
1
Proyecto Genoma Humano
1 Génesis y desarrollo del proyecto2 Estrategias de secuenciación del genoma:
secuenciación aleatoria y jerárquica3 Contenido en CG4 Islas CpG5 Tasas de recombinación6 Elementos transponibles7 Contenido en genes8 El Proyecto ENCODE
Referencias: Brown 2007, págs. 82-95 y 104-124.Lander et al. 2001, Nature 409:860-875.
1. Consorcio de 20 laboratorios públicos pertenecientes a 6 países. Liderado por F. S. Collins y E. Lander.
2. Discusión y debate en la comunidad científica 1984-1990. Iniciativa: Departament of Energy y National Institutes ofHealth (US). Comienzo del proyecto: 1990. Borrador: Oct 2000. Publicación: Feb 2001. Finalización: Octubre 2004.
3. Secuenciación aleatoria jerárquica (Hierarchical ShotgunSequencing) o secuenciación clon a clon.
4. Material: DNA obtenido de donantes anónimos. La identidad de los donantes no es conocida (ni siquiera por ellos mismos).
5. Los datos se han hecho públicos a través de los bancos públicos de datos sin ninguna restricción a medida que se progresaba en el proyecto.
6. Publicación: Nature 409: 860-921 (15 febrero 2001); Nature431: 931-945 (21 Octubre 2004).
El Proyecto Genoma Humano

2
1. Celera Genomics. Empresa privada de biotecnología que dirige J. Craig Venter y cotiza en bolsa.
2. Anuncio del proyecto: 1998. Comienzo de la secuenciación: 8 Sep 1999. Finalización de la secuenciación: 17 Jun 2000. Ensamblaje del borrador: 1 Oct 2000.
3. Estrategia: Secuenciación aleatoria del genoma (Whole-genome shotgun sequencing).
4. Material: Se reclutaron 21 donantes voluntarios. De ellos se seleccionaron 5 sujetos (dos hombres y tres mujeres): 2 caucásicos, un afroamericano, un asiático (chino) y un hispano (mejicano).
5. Condiciones para el acceso a los datos mediante acuerdo entre Science y Celera Genomics. Los datos está a disposición de los investigadores a partir de la fecha de publicación a través de la Web de Celera y con ciertas restricciones.
6. Publicación: Science 291: 1304-1351 (16 febrero 2001).
Celera Sequencing Project
Eric Lander
Francis Collins

3
Graig Venter
Hierarchical Shotgun Sequencing

4
Whole-genome shotgun sequencing
Características de una genoteca genómica
• Número de clones (N).• Tamaño promedio (a) y varianza del inserto.• Redundancia teórica (R = Na/b).• Aleatoriedad (randomness): representación
de secuencias genómicas diana.• Completa (completeness): si todas las
secuencias diana en el genoma estárepresentadas en la genoteca.
• Fidelidad (fidelity): medida en que los insertos de los clones son copias fieles de las secuencias del genoma.

5
P = 1 - (1 – a/b)N 1 – e-Na/b = 1 – e-R
N = ln (1 – P)/ln (1 – a/b)
R - ln (1 – P)
donde N = número de clones; a = tamaño promedio del inserto; b = tamaño del genoma; R = redundancia teórica (Na/b); P = probabilidad de que una región dada esté incluida en la genoteca.
RPCI = Roswell Park Cancer Institute, Buffalo (NY)
Caltech = California Institute of Technology, Pasadena (California)

6
Dos métodos de cartografía física para detectar solapamiento entre clones genómicos
Ensamblaje de clones mediante fingerprinting

7
Secuenciación de un clon 100-200 kb
• Fase 1. Secuenciación aleatoria– Construcción de una genoteca aleatoria en
plásmido -> colección de clones 1-2 kb de tamaño promedio.
– Secuenciación de uno o ambos extremos de un cierto número de clones -> colección de lecturas (“reads”).
– Ensamblaje de las lecturas -> un cierto número de “contigs” con huecos (“gaps”) entre ellos.
• Fase 2. Finalización (corrección de errores y rellenado de huecos mediante secuenciación dirigida)
Calidad de una secuencia (PHRAP score)
• P = Probabilidad de error de cada base
• Q = Calidad de una base
• Q = - 10 log10 P
• Al iniciar un proyecto de secuenciación es conveniente fijar cual es el objetivo: la calidad de la secuencia final a obtener. De ella depende la redundancia necesaria.
• Q = 20, 30 y 40 corresponden a P = 1%, 0,1% y 0,01%.

8
Secuenciación de un clon 100-200 kb
• Base-calling. PHRED permite obtener la probabilidad de error de cada base.
• Ensamblaje. PHRAP permite ensamblar las lecturas en contigs.
• Edición. CONSED permite visualizar el ensamblaje y la secuencia consenso asícomo calcular la probabilidad de error de cada base en la secuencia consenso.
• Finalización. AUTOFINISH permite dirigir toda la operación de finalización basándose en la calidad de cada base.
Borrador 7 Octubre 2000
• Cartografía. Ensamblaje de los clones BAC (por fingerprinting) en 1246 contigs.
• Secuenciación y ensamblaje de 29.298 clones que representan un total de 4,26 Gb de secuencia.
• Las secuencias brutas subyacentes suponen un total de 23 Gb (promedio 7.5x).

9
Fingerprint Sequenced- Gaps between
clone contigs clone contigs sequence contigs
El borrador del genoma humano (7-10-2000) representa el 88% de todo el genoma y un 93% de la eucromatina

10
Range: 31-65% (20-kb windows)
Mean: 41%
SD: 5.2%
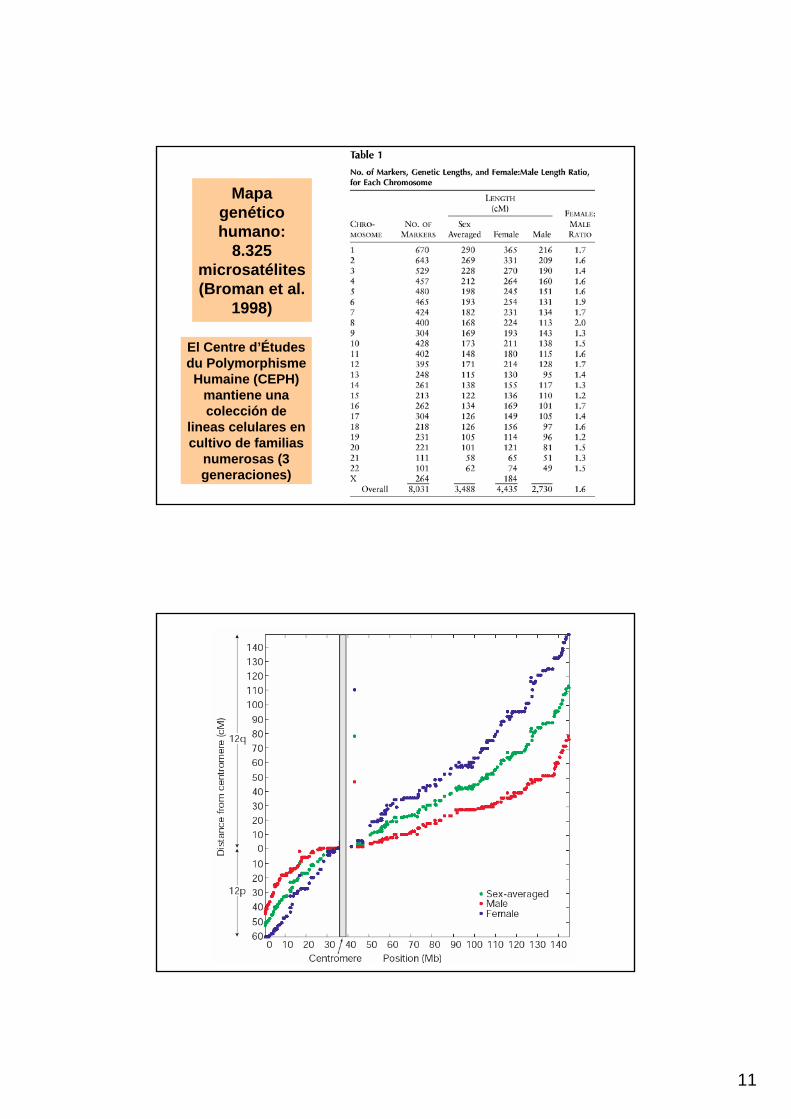
11
Mapagenético humano:
8.325 microsatélites(Broman et al.
1998)
El Centre d’Étudesdu PolymorphismeHumaine (CEPH)
mantiene una colección de
lineas celulares en cultivo de familias
numerosas (3 generaciones)
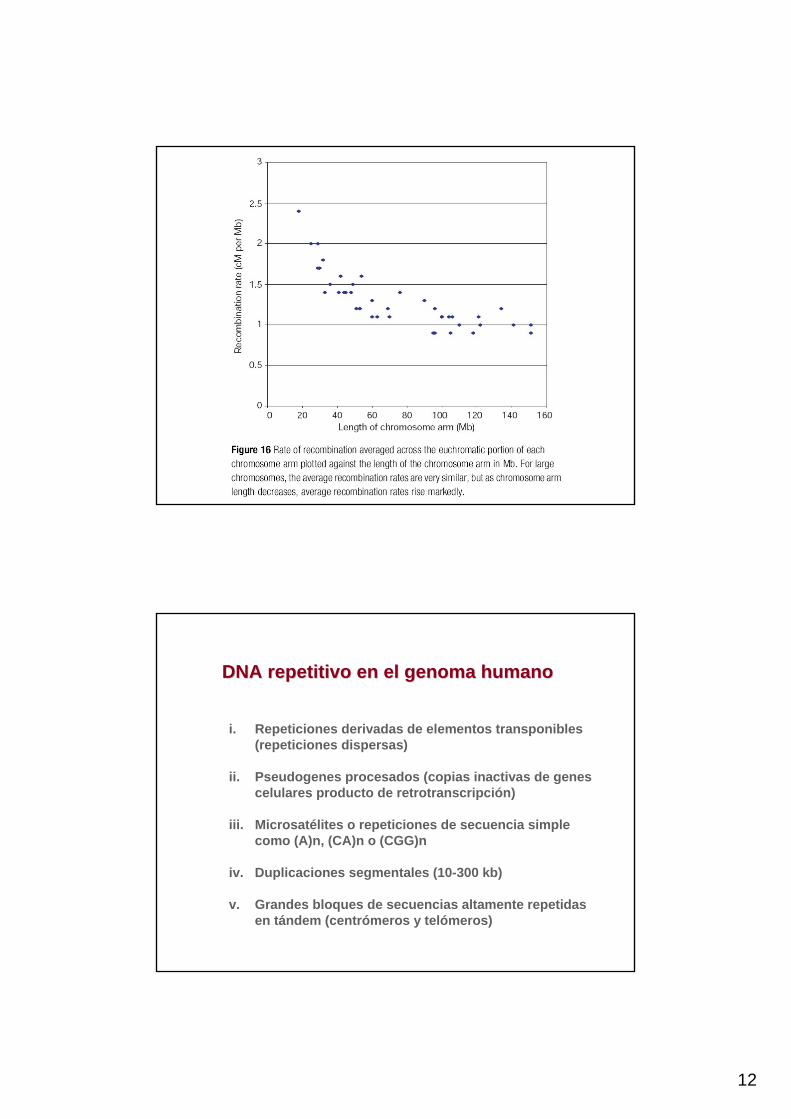
12
DNA repetitivo en el genoma humanoDNA repetitivo en el genoma humano
i. Repeticiones derivadas de elementos transponibles (repeticiones dispersas)
ii. Pseudogenes procesados (copias inactivas de genes celulares producto de retrotranscripción)
iii. Microsatélites o repeticiones de secuencia simple como (A)n, (CA)n o (CGG)n
iv. Duplicaciones segmentales (10-300 kb)
v. Grandes bloques de secuencias altamente repetidas en tándem (centrómeros y telómeros)

13
Elementos transponibles en el genoma humano
80-3000 bp
100-300 bp
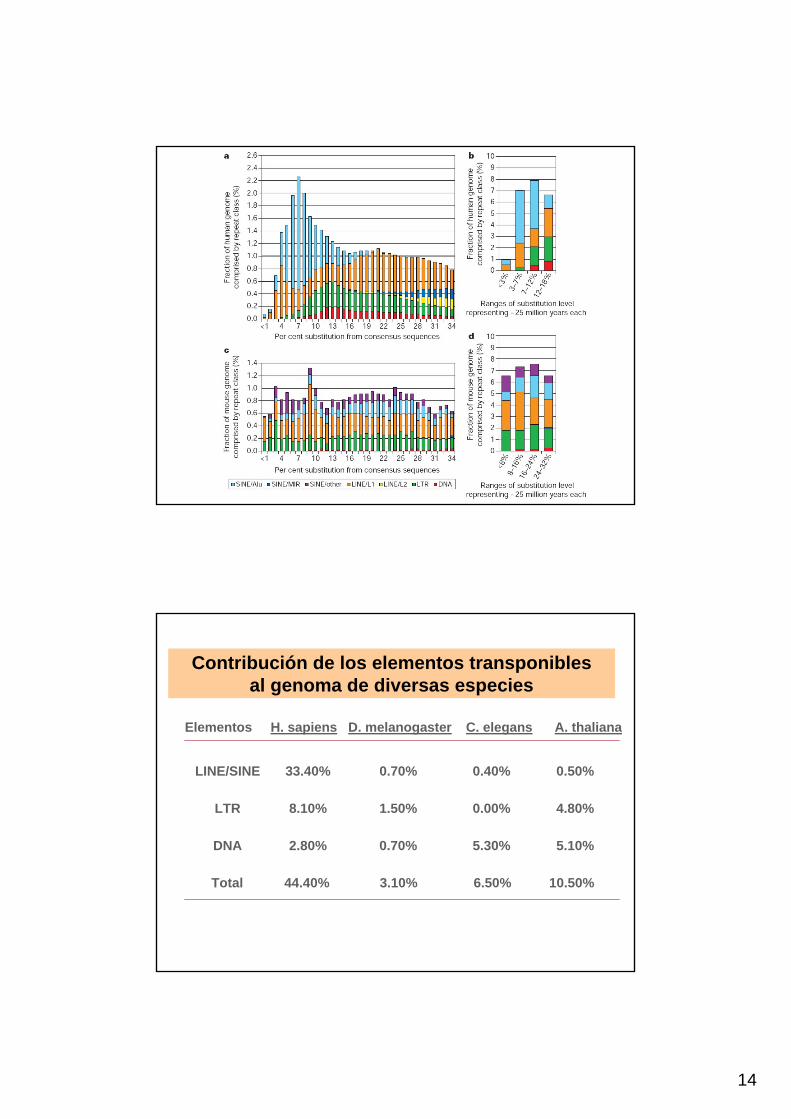
14
Contribución de los elementos transponibles al genoma de diversas especies
Elementos H. sapiens D. melanogaster C. elegans A. thaliana
LINE/SINE 33.40% 0.70% 0.40% 0.50%
LTR 8.10% 1.50% 0.00% 4.80%
DNA 2.80% 0.70% 5.30% 5.10%
Total 44.40% 3.10% 6.50% 10.50%

15
Mutaciones causadas por la inserción de elementos transponibles
• En Drosophila, ~50% de las mutacionesmorfológicas clásicas (por ejemplo, white) están causadas por la inserción de elementos transponibles.
• En el raton, 10% de las mutacionesespóntáneas están causadas portransposiciones.
• En humanos, 1/600 de las mutacionesespontáneas están causadas por la inserciónde elementos transponibles.
Genes que cifran RNA no Genes que cifran RNA no codificantecodificante en el genoma humano.en el genoma humano.
Genes deRNA
Número esperado
Número Observado
Genes relacionados (pseudogenes, fragmentos,
parálogos)
tRNA 1.310 497 324
18S rRNA 150-200 0 40
5.8S rRNA 150-200 1 11
28S rRNA 150-200 0 181
5S rRNA 200-300 4 520
snoRNA 97 84 645
snRNA (U1-U12) ?? 78 1542
7SL RNA 4 3 773

16
Características de los genes humanos que codifican proteínas(10.272 mRNAS en la base de datos RefSeq alineados
con el borrador del genoma, Lander et al. 2001)
Característica Mediana Promedio Tamaño muestra
Tamaño exones 122 bp 145 bp 43.317
Número exones 7 8.8 3.501
Tamaño intrones 1.023 bp 3.365 bp 27.238
3’ UTR 400 bp 770 bp 689 (crom. 22)
5’ UTR 240 300 463 (crom. 22)
Secuencia codificadora 1.100 bp 1.340 bp 1.804
(CDS) 367 aa 447 aa
Extensión genómica 14 kb 27 kb 1.804
Comparación de los genes humanos con los de Caenorhabditis y Drosophila
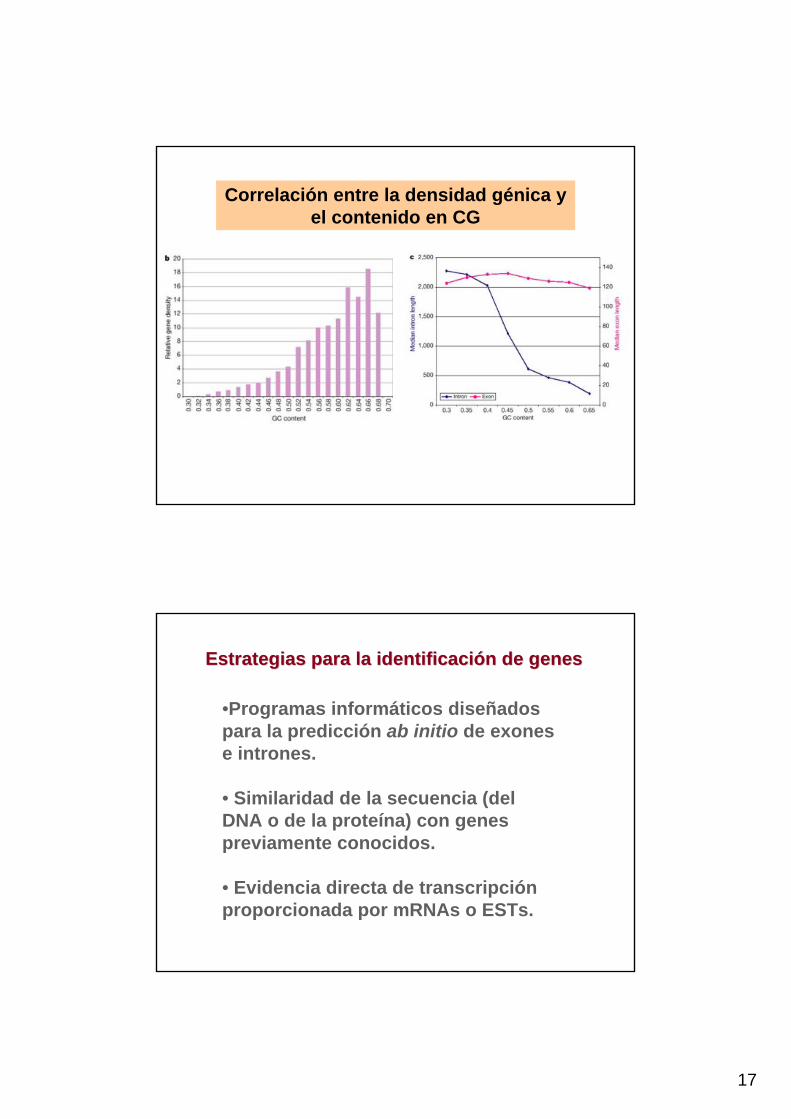
17
Correlación entre la densidad génica y el contenido en CG
•Programas informáticos diseñados para la predicción ab initio de exones e intrones.
• Similaridad de la secuencia (del DNA o de la proteína) con genes previamente conocidos.
• Evidencia directa de transcripción proporcionada por mRNAs o ESTs.
Estrategias para la identificaciEstrategias para la identificacióón de genesn de genes

18
Genes detectados por el Proyecto Genoma Humano Genes detectados por el Proyecto Genoma Humano (IGI/IPI ver 1)(IGI/IPI ver 1)
MétodoNúmero
de genesLongitud
promedio (aa)
Genes conocidos (RefSeq/SwissProt/TrEMBL)
14.882 469
Ensembl (Genscan + similaridad con prot, EST y mRNA de cualquier organismo) + Genie
4.057 443
Ensembl 12.839 187
Total 31.778 352
Evaluación de IGI/IPI• IGI contains 19/31 (61%) novel genes in the human
genome. Fragmentation rate = 1.4.• 81% of 15.294 mouse cDNAS showed similarity to the
human genome seq whereas only 69% showed similarity to IGI suggesting a sensitivity 69/81 = 85%.
• 477 IGI gene predictions were compared to 539 confirmed genes and 133 pseudogenes on chr. 22. 43 (9%) hit 34 pseudogenes (fragmentation rate = 1.2). 63 (13%) did not overlap any current annotation.
• Assuming a rate of overprediction of 20% and a rate of fragmentation of 1.4, the IGI would be estimated to contain 24.500 actual human genes.
• Assuming that the gene predictions contains only 60% of previously unknown human genes, the total number of genes in the human genome would be 31.000.

19
Estimas del nEstimas del núúmero de genes existentes en el genoma humano (2001)mero de genes existentes en el genoma humano (2001)
Método Estima Autores
Cinética de reasociación 40.000 Lewin 1980
Tamaño del genoma/tamaño del gen 100.000 W. Gilbert (Lewin 1990)
Número de islas CpG 70.000-80.000
Antequera and Bird 1993
Número de EST 35.000-120.000
Ewing and Green 2000; Liang et al. 2000
Asociación de EST e islas CpG 142.000 Dickson 1999
Comparación del genoma humano con el de Tetraodon nigroviridis
30.000 Roest Crollius et al. 2000
Extrapolación de los cromosomas 21 y 22
30.500-35.000
Dunham 2000
Análisis inicial del genoma humano 32.000 Lander et al. 2001
Análisis inicial del genoma humano 38.000 Venter et al. 2001
Genoma Humano (versión 21-Oct-2004)
• Se han conseguido secuenciar 2.850 Mb (99% de la eucromatina).
• La tasa de error es 1/100.000 bases (Q = 50).
• Se ha reducido el número de “gaps” (huecos) de ~150.000 a sólo 341.
• De ellos, 33 (total ~198 Mb) en la heterocromatina y 308 (total ~28 Mb) en la eucromatina.
• Tamaño total: 2.850 + 198 + 28 = 3.080 Mb.
• Número de genes: 20.000-25.000 (19.600 genes conocidos + 2.200 predicciones).
• Pseudogenes: ~20.000.

20
Segmental duplications comprise~5.3% of the euchromatic genome
El proyecto ENCODE• El proyecto ENCODE (Encyclopedia od
DNA elements) pretende identificar TODOS los elementos funcionales de la secuencia del genoma humano.
• Fase piloto. Análisis detallado de 44 regiones discretas repartidas por todo el genoma que suman ~30 Mb (~1%).
• Fase de desarrollo tecnológico.• Fase de producción. Aplicación de las
técnicas desarrolladas en la fase anterior al restante 99% del genoma.







