clase termoquimica 2
DESCRIPTION
termoquimicaTRANSCRIPT

Termodinámica

La Termodinámica estudia los intercambios energéticos que acompañan a los fenómenos físico-químicos.
Al estudiar el intercambio de energía entre un sistema y su entorno, se puede predecir en qué sentido puede ocurrir el cambio químico o físico.
Termodinámica

En ese aspecto, la Termodinámica predice:
si los reaccionantes se transforman en productos, o sea, si la reacción es espontánea o no.
en qué medida ocurre el cambio, o sea, las cantidades de productos que se obtienen y la cantidad de reaccionantes que quedan sin reaccionar una vez terminada la reacción, o sea, cuando se alcanza el estado de equilibrio.
Termodinámica

A la Termodinámica:
sólo le interesa el estado inicial y el estado final (no le importa cómo ocurre la reacción).
no le interesa el tiempo que demora en ocurrir el proceso.
para estudiar el proceso mide propiedades macroscópicas, tales como:
temperatura, presión, volumen.
Termodinámica

Sistemas• Parte pequeña del universo que se aísla para
someter a estudio.• El resto se denomina ENTORNO.• Pueden ser los Sistemas:
o Abiertos (intercambia materia y energía).o Cerrados (no intercambia materia y sí energía).o Aislados (no intercambia ni materia ni energía).
• En reacciones químicas...SISTEMAS = Sustancias químicas
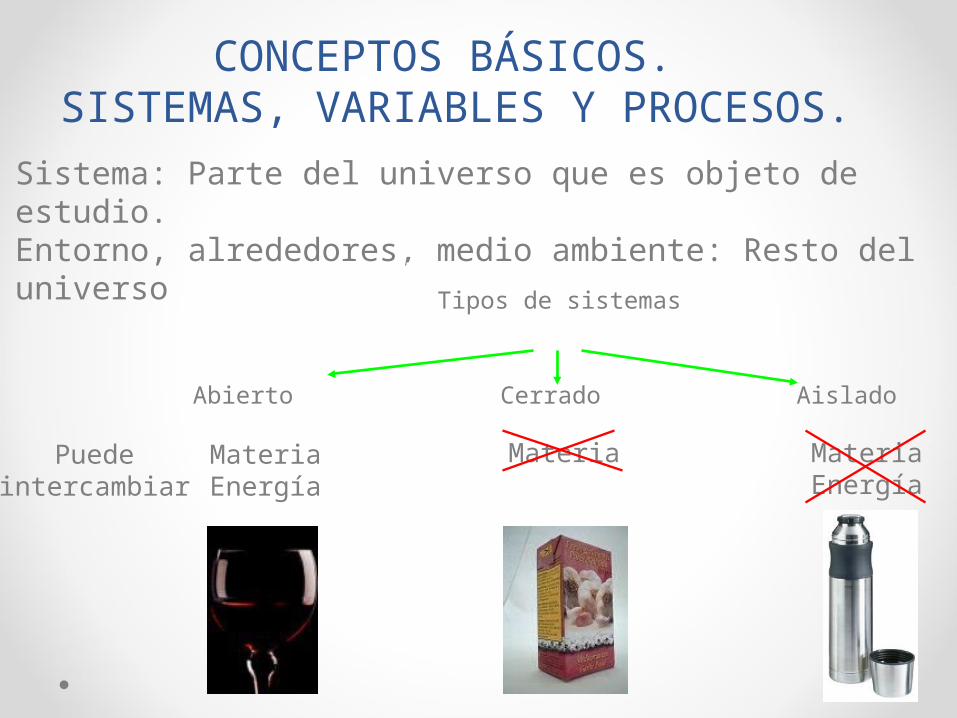
CONCEPTOS BÁSICOS. SISTEMAS, VARIABLES Y PROCESOS.
Sistema: Parte del universo que es objeto de estudio.Entorno, alrededores, medio ambiente: Resto del universo
Abierto Cerrado Aislado
Tipos de sistemas
Puedeintercambiar
MateriaEnergía
Materia MateriaEnergía

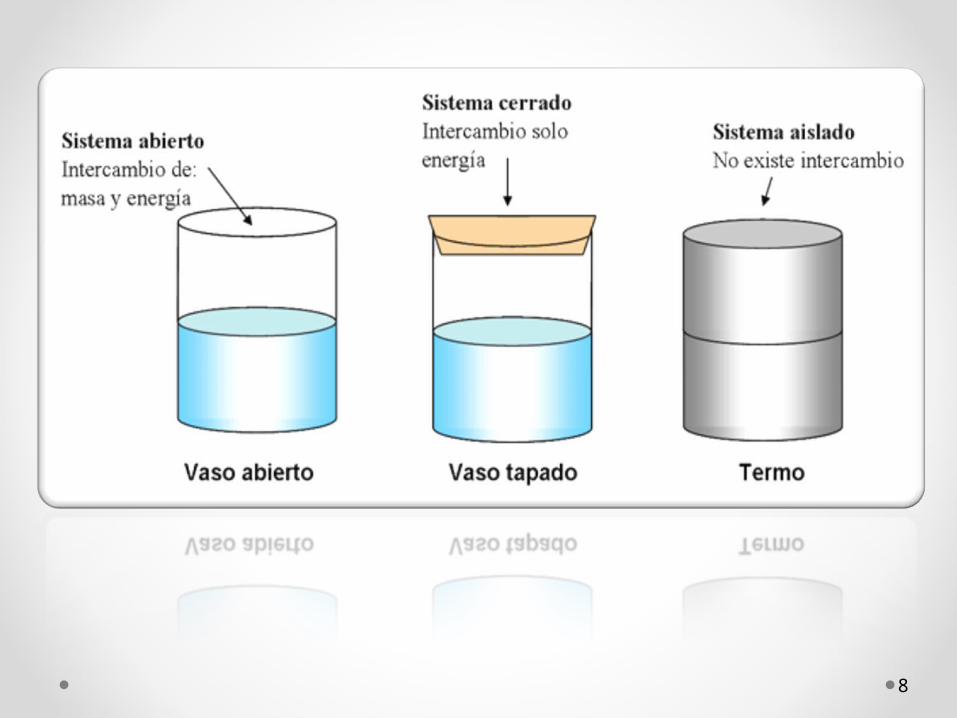
sistema abierto: intercambia materia y energía con el medio . Ej: la célula.
sistema cerrado: sólo intercambia energía con el medio. Ej: una estufa.
sistema aislado: no intercambia materia ni energía. Ej: café caliente en el interior de un termo aislado.
8
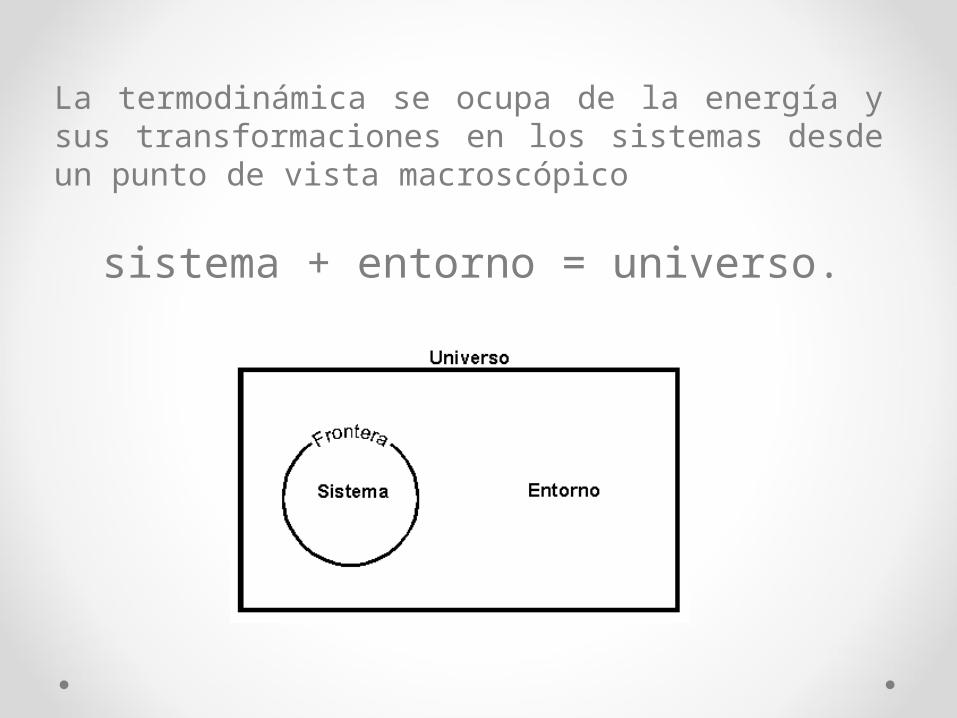
La termodinámica se ocupa de la energía y sus transformaciones en los sistemas desde un punto de vista macroscópico
sistema + entorno = universo.
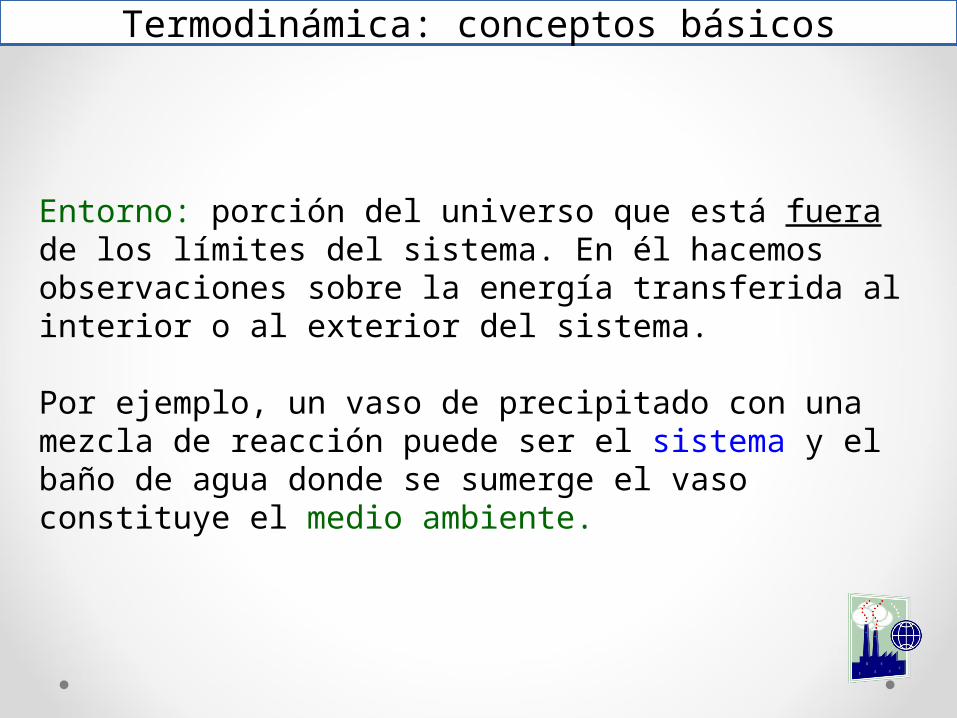
Entorno: porción del universo que está fuera de los límites del sistema. En él hacemos observaciones sobre la energía transferida al interior o al exterior del sistema.
Por ejemplo, un vaso de precipitado con una mezcla de reacción puede ser el sistema y el baño de agua donde se sumerge el vaso constituye el medio ambiente.
Termodinámica: conceptos básicos

Definición de Termoquímica.
• Es la parte de la Química que se encarga del estudio del intercambio energético de un sistema químico con el exterior.
• Hay sistemas químicos que evolucionan de reactivos a productos desprendiendo energía. Son las reacciones exotérmicas.
• Otros sistemas químicos evolucionan de reactivos a productos precisando energía. Son las reacciones endotérmicas.

Para definir un proceso termodinámico basta establecer la diferencia entre el estado final y el estado inicial de sus propiedades macroscópicas, las cuales se llaman funciones de estado, como
temperatura presión volumen
Termodinámica: conceptos básicos
Estado termodinámico: es la condición en la que se encuentra el sistema. Cada estado termodinámico se define por un conjunto de sus propiedades macroscópicas llamadas funciones de estado.

Las funciones de estado sólo dependen del estado inicial y del estado final y no dependen de cómo ocurrió el proceso. Las funciones de estado son:
T = temperatura P = presión V = volumen E = energía interna H = entalpía S = entropía G = energía libre
Funciones de estado
Las funciones de estado se escriben con mayúsculas. Otras funciones que dependen de cómo se realice el proceso no son termodinámicas y se escriben con minúsculas. Estas son: q = calorw = trabajo

Energía interna y temperatura
Energía interna: es la capacidad de un sistema para realizar un trabajo. Tiene que ver con la estructura del sistema. Se debe a la energía cinética de las moléculas, la energía de vibración de los átomos y a la energía de los enlaces. No se puede conocer su valor absoluto, sólo la diferencia al ocurrir un cambio en el sistema: DE. Es una función de estado.Temperatura (T): es una función de estado y corresponde a la medida de la energía cinética de las moléculas de un sistema.

15
Temperatura (T)
30 °C30 °C
q1 q2
20 °C 20 °C

Calor y trabajo
Calor (q): es la energía transferida entre el sistema y su ambiente debido a que existe entre ambos una diferencia de temperatura. No es una función de estado.

Calor y trabajo
Trabajo (w): es la energía transferida entre el sistema y su ambiente a través de un proceso equivalente a elevar un peso. No es una función de estado.
Tipos de trabajo: expansión, extensión, elevación de un peso, eléctrico, etc.
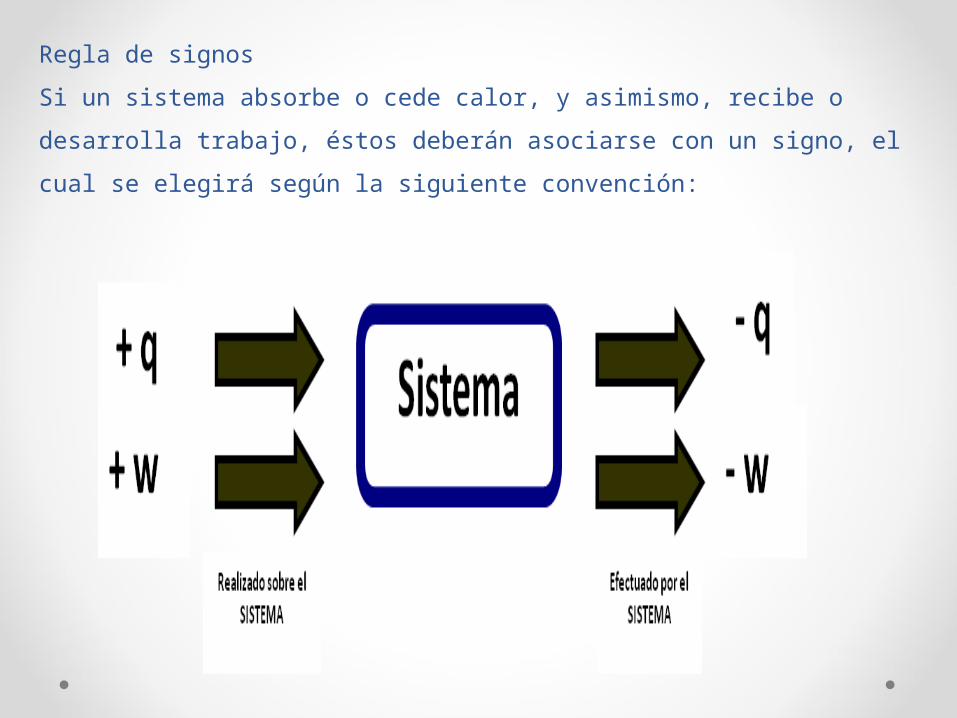
Regla de signos
Si un sistema absorbe o cede calor, y asimismo, recibe o desarrolla
trabajo, éstos deberán asociarse con un signo, el cual se elegirá según
la siguiente convención:
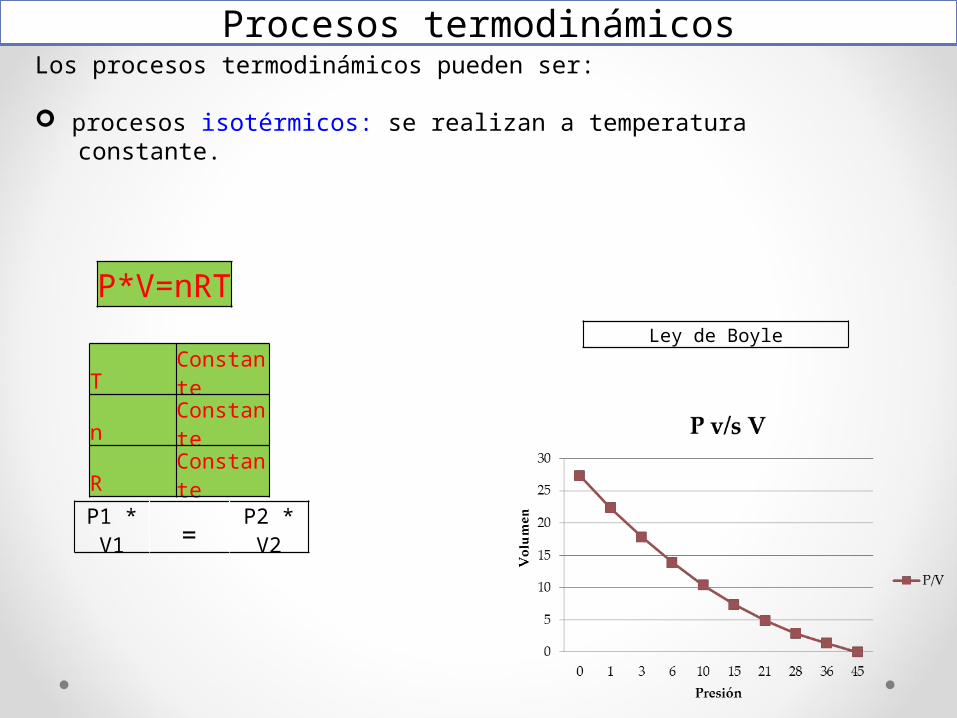
Los procesos termodinámicos pueden ser:
procesos isotérmicos: se realizan a temperatura constante.
Procesos termodinámicos
P1 * V1 = P2 * V2
P*V=nRT
T Constanten ConstanteR Constante
Ley de Boyle
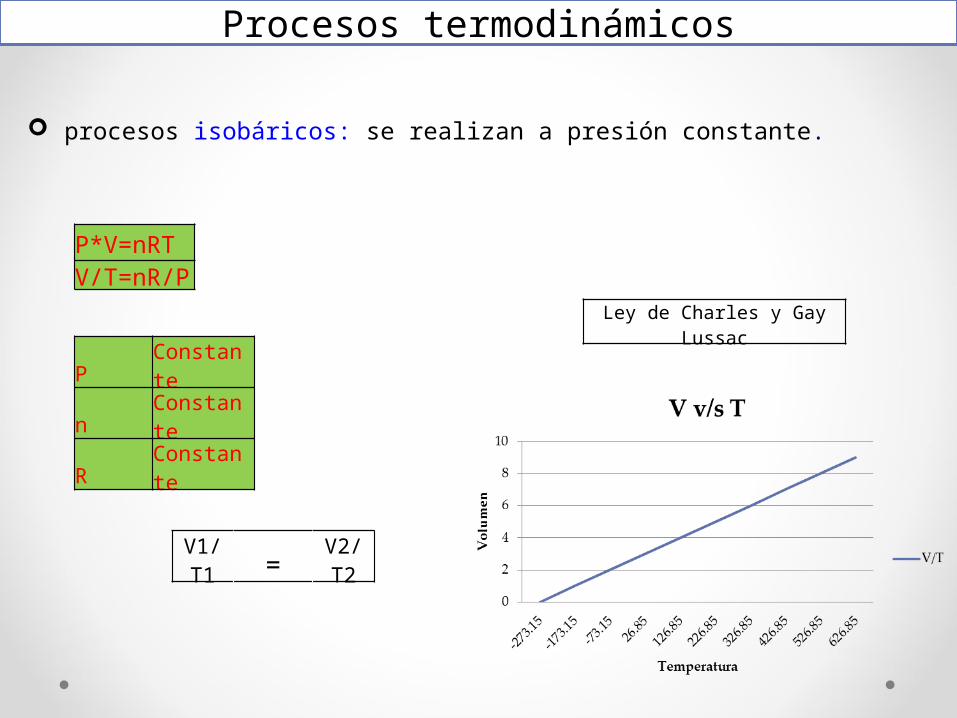
procesos isobáricos: se realizan a presión constante.
Procesos termodinámicos
P*V=nRTV/T=nR/P
P Constanten ConstanteR Constante
V1/T1 = V2/T2
Ley de Charles y Gay Lussac
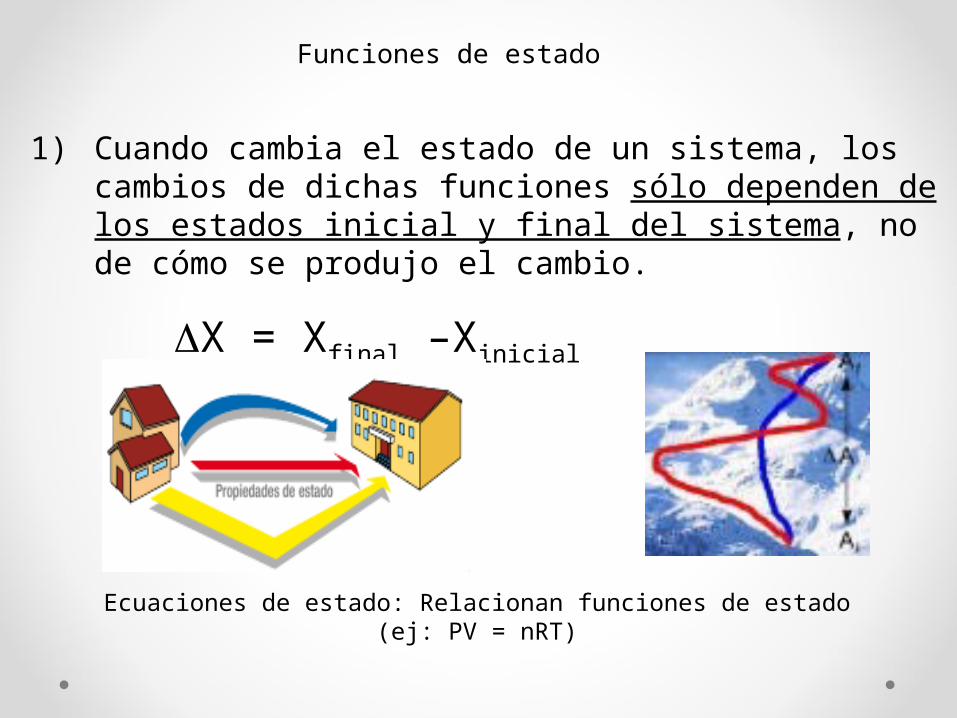
Funciones de estado
1) Cuando cambia el estado de un sistema, los cambios de dichas funciones sólo dependen de los estados inicial y final del sistema, no de cómo se produjo el cambio.
ΔX = Xfinal –Xinicial
Ecuaciones de estado: Relacionan funciones de estado(ej: PV = nRT)

Cuando alguna de las variables de estado cambia con el tiempo⇓
PROCESO termodinámico
Tipos deprocesos
• Isotermo (T = cte)• Isóbaro (P = cte)• Isócoro (V = cte)• Adiabático (Q = 0)• Cíclico (estado final = estado inicial)
• Reversible (sistema siempre infinitesimalmente próximo al equilibrio; un cambio infinitesimal en las condiciones puede invertir el proceso)• Irreversible (Un cambio infinitesimal en las condiciones no produce un cambio de sentido en la transformación).
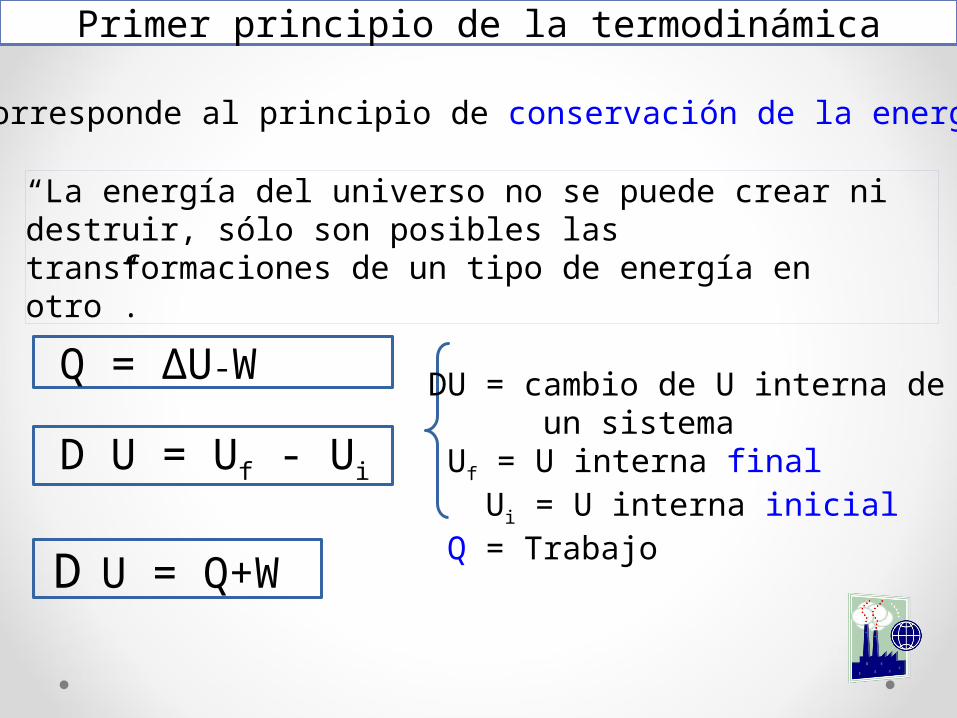
Primer principio de la termodinámica
Corresponde al principio de conservación de la energía.
“La energía del universo no se puede crear ni destruir, sólo son posibles las transformaciones de un tipo de energía en otro”.
D U = Q+W
D U = Uf - Ui
DU = cambio de U interna de un sistema Uf = U interna final Ui = U interna inicial Q = Trabajo
Q = ∆U-W
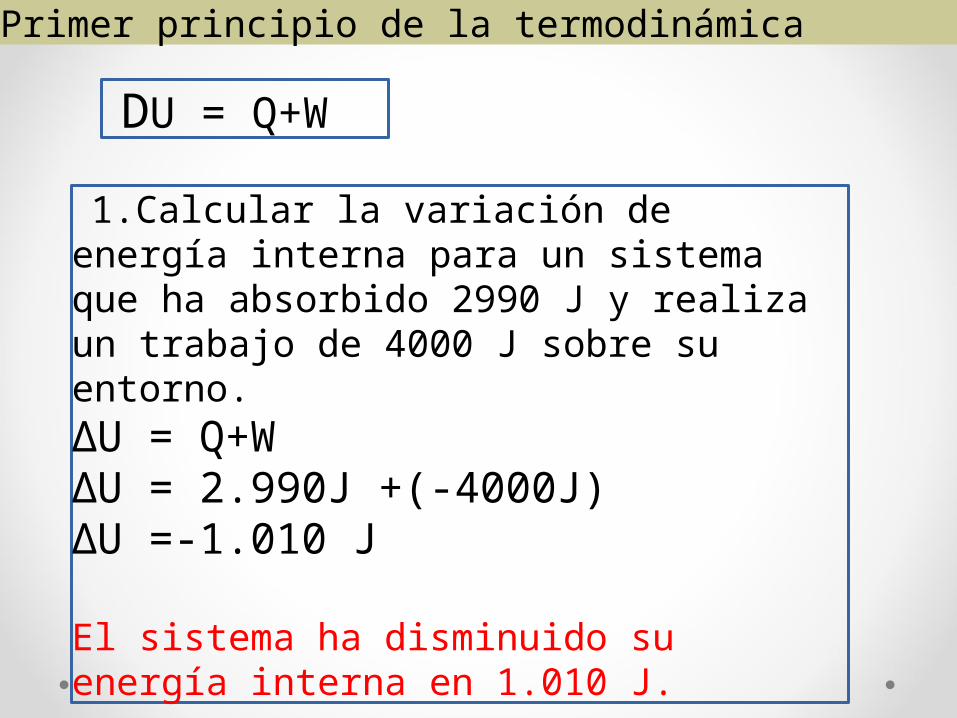
Primer principio de la termodinámica
DU = Q+W
1.Calcular la variación de energía interna para un sistema que ha absorbido 2990 J y realiza un trabajo de 4000 J sobre su entorno.∆U = Q+W∆U = 2.990J +(-4000J)∆U =-1.010 J
El sistema ha disminuido su energía interna en 1.010 J.

1.Calcular la variación de energía interna para un sistema que ha absorbido 5000 J y realiza un trabajo de 3000 J sobre su entorno.
∆U = Q+W ∆U = 5.000J +(-3000J) ∆U = 2000 J
Primer principio de la termodinámica

2.Calcular la variación de energía interna para un sistema que ha liberado 2.590 J y el trabajo es realizado por las fuerzas exteriores sobre el sistema,siendo el valor del trabajo 3.560 J.
∆U = Q+W ∆U = -2590J +(+3560J) ∆U = +978 J
Primer principio de la termodinámica

Describe los cambios térmicos que se llevan a presión cte. :
Entalpía
ΔU = qp + P x ΔV donde,QP es calor a presión cte
Por lo que, para el calor intercambiado en estas condiciones se cumple:Qp =(U2+ P xV2)-(U1+ P x V1)
El término U+ P xV recibe el nombre de entalpía(H)
QP= H2-H1=∆H

La variación de entalpía(∆H) es igual a la diferencia entre la entalpía de los productos y la de los reactantes:
∆H = Hproductos-Hreactantes
Dependiendo del calor puesto en juego en un proceso químico, las reacciones
pueden ser endotérmicas o exotérmicas.
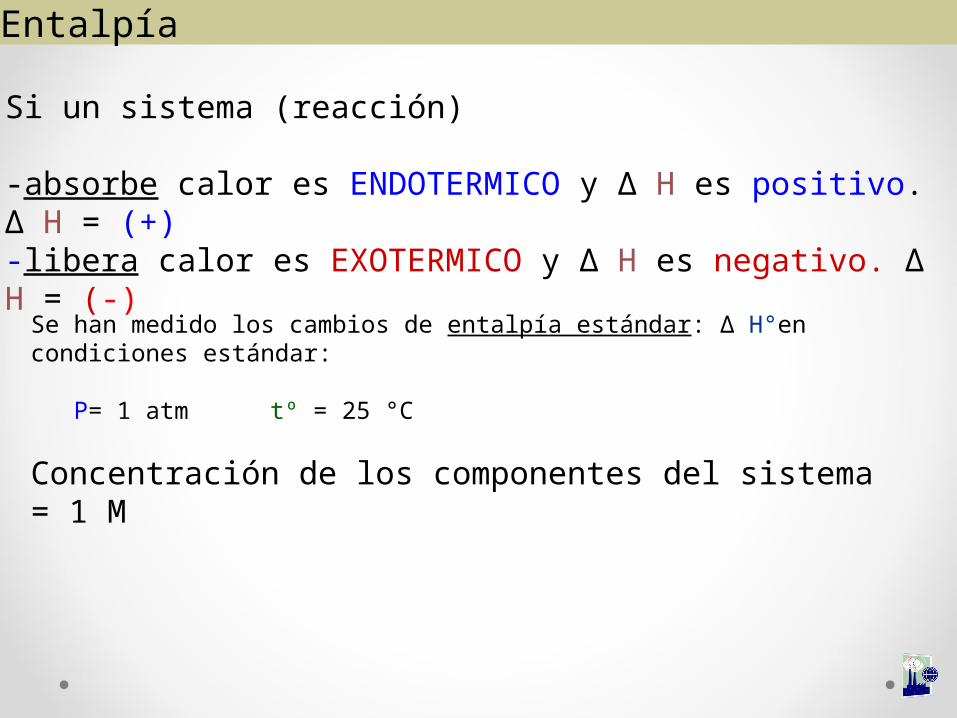
Si un sistema (reacción)
-absorbe calor es ENDOTERMICO y ∆ H es positivo. ∆ H = (+)-libera calor es EXOTERMICO y ∆ H es negativo. ∆ H = (-)
Entalpía
Se han medido los cambios de entalpía estándar: ∆ H°en condiciones estándar:
P= 1 atm tº = 25 °C
Concentración de los componentes del sistema = 1 M
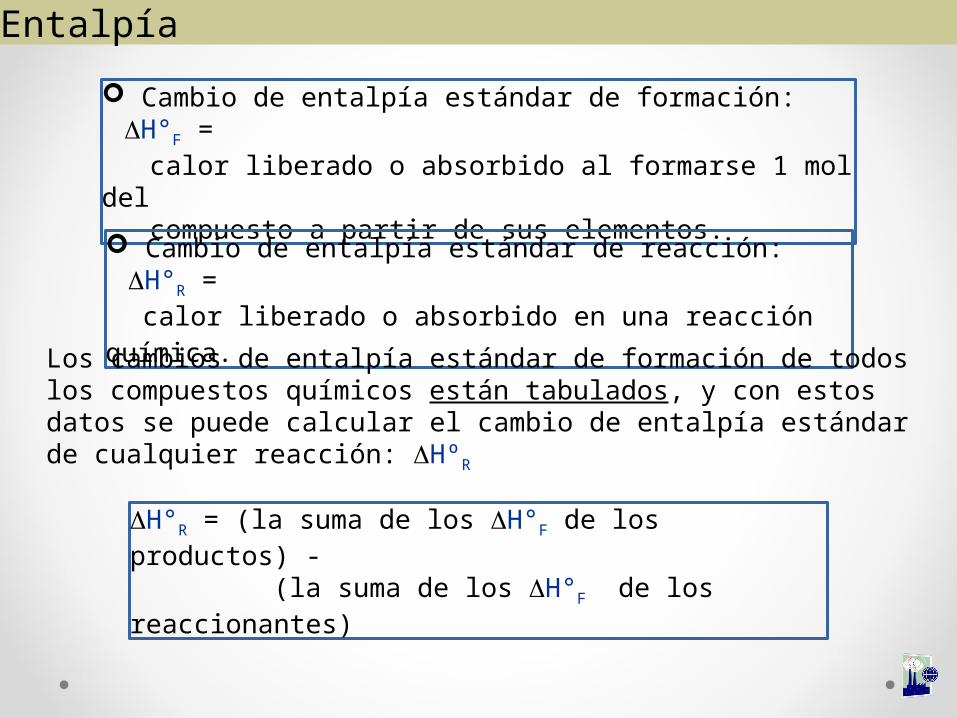
Cambio de entalpía estándar de formación: ΔH°F = calor liberado o absorbido al formarse 1 mol del compuesto a partir de sus elementos.
Cambio de entalpía estándar de reacción: ΔH°R =
calor liberado o absorbido en una reacción química.
Entalpía
Los cambios de entalpía estándar de formación de todos los compuestos químicos están tabulados, y con estos datos se puede calcular el cambio de entalpía estándar de cualquier reacción: ΔHºR
ΔH°R = (la suma de los ΔH°F de los productos) - (la suma de los ΔH°F de los reaccionantes)

Ejemplo: Determinar la variación de energía interna para el proceso de combustión de 1 mol de propano a 25ºC y 1 atm, si la variación de entalpía, en estas condiciones, vale – 2219,8 kJ.
C3H8 (g) + 5 O2 (g) ⎯→ 3 CO2 (g) + 4 H2O (l) Δ H = –2219,8 kJ
n reactivos = 1+5 = 6 ; n productos = 3+4=7 ⇒ Δ n =7 – 6=1
Despejando en Δ U = Δ H – Δ n · R · T =– 2219,8 kJ - 1 mol · (8,3 J/mol.K) · 298 K = –2222,3 kJ
Δ U = – 2222,3 kJ

Ejemplo:
1.Calcular la variación de entalpia(∆H) que se produce en la reacción de transformación del diamante en grafito:
C(diamante)→ C(grafito)
(1) C(diamante) +O2(g)→CO2(g) ∆H=-395.4 KJ/mol
(2) C(grafito) +O2(g) →CO2(g) ∆H=-393.5 KJ/mol
(1)C(diamante) +O2(g)→CO2(g) ∆H=-395.4 KJ/mol
(3) CO2(g) → C(grafito) +O2(g) ∆H=+393.5 KJ/mol
C(diamante) → C(grafito) ∆H=-1.9 KJ/mol

2. Calcular ∆Hf para la formación de la acetona (CH3COCH3)según la reacción:
3C(s)+3H2(g)+1/2O2 (g)→ CH3COCH3(l), conociendo los valores de calor de combustión:
(1) H2(g) +1/2 O2→ H2O(l) ∆H=-285.8 KJ/mol /x 3
(2) C (S) + O2→ CO2 (l) ∆H=-393.5 KJ/mol /x 3(3) CH3COCH3(l) +4 O2→3CO2(g)+ 3H2O(l) ∆H=-1.786KJ/mol
(1) H2(g) +3/2 O2→ 3H2O(l) ∆H=-857.4 KJ/mol
(2) 3C (S) + 3O2→ 3CO2 (l) ∆H=-1.180,5 KJ/mol
(3) 3CO2(g)+ 3H2O(l) → CH3COCH3(l) +4 O2 ∆H= +1.786KJ/mol
3H2(g)+3C(s)+ 1/2 O2→ CH3COCH3(l) ∆H= -251.9 KJ/mol

Ley de Hess• ΔH en una reacción química es constante con
independencia de que la reacción se produzca en una o más etapas.
• Recuerda que H es función de estado.• Por tanto, si una ecuación química se puede
expresar como combinación lineal de otras, podremos igualmente calcular ΔH de la reacción global combinando los ΔH de cada una de las reacciones.
MUY IMPORTANTE
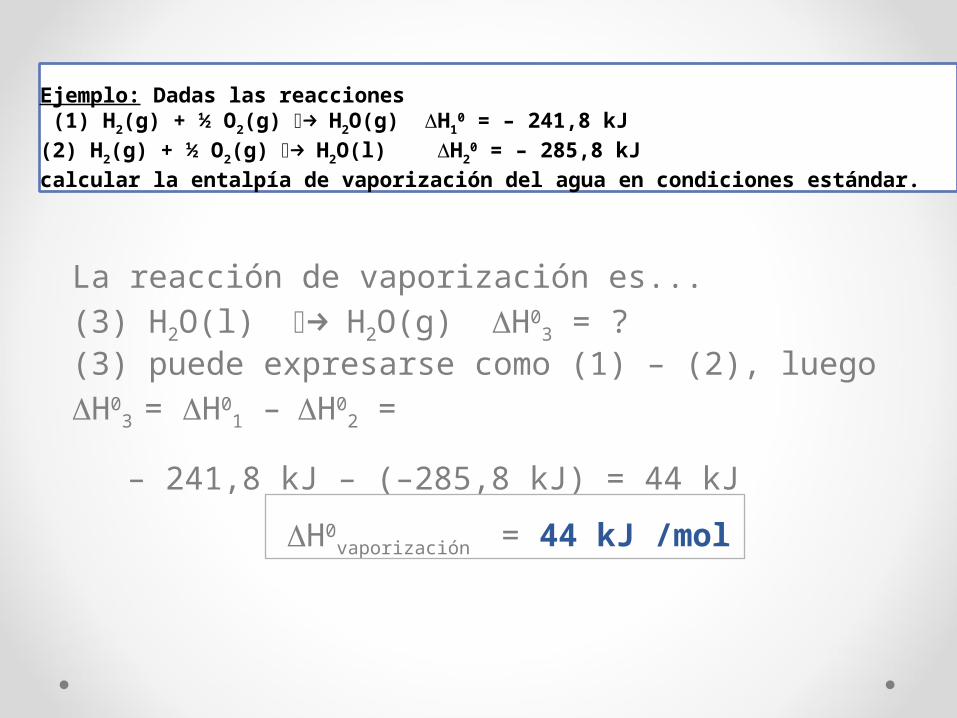
Ejemplo: Dadas las reacciones (1) H2(g) + ½ O2(g) ⎯→ H2O(g) ΔH1
0 = – 241,8 kJ (2) H2(g) + ½ O2(g) ⎯→ H2O(l) ΔH2
0 = – 285,8 kJcalcular la entalpía de vaporización del agua en condiciones estándar.
La reacción de vaporización es...(3) H2O(l) ⎯→ H2O(g) ΔH0
3 = ?(3) puede expresarse como (1) – (2), luegoΔH0
3 = ΔH01 – ΔH0
2 =
– 241,8 kJ – (–285,8 kJ) = 44 kJ
ΔH0vaporización = 44 kJ /mol

Esquema de la ley de Hess
ΔH10 = – 241’8
kJΔH2
0 = – 285’8 kJ
ΔH30 = 44 kJ
HH2(g) + ½ O2(g)
H2O(g)
H2O(l)

Ejercicio B: Conocidas las entalpías estándar de formación del butano (C4H10), agua líquida y CO2, cuyos valores son respectivamente –124,7 –285,8 y –393,5 kJ/mol, calcular la entalpía estándar de combustión del butano.
• Si utilizamos la ley de Hess, la reacción:
• (4) C4H10(g) +13/2O2(g) → 4 CO2(g) + 5H2O(l) ΔH0comb=?
• Puede obtenerse a partir de:• (1) H2(g) + ½ O2(g) ⎯→ H2O(l) ΔH1
0 = – 285,8 kJ (2) C(s) + O2(g) ⎯→ CO2(g) ΔH2
0 = – 393,5 kJ(3) 4 C(s) + 5 H2(g) ⎯→ C4H10(g) ΔH3
0 = – 124,7 kJ
• (4) = 4 · (2) + 5 · (1) – (3)• 4 C(s) + 4 O2(g) +5 H2(g) + 5/2 O2(g) + C4H10(g) ⎯→
4 CO2(g) + 5H2O(l) + 4 C(s) + 5 H2(g)
• Δ H04 = 4 mol(–393,5 kJ/mol) + 5 mol(–285,8 kJ/mol) –1 mol(– 124,7
kJ/mol) = – 2878,3 kJ

Ejercicio C: Determinar Δ Hf0 del eteno (C2H4) a
partir de los calores de reacción de las siguientes reacciones químicas:(1) H2(g) + ½ O2(g) → H2O(l) ΔH1
0 = – 285’8 kJ (2) C(s) + O2(g) → CO2(g) ΔH2
0 = – 393’13 kJ(3) C2H4(g) + 3O2(g) → 2CO2(g) + 2 H2O(l)
ΔH30 = – 1422 kJ
• La reacción de formación del eteno C2H4(g) a partir de sus constituyentes en estado normal es:
• (4) 2 C(s) + 2 H2(g) ⎯→ C2H4(g)
• (4) se puede expresar como 2·(2) + 2·(1) – (3)

Ejercicio C: Determinar ΔHf0 del eteno (C2H4) a partir
de los calores de reacción de las siguientes reacciones químicas:
(1) H2(g) + ½ O2(g) ⎯→ H2O(l) ΔH10 = – 285’8 kJ
(2) C(s) + O2(g) ⎯→ CO2(g) ΔH20 = – 393’13 kJ
(3) C2H4(g) + 3O2(g) → 2CO2(g) + 2 H2O(l) ΔH30 = – 1422 kJ
(4) 2 C(s) + 2 H2(g) ⎯→ C2H4(g)(4) = 2·(2) + 2·(1) – (3)luego ΔH4
0 = 2·ΔH20 + 2·ΔH1
0 – ΔH30 =
= 2 · (–393’13 kJ) + 2 · (– 285’8 kJ) – (– 1422 kJ) = = 64’14 kJ
es decir ΔHf0 (eteno) = 64’14 kJ/mol
Se trata, pues, de una reacción endotérmica.

Ejercicio D: Las entalpías de combustión de la glucosa (C6H12O6) y del etanol (C2H5OH) son –2815 kJ/mol y –1372 kJ/mol, respectivamente. Con estos datos determina la energía intercambiada en la fermenta– ción de un mol de glucosa, reacción en la que se produce etanol y CO2. ¿Es exotérmica la reacción?
• Las reacciones de combustión son, respectivamente:(1) C6H12O6 + 6 O2 ⎯→ 6 CO2 + 6 H2O ; ΔH1 = – 2815 kJ
(2) C2H5OH + 3 O2 ⎯→ 2 CO2 + 3 H2O ; ΔH2 = – 1372 kJ
• La reacción de fermentación de la glucosa es: (3) C6H12O6 ⎯→ 2 C2H5OH +2 CO2 ΔH3 = ?
• (3) puede expresarse como (1) – 2· (2), luegoΔH3 = ΔH1 – 2·ΔH2 = – 2815 kJ – 2· (– 1372 kJ) = – 71 kJ
• y la reacción es exotérmica.

Energía de enlace.• “Es la energía necesaria para romper un enlace
de un mol de sustancia en estado gaseoso”• En el caso de moléculas diatómicas es igual que la
energía de disociación:• A—B(g) ⎯→ A(g) + B(g) ΔHdis = Eenlace= Ee
• Ejemplo: H2(g) ⎯→ 2 H(g) ΔH = 436 kJ
• Es positiva (es necesario aportar energía al sistema)• Es difícil de medir.• Se suele calcular aplicando la ley de Hess.

Ejemplo: Calcular la energía del enlace H—Cl en el cloruro de hidrógeno conociendo ΔHf
0(HCl) cuyo valor es –92,3 kJ/mol y las entalpías de disociación del H2 y del Cl2 que son 436,0 kJ/mol y 243,4 kJ/mol, respectivamente.
• La reacción de disociación del HCl será:• (4) HCl(g) ⎯→ H(g) + Cl(g) ΔH0
4= ?• (1) ½H2(g) + ½Cl2(g) → HCl(g) ΔH0
1 = –92,3 kJ• (2) H2(g) ⎯→ 2H(g) ΔH0
2 = 436,0 kJ• (3) Cl2(g) ⎯→ 2Cl(g) ΔH0
3 = 243,4 kJ• (4) = –(1) + ½(2) + ½(3)• ΔH0
4 = –(– 92,3 kJ ) + ½(436,0 kJ) + ½(243,4 kJ) =
= 432,0 kJ Ee(HCl) = 432,0 kJ/mol

1.Cálculo de ΔH0 a partir de las Energía de enlace (disociación).
• Aplicando la ley de Hess en cualquier caso se obtiene la siguiente fórmula:
ΔH0 = Σ ni · Ee(enl. rotos) – Σ nj · Ee(enl. formados)
en donde ni representa el número de enlaces rotos y formados de cada tipo.
MUY IMPORTANTE

Ejemplo: Sabiendo que las energía de los siguientes enlaces (kJ/mol): C=C : 611; C–C : 347; C–H : 413 y H–H : 436, calcular el valor de ΔH0 de la reacción de hidrogenación del eteno.
• Reacción: CH2=CH2(g) + H2(g) → CH3–CH3(g)
• En el proceso se rompe un enlace C=C y otro H–H y se forman 2 enlaces C–H nuevos (el etano tiene 6 mientras que el eteno tenía sólo 4) y un enlace C–C.
• Δ H0 = Σ Ee(enl. rotos) – Σ Ee(enl. formados) =
• Δ H0 = 1Ee(C=C) + 1 Ee(H–H) – 1Ee(C–C) – 2 Ee(C–H)
• Δ H0 = 1 mol · 611 kJ/mol + 1mol · 436 kJ/mol – (1 mol · 347 kJ/mol + 2 mol · 413 kJ/mol) = –126 kJ
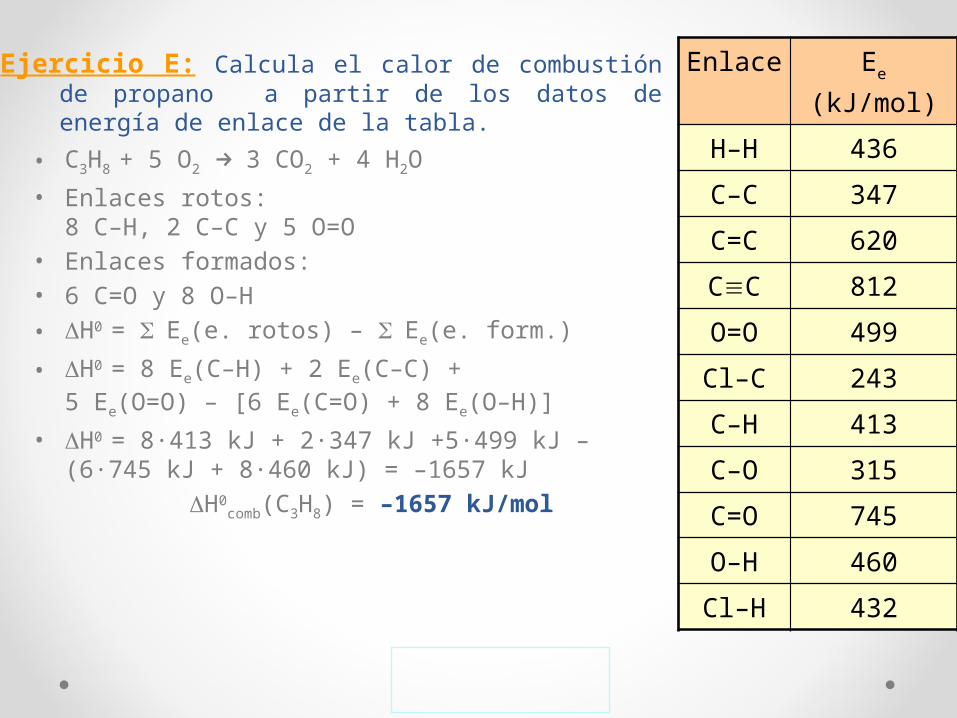
Ejercicio E: Calcula el calor de combustión de propano a partir de los datos de energía de enlace de la tabla.
• C3H8 + 5 O2 → 3 CO2 + 4 H2O
• Enlaces rotos: 8 C–H, 2 C–C y 5 O=O
• Enlaces formados:• 6 C=O y 8 O–H• ΔH0 = Σ Ee(e. rotos) – Σ Ee(e. form.)
• ΔH0 = 8 Ee(C–H) + 2 Ee(C–C) + 5 Ee(O=O) – [6 Ee(C=O) + 8 Ee(O–H)]
• ΔH0 = 8·413 kJ + 2·347 kJ +5·499 kJ – (6·745 kJ + 8·460 kJ) = –1657 kJ
ΔH0comb(C3H8) = –1657 kJ/mol
Enlace Ee (kJ/mol)
H–H 436
C–C 347
C=C 620
C≡C 812
O=O 499
Cl–C 243
C–H 413
C–O 315
C=O 745
O–H 460
Cl–H 432

Entropía (S)• Es una medida del desorden del sistema que sí
puede medirse y tabularse.
ΔS = Sfinal – Sinicial • Existen tablas de S0 (entropía molar estándar) de
diferentes sustancias. • En una reacción química: ΔS0 = Σ np· S0
productos – Σ nr· S0reactivos
• La entropía es una función de estado.

Ejemplo: Calcula ΔS0 para las siguientes reacciones químicas: a) N2(g) + O2(g) → 2 NO(g); b) 3 H2(g) + N2(g) → 2 NH3(g). Datos: S0 (J·mol–1·K–1): H2(g) = 130,6; O2(g) =205; N2(g) = 191,5; NO(g) = 210,7; NH3(g) =192,3
ΔS0 = Σ np· S0productos – Σ nr· S0
reactivos
a) ΔS0 = 2 mol · 210,7 J ·mol–1 ·K–1 –
(191,5 J·K–1 + 205 J·K–1 ) = 24,9 J·K–1
b) ΔS0 = 2·192,3 J·K–1 –
(3 mol ·130,6 J· mol–1·K–1 + 191,5 J·K–1 ) = –198,7 J·K–1

Segundo principio de la Termodinámica.
• “En cualquier proceso espontáneo la entropía total del universo tiende a aumentar siempre”.
• ΔSuniverso = ΔSsistema + ΔSentorno ≥ 0
• A veces el sistema pierde entropía (se ordena) espontáneamente. En dichos casos el entorno se desordena.

Tercer principio de la Termodinámica
• “La entropía de cualquier sustancia a 0 K es igual a 0” (máximo orden).
• Equivale a decir que no se puede bajar de dicha temperatura.
• ¡CUIDADO! Las S de los elementos en condiciones estándar no son 0 sino que es positiva.

• En procesos reversibles y a temperatura constante se puede calcular ΔS de un sistema como:
Q ΔS = — T
• y si el proceso químico se produce a presión constante:
• ΔHsistema – ΔHsistema ΔSsistema = ——— ; ΔSentorno= ———— T T
• S0 (entropía molar estándar) se mide en J·mol–1·K–1.
• ΔSreacción se mide en J·K–1.