
Dr. Eduardo Fernández Quintana
Centro de Farmacovigilancia e Información Terapéutica de Canarias de Canarias.
Registro y Notificación de acontecimientos y reacciones adversas durante el Ensayo
Clínico.

Un objetivo constante de las Agencias Reguladoras: garantizar la seguridad de los medicamentos que autorizan.
CONCEPTO SEGURIDAD DE UN MEDICAMENTO
No absoluto……… es relativo
No estático……… es dinámico

¿por qué es relativa la seguridad de un medicamento?
• Depende de la relación beneficio / riesgo : un medicamento se considera seguro, cuando los beneficios que se obtienen con su utilización superan los riesgos que supone la administración del fármaco.
Beneficio
Riesgo
Seguridad relativa

¿por qué es dinámica la seguridad de un medicamento?
• Depende del tipo de pacientes y de las condiciones de uso.
• Varía con el tiempo.
Seguridad dinámica
=
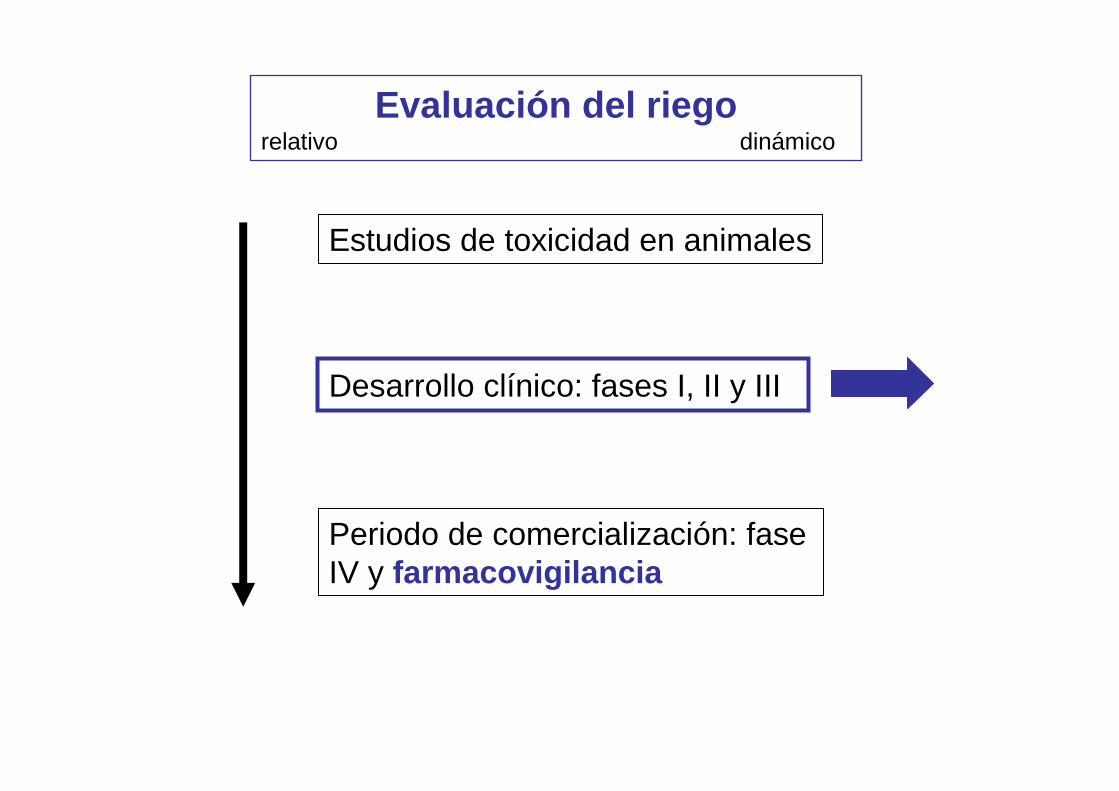
Evaluación del riegorelativo dinámico
Estudios de toxicidad en animales
Desarrollo clínico: fases I, II y III
Periodo de comercialización: fase IV y farmacovigilancia

Protocolos de ensayos clínicosAspectos generales de la seguridad de un
medicamento durante un EC
1. Deben contribuir a reunir la máxima información posible sobre la seguridad de los medicamentos evaluados.
2. Deben ajustarse a las regulaciones nacionales y europeas: Marco Legal.

Protocolos de Ensayo Clínico
1. Para contribuir a reunir la información sobre el riesgo de los medicamentos.Un protocolo de EC debe incluir los criterios, directrices y procedimientos de actuación , en materia de seguridad, de todos los participantes en el ensayo: investigador , promotor , pacientes , CEICs, Autoridad Sanitaria .
Apartado de seguridad.
• Aspectos técnicos:- Definiciones: AA; AAG; RA; RAG: - Procedimiento de detección*.- Procedimiento de registro.- Causalidad y gravedad.
• Aspectos administrativos:- Criterios de notificación de los AA entre participantes: normativa vigente
• Aspectos éticos:- Medidas a tomar como consecuencia de la aparición de problemas de seguridad.
. Modificación de la hoja de información destinada al paciente.. Retirada del sujeto.. Suspensión del ensayo.
¿qué hay que notificar?* ICB nº 11 (jul-sept-94). Registro de acontecimientos adversos en ensayos clínicos.
¿quién tiene que notificar? ¿a dónde hay que notificar y en qué tiempo?

Protocolos de ensayos clínicos
RD 223/2004, de 6 de febrero, por el que se regulan los ensayos clínicos con medicamentos.
Ley 29/2006, de 26 de julio , de garantías y uso de uso racional de los medicamentos y productos sanitarios.
ORDEN SCO/256/2007, de 5 de febrero, por las que se establecen los principios y las directrices detalladas de buena práctica clínica y los requisitos para autorizar la fabricación o importación de medicamentos en investigación de uso humano.
Directiva 75/318/EEC : Gudelines Good Clinicla Practice: versión julio 1996.
Directiva 2005/28/CE DE LA COMISIÓN, de 8 de abril de 2005, por la que se establecen los principios y las directrices detalladas de las buenas prácticas clínicas respecto a los medicamentos en investigación de uso humano…
Directiva 2001/20/CE … de aplicación de buenas prácticas clínica…
Aclaraciones de la AEMyPS sobre la aplicación de la normativa europea de ensayos clínicos con medicamentos de uso humano. Versión nº 6 mayo de 2008.
2. Para ajustarse a la regulación nacional y europea.

Objetivo.
• Conocer cuáles son los procedimientos que tienen que seguir los investigadores y los promotores de los ensayos clínicos a la hora de registrar y notificar un acontecimiento o una reacción adversa ocurridos con medicamentos en fase de investigación .

Objetivo.
• ¿Quién notifica?• ¿Qué se notifica?• ¿A quién se notifica?• ¿Cómo se notifica?• ¿Cuándo se notifica?

Marco Legal. Procedimientos de notificación
• Procedimientos de notificación de sospechas de reacciones adversas a medicamentos comercializados .
• Procedimientos de notificación de sospechas de acontecimientos adversos y reacciones adversas a medicamentos en fase de investigación .
- Prof. Sanitarios.- Notificación espontánea.- S.E.F.V.
- Investigadores y promotores.- Notificación expeditiva.- I.P.S

RD 1344/2007 de RD 1344/2007 de farmacovigilanciafarmacovigilancia
ENSAYOS CLÍNICOS fuente de información en ENSAYOS CLÍNICOS fuente de información en farmacovigilanciafarmacovigilancia
… los investigadores de los EC están comprometidos a notificar al SEFV aquellas
Reacciones Adversas relacionadas con medicamentos autorizados que no están
siendo objeto de estudio en el contexto de un ensayo clínico.

Medicamentos comercializados.
• Sistema Español de Farmacovigilancia de Medicamentos de Uso Humano.
• Programa de Farmacovigilancia de Canarias. RA a medicación autorizada en
práctica clínica habitual.
RA medicación concomitante de un EC no objeto de estudio en el contexto de un ensayo clínico.

Programa de Farmacovigilancia de Canarias.
Órgano Competente en materia de Farmacovigilancia.
Dirección General de Farmacia.
Servicio de Farmacología Clínica.
Unidad o Centro de Farmacovigilancia.
Agencia Española de Medicamentos y ps.
Centro Coordinador.

Programa de Farmacovigilancia de Canarias.
STAF• Director del Programa de F: Dr. José
N. Boada.• Adjuntos del Servicio:
– Dra. Maria del Mar García Saiz.– Dra. Consuelo Rodríguez Jiménez.
• Técnicos del SEFV– Dr. Marcelino García S-Colomer– Dr. Eduardo Fernández Quintana– Dr. Carlos Boada Fedz-del Campo
• Residentes MIR.

Sistema Español de Farmacovigilancia.
Estructura descentralizada.
17 Centros de Farmacovigilancia en CCAA.
Centro Coordinador: División de Farmacoepidemiología y Farmacovigilanica de la AEMyPS.
Programa de Notificación Espontánea.
Programa de la Tarjeta Amarilla.

TARJETA AMARILLA:
• Formulario de notificación de sospechas de RAM del SEFV.
PROCEDIMEINTO

HUC-FC 85 73SAP: farmaclinCRFV 922 319 341FAX: 922 65 59 95http://www.fitec.ull.es/

Profesional SanitarioProfesional Sanitario TarjetaTarjeta AmarillaAmarilla
Centros Autonómicos de Centros Autonómicos de FarmacovigilanciaFarmacovigilancia
F.E.D.R.A.F.E.D.R.A.Revista MédicaRevista Médica
Formato “C.I.O.M.S.”Formato “C.I.O.M.S.”
Compañías Compañías FarmacéuticasFarmacéuticas
Centro Coordinador Centro Coordinador Div. Div. FepiFepi y FV, AEMy FV, AEM
XML
Vías de Comunicación de notificaciones individuales de sospechasVías de Comunicación de notificaciones individuales de sospechas de de RAM del SEFV.RAM del SEFV.
Práctica clínica habitualPráctica clínica habitual
Pre-FEDRA
Ensayo ClínicoEnsayo Clínico

PROCEDIMIENTO PARA EL REGISTRO Y NOTIFICACIÓN
DE ACONTECIMIENTOS ADVERSOS DURANTE EL
ENSAYOS CLÍNICOSCLÍNICOS.

• ¿Quién notifica?• ¿Qué se notifica?• ¿A quién se notifica?• ¿Cómo se notifica?• ¿Cuándo se notifica?
Objetivo.

¿Quién notifica?

¿quién notifica?
– INVESTIGADORES, responsables de la realización del ensayo clínico en un centro.
• Registro y envío de Acontecimientos Adversos de los medicamentos en investigación.
– PROMOTORES, responsables del inicio, gestión y/o financiación de un ensayo clínico.
• Procesamiento y notificación de Reacciones Adversas a los medicamentos en investigación.

¿Qué se notifica?

Acontecimiento Adverso:AAAcontecimiento Adverso:AA
Reacción Adversa:RAReacción Adversa:RA
… cualquier incidencia perjudicial para la salud del sujeto del ensayo clínico tratado con un medicamento sin determinar necesariamente su relación causal con dicho tratamiento.
… toda reacción nociva y no intencionada a un medicamento en investigación, independientemente de la dosis administrada.
DEFINICIONES INormativa
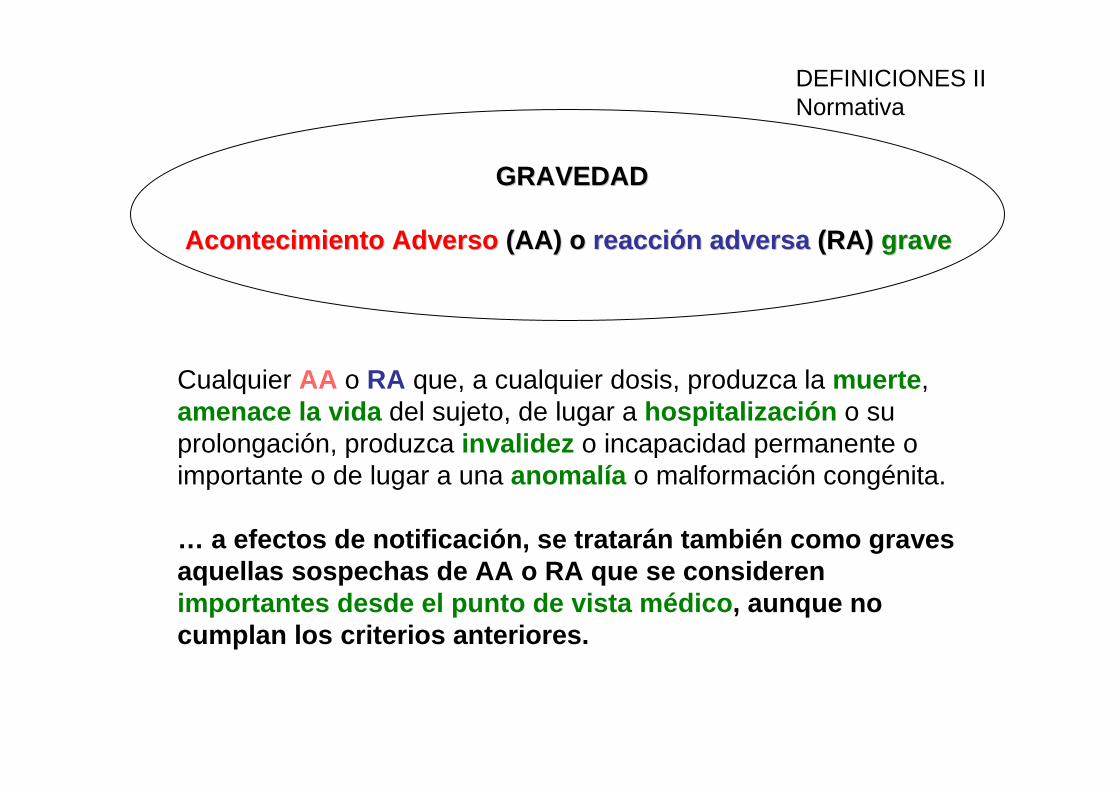
GRAVEDADGRAVEDAD
Acontecimiento AdversoAcontecimiento Adverso (AA) o (AA) o reacción adversareacción adversa (RA) (RA) gravegrave
Cualquier AA o RA que, a cualquier dosis, produzca la muerte , amenace la vida del sujeto, de lugar a hospitalización o su prolongación, produzca invalidez o incapacidad permanente o importante o de lugar a una anomalía o malformación congénita.
… a efectos de notificación, se tratarán también com o graves aquellas sospechas de AA o RA que se consideren importantes desde el punto de vista médico , aunque no cumplan los criterios anteriores.
DEFINICIONES IINormativa

CONOCIMIENTO PREVIOCONOCIMIENTO PREVIO
Reacción adversa inesperada. Reacción adversa inesperada.
… RA de naturaleza o gravedad desconocida , que no se corresponde con la información referente al producto.
1. Manual del Investigador: para medicamentos en investigación no autorizados para su comercialización.
2. Ficha Técnica: para medicamentos en investigación que posean ya una autorización de comercialización
DEFINICIONES IIINormativa

Manual del Investigador : conjunto de datos clínicos y no clínicos del medicamento en investigación pertinente para el estudio de dicho medicamento en seres humanos (PROMOTOR).
Ficha Técnica: Ficha normalizada en la que se recoge la información científica esencial sobre la especialidad farmacéuticaa que se refiere, para su difusión a los profesionales sanitarios por el Titular de Autorización de Comercialización (TAC). Aprobada por las Autoridades Sanitarias (AEMPS, EMEA).
DEFINICIONES IVNormativa

Acontecimiento Adverso:AAAcontecimiento Adverso:AA
… es obligación del investigador recoger, registrar y comunicar de manera inmediata los AA graves o inesperadosinesperados .
COMPROMISOINVESTIGADOR
¿qué se notifica?

Criterios básicos de notificación inmediata.Criterios básicos de notificación inmediata.
1. AA grave:Produzca la muerte del paciente.Amenace la vida del sujeto.Hospitalización o prolongación de hospitalización.Invalidez o incapacidad permanente o importante.Anomalía o malformación congénita.
Cualquier AA que el investigador considere clínicamente relevante.
2. AA inesperada:De naturaleza o gravedad no correspondiente con la información del producto.
Fuente de Información
¿qué se notifica?INVESTIGADOR
Manual del Investigador (PEI)
Ficha Técnica (med. autorizados).
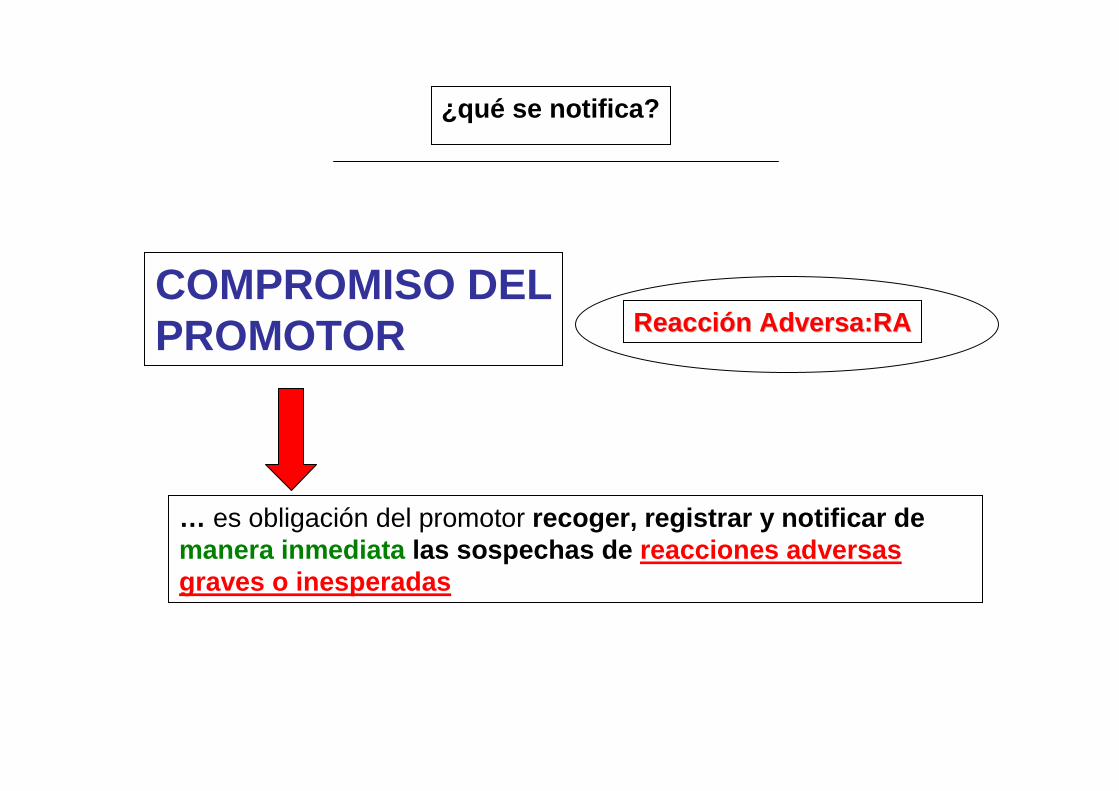
Reacción Adversa:RAReacción Adversa:RA
… es obligación del promotor recoger, registrar y notificar de manera inmediata las sospechas de reacciones adversas graves o inesperadas
COMPROMISO DELPROMOTOR
¿qué se notifica?

Criterios para la notificación expeditiva.Criterios para la notificación expeditiva.
1. RA grave:Produzca la muerte del paciente.Amenace la vida del sujeto.Hospitalización o prolongación de hospitalización.Invalidez o incapacidad permanente o importante.Anomalía o malformación congénita.
Cualquier AA que el investigador considere importante.
2. RA inesperada:De naturaleza o gravedad no correspondiente con la información del producto (Manual del Investigador o Ficha Técnica).
¿qué se notifica?PROMOTOR

¿A quién se notifica?
¿Cómo se notifica?

INVESTIGADOR
” el investigador de un ensayo clínico deberá notificar inmediatamenteal promotor todos los acontecimientos adversos (AA) graves
PROMOTOR
AA graves o inesperadas
¿a quién se notifica?
INVESTIGADOR
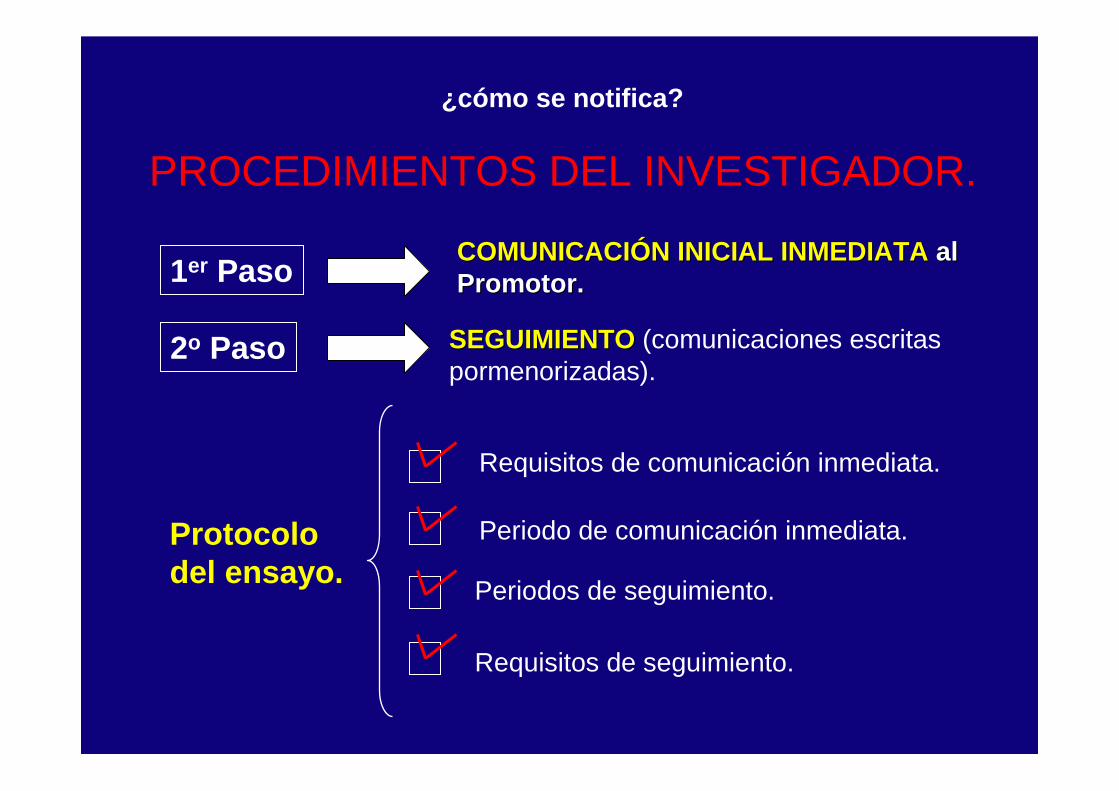
PROCEDIMIENTOS DEL INVESTIGADOR.
1er PasoCOMUNICACIÓN INICIAL INMEDIATA COMUNICACIÓN INICIAL INMEDIATA al al Promotor.Promotor.
2o Paso SEGUIMIENTOSEGUIMIENTO (comunicaciones escritas pormenorizadas).
Protocolo del ensayo.
Periodo de comunicación inmediata.
Requisitos de comunicación inmediata.
Periodos de seguimiento.
Requisitos de seguimiento.
¿cómo se notifica?
…” el promotor notificará , en el plazo menor posible , a la Autoridades Sanitarias , CEICS e Investigadores , las sospechas reacciones adversas graves e inesperadas que surjan a lo largo
del ensayo…”
• Autoridades Sanitarias• CEICs• Investigadores
NOTIFICACIÓN EXPEDITIVARAM graves e inesperadas:
¿a quién se notifica?
PROMOTOR
PROMOTOR I

PROMOTOR II
…” Deberá de mantener un registro detallado de todos los acontecimientos adversos que le sean comunicados: el promotor
enviará Informes Periódicos de Seguridad (IPS)
AEMPSCCAACEICS
IPS
¿a quién se notifica?
PROMOTOR

¿a quién se notifica?
PROMOTOR
Procedimiento IPS
Documento de Evaluación de Seguridad con toda la in formación dis ponible.
AEMPS; CCAA y CEICS.
ANUAL : hasta que finalice el ensayo.
SIEMPRE: que lo soliciten las Autoridades Sanitarios o los CEICS.
PERIODO DE NOTIFICACIÓNPERIODO DE NOTIFICACIÓN..
Formulario: determinado por la AEMPS.

¿a quién se notifica?
Resumen de los Principios básicos en la vigilancia de la seguridad de medicamentos en investigación.
INVESTIGADOR PROMOTOR
• Notificación inmediata al promotor de los AAgraves / inesperados.
• Notificación de las RA graves / inesperadas.
Autoridad Sanitaria.
Investigador . CEICSPromotor .
N. ExpeditivaIPS

PROCEDIMIENTO:NOTIFICACIÓN EXPEDITIVA
¿a quién se notifica?
PROMOTOR

NOTIFICACIÓN EXPEDITIVA A LA AEMPS (I).
notificación de las RA graves / inesperadas ocurridas en España u otro Estado.
notificación de casos del medicamento control y del tratamiento concomitante contemplado en el EC.
Plazo máximo de notificación. 15 días naturales .
RA mortales o comprometan la vida: 7 días naturales .
Información de seguimiento a los 8 días .
PERIODO DE NOTIFICACIÓN.PERIODO DE NOTIFICACIÓN.
¿cómo se notifica?

NOTIFICACIÓN EXPEDITIVA A LA AEMPS (II).
Formato : XML -FEDRA (Pre-FEDRA) / EUDRAVIGILANCE-CT.
Idioma: Lengua española oficial del Estado: “ESPAÑO L”
E.C. placebo.E.C. placebo.
las RA a placebo no están sujetas a este procedimiento de notificaci ón individualizada. Comunicación en los IPS.
E.C. doble ciego.E.C. doble ciego.
siempre que sea posible se mantendrá el carácter ci ego para el investigador.
Formulario CIOMs: VALIDACIÓN en FEDRA por e CC.
¿cómo se notifica?

NOTIFICACIÓN EXPEDITIVA A LAS CCAA.PROCEDIMIENTO DE NOTIFICACIÓN.PROCEDIMIENTO DE NOTIFICACIÓN.
notificación de las RA graves / inesperadas ocurridas en la CCAA.
notificación de casos con med. control y tratamiento concomitante .
Plazo máximo de notificación. 15 días naturales .
RA mortales o graves: 7 días naturales .
Información de seguimiento a los 8 días.
PERIODO DE NOTIFICACIÓN.PERIODO DE NOTIFICACIÓN.
Formulario CIOMs al CRFV de Canarias.
Idioma: Lengua española oficial del Estado: “ESPAÑO L”
¿cómo se notifica?

NOTIFICACIÓN EXPEDITIVA A LOS CEICS.PROCEDIMIENTO DE NOTIFICACIÓNPROCEDIMIENTO DE NOTIFICACIÓN ..
notificación de las RA graves / inesperadas en el ámbito del CEIC.
notificación de casos con med. control y tratamiento concomitante .
Plazo máximo de notificación. 15 días naturales.
RA mortales o graves: 7 días naturales.
Los CEICS podrán solicitar informes periódicos.
PERIODO DE NOTIFICACIÓN.PERIODO DE NOTIFICACIÓN.
Formulario (obligatorio): CIOMs
Idioma: Lengua española oficial del Estado: “ESPAÑO L”
¿cómo se notifica?

ENVÍO Y CUMPLIMENTACIÓN DE UN FORMULARIO DE NOTIFICACIÓN DE
REACCIONES ADVERSAS.
Notificación expeditiva.







