enfermedades-mutacionales
TRANSCRIPT

INTEGRANTES :
Contreras Villegas RosaCotrina Cerquera María Elena
Cotrina Morán CarlosCuro Bancayán Diego
Dávila Burga Jhonatan
Docente:
Monge Moyano Aníbal
Curso:
Biología Celular
Tema:
“Enfermedades mutacionales”
Ciclo:
2015-I
UNIVERSIDAD NACIONAL PEDRO RUIZ
GALLO

Seminario Biología Celular
INTRODUCCIÓN
El botánico Hugo de Vries, en sus estudios sobre herencia mendeliana en la
planta, encontró que la herencia en la mayoría de las especies generalmente
era ordenada y predecible, como ocurría en el poroto. Sin embargo, pudo
observar que cada tanto aparecía alguna variante que no estaba presente ni en
los progenitores ni en ningún antecesor de estas mismas plantas.
A partir de lo ocurrido De Vries planteo una hipótesis, donde determinó que
estas variantes surgían como resultado de cambios súbitos en los genes y que
la variante producida por un gen cambiado se transmitía luego a la progenie,
como lo hace cualquier otra característica hereditaria. De Vries denominó
mutaciones a estos cambios hereditarios repentinos, y a los organismos que
exhibían estos cambios, mutantes. Los conceptos propuestos por de Vries no
resultaron tan erróneos, el concepto de mutación como fuente de la variación
genética demostró ser de suma importancia, aunque la mayoría de sus
ejemplos no eran muy válidos.
Hoy se sabe que las mutaciones son cambios abruptos en el material genético.
Como resultado de las mutaciones, existe una amplia gama de variabilidad en
las poblaciones naturales. En un ambiente heterogéneo o cambiante, una
variación determinada puede darle a un individuo o a su progenie una ligera
ventaja, como así también un efecto negativo provocando la extinción de una
especie. En consecuencia, aunque las mutaciones no determinan la dirección
del cambio evolutivo, constituyen la fuente primaria y constante de las
variaciones hereditarias que hacen posible la evolución.
2

Seminario Biología Celular
MUTACIONES
La mutación en genética y biología, es una alteración o cambio en la
información genética (genotipo) de un ser vivo y que, por lo tanto, va a producir
un cambio de características, que se presenta súbita y espontáneamente, y
que se puede transmitir o heredar a la descendencia. La unidad genética capaz
de mutar es el gen que es la unidad de información hereditaria que forma parte
del ADN. En los seres multicelulares, las mutaciones sólo pueden ser
heredadas cuando afectan a las células reproductivas. Una consecuencia de
las mutaciones puede ser una enfermedad genética, sin embargo, aunque en el
corto plazo puede parecer perjudicial, a largo plazo las mutaciones son
esenciales para nuestra existencia. Sin mutación no habría cambio y sin
cambio la vida no podría evolucionar.
Las mutaciones fueron descritas por primera vez en 1901 por uno de los
redescubridores de Méndez, el botánico holandés Hugo De Vries.
Si esto se produce en la secuencia de nucleótidos que codifica un polipéptido
particular, éste puede presentar un aminoácido cambiado en la cadena
polipeptídica. Esta modificación puede alterar seriamente las propiedades de la
proteína resultante. Cuando se produce una mutación durante la formación de
los gametos, ésta se transmitirá a las siguientes generaciones. La mayoría de
las mutaciones genéticas son perjudiciales para el organismo que las porta.
Una modificación aleatoria es más fácil que deteriore y que no mejore la
función de un sistema complejo como el de una proteína. Por esta razón, en
cualquier momento, el número de sujetos que portan un gen mutante
determinado se debe a dos fuerzas opuestas: la tendencia a aumentar debido a
la propagación de individuos mutantes nuevos en una población, y la tendencia
a disminuir debido a que los individuos mutantes no sobreviven o se
reproducen menos que sus semejantes.
3

Seminario Biología Celular
AGENTES MUTÁGENOS
Son agentes mutágenos aquellos factores que aumentan la frecuencia normal
de mutación.
A. AGENTES FISICOS
1. Fluctuaciones térmicas:
Despurinización : pérdida de bases púricas
Desaminación: modificaciones en las bases
2. Radiaciones no ionizantes ultravioleta: Formación de dímeros de T y
dímeros de C desorganizándose la doble hélice de ADN
3. Radiaciones ionizantes: Radiaciones electromagnéticas de longitud de
onda muy corta (muy energéticas y alto poder de penetración. Rayos gamma,
rayos X y flujos de electrones y protones
4. Radiaciones ionizantes corpusculares: partículas alfa y beta. Sus efectos
son de rotura de cadenas de ADN
B. AGENTES QUÍMICOS
1. Metabolitos reactivos: radicales libres derivados del oxígeno.
2. Sustancias químicas: Ac. Nitroso, gas mostaza, Nitrosamina del humo del
tabaco o de los alimentos ahumados; nitritos benzopirenos del humo del tabaco
y de alimentos quemados.
C. AGENTES BIOLÓGICOS
1. Virus: Retrovirus, adenovirus, hepatitis B
2. Transposones: Segmentos móviles de ADN
4

Seminario Biología Celular
CLASIFICACIÓN LAS MUTACIONES
A. SEGÚN LAS CELULAS AFECTADAS
Puede ocurrir en células somáticas: modificaciones no heredables.
Afectan al soma del individuo desapareciendo con él
Pueden ocurrir en células germinales: Afecta a los gametos. Se
transmite a la descendencia si el gameto es fecundado.
B. SEGÚN LA CAUSA
Espontánea
Provocada por distintos agentes mutágenos.
C. SEGÚN LOS EFECTOS
Neutras
Beneficiosas
Perjudiciales letales (muere el 90%)
Perjudiciales subletales (muere menos del 10%)
Perjudiciales patológicas (producen alguna enfermedad)
D. SEGÚN EL TIPO DE EXPRESION GENETICA
Dominantes (respecto del alelo normal no mutado)
Recesivas (respecto del alelo normal no mutado)
E. SEGÚN LA ALTERACION PROVOCADA
1. MUTACIÓN GÉNICA.- Puntual o molecular. No detectable. Cambios
químicos en el ADN que afectan a la secuencia de nucleótidos.
5

Seminario Biología Celular
Por sustitución de bases (solo cambia un triplete): transiciones y
transversiones, desaminaciones, despurizaciones, formación de dímeros
de timina
Perdida o inserción de bases (cambia toda la secuencia de bases)
Translocaciones
2. MUTACIÓN ESTRUCTURAL: Cromosómicas. Detectable citológicamente
en la meiosis. Alteración de la secuencia normal de los fragmentos
transversales de un cromosoma.
Por numero incorrecto de genes : Deficiencia o delección y Duplicación
Por alteraciones en el orden de los genes: Translocación, Inversión
3. MUTACIONES NUMÉRICAS: Genómicas. Detectable citológicamente en
los cariotipos. Alteración del número de cromosomas.
Euploidias: Alteración en el nº de juegos cromosómicos: Monoploidia y
poliploidia
Aneuploidia: Falta o sobra algún cromosoma: Nulisomía, monosomía,
trisomía, tetrasomía
6
MUTACIONES
GÉNICAS O PUNTUALES
GENÓMICAS O CARIOTÍPICAS
CROMOSÓMICASESTRUCTURALES
Aneuploidias Poliploidias

Seminario Biología Celular
I. MUTACIONES GÉNICAS
7

Seminario Biología Celular
I. MUTACIONES GÉNICAS
Son las mutaciones que alteran la secuencia de nucleótidos del ADN.
1.1 Mutaciones por sustitución de bases
Se producen al cambiar en una posición un par de bases, es decir hay una
variación en alguna de las bases nitrogenadas y por tanto en el nucleótido
correspondiente
Existen dos tipos de estas mutaciones por sustitución
a. Mutaciones transicionales o transiciones: Cuando un par de bases
es sustituido por otra del mismo tipo. Las dos bases púricas son
adenina (A) y guanina (G), y las dos pirimídicas son citosina (C) y
timina (T). La sustitución de un par AT, por ejemplo, por un par GC,
sería una mutación de este tipo.
b. Mutaciones transversionales o transversiones: Cuando un par de
bases es sustituida por otra del otro tipo. Por ejemplo, la sustitución del
par AT por TA o por CG.
1. 2 Mutaciones de corrimiento estructural
Cuando se añaden o se quitan pares de nucleótidos alterándose la longitud de
la cadena. Hay dos casos:
a. Mutación por pérdida o deleción de nucleótidos: En la secuencia de
nucleótidos se pierde uno y la cadena se acorta en una unidad.
b. Mutación por inserción de nuevos nucleótidos: Dentro de la
secuencia del ADN se introducen nucleótidos adicionales, interpuestos
entre los que ya había, alargándose correspondientemente la cadena.
8

Seminario Biología Celular
9

Seminario Biología Celular
1. TALASEMIA
1.1 Definición
Llamada también anemia mediterránea o anemia de Cooley y es un transtorno
hereditario caracterizados por la producción anormal de hemoglobina, que
ocasiona disminución en su producción y destrucción excesiva de los glóbulos
rojos.
1.2 Etiología
La hemoglobina contiene dos cadenas, la globina alfa y beta. Las anomalías
genéticas, que causan un desequilibrio en la producción de cualquiera de las
cadenas, pueden ser hereditarias.
Las talasemias beta son causadas por una mutación en la cadena de la globina
beta. Para adquirir la forma mayor de esta enfermedad, los genes mutados se
deben heredar de ambos padres. Si se hereda un solo gen mutado, la persona
será portadora de la enfermedad, pero no experimenta los síntomas, lo cual
corresponde a la forma menor de la enfermedad.
En la forma mayor, los niños son normales al nacer, pero desarrollan anemia
durante el primer año de vida. Algunos problemas que se pueden presentar
son: insuficiencia en el crecimiento, deformidades de los huesos, y
agrandamiento del hígado y del bazo. Las transfusiones de sangre podrían
modificar algunos de los signos de la enfermedad, pero la sobrecarga de hierro
por las transfusiones puede causar daño a los sistemas cardíaco, hepático y
endocrino.
10

Seminario Biología Celular
1.3 Síntomas
Fatiga
Dificultad respiratoria
Ictericia
Deformidades en los huesos de la cara.
1.4 Tipos
La talasemia incluye diferentes tipos de anemia (insuficiencia de glóbulos
rojos). Las dos clasificaciones principales de esta enfermedad son la talasemia
“alfa” y la talasemia “beta”, según cuál sea la parte de la proteína encargada de
llevar el oxígeno (llamada hemoglobina) que falta en los glóbulos rojos.
Los tipos más graves de talasemia alfa, que afecta principalmente a individuos
de ascendencia china, filipina y del sudeste asiático, resultan en la muerte del
feto o del neonato. La mayoría de los individuos con talasemia alfa presentan
alguno de los tipos más leves de la enfermedad y diversos grados de anemia.
La talasemia mayor, el tipo más grave, también se denomina “anemia de
Cooley” en conmemoración del primer doctor que la describió en el año
1925.
La talasemia intermedia es un tipo más leve de anemia de Cooley.
La talasemia menor (también llamada “rasgo de talasemia”) puede no
causar síntomas a pesar de que ocurren cambios en la sangre.
A. BETA-TALASEMIA HOMOCIGÓTICA
11

Seminario Biología Celular
(ANEMIA DE COOLEY O TALASEMIA MAYOR)
Severa anemia microcítica, hipocrómica y hemolítica (se destruyen los
hematíes), que es rápidamente progresiva durante el segundo semestre de
la vida. Falla la síntesis de la cadena beta y de forma característica tienen
aumentada la HbF.
Requieren repetidas transfusiones para mantener la Hb por encima de
10g/dl) para evitar la gran debilidad y la descompensación cardíaca.
Frecuentemente requieren extirpación del bazo.
Presentan hipertrofia del tejido hematopoyético (formador de las células
sanguíneas)
La cortical ósea se adelgaza (porque se expande la médula ósea) lo que
conduce a fracturas óseas patológicas
Falta de neumatización de los senos maxilares
Los huesos de la cara y el cráneo se deforman dando lugar a una cara
peculiar (prominencia de los dientes incisivos superiores, separación de las
órbitas)
Aumenta el tamaño del hígado y el bazo.
B. ALFA-TALASEMIA
12

Seminario Biología Celular
La cadena alfa de la Hb viene determinada por 4 genes y por lo tanto
existen 4 formas de alfa-talasemia según falte 1, 2, 3 ó 4 genes.
Si falta un solo gen da lugar a un portador oculto. Éstos tan sólo
presentan microcitosis (volumen corpuscular medio pequeño). Es el
caso del 25% de los norteamericanos de origen africano.
Si faltan 2 genes aparece el rasgo talasémico que presenta anemia
microcítica leve, frecuente en poblaciones mediterráneas y asiáticas.
Cuando faltan 3 genes se produce una talasemia intermedia y si faltan
los 4 genes se produce la forma más grave, nacen muertos o si no
mueren poco después de nacer, precisan de muchas transfusiones.
Clínicamente es similar a la beta-talasemia.
Talasemia menor Talasemia menor
13

Seminario Biología Celular
2. ANEMIA FALCIFORME
2.1 Definición
Es una enfermedad hereditaria de los glóbulos rojos. Las personas con anemia
falciforme tienen hemoglobina anormal. La hemoglobina anormal hace que los
glóbulos rojos adopten la forma de una letra C (como una hoz) y que se
endurezcan.
Normalmente, los glóbulos rojos son redondos y flexibles y se desplazan
fácilmente por los vasos sanguíneos. Los glóbulos rojos endurecidos se
atascan en los vasos sanguíneos pequeños, interrumpiendo la irrigación
sanguínea y causando dolor y, a veces, daños en los órganos. Los glóbulos
rojos falciformes mueren y se descomponen más rápidamente que los glóbulos
normales, lo cual produce anemia.
2.2 Etiología
La anemia de células falciformes es una enfermedad genética autosómica
recesiva resultado de la sustitución de adenina por timina en el gen de la
globina beta, ubicado en el cromosoma 11, lo que conduce a una mutación de
ácido glutámico por valina en la posición 6 de la cadena polipeptídica de
globina beta y a la producción de una hemoglobina funcionalmente defectuosa,
la hemoglobina S. Debido al cambio de ese aminoácido, las moléculas de
hemoglobina se agregan formando fibras y dándole al hematíe esa forma de hoz.
La transformación del eritrocito se produce cuando no transporta oxígeno, pues
con oxihemoglobina, el glóbulo tiene la forma clásica bicóncava.
14

Seminario Biología Celular
2.3Tipos
Existen varios tipos comunes de anemia falciforme:
Hemoglobina SS. La persona hereda un gen de glóbulos falciformes de
cada padre. Ésta es la forma más común.
Hemoglobina SC. La persona hereda un gen de glóbulos falciformes y
un gen de otro tipo anormal de hemoglobina llamado "C".
Hemoglobina S con beta-talasemia. La persona hereda un gen de
glóbulos falciformes y un gen de beta-talasemia, otro tipo de anemia
hereditaria.
2.4 Síntomas
Aumento del número de pulsaciones y de la longitud de las fibras cardíacas, lo que conlleva riesgo de insuficiencia cardíaca.
Mareos frecuentes.
Disminución del número de hematíes, debido a su extrema fragilidad.
Anoxia de tejidos, provocada por el esfuerzo.
Obstrucción y desgarro de venas.
Disminución de hemoglobina.
En niños es más común la Dactilitis
Crisis vaso-oclusivas.
Problemas de visión.
15

Seminario Biología Celular
3. RETINOBLASTOMA
3.1 Definición
Es un tumor maligno de la retina que generalmente afecta a niños menores de
6 años. Es llamada también cáncer o tumor de retina.
3.2 Causas, Incidencia Y Factores De Riesgo
El retinoblastoma se presenta cuando una célula de la retina en crecimiento
sufre una mutación, haciendo que dicha célula crezca sin control y se vuelva
cancerosa.
Algunas veces, esta mutación se desarrolla en un niño cuya familia nunca ha
tenido cáncer en el ojo, pero otras veces, la mutación está presente en varios
miembros de la familia. Si la mutación es un mal de familia, existe un 50% de
probabilidades de que los hijos de la persona afectada también tengan la
mutación y, por lo tanto, tendrán un alto riesgo de desarrollar retinoblastoma.
Uno o ambos ojos pueden estar afectados y se puede presentar una blancura
visible en la pupila. La ceguera puede ocurrir en el ojo afectado y los ojos
pueden parecer bizcos o con estrabismo convergente. Este tumor se puede
diseminar a la órbita a través del nervio óptico e igualmente al cerebro, los
pulmones y los huesos. Se trata de un tumor poco común, excepto en las
familias portadoras de la mutación del gen RB.
3.3 Síntomas
Manchas blancas en la pupila
Estrabismo convergente
Enrojecimiento y dolor en el ojo
Visión deficiente
Iris que puede ser de diferente color en cada ojo
16

Seminario Biología Celular
4. ACONDROPLASIA
4.1 Definición
La acondroplasia es un trastorno óseo genético (hereditario) que se presenta
en uno de cada 25.000 niños que nacen vivos. La acondroplasia es el tipo más
frecuente de enanismo, en la cual los brazos y las piernas del niño son cortas
en proporción a la longitud corporal. Además, con frecuencia, la cabeza es de
un tamaño mayor y el tronco, de tamaño normal. La estatura promedio de los
adultos hombres con acondroplasia es de 1,32m. La estatura promedio de las
mujeres adultas con acondroplasia es de 1,25 m.
4.2 Etiología
La acondroplasia se hereda mediante un gen autosómico dominante que causa
una formación anormal de los cartílagos. La herencia autosómica dominante
significa que el gen está ubicado en uno de los autosomas (pares de
cromosomas 1 a 22). Esto significa que afecta a hombres y mujeres por igual.
Dominante significa que un solo gen es necesario para tener el rasgo. Cuando
uno de los padres tiene la característica dominante, hay un 50 por ciento de
posibilidades de que cualquiera de sus hijos también herede ese rasgo. Por lo
tanto, en algunos casos, el niño hereda la acondroplasia de un padre con
acondroplasia. La mayoría de los casos de acondroplasia (80 por ciento) son
consecuencia de una nueva mutación en la familia, los padres tienen una
estatura promedio y no poseen el gen anormal.
Los genetistas han descubierto que los padres mayores de 45 años tienen una
mayor posibilidad de tener hijos con ciertas condiciones autosómicas
dominantes tales como la acondroplasia, pero aún no se ha descubierto la
causa que origina las nuevas mutaciones en el esperma.
El gen responsable de la acondroplasia se descubrió en 1994 y, gracias a esto,
se pueden hacer diagnósticos prenatales precisos, en la mayoría de los casos.
17

Seminario Biología Celular
4.3 Síntomas
Brazos y piernas cortos, con la parte superior de los brazos más cortos
que los antebrazos y los muslos más cortos que la parte inferior de las
piernas.
Cabeza de gran tamaño, frente prominente y tabique nasal aplanado.
Dientes mal alineados o montados.
Parte baja de la columna vertebral curvada - condición también
denominada lordosis (o "corcova") que puede ocasionar cifosis o la
formación de una pequeña corcova cerca de los hombros que
generalmente desaparece una vez que el niño comienza a caminar.
Parte inferior de las piernas curvada.
Pie plano, corto y ancho.
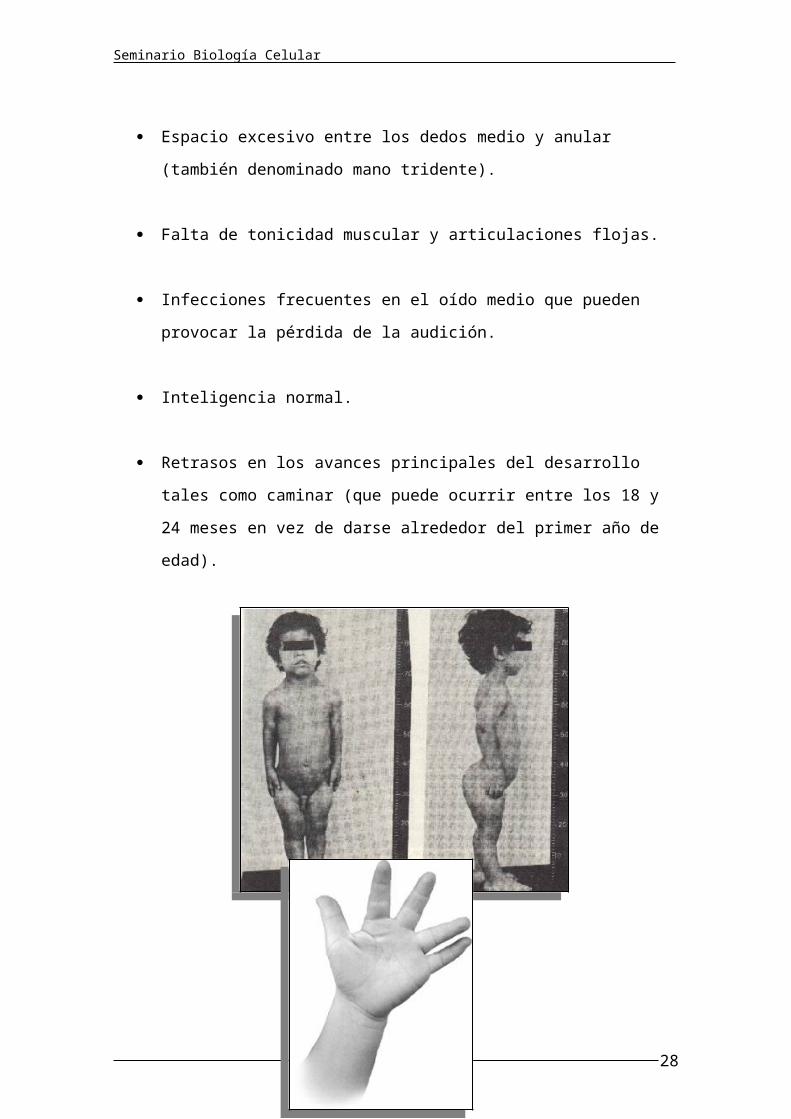
Espacio excesivo entre los dedos medio y anular (también denominado
mano tridente).
18

Seminario Biología Celular
Falta de tonicidad muscular y articulaciones flojas.
Infecciones frecuentes en el oído medio que pueden provocar la pérdida
de la audición.
Inteligencia normal.
Retrasos en los avances principales del desarrollo tales como caminar
(que puede ocurrir entre los 18 y 24 meses en vez de darse alrededor
del primer año de edad).
19

Seminario Biología Celular
5. ALBINISMO
5.1 Definición
El albinismo es un conjunto de condiciones congénitas (heredadas) que afectan
a los humanos (y al resto de animales) y que globalmente se caracterizan por
la ausencia o disminución de pigmento (melanina) en la piel, los ojos o el pelo.
Una de cada 17,000 personas en los Estados Unidos tiene algún tipo de
albinismo. El albinismo afecta a personas de todas las razas.
5.2 Etiología
Los Genes del Albinismo para casi todos los tipos de albinismo, los dos padres
tienen que tener un gen para albinismo para tener un hijo con albinismo. Como
el cuerpo tiene dos pares enteros de genes, una persona puede que se vea
normal, pero puede contener los genes para el albinismo. Si una persona tiene
un par de genes normales y un par de genes con albinismo, el ó ella tienen la
información genética suficiente para hacer pigmento normal. El gen del
albinismo es "recesivo" y no resultará en una persona con albinismo a menos
que los dos pares de genes contengan albinismo y no hay copia del gen que
tiene pigmento normal.
Cuando los dos padres tienen el gen pero ninguno de los dos tienen albinismo,
existe una probabilidad de 25% en cada embarazo de que el bebé nazca con
albinismo. Este tipo de herencia se llama " herencia recesiva autosomal".
5.2 Tipos
a) ALBINISMO OCULOCUTÁNEO (OCA):
Se presenta principalmente con la disminución o ausencia de pigmento que
afecte a la piel, el pelo y a los ojos.
20

Seminario Biología Celular
Albinismo Oculocutáneo de tipo 1 (OCA1): Eestá producido por
mutaciones o alteraciones en el gen de la tirosinasa (situado en el
cromosoma 11 humanos) que lleva la información genética de una de
las principales enzimas responsable del primer paso de la ruta de
síntesis de la melanina.
Albinismo Oculocutáneo de tipo 2 (OCA2): Es el tipo más frecuente
de albinismo, principalmente en personas de raza negra. y está
producido por mutaciones o alteraciones en el gen P (situado en el
cromosoma 15 humano) que ver con la formación de melanosomas, los
orgánulos subcelulares que sintetizan y acumulan la melanina.
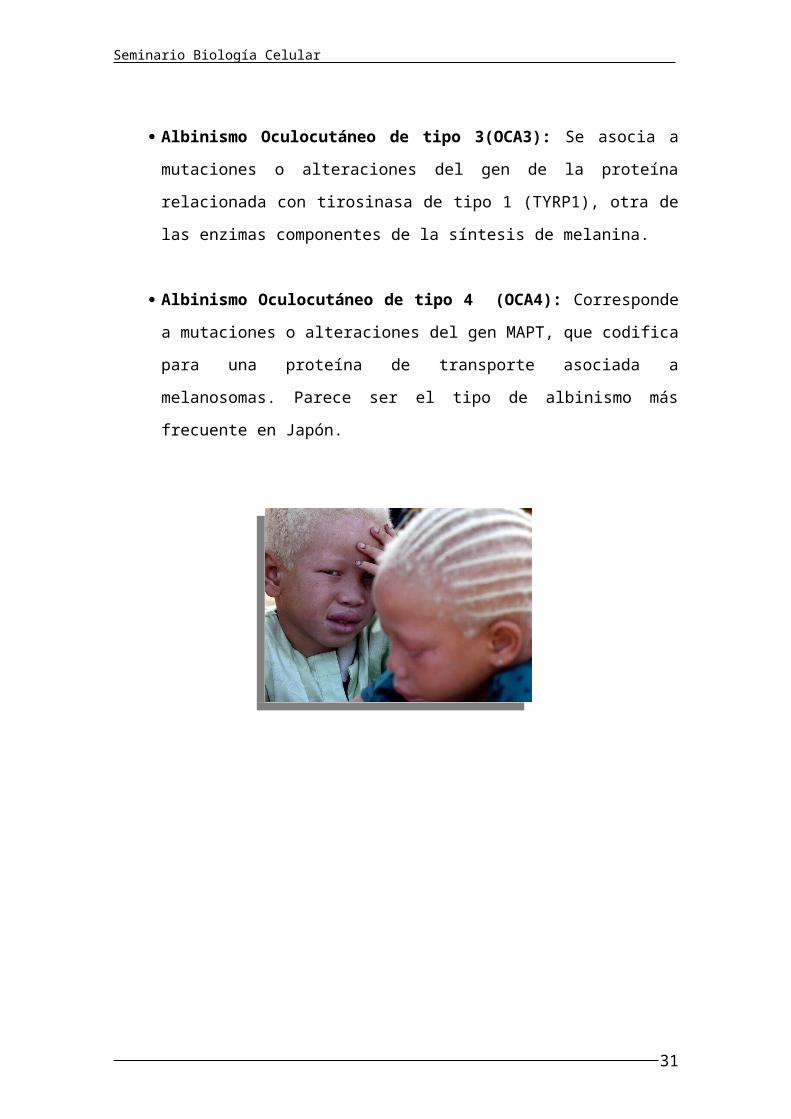
Albinismo Oculocutáneo de tipo 3(OCA3): Se asocia a mutaciones o
alteraciones del gen de la proteína relacionada con tirosinasa de tipo 1
(TYRP1), otra de las enzimas componentes de la síntesis de melanina.
Albinismo Oculocutáneo de tipo 4 (OCA4): Corresponde a
mutaciones o alteraciones del gen MAPT, que codifica para una proteína
de transporte asociada a melanosomas. Parece ser el tipo de albinismo
más frecuente en Japón.
21

Seminario Biología Celular
Adicionalmente existen otros tipos de albinismo oculocutáneos, mucho menos
frecuentes, en los que la disminución o ausencia de pigmento en piel, pelo y
ojos se manifiesta de forma combinada con otros síntomas, dentro de
síndromes más complejos. Es el caso del síndrome de Hermansky-Pudlak
(HPS),que se manifiesta adicionalmente con hemorragias, problemas
respiratorios y digestivos y que puede causar la muerte temprana de las
personas afectadas; o el síndrome de Chediak-Higashi (CHS) que con
síntomas parcialmente similares a HPS presenta adicionalmente problemas del
sistema inmunológico y susceptibilidad aumentada frente a infecciones y
también puede ser mortal.
b) ALBINISMO OCULAR:
Disminución o ausencia de pigmento que principalmente afecte a los ojos.
Albinismo Ocular de tipo 1 (OA1): se caracteriza por la falta o
disminución de pigmentación en los ojos, aunque también puede
manifestarse con un déficit pigmentario ligero en la piel y el pelo. Se
asocia a mutaciones o alteraciones al gen OA1 (situado en el
cromosoma X) una proteína asociada a la membrana de los
melanosoma cuya función anómala se traduce en el acúmulo de
melanosomas anormalmente grandes dentro de la célula.
Albinismo Ocular de tipo 2 (OA2): no asociado a mutaciones o
alteraciones de genes en el cromosoma X, cuyo gen implicado no se
conoce con exactitud.
22

Seminario Biología Celular
5.3 Signos y Síntomas
Nistagmus, movimiento irregular del ojo.
Estrabismo, descontrol de los músculos de los ojos ("ojos cruzados" o
sin coordinación).
Sensibilidad a luces brillantes ó claras. Las personas con albinismo
pueden tener miopía (mala vista de lejos ó de cerca) y frecuentemente
tienen astigmatismo (imágenes distorsionadas).
Estos problemas resultan de un desarrollo anormal del ojo porque no hay
suficiente pigmento. La retina, la superficie dentro del ojo que recibe luz, no
se desarrolla normalmente antes de nacer y en la infancia. Las señales del
nervio de la retina al cerebro no siguen los caminos usuales. El iris, la parte
de color en el centro del ojo, no tiene pigmento suficiente para protegerse
de los rayos de luz que entran al ojo.
23

Seminario Biología Celular
6. GALACTOSEMIA
6.1 Definición
Es la incapacidad del organismo para utilizar (metabolizar) el azúcar simple
galactosa, ocasionando la acumulación de galactosa 1-fosfato en el cuerpo, lo
cual causa daño al hígado, al sistema nervioso central y a otros sistemas del
organismo.
6.2 Etiología
La galactosemia es una enfermedad enzimática hereditaria, transmitida como
un rasgo autosómico recesivo y cuya ocurrencia es aproximadamente de 1 por
cada 60.000 nacimientos entre personas de raza blanca, mientras que la tasa
es diferente para otros grupos.
Existen 3 formas de la enfermedad: deficiencia de galactosa-1-fosfatouridil
transferasa (galactosemia clásica, la forma más común y la más grave),
deficiencia de galactosa cinasa y deficiencia de galactosa-6-fosfato epimerasa.
6.3 Características
Las personas con galactosemia son incapaces de descomponer
completamente el azúcar simple galactosa, que compone la mitad de la
lactosa, el azúcar que se encuentra en la leche. La lactosa es un disacárido (di
significa 2 y sacárido significa azúcar) debido a que está compuesto de dos
azúcares, galactosa y glucosa, enlazados.
Si a un bebé con galactosemia se le da leche, los derivados de la galactosa se
acumulan en el sistema del bebé, causando daño al hígado, al cerebro, a los
riñones y a los ojos. Los individuos con galactosemia no pueden tolerar
ninguna forma de leche (ni humana ni animal) y deben vigilar cuidadosamente
la ingesta de otros alimentos que contengan galactosa. La exposición a los
productos lácteos puede ocasionar daño hepático, retardo mental, formación de
cataratas e insuficiencia renal.
24

Seminario Biología Celular
Después de tomar leche durante algunos días, un neonato con galactosemia se
rehusará a comer y desarrollará ictericia, vómitos, letargo, irritabilidad y
convulsiones. Asimismo, se presentará agrandamiento del hígado y el azúcar
puede estar bajo. La alimentación continua con productos lácteos lleva a que
se presente cirrosis hepática, formación de cataratas en el ojo (que puede
ocasionar ceguera parcial) y retardo mental.
6.4 Síntomas
Ictericia (coloración amarillenta de la piel y de la esclerótica).
Vómitos
Alimentación deficiente (el bebé se niega a beber fórmula que contenga
leche)
Poco aumento de peso
Letargo
Irritabilidad
Convulsiones
25

Seminario Biología Celular
7. FIBROSIS QUÍSTICA
7.1 Definición
Es una enfermedad hereditaria que provoca la acumulación de moco espeso y
pegajoso en los pulmones y el tubo digestivo. Es el tipo de enfermedad
pulmonar crónica más común en niños y adultos jóvenes, y puede ocasionar la
muerte prematura.
7.2 Etiología
La fibrosis quística (FQ) es causada por un gen defectuoso que le indica al
cuerpo que produzca un fluido anormalmente espeso y pegajoso llamado
moco. Este moco se acumula en las vías respiratorias de los pulmones y en el
páncreas, el órgano que ayuda a descomponer y absorber los alimentos.
Esta acumulación de moco pegajoso ocasiona infecciones pulmonares
potencialmente mortales y serios problemas digestivos. Esta enfermedad
también puede afectar las glándulas sudoríparas y el aparato reproductor
masculino.
Millones de estadounidenses llevan el gen defectuoso de la fibrosis quística,
pero no manifiestan ningún síntoma. Esto se debe a que una persona que
padece esta enfermedad debe heredar dos genes defectuosos para fibrosis
quística: uno de cada padre. Se estima que 1 de cada 29 estadounidenses de
raza blanca tiene el gen de la fibrosis quística. La enfermedad es el trastorno
hereditario y mortal más común que afecta a las personas de raza blanca en
los Estados Unidos y es más común entre aquellas personas descendientes de
europeos del centro y norte.
A la mayoría de los niños se les diagnostica fibrosis quística a los dos años. Sin
embargo, a un pequeño número no se le diagnostica la enfermedad hasta los
18 años o más. Estos pacientes generalmente padecen una forma más leve de
la enfermedad.
26

Seminario Biología Celular
7.3 Síntomas
Dado que existen más de 1.000 mutaciones del gen de la fibrosis
quística, los síntomas son distintos de persona a persona. Pero, en
general, incluyen:
Ausencia de deposiciones durante las primeras 24 a 48 horas de vida
Heces pálidas o color arcilla y con olor fétido o heces flotantes
Es posible que los bebés tengan la piel salada
Infecciones respiratorias recurrentes, como neumonía o sinusitis y tos
Pérdida de peso o ausencia de aumento de peso normal en la niñez
Diarrea
Retraso en el crecimiento
Fatiga
27
Seminario Biología Celular
8. OSTEOGENESIS IMPERFECTA
8.1 Definición
Es una afección que ocasiona huesos extremadamente frágiles por ello se le
llama también Enfermedad de los huesos frágiles
8.2 Etiología
La osteogénesis u osteogenia imperfecta (OI) es una enfermedad congénita, lo
que quiere decir que está presente al nacer. Con frecuencia es causada por un
defecto en un gen que produce el colágeno tipo 1, un pilar fundamental del
hueso. Existen muchos defectos diferentes que pueden afectar este gen y la
gravedad de esta enfermedad depende del defecto específico de dicho gen. La
osteogénesis imperfecta es una enfermedad autonómica dominante, lo que
quiere decir que la persona la padecerá si tiene una copia del gen. La mayoría
de los casos de OI se heredan de uno de los padres, aunque algunos casos
son el resultado de nuevas mutaciones genéticas. Una persona con
osteogénesis imperfecta tiene un 50% de posibilidades de transmitirles el gen y
la enfermedad a sus hijos.
28

Seminario Biología Celular
8.3 Síntomas
Todas las personas con osteogénesis imperfecta (OI) tienen huesos débiles, lo
cual las hace susceptibles a sufrir fracturas. Las personas con OI generalmente
tienen una estatura por debajo del promedio (estatura baja). Sin embargo, la
gravedad de la enfermedad varía enormemente. Los síntomas clásicos
abarcan:
Tinte azul en la parte blanca de los ojos (esclerótica azul)
Fracturas óseas múltiples
Pérdida temprana de la audición (sordera)
Los síntomas de las formas más severas de OI pueden abarcar:
Brazos y piernas arqueadas
Cifosis (curvatura de la columna en forma de "S")
Escoliosis
29

Seminario Biología Celular
9. SINDROME DE MARFAN
9.1 Definición
Se trata de un trastorno hereditario del tejido conectivo, que es el encargado de
mantener unidos los tejidos del cuerpo. Se pueden producir un número variable
de alteraciones que pueden afectar al corazón, los vasos sanguíneos, los
pulmones, a los huesos y a los ligamentos.
En general, son personas muy altas, con los miembros desproporcionadamente
largos con respecto al torso.
9.2 Etiología
El Síndrome de Marfan es de origen genético y se debe a un anormal
comportamiento del gen FBN1, que determina la producción de una proteína
denominada fibrilina. Este gen reside en el cromosoma 15; hay gran variedad
de cambios (mutaciones) en este gen que puede causar el síndrome, lo que
explicaría la gran variabilidad de afectación de las personas.
El síndrome de Marfan se hereda como rasgo autosómico dominante y, por
tanto, cada niño con un padre con el gen tienen el 50% de probabilidades de
heredarlo. Sin embargo, hasta el 30% de los casos no tienen historia familiar y
se les denominan casos "esporádicos". La fibrilina es un componente
fundamental del tejido conectivo contribuyendo a su fuerza y elasticidad. La
fibrilina es especialmente abundante en la arteria aorta, en los elementos de
sostén del ojo y en los huesos. Las personas con Síndrome de Marfan tienen
poca cantidad de fibrilina o ésta es de escasa calidad.
30
Seminario Biología Celular
9.3 Síntomas
Hay una importante hiperlaxitud, con escasa masa muscular en la
mayoría de las personas.
Articulaciones muy flexibles.
Los brazos y las piernas suelen ser más largos de lo normal en relación
con el torso.
Con frecuencia pies planos
Curvatura anormal de la columna (escoliosis). En los casos extremos se
presenta en el nacimiento Espina Bífida.
El esternón puede sobresalir (tórax en quilla) o estar hundido (tórax en
embudo).
Dedos largos, como de araña (aracnodactilia).
Cara fina y delgada.
Mandíbula pequeña.
Problemas oftalmológicos
Luxación o subluxación del cristalino
Desprendimiento de retina: amenaza gravemente la vista.
Catarata.
31

Seminario Biología Celular
II. MUTACIONES
CROMOSÓMICAS ESTRUCTURALES
32

Seminario Biología Celular
II. MUTACIONES CROMOSÓMICAS ESTRUCTURALES
Mutaciones que producen cambios estructurales que eliminan, añaden o
reordenan partes sustanciales de uno o más cromosomas.
Los cambios estructurales se deben a una o más roturas distribuidas a lo largo
del cromosoma, seguidas por la pérdida o la reordenación del material
genético. Los cromosomas pueden romperse espontáneamente, pero la tasa
de roturas puede aumentar en celulas expuestas a sustancias químicas o a
radiación. Aunque los extremos normales de los cromosomas, los telómeros,
no se fusionan fácilmente con extremos nuevos de cromosomas rotos o con
otros telómeros, los extremos producidos en los puntos de rotura son
“pegajosos” y pueden reunirse con otros extremos rotos. Si la rotura y reunión
no restablece las relaciones originales y si la alteración se produce en el
plasma germinal, los gametos tendrán una reordenación estructural que será
heredable. Si la aberración se encuentra en un homologo, pero no en el otro,
se dice que los individuos son heterocigotos para la aberración. En tales casos
se producen configuraciones raras en el apareamiento durante la sinapsis
meiótica. Si no hay pérdida o ganancia de material genético, los individuos que
llevan la aberración en heterocigosis en uno de los dos homólogos
probablemente no quedaran afectados en su fenotipo.
Estas mutaciones pueden ser por:
Delección
Duplicación
Inversión
Translocación
33

Seminario Biología Celular
1. DELECCIÓN
Mutaciones en la que una parte del cromosoma se elimina. Una pieza muy
pequeña de un cromosoma puede contener muchos genes diferentes y si la
parte perdida es muy grande, la situación es incompatible con la vida.
1.1 Deleción Terminal: Ocurre cerca de un extremo del cromosoma.
1.2 Delección intersticial: Se produce en la parte interna del cromosoma.
Enfermedades por delección:
Síndrome del maullido de gato
El síndrome de cromosoma 15 en anillo
síndrome de Prader-Willi
Síndrome de George
SÍNDROME DEL MAULLIDO DE GATO
Del francés Cri du Chat o síndrome de Lejeune, es una enfermedad congénita
infrecuente con alteración cromosómica provocada por un tipo de delección
estructural de parte o de todo el brazo corto del cromosoma 5, caracterizada
por un llanto que se asemeja al maullido de un gato y que se va modificando
con el tiempo.
El síndrome del maullido fue descrito inicialmente por Lejeune en 1963. Tiene
una prevalencia estimada de aproximadamente de 1/20.000-50.000
nacimientos y predomina en las niñas.
34

Seminario Biología Celular
Características
Inclinación de los ojos hacia abajo
Bajo peso al nacer y crecimiento lento
Orejas de implantación baja o de forma anormal
Retardo mental
Dedos de las manos y pies parcialmente unidos por membranas.
(Sindactilia parcial).
Una sola línea en la palma de la mano (pliegue simiesco)
Bajo tono muscular.
Pliegues del epicanto (pliegue extra de piel sobre el ángulo interior del ojo).
2. TRANSLOCACIÓN
Es el desplazamiento de un segmento de un cromosoma a un nuevo lugar en el
genoma.
2.1 Translocaciones reciprocas
Estas translocaciones tienen lugar cuando existen roturas en dos cromosomas
y se produce un intercambio mutuo de material.
Se les llama FRATERNAS cuando se produce en cromosomas homólogos
Y EXTERNAS entre cromosomas no homólogos.
35

Seminario Biología Celular
LEUCEMIA PROMIELOCÍTICA AGUDA
Es un tipo de cáncer conocido como leucemia mieloide 3 que se caracteriza por
un predominio de promielocitos malignos que muestran una translocación
recíproca entre los brazos largos de los cromosomas 15 y 17, t(15;17)
(q22;q11.2-q12) que producen la proteína RARα, la que únicamente puede
responder a acido retinoico.
Características y síntomas
Se caracteriza por promielocitos (una forma de glóbulos blancos)
anormales, fuertemente granulados. Esta enfermedad favorece a una
acumulación de estos promielocitos atípicos en la médula ósea y la
sangre periférica, y reemplaza los glóbulos normales.
Una de las principales características es la presencia de un síndrome
hemorrágico grave.
Con menor frecuencia fiebre y leucocitosis.
También son frecuentes los síntomas asociados a anemia y
trombocitopenia.
2.2 Translocaciones Robertsonianas o Fusión Céntrica
Esta mutación implica roturas en el extremo final de los brazos cortos de dos
cromosomas acrocéntricos no homólogos. Los pequeños fragmentos
acéntricos se pierden y los segmentos cromosómicos grandes se fusionan por
sus regiones centroméricas, dando lugar a un nuevo cromosoma grande,
metacéntrico o submetacéntrico
Este tipo de translocación se limita a los cromosomas 13,14,15,21y 22 pues
los brazos cortos de estos cromosomas son muy pequeños y contienen
material genético no esencial .Cuando se produce una translocacion
robertsoniana los brazos cortos suelen perderse en divisiones celulares
posteriores .Dado que los portadores de las translocaciones robertsonianas
pierden material genético que no es esencial son fenotipicamente normales ,
36

Seminario Biología Celular
pero solo tienen 45 cromosomas en cada célula. Su descendencia, sin
embargo puede heredar un brazo largo de un cromosoma acrocentrico de más
o menos. La fusión de los centrómeros representa la anomalía cromosómica
más común; estas translocaciones han jugado un importante papel en la
especiación. Las translocaciones Robertsonianas que afectan a los
cromosomas 13 y/o 21 producen embriones viables con trisomías 13 o 21.
SÍNDROME DE DOWN POR TRANSLOCACIÓN
La característica principal de las translocaciones robertsonianas es su
predisposición a originar síndromes de Down como resultado de heredar dos
cromosomas 21 normales (uno de cada parental) y un cromosoma portador de
una translocación 21. Las consecuencias clínicas son exactamente las mismas
que se observan en casos de trisomía 21. Sin embargo, a diferencia de la
trisomía 21, los padres de un niño con síndrome de Down por translocación
tienen un riesgo relativamente mayor de tener otro hijo afectado si uno de ellos
es portador de una translocación equilibrada. El cromosoma 21 también está
presente en el triplicado. La diferencia es que el cromosoma adicional se une
con otro cromosoma, generalmente con el cromosoma número 14 o con el otro
cromosoma número 21. Aproximadamente la cuarta parte de las
translocaciones ocurre de manera espontánea durante la fertilización. Las
translocaciones restantes son heredadas de uno de los progenitores. Esta es la
única forma del síndrome de Down que es consecuencia de alguna condición
genética de los padres, y se presenta en un 5 a 7 % de los casos de síndrome
de Down
37

Seminario Biología Celular
3. DUPLICACIÓN
Las duplicaciones surgen cuando un segmento cromosómico se replica más de
una vez por error en la duplicación del DNA
3.1 Duplicación en Tándem: La región duplicada se encuentra
inmediatamente adyacente al segmento original y
Tándem directo: Si la región duplicada se encuentra en el mismo orden
que la región original.
Tándem inverso: Si encuentra en orden inverso a la región original
3.2 Duplicación Desplazada: Si el segmento duplicado se localiza a cierta
distancia del segmento original, sea en el mismo cromosoma o en un
cromosoma diferente.
Desplazada directa: Si la región duplicada se encuentra en el mismo
orden que la original
Duplicaron Inversa: Se da cuando el segmento duplicado gira en 180
grados.
38

Seminario Biología Celular
4. INVERSIÓN
Es un cambio estructural por el cual un segmento cromosómico cambia de
sentido dentro del propio cromosoma.
4.1 Inversiones simples: Cuando en un cromosoma sólo tenemos un
fragmento invertido.
Pericéntricas: Si el segmento afectado incluye al centrómero. Se
detectan fácilmente al microscopio óptico pues implican un cambio en la
forma del cromosoma.
Paracéntricas: No incluyen al centrómero
4.2 Inversiones Complejas: Cuando intervienen simultáneamente diversos
segmentos de un mismo cromosoma.
Solapantes: Que se producen cuando una parte de un segmento
incluido en una inversión, se ve afectado por otra inversión.
Independientes: Cuando entre cada segmento invertido, tenemos una
zona que no ha experimentado inversión.
En tándem: Cuando los dos segmentos invertidos, se presentan
adyacentes.
Incluidas: Cuando dentro de un fragmento invertido, se produce la
inversión de un fragmento menor.
39

Seminario Biología Celular
40

Seminario Biología Celular
III. MUTACIONES GENÓMICAS
41

Seminario Biología Celular
II. MUTACIONES GENÓMICAS
Cuando producen variaciones numéricas en la dotación cromosómica
(CARIOTIPO), es decir si el número de cromosomas presentes en un
organismo es diferente al típico de su especie.
Existen dos posibilidades
2.1 Poliploidias
Son dotaciones superiores a lo normal en un número múltiplo del característico
de la especie
2.2 Aneuploidías
Indican la presencia de uno o más cromosomas por exceso o por defecto del
característico de la especie, afectando tanto a cromosomas somáticos como
sexuales.
42

Seminario Biología Celular
A. ANEUPLOIDIAS EN CROMOSOMAS SOMÁTICOS
Son alteraciones en el número de copias de alguno de los cromosomas no
sexuales. En humanos, no todas las aneuploidias numéricas son viables, pero
existen y generan alteraciones en el fenotipo de los humanos. Entre las más
frecuentes destacan:
Síndrome de Down: Trisomía del cromosoma 21
Síndrome de Edwards: Trisomía del cromosoma 18
Síndrome de Patau: Trisomía del cromosoma 13
43

Seminario Biología Celular
1. SÍNDROME DE DOWN
1.1 Definición
El Síndrome de Down es un grave trastorno genético que ocasiona
retraso mental al igual que ciertas deformidades físicas. En este
síndrome, la cara tiene algunos rasgos semejantes a los grupos
mongoles, de ahí que en el pasado se le llamara, incorrectamente
mongolismo.
1.2 Formas De Trisomía 21
A. TRISOMÍA LIBRE O SIMPLE
Cuando se forman los óvulos y los espermatozoides, lo hacen a partir de
células originarias en las que, al dividirse, sus 46 cromosomas se separan: 23
van a una célula y sus correspondientes parejas se van a otra; por eso cada
una tiene 23 cromosomas. Pero a veces ocurre que esta división y separación
de las parejas de cromosomas no se realizan correctamente; es decir, una de
las parejas de cromosomas (en nuestro caso la pareja 21) no se separa sino
que los dos cromosomas 21 permanecen unidos y se quedan en una de las
células (óvulo o espermatozoide) divididas. Ha ocurrido lo que los técnicos
llaman "no-disyunción" o "no-separación". Con lo cual esa célula tiene ya 24
cromosomas, dos de ellos de la pareja 21; al unirse con la otra célula germinal
normal que aporta sus 23 cromosomas, la nueva célula resultante de la fusión
en el momento de la concepción tendrá 47 cromosomas, tres de los cuales
serán 21, y a partir de ella se originarán todas las demás células del nuevo
organismo que poseerán también los 47 cromosomas.
Esta circunstancia es la más frecuente en el síndrome de Down. El 95 % de las
personas con síndrome de Down poseen esta trisomía simple: 47 cromosomas
de los que tres completos corresponden al par 21.
44

Seminario Biología Celular
Cromosomas humanos normal (varón) Cromosomas en el síndromeDown. (Varón
B. MOSAICISMO
Aparece en el 1,5 % de los niños con síndrome de Down. Corresponde a la
situación en que óvulo y espermatozoide poseen los 23 cromosomas normales,
y por tanto la primera célula que se forma de la fusión de ambos es normal y
posee sus 46 cromosomas. Pero a lo largo de las primeras divisiones de esa
célula y de sus hijas surge en alguna de ellas el mismo fenómeno de la no-
disyunción o no-separación de la pareja de cromosomas 21 que antes
comentábamos, de modo que una célula tendrá 47 cromosomas, tres de los
cuales serán del par 21. A partir de ahí, todos los millones de células quede
deriven de esa célula anómala tendrán 47 cromosomas (serán trisómicas),
mientras que los demás millones de células que se deriven de las células
normales tendrán 46, serán también normales.
Dependiendo de cuándo haya aparecido la no-disyunción en el curso de
divisiones sucesivas, así será el porcentaje final de células trisómicas y
normales que el individuo posea. Cuanto más inicialmente aparezca la
anomalía, mayor será el porcentaje de trisómicas y viceversa. Como se
entiende fácilmente, si las trisómicas están en escasa proporción, la afectación
patológica resultante será menos intensa.
C. POR TRANSLOCACIÓN ROBERTSONIANAS
45

Seminario Biología Celular
Expuestas anteriormente
1.3 Características
Disminución del tono muscular al nacer
Exceso de piel en la nuca
Nariz achatada
Suturas separadas (articulaciones entre los huesos del cráneo)
Cabeza anormalmente grande, pequeña o deformada
Pliegue único en la palma de la mano
Orejas pequeñas
Boca pequeña
Ojos inclinados hacia arriba
Manos cortas y anchas con dedos cortos
Manchas blancas en la parte coloreada del ojo (manchas de Brushfield)
El retraso mental puede variar entre leve y moderado, con un
coeficiente intelectual (IQ) de 50 como promedio. Cerca de la tercera
parte de quienes nacen con Síndrome de Down tienen graves
defectos cardiacos, lo que ocasiona muertes prematuras. Otros
sobreviven gracias a una cirugía correctiva.
46

Seminario Biología Celular
Debe mencionarse que existen pruebas para detectar durante el
embarazo posibles anomalías en el embrión o feto, como la
amniocentesis, que es el estudio del líquido amniótico, que permite
determinar el adecuado desarrollo del feto.
1.4 Consideraciones importantes
Es importante informar a los padres que tener un hijo con Síndrome
de Down no es un castigo ni un estigma, es simplemente un trastorno
generado por la naturaleza. Por lo que, estos niños no deben de ser
aislados porque esto disminuye sus posibilidades de desarrollo e
integración sociolaboral a través de cursos especiales o programas de
inserción laboral, entre otros.
1.5 Tratamiento
El tratamiento depende del grado de retraso y de los problemas
relacionados. Los defectos cardiacos, por ejemplo, requieren una
corrección quirúrgica. Más allá de los problemas físicos, el niño
necesita educación especial. Muchos padres encuentran que es fácil
proveer esto, ya que los niños con Síndrome de Down y otros retrasos
tienden a ser calmados y tratables cuando son jóvenes. Por lo general
estos niños son plácidos, agradables y rara vez lloran o se quejan.
47

Seminario Biología Celular
2. SÍNDROME DE PATAU
2.1 Definición
El síndrome de Patau, trisomía en el par 13 o trisomía D es una enfermedad
genética que resulta de la presencia de un cromosoma 13 suplementario. El
cariotipo da 47 cromosomas y sirve de diagnóstico prenatal por amniocentesis
o cordiocentesis sobre todo si los padres optan por el aborto eugenésico. Se
trata de la trisomía menos frecuente, descubierta en 1960 por Patau. Los
afectados mueren poco tiempo después de nacer, la mayoría a los 3 meses,
como mucho llegan al año. Se cree que entre el 80-90% de los fetos con el
síndrome no llegan a término.
2.2 Características
Crecimiento: Retraso de crecimiento pre- y postnatal
Sistema nervioso central: Retraso psicomotor/mental profundo,
microcefalia, holoprosencefalia, episodios de apnea,hipotonía/hipertonía.
Area craneofacial: Anomalías oculares (microftalmia, coloboma del iris),
Micrognatia, hipotelorismo ocular, pabellones auriculares malformados ,
defectos en cuero cabelludo, paladar ojival , hemangiomas capilares,
labio leporino más fisura palatina, epicanto
Sistema cardiovascular: Comunicación interauricular , persistencia del
ductus arteriosus, comunicación interventricular
Aparato genitourinario: Criptorquidia (varones), riñón poliquístico,
útero bicorne (mujeres), hidronefrosis
48

Seminario Biología Celular
Extremidades: Polidactilia, dedos en flexión y superpuestos, uñas
hiperconvexas, surco de los 4 dedos en palmas, calcáneo prominente
(menos común).
Mamilas hipoplásicas
Fragmentaciones nucleares en polimorfonucleares
Hernia inguinal/umbilical
49

Seminario Biología Celular
3. SINDROME DE EDWARDS
3.1 Definición
El Sindrome de Edwards, más conocida como trisomía 18, es una aneuploidía
humana que se caracteriza usualmente por la presencia de un cromosoma
adicional completo en el par 18. También se puede presentar por la presencia
parcial del cromosoma 18 o por mosaicismo en las células fetales.
Su frecuencia se calcula entre 1/6000-1/13000 nacidos vivos.
3.2 Características
Faciales: Microcefalia, occipucio prominente, frente estrecha. orejas
bajas y malformadas, micrognatia, labio leporino
Músculo-Esqueléticos: Cuello alado, esternón corto, areolas
separadas, pelvis estrecha, dislocación de caderas, focomelia, pie en
mecedora, dedos sobrepuestos, uñas hipoplásicas.
Anomalías del SNC y cráneo: Ventriculomegalia, megacisterna magna,
quiste de fosa posterior, quiste de plexo coroídeo, microcefalia,
mielomeningocele
Anomalías del cuello: Higroma quístico/edema nucal,
Hidrops/linfangectasia
Anomalías cardiovasculares: Defectos septales, coartación aórtica,
ductus arteriosus persistente, transposición de grandes vasos,
dextrocardia, lesiones valvulares
Anomalías gastrointestinales: Hernia diafragmática, onfalocele, hernia
inguinal o umbilical, intestino ecogénico
50

Seminario Biología Celular
Anomalías genitourinarias: Riñón poliquístico, hidronefrosis, riñón en
herradura, riñón ectópico, criptorquidia, micropene.
Anomalías de extremidades: Polidactilia, pie bott, clinodactilia
51

Seminario Biología Celular
B. ANEUPLOIDÍAS EN CROMOSOMAS SEXUALES
Son alteraciones en el número de copia de alguno de los dos cromosomas
sexuales humanos. Las aneuploidias en este caso suelen ser viables.
Entre las más frecuentes destacan:
Síndrome de Klinefelter (trisomía de los cromosomas sexuales: 47,XXY)
Síndrome de Turner (monosomía de los cromosomas sexuales: 45,X)
Síndrome del doble Y (llamado síndrome del supermacho: 47,XYY)
Síndrome del triple X (llamado síndrome de la superhembra: 47,XXX)
52

Seminario Biología Celular
1. SINDROME DE TURNER (XO)
1.1 Definición:
Trastorno de la diferenciación sexual, derivado de la ausencia de un
cromosoma X. A esta anomalía cromosómica también se la denomina
Bonnevie-Ullrich o disgenesia gonadal y está presente en aproximadamente 1
de cada 2.500-3.000 nacimientos de niñas vivas.
1.2 Caracterísiticas:
El cariotipo muestra 45 cromosomas con un modelo de 44 X, o es decir,
un cromosoma sexual ausente.
Genitales y mamas subdesarrollados. Casi siempre estériles
Cuello corto
baja estatura
Caja torácica y hombros anchos
El ultrasonido puede revelar órganos reproductores femeninos pequeños
o subdesarrollados.
El examen ginecológico puede revelar sequedad del recubrimiento de la
vagina.
La hormona luteinizante sérica se encuentra elevada.
La hormona foliculoestimulante sérica se encuentra elevada.
53

Seminario Biología Celular
54

Seminario Biología Celular
2. SINDROME DE KLINEFELTER (XXY)
1.1 Definición
El síndrome de Klinefelter (SK) es una forma de hipogonadismo masculino debido a
una anomalía de los cromosomas sexuales, de hecho, la primera que fue descrita en
humanos, y que tiene una incidencia de 1 de cada 1000 varones nacidos.
1.2 Características
Se debe tener en cuenta que no todos los varones con cariotipo 47,XXY
manifestarán todas estas características.
Sistema Musculoesquelético: La masa muscular es poco desarrollada,
por lo que el cansancio es mas fácil. Pueden presentar una displasia
leve a nivel de la articulación del codo, y clinodactilia del 5º dedo de las
manos. La osteoporosis aparece sobretodo en los indivuduos que no
reciben testosterona. Los adolescentes presentan escoliosis con mas
frecuencia que la población general. Los individuos con cariotipo 48,
XXXY pueden tener talla baja y sinóstosis radio-cubital.
Desarrollo Sexual: La pubertad aparece a una edad normal, pero los
testículos no se desarrollan y permanecen pequeños. Los caracteres
sexuales secundarios se desarrollan poco. El vello corporal es escaso y
la distribución puede ser ginecoide. El tejido celular subcutáneo también
puede adoptar una distribución femenina sobretodo a nivel de las
caderas, y pueden presentar ginecomastia. La actividad sexual
generalmente es normal o levemente deprimida. Debido al exceso de
gonadotropina se produce de forma progresiva una hialinización y
fibrosis de los túbulos seminíferos, con una inadecuada producción de
testosterona y azoospermia en la mayoría de casos, requiriendo por ello
55

Seminario Biología Celular
tratamiento con testosterona a largo plazo. La mayoría de ellos son
infértiles. Ocasionalmente pueden presentar criptorquídia e hipospadias.
Capacidad Intelectual: El coeficiente intelectual de estos individuos es,
ligera pero significativamente, inferior que el de los varones con
cromosomas normales. Dos tercios tienen problemas de aprendizaje,
especialmente dislexia. El lenguaje expresivo, la capacidad de
procesamiento auditivo y la memoria auditiva son deficientes, lo cual
conlleva una menor habilidad para leer y escribir. CARÁCTER Los
trastornos del comportamiento son frecuentes, especialmente
inmadurez, inseguridad, timidez, y poca capacidad de juicio. Les cuesta
relacionarse con individuos de su grupo de edad y pueden tener
problemas de adaptación social. La depresión es frecuente en estos
individuos.
Sistema Venoso: La enfermedad varicosa y las úlceras de
extremidades inferiores pueden ser los primeros síntomas de los
varones 47,XXY.
Enfermedades autoinmunes: Existe un mayor riesgo de desarrollar
enfermedades autoinmunes como diabetes, artritis reumatoide, tiroiditis
y el lupus eritematoso.
Neoplasias: Los varones XXY con ginecomastia tienen mayor riesgo de
cáncer de mama. Se ha descrito una mayor incidencia de tumores
germinales extragonadales con afectación principalmente mediastínica.
56

Seminario Biología Celular
57

Seminario Biología Celular
3. SINDROME XYY
3.1 Definición:
El síndrome XYY (también llamado síndrome del superhombre, entre otros
nombres) es una trisomía de los cromosomas sexuales donde el hombre recibe
un cromosoma Y extra, produciendo el cariotipo 47,XYY. Algunos médicos
genetistas cuestionan si el uso del término «síndrome» es apropiado para ésta
condición, porque el fenotipo es normal, ya que la gran mayoría de hombres
con 47,XYY no conocen su cariotipo.
La incidencia es cerca de 1 de cada 1.000 niños
3.2 Características
Con gran frecuencia, esta alteración cromosómica no causa características
físicas inusuales o problemas médicos. Los jóvenes y adultos con 47,XYY son
regularmente algunos centímetros más altos que sus padres y hermanos. En
muy pocos casos se ha reportado acné severo, pero dermatólogos
especialistas en este campo manifiestan que no existe evidencia que se
relacione con 47,XYY
Los niveles de testosterona (prenatal y postnatal) son normales en hombres
con 47,XYY.La mayoría de los hombres con 47,XYY tienen un desarrollo
sexual normal y por lo regular son fértiles. El XYY no ha sido identificado por
las características físicas, la condición es usualmente detectada sólo durante el
análisis genético, solicitado por razones distintas.
58

Seminario Biología Celular
59

Seminario Biología Celular
4. SINDROME XXX
4.1 Definición
El síndrome XXX o triple X, es una anomalía numérica que se presenta en las
mujeres que poseen un cromosoma X extra. Aproximadamente una de cada
1000 a 1200 mujeres tienen este síndrome.
4.2Caracteristicas
Rasgos físicos
Las recién nacidas y las niñas con síndrome 47, XXX se parecen a otras niñas
de su edad. Suelen ser más altas que el resto de las niñas en su familia y
pueden tener menos coordinación. Las mujeres con síndrome 47, XXX
usualmente son capaces de tener hijos (son fértiles).
Rasgos mentales y sociales
De todas las condiciones de cromosomas del sexo, el síndrome 47, XXX es
uno de los que se asocian más con problemas mentales y de comportamiento.
Una probabilidad alta de tener problemas en el lenguaje y el habla pueden
causar retrasos en las habilidades sociales y de aprendizaje. Por consiguiente,
estas niñas suelen necesitar ayuda adicional para tener éxito en la escuela. En
un pequeño estudio llevado a cabo en 11 niñas que fueron diagnosticadas con
síndrome 47, XXX al nacer y que mantuvieron un seguimiento para ver cómo
se desarrollaban, se descubrió que menos de la mitad se graduaron de la
secundaria. Aunque estas niñas tenían amigos en la escuela, tendían a
comportarse con menos madurez que otros niños de su edad. No les gustaba
participar en las actividades en grupo y tenían más tendencia que sus
hermanas a sufrir depresiones. De este pequeño grupo que fue estudiado, una
de ellas asistió a la universidad.
60

Seminario Biología Celular
61