desarrollo de una técnica analítica
TRANSCRIPT

Facultad de Química Farmacia Centro de Bioactivos Químicos
Desarrollo de una técnica analítica para la cuantificación de piridina en
residuales complejos
Autora: Lilibét Pérez Brito
Tutores: Lic. Teófilo Exiquio Gaitán Placeres Lic. Blanca Nieves Hernández Martínez
Curso: 2005-2006 “Año de la Revolución Energética en Cuba”

Resumen
Se ha realizado un estudio de la formación y extracción del complejo mezclado
Cu (II)- (SCN- )2 - (piridina)2 , para desarrollar una técnica extractofotométrica
que permita determinar el contenido de esta amina, seleccionando la longitud
de onda de trabajo, el pH de equilibrio de la fase acuosa, la cantidad de
reactivos a emplear, el camino óptico, el tiempo de medida y el rango de
concentraciones en que se cumple de la ley de Bouguer Lambert Beer,
correspondiente a: 75-300 ppm.
Además se comprobaron algunos parámetros analíticos como: repetibilidad,
reproducibilidad, linealidad y exactitud obteniendo resultados satisfactorios.
También se determinó el límite de cuantificación de la técnica tanto por el
método de la curva de calibrado como por adición de estándar.
Por otra parte fueron evaluadas, varias especies potencialmente interferentes
presentes en las matrices de los residuales del proceso productivo de G-1, tales
como: bromo, bromuro de potasio, disulfuro de carbono, anhídrido acético,
nitrometano, furfural, isobutilamina, G-1 y G-0, encontrando que solo estos dos
últimos causan interferencia en la determinación de piridina por la técnica
propuesta.
Para realizar la aplicación de la técnica a residuales parcialmente tratados del
proceso productivo de G-1 se investigo la composición de las matrices
correspondientes comprobando la ausencia de especies interferentes. Los
métodos analíticos empleados para ello fueron: CCD, HPLC y ensayos
cualitativos. Finalmente se determino el contenido de piridina en dichos
residuales tanto por la técnica desarrollada en el trabajo como por UV.
Palabras claves: Complejo mezclado, extractofotometría, piridina.

Summary
A study has been carried out about the formation and extraction of the mixed
complex Cu(II)-(SCN - )2 - (pyridine)2, to develop a extractophotometric technique
that allows to determine the content of this amine. We select the wave length
work, the equilibrium pH of the aqueous phase, the quantity of all the reagents to
use, the time contact of measure and the range of concentrations in what the of
Bouguer Lambert Beer law Is obeyed ; the range , correspond to 75-300 ppm.
Some analytic parameters as repeatability, reproducibility, linialirity and accuracy
was assayed obtaining satisfactory results. The limit of quantification of that
technique was also determined by the method of the calibrating curve and by
standard addition methods.
On the other hand they were evaluated, several species potentially present as
interfering ones in the examined samples of waste water from G-1 production
plant. They include bromine, potassium bromide, carbon disulphide, acetic
anhydride, nitrometane, furfurol, isobutilamine, G-1 and G-0, finding that within
these compounds only two last cause interference in the pyridine determination
by the proposed technique
To carry out the application of the technique a pre treatment must by applied to
the waste water samples in order to check absence of interfering species. Other
analytic techniques employees were TLC, HPLC. Finally we determine the
pyridine content in this waste water resulting prom de G-1 production plant.
Key words: Mixed complex, extractophotometric method, pyridine.

Índice
Índice
Contenido Pág.
Introducción................................................................................................... 1
Capítulo I: Fundamentación teórica............................................................... 4
1.1 Extracción con solventes orgánicos: Fundamento............................. 4
1.2 Equilibrio de extracción...................................................................... 4
1.2.1 Extracción física............................................................................ 4
1.2.2 Extracción por vía química............................................................
1.3 Extracción y su combinación con técnicas analíticas....................... 9
1.3.1 Generalidades............................................................................... 9
1.3.2 Extractofotometría.......................................................................... 9
1.4 Características de la piridina.............................................................. 10
1.5 Peligrosidad de la piridina................................................................... 13
1.6 Efectos sobre la salud........................................................................ 15
1.7 Características toxicológicas de la piridina........................................ 16
1.8 Influencia ecológica de la piridina..................................................... 17
1.9Degradación, producto de la descomposición y el tiempo de vida
medio...............................................................................................................
18
1.10 Técnica analítica desarrollada en el trabajo..................................... 18
1.10.1 Antecedentes................................................................................ 18
1.10.2 Fundamento de la técnica extractofotométrica empleada en el
trabajo...........................................................................................................
22
1.11 Técnica cromatográfica utilizada en el trabajo................................ 23
1.11.1 Cromatografía de capa delgada (CCD).....................................
23
1.11.2 Cromatografía liquida de alta resolución, HPLC......................... 24
1.12 Consideraciones acerca del diseño de experimento........................ 27
1.12.1 Generalidades............................................................................. 27
1.12.2 Algunos conceptos generales relacionados con el diseño factorial
fraccionado......................................................................................................
29
1.13 Características fundamentales de los métodos analíticos.................. 30

Índice
Capitulo II: Parte experimental.......................................................................... 34
2.1 Reactivos y disoluciones...................................................................... 34
2.2 Selección de las condiciones de trabajo.............................................. 35
2.3 Procedimiento para realizar la curva de calibración............................ 36
2.4 Comprobación de algunos parámetros analíticos que caracterizan la
técnica...............................................................................................................
36
2.5 Adición de estándar.............................................................................. 38
2.6 Aplicaciones......................................................................................... 38
2.6.1Características cromatográficas (HPLC) de la piridina e impurezas
presentes........................................................................................................
38
2.6.2 Estudio de la composición de las matrices de aplicación............... 39
2.6.3 Determinación del contenido de piridina
en los residuales..............................................................................................
41
Capitulo III: Presentación y discusión de los resultados................................. 43
3.1 Estudio de las condiciones de trabajo.................................................. 43
3.2 Confección de la curva de calibración.................................................. 47
3.3 Parámetros que caracterizan la técnica
analítica..............................................................................................................
49
3.4 Adición de estándar................................................................................ 55
3.5 Aplicación de la técnica........................................................................... 56
Conclusiones...................................................................................................... 68
Recomendaciones............................................................................................. 69
Bibliografía......................................................................................................... 70
Anexos................................................................................................................ 76

Introducción
Introducción
El método de extracción es quizás la técnica analítica más versátil de todas las
que se conocen. Esto se debe al campo tan amplio de aplicación que posee y al
hecho de que se relaciona con la mayor parte de los principios físicos-químicos
que se utilizan en la Química Analítica.
Este método es muy simple, rápido y sencillo por lo que se emplea para la
separación, purificación, concentración y análisis de muestras en todas las
escalas de trabajo, desde el microanálisis hasta en los proceso de producción;
se combina de manera muy útil con otras técnicas analíticas de determinación
cuantitativas y cualitativas (1-5).
Actualmente se conocen sistemas de extracciones en los que intervienen
diferentes fases tales como: extracción líquido-líquido, sólido-líquido y sólido-
sólido.
La extracción líquido-líquido se caracteriza por el establecimiento del equilibrio
de extracción entre dos fases líquidas inmiscibles entre si, una de naturaleza
orgánica y otra generalmente acuosa. Este tipo de sistema constituye una
herramienta de trabajo que posibilitan separaciones extremadamente selectivas
de muchos iones y sustancias moleculares, utilizando agentes formadores de
complejos y solventes apropiados.
También se emplea para obtener elementos o compuestos con elevado grado
de pureza a escala industrial.
La utilización de sistemas de extracción con solventes orgánicos con fines
analíticos ha permitido desarrollar numerosas técnicas para cuantificar especies
químicas en diferentes matrices. Dentro de estas se destacan las técnicas
extractofotométricas, debido al empleo de un equipamiento relativamente barato
(6 y 7).
En el Centro de Bioactivos Químicos en la Universidad central de las Villas se
obtienen principios activos, por vía sintética a partir de los desechos de la caña
1

Introducción
de azúcar. El producto principal allí obtenido es el 2-bromo-5-(2-bromo-2-
nitrovinil)-furano (G-1) que presenta una potente acción bactericida fungicida.
En el proceso de obtención de este principio activo se emplea piridina y otras
sustancias tóxicas que se incorporan a los residuales siendo necesario
desarrollar las técnicas analíticas que permitan su cuantificación (8-10). El
siguiente esquema muestra las corrientes residuales del proceso de producción
de G-1:
G-O puro
Taller de G-1
G-1 puro
Carbón activado Carbón impuro (Br2, CS2, py, G-1 etc. )
Anhídrido acético
Br2, CS2 Etanol Impuro ( Br2, CS2, py, G-1 etc. )
Piridina( py) Colas crudas (Br2, CS2, py, G-1 etc.)
Gases (Br2, CS2, py, G-1 etc. )
Etanol
2

Introducción
Problema científico En la Planta de Producción de Bioactivos Químicos se emplea piridina en la
síntesis de G-1, siendo esta una sustancia altamente tóxica y no existe una
técnica analítica que permita determinar el contenido de dicha amina en los
distintos residuales del proceso productivo.
Hipótesis Es posible utilizar la formación de complejos mezclados en los que participen la
piridina y el SCN- como ligandos, en combinación con iones metálicos para
desarrollar una técnica extractofotométrica de determinación de la misma.
Objetivo General
Proponer una técnica analítica extractofotométrica para la determinación de
piridina, que cumpla con los parámetros analíticos establecidos para la
cuantificación de esta amina, potencialmente aplicable a los residuales de la
planta de producción del CBQ.
Objetivos Específicos
1. Investigar las condiciones de formación y extracción del complejo
mezclado formado en el sistema SCN-—piridina- Cu (II). 2. Estudiar el cumplimiento de la ley de Bouguer Lambert Beer del
complejo formado en el extracto clorofórmico. 3. Comprobar la linealidad, precisión, límite de cuantificación y exactitud
de la técnica analítica propuesta.
4. Investigar la especificidad de la técnica analítica estudiada, teniendo
en cuenta la composición de las matrices de aplicación.
5. Determinar el contenido de piridina en residuales de la PPBQ
aplicando la técnica desarrollada.
3

Introducción
4

Capítulo I: Fundamentación Teórica
Capitulo I: Fundamento teórico
1.1 Extracción con solventes orgánicos: Fundamento La extracción con solventes orgánicos o extracción líquido-líquido, se basa en
el principio de que un soluto puede distribuirse en determinada relación entre
dos solventes inmiscibles o ligeramente miscibles entre sí, uno de los cuales es
agua y otro un solvente orgánico (11).
1.2 Equilibrio de extracción: Los equilibrios de extracción líquido- líquido pueden clasificarse de la forma
siguiente, atendiendo al tipo de interacción que ocurre en las fases que
intervienen en el mismo:
• Extracción física.
• Extracción química.
1.2.1 Extracción física:
El proceso de extracción por vía física se caracteriza por interacciones debidas
a fuerzas de van der Waals, la sustancia a distribuir no forma compuestos de
composición definida con el reactivo extractor.
Las moléculas covalentes de las sustancias que se distribuyen prefieren el
disolvente orgánico, ya que éstas entran en el mismo con una pequeña barrera
energética o sin ella, en comparación con la fase acuosa altamente ordenada. El
aumento de tamaño de la molécula a extraer provoca un incremento del
coeficiente de distribución.
1.2.2 Extracción por vía química:
La extracción por vía química es un proceso de transferencia de masa entre las
fases, acompañado de transformaciones químicas. En el mismo se cumple la
ley de reparto en la primera parte de la isoterma de Langmuir (log D vs pH) (I), si
4
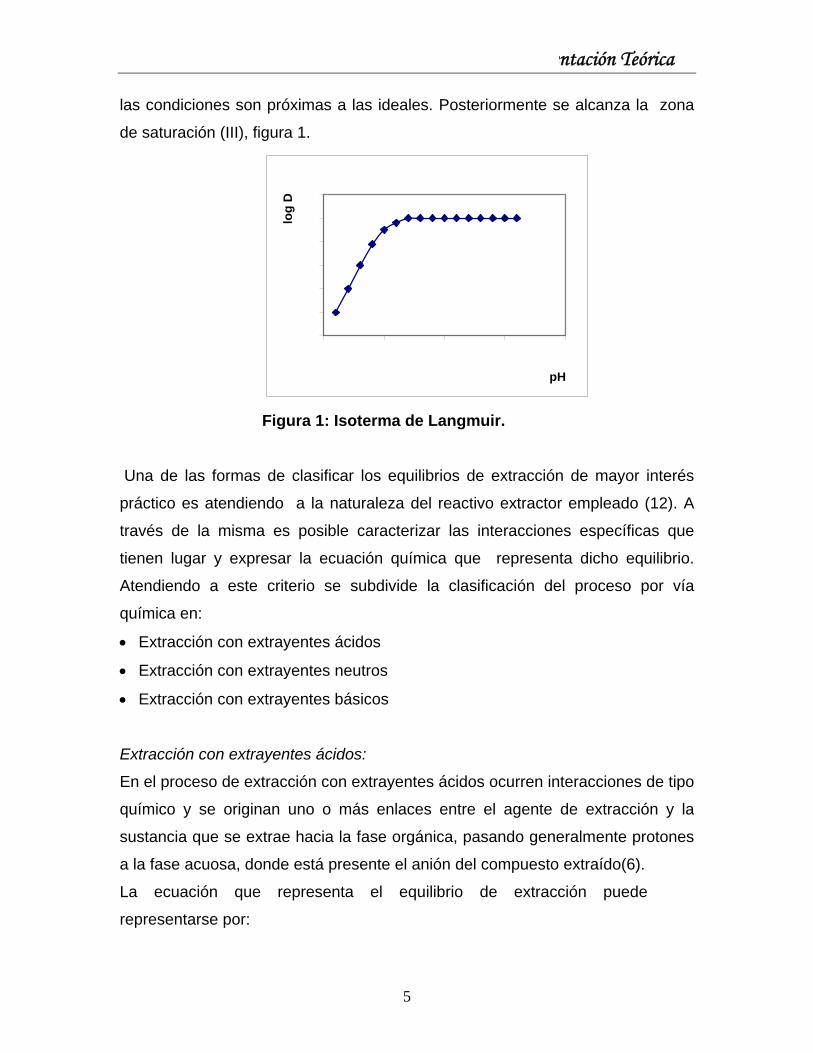
Capítulo I: Fundamentación Teórica
las condiciones son próximas a las ideales. Posteriormente se alcanza la zona
de saturación (III), figura 1.
0
10
20
30
40
50
60
0 50 100 150 200pH
log
D
Figura 1: Isoterma de Langmuir.
Una de las formas de clasificar los equilibrios de extracción de mayor interés
práctico es atendiendo a la naturaleza del reactivo extractor empleado (12). A
través de la misma es posible caracterizar las interacciones específicas que
tienen lugar y expresar la ecuación química que representa dicho equilibrio.
Atendiendo a este criterio se subdivide la clasificación del proceso por vía
química en:
• Extracción con extrayentes ácidos
• Extracción con extrayentes neutros
• Extracción con extrayentes básicos
Extracción con extrayentes ácidos:
En el proceso de extracción con extrayentes ácidos ocurren interacciones de tipo
químico y se originan uno o más enlaces entre el agente de extracción y la
sustancia que se extrae hacia la fase orgánica, pasando generalmente protones
a la fase acuosa, donde está presente el anión del compuesto extraído(6).
La ecuación que representa el equilibrio de extracción puede
representarse por:
5

Capítulo I: Fundamentación Teórica
Mn+(ac) + nHRo = MRn(o) + nH+
(ac)
Corresponden a este tipo de reactivos extractores los siguientes:
• Ácidos carboxílicos: RCOOH
• Ácidos alquilfosfóricos: (RO)2POOH (Ej. di-2-etilexilfosfórico DEHF )
• Ácidos derivados del arsénico ROAsO(OH)2
• Fenoles y sus derivados
• Oximas, las que existen en dos formas tautoméricas ( =C=N-OH y
=C=NH →O)
En el caso de ligandos que presentan además del grupo acídico otro que
posea pares de electrones libres pueden formar más de un tipo de enlace,
pues no sólo intercambian protones sino que forman enlaces donante-
aceptores, dando lugar a ciclos estables ”quelatos”. Las oximas y otros
ligandos capaces de formar quelatos tienen importantes aplicaciones
relacionadas con el análisis químico.
En algunos extrayentes ácidos se presenta también el mecanismo de
solvatación. Así ocurre por ejemplo en la extracción de ácidos minerales
con ácidos alquilfosfóricos:
(RO)2POHO ……HX
En el mismo tiene lugar una interacción electrostática entre el oxígeno del
grupo fosfórico y el hidrógeno acídico. El equilibrio de extracción puede
representarse de la forma siguiente:
mHI(ac) + n(RO)2POOH(ac) = n(RO)2 POOH.mHI(o)
Extracción con extrayentes neutros:
Si el proceso de extracción del soluto se realiza empleando extrayentes
neutros, las interacciones químicas que ocurren dan lugar a enlaces
donante-aceptores. Los reactivos extractores de este tipo incluyen:
Cetonas, aldehídos, éteres, ésteres, sulfóxidos, óxidos etc.,
6

Capítulo I: Fundamentación Teórica
Los grupos funcionales, entre otros, son:
| | | | | | | ⎜
−C−O−C− ; −C−S−C− ; − C=S; − C=O ; −P=O; S=O
| | | | | ⎜
El equilibrio de extracción puede representarse de la forma siguiente:
MXq (ac) + mL(o) = MXq .mL(o)
Los extrayentes neutros también se han utilizado para la extracción de
ácidos minerales. Este tipo de mecanismo se le llama ónicos
TBP(o) + HA (ac) ↔ HA:TBP(o)
⎜
⎯ P→O……HA
⏐
Como se observa el reactivo extractor posee grupos funcionales con
pares de electrones libres a través de los cuales se produce el enlace
donante-aceptor. Entre las cetonas se destaca el empleo de
metilisobutilcetona (MIBC) en la extracción de metales (6).
Extracción con extrayentes básicos.
Los llamados extrayentes básicos corresponden a aminas de alta masa
molecular. Estas pueden ser primarias, secundarias, terciarias y bases de
amonio cuaternarias según el número de radicales orgánicos que posean
(RH2N:, R2HN:, R3HN:, R3NH+).
7

Capítulo I: Fundamentación Teórica
Las aminas constituyen un grupo muy estudiado debido a sus múltiples
aplicaciones industriales (13). Estas bases se protonan en medio ácido
según el siguiente equilibrio:
R3N:(ac) + HA (ac) = R3NH+(ac) + A-
(ac)
Las bases protonadas así obtenida participan en procesos químicos tales
como:
• Intercambio aniónico:
n[(R3NH+)A-] (o) + MBm-n
(ac) = [(R3NH+)nMBm-n] (o) +nA-
• Adición:
n[(R3NH+)A-] (o) + MAp(ac) = [(R3NH+)nMA-(p+n)] (o)
En la práctica no siempre se utiliza una sola amina o su sal como reactivo
extractor, sino mezclas de ellas que se conocen como mezclas técnicas.
También se han empleado con excito mezclas de ácidos y aminas u otros
reactivos extractores, las cuales presentas ventajas relacionadas con la
zona de pH de extracción (14).
Ejemplo:
Co2+ (ac) + 2RCOOH(o) + Am(o) = Co(OOCR)2 Am.xH2O(o) +2H+
(ac)
8

Capítulo I: Fundamentación Teórica
1.3 Extracción y su combinación con técnicas analíticas. 1.3.1 Generalidades
El método de extracción es una de las herramientas de trabajo más importante
para el químico. La extracción líquido-líquido presenta posibilidades para la
separación y purificación de diversas sustancias, tanto a escala industrial como
de laboratorio, pudiendo ser empleada como:
• Método de separación, donde es posible extraer una especie individual o
un conjunto de ellas. Esta variante es de gran importancia práctica puesto
que permite separar impurezas, extraer grupos de elementos afines, etc.
(14-16).
• Método de determinación analítica combinado con otras técnicas.
La combinación del procedimiento de extracción con diferentes técnicas de
cuantificación de la sustancia de interés analítico, constituye un campo amplio
dentro de las técnicas combinadas de análisis que ofrece múltiples posibilidades
de aplicación y se encuentra en un proceso de desarrollo continuo. De particular
interés resultan aquellas en las que la determinación se realiza directamente en
la fase orgánica. Dentro de las técnicas analíticas empleadas con este fin se
encuentran: Extractofotometría, extractopolarografía, extractopotenciometría,
extractocromatografia y otras. Así por ej. T. Eldem, N. Arican-Cellat
recientemente realizaron la determinación analítica de amfotericin B en plasma,
empleando extracción en fase sólida combinada con HPLC y R. Draisci, C.
Marchiafava investigaron la determinación de residuos cortiesteroides mediante
técnicas combinadas de análisis, mientras Draisci y otros colaboradores
determinaron pequeñas cantidades de nitrofuranos en huevos empleando
técnicas cromatográficas combinadas con espectrofotometría y espectrometría
de masa (3-5).
• Método de concentración:
El método de extracción líquido-líquido permite elevar la concentración de uno o
más componentes de manera absoluta o relativa cambiando la relación de
volúmenes de fases. Este procedimiento conlleva a la concentración de la
9

Capítulo I: Fundamentación Teórica
especie a determinar en fase orgánica, disminuyendo el límite de detección de
la técnica combinada en cuestión (17 y 18).
Una ventaja importante de ésta técnica que la distingue de otras clásicas como
la precipitación, recristalización fraccionada, etc., es el hecho de poder organizar
el proceso de extracción de manera continua y por tanto con mayor
productividad y economía, siendo además un método rápido y sencillo.
1.3.2 Extractofotometría
La extractofotometría es una de las técnicas combinadas que más se emplean
en la práctica, puesto que no requiere de equipos costosos. Este método se
basa en la extracción de quelatos hacia la fase orgánica y la medición de su
absorción en la región (UV-VIS) del espectro. Esta propiedad se mide
directamente en el extracto a la longitud de onda seleccionada, siendo
numerosos los artículos reportados que aplican el mismo (19 y 20). Para realizar
la determinación analítica de la especie química correspondiente, es necesario
comprobar previamente el cumplimiento de la ley fundamental de la
espectrofotometría, denominada ley de Bouguer – Lambert – Beer. La misma se
basa en dos postulados esenciales:
1. Cuando un haz de luz, cuyos rayos son paralelos y monocromáticos incide
perpendicularmente sobre una superficie en un medio homogéneo, cada
capa o segmento infinitesimal de ese medio hace decrecer la intensidad de
la radiación incidente en una fracción constante.
10

Capítulo I: Fundamentación Teórica
El decrecimiento infinitesimal dI con el espesor del medio absorbente también
infinitesimal db, puede ser denotado mediante la expresión:
- dI =I
Kdb (1)
Donde:
K es una constante que depende de la longitud de onda y de la naturaleza del
medio, I es la intensidad de la radiación incidente y b es el camino óptico.
Una expresión análoga que relaciona la disminución de la intensidad de la
radiación, en condiciones similares a las anteriormente citadas, con la
concentración del medio absorbente, corresponde a la siguiente ecuación:
-dI/I = Kdc (2)
La combinación de las expresiones 1 y 2 integrada y expresada en forma
logarítmica es conocida clásicamente como ley de Bouguer- Lambert –Beer:
I0 =I
KcLog (3)
Para un camino óptico dado la tramitancia disminuye en progresión geométrica,
cuando la concentración aumenta en progresión aritmética.
La constante K depende de la longitud de onda, la naturaleza del medio y del
camino óptico.
Otra forma de expresar la ley anterior es:
A = abc (4)
Donde: A – absorbancia de la disolución.
a – constante para cada especie en particular que recibe el nombre de
absortividad molar o específica.
c – la concentración de la disolución, mol.L-1.
b – camino óptico
La absorbancia A, no es otra cosa que el logaritmo base 10 de la magnitud
conocida como transmitancia:
11

Capítulo I: Fundamentación Teórica
IoILogA −=
(5)
La interacción de la radiación monocromática con las especies que absorben
energía radiante, permite el establecimiento de las relaciones funcionales (curva
de calibrado) entre las magnitudes A y C(x), en determinado rango de
concentración en el que se cumple la ley, lo que constituye el basamento de la
extractofotometría si la medida de la absorbancia se realiza en el extracto (17).
Ventajas de la extractofotometría:
• Generalmente resulta más sensible que el método fotométrico en fase
acuosa, lo que puede incrementar por la reducción del volumen de la fase
orgánica respecto a la fase acuosa.
• Incrementa la selectividad con relación a la determinación de la
absorbancia en medio acuoso. Esta propiedad puede ser mejorada por
diferentes vías tales como: selección del solvente y condiciones de pH,
así como el empleo de agentes enmascarantes (5).
• Requiere de un equipamiento sencillo y puede ser automatizado.
Las ventajas anteriormente señaladas son de gran importancia para el análisis
de trazas, lo cual es de mucho interés en diversas ramas como la Industria
Electrónica, el control de contaminantes del medio ambiente, la determinación
de trazas de metales en agua, etc.
Efecto del reactivo
Al aplicar el método extractofotométrico es necesario comprobar la posible
absorción del reactivo extractor en la región del espectro considerada. Si el
ligando no absorbe en dicha región no existen interferencias espectrales
respecto al complejo, pero cuando este absorbe hay que tener en cuenta la
posición de la banda correspondiente. En el caso en que ambas absorbancias
12

Capítulo I: Fundamentación Teórica
sean cercanas, resulta necesario reextraer el exceso del reactivo extractor, lo
que frecuentemente se realiza en medio básico (7).
Estabilidad del color de extracto.
Al estudiar un método extractofotométrico es necesario tener en cuenta la
estabilidad del color en el extracto, investigando su posible variación al
transcurrir el tiempo.
Puesto que la extracción se realiza en un sistema heterogéneo, la estabilidad del
compuesto a extraer depende de varios factores, relacionados tanto con la
naturaleza de la fase acuosa como con la fase orgánica. Así por ejemplo en la
fase orgánica pueden ocurrir cambios en el estado de oxidación del metal, así
como interacciones con el solvente orgánico, descomposición del reactivo
extractor etc., cuyos productos pueden afectar la estabilidad del complejo.
Por otra parte hay que tener en cuenta factores externos como la luz, el oxígeno,
el recipiente en que se almacena, entre otros (6).
1.4 Características de la piridina: La piridina en condiciones ambientales es un líquido incoloro de olor
característico y su fórmula estructural corresponde a la siguiente:
Es una sustancia muy higroscópica que forma hidratos tales como el
C5H5N.3H2O, de temperatura de ebullición 115 oC (21 y 22).
Las principales características químico- físicas de dicha amina se refieren en la
tabla siguiente:
13
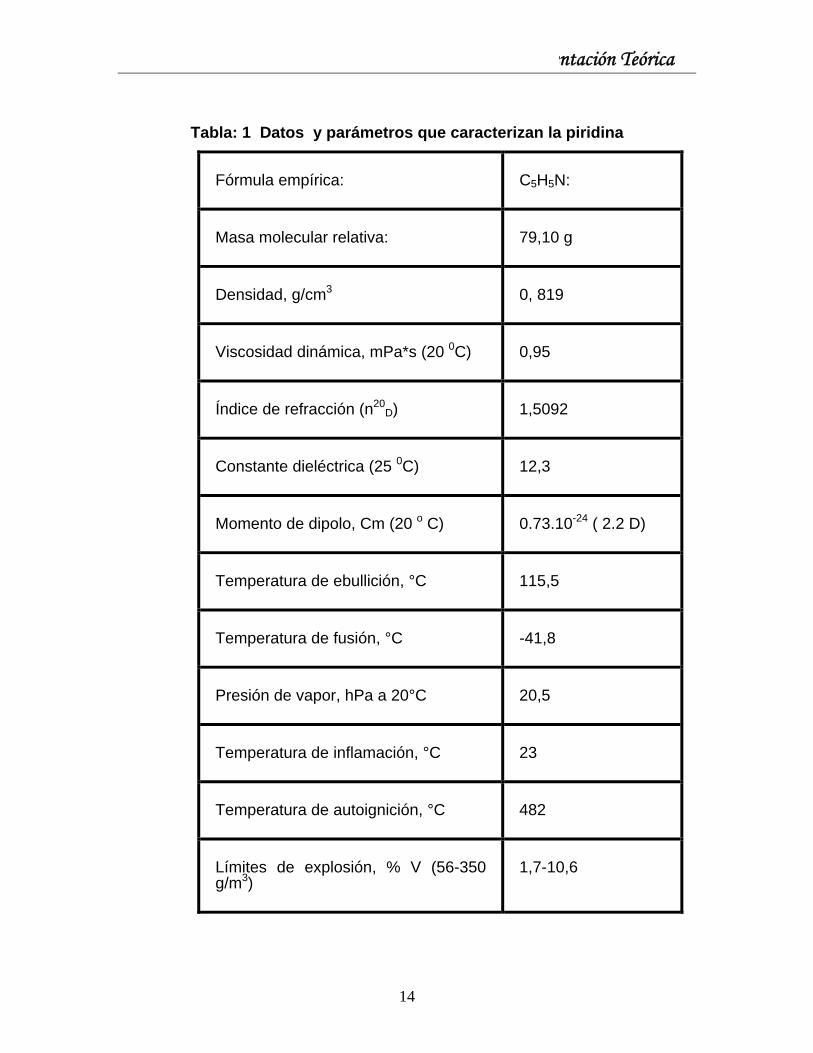
Capítulo I: Fundamentación Teórica
Tabla: 1 Datos y parámetros que caracterizan la piridina
Fórmula empírica: C5H5N:
Masa molecular relativa: 79,10 g
Densidad, g/cm3 0, 819
Viscosidad dinámica, mPa*s (20 0C) 0,95
Índice de refracción (n20D) 1,5092
Constante dieléctrica (25 0C) 12,3
Momento de dipolo, Cm (20 o C) 0.73.10-24 ( 2.2 D)
Temperatura de ebullición, °C 115,5
Temperatura de fusión, °C -41,8
Presión de vapor, hPa a 20°C 20,5
Temperatura de inflamación, °C 23
Temperatura de autoignición, °C 482
Límites de explosión, % V (56-350 g/m3)
1,7-10,6
14

Capítulo I: Fundamentación Teórica
La amina ya mencionada presenta un parecido más acentuado con el
benceno que con los heterociclos de 5 átomos: la molécula es hexagonal y
plana. Ésta tiene, como el benceno, tres pares de electrones Π deslocalizados.
La piridina es una base muy débil con una Kb = 2,3 10-9, lo que es
consecuencia de la presencia del par de electrones no compartidos retenidos
fuertemente en un orbital sp2, resultando menos disponibles que en las aminas
alifáticas (22).
La reactividad química de la amina antes mencionada se manifiesta en su
reacción con agentes oxidantes atacando materiales plásticos y diversas
gomas.
La piridina técnica está mezclada con picolinas y otras sustancias. Se utiliza
como desnaturalizante del etanol, como solvente en el laboratorio y en la
industria para la obtención de sales orgánicas y sustancias químicas. La misma
forma parte de la síntesis de gran cantidad de medicamentos, alcaloides,
colorantes, desinfectantes, herbicidas e insecticidas.
1.5 Peligrosidad de la piridina. Esta amina es relativamente inflamable como se observa de los datos
reportados en la tabla 1. Resulta nociva por inhalación, ingestión y contacto con
la piel (23 y 24). En estado gaseoso o en forma de vapor origina mezclas
explosivas con el aire. En caso de incendio puede producir gases nitrosos por
descomposición. Esta amina no debe ponerse en contacto con materiales como:
fluor, halogenuros de halógenos, cromatos, percromatos, óxido de cromo (VI),
ácido nítrico, peróxidos, óxidos de nitrógeno, sulfóxidos y anhídridos.
En el Anexo 13 se refieren las medidas de protección e higiene así como la
forma de proceder en caso de accidente.
1.6 Efectos sobre la salud de los seres humanos. La piridina es un tóxico nervioso e irritante local que afecta especialmente los
ojos y mucosas. Dentro de los síntomas típicos de la intoxicación con esta amina
15

Capítulo I: Fundamentación Teórica
se encuentran: mareos, dolor de cabeza, vómitos, enrojecimiento de la piel y
parálisis de los nervios de la cabeza. En los mamíferos se presentan efectos
adversos cuando hay exposición prolongada, ya que se inhibe el metabolismo
del amoníaco en el cerebro, el hígado y los riñones. La concentración máxima
permisible en el aire es de 0.05 μg/mL (25).
Los vapores de piridina actúan como irritantes de las mucosas, principalmente
en los ojos y vías respiratorias. Además este compuesto puede producir
dermatitis.
1.7 Características toxicológicas de la piridina. Toxicidad aguda
La dosis letal media DL50 expresada en mg/Kg de peso es de 1580 para ratones
y 891 para ratas. La CL50 (inhalativo en ratas) es de 4000 ppm (V)/ 4h y la DL50
(dérmica, conejos) de 1121 mg/ kg(26).
Síntomas específicos, en ensayos sobre animales:
• Test de sensibilización (cobaya): sin efecto sensibilizante.
• No teratógenico en experiencias con animales.
• Mutagenesidad bacteriana.
• Test de Ames: negativo.
Informaciones adicionales sobre toxicidad:
• Test de inhalación: irritaciones leves.
• En contacto con la piel: irritaciones leves y riesgos de absorción.
• Ingestión: nauseas, cefaleas, intranquilidad, insomnio. Cuando la dosis es
elevada produce efectos sobre el sistema cardiovascular, colapso y
narcosis.
• La administración crónica lesiona al hígado y los riñones.
16

Capítulo I: Fundamentación Teórica
1.8 Influencia ecológica de la piridina. Comportamiento en el medio ambiente:
Agua:
La piridina se disuelve por completo en el agua y forma mezclas tóxicas,
incluso estando muy diluida, constituyendo una amenaza para el agua. La
absorción continua de piridina puede incrementar el metabolismo de la
microflora, pero 0,5 mg/L ya es suficiente para suprimir los procesos que
generan nitrato y amoniaco. También la oxidación disminuye sensiblemente
alrededor de los 5 mg/L. El compuesto es estable en agua porque no se
produce hidrólisis. El límite de vertimiento en soluciones de suelos o cuerpos
de agua es de 10 μg/mL (27).
Efectos ecotóxicos:
• Efectos biológicos: perjudicial en organismos acuáticos.
• Toxicidad para peces: P promelas CL50: 93.8mg/L/96h (agua blanda).
• Toxicidad de dafnia: Dafnia magna CE50: 940 mg/L/ 48 h
• Toxicidad de bacterias: photobacterium phosphoreum, CE50: 210-740
mg/L/30 min test microtoxicidad.
Concentración tóxica límite:
• Toxicidad para las algas: Sc. Cuadricaud CL5 : 340 mg/L/ 16 h; 7d;
• Toxicidad de bacterias: Ps. Putida CE5: 340 mg/L/ 16 h;
• M. aeruginosa CE5: 28 mg/ mL/ 16 h;
• Protozoos: E. Salcatum CE5: 3.5 mg/ 72 h
Aire:
La piridina es un líquido tóxico y combustible que se evapora fácilmente
generando vapores inflamables más densos que el aire.
• Degradación abiótica. En el aire la degradación es lenta.
17

Capítulo I: Fundamentación Teórica
Comportamiento en sistemas ecológicos:
Suelo:
La piridina tiene gran movilidad. La aplicación combinada de esta amina y
fenol favorece la estabilidad de la primera en el suelo. Después de una
inhibición inicial del crecimiento de las bacterias, éstas se adaptan tanto en el
suelo como en los sistemas acuáticos. Concentraciones de 750 mg/kg en el
suelo pueden desaparecer al cabo de 4 meses.
1.9 Degradación, productos de la descomposición y tiempo de vida media. La piridina es notablemente móvil y se dispersa ampliamente en la hidrosfera y
atmósfera, debido a su hidrosolubilidad y la volatilidad; tiene poca tendencia a la
bioacumulación y geoacumulación Una vez absorbida, esta se distribuye
rápidamente por todo el cuerpo. La degradación metabólica se produce
fundamentalmente por metilación y oxidación a través del par de electrones
libres del átomo de nitrógeno. La N-oximetilpiridina ha sido identificada como
metabolito. Además, la sustancia es expulsada rápidamente del organismo:
concentraciones de 0,4 g/kg de peso corporal se eliminan completamente en el
transcurso de 3 días (27).
1.10 Técnica analítica desarrollada en el trabajo. 1.10.1 Antecedentes.
En la literatura se reportan numerosas técnicas analíticas para la determinación
de piridina entre las cuales se encuentran:
1. Por valoración ácido-base con detección visual en un medio salino
concentrado.
La piridina puede ser determinada por valoración con un ácido fuerte en
presencia de cloruro de litio de concentración correspondiente a 8 mol/L,
empleando azul timol como indicador (7).
18

Capítulo I: Fundamentación Teórica
2. Valoración potenciométrica.
En la literatura se refieren numerosas variantes para la determinación
potenciométrica de piridina en medio no acuoso que en agua, en el cual dicha
sustancia revela propiedades básicas más fuertes (28).
Las bases heterocíclicas mono-funcionales pueden ser valoradas con ácido
perclórico en medio acético, si su constante de disociación en agua no es menor
que 10-12. Gyenes y colaboradores recomiendan una mezcla de clorobenceno
con ácido acético como solvente más adecuado para la valoración de piridina,
alquilpiridina, quinoleína, isoquinoleína y otras, donde el punto de equivalencia
es determinado potenciométricamente o de manera visual con cristal violeta o
1-naftolbenceno (29).
Otro solvente que también se recomienda es el nitrometano, el que fue
seleccionado por investigadores del Centro de Bioactivos Químicos para ajustar
una técnica potenciométrica con el fin de cuantificar esta amina en muestras del
proceso de producción que allí se realiza; sin embargo no resultó una buena
variante en las condiciones empleadas (30).
3 .Determinación de piridina por gravimetría.
La reacción del reactivo de Dragendorff con la piridina conduce a la formación de
un precipitado rojo en medio ácido de fórmula NHCHBiI 554 ⋅ . La formación de
este precipitado puede ser empleada para la detección analítica de la base
mencionada.
Esta reacción también puede ser empleada para la determinación cuantitativa
de piridina si una muestra de la base es tratada con un exceso de reactivo, el
residuo es filtrado, secado convenientemente y pesado (31).
4. Determinaciones espectrofotométricas
Región ultravioleta del espectro.
Se fundamenta en la mediada de la absorbancia de esta amina a la longitud de
onda correspondiente a la máxima absorbancia de la misma, mediante la
confección de una curva de calibrado (32).
19

Capítulo I: Fundamentación Teórica
Región visible
En la literatura se reportan varias técnicas analíticas para la determinación de
piridina por espectrofotometría en la región visible. Así por ejemplo el p-N-
dimetilaminobenzaldehido reacciona con piridina originando un producto
coloreado, lo que varía en función de los componentes del sistema (33-35). Este
reactivo mezclado con ácido sulfúrico-tolueno resulta púrpura brillante, mientras
que en presencia de ácido sulfúrico-tolueno-alcohol etílico se torna incoloro lo
que sirve de base al desarrollo de técnicas espectrofotométricas si se cumple la
ley de Bouguer – Lambert – Beer en las condiciones de trabajo.
5. Extractofotométrica por formación de complejos mezclados
Indirecta
Determinación indirecta de piridina mediante la determinación
extractofotomètrica del exceso de Ni2+, con pirazolina no metilada.
Esta técnica se basa fundamentalmente en la adición de tiocianato de potasio y
exceso de Ni(II) a una solución que contiene piridina, separando el complejo
formado por filtración. El Ni (II) presente en el filtrado se extrae de forma
sucesiva con disolución de pirazolina no metilada en cloroformo a pH constante
y se mide la absorbancia directamente en el extracto a 595 nm (36).
Directas
Determinación de piridina empleando como extrayente ácidos carboxílicos en
cloroformo.
En la literatura se reporta la extracción de cobre (II) con piridina y ácidos
carboxílicos tales como el octanóico y el salicílico (37 y 38). En el primer caso se
propone una técnica extractofotométrica para determinar el contenido de cobre
en aceros empleando como extrayente α-picolina y ácido octanóico disueltos en
cloroformo (37), mientras en el segundo se estudia la distribución del complejo
formado en el sistema: Cu (II)- piridina – ácido salicílico, recomendando las
condiciones de pH en las que se logra una buena extracción de Cu (II). Más
recientemente se reporta el desarrollo de una técnica analítica para la
20

Capítulo I: Fundamentación Teórica
determinación de piridina con ácido salicílico en cloroformo basada en la
formación del complejo mixto ya referido, que puede ser extraído de manera
sucesiva con cloroformo o tetracloruro de carbono, en condiciones de pH de la
fase acuosa constante(38). El equilibrio de extracción para este sistema puede
representarse mediante la siguiente ecuación:
Cu2+ (ac) + p PyC6H5OCOOH(o) + (n-p) C6H5OCOOH(O) = CuPyp (OOC
C6H5O)n (o) + nH+
Donde: Py = piridina
El rango de concentraciones de piridina donde se cumple la ley de Bouguer
Lambert Beer es de 150-450 ppm y se pueden determinar concentraciones
hasta de 60 ppm mediante el método de adición de estándar. Esta técnica fue
aplicada a la determinación de piridina en etanol de purificación de 2-bromo-5-
(2-bromo-2-nitrovinil)-furano (G-1)(38).
Por otra parte en la literatura se reportan datos sobre la determinación de
cationes divalentes por métodos colorimétricos, nefelométricos y turbidimétricos,
basados en la formación de complejos escasamente solubles con tiocianato y
bases de alta masa molecular (metil violeta y rodamina) (39).
La piridina forma un gran número de compuestos metalorgánicos de
composición estable que pueden ser empleados para la detección cualitativa y
cuantitativa de esta amina y de los cationes correspondientes. En la tabla
siguiente se muestran algunos ejemplos de estos complejos mezclados (35):
21
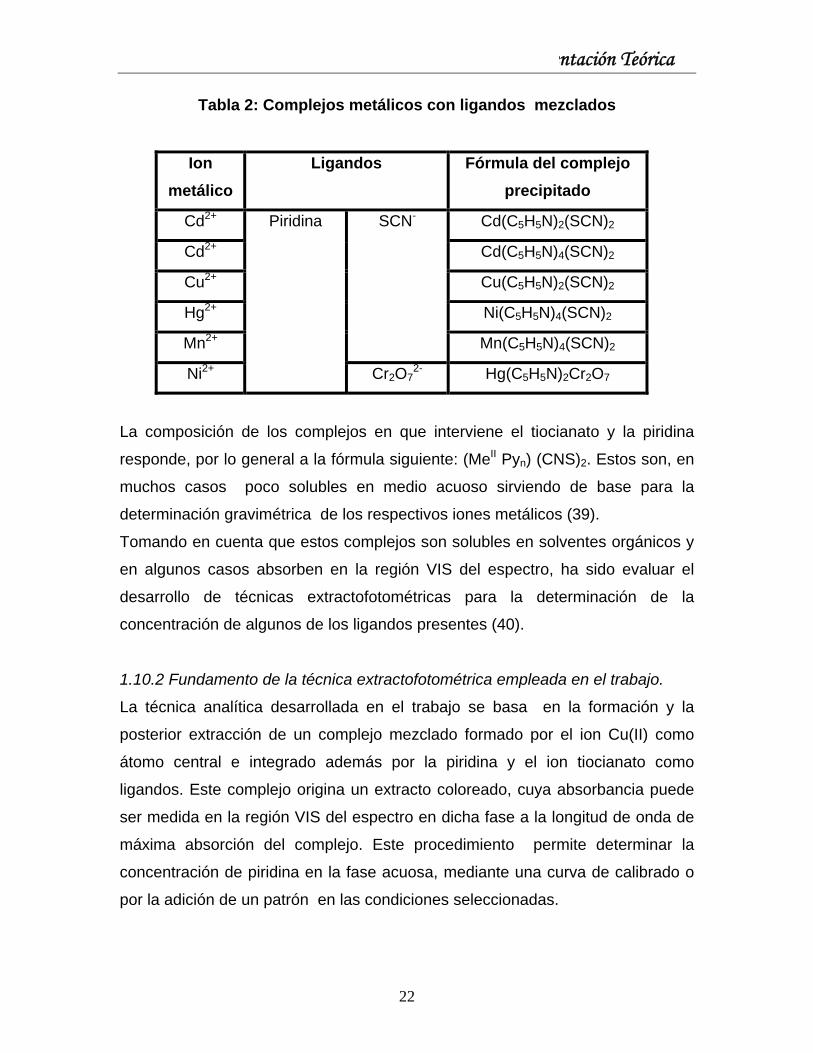
Capítulo I: Fundamentación Teórica
Tabla 2: Complejos metálicos con ligandos mezclados
Ion metálico
Ligandos Fórmula del complejo precipitado
Cd2+ Cd(C5H5N)2(SCN)2
Cd2+ Cd(C5H5N)4(SCN)2
Cu2+ Cu(C5H5N)2(SCN)2
Hg2+ Ni(C5H5N)4(SCN)2
Mn2+
SCN-
Mn(C5H5N)4(SCN)2
Ni2+
Piridina
Cr2O72- Hg(C5H5N)2Cr2O7
La composición de los complejos en que interviene el tiocianato y la piridina
responde, por lo general a la fórmula siguiente: (MeII Pyn) (CNS)2. Estos son, en
muchos casos poco solubles en medio acuoso sirviendo de base para la
determinación gravimétrica de los respectivos iones metálicos (39).
Tomando en cuenta que estos complejos son solubles en solventes orgánicos y
en algunos casos absorben en la región VIS del espectro, ha sido evaluar el
desarrollo de técnicas extractofotométricas para la determinación de la
concentración de algunos de los ligandos presentes (40).
1.10.2 Fundamento de la técnica extractofotométrica empleada en el trabajo.
La técnica analítica desarrollada en el trabajo se basa en la formación y la
posterior extracción de un complejo mezclado formado por el ion Cu(II) como
átomo central e integrado además por la piridina y el ion tiocianato como
ligandos. Este complejo origina un extracto coloreado, cuya absorbancia puede
ser medida en la región VIS del espectro en dicha fase a la longitud de onda de
máxima absorción del complejo. Este procedimiento permite determinar la
concentración de piridina en la fase acuosa, mediante una curva de calibrado o
por la adición de un patrón en las condiciones seleccionadas.
22

Capítulo I: Fundamentación Teórica
La siguiente ecuación química representa el equilibrio de extracción del sistema
considerado:
Cu2+ (ac) + 2 Py ( ac) + 2SCN-
(ac) = [Cu(SCN)2 (Py)2 ] (o)
Donde: Py = C5 H5N:
1.11 Técnicas cromatográficas utilizadas en el trabajo. Las técnicas cromatográficas en general se basan en la separación de
moléculas por procesos de migración diferencial (diferentes velocidades de
transportación de moléculas) de una mezcla arrastradas en una fase móvil a lo
largo de una fase estacionaria (7).
Hay diferentes tipos de técnicas cromatográficas entre las que se encuentran:
Cromatografía de papel, de capa delgada (CCD), cromatografía líquida de alta
resolución (HPLC), cromatografía gaseosa (CG) etc.
1.11.1 Cromatografía de capa delgada (CCD). Las primeras aplicaciones de la cromatografía con fines analíticos fueron la
correspondiente a papel y capa delgada y actualmente continúan siendo
herramientas de trabajo indispensable para el químico. Así por ejemplo se
emplea mucho en química orgánica para seguir el curso de una reacción, el
agotamiento de las materias primas, los productos de reacción etc. (41-43).
También se utiliza ampliamente en el campo del análisis para el desarrollo de
técnicas cuantitativas en matrices complejas, la determinación de la composición
de las matrices objeto de análisis etc. (44-51). Recientemente se han
desarrollado nuevas técnicas analíticas para cuantificar principios activos como
el G-1 y el G-O (2(-2- nitrovinil furano) en residuales y muestras del proceso
productivo de la Planta de Producción de Bioactivos Químicos, de la región
central de Cuba, utilizando CCD (52 y 53).
Dentro de las ventajas que presenta la cromatografía de capa delgada frente a
otros métodos cromatográficos está la simplicidad del equipamiento, consumo
23

Capítulo I: Fundamentación Teórica
de reactivos de menor coste, el tiempo necesario para conseguir la separación
que es mucho menor que el empleado en cromatografía de papel, columna etc.
Con esta técnica se logra excelente nitidez y alta sensibilidad.
En el método de CCD la separación de las sustancias se lleva a cabo sobre una
pequeña superficie de la fase estacionaria adherida sobre un soporte de
sustancias porosas tales como: silicagel, alúmina, poliamidas etc. En
dependencia de la composición del absorbente y del disolvente empleado la
separación de las sustancias puede ocurrir por diferentes mecanismos tales
como: adsorción, distribución, intercambio iónico, filtración gel etc.
Los campos fundamentales de aplicación de la CCD se corresponden con la
cromatografía de adsorción. La toma de la muestra y la introducción de la fase
móvil se realizan de modo análogo que en el método de la cromatografía de
papel.
Descripción del proceso de adsorción
La muestra aplicada sobre la capa es adsorbida en la superficie del material por
la acción de fuerzas electrostáticas (fuerzas de van der Waals, puentes de
hidrógeno, efectos inductivos, etc.). Luego, cuando la capa es expuesta a un
flujo por acción capilar, se inicia una competencia entre los sitios activos del
adsorbente y la sustancia con el solvente.
1.11. 2 Cromatografía líquida de alta resolución, HPLC.
La cromatografía líquida de alta resolución es actualmente la técnica de
separación más ampliamente utilizada en los laboratorios modernos, siendo una
de las herramientas analíticas más importantes para separar y detectar
compuestos químicos (54-58). Se caracteriza por su versatilidad y amplio
campo de aplicación.
Los componentes de la muestra, previamente disueltos en un disolvente
adecuado (fase móvil), son forzados a atravesar la columna cromatográfica
gracias a la aplicación de altas presiones. El material interno de la columna (fase
estacionaria) está constituido por un relleno capaz de retener de forma selectiva
los componentes de la mezcla. La resolución de esta separación depende de la
24

Capítulo I: Fundamentación Teórica
interacción entre la fase estacionaria y la fase móvil, pudiendo ser variada a
través de la elección de diferentes mezclas disolventes y distintos tipos de
rellenos. Como resultado final, los componentes de la mezcla salen de la
columna separados en función de sus tiempos de retención en lo que constituye
el cromatograma. A través del cromatograma se puede realizar la identificación
cualitativa y cuantitativa de las especies separadas.
Tipos de cromatografía líquida de alta resolución.
Los tipos de cromatografía líquida de alta resolución más conocidos son: de
fase normal, de fase reversa ( RP), RP de supresión iónica, RP par iónico, de
intercambio iónico, SEC( Sieve exclusión chromatography ) de filtración en gel,
permeación a través de un gel y cromatografía de afinidad.
Cromatografía de fase reversa
Este tipo de cromatografía de alta resolución es la más utilizada, alrededor del
60 % de las separaciones son realizadas por este modo cromatográfico.
En este modo la fase móvil es más polar que la fase estacionaria y las fuerzas
de atracción son dominantes entre ellas. La fase estacionaria está compuesta
por un hidrocarburo inerte, siendo la única interacción con el soluto hidrofóbica
y la selectividad está dominada por el efecto del solvente.
Esquema del cromatógrafo de HPLC:
25

Capítulo I: Fundamentación Teórica
Breve descripción de las partes del equipo.
1. Recipiente para la fase móvil: Reservorio para el sistema de solventes
puros.
2. Bomba: Aditamento que garantiza el flujo constante y adecuado de
solvente. De no ser constante se producirían pérdidas de
reproducibilidad.
3. Inyector: Dispositivo para introducir la muestra en el cromatógrafo
4. Termostato: Aditamento que tiene como función regular la
temperatura de la muestra.
5. Columna: En las mismas se produce la separación de las sustancias.
Existen diferentes tipos según el objetivo del trabajo, ejemplo.
• Analíticas: para separar
• Preparativas: para purificar
6. Detector: Proporciona una señal que indica que la sustancia ha sido
separada. Existen muchos tipos y se eligen según la naturaleza de la
muestra.
• UV
• UV-Visible
• Fluorescencia
• Electroquímicos
• Índice de refracción (carbohidratos)
• Arreglo de diodos (DAD)
9. Colector de fracciones: se usa con fines preparativos
10. Medidor de flujo: Es un instrumento de medición que proporciona datos
sobre el flujo de la fase móvil.
26

Capítulo I: Fundamentación Teórica
Solventes en HPLC (59).
El sistema de solventes empleados constituyen la fase móvil FM y debe disolver
totalmente la muestra sin destruirla. Además debe ser compatible con la fase
estacionaria (FE) y el sistema de detección. Las mismas son típicamente agua o
mezclas de ellas con algunos modificadores como el acetonitrilo, metanol y el
tetrahidrofurano. Los flujos de fase móvil más empleados generalmente se
encuentran entre 0.5 y 2 mL/min.
Requisitos generales para los solventes empleados en cromatografía:
Para seleccionar el solvente a emplear deben tenerse en cuenta diversos
factores tales como:
• Elevada pureza y estabilidad.
• Capacidad para disolver la muestra
• Que sea miscible con otros solventes para formar mezclas útiles
• Que no degrade o disuelva la fase estacionaria
• Baja toxicidad.
• Debe ser compatible con el detector utilizado. Así cuando se emplea un
detector UV debe presentar transparencia óptica.
• Baja viscosidad para reducir las caídas de presión
• Elevado poder de elusión para lograr una buena separación.
• Disponibilidad comercial
• Precio
1.12 Consideraciones acerca del diseño experimental. 1.12.1 Generalidades
El diseño de un experimento es el procedimiento de selección del número de
vías y condiciones suficientes y esenciales para dar solución a un problema
planteado con la precisión requerida y un error en la determinación de los
efectos de interés menor que otros métodos.
Es frecuente que los químicos necesiten enfrentarse a numerosos problemas
relacionados con la realización de experimentos más o menos costosos y
27

Capítulo I: Fundamentación Teórica
complejos con el objetivo de obtener información sobre el sistema en estudio.
Muchos son los ejemplos que pueden citarse al respecto: la síntesis de una
reacción, las condiciones de realización de un experimento, la influencia de
factores sobre las propiedades químico-físicas de un producto y otras. En la
mayoría de estos problemas químicos, se investiga cómo influyen diferentes
condiciones de realización sobre una propiedad o característica del sistema
investigado.
Los métodos de diseño de experimento permiten sistematizar la forma de
realización de las corridas experimentales y obtener la máxima información
posible con la mínima cantidad de experimentos.
La importancia de un diseño de experimento radica en que disminuye, de forma
considerable, la inversión de tiempo, recursos materiales y humanos, estudia la
variación simultánea de las variables determinantes del proceso, utiliza un
aparato matemático que formaliza muchas acciones del experimento
(planificación, preparación y realización) y brinda estrategias claras, luego de
tomar decisiones sustentadas a partir de cada serie de corridas experimentales.
En Química y Tecnología Química, el diseño experimental se utiliza
fundamentalmente en dos direcciones:
• Para el estudio de los mecanismos de procesos complejos y de las
propiedades de sistemas multicomponentes.
• Para la optimización de los procesos y propiedades de los sistemas
multicomponentes.
Para realizar un diseño de experimento es necesario conocer el objeto de
investigación, para lo cual se establece un método cibernético que consta de los
parámetros de optimización y de los factores.
Un parámetro de optimización debe ser: efectivo desde el punto de vista
investigativo, de naturaleza universal, cuantitativo y expresado mediante un valor
único así como efectivo estadísticamente.
28

Capítulo I: Fundamentación Teórica
1.12.2 Algunos conceptos generales relacionados con el diseño factorial
experimental (60-62):
Factor:
Las variables independientes que influyen o pueden influir sobre un proceso
investigativo determinado son conocidas con el nombre de factores.
En un proceso químico los factores pueden ser: la temperatura, la presión, el pH,
la concentración de un reactivo, el tiempo de reacción, etc. Las variables son
designados con la letra X, o sea: X1, X2,... Xn, correspondientes a los factores 1,
2,... n respectivamente.
Función respuesta:
Cuando se realiza un experimento, los resultados se expresan a través de una o
más variables dependientes, por ejemplo en Química: el rendimiento de una
síntesis, la pureza de un reactivo que se obtiene o se purifica, el costo de un
proceso de síntesis, entre otros. Estas propiedades que generalmente
constituyen el blanco u objeto de estudio, son conocidas como función respuesta
y se representan con la letra Y.
La función respuesta depende de los factores y puede expresarse como:
Y = f(X1, X2,...., X n)
Nivel del factor:
Es el valor que puede tomar un factor; y el conjunto de factores que condicionan
una vía.
Superficie de nivel:
La forma geométrica de la función respuesta como función de los factores, es
conocida como superficie de nivel.
Espacio factorial:
Se denomina así al espacio comprendido por los ejes del sistema de
coordenadas en que se representan los valores de los factores.
29

Capítulo I: Fundamentación Teórica
1.13 Características fundamentales de los métodos analíticos (63-65).
Las características de funcionamiento de un método analítico comprenden todos
los datos y resultados experimentales que demuestran su aptitud para el uso al
que se destine, siendo considerados los siguientes grupos:
Características de practicabilidad: Son las que deciden si el procedimiento es
fácil o difícilmente realizable en la práctica. Los parámetros de practicabilidad se
evalúan en la fase de desarrollo del método analítico: tiempo, costo, tamaño de
la muestra, calificación del personal, tipo de equipo e instrumentación,
condiciones de seguridad, etc.
Características de idoneidad: Son el conjunto de parámetros que garantizan que
el sistema responde, en el momento de los análisis, a los requisitos fijados en la
validación del método. La idoneidad verifica el buen funcionamiento del sistema
(instrumento y método) en el momento de uso.
Características de fiabilidad: Son las que demuestran la capacidad de un método
analítico para mantener a lo largo del tiempo los criterios fundamentales de
validación, los parámetros que expresan la fiabilidad de los métodos analíticos
son: linealidad, precisión, exactitud, sensibilidad y especificidad.
Linealidad.
Se entiende como linealidad, la capacidad de un método analítico de obtener
resultados linealmente proporcionales a la concentración del analito en la
muestra, en un intervalo determinado. Dentro de este término se incluye la
proporcionalidad entre la concentración y la respuesta, así como el intervalo o
rango de concentraciones de la sustancia para las cuales el método es
satisfactorio.
Test de linealidad.
- Coeficiente de variación de los factores de respuesta ( CVf).
El factor de respuesta (f) es la relación entre la lectura y la concentración. Para
una concentración determinada el factor respuesta puede tomarse como una
expresión aproximada de la sensibilidad de calibrado a esta concentración. En
una calibración lineal, los factores respuesta deben ser semejantes entre sí y
30

Capítulo I: Fundamentación Teórica
cercanos al valor de la pendiente, por este motivo se puede tomar el CVf como
una expresión de linealidad.
Se considera que valores del CVf superiores al 5% indican falta de linealidad.
El coeficiente de variación de los factores respuestas se obtienen según:
CVf Sff
= ⋅100
- Significación estadística de la varianza de la pendiente.
La pendiente se conoce también como coeficiente de regresión, a mayor
pendiente mayor sensibilidad.
Para expresar la linealidad, se utiliza el coeficiente de variación de la pendiente
el cual debe ser menor que el 3%.
Este parámetro se determina según:
CVm Smm
= ⋅100
Test de proporcionalidad.
El valor de b, intersección con el eje de las coordenadas, indica el error
sistemático del método, en el caso ideal debe ser cero.
Parámetros analíticos que caracterizan la precisión de la técnica
La precisión es el grado de concordancia entre los valores de una serie repetida
de ensayos analíticos, efectuado sobre una muestra homogénea. La precisión
se expresa matemáticamente por el coeficiente de variación (CV), a través de la
repetibilidad y de la reproducibilidad de la técnica.
Del término precisión del método se pueden distinguir dos tipos de estudios:
Repetibilidad: Es la medida de la precisión de un método efectuado en las
mismas condiciones, sobre la misma muestra, por un mismo analista, en un
mismo laboratorio, con los mismos aparatos y reactivos y en el curso de la
misma serie de análisis efectuados en un corto intervalo de tiempo
(generalmente el mismo día).
El ensayo de repetibilidad del método se efectúa sobre una serie de porciones
de una muestra homogénea que se analiza desde el principio (preparación de la
31

Capítulo I: Fundamentación Teórica
muestra) hasta el final (lectura de los resultados) por el mismo analista y el
mismo instrumento. El numero de repeticiones del análisis deberá ser superior a
5 y la concentración del analito en la muestra problema suele ser similar a la
nominal o declarada.
El criterio seleccionado para evaluar la repetibilidad depende del tipo de muestra
a ensayar, del criterio del investigador etc, .En este caso fue seleccionado el
valor de: CV≤ 3%.
Reproducibilidad: Es la medida de la precisión de los resultados de un método
analítico efectuado sobre la misma muestra pero en condiciones diferentes
(diferentes analistas, aparatos, días, etc.). Cuando además, los laboratorios son
distintos se habla de precisión ínter laboratorios.
La reproducibilidad global se determina por el coeficiente de variación. Si se
desea estudiar el efecto de cada uno de los tres factores (día, analista,
instrumento) por separado, deberá realizarse un análisis de varianza.
Para este tipo prueba, al igual que para la repetibilidad el criterio del coeficiente
de variación no siempre es el mismo, siendo seleccionado en este caso el valor
de: CV≤ 5%( 64).
Expresión de la precisión de un método analítico.
Desviación estándar y coeficiente de variación.
La precisión se expresa matemáticamente por la desviación estándar o
preferiblemente por el coeficiente de variación (desviación estándar relativa). El
valor aceptable de precisión de un método depende de la concentración del
analito y del número de repeticiones del análisis.
Límites de confianza.
I.- De los resultados individuales.
El límite de confianza en este caso es igual a la media ± la desviación estándar
multiplicada por la t de student (X ± tS), siendo t el valor de la t de student
tabulada para n-1 grados de libertad y una significación generalmente del 95%.
II.- De los resultados promedios.
Los límites de confianza de la media de una serie de resultados son X ± t Sx
siendo Sx la desviación estándar de la media.
32

Capítulo I: Fundamentación Teórica
Exactitud
La exactitud indica la capacidad de un método analítico para dar resultados lo
más próximo posible al valor verdadero.
Matemáticamente, la exactitud se expresa en forma del porcentaje de
recuperación de la cantidad de analito presente en la muestra, o bien, en forma
de la diferencia entre el valor hallado y el verdadero. Estadísticamente se
efectúa un test de Student.
donde:
S
nXXvt
−=exp
En esta fórmula Xv es el valor verdadero, X el valor medio, n es el número de
determinaciones y S es la desviación estándar.
Si texp < tab para la probabilidad escogida (p = 0.05) y n-1 grados de libertad,
significa que ambos valores son estadísticamente semejantes y que el método
analítico tiene la exactitud requerida.
Otra vía para la comprobación de la exactitud es mediante la comparación
estadística de los valores medios obtenidos por dos técnicas, una de las cuales
es la que se quiere validar.
33

Capítulo II: Parte experimental
Capítulo II: Parte experimental
2.1 Reactivos y disoluciones.
Reactivos:
Sulfato de cobre anhidro.
Tiocianato de sodio.
Cloroformo.
Piridina.
Agua destilada.
Todos los reactivos empleados son de calidad puros para análisis (p.a).
Disoluciones:
Disolución de CuSO4 anhidro 0.02 mol/L.
Disolución de NaSCN 0.02 mol/L.
Disolución de piridina de concentración 1500 ppm.
Equipos:
Espectrofotómetro marca SPEKOL, equipado con cubetas de 5 cm de
espesor de la capa absorbente A.
pH-metro, Metrhom E-510.
Procedimiento general.
La metodología general consiste en tomar alícuotas de la disolución de piridina
(py) en embudos separadores de 150 mL de capacidad, a los cuales se añaden
cantidades adecuadas de agua destilada y de disoluciones de Cu (II) y de SCN–
respectivamente. El volumen de la fase acuosa es constante e igual a 20 mL.
El complejo formado se extrae de manera sucesiva, mediante la adición de
porciones de 5 mL de cloroformo agitando en cada caso durante 15 min. Los
extractos orgánicos son colectados en matraces aforados de 10 mL de
capacidad, completando el volumen con el disolvente puro y midiendo
posteriormente la absorbancia a la longitud de onda seleccionada.
34

Capítulo II: Parte experimental
2.2 Selección de las condiciones de trabajo. Longitud de onda:
Para seleccionar la longitud de onda de trabajo fueron registrados los espectros de
los extractos obtenidos al contactar cloroformo con soluciones acuosas de la
siguiente composición:
• Cu2+, SO42-
, piridina, Na+ y SCN- .
• Na+ y SCN –.
• Cu2+ y SO42- .
• Piridina.
• Piridina, Na+ y SCN-.
Después de extraer durante 15 min con agitación constante y separar las fases, se
procedió a registrar el espectro de absorción del complejo extraído en cloroformo,
en un intervalo de 200 – 500 nm, utilizando como blanco cloroformo.
Estudio del pH:
Para estudiar la influencia del pH se varió la acidez de la solución acuosa
inicial desde 4.5-7.5 unidades, empleando para ello hidróxido de sodio y ácido
clorhídrico; después de la extracción y separación de las fases, se midió la
absorbancia del extracto y el pH de equilibrio de la fase acuosa.
Estudio de estabilidad del complejo extraído:
Con el fin de seleccionar el tiempo adecuado par la medida de la señal analítica,
fue investigado el comportamiento del extracto clorofórmico al transcurrir el
tiempo, midiendo la absorbancia de este a intervalos de cinco minutos durante
cuarenta minutos.
Efecto de la cantidad de reactivos a emplear sobre la absorbancia del complejo:
Con el objetivo de analizar la influencia de las cantidades de reactivos a emplear
incluyendo, el ion acetato empleado en la preparación de sistemas buffer, se
utilizó un diseño factorial 23, cuyos factores y niveles se muestran a
continuación.
35

Capítulo II: Parte experimental
Tabla 3: Factores y niveles de diseño 23.
Niveles /V(x), mL Cu(II) ( SCN-) (Acetato)
Inferior 2 4 0.5
Superior 3 6 1
2.3 Procedimiento para realizar la curva de calibración: En una serie de 5 matraces aforados se miden exactamente, 1.0; 1.5; 2.25; 2.5;
3.0; 3.5 y 4.0 mL de la solución inicial de piridina; añadir en cada caso 6 mL de
disolución de tiocianato de sodio y 2 mL de la disolución de sulfato de cobre (II)
y se ajusta el volumen con agua destilada a un valor fijo.
Las disoluciones acuosas antes preparadas se contactan con 5.0 mL de
cloroformo, durante 15 minutos, agitando mecánicamente. La extracción se realiza
de manera sucesiva hasta que el extracto clorofórmico resulte incoloro; la fase
orgánica extraída es llevada a matraces de 10 mL y enrasada con cloroformo p.a,
leyendo la absorbancia en cubetas de 5cm de espesor, a la longitud de onda de
410 nm. El blanco utilizado fue cloroformo.
Con las lecturas de absorbancia y la concentración de piridina de cada
disolución se construye la curva de calibración.
2.4 Comprobación de algunos parámetros analíticos que caracterizan la técnica. Precisión y exactitud
Repetibilidad:
Para determinar la repetibilidad de la técnica fueron realizadas cinco réplicas de
un punto central de la curva en un mismo día y bajo las mismas condiciones.
Reproducibilidad
En este caso se realizaron réplicas correspondientes al punto medio de la
curva, en diferentes días, bajo las mismas condiciones.
Los criterios seleccionados para la aceptación de la repetibilidad y
reproducibilidad fue el siguiente: CV≤3% y el 5% respectivamente (63-65).
36

Capítulo II: Parte experimental
La exactitud fue evaluada mediante el cálculo del recobrado, tomando como
criterio de aceptación CV≤ 3% y 97 %≥ R≤ 103 %( 63-65).
Límite de cuantificación:
Para determinar el límite de cuantificación de la técnica fue evaluada la
precisión y la exactitud de la misma en los puntos correspondientes a las
menores concentraciones de piridina en la curva de calibrado en el rango de
18.75-105 ppm.
Estudio de interferencia
Taller de G-1
Se realizó un estudio de interferencia considerando las variables relacionadas
en la tabla siguiente, donde aparecen los niveles inferiores (NI) y superiores
(NS) de las mismas. El diseño experimental es factorial fraccionado 2 5-2 con los
siguientes contrastes para generar la matriz del diseño: X4 = X1 X2 ; X5 = X1
X2X3.
Tabla 4: Factores y niveles del diseño de interferencia
Niveles/V(x),mL Br2 KBr
CS2 G -1
Anhídrido acético(Anh A)
Factores X1 X2 X3 X4 X5
N.I 0.1 2 0 0 0.1
N.S 1 4 0.4 4 0.2
Taller de G-0
Se realizó el estudio de interferencias considerando las variables relacionadas
en la tabla siguiente donde aparecen los niveles inferiores (NI) y superiores (NS)
de las mismas. El diseño experimental corresponde a un factorial fraccionado
2 4-1 .
37

Capítulo II: Parte experimental
Tabla 5: Factores y niveles del diseño de interferencias.
Niveles/V(x),mL G-0 Nitro metano
furfural Isobutil amina
Factores X1 X2 X3 X4
N.I 0 0.57 0.57 0
N.S 2.28 1.42 1.42 1.42
2.5 Adición de estándar. Otra variante investigada fue la capacidad de la técnica para determinar
concentraciones de piridina inferiores a 105 ppm, para lo cual se tomó una
alícuota de la solución patrón de esta amina (1500 ppm), que contenga una
cantidad de la misma correspondiente a la región del centro de la curva y se
realizaron adiciones de la solución estándar de piridina en el intervalo de 10-90
ppm.
2.6 Aplicaciones. 2.6.1 Características cromatográficas (HPLC) de la piridina e impurezas
presentes.
Para determinar la concentración de piridina en los residuales objeto de
investigación primeramente se realizó un estudio cromatográfico tanto a la
solución patrón de piridina como a los posibles interferentes mediante HPLC,
determinando el tiempo de retención de cada uno de ellos cuando se emplea
acetonitrilo-agua como sistema de solventes. Las condiciones de operación del
cromatógrafo son las siguientes:
• Velocidad de flujo 0.3 mL/min.
• Tiempo de registro 15 min.
• Tipo de columna Hypersil BDC 150 mm de longitud.
38

Capítulo II: Parte experimental
• Diámetro 4.6 mm.
• Longitud de onda 255 nm.
• Fase móvil acetonitrilo: agua 80:20.
La metodología empleada en este caso consistió en:
1- Preparar una solución de piridina a una concentración de 20 ppm en un
matraz de 10 mL, enrazando con una mezcla de acetonitrilo-agua 80:20 y
registrando el cromatograma correspondiente por HPLC.
2- Preparar 6 soluciones de dichas interferencias por separado en las
condiciones anteriormente señaladas y registrar los cromatogramas
respectivos.
3- Preparar soluciones de piridina 20 ppm en presencia de posibles
interferencias, tales como nitrometano(NM), furfural(ff), G-1, G-0, disulfuro
de carbono y anhídrido acético en un matraz de 25 mL, completando el
volumen con la fase móvil descrita en el1. Seguidamente se corrieron los
cromatogramas correspondientes. 2.6.2 Estudio de la composición de las matrices de aplicación.
Una vez conocido el comportamiento cromatográfico del contaminante objeto de
estudio y los posibles interferentes, se estudió la composición del residual
compuesto por las corrientes procedentes de los diferentes talleres en fase
inicial y después de cada etapa del tratamiento aplicado para su desactivación,
mediante HPLC.
Posteriormente el residual fue sometido a un proceso de desactivación que
consta de las siguientes etapas :
• Pretratamienro.
• Tratmiento primario.
• Tratamiento secundario.
• Tratamiento terciario.
39

Capítulo II: Parte experimental
El siguiente esquema representa dicho proceso.
Pretratamiento 1
Tratamiento Primario 2
Tratamiento Secundario 3
Tratamiento Terciario 4
Filtración 5
ColasLicores
Aguas de lavado NaOH
Fe (III)Al (III);CaO; ClO -
CaO;ClO - Al (III)
Estabilización 6
Sedimentación 8
Lodos 9
Líquido de Vertimiento 7
,
Como se observa en el esquema el pretratamiento consiste en la adición de
NaOH ( con) al residual; mientras que en el tratamiento primario se adiciona de
CaO, alumbre de hierro (III) , Al2 (SO4 )3 e hipoclorito de sodio, originando un
precipitado que se filtra y se separa. El tratamiento secundario por su parte
consiste en adicionar solución acuosa de CaO , Al2 (SO4 )3 e hipoclorito de
sodio. A continuación se filtra y se le regula el pH con Al2 (SO4 )3 de 5-9
unidades, constituyendo este último paso, el tratamiento terciario.
Después de cada etapa de tratamiento se realizaron los siguientes experimentos
para conocer la composicion de la matriz:
40

Capítulo II: Parte experimental
• Registro del espectro en la región correspondiente a 200-500 nm (este
experimento se realizó después del tratamiento primario y terciario).
• Registro de un cromatograma por capa delgada empleando una placa de
sílica gel-60 para investigar la posible presencia de furfural, G-O y G-1(los
componentes señalados fueron investigados por esta vía después del
pretratamiento). El solvente utilizado fue cloroformo y el revelado se
realizó con vapores de yodo. El procedimiento empleado consistió en
aplicar un patrón de cada interferente investigado y la muestra en la línea
base de la placa y correr la misma con el solvente mencionado.
• Realización del ensayo de Meyer para investigar la posible presencia de
nitrometano en el residual antes y después del pretratamiento. Este
experimento consistió en tratar cada muestra con disolución alcalina de
nitrito de sodio, acidificar después con H2SO4 diluido y llevar a pH básico
con NaOH.
2.6.3 Determinación del contenido de piridina en los residuales.
Una vez conocida la composición de las matrices se procedió a realizar la
determinación analítica de piridina en el residual compuesto, después de cada
una de las diferentes etapas de tratamiento.
Procedimiento empleado en el pretratamiento.
Se prepararon 6 muestras del residual compuesto de 25 mL cada una, a las
cuales se le aplica el pretratamiento, se llevan a 50 mL en un matraz aforado, se
toman 14 mL en cada caso, se regula el pH a 5.5 unidades, empleando HCl de
concentración correspondiente a 1.2 mol.L- 1 y se procede a determinar la
concentración de piridina por extractofotometría y espectrofotometría
respectivamente. La longitud de onda de trabajo al aplicar la última técnica fue
de 240 nm. Simultáneamente se preperaron 6 patrones de piridina para lo cual
fueron tomados 23 mL de la solución de 1500 ppm y se procedió de la forma
descrita para la muestra, solo que tomando 4.79 mL de la solución obtenida
después del pretratamiento.
41

Capítulo II: Parte experimental
Procedimiento empleado después del tratamiento primario.
En el caso de la determinación de piridina después del tratamiento primario se
procedió de la siguiente forma: a una muestra de 50 mL del residual se le
realizó el pretratamiento y el tratamiento primario de la forma ya descrita, se
llevó a un volumen de 100 mL con agua destilada. De esta disolución se
tomaron 25 mL para cada determinación analítica, se le reguló el pH a 5.5
unidades y se realizó una adición de estándar de piridina correspondiente a
131.25 ppm, después se aplicó la técnica extractofotométrica desarrollada en el
trabajo, siguiendo el procedimiento descrito en el epígrafe 2.1. También se
realizó la determinación por espectrofotometría UV midiendo la absorbancia a
240 nm con el mismo número de réplicas .
Procedimiento empleado después del tratamiento terciario.
Una vez concluido el tratamiento treciario se determinó la concentracion de
piridina por espectrofotometría UV, empleando el método de adición de
estándar. La longitud de onda fijada para la medida de la absorbancia fue de
240 nm.
El procedimiento consistió en tomar 5 mL del residual tratado, adicionarle 1.3
mL de piridina de concentración correspondiente a 1500 ppm(equivalente a 40
ppm de esta amina) y transvasarlo a un matraz de 50 mL, realizando
posteriormente la medida de la absorbancia. Paralelamente se prepararon 5
patrones de piridina de 40 ppm y se midió la absorbancia a 240 nm con el
objetivo de estimar la concentración de piridina en el residual.
42

Capítulo III: Presentación y discusión de los resultados
Capítulo III: Presentación y discusión de los resultados.
3.1 Estudio de las condiciones de trabajo.
Selección de la longitud de onda.
La figura 2 muestra el espectro del extracto clorofórmico del complejo y sus
componentes.
igura 2: Espectro de los reactivos y el complejo.
l espectro del complejo mezclado presenta dos zonas de absorción, una entre
0
0.4
0.8
1.2
1.6
200 300 400 500
λ, nm
Abs
Cobre (II) SCN- y Pird
Complejo PiridinaSCN-
F E
230-300 nm y la otra alrededor de 400nm. En la primera absorben además del
complejo, las restantes especies presentes, por lo que la longitud de onda
seleccionada resultó de 410 nm. Los datos primarios correspondientes a los
espectros anteriormente representados se refieren en el anexo 1.
43

Capítulo III: Presentación y discusión de los resultados
Efecto del pH
relacionan los valores de absorbancia obtenidos al variar el pH
Tabla 6: Estudio del pH.
En la tabla 6 se
de equilibrio.
pH(eq) Abs
4 0.04
4.5 0.062
5 0.152
5.5 0.166
6 0.165
6.5 0.164
7 0.064
l representar gráficamente dichos valores en función del pH se obtiene la
A
curva de distribución característica de este tipo de sistema, figura 3.
Figura 3: Estudio del pH de equilibrio.
0
0.1
0.2
3 5 7
pH
Asb
44

Capítulo III: Presentación y discusión de los resultados
Como se observa de la figura anterior inicialmente se produce un incremento de
stabilidad del complejo extraído:
iar el comportamiento de la absorbancia del
Tabla 7: Datos del estudio de la estabilidad del complejo
la absorbancia del complejo hasta alcanzar una zona de saturación y
posteriormente una disminución de la misma. A partir de este estudio se
selecciona el pH de 5.5 unidades.
E
Los resultados obtenidos al estud
complejo en el tiempo se muestran en la tabla 7 y figura 4.
extraído.
Absorbancia Tiempo, min
2.073 5
2.070 10
2.072 15
2.068 20
2.074 25
2.077 30
2.099 35
45

Capítulo III: Presentación y discusión de los resultados
2
2.05
2.1
2.15
2.2
0 10 20 30 40
t, min
Asb
Figura 4: Estabilidad de complejo extraído.
Como se observa la absorbancia del complejo extraído presenta un
comportamiento estable aproximadamente hasta los 30 min, por lo que las
medidas fueron realizadas antes de este tiempo.
Efecto de la cantidad de reactivos a emplear sobre la absorbancia del complejo:
Los resultados del diseño aplicado para el estudio de la evaluación de la
influencia de cobre (II), tiocianato y acetato sobre a absorbancia del complejo
extraído fueron procesados mediante el programa estadístico statgraphic plus
4.1, anexo2. Los datos correspondientes al análisis de varianzas se muestran a
continuación.
Tabla 8: Resultados del análisis de varianza.
Fuente de
variación
Suma de cuadrados
Grados de libertad
(g)
Cuadrado medio
Razón F
Probabilidad,
p(α= 0.05)
Acetato 0.0401861 1 0.0401841 1.73 0.2583
CNS- 0.103285 1 0.103285 4.46 0.1024
Cobre(II) 0.0007031 1 0.0007031 0.03 0.8702
Residual 0.0927045 4 0.021761
Total 0.236879 7
46

Capítulo III: Presentación y discusión de los resultados
omo se puede apreciar de estos datos ninguno de los factores estudiados
ondiciones seleccionadas y en
.2 Confección de la curva de calibración. ondientes a la curva de calibrado y
C
influye significativamente en la función respuesta, ya que las probabilidades son
mayores que 0.05. Sin embargo, los intervalos LSD revelan que un incremento
de la concentración de tiocianato, permite lograr mejores valores de
absorbancia del complejo extraído, siendo seleccionadas las siguientes
condiciones respecto a los reactivos estudiados: 2 mL de solución de Cu (II), y 6
mL de la correspondiente al Na SCN , anexo 2.
El espectro de absorción del complejo en las c
cubetas de 5 cm es el siguiente, figura 5:
0.2
0.25
0.3
0.35
0.4
300 400 500 600λ, nm
Abs
Figura 5: Espectro de absorción del complejo 3Las tablas 9 y 10 muestran los datos corresp
el tratamiento estadístico de los mismos:
47

Capítulo III: Presentación y discusión de los resultados
Tabla 9: Variación de la absorbancia al cambiar la concentración de
Absorbancia C(Piridina), F(y/x).102
piridina.
ppm
0.213 75 0.00284
0.362 1 12.5 0.32178
0.487 150 0.32467
0.543 1 68.75 0.32178
0.608 187.5 0.32427
0.73 225 0.32444
0.85 262.5 0.32381
0.98 300 0. 32667
Tabla 10: Resultados estadísticos del estudio de linealidad.
Parámetros Valor experimental Criterio
# Total de datos (n) 8 -
Coeficiente de correlación 0.99957 r ≥ 0.99 (r)
Pendiente (b) 0.003351 Desigual de cero
S relativa de b (Sb) 0.00004125 -
C V(b), % 1.23 % ≤ 3 %
C V f(y/x) % 4.45% ≤ 5%
48

Capítulo III: Presentación y discusión de los resultados
De los resultados presentados en la tabla 10 y el anexo3, se infiere que la
ra
ión de regresión correspondiente a la curva de calibración obtenida se
.3 Parámetros que caracterizan la técnica analítica.
se muestran los resultados del estudio de repetibilidad y
Tabla 11: Evaluación de datos de repetibilidad.
os
función C (piridina) vs absorbancia es lineal en el rango de concentración de
75 – 300 ppm, ya que cumple con los parámetros establecidos en la literatu
(63-65). La ecuac
ofrece a continuación:
A = -0.023969 + 0.00335173*C (piridina), ppm
3Precisión y exactitud:
En las tablas 11 y 12
reproducibilidad. El tratamiento estadístico completo se describe en el anexo 4.
Cpy =187.5 ppm Parámetrestadísticos
Abs C(py)exp, ppm
0.609 187.58
0.610 187.89
0.611 188.2
0.610 187.89
n = 5
Cpy = 187.812 ppm
S =
9%
0.280
C V. = 0.14
0.608 187.5 R = 100.16 %
49

Capítulo III: Presentación y discusión de los resultados
Tabla 12: Evaluación de reproducibilidad.
tros
Cpy =405 ppm Parámeestadísticos
Abs C(py)exp, ppm
0.609 187.58
0.610 187.89
0.611 188.2
0.610 187.89
0.608 187.5
0.608 187.5
0.609 187.58
0.610 187.89
0.609 187.58
Cpy =187.73
n =
243
9
S = 0.
C.V:=0.12%
R= 100.12 %
Los datos ofrecidos en las tablas anteriores y el anexo 4 demuestran la
ímite de cuantificación.
y el coeficiente de variación correspondientes a cada
precisión y exactitud de la técnica en las condiciones estudiadas, ya que el
coeficiente de variación es menor que el 3 % y el recobrado esta entre 97-
103.7%.
L
Al analizar el recobrado
nivel de concentración de piridina, se observa que los mismos se corresponden
con los parámetros establecidos, para valores ≥ 105 ppm, siendo este el límite
inferior de cuantificación de la técnica, tabla 13.
50

Capítulo III: Presentación y discusión de los resultados
Tabla13: Determinación del límite de cuantificación.
97.5 105 C(py), ppm 18.75 37.5 52.5 75 90
S 0.246 0.359 8 7 0.288 0.28 0.38 0.153 0.512
C.V(%) 1.32 1.30 0.61 0.38 0.35 0.49 0.19
%R 115.9 93.8 93 104 94 93.4 99.3
studio de interferencias. E
Taller de G-1
El resultado correspondiente a la función respuesta del diseño 25-2 se presenta
la 14: Matriz del diseño factorial fraccionado 25-2 .
en la tabla 14.
Tab
Exp. X1 X2 X3 X4 X5 A
1 - - - + - 2.983
2 + - - - + 0.106
3 - + - - + 0.084
4 + + - + - 3.150
5 - - + + + 3.555
6 + - + - - 0.156
7 - + + - - 0.141
8 + + + + + 2.646
Los datos anteriores se procesaron empleando un programa estadístico
is de varianza se deriva que los valores de la probabilidad están por
profesional statgraphycs plus 4.1 y los resultados obtenidos se recogen en los
anexo 5.
Del anális
encima de 0.05 a excepción del G-1, por lo que es el único de los componentes
51

Capítulo III: Presentación y discusión de los resultados
estudiados que influye significativamente en el valor de la absorbancia y tabla 15.
Los gráficos LSD confirman este criterio, anexo5.
Tabla 15: Probabilidad de significación de los coeficientes. Especie
Probabilidad(α= 0.05 )
Anh.
Acético
0.6277
Br2 0.3773 CS2 0.6830
G-1
0.0143
KBr 0.9872
Del análisis de regresión se obtiene la ecuación que representa el modelo, que
relaciona la absorbancia con las variables estudiadas.
Abs = 1.702 + 0.0945*AnhA - 0.187*Br2 - 0.0787*CS2 + 1.3802*G-1 +
0.00325*KBr
Taller de G-0
El estudio de interferencias realizado en correspondencia con la composición de
los residuales procedentes del taller de G-O contempló las variables y niveles
relacionadas en la tabla siguiente. El diseño experimental corresponde a un 24-1 .
52

Capítulo III: Presentación y discusión de los resultados
Tabla 16: Matriz del diseño 24-1.
Exp. X1 X2 X3 X4 A
1 - - - - 0.362
2 + - - + 2.355
3 - + - + 1.012
4 + + - - 3.453
5 - - + + 0.974
6 + - + - 2.915
7 - + + - 0.380
8 + + + + 3.202
El análisis de varianzas correspondiente a los resultados obtenidos al procesar
el diseño anterior, permite apreciar la probabilidad referida a la influencia de
cada variable para un 95 % de confiabilidad.
Tabla 17: Probabilidad de significación de los coeficientes.
Especie
Probabilidad( α= 0.05 ) Furfural
0.8527
G-O
0.0075
IBA 0.7818
Nitrometano 0.3864
La tabla anterior refleja que solo en el caso del G-O la probabilidad resulta inferior
a 0.05, siendo este el factor que influye significativamente en la absorbancia para
la confiabilidad referida, anexo 6.
53

Capítulo III: Presentación y discusión de los resultados
Del análisis de regresión se obtiene la ecuación que representa el modelo, que
relaciona la absorbancia con las variables estudiadas.
A = 1.83263 + 0.035125*FUR + 1.14863*Go + 0.055125*IBA + 0.181125*NM
Al representar gráficamente la variación de la concentración de piridina en las
corridas experimentales del diseño arriba descrito, se aprecia que las restantes
variables también afectan en alguna medida la señal analítica.
Figura 6: Concentración de piridina en las corridas experimentales.
0
200
400
600
800
1000
1200
C(py), ppm
1 2 3 4 5 6 7 8 piridina
Corridas experimentales
54

Capítulo III: Presentación y discusión de los resultados
3.4 Adición de estándar. Teniendo en cuenta que en los residuales de salida la concentración de piridina es
muy baja, en correspondencia con el límite de cuantificación de la técnica se
investigó otra vía para cuantificar esta amina, buscando una mayor sensibilidad.
Los resultados obtenidos al realizar adiciones de estándar al patrón de piridina se
reflejan en la tabla siguiente:
Tabla18: Parámetros de adición de estándar.
Adición py ,ppm 10 20 40 50 60 80 90
Cpy ,ppm 5.30 16.638 43.185 51.692 61.615 79.740 84.58
5
5.3 1.71 0.66 0.54 0.39 0.35 0.28 C.V., %
53 83.19 107.9 103.38 102.69 %R
99.67 93.98
Como se puede apreciar por esta vía es posible determinar el contenido de
piridina con una adecuada precisión y exactitud hasta valores de 50 ppm con un
coeficiente de variación menor que el 3%.
55

Capítulo III: Presentación y discusión de los resultados
3.5 Aplicación de la técnica. Los tiempos de retención correspondiente a la piridina y los posibles
interferentes se muestran en la tabla 19 y anexo 7.
Tabla 19: Tiempo de retención de especies presentes en los residuales.
Compuesto Tiempo de retención, min
Piridina 6.05
Anhídrido acético 6.017
Disulfuro de carbono 5.082
Furfural 5.850
G-O 6.782
NM 6.05
G-1 9.417
La figura siguiente representa el comportamiento cromatográfico de una solución
que contiene piridina en presencia del efecto matriz.
Figura 7: Cromatograma piridina en presencia de impurezas.
56

Capítulo III: Presentación y discusión de los resultados
Tanto los tiempos de retención de los componentes puros como el cromatograma
de la matriz simulada evidencian que la señal de la piridina puede quedar
enmascarada por la presencia de nitrometano y furfural; cuestión por la cual se
justifica un pretratamiento químico de la muestra.
Por otra parte los compuestos G-0 y G-1 diferenciables en HPLC pueden mostrar
interferencias espectrales al aplicar la técnica extractofotométrica, ya que absorben
en una región cercana a 410 nm.
El cromatograma del residual compuesto se muestra a continuación:
Señales • 4.082 • 4.650 • 5.800 • 6.000 • 6.817 • 11.822 • 12.200
Figura 8: Cromatograma del residual compuesto.
57

Capítulo III: Presentación y discusión de los resultados
El pretratamiento alcalino elimina o disminuye la intensidad de las señales del
G-0 y el G-1como muestra el siguiente cromatograma, figura 9.
Señales • 5.750 • 6.617 • 6.880 • 8.767
Figura 9: Cromatograma en el pretratamiento. La presencia de probables trazas de G-0 se comprobó con la adición de una
pequeña cantidad de solución patrón de este principio activo al residual
pretratado y el posterior registro del cromatograma, figura 10.
58

Capítulo III: Presentación y discusión de los resultados
Figura 10: Cromatograma en el pretratamiento más la adición de G-0.
on el objetivo de corroborar que el G-O se encuentra en cantidades de trazas
Señales • 5.410 • 6.850 • 8.967 • 10.46
C
en el residual pretratado, así como la ausencia de cantidades detectables de G-
O y G-1 en el mismo se registró un cromatograma de capa delgada en las
condiciones cromatográficas descritas en el epígrafe 2.6.1, figura 11.
59

Capítulo III: Presentación y discusión de los resultados
M
F G-0 G-1
Figura 11: Cromatograma de capa delgada en el pretratamiento. tra a diferencia Al correr el cromatograma no se observa desplazamiento de la mues
de los patrones, por lo que no resultan detectables por esta vía las impurezas
ensayadas. Teniendo en cuenta este resultado se procede a determinar el
contenido de piridina mediante extractofotometría y espectrofotometría UV, tabla 20.
60

Capítulo III: Presentación y discusión de los resultados
Tabla 20: Contenido de piridina en el residual compuesto
e la tabla anterior se infiere que en el patrón no se destruye la piridina con el
Técnicas Parámetro Valor Muestra Número de replicas(n) 6
874.1 C (piridina)
D
pretratamiento obteniéndose un recobrado entre el 97 y 103.7% y un coeficiente
de variación menor que el 3%. La concentración de piridina determinada en la
muestra por ambas técnicas es del mismo orden aunque difieren ligeramente.
Para verificar la correspondencia de los valores obtenidos se realiza una
comparación de medias, tabla 21 y anexo 8.
S 0.363 CV, % 0.178 Patrón Número de replicas(n) 6C (piridina) 115.15 S 0.225 CV, % 0.196
Extractofotometría
R, % 100.08Muestra Número de replicas(n) 6C (piridina) 915.35 S 13.75
spectrofotometría
, %
E
CV 1.5
61

Capítulo III: Presentación y discusión de los resultados
Tabla 21: Comparación de medias.
metría Espectrofotometría Parámetros Extractofotoestadísticos ( EF) UV n 6 6 Cpy 4.1 5.32 87 91
S2 9 2.0225 188.804S 1.42218 13.7406 Mínimo 871.97 902.78 Máximo 875.82 927.9 Rango 3.85 25.12 Asimetría 538 289844 0.380 0.0000Curtosis 0.380538 0.0000289844 CV, % 0.178 138
os valores de la asimetría y la curtosis estandarizados están dentro del rango
ación de medias tomando en cuenta las siguientes
la: Ho 1 = H02; σ1 = σ2
2; σ1 = σ2
258081
p es menor que 0.05, si
el pretratamiento debe hacerse en un tiempo igual
to primario a una muestra del residual
L
esperado, correspondiente a una distribución normal, lo que permite realizar la
comparación de las medias.
Al aplicar el test t de compar
hipótesis:
Hipótesis nu
Hipótesis alternativa: Ho 1≠ Ho
Asumiendo que: t = 7.30675 valor de p= 0.0000
Puesto que el intervalo no contiene al cero y el valor de
hay diferencias significativas entre las medias de las dos muestras, para un 95
% de confiabilidad, anexo 8.
Un aspecto a destacar es que
o mayor a cinco días y con una elevada concentración de hidróxido de sodio
para lograr eliminar las interferencias.
Por otra parte al aplicar el tratamien
pretratado la concentración de piridina disminuye alrededor de 10 veces lo cual
se puede apreciar en los cromatogramas donde aparece una señal mas limpia al
62

Capítulo III: Presentación y discusión de los resultados
tiempo de retención de la piridina, figura 13,aspecto sobre el cual deberá
profundizarse en estudios posteriores.
Señales • 5.586 • 5.719 • 6.886
igura 12: Cromatograma del tratamiento primario. espectro en la región de
FPara corroborar la presencia de piridina se registró un
200-500 nm, figura 14, (tabla II, anexo 9).
63

Capítulo III: Presentación y discusión de los resultados
0
0.5
1
1.5
2
2.5
150 250 350 450 550λ,nm
Asb
Figura 13: Espectro del residual real con el tratamiento primario. En estas condiciones también se realizó la aplicación de la técnica
extractofotométrica conjuntamente con la espectrofotométrica, los resultados se
muestran en la tabla 22.
Tabla 22: Concentración de piridina en el residual real (T. 1rio ).
C(py), ppm UV EF-VIS 92.6 91.2 92.34 91.68 92.47 92.62 92.34 93.104 92.6 92.89 Parámetros estadísticos Cpy = 92.47 Cpy = 92.28 ppm S = 0.13 S = 0.80 CV= 0.14% CV = 0.87%
Los resultados de la comparación de medias de de la concentración de piridina
por ambas técnicas en el residual que ha recibido el tratamiento primario se
muestra en la tabla 23, figura 14 y anexo10.
64

Capítulo III: Presentación y discusión de los resultados
Tabla 23: Comparación de medias.
Parámetros estadísticos
Extractofotometría ( EF)
Espectrofotometría UV
n 5 5 Cpy 92.299 92.470
S2 0.673 0.017 S 0. 820 0.130 Mínimo 91.2 92.34 Máximo 93.104 92.60 Rango 1.904 0.260 Asimetría -0.605 0.00 Curtosis -2.043 -3.00 CV, % 0.888 0.140
Los valores de la asimetría y la curtosis estandarizados están dentro del rango
esperado, correspondiente a una distribución normal, lo que permite realizar la
comparación de las medias.
Al aplicar el test t de comparación de medias tomando en cuenta las siguientes
hipótesis:
Hipótesis nula: Ho 1 = H02; σ1 = σ2
Hipótesis alternativa: Ho 1≠ Ho 2; σ1 = σ2
Asumiendo que: t = -0.460946 valor de p = 0.65711
Puesto que el intervalo contiene al cero y el valor de p no es menor que 0.05, no
hay diferencias significativas entre las medias de las dos muestras, para un 95%
de confiabilidad, anexo 10.
El siguiente gráfico refleja el comportamiento descrito en el párrafo anterior.
65

Capítulo III: Presentación y discusión de los resultados
VariablesEFVISUV
Density Traces
91 91.4 91.8 92.2 92.6 93 93.40
0.3
0.6
0.9
1.2
1.5de
nsity
Figura 14: Comparación de medias.
El tratamiento terciario da lugar a una disminución apreciable de la
concentración de piridina así como de las restantes impurezas, como se aprecia
en el cromatograma y espectro UV, figuras 15 y 16.
Señales • 6.100 • 7.117
Figura 15: Cromatograma en el tratamiento terciario.
66

Capítulo III: Presentación y discusión de los resultados
0
0.1
0.2
0.3
0 200 400 600 λ,nm
Abs
Figura 16: Espectro de la piridina en el tratamiento terciario. Los datos primarios del espectro se relacionan en el anexo 9.
La tabla siguiente refleja la cuantificación de esta amina realizada mediante
espectrofotometría.
Tabla 24: Concentración de piridina en el residual real (T. 3rio).
C(py), ppm Parámetros estadísticos 18.18 Cpy =17.69 18.2 S = 0.621 17.5 CV = 3.5 % 16.9
De la tabla anterior se puede concluir que la concentración de piridina en el
residual de salida corresponde a 17.69 ppm para un coeficiente de variación
menor que el 5 %, lo que está por encima del límite de vertimiento.
67

Conclusiones
Conclusiones
1. Ha sido desarrollada una técnica extractofotométrica para la
cuantificación de piridina en muestras de residuales del proceso de
producción de G-1, basada en la extracción del complejo coloreado
Cu(II)-SCN - - piridina en cloroformo .
2. La determinación analítica de piridina aplicando la técnica estudiada,
resulta repetible, lineal y exacta en las condiciones empleadas para un
rango de 75-300 ppm, con un límite de cuantificación de 105 ppm y por
adición de estándar hasta 50 ppm.
3. Fue investigado el efecto de las especies químicas presentes en los
residuales de los talleres de G-O y G-1 en la función respuesta,
comprobando que aún cuando sólo interfieren estadísticamente el G-O y
el G-1, para α= 0.05, la mayoría de las especies presentes afectan la
señal analítica.
4. El estudio cromatográfico y el ensayo de Meyer demostraron que los
residuales pretratados en medio básico presentan principalmente piridina
y solo trazas de otras impurezas.
5. El contenido de piridina en el residual compuesto por las corrientes
residuales de los talleres de producción de G-O y G-1disminuye 10 veces
por efecto del tratamiento primario y cinco veces por los tratamientos
secundarios y terciario.
68

Recomendaciones
Recomendaciones. 1. Aplicar la técnica desarrollada para la cuantificacion de piridina en
residuales con alto contenido de esta base.
2. Ampliar el estudio cromatográfico por HPLC con el fin de aplicar esa
metodología a l a determinacion de piridina en residuales de salida.
69

Bibliografía
Bibliografía 1. Molins-Legua , C. and Campins-Falcó, P.: “ Solid phase extraction of
amines”. Analytica Chimica Acta, Volume 546, Issue 2 , pp. 206-220, (
2005).
2. Thelander, G. Jonsson , J. and Schuberth J.: “Is urine a suitable material
for the preliminary screening of drugs in autopsy cases?” .Forensic
Science International, Volume 22, Issues 2-3 , August-September Pages
189-194 (1983).
3. Eldem, T.; Arican-Cellat, N.: “Determination of amphotericin B in human
plasma using solid-phase extraction and high-performance liquid
chromatography”. Journal of Pharmaceutical and Biomedical Analysis, 25
(1),pp53-64, (2001).
4. Draisci, R.; Marchiafava, C.; Palleschi, L.; Cammarata, P.; Cavalli, S.:
“Accelerated solvent extraction and liquid chromatography-tandem mass
spectrometry quantitation of corticosteroid residues in bovine liver”.
Journal of Chromatography B: Biomedical Sciences and Applications,
753(2), pp 217- 223, (2001).
5. Draisci, R.; Giannetti, L.; Lucentini L. et-al. : “ Determination of nitrofuran
residues in avian eggs by liquid chromatography–UV photodiode array
detection and confirmation by liquid chromatography–ionspray mass
spectrometry” . Journal of Chromatography A Volume 777, Issue 1 , pp.
201-211 (1997).
6. Zolotov,Yu. A:”Extraction of Quelate Compounds”. Ed. Khimia Moscú,
Tésisde Maestría, C.B.Q., U.C.L.V. pp11-13 y 178, (1967).
7. Kreshkov, P.: “Ocnovy Ananliticcheskoi Kjimii”. Izd. Kjimiya. Moskwa,
(1971).
8. Castañedo, N.; et-al.: “Registro de Queratofural”, No211, C.B.Q., U.C.L.V.,
Cuba, 1993
9. Castañedo, N.; et. al.: “Registro de Vitrofural”, No 013, C.B.Q., U.C.L.V. ,
Cuba, 1999.
70

Bibliografía
10. Gaytan T.E, Castañedo N.R. “Nuevo procedimiento para la extención del
2-bromo-5-(2-bromo-2-nitrovinil)-furano. No.22932, 30.10.2003.USA; No
1249449, 17,9 ,03 referencia 64/ 2002. Europa.
11. Glastone. S : “ Tratado de Química Física ”.Ediciones
Revolucionarias,1972.
12. Kliachko Yu.A. Manual de química analítica. U2d. MiR. Moskua 1975.
13. Mezhov, E.A.: “Extracción con aminas, sales de aminas y bases de
amoniocuaternarias”. Tomo II. Atomizdat, Moscú, p110, (1977).
14. Hernández, B.N.; Peralta, G.: “Separación de Cu(II) y Co(II) con ácido
octanóico y 2-metilpiridina”. Revista Cubana de Química, Vol.8, No.2,
(1996).
15. Mustafa, Z.Ö.; Keith, D.B.; Anthony A.C.; Mark D.B.: “Extraction, solubility
and stability of metal complexes using stainless steel supercritical fluid
extraction system”. Analytica Chimica Acta, 417 (2), pp177 – 184,
(2000).
16. Liu, J.; Wang, W.; Li, G.: “A new strategy for supercritical fluid extraction
ofcopper ions”. Talanta, 53(6), pp 1149-1154, (2001).
17. Hengwu, C.; Jincao, J.; Yufeng, W.: "Flow injection on-line coprecipitation
preconcentration system using copper(II) diethyldithiocarbamate as
carrier for flame atomic absorption spectrometric determination of
cadmium, lead and nickel in environmental samples. Analytica Chimica
Acta, 353 (2-3) , pp181- 188, (1997).
18. Hirata, S.; Ishida, Y.; Aihara, M.; Honda, K.; Shikino, O.: " Determination
of trace metals in seawater by on-line column preconcentration
inductively coupled plasma mass spectrometry". Analytica Chimica Acta,
438(1-2), pp 205-214, (2001).
19. Romberg, H. Muller: “Photometric screening-test for heavy metals under
flow injection conditions using extractive determination with dithizone”,
Analytica Chimica Acta, 353(2-3), pp165-172, (1997).
71

Bibliografía
20. Perez-Ruiz, T.; Martinez-Lozano, C.; Tomas, V.; Sanz, A.; Sahuquillo, E.:
"Flow-injection extraction-spectrophotometric method for the
determination of ranitidine in pharmaceutical preparations". Journal
of Pharmaceutical and Biomedical Analysis, 26(4), pp609-615,
(2001).
21. Ruso, R.; Ramos, Y.; Borodulia, L; Lezcano, M.: “Química Orgánica
Experimental,T. 1”, Universidad Central de Las Villas, p. 93, 1971.
22. Morrison R. T. y Boyd R. N.: “Química Orgánica”, Universidad de New
York, Versión en español, 4 ta edición. Fondo Educativo Interamericano,
México,PP. 1275-1276, 1983.
23. “Grupo de Trabajo de Información a la población en materia de
contaminación atmosférica, Índice de calidad del aire”, p 65, (2000).
24. Kovacs M. Pollution control and conservation. Budapest 1985
25. Dangeruos properties of organic compounds. NIOSH Pocket Guide to
Chemical Hazards ( www.cdc.gov/niosh/npg./npg.htlm).
26. Cesard: “Chemical Evaluation Search And Retrieval, Canadian Centre
for Occupational Health and Safety”, Issue, (2001).
27. Colectivo de autores: “Rtecs- Registry of Toxic Effects of Chemical
Substances, On-line search, Canadian Centre for Occupational
Healthand Safety RTECS database”, (2001).
28. Siggia, S.: “Quantitave Organic Analysis via functional group”. John
Wiley,P 428, (1963).
29. Gyenes, I.: " Titration in Non-Aqueous Media”, First Ed., Akademia
Kiadó, Budapest, pp270-272 y 326-327, (1963).
30. Pérez, C.J.: “Montaje de una técnica para la determinación de la piridina
enmedio no acuoso”. Trabajo de Curso, Centro de Bioactivos Químicos,
Universidad Central de Las Villas, (2000).
31. Villavecchia, V.: “Tratado de Química Analítica”. Editorial Gustavo Gili,
S.A. Tomo II, p331, (1963).
32. Blumenthal, Bchemi. 2.19.521 , (1909).
72

Bibliografía
33. Burr, G.O.; Grtne, R.A.; Ame, J.Chem. Soc. 46, pp1224-1246, (1924).
34. Bmistrov, I. J. Chem. 19, pp1511-1514, (1949).
35. Sandell, E.B.: “Colorimetric Determination of Traces of Metals”.
Interscience publishers, New York, 3 rd ed., p198, (1959).
36. Alonso L.: “Extracción de metales divalentes y determinación de piridina
con un nuevo extrayente de base pirazolínica”. Trabajo de Diploma.
Centro de Bioactivos Químicos. Universidad Central de Las Villas. 1995.
37. Hernández, B.N.; Peralta, G.: “Determinación extractofotométrica de
cobre en aceros con ácido octanóico-2-metilpiridina”. Revista Cubana
de Química, Vol.VI, No.2, (1990).
38. Betancort, E. Determinación extractofotométrica de piridina por su
acción complejante en el sistema Cu(II) - ácido salicílico. Trabajo de
diploma, UCLV, Cuba, 2003-04.
39. Bankowski, Z. y otros: “ Maly Kalendarz Chemika”. P. W. T. Varsovia,
pp851- 855, (1958).
40. Gaitán, E.; Hernández, B.N.; Pérez, L.: “Estudio de la extarccion del
complejo C(Py)2(SCN)2 para la determinación extractofotométrica
de piridina”. Trabajo de curso , UCLV, (2004).
41. Scott E. Collibee and Jiaxin Yu.: “ A facile and convenient synthesis of
functionalized ortho-nitrophenylboronic acids “.Tetrahedron Letters
,Volume 46, Issue 26 , pp 4453-4455, (2005).39. Michael C. Harlow;
Frank R. Stermitz and Richard D. Thomas :”
42. Isolation of nitro compounds from Astragalus species.Phytochemistry
Volume 14, Issues 5-6 , June 1975, Pages 1421-1423.
43. Fujinori Hanawa, Satoshi Tahara and G. H. Neil Towers:” Antifungal nitro
compounds from Skunk Cabbage (Lysichitum americanum) leaves
treated with cupric chloride. Phytochemistry Volume 53, Issue 1 ,
January 2000, Pages 55-58
44. Sohr, J. Janes y W. Bongartz, A.: “ TLC analysis of nitro compounds in
residual warfare site contamination”, Analusis, Mar 1995. Phytochemistry
,Volume 14, Issues 5-6 , pp.1421-1423, (1975).
73

Bibliografía
45. Patravale VB, Nair VB, Gore SP. High performance thin layer
chromatographic determination of nifedipine from bulk drug and from
pharmaceuticals. J Pharm Biomed Anal. 23(4),pp.623-627.(2000).
46. Alemany G, Akaarir M, Gamundi: ” AThin-layer chromatographic
determination of catecholamines, 5-hydroxytryptamine, and their
metabolites in biological samples” review., J AOAC Int. Jan-Feb;82(1),
pp17-24 (1999).
47. Pisternick W, Kovar KA, Ensslin H.: “ High-performance thin-layer
chromatographic determination of N-ethyl-3,4-
methylenedioxyamphetamine and its major metabolites in urine and
comparison with high-performance liquid chromatography”. J
Chromatogr B Biomed Sci 10, pp.688 (1):63-9(1997).
48. A lemany G; Akaarir M.; Rossello C, Gamundi A : “Thin-layer
chromatographic determination of catecholamines in rat plasma”.
Biomed Chromatogr.;10(5):pp. 225-7. (1996).
49. N. Sistovaris, E. E. Dagrosa and A. Keller : “Thin-layer
chromatographic determination of imipramine and desipramine in human
plasma and urine at single—dose levels.”. Journal of Chromatography B:
Biomedical Sciences and Applications Volume 277 , pp.273-281(1983).
50. Thin-layer chromatographic determination of brain catecholamines and
5-hydroxytryptamine.Alemany G, Nicolau MC, Gamundi A, Rial R.
Biomed Chromatography. 7(6), pp.315-6. (1993).
51. Trondina GA.: “ Chromatographic determination of methylnitrophos and
para- nitrocresol poisonings”. Veterinariia., 4 pp.84-5 (1972).
52. López M. E. “ Desarrollo y aplicación de una técnica cromatográfica para
la cuantificación de G-1 en el taller de crudo – 3.
53. Torres L.K. Trabajo diploma: “Desarrollo de una técnica combinada por
CCD-UV para la cuantificación del 2-(2-nitrovinil)- furano” (2003).
54. Lapa-Guimaraes J, Pickova J.: “New solvent systems for thin-layer
chromatographic determination of nine biogenic amines in fish and
squid”. J Chromatogr A. 2004 Aug 6;1045(1-2):223-32.
74

Bibliografía
55. Barnes, A. R.Determination of ceftazidime and pyridine by HPLC:
Application to a viscous eye drop formulation. J. Liq.
Chromatography Volume: 18(15), pp. 3117(1995).
56. Kalchenko OI, Golub VA, Zavatskaja IV. : “HPLC and GLC determination
of residual solvents in busulphan. J Pharm Biomed Anal. “Dec;14(1-2),
pp. 107-11( 1995).
57. Wu AT, Massey IJ, Kushinsky S.A : “High-performance liquid
chromatographic method for the simultaneous determination of
nicardipine and its pyridine metabolite II in plasma”. J Pharm Sci.
Oct;73(10), pp.:444-7( 1984).
58. Stephan M. C. Buckenmaier David V. McCalley, and Melvin R. Euerby
Determination of pKa values of organic bases in aqueous acetonitrile
solutions using capillary electrophoresis. Journal of Chromatography A
.Volume 1004, Issues 1-2 , Pages 71-79 (2003).
59. Quattrochi, O.: Abelaira, S. y Laba, R.: “Introducción a la HPLC”.
Aplicación y práctica”.Ed. Artes gráficas Farro S.A., Buenos Aires(
1992).
60. Hald, A.: “ Statistical theory with engineering aplications”. John Wiley and
Sons, (1952).
61. Freud, J.E.: “ Estadística elemental moderna”. 3a Edición. Editorial
Pueblo y Educación, p466, (1983).
62. Comat 99.: “4O Evento Internacional Científico Metodológico de la
Matemática y la Computación”, (1999).
63. Masert, D.L.; Semyers-verbeke, J.; Vandeginste, B.: “An Introduction to
method validation”. Anal. Magazine (USA), 22(5), M14-M15, (1994).
64. Castro C,M. “Validación de metodos analiticos”. Monografía , Asociación
española de Farmaceuticos de l;a industria , 1990. validación (Castro).
65. Calpena Campnam , A.C. “Validación de los Metodos Analiticos “.
Universidad de Barcelona, 1990.
75

Bibliografía
76

Anexos
Anexo 1. Tabla I. Datos de los espectros del complejo y los componentes
del sistema.
Landa Cobre(II)SCN -
Piridina Complejo Piridina SCN-
200 0 0,009 0,033 0,013 0,01
210 0 0,011 0,04 0,01 0,012
220 0 0,011 0,058 0,02 0,012
230 0,006 0,02 0,381 0,025 0,022
240 0,11 0,275 1,503 0,436 0,356
250 0,222 1,306 1,554 1,418 0,559
260 0,241 1,389 1,431 1,466 0,42
270 0,162 1,242 1,159 1,118 0,376
280 0,121 0,382 0,748 0,231 0,351
290 0,085 0,055 0,58 0,068 0,177
300 0,06 0,018 0,451 0,055 0,114
310 0,035 0,004 0,334 0,05 0,092
320 0,009 0 0,288 0,042 0,064
330 0,005 0 0,256 0,038 0,048
340 0 0 0,252 0,034 0,039
350 0 0 0,256 0,032 0,03
360 0 0 0,293 0,028 0,02
370 0 0 0,314 0,026 0,016
380 0 0 0,343 0,026 0,015
390 0 0 0,362 0,025 0,013
400 0 0 0,372 0,023 0,011
410 0 0 0,364 0,021 0,01
420 0 0 0,345 0,021 0,008
430 0 0 0,314 0,02 0,008
440 0 0 0,278 0,019 0,007
450 0 0 0,256 0,018 0,006
460 0 0 0,236 0,017 0
470 0 0 0,222 0,017 0
480 0 0 0,202 0,016 0
490 0 0 0,202 0,016 0
500 0 0 0,203 0,014 0
76

Anexos
Anexo 2. Estudio de la influencia de algunos reactivos en la absorbancia del complejo extraído.
Analysis of Variance for Absorbancia - Type III Sums of Squares
--------------------------------------------------------------------------------
Source Sum of Squares Df Mean Square F-Ratio P-Value
--------------------------------------------------------------------------------
MAIN EFFECTS
A:Acetato 0.0401861 1 0.0401861 1.73 0.2583
B:CNS 0.103285 1 0.103285 4.46 0.1024
C:Cobre2 0.000703125 1 0.000703125 0.03 0.8702
RESIDUAL 0.0927045 4 0.0231761
--------------------------------------------------------------------------------
TOTAL (CORRECTED) 0.236879 7
--------------------------------------------------------------------------------
All F-ratios are based on the residual mean square error.
The StatAdvisor
---------------
The ANOVA table decomposes the variability of Absorbancia into
contributions due to various factors. Since Type III sums of squares
(the default) have been chosen, the contribution of each factor is
measured having removed the effects of all other factors. The
P-values test the statistical significance of each of the factors.
Since no P-values are less than 0.05, none of the factors have a
statistically significant effect on Absorbancia at the 95.0%
confidence level.
77

Anexos
Influencia del cobre:
Means and 95.0 Percent LSD Intervals
Cobre2
Abs
orba
ncia
-1 10.77
0.87
0.97
1.07
1.17
Influencia del ion acetato:
Means and 95.0 Percent LSD Intervals
Acetato
Abs
orba
ncia
-1 10.71
0.81
0.91
1.01
1.11
1.21
Influencia del ion CNS:
Means and 95.0 Percent LSD Intervals
CNS
Abs
orba
ncia
-1 10.66
0.76
0.86
0.96
1.06
1.16
1.26
78

Anexos
Anexo 3. Estudio de la linealidad de la técnica Regression Analysis - Linear model: Y = a + b*X
-----------------------------------------------------------------------------
Dependent variable: A
Independent variable: CBr2ppm
-----------------------------------------------------------------------------
Standard T
Parameter Estimate Error Statistic P-Value
-----------------------------------------------------------------------------
Intercept -0.023969 0.00817311 -2.93266 0.0262
Slope 0.00335173 0.0000412577 81.2388 0.0000
-----------------------------------------------------------------------------
Analysis of Variance
-----------------------------------------------------------------------------
Source Sum of Squares Df Mean Square F-Ratio P-Value
-----------------------------------------------------------------------------
Model 0.445799 1 0.445799 6599.75 0.0000
Residual 0.000405287 6 0.0000675478
-----------------------------------------------------------------------------
Total (Corr.) 0.446204 7
Correlation Coefficient = 0.999546
R-squared = 99.9092 percent
Standard Error of Est. = 0.00821875
The StatAdvisor
---------------
The output shows the results of fitting a linear model to describe
the relationship between A and CBr2ppm. The equation of the fitted
model is
79

Anexos
A = -0.023969 + 0.00335173*C(piridina) ppm
Since the P-value in the ANOVA table is less than 0.01, there is a
statistically significant relationship between A and C(piridina) ppm at the
99% confidence level.
The R-Squared statistic indicates that the model as fitted explains
99.9092% of the variability in A. The correlation coefficient equals
0.999546, indicating a relatively strong relationship between the
variables. The standard error of the estimate shows the standard
deviation of the residuals to be 0.00821875. This value can be used
to construct prediction limits for new observations by selecting the
Forecasts option from the text menu.
80

Anexos
Anexo 4. Tratamiento estadístico de los datos de repetibilidad. Summary Statistics for Col_3
Count = 5
Average = 187.812
Variance = 0.07847
Standard deviation = 0.280125
Minimum = 187.5
Maximum = 188.2
Range = 0.7
Skewness = 0.312796
Stnd. skewness = 0.285543
Kurtosis = -0.872479
Stnd. kurtosis = -0.39823
Coeff. of variation = 0.149152%
The StatAdvisor
---------------
This table shows summary statistics for Col_3. It includes
measures of central tendency, measures of variability, and measures of
shape. Of particular interest here are the standardized skewness and
standardized kurtosis, which can be used to determine whether the
sample comes from a normal distribution. Values of these statistics
outside the range of -2 to +2 indicate significant departures from
normality, which would tend to invalidate any statistical test
regarding the standard deviation. In this case, the standardized
skewness value is within the range expected for data from a normal
distribution. The standardized kurtosis value is within the range
expected for data from a normal distribution
81

Anexos
Anexo 5. Análisis del diseño de interferencia 25-2.
Analysis of Variance for Abs - Type III Sums of Squares
--------------------------------------------------------------------------------
Source Sum of Squares Df Mean Square F-Ratio P-Value
--------------------------------------------------------------------------------
MAIN EFFECTS
A:AnhA 0.071442 1 0.071442 0.32 0.6277
B:Br2 0.28125 1 0.28125 1.27 0.3773
C:CS2 0.0496125 1 0.0496125 0.22 0.6830
D:G1 15.2407 1 15.2407 68.66 0.0143
E:KBr 0.0000845 1 0.0000845 0.00 0.9862
RESIDUAL 0.443973 2 0.221986
--------------------------------------------------------------------------------
TOTAL (CORRECTED) 16.0871 7
--------------------------------------------------------------------------------
All F-ratios are based on the residual mean square error.
The StatAdvisor
---------------
The ANOVA table decomposes the variability of Abs into
contributions due to various factors. Since Type III sums of squares
(the default) have been chosen, the contribution of each factor is
measured having removed the effects of all other factors. The
P-values test the statistical significance of each of the factors.
Since one P-value is less than 0.05, this factor has a statistically
significant effect on Abs at the 95.0% confidence level.
Table of Least Squares Means for Abs
82

Anexos
with 95.0 Percent Confidence Intervals
--------------------------------------------------------------------------------
Stnd. Lower Upper
Level Count Mean Error Limit Limit
--------------------------------------------------------------------------------
GRAND MEAN 8 1.702
AnhA
-1 4 1.6075 0.235577 0.593894 2.62111
1 4 1.7965 0.235577 0.782894 2.81011
Br2
-1 4 1.8895 0.235577 0.875894 2.90311
1 4 1.5145 0.235577 0.500894 2.52811
CS2
-1 4 1.78075 0.235577 0.767144 2.79436
1 4 1.62325 0.235577 0.609644 2.63686
G1
-1 4 0.32175 0.235577 -0.691856 1.33536
1 4 3.08225 0.235577 2.06864 4.09586
KBr
-1 4 1.69875 0.235577 0.685144 2.71236
1 4 1.70525 0.235577 0.691644 2.71886
--------------------------------------------------------------------------------
The StatAdvisor
---------------
This table shows the mean Abs for each level of the factors. It
also shows the standard error of each mean, which is a measure of its
sampling variability. The rightmost two columns show 95.0% confidence
intervals for each of the means. You can display these means and
intervals by selecting Means Plot from the list of Graphical Options.
83

Anexos
-1 1
Means and 95.0 Percent LSD Intervals
AnhA
0.8
1.1
1.4
1.7
2
2.3
2.6A
bs
Means and 95.0 Percent LSD Intervals
CS2
Abs
-1 10.9
1.3
1.7
2.1
2.5
eans and 95.0 Percent LSD Interv
G1
Abs
-1 1-0.4
0.6
1.6
2.6
3.6
4.6
eans and 95.0 Percent LSD Interv
KBr
Abs
-1 10.9
1.3
1.7
2.1
2.5
84

Anexos
• Análisis de regresión.
Multiple Regression Analysis
-----------------------------------------------------------------------------
Dependent variable: Abs
-----------------------------------------------------------------------------
Standard T
Parameter Estimate Error Statistic P-Value
-----------------------------------------------------------------------------
CONSTANT 1.702 0.166578 10.2174 0.0094
AnhA 0.0945 0.166578 0.567301 0.6277
Br2 -0.1875 0.166578 -1.1256 0.3773
CS2 -0.07875 0.166578 -0.472751 0.6830
G1 1.38025 0.166578 8.2859 0.0143
KBr 0.00325 0.166578 0.0195104 0.9862
-----------------------------------------------------------------------------
Analysis of Variance
-----------------------------------------------------------------------------
Source Sum of Squares Df Mean Square F-Ratio P-Value
-----------------------------------------------------------------------------
Model 15.6431 5 3.12862 14.09 0.0676
Residual 0.443972 2 0.221986
-----------------------------------------------------------------------------
Total (Corr.) 16.0871 7
R-squared = 97.2402 percent
R-squared (adjusted for d.f.) = 90.3407 percent
Standard Error of Est. = 0.471154
Mean absolute error = 0.233
Durbin-Watson statistic = 0.751726
85

Anexos
The StatAdvisor
---------------
The output shows the results of fitting a multiple linear
regression model to describe the relationship between Abs and 5
independent variables. The equation of the fitted model is
Abs = 1.702 + 0.0945*AnhA - 0.1875*Br2 - 0.07875*CS2 + 1.38025*G1 +
0.00325*KBr
Since the P-value in the ANOVA table is less than 0.10, there is a
statistically significant relationship between the variables at the
90% confidence level.
The R-Squared statistic indicates that the model as fitted
explains 97.2402% of the variability in Abs. The adjusted R-squared
statistic, which is more suitable for comparing models with different
numbers of independent variables, is 90.3407%. The standard error of
the estimate shows the standard deviation of the residuals to be
0.471154. This value can be used to construct prediction limits for
new observations by selecting the Reports option from the text menu.
The mean absolute error (MAE) of 0.233 is the average value of the
residuals. The Durbin-Watson (DW) statistic tests the residuals to
determine if there is any significant correlation based on the order
in which they occur in your data file. Since the DW value is less
than 1.4, there may be some indication of serial correlation. Plot
the residuals versus row order to see if there is any pattern which
can be seen.
In determining whether the model can be simplified, notice that the
highest P-value on the independent variables is 0.9862, belonging to
KBr. Since the P-value is greater or equal to 0.10, that term is not
statistically significant at the 90% or higher confidence level.
Consequently, you should consider removing KBr from the model.
86

Anexos
Anexo 6. Análisis estadístico del diseño de interferencia 24-1 .
Analysis of Variance for Error - Type III Sums of Squares -------------------------------------------------------------------------------- Source Sum of Squares Df Mean Square F-Ratio P-Value -------------------------------------------------------------------------------- MAIN EFFECTS A:FUR 0.0280845 1 0.0280845 0.04 0.8527 B:Go 28.5995 1 28.5995 41.64 0.0075 C:IBA 0.0630125 1 0.0630125 0.09 0.7818 D:NM 0.702112 1 0.702112 1.02 0.3864 RESIDUAL 2.06041 3 0.686802 -------------------------------------------------------------------------------- TOTAL (CORRECTED) 31.4531 7 -------------------------------------------------------------------------------- All F-ratios are based on the residual mean square error. The StatAdvisor --------------- The ANOVA table decomposes the variability of Error into contributions due to various factors. Since Type III sums of squares (the default) have been chosen, the contribution of each factor is measured having removed the effects of all other factors. The P-values test the statistical significance of each of the factors. Since one P-value is less than 0.05, this factor has a statistically significant effect on Error at the 95.0% confidence level. Table of Least Squares Means for A with 95.0 Percent Confidence Intervals -------------------------------------------------------------------------------- Stnd. Lower Upper Level Count Mean Error Limit Limit -------------------------------------------------------------------------------- GRAND MEAN 8 1.83262 FUR -1 4 1.7975 0.252376 0.994327 2.60067 1 4 1.86775 0.252376 1.06458 2.67092 Go -1 4 0.684 0.252376 -0.119173 1.48717 1 4 2.98125 0.252376 2.17808 3.78442 IBA -1 4 1.7775 0.252376 0.974327 2.58067 1 4 1.88775 0.252376 1.08458 2.69092 NM -1 4 1.6515 0.252376 0.848327 2.45467 1 4 2.01375 0.252376 1.21058 2.81692 --------------------------------------------------------------------------------
87

Anexos
The StatAdvisor --------------- This table shows the mean A for each level of the factors. It also shows the standard error of each mean, which is a measure of its sampling variability. The rightmost two columns show 95.0% confidence intervals for each of the means. You can display these means and intervals by selecting Means Plot from the list of Graphical
Means and 95.0 Percent LSD Intervals
FUR
A
-1 11.2
1.5
1.8
2.1
2.4
2.7
Means and 95.0 Percent LSD Intervals
Go
A
-1 10
1
2
3
4
Means and 95.0 Percent LSD Intervals
IBA
A
-1 11.2
1.5
1.8
2.1
2.4
2.7
Means and 95.0 Percent LSD Intervals
NM
A
-1 11
1.4
1.8
2.2
2.6
88

Anexos
Analysis of Variance for Error - Type III Sums of Squares -------------------------------------------------------------------------------- Source Sum of Squares Df Mean Square F-Ratio P-Value -------------------------------------------------------------------------------- MAIN EFFECTS A:FUR 0.0280845 1 0.0280845 0.04 0.8527 B:Go 28.5995 1 28.5995 41.64 0.0075 C:IBA 0.0630125 1 0.0630125 0.09 0.7818 D:NM 0.702112 1 0.702112 1.02 0.3864 RESIDUAL 2.06041 3 0.686802 -------------------------------------------------------------------------------- TOTAL (CORRECTED) 31.4531 7 -------------------------------------------------------------------------------- All F-ratios are based on the residual mean square error. The StatAdvisor --------------- The ANOVA table decomposes the variability of Error into contributions due to various factors. Since Type III sums of squares (the default) have been chosen, the contribution of each factor is measured having removed the effects of all other factors. The P-values test the statistical significance of each of the factors. Since one P-value is less than 0.05, this factor has a statistically significant effect on Error at the 95.0% confidence level. Table of Least Squares Means for A with 95.0 Percent Confidence Intervals -------------------------------------------------------------------------------- Stnd. Lower Upper Level Count Mean Error Limit Limit -------------------------------------------------------------------------------- GRAND MEAN 8 1.83262 FUR -1 4 1.7975 0.252376 0.994327 2.60067 1 4 1.86775 0.252376 1.06458 2.67092 Go -1 4 0.684 0.252376 -0.119173 1.48717 1 4 2.98125 0.252376 2.17808 3.78442 IBA -1 4 1.7775 0.252376 0.974327 2.58067 1 4 1.88775 0.252376 1.08458 2.69092 NM
89

Anexos
Multiple Regression Analysis ----------------------------------------------------------------------------- Dependent variable: A ----------------------------------------------------------------------------- Standard T Parameter Estimate Error Statistic P-Value ----------------------------------------------------------------------------- CONSTANT 1.83263 0.178457 10.2693 0.0020 FUR 0.035125 0.178457 0.196827 0.8565 Go 1.14863 0.178457 6.43644 0.0076 IBA 0.055125 0.178457 0.308899 0.7776 NM 0.181125 0.178457 1.01495 0.3849 ----------------------------------------------------------------------------- Analysis of Variance ----------------------------------------------------------------------------- Source Sum of Squares Df Mean Square F-Ratio P-Value ----------------------------------------------------------------------------- Model 10.8513 4 2.71284 10.65 0.0405 Residual 0.764322 3 0.254774 ----------------------------------------------------------------------------- Total (Corr.) 11.6157 7 R-squared = 93.4199 percent R-squared (adjusted for d.f.) = 84.6464 percent Standard Error of Est. = 0.504752 Mean absolute error = 0.257875 Durbin-Watson statistic = 1.54994 The StatAdvisor --------------- The output shows the results of fitting a multiple linear regression model to describe the relationship between A and 4 independent variables. The equation of the fitted model is A = 1.83263 + 0.035125*FUR + 1.14863*Go + 0.055125*IBA + 0.181125*NM Since the P-value in the ANOVA table is less than 0.05, there is a statistically significant relationship between the variables at the 95% confidence level. The R-Squared statistic indicates that the model as fitted explains 93.4199% of the variability in A. The adjusted R-squared statistic, which is more suitable for comparing models with different numbers of independent variables, is 84.6464%. The standard error of the estimate shows the standard deviation of the residuals to be 0.504752. This value can be used to construct prediction limits for new observations by selecting the Reports option from the text menu.
90

Anexos
The mean absolute error (MAE) of 0.257875 is the average value of the residuals. The Durbin-Watson (DW) statistic tests the residuals to determine if there is any significant correlation based on the order in which they occur in your data file. Since the DW value is greater than 1.4, there is probably not any serious autocorrelation in the residuals. In determining whether the model can be simplified, notice that the highest P-value on the independent variables is 0.8565, belonging to FUR. Since the P-value is greater or equal to 0.10, that term is not statistically significant at the 90% or higher confidence level. Consequently, you should consider removing FUR from the model.
91

Anexos
Anexo 7.
Cromatograma de la piridina por HPLC
Cromatogramas registrados a los interferentes estudiados
Anhídrido Acético.
92

Anexos
Furfural
Nitrometano
93

Anexos
G-0
G-1
94

Anexos
Disulfuro de carbono
95

Anexos
Anexo 8. Comparación de medias en el pretratamiento. Summary Statistics Espf Extf ------------------------------------------------------------ Count 6 6 Average 915.323 874.117 Variance 188.804 2.02259 Standard deviation 13.7406 1.42218 Minimum 902.78 871.97 Maximum 927.9 875.82 Range 25.12 3.85 Stnd. skewness 0.0000289844 -0.380538 Stnd. kurtosis -1.66663 -0.331121 ------------------------------------------------------------ The StatAdvisor --------------- This table shows summary statistics for the two samples of data. Other tabular options within this analysis can be used to test whe ther differences between the statistics from the two samples are statistically significant. Of particular interest here are the standardized skew ness and standardized kurtosis, which can be used to determine whether the samples come from normal distributions. Values of these statistics outside the range of -2 to +2 indicate significant departures from normality, which would tend to invalidate the tests which compare the standard deviations. In this case, both standardized skew ness values are within the range expected. Both standardized kurtosis values are within the range expected.
96

Anexos
Comparison of Means ------------------- 95.0% confidence interval for mean of Espf: 915.323 +/- 14.4199 [900.903,929.743] 95.0% confidence interval for mean of Extf: 874.117 +/- 1.49249 [872.624,875.609] 95.0% confidence interval for the difference between the means assuming equal variances: 41.2067 +/- 12.5657 [28.641,53.7724] t test to compare means Null hypothesis: mean1 = mean2 Alt. hypothesis: mean1 NE mean2 assuming equal variances: t = 7.30675 P-value = 0.0000258081 The StatAdvisor --------------- This option runs a t-test to compare the means of the two samples. It also constructs confidence intervals or bounds for each mean and for the difference between the means. Of particular interest is the confidence interval for the difference between the means, which extends from 28.641 to 53.7724. Since the interval does not contain the value 0.0, there is a statistically significant difference between the means of the two samples at the 95.0% confidence level. A t-test may also be used to test a specific hypothesis about the difference between the means of the populations from which the two samples come. In this case, the test has been constructed to determine whether the difference between the two means equals 0.0 versus the alternative hypothesis that the difference does not equal 0.0. Since the computed P-value is less than 0.05, we can reject the null hypothesis in favor of the alternative. NOTE: these results assume that the variances of the two samples are equal. In this case, that assumption is questionable since the results of an F-test to compare the standard deviations suggests that there may be a significant difference between them. You can see the results of that test by selecting Comparison of Standard Deviations from the Tabular Options menu.
97

Anexos
Anexo 9. Tabla II. Espectro de la piridina en el tratamiento primario
Longitud de onda (nm) Absorbancia 200 0.734 210 1.166 220 1.668 230 2.154 240 2.154 250 1.913 260 1.749 270 1.595 280 1.556 290 1.612 300 1.659 310 1.495 320 1.175 330 0.902 340 0.673 350 0.544 360 0.451 370 0.364 380 0.291 390 0.231 400 0.181 410 0.139 420 0.111 430 0.090 440 0.074 450 0.061 460 0.049 470 0.049 480 0.046 490 0.040 500 0.037
98

Anexos
Tabla III: Espectro del tratamiento secundario Longitud de onda, nm Absorbancia
200 0.032 210 0.066 220 0.127 230 0.151 240 0.225 250 0.277 260 0.211 270 0.21 280 0.192 290 0.19 300 0.16 310 0.141 320 0.14 330 0.125 340 0.115 350 0.099 360 0.099 370 0.096 380 0.097 390 0.079 400 0.051 410 0.035 420 0.029 430 0.025 440 0.022 450 0.02 460 0.017 470 0.016 480 0.016 490 0.015 500 0.014
99

Anexos
Anexo 10. Comparación de medias en tratamiento primario. Summary Statistics EFVIS UV ------------------------------------------------------------ Count 5 5 Average 92.2988 92.47 Variance 0.672827 0.0169 Standard deviation 0.82026 0.13 Minimum 91.2 92.34 Maximum 93.104 92.6 Range 1.904 0.26 Skewness -0.60532 0.0 Kurtosis -2.04263 -3.0 Coeff. of variation 0.888701% 0.140586% ------------------------------------------------------------ The StatAdvisor --------------- This table shows summary statistics for the two samples of data. Other tabular options within this analysis can be used to test whether differences between the statistics from the two samples are statistically significant. Of particular interest here are the standardized skewness and standardized kurtosis, which can be used to determine whether the samples come from normal distributions. Values of these statistics outside the range of -2 to +2 indicate significant departures from normality, which would tend to invalidate the tests which compare the standard deviations. In this case, both standardized skewness values are within the range expected. Both standardized kurtosis values are within the range expected. Comparison of Means ------------------- 95.0% confidence interval for mean of EFVIS: 92.2988 +/- 1.01849 [91.2803,93.3173] 95.0% confidence interval for mean of UV: 92.47 +/- 0.161417 [92.3086,92.6314] 95.0% confidence interval for the difference between the means assuming equal variances: -0.1712 +/- 0.856475 [-1.02768,0.685275] t test to compare means Null hypothesis: mean1 = mean2 Alt. hypothesis: mean1 NE mean2
100

Anexos
assuming equal variances: t = -0.460946 P-value = 0.65711 The StatAdvisor --------------- This option runs a t-test to compare the means of the two samples. It also constructs confidence intervals or bounds for each mean and for the difference between the means. Of particular interest is the confidence interval for the difference between the means, which extends from -1.02768 to 0.685275. Since the interval contains the value 0.0, there is not a statistically significant difference between the means of the two samples at the 95.0% confidence level. A t-test may also be used to test a specific hypothesis about the difference between the means of the populations from which the two samples come. In this case, the test has been constructed to determine whether the difference between the two means equals 0.0 versus the alternative hypothesis that the difference does not equal 0.0. Since the computed P-value is not less than 0.05, we cannot reject the null hypothesis. NOTE: these results assume that the variances of the two samples are equal. In this case, that assumption is questionable since the results of an F-test to compare the standard deviations suggests that there may be a significant difference between them. You can see the results of that test by selecting Comparison of Standard Deviations from the Tabular Options menu.
101

Anexos
Anexo 11. LA BIOSEGURIDAD EN LABORATORIOS Y PLANTAS DE PRODUCCIÓN
QUÍMICA ( 10 y 11)
Un concepto más amplio que el de medidas de proyección e higiene del
trabajo, primeros auxilios, o el propio tema de protección contra incendio,
catástrofe, están recogidos bajo el termino bioseguridad.
En el centro de biactivos químicos esta constituido un comité de
bioseguridad el cual cuenta con personal dedicado a cada uno de los
aspectos más relevantes de este ámbito como lo son la seguridad biológica,
la seguridad en el manejo y uso de productos químico- tóxico, la protección e
higiene del trabajo etc.
La bioseguridad esta destinada a la seguridad del medio ambiente o el
entorno entendiéndose por esto la protección del suelo, la fauna, la flora y al
hombre que forma parte de ese habitad. En el marco de las nuevas
tendencias medioambientales especial énfasis se la dedica al tratamiento
manejo y disposición de residuales, tendiendo a lo que se llama producción
limpia.
En el caso de los laboratorios de integración o producción todo el personal
debe estar adecuado para cualquier catástrofe que ocurra y mantener en
cada puesto de trabajo y alcance de todos los medios de seguridad para
prevenir o actuar adecuadamente en caso de accidentes.
En el caso de nuestro trabajo se debe tener presente algunas medidas para
con el manejo de sustancias tóxicas y/o peligrosas.
También es imprescindible el conocimiento que aporta el etiquetado de cada
frasco de reactivo en el que se describe el riesgo químico y las medidas a
adoptar en caso de accidentes. Este aspecto no es dominado incluso por
muchos químicos y está bien establecido en el CBQ.
102

Anexos
Sustancias tóxicas manipuladas durante este trabajo.
Sustancia Tóxica Inflamable Tipo de
riesgo
Consejo de
prudencia
CS2 X X R3, R7, R23 S7, 14, 24,
25, 29, 33
Br2 X - R36, 37, 38,
48, 39,23,
24, 25
S9, 24, 25,
26, 35
CHCl3 X - R10, 20, 25,
45
S46, 51, 56
NH3 X - R20, 36, 37 S23, 26, 3, 9,
7
En la tabla anterior se especifica la sustancia, si es tóxica, inflamable y el
tipo de riesgo (R) y el consejo de prudencia (S). Los riesgos y los consejos
de prudencia aparecen tratados en el texto ″Trabajar con productos
peligrosos″ en la comisión de la comunidad europea y otros materiales. Las
frases de riesgo y consejos de prudencia aparecen en las etiquetas de los
frascos de reactivos o los del fabricante en casos específicos.
Las frases de riesgo que incluimos fueron:
R3.- Elevado riesgo de explosión como consecuencia de choques, fricción o
fuego.
R7.- Puede provocar incendios.
R23.- Tóxico en caso de inhalación.
R25.- Tóxico en caso de ingestión.
R24.- Tóxico el caso de contacto con la piel.
R48.- Riesgo de efectos graves para la salud en caso de exposición
prolongada.
R38.- Irritante para la piel.
R37.- Irritante para las vías respiratorias.
R36.- Irritante para los ojos.
103

Anexos
R10.- Inflamable.
R45.- Puede producir cáncer.
Existe un numero mayor de frases de riesgo que no hemos citado aquí
como ejemplo.
Las frases o consejos de prudencia que incluimos y que responden a las normas
tomadas del documento citado, se expone solo a modo de ejemplo son:
S7.- Manténgase en recipientes bien cerrados.
S9.- Manténgase en un lugar bien ventilado.
S23.- No respirar el gas, vapores, humo, aerosol.
S14.- Manténgase apartado.
S24.- Evite el contacto con la piel
S25.- Evite el contacto con los ojos.
S29.- No echar los residuos a la alcantarilla.
S33.- Evite la acumulación de capas electrostática.
S18.- Manipule y abra el recipiente con prudencia.
S26.- En caso de contacto con los ojos, lávese enseguida y abundantemente
con agua y acudir a un especialista.
S35.- Para deshacerse de este producto y de su recipiente es preciso tomar
las precauciones que exige su uso.
S46.- En caso de ingestión consulte inmediatamente a un medico y enséñale
el frasco o la etiqueta.
S51.- Utilice únicamente en zonas bien ventiladas.
S3.- Manténgase en lugares frescos.
Muchos otros consejos de prudencia hasta un total de 60 aparecen
reportados, además de combinaciones de estas como por ejemplo:
S36 / 39 / 37.- Utilice la ropa de protección apropiada, guantes y un aparato de
protección de los ojos / de la cara.
104

Anexos
Medidas para el trabajo con piridina
• No inhalar los vapores de piridina.
• Evitar el contacto con la piel y los ojos.
• Ventilar los locales donde se encuentre.
• Mantener alejado de las fuentes de ignición.
• Evitar la carga electrostática.
Protección personal
• Protección respiratoria en presencia de vapores.
• Protección de los ojos.
• Protección de las manos.
Medidas de higiene
• Trabajar bajo campana de extracción. No inhalar la sustancia.
• No inhalar la sustancia.
• Sustituir la ropa contaminada.
• Lavar la cara y las manos al terminar el trabajo.
Medidas a tomar en caso de accidente.
Primeros auxilios
Los vapores de piridina actúan como irritantes de las mucosas,
principalmente en los ojos y vías respiratorias, por ello cuando se inhalan
vapores de piridina se recomienda trasladarse al aire fresco y de ser
necesario recibir respiración artificial (2-3) .
Por otra parte este compuesto puede producir dermatitis por lo que en el
caso de contacto con la piel se debe lavar con abundante agua y cambiar la
ropa contaminada. Si el contacto es con los ojos el lavado debe realizarse
con los párpados abiertos al menos durante 10 minutos. Debe consultarse
al oftalmólogo.
Cuando se produce la ingestión de esta sustancia se debe beber abundante
agua, provocar vómitos y acudir al médico.
Medidas de lucha contra incendio.
Cuando se produce incendio en lugares donde esta presente piridina se
recomienda usar extintores de CO2., espuma o polvo. El personal que
participa en esta actividad debe protegerse con ropas adecuadas y con
sistema de respiración artificial e independiente del ambiente.
105

Anexos
Los vapores de piridina deben condensarse con agua evitando la extensión
de esta a acuíferos superficiales o subterráneos.
106