el dolç fenotip de les cèl·lules tumorals: buscant ... · el dolç fenotip de les cèl·lules...
TRANSCRIPT

Institutd’Estudis Catalans
SECCIÓ DE CIÈNCIESBIOLÒGIQUES
El dolç fenotip de les cèl·lules tumorals: buscant dianes terapèutiquesDiscurs de recepció de Ramon Bartrons i Bachcom a membre numerari de la Secció de Ciències Biològiques, llegit el dia 18 de febrer de 2013
9 7 8 8 4 9 9 6 5 1 5 3 8
021-Coberta EL DOLÇ FENOTIP.indd 1 09/01/13 09:57

El dolç fenotip de les cèl·lules tumorals:buscant dianes terapèutiques
021-EL DOLÇ FENOTIP.indd 1 09/01/13 10:03

021-EL DOLÇ FENOTIP.indd 2 09/01/13 10:03

El dolç fenotipde les cèl·lules tumorals: buscant dianes terapèutiquesDiscurs de recepció de Ramon Bartrons i Bachcom a membre numerari de la Secció de Ciències Biològiques, llegit el dia 18 de febrer de 2013
Barcelona, 2013
Institutd’Estudis Catalans
SECCIÓ
DE CIÈNCIES
BIOLÒGIQUES
021-EL DOLÇ FENOTIP.indd 3 09/01/13 10:03

Biblioteca de Catalunya. Dades CIP
Bartrons Bach, RamonEl Dolç fenotip de les cèl·lules tumorals : buscant dianes terapèutiquesBibliografiaISBN 9788499651538I. Institut d’Estudis Catalans. Secció de Ciències Biològiques II. Títol1. Cèl·lules canceroses 2. Fenotip576.31
Il·lustració de la coberta: cèl·lules HeLa. Verd: tubulina; vermell: aparell de Golgi; blau: nuclis. Doctor Benjamin Torrejón i doctora Esther Castaño. Campus de Ciències de la Salut. Centres Científics i Tecnològics de la Universitat de Barcelona (CCiTUB).
Disseny de la coberta: Azcunce | Ventura
© Ramon Bartrons i Bach© 2013, Institut d’Estudis Catalans, per a aquesta edicióCarrer del Carme, 47. 08001 Barcelona
Primera edició: gener del 2013Tiratge: 210 exemplars
Text revisat lingüísticament per la Unitat de Correcció del Servei Editorial de l’IEC
Compost per Víctor Igual, SLImprès a Service Point FMI, SA
ISBN: 978-84-9965-153-8Dipòsit Legal: B-788-2013
Són rigorosament prohibides, sense l’autorització escrita dels titulars del copyright, la reproducció total o par-cial d’aquesta obra per qualsevol procediment i suport, incloent-hi la reprografia i el tractament informàtic, la distribució d’exemplars mitjançant lloguer o préstec comercial, la inclusió total o parcial en bases de dades i la consulta a través de xarxa telemàtica o d’Internet. Les infraccions d’aquests drets estan sotmeses a les sancions establertes per les lleis.
021-EL DOLÇ FENOTIP.indd 4 09/01/13 10:03

5
En el foc de la joventut em semblava que tot havia de trobar fàcil explicació, que les lleis de l’experiència serien vàlides universalment i, per altra part, que aquí tot fóra explicable per determinisme. Però, per què una flor creix i exhala el seu perfum, i una papallona mostra la seva delicada bellesa?
August Pi i Sunyer
1. IntroduccIó
El meu periple científic, durant la dècada dels setanta, s’inicià als laboratoris que havien acollit l’Institut de Fisiologia de Barcelona que havia creat el professor August Pi i Sunyer. Al Departament de Fisiologia de la Facultat de Medicina en-cara hi havia intactes les estructures d’alguns dels seus laboratoris amb aparells, gàbies i material, i la biblioteca amb llibres i força col·leccions de revistes de l’èpo-ca. A l’entrada del departament, sobre la porta del passadís, es notava el pas del franquisme amb una empremta de vidre amb l’escut de l’àguila i les fletxes, cosa que donava compte del càstig a què havia estat sotmesa aquesta disciplina cientí-fica pel paper polític rellevant que varen tenir alguns fisiòlegs en la defensa de la República; entre d’altres, August Pi i Sunyer a Barcelona o Juan Negrín a Madrid. Pi i Sunyer havia estat el fundador de la primera societat filial de l’Institut d’Estu-dis Catalans, la Societat Catalana de Biologia, que vaig tenir l’honor de presidir durant els anys 1994 a 2000.
021-EL DOLÇ FENOTIP.indd 5 09/01/13 10:03

6
Curiosament, la tesi doctoral del professor August Pi i Sunyer va versar sobre «La vida anaeròbia» (Pi i Sunyer, 1901). La tesi es va presentar a Madrid, l’any 1900, quan just tenia vint-i-un anys i havia acabat els estudis de medicina, amb un con-tingut molt limitat de fisiologia i gairebé inexistent de química biològica. La tesi representava un esforç jovenívol més ric en erudició i intuïció que no pas en apor-tacions experimentals (Pi i Sunyer Bayo, 1966).
Uns quants anys abans, Louis Pasteur havia demostrat el concepte de vida anaeròbia, en confirmar l’existència de microorganismes capaços de viure sense oxigen, i, també, havia suggerit que el metabolisme primitiu devia ser sense oxi-gen («vie sans air») (Pasteur, 1861). Pasteur distingia dues classes de processos químics vitals: oxidatius o aerobis i fermentatius o anaerobis. Quan coexisteixen, la intensitat de la fermentació varia en relació inversa respecte de la concentració d’oxigen en el medi. En condicions anaeròbies el llevat fermenta el sucre i pro-dueix alcohol, però la presència d’oxigen redueix la fermentació i estimula les reaccions oxidatives. Així, el consum de glucosa és inversament proporcional a la disponibilitat d’oxigen. És el que es coneix com a efecte Pasteur.
En aquella època no es coneixien les vies metabòliques, ni els enzims que hi participaven, ni els mecanismes que seguien. Caldria un gran esforç que liderarien investigadors com Buchner, Meyerhof, Hill, Harden, Young, Embden, Lipmann, Krebs, Cori, Theorell i molts d’altres per anar avançant en la comprensió i la re-gulació de les vies metabòliques. Actualment, sabem que l’efecte Pasteur s’explica a partir de les propietats al·lostèriques de la 6-fosfofructo-1-cinasa (PFK-1) i la seva inhibició pel citrat (Sols, 1981; Krebs, 1972) (figura 1).
2. Warburg I el metabolIsme de les cèl·lules tumorals
Els primers estudis sobre el metabolisme de les cèl·lules tumorals varen ser publicats pel bioquímic alemany Otto Warburg al llibre Über den Stoff-wechsel der Tumoren («El metabolisme dels tumors»), en què plantejava la possible relació entre l’origen dels tumors i els canvis metabòlics de les cèl·lules tumo-rals (Warburg, 1926 i 1956). Warburg va posar a punt un aparell (manòmetre de War burg) que li permetia mesurar el consum d’oxigen de cèl·lules i talls de teixits. Amb aquest nou mètode, va demostrar que les cèl·lules tumorals regis-traven una ràpida i intensa glicòlisi, en la qual la glucosa s’oxidava fins a lactat, encara que hi hagués oxigen abundant en el medi (figura 1). Aquest fet, ano-menat efecte Warburg, és característic de cèl·lules proliferants i transformades. Warburg considerava que el càncer es produïa com a conseqüència de defectes en la respiració mitocondrial, que forçava la cèl·lula a canviar a una forma més primitiva d’obtenció d’energia, la glicòlisi. Des del seu punt de vista, aquest canvi provocava la desdiferenciació de les cèl·lules. Warburg va pensar que
021-EL DOLÇ FENOTIP.indd 6 09/01/13 10:03

7
aquesta «glicòlisi aeròbia» era una propietat universal de les cèl·lules cancero-ses (Warburg, 1956).
Aquest canvi metabòlic de les cèl·lules tumorals provoca que la cèl·lula única-ment obtingui un rendiment del 6 % de l’energia total a què podria optar si la glu-cosa s’oxidés en entrar en el cicle de Krebs i utilitzar la cadena respiratòria i la fos-forilació oxidativa. Malgrat això, permet a les cèl·lules obtenir prou energia i sintetitzar les biomolècules essencials, com ara àcids nucleics, proteïnes i lípids, necessàries per duplicar els components cel·lulars durant la divisió.
En la mateixa època que Otto Warburg, Herbert G. Crabtree va descriure un fenomen semblant. Va observar que alguns tumors presentaven una disminució de la respiració cel·lular en presència de glucosa (Crabtree, 1929). Aquest fet, anomenat efecte Crabtree, s’ha descrit en cèl·lules proliferants i tumorals (Guppy et al., 1993).
L’efecte Crabtree i l’efecte Warburg s’explicaven a partir d’alteracions de la ca-
Figura 1. Regulació del metabolisme de la glucosa. En presència d’oxigen i glucosa la concentra-ció de citrat i ATP augmenten i inhibeixen al·lostèricament la PFK-1 i la glicòlisi. GLUT: transpor-tador de glucosa; HK: hexocinasa; PFK-1: 6-fosfofructo-1-cinasa; MCT: transportador de monocar-boxilats.
021-EL DOLÇ FENOTIP.indd 7 09/01/13 10:03

8
dena respiratòria, però alguns van qüestionar la hipòtesi que el canvi metabòlic d’activació de la glicòlisi fos degut a un defecte en la respiració mitocondrial. Al-guns tumors no tenen defectes en la respiració i, a més, aquesta exerceix un efecte regulador de la glicòlisi (Chance, 2005). La teoria de Warburg sobre el càncer es va anar oblidant en descobrir que algunes cèl·lules tumorals no experimentaven can-vis en la respiració i, sobretot, pel protagonisme que anava prenent la biologia molecular, que donava pas a la idea predominant que el càncer era una malaltia produïda per mutació de gens relacionats amb la proliferació. Encara que algunes cèl·lules canceroses no tenen una alta activitat glicolítica (Zu i Guppy, 2004), l’efec-te Warburg ha estat consistentment observat en una àmplia varietat de càncers humans. Els mecanismes moleculars per explicar l’efecte Warburg són probable-ment múltiples i inclouen: mal funcionament mitocondrial (Wallace, 2005), alte-racions oncogèniques (Dang i Semenza, 1999; Hanahan i Weinberg, 2000), així com respostes d’adaptació al microambient del tumor (Gatenby i Gillies, 2004; Tennant et al., 2010).
3. HabIlItats de les cèl·lules tumorals
La cèl·lula, en condicions normals, disposa de múltiples mecanismes que en controlen el metabolisme, la proliferació, la diferenciació i la mort. Aquests, en funció dels nutrients i dels diferents senyals externs i interns que li arriben, perme-ten que la cèl·lula sobrevisqui, proliferi o bé entri en un procés de senescència o d’apoptosi. En els processos tumorals hi ha una pèrdua del control d’aquests me-canismes, cosa que provoca la proliferació cel·lular descontrolada, la invasió del teixit que l’envolta i la metàstasi. En aquesta formació i progressió tumoral hi es-tan implicats gens que experimenten alteracions que comporten un guany de fun-ció, com és el cas dels oncogens (Land et al., 1983), o bé una pèrdua d’aquesta, com passa amb els gens supressors o els gens mutadors (Vogelstein i Kinzler, 1993; Levine i Puzio-Kuter, 2010). En qualsevol cas, aquestes alteracions suposen un avantatge per al creixement i el desenvolupament tumoral.
Davant la gran diversitat de cèl·lules canceroses, Robert Weinberg i Douglas Hanahan van sistematitzar sis característiques comunes que defineixen les cèl·lu les tumorals (Hanahan i Weinberg, 2000). Aquests mecanismes, que han esdevingut clàssics, són els següents: 1) activació de senyals de proliferació; 2) insensibilitat a senyals antiproliferatius; 3) evasió de la mort cel·lular programada o apoptosi; 4) potencial replicatiu il·limitat; 5) alta capacitat angiogènica, i 6) invasió de teixits pròxims i me tàstasi en teixits llunyans. Posteriorment i com a resultat de les noves dades, n’hi han afegit uns quants més, com evasió de l’atac immunològic, inestabi-litat genòmica, inflamació o desregulació metabòlica (Hanahan i Weinberg, 2011). En tots aquests processos hi estan implicats oncogens i gens supressors.
021-EL DOLÇ FENOTIP.indd 8 09/01/13 10:03

9
Les cèl·lules normals evolucionen cap a un estat neoplàstic quan adquireixen progressivament aquestes propietats, de manera que segueixen un procés de pas-sos successius que proporciona a les cèl·lules transformades característiques que els permeten reeixir en ambients poc propicis i esdevenir tumorals (Vogelstein i Kinzler, 1993).
4. el fenotIp glIcolítIc
El ressorgiment del paper de la bioenergètica en el càncer s’inicia a comença-ments dels anys noranta, quan els radiòlegs van mostrar que la tomografia per emissió de positrons (PET) permetia detectar la majoria de tumors i metàstasis. La tècnica aplicada utilitzava la desoxiglucosa unida amb fluor radioactiu com a marcador (fluoro-desoxiglucosa, FDG).
La tomografia per emissió de positrons és una tècnica de medicina nuclear que permet obtenir imatges tridimensional dels processos metabòlics de l’organisme. El sistema detecta parells de raigs gamma emesos indirectament per un radionú-
Figura 2. Captació cel·lular de 18fluoro-desoxiglucosa. La 18fluoro-desoxiglucosa és transportada a l’interior de la cèl·lula pels transportadors de glucosa (GLUT), on és fosforilada per l’hexocinasa (HK) a 18fluoro-desoxiglucosa-6-fosfat (FDG-6-P).
021-EL DOLÇ FENOTIP.indd 9 09/01/13 10:03

10
clid emissor de positrons, el qual és introduït al cos en una molècula biològica-ment activa. S’obtenen imatges tridimensionals d’acord amb la concentració de la molècula traçadora incorporada. Si la molècula biològicament activa escollida és la fluoro-desoxiglucosa, aquesta pot ser captada per les cèl·lules a través dels trans-portadors de glucosa (GLUT) i, una vegada dins, fosforilada per l’hexocinasa (HK) a fluoro-desoxiglucosa-6-fosfat (FDG-6-P). Com que la desoxiglucosa no pot continuar oxidant-se, s’acumula dins la cèl·lula i les concentracions que s’obtenen reflecteixen l’activitat metabòlica, en termes de captació i fosforilació de glucosa (figura 2). Aquesta tècnica, àmpliament utilitzada en la detecció de tumors i me-tàstasis, ha posat en relleu l’avidesa de les cèl·lules canceroses per la glucosa.
La tomografia per emissió de positrons ha demostrat que la majoria de tu-mors presenten una captació de glucosa que és aproximadament un ordre de magnitud superior que la del teixit normal (Gatenby i Gillies, 2004). Més del 90 % de tumors primaris i metastàtics tenen un elevat consum de glucosa i hi ha una bona correlació entre l’agressivitat del tumor i la taxa d’utilització de glucosa (Smallbone et al., 2007).
La majoria de cèl·lules tumorals, de manera semblant a les proliferants, presen-ten un metabolisme energètic marcadament modificat en comparació de les cèl·lules diferenciades. En concret, s’ha observat de manera consistent un canvi d’un meta-bolisme basat en la respiració a un altre d’eminentment glicolític, reconegut com a fenotip glicolític (Bartrons i Caro, 2007; DeBerardinis et al., 2008). Diversos gens glicolítics o relacionats amb la glicòlisi estan sobreexpressats sistemàticament en molts tipus de càncer i formen part d’aquest fenotip, independent del tipus de càn-cer, que es pot utilizar per diagnosticar la malignitat d’un tumor i la possible respos-ta terapèutica per aplicar-hi (Cuezva et al., 2002). Aquestes dades han fet considerar el fenotip glicolític i l’avidesa per la glucosa com un signe distintiu de la cèl·lula tu-moral (Bartrons i Caro, 2007; Vander Heiden et al., 2009; Koppenol et al., 2011).
Actualment, comencem a conèixer els factors i les vies responsables de l’apa-rició d’aquest fenotip glicolític i de la reprogramació metabòlica que apareix en les cèl·lules tumorals. S’han estudiat molts gens que hi són implicats: RAS (Shaw i Cantley, 2006), TP53 (Vousden i Prives, 2009), HIF-1 (hypoxia inducible factor-1) (Semenza, 2002; Semenza 2012), c-Myc (Dang et al., 2009; Dang, 2010) i d’altres (Gatenby i Gillies, 2004). Encara que el canvi al fenotip glicolític de les cèl·lules canceroses no fos un requisit imprescindible per a la transformació maligna, la gran majoria dels estudis indiquen que és un fenomen important en la transfor-mació, associat a un avantatge de supervivència per a la cèl·lula en l’entorn tumo-ral i a un elevat potencial metastàtic.
Un dels descobriments importants que han permès explicar l’aparició del fe-notip glicolític el va fer el grup de Greg Semenza (Semenza, 2002; Semenza, 2012), en caracteritzar el factor induïble per hipòxia (HIF), que exerceix una funció es-
021-EL DOLÇ FENOTIP.indd 10 09/01/13 10:03

11
sencial en el manteniment de l’homeòstasi respiratòria en situacions de baixa pres-sió parcial d’oxigen. HIF controla l’expressió de molts gens i indueix canvis fisio-lògics que permeten que les cèl·lules sobrevisquin en condicions d’hipòxia. Aquests gens estan involucrats en el transport d’oxigen, la captació de glucosa, la glicòlisi, l’angiogènesi, la senyalització per factors de creixement, l’apoptosi, la invasió i la metàstasi (figura 3) (Semenza, 2002; Pouyssegur et al., 2006; Semenza, 2012). Molts tumors estan en una situació d’hipòxia i el factor està freqüentment sobre-expressat en tumors, de manera que és un element clau per explicar el fenotip gli-colític (Pouyssegur et al., 2006; Denko, 2008; Semenza, 2012). Hi ha diferents iso-formes de HIF, tot i que en aquesta revisió només analitzarem HIF-1, ja que és la isoforma més directament relacionada amb la inducció del fenotip glicolític.
HIF-1 és un complex proteic heterodimèric compost de dues subunitats: HIF-1β, que s’expressa constitutivament, i HIF-1α, els nivells de la qual depenen de la concentració d’oxigen als teixits. Ambdues proteïnes són membres de la fa-mília de factors de transcripció bàsics amb estructura hèlix-loop-hèlix (bHLH-PAS). HIF-1α dimeritza amb HIF-1β, per via del domini bHLH, i una vegada el
Figura 3. El factor de transcripció HIF-1 reconeix l’element de resposta a hipòxia (HRE) dels seus gens diana i en regula la transcripció. Quan hi ha oxigen, HIF-1α és hidroxilat, ubiqüitinitzat i de-gradat per via del proteasoma. En condicions d’hipòxia, HIF-1α s’acumula i es transloca al nucli, on s’hibrida amb la subunitat HIF-1β i reconeix la seqüència consens d’unió en els gens diana (HRE). PHD: prolil hidroxilasa; VHL: proteïna Von Hippel-Lindau.
021-EL DOLÇ FENOTIP.indd 11 09/01/13 10:03

12
complex està format s’uneix al DNA per via d’aquest mateix domini, de manera que activa la transcripció dels gens diana. Aquests dominis de transactivació per-meten també la interacció amb coactivadors. La formació del complex HIF-1 és un procés regulat principalment pels nivells de la subunitat HIF-1α (Semen-za, 2002; Denko, 2008; Semenza, 2012). En presència d’oxigen, HIF-1α té una vida mitjana molt curta a causa de la seva ràpida degradació pel sistema ubiqüiti-na-proteasoma. Així, en condicions aeròbies, les concentracions de HIF-1α són molt baixes i, en canvi, augmenten ràpidament durant l’exposició a hipòxia. La degradació de HIF-1α depèn de la presència d’una seqüència d’aminoàcids cone-guda com el domini ODDD (oxygen dependent degradation domain), ubicat al centre de la proteïna. El domini ODDD conté dos residus de prolina que, en pre-sència d’oxigen, són hidroxilats per enzims específics, anomenats HIF-prolil hidroxilases (HIF-PHD) (Semenza, 2002; Denko, 2008; Semenza, 2012). Un cop hi-droxilats, HIF-1α és reconegut per la proteïna Von Hippel-Lindau (VHL), prote-ïna supressora de tumors, que actua com una E3 ubiqüitina lligasa (Kaelin, 2005). Posteriorment, es degrada ràpidament per via del proteosoma 26S (figura 4). Com que les reaccions d’hidroxilació necessiten la presència d’oxigen, ferro i 2-ce-toglutarat, les HIF-1α hidroxilases no funcionen en condicions d’hipòxia i VHL no pot hidroxilar HIF-1α perquè sigui reconegut i ubiqüitinitzat. Això permet, en condicions d’hipòxia, una acumulació d’aquesta subunitat a la cèl·lula, la translo-cació al nucli i la dimerització amb HIF-1β, i, seguidament, la unió als elements de resposta a HIF (HRE) que es troben en la seqüència de DNA dels gens diana (Semenza, 2002; Pouyssegur et al., 2006; Denko, 2008; Semenza, 2012).
Els estudis inicials de Gregg Semenza i col·laboradors van demostrar que la incubació de cèl·lules d’hepatoblastoma humà (Hep3B) a baixes concentracions d’oxigen (1 %) activava la transcripció del gen de l’eritropoetina (EPO) (Goldberg et al., 1991) per acció del factor HIF-1. Van comprovar també que cèl·lules que no expressaven EPO també podien induir l’activitat de HIF-1 com a resposta a l’ex-posició a hipòxia (1 % d’oxigen), com per exemple les cèl·lules HeLa (cèl·lules de carcinoma cervical). Tot això els va fer pensar que HIF-1 podia transactivar altres gens, a més del de l’eritropoetina; així que van estudiar l’expressió de tres gens glicolítics: aldolasa A (ALD-A), fosfoglicerat cinasa (PGK-1) i piruvat cinasa (PKM), i van corroborar la seva inducció per hipòxia (Semenza et al., 1994). Al cap de dos anys, van determinar que HIF-1 activava la transcripció d’enolasa-1 (ENO-1) i lactat deshidrogenasa A (LDH-A). Posteriorment, es varen trobar al-tres gens que s’expressen a través de HIF: els transportadors de glucosa (GLUT-1 i 3) (Dang i Semenza, 1999) o els gens que codifiquen alguns isoenzims bifuncio-nals 6-fosfofructo-2-cinasa / fructosa 2,6-bisfosfatasa (Obach et al., 2004). Tots aquests gens d’enzims glicolítics són estimulats per la hipòxia a través d’elements de resposta a hipòxia (hypoxia response elements, HRE) en els seus promotors.
021-EL DOLÇ FENOTIP.indd 12 09/01/13 10:03

13
La hipòxia produeix una disminució de la respiració mitocondrial a causa dels baixos nivells d’oxigen (Esteban i Maxwell, 2005). S’ha demostrat que l’adap-tació metabòlica a la hipòxia produeix una inhibició del metabolisme mitocon-drial del piruvat i el consum d’oxigen a través de la regulació transcripcional de HIF-1 (Kim et al., 2006). HIF-1 suprimeix tant el cicle de Krebs com la respira-ció mitocondrial mitjançant la inducció de la piruvat-deshidrogenasa cinasa 1 (PDK1), que inhibeix l’activitat de la piruvat deshidrogenasa per fosforilació de la subunitat E1 (figura 5). D’aquesta manera, l’entrada de piruvat, en el cicle de Krebs queda limitada i les cèl·lules hipòxiques acumulen piruvat, que es conver-teix en lactat a través de la lactat deshidrogenasa (LDH), cosa que regenera el NAD+ necessari per a la glicòlisi. El lactat, al seu torn, es transporta a l’espai extracel·lular a través dels transportadors de monocarboxilats (MCT). Aquesta regulació, dependent de HIF-1, permet la supervivència cel·lular quan la privació d’oxigen és important i perllongada. La inhibició del complex piruvat deshidro-genasa per la PDK1 no només frena la respiració mitocondrial, sinó també la producció mitocondrial d’espècies reactives d’oxigen (ROS) en les cèl·lules hipò-xiques, i això promou la supervivència cel·lular (Kim et al., 2006).
HIF-1α es pot estabilitzar, també, en addició a la hipòxia a través d’alguns oncogens com v-src, HER2 i H-RAS, o bé per inactivació d’alguns supressors tu-morals com TP53 o PTEN (phosphatase and tensin homdog) (Kim et al., 2007). Igualment, HIF-1α pot estabilitzar-se per altes concentracions de piruvat o lactat generades per la glicòlisi. Aquests metabòlits monocarboxilats, juntament amb l’oxaloacetat, inhibeixen l’activitat de les prolil hidroxilases (PHD) i competeixen amb el 2-cetoglutarat. Igualment, les mutacions que disminueixen l’activitat de la succinat deshidrogenasa i la fumarat hidratasa produeixen acumulacions de suc-cinat i fumarat que inhibeixen les PHD i estabilitzen HIF-1α, i s’associen amb el desenvolupament de diferents tipus de carcinomes (Pouyssegur et al., 2006). Aquestes mutacions de gens codificants per enzims metabòlics fan que es com-portin com a veritables oncogens.
HIF-1α està implicat en l’activació de nombrosos processos cel·lulars, in-cloent-hi la resistència a l’apoptosi, la sobreexpressió de bombes expulsores de drogues, la remodelació i l’angiogènesi vascular, així com la metàstasi. En les cèl-lules canceroses, HIF-1α indueix sobreexpressió i augment d’activitat d’isoen-zims glicolítics específics que difereixen dels que es troben en les cèl·lules diferen-ciades (taula 1). Aquests isoenzims mostren patrons cinètics característics, amb una alta afinitat pels substrats i una baixa afinitat pels productes, amb una alta capacitat catalítica (Vmax), amb una disminució de la sensibilitat envers els seus inhibidors fisiològics i amb una capacitat de regulació per diferents proteïnes ci-nases (Colomer et al., 1987; Bartrons i Caro, 2007).
021-EL DOLÇ FENOTIP.indd 13 09/01/13 13:33

14
5. oncogens I efecte Warburg
Sembla evident que no totes les àrees d’un tumor que són altament glicolíti-ques han de ser necessàriament hipòxiques (Gatenby i Gillies, 2004; Smallbone et al., 2007). Per tant, hi ha d’haver altres mecanismes de regulació per induir el fe-notip glicolític. Moltes cèl·lules tumorals creixen en condicions normals de pres-sió parcial d’oxigen i mostren també elevades taxes glicolítiques que es correla-cionen amb increments en l’expressió d’enzims glicolítics i transportadors de glucosa (Dang i Semenza, 1999; Dang et al., 2009). Això podria ser degut a l’esta-bilització de HIF-1α per factors diferents a la hipòxia; per exemple, la inactivació de la ubiqüitina lligasa (VHL) (Kaelin, 2005) o l’activació de HIF per factors de creixement, per diferents oncogens o per metabòlits cel·lulars (Zhou et al., 2006; Semenza, 2012). S’ha demostrat que hi ha oncogens que activen la glicòlisi i in-dueixen el fenotip glicolític (Hsu i Sabatini, 2008). Així, l’oncogèn RAS pot in duir els transportadors de glucosa i la glicòlisi en cèl·lules transformades (Dang i Se-menza, 1999; Mazurek, 2011). Una altra de les dianes d’aquest oncogèn s’ha de-mostrat que és la 6-fosfofructo-1-cinasa (Kole et al., 1991) (figura 5). D’altra ban-da, és interessant ressaltar que la majoria dels enzims glicolítics contenen evolutivament conservades seqüències consens d’enllaç a Myc (E-boxes) en els seus promotors. L’oncogèn Myc s’uneix a aquests promotors i els activa en con-dicions de pressió parcial d’oxigen normals, cosa que indueix el fenotip glicolític (Dang et al., 2009). Myc coopera amb HIF per induir la transcripció de transpor-
Fe2+
HIF-β
HIF-α
Fumarat
Succinat
Degradacióproteosomal
2-cetoglutarat
Hipòxia
ROS (?)
OH OH
UbUb
Ub
pVHLpVHL
O2
PHD
Figura 4. Prolil hidroxilasa. Aquest enzim, en presència d’oxigen, hidroxila HIF-1α, que és ubi-qüitinitzat per la proteïna Von Hippel-Lindau i és degradat per via del proteasoma. En condicions d’hipòxia, HIF-1α s’acumula i es transloca al nucli, on s’hibrida amb la subunitat HIF-1β i reconeix la seqüència consens d’unió dels gens diana (HRE). PHD: prolil hidroxilasa.
021-EL DOLÇ FENOTIP.indd 14 09/01/13 10:03

15
Tau
la 1
Isoe
nzim
s que
con
figur
en e
l fen
otip
glic
olít
ic c
arac
terí
stic
de
les c
èl·lu
les t
umor
als
Enzi
mG
ens
Isoe
nzim
sEs
tat
olig
omèr
icIs
oenz
ims
en tu
mor
sC
arac
terí
stiq
ues
Reg
ulac
ió
Hex
ocin
asa
4H
K-1
, 2, 3
i 4
HK
-1,
HK
-2A
lta a
finita
t glu
cosa
Uni
ó m
embr
ana
mito
cond
rial
HIF
, Myc
i PI
3K
Fosf
oglu
cosa
isom
eras
a1
PGI-
A i
BPG
I-B
6-fo
sfof
ruct
o-1-
cina
sa3
PFK
-L, P
FK-M
, PFK
-BPF
K-L
Men
ys se
nsib
le a
inhi
bido
rsM
és se
nsib
le a
act
ivad
ors
HIF
, RA
S i S
RC
6-fo
sfof
ruct
o-2-
cina
sa4
PFK
FB1-
4PF
KFB
3
PFK
FB4
Alta
act
ivita
t cin
asa
Bai
xa a
ctiv
itat
cin
asa
HIF
, LPS
, ade
nosi
na, P
I3K
, in
sulin
a, E
GF,
pro
gest
eron
aH
IF
TIG
AR
1T
IGA
RT
IGA
RA
ctiv
itat
Fru
-2,6
-P2
bisf
osfa
tasa
p53
Ald
olas
a3
ALD
-A, B
i C
ALD
-A i
CH
IF
Tri
osa-
fosf
at is
omer
asa
1N
o té
isoe
nzim
sT
PIG
licer
alde
hid-
3-fo
sfat
desh
idro
gena
sa1
No
té is
oenz
ims
GA
PDH
HIF
Fosf
oglic
erat
cin
asa
2PG
K-1
i 2
PGK
-1H
IFFo
sfog
licer
at m
utas
a2
PGA
M-M
, B i
MB
PGA
M-B
Alta
afin
itat
3-P
GA
HIF
Enol
asa
3EN
O-1
(αα
), 2
(γγ
) i 3
(ββ
)EN
O-1
Act
ua ta
mbé
com
a r
ecep
tor
del p
lasm
inog
enH
IF
Piru
vat c
inas
a2
PK-R
, PK
-L, P
K-M
1 i
PK-M
2
PK-M
2D
imèr
ica
(bai
xa a
finit
at P
EP)
Fru-
1,6-
P 2 in
duei
x la
form
ate
tram
èric
a (a
lta a
finit
at P
EP)
HIF
Lact
at d
eshi
drog
enas
a3
LDH
-A, B
i C
LDH
-AA
lta a
finit
at p
iruv
atH
IF, M
yc
Tra
nspo
rtad
ors
Glu
cosa
4G
LUT
-1, 2
, 3, 4
GLU
T-1
i G
LUT
-3A
lta a
finit
at g
luco
saH
IF, R
AS,
AK
T
Mon
ocar
boxi
lats
4M
CT
1-14
MC
T1
i 4A
lta a
finit
at la
ctat
HIF
021-EL DOLÇ FENOTIP.indd 15 09/01/13 10:03

16
tadors de glucosa o gens glicolítics, com hexocinasa-2 (HK-2), PFK-1, enolasa-1 o lactat deshidrogenasa A. Igualment, pot induir enzims de la síntesi de purines i pirimidines, i generar addicció a la glutamina (Wise et al., 2008; Dang, 2010). Myc també indueix els transportadors de glutamina (ASCT2 i SLC7A25) i glutaminasa mitocondrial (GLS1), cosa que permet la incorporació de la glutamina, en forma de glutamat i α-cetoglutarat, com a intermediari en el cicle de Krebs. Així, s’ha demostrat que les cèl·lules que sobreexpressen Myc tenen addicció a la glutamina i que en la seva absència entren en apoptosi (Dang et al., 2009; Dang, 2010). Aquests treballs demostren que amb diferents senyals oncogènics es poden obte-nir canvis metabòlics semblants, i incrementar la glutaminasa i afavorir la trans-formació. Així, les cèl·lules poden, també, utilizar la glutamina com a substrat, que pot acabar produint lactat o bé incorporar-se a la síntesi de biomolècules com a nucleòtids, aminoàcids o àcids grassos. Contràriament, la proteïna p53 pot in-
Figura 5. Regulació del metabolisme en cèl·lules tumorals. Els factors de creixement, per via de PI3K i AKT, o bé la hipòxia activen HIF, que, al seu torn, indueix el fenotip glicolític. La proteïna p53 té efectes oposats a través de la inducció dels enzims TIGAR (inhibidor glicolític) i COX (acti-vador de la cadena respiratòria mitocondrial).
021-EL DOLÇ FENOTIP.indd 16 09/01/13 10:03

17
duir la glutaminasa de tipus hepàtic (GLS2), que redueix la formació de tumors. Aquest enzim promou el metabolisme de la glutamina, la respiració mitocon-drial, la síntesi de glutatió i la disminució de ROS (Lu et al., 2010). Els autors suggereixen que l’augment de la capacitat antioxidant, a causa de l’augment de glutatió, podria contribuir a la funció supressora tumoral de p53.
AKT (proteïna cinasa B) és un element cabdal en la transmissió de senyals a partir de factors de creixement i citocines. Promou el metabolisme anabòlic, cosa que activa vies de supervivència cel·lular i inhibeix les que condueixen a apoptosi. AKT, activada a través del fosfatidilinositol-trifosfat (PIP3) sintetitzat per la fosfa-tidilinositol-3-cinasa (PI3K), indueix HIF-1 i és una proteïna fonamental en la senyalització de les vies proliferatives i la inducció del fenotip glicolític. Ha estat implicada com un factor important en diferents tipus de càncer (Manning i Cant-ley, 2007). AKT indueix transportadors de glucosa i enzims glicolítics, com l’he-xocinasa, que s’uneix a la membrana dels mitocondris, de manera que té accés preferencial a l’ATP mitocondrial i, al mateix temps, evita l’efecte inhibitori de la glucosa-6-fosfat (Manning i Cantley, 2007). Contràriament, PTEN, gen supressor tumoral que apareix freqüentment mutat en tumors, catalitza la hidròlisi del fos-fatidilinositol-trifosfat i produeix la inhibició del senyal. Recentment, s’ha de-mostrat que la sobreexpressió de PTEN indueix resistència a la transformació oncogènica a través de la reducció dels gens de glutaminasa, GLUT i PFKFB3, de manera que disminueix la captació i oxidació de glucosa i glutamina, i augmenta la respiració mitocondrial (García-Cao et al., 2012).
6. la fructosa 2,6-bIsfosfat I la regulacIó de la glIcòlIsI
Tal com s’ha comentat anteriorment, en les cèl·lules tumorals es registra un augment de la transcripció dels gens codificants dels enzims glicolítics i transpor-tadors de glucosa. És important ressaltar que en aquest procés de canvi fenotípic s’activa l’expressió de gens que codifiquen isoenzims característics de les cèl·lules embrionàries i proliferants (taula 1).
El nostre grup de recerca ha estat estudiant durant els darrers anys els gens i els enzims responsables de la síntesi i la degradació d’un metabòlit regulador de la glicòlisi, la fructosa 2,6-bisfosfat (Fru-2,6-P2), descobert al laboratori del profes-sor Henry G. Hers a principis de la dècada dels vuitanta, durant l’estudi dels me-canismes d’acció del glucagó en la gluconeogènesi hepàtica. Aquest metabòlit és l’activador al·lostèric més potent de la 6-fosfofructo-1-cinasa, enzim regulador de la via (Hers i Van Schaftingen, 1982; Rider i Bartrons, 2010) (figura 5). Els canvis en la concentració d’aquest metabòlit regulen l’activitat de la PFK-1 i el flux gli-colític.
La PFK-1 catalitza la fosforilació de la fructosa-6-fosfat a fructosa-1,6-bisfosfat,
021-EL DOLÇ FENOTIP.indd 17 09/01/13 10:03

18
en presència d’ATP, que es converteix en ADP. Clàssicament, ha estat considerat un dels enzims reguladors de la glicòlisi. És una proteïna tetramèrica (homotetramèri-ca o heterotetramèrica) de 380 kDa amb subunitats generades per tres gens que codifiquen les subunitats M (muscle), L (liver) i B (brain). És un dels enzims més ben regulats i les seves característiques cinètiques i reguladores depenen de la seva composició a partir de les diferents subunitats. La PFK-1 és un enzim multimodulat que presenta regulació al·lostèrica per múltiples efectors i metabòlits de la via. Aquesta regulació la realitzen un seguit d’efectors negatius (citrat, glicerol-3-P i [H+]) i positius (com l’AMP), que reforcen l’acció dels substrats. L’ATP actua com un efector al·lostèric negatiu, mentre que la fructosa-6-P allibera l’enzim d’aquesta inhibició per l’ATP (Sols, 1981). La Fru-2,6-P2 exerceix una doble funció: augmen-ta l’afinitat de la PFK-1 per la Fru-6-P i allibera l’enzim de la inhibició per l’ATP. També augmenta sinèrgicament l’afinitat de la PFK-1 per l’AMP, efector al·lostèric positiu de l’enzim. L’AMP activa la PFK-1 i inhibeix la fructosa 1,6-bisfosfatasa (FBPasa-1), que catalitza el pas de Fru-1,6-P2 a Fru-6-P. Curiosament, la Fru-2,6-P2 inhibeix també la FBPasa-1 i té efectes sinèrgics amb l’AMP (Hers i Van Schaftin-gen, 1982; Rider i Bartrons, 2010). Així doncs, els canvis en la concentració d’aquest metabòlit regulen l’activitat de la PFK-1 en tots els teixits. Indirectament, regulen la intensitat i la direcció de la glicòlisi i la gluconeogènesi a través del control de la PFK-1 i la FBPasa-1. Des que es va descobrir al fetge, s’ha trobat en totes les espè cies eucariotes estudiades.
La forma majoritària de PFK-1 en les cèl·lules tumorals és la L (L4, liver). Té com a característiques principals: registra activitats més altes en tumors, és menys sensible a la inhibició per ATP i citrat, i és més sensible a l’activació per Fru-2,6-P2 i AMP (Colomer et al., 1987). A més, l’activitat de la PFK-1 s’incrementa en línies cel·lulars transformades i en cèl·lules tumorals, i es pot activar per sobreexpressió dels oncogens RAS i SRC (Bosca et al., 1985; Kole et al., 1991).
La concentració de Fru-2,6-P2 en cèl·lules tumorals és clarament superior que en les cèl·lules normals (Colomer et al., 1987; Riera et al., 2002). S’han dut a terme molts estudis per investigar el paper de la Fru-2,6-P2 en l’activació del flux glico-lític de cèl·lules proliferants, tumorals o tractades amb agents mitogènics, s’ha corroborat el paper de la Fru-2,6-P2 en la regulació del flux glicolític en diferents condicions experimentals en què s’estimula la proliferació cel·lular.
La concentració de Fru-2,6-P2 depèn de l’activitat d’un grup d’enzims bifuncio-nals anomenats 6-fosfofructo-2-cinasa / fructosa 2,6-bisfosfatasa (PFK-2/FBPasa-2), que catalitzen la síntesi i la degradació de Fru-2,6-P2 (Rider et al., 2004; Rider i Bar-trons, 2010). El balanç entre l’activitat 6-fosfofructo-2-cinasa (PFK-2), que sintetitza la Fru-2,6-P2 a partir de Fru-6-P i ATP, i l’activitat fructosa 2,6-bisfosfatasa, que hi-drolitza la Fru-2,6-P2 en Fru-6-P i fosfat inorgànic, és el que acaba determinant la concentració d’aquest metabòlit.
021-EL DOLÇ FENOTIP.indd 18 09/01/13 10:03

19
La PFK-2/FBPasa-2 és un enzim bifuncional homodimèric de 110 kDa amb subunitats de 55 kDa. Cada monòmer presenta les dues activitats (cinasa/bisfos-fatasa) en la mateixa cadena polipeptídica, i conté el domini cinasa a l’extrem N-terminal de la proteïna i el domini bisfosfatasa, al C-terminal (Okar et al., 2001; Rider i Bartrons, 2010). Els aminoàcids localitzats prop dels extrems N-terminals i C-terminals de la proteïna són residus fosforilables i, per tant, responsables de part de la seva regulació posttraduccional. Aquesta proteïna deriva de la fusió de dos gens que expressen un domini cinasa, evolutivament relacionat amb la família de proteïnes que enllacen mononucleòtids, com l’adenilat cinasa, i un do-mini bisfosfatasa, relacionat amb la família de les fosfoglicerat mutases i fosfatases àcides (Okar et al., 2001). La funció reguladora de la Fru-2,6-P2 en el control del metabolisme dels carbohidrats fa que la modulació de les dues activitats encarre-gades de sintetitzar-la i degradar-la hagin d’estar molt ben compenetrades per adaptar les concentracions de Fru-2,6-P2 a les necessitats de la cèl·lula. La concen-tració de Fru-2,6-P2 depèn de l’activitat relativa de les activitats cinasa/bisfosfata-
Figura 6. 6-fosfofructo-2-cinasa / fructosa-2,6-bisfosfatasa. Enzim amb dues subunitats de 55 kDa, cadascuna amb un domini que conté un centre actiu cinasa (PFK-2) i un altre bisfosfatasa (FBPasa-2). En l’isoenzim ubic PFKFB3 predomina l’activitat cinasa respecte de la bisfosfatasa (quocient C/B: 700), cosa que afavoreix altes concentracions de fructosa 2,6-bisfosfat (Fru-2,6-P2).
021-EL DOLÇ FENOTIP.indd 19 09/01/13 10:03

20
sa de la PFK-2/FBPasa-2, i la importància d’aquest metabòlit en el control del metabolisme dels carbohidrats no és igual en tots els teixits. Així, trobem que la relació entre la concentració de Fru-2,6-P2 i la regulació del flux glicolític i gluco-neogènic és ben evident al fetge. En canvi, en teixits com el múscul esquelètic, el testicle o el cervell, passa a tenir un paper permissiu, de manera que manté la PFK-1 en estat actiu, tot i la presència d’inhibidors al·lostèrics (ATP). En els teixits on la glicòlisi és la font principal d’energia (cervell), la funció prioritària d’aquest metabòlit és mantenir la via permanentment activa.
El grau de complexitat que suposa la regulació dels nivells de Fru-2,6-P2 a cada teixit i condició fisiològica explica el fet que hi hagi diferents isoenzims de la PFK-2/FBPasa-2 capaços d’adaptar-se a les diferents condicions en les diferents cèl·lules o teixits.
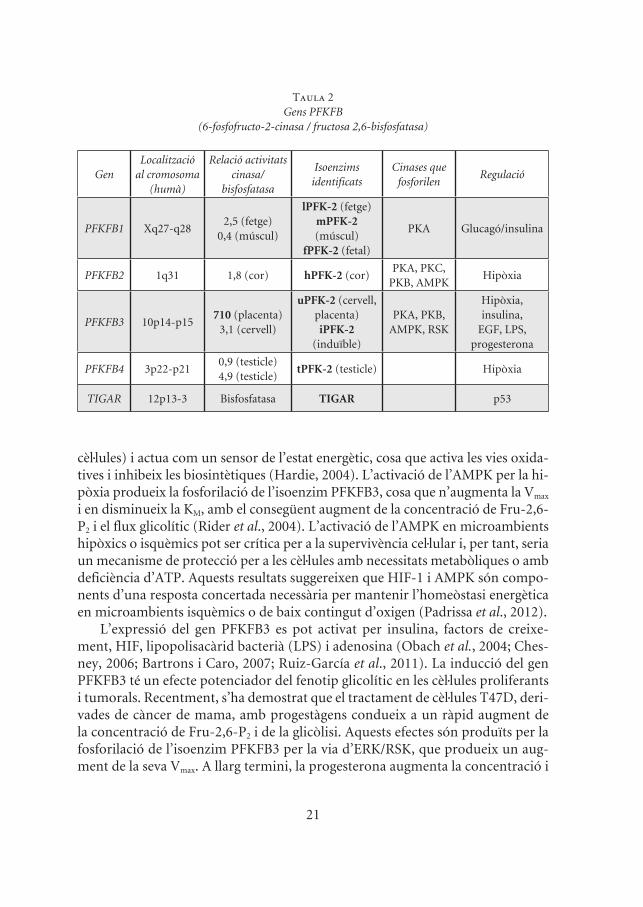
Des del descobriment de l’enzim PFK-2/FBPasa-2 del fetge, s’han descrit altres isoenzims localitzats originalment al múscul esquelètic, al cor, al cervell, al testicle, així com un isoenzim fetal, molt semblant a les isoformes de fetge i de múscul (Rider et al., 2004). Aquests isoenzims mostren diferències pel que fa a la distribució en les cèl·lules i els teixits, i les seves propietats cinètiques varien en resposta als efectors al·lostèrics, a les hormones i als factors de creixement (Okar et al., 2001). Els diferents isoenzims són el resultat de l’expressió de quatre gens (PFKFB1-PFKFB4) (Okar et al., 2001). El gen PFKFB1 codifica per les isoformes originalment identificades al teixit hepàtic i al muscular. El gen PFKFB2 codifica per l’isoenzim identificat al cor i al ronyó. El gen PFKFB3 codifica pels isoenzims del cervell, la placenta i el teixit adipós, i és el més abundant a les cèl·lules prolife-rants i canceroses. I per acabar, el gen PFKFB4 codifica per l’isoenzim del testicle, tot i que també s’ha detectat en diverses línies tumorals (Minchenko et al., 2005) (taula 2). L’expressió d’aquests gens depèn del tipus cel·lular, del teixit i de la fase de desenvolupament (Goren et al., 2000). Aquest patró d’expressió suggereix que cada isoenzim desenvolupa una funció específica en diferents condicions fisiolò-giques i en resposta a diferents estímuls.
El gen més abundant a les cèl·lules canceroses és el PFKFB3, que es troba, tam-bé, en totes les cèl·lules proliferants (Hamilton et al., 1997; Chesney et al., 1999; Riera et al., 2002; Peña-Rico et al., 2011). Es va clonar a partir d’una llibreria de cDNA de cervell fetal i es va veure que s’expressava en cèl·lules de tots els teixits estudiats (Manzano et al., 1998). Aquest gen expressa un isoenzim ubic que regis-tra una alta activitat cinasa i que permet explicar l’alta concentració de Fru-2,6-P2 en les cèl·lules proliferants i tumorals (Bartrons i Caro, 2007).
L’isoenzim ubic PFKFB3 conté un centre de fosforilació per la cinasa que de-pèn d’AMP (AMPK) (Rider et al., 2004). L’AMPK és un regulador metabòlic que es troba en totes les cèl·lules eucariotes. S’activa quan augmenta la relació AMP/ATP (com passa durant la hipòxia o durant la falta d’aportació de nutrients a les
021-EL DOLÇ FENOTIP.indd 20 09/01/13 10:03
21
cèl·lules) i actua com un sensor de l’estat energètic, cosa que activa les vies oxida-tives i inhibeix les biosintètiques (Hardie, 2004). L’activació de l’AMPK per la hi-pòxia produeix la fosforilació de l’isoenzim PFKFB3, cosa que n’augmenta la Vmax i en disminueix la KM, amb el consegüent augment de la concentració de Fru-2,6-P2 i el flux glicolític (Rider et al., 2004). L’activació de l’AMPK en microambients hipòxics o isquèmics pot ser crítica per a la supervivència cel·lular i, per tant, seria un mecanisme de protecció per a les cèl·lules amb necessitats metabòliques o amb deficiència d’ATP. Aquests resultats suggereixen que HIF-1 i AMPK són compo-nents d’una resposta concertada necessària per mantenir l’homeòstasi energètica en microambients isquèmics o de baix contingut d’oxigen (Padrissa et al., 2012).
L’expressió del gen PFKFB3 es pot activat per insulina, factors de creixe-ment, HIF, lipopolisacàrid bacterià (LPS) i adenosina (Obach et al., 2004; Ches-ney, 2006; Bartrons i Caro, 2007; Ruiz-García et al., 2011). La inducció del gen PFKFB3 té un efecte potenciador del fenotip glicolític en les cèl·lules proliferants i tumorals. Recentment, s’ha demostrat que el tractament de cèl·lules T47D, deri-vades de càncer de mama, amb progestàgens condueix a un ràpid augment de la concentració de Fru-2,6-P2 i de la glicòlisi. Aquests efectes són produïts per la fosforilació de l’isoenzim PFKFB3 per la via d’ERK/RSK, que produeix un aug-ment de la seva Vmax. A llarg termini, la progesterona augmenta la concentració i
Taula 2Gens PFKFB
(6-fosfofructo-2-cinasa / fructosa 2,6-bisfosfatasa)
GenLocalització
al cromosoma (humà)
Relació activitats cinasa/
bisfosfatasa
Isoenzims identificats
Cinases que fosforilen
Regulació
PFKFB1 Xq27-q282,5 (fetge)
0,4 (múscul)
lPFK-2 (fetge)mPFK-2 (múscul)
fPFK-2 (fetal)
PKA Glucagó/insulina
PFKFB2 1q31 1,8 (cor) hPFK-2 (cor)PKA, PKC,
PKB, AMPKHipòxia
PFKFB3 10p14-p15710 (placenta)
3,1 (cervell)
uPFK-2 (cervell, placenta)iPFK-2
(induïble)
PKA, PKB, AMPK, RSK
Hipòxia, insulina,
EGF, LPS, progesterona
PFKFB4 3p22-p210,9 (testicle)4,9 (testicle)
tPFK-2 (testicle) Hipòxia
TIGAR 12p13-3 Bisfosfatasa TIGAR p53
021-EL DOLÇ FENOTIP.indd 21 09/01/13 10:03

22
l’activitat enzimàtica de la PFKFB3 a través de la inducció transcripcional del gen PFKFB3, com a conseqüència de la unió de la progesterona i el receptor a les seqüències diana localitzades en el seu promotor. Aquest mecanisme, que depèn de la progesterona, assegura el fenotip glicolític a les cèl·lules de càncer de mama (Novellasdemunt et al., 2012).
La regulació de l’isoenzim ubic PFKFB3 no només es porta a terme a través de mecanismes de fosforilació i regulació de la transcripció. El nostre grup ha de-mostrat que és també regulable a través del sistema ubiqüitina-proteasoma. Aquest procés de degradació de proteïnes requereix dos passos successius: la con-jugació de múltiples molècules d’ubiqüitina a la proteïna i la degradació d’aques-ta pel proteasoma 26S (Riera et al., 2003). L’isoenzim PFKFB3 conté una caixa KEN que és substrat d’ubiqüitinització pel complex E3 ubiqüitina lligasa promo-tora de l’anafase (APC/C), activada per Cdh1, de manera semblant a altres pro-teïnes del cicle cel·lular. Els resultats publicats indiquen que la resposta prolifera-tiva depèn de la disminució de l’activitat APC/C-Cdh1 que activa la proliferació i la glicòlisi (Almeida et al., 2010). La silenciació del gen que expressa l’isoenzim PFKFB3, que normalment apareix a mitjan fase G1, impedeix la progressió en el cicle i demostra que és essencial per a la divisió cel·lular. Així, aquest isoenzim PFKFB3 està estretament controlat, cosa que assegura la regulació de la gli còlisi en un punt específic de la fase G1 i suggereix que aquesta regulació representa un punt de restricció, sensible a nutrients, en les cèl·lules dels mamífers (Tudzarova et al., 2011). En aquest mateix sentit, s’ha demostrat que l’isoenzim PFKFB3 està incrementat en les cèl·lules proliferants (Durán et al., 2009) i es pot localitzar al nucli cel·lular, on regula la proliferació a través de cinases dependents de ciclines (Yalcin et al., 2009). L’augment de l’activitat de la PFKFB3 i la glicòlisi estaria, així, coordinada amb la proliferació cel·lular, per assegurar el subministrament d’energia i substrats biosintètics.
Un altre gen important en la regulació del fenotip glicolític és el gen supressor tumoral TP53, responsable de la síntesi de la proteïna p53. En resposta a l’estrès cel·lular, a l’activació de determinats oncogens o al dany en el DNA, la proteïna p53 s’estabilitza i activa l’expressió de nombrosos gens. Aquests gens diana in dueixen una gran varietat de respostes cel·lulars, incloent-hi l’aturada del cicle cel·lular, la reparació del DNA, la senescència o l’apoptosi (Vousden i Prives, 2009). Un dels gens induïts per la proteïna p53 el van clonar i el van caracteritzar, l’any 2006, la doctora Karen Vousden i el nostre grup, i es va anomenar TIGAR (TP53-induced glycolysis and apoptosis regulator) (Bensaad et al., 2006).
TIGAR és un nou isoenzim de la família de gens PFKFB que comparteix si-milituds estructurals i funcionals amb els isoenzims PFK-2/FBPasa-2, amb una particularitat important: conté una seqüència semblant a la del domini bisfos-fatasa de l’enzim bifuncional, però no té el domini cinasa (taula 2). Així, la
021-EL DOLÇ FENOTIP.indd 22 09/01/13 10:03

23
proteïna TIGAR, amb l’activitat Fru-2,6-bisfosfatasa, disminueix la concentra-ció de Fru-2,6-P2 i frena la glicòlisi a nivell de la PFK-1, de manera que rediri-geix l’oxidació de glucosa cap a la via de les pentoses fosfat, fonamental per produir NADPH, energia en forma de poder reductor (figura 5). Una conse-qüència de la derivació de l’oxidació de glucosa cap a la via de les pentoses-fosfat i l’augment consecutiu de generació de NADPH és l’augment del glutatió reduït (GSH), substrat reductor necessari per a la reconversió de les espècies reactives de l’oxigen. L’expressió del gen TIGAR protegeix les cèl·lules davant els ROS i l’apoptosi in duïda pels danys oxidatius en el DNA. D’aquesta manera, la inducció de TIGAR, a través de p53, pot aportar estabilitat genòmica en dismi-nuir l’oxidació del DNA (Vousden i Prives, 2009; Mor i Vousden, 2011). La funció del gen TIGAR serviria per desviar l’oxidació de glucosa cap a la via de les pentoses, on, a més de sintetitzar NADPH, pot dirigir l’esquelet carbonat de la glucosa cap a la síntesi de nu cleòtids i altres productes que són essencials per a la biosíntesi i la reparació de lesions del DNA i per a la proliferació cel-lular. D’altra banda, Matoba i col·laboradors (Matoba et al., 2006) han demos-
Figura 7. TIGAR (TP53-induced glycolysis and apoptosis regulator). L’enzim TIGAR és una pro-teïna de 29 kDa amb un centre actiu que conté activitat fosfatasa que hidrolitza la fructosa 2,6-bis-fosfat a fructosa-6-fosfat i pi, cosa que disminueix les concentracions d’aquest metabòlit.
021-EL DOLÇ FENOTIP.indd 23 09/01/13 10:03

24
trat que la proteïna p53 indueix també el gen SCO2 (citocrom C-oxidasa-2), que participa en el complex de la citocrom C-oxidasa (COX) mitocondrial, de mane-ra que catalitza la transferència d’electrons del citocrom C a l’oxigen molecular i bombeja protons a través de la membrana mitocondrial interna. El bloqueig del gen SCO2 en cèl·lules recondueix cap al fenotip glicolític. Les cèl·lules que no te-nen p53 o que tenen p53 mutada, no funcional, registren un baix consum d’oxi-gen, cosa que es pot restaurar activant l’expressió de SCO2. En condicions en què el consum d’oxigen mitocondrial és limitant, la generació de ROS pot augmentar, de manera que activa TP53 i TIGAR, i frena la glicòlisi, de manera que disminueix l’oxidació de piruvat mitocondrial i la transferència d’electrons a la cadena respi-ratòria mitocondrial. Durant l’oncogènesi, aquests passos queden alterats, ja que en les cèl·lules transformades solen acumular-se defectes en la via de p53, i això fa que no s’indueixin TIGAR i COX, i afavoreix el fenotip glicolític.
Molts altres isoenzims, com l’hexocinasa-2, el piruvat cinasa M2 o el lactat deshidrogenasa A, tenen també papers importants en el desenvolupament del fe-notip glicolític (Mathupala et al., 2009; Mazurek, 2011; Le et al., 2010).
7. avantatges del fenotIp glIcolítIc
El fenotip glicolític, característic de les cèl·lules tumorals, té avantatges selec-tius pel que fa a la proliferació i la supervivència de les cèl·lules, cosa que explica per què aquesta estratègia metabòlica es detecta en la majoria de tumors. Els efec-tes d’aquest fenotip són múltiples i probablement engloben avantatges evolutius que es troben en altres cèl·lules proliferants, com les embrionàries. Podem dir que hi ha diferents raons per les quals el fenotip glicolític de les cèl·lules tumorals re-sulta avantatjós per al desenvolupament del càncer. El fenotip glicolític dóna a la cèl·lula la capacitat de captar quantitats de glucosa un ordre de magnitud superior respecte de la cèl·lula normal, a causa de l’augment de transportadors de glucosa. A més, la sobreexpressió d’enzims glicolítics permet obtenir un alt flux d’oxidació de glucosa a lactat i generar metabòlits fosforilats, cosa que assegura concen-tracions d’ATP i intermediaris biosintètics compatibles amb les exigències de la ràpida proliferació de les cèl·lules tumorals (Bartrons i Caro, 2007; Smallbone et al., 2007). Aquest fenotip glicolític es caracteritza per un alt flux però amb baix rendiment, ja que la producció d’ATP és molt més baixa que en condicions d’oxi-dació de glucosa a CO2, i utilitza la respiració mitocondrial, limitada en aquest fenotip. La condició necessària per mantenir aquest alt flux és que hi hagi prou aportació de glucosa i que les cèl·lules puguin desprendre lactat. Per això és molt important l’activitat dels transportadors d’aquest metabòlit (MCT1 i MCT4). En-cara que el rendiment en la producció d’ATP per la glicòlisi és baix, si el flux gli-colític és prou alt, el percentatge d’ATP produït per la glicòlisi pot arribar a supe-
021-EL DOLÇ FENOTIP.indd 24 09/01/13 10:03

25
rar el de la respiració mitocondrial (DeBerardinis et al., 2008). La producció glicolítica d’ATP té lloc a una velocitat 100 vegades superior respecte de l’oxidació mitocondrial. La glicòlisi és tan ràpida que supera la velocitat d’oxidació del piru-vat, de manera que les cèl·lules han d’eliminar aquest piruvat usant una altra via que tingui també un alt flux, la conversió en lactat per la LDH-A. La LDH-A s’in-dueix per oncogens i estímuls mitogènics, a part que permet regenerar NAD+ i que el seu producte (lactat) és fàcilment exportat pels transportadors MCT (De-Berardinis et al., 2008; Draoui i Feron, 2011). A més, la simbiosi entre els diferents tipus de cèl·lules tumorals i de l’estroma, en secretar lactat (MCT4) o utilitzar-lo (MCT1), permetria un rendiment òptim en les condicions relatives d’hipòxia tu-moral (Draoui i Feron, 2011). D’altra banda, el gradient en la concentració de lactat pot afectar, també, les cèl·lules dels teixits veïns. S’ha demostrat que l’expo-sició perllongada de les cèl·lules normals a un microambient àcid in dueix apop-tosi (mort cel·lular programada), a través de mecanismes dependents de p53 i caspasa-3, contràriament al que succeeix en les cèl·lules tumorals (Gatenby i Gi-llies, 2004). L’acidificació del microambient en el teixit a l’entorn del tumor pro-porcionaria un avantatge de creixement selectiu de les cèl·lules que han perdut la funció del gen TP53, cosa que dóna lloc a l’expansió clonal de pobla cions de cèl-lules aberrants. A més, l’associació de l’hexocinasa-2 als mitocondris, induïda per l’AKT, és un dels mecanismes que protegeix les cèl·lules de l’apoptosi (Mathupala et al., 2009). Aquesta estratègia es considera que proporciona un avantatge de creixement a la cèl·lula tumoral, tot i que la fa en gran mesura dependent de la disponibilitat de glucosa per a la seva supervivència.
Altres beneficis potencials són que alguns metabòlits de la glicòlisi (piruvat i lactat) poden inhibir les prolil hidroxilases i, al seu torn, activar HIF, cosa que indueix el fenotip glicolític i estimula l’angiogènesi i la invasivitat (López-Láza-ro, 2006). El canvi a fenotip glicolític no és absolutament necessari per a la trans-formació però aporta a les cèl·lules autonomia metabòlica, tolerància a l’acidosi i un major potencial proliferatiu i invasiu.
A més d’ATP i nucleòtids, la cèl·lula proliferant necessita macromolècules, com àcids grassos, lípids i proteïnes, i la reprogramació metabòlica serveix per proveir aquestes necessitats (Hsu i Sabatini, 2008; Mazurek, 2011). Per aconse-guir-ho, la cèl·lula capta del medi dos nutrients abundants, glucosa i glutamina, i els transforma en energia i precursors biosintètics (DeBerardinis et al., 2008). En concret, la glucosa contribueix a la generació de metabòlits com dihidroxiacetona fosfat (DHAP) per a la síntesi de triacilglicèrids i fosfolípids; 3-fosfoglicerat per a la síntesi de serina, cisteïna i glicina, o piruvat per a la síntesi de malat i alanina; o bé riboses-5-P, a través de la via de les pentoses (DeBerardinis et al., 2008). La utilització de trioses per a biosíntesi es veu afavorida per la presència en les cèl-lules tumorals de l’isoenzim de la piruvat cinasa específica de tumors (PKM2).
021-EL DOLÇ FENOTIP.indd 25 09/01/13 10:03

26
Aquest isoenzim té disminuïda l’afinitat pel substrat (PEP), la qual cosa afavoreix que els intermediaris glicolítics per sobre del PEP es dirigeixin cap a processos biosintètics (Vander Heiden et al., 2009; Mazurek, 2011). Recentment, s’ha de-mostrat que en alguns tipus cel·lulars de càncer una quantitat relativament gran dels intermediaris glicolítics es desvia cap al metabolisme de la serina i la glicina a través de la fosfoglicerat deshidrogenasa (PHGDH). Aquest enzim està amplificat en una proporció alta de càncers, i és freqüent en el melanoma. La disminució de l’expressió de la PHGDH impedeix la proliferació, mentre que la sobreexpressió predisposa a la transformació i s’associa amb alguns subtipus de càncer de mama (Locasale et al., 2011). Aquests resultats mostren que la desviació del flux glicolític cap a una via alternativa específica pot afavorir el desenvolupament tumoral. En aquest escenari, les cèl·lules tumorals poden també redirigir l’excés de piruvat gli-colític cap a la síntesi de novo d’àcids grassos, necessaris per a la biosíntesi de membranes i altres components. De fet, en els anys 1950, mitjançant el marcatge amb 14C-glucosa, es va demostrar, en models de tumors, que pràcticament tots els àcids grassos esterificats derivaven de la síntesi de novo. En les cèl·lules tumo-rals és característic trobar-hi augments de l’activitat i l’expressió de diversos en-zims lipogènics. Per exemple, l’augment de l’àcid gras sintasa (FAS) representa una de les alteracions fenotípiques més freqüents en càncer i s’ha relacionat amb l’alt risc de recurrència i mort. La inducció d’aquests enzims està relaciona-da amb factors de creixement, insulina, progestàgens o oncogens (Menéndez i Lupu, 2007).
Les reaccions de biosíntesi expliquen l’ús incrementat de glucosa en les cèl-lules tumorals. És evident que aquestes cèl·lules tenen un alt cost energètic i utilit-zen altes concentracions d’intermediaris glicolítics i també del cicle de Krebs per a processos biosintètics. Aquesta situació metabòlica no ajuda a explicar l’incre-ment de la producció de lactat que va observar originàriament Warburg, ni el manteniment de l’activitat del cicle de Krebs. La glutamina, segon substrat de les cèl·lules tumorals, apareix també com un actor principal, ja que facilita l’aporta-ció de nitrogen per a la biosíntesi d’aminoàcids i nucleòtids (Mazurek, 2011; Vander Heiden et al., 2009). La glutamina és l’aminoàcid més abundant en el plasma. Les cèl·lules proliferants poden metabolitzar la glutamina en un procés anomenat glutaminòlisi, que utilitza diversos passos del cicle de Krebs, produeix NAD+, NADPH i α-ceto-glutarat, i reomple els intermediaris del cicle de Krebs (anaplerosi). Finalment, pot convertir-se en lactat en el citosol, o bé en àcids gras-sos, aminoàcids, nucleòtids o glutatió (DeBerardinis et al., 2008). La metabolit-zació de la glutamina s’inicia amb la glutaminasa, que la converteix en glutamat i amoni. L’amoni pot actuar com un inductor d’autofàgia, que allibera més glu-tamina i, al seu torn, el seu metabolisme estimula l’autofàgia (Martínez-Out-schoorn et al., 2011). Hi ha dos isoenzims de glutaminasa (GLS1 i GLS2) i llur
021-EL DOLÇ FENOTIP.indd 26 09/01/13 10:03

27
expressió té efectes oposats en la proliferació cel·lular. L’isoenzim GLS1 és activat per l’oncogèn c-Myc i promou l’entrada de la glutamina en el cicle de Krebs i l’addicció de les cèl·lules a la glutamina. Si s’inhibeix la GLS1, s’alenteix el creixe-ment de les cèl·lules canceroses (Martínez-Outschoorn et al., 2011; Dang, 2010). En canvi, l’isoenzim GLS2 és induït per p53, en resposta a estrès oxidatiu i dany al DNA, i regula la síntesi de glutatió i l’estat redox cel·lular. La sobreexpressió de GLS2 redueix el creixement de tumors (Suzuki et al., 2010).
En cèl·lules normals, l’estimulació mitogènica condueix a un pic d’expressió de c-Myc en la fase G1 que facilita l’entrada a la fase S (DeBerardinis et al., 2008). Entre les dianes de c-Myc hi ha ciclines i CDK, i enzims implicats en el metabolis-me de nucleòtids. També, diversos enzims glicolítics, com la LDH-A (Dang et al., 2009). S’ha vist que les cèl·lules transformades amb c-Myc tenen una necessitat absoluta de glutamina per a la biosíntesi de biomolècules (Dang, 2010).
En resum, podem dir que hi ha molts tipus de cèl·lules tumorals i que, fins i tot dins un mateix tumor, apareixen cèl·lules amb variacions metabòliques específi-ques (Martínez-Outschoorn et al., 2011). La potenciació de la glicòlisi no implica necessàriament que la respiració mitocondrial estigui bloquejada (Chance, 2005). Poden coexistir ambdues vies, ja sigui per a la producció preferent d’ATP o per proveir de substrats biosintètics la cèl·lula. Tot i així, mentre que moltes cèl·lules tumorals conserven la capacitat d’utilitzar la cadena respiratòria i la fosforilació oxidativa mitocondrial, quan la via glicolítica és suprimida es frena el creixement (Tennant et al., 2010). La limitació de la respiració protegeix les cèl·lules del dany oxidatiu mitocondrial que es produeix quan la respiració cel·lular funciona anor-malment en condicions hipòxiques (Dang, 2010).
En aquests darrers anys s’ha anat fent evident el paper del microambient tu-moral en la progressió del fenotip glicolític. Les cèl·lules canceroses no existeixen com a poblacions homogènies pures. Estan integrades en colònies envoltades per les cèl·lules de l’estroma. Per tant, és raonable sospitar que aquestes influeixen en el metabolisme de les cèl·lules canceroses adjacents. De fet, quan les cèl·lules can-ceroses són cocultivades amb fibroblasts, les cèl·lules canceroses augmenten la massa mitocondrial, mentre que els fibroblasts perden els mitocondris. L’anàlisi d’aquest fenomen revela que les cèl·lules agressives del càncer es comporten de manera semblant als paràsits intracel·lulars, de manera que utilitzen l’estrès oxi-datiu com a mecanisme per usurpar els nutrients de les cèl·lules de l’estroma cir-cumdant. L’estrès oxidatiu indueix HIF, NFkB i altres factors que activen l’auto-fàgia d’organel·les i mitocondris (mitofàgia) de les cèl·lules de l’estroma, i recluten macròfags, cosa que produeix citocines i inflamació. Com a conseqüència dels ROS i factors com HIF i NFkB s’indueix, també, el fenotip glicolític amb el resul-tat de sobreproduir lactat, glutamina i cossos cetònics, que són utilitzats per les cèl·lules canceroses (Martínez-Outschoorn et al., 2011). Aquest mecanisme per-
021-EL DOLÇ FENOTIP.indd 27 09/01/13 10:03

28
met a les cèl·lules canceroses metastatitzar en qualsevol lloc, sense necessitat d’an-giogènesi inicial, ja que pot induir estrès oxidatiu i parasitar de nou les cèl·lules de l’estroma. Aquesta hipòtesi, anomenada efecte Warburg invertit, indueix, per via de ROS i citocines inflamatòries, el catabolisme de les cèl·lules de l’estroma, a tra-vés de l’autofàgia, i això facilita nutrients i fomenta el creixement anabòlic de les cèl·lules tumorals, la promoció de la progressió del tumor i la metàstasi (Pavlides et al., 2009; Martínez-Outschoorn et al., 2011). D’acord amb aquestes dades, no ens ha de sorprendre que algunes drogues antiparasitàries i inhibidores d’autofà-gia, com la cloroquina, siguin també potents agents antitumorals.
8. ImplIcacIons per al tractament
La dependència que les cèl·lules canceroses mostren respecte del consum de glucosa ha permès dissenyar diferents propostes terapèutiques. Des del punt de vis-ta farmacològic, les drogues capaces de pertorbar el metabolisme, específicament la glicòlisi, han esdevingut cada vegada més interessants. En primer lloc, s’ha de-mostrat que la sobreexpressió de HIF-1, present en molt tumors, genera resistèn-cia a la quimioteràpia i a la radiació (Semenza, 2002; Semenza, 2012). La inhibició de l’activitat de HIF-1, per tant, pot ser un component important en la teràpia contra el càncer. Les cèl·lules transformades poden ser particularment sensibles a la inhibició del flux glicolític quan hi ha limitacions d’oxigen i glucosa. Això s’ha demostrat en experiments que empraven inhibidors de la glicòlisi o bé que inhi-beixen l’expressió de diferents enzims glicolítics (Tennant et al., 2010).
Un dels inhibidors més estudiats, és la 2-desoxi-D-glucosa (2-DG), molècula de glucosa en la qual el grup 2-hidroxil ha estat substituït per hidrogen. L’hexoci-nasa pot fosforilar aquesta hexosa a 2-DG-6-fosfat, que no pot ser metabolitzada per la fosfohexosa-isomerasa. Per tant, la seva acumulació intracel·lular produeix una inhibició competitiva de les hexocinases. Els estudis experimentals han de-mostrat clarament que la 2-DG té efectes citotòxics en cèl·lules de diferents tipus de càncer, especialment els que sobreexpressen HIF i tenen defectes en el metabo-lisme mitocondrial (Pelicano et al., 2006). D’altra banda, la 2-DG augmenta sig-nificativament la resposta al tractament amb adriamicina i paclitaxel en ratolins portadors d’osteosarcoma humà, així com el de cèl·lules petites de càncer de pul-mó (Pelicano et al., 2006).
L’àcid bromopirúvic o bromopiruvat (3-BrPA) és un potent derivat haloge-nat de l’àcid pirúvic. Pot inhibir diferents enzims de la glicòlisi (hexocinasa, gli-ceraldehid-3-fosfat deshidrogenasa) (Pelicano et al., 2006). Un estudi experimen-tal en cèl·lules de leucèmia de limfoma humà i cèl·lules humanes de càncer de còlon ha mostrat que el tractament amb 3-BrPA inhibeix la glicòlisi i activa l’apoptosi (Scatena et al., 2008).
021-EL DOLÇ FENOTIP.indd 28 09/01/13 10:03

29
També, s’ha demostrat que bloquejant l’expressió del gen PFKFB3 es dismi-nueix la glicòlisi, la proliferació cel·lular i el creixement independent d’ancoratge (Calvo et al., 2006). El coneixement de l’estructura cristal·lina de la proteïna PFKFB3 ha permès desenvolupar molècules terapèutiques amb una alta especifi-citat, i s’ha demostrat que un inhibidor de la proteïna PFKFB3 suprimeix el flux glicolític i té un efecte citostàtic per a les cèl·lules neoplàstiques (Chesney, 2006). El fet que el gen PFKFB3 estigui sobreexpressat en diferents tumors i s’activi per hipòxia i/o oncogens indica que té un paper essencial en el desenvolupament del fenotip glicolític, cosa que facilita l’adaptació i la supervivència de les cèl·lules tu-morals en microambients hipòxics. També, recentment (Peña-Rico et al., 2011), el nostre grup ha demostrat que la silenciació del gen TIGAR fa que les cèl·lules de glioblastoma siguin més sensibles a la radioteràpia.
La capacitat de modular selectivament el metabolisme de les cèl·lules cance-roses és un tema amb una alta potencialitat terapèutica. Les característiques de les cèl·lules tumorals que contenen oncogens actius (per exemple, RAS, HER2, AKT) i/o defectes en supressors tumorals (per exemple, TSC1/2, PTEN, LKB1, p53) fan que aquestes cèl·lules entrin en apoptosi en condicions de limitació d’oxidació de glucosa. Curiosament, aquestes mateixes cèl·lules sovint mostren resistència a altres formes d’estímuls apoptòtics (radiació, quimioteràpia). L’ús d’inhibidors de la glicòlisi sensibilitza les cèl·lules davant aquests estímuls i en facilita la mort.
Finalment, és evident que la glicòlisi de les cèl·lules tumorals és un fenomen complex en el qual aquesta i altres vies metabòliques es reprogramen per tal de millorar l’obtenció d’energia i la síntesi de biomolècules, necessàries per a la pro-liferació cel·lular. La comprensió de la regulació dels gens i els isoenzims glicolítics ha de tenir, ben segur, implicacions en el diagnòstic, el pronòstic i el desenvolu-pament de teràpies més selectives contra el càncer.
agraïments
A Àurea Navarro, Anna Manzano i Laura Novellasdemunt pels valuosos co-mentaris i per la discussió del treball. Al doctor B. Torrejón i a la doctora Esther Castaño dels Centres Científics i Tecnològics de la Universitat de Barcelona (CCiTUB) per la cessió de la fotografia de la portada. El treball experimental ha rebut el suport del Ministeri de Ciència i Tecnologia (BMC 2003/01442), el Mi-nisteri d’Educació i Ciència (BFU2006-02412/BMC), el Ministeri de Ciència i Innovació (BFU2009-07380) i la Generalitat de Catalunya (2005SGR-022 i 2009 SGR 1059).
021-EL DOLÇ FENOTIP.indd 29 09/01/13 10:03

30
referèncIes
Almeida, A.; Bolaños, J. P.; Moncada, S. «E3 ubiquitin ligase APC/C-Cdh1 accounts for the Warburg effect by linking glycolysis to cell proliferation». Proc. Natl. Acad. Sci. USA, vol. 107 (2010), p. 738-741.
Bartrons, R.; Caro, J. «Hypoxia, glucose metabolism and the Warburg’s effect». J. Bio-energ. Biomembr., vol. 39 (2007), p. 223-229.
Bensaad, K.; Tsuruta, A.; Selak, M. A.; Vidal, M. N.; Nakano, K.; Bartrons, R.; Gottlieb, E.; Vousden, K. H. «TIGAR, a p53-inducible regulator of glycolysis and apoptosis». Cell, vol. 126 (2006), p. 107-120.
Bosca, L.; Rousseau, G. G.; Hue, L. «Phorbol 12-myristate 13-acetate and insulin in-crease the concentration of fructose 2,6-bisphosphate and stimulate glycolysis in chicken embryo fibroblasts». Proc. Natl. Acad. Sci. USA, vol. 82 (1985), p. 6440-6444.
Calvo, M. N.; Bartrons, R.; Castaño, E.; Perales, J. C.; Navarro-Sabaté, A.; Man-zano, A. «PFKFB3 gene silencing decreases glycolysis, induces cell-cycle delay and inhibits anchorage-independent growth in HeLa cells». FEBS Lett., vol. 580 (2006), p. 3308-3314.
Chance, B. «Was Warburg right? Or was it that simple?». Cancer Biol. Ther., vol. 4 (2005), p. 125-126.
Chesney, J. «6-phosphofructo-2-kinase / fructose-2,6-bisphosphatase and tumor cell glycolysis». Curr. Opin. Clin. Nutr. Metab. Care, vol. 9 (2006), p. 535-539.
Chesney, J.; Mitchell, R.; Benigni, F.; Bacher, M.; Spiegel, L.; Al-Abed, Y.; Metz, C.; Bucala, R. «An inducible gene product for 6-phosphofructo-2-kinase with an AU-rich instability element: role in tumor cell glycolysis and the Warburg effect». Proc. Natl. Acad. Sci. USA, vol. 96 (1999), p. 3047-3052.
Colomer, D.; Vives-Corrons, J. L.; Pujades, A.; Bartrons, R. «Control of phospho-fructokinase by fructose 2,6-bisphosphate in B-lymphocytes and B-chronic lympho-cytic leukemia cells». Cancer Res., vol. 47 (1987), p. 1859-1862.
Crabtree, H. G. «Observations on the carbohydrate metabolism of tumours». Biochem. J., vol. 23 (1929), p. 536-545.
Cuezva, J. M.; Krajewska, M.; Heredia, M. L. de; Krajewski, S.; Santamaria, G.; Kim, H.; Zapata, J. M.; Marusawa, H.; Chamorro, M.; Reed, J. C. «The bioener-getic signature of cancer: a marker of tumor progression». Cancer Res., vol. 62 (2002), p. 6674-6681.
Dang, C. V. «Rethinking the Warburg effect with Myc micromanaging glutamine me-tabolism». Cancer Res., vol. 70 (2010), p. 859-862.
Dang, C. V.; Le, A.; Gao, P. «Myc-induced cancer cell energy metabolism and therapeu-tic opportunities». Clin. Cancer Res., vol. 15 (2009), p. 6479-6483.
Dang, C. V.; Semenza, G. L. «Oncogenic alterations of metabolism». Trends Biochem. Sci., vol. 24 (1999), p. 68-72.
DeBerardinis, R. J.; Lum, J. J.; Hatzivassiliou, G.; Thompson, C. B. «The biology of cancer: metabolic reprogramming fuels cell growth and proliferation». Cell Metab., vol. 7 (2008), p. 11-20.
Denko, N. C. «Hypoxia, HIF1 and glucose metabolism in the solid tumour». Nat. Rev. Cancer, vol. 8 (2008), p. 705-713.
021-EL DOLÇ FENOTIP.indd 30 09/01/13 10:03

31
Draoui, N.; Feron, O. «Lactate shuttles at a glance: from physiological paradigms to anti-cancer treatments». Dis. Model Mech., vol. 4 (2011), p. 727-732.
Durán, J.; Obach, M.; Navarro-Sabaté, A.; Manzano, A.; Gómez, M.; Rosa, J. L.; Ventura, F.; Perales, J. C.; Bartrons, R. «PFKFB3 is transcriptionally upregulated in diabetic mouse liver through proliferative signals». FEBS J., vol. 276 (2009), p. 4555-4568.
Esteban, M. A.; Maxwell, P. H. «HIF, a missing link between metabolism and cancer». Nat. Med., vol. 11 (2005), p. 1047-1048.
García-Cao, I.; Song, M.; Hobbs, R. M.; Laurent, G.; Giorgi, C.; Boer, V. C. J. de; Anastasiou, D.; Ito, K.; Sasaki, A. T.; Rameh, L.; Carracedo, A.; Vander Hei-den, M. G.; Cantley, L. C.; Pinton, P.; Haigis, M. C.; Pandolfi, P. P. «Systemic elevation of PTEN induces a tumor-suppressive metabolic state». Cell, vol. 149 (2012), p. 1-14.
Gatenby, R. A.; Gillies, R. J. «Why do cancers have high aerobic glycolysis?». Nat. Rev. Cancer, vol. 4 (2004), p. 891-899.
Goldberg, M. A.; Gaut, C. C.; Bunn, H. F. «Erythropoietin mRNA levels are governed by both the rate of gene transcription and posttranscriptional events». Blood, vol. 77 (1991), p. 271-277.
Goren, N.; Manzano, A.; Riera, L.; Ambrosio, S.; Ventura, F.; Bartrons, R. «6-phos-phofructo-2-kinase / fructose-2,6-bisphosphatase expression in rat brain during de-velopment». Brain Res. Mol. Brain Res., vol. 75 (2000), p. 138-142.
Guppy, M.; Greiner, E.; Brand, K. «The role of the Crabtree effect and an endogenous fuel in the energy metabolism of resting and proliferating thymocytes». Eur. J. Bio-chem., vol. 212 (1993), p. 95-99.
Hamilton, J. A.; Callaghan, M. J.; Sutherland, R. L.; Watts, C. K. «Identification of PRG1, a novel progestin-responsive gene with sequence homology to 6-phosphofruc-to-2-kinase/fructose-2,6-bisphosphatase». Mol. Endocrinol., vol. 11 (1997), p. 490-502.
Hanahan, D.; Weinberg, R. A. «The hallmarks of cancer». Cell, vol. 100 (2000), p. 57-70.
— «Hallmarks of cancer: the next generation». Cell, vol. 144 (2011), p. 646-674.Hardie, D. G. «The AMP-activated protein kinase pathway-new players upstream and
downstream». J. Cell Sci., vol. 117 (2004), p. 5479-5487.Hers, H. G.; Schaftingen, E. van. «Fructose 2,6-bisphosphate 2 years after its dis covery».
Biochem. J., vol. 206 (1982), p. 1-12.Hsu, P. P.; Sabatini, D. M. «Cancer cell metabolism: Warburg and beyond». Cell,
vol. 134 (2008), p. 703-707.Kaelin, W. G. «Proline hydroxylation and gene expression». Annu. Rev. Biochem., vol. 74
(2005), p. 115-128.Kim, J. W.; Gao, P.; Dang, C. V. «Effects of hypoxia on tumor metabolism». Cancer Me-
tastasis Rev., vol. 26 (2007), p. 291-298.Kim, J. W.; Tchernyshyov, I.; Semenza, G. L.; Dang, C. V. «HIF-1-mediated expression
of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia». Cell Metab., vol. 3 (2006), p. 177-185.
Kole, H. K.; Resnick, R. J.; Doren, M. van; Racker, E. «Regulation of 6-phosphofruc to-1-kinase activity in ras-transformed rat-1 fibroblasts». Arch. Biochem. Biophys., vol. 286 (1991), p. 586-590.
021-EL DOLÇ FENOTIP.indd 31 09/01/13 10:03

32
Koppenol, W. H.; Bounds, P. L.; Dang, C. V. «Otto Warburg’s contributions to current concepts of cancer metabolism». Nat. Rev. Cancer, vol. 11 (2011), p. 325-337.
Krebs, H. A. «The Pasteur effect and the relations between respiration and fermentation». Essays Biochem., vol. 8 (1972), p. 1-34.
Land, H.; Parada, L. F.; Weinberg, R. A. «Tumorigenic conversion of primary embryo fibroblasts requires at least two cooperating oncogenes». Nature, vol. 304 (1983), p. 596-602.
Le, A.; Cooper, C. R.; Gouw, A. M.; Dinavahi, R.; Maitra, A.; Deck, L. M.; Royer, R. E.; Vander Jagt, D. L.; Semenza, G. L.; Dang, C. V. «Inhibition of lactate dehydro-genase A induces oxidative stress and inhibits tumor progression». Proc. Natl. Acad. Sci. USA, vol. 107 (2010), p. 2037-2042.
Levine, A. J.; Puzio-Kuter, A. M. «The control of the metabolic switch in cancers by oncogenes and tumor suppressor genes». Science, vol. 330 (2010), p. 1340-1344.
Locasale, J. W.; Grassian, A. R.; Melman, T.; Lyssiotis, C. A.; Cantley, L. C.; Van-der Heiden, M. G. «Phosphoglycerate dehydrogenase diverts glycolytic flux and contributes to oncogenesis». Nat. Genet., vol. 43 (2011), p. 869-874.
López-Lázaro, M. «Hypoxia-inducible factor 1 as a possible target for cancer chemopre-vention». Cancer Epidemiol. Biomarkers Prev., vol. 15 (2006), p. 2332-2335.
Lu, W.; Pelicano, H.; Huang, P. «Cancer metabolism: is glutamine sweeter than glu-cose?». Cancer Cell, vol. 18 (2010), p. 199-200.
Manning, B. D.; Cantley, L. C. «AKT/PKB signaling: navigating downstream». Cell, vol. 129 (2007), p. 1261-1274.
Manzano, A.; Rosa, J. L.; Ventura, F.; Pérez, J. X.; Nadal, M.; Estivill, X.; Am-brosio, S.; Gil, J.; Bartrons, R. «Molecular cloning, expression, and chromoso-mal localization of a ubiquitously expressed human 6-phosphofructo-2-kinase / fructose-2, 6-bisphosphatase gene (PFKFB3)». Cytogenet. Cell Genet., vol. 83 (1998), p. 214-217.
Martínez-Outschoorn, U. E.; Pavlides, S.; Howell, A.; Pestell, R. G.; Tanowitz, H. B.; Sotgia, F.; Lisanti, M. P. «Stromal-epithelial metabolic coupling in cancer: integrating autophagy and metabolism in the tumor microenvironment». Int. J. Bio-chem. Cell Biol., vol. 43 (2011), p. 1045-1051.
Mathupala, S. P.; Ko, Y. H.; Pedersen, P. L. «Hexokinase-2 bound to mitochondria: cancer’s stygian link to the “Warburg effect” and a pivotal target for effective thera-py». Semin. Cancer Biol., vol. 19 (2009), p. 17-24.
Matoba, S.; Kang, J. G.; Patino, W. D.; Wragg, A.; Boehm, M.; Gavrilova, O.; Hur-ley, P. J.; Bunz, F.; Hwang, P. M. «P53 regulates mitochondrial respiration». Science, vol. 312 (2006), p. 1650-1653.
Mazurek, S. «Pyruvate kinase type M2: a key regulator of the metabolic budget system in tumor cells». Int. J. Biochem. Cell Biol., vol. 43 (2011), p. 969-980.
Menéndez, J. A.; Lupu, R. «Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis». Nat. Rev. Cancer, vol. 7 (2007), p. 763-777.
Minchenko, O. H.; Ochiai, A.; Opentanova, I. L.; Ogura, T.; Minchenko, D. O.; Caro, J.; Komisarenko, S. V.; Esumi, H. «Overexpression of 6-phosphofructo-2-kinase / fructose-2,6-bisphosphatase-4 in the human breast and colon malignant tu-mors». Biochimie, vol. 87 (2005), p. 1005-1010.
Mor, I.; Cheung, E. C.; Vousden K. H. «Control of glycolysis through regulation of
021-EL DOLÇ FENOTIP.indd 32 09/01/13 10:03

33
PFK1: old friends and recent additions». Cold Spring Harb. Symp. Quant. Biol., vol. 76 (2011), p. 211-216.
Novellasdemunt, L.; Millán, L.; Manzano, A.; Ventura, F.; Rosa, J. L.; Jordan, A.; Navarro-Sabaté, A.; Bartrons, R. «Progestins activate 6-phosphofructo-2-kinase / fructose-2,6-bisphosphatase (PFKFB3) in breast cancer cells». Biochem. J., vol. 442 (2012), p. 345-356.
Obach, M.; Navarro-Sabaté, A.; Caro, J.; Kong, X.; Durán, J.; Gómez, M.; Perales, J. C.; Ventura, F.; Rosa, J. L.; Bartrons, R. «6-phosphofructo-2-kinase (PFKFB3) gene promoter contains hypoxia-inducible factor-1 binding sites necessary for trans-activation in response to hypoxia». J. Biol. Chem., vol. 279 (2004), p. 53562-53570.
Okar, D. A.; Manzano, A.; Navarro-Sabaté, A.; Riera, L.; Bartrons, R.; Lange, A. J. «PFK-2/FBPase-2: maker and breaker of the essential biofactor fructose-2,6-bis-phosphate». Trends Biochem. Sci., vol. 26 (2001), p. 30-35.
Padrissa-Altés, S.; Zaouali, M.; Bartrons, R.; Rosselló, J. «The use of a reversible proteasome inhibitor in a model of reduced-size orthotopic liver transplantation in rats». Exp. Mol. Pathol., vol. 93 (2012), p. 99-110.
Pasteur, L. «Experiences et vues nouvelles sur la nature des fermentations». Comp. Rend. Acad. Sci., vol. 52 (1861), p. 1260-1264.
Pavlides, S.; Whitaker-Menezes, D.; Castello-Cros, R.; Flomenberg, N.; Witkie-wicz, A. K.; Frank, P. G.; Casimiro, M. C.; Wang, C.; Fortina, P.; Addya, S.; Pestell, R. G.; Martínez-Outschoorn, U. E.; Sotgia, F.; Lisanti, M. P. «The re-verse Warburg effect: aerobic glycolysis in cancer associated fibroblasts and the tumor stroma». Cell Cycle, vol. 8 (2009), p. 3984-4001.
Pelicano, H.; Martin, D. S.; Xu, R. H.; Huang, P. «Glycolysis inhibition for anticancer treatment». Oncogene, vol. 25 (2006), p. 4633-4646.
Peña-Rico, M. A.; Calvo-Vidal, M. N.; Villalonga-Planells, R.; Martínez-Soler, F.; Giménez-Bonafé, P.; Navarro-Sabaté, A.; Tortosa, A.; Bartrons, R.; Manza-no, A. «TP53 induced glycolysis and apoptosis regulator (TIGAR) knockdown results in radiosensitization of glioma cells». Radiother. Oncol., vol. 101 (2011), p. 132-139.
Pi i Sunyer, A. «La vida anaerobia». Gaceta Médica Catalana (1901), p. 15 -112.Pi i Sunyer Bayo, J. «August Pi i Sunyer: l’home i l’obra». Treballs de la Societat Catalana
de Biologia (1966), p. 33-53.Pouyssegur, J.; Dayan, F.; Mazure, N. M. «Hypoxia signalling in cancer and approach-
es to enforce tumour regression». Nature, vol. 441 (2006), p. 437-443.Rider, M. H.; Bartrons, R. «Fructose 2,6-bisphosphate: the last milestone of the 20th
century in metabolic control». Biochem. J., Classic Paper (2010), p. 1-10.Rider, M. H.; Bertrand, L.; Vertommen, D.; Michels, P. A.; Rousseau, G. G.; Hue, L.
«6-phosphofructo-2-kinase / fructose-2,6-bisphosphatase: head-to-head with a bi-functional enzyme that controls glycolysis». Biochem. J., vol. 381 (2004), p. 561-79.
Riera, L.; Manzano, A.; Navarro-Sabaté, A.; Perales, J. C.; Bartrons, R. «Insulin induces PFKFB3 gene expression in HT29 human colon adenocarcinoma cells». Bio-chim. Biophys. Acta, vol. 1589 (2002), p. 89-92.
Riera, L.; Obach, M.; Navarro-Sabaté, A.; Durán, J.; Perales, J. C.; Viñals, F.; Rosa, J. L.; Ventura, F.; Bartrons, R. «Regulation of ubiquitous 6-phosphofructo-2-ki-nase by the ubiquitin-proteasome proteolytic pathway during myogenic C2C12 cell differentiation». FEBS Lett., vol. 550 (2003), p. 23-29.
021-EL DOLÇ FENOTIP.indd 33 09/01/13 10:03

34
Ruiz-García, A.; Monsalve, E.; Novellasdemunt, L.; Navarro-Sabaté, A.; Manza-no, A.; Rivero, S.; Castrillo, A.; Casado, M.; Laborda, J.; Bartrons, R.; Díaz-Guerra, M. J. «Cooperation of adenosine with macrophage Toll-4 receptor agonists leads to increased glycolytic flux through the enhanced expression of PFKFB3 gene». J. Biol. Chem., vol. 286 (2011), p. 19247-19258.
Scatena, R.; Bottoni, P.; Pontoglio, A.; Mastrototaro, L.; Giardina, B. «Glycolytic enzyme inhibitors in cancer treatment». Expert Opin. Investig. Drugs, vol. 17 (2008), p. 1533-1545.
Semenza, G. L. «HIF-1 and tumor progression: pathophysiology and therapeutics». Trends Mol. Med., vol. 8 (2002), p. 62-67. [Suplement]
— «Hypoxia-inducible factors in physiology and medicine». Cell, vol. 148 (2012), p. 399-408.
Semenza, G. L.; Roth, P. H.; Fang, H. M.; Wang, G. L. «Transcriptional regulation of genes encoding glycolytic enzymes by hypoxia-inducible factor 1». J. Biol. Chem., vol. 269 (1994), p. 23757-23763.
Shaw, R. J.; Cantley, L. C. «RAS, PI(3)K and mTOR signalling controls tumour cell growth». Nature, vol. 441 (2006), p. 424-430.
Smallbone, K.; Gatenby, R. A.; Gillies, R. J.; Maini, P. K.; Gavaghan, D. J. «Meta-bolic changes during carcinogenesis: potential impact on invasiveness». J. Theor. Biol., vol. 244 (2007), p. 703-713.
Sols, A. «Multimodulation of enzyme activity». Curr. Top Cell Regul., vol. 19 (1981), p. 77-101.
Suzuki, S.; Tanaka, T.; Poyurovsky, M. V.; Nagano, H.; Mayama, T.; Ohkubo, S.; Lokshin, M. «Phosphate-activated glutaminase (GLS2), a p53-inducible regulator of glutamine metabolism and reactive oxygen species». Proc. Natl. Acad. Sci. USA, vol. 107 (2010), p. 7461-7466.
Tennant, D. A.; Durán, R. V.; Gottlieb, E. «Targeting metabolic transformation for cancer therapy». Nat. Rev. Cancer, vol. 10 (2010), p. 267-277.
Tudzarova, S.; Colombo, S. L.; Stoeber, K.; Carcamo, S.; Williams, G. H.; Monca-da, S. «Two ubiquitin ligases, APC/C-Cdh1 and SKP1-CUL1-F (SCF)-beta-TrCP, sequentially regulate glycolysis during the cell cycle». Proc. Natl. Acad. Sci. USA, vol. 108 (2011), p. 5278-5283.
Vander Heiden, M. G.; Cantley, L. C.; Thompson, C. B. «Understanding the Warburg effect: the metabolic requirements of cell proliferation». Science, vol. 324 (2009), p. 1029-1033.
Vogelstein, B.; Kinzler, K. W. «The multistep nature of cancer». Trends Genet., vol. 9 (1993), p. 138-141.
Vousden, K. H.; Prives, C. «Blinded by the light: the growing complexity of p53». Cell, vol. 137 (2009), p. 413-431.
Wallace, D. C. «Mitochondria and cancer: Warburg addressed». Cold Spring Harb. Symp. Quant. Biol., vol. 70 (2005), p. 363-374.
Warburg, O. Über den Stoff-wechsel der Tumoren. Berlín: J. Springer, 1926.— «On the origin of cancer cells». Science, vol. 123 (1956), p. 309-314.Wise, D. R.; DeBerardinis, R. J.; Mancuso, A.; Sayed, N.; Zhang, X. Y.; Pfeiffer, H.
K.; Nissim, I.; Daikhin, E.; Yudkoff, M.; McMahon, S. B.; Thompson, C. B. «Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis
021-EL DOLÇ FENOTIP.indd 34 09/01/13 10:03

35
and leads to glutamine addiction». Proc. Natl. Acad. Sci. USA, vol. 105 (2008), p. 18782-18787.
Yalcin, A.; Clem, B. F.; Simmons, A.; Lane, A.; Nelson, K.; Clem, A. L.; Brock, E.; Siow, D.; Wattenberg, B.; Telang, S.; Chesney, J. «Nuclear targeting of 6-phos-phofructo-2-kinase (PFKFB3) increases proliferation via cyclin-dependent kinases». J. Biol. Chem., vol. 284 (2009), p. 24223-24232.
Zhou, Y. F.; Bosch-Marce, M.; Okuyama, H.; Krishnamachary, B.; Kimura, H.; Zhang, L.; Huso, D. L.; Semenza, G. L. «Spontaneous transformation of cultured mouse bone marrow-derived stromal cells». Cancer Res., vol. 66 (2006), p. 10849-10854.
Zoncu, R.; Efeyan, A.; Sabatini, D. M. «mTOR: from growth signal integration to can-cer, diabetes and ageing». Nat. Rev. Mol. Cell Biol., vol. 12 (2011), p. 21-35.
Zu, X. L.; Guppy, M. «Cancer metabolism: facts, fantasy, and fiction». Biochem. Biophys. Res. Commun., vol. 313 (2004), p. 459-465.
021-EL DOLÇ FENOTIP.indd 35 09/01/13 10:03

021-EL DOLÇ FENOTIP.indd 36 09/01/13 10:03

Institutd’Estudis Catalans
SECCIÓ DE CIÈNCIESBIOLÒGIQUES
El dolç fenotip de les cèl·lules tumorals: buscant dianes terapèutiquesDiscurs de recepció de Ramon Bartrons i Bachcom a membre numerari de la Secció de Ciències Biològiques, llegit el dia 18 de febrer de 2013
9 7 8 8 4 9 9 6 5 1 5 3 8
021-Coberta EL DOLÇ FENOTIP.indd 1 09/01/13 09:57