· web viewmeta-assembly and identification of novel qtl for milk production traits in sheep....
TRANSCRIPT

Validación de herramientas
bioinformáticas para la detección de
señales de selección en el genoma de
Ovis aries.
Autora: Marta Alfaro*
Supervisoras: Dra. Otsanda Ruiz-Larrañaga* y Dra. Andone Estonba*
*Departamento de Genética, Antropología Física y Fisiología Animal. Universidad del País Vasco-
EuskalHerrikoUnibertsitatea (UPV/EHU), C/Barrio Sarriena s/n, CP48940, Leioa, Bizkaia.
Contacto: [email protected]
1

ÍNDICE
Abstract-Resumen 3
Introducción 4
Efectos de la selección en el genoma 4
Antecedentes del trabajo 7
Hipótesis y objetivos 11
Materiales y Métodos 12
Simulación del experimento PoolSeq y tratamiento de secuencias 12
Detección de señales de selección en el genoma 15
Identificación de regiones CCR 16
Validación de regiones CCR 16
Resultados 17
Discusión 23
Conclusión 27
Agradecimientos 28
Bibliografía 29
ANEXO 1 32
ANEXO 2 35
ANEXO 3 36
2

ABSTRACT
The adaptation and specialization of sheep to the human environment has resulted in phenotypically highly
diverse breeds. Artificial selection causes “selection sweep” in the genome of species by: a) increasing the
linkage disequilibrium between polymorphisms, b) distorting the frequency spectrum of them, and c)
increasing between-breed differentiation. The aim of this study is the validation of a bioinformatic tool
designed for the identification of selection signatures in the genome. With this purpose we applied the
Popoolation software to simulated PoolSeq experimental data. Popoolation is a computational program that
allows an intra-population analysis by the estimation of Tajima´s D values and an inter-population analysis
by Fst calculation. In all, 66 CCR (Convergent Candidate Regions) were identified as candidate regions for
being selection signatures in the ovine genome. From them, 36 were previously related with production
traits; therefore, Popoolation software has been validated and will be used in the ongoing study focused on
Sasi-ardi and Latxa sheep breeds. Further studies focused on the new 30 CCR identified in the present work
could provide relevant information to be applied in breeding and improvement programs of the species.
RESUMEN
La adaptación y la especialización de las ovejas a ambientes humanizados ha dado lugar a especies fenotípi -
camente muy diversas. La selección artificial provoca diversos cambios en el genoma de las especies, fenó-
meno conocido como "selective sweep", mediante: a) el aumento del desequilibrio de ligamiento entre poli -
morfismos, b) la distorsión del espectro de frecuencia de los mismos, y c) el aumento de la diferenciación en-
tre razas. El objetivo de este estudio es la validación de una herramienta bioinformática diseñada para la
identificación de señales de selección en el genoma. Hemos utilizado con ese propósito el software Popoola-
tion a datos experimentales simulados de PoolSeq. El Popoolation es un programa bioinformático que per-
mite el análisis intra-poblacional mediante la estima del estadístico D de Tajimay el análisis inter-poblacional
mediante el cálculo del parámetro Fst. En total, se han identificado 66 regiones CCR (Convergent Candidate
Regions) definidas como aquellas regiones candidatas a ser señales de selección en el genoma ovino. De
ellas, 36 han podido ser validadas en las bases de datos de QTL existentes para la especie, así como a través
de la bibliografía. Por tanto, se puede decir que el software Popoolation queda validado para este tipo de es-
tudios y será utilizado para los propósitos de un proyecto similar en marcha enfocado a las razas ovinas loca-
les Sasi-ardi y Latxa. Un análisis más exhaustivo de las 30 nuevas regiones CCR identificadas en el presente
trabajo podría proporcionar información de utilidad en los programas de selección y mejora de la especie.
3

INTRODUCCIÓN
EFECTOS DE LA SELECCIÓN EN EL GENOMA
La historia de la civilización está estrechamente relacionada con los animales domésticos. La
domesticación hizo posible la transición de la vida nómada a una sedentaria, que supuso una profunda
reorientación de la sociedad humana, aumentando la estabilidad de la subsistencia y alimentando el
crecimiento demográfico y la expansión [1]. Hay más de 40 especies que han sido domesticadas durante los
últimos 10.000 años, contribuyendo directa o indirectamente a la producción agrícola. Estos recursos
zoogenéticos son vitales para el desarrollo económico de la mayoría de los países en el mundo
desempeñando un papel fundamental en la subsistencia de muchas comunidades y en la sostenibilidad de los
sistemas agropecuarios [2].
La adaptación de las especies a nuevos ambientes se ha llevado a cabo mediante selección natural
(que es la capacidad de sobrevivir y reproducirse en cautiverio) y selección artificial (que es la selección
controlada por los seres humanos) y ha dado como resultado una amplia diversidad fenotípica dentro de estas
especies [3]. Esta diversidad provoca ciertos cambios en el genoma de las especies [4].Por un lado, cuando
en un determinado locus el alelo favorable para un rasgo es seleccionado, su prevalencia en la población
aumenta, persistiendo durante muchas generaciones. Esta selección puede ser bien parcial o incompleta, o
bien completa cuando el alelo favorable acaba fijándose en la población (Figura 1a). A su vez, dado el
desequilibrio de ligamiento (LD) existente entre el alelo seleccionado y los que le rodean, la frecuencia de
estos últimos también se ve modificada, arrastre que se conoce como efecto “Hitchhicking” [5]. Este hecho
provoca una distorsión en el espectro de frecuencias alélicas o SFS (Site Frequency Spectrum) de la región
seleccionada. Al mismo tiempo, este LD entre el alelo seleccionado y los adyacentes persiste en el tiempo
hasta que la recombinación lo descompone. Todo ello trae como consecuencia una reducción en la diversidad
genética o heterocigosidad de la población, lo que se conoce como “selective sweep”. Por último, la
selección de diferentes regiones genómicas en una población y en otra dentro de una misma especie (por
ejemplo, debido al interés por diferentes rasgos) produce un distanciamiento genético entre ellas dado que las
frecuencias alélicas diferirán en una y en otra.
La identificación de estos cambios o señales de selección en el genoma puede proporcionarnos
información relativa al proceso de domesticación de las especies. Existen diversos métodos para la detección
de estas regiones (Figura 1).
4

1-Métodos basados en el SFS
Estos métodos son los que se basan en la búsqueda de aquellas distorsiones en las frecuencias de los
polimorfismos o alelos seleccionados y los que le rodean (Figura 1b). Para detectar este tipo de cambios en el
genoma contamos con diferentes estadísticos. La D de Tajima [6] fue la primera en utilizarse para detectar
este tipo de señales, y es, hoy por hoy, el estadístico más extendido para este tipo de análisis. Este parámetro
compara el número de diferencias entre pares de individuos respecto al número total de polimorfismos en
una determinada región genómica. Debido a que los alelos de baja frecuencia contribuyen menos al número
de diferencias por pares, un excedente de alelos raros disminuye el último valor de manera desproporcionada
al primero. Por lo tanto, bajos valores de este parámetro pueden ser indicativos de selección positiva en la
región que estamos analizando.
Hay otros parámetros que utilizan un enfoque similar al de la D de Tajima, como es la H de Fay y
Wu que compara también el número de diferencias entre pares de individuos pero respecto al número de
individuos homocigotos para el alelo derivado. Los valores bajos de H indican un exceso de alelos derivados
de alta frecuencia, que sugieren que ha habido una selección positiva en la región examinada (revisado en
ref. 4).
2-Métodos basados en el desequilibrio de ligamiento
Tal y como se ha mencionado, cuando el alelo de un polimorfismo concreto es seleccionado se
genera un fuerte LD con los alelos que lo rodean hasta que la recombinación descompone estas asociaciones.
Por lo tanto se puede dirigir el análisis hacia la detección de regiones con un alto LD en relación con su
prevalencia dentro de una población (Figura 1c). Los métodos basados en el LD son particularmente útiles
para la identificación de variantes o alelos que han sufrido una selección parcial en una región del genoma.
Ejemplos de estos métodos podrían ser el test LHR (Long-RangeHaplotype), que compara la
frecuencia de los diferentes haplotipos existentes en una determinada región con su homocigosidad para
poder así identificar aquellos haplotipos extendidos que sugieran una alta prevalencia, o el iHS (Integrated
Haplotype Score) que compara el área de debajo de la curva definida por la homocigosidad de las variantes
derivadas y ancestrales (revisado en ref.4).
5

3-Métodos basados en la diferenciación de poblaciones
Como ya se ha comentado anteriormente, diferentes poblaciones pueden estar sujetas a diferentes
presiones ambientales y humanas y como resultado las frecuencias de los alelos seleccionados en una y otra
pueden diferir significativamente si comparamos ambas poblaciones (Figura 1d).
La métrica más utilizada para detectar estas diferencias inter-poblacionales es el índice de fijación de
Wright, Fst [7]. Grandes valores de Fst en un locus indican una diferenciación mayor entre las poblaciones, lo
que sugiere que dicho locus ha sufrido un proceso de selección en alguna de las poblaciones analizadas.
4-Métodos compuestos
Dados los diferentes tipos de cambios que la selección provoca en el genoma, y los diferentes
estadísticos específicos desarrollados para su detección, hoy en día numerosos autores combinan múltiples
parámetros con el objetivo de proporcionar una mayor potencia y/o resolución espacial a la hora de
identificar las señales de selección a lo largo del genoma(Figura 1e). Esto contribuye, a su vez, a distinguir
entre los efectos causados en el genoma por una selección real en lugar de por eventos demográficos tales
como la expansión o contracción de la población.
Un ejemplo es la prueba de CLR (Composite Likelihood Ratio) que evalúa la probabilidad de que
un evento selectivo sea responsable de un exceso de alelos raros. Éste método utiliza la distribución espacial
de las frecuencias de las mutaciones en una población dada para detectar las señales de selección. Existen
variaciones posteriores de esta prueba que incluyen datos basados en LD, así como otros basados en la
diferenciación de la población en relación a la frecuencia de los alelos para generar un espectro
bidimensional de frecuencias [revisado en ref. 4].
6

Figura 1: Métodos para la detección de señales de selección: a) Diversidad poblacional. b) Distorsiones del espectro de frecuencia al
seleccionarse un polimorfismo determinado. c) Incrementos del desequilibrio de ligamiento entre los polimorfismos seleccionados y
los adyacentes. d) Diferenciación entre dos poblaciones. e) Métodos compuestos (tomada de ref. 4).
ANTECENDENTES DEL TRABAJO
El grupo de investigación liderado por la Dra. Andone Estonba trabaja actualmente en un proyecto
basado en la detección de este tipo de señales de selección en el genoma de la especie ovina. Esta especie fue
domesticada junto con otras como la porcina o bovina hace aproximadamente 9000 años, dando lugar a una
vida sedentaria dedicada a la agricultura y a la cría animal y convirtiéndose en el sistema principal de
7

producción de comida. Desde su domesticación en Asia Central y Oriente Medio, la especie se ha establecido
en una amplia distribución geográfica debido a su capacidad de adaptación a las dietas de nutrición pobre, la
tolerancia a las condiciones climáticas extremas y a su tamaño manejable [8] Se trata de una especie de
comportamiento dócil y que vive en manadas pudiendo un solo pastor controlar un gran rebaño de ovejas.
Constituye además un animal de vital importancia económica en nuestra sociedad como animal ecológico y
como fuente de lana, carne y leche. Es por ello que la selección de las diferentes poblaciones ovinas se ha
dirigido principalmente a la producción lechera, lanera y cárnica [9].
En concreto, el estudio que se está llevando a cabo se centra en dos razas ovinas locales. La primera
es la raza Sasi-ardi; una raza local semisalvaje de la región pirenaica occidental que se utiliza principalmente
como animal ecológico ya que presenta una serie de cualidades idóneas para adaptarse a terrenos abruptos y
de alta pluviosidad como son una gran agilidad de movimientos y elevada rusticidad. Actualmente la raza se
encuentra en un estado crítico de conservación [10], estando incluida en el Grupo de Razas Autóctonas de
Protección Especial del Catálogo Oficial de Razas de Ganado de España. El principal interés de esta raza
estriba en que puede ser considerada como descendiente actual semisalvaje del ovino doméstico local, ya que
las restantes razas ovinas de la región han estado bajo intensa selección artificial. Tal es el caso de la raza
Latxa, la segunda raza en estudio. Esta es una raza propia del Norte de España, que tiene dos sub-razas como
son la Latxa de Cara Negra (predominante en Navarra y Guipúzcoa) y la de Cara Rubia (predominante en
Álava). Se trata en general de una raza ovina de lana áspera que ha estado bajo una fuerte selección en los
últimos treinta años y que se cría principalmente por su leche para la producción de queso de tipo Idiazábal y
para la venta de sus corderos.
La comparación entre el genoma de la raza Sasi-ardi, representante de la población ovina primigenia,
rústica y adaptada a entornos desfavorables, y de la raza Latxa, representante de nuestro ovino actual tras la
domesticación y bajo intensa selección artificial, se presenta como un escenario idóneo para la evaluación
del efecto de la selección en el genoma del ovino de la región pirenaica occidental. Los resultados que se
obtengan de este estudio podrían aportar información relevante sobre los efectos de la domesticación y la
selección en estas razas complementando así la información existente sobre QTLs (Quantitative Trait Loci) y
genes asociados a caracteres de interés en la especie. Esto, a su vez, podría dar lugar a futuros estudios
genéticos con una aplicación directa en los programas de selección y mejora.
El estudio del efecto de la selección en razas ganaderas ha venido realizándose desde hace escasos
años mediante genotipado de miles de SNP, generalmente, o al menos en la especie ovina y bovina,
recogidos en chips de alta densidad. En este sentido, el estudio más reciente en la especie ovina siguiendo
esta estrategia es el llevado a cabo por Gutiérrez-Gil et al. [10]. En él, los autores identifican diversas señales
de selección a lo largo del genoma de la especie específicamente relacionadas con la producción lechera
mediante la comparación de un pool de razas seleccionadas con otro pool de razas no seleccionadas. Para
8

ello, hicieron uso del chip de genotipado OvineSNP50 BeadChip de Illumina que consta de 49.034
marcadores SNP (Single Nucleotide Polymorphism). Estos marcadores fueron analizados siguiendo dos
aproximaciones; por un lado, los autores han identificado las regiones genómicas con diferencias extremas
en cuanto a frecuencias alélicas (Fst) entre ambas poblaciones y regiones con una reducida heterocigosidad.
De este modo, en dicho estudio se identifican 6 regiones significativas siguiendo ambos métodos. Además,
los mismos autores proponen algunos genes candidatos en estas regiones como responsables del posible
efecto en caracteres de producción, como son, el gen ABCG2 (ATP-Binding Cassette) y el gen SPP1
(Secreted Phosphoprotein 1) localizados en el cromosoma 6.
Sin embargo, a día de hoy, las tecnologías NGS (Next Generation Sequencing) resultan ser una
aproximación más rentable al uso de estos chips. Estas tecnologías son efectivas para el descubrimiento y
genotipado de cientos de marcadores polimórficos a lo largo del genoma completo de cualquier organismo.
Además la información para realizar estudios de genómica de poblaciones puede ser extraída del análisis del
DNA de un pool de individuos de forma conjunta, estrategia denominada PoolSeq [11], que resulta ser un
método efectivo de reducir los costes de secuenciación sin reducir el tamaño de muestra ni el poder
estadístico. Es por ello que el grupo de investigación ha escogido esta aproximación metodológica para
acometer el mencionado estudio
Si bien en ovino no existen trabajos publicados en esta línea, algunos autores ya han aplicado de
forma efectiva esta aproximación PoolSeq en otras especies. Rubin et al. [12] llevaron a cabo un estudio
donde resecuenciaron el genoma de la gallina, tanto de la especie doméstica como de su ancestro salvaje-
Mediante la estima de la heterocigosidad a lo largo del genoma, utilizando para el análisis los más de5M de
SNPs detectados, identificaron numerosas regiones candidatas a estar bajo el efecto de la selección artificial
relativa a la domesticación. Uno de los hallazgos más relevantes es el del gen TSHR (Tyroid stimulating
Hormone Receptor), implicado en la regulación metabólica y el calendario reproductivo como respuesta a
cambios en la duración del día. Los mismos autores trasladaron esta metodología a la especie porcina [13],
donde tras la resecuenciación del genoma de 8 poblaciones de cerdos y jabalíes, detectaron fuertes signos de
selección en tres loci valiéndose también del cálculo de la heterocigosidad. Estos loci albergan,
precisamente, 3 QTL previamente descritos en porcino involucrados en uno de los cambios morfológicos
más característicos en la especie doméstica, la elongación dorsal por el aumento del número de vértebras.
Posteriormente, Axelsson et al. [14] confirmaron el alto poder de resolución de la secuenciación de pools
para el estudio de los efectos de la domesticación. Su trabajo, basado en la resecuenciación del genoma
completo de poblaciones de perros y lobos, identifica, mediante el análisis de la Fst y la heterocigosidad de
más de 3M de variantes genéticas, 36 regiones genómicas posiblemente seleccionadas durante el proceso de
domesticación de la especie canina. Resalta, además, la localización en estas regiones de genes involucrados
en el desarrollo del sistema nervioso, relacionados probablemente con cambios de comportamiento
fundamentales para la domesticación del perro.
9

En los últimos años se han desarrollado diversos software bioinformáticos diseñados para la
detección de señales de selección a partir de datos generados de experimentos PoolSeq. Uno de ellos es el
SweeD [15], una herramienta para la detección de estas señales utilizando el método CLR mencionado
anteriormente. El Pool-hmm [16], por su parte, implementa el modelo HMM (Hidden Markov Model) en el
que se estima la probabilidad de que cada posición del genoma haya sido seleccionada basándose en el
espectro de frecuencias alélicas. Por último, el programa Popoolation, tiene como ventaja frente a los
anteriores el hecho de que permite realizar, por un lado, un análisis intra-poblacional mediante métodos
basados en los cambios en el SFS, y por otro, un análisis inter-poblacional [17,18]. Ninguna de estas tres
herramientas ha sido hasta el momento utilizada para la detección de señales de selección en especies
domésticas.
10

HIPÓTESIS Y OBJETIVOS
La selección artificial a la que la especie ovina ha sido sometida con diferentes objetivos ha
provocado diversos cambios en el genoma de los individuos. La hipótesis del presente trabajo es que el
software Poopolation, diseñado para la detección de este tipo cambios o señales de selección, permite
detectar aquellas regiones que han sido seleccionadas para un determinado carácter. Por lo tanto, nos
planteamos como objetivo la validación del software Poopolation, como herramienta bioinformática,
mediante la detección de señales de selección asociadas a la producción lechera y a la producción cárnica en
la especie ovina simulando un experimento PoolSeq.
11

MATERIALES Y MÉTODOS
SIMULACIÓN DEL EXPERIMENTO POOLSEQ Y TRATAMIENTO DE SECUENCIAS
En la Figura 2 se describen los pasos que se han seguido para simular el experimento PoolSeq
llevado a cabo en el laboratorio.
Figura 2: Esquema general de trabajo para la simulación del experimento PoolSeq.
Como datos de partida se han utilizado las secuencias relativas al genoma de 20 individuos
(hembras) de la especie ovina disponibles en la base de datos NCBI(http://www.ncbi.nlm.nih.gov/sra).
Estos datos pertenecen a un proyecto llevado a cabo por el ISGC (Internation Sheep Genomic Consortium)
en el que se ha secuenciado el genoma de 75 individuos de 45 razas ovinas mediante la tecnología de
Illumina. Se han seleccionado las secuencias de 10 individuos de 4 razas de producción cárnica y de otros 10
individuos de 6 razas de producción lechera razas en su mayoría de origen Europeo (Tabla 1).
12

Tabla 1. Individuos seleccionados para la simulación del experimento PoolSeq
Raza Nº Individuos Producción
lechera
Producción
cárnica
Número de acceso
en el NCBI
Ojalada 2 x SRX150351
SRX150340
Churra 2 x SRX150349
SRX150288
Castellana 2 x SRX150344
SRX150323
Lacaune 1 x SRX150291
Merino 3 x SRX150327
SRX150308
SRX150292
Swissalpine 4 x SRX150348
SRX150324
SRX150314
SRX150302
TurkishAwassi 2 x SRX150333
SRX150312
Awassi 1 x SRX150330
Swissmirror 1 x SRX150304
Sakiz 2 x SRX150318
SRX150283
TOTAL 20 10 10
Una vez descargadas las secuencias se ha realizado un control de calidad de las mismas en cuanto a
dos parámetros: calidad de las bases asignadas y longitud de las secuencias. Para ello se ha utilizado el
software FastQC (http://www.bioinformatics.babraham.ac.uk/projects/download.html#fastqc) y los scripts
descritos en el ANEXO-1-A. Posteriormente, y mediante el programa Trimmomatic-v0.22 (19;
http://www.usadellab.org/cms/?page=trimmomatic), se han eliminado por un lado, los adaptadores propios
de la tecnología Illumina que se usan en el proceso de secuenciación (ANEXO 1-B-C) y, por otro, aquellas
secuencias que no alcanzan los mínimos establecidos respecto a la calidad ,siendo 20 la calidad mínima de
las bases y 31pb la longitud mínima de las secuencias (ANEXO-1C). Finalmente, se comprueba de nuevo la
13

calidad de las secuencias con el FastQC (Figura 3).
Figura 3: Imagen obtenida del análisis FastQC a lo largo del genoma de los individuos de razas lecheras y cárnicas tras utilizar el
Trimmomatic.
Dada la naturaleza del cromosoma X y del ADN mitocondrial es conveniente analizar su variabilidad
genética de forma separada, por lo que, en un siguiente paso se han eliminado las secuencias relativas al
cromosoma X y al ADN mitocondrial. Para ello, se ha realizado un mapeo de nuestras secuencias contra el
genoma de referencia de Ovis aries, OAR_v3.1 mediante el programa Bowtie2 (20;
http://sourceforge.net/projects/bowtie-bio/files/bowtie2/2.2.1/) diseñado para el mapeo de lecturas cortas
contra largas secuencias de referencia (ANEXO 1-D). Tras esta eliminación de esta fracción del genoma se
han creado los pooles, uniendo en un mismo set de datos, por un lado, las secuencias de los 10 individuos
correspondientes a razas de producción cárnica y por el otro las de los 10 individuos de razas de producción
lechera. Estos pooles se han llevado a una cobertura de 10X (ANEXO-1E y ANEXO-2), teniendo en cuenta
el número de individuos del que disponíamos y lo aconsejado en la literatura [11].
Por último, se ha realizado el mapeo de ambos pooles usando nuevamente el Bowtie2 (ANEXO-1F).
Este mapeo permite detectar aquellas bases nucleotídicas que difieren entre el genoma de referencia y el
genoma de los individuos analizados, permitiendo identificar así los polimorfismos tipo SNP (Single
Nucleotide Polymorphism) que se usarán para el análisis posterior. Los archivos de salida de este mapeo son
finalmente modificados en cuanto a su formato mediante los software Picardtools
(http://sourceforge.net/projects/picard/files/) y SAMtools (http://sourceforge.net/projects/samtools/files/) [21], para su posterior uso en el software Popoolation (ANEXO-1G).Lo que se consigue con el uso de estos
14

programas es; por un lado eliminar los duplicados y las secuencias no alineadas tras el mapeo, y por el otro
modificar la extensión de salida SAM a la usada por el programa a validar que ese la extensión MPILEUP.
DETECCIÓN DE SEÑALES DE SELECCIÓN EN EL GENOMA
PoPoolationv 1.2.2 es un programa para el análisis de datos obtenidos mediante experimentos
PoolSeq (http://popoolation.googlecode.com/files/teaching-data.zip). Este programa permite calcular
diferentes estadísticos para la detección de señales selección a lo largo de un genoma utilizando un enfoque
de ventana deslizante: la D de Tajima (adaptada para el análisis de datos provenientes de pooles ya que el
estadístico original estima las diferencias inter-individuales) para un análisis intra-poblacional y el F st para un
análisis inter-poblacional [17,18].
Para el análisis con este programa son varios los parámetros que se deben definir. En primer
lugar, se debe ajustar el “mínimum count”, que es el mínimo número de veces que debe aparecer una
variante alélica para considerarlo como SNP. En el caso de este estudio se ha establecido un “mínimum
count” de 3 en base a estudios previos [14]. El tamaño de las ventanas a analizar es otro de los parámetros
variables considerados en el cálculo de los estadísticos. Para determinar el tamaño de ventana apropiado en
este estudio se ha realizado una comparativa de la distribución del número de SNP/ventana a lo largo del
genoma utilizando los tamaños de 50Kb, 100Kb, 150Kb y 200Kb. Se ha establecido que el tamaño de
ventana más apropiado será aquel que resulte en un menor porcentaje de ventanas con un número de SNPs
inferior a diez, evitando así estimas poco precisas de los parámetros a analizar en la búsqueda de señales de
selección [13,14].
En los ANEXOS 4A-H se muestran los script finales utilizados para el cálculo de la D de Tajima y el
Fst tanto para el pool de razas lecheras como el de cárnicas.
IDENTIFICACIÓN DE REGIONES CCR (Convergent Candidate Regions)
Una vez obtenidos los resultados para los dos parámetros, D de Tajima y Fst a lo largo del genoma, se
han considerado como regiones significativas o “outliers” aquellas en las que el valor obtenido se posiciona
en el extremo 2% del total de los valores para cada parámetro. Por otra parte, debido a las dificultades para
distinguir entre los efectos causados en el genoma por un evento selectivo en lugar de por eventos
demográficos tales como la expansión o contracción de la población [8], se definieron como regiones
candidatas a sufrir selección o CCR (Convergent Candidate Regions) aquellas para las cuales se han
obtenido valores significativos tanto para la D de Tajima como para el F st. De esta forma, sólo consideramos
15

para un estudio más exhaustivo las regiones que se encuentran dentro de ese 2% y que son convergentes en
ambos métodos siendo pues las regiones CCR de mayor fiabilidad.
VALIDACIÓN DE REGIONES CCR
Con el fin de dar validez a los resultados obtenidos, y por consiguiente, al software Popoolation
utilizado en el presente estudio, se han contrastado las regiones CCR identificadas con aquellas regiones
donde previamente se haya descrito un QTL (Quantitative Trait Loci) relacionado bien con la producción
cárnica, bien con la producción lechera, en la especie. Para ello recurrimos a la base de datos SheepQTL:
http://www.animalgenome.org/cgi-bin/QTLdb/OA/index. Además, también se han contrastado los resultados
con la bibliografía existente tratando de identificar los genes candidatos que se hayan descrito relacionados
con caracteres de producción.
16

RESULTADOS
En las Tablas 2 y 3 y en las Figuras 4 y 5 se muestran el número de SNPs por ventana en base a los
diferentes tamaños testados. Se ha determinado que el tamaño de ventana más adecuado para nuestros
propósitos es el de 200Kb.
Tabla 2: Resultados obtenidos respecto al número de SNPs por ventana y ventanas a
eliminar (< 10SNPs) considerando los tamaños de 50Kb, 100Kb, 150Kb y 200Kb en el
pool de razas lecheras.
Tamaño de ventana
Ventanas con presencia de
SNPs
Ventanas con más de
10 SNPs
Ventanas Eliminadas
50Kb 49054 48848 206
100Kb 24534 24517 17
150Kb 16362 16356 6
200Kb 12276 12272 4
Tabla 3: Resultados obtenidos respecto al número de SNPs por ventana y ventanas a
eliminar (< 10SNPs) considerando los tamaños de 50Kb, 100Kb, 150Kb y 200Kb en el
pool de razas cárnicas.
Tamaño de ventana
Ventanas con presencia de
SNPs
Ventanas con más de 10
SNPs
Ventanas Eli-minadas
50Kb 49054 48886 168
100Kb 24534 24522 12
150Kb 16362 16355 7
200Kb 12276 12272 4
17

Figura 4: Distribución gráfica del número de SNPs por ventana para los tamaños de ventana testados de 50Kb, 150Kb, 100Kb y
200Kb en el pool de razas lecheras. En la fila inferior se representa la región de 0 a 150 SNPs/ventana ampliada
Figura 5: Distribución gráfica del número de SNPs por ventana para los tamaños de ventana testados de 50Kb, 150Kb, 100Kb y
200Kb en el pool de razas cárnicas. En la fila inferior se representa la región de 0 a 150 SNPs/ventana ampliada
18

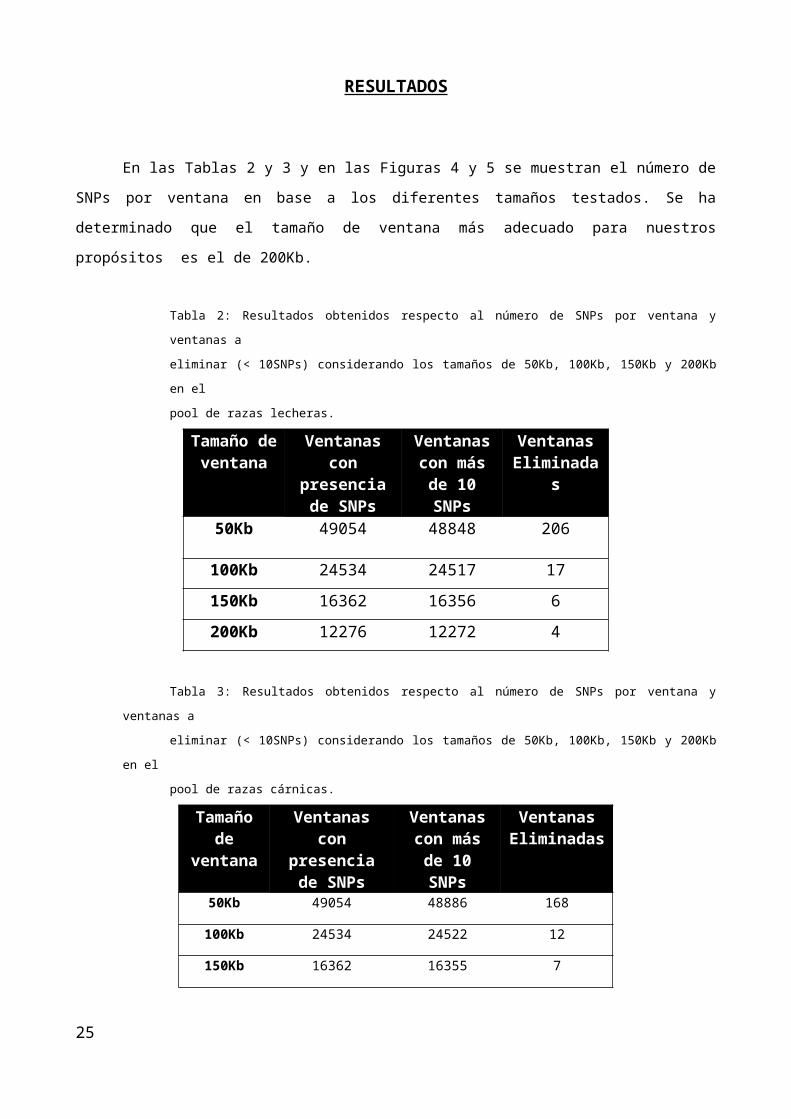
En la Figura 6 se muestran los valores de la D de Tajima obtenidos para cada una de las ventanas
analizadas a lo largo del genoma ovino del pool de razas lecheras (a) y razas cárnicas (b)
a) Pool de razas lecheras:
b) Pool de razas cárnicas:
Figura. 6: Valores de la D de Tajima a lo largo del genoma del a) pool de razas lecheras y b) pool de razas cárnicas. Los cambios en el
color representan cambios de un cromosoma a otro.
19

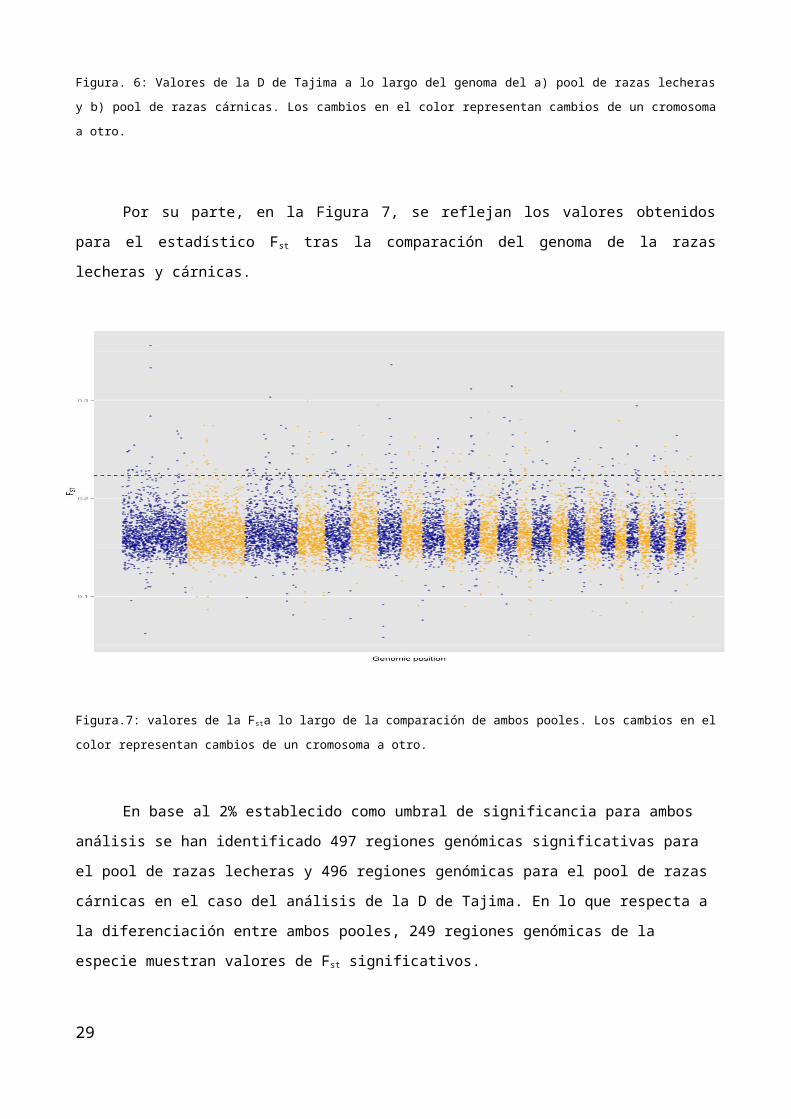
Por su parte, en la Figura 7, se reflejan los valores obtenidos para el estadístico Fst tras la
comparación del genoma de la razas lecheras y cárnicas.
Figura.7: valores de la Fsta lo largo de la comparación de ambos pooles. Los cambios en el color representan cambios de un
cromosoma a otro.
En base al 2% establecido como umbral de significancia para ambos análisis se han identificado 497
regiones genómicas significativas para el pool de razas lecheras y 496 regiones genómicas para el pool de ra-
zas cárnicas en el caso del análisis de la D de Tajima. En lo que respecta a la diferenciación entre ambos
pooles, 249 regiones genómicas de la especie muestran valores de Fst significativos.
IDENTIFICACIÓN DE LAS REGIONES CCR
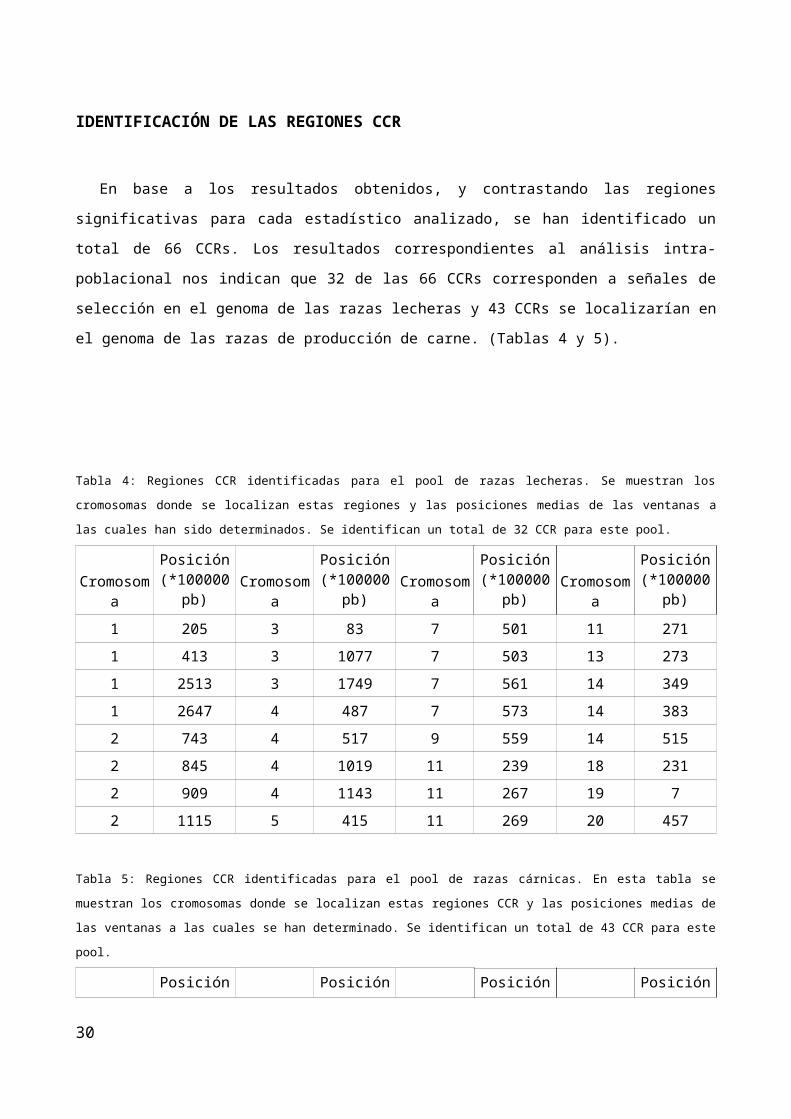
En base a los resultados obtenidos, y contrastando las regiones significativas para cada estadístico
analizado, se han identificado un total de 66 CCRs. Los resultados correspondientes al análisis intra-
poblacional nos indican que 32 de las 66 CCRs corresponden a señales de selección en el genoma de las
razas lecheras y 43 CCRs se localizarían en el genoma de las razas de producción de carne. (Tablas 4 y 5).
20

Tabla 4: Regiones CCR identificadas para el pool de razas lecheras. Se muestran los cromosomas donde se localizan estas regiones y
las posiciones medias de las ventanas a las cuales han sido determinados. Se identifican un total de 32 CCR para este pool.
Cromosoma
Posición (*100000pb
)Cromosom
a
Posición (*100000pb
)Cromosom
a
Posición (*100000pb
)Cromosom
a
Posición (*100000pb
)
1 205 3 83 7 501 11 271
1 413 3 1077 7 503 13 273
1 2513 3 1749 7 561 14 349
1 2647 4 487 7 573 14 383
2 743 4 517 9 559 14 515
2 845 4 1019 11 239 18 231
2 909 4 1143 11 267 19 7
2 1115 5 415 11 269 20 457
Tabla 5: Regiones CCR identificadas para el pool de razas cárnicas. En esta tabla se muestran los cromosomas donde se localizan
estas regiones CCR y las posiciones medias de las ventanas a las cuales se han determinado. Se identifican un total de 43 CCR para
este pool.
Cromosoma
Posición (*100000pb
)Cromosom
a
Posición (*100000pb
)Cromosom
a
Posición (*100000pb
)Cromosom
a
Posición (*100000pb
)
1 205 4 471 9 615 14 345
1 1957 4 479 9 639 14 515
1 2343 4 653 9 665 15 657
1 2513 5 227 11 185 17 203
1 2647 5 435 11 247 17 297
2 1115 5 1073 11 271 17 447
2 2173 6 361 11 383 20 183
3 1007 7 501 12 395 25 75
3 1077 8 81 12 393 25 73
3 1543 8 317 13 273 26 97
4 427 8 319 13 505
En la Tabla 6 se muestra la correspondencia encontrada entre las regiones CCR identificadas en el
presente estudio y los QTLs hasta el momento descritos según la base de datos SheepQTL, así como los ge-
nes asociados con caracteres de producción en base a la bibliografía existente.
21

Tabla 6. Correspondencia entre las regiones CCR identificadas y los QTL y genes asociados descritos previamente. Se muestran los cromosomas en los cuales se identifican estas regiones CCR y las ventanas medias de la posición en la cual se han determinado.
CROMOSOMA POSICIÓN CCR (*100.000 pb) LECHE/CARNE QTL GEN CANDIDATO
1
205,1957,2343,2513, 2647 Carne Producción cárnica [26] -
2513, 2647 Leche Contenido de PUFA* en la leche PCCB [22]
2
1115, 2173 Carne Crecimiento muscular GDF8 [27]
743, 845, 909 Leche CLA** en leche LPL [22]
3
1007, 1077, 1543 Carne CLA en la carne [28] -
1077 Leche CLA en la leche IGF1 [22]
5 227, 435, 1073 Carne MUFA*** en carne [28] -
7 501, 503, 561, 573 Leche Lactancia [23] -
8 81, 317, 319 Carne Grasa corporal [26] -
9 615, 639, 665 Carne Peso y composición corporal [26] -
11
383 Carne Composición de la carne GIP [29]
239, 267, 269, 271 Leche Lactancia [24] -
13 505 Carne Peso muscular [26] -
14
345, 515 Carne Grasa corporal [26] -
349, 383 Leche Contenido de PUFA y CLA en leche LCAT [22]
18 231 Leche Producción de leche [25] -
20 457 Leche Lactancia [23] -
*PUFA: ácidos grasos poliinsaturados,
**CLA: ácido linoleico conjugado,
22

***MUFA: ácidos grasos monoinsaturados.
23

DISCUSIÓN
El principal objetivo de este estudio es la validación del programa Popoolation diseñado para la
detección de señales de selección en el genoma a partir de datos obtenidos mediante la estrategia PoolSeq. El
estudio se engloba dentro de un proyecto que se está llevando a cabo en el grupo de investigación liderado
por la Dra. Andone Estonba enfocado a la detección de señales de selección en el genoma de las dos razas
ovinas locales Sasi-ardi y Latxa mediante esta misma aproximación. En el presente estudio se ha realizado
una simulación del experimento que se está llevando a cabo en el laboratorio utilizando secuencias
disponibles de razas ovinas de producción de leche y de carne creando un pool de secuencias para cada una
de ellas.
El programa Popoolation nos ha permitido la identificación de señales de selección en los genomas de
estas razas ovinas mediante dos aproximaciones. Por un lado, con la estima de la D de Tajima se ha realizado
un análisis intra-poblacional en cada uno de los pooles creados identificando aquellas regiones que destacan
sobre el resto a lo largo de un mismo genoma. Por otro, el estadístico F st utilizado para un análisis inter-
poblacional nos ha proporcionado información relativa a la diferenciación entre ambos pooles identificando
aquellas regiones susceptibles a sufrir selección en unas razas a diferencia de las otras. El realizar el análisis
mediante la estimación de dos parámetros nos ayuda a distinguir entre los efectos causados en el genoma por
eventos selectivos en lugar de por eventos demográficos.
La consideración simultánea de ambos parámetros analizados nos ha llevado a la identificación de 66
CCRs, 43 de los cuales serían reflejo de la selección artificial a la que se han sometido las razas cárnicas y 32
de la selección a favor a la producción de leche. Más de la mitad de las regiones CCRs identificadas en este
trabajo concuerdan de algún modo con regiones QTL previamente descritas para diversas características de
producción lechera y cárnica. Incluso en algunos casos se ha llegado a la identificación en esas regiones de
los genes candidatos a ser responsables del efecto en dichas características. A continuación, se discuten las
regiones CCR identificadas para cada uno de los conjuntos de razas estudiadas.
1- CCRs DE PRODUCCIÓN DE LECHE
- OAR1: Los dos parámetros estimados en este estudio nos han llevado a la identificación de
4regiones CCR en este cromosoma. Según la base de datos SheepQTL dos de ellas se localizan
dentro de un QTL previamente descrito para características de producción lechera. Además, en esta
región QTL se localiza el gen PCCB (propionyl CoA carboxylase), el cual parece tener un efecto
positivo en las propiedades nutracéuticas de la leche al contribuir al aumento de todos los ácidos
24
grasos poli-insaturados, y a la disminución del contenido de ácido palmítico [22].
- OAR2: Las 3 CCRs detectadas en este cromosoma se encuentran dentro de un QTL descrito con
anterioridad en relación a la producción de leche. Según lo descrito en la literatura, en esta región se
localiza el gen LPL (lipoprotein lipase). Este gen, desempeña un papel fundamental durante la
lactancia permitiendo la absorción y secreción de ácidos grasos de cadena larga en la leche, y
además provoca un efecto positivo en el contenido de CLA (conjugated linoleic acid) en la misma
[22].
- OAR3: Una de las tres regiones CCR identificadas en este cromosoma se localiza también dentro de
un QTL en el que se encuentra el gen IGF1 (insulin-likegrowth factor 1). Este gen tiene un papel
importante en la disminución del contenido de CLA vinculado a la fertilidad ovina y la lactancia
temprana [22].
- OAR7: Las cuatro regiones CCR que hemos descrito para este cromosoma, relativamente próximas
entre ellas, quedan englobadas dentro de una región QTL previamente determinada como influyente
en el porcentaje de proteínas de la leche, rasgo asociado a la lactancia [23].
- OAR11: Basándonos en los QTLs relacionados con la producción de leche previamente descritos en
otros estudios podemos determinar que estas 4 regiones CCR están localizadas dentro de una región
asociada a la persistencia a la lactancia y a la lactancia extendida [24]
- OAR14: De las 3 CCRs identificadas en este cromosoma, dos de ellas se localizan dentro de una
región QTL previamente descrita para características de producción lechera. Ésta región incluye
además al gen LCAT (lecithin-cholesterol acyltransferase), que influye en los niveles de CLA y
PUFA en la leche [22].
- OAR18: La única región CCR identificada en este cromosoma se localiza dentro de un QTL
relacionado con la producción de leche, en concreto con el contenido de ácidos grasos poli-
insaturados en la leche [25].
- OAR20: al igual que en el caso anterior, solo se ha identificado una región CCR en este cromosoma.
Esta región se encuentra dentro de un QTL asociado a la lactancia y al porcentaje de lactosa en la
leche [23].
25

2- CCRs DE PRODUCCIÓN DE CARNE
- OAR1: Las 5 regiones CCR identificadas en este cromosoma se engloban dentro de un QTL
previamente descrito para la producción cárnica que parece influir en el porcentaje de grasa de la
carcasa de los animales [26].
- OAR2: En este cromosoma se han identificado 2 regiones CCR. Una de ellas (posición 217.300Kb),
está localizada dentro de una región QTL asociada con el contenido de ácidos grasos poli-
insaturados de la carne. La segunda CCR (posición 111.500Kb),se encuentra muy próxima al gen
GDF8 (myostatin), que tiene un rol principal en el crecimiento muscular. Esta región se encuentra
dentro de un QTL descrito para la producción cárnica [27].
- OAR3: Las 3 regiones CCR identificadas en este cromosoma forman parte de un QTL descrito con
anterioridad en relación al contenido de CLA en el músculo. El contenido de estos ácidos en el
músculo es importante para la nutrición humana porque se ha relacionado con una multitud de
efectos metabólicos, incluyendo la inhibición a la carcinogénesis o la reducción de la deposición de
grasa [28].
- OAR5: Las 3 regiones CCR identificadas en el cromosoma 5 concuerdan con un QTL relacionado
con el contenido de MUFA en la carne [28].
- OAR8: Según Clop et al. [26], una de las 3 regiones CCR identificadas, (la localizada en la posición
8.100Kb) se sitúa dentro de un QTL asociado a la composición de la carcasa y las otras dos, son
parte de otro QTL descrito para el contenido de grasa corporal.
- OAR9: Las 3 regiones CCR que hemos identificado están localizadas dentro de un QTL previamente
descrito para la composición y peso corporal en ovinos [26].
- OAR11: La región CCR identificada en la posición 38.300 Kb de este cromosoma se encuentra
dentro de un QTL, donde, a su vez, se localiza el gen GIP (Gastric inhibitor ypolypeptide) [28]. Este
gen tiene un rol principal en la acumulación de grasa en el tejido adiposo y un estudio reciente de
asociación ha mostrado una relación entre GIP y la composición de los ácidos grasos en los cerdos
[29].
- OAR13: Una de las dos regiones CCR de este cromosoma forma parte de un QTL previamente
relacionado con el peso muscular [26].
26

- OAR14: Está descrito un QTL en relación al contenido de grasa corporal en la especie ovina,
característica relacionada con la calidad y la producción de la carne, que engloba las dos regiones
CCR de este cromosoma [26].
Cabe mencionar la identificación de las mismas regiones CCR tanto para el pool de razas lecheras como
para el de cárnicas como puede ser por ejemplo la región CCR a 251.300Kb en el cromosoma 1 de ambos
pooles. Estas coincidencias se pueden deber principalmente al hecho de que el análisis de ambos parámetros
se ha realizado con tamaños de ventana muy grandes (200.000Kb) por lo que no es de extrañar que en
regiones tan amplias puedan existir zonas relativas tanto a la producción cárnica como a la lechera.
Además de las CCRs descritas hasta el momento, en el presente trabajo se han identificado también otras
30 regiones CCRs (14 en el pool de razas lecheras y 20 en el pool de razas cárnicas), para las que no se han
encontrado concordancia alguna ni con los QTLs recogidos en las bases de datos ni con lo descrito en la
literatura a día de hoy. A priori, podríamos considerar estas 30 regiones CCR como nuevas regiones
asociadas a la producción cárnica o lechera en la especie ovina. Sin embargo, debemos tener presente la
posibilidad de que estas regiones sean reflejo de falsos positivos en nuestro estudio. Por lo tanto, se hace
necesario un análisis más exhaustivo de estas regiones para llegar a identificar los genes candidatos que se
localizan en las mismas y estudiar la posible relación entre estos y los múltiples caracteres que pudieran estar
afectando a la producción tanto de leche como de carne.
27

CONCLUSIÓN
-Este estudio presenta el primer mapeo de señales de selección en el genoma ovino utilizando la
aproximación PoolSeq, y demuestra la eficacia del programa Popoolation para detectar señales de selección
o De las 66 regiones CCR identificadas mediante el uso del programa Popoolation para la
producción lechera y cárnica, 36 corresponden a regiones QTL previamente descritas en la
especie en relación a dichos caracteres. Además, para algunos de estos caracteres están
descritos ya los genes candidatos en la región y responsables del efecto en los mismos.
o En lo que respecta a las 30 nuevas regiones CCR identificadas en este trabajo, es necesario
un análisis en profundidad de las mismas en busca de los posibles genes candidatos que
pudieran estar asociados a caracteres de producción tanto lechera como cárnica y obtener así
información de utilidad para programas de selección y mejora de la especie.
-La validación del programa Popoolation hace que éste sea una herramienta útil para los propósitos
en el estudio que se está llevando a cabo en torno a las razas ovinas locales Sasi-ardi y Latxa.
28
AGRADECIMIENTOS
La autora de este trabajo agradece a la Dra.Otsanda Ruiz-Larrañaga y la Dra. Andone Estonba,
tutoras de este Trabajo de Fin de Máster, la orientación y el apoyo para la realización del presente estudio.
Debe además un agradecimiento singular a Jorge Langa por toda la ayuda prestada a la hora de
realizar los análisis bioinformáticos.
El presente estudio se engloba dentro de un proyecto financiado por la Universidad del País
Vasco/EuskalHerrikoUnibertsitatea (UPV/EHU) dentro de la convocatoria Universidad-Sociedad 2013
(US13/29) y por el Conservatoire des Races d'Aquitaine (CRA, Francia). El proyecto cuenta también con la
colaboración del Instituto Navarro de Tecnologías e Infraestructuras Agroalimentarias (INTIA).
29
BIBLIOGRAFÍA
1-Rege JEO, and Gibson. JP. (2003). Animal genetic resources and economic development: issues in relation
to economic valuation. Ecological Economics, 45(3):319-330.
2-Fay JC, and Wu CI. (2000). Hitchhicking under positive Darwinian selection. Genetics, 155(3):1405-13.
3-Andersson L. (2011). How selective sweeps in domestic animals provide new insight into biological mech-
anisms. Journal of internal medicine, 271(1):1-14.
3-Vitti JJ, Grossman SR, and Sabeti PC. (2013). Detecting Natural Selection in Genomic Data. Annual Re-
view of Genetics, 49:97-120.
5-Hedrick PW. (1980).Hitchhiking: a comparison of linkage and partial selfing. Genetics, (3):791-808.
6- Tajima F. (1989). Statistical method for testing the neutral mutation hypothesis by DNA polymorphism.
Genetics, 123(2):858-95
7-Holsinger KE, and Weir BS. (2009). Genetics in geographically structured populations: defining, estimat-
ing and interpreting F (ST). Nature Reviews Genetics, 10(9):639–50.
8- Gutiérrez-Gil B, Arranz JJ, Pong-Wong R, García-Gámez E, Kijas J, et al. (2014). Application of Selec-
tion Mapping to Identify Genomic Regions Associated with Dairy Production in Sheep. Plos ONE, 9(5):
e94623.
9-Chessa B, Pereira F, Arnaud F, Amorim A, Goyache F, Mainland I, Kao RR, Pemberton JM, Beraldi D,Stear MJ,Alberti A, Pittau M,Iannuzzi L,Banabazi MH, Kazwala RR,Zhang YP,Arranz JJ,Ali BA,Wang Z,Uzun M,Dione MM,Olsaker I,Holm LE,Saarma U,Ahmad S,Marzanov N,Eythorsdottir E,Holland MJ,Ajmone-Marsan P,Bruford MW,Kantanen J,Spencer TE, and Palmarini M. (2009). Revealing the history of sheep domestication using retrovirus integrations. Science, 324(5926):532-6.
10- Food and Agricultural Organization of United Nations, 2007. http://www.fao.org/dad-is
11-Ferreti.L, Ramos-Onsins SE, and Pérez-Enciso.M. (2013). Population genomics from pool sequencing.
Molecular Ecology, 22:5564-5576.
12- Rubin CJ, Zody MC, Eriksson J, Meadows JR, Sherwood E, Webster MT, Jiang L, Ingman M, Sharpe T,
Ka S, Hallböök F, Besnier F, Carlborg O, Bed'homB,Tixier-Boichard M, Jensen P, Siegel P, Lindblad-Toh K,
and Andersson L. (2010). Whole-genome resequencing reveals loci under selection during chicken domesti-
cation. Nature, 464(7288):587-91.
30

13- Rubin CJ, Megens HJ, Martinez Barrio A, Maqbool K, Sayyab S, Schwochow D, Wang C, Carlborg Ö,
Jern P, Jørgensen CB, Archibald AL, FredholmM, Groenen MA, and Andersson L. (2012).Strong signatures
of Selection in the domestic pig genome. Proceedings of the Natural Academy of Sciences, 109(48):19529-
36.
14- Axelsson E, Ratnakumar A, Arendt ML, Maqbool K, Webster MT, Perloski M, Liberg O, Arnemo JM,
HedhammarA, and Lindblad-Toh K. (2013). The Genomic Signatures of dog domestication reveals adapta-
tion to a starch-rich diet. Nature, 495(7441):360-4.
15- Pavlidis P, Živkovic D, Stamatakis A, and Alachiotis N. (2013). SweeD: likelihood-based detection of selective sweeps in thousands of genomes. Molecular Biology and Evolution, 30(9):2224-34.
16- Boitard S, Kofler R, FranÇoise P, Robelin D, Schlottere C, andFutschik A. (2013). Pool-hmm: a Phyton
program for estimating the allele frequency spectrum and detecting selective sweeps from next generation
sequencing of pooled samples. Molecular Ecology Resources, 13(2):337-340.
17- Kofler R, Orozco TR, Wengel P, De Maio N, Pandey RV, Nolte V, et al. (2011). PoPoolation: A Toolbox
for Population Genetic Analysis of Next Generation Sequencing Data from Pooled Individuals. PLoS ONE,
6(1):e15925.
18- Kofler R, VinayPandey, R, and Schloetterer, C. (2011). PoPoolation2: Identifying differentiation between
populations using sequencing of pooled DNA samples (Pool-Seq). Bioinformatics, 27(24):3435–3436.
19-Lohse M, Bolger AM, Nagel A, Fernie AR, Lunn JE, Stitt M, and Usadel B.(2012). RobiNA: a user-
friendly, integrated software solution for RNA-Seq-based transcriptomics. Nucleic Acids Research, 40:622-
7.
20-Trapnell C., Pop M., and Salzberg S.L. (2009). Ultrafast and memory-efficient alignment of short DNA
sequences to the human genome. Genome Biology, 10:R25.
21- Li H., Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R and 1000
Genome Project Data Processing Subgroup. (2009). The Sequence alignment/map (SAM) format and SAM-
tools. Bioinformatics, 25:2078-9.
22- Crisà A, Marchitelli C, Pariset L, Contarini G, Signorelli F, Napolitano F, Catillo G, Valentini A, and
Moioli B. (2010). Exploring polymorphisms and effects of candidate genes on milk fat quality in dairy
sheep. Journal of Dairy Science. 93(8):3834-45.
31

23- Raadsma HW, Jonas E, McGill D, Hobbs M, Lam MK, and Thomson PC. (2009). Mapping quantitative
trait loci (QTL) in sheep. II. Meta-assembly and identification of novel QTL for milk production traits in
sheep. Genetic Selection Evolution. 22; 41:45.
24- Jonas E, Thomson PC, Hall EJ, McGill D, Lam MK, and Raadsma HW. (2011). Mapping quantitative
trait loci (QTL) in sheep. IV. Analysis of lactation persistency and extended lactation traits insheep. Genetic
Selection Evolution. 21; 43:22.
25- Mateescu RG andThonney ML. (2010).Genetic mapping of quantitative trait loci for milk production in
sheep. Animal Genetics, 41(5):460-6.
26-Cavanagh CR, Jonas E, Hobbs M, Thomson PC, Tammen I, and Raadsma HW.(2010). Mapping Quantita-
tive Trait Loci (QTL) in sheep. III. QTL for carcass composition traits derived from CT scans and aligned
with a meta-assembly for sheep and cattle carcass QTL. Genetic Selection Evolution, 16:422-36
27- Clop A, Marcq F, Takeda H, Pirottin D, Tordoir X, Bibé B, Bouix J, Caiment F, Elsen JM, Eychenne F,
Larzul C, Laville E, Meish F, Milenkovic D, Tobin J, Charlier C, and Georges M.(2006). A mutation creating
a potential illegitimate microRNA target site in the myostatin gene affects muscularity in sheep. Natural Ge-
netics, 38(7):813-8.
28- García-Fernández M, Gutiérrez-Gil B, García-Gámez E, Sánchez JP, and Arranz JJ. (2010). The identifi-
cation of QTL that affect the fatty acid composition of milk on sheep chromosome 11.Animal Genetics,
41(3):324-8.
29-Muñoz G,Alves E, Fernández A, Ovilo C, Barragán C, Estellé J, Quintanilla R, Folch JM, Silió L, Ro-dríguez MC, and Fernández AI. (2007). QTL detection on porcine chromosome 12 for fatty-acid composition and association analyses of the fatty acid synthase, gastric inhibitory polypeptide and acetyl-coenzyme A carboxylase alpha genes. Animal Genetics, 38(6):639-46.
32

ANEXO 1- SCRIPTS GENERALES
A) Script para realizar el análisis de calidad de las secuencias originales:
ssh –X medusa
fastqc –t 24 –nogroup *.fastq.gz
B) Script para comprobar que adaptadoresse han usado en la secuenciación:
foriin *.fastq.gz
do
parallel --tag –k zgrep–c {} $i :::(cat/home/SHARE/oari/adapters/) > ${i%.fq.gz}.results
done
C) Script para usar el Trimmomatic con todas las variables que nos son de utilidad:
foriinAWAS CAS1 CAS2 CHU1 CHU2…..
do
java –jar /opt/bioinfo/trimmomatic/trimmomatic-0.30.jar PE\
-threads 24\
-phred33\
-trimlog ${i}_log
../originseq/${i}_1.fastq.gz
../originseq/${i}_2.fastq.gz
${i}_1.fastq.gz\
${i}_3.fastq.gz\
${i}_2.fastq.gz\
${i}_4.fastq.gz\
AVGQUAL:3\
ILLUMINACLIP:/opt/bioinfo/trimmomatic/adapters/TruSeq3-SE.fa:2:30:10\
MINLEN: 31\
33

LEADING: 19\
TRAILING: 19\
MINLEN :31 2> ${I}.results
done
D) Scripts para eliminar el DNA mitcondrial:
1- Crear un índice:
bowtie2-build …/…/refseq/dna/Ovis_aries.Oar_v3.1.74.dna.chromosome.MT.fa oariMT
2- Lanzar el programa:
fori inAWAS
do
bowtie2\
-xoariMT\
-p 24\
-1../../trimmed/ ${i}_1.fastq.gz
-2../../trimmed/ ${i}_2.fastq.gz
--un-gz ${i}_U.nomito.fastq.gz
--un -conc –gz ${i}_%.nomito.fastq.gz
--al-gz ${i}_U.mito.fastq.gz
--al -conc –gz ${i}_%.mito.fastq.gz
-S/dev/null
E) Script de recorte de secuencias para generar archivos de cobertura 10x:
foriin*.fastq.gz
do
34

pigz –dc ${i} | head – 51198872 | pigz –best >../recortadas/ ${i}
done
F) Script para el mapeo con las secuencias a 10x y recortadas:
1) Generar el índice:
bowtie2-build…/../refseq/dna/dnabruto.fa oariDNA
2) Lanzar el Bowtie:
Bowtie2 \
--quiet \
--no-unal \
-p 24 \
-x oariDNA \
-1../../recortadas/archivo_1.fatq.gz
-2../../recortadas/archivo_2.fatq.gz
-S archivo.sam \
G) Script para modificar la extensión de SAM a BAM:
samtools -@ 24 -Sbh archivo.sam-oarchivo.bam
35

ANEXO 2
Para determinar la longitud a recortar de cada genoma de los individuos que constituyen el pool se usa la
ecuación de la cobertura C= (N*L)/G, dónde C es igual a uno en este caso, G es la longitud del genoma de
referencia (2534344180 pb) y L (longitud de las lecturas). La L se puede calcular haciendo la media
ponderada de la información de la longitud de las secuencias que se puede obtener de los análisis de calidad
realizados:
Con estos datos calculamos la media ponderada que sería:
Y sabiendo L se puede calcular el número de lecturas que es necesario recortar de los genomas:
36

ANEXO 3- SCRIPTS PARA EL POPOOLATION
A) Script para ordenar los archivos BAM:
Picard-tools SortSam I=archivo.bam O=archivo.sort.bamVALIDATION_STRINGENCY=SILENT
SO=coordinate
B) Script para eliminar los duplicados de los pool:
picard-tools MarkDuplicates I=archivo.sort.bam O=archivo.rmd.sort.bam
M=dupstat.txtVALIDATION_STRINGENCY=SILENT REMOVE_DUPLICATES=true
C) Script para eliminar las alienaciones de baja calidad y no alineadas con el genoma de referencia en PE:
Samtoolsview -q 20 -f 0x0002 -F 0x0004 -F 0x0008 -b archivo.rmd.sort.bam>archivo.q20.rmd.sort.bam
D) Convertir los archivos BAM en mpileup:
Samtoolsmpileup -B -Q 0 -f secuenciadereferencia.faarchivo.q20.rmd.sort.bam >archivo.mpileup
E)Eliminar los indels de las secuencias:
perl popoolation/basic-pipeline/identify-genomic-indel-regions.pl --indel-window 5 --min-count 2 --
inputarchivo.mpileup --output indels.gtf
perl popoolation/basic-pipeline/filter-pileup-by-gtf.pl --inputarchivo.mpileup--gtfindels.gtf--
outputarchivo.idf.mpileup
37

F) Script para realizar el subsampling para el popoolation:
perl popoolation/basic-pipeline/subsample-pileup.pl –min-qual 20 –method withoutreplace –max-
coverage 50 –fastq-type sanger –target-coverage 20 –inputarchivo.idf.mpileup –
outputarchivo.ss10.idf.mpileup
G)Script para lanzar el análisis inter-poblacional:
perl popoolation/Variance-sliding.pl –fastq-type sanger –measure D –input archivo.idf.mpileup –min-
count 2 –min-coverage 4 –max-coverage 11 –min-covered-fraction 0.5 –pool-size 20 –window-size
10000 –step-size 10000 –outputarchivo.D–snp-output archivo.snps
H) Eliminar el cromosoma X:
grep -v -e ^X archivo.D>arcvhivosinX.D
Análisis intra-poblaciona:
I) Generar un mpileup que contenga tanto las secuencias cárnicas como las lecheras:
samtoolsmpileup -B -Q 0 -f referenciaarchivo1.bamarchivo2.bam >todasjuntas.mpileup
J) Script para generar un archivo sincronizado:
perl popoolation2/mpileup2sync.pl –inputtodasjuntas.mpileup -outputtodasjuntas.sync –fastq-
typesanger –min-qual 20
38

K) Lanzar el programa:
perl popoolation2/fst-sliding –window-size 10000 –step-size 10000 –suppress-noninformative –
inputtodasjuntas.sync -min-covered-fraction 1.0 –min-coverage 4 –max-coverage 10 –min-count 3 –
outputtodasjuntas.txt–pool-size 20
39