dinámica molecular de mezclas multicomponentes
TRANSCRIPT

Dinámica Molecular de Mezclas Multicomponentes
F,z,iI
Gustavo Adolfo Chapela C as tañares *
Trabajo presentado en ocación de su ingreso a la Academia Mexicana de México D. F. , a 23 de junio de

Dinámica Molecular de Mezclas Multicomponentes 1. Introdución
1. Introdución.
La Dinámica Molecular es un método de simulación por computadora de
átomos, moléculas y sus mezclas que se desarrolló durante la década de los
años sesentas, utilizando la solución numérica de las ecuaciones de
movimiento de un sistema de pocos átomos (algunos cientos) interaccionando
con un potencial definido.
Los potenciales impulsivos de esferas duras y pozos cuadrados son hasta la
fecha muy populares, pero los potenciales tipo Lennard-Jones, siendo mas
realistas, permiten el uso de un potencial continuo que representa bien
algunas sustancias simples tales como los gases nobles.
Las restricciones de la interacción por parejas y las condiciones periódicas a
la frontera se imponen para poder simular el comportamiento de sistemas
realistas. Un sistema que contenga una mol de argón a condiciones normales,
tendrá 6.023xE23 partículas, que obiamente son mucho mas que algun
centenar. También sabemos que la interacción de tres cuerpos contribuye de
manera significativa ( entre un 10 a 20 %).
Para lograr la simulación de sistemas de moléculas complejas, se han utilizado
varias formas de representarlas. Una de las mas exitosas y a la vez sencilla,
es la de pensar que las moléculas estan formadas por átomos o pseudo
.19

Dinámica Molecular de Mezclas Multicomponentes 1. Introdución
átomos que interaccionan entre si (intermolecularmente) con potenciales de
Lennard-Jones y en algunos casos con una carga puntual situada en el centro
de la partícula correspondiente. Esta es la forma que emplearemos en este
trabajo para representar a las moléculas.
Asi, el agua estará representada por un átomo central de oxígeno y dos
átomos de hidrógeno colocados a 1.7 Á de distancia y formando un ángulo de
109 0 , que corresponden con las mediciónes experimentales. Se asignan las
masas de cada uno de ellos, posiciones y velocidades iniciales y se procede a
resolver numéricamente las ecuaciones de movimiento. El método de
integración es un simple Euler o método trapesoidal, ya que se ha
comprobado que los métodos mas sofisticados no dan una gran ventaja, dado
el gran número de partículas, y por tanto ecuaciones diferenciales, que hay
que considerar.
Los sistemas que reportarmos tienen, típicamente, entre 256 y 500
moléculas. Se acomodan en una caja cúbica cuyos lados se ajustan para
obtener la densidad deseada, formando un arreglo cristalino fcc, es decir un
arreglo cúbico centrado en las caras del cubo.
Las velocidades se asignan para obtener una temperatura cercana a la
deseada y vigilando que los momentos lineal y angular sean cero. Dadas las
condiciones periódicas, estos momentos deberán conservarse durante la
corrida. La energía total del sistema deberá ser constante, ya que estamos
wi

Dinámica Molecular de Mezclas Multicomponentes 1. Introdución
usando un ensamble micro canónico donde, por definición, la energía, el
volumen y el número de partículas se mantienen constantes.
Una vez asignadas las posiciones inicales de las partículas, la caja se agranda
en una de sus dimensiones, al doble o al triple, para formar una caja alargada
con una muestra de material cristalino en el centro. La caja se agranda para
producir una lámina infinita en las direcciones que permanecen constantes.
La dinámica se inicia a condiciones de temperatura y densidad que nos
aseguren que el sistema esta dentro de la curva ortobárica y también dentro
de la curva espinodal, o sea dentro de la zona de inestabilidad del fluído. Si
estamos en esta zona de inestabilidad el fluído perderá su estado cristalino y
se descompondrá en un líquido en equilibrio con su vapor a la temperatura
del sistema. Esto se denomina descomposición espinodal y es un fenómeno
que se observa en la naturaleza, aun a escala macroscópica.
Las condiciones de inestabilidad en un fluído, se dan cuando la pendiente de
una isoterma en un diagrama de volumen contra presión es positiva.
Es así que al usar una de las propiedades intrínsecas de los sistemas
termodinámicos se pueden preparar muestras para medir las densidades del
líquido y el vapor en equilibrio, asi como las tensiones superficiales y las
presiones correspondientes.
me

Dinámica Molecular de Mezclas Multicomponentes 1. Introdución
El fenómeno de descomposición espinodal es un fenómeno que ocurre con
gran rapidez, del orden de pico segundos, permitiendo asi la formación de
esta muestras en una escala de tiempo de simulación rasonable y por ende de
tiempo de máquina. Entre unos 1000 a 5000 pasos de integración de 3
fanthom segundos son necesarios para obtener una muestra de Lennard-
Jonesium. Para sistemas como agua y sus mezclas puede tomar mas tiempo.
Se utilizan entre 10,000 y 100,000 pasos de integración para obtener los
promedios que se exponen en este estudio.
En las siguientes dos secciones se presentan, de manera resumida, los
métodos de Dinámica Molecular y Monte Carlo, y en la sección 4 se
ejemplifica el uso de los potenciales de interacción molécular. En la sección 5
se describe la forma de obtener las propiedades que nos interesan en este
trabajo: la presión, la densidad y su perfil, la función de correlación radial y
la tensión superficial. En la sección 6 se extiende la explicación del fenómeno
de descomposición espinodal y en la sección 7 se dan como ejemplo algunos
de los resultados obtenidos para componentes puros y sus mezclas. Las
mezclas, en todos los casos son acuosas. En esta sección final se avanzan
algunas concluciones y se habla del trabajo futuro en este campo.
vi

Dinámica Molecular de Mezclas Multicomponentes 2. Dinámica Molecular
2. Dinámica Molecular.
El método de Dinámica Molecular para potenciales continuos fué desarrollado
por Verlet 121 y se basa en la solución numérica de las ecuaciones de
movimiento de un conjunto de partículas. Se asignan posiciones y velocidades
inicales a las partículas que interaccionan con un potencial continuo tipo
Lennard-Jones. Se imponen condiciones periódicas a la frontera para simular
un sistema macroscópico. La caja típica es un cubo rodeado por réplicas de si
misma.
Si la velocidad de la partícula 1 esta dada como LI. y la fuerza que se ejerce
sobre ella es F. y tiene una masa mi la ecuación de movimiento es:
F.
dt(4.1)
Si se utiliza un esquema de diferencias centrales la ecuación (4.1) se puede
escribir, despejando las posiciones en el tiempo t +h queda:
2 r1 (t +h )=2r 1 (t )_r 1 (t—h )+h —f1 (4.2)
m
Las velocidades se pueden calcular empleando la siguiente relación:

Dinámica Molecular de Mezclas Multicomponentes 2. Dinámica Molecular
r. (t-r. (t +h)
L1 (t ¡ '
(4.3)
1\ h
Las ecuaciones (4.1)-(4.3) son las requeridas para el procedimiento de
Dinámica Molecular.
Oj

Dinámica Molecular de Mezclas Multicomponentes 3. Monte Carlo.
3. Monte Carlo.
El método propuesto por Panagiotopoulos [1] se emplea para calcular el
equilibrio entre fases, normalmente líquido-vapor, empleando lo que el
denomina el ensamble de Gibbs, debido a que se aplican las condiciones de
equilibrio entre fases propuestas por él hace mas de 100 años. Estas
condiciones son:
p l =P,J (3.1)
/L1 =111) (3.2)
donde p y /1 son la presión y el potencial químico, respectivamente, de las
fases líquida ,I , y vapor ,v.
El método consiste en una serie de pasos que implican, primero, una
simulacion de Monte Carlo en el ensamble canónico (N, Y, T) para cada una
de las fases, y para asegurar que la condición (3.1) de equilibrio entre dichas
fases se aplica, se procede a efectuar un paso en un ensamble a presión y
temperatura constantes (N, P, T), que consiste en cambiar el volumen de
ambas cajas, manteniendo el volumen total constante. El tercer paso se realiza
en un ensamble macro canónico (1_L, Y, T), es decir a potencial químico,
volumen y temperatura constantes, para garantizar que la condición (3.2) de
10

Dinámica Molecular de Mezclas Multicomponentes 3. Monte Carlo.
equilibrio se cumple.
En realidad cada caja, la que contiene el líquido y la que contiene el vapor,
son sistemas representativos del ensamble generalizado de Hill 131 a (/1, P, T)
constantes. Las probabilidades para aceptar cada uno de los movimientos en
cada uno de los pasos es
Desplazamiento de partículas, ensamble canónico (N, Y, T).
desp =exp(_/3AE)
(3.3)
donde la probabilidad PdL25P de aceptación del desplazaminto depende de
/3=1/kbr , con k la constante de Boltzman y T la temperatura absoluta, y de
cambio de energía del sistema AE.
Cambio de volumen, ensamble a presión y temperatura constantes
(N, P, T).
II uoI =eHP_P[AE1 +AE _NbTIn + I _N
V k
b T 1U
11L1 -Av
_i (3.4)
11

Dinámica Molecular de Mezclas Multicomponentes 3. Monte Carlo.
donde la probabilidad de la aceptación del cambio de volumen kjuol
depende del cambio de las energías y AEU , de los números de
partículas N' y NL! y de los volúmenes ¡11 y 110 del líquido y del vapor. El
cambio de volumen LLI del sistema también influye. ¿Ch y T significan lo
mismo que en la ecuación (3.3).
Intercambio de partículas, ensamble macro canónico/.L Y, T).
¡jU
(N i + i)l go jfl =exP[_P[AEI +E11 +kbT In VINO
jJ (3.5)
donde la probabilidad de aceptar un intercambio de partículas 9jnt
depende
de las cantidades que se muestran en la ecuación (3.5) y que significan lo
mismo que en las anteriores.
12

Dinámica Molecular de Mezclas Multicomponentes 4. Potenciales de Interacción.
4. Potenciales de Interacción.
Los potenciales de interaccion empleados en este trabajo son, básicamente, el
de Lennard-Jones y el potencial jónico; ambos centrados en cada uno de los
átomos o pseudo-átomos que componen una molécula.
El potencial de Lennard-Jones UU entre dos átomos de especies i y j, se
describe con la
siguiente relación:
U¡j(r¡j u [(a 12 íaii]6]
(4.1) [HJ
donde E. y G, son el tamaño del pozo del potencial promedio y el diámetro
atómico promedio de la sustancias i y j de que se trate y r es la distancia
entre los centros de dichos átomos.
El potencial iónico Ujj entre dos iones de especies i y j esta representado por
la relación:
q1q1 (4.2)
Ii
13

Dinámica Molecular de Mezclas Multicomponentes 4. Potenciales de Interacción.
Donde q1 y q- son las cargas centradas en los iones de especies i y j y r-1 es
la distancia entre los sus centros.
Para ilustrar el tipo de potenciales de interacción que se han utilizado se
presenta el empleado en el caso del agua. Se trata del potencial SPCE [51
(por
sus siglas en ingles Simple Point Çharged Extended potential) que se basa en
un potencial de Lennard-Jones centrado en el oxígeno y una carga negativa
también centrada en este mismo átomo. Los átomos de hidrógeno presentan
solo una carga cada uno de ellos. Los parámetros de este potencial son:
U O = 3.166 A E O IkJJ = 77.34 °K
q 0 = -0.8476 q 11 = 0.4238. (4.3)
r 0 = 1.0 A aHOH = 109.5°.
Con rOH la distancia entre el oxígeno y los hidrógenos y aH011 el ángulo que
forma la terna HOH.
La primera simulación de moléculas diatómicas con potenciales de Lennard-
Jones en los centros de los átomos fue realizada en 1977 por Singer et al. 161,
donde simularon moléculas como C12, Br2, F2 y CO2.
Para el caso de moléculas de mayor tamaño, como es el caso del butano, se
14

Dinámica Molecular de Mezclas Multicomponentes 4. Potenciales de Interacción.
requiere tomar en cuenta el potencial intramolecular que proviene del
mantenimiento de la estructura de la molécula. Así, se tendrán que tomar en
cuenta los potenciales que mantienen los ángulos entre los átomos de la
molécula y también los que mantienen a los átomos en un mismo plano, es
decir potenciales de torsión o también llamados del ángulo diedro.
Para el caso del butano 71 se emplean dos pseudo-átomos, el CH3 y el CH2 con
la molécula formada por cuatro partículas como: CH3-CH2-CH2-CH3
El potencial que considera los factores mencionados se ve como sigue:
II = (aj -aj + Ck,Ø+(Ck,J CoSIEk) angulos angulos / =1
diedros
(4.4)
Para el caso del butano los parámetros son:
aCH3 = 3.740 Á ECH3/kb
aCH2 = 3.740 A 8CH2b
qc113 = 0.0 qc112
rcC =1.53 A accC
= 111.4 °K.
= 111.4 °K. (4.5)
= 0.0.
= 111.00.
15

Dinámica Molecular de Mezclas Multicomponentes 4. Potenciales de Interacción.
Las constantes para el caso de los ángulos diedros, en (kjlmol) son:
C 0 = 209.0 C 1 = 0.0 C 2 = 0.0 C 3 = 209.0.
(4.6)
Las fuerzas desde luego, por definición, se calculan evaluando el gradiente del
potencial.
Las constantes de interacción cruzadas se obtienen con las siguientes
relaciones:
a. +a.
Ii 2 J (4.7)
su = \JSj Sj
16

Dinámica Molecular de Mezclas Multicomponentes 5. Propiedades
S. Propiedades.
Las propiedades que se calculan en este trabajo son: la temperatura, la
presión, el perfil de densidad, la función de correlación radial y la tensión
superficial. A continuación describiremos brevemente como se calcula cada
una de ellas.
La temperaturaT , se calcula empleando la velocidad cuadrática media en
unidades de temperatura.
m (5.1)
3Nk.(t
b ¡ =1
Donde m es la masa, N el número de partículas, u (t ) la velocidad al tiempo
' b la constante de Boltzman.
La presión, p, se obtiene con el teorema del vinal de Clausius, como sigue:
N P4-1 N P m.u. U. + F.. r.. (5.2)
ab y . ¡ la ¡/3 . - ¡ja ij/3 ¡=1 I=1j =l+1
donde pab es un elemento del tensor de presiones,a y b son los componentes
(x, y, z), V el volumen, mi es la masa de la partícula i, via es la velocidad en la
dirección a, Fija es el componente a de la fuerza total ejercida sobre la
17

Dinámica Molecular de Mezclas Multicomponentes 5. Propiedades
partícula i por la j, y rijb es el componente b del vector ri-rj. La contribución
cinética a la presión es el primer término de la ecuación (5.2) y la
contribución vinal está dada por el segundo. Las tres componentes diagonales
en el tensor de presiones representan los componentes relevantes de la
presión.
La tensión superficial, g, esta definida cuando la interface es
perpendicular al eje z, como:
(
ra
y = - f P 7Z) - p ~z (5.3)
donde p TZ) es la presión lateral, p es la presión del bulto y la integral está
definida a lo largo de la interface. La integral se piede extrender al infinito,
pues pz) = ¡.1 en el bulto. Con dos interfaces perpendiculares, tal y como es
nuestro caso, esto da:
y--- 2 2 y
-
PLz (5.4)
en donde p = P (a=x, y, z) y Lz es la longitud de la caja en la dirección a aa

Dinámica Molecular de Mezclas Multicomponentes 6. Resultados
6. Resultados.
Los resultados que se presentan aqui son algunos ejemplos de lo que se ha
obtenido del método descito. En primer lugar se dan la ortobárica, el perfil de
densidad y la tensión superficial del agua con el potencial de SPCE. Es claro
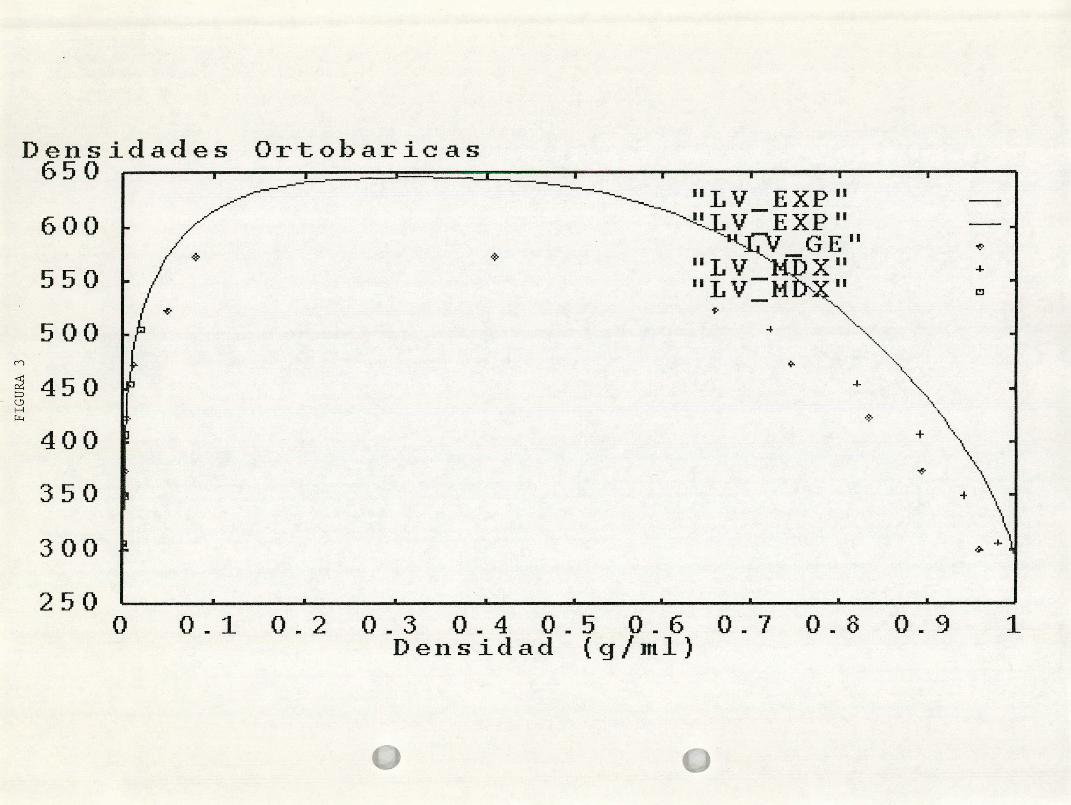
que los resultados obtenidos con Dinámica molecular son mejores que los del
ensamble de Gibbs para el caso de las densidades de coexistencia. Las
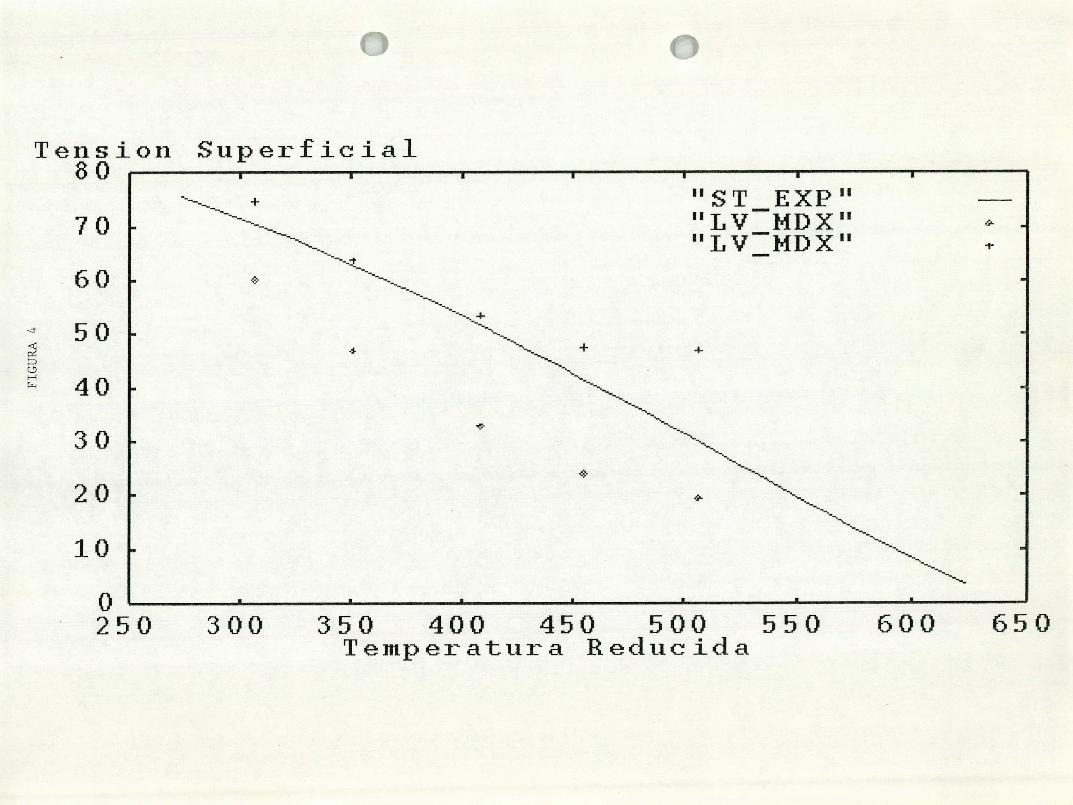
tensiones superficiales estan dentro de un 9% alejadas de la curva
experimental. Datos obtenidos anteriormente ni siquiera caen dentro de la
gráfica que se muestra.
En segundo lugar se muestran los perfiles de densidad del butano, de las
mezclas metanol-agua y tetracloruro de carbono-agua. Al final se dan
mustran algunas fotografías de configuraciones, inicial y de equilibrio, del
agua y de las mezclas de metanol-agua y tetracloruo de carbono-agua.
El trabajo futuro seguirá con el estudio de mezclas mas complicadas y la
obtención de los diagramas de fases para éstas. La prueba que este método
implica para los potenciales de interacción y para los métodos de simulación
con fuerzas de largo alcance es verdaderamente pesada.
19

Dinámica Molecular de Mezclas Multicomponentes Referencias
Referencias.
1 Panagiotopolous, A. Z., Molec. Phys., 1987, 61, 813.
2 Verlet, L., Phys. Rey., 1964, 159, 99.
3 Hill, T. L., Statistical Mechanics, 1956, (McGraw-Hill).
4 Panagiotopolous, A. Z., Quirke, N., Stapleton, M. and Tildesley, D. J., Molec. Phys.,1988, 63, 527.
5 Berendsen, H. J. C., Grigera, J. R. and Straatma J. P., J. Phys. Chem., 1987, 91, 6269.
6 Singer, K., Taylor, A. and Singer, J. V. L., Molec. Phys., 1977, 33, 1757.
7 Ryckaert, J. P. and Bellemans A., Faraday Discuss. Chem. Soc.,1978, 66, 95.
20

Dinámica Molecular de Mezclas Multicomponentes Pies de las figuras.
Pies de las figuras.
Figura 1. El perfil de densidad del h2o a 350 ° K.
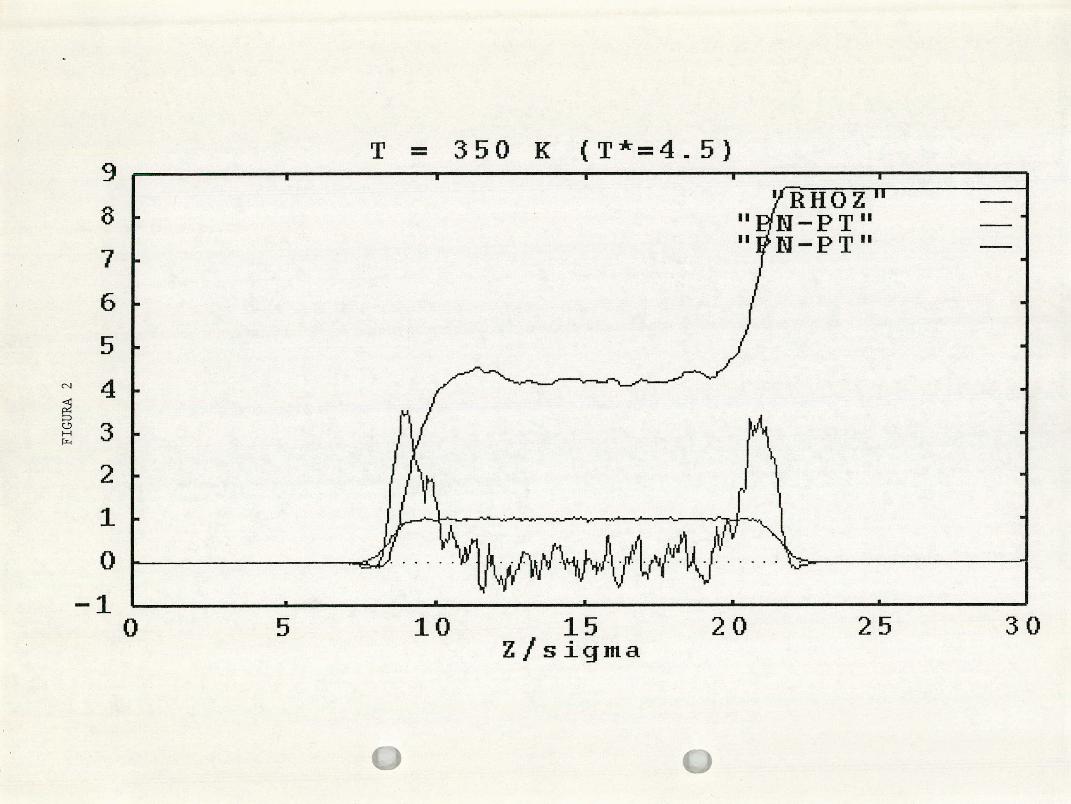
Figura 2. El mismo perfil graficado con las componentes normal y
transversal del tensor de presiones.
Figura 3. La curva experimental de densidades ortobáricas para el agua, en
comparación de los datos obtenidos con el presente método (+) y con el del
ensamble de Gibbs ( ).
Figura 4. La curva de tensión superficial experimental para el agua, en
comparación con los datos obtenidos en el presente trabajo. Los ( )
representan las tensiones superficiales antes de la correción por las fuezas de
largo alcance. Cuando esta correción se aplica se obtienen las (+).
Figura S. Configuración inical de la mezcla de cc14-h2o.
Figura 6. Configuración de equilibrio de la mezcla de cc14-h2o.
Figura 7. Configuración de equilibrio del butano.
21
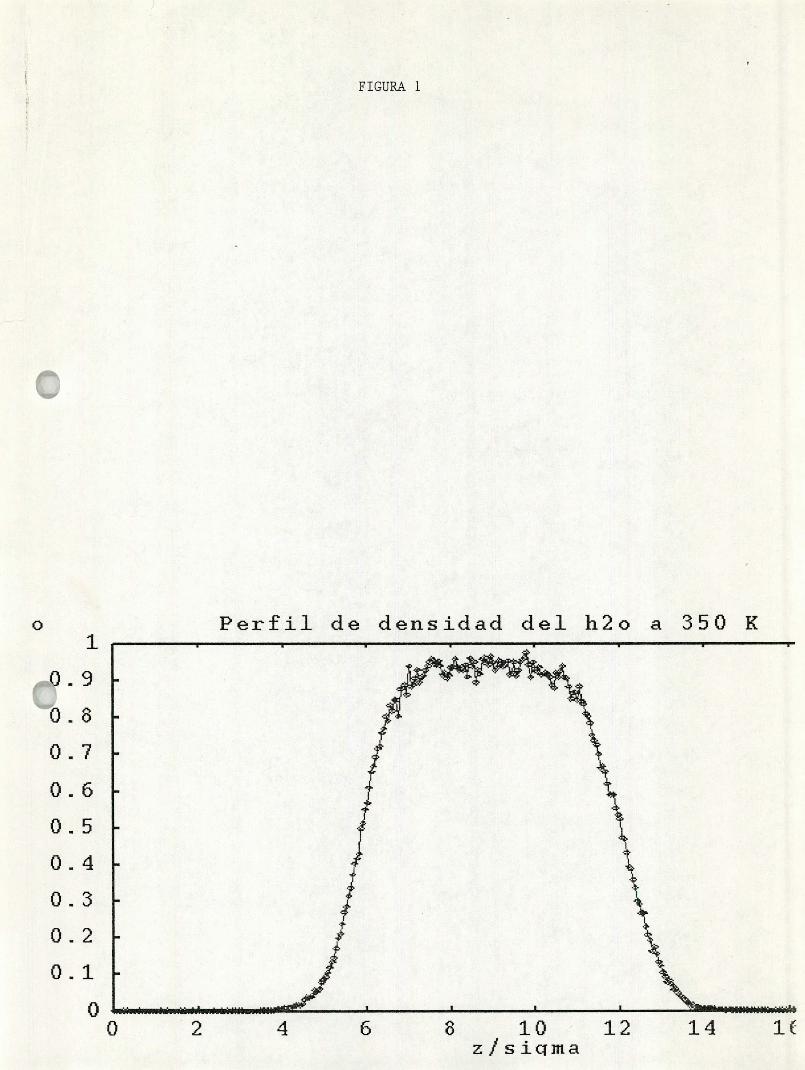
FIGURA 1
cm
o Perfil de densidad del h2o a 350 K
2 4 6 6 10 12 14 1 z/siqina
1
0.9
0.8
0.7
0.6
05
0.4
0.3
02
0.1
9
8
7
6
5
ji4
3
T = 350 K (T*=45) u
RHOZ II N-PT"
0 5 10 15
20 25 30 Z/siqnia
Dens idades Ortobarie as
600
550
500
450
400
350
300
"LV EXP" TIVEXP
]V_GE II "LV tPX 4-
V EM brX"
O 0- 1 02 03 04 0- 5 06 07 0- 8` 09
1 Densidad (q/inl)
Tension Superficial 0V
70
60
50
40
30
20
10
fb
+ " 5 T E XP ulJVMDXII "LV MDX" -
3 + +
-
•
250 300 350 400 450 500 550
600 650 Temperatura Recluc ida
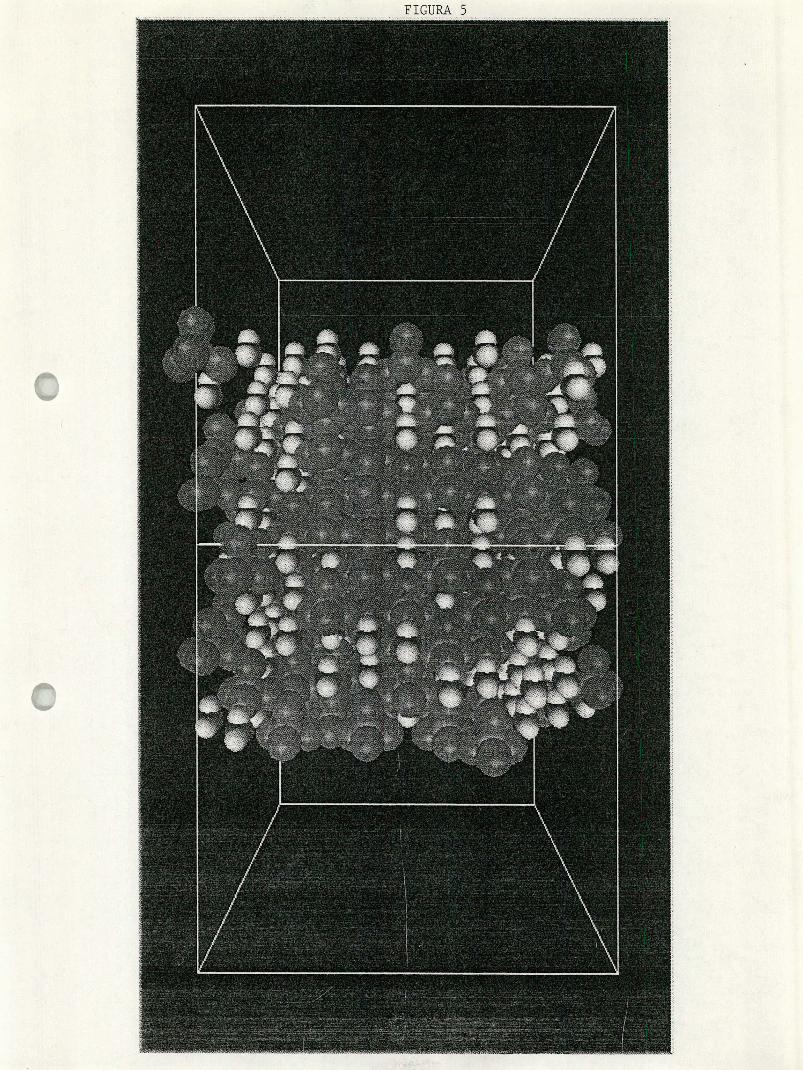
FIGURA 3
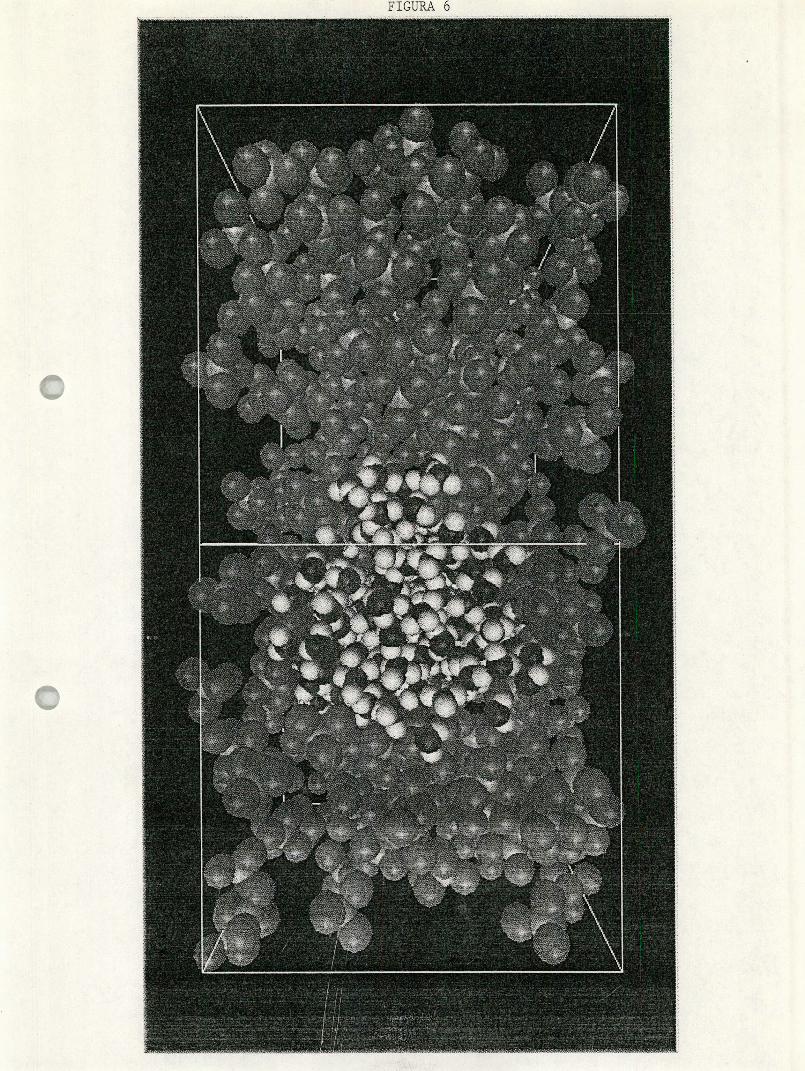
FIGURA 6

Dinámica Molecular de Mezclas Multicomponentes Resumen
Resumen.
La importancia del cálculo del equilibrio líquido-vapor de mezclas
multicomponentes en el cálculo de equipos de proceso químico es innegable.
Las equaciones de estado han ocupado un lugar preponderante en este
campo. Los avances en la capacidad de cómputo han permitido que cada vez
dichas ecuaciones sean mas sofisticadas y por tanto representen mejor el
comportamiento de las sustancias químicas mas utilizadas y de sus mezclas.
Sin embargo al desear obtener las propiedades de estas mezclas cuando se
trata de componentes polares, jónicos o altamente no esféricos, aun las
ecuaciones mas modernas requieren de la ayuda de los datos obtenidos en el
laboratorio.
Los desarrollos recientes ( de los últimos 20 años) en el cálculo de
propiedades termodinámicas por la via de la simulación por computadora de
sustancias puras y sus mezclas, nos permiten avisorar un futuro cercano
cuando podamos obtener datos precisos del comportamiento de mezclas de
componentes polares, no esféricos o iónicos. Dichos cálculos serán de mucha
utilidad en la industria de procesos de sustancias complejas y sus mezclas.
En el presente trabajo mencionamos los resultados obtenidos con una técnica
nueva, la descomposición espinodal, que nos permite obtener resultados, por
la vía de la Dinámica Molecular, del equilibrio líquido-vapor de sustancias

Dinámica Molecular de Mezclas Multicomponentes Resumen
complejas y sus mezclas. Los valores calculados se contrastan con aquellos
que arroja el método del ensamble de Gibbs de Monte CarloJ 11 Entre las
ventajas del método que aqui se reporta estan la de tener una superficie
líquido-vapor con la cual se puede obtener el valor de la tensión superficial,
ademas de no presentar el problema de insertar moléculas altamente no
esféricas ( butano por ejemplo) en sistemas de alta densidad como lo son los
líquidos.
Se presentan resultados de las densidades en la curva ortobárica para
sustancias puras como: agua, butano, cloro y metanol, asi como de algunas de
sus mezclas. Los valores de la tensión superficial también se incluyen.
3

~ NG, ERNESTO RIOS MONTERO
QU 5 LRA ACRADECLR AL DR. CUSAVO CHAPELA, LA lWIlAC I ON QUL ML
HE lIZO PARA COMENTAR SU TRABAJO DL INGRESO A LA ACADEMA,
ITULADO D NAMCA MOLECULAR DE MEZCLAS MULTCOMPONENTES,
EN SU TRABAJO, SE ANALZA UNA NUEVA TECNCA QUE PERMTE
OBTENER RESULTADOS POR LA VA DELA DNAMCA MOLECULAR DE
LQULBRO OQUDO-VAPOR DE SUSTANCAS COMPLEJAS Y SUS MEZCLAS, CONSDERANDO QUE EN LA INDUSTR I A DE PROCESO, EL
COSTO DE SEPARACON CELOS PRODUCTOS DE REACCON ES UNA
PARTE SCN FJCATVA DEL COSTO TOTAL DE ELABORACON DE D CHOS
PRODUCTOS. FORMAS EFJC ENTES Y BARATAS DE PREDEC R EL
COMPOR AMENTO DE LAS SUSTANCAS QUE INTERV I ENEN EN DCHOS PROCESOS, ES UN PUNTO NODAL PARA DSM NUR EL COSTO DEL
DESARROLLO DE NUEVOS PROCESOS Y CELA OPTMZAC ON DE LOS YA LXSTENTES. EN LA ACTUAL DAD, ES IND I SPENSABLE QUE EN NUESTRO
PAS SE CUENTE CON LA TECNOLOCA Y HLRRAM ENTAS QUE PERMTAN
EFECTUAR LA INGEN I ER I A BASCA DE LS PROYECTOS EN L AS EMLESAS
NACONALES DE NCENERA QUE ACTUALMENTE EJECUTAN LOS
PROYECTOS DELA INDUSTR I A PETROLERA, PETROQUMCA Y QUMCA DEL
PAS, EL TRABAJO QUE COMENTAMOS, PROR CA EL DSPONER DE
HERRAMENTAS QUE PERMIAN REALZAR LO ANTES MLNC ONADO.
EL PROCEDMENTO DE D NAMCA MOILICULAR QUE NOS DESCPBE EL
TRABAJO DEL DR. CHAPELA, ESTA PROXMO /\ SER UT I L PARA ESTOS
F NES, EL CA CULO DE LAS PROP EDADES TLRMODNAMCAS DE
COMPONEN ES COMPLEJOS, POLARE 5, ION I COS 0 CON FORMAS

N. N RIO RFOS MONTERO
ULRTLV HE NO ESPERA, DL AUU LA H OHANA DEL HABAJO
E PESEN lA DO.
EL PROCEDMENTO EMPLEADO SE BASE EN EL PRNCP O DE
DESCOMPOSHON ESE NODAL QUE PERMTE PRODUCR MUESTRAS DE
L QU DO EN EQULHRO CON SU VAPOR EN TEM POS RASONABLES Y LOS
RESULTADOS QUE EMPEZA A ARROJAR SON MEJORES QUE LOS DE
MONTE CARLO DEL CBBS ENSAMBLE, ADEMAS DE SER UN METODO QUE
PUEDE DAN RESULTADOS A ALTAS DENSDADES.
AS HSMO, DEBEMOS COMENTAR QUE LA EACHDAD DEL NUEVO CENTRO
DE SUPERCOMPLJTO Y VSUALZACON Dr LA UAM/ZTAPALAPA, PERMHO
LLEVAR A CABO ESTE ESIUDO, SN ESTO NO HUBERA SDO POSHLE EL
REALZAR EL TRABAJO QUE HOY NOS PRESENTA EL DR. CHAPELA.
QUHERA TERHNAR MS COMENTAR OS MENCONANDO QUE EL TRABAJO
QUE HOY PRESENTA EL DR. CHAPELA, ES DE UNA CRAN CA.UDAD
e TECNCA, RCLJROSO EN SUS PLAN EAMENTOS Y ORCNAL EN SU
PRESENIACON, POR TANTO, FELCTO AL DR. CHAPELA POR EL TRABAJO
PRESEN1ADO QUE SN DUDA, ES UNA CONIRBUCON A LA NCEN ERA
QUMCA Y UNA PATHCACON A LA CAPAC DAD DE LOS INGENIEROS
MEX CANOS. DEBEMOS RESALTAR POR CONSCU ENTE, LA IMPORTANCIA
QUE TENE r CONTAR EN NUESTRO RAS CON INSTALACIONIES
MODERNAS Y FK.4NTES, QUE PERMTAN REALZAR INVESTIGACIONES Y
DESARROLLOS ACORDES AL AVANCE TECNOLOCCO DE NUESTRO PAS.
MUCHAS CRACAS JUNO 23, 1994

PALABRAS DE BIENVENIDA DEL llG. EDUARDO ROJO Y DE REGIL, CON MOTIVO DEL IIGRESO A LA ACADEMIA MEXICANA DE DGENIERIA DEL DR. GUSTAVO ADOLFO CHAPELA CASTAÑARES EL 23 DE JUNIO DE 1994.
Señor Presidente de la Acedemia Mexicana de Ingeniería, compañeros académicos, señoras y señores:
Es un honor para la Comisión de Especialidad de Ingeniería Química, recibir el día de hoy como Académico de Número al Dr. Gustavo Adolfo Chapela Castañares, quien presentó el trabajo "Dinámica Molecular de Mezclas Multicomponentes", el cual fue comentado por los académicos:
Ernesto Ríos Montero Carlos Mena Brito.
Resulta de suma importancia para el diseño de equipos utilizados en la industria de proceso, poder contar con la información relativa al equilibrio líquido-vapor de mezclas multicomponentes.
Es fundamental, la aportación que hace el Dr. Chapela con una nueva técnica que es la DESCOMPOSICION ESPINODAL, la cual permite la obtención de resultados por la vía de la dinámica molecular, del equilibrio líquido-vapor de sustancias complejas y sus mezclas.
Es nuestro interés en la Comisión de Especialidad de Ingeniería Química, que se continúen realizando trabajos especializados de este tipo, ya que son un fuerte respaldo a la carrera profesional, así como también son fuentes de enriquecimiento para la categoría y prestigio de nuestra academia.
Espero que el Dr. Chapela se incorpore de inmediato a colaborar en las actividades de esta Comisión de Especialidad, así como las Coordinaciones de Programas.