caracterizaciónclínicayfactorespronósticosensubgrupos de ... · id4’...
TRANSCRIPT

Caracterización clínica y factores pronósticos en subgrupos
de pacientes pediátricos con leucemia linfoblástica aguda
de alto riesgo de recaída
Tesis presentada por:
Susana Rives Solà
para aspirar al grado de Doctora en Medicina
Directores de la tesis: Dra. Mireia Camós Guijosa y Dr. Jordi Esteve Reyner
Facultat de Medicina
Universitat de Barcelona
Septiembre 2013

II

III
Dedicatoria
A mis padres, que tanto me enseñaron y a quienes extraño
A Alfonso, Toni y Anna

IV

V
ÍNDICE I.-‐ AGRADECIMIENTOS VII II.-‐ ABREVIATURAS O ACRÓNIMOS XIII III.-‐GLOSARIO DE GENES XVII IV.-‐INTRODUCCIÓN 1
1. Leucemia linfoblástica aguda (LLA) 1.1 Incidencia 3 1.2 Etiología 4 1.3 Fisiopatología 7 1.4 Clínica 9 1.5 Diagnóstico diferencial 11 1.6 Diagnóstico clínico y biológico 12 1.7 Biología 17 Clasificaciones 17 Subtipos genéticos 19 Alteraciones primarias 21 Alteraciones secundarias 28 1.8 Tratamiento 34 Contribución de los grupos cooperativos 34 Tratamiento de primera línea de la LLA 39 Trasplante alogénico de progenitores hemopoyéticos 42 Seguimiento y efectos secundarios
a largo plazo 43 LLA recaída 44 Nuevos tratamientos 45
1.9 Tratamiento en subgrupos de pacientes que requieren un abordaje específico 45
LLA con cromosoma Philadelphia (LLA Ph+) 45 LLA del lactante 45 LLA en pacientes con Síndrome de Down 46 1.10 Factores pronósticos 47
2. LLA con cromosoma Philadelphia (LLA Ph+) 53
2.1 Perspectiva histórica 53 2.2 Cromosoma Ph y gen de fusión BCR-‐ABL 56

VI
2.3 Heterogeneidad clínica y biológica dentro de la LLA Ph+ 57
2.4 Pronóstico de la LLA Ph+ antes de la introducción del tratamiento con inhibidores de tirosín-‐quinasas (TKI) (era pre-‐TKI) 58
2.5 Pronóstico de la LLA Ph+ en la era TKI 59 2.6 Protocolos de tratamiento en la LLA Ph+ 62
3. LLA-‐T 65 3.1 Incidencia 65 3.2 Clínica 65 3.3 Biología 66 3.4 Factores pronósticos clínicos 72 3.5 Factores pronósticos biológicos 74 3.6 Tratamiento 75
Quimioterapia 75 Trasplante alogénico de progenitores hemopoyéticos 77 Nuevos fármacos 78
V.-‐ HIPÓTESIS Y OBJETIVOS 81 2.1 Hipótesis 83 2.2 Objetivos 84 VI.-‐ RESULTADOS 87 Trabajo 1 89 Trabajo 2 107 Trabajo 3 115 Trabajo 4 121 VII.-‐ DISCUSIÓN 137 LLA Ph+ 139 LLA-‐T 151 Limitaciones del estudio 158 Beneficios de la investigación 159 Perspectivas de futuro 160 VIII.-‐ CONCLUSIONES 163 IX.-‐ FINANCIACIÓN 167 X.-‐ BIBLIOGRAFÍA 171

VII
Agradecimientos

VIII

IX
AGRADECIMIENTOS A mis directores de tesis, la Dra. Mireia Camós y el Dr. Jordi Esteve. Ha sido
un privilegio y un placer tenerlos como directores.
A la Dra. Isabel Badell, que me animó a escribir la tesis y me sugirió los temas
de investigación que conforman este trabajo de tesis.
A todos los hemato-‐oncólogos pediátricos del grupo SHOP que han tratado a
los pacientes incluidos en este trabajo y que han colaborado en el registro de
sus datos.
Al equipo de Clever Instruments por su ayuda en la recogida de datos y en el
análisis estadístico: Dr. Agustí Martí, Carmen Romero y Rocío Sánchez.
A mi jefe, el Dr. Jesús Estella, por todo lo que me ha enseñado y me enseña.
A la Dra. Marina Mateo, anterior jefa de Hematología y con quien empecé a
trabajar en el Hospital Sant Joan de Déu y al Dr. Iñaki Alcorta, de quien
también aprendí mucho. A la Dra. Teresa Toll, por todo lo que me enseña.

X
A todos mis compañeros del equipo de hematología pediátrica, porque voy a
trabajar a gusto cada día gracias a ellos y de quienes aprendo. También les
agradezco el esfuerzo adicional que han tenido que hacer a causa de mi
reducción de jornada: la Dra. Teresa Toll, el Dr. Albert Català, el Dr. Rubén
Berrueco, la Dra. Montserrat Torrebadell y la Dra. Mireia Camós.
A los médicos del máster de Hematología Pediátrica: Anna Ruiz Llobet,
Gabriela Corbalán, Núria Conde, Raquel Castro, María Trabazo, Anna Alonso,
Montse Mesegué.
A los médicos y otros profesionales de mi hospital que forman parte del
equipo multidisciplinar para el diagnóstico y tratamiento de los niños con
leucemia: Dra. Mar Pérez (Genética), Joan Vinent (Farmacia), Dra. Natalia
Rodríguez y Gemma Calaf (Rehabilitación), Dra. Ester Gean (Genética clínica),
Núria Carsí, Laia Jané y Marta Albert (Psicología), Mª Àngels Claramonte y
Ramon Badosa (Trabajo Social), Rocío Escobar, Marta Palomares, Dra. Jessica
Ortiz (Equipo de Paliativos), Dra. Margarita Vancells y Dra. Rosalía Carrasco
(Cirugía), Dr. Ferran Torné y Dr. Ramon Huguet (Traumatología), Roberta
Malatesta, Núria Vega (biólogas e investigadoras pre-‐doctorales) y muchos
profesionales más.

XI
Al equipo de enfermería del laboratorio de hematología: Camino, Araceli,
Rosa y Justo.
A las enfermeras, auxiliares y secretarias de la planta 8ª y de Hospital de Día,
por su profesionalidad y por su cariño hacia los niños oncológicos. A Puri
Gonzalo, por su gran humanidad.
A los médicos y profesores del Hospital Clínic que me enseñaron durante mi
residencia, en especial a los doctores: Montserrat Rovira, Arturo Pereira,
Jordi Esteve, Armando López-‐Guillermo, Francisco Cervantes, Joan Bladé,
Benet Nomdedéu, Francesc Bosch, Enric Carreras, Joan Carles Reverter, Neus
Villamor, Dolors Colomer y Elías Campo.
Al Dr. Josep Maria Ribera, por lo que me ha enseñado y porque constituye
un ejemplo a seguir.
A mis compañeros de residencia porque disfruté y aprendí con ellos: Juan
Carlos Hernández-‐Boluda, Joan Cid, Silvia Montoto, Llúcia Sanz , María
Perales, Anna Ferrer, Mireia Camós, Alberto Álvarez-‐Larrán.

XII
A mis amigas, por su apoyo y cariño: Mónica, Cristina, Nati y Myriam.
A mi amiga Paloma, a quien extraño, por su alegría y generosidad.
A Alfonso, Toni y Anna, por vuestro amor y por el tiempo que esta tesis os ha
robado.
A mi padres por su amor y por transmitirme entusiasmo por aprender en la
medicina y en tantas otras disciplinas.
A mis hermanas María y Mercè, a mis abuelos, a la madrina, a Rosa y a todos
mis tíos, a mis suegros y a toda mi familia, por su cariño y apoyo.
A los niños con leucemia y sus padres, porque son la principal motivación
para seguir trabajando.

XIII
Abreviaturas

XIV

XV
ABREVIATURAS o ACRÓNIMOS Ara-‐G: análogo del arabinósido de guanina Ara-‐GTP: arabinósido de guanosín trifosfato BFM: Berlin Frankfurt Münster CARTs: Chimeric antigen receptor-‐modified T cells CNA: copy number alterations o alteración en el número de copias de DNA COG: Children Oncology Group DCFI: Dana Farber Cancer Institute DNA: deoxyribonucleic acid o ADN (ácido desoxirribonucleico) EGIL: European Group of Immunological classification of Leukemias ERM: enfermedad residual mínima EsPhALL: European Intergroup Study on Post Induction Treatment of Philadelphia Positive Acute Lymphoblastic Leukaemia with Imatinib. ETP: Early-‐T cell Precursor o precursor inmaduro de las células T FAB: clasificación de leucemias French American British FISH: fluorescence in situ hybridization o hibridación in situ fluorescente FRALLE: French Acute Lymphoblastic Leukaemia Study Group GMALL German Multicenter Acute Lymphoblastic Leukemia HLA: Human Leukocyte Antigen I-‐BFM: grupo internacional BFM LCR: líquido cefalorraquídeo LDH: lactato deshidrogenasa LLA: leucemia linfoblástica aguda LLA-‐B: leucemia linfoblástica aguda de fenotipo B LLA Ph+: leucemia linfoblástica aguda con cromosoma Philadelphia LLA-‐T: leucemia linfoblástica aguda de fenotipo T LMA: leucemia mieloblástica aguda LMC: leucemia mieloide crónica MPAL: mixed phenotype acute leukemia o leucemias de fenotipo mixto MTHFR: enzima metilen-‐tetrahidrofolato reductasa NCI: National Cancer Institute PCR: polymerase chain reaction o reacción en cadena de la polimerasa PETHEMA: Protocolo para el Estudio y Tratamiento de las Hemopatías Malignas PL: punción lumbar PNP: Purine Nucleoside Phosphorylase

XVI
RC: remisión completa SG: supervivencia global SHOP / SEHOP: Sociedad Española de Hemato-‐Oncología Pediátrica SLE: supervivencia libre de evento SNC: sistema nervioso central SEER: US National Cancer Institute’s Surveillance, Epidemiology and End Results (SEER) program SJCRH: Saint Jude Children’s Research Hospital TCR: T-‐cell receptor o RCT, receptor de células T TKI: Tyrosin Kinase Inhbitor o inhibidor de tirosín-‐quinasas TPH: trasplante de progenitores hematopoyéticos TPMT: enzima tiopurin-‐metiltransferasa UKALL: United Kingdom Acute Lymphoblastic Leukemia Group WHO: World Health Organization (Organización Mundial de la Salud, OMS)

XVII
Glosario de genes

XVIII

XIX
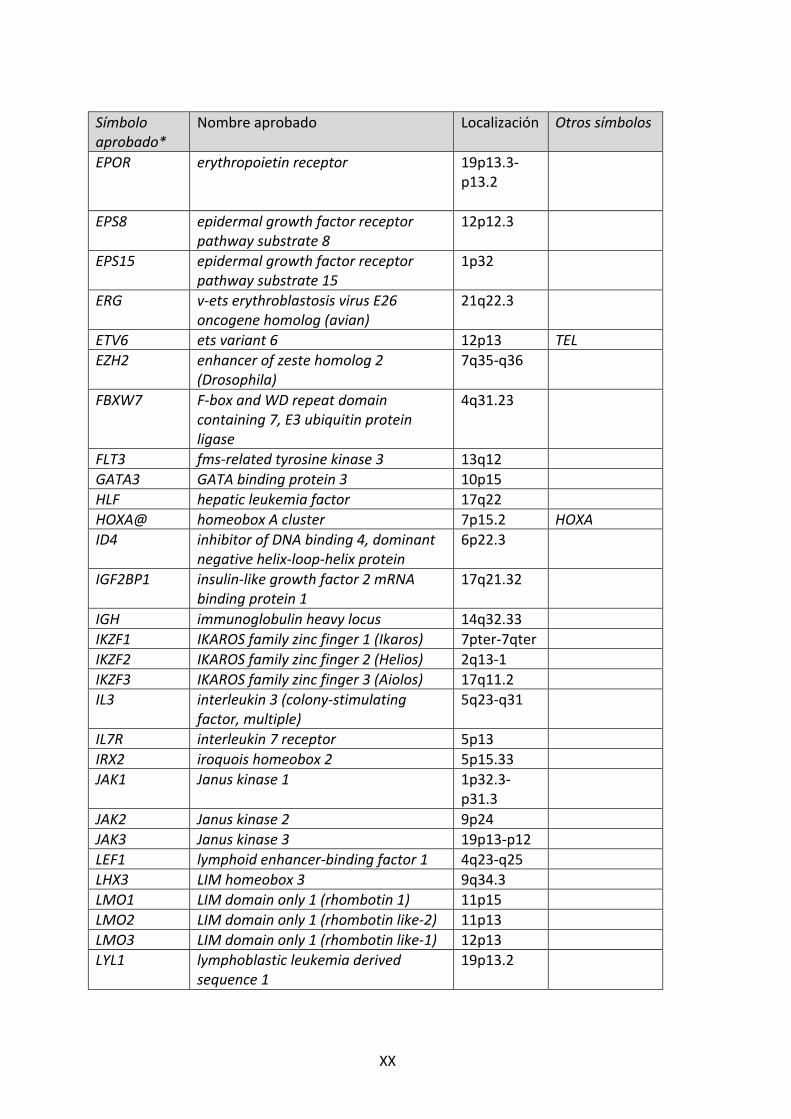
GLOSARIO DE GENES Símbolo aprobado*
Nombre aprobado Localización Otros símbolos
ABL1 v-‐abl Abelson murine leukemia viral oncogene homolog1
9q34.1 ABL, JTK7, C-‐ABL, p150, v-‐abl
AKT1 v-‐akt murine thymoma viral oncogene homolog 1
14q32.32-‐q32.33
AKT
ARID5B AT rich interactive domain 5B (MRF1-‐like)
10q11.22
BCL11B B-‐cell CLL/lymphoma 11B (zinc finger protein)
14q32
BCL2 B-‐cell CLL/lymphoma 2 18q21.3 BCL9 B-‐cell CLL/lymphoma 9 1q21 BCR Break cluster region 22q11 BMI1 BMI1 polycomb ring finger oncogene 10p13 CDKN1B cyclin-‐dependent kinase inhibitor 1B
(p27, Kip1) 12p13.1-‐p12
CDKN2A cyclin-‐dependent kinase inhibitor 2A 9p21 CDK4 inhibitor p16INK4, ARF, p14ARF
CDKN2B cyclin-‐dependent kinase inhibitor 2B (p15, inhibits CDK4)
9p21 CDK4 inhibitor p15INK4b, p15INK4
CEBPE CCAAT /enhancer binding protein (C/EBP), epsilon
14.q11.2
CEBPA CCAAT/enhancer binding protein (C/EBP), alpha
19q13.1
CEBPB CCAAT/enhancer binding protein (C/EBP), beta
20q13.1
CEBPD CCAAT/enhancer binding protein (C/EBP), delta
8p11.2-‐p11.1
CEBPG CCAAT/enhancer binding protein (C/EBP), gamma
19q13.2
CREBBP CREB binding protein 16p13.3 CRLF2 cytokine receptor-‐like factor 2 Xp22.3 e
Yp11.3
CTGF connective tissue growth factor 6q23.2 CCN2 EBF1 connective tissue growth factor 5q34 EBF EED embryonic ectoderm development 11q14.2-‐
q22.3
EML1 echinoderm microtubule associated protein like 1
14q32
XX
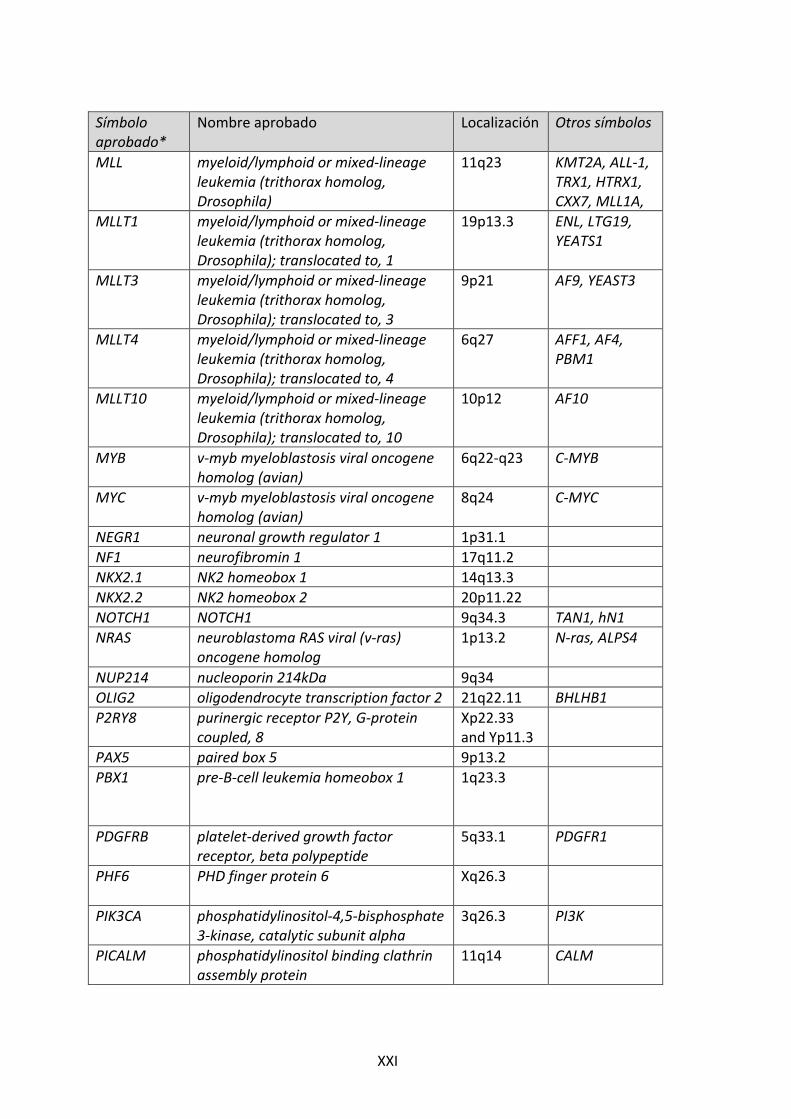
Símbolo aprobado*
Nombre aprobado Localización Otros símbolos
EPOR erythropoietin receptor 19p13.3-‐p13.2
EPS8 epidermal growth factor receptor pathway substrate 8
12p12.3
EPS15 epidermal growth factor receptor pathway substrate 15
1p32
ERG v-‐ets erythroblastosis virus E26 oncogene homolog (avian)
21q22.3
ETV6 ets variant 6 12p13 TEL EZH2 enhancer of zeste homolog 2
(Drosophila) 7q35-‐q36
FBXW7 F-‐box and WD repeat domain containing 7, E3 ubiquitin protein ligase
4q31.23
FLT3 fms-‐related tyrosine kinase 3 13q12 GATA3 GATA binding protein 3 10p15 HLF hepatic leukemia factor 17q22 HOXA@ homeobox A cluster 7p15.2 HOXA ID4 inhibitor of DNA binding 4, dominant
negative helix-‐loop-‐helix protein 6p22.3
IGF2BP1 insulin-‐like growth factor 2 mRNA binding protein 1
17q21.32
IGH immunoglobulin heavy locus 14q32.33 IKZF1 IKAROS family zinc finger 1 (Ikaros) 7pter-‐7qter IKZF2 IKAROS family zinc finger 2 (Helios) 2q13-‐1 IKZF3 IKAROS family zinc finger 3 (Aiolos) 17q11.2 IL3 interleukin 3 (colony-‐stimulating
factor, multiple) 5q23-‐q31
IL7R interleukin 7 receptor 5p13 IRX2 iroquois homeobox 2 5p15.33 JAK1 Janus kinase 1 1p32.3-‐
p31.3
JAK2 Janus kinase 2 9p24 JAK3 Janus kinase 3 19p13-‐p12 LEF1 lymphoid enhancer-‐binding factor 1 4q23-‐q25 LHX3 LIM homeobox 3 9q34.3 LMO1 LIM domain only 1 (rhombotin 1) 11p15 LMO2 LIM domain only 1 (rhombotin like-‐2) 11p13 LMO3 LIM domain only 1 (rhombotin like-‐1) 12p13 LYL1 lymphoblastic leukemia derived
sequence 1 19p13.2
XXI
Símbolo aprobado*
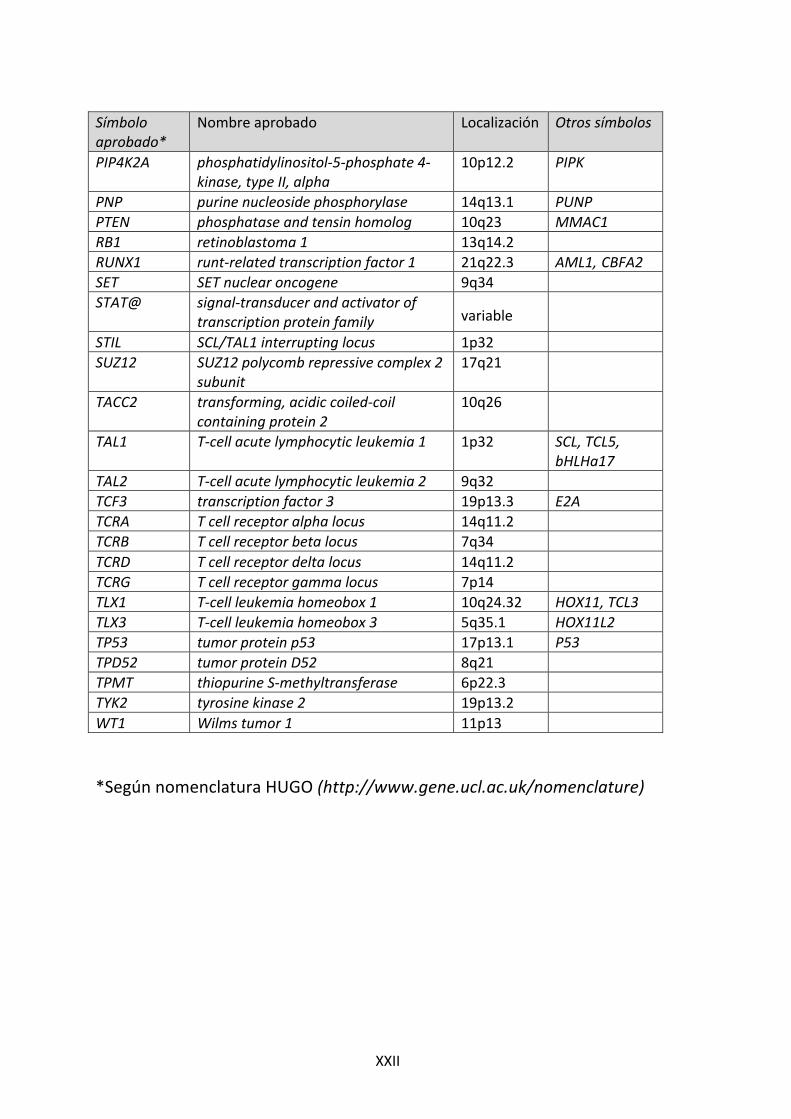
Nombre aprobado Localización Otros símbolos
MLL myeloid/lymphoid or mixed-‐lineage leukemia (trithorax homolog, Drosophila)
11q23 KMT2A, ALL-‐1, TRX1, HTRX1, CXX7, MLL1A,
MLLT1 myeloid/lymphoid or mixed-‐lineage leukemia (trithorax homolog, Drosophila); translocated to, 1
19p13.3 ENL, LTG19, YEATS1
MLLT3 myeloid/lymphoid or mixed-‐lineage leukemia (trithorax homolog, Drosophila); translocated to, 3
9p21 AF9, YEAST3
MLLT4 myeloid/lymphoid or mixed-‐lineage leukemia (trithorax homolog, Drosophila); translocated to, 4
6q27 AFF1, AF4, PBM1
MLLT10 myeloid/lymphoid or mixed-‐lineage leukemia (trithorax homolog, Drosophila); translocated to, 10
10p12 AF10
MYB v-‐myb myeloblastosis viral oncogene homolog (avian)
6q22-‐q23 C-‐MYB
MYC v-‐myb myeloblastosis viral oncogene homolog (avian)
8q24 C-‐MYC
NEGR1 neuronal growth regulator 1 1p31.1 NF1 neurofibromin 1 17q11.2 NKX2.1 NK2 homeobox 1 14q13.3 NKX2.2 NK2 homeobox 2 20p11.22 NOTCH1 NOTCH1 9q34.3 TAN1, hN1 NRAS neuroblastoma RAS viral (v-‐ras)
oncogene homolog 1p13.2 N-‐ras, ALPS4
NUP214 nucleoporin 214kDa 9q34 OLIG2 oligodendrocyte transcription factor 2 21q22.11 BHLHB1 P2RY8 purinergic receptor P2Y, G-‐protein
coupled, 8 Xp22.33 and Yp11.3
PAX5 paired box 5 9p13.2 PBX1 pre-‐B-‐cell leukemia homeobox 1
1q23.3
PDGFRB platelet-‐derived growth factor receptor, beta polypeptide
5q33.1 PDGFR1
PHF6 PHD finger protein 6
Xq26.3
PIK3CA phosphatidylinositol-‐4,5-‐bisphosphate 3-‐kinase, catalytic subunit alpha
3q26.3 PI3K
PICALM phosphatidylinositol binding clathrin assembly protein
11q14 CALM
XXII
Símbolo aprobado*
Nombre aprobado Localización Otros símbolos
PIP4K2A phosphatidylinositol-‐5-‐phosphate 4-‐kinase, type II, alpha
10p12.2 PIPK
PNP purine nucleoside phosphorylase 14q13.1 PUNP PTEN phosphatase and tensin homolog 10q23 MMAC1 RB1 retinoblastoma 1 13q14.2 RUNX1 runt-‐related transcription factor 1 21q22.3 AML1, CBFA2 SET SET nuclear oncogene 9q34 STAT@ signal-‐transducer and activator of
transcription protein family variable
STIL SCL/TAL1 interrupting locus 1p32 SUZ12 SUZ12 polycomb repressive complex 2
subunit 17q21
TACC2 transforming, acidic coiled-‐coil containing protein 2
10q26
TAL1 T-‐cell acute lymphocytic leukemia 1 1p32 SCL, TCL5, bHLHa17
TAL2 T-‐cell acute lymphocytic leukemia 2 9q32 TCF3 transcription factor 3 19p13.3 E2A TCRA T cell receptor alpha locus 14q11.2 TCRB T cell receptor beta locus 7q34 TCRD T cell receptor delta locus 14q11.2 TCRG T cell receptor gamma locus 7p14 TLX1 T-‐cell leukemia homeobox 1 10q24.32 HOX11, TCL3 TLX3 T-‐cell leukemia homeobox 3 5q35.1 HOX11L2 TP53 tumor protein p53 17p13.1 P53 TPD52 tumor protein D52 8q21 TPMT thiopurine S-‐methyltransferase 6p22.3 TYK2 tyrosine kinase 2 19p13.2 WT1 Wilms tumor 1 11p13 *Según nomenclatura HUGO (http://www.gene.ucl.ac.uk/nomenclature)

Introducción
1
Introducción

Introducción
2

Introducción
3
1. Leucemia linfoblástica aguda pediátrica
Las leucemias agudas constituyen un grupo heterogéneo de enfermedades
neoplásicas caracterizadas por la expansión clonal de células derivadas de un
progenitor hematopoyético inmaduro de línea linfoide (leucemia
linfoblástica aguda, LLA) o mieloide (leucemia mieloblástica aguda, LMA).
Dentro de las LLA podemos distinguir las leucemias de línea B (LLA-‐B
precursoras) y las de línea T (LLA-‐T).
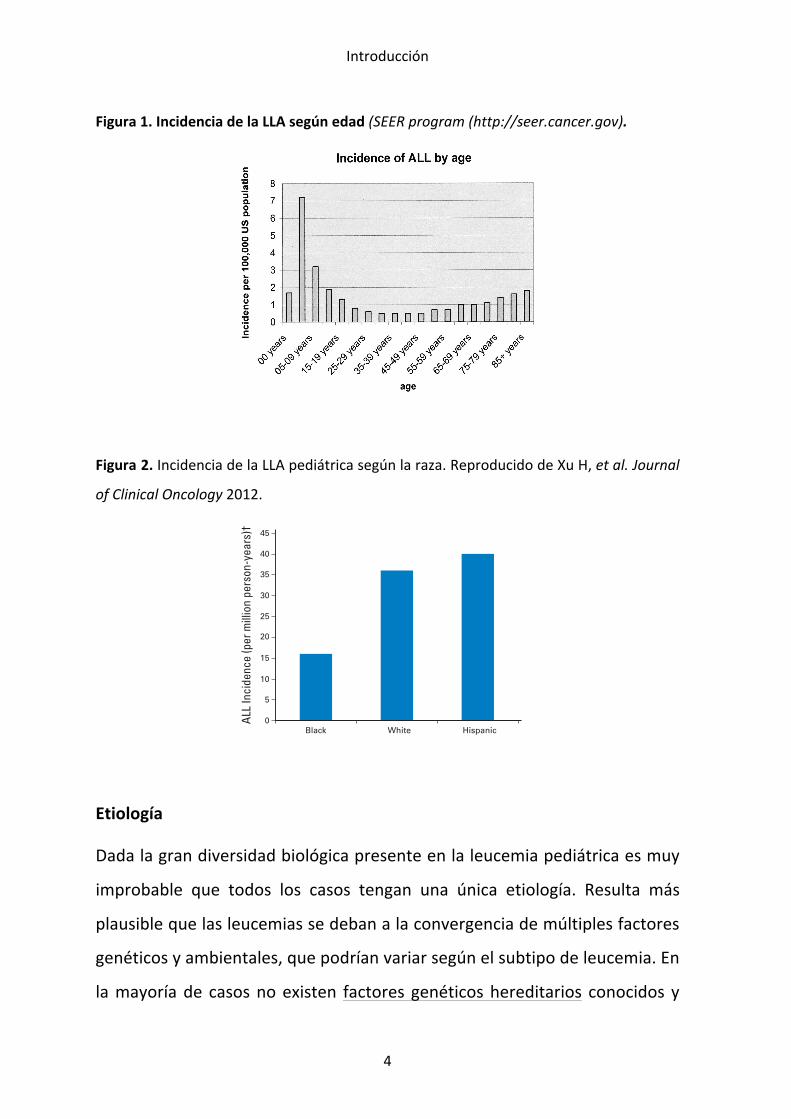
Incidencia
La leucemia aguda es el cáncer más frecuente en la edad pediátrica y
representa la tercera parte de todos los cánceres en esta edad. En niños, a
diferencia de lo que sucede en la edad adulta, la LLA es mucho más
frecuente que la LMA, con una proporción de 4 a 1. La incidencia de la LLA
en niños menores de 15 años es de 35 casos por millón de habitantes, con un
pico de incidencia entre los 2 y 5 años de edad y oscila según la raza y área
geográfica (US National Cancer Institute’s Surveillance, Epidemiology and End
Results (SEER) program, ver figura 1) (Gurney et al., 1995). Así, es menos
frecuente en los niños de raza negra y más frecuente entre los hispano-‐
americanos que en los niños de origen caucásico (Glazer et al., 1999) (Pui,
2012). Ver figura 2 (Xu et al., 2012).
Introducción
4
Figura 1. Incidencia de la LLA según edad (SEER program (http://seer.cancer.gov).
Figura 2. Incidencia de la LLA pediátrica según la raza. Reproducido de Xu H, et al. Journal
of Clinical Oncology 2012.
Etiología
Dada la gran diversidad biológica presente en la leucemia pediátrica es muy
improbable que todos los casos tengan una única etiología. Resulta más
plausible que las leucemias se deban a la convergencia de múltiples factores
genéticos y ambientales, que podrían variar según el subtipo de leucemia. En
la mayoría de casos no existen factores genéticos hereditarios conocidos y
causative polymorphisms and the exact nature of the racial differencesin ALL that are explained by ARID5B. No ARID5B SNPs were signif-icantly associated with genomic loci previously linked to ALL suscep-tibility (ie, IKZF1; Data Supplement), suggesting that these genesindependently contribute to ALL pathobiology.
Interestingly, our multivariate analyses identified two ARID5BSNPs that independently contributed to ALL susceptibility in Hispan-ics, although a single SNP (rs10821936) almost entirely accounted forthe association signal at this locus in whites. These observations implythat relationships between genetic variations and ALL susceptibilityshould be examined in the context of genetic ancestry. The C allele atrs10821936 was significantly over-represented in patients with a highpercentage of NA ancestry at this locus and was surprisingly commonin indigenous NA populations, raising the question of whether NAsare particularly susceptible to ALL. Although there is a paucity of dataspecifically assessing the incidence of ALL in NAs, a recent analysis ofSurveillance, Epidemiology, and End Results (SEER) data from fivestates in the United States reported a 1.63-fold greater incidence ofALL in NA children than in whites, although the difference did notreach statistical significance because of small sample size.4 Interna-tionally, one of highest incidence rates of childhood ALL occurs inCosta Rica,36,37 where Mestizos (admixed groups with NA ances-try) constitute a high proportion of the population. These obser-vations are of interest but should be interpreted with caution forseveral reasons: race/ethnic classification by self-reports, relativelysmall size of NA samples, and possible incompleteness of thecancer registries (particularly in developing countries). It is in-triguing that the population frequency of this allele somewhatparallels the order of human migration out of Africa38: the fre-quency is lowest in blacks (Africa), intermediate in whites andAsians (Europe and Asia), and highest in Native Americans (the Ameri-cas). Whether the ARID5B locus has been subject to selection pressureduring human evolution warrants further investigation.39
Finally, one of the most significant findings from this study is thatARID5B germline SNPs related to ALL susceptibility were also associ-
ated with ALL outcome. To the best of our knowledge, this is the firstreport describing the relation between ARID5B and ALL treatmentresponse in the context of a frontline ALL clinical trial. Further exam-ination of ARID5B variation in the context of different ALL treatmentregimens is warranted to refine its value as a prognostic marker.Interestingly, we previously observed that ARID5B SNP genotype isassociated with leukemic cell accumulation of methotrexate metabo-lites (ie, polyglutamated methotrexate),18 offering a plausible mecha-nism by which ARID5B is linked to ALL relapse. Together, theseresults point to the possibility that leukemogenesis and antileukemicdrug response mechanisms may converge on common pathways.
AUTHORS’ DISCLOSURES OF POTENTIAL CONFLICTSOF INTEREST
Although all authors completed the disclosure declaration, the followingauthor(s) indicated a financial or other interest that is relevant to the subjectmatter under consideration in this article. Certain relationships markedwith a “U” are those for which no compensation was received; thoserelationships marked with a “C” were compensated. For a detaileddescription of the disclosure categories, or for more information aboutASCO’s conflict of interest policy, please refer to the Author DisclosureDeclaration and the Disclosures of Potential Conflicts of Interest section inInformation for Contributors.Employment or Leadership Position: Mary V. Relling, St. JudeChildren’s Research Hospital (C) Consultant or Advisory Role: NoneStock Ownership: None Honoraria: None Research Funding: Mary V.Relling, Sigma Tau Pharmaceuticals Expert Testimony: None OtherRemuneration: None
AUTHOR CONTRIBUTIONS
Conception and design: Cheng Cheng, Naomi J. Winick, Cheryl L. Willman,Gregory H. Reaman, William L. Carroll, Mignon Loh, William E. Evans,Ching-Hon Pui, Stephen P. Hunger, Mary V. Relling, Jun J. YangAdministrative support: Celeste Eng, Gregory H. Reaman
B
ALL
Inci
denc
e (p
er m
illio
n pe
rson
-yea
rs)†
15
10
5
0
20
30
25
40
35
45
18
33
47
33
48
62
ANon-ALL controlsALL cases
Black*n = 112 n = 93
P = .0015
Whiten = 1,046 n = 978
P = 8.38 ! 10-20
Hispanicn = 541 n = 330
P = 1 ! 10-6
Black White Hispanic
Freq
uenc
y of
the
Risk
Alle
le
at rs
1082
1936
(%)
20
10
0
30
40
60
50
70
Fig 3. Racial differences in the risk allele frequency at rs10821936 and in acute lymphoblastic leukemia (ALL) incidence. (A) Genotype of rs10821936 is associated with ALLsusceptibility in all three race groups. (B) Frequency of the risk allele (allele C) increases in order for blacks, whites, and Hispanics, consistent with the racial differences in ALL incidence.*Association of rs10821936 with ALL in blacks is based on a previous report by Yang et al.20 †ALL incidence by race is based on the report by Linabery et al.3
Xu et al
756 © 2012 by American Society of Clinical Oncology JOURNAL OF CLINICAL ONCOLOGY

Introducción
5
sólo en un porcentaje muy pequeño de casos la LLA se desarrolla en
pacientes con una enfermedad genética subyacente con predisposición a la
leucemia, como sería el Síndrome de Down o la ataxia-‐telangiectasia (Pui,
2012). Sin embargo, recientemente se ha descrito la asociación entre ciertos
polimorfismos genéticos en la población general y el riesgo aumentado de
desarrollar LLA pediátrica. Así, determinados polimorfismos en los genes
ARID5B (como por ejemplo el rs10821936), IKZF1, CEBPE, BMI1-‐PIP4K2A y
CDKN2A, se asocian a una mayor predisposición a padecer una LLA (Xu et al.,
2013). Algunos polimorfismos en el gen ARID5B asociados a mayor riesgo de
desarrollar LLA son menos frecuentes en la raza negra y más frecuentes en
los hispano-‐americanos, lo que podría explicar las diferencias en la incidencia
de LLA entre razas (Yang et al., 2012; Xu et al., 2012). Además, determinados
polimorfismos genéticos se asocian específicamente a ciertos subgrupos
biológicos de LLA, como sería el caso de la LLA hiperdiploide. Así,
determinadas variantes polimórficas en los genes ARID5B e IKZF1 se asocian
a la LLA con hiperdiploidía (Papaemmanuil et al., 2009).
En cuanto a los factores ambientales, hay estudios epidemiológicos que han
mostrado asociaciones con algunas exposiciones a agentes físicos o químicos
y la LLA, como radiaciones ionizantes y electromagnéticas, exposiciones
laborales de los padres, consumo de alcohol durante la gestación y otros. Sin
embargo, estas asociaciones son débiles y en algunos casos no se han
confirmado (Malagoli et al., 2010; Bailey et al., 2011; Hsu et al., 2013). En el
caso de la leucemia del lactante, por el contrario, sí que parece que la
exposición transplacentaria a inhibidores de topoisomerasa-‐II tenga un papel
importante en el origen de la leucemia. En este sentido, se ha observado que
tanto las leucemias del lactante como las leucemias secundarias a

Introducción
6
inhibidores de topoisomerasa-‐II con reordenamiento del gen MLL tienen con
mayor frecuencia el punto de rotura localizado en el intrón 11 (Meyer et al.,
2013). Dicho intrón contiene múltiples dominios de unión a topoisomerasa-‐
II, por lo que sería muy sensible a la acción de sustancias citotóxicas. Existen
componentes naturales y sintéticos con actividad frente a topoisomerasa-‐II
(quinolonas, flavonoides presentes en alimentos y bebidas como el té).
Aunque esta actividad sea mucho más débil que la de los quimioterápicos, la
capacidad detoxificadora podría ser menor en el feto o en algunas gestantes
que tuvieran polimorfismos genéticos asociados a una menor actividad
enzimática (Biondi et al., 2000).
En la mayoría de las LLA en niños mayores de 1 año, sin embargo, se
desconocen las causas ambientales que pueden originarla. Entre las
diferentes hipótesis destaca la teoría infecciosa, que podría explicar el pico
de incidencia de LLA entre los 2 y 5 años. En esta teoría se postula que la
exposición a infecciones en ciertos momentos del desarrollo y la respuesta
inmunológica a dichas infecciones podría desempeñar un papel importante
en la etiología de la leucemia. Se han propuesto dos hipótesis, una que se
basa en la mezcla de poblaciones (Kinlen, 1997) y otra basada en la infección
tardía (Greaves, 1988). La primera hipótesis se ve apoyada por estudios
epidemiológicos en los que la mezcla de poblaciones y el contacto con
ciertos virus frente a los que no se tenía inmunidad, se asociaron a un
aumento en la incidencia de leucemia infantil (Eden, 2010). La hipótesis de la
“infección tardía” postula que el sistema inmune está programado durante
el primer año de vida a anticipar determinadas infecciones y estas
exposiciones serían necesarias para el desarrollo posterior de respuestas
inmunológicas eficaces. La exposición tardía a las infecciones comunes por

Introducción
7
un exceso de higiene y por la falta de contacto con otros niños que
transmiten estas infecciones podría favorecer la leucemia. En esta teoría se
contempla este retraso en la infección como un evento secundario que sería
el desencadenante sólo en aquellos individuos que ya tienen un evento
primario (por ejemplo, el reordenamiento ETV6-‐RUNX1) y que además
tienen cierta predisposición genética añadida, como podría ser la presencia
de determinados polimorfismos en genes que modulan la respuesta inmune.
Esta teoría la apoyan varios estudios epidemiológicos que describen un
aumento de casos de LLA en niños que no acuden a la guardería (Perrillat et
al., 2002) (Ma et al., 2002) (Gilham et al., 2005) (Ma et al., 2009).
Fisiopatología – Origen clonal de la LLA
Si bien nuestro conocimiento sobre la etiología de la leucemia es aún muy
limitado, el conocimiento sobre la célula leucémica y los mecanismos de
leucemogénesis ha experimentado un gran avance en las últimas décadas.
Ello se debe en gran medida a la facilidad en la obtención de células
leucémicas a partir de muestras de sangre periférica o de médula ósea. El
estudio de estas células ha identificado alteraciones citogenéticas y
moleculares recurrentes que definen distintos subtipos de LLA. Algunas de
estas lesiones se consideran las lesiones iniciales o primarias (founding),
como la traslocación t(9;22)(q34;q11)/BCR-‐ABL o los reordenamientos del
gen MLL, y otras son secundarias y cooperan en el proceso de la
leucemogénesis, como las deleciones del gen IKZF1.
Con el fin de comprender mejor este proceso el grupo de Mel Greaves y
otros grupos de investigadores realizaron estudios para identificar el origen
de la célula leucémica (Greaves et al., 2003). Para ello estudiaron la

Introducción
8
secuencia temporal en el proceso de la leucemogénesis, comparando las
células preleucémicas (portadoras sólo del evento primario) con las células
leucémicas (con alteraciones genéticas añadidas o secundarias). Realizaron,
por un lado, estudios en gemelos univitelinos en los que sólo uno de ellos
desarrolló la leucemia y, por otro, estudios en pacientes con LLA de los que
se disponía de muestra de sangre en el momento de nacer (obtenida de las
cartas de Guthrie o de células criopreservadas de sangre de cordón
umbilical). Así, pudieron demostrar no sólo el origen in utero de una gran
proporción de casos de LLA pediátrica, donde tiene lugar el primer evento,
sino también cómo algunas células adquieren posteriormente, en el periodo
post-‐natal, lesiones genéticas secundarias que desencadenan la leucemia. El
estudio de gemelos con LLA y reordenamiento ETV6-‐RUNX1 ha permitido
inferir la historia natural de este subtipo de LLA. Ambos gemelos
compartirían el clon pre-‐leucémico (con el reordenamiento ETV6-‐RUNX1)
debido al paso transplacentario in utero. Sin embargo, en un gemelo se
desarrollaría la leucemia mediante la adquisición de anomalías genéticas
secundarias y en el otro gemelo (sano), el clon preleucémico podría no
adquirir nuevas lesiones y quedarse detenido en su trayectoria evolutiva
(Wiemels et al., 1999) (Greaves et al., 2003) (Greaves, 2009). En el caso de
las LLA con reordenamiento de MLL el grado de concordancia en el
desarrollo de la LLA entre gemelos es mayor, lo que sugiere que la capacidad
oncogénica de MLL es superior.
Recientemente se ha revisado el concepto de la leucemogénesis y la
linealidad en la evolución de las leucemias (Greaves, 2010). La teoría clásica
de una única clona inicial con una evolución lineal, en la que aparecen
subclones que adquieren nuevas mutaciones, se va viendo sustituida por una

Introducción
9
explicación darwiniana, en la que la evolución clonal tendría una
arquitectura ramificada. De esta manera, al diagnóstico de la leucemia ya
existirían diversos subclones de distintas células madre leucémicas o células
iniciadoras de leucemia (leukemic initiating cells) con diferentes alteraciones
genotípicas (Notta et al., 2011). En las recaídas se pueden hallar clones
derivados de un clon ancestral diferente al clon mayoritario que originó la
leucemia (Mullighan et al., 2008). Esta teoría tiene importantes
implicaciones en el seguimiento y en el tratamiento dirigido frente a dianas
terapéuticas.
Clínica
La presentación clínica de la LLA es variable y por lo general los síntomas y
signos al diagnóstico son secundarios a la infiltración de las células
leucémicas en médula ósea y en otros órganos. Aunque puede presentarse
de forma insidiosa, la LLA infantil suele hacerlo de forma aguda, con una
historia de menos de tres meses desde el inicio de la clínica hasta el
diagnóstico (Pui, 2012). Uno de los hallazgos más frecuentes es la fiebre, que
ocurre en más de la mitad de los casos. Si bien la fiebre puede ser de causa
infecciosa, a menudo se debe a la propia leucemia y se resuelve en 24-‐72
horas desde el inicio del tratamiento con glucocorticoides y/o quimioterapia.
En ocasiones, el niño presenta malestar general y astenia. Si la infiltración
leucémica da lugar a insuficiencia medular puede haber un síndrome
anémico y/o hemorragia por la plaquetopenia. En una tercera parte de los
casos el principal síntoma es el dolor óseo o articular, que puede ser
migratorio o localizado en una extremidad. En niños pequeños, este dolor
puede manifestarse únicamente en forma de cojera. Los casos que cursan

Introducción
10
con dolor óseo con frecuencia tienen inicialmente pocas alteraciones en el
hemograma y esto dificulta y puede retrasar el diagnóstico. La LLA-‐T suele
darse en niños más mayores y frecuentemente se presenta con una carga
tumoral elevada, masa mediastínica, hepato-‐esplenomegalia e
hiperleucocitosis. Debido a la masa mediastínica, algunos niños tienen
disnea y ortopnea.
En la exploración física los hallazgos más frecuentes son la palidez, las
equimosis y petequias, las adenopatías y/o hepato-‐esplenomegalia. En los
varones se deben explorar los testes, que pueden estar infiltrados. En menos
de un 5% de los casos se halla infiltración del sistema nervioso central (SNC)
al diagnóstico. Ésta es más frecuente en las LLA-‐T, en los niños menores de 2
años y en las leucemias con reordenamiento BCR-‐ABL. Puede manifestarse
con clínica de hipertensión endocraneal o con parálisis de un par craneal.
Más rara vez, puede observarse infiltración ocular, en forma de hemorragia
o infiltración leucocitaria en la retina o en el nervio óptico. Se han descrito
otras manifestaciones clínicas secundarias a infiltración extramedular como
los nódulos subcutáneos, el priapismo o la compresión de médula espinal
por masas epidurales, pero son mucho menos frecuentes en la LLA que en
las LMA. Los lactantes con reordenamiento del gen MLL son un caso
particular, con frecuente hiperleucocitosis, lesiones cutáneas,
organomegalias e infiltración del SNC.
El hemograma suele ser diagnóstico por la presencia de blastos en sangre
periférica. Generalmente se observa anemia, plaquetopenia y leucopenia o
leucocitosis. Hasta en un 10% de los casos al diagnóstico el hemograma es
normal o con alteraciones mínimas, por lo que si la clínica es sugestiva, se
debe realizar un aspirado de médula ósea para descartar con seguridad la

Introducción
11
leucemia. Con menor frecuencia que la LMA, la LLA al diagnóstico puede
ocasionar situaciones clínicas de emergencia, con compromiso vital que
requieren un tratamiento urgente. La presentación con alteraciones en la
coagulación o con leucostasis, a diferencia de lo que ocurre en la LMA ocurre
rara vez (ver tabla 1).
Diagnóstico diferencial
En la mayoría de los casos el diagnóstico es sencillo con una anamnesis
minuciosa, una exploración física completa y una analítica general. Sin
embargo, en un 10% de los casos la LLA puede ser más difícil de distinguir de
otros procesos y debe siempre excluirse antes de establecer el diagnóstico
de enfermedades tales como la plaquetopenia inmune, la artritis reumatoide
juvenil, el neuroblastoma (con células tumorales infiltrando médula ósea que
pueden parecerse morfológicamente a los linfoblastos), la aplasia medular y
la mononucleosis infecciosa. Antes de iniciar tratamiento con
glucocorticoides en determinados casos de niños con orientación diagnóstica
de plaquetopenia inmune y de artritis reumatoide juvenil debe realizarse un
aspirado de médula ósea para descartar la leucemia. En los casos de LLA con
dolores óseos la radiografía puede ser normal, pero también puede mostrar
signos característicos, como líneas transversas metafisarias. Otros hallazgos,
como las lesiones líticas o formación ósea subperióstica, obligan a establecer
el diagnóstico diferencial con sarcomas óseos.

Introducción
12
Tabla 1. Situaciones clínicas de emergencia al diagnóstico de la LLA.
Situaciones de emergencia
Clínica Frecuencia Tratamiento
Infección Fiebre y síntomas según el foco infeccioso
Frecuente Antibióticos de amplio espectro y según foco y germen
Masa mediastínica Disnea, ortopnea, tos, síndrome vena cava superior
Frecuente en la LLA-‐T
Evitar anestesia general. Glucocorticoides y/o quimioterapia. Radioterapia en algunos casos
Síndrome de lisis tumoral
Insuficiencia renal aguda, convulsiones, arritmia.
Más frecuente en las LLA con hiperleucocitosis y masa tumoral elevada (lactante, LLA-‐T), en especial tras iniciar tratamiento con glucocorticoides y/o quimioterapia.
Hiperhidratación, alopurinol, rasburicasa. No corregir hipocalcemia si es asintomática.
Coagulación intravascular diseminada
Sangrado
Infrecuente. Se observa más frecuentemente en la LLA con t(17;19).
Hemoderivados, glucocorticoides y/o quimioterapia.
Leucostasis
Disnea, confusión, focalidad neurológica (hemiparesia y otras).
Infrecuente incluso en casos con hiperleucocitosis. Más frecuente en lactantes.
Glucocorticoides y/o quimioterapia. Evitar en lo posible transfusión de hematíes (no incrementar la viscosidad sanguínea).
Hipercalcemia
Confusión, coma, convulsiones, debilidad, alteraciones ritmo cardiaco.
Muy infrecuente. Se ha descrito con mayor frecuencia en la LLA con t(17;19).
Hiperhidratación y diuréticos, glucocorticoides y en algún caso bifosfonatos. Quimioterapia.
Masa que comprime médula espinal
Síndrome de compresión medular (paresia, parálisis, alteración sensibilidad).
Muy infrecuente
Glucocorticoides y/o quimioterapia. Rara vez requiere descompresión quirúrgica dada la gran quimio-‐sensibilidad de la LLA al diagnóstico.
Diagnóstico clínico y biológico
Además de la exploración física, se debe realizar una serie de pruebas
complementarias de imagen y de laboratorio, así como una punción lumbar
para descartar la infiltración del SNC. Se ha demostrado que la punción

Introducción
13
lumbar traumática, debida al paso de blastos de sangre periférica al líquido
cefalorraquídeo (LCR), se asocia a una peor supervivencia del paciente (Pui
et al., 2009). Por ello, resulta muy importante que las punciones lumbares al
diagnóstico las realicen médicos con experiencia en este procedimiento y
que se asegure un recuento plaquetario adecuado previo a su realización. El
estado de afectación del SNC puede definirse según parámetros clínicos o
pruebas de imagen y/o el recuento celular y la citomorfología de LCR:
• SNC-‐1: ausencia de blastos en el LCR
• SNC-‐2: blastos en LCR con menos de 5 leucocitos/µl
• SNC-‐3: blastos en el LCR con 5 o más leucocitos/µl y/o afectación de
pares craneales y/o masa tumoral en cerebro o meninges detectada
por imagen.
• PL traumática sin blastos: más de 10 hematíes/µl sin blastos en LCR
• PL traumática con blastos: más de 10 hematíes/µl con blastos en LCR
El diagnóstico de LLA se basa en la observación morfológica de ≥25% de
blastos de línea linfoide en médula ósea. Si bien con frecuencia el
diagnóstico de LLA se puede realizar con el estudio de las células leucémicas
en sangre periférica, es preferible confirmar el diagnóstico de la LLA
mediante un aspirado de médula ósea. En algunos casos la obtención de
células leucémicas es difícil y se precisan múltiples aspirados o la realización
de una biopsia de médula ósea debido a una infiltración medular importante
(médula empaquetada) o por fibrosis. Además de la microscopía óptica
mediante la que se realizan los estudios morfológicos y citoquímicos (ver
figura 3), en el diagnóstico de la LLA se utilizan otras técnicas que ayudan a
confirmar el linaje y a caracterizar mejor las células leucémicas:

Introducción
14
• Citometría de flujo: establece el linaje de la leucemia y el estadio de
diferenciación de los blastos a través del estudio inmunofenotípico de
antígenos de membrana e intracelulares. Además, facilita el
seguimiento de la enfermedad residual mínima (ERM) por medio del
estudio del fenotipo aberrante de las células leucémicas.
• La citometría de flujo también estima la ploidía celular mediante el
cálculo del índice de DNA, que permite distinguir grupos pronósticos
como la hipodiploidía o la hiperdiploidía alta.
• Citogenética: el estudio del cariotipo permite la detección de
alteraciones numéricas y estructurales (traslocaciones, deleciones y
otras) en las células leucémicas. Mediante esta técnica se pueden
identificar alteraciones genéticas con gran implicación pronóstica (ver
apartado “subtipos genéticos”, más adelante). La técnica de FISH
(Fluorescent in Situ Hybridization) ayuda a la detección de anomalías
genéticas no detectadas por citogenética convencional, bien sea por
falta de obtención de metafases o por tratarse de alteraciones de un
tamaño menor a la resolución de la citogenética convencional
(alteraciones crípticas). Además de la técnica de FISH existen otras
técnicas de citogenética molecular que permiten aumentar la
resolución de las alteraciones genéticas a analizar. Entre ellas se hallan
la técnica SKY (Spectral Karyotiping), que es una variante del FISH que
permite teñir los 23 cromosomas simultáneamente con distintas
sondas, la CGH (Comparative Genomic Hybridization) y los SNParrays
(Single polymorphisms arrays).

Introducción
15
• Biología molecular: mediante la técnica de PCR (reacción en cadena de
la polimerasa) pueden detectarse diferentes alteraciones, como los
genes de fusión con implicación pronóstica en la LLA, así como
estudiar los reordenamientos específicos para cada paciente del gen
de la cadena pesada de las inmunoglobulinas y/o del receptor de
células T (IGH/TCR)(técnicas clonoespecíficas), de gran utilidad para el
seguimiento de la ERM.

Introducción
16
Figura 3. Estudio diagnóstico de un caso de LLA Ph+. A. Morfología de sangre periférica donde se observan blastos linfoides. B. Estudio por citometría de flujo del inmunofenotipo de las células de médula ósea (en rojo los blastos), al diagnóstico (recuadro superior derecha) y al final de inducción, donde se detecta enfermedad residual mínima de 0,08% (recuadro inferior). Los blastos son CD45 débiles y coexpresan de forma aberrante marcadores de línea B y de línea mieloide (CD19++/CD123+). C. Estudio citogenético de las células leucémicas al diagnóstico, en el que se objetiva el cromosoma Ph+. D. Estudio por FISH en el que se confirma la presencia del reordenamiento BCR-‐ABL. E. RT-‐PCR cuantitativa del transcrito BCR-‐ABL. La curva de la izquierda es la muestra al diagnóstico y la de la derecha es la muestra al final de inducción, en la que se objetiva presencia de ERM. Cortesía de la Dra. Toll, Dra. Camós, Dra. Torrebadell, Dra. Pérez-‐Iribarne y Dra. Carrió. A B
C D E
BCR-ABL Diagnóstico ! BCR-ABL
Seguimiento ERM+ !!
RT-PCR cuantitativa

Introducción
17
Biología
Clasificaciones
Las clasificaciones de la LLA han ido cambiando con el mejor conocimiento
de la biología de esta enfermedad (ver tablas 2 y 3).
La clasificación morfológica y citoquímica o clasificación de la FAB (French-‐
American-‐British) (Bennett et al., 1976; Bennett et al., 1981) no se usa
actualmente en la estratificación de los pacientes. Las clasificaciones que
mayor utilidad tienen en la actualidad son la clasificación inmunológica
según el grupo EGIL (European Group of immunological classification of
leukemias) (Bene et al., 1995) y la clasificación de la WHO (World Health
Organization) (Swerdlow et al., 2008; Campo et al., 2011). En la clasificación
EGIL se sistematiza la LLA según el grado de madurez de la célula leucémica.
El fenotipo más frecuente en la LLA pediátrica es el B común, definido por la
positividad de CD10. La clasificación de la WHO integra aspectos clínicos,
morfológicos, inmunofenotípicos y genéticos y reconoce como entidades
diferentes subgrupos de LLA (Swerdlow et al., 2008) (Campo et al., 2011).

Introducción
18
Tabla 2. Clasificación inmunológica (EGIL) (Bene et al., 1995)
Clasificación inmunológica de la LLA según grupo EGIL
LLAde línea B: CD22+ y/ó CD79a+ y/ó CD19+
Pro-‐B (B-‐I): TdT+, CD10-‐, Ig citoplasma-‐, Ig membrana-‐, CD38+.
Común (B-‐II): TdT+, CD10+, Ig citoplasma-‐, Ig membrana-‐, CD38+
Pre-‐B (B-‐III): TdT+, CD10+/-‐, Ig citoplasma +, Ig membrana-‐, CD38+/-‐
B madura (B-‐IV): CD20+, TdT-‐, CD10-‐, Ig citoplasma-‐, cadenas ligeras de superficie o citoplasmáticas +, CD38-‐
LLAde línea T: CD3 de citoplasma +
Pro-‐T (T-‐I): CD7+, CD2-‐, CD5-‐, CD8-‐, CD1a-‐
Pre-‐T (T-‐II): CD2+ y/o CD5+ y/o CD8+, CD1a-‐, CD71+
T cortical: CD1a+, CD3 de superficie + o -‐, CD71-‐
T madura: CD3 de superficie+, CD1a-‐, CD2+, CD5+, CD4/8+
Tabla 3. Clasificación WHO (World Health Organization) 2008 (Campo et al., 2011) (Swerdlow et al., 2008) Clasificación WHO de las neoplasias de precursores linfoides
Leucemia/linfoma linfoblástico B, no especificado
Leucemia/linfoma linfoblástico B con alteraciones genéticas recurrentes
Leucemia linfoblástica B con t(9;22)(q34;q11); BCR-‐ABL
Leucemia linfoblástica B con t(v;11q23); reordenamiento del gen MLL
Leucemia linfoblástica B con t(12;21)(p13;q22); ETV6-‐RUNX1 (TEL-‐AML1)
Leucemia linfoblástica B con hiperdiploidía
Leucemia linfoblástica B con hipodiploidía
Leucemia linfoblástica B con t(5;14)(q31;q32); IL3-‐IGH
Leucemia linfoblástica B con t(1;19)(q23;p13.3)
Leucemia/linfoma linfoblástico T

Introducción
19
Subtipos genéticos
Las alteraciones genéticas presentes en la LLA difieren según la edad de
presentación, de manera que en la edad pediátrica predominan las
alteraciones de buen pronóstico (hiperdiploidía, reordenamiento ETV6-‐
RUNX1), mientras que en la edad adulta son más frecuentes las alteraciones
de mal pronóstico, como el reordenamiento BCR-‐ABL (figuras 4 y 5). En las
últimas décadas ha habido un gran avance en el conocimiento de las
alteraciones genéticas subyacentes en la LLA y la comprensión de los
mecanismos de la leucemogénesis. Mediante técnicas genéticas y
moleculares podemos detectar alteraciones en el 80% de los casos de LLA.
Así, se han podido identificar subtipos genéticos y moleculares con entidad
clínica y biológica diferenciada con valor pronóstico (ver figura 6).
Figura 4. Frecuencias de los subtipos genéticos en LLA pediátrica. Reproducido de Pui et al. Blood 2012 (Pui et al., 2012). Frecuencia estimada de los distintos genotipos en la LLA pediátrica. En dorado se indican aquellas lesiones observadas en LLA-‐T y en gris-‐azul las observadas en LLA B precursoras. En tonalidades más oscuras se representan a aquéllas de peor pronóstico.
patients treated with triple intrathecal therapy compared with thosetreated with intrathecal methotrexate therapy20 (as discussed in“The case for complete omission of prophylactic cranialirradiation”).
Primary genetic abnormalities can be identified in 75% to80% of childhood ALL cases with standard chromosomal andmolecular genetic analyses,9 but in virtually all cases with theaddition of genome-wide analyses (Figure 1). Of several newlydiscovered subtypes, one is characterized by increased CRLF2expression with or without a corresponding genomic lesion(IGH@-CRLF2, P2RY8-CRLF2, and CRLF2 F232C) and com-
monly with a concomitant JAK1/2 sequence mutation, and occursin 5% to 7% of children with precursor B-cell ALL and, remark-ably, in approximately 50% of the cases with Down syndrome.21-26
The non-Down syndrome patients with this genotype probablyrequire more intensive therapy because they generally have a pooroutcome in the reported series (particularly among the NationalCancer Institute high-risk patients).26 The prognostic impact of thisgenotype among patients with Down syndrome remains to bedetermined.
Another novel high-risk subtype, termed “BCR-ABL1–like”ALL, also has a precursor B-cell phenotype, exhibits a gene
Table 1. Patient characteristics and treatment results from selected clinical trials enrolling children with ALL
5-y outcome, %
Study groupYears of
studyNo. of
patientsAge, y,range
T-cellALL, %
Cumulative CNSrelapse rate EFS Survival Data source
AIEOP-95 1995-2000 1743 0-18 11 1.2 ! 0.3 75.9 ! 1.0 85.5 ! 0.8 Conter et al1
BFM-95 1995-1999 2169 0-18 13 4.0 ! 0.4 79.6 ! 0.9 87.0 ! 0.7 Möricke et al2
COG 2000-2005 7153 0-21 7 NA NA 90.4 ! 0.5 Hunger et al3
DCOG-9 1997-2004 859 1-18 11 2.6 ! 0.6 80.6 ! 1.4 86.4 ! 1.2 Veerman et al4
DFCI 00-01 2000-2004 492 1-18 11 NA 80.0 ! 2 91 ! 1 Vrooman et al5
NOPHO-2000 2002-2007 1023 1-15 11 2.7 ! 0.6 79.4 ! 1.5 89.1 ! 11 Schmiegelow et al6
SJCRH 15 2000-2007 498 1-18 15 2.7 ! 0.8 85.6 ! 2.9 93.5 ! 1.9 Pui et al7
UKALL 97/99 1999-2002 938 1-18 11 3.0 ! 0.6 80.0 ! 1.3 88.0 ! 1.1 Mitchell et al8
EFS indicates event-free survival; AIEOP, Associazione Italiana di Ematologia ed Oncologia Pediatrica; BFM, Berlin-Frankfurt-Münster; NA, not available; DCOG, DutchChildhood Oncology Group; DFCI, Dana-Farber Cancer Institute consortium; NOPHO, Nordic Society of Pediatric Hematology and Oncology; SJCRH, St Jude Children’sResearch Hospital; and UKALL, United Kingdom Medical Research Council Working Party on Childhood Leukaemia.
Figure 1. Estimated frequency of specific genotypes in childhood ALL. Data were modified from Pui et al9 by including recently identified genotypes. The genetic lesionsthat are exclusively seen in cases of T-cell ALL are indicated in gold and those commonly associated with precursor B-cell ALL in blue. The darker gold or blue color indicatesthose subtypes generally associated with poor prognosis. BCR-ABL1–like cases can be separated into one group with CRLF2 dysregulation and the other with activatingcytokine receptor and kinase signaling.
1166 PUI et al BLOOD, 9 AUGUST 2012 ! VOLUME 120, NUMBER 6
For personal use only. by guest on January 21, 2013. bloodjournal.hematologylibrary.orgFrom

Introducción
20
Figura 5. Frecuencias de los distintos subgrupos genéticos según la edad. Reproducido de Moorman. Blood Review 2012 (Moorman, 2012)
Figura 6. Supervivencia libre de evento según el subgrupo genético. Reproducido de Moorman, Blood Review 2012 (Moorman, 2012)
and adult studies have shown it to be associated with a favourableoutcome (Fig. 2 and Fig. 3).23,25,38,41–43 In vitro studies have sug-gested that this favourable response to chemotherapy is driven bythe fact that HeH cells are particularly sensitive to methotrexatewhich is one of the mainstays of contemporary treatmentprotocols.44–47
Even though HeH patients have an excellent chance of a curerelative to other patients, the high prevalence of the subgroup, par-ticularly among children, means that treating relapsed HeH patientsis still a clinical challenge. Thus the identification of risk factorswithin this genetic subgroup is a high priority. Over the past two de-cades, several research groups have subdivided this group accordingto gain of specific chromosome(s), modal number and the presenceor absence of structural abnormalities in the search for risk fac-tors.39,41,42,48,49 Unfortunately a consensus has not emerged. TheChildren's Oncology Group (COG) currently use the presence of thetriple trisomy (simultaneous gain of chromosomes 4, 10 and 17) toidentify a subgroup of HeH with a very low risk of relapse.42 Incontrast, we found that trisomy 18 and not triple trisomy to be thebetter indicator of improved survival.23
6. t(1;19)(q23;p13)/TCF3-PBX1
In 90–95% of cases the chromosomal translocation, t(1;19)(q23;p13),results in the fusion of the TCF3 (formerly E2A) gene with the PBX1gene.50 In the remaining 5–10% of cases neither of these genes is in-volved and while a few cases appear to harbour alternative gene fu-sions the subgroup does not appear to be homogeneous.51–53 Thistranslocation is unusual because it can present either as a balancedtranslocation, t(1;19), or unbalanced where just the der(19)t(1;19)is present. The ratio of balanced to unbalanced is approximately50:50.51 The balanced form of the translocation almost always resultsin TCF3-PBX1 fusion and thus most TCF3-PBX1-negative cases presentas der(19)t(1;19).51 TCF3-PBX1-negative cases are also known to coex-ist with high hyperdiploidy and, more rarely, t(9;22)(q34;q11).51 Pa-tients with TCF3-PBX1 usually have a pre-B immunophenotypeexpressing cytoplasmic μ.50 Interestingly it is one of the few genetic
abnormalities which does not appear to vary in frequency with age;being present in 3–5% of cases at all ages, with the exception of infants(Fig. 1). In paediatric ALL, patients with t(1;19) have been reported tohave an adverse outcome. However, these data were largely from theUSA and prior to the use of modern intensive protocols.50 On contem-porary paediatric protocols t(1;19) patients have an intermediate/goodprognosis.23,25,54–56 In adult ALL, there is a great deal of debate overthe prognosis of t(1;19) patients with French and Italian studiesreporting poor outcomes while the UKALLXA and UKALLXII/ECOG2993 trials finding no strong association with outcome.38,57–59
Although some studies have reported a differential outcome be-tween the balanced and unbalanced this is likely to reflect differ-ences between TCF3-PBX1 positive and negative patients.60 Thedifferential outcome in adult ALL is likely to reflect variation in treat-ment protocols and be mirroring historical differences observed inpaediatric cohorts.
Fig. 1. Estimated age-specific frequency of selected chromosomal abnormalities in ALL. The incidence of each abnormality in each age group was calculated using a denominatorthat only included cases appropriately tested by cytogenetic, FISH or MLPA for the abnormality in question. Mutual exclusivity was assumed for the abnormalities listed. A minimumof 80 cases were tested for each abnormality in each age group, with the exception of the infant (b1 year) age group. The total cohort comprised 7113 patients aged as follows: b1 yn=142, 1–4 y n=3128, 5–9 y n=1583, 10–14 y n=915, 15–19 y n=452, 20–24 y 169, 25–39 y n=350, 40–59 y n=374.
0.0
00
.25
0.5
00
.75
1.0
0
Pro
po
rtio
n S
urv
ivn
g E
ven
t F
ree
0 2 4 6 8 10 12Years from diagnosis
ETV6
High hyperdiploidy
87%
83%
74%
50%
41%40%
24%
Other abnormalities
MLL translocations
<40 chromosomest(9;22)(q34;q11)
iAMP21
RUNX1
Fig. 2. Event free survival of childhood BCP-ALL by primary genetic subgroup. Data isfrom ALL97 trial and updated from Moorman et al. (2010) (Ref. [22]). Percentagesare event free survival at 7 years after a median follow-up time of 8.2 years.
4 A.V. Moorman / Blood Reviews xxx (2012) xxx–xxx
Please cite this article as: Moorman AV, The clinical relevance of chromosomal and genomic abnormalities in B-cell precursor acute lympho-blastic leukaemia, Blood Rev (2012), doi:10.1016/j.blre.2012.01.001and adult studies have shown it to be associated with a favourable
outcome (Fig. 2 and Fig. 3).23,25,38,41–43 In vitro studies have sug-gested that this favourable response to chemotherapy is driven bythe fact that HeH cells are particularly sensitive to methotrexatewhich is one of the mainstays of contemporary treatmentprotocols.44–47
Even though HeH patients have an excellent chance of a curerelative to other patients, the high prevalence of the subgroup, par-ticularly among children, means that treating relapsed HeH patientsis still a clinical challenge. Thus the identification of risk factorswithin this genetic subgroup is a high priority. Over the past two de-cades, several research groups have subdivided this group accordingto gain of specific chromosome(s), modal number and the presenceor absence of structural abnormalities in the search for risk fac-tors.39,41,42,48,49 Unfortunately a consensus has not emerged. TheChildren's Oncology Group (COG) currently use the presence of thetriple trisomy (simultaneous gain of chromosomes 4, 10 and 17) toidentify a subgroup of HeH with a very low risk of relapse.42 Incontrast, we found that trisomy 18 and not triple trisomy to be thebetter indicator of improved survival.23
6. t(1;19)(q23;p13)/TCF3-PBX1
In 90–95% of cases the chromosomal translocation, t(1;19)(q23;p13),results in the fusion of the TCF3 (formerly E2A) gene with the PBX1gene.50 In the remaining 5–10% of cases neither of these genes is in-volved and while a few cases appear to harbour alternative gene fu-sions the subgroup does not appear to be homogeneous.51–53 Thistranslocation is unusual because it can present either as a balancedtranslocation, t(1;19), or unbalanced where just the der(19)t(1;19)is present. The ratio of balanced to unbalanced is approximately50:50.51 The balanced form of the translocation almost always resultsin TCF3-PBX1 fusion and thus most TCF3-PBX1-negative cases presentas der(19)t(1;19).51 TCF3-PBX1-negative cases are also known to coex-ist with high hyperdiploidy and, more rarely, t(9;22)(q34;q11).51 Pa-tients with TCF3-PBX1 usually have a pre-B immunophenotypeexpressing cytoplasmic μ.50 Interestingly it is one of the few genetic
abnormalities which does not appear to vary in frequency with age;being present in 3–5% of cases at all ages, with the exception of infants(Fig. 1). In paediatric ALL, patients with t(1;19) have been reported tohave an adverse outcome. However, these data were largely from theUSA and prior to the use of modern intensive protocols.50 On contem-porary paediatric protocols t(1;19) patients have an intermediate/goodprognosis.23,25,54–56 In adult ALL, there is a great deal of debate overthe prognosis of t(1;19) patients with French and Italian studiesreporting poor outcomes while the UKALLXA and UKALLXII/ECOG2993 trials finding no strong association with outcome.38,57–59
Although some studies have reported a differential outcome be-tween the balanced and unbalanced this is likely to reflect differ-ences between TCF3-PBX1 positive and negative patients.60 Thedifferential outcome in adult ALL is likely to reflect variation in treat-ment protocols and be mirroring historical differences observed inpaediatric cohorts.
Fig. 1. Estimated age-specific frequency of selected chromosomal abnormalities in ALL. The incidence of each abnormality in each age group was calculated using a denominatorthat only included cases appropriately tested by cytogenetic, FISH or MLPA for the abnormality in question. Mutual exclusivity was assumed for the abnormalities listed. A minimumof 80 cases were tested for each abnormality in each age group, with the exception of the infant (b1 year) age group. The total cohort comprised 7113 patients aged as follows: b1 yn=142, 1–4 y n=3128, 5–9 y n=1583, 10–14 y n=915, 15–19 y n=452, 20–24 y 169, 25–39 y n=350, 40–59 y n=374.
0.00
0.25
0.50
0.75
1.00
Pro
port
ion
Sur
vivn
g E
vent
Fre
e
0 2 4 6 8 10 12Years from diagnosis
ETV6
High hyperdiploidy
87%
83%
74%
50%
41%40%
24%
Other abnormalities
MLL translocations
<40 chromosomest(9;22)(q34;q11)
iAMP21
RUNX1
Fig. 2. Event free survival of childhood BCP-ALL by primary genetic subgroup. Data isfrom ALL97 trial and updated from Moorman et al. (2010) (Ref. [22]). Percentagesare event free survival at 7 years after a median follow-up time of 8.2 years.
4 A.V. Moorman / Blood Reviews xxx (2012) xxx–xxx
Please cite this article as: Moorman AV, The clinical relevance of chromosomal and genomic abnormalities in B-cell precursor acute lympho-blastic leukaemia, Blood Rev (2012), doi:10.1016/j.blre.2012.01.001

Introducción
21
Además, en algunos casos la identificación de una alteración genética
específica permite no sólo una mejor estratificación en grupos de riesgo para
guiar la intensidad del tratamiento, sino también el seguimiento de la
respuesta al mismo mediante el estudio de la ERM de forma específica. Por
último, dicho conocimiento se ha traducido en un tratamiento dirigido
contra dianas moleculares específicas en algún subtipo de LLA, como la LLA
con cromosoma Philadelphia y presencia del gen de fusión BCR-‐ABL (LLA
Ph+). Es previsible que en los próximos años puedan diseñarse nuevas
terapias dianas frente a otros subgrupos moleculares.
Dentro de las alteraciones genéticas podemos distinguir unas alteraciones
primarias, que generalmente se excluyen mutuamente y unas alteraciones
secundarias, que se adquieren con posterioridad al evento inicial y que
cooperan en el desarrollo de la leucemogénesis. En las tablas 4 y 5 se
describen algunas de estas alteraciones junto con su repercusión clínica. La
descripción de los subgrupos genéticos en la LLA-‐T se realiza más adelante
(ver apartado de biología en la LLA-‐T). A continuación se destacan algunas de
las alteraciones genéticas recurrentes primarias y secundarias en la LLA B
precursora:
Alteraciones primarias:
Hiperdiploidía alta (51-‐65 cromosomas): constituye el grupo genético más
frecuente de LLA pediátrica con alrededor del 30% de los casos y se asocia a
características clínicas favorables, como cifras bajas de leucocitos, edad
entre 2 y 5 años y buen pronóstico, con supervivencias superiores al 90% con
los protocolos de tratamiento actuales. Suelen tener una buena respuesta al
tratamiento con antimetabolitos y presentan mayor sensibilidad al

Introducción
22
tratamiento con metotrexato, ya que acumulan mayores concentraciones de
metotrexato intracelular en relación con el aumento de dosis del gen
transportador de tetrahidofolatos que se halla en el cromosoma 21 (Kager et
al., 2005). Se caracteriza por la ganancia de cromosomas, que suelen ser +X,
+4, +6, +10, +14, +17, +18, y +21, +21. Dentro de este grupo se ha descrito
que algunas trisomías, como la trisomía 18 o la triple trisomía +4, +10 y +17
(Paulsson et al., 2013) y los casos con mayor número modal parecen tener
mejor pronóstico (Dastugue et al., 2013). En ocasiones se asocian
alteraciones estructurales, que se consideran eventos secundarios (Paulsson
et al., 2010) y cuyo valor pronóstico es controvertido. En tan sólo 1-‐4% de los
casos la hiperdiploidía alta se asocia a traslocaciones recurrentes
características de la LLA como son la t(1;19)(q23;p13), la t(9;22) (q34;q11) ó
la t(12;21)(p13;q22) y el pronóstico en estos casos se considera que viene
dado por la traslocación.
Hipodiploidía (<46 cromosomas): son infrecuentes y se asocian a mal
pronóstico. Se pueden distinguir 3 subgrupos (Moorman, 2012): la
hipodiploidía alta (40-‐45 cromosomas), la hipodiploidía baja (30-‐39
cromosomas) y la casi haploidía (<30 cromosomas). En la mayoría de
protocolos se considera de mal pronóstico y se tratan con protocolos de alto
riesgo cuando tienen menos de 44 cromosomas.
1) Hipodiploidía alta (40-‐45 cromosomas): representa menos del 1% de la
LLA. Puede asociarse a las traslocaciones (t(9;22), t(1;19) o t(12;21)).
2) Hipodiploidía baja (30-‐39 cromosomas): constituye el 3-‐5% de LLA de
adolescentes y adultos y es menos frecuente todavía en niños. Se presenta
con monosomías de los cromosomas 3, 7, 15, 16 y 17. Puede presentar un

Introducción
23
fenómeno de endoduplicación y aparecer clonas con casi triploidía (60-‐78
cromosomas). Se asocia a mal pronóstico, por lo que es importante
distinguirlo de la hiperdiploidía alta.
3) Casi haploidía (<30 cromosomas): se observa en el 1% de LLA en niños,
nunca se ha descrito en lactantes o adultos. Habitualmente se pierden los
cromosomas sexuales (X/Y) y se mantienen los cromosomas 10, 14, 18 y 21.
Es frecuente la endoduplicación del clon primario (falsa hiperdiploidía alta).
Se han descrito una frecuencia elevada de mutaciones y deleciones que
activan la vía RAS. Tanto en este subgrupo como en el de hipodiploidía baja
se han descrito alteraciones que inactivan a los genes IKZF2 (HELIOS) e IKZF3
(AIOLOS), que son muy infrecuentes en otros tipos de LLA (Moorman, 2012;
Mullighan, 2012). Se ha descrito recientemente que los inhibidores de PI3K y
de mTOR inhiben con gran potencia la proliferación de células con
hipodiploidía baja y casi haploides, por lo que podrían investigarse como una
nueva modalidad terapéutica en estos subtipos de leucemia de muy mal
pronóstico (Holmfeldt et al., 2013).
t(12;21)(p13;q22)/ETV6-‐RUNX1 (TEL-‐AML1): representa alrededor de un 25%
de casos de LLA pediátrica. En la mayoría de casos se trata de una
traslocación críptica, que precisa de técnicas de genética molecular (FISH o
PCR) para su diagnóstico. Se ha demostrado su frecuente origen prenatal,
con la presencia de la traslocación en sangre obtenida al nacimiento. En
algunos casos la leucemia se presenta en la adolescencia tras una larga
latencia, lo que sugiere el requerimiento de eventos secundarios para la
leucemogénesis. Los pacientes con esta traslocación presentan una mayor
sensibilidad al tratamiento con asparraginasa y un pronóstico favorable, con
supervivencias de más del 90% con los protocolos terapéuticos actuales. El

Introducción
24
pronóstico de la presencia de alteraciones estructurales secundarias
añadidas es controvertido (Barbany et al., 2012; Enshaei et al., 2013).
t(1;19)(q23;p13)/TCF3-‐PBX1 (E2A-‐PBX1): se halla en el 5% de la LLA
pediátrica y se asocia a fenotipo pre-‐B, aunque en niños no es infrecuente el
fenotipo B común. En este subgrupo de pacientes se demuestra la
importancia de la estratificación en grupos de riesgo y del protocolo de
tratamiento aplicado; se trataba de un subgrupo de niños con mal
pronóstico que ha pasado a tener buen pronóstico gracias a la intensificación
del tratamiento y su exclusión del grupo riesgo bajo, con supervivencias en la
actualidad superiores al 80% (Andersen et al., 2011). Se ha descrito una
mayor frecuencia de recaídas en SNC en estos pacientes (Pui et al., 2009),
aunque este hecho no se confirma en otra serie de 49 pacientes con esta
traslocación (Andersen et al., 2011).
t(17;19)(q22;p13)/TCF3-‐HLF: se trata de una alteración muy infrecuente
(<1%) en la LLA pediátrica. TCF3-‐HLF regula genes que controlan la muerte
celular en los progenitores linfoides, incluyendo LMO2 y BCL2 (Mullighan,
2012). Este tipo de leucemia se asocia a mayor edad, a coagulación
intravascular diseminada y a hipercalcemia y constituye un subgrupo de muy
mal pronóstico. En una revisión reciente se analizaron 21 casos publicados y
todos los pacientes, salvo uno con poco seguimiento, fallecieron,
mayoritariamente tras recaída (Minson et al., 2013).
t(9;22)(q34;q11)/BCR-‐ABL (LLA Ph+): se observa en el 3-‐5% de la LLA
pediátrica y es mucho más frecuente en el adulto. Este subtipo de LLA se
describe con más detalle en el capítulo 2.

Introducción
25
t(4;11)(q21;q23) y otros reordenamientos del gen MLL: el gen MLL puede
reordenarse con más de 100 genes y se presenta como LLA, LMA o con
marcadores de diferentes líneas (mixed phenotype acute leukemia, MPAL)
(Meyer et al., 2013). Los reordenamientos del gen MLL se observan en
leucemias de novo y en leucemias secundarias a tratamientos con
inhibidores de topoisomerasa-‐II. En la LLA pediátrica representa el 5% de los
casos, aunque en los lactantes su frecuencia supera el 80%. Las
traslocaciones más frecuentes en LLA son la t(4;11) y la
t(11;19)(q23;p13.3)/MLL-‐ENL. El fenotipo suele ser pro-‐B y con frecuencia
sobreexpresan FLT3. Se asocian a un pronóstico muy desfavorable en
lactantes, en especial cuando son menores de 6 meses, aunque el pronóstico
de las diferentes traslocaciones en otros grupos de edad está menos
definido (Pui et al., 2003; Emerenciano et al., 2013).
Amplificación intracromosómica del cromosoma 21 (iAMP21): se observa en
un 3-‐5% de la LLA pediátrica, con una mediana de edad de 9 años y una cifra
de leucocitos baja al diagnóstico. Se diagnostica por la presencia de un
cromosoma 21 anómalo con ≥3 señales extras del gen RUNX1 en los estudios
de FISH. La iAMP21 se caracteriza por la amplificación de 5.1 a 24 Mb de una
región del cromosoma 21, que incluye RUNX1 entre otros genes (Moorman,
2012). Se asocia a un mayor riesgo de recaídas y a un mal pronóstico si no se
trata con protocolos intensivos. La respuesta al tratamiento mediante
determinación de la ERM parece discriminar aquellos pacientes con menor
riesgo de recaída (Attarbaschi et al., 2008).
Sobreexpresión del gen CRLF2: se presenta en el 5-‐15% de la LLA pediátrica y
en más de la mitad de los casos de LLA en pacientes con síndrome de Down
(Mullighan et al., 2009; Moorman et al., 2012; Harvey et al., 2010). También

Introducción
26
se ha visto con mayor frecuencia en los pacientes de origen hispano-‐
americano. La sobreexpresión de CRLF2 se produce por tres mecanismos: su
reordenamiento con el gen de la cadena pesada de las inmunoglobulinas
(IGH-‐CRLF2); el gen de fusión P2RY8-‐CRLF2, que se produce por una deleción
intersticial en la región pseudo-‐autosómica de los cromosomas X o Y o, más
raramente, por la mutación puntual F232C en el gen CRLF2. Este subgrupo
de LLA activa la vía JAK-‐STAT y se asocia a mutaciones de JAK2 en el 50% de
casos, a deleciones de IKZF1 y a una peor evolución en adultos, aunque el
pronóstico en niños es más controvertido y puede depender del protocolo
terapéutico aplicado (Harvey et al., 2010) (Attarbaschi et al., 2012; Palmi et
al., 2013) (van der Veer et al., 2013).
Reordenamiento con IGH: además de las traslocaciones de C-‐MYC en las
leucemias B maduras tipo Burkitt y linfomas, las traslocaciones con el gen de
la cadena pesada de las inmunoglobulinas (IGH@) se han descrito
recientemente como un subgrupo clínicamente relevante en la LLA-‐B
precursora. Se observan en el 5% de LLA en adolescentes y adultos jóvenes y
en algunos casos se asocian a mal pronóstico (Moorman et al., 2012). Entre
ellas se hallan la t(6;14)(p22.3;q32) con reordenamiento ID4-‐IGH, que se
asocia a buena respuesta al tratamiento (Russell et al., 2008), o la
t(5;14)(q23-‐q31;q32), en la que IGH se reordena con IL3 y se asocia a
eosinofilia (Meeker et al., 1990). Otros genes traslocados con IGH son PAX5,
ETV6 (TEL), EPOR y CEPBA (Pui, 2012).

Introducción
27
Tabla 4. Eventos genéticos primarios en la leucemia linfoblástica aguda de línea B.
Adaptada de Moorman, Blood Reviews 2012 (Moorman, 2012)
Alteración citogenética/molecular Genes implicados
Características demográficas y clínicas Pronóstico
t(12;21)(p12;q22)* ETV6-‐RUNX1 (TEL-‐AML1) 2% LLA adultos/ 25% LLA
pediátrica Favorable
Hiperdiploidía (51-‐65 cromosomas)* -‐
7% LLA adultos/ 25-‐30% LLA pediátrica Favorable
Hipodiploidía (<45 cromosomas)* -‐
2% LLA adultos/ 1% LLA pediátrica Desfavorable
Baja Hipodiploidía/ casi triploidía (30-‐39/60-‐78 cromosomas)
-‐ 3-‐5% adolescentes y
jóvenes Muy desfavorable
Casi haploidía -‐ 1% LLA pediátrica Muy desfavorable
t(1;19)(q23;p13)* TCF3-‐PBX1 (E2A-‐PBX1) 3% LLA adultos/ 5% LLA pediátrica
Intermedio
t(17;19)(q22;p13) TCF3-‐HLF (E2A-‐HLF) 0,1% LLA pediátrica Muy desfavorable
t(9;22)(q34;q11)* BCR-‐ABL 25-‐30% LLA adultos/ 2-‐
5% LLA pediátrica Desfavorable
t(4,11)(q21;q23)* MLL-‐AFF1 (MLL-‐AF4) 5-‐10% LLA adultos y LLA
pediátrica 40-‐60% LLA lactantes
Muy desfavorable
Otras traslocaciones de MLL
6q27 (MLLT4/AF6), 9p21 (MLLT3/AF9), 10p12
(MLLT10/AF10), 1p32(EPS15)
5-‐10% LLA adultos y LLA pediátrica
40% LLA lactantes
Desfavorable (muy desfavorable en
lactantes)
t(5;14)(q31;q32)* IL3-‐IGH <1% LLA adultos y niños Indeterminado
Otras traslocaciones con IGH
CEBPA, CEBPG, CEBPB, CEBPE, CEBPD
BCL9, LHX3, ID4, TCRG,BCL1, IGF2BP1, EPOR
Adolescentes y adultos jóvenes Desfavorable
iAMP21 Amplificación 5.1Mb en 21q22.11-‐
21q22.12 (incluye RUNX1) y deleción <1Mb zona telomérica
3-‐5% niños 7-‐13 años Desfavorable si
tratamiento de riesgo estándar
Sobreexpresión de CRLF2
t(X;14)(p22;q32)/ t(Y;14)(p11;q32) IGH-‐CRLF2
del(X)(p22.33p22.33)/del(Y)(p11.32p11.32) P2RY8-‐CRLF2 /mut
CRLF2-‐F232C
5-‐15% LLA >50% LLA asociada a S. de Down
Desfavorable en adultos
(controvertido en niños)
* Subtipos reconocidos por la clasificación WHO 2008.

Introducción
28
Alteraciones secundarias:
Para el desarrollo de la leucemia se requieren unas alteraciones o eventos
primarios a la que se añaden lesiones genéticas y epigenéticas que
constituyen eventos secundarios. Los eventos secundarios no son exclusivos
de un subtipo de LLA y, aunque algunos se asocian con más frecuencia a un
tipo de alteración primaria, los podemos encontrar en diversos subgrupos
genéticos. El conocimiento de estas lesiones se ha visto facilitado por las
tecnologías de análisis genómico masivo. En un estudio realizado por
Mullighan y cols. se analizó el número de copias génicas (amplificaciones y
deleciones, copy number alterations, CNA) en muestras de pacientes
pediátricos con LLA y se observó la presencia de entre 6 y 8 CNA de media
por caso. En el caso de las LLA con reordenamiento de MLL el número de
CNA era mucho menor, lo que sugería que el gen MLL es un oncogén muy
potente que requiere pocos eventos secundarios para el desarrollo de la
leucemia. Este hecho podría explicar la corta latencia de las LLA del lactante
en las que mayoritariamente se halla el reordenamiento MLL. En la mayoría
de casos de LLA, las CNA se dieron en genes implicados en la maduración
linfoide B (PAX5, IKZF1, EBF, LEF1 y otros) o en genes reguladores del ciclo
celular y supresores de tumores (CDKN2A/B, PTEN, RB1) y genes de factores
de transcripción (ETV6, ERG y otros). En la tabla 5 se describen estas
alteraciones y su implicación clínica.
CDKN2A/B
En el locus CDKN2A/B, en 9p21, se encuentran genes supresores de tumores
que codifican las proteínas p15INK4B, p16INK4A y p14ARF, implicadas en la
estabilización de p53. Se ha observado la deleción de CDKN2A y CDKN2B en
alrededor del 40% de la LLA pediátrica, tanto en LLA B precursora como en la

Introducción
29
LLA-‐T (Pui, 2012). Se asocian a peor pronóstico en la LLA Ph+ (Iacobucci et
al., 2011), pero su pronóstico en otros tipos de LLA es controvertido
(Iacobucci et al., 2012).
IKZF1 (Ikaros)
Ikaros es un factor de transcripción importante desde las fases precoces del
desarrollo de los progenitores linfoides. Se codifica en el gen IKZF1,
localizado en el brazo corto del cromosoma 7 (7p12). Las alteraciones de
Ikaros se observaron inicialmente en el 80% de pacientes con LLA Ph+ y en el
66% de casos de crisis blástica linfoide de la leucemia mieloide crónica (LMC)
(Mullighan et al., 2008) y posteriormente se detectaron en el 15% de LLA-‐B
precursora pediátrica (Kuiper et al., 2010) (Palmi et al., 2013). Las
alteraciones en IKZF1 en la LLA de línea B tienen un valor pronóstico
desfavorable, aunque su aplicación en la estratificación de los pacientes en
protocolos asistenciales es controvertida (Waanders et al., 2011) (Palmi et
al., 2013).
PAX5
PAX5 es un gen localizado en el cromosoma 9 (9p13.2) que codifica un factor
de transcripción implicado en la diferenciación linfoide normal. En alrededor
de un 30% de niños y adultos con LLA se han identificado deleciones,
mutaciones o traslocaciones de PAX5. Entre las alteraciones que implican al
gen PAX5 se halla la dic(9;20)(p13.2;q11.2). Estos casos con frecuencia se
asocian a leucocitosis e infiltración del SNC, pero tienen buen pronóstico. A
menudo se observa en el cariotipo una monosomía del cromosoma 20 y la
alteración genética debe confirmarse por FISH. Por lo general, las
alteraciones de PAX5 no influyen en la evolución de los pacientes pediátricos
y tampoco en la de los paciente adultos (Pui, 2012) (Iacobucci et al., 2012).

Introducción
30
ETV6 (TEL)
Alrededor de un 70% de los casos de LLA con ETV6-‐RUNX1 tienen como
alteración secundaria la deleción del alelo no reordenado de ETV6, que
también puede observarse en otros subtipos de leucemias (Moorman, 2012).
JAK1/2
En un 20% de niños con síndrome de Down y en alrededor de un 10% de
niños mayores y adolescentes sin este síndrome, se hallan mutaciones en
JAK1 (1p32), JAK2 (9p24) o JAK3 (19p13). Las mutaciones que afectan a JAK2
se producen con frecuencia en la posición R683 y nunca en V617, a
diferencia de lo que ocurre en los síndromes mieloproliferativos crónicos.
Las mutaciones de JAK2 se asocian a sobreexpresión de CRLF2 y ambas
cooperan en la leucemogénesis (Mullighan et al., 2009). Estas mutaciones
también son frecuentes en la LLA BCR-‐ABL like (ver abajo) y se asocian a peor
pronóstico (Mullighan et al., 2009).
CREBBP
CREBBP es un gen que codifica una proteína con actividad histona acetil-‐
transferasa. Se han descrito alteraciones de CREBBP en la LMA (Camós et al.,
2006), en linfomas y en el 19% de LLA-‐B hiperdiploides en recaída (Mullighan
et al., 2011; Inthal et al., 2012).
LLA-‐B precursora BCR-‐ABL-‐ like
Mediante las técnicas de estudios de expresión génica de alta densidad
(microarrays), se ha identificado recientemente un subgrupo de LLA que
presenta un perfil de expresión génica muy similar al de las LLA Ph+, a pesar
de carecer del reordenamiento BCR-‐ABL (ver figura 7), y que posee una
activación aberrante de la vía de señalización de tirosín-‐quinasas (Den Boer

Introducción
31
et al., 2009). Este subgrupo de LLA, denominado BCR-‐ABL like, representa
alrededor del 15% de las LLA-‐B precursoras pediátricas. Se asocia con
frecuencia a alteraciones de mal pronóstico como son deleciones de IKZF1,
sobreexpresión de CRLF2, mutaciones de JAK1/2 o reordenamientos de ABL,
PDGFRB, EPOR o ILR7. Algunas de las alteraciones genéticas subyacentes
podrían ser dianas terapéuticas (imatinib u otros inhibidores de tirosín-‐
quinasas (TKI) en el caso de alteraciones en ABL o PDGFR, inhibidores de
JAK2, etc.).
TP53
P53 es una proteína que juega un papel central en la regulación del ciclo
celular y la apoptosis tras el daño producido en el DNA. Su papel en el cáncer
es bien conocido, tanto en tumores sólidos como en neoplasias
hematológicas como la LMA y la leucemia linfática crónica, en los que la
pérdida de su regulación confiere mal pronóstico. En la LLA pediátrica la
incidencia de mutaciones o deleciones en TP53 es baja en el momento del
diagnóstico, pero aumenta en las recaídas y se asocia a mal pronóstico (Hof
et al., 2011). En la LLA del adulto tiene una incidencia al diagnóstico del 14%
y se asocia a mala respuesta al tratamiento de inducción (Chiaretti et al.,
2013).
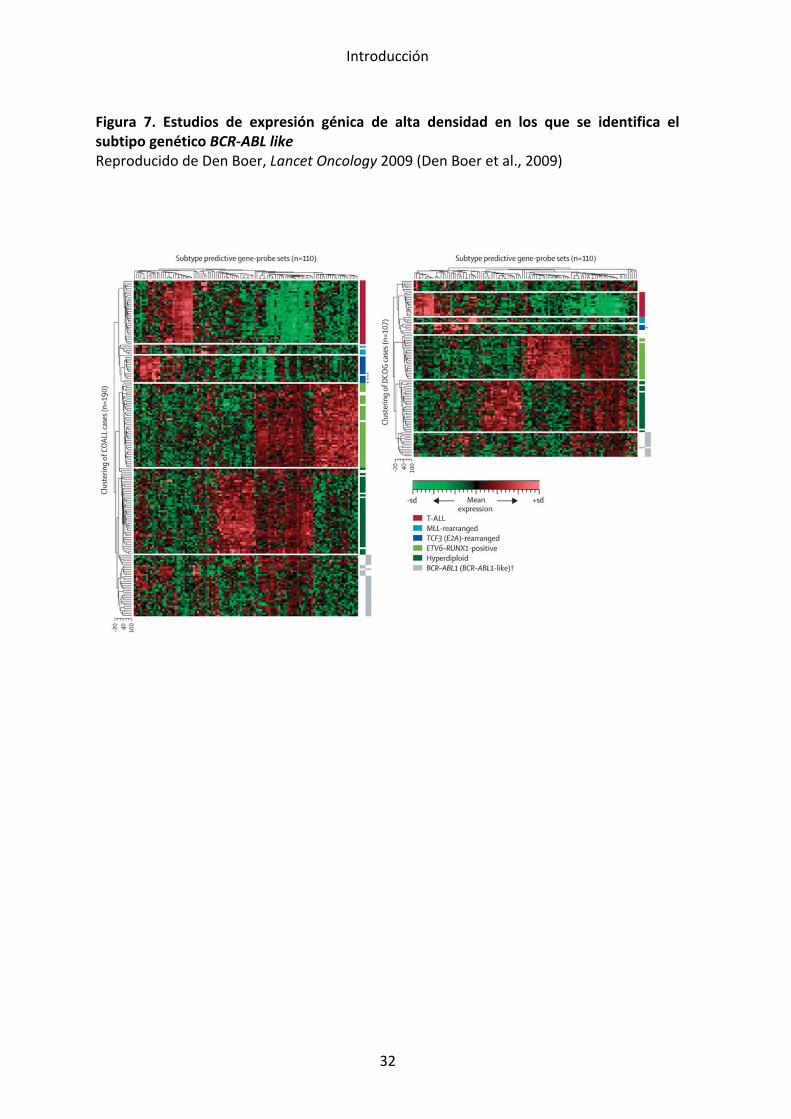
Introducción
32
Figura 7. Estudios de expresión génica de alta densidad en los que se identifica el subtipo genético BCR-‐ABL like Reproducido de Den Boer, Lancet Oncology 2009 (Den Boer et al., 2009)

Introducción
33
Tabla 5. Eventos genéticos secundarios en la leucemia linfoblástica aguda de línea B. Adaptado de Iacobucci, Curr Hematol Malig Rep (2012) (Iacobucci et al., 2012)
Gen Alteración Características demográficas
y clínicas Pronóstico
CDKN2A/B Deleciones 30% LLA adultos y niños / 47%
de LLA Ph+ recaída
Desfavorable en LLA Ph+ / controvertido en otros
subtipos
IKZF1 (Ikaros) Deleciones / mutaciones 80% LLA Ph+ / 40% LLA BCR-‐
ABL like / 15% LLA pediátrica
Desfavorable
PAX5 Deleciones, mutaciones, traslocaciones 30% LLA adultos y niños No asociación con
pronóstico
ETV6 Deleciones 60% LLA ETV6-‐RUNX1+ / 10%
del resto de LLA-‐B Controvertido
JAK1/2 Mutaciones Hasta 50% LLA BCR-‐ABL like / >30% LLA síndrome de Down
Desfavorable
CREBBP Mutaciones 19% LLA pediátrica recaída Desfavorable
TP53 Deleciones, mutaciones 12% LLA Desfavorable
CRLF2 Sobreexpresión por traslocación, deleción intersticial o mutación
16% LLA adultos y niños, >50% LLA en síndrome de Down
Desfavorable

Introducción
34
Tratamiento
Contribución de los grupos cooperativos
Una de las claves de los avances en la curación de la LLA ha residido en el
tratamiento de los pacientes por instituciones o grupos cooperativos que
aplican protocolos de tratamiento en el contexto de ensayos clínicos. Ello ha
permitido identificar factores pronósticos y adaptar la intensidad del
tratamiento según el grupo de riesgo, así como aplicar en los protocolos
sucesivos las mejores combinaciones y formas de administración de los
quimioterápicos. Así, se ha conseguido pasar de una supervivencia inferior al
10% a principios de la década de los años 60 al 85-‐90% de curación en la
actualidad y ello con quimioterápicos en su mayoría conocidos desde hace
más de 40 años (ver figuras 8 y 9) (Pui y Evans, 2006) (Pui et al., 2011).
Otra de las contribuciones importantes en el tratamiento de la LLA que han
realizado los distintos grupos cooperativos ha sido la modulación de la
intensidad de la quimioterapia según la respuesta precoz al tratamiento,
evaluada en sangre periférica tras 1 semana de prednisona o en médula ósea
en el día +8 o +15 (Nachman et al., 1997). Posteriormente, distintos grupos
cooperativos mostraron la importancia de la ERM como factor pronóstico
(Cavé et al., 1998; van Dongen et al., 1998) (Coustan-‐Smith et al., 1998)
(Möricke et al., 2008) (Vidriales et al., 2003) (Schrappe et al., 2011) y en la
actualidad es uno de los principales factores que determinan el tratamiento
de los pacientes en la mayoría de los protocolos terapéuticos.

Introducción
35
Figura 8. Supervivencia global de pacientes pediátricos con LLA según el año de tratamiento en la institución SJCRH. Reproducido de Pui y Evans, New England Journal of Medicine 2006 (Pui y Evans, 2006)
Figura 9. Supervivencia global de pacientes pediátricos con LLA según el año de tratamiento obtenida con los protocolos sucesivos del grupo cooperativo SHOP. Cortesía de la Dra. Badell, en representación del grupo de LLA de la SEHOP.
Una de las principales instituciones es St Jude Children’s Research Hospital
(SJCRH), pionera en la investigación y el tratamiento de la leucemia
pediátrica, que se fundó en 1962 con el fin de investigar en la leucemia y su
curación. Una de sus grandes contribuciones fue la demostración de que la
drug ther apy
n engl j med 354;2 www.nejm.org january 12, 2006 169
associated with a reduced risk of relapse.45 A tan-dem-repeat polymorphism within the enhancer region of the thymidylate synthase gene, one of the major targets of methotrexate, has been linked to increased expression of the enzyme and an in-creased risk of relapse.45,46 However, the prognos-tic importance of individual pharmacogenetic variables is also influenced by the treatment.47,48 Moreover, multiple genetic polymorphisms prob-ably interact to influence the treatment response so that polygenic pharmacogenetic models may be necessary to predict treatment outcome reli-ably.45 Finally, the acquisition of additional chro-mosomes in leukemia cells can create discordance between germ-line genotypes and leukemia-cell phenotypes, including pharmacogenomics.49
global gene-expression patternsGlobal gene-expression profiling has the potential to reveal new dimensions of the pathologic fea-tures of ALL and to identify novel therapeutic targets. Several studies have revealed distinct gene-expression patterns in specific subtypes of leukemia.37,50-52 In TEL-AML1–positive ALL, the erythropoietin receptor was found to be highly expressed in leukemia cells, potentially affecting proliferation and survival signals.51 In T-cell ALL, the levels of expression of several known T-cell oncogenes were related to the expression of many genes that define discrete stages of T-cell differ-entiation.37,53 Cases in the HOX11+ cluster showed an increased expression of genes that promote pro-liferation and a lack of expression of antiapoptotic genes, which may explain the better response of these patients to therapy.37 In T-cell ALL in adults, TTK, a gene with kinase activity that is overex-pressed in proliferating cells, was found to have decreased expression in the blast cells of patients whose disease ultimately relapsed.53 Consistent with the idea that impaired cell proliferation may be a mechanism for resistance to treatment, ex-pression of this gene was also reduced among children with B-cell precursor ALL who had poor-er responses to remission-induction therapy.54 Gene-expression profiling that was performed in patients with rearrangements of the MLL gene iden-tified two subgroups of patients with prognostic significance that was independent of the gene-fusion partner and of clinical factors.55 The iden-tification of high levels of FLT3 expression in cases with MLL rearrangement or hyperdiploidy 50,56 has led to the development of FLT3 inhibitors.57
The gene-expression profiles of leukemia cells have also been used to identify genes related to the intracellular disposition of antileukemia agents in vivo58-60 and to reveal distinct sets of genes associated with resistance to each of four struc-turally different and widely used medications (prednisolone, vincristine, asparaginase, and dau-norubicin).61 The independent prognostic signifi-cance of the resistance patterns was validated in a separate cohort of patients treated with the same agents at a different institution.61 A sub-
Prob
abili
ty o
f Eve
nt-fr
ee S
urvi
val (
%)
80
90
70
60
40
30
10
50
20
00 5 10 15 20 25 30 35 40 45
Years after Diagnosis
Study 15, 2000–2005 (N=274)
Studies 13A, 13B, and 14, 1991–1999 (N=465) Studies 11 and 12, 1984–1991 (N=546)
Study 10, 1979–1983 (N=428)
Studies 5 to 9, 1967–1979 (N=825)
Studies 1 to 4, 1962–1966 (N=90)
Study 15, 2000–2005 (N=274)
Studies 13A, 13B, and 14, 1991–1999 (N=465)
Studies 11 and 12, 1984–1991 (N=546)
Study 10, 1979–1983 (N=428)
Studies 5 to 9, 1967–1979 (N=825)
Studies 1 to 4, 1962–1966 (N=90)
100
Prob
abili
ty o
f Ove
rall
Surv
ival
(%)
80
90
70
60
40
30
10
50
20
00 5 10 15 20 25 30 35 40 45
Years after Diagnosis
96±384±2
81±2
74±2
48±2
21±4
100
79±2
70±2
92±4
53±2
9±3
36±2
A
B
Figure 1. Kaplan–Meier Analyses of Event-free Survival (Panel A) and Over-all Survival (Panel B) in 2628 Children with Newly Diagnosed ALL.
The patients participated in 15 consecutive studies conducted at St. Jude Children’s Research Hospital from 1962 to 2005. The five-year event-free and overall survival estimates (±SE) are shown, except for Study 15, for which preliminary results at four years are provided. The results demon-strate steady improvement in clinical outcome over the past four decades. The difference in event-free and overall survival rates has narrowed in the recent periods, suggesting that relapses or second cancers that occur after contemporary therapy are more refractory to treatment.
Copyright © 2006 Massachusetts Medical Society. All rights reserved. Downloaded from www.nejm.org at HOSP SANT JOAN DE DEU on February 20, 2006 .

Introducción
36
profilaxis del SNC, inicialmente mediante irradiación cráneo-‐espinal, evita un
gran número de recaídas y aumenta la curación. También estableció cuatro
componentes del tratamiento de la LLA que aún se utilizan en la actualidad:
inducción a la remisión, tratamiento profiláctico del SNC, intensificación
post-‐inducción o consolidación y tratamiento de mantenimiento (Pui, 2012).
Entre los principales grupos cooperativos se halla el grupo alemán BFM
(Berlin-‐Frankfurt-‐Münster), que empezó a aplicar protocolos de tratamiento
a principios de la década de los 80 y que posteriormente se expandió e
incluyó a otros países europeos (Austria, Italia, Suiza) y también a países con
menos recursos como Chile y Argentina, en los que se adaptó la intensidad
del tratamiento a las infraestructuras y recursos disponibles. En la
actualidad, el grupo internacional I-‐BFM está formado por la mayoría de los
países europeos, entre los que se incluye España desde el año 2011 y
algunos países latinoamericanos y asiáticos. Muchos de los países utilizan sus
propios protocolos terapéuticos pero contribuyen al grupo mediante una
estrecha colaboración entre los grupos clínicos y biológicos de los distintos
países. Ello ha permitido un gran avance en el conocimiento de la biología de
la LLA y en los resultados de los tratamientos aplicados.
Con los distintos protocolos BFM se ha obtenido una mejoría en la
supervivencia y se han podido realizar cambios destinados a la reducción de
la toxicidad a corto y a largo plazo, gracias a una progresiva mejora en la
selección y estratificación de pacientes según el riesgo de recaída (Schrappe
et al., 2013). Los principales hallazgos de estos protocolos han sido: 1) es
seguro omitir la radioterapia craneal en los niños con bajo riesgo de recaída
en SNC y reducir la dosis de radioterapia en aquéllos con más riesgo si se
sustituye por tratamiento intratecal intensivo y dosis altas de metotrexato;

Introducción
37
2) la reinducción tardía es necesaria incluso en los pacientes de bajo riesgo,
porque su omisión resultó en un empeoramiento en la supervivencia de
estos pacientes; 3) no se puede acortar el tratamiento de mantenimiento a
menos de 2 años, porque la reducción en 6 meses causó un aumento de
recaídas sistémicas; 4) la respuesta lenta a una semana de tratamiento con
prednisona es un factor pronóstico adverso con gran valor que ayuda en la
estratificación de los pacientes en grupos de riesgo; 5) la dosis de
daunorrubicina en inducción puede reducirse sin empeorar los resultados en
los pacientes de bajo riesgo; 6) la consolidación e intensificación del
tratamiento ha conseguido mejorar de forma importante la supervivencia en
los pacientes de alto riesgo y 7) el estudio de la ERM permite refinar la
estratificación de los pacientes y mejorar los resultados en algunos
subgrupos de alto riesgo así como disminuir la intensidad de tratamiento en
otros (Schrappe et al., 2013).
El grupo cooperativo del Dana Farber Cancer Institute (DCFI) mostró ya a
principios de la década de los 90 que los niños con LLA-‐T podían alcanzar una
supervivencia similar a los de fenotipo B con un tratamiento intensivo y
prolongado con asparraginasa.
Otros grupos cooperativos norteamericanos (Children Oncology Group,
formado por los dos grupos Children Cancer Group y Pediatric Oncology
Group) y europeos han colaborado también en las mejoras en el tratamiento
de la LLA.
En España, recientemente se han fusionado los dos principales grupos
cooperativos para el tratamiento de la LLA pediátrica: el grupo SHOP, de la
Sociedad Española de Hemato-‐Oncología Pediátrica y el grupo PETHEMA
(Protocolo para el Estudio y Tratamiento de las Hemopatías Malignas).

Introducción
38
En cuanto al grupo SHOP, las principales conclusiones que se obtuvieron en
sus protocolos sucesivos fueron: 1) la quimioterapia de consolidación tardía
en pacientes de alto riesgo no ofrecía ningún beneficio (SHOP/LLA-‐89) y 2)
con la intensificación del protocolo, la ampliación del grupo de pacientes de
alto riesgo y la creación de un grupo de muy alto riesgo se consiguió una
mejoría significativa de la supervivencia libre de evento o SLE (SHOP/LLA-‐94)
(Badell et al., 2008). En el protocolo SHOP/LLA-‐99 se estableció como
objetivo principal mejorar la supervivencia en el grupo de pacientes con LLA-‐
T, mediante la intensificación del tratamiento con asparraginasa y
ciclofosfamida. En el protocolo SHOP/LLA-‐2005 se intensificó el tratamiento
aumentando la dosis de metotrexato sistémico de 3 a 5 g/m2 y se incorporó
la determinación de la ERM para la estratificación de los pacientes. Los
resultados obtenidos en dos subgrupos de pacientes de alto y muy alto
riesgo en estos dos protocolos se analizan en el presente trabajo de tesis.
Respecto al grupo PETHEMA, los resultados del protocolo PETHEMA LAL-‐
2001-‐BR fueron muy favorables en el grupo de pacientes de bajo riesgo y se
mostró la seguridad de la reducción de dosis de antraciclínicos en estos
pacientes. En el protocolo PETHEMA LAL 96-‐RI para pacientes de riesgo
intermedio se intensificó la inducción con ciclofosfamida durante la primera
semana de tratamiento y se mostró que la omisión de radioterapia fue
segura, con una SLE a los 5 años del 80%. El protocolo PETHEMA LAL-‐2005-‐
AR para niños de alto riesgo tuvo entre sus objetivos disminuir las
indicaciones del trasplante alogénico y excluyó del trasplante a aquellos
pacientes con buena respuesta evaluada por el estudio de ERM.

Introducción
39
Tratamiento de primera línea de la LLA
Todos los protocolos actuales de tratamiento de la LLA siguen los mismos
principios básicos y constan de las siguientes fases de tratamiento:
inducción, consolidación o intensificación, profilaxis del SNC y
mantenimiento. No obstante, existen algunas diferencias en la selección de
ciertos fármacos, en la estratificación de los pacientes y en la intensidad o en
los esquemas de administración y duración de las fases de tratamiento.
Inducción a la remisión:
Tiene como objetivo la máxima reducción posible de la carga tumoral con
obtención de la remisión completa (RC). Esta fase consta de la combinación
de glucocorticoides (prednisona o dexametasona) y vincristina y al menos un
tercer fármaco (asparraginasa, un antraciclínico o ambos). En algunos
protocolos se omiten o se reducen las dosis de los antraciclínicos en los
pacientes de bajo riesgo. En cuanto a la elección del glucocorticoide en la
inducción, la dexametasona podría tener mayor potencia antileucémica y
proteger mejor frente a las recaídas en SNC, pero se ha asociado a mayor
toxicidad (alteraciones neuro-‐psiquiátricas, mayor incidencia de sepsis e
infecciones fúngicas). Se ha descrito una mayor mortalidad en inducción por
infecciones en los pacientes tratados con dexametasona, en especial en
niños mayores de 10 años. En algún estudio se ha mostrado mayor
supervivencia con dexametasona (Mitchell et al., 2005), pero esta ventaja se
pierde si se aumenta el ratio de conversión entre la prednisona y
dexametasona (Igarashi et al., 2005) (Pui, 2012). En cuanto a la asparraginasa
empleada, existen diversas formulaciones (nativa de E.coli, la obtenida de
Erwinia y la pegilada de E. coli) que difieren en su vida media, en la
inmunogenicidad y en su coste. Hasta la fecha no se han demostrado

Introducción
40
diferencias en la supervivencia cuando se utilizan a dosis equipotentes; sin
embargo, en la actualidad la mayoría de los protocolos en países con más
recursos utilizan la forma pegilada por su mayor comodidad de
administración (vida media mucho mayor) y por la menor frecuencia de
reacciones inmunoalérgicas (Avramis et al., 2002; Rizzari et al., 2013).
Consolidación o intensificación
Tiene como objetivo la eliminación de la enfermedad residual. Si bien es una
fase de importancia indiscutible, no existe consenso sobre la mejor
combinación de fármacos y su duración. Uno de los esquemas más utilizados
consiste en la denominada reinducción, que consiste en la aplicación de los
mismos fármacos o similares a los utilizados en inducción, alrededor de tres
meses tras la remisión. En la mayoría de protocolos, durante esta fase se
intensifica el tratamiento a los pacientes con respuesta lenta durante la
inducción (Nachman et al., 1997). Algunos protocolos utilizan más de una vez
este esquema en los pacientes de riesgo intermedio o alto (Lange et al.,
2002). En esta fase se utilizan además otros fármacos no utilizados en la
inducción, como metotrexato a dosis altas o mercaptopurina, entre otros. El
uso de diversos esquemas de intensificación ha permitido identificar algunos
tratamientos eficaces en distintos subgrupos de LLA. De esta forma, el uso
intensivo de asparraginasa utilizado en los protocolos de DCFI permitió
identificar la importancia de este fármaco en la LLA-‐T y en la LLA con
reordenamiento ETV6-‐RUNX1 (TEL-‐AML1) (Moghrabi et al., 2007) (Pui et al.,
2009; Loh et al., 2006; Silverman et al., 2010).
Profilaxis del SNC:
Tiene como finalidad evitar las recaídas, mediante la eliminación de las
células leucémicas que puedan infiltrar el SNC y a las que no se puede

Introducción
41
acceder mediante el tratamiento sistémico por la mala penetración de
muchos de los citostáticos en SNC. La primera de las estrategias utilizadas
fue la irradiación cráneo-‐espinal, que constituyó uno de los grandes avances
en la curación de estos niños. La radioterapia es muy eficaz pero se asocia a
secuelas graves y se ha ido sustituyendo progresivamente por el tratamiento
precoz e intensivo con quimioterapia intratecal y sistémica con fármacos que
atraviesan la barrera hemato-‐encefálica (Richards et al., 2013), como
dexametasona o metotrexato a dosis altas. Algunos protocolos han
conseguido obtener muy buenos resultados aun con la eliminación de la
radioterapia craneal, incluso en pacientes con infiltración del SNC al
diagnóstico (Pui et al., 2009; Veerman et al., 2009).
Mantenimiento:
Es una fase del tratamiento prolongada (entre 2 y 3 años tras el diagnóstico)
con citostáticos a dosis bajas y que tiene como objetivo la reducción de las
recaídas tras el tratamiento de intensificación. Los intentos en el pasado de
reducción de la duración de esta fase resultaron en un aumento de las
recaídas (Childhood ALL Collaborative Group, 1996), con la excepción de los
pacientes con LLA-‐B madura (tipo Burkitt) que siguen protocolos específicos.
Los principales citostáticos utilizados en el mantenimiento son
antimetabolitos: mercaptopurina diaria oral y metotrexato a dosis bajas
semanal, administrado de forma oral o intramuscular. En muchos protocolos
se intensifica el tratamiento de mantenimiento con la administración de
forma intermitente de prednisona o dexametasona y vincristina. La utilidad
de esta intensificación es controvertida y sólo en algunos protocolos de
tratamiento se ha demostrado su eficacia (De Moerloose et al., 2010)

Introducción
42
(Conter et al., 2007), por estar probablemente condicionada al tratamiento
recibido en las fases previas al mantenimiento (Eden et al., 2010).
La mejoría progresiva en los resultados obtenidos en estos pacientes se debe
también en gran medida al tratamiento de soporte, como las transfusiones y
la profilaxis y tratamiento de las infecciones. La transfusión de plaquetas que
pudo utilizarse de forma rutinaria a principios de los 70 y la profilaxis frente
a P. jirovecii (P. carinii) constituyen dos ejemplos importantes en la
disminución de la mortalidad de estos pacientes (Hughes et al., 1977)
(Blajchman, 2008).
Trasplante alogénico de progenitores hemopoyéticos (TPH) en primera RC
El TPH en primera RC se indica en aquellos pacientes de muy alto riesgo de
recaída en los que las probabilidades de curación sólo con quimioterapia son
muy bajas (Fagioli et al., 2013). El trasplante aumenta la intensidad del
tratamiento mediante la administración de un acondicionamiento con
quimioterapia a dosis altas (con un posible efecto dosis/respuesta) asociado
frecuentemente a radioterapia (irradiación corporal total). Además, el
trasplante aporta como mecanismo antitumoral el efecto del injerto contra
la leucemia. Ello se traduce en una reducción del riesgo de recidiva, aunque
se asocia a una mayor morbi-‐mortalidad. Las indicaciones del TPH deben
revisarse periódicamente, ya que tanto la quimioterapia como el TPH han
mejorado sus resultados. A la hora de escoger el tratamiento idóneo se
deben tener en cuenta no sólo la supervivencia sino también las secuelas,
que son mayores con el trasplante. En la actualidad, los pacientes pediátricos
con LLA en primera RC con indicación de TPH son aquéllos con fallo de
inducción (Schrappe et al., 2012), con ERM >1% tras la inducción o ERM

Introducción
43
persistente tras la consolidación. Existen otras indicaciones en las que hay
menos consenso, como son la hipodiploidía, la presencia de reordenamiento
del gen MLL, en especial la t(4;11) o la LLA Ph+ (Oliansky et al., 2012). Puesto
que en la última década, desde la disponibilidad de métodos moleculares de
tipaje HLA de alta resolución que aseguran una mejor elección de los
donantes no emparentados, los resultados con donante familiar y no
emparentado con alto nivel de compatibilidad son similares, las indicaciones
de trasplante son las mismas tanto si el donante HLA-‐idéntico es un hermano
como si es un donante no familiar. La fuente de progenitores hemopoyéticos
preferida en niños con LLA es la médula ósea frente a la sangre periférica,
por la mayor mortalidad relacionada con el trasplante cuando se utiliza esta
última (Fagioli et al., 2013). El régimen de acondicionamiento recomendado,
por el momento, incluye la irradiación corporal total en niños mayores de 2-‐
3 años. En cuanto a los factores pronósticos en la supervivencia tras el
trasplante, uno de los principales factores es el nivel de ERM pre-‐trasplante
(Bader et al., 2009; Campana y Leung, 2013).
Seguimiento y efectos secundarios a largo plazo
Tras completar el tratamiento quimioterápico se debe realizar un
seguimiento de los pacientes, no sólo para detectar las posibles recaídas,
sino para diagnosticar y tratar los posibles efectos secundarios a largo plazo.
Dada la elevada tasa de curación de estos niños se ha incrementado el
número de largos supervivientes a riesgo de desarrollar complicaciones
derivadas del tratamiento y su prevención y monitorización han pasado a ser
un tema prioritario. Entre las principales complicaciones se hallan la
cardiotoxicidad (Lipshultz et al., 2012; Bosch et al., 2013) , la osteonecrosis

Introducción
44
avascular y osteopenia (Kawedia et al., 2011), alteraciones psicológicas y
sociales, obesidad y anomalías endocrinológicas (Dvorak et al., 2011),
alteraciones neurocognitivas y déficits de atención y segundas neoplasias
(más frecuentes en niños tratados con radioterapia) (Schmiegelow et al.,
2013). En el caso de los niños trasplantados las secuelas son más frecuentes
y hay que añadir la esterilidad y las derivadas de la irradiación corporal total
(si se incluye en el régimen de acondicionamiento) y de la enfermedad del
injerto contra el huésped crónica.
LLA recaída
La principal causa de muerte en los pacientes con LLA es la recaída, que tiene
lugar en el 15-‐20% de los niños. La probabilidad de supervivencia en la
recaída depende del fenotipo (la LLA-‐T tiene peor pronóstico), de la duración
de la respuesta (<30 meses desde el diagnóstico, peor pronóstico) y de la
localización (recaída extramedular aislada, mejor pronóstico). Del mismo
modo que al diagnóstico, la evaluación de la respuesta mediante la
determinación de niveles de ERM también es un factor pronóstico de gran
importancia. El tratamiento se adapta según estos factores de riesgo y sólo
los de alto riesgo tienen indicación de trasplante alogénico. Así, el TPH en
segunda RC está indicado en todas las recaídas medulares de pacientes con
LLA-‐T y en las recaídas medulares precoces en las de línea B. También está
indicado en 3ª o posterior RC. Los casos refractarios o con ERM elevada tras
el tratamiento de rescate presentan mal pronóstico aun con trasplante, por
lo que en estos casos estarían indicados tratamientos experimentales con
nuevos fármacos o nuevas estrategias de inmunoterapia.

Introducción
45
Nuevos tratamientos
En los últimos años se han desarrollado nuevos fármacos y estrategias de
inmunoterapia. Entre los nuevos fármacos se hallan análogos de los
nucleósidos como la clofarabina y nelarabina (ver apartado de LLA-‐T, más
adelante) y formulaciones nuevas de citostáticos como la vincristina,
daunorrubicina y citarabina liposómicas. También se están realizando
ensayos clínicos con anticuerpos monoclonales conjugados con
inmunotoxinas (como inotuzumab), anticuerpos biespecíficos
(blinatumomab) y otros (Locatelli et al., 2012). Por otro lado, se están
desarrollando nuevas estrategias de inmunoterapia que dirigen a células T
autólogas frente a la célula tumoral e inducen su destrucción mediante la
transfección de un receptor antigénico quimérico, con especificidad anti-‐
CD19, denominados CART19 (Chimeric antigen receptor-‐modified T cells)
(Brentjens et al., 2013; Grupp et al., 2013).
Tratamiento en subgrupos de pacientes que requieren un abordaje
específico:
LLA Philadelphia positiva (LLA Ph+): ver apartado de LLA Ph+, más adelante.
Leucemia del lactante:
Este subgrupo de pacientes tiene peor pronóstico, con tasas de curación
entre 50 y 60% (Mann et al., 2010). En gran medida el mal pronóstico de
estos niños se debe a la presencia en el 80% de los casos de reordenamiento
del gen MLL. Muchos grupos cooperativos emplean protocolos específicos
con quimioterapia intensiva en la que se mezclan estrategias de tratamiento
para el componente mieloide y linfoide de la leucemia y en los que se
administra citarabina a dosis altas. El TPH se indica en el subgrupo de

Introducción
46
pacientes con MLL reordenado y que además tiene otros factores de riesgo
(Mann et al., 2010). La edad inferior a 6 meses, la mala respuesta a la
prednisona, la hiperleucocitosis superior a 300 x 109/L, la presencia de
reordenamiento MLL y la ERM elevada son factores pronósticos adversos
(van der Linden et al., 2009; Mann et al., 2010; Van der Velden et al., 2009).
El perfil de expresión génica en estos pacientes ha mostrado patrones
distintos entre los lactantes de menos y más de 3 meses de edad y ha
detectado, más allá de MLL, algunos genes con una fuerte correlación con la
supervivencia (FLT3, NEGR1, IRX2, TACC2, EPS8, TPD52) en dos cohortes
independientes de pacientes (Kang et al., 2012).
Leucemia en niños con Síndrome de Down:
Los niños con Síndrome de Down tienen una incidencia 14 veces superior de
LLA que los demás niños (Meyr et al., 2013). La LLA en estos niños tiene
características biológicas distintas. Son menos frecuentes tanto las
alteraciones de buen pronóstico (hiperdiploidía y reordenamiento ETV6-‐
RUNX1) como las de muy mal pronóstico como la LLA Ph+ o el
reordenamiento de MLL. Sin embargo, hasta un 50% de casos presentan
sobreexpresión de CRLF2 y mutaciones de JAK2, que podrían conferir peor
pronóstico (Mullighan et al., 2009). La supervivencia de estos pacientes es
algo inferior a la de los niños sin Síndrome de Down, tanto por una mayor
tasa de recaídas como por mayor mortalidad tóxica (debida a infecciones,
mucositis graves y toxicidad cardíaca principalmente). En estos niños se
deben intensificar las medidas de soporte, ajustar las dosis de metotrexato e
intentar, en la medida de lo posible, no disminuir la intensidad del
tratamiento (Maloney, 2011) (Goto et al., 2011).

Introducción
47
Factores pronósticos
La supervivencia global (SG) y la libre de evento (SLE) de la LLA son
superiores en niños respecto a las de pacientes adultos (SG a los 5 años de
85-‐90% en niños frente a 40-‐50% en adultos) y dentro de la LLA pediátrica
también varía según la edad.
El análisis de factores pronósticos constituye una parte fundamental en el
tratamiento de la LLA para la evaluación individualizada del riesgo de
recaída. Esta evaluación permite la estratificación de pacientes en grupos de
riesgo que dirigirán la intensidad del tratamiento del paciente. De esta
manera se intensifica el tratamiento en los pacientes de alto riesgo y se
reduce la intensidad de aquéllos de bajo riesgo. Dicha estratificación ha sido
uno de los pilares principales en la mejoría progresiva de los resultados
obtenidos en el tratamiento de la LLA pediátrica. En algunos casos la
adaptación de la intensidad del tratamiento ha permitido que algunos
factores de mal pronóstico pierdan incluso su valor adverso. A modo de
ejemplos, el valor pronóstico adverso del fenotipo T o incluso la edad
(adolescentes) han dejado de ser factores pronósticos adversos en algunos
protocolos de tratamiento (Barry et al., 2007) (Pui et al., 2011).
Entre los factores pronósticos que se utilizan en la mayoría de los protocolos
para la estratificación de los pacientes con LLA pediátrica se hallan factores
clínicos y biológicos presentes al diagnóstico y otros evolutivos que evalúan
la respuesta al tratamiento.
El principal factor clínico al diagnóstico es la edad, con mejor pronóstico en
los niños entre 1 y 9 años y un pronóstico adverso en aquellos menores de 1
año. Entre los factores biológicos destaca la cifra de leucocitos al
diagnóstico, con mejor pronóstico si es <50 x 109/L y mejor aún si es <20 x

Introducción
48
109/L. En muchos protocolos se emplea una clasificación que agrupa la edad
y la cifra de leucocitos, y que resulta útil para la comparación de los
resultados entre los distintos protocolos de tratamiento. Esta clasificación
del US NCI (United States National Cancer Institute) distingue un grupo de
bajo riesgo, con edad entre 1 y 9 años y leucocitos <50 x 109/L, que agrupa a
dos terceras partes de los pacientes y un grupo de alto riesgo en el que se
hallan el resto de pacientes (Smith et al., 1996).
Otros factores biológicos importantes son: 1) el inmunofenotipo de la célula
leucémica, con peor pronóstico en la LLA-‐T, aunque si se trata con intensidad
adecuada pierde su valor adverso y 2) la genética de la célula tumoral (ver
tabla 6). Algunas de las alteraciones genéticas (hiperdiploidía y
reordenamiento ETV6-‐RUNX1) se asocian a buen pronóstico y en algún
protocolo se utilizan para disminuir la intensidad del tratamiento y otras, en
cambio, determinan la estratificación de los pacientes en grupos de alto o
muy alto riesgo. Entre estas últimas se hallan la presencia del
reordenamiento BCR-‐ABL (LLA Ph+), la hipodiploidía <44 cromosomas y el
reordenamiento del gen MLL. En los últimos años se han descrito nuevos
subgrupos biológicos con pronóstico adverso que, por el momento, sólo se
han incluido en algunos protocolos de tratamiento en la estratificación de
pacientes. Entre ellas se encuentran la amplificación intracromosómica del
cromosoma 21 (iAMP21), el subtipo inmaduro de LLA-‐T “early T precursor”
(ETP), el reordenamiento TCF3-‐HLF, fruto de la traslocación
t(17;19)(q22;p13) o las alteraciones del gen IKZF1.
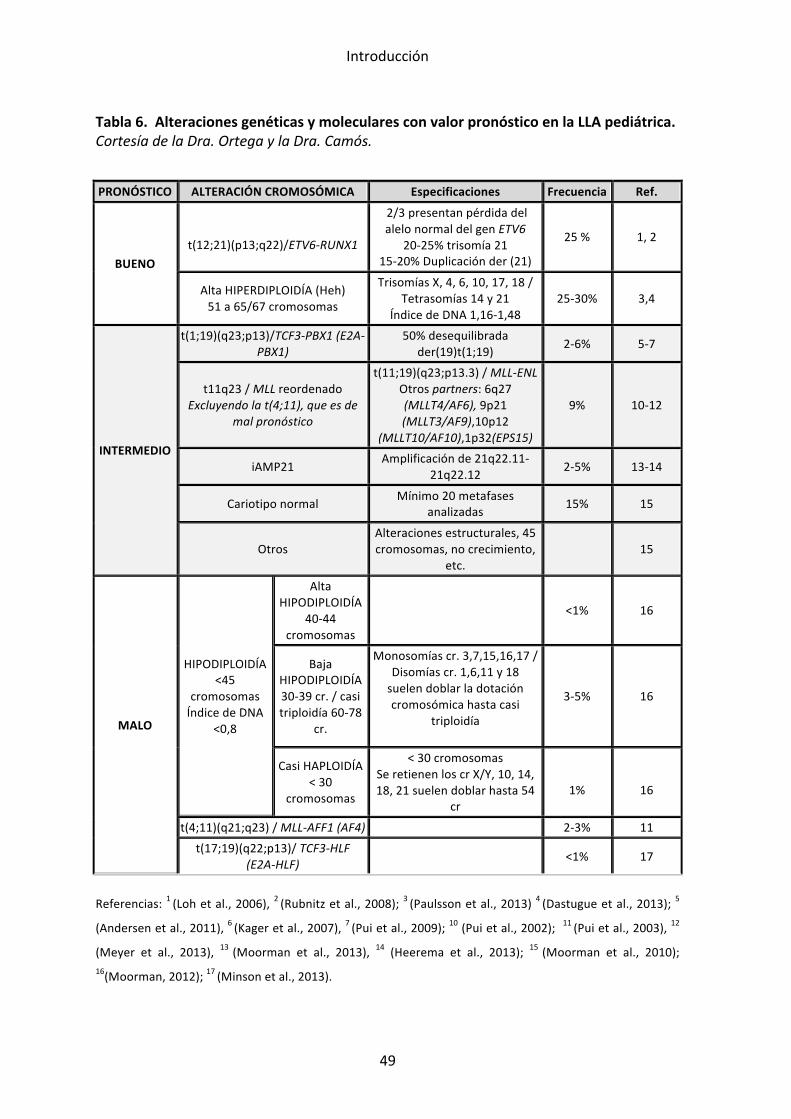
Introducción
49
Tabla 6. Alteraciones genéticas y moleculares con valor pronóstico en la LLA pediátrica. Cortesía de la Dra. Ortega y la Dra. Camós.
PRONÓSTICO ALTERACIÓN CROMOSÓMICA Especificaciones Frecuencia Ref.
BUENO t(12;21)(p13;q22)/ETV6-‐RUNX1
2/3 presentan pérdida del alelo normal del gen ETV6
20-‐25% trisomía 21 15-‐20% Duplicación der (21)
25 % 1, 2
Alta HIPERDIPLOIDÍA (Heh) 51 a 65/67 cromosomas
Trisomías X, 4, 6, 10, 17, 18 / Tetrasomías 14 y 21
Índice de DNA 1,16-‐1,48 25-‐30% 3,4
INTERMEDIO
t(1;19)(q23;p13)/TCF3-‐PBX1 (E2A-‐PBX1)
50% desequilibrada der(19)t(1;19) 2-‐6% 5-‐7
t11q23 / MLL reordenado Excluyendo la t(4;11), que es de
mal pronóstico
t(11;19)(q23;p13.3) / MLL-‐ENL Otros partners: 6q27 (MLLT4/AF6), 9p21 (MLLT3/AF9),10p12
(MLLT10/AF10),1p32(EPS15)
9% 10-‐12
iAMP21 Amplificación de 21q22.11-‐
21q22.12 2-‐5% 13-‐14
Cariotipo normal Mínimo 20 metafases analizadas 15% 15
Otros Alteraciones estructurales, 45 cromosomas, no crecimiento,
etc. 15
MALO
HIPODIPLOIDÍA <45
cromosomas Índice de DNA
<0,8
Alta HIPODIPLOIDÍA
40-‐44 cromosomas
<1% 16
Baja HIPODIPLOIDÍA 30-‐39 cr. / casi triploidía 60-‐78
cr.
Monosomías cr. 3,7,15,16,17 / Disomías cr. 1,6,11 y 18 suelen doblar la dotación cromosómica hasta casi
triploidía
3-‐5% 16
Casi HAPLOIDÍA < 30
cromosomas
< 30 cromosomas Se retienen los cr X/Y, 10, 14, 18, 21 suelen doblar hasta 54
cr 1% 16
t(4;11)(q21;q23) / MLL-‐AFF1 (AF4) 2-‐3% 11
t(17;19)(q22;p13)/ TCF3-‐HLF (E2A-‐HLF) <1% 17
Referencias: 1 (Loh et al., 2006), 2 (Rubnitz et al., 2008); 3 (Paulsson et al., 2013) 4 (Dastugue et al., 2013); 5
(Andersen et al., 2011), 6 (Kager et al., 2007), 7 (Pui et al., 2009); 10 (Pui et al., 2002); 11 (Pui et al., 2003), 12
(Meyer et al., 2013), 13 (Moorman et al., 2013), 14 (Heerema et al., 2013); 15 (Moorman et al., 2010); 16(Moorman, 2012); 17 (Minson et al., 2013).

Introducción
50
La edad y en menor medida la cifra de leucocitos son factores cuyo valor
pronóstico proviene de su asociación con subgrupos genéticos concretos.
Así, el 70% de los niños con edad entre 1 y 9 años tiene alteraciones de buen
pronóstico como la hiperdiploidía y el reordenamiento ETV6-‐RUNX1
mientras que, por el contrario, la edad inferior a 1 año se asocia en un 80%
de los casos a reordenamientos de MLL. Sin embargo, cabe destacar que las
alteraciones genéticas de la célula tumoral no determinan por completo la
probabilidad de curación de los pacientes, ya que ésta depende en gran
medida del tratamiento recibido. Así, los pacientes adolescentes con LLA
tratados con protocolos de tratamiento pediátrico tuvieron tasas de curación
de hasta un 20% superior a aquellos tratados con protocolos de adultos
(Boissel et al., 2003) (Ribera et al., 2008) y estas diferencias han disminuido
tras la aplicación a estos pacientes de protocolos pediátricos también en los
centros de adultos (Pole et al., 2013). Además del tratamiento recibido,
también contribuyen factores genéticos del huésped, como son
polimorfismos en enzimas que intervienen en el metabolismo de los
citostáticos (farmacogenómica). Estos polimorfismos pueden influir no sólo
en las tasas de curación, sino también en el riesgo de desarrollar toxicidad a
corto y a largo plazo. Un ejemplo lo constituyen los polimorfismos de la
enzima tiopurin-‐metiltransferasa (TPMT), implicada en el metabolismo de la
mercaptopurina y tioguanina. Los individuos homocigotos y heterocigotos
para polimorfismos con menor actividad TPMT tienen por un lado una
supervivencia mayor y por otro presentan mayor toxicidad aguda
(mielosupresión) y a largo plazo (leucemias mieloides secundarias y tumores
en SNC) (Schmiegelow et al., 2009) (Relling et al., 1999) (Relling et al., 1998).
De igual modo, determinados polimorfismos en otros genes como los

Introducción
51
implicados en el metabolismo de metotrexato o en el metabolismo óseo
pueden traducirse en distinta respuesta al tratamiento o toxicidad (Salazar et
al., 2012) (Kawedia et al., 2011; Radtke et al., 2013).
Uno de los factores pronósticos más importantes es la evaluación in vivo de
la respuesta al tratamiento, puesto que en ella se reflejan tanto los factores
genéticos de la célula leucémica como los factores genéticos del huésped y
el tratamiento recibido. Esta respuesta se puede evaluar mediante estudio
morfológico en sangre periférica tras 1 semana de tratamiento con
prednisona o en médula ósea a los 8 o 15 días del tratamiento
quimioterápico. Otra forma de evaluar la respuesta al tratamiento con
mayor sensibilidad y especificidad que la evaluación morfológica y que ha
demostrado tener un gran valor pronóstico es el estudio de los niveles de
ERM. La ERM puede medirse por citometría de flujo o por biología molecular
al final de inducción, al final de la consolidación o en momentos más tardíos.
En la actualidad, la determinación de la ERM se utiliza en la mayoría de los
protocolos de tratamiento para la estratificación de los pacientes. Además
de tener valor pronóstico, la utilización de la ERM para dirigir la intensidad
del tratamiento ha logrado aumentar la tasa de curación de los pacientes
con niveles de ERM entre 0,1% y 0,01% al final de la inducción (Pui et al.,
2009).
La modulación del tratamiento en función de los factores pronósticos ha
permitido obtener la curación en la mayoría de los niños con LLA. Sin
embargo, todavía fallecen por la enfermedad entre un 10-‐15% de los
pacientes. En determinados subgrupos de pacientes las tasas de curación
son muy inferiores y los factores pronósticos no están bien definidos. Dentro

Introducción
52
de estos subgrupos se hallan los pacientes con algunas alteraciones
genéticas de alto riesgo descritas anteriormente (LLA Ph+, reordenamientos
del gen MLL, hipodiploidía, iAMP21) y algunos subgrupos de pacientes con
LLA-‐T. El presente trabajo de tesis estudia dos de estos subgrupos de
pacientes de alto riesgo, la LLA Ph+ y la LLA-‐T y analiza factores pronósticos y
el impacto en la supervivencia de las modificaciones en el tratamiento de
estos niños.

Introducción
53
2. LLA cromosoma Philadelphia +
La leucemia linfoblástica aguda con traslocación t(9;22)(q34;q11), o
cromosoma Philadelphia positiva (LLA Ph+) constituye uno de los subtipos de
leucemias de alto riesgo más característicos. Tiene una incidencia del 2-‐5%
en la edad infantil y del 25-‐30% en la edad adulta (Ribera, 2013).
Perspectiva histórica
El cromosoma Philadelphia fue el primer ejemplo de una anomalía
cromosómica asociada a un cáncer, la leucemia mieloide crónica (LMC).
Peter Nowell y David Hungerford observaron la presencia de un cromosoma
minúsculo en cultivos de células de pacientes con LMC y lo publicaron en la
revista Science en 1960 (Nowell y Hungerford, 1960). A dicho cromosoma,
que se identificó como cromosoma 22, se le denominó cromosoma
Philadelphia por ser la ciudad en la que se describió (ver figura 10). Durante
la siguiente década, se cuestionó la importancia de este hallazgo y se
consideró un epifenómeno. En la década de los 70s, con la mejoría en las
técnicas de bandeo de cromosomas y en los 80s con las técnicas
moleculares, se describieron otras alteraciones citogenéticas recurrentes
asociadas a leucemias y a otros tumores. Finalmente, se aceptó su papel
causal en la génesis del cáncer y se identificaron otros oncogenes implicados
en diversos tipos de tumores (Nowell, 2007).

Introducción
54
Figura 10. Fotografía de Peter Nowell (izquierda) y David Hungerford (derecha). Al lado, cariograma en el que se observa la traslocación t(9;22)
En 1973 Janet Rowley y cols. describieron que el cromosoma Philadelphia
era el producto de la traslocación de los cromosomas 9 y 22 (Rowley, 1973) y
en 1982 Annelies de Klein y cols. descubrieron que los oncogenes implicados
en esta traslocación eran el oncogén homólogo humano del virus de la
leucemia murina Abelson, c-‐ABL, en el cromosoma 9 y el gen BCR (Break
Point Cluster region) en el cromosoma 22 (De Klein et al., 1982) (ver figuras
11 y 12).
Figura 11. Esquema de la traslocación t(9;22)(q34;q11)
Cromosomas'normales''''''Rotura'de'cromosomas''''Cromosomas'reordenados'
Cromosoma'Phildelphia'

Introducción
55
Figura 12. Gen de fusión BCR-‐ABL y los distintos puntos de rotura. Deininger y Druker. Pharmacological Reviews, 2013 (Deininger y Druker, 2003)
En 1995 Brian Druker diseñó en el laboratorio un fármaco (imatinib) dirigido
a inhibir la actividad tirosín-‐quinasa de la proteína quimérica BCR-‐ABL
(Druker et al., 1996) y en 2001 se demostró su eficacia en el tratamiento de
la LMC y en LLA Ph+ (Druker et al., 2001) (ver figura 13).
Así pues, el cromosoma Philadelphia ha sido uno de los primeros hallazgos
que demostraron el papel de las alteraciones genéticas en el cáncer. Casi 40
años después, dicho conocimiento se ha traducido en una nueva manera de
abordar el tratamiento frente al cáncer: la terapia frente a dianas
moleculares. Imatinib es un fármaco dirigido específicamente frente a la
lesión genética que origina la leucemia Ph+ y ha sido paradigmático en el
diseño de otros fármacos dirigidos frente a diversos oncogenes en distintos
tumores.
conventional cytogenetics. Moreover, it is useful for sam-ples in which metaphases cannot be obtained. Its mainlimitation is the fact that it does not detect abnormali-ties other than the BCR-ABL translocation.
Reverse transcription-polymerase chain reaction (RT-PCR) is used for both diagnostic purposes and follow-up,particularly after allogeneic stem cell transplantation(Cross et al., 1993). It is the most sensitive method formonitoring residual disease. Improved methods of quan-tification enable reliable monitoring of leukemic burdenand allow for therapeutic interventions before cytoge-netic or hematological relapse occurs (Dazzi and Gold-man, 1999).
B. Bcr and Abl Proteins
The function of the various structural motifs in theBcr and Abl proteins have recently been reviewed (Dein-inger et al., 2000a). Both Bcr and Abl are multidomainproteins. The physiological function of Bcr is not wellunderstood. The N terminus of the protein has serine/threonine kinase activity (Maru and Witte, 1991) and adimerization domain (McWhirter et al., 1993). The cen-tral portion of the protein contains dbl-like and pleck-strin homology domains that stimulate GDP-GTP ex-change on rho guanidine exchange factors (Ron et al.,1991) (Fig. 3A). The C terminus has GTPase activity forRac, a Ras-family protein that activates an NADPHoxidase in neutrophils (Diekmann et al., 1991). Alto-gether, this suggests a function for Bcr in signal trans-duction. However, apart from an increased neutrophilicburst, the phenotype of mice with homozygous deletionof BCR is normal (Voncken et al., 1995).
The Abl protein contains several domains such as theSrc homology domains 2 and 3 (SH2 and SH3), proline-rich regions in the center, and the actin-binding domainat the C terminus, which allow for interactions withother proteins (Fig. 3B). In addition, there is a DNA-binding domain as well as nuclear localization signals.The SH1 domain has the PTK activity that may beregulated by the NH2-terminal SH3 domain. The dataregarding the physiological function of Abl are complex(recently reviewed in Van Etten, 1999). The nuclearfraction appears to play an inhibitory role in cell cycleregulation, which led to the notion that ABL is a tumorsuppressor gene (Sawyers et al., 1994). The cytoplasmicpool may function in the transmission of integrin-medi-ated signals from the cellular environment (Lewis andSchwartz, 1998). Importantly, Abl interacts with severalproteins involved in DNA repair such as ataxia telangi-ectasia-mutated (Atm) (Baskaran et al., 1997; Shafmanet al., 1997), DNA-PK (Kharbanda et al., 1997), andRad51 (Yuan et al., 1998; Chen et al., 1999). It appearsthat Abl kinase activity is important for the induction ofapoptosis in response to genotoxic stress such as ionizingradiation (Yuan et al., 1997). ABL null mice have a highneonatal mortality, and the survivors exhibit a variety ofdefects such as disturbed immune function, bone de-fects, and a rather ill-defined wasting syndrome(Schwartzberg et al., 1991; Tybulewicz et al., 1991).There is, however, no increased incidence of tumors inthese mice, which argues against the concept of ABL asa tumor suppressor. It is possible that ARG (ABL-re-lated gene), a close homolog of ABL, is capable of com-pensating for the loss of some of the functions of ABL.
FIG. 2. Genomic organization of the BCR and ABL loci. The arrows indicate breakpoints within ABL, and the location of the minor (m-BCR), major(M-BCR) and micro breakpoint cluster region (!-BCR) are shown. Regardless of the specific breakpoint in ABL, mRNAs are produced that fuse BCRsequences to the second ABL exon. Fusions between BCR exons e13 (previously b2) or e14 (previously b3) and ABL exon a2 produce p210BCR-ABL thatis characteristic of CML, whereas fusions between BCR exon e1 and ABL exon a2 give rise to p190BCR-ABL (found in two of three of patients withPh-positive acute lymphoblastic leukemia). Rare CML patients have a breakpoint in the so-called micro breakpoint cluster region (!-BCR) and producep230BCR-ABL. Note that there are two alternative 1st exons in ABL (Ia and Ib) and two alternative 2nd exons in BCR (e1! and e2!).
MOLECULAR TARGETED THERAPY OF CML 405
by guest on July 2, 2013pharm
rev.aspetjournals.orgD
ownloaded from

Introducción
56
Figura 13. A. Esquema de la quinasa ABL con la molécula de imatinib unida al dominio quinasa de ABL. B. Molécula de imatinib Reproducido de Deininger y Druker, Pharmacological Review 2003 (Deininger y Druker, 2003)
Cromosoma Philadelphia y gen de fusión BCR-‐ABL
El cromosoma Philadelphia es producto de la traslocación t(9;22)(q34;q11) y
origina el gen de fusión BCR-‐ABL (ver figuras 11 y 12). Esta traslocación
puede hallarse en leucemias crónicas como la leucemia mieloide crónica
(LMC) y en leucemias agudas, mayoritariamente de línea linfoide (LLA),
aunque pueden presentar un fenotipo mixto (MPAL) y muy raramente de
línea mieloide (LMA). Este gen codifica una proteína quimérica con actividad
tirosín-‐quinasa incrementada. A nivel molecular, los puntos de rotura
pueden variar y esta traslocación puede dar lugar a 3 transcritos, que
codificarán 3 proteínas diferentes: p190, p210 e infrecuentemente p230. La
proteína de 190 kDa es más frecuente en la LLA Ph+, mientras que la
commodates the phosphate groups of ATP and is there-fore referred to as the P-loop. The P-loop is a glycine-richand highly flexible structure, which folds down uponbinding of imatinib, resulting in increased surfacecomplementarity. This change in position is stabilizedby a newly formed hydrogen bond between Tyr-253 andAsn-322 (Schindler et al., 2000). A consequence of theinduced fit is the formation of a hydrophobic cage thatsurrounds imatinib, engaging van der Waals interac-tions with residues Tyr-253, Leu-370, and Phe-382 (Fig.8A) (Nagar et al., 2002). Moreover, imatinib forms anumber of hydrogen bonds with the kinase domain. Me-thionine 318 [which normally binds N1 of ATP (Schin-dler et al., 2000)], threonine 315, methionine 290, glu-tamine 286, lysine 270, and asparagine 381, togetherwith water molecules, form a network of hydrogen bondsaround the imatinib molecule (Fig. 8B). Given this ex-tremely tight fit, it is not surprising that changes ofsingle amino acids can affect the binding of imatinib. Forexample, threonine 315 is replaced by methionine in theinsulin receptor kinase (Schindler et al., 2000), which iscompletely insensitive to imatinib (Table 1) (Nagar etal., 2002).
In agreement with these predictions, Abl phosphory-lated on tyrosine 393 is much less sensitive to imatinib,since phosphorylation of tyrosine 393 stabilizes the ac-tive, open conformation of the A-loop (Schindler et al.,2000; Roumiantsev et al., 2002), to which imatinib doesnot bind. Bcr-Abl is constitutively tyrosine-phosphory-lated and will thus be in a conformation that is unable tobind imatinib. From experiments with BCR-ABL-posi-tive cell lines treated with imatinib, it is known thatexposure to the compound results in inhibition of kinaseactivity within minutes. This indicates that there israpid turnover between the phosphorylated form (con-stituting the bulk of the protein) and the unphosphory-lated form, which is capable of binding imatinib. Thispoints to an important role of tyrosine phosphatases.
IX. Resistance to ImatinibA. In Vitro Models
Several groups have generated cell lines that are re-sistant to imatinib (le Coutre et al., 2000; Mahon et al.,2000; Weisberg and Griffin, 2000). In most cases, theselines were generated by continuous growth of cells inincreasing concentrations of the inhibitor. Both humanCML lines and murine lines engineered to express aBCR-ABL construct were used. The most common mech-anism of resistance was increased expression of BCR-ABL mRNA and protein, caused by gene amplification orincreased transcription. In one line, overexpression ofthe MDR-1 gene was demonstrated in addition to over-expression of Bcr-Abl (Mahon et al., 2000), suggestingthat drug efflux may contribute to resistance. In severalcases, it was not possible to define a specific mechanismof resistance. In contrast to clinical resistance (see be-
FIG. 8. A, structure of imatinib bound to the kinase domain of Abl.Arrow, P-loop; arrowhead, helix !C. The activation loop is shown in blue,with the conserved DFG motif in gold. Imatinib penetrates the center ofthe kinase, stabilizing the inactive conformation of the activation loop.Binding to the activation loop occurs without major steric clashes,whereas binding to the P-loop involves an induced fit mechanism. B,amino acids contacting imatinib. Imatinib: carbon is shown in green,nitrogen in blue, and oxygen in red. Carbons of the protein backbone areshown in orange. Hydrogen bonds are indicated as dashed lines. Numbersrepresent distances in Angstrom units. Residues that form hydrophobicinteractions are circled. Imatinib contacts 21 amino acids within the Ablkinase domain (adapted from Nagar et al., 2002 with permission from theAmerican Association for Cancer Research).
416 DEININGER AND DRUKER
by guest on July 2, 2013pharm
rev.aspetjournals.orgD
ownloaded from
commodates the phosphate groups of ATP and is there-fore referred to as the P-loop. The P-loop is a glycine-richand highly flexible structure, which folds down uponbinding of imatinib, resulting in increased surfacecomplementarity. This change in position is stabilizedby a newly formed hydrogen bond between Tyr-253 andAsn-322 (Schindler et al., 2000). A consequence of theinduced fit is the formation of a hydrophobic cage thatsurrounds imatinib, engaging van der Waals interac-tions with residues Tyr-253, Leu-370, and Phe-382 (Fig.8A) (Nagar et al., 2002). Moreover, imatinib forms anumber of hydrogen bonds with the kinase domain. Me-thionine 318 [which normally binds N1 of ATP (Schin-dler et al., 2000)], threonine 315, methionine 290, glu-tamine 286, lysine 270, and asparagine 381, togetherwith water molecules, form a network of hydrogen bondsaround the imatinib molecule (Fig. 8B). Given this ex-tremely tight fit, it is not surprising that changes ofsingle amino acids can affect the binding of imatinib. Forexample, threonine 315 is replaced by methionine in theinsulin receptor kinase (Schindler et al., 2000), which iscompletely insensitive to imatinib (Table 1) (Nagar etal., 2002).
In agreement with these predictions, Abl phosphory-lated on tyrosine 393 is much less sensitive to imatinib,since phosphorylation of tyrosine 393 stabilizes the ac-tive, open conformation of the A-loop (Schindler et al.,2000; Roumiantsev et al., 2002), to which imatinib doesnot bind. Bcr-Abl is constitutively tyrosine-phosphory-lated and will thus be in a conformation that is unable tobind imatinib. From experiments with BCR-ABL-posi-tive cell lines treated with imatinib, it is known thatexposure to the compound results in inhibition of kinaseactivity within minutes. This indicates that there israpid turnover between the phosphorylated form (con-stituting the bulk of the protein) and the unphosphory-lated form, which is capable of binding imatinib. Thispoints to an important role of tyrosine phosphatases.
IX. Resistance to ImatinibA. In Vitro Models
Several groups have generated cell lines that are re-sistant to imatinib (le Coutre et al., 2000; Mahon et al.,2000; Weisberg and Griffin, 2000). In most cases, theselines were generated by continuous growth of cells inincreasing concentrations of the inhibitor. Both humanCML lines and murine lines engineered to express aBCR-ABL construct were used. The most common mech-anism of resistance was increased expression of BCR-ABL mRNA and protein, caused by gene amplification orincreased transcription. In one line, overexpression ofthe MDR-1 gene was demonstrated in addition to over-expression of Bcr-Abl (Mahon et al., 2000), suggestingthat drug efflux may contribute to resistance. In severalcases, it was not possible to define a specific mechanismof resistance. In contrast to clinical resistance (see be-
FIG. 8. A, structure of imatinib bound to the kinase domain of Abl.Arrow, P-loop; arrowhead, helix !C. The activation loop is shown in blue,with the conserved DFG motif in gold. Imatinib penetrates the center ofthe kinase, stabilizing the inactive conformation of the activation loop.Binding to the activation loop occurs without major steric clashes,whereas binding to the P-loop involves an induced fit mechanism. B,amino acids contacting imatinib. Imatinib: carbon is shown in green,nitrogen in blue, and oxygen in red. Carbons of the protein backbone areshown in orange. Hydrogen bonds are indicated as dashed lines. Numbersrepresent distances in Angstrom units. Residues that form hydrophobicinteractions are circled. Imatinib contacts 21 amino acids within the Ablkinase domain (adapted from Nagar et al., 2002 with permission from theAmerican Association for Cancer Research).
416 DEININGER AND DRUKER
by guest on July 2, 2013pharm
rev.aspetjournals.orgD
ownloaded from

Introducción
57
proteína p210 suele hallarse en la LMC. En un 3-‐5% de las muestras, la
citogenética convencional no detecta el cromosoma Philadelphia, pero el
reordenamiento BCR-‐ABL puede entonces identificarse por FISH y/o por PCR.
En alrededor de la mitad de los casos se asocian alteraciones citogenéticas
añadidas, algunas de ellas recurrentes, como son hiperdiploidías, deleción
del cromosoma 7 o de 9p (Heerema et al., 2004). El valor pronóstico de estas
alteraciones se discute más adelante.
Heterogeneidad clínica y biológica dentro de la LLA Ph+
Los pacientes con LLA Ph+ constituyen un grupo con características clínicas y
biológicas propias y recientemente se han aceptado como una entidad
diferenciada en la clasificación de la WHO (Swerdlow et al., 2008).
Clínicamente, los pacientes pediátricos con LLA Ph+ se caracterizan por tener
una mediana de edad y una cifra de leucocitos superiores a las de los demás
niños con LLA, así como por presentar con mayor frecuencia infiltración del
SNC y resistencia al tratamiento quimioterápico convencional. No obstante,
los pacientes con LLA Ph+ constituyen un grupo biológicamente heterogéneo
(diferentes transcritos de fusión, distintas alteraciones citogenéticas
añadidas) y en los que la respuesta al tratamiento es variable (Schrappe et
al., 1998) (Heerema et al., 2004) (Mullighan et al., 2008) (Cazzaniga et al.,
2011). La identificación de subgrupos pronósticos dentro de esta entidad
podría permitir una mejor adecuación de la intensidad de tratamiento y una
mejoría en las opciones curativas de estos pacientes.

Introducción
58
Pronóstico de la LLA Ph+ antes de la introducción del tratamiento con
inhibidores de tirosín-‐quinasas (TKI) (era pre-‐TKI)
La presencia del reordenamiento BCR-‐ABL en la era pre-‐TKI confería un valor
pronóstico muy adverso en los pacientes con LLA, tanto si se trataba de
adultos como de niños. En niños, la SLE a los 5 años era inferior al 40% y en
adultos inferior al 20% (Aricò et al., 2010) (Ribera et al., 2002). En niños la
LLA Ph+ constituía una de las pocas indicaciones de TPH alogénico en
primera RC y estaba indicado incluso en aquellos pacientes con buena
respuesta al tratamiento inicial. Si bien en todas las series publicadas había
un pequeño grupo de niños que podían curarse sólo con quimioterapia, el
TPH, tanto familiar como no relacionado, ofrecía mayor probabilidad de
supervivencia a largo plazo (Aricò et al., 2010).
Se describieron factores biológicos con valor pronóstico, tales como la
presencia de alteraciones citogenéticas recurrentes añadidas al cromosoma
Philadelphia. Así, se describió que los pacientes con pérdida de cromosoma
7, 7p o de 9p tenían peor pronóstico que aquéllos con ganancia de un
segundo cromosoma Philadelphia o con hiperdiploidía. Este valor pronóstico,
sin embargo, perdía significación estadística cuando se analizaban
conjuntamente con otros factores clínicos (Heerema et al., 2004) (Li et al.,
2009). También se identificaron factores clínicos con valor pronóstico. La
edad entre 1 y 10 años, una cifra de leucocitos inferior a 25, 50 ó 100 x 109/L
y una buena respuesta precoz al tratamiento (medida por la respuesta a una
semana de prednisona o por la presencia de <5% de blastos en la médula
ósea del día +15 o +21) eran factores pronósticos asociados a mayor
probabilidad de curación (Ribeiro et al., 1997) (Schrappe et al., 1998). Así, en
un estudio retrospectivo colaborativo intergrupo los pacientes de menor

Introducción
59
riesgo tuvieron una SLE a los 5 años de 49% frente a un 20% en aquéllos de
alto riesgo (p<0,001) (Aricò et al., 2000). En la serie francesa de pacientes
Ph+ tratados según el protocolo FRALLE 93 se describió un índice pronóstico
en el que se tenía en cuenta la edad, la cifra de leucocitos y la respuesta
precoz (<5% de blastos en médula ósea en el día +21). Este índice discriminó
claramente dos grupos pronósticos con distinta supervivencia (SG a los 5
años de 79% vs. 27% (p < 0,005)) (Gandemer et al., 2009). La valoración de la
respuesta al tratamiento mediante el estudio de ERM también pudo
identificar a pacientes adultos y pediátricos con menor riesgo de recaída
(Pane et al., 2005). Cazzaniga y cols. estudiaron en pacientes pediátricos la
cinética de la reducción del clon Ph+ mediante PCR cualitativa del gen de
fusión BCR-‐ABL y mostraron que la negativización precoz de la ERM se asoció
a menor riesgo de recaída. Plantearon la posibilidad de que el subgrupo de
pacientes buenos respondedores se tratara únicamente con quimioterapia
intensiva y se omitiera el trasplante (Cazzaniga et al., 2002).
Pronóstico de la LLA Ph+ en la era TKI
A finales de la década de los años 90 se sintetizó un nuevo fármaco, imatinib,
diseñado para unirse a la proteína quimérica BCR-‐ABL e inhibir su actividad
tirosín-‐quinasa. En la última década se han incorporado imatinib y otros
inhibidores tirosín-‐quinasa (TKI) de segunda generación (dasatinib, nilotinib)
y, más recientemente, de tercera generación (ponatinib) en el tratamiento
de la LLA Ph+ del adulto, con mejoría de la supervivencia (Druker et al., 2001;
de Labarthe et al., 2007). La utilización de imatinib como agente único en
pacientes de edad adulta y pediátrica con LLA Ph+ en fases avanzadas
obtuvo remisiones completas que fueron, sin embargo, poco duraderas

Introducción
60
(Druker et al., 2001) (Champagne et al., 2004). Sin embargo, cuando se
administró imatinib en combinación con quimioterapia intensiva en
pacientes adultos se consiguió una mejoría importante de los resultados. Así,
se obtuvo un aumento tanto de la tasa de remisión completa como de la
tasa de pacientes que llegaron a trasplante y una mejoría significativa de la
supervivencia a corto y a largo plazo en comparación con controles históricos
de pacientes no tratados con imatinib (Thomas et al., 2004) (Yanada et al.,
2006) (Ribera et al., 2010) (Ribera, 2013). La supervivencia global a largo
plazo (4-‐5 años) en pacientes adultos pasó de ser inferior al 20% al 30-‐40%
(Ribera et al., 2010) (Bassan et al., 2010). Entre los pacientes que llegaron a
trasplante la SLE a 5 años alcanzó el 50-‐63% (Lee et al., 2012) (Tanguy-‐
Schmidt et al., 2013). Debido a los mejores resultados obtenidos con
imatinib asociado a quimioterapia, en la actualidad se incluye este fármaco u
otro TKI en el tratamiento de los pacientes adultos con LLA Ph+. La mejoría
de los resultados obtenidos en los pacientes adultos al añadir un TKI al
tratamiento quimioterápico seguido de TPH hace que incluso se cuestione el
valor pronóstico adverso de la presencia del cromosoma Philadelphia en la
LLA del adulto, ya que la supervivencia alcanzada no es inferior a la obtenida
en la LLA Ph negativa (Ribera et al., 2012).
En la población adulta, en concordancia con los resultados hallados en otros
subgrupos de leucemia, la respuesta al tratamiento medida por la
disminución de los niveles de ERM podría ser un factor pronóstico. El estudio
de ERM en pacientes adultos tratados con un TKI ha dado resultados
distintos según el protocolo e intensidad de tratamiento administrados.
Además, los distintos estudios difieren en qué niveles de ERM y cuándo
pueden tener valor pronóstico (Wassmann et al., 2005). (Yanada et al., 2008)

Introducción
61
(Mizuta et al., 2012) (Lee et al., 2012). En algunas series, la cinética de la
ERM pre-‐trasplante tiene valor pronóstico (Lee et al., 2012), mientras que en
otros la ERM pre-‐trasplante pierde valor pronóstico si ésta consigue
negativizarse tras el TPH (Yanada et al., 2008). Recientemente, se ha descrito
el valor pronóstico de la detección de ERM en una serie de pacientes con LLA
Ph+ que recibieron tratamiento con TKIs (imatinib o dasatinib) en
combinación con quimioterapia y a los que no se realizó un TPH. La ERM,
medida tanto por citometría de flujo como por RT-‐PCR cuantitativa del
transcrito de fusión BCR-‐ABL, tuvo valor pronóstico a los 3 meses del inicio
del tratamiento y en momentos más tardíos (Ravandi et al., 2013).
En niños se dispone de mucha menos información acerca de la utilización de
imatinib y quimioterapia en primera línea en pacientes con LLA Ph+. De
hecho, previo a la publicación del presente trabajo de tesis, únicamente se
había publicado un estudio en el que se incluyó imatinib en combinación con
quimioterapia en pacientes pediátricos (Schultz et al., 2009). Estos pacientes
presentaron una mejoría espectacular en la supervivencia a corto plazo,
incluso aquéllos que no recibieron trasplante alogénico. Con estos resultados
se sugirió que, en la era imatinib, los niños con LLA Ph+ tal vez podrían
curarse sin necesidad de realizar un TPH alogénico. Dada la heterogeneidad
clínica y biológica de estos pacientes y con el fin de plantearse una reducción
en la intensidad del tratamiento, sería muy importante conocer qué factores
pronósticos segregarían a aquellos pacientes con mayor probabilidad de
curación sin trasplante. En la era TKI se dispone de muy poca información
sobre los factores pronósticos en la LLA Ph+ en esta población de pacientes.
En el único estudio realizado antes de la publicación del presente trabajo de
tesis sobre ERM en LLA Ph+ en la edad pediátrica, el valor pronóstico de la

Introducción
62
ERM varió según la intensidad recibida del tratamiento con imatinib. Así, en
pacientes tratados con imatinib intermitente, la ERM al final del tratamiento
de inducción tuvo valor pronóstico, mientras que en aquéllos tratados con
imatinib continuo y mayores dosis acumuladas de este fármaco, la ERM al
final de inducción perdió su valor pronóstico, ya que la intensificación del
tratamiento permitió rescatar a aquellos pacientes con respuesta más lenta
(Schultz et al., 2009).
Protocolos de tratamiento de la LLA Ph+
Antes de la inclusión de imatinib u otro TKI en el tratamiento de la LLA Ph+,
los pacientes con este tipo de leucemia recibían protocolos de tratamiento
para pacientes de alto riesgo con quimioterapia muy intensiva seguida de
TPH alogénico, siempre que fuera posible. El TPH alogénico había
demostrado su superioridad frente a la quimioterapia tanto en niños como
en adultos (Fielding et al., 2009) (Ribera, 2013).
En el tratamiento actual de la LLA Ph+ del adulto se incluye un TKI en
combinación con la quimioterapia ya en el tratamiento de inducción y éste
se mantiene durante la consolidación y hasta el trasplante. Se recomienda la
realización de un TPH alogénico en todos aquellos pacientes en que sea
posible por su edad y comorbilidad porque aumenta las probabilidades de
supervivencia a largo plazo (Ribera, 2013).
Quedan aún muchos aspectos no resueltos en el tratamiento de estos
pacientes. Uno de ellos radica en hallar la intensidad idónea de tratamiento
quimioterápico y del TKI. En algunos estudios se ha observado que la
reducción de la intensidad del tratamiento quimioterápico asociada a una
intensificación del tratamiento con TKI redujo la toxicidad relacionada con el

Introducción
63
tratamiento pre-‐trasplante y aumentó la proporción de pacientes que
pudieron recibir trasplante (Chalandon et al., 2008; Ribera et al., 2012). De
igual manera, se desconoce el papel del trasplante de donantes alternativos
no HLA-‐idénticos (cordón, haploidéntico, mismatched no emparentado) así
como de los trasplantes de intensidad reducida (Ram et al., 2011) (Ribera,
2013). Otro aspecto controvertido radica en el tratamiento post-‐TPH, sobre
si debe administrarse imatinib (u otro TKI) de forma profiláctica a todos los
pacientes (upfront) o sólo a aquéllos en los que se detecta positivización de
la ERM (pre-‐emptive) (Burke et al., 2009) (Nishiwaki et al., 2010) (Pfeifer et
al., 2011) (Chen et al., 2012) (Kebriaei et al., 2012). Por otro lado, queda por
demostrar si la utilización de TKI de nueva generación (dasatinib, nilotinib),
activos frente algunas mutaciones en ABL que confieren resistencia a
imatinib y con un espectro de dianas moleculares más amplio en el caso de
dasatinib, podría mejorar los resultados obtenidos con imatinib en la LLA
Ph+, tal como ha sucedido en la LMC (Saglio et al., 2010) (Kantarjian et al.,
2010; Kantarjian et al., 2012). Hasta la fecha, existen muy pocos datos y no
ha habido ningún estudio que demuestre su superioridad frente a imatinib
(Ravandi et al., 2010) (Ribera, 2013).
En cuanto a los pacientes pediátricos con LLA Ph+, en el momento del inicio
del presente trabajo de tesis, no se disponía de estudios que analizasen las
indicaciones, dosis, tolerabilidad y eficacia de imatinib en combinación con
quimioterapia. Los estudios fase I de imatinib en monoterapia en niños con
LLA Ph+ recaída o refractaria obtuvieron buenos resultados con un perfil de
toxicidad aceptable (Champagne et al., 2004). En vista de los buenos
resultados observados en la LLA Ph+ en adultos con imatinib en combinación
con quimioterapia intensiva, en 2005 el grupo cooperativo SHOP elaboró un

Introducción
64
protocolo para el tratamiento de la LLA Ph+ pediátrica utilizando este
tratamiento combinado y seguido de TPH alogénico en primera RC. De igual
modo, recientemente dos grandes grupos cooperativos han publicado sus
resultados con pacientes pediátricos afectos de LLA Ph+ tratados con
imatinib en combinación con quimioterapia intensiva en los que se
obtuvieron buenos resultados a corto plazo (Biondi et al., 2012) (Schultz et
al., 2009).
El análisis de los resultados del protocolo SHOP/LLA-‐2005, que incluyó
imatinib a dosis intermedias y de forma continua desde el día +15 del
tratamiento de inducción y hasta el momento del TPH a los pacientes
pediátricos con LLA Ph+, constituye uno de los trabajos que conforman el
presente proyecto de tesis.

Introducción
65
3. Leucemia linfoblástica de línea T
Incidencia
La LLA de línea T (LLA-‐T) representa el 10-‐15% de leucemias linfoblásticas en
la edad infantil y alrededor del 25% en la edad adulta (Van Vlierberghe y
Ferrando, 2012).
Clínica
En la edad pediátrica, la LLA-‐T se caracteriza por ser más frecuente en
varones y tener una mediana de edad mayor que la de los pacientes con LLA
de línea B. Suele presentarse con mayor carga tumoral, que se refleja en un
cifra mayor de leucocitos y elevación de la lactato deshidrogenasa (LDH), así
como presencia de masa mediastínica y/o gran hepato-‐esplenomegalia. Los
pacientes con masa mediastínica pueden no tener síntomas pero a menudo
refieren ortopnea o tos. En algunos pacientes la masa mediastínica
constituye una de las pocas emergencias médicas en la LLA pediátrica, ya
que puede comprimir las vías aéreas y requerir un tratamiento urgente con
corticoides, radioterapia y/o quimioterapia. En estos casos, es preciso
realizar los estudios diagnósticos con urgencia antes de iniciar el tratamiento
y se ha de tener en cuenta que conviene evitar una anestesia general y el
decúbito supino porque pueden agravar el compromiso de la vía aérea. Por
otro lado, la infiltración del SNC, tanto en el momento del diagnóstico como
en la recaída, es más frecuente en la LLA-‐T que en la LLA de línea B.
Algunos pacientes presentan un cuadro clínico similar pero sin células
leucémicas en sangre periférica y un grado menor de infiltración en médula
ósea o sin infiltración alguna y se diagnostican de linfoma linfoblástico T.

Introducción
66
Este tipo de linfoma se considera la misma entidad que la LLA-‐T en la
clasificación de la WHO y se trata con los mismos protocolos o con
protocolos muy parecidos a los de la LLA-‐T. A nivel biológico comparte
muchas alteraciones genéticas con la LLA-‐T, aunque parecen existir algunas
diferencias que podrían explicar el distinto fenotipo de estos pacientes
(Sekimizu et al., 2011).
Biología
La LLA-‐T constituye un tipo de leucemia particularmente heterogéneo y
complejo que resulta de la combinación de múltiples lesiones genéticas que
implican a numerosos oncogenes y genes supresores de tumores, así como a
la alteración en la regulación epigenética. Algunas de las alteraciones
genéticas se excluyen mutuamente y se correlacionan con la detención del
desarrollo del linfocito T en un estadio madurativo específico (inmaduro,
cortical, maduro). Con frecuencia, se produce la yuxtaposición de un
oncogén junto a los genes del receptor de células T (TCR), lo que da lugar a
su expresión aberrante en los progenitores T. Entre los factores de
transcripción oncogénicos sobreexpresados mediante éste u otros
mecanismos en la LLA-‐T se hallan genes de la familia bHLH (basic helix-‐loop-‐
helix), como TAL1 o LYL1, genes de la familia homeobox como HOXA,
TLX1/HOX11, TLX3/HOX11L2 y genes LMO (LIM only domain) como LMO2
(ver tabla 7).
Otras alteraciones genéticas observadas en la LLA-‐T de forma recurrente se
comparten en los distintos subgrupos y alteran los mecanismos normales del
control del crecimiento celular, proliferación, supervivencia y diferenciación
del timocito durante su desarrollo. Entre ellas cabe destacar las mutaciones

Introducción
67
de NOTCH1 y la deleción de CDKN2A/B, que se hallan en la mayoría de los
pacientes y que son fundamentales en la patogenia de esta enfermedad. Ver
tabla 8.
Tabla 7. Lesiones genéticas que definen subgrupos genéticos-‐moleculares en la LLA-‐T. Adaptado de Van Vlierberghe P y Ferrando A, Journal of Clinical Investigation 2012 (Van Vlierberghe y Ferrando, 2012)
1Frecuencia entre pacientes con LLA-‐T 2Según el protocolo de tratamiento se ha asociado a bueno, malo o sin valor pronóstico
Categoría genética Genes implicados Reordenamiento genético Pronóstico Frecuencia1
Genes de la familia bHLH
TAL1 t(1;14)(p32;q11) t(1;7)(p32;q34) deleción 1p32
Favorable 3% 3%
16-‐30%
TAL2 t(7;9)(q34;q32) Indeterminado 1%
LYL1 t(7;19)(q34;p13) Indeterminado 1%
BHLHB1 t(14;21)(q11.2;q22) Indeterminado 1%
Genes de la familia LMO
LMO1 t(11;14)(p15;q11) t(7;11)(q34;p15) Indeterminado 1%
1%
LMO2 t(11;14)(p13;q11) t(7;11)(q34;p13) deleción 11p13
Indeterminado 6% 6% 3%
LMO3 t(7;12)(q34;p12) Indeterminado <1%
Genes de la familia
Homeobox
TLX1 t(11;14)(p15;q11) Favorable 5-‐30%
TLX3 t(11;14)(p15;q11) Controvertido2 5-‐20%
HOXA Inv(7)(p15;q34) t(7;7)(p15;q34) Indeterminado 3%
3%
HOXA (CALM-‐AF10) t(10;11)(p13;q14) Desfavorable 5-‐10%
HOXA (MLL-‐ENL) t(11;19)(q23;p13) Indeterminado 1%
HOXA (SET-‐NUP214) Deleción 9q34 Sin impacto 3%
NKX2.1 inv14(q11.2;q13) inv14(q13;q32.33) t(7;14)(q34;q13)
Indeterminado 5%
NKX2.2 t(14;20)(q11;p11) Indeterminado 1%
Proto-‐oncogenes c-‐MYB t(6;7)(q23;q34) Indeterminado 3%
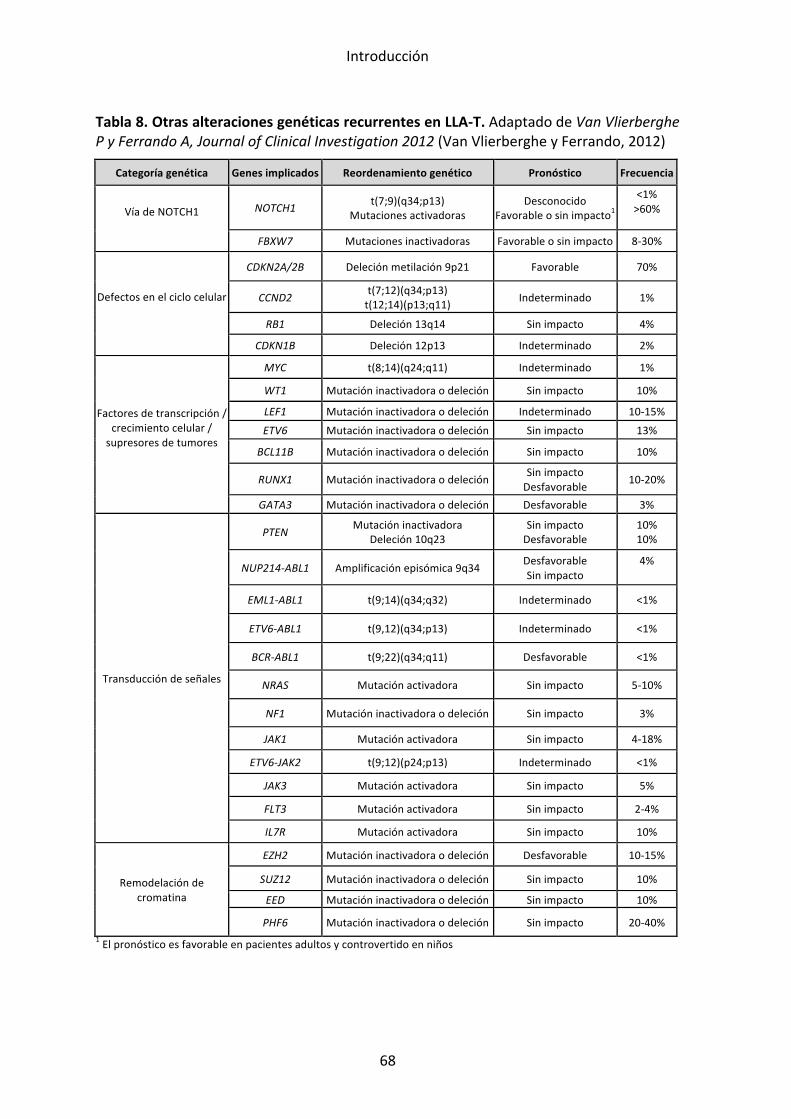
Introducción
68
Tabla 8. Otras alteraciones genéticas recurrentes en LLA-‐T. Adaptado de Van Vlierberghe P y Ferrando A, Journal of Clinical Investigation 2012 (Van Vlierberghe y Ferrando, 2012)
1 El pronóstico es favorable en pacientes adultos y controvertido en niños
Categoría genética Genes implicados Reordenamiento genético Pronóstico Frecuencia
Vía de NOTCH1
NOTCH1 t(7;9)(q34;p13) Mutaciones activadoras
Desconocido Favorable o sin impacto1
<1% >60%
FBXW7 Mutaciones inactivadoras Favorable o sin impacto 8-‐30%
Defectos en el ciclo celular
CDKN2A/2B Deleción metilación 9p21 Favorable 70%
CCND2 t(7;12)(q34;p13) t(12;14)(p13;q11) Indeterminado 1%
RB1 Deleción 13q14 Sin impacto 4%
CDKN1B Deleción 12p13 Indeterminado 2%
Factores de transcripción / crecimiento celular / supresores de tumores
MYC t(8;14)(q24;q11) Indeterminado 1%
WT1 Mutación inactivadora o deleción Sin impacto 10%
LEF1 Mutación inactivadora o deleción Indeterminado 10-‐15%
ETV6 Mutación inactivadora o deleción Sin impacto 13%
BCL11B Mutación inactivadora o deleción Sin impacto 10%
RUNX1 Mutación inactivadora o deleción Sin impacto Desfavorable 10-‐20%
GATA3 Mutación inactivadora o deleción Desfavorable 3%
Transducción de señales
PTEN Mutación inactivadora Deleción 10q23
Sin impacto Desfavorable
10% 10%
NUP214-‐ABL1 Amplificación episómica 9q34 Desfavorable Sin impacto
4%
EML1-‐ABL1 t(9;14)(q34;q32) Indeterminado <1%
ETV6-‐ABL1 t(9,12)(q34;p13) Indeterminado <1%
BCR-‐ABL1 t(9;22)(q34;q11) Desfavorable <1%
NRAS Mutación activadora Sin impacto 5-‐10%
NF1 Mutación inactivadora o deleción Sin impacto 3%
JAK1 Mutación activadora Sin impacto 4-‐18%
ETV6-‐JAK2 t(9;12)(p24;p13) Indeterminado <1%
JAK3 Mutación activadora Sin impacto 5%
FLT3 Mutación activadora Sin impacto 2-‐4%
IL7R Mutación activadora Sin impacto 10%
Remodelación de cromatina
EZH2 Mutación inactivadora o deleción Desfavorable 10-‐15%
SUZ12 Mutación inactivadora o deleción Sin impacto 10%
EED Mutación inactivadora o deleción Sin impacto 10%
PHF6 Mutación inactivadora o deleción Sin impacto 20-‐40%

Introducción
69
NOTCH1 es un receptor que tiene un papel principal en el compromiso de
linaje celular durante el desarrollo. En más del 50% de las LLA-‐T se han
observado mutaciones activadoras en distintos niveles de NOTCH1 o
mutaciones inactivadoras en FBXW7, que codifica una proteína que
interviene en su degradación. En ambos casos se activa la señalización de
NOTCH1. En cuanto a CDKN2A, es un locus que engloba los genes supresores
de tumores p16INK4A y p14ARF. La pérdida de función de estos genes altera la
regulación del ciclo celular y contribuye a la leucemogénesis en una
proporción elevada de LLA-‐T. La deleción de p16INK4A altera la actividad tanto
del gen de retinoblastoma (RB1) como de p53 y predispone al desarrollo de
una LLA-‐T (Volanakis et al., 2009).
Entre otras alteraciones genéticas descubiertas recientemente, se hallan las
mutaciones de la vía PI3K/AKT, descritas en un 10% de los casos. La vía
PI3K/AKT controla múltiples funciones celulares como la diferenciación y el
crecimiento celular. La fosfatasa PTEN es un regulador negativo de esta vía.
Se han descrito mutaciones en PI3K (PIK3CA), en AKT y en PTEN que,
mediante la desregulación de la vía PI3K/AKT, contribuyen a la patogenia de
la LLA-‐T (Palomero et al., 2007) (Gutiérrez et al., 2009). Por otro lado, las
deleciones y mutaciones que inactivan PHF6 se han descrito en un 20-‐40%
de los pacientes con LLA-‐T (Van Vlierberghe et al., 2010). PHF6 es un gen
supresor de tumores que se encuentra localizado en la región autosómica
del cromosoma X, Xq26. Las mutaciones en este gen se han descrito casi
exclusivamente en varones. De hecho, su identificación se realizó
precisamente en la búsqueda de genes supresores de tumores que se
hallaran en el cromosoma X y que explicaran el predominio de este tipo de

Introducción
70
leucemia en varones (Van Vlierberghe et al., 2010).
Otras alteraciones recurrentes en las que se altera la transducción de señales
resultan de traslocaciones que dan lugar a proteínas quiméricas con
actividad quinasa, como NUP214-‐ABL o ETV6–JAK2, o bien de la activación
de estas vías mediante mutaciones o mecanismos epigenéticos. La
identificación de los casos que presentan el gen de fusión NUP214-‐ABL
puede ser muy importante ya que pueden responder a tratamiento con
imatinib u otros TKI con actividad frente a ABL. La búsqueda de otras dianas
terapéuticas en la LLA-‐T ha puesto de manifiesto otras vías de quinasas,
como la vía de TYK2-‐STAT1-‐BCL2. Se ha mostrado que los linfoblastos T
precisan de su activación para su supervivencia y esto apoyaría el desarrollo
de tratamientos diana dirigidos frente a TYK2 u otros componentes de esta
vía (Sanda et al., 2013).
A pesar del gran número y heterogeneidad de alteraciones genéticas
halladas en la LLA-‐T, los estudios de expresión génica de alta densidad
muestran un número limitado de subgrupos moleculares en esta
enfermedad, caracterizados por la detención del desarrollo del linfocito T en
distintos estadios madurativos: 1) el subgrupo con sobreexpresión de TLX1
(HOX11), en el que predominan LLA-‐T de fenotipo cortical; 2) el subgrupo
TAL/LMO2; 3) el subgrupo inmaduro (doble negativos CD4 y CD8), que en un
número elevado de casos sobreexpresa LYL1 y en el que se hallan la mayoría
de los casos de leucemia “early T-‐cell precursor” o ETP (ver más adelante); 4)
el subgrupo TLX3 (HOX11L2) y 5) el subgrupo HOXA, entre otros (Meijerink et
al., 2009) (Homminga et al., 2011) (Ver figura 14).

Introducción
71
Figura 14. Subgrupos moleculares en LLA-‐T. Reproducido de Homminga et al., Cancer cell 2011 (Homminga et al., 2011)
Recientemente se ha identificado un nuevo subtipo de LLA-‐T denominado
“early T-‐cell precursor leukemia” (ETP), que representa alrededor del 15% de
las LLA-‐T pediátricas y del 7% de la LLA-‐T del adulto. Este subtipo se
caracteriza por la detención del progenitor T en un estadio muy inmaduro de
su desarrollo, por presentar un inmunofenotipo característico (negatividad
para CD1a y CD8, con expresión débil de CD5 y expresión de antígenos de
célula madre y/o de línea mieloide) y por tener un perfil transcripcional y
mutacional similar al de las células madre hematopoyéticas normales y al de
las células mieloides más inmaduras. A diferencia de lo que ocurre en otros
subgrupos de LLA-‐T, las mutaciones en NOTCH1 y en CDKN2A/B son
cluster. High microarray expression levels of NKX2-1 andMEF2Cwere validated by RQ-PCR (Figure 2) for the proliferativeand immature cluster cases, respectively, that lack known onco-genic rearrangements. These cases form separate clusters in thesupervised analysis (Figure 1C). NKX2-1 or MEF2C was eitherabsent or expressed at relative low levels in most casesbelonging to other supervised clusters. However, some TLX1-positive patient samples that are part of the proliferative clusterin the unsupervised analyses express NKX2-1. Also, theCALM-AF10-positive HOXA-activated patient sample No. 1509that highly expresses MEF2C has an immature phenotype andco-clusters in the immature cluster in unsupervised analyses.
Molecular-Cytogenetic Identification of NKX2-1RearrangementsThese data formed the start of detailed molecular-cytogeneticanalyses on the 12 immature cluster and the 12 proliferativecluster samples that seemed to form two genetic T-ALL entities(Figure 1C), and for which driving oncogenic hits were unknown.We used a variety of molecular-cytogenetic techniques includingFISH, array-comparative genomic hybridization (array-CGH),and Chromosome Conformation Capture on Chip (4C) (Simoniset al., 2009) to identify potential deletions, amplifications, andT cell receptor- or BCL11B-driven oncogenic events (Table 2;Table S6). The 4C method was originally developed to study
Figure 1. Identification of Two Entities in Pediatric T-ALL that Lack Known Driving Oncogenic Hits(A) Unsupervised hierarchical cluster analysis by the average linkage method in dChip based on 435 probe sets (Table S3) for RMA-solo (Soulier et al., 2005)
normalizedU133 plus two Affymetrix data from 117 pediatric T-ALL samples and seven normal bonemarrow controls. Cytogenetic rearrangements indicated are:
S, SIL-TAL1; T, TAL1; t, TAL2; O, LMO1; L, LMO2 (includes del(11)(p12p13)); $, TAL2/LMO1; N, SET-NUP214; C, CALM-AF10; M, MYB; A, Inv(7)(p15q34); 1,
TLX1; 3, TLX3; and n, normal bonemarrow controls. The 50th and/or the 25th percentiles of samples with the highest TAL1 or LYL1 expression, positivity for TLX1
and TLX3 expression as measured by RQ-PCR, and expression of the immunophenotypic markers CD13 and/or CD33, CD4 or CD8 are indicated; u, no data
available.
(B) Pearson correlation plot for the patient samples belonging to the four unsupervised TAL/LMO, TLX, proliferative, and immature clusters.
(C) PCA of patients with pediatric T-ALL based upon the top 100 most significant differentially expressed probe sets among major T-ALL subgroups (i.e., TAL1/
LMO2, HOXA, TLX1, and TLX3 [Table S3]). The immature cluster (12 cases) and the proliferative cluster (12 cases) are indicated by green and purple
dots, respectively. Samples repeatedly assigned to the proliferative or immature clusters (i.e., the core samples) in multiple unsupervised analyses on RMA-solo
(A), RMA, or VSN normalized data sets (not shown) or the supervised cluster analysis (C) are visualized by dark-green or purple dots. See also Figure S1 and
Tables S1–S4.
Cancer Cell
NKX2-1 and MEF2C as Potential Oncogenes in T-ALL
486 Cancer Cell 19, 484–497, April 12, 2011 ª2011 Elsevier Inc.

Introducción
72
infrecuentes y, en cambio, son frecuentes las mutaciones en FLT3 (Neumann
et al., 2012) y en otros genes como IDH1, IDH2 o DNMT3A, mutaciones
recurrentes en la LMA en adultos (Van Vlierberghe et al., 2011; Haydu y
Ferrando, 2013; Zhang et al., 2012; Neumann et al., 2012). El hallazgo de
este subgrupo biológico es clínicamente relevante porque se asocia a muy
mal pronóstico, tanto en niños como en adultos, y porque puede
identificarse por citometría de flujo, dado su inmunofenotipo característico
(Coustan-‐Smith et al., 2009). La presencia de alteraciones genéticas comunes
a otras leucemias de estirpe mieloide sugiere que los pacientes con LLA-‐T
ETP podrían beneficiarse de añadir fármacos dirigidos a la línea mieloide, y/o
inhibidores de FLT3 y/o de la vía JAK-‐STAT (Neumann et al., 2012) (Zhang et
al., 2012).
Factores pronósticos clínicos
Por lo general, en la mayoría de los protocolos cooperativos pediátricos, los
pacientes con fenotipo T tienen peor pronóstico que los de fenotipo B
precursor (Pui et al., 2009) (Möricke et al., 2009). Entre los factores
pronósticos utilizados para clasificar a los pacientes en grupos de riesgo
están la edad, la cifra de leucocitos, la citogenética, el fenotipo (T o B) y la
respuesta precoz al tratamiento (Pui et al., 2009). En la última década ha
adquirido gran importancia pronóstica la valoración de la respuesta a la
quimioterapia mediante la determinación de la ERM en distintos momentos
del tratamiento (Conter et al., 2010) (Cavé et al., 1998). En la LLA-‐T, con la
excepción de la respuesta al tratamiento (citológica y por ERM), los factores
pronósticos mencionados anteriormente carecen del valor predictivo que

Introducción
73
tienen en los demás subgrupos (Riehm et al., 1987) (Pullen et al., 1999),
(Schrappe et al., 2011) (Lauten et al., 2012).
Así, en la LLA-‐T el valor pronóstico de la edad varía según las series, con la
edad mayor a 15 años asociada a peor pronóstico en alguna serie y sin valor
pronóstico en otras. De igual forma sucede con la cifra de leucocitos y con la
presencia de masa mediastínica o de infiltración del SNC al diagnóstico (Pui
et al., 1990) (Pullen et al., 1999) (Goldberg et al., 2003) (Schrappe et al.,
2011). En cuanto al estadio madurativo del linfocito T, definido por su
inmunofenotipo, tampoco ha resultado de utilidad para estratificar a los
pacientes, con excepción del subgrupo de LLA-‐T ETP (Babusikova et al., 2009)
(Shimizu et al., 2013) (Pullen et al., 1999) (Schrappe et al., 2011).
Probablemente, la falta de resultados congruentes en todos los estudios en
cuanto a su valor pronóstico se deba a que el mismo inmunofenotipo
engloba subtipos biológicos distintos. Respecto a las alteraciones
citogenéticas o moleculares, que en la LLA de línea B son factores
pronósticos muy importantes y que se contemplan en la estratificación de
los pacientes, en la LLA-‐T no se han incorporado aún a los protocolos de
tratamiento (ver más adelante, en factores pronósticos biológicos).
El principal factor clínico con valor pronóstico en la LLA-‐T y el único que se
utiliza en la actualidad para estratificar a los pacientes en grupos de riesgo es
la respuesta precoz al tratamiento. Así, la respuesta a una semana de
prednisona, evaluada en sangre periférica, es un factor pronóstico de gran
importancia y, en el caso de la LLA-‐T, mantiene su valor incluso cuando se
estratifica a los pacientes según su nivel de ERM (Schrappe et al., 2011). La
respuesta evaluada en médula ósea en el día +15 del tratamiento de
inducción también tiene valor pronóstico, aunque menor en los pacientes

Introducción
74
con fenotipo T cuando se comparan con los de línea B (Lauten et al., 2012)
(Schrappe et al., 2011). Otra forma de evaluar la respuesta precoz al
tratamiento, la ERM, también ha resultado muy informativa en la LLA-‐T y
sirve para guiar los protocolos de tratamiento (Pui et al., 2009) (Conter et al.,
2010) (Schrappe et al., 2011).
Factores pronósticos biológicos
En la LLA de precursores B, la capacidad de identificar factores biológicos
que se asocian a mayor riesgo de recaída ha permitido la aplicación de
tratamientos más intensivos o más específicos y ello se ha traducido en un
aumento en la supervivencia de los pacientes. En la LLA-‐T, sin embargo, si
bien se han producido avances importantes en su conocimiento biológico,
éstos no han tenido una traducción en la estratificación de los pacientes en
grupos de riesgo. La única excepción la constituye la reciente identificación
del subgrupo LLA-‐T ETP, para el que grupos cooperativos independientes han
hallado un pronóstico muy adverso (Coustan-‐Smith et al., 2009) (Ma et al.,
2012) (Inukai et al., 2012) (Neumann et al., 2012). Aun así, aunque algunos
grupos cooperativos indican un TPH alogénico en primera RC a estos
pacientes, se desconoce por el momento si dicha modificación del
tratamiento mejorará la supervivencia de estos pacientes.
El estudio de otros factores biológicos en los pacientes con LLA-‐T ha dado
resultados discordantes en los distintos trabajos realizados. Así, la presencia
de mutaciones de NOTCH1 se ha asociado a buen pronóstico en algunos
estudios y carece de valor en otros (Asnafi et al., 2009) (Kox et al., 2010)
(Zuurbier et al., 2010). Esto podría deberse a diferencias en los protocolos de
tratamiento utilizados. También podría deberse a la mutación específica de

Introducción
75
NOTCH1 y/o FBXW7, ya que pueden diferir en el grado de activación de la vía
de NOTCH1. En este sentido, en un estudio del grupo británico MRC (Medical
Research Council), sólo las mutaciones dobles en el receptor NOTCH1 o en
dicho receptor asociadas a mutaciones en FBXW7 se asociaron a un
pronóstico favorable. Este hallazgo se atribuyó al posible efecto sinérgico de
las dos mutaciones (Jenkinson et al., 2013). Las diferencias encontradas
también podrían explicarse por la asociación de mutaciones de NOTCH1 con
mutaciones o alteración de la expresión de otros genes (Palomero et al.,
2007) (Bandapalli et al., 2013).
Se han identificado otros grupos con distintos perfiles de expresión génica,
así como nuevas alteraciones moleculares en la LLA-‐T, pero su valor
pronóstico no se ha validado y su resultados son controvertidos (van Grotel
et al., 2006) (Ballerini et al., 2002). Ver tablas 7 y 8.
Tratamiento
Quimioterapia
Por lo general, en la mayoría de los protocolos cooperativos pediátricos los
pacientes con fenotipo T tienen peor pronóstico que los B, aunque las
diferencias se han ido acortando y han llegado a desaparecer con algunos
protocolos de tratamiento. Para obtener esta mejoría, sin embargo, a estos
pacientes nunca se les estratifica en el grupo de bajo riesgo y reciben, por
tanto, un tratamiento más intensivo (Pui et al., 2009), (Goldberg et al.,
2003). Además de la intensificación en el tratamiento, en algunos protocolos
existen modificaciones específicas para pacientes de fenotipo T. Así, en el
protocolo actual del grupo alemán BFM (BFM/ALL-‐2009) un subgrupo de
pacientes de pacientes con LLA-‐T recibe tratamiento con dexametasona en

Introducción
76
la inducción, a diferencia de los de línea B, que reciben prednisona. En otros
protocolos, como el del SJCRH (Total XV y Total XVI) (Pui et al., 2009) o el
protocolo internacional ALLIC-‐BFM, se aumenta la dosis de metotrexato
sistémico a 5 g/m2 en todos los pacientes con LLA-‐T, mientras que en
algunos subgrupos de menor riesgo de línea B se administra entre 2 y 3
g/m2. El aumento de dosis de metotrexato en estos pacientes se basa en que
los linfoblastos T requieren concentraciones superiores de metotrexato
extracelular para alcanzar los mismos niveles intracelulares que los blastos
de línea B (Kager et al., 2005). Otro fármaco que ha demostrado ser muy
importante en la mejoría de los resultados en los pacientes con LLA-‐T ha sido
la asparraginasa. El grupo norteamericano DFCI consiguió por primera vez,
ya en la década de los 90, equiparar los resultados obtenidos en pacientes
pediátricos con LLA-‐T a los de línea B. Uno de los motivos del éxito de su
protocolo de tratamiento fue precisamente la administración muy intensiva
de asparraginasa (Silverman et al., 2001) (Goldberg et al., 2003).
Posteriormente, otros grupos aplicaron dicha intensificación del tratamiento
con asparraginasa en sus protocolos y también consiguieron mejorar sus
resultados en los pacientes con LLA-‐T (Pui et al., 2009) (Al-‐Khabori et al.,
2010). Otra medida terapéutica que puede tener importancia en el control
de las recaídas de los pacientes con LLA-‐T es la radioterapia craneal
profiláctica. Si bien en varios grupos cooperativos se ha conseguido eliminar
la radioterapia craneal profiláctica en todos los pacientes con LLA (Veerman
et al., 2009) (Pui et al., 2009), todavía se indica en algunos pacientes con
mayor riesgo de recaída en SNC en muchos protocolos y uno de los
subgrupos en los que se mantiene esta indicación es precisamente el grupo
de pacientes con LLA-‐T e hiperleucocitosis. Otros agentes con importancia en

Introducción
77
la LLA-‐T son la citarabina y los alquilantes como la ciclofosfamida. La mejoría
alcanzada en la última década en los resultados del tratamiento en la LLA-‐T
se ha debido a la intensificación del mismo y a la utilización de ciertos
citostáticos, mencionados anteriormente, con o sin radioterapia. Sin
embargo, pese que éstos han demostrado su eficacia en la LLA-‐T, ninguno de
ellos resulta indispensable ya que se han alcanzado resultados similares en
los distintos grupos cooperativos con protocolos que utilizan distintos
fármacos. A modo de ejemplo, los protocolos UKALL, del Reino Unido, y los
del grupo norteamericano DCFI utilizan dexametasona y asparraginasa
pegilada intensiva y en cambio no administran metotrexato a dosis altas.
Éste, sin embargo, sí se administra en el protocolo alemán BFM o el
norteamericano del SJCRH. En todos los protocolos se utiliza asparraginasa
pero la dosis total es muy variable, entre 100.000 UI/m2 y 500.000 UI/m2. De
igual manera, la profilaxis del SNC también es variable, en algunos se utiliza
metotrexato a dosis altas, en otros se aplica la radioterapia y otros se basan
más en la dexametasona y en el tratamiento intensivo con asparraginasa
que, aunque no atraviesa la barrera hemato-‐encefálica, puede producir
depleción de asparragina en el LCR.
Trasplante alogénico de progenitores hemopoyéticos
El TPH alogénico en primera RC está indicado en el subgrupo de pacientes
con LLA-‐T de alto riesgo por mala respuesta precoz al tratamiento, medida
por la respuesta a la prednisona (Schrauder et al., 2006) o por la ERM.
Desafortunadamente, en la LLA-‐T pediátrica, la intensificación del
tratamiento con el TPH alogénico en primera RC en aquellos pacientes con
niveles elevados de ERM en momentos tardíos del tratamiento no se tradujo

Introducción
78
en una mejoría de la supervivencia de estos niños (Schrappe et al., 2011).
Por ello, los pacientes con niveles altos de ERM tardía serían candidatos a
recibir tratamientos experimentales con nuevos fármacos antileucémicos
con el fin de intentar reducir la ERM antes de la realización del trasplante.
Otra indicación del TPH en primera RC que se ha incluido recientemente en
algún protocolo de tratamiento la constituye la LLA-‐T ETP, aunque no está
claro si el trasplante puede mejorar los resultados. En estos pacientes, se ha
sugerido la posibilidad de aplicar protocolos terapéuticos propios de LMA,
dado su perfil de expresión genética similar al de leucemias mieloides (Zhang
et al., 2012) o de añadir inhibidores de FLT3, ya que FLT3 con frecuencia está
mutado (Zaremba et al., 2012).
A diferencia de la LLA-‐B precursora, en la que el TPH se reserva para las
recaídas medulares precoces, en la LLA-‐T se indica un trasplante en todas las
recaídas medulares. Ello es debido al mal pronóstico que presentan todas las
recaídas medulares en la LLA-‐T (Tallen et al., 2010).
Nuevos fármacos
En los últimos años han aumentado las opciones terapéuticas en la LLA-‐T,
con aprobación de fármacos nuevos para los pacientes refractarios o en
recaída. Algunos de estos fármacos se están estudiando en ensayos clínicos,
incluso en primera línea en aquellos pacientes con mala respuesta precoz al
tratamiento, dado su mal pronóstico con el tratamiento estándar. Entre ellos
se hallan análogos de los nucleósidos como la clofarabina y la nelarabina, la
forodesina, inhibidores de tirosín-‐quinasas e inhibidores de las
gammasecretasas.

Introducción
79
La nelarabina es un profármaco de Ara-‐G (análogo del arabinósido de
guanina) que tiene una citotoxicidad selectiva para los linfoblastos T. Se cree
que esta selectividad se debe a que los linfoblastos T expresan menos la
enzima de la vía de las purinas PNP (Purine Nucleoside Phosphorylase). El
Ara-‐G en el interior de la célula se fosforila y se convierte en Ara-‐GTP, cuya
incorporación en el DNA bloquea la síntesis de DNA. La enzima PNP es
necesaria para metabolizar el Ara-‐GTP y la menor expresión de PNP en el
linfocito T podría explicar la mayor actividad citotóxica de nelarabina en la
LLA de línea T (Homminga et al., 2011). En un estudio fase II con nelarabina
en pacientes con LLA-‐T en recaída se objetivó una buena actividad
antileucémica, aunque con una toxicidad neurológica elevada (Berg et al.,
2005). Dados los resultados prometedores obtenidos en pacientes con LLA-‐T
en recaída, el grupo cooperativo norteamericano COG incluyó nelarabina en
primera línea en pacientes con LLA-‐T de alto riesgo. Los pacientes incluidos
en este ensayo clínico presentaron una tolerancia aceptable al tratamiento y
buenos resultados, con obtención de SLE de alrededor del 70% en pacientes
con mala respuesta precoz al tratamiento (Dunsmore et al., 2012).
Otros tratamientos que se están investigando en pacientes con LLA-‐T que
presentan mutaciones en NOTCH1 son los inhibidores de la gamma-‐
secretasa, que impiden la activación de este receptor. Estos tratamientos se
administran con glucocorticoides para evitar la toxicidad intestinal
secundaria a la inhibición de NOTCH en las células intestinales (Samon et al.,
2012), (Shih y Wang, 2007). Además de los inhibidores de la gamma-‐
secretasa, se están estudiando a nivel pre-‐clínico otros fármacos que inhiben
NOTCH1 por mecanismos distintos, como anticuerpos frente a NOTCH1 (Wu

Introducción
80
et al., 2010) o péptidos que inhiben el complejo de transcripción de NOTCH1
(Moellering et al., 2009).
Una minoría de pacientes con LLA-‐T (6-‐8%), en los que se detecta el
reordenamiento NUP214-‐ABL u otros reordenamientos o amplificaciones de
ABL (BCR-‐ABL, ETV6-‐ABL y EML-‐ABL), podrían ser candidatos a recibir
tratamiento con un TKI como imatinib u otros, por su acción inhibitoria
frente a ABL (Hagemeijer y Graux, 2010) (De Keersmaecker et al., 2005)
(Crombet et al., 2012). De forma similar, los pacientes con mutaciones que
activan JAK1, JAK3 o IL7R se podrían beneficiar de inhibidores de la vía
JAK/STAT que se están desarrollando para el tratamiento de trastornos
mieloproliferativos Philadelphia negativos (Van Vlierberghe y Ferrando,
2012).

Hipótesis y objetivos
81
Hipótesis y objetivos

Hipótesis y objetivos
82

Hipótesis y objetivos
83
HIPÓTESIS DE TRABAJO Y OBJETIVOS
1. Hipótesis
En la LLA existen subgrupos de pacientes de alto riesgo como aquéllos con
LLA Ph+ y LLA-‐T, en los que los factores clínicos y biológicos clásicos no son
útiles para predecir el riesgo de recaída. El hecho de que estos subgrupos
sean enfermedades infrecuentes dificulta el estudio de factores pronósticos.
Por tanto, es necesario disponer de series con un gran número de pacientes
para poder identificar variables con impacto pronóstico. Por otro lado, en el
subgrupo de pacientes con LLA Ph+ del adulto se ha demostrado que el
tratamiento con inhibidores de tirosín-‐quinasas (TKI) ha tenido un impacto
favorable en su supervivencia. La experiencia de estos tratamientos en la LLA
Ph+ pediátrica es escasa y se hace necesario analizar su impacto pronóstico
en los pacientes de esta edad.
-‐El análisis de series amplias de pacientes tratados de forma homogénea con
protocolos del grupo SHOP, que reúne la mayoría de pacientes pediátricos
con LLA en España, permitirá reconocer factores pronósticos predictores de
recaída que pueden ser aplicables en futuros protocolos de tratamiento.
-‐El análisis de las variables pronósticas identificadas permitirá comprobar si
los cambios en el tratamiento aplicados en protocolos sucesivos se traducen
en un aumento de la supervivencia en estos pacientes de alto riesgo.
-‐La eficacia que han mostrado en los pacientes adultos los tratamientos con
TKI frente a una diana biológica compartida (LMC y LLA Ph+) sugiere que
estos tratamientos podrían tener un impacto favorable en la supervivencia
de los pacientes pediátricos con LLA Ph+.

Hipótesis y objetivos
84
2. Objetivos generales
2.1. Identificar factores pronósticos en una serie amplia de pacientes con LLA
Ph+ tratados de forma homogénea según protocolos sucesivos del grupo
SHOP.
2.2. Analizar el efecto de la adición del TKI imatinib a la quimioterapia
intensiva sobre la tasa de remisión completa y la supervivencia de pacientes
pediátricos con LLA Ph+.
2.3. Identificar variables con impacto pronóstico en una serie amplia de
pacientes con LLA-‐T tratados de forma homogénea según protocolos
sucesivos del grupo SHOP.
3.-‐ Objetivos específicos
LLA Ph+
3.1. Analizar las diferentes características clínicas al diagnóstico y la
supervivencia de los pacientes pediátricos con LLA Ph+.
3.2. Analizar la tasa de remisión molecular y los niveles de ERM en los
pacientes tratados con imatinib y quimioterapia intensiva.
3.3. Evaluar la toxicidad de la adición de imatinib al tratamiento con
quimioterapia intensiva.
LLA-‐T
3.4. Analizar las diferentes características clínicas al diagnóstico y la
supervivencia de los pacientes pediátricos con LLA-‐T.
3.5. Comparar los resultados obtenidos con los protocolos del grupo SHOP
con los obtenidos por otros grupos cooperativos internacionales.

Hipótesis y objetivos
85
3.6. Analizar el impacto en la supervivencia de cambios en el tratamiento
destinados a mejorar la respuesta de este grupo de pacientes, tales como el
aumento de la dosis de metotrexato, la intensificación del tratamiento con
asparraginasa y el aumento de la dosis acumulada de ciclofosfamida.

Hipótesis y objetivos
86

Resultados
87
Resultados

Resultados
88

Resultados
89
Trabajo 1 (artículo original)
Susana Rives, Jesús Estella, Pedro Gómez, Mónica López-‐Duarte, Purificación
García de Miguel, Amparo Verdeguer, María José Moreno, José Luis Vivanco,
José Miguel Couselo, Rafael Fernández-‐Delgado, Marisol Maldonado, María
Tasso, Blanca López-‐Ibor, Francisco Lendínez, Ricardo López-‐Almaraz, Javier
Uriz, Montserrat Melo, Ana Fernández-‐Teijeiro, Isidro Rodríguez, Isabel
Badell. En representación de la Sociedad Española de Hemato-‐Oncología
Pediátrica (SHOP/SEHOP)
British Journal of Haematology 2011; 54:600-‐11.
Intermediate dose of imatinib in combination with chemotherapy
followed by allogeneic stem cell transplantation improves early outcome
in paediatric Philadelphia chromosome-‐positive acute lymphoblastic
leukaemia (ALL): results of the Spanish Cooperative Group SHOP studies
ALL-‐94, ALL-‐99 and ALL-‐2005

Resultados
90

Resultados
91
Imatinib a dosis intermedias administrado en combinación con
quimioterapia y seguido de trasplante alogénico de progenitores
hematopoyéticos mejora la supervivencia a corto plazo en la leucemia
linfoblástica aguda Philadelphia positiva: resultados de los estudios del
grupo español cooperativo SHOP/LLA-‐94, LLA-‐99 y LLA-‐2005
La LLA Ph+, tanto en niños como en adultos, tiene peor pronóstico incluso
con tratamientos más intensivos que incluyen el TPH alogénico en primera
remisión completa. La adición de imatinib u otros TKI a la quimioterapia en
pacientes adultos con LLA Ph+ ha aumentado la supervivencia de estos
pacientes. Estos factores motivaron la incorporación de imatinib al
tratamiento quimioterápico en el protocolo de la SEHOP para pacientes
pediátricos con LLA Ph+. El primer artículo del presente trabajo de tesis versa
sobre el análisis de los resultados de tolerabilidad, toxicidad, factores
pronósticos y supervivencia de estos pacientes, en comparación con los
obtenidos con una cohorte histórica de pacientes pediátricos tratados con
quimioterapia intensiva similar pero sin imatinib.
Características de los pacientes
Se incluyeron 43 pacientes pediátricos con LLA Ph+ durante el periodo
comprendido entre febrero de 1994 y abril de 2010. De ellos, 27 recibieron
tratamiento quimioterápico sin imatinib (8 según protocolo SHOP/LLA-‐94 y
19 según protocolo SHOP/LLA-‐99). Dieciséis pacientes recibieron
tratamiento quimioterápico con imatinib, según protocolo SHOP/LLA-‐2005.
La quimioterapia en los 3 protocolos fue similar y la principal diferencia fue
la incorporación en el último protocolo de tratamiento (SHOP/LLA-‐2005) de

Resultados
92
imatinib de forma continua desde el día +15 del tratamiento de inducción.
En todos los protocolos se indicó la realización de TPH alogénico de donante
HLA-‐idéntico en primera RC, tanto familiar como de donante no relacionado.
Las características al diagnóstico de los pacientes que recibieron imatinib y
de los del grupo pre-‐imatinib fueron comparables y se describen en la tabla II
del artículo.
Respuesta
La tabla II resume la respuesta al tratamiento. No hubo diferencias en ambos
grupos en cuanto a la respuesta precoz en el día +15, antes del inicio de
imatinib. La tasa de RC al final del tratamiento de inducción fue del 100% en
el grupo de imatinib y del 89% en el grupo pre-‐imatinib. Los tres pacientes
con fallo de inducción alcanzaron la RC tras recibir quimioterapia de rescate.
En cuanto a la ERM determinada mediante PCR cuantitativa para el
reordenamiento BCR-‐ABL, todos los pacientes analizados del grupo de
imatinib estaban en remisión molecular en el momento del trasplante.
Evolución
Ningún paciente falleció durante la fase de inducción. Un paciente de la
cohorte que recibió imatinib murió debido a una complicación infecciosa
durante la consolidación previa a la realización del trasplante.
El trasplante alogénico se realizó en 32 pacientes en primera RC: 17 de 24
pacientes de la cohorte pre-‐imatinib (71%) y 15 de 16 del grupo de imatinib
(94%). Ocho niños fallecieron en primera RC debido a complicaciones
relacionadas con el trasplante, siete del grupo pre-‐imatinib y uno del grupo
de imatinib.

Resultados
93
Recaídas
Nueve pacientes de la cohorte pre-‐imatinib y uno del grupo de imatinib
recayeron, con una incidencia acumulada de recaída a los 2 años de 33,3 ±
9,6% en la cohorte pre-‐imatinib y de 9,5 ± 9% en la cohorte de imatinib
(p=0,155). Antes del trasplante un niño recayó en el protocolo SHOP/LLA-‐99
y ninguno en el protocolo SHOP/LLA-‐2005. Seis pacientes recayeron tras el
trasplante con un tiempo mediano desde el trasplante hasta la recaída de
7,3 meses (extremos 2,3-‐28), 5 en la cohorte pre-‐imatinib y 1 en el grupo de
imatinib. Entre los pacientes que no recibieron TPH alogénico, 3 recayeron
entre 15,4 y 20,7 meses desde la RC.
No se observó ninguna segunda neoplasia en el momento de la publicación
de este trabajo.
Globalmente, la mortalidad no relacionada con la recaída a los 2 años de
haber alcanzado la RC fue de 29,2 ± 9,3% y de 14,3 ± 9,3 en las cohortes pre-‐
imatinib e imatinib respectivamente (p=0,465).
Supervivencia
En el momento del análisis, de los 43 pacientes de la serie global, 21
permanecían vivos en primera RC. La mediana de seguimiento fue de 109
meses (extremos 61-‐184) para la cohorte pre-‐imatinib y de 39 meses
(extremos 7-‐55) para el grupo de imatinib. La probabilidad de SLE a los 3
años fue de 29,6% (± 9%) para la cohorte pre-‐imatinib y de 78,7% (±11%)
para el grupo de imatinib, respectivamente (p=0,01) (ver figura 2). La
probabilidad de SG a los 3 años fue de 34,6% (±9%) y de 86,5% (±9%) para
los grupos pre-‐imatinib e imatinib respectivamente (p<0,01) (ver figura 3).

Resultados
94
Toxicidad
Por lo general, los pacientes presentaron buena tolerancia a imatinib, a dosis
intermedias de 260 mg/m2/día administrado desde el día +15 del
tratamiento de inducción. Los pacientes que recibieron imatinib presentaron
con mayor frecuencia naúseas y mucositis grado III-‐IV durante la inducción,
pero dichas diferencias no fueron estadísticamente significativas. Además, se
observó con más frecuencia en el grupo que recibió imatinib una elevación
importante (grado III-‐IV) de las transaminasas durante el tratamiento de
consolidación. Esta elevación fue transitoria sin interrupción o ajuste de osis
del tratamiento con imatinib y no se asoció en ningún caso a insuficiencia
hepática o a elevación grado III-‐IV de la bilirrubina. Tanto la mucositis como
la elevación de las transaminasas podrían tener relación con la
administración concomitante de imatinib y metotrexato a dosis altas
(5g/m2), que se administró durante la fase de consolidación en los pacientes
del protocolo SHOP/LLA-‐2005. Esta dosis de metotrexato fue superior a la
dosis administradas en los protocolos pre-‐imatinib SHOP/LLA-‐94 y
SHOP/LLA-‐99, que fue de 3 g/m2.
Un paciente del grupo que recibió imatinib falleció por sepsis por Aeromonas
hydrophila caviae de origen intestinal durante la fase de aplasia medular tras
la administración del bloque C de quimioterapia, previo a recibir el TPH
previsto.
En conclusión, la adición de imatinib a la quimioterapia intensiva y seguida
de TPH alogénico fue bien tolerada en la población pediátrica y consiguió
una mejoría muy importante en la supervivencia a corto plazo, tanto la libre
de enfermedad como la supervivencia global.

Resultados
95
Intermediate dose of imatinib in combination with chemotherapyfollowed by allogeneic stem cell transplantation improves earlyoutcome in paediatric Philadelphia chromosome-positive acutelymphoblastic leukaemia (ALL): results of the SpanishCooperative Group SHOP studies ALL-94, ALL-99 and ALL-2005
Susana Rives,1,2 Jesus Estella,1 Pedro
Gomez,3 Monica Lopez-Duarte,4
Purificacion Garcıa de Miguel,5 Amparo
Verdeguer,6 Maria Jose Moreno,7 Jose
Luis Vivanco,8 Jose Miguel Couselo,9
Rafael Fernandez-Delgado,10 Marisol
Maldonado,11 Marıa Tasso,12 Blanca
Lopez-Ibor,13 Francisco Lendınez,14
Ricardo Lopez-Almaraz,15 Javier Uriz,16
Montserrat Melo,17 Ana Fernandez-
Teijeiro,18 Isidoro Rodrıguez19 and Isabel
Badell20
1Paediatric Haematology Department, Hospital
Sant Joan de Deu de Barcelona, 2Universitat de
Barcelona, Barcelona, 3Hospital Reina Sofıa,
Cordoba, 4Hematologıa, Hospital Universitario
Marques de Valdecilla, Santander, 5Hemato-
Oncologıa Pediatrica, Hospital Universitario La
Paz, Madrid, 6Hemato-Oncologıa Pediatrica,
Hospital Universitario La Fe, Valencia, 7Hospital
Virgen de las Nieves, Granada, 8Hospital Doce de
Octubre, Madrid, 9Hospital Xeral de Galicia,
Santiago de Compostela, 10Hospital Clınico de
Valencia, Valencia, 11Hospital Ramon y Cajal,
Madrid, 12Hospital Universitario de Alicante,
Alicante, 13Hospital Monteprıncipe, Madrid,14Hospital Torrecardenas, Almerıa, 15Hospital
Universitario de Canarias, San Cristobal de la
Laguna Tenerife, 16Hospital de Donostia, San
Sebastian, 17Hospital Parc Taulı, Sabadell,18Hospital Virgen de la Macarena, Sevilla,19Hospital Marıa Teresa Herrera/Juan Canalejo,
La Coruna, and 20Hospital de la Santa Creu i Sant
Pau, Universitat Autonoma de Barcelona, Spain
Received 10 March 2011; accepted for
publication 15 May 2011
Correspondence: Susana Rives, Haematology
Department, Hospital Sant Joan de Deu de
Barcelona, Passeig Sant Joan de Deu 2, 08950
Esplugues de Llobregat, Barcelona, Spain.
E-mail: [email protected]
Summary
Philadelphia-chromosome acute lymphoblastic leukaemia (Ph+ ALL) is asubgroup of ALL with very high risk of treatment failure. We report here theresults of the Sociedad Espanola de Hematologıa y Oncologıa Pediatricas(SEHOP/SHOP) in paediatric Ph+ ALL treated with intermediate-doseimatinib concurrent with intensive chemotherapy. The toxicities andoutcome of these patients were compared with historical controls notreceiving imatinib. Patients with Ph+ ALL aged 1–18 years were enrolled inthree consecutive ALL/SHOP trials (SHOP-94/SHOP-99/SHOP-2005). In theSHOP-2005 trial, imatinib (260 mg/m2 per day) was given on day-15 ofinduction. Allogeneic haematopoietic stem-cell transplantation (HSCT) froma matched related or unrelated donor was scheduled in first completeremission (CR1). Forty-three patients were evaluable (22 boys, median age6Æ8 years, range, 1Æ2–15). Sixteen received imatinib whereas 27 receivedsimilar chemotherapy without imatinib. Seventeen of 27 and 15 of 16 patientsin the non-imatinib and imatinib cohort, respectively, underwent HSCT inCR1. With a median follow-up of 109 and 39 months for the non-imatiniband imatinib cohorts, the 3-year event-free survival (EFS) was 29Æ6% and78Æ7%, respectively (P = 0Æ01). These results show that, compared tohistorical controls, intermediate dose of imatinib given concomitantly withchemotherapy and followed by allogeneic HSCT markedly improved earlyEFS in paediatric Ph+ ALL.
Keywords: Philadelphia-chromosome acute lymphoblastic leukaemia, BCR-ABL1, children, imatinib, stem cell transplantation.
research paper
First published online 28 June 2011doi:10.1111/j.1365-2141.2011.08783.x ª 2011 Blackwell Publishing Ltd, British Journal of Haematology, 154, 600–611

Resultados
96
Childhood acute lymphoblastic leukaemia (ALL) can be cured
in more than 80% of cases with current intensive chemother-
apy protocols (Pui & Evans, 2006). However, there are some
minor subgroups of children with very high risk ALL in whom
cure rates are <50%. Philadelphia positive ALL (Ph+ ALL) is a
subtype of very high risk ALL in which allogeneic haemato-
poietic stem cell transplantation (HSCT) in first complete
remission (CR1) is considered the standard treatment (Arico
et al, 2000, 2010; Satwani et al, 2007). Despite this intensive
treatment, relapses prior to and after HSCT are frequent and
only 30–50% of these children can be cured with this approach
(Schrappe et al, 1998; Arico et al, 2000; Roy et al, 2005;
Gandemer et al, 2009).
Imatinib is a tyrosine kinase inhibitor (TKI) that targets the
BCR-ABL1 fusion oncogenic protein, product of the trans-
location t(9;22) characteristic of Ph+ ALL. In the last decade,
imatinib has been incorporated into the treatment of Ph+ ALL
with promising results. Several studies have demonstrated that
imatinib in combination with intensive chemotherapy
increases complete remission (CR) and allogeneic HSCT rates
and improves early outcome in adult Ph+ ALL (Thomas et al,
2004; Towatari et al, 2004; Lee et al, 2005; Wassmann et al,
2006; Yanada et al, 2008). Its efficacy regarding long-term
outcome has also recently been reported and is currently
accepted as standard first-line treatment in adult Ph+ ALL
(Ribera et al, 2010; Bassan et al, 2010; Fielding et al, 2010;
Pfeifer et al, 2010). Second generation TKI and minimal
residual disease (MRD) determination to guide treatment are
now being tested to further improve outcome in these patients
(Cazzaniga et al, 2002; Pane et al, 2005; Yanada et al, 2008; Lee
et al, 2009; Castillo et al, 2010; Ravandi et al, 2010a,b). Studies
focusing on Ph+ ALL in children are scarce due to its rarity
(only 3–5% of childhood ALL) (Schrappe et al, 1998; Arico
et al, 2000, 2010; Heerema et al, 2004; Roy et al, 2005;
Bhojwani et al, 2009; Gandemer et al, 2009; Schultz et al,
2009) and additional information on indications, dosage,
tolerability and efficacy of imatinib in combination with
chemotherapy is needed (Barr, 2010). Phase I studies with
imatinib as a sole agent in refractory and relapsed Ph+ ALL
were promising with a good toxicity profile (Champagne et al,
2004). The Children’s Oncology Group published the results of
their trial of intensive chemotherapy in combination with
imatinib given at a dose of 340 mg/m2 per day after induction
treatment and followed by allogeneic HSTC only if a matched
related donor were available (Schultz et al, 2009). They
reported an impressive improvement in early outcome [81%
3-year event-free survival (EFS)] with an acceptable toxicity
profile.
In view of the good results observed in adults with imatinib
in combination with chemotherapy, in 2005 the Sociedad
Espanola de Hematologıa y Oncologıa Pediatricas (SEHOP/
SHOP) designed a protocol in paediatric Ph+ ALL using this
approach followed by allogeneic HSCT from a matched or an
unrelated donor in CR1. The objective of the study was to
assess the efficacy in terms of CR rate, allogeneic HSCT rate in
CR1 and survival of children newly diagnosed with Ph+ ALL
who were treated with imatinib at an intermediate dose
(260 mg/m2 per day) concurrently with chemotherapy. We
present here the results of the ALL/SHOP-2005 study and
compare these preliminary results with those obtained with
our historical control (pre-imatinib cohort).
Patients and methods
Patients
Newly diagnosed Ph+ ALL patients aged 1–18 years enrolled in
the three consecutive SHOP protocols ALL/SHOP-94, ALL/
SHOP-99 and ALL/SHOP-2005 between February 1994 and
April-2010 entered the study. All patients had the Philadelphia
chromosome t(9;22)(q34;q11.2) detected by conventional or
molecular cytogenetics, or BCR-ABL1 fusion transcript
detected by reverse transcription polymerase chain reaction
(RT-PCR). Patients enrolled in protocol ALL/SHOP 2005,
from April 2005 to April 2010 constitute the study group and
received imatinib concurrently with chemotherapy.
Patients enrolled in protocols ALL/SHOP-94 and ALL/
SHOP-99, from February 1994 to March 2005 received a
similar chemotherapy protocol without imatinib and consti-
tute the historical control group. Patients treated according
protocols ALL/SHOP-94 and ALL/SHOP-99 were analysed
together and had no differences in clinical characteristics at
presentation and no significant differences in outcome.
Diagnostic and follow-up studies
Pre-treatment evaluations included clinical history and phys-
ical examination, complete blood count with differential, bone
marrow aspiration for cytology, flow cytometry, cytogenetic
and molecular studies and cytological study of cerebrospinal
fluid.
Flow cytometry studies
Immunophenotypic study of blast population by flow cyto-
metry was performed in bone marrow and/or peripheral blood
samples at diagnosis. In protocol ALL/SHOP-2005 flow
cytometry was also used for detection of minimal residual
disease (MRD) in all patients at different time points. The
MRD studies were performed at the site of diagnosis and were
not centralized.
Cytogenetic studies
Bone marrow samples were studied at the site of diagnosis with
standard banding techniques at diagnosis and follow-up.
Karyotypes were classified according to the International
System for Human Cytogenetic Nomenclature (Mitelman,
1995). BCR-ABL1 fusion gene was screened at diagnosis with
the fluorescence in situ hybridization (FISH) technique in cases
Imatinib in Paediatric Ph+ ALL
ª 2011 Blackwell Publishing Ltd, British Journal of Haematology, 154, 600–611 601

Resultados
97
in which no metaphases were obtained or as an initial
screening method for the presence of the BCR-ABL1 rear-
rangement in some centres.
Molecular studies
Qualitative and quantitative RT-PCR for BCR-ABL1 transcript
detection was performed on bone marrow samples at diagnosis
according to the guidelines established by the Europe Against
Cancer Programme (van Dongen et al, 1999; Gabert et al, 2003).
In ALL/SHOP-2005 protocol, quantitative RT-PCR was recom-
mended for detection of MRD at the time points described
below. These studies were not centralized but performed in
several reference laboratories in which national quality control
programmes were implemented on a regular basis.
Response evaluation and study of MRD
Response evaluation as assessed by morphology was performed
on day-15 bone marrow and at the end of induction. Good and
poor early responses were defined by less or more than 5% of
blasts on day-15 bone marrow, respectively. CR was defined by
<5% blasts with normal cellularity on bone marrow smears,
normal peripheral blood cell counts and no evidence of
extramedullary disease. In protocol ALL/SHOP 2005, bone
marrow aspirates with assessment of MRD by flow cytometry
and/or quantitative RT-PCR were performed at the end of
induction, at the beginning of consolidation block A and prior
to HSCT.
Study design, therapy and patient stratification
ALL/SHOP-94, ALL/SHOP-99 and ALL/SHOP 2005 were
three consecutive treatment protocols conducted by SHOP
(SEHOP). The protocols were reviewed and approved by the
institutional review boards of each of the participating centres
and were conducted in accordance with the Declaration of
Helsinki. Patients, their parents or guardians provided written
consent before entering the study.
All Philadelphia chromosome and/or BCR-ABL1 positive
patients were assigned to the very high risk group and were
allocated to intensive treatment followed by allogeneic HSCT if
a suitable human leucocyte antigen (HLA)-identical donor
(related or unrelated) were available. The ALL/SHOP-94
treatment schedule has been described elsewhere (Badell et al,
2008) and the ALL/SHOP-99 and ALL/SHOP 2005 treatment
schedules are shown in Table I. In the subset of Philadelphia –
chromosome positive patients, the main difference between the
ALL/SHOP 2005 and the prior ALL/SHOP-99 and ALL/
SHOP-94 protocols was the use of imatinib at an intermediate
dose (260 mg/m2 per day) from day 15 of induction treatment
and until HSCT in the ALL/SHOP 2005.
Patients not attaining CR at the end of induction were
excluded from the protocol and underwent second-line
therapy. Patients achieving CR received three consolidation
cycles, consisting of high-dose methotrexate and mercaptop-
urine and a fourth cycle of consolidation with high-dose
cytarabine. These consolidation cycles were followed by three
intensive consolidation blocks. Patients with an HLA-identical
matched sibling or unrelated donor were submitted to
allogeneic HSCT, whereas those without a suitable donor
received two additional blocks of consolidation and underwent
an autologous HSCT or continued with maintenance chemo-
therapy according to their physicians’ choice.
The HSCT procedure was not standardized and varied
according to the transplant policies of each Bone Marrow
Transplant Unit. The preparative regimen for autologous or
allogeneic HSCT (either from an HLA-identical sibling or from
an unrelated donor) was decided locally, although in most
cases it consisted of fractionated total body irradiation (total
dose 13 Gy), cyclophosphamide (120 mg/kg) and etoposide
(60 mg/kg). In children <3 years old, total body irradiation
was not indicated and oral busulfan at a dose of 16 mg/kg
(with dose adjustments according to age) was recommended.
Parenteral busulfan was used in some centres at equivalent
doses and adjusted to age. Recommended graft-versus-host
disease prophylaxis was standard cyclosporine and short-
course methotrexate.
Imatinib therapy
Imatinib was administered in the ALL/SHOP-2005 protocol,
daily from day 15 of induction (260 mg/m2 per day) until
HSCT. The protocol did not contemplate the use of imatinib
as prophylactic treatment after HSCT to prevent relapses.
Nevertheless, the use of imatinib and/or donor lymphocyte
infusions was allowed as pre-emptive treatment in patients
with molecular relapse (MRD positive after HSCT), the
therapeutic decision being left to the treating physicians.
Toxicity assessment
Toxicity was evaluated on the basis of the World Health
Organization Toxicity Criteria (Miller et al, 1981). Haemato-
logical recovery, defined as a neutrophil count of at least
1 · 109/l and a platelet count of at least 75 · 109/l, was
required for the initiation of each course of consolidation. In
addition, normal creatinine (adjusted to age) as well as
bilirubin <34Æ2 lmol/l were required before administration of
chemotherapy.
Statistical analysis
All patients were centrally registered at the SHOP operation
office (Clever Instruments, Barcelona, Spain). Overall survival
(OS) was measured from the date of start of therapy until
death by any cause. EFS was defined as the time from start of
treatment to relapse or death. Induction failures were also
considered events. Transplant-related mortality (TRM) was
defined as death occurring after HSCT in relapse-free patients.
S. Rives et al
602 ª 2011 Blackwell Publishing Ltd, British Journal of Haematology, 154, 600–611
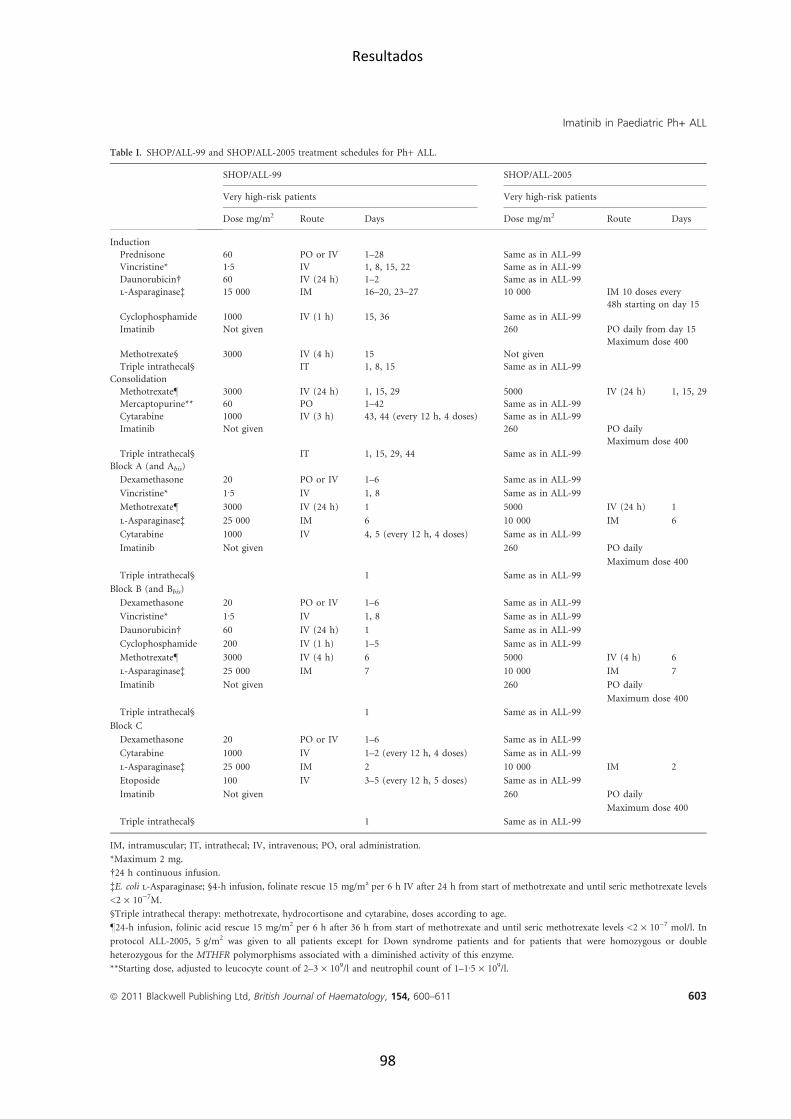
Resultados
98
Table I. SHOP/ALL-99 and SHOP/ALL-2005 treatment schedules for Ph+ ALL.
SHOP/ALL-99 SHOP/ALL-2005
Very high-risk patients Very high-risk patients
Dose mg/m2 Route Days Dose mg/m2 Route Days
InductionPrednisone 60 PO or IV 1–28 Same as in ALL-99Vincristine* 1Æ5 IV 1, 8, 15, 22 Same as in ALL-99Daunorubicin! 60 IV (24 h) 1–2 Same as in ALL-99l-Asparaginase" 15 000 IM 16–20, 23–27 10 000 IM 10 doses every
48h starting on day 15Cyclophosphamide 1000 IV (1 h) 15, 36 Same as in ALL-99Imatinib Not given 260 PO daily from day 15
Maximum dose 400Methotrexate§ 3000 IV (4 h) 15 Not givenTriple intrathecal§ IT 1, 8, 15 Same as in ALL-99
ConsolidationMethotrexate– 3000 IV (24 h) 1, 15, 29 5000 IV (24 h) 1, 15, 29Mercaptopurine** 60 PO 1–42 Same as in ALL-99Cytarabine 1000 IV (3 h) 43, 44 (every 12 h, 4 doses) Same as in ALL-99Imatinib Not given 260 PO daily
Maximum dose 400Triple intrathecal§ IT 1, 15, 29, 44 Same as in ALL-99
Block A (and Abis)
Dexamethasone 20 PO or IV 1–6 Same as in ALL-99
Vincristine* 1Æ5 IV 1, 8 Same as in ALL-99
Methotrexate– 3000 IV (24 h) 1 5000 IV (24 h) 1
l-Asparaginase" 25 000 IM 6 10 000 IM 6
Cytarabine 1000 IV 4, 5 (every 12 h, 4 doses) Same as in ALL-99
Imatinib Not given 260 PO daily
Maximum dose 400
Triple intrathecal§ 1 Same as in ALL-99
Block B (and Bbis)
Dexamethasone 20 PO or IV 1–6 Same as in ALL-99
Vincristine* 1Æ5 IV 1, 8 Same as in ALL-99
Daunorubicin! 60 IV (24 h) 1 Same as in ALL-99
Cyclophosphamide 200 IV (1 h) 1–5 Same as in ALL-99
Methotrexate– 3000 IV (4 h) 6 5000 IV (4 h) 6
l-Asparaginase" 25 000 IM 7 10 000 IM 7
Imatinib Not given 260 PO daily
Maximum dose 400
Triple intrathecal§ 1 Same as in ALL-99
Block C
Dexamethasone 20 PO or IV 1–6 Same as in ALL-99
Cytarabine 1000 IV 1–2 (every 12 h, 4 doses) Same as in ALL-99
l-Asparaginase" 25 000 IM 2 10 000 IM 2
Etoposide 100 IV 3–5 (every 12 h, 5 doses) Same as in ALL-99
Imatinib Not given 260 PO daily
Maximum dose 400
Triple intrathecal§ 1 Same as in ALL-99
IM, intramuscular; IT, intrathecal; IV, intravenous; PO, oral administration.
*Maximum 2 mg.
!24 h continuous infusion.
"E. coli l-Asparaginase; §4-h infusion, folinate rescue 15 mg/m2 per 6 h IV after 24 h from start of methotrexate and until seric methotrexate levels
<2 · 10)7M.
§Triple intrathecal therapy: methotrexate, hydrocortisone and cytarabine, doses according to age.
–24-h infusion, folinic acid rescue 15 mg/m2 per 6 h after 36 h from start of methotrexate and until seric methotrexate levels <2 · 10)7 mol/l. In
protocol ALL-2005, 5 g/m2 was given to all patients except for Down syndrome patients and for patients that were homozygous or double
heterozygous for the MTHFR polymorphisms associated with a diminished activity of this enzyme.
**Starting dose, adjusted to leucocyte count of 2–3 · 109/l and neutrophil count of 1–1Æ5 · 109/l.
Imatinib in Paediatric Ph+ ALL
ª 2011 Blackwell Publishing Ltd, British Journal of Haematology, 154, 600–611 603
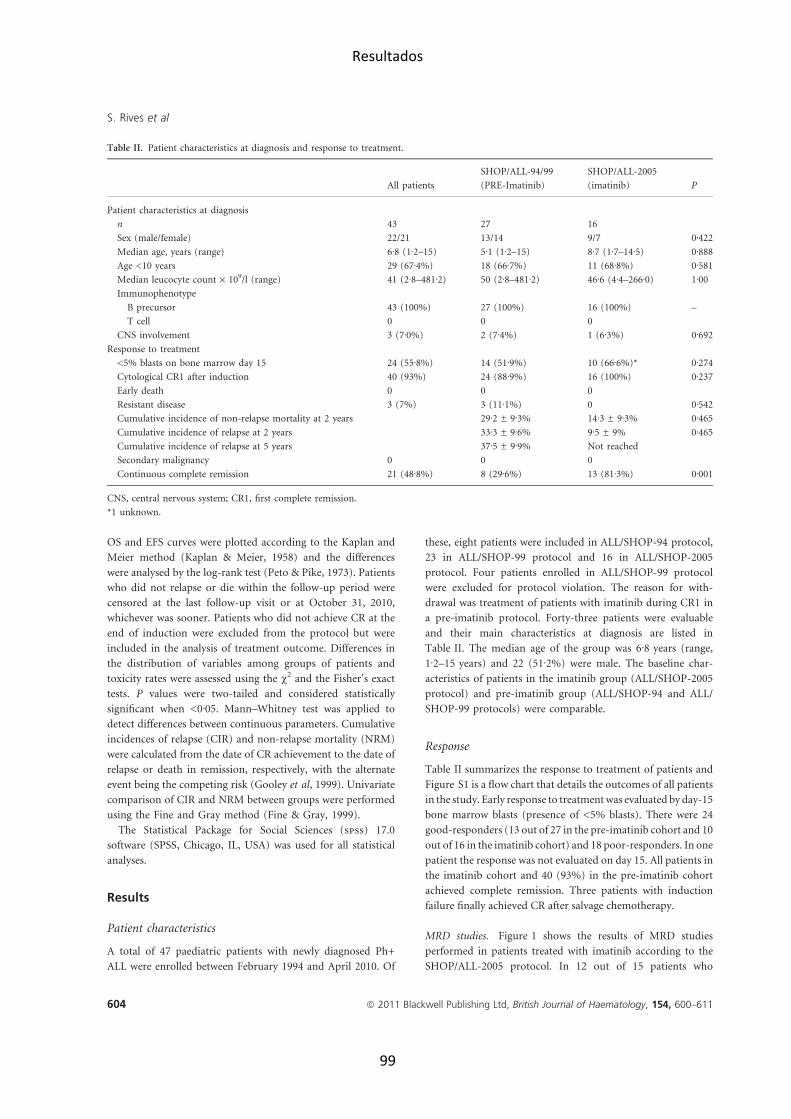
Resultados
99
OS and EFS curves were plotted according to the Kaplan and
Meier method (Kaplan & Meier, 1958) and the differences
were analysed by the log-rank test (Peto & Pike, 1973). Patients
who did not relapse or die within the follow-up period were
censored at the last follow-up visit or at October 31, 2010,
whichever was sooner. Patients who did not achieve CR at the
end of induction were excluded from the protocol but were
included in the analysis of treatment outcome. Differences in
the distribution of variables among groups of patients and
toxicity rates were assessed using the v2 and the Fisher’s exact
tests. P values were two-tailed and considered statistically
significant when <0Æ05. Mann–Whitney test was applied to
detect differences between continuous parameters. Cumulative
incidences of relapse (CIR) and non-relapse mortality (NRM)
were calculated from the date of CR achievement to the date of
relapse or death in remission, respectively, with the alternate
event being the competing risk (Gooley et al, 1999). Univariate
comparison of CIR and NRM between groups were performed
using the Fine and Gray method (Fine & Gray, 1999).
The Statistical Package for Social Sciences (spss) 17.0
software (SPSS, Chicago, IL, USA) was used for all statistical
analyses.
Results
Patient characteristics
A total of 47 paediatric patients with newly diagnosed Ph+
ALL were enrolled between February 1994 and April 2010. Of
these, eight patients were included in ALL/SHOP-94 protocol,
23 in ALL/SHOP-99 protocol and 16 in ALL/SHOP-2005
protocol. Four patients enrolled in ALL/SHOP-99 protocol
were excluded for protocol violation. The reason for with-
drawal was treatment of patients with imatinib during CR1 in
a pre-imatinib protocol. Forty-three patients were evaluable
and their main characteristics at diagnosis are listed in
Table II. The median age of the group was 6Æ8 years (range,
1Æ2–15 years) and 22 (51Æ2%) were male. The baseline char-
acteristics of patients in the imatinib group (ALL/SHOP-2005
protocol) and pre-imatinib group (ALL/SHOP-94 and ALL/
SHOP-99 protocols) were comparable.
Response
Table II summarizes the response to treatment of patients and
Figure S1 is a flow chart that details the outcomes of all patients
in the study. Early response to treatment was evaluated by day-15
bone marrow blasts (presence of <5% blasts). There were 24
good-responders (13 out of 27 in the pre-imatinib cohort and 10
out of 16 in the imatinib cohort) and 18 poor-responders. In one
patient the response was not evaluated on day 15. All patients in
the imatinib cohort and 40 (93%) in the pre-imatinib cohort
achieved complete remission. Three patients with induction
failure finally achieved CR after salvage chemotherapy.
MRD studies. Figure 1 shows the results of MRD studies
performed in patients treated with imatinib according to the
SHOP/ALL-2005 protocol. In 12 out of 15 patients who
Table II. Patient characteristics at diagnosis and response to treatment.
All patients
SHOP/ALL-94/99
(PRE-Imatinib)
SHOP/ALL-2005
(imatinib) P
Patient characteristics at diagnosis
n 43 27 16
Sex (male/female) 22/21 13/14 9/7 0Æ422
Median age, years (range) 6Æ8 (1Æ2–15) 5Æ1 (1Æ2–15) 8Æ7 (1Æ7–14Æ5) 0Æ888
Age <10 years 29 (67Æ4%) 18 (66Æ7%) 11 (68Æ8%) 0Æ581
Median leucocyte count · 109/l (range) 41 (2Æ8–481Æ2) 50 (2Æ8–481Æ2) 46Æ6 (4Æ4–266Æ0) 1Æ00
Immunophenotype
B precursor 43 (100%) 27 (100%) 16 (100%) –
T cell 0 0 0
CNS involvement 3 (7Æ0%) 2 (7Æ4%) 1 (6Æ3%) 0Æ692
Response to treatment
<5% blasts on bone marrow day 15 24 (55Æ8%) 14 (51Æ9%) 10 (66Æ6%)* 0Æ274
Cytological CR1 after induction 40 (93%) 24 (88Æ9%) 16 (100%) 0Æ237
Early death 0 0 0
Resistant disease 3 (7%) 3 (11Æ1%) 0 0Æ542
Cumulative incidence of non-relapse mortality at 2 years 29Æ2 ± 9Æ3% 14Æ3 ± 9Æ3% 0Æ465
Cumulative incidence of relapse at 2 years 33Æ3 ± 9Æ6% 9Æ5 ± 9% 0Æ465
Cumulative incidence of relapse at 5 years 37Æ5 ± 9Æ9% Not reached
Secondary malignancy 0 0 0
Continuous complete remission 21 (48Æ8%) 8 (29Æ6%) 13 (81Æ3%) 0Æ001
CNS, central nervous system; CR1, first complete remission.
*1 unknown.
S. Rives et al
604 ª 2011 Blackwell Publishing Ltd, British Journal of Haematology, 154, 600–611

Resultados
100
underwent HSCT, molecular MRD assessment was performed
pre-transplant. Of note, all of them were in molecular CR
(negative for the BCR-ABL1 rearrangement) at that time point.
Outcome
There were no induction deaths. One patient in the imatinib
group died from an infectious complication after a chemo-
therapy consolidation block before planned HSCT.
Allogeneic HSCT was performed in 32 patients in CR1 (17
of 24 in the pre-imatinib group and 15 out of 16 in the
imatinib group). Reasons for not undergoing allogeneic
transplantation were: (i) in the pre-imatinib group, two
patients treated according ALL/SHOP-94 protocol did not
proceed to HSCT due to their initial leucocyte count of
<25 · 109/l. In that protocol, patients with Ph+ and leucocyte
count of <25 · 109/l were not candidates for an allogeneic
HSCT in CR1. In one patient the reason for not undergoing
HSCT is unknown. One child relapsed before HSCT and
underwent HSCT in second CR (CR2). Three patients did not
have an HLA-identical donor (related or unrelated), two of
them underwent an autologous HSCT in CR1 and one received
intensive chemotherapy and (ii) in the imatinib group, one
patient died from an infectious complication after a chemo-
therapy block prior to planned allogeneic HSCT.
The median time from CR to HSCT was 5Æ6 months (range,
3–14Æ8 months), being 5Æ2 months (range, 3–14Æ8 months) for
the pre-imatinib group and 6Æ5 months (range, 4Æ8–
14Æ7 months) for the imatinib group. Conditioning regimens
were cyclophosphamide, etoposide and total body irradiation
in most patients. The donor was an HLA-identical sibling in 11
patients, and unrelated donor in 21. Eight patients died in CR1
due to transplant-related complications, seven in the pre-
imatinib group and one in the imatinib group.
Relapses
Nine patients in the pre-imatinib group and only one patient
in the imatinib cohort relapsed after CR achievement, resulting
in a cumulative incidence of relapse (CIR) at 2 and 5 years of
33Æ3 ± 9Æ6% and 37Æ5 ± 9Æ9% in the pre-imatinib and
9Æ5 ± 9% (2 and 5-year CIR) in the imatinib group, respec-
tively (P = 0Æ155). One child relapsed before HSCT in ALL/
SHOP-99 and none in ALL/SHOP-2005.
Six patients relapsed after allogeneic HSCT with a median
time to relapse of 7Æ3 months (range, 2Æ3–28 months), 5 in the
pre-imatinib group and 1 in the imatinib group. Among the
Event-free survival (months)
Non imatinib cohort n = 27 3 year-pEFS 29·6% (+/–9)
Imatinib cohort n = 16 3 year-pEFS 78·7% (+/–11)
P = 0·01
Fig 2. Event-Free Survival in the imatinib and pre-imatinib cohorts.Probabilities of Event-Free Survival of the patients included in ALL/SHOP-94-99 trials (pre-imatinib group) and ALL/SHOP-2005 trial(imatinib group).
Non imatinib cohort n = 27 3 year-pOS 34·6% (+/–9)
Imatinib cohort n = 16 3 year-pOS 86·5% (+/–9)
P < 0·01
Fig 3. Overall Survival in the imatinib and pre-imatinib cohorts.Probabilities of Overall Survival of the patients included in ALL/SHOP-94-99 trials (pre-imatinib group) and ALL/SHOP-2005 trial(imatinib group).
BCR-ABL1 BCR-ABL1
n n n–
n n n
Fig 1. BCR-ABL1 status of patients treated as per protocol ALL/SHOP-2005 (imatinib group) at different time-points*. The number ofpatients analysed at each time-point is indicated in brackets.
Imatinib in Paediatric Ph+ ALL
ª 2011 Blackwell Publishing Ltd, British Journal of Haematology, 154, 600–611 605
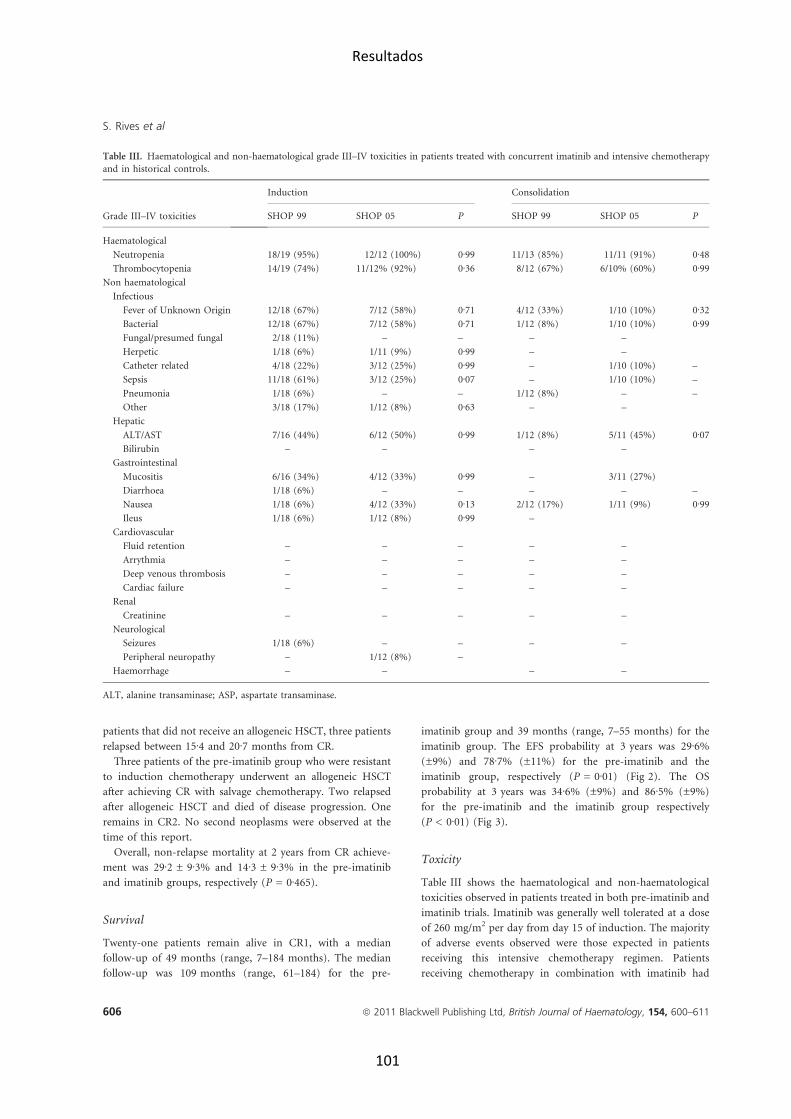
Resultados
101
patients that did not receive an allogeneic HSCT, three patients
relapsed between 15Æ4 and 20Æ7 months from CR.
Three patients of the pre-imatinib group who were resistant
to induction chemotherapy underwent an allogeneic HSCT
after achieving CR with salvage chemotherapy. Two relapsed
after allogeneic HSCT and died of disease progression. One
remains in CR2. No second neoplasms were observed at the
time of this report.
Overall, non-relapse mortality at 2 years from CR achieve-
ment was 29Æ2 ± 9Æ3% and 14Æ3 ± 9Æ3% in the pre-imatinib
and imatinib groups, respectively (P = 0Æ465).
Survival
Twenty-one patients remain alive in CR1, with a median
follow-up of 49 months (range, 7–184 months). The median
follow-up was 109 months (range, 61–184) for the pre-
imatinib group and 39 months (range, 7–55 months) for the
imatinib group. The EFS probability at 3 years was 29Æ6%
(±9%) and 78Æ7% (±11%) for the pre-imatinib and the
imatinib group, respectively (P = 0Æ01) (Fig 2). The OS
probability at 3 years was 34Æ6% (±9%) and 86Æ5% (±9%)
for the pre-imatinib and the imatinib group respectively
(P < 0Æ01) (Fig 3).
Toxicity
Table III shows the haematological and non-haematological
toxicities observed in patients treated in both pre-imatinib and
imatinib trials. Imatinib was generally well tolerated at a dose
of 260 mg/m2 per day from day 15 of induction. The majority
of adverse events observed were those expected in patients
receiving this intensive chemotherapy regimen. Patients
receiving chemotherapy in combination with imatinib had
Table III. Haematological and non-haematological grade III–IV toxicities in patients treated with concurrent imatinib and intensive chemotherapyand in historical controls.
Grade III–IV toxicities
Induction Consolidation
SHOP 99 SHOP 05 P SHOP 99 SHOP 05 P
Haematological
Neutropenia 18/19 (95%) 12/12 (100%) 0Æ99 11/13 (85%) 11/11 (91%) 0Æ48
Thrombocytopenia 14/19 (74%) 11/12% (92%) 0Æ36 8/12 (67%) 6/10% (60%) 0Æ99
Non haematological
Infectious
Fever of Unknown Origin 12/18 (67%) 7/12 (58%) 0Æ71 4/12 (33%) 1/10 (10%) 0Æ32
Bacterial 12/18 (67%) 7/12 (58%) 0Æ71 1/12 (8%) 1/10 (10%) 0Æ99
Fungal/presumed fungal 2/18 (11%) – – – –
Herpetic 1/18 (6%) 1/11 (9%) 0Æ99 – –
Catheter related 4/18 (22%) 3/12 (25%) 0Æ99 – 1/10 (10%) –
Sepsis 11/18 (61%) 3/12 (25%) 0Æ07 – 1/10 (10%) –
Pneumonia 1/18 (6%) – – 1/12 (8%) – –
Other 3/18 (17%) 1/12 (8%) 0Æ63 – –
Hepatic
ALT/AST 7/16 (44%) 6/12 (50%) 0Æ99 1/12 (8%) 5/11 (45%) 0Æ07
Bilirubin – – – –
Gastrointestinal
Mucositis 6/16 (34%) 4/12 (33%) 0Æ99 – 3/11 (27%)
Diarrhoea 1/18 (6%) – – – – –
Nausea 1/18 (6%) 4/12 (33%) 0Æ13 2/12 (17%) 1/11 (9%) 0Æ99
Ileus 1/18 (6%) 1/12 (8%) 0Æ99 –
Cardiovascular
Fluid retention – – – – –
Arrythmia – – – – –
Deep venous thrombosis – – – – –
Cardiac failure – – – – –
Renal
Creatinine – – – – –
Neurological
Seizures 1/18 (6%) – – – –
Peripheral neuropathy – 1/12 (8%) –
Haemorrhage – – – –
ALT, alanine transaminase; ASP, aspartate transaminase.
S. Rives et al
606 ª 2011 Blackwell Publishing Ltd, British Journal of Haematology, 154, 600–611

Resultados
102
more grade III–IV nausea in induction treatment and more
grade III–IV mucositis during the consolidation phase,
compared to patients receiving chemotherapy alone, but these
differences did not reach statistical significance. In addition,
serum transaminase elevation was observed more frequently in
consolidation phase in patients receiving imatinib. The latter
toxicities (mucositis and transaminase elevation) might be
related to the dose of systemic methotrexate (5 g/m2) admin-
istered in the consolidation phase in the ALL/SHOP-2005 trial,
which was higher than in the ALL/SHOP-94 and ALL/SHOP-
99 protocols (3 g/m2). Hepatic failure or bilirubin elevation
greater than grade I was not observed in these cases and
transaminase elevations were transient without imatinib
discontinuation or dose-adjustment.
One patient in the imatinib group died of treatment-related
toxicity after block C chemotherapy, before planned HSCT.
This patient was a 4-year-old girl that died during severe
neutropenia after the third consolidation block of chemother-
apy. She developed septic shock and disseminated intravascu-
lar coagulation caused by Aeromonas hydrophila caviae of
intestinal origin. Intestinal necrosis was observed in the
necropsy and Aeromonas hydrophila caviae was isolated from
intestinal, spleen and peritoneal fluid.
Discussion
The results of this study confirm that the use of intermediate
dose imatinib in front-line therapy, concurrently with inten-
sive chemotherapy and followed by allogeneic HSCT, is an
effective treatment in paediatric patients with Ph+ ALL and
greatly improves early EFS and OS as compared to historical
controls.
The last SHOP protocol for children and adolescents with
ALL (ALL/SHOP-2005) included imatinib in combination
with front-line intensive chemotherapy in patients with newly
diagnosed Ph+ ALL. As stated in the introduction section, at
that time the use of imatinib in combination with intensive
chemotherapy had not yet been reported as first-line treatment
in children. It was decided to give it early, from day 15 of
induction, and concurrently with chemotherapy with the aim
of reducing disease burden and increasing the CR rate after
induction treatment. Imatinib was scheduled on day 15 to
ensure that the results of BCR-ABL1 rearrangement and/or
Philadelphia chromosome were obtained. An intermediate
dose of imatinib (260 mg/m2 per day) was chosen in order to
avoid excessive toxicity when given concomitantly with
intensive chemotherapy.
Allogeneic HSCT was offered to all patients with an identical
HLA donor, matched related or unrelated in CR1 after
completion of consolidation chemotherapy blocks. Although
at that time the superiority of allogeneic HSCT to chemotherapy
in paediatric Ph+ ALL had only been demonstrated for matched
related donor HSCT, it was decided to also indicate a matched
unrelated donor HSCT, given the improved outcome of this
type of transplant with recent advances in supportive care as
well as better donor selection with high-resolution HLA typing
(Baron & Storb, 2004). Recently, the early use of imatinib and its
continuous administration concurrently with intensive chemo-
therapy has been reported (Wassmann et al, 2006; Schultz et al,
2009; Ribera et al, 2010). This approach was well tolerated and
seemed to have superior antileukaemic activity than imatinib
given in a less intensive schedule (Wassmann et al, 2006; Schultz
et al, 2009). Likewise, the advantage of HSCT over chemother-
apy in paediatric Ph+ ALL has very recently been reported for
matched unrelated transplants in paediatric patients treated
without a TKI (Arico et al, 2010).
Patients with Ph+ ALL treated with pre-imatinib SHOP
protocols (ALL/SHOP-94 and ALL/SHOP-99) had a similar
outcome to those reported in other contemporary protocols
from collaborative cooperative groups (Roy et al, 2005;
Gandemer et al, 2009; Arico et al, 2010). The disease outcome
in these patients was heterogeneous, depending on the clinical
features at diagnosis and early response to treatment, in line
with other series of children with Ph+ ALL (Ribeiro et al, 1997;
Schrappe et al, 1998; Arico et al, 2000, 2010; Roy et al, 2005;
Gandemer et al, 2009). The median age of this series of
patients was slightly younger than that of other paediatric Ph+
ALL and this may have impact on outcome as older age is a
known risk factor in this subtype of ALL.
In patients treated with the imatinib-containing regimen
(ALL/SHOP-2005) the CR rate after induction treatment was
100%. Of note, all patients that were poor early responders
(more than 5% blasts in day-15 bone marrow) before imatinib
was started on day 15, achieved CR at the end of induction. We
have, thus, not observed any induction failure among the 16
patients treated with the imatinib-containing regimen. This
contrasts with the reported induction failures of more than
10% in other series of paediatric Ph+ ALL patients treated
without a TKI. (Arico et al, 2000, 2010; Roy et al, 2005;
Gandemer et al, 2009). There were no relapses before sched-
uled allogeneic HSCT, thus all patients but one who died of an
infectious complication before HSCT proceeded to transplant.
Importantly, all assessed patients underwent HSCT in molec-
ular remission. Regarding toxicity, the treatment was generally
well tolerated. With this intermediate dose of imatinib in
combination with intensive chemotherapy, worsening of
myelosuppression was not observed when compared to
historical controls of Ph+ ALL who received similar chemo-
therapy treatment without imatinib. Extramedullary toxicity
occurred but was manageable. As reported in adult patients, we
observed more nausea and transaminase elevation in patients
receiving imatinib. Transaminase elevation was not associated
with liver failure or grade III–IV bilirubin elevation and was
transient without requiring imatinib discontinuation. One
patient in the imatinib group died during the aplasia of a
consolidation block with a septic shock as a result of
Aeromonas infection. This infection had been described in
ALL patients not receiving imatinib and has a high mortality
rate in severely neutropenic patients such as ours (Martino
et al, 1997; Tsai et al, 2006).
Imatinib in Paediatric Ph+ ALL
ª 2011 Blackwell Publishing Ltd, British Journal of Haematology, 154, 600–611 607

Resultados
103
To the best of our knowledge, only one series of paediatric
patients with newly diagnosed Ph+ ALL treated with imatinib
in combination with chemotherapy has been reported to date
(Schultz et al, 2009). Our series confirms the impressive
improvement in early EFS and OS in patients receiving
imatinib concurrently with chemotherapy as compared to
historical controls, in whom a very similar chemotherapy
backbone was administered. In spite of the low number of
patients in our cohort, this difference in early outcome was
statistically significant. In this study we have to consider the
inherent limitations of historical comparisons. This has
particular relevance in this series in which TRM was higher
in the historical control (pre-imatinib cohort). Although this
difference in TRM may have contributed to the better results
obtained in the imatinib group, it is unlikely to account for
the major improvement in early outcome. Rather, the results
obtained in our imatinib cohort compare favourably with all
other pre-imatinib series of Ph+ ALL (Roy et al, 2005;
Gandemer et al, 2009; Arico et al, 2010), regardless of
whether they had a lower TRM. On the other hand, given
the dramatic improvement in outcome obtained by the
Children’s Oncology Group in paediatric patients with de
novo Ph+ ALL treated with imatinib (Schultz et al, 2009), it
would be very difficult to justify, from an ethical point of
view, a trial that randomizes the use of a TKI against
placebo.
The good results obtained in this study with targeted
therapy in combination with intensive chemotherapy may
contribute to redefine the role of allogeneic HSCT in CR1 in
paediatric Ph+ ALL, particularly when considering the out-
standing early outcome reported by the Children’s Oncology
Group even in patients not receiving allogeneic HSCT.
Nevertheless, long-term follow-up is needed to determine
whether these results are maintained over time, as late relapses
may occur, especially among those not undergoing transplant.
Indeed, chronic graft-versus-host disease has a role in
protecting against relapses in Ph+ ALL (Esperou et al, 2003;
Lee et al, 2005; Bhojwani et al, 2009; Burke et al, 2009). Of
note, in adult series of Ph+ ALL, imatinib-containing regimens
improved long-term survival (Fielding et al, 2010; Rambaldi
et al, 2010) and allogeneic transplant seemed to keep its
beneficial effect in reducing relapses and improving outcome
also in the imatinib era (Ottmann & Pfeifer, 2009; Fielding
et al, 2010; Pfeifer et al, 2010; Rambaldi et al, 2010). It is
crucial to identify which children could be spared HSCT. The
good-risk group of patients, as defined by presenting features
and early response, could be candidates for omitting allogeneic
HSCT. In this regard, MRD measurement could help to refine
the identification of patients for whom transplantation could
be avoided.
In conclusion, in our series of patients treated in the
imatinib era, we have shown an impressive improvement in
early outcome using imatinib in combination with intensive
chemotherapy followed by allogeneic HSCT. The results of this
series of patients treated with imatinib and allogeneic HSCT
may contribute to future comparisons of patients treated
without allogeneic HSCT in the TKI era.
Overall, longer follow-up and the analysis of a larger
number of patients included in international trials, such as the
ongoing clinical trial EsPhALL (Biondi, 2006) are needed and
may help to address the role of targeted therapy in this subset
of very high risk ALL.
Acknowledgements
We would like to thank Rocıo Sanchez, Carme Romero,
Dr. Agustı Martı, for their help in data collection and statistical
analysis. We would also like to thank Dr Mireia Camos,
Dr. Jordi Esteve and Dr. Arturo Pereira for their help in the
statistical analysis.
This work was supported in part by a grant from the
Fundacion de Oncologıa Infantil Enriqueta Villavecchia.
Authorship and disclosures
A complete list of the institutions and physicians participating
in the protocols SHOP/ALL-94, ALL-99 and ALL-2005 are
listed in the ‘Appendix I’.
SR was the principal investigator and takes primary
responsibility for the paper. SR, JE, PG, ML, PGM, AV,
MJM, JLV, JMC, RF, MM, MT, BL, FL, RL, JU, MM, AF, IR
and IB recruited the patients. SR participated in the statistical
analysis and wrote the paper. The authors reported no
potential conflicts of interest.
The preliminary results of this study were presented at the
51th Annual Meeting of the American Society of Haematology,
December 2010, Orlando, USA.
Conflict of interest
The authors have no competing interests.
Supporting Information
Additional Supporting Information may be found in the
online version of this article
Fig S1. (A) Flow-chart of patients treated according ALL/
SHOP-2005 (imatinib cohort). (B) Flow-chart of patients
treated according ALL/SHOP-94–99 (non-imatinib cohort).
Please note: Wiley-Blackwell are not responsible for the
content or functionality of any supporting materials supplied
by the authors. Any queries (other than missing material)
should be directed to the corresponding author for the article.
Appendix I
The following institutions and physicians participated in the
protocols SHOP/ALL-94, ALL-99 and ALL-2005: Hospital
Sant Joan de Deu de Barcelona (Jesus Estella, Susana Rives,
Mireia Camos, Albert Catala, Teresa Toll, Ruben Berrueco,
S. Rives et al
608 ª 2011 Blackwell Publishing Ltd, British Journal of Haematology, 154, 600–611

Resultados
104
Montserrat Torrebadell), Hospital Reina Sofıa de Cordoba
(Pedro Gomez, Antonia Rodrıguez), Hospital Universitario
Marques de Valdecilla, Santander (Encarna Bureo, Monica
Lopez-Duarte), Hospital Universitario La Paz, Madrid (Puri-
facion Garcıa de Miguel), Hospital Universitario La Fe,
Valencia (Amparo Verdeguer, Jose Marıa Fernandez-Navar-
ro), Hospital Virgen de las Nieves, Granada (Marıa Jose
Moreno, Emilia Urrutia), Hospital Doce de Octubre, Madrid
(Jose Luis Vivanco, Carmen Melero), Hospital Xeral de
Galicia, Santiago de Compostela (Jose Miguel Couselo),
Hospital Clınico de Valencia (Rafael Fernandez-Delgado),
Hospital Ramon y Cajal, Madrid (Marisol Maldonado),
Hospital Universitario de Alicante (Marıa Tasso), Hospital
Monteprıncipe, Madrid (Blanca Lopez-Ibor, Marta Villa),
Hospital Torrecardenas, Almerıa (Francisco Lendınez, Mª
Angeles Vazquez), Hospital Universitario de Canarias, San
Cristobal de la Laguna Tenerife (Ricardo Lopez-Almaraz, Jose
Cayetano Rodrıguez-Luis), Hospital de Donostia, San Sebas-
tian (Javier Uriz), Hospital Parc Taulı, Sabadell (Montserrat
Melo), Hospital Virgen de la Macarena, Sevilla (Ana Fernan-
dez-Teijeiro), Hospital Marıa Teresa Herrera/Juan Canalejo,
La Coruna (Isidoro Rodrıguez), Hospital Virgen del Camino
(Javier Molina), Hospital de la Santa Creu i Sant Pau (Isabel
Badell, Montserrat Torrent, Marta Garcıa-Bernal, Nuria
Pardo), Hospital Son Espases/Hospital Son Dureta, Mallorca
(Isabel Hernandez, Mercedes Guibelalde), Hospital Materno-
Infantil de Badajoz (Jose Manuel Vagace), Hospital Gregorio
Maranon, Madrid (Elena Cela, Cristina Belendez), Hospital
Universitario de Salamanca (Dorotea Fernandez, Manuela
Muriel, Gabriel Mateos), Hospital de Jaen (Irene Pelaez, Ana
Belen Lopez), Hospital Virgen de la Salud, Toledo (Marıa
Rosario Velasco, Marcos Zamora), Hospital Germans Trias i
Pujol, Badalona (Francisco Almazan, Javier German), Hospital
Universitario de Albacete (Miguel Lillo), Hospital Universi-
tario de Valladolid (Carmen Valbuena, Blanca Quiros),
Hospital de Ourense (Arturo Fuentes), Hospital de Basurto
(Jose Marıa Indiano), Hospital Sant Joan, Alicante (Raul
Gonzalez, Cesar Gavilan), Hospital Central de Asturias,
Oviedo (Soledad Gonzalez), Hospital Xeral Cies, Vigo (Man-
uel Fernandez-Sanmartın), Hospital Materno-Infantil de las
Palmas, Gran Canaria (Antonio Molines), Hospital de Cruces,
Bilbao (Aurora Navajas).
This paper is presented by Susana Rives as part of a PhD
thesis at the Universitat de Barcelona.
References
Arico, M., Valsecchi, M.G., Camitta, B., Schrappe,
M., Chessells, J., Baruchel, A., Gaynon, P., Silv-
erman, L., Janka-Schaub, G., Kamps, W., Pui,
C.H. & Masera, G. (2000) Outcome of treatment
in children with Philadelphia chromosome-po-
sitive acute lymphoblastic leukemia. New England
Journal of Medicine, 342, 998–1006.
Arico, M., Schrappe, M., Hunger, S.P., Carroll,
W.L., Conter, V., Galimberti, S., Manabe, A.,
Saha, V., Baruchel, A., Vettenranta, K., Horibe,
K., Benoit, Y., Pieters, R., Escherich, G., Silver-
man, L.B., Pui, C.H. & Valsecchi, M.G. (2010)
Clinical outcome of children with newly diag-
nosed Philadelphia chromosome-positive acute
lymphoblastic leukaemia treated between 1995
and 2005. Journal of Clinical Oncology, 28, 4755–
4761.
Badell, I., Munoz, A., Estella, J., Fernandez-Delgado,
R., Javier, G., Verdeguer, A. & Cubells, J. (2008)
Long-term results of two consecutive trials in
childhood acute lymphoblastic leukaemia
performed by the Spanish Cooperative Group for
Childhood Acute Lymphoblastic Leukemia
Group (SHOP) from 1989 to 1998. Clinical and
Translational Oncology, 10, 117–124.
Baron, F. & Storb, R. (2004) Allogeneic hemato-
poietic cell transplantation as treatment for
hematological malignancies: a review. Seminars in
Immunopathology, 26, 71–94.
Barr, R.D. (2010) Imatinib mesylate in children and
adolescents with cancer. Pediatric Blood and
Cancer, 55, 18–25.
Bassan, R., Rossi, G., Pogliani, E.M., Di, B.E.,
Angelucci, E., Cavattoni, I., Lambertenghi-
Deliliers, G., Mannelli, F., Levis, A., Ciceri, F.,
Mattei, D., Borlenghi, E., Terruzzi, E., Borghero,
C., Romani, C., Spinelli, O., Tosi, M., Oldani, E.,
Intermesoli, T. & Rambaldi, A. (2010) Chemo-
therapy-phased imatinib pulses improve long-
term outcome of adult patients with Philadelphia
chromosome-positive acute lymphoblastic leuke-
mia: Northern Italy Leukemia Group protocol 09/
00. Journal of Clinical Oncology, 28, 3644–3652.
Bhojwani, D., Howard, S.C. & Pui, C.H. (2009)
High-risk childhood acute lymphoblastic leuke-
mia. Clinical Lymphoma and Myeloma, 9(Suppl.
3), S222–S230.
Biondi, A. (2006). 38th Congress of the Interna-
tional Society of Paediatric Oncology. Geneva,
Switzerland September 17-21, 2006-SIOP Edu-
cation Book 2006, pp 55–58.
Burke, M.J., Cao, Q., Trotz, B., Weigel, B., Kumar,
A., Smith, A. & Verneris, M.R. (2009) Allogeneic
hematopoietic cell transplantation (allogeneic
HCT) for treatment of pediatric Philadelphia
chromosome-positive acute lymphoblastic
leukemia (ALL). Pediatric Blood and Cancer, 53,
1289–1294.
Castillo, E., Al-Rajabi, R., Pandya, D.M.,
Varadarajan, P., Kelly, K.R., Swords, R., Tharian,
A., Kraus, M.K. & Padmanabhan, S. (2010) A
pilot study of the combination of nilotinib and
Hyper-CVAD for Philadelphia chromosome
positive acute lymphocytic leukemia and
lymphoid blast crisis chronic myelogenous leu-
kemia. Blood (ASH Annual Meeting Abstracts),
116, 2144.
Cazzaniga, G., Lanciotti, M., Rossi, V., Di, M.D.,
Arico, M., Valsecchi, M.G., Basso, G., Masera, G.,
Micalizzi, C. & Biondi, A. (2002) Prospective
molecular monitoring of BCR/ABL transcript in
children with Ph+ acute lymphoblastic leukaemia
unravels differences in treatment response. British
Journal of Haematology, 119, 445–453.
Champagne, M.A., Capdeville, R., Krailo, M., Qu,
W., Peng, B., Rosamilia, M., Therrien, M., Zo-
ellner, U., Blaney, S.M. & Bernstein, M. (2004)
Imatinib mesylate (STI571) for treatment of
children with Philadelphia chromosome-positive
leukemia: results from a Children’s Oncology
Group phase 1 study. Blood, 104, 2655–2660.
van Dongen, J.J., Macintyre, E.A., Gabert, J.A.,
Delabesse, E., Rossi, V., Saglio, G., Gottardi, E.,
Rambaldi, A., Dotti, G., Griesinger, F., Parreira,
A., Gameiro, P., Diaz, M.G., Malec, M., Langerak,
A.W., San Miguel, J.F. & Biondi, A. (1999)
Standardized RT-PCR analysis of fusion gene
transcripts from chromosome aberrations in
acute leukemia for detection of minimal residual
disease. Report of the BIOMED-1 Concerted
Action: investigation of minimal residual disease
in acute leukemia. Leukemia, 13, 1901–1928.
Esperou, H., Boiron, J.M., Cayuela, J.M., Blanchet,
O., Kuentz, M., Jouet, J.P., Milpied, N., Cahn,
J.Y., Faucher, C., Bourhis, J.H., Michallet, M.,
Tanguy, M.L., Vernant, J.P., Gabert, J., Bordig-
oni, P., Ifrah, N., Baruchel, A. & Dombret, H.
(2003) A potential graft-versus-leukemia effect
after allogeneic hematopoietic stem cell trans-
plantation for patients with Philadelphia chro-
mosome-positive acute lymphoblastic leukemia:
results from the French Bone Marrow Trans-
plantation Society. Bone Marrow Transplantation,
31, 909–918.
Fielding, A.K., Buck, G., Lazarus, H.M., Litzow,
M.R., Luger, S., Marks, D.I., Luger, S., Marks,
D.I., McMillan, A., Moorman, A.V., Paietta, E.,
Richards, S.M., Tallman, M.S., Rowe, J.M. &
Goldstone, A.H. (2010) Imatinib significantly
Imatinib in Paediatric Ph+ ALL
ª 2011 Blackwell Publishing Ltd, British Journal of Haematology, 154, 600–611 609

Resultados
105
enhances long-term outcomes in Philadelphia
positive acute lymphoblastic leukaemia; final
results of the UKALLXII/ECOG2993 trial. Blood,
116, 169.
Fine, J.P. & Gray, R.J. (1999) A proportional hazards
model for subdistribution of a competing risk.
Journal of the American Statistical Association, 94,
496–509.
Gabert, J., Beillard, E., van der Velden, V.H., Bi, W.,
Grimwade, D., Pallisgaard, N., Barbany, G.,
Cazzaniga, G., Cayuela, J.M., Cave, H., Pane, F.,
Aerts, J.L., De, M.D., Thirion, X., Pradel, V.,
Gonzalez, M., Viehmann, S., Malec, M., Saglio,
G. & van Dongen, J.J. (2003) Standardization and
quality control studies of ‘real-time’ quantitative
reverse transcriptase polymerase chain reaction of
fusion gene transcripts for residual disease
detection in leukemia – a Europe Against Cancer
program. Leukemia, 17, 2318–2357.
Gandemer, V., Auclerc, M.F., Perel, Y., Vannier, J.P.,
Le, G.E., Demeocq, F., Schmitt, C., Piguet, C., Ste-
phan, J.L., Lejars, O., Debre, M., Jonveaux, P.,
Cayuela, J.M., Chevret, S., Leverger, G. & Baruchel,
A. (2009) Impact of age, leukocyte count and day
21-bone marrow response to chemotherapy on the
long-term outcome of children with philadelphia
chromosome-positive acute lymphoblastic leuke-
mia in the pre-imatinib era: results of the FRALLE
93 study. BMC Cancer, 9, 14.
Gooley, T.A., Leisenring, W., Crowley, J. & Storer,
B.E. (1999) Estimation of failure probabilities in
the presence of competing risks: new represen-
tations of old estimators. Statistics in Medicine,
18, 695–706.
Heerema, N.A., Harbott, J., Galimberti, S., Camitta,
B.M., Gaynon, P.S., Janka-Schaub, G., Kamps,
W., Basso, G., Pui, C.H., Schrappe, M., Auclerc,
M.F., Carroll, A.J., Conter, V., Harrison, C.J.,
Pullen, J., Raimondi, S.C., Richards, S., Riehm,
H., Sather, H.N., Shuster, J.J., Silverman, L.B.,
Valsecchi, M.G. & Arico, M. (2004) Secondary
cytogenetic aberrations in childhood Philadelphia
chromosome positive acute lymphoblastic leu-
kemia are nonrandom and may be associated
with outcome. Leukemia, 18, 693–702.
Kaplan, E.L. & Meier, P. (1958) Nonparametric
estimation from incomplete observations. Journal
of the American Statistical Association, 53, 457–
481.
Lee, S., Kim, Y.J., Min, C.K., Kim, H.J., Eom,
K.S., Kim, D.W., Lee, J.W., Min, W.S. & Kim,
C.C. (2005) The effect of first-line imatinib
interim therapy on the outcome of allogeneic
stem cell transplantation in adults with newly
diagnosed Philadelphia chromosome-positive
acute lymphoblastic leukemia. Blood, 105,
3449–3457.
Lee, S., Kim, Y.J., Chung, N.G., Lim, J., Lee, D.G.,
Kim, H.J., Min, C.K., Lee, J.W., Min, W.S. &
Kim, C.C. (2009) The extent of minimal residual
disease reduction after the first 4-week imatinib
therapy determines outcome of allogeneic stem
cell transplantation in adults with Philadelphia
chromosome-positive acute lymphoblastic leu-
kemia. Cancer, 115, 561–570.
Martino, R., Santamaria, A., Pericas, R., Sureda, A.
& Brunet, S. (1997) Acute rhabdomyolysis and
myonecrosis complicating aeromonas bacteremia
in neutropenic patients with hematologic malig-
nancies: report of two cases. Haematologica, 82,
692–694.
Miller, A.B., Hoogstraten, B., Staquet, M. & Win-
kler, A. (1981) Reporting results of cancer treat-
ment. Cancer, 47, 207–214.
Mitelman, F (ed.) (1995) ISCN 1995: An Interna-
tional System for Human Cytogenetic Nomencla-
ture. S Karger, Basel, Switzerland.
Ottmann, O.G. & Pfeifer, H. (2009) First-line
treatment of Philadelphia chromosome-positive
acute lymphoblastic leukaemia in adults. Current
Opinion in Oncology, 21(Suppl. 1), S43–S46.
Pane, F., Cimino, G., Izzo, B., Camera, A., Vitale, A.,
Quintarelli, C., Picardi, M., Specchia, G., Man-
cini, M., Cuneo, A., Mecucci, C., Martinelli, G.,
Saglio, G., Rotoli, B., Mandelli, F., Salvatore, F. &
Foa, R. (2005) Significant reduction of the hybrid
BCR/ABL transcripts after induction and con-
solidation therapy is a powerful predictor of
treatment response in adult Philadelphia-positive
acute lymphoblastic leukemia. Leukemia, 19,
628–635.
Peto, R. & Pike, M.C. (1973) Conservatism of the
approximation sigma (O-E)2-E in the logrank
test for survival data or tumor incidence data.
Biometrics, 29, 579–584.
Pfeifer, H., Goekbuget, N., Volp, C., Huttmann, A.,
Lubbert, M., Stuhlmann, R., Dengler, J., Kreuzer,
K.A., Beck, J., Stelljes, M., Schmid, M., Wass-
mann, B., Leimer, L., Spiekermann, K., Terwey,
T.H., Schafer-Eckart, K., Ditz, D., Lindemann,
H., Morgner, A., Schaich, M., Burmeister, T.,
Binckebanck, A., Hoelzer, D. & Ottmann, O.G.
(2010) Long-term outcome of 335 adult patients
receiving different schedules of imatinib and
chemotherapy as front-line treatment for Phila-
delphia-positive acute lymphoblastic leukemia
(Ph+ ALL). Blood, 116, 173.
Pui, C.H. & Evans, W.E. (2006) Treatment of acute
lymphoblastic leukemia. New England Journal of
Medicine, 354, 166–178.
Rambaldi, A., Spinelli, O., Oldani, E., Grassi, A.,
Rossi, G., Pogliani, E.M., Di Bona, E., Angelucci,
E., Cavattoni, I., Lambertenghi-Deliliers, G.,
Mannelli, F., Levis, A., Ciceri, F., Mattei, D.,
Borlenghi, E., Terruzzi, E., Borghero, C., Romani,
C., Tosi, M., Bussini, A., Intermesoli, T. & Bas-
san, R. (2010) Improved clinical outcome of
adult patients with Ph+ ALL after a combined
imatinib-chemotherapy induction/consolidation
program followed by allogeneic hematopoietic
stem cell transplantation: results from a pro-
spective study of the Northern Italy Leukemia
Group. Blood, 116, 682.
Ravandi, F., O’Brien, S., Thomas, D., Faderl, S.,
Jones, D., Garris, R., Dara, S., Jorgensen, J.,
Kebriaei, P., Champlin, R., Borthakur, G., Burger,
J., Ferrajoli, A., Garcia-Manero, G., Wierda, W.,
Cortes, J. & Kantarjian, H. (2010a) First report of
phase 2 study of dasatinib with hyper-CVAD for
the frontline treatment of patients with Phila-
delphia chromosome-positive (Ph+) acute lym-
phoblastic leukemia. Blood, 116, 2070–2077.
Ravandi, F., Thomas, D.A., O’Brien, S., Faderl, S.,
Jorgensen, J., Luthra, R., Jones, D., Garris, R.,
Cortes, J.E. & Kantarjian, H. (2010b) Dynamics
of minimal residual leukemia after combinations
of the HyperCVAD regimen with imatinib or
dasatinib in patients with Philadelphia-chromo-
some positive acute lymphoblastic leukemia.
Blood, 116, 2127.
Ribeiro, R.C., Broniscer, A., Rivera, G.K., Hancock,
M.L., Raimondi, S.C., Sandlund, J.T., Crist, W.,
Evans, W.E. & Pui, C.H. (1997) Philadelphia
chromosome-positive acute lymphoblastic leu-
kemia in children: durable responses to chemo-
therapy associated with low initial white blood
cell counts. Leukemia, 11, 1493–1496.
Ribera, J.M., Oriol, A., Gonzalez, M., Vidriales, B.,
Brunet, S., Esteve, J., Del, P.E., Rivas, C., Moreno,
M.J., Tormo, M., Martin-Reina, V., Sarra, J.,
Parody, R., de Oteyza, J.P., Bureo, E. & Bernal,
M.T. (2010) Concurrent intensive chemotherapy
and imatinib before and after stem cell trans-
plantation in newly diagnosed Philadelphia
chromosome-positive acute lymphoblastic leu-
kemia. Final results of the CSTIBES02 trial.
Haematologica, 95, 87–95.
Roy, A., Bradburn, M., Moorman, A.V., Burrett, J.,
Love, S., Kinsey, S.E., Mitchell, C., Vora, A.,
Eden, T., Lilleyman, J.S., Hann, I. & Saha, V.
(2005) Early response to induction is predictive
of survival in childhood Philadelphia chromo-
some positive acute lymphoblastic leukaemia:
results of the Medical Research Council ALL 97
trial. British Journal of Haematology, 129, 35–44.
Satwani, P., Sather, H., Ozkaynak, F., Heerema,
N.A., Schultz, K.R., Sanders, J., Kersey, J., Dav-
enport, V., Trigg, M. & Cairo, M.S. (2007)
Allogeneic bone marrow transplantation in first
remission for children with ultra-high-risk fea-
tures of acute lymphoblastic leukemia: a chil-
dren’s oncology group study report. Biology of
Blood and Marrow Transplantation, 13, 218–227.
Schrappe, M., Arico, M., Harbott, J., Biondi, A.,
Zimmermann, M., Conter, V., Reiter, A., Val-
secchi, M.G., Gadner, H., Basso, G., Bartram,
C.R., Lampert, F., Riehm, H. & Masera, G. (1998)
Philadelphia chromosome-positive (Ph+) child-
hood acute lymphoblastic leukemia: good initial
steroid response allows early prediction of a
favorable treatment outcome. Blood, 92, 2730–
2741.
Schultz, K.R., Bowman, W.P., Aledo, A., Slayton,
W.B., Sather, H., Devidas, M., Wang, C., Davies,
S.M., Gaynon, P.S., Trigg, M., Rutledge, R.,
Burden, L., Jorstad, D., Carroll, A., Heerema,
N.A., Winick, N., Borowitz, M.J., Hunger, S.P.,
Carroll, W.L. & Camitta, B. (2009) Improved
early event-free survival with imatinib in Phila-
delphia chromosome-positive acute lymphoblas-
tic leukemia: a children’s oncology group study.
Journal of Clinical Oncology, 27, 5175–5181.
Thomas, D.A., Faderl, S., Cortes, J., O’Brien, S.,
Giles, F.J., Kornblau, S.M., Garcia-Manero, G.,
Keating, M.J., Andreeff, M., Jeha, S., Beran, M.,
S. Rives et al
610 ª 2011 Blackwell Publishing Ltd, British Journal of Haematology, 154, 600–611

Resultados
106
Verstovsek, S., Pierce, S., Letvak, L., Salvado, A.,
Champlin, R., Talpaz, M. & Kantarjian, H. (2004)
Treatment of Philadelphia chromosome-positive
acute lymphocytic leukemia with hyper-CVAD
and imatinib mesylate. Blood, 103, 4396–4407.
Towatari, M., Yanada, M., Usui, N., Takeuchi, J.,
Sugiura, I., Takeuchi, M., Yagasaki, F., Kawai, Y.,
Miyawaki, S., Ohtake, S., Jinnai, I., Matsuo, K.,
Naoe, T. & Ohno, R. (2004) Combination of
intensive chemotherapy and imatinib can rapidly
induce high-quality complete remission for a
majority of patients with newly diagnosed BCR-
ABL-positive acute lymphoblastic leukemia.
Blood, 104, 3507–3512.
Tsai, M.S., Kuo, C.Y., Wang, M.C., Wu, H.C.,
Chien, C.C. & Liu, J.W. (2006) Clinical features
and risk factors for mortality in Aeromonas
bacteremic adults with hematologic malignancies.
Journal of Microbiology, Immunology, Infection,
39, 150–154.
Wassmann, B., Pfeifer, H., Goekbuget, N., Beelen,
D.W., Beck, J., Stelljes, M., Bornhauser, M.,
Reichle, A., Perz, J., Haas, R., Ganser, A., Sch-
mid, M., Kanz, L., Lenz, G., Kaufmann, M.,
Binckebanck, A., Bruck, P., Reutzel, R.,
Gschaidmeier, H., Schwartz, S., Hoelzer, D. &
Ottmann, O.G. (2006) Alternating versus con-
current schedules of imatinib and chemotherapy
as front-line therapy for Philadelphia-positive
acute lymphoblastic leukemia (Ph+ ALL). Blood,
108, 1469–1477.
Yanada, M., Sugiura, I., Takeuchi, J., Akiyama, H.,
Maruta, A., Ueda, Y., Usui, N., Yagasaki, F.,
Yujiri, T., Takeuchi, M., Nishii, K., Kimura, Y.,
Miyawaki, S., Narimatsu, H., Miyazaki, Y., Oh-
take, S., Jinnai, I., Matsuo, K., Naoe, T. & Ohno,
R. (2008) Prospective monitoring of BCR-ABL1
transcript levels in patients with Philadelphia
chromosome-positive acute lymphoblastic
leukaemia undergoing imatinib-combined che-
motherapy. British Journal of Haematology, 143,
503–510.
Imatinib in Paediatric Ph+ ALL
ª 2011 Blackwell Publishing Ltd, British Journal of Haematology, 154, 600–611 611

Resultados
107
Trabajo 2 (carta al editor)
Susana Rives, Mireia Camós, Jesús Estella, Pedro Gómez, María José Moreno,
José Luis Vivanco, Montserrat Melo, Rafael Fernández-‐Delgado, Amparo
Verdeguer, Ana Fernández-‐Teijeiro, Francisco Lendínez, Ricardo López-‐
Almaraz, José Javier Uriz, Isidro Rodríguez, Isabel Badell. En representación
de la Sociedad Española de Hemato-‐Oncología Pediátrica (SHOP/SEHOP)
British Journal of Haematology 2013; 162:419-‐21.
Longer follow-‐up confirms major improvement in outcome in
children and adolescents with Philadelphia chromosome acute
lymphoblastic leukaemia treated with continuous imatinib and
haematopoietic stem cell transplantation. Results from the
Spanish Cooperative Study SHOP/ALL-‐2005

Resultados
108

Resultados
109
Confirmación con mayor seguimiento de la gran mejoría en la
supervivencia en niños y adolescentes con LLA Ph+ tratados con imatinib
continuo y TPH alogénico. Resultados del grupo cooperativo español
SHOP/LLA-‐2005
Con posterioridad a la publicación de la serie, en la que se había analizado a
los pacientes incluidos hasta abril de 2010, se realizó un análisis de la serie
de pacientes tratados con imatinib con mayor seguimiento (hasta enero
2013) para verificar si los buenos resultados obtenidos a corto plazo (SLE y
SG a 3 años) se mantenían en el tiempo. Este análisis resultaba de especial
interés porque los pacientes pediátricos con LLA Ph+ pueden presentar
recaídas tardías.
Dichos resultados se publicaron en forma de carta al editor (trabajo 2) y
confirman, con una mediana de seguimiento de más de 5 años, que los niños
y adolescentes con LLA Ph+ tratados con imatinib continuo y quimioterapia
intensiva seguidos de trasplante alogénico (según el protocolo SHOP/LLA-‐
2005) obtuvieron resultados muy superiores en la supervivencia tanto libre
de evento como global que la cohorte histórica de pacientes tratados sin
imatinib.
Así, con una mediana de seguimiento de 65 meses (34-‐81), no se produjeron
nuevos eventos en la cohorte de pacientes tratados con imatinib. De los 16
pacientes, en el momento del análisis 14 estaban vivos, 13 en RC continuada
y uno en segunda RC. Todos ellos en remisión molecular. Todos fueron al
TPH en primera RC salvo un paciente que falleció antes de llegar al trasplante
por una complicación infecciosa.

Resultados
110
Sólo tres pacientes recibieron imatinib post-‐trasplante: uno tras tratamiento
de rescate por recaída post-‐trasplante, uno como parte del tratamiento de
una enfermedad del injerto contra el huésped crónica extensa y otro
paciente como tratamiento profiláctico, sin evidencia de recaída molecular.
La SLE a los 5 años fue de 81,3% (+/-‐10) y de 29,6% (+/-‐8) para las cohortes
de imatinib y pre-‐imatinib respectivamente (p=0,004) y la SG fue de 87,5%
(+/-‐8) y de 30,8% (+/-‐9) para las cohortes imatinib y pre-‐imatinib
respectivamente (p=0,002), ver figuras 1 y 2 del artículo.
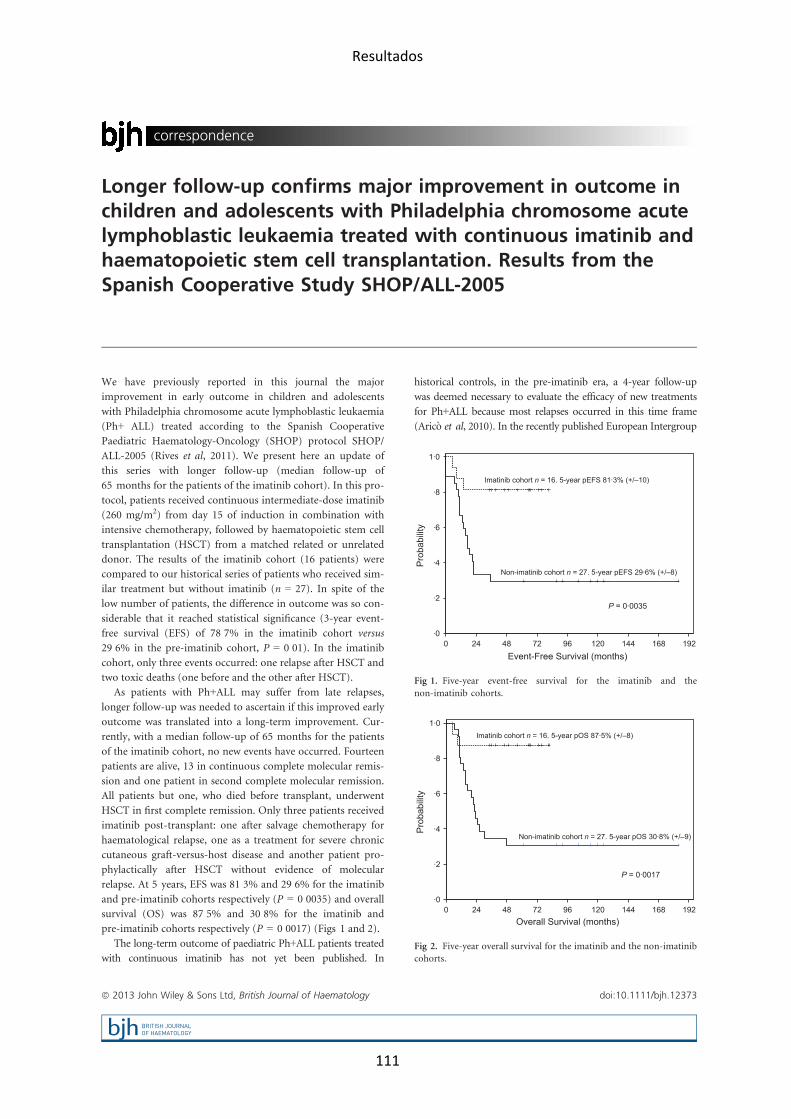
Resultados
111
Longer follow-up confirms major improvement in outcome inchildren and adolescents with Philadelphia chromosome acutelymphoblastic leukaemia treated with continuous imatinib andhaematopoietic stem cell transplantation. Results from theSpanish Cooperative Study SHOP/ALL-2005
We have previously reported in this journal the major
improvement in early outcome in children and adolescents
with Philadelphia chromosome acute lymphoblastic leukaemia
(Ph+ ALL) treated according to the Spanish Cooperative
Paediatric Haematology-Oncology (SHOP) protocol SHOP/
ALL-2005 (Rives et al, 2011). We present here an update of
this series with longer follow-up (median follow-up of
65 months for the patients of the imatinib cohort). In this pro-
tocol, patients received continuous intermediate-dose imatinib
(260 mg/m2) from day 15 of induction in combination with
intensive chemotherapy, followed by haematopoietic stem cell
transplantation (HSCT) from a matched related or unrelated
donor. The results of the imatinib cohort (16 patients) were
compared to our historical series of patients who received sim-
ilar treatment but without imatinib (n = 27). In spite of the
low number of patients, the difference in outcome was so con-
siderable that it reached statistical significance (3-year event-
free survival (EFS) of 78!7% in the imatinib cohort versus
29!6% in the pre-imatinib cohort, P = 0!01). In the imatinib
cohort, only three events occurred: one relapse after HSCT and
two toxic deaths (one before and the other after HSCT).
As patients with Ph+ALL may suffer from late relapses,
longer follow-up was needed to ascertain if this improved early
outcome was translated into a long-term improvement. Cur-
rently, with a median follow-up of 65 months for the patients
of the imatinib cohort, no new events have occurred. Fourteen
patients are alive, 13 in continuous complete molecular remis-
sion and one patient in second complete molecular remission.
All patients but one, who died before transplant, underwent
HSCT in first complete remission. Only three patients received
imatinib post-transplant: one after salvage chemotherapy for
haematological relapse, one as a treatment for severe chronic
cutaneous graft-versus-host disease and another patient pro-
phylactically after HSCT without evidence of molecular
relapse. At 5 years, EFS was 81!3% and 29!6% for the imatinib
and pre-imatinib cohorts respectively (P = 0!0035) and overall
survival (OS) was 87!5% and 30!8% for the imatinib and
pre-imatinib cohorts respectively (P = 0!0017) (Figs 1 and 2).
The long-term outcome of paediatric Ph+ALL patients treated
with continuous imatinib has not yet been published. In
historical controls, in the pre-imatinib era, a 4-year follow-up
was deemed necessary to evaluate the efficacy of new treatments
for Ph+ALL because most relapses occurred in this time frame
(Aric!o et al, 2010). In the recently published European Intergroup
Event-Free Survival (months)192168144120967248240
Prob
abilit
y
Imatinib cohort n = 16. 5-year pEFS 81·3% (+/–10)
Non-imatinib cohort n = 27. 5-year pEFS 29·6% (+/–8)
P = 0·0035
1·0
·8
·6
·4
·2
·0
Fig 1. Five-year event-free survival for the imatinib and thenon-imatinib cohorts.
Overall Survival (months)192168144120967248240
Prob
abilit
y
Imatinib cohort n = 16. 5-year pOS 87·5% (+/–8)
Non-imatinib cohort n = 27. 5-year pOS 30·8% (+/–9)
P = 0·0017
1·0
·8
·6
·4
·2
·0
Fig 2. Five-year overall survival for the imatinib and the non-imatinibcohorts.
ª 2013 John Wiley & Sons Ltd, British Journal of Haematology doi:10.1111/bjh.12373
correspondence

Resultados
112
Study on Post-Induction Treatment with Imatinib followed by
HSCT in childhood Ph+ALL (EsPhALL), all the relapses
occurred during the first 3 years (Biondi et al, 2012). Thus, we
believe that the 5-year EFS and OS reported in the present ser-
ies of patients may probably be close to their long-term out-
come.
The American Children’s Oncology Group (COG) had previ-
ously reported 45 Ph+ALL patients treated with continuous
imatinib after remission induction (cohort 5), followed by HSCT
only if a matched sibling donor was available (Schultz et al,
2009). The results in their cohort of patients also showed an
impressive improvement in early outcome (3-year EFS of 80%).
Noteably, these results were achieved even in patients treated
with chemotherapy only, without transplantation. Longer fol-
low-up of this series is needed to confirm that patients treated
with chemotherapy do not suffer from late relapses. In the adult
setting, a clear benefit of HSCT in PhALL+ patients treated with
imatinib only emerged with longer follow-up (Thomas, 2012).
In the EsPhALL study (Biondi et al, 2012), which included
a large series of patients (n = 178), the impact of adding
imatinib to chemotherapy compared favourably to historical
controls, with better 4-year disease-free survival [61!9% in
the EsPhALL study versus 42!3% in the historical cohort
(Aric!o et al, 2010)]. The improvement in the outcome of
these patients, although statistically significant, was inferior
to the results achieved with the COG and the SHOP proto-
cols, probably due to the less intensive and intermittent use
of imatinib. In addition, good risk patients were randomized
to receive or not imatinib. Following the publication of the
COG results (Schultz et al, 2009), the EsPhALL protocol was
amended in December 2009 in which imatinib was to be
given in all patients (good and poor risk patients) in a con-
tinuous fashion. The results of this amendment will be of
great interest to confirm the major improvement obtained
when imatinib or another tyrosine-kinase inhibitor (TKI) is
added in a continuous manner early in induction in paediat-
ric patients with Ph+ALL in a larger series of patients.
An unresolved question in paediatric Ph+ALL is whether
the use of TKI following HSCT should be used prophylactically
or pre-emptively (triggered by minimal residual disease) to
prevent relapses. Twelve out of 15 patients of the SHOP series
who underwent HSCT did not receive imatinib post-HSCT
and remain in complete molecular remission. Three patients
received imatinib post-HSCT, one after relapse, one prophy-
lactically and one as part of the treatment of severe cutaneous
graft-versus-host disease. Although our series of patients is too
small to answer this question, it may suggest that prophylactic
treatment with imatinib after HSCT might not be necessary.
The pre-emptive versus prophylactic use of imatinib after
HSCT in the adult setting is not yet clear (Nishiwaki et al,
2010; Ribera et al, 2010) and is currently being addressed in a
randomized study conducted by the German Multicentre ALL
group, GMALL (Pfeifer et al, 2011).
In conclusion, the results of our series of paediatric
Ph+ALL patients treated with continuous imatinib
concurrently with intensive chemotherapy and followed by
HSCT, indicate that major improvement in outcome is
maintained with a longer follow-up, with 5-year EFS and OS
of 81!3% and 87!5%, respectively.
Author contributions
SR was the principal investigator and takes primary responsi-
bility for the paper. SR, JE, PG, AV, MJM, JLV, RF, MM, FL,
RL, JU, AF and IB recruited the patients. SR and MC partici-
pated in the statistical analysis and SR wrote the paper.
Author disclosures
The authors report no potential conflicts of interest.
Susana Rives1
Mireia Camos1,2
Jesus Estella1
Pedro Gomez3
Mª Jose Moreno4
Jose Luis Vivanco5
Montserrat Melo6
Rafael Fernandez-Delgado7
Amparo Verdeguer8
Ana Fernandez-Teijeiro9
Francisco Lend"ınez10
Ricardo Lopez-Almaraz11
Jose Javier Uriz12
Isabel Badell13
on behalf of the Spanish Paediatric Haemato-Oncology
Group (SHOP/ SEHOP)1Paediatric Haemato-oncology, Hospital Sant Joan de Deu de Barce-
lona, University of Barcelona, 2Laboratory of Haematology, Hospital
Sant Joan de Deu de Barcelona, University of Barcelona, 3Paediatric
Haemato-oncology, Hospital Universitario Reina Sof"ıa, Cordoba,4Paediatric Haemato-oncology, Hospital Universitario Virgen de las
Nieves, Granada, 5Paediatric Haemato-oncology, Hospital Universitario
Doce de Octubre, Madrid, 6Paediatric Haemato-oncology, Hospital de
Sabadell. C.S. Parc Taul"ı, Sabadell, 7Paediatric Haemato-oncology,
Hospital Cl"ınic Universitari Val!encia, 8Paediatric Haemato-oncology,
Hospital Universitari La Fe, Valencia, 9Paediatric Haemato-oncology,
Hospital Universitario Virgen de la Macarena, Sevilla, 10Paediatric
Haemato-oncology, Hospital Universitario Torrecardenas, Almer"ıa,11Paediatric Haemato-oncology, Hospital Universitario de Canarias,
Santa Cruz de la Laguna Tenerife, 12Paediatric Haemato-oncology,
Hospital Universitario de Donostia, Donostia, and 13Paediatric Haema-
tology, Hospital de la Santa Creu i Sant Pau, Universitat Aut!onoma de
Barcelona, Barcelona, Spain
E-mail: [email protected]
Keywords: Philadelphia chromosome, acute lymphoblastic leukae-
mia, children, imatinib, tyrosine kinase inhibitor
Correspondence
2 ª 2013 John Wiley & Sons Ltd, British Journal of Haematology

Resultados
113
References
Aric!o, M., Schrappe, M., Hunger, S.P., Carroll,
W.L., Conter, V., Galimberti, S., Manabe, A.,
Saha, V., Baruchel, A., Vettenranta, K., Horibe,
K., Benoit, Y., Pieters, R., Escherich, G., Silver-
man, L.B., Pui, C.H. & Valsecchi, M.G. (2010)
Clinical outcome of children with newly diag-
nosed Philadelphia chromosome-positive acute
lymphoblastic leukaemia treated between 1995
and 2005. Journal of Clinical Oncology, 28,
4755–4761.
Biondi, A., Schrappe, M., De Lorenzo, P., Castor,
A., Lucchini, G., Gandemer, V., Pieters, R.,
Stary, J., Escherich, G., Campbell, M., Li, C.K.,
Vora, A., Aric!o, M., R"ottgers, S., Saha, V. &
Valsecchi, M.G. (2012) Imatinib after induction
for treatment of children and adolescents with
Philadelphia-chromosome-positive acute lym-
phoblastic leukaemia (EsPhALL): a randomised,
open-label, intergroup study. The Lancet
Oncology, 13, 936–945.
Nishiwaki, S., Miyamura, K., Kato, C., Terakura,
S., Ohashi, K., Sakamaki, H., Nakao, S., Harigae,
H. & Kodera, Y. (2010) Impact of post-trans-
plant imatinib administration on Philadelphia
chromosome-positive acute lymphoblastic
leukaemia. Anticancer Research, 30, 2415–2418.
Pfeifer, H., Wassmann, B., Bethge, W., Dengler, J.,
Bornhauser, M. & Stadler, M. (2011) Updated
long-term results of a randomized comparison
of prophylactic and pre-emptive imatinib
following allogeneic stem cell transplantation for
Philadelphia chromosome positive acute
lymphoblastic leukemia (Ph+ ALL). Blood (ASH
Annual Meeting Abstracts), 118, 247.
Ribera, J.M., Oriol, A., Gonzalez, M., Vidriales,
B., Brunet, S., Esteve, J., Del Potro, E., Rivas,
C., Moreno, M.J., Tormo, M., Mart#ın-Reina,
V., Sarra, J., Parody, R., Perez de Oteyza, J.,
Bureo, E., Bernal, M.T. & Grupo Espa~nol de
Trasplante Hemopoyetico Groups. (2010)
Concurrent intensive chemotherapy and imati-
nib before and after stem cell transplantation
in newly diagnosed Philadelphia chromosome-
positive acute lymphoblastic leukemia. Final
results of the CSTIBES02 trial. Haematologica,
95, 87–95.
Rives, S., Estella, J., Gomez, P., Lopez-Duarte,
M., Garc#ıa de Miguel, P., Verdeguer, A.,
Moreno, M.J., Vivanco, J.L., Couselo, J.M.,
Fernandez-Delgado, R., Maldonado, M., Tasso,
M., Lopez-Ibor, B., Lend#ınez, F., Lopez-Alma-
raz, R., Uriz, J., Melo, M., Fernandez-Teijeiro,
A., Rodr#ıguez, I. & Badell, I. (2011) Intermedi-
ate dose of imatinib in combination with che-
motherapy followed by allogeneic stem cell
transplantation improves early outcome in pae-
diatric Philadelphia chromosome-positive acute
lymphoblastic leukaemia (ALL): results of the
Spanish Cooperative Group SHOP studies
ALL-94, ALL-99 and ALL-2005. British Journal
of Haematology, 154, 600–611.
Schultz, K.R., Bowman, W.P., Aledo, A., Slayton,
W.B., Sather, H., Devidas, M., Wang, C.,
Davies, S.M., Gaynon, P.S., Trigg, M., Rutledge,
R., Burden, L., Jorstad, D., Carroll, A., Heer-
ema, N.A., Winick, N., Borowitz, M.J., Hunger,
S.P., Carroll, W.L. & Camitta, B. (2009)
Improved early event-free survival with imati-
nib in Philadelphia chromosome-positive acute
lymphoblastic leukemia: a Children’s Oncology
Group study. Journal of Clinical Oncology, 27,
5175–5181.
Thomas, D. (2012) Childhood Philadelphia-
chromosome-positive B-lymphoblastic leukae-
mia. The Lancet Oncology, 13, 860–862.
Correspondence
ª 2013 John Wiley & Sons Ltd, British Journal of Haematology 3

Resultados
114

Resultados
115
Trabajo 3 (carta al editor)
Susana Rives, Mireia Camós, Jesús Estella, Pedro Gómez, Mónica López
Duarte, Aurora Navajas, Isabel Badell. En representación de la Sociedad
Española de Hemato-‐Oncología Pediátrica (SHOP/SEHOP)
British Journal of Haematology 2012; 156:284-‐6.
Validation of the ‘French Acute Lymphoblastic Leukaemia Study
Group FRALLE prognostic index’ for paediatric Philadelphia-‐
chromosome acute lymphoblastic leukaemia

Resultados
116

Resultados
117
Factores pronósticos en pacientes pediátricos con LLA Ph+ tratados en la
era pre-‐imatinib
Con anterioridad a la publicación de los resultados de nuestra serie
pediátrica de pacientes con LLA Ph+ tratados según los protocolos SHOP/LLA,
el grupo norteamericano COG publicó sus excelentes resultados en pacientes
pediátricos con LLA Ph+ tratados con quimioterapia muy intensiva en
combinación con imatinib. Sus pacientes recibieron trasplante alogénico sólo
si tenían un hermano HLA-‐idéntico. Su protocolo contemplaba la
administración de quimioterapia intensiva seguida de tratamiento de
mantenimiento, en el que también se incluyó imatinib, a aquellos pacientes
en primera RC que carecieran de donante familiar. Los resultados de los
pacientes tratados con quimioterapia e imatinib sin TPH no fueron inferiores
a los de los pacientes trasplantados y cuestionaron la necesidad del
trasplante en primera RC a todos los pacientes pediátricos con LLA Ph+. Por
ello resulta necesario poder identificar a los pacientes con LLA Ph+ de mejor
pronóstico en los que tal vez podría omitirse el trasplante.
Con anterioridad a nuestro análisis, el grupo cooperativo francés FRALLE,
analizó su serie de pacientes tratados en la era pre-‐imatinib y describió un
índice pronóstico que pudo discriminar a un subgrupo de pacientes
pediátricos con LLA Ph+ con mejor supervivencia. Este índice incluyó dos
características presentes en el diagnóstico (edad <10 años y leucocitos <100
x 109/l) y una evolutiva (respuesta precoz en médula ósea) para definir un
grupo de buen pronóstico. Una tercera parte de sus pacientes fueron de
buen pronóstico según este índice y tuvieron una SLE a los 5 años del 79%
(Gandemer et al., 2009).

Resultados
118
En el presente trabajo se realizó un análisis retrospectivo de nuestra serie de
pacientes tratados sin imatinib para identificar factores pronósticos que
pudieran ayudar a seleccionar a aquellos niños en los que podría plantearse
evitar el TPH alogénico en primera RC. Para ello, aplicamos el índice
pronóstico FRALLE en los pacientes tratados con protocolos SHOP pre-‐
imatinib (SHOP/LLA-‐94 y SHOP/LLA-‐99) y confirmamos su valor pronóstico en
una serie independiente de pacientes.
Entre febrero de 1994 y marzo de 2005 se trataron 27 pacientes con
quimioterapia intensiva seguida de TPH alogénico de donante familiar o no
relacionado. Los resultados de supervivencia de los pacientes fueron
heterogéneos. La aplicación del índice pronóstico FRALLE en nuestra cohorte
de pacientes permitió identificar a un subgrupo de pacientes con mejor
pronóstico. Así, el análisis de factores pronósticos reveló que los niños de
menor edad (entre 1 y 9 años) con recuento de leucocitos inferior a 100 x
109/l y una buena respuesta inicial al tratamiento, analizada mediante la
evaluación en médula ósea en el día +15 (menos de 5% de blastos) tuvieron
una supervivencia muy superior a los demás pacientes (p=0,018), con
alrededor de 70% de pacientes supervivientes a largo plazo frente a 15% del
resto de los pacientes (figura 1 , tabla I del artículo).
En conclusión, pudimos validar en nuestra cohorte de pacientes tratados con
protocolos pre-‐imatinib un índice pronóstico descrito con anterioridad, que
permite discriminar a un subgrupo de pacientes pediátricos con LLA Ph+ con
un pronóstico significativamente mejor.

Resultados
119
Validation of the ‘French Acute Lymphoblastic Leukaemia StudyGroup FRALLE prognostic index’ for paediatric Philadelphia-chromosome acute lymphoblastic leukaemia
We recently reported the Spanish Paediatric Haematology
Oncology Group (SHOP) experience in treating children and
adolescents with Philadelphia-chromosome positive acute
lymphoblastic leukaemia (Ph+ ALL) with concurrent imatinib
and chemotherapy before stem cell transplantation, and
showed an impressive improvement in the early outcome of
these children when treated with this approach (Rives et al,
2011). Previously, the Children’s Oncology Group (COG)
reported an excellent outcome in Ph+ ALL paediatric patients
also treated with imatinib in combination with chemotherapy.
Their patients underwent haematopoietic stem cell transplant
(HSCT) only when a matched sibling donor was available
(Schultz et al, 2009). The early outcome of patients undergo-
ing HSCT was not superior to those that had received only
chemotherapy. The major improvement achieved with tyro-
sine-kinase inhibitors (TKI) in combination with chemother-
apy in paediatric Ph+ ALL challenges the need for HSCT in all
children with this subgroup of leukaemia. Therefore, it is
necessary to identify a good-risk group of patients for whom
allogeneic HSCT could be omitted. We retrospectively anal-
ysed our pre-imatinib cohort of patients with the aim of
identifying prognostic factors that might help to select those
patients who could be spared HSCT.
Patients with Ph+ALL treated with pre-imatinib SHOP
protocols (SHOP/ALL-94 and SHOP/ALL-99) were analysed.
From February 1994 to March 2005, 27 patients were treated
with intensive chemotherapy and HSCT from a matched
related or unrelated donor (Rives et al, 2011). The disease
outcome in our patients was heterogeneous, depending on
clinical features at diagnosis and early response to treatment, in
line with other series of children with Ph+ALL (Ribeiro et al,
1997; Schrappe et al, 1998; Arico et al, 2000, 2010; Roy et al,
2005; Gandemer et al, 2009).
Recently, the French Acute Lymphoblastic Leukaemia Study
Group (FRALLE) analysed prognostic factors in their pre-
imatinib series of paediatric patients with Ph+ ALL and
described a prognostic index that could discriminate a
subgroup of patients with a better prognosis. This prognostic
index included two presenting features (age < 10 years and
white blood cell [WBC] count <100 · 109/l) and early
response (less than 5% of blasts in bone marrow on day 21
of induction treatment) for good risk. One third of their
patients belonged to the good-risk group and had a 5-year
event-free survival (EFS) of 79% (Gandemer et al, 2009). In
order to test this prognostic index in an independent cohort of
patients we analysed the impact of the FRALLE index in our
series of paediatric Ph+ ALL patients. With this index it was
possible to identify a subgroup of patients with better
outcome. Indeed, the analysis of prognostic factors revealed
that younger children (1–9 years) with WBC count
<100 · 109/l count and a good early response, as per bone
marrow blast count on day-15, had a significantly superior
outcome (P = 0Æ018) with around 70% of long-term survivors
(Table I, Fig. 1). Our series validated, in an independent
cohort of paediatric Ph+ ALL, a strong prognostic factor that
can be easily estimated early in the course of the disease and
enables a good risk group to be defined among Ph+ALL
patients. In this regard, minimal residual disease measurement
could also help to refine the identification of good-risk patients
Table I. Outcome of patients as a function of the main patient char-acteristics of the pre-imatinib group
All patients
n 5-year EFS 5-year OS
27 30 (±9) P 31 (±9) P
Sex
Male 13 31 (±13) 33 (±14)
Female 14 29 (±12) 0Æ8 29 (±12) 0Æ44
Age
<10 years 18 33 (±11) 33 (±11)
‡10 years 9 22 (±14) 0Æ22 25 (±15) 0Æ56
Leucocyte count (·109/l)
<25 11 55 (±15) 55 (±15)
‡25 16 13 (±8) 0Æ055 13 (±9) 0Æ16
<50 15 40 (±13) 40 (±13)
‡50 12 17 (±11) 0Æ33 18 (±12) 0Æ7<100 20 40 (±11) 40 (±11)
‡100 7 0 0Æ21 0 0Æ39
Blasts on day 15 bone marrow
<5% 14 43 (±13) 43 (±13)
‡5% 13 15 (±10) 0Æ09 17 (±11) 0Æ18
FRALLE index
Good* 7 71 (±17) 71 (±17)
Others 20 15 (±8) 0Æ018 16 (±8) 0Æ032
FRALLE, French Acute Lymphoblastic Leukaemia Study Group.
*Age < 10 years. leucocyte count < 100 · 109/l and <5% blasts on
bone marrow on day 15–21 of induction treatment.
correspondence
ª 2011 Blackwell Publishing Ltd, British Journal of Haematology doi:10.1111/j.1365-2141.2011.08860.x

Resultados
120
who might be considered for treatment de-escalation, e.g. omit
allogeneic HSCT in first complete remission.
In conclusion, we have confirmed in our pre-imatinib
cohort of patients, a previously described good prognostic
index to identify a subset of patients with Ph+ALL with
significantly better outcome. This index may help to identify
children in whom allogeneic HSCT could be omitted in the
present TKI era.
Susana Rives1
Mireia Camos2
Jesus Estella1
Pedro Gomez3
Monica Lopez-Duarte4
Aurora Navajas5
Isabel Badell6
1Paediatric Haematology, 2Haematology Laboratory, Paediatric
Haemato-Oncology Hospital Sant Joan de Deu de Barcelona, Barcelona,3Paediatric Haemato-Oncology, Hospital Reina Sofıa, Cordoba,4Department of Haematology, Hospital Universitario Marques de
Valdecilla, Santander, 5Paediatric Haemato-Oncology, Hospital de
Cruces, Bilbao, and 6Paediatric Haemato-Oncology, Hospital de la Santa
Creu i Sant Pau, Universitat Autonoma de Barcelona, Spain
E-mail: [email protected]
Keywords: acute leukaemia, paediatric haematology, BCR-ABL1,
Philadelphia chromosome, prognosis.
References
Arico, M., Valsecchi, M.G., Camitta, B., Schrappe,
M., Chessells, J., Baruchel, A., Gaynon, P., Silv-
erman, L., Janka-Schaub, G., Kamps, W., Pui,
C.H. & Masera, G. (2000) Outcome of treatment
in children with Philadelphia chromosome-
positive acute lymphoblastic leukemia. New
England Journal of Medicine, 342, 998–1006.
Arico, M., Schrappe, M., Hunger, S.P., Carroll,
W.L., Conter, V., Galimberti, S., Manabe, A.,
Saha, V., Baruchel, A., Vettenranta, K., Horibe,
K., Benoit, Y., Pieters, R., Escherich, G., Silver-
man, L.B., Pui, C.H. & Valsecchi, M.G. (2010)
Clinical outcome of children with newly diag-
nosed Philadelphia chromosome-positive acute
lymphoblastic leukaemia treated between 1995
and 2005. Journal of Clinical Oncology, 28, 4755–
4761.
Gandemer, V., Auclerc, M.F., Perel, Y., Vannier, J.P.,
Le, G.E., Demeocq, F., Schmitt, C., Piguet, C.,
Stephan, J.L., Lejars, O., Debre, M., Jonveaux, P.,
Cayuela, J.M., Chevret, S., Leverger, G. & Baru-
chel, A. (2009) Impact of age, leukocyte count
and day 21-bone marrow response to chemo-
therapy on the long-term outcome of children
with Philadelphia chromosome-positive acute
lymphoblastic leukemia in the pre-imatinib era:
results of the FRALLE 93 study. BioMed Central
Cancer, 9, 14.
Ribeiro, R.C., Broniscer, A., Rivera, G.K., Hancock,
M.L., Raimondi, S.C., Sandlund, J.T., Crist, W.,
Evans, W.E. & Pui, C.H. (1997) Philadelphia
chromosome-positive acute lymphoblastic leu-
kemia in children: durable responses to chemo-
therapy associated with low initial white blood
cell counts. Leukemia, 11, 1493–1496.
Rives, S., Estella, J., Gomez, P., Lopez-Duarte, M.,
Garcıa de Miguel, P., Verdeguer, A., Moreno,
M.J., Vivanco, J.L., Couselo, J.M., Fernandez-
Delgado, R., Maldonado, M., Tasso, M., Lopez-
Ibor, B., Lendınez, F., Lopez-Almaraz, R., Uriz, J.,
Melo, M., Fernandez-Teijeiro, A., Rodrıguez, I. &
Badell, I. (2011) Intermediate dose of imatinib in
combination with chemotherapy followed by
allogeneic stem cell transplantation improves
early outcome in paediatric Philadelphia chro-
mosome-positive acute lymphoblastic leukaemia
(ALL): results of the Spanish Cooperative Group
SHOP studies ALL-94, ALL-99 and ALL-2005.
British Journal of Haematology, 154, 600–611.
Roy, A., Bradburn, M., Moorman, A.V., Burrett, J.,
Love, S., Kinsey, S.E., Mitchell, C., Vora, A.,
Eden, T., Lilleyman, J.S., Hann, I. & Saha, V.
(2005) Early response to induction is predictive
of survival in childhood Philadelphia chromo-
some positive acute lymphoblastic leukaemia:
results of the Medical Research Council ALL
97 trial. British Journal of Haematology, 129, 35–
44.
Schrappe, M., Arico, M., Harbott, J., Biondi, A.,
Zimmermann, M., Conter, V., Reiter, A., Val-
secchi, M.G., Gadner, H., Basso, G., Bartram,
C.R., Lampert, F., Riehm, H. & Masera, G. (1998)
Philadelphia chromosome-positive (Ph+) child-
hood acute lymphoblastic leukemia: good initial
steroid response allows early prediction of a
favorable treatment outcome. Blood, 92, 2730–
2741.
Schultz, K.R., Bowman, W.P., Aledo, A., Slayton,
W.B., Sather, H., Devidas, M., Wang, C., Davies,
S.M., Gaynon, P.S., Trigg, M., Rutledge, R.,
Burden, L., Jorstad, D., Carroll, A., Heerema,
N.A., Winick, N., Borowitz, M.J., Hunger, S.P.,
Carroll, W.L. & Camitta, B. (2009) Improved
early event-free survival with imatinib in Phila-
delphia chromosome-positive acute lympho-
blastic leukemia: a children’s oncology group
study. Journal of Clinical Oncology, 27, 5175–
5181.
Fig 1. Event-free survival according FRALLE index in the pre-imatinibcohort.
Correspondence
2 ª 2011 Blackwell Publishing Ltd, British Journal of Haematology

Resultados
121
Trabajo 4 (artículo original)
Susana Rives, Jesús Estella, Mireia Camós, Purificación García de Miguel,
Amparo Verdeguer, José Miguel Couselo, María Tasso, Javier Molina, Pedro
Gómez, Rafael Fernández Delgado, Aurora Navajas, Isabel Badell, en
representación del grupo cooperativo de la Sociedad Española de Hemato-‐
Oncología Pediátrica (SHOP/SEHOP)
Medicina Clínica 2012; 139:141-‐9.
Leucemia linfoblástica aguda T pediátrica: análisis de
supervivencia y factores pronósticos en 4 protocolos
consecutivos del grupo cooperativo multicéntrico SHOP

Resultados
122

Resultados
123
Leucemia linfoblástica aguda T pediátrica: análisis de supervivencia y
factores pronósticos en 4 protocolos consecutivos del grupo cooperativo
multicéntrico SHOP
Los pacientes pediátricos con LLA-‐T constituyen un subgrupo minoritario (10-‐
15% de la LLA pediátrica), en el que la mayoría de grupos cooperativos
obtienen peores resultados que los obtenidos en pacientes con LLA de
estirpe B. En la LLA-‐T, a diferencia de lo que ocurre con la LLA de estirpe B, se
desconocen los factores pronósticos, con excepción de la respuesta precoz al
tratamiento. Ello hace difícil identificar a aquellos pacientes con aumento de
riesgo de recidiva. Se hace necesario, pues, estudiar series amplias de
pacientes con LLA-‐T tratados de forma homogénea con el fin de identificar
factores pronósticos específicos para este subgrupo de LLA.
En el presente estudio analizamos una serie importante de pacientes
pediátricos con LLA-‐T, tratados de forma homogénea según los protocolos
cooperativos de la Sociedad Española de Hematología Pediátrica (SHOP)
durante un periodo de más de 20 años (1989-‐2009).
Se incluyeron 218 pacientes con LLA-‐T sobre un total de 1.652 casos con LLA,
con edades comprendidas entre 1 y 18 años. No hubo diferencias
significativas en los protocolos sucesivos en cuanto a las principales
características clínicas al diagnóstico. En cuanto a la respuesta al tratamiento
de inducción, 150 (69%) pacientes tenían menos de 5% de blastos en médula
ósea en el día +15 de tratamiento y 199 (91%) alcanzaron la RC al final de la
inducción. Once pacientes fallecieron durante esta fase del tratamiento por
causas infecciosas o tóxicas. Ocho pacientes no alcanzaron la RC al final de la
inducción. De los 199 que alcanzaron la RC, 62 recayeron, 8 murieron en

Resultados
124
primera RC y 3 fallecieron por segundas neoplasias.
La SG y la SLE a los 5 años de la serie global de LLA-‐T fueron de 59% (+/-‐4) y
de 53% (+/-‐4), respectivamente, con una mediana de seguimiento de 67
meses. En cuanto a la SLE a los 5 años de los pacientes tratados en los
protocolos sucesivos, fue de 43% (+/-‐8), 44% (+/-‐6) y 61% (+/-‐6) en los
protocolos SHOP/LLA-‐89, LLA-‐94 y LLA-‐99, respectivamente. La SLE a los 48
meses para el protocolo SHOP/LLA-‐2005 (protocolo en curso en el momento
del análisis) fue de 73% (+/-‐7) (figura 1 del artículo). La mediana de
seguimiento fue de 206, 152, 74 y 17 meses para los protocolos SHOP/LLA-‐
89, LLA-‐94, LLA-‐99 y LLA-‐2005, respectivamente.
El análisis de factores pronósticos no mostró diferencias significativas en
cuanto a sexo ni edad. La cifra de leucocitos al diagnóstico no resultó
significativa tomando los puntos de corte utilizados en la mayoría de
protocolos de LLA para estratificar a los pacientes en grupos de riesgo (20 o
50 x109/l). Sin embargo, el punto de corte de 200 x 109/l, en el que se hallan
el 27% de los pacientes de esta serie, obtuvo valor pronóstico, tanto para la
SG como para la SLE (p = 0,028). Otro parámetro al diagnóstico que resultó
significativo fue la infiltración del SNC, tanto para la SG como para la SLE (p <
0,006). Si bien se observaron más casos de hiperleucocitosis entre los
pacientes con infiltración del SNC, ésta mantuvo el valor pronóstico adverso
en el grupo de pacientes con cifras de leucocitos inferiores a 200 x 109/l.
Respecto a la respuesta al tratamiento, fueron significativas tanto la
respuesta precoz al tratamiento como la consecución de RC al final de
inducción, tanto para la SG como para la SLE. La SLE a 5 años en los pacientes
que alcanzaron la RC al final de inducción fue del 58% (+/-‐4), mientras que
ningún paciente sobrevivió entre los que no alcanzaron la RC al final de la

Resultados
125
inducción (p = 0,0000) (figura 2 del artículo).
Por último, se analizó el efecto del protocolo de tratamiento sobre la
supervivencia. Para ello se agrupó a los pacientes tratados con los 2 primeros
protocolos, por un lado, y a los tratados con los 2 últimos, por otro. Con los 2
protocolos más recientes se consiguió una mejoría significativa en la
supervivencia de los pacientes con LLA-‐T, con una SLE a los 5 años de 63%
(+/-‐5) en los protocolos SHOP/LLA-‐99 + LLA-‐2005 frente a 44% (+/-‐5) en los
protocolos SHOP/LLA-‐89 + LLA-‐94 (p = 0,003). De igual modo, también se
mejoró la SG. Esta mejoría se debió principalmente a un menor índice de
recaídas en los 2 últimos protocolos (figura 3 y tabla 3 del artículo).
En conclusión, en este trabajo se describe una serie amplia de pacientes con
LLA-‐T tratados con 4 protocolos sucesivos del grupo SHOP, en la que se
obtuvo una mejoría significativa de los resultados con los 2 últimos
protocolos. Asimismo, se identificaron factores asociados a mal pronóstico:
leucocitosis superior a 200 x 109/l, infiltración en SNC, presencia de más de
5% de blastos en médula ósea en el día +15 de tratamiento de inducción y la
falta de obtención de la RC al finalizar dicho tratamiento. Estos factores
pronósticos, junto con la posible identificación de factores biológicos en
próximos estudios, podrán contribuir a una mejor estratificación de este
subgrupo minoritario de pacientes en los próximos protocolos de
tratamiento.

Resultados
126

Resultados
127
Original
Leucemia linfoblastica aguda T pediatrica: analisis de supervivencia y factorespronosticos en 4 protocolos consecutivos del grupo cooperativo multicentricoSHOP§
Susana Rives a,*, Jesus Estella a, Mireia Camos b, Purificacion Garcıa-Miguel c, Amparo Verdeguer d,Jose Miguel Couselo e, Marıa Tasso f, Javier Molina g, Pedro Gomez h, Rafael Fernandez-Delgado i,Aurora Navajas j e Isabel Badell k, en representacion del grupo cooperativo SHOP (Sociedad Espanolade Hemato-Oncologıa Pediatrica)^
a Hematologıa Pediatrica, Hospital Sant Joan de Deu, Universitat de Barcelona, Barcelona, Espanab Laboratorio de Hematologıa, Hospital Sant Joan de Deu, Universitat de Barcelona, Barcelona, Espanac Hemato-Oncologıa Pediatrica, Hospital La Paz, Madrid, Espanad Departamento de Hemato-oncologıa Pediatrica, Hospital La Fe, Valencia, Espanae Departamento de Hemato-oncologıa Pediatrica, Hospital Xeral de Galicia, Santiago de Compostela, La Coruna, Espanaf Departamento de Hemato-oncologıa Pediatrica, Hospital Universitario de Alicante, Alicante, Espanag Departamento de Hemato-oncologıa Pediatrica, Hospital Virgen del Camino, Pamplona, Navarra, Espanah Departamento de Hemato-oncologıa Pediatrica, Hospital Reina Sofıa, Cordoba, Espanai Departamento de Hemato-oncologıa Pediatrica, Hospital Clınico de Valencia, Valencia, Espanaj Departamento de Hemato-oncologıa Pediatrica, Hospital de Cruces, Bilbao, Bizkaia, Espanak Departamento de Hemato-oncologıa Pediatrica, Hospital de la Santa Creu i Sant Pau, Universitat Autonoma de Barcelona, Barcelona, Espana
Med Clin (Barc). 2012;139(4):141–149
I N F O R M A C I O N D E L A R T I C U L O
Historia del artıculo:Recibido el 7 de septiembre de 2011Aceptado el 20 de diciembre de 2011On-line el 28 de marzo de 2012
Palabras clave:Leucemia linfoblastica aguda TPediatricaFactores pronosticos
R E S U M E N
Fundamento y objetivo: La leucemia linfoblastica aguda (LLA) es el cancer mas frecuente en la edadpediatrica, con tasas de curacion del 80-85%. En la LLA de fenotipo T (LLA-T, 15% de casos) los factorespronosticos no estan bien definidos. Nuestro objetivo es analizar la supervivencia y los factorespronosticos clınicos en una serie de pacientes con LLA-T.Pacientes y metodo: Se analizaron los ninos con LLA-T (1-18 anos) tratados segun los protocolos SHOP/LLA-89/LLA-94/LLA-99/LLA-2005 (desde febrero de 1989 hasta noviembre de 2009) en 37 instituciones.Resultados: Se incluyeron 218 pacientes con LLA-T sobre un total de 1.652 LLA pediatricas. Deellos, 164 (75%) eran varones. La edad mediana fue de 7,8 anos (extremos 1,3-18,6). La medianade leucocitos fue 78,2 ! 109/l (extremos 0,8-930). Quince ninos (6,8%) tuvieron infiltracion delsistema nervioso central (SNC). En cuanto a la respuesta al tratamiento de induccion, 150 (75%) pacientestenıan menos de 5% de blastos en medula osea del dıa +14 y 199 alcanzaron la remision completa. Lasupervivencia global (SG) media (DE) a 60 meses para los protocolos SHOP/LLA-89, LLA-94 y LLA-99 fuedel 48 (8), 49 (6) y 70 (6) %, respectivamente, y la SG a 48 meses para el protocolo SHOP/LLA-05(protocolo en curso) del 74 (8) %. La mediana de seguimiento fue de 206, 152, 74 y 17 meses,respectivamente. El analisis de factores pronosticos no mostro diferencias significativas en cuanto a sexoni edad. Resultaron significativos la cifra de leucocitos mayor o igual a 200 ! 109/l (p = 0,024), lainfiltracion del SNC al diagnostico (p < 0,006), la respuesta al tratamiento (medula osea dıa +14)(p = 0,005) y la remision completa al final de la induccion (p = 0,0000).Conclusiones: Los resultados obtenidos en la LLA-T con los protocolos SHOP/LLA-89 y SHOP/LLA-94 fueroninferiores a otros protocolos contemporaneos, pero la supervivencia mejoro en los 2 ultimos protocolos. Enconcordancia con otras series de LLA-T, la respuesta al tratamiento fue el principal factor pronostico.
! 2011 Elsevier Espana, S.L. Todos los derechos reservados.
§ La primera firmante, Susana Rives, presenta este trabajo como parte de la investigacion realizada para la obtencion del tıtulo de Doctor en Medicina por la Universidadde Barcelona.
* Autor para correspondencia.Correo electronico: [email protected] (S. Rives).
^ En el Anexo 1 se incluye los componentes del grupo.
ww w.els evier .es /med i c in ac l in i c a
0025-7753/$ – see front matter ! 2011 Elsevier Espana, S.L. Todos los derechos reservados.doi:10.1016/j.medcli.2011.12.019

Resultados
128
Introduccion
La leucemia linfoblastica aguda (LLA) es el cancer mas frecuenteen la edad pediatrica. Esta enfermedad constituye uno de los exitosdel tratamiento oncologico, con tasas de curacion mayores al80% en la actualidad1. Esta mejora se debe en gran medida a laestratificacion de los pacientes en grupos de riesgo de recaıda yadaptacion de la intensidad de tratamiento en cada subgrupo.
Si bien la tasa de curacion es elevada, el pronostico no es igualen todos los casos y todavıa fallecen por la enfermedad un 20%. Endeterminados subgrupos de LLA, el ındice de fracaso deltratamiento es mayor. Ası, la mayorıa de grupos cooperativosobtienen peores resultados en las LLA de estirpe T (LLA-T) que enlas LLA de estirpe B1–3. Entre los factores utilizados para clasificar alos pacientes en grupos de riesgo estan la edad, la cifra deleucocitos, la citogenetica, el fenotipo (T o B) y la respuesta precozal tratamiento1. En la ultima decada, ha adquirido gran impor-tancia pronostica la valoracion de la respuesta a la quimioterapiamediante la determinacion de la enfermedad residual mınima(ERM) en distintos momentos del tratamiento4,5. En la LLA-T(10-15% de la LLA pediatrica), con la excepcion de la respuesta altratamiento (citologica y por ERM), los factores pronosticosmencionados anteriormente carecen del valor predictivo quetienen en los demas subgrupos6,7. La LLA-T tiene una marcadaheterogeneidad clınica y biologica. Pese a que ha habido un granavance en el conocimiento de la biologıa de este tipo deleucemias8–10, no ha habido una traduccion de dichos conoci-mientos a los protocolos de tratamiento. Resulta, por tanto, difıcilidentificar que pacientes con LLA-T tienen un riesgo aumentado derecidiva. Ademas, la recaıda en estos pacientes tiene muy malpronostico, con una supervivencia a los 5 anos inferior al 25%11. Sehace necesario, pues, estudiar series amplias de pacientes con LLA-T tratados de forma homogenea para identificar factores pronos-ticos especıficos de este subgrupo de LLA.
En el presente estudio, analizamos una serie importantede pacientes con LLA-T, tratados de forma homogenea segunlos protocolos cooperativos de la Sociedad Espanola de Hemato-logıa Pediatrica (SHOP) durante un perıodo de mas de 20 anos(1989-2009). Se describen las caracterısticas clınicas al diagnostico
y se analizan la supervivencia y los factores pronosticos de recaıday de fracaso del tratamiento en pacientes con LLA-T tratados en4 protocolos sucesivos.
Pacientes y metodo
Caracterısticas del estudio
Se trata de un estudio retrospectivo en el que se analiza elsubgrupo de pacientes con LLA-T del total de pacientes con LLApediatrica incluidos en 4 protocolos multicentricos consecutivosde la SHOP.
Criterios de inclusion
Los criterios de inclusion fueron: edad entre 1 y 18 anos,diagnostico de LLA de fenotipo T, no haber recibido tratamientocitostatico previo o corticoides durante mas de una semana yrecibir tratamiento segun protocolos de la SHOP en alguno de losdistintos centros del grupo cooperativo.
Pacientes
Durante el perıodo comprendido entre febrero de 1989 ynoviembre de 2009 se diagnosticaron de LLA 1.652 pacientespediatricos en 37 centros distintos (la lista de los centros se detallaen el anexo). En 218 pacientes la leucemia fue de fenotipo T. Losresultados globales (LLA de estirpe B y T) obtenidos en lospacientes incluidos en los protocolos SHOP/LLA-89 y LLA-94 sepublicaron con anterioridad12.
Estudios diagnosticos y de seguimiento
Estudio morfologicoEl diagnostico de LLA se baso en la presencia de blastos de
morfologıa linfoide en medula osea. Se realizaron las principalestecnicas citoquımicas: mieloperoxidasa, fosfatasa acida y periodicacid-Schiff (PAS, «acido periodico Schiff»).
T-cell pediatric acute lymphoblastic leukemia: analysis of survival and prognosticfactors in 4 consecutive protocols of the Spanish cooperative study group SHOP
Keywords:T-cell acute lymphoblastic leukemiaPediatricPrognostic factors
A B S T R A C T
Background and objectives: Acute lymphoblastic leukemia (ALL) is the most frequent cancer in childhood,with cure rates of 80-85%. In T-cell ALL (15% of ALL), prognostic factors are ill defined. We aimed todescribe the event-free survival (EFS) and analyze clinical prognostic factors in a series of pediatric T-ALLof 4 consecutive clinical trials.Patients and methods: Children with T-ALL aged 1-18 years treated in 37 institutions in Spain wereenrolled in 4 consecutive trials from February-1989 to November-2009.Results: A total of 218 T-ALL patients out of 1,652 pediatric ALL were evaluable during the study period(SHOP/ALL-89: 35, ALL-94: 63, ALL-99: 62, ALL-2005: 58). There were 164 boys (75%). Median age(years) was 7.8 range (1.3-18.6). Median leukocytes (109/L) was 78.2, range 0.8-930. Fifteen (6.8%)children had central nervous system (CNS) involvement at diagnosis. Regarding response to inductiontreatment, 150 (75%) patients had less than 5% blasts on day-14 bone marrow and 199 achievedcomplete remission at the end of induction. Overall survival (OS) at 60 months for SHOP/ALL-89, ALL-94,ALL-99 was 48 (8), 49 (6), 70 (6) %, respectively, and at 48 months for SHOP/ALL-2005 (ongoing protocol)was 74 (8) %. Median follow-up (months) was 206, 152, 74 and 17 respectively. Analysis of prognosticfactors revealed no statistical differences regarding sex or age. Leukocyte count over 200 ! 109/l(P = .024), CNS infiltration at diagnosis (P < .006) and treatment response had prognostic significance(end-induction complete remission) (P = .0000), day 14-bone marrow (P = .005).Conclusions: Results for the SHOP/ALL-89 and ALL-94 protocols were inferior to other contemporaryprotocols but there has been an improvement in survival in the 2 last trials. In line with other T-ALLseries, response to treatment had the strongest prognostic impact.
! 2011 Elsevier Espana, S.L. All rights reserved.
S. Rives et al / Med Clin (Barc). 2012;139(4):141–149142

Resultados
129
Estudio inmunofenotıpicoSe estudio mediante citometrıa de flujo el inmunofenotipo de la
poblacion blastica con marcadores de las 3 lıneas hematopoyeticas.La positividad se definio como la deteccion de un marcador en un20% o mas de los blastos.
Estudio citogeneticoEl analisis citogenetico sobre las muestras al diagnostico se
realizo en medula osea y/o sangre periferica. Inmediatamente trassu obtencion, las muestras se procesaron para su cultivo. Lasmetafases obtenidas se tineron segun la tecnica de bandeo deGiemsa (bandas G).
Estudios geneticos molecularesEn los 2 ultimos protocolos se realizo de forma sistematica el
estudio de reordenamiento del gen MLL por fluorescence in situhybridization (FISH, «hibridacion in situ con fluorescencia») y delgen de fusion BCR-ABL1 por reverse transcriptase polymerase chainreaction (RT-PCR, «reaccion en cadena de la polimerasa en tiemporeal») cualitativa, RT-PCR cuantitativa y/o por FISH. Estos estudiosse realizaron en distintos centros y de forma no centralizada. Demanera optativa, en algunos centros se realizo, en los casos deLLA-T, el estudio de reordenamiento del receptor de celulas T.
Estudio de enfermedad residual mınimaDesde el protocolo SHOP/LLA-2005, la realizacion de estudios en
medula osea de ERM por citometrıa de flujo fue obligatoria. LaERM se analizo en los siguientes momentos del tratamiento: dıa+14, final de induccion, final de consolidacion, final de intensifi-cacion, final de primer ano de mantenimiento y final detratamiento. En los casos de muy alto riesgo, la ERM se analizotambien despues de 3 bloques de intensificacion y antes de larealizacion de un trasplante de progenitores hematopoyeticos. Elanalisis de la ERM por citometrıa de flujo no se realizo de formacentralizada, si bien se establecio un numero mınimo demarcadores (anticuerpos monoclonales) y se escogieron unoslaboratorios como centros de referencia para la cuantificacion de laERM. A todos los centros del grupo SHOP se les ofrecio la posibilidadde envıo de muestras a los laboratorios de referencia (anexo).
Tratamiento
Todos los pacientes recibieron tratamiento quimioterapicosegun los protocolos SHOP/LLA-89 (febrero 1989-febrero 1994),SHOP/LLA-94 (marzo 1994-julio 1999), SHOP/LLA-99 (agosto1999-febrero 2005) y SHOP/LLA-2005 (desde marzo 2005,protocolo en activo).
Tabla 1Protocolo de tratamiento de SHOP/LLA-99 y SHOP/LLA-2005 para pacientes de riesgo alto
SHOP/LLA-99 SHOP/LLA-2005
mg/m2 Dıas mg/m2 Dıas
InduccionPrednisona 60 1 al 28 60 1 al 28Vincristina* 1,5 1, 8, 15, 22 1,5 1, 8, 15, 22Daunorrubicinay 60 1 y 2 60 1 y 2Asparaginasaz 15.000 16 al 20 10.000 15, 17, 19, 21, 23
23 al 27 27, 29, 31, 33 y 35Ciclofosfamida 1.000 15 y 36 1.000 15 y 36Metotrexato§ 3.000 15 No administrado en fase de induccionTITjj 1,8 y 15 1,8 y 15
ConsolidacionMetotrexato! 3.000 1, 15, 29 5.000 1, 15, 29Mercaptopurina** 60 Diaria 60 DiariaCitarabina 1.000 43 y 44 (cada 12 h, 4 dosis) 1.000 43 y 44 (cada 12 h, 4 dosis)TITjj 1, 15, 29 y 43 1, 15 29 y 43
IntensificacionDexametasona 10 1 al 15 y descenso en 7 dıas 8 1 al 15 y descenso en 7 dıasVincristina* 1,5 1, 8, 15, 22 1,5 1, 8, 15, 22Epiadriamicina 25 1, 8, 15 25 1 y 8Asparaginasaz 15.000 9 al 13 y 16 al 20 10.000 1, 3, 5,7, 9, 11, 13, 15, 17,19Ciclofosfamida 1.000 22 1.000 22Metotrexato§ 3.000 22 5.000 22Citarabina 200 30 al 34 200 30 al 34TITjj 1,8 y 22 1, 8 y 22
MantenimientoMercaptopurina** 60 Diaria (oral) 60 Diaria (oral)Metotrexato 20 Semanal (intramuscular) 20 Semanal (intramuscular)TITjj yy
Radioterapia craneoespinalzz
Reinducciones§§
Prednisona 40 1 al 7 40 1 al 7Vincristina* 1,5 1 1,5 1Asparaginasaz 20.000 Cada 8 semanas (total 5 dosis) 10.000 Cada 8 semanas (total 4 dosis)Ciclofosfamida 1.000 Cada 8 semanas (total 4 dosis) 1.000 Cada 8 semanas (total 4 dosis)
Todos los citostaticos se administran por vıa intravenosa salvo si se especifica otra vıa. *Dosis maxima 2 mg. yInfusion 24 h. zAsparaginasa de E. coli, intramuscular.§Administrado en 4 h, rescate con folinato 15 mg/m2/6 h desde 24 h del inicio de metotrexato hasta alcanzar niveles sericos < 2 ! 10"7M. jjTratamiento intratecal triple:metotrexato, hidrocortisona y citarabina, dosis adaptada segun edad. !Administrado en 24 h, rescate con folinato 15 mg/m2/6 h desde 36 h del inicio de metotrexato hastaalcanzar valores sericos < 2 ! 10"7M. **Dosis inicial, despues ajustada a leucocitos 2-3 ! 109/l. yyTratamiento intratecal triple mensual durante 4 meses. zzRadioterapiacraneoespinal solo en el tratamiento de pacientes con infiltracion del SNC (18 Gy craneal y 6 Gy espinal). Radioterapia craneal profilactica (12 Gys) en LLA-T con leucocitos> 50 ! 109/l y LLA-lınea B con leucocitos > 100.000 ! 109/l en el protocolo SHOP/LLA-99 o en LLA-T y leucocitos > 100 ! 109/l en el protocolo SHOP/LLA-2005. Inicio de laradioterapia a principio del mantenimiento y suspension posterior de las dosis de TIT.§§Total de 9 ciclos de reinduccion en el protocolo LLA/SHOP-99 y de 8 en el protocolo LLA/SHOP-2005.
S. Rives et al / Med Clin (Barc). 2012;139(4):141–149 143

Resultados
130
Los detalles del tratamiento y de los grupos de riesgo de los2 primeros protocolos (SHOP/LLA-89 y SHOP/LLA-94) se hanpublicado12. El tratamiento de los 2 ultimos protocolos (SHOP/LLA-99 y SHOP/LLA-2005) se describe en las tablas 1 y 2. Los pacientesrecibıan tratamiento de distinta intensidad segun el grupo deriesgo al que pertenecieran. En los 2 ultimos protocolos, los gruposde riesgo se definıan segun los siguientes criterios:
1) Riesgo estandar: edad entre 1 y 9 anos, leucocitos al diagnosticoinferiores a 20 ! 109/l, ausencia de afectacion extramedular,fenotipo B comun, citogenetica favorable, buena respuesta en eldıa +14 (menos del 5% de blastos en medula osea) y al final deltratamiento (obtencion de remision completa [RC] al final de lainduccion). Ademas, en el protocolo 2005 la ERM debıa serinferior a 0,1% al final de la induccion. Se debıan cumplir todaslas condiciones para permanecer en este grupo.
2) Riesgo alto: pacientes de edad igual o mayor a 10 anos,inmunofenotipo distinto al B comun, leucocitos entre 20 y200 ! 109/l, citogenetica desfavorable, afectacion extramedular(sistema nervioso central [SNC] o testes) y aquellos pacientes deriesgo estandar con mas de un 5% de blastos en medula osea enel dıa +14. En el protocolo 2005 pasaban a este grupo tambienaquellos pacientes de riesgo estandar que al final de la inducciontenıan una ERM superior o igual a 0,1%.
3) Riesgo muy alto: aquellos pacientes que presentaran al menosuno de los siguientes criterios: a) leucocitos superiores a200 ! 109/l; b) citogenetica muy desfavorable, y c) pacientes dealto riesgo con mas de un 5% de blastos en medula osea en el dıa+14. Los pacientes que no alcanzaran la RC al final detratamiento salıan de protocolo. En el protocolo 2005 pasabana muy alto riesgo aquellos pacientes en los que la ERM fuerasuperior a 0,1% al final de la consolidacion.
La citogenetica se definio como favorable, desfavorable o muydesfavorable segun se cumplieran los siguientes criterios:
1) Citogenetica favorable: citogenetica normal, traslocaciont(12;21) (o reordenamiento TEL-RUNX1) e hiperdiploidıa alta(51 a 81 cromosomas o ındice de ADN superior o igual a 1,16),y en ausencia de citogenetica desfavorable o muy desfavorable.
2) Citogenetica desfavorable: hiperdiploidıa baja, de 47 a50 cromosomas (o ındice de ADN entre 1 y 1,15), hipodiploidıade 30 a 45 cromosomas (o ındice de ADN entre 0,6 y 0,99), casitetraploidıa de 82 a 94 cromosomas (o ındice superior a 1,78),cualquier alteracion estructural excepto la traslocaciont(12;21), que es favorable, y las traslocaciones t(9;22) yt(4;11), que son muy desfavorables.
3) Citogenetica muy desfavorable: casi haploidıa de 23 a28 cromosomas (o ındice de ADN inferior a 0,6), presencia delcromosoma Filadelfia (o traslocacion t[9;22] o reordenamientoBCR-ABL1) y traslocacion t(4;11) con reordenamiento del genMLL.
Definiciones empleadas
- Infiltracion del SNC, definida como: a) presencia de mas de5 leucocitos por microlitro en una muestra de lıquido cefalorra-quıdeo en la que se identifican por citologıa linfoblastos;b) presencia de paralisis de par craneal sin otra causa aparenteque la propia enfermedad, o c) presencia de infiltrados cerebraleso de medula espinal documentados por pruebas de imagen(resonancia magnetica o tomografıa computarizada).
- RC: infiltracion blastica medular inferior al 5% en presencia de unaspirado medular valorable, con celularidad normal juntocon recuperacion de cifras hemoperifericas. Estos criterios deben
Tabla 2Protocolo de tratamiento de SHOP/LLA-99 y SHOP/LLA-2005 para pacientes de riesgo muy alto
SHOP/LLA-99 SHOP/LLA-2005
InduccionIgual que la induccion de riesgo alto Igual que la induccion de riesgo alto
ConsolidacionIgual que la consolidacion de riesgo alto, seguida de 3 a 5 bloques Igual que la consolidacion de riesgo alto, seguida de 3 a
5 bloquesmg/m2 Dıas mg/m2 Dıas
Bloques A (y Abis)Dexametasona 20 1 al 6 20 1 al 6Vincristina* 1,5 1 y 8 1,5 1 y 8Metotrexatoy 3.000 1 5.000 1Asparaginasaz 25.000 6 10.000 6Citarabina 1.000 4 y 5 (cada 12 h, 4 dosis) 1.000 4 y 5 (cada 12 h, 4 dosis)TIT§ 1 1
Bloque B (y Bbis)Dexametasona 20 1 al 6 20 1 al 6Vincristina* 1,5 1 y 8 1,5 1 y 8Daunorrubicinajj 60 1 60 1Ciclofosfamida 200 2 al 6 200 2 al 6Metotrexato! 3.000 6 5.000 6Asparaginasaz 25.000 7 10.000 7TIT§ 1 1
Bloque CDexametasona 20 1 al 6 20 1 al 6Citarabina 1.000 1 y 2 (cada 12 h, 4 dosis) 1.000 1 y 2 (cada 12 h, 4 dosis)Asparaginasaz 25.000 2 10.000 2Etoposido 100 3 al 5 (cada 12 h, 5 dosis) 100 3 al 5 (cada 12 h, 5 dosis)TIT§ 1 1
Todos los citostaticos se administran por vıa intravenosa salvo si se especifica otra vıa. *Dosis maxima 2 mg. yAdministrado en 24 h, rescate con folinato 15 mg/m2/6 h desde36 h del inicio de metotrexato hasta alcanzar niveles sericos < 2 ! 10"7M. zAsparaginasa de E. coli, administrada intramuscular. §Tratamiento intratecal triple: metotrexato,hidrocortisona y citarabina, dosis adaptada segun edad. jjAdministrada en 24 h. !Administrado en 4 horas, rescate con folinato 15 mg/m2/6 h desde 24 h del inicio demetotrexato hasta alcanzar valores sericos < 2 ! 10"7M.
S. Rives et al / Med Clin (Barc). 2012;139(4):141–149144

Resultados
131
cumplirse en ausencia de infiltrados extramedulares y desintomatologıa clınica de la enfermedad.
- Recidiva: reaparicion de infiltracion blastica medular citologica y/oen localizacion extramedular, como SNC, testes, cutanea u otras.
- Supervivencia global (SG): tiempo transcurrido desde el diag-nostico hasta el fallecimiento o fecha de la ultima visita.
- Supervivencia libre de evento (SLE): tiempo transcurrido desde eldiagnostico hasta la ocurrencia de fallecimiento por cualquiercausa, recaıda o segunda neoplasia, o hasta la fecha de la ultimavisita.
- Supervivencia libre de recaıda (SLR): tiempo transcurrido desdela obtencion de la remision completa hasta la ocurrenciade recaıda o fallecimiento por cualquier causa o hasta lafecha de la ultima visita.
Variables
En este trabajo se analizaron variables clınico-analıticas aldiagnostico: edad (puntos de corte de 10 y 15 anos), sexo,presencia de masa mediastınica y cifra de leucocitos (distintospuntos de corte: 20, 50, 100, 200 y 300 ! 109/l). Tambien seanalizaron variables evolutivas: respuesta al tratamiento en el dıa+14, RC al final de la induccion y supervivencia (SG, SLE y SLR).
Analisis estadıstico
Para el analisis estadıstico de los datos se utilizo el paqueteinformatico SPSS1 para Windows1, version 17.0. Se utilizaron,ademas del analisis descriptivo, las pruebas de analisis desupervivencia de Kaplan-Meier, con el test estadıstico Log-rank.Las variables categoricas se compararon mediante la prueba de lachi al cuadrado o prueba exacta de Fisher y las continuas mediantet de Student o U de Mann-Whitney.
Aspectos eticos
Los protocolos se aprobaron por el Comite Etico del centrocoordinador del estudio (Hospital de Sant Pau i la Santa Creu) yse llevaron a cabo de acuerdo con la Declaracion de Helsinki. Seobtuvo el consentimiento informado de los pacientes, padres otutores en todos los casos del estudio.
Resultados
Durante el perıodo de estudio cumplieron los criterios deinclusion 218 pacientes con LLA-T sobre un total de 1.652 casos con
LLA en edades comprendidas entre 1 y 18 anos. De ellos, 164 (75%)eran varones. La edad mediana fue de 7,8 anos (extremos 1,3-18,6).La mediana de leucocitos fue 78,2 ! 109/l (extremos 0,8-930).Ciento veintitres pacientes (56%) tenıan masa mediastınica y15 ninos (6,8%), infiltracion del SNC. No hubo diferenciassignificativas en los protocolos sucesivos en cuanto a lasprincipales caracterısticas clınicas de los pacientes al diagnostico.En cuanto a la respuesta al tratamiento de induccion, 150 (69%)pacientes tenıan menos de 5% de blastos en medula osea en el dıa+14 de tratamiento y 199 alcanzaron la RC al final de la induccion.Once pacientes fallecieron durante esta fase del tratamiento. En 3de ellos la causa de la muerte fue sepsis y en un paciente en cadacaso las causas fueron aspergilosis pulmonar invasiva, miocardio-patıa, muerte subita, distres respiratorio agudo, fracaso multior-ganico por toxicidad, fallo hepatico fulminante, candidiasissistemica y hemorragia cerebral. Ocho pacientes no alcanzaronla RC al final de la induccion. De los 199 que alcanzaron la RC,62 recayeron, 8 murieron en primera RC y 3 fallecieron porsegundas neoplasias (histiocitosis de celulas de Langerhans,linfoma cutaneo y tumor en SNC) (tabla 3). Las causas defallecimiento en primera RC fueron toxicidad relacionada con eltrasplante de progenitores hemopoyeticos (4 pacientes), sepsis(2 pacientes), neumopatıa intersticial (un paciente) y hemorragiacerebral (un paciente).
Las probabilidades medias (DE) de SG y de SLE a los 60 meses dela serie global de LLA-T fueron del 59 (4) y del 53 (4) %,respectivamente, con una mediana de seguimiento de 67 meses(figura 1A). En cuanto a la SG y la SLE a los 60 meses en lospacientes tratados en los distintos protocolos sucesivos, fueron enel protocolo SHOP/LLA-89 del 48 (8) y 43 (8) %, respectivamente, enel SHOP/LLA-94 del 49 (6) y 44 (6) %, respectivamente, y en elSHOP/LLA-99 del 70 (6) y 61 (6) %, respectivamente. La SG y la SLE alos 48 meses para el protocolo SHOP/LLA-2005 (protocolo en curso)fueron del 74 (8) y 73 (7) %, respectivamente (fig. 1B). La medianade seguimiento en el momento del analisis fue de 206, 152, 74 y17 meses para los protocolos SHOP/LLA-89, LLA-94, LLA-99 y LLA-2005, respectivamente.
El analisis de factores pronosticos no mostro diferenciassignificativas en cuanto a sexo ni edad (figura 2). La cifra deleucocitos al diagnostico no resulto significativa tomando lospuntos de corte utilizados en la mayorıa de protocolos de LLA paraestratificar a los pacientes en grupos de riesgo (20 o 50 ! 109/l). Sinembargo, el punto de corte de 200 ! 109/l, en el que se hallan el27% de los pacientes de esta serie, obtuvo valor pronostico, tantopara la SG como para la SLE (figura 2). Ası, la probabilidad media(DE) de SLE a los 60 meses fue del 57 (4) y 44 (7) % para los
Tabla 3Resultados del tratamiento en LLA-T en los protocolos SHOP (LLA-89, LLA-94, LLA-99 y LLA-2005)
Tratamiento de induccion Total n = 218 SHOP 89 + 94, n= 98 SHOP 99 + 2005, n = 120
Medula osea dıa +14 (< 5% blastos) 150 (69%) 57 (58%) 93 (78%)RC final induccion 199 (91%) 86 (88%) 113 (94%)Muerte en induccion 11 (5%) 6 (6,1%) 5 (4%)No respuesta (refractariedad) 8 (3,7%) 6 (6,1%) 2 (1,7%)
Estado de las RCRC continuada 120 (55%) 41 (42%) 79 (66%)Recaıda 62 (28%) 39 (40%) 23 (19%)Recaıda medular 37 (17%) 24 (25%) 13 (11%)Recaıda SNC
Aislada 7 (3,2%) 5 (5,1%) 2 (1,6%)Combinada 6 (2,7%) 6 (6,1%) 0
Recaıda testesAislada 4 (1,8%) 1 (1%) 3 (2,5%)Combinada 5 (2,2%) 2 (2,1%) 3 (2,5%)
Segundos tumores 3 (1,4%) 2 (2%) 1 (0,8%)Muerte en RC 14 (6,4%) 4 (4,1%) 10 (8,3%)
LLA-T: leucemia linfoblastica aguda de fenotipo T; RC: remision completa; SHOP: Sociedad Espanola de Hemato-Oncologıa Pediatrica; SNC: sistema nervioso central.
S. Rives et al / Med Clin (Barc). 2012;139(4):141–149 145
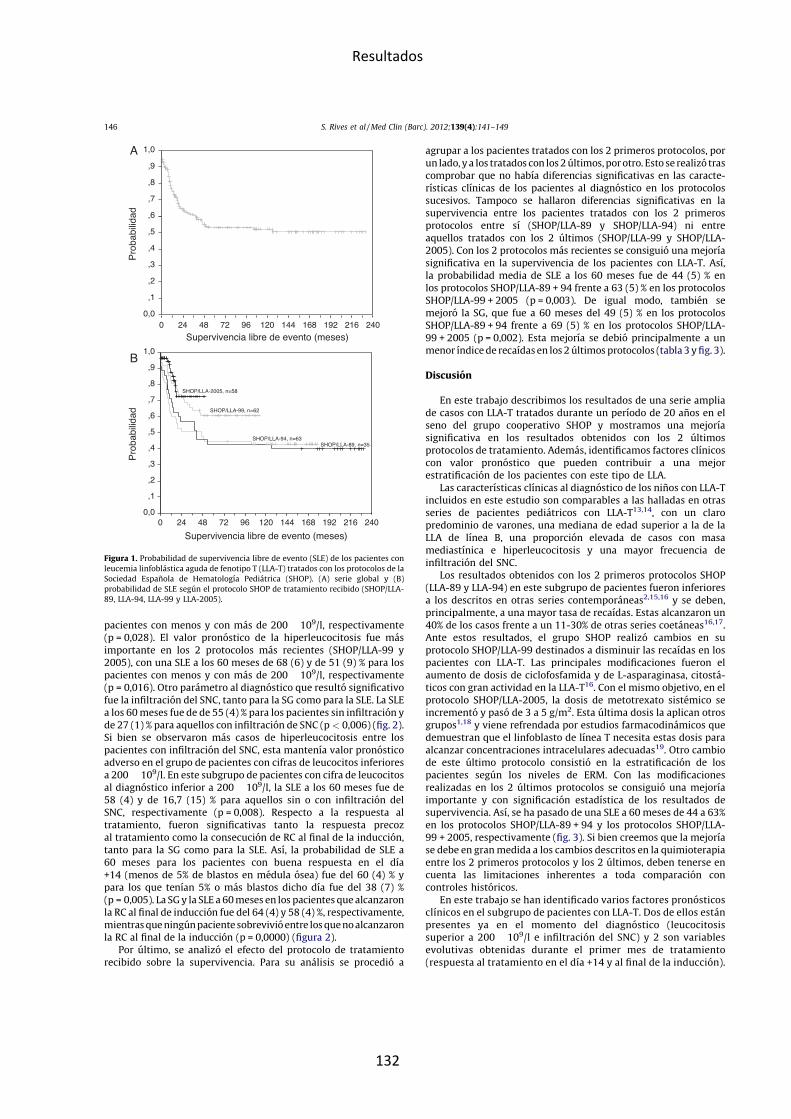
Resultados
132
pacientes con menos y con mas de 200 ! 109/l, respectivamente(p = 0,028). El valor pronostico de la hiperleucocitosis fue masimportante en los 2 protocolos mas recientes (SHOP/LLA-99 y2005), con una SLE a los 60 meses de 68 (6) y de 51 (9) % para lospacientes con menos y con mas de 200 ! 109/l, respectivamente(p = 0,016). Otro parametro al diagnostico que resulto significativofue la infiltracion del SNC, tanto para la SG como para la SLE. La SLEa los 60 meses fue de de 55 (4) % para los pacientes sin infiltracion yde 27 (1) % para aquellos con infiltracion de SNC (p < 0,006) (fig. 2).Si bien se observaron mas casos de hiperleucocitosis entre lospacientes con infiltracion del SNC, esta mantenıa valor pronosticoadverso en el grupo de pacientes con cifras de leucocitos inferioresa 200 ! 109/l. En este subgrupo de pacientes con cifra de leucocitosal diagnostico inferior a 200 ! 109/l, la SLE a los 60 meses fue de58 (4) y de 16,7 (15) % para aquellos sin o con infiltracion delSNC, respectivamente (p = 0,008). Respecto a la respuesta altratamiento, fueron significativas tanto la respuesta precozal tratamiento como la consecucion de RC al final de la induccion,tanto para la SG como para la SLE. Ası, la probabilidad de SLE a60 meses para los pacientes con buena respuesta en el dıa+14 (menos de 5% de blastos en medula osea) fue del 60 (4) % ypara los que tenıan 5% o mas blastos dicho dıa fue del 38 (7) %(p = 0,005). La SG y la SLE a 60 meses en los pacientes que alcanzaronla RC al final de induccion fue del 64 (4) y 58 (4) %, respectivamente,mientras que ningun paciente sobrevivio entre los que no alcanzaronla RC al final de la induccion (p = 0,0000) (figura 2).
Por ultimo, se analizo el efecto del protocolo de tratamientorecibido sobre la supervivencia. Para su analisis se procedio a
agrupar a los pacientes tratados con los 2 primeros protocolos, porun lado, y a los tratados con los 2 ultimos, por otro. Esto se realizo trascomprobar que no habıa diferencias significativas en las caracte-rısticas clınicas de los pacientes al diagnostico en los protocolossucesivos. Tampoco se hallaron diferencias significativas en lasupervivencia entre los pacientes tratados con los 2 primerosprotocolos entre sı (SHOP/LLA-89 y SHOP/LLA-94) ni entreaquellos tratados con los 2 ultimos (SHOP/LLA-99 y SHOP/LLA-2005). Con los 2 protocolos mas recientes se consiguio una mejorıasignificativa en la supervivencia de los pacientes con LLA-T. Ası,la probabilidad media de SLE a los 60 meses fue de 44 (5) % enlos protocolos SHOP/LLA-89 + 94 frente a 63 (5) % en los protocolosSHOP/LLA-99 + 2005 (p = 0,003). De igual modo, tambien semejoro la SG, que fue a 60 meses del 49 (5) % en los protocolosSHOP/LLA-89 + 94 frente a 69 (5) % en los protocolos SHOP/LLA-99 + 2005 (p = 0,002). Esta mejorıa se debio principalmente a unmenor ındice de recaıdas en los 2 ultimos protocolos (tabla 3 y fig. 3).
Discusion
En este trabajo describimos los resultados de una serie ampliade casos con LLA-T tratados durante un perıodo de 20 anos en elseno del grupo cooperativo SHOP y mostramos una mejorıasignificativa en los resultados obtenidos con los 2 ultimosprotocolos de tratamiento. Ademas, identificamos factores clınicoscon valor pronostico que pueden contribuir a una mejorestratificacion de los pacientes con este tipo de LLA.
Las caracterısticas clınicas al diagnostico de los ninos con LLA-Tincluidos en este estudio son comparables a las halladas en otrasseries de pacientes pediatricos con LLA-T13,14, con un claropredominio de varones, una mediana de edad superior a la de laLLA de lınea B, una proporcion elevada de casos con masamediastınica e hiperleucocitosis y una mayor frecuencia deinfiltracion del SNC.
Los resultados obtenidos con los 2 primeros protocolos SHOP(LLA-89 y LLA-94) en este subgrupo de pacientes fueron inferioresa los descritos en otras series contemporaneas2,15,16 y se deben,principalmente, a una mayor tasa de recaıdas. Estas alcanzaron un40% de los casos frente a un 11-30% de otras series coetaneas16,17.Ante estos resultados, el grupo SHOP realizo cambios en suprotocolo SHOP/LLA-99 destinados a disminuir las recaıdas en lospacientes con LLA-T. Las principales modificaciones fueron elaumento de dosis de ciclofosfamida y de L-asparaginasa, citosta-ticos con gran actividad en la LLA-T16. Con el mismo objetivo, en elprotocolo SHOP/LLA-2005, la dosis de metotrexato sistemico seincremento y paso de 3 a 5 g/m2. Esta ultima dosis la aplican otrosgrupos1,18 y viene refrendada por estudios farmacodinamicos quedemuestran que el linfoblasto de lınea T necesita estas dosis paraalcanzar concentraciones intracelulares adecuadas19. Otro cambiode este ultimo protocolo consistio en la estratificacion de lospacientes segun los niveles de ERM. Con las modificacionesrealizadas en los 2 ultimos protocolos se consiguio una mejorıaimportante y con significacion estadıstica de los resultados desupervivencia. Ası, se ha pasado de una SLE a 60 meses de 44 a 63%en los protocolos SHOP/LLA-89 + 94 y los protocolos SHOP/LLA-99 + 2005, respectivamente (fig. 3). Si bien creemos que la mejorıase debe en gran medida a los cambios descritos en la quimioterapiaentre los 2 primeros protocolos y los 2 ultimos, deben tenerse encuenta las limitaciones inherentes a toda comparacion concontroles historicos.
En este trabajo se han identificado varios factores pronosticosclınicos en el subgrupo de pacientes con LLA-T. Dos de ellos estanpresentes ya en el momento del diagnostico (leucocitosissuperior a 200 ! 109/l e infiltracion del SNC) y 2 son variablesevolutivas obtenidas durante el primer mes de tratamiento(respuesta al tratamiento en el dıa +14 y al final de la induccion).
1,0A
B
,9,8,7,6,5,4,3
Supervivencia libre de evento (meses)240216192168144120967248240
Prob
abilid
adPr
obab
ilidad
,2,1
0,0
1,0,9,8,7,6,5,4
SHOP/LLA-2005, n=58
SHOP/LLA-99, n=62
SHOP/LLA-94, n=63SHOP/LLA-89, n=35
240216192168144120967248240
,3,2,1
0,0
Supervivencia libre de evento (meses)
Figura 1. Probabilidad de supervivencia libre de evento (SLE) de los pacientes conleucemia linfoblastica aguda de fenotipo T (LLA-T) tratados con los protocolos de laSociedad Espanola de Hematologıa Pediatrica (SHOP). (A) serie global y (B)probabilidad de SLE segun el protocolo SHOP de tratamiento recibido (SHOP/LLA-89, LLA-94, LLA-99 y LLA-2005).
S. Rives et al / Med Clin (Barc). 2012;139(4):141–149146

Resultados
133
La edad no tuvo valor pronostico. Precisamente, el valorpronostico de la edad en los pacientes con LLA-T, a diferencia delo que ocurre en la LLA de lınea B, varıa segun las seriespublicadas6,16. Lo mismo sucede con los demas factores pronosticosidentificados14,20, con excepcion de la respuesta precoz al trata-miento21. Estas discrepancias entre series podrıan deberse adiferencias en el tratamiento que neutralizaran el efecto adversode algunos de ellos. Sin embargo, el factor pronostico mas relevantede nuestra serie, la respuesta precoz al tratamiento, sı concuerda conlo hallado en otros grupos de trabajo2,21. Ası, resultaron factorespronosticos significativos tanto la respuesta precoz en el dıa +14 (SGa 60 meses de 69 frente a 38%, p = 0,005), como al final de lainduccion (SG a 60 meses de 64 frente a 0%, p = 0,0000). Otrasvariables de respuesta al tratamiento con gran importanciapronostica en otras series son la respuesta a una semana deprednisona (mas una dosis de metotrexato intratecal) y la ERM7. Larespuesta a una semana de prednisona permite la identificacion dealrededor de un 25-30% de pacientes con LLA-T que son malosrespondedores (> 1.000 blastos/microlitro en sangre periferica en eldıa +7) y que tienen un elevado riesgo de recaıda13. Este parametrono se pudo analizar en nuestra serie por carecer los protocolosSHOP/LLA de esta prefase de tratamiento con prednisona. Respecto ala ERM, esta se analiza desde el ultimo protocolo (SHOP/LLA-2005) yel analisis de su valor pronostico debe esperar a alcanzar una mayor
mediana de seguimiento, por ahora muy corta. Si bien todavıa nopodemos analizar el valor pronostico de este parametro en nuestraserie, este es en la actualidad uno de los principales factorespronosticos en la LLA. La ERM no solo tiene valor pronostico, sino quese ha incorporado en los protocolos terapeuticos para estratificar alos pacientes y dirigir la intensidad de tratamiento, de acuerdo conlos niveles alcanzados de ERM en distintos momentos deltratamiento1,15.
En cuanto a factores biologicos con valor pronostico, adiferencia de la LLA de lınea B, en la LLA-T estos estan pocodefinidos y dependen del protocolo de tratamiento en el quese analicen. Esto sucede tanto para parametros favorables(mutaciones en NOTCH1)22,23 como para otros adversos (sobreex-presion de TLX3/HOX11L2)24,25. Recientemente, se ha descrito unsubgrupo de LLA-T con muy mal pronostico, que corresponde a unestadio madurativo muy precoz del linfoblasto T (early Tprecursor) y que se puede identificar facilmente por su inmuno-fenotipo26. Con excepcion de este subgrupo (12% de las LLA-T), elinmunofenotipo y el estadio madurativo del linfoblasto T nopermite distinguir claros grupos pronosticos27,28. Se hace, pues,necesario identificar factores biologicos en la LLA-T que con-tribuyan a estratificar mejor a los pacientes o incluso quepermitan aplicar tratamientos dirigidos a las lesiones molecula-res subyacentes, como ya sucede en la LLA Filadelfia positiva29,30.
1,0,9
1,0,9
BA
,8,7
,6,5
,4
,3,2
Sexo femenino, n=164
Sexo masculino, n=54
,8,7
,6,5
,4
,3,2
Edad 1-9 años n=139
Edad > 10 años n= 79
Supervivencia libre de evento (meses)
240216192168144120967248240
Pro
babi
lidad
Pro
babi
lidad
Pro
babi
lidad
Pro
babi
lidad
Pro
babi
lidad
Pro
babi
lidad
,10,0
p= 0,1
Supervivencia libre de evento (meses)
240216192168144120967248240
,10,0
p= 0,09
DC 1,0,9
,8,7
,6,5
,4
Leucocitos < 200 x 10 /L n=160
Leucocitos > 200 x 10 /L n=58
1,0,9
,8,7
,6,5
,4
SNC no infiltrado n=203
Supervivencia libre de evento (meses)
240216192168144120967248240 24021619216814412096724824 0
240216192168144120967248240240216192168144120967248240
,4
,3,2
,10,0
p= 0,028
Supervivencia libre de evento (meses)
,4
,3,2
,10,0
SNC infiltrado n=15
p<0,006
1,0
,9
,8
7
1,0
,9
,8
FE
,6
,5
,4
,3
,2
,1
Médula ósea día+14 <5% blastosn=150
Médula ósea día+14 !5% blastos n=51
p= 0,005
,6
,5
,4
,3
,2
1p= 0,0000
RC al final de inducción n=199
No RC al final de inducción n=19
Supervivencia libre de evento (meses)
0,0
Supervivencia libre de evento (meses)
0,0
Figura 2. Probabilidad de supervivencia libre de evento (SLE) segun variables al diagnostico: (A) sexo, (B) edad, (C) leucocitos y (D) infiltracion del SNC, y probabilidad de SLEsegun respuesta al tratamiento: (E) respuesta precoz (medula osea dıa +14) y (F) remision completa al final de la induccion.
S. Rives et al / Med Clin (Barc). 2012;139(4):141–149 147

Resultados
134
Los resultados de este trabajo pueden servir de plataforma para elestudio de factores biologicos y su correlacion clınica.
En conclusion, en este trabajo se describe una serie ampliade pacientes con LLA-T tratados con 4 protocolos sucesivos delgrupo SHOP, en la que se obtuvo una mejorıa significativa en losresultados con los 2 ultimos protocolos. Asimismo, se identifi-caron factores clınicos asociados a mal pronostico: leucocitosissuperior a 200 ! 109/l, infiltracion en SNC, presencia de masde 5% de blastos en medula osea en el dıa +14 de tratamiento deinduccion y la falta de obtencion de la RC al finalizar dichotratamiento. Estos factores pronosticos, junto con la posibleidentificacion de factores biologicos en proximos estudios,podran contribuir a una mejor estratificacion de este subgrupominoritario de pacientes en los proximos protocolos detratamiento.
Conflicto de intereses
Los autores declaran no tener ningun conflicto de intereses.
Anexo 1
Listado de los centros integrantes del grupo SHOP/SEHOP(Sociedad Espanola de Hemato-Oncologıa Pediatrica) y los medicosresponsables en cada uno de ellos.
Hospital Sant Joan de Deu de Barcelona (Jesus Estella, SusanaRives, Mireia Camos, Albert Catala, Teresa Toll, MontserratTorrebadell, Ruben Berrueco), Hospital Reina Sofıa de Cordoba(Pedro Gomez, Antonia Rodrıguez), Hospital Universitario Marquesde Valdecilla, Santander (Encarna Bureo, Monica Lopez-Duarte),Hospital Universitario La Paz, Madrid (Purifacion Garcıa deMiguel), Hospital Universitario La Fe, Valencia (Amparo Verdeguer,Jose Marıa Fernandez-Navarro), Hospital Virgen de las Nieves,Granada (Marıa Jose Moreno, Emilia Urrutia), Hospital Universi-tario 12 de Octubre, Madrid (Jose Luis Vivanco, Carmen Melero),Hospital Xeral de Galicia, Santiago de Compostela (Jose MiguelCouselo), Hospital Clınico de Valencia (Rafael Fernandez-Delgado),Hospital Ramon y Cajal, Madrid (Marisol Maldonado, ArturoMunoz), Hospital Universitario de Alicante (Marıa Tasso), HospitalMonteprıncipe, Madrid (Blanca Lopez-Ibor, Marta Villa), HospitalTorrecardenas, Almerıa (Francisco Lendınez, Marıa Angeles Vaz-quez), Hospital Universitario de Canarias, San Cristobal de laLaguna, Tenerife (Ricardo Lopez-Almaraz, Jose Cayetano Rodrı-guez-Luis), Hospital Universitario de Donostia, San Sebastian(Javier Uriz), Hospital Parc Taulı, Sabadell, Barcelona (MontserratMelo), Hospital Virgen de la Macarena, Sevilla (Ana Fernandez-Teijeiro), Hospital Marıa Teresa Herrera/Juan Canalejo, La Coruna(Isidoro Rodrıguez),Hospital Virgen del Camino (Javier Molina),Hospital de la Santa Creu i Sant Pau, Barcelona (Isabel Badell,Montserrat Torrent, Marta Garcıa-Bernal, Nuria Pardo), HospitalSon Espases/Hospital Son Dureta, Mallorca (Isabel Hernandez,Mercedes Guibelalde), Hospital Materno-Infantil de Badajoz (JoseManuel Vagace), Hospital Gregorio Maranon, Madrid (Elena Cela,Cristina Belendez, Paloma Galaron), Hospital Universitario deSalamanca (Dorotea Fernandez, Manuela Muriel, Gabriel Mateos),Hospital de Jaen (Irene Pelaez, Ana Belen Lopez), Hospital Virgen dela Salud, Toledo (Marıa Rosario Velasco, Marcos Zamora), HospitalGermans Trias i Pujol, Badalona, Barcelona (Francisco Almazan,Javier German), Hospital Universitario de Albacete (Miguel Lillo),Hospital Clınico Universitario de Valladolid (Carmen Valbuena,Blanca Quiros, A. Blanco), Hospital Xeral de Ourense (ArturoFuentes), Hospital de Basurto (Jose Marıa Indiano), Hospital SantJoan, Alicante (Raul Gonzalez, Cesar Gavilan), Hospital Central deAsturias, Oviedo (Soledad Gonzalez), Hospital Xeral Cies, Vigo,Pontevedra (Manuel Fernandez-Sanmartın), Hospital Materno-Infantil de Las Palmas, Gran Canaria (Antonio Molines), Hospital deBasurto, Bilbao, Bizkaia (Jose Marıa Indiano), Hospital de Cruces,Bilbao, Bizkaia (Aurora Navajas).
Centros de referencia para el estudio de la enfermedad residualmınima en el protocolo SHOP/LLA-2005:
Laboratorio de Hematologıa, Hospital de la Santa Creu i Sant Pau,Barcelona (Dr. J. Nomdedeu), Laboratorio de Hematologıa, HospitalPediatrico Sant Joan de Deu, Barcelona (Dra. E. Tuset, Dra. M. Camos),Hospital Universitario de Salamanca (Dra. B. Vidrales), Unidad deHistocompatibilidad y Biologıa Molecular, Centro de Transfusion deMadrid (Dr. F. Garcıa Sanchez), Hospital Universitario La Fe, Valencia(Dra. A. Sempere), Hospital de Cruces, Bilbao, Bizkaia (Dra. M. RinonMartınez Gallo), Hospital Marques de Valdecilla, Santander (Dra.M.A. Cuadrado), Seccion de Inmunologıa, Hospital General deAlicante (Dra. M.L. de la Sen y Dr. C. Munoz), Hospital de Murcia
1,0
,9
,8
,7 SHOP/LLA99+2005(n=120)
A
B
C
,6
,5
,4
,3
,2
SHOP/LLA 89+ 94 (n=98)
1,0
Supervivencia libre de evento (meses)240216192168144120967248240
Pro
babi
lidad
Pro
babi
lidad
Pro
babi
lidad
,1
0,0
p=0,003
,8
,6
SHOP/LLA 99+2005 (n= 120)
SHOP/LLA 89+94 (n= 98)
240216192168144120967248240
4,
,2
0,0
p=0,002
1,0
,9
,8
Supervivencia global (meses)
,7
,6
,5
,4
,3
SHOP/LLA 99+2005 (n= 113)
SHOP/LLA 89+94 (n=86)
Supervivencia libre de recaída (meses)240216192168144120967248240
,2
,1
0,0
p=0,013
Figura 3. Probabilidad de supervivencia libre de evento (SLE), supervivencia global(SG) y supervivencia libre de recaıda (SLR) en la leucemia linfoblastica aguda defenotipo T (LLA-T). Comparacion entre protocolos SHOP/LLA-89 + 94 y SHOP/LLA-99 + 2005. (A) SLE; (B) SG; (C) SLR.
S. Rives et al / Med Clin (Barc). 2012;139(4):141–149148

Resultados
135
(Dr. F. Ortuno), Hospital Clınico, Santiago de Compostela, La Coruna(Dra. I. Abuın), Servicio de Inmunologıa, Hospital Ramon y Cajal,Madrid (Dr. E. Roldan).
Bibliografıa
1. Pui CH, Campana D, Pei D, Bowman WP, Sandlund JT, Kaste SC, et al. Treatingchildhood acute lymphoblastic leukemia without cranial irradiation. N Engl JMed. 2009;360:2730–41.
2. Moricke A, Zimmermann M, Reiter A, Henze G, Schrauder A, Gadner H, et al.Long-term results of five consecutive trials in childhood acute lymphoblasticleukemia performed by the ALL-BFM study group from 1981 to 2000. Leukemia.2010;24:265–84.
3. Schmiegelow K, Forestier E, Hellebostad M, Heyman M, Kristinsson J, Soderhall S,et al. Long-term results of NOPHO ALL-92 and ALL-2000 studies of childhoodacute lymphoblastic leukemia. Leukemia. 2010;24:345–54.
4. Cave H, van der Werff ten Bosch J, Suciu S, Guidal C, Waterkeyn C, Otten J, et al.Clinical significance of minimal residual disease in childhood acute lympho-blastic leukemia. European Organization for Research and Treatment of Cancer–Childhood Leukemia Cooperative Group. N Engl J Med. 1998;339:591–8.
5. Conter V, Bartram CR, Valsecchi MG, Schrauder A, Panzer-Grumayer R,Moricke A, et al. Molecular response to treatment redefines all prognosticfactors in children and adolescents with B-cell precursor acute lymphoblasticleukemia: results in 3184 patients of the AIEOP-BFM ALL 2000 study. Blood.2010;115:3206–14.
6. Pullen J, Shuster JJ, Link M, Borowitz M, Amylon M, Carroll AJ. Significance ofcommonly used prognostic factors differs for children with T cell acute lym-phocytic leukemia (ALL), as compared to those with B-precursor ALL. A PediatricOncology Group (POG) study. Leukemia. 1999;13:1696–707.
7. Willemse MJ, Seriu T, Hettinger K, d’Aniello E, Hop WC, Panzer-Grumayer ER,et al. Detection of minimal residual disease identifies differences in treatmentresponse between T-ALL and precursor B-ALL. Blood. 2002;99:4386–93.
8. Ferrando AA, Neuberg DS, Staunton J, Loh ML, Huard C, Raimondi SC. Geneexpression signatures define novel oncogenic pathways in T cell acute lympho-blastic leukemia. Cancer Cell. 2002;1:75–87.
9. Graux C, Cools J, Melotte C, Quentmeier H, Ferrando A, Levine R, et al. Fusion ofNUP214 to ABL1 on amplified episomes in T-cell acute lymphoblastic leukemia.Nat Genet. 2004;36:1084–9.
10. Sanda T, Li X, Gutierrez A, Ahn Y, Neuberg DS, O’Neil J, et al. Interconnectingmolecular pathways in the pathogenesis and drug sensitivity of T-cell acutelymphoblastic leukemia. Blood. 2010;115:1735–45.
11. Tallen G, Ratei R, Mann G, Kaspers G, Niggli F, Karachunsky A, et al. Long-termoutcome in children with relapsed acute lymphoblastic leukemia after time-point and site-of-relapse stratification and intensified short-course multidrugchemotherapy: results of trial ALL-REZ BFM 90. J Clin Oncol. 2010;28:2339–47.
12. Badell I, Munoz A, Estella J, Fernandez-Delgado R, Javier G, Verdeguer A, et al.Long-term results of two consecutive trials in childhood acute lymphoblasticleukaemia performed by the Spanish Cooperative Group for Childhood AcuteLymphoblastic Leukemia Group (SHOP) from 1989 to 1998. Clin Transl Oncol.2008;10:117–24.
13. Arico M, Basso G, Mandelli F, Rizzari C, Colella R, Barisone E, et al. Good steroidresponse in vivo predicts a favorable outcome in children with T-cell acutelymphoblastic leukemia. The Associazione Italiana Ematologia OncologiaPediatrica (AIEOP). Cancer. 1995;75:1684–93.
14. Pui CH, Behm FG, Singh B, Schell MJ, Williams DL, Rivera GK, et al. Heterogeneityof presenting features and their relation to treatment outcome in 120 childrenwith T-cell acute lymphoblastic leukemia. Blood. 1990;75:174–9.
15. Conter V, Arico M, Basso G, Biondi A, Barisone E, Messina C, AssociazioneItaliana di Ematologia ed Oncologia Pediatrica. Long-term results of the ItalianAssociation of Pediatric Hematology and Oncology (AIEOP) Studies 82, 87, 88,
91 and 95 for childhood acute lymphoblastic leukemia. Leukemia. 2010;24:255–64.
16. Goldberg JM, Silverman LB, Levy DE, Dalton VK, Gelber RD, Lehmann L, et al.Childhood T-cell acute lymphoblastic leukemia: the Dana-Farber Cancer Insti-tute acute lymphoblastic leukemia consortium experience. J Clin Oncol.2003;21:3616–22.
17. Stark B, Avrahami G, Nirel R, Abranov A, Attias D, Bielorai B, et al. Extendedtriple intrathecal therapy in children with T-cell acute lymphoblastic leuke-mia: a report from the Israeli National ALL-Studies. Br J Haematol. 2009;147:113–24.
18. Moricke A, Reiter A, Zimmermann M, Gadner H, Stanulla M, Dordelmann M,et al. Risk-adjusted therapy of acute lymphoblastic leukemia can decreasetreatment burden and improve survival: treatment results of 2169 unselectedpediatric and adolescent patients enrolled in the trial ALL-BFM 95. Blood.2008;111:4477–89.
19. Kager L, Cheok M, Yang W, Zaza G, Cheng Q, Panetta JC, et al. Folate pathwaygene expression differs in subtypes of acute lymphoblastic leukemia andinfluences methotrexate pharmacodynamics. J Clin Invest. 2005;115:110–7.
20. Sancho JM, Morgades M, Arranz R, Fernandez-Abellan P, Deben G, Alonso N,et al. Practice of central nervous system prophylaxis and treatment in acuteleukemias in Spain. Prospective registry study. Med Clin (Barc). 2008;131:401–5.
21. Schultz KR, Massing B, Spinelli JJ, Gaynon PS, Wadsworth L. Importanceof the day 7 bone marrow biopsy as a prognostic measure of the outcomein children with acute lymphoblastic leukemia. Med Pediatr Oncol. 1997;29:16–22.
22. Park MJ, Taki T, Oda M, Watanabe T, Yumura-Yagi K, Kobayashi R, et al. FBXW7and NOTCH1 mutations in childhood T cell acute lymphoblastic leukaemia andT cell non-Hodgkin lymphoma. Br J Haematol. 2009;145:198–206.
23. Larson Gedman A, Chen Q, Kugel Desmoulin S, Ge Y, LaFiura K, Haska CL, et al.The impact of NOTCH1, FBW7 and PTEN mutations on prognosis and down-stream signaling in pediatric T-cell acute lymphoblastic leukemia: a reportfrom the Children’s Oncology Group. Leukemia. 2009;23:1417–25.
24. Cave H, Suciu S, Preudhomme C, Poppe B, Robert A, Uyttebroeck A, et al. Clinicalsignificance of HOX11L2 expression linked to t(5;14)(q35;q32), of HOX11expression, and of SIL-TAL fusion in childhood T-cell malignancies: results ofEORTC studies 58881 and 58951. Blood. 2004;103:442–50.
25. Ballerini P, Landman-Parker J, Cayuela JM, Asnafi V, Labopin M, Gandemer V,et al. Impact of genotype on survival of children with T-cell acute lymphoblasticleukemia treated according to the French protocol FRALLE-93: the effect ofTLX3/HOX11L2 gene expression on outcome. Haematologica. 2008;93:1658–65.
26. Coustan-Smith E, Mullighan CG, Onciu M, Behm FG, Raimondi SC, Pei D, et al.Early T-cell precursor leukaemia: a subtype of very high-risk acute lympho-blastic leukaemia. Lancet Oncol. 2009;10:147–56.
27. Xicoy B, Ribera JM, Oriol A, Sanz MA, Abella E, Tormo M, et al. Significadopronostico de los subtipos inmunologicos de la leucemia aguda linfoblastica Tdel adulto. Estudio de 81 pacientes. Med Clin (Barc). 2006;126:41–6.
28. Marks DI, Paietta EM, Moorman AV, Richards SM, Buck G, DeWald G, et al. T-cellacute lymphoblastic leukemia in adults: clinical features, immunophenotype,cytogenetics, and outcome from the large randomized prospective trial (UKALLXII/ECOG 2993). Blood. 2009;114:5136–45.
29. Schultz KR, Bowman WP, Aledo A, Slayton WB, Sather H, Devidas M, et al.Improved early event-free survival with imatinib in Philadelphia chromosome-positive acute lymphoblastic leukemia: a children’s oncology group study. J ClinOncol. 2009;27:5175–81.
30. Rives S, Estella J, Gomez P, Lopez-Duarte M, de Miguel PG, Verdeguer A, et al.Intermediate dose of imatinib in combination with chemotherapy followed byallogeneic stem cell transplantation improves early outcome in paediatricPhiladelphia chromosome positive acute lymphoblastic leukaemia (ALL):results of the Spanish Cooperative Group SHOP studies ALL-94, ALL-99 andALL-2005. Br J Haematol. 2011;154:600–11.
S. Rives et al / Med Clin (Barc). 2012;139(4):141–149 149

Resultados
136

Resultados
137

Discusión
138
Discusión

Discusión
139

Discusión
140
DISCUSIÓN
Los trabajos planteados en esta tesis tenían como objetivo confirmar la
utilidad del análisis de grandes series de pacientes incluidos en grupos
cooperativos para identificar factores pronósticos en grupos infrecuentes de
LLA pediátrica de alto riesgo, así como evaluar el impacto sobre la
supervivencia de determinadas estrategias terapéuticas. En estos estudios
hemos identificado variables pronósticas en dos subgrupos minoritarios de
LLA de alto riesgo (LLA Ph+ y LLA-‐T) y mostramos el impacto positivo de los
cambios sucesivos en el tratamiento sobre la supervivencia de los pacientes.
Se discuten a continuación los resultados obtenidos en cada uno de los dos
subgrupos de pacientes.
LLA Ph+
Los resultados de nuestros estudios confirman que el uso continuo de
imatinib a dosis intermedias en el tratamiento de primera línea, asociado a
quimioterapia intensiva y seguido de TPH alogénico, constituye un
tratamiento eficaz en los pacientes pediátricos con LLA Ph+. Los resultados
obtenidos con este tratamiento combinado son muy superiores a los
obtenidos en los controles históricos, en series de pacientes tratados de
forma similar pero sin la adición de imatinib. Esta mejoría se traduce en un
aumento muy importante tanto en la supervivencia libre de evento como en
la supervivencia global a corto y a largo plazo.
Protocolo SHOP/LLA-‐2005: fundamentos para su diseño
El último protocolo SHOP para el tratamiento de niños y adolescentes con
LLA (SHOP/LLA-‐2005) incluyó imatinib en combinación con quimioterapia

Discusión
141
intensiva en los pacientes diagnosticados con LLA Ph+ de novo. Tal como se
ha comentado en la introducción, en aquel momento el uso de imatinib en
combinación con quimioterapia intensiva no se había descrito como
tratamiento de primera línea en la población pediátrica. Se decidió
administrarlo de forma precoz, desde el día +15 del tratamiento de
inducción y simultáneamente a la quimioterapia, con el objetivo de reducir la
masa tumoral y aumentar la tasa de remisión completa. Se optó por el día
+15 para asegurar que en todos los pacientes se dispusiera de los resultados
del reordenamiento BCR-‐ABL y/o la presencia del cromosoma Philadelphia.
La dosis intermedia de imatinib (260 mg/m2 al día, que corresponde a una
dosis de 400 mg/día en el adulto) se escogió con el fin de evitar una
toxicidad excesiva al administrarse de forma concomitante con la
quimioterapia.
Se indicó el TPH alogénico de donante HLA-‐idéntico, tanto familiar como no
emparentado, en primera remisión completa tras finalizar los bloques de
quimioterapia de consolidación. En el momento de diseñar el protocolo la
superioridad del trasplante frente a la quimioterapia en pacientes
pediátricos con LLA Ph+ se había demostrado sólo en el caso del TPH familiar
HLA-‐idéntico. Sin embargo, se decidió incluir el TPH de donante no
relacionado por la mejoría de los resultados obtenidos en los años previos
con este tipo de trasplante, gracias a los avances en el tratamiento de
soporte y a la mejoría en la selección del donante con las técnicas de tipaje
HLA por alta resolución (Baron y Storb, 2004)
En los últimos años se ha descrito en pacientes adultos con LLA Ph+ el uso de
imatinib continuo administrado simultáneamente con quimioterapia

Discusión
142
(Wassmann et al., 2006) (Ribera et al., 2010). Este modo de administración
se toleró bien y parece que tuvo una actividad antileucémica superior que
administrado de forma intermitente o con menor intensidad de tratamiento
de imatinib (Wassmann et al., 2006). Paralelamente, en pacientes
pediátricos con LLA Ph+ se demostró la ventaja del trasplante alogénico,
tanto de donante familiar como no emparentado, sobre la quimioterapia en
los pacientes tratados en la era pre-‐TKI (Aricò et al., 2010).
Resultados de los protocolos SHOP en la era pre-‐TKI
En los pacientes con LLA Ph+ tratados con protocolos SHOP pre-‐imatinib
(SHOP/LLA-‐94 y LLA-‐99) se obtuvieron unos resultados pobres, con una
supervivencia a los 4 años inferior al 40% y SLE inferior al 30%. Estos
resultados son similares a los publicados por otros grupos cooperativos con
protocolos de tratamiento contemporáneos (Roy et al., 2005) (Gandemer et
al., 2009) (Aricò et al., 2010). No obstante, el pronóstico de estos pacientes
fue heterogéneo y dependió de las características clínicas al diagnóstico y de
la respuesta precoz al tratamiento, de forma similar a lo hallado en otras
series pediátricas de LLA Ph+ (Ribeiro et al., 1997) (Schrappe et al., 1998)
(Aricò et al., 2000) (Roy et al., 2005) (Gandemer et al., 2009) (Aricò et al.,
2010). La mediana de edad de nuestra serie de pacientes fue ligeramente
menor que la de otras series pediátricas con LLA Ph+ y esto puede haber
tenido impacto en la supervivencia, ya que la edad es un factor pronóstico
conocido en este subtipo de LLA.
Protocolo SHOP/LLA-‐2005 con imatinib: gran eficacia antileucémica
En los pacientes tratados con el protocolo de tratamiento que contiene
imatinib (LLA/SHOP-‐2005) la tasa de remisión completa tras el tratamiento

Discusión
143
de inducción fue del 100%. Cabe destacar que todos los pacientes que
presentaron una mala respuesta precoz, antes de que se hubiera iniciado
imatinib en el día +15, alcanzaron la remisión completa morfológica al final
de la inducción. No se observó, por tanto, ningún fallo de inducción entre los
16 pacientes tratados con el régimen quimioterápico que incluía imatinib.
Este hallazgo contrasta con las tasas de alrededor del 10% de fallo de
inducción tanto en nuestra propia serie de pacientes tratados sin imatinib
como en otras series pediátricas de LLA Ph+ en pacientes tratados sin un TKI
(Aricò et al., 2000) (Aricò et al., 2010) (Roy et al., 2005) (Gandemer et al.,
2009). Ningún paciente de la cohorte de imatinib recayó antes del
trasplante, por lo que en todos se pudo realizar el trasplante con excepción
de un paciente, que falleció en RC por una complicación infecciosa previa al
trasplante (tasa de trasplante 94%). Cabe destacar que todos los pacientes
realizaron el trasplante en remisión molecular.
Toxicidad asociada al uso de imatinib
En cuanto a la toxicidad, la mayoría de los pacientes toleró bien el
tratamiento. Con estas dosis intermedias de imatinib, en combinación con
quimioterapia intensiva, no se observó un incremento de la mielosupresión
en comparación con los controles históricos de pacientes con LLA Ph+ que
recibieron un régimen similar de quimioterapia pero sin imatinib. La
toxicidad extramedular fue superior a la observada en los pacientes que no
recibieron imatinib, pero fue tolerable. De forma similar a lo descrito en
pacientes adultos, observamos mayor incidencia de náuseas y elevación de
las transaminasas en los pacientes que recibieron imatinib. La elevación de
transaminasas no se asoció a elevación de la bilirrubina grado III-‐IV ni a

Discusión
144
insuficiencia hepática, fue reversible en todos los casos y no precisó de
interrupción de imatinib. Un paciente en el grupo que recibió imatinib
falleció durante la aplasia secundaria a un ciclo de quimioterapia intensiva
de consolidación. Este niño sufrió una sepsis por Aeromonas. Esta infección
se ha descrito en pacientes con LLA que no habían recibido imatinib y tiene
una mortalidad elevada en pacientes con neutropenia grave como el nuestro
(Martino et al., 1997) (Tsai et al., 2006).
Supervivencia
En el momento de la publicación de los resultados del presente trabajo de
tesis, sólo se había descrito una serie de pacientes pediátricos con LLA Ph+
tratados con imatinib en combinación con quimioterapia intensiva (Schultz
et al., 2009), en la que se mostró un aumento muy importante en la
supervivencia de estos pacientes. Estos pacientes recibieron el protocolo de
tratamiento del grupo norteamericano Children Oncology Group (COG) COG-‐
AALL0031. Incluyeron 93 pacientes pediátricos con LLA Ph+ que
estratificaron en 5 cohortes, en las que de forma progresiva se aumentó la
exposición a imatinib. La cohorte 5, con 44 pacientes, recibió imatinib
continuo y fue la que obtuvo los mejores resultados, con una SLE a 3 años
del 80%, muy superior a la comparación con su cohorte histórica, que era del
35% (p<0,0001). En dicho protocolo de tratamiento sólo se contempló la
realización de TPH en el caso de que hubiera un donante familiar HLA-‐
idéntico. En aquellos pacientes que no dispusieran de donante familiar se
indicó tratamiento sólo con quimioterapia e imatinib. La SLE en los 25
pacientes tratados sólo con quimioterapia e imatinib no fue inferior a la de
los pacientes que recibieron además un TPH. Con estos datos los autores

Discusión
145
concluyeron que el trasplante en primera RC tal vez pudiera evitarse en los
pacientes pediátricos con LLA Ph+ tratados con quimioterapia intensiva en
combinación con un TKI.
Nuestra serie confirmó el gran aumento en la supervivencia libre de evento y
en la supervivencia global de los niños con LLA Ph+ tratados con imatinib, en
comparación con los resultados obtenidos en una serie histórica de
pacientes que habían recibido tratamiento quimioterápico muy similar pero
sin imatinib. A pesar del bajo número de pacientes de nuestra cohorte, esta
diferencia en la supervivencia a corto plazo resultó ser estadísticamente
significativa. Estos resultados favorables asociados al empleo de imatinib en
la LLA Ph+ se mantuvieron con mayor seguimiento, de más de 5 años, con
una SLE y SG a 5 años del 81% y 87% respectivamente (trabajo 2).
En este estudio debemos considerar las limitaciones inherentes a las
comparaciones históricas. Este hecho tiene especial relevancia en esta serie,
en la cual la mortalidad relacionada con el trasplante es mayor en el control
histórico (cohorte pre-‐imatinib). Aunque esta diferencia podría contribuir a
los mejores resultados obtenidos en el grupo de imatinib, es muy
improbable que este hecho explique el aumento tan importante en la
supervivencia de estos pacientes. Así, los resultados obtenidos en nuestra
cohorte de imatinib son claramente superiores a los obtenidos en todas las
otras series de pacientes pediátricos con LLA Ph+ en la era pre-‐imatinib (Roy
et al., 2005) (Gandemer et al., 2009) (Aricò et al., 2010),
independientemente de que en dichas series la mortalidad relacionada con
el trasplante era menor a la que presentaron nuestros pacientes de la

Discusión
146
cohorte pre-‐imatinib. Por otro lado, debido al marcado aumento en la
supervivencia obtenido por el grupo norteamericano COG en pacientes
tratados con imatinib (Schultz et al., 2009) y los resultados obtenidos en
pacientes adultos hubiera sido muy difícil de justificar, desde un punto de
vista ético, un ensayo clínico en el que se aleatorizara la administración de
imatinib u otro TKI frente a placebo.
Con posterioridad a la publicación del primer artículo del presente trabajo de
tesis, se publicaron los resultados de un protocolo internacional específico
para pacientes pediátricos con LLA Ph+, EsPhALL (Biondi et al., 2012). En
dicho protocolo se quiso evaluar el impacto de añadir imatinib a la
quimioterapia intensiva seguida de TPH. Se decidió aleatorizar la
administración o no de imatinib en el subgrupo de pacientes con buena
respuesta inicial al tratamiento quimioterápico, mientras que a todos los
pacientes con mala respuesta se les administró imatinib. A finales de 2009 se
realizó una enmienda a dicho protocolo motivada por la publicación de los
buenos resultados a corto plazo obtenidos por el grupo COG en los pacientes
tratados con imatinib continuo y quimioterapia intensiva, junto a la negativa
de algunos médicos y familiares de los pacientes a participar en la
aleatorización de imatinib. Se decidió que todos los pacientes recibieran
imatinib y se pasó a administrar de forma continua en lugar de intermitente,
tal como se hizo en la cohorte 5 del protocolo del COG y en nuestra serie de
pacientes tratados con el protocolo LLA/SHOP-‐2005. Los resultados
obtenidos con el protocolo EsPhALL antes de la enmienda en una serie
amplia de pacientes (n=178) fueron significativamente mejores que los
alcanzados con su cohorte histórica. Así, la SLE a los 4 años fue de 61,9% en

Discusión
147
su cohorte de imatinib frente a 42,3% en su cohorte histórica (pre-‐imatinib)
(Aricò et al., 2010; Biondi et al., 2012). La mejoría en la supervivencia
obtenida con su protocolo, pese a ser estadísticamente significativa, fue
inferior a la obtenida con los protocolos del grupo COG y de nuestro grupo
SHOP, probablemente debido al uso intermitente y menos intensivo de
imatinib, así como a la no administración de imatinib en un subgrupo de
pacientes de buen pronóstico. Los resultados que se obtengan con el
protocolo EsPhALL tras la realización de la enmienda tendrán gran
importancia para corroborar la mejoría tan importante en la supervivencia
cuando imatinib se administra de forma continua y precoz durante el
tratamiento de inducción en una serie amplia de pacientes pediátricos.
Papel del TPH en primera RC en la LLA Ph+ pediátrica
Tras los buenos resultados obtenidos en el subgrupo de pacientes del
protocolo COG que no recibió trasplante, se plantea la necesidad de
trasplante en estos niños. Este aspecto no se puede analizar ni en la serie del
protocolo EsPhALL ni en la nuestra, puesto que la gran mayoría de pacientes
recibieron un TPH. En cuanto a los resultados obtenidos por el grupo COG en
pacientes tratados sin trasplante, se necesita un seguimiento a largo plazo
para ver si dichos resultados se mantienen en el tiempo. Esto es así porque
los pacientes con LLA Ph+ pueden presentar recaídas tardías, en especial los
que no han recibido un trasplante. De hecho, la enfermedad del injerto
contra el huésped tiene un papel demostrado en la protección frente a las
recaídas en la LLA Ph+ (Esperou et al., 2003) (Lee et al., 2005) (Bhojwani et
al., 2009) (Burke et al., 2009). En este sentido, en series de pacientes adultos
con LLA Ph+ los tratamientos quimioterápicos que incluyeron imatinib

Discusión
148
aumentaron la supervivencia a largo plazo (Fielding et al., 2010) (Bassan et
al., 2010) y el trasplante alogénico parece que mantuvo su efecto
beneficioso reduciendo las recaídas y mejorando la supervivencia de estos
pacientes también en la era de los TKI (Ottmann y Pfeifer, 2009) (Fielding et
al., 2010) (Pfeifer et al., 2010) (Bassan et al., 2010). Resulta de gran
importancia identificar en qué niños se podría evitar el trasplante por la
mayor morbi-‐mortalidad de este procedimiento frente a la quimioterapia
tanto a corto como a largo plazo. El grupo de pacientes de buen pronóstico,
definido por las características al diagnóstico y por la respuesta precoz al
tratamiento, podría ser candidato a omitir el TPH alogénico. En este sentido,
el índice pronóstico descrito por el grupo francés FRALLE (Gandemer et al.,
2009) y validado con nuestra serie de pacientes (ver trabajo 3 de esta tesis),
podría ser de utilidad para este fin.
Habrá que esperar a nuevos protocolos cooperativos internacionales de
tratamiento de LLA Ph+ con mayor número de pacientes que el de la serie
norteamericana, en el que se reevalúe la posibilidad de eliminar el trasplante
alogénico en primera remisión en aquellos pacientes pediátricos con menor
riesgo de recaída tratados con quimioterapia y un TKI. El análisis de factores
pronósticos en pacientes tratados con quimioterapia y un TKI y que no
reciban un trasplante alogénico podría ayudar a identificar en qué pacientes
puede ser segura la omisión del trasplante en primera RC.
Factores pronósticos en la era TKI
Debemos tener en cuenta que los factores pronósticos hallados en los
pacientes tratados sin TKIs no necesariamente han de reproducirse en
aquéllos tratados con TKIs. Por ello, se hace necesario el análisis de factores

Discusión
149
pronósticos en la era de los TKIs. Así se podrían identificar grupos de
pacientes de bajo riesgo de recaída, en los que tal vez fuera posible disminuir
la intensidad del tratamiento. Por otro lado, la identificación de grupos con
pronóstico desfavorable permitiría la aplicación de otras intervenciones,
tales como TKIs de nueva generación o el tratamiento profiláctico con TKI
tras el trasplante.
En este sentido, la evaluación de la respuesta mediante la determinación de
la ERM y su evaluación continuada podría ayudar a refinar la identificación
de grupos de riesgo. Resulta pues importante el estudio de la cinética del
aclaramiento de la ERM en los pacientes pediátricos tratados con TKIs. Dada
la elevada tasa de respuesta con los TKIs, es probable que el momento de
estudio de la ERM que aporte mayor valor pronóstico no sea en las fases
iniciales del tratamiento sino en momentos más tardíos. Recientemente, en
la población adulta con LLA Ph+ se ha mostrado como la determinación de la
ERM a los 3 meses del inicio del tratamiento y en momentos más tardíos
tuvo valor pronóstico (Ravandi et al., 2013).
TKIs de nueva generación
Los TKI de nueva generación (dasatinib, nilotinib y otros), por su posible
mayor eficacia, podrían contribuir a mejorar los resultados, aunque todavía
no se ha demostrado que sean superiores a imatinib en la LLA Ph+ en adultos
(Ravandi et al., 2010; Lee et al., 2011) (Kim DY et al., 2011; Ribera, 2013). En
niños, la experiencia con estos fármacos es escasa, pero los estudios de fase
I-‐II con dasatinib en pacientes con LMC y con LLA Ph+ refractaria o recaída
han mostrado una farmacocinética similar a la de los adultos y un perfil de
eficacia y de seguridad mejores que en la población adulta (Zwaan et al.,

Discusión
150
2013). En la actualidad dos grupos cooperativos (COG y EsPhALL) están
realizando un estudio fase II con dasatinib en combinación con
quimioterapia en pacientes con LLA Ph+ de novo (ClinicalTrials.gov
Identifier:NCT01460160).
Beneficios del uso de los TKIs
El uso de TKIs tal vez permita reducir la intensidad del tratamiento en estos
niños, no tan sólo mediante la reducción de las indicaciones de trasplante
sino también mediante la disminución de la intensidad de la quimioterapia
que se les administra, que es muy importante por tratarse de pacientes en el
grupo de muy alto riesgo. Esta reducción de la intensidad ya ha demostrado
ser eficaz, al menos a corto plazo, en pacientes adultos (Ribera et al., 2012).
Entre los beneficios potenciales de la adición de un TKI en los pacientes con
LLA Ph+, además de la mayor eficacia antileucémica se ha sugerido que estos
fármacos podrían reducir la incidencia de la enfermedad crónica del injerto
contra el huésped (EICH) crónica (Sánchez-‐Ortega et al., 2012).
En cuanto a la eficacia antileucémica el uso de TKI se ha asociado a un
aumento en la tasas de remisión completa y en la tasa de pacientes que
pueden realizar un trasplante. Además, su uso pre-‐trasplante podría tener
un efecto protector frente a las recaídas. En este sentido, en una serie
amplia de pacientes adultos del registro de la European Blood and Bone
Marrow Transplantation (EBMT), publicada recientemente en forma de
abstract, se observó una mayor tasa de recaídas en aquellos pacientes que
no recibieron un TKI previo al TPH alogénico en primera RC. Así, la incidencia
de recaídas a los 3 años fue de 43% en los pacientes que no recibieron un TKI
pre-‐trasplante frente al 35% en aquéllos que sí lo recibieron (p=0,04),

Discusión
151
(Brissot E et al., 2013). Este efecto citorreductor del TKI pre-‐trasplante es
controvertido y no ha resultado estadísticamente significativo en otras series
con menor número de pacientes (Kebriaei et al., 2012) (Burke et al., 2009).
En nuestra serie de pacientes pediátricos tratados con imatinib, de 15
pacientes que recibieron un TPH en primera RC sólo 1 recayó tras el
trasplante, frente a 5 recaídas tras el trasplante en 18 pacientes de la
cohorte pre-‐imatinib. Estos datos podrían sugerir un efecto beneficioso de
imatinib previo al trasplante. Sin embargo, dado el número pequeño de
pacientes y la elevada tasa de mortalidad relacionada con el trasplante de
nuestra cohorte de pacientes de la era pre-‐imatinib, este aspecto no se
puede afirmar.
Respecto al posible efecto protector de los TKIs frente a la EICH crónica, en la
serie de pacientes de la EBMT mencionada anteriormente, se observó que el
uso de TKI post-‐trasplante redujo la incidencia de EICH crónica (Brissot E et
al., 2013).
Uso de los TKIs post-‐trasplante
Otra cuestión no resuelta en los pacientes pediátricos con LLA Ph+ es si
imatinib u otro TKI debe usarse tras el trasplante de forma profiláctica o sólo
en caso de detección de ERM. En nuestra cohorte de pacientes tratados con
imatinib, de los 15 pacientes que recibieron un TPH alogénico, 12 no
recibieron imatinib post-‐trasplante y permanecían en RC molecular en el
momento del análisis. Tres pacientes recibieron imatinib post-‐trasplante,
uno tras recaída hematológica, otro de forma profiláctica y otro como parte
del tratamiento de una enfermedad de injerto contra el huésped extensa.
Aunque el número de pacientes de nuestra serie es demasiado bajo para

Discusión
152
contestar esta pregunta, nuestros resultados podrían sugerir que el
tratamiento profiláctico con imatinib no es necesario. En el contexto de la
LLA Ph+ en adultos, tampoco está claro si es mejor el uso profiláctico de
imatinib versus su uso en caso de detección de ERM tras el trasplante (pre-‐
emptive) (Ribera et al., 2010; Ribera, 2013; Nishiwaki et al., 2010). Este
aspecto se analizó en un estudio aleatorizado realizado por el grupo alemán
German Multicenter Acute Lymphoblastic Leukmia GMALL y aunque se
observaron menos recaídas moleculares en el grupo de pacientes tratado
con imatinib profiláctico, no se detectaron diferencias en su supervivencia
(Pfeifer et al., 2013).
En conclusión, en nuestra serie de pacientes tratados en la era de los TKI
hemos demostrado una mejoría muy importante de la supervivencia a los 5
años utilizando imatinib en combinación con tratamiento quimioterápico
intensivo, seguido de trasplante alogénico. Los resultados de nuestra serie
de pacientes tratados con imatinib y trasplante alogénico pueden contribuir
a realizar comparaciones en un futuro con pacientes tratados sólo con
quimioterapia y TKI sin trasplante. Para ello, se necesita analizar un mayor
número de pacientes que se incluyan en protocolos internacionales para
poder redefinir el papel del trasplante en este subgrupo de pacientes con
LLA de muy alto riesgo.
LLA-‐T
En este trabajo describimos los resultados de una serie amplia de casos con
LLA-‐T tratados durante un período de 20 años en el seno del grupo
cooperativo SHOP. El estudio demuestra que los cambios aplicados en los

Discusión
153
dos últimos protocolos de tratamiento han resultado en una mejoría
significativa en la evolución de este subgrupo de pacientes. Además, hemos
identificado factores clínicos con valor pronóstico que pueden contribuir a
una mejor estratificación de los pacientes con este tipo de LLA.
Las características clínicas al diagnóstico de los niños con LLA-‐T incluidos en
este estudio son comparables a las halladas en otras series de pacientes
pediátricos con LLA-‐T (Pui et al., 1990) (Aricò et al., 1995), con un claro
predominio de varones, una mediana de edad superior a la de la LLA de línea
B, una proporción elevada de casos con masa mediastínica e
hiperleucocitosis y una mayor frecuencia de infiltración del SNC.
Los resultados obtenidos con los dos primeros protocolos SHOP (SHOP/LLA-‐
89 y LLA-‐94) en este subgrupo de pacientes fueron inferiores a los descritos
en otras series contemporáneas (Möricke et al., 2009) (Conter et al., 2010)
(Goldberg et al., 2003) y se deben, principalmente, a una mayor tasa de
recaídas. Éstas alcanzaron un 40% de los casos frente a un 11-‐30% de otras
series coetáneas (Goldberg et al., 2003) (Stark et al., 2009). En la década de
los 90, las principales instituciones y grupos cooperativos obtuvieron
resultados en la supervivencia del subgrupo de pacientes con LLA-‐T que
difirieron de forma importante. Así, la SLE a los 10 años en la primera mitad
de la década de los 90 osciló entre un 79% en el grupo de DCFI y un 39% en
el grupo italiano AIEOP o 42% del grupo SHOP. En la segunda mitad de dicha
década los resultados obtenidos en la supervivencia de este subgrupo de
pacientes mejoraron de forma significativa en la mayoría de los grupos y en
cambio en el grupo SHOP permanecieron sin cambios significativos (ver
tabla 9).
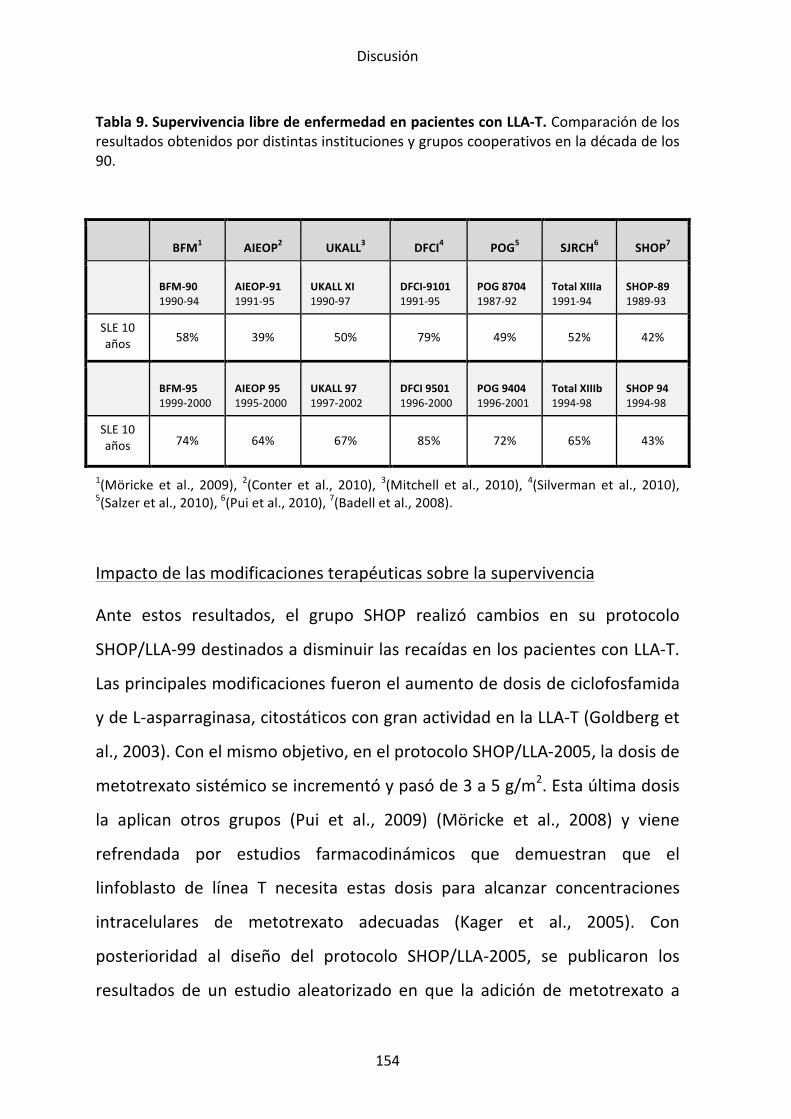
Discusión
154
Tabla 9. Supervivencia libre de enfermedad en pacientes con LLA-‐T. Comparación de los resultados obtenidos por distintas instituciones y grupos cooperativos en la década de los 90.
BFM1 AIEOP2 UKALL3 DFCI4 POG5 SJRCH6 SHOP7
BFM-‐90 1990-‐94
AIEOP-‐91 1991-‐95
UKALL XI 1990-‐97
DFCI-‐9101 1991-‐95
POG 8704 1987-‐92
Total XIIIa 1991-‐94
SHOP-‐89 1989-‐93
SLE 10 años 58% 39% 50% 79% 49% 52% 42%
BFM-‐95 1999-‐2000
AIEOP 95 1995-‐2000
UKALL 97 1997-‐2002
DFCI 9501 1996-‐2000
POG 9404 1996-‐2001
Total XIIIb 1994-‐98
SHOP 94 1994-‐98
SLE 10 años 74% 64% 67% 85% 72% 65% 43%
1(Möricke et al., 2009), 2(Conter et al., 2010), 3(Mitchell et al., 2010), 4(Silverman et al., 2010), 5(Salzer et al., 2010), 6(Pui et al., 2010), 7(Badell et al., 2008).
Impacto de las modificaciones terapéuticas sobre la supervivencia
Ante estos resultados, el grupo SHOP realizó cambios en su protocolo
SHOP/LLA-‐99 destinados a disminuir las recaídas en los pacientes con LLA-‐T.
Las principales modificaciones fueron el aumento de dosis de ciclofosfamida
y de L-‐asparraginasa, citostáticos con gran actividad en la LLA-‐T (Goldberg et
al., 2003). Con el mismo objetivo, en el protocolo SHOP/LLA-‐2005, la dosis de
metotrexato sistémico se incrementó y pasó de 3 a 5 g/m2. Esta última dosis
la aplican otros grupos (Pui et al., 2009) (Möricke et al., 2008) y viene
refrendada por estudios farmacodinámicos que demuestran que el
linfoblasto de línea T necesita estas dosis para alcanzar concentraciones
intracelulares de metotrexato adecuadas (Kager et al., 2005). Con
posterioridad al diseño del protocolo SHOP/LLA-‐2005, se publicaron los
resultados de un estudio aleatorizado en que la adición de metotrexato a

Discusión
155
dosis de 5 g/m2 mejoró la supervivencia en pacientes con LLA-‐T (Asselin et
al., 2011). Por ello, en cierta medida, la mejora de los resultados obtenidos
con los dos últimos protocolos SHOP en los pacientes con LLA-‐T podría
deberse al aumento de la dosis de este fármaco en el último protocolo. En el
momento del análisis del presente trabajo de tesis, el seguimiento de la serie
de pacientes tratados con el último protocolo (SHOP/LLA-‐2005), era corto y
no permitió la comparación de los resultados obtenidos con respecto al
anterior (SHOP/LLA-‐99). Sin embargo, resultará de gran interés analizar el
impacto de esta modificación en los resultados obtenidos en pacientes con
LLA-‐T entre ambos protocolos cuando dispongamos de mayor seguimiento.
En este sentido, con posterioridad a la publicación del presente trabajo de
tesis, nuestro grupo participó en el análisis de los resultados obtenidos a
corto plazo en los pacientes tratados según el protocolo SHOP/LLA-‐2005 de
acuerdo con la dosis de metotrexato administrada (Salazar et al., 2012). En
este análisis se comparó la supervivencia entre dos grupos de pacientes que
recibieron dosis distintas de metotrexato: un grupo recibió dosis de 5 g/m2,
mientras que en otro grupo se administraron dosis de 3 g/m2. La dosis
inferior de 3 g/m2 se administró al subgrupo minoritario de pacientes que
tenían polimorfismos genéticos en la enzima metilen-‐tetrahidrofolato
reductasa (MTHFR) asociados a menor actividad de dicha enzima. Para estos
pacientes se pensó que el aumento de dosis de metotrexato a 5 g/m2podría
asociarse a mayor toxicidad debido a la menor actividad de MTHFR. Por
tanto, se decidió mantener en estos pacientes la dosis del anterior protocolo
(3 g/m2) con el fin de evitar una toxicidad excesiva. Sin embargo, se observó
que los pacientes que recibieron dosis de metotrexato de 5 g/m2 obtuvieron

Discusión
156
una mejor SLE a corto plazo que aquellos que recibieron 3 g/m2 (Salazar et
al., 2012).
Otro de los cambios realizados en el último protocolo consistió en la
estratificación de los pacientes según los niveles de ERM. Con las
modificaciones realizadas en los dos últimos protocolos se consiguió una
mejoría importante y con significación estadística de los resultados de
supervivencia. Así, se pasó de una SLE a 60 meses de 44% a 63% en los
protocolos SHOP/LLA-‐89 + 94 y los protocolos SHOP/LLA-‐99 + 2005,
respectivamente (figura 3 del trabajo 4). Si bien creemos que la mejoría se
debe en gran medida a los cambios descritos en la quimioterapia entre los
dos primeros protocolos y los dos últimos, deben tenerse en cuenta las
limitaciones inherentes a toda comparación con controles históricos.
Identificación de factores pronósticos en la LLA-‐T
En este trabajo se han identificado varios factores pronósticos en el
subgrupo de pacientes con LLA-‐T. Dos de ellos están presentes ya en el
momento del diagnóstico (leucocitosis superior a 200 x 109/L e infiltración
del SNC) y dos son variables evolutivas obtenidas durante el primer mes de
tratamiento (respuesta al tratamiento en el día +15 y al final de la
inducción).
La edad no tuvo valor pronóstico. Precisamente, el valor pronóstico de la
edad en los pacientes con LLA-‐T, a diferencia de lo que ocurre en la LLA de
línea B, varía según las series publicadas (Pullen et al., 1999) (Goldberg et al.,
2003). Lo mismo sucede con los demás factores pronósticos identificados
(Pui et al., 1990) (Sancho et al., 2008), con excepción de la respuesta precoz

Discusión
157
al tratamiento (Schultz et al., 1997). Estas discrepancias entre series podrían
deberse a diferencias en el tratamiento que neutralizaran el efecto adverso
de algunos de ellos. Sin embargo, el factor pronóstico más relevante de
nuestra serie, la respuesta precoz al tratamiento, sí concuerda con lo hallado
en otros grupos de trabajo (Schultz et al., 1997) (Möricke et al., 2009). Así,
resultaron factores pronósticos significativos tanto la respuesta precoz en el
día +15 (SG a 60 meses de 69% frente a 38%, p=0,005), como al final de la
inducción (SG a 60 meses de 64% frente a 0%, p=0,0000). Otras variables de
respuesta al tratamiento con gran importancia pronóstica en otras series son
la respuesta a una semana de prednisona (más una dosis de metotrexato
intratecal) y la ERM (Willemse et al., 2002). La respuesta a una semana de
prednisona permite la identificación de alrededor de un 25-‐30% de pacientes
con LLA-‐T que son malos respondedores (>1.000 blastos/μL en sangre
periférica en el día +8) y que tienen un elevado riesgo de recaída (Aricò et al.,
1995). Este parámetro no se pudo analizar en nuestra serie por carecer los
protocolos SHOP/LLA de esta prefase de tratamiento con prednisona.
Respecto a la ERM, ésta se analiza desde el último protocolo (SHOP/LLA-‐
2005) y el análisis de su valor pronóstico debe esperar a alcanzar una mayor
mediana de seguimiento, por ahora muy corta. Si bien no pudimos analizar
el valor pronóstico de la ERM en nuestra serie por el corto seguimiento de la
cohorte tratada con el protocolo SHOP/LLA-‐2005, éste es en la actualidad
uno de los principales factores pronósticos en la LLA. La ERM no solo tiene
valor pronóstico, sino que se ha incorporado en los protocolos terapéuticos
para estratificar a los pacientes y dirigir la intensidad de tratamiento, de
acuerdo con los niveles alcanzados de ERM en distintos momentos del
tratamiento (Pui et al., 2009) (Conter et al., 2010) (Schrappe et al., 2011).

Discusión
158
En cuanto a factores biológicos con valor pronóstico, a diferencia de la LLA
de línea B, en la LLA-‐T están poco definidos y dependen del protocolo de
tratamiento en el que se analicen. Esto sucede tanto para parámetros
favorables (mutaciones en NOTCH1) (Larson Gedman et al., 2009) (Park et
al., 2009), como para otros adversos (sobreexpresión de TLX3/HOX11L2,
subgrupo early-‐T cell precursor, ETP) (Zuurbier et al., 2010) (Cavé et al.,
2004) (Coustan-‐Smith et al., 2009). Con excepción del subgrupo ETP, el
inmunofenotipo y el estadio madurativo del linfoblasto T no permite
distinguir claros grupos pronósticos (Marks et al., 2009) (Xicoy et al., 2006).
Se hace, pues, necesario identificar factores biológicos en la LLA-‐T que
contribuyan a estratificar mejor a los pacientes o incluso que permitan
aplicar tratamientos dirigidos a las lesiones moleculares subyacentes, como
ya sucede en la LLA Ph+ (Schultz et al., 2009). En este sentido, los resultados
de nuestro trabajo pueden servir de plataforma para el estudio de factores
biológicos y su correlación clínica.
En conclusión, en este trabajo se describe una serie amplia de pacientes con
LLA-‐T tratados con 4 protocolos sucesivos del grupo SHOP, en la que se
obtuvo una mejoría significativa de los resultados con los 2 últimos
protocolos. Asimismo, se identificaron factores clínicos asociados a mal
pronóstico: leucocitosis superior a 200 x 109/l, infiltración en SNC, presencia
de más de 5% de blastos en médula ósea en el día +15 de tratamiento de
inducción y la falta de obtención de la remisión completa al finalizar dicho
tratamiento. Estos factores pronósticos, junto con la posible identificación
de factores biológicos en próximos estudios, podrán contribuir a una mejor

Discusión
159
estratificación de este subgrupo minoritario de pacientes en los próximos
protocolos de tratamiento.
Análisis de la LLA Ph+ y la LLA-‐T
Con los resultados obtenidos en los trabajos presentados en esta tesis
hemos demostrado que el análisis individualizado de series amplias de
pacientes con diferentes subgrupos de LLA pediátrica de alto riesgo (LLA Ph+
y LLA-‐T) permite un mejor conocimiento de sus características clínicas y la
identificación de factores pronósticos específicos. Dicho conocimiento
contribuye a una mejor estratificación de los pacientes según su riesgo
pronóstico y a la aplicación de tratamientos más específicos, lo que redunda
en una mejoría de la supervivencia de estos pacientes.
Limitaciones del estudio
Los estudios que conforman el trabajo actual presentan las siguientes
limitaciones:
• Se trata de estudios retrospectivos en los que se realizan
comparaciones con cohortes históricas de pacientes, por lo que se
asocian a sesgos inherentes a dichas comparaciones.
• Por otro lado, al tratarse de dos estudios multicéntricos con casos
tratados en más de 30 centros de todo el Estado, se añade una
dificultad importante debida a la heterogeneidad en la forma de la
recogida de datos. Ello ha implicado mayor trabajo de la doctoranda a
la hora de homogeneizar, comprobar y completar los datos obtenidos.

Discusión
160
• La leucemia pediátrica es una enfermedad infrecuente y, en concreto,
los dos subgrupos analizados en los dos estudios presentes son
minoritarios (constituyen un 15% de los pacientes en el caso de la LLA-‐
T y tan sólo un 2-‐3% en el caso de la LLA Ph+). Este hecho reduce el
número de casos a analizar, aunque el hecho de disponer de los datos
de todos los pacientes de una serie amplia ha minimizado esta
limitación y ha logrado reunir un número suficiente de casos para
obtener datos estadísticamente significativos.
Beneficios de la investigación
El trabajo de investigación presentado, asociado a la experiencia clínica en
esta enfermedad, ha impulsado a la doctoranda a formar parte del comité
elaborador del nuevo protocolo asistencial de estudio y tratamiento de la
LLA pediátrica a nivel nacional. En este nuevo protocolo se fusionan los dos
grupos cooperativos SHOP y PETHEMA, que tratan en la actualidad a los
pacientes pediátricos con LLA en España.
Por otro lado, gracias al estudio realizado sobre LLA Ph+ y a su publicación,
así como a los contactos establecidos a través del grupo I-‐BFM Study Group,
la doctoranda es la representante de España en la elaboración del nuevo
protocolo internacional para pacientes pediátricos con LLA Ph+. En este
protocolo participan, además de miembros del I-‐BFM Study Group, otros
grupos cooperativos entre los que se halla el principal grupo cooperativo
norteamericano en leucemia pediátrica, el Children Oncology Group. Este
tipo de colaboración es fundamental para poder estudiar y avanzar en el
conocimiento de enfermedades tan infrecuentes como el subgrupo de LLA
Ph+ en la edad pediátrica. En este protocolo se va a estudiar de forma

Discusión
161
prospectiva la opción de tratar sólo con quimioterapia y TKI (imatinib,
dasatinib o nilotinib) y evitar el trasplante alogénico en el subgrupo de LLA
Ph+ con buena respuesta al tratamiento.
Perspectivas de futuro
Los trabajos presentados pueden ser el origen de nuevas líneas de trabajo.
La constitución del nuevo grupo SEHOP-‐PETHEMA y la reciente creación de
un grupo biológico de estudio de la LLA pediátrica permitirán la coordinación
a nivel nacional de un mayor número de casos de LLA, lo que facilitará la
realización de nuevos proyectos clínicos y biológicos.
Además, la doctoranda coordinará el nuevo protocolo para la LLA Ph+ a nivel
nacional. Este protocolo estará adherido al protocolo internacional, en el
que está previsto el uso de dasatinib de forma concomitante a la
quimioterapia.
A continuación se describen algunos de los posibles estudios que podrían
generarse a partir de los resultados de los trabajos de esta tesis.
LLA Ph+
1) Valorar de forma prospectiva si es seguro eliminar el trasplante en
aquellos pacientes con LLA Ph+ con buena respuesta al tratamiento
con quimioterapia y un TKI en el seno del nuevo protocolo
internacional. La cohorte tratada con el protocolo SHOP/LLA-‐2005,
que ha sido analizada en el presente trabajo de tesis, serviría para
realizar una comparación histórica como grupo control.
2) Estudiar factores pronósticos en la era TKI. El análisis de factores
pronósticos en pacientes con LLA Ph+ que hayan recibido tratamiento

Discusión
162
quimioterápico asociado a TKI y que no hayan recibido un trasplante
alogénico podría contribuir a identificar a aquellos pacientes en los
que podría evitarse el trasplante.
3) Analizar, en el seno del nuevo protocolo internacional, si el
tratamiento con un TKI de nueva generación (dasatinib o nilotinib)
mejora los resultados obtenidos con imatinib.
4) Análisis del valor pronóstico de la ERM: establecer qué niveles y
medidos en qué puntos del tratamiento son pronósticos en el seno de
un tratamiento continuo con un TKI. Comprobar el valor de la ERM en
el pre y post-‐trasplante en el seno del protocolo internacional.
5) Estudio de ERM por distintas técnicas: citometría de flujo
multiparamétrica de 8 fluorescencias, RT-‐PCR cuantitativa del gen
BCR-‐ABL, PCR clonoespecífica del reordenamiento de IgH/TCR. Valorar
la sensibilidad y el impacto pronóstico de la detección de la ERM por
las diferentes técnicas.
6) Estudio de nuevos factores biológicos que den información sobre los
mecanismos de resistencia a los TKIs.
LLA-‐T
7) Analizar el impacto de la ERM en nuestra cohorte de pacientes con
LLA-‐T tratados con el protocolo SHOP/LLA-‐2005.
8) Analizar el impacto de la dosis de metotrexato de 5 g/m2 (cohorte de
pacientes tratados con SHOP/LLA-‐2005) frente a la de 3 g/m2 (cohorte
de pacientes tratados con SHOP/LLA-‐99), específicamente en el

Discusión
163
subgrupo de pacientes con LLA-‐T.
9) Analizar en el seno del nuevo protocolo LLA SEHOP-‐PETHEMA 2013 el
valor pronóstico de diferentes subtipos de LLA-‐T, como el early-‐T cell
Precursor, ETP.

Conclusiones
164
Conclusiones

Conclusiones
165

Conclusiones
166
Conclusiones
LLA Ph+
1. La administración de imatinib a dosis intermedias en combinación con
quimioterapia intensiva a pacientes pediátricos con LLA Ph+ fue factible y no
se asoció a un aumento significativo del perfil de toxicidad.
2. La adición de imatinib administrado de forma continua y precoz en
combinación con quimioterapia en niños y adolescentes con LLA Ph+
aumentó la tasa de remisión completa y la tasa de pacientes que llegaron a
realizar un TPH alogénico, en comparación con una cohorte histórica de
pacientes tratados sin imatinib.
3. La adición de imatinib al tratamiento quimioterápico seguido de TPH
alogénico se asoció a un aumento muy importante en la supervivencia global
y libre de evento a los 5 años de los pacientes con LLA Ph+ pediátrica, en
comparación con una cohorte histórica de pacientes tratados sin imatinib.
4. El índice pronóstico FRALLE fue útil para identificar en nuestra serie de
pacientes tratados en la era pre-‐imatinib un subgrupo de pacientes
pediátricos con LLA Ph+ con un pronóstico significativamente mejor al resto
de pacientes.

Conclusiones
167
LLA-‐T
5. La leucocitosis superior a 200 x 109/L, la infiltración del SNC y la mala
respuesta al tratamiento de inducción en el día +15 y al final de inducción se
asociaron a un pronóstico adverso en los pacientes pediátricos con LLA-‐T
tratados con los protocolos sucesivos del grupo SHOP.
6. Las modificaciones en el tratamiento realizadas en los dos últimos
protocolos del grupo SHOP, tales como el aumento de dosis de
asparraginasa, ciclofosfamida y metotrexato, consiguieron mejorar la
supervivencia global y libre de evento de estos pacientes, en comparación
con una cohorte histórica de pacientes tratados con protocolos anteriores.

Financiación
168
Financiación

Financiación
169

Financiación
170
FINANCIACIÓN
Este estudio se ha financiado en parte gracias a una beca de la Fundación de
Oncología Infantil Enriqueta Villavecchia y al proyecto “Força Miquel”.

Bibliografía
171

Bibliografía
172
Bibliografía

Bibliografía
173

Bibliografía
174
Bibliografía
Al-‐Khabori, M., Minden, M.D., Yee, K.W., Gupta, V., Schimmer, A.D., Schuh, A.C., Xu, W., and Brandwein, J.M. (2010). Improved survival using an intensive, pediatric-‐based chemotherapy regimen in adults with T-‐cell acute lymphoblastic leukemia. Leuk Lymphoma 51, 61-‐65.
Andersen, M.K., Autio, K., Barbany, G., Borgström, G., Cavelier, L., Golovleva, I., Heim, S., Heinonen, K., Hovland, R., and Johannsson, J.H. (2011). Paediatric B-‐cell precursor acute lymphoblastic leukaemia with t (1; 19)(q23; p13): clinical and cytogenetic characteristics of 47 cases from the Nordic countries treated according to NOPHO protocols. Br J Haematol 155, 235-‐243.
Aricò, M., Basso, G., Mandelli, F., Rizzari, C., Colella, R., Barisone, E., Zanesco, L., Rondelli, R., Pession, A., and Masera, G. (1995). Good steroid response in vivo predicts a favorable outcome in children with T-‐cell acute lymphoblastic leukemia. The Associazione Italiana Ematologia Oncologia Pediatrica (AIEOP). Cancer 75, 1684-‐693.
Aricò, M., Schrappe, M., Hunger, S.P., Carroll, W.L., Conter, V., Galimberti, S., Manabe, A., Saha, V., Baruchel, A., et al. (2010). Clinical outcome of children with newly diagnosed Philadelphia chromosome-‐positive acute lymphoblastic leukemia treated between 1995 and 2005. J Clin Oncol 28, 4755-‐761.
Aricò, M., Valsecchi, M.G., Camitta, B., Schrappe, M., Chessells, J., Baruchel, A., Gaynon, P., Silverman, L., Janka-‐Schaub, G., et al. (2000). Outcome of treatment in children with Philadelphia chromosome-‐positive acute lymphoblastic leukemia. N Engl J Med 342, 998-‐1006.
Asnafi, V., Buzyn, A., Le Noir, S., Baleydier, F., Simon, A., Beldjord, K., Reman, O., Witz, F., Fagot, T., and Tavernier, E. (2009). NOTCH1/FBXW7 mutation identifies a large subgroup with favorable outcome in adult T-‐cell acute lymphoblastic leukemia (T-‐ALL): a Group for Research on Adult Acute Lymphoblastic Leukemia (GRAALL) study. Blood 113, 3918-‐924.
Asselin, B.L., Devidas, M., Wang, C., Pullen, J., Borowitz, M.J., Hutchison, R., Lipshultz, S.E., and Camitta, B.M. (2011). Effectiveness of high-‐dose methotrexate in T-‐cell lymphoblastic leukemia and advanced-‐stage lymphoblastic lymphoma: a randomized study by the Children's Oncology Group (POG 9404). Blood 118, 874-‐883.

Bibliografía
175
Attarbaschi, A., Mann, G., Panzer-‐Grümayer, R., Röttgers, S., Steiner, M., König, M., Csinady, E., Dworzak, M.N., Seidel, M., and Janousek, D. (2008). Minimal Residual Disease Values Discriminate Between Low and High Relapse Risk in Children With B-‐Cell Precursor Acute Lymphoblastic Leukemia and an Intrachromosomal Amplification of Chromosome 21: The Austrian and German
Acute Lymphoblastic Leukemia Berlin-‐Frankfurt-‐Münster (ALL-‐BFM) Trials. J Clin Oncol 26, 3046-‐050.
Attarbaschi, A., Morak, M., Cario, G., Cazzaniga, G., Ensor, H.M., te Kronnie, T., Bradtke, J., Mann, G., Vendramini, E., et al. (2012). Treatment outcome of CRLF2-‐rearranged childhood acute lymphoblastic leukaemia: a comparative analysis of the AIEOP-‐BFM and UK NCRI-‐CCLG study groups. Br J Haematol 158, 772-‐77.
Avramis, V.I., Sencer, S., Periclou, A.P., Sather, H., Bostrom, B.C., Cohen, L.J., Ettinger, A.G., Ettinger, L.J., Franklin, J., et al. (2002). A randomized comparison of native Escherichia coli asparaginase and polyethylene glycol conjugated asparaginase for treatment of children with newly diagnosed standard-‐risk acute lymphoblastic leukemia: a Children's Cancer Group study. Blood 99, 1986-‐994.
Babusikova, O., Stevulova, L., and Fajtova, M. (2009). Immunophenotyping parameters as prognostic factors in T-‐acute leukemia patients. Neoplasma 56, 508-‐513.
Badell, I., Muñoz, A., Estella, J., Fernández-‐Delgado, R., Javier, G., Verdeguer, A., and Cubells, J. (2008). Long-‐term results of two consecutive trials in childhood acute lymphoblastic leukaemia performed by the Spanish Cooperative Group for Childhood Acute Lymphoblastic Leukemia Group (SHOP) from 1989 to 1998. Clin Transl Oncol 10, 117-‐124.
Bader, P., Kreyenberg, H., Henze, G.H., Eckert, C., Reising, M., Willasch, A., Barth, A., Borkhardt, A., Peters, C., and Handgretinger, R. (2009). Prognostic value of minimal residual disease quantification before allogeneic stem-‐cell transplantation in relapsed childhood acute lymphoblastic leukemia: the ALL-‐REZ BFM Study Group. J Clin Oncol 27, 377-‐384.
Bailey, H.D., Armstrong, B.K., de Klerk, N.H., Fritschi, L., Attia, J., Scott, R.J., Smibert, E., Milne, E., and Aus-‐ALL Consortium (2011). Exposure to professional pest control treatments and the risk of childhood acute lymphoblastic leukemia. Int J Cancer 129, 1678-‐688.
Ballerini, P., Blaise, A., Busson-‐Le Coniat, M., Su, X.Y., Zucman-‐Rossi, J., Adam, M., van den Akker, J., Perot, C., Pellegrino, B., et al. (2002). HOX11L2 expression defines a clinical subtype of pediatric T-‐ALL associated with poor prognosis. Blood 100, 991-‐97.

Bibliografía
176
Bandapalli, O.R., Zimmermann, M., Kox, C., Stanulla, M., Schrappe, M., Ludwig, W.D., Koehler, R., Muckenthaler, M.U., and Kulozik, A.E. (2013). NOTCH1 activation clinically antagonizes the unfavorable effect of PTEN inactivation in BFM-‐treated children with precursor T-‐cell acute lymphoblastic leukemia. Haematologica 98, 928-‐936.
Barbany, G., Andersen, M.K., Autio, K., Borgström, G., Franco, L.C., Golovleva, I., Heim, S., Heinonen, K., Hovland, R., and Johansson, B. (2012). Additional aberrations of the ETV6 and RUNX1 genes have no prognostic impact in 229 t (12; 21)(p13; q22)-‐positive B-‐cell precursor acute lymphoblastic leukaemias treated according to the NOPHO-‐ALL-‐2000 protocol. Leuk Res 36, 936.
Baron, F., and Storb, R. (2004). Allogeneic hematopoietic cell transplantation as treatment for hematological malignancies: a review. Springer seminars in immunopathology 26, 71-‐94.
Barry, E., DeAngelo, D.J., Neuberg, D., Stevenson, K., Loh, M.L., Asselin, B.L., Barr, R.D., Clavell, L.A., Hurwitz, C.A., et al. (2007). Favorable outcome for adolescents with acute lymphoblastic leukemia treated on Dana-‐Farber Cancer Institute Acute Lymphoblastic Leukemia Consortium Protocols. J Clin Oncol 25, 813-‐19.
Bassan, R., Rossi, G., Pogliani, E.M., Di Bona, E., Angelucci, E., Cavattoni, I., Lambertenghi-‐Deliliers, G., Mannelli, F., Levis, A., et al. (2010). Chemotherapy-‐phased imatinib pulses improve long-‐term outcome of adult patients with Philadelphia chromosome-‐positive acute lymphoblastic leukemia: Northern Italy Leukemia Group protocol 09/00. J Clin Oncol 28, 3644-‐652.
Bene, M.C., Castoldi, G., Knapp, W., Ludwig, W.D., Matutes, E., Orfao, A., and Van't Veer, M.B. (1995). Proposals for the immunological classification of acute leukemias. European Group for the Immunological Characterization of Leukemias (EGIL). Leukemia 9, 1783-‐86.
Bennett, J.M., Catovsky, D., Daniel, M.T., Flandrin, G., Galton, D.A., Gralnick, H.R., and Sultan, C. (1976). Proposals for the classification of the acute leukaemias. French-‐American-‐British (FAB) co-‐operative group. Br J Haematol 33, 451-‐58.
Bennett, J.M., Catovsky, D., Daniel, M.T., Flandrin, G., Galton, D.A., Gralnick, H.R., and Sultan, C. (1981). The morphological classification of acute lymphoblastic leukaemia: concordance among observers and clinical correlations. Br J Haematol 47, 553-‐561.
Berg, S.L., Blaney, S.M., Devidas, M., Lampkin, T.A., Murgo, A., Bernstein, M., Billett, A., Kurtzberg, J., Reaman, G., et al. (2005). Phase II study of nelarabine (compound 506U78) in children and young adults with refractory T-‐cell malignancies: a report from the Children's Oncology Group. J Clin Oncol 23, 3376-‐382.

Bibliografía
177
Bhojwani, D., Howard, S.C., and Pui, C.H. (2009). High-‐risk childhood acute lymphoblastic leukemia. Clin Lymphoma Myeloma 9 Suppl 3, S222-‐230.
Biondi, A., Cimino, G., Pieters, R., and Pui, C.-‐H. (2000). Biological and therapeutic aspects of infant leukemia. Blood 96, 24-‐33.
Biondi, A., Schrappe, M., De Lorenzo, P., Castor, A., Lucchini, G., Gandemer, V., Pieters, R., Stary, J., Escherich, G., et al. (2012). Imatinib after induction for treatment of children and adolescents with Philadelphia-‐chromosome-‐positive acute lymphoblastic leukaemia (EsPhALL): a randomised, open-‐label, intergroup study. Lancet Oncol 13, 936-‐945.
Blajchman, M.A. (2008). Platelet Transfusions: An Historical Perspective. ASH Education Program Book 2008, 197-‐7.
Boissel, N., Auclerc, M.-‐F., Lhéritier, V., Perel, Y., Thomas, X., Leblanc, T., Rousselot, P., Cayuela, J.-‐M., Gabert, J., and Fegueux, N. (2003). Should adolescents with acute lymphoblastic leukemia be treated as old children or young adults? Comparison of the French FRALLE-‐93 and LALA-‐94 trials. J Clin Oncol 21, 774-‐780.
Bosch, X., Rovira, M., Sitges, M., Domènech, A., Ortiz-‐Pérez, J.T., de Caralt, T.M., Morales-‐Ruiz, M., Perea, R.J., Monzó, M., and Esteve, J. (2013). Enalapril and Carvedilol for Preventing Chemotherapy-‐Induced Left Ventricular Systolic Dysfunction in Patients With Malignant Hemopathies: The OVERCOME Trial (preventiOn of left Ventricular dysfunction with Enalapril and carvedilol in patients submitted to intensive ChemOtherapy for the treatment of Malignant hEmopathies). J Am Coll Cardiol 61, 2355-‐362.
Brentjens, R.J., Davila, M.L., Riviere, I., Park, J., Wang, X., Cowell, L.G., Bartido, S., Stefanski, J., Taylor, C., et al. (2013). CD19-‐targeted T cells rapidly induce molecular remissions in adults with chemotherapy-‐refractory acute lymphoblastic leukemia. Sci Transl Med 5, 177ra38.
Brissot, E., Labopin, M., Beckers, M., Socie, G., Volin, L., Rambaldi, A., Finke, J., Lenhoff, S., Kröger, N. et al., on behalf of the Acute Leukemia Working Party of EBMT. Tyrosine kinase inhibitors (TKIs) improve outcome of allogeneic haematopoietic stem cell transplantation (allo-‐HCST) for Philadelphia chromosome acute lymphoblastic leukaemia (Ph+ALL) in first complete remission (CR1): a study from the ALWP of EBMT. Bone Marrow Transplant, 48:S2 Abstract O96.
Burke, M.J., Trotz, B., Luo, X., Baker, K.S., Weisdorf, D.J., Wagner, J.E., and Verneris, M.R. (2009). Allo-‐hematopoietic cell transplantation for Ph chromosome-‐positive ALL: impact of imatinib on relapse and survival. Bone Marrow Transplant 43, 107-‐113.

Bibliografía
178
Camós, M., Esteve, J., Jares, P., Colomer, D., Rozman, M., Villamor, N., Costa, D., Carrió, A., Nomdedéu, J., and Montserrat, E. (2006). Gene expression profiling of acute myeloid leukemia with translocation t (8; 16)(p11; p13) and MYST3-‐CREBBP rearrangement reveals a distinctive signature with a specific pattern of HOX gene expression. Cancer Res 66, 6947-‐954.
Campana, D., and Leung, W. (2013). Clinical significance of minimal residual disease in patients with acute leukaemia undergoing haematopoietic stem cell transplantation. Br J Haematol 162, 147-‐161.
Campo, E., Swerdlow, S.H., Harris, N.L., Pileri, S., Stein, H., and Jaffe, E.S. (2011). The 2008 WHO classification of lymphoid neoplasms and beyond: evolving concepts and practical applications. Blood 117, 5019-‐032.
Cavé, H., Suciu, S., Preudhomme, C., Poppe, B., Robert, A., Uyttebroeck, A., Malet, M., Boutard, P., Benoit, Y., and Mauvieux, L. (2004). Clinical significance of HOX11L2 expression linked to t (5; 14)(q35; q32), of HOX11 expression, and of SIL-‐TAL fusion in childhood T-‐cell malignancies: results of EORTC studies 58881 and 58951. Blood 103, 442-‐450.
Cavé, H., van der Werff ten Bosch, J., Suciu, S., Guidal, C., Waterkeyn, C., Otten, J., Bakkus, M., Thielemans, K., Grandchamp, B., and Vilmer, E. (1998). Clinical significance of minimal residual disease in childhood acute lymphoblastic leukemia. European Organization for Research and Treatment of Cancer-‐Childhood Leukemia Cooperative Group. N Engl J Med 339, 591-‐98.
Cazzaniga, G., Lanciotti, M., Rossi, V., Di Martino, D., Aricò, M., Valsecchi, M.G., Basso, G., Masera, G., Micalizzi, C., and Biondi, A. (2002). Prospective molecular monitoring of BCR/ABL transcript in children with Ph+ acute lymphoblastic leukaemia unravels differences in treatment response. Br J Haematol 119, 445-‐453.
Cazzaniga, G., van Delft, F.W., Lo Nigro, L., Ford, A.M., Score, J., Iacobucci, I., Mirabile, E., Taj, M., Colman, S.M., et al. (2011). Developmental origins and impact of BCR-‐ABL1 fusion and IKZF1 deletions in monozygotic twins with Ph+ acute lymphoblastic leukemia. Blood 118, 5559-‐564.
Chalandon, Y., Thomas, X., Hayette, S., Cayuela, J.M., Abbal, C., Escoffre-‐Barbe, M., Maury, S., Lepretre, S., Witz, F., and Benboubker, L. (2008). First results of the GRAAPH-‐2005 study in younger adult patients with de novo Philadelphia positive acute lymphoblastic leukemia. Blood 112 (Suppl1), Abstract 12.
Champagne, M.A., Capdeville, R., Krailo, M., Qu, W., Peng, B., Rosamilia, M., Therrien, M., Zoellner, U., Blaney, S.M., et al. (2004). Imatinib mesylate (STI571) for treatment of children with Philadelphia chromosome-‐positive leukemia: results from a Children's Oncology Group phase 1 study. Blood 104, 2655-‐660.

Bibliografía
179
Chen, H., Liu, K.Y., Xu, L.P., Liu, D.H., Chen, Y.H., Zhao, X.Y., Han, W., Zhang, X.H., Wang, Y., et al. (2012). Administration of imatinib after allogeneic hematopoietic stem cell transplantation may improve disease-‐free survival for patients with Philadelphia chromosome-‐positive acute lymphobla stic leukemia. J Hematol Oncol 5, 29.
Chiaretti, S., Brugnoletti, F., Tavolaro, S., Bonina, S., Paoloni, F., Marinelli, M., Patten, N., Bonifacio, M., Kropp, M.G., et al. (2013). TP53 mutations are frequent in adult acute lymphoblastic leukemia cases negative for recurrent fusion genes and correlate with poor response to induction therapy. Haematologica 98, e59-‐e61.
Childhood ALL Collaborative Group (1996). Duration and intensity of maintenance chemotherapy in acute lymphoblastic leukaemia: overview of 42 trials involving 12 000 randomised children. Childhood ALL Collaborative Group. Lancet 347, 1783-‐88.
Conter, V., Aricò, M., Basso, G., Biondi, A., Barisone, E., Messina, C., Parasole, R., De Rossi, G., Locatelli, F., et al. (2010). Long-‐term results of the Italian Association of Pediatric Hematology and Oncology (AIEOP) Studies 82, 87, 88, 91 and 95 for childhood acute lymphoblastic leukemia. Leukemia 24, 255-‐264.
Conter, V., Bartram, C.R., Valsecchi, M.G., Schrauder, A., Panzer-‐Grümayer, R., Möricke, A., Aricò, M., Zimmermann, M., Mann, G., et al. (2010). Molecular response to treatment redefines all prognostic factors in children and adolescents with B-‐cell precursor acute lymphoblastic leukemia: results in 3184 patients of the AIEOP-‐BFM ALL 2000 study. Blood 115, 3206-‐214.
Conter, V., Valsecchi, M.G., Silvestri, D., Campbell, M., Dibar, E., Magyarosy, E., Gadner, H., Stary, J., Benoit, Y., et al. (2007). Pulses of vincristine and dexamethasone in addition to intensive chemotherapy for children with intermediate-‐risk acute lymphoblastic leukaemia: a multicentre randomised trial. Lancet 369, 123-‐131.
Coustan-‐Smith, E., Behm, F.G., Sanchez, J., Boyett, J.M., Hancock, M.L., Raimondi, S.C., Rubnitz, J.E., Rivera, G.K., Sandlund, J.T., et al. (1998). Immunological detection of minimal residual disease in children with acute lymphoblastic leukaemia. Lancet 351, 550-‐54.
Coustan-‐Smith, E., Mullighan, C.G., Onciu, M., Behm, F.G., Raimondi, S.C., Pei, D., Cheng, C., Su, X., Rubnitz, J.E., et al. (2009). Early T-‐cell precursor leukaemia: a subtype of very high-‐risk acute lymphoblastic leukaemia. Lancet Oncol 10, 147-‐156.

Bibliografía
180
Crombet, O., Lastrapes, K., Zieske, A., and Morales-‐Arias, J. (2012). Complete morphologic and molecular remission after introduction of dasatinib in the treatment of a pediatric patient with t-‐cell acute lymphoblastic leukemia and ABL1 amplification. Pediatr Blood Cancer 59, 333-‐34.
Dastugue, N., Suciu, S., Plat, G., Speleman, F., Cavé, H., Girard, S., Bakkus, M., Pagès, M.P., Yakouben, K., et al. (2013). Hyperdiploidy with 58-‐66 chromosomes in childhood B-‐acute lymphoblastic leukemia is highly curable: 58951 CLG-‐EORTC results. Blood 121, 2415-‐423.
Deininger, M.W., and Druker, B.J. (2003). Specific targeted therapy of chronic myelogenous leukemia with imatinib. Pharmacol Rev 55, 401-‐423.
Den Boer, M.L., van Slegtenhorst, M., De Menezes, R.X., Cheok, M.H., Buijs-‐Gladdines, J.G., Peters, S.T., Van Zutven, L.J., Beverloo, H.B., Van der Spek, P.J., et al. (2009). A subtype of childhood acute lymphoblastic leukaemia with poor
treatment outcome: a genome-‐wide classification study. Lancet Oncol 10, 125-‐134.
Den Boer, M.L., van Slegtenhorst, M., De Menezes, R.X., Cheok, M.H., Buijs-‐Gladdines, J.G., Peters, S.T., Van Zutven, L.J., Beverloo, H.B., Van der Spek, P.J., et al. (2009). A subtype of childhood acute lymphoblastic leukaemia with poor treatment outcome: a genome-‐wide classification study. Lancet Oncol 10, 125-‐134.
Druker, B.J., Sawyers, C.L., Kantarjian, H., Resta, D.J., Reese, S.F., Ford, J.M., Capdeville, R., and Talpaz, M. (2001). Activity of a specific inhibitor of the BCR-‐ABL tyrosine kinase in the blast crisis of chronic myeloid leukemia and acute lymphoblastic leukemia with the Philadelphia chromosome. New Engl J Med 344, 1038-‐042.
Druker, B.J., Tamura, S., Buchdunger, E., Ohno, S., Segal, G.M., Fanning, S., Zimmermann, J., and Lydon, N.B. (1996). Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-‐Abl positive cells. Nat Med 2, 561-‐66.
Dunsmore, K.P., Devidas, M., Linda, S.B., Borowitz, M.J., Winick, N., Hunger, S.P., Carroll, W.L., and Camitta, B.M. (2012). Pilot study of nelarabine in combination with intensive chemotherapy in high-‐risk T-‐cell acute lymphoblastic leukemia: a report from the Children's Oncology Group. J Clin Oncol 30, 2753-‐59.
Dvorak, C.C., Gracia, C.R., Sanders, J.E., Cheng, E.Y., Baker, K.S., Pulsipher, M.A., and Petryk, A. (2011). NCI, NHLBI/PBMTC first international conference on late effects after pediatric hematopoietic cell transplantation: endocrine challenges-‐thyroid dysfunction, growth impairment, bone health, & reproductive risks. Biol Blood Marrow Transplant 17, 1725-‐738.

Bibliografía
181
Eden, T. (2010). Aetiology of childhood leukaemia. Cancer Treat Rev 36, 286-‐297.
Eden, T.O., Pieters, R., and Richards, S. (2010). Systematic review of the addition of vincristine plus steroid pulses in maintenance treatment for childhood acute lymphoblastic leukaemia-‐an individual patient data meta-‐analysis involving 5,659 children. Br J Haematol 149, 722.
Emerenciano, M., Meyer, C., Mansur, M.B., Marschalek, R., Pombo-‐de-‐Oliveira, M.S., and Brazilian Collaborative Study Group of Infant Acute Leukaemia (2013). The distribution of MLL breakpoints correlates with outcome in infant acute leukaemia. Br J Haematol 161, 224-‐236.
Enshaei, A., Schwab, C.J., Konn, Z.J., Mitchell, C.D., Kinsey, S.E., Wade, R., Vora, A., Harrison, C.J., and Moorman, A.V. (2013). Long-‐term follow-‐up of ETV6-‐-‐RUNX1 ALL reveals that NCI risk, rather than secondary genetic abnormalities, is the key risk factor. Leukemia doi: 10.1038/leu.2013.136
Esperou, H., Boiron, J.M., Cayuela, J.M., Blanchet, O., Kuentz, M., Jouet, J.P., Milpied, N., Cahn, J.Y., Faucher, C., and Bourhis, J.H. (2003). A potential graft-‐versus-‐leukemia effect after allogeneic hematopoietic stem cell transplantation for patients with Philadelphia chromosome-‐positive acute lymphoblastic leukemia: results from the French Bone Marrow Transplantation Society. Bone Marrow Transplant 31, 909-‐918.
Fagioli, F., Quarello, P., Zecca, M., Lanino, E., Rognoni, C., Balduzzi, A., Messina, C., Favre, C., Foà, R., et al. (2013). Hematopoietic stem cell transplantation for children with high-‐risk acute lymphoblastic leukemia in first complete remission: a report from the AIEOP registry. Haematologica 98, 1273-‐281.
Fagioli, F., Quarello, P., Zecca, M., Lanino, E., Rognoni, C., Balduzzi, A., Messina, C., Favre, C., Ripaldi, M., and Rutella, S. (2013). Hematopoietic stem cell transplantation for children with high risk acute lymphoblastic leukemia in first complete remission: a report from the AIEOP registry. Haematologica doi:10.3324/haematol.2012.079707
Fielding, A.K., Buck, G., Lazarus, H.M., Litzow, M.R., Luger, S., Marks, D.I., McMillan, A., Moorman, A.V., Paietta, E., and Richards, S.M. (2010). Imatinib significantly enhances long-‐term outcomes in Philadelphia positive acute lymphoblastic leukaemia; final results of the UKALLXII/ECOG2993 Trial.
Fielding, A.K., Rowe, J.M., Richards, S.M., Buck, G., Moorman, A.V., Durrant, I.J., Marks, D.I., McMillan, A.K., Litzow, M.R., et al. (2009). Prospective outcome data on 267 unselected adult patients with Philadelphia chromosome-‐positive acute lymphoblastic leukemia confirms superiority of allogeneic transplantation over chemotherapy in the pre-‐imatinib era: results from the International ALL Trial MRC UKALLXII/ECOG2993. Blood 113, 4489-‐496.

Bibliografía
182
Gandemer, V., Auclerc, M.F., Perel, Y., Vannier, J.P., Le Gall, E., Demeocq, F., Schmitt, C., Piguet, C., Stephan, J.L., et al. (2009). Impact of age, leukocyte count and day 21-‐bone marrow response to chemotherapy on the long-‐term outcome of children with philadelphia chromosome-‐positive acute lymphoblastic leukemia in the pre-‐imatinib era: results of the FRALLE 93 study. BMC Cancer 9, 14.
Gilham, C., Peto, J., Simpson, J., Roman, E., Eden, T.O., Greaves, M.F., Alexander, F.E., and UKCCS Investigators (2005). Day care in infancy and risk of childhood acute lymphoblastic leukaemia: findings from UK case-‐control study. BMJ 330, 1294.
Glazer, E.R., Perkins, C.I., Young, J.L., Schlag, R.D., Campleman, S.L., and Wright, W.E. (1999). Cancer among Hispanic children in California, 1988-‐1994: comparison with non-‐Hispanic white children. Cancer 86, 1070-‐79.
Goldberg, J.M., Silverman, L.B., Levy, D.E., Dalton, V.K., Gelber, R.D., Lehmann, L., Cohen, H.J., Sallan, S.E., and Asselin, B.L. (2003). Childhood T-‐cell acutelymphoblastic leukemia: the Dana-‐Farber Cancer Institute acute lymphoblastic leukemia consortium experience. J Clin Oncol 21, 3616-‐622.
Goto, H., Inukai, T., Inoue, H., Ogawa, C., Fukushima, T., Yabe, M., Kikuchi, A., Koike, K., Fukushima, K., and Isoyama, K. (2011). Acute lymphoblastic leukemia and Down syndrome: the collaborative study of the Tokyo Children’s Cancer Study Group and the Kyushu Yamaguchi Children’s Cancer Study Group. Int J Hematol 93, 192-‐98.
Greaves, M. (2009). Darwin and evolutionary tales in leukemia. Hematology Am Soc Hematol Educ Program 2009, 3-‐12.
Greaves, M. (2010). Cancer stem cells: back to Darwin? Semin Cancer Biol 20, 65-‐70.
Greaves, M.F. (1988). Speculations on the cause of childhood acute lymphoblastic leukemia. Leukemia 2, 120-‐25.
Greaves, M.F., Maia, A.T., Wiemels, J.L., and Ford, A.M. (2003). Leukemia in twins: lessons in natural history. Blood 102, 2321-‐333.
Grupp, S.A., Kalos, M., Barrett, D., Aplenc, R., Porter, D.L., Rheingold, S.R., Teachey, D.T., Chew, A., Hauck, B., et al. (2013). Chimeric Antigen Receptor-‐Modified T Cells for Acute Lymphoid Leukemia. N Engl J Med 368, 1509-‐518.
Gurney, J.G., Severson, R.K., Davis, S., and Robison, L.L. (1995). Incidence of cancer in children in the United States. Sex-‐, race-‐, and 1-‐year age-‐specific rates by histologic type. Cancer 75, 2186-‐195.
Gutiérrez, A., Sanda, T., Grebliunaite, R., Carracedo, A., Salmena, L., Ahn, Y., Dahlberg, S., Neuberg, D., Moreau, L.A., et al. (2009). High frequency of PTEN, PI3K, and AKT abnormalities in T-‐cell acute lymphoblastic leukemia. Blood 114, 647-‐650.

Bibliografía
183
Hagemeijer, A., and Graux, C. (2010). ABL1 rearrangements in T-‐cell acute lymphoblastic leukemia. Genes Chromosomes Cancer 49, 299-‐308.
Harvey, R.C., Mullighan, C.G., Chen, I.M., Wharton, W., Mikhail, F.M., Carroll, A.J., Kang, H., Liu, W., Dobbin, K.K., et al. (2010). Rearrangement of CRLF2 is associated with mutation of JAK kinases, alteration of IKZF1, Hispanic/Latino ethnicity, and a poor outcome in pediatric B-‐progenitor acute lymphoblastic leukemia. Blood 115, 5312-‐321.
Haydu, J.E., and Ferrando, A.A. (2013). Early T-‐cell precursor acute lymphoblastic leukaemia. Curr Opin Hematol 20, 369-‐373.
Heerema, N.A., Carroll, A.J., Devidas, M., Loh, M.L., Borowitz, M.J., Gastier-‐Foster, J.M., Larsen, E.C., Mattano, L.A., Maloney, K.W., et al. (2013). Intrachromosomal Amplification of Chromosome 21 Is Associated With Inferior Outcomes in Children With Acute Lymphoblastic Leukemia Treated in Contemporary Standard-‐Risk Children's Oncology Group Studies: A Report From the Children's Oncology Group. J Clin Oncol doi: 10.1200/ JCO.2013.49.1308
Heerema, N.A., Harbott, J., Galimberti, S., Camitta, B.M., Gaynon, P.S., Janka-‐Schaub, G., Kamps, W., Basso, G., Pui, C.H., and Schrappe, M. (2004). Secondary cytogenetic aberrations in childhood Philadelphia chromosome positive acute lymphoblastic leukemia are nonrandom and may be associated with outcome. Leukemia 18, 693-‐702.
Hof, J., Krentz, S., van Schewick, C., Körner, G., Shalapour, S., Rhein, P., Karawajew, L., Ludwig, W.D., Seeger, K., et al. (2011). Mutations and deletions of the TP53 gene predict nonresponse to treatment and poor outcome in first relapse of childhood acute lymphoblastic leukemia. J Clin Oncol 29, 3185-‐193.
Holmfeldt, L., Wei, L., Diaz-‐Flores, E., Walsh, M., Zhang, J., Ding, L., Payne-‐Turner, D., Churchman, M., Andersson, A., et al. (2013). The genomic landscape of hypodiploid acute lymphoblastic leukemia. Nat Genet 45, 242-‐252.
Homminga, I., Zwaan, C.M., Manz, C.Y., Parker, C., Bantia, S., Smits, W.K., Higginbotham, F., Pieters, R., and Meijerink, J.P. (2011). In vitro efficacy of forodesine and nelarabine (ara-‐G) in pediatric leukemia. Blood 118, 2184-‐190.
Homminga, I., Pieters, R., Langerak, A.W., de Rooi, J.J., Stubbs, A., Verstegen, M., Vuerhard, M., Buijs-‐Gladdines, J., Kooi, C., et al. (2011). Integrated transcript and genome analyses reveal NKX2-‐1 and MEF2C as potential oncogenes in T cell acute lymphoblastic leukemia. Cancer Cell 19, 484-‐497.

Bibliografía
184
Hsu, W.L., Preston, D.L., Soda, M., Sugiyama, H., Funamoto, S., Kodama, K., Kimura, A., Kamada, N., Dohy, H., et al. (2013). The incidence of leukemia, lymphoma and multiple myeloma among atomic bomb survivors: 1950-‐2001. Radiat Res 179, 361-‐382.
Hughes, W.T., Kuhn, S., Chaudhary, S., Feldman, S., Verzosa, M., Aur, R.J., Pratt, C., and George, S.L. (1977). Successful chemoprophylaxis for Pneumocystis carinii pneumonitis. N Engl J Med 297, 1419.
Iacobucci, I., Ferrari, A., Lonetti, A., Papayannidis, C., Paoloni, F., Trino, S., Storlazzi, C.T., Ottaviani, E., Cattina, F., et al. (2011). CDKN2A/B alterations impair prognosis in adult BCR-‐ABL1-‐positive acute lymphoblastic leukemia patients. Clin Cancer Res 17, 7413-‐423.
Iacobucci, I., Papayannidis, C., Lonetti, A., Ferrari, A., Baccarani, M., and Martinelli, G. (2012). Cytogenetic and molecular predictors of outcome in acute lymphocytic leukemia: recent developments. Curr Hematol Malig Rep 7, 133-‐143.
Igarashi, S., Manabe, A., Ohara, A., Kumagai, M., Saito, T., Okimoto, Y., Kamijo, T., Isoyama, K., Kajiwara, M., and Sotomatsu, M. (2005). No advantage of dexamethasone over prednisolone for the outcome of standard-‐and intermediate-‐risk childhood acute lymphoblastic leukemia in the Tokyo Children's Cancer Study Group L95-‐14 protocol. J Clin Oncol 23, 6489-‐498.
Inthal, A., Zeitlhofer, P., Zeginigg, M., Morak, M., Grausenburger, R., Fronkova, E., Fahrner, B., Mann, G., Haas, O.A., and Panzer-‐Grümayer, R. (2012). CREBBP HAT domain mutations prevail in relapse cases of high hyperdiploid childhood acute lymphoblastic leukemia. Leukemia 26, 1797-‐1803.
Inukai, T., Kiyokawa, N., Campana, D., Coustan-‐Smith, E., Kikuchi, A., Kobayashi, M., Takahashi, H., Koh, K., Manabe, A., et al. (2012). Clinical significance of early T-‐cell precursor acute lymphoblastic leukaemia: results of the Tokyo Children's Cancer Study Group Study L99-‐15. Br J Haematol 156, 358-‐365.
Jenkinson, S., Koo, K., Mansour, M.R., Goulden, N., Vora, A., Mitchell, C., Wade, R., Richards, S., Hancock, J., et al. (2013). Impact of NOTCH1/FBXW7 mutations on outcome in pediatric T-‐cell acute lymphoblastic leukemia patients treated on the MRC UKALL 2003 trial. Leukemia 27, 41-‐47.
Kager, L., Cheok, M., Yang, W., Zaza, G., Cheng, Q., Panetta, J.C., Pui, C.H., Downing, J.R., Relling, M.V., and Evans, W.E. (2005). Folate pathway gene expression differs in subtypes of acute lymphoblastic leukemia and influences methotrexate pharmacodynamics. J Clin Invest 115, 110-‐17.

Bibliografía
185
Kager, L., Lion, T., Attarbaschi, A., Koenig, M., Strehl, S., Haas, O.A., Dworzak, M.N., Schrappe, M., Gadner, H., and Mann, G. (2007). Incidence and outcome of TCF3-‐PBX1-‐positive acute lymphoblastic leukemia in Austrian children. Haematologica 92, 1561-‐64.
Kang, H., Wilson, C.S., Harvey, R.C., Chen, I.M., Murphy, M.H., Atlas, S.R., Bedrick, E.J., Devidas, M., Carroll, A.J., et al. (2012). Gene expression profiles predictive of outcome and age in infant acute lymphoblastic leukemia: a Children's Oncology Group study. Blood 119, 1872-‐881.
Kantarjian, H., Shah, N.P., Hochhaus, A., Cortes, J., Shah, S., Ayala, M., Moiraghi, B., Shen, Z., Mayer, J., and Pasquini, R. (2010). Dasatinib versus imatinib in newly diagnosed chronic-‐phase chronic myeloid leukemia. New Engl J Med 362, 2260-‐270.
Kantarjian, H.M., Shah, N.P., Cortes, J.E., Baccarani, M., Agarwal, M.B., Undurraga, M.S., Wang, J., Ipiña, J.J., Kim, D.W., et al. (2012). Dasatinib or imatinib in newly diagnosed chronic-‐phase chronic myeloid leukemia: 2-‐year follow-‐up from a randomized phase 3 trial (DASISION). Blood 119, 1123-‐29.
Kawedia, J.D., Kaste, S.C., Pei, D., Panetta, J.C., Cai, X., Cheng, C., Neale, G., Howard, S.C., Evans, W.E., et al. (2011). Pharmacokinetic, pharmacodynamic, and pharmacogenetic determinants of osteonecrosis in children with acute lymphoblastic leukemia. Blood 117, 2340-‐47.
Kebriaei, P., Saliba, R., Rondon, G., Chiattone, A., Luthra, R., Anderlini, P., Andersson, B., Shpall, E., Popat, U., et al. (2012). Long-‐term follow-‐up of allogeneic hematopoietic stem cell transplantation for patients with Philadelphia chromosome-‐positive acute lymphoblastic leukemia: impact of tyrosine kinase inhibitors on treatment outcomes. Biol Blood Marrow Transplant 18, 584-‐592.
Kebriaei, P., Saliba, R., Rondon, G., Chiattone, A., Luthra, R., Anderlini, P., Andersson, B., Shpall, E., Popat, U., et al. (2012). Long-‐Term Follow-‐up of Allogeneic Hematopoietic Stem Cell Transplantation for Patients with Philadelphia Chromosome-‐Positive Acute Lymphoblastic Leukemia: Impact of Tyrosine Kinase Inhibitors on Treatment Outcomes. Biol Blood Marrow Transplant 18, 584-‐592.
De Keersmaecker, K., Graux, C., Odero, M.D., Mentens, N., Somers, R., Maertens, J., Wlodarska, I., Vandenberghe, P., Hagemeijer, A., et al. (2005). Fusion of EML1 to ABL1 in T-‐cell acute lymphoblastic leukemia with cryptic t(9;14)(q34;q32). Blood 105, 4849-‐852.
Kim DY, Joo DJ, Lee JH, and et al. (2011). Nilotinib combined with multi-‐ agent chemotherapy for adult patients with newly diagnosed Phila-‐ delphia chromosome-‐positive acute lymphoblastic leukemia: interim results of Korean adult ALL working party phase 2 study. Blood 118 (Suppl1), Abstract 1517.

Bibliografía
186
Kinlen, L.J. (1997). High-‐contact paternal occupations, infection and childhood leukaemia: five studies of unusual population-‐mixing of adults. Br J Cancer 76, 1539-‐545.
De Klein, A., van Kessel, A.G., Grosveld, G., Bartram, C.R., Hagemeijer, A., Bootsma, D., Spurr, N.K., Heisterkamp, N., Groffen, J., and Stephenson, J.R. (1982). A cellular oncogene is translocated to the Philadelphia chromosome in chronic myelocytic leukaemia. Nature 300, 765-‐67.
Kox, C., Zimmermann, M., Stanulla, M., Leible, S., Schrappe, M., Ludwig, W.D., Koehler, R., Tolle, G., Bandapalli, O.R., et al. (2010). The favorable effect of activating NOTCH1 receptor mutations on long-‐term outcome in T-‐ALL patients treated on the ALL-‐BFM 2000 protocol can be separated from FBXW7 loss of function. Leukemia 24, 2005-‐013.
Kuiper, R.P., Waanders, E., van der Velden, V.H., van Reijmersdal, S.V., Venkatachalam, R., Scheijen, B., Sonneveld, E., van Dongen, J.J., Veerman, A.J., et al. (2010). IKZF1 deletions predict relapse in uniformly treated pediatric precursor B-‐ALL. Leukemia 24, 1258-‐264.
de Labarthe, A., Rousselot, P., Huguet-‐Rigal, F., Delabesse, E., Witz, F., Maury, S., Réa, D., Cayuela, J.M., Vekemans, M.C., et al. (2007). Imatinib combined with induction or consolidation chemotherapy in patients with de novo Philadelphia chromosome-‐positive acute lymphoblastic leukemia: results of the GRAAPH-‐2003 study. Blood 109, 1408-‐413.
Lange, B.J., Bostrom, B.C., Cherlow, J.M., Sensel, M.G., La, M.K., Rackoff, W., Heerema, N.A., Wimmer, R.S., Trigg, M.E., et al. (2002). Double-‐delayed intensification improves event-‐free survival for children with intermediate-‐risk acute lymphoblastic leukemia: a report from the Children's Cancer Group. Blood 99, 825-‐833.
Larson Gedman, A., Chen, Q., Kugel Desmoulin, S., Ge, Y., LaFiura, K., Haska, C.L., Cherian, C., Devidas, M., Linda, S.B., et al. (2009). The impact of NOTCH1, FBW7 and PTEN mutations on prognosis and downstream signaling in pediatric T-‐cell acute lymphoblastic leukemia: a report from the Children's Oncology Group. Leukemia 23, 1417-‐425.
Lauten, M., Möricke, A., Beier, R., Zimmermann, M., Stanulla, M., Meissner, B., Odenwald, E., Attarbaschi, A., Niemeyer, C., et al. (2012). Prediction of outcome by early bone marrow response in childhood acute lymphoblastic leukemia treated in the ALL-‐BFM 95 trial: differential effects in precursor B-‐cell and T-‐cell leukemia. Haematologica 97, 1048-‐056.

Bibliografía
187
Lee, S., Kim, D.W., and Kim, H.J. (2011). First-‐Line Dasatinib Plus Conventional Chemotherapy in Adults with Newly Diagnosed Philadelphia Chromosome-‐Positive Acute Lymphoblastic Leukemia (Ph+ ALL): Interim Analysis of the Korean Prospective Phase II Study. Blood (ASH Annual Meeting Abstracts) 118 (Suppl1), Abstract 1516.
Lee, S., Kim, D.W., Cho, B.S., Yoon, J.H., Shin, S.H., Yahng, S.A., Lee, S.E., Eom, K.S., Kim, Y.J., et al. (2012). Impact of minimal residual disease kinetics during imatinib-‐based treatment on transplantation outcome in Philadelphia chromosome-‐positive acute lymphoblastic leukemia. Leukemia 26, 2367-‐374.
Lee, S., Kim, Y.J., Min, C.K., Kim, H.J., Eom, K.S., Kim, D.W., Lee, J.W., Min, W.S., and Kim, C.C. (2005). The effect of first-‐line imatinib interim therapy on the outcome of allogeneic stem cell transplantation in adults with newly diagnosed Philadelphia chromosome-‐positive acute lymphoblastic leukemia. Blood 105, 3449-‐457.
Li, Y., Qiu, L., Zou, D., Zhao, Y., Mi, Y., and Wang, J. (2009). Additional chromosomal abnormalities and their prognostic significance in adult Philadelphia-‐positive acute lymphoblastic leukemia: with or without imatinib in chemotherapy. Ann Hematol 88, 1069-‐077.
Lipshultz, S.E., Miller, T.L., Lipsitz, S.R., Neuberg, D.S., Dahlberg, S.E., Colan, S.D., Silverman, L.B., Henkel, J.M., Franco, V.I., et al. (2012). Continuous Versus Bolus Infusion of Doxorubicin in Children With ALL: Long-‐term Cardiac Outcomes. Pediatrics 130, 1003-‐011.
Locatelli, F., Schrappe, M., Bernardo, M.E., and Rutella, S. (2012). How I treat relapsed childhood acute lymphoblastic leukemia. Blood 120, 2807-‐816.
Loh, M.L., Goldwasser, M.A., Silverman, L.B., Poon, W.M., Vattikuti, S., Cardoso, A., Neuberg, D.S., Shannon, K.M., Sallan, S.E., and Gilliland, D.G. (2006). Prospective analysis of TEL/AML1-‐positive patients treated on Dana-‐Farber Cancer Institute Consortium Protocol 95-‐01. Blood 107, 4508-‐513.
Ma, M., Wang, X., Tang, J., Xue, H., Chen, J., Pan, C., Jiang, H., and Shen, S. (2012). Early T-‐cell precursor leukemia: a subtype of high risk childhood acute lymphoblastic leukemia. Front Med 6, 416-‐420.
Ma, X., Buffler, P.A., Selvin, S., Matthay, K.K., Wiencke, J.K., Wiemels, J.L., and Reynolds, P. (2002). Daycare attendance and risk of childhood acute lymphoblastic leukaemia. Br J Cancer 86, 1419-‐424.
Ma, X., Urayama, K., Chang, J., Wiemels, J.L., and Buffler, P.A. (2009). Infection and pediatric acute lymphoblastic leukemia. Blood Cells Mol Dis 42, 117-‐120.

Bibliografía
188
Malagoli, C., Fabbi, S., Teggi, S., Calzari, M., Poli, M., Ballotti, E., Notari, B., Bruni, M., Palazzi, G., et al. (2010). Risk of hematological malignancies associated with magnetic fields exposure from power lines: a case-‐control study in two municipalities of northern Italy. Environ Health 9, 16.
Maloney, K.W. (2011). Acute lymphoblastic leukaemia in children with Down syndrome: an updated review. Br J Haematol 155, 420-‐25.
Mann, G., Attarbaschi, A., Schrappe, M., De Lorenzo, P., Peters, C., Hann, I., De Rossi, G., Felice, M., Lausen, B., and LeBlanc, T. (2010). Improved outcome with hematopoietic stem cell transplantation in a poor prognostic subgroup of infants with mixed-‐lineage-‐leukemia (MLL)-‐-‐rearranged acute lymphoblastic leukemia: results from the Interfant-‐99 Study. Blood 116, 2644-‐650.
Marks, D.I., Paietta, E.M., Moorman, A.V., Richards, S.M., Buck, G., DeWald, G., Ferrando, A., Fielding, A.K., Goldstone, A.H., et al. (2009). T-‐cell acute lymphoblastic leukemia in adults: clinical features, immunophenotype, cytogenetics, and outcome from the large randomized prospective trial (UKALL XII/ECOG 2993). Blood 114, 5136-‐145.
Martino, R., Santamaría, A., Pericas, R., Sureda, A., and Brunet, S. (1997). Acute rhabdomyolysis and myonecrosis complicating aeromonas bacteremia in neutropenic patients with hematologic malignancies: report of two cases. Haematologica 82, 692-‐94.
Meeker, T.C., Hardy, D., Willman, C., Hogan, T., and Abrams, J. (1990). Activation of the interleukin-‐3 gene by chromosome translocation in acute lymphocytic leukemia with eosinophilia. Blood 76, 285-‐89.
Meijerink, J.P., den Boer, M.L., and Pieters, R. (2009). New genetic abnormalities and treatment response in acute lymphoblastic leukemia. Semin Hematol 46, 16-‐23.
Meyer, C., Hofmann, J., Burmeister, T., Gröger, D., Park, T.S., Emerenciano, M., Pombo de Oliveira, M., Renneville, A., Villarese, P., et al. (2013). The MLL recombinome of acute leukemias in 2013. Leukemia doi: 10.1038/leu.2013.135.
Meyr, F., Escherich, G., Mann, G., Klingebiel, T., Kulozik, A., Rossig, C., Schrappe, M., Henze, G., von Stackelberg, A., and Hitzler, J. (2013). Outcomes of treatment for relapsed acute lymphoblastic leukaemia in children with Down syndrome. Br J Haematol 162, 98-‐106.
Minson, K.A., Prasad, P., Vear, S., Borinstein, S., Ho, R., Domm, J., and Frangoul, H. (2013). t (17; 19) in Children with Acute Lymphocytic Leukemia: A Report of 3 Cases and a Review of the Literature. Case Rep Hematol 2013:563291.

Bibliografía
189
Mitchell, C., Richards, S., Harrison, C.J., and Eden, T. (2010). Long-‐term follow-‐up of the United Kingdom medical research council protocols for childhood acute lymphoblastic leukaemia, 1980-‐2001. Leukemia 24, 406-‐418.
Mitchell, C.D., Richards, S.M., Kinsey, S.E., Lilleyman, J., Vora, A., Eden, T.O., and Medical Research Council Childhood Leukaemia Working Party (2005). Benefit of dexamethasone compared with prednisolone for childhood acute lymphoblastic leukaemia: results of the UK Medical Research Council ALL97 randomized trial. Br J Haematol 129, 734-‐745.
Mizuta, S., Matsuo, K., Maeda, T., Yujiri, T., Hatta, Y., Kimura, Y., Ueda, Y., Kanamori, H., Usui, N., et al. (2012). Prognostic factors influencing clinical outcome of allogeneic hematopoietic stem cell transplantation following imatinib-‐based therapy in BCR-‐ABL-‐positive ALL. Blood Cancer J 2, e72.
Moellering, R.E., Cornejo, M., Davis, T.N., Del Bianco, C., Aster, J.C., Blacklow, S.C., Kung, A.L., Gilliland, D.G., Verdine, G.L., and Bradner, J.E. (2009). Direct inhibition of the NOTCH transcription factor complex. Nature 462, 182-‐88.
De Moerloose, B., Suciu, S., Bertrand, Y., Mazingue, F., Robert, A., Uyttebroeck, A., Yakouben, K., Ferster, A., Margueritte, G., et al. (2010). Improved outcome with pulses of vincristine and corticosteroids in continuation therapy of children with average risk acute lymphoblastic leukemia (ALL) and lymphoblastic non-‐Hodgkin lymphoma (NHL): report of the EORTC randomized phase 3 trial 58951. Blood 116, 36-‐44.
Moghrabi, A., Levy, D.E., Asselin, B., Barr, R., Clavell, L., Hurwitz, C., Samson, Y., Schorin, M., Dalton, V.K., and Lipshultz, S.E. (2007). Results of the Dana-‐Farber Cancer Institute ALL Consortium Protocol 95-‐01 for children with acute lymphoblastic leukemia. Blood 109, 896-‐904.
Moorman, A.V. (2012). The clinical relevance of chromosomal and genomic abnormalities in B-‐cell precursor acute lymphoblastic leukaemia. Blood Rev 26, 123-‐135.
Moorman, A.V., Ensor, H.M., Richards, S.M., Chilton, L., Schwab, C., Kinsey, S.E., Vora, A., Mitchell, C.D., and Harrison, C.J. (2010). Prognostic effect of chromosomal abnormalities in childhood B-‐cell precursor acute lymphoblastic leukaemia: results from the UK Medical Research Council ALL97/99 randomised trial. Lancet Oncol 11, 429-‐438.
Moorman, A.V., Robinson, H., Schwab, C., Richards, S.M., Hancock, J., Mitchell, C.D., Goulden, N., Vora, A., and Harrison, C.J. (2013). Risk-‐Directed Treatment Intensification Significantly Reduces the Risk of Relapse Among Children and Adolescents With Acute Lymphoblastic Leukemia and Intrachromosomal Amplification of Chromosome 21: A Comparison of the MRC ALL97/99 and UKALL2003 Trials. J Clin Oncol 20, 31:3389-‐96.

Bibliografía
190
Moorman, A.V., Schwab, C., Ensor, H.M., Russell, L.J., Morrison, H., Jones, L., Masic, D., Patel, B., Rowe, J.M., et al. (2012). IGH@ translocations, CRLF2 deregulation, and microdeletions in adolescents and adults with acute lymphoblastic leukemia. J Clin Oncol 30, 3100-‐08.
Möricke, A., Reiter, A., Zimmermann, M., Gadner, H., Stanulla, M., Dördelmann, M., Löning, L., Beier, R., Ludwig, W.D., et al. (2008). Risk-‐adjusted therapy of acute lymphoblastic leukemia can decrease treatment burden and improve survival: treatment results of 2169 unselected pediatric and adolescent patients enrolled in the trial ALL-‐BFM 95. Blood 111, 4477-‐489.
Möricke, A., Zimmermann, M., Reiter, A., Henze, G., Schrauder, A., Gadner, H., Ludwig, W.D., Ritter, J., Harbott, J., and Mann, G. (2009). Long-‐term results of five consecutive trials in childhood acute lymphoblastic leukemia performed by the ALL-‐BFM study group from 1981 to 2000. Leukemia 24, 265-‐284.
Mullighan, C.G. (2012). Molecular genetics of B-‐precursor acute lymphoblastic leukemia. J Clin Invest 122, 3407-‐415.
Mullighan, C.G., Collins-‐Underwood, J.R., Phillips, L.A., Loudin, M.G., Liu, W., Zhang, J., Ma, J., Coustan-‐Smith, E., Harvey, R.C., et al. (2009). Rearrangement of CRLF2 in B-‐progenitor-‐ and Down syndrome-‐associated acute lymphoblastic leukemia. Nat Genet 41, 1243-‐46.
Mullighan, C.G., Miller, C.B., Radtke, I., Phillips, L.A., Dalton, J., Ma, J., White, D., Hughes, T.P., Le Beau, M.M., et al. (2008). BCR-‐ABL1 lymphoblastic leukaemia is characterized by the deletion of Ikaros. Nature 453, 110-‐14.
Mullighan, C.G., Phillips, L.A., Su, X., Ma, J., Miller, C.B., Shurtleff, S.A., and Downing, J.R. (2008). Genomic analysis of the clonal origins of relapsed acute lymphoblastic leukemia. Science 322, 1377-‐380.
Mullighan, C.G., Zhang, J., Harvey, R.C., Collins-‐Underwood, J.R., Schulman, B.A., Phillips, L.A., Tasian, S.K., Loh, M.L., Su, X., et al. (2009). JAK mutations in high-‐risk childhood acute lymphoblastic leukemia. Proc Natl Acad Sci U S A 106, 9414-‐18.
Mullighan, C.G., Zhang, J., Kasper, L.H., Lerach, S., Payne-‐Turner, D., Phillips, L.A., Heatley, S.L., Holmfeldt, L., Collins-‐Underwood, J.R., and Ma, J. (2011). CREBBP mutations in relapsed acute lymphoblastic leukaemia. Nature 471, 235-‐39.
Nachman, J., Sather, H.N., Gaynon, P.S., Lukens, J.N., Wolff, L., and Trigg, M.E. (1997). Augmented Berlin-‐Frankfurt-‐Munster therapy abrogates the adverse prognostic significance of slow early response to induction chemotherapy for children and adolescents with acute lymphoblastic leukemia and unfavorable presenting features: a report from the Children's Cancer Group. J Clin Oncol 15, 2222-‐230.

Bibliografía
191
Neumann, M., Heesch, S., Gökbuget, N., Schwartz, S., Schlee, C., Benlasfer, O., Farhadi-‐Sartangi, N., Thibaut, J., Burmeister, T., et al. (2012). Clinical and molecular characterization of early T-‐cell precursor leukemia: a high-‐risk subgroup in adult T-‐ALL with a high frequency of FLT3 mutations. Blood Cancer J 2, e55.
Nishiwaki, S., Miyamura, K., Kato, C., Terakura, S., Ohashi, K., Sakamaki, H., Nakao, S., Harigae, H., and Kodera, Y. (2010). Impact of post-‐transplant imatinib administration on Philadelphia chromosome-‐positive acute lymphoblastic leukaemia. Anticancer Res 30, 2415-‐18.
Notta, F., Mullighan, C.G., Wang, J.C., Poeppl, A., Doulatov, S., Phillips, L.A., Ma, J., Minden, M.D., Downing, J.R., and Dick, J.E. (2011). Evolution of human BCR-‐ABL1 lymphoblastic leukaemia-‐initiating cells. Nature 469, 362-‐67.
Nowell, P.C. (2007). Discovery of the Philadelphia chromosome: a personal perspective. J Clin Invest 117, 2033-‐35.
Nowell, P.C., and Hungerford, D. (1960). A minute chromosome in human chronic granulocytic leukemia. Science 132, 1497.
Oliansky, D.M., Camitta, B., Gaynon, P., Nieder, M.L., Parsons, S.K., Pulsipher, M.A., Dillon, H., Ratko, T.A., Wall, D., et al. (2012). Role of cytotoxic therapy with hematopoietic stem cell transplantation in the treatment of pediatric acute lymphoblastic leukemia: update of the 2005 evidence-‐based review. Biol Blood Marrow Transplant 18, 505-‐522.
Ottmann, O.G., and Pfeifer, H. (2009). Management of Philadelphia chromosome-‐positive acute lymphoblastic leukemia (Ph+ ALL). Hematology Am Soc Hematol Educ Program , 371-‐381.
Palmi, C., Valsecchi, M.G., Longinotti, G., Silvestri, D., Carrino, V., Conter, V., Basso, G., Biondi, A., Kronnie, G.T., and Cazzaniga, G. (2013). What is the relevance of Ikaros gene deletions as a prognostic marker in pediatric Philadelphia-‐negative B-‐cell precursor acute lymphoblastic leukemia? Haematologica 98, 1226-‐231.
Palomero, T., Sulis, M.L., Cortina, M., Real, P.J., Barnes, K., Ciofani, M., Caparros, E., Buteau, J., Brown, K., and Perkins, S.L. (2007). Mutational loss of PTEN induces resistance to NOTCH1 inhibition in T-‐cell leukemia. Nat Med 13, 1203-‐210.
Pane, F., Cimino, G., Izzo, B., Camera, A., Vitale, A., Quintarelli, C., Picardi, M., Specchia, G., Mancini, M., et al. (2005). Significant reduction of the hybrid BCR/ABL transcripts after induction and consolidation therapy is a powerful predictor of treatment response in adult Philadelphia-‐positive acute lymphoblastic leukemia. Leukemia 19, 628-‐635.

Bibliografía
192
Papaemmanuil, E., Hosking, F.J., Vijayakrishnan, J., Price, A., Olver, B., Sheridan, E., Kinsey, S.E., Lightfoot, T., Roman, E., et al. (2009). Loci on 7p12.2, 10q21.2 and 14q11.2 are associated with risk of childhood acute lymphoblastic leukemia. Nat Genet 41, 1006-‐010.
Park, M.J., Taki, T., Oda, M., Watanabe, T., Yumura-‐Yagi, K., Kobayashi, R., Suzuki, N., Hara, J., Horibe, K., and Hayashi, Y. (2009). FBXW7 and NOTCH1 mutations in childhood T cell acute lymphoblastic leukaemia and T cell non-‐Hodgkin lymphoma. Br J Haematol 145, 198-‐206.
Paulsson, K., Forestier, E., Andersen, M.K., Autio, K., Barbany, G., Borgström, G., Cavelier, L., Golovleva, I., Heim, S., et al. (2013). High modal number and triple trisomies are highly correlated favorable factors in childhood B-‐cell precursor high hyperdiploid acute lymphoblastic leukemia treated according to the NOPHO ALL 1992/2000 protocols. Haematologica 98, 1424-‐32.
Paulsson, K., Forestier, E., Lilljebjörn, H., Heldrup, J., Behrendtz, M., Young, B.D., and Johansson, B. (2010). Genetic landscape of high hyperdiploid childhood acute lymphoblastic leukemia. Proc Natl Acad Sci U S A 107, 21719-‐724.
Perrillat, F., Clavel, J., Auclerc, M.-‐F., Baruchel, A., Leverger, G., Nelken, B., Philippe, N., Schaison, G., Sommelet, D., and Vilmer, E. (2002). Day-‐care, early common infections and childhood acute leukaemia: a multicentre French case-‐-‐control study. Br J Cancer 86, 1064-‐69.
Pfeifer, H., Goekbuget, N., Volp, C., Hüttmann, A., Lübbert, M., and Stuhlmann, R. (2010). Long-‐term outcome of 335 adult patients receiving different schedules of imatinib and chemotherapy as front-‐line treatment for Philadelphia-‐positive acute lymphoblastic leukemia (Ph+ ALL). Blood 116, 173.
Pfeifer, H., Wassmann, B., Bethge, W., Dengler, J., Bornhauser, M., and Stadler, M. (2011). Updated Long-‐Term Results of a Randomized Comparison of Prophylactic and Pre-‐Emptive Imatinib Following Allogeneic Stem Cell Transplantation for Philadelphia Chromosome Positive Acute Lymphoblastic Leukemia (Ph+ ALL). Blood 118 (Suppl1), Abstract 247.
Pfeifer, H., Wassmann, B., Bethge, W., Dengler, J., Bornhäuser, M., Stadler, M., Beelen, D., Vucinic, V., Burmeister, T., et al. (2013). Randomized comparison of prophylactic and minimal residual disease-‐triggered imatinib after allogeneic stem cell transplantation for BCR-‐ABL1-‐positive acute lymphoblastic leukemia. Leukemia 27, 1254-‐262.
Pole, J.D., Alibhai, S.M., Ethier, M.C., Teuffel, O., Portwine, C., Zelcer, S., Johnston, D.L., Silva, M., Alexander, S., et al. (2013). Adolescents with acute lymphoblastic leukemia treated at pediatric versus adult hospitals. Ann Oncol 24, 801-‐06.

Bibliografía
193
Pui, C.H. (2012). Childhood leukemias (Cambridge: Cambridge University Press).
Pui, C.H., and Evans, W.E. (2006). Treatment of acute lymphoblastic leukemia. New Engl J Med 354, 166-‐178.
Pui, C.H., Behm, F.G., Singh, B., Schell, M.J., Williams, D.L., Rivera, G.K., Kalwinsky, D.K., Sandlund, J.T., Crist, W.M., and Raimondi, S.C. (1990). Heterogeneity of presenting features and their relation to treatment outcome in 120 children with T-‐cell acute lymphoblastic leukemia. Blood 75, 174-‐79.
Pui, C.H., Campana, D., Pei, D., Bowman, W.P., Sandlund, J.T., Kaste, S.C., Ribeiro, R.C., Rubnitz, J.E., Raimondi, S.C., and Onciu, M. (2009). Treating childhood acute lymphoblastic leukemia without cranial irradiation. New Engl J Med 360, 2730-‐741.
Pui, C.H., Carroll, W.L., Meshinchi, S., and Arceci, R.J. (2011). Biology, risk stratification, and therapy of pediatric acute leukemias: an update. J Clin Oncol 29, 551-‐565.
Pui, C.H., Chessells, J.M., Camitta, B., Baruchel, A., Biondi, A., Boyett, J.M., Carroll, A., Eden, O.B., Evans, W.E., et al. (2003). Clinical heterogeneity in childhood acute lymphoblastic leukemia with 11q23 rearrangements. Leukemia 17, 700-‐06.
Pui, C.H., Gaynon, P.S., Boyett, J.M., Chessells, J.M., Baruchel, A., Kamps, W., Silverman, L.B., Biondi, A., Harms, D.O., et al. (2002). Outcome of treatment in childhood acute lymphoblastic leukaemia with rearrangements of the 11q23 chromosomal region. Lancet 359, 1909-‐915.
Pui, C.H., Mullighan, C.G., Evans, W.E., and Relling, M.V. (2012). Pediatric acute lymphoblastic leukemia: where are we going and how do we get there? Blood 120, 1165-‐174.
Pui, C.H., Pei, D., Campana, D., Bowman, W.P., Sandlund, J.T., Kaste, S.C., Ribeiro, R.C., Rubnitz, J.E., Coustan-‐Smith, E., et al. (2011). Improved prognosis for older adolescents with acute lymphoblastic leukemia. J Clin Oncol 29, 386-‐391.
Pui, C.H., Pei, D., Sandlund, J.T., Ribeiro, R.C., Rubnitz, J.E., Raimondi, S.C., Onciu, M., Campana, D., Kun, L.E., et al. (2010). Long-‐term results of St Jude Total Therapy Studies 11, 12, 13A, 13B, and 14 for childhood acute lymphoblastic leukemia. Leukemia 24, 371-‐382.
Pullen, J., Shuster, J.J., Link, M., Borowitz, M., Amylon, M., Carroll, A.J., Land, V., Look, A.T., McIntyre, B., and Camitta, B. (1999). Significance of commonly used prognostic factors differs for children with T cell acute lymphocytic leukemia (ALL), as compared to those with B-‐precursor ALL. A Pediatric Oncology Group (POG) study. Leukemia 13, 1696.

Bibliografía
194
Radtke, S., Zolk, O., Renner, B., Paulides, M., Zimmermann, M., Möricke, A., Stanulla, M., Schrappe, M., and Langer, T. (2013). Germline genetic variations in methotrexate candidate genes are associated with pharmacokinetics, toxicity, and outcome in childhood acute lymphoblastic leukemia. Blood 121, 5145-‐153.
Ram, R., Storb, R., Sandmaier, B.M., Maloney, D.G., Woolfrey, A., Flowers, M.E., Maris, M.B., Laport, G.G., Chauncey, T.R., and Lange, T. (2011). Non-‐myeloablative conditioning with allogeneic hematopoietic cell transplantation for the treatment of high-‐risk acute lymphoblastic leukemia. Haematologica 96, 1113-‐120.
Ravandi, F., Jorgensen, J.L., Thomas, D.A., O'Brien, S., Garris, R., Faderl, S., Huang, X., Wen, S., Burger, J.A., et al. (2013). Detection of MRD may predict the outcome of patients with Philadelphia chromosome-‐positive ALL treated with tyrosine kinase inhibitors plus chemotherapy. Blood 122, 1214-‐221.
Ravandi, F., O'Brien, S., Thomas, D., Faderl, S., Jones, D., Garris, R., Dara, S., Jorgensen, J., Kebriaei, P., et al. (2010). First report of phase 2 study of dasatinib with hyper-‐CVAD for the frontline treatment of patients with Philadelphia chromosome-‐positive (Ph+) acute lymphoblastic leukemia. Blood 116, 2070-‐77.
Relling, M.V., Hancock, M.L., Rivera, G.K., Sandlund, J.T., Ribeiro, R.C., Krynetski, E.Y., Pui, C.H., and Evans, W.E. (1999). Mercaptopurine therapy intolerance and
heterozygosity at the thiopurine S-‐methyltransferase gene locus. J Natl Cancer Inst 91, 2001-‐08.
Relling, M.V., Yanishevski, Y., Nemec, J., Evans, W.E., Boyett, J.M., Behm, F.G., and Pui, C.H. (1998). Etoposide and antimetabolite pharmacology in patients who develop secondary acute myeloid leukemia. Leukemia 12, 346-‐352.
Ribeiro, R.C., Broniscer, A., Rivera, G.K., Hancock, M.L., Raimondi, S.C., Sandlund, J.T., Crist, W., Evans, W.E., and Pui, C.H. (1997). Philadelphia chromosome-‐positive acute lymphoblastic leukemia in children: durable responses to chemotherapy associated with low initial white blood cell counts. Leukemia 11, 1493-‐96.
Ribera, J.M. (2013). Optimal approach to treatment of patients with Philadelphia chromosome-‐positive acute lymphoblastic leukemia: how to best use all the available tools. Leuk Lymphoma 54, 21-‐27.
Ribera, J.M., García, O., Fernández-‐Abellán, P., Lavilla, E., Bernal, M.T., González-‐Campos, J., Brunet, S., Monteserín, M.C., Montesinos, P., and Sarrá, J. (2012). Lack of negative impact of Philadelphia chromosome in older patients with acute lymphoblastic leukaemia in the thyrosine kinase inhibitor era: comparison of two prospective parallel protocols. Br J Haematol 159, 485-‐88.

Bibliografía
195
Ribera, J.M., García, O., Montesinos, P., Brunet, S., Abella, E., Barrios, M., González-‐Campos, J., Bravo, P., Amigo, M.L., and Hernández-‐Rivas, J.M. (2012). Treatment of young patients with Philadelphia chromosome-‐positive acute lymphoblastic leukaemia using increased dose of imatinib and deintensified chemotherapy before allogeneic stem cell transplantation. Br J Haematol 159, 78-‐81.
Ribera, J.M., Oriol, A., González, M., Vidriales, B., Brunet, S., Esteve, J., Del Potro, E., Rivas, C., Moreno, M.J., et al. (2010). Concurrent intensive chemotherapy and imatinib before and after stem cell transplantation in newly diagnosed Philadelphia chromosome-‐positive acute lymphoblastic leukemia. Final results of the CSTIBES02 trial. Haematologica 95, 87-‐95.
Ribera, J.M., Oriol, A., Sanz, M.-‐A., Tormo, M., Fernández-‐Abellán, P., del Potro, E., Abella, E., Bueno, J., Parody, R., and Bastida, P. (2008). Comparison of the results of the treatment of adolescents and young adults with standard-‐risk acute lymphoblastic leukemia with the Programa Espanol de Tratamiento en Hematologia pediatric-‐based protocol ALL-‐96. J Clin Oncol 26, 1843-‐49.
Ribera, J.M., Ortega, J.J., Oriol, A., Granada, I., Hernández-‐Rivas, J.M., Parody, R., Bethencourt, C., Rivas, C., Bastida, P., et al. (2002). Prognostic value of karyotypic analysis in children and adults with high-‐risk acute lymphoblastic leukemia included in the PETHEMA ALL-‐93 trial. Haematologica 87, 154-‐166.
Richards, S., Pui, C.H., Gayon, P., and Childhood Acute Lymphoblastic Leukemia Collaborative Group (CALLCG) (2013). Systematic review and meta-‐analysis of randomized trials of central nervous system directed therapy for childhood acute lymphoblastic leukemia. Pediatr Blood Cancer 60, 185-‐195.
Riehm, H., Reiter, A., Schrappe, M., Berthold, F., Dopfer, R., Gerein, V., Ludwig, R., Ritter, J., Stollmann, B., and Henze, G. (1987). [Corticosteroid-‐dependent reduction of leukocyte count in blood as a prognostic factor in acute lymphoblastic leukemia in childhood (therapy study ALL-‐BFM 83)]. Klin Padiatr 199, 151-‐160.
Rizzari, C., Conter, V., Starý, J., Colombini, A., Moericke, A., and Schrappe, M. (2013). Optimizing asparaginase therapy for acute lymphoblastic leukemia. Curr Opin Oncol 25 Suppl 1, S1-‐S9.
Rowley, J.D. (1973). Letter: A new consistent chromosomal abnormality in chronic myelogenous leukaemia identified by quinacrine fluorescence and Giemsa staining. Nature 243, 290-‐93.

Bibliografía
196
Roy, A., Bradburn, M., Moorman, A.V., Burrett, J., Love, S., Kinsey, S.E., Mitchell, C., Vora, A., Eden, T., et al. (2005). Early response to induction is predictive of survival in childhood Philadelphia chromosome positive acute lymphoblastic leukaemia: results of the Medical Research Council ALL 97 trial. Br J Haematol 129, 35-‐44.
Rubnitz, J.E., Wichlan, D., Devidas, M., Shuster, J., Linda, S.B., Kurtzberg, J., Bell, B., Hunger, S.P., Chauvenet, A., et al. (2008). Prospective analysis of TEL gene rearrangements in childhood acute lymphoblastic leukemia: a Children's Oncology Group study. J Clin Oncol 26, 2186-‐191.
Russell, L.J., Akasaka, T., Majid, A., Sugimoto, K.-‐J., Karran, E.L., Nagel, I., Harder, L., Claviez, A., Gesk, S., and Moorman, A.V. (2008). t (6; 14)(p22; q32): a new recurrent IGH@ translocation involving ID4 in B-‐cell precursor acute lymphoblastic leukemia (BCP-‐ALL). Blood 111, 387-‐391.
Saglio, G., Kim, D.-‐W., Issaragrisil, S., Le Coutre, P., Etienne, G., Lobo, C., Pasquini, R., Clark, R.E., Hochhaus, A., and Hughes, T.P. (2010). Nilotinib versus imatinib for newly diagnosed chronic myeloid leukemia. New Engl J Med 362, 2251-‐59.
Salazar, J., Altés, A., del Río, E., Estella, J., Rives, S., Tasso, M., Navajas, A., Molina, J., Villa, M., et al. (2012). Methotrexate consolidation treatment according to pharmacogenetics of MTHFR ameliorates event-‐free survival in childhood acute lymphoblastic leukaemia. Pharmacogenomics J 12, 379-‐385.
Salzer, W.L., Devidas, M., Carroll, W.L., Winick, N., Pullen, J., Hunger, S.P., and Camitta, B.A. (2010). Long-‐term results of the pediatric oncology group studies for childhood acute lymphoblastic leukemia 1984-‐2001: a report from the children's oncology group. Leukemia 24, 355-‐370.
Samon, J.B., Castillo-‐Martin, M., Hadler, M., Ambesi-‐Impiobato, A., Paietta, E., Racevskis, J., Wiernik, P.H., Rowe, J.M., Jakubczak, J., et al. (2012). Preclinical analysis of the γ-‐secretase inhibitor PF-‐03084014 in combination with glucocorticoids in T-‐cell acute lymphoblastic leukemia. Mol Cancer Ther 11, 1565-‐575.
Sancho, J.M., Morgades, M., Arranz, R., Fernández-‐Abellán, P., Deben, G., Alonso, N., Blanes, M., Rodríguez, M.J., Nicolás, C., et al. (2008). Practice of central nervous system prophylaxis and treatment in acute leukemias in Spain. Prospective registry study. Med Clin (Barc) 131, 401-‐05.
Sanda, T., Tyner, J.W., Gutiérrez, A., Ngo, V.N., Glover, J., Chang, B.H., Yost, A., Ma, W., Fleischman, A.G., et al. (2013). TYK2-‐STAT1-‐BCL2 pathway dependence in T-‐cell acute lymphoblastic leukemia. Cancer Discov 3, 564-‐577.

Bibliografía
197
Sánchez-‐Ortega, I., Servitje, O., Arnan, M., Ortí, G., Peralta, T., Manresa, F., and Duarte, R.F. (2012). Dasatinib as salvage therapy for steroid refractory and imatinib resistant or intolerant sclerotic chronic graft-‐versus-‐host disease. Biol Blood Marrow Transplant 18, 318-‐323.
Schmiegelow, K., Al-‐Modhwahi, I., Andersen, M.K., Behrendtz, M., Forestier, E., Hasle, H., Heyman, M., Kristinsson, J., Nersting, J., et al. (2009). Methotrexate/6-‐mercaptopurine maintenance therapy influences the risk of a second malignant neoplasm after childhood acute lymphoblastic leukemia: results from the NOPHO ALL-‐92 study. Blood 113, 6077-‐084.
Schmiegelow, K., Levinsen, M.F., Attarbaschi, A., Baruchel, A., Devidas, M., Escherich, G., Gibson, B., Heydrich, C., Horibe, K., et al. (2013). Second malignant neoplasms after treatment of childhood acute lymphoblastic leukemia. J Clin Oncol 31, 2469-‐476.
Schrappe, M., Aricò, M., Harbott, J., Biondi, A., Zimmermann, M., Conter, V., Reiter, A., Valsecchi, M.G., Gadner, H., et al. (1998). Philadelphia chromosome-‐positive (Ph+) childhood acute lymphoblastic leukemia: good initial steroid response allows early prediction of a favorable treatment outcome. Blood 92, 2730-‐741.
Schrappe, M., Hunger, S.P., Pui, C.H., Saha, V., Gaynon, P.S., Baruchel, A., Conter, V., Otten, J., Ohara, A., et al. (2012). Outcomes after induction failure in childhood acute lymphoblastic leukemia. N Engl J Med 366, 1371-‐381.
Schrappe, M., Möricke, A., Reiter, A., Henze, G., Welte, K., Gadner, H., Ludwig, W.D., Ritter, J., Harbott, J., and Mann, G. (2013). Key Treatment Questions in Childhood Acute Lymphoblastic Leukemia: Results in 5 Consecutive Trials Performed by the ALL-‐BFM Study Group From 1981 to 2000. Klin Padiatr 225, S62-‐S72.
Schrappe, M., Valsecchi, M.G., Bartram, C.R., Schrauder, A., Panzer-‐Grümayer, R., Möricke, A., Parasole, R., Zimmermann, M., Dworzak, M., et al. (2011). Late MRD response determines relapse risk overall and in subsets of childhood T-‐cell ALL: results of the AIEOP-‐BFM-‐ALL 2000 study. Blood 118, 2077-‐084.
Schrauder, A., Reiter, A., Gadner, H., Niethammer, D., Klingebiel, T., Kremens, B., Peters, C., Ebell, W., Zimmermann, M., et al. (2006). Superiority of allogeneic hematopoietic stem-‐cell transplantation compared with chemotherapy alone in high-‐risk childhood T-‐cell acute lymphoblastic leukemia: results from ALL-‐BFM 90 and 95. J Clin Oncol 24, 5742-‐49.
Schultz, K.R., Bowman, W.P., Aledo, A., Slayton, W.B., Sather, H., Devidas, M., Wang, C., Davies, S.M., Gaynon, P.S., et al. (2009). Improved early event-‐free survival with imatinib in Philadelphia chromosome-‐positive acute lymphoblastic leukemia: a children's oncology group study. J Clin Oncol 27, 5175-‐181.

Bibliografía
198
Schultz, K.R., Massing, B., Spinelli, J.J., Gaynon, P.S., and Wadsworth, L. (1997). Importance of the day 7 bone marrow biopsy as a prognostic measure of the outcome in children with acute lymphoblastic leukemia. Med Pediatr Oncol 29, 16-‐22.
Sekimizu, M., Sunami, S., Nakazawa, A., Hayashi, Y., Okimoto, Y., Saito, A.M., Horibe, K., Tsurusawa, M., and Mori, T. (2011). Chromosome abnormalities in advanced stage T-‐cell lymphoblastic lymphoma of children and adolescents: a report from Japanese Paediatric Leukaemia/Lymphoma Study Group (JPLSG) and review of the literature. Br J Haematol 154, 612-‐17.
Shih, I.M., and Wang, T.L. (2007). Notch signaling, γ-‐secretase inhibitors, and cancer therapy. Cancer Res 67, 1879-‐882.
Shimizu, H., Handa, H., Hatsumi, N., Takada, S., Saitoh, T., Sakura, T., Miyawaki, S., and Nojima, Y. (2013). Distinctive disease subgroups according to differentiation stages in adult patients with T-‐cell acute lymphoblastic leukemia. Eur J Haematol 90, 301-‐07.
Silverman, L.B., Gelber, R.D., Dalton, V.K., Asselin, B.L., Barr, R.D., Clavell, L.A., Hurwitz, C.A., Moghrabi, A., Samson, Y., et al. (2001). Improved outcome for children with acute lymphoblastic leukemia: results of Dana-‐Farber Consortium Protocol 91-‐01. Blood 97, 1211-‐18.
Silverman, L.B., Stevenson, K.E., O'Brien, J.E., Asselin, B.L., Barr, R.D., Clavell, L., Cole, P.D., Kelly, K.M., Laverdiere, C., et al. (2010). Long-‐term results of Dana-‐Farber Cancer Institute ALL Consortium protocols for children with newly diagnosed acute lymphoblastic leukemia (1985-‐2000). Leukemia 24, 320-‐334.
Smith, M., Arthur, D., Camitta, B., Carroll, A.J., Crist, W., Gaynon, P., Gelber, R., Heerema, N., Korn, E.L., et al. (1996). Uniform approach to risk classification and treatment assignment for children with acute lymphoblastic leukemia. J Clin Oncol 14, 18-‐24.
Stark, B., Avrahami, G., Nirel, R., Abramov, A., Attias, D., Ballin, A., Bielorai, B., Burstein, Y., Gavriel, H., et al. (2009). Extended triple intrathecal therapy in children with T-‐cell acute lymphoblastic leukaemia: a report from the Israeli National ALL-‐Studies. Br J Haematol 147, 113-‐124.
Swerdlow, S.H., Campo, E., Harris, N., Jafe, E., Pileri, S., Stein, H., Tiele, J., and Vardiman, J. (2008). WHO classification of tumours of haematopoietic and lymphoid tissues (Lyon, France: International Agency for Research on Cancer).

Bibliografía
199
Tallen G, Ratei R, Mann G, Kaspers G, Niggli F, Karachunsky A, Ebell W,Escherich G, Schrappe M, et al. (2010). A.Long-‐term outcome in children with relapsed acute lymphoblastic leukemia aftertime-‐point and site-‐of-‐relapse stratification and intensified short-‐course multidrug chemotherapy: results of trial ALL-‐REZ BFM 90. J Clin Oncol. 10;28(14):2339-‐47.
Tanguy-‐Schmidt, A., Rousselot, P., Chalandon, Y., Cayuela, J.M., Hayette, S., Vekemans, M.C., Escoffre, M., Huguet, F., Réa, D., et al. (2013). Long-‐term follow-‐up of the imatinib GRAAPH-‐2003 study in newly diagnosed patients with de novo Philadelphia chromosome-‐positive acute lymphoblastic leukemia: a GRAALL study. Biol Blood Marrow Transplant 19, 150-‐55.
Thomas, D.A., Faderl, S., Cortes, J., O'Brien, S., Giles, F.J., Kornblau, S.M., Garcia-‐Manero, G., Keating, M.J., Andreeff, M., et al. (2004). Treatment of Philadelphia chromosome-‐positive acute lymphocytic leukemia with hyper-‐CVAD and imatinib mesylate. Blood 103, 4396-‐4407.
Tsai, M.S., Kuo, C.Y., Wang, M.C., Wu, H.C., Chien, C.C., and Liu, J.W. (2006). Clinical features and risk factors for mortality in Aeromonas bacteremic adults with hematologic malignancies. J Microbiol Immunol Infect 39, 150-‐54.
van der Linden, M.H., Valsecchi, M.G., De Lorenzo, P., Möricke, A., Janka, G., Leblanc, T.M., Felice, M., Biondi, A., Campbell, M., et al. (2009). Outcome of congenital acute lymphoblastic leukemia treated on the Interfant-‐99 protocol. Blood 114, 3764-‐68.
van der Veer, A., Waanders, E., Pieters, R., Willemse, M.E., Van Reijmersdal, S.V., Russell, L.J., Harrison, C.J., Evans, W.E., van der Velden, V.H., and Hoogerbrugge, P.M. (2013). Independent prognostic value of BCR-‐ABL1-‐like signature and IKZF1 deletion, but not high CRLF2 expression, in children with B-‐cell precursor ALL. Blood doi: 10.1182/blood-‐2012-‐10-‐462358.
Van der Velden, V.H., Corral, L., Valsecchi, M.G., Jansen, M.W., De Lorenzo, P., Cazzaniga, G., Panzer-‐Grümayer, E.R., Schrappe, M., Schrauder, A., et al. (2009). Prognostic significance of minimal residual disease in infants with acute lymphoblastic leukemia treated within the Interfant-‐99 protocol. Leukemia 23, 1073-‐79.
van Dongen, J.J., Seriu, T., Panzer-‐Grümayer, E.R., Biondi, A., Pongers-‐Willemse, M.J., Corral, L., Stolz, F., Schrappe, M., Masera, G., and Kamps, W.A. (1998). Prognostic value of minimal residual disease in acute lymphoblastic leukaemia in childhood. The Lancet 352, 1731-‐38.

Bibliografía
200
van Grotel, M., Meijerink, J.P., Beverloo, H.B., Langerak, A.W., Buys-‐Gladdines, J.G., Schneider, P., Poulsen, T.S., den Boer, M.L., Horstmann, M., and Kamps, W.A. (2006). The outcome of molecular-‐cytogenetic subgroups in pediatric T-‐cell acute lymphoblastic leukemia: a retrospective study of patients treated according to DCOG or COALL protocols. Haematologica 91, 1212-‐221.
Van Vlierberghe, P., Ambesi-‐Impiombato, A., Pérez-‐García, A., Haydu, J.E., Rigo, I., Hadler, M., Tosello, V., Della Gatta, G., Paietta, E., and Racevskis, J. (2011). ETV6 mutations in early immature human T cell leukemias. J Exp Med 208, 2571-‐79.
Van Vlierberghe, P., and Ferrando, A. (2012). The molecular basis of T cell acute lymphoblastic leukemia. J Clin Invest 122, 3398.
Van Vlierberghe, P., Palomero, T., Khiabanian, H., Van der Meulen, J., Castillo, M., Van Roy, N., De Moerloose, B., Philippé, J., González-‐García, S., and Toribio, M.L. (2010). PHF6 mutations in T-‐cell acute lymphoblastic leukemia. Nat Genet 42, 338-‐342.
Veerman, A.J., Kamps, W.A., van den Berg, H., van den Berg, E., Bökkerink, J.P., Bruin, M.C., van den Heuvel-‐Eibrink, M.M., Korbijn, C.M., Korthof, E.T., et al. (2009). Dexamethasone-‐based therapy for childhood acute lymphoblastic leukaemia: results of the prospective Dutch Childhood Oncology Group (DCOG) protocol ALL-‐9 (1997-‐2004). Lancet Oncol 10, 957-‐966.
Vidriales, M.B., Pérez, J.J., López-‐Berges, M.C., Gutiérrez, N., Ciudad, J., Lucio, P., Vazquez, L., García-‐Sanz, R., del Cañizo, M.C., et al. (2003). Minimal residual disease in adolescent (older than 14 years) and adult acute lymphoblastic leukemias: early immunophenotypic evaluation has high clinical value. Blood 101, 4695-‐4700.
Volanakis, E.J., Williams, R.T., and Sherr, C.J. (2009). Stage-‐specific Arf tumor suppression in Notch1-‐induced T-‐cell acute lymphoblastic leukemia. Blood 114, 4451-‐59.
Waanders, E., van der Velden, V.H., van der Schoot, C.E., van Leeuwen, F.N., van Reijmersdal, S.V., de Haas, V., Veerman, A.J., van Kessel, A.G., Hoogerbrugge, P.M., et al. (2011). Integrated use of minimal residual disease classification and IKZF1 alteration status accurately predicts 79% of relapses in pediatric acute lymphoblastic leukemia. Leukemia 25, 254-‐58.
Wassmann, B., Pfeifer, H., Goekbuget, N., Beelen, D.W., Beck, J., Stelljes, M., Bornhäuser, M., Reichle, A., Perz, J., and Haas, R. (2006). Alternating versus concurrent schedules of imatinib and chemotherapy as front-‐line therapy for Philadelphia-‐positive acute lymphoblastic leukemia (Ph+ ALL). Blood 108, 1469-‐477.

Bibliografía
201
Wassmann, B., Pfeifer, H., Stadler, M., Bornhaüser, M., Bug, G., Scheuring, U.J., Brück, P., Stelljes, M., Schwerdtfeger, R., et al. (2005). Early molecular response to posttransplantation imatinib determines outcome in MRD+ Philadelphia-‐positive acute lymphoblastic leukemia (Ph+ ALL). Blood 106, 458-‐463.
Wiemels, J.L., Cazzaniga, G., Daniotti, M., Eden, O.B., Addison, G.M., Masera, G., Saha, V., Biondi, A., and Greaves, M.F. (1999). Prenatal origin of acute lymphoblastic leukaemia in children. The Lancet 354, 1499-‐1503.
Willemse, M.J., Seriu, T., Hettinger, K., d'Aniello, E., Hop, W.C., Panzer-‐Grümayer, E.R., Biondi, A., Schrappe, M., Kamps, W.A., et al. (2002). Detection of minimal residual disease identifies differences in treatment response between T-‐ALL and precursor B-‐ALL. Blood 99, 4386-‐393.
Wu, Y., Cain-‐Hom, C., Choy, L., Hagenbeek, T.J., de Leon, G.P., Chen, Y., Finkle, D., Venook, R., Wu, X., et al. (2010). Therapeutic antibody targeting of individual Notch receptors. Nature 464, 1052-‐57.
Xicoy, B., Ribera, J.M., Oriol, A., Sanz, M.A., Abella, E., Tormo, M., del Potro, E., Bueno, J., Grande, C., and Fernández-‐Calvo, J. (2006). Significado pronóstico de los subtipos inmunológicos de la leucemia aguda linfoblástica T del adulto. Estudio de 81 pacientes. Med Clin (Barc) 126, 41-‐46.
Xu, H., Cheng, C., Devidas, M., Pei, D., Fan, Y., Yang, W., Neale, G., Scheet, P., Burchard, E.G., et al. (2012). ARID5B genetic polymorphisms contribute to racial disparities in the incidence and treatment outcome of childhood acute lymphoblastic leukemia. J Clin Oncol 30, 751-‐57.
Xu, H., Yang, W., Pérez-‐Andreu, V., Devidas, M., Fan, Y., Cheng, C., Pei, D., Scheet, P., Burchard, E.G., et al. (2013). Novel susceptibility variants at 10p12.31-‐12.2 for childhood acute lymphoblastic leukemia in ethnically diverse populations. J Natl Cancer Inst 105, 733-‐742.
Yanada, M., Sugiura, I., Takeuchi, J., Akiyama, H., Maruta, A., Ueda, Y., Usui, N., Yagasaki, F., Yujiri, T., et al. (2008). Prospective monitoring of BCR-‐ABL1 transcript levels in patients with Philadelphia chromosome-‐positive acute lymphoblastic leukaemia undergoing imatinib-‐combined chemotherapy. Br J Haematol 143, 503-‐510.
Yanada, M., Takeuchi, J., Sugiura, I., Akiyama, H., Usui, N., Yagasaki, F., Kobayashi, T., Ueda, Y., Takeuchi, M., and Miyawaki, S. (2006). High complete remission rate and promising outcome by combination of Imatinib and chemotherapy for newly diagnosed BCR-‐ABL-‐-‐Positive acute lymphoblastic leukemia: a phase II study by the Japan adult leukemia study group. Journal of clinical oncology 24, 460-‐66.

Bibliografía
202
Yang, J.J., Cheng, C., Devidas, M., Cao, X., Campana, D., Yang, W., Fan, Y., Neale, G., Cox, N., et al. (2012). Genome-‐wide association study identifies germline polymorphisms associated with relapse of childhood acute lymphoblastic leukemia. Blood 120, 4197-‐4204.
Zaremba, C.M., Oliver, D., Cavalier, M., Fuda, F., Karandikar, N.J., and Chen, W. (2012). Distinct immunophenotype of early T-‐cell progenitors in T lymphoblastic leukemia/lymphoma may predict FMS-‐like tyrosine kinase 3 mutations. Ann Diagn Pathol 16, 16-‐20.
Zhang, J., Ding, L., Holmfeldt, L., Wu, G., Heatley, S.L., Payne-‐Turner, D., Easton, J., Chen, X., Wang, J., et al. (2012). The genetic basis of early T-‐cell precursor acute lymphoblastic leukaemia. Nature 481, 157-‐163.
Zuurbier, L., Homminga, I., Calvert, V., te Winkel, M.L., Buijs-‐Gladdines, J.G., Kooi, C., Smits, W.K., Sonneveld, E., Veerman, A.J., et al. (2010). NOTCH1 and/or FBXW7 mutations predict for initial good prednisone response but not for improved outcome in pediatric T-‐cell acute lymphoblastic leukemia patients treated on DCOG or COALL protocols. Leukemia 24, 2014-‐022.
Zwaan, C.M., Rizzari, C., Mechinaud, F., Lancaster, D.L., Lehrnbecher, T., van der Velden, V.H., Beverloo, B.B., den Boer, M.L., Pieters, R., et al. (2013). Dasatinib in children and adolescents with relapsed or refractory leukemia: results of the CA180-‐018 phase I dose-‐escalation study of the Innovative Therapies for Children with Cancer Consortium. J Clin Oncol 31, 2460-‐68.