (Kuntz et al., 1999).However, the cytotoxicity of quercetin 4was greatly improved
by acetylation and methylation of the free hydroxyl groups in allcell lines tested. In HL-60 the cytotoxic potency increased tentimes. Since acetylated/methylated derivatives compounds canpass through biological membranes more easily than quercetin, apossible explanation of the difference in IC50 could be the higherintracellular concentration reached. However, there are probablyadditional factors that play important roles to determine the finalbiological response in a particular tumor cell line. For example,although the acetylated derivative of eriodictyol 20 greatlyimproved the antiproliferative activity in most cell lines (HL-60,A431, HeLa and SK-OV-3), the increase in lypophilicity does not
Fig. 8. Differential effect of betuletol derivatives on proliferation of normalperipheral blood mononuclear cells (PBMC) vs. HL-60 cells. Proliferation ofHL-60 cells, quiescent PBMC and phytohemagglutinine (PHA)-activatedhealthy human PBMC cultured in presence of indicated concentrations ofcompounds 1 and 2 for 24 h. Values represent means±S.E. These histograms arerepresentative of two independent experiments each performed in triplicate.⁎Pb0.05, significantly different from the untreated control.
18 S. Rubio et al. / European Journal of Pharmacology 548 (2006) 9–20

explain its ineffectiveness in HOS cells. Also, among the 22compounds presented in Table 1, betuletol 3-methyl ether 1 andits diacetate-derivative 2 exhibited the highest cytotoxic activitiesagainst all human cancer cell lines evaluated. Inmost cell lines theIC50 values for these betuletol derivatives were lower than 10μM,although in HL-60 cells these values were about 1 μM.As for all of the agents used or developed for cancer
treatment, selectivity toward cancer cells is an importantcriterion. We therefore compared the effects of compounds 1and 2 between HL-60 cells and human peripheral bloodmononuclear cells (PBMC). Interestingly, dose-response stud-ies revealed that quiescent PBMC and proliferating PBMCwereresistant toward both betuletol derivatives 1 and 2.In this work, we demonstrate that the exposure of HL-60
cells to betuletol 3-methyl ether and its diacetyl derivativeexerts a strong antiproliferative effect and both compoundsdisplayed similar IC50 values. Among the number of pathwaysthat can be exploited for cancer prevention and treatment,apoptosis is a physiological process by which cells are removedwhen an agent damages their DNA, therefore compounds thatcan induce apoptosis may be useful in management and therapyof cancer. Although previous studies have documented theinduction of apoptotic cell death and DNA fragmentation byphenylbenzo-γ-pirones derivatives in the human myeloidleukemia HL-60 cells (Lee et al., 2002) betuletol derivativeshave yet to be assessed. Our results show that betuletol 3-methylether and its diacetyl derivative induced apoptosis in thehematopoietic cell line HL-60.Both betuletol derivatives promote the formation of apoptotic
bodies and the internucleosomal degradation of DNA, resulting inthe formation and eventual release of oligonucleosomal DNAfragments. The apoptosis induction was accompanied by poly(ADP-ribose) polymerase cleavage which is considered to be oneof the hallmarks of apoptosis (Nicholson and Thornberry, 1997).We report in the present study that z-VAD-fmk, a broad-
spectrum caspase inhibitor, completely abrogated the morpho-logical changes associated with apoptosis, DNA fragmentationand the cleavage of poly(ADP-ribose) polymerase, whichindicated that caspases are essential components in betuletol3-methyl ether-induced apoptosis.Caspase-3 is the most active effector caspase, in both the
intrinsic and extrinsic pathways where it is processed andactivated by caspase-9 and caspase-8 respectively. Caspase-3has been found to be involved in leukemia-cell apoptosis inducedby cytotoxic agents such as 1-β-D-arabinofuranosylcytosine,mitoxantrone, etoposide and CPT-11 (Motwani et al., 1999). Ahigh level of caspase-3 activation and processing was found aftertreatment of HL-60 cells with betuletol derivatives 1 and 2.Previous studies have already demonstrated that most of
cytotoxic agents induce the release of mitochondrial cyto-chrome c (Szewczyk and Wojtczak, 2002), and this proteintriggers a caspase-dependent assembly of the apoptosome (VanGurp et al., 2003). Although we demonstrate that betuletolderivatives initiated redistribution of cytochrome c into thecytosol and procaspase-9 processing, our results indicate thatthe role of caspase-9 in the betuletol induced-apoptosis overallis, at most, minimal. Moreover, we analyzed the kinetic patterns
of cytochrome c release in time course experiments afterbetuletol derivatives treatments. The cytochrome c release wasnot an early event in the apoptotic cell death induced by bothbetuletol derivatives (results not shown). Although the exposureof HL-60 cells to both betuletol derivatives resulted in increasedcytosolic cytochrome c, this response was not detected untilafter 12 h of treatment. Consistently with this finding, theactivation of caspase-9 (as determined by a colorimetric assayand western blot), was not observed until the same time period.Furthermore, there have been ample examples of cross-talkbetween cell death receptor-mediated signaling through thecaspase-8 and the apoptosome cascade. Caspase-8 directlyactivates downstream effector caspases and also cleaves Bid andtriggers mitochondrial damage that in turn leads to cytochromec release. Studies have demonstrated that the intrinsic pathwayfor apoptosis initiation is controlled by members of the Bcl-2family (Marsden and Strasser, 2003; Borner, 2003), which arecrucial regulators of apoptosis in mammalian cells. However,we were unable to demonstrate alterations in the expression orin the mobility of Bcl-2 on sodium dodecyl sulfate-polyacryl-amide gel electrophoresis, a phenomenon that generally(although not invariably) accompanies perturbations in phos-phorylation state (Haldar et al., 1995) (data not shown).Previous studies have shown that apoptosis caused by anti-
cancer drugsmay bemediated via theCD95 system (Friesen et al.,1996), although many drugs seem to initiate the mitochondrialpathway directly. An ample variety of mechanisms of resistanceto apoptosis that interfere at different levels of apoptosissignalling have been described (Igney and Krammer, 2002).Thus a search for novel agents that target aberrant apoptosispathways or induce alternative death pathways may be suited toovercome treatment resistance.Using irreversible inhibitors for selected caspases, we
attempted to further delineate the pathways of caspase activationand their relationship to poly(ADP-ribose) polymerase cleavageand DNA fragmentation induced by betuletol derivatives. Theeffective blockage of apoptosis induction by a cell-permeableinhibitor of caspase-8, z-IETD-fmk, but not by a cell-permeableinhibitor of caspase-9, z-LEHD-fmk (also confirmed by kineticstudies of caspases activation) supports a model in whichbetuletol 3-methyl ether induces apoptosis by a caspase-8 mediated mechanism.
5. Conclusion
Our results indicate that betuletol 3-methyl ether inhibitshuman HL-60 cell growth and induces apoptotic cell deathinvolving the extrinsic pathway and could be useful in thedevelopment of novel anticancer agents.
Acknowledgments
This work was supported by Grants from the Ministerio deEducación y Ciencia of Spain and FEDER (SAF2004-07928),from Consejería de Educación, Cultura y Deportes (CanaryIslands Government) and FEDER (GRUP2004/44) and Insti-tuto Canario de Investigación del Cáncer (08/2004). We thank J.
19S. Rubio et al. / European Journal of Pharmacology 548 (2006) 9–20

Estévez (Hospital Universitario Insular de Gran Canaria) for hiscollaboration in the Western blot assays, J.C. Hernández fortechnical assistance as well as the encouragement and support ofLennart Loven and Dr. Jaime Bermejo.
References
Block, G., Patterson, B., Subar, A., 1992. Fruit, vegetables and cancerprevention: a review of the epidemiological evidence. Nutr. Cancer 18,1–29.
Boldin, M., Goncharov, T., Goltsev, Y., Wallach, D., 1996. Involvement ofMACH, a novel MORT1/FADD-interacting protease, in Fas/APO-1- andTNF receptor-induced cell death. Cell 85, 803–815.
Borner, C., 2003. The Bcl-2 protein family: sensors and checkpoints for life-or-death decisions. Mol. Immunol. 39, 615–647.
Budihardjo, I., Oliver, H., Lutter, M., Luo, X., Wang, X., 1999. Biochemicalpathways of caspase activation during apoptosis. Annu. Rev. Cell Dev. Biol.15, 269–290.
Byrne, L.T., Cannon, J.R., Gawad, D.H., Joshi, B.S., Skelton, B.W., Toia, R.F.,White, A.H., 1982. The crystal structure of (S)-(−)-6-Bromo-5,7-dihydroxy-8-methyl-2-phenyl-2,3-dihydro-4H-1-benzopyran-4-one [(−)-6-bromocryp-tostrobin] and a 13C N.M.R. Study of (±)-cryptostrobin and relatedsubstances. Revision of the structures of the natural products (±)-lawinal,unonal, 7-O-methylunonal and isounonal. Aust. J. Chem. 35, 1851–1858.
Chipuk, J.E., Green, D.R., 2005. Do inducers of apoptosis trigger caspase-independent cell death? Nat. Rev., Mol. Cell Biol. 6, 268–275.
Cohen, G.M., 1997. Caspases: the executioners of apoptosis. Biochem. J. 326,1–16.
Friesen, C., Herr, I., Krammer, P.H., Debatin, K.M., 1996. Involvement of theCD95 (APO-1/FAS) receptor/ligand system in drug-induced apoptosis inleukemia cells. Nat. Med. 2, 574–577.
González, A.G., Bermejo, J., Triana, J., López, M., Eiroa, J.L., 1990.Sesquiterpene lactones from Tanacetum ferulaceum. Phytochemistry 29,2339–2341.
González, A.G., Bermejo, J., Triana, J., Eiroa, J.L., López, M., 1992a.Germacranolides from Allagopappus viscosissimus. Phytochemistry 31,330–331.
González, A.G., Bermejo, J., Triana, J., López, M., Eiroa, J.L., 1992b.Sesquiterpene lactones and other constituents of Tanacetum species.Phytochemistry 31, 1821–1822.
González, A.G., Bermejo, J., Triana, J., Eiroa, J.L., López,M., 1995. Sesquiterpenelactones and other constituents of Allagopappus species. J. Nat. Prod. 58,432–437.
Haldar, S., Jena, N., Croce, C.M., 1995. Inactivation of Bcl-2 by phosphory-lation. Proc. Natl. Acad. Sci. U. S. A. 92, 4507–4511.
Igney, F.H., Krammer, P.H., 2002. Death and anti-death: tumour resistance toapoptosis. Nat. Rev., Cancer 2, 277–288.
Kandaswami, C., Perkins, E., Soloniuk, D.S., Drzewiecki, G., Middleton, E.,1993. Ascorbic acid-enhanced antiproliferative effect of flavonoids onsquamous cell carcinoma in vitro. Anticancer Drugs 4, 91–96.
Kawaii, S., Tomono, Y., Katase, E., Ogawa, K., Yano, M., 1999. Antiprolifera-tive activity of flavonoids on several cancer cell lines. Biosci. Biotechnol.Biochem. 63, 896–899.
Kim, K.M., Lee, Y.J., 2005. Role of HER-2/neu signaling in sensitivity to tumornecrosis factor-related apoptosis-inducing ligand: enhancement of TRAIL-mediated apoptosis by amiloride. J. Cell. Biochem. 96, 376–389.
Kluck, R.M., Bozy-Wetzel, E., Green, D.R., Newmeyer, D.D., 1997. The releaseof cytochrome c from mitochondria: a primary site for Bcl-2 regulation ofapoptosis. Science 275, 1132–1136.
Ko, C.H., Shen, S.C., Lin, H.Y., Hou, W.C., Lee, W.R., Yang, L.L., Chen, Y.C.,2002. Flavanones structure related inhibition on TPA-induced tumorpromotion through suppression of extracellular signal-regulated proteinkinases: involvement of prostaglandin E2 in anti-promotive process. J. Cell.Physiol. 193, 93–102.
Krammer, P.H., 2000. CD95's deadly mission in the immune system. Nature407, 789–795.
Kuntz, S., Wenzel, U., Daniel, H., 1999. Comparative analysis of the effects offlavonoids on proliferation, cytotoxicity, and apoptosis in human coloncancer cell lines. Eur. J. Nutr. 38, 133–142.
Lee, W.R., Shen, S.C., Lin, H.Y., Hou, W.C., Yang, L.L., Chen, Y.C., 2002.Wogonin and fisetin induce apoptosis in human promyeloleukemic cells,accompanied by a decrease of reactive oxygen species, and activation ofcaspase 3 and Ca2+-dependent endonuclease. Biochem. Pharmacol. 63,225–236.
Li, P., Nijhawan, D., Budihardjo, I., Srinivasula, S.M., Ahmad, M., Alnemri, E.S.,Wang, X., 1997. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell 91, 479–489.
Liu, X., Kim, C., Yang, J., Jemmerson, R., Wang, X., 1996. Induction ofapoptotic program in cell-free extracts: requirement for dATP andcytochrome c. Cell 86, 147–157.
Marsden, V.S., Strasser, A., 2003. Control of apoptosis in the immune system:Bcl-2, BH3-only proteins and more. Annu. Rev. Immunol. 21, 71–105.
Middleton, E., Kandaswami, C., Theoharides, T.C., 2000. The effects of plantflavonoids on mammalian cells: implications for inflammation, heartdisease, and cancer. Pharmacol. Rev. 52, 673–751.
Motwani, M., Delohery, T.M., Schwartz, G.K., 1999. Sequential dependentenhancement of caspase activation and apoptosis by flavopiridol onpaclitaxel-treated human gastric and breast cancer cells. Clin. Cancer Res.5, 1876–1883.
Muzio,M., Chinnaiyan, A.M., Kischkel, F.C., O'Rourke,K., Schevchenko,A., Ni,J., Scaffidi, C., Bretz, J.D., Zhang, M., Gentz, R., Mann, M., Krammer, P.H.,Peter, M.E., Dixit, V.M., 1996. FLICE, a novel FADD-homologous ICE/CED-3-like protease, is recruited to theCD95 (Fas/APO-1) death-inducing signallingcomplex. Cell 85, 817–827.
Nicholson, D.W., Thornberry, N.A., 1997. Caspases: killer proteases. TrendsBiochem. Sci. 22, 299–306.
Rivero, A., Quintana, J., Eiroa, J.L., López, M., Triana, J., Bermejo, J., Estévez,F., 2003. Potent induction of apoptosis by germacranolide sesquiterpenelactones on human myeloid leukemia cells. Eur. J. Pharmacol. 482, 77–84.
Srinivasula, S., Ahmad, M., Alnemri, T., Alnemri, E., 1998. Autoactivation ofprocaspase-9 by Apaf-1 mediated oligomerization. Mol. Cell 1, 949–957.
Stennicke, H.R., Jurgensmeier, J.M., Shin, H., Deveraux, Q., Wolf, B.B., Yang,X., Zhou, Q., Ellerby, H.M., Ellerby, L.M., Bredesen, D., Green, D.R., Reed,J.C., Froelich, C.J., Salvesen, G.S., 1998. Procaspase-3 is a majorphysiologic target of caspase-8. J. Biol. Chem. 273, 27084–27090.
Szewczyk, A., Wojtczak, L., 2002. Mitochondria as a pharmacological target.Pharmacol. Rev. 54, 101–127.
Thornberry, N.A., Lazebnik, Y., 1998. Caspases: enemies within. Science 281,1312–1316.
Triana, J., López, M., Pérez, F.J., González-Platas, J., Quintana, J., Estévez, F.,León, F., Bermejo, J., 2005. Sesquiterpenoids from Pulicaria canariensisand their cytotoxic activities. J. Nat. Prod. 68, 523–531.
Van Gurp, M., Festjens, N., van Loo, G., Saelens, X., Vandenabeele, P., 2003.Mitochondrial intermembrane proteins in cell death. Biochem. Biophys.Res. Commun. 304, 487–497.
Yang, J., Liu, X., Bhalla, K., Kim, C.N., Ibrado, A.M., Cai, J., Peng, T.I., Jones,D.P., Wang, X., 1997. Prevention of apoptosis by Bcl-2: release ofcytochrome c from mitochondria blocked. Science 275, 1129–1132.
20 S. Rubio et al. / European Journal of Pharmacology 548 (2006) 9–20

Carcinogenesis vol.28 no.10 pp.2105–2113, 2007doi:10.1093/carcin/bgm131Advance Access publication June 4, 2007
Acetyl derivative of quercetin 3-methyl ether-induced cell death in human leukemiacells is amplified by the inhibition of ERK
Sara Rubio, Jose Quintana, Jose L. Eiroa1,Jorge Triana1 and Francisco Estevez�
Department of Biochemistry, University of Las Palmas de Gran Canaria, PlazaDr. Pasteur s/n, 35016 Las Palmas de Gran Canaria, Spain and 1Department ofChemistry, Instituto Canario de Investigacion del Cancer, University of LasPalmas de Gran Canaria, Campus Universitario de Tafira, 35017 Las Palmasde Gran Canaria, Spain
�To whom correspondence should be addressed. Tel: þ34 928 451443;Fax: þ34 928 451441;Email: [email protected]
Flavonoids are polyphenolic compounds that are ubiquitously inplants and display a vast array of biological activities. Here wehave studied the effect of the phenylbenzo-g-pyrone-derivativequercetin 3-methyl ether tetracetate (QD), obtained by acetyla-tion of the natural product quercetin 3-methyl ether, on cell via-bility of human leukemia HL-60 and U937 cell lines. The resultsshow that QD was cytotoxic and induced G2–M phase cell cyclearrest on both cell lines and it was a potent apoptotic inducer onHL-60 cells. QD-induced apoptosis is (i) mediated by caspaseactivation, since it was prevented by the non-specific caspaseinhibitor z-VAD-fmk, (ii) associated with cytochrome c releaseand (iii) triggered in Bcl-2 over-expressing U937 cells. The treat-ment of HL-60 and U937 cells with QD also induces the activationof the mitogen-activated protein kinases (MAPKs) pathway, in-cluding c-Jun N-terminal kinase, p38 mitogen-activated proteinkinase and extracellular signal-regulated kinases (ERK) 1/2. In-hibition of c-Jun N-terminal kinase by SP600125 and of p38 mi-togen-activated protein kinase by SB203580 had no influence onQD-mediated apoptosis. In contrast, inhibition of ERK1/2 withthe pharmacologic inhibitors U0126 or PD98059, together withQD, resulted in an important enhancement of apoptosis. Cells aresensitized to QD-mediated apoptosis after blocking ERK1/2,which suggests that inhibition of this pathway is a valuable strat-egy to increase the sensitivity of human leukemia HL-60 cellstoward QD.
Introduction
In recent years, the use of naturally occurring agents to prevent thedevelopment or recurrence of cancers has become widely accepted asa realistic option for fighting the disease. Flavonoids are phenylbenzo-c-pyrones derivatives which comprise a very large class of naturallyoccurring polyphenol plant compounds (1). Flavonoids are ubiquitousin plant foods and drinks and therefore consumed in significant quan-tities in our daily diet (2). These polyphenolic compounds displaya remarkable spectrum of biological activities and they are amongthe most promising anticancer agents (3). Quercetin (3,3#,4#,5,7-pen-tahydroxyflavone) is one of the best-studied bioflavonoid and it iswidely distributed in nature. This compound potentiates the cytotoxicaction of 1-b-D-arabinofuranosylcytosine (ara-C) (4) and inhibits cellinvasion and induces apoptosis (5). Apoptosis plays a crucial role innormal development, homeostasis and in the defense response againstpathogens. This kind of cell death is thought to be an importantresponse to most chemotherapeutic agents in leukemia cells (6–8).
Essential executioners of apoptosis are the caspases, a family of con-served cysteine aspartate-specific proteases (9). Caspases are generallysynthesized as inactive proenzymes or zymogens, which are activatedby proteolytic cleavage. The apoptotic caspases are classified as ini-tiators or executioners, depending on their point of entry into theapoptotic cascade. The initiator caspases are the first to be activatedin a particular death pathway, and they constitute the first step ina minimal two-step cascade by activating the executioner caspases.Two pathways of caspase activation during apoptosis have been de-scribed (10). The extrinsic pathway involves apoptosis mediated bydeath receptors, such as Fas or tumor necrosis factor receptors (11),involving caspase-8 activation, which can process effector caspases(caspase-3, -6 and -7), inducing a cascade of caspases. In the intrinsicpathway, diverse pro-apoptotic signals provoke the translocation ofcytochrome c from mitochondria to cytoplasm and caspase-9 activa-tion, which cleaves and activates pro-caspase-3. Caspase-3 is respon-sible for cleaving specific cellular proteins during apoptosis (12).However, nowadays, apoptosis is described as being mediated notonly by the death receptor (or extrinsic pathway) and the mitochon-drial (or intrinsic pathway) pathways but also by the endoplasmicreticulum signaling pathway (13). Moreover, apoptosis can also occurindependently of caspase activation (14).Cell responses to apoptotic-inducing drugs have been associated
with the inactivation of survival kinases and the activation of apopto-tic kinases. One of the most relevant aspects in the regulation ofapoptosis is the involvement of mitogen-activated protein kinases(MAPKs), a family of proline-directed serine/threonine proteinkinases that mediate intracellular signal transduction in response tovarious stimuli (15). The MAPKs are activated by phosphorylation onboth a threonine and tyrosine in a TXY motif found in an activationloop proximal to the ATP- and substrate-binding sites (16). To date,three major MAPKs have been identified: the c-Jun N-terminalkinases/stress-activated protein kinases (JNKs/SAPKs), the p38mitogen-activated protein kinases (p38MAPK) and the extracellularsignal-regulated kinases (ERK) 1/2. The ERKs are mainly, but notexclusively, activated by growth factors and involved in the regulationof mitogenesis (17,18). On the other hand, JNK and p38MAPK areactivated mainly by cytotoxic insults and are often associated withapoptosis. However, there are multiple exceptions to this rule.ERK activation can exert either anti-apoptotic or pro-apoptotic
effects, depending upon the stimuli and cell type. JNK/SAPK ispreferentially activated by stressful stimuli including UV irradiation,hyperthermia, inflammatory cytokines, chemotherapeutic agents andthe translational inhibitors such as anisomycin and cycloheximide.Activation of p38MAPK has been implicated in the cellular responseto osmotic shock, physiological stress, UV radiation and treatmentwith chemotherapeutic drugs (19).Here we have studied the effect of tetracetyl derivative of quercetin
3-methyl ether (QD), obtained by acetylation of the natural productquercetin 3-methyl ether, on cell viability of human leukemia celllines. QD was relatively cytotoxic on both cell lines studied, humanmyeloid leukemia HL-60 and U937 cells. This compound inducedG2–M arrest on both cells and it was also a potent apoptotic induceron human myeloid leukemia HL-60 cells. The results also demon-strate that QD-induced apoptosis is mediated by caspase activationand was also associated by cytochrome c release. The present studiesdemonstrate that treatment of human HL-60 and U937 cells with QDalso induces the activation of the MAPKs JNK/SAPK, p38MAPK andERK1/2. Inhibition of JNK by SP600125 and of p38MAPK bySB203580 had no influence on QD-mediated apoptosis. In contrast,inhibition of ERK1/2 with the pharmacologic inhibitors U0126 orPD98059 together with QD resulted in an important enhancementof apoptosis.
Abbreviations: ara-C, 1-b-D-arabinofuranosylcytosine; ERK, extracellularsignal-regulated kinase; IC50, 50% inhibition of cell growth; JNK, c-Jun N-terminal kinase; MAPK, mitogen-activated protein kinase; MEK, mitogen-activated extracellular kinase; PARP, poly (ADP-ribose) polymerase; PBMC,peripheral blood mononuclear cell; p38MAPK, p38 mitogen-activated proteinkinase; ROS, reactive oxygen species; SAPK, stress-activated protein kinase;QD, tetracetyl derivative of quercetin 3-methyl ether.
� The Author 2007. Published by Oxford University Press. All rights reserved. For Permissions, please email: [email protected] 2105
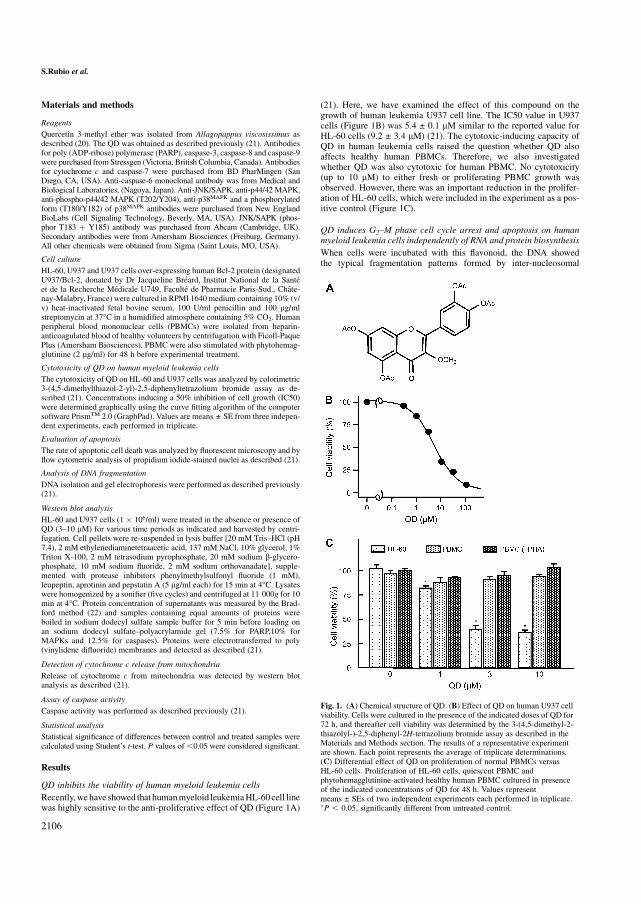
Materials and methods
Reagents
Quercetin 3-methyl ether was isolated from Allagopappus viscosissimus asdescribed (20). The QD was obtained as described previously (21). Antibodiesfor poly (ADP-ribose) polymerase (PARP), caspase-3, caspase-8 and caspase-9were purchased from Stressgen (Victoria, British Columbia, Canada). Antibodiesfor cytochrome c and caspase-7 were purchased from BD PharMingen (SanDiego, CA, USA). Anti-caspase-6 monoclonal antibody was from Medical andBiological Laboratories, (Nagoya, Japan). Anti-JNK/SAPK, anti-p44/42 MAPK,anti-phospho-p44/42 MAPK (T202/Y204), anti-p38MAPK and a phosphorylatedform (T180/Y182) of p38MAPK antibodies were purchased from New EnglandBioLabs (Cell Signaling Technology, Beverly, MA, USA). JNK/SAPK (phos-phor T183 þ Y185) antibody was purchased from Abcam (Cambridge, UK).Secondary antibodies were from Amersham Biosciences (Freiburg, Germany).All other chemicals were obtained from Sigma (Saint Louis, MO, USA).
Cell culture
HL-60, U937 and U937 cells over-expressing human Bcl-2 protein (designatedU937/Bcl-2, donated by Dr Jacqueline Breard, Institut National de la Santeet de la Recherche Medicale U749, Faculte de Pharmacie Paris-Sud., Chate-nay-Malabry, France) were cultured in RPMI 1640 medium containing 10% (v/v) heat-inactivated fetal bovine serum, 100 U/ml penicillin and 100 lg/mlstreptomycin at 37�C in a humidified atmosphere containing 5% CO2. Humanperipheral blood mononuclear cells (PBMCs) were isolated from heparin-anticoagulated blood of healthy volunteers by centrifugation with Ficoll-PaquePlus (Amersham Biosciences). PBMC were also stimulated with phytohemag-glutinine (2 lg/ml) for 48 h before experimental treatment.
Cytotoxicity of QD on human myeloid leukemia cells
The cytotoxicity of QD on HL-60 and U937 cells was analyzed by colorimetric3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay as de-scribed (21). Concentrations inducing a 50% inhibition of cell growth (IC50)were determined graphically using the curve fitting algorithm of the computersoftware PrismTM 2.0 (GraphPad). Values are means ± SE from three indepen-dent experiments, each performed in triplicate.
Evaluation of apoptosis
The rate of apoptotic cell death was analyzed by fluorescent microscopy and byflow cytometric analysis of propidium iodide-stained nuclei as described (21).
Analysis of DNA fragmentation
DNA isolation and gel electrophoresis were performed as described previously(21).
Western blot analysis
HL-60 and U937 cells (1 � 106/ml) were treated in the absence or presence ofQD (3–10 lM) for various time periods as indicated and harvested by centri-fugation. Cell pellets were re-suspended in lysis buffer [20 mM Tris–HCl (pH7.4), 2 mM ethylenediaminetetraacetic acid, 137 mM NaCl, 10% glycerol, 1%Triton X-100, 2 mM tetrasodium pyrophosphate, 20 mM sodium b-glycero-phosphate, 10 mM sodium fluoride, 2 mM sodium orthovanadate], supple-mented with protease inhibitors phenylmethylsulfonyl fluoride (1 mM),leupeptin, aprotinin and pepstatin A (5 lg/ml each) for 15 min at 4�C. Lysateswere homogenized by a sonifier (five cycles) and centrifuged at 11 000g for 10min at 4�C. Protein concentration of supernatants was measured by the Brad-ford method (22) and samples containing equal amounts of proteins wereboiled in sodium dodecyl sulfate sample buffer for 5 min before loading onan sodium dodecyl sulfate–polyacrylamide gel (7.5% for PARP,10% forMAPKs and 12.5% for caspases). Proteins were electrotransferred to poly(vinylidene difluoride) membranes and detected as described (21).
Detection of cytochrome c release from mitochondria
Release of cytochrome c from mitochondria was detected by western blotanalysis as described (21).
Assay of caspase activity
Caspase activity was performed as described previously (21).
Statistical analysis
Statistical significance of differences between control and treated samples werecalculated using Student’s t-test. P values of,0.05 were considered significant.
Results
QD inhibits the viability of human myeloid leukemia cells
Recently,wehave showed that humanmyeloid leukemiaHL-60cell linewas highly sensitive to the anti-proliferative effect of QD (Figure 1A)
(21). Here, we have examined the effect of this compound on thegrowth of human leukemia U937 cell line. The IC50 value in U937cells (Figure 1B) was 5.4 ± 0.1 lM similar to the reported value forHL-60 cells (9.2 ± 3.4 lM) (21). The cytotoxic-inducing capacity ofQD in human leukemia cells raised the question whether QD alsoaffects healthy human PBMCs. Therefore, we also investigatedwhether QD was also cytotoxic for human PBMC. No cytotoxicity(up to 10 lM) to either fresh or proliferating PBMC growth wasobserved. However, there was an important reduction in the prolifer-ation of HL-60 cells, which were included in the experiment as a pos-itive control (Figure 1C).
QD induces G2–M phase cell cycle arrest and apoptosis on humanmyeloid leukemia cells independently of RNA and protein biosynthesis
When cells were incubated with this flavonoid, the DNA showedthe typical fragmentation patterns formed by inter-nucleosomal
Fig. 1. (A) Chemical structure of QD. (B) Effect of QD on human U937 cellviability. Cells were cultured in the presence of the indicated doses of QD for72 h, and thereafter cell viability was determined by the 3-(4,5-dimethyl-2-thiazolyl-)-2,5-diphenyl-2H-tetrazolium bromide assay as described in theMaterials and Methods section. The results of a representative experimentare shown. Each point represents the average of triplicate determinations.(C) Differential effect of QD on proliferation of normal PBMCs versusHL-60 cells. Proliferation of HL-60 cells, quiescent PBMC andphytohemagglutinine-activated healthy human PBMC cultured in presenceof the indicated concentrations of QD for 48 h. Values representmeans ± SEs of two independent experiments each performed in triplicate.�P , 0.05, significantly different from untreated control.
S.Rubio et al.
2106
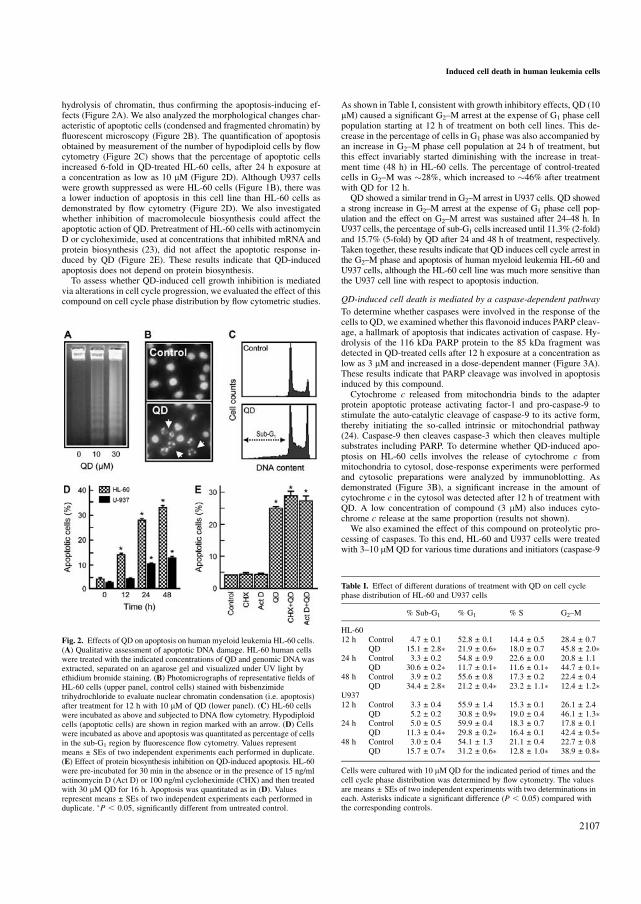
hydrolysis of chromatin, thus confirming the apoptosis-inducing ef-fects (Figure 2A). We also analyzed the morphological changes char-acteristic of apoptotic cells (condensed and fragmented chromatin) byfluorescent microscopy (Figure 2B). The quantification of apoptosisobtained by measurement of the number of hypodiploid cells by flowcytometry (Figure 2C) shows that the percentage of apoptotic cellsincreased 6-fold in QD-treated HL-60 cells, after 24 h exposure ata concentration as low as 10 lM (Figure 2D). Although U937 cellswere growth suppressed as were HL-60 cells (Figure 1B), there wasa lower induction of apoptosis in this cell line than HL-60 cells asdemonstrated by flow cytometry (Figure 2D). We also investigatedwhether inhibition of macromolecule biosynthesis could affect theapoptotic action of QD. Pretreatment of HL-60 cells with actinomycinD or cycloheximide, used at concentrations that inhibited mRNA andprotein biosynthesis (23), did not affect the apoptotic response in-duced by QD (Figure 2E). These results indicate that QD-inducedapoptosis does not depend on protein biosynthesis.To assess whether QD-induced cell growth inhibition is mediated
via alterations in cell cycle progression, we evaluated the effect of thiscompound on cell cycle phase distribution by flow cytometric studies.
As shown in Table I, consistent with growth inhibitory effects, QD (10lM) caused a significant G2–M arrest at the expense of G1 phase cellpopulation starting at 12 h of treatment on both cell lines. This de-crease in the percentage of cells in G1 phase was also accompanied byan increase in G2–M phase cell population at 24 h of treatment, butthis effect invariably started diminishing with the increase in treat-ment time (48 h) in HL-60 cells. The percentage of control-treatedcells in G2–M was �28%, which increased to �46% after treatmentwith QD for 12 h.QD showed a similar trend in G2–M arrest in U937 cells. QD showed
a strong increase in G2–M arrest at the expense of G1 phase cell pop-ulation and the effect on G2–M arrest was sustained after 24–48 h. InU937 cells, the percentage of sub-G1 cells increased until 11.3% (2-fold)and 15.7% (5-fold) by QD after 24 and 48 h of treatment, respectively.Taken together, these results indicate that QD induces cell cycle arrest inthe G2–M phase and apoptosis of human myeloid leukemia HL-60 andU937 cells, although the HL-60 cell line was much more sensitive thanthe U937 cell line with respect to apoptosis induction.
QD-induced cell death is mediated by a caspase-dependent pathway
To determine whether caspases were involved in the response of thecells to QD, we examined whether this flavonoid induces PARP cleav-age, a hallmark of apoptosis that indicates activation of caspase. Hy-drolysis of the 116 kDa PARP protein to the 85 kDa fragment wasdetected in QD-treated cells after 12 h exposure at a concentration aslow as 3 lM and increased in a dose-dependent manner (Figure 3A).These results indicate that PARP cleavage was involved in apoptosisinduced by this compound.Cytochrome c released from mitochondria binds to the adapter
protein apoptotic protease activating factor-1 and pro-caspase-9 tostimulate the auto-catalytic cleavage of caspase-9 to its active form,thereby initiating the so-called intrinsic or mitochondrial pathway(24). Caspase-9 then cleaves caspase-3 which then cleaves multiplesubstrates including PARP. To determine whether QD-induced apo-ptosis on HL-60 cells involves the release of cytochrome c frommitochondria to cytosol, dose-response experiments were performedand cytosolic preparations were analyzed by immunoblotting. Asdemonstrated (Figure 3B), a significant increase in the amount ofcytochrome c in the cytosol was detected after 12 h of treatment withQD. A low concentration of compound (3 lM) also induces cyto-chrome c release at the same proportion (results not shown).We also examined the effect of this compound on proteolytic pro-
cessing of caspases. To this end, HL-60 and U937 cells were treatedwith 3–10 lMQD for various time durations and initiators (caspase-9
Fig. 2. Effects of QD on apoptosis on human myeloid leukemia HL-60 cells.(A) Qualitative assessment of apoptotic DNA damage. HL-60 human cellswere treated with the indicated concentrations of QD and genomic DNAwasextracted, separated on an agarose gel and visualized under UV light byethidium bromide staining. (B) Photomicrographs of representative fields ofHL-60 cells (upper panel, control cells) stained with bisbenzimidetrihydrochloride to evaluate nuclear chromatin condensation (i.e. apoptosis)after treatment for 12 h with 10 lM of QD (lower panel). (C) HL-60 cellswere incubated as above and subjected to DNA flow cytometry. Hypodiploidcells (apoptotic cells) are shown in region marked with an arrow. (D) Cellswere incubated as above and apoptosis was quantitated as percentage of cellsin the sub-G1 region by fluorescence flow cytometry. Values representmeans ± SEs of two independent experiments each performed in duplicate.(E) Effect of protein biosynthesis inhibition on QD-induced apoptosis. HL-60were pre-incubated for 30 min in the absence or in the presence of 15 ng/mlactinomycin D (Act D) or 100 ng/ml cycloheximide (CHX) and then treatedwith 30 lM QD for 16 h. Apoptosis was quantitated as in (D). Valuesrepresent means ± SEs of two independent experiments each performed induplicate. �P , 0.05, significantly different from untreated control.
Table I. Effect of different durations of treatment with QD on cell cyclephase distribution of HL-60 and U937 cells
% Sub-G1 % G1 % S G2–M
HL-6012 h Control 4.7 ± 0.1 52.8 ± 0.1 14.4 ± 0.5 28.4 ± 0.7
QD 15.1 ± 2.8� 21.9 ± 0.6� 18.0 ± 0.7 45.8 ± 2.0�24 h Control 3.3 ± 0.2 54.8 ± 0.9 22.6 ± 0.0 20.8 ± 1.1
QD 30.6 ± 0.2� 11.7 ± 0.1� 11.6 ± 0.1� 44.7 ± 0.1�48 h Control 3.9 ± 0.2 55.6 ± 0.8 17.3 ± 0.2 22.4 ± 0.4
QD 34.4 ± 2.8� 21.2 ± 0.4� 23.2 ± 1.1� 12.4 ± 1.2�U93712 h Control 3.3 ± 0.4 55.9 ± 1.4 15.3 ± 0.1 26.1 ± 2.4
QD 5.2 ± 0.2 30.8 ± 0.9� 19.0 ± 0.4 46.1 ± 1.3�24 h Control 5.0 ± 0.5 59.9 ± 0.4 18.3 ± 0.7 17.8 ± 0.1
QD 11.3 ± 0.4� 29.8 ± 0.2� 16.4 ± 0.1 42.4 ± 0.5�48 h Control 3.0 ± 0.4 54.1 ± 1.3 21.1 ± 0.4 22.7 ± 0.8
QD 15.7 ± 0.7� 31.2 ± 0.6� 12.8 ± 1.0� 38.9 ± 0.8�
Cells were cultured with 10 lM QD for the indicated period of times and thecell cycle phase distribution was determined by flow cytometry. The valuesare means ± SEs of two independent experiments with two determinations ineach. Asterisks indicate a significant difference (P , 0.05) compared withthe corresponding controls.
Induced cell death in human leukemia cells
2107
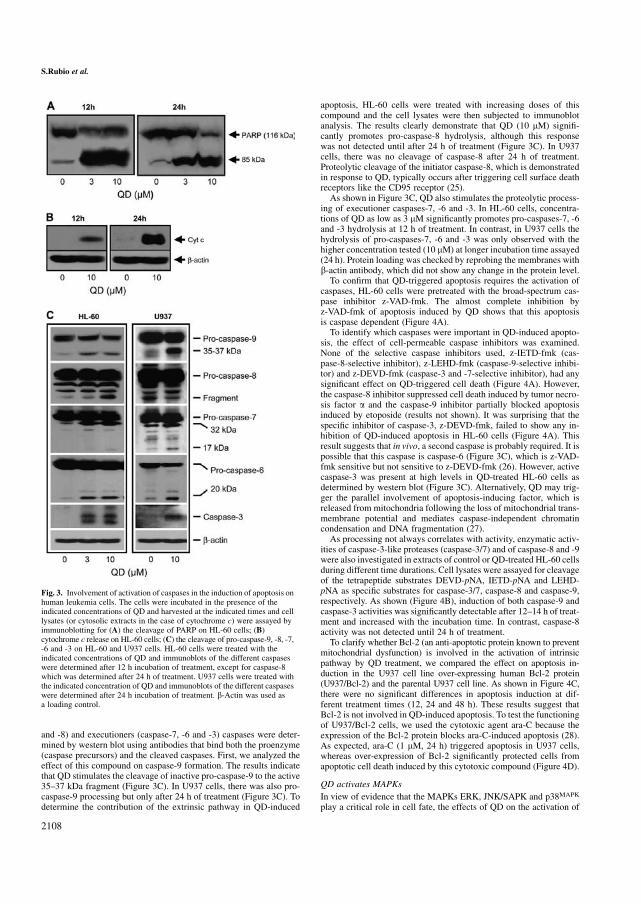
and -8) and executioners (caspase-7, -6 and -3) caspases were deter-mined by western blot using antibodies that bind both the proenzyme(caspase precursors) and the cleaved caspases. First, we analyzed theeffect of this compound on caspase-9 formation. The results indicatethat QD stimulates the cleavage of inactive pro-caspase-9 to the active35–37 kDa fragment (Figure 3C). In U937 cells, there was also pro-caspase-9 processing but only after 24 h of treatment (Figure 3C). Todetermine the contribution of the extrinsic pathway in QD-induced
apoptosis, HL-60 cells were treated with increasing doses of thiscompound and the cell lysates were then subjected to immunoblotanalysis. The results clearly demonstrate that QD (10 lM) signifi-cantly promotes pro-caspase-8 hydrolysis, although this responsewas not detected until after 24 h of treatment (Figure 3C). In U937cells, there was no cleavage of caspase-8 after 24 h of treatment.Proteolytic cleavage of the initiator caspase-8, which is demonstratedin response to QD, typically occurs after triggering cell surface deathreceptors like the CD95 receptor (25).As shown in Figure 3C, QD also stimulates the proteolytic process-
ing of executioner caspases-7, -6 and -3. In HL-60 cells, concentra-tions of QD as low as 3 lM significantly promotes pro-caspases-7, -6and -3 hydrolysis at 12 h of treatment. In contrast, in U937 cells thehydrolysis of pro-caspases-7, -6 and -3 was only observed with thehigher concentration tested (10 lM) at longer incubation time assayed(24 h). Protein loading was checked by reprobing the membranes withb-actin antibody, which did not show any change in the protein level.To confirm that QD-triggered apoptosis requires the activation of
caspases, HL-60 cells were pretreated with the broad-spectrum cas-pase inhibitor z-VAD-fmk. The almost complete inhibition byz-VAD-fmk of apoptosis induced by QD shows that this apoptosisis caspase dependent (Figure 4A).To identify which caspases were important in QD-induced apopto-
sis, the effect of cell-permeable caspase inhibitors was examined.None of the selective caspase inhibitors used, z-IETD-fmk (cas-pase-8-selective inhibitor), z-LEHD-fmk (caspase-9-selective inhibi-tor) and z-DEVD-fmk (caspase-3 and -7-selective inhibitor), had anysignificant effect on QD-triggered cell death (Figure 4A). However,the caspase-8 inhibitor suppressed cell death induced by tumor necro-sis factor a and the caspase-9 inhibitor partially blocked apoptosisinduced by etoposide (results not shown). It was surprising that thespecific inhibitor of caspase-3, z-DEVD-fmk, failed to show any in-hibition of QD-induced apoptosis in HL-60 cells (Figure 4A). Thisresult suggests that in vivo, a second caspase is probably required. It ispossible that this caspase is caspase-6 (Figure 3C), which is z-VAD-fmk sensitive but not sensitive to z-DEVD-fmk (26). However, activecaspase-3 was present at high levels in QD-treated HL-60 cells asdetermined by western blot (Figure 3C). Alternatively, QD may trig-ger the parallel involvement of apoptosis-inducing factor, which isreleased from mitochondria following the loss of mitochondrial trans-membrane potential and mediates caspase-independent chromatincondensation and DNA fragmentation (27).As processing not always correlates with activity, enzymatic activ-
ities of caspase-3-like proteases (caspase-3/7) and of caspase-8 and -9were also investigated in extracts of control or QD-treated HL-60 cellsduring different time durations. Cell lysates were assayed for cleavageof the tetrapeptide substrates DEVD-pNA, IETD-pNA and LEHD-pNA as specific substrates for caspase-3/7, caspase-8 and caspase-9,respectively. As shown (Figure 4B), induction of both caspase-9 andcaspase-3 activities was significantly detectable after 12–14 h of treat-ment and increased with the incubation time. In contrast, caspase-8activity was not detected until 24 h of treatment.To clarify whether Bcl-2 (an anti-apoptotic protein known to prevent
mitochondrial dysfunction) is involved in the activation of intrinsicpathway by QD treatment, we compared the effect on apoptosis in-duction in the U937 cell line over-expressing human Bcl-2 protein(U937/Bcl-2) and the parental U937 cell line. As shown in Figure 4C,there were no significant differences in apoptosis induction at dif-ferent treatment times (12, 24 and 48 h). These results suggest thatBcl-2 is not involved in QD-induced apoptosis. To test the functioningof U937/Bcl-2 cells, we used the cytotoxic agent ara-C because theexpression of the Bcl-2 protein blocks ara-C-induced apoptosis (28).As expected, ara-C (1 lM, 24 h) triggered apoptosis in U937 cells,whereas over-expression of Bcl-2 significantly protected cells fromapoptotic cell death induced by this cytotoxic compound (Figure 4D).
QD activates MAPKs
In view of evidence that the MAPKs ERK, JNK/SAPK and p38MAPK
play a critical role in cell fate, the effects of QD on the activation of
Fig. 3. Involvement of activation of caspases in the induction of apoptosis onhuman leukemia cells. The cells were incubated in the presence of theindicated concentrations of QD and harvested at the indicated times and celllysates (or cytosolic extracts in the case of cytochrome c) were assayed byimmunoblotting for (A) the cleavage of PARP on HL-60 cells; (B)cytochrome c release on HL-60 cells; (C) the cleavage of pro-caspase-9, -8, -7,-6 and -3 on HL-60 and U937 cells. HL-60 cells were treated with theindicated concentrations of QD and immunoblots of the different caspaseswere determined after 12 h incubation of treatment, except for caspase-8which was determined after 24 h of treatment. U937 cells were treated withthe indicated concentration of QD and immunoblots of the different caspaseswere determined after 24 h incubation of treatment. b-Actin was used asa loading control.
S.Rubio et al.
2108
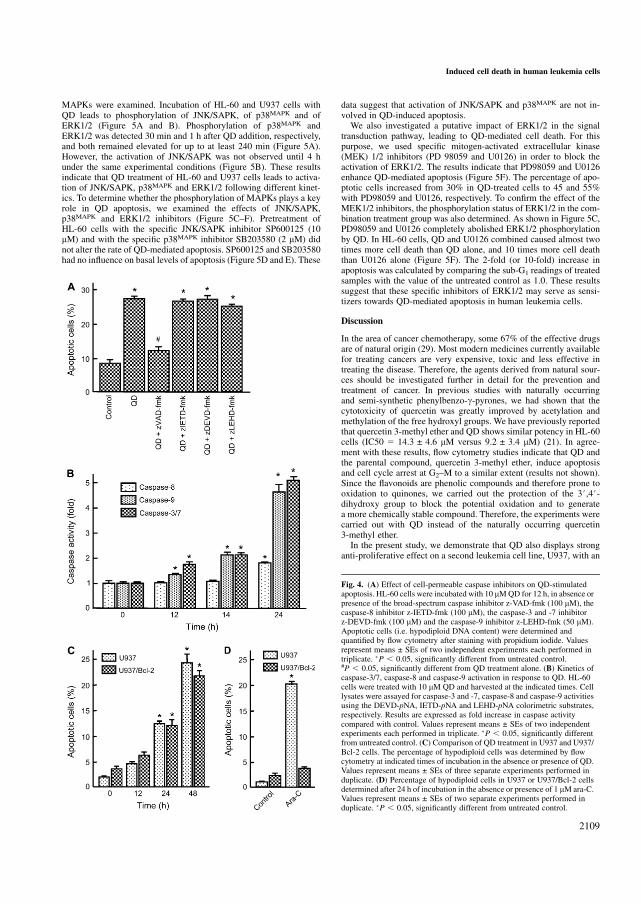
MAPKs were examined. Incubation of HL-60 and U937 cells withQD leads to phosphorylation of JNK/SAPK, of p38MAPK and ofERK1/2 (Figure 5A and B). Phosphorylation of p38MAPK andERK1/2 was detected 30 min and 1 h after QD addition, respectively,and both remained elevated for up to at least 240 min (Figure 5A).However, the activation of JNK/SAPK was not observed until 4 hunder the same experimental conditions (Figure 5B). These resultsindicate that QD treatment of HL-60 and U937 cells leads to activa-tion of JNK/SAPK, p38MAPK and ERK1/2 following different kinet-ics. To determine whether the phosphorylation of MAPKs plays a keyrole in QD apoptosis, we examined the effects of JNK/SAPK,p38MAPK and ERK1/2 inhibitors (Figure 5C–F). Pretreatment ofHL-60 cells with the specific JNK/SAPK inhibitor SP600125 (10lM) and with the specific p38MAPK inhibitor SB203580 (2 lM) didnot alter the rate of QD-mediated apoptosis. SP600125 and SB203580had no influence on basal levels of apoptosis (Figure 5D and E). These
data suggest that activation of JNK/SAPK and p38MAPK are not in-volved in QD-induced apoptosis.We also investigated a putative impact of ERK1/2 in the signal
transduction pathway, leading to QD-mediated cell death. For thispurpose, we used specific mitogen-activated extracellular kinase(MEK) 1/2 inhibitors (PD 98059 and U0126) in order to block theactivation of ERK1/2. The results indicate that PD98059 and U0126enhance QD-mediated apoptosis (Figure 5F). The percentage of apo-ptotic cells increased from 30% in QD-treated cells to 45 and 55%with PD98059 and U0126, respectively. To confirm the effect of theMEK1/2 inhibitors, the phosphorylation status of ERK1/2 in the com-bination treatment group was also determined. As shown in Figure 5C,PD98059 and U0126 completely abolished ERK1/2 phosphorylationby QD. In HL-60 cells, QD and U0126 combined caused almost twotimes more cell death than QD alone, and 10 times more cell deaththan U0126 alone (Figure 5F). The 2-fold (or 10-fold) increase inapoptosis was calculated by comparing the sub-G1 readings of treatedsamples with the value of the untreated control as 1.0. These resultssuggest that these specific inhibitors of ERK1/2 may serve as sensi-tizers towards QD-mediated apoptosis in human leukemia cells.
Discussion
In the area of cancer chemotherapy, some 67% of the effective drugsare of natural origin (29). Most modern medicines currently availablefor treating cancers are very expensive, toxic and less effective intreating the disease. Therefore, the agents derived from natural sour-ces should be investigated further in detail for the prevention andtreatment of cancer. In previous studies with naturally occurringand semi-synthetic phenylbenzo-c-pyrones, we had shown that thecytotoxicity of quercetin was greatly improved by acetylation andmethylation of the free hydroxyl groups. We have previously reportedthat quercetin 3-methyl ether and QD shows similar potency in HL-60cells (IC50 5 14.3 ± 4.6 lM versus 9.2 ± 3.4 lM) (21). In agree-ment with these results, flow cytometry studies indicate that QD andthe parental compound, quercetin 3-methyl ether, induce apoptosisand cell cycle arrest at G2–M to a similar extent (results not shown).Since the flavonoids are phenolic compounds and therefore prone tooxidation to quinones, we carried out the protection of the 3#,4#-dihydroxy group to block the potential oxidation and to generatea more chemically stable compound. Therefore, the experiments werecarried out with QD instead of the naturally occurring quercetin3-methyl ether.In the present study, we demonstrate that QD also displays strong
anti-proliferative effect on a second leukemia cell line, U937, with an
Fig. 4. (A) Effect of cell-permeable caspase inhibitors on QD-stimulatedapoptosis. HL-60 cells were incubated with 10 lMQD for 12 h, in absence orpresence of the broad-spectrum caspase inhibitor z-VAD-fmk (100 lM), thecaspase-8 inhibitor z-IETD-fmk (100 lM), the caspase-3 and -7 inhibitorz-DEVD-fmk (100 lM) and the caspase-9 inhibitor z-LEHD-fmk (50 lM).Apoptotic cells (i.e. hypodiploid DNA content) were determined andquantified by flow cytometry after staining with propidium iodide. Valuesrepresent means ± SEs of two independent experiments each performed intriplicate. �P , 0.05, significantly different from untreated control.#P , 0.05, significantly different from QD treatment alone. (B) Kinetics ofcaspase-3/7, caspase-8 and caspase-9 activation in response to QD. HL-60cells were treated with 10 lM QD and harvested at the indicated times. Celllysates were assayed for caspase-3 and -7, caspase-8 and caspase-9 activitiesusing the DEVD-pNA, IETD-pNA and LEHD-pNA colorimetric substrates,respectively. Results are expressed as fold increase in caspase activitycompared with control. Values represent means ± SEs of two independentexperiments each performed in triplicate. �P , 0.05, significantly differentfrom untreated control. (C) Comparison of QD treatment in U937 and U937/Bcl-2 cells. The percentage of hypodiploid cells was determined by flowcytometry at indicated times of incubation in the absence or presence of QD.Values represent means ± SEs of three separate experiments performed induplicate. (D) Percentage of hypodiploid cells in U937 or U937/Bcl-2 cellsdetermined after 24 h of incubation in the absence or presence of 1 lM ara-C.Values represent means ± SEs of two separate experiments performed induplicate. �P , 0.05, significantly different from untreated control.
Induced cell death in human leukemia cells
2109
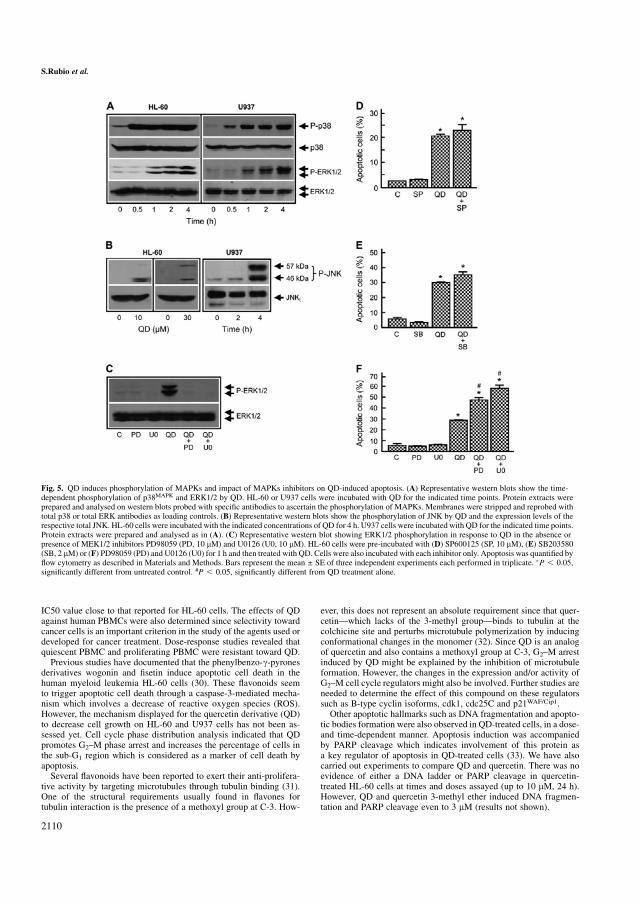
IC50 value close to that reported for HL-60 cells. The effects of QDagainst human PBMCs were also determined since selectivity towardcancer cells is an important criterion in the study of the agents used ordeveloped for cancer treatment. Dose-response studies revealed thatquiescent PBMC and proliferating PBMC were resistant toward QD.Previous studies have documented that the phenylbenzo-c-pyrones
derivatives wogonin and fisetin induce apoptotic cell death in thehuman myeloid leukemia HL-60 cells (30). These flavonoids seemto trigger apoptotic cell death through a caspase-3-mediated mecha-nism which involves a decrease of reactive oxygen species (ROS).However, the mechanism displayed for the quercetin derivative (QD)to decrease cell growth on HL-60 and U937 cells has not been as-sessed yet. Cell cycle phase distribution analysis indicated that QDpromotes G2–M phase arrest and increases the percentage of cells inthe sub-G1 region which is considered as a marker of cell death byapoptosis.Several flavonoids have been reported to exert their anti-prolifera-
tive activity by targeting microtubules through tubulin binding (31).One of the structural requirements usually found in flavones fortubulin interaction is the presence of a methoxyl group at C-3. How-
ever, this does not represent an absolute requirement since that quer-cetin—which lacks of the 3-methyl group—binds to tubulin at thecolchicine site and perturbs microtubule polymerization by inducingconformational changes in the monomer (32). Since QD is an analogof quercetin and also contains a methoxyl group at C-3, G2–M arrestinduced by QD might be explained by the inhibition of microtubuleformation. However, the changes in the expression and/or activity ofG2–M cell cycle regulators might also be involved. Further studies areneeded to determine the effect of this compound on these regulatorssuch as B-type cyclin isoforms, cdk1, cdc25C and p21WAF/Cip1.Other apoptotic hallmarks such as DNA fragmentation and apopto-
tic bodies formation were also observed in QD-treated cells, in a dose-and time-dependent manner. Apoptosis induction was accompaniedby PARP cleavage which indicates involvement of this protein asa key regulator of apoptosis in QD-treated cells (33). We have alsocarried out experiments to compare QD and quercetin. There was noevidence of either a DNA ladder or PARP cleavage in quercetin-treated HL-60 cells at times and doses assayed (up to 10 lM, 24 h).However, QD and quercetin 3-methyl ether induced DNA fragmen-tation and PARP cleavage even to 3 lM (results not shown).
Fig. 5. QD induces phosphorylation of MAPKs and impact of MAPKs inhibitors on QD-induced apoptosis. (A) Representative western blots show the time-dependent phosphorylation of p38MAPK and ERK1/2 by QD. HL-60 or U937 cells were incubated with QD for the indicated time points. Protein extracts wereprepared and analysed on western blots probed with specific antibodies to ascertain the phosphorylation of MAPKs. Membranes were stripped and reprobed withtotal p38 or total ERK antibodies as loading controls. (B) Representative western blots show the phosphorylation of JNK by QD and the expression levels of therespective total JNK. HL-60 cells were incubated with the indicated concentrations of QD for 4 h. U937 cells were incubated with QD for the indicated time points.Protein extracts were prepared and analysed as in (A). (C) Representative western blot showing ERK1/2 phosphorylation in response to QD in the absence orpresence of MEK1/2 inhibitors PD98059 (PD, 10 lM) and U0126 (U0, 10 lM). HL-60 cells were pre-incubated with (D) SP600125 (SP, 10 lM), (E) SB203580(SB, 2 lM) or (F) PD98059 (PD) and U0126 (U0) for 1 h and then treated with QD. Cells were also incubated with each inhibitor only. Apoptosis was quantified byflow cytometry as described in Materials and Methods. Bars represent the mean ± SE of three independent experiments each performed in triplicate. �P , 0.05,significantly different from untreated control. #P , 0.05, significantly different from QD treatment alone.
S.Rubio et al.
2110

Previous work described by Nguyen et al. (34) indicates that theconcentrations of quercetin and its metabolites in rats were 25.1, 43.3and 54.3 lM after 10 days of treatment with 50, 100 and 150 lg ofquercetin per kg body wt, respectively, suggesting the possibility thatthe concentrations of QD employed in the present study might beachieved in vivo. In this regard, we have used concentrations 5-foldlower (10 lM versus 54.3 lM) and even almost 20-fold lower (3 lMversus 54.3 lM) than that detected by Nguyen et al. In vitro studieshave shown that quercetin is bound to human plasma proteins andalbumin is the primary protein responsible for the binding. However,substitution of 3-OH in quercetin markedly weakens binding to albu-min (35). This suggests that the introduction of a methyl group in C-3might allow weak binding to albumin and therefore increase its de-livery to cells.It is interesting to note that a low concentration (3 lM) of QD was
sufficient to induce cytochrome c release, hydrolysis of caspases(caspases-3, -6, -7 and -9) and also PARP cleavage in HL-60 cellsin similar extension to 10 lM concentration. Recent studies have alsoindicated that liposomal quercetin can significantly improve the sol-ubility and bioavailability of quercetin and might enhance antitumorefficacy (36) and these results could be applicable to other flavonoidsand especially to QD. Further studies are needed to investigate thesafety, the bioavailability and the bioefficacy of quercetin 3-methylether and QD.Although QD-induced cell cycle perturbation showed a similar pat-
tern in HL-60 and U937 cells, it is important to note that the U937 cellline demonstrated to be more resistant to entry in apoptosis whichsuggest that there is heterogeneity in the response to QD. The induc-tion of apoptosis by QD does not involve protein biosynthesis, andtherefore, the target tumor cells already contain the complete set ofproteins required for QD-induced apoptosis.The experiments shown here demonstrate proteolytic processing of
the effector caspases-3, -6 and -7 and the initiator caspases-8 and -9,suggesting that QD activates these proteases in HL-60 cells. However,proteolytic cleavage of caspase-8 was not detected until after 24 h oftreatment and then only with the higher concentration tested. Similarresults were obtained in U937 cells, but at longer times (24 h), withthe only exception of caspase-8 which was not activated.The evidence indicates that caspase-independent cell death is usu-
ally initiated along with an apoptotic cascade, although the apoptoticmediators inhibit the caspase-independent cell death pathway (14).Our results indicate that the cell death induced by QD was dependenton caspase activation since apoptosis was pharmacologically in-hibited by the general caspase inhibitor z-VAD-fmk.Previous studies have already demonstrated that many cytotoxic
agents induce the release of mitochondrial cytochrome c (37), whichtriggers a caspase-dependent assembly of the apoptosome (38). Here,we have shown that QD induces the release of cytochrome c frommitochondria into the cytosol, and thus the activation of caspase-9 andcaspase-3. Although we have shown that QD-induced apoptosis wasprevented by a broad-spectrum caspase inhibitor, the use of selectiveinhibitors against caspase-8 (z-IETD-fmk), caspase-3 (z-DEVD-fmk)and caspase-9 (z-LEHD-fmk) were unable to block the cell death.We have previously demonstrated on HL-60 cells, an effective
blockage of betuletol 3-methyl ether-induced apoptosis by z-IETD-fmk but not by z-LEHD-fmk, supporting a caspase-8-mediated mech-anism (21). Therefore, different apoptotic pathways are activatedin this cell line in response to the phenylbenzo-c-pyrones QD andbetuletol 3-methyl ether. A possible role of Bid can be ruled out inQD-induced apoptosis since Bid requires its previous proteolytic ac-tivation by caspase-8, and inhibition of this caspase with z-IETD-fmkdid not prevent cell death.Our data clearly demonstrate that caspases-3 and -7 are activated in
HL-60 cells in response to QD. However, the lack of any effect ofz-DEVD-fmk, a caspases-3 and -7-selective inhibitor, on QD-inducedapoptosis indicates that cell death can occur in absence of caspaseactivity. Consequently, it appears that other factors may be able tocompensate for a lack of caspase-3 and caspase-7 activity. Sincez-DEVD-fmk is a more potent inhibitor for caspase-3 than for
caspase-7 (39), one possibility is that other executioner caspases, suchas caspase-7 or -6, also contribute to apoptosis caused by QD.The lack of a significant effect of z-LEHD-fmk on QD-induced cell
death was also surprising, given the presence of proteolytic processingof caspase-9 and suggests that caspase-9 may not be involved in QD-mediated cell death. Since caspase-9 activation does not require pro-teolytic processing (40,41), we also performed kinetic studies. Theresults clearly demonstrate that caspase-9 is activated by QD, suggest-ing that QD-induced apoptosis is linked with cytochrome c but notwith a receptor-mediated mechanism. The improved protection of-fered by z-VAD-fmk versus z-LEHD-fmk is probably caused by itsbetter accessibility to cellular caspase-9 (42). Although it is oftenextremely difficult to determine the order of caspase activation, ki-netic studies shown in this paper indicate inductions of both caspases-9and -3 activities were significantly detectable at 12–14 h of treatment.In contrast, the increase in caspase-8 activity was not detectable until24 h of treatment.Increased expression of Bcl-2 has been associated with chemore-
sistance, especially in the case of hematologic malignancies (43).Therefore, the finding and development of compounds capable ofinducing cell death in tumor cells over-expressing Bcl-2 is of greattherapeutic importance. The present results suggest that QD treatmenteffectively induces cell death in leukemic cells over-expressing Bcl-2.In this regard, it has been shown that flavopiridol induces mito-chondrial injury in various neoplastic cells, including those over-expressing Bcl-2 (44). Thus, although it is tempting to speculate thatthe activation of the extrinsic apoptotic pathway might be responsiblefor QD cell death in Bcl-2 over-expressing cells, another possibilitycould be that QD would be able to overcome blockade of cytochrome crelease and mitochondrial injury conferred by anti-apoptotic Bcl-2. Inthis regard, previous works have described how the inactivation ofBcl-2 can be achieved by different mechanisms including cleavage byactivated caspases and hyperphosphorylation (45,46).MAPKs are essential parts of the signal transduction machinery and
play central roles in cell growth, differentiation and programmed celldeath. The JNK/SAPK and p38MAPK have generally been associatedwith pro-apoptotic actions, whereas ERK1/2 in most cases exertscytoprotective effects (47). The ability of ERK1/2 inhibitors has beenreported to potentiate the antitumour effects of cytotoxic agents suchas ara-C in a substantial increase in release of cytochrome c (48).Another hierarchical model of activation of JNK/SAPK by endoplas-mic reticulum stress suggests induction of this pathway in Jurkat cellsdownstream of cytochrome c release and caspase-3 (49). Althoughmost reports support the idea that JNK/SAPK contributes to stress-induced apoptosis, its effect appears to depend on cell type and thecontext of other signals received by the cell (49). Here, we show thatQD induces JNK/SAPK activation in HL-60 and U937 cells. How-ever, the selective JNK/SAPK inhibitor SP600125 did not influenceQD-induced apoptosis, indicating that JNK/SAPK is not required forcell death.The p38MAPK signaling has been shown not only to promote cell
death in some cell lines but also to enhance survival, cell growth anddifferentiation. Therefore, the role of p38MAPK in apoptosis is depen-dent on cell types and stimuli (50). In the present study, we show thatp38MAPK phosphorylation occurred prior to caspase activation interms of time (30 min). However, the p38MAPK inhibitor SB203580did not attenuate apoptosis, suggesting that activation of p38MAPK isnot required for QD-induced apoptosis. Activation of p38MAPK hasbeen reported to be involved in apoptosis in a variety of cell types. Forexample, the activation of p38MAPK is involved in cadmium-inducedapoptosis on U937 cells (51) and resveratrol-induced apoptosis ofhuman malignant B cells (52); however, this activation is not involvedin apoptosis in UV-treated U937 cells (53) and/or in Fas- andUV-treated Jurkat T cells (54).Previous studies have shown that quercetin treatment results in high
and sustained activation of ERK in A549 lung cancer epithelial cells(34). However, the inhibition of MEK–ERK activation utilizingU0126 or PD98059 abolishes quercetin-induced apoptosis (34). Treat-ment of ovarian carcinoma cells (55,56) and the C8161 melanoma cell
Induced cell death in human leukemia cells
2111

line (57) with cisplatin caused ERK activation and cell death, and thislatter effect was potentiated by ERK inhibitors, indicating that ERKsbehave as survival-inducing kinases in these cells.One of the findings described here is that QD also enhances the
activation of the MEK/ERK pathway, which is expected to increasecell proliferation and survival, and may compromise the efficacy ofQD in potential cancer treatment. Our data indicated that ERK inhib-itors PD98059 and U0126 potentiated the apoptotic effects of QD.The combination of QD plus PD98059 or QD plus U0126 enhancedcell death. The potential use of low-dose chemotherapy is importantbecause lower dosages are more attainable during cancer therapy andprobably to be less toxic to patients. These results have importantclinical implications for the use of QD in combination of ERK inhib-itors as potential therapeutic agents. Although it has been shown thatincreased ROS production in leukemic cells leads to the activation ofMAPKs and cell death (58–61), however, we were unable to demon-strate intracellular ROS generation after exposure to QD (resultsnot shown). So, the mechanism of cell death triggered by QD seemsto be different to wogonin and fisetin which involves a decrease inROS (30).In summary, our results show that QD displays cytotoxic properties,
induces G2–M phase cell cycle arrest and apoptosis on human mye-loid leukemia HL-60 and U937 cells. Since these cells are p53 null,our results clearly demonstrate that apoptosis induced by QD couldoccur independently of p53-mediated cellular events. Given the factthat �50% of human cancers harbor p53 mutations (62,63), theseresults have important implications for developing QD as a chemo-preventive or chemotherapeutic agent. QD-induced apoptosis is ac-companied by the activation of multiple caspases, mitochondrialrelease of cytochrome c, PARP cleavage and DNA fragmentation.Although cleavage of pro-caspase-8, -9, -3 and -7 is detected inQD-treated cells by immunobloting, the inhibition data indicate thatthese caspases do not fully account for cell death as a result of expo-sure to QD. These results, when considered together with the com-plete blockage of apoptosis by z-VAD-fmk, suggest that additionalcaspases may contribute to QD-induced apoptosis. One possibility isthat other executioner caspases, such as caspase-6, are involved in thisprocess as it is shown in this paper. The mitochondria-protecting pro-tein Bcl-2 was unable to prevent QD-induced apoptotic cell death.This result suggests that QD might trigger an alternative pathway thatbypasses mitochondria and/or that QD might be able to inactivate themitochondrial protection conferred by Bcl-2 protein. QD enhances theactivation of the MEK/ERK pathway, and the combined treatment ofQD and inhibitors of MEK1/2 kinase leads to enhanced cell death.The findings of this study suggest that QD could be useful in thedevelopment of novel anticancer agents.
Funding
Ministerio de Educacion y Ciencia of Spain; Fondo Europeo de De-sarrollo Regional (SAF2004-07928); Fundacion Canaria Universita-ria de Las Palmas (Clinica San Roque and La Caja de Canarias).
Acknowledgements
We thank Dr Jacqueline Breard (INSERM U749, Faculte de Pharmacie Paris-Sud., Chatenay-Malabry, France) for supplying U937/Bcl-2 cells.
Conflict of Interest Statement: None declared.
References
1.Middleton,E. et al. (2000) The effects of plant flavonoids on mammaliancells: implications for inflammation, heart disease, and cancer. Pharmacol.Rev., 52, 673–751.
2.Havsteen,B.H. (2002) The biochemistry and medical significance of theflavonoids. Pharmacol. Ther., 96, 67–202.
3.Ren,W. et al. (2003) Flavonoids: promising anticancer agents. Med. Res.Rev., 23, 519–534.
4.Teofili,L. et al. (1992) The combination of quercetin and cytosine arabino-side synergistically inhibits leukemic cell growth. Leuk. Res., 16, 497–503.
5.Wei,Y.Q. et al. (1994) Induction of apoptosis by quercetin: involvement ofheat shock protein. Cancer Res., 54, 4952–4957.
6.Dive,C. et al. (1991) Drug-target interactions:only the first step in thecommitment to a programmed cell death? Br. J. Cancer, 64, 192–196.
7.Fisher,D.E. (1994) Apoptosis in cancer therapy: crossing the threshold.Cell, 78, 539–542.
8.Hickman,J.A. (1992) Apoptosis induced by anticancer drugs. CancerMetastasis Rev., 11, 121–139.
9.Thornberry,N.A. et al. (1998) Caspases: enemies within. Science, 281,1312–1316.
10.Boatright,K.M. et al. (2003) Mechanisms of caspase activation. Curr. Opin.Cell Biol., 15, 725–731.
11.Nagata,S. (1997) Apoptosis by death factor. Cell, 88, 355–365.12.Cohen,G.M. (1997) Caspases: the executioners of apoptosis. Biochem. J.,
326, 1–16.13.Breckenridge,D.G. et al. (2003) Regulation of apoptosis by endoplasmic
reticulum pathways. Oncogene, 22, 8608–8618.14.Chipuk,J.E. et al. (2005) Do inducers of apoptosis trigger caspase-
independent cell death? Nat. Rev. Mol. Cell Biol., 6, 268–275.15.Tibbles,L.A. et al. (1999) The stress-activated protein kinases pathways.
Cell. Mol. Life Sci., 55, 1230–1254.16.Cobb,M.H. et al. (1995) How MAP kinases are regulated. J. Biol. Chem.,
270, 14843–14846.17.Seger,R. et al. (1995) The MAPK signaling cascade. FASEB J., 9, 726–735.18.Xia,Z. et al. (1995) Opposing effects of ERK and JNK-p38MAP kinases on
apoptosis. Science, 270, 1326–1331.19.Olson,J.M. et al. (2004) p38 MAP kinase: a convergence point in cancer
therapy. Trends Mol. Med., 10, 125–129.20.Gonzalez,A.G. et al. (1992) Germacranolides from Allagopappus viscosis-
simus. Phytochemistry, 31, 330–331.21.Rubio,S. et al. (2006) Phenylbenzopyrones structure-activity studies iden-
tify betuletol derivatives as potential antitumoral agents. Eur. J. Pharma-col., 548, 9–20.
22.Bradford,M.M. (1976) A rapid and sensitive method for the quantitation ofmicrogram quantities of protein utilizing the principle of protein-dye bind-ing. Anal. Biochem., 72, 248–254.
23.Perez-Sala,D. et al. (1995) Intracellular alkalinization suppresses lova-statin-induced apoptosis in HL-60 cells through the inactivation of a pH-dependent endonuclease. J. Biol. Chem., 270, 6235–6242.
24.Riedl,S.J. et al. (2004) Molecular mechanisms of caspase regulation duringapoptosis. Nat. Rev. Mol. Cell Biol., 5, 897–907.
25. Igney,F.H. et al. (2002) Death and anti-death: tumour resistance to apopto-sis. Nat. Rev. Cancer, 2, 277–288.
26.Srinivasula,S.M. et al. (1996) The Ced-3/interleukin 1b convertingenzyme-like homolog Mch6 and the lamin-cleaving enzyme Mch2a aresubstrates for the apoptotic mediator CPP32. J. Biol. Chem., 271, 27099–27106.
27.Lorenzo,H.K. et al. (1999) Apoptosis inducing factor (AIF): a phylogenet-ically old, caspase-independent effector of cell death. Cell Death Differ., 6,516–524.
28. Ibrado,A.M. et al. (1996) Overexpression of Bcl-2 or Bcl-xL inhibits ara-C-induced CPP32/Yama protease activity and apoptosis of human myeloge-nous leukaemia HL-60 cells. Cancer Res., 56, 4743–4748.
29.Cragg,G.M. et al. (2005) Anticancer Agents from Natural Products. Taylor& Francis Group, Boca Raton, FL.
30.Lee,W.R. et al. (2002) Wogonin and fisetin induce apoptosis in humanpromyeloleukemic cells, accompanied by a decrease of reactive oxygenspecies, and activation of caspase 3 and Ca2þ-dependent endonuclease.Biochem. Pharmacol., 63, 225–236.
31.Beutler,J.A. et al. (1998) Structure-activity requirements for flavone cyto-toxicity and binding to tubulin. J. Med. Chem., 41, 2333–2338.
32.Gupta,K. et al. (2002) Perturbation of microtubule polymerization by quer-cetin through tubulin binding: a novel mechanism of its antiproliferativeactivity. Biochemistry, 41, 13029–13038.
33.Nicholson,D.W. et al. (1997) Caspases: killer proteases. Trends Biochem.Sci., 22, 299–306.
34.Nguyen,T.T.T. et al. (2004) The role of activated MEK-ERK pathway inquercetin-induced growth inhibition and apoptosis in A549 lung cancercells. Carcinogenesis, 25, 647–659.
35.Dangles,O. et al. (1999) Flavonol-serum albumin complexation. Two-elec-tron oxidation of flavonols and their complexes with serum albumin.J. Chem. Soc. Perkin Trans, 2, 737–744.
36.Yuan,Z.P. et al. (2006) Liposomal quercetin efficiently suppresses growthof solid tumors in murine models. Clin. Cancer Res., 12, 3193–3199.
S.Rubio et al.
2112

37.Szewczyk,A. et al. (2002) Mitochondria as a pharmacological target. Phar-macol. Rev., 54, 101–127.
38.Van Gurp,M. et al. (2003) Mitochondrial intermembrane proteins in celldeath. Biochem. Biophys. Res. Commun., 304, 487–497.
39.Margolin,N. et al. (1997) Substrate and inhibitor specificity of interleukin-1b-converting enzyme and related caspases. J. Biol. Chem., 272, 7223–7228.
40.Rodriguez,J. et al. (1999) Caspase-9 and APAF-1 form an active holoen-zyme. Genes Dev., 13, 3179–3184.
41.Stennicke,H.R. et al. (1999) Caspase-9 can be activated without proteolyticprocessing. J. Biol. Chem., 274, 8359–8362.
42.Scoltock,A.B. et al. (2004) Activation of intrinsic and extrinsic pathways inapoptotic signaling during UV-C-induced death of Jurkat cells: the role ofcaspase inhibition. Exp. Cell Res., 297, 212–223.
43.Campos,L. et al. (1993) High expression of Bcl-2 protein in acute myeloidleukaemia cells is associated with poor response to chemotherapy. Blood,81, 3091–3096.
44.Achenbach,T.V. et al. (2000) Bcl-2 independence of flavopiridol-inducedapoptosis. Mitochondrial depolarization in the absence of cytochromec release. J. Biol. Chem., 275, 32089–32097.
45.Fadeel,B. et al. (1999) Cleavage of Bcl-2 is an early event in chemotherapy-induced apoptosis of human myeloid leukemia cells. Leukemia, 13,719–728.
46.Yamamoto,K. et al. (1999) BCL-2 is phosphorylated and inactivated by anASK1/Jun N-terminal protein kinase pathway normally activated at G2/M.Mol. Cell. Biol., 19, 8469–8478.
47.Cross,T.G. et al. (2000) Serine/threonine protein kinases and apoptosis.Exp. Cell Res., 256, 34–41.
48.Yu,C. et al. (2001) MEK1/2 inhibitors promote Ara-C-induced apoptosisbut not loss of DWm in HL-60 cells. Biochem. Biophys. Res. Commun., 286,1011–1018.
49.Herr,I. et al. (2001) Cellular stress response and apoptosis in cancer ther-apy. Blood, 98, 2603–2614.
50.Zarubin,T. et al. (2005) Activation and signaling of the p38 MAP kinasepathway. Cell Res., 15, 11–18.
51.Galan,A. et al. (2000) Stimulation of p38 mitogen-activated protein kinaseis an early regulatory event for the cadmium-induced apoptosis in humanpromonocytic cells. J. Biol. Chem., 275, 11418–11424.
52.Shimizu,T. et al. (2006) Resveratrol induces apoptosis of human malignantB cells by activation of caspase-3 and p38 MAP kinase pathways. Biochem.Pharmacol., 71, 742–750.
53.Franklin,C.C. et al. (1998) Conditional expression of mitogen-activatedprotein kinase phosphatase-1, MKP-1, is cytoprotective against UV-induced apoptosis. Proc. Natl Acad. Sci. USA, 95, 3014–3019.
54. Juo,P. et al. (1997) Fas activation of the p38 mitogen-activated proteinkinase signalling pathway requires ICE/CED-3 family proteases.Mol. Cell.Biol., 17, 24–35.
55.Hayakawa,J. et al. (1999) Inhibition of extracellular signal-regulated pro-tein kinase or c-Jun N-terminal protein kinase cascade, differentially acti-vated by cisplatin, sensitizes human ovarian cancer cell line. J. Biol. Chem.,274, 31648–31654.
56.Persons,D.L. et al. (1999) Cisplatin-induced activation of mitogen-activated protein kinases in ovarian carcinoma cells: inhibition of extracel-lular signal-regulated kinase activity increases sensitivity to cisplatin. Clin.Cancer Res., 5, 1007–1014.
57.Mandic,A. et al. (2001) The MEK1 inhibitor PD98059 sensitizes C8161melanoma cells to cis-platin-induced apoptosis.Melanoma Res., 11, 11–19.
58.Chen,Y.R. et al. (1998) Molecular mechanisms of c-Jun N-terminal kinase-mediated apoptosis induced by anticarcinogenic isothiocyanates. J. Biol.Chem., 273, 1769–1775.
59.Shiah,S.G. et al. (1999) Activation of c-Jun NH2-terminal kinase and sub-sequent CPP32/Yama during topoisomerase inhibitor b-lapachone-inducedapoptosis through an oxidation-dependent pathway.Cancer Res., 59, 391–398.
60.Watabe,M. et al. (1999) Requirement of protein kinase (Krs/MST) activa-tion for MT-21-induced apoptosis. Oncogene, 18, 5211–5220.
61.Zhuang,S. et al. (2000) p38 mitogen-activated protein kinase mediates bidcleavage, mitochondrial dysfunction, and caspase-3 activation during apo-ptosis induced by singlet oxygen but not by hydrogen peroxide. J. Biol.Chem., 275, 25939–25948.
62.Hollstein,M. et al. (1994) Database of p53 gene somatic mutations inhuman tumors and cell lines. Nucleic Acids Res., 22, 3551–3555.
63.Hollstein,M. et al. (1991) p53 mutations in human cancers. Science, 253,49–53.
Received January 24, 2007; revised May 22, 2007; accepted May 23, 2007
Induced cell death in human leukemia cells
2113


