6.3. grados comerciales del glicerol -...
TRANSCRIPT

Biogl icerol como Mater ia Prima para la Obtención de Productos de Valor Agregado
de poliéster/polioles (14%), alimentos (11%), triacetinas (10%), resinas alquídicas (8%) tabaco (6%), detergentes (2%), celofán (2%) y explosivos (2%), la porción restante (11%' es usada en la manufactura de lacas, barnices, tintas, adhesivos, plásticos sintéticos, celulosí regenerada y otros usos industriales. Además el uso del glicerol también está aumentando como sustituto del propilenglicol [4].
El precio de la glicerina refinada ha permanecido casi sin cambios durante los últimos años mientras que la venta de glicerina cruda por parte de los productores de biodiesel ha genera do una vertiginosa caída del precio en la glicerina de esta calidad. El suministro de glicerin; cruda casi se ha duplicado, mientras que la demanda del producto se ha mantenido en grai medida sin cambios. Este exceso de oferta y la limitada demanda ha causado que los precio: de la glicerina se mantengan en un nivel muy bajo. Los precios de la glicerina cruda cayeroi a su punto más bajo a partir del 2006, debido a que la sobreoferta obligó a los productor de biodiesel a que recibieran precios de venta de hasta 2 centavos por libra o incluso meno res por el subproducto crudo, pero desde entonces se ha notado un alza gradual y constanti en los precios del mercado debido a un aumento en la demanda de la glicerina cruda, y y: para mediados del 2007 se alcanzaron precios de venta entre 6 y 10 centavos de dólar po libra de glicerina sin refinar. Por otro lado, el mercado y los precios de la glicerina refinad: se han mantenido fuertes, registrando entre 30 y 40 centavos por libra, dependiendo de 1 calidad, grado y pureza.
6.3. Grados comerciales del glicerol
Comercialmente se conocen varios grados de glicerol. La glicerina grado "USP" (U.S. Pha macopeia) tiene un contenido de glicerol superior al 95% peso. La glicerina grado "CP" < glicerina "químicamente pura" generalmente tiene la misma calidad que la glicerina gradi USP, pero este término es considerado genérico en los Estados Unidos debido a la falta d certificación por parte de organismos oficiales. Aunque en Europa la glicerina USP con firma las especificaciones para el Chemically Pure Glycerolbs 2625:1979, publicado pori: British Standards Institution, la glicerina "grado alimenticio" cumple con los requerimiento establecidos para glicerina en el Food Chemicals Codex del Committee on Food Protection Oj the NationalKesearch Councilde los Estados Unidos y tiene características similares al estánd USP. Mientras que en Europa la glicerina para ser usada en productos alimenticios de cumplir con las Council Directive 78/663/EEC, que especifica los estándares para agente emulsificantes, estabilizantes, espesantes y gelificantes para el uso en alimentos.
La glicerina de "alta gravedad" confirma las especificaciones estándar establecidas por 1 norma D-1257 de la A.merican Society for Testing and Materials (ASTM). Este grado debe con tener no menos de 987% peso de glicerol, pero es comúnmente proporcionado con un concentración superior a 99.0% peso. Esta misma norma (ASTM D-1257) es reconocida ei
^ m | 1061 mrnmmmmmmmmmmmmamma^mà

Avances Invesrigativos en la Producción de Biocombiistibles
Europa para definir el grado de glicerina para propósitos comerciales. La glicerina "grado dinamita" reúne las mismas especificaciones de la glicerina de alta gravedad, excepto el co-lor. En Europa la glicerina grado dinamita es definida por la especificación 21D establecida por la Nobel Explosives Company Etd. Además la British Standards Institution también tiene una especificación estándar para este grado de glicerina, llamado British Standard. Specflcation for Dynamite Glycerot, BS 2624: 1979. En algunas ocasiones se utilizan términos genéricos como "glicerina de saponificación 88%" y "lejía de jabón 80%" para designar diferentes grados de glicerinas crudas [7].
La habilidad para generar valor agregado a las corrientes de producto es un factor im-portante para la rentabilidad del proceso de producción de biodiesel y en general para los procesos de transformación química. Investigadores de todo el mundo se esfuerzan para desarrollar nuevos usos y tecnologías para el aprovechamiento de la glicerina cruda. Una vez estas tecnologías puedan ser comercializadas tienen el potencial para mejorar la economía de la producción del biodiesel. Es así como la gran diferencia de precios entre la glicerina cruda y refinada, sumada a la versatilidad química del glicerol ha creado una gran cantidad de investigación en un esfuerzo por encontrar soluciones prácticas y usos alternativos de este abundante biomaterial. Esto ha llevado a encontrar varias rutas de transformación de glicerol hacia productos de valor agregado como son los de oxidación, hidrogenólisis a gli-coles, eterificación a poligliceroles, pirólisis y la gasificación a hidrógeno o gases de síntesis, además de las rutas de transformación biotecnológicas con bacterias o enzimas, como se presenta en las siguientes secciones.
6.4. Oxidación con catalizadores metálicos
La oxidación de glicerol con catalizadores metálicos se lleva a cabo por medio del meca-nismo de deshidrogenación oxidativa sobre la superficie del metal [8], en donde el primer paso es la deshidrogenación del alcohol, seguida por la oxidación del intermediario formado [9], Un punto clave del proceso de oxidación es el control sobre la selectividad del sistema reactivo debido a la potencial complejidad de la distribución de productos [10-11], como muestra la Figura 6.1. Entre los derivados oxigenados que se pueden encontrar del glicerol (GLY), están el ácido glicérico (GUYAC), dihidroxiacetona (DHA), ácido hidroxipirúvi-co (HYZMC), ácido tartrónico (TARAQ, ácido mesoxálico (MESOXAC) , ácido oxálico (OXALAQ, además de algunos intermediarios como gliceraldehído (GLYAE), ácido gli-cólico (GLYCAQ y ácido glioxílico (GLYOXAQ.
1 1 0 7 1 «

Btogiiceroi como Maten; ' Prima pa r a l a O b ' c n n o n di- P j e Valor Agregado
Figura 6.1. Posibles productos para la oxidación de glicerol
Los catalizadores metálicos más estudiados son paladio (Pd), platino (Pt) y oro (Au), aun-que la principal desventaja de utilizar los dos primeros, Pd y Pt, es su desactivación con el incremento del tiempo de reacción [9], Para mejorar la actividad, selectividad y estabilidad se usan promotores con deposiciones de átomos p-electrón, particularmente átomos de metales pesados de los grupos IV (plomo, Pb) y V (bismuto, Bi) [12], sobre todo en Pt y Au para reacciones redox, lo que permite prevenir la sobre-oxidación de los productos en la su-perficie del metal, evitando la degradación del producto hasta la oxidación total a dióxido de carbono, además el promotor también favorece la oxidación de alcoholes secundarios. Los alcoholes primarios en ausencia de Bi a pH básico se oxidan a ácidos carboxílicos (GLYAC, TARAC y H\TAC vía DHA), los alcoholes secundarios son selectivamente oxidados en el catalizador metal de Pt con Bi como promotor a pH ácido (DHA, HYPAC vía GLYACi MESAQ [8, 9],
Investigadores como Gallezot [12-15, 26], Hutchings [15-17], Prati [18-25], Claus [9,27] y Davis [28-30], han estudiado la oxidación selectiva de glicerol sobre catalizadores mono-o bi- metálicos de Pd, Pt y Au, con presencia o no de promotores usando oxígeno como agente oxidante.
Los trabajos realizados por Gallezot el. al., muestran que para un pH básico se obtiene GLYAC y TARAC, para un pH poco ácido, HITMCvía DHA y para pH ácido se obtiene DHA, HYTAC vía GLYACy MESAC [8, 12-15], Para los catalizadores de Pd y Pt se logra una conversión del 100% en los dos casos, con selectividades de 70% y 35% hacia GLYAC y HYTAC respectivamente. Además para el catalizador de Pt-Bi se logran selectividades de 83%, 74%, 37% y 39% hacia TARAC, HYPAC, DHAj MESOXAC respectivamente con
— ^ ^ L M J —

rices invest igamos en i a Producción de fíi »combustibles . - I I M I L — A É I — — — — — — — — ^ —
conversiones superiores al 75%, excepto para MESOXAC que fue del 53%. Otras pruebas realizadas con Pt y Pd soportado sobre carbón activado (CA) en presencia de Bi (Pt-Bi/CA o Pd-Bi/CA), muestran que para el caso de 5%Pd/CA a pH de 7, 9 y 11, las máximas se-lectividades hacia GLYAC son 30%, 55% y 77%, respectivamente. Menores selectividades son reportadas para el catalizador de 5%Pt/CA atribuida a un menor potencial redox del Pt [12]. La oxidación sobre 5%Pt-Bi/CA genera DHA a una selectividad de 50% y una conversión del 60%. Sin embargo, para mayor tiempo de contacto, las funciones del alcohol primario son lentamente oxidadas para producir HYPAC. Las condiciones óptimas para la conversión de GLYAC a TARAC y HYPAC [26] muestran un rendimiento de 61% a una conversión de 94% y un pH entre 10-11 sobre un catalizador de Pt y para el mismo pH el rendimiento es de 83% a un nivel de conversión del 90% sobre un catalizador de Pt-Bi, esto para TARAC. En cuanto al HYPAC, con el catalizador Pt-Bi se obtuvo un rendimiento de 64% a un nivel de conversión del 75% para un pH de entre 3-4. Por otro lado, para el cata-lizador de Au se encontró que las partículas de menor diámetro son las más activas, además la selectividad también es afectada por el tamaño de la partícula catalítica.
El catalizador de Au también fue estudiado por Hutchings et al. [10, 16-18] y Prati et al [11, 19-25], Cuando el Au es soportado sobre carbono (Au/C) es extremadamente selectivo para la reacción hacia GLYAC (>82%), además no se presentan productos C, y C, y la conversión es superior al 60% [10]. Hutchings et al comparó la selectividad y la conversión del GLY para diferentes relaciones de carga de catalizador a GLY, presiones de oxígeno y pH. A mayores cargas de catalizador es mayor la conversión de GLY, pero la selectividad no es igualmente beneficiada. Para sistemas a pH básico la selectividad hacia GLYAC au-menta con el pH, lo mismo ocurre cuando se aumenta la presión de oxígeno. Entre tanto Prati et al. evaluaron los catalizadores bimetalitos de Pd y Au, y éstos reportaron un buen grado de conversión de GLY. (100%) y una alta selectividad hacia el GLYAC (>45%). Los catalizadores metálicos fueron soportados sobre grafito inmovilizado y se encontró que la actividad de los catalizadores y la selectividad hacia GLYAC aumentan con la temperatura, además se recomienda una preparación con 1% fósforo (P)-metal/grafito, para aumentar la selectividad de GLYAC.
Zlaus et al, también reportan la oxidación de GLY sobre catalizadores de Au soportados so-are grafito, carbón activado y nanopartículas de carbono, siendo esta última la más activa [9, >7], Al igual que el resultado encontrado por Hutchings et al, se estableció que la selectividad lacia GLYAC es dependiente del tamaño de la nanopartícula, alcanzando una selectividad leí 75% para el tamaño óptimo de la nanopartícula. Por otro lado, un catalizador bimetálico /Vu-Pt/C genera un incremento tanto en la actividad del catalizador como en la selectividad hacia DHA.
Otras nanopartículas mono- y bi-metálicas de Au y Pd fueron evaluadas en la oxidación en
m 1109 |

^ u ^ i c e n ^ ^ u m ^ ^ a ^ n ^ P ^ m ^ ^ m ^ de Valor Anreeado
fase líquida de GLYpor Davis et al [28-30], A partir de medidas de la velocidad de oxidación de GUf encontraron que la mayor frecuencia de cambio (turnover frequency, TOF) la ex-hibe el catalizador monometálico de Au, mientras que la mayor selectividad hacia GIYAC es alcanzada por el Pd. Además la actividad y selectividad del catalizador bimetálico Au-Pd es dependiente de la cantidad de Au presente en la partícula, el TOF no es mayor que el del catalizador monometálico de Au, mientras que la selectividad hacia GEYAC sí lo es.
6.5. Reducción a glicoles
Los principales productos de la reducción de GUí son el 1,2- y 1,3-propilenglicol (12-PG- o 13-PG), el etilenglicol ETGEYj otros subproductos como ácido láctico (L.4CAQ, acetol (ACET), acroleína (ACRO), además de los productos de degradación como son propanol (PROPOH), metanol (METOH) , metano (MET) y dióxido de carbono (CO„) [31], Aunque la mayoría de los trabajos se centran en la producción del propilenglicol debido a su alta de-manda, con una producción superior a 1 billón de libras por año en Estados Unidos con un crecimiento anual en el mercado de 4% [31], La alta producción del propilenglicol es dada por su funcionalidad, este componente puede ser usado en resinas de poliéster insaturadas, en fluidos funcionales (anticongelantes, descongelantes y transferencia de calor), farmacéu-ticos, alimentos, cosméticos, detergentes líquidos, humectantes de tabaco, saborizantes y fragancias, cuidado personal, pinturas y alimento animal. Comercialmente las rutas para k producción de propilenglicol son la hidratación de óxido de propileno, derivado del propi-leno por dos procesos, la clorhidrina o hidroperóxido [32,33],
Varias patentes describen múltiples esquemas para la hidrogenación de glicerol a propilen-glicol [33-37], en donde se puede encontrar una gran variedad de catalizadores tales como cobre, zinc, rutenio, cobalto, magnesio, molibdeno, níquel, paladio y platino. Igualmente s cumple esta variedad para las condiciones de reacción en donde se pueden encontrar pre-siones mayores a 2000 y cercanas a 5000 psi, además de temperaturas entre 200 y 350 °C.
Shanks y Lahr [38,39], estudiaron las interacciones entre los reactantes y los catalizadores para los dos casos, dehidrogenación/hidrogenación, así como los efectos de pH, absorció competitiva, degradación de productos (como etilenglicol y propilenglicol), además de 1 influencia de la base utilizada como catalizador. Las condiciones de la reacción bajo la cuales trabajaron Shanks y Lahr [38], son de alta presión, temperatura media y alta relació de dilución (1450 psi, 205 °C y 10 %Peso solución de glicerol, respectivamente). Adema Rutenio (Ru) fue utilizado como catalizador soportado sobre carbón activado para cargas a sistema de 5% PesoRu/CA (1.5 mMRu).
Debido a la alta actividad del catalizador de Ru en la hidrogenólisis y en la subsecuent degradación de productos, mostró que la velocidad de degradación tanto para el etilengli-
I 110 I

Avances Inyesti^atiyos en la Producción de Biocombustibh
col como para el propilenglicol es independiente de la concentración inicial del glicol, sin embargo, el propilenglicol es menos competitivo por los sitios activos en el catalizador que el etilenglicol. Por otro lado, los mismos investigadores demostraron que la presencia de etilenglicol en la hidrogenólisis de glicerol reduce la velocidad de reacción, en contraste con el propilenglicol que tiene un mínimo efecto en la velocidad de reacción.
La Tabla 6.2 presenta una serie de datos recopilados desde diferentes fuentes para el proce-so de reducción de glicerol hacia glicoles. La selectividad hacia propilenglicol no cambia con el pH, a diferencia del etilenglicol que además se ve favorecida bajo condiciones menos bá-sicas. La selectividad hacia propilenglicol aumentó a partir de la adición de un sulfuro [39], aunque esto implicó una reducción en la velocidad global de reacción, ya que el azufre es bien conocido como un veneno para catalizadores metálicos. También se notó un segundo efecto sobre los polioles producidos y es la relación directa entre la selectividad hacia pro-pilenglicol y la temperatura de reacción. Caso contrario ocurre con el etilenglicol para quien la selectividad no se ve afectada por la carga del sulfuro ni por la temperatura.
El modelo de reacción para la hidrogenólisis de glicerol propuesto por Shanks y Lahr su-giere que el glicerol inicialmente es adsorbido y deshidrogenado reversiblemente en el cata-lizador metálico para formar gliceraldehído, entonces éste es desorbido y puede reaccionar a través de cuatro caminos diferentes en un medio básico, (I) el mecanismo retro-aldol para formar el precursor de etilenglicol (glicolaldehído), (II) la oxidación y la subsiguiente descarboxilación para formar también glicolaldehído, (III) la deshidrogenación al precursor de propilenglicol (2-hidroxipropionaldehído) o (IV) la degradación a un subproducto no deseado, que también es un camino posible para los precursores de los glicoles. Finalmente los precursores son hidrogenados a los respectivos glicoles [39].
Suppes et al [31] estudiaron la hidrogenólisis de glicerol a propilenglicol bajo condiciones de operación moderadas, además ellos proponen un nuevo mecanismo de reacción en donde la hidroxiacetona es formada por la deshidrogenación de una molécula de glicerol, la cual después reacciona con hidrógeno para formar el propilenglicol y agua.
Los catalizadores de Ru y Pd muestran bajas selectividades, generalmente menores que 50% (Tabla 6.2) debido a la hidrogenólisis competitiva que llevan los enlaces C—C y C—O a una excesiva degradación de glicerol para formar alcoholes menores y gases. Por otro lado, el Cu o los catalizadores basados en Cu exhiben mayor selectividad hacia propilenglicol con reducidas selectividades hacia etilenglicol y otros subproductos de degradación.
Para las condiciones de operación en la hidrogenólisis del glicerol se puede decir a partir de la información recopilada en la Tabla 6.2 que la temperatura aumenta significativamente la conversión de glicerol, mientras que el rendimiento hacia propilenglicol tiene un máximo
H H H H M H M H n » [ 111 1 » • • • • • • • H M M H
i'.jj y ha/i-. ? c o m o M a i e r l a ' o r ; ; pa ra la O b i ' nu<)n^ac r
alrededor de 200°C. Esta característica podría ser explicada por la degradación de productos que ocurre a mayores temperaturas, lo que también puede evitarse con el aumento en la presión de H„ porque como puede verse (Tabla 6.2) la conversión y el rendimiento aumenta con la presión.
Con respecto al contenido de agua en la mezcla con glicerol, ha sido reportada una relación inversa con la conversión de glicerol y directa con la selectividad hacia propilenglicol. El cambio global genera un incremento sustantivo en el rendimiento hacia propilenglicol.
6.6. Eterificación a poligliceroles
Los poligliceroles son componentes oxigenados que están ganando prominencia en nuevo; productos como surfactantes, lubricantes, cosméticos y aditivos de alimentos. Estos com-ponentes tienen un bajo grado de polimerización y pueden ser obtenidos en cadenas linea les, cíclicas o ramificadas, aunque los esfuerzos de los investigadores se han centrado en 1; producción de selectiva de d i - y/o trigliceroles. La selectividad de la eterificación de glicero es similar a una pseudo-polimerización y en general como producto de reacción se obtiem una mezcla de poligliceroles lineales y cíclicos, especialmente en la presencia de catalizado res homogéneos como son hidróxido de sodio, potasio o carbonato [40-42],
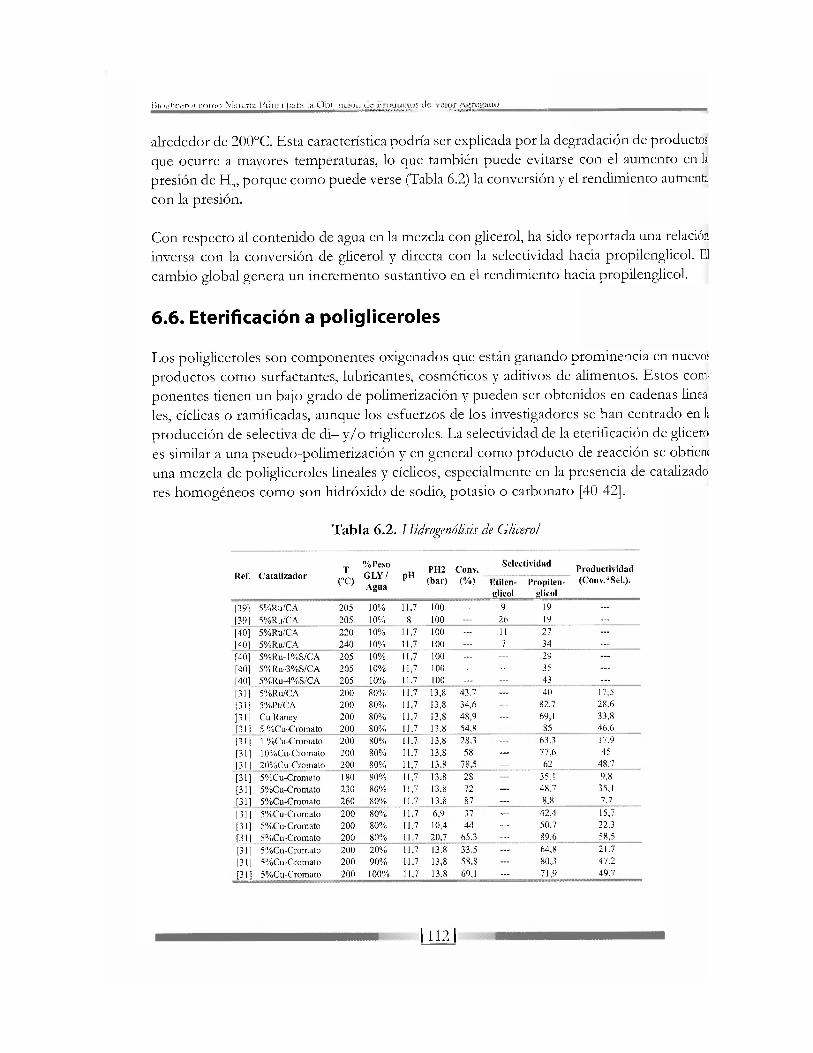
Tabla 6.2. Hidrogenólisis de Glicerol
Ref . C a t a l i z a d o r T
% P e s o
G L Y /
A g u a
pH PH2 Conv ,
Se lect iv idad Product iv idad
Ref . C a t a l i z a d o r (°C)
% P e s o
G L Y /
A g u a
pH (bar ) (%) Etilen- Propi len- (Conv.*Sel . ) .
% P e s o
G L Y /
A g u a glicol glicol
1391 5%Ru/CA 205 10% 11.7 100 — 9 19 —
[39] 5%Ru/CA 205 10% 8 100 26 19 —
|401 5%Ru/CA 220 10% 11,7 100 . . . 11 27 . . .
|401 5%Ru/CA 240 10% 11,7 100 . . . 7 34 . . .
[401 5%Ru-l%S/CA 205 10% 11,7 100 . . . . . . 29 . . .
[401 5%Ru-3%S/CA 205 10% 11,7 100 . . . . . . 35 —
[401 5%Ru-4%S/CA 205 10% 11,7 100 . . . . . . 43 . . .
[311 5%Ru/CA 200 80% 11,7 13,8 43.7 . . . 40 17,5
[311 5%Pt/CA 200 80% 11,7 13,8 34,6 . . . 82.7 28.6
1311 Cu Raney 200 80% 11,7 13,8 48,9 . . . 69,1 33,8
[311 5 %Cu-Cromato 200 80% 11,7 13.8 54,8 . . . 85 46.6
[311 1 %Cu-Cromato 200 80?-:, 11.7 13,8 28,3 . . . 63.3 17.9
1311 10%Cu-Cromato 200 80% 11.7 13,8 58 . . . 77,6 45
1311 20%Cu-Cromato 200 80% 11,7 13,8 78.5 . . . 62 48.7
[311 5%Cu-Cromato 180 80% 11,7 13.8 28 35,1 9,8
[311 5%Cu-Cromato 230 80% 11,7 13.8 72 . . . 48.7 35,1
[311 5%Cu-Cromato 260 80% 11.7 13.8 87 — 8.8 7.7
[311 5%Cu-Cromato 200 80% 11.7 6,9 37 . . . 42,4 15,7
[311 5%Cu-Cromato 200 80% 11,7 10,4 44 . . . 50,7 22.3
[311 5%Cu-Cromato 200 80% 11,7 20.7 65,3 . . . 89,6 58,5
[311 5%Cu-Cromato 200 20% 11,7 13.8 33,5 . . . 64,8 21,7
[311 5%Cu-Cromato 200 90% 11.7 13,8 58,8 . . . 80,3 47,2
[31] 5%Cu-Cromato 200 100% 11,7 13.8 69,1 . . . 71,9 49,7
| 112 |

Avances jnvesti^ativos en la Producción de Biocombustiblt
La selectividad del primer paso de la eterificación sobre catalizadores ácidos no es real-mente controlada y se obtienen mezclas de di- a hexa-gliceroles (lineales y cíclicos), además de los ésteres de poligliceroles y acroleína como subproductos. Pero la selectividad hacia poligliceroles se puede mejorar, por ejemplo, por medio de la modificación del tamaño del pseudo-poro en materiales mosoporosos [43].
Las primeras investigaciones sobre la eterificación del glicerol que trataron de mejorar la selectividad hacia d i - y trigliceroles se realizaron sobre Na,C0 3 , con altos valores de con-versión pero bajas selectividades. Ese trabajo se continuó con catalizadores básicos usando zeolitas de intercambio alcalino, quienes tienen una mejor selectividad [40],
Las zeolitas intercambiadas son realmente selectivas a d i - y trigliceroles, debido a la variación del tamaño de los canales por la relación Si/Al. La incorporación de elementos como Al, Mg y La, en la estructura de los catalizadores mesoporosos en general modifican la actividad pero no influyen significativamente sobre la selectividad [40]. Clacens [43] determinó que el método químico de impregnación para los materiales básicos que evaluó es más selectivo y estable que los materiales modificados por el método de incorporación. En cuanto al tipo de material impregnado el La es muy activo pero a su vez es poco selectivo, caso opuesto al Mg que si es selectivo pero poco activo. Por otro lado el Cs impregnado presentó tanto una buena conversión del glicerol como una alta selectividad hacia di y tri-glicoles.
6.7. Pirólisis y gasificación
El proceso de pirólisis produce combustibles líquidos a bajas temperaturas (400 a 600°C) y productos gaseosos a mayores temperaturas (>750 °C). La gasificación es un proceso simi-lar a la pirólisis, pero la mayor diferencia es que la gasificación es llevada a cabo en presencia de oxígeno ya sea en forma de aire, oxígeno puro o como vapor.
La variabilidad tanto de la constante dieléctrica como del producto iónico del agua con respecto a su temperatura y presión, abren una gama de posibilidades para las reacciones de síntesis, las cuales se pueden llevar a cabo en condiciones de agua supercrítica o cercanas a este punto (P > 22.1 MPa y T > 647 K). Es así como reacciones catalizadas por protones o iones hidróxilo pueden ser llevadas a estas condiciones, ya que el agua no es solamente un solvente, sino también un catalizador debido al fenómeno de auto-disociación, a partir del cual se forman iones hidróxilo y protones.
Bajo estas condiciones se identifican dos caminos de reacción competitivos. Uno consiste en una serie de pasos de reacciones iónicas, que ocurre a presiones altas y/o bajas tempera-turas. El otro camino es una reacción de degradación de radicales libres, que domina a bajas presiones y/o altas temperaturas. Con respecto a la velocidad de reacción, ésta generalmen-
m-- | 113 |

Bi ^iiceroi coi ' •: ' ^ .. .• p . . a v j p t e n o u " 'c . ! • "i. >s V . 1 ;
te incrementa con la temperatura hasta que se alcanza la temperatura critica, en este punto la velocidad de reacción decrece drásticamente comparada con las condiciones subcríticas y la velocidad nuevamente se incrementa con la temperatura pero sólo hasta cierto valor.
Los principales productos de la degradación del glicerol son: metanol, acetaldehído, pro-pionaldehído, acroleína, alcohol alílico, etanol, formaldehído, monóxido de carbón, dióxido de carbón e hidrógeno. La formación de acetaldehído y formaldehído incrementan con la presión, indicando que estos compuestos son principalmente formados por la reacción ió-nica, favorecida a altas densidades. Por otra parte, la formación de metanol y alcohol alílico decrece con la presión, indicando que se forman por la vía de radicales libres preferida a bajas densidades [44], La formación de productos gaseosos ocurre a mayores temperaturas que los anteriores, además la formación de los gases disminuye con la presión, lo que indica que éstos son producidos por un mecanismo de reacción de radicales libres.
Eder y Kruse [44] asumieron para el mecanismo iónico una base de reacciones elementales como son protonación, desprotonación por iones hidróxilo, deshidratación, tautomeriza-ción ceto—enol, acetilación y condensaciones aldol.
El producto gaseoso más importante de los procesos de pirólisis y gasificación son los gases de síntesis, una mezcla de hidrógeno (H„) y monóxido de carbono (C0). La literatura reporta conversiones y selectividades muy diferentes dependiendo de las condiciones de operación como son temperatura, presión, relación de dilución del glicerol [44-50], y presencia de algunos contaminantes para el caso del glicerol crudo, entre los que se destaca el metanol y el KOH [50]. Para condiciones donde el glicerol está muy diluido y a altas temperaturas, ge-neralmente se obtienen grandes concentraciones de C 0 9 en el producto gaseoso y además la mayoría de productos quedan presentes en la fase líquida [45], El aumento en la tempe-ratura de gasificación genera un aumento en la producción de H, pero también incrementa la producción de CO„ lo que implica una disminución de la concentración de CO en el gas de producto.
El proceso de pirólisis se lleva a cabo en un medio inerte acarreador del glicerol, se utiliza generalmente el nitrógeno (N2). Una característica de este medio de reacción es que a medi-da que aumenta la proporción de N2, también aumenta el rendimiento hacia la fase líquida y disminuye apreciablemente la producción de gases. También el aumento en la temperatura del sistema mejora el rendimiento a H2, pero la formación de todos los demás componen-tes decrece. La pirólisis puede tener un rendimiento del 93% a gases de síntesis (H2+CO) a 800 °C como lo muestra Valliyappan [50], debido al cracking térmico de los hidrocarburos a hidrógeno.
La gasificación se hace con vapor y sin presencia de gas acarreador. Una conversión total de
1 1 1 4 - 1 «

Avances Invesñffiidvos en la Producción de Bjocombjastibles
»licerol a gas y carbón para una alimentación de 50%Peso de glicerol con vapor ha sido re-criada, además del uso de glicerol crudo como materia prima para su gasificación [50]. En il segundo caso se reporta una composición para el glicerol crudo de 60%Peso de glicerol, il%Peso de metanol, 7,5%Peso de agua y l,5%Peso de hidróxido de potasio. El metanol :n la mezcla representa una buena oportunidad para gasificar el glicerol ya que el metanol )uro puede ser completamente gasificado para producir gases y carbón, siendo alcanzado íasta un 97%mol de gas de síntesis, con 65,7%mol de H2.
5.8. Esterificación e interesterificación
En los úldmos años, la esterificación e interesterificación de glicerol ha sido área particu-armente activa de investigación. Han sido reportados procesos que emplean catalizadores químicos y enzimáticos como lipasas.
.8.1. Esterificación de ácidos carboxílicos e interesterificación de triglicéridos Producción de Monoglicéridos. La producción de monoglicéridos tiene gran interés da-das sus propiedades emulsificantes. En 1992, el mercado de emulsificantes en el mundo fue estimado en 180.000 toneladas y el 75% de la producción fue de monoglicéridos, digüeé-ridos y mezcla de los dos [51]. Los monoglicéridos se forman cuando en una molécula de glicerol, un grupo hidróxilo es reemplazado por un ácido graso, pueden ser l(o alfa)-mo-noglicéridos o 2(o beta)-monoglicéridos dependiendo de la posición del ácido graso [52], como se muestra a continuación en la Figura 6.2.
IGlicerinal
H I
H—C—O H
H— C—O—H O
H—O—O— ácido graso
n l (o alfa)-monoglkérido
IGlicerinal
H i
H—C—rO—H I ? H - C — O —
ácido graso
H— C—iO—H I H
2(o beta)-monoglicérido
Figura 6.2. Estructura Monoglicéridos
En la industria farmacéutica los monoglicéridos son usados como cohesores en tabletas y como emolientes para drogas de absorción lenta. En la industria alimenticia los monoglicé-
m-- | 115 |

ridos son los emulsificantes más comunes empleados en productos de panadería, margari-nas, salsas, etc. En la industria cosmética, son usados como agentes texturizantes para me-jorar la consistencia de cremas y lociones. Debido a sus excelentes propiedades lubricantes y plastificantes, los monoglicéridos son usados en procesos textiles, producción de plásticos y formulación de aceites para diferentes tipos de maquinaria [53]. Recientemente nuevos productos también usan monoglicéridos, como los nutracéuticos que son de interés porque pueden prevenir la obesidad, arterioesclerosis y ayudar al tratamiento de la diabetes.
Generalmente los monoglicéridos (monoésteres de glicerol) son obtenidos a escala indus-trial dependiendo de la materia prima empleada, por dos rutas sintéticas: partiendo de ácidos grasos, de la esterificación directa con glicerina, y partiendo de aceite puro (triglicéridos), de la interesterificación con glicerina [54],
En la esterificación directa ocurren tres reacciones simultáneas, en la primera reacción, el ácido graso y la glicerina se combinan para producir los monoglicéridos, sin embargo, en la reacción secundaria se reduce la concentración de éstos debido a que reaccionan a su vez con el ácido graso para producir diglicéridos, los cuales en compañía del ácido graso gene-ran la tercera reacción en la que se obtienen triglicéridos. El mecanismo de reacción de la esterificación directa se muestra en la Figura 6.3.
ESTERIFICACIÓN DIRECTA (Á«do Graso yGhcenna)
i. ECOÍH + 1 CiHsOs - 1 RCO>CtH-C>. +• 1 H » 0 Acido Glicerina MonogSicérido Agua
1 RCQ2GH7G2 + 1 RCO2H 1 (RCÜ'V.OjHbO + l H í O Monogíieérido Ácido Dtgiscérïdo Agua
1 (RCGateCíH&O + 1 R C 0 2 H 1 <RCO?)JCSH5 + IHaO Dighcerido Ácido Tngíscérido Agua
Figura 6.3. Mecanismo de reacción por esterificación directa
La interesterificación consta de dos reacciones, en la primera el triglicérido y la glicerina s combinan para obtener monoglicéridos y diglicéridos, y en la secundaria los diglicéridos y la glicerina reaccionan para producir nuevamente monoglicéridos [54], El mecanismo d reacción de la interesterificación se muestra en la Figura 6.4.

Avances investigativos en la Producción de Biocombustibles
INTERESTERIEtCACIÓN (Aceite Puro y Glicerina) 1 (RCO/hCiIIs + 1 CIHSOJ
Triglicéndo Glicenna Glicenna « 1 RC0?CJH-0» + 1 íRCO.bCjHéO
MonogSicendo Dighcérido
Figura 6.4. Mecanismo de Reacción por Interesterificación
En ambos tipos de síntesis para mantener una alta calidad en el producto final, se debe li-mitar la temperatura aproximadamente entre 220°C-260°C y el tiempo de residencia en este rango de temperatura de proceso no debe exceder una hora y media, la concentración de monoglicéridos al final de la reacción está entre 40 y 60% [54].
Normalmente en estos procesos tradicionales se emplean catalizadores homogéneos inor-gánicos de notable impacto ambiental y baja selectividad hacia monoglicéridos [55]; para la esterificación directa se utiliza un catalizador ácido (sulfúrico, fosfórico o ácido sulfónico orgánico) y para la interesterificación se emplea un catalizador básico fuerte como KOH y Ca(OH)2 [56], En la literatura de los últimos años se reportan aluminosilicatos, zeolitas, resinas de intercambio y óxidos, básicos y ácidos, como catalizadores de la reacción de esterificación [57], Comúnmente para el proceso llevado a cabo por cualquiera de las dos rutas, los reactivos deben ser vigorosamente agitados durante toda la reacción, y al final los catalizadores deben ser neutralizados y la mezcla de reacción debe ser enfriada rápidamente para prevenir la reversión de las reacciones [56]. En algunos casos la destilación molecular es necesaria para separar las productos, porque los monoglicéridos requieren una alta pure-za para ser aplicados en las industrias alimenticia y farmacéutica, debido a que puros tienen mejores propiedades emulsificantes que una mezcla de diferentes glicéridos (mono-, di- y tri-glicéridos) [53],
La búsqueda de nuevos catalizadores adecuados para el ambiente, que sean económicos y con un rendimiento comparable a los catalizadores tradicionales es de gran interés. En contraste a los procesos químicos, la glicerólisis de aceites y ácidos grasos empleando cata-lizadores de origen biológicos o biocatalizadores (enzimas), permite obtener rendimientos comparables o incluso más altos, y mejor calidad de producto en cuanto a sabor, color y textura [51]. Las enzimas son proteínas capaces de incrementar las tasas de reacción y son catalizadores promisorios dadas las suaves condiciones de proceso que manejan (presión, temperatura, tiempo) y su buena conversión, selectividad y estereoespecificidad [58], Exis-ten diversos artículos científicos acerca de la síntesis enzimática de monoglicéridos usando como catalizador diferentes tipos de lipasas [51, 53, 59] tales como Pseudomonas sp., Candida rugosa, Rhi^opus delegar; Mucorjavanicus, Akaligenes sp.j Chromobacterium viscosum, en los cuales
|117 | •

Biog]Icte¡>l como Mater ia Prima para la Obtención de Productos de Vg .... •. .-ido
se ratifica su capacidad de igualar o mejorar el rendimiento del proceso y la selectividad ;
hacia monoglicéridos en comparación con los procesos químicos. Sin embargo, la principal desventaja de usar lipasas en las aplicaciones industriales para producción de monoglicéri-dos es el alto costo de la enzima. Para evitar este problema, se emplean las lipasas en forma inmovilizada debido a que esto permite la reutilización de la enzima. Al utilizar la enzima inmovilizada es posible operar el proceso en continuo. Muchos soportes tales como el car-bonato de calcio (CaCO,), Zeolitas y resinas de intercambio iónico han sido usados para la inmovilización de las lipasas [53],
Debido a que en el proceso de obtención de monoglicéridos algunos de los reactivos (aceite y ácidos grasos) son componentes lipofíücos o insolubles en agua, es necesario introducir solventes orgánicos en los sistemas de reacción para mejorar la solubilidad de estos reac-tivos. Además, el uso de solventes orgánicos es benéfico para la construcción de sistemas de reacción homogéneos y para facilitar el proceso en reactor continuo. Sin embargo, los solventes orgánicos producen varios efectos fisicoquímicos en las moléculas de la enzima, y los efectos difieren dependiendo de la clase de solventes y enzimas usadas [53].
Comúnmente se ha utilizado hexano puro como solvente para la obtención de monogli-céridos a partir de la esterificación directa de glicerol y ácidos grasos catalizada por lipasa, pero debido a que el glicerol es muy poco soluble en este solvente se ha presentado la obs-trucción del catalizador [51]. Para contrarrestar este problema fue propuesto inicialmente adsorber el glicerol sobre sílica gel seca [60], Analizando el esquema global de reacción, se observó que empleando como solvente n-hexano puro, la reacción no es selectiva y deja a los triglicéridos como el producto principal, el monoglicérido es únicamente el 6% molar de los productos totales y la conversión en el equilibrio es de 34% para diglicéridos y 60% para triglicéridos. Se demostró que un incremento en la polaridad del solvente, usando mezclas de n-hexano y 2-metil-2-butanol, mejora drásticamente la selectividad hacia la formación del monoglicérido. El uso de una mezcla equimolar de n-hexano y 2-metil-2-butanol, per-mite obtener un producto de 94% de monoglicéridos, 2,4% de diglicéridos y 0% de triglicé-ridos en el equilibrio. Este efecto positivo es opacado por un decremento en las velocidades iniciales y en la conversión del sustrato en equilibrio termodinámico [51].
En otros casos más específicos como la obtención de monoglicéridos a partir de aceite de palma y glicerol catalizada por lipasa Vseudomonas sp., se realizó una investigación [53] con diferentes solventes: hexano, isooctano y acetona, y la combinación de éstos, para escoger el mejor medio de reacción. Los rendimientos más altos se encontraron empleando acetona y mezcla de acetona/isooctano (3:1,v/v).
Producción de Diglicéridos. Diversos métodos han sido reportados para la producción de 1,3-DG usando lipasas, incluyendo la hidrólisis de trioleína [61], la glicerólisis de trigli-
R . | 11S |

ees Jnvcsngat ivos en la l-'rouuccton <L<: muconiuust ip ies
céridos [62], la esterificación de ácidos grasos y glicerol en solventes orgánicos [63, 64] y en sistemas libres de solvente [65], En la esterificación en sistemas libres de solvente reportada por Rosu et al., se requiere un gran tiempo de reacción (por ejemplo 12 h) para obtener un contenido suficientemente alto de dilinoleína (71,7%) llevando la reacción a baja tem-peratura de 25°C. Un alto rendimiento de 84% con una pureza de diglicérido del 90%, se obtuvieron reaccionando 1.29 mM de glicerol y 2.59 mM de ácido graso en un sistema libre de solvente a 50°C usando lipasa inmovilizada 1,3-regioselectiva con remoción de agua si-multánea en un biorreactor agitado [66].
6.8.2. Carboxilación de glicerol a carbonato de glicerol El carbonato de glicerol es usado como solvente para plásticos y resinas, tales como acetato de celulosa, nylon, nitrocelulosa y poliacronitrilo. Reacciona rápidamente con fenoles, al-coholes y ácidos carboxíücos para producir ésteres de glicerol o esteres de estos materiales, por ejemplo, poliésteres, poücarbonatos, poliuretanos y poliamidas. Un método típico para obtener carbonato de glicerol es su transesterificación con carbonato de etileno o con car-bonato dialquílico [67]. En una reacción con carbonato de etileno conducida a 125°C en la presencia de bicarbonato de sodio, los productos se obtuvieron con un rendimiento del 81%.Una patente reciente reporta un proceso en el cual una reacción entre úrea y glicerol produce carbonato de glicerol con una buena selectividad 92% [68].
Otro promisorio método para la preparación de carbonato de glicerol utiliza la reacción de glicerol con CO, o monóxido de carbono y oxígeno en la presencia de catalizador de Cu(I) [69], Los métodos anteriormente mencionados requieren que el carbonato de glicerol sea subsecuentemente purificado por medio de destilación a baja presión.
6.8.3 Nitración de glicerol a nitrato de glicerol El glicerol puede ser tratado con agentes de nitrato para formar una solución que contenga dinitroglicerol. Posteriormente la solución se trata con un agente de ciclación para convertir el dinitroglicerol en nitrato glicidílico, el cual es polimerizado en poli(nitrato gücidílico) [70], El poü(nitrato glicidíüco) ha sido reconocido como un polímero energético con potencial para ser usado en explosivos, generadores de gas y compuestos pirotécnicos. La síntesis in-dustrial de poli(nitrato glicidílico) típicamente sigue un mecanismo de tres pasos. El primer paso involucra la nitración de epiclorohidrina, seguido por un segundo paso en el cual la epiclorohidrina hidratada es ciclada con una base para formar nitrato glicidílico. El nitrato glicidílico es entonces polimerizado en un tercer paso por polimerización catiónica para formar poli(nitrato glicidílico) En este proceso la materia prima es derivada de fuentes pe-troquímicas no renovables. Sin embargo, es posible producir poli(nitrato glicidílico) usando glicerol obtenido de recursos renovables.
m-- | 119 |

! >;i.ra .la Obtención de '-'roriiictns de Va.'.or A g r e e a á
6.9. Eterifìcación
Los éteres de glicerol de interés incluyen los componentes resultantes de reacciones de gli-cerol con isobutileno o tert-butanol, poligliceroles y glicosil glicerol. Los éteres de glicerol son excelentes oxigenantes para combustibles diesel [71-73], Los combustibles diesel oxi-genados son convenientes tanto para evitar contaminación como para mejorar la eficiencia del motor [74].
6.9.1 Butilación de glicerol Entre las aplicaciones potenciales del glicerol, se encuentra la preparación de éteres alquíli-cos de glicerol mediante eterifìcación de glicerol por alquenos, particularmente isobutileno. Esta reacción puede ser conducida con una relación molar de glicerol/isobutileno de 1:2 o mayor, y a una temperatura de 50 a 150°C, obteniendo una mezcla de éteres alquílicos. Estos éteres alquílicos se pueden adicionar a una mezcla de diesel original con biodiesel, o a cada uno por separado. Se ha logrado una gran reducción de dióxido de carbono, hidro-carburos, aldehidos y materias particuladas cuando se adicionan éteres alquílicos de glicerol al combustible diesel [71-73], El p-tolueno sulfónico es empleado como catalizador parala eterifìcación de glicerol. Klepacova et al. [75-77] reportaron la eterifìcación de glicerol con tert-butanol empleando fuerte intercambio iónico, como catalizadores se usaron resinas tipo Amberlite y dos zeolitas de poros grandes HY y H¡3. La conversión máxima de glicerol 100% fue alcanzada sobre una resina Amberlyst 35 de intercambio iónico macro-reticulaf fuertemente àcida a una temperatura de 60°C. Una más alta temperatura (90°C) causa una considerable caída en la conversión y rendimiento del producto deseado di- y triéteres, prin-cipalmente en el caso de resinas de intercambio iónico Amberlyst-35 (39.2%).
Recientemente, Karinen et al. [78] han estudiado la butilación de glicerol usando isobutano en fase líquida empleando como catalizador resina de intercambio iónico acídica. Cinco tipos de éteres fueron obtenidos mediante este proceso. Se estudió el efecto de las condi-ciones de reacción, encontrando las condiciones para óptima selectiva hacia éteres en una relación molar isobuteno/glicerol de 3 a 80°C.
6.9.2. Glicosilación de glicerol El O-a-D-Glucosil Glicerol (Glc-GL) es un compuesto encontrado en alimentos tradicio-nales fermentados de Japón tales como sake, miso y mirin [79], El Glc-GL es un glucósido no reducido que presenta alrededor de la mitad de la dulzura de la sacarosa, tiene alta estabi-lidad térmica, baja higroscopicidad, alta capacidad de retención de agua, no es cancerígeno y baja reactividad Maillard [79-81]. El Glc-GL es producido por un proceso enzimàtico pri-mero descrito en detalle por Sawai y Henre [82], El proceso usa Candida tropicalis R-glucosi-dasa para transferir el residuo Glc de almidón y dextrinas a la posición 1- (o3-) del glicerol.
m--,. I 12( ) I

v :es Invesrigativos en la Producción de Biocom bus tibies
>.10. Halogenación
Las investigaciones en halogenación de glicerol se han enfocado en la producción de 1,3-di-doropropanol, [83-94] un intermediario en la síntesis de epiclorohidrina. La epiclorohidrina es una importante materia prima para la producción de compuestos como resinas epóxidas, elastómeros sintéticos y agentes adherentes para la industria de papel.
En el mecanismo de reacción global para producir 1,3-dicloropropanol de glicerol la pri-mera reacción que ocurre es la clorinación del glicerol, obteniendo en un principio 1-mo-nocloropropanodiol y agua, con pequeñas cantidades de 2-monocloropropanodiol. Luego ocurre una segunda clorinación de la cual se obtiene 1,3-dicloropropanol, combinado con pequeñas cantidades de 1,2-dicloropropanol como coproducto. Los procesos tradicionales [88-94] a partir de glicerol, consisten de la reacción de glicerol con ácido clorhídrico en solu-ción, en la presencia de ácido acético como catalizador, a temperatura de aproximadamente 80-100°C. Numerosas patentes describen procesos usando un solvente orgánico inerte in-soluble en agua y soluble en 1,3-dicloropropanol. La reacción es realizada a la temperatura de ebullición de la mezcla, por lo tanto, la temperatura varía dependiendo del solvente uti-lizado. Otras patentes describen reacciones seguidas por complicadas neutralizaciones, ex-tracciones y destilaciones para recuperar 1,3-dicloropropanol. Estos procesos tradicionales a partir de glicerol, presentan considerables desventajas: la pérdida de catalizador durante la reacción, debido al bajo punto de ebullición del ácido acético, el extenso tiempo de reacción causado por la introducción de agua en la mezcla de reacción en forma de soluciones acuo-sas de ácido clorhídrico y la complejidad de retirar el agua que se forma como consecuen-cia de la reacción, y la difícil separación del 1,3-dicloropropanol de la mezcla de reacción. Estas desventajas, junto con el alto precio del glicerol como materia prima, han impedido el establecimiento de este proceso. El método normalmente emplea propileno como una materia prima y el rendimiento alcanzado es una mezcla de 30% de 1,2-dicloropropanol y 70% de 1,3-dicloropropanol. El alto porcentaje de 1,2-dicloropropanol causa dificultades, debido a que esta sustancia reacciona mucho más lentamente que el 1,3-dicloropropanol en la producción de epiclorohidrina y en consecuencia en la reacción de dehidroclorinación. Esto tiene repercusiones sobre el dimensionamiento de plantas y reduce el rendimiento por la formación de coproductos. Recientemente, Siano et al. [83] patentaron un proceso para producción de 1,3-dicloropropanol de glicerol empleando ácido clorhídrico anhidro gaseo-so en la presencia de ácidos orgánicos de baja volatilidad como catalizador con un punto de ebullición mayor de 120°C. El proceso para producir 1,3-dicloropropanol es realizado partiendo de glicerol puro o crudo, como el obtenido como coproducto en el proceso de producción de biodiesel, con ácido clorhídrico gaseoso, en la presencia de varios ácidos orgánicos (ácidos monocarboxílicos con 3-10 átomos de carbono o ácidos ditricarboxílicos con un número de carbonos entre 2 y 10) como catalizador. La temperatura de reacción es controlada entre 80°C y 180°C y la presión del ácido clorhídrico gaseoso debajo de 0.5
m-- | 121 |

11 r L i1 •! -nn . j
MPa. El incremento en la producción de biodiesel permite que esta reacción partiendo de
glicerol sea part icularmente ventajosa, dada la gran cantidad de glicerol que esta reacción
podría consumir.
Referencias 1. M. Ying, T. Hu, Z. Dai-Jia, W. Wei, X. Zhi-Long, "Microbial production of 1,3-propane-
diol by Klebsiella pneumoniae using crude glycerol from biodiesel preparations", Biotechnology letters. Vol. 28, 2006. pp. 1755-1759.
2. C. W. Chiu, M. A. Dasari, W. R. Sutterlin, G. J. Suppes, "Removal of residual catalyst from simulated biodiesel crude glycerol for glycerol hydrogenolysis to propylene glycol," Ind. Eng. Chem. Res. Vol. 45, 2006. pp. 791.
3. S. Demirel, P. Kern, M. Lucas, P. Claus. "Oxidation of mono- and polyalcohols with gold: Comparison of carbon and ceria supported catalysts," Catalysis Today. Vol. 122, 2007. pp. 292-300.
4. M. Pagliario M. Rossi. "The future of Glycerol: New usages for a versatile raw Material", RSCPublishing, Cambridge, p.p. 128. 2008.
5. C.S. Miner, N.N. Dalton, Editors. Glycerol, American Chemical Society Monograph Series. Reinhold Publishing Company, New York. 1953. p. 1.
6. W.L.Carpenter, A Yeatise, "On the Manufacture of Soap and Candles, Lubticants and Gly-cerin". E. &>E Spon, London. 1885, pp. 307-8.
7. "Glycerine: an overview. The Soap and Detergent Association Glycerine & Oleochemical Division", 1990, p: 27.
8. P. Gallezot, "Selective Oxidation with Air on Metal Catalysts," Catalysis Today. Vol. 37. 1997. pp. 405 - 418.
9. S. Demirel-Gulen, M. Lucas, P. Claus, "Liquid Phase Oxidation of Glycerol over Carbon Supported Gold Catalysts," Catalysis Today. Vol. 102.-103. 2005. pp. 166-172.
10. G. J. Hutchings, "Catalysis by Gold," Catalysis Today. Vol. 100. 2005. pp. 55-61. 11. F. Porta, L. Prati. "Selective Oxidation of Glycerol to Sodium Glycerate with Gold-On-
Carbon Catalyst: An Insight into Reaction Selectivity," Journal of Catalysis. Vol. 224. 2004. p.p. 397-403.
12. R. García, M. Besson, P. Gallezot, "Chemoselective Catalytic Oxidation of Glycerol with Air on Platinum Metals," Applied Catalysis General. Vol. 127. 1995. pp. 165-176
13. P. Fordham, R. García, M. Besson, P. Gallezot, "Selective catalytic oxidation with air of glycerol and oxygenated derivatives on platinum metals," Stud. Surf. Sci. Catal. Vol. 101. 1996. pp. 161-170.
14. P. Fordham, M. Besson, P. Gallezot, "Catalytic oxidation with air of tartronic acid to mesoxalic acid on bismuth-promoted platinum". Catal. Lett. 1997. Vol. 46. pp. 195-199.
15. P. Fordham, M. Besson, P. Gallezot, "Selective oxidation with air of glyceric to hydroxypyru-vic acid and tartronic to mesoxalic acid on PtBi/C catalysts" Stud Surf. Sci. Catal Vol. 108. 1997. pp. 429-436.
16. S. Carrettin, P. McMorn, P. Johnston, K Griffin, G. J Hutchings, "Selective oxidation of
| 122| ma

': Lme^ngal¡vor. en I:, Prntiueciun de Biocumbusnbles
glycerol to glyceric acid using a gold catalyst in aqueous sodium hydroxide," Cbem. Commun. Vol. 7. 2002. pp. 696-697.
17. S. Carrettin, P. McMorn, P. Johnston, K. Griffin, C. J. Kiely, G. A. Attard, G. J Hut-chings, "Oxidation of glycerol using supported gold catalysts," Top. Catal, 2004. Vol. 27. pp. 131-136.
18. S. Carrettin, P. McMorn, P. Johnston, K. Griffin, G. J Hutchings, "Oxidation of glyce-rol using supported Pt, Pd and Au catalysts," Phys. Cbem. Cbem. Pbys. 2003. Vol. 5. pp. 1329-1336.
19. N. Dimitratos, F. Porta, L. Prati. "Au, Pd (Mono and Bimetallic) Catalysts Supported on Graphite Using the Immobilization Method Synthesis and Catalytic Testing for Liquid Phase Oxidation of Glycerol". Applied Catalysis A: General. Vol. 291. 2005. pp. 210-214.
0. C. L. Bianchi, P. Canton, N. Dimitratos, F. Porta, L. Prati, "Selective oxidation of glyce-rol with oxygen using mono and bimetallic catalysts based on Au, Pd and Pt metals," Catalysis Today. 2005. Vol. 102-103. pp. 203-212.
1. N. Dimitratos, A. Villa, D. Wang, F. Porta, D. Su, L. Prati, "Pd and Pt catalysts modified by alloying with Au in the selective oxidation of alcohols", journal of Catalysis. Vol. 244. pp. 113-121.
2. N. Dimitratos, J.A. Lopez-Sanchez, D. Lennon, F Porta, L. Prati, A. Villa, "Effect of particle size on monometallic and bimetallic (Au, Pd)/C on the liquid phase oxidation of glicerol," Catal. Lett. 2006. Vol. 108. pp. 147.
3. L. Prati, F. Porta, "Oxidation of alcohols and sugars using Au/C catalysts - Part 1. Alco-ho l s , "^/ . Catal., A. 2005. Vol. 291. pp. 199-203.
24. L. Prati, M. Rossi, "Chemoselective catalytic oxidation of polvols with dioxygen on gold supported catalysts", Stud. Surf. Sci. Catal, 1997. Vol. 110. pp. 509-516.
5. D. Wang, A. Villa, F. Porta, D. Su, L. Prati, "Single-phase bimetallic system for the selecti-ve oxidation of glycerol to glycerate," Cbem. Commun. 2006. Vol. 18. pp. 1956-1958.
26. P. Fordham, M. Besson, P. Gallezot, "Selective Catalytic Oxidation of Glyceric Acid to Tar-tronic and Hydroxypyruvic Acids." Applied Catalysis A: General. 1995. Vol. 133. pp. L179-L184.
27. S. Demirel, K. Lehnert, M. Lucas, P. Claus, "Use of Renewables for the Production of Chemicals: Glycerol Oxidation over Carbon Supported Gold Catalysts," Appl. Catal, B: Envi-ronmental. 2007. Vol. 70. pp. 637-643.
28. W. C. Ketchie, M. Murayama, R. J. Davis, "Selective oxidation of glycerol over carbon-supported AuPd catalystsJournal of Catalysis. 2007. Vol. 250. pp. 264-273.
9. W.C. Ketchie, M. Murayama, R.J. Davis, "Promotional Effect of Hydroxyl on the Aqueous Phase Oxidation of Carbon Monoxide and Glycerol over Supported Au Catalysts", Topics in Catalysis. 2007. Vol.44, pp. 307-317.
30. W. C. Ketchie, Y-L. Fang, M. S. Wong, M. Murayama, R. J. Davis, "Influence of gold particle size on the aqueous-phase oxidation of carbon monoxide and glycerol," journal of Catalysis. 2007. Vol. 250. pp. 94-101.
31. M. A. Dasari, P-P. Kiatsimkul, W. R. Sutterlin, G. J. Suppes, "Low-Pressure Hydroge-nolysis of Glycerol to Propylene Glycol," Applied, Catalysis A: General, 2005. Vol. 281. pp. 225-231.
32. A.E. Martin, F.H. Murphy, Krik-Othmer, "Encyclopedia of Chemical Technology", 4th edition, vol. 17, Wiley, New York, 1994, p. 715.
^ a m m m m m m m m m m m m m I J23 J —

Eiuffliccrol cornil Matet ia ¡ 'nn ia para ia 1
33. D. T. Trent, Krik-Othmer, "Encyclopedia of Chemical Technology", 4th edition, vol. 20, Wiley, New York, (1996), p. 271.
34. C. Tessie, "Production of propanediols". U.S. Patent N.° 4,642,394. 1987. 35. B. Casale, A.M. Gómez, "Method of hydrogenating glycerol". U.S. Patent N°. 5,214,219,
1993. 36. B. Casale, A.M. Gómez, "Catalytic method of hydrogenating glycerol". U.S. Patent N°
5,276,181. 1994. 37. S. Ludwig, E. Manfred, "Preparation of 1,2-propaned". U.S. Patent N°. 5,616,817. 1997. 38. D. G. Lahr, B. H. Shanks, "Kinetic Analysis of the Hydrogenolysis of Lower Polyhydric
Alcohols: Glycerol to Glycols," Ind. Eng. Chem. Res. 2003. Vol. 42. pp. 5467-5472. 39. D. G. Lahr, B. H. Shanks, "Effect of Sulfur and Temperature on Ruthenium-Catalyzed
Glycerol Hydrogenolysis to Glycols," J ournal of Catalysis. 2005. Vol. 232. pp. 386-394. 40. J. Barrault, Y. Pouilloux, J.M. Clacens, C. Vanhove, S. Bancquart, "Catalysis and Fine
Chemistry," Catalysis Today. 2002. Vol. 75. pp. 177-181. 41. J. Gerald, S. Werner, D. Helmut, Solvay Werke GMBH, "Process for the Preparation of
Diglycerol and/or Polyglycerol". U.S. Patent N°. 5 243 086. 1993. 42. J. Lutz, G. Bernhard, B. Reinhard, J. Volkmar, Henkel. KGAA, "Process for the produc
tion of diglycerol". U.S. Patent N°. 5 710 350. 1998. 43. J. -M. Clacens, Y. Pouilloux, J. Barraultn, "Selective Etherification of Glycerol to Po-
lyglycerols over Impregnated Basic MCM-41 type Mesoporous Catalysts," Applied Catalysis A: General, 2002. Vol. 227. pp. 181-190.
44. W. Buhler, E. Dinjus, H.J. Ederer, A. Kruse, C. Mas, "Ionic Reactions and Pyrolysis of Glycerol as Competing Reaction Pathways In Near- and Supercritical Water," Journal of Super-critical Fluids. 2002. Vol. 22. pp. 37-53.
45. Technical Feasibility of Biomass Gasification in Fluidized Bed with Supercritical Water. Re-search Coordinator: Willibrordus P. M. van Swaaij Duration April, 2000 ~ March, 2003.
46. Y. Matsumura, T. Minowa, B. Potic, S. R. A. Kersten, W. Prins, W. P. M. van Swaaij, B. van de Beld, D. C. Elliott, G. G. Neuenschwander, A. Kruse, M. J. Antal Jr, "Biomass Gasification in Near- and Super-Critical Water: Status and Prospects (Review)," Biomass ani Bioenergy. 2005. Vol. 29. pp. 269-292.
47. M. Mozaffarian, E.P. Deurwaarder. S.R.A. Kersten, "Green Gas" (SNG) Production by Supercritical Gasification of Biomass," ECN-C-04-081. November 2004.
48. M. J Antal Jr, S.G. Allen, D. Schulman, X. Xu, R.J. Divilio, "Biomass Gasification in Su percritical Water," Ind. Eng. Chem. Res. 2000. Vol. 39. pp. 4040-4053.
49. X. Xu, Y. Matsumura, J. Stenberg, M.J. Antal Jr, "Carbon-Catalysed Gasification of Or-ganic Feedstocks in Supercritical Water," Ind. Eng. Chem. Res. 1996. Vol. 35. pp. 2522-2530.
50. T. Valliyappan, "Hydrogen or Syn Gas Production from Glycerol Using Pyrolysis and Steam Gasification Processes". Thesis for the Degree of Master of Science in the Department of Chemical Engineering.
51. J. C. Bellot, L. Choisnard, E. Castillo, A. Marty, "Combining solvent engineering and thermodynamic modeling to enhance selectivity during monoglyceride synthesis by lipase-catalyzed esterification," Enzyme and Microbial Technology. 2001. Vol. 28. pp. 362-369.
52. H. Lawson, "Aceites y Grasas Alimentarias: Tecnología, Utilización y Nutrición", Editorial Acribia, S.A. 2000.
I 124 | — —

Avances lnvest igat ivos en ja Producción de Biocombust ib les
53. W. Kaewthong, S. Sirisansaneeyakul, P. Prasertsan, A. H-Kittikun, "Continuous pro-duction of monoacylglycerols by glycerolysis of palm olein with immobilized lipase," Process Biochemistry. 2005. Vol. 40. pp. 1525-1530.
54. W. Fisher, "Production of High Concentrated Monoglyceride". Lecture given on occasion of the DGF-Symposium in Magdeburg / Germany, October, 1998.
55. A. M. Cortez, R. Mora, J. C. Vargas, "Inmovilización de lipasas: estudio de la actividad catalítica en la Esterificación de ácidos grasos," Departamento de ingeniería química, Univer-sidad Nacional de Colombia. Sede Bogotá. (2000)
56. A. Corma, S. Bee Abd Hamid, S. Iborra, A. Velty, "Lewis and Brónsted basic active sites on solid catalysts and their role in the synthesis of monoglycerides," J o u m a l o f Catalysis. 2005. Vol. 234, pp. 340-347.
57. P. H. L. Moquina, F. Temelli, H. Sovová, M. Saldaña, D. A. Marleny, "Kinetic modeling of glycerolysis—hydrolysis of canola oil in supercritical carbon dioxide media using equili-brium data " Journal of Supercritical Fluids. 2006. Vol. 37. pp. 417—424.
58. K. N. Kilcawley, M. G. Wilkinson, P. F. Fox, "Determination of key enzyme activities in commercial peptidase and lipase preparations from microbial or animal sources," Enzyme and Microbial Technology. 2002. Vol. 31. pp. 310-320.
59. S. D. Ferreira, A. C. Correia, M. M. R. Fonseca, "Response surface modeling of glycero-lysis catalyzed by Candida rugosa lipase immobilized in different polyurethane foams for the production of partial glycerides," journal of Molecular Catalysis B: Enzymatic. 2003. Vol. 21. pp. 71-80.
60. H. A. Aksoy, M. Tuter, "Enzymatic Glycerolysis of Palm and Palm Kernel Oils," Chem. Eng. Comm. 2005. Vol. 192. pp. 14-17.
61. F. J. Plou, M. Barandiarán, M. V. Calvo, A. Ballesteros, E. Pastor, "High-yield produc-tion of mono- and di-oleylglycerol by lipase-catalyzed hydrolysis of triolein", Enzyme and Microbial Technology. 1996 Vol. 18. pp. 66-71.
62. A. Coteron, M. Martínez, J. Aracil, "Reactions of olive oil and glycerol over immobilized lipases "J. Am. Oil Chem. Soc. 1998. Vol. 75. pp. 657.
63. M. Berger, K. Laumen, M. P Schneider, "Enzymatic esterification of Glycerol I. Lipaseca-talyzed synthesis of regioisomerically pure 1,3-sn-diacylglycerols," J. Am. Oil Chem. Soc. 1992. Vol. 69. pp. 955-960.
64. M. Berger, M.P Schneider, "Enzymatic esterification of Glycerol II. Lipase-catalyzed syn-thesis of regioisomerically pure 1 (3)-rac-monoacylglycerols," J. Am. Oil Chem. Soc. 1992. Vol. 69. pp. 961-965.
65. R. Rosu, M. Yasui, Y. Iwasaki, T. Yamane, "Enzymatic Synthesis of Symmetrical 1,3-Dia-cylglycerols by Direct Esterification of Glycerol in Solvent-Free System ,"/. Am. Oil Chem. Soc. 1999. Vol. 76. pp. 839.
66. T. Watanabe, M. Shimizu, M. Sugiura, M. Sato, J. Kohori, N. Yamada, K. Nakanishi, "Optimization of Reaction Conditions for the Production of DAG Using Immobilized 1,3-Regiospecific Lipase Lipozyme RM IM,"/. Am. Oil Chem. Soc. 2003. Vol. 80. pp. 1201-1207.
67. J. B. Bell, L. Silver, V. Arthur, "Method for preparing glycerin carbonate". U.S. Patent N°. 2,915,529. 1959.
68. M. Okutsu, T. Kitsuki, "Process for the preparation of glycerol carbonate". U.S. Patent N°. 6,495,703. 2002.
I 125 I

B ^ o ^ i ^ ^ h ^ o m ^ ^ ^ ^ ^ ^ n m ^ ^ ^ a Cprenc ion at: J ^
69. J. H. Teles, N. Rieber, "Harder W. Preparation of glycerol carbonate". U.S. Patent N°. 5,359,094. 1994.
70. T. K. Highsmith, "Continuous process and system for production of glycidyl nitrate from glycerin, nitric acid and caustic and conversion of glycidyl nitrate to poly(glycidyl nitrate)", U.S. Patent N°. 2004138481. 2004.
71. D. J. Morgans Jr, "Alkoxymethyl ethers and alkoxymethyl esters of glycerol". European Pa-tent N°. 187297. 1986.
72. V. P. Gupta, "Preparation of glycerol di-tertiary-butyl ether from isobutylene and glycerol", U.S. Patent N°. 5476971. 1995.
73. F. Ancillotti, V. Fattore, "Oxygenate fuels: Market expansion and cata- lytic aspect of syn-thesis" Fue/Proc. Techno!, 1998. Vol. 57. pp. 163-194.
74. H. Noureddini, "Process for producing biodiesel fuel with reduced viscosity and a cloud point below 32 °F". U.S. Patent N°. 6015440. 2000.
75. K. Klepacova, D. Mravec, M. Bajus, "Tert-Butylation of glycerol by ion-exchange resins," Appl. Catal,, A. 2005. Vol, 294. pp. 141.
76. K. Klepacova, D. Mravec, M. Bajus, "Etherification of glycerol with tert-butyl alcoho catalysed by ion-exchange resins," Chem. Pap. 2006, Vol. 60. pp. 224.
77. K Klepacova, D Mravec, A. Kaszonyi, M. Bajus, "Etherification of glycerol and ethylene glycol by isobutylene," Appl, Catal., A. 2007. Vol. 328. pp. 1-13.
78. R.S. Karinen, A.O.I. Krause, "New biocomponents from glycerol," Appl. Catal. A-Gen. Vol. 306. 2006. pp. 128-133.
79. F. Takenaka, H. Uchiyama, T. Imamura, "Identification of a-D-glucosyiglycerol in sake," Biosci. Biotechnol. Biochem. 2000. Vol. 64. pp. 378-385.
80. F. Takenaka, H. Uchiyama, "Effects of _-D-glucosylglycerol on the in vitro digestion of disaccharides by rat intestinal enzymes," Biosci. Biotechnol. Biochem. 2001. Vol. 65. pp. 1458—63.
81. F. Takenaka, H. Uchiyama, "Synthesis of a-D-glucosylglycerol by a-glucosidase and som of its characteristics," Biosci, Biotechnol. Biochem. 2000. Vol. 64. pp. 1821-1826.
82. T. Sawai, E. J. Hehre, "A novel amvlase (Candida transglycosyl-amylase) that catalyzes glu-cosvl transfer from starch and dextrins,"/. Biol. Chem. 1962. Vol. 237. pp. 2047-2052.
83. D. Siano, E. Santacesaria, V. Fiandra, R. Tesser, G. Di Nuzzi, M. Di Serio, M. Nasta-si, "Continuous regioselective process for the production of l,3-dichloro-2-propanol fro glycerin and hvdrochloric acid in the presence of organic carboxylic acid catalysts". Worl Organization Patent N°. 2006111810. 2006.
84. P. Gilbeau, "Catalytic chlorination method for making a chlorohydrins from a polyhydnt alcohol or polyhydric alcohol esters". \Xorld Organization Patent N°. 2006106154. 2006.
85. P. Gilbeau, "Method for making an epoxide with removal of byproducts". WO Patent N° 2006100311. 2006.
86. T. Aoki, T. Ohe, H. Ishikami, "Production of epichlorohydrin and dichloropropanol inter mediate". European Patent N°. 1059278. 2000.
87. M. Spadlo, M. Adamczyk, A. Brzezicki, J. Dula, K. Giza, A. Gorzka, W. Madej, G Masztalerz, E. Okninska, "Manufacture of l,3-dichloro-2-propanol". Polish Patent N° 163256, 1994.
88. F. N. Grimsby, "Dichlorohydrin". U.S. Patent N°. 4620912. 1986. 89. A. Lauer, E. Okninska, G. Lewandowski, W. Madej, M. Spadlo, J. Wasilewski, Z
!
mm,, | 126 | » m m m b h b i ^ M M M ^ ^ ^ ^ h h h h


Avances ínvesrigat ivos en 3a Producción de Biocombust ib ies
CAPÍTULO 7
EFECTO DEL USO DE BIODIESEL EN LA EMISIÓN DE CONTAMINANTES ATMOSFÉRICOS
Franz Edwin López1, Agustín Bueno1,
María José lllánT, Óscar Hernán Giraldo2
Introducción Actualmente, el 95% del transporte mundial se basa en el uso de combustibles de origen fó-sil como fuente de energía [1], Sin embargo, la necesidad de reducir la dependencia de estos combustibles y de controlar el cambio climático, está generando un interés creciente por fuentes de energía alternativas [2, 3]. El uso de biocombustibles ha despertado un gran inte-rés en los últimos años a escala mundial con el objetivo de: I) reducir las emisiones de gases que causan el efecto invernadero, II) asegurar el abastecimiento energético y III) disminuir la dependencia de combustibles importados desde zonas geopolíticamente en conflicto.
En este sentido, el constante aumento en la concentración de sustancias contaminantes en el aire está originando que los gobiernos de diferentes regiones del planeta impongan regula-ciones cada vez más estrictas con el fin de proteger la población y mejorar la calidad de vida. Como ejemplo, según directivas de la Unión Europea (2003/30/EC), los países de la comu-nidad deben sustituir progresivamente el uso de gasolina y diesel por otras fuentes de energía renovable, de tal forma que, en el año 2010, el 5,75% de la energía provendrá de fuentes renovables (EU 2003). Como consecuencia de ello, el interés en la producción y uso de bio-diesel y de otros combustibles derivados de la biomasa está aumentando rápidamente.
Por otro lado, el fenómeno de globalización mundial está originando un aumento de la de-manda de movilidad de la población. Este aumento de la movilidad es responsable de que el transporte sea el sector para el que se prevé mayor crecimiento porcentual de emisiones de gases de efecto invernadero para la próxima década. Como consecuencia, la contamina-
' Universidad de Alicante, Departamento de Química Inorgánica. Ap. de correos, 99 03080, Alicante, España. * E-mail: [email protected] 1 Departamento de Física y Química. Universidad Nacional de Colombia-Sede Maníjales,
1129 |

ecto dci uso e Biociicscl en Emis ión de Contaminantes Atmosfér icos
ción atmosférica generada por fuentes móviles es un problema de interés general. Desde una perspectiva de sostenibilidad es necesario alcanzar un equilibrio entre los combustibles fósiles (como el petróleo), y otros renovables. Para ello, se requiere potenciar la utilización de biocombustibles que sean eficientes para la reducción de emisiones y que presenten un fuerte impacto en la reducción de importación de crudo o productos petrolíferos.
El biodiesel es un combustible renovable, con un efecto beneficioso en el control de las emisiones de CO„ ya que su producción y consumo permiten alcanzar un equilibrio neto en el balance de C 0 2 atmosférico. Es por ello una fuente de energía de interés para contribuir al cumplimiento de los objetivos del protocolo de Kyoto, así como también para asegurar un suministro continuo capaz de cubrir la demanda y reducir el consumo de materias ener-géticas fósiles.
El biodiesel se utiliza como aditivo del combustible diesel convencional, en mezclas del 10% (B10) y 20% (B20). En algunos casos, dependiendo de su pureza, también se emplea directamente en motores de ignición. El uso de biodiesel está creciendo rápidamente a es-cala mundial y, por ello, es imperativo determinar el impacto de su uso sobre el proceso de combustión, en la formación de contaminantes y en las tecnologías de post-tratamiento uti-lizadas para la eliminación de los contaminantes. En este capítulo se analiza el impacto del uso de biodiesel en la generación de contaminantes y en las tecnologías de purificación de los gases emitidos. Específicamente, se analizan las características del material particulado generado por este combustible, para determinar su efecto en el proceso de regeneración de los filtros de partículas por oxidación catalítica.
7.1. Composición de los gases de escape de los motores diesel y gasolina
Los motores de combustión interna más utilizados son los motores Ottoj Diese/, que em-plean como combustible gasolina y diesel, respectivamente. Los principales productos de la oxidación de estos combustibles son CO, y H 20, siendo el CO2 uno de los gases respon-sables del calentamiento global del planeta debido a su contribución al efecto invernadero. Adicionalmente, se generan otros agentes contaminantes que presentan efectos perjudicia-les para el medio ambiente y la salud humana. Estos agentes contaminantes incluyen el mo-nóxido de carbono (CO), los hidrocarburos no quemados (HC), los óxidos del nitrógeno (NOx) y el material particulado, también denominado hollín o carbonilla [1, 4-9], El motor diesel emite valores más bajos de CO e HC que el de gasolina, pero genera mucha mayor cantidad de carbonilla, contaminante que afecta las vías respiratorias y contiene hidrocar-buros adsorbidos que son altamente cancerígenos. Las emisiones de NOx son del mismo orden de magnitud en los dos tipos de motores.
isfe | 130 |

Avancc !•-• estipas• I' ¡V • iuc< ón - ; • . »mbuatihj
El control de las emisiones en motores a gasolina se lleva a cabo utilizando los conocidos catalizadores de tres vías (TWC1) . Sin embargo, para los motores diesel, el problema de las emisiones no está resuelto, lo que supone un importante inconveniente que enmascara las ventajas que presentan estos motores. La combustión diesel es ampliamente utilizada tanto en fuentes fijas como en móviles, especialmente donde es necesaria una elevada salida de potencia. Dentro del sector automovilístico, la mayor ventaja de los motores diesel frente a los motores de gasolina es el alto torque a velocidades bajas del motor, lo que equivale a un mayor rendimiento y eficiencia y, por lo tanto, a una disminución en la generación de CO, por unidad de potencia producida. Sin embargo, esta ventaja de los motores diesel no podrá ser aprovechada a menos que las emisiones de carbonilla y NOx cumplan con la legislación. Así, existe un renovado interés sobre la tecnología diesel para los automóviles motivada por el deseo de mejorar la eficiencia del combustible y disminuir las emisiones contaminantes
[4]-
El biodiesel presenta propiedades físicas y una composición química muy diferente del combustible diesel convencional, lo que implica que la composición de las emisiones pueda ser considerablemente distinta. Los cambios más significativos en este sentido, se presen-tan en las emisiones de óxidos de nitrógeno (NOx) y en el material particulado. Muchos autores concluyen que el uso de biodiesel implica una reducción en la cantidad de material particulado generado, que, además, presenta características físico-químicas diferentes. Por el contrario, hay autores que sugieren que la concentración de óxidos de nitrógeno en los gases emitidos se ve incrementada al usar biodiesel ¡5,6], siendo ésta una de sus desventajas. En estos estudios se ha llegado a la conclusión de que la emisión de contaminantes depende de variables como el tipo de motor, el modo de operación (velocidad y carga), condiciones ambientales y calidad del biodiesel, entre otras.
A continuación se presenta una breve revisión de los sistemas de post-combustión que se han propuesto para la purificación de los gases emitidos por los motores diesel. Asimismo se comenta el posible efecto del uso de biodiesel como sustituto parcial o total del diesel convencional.
7.2. Sistemas de eliminación de contaminantes en motores diesel
El funcionamiento de un motor diesel es diferente al de gasolina y, como consecuencia, los requisitos del sistema de post-combustión para los motores diesel son diferentes de los adoptados para los vehículos de gasolina. El motor de gasolina emplea la chispa generada
Dei inglés, Three Way Cataysts
|131 | i

Electo del usu de Biodicscl en la Emis ión de Contamíname; , Atmosret i
por una bujía para producir la ignición del combustible, el cual se introduce en el carburadoi junto con la cantidad de aire estequiométricamente necesaria para llevar a cabo la combus-" tión. Este modo de operar tiene como consecuencia que la concentración en el escape de especies oxidantes (O, y NOx) y reductoras (CO e HC) también sea próxima a la estequio-métrica. El papel de los catalizadores de tres vías es conseguir que las especies oxidantes y reductoras reaccionen entre sí produciendo gases no tóxicos, como son N?, C 0 2 y H20.
Sin embargo, el motor diesel emplea un proceso de compresión para conseguir la combus-tión. En el motor diesel, el combustible se inyecta dentro de un cilindro en el que hay una carga altamente comprimida de aire, y dicha presión produce la energía necesaria para que ocurra la ignición [7]. Este modo de operar de los motores diesel, por un lado, origina la formación de carbonilla, es decir, de pequeñas partículas de carbón formadas por conden-sación de hidrocarburos que no reaccionan. Por otro lado, en los motores diesel el oxígeno está siempre en exceso y, por ello, siempre está presente en elevada concentración en los gases de escape, pudiendo alcanzar valores hasta del 18% [8]. Estas dos diferencias impi-den la utilización de los catalizadores de tres vías. En primer lugar la carbonilla, al ser un contaminante sólido, requiere un sistema específico de eliminación que incorpore un filtro. En segundo lugar, la elevada concentración de oxígeno dificulta la reducción de los NOx en una atmósfera netamente oxidante. Teniendo esto en cuenta queda claro que el objetivo principal de los sistemas de control de emisiones diesel es la reducción de los NOx y la eliminación de carbonilla [9, 10].
La catálisis es una de las tecnologías claves para el control de la contaminación atmosférica. La eliminación catalítica de compuestos contaminantes presentes en los gases de escape se utiliza para reducir las emisiones de los automóviles a gasolina desde los años setenta. Des-pués de la introducción de los catalizadores de tres vías (TWC) para los motores a gasolina en los años ochenta, la atención se ha centrado en el control de las emisiones de los motores diesel. Tal y como se indicó, la carbonilla y los NOx son los principales contaminantes ge-nerados por este tipo de motores. Consecuentemente, el control de los NOx y la carbonilla emitidos por motores diesel, es uno de los grandes desafíos del sector automovilístico para la sociedad del siglo XXI. El tratamiento de post-combustión de los gases de escape en motores diesel aborda la eliminación de los contaminantes mediante tres procesos com-plementarios: oxidación catalítica de HC y CO, sistemas de control de NOx en exceso de oxígeno y filtros para partículas [11],
Oxidación catalítica de HC y CO
La oxidación catalítica fue uno de los primeros procesos desarrollados para eliminar hidro-carburos y CO de las emisiones procedentes del escape de los automóviles. Los catalizado-res de oxidación diesel se utilizan para oxidar HC, CO, aldehidos y compuestos aromático
| 132 | i

Avances mvesuiíativos en la Producción de Biocombusubles
[12], y suelen incorporar Pt en su formulación. Como ya se ha comentado, las emisiones de CO disminuyen cuando se usa biodiesel en vez del diesel convencional.
Sistemas de control de NOx en exceso de oxígeno
Una forma de reducir las emisiones de NOx es reducir la formación de estos óxidos de nitrógeno mediante modificaciones en el propio proceso de combustión. Esto ha motivado el que se introduzcan modificaciones en el propio motor, como son el reajuste de la sinto-nización del motor, específicamente en el sistema de inyección, o la implementación de un sistema de recirculación de gases (EGR2) [12].
Como ya se ha comentado, el uso de biodiesel desafortunadamente incrementa la pro-porción de NOx emitida debido a propiedades del biodiesel tales como son el número de cetano y/o al grado de instauración del biodiesel empleado. Así, se ha observado que a medida que aumenta el grado de insaturación aumentan las emisiones de NOx. En general, está aceptado que el aumento en las emisiones de NOx es proporcional a la cantidad de biodiesel usado (B10, B20, etc.) siendo el incremento máximo del 10% respecto al nivel de emisión de NOx generados con el combustible diesel convencional.
Para disminuir la producción de NOx atribuible al uso de biodiesel se han propuesto varias posibles soluciones, como son la selección de biodiesel con un mayor grado de saturación o hidrogenar el biocombustible mediante la adición de metil ésteres de cadenas cortas. Para reducir el grado de instauración es posible adicionar aditivos para aumentar el número de cetano (di-ter-butyl peróxido (DTBP), etil hexil nitrato (EHN)) o aditivos antioxidantes (ter -butil - hidroquinona (TBHQ) y butil hidroxianisol (BHA)).
Sin embargo, a pesar de estas posibles mejoras tanto en el motor como en el combustible empleado, el uso de sistemas de post—combustión continúa siendo necesario para cumplir con la normativa respecto a NOx y carbonilla.
La descomposición de NO en sus elementos según la reacción
2NO<r> N2 +02
es termodinàmicamente favorable tanto a temperatura ambiente como a temperatura rela-tivamente alta [13]. El NO es un compuesto termodinàmicamente inestable (entalpia de la formación AHf = 90 kj/mol), y un radical libre bajo condiciones prácticas en el escape de una automóvil. Sin embargo, en presencia de oxígeno no es posible lograr su disociación en O, y N, por restricciones cinéticas [11]. En presencia de un catalizador, los sitios activos
1 Del inglés, "Exhanst Gas Reàrcuìation "
| 133 |

Efec to del uso de Biodiesel en la Emis ión de Contaminantes Atmosfér icos
del metal se cubren por oxígeno fuertemente adsorbido, evitando así la adsorción de NO Para eliminar el oxígeno de la superficie y permitir la adsorción y disociación del NO es necesario añadir especies reductoras [8]. Así, para la reducción de NOx se han propuesto dos procesos: la reducción catalítica selectiva (SCR3), y las trampas de almacenamiento) reducción de NOx (NSRC4) [8].
En la reducción catalítica selectiva de NOx se pueden usar diferentes agentes reductores como alcoholes, hidrocarburos, amoníaco (NH.;) o urea ((NH,) CO), y CO o H, [12, 14], El uso de hidrocarburos como agente reductor supone una ventaja comparada con otros reductores, ya que el mismo combustible podría ser empleado como tal. La reactividad de los hidrocarburos en condiciones de exceso de oxígeno depende de la temperatura de ope-ración y de la naturaleza del catalizador. Se están estudiando diferentes formulaciones pata el catalizador basados en metales nobles y de transición soportados en óxidos, así como ca-talizadores basados en zeolitas [15-17], La utilización de urea o amoníaco es una tecnología bien establecida que se está empleado con éxito en la reducción selectiva de NOx en fuentes estacionarias como, por ejemplo, centrales térmicas de combustión, y ya existen prototipos de este proceso que se están utilizando en vehículos pesados. Para catalizar este proceso, los catalizadores más utilizados son los de pentóxido de vanadio soportado en titanio.
Los catalizadores para el almacenamiento y reducción de NOx (NSRC) operan en dos eta-pas que se repiten periódicamente. En una primera etapa, que ocurre durante el período d conducción con condiciones ricas en oxígeno, se lleva a cabo la retención de los NOx como nitrato, empleando un óxido metálico básico (por ejemplo BaO). Periódicamente, cuando el material adsorbente está saturado, se introduce una cierta cantidad de gas reductor en el escape. Al cambiar la composición de los gases de escape a una atmósfera reductora poi un periodo corto de tiempo, el NOx almacenado es desorbido y reducido a N . Se han propuesto distintos gases reductores, siendo el H, uno de los más estudiados. Para cataliza! esta reducción, los catalizadores suelen incorporar algún metal como, por ejemplo, platino,
Filtros para partículas
Como ya se ha comentado, en la mayoría de estudios realizados se concluye que existe una disminución en la cantidad de carbonilla emitida en los gases de escape cuando se reemplaza el combustible diesel por biodiesel. Algunos autores atribuyen esta reducción a un conteni-do más alto de oxígeno en las moléculas de biodiesel que favorece la combustión completa^ promueve la oxidación de la carbonilla formada. Otros autores argumentan que el biodiese acelera la combustión al aumentar el tiempo de residencia en la cámara (debido a la mayoi
' Del inglés, "Selective Catalytic. Reduction ". 4 Del inglés, "NOx Storage Reduction Catalysts".
|134|

,/wances jnv.;seií!-]ic>voh i í;í f r o a u c c i o n ue n iocomousuu ies
viscosidad del combustible), lo que disminuye la cantidad final de carbonilla en los gases de escape. Otra explicación se basa en la diferencia entre las estructuras de la carbonilla diesel y biodiesel, que tiene como consecuencia una oxidación hasta seis veces más rápida para la carbonilla biodiesel. Finalmente, se ha indicado que la baja o casi nula concentración de sulfatos en el combustible biodiesel también contribuye a formar una menor cantidad de carbonilla. Los sulfatos, que suelen estar presentes en el combustible diesel convencional, quedan adsorbidos en las partículas de carbonilla contribuyendo a su masa.
Las modificaciones en el proceso de operación del motor diesel han dado lugar a una reduc-ción significativa de la carbonilla generada, aunque los sistemas de post-combustión siguen siendo necesarios para cumplir con las nuevas regulaciones. La oxidación de la carbonilla es una de las soluciones propuestas para su eliminación de los escapes diesel. Así, el sistema más prometedor consiste en la captura de las partículas y su oxidación, y para ello lo que se está implementando con mayor eficacia es el uso de filtros de partículas diesel [11].
Un filtro de partículas diesel tiene que retener las partículas de los gases de escape, y los filtros disponibles actualmente permiten la captura de hasta el 95% de la carbonilla seca. Al quedar partículas retenidas en el filtro, la presión de los gases de combustión aumenta. Así, para evitar caídas de presión en el sistema debido a la acumulación de partículas y mantener la eficiencia del motor, se utiliza un sistema de control electrónico que advierte cuándo el filtro necesita ser regenerado. Debido a que las partículas diesel están compuestas principal-mente por carbón y una fracción de hidrocarburos adsorbidos, la forma más adecuada de regeneración es su oxidación a CO? y HzO. Este proceso requiere una temperatura superior a los 600°C [18], que no se suele alcanzar en los gases de combustión. En general, la tem-peratura de los gases producidos en los diferentes regímenes de operación del motor suele ser inferior a 450°C, por lo que la oxidación no tiene lugar en estas condiciones. Para llevar a cabo la regeneración existen dos métodos, denominados regeneración activa y regeneración pasiva. Los métodos que requieren un aporte externo de energía para aumentar la tempe-ratura del filtro se denominan métodos de "regeneración activa" y los métodos basados en la disminución de la temperatura de combustión se denominan métodos de "regeneración pasiva". En la regeneración activa la energía térmica necesaria para la oxidación se puede obtener de diversas maneras, como, por ejemplo, con un quemador diesel, transformando energía eléctrica en calor o inyectando una pequeña cantidad de combustible en el escape cuva combustión originaría el aumento de temperatura. La gran ventaja de la regeneración activa es la independencia de la temperatura del gas de escape. Aunque son procesos relati-vamente simples, son ineficientes, incontrolados y presentan un coste económico al requerir un consumo adicional de combustible para realizar la regeneración. Por otro lado, en la rege-neración pasiva, se trata de reducir la temperatura de ignición de la carbonilla, y para ello se incluyen aditivos catalíticos al combustible y/o especies catalíticas antes y/o dentro de los fil-tros [12], Estos métodos evitan total o parcialmente el consumo adicional de combustible.
— ^ ^ ^ — m m m m m m m 1351 m m m m m — ^ ^ ^ — m m — ^

{ *• > < 1 " ' He Biodiesei en la [ tmis i im de Contaminantes Amioster i
En condiciones reales, uno de los principales inconvenientes de la oxidación catalizada es el contacto entre el catalizador y las partículas de carbonilla. Se trata de un contacto sólido — sólido y, por lo tanto, es débil, lo que implica que se requieren temperaturas elevadas pan activar la regeneración del filtro. El problema del contacto entre el catalizador y la carbonilla puede ser solucionado incorporando un precursor del catalizador al combustible ya que, en estas condiciones, el catalizador se incorpora a la partícula de carbonilla. Para este objetivo se han utilizado catalizadores basados en Mn, Cu, Fe, Sr, Ce y Pt [19-22] ya que son metales activos para la oxidación del carbón y se consigue una disminución de la temperatura en unos 200°C aproximadamente, comparado con la reacción sin catalizar [22], En estos siste-mas, el metal presente en el motor después del proceso de combustión sirve como núcleo para el crecimiento de la carbonilla a partir de la condensación de los hidrocarburos no quemados.
Otros sistemas alternativos se basan en el uso de uno o varios filtros con metales. En las condiciones actuales de funcionamiento, se puede usar el NO presente en los escapes de los motores diesel para oxidar continuamente la carbonilla atrapada. Este planteamiento se basa en que el N 0 2 , generado por la oxidación catalizada de NO por Pt, es un compuesto mucho más oxidante que el 0 2 y que, si está en concentraciones adecuadas en el escape del automóvil, puede oxidar la carbonilla depositada en el filtro a una menor temperatura [21], El papel de platino es acelerar la oxidación de NO a N02 . Por otra parte, también s han desarrollado catalizadores de oxidación para facilitar el quemado del combustible extra que es adicionado al escape para elevar la temperatura del filtro de partículas y mejorarla combustión de la carbonilla con oxígeno (usualmente sobre 550°C).
Finalmente, se han propuesto sistemas capaces de eliminar simultáneamente NOx y carbo-nilla haciendo reaccionar ambos contaminantes en un único sistema catalítico. Estos siste-mas están basados en el carácter reductor de los materiales carbonosos que pueden reducif NOx a N2 en presencia de oxígeno y, por lo tanto, en lo que carbonilla podría actuar com agente reductor. Así, se ha demostrado que algunos metales como K, Cr, Fe, Co, Ni y Cu pueden ser catalizadores prometedores para la reducción de NOx con carbonilla [22-24], Sin embargo, debido a la complejidad técnica y la dificultad del contacto entre el catalizada y la carbonilla, estos sistemas no son utilizados comercialmente.

Avances liivestigaávos en la Producción de Biocombusribles
7.3. Estudio experimental del efecto del uso de biodiesel comercial en la generación de contaminantesy en su eliminación en sistemas de post-combustión
Para analizar el efecto del uso del biodiesel comercial en la generación de contaminantes se ha llevado a cabo un estudio experimental utilizando un motor diesel de inyección directa (Nisan 2.0) montado en un banco de pruebas. Este dispositivo experimental está disponi-ble en los laboratorios del Grupo de Materiales Carbonosos y Medio Ambiente (MCMA) del departamento de Química Inorgánica de la Universidad de Alicante. Se han simulado condiciones normales de encendido, operado a una a velocidades entre 1.800 y 2.000 rpm en condiciones de ausencia carga. Para llevar a cabo el estudio se ha empleado tanto diesel como biodiesel comercial. Así, se determinó la composición de los gases de escape (CO, C02, NO, NO„ N,0 , 0 2 e hidrocarburos totales) y la velocidad de generación de carboni-lla. Las carbonillas generadas con diesel y biodiesel (100%) fueron recolectadas y se estudió su combustión en condiciones de laboratorio. Se llevaron a cabo experimentos de oxidación no catalizada y en presencia de un catalizador Cu/Al,,0, [25] utilizando una mezcla gaseosa de 500 ppm NOx + 5%On, simulando la composición de los gases del escape. Los ensayos se llevaron a cabo en un reactor cilindrico de cuarzo de 1 cm de diámetro interno totalmen-te automatizado y conectado a los analizadores de gases NDIR - UV para determinar la composición de los gases de combustión o reacción NO, NOz, CO, C 0 2 y O,. Además de las carbonillas diesel y biodiesel, como referencia, se han estudiado dos negros de carbón comerciales (Printex - U y Vulcan XC72) que son habitualmente utilizados como carboni-llas modelo ya que la homogeneidad de sus propiedades facilita la realización de estudios sistemáticos.
Para la caracterización de las cuatro carbonillas se utilizaron diferentes técnicas: análisis ele-mental e inmediato, microscopía electrónica de transmisión (TEM), difracción de rayos X (DRX), espectroscopia Raman, espectroscopia infrarroja de reflectancia difusa con Trans-formada de Fourier (DRIFT) y espectroscopia infrarroja (DRIFTS).
7.3.1. Efecto del uso de biodiesel en la emisión de contaminantes Los resultados de los gases generados por el motor diesel, utilizando los dos combustibles -diesel y biodiesel comercial-, se venn en la Figura 7.1. Se muestra que las cantidades de NOx son comparables, las concentraciones de CO, son similares, la cantidad de carbonilla disminuye de forma notable y la concentración de monóxido de carbono aumenta. El au-mento en la concentración de CO en los gases emitidos cuando se usa biodiesel difiere de los resultados presentados en la literatura, lo que puede estar relacionado con las condicio-nes de funcionamiento del motor (ausencia de carga).
1137 |
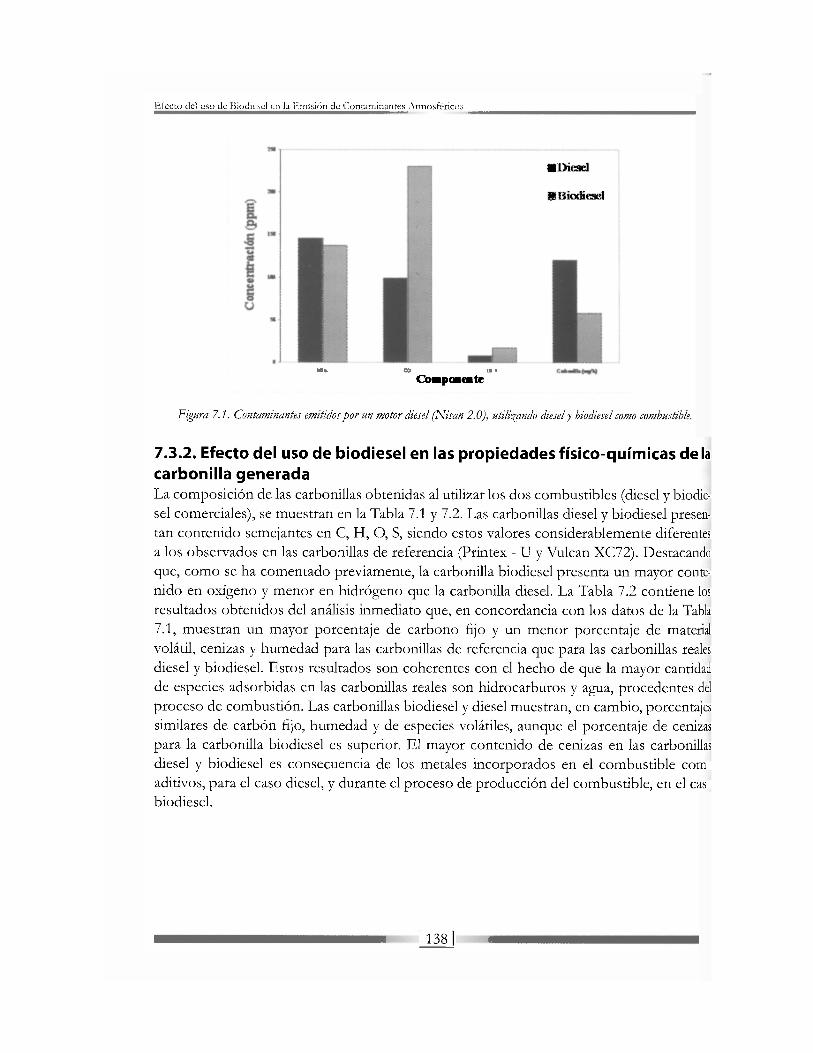
Efecto del uso de Biodiesel en Ja Emisión de Contaminantes Atmosfér icos
•fc CO ni CoMpaacate
Figura 7.1. Contaminantes emitidos por un motor diesel (Nisan 2.0), utilizando diesel y biodiesel como combustible.
7.3.2. Efecto del uso de biodiesel en las propiedades físico-químicas déla carbonilla generada La composición de las carbonillas obtenidas al utilizar los dos combustibles (diesel y biodie-sel comerciales), se muestran en la Tabla 7.1 y 7.2. Las carbonillas diesel y biodiesel presen-tan contenido semejantes en C, H, O, S, siendo estos valores considerablemente diferentes a los observados en las carbonillas de referencia (Printex - U y Vulcan XC72). Destacando que, como se ha comentado previamente, la carbonilla biodiesel presenta un mayor conte-nido en oxígeno y menor en hidrógeno que la carbonilla diesel. La Tabla 7.2 contiene los resultados obtenidos del análisis inmediato que, en concordancia con los datos de la Tabla 7.1, muestran un mayor porcentaje de carbono fijo y un menor porcentaje de material volátil, cenizas y humedad para las carbonillas de referencia que para las carbonillas reales diesel y biodiesel. Estos resultados son coherentes con el hecho de que la mayor cantidad de especies adsorbidas en las carbonillas reales son hidrocarburos y agua, procedentes del proceso de combustión. Las carbonillas biodiesel y diesel muestran, en cambio, porcentajes similares de carbón fijo, humedad y de especies volátiles, aunque el porcentaje de cenizas para la carbonilla biodiesel es superior. El mayor contenido de cenizas en las carbonillas diesel y biodiesel es consecuencia de los metales incorporados en el combustible com aditivos, para el caso diesel, y durante el proceso de producción del combustible, en el cas biodiesel.
138 |

Avances Imesaga t i yn s en la. Producción de Bio«>mbustiblt:s
Tabla 7.1. Análisis elemental de las carbonillas.
Componente Diesel Biodiesel Printex - U Vulcan C (%) 86,08 82,15 91,22 96,93 H (%) 3,73 2,70 0,70 0,12 N (%) 0,63 0,69 0,33 0,25 S (%) 0,14 0,25 0,12 0,37 O* (%) 9,41 14,22 7,63 2,33
*Obtenido por diferencia.
Tabla 7.2. Análisis inmediato de las carbonillas.
Componente Diesel Biodiesel Vulcan Carbón Fijo (% peso) 64,3 65,8 91,8 96,0 Voláatiles (% peso) 34,5 30,4 8,1 3,5 Humedad (% peso) 1,6 2,2 0,9 0,5 Cenizas* (% peso) 1,2 4,3 0,1 0
*Expresada en base seca
Los difractogramas de rayos X incluidos en la Figura 7.2 muestran, en todos los casos, dos bandas de difracción con intensidad máxima a 24.9 y 43.6°, correspondiente a picos carac-terísticos de grafito. La menor intensidad de las bandas para las carbonillas diesel y biodiesel indica que la estructura de los planos grafiticos es más imperfecta (menos cristalina). Sin embargo, los espectros Raman incluidos en la Figura 7.3, deconvolucionados en cuatro bandas habitualmente denominadas T, D, A y G, no revelan diferencias significativas en la estructura de las cuatro carbonillas.
||,-<|| Vulcan
Printac-U
Biodiesel
j — , , I
10 20 30 40 50 60 2 « 0
Figura 7.2. Difractogramas de rayos X de las carbonillas.
M H H i ' ' | 139 | M M H H H H i ^ M
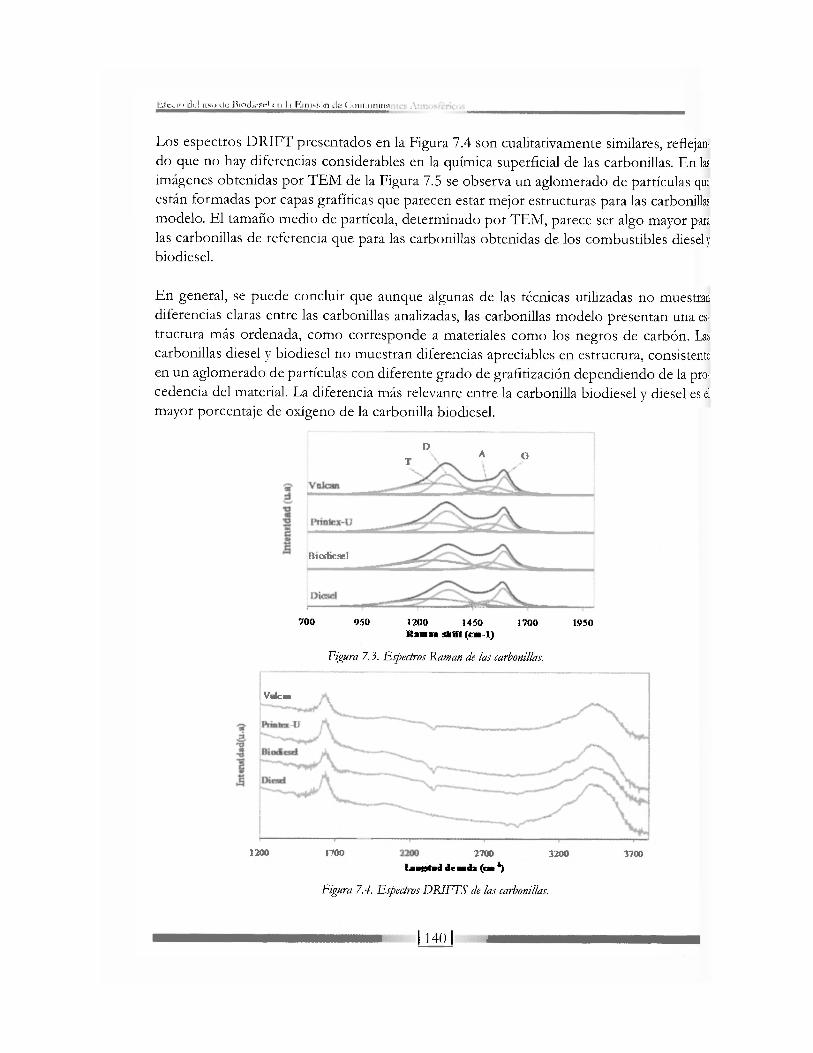
Los espectros DRIFT presentados en la Figura 7.4 son cualitativamente similares, reflejan-do que no hay diferencias considerables en la química superficial de las carbonillas. En las imágenes obtenidas por TEM de la Figura 7.5 se observa un aglomerado de partículas que están formadas por capas grafiticas que parecen estar mejor estructuras para las carbonillas modelo. El tamaño medio de partícula, determinado por TEM, parece ser algo mayor pata las carbonillas de referencia que para las carbonillas obtenidas de los combustibles diesel y biodiesel.
En general, se puede concluir que aunque algunas de las técnicas utilizadas no muestran diferencias claras entre las carbonillas analizadas, las carbonillas modelo presentan una es-tructura más ordenada, como corresponde a materiales como los negros de carbón. Las carbonillas diesel y biodiesel no muestran diferencias apreciables en estructura, consistente en un aglomerado de partículas con diferente grado de grafitización dependiendo de la pro-cedencia del material. La diferencia más relevante entre la carbonilla biodiesel y diesel es el mayor porcentaje de oxígeno de la carbonilla biodiesel.
D A O T
Biodiesel
700 950 1200 1450 1700 1950 Rnm sfciTt (CM I)
Figura 7.3. Espectros Raman de las carbonillas.
Volca.
1200 1700 2700 3200 3700 Lugted deaada (ca1)
Figura 7.4. Espectros DRIFTS de las carbonillas.
| 140 |