biología celular y sistémica 2011 teórico : aparato de … · 2014-08-13 · 14 proteasas y...
TRANSCRIPT

Biología Celular y Sistémica 2011
Teórico : Aparato de Golgi, Lisosomas y
Peroxisomas.
Dr. Roberto Najle
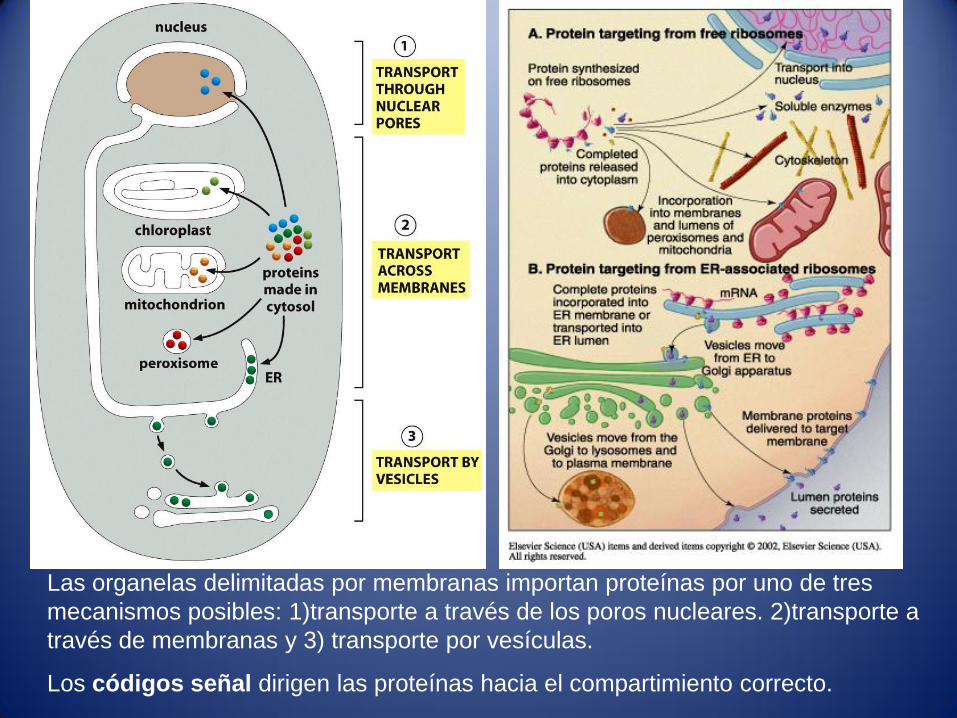
Las organelas delimitadas por membranas importan proteínas por uno de tres
mecanismos posibles: 1)transporte a través de los poros nucleares. 2)transporte a
través de membranas y 3) transporte por vesículas.
Los códigos señal dirigen las proteínas hacia el compartimiento correcto.
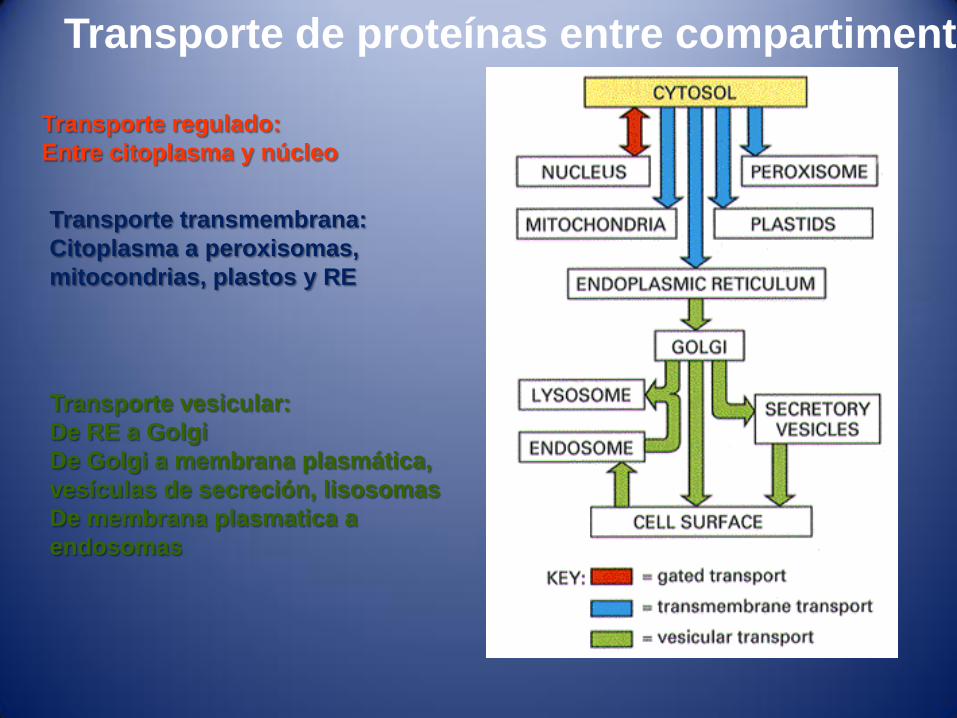
Transporte de proteínas entre compartimentos
Transporte regulado:
Entre citoplasma y núcleo
Transporte transmembrana:
Citoplasma a peroxisomas,
mitocondrias, plastos y RE
Transporte vesicular:
De RE a Golgi
De Golgi a membrana plasmática,
vesículas de secreción, lisosomas
De membrana plasmatica a
endosomas
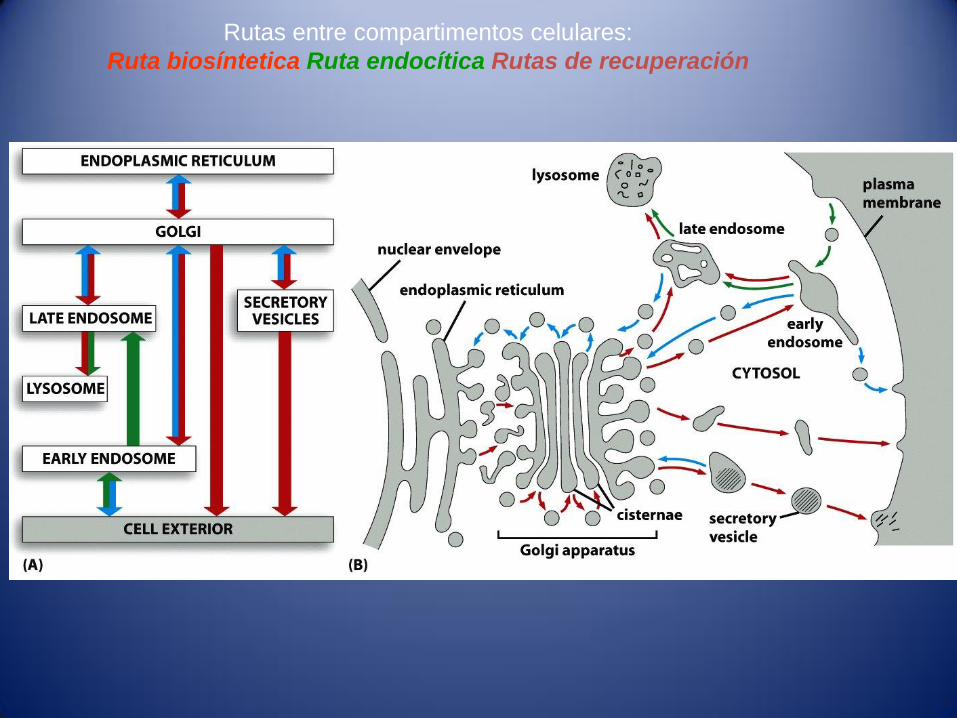
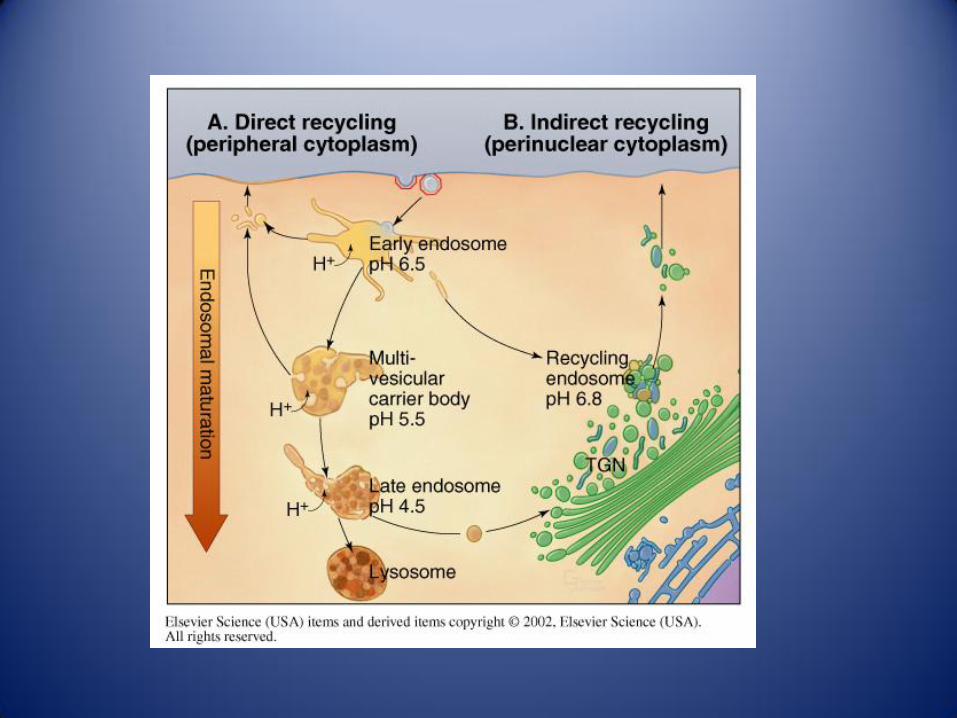
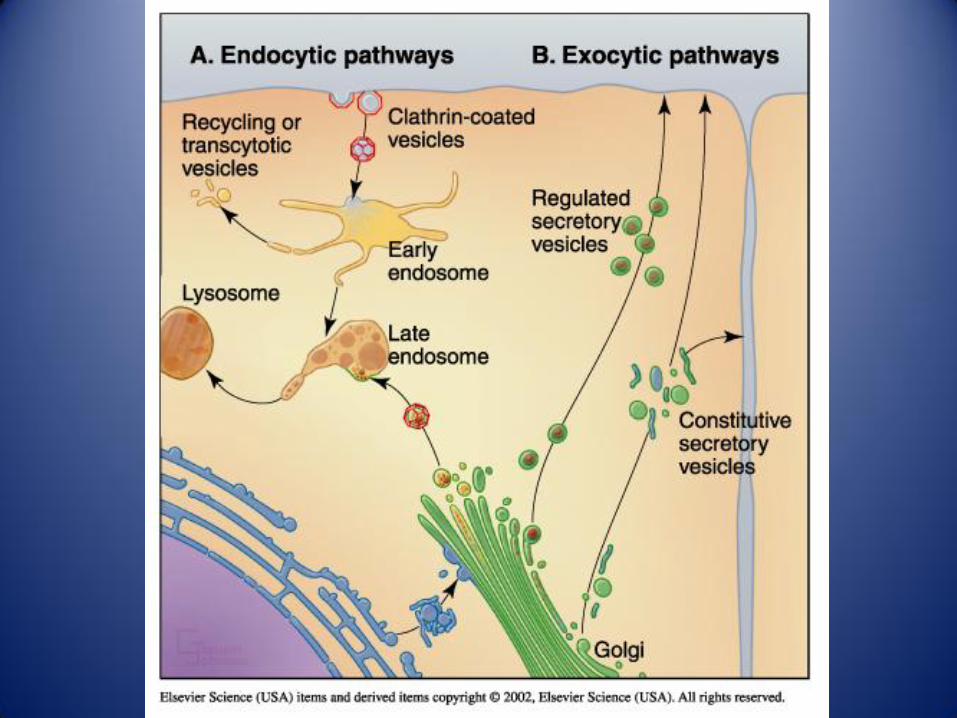
Rutas entre compartimentos celulares:
Ruta biosíntetica Ruta endocítica Rutas de recuperación
Vesículas de transición llegando desde el RE
Red cis-Golgi(RCG)
Red trans-Golgi (RTG)
Cisternas intermedias
Vesículas de transporte
partiendo de la red
trnasGolgi RTG
Red trans-Golgi (RTG)
Red cis-Golgi (RCG)
Envoltura nuclear
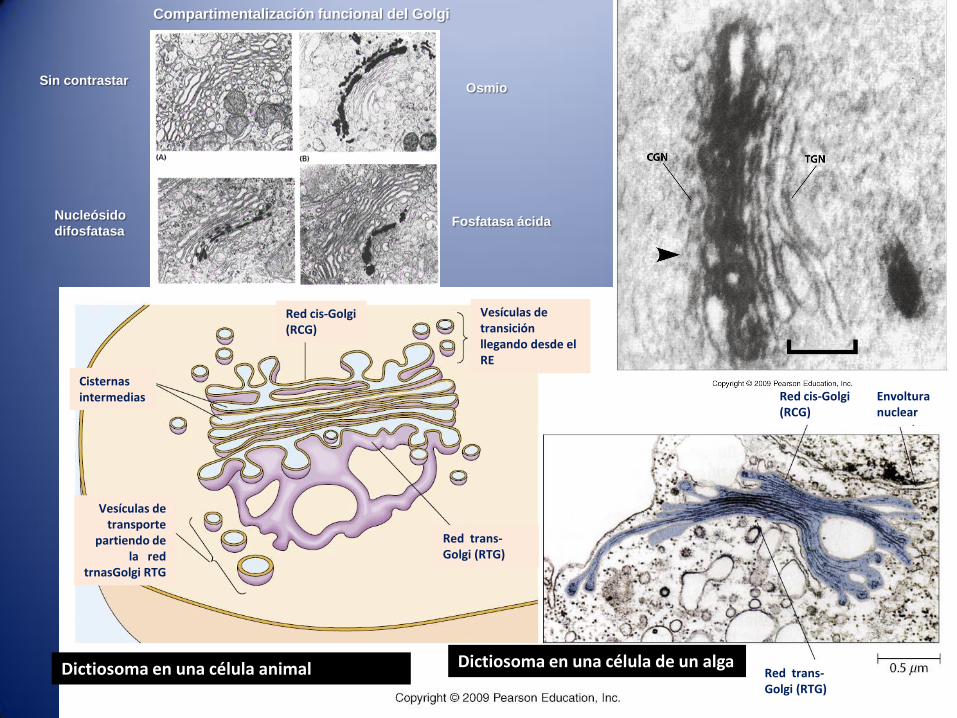
Dictiosoma en una célula animal Dictiosoma en una célula de un alga
Compartimentalización funcional del Golgi
Sin contrastarOsmio
Nucleósido
difosfatasaFosfatasa ácida

La cara cis (o de formacion): mira
hacia los elementos de
transición del RE.
El compartimento del Golgi mas
próximo a los elementos de
transición es una red tubular
denominada red cis- Golgi
(RCG).
cara opuesta La cara trans (o de
maduración) los compartimentos
de este lado forman red de
túbulos red trans-Golgi (RTG).
Las cisternas cis, intermedias y trans son bioquímica y funcionalmente
diferentes, cada compartimiento tiene su propia dotación de enzimas

Tanto el RE como el complejo de Golgi están rodeados de multitud de
vesículas de transporte, que portan lípidos y proteínas desde los elementos
de transición del RE al complejo de Golgi, entre las cisternas del propio
dictiosoma y desde el complejo de Golgi hasta varios destinos celulares(como
gránulos de secreción, los endosomas, los lisosomas y membrana
plasmática).
La mayoría de las vesículas implicadas en la transferencia de lípidos y
proteínas , se consideran vesículas cubiertas (Capas de proteínas), son
responsables de encurvar la membrana, facilitando la vesiculacion y
desaparecen de la vesícula antes de que esta se fusione con la membrana de
destino.
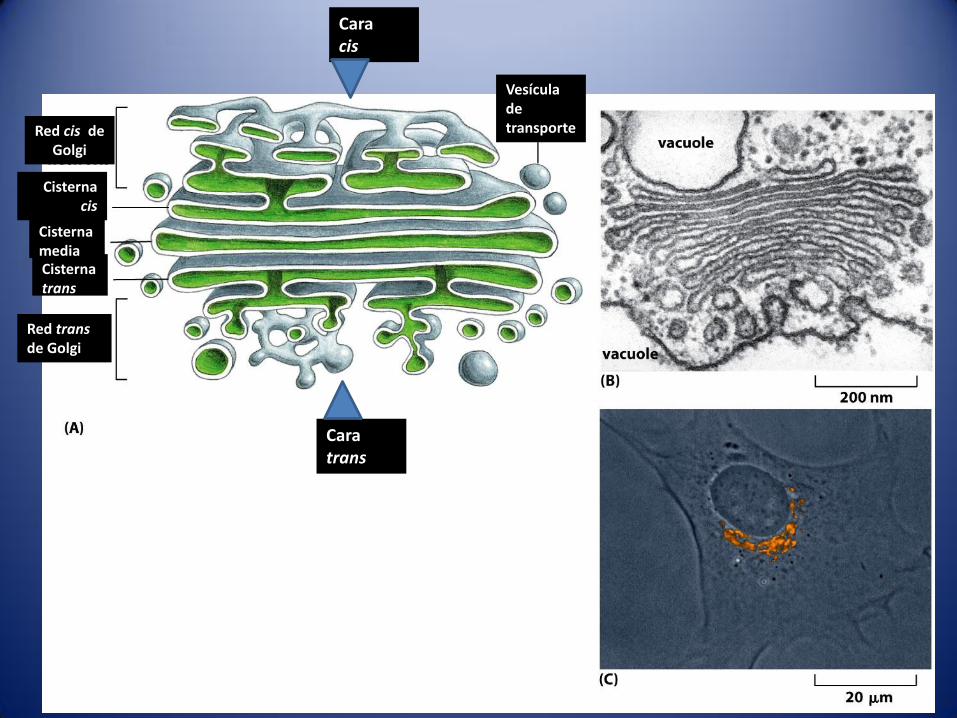
Red cis de Golgi
Cisterna cis
Cara cis
Vesícula de transporte
Red transde Golgi
Cisterna trans
Cisterna media
Cara trans

MODELO DE MADURACION DE CISTERNAMODELO DE TRANSPORTE VESICULAR
cisterna
RCG RTG
Grupo
vesícular
tubular

Papel del RE y el complejo de Golgi en la glicosilación de proteínas
Glicosilación : es la adición de cadenas laterales de hidratos de carbono, a
residuos aminoacílicos específicos de proteínas , para formar las Glicoproteínas.
Existen dos tipos de glicosilaciones:
a) La glicosilacion asociada a N: adicion de una unidad especifica de
oligosacarido al grupo amino terminal de ciertos residuos de aspargina
b) La glicosilacion asociada a O: adicion del oligosacarido al grupo hidroxilo de
residuos de serinas o treoninas.
La primera etapa es la glicosilacion central se verifica en el RE.
Oligosacarido: dos unidades de GlcNAc, nueve de manosa y tres de glucosa.


•Biosíntesis del oligosacarido
central para la glicosilacion en N
de residuos de aspargina .
•Procesamiento inicial del
oligosacarido central.
•Identificación y eliminación de
proteínas mal plegadas.
•Unión de N-acetilgalactosamina
a serina o treonina.
•Primera etapa en la fosforilacion
de proteínas lisosomales.
•Adición de galactosa.
•Adición de ácido siálico.
•Adición de acido sialico.
•Sulfatación de tirosina.
•Eliminación de manosa.•Segunda etapa en la fosforilacion
de proteínas lisosomales.
•Eliminación de manosa
•Unión de N-acetilglucosamina
RCG
RTG
Mig
ració
n a
tra
vé
s d
el C
om
ple
jo d
e G
olg
i


Tipos de proteínas Ejemplos Sitio de síntesis
Proteínas séricas Prot. Matriz extcel. Hormonas peptídicas Enzimas digestivas Proteinas de la leche
Albúmina, transferrina Lipoproteínas Inmunoglobulinas Colágeno, fibronectina Proteoglicanos Insulina, glucagón Endorfinas, encefalinas Tripsina, amilasa, ribonucleasa Caseina, lactoalbúmina
Hígado Linfocitos fibroblastos pancreas cel.neurosecret. pancreas gland.mamarias
Clases de proteínas secretorias en mamíferos
Tipos de proteínas Ejemplos Sitio de síntesis
Proteínas séricas Prot. Matriz extcel. Hormonas peptídicas Enzimas digestivas Proteinas de la leche
Albúmina, transferrina Lipoproteínas Inmunoglobulinas Colágeno, fibronectina Proteoglicanos Insulina, glucagón Endorfinas, encefalinas Tripsina, amilasa, ribonucleasa Caseina, lactoalbúmina
Hígado Linfocitos fibroblastos pancreas cel.neurosecret. pancreas gland.mamarias
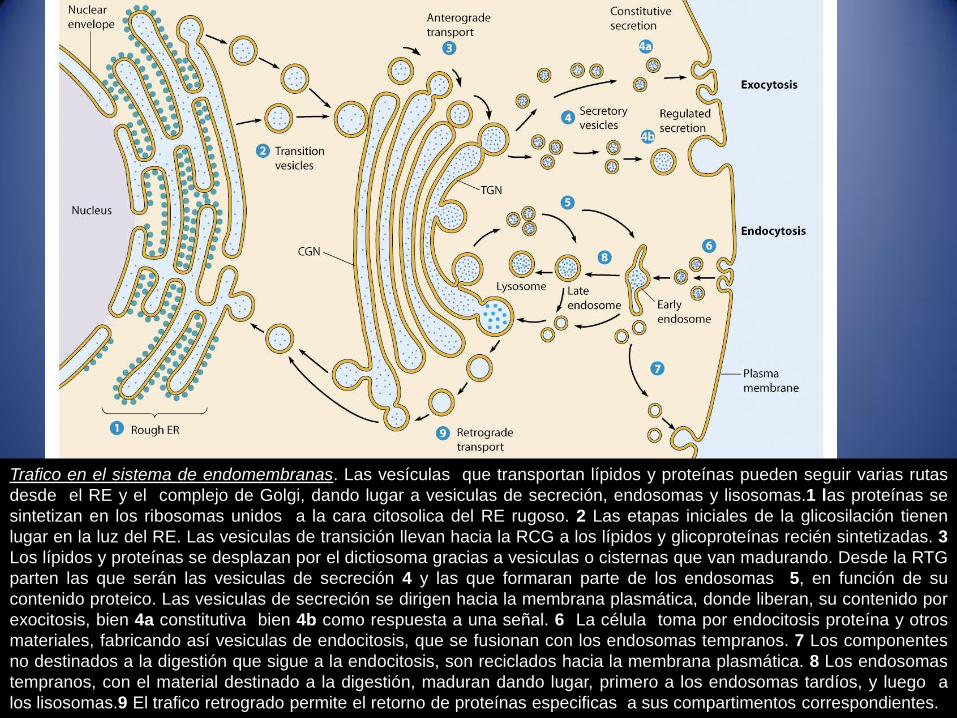
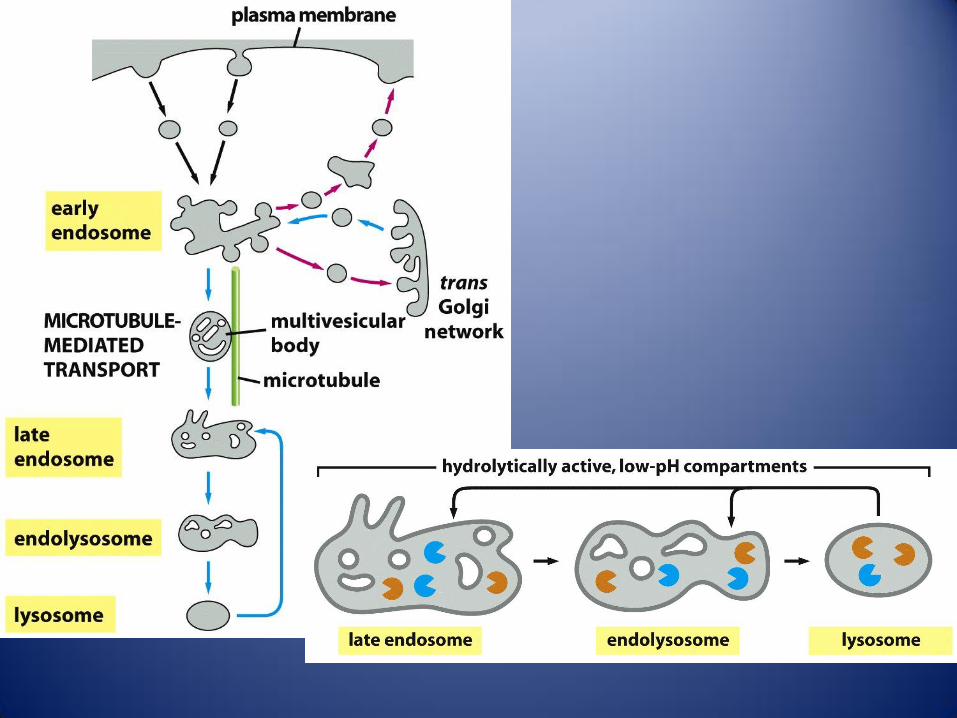
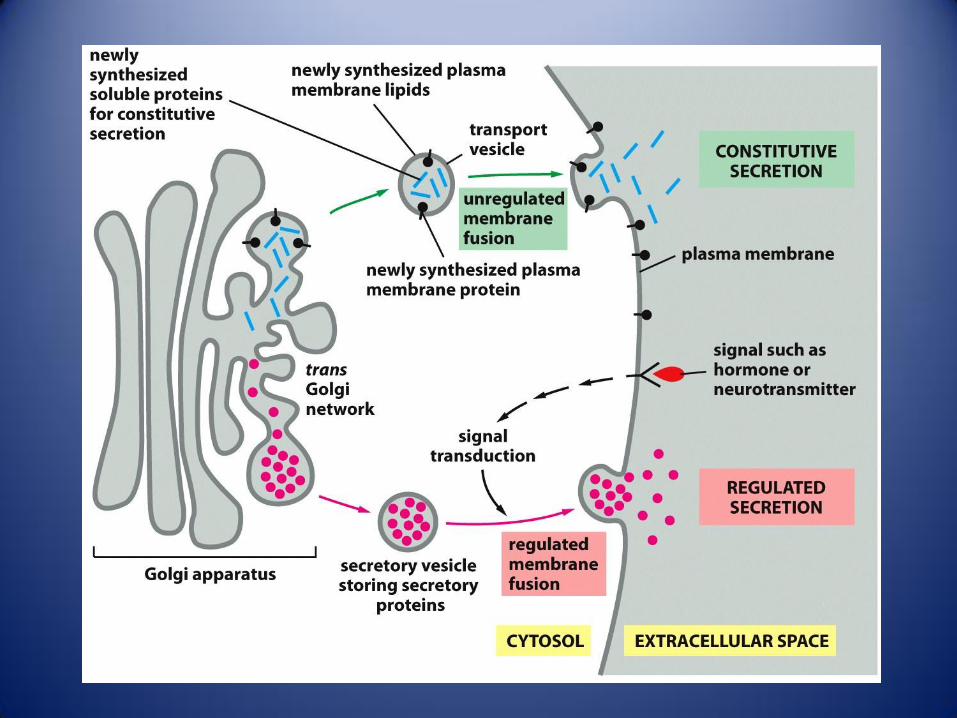
Trafico en el sistema de endomembranas. Las vesículas que transportan lípidos y proteínas pueden seguir varias rutas
desde el RE y el complejo de Golgi, dando lugar a vesiculas de secreción, endosomas y lisosomas.1 las proteínas se
sintetizan en los ribosomas unidos a la cara citosolica del RE rugoso. 2 Las etapas iniciales de la glicosilación tienen
lugar en la luz del RE. Las vesiculas de transición llevan hacia la RCG a los lípidos y glicoproteínas recién sintetizadas. 3
Los lípidos y proteínas se desplazan por el dictiosoma gracias a vesiculas o cisternas que van madurando. Desde la RTG
parten las que serán las vesiculas de secreción 4 y las que formaran parte de los endosomas 5, en función de su
contenido proteico. Las vesiculas de secreción se dirigen hacia la membrana plasmática, donde liberan, su contenido por
exocitosis, bien 4a constitutiva bien 4b como respuesta a una señal. 6 La célula toma por endocitosis proteína y otros
materiales, fabricando así vesiculas de endocitosis, que se fusionan con los endosomas tempranos. 7 Los componentes
no destinados a la digestión que sigue a la endocitosis, son reciclados hacia la membrana plasmática. 8 Los endosomas
tempranos, con el material destinado a la digestión, maduran dando lugar, primero a los endosomas tardíos, y luego a
los lisosomas.9 El trafico retrogrado permite el retorno de proteínas especificas a sus compartimentos correspondientes.

Los Lisosomas son organelas acidas que contienen una batería de enzimas
degradativas.
Las plantas y las células fungicidas contienen la mayor parte de los organelas
encontrados en una célula animal pero carecen de lisosomas. En lugar,
contienen una vacuola central grande que favorece muchas de las funciones
de lisosoma.
LISOSOMAS
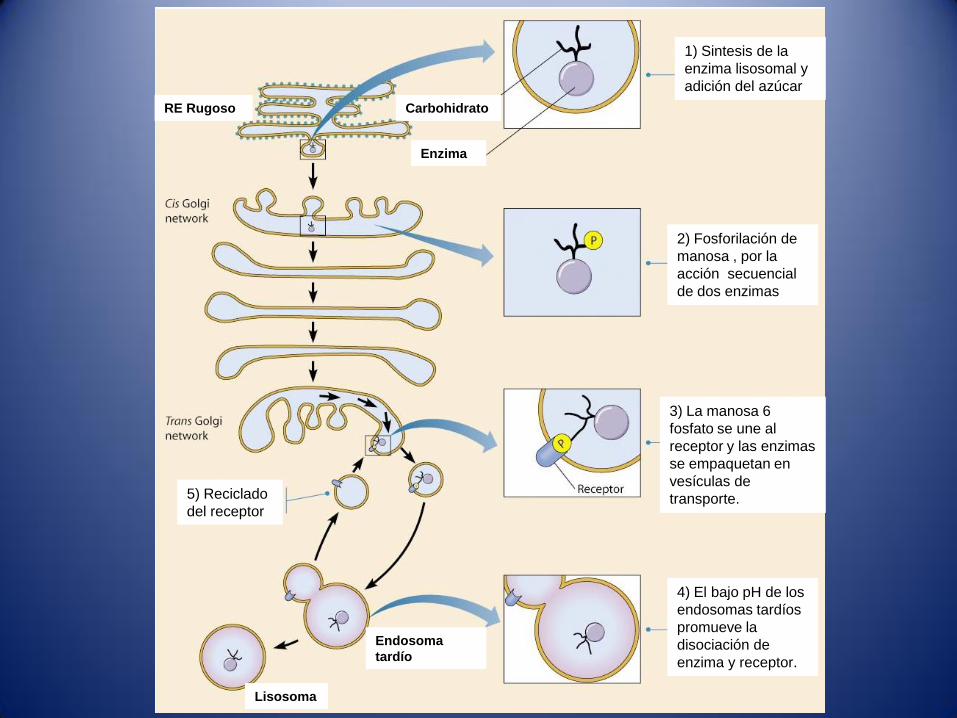
1) Sintesis de la
enzima lisosomal y
adición del azúcar
2) Fosforilación de
manosa , por la
acción secuencial
de dos enzimas
3) La manosa 6
fosfato se une al
receptor y las enzimas
se empaquetan en
vesículas de
transporte.
4) El bajo pH de los
endosomas tardíos
promueve la
disociación de
enzima y receptor.
5) Reciclado
del receptor
Endosoma
tardío
Lisosoma
RE Rugoso Carbohidrato
Enzima

1) En la superficie interna RTG (pH 6,4) hay receptores manosa 6 fosfato el
pH favorece la unión de enzimas solubles lisosomales.
2) Los complejos receptor-ligando se empaquetan en vesículas de
transporte y se distribuyen hacia los endosomas.
3) Las enzimas lisosomales necesarias para la degradación del material
tomado por endocitosis se transportan desde RTG hasta los endosomas
tardíos(pH5,5).
4) Los endosomas tardíos son la evolución de los endosomas tempranos
formados por vesículas originadas en la RTG y en la membrana
plasmática.
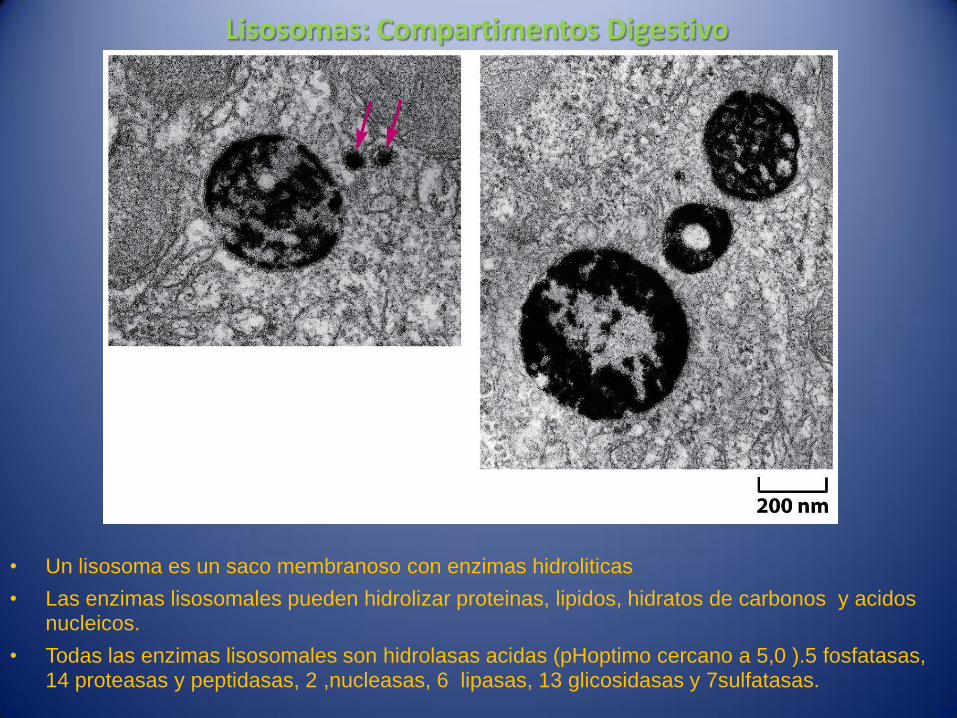
Lisosomas: Compartimentos Digestivo
• Un lisosoma es un saco membranoso con enzimas hidroliticas
• Las enzimas lisosomales pueden hidrolizar proteinas, lipidos, hidratos de carbonos y acidos nucleicos.
• Todas las enzimas lisosomales son hidrolasas acidas (pHoptimo cercano a 5,0 ).5 fosfatasas, 14 proteasas y peptidasas, 2 ,nucleasas, 6 lipasas, 13 glicosidasas y 7sulfatasas.

Enfermedades lisosomales•Exceso de actividad lítica debido al aumento y a la falta de control de la autofagia.
•Daño y cambios en la permeabilidad de la membrana del lisosoma
•Excesiva liberación de hidrolasas hacia el exterior de la célula. las enzimas se
sintetizan en forma normal , pero en lugar de ser dirigidas hacia los lisosomas , son
segregadas hacia el medio extracelular.
•Actividad lítica inadecuada: en Silicosis y ASBETOSIS , GOTA
•Silicosis o enfermedad de los mineros se debe a la captación de fibras de silice por las
celulas fagocitarias de los pulmones. Las fibras quedan encerradas en los lisosomas ,
pero no pueden degradarse, provocando fugas en la membrana lisosomica ,derrame del
contenido de enzimas dentro de la célula y daño al tejido pulmonar.
•Ocurre algo similar cuando las células carroñeras captan fibras de asbesto y ocasionan
la enfermedad llamada asbestosis, ambos padecimientos son debilitantes e incluso
pueden causar muerte.

•Enfermedades de almacenaje debidas a anormalidades genéticas de los lisosomas: se
conocen unas 40 enfermedades lisosomales de acumulación ,de carácter hereditario ,
cada una de ellas caracterizada por la acumulación anómala de una o de múltiples
sustancias, habitualmente polisacáridos o lípidos, que en condiciones normales son
catalizados por hidrolasas lisosomales.
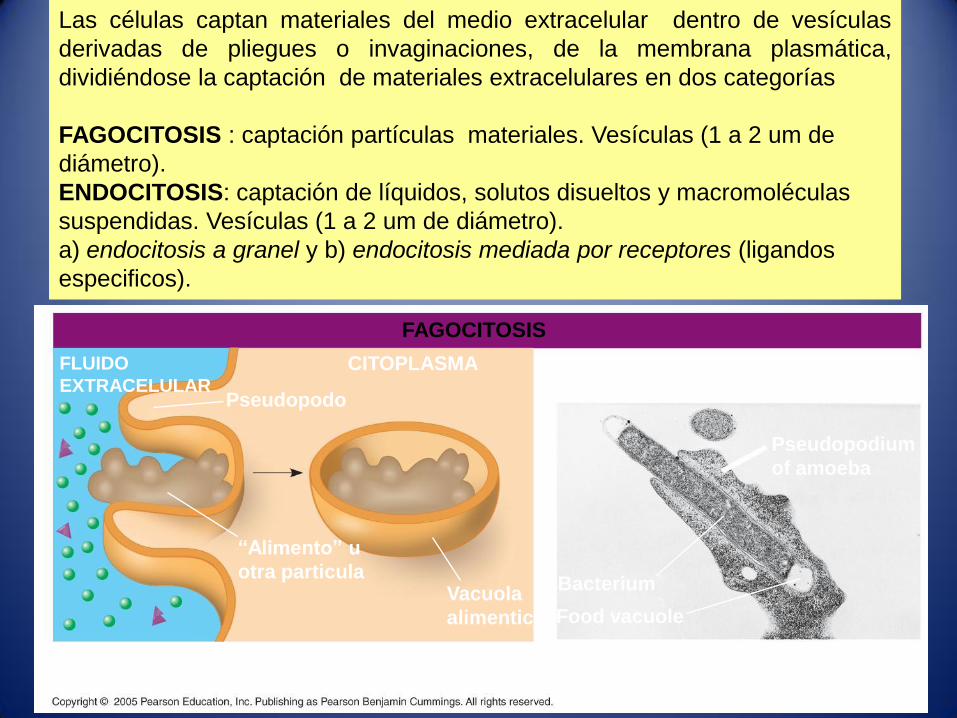
Las células captan materiales del medio extracelular dentro de vesículas
derivadas de pliegues o invaginaciones, de la membrana plasmática,
dividiéndose la captación de materiales extracelulares en dos categorías
FAGOCITOSIS : captación partículas materiales. Vesículas (1 a 2 um de
diámetro).
ENDOCITOSIS: captación de líquidos, solutos disueltos y macromoléculas
suspendidas. Vesículas (1 a 2 um de diámetro).
a) endocitosis a granel y b) endocitosis mediada por receptores (ligandos
especificos).
CITOPLASMA
Pseudopodo
“Alimento” u
otra particula
FLUIDO
EXTRACELULAR
Bacterium
Food vacuole
An amoeba engulfing a bacterium via
phagocytosis (TEM)
Pseudopodium
of amoeba
1 µm
Vacuola
alimenticia
FAGOCITOSIS

Plasmamembrane Pinocytosis
vesicles forming
(arrows) in a cell
lining a small
blood vessel
(TEM).
0.5 µm
Vesicle
ENDOCITOSIS a GRANEL
Receptor
ENDOCITOSIS MEDIADA POR RECEPTORES
Ligando
Coated
pit
Coated
vesicle
Coat protein
Coat
protein
Plasma
membrane
0.25 µm
A coated pit
and a coated
vesicle formed
during
receptor-
mediated
endocytosis
(TEMs).

Los lisosomas son necesarios para
actividades celulares tan variadas como la
nutrición, defensa, reciclado de
componentes celulares y diferenciación.
Aunque la digestión es, casi siempre,
intracelular, en algunos casos las enzimas
lisosomales son segregadas por exocitosis.
Con respecto al origen del material si
proviene del exterior se habla de lisosomas
heterofágicos, si es de origen intracelular se
denominan lisosomas autofágicos.
ial
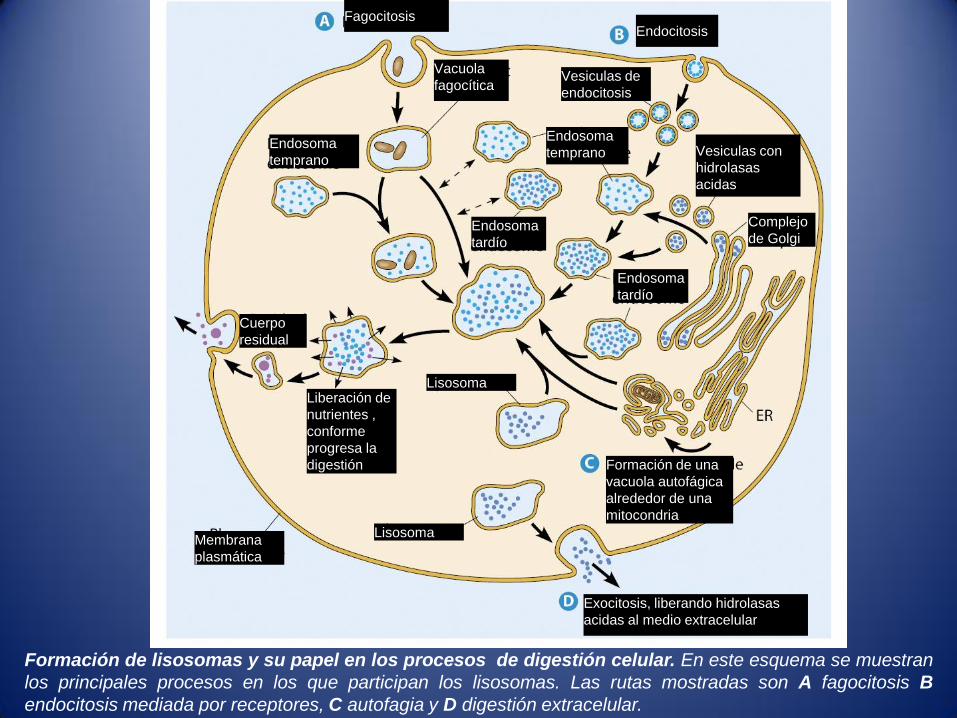
Formación de lisosomas y su papel en los procesos de digestión celular. En este esquema se muestran
los principales procesos en los que participan los lisosomas. Las rutas mostradas son A fagocitosis B
endocitosis mediada por receptores, C autofagia y D digestión extracelular.
Vesiculas con
hidrolasas
acidas
Endosoma
temprano
Endosoma
temprano
Liberación de
nutrientes ,
conforme
progresa la
digestión
Vacuola
fagocítica
Cuerpo
residual
Endosoma
tardío
Lisosoma
Lisosoma
Endosoma
tardío
Formación de una
vacuola autofágica
alrededor de una
mitocondria
Complejo
de Golgi
Vesiculas de
endocitosis
EndocitosisFagocitosis
Exocitosis, liberando hidrolasas
acidas al medio extracelular
Membrana
plasmática

Peroxisomas: Oxidación
• Peroxisomas son compartimentos metabólicos especializados rodeados deuna simple membrana. Aparecen en células de vegetales superiores yanimales así como hongos, protozoos y algas. En los animales , losperoxisomas se encuentran en la mayoría de las células , pero sonespecialmente abundantes en la del hígado y riñón.
• Además de su papel en la detoxificación del peróxido de hidrogeno, losperoxisomas animales cumplen otras funciones, incluyendo la neutralizaciónde los compuestos peligrosos (como el metanol, el etanol, el formol y elformaldehido ) y el catabolismo de sustancias extrañas (tales como los D-aminoacidos).
• Los peroxisomas animales intervienen también en la degradación oxidativade los ácidos grasos , que son componentes de los traiacilgliceroles, losfosfolípidos y los glicolípidos.
• Peroxisomas son pequeños organelas que contienen enzimas que oxidavarios compuestos orgánicos sin la producción de ATP. Los subproductos de laoxidación se utilizan dentro reacciones biosintéticas.
• Peroxisomas producen peróxido de hidrogeno y convierte a este en agua

Vesículas importantes que se encuentran en el citoplasmade las células eucarióticas.
Miden: 0,5 a 1um de diámetro y presentan en algunascélulas un nucleoide cristalino
Urato de oxidasa, d-aminoácido-oxidasa y oxidasa del ácidoa-hidroxílico oxidan los sutratos y reducen el oxígeno aperóxido de hidrógeno y la CATALASA lo descompone enagua y oxígeno
Peroxisomas

Peroxisomas degradan los ácidos grasos y compuestos
tóxicos. Todas las células animales (excepto eritrocitos) y
muchas células de la planta contienen peroxisomas, una
clase de organelas áspero esféricos, 0.2-1.0 m de diámetro .
Peroxisomas contienen varias enzimas-oxidasas- que
utilizan el oxígeno molecular para oxidar sustancias
orgánicas, en el proceso se forma peróxido de hidrógeno
(H2O2), una sustancia corrosiva.
Peroxisomas también contienen cantidades de copias de
la enzima catalasa que degrada el peróxido de hidrógeno
para rendir el agua y el oxígeno:
2 H2O2 → 2 H2O + O2
Al contrario de la oxidación de ácidos grasos en
mitocondrias, que produce el CO2 y esta acoplada a la
generación de ATP, oxidación peroxisomal de ácidos grasos
produce grupos acetilos y no se ligan a la formación del
ATP

FUNCIONES DEL PEROXISOMA•Catabolismo de ácidos grasos de cadena larga•Metabolismo de radicales libres de oxígeno•Síntesis de lípidos de colesterol y éter•Formación de ácidos biliares•Catabolismo de purinas, prostaglandinas,leucotrienos•Detoxificación de alcohol en el hígado•Metabolismo de estradiol

• Algunas enfermedades congénitas humanas se asocian con laausencia de peroxisomas y/o con la disfunción de sus enzimascomo es:
• Adrenoleucodistrofia neonatal (NALD)alteracion ligada al sexosolo en los varones alteraciones neurologicas profundas yfinalmente la muerte. falla en la b-oxidación de los ácidosgrasos, almacenamiento anormal de lípidos en cerebro, médula,glándulas adrenales)
• Diversas sustancias químicas (drogas, contaminantes
industriales) inducen una proliferación marcada de peroxisomas.
• El estradiol parece tener un efecto depresivo sobre losperoxisomas (por lo menos en hepatocitos de pez).