equilibrio de fases en sistemas de multicomponentes
TRANSCRIPT

Greyssy BarrazaJessica Caicedo
Aida Picón
EQUILIBRIO DE FASES EN SISTEMAS MULTICOMPONENTE
S

CONCEPTOS BÁSICOS.FASES Y TRANSICIONES DE FASE.
Fase: Porción homogénea de un sistema. Las propiedades macroscópicas intensivas son idénticas en cualquier punto del sistema
Sistema homogéneoFormado por una fase.
Sistema heterogéneoFormado por más de una fase.
Varios componentes Un solo componente(sustancia pura)
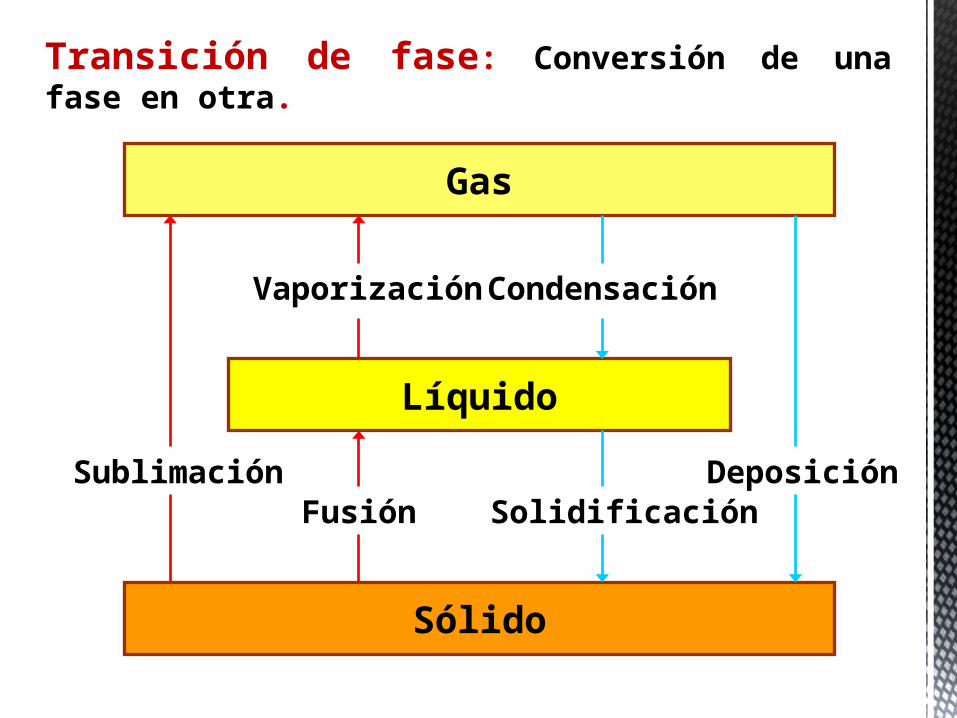
Transición de fase: Conversión de una fase en otra.
Gas
Líquido
Sólido
Sublimación
Vaporización
Fusión
Condensación
SolidificaciónDeposición

Los dos tipos de equilibrio material son el equilibrio químico y el equilibrio de fases.
La condición de equilibrio de fases indica que el potencial químico de cada una de las especies debe ser el mismo en cada fase en la que se presenta cada especie.
El equilibrio de fases y las transiciones de fases aparecen con mucha frecuencia en nuestro entorno, desde la ebullición del agua en una tetera a la fusión de los glaciares en la Antártida.

Para describir el estado de equilibrio de un sistema de varias fases y diversas especies químicas deberemos conocer el número de variables intensivas independientes que definen el sistema. Para conocer este número se aplica la regla de las fases: L=C-F+2; donde L es número de variables intensivas independientes (grados de libertad), C el número de componentes químicos del sistema, y F el número de fases presentes en el sistema
Cuando en el sistema pueden ocurrir una o varias reacciones químicas (r), entonces el número de variables intensivas independientes se reduce en el número de reacciones que ocurren y la regla de las fases se transforma en: L=C-F+2-r
Pero además si en el sistema existen relaciones debidas a la estequiometria o de conservación de la electroneutralidad del sistema, el número de variables intensivas independientes se reduce en un número correspondiente a estas relaciones que llamaremos a. La regla de las fases con todas estas restricciones queda definida por la siguiente ecuación: L=C-F+2-r-a
REGLA DE LAS FASES

1, Calcular el número de grados de libertad que definen un sistema compuesto por sacarosa sólida en equilibrio con una disolución acuosa de sacarosa.
El número de componentes = número de especies químicas diferentes, en este caso será 2, la sacarosa y el agua. Por lo tanto C = 2
El número de fases = tendremos dos fases, la disolución de sacarosa que será una fase líquida y la sacarosa sólida. Por lo tanto F = 2
No existen ni reacciones ni relaciones entre los componentes por lo tanto el número de grados de libertad será: L=2-2+2=2
Es decir con dos variables independientes podremos definir el sistema, estas dos variables pueden ser por ejemplo la presión y la temperatura, ya que a una (P,T) dada la solubilidad de la sacarosa sólo tiene un valor posible, y es el que determina la concentración de sacarosa en agua.
Ejemplos de la
aplicación de la
regla de las fases

2, Calcular el número de grados de libertad para una mezcla gaseosa de N2, H2 y NH3 que no reaccionan entre sí.
C = 3, las tres especies químicas diferentes. F = 1 una única fase gaseosa No existen relaciones entre los componentes del sistema, luego L=3-
1+2=4 La misma mezcla gaseosa de N2, H2 y NH3 pero en este caso añadimos
un catalizador para que se produzca la reacción. La reacción que tendrá lugar será C = 3 especies diferentes; F = 1 fase gaseosa; r = 1, existe una reacción
que relaciona las especies del sistema, luego L=3-1+2-1=3; son necesarias 3 variables intensivas independientes para definir el sistema.
3, Calcular el número de grados de libertad para definir un sistema formado por NH3 al que se le adiciona un catalizador para establecer el equilibrio
Este caso difiere ligeramente con respecto al anterior ya que además de existir una reacción química que relaciona las especies, existe también una relación estequiométrica entre las especies que es que la fracción molar de hidrógeno es 3 veces la fracción molar de nitrógeno por lo tanto a en este caso es 1
L=3-1+2-1-1=2; en este caso con 2 variables intensivas independientes se define totalmente el sistema.

Para especificar el estado termodinámico de un sistema formado por una sustancia pura el número variables intensivas independientes que hay conocer (grados de libertad) es:
Si hay presente una fase, L=1componente-1fase+2=2 variables, es necesario especificar por ejemplo la P y la T
Si hay presente dos fases, L=1componente-2fases+2=1 variable, es necesario especificar sólo P o T
Si hay presente tres fases, L=1componente-3fases+2=0 variables
Por tanto podemos representar cualquier estado de equilibrio del sistema formado por una sustancia pura mediante un punto en un diagrama bidimensional de presión-temperatura. Este diagrama se denomina diagrama de fases.
DIAGRAMA DE FASES
PARA SISTEMAS
DE UN COMPONE
NTE
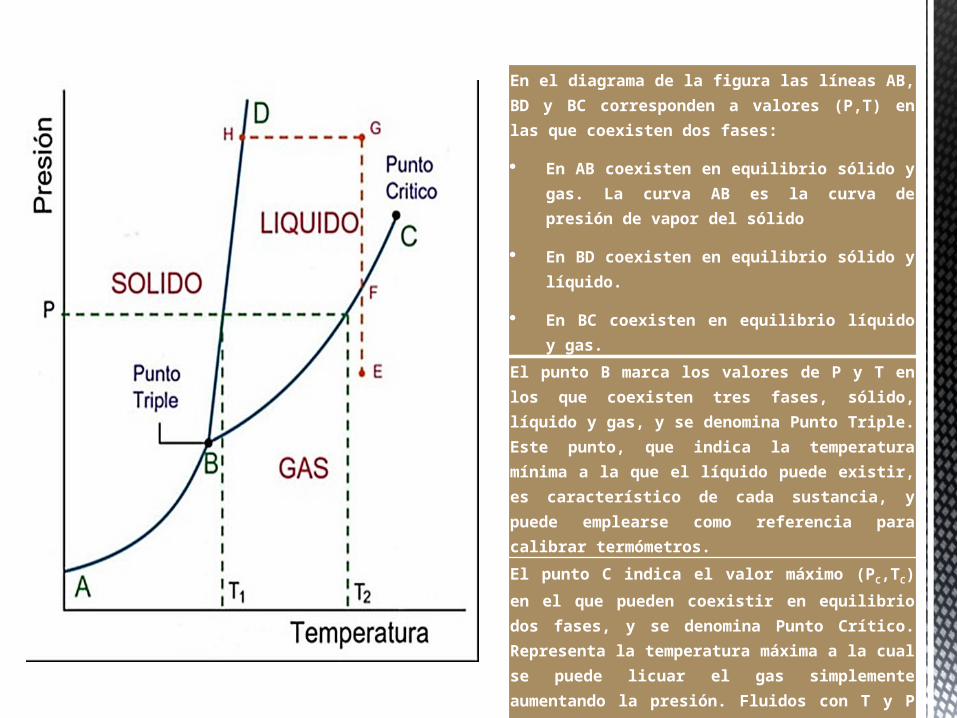
En el diagrama de la figura las líneas AB, BD y BC corresponden a valores (P,T) en las que coexisten dos fases:
En AB coexisten en equilibrio sólido y gas. La curva AB es la curva de presión de vapor del sólido
En BD coexisten en equilibrio sólido y líquido.
En BC coexisten en equilibrio líquido y gas.
El punto B marca los valores de P y T en los que coexisten tres fases, sólido, líquido y gas, y se denomina Punto Triple. Este punto, que indica la temperatura mínima a la que el líquido puede existir, es característico de cada sustancia, y puede emplearse como referencia para calibrar termómetros.
El punto C indica el valor máximo (PC,TC) en
el que pueden coexistir en equilibrio dos fases, y se denomina Punto Crítico. Representa la temperatura máxima a la cual se puede licuar el gas simplemente aumentando la presión. Fluidos con T y P mayores que TC y PC se denominan fluidos
supercríticos

Efecto de la presión
y de la temperatur
a Ecuación
de Clapeyron
.
La ecuación de Clapeyron permite calcular la pendiente de una línea de equilibrio entre dos fases en el diagrama de fases P- T de un sistema de un componente.
Deducción
Consideremos un punto cualquiera sobre una línea de equilibrio entre dos las fases, que llamaremos α y β. La condición para que exista equilibrio de
fases es que: , pero para una sustancia pura , por
tanto en un punto sobre la curva de equilibrio de dos fases , y cualquier variación infinitesimal que suponga un desplazamiento sobre la
curva de equilibrio implica que . O lo que es lo mismo,
, y reagrupando términos .
Por otra parte si se considera que en un cambio de fase reversible a T y P
constantes , se tiene que
Ecuación de Clapeyron
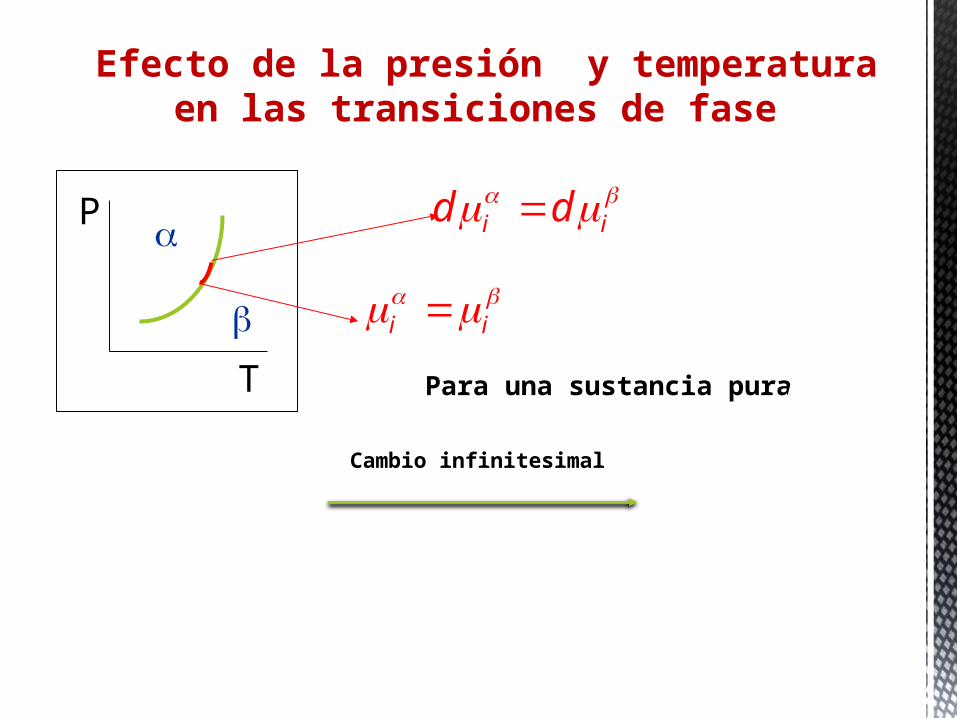
Efecto de la presión y temperatura en las transiciones de fase
G G
dG dG
Cambio infinitesimal
S dT V dP S dT V dP
P
P
P
T
i i
i id d
Para una sustancia pura G
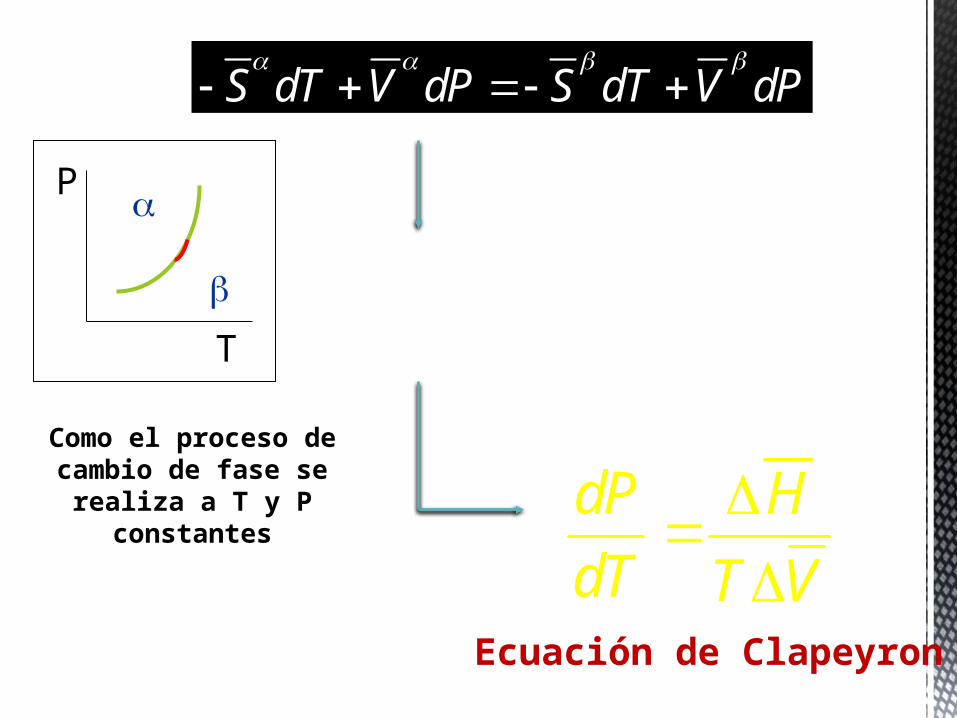
Como el proceso de cambio de fase se
realiza a T y P constantes
HS
T
S dT V dP S dT V dP
( ) ( ) S S dT V V dP
dP S
dT V
dP H
dT T VEcuación de Clapeyron
P
P
T

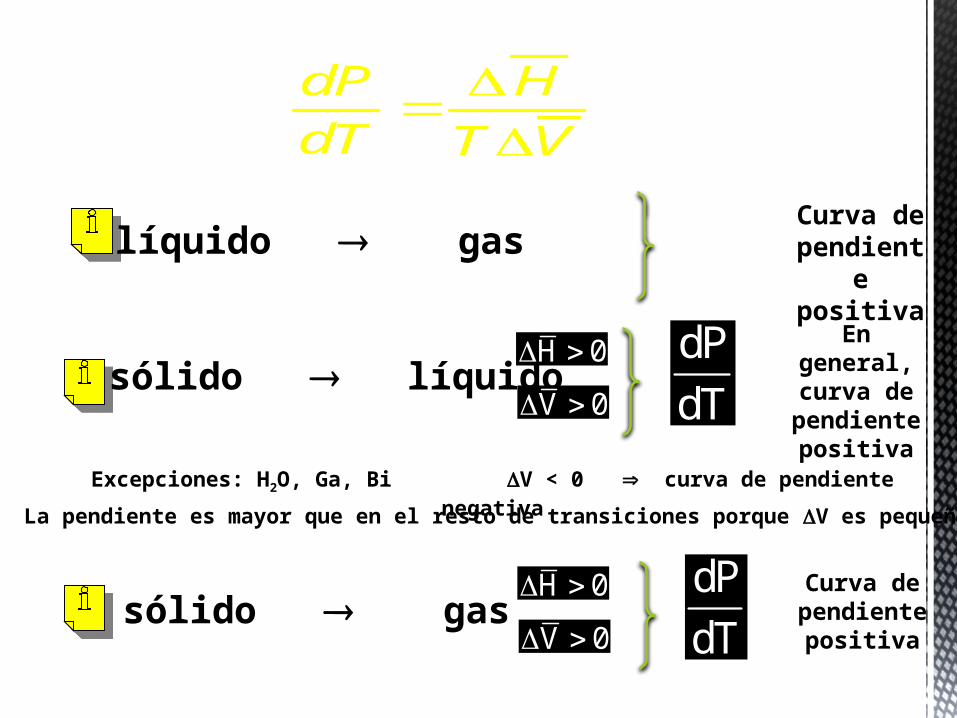
En un cambio de fase líquido-vapor, tanto ΔH como ΔV son positivos, por tanto la pendiente de la línea de equilibrio líquido-vapor es positiva. Lo mismo sucede con la línea sólido-vapor.
En un cambio de fase sólido-líquido, ΔH es positivo y en general ΔV también, por lo tanto la pendiente de esta línea también será positiva. Existen sin embargo algunas excepciones como el H2O, Ga y Bi debido a una disminución de volumen que sufren estos componentes al fundirse, en estos casos la pendiente de la línea de equilibrio sólido-líquido será negativa.
En el cambio de fase sólido-líquido ΔV es mucho menor que en los cambios de fase sólido-gas o líquido-gas. Por esta razón la pendiente en el primer caso es mucho mayor que en los últimos.
Consideraciones sobre la ecuación de
Clapeyron
dP H
dT T V
líquido gasH 0
V 0
Curva de pendient
e positiva
dP
dT
sólido líquidoH 0
V 0
En general,curva de
pendiente positiva
dP
dT
Excepciones: H2O, Ga, Bi V < 0 curva de pendiente negativa
sólido gasH 0
V 0
Curva de pendiente positiva
dP
dT
La pendiente es mayor que en el resto de transiciones porque V es pequeño
APLICACIÓN DE LA ECUACIÓN DE CLAPEYRON
Equilibrio líquido-vapor y sólido-vapor
g l gV V V V
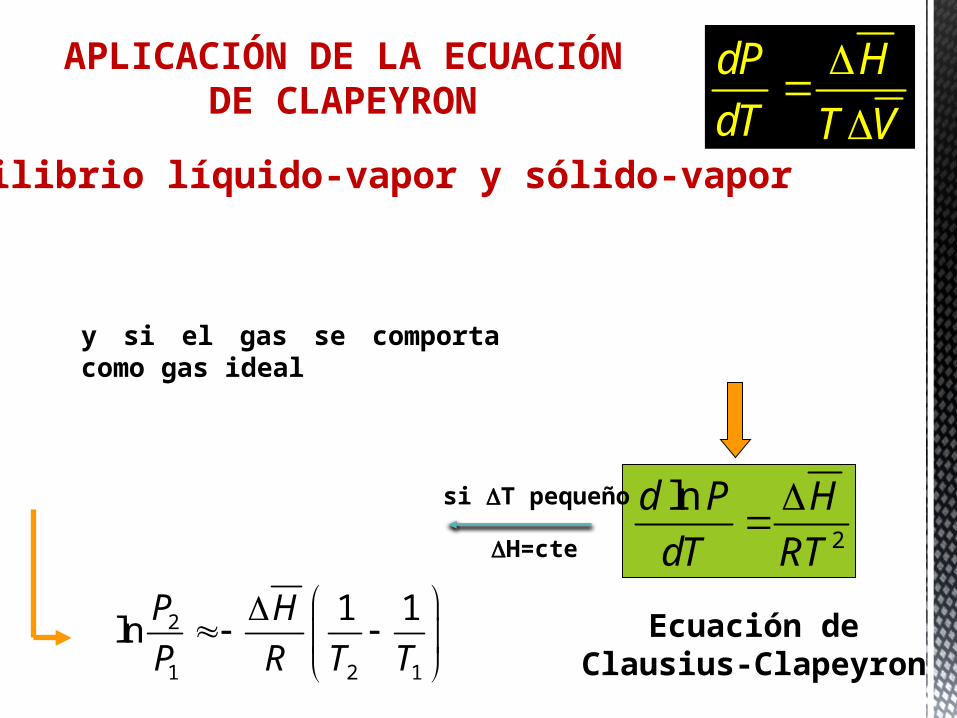
y si el gas se comporta como gas ideal
2
g
dP H H P H
dT RTT V TV
2
lnd P H
dT RT
Ecuación de Clausius-Clapeyron
dP H
dT T V
2 2
1 12
ln P T
P T
dTd P H
RTsi T pequeño
H=cte
2
1 2 1
1 1lnP H
P R T T

Equilibrio sólido-líquido
ImportanteNo puede aplicarse la ec. de Clausius-Clapeyron,
Si la ecuación de Clapeyron (VVg)
dP H
dT T V
2 2
1 1
P T
P T
HdP dT
T V
La elevada pendiente de esta línea, implica que si P no cambia de forma considerable, la variación de T será muy pequeña
22 1
1
lnfus
fus
THP P
TV
si H y V cte
en el rango de T y P

Muchas sustancias presentan más de una forma sólida. Cada una de ellas tiene una estructura cristalina diferente que es estable termodinámicamente en un intervalo de P y T. En general, a este fenómeno se le denomina polimorfismo, y a las diferentes estructuras cristalinas posibles, formas polimórficas. En el caso particular de que la sustancia sea un elemento, al polimorfismo se le denomina alotropía. La forma estable a una determinada P y T es la de mínima G, pero en ocasiones se puede mantener durante cierto tiempo la fase denominada metaestable (inestable termodinámicamente), si la velocidad del proceso espontáneo de conversión de fase es lo suficientemente lento: por ejemplo el C diamante es una fase metaestable a 25ºC y 1bar, la fase estable es el C grafito, pero ambas se pueden tener en esas condiciones de P y T dado que la cinética del cambio de fase es muy lenta.
Transiciones de fase
sólido-sólido

EQUILIBRIO DE
FASES EN SISTEMAS MULTICOMPONENTES
Equilibrio líquido-líquido en mezclas binarias.
Si consideramos la mezcla a P y T constante de dos líquidos A y B, en cantidades nA y nB, esta se producirá cuando G disminuya, es decir cuando la energía libre de la mezcla sea menor que la energía libre de los dos componentes puros. Así si definimos la energía libre de mezcla ΔGmezcla como el cambio en la energía libre del sistema al llevar a cabo el proceso de mezclar ambos
líquidos: , y por mol de mezcla:
, que debe ser < 0 para que el proceso tenga lugar.
La ΔGmezcla, a T y P constantes, puede variar con la composición del sistema (con la fracción molar de sus componentes), según se representa esquemáticamente en la figura:
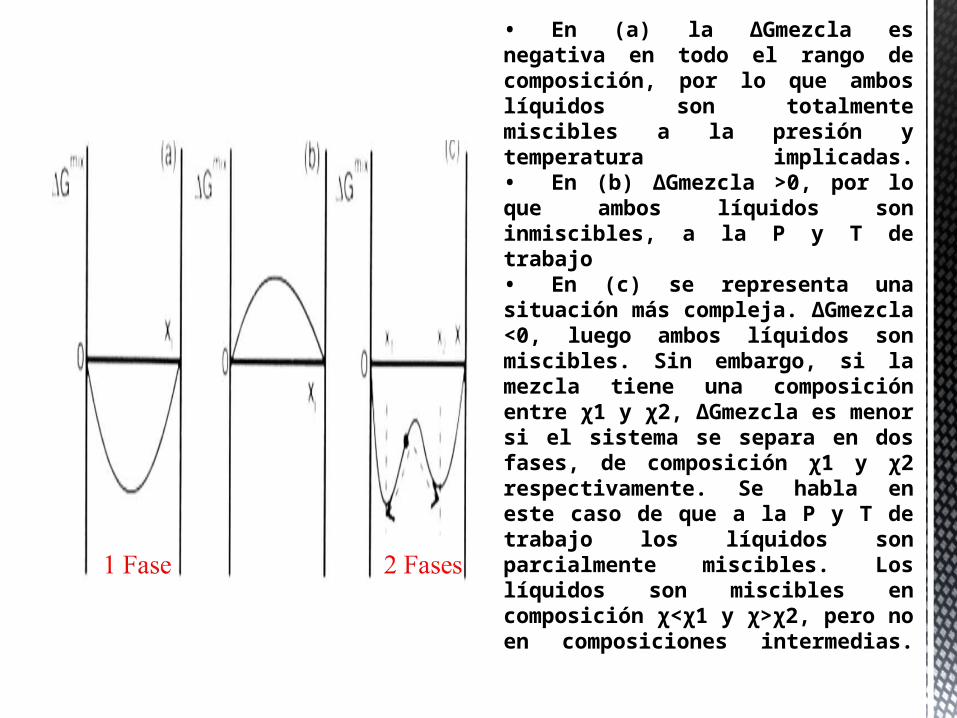
• En (a) la ΔGmezcla es negativa en todo el rango de composición, por lo que ambos líquidos son totalmente miscibles a la presión y temperatura implicadas.• En (b) ΔGmezcla >0, por lo que ambos líquidos son inmiscibles, a la P y T de trabajo• En (c) se representa una situación más compleja. ΔGmezcla <0, luego ambos líquidos son miscibles. Sin embargo, si la mezcla tiene una composición entre χ1 y χ2, ΔGmezcla es menor si el sistema se separa en dos fases, de composición χ1 y χ2 respectivamente. Se habla en este caso de que a la P y T de trabajo los líquidos son parcialmente miscibles. Los líquidos son miscibles en composición χ<χ1 y χ>χ2, pero no en composiciones intermedias.
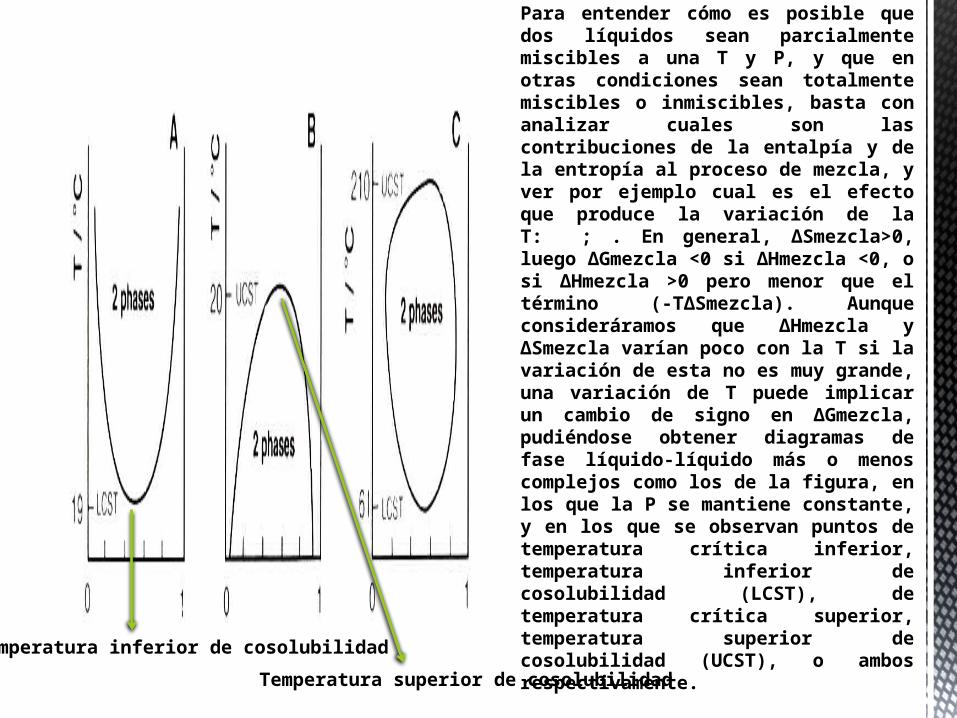
Para entender cómo es posible que dos líquidos sean parcialmente miscibles a una T y P, y que en otras condiciones sean totalmente miscibles o inmiscibles, basta con analizar cuales son las contribuciones de la entalpía y de la entropía al proceso de mezcla, y ver por ejemplo cual es el efecto que produce la variación de la T: ; . En general, ΔSmezcla>0, luego ΔGmezcla <0 si ΔHmezcla <0, o si ΔHmezcla >0 pero menor que el término (-TΔSmezcla). Aunque consideráramos que ΔHmezcla y ΔSmezcla varían poco con la T si la variación de esta no es muy grande, una variación de T puede implicar un cambio de signo en ΔGmezcla, pudiéndose obtener diagramas de fase líquido-líquido más o menos complejos como los de la figura, en los que la P se mantiene constante, y en los que se observan puntos de temperatura crítica inferior, temperatura inferior de cosolubilidad (LCST), de temperatura crítica superior, temperatura superior de cosolubilidad (UCST), o ambos respectivamente.
Temperatura inferior de cosolubilidad
Temperatura superior de cosolubilidad

Equilibrio líquido-gas de mezclas
binarias
Equilibrio líquido-gas de mezclas binarias.
Hemos visto que a una T y P dadas, en un sistema constituido por dos fases (ej. líquido y gas) de una sustancia pura, el equilibrio termodinámico se produce cuando los potenciales
químicos de la sustancia son iguales en ambas fases: . En el caso de una mezcla homogénea de varias sustancias, disolución, el equilibrio material se produce igualmente cuando los potenciales químicos de cada una de las sustancias son iguales en ambas fases.
Por sencillez, vamos a considerar que las sustancias que forman la mezcla binaria en fase gaseosa se comportan como gases ideales, y que en fase líquida constituyen una disolución ideal (los dos componentes líquidos son estructuralmente y químicamente muy similares, de forma que las interacciones moleculares soluto- disolvente, soluto- soluto y disolvente-disolvente son iguales).
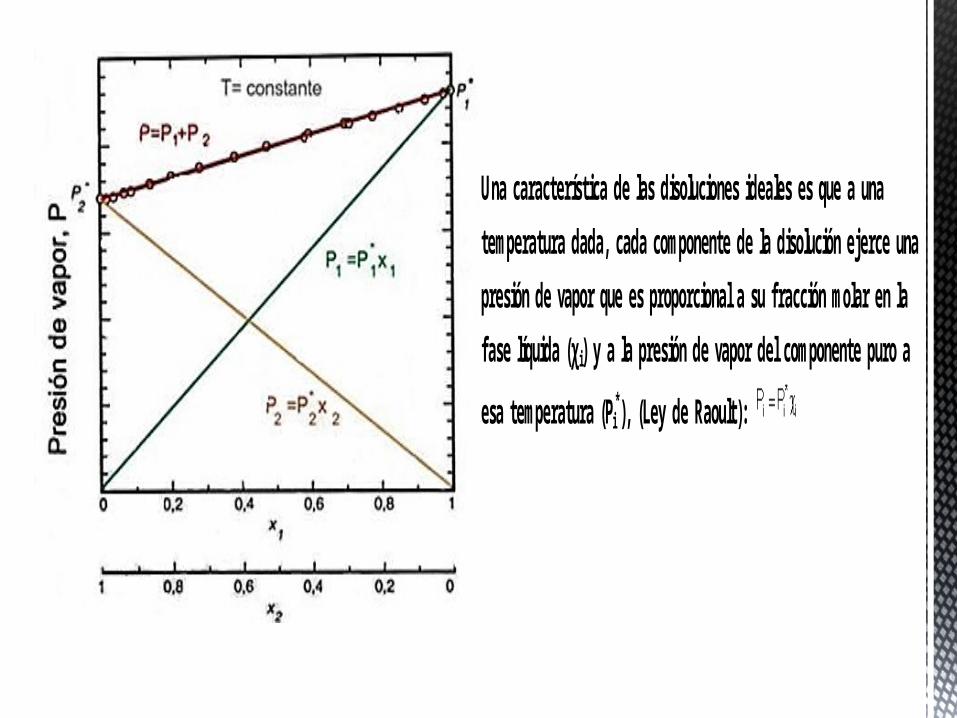
Una característica de las disoluciones ideales es que a una
temperatura dada, cada componente de la disolución ejerce una
presión de vapor que es proporcional a su fracción molar en la
fase líquida (χi) y a la presión de vapor del componente puro a
esa temperatura (Pi*), (Ley de Raoult):

Recordando que el potencial químico de un gas ideal (i) en una
mezcla de gases es: ; y teniendo en cuenta que en
equilibrio, para cada componente se debe cumplir , se obtiene que el potencial químico de cada componente de la disolución ideal en la fase líquida a una T dada, es:
.
Si χi=1, se tiene el líquido puro, pudiéndose definir el potencial químico de una sustancia pura en fase líquida como
, que depende de la temperatura y de la presión de vapor del compuesto puro a dicha temperatura. Así, en el caso de disoluciones ideales, el potencial químico de cada componente en la fase líquida vendrá dado por la expresión:
.
En la formación de una disolución ideal a T y P constantes,
. Si se considera la formación de
un mol de disolución ideal: . Por tanto la formación de una disolución ideal es siempre un proceso espontáneo. Además, por ser muy similares las moléculas de soluto y disolvente, y las interacciones entre ellas, no hay cambio en el volumen al realizar la disolución ideal, ΔVmezcla=0, ni cambio en la entalpía, ΔHmezcla=0. En consecuencia, la
variación de entropía en el proceso será:

Relaciones presión-
composición y
temperatura-
composición en
disoluciones ideales
Si la disolución es ideal, ambos componentes obedecen la ley de Raoult, luego la presión total es una función lineal de la fracción molar de cualquiera de los dos componentes en la fase líquida (χi):
Para conocer la relación entre la presión total del sistema y la composición del vapor, basta suponer el comportamiento ideal del gas: la presión parcial de cada gas (Pi) será igual a la presión total (P) por su fracción molar (yi), es decir, se cumple
la ley de Dalton , por tanto la composición del gas se relaciona con la composición del líquido por la expresión:
. Por lo que la relación entre la presión del gas y su composición vendrá dada por:
El conocimiento de la relación entre la P del sistema y la composición de las dos fases, líquida y gas, permite dibujar el diagrama de fases a T constante:
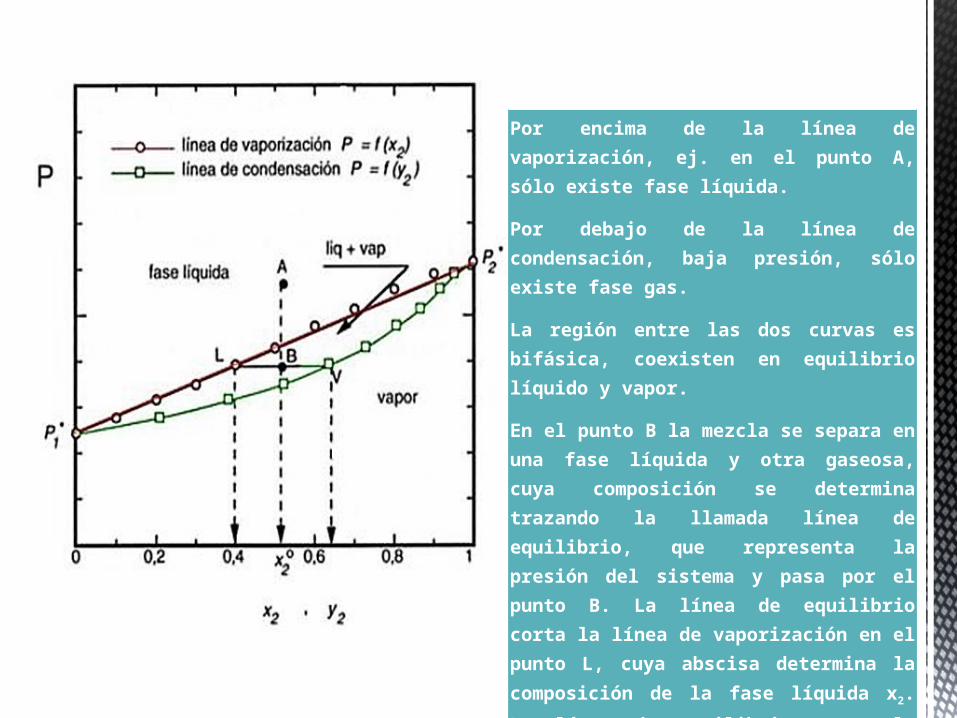
Por encima de la línea de vaporización, ej. en el punto A, sólo existe fase líquida.
Por debajo de la línea de condensación, baja presión, sólo existe fase gas.
La región entre las dos curvas es bifásica, coexisten en equilibrio líquido y vapor.
En el punto B la mezcla se separa en una fase líquida y otra gaseosa, cuya composición se determina trazando la llamada línea de equilibrio, que representa la presión del sistema y pasa por el punto B. La línea de equilibrio corta la línea de vaporización en el punto L, cuya abscisa determina la composición de la fase líquida x2. La línea de equilibrio corta la línea de condensación en el punto V, cuya abscisa determina la composición de la fase gas y2.
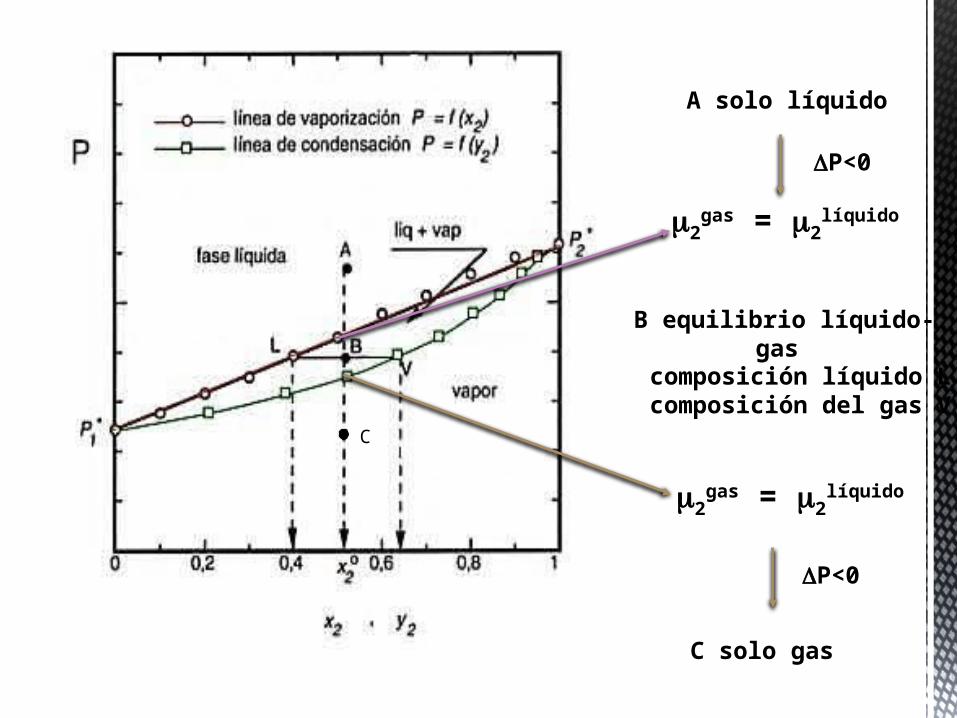
A solo líquido
P<0
2gas = 2
líquido
B equilibrio líquido-gas
composición líquido Lcomposición del gas V
2gas = 2
líquido
C
P<0
C solo gas
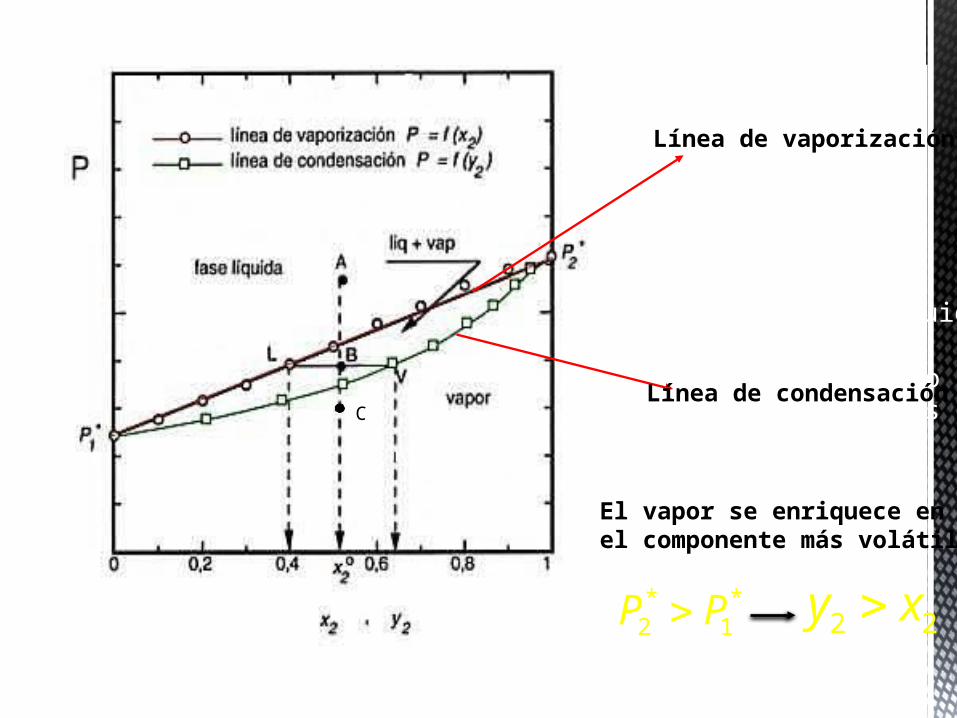
B equilibrio líquido-gas
composición Líquido Lcomposición del gas VC
C solo gas
Línea de vaporización
Línea de condensación
El vapor se enriquece enel componente más volátil
* *2 1P P 2 2y x
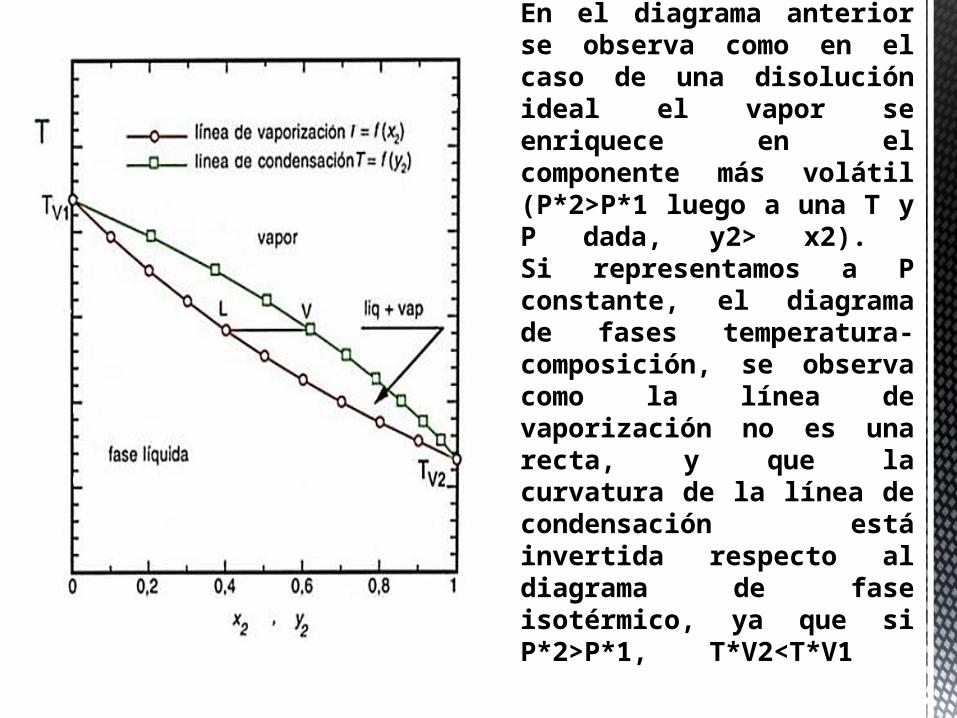
En el diagrama anterior se observa como en el caso de una disolución ideal el vapor se enriquece en el componente más volátil (P*2>P*1 luego a una T y P dada, y2> x2). Si representamos a P constante, el diagrama de fases temperatura-composición, se observa como la línea de vaporización no es una recta, y que la curvatura de la línea de condensación está invertida respecto al diagrama de fase isotérmico, ya que si P*2>P*1, T*V2<T*V1
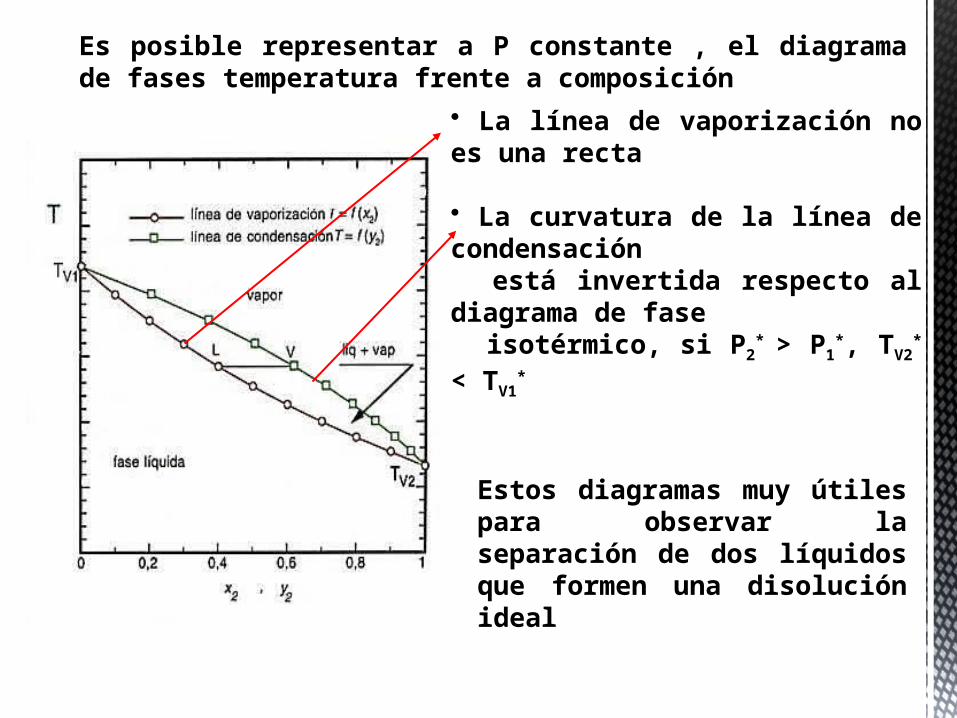
Es posible representar a P constante , el diagrama de fases temperatura frente a composición
Estos diagramas muy útiles para observar la separación de dos líquidos que formen una disolución ideal
• La línea de vaporización no es una recta
• La curvatura de la línea de condensación está invertida respecto al diagrama de fase isotérmico, si P2
* > P1
*, TV2* <
TV1*

Relaciones presión-
composición y
temperatura-
composición en
disoluciones no-
ideales
Muy pocos sistemas forman disoluciones ideales en todo el rango de composición, aunque todas se aproximan al comportamiento ideal cuando están suficientemente diluidas. De hecho se habla de disoluciones diluidas ideales (o idealmente diluidas) cuando la fracción molar del disolvente se aproxima a la unidad, de forma que las moléculas de soluto sólo interaccionan con el disolvente dada su dilución.
En el caso de disoluciones no- ideales el potencial químico se define en función de la actividad (a), que podríamos considerar como una
"concentración efectiva" : ; si el μi0es el potencial químico
de la sustancia en su estado normal.
o a = P/P0 en el caso de gases ideales , siendo P0 = 1bar o a = Pi en el caso de gases ideales en una mezcla o a = 1 en el caso de sólidos o líquidos puros, ya que por
definición μi=μi0
o a = χi en el caso de disoluciones ideales o a = γiχi en el caso de disoluciones reales; el coeficiente
de actividad, γi es una medida de la discrepancia del comportamiento de la sustancia i respecto a la idealidad.
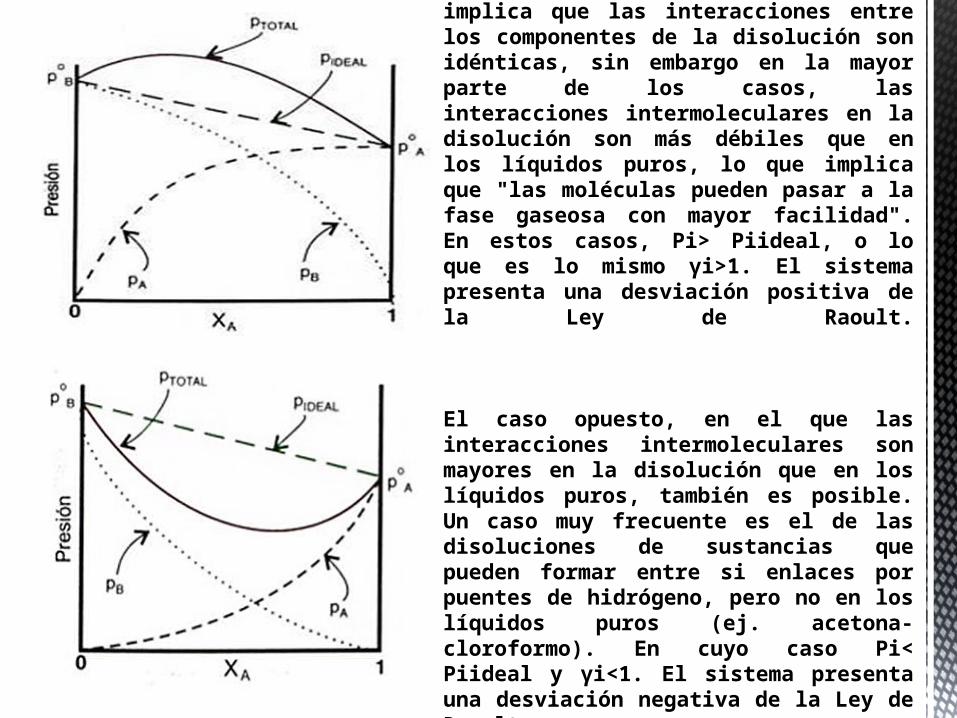
El modelo de disolución ideal implica que las interacciones entre los componentes de la disolución son idénticas, sin embargo en la mayor parte de los casos, las interacciones intermoleculares en la disolución son más débiles que en los líquidos puros, lo que implica que "las moléculas pueden pasar a la fase gaseosa con mayor facilidad". En estos casos, Pi> Piideal, o lo que es lo mismo γi>1. El sistema presenta una desviación positiva de la Ley de Raoult.
El caso opuesto, en el que las interacciones intermoleculares son mayores en la disolución que en los líquidos puros, también es posible. Un caso muy frecuente es el de las disoluciones de sustancias que pueden formar entre si enlaces por puentes de hidrógeno, pero no en los líquidos puros (ej. acetona-cloroformo). En cuyo caso Pi< Piideal y γi<1. El sistema presenta una desviación negativa de la Ley de Raoult.
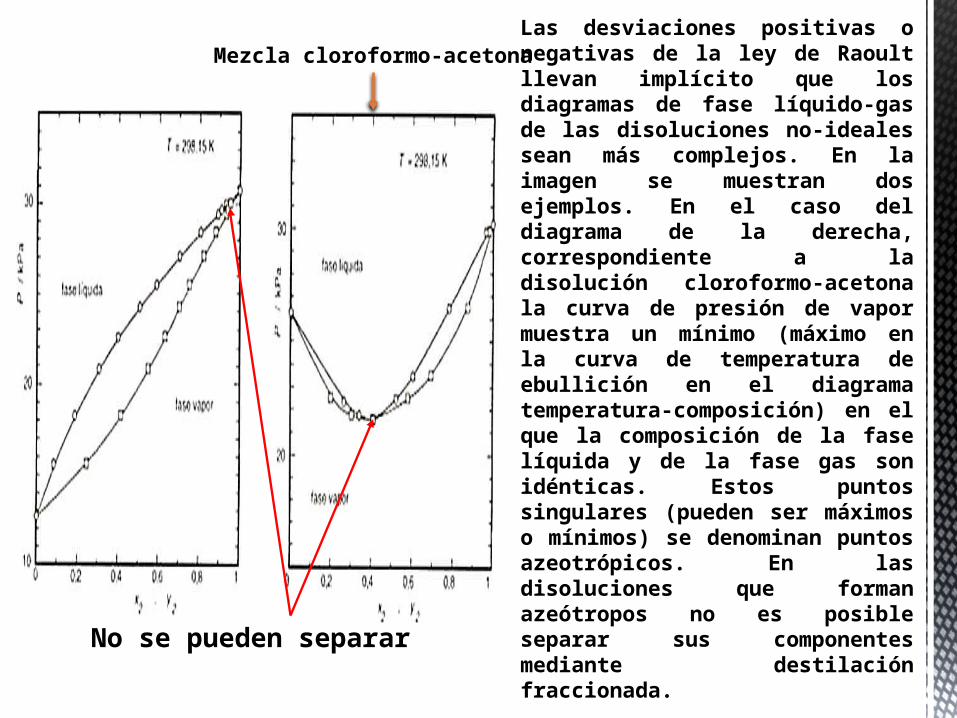
Las desviaciones positivas o negativas de la ley de Raoult llevan implícito que los diagramas de fase líquido-gas de las disoluciones no-ideales sean más complejos. En la imagen se muestran dos ejemplos. En el caso del diagrama de la derecha, correspondiente a la disolución cloroformo-acetona la curva de presión de vapor muestra un mínimo (máximo en la curva de temperatura de ebullición en el diagrama temperatura-composición) en el que la composición de la fase líquida y de la fase gas son idénticas. Estos puntos singulares (pueden ser máximos o mínimos) se denominan puntos azeotrópicos. En las disoluciones que forman azeótropos no es posible separar sus componentes mediante destilación fraccionada.
No se pueden separar
Mezcla cloroformo-acetona
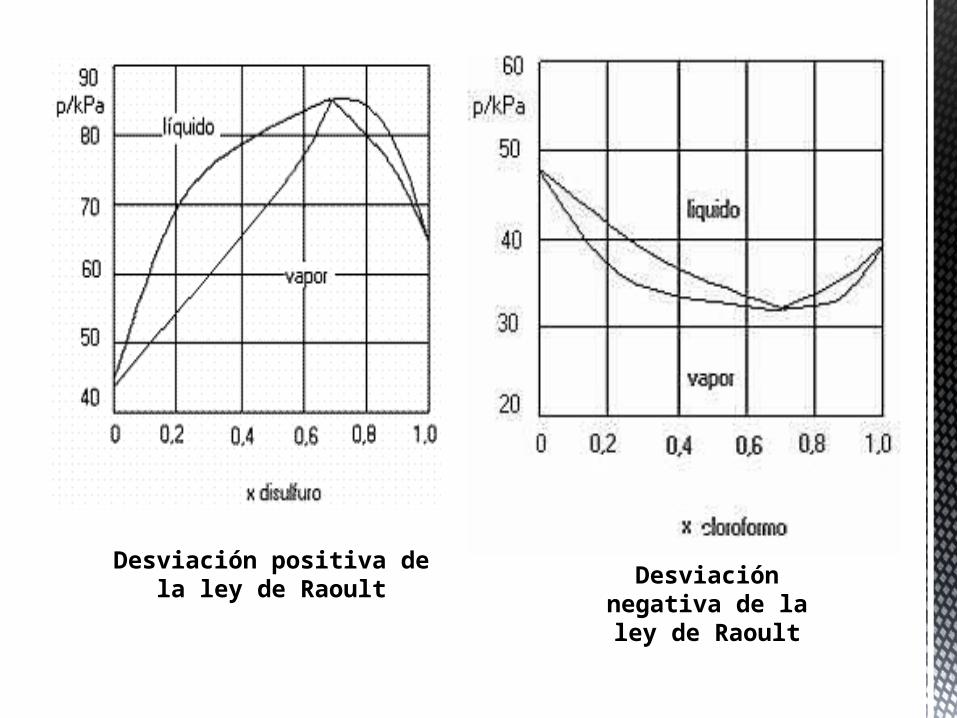
Desviación positiva de la ley de Raoult Desviación
negativa de la ley de Raoult

Por simplicidad, y como en los casos anteriores, supondremos que el sistema está formado por dos componentes totalmente miscibles, y que ninguno de ellos es un electrolito. Convencionalmente llamaremos disolvente (B) al componente más abundante de la disolución, y soluto (A) al componente minoritario, si bien desde un punto de vista termodinámico no sería necesario.
Al enfriar la disolución a presión constante se producirá la solidificación, pero la experiencia diaria muestra que son posibles dos situaciones:
• Sólido A ←→ líquido (A+B). Al descender la temperatura se alcanza el límite de solubilidad del soluto en el disolvente, dando como resultado A sólido en equilibrio con la disolución saturada de A en B.
• Sólido B ←→ líquido (A+B). A una determinada temperatura solidifica el disolvente, se ha alcanzado el punto de solidificación de la disolución.
Equilibrio de fases
sólido-líquido en sistemas
de dos component
es
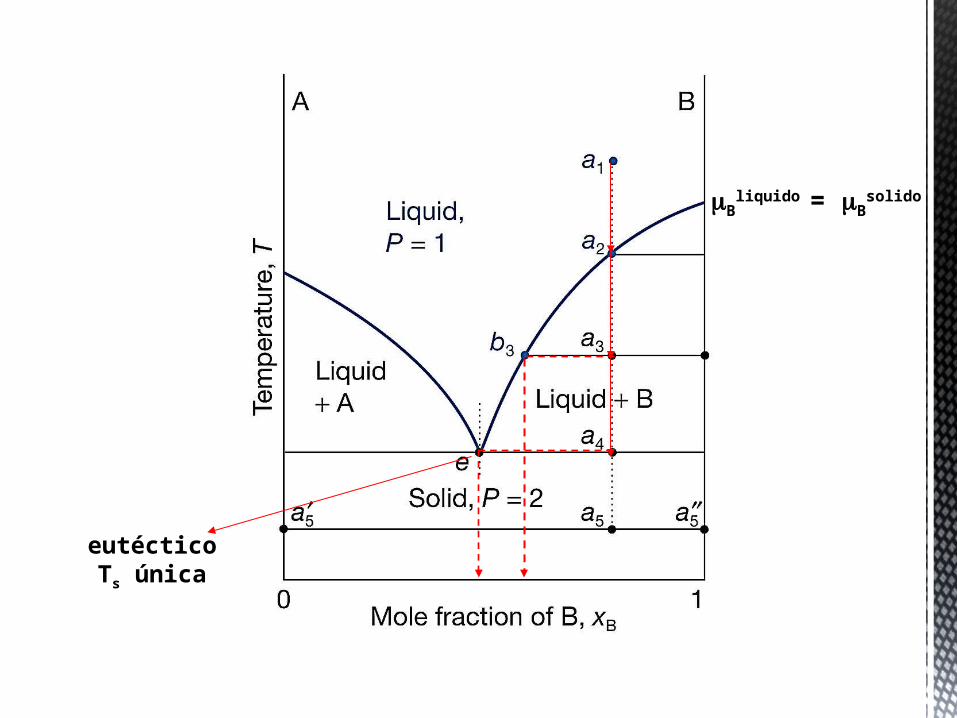
Bliquido = B
solido
eutécticoTs única

El modelo empleado para tratar ambas situaciones es el mismo. Así, si tomamos por ejemplo el primer caso: en el equilibrio Sólido A ←→ A en disolución, debe cumplirse que los potenciales químicos de A en ambas
fases sean iguales, es decir: , o lo
que es lo mismo: . En esta expresión, el término de la izquierda corresponde a la diferencia entre la energía libre de Gibbs molar del sólido y del líquido puros en el punto de fusión (Tf), es decir, es la energía libre de fusión ΔGfusión, y por tanto:
. Al reagrupar términos se
tiene que: ; o en su forma diferencial
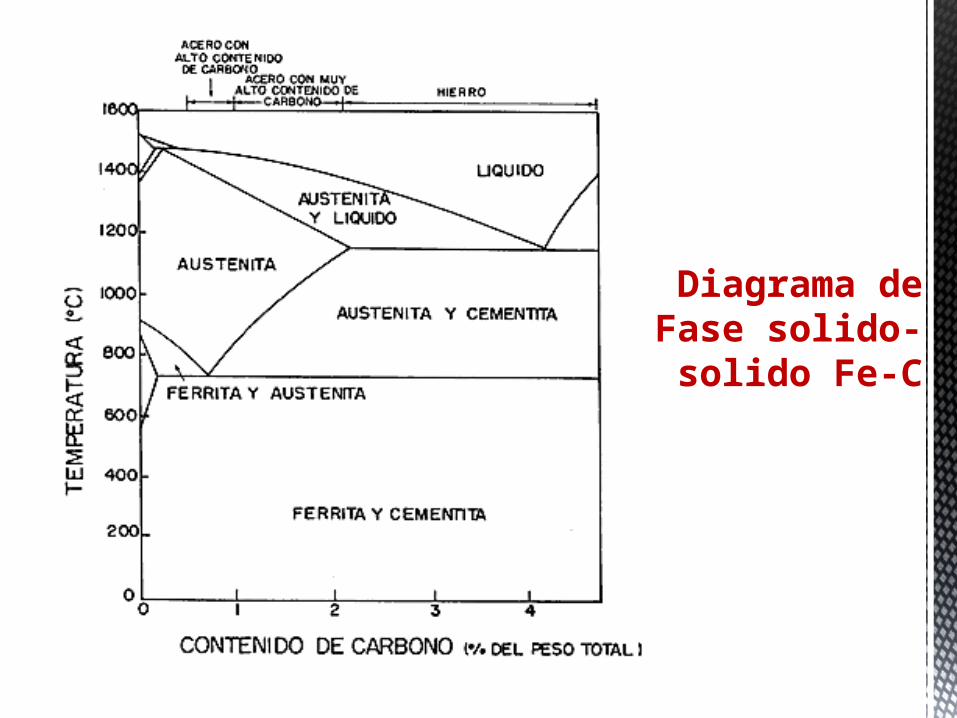
Diagrama de Fase solido-solido Fe-C
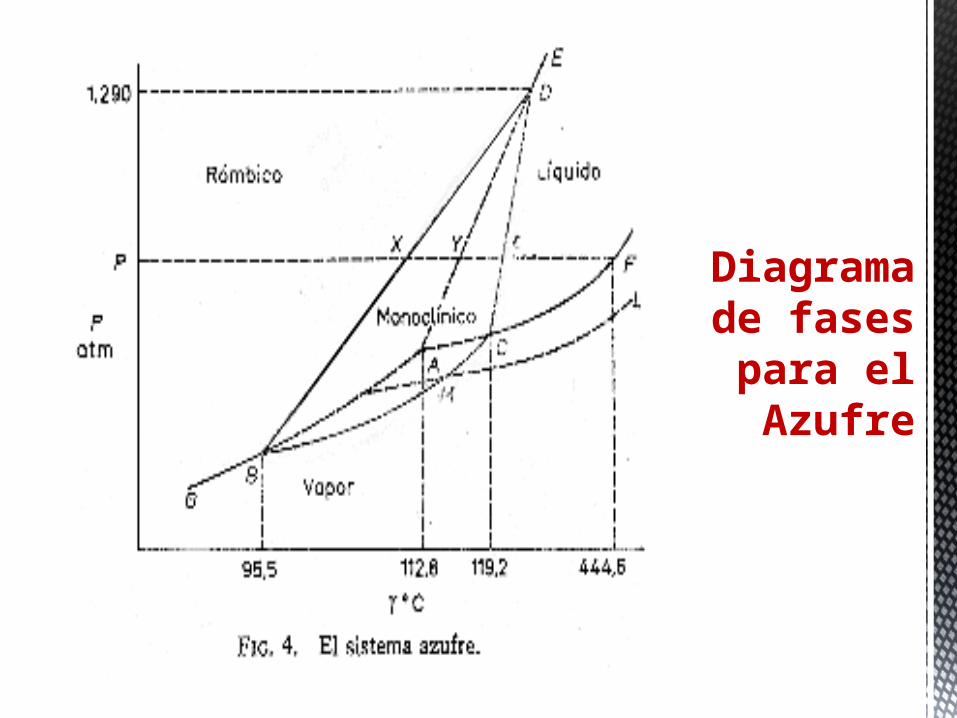
Diagrama de fases
para el Azufre
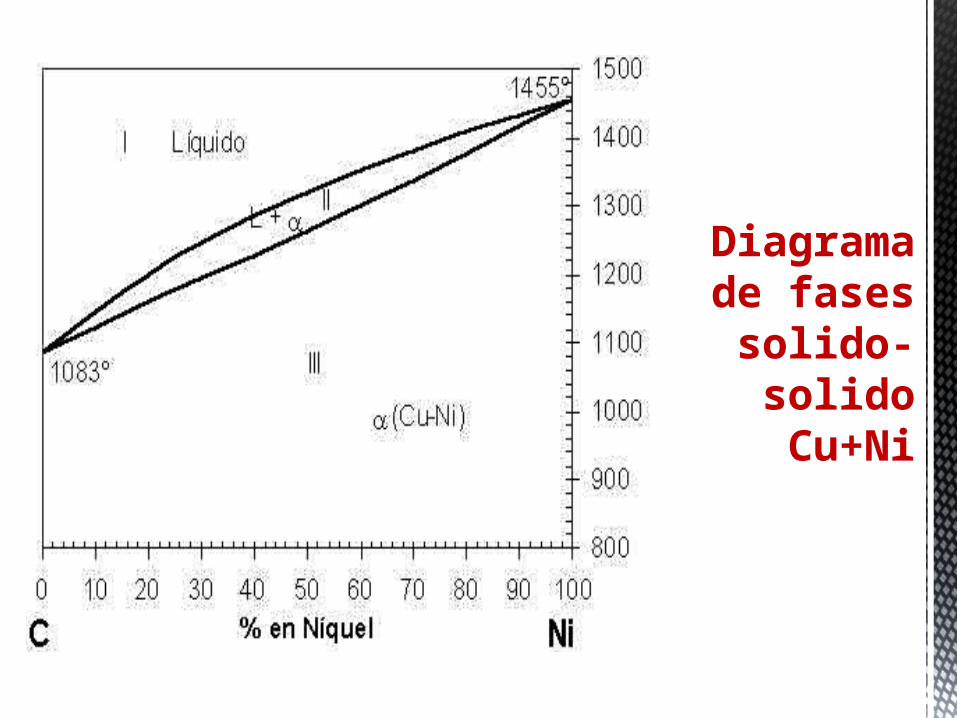
Diagrama de fases
solido-solido Cu+Ni
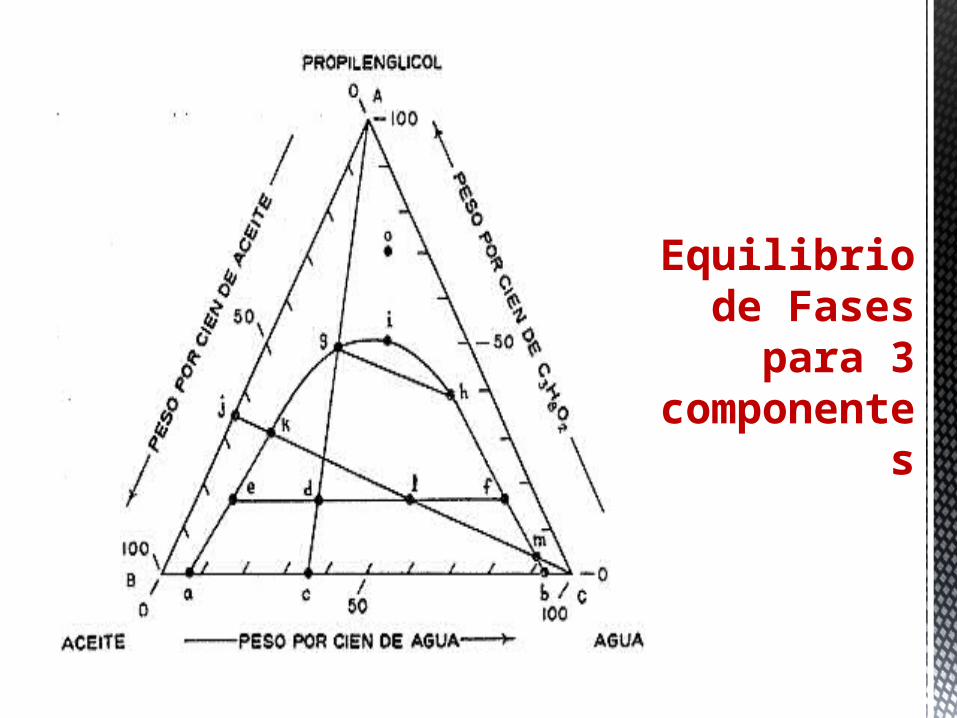
Equilibrio de Fases
para 3 component
es
